ES2860298T3 - Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso - Google Patents
Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso Download PDFInfo
- Publication number
- ES2860298T3 ES2860298T3 ES18182695T ES18182695T ES2860298T3 ES 2860298 T3 ES2860298 T3 ES 2860298T3 ES 18182695 T ES18182695 T ES 18182695T ES 18182695 T ES18182695 T ES 18182695T ES 2860298 T3 ES2860298 T3 ES 2860298T3
- Authority
- ES
- Spain
- Prior art keywords
- combination
- epilepsy
- compound
- levetiracetam
- comp
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- IPCYZQQFECEHLI-UHFFFAOYSA-N Cc1cc(-c2nc(CN3CCNCC3)n[o]2)cc(CN2Cc(cc3)ccc3OC(F)(F)F)c1C2=O Chemical compound Cc1cc(-c2nc(CN3CCNCC3)n[o]2)cc(CN2Cc(cc3)ccc3OC(F)(F)F)c1C2=O IPCYZQQFECEHLI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/4015—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil having oxo groups directly attached to the heterocyclic ring, e.g. piracetam, ethosuximide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/177—Receptors; Cell surface antigens; Cell surface determinants
- A61K38/1787—Receptors; Cell surface antigens; Cell surface determinants for neuromediators, e.g. serotonin receptor, dopamine receptor
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Cell Biology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Una combinación que comprende (a) un ligando de una proteína de la vesícula sináptica 2A ("SV2A") seleccionado del grupo que consiste en levetiracetam, brivaracetam y seletracetam; y (b) un compuesto de modulador alostérico positivo ("PAM") del receptor glutamatérgico metabotrópico de subtipo 2 ("mGluR2") seleccionado de **(Ver fórmula)** , o una sal o un solvato farmacéuticamente aceptable del mismo.
Description
d e s c r ip c ió n
Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso
c a m p o de la in ven c ió n
La presente invención se refiere a combinaciones que comprenden un modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) o una sal o un solvato farmacéuticamente aceptable del mismo, y un ligando de una proteína de la vesícula sináptica 2A (“SV2A”).
a n t e c e d e n t e s de la in v e n c ió n
El término "epilepsia" describe una afección en la que una persona presenta convulsiones recurrentes debidas a un proceso subyacente crónico. La epilepsia se refiere a un fenómeno clínico más que a una sola entidad patológica, ya que existen muchas formas y causas de la epilepsia. Si se define la epilepsia como dos o más convulsiones no provocadas, se estima que la incidencia de la epilepsia está comprendida entre aproximadamente un 0.3 y un 0.5 por ciento para diferentes poblaciones en todo el mundo, estimándose la prevalencia de la epilepsia entre 5 y 10 personas de cada 1000.
Un paso esencial en la evaluación y la gestión de un paciente con una convulsión consiste en determinar el tipo de convulsión que ha tenido lugar. La característica principal que distingue las diferentes categorías de convulsiones consiste en si la actividad convulsiva es parcial (sinónimo de focal) o generalizada.
Las convulsiones parciales son aquellas en las que la actividad convulsiva se restringe a áreas concretas de la corteza cerebral. Si se mantiene la consciencia completamente durante la convulsión, se considera que las manifestaciones clínicas son relativamente simples y la convulsión se denomina convulsión parcial-simple. Si se ve afectada la consciencia, la convulsión se denomina convulsión parcial-compleja. Un subgrupo adicional importante comprende aquellas convulsiones que empiezan como convulsiones parciales y a continuación se dispersan de forma difusa en toda la corteza, que se conocen como convulsiones parciales con generalización secundaria.
Las convulsiones generalizadas implican regiones difusas del cerebro simultáneamente de manera bilateralmente simétrica. Las convulsiones de tipo pequeño mal o de ausencia se caracterizan por pérdidas breves y repentinas de la consciencia sin pérdida del control de la postura. Las convulsiones de ausencia atípicas normalmente incluyen una duración más prolongada de la pérdida de consciencia, un inicio y un cese menos abruptos y unos signos motores más evidentes que pueden incluir características focales o lateralizantes.
Las convulsiones de tipo gran mal o tónico-clónicas generalizadas, que representan el tipo principal de convulsiones generalizadas, se caracterizan por un inicio abrupto, sin previo aviso. Normalmente, la fase inicial de la convulsión consiste en una contracción tónica de los músculos, dificultad para respirar y una intensificación significativa del tono simpático que provoca un incremento de la frecuencia cardíaca, la presión sanguínea y el tamaño de las pupilas. Después de 10-20 s, la fase tónica de la convulsión normalmente evoluciona hacia la fase clónica, producida por la superposición de periodos de relajación muscular sobre la contracción muscular tónica. Los periodos de relajación se incrementan progresivamente hasta el final de la fase ictal, que habitualmente no dura más de 1 min. La fase posictal se caracteriza por falta de respuesta, flacidez muscular y salivación excesiva, que puede provocar una respiración estridente y la obstrucción parcial de las vías respiratorias. Las convulsiones atónicas se caracterizan por una pérdida repentina del tono muscular postural que dura 1-2 s. Se pierde la consciencia momentáneamente, pero normalmente no se observa confusión posictal. Las convulsiones mioclónicas se caracterizan por una contracción muscular breve y repentina que puede implicar una parte del cuerpo o todo el cuerpo.
Se ha identificado la proteína de la vesícula sináptica 2A (“SV2A”) como una diana anticonvulsiva de amplio espectro en modelos de epilepsia parcial y generalizada. Algunos estudios realizados en modelos con animales y tejido humano sugieren que ciertos cambios en la expresión de SV2A están relacionados con la epilepsia (para una revisión véase, por ejemplo: (a) Mendoza-Torreblanca et al. "Synaptic vesicle protein 2A: basic facts and role in synaptic function" European Journal of Neuroscience 2013, págs. 1-11; (b) Kaminski RM, et al. "Targeting SV2A for Discovery of Antiepileptic Drugs". En: Noebels JL, Avoli M, Rogawski MA et al., editores. Jasper's Basic Mechanisms of the Epilepsies [internet]. 4a edición. Bethesda (MD): Centro Nacional para la Información Biotecnológica (EE. UU.); 2012. Disponible de: http://www.ncbi.nlm.nih.gov/books/NBK98183/).
El papel exacto de SV2A permanece incierto, pero estudios sugieren que cambios en la expresión de SV2A afectan a la función sináptica (Nowack et al. "Levetiracetam reverses synaptic deficits produced by overexpression of SV2A" PLoS One 2011, volumen 6 (12), e29560). También se ha sugerido que SV2A es un elemento clave en la exocitosis y está imlicada en la neurotransmisión (Crowder et al. "Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A)" Proc Nat Acad Sci USA 1999, 96, págs. 15268-15273) y estudios con ratones deficientes sugieren que la falta de SV2A da como resultado un desequilibrio entre neurotransmisión glutamatérgica y GABAérgica (Venkatesan et al. "Altered balance between excitatory and inhibitory inputs onto CA pyramidal neurons from SV2A-deficient but not SV2B-deficient mice" J Neurosci Res 2012, 90, págs. 2317-2327). La expresión disminuida de SV2A puede ser una consecuencia de actividad convulsiva y puede estar implicada en la evolución de la epilepsia (van Vliet
et al. "Decreased expression of synaptic vesicle protein 2A, the binding site for levetiracetam, during epileptogenesis and chronic epilepsy" Epilepsia 2009, 50, págs. 422-433; Feng et al. "Down-regulation of synaptic vesicle protein 2A in the anterior temporal neocortex of patients with intractable epilepsy" J Mol Neurosci 2009, 39, págs. 354-359; Toering et al. "Expression patterns of synaptic vesicle protein 2A in focal cortical dysplasia and TSC-cortical tubers" Epilepsia 2009, 50, págs. 1409-1418) y epileptogénesis en pacientes con tumores cerebrales (de Groot et al. "Expression of synaptic vesicle protein 2A in epilepsy-associated brain tumors and in the peritumoral cortex" Neuro-Oncology 2010, 12, págs. 265-273).
Los ligandos de SV2A incluyen levetiracetam (Lynch et al. "The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam" Proc. Natl. Acad. Sci. USA 2004, vol. 101, págs. 9861-9866), brivaracetam y seletracetam (Kaminski RM, et al. "Targeting SV2A for Discovery of Antiepileptic Drugs". En: Noebels JL, Avoli M, Rogawski MA et al., editores. Jasper's Basic Mechanisms of the Epilepsies [internet]. 4a edición. Bethesda (MD): Centro Nacional para la Información Biotecnológica (EE. UU.); 2012. Disponible de: http://www.ncbi.nlm.nih.gov/books/NBK98183/; Nowack et al. "Levetiracetam reverses synaptic deficits produced by overexpression of SV2A" PLoSone, diciembre de 2011, vol. 6(12), e29560).
El levetiracetam, (-)-(S)-a-etil-2-oxo-1-pirrolidinacetamida o (S)-2-(2-oxopirrolidin-1-il)butanamida,

es un fármaco contra la epilepsia. No ha mostrado ninguna actividad en modelos agudos tradicionales (pruebas de convulsiones con pentilenotetrazol y por electrochoque máximo), pero se ha encontrado que es potente en modelos de epilepsia crónica y en modelos genéticos de epilepsia generalizada. Ha mostrado un margen de seguridad elevado en comparación con otros fármacos contra la epilepsia (Klitgaard “Levetiracetam: the preclinical profile of a new class of antiepileptic drugs” Epilepsia 2001, 42 (Suplemento 4), págs. 13-18). Se comercializa con la marca Keppra®, disponible en forma de comprimidos, como disolución oral y como un concentrado que se convierte en una disolución para infusión. Keppra® ha sido aprobado en Europa como monoterapia en pacientes de más de 16 años de edad a los que se les ha diagnosticado epilepsia recientemente, en el tratamiento de convulsiones (ataques) de inicio parcial con o sin generalización secundaria y como una terapia adyuvante para su uso con otros fármacos contra la epilepsia en el tratamiento de convulsiones de inicio parcial con o sin generalización en pacientes de más de 1 mes de edad; convulsiones mioclónicas en pacientes de más de 12 años de edad con epilepsia mioclónica juvenil; y convulsiones tónico-clónicas generalizadas primarias en pacientes de más de 12 años de edad con epilepsia generalizada idiopática (www.ema.europa.eu). Keppra® también ha sido aprobado en los EE. UU. como terapia adyuvante para el tratamiento de convulsiones de inicio parcial en pacientes de más de 1 mes de edad; convulsiones mioclónicas en pacientes de 12 años de edad y mayores con epilepsia mioclónica juvenil; y convulsiones tónico-clónicas generalizadas primarias en pacientes de 6 años de edad y mayores con epilepsia generalizada idiopática. Keppra XR®, disponible en forma de comprimidos de liberación prolongada, ha sido aprobado en los EE. UU. para el tratamiento adyuvante de convulsiones de inicio parcial en pacientes de 16 años de edad y mayores con epilepsia (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm).
El brivaracetam, el análogo 4-n-propílico de levetiracetam, (2S)-2-[(4R)-oxo-4-propilpirrolidin-1-il]butanamida,

se encuentra en ensayos clínicos y está siendo investigado como monoterapia en convulsiones de inicio parcial y neuralgia posherpética, y como terapia adyuvante en convulsiones de inicio parcial refractarias, la enfermedad de Unverricht-Lundborg en adolescentes y adultos y en epilepsia fotosensible (www.clinicaltrials.gov).
El seletracetam, (2S)-2-[(4S)-4-(2,2,-difluorovinil)-2-oxo-pirrolidin-1 -iljbutanamida,

se ha sometido a prueba en ensayos clínicos.
En la bibliografía se describen procedimientos para preparar los tres compuestos. Por ejemplo, en los documentos EP 0 162036 y GB 2225 322 se describen procesos para preparar levetiracetam. En el documento WO 01/62726, por ejemplo, se describe un proceso para la preparación de brivaracetam. A partir del documento WO2005/121082, por ejemplo, se conoce un proceso para la preparación de seletracetam. En el documento EP1806339 se describen procesos alternativos para preparar los tres compuestos.
Los fármacos contra la epilepsia han encontrado utilidad en trastornos neurológicos y psiquiátricos, que incluyen dolor neuropático, migraña, temblores esenciales, y en ansiedad, esquizofrenia y trastorno bipolar (Landmarck “Antiepileptic drugs in non-epilepsy disorders. Relations between mechanisms of action and clinical efficacy" CNS Drugs 2008, vol.
22(1), págs. 27-47; Calabresi et al. "Antiepileptic drugs in migraine: from clinical aspects to cellular mechanisms" Trends in Pharmacological Sciences 2007, vol. 28(4), págs. 188-195; Rogawski y Loscher "The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions" Nat Med 2004, vol. 10, págs. 685-692).
Se ha encontrado que levetiracetam es eficaz o potencialmente eficaz en una amplia gama de trastornos neuropsiquiátricos, que incluyen trastornos del estado de ánimo (Muralidharan y Bhagwagar “Potential of levetiracetam in mood disorders: a preliminary review” CNS Drugs 2006, vol. 20, págs. 969-979; Mula et al. "The role of anticonvulsant drugs in anxiety disorders: a critical review of the evidence" J Clin Pshycopharmacol 2007, vol. 27, págs. 263-272), trastornos de ansiedad (Kinrys et al. "Levetiracetam as adjunctive therapy for refractory anxiety disorders" J Clin Psychiatry 2007, vol. 68, págs. 1010-1013; Zhang et al. "Levetiracetam in social phobia: a placebo controlled pilot study" J Psychopharmacol 2005, vol. 19, págs. 551-553; Kinrys et al. "Levetiracetam for treatment-refractory posttraumatic stress disorder" J Clin Psychiatry 2006, vol. 67, págs. 211-214), dolor (Enggaard et al. "Specific effect of levetiracetam in experimental human pain models" Eur J Pain 2006, vol. 10, págs. 193-198; Dunteman "Levetiracetam as an adjunctive analgesic in neoplastic plexopathies: case series and commentary" J Pain Palliative Care Pharmacother 2005, vol. 19, págs. 35-43; Price "Levetiracetam in the treatment of neuropathic pain: three case studies" Clin J Pain 2004, vol. 20, págs. 33-36), trastornos del movimiento (Bushara et al. "The effect of levetiracetam on essential tremor" Neurology 2005, vol. 64, págs. 1078-1080; McGavin et al. "Levetiracetam as a treatment for tardive dyskinesia: a case report" Neurology 2003, vol. 61, págs. 419; Woods et al. "Effects of levetiracetam on tardive dyskinesia: a randomized, double-blind, placebo-controlled study" J Clin Psychiatry 2008, vol. 69, págs. 546-554; Zivkovic et al. "Treatment of tardive dyskinesia with levetiracetam in a transplant patient" Acta Neurol Scand 2008, vol.
117, págs. 351-353; Striano et al. "Dramatic response to levetiracetam in post-ischaemic Holmes' tremor" J Neurol Neurosurg Psychiatry 2007, vol. 78, págs. 438-439) y se sospecha que muestra efectos potencialmente beneficiosos en la función cognitiva (Piazzini et al. "Levetiracetam: An improvement of attention and of oral fluency in patients with partial epilepsy” Epilepsy Research 2006, vol. 68, págs. 181-188; de Groot et al. "Levetiracetam improves verbal memory in high-grade glioma patients" Neuro-oncology 2013, vol. 15(2), págs. 216-223; Bakker et al. "Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment" Neuron 2012, vol. 74, págs. 467 474; para una revisión: Eddy et al. “The cognitive impact of antiepileptic drugs” Ther Adv Neurol Disord 2011, vol. 4(6), págs. 385-407 y las referencias citadas en el mismo; Wheless “Levetiracetam in the treatment of childhood epilepsy” Neuropsychiatric Disease and Treatment 2007, vol. 3(4), págs. 409-421) y los síntomas de comportamiento en la demencia (Dolder y Nealy “The efficacy and safety of newer anticonvulsants in patients with dementia” Drugs Aging 2012, vol. 29(8), págs. 627-637). Los datos de estudios con animales y algunos ensayos clínicos preliminares sugieren que levetiracetam puede presentar cierto potencial para restringir la epilepsia postraumática, tal como la provocada por un estado epiléptico, una lesión cerebral traumática y un accidente cerebrovascular isquémico, y al parecer presenta efectos neuroprotectores. El potencial de levetiracetam para aliviar la epileptogénesis o disfunción cognitiva todavía no ha sido demostrado mediante estudios clínicos y con animales concluyentes (para revisiones: Loscher y Brandt “Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research” Pharmacol Rev 2010, vol. 62, 668-700; Shetty “Prospects of levetiracetam as a neuroprotective drug against status epilepticus, traumatic brain injury and stroke” Front. Neur. 2013, 4:172. Doi: 10.3389/fneur.2013.00172) pero ha presentado actividad antiepileptogénica en el modelo de estimulación en ratas y ratones. También se ha sugerido que levetiracetam inhibe la liberación de glutamato (Lee et al. "Levetiracetam inhibits glutamate transmission through presynaptic P/Q-type calcium channels on the granule cells of the dentate gyrus" British Journal of Pharmacology 2009, vol. 158, págs. 1753-1762).
Se ha encontrado que seletracetam y brivaracetam reducen la gravedad de distonía en el modelo de hámster mutante dtsz y pueden ser útiles en algunos pacientes que padecen trastornos del movimiento discinéticos y distónicos (Hamann etal. "Brivaracetam and seletracetam, two new SV2A ligands, improve paroxysmal dystonia in the dtsz mutant hamster" European Journal of Pharmacology 2008, vol. 601, págs. 99-102).
Los moduladores alostéricos positivos de mGluR2 han surgido recientemente como estrategias terapéuticas novedosas prometedoras para el tratamiento de varios trastornos del SNC, incluyendo la epilepsia, y algunos PAM de mGluR2 están sometiéndose actualmente a ensayos clínicos para el tratamiento de esquizofrenia y ansiedaddepresión (www.clinicaltrials.gov, véase, por ejemplo: JNJ-40411813/ADX71149 por Addex Therapeutics y Janssen Pharmaceuticals, Inc.). La sugerencia inicial de que los fármacos que reducen la transmisión glutamatérgica pueden ser eficaces en el tratamiento de la epilepsia procede de estudios no clínicos agudos con agonistas de receptores mGlu2/3 mixtos (Moldrich et al. ''Glutamate metabotropic receptors as targets for drug therapy in epilepsy" Eur J Pharmacol. 2003, vol.476, págs. 3-16). Se encontró que LY379268 y LY389795, dos agonistas de receptores mGlu2/3, eran ineficaces bloqueando convulsiones MES hasta dosis que producían alteración motora, pero se encontraron eficaces en el modelo de 6 Hz de una manera dependiente de la dosis (Barton et al. "Comparison of the effect of glutamate receptor modulators in the 6 Hz and maximal electroshock seizure models" Epilepsy Research 2003, vol.
56, págs. 17-26). La administración continuada de un agonista de mGlu2/3 indujo, paradójicamente, actividad convulsiva en estudios de toxicología a largo plazo (Dunayevich et al. "Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder" Neuropsychopharmacology. 2008, vol. 33(7), págs. 1603-10). Este efecto paradójico podría estar relacionado con cambios inducidos por el agonista en la sensibilidad del sistema receptor (taquifilaxia), pero sin embargo no se ha notificado en modelos preclínicos de epilepsia. En cambio, los moduladores alostéricos positivos modulan la neurotransmisión en curso, pero no son directamente estimulantes, con lo que se reduce el riesgo de taquifilaxia.
Antes de la actividad convulsiva, se registran aumentos de glutamato extracelular en el hipocampo humano y el aumento se sostiene durante la actividad epileptogénica (During y Spencer “Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain” Lancet 1993, vol. 341(8861), págs. 1607-10), lo que respalda la idea de que una reducción de los niveles de glutamato podría ser beneficiosa para el tratamiento de la epilepsia. De hecho, durante la actividad convulsiva, los niveles de glutamato aumentan hasta niveles potencialmente neurotóxicos. La actividad convulsiva da como resultado daños estructurales progresivos en el cerebro humano induciendo anomalías adicionales en el metabolismo de glutamato (Petroff et al. "Glutamate-glutamine cycling in the epileptic human hippocampus" Epilepsia 2002, vol. 43(7), págs. 703-10). Por tanto, puede esperarse que un modulador alostérico positivo de mGluR2 o un agonista ortostérico de mGluR2 ofrezcan protección contra los daños neurológicos inducidos por las convulsiones.
Los documentos WO2009/033704 y WO2010/130424 dan a conocer moduladores alostéricos positivos de mGluR2, usos de los mismos y procedimientos para sintetizar los compuestos. Los documentos WO1997/18199 y WO2003/104217 dan a conocer compuestos de modulador de receptor de aminoácido excitatorios que más tarde mostraron tener actividad de agonista ortostérico de mGlu2/3 (véase por ejemplo Rorick-Kehn et al. (2007) The Journal of Pharmacology and Experimental therapeutics vol. 321, n.° 1, págs. 308-317), la bibliografía de patentes y científica adicional da a conocer ejemplos adicionales de compuestos que tienen actividad de agonista ortostérico de mGlu2/3, y el documento WO2008/150233 da a conocer compuestos con actividad de activador alostérico de mGluR2.
Myhrer, T. et al. “Capacities of metabotropic glutamate modulators in counteracting soman-induced seizures in rats” Eur. J. Pharmacol. 718 (2013) 253-260 divulga el efecto de combinaciones de DCG-IV contra convulsiones inducidas por somano en ratas. Si bien la combinación de HI-6 y prociclidina con DCG-IV mostró eficacia como antídoto, la combinación de HI-6 y levetiracetam con DCG-IV no cesó las convulsiones ni preservó vidas, y la eficacia como antídoto observada fue inferior.
Klitgaard, H. “Antiepileptic drug Discovery: lessons from the past and future challenges” Acta Neurol Scand 2005: 112 (Suppl. 181): 68-72 proporciona un breve contexto histórico sobre la investigación de fármacos antiepilépticos, y describe el descubrimiento de levetiracetam y la posterior identificación de brivaracetam y seletracetam, que tiene un perfil de actividad anticonvulsiva único y potencialmente un mejor perfil de seguridad en comparación con las terapias existentes.
Piazzini et al. “Levetiracetam: An improvement of attention and of oral fluency in patients with partial epilepsy” Epilepsy Research 2006, Vol. 68, págs. 181 -188 divulga resultados preliminares que muestran una mejora cognitiva significativa en la atención y la fluidez oral en los pacientes diagnosticados con epilepsia parcial resistente a fármacos que reciben levetiracetam como terapia complementaria en comparación con los controles.
Johannessen Landmark, C and Johannessen, S. I. “Modifications of Antiepileptic Drugs for Improved Tolerability and Efficacy” Perspect Medicin Chem 2008:2 21-39 divulga una revisión sobre productos de investigación y fármacos de diseño y comercializados en desarrollo en 2008, incluido levetiracetam.
Price, M.J. “Levetiracetam in the treatment of neuropathic pain: three case studies” Clin J Pain 2004, Vol. 20, págs.
33-36 describe tres estudios de un caso de pacientes que presentan dolor neuropático que responden favorablemente al tratamiento con levetiracetam, sin eventos adversos importantes.
El documento WO 01/39779 (UCB, S.A) divulga el uso de levetiracetam en trastornos que incluyen migraña, trastornos bipolares y dolor crónico o neuropático solo o combinado con compuestos que incluyen inhibición neural mediada por receptores GABAa .
El documento EP 2462990 (UCB Pharma GmbH) describe una composición farmacéutica que comprende lacosamida y levetiracetam para la prevención, alivio y/o tratamiento de convulsiones epilépticas.
El documento WO 2014/078568 (Univ Johns Hopkins) se refiere a grandes rasgos a métodos para tratar la esquizofrenia o el trastorno bipolar administrando una combinación de un inhibidor de una proteína de la vesícula sináptica 2A y un antipsicótico. Los ejemplos divulgan, sin embargo, levetiracetam, brivaracetam o valproato en estudios relacionados con la cognición (aprendizaje espacial y retención de memoria, ensayos en pacientes con afectación cognitiva amnésica leve).
El documento WO2015/032790 (Janssen Pharmaceutica NV) describe compuestos de 1,2,4-triazolo[4,3-a]piridina como moduladores alostéricos positivos del subtipo 2 del receptor metabotrópico de glutamato.
Los fármacos contra la epilepsia disponibles actualmente no afectan únicamente a la transmisión glutamatérgica. Su mecanismo de acción se concibe en general como una alteración del equilibrio entre la transmisión de estímulos excitatorios (mediada por glutamato) y la transmisión de estímulos inhibidores (mediada por GABA) (Johannessen Landmark “Antiepileptic drugs in non-epilepsy disorders: relations between mechanisms of action and clinical efficacy” CNS Drugs 2008, Vol. 22(1), págs. 27-47).
Un factor limitante significativo para el uso de ligandos de SV2A es la tolerabilidad y el perfil de efectos secundarios. Por ejemplo, la dosis eficaz de levetiracetam para convulsiones de inicio parcial es de 1000 mg, 2000 mg y 3000 mg, administrada dos veces al día. Los efectos secundarios descritos para levetiracetam incluyen un comportamiento irritable o agresivo, ansiedad, cambios de personalidad, escalofríos, tos o ronquera, llantos, despersonalización, diarrea, sequedad bucal, euforia, fiebre, sensación general de malestar o enfermedad, cefalea, hiperventilación, latidos del corazón irregulares, irritabilidad, dolor de las articulaciones, pérdida de apetito, lumbalgia o dolor de los costados, depresión mental, dolores y picores musculares, náusea, dolor o dificultad al orinar, paranoia, reacciones rápidas o desmesuradas a nivel emocional, cambios rápidos del estado de ánimo, inquietud, temblores, estremecimientos, dificultad para respirar, insomnio o somnolencia inusual, dolor de garganta, congestión o secreción nasal, sudores, problemas para conciliar el sueño, cansancio o debilidad inusual y vómitos. Por lo tanto, sigue siendo necesario disponer de un tratamiento eficaz con una dosis eficaz de levetiracetam más baja y un perfil de efectos secundarios más favorable para el tratamiento de la epilepsia y trastornos relacionados, no solamente en la población adulta sino también en la pediátrica.
b r eve d e s c r ip c ió n de la s f ig u r a s
Figura 1: Respuesta a la dosis para la determinación de DE50 a 6 Hz, 44 mA para el comp. de referencia n.° 2 y LEV solo y en combinación.
Figura 2: Análisis isobolográfico para la combinación del comp. de referencia n.° 1 con levetiracetam (LEV) in el ensayo de 6 Hz (44 mA). Se determinaron los valores iniciales de DE50 (mostrados a continuación) tanto para el comp. de referencia n.° 1 como para LEV (puntos de datos sobre ejes x e y; rombos rellenos). La línea teórica de aditividad conecta los valores de DE50 calculados para los dos compuestos (línea continua negra). Se representan gráficamente los DE50 teóricos (+ EEM) para las tres combinaciones de razón de dosis fija (LEV: comp. de referencia n.° 1): 1:3 - cuadrados rellenos/línea continua negra, 1:1 - triángulos hacia arriba rellenos/línea continua negra, y 3:1 - triángulos hacia abajo rellenos/línea continua negra. Inicialmente se derivaron las dosis de tratamiento experimentales de valores teóricos y se ajustaron según los efectos observados. También se muestran valores de DE50 (+ EEM) determinados experimentalmente para cada combinación de razón de dosis fija: 1:3' - cuadrados vacíos/línea de puntos, 1:1' - triángulos hacia arriba vacíos/línea de puntos, y 3:1' - triángulos hacia abajo vacíos/línea de puntos. Se compararon comparaciones entre valores de DE50 determinados experimentalmente y teóricos usando la prueba de la t (***P<0.001). N=8 por groupo. En la figura 2, se representa la razón de LEV con respecto al comp. de referencia n.° 1 como sigue:

Figura 3 : Estudios de combinación para el comp. de referencia n.° 25-a con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA). A una dosis de 10 mg/kg s.c., el comp. de referencia n.° 25-a aumenta la potencia de LEV, conduciendo a un desplazamiento de aproximadamente 70 veces en la DE50. Esto sugiere una relación farmacodinámica positiva.
Figura 4: Estudios de combinación para el comp. de referencia n.° 2-a con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA). A una dosis de 10 mg/kg s.c., el comp. de referencia n.° 2-a aumenta la potencia de LEV, conduciendo a un desplazamiento de aproximadamente 35 veces en la DE50. Esto sugiere una relación farmacodinámica positiva. Figura 5 : Estudios de combinación para el comp. n.° 6-b con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA). A una dosis de 10 mg/kg p.o., el comp. n.° 6-b aumenta la potencia de LEV, conduciendo a un desplazamiento de aproximadamente 100 veces en la DE50. Esto sugiere una relación farmacodinámica positiva.
Figura 6 : Estudios de combinación para el compuesto de referencia LY-404039 con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA). A una dosis de 5 mg/kg s.c., el compuesto de referencia LY-404039 aumenta la potencia de LEV, conduciendo a un desplazamiento de aproximadamente 27 veces en la DE50. Esto sugiere una relación farmacodinámica positiva.
d e s c r ip c ió n de la in v e n c ió n
La presente invención se refiere a una combinación que comprende
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo.
En una realización particular, la invención tal como se describe en la presente se refiere a una combinación farmacéutica, en particular a un producto farmacéutico combinado, que comprende
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo; y (c) al menos un portador farmacéuticamente aceptable.
En una realización adicional, la invención se refiere a la combinación descrita en la presente para su uso como medicamento.
Una realización adicional de esta invención se refiere al uso de la combinación descrita en la presente para la elaboración de un medicamento o un producto farmacéutico para el tratamiento o la prevención de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente y trastornos bipolares y trastornos relacionados.
Una realización adicional de esta invención se refiere al uso de la combinación descrita en la presente para la elaboración de un medicamento o un producto farmacéutico para la neuroprotección.
Una realización adicional de esta invención se refiere al uso de la combinación descrita en la presente para la elaboración de un medicamento o un producto farmacéutico para la prevención de la epileptogénesis.
Una realización adicional se refiere al tratamiento o la prevención de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; y trastornos bipolares y trastornos relacionados de un sujeto que comprende administrar de forma concurrente o secuencial al sujeto que lo necesita un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entree

o una sal o un solvato farmacéuticamente aceptable del mismo, en cantidades que serían terapéuticamente eficaces cuando el ligando de SV2A y el compuesto de mGluR2 se administran conjuntamente.
Una realización adicional se refiere a una combinación tal como se describe en la presente para la neuroprotección; o a una combinación tal como se describe en la presente para su uso en la neuroprotección.
Una realización adicional se refiere a una combinación tal como se describe en la presente para la prevención de la epileptogénesis; o a una combinación tal como se describe en la presente para su uso en la prevención de la epileptogénesis.
En una realización adicional, la invención se refiere a una combinación de dosis fija de
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo,
en cantidades que serían terapéuticamente eficaces cuando el ligando de SV2A y el compuesto de tipo mGluR2 se administran conjuntamente a pacientes, para su uso en el tratamiento o la prevención de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados.
Una divulgación se refiere a un método para el tratamiento o la prevención de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados, comprendiendo dicho método la administración de una cantidad terapéuticamente eficaz de una combinación o un producto de combinación que comprende
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre
o una sal o un solvato farmacéuticamente aceptable del mismo,
a un sujeto que lo necesita, tal como un animal de sangre caliente, en particular un ser humano.
Una divulgación adicional se refiere a un método de neuroprotección, comprendiendo dicho método la administración de una cantidad terapéuticamente eficaz de una combinación o un producto de combinación que comprende (a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo,
a un sujeto que lo necesita, tal como un animal de sangre caliente, en particular un ser humano.
Una divulgación adicional se refiere a un método contra la epileptogénesis, comprendiendo dicho método la administración de una cantidad terapéuticamente eficaz de una combinación o un producto de combinación que comprende
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo, a un sujeto que lo necesita, tal como un animal de sangre caliente, en particular un ser humano.
En una realización adicional, la presente invención se refiere a un producto farmacéutico o un envase comercial que comprende una combinación según la invención tal como se describe en la presente, en particular junto con instrucciones, para su uso simultáneo, secuencial o por separado en el tratamiento o la prevención de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados. En una realización adicional, la presente invención se refiere a un producto farmacéutico o un envase comercial que comprende una combinación según la invención tal como se describe en la presente, en particular junto con instrucciones, para su uso simultáneo, secuencial o por separado en la neuroprotección.
En una realización adicional, la presente invención se refiere a un producto farmacéutico o un envase comercial que comprende una combinación según la invención tal como se describe en la presente, en particular junto con instrucciones, para su uso simultáneo, secuencial o por separado contra la epileptogénesis.
En una realización adicional, la invención se refiere a una combinación que comprende una cantidad que es de manera conjunta terapéuticamente eficaz contra la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados; de
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo, y al menos un portador farmacéuticamente aceptable. En una realización adicional, la invención se refiere a una combinación que comprende una cantidad que es de manera conjunta terapéuticamente eficaz como neuroprotector, de
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo, y al menos un portador farmacéuticamente aceptable. En una realización adicional, la invención se refiere a una combinación que comprende una cantidad que es de manera conjunta terapéuticamente eficaz en la prevención de la epileptogénesis, de
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo, y al menos un portador farmacéuticamente aceptable. En una realización adicional, la invención se refiere al uso de
(a) un ligando de una proteína de la vesícula sináptica 2A (“SV2A”) seleccionado entre levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado entre

o una sal o un solvato farmacéuticamente aceptable del mismo,
para la preparación de un producto combinado según la presente invención
El componente (b) de la combinación de la invención se denomina en general en la presente "compuesto de mGluR2" o "compuesto de PAM de mGluR2", o "compuesto de modulador alostérico positivo de mGluR2" lo que significa que el compuesto tiene principalmente actividad en el receptor glutamatérgico metabotrópico de subtipo 2, y en particular es un modulador alostérico positivo (PAM) del receptor glutamatérgico metabotrópico de subtipo 2. Un experto en la técnica estará familiarizado con la gran homología de mGluR2 y mGluR3, a la que se debe que algunos agonistas
ortostérico de mGluR2 también muestren actividad como agonistas ortostéricos de mGluR3. Tal es el caso por ejemplo, del ácido (-)-(1R,4S,5S,6S)-4-amino-2-sulfonilbiciclo[3.1.0]-hexano-4,6-dicarboxílico (también conocido como LY-404,039 [CAS 635318-11 -5]), con un Ki = 149 nM (receptor de mGlu2) y Ki = 92 nM (receptor de mGlu3), selectividad de 100 vaces para mGlu2 y mGlu3 con respecto a mGlu4a, -6, -7a, y -8a, y sin actividad en mGlu1a y mGlu5a (Rorick-Kehn et al. (2007) The Journal of Pharmacology and Experimental Therapeutics vol. 321, n.° 1, págs. 308-317). El término "compuesto de mGluR2" o "compuesto de PAM de mGluR2", o "compuesto de modulador alostérico positivo de mGluR2" no excluye por tanto compuestos que muestren alguna otra actividad menor in vitro o in vivo.
Los compuestos de referencia de PAM de mGluR2 de la combinación de la divulgación se seleccionan en particular de aquellos dados a conocer en el documento WO2010/130424 y pueden prepararse según los procedimientos descritos en ese documento. Los compuestos de referencia particulares incluyen

Los compuestos adicionales de referencia de PAM de mGluR2 de la combinación de la divulgación también se seleccionan en particular de aquellos dados a conocer en el documento WO2009/033704 y pueden prepararse según los pocedimientos descritos en ese documento. Los compuestos particulares incluyen

o una sal o un solvato farmacéuticamente aceptable de los mismos.
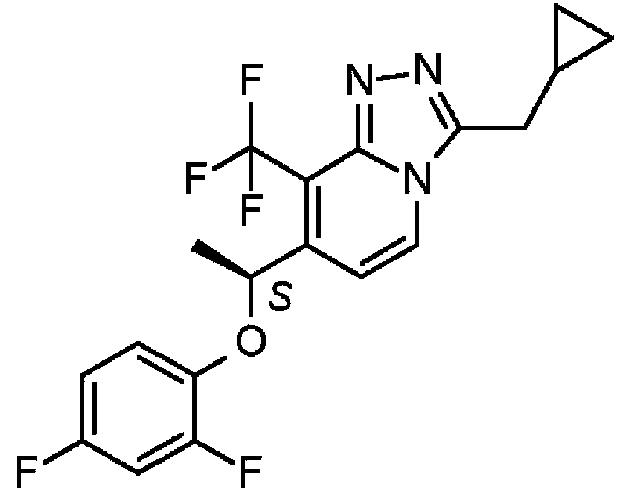
El compuesto de PAM de mGluR2 de la combinación de la invención se selecciona en particular entre los divulgados en el documento PCT/EP2014/068676. El compuesto particular es 3-(ciclopropilmetil)-7-[(1 S)-1 -(2,4-difluorofenoxi)etil]-8-(trifluorometil)[1,2,4]triazolo[4,3-a]piridina; incluidas las sales y los solvatos farmacéuticamente aceptables de los mismos.
En una realización adicional, el compuesto se puede seleccionar entre la sal de clorhidrato de 3-(ciclopropilmetil)-7-[(1 S)-1 -(2,4-difluorofenoxi)etil]-8-(trifluorometil)[1,2,4]triazolo[4,3-a]piridina.
Los nombres de los compuestos de la presente invención se generaron según las reglas de nomenclatura que se acordaron por el Chemical Abstracts Service (C.A.S.) usando el software (ACD/Name product, versión 10.01.0.14105, octubre de 2006) de Advanced Chemical Development, Inc.
Tal como se usa en la presente, y a menos que se indique lo contrario, el término “agente contra la epilepsia” y la abreviatura “AED”, que corresponde a sus siglas en inglés, se emplearán de manera intercambiable con el término “agente contra las convulsiones” y tal como se usan en la presente, se refieren a un agente capaz de tratar, inhibir o prevenir la actividad convulsiva o ictogénesis cuando el agente se administra a un sujeto o paciente.
Tal como se usa en la presente, a menos que se indique lo contrario, el término “ligando de una proteína de la vesícula sináptica 2A” y la abreviatura “ligando de SV2A”, se emplearán de manera intercambiable. Los ejemplos de ligandos de SV2A incluyen, pero no se limitan a, los compuestos incluidos en las publicaciones GB 1,039,113; GB 1,309,692; EP 1262 036; EP 1806 339; WO 2001/062726; US 2002/094787; WO 2004/087658; WO 2005/121082; WO 2005/054188; WO 2006/128692; WO 2006/128693; WO 2007/065595; WO 2008/132139, and WO 2008/132142; WO 2011/047860; WO 2012/143116; and WO 2012/143117. Ejemplos particulares adecuados de ligandos de SV2A incluyen, pero no se limitan a: levetiracetam, brivaracetam y seletracetam.
En una realización particular, el ligando de SV2A es levetiracetam.
En una realización particular, el ligando de SV2A es brivaracetam.
Se conocen procedimientos para la preparación de los ligandos de SV2A anteriores a partir de la bibliografía y se describen, por ejemplo, en el documento EP 1806339; en el documento EP 0 162036 y en el documento GB 2225 322 (levetiracetam); en el documento WO 01/62726 (brivaracetam); y en el documento WO 2005/121082 (seletracetam).
En una realización adicional, la combinación según la invención comprende (a) una cantidad farmacéuticamente eficaz de levetiracetam o brivaracetam; y (b) una cantidad farmacéuticamente eficaz de

o una sal farmacéuticamente aceptable del mismo, en particular la sal de clorhidrato del mismo, o un solvato del mismo.
En una realización adicional, la composición farmacéutica según la invención comprende (a) una cantidad farmacéuticamente eficaz de levetiracetam o brivaracetam; y (b) una cantidad farmacéuticamente eficaz de
o una sal farmacéuticamente aceptable del mismo, en particular la sal de clorhidrato del mismo, o un solvato del mismo.
El producto de combinación de la presente invención, en particular, la composición farmacéutica según la invención, es especialmente adecuado para el tratamiento de la epilepsia y trastornos relacionados.
Se apreciará que algunos de los compuestos de mGluR2, en particular el compuesto de PAM de mGluR2 de la invención y sus sales de adición y solvatos farmacéuticamente aceptables del mismo pueden contener uno o más centros de quiralidad y existir como formas estereoisoméricas.
La expresión “compuesto de la invención” tal como se usa en la presente, se pretende que incluya el compuesto 6-b como se divulgan la presente, y las sales y solvatos del mismo.
Tal como se usa en la presente, cualquier fórmula química con enlaces que se muestran solo como líneas continuas y no como enlaces en cuña continuos o enlaces en cuña discontinuos, o que se indique otra manera que tienen una configuración particular (por ejemplo, R, S) alrededor de uno o más átomos, contempla cada estereoisómero posible, o mezcla de dos o más estereoisómeros.
Anteriormente o a continuación en la presente, la configuración absoluta se especifica de acuerdo con el sistema de Cahn-Ingold-Prelog. La configuración en un átomo asimétrico está especificada por R o S.
Cuando se identifica un estereoisómero específico, esto significa que dicho estereoisómero está sustancialmente exento, es decir, asociado con menos de un 50%, preferiblemente menos de un 20%, más preferiblemente menos de un 10%, incluso más preferiblemente menos de un 5%, en particular menos de un 2% y de la manera más preferible menos de un 1%, de los otros isómeros. Por lo tanto, por ejemplo, cuando se especifica que un compuesto de mGluR2 es (R), esto significa que el compuesto está sustancialmente exento del isómero (S); por ejemplo, cuando se especifica que un compuesto de mGluR2 es E, eso significa que el compuesto está sustancialmente exento del isómero Z; por ejemplo, cuando se especifica que un compuesto de mGluR2 es cis, esto significa que el compuesto está sustancialmente exento del isómero trans.
Para su uso en medicina, las sales del compuesto de esta invención se refieren a "sales farmacéuticamente aceptables" no tóxicas (sales de los compuestos de la presente invención en las que el contraión es farmacéuticamente aceptable). Sin embargo, otras sales pueden ser útiles en la preparación o purificación de los compuestos según esta invención o de sus sales farmacéuticamente aceptables, y pueden abarcar ácidos y bases que no son farmacéuticamente aceptables. Todas las sales, ya sean farmacéuticamente aceptables o no, quedan incluidas dentro del ámbito de la presente invención.
Se pretende que las sales de adición de ácidos y bases farmacéuticamente aceptables tal como se mencionan anteriormente o a continuación en la presente comprendan las formas de sal de adición de ácidos y bases no tóxicas terapéuticamente activas que los compuestos de la invención pueden formar. Las sales farmacéuticamente aceptables de los compuestos incluyen sales de adición de ácidos que pueden formarse, por ejemplo, mezclando una disolución del compuesto con una disolución de un ácido farmacéuticamente aceptable tal como por ejemplo, ácidos inorgánicos tales como hidrácidos halogenados, p. ej. ácido clorhídrico o bromhídrico, ácidos sulfúrico, nítrico o fosfórico; o ácidos orgánicos tales como, por ejemplo, ácidos acético, propanoico, hidroxiacético, láctico, pirúvico, oxálico (es decir etanodióico), malónico, succínico (es decir ácido butanodióico), maléico, fumárico, málico, tartárico, cítrico, metanosulfónico, etanosulfónico, bencenosulfónico, p-toluenosulfónico, ciclámico, salicílico, p-aminosalicílico y pamoicos. A la inversa, dichas formas de sales pueden convertirse en la forma de base libre mediante tratamiento con una base adecuada. Además, cuando los compuestos de la invención portan un resto ácido, las sales farmacéuticamente aceptables de los mismos pueden incluir bases orgánicas e inorgánicas. Las formas salinas de bases adecuadas comprenden, por ejemplo, las sales de amonio, las sales de metales alcalinos y alcalinotérreos, p. ej., sales de litio, sodio, potasio, magnesio y calcio, sales con bases orgánicas, p. ej., aminas primarias, secundarias y terciarias alifáticas y aromáticas tales como metilamina, etilamina, propilamina, isopropilamina, los cuatro isómeros de la butilamina, dimetilamina, dietilamina, dietanolamina, dipropilamina, diisopropilamina, di-n-butilamina, pirrolidina, piperidina, morfolina, trimetilamina, trietilamina, tripropilamina, quinuclidina, piridina, quinolina e isoquinolina; la benzatina, N-metil-D-glucamina, sales de hidrabamina y sales de aminoácidos tales como, por ejemplo, arginina y lisina. A la inversa, la forma salina puede convertirse en la forma de ácido libre mediante tratamiento con ácido.
El término "solvato" comprende las formas de adición de disolventes, así como las sales de los mismos, que el compuesto 6-b puede formar. Ejemplos de tales formas de adición de disolventes son, p. ej., hidratos y alcoholatos.
Preparación del compuesto
El compuesto de acuerdo con la invención se puede preparar por lo general mediante una sucesión de pasos, cada uno de los cuales conocido por el experto. En particular, el compuesto se puede preparar de acuerdo con los siguientes métodos de síntesis.
El compuesto se puede sintetizar en forma de mezclas racémicas de enantiómeros que se pueden separar entre sí siguiendo procedimientos de resolución conocidos en la técnica. El compuesto racémico se puede convertir en las formas salinas diastereoméricas correspondientes mediante reacción con un ácido quiral adecuado. Dichas formas salinas diasteroméricas se separan posteriormente, por ejemplo, mediante cristalización selectiva o fraccionada y los enantiómeros se liberan de las mismas con álcali. Una manera alternativa para separar las formas enantioméricas del compuesto conlleva cromatografía líquida o cromatografía con fluidos supercríticos (SFC) utilizando una fase estacionaria quiral. Dichas formas esteroquímicamente isoméricas puras también se pueden derivar de las formas estereoquímicamente isoméricas puras correspondientes de los materiales de partida apropiados, siempre que la reacción transcurra de manera estereoespecífica.
Tal como se usa en la presente, se pretende que el término "composición” abarque un producto que comprende los componentes especificados en las cantidades especificadas, así como cualquier producto que resulte, directa o indirectamente, de combinaciones de los componentes especificados en las cantidades especificadas.
Tal como se usa en la presente, el término "sujeto" se refiere a un animal, preferiblemente un mamífero, más preferiblemente un ser humano adulto, niño o bebé, que es o que ha sido objeto de tratamiento, observación o experimentación.
El término "cantidad terapéuticamente eficaz", tal como se usa en la presente, significa la cantidad de compuesto o agente farmacéutico activo que provoca la respuesta biológica o medicinal en un sistema tisular, animal o humano que busca un investigador, veterinario, médico u otro profesional sanitario, que incluye el alivio de los síntomas de la enfermedad o trastorno que está tratándose y/o la reducción de la gravedad de uno o más de los síntomas de la enfermedad que está tratándose.
La combinación de (a) ligando de SV2A y (b) modulador alostérico positivo ("PAM") del receptor glutamatérgico metabotrópico de subtipo 2 ("mGluR2") o una sal o un solvato farmacéuticamente aceptable del mismo tanto si los compuestos (a) and (b) se administran simultáneamente, por separado o de manera secuancial, puede ser beneficiosa en comparación con el efecto de los compuestos (a) o (b) administrados solos. En particular, se puede producir al menos un efecto beneficioso, p. ej., una mejora mutua del efecto de los compuestos (a) y (b), un efecto más que aditivo, en particular un efecto sinérgico; otros efectos beneficiosos incluyen, por ejemplo, una dosis eficaz significativamente reducida para la combinación de (a) y (b); un efecto terapéutico adicional no observado para ninguno de los compuestos (a) o (b) por sí solos, un perfil de efectos secundarios más beneficioso o un efecto terapéutico combinado con una dosis no eficaz de uno de los compuestos (a) y (b) o de ambos.
Cuando la presente invención se refiera a la coterapia o terapia de combinación, que comprende la administración de (a) ligando de proteína de la vesícula sináptica 2A (“SV2A”) y (b) compuesto 6-b tal como se define en la presente, cantidad farmacéutica o terapéuticamente eficaz significará la cantidad de la combinación de agentes tomados conjuntamente, de manera que el efecto combinado provoque la respuesta medicinal o biológica deseada. Por ejemplo, la cantidad terapéuticamente eficaz de una coterapia que comprende la administración de (a) un ligando de SV2A tal como se define en la presente y (b) compuesto 6-b tal como se define en la presente sería la cantidad del (a) ligando de SV2A tal como se describe en la presente y la cantidad de (b) un compuesto 6-b que cuando se administran conjuntamente o de manera secuencial tiene un efecto combinado que es terapéuticamente eficaz. Además, un experto en la técnica reconocerá que en el caso de coterapia con una cantidad terapéuticamente eficaz, como en el ejemplo anterior, la cantidad del compuesto 6-b, y/o la cantidad del ligando de SV2A adecuado puede ser o puede no ser, de manera individual, terapéuticamente eficaz.
La presente invención proporciona métodos de prevención o tratamiento que comprenden administrar a un sujeto que lo necesita una coterapia con una cantidad terapéuticamente eficaz de un ligando de SV2A y una cantidad terapéuticamente eficaz de un compuesto 6-b, tal como se describe en la presente. Con el fin de lograr este objetivo, los compuestos o composiciones de esta invención deben utilizarse en la cantidad o dosis terapéuticamente eficaz correcta, tal como se describe a continuación.
Las dosis y los programas óptimos que deben administrarse pueden determinarse fácilmente por los expertos en la técnica y variarán dependiendo del compuesto particular usado, el modo de administración, la concentración de la preparación, el modo de administración y el avance del estado patológico. Además, factores asociados con el paciente particular que está tratándose, que incluyen edad del paciente, peso, dieta y tiempo de administración, darán como resultado la necesidad de ajustar las dosificaciones.
Un experto en la técnica reconocerá que una dosis terapéuticamente eficaz de los compuestos de la presente invención puede incluir dosis repetidas dentro de un régimen de tratamiento prolongado que proporcionará resultados clínicamente significativos.
Las cantidades del compuesto 6-b en las combinaciones de la invención que se administran diariamente pueden variar de desde aproximadamente 0.01 hasta aproximadamente 2000 mg. Ejemplos de cantidades diarias del compuesto 6-b son 0.01,0.05, 0.1,0.5, 1.0, 2.5, 5.0, 10.0, 15.0, 25.0, 50.0, 100, 150, 200, 250, 300, 400, 500, 750 y 1000 miligramos para el ajuste sintomático de la dosis al paciente que va a tratarse. Una cantidad eficaz del fármaco se administra normalmente con un nivel de dosificación de desde aproximadamente 0.01 mg/kg hasta aproximadamente 150.0 mg/kg de peso corporal por día o cualquier intervalo dentro del mismo. Preferiblemente, el intervalo es de desde aproximadamente 0.1 hasta aproximadamente 100.0 mg/kg de peso corporal por día, más preferiblemente desde aproximadamente 0.5 mg/kg hasta aproximadamente 50 mg/kg, más preferiblemente desde aproximadamente 1.0 hasta aproximadamente 25.0 mg/kg de peso corporal por día. Los compuestos pueden administrarse siguiendo un régimen de 1, 2, 3 ó 4 veces por día. Las cantidades de ligando de SV2A que se administran diariamente pueden variar desde aproximadamente 0.01 hasta aproximadamente 7000 mg, preferiblemente estarán entre 250 y 5000 mg y más preferiblemente estarán entre 500 y 3000 mg. Ejemplos de cantidades diarias del ligando de SV2A son 2.5, 5.0, 10.0, 15.0, 25.0, 50.0, 100, 150, 200, 250, 500, 750, 1000, 1500 y 3000 miligramos para el ajuste sintomático de la dosis al paciente que va a tratarse. Normalmente, una cantidad eficaz del fármaco se administra con un nivel de dosificación de desde aproximadamente 0.01 mg/kg hasta aproximadamente 150.0 mg/kg de peso corporal por día o cualquier intervalo dentro del mismo. Preferiblemente, el intervalo es de desde aproximadamente 0.1 hasta aproximadamente 100.0 mg/kg de peso corporal por día, más preferiblemente desde aproximadamente 0.5 mg/kg hasta aproximadamente 50 mg/kg, más preferiblemente desde aproximadamente 1.0 hasta aproximadamente 25.0 mg/kg de peso corporal por día. Los compuestos pueden administrarse siguiendo un régimen de 1,2, 3 ó 4 veces por
día. Todas las cantidades mencionadas en este párrafo y en los siguientes se refieren a la forma libre (es decir, la forma no salina). Los valores anteriores representan equivalentes de la forma libre, es decir, cantidades como si se administrase la forma libre. Si se administran sales, deben calcularse las cantidades en función de la razón de pesos moleculares entre la sal y la forma libre.
Las dosis diarias mencionadas anteriormente se calculan para un peso corporal promedio de aproximadamente 70 kg y deben recalcularse en el caso de aplicaciones pediátricas o cuando se utilicen en pacientes con un peso corporal que sea sustancialmente diferente.
Las dosificaciones pueden presentarse en forma de una, dos, tres, cuatro o más subdosis administradas en intervalos adecuados a lo largo del día. La dosificación usada corresponde preferiblemente a la cantidad diaria del compuesto 6-b, o del ligando de SV2A, mencionado anteriormente, o una subdosis de la misma, tal como 1/2, 1/3, 1/4 de la misma. Una forma de dosificación puede contener el compuesto 6-b, o el ligando de SV2A o ambos juntos, en una cantidad equivalente a los intervalos o las cantidades mencionados en los párrafos anteriores, por ejemplo una forma de dosificación puede contener 10 mg, 25 mg, 50 mg, 100 mg, 150 mg, o 200 mg del compuesto 6-b, 10 mg, 25 mg, 50 mg, 100 mg o 250 mg, del ligando de SV2A, tanto en formulaciones separadas como en una formulación combinada. En una realización, el compuesto 6-b se administra una vez al día (q.d.), en particular como una dosis al día, y el ligando de SV2A se administra una o dos veces al día (q.d. o b.i.d.), en particular como una o dos dosis al día. En el caso de que ambos compuestos deban administrarse una vez al día, esto puede lograrse administrando dos dosis por separado, una con el compuesto 6-b, una con el ligando de SV2A, o administrando una dosis combinada que contiene el compuesto 6-b y el ligando de SV2A.
Las combinaciones de la invención pueden administrarse una, dos, tres, cuatro o, si se desea, múltiples veces al día. En una realización, la combinación se administra una vez al día. En otra realización, la combinación se administra dos veces al día o tres veces al día. La administración de las dosis puede realizarse mediante formas de dosificación separadas, es decir, formas de dosificación que contienen solo el compuesto 6-b o solo el ligando de SV2A; o mediante formas de dosificación combinadas que contienen los principios activos compuesto 6-b y ligando de SV2A. Además, puede usarse una mezcla del uso de una forma de dosificación combinada y de formas de dosificación separadas. Más adelante en la presente se describen las formas de dosificación que pueden administrarse, siendo las preferidas las formas de dosificación orales, en particular los comprimidos o las cápsulas.
Los principios activos pueden formularse en composiciones farmacéuticas o bien por separado o bien como una composición farmacéutica combinada. En el último caso, se proporciona una composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto 6-b, o una sal farmacéuticamente aceptable del mismo, y del ligando de SV2A, siendo el anterior tal como se especifica en la presente, y un portador farmacéuticamente aceptable.
En un aspecto adicional, esta invención se refiere a un proceso para preparar una composición farmacéutica tal como se especifica en la presente, que comprende mezclar de forma íntima un portador farmacéuticamente aceptable con una cantidad terapéuticamente eficaz del compuesto 6-b o un solvato o una sal farmacéuticamente aceptable del mismo, y una cantidad terapéuticamente eficaz de al menos un ligando de SV2A.
Las combinaciones proporcionadas en la presente también pueden formularse como una preparación combinada para su uso simultáneo, secuencial o por separado en la prevención y el tratamiento de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados; en neuroprotección; o en la prevención de la epileptogénesis. En tal caso, el compuesto 6-b se formula en una composición farmacéutica que contiene otros excipientes farmacéuticamente aceptables, y el ligando de SV2A se formula por separado en una composición farmacéutica que contiene otros excipientes farmacéuticamente aceptables. De manera conveniente, estas composiciones farmacéuticas separadas pueden ser parte de un kit para su uso simultáneo, secuencial o por separado.
Los componentes individuales de la combinación de la presente invención pueden administrarse simultáneamente o por separado en diferentes momentos durante el transcurso de una terapia o de forma concurrente en formas de combinación únicas o divididas.
Por tanto, el compuesto 6-b y el ligando de SV2A, individualmente o combinados, pueden formularse en diversas composiciones farmacéuticas adecuadas para fines de administración. En estas, una cantidad terapéuticamente eficaz del compuesto particular, o de los dos compuestos, se combina con un portador farmacéuticamente aceptable, que puede tomar una gran variedad de formas dependiendo de la forma de preparación deseada para la administración. Las composiciones farmacéuticas pueden prepararse como medicamentos que deben administrarse por vía oral, parenteral (incluyendo la vía subcutánea (s.c.), intramuscular (i.m.) e intravenosa (i.v.)), rectal, transdérmica, bucal o nasal. Las composiciones farmacéuticas también pueden prepararse para administrarse directamente al sistema nervioso por vías que incluyen la vía intracerebral, intraventricular, intracerebroventricular, intratecal, intracisternal, intraespinal y/o periespinal mediante la administración con agujas y/o catéteres por vía intracraneal o intravertebral con o sin dispositivos de bombeo. Las composiciones adecuadas para la administración oral incluyen polvos, materiales granulados, agregados, comprimidos, pastillas recubiertas o prensadas, grajeas, sobres, cápsulas duras o de gelatina, jarabes y suspensiones. Las composiciones adecuadas para la administración parenteral incluyen
soluciones o emulsiones acuosas o no acuosas, mientras que para la administración rectal las composiciones adecuadas incluyen supositorios con un vehículo hidrófilo o hidrófobo. Para la administración tópica, pueden usarse sistemas de liberación transdérmica adecuados y, para el suministro nasal, pueden usarse sistemas de liberación de aerosol adecuados.
Por ejemplo, en la preparación de las composiciones para administración oral, puede emplearse cualquiera de los medios farmacéuticos habituales tales como, por ejemplo, agua, glicoles, aceites, alcoholes en el caso de composiciones líquidas orales tales como suspensiones, jarabes, elixires, emulsiones y disoluciones; o portadores sólidos tales como almidones, azúcares, caolín, lubricantes, aglutinantes, agentes disgregantes en el caso de composiciones sólidas. Para las composiciones parenterales, el portador normalmente comprenderá agua estéril, al menos en gran parte, aunque se le pueden añadir otros componentes, tales como solubilizantes, emulsionantes o auxiliares adicionales. Pueden prepararse disoluciones inyectables en las que el portador comprende solución salina, disolución de glucosa o una mezcla de ambas. También pueden prepararse suspensiones inyectables, en cuyo caso pueden emplearse portadores líquidos, agentes de suspensión adecuados. También se incluyen preparaciones en forma sólida previstas para convertirse, poco antes de su uso, en preparaciones en forma líquida tales como polvos para reconstitución. En las composiciones adecuadas para la administración percutánea, el portador comprende opcionalmente un agente potenciador de la penetración en la piel y/o un agente humectante, opcionalmente combinados con aditivos compatibles con la piel adecuados en proporciones minoritarias. El compuesto 6-b o el ligando de SV2A, o combinaciones de los mismos, también pueden administrarse por inhalación o insuflación oral mediante formulaciones adecuadas para este tipo de administración tales como una disolución, una suspensión o un polvo seco. Algunas composiciones farmacéuticas adecuadas para su administración en forma de aerosoles o pulverizadores son, por ejemplo, suspensiones del compuesto 6-b o del ligando de SV2A, o de ambos, en un portador líquido farmacéuticamente aceptable tal como etanol o agua o una mezcla de los mismos. Si se requiere, la formulación también puede contener adicionalmente otros auxiliares farmacéuticos tales como tensioactivos, emulsionantes y estabilizantes, así como un propelente. Una preparación de este tipo contiene el compuesto activo con una concentración de desde aproximadamente el 0.1 hasta el 50%, en particular desde aproximadamente el 0.3 hasta el 3% en peso.
Las composiciones farmacéuticas pueden contener el principio activo del compuesto 6-b o el ligando de SV2A, o ambos combinados en una concentración de aproximadamente el 0.1% a aproximadamente el 50%, o de aproximadamente el 1% a aproximadamente el 30%, o de aproximadamente el 3% a aproximadamente el 20%, o de aproximadamente el 5% a aproximadamente el 20%, estando todos los porcentajes en peso, en las que el total de todos los componentes en dichas composiciones farmacéuticas no excede el 100%. En las composiciones que contienen los dos compuestos, el compuesto 6-b y el ligando de SV2A, el compuesto 6-b está presente en una concentración de aproximadamente el 0.1% a aproximadamente el 50%, o de aproximadamente el 1% a aproximadamente el 30%, o de aproximadamente el 3% a aproximadamente el 20%, o de aproximadamente el 5% a aproximadamente el 20%; y el ligando de SV2A está presente con una concentración de aproximadamente el 3% a aproximadamente el 50%, o de aproximadamente el 5% a aproximadamente el 50%, o de aproximadamente el 10% a aproximadamente el 50%, o de aproximadamente el 10% a aproximadamente el 40%, o de aproximadamente el 10% a aproximadamente el 30%, en las que el total de todos los componentes en dichas composiciones farmacéuticas no excede el 100%.
Las composiciones farmacéuticas pueden presentarse convenientemente en una forma de dosificación unitaria para facilidad de administración y uniformidad de dosificación. Los ejemplos incluyen comprimidos (incluyendo los comprimidos recubiertos o ranurados), cápsulas, pastillas, supositorios, sobres de polvos, obleas, disoluciones o suspensiones inyectables, y múltiplos segregados de los mismos. Son de interés las formas de dosificación sólidas para administración oral tales como comprimidos o cápsulas.
Las formas de dosificación sólidas en una forma de dosis unitaria pueden envasarse en cualquier envase conocido, prefiriéndose los envases de tipo blíster, en particular para comprimidos y cápsulas. Cuando el compuesto 6-b y el ligando de SV2A se formulan por separado, podrían envasarse en blísteres por separado, pero un blíster también podría comprender formas de dosis unitarias del compuesto 6-b y del ligando de SV2A, por ejemplo, una fila con unidades del compuesto 6-b y otra con el ligando de SV2A. También pueden existir otras posibilidades.
Las combinaciones de esta invención pueden usarse para tratar o prevenir la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados; o pueden usarse como un neuroprotector o para prevenir la epileptogénesis.
Tal como se usa en la presente, se pretende que el término "tratamiento" se refiera a todos los procesos en los cuales pueda producirse una ralentización, interrupción, detención o finalización del avance de una enfermedad o un alivio de sus síntomas, pero no indica necesariamente una eliminación total de todos los síntomas.
Tal como se usan en la presente y a menos que se indique lo contrario, los términos "epilepsia y trastornos relacionados" o "epilepsia o trastornos relacionados", significarán cualquier trastorno en el cual un sujeto (preferiblemente un ser humano adulto, niño o bebé) experimenta una o más convulsiones y/o temblores. Los ejemplos adecuados incluyen epilepsia (que incluye epilepsias relacionadas con la localización, epilepsias generalizadas, epilepsias con convulsiones tanto generalizadas como locales), convulsiones de inicio parcial con o sin generalización,
convulsiones mioclónicas, convulsiones tónico-clónicas generalizadas primarias, en particular en pacientes con epilepsia generalizada idiopática, convulsiones asociadas con el síndrome de Lennox-Gastaut, convulsiones como una complicación de una enfermedad o afección (tales como las convulsiones asociadas con una encefalopatía, fenilcetonuria, enfermedad de Gaucher juvenil, epilepsia mioclónica progresiva de Lundborg, accidente cerebrovascular, traumatismo craneal, estrés, cambios hormonales, uso o abstinencia de drogas, uso o abstinencia del alcohol, privación del sueño, fiebre, infección) estados epilépticos (convulsivos o no convulsivos), temblor esencial, síndrome del miembro inquieto. Preferiblemente, el trastorno se selecciona de epilepsia (independientemente del tipo, causa subyacente u origen), temblor esencial o síndrome del miembro inquieto. Más preferiblemente, el trastorno es epilepsia (independientemente del tipo, causa subyacente u origen) o temblor esencial. Un ejemplo particular de epilepsia es la epilepsia refractaria, también denominada epilepsia resistente a la terapia o al tratamiento. Este término se usa a menudo cuando en los pacientes han fracasado tres o más fármacos antiepilépticos (FAE). La epilepsia refractaria también incluye la epilepsia parcial refractaria y la epilepsia generalizada refractaria (incluyendo la idiopática o sintomática).
Tal como se usa en la presente, el término "dolor neuropático", incluye el dolor resultante de estados o trastornos crónicos o debilitantes. Los estados o trastornos crónicos o debilitantes que pueden conducir a dolor neuropático incluyen neuropatía periférica diabética dolorosa, neuralgia posherpética, neuralgia trigeminal, dolor posterior a accidente cerebrovascular, dolor asociado con esclerosis múltiple, dolor asociado con neuropatías tal como el que se produce en mononeuritis y neuropatía postraumática o idiopática, dolor neuropático asociado con VIH, dolor neuropático asociado con cáncer, dolor neuropático asociado con túnel carpiano, dolor asociado con una lesión de la médula espinal, síndrome de dolor regional complejo, dolor neuropático asociado con fibromialgia, dolor cervical y lumbar, distrofia simpática refleja, síndrome del miembro fantasma y síndromes de dolor asociados con otros estados debilitantes y crónicos.
Tal como se usa en la presente, el término "migraña” significará una afección clínica crónica, episódica y debilitante que se diagnostica por la presencia de cefaleas pulsantes unilaterales de moderadas a graves que perduran entre 4 y 72 h, que incluye migraña con aura y migraña sin aura. Tal como se usa en la presente, "migraña sin aura", significará al menos cinco ataques que cumplen los siguientes criterios: (a) el ataque de cefalea perdura durante 4-72 horas, presentando la cefalea al menos dos de las siguientes características: localización unilateral, calidad pulsante, intensidad de moderada a grave con una influencia directa sobre actividades de la vida diaria y que se agrava al subir escaleras o realizar rutinas similares; y (b) durante la cefalea, se produce al menos uno de los siguientes fenómenos: náusea y/o vómitos, fotofobia y fonofobia. Tal como se usa en la presente, "migraña con aura", significará al menos dos ataques acompañados por al menos 3 de las 4 características siguientes: (a) uno o más síntomas de aura completamente reversibles; (b) al menos un síntoma de aura que se desarrolla de forma gradual a lo largo de más de cuatro minutos o dos o más síntomas que se producen de forma sucesiva; (c) ningún síntoma de aura que perdure más de 60 minutos; (d) se produce una cefalea antes, de manera simultánea con o después del aura, con un intervalo libre entre el aura y la cefalea de menos de aproximadamente 60 minutos.
Tal como se usa en la presente, el término "trastornos bipolares y trastornos relacionados" incluirá trastorno bipolar de tipo I (p. ej., episodio maníaco único, episodio hipomaníaco más reciente, episodio maníaco más reciente, episodio mixto más reciente, episodio depresivo más reciente y episodio no especificado más reciente), trastorno bipolar de tipo II, trastorno ciclotímico y trastorno bipolar no especificado de otro modo (tal como estos términos se definen según sus criterios de diagnóstico, en el Diagnostic and Statistical manual of Mental Disorders, 4a edición, revisión de texto, Asociación Psiquiátrica Americana, 2000 (DSM-IV-TR) o en la 5a edición, revisión de texto, Asociación Psiquiátrica Americana, 2013 (DSM-5TM). Preferiblemente, el trastorno bipolar se caracteriza por fases depresivas y maníacas (o hipomaníacas), en el que las fases son cíclicas. Preferiblemente, el trastorno bipolar es un trastorno bipolar de tipo I o un trastorno bipolar de tipo II. Tal como se usa en la presente "manía” debe incluir manía o una fase de estado de ánimo maníaco, independientemente de la causa subyacente. Tal como se usa en la presente, se pretende que el término "manía bipolar" signifique la manía asociada con una característica de o sintomática de un trastorno bipolar. Por tanto, los métodos para tratar la manía bipolar de la presente invención se refieren a métodos que tratan la manía y/o la fase maníaca de los trastornos bipolares. Tal como se usa en la presente, se pretende que el término "depresión bipolar", signifique la depresión asociada con una característica de o sintomática de un trastorno bipolar. Por tanto, métodos para tratar la depresión bipolar de la presente invención se refieren a métodos que tratan la depresión y/o la fase depresiva de los trastornos bipolares. Tal como se usan en la presente, a menos que se indique lo contrario, los términos "ciclo" o "ciclo bipolar" se referirán a la alteración del estado de ánimo entre las fases depresiva y maníaca características de los trastornos bipolares. Por tanto, la presente invención incluye métodos para la estabilización de dicho ciclo, que incluyen la reducción de la frecuencia del ciclo y/o la reducción de la magnitud de las fases maníaca y/o depresiva.
Por tanto, en una realización, la composición farmacéutica de la presente invención puede utilizarse para estabilizar el estado de ánimo, en particular estabilizar el estado de ánimo durante una depresión maníaca.
Tal como se usa en la presente, el término "epileptogénesis", se refiere al proceso gradual mediante el cual se desarrolla la epilepsia. Este proceso puede suceder tras una lesión cerebral o después de una serie de estados, que incluyen enfermedades neurodegenerativas, lesión cerebral traumática, accidente cerebrovascular, tumor cerebral, infecciones del sistema nervioso central, estado epiléptico, o puede suceder después de que se produzcan mutaciones génicas.
Tal como se usa en la presente, el término "ansiedad", se refiere en particular al trastorno de ansiedad generalizado.
Tal como se usa en la presente, el término "aproximadamente", tiene su significado convencional. En realizaciones particulares, cuando se refiere a un valor numérico, puede interpretarse que significa el valor numérico ± 10%, o ± 5%, o ± 2%, o ± 1%, o ± 0.5 % o ± 0.1%. En otras realizaciones, se hace referencia al valor exacto, es decir, no se menciona el término "aproximadamente".
"Y/o" significa que cada uno de o ambos o todos los componentes o características de una lista, son posibles variantes, especialmente dos o más de los mismos de un modo alternativo o acumulativo.
Tal como se usan en la presente, los términos "un/a", "el/la" y términos similares usados en el contexto de la presente invención (especialmente en el contexto de las reivindicaciones), deben interpretarse de manera que abarquen tanto el singular como el plural, a menos que se indique de otro modo en la presente o que el contexto indique claramente lo contrario.
Ejemplos
Los siguientes ejemplos se exponen para ayudar en la comprensión de la invención.
a ) c o m p u e s t o s - q u ím ic a y p r u e b a s in v it r o
A menos que se indique lo contrario, todos los materiales de partida se obtuvieron de proveedores comerciales y se utilizaron sin purificación adicional.
A continuación, “ac.” significa acuoso; “DCE” significa 1,2-dicloroetano, “DCM” significa diclorometano; “DIPE” significa éter diisopropílico; “DIPEA” significa W,W-diisopropiletilamina; “DMF” significa W,W-dimetilformamida; “ES” significa electrospray; “Et3N” significa trietilamina; “Et2Ü” significa éter dietílico; “EtOAc” significa acetato de etilo; “h” significa horas; “HPLC” significa cromatografía líquida de alta resolución; “HRMS” significa espectros/espectrometría de masas de alta resolución; “l” o “L” significa litro; “LRMS” significa espectrometría/espectros de masas de baja resolución; “MeOH” significa metanol; “min” significa minuto(s); “pf” significa punto de fusión; “Pd(PPh3)4” significa tetrakis(trifenilfosfina)paladio (0); “FI” significa fase inversa; “t.a.” significa temperatura ambiente; “s” significa segundos; “sat.” significa saturado; “SFC” significa cromatografía de fluidos supercríticos; “sol.” significa solución; “THF” significa tetrahidrofurano.
Las reacciones asistidas por microondas se realizaron en un reactor monomodal; reactor de microondas Initiator™ Sixty EXP (Biotage AB) o en un reactor multimodal: MicroSYNTH Labstation (Milestone, Inc.).
La cromatografía en capa fina (TLC) se llevó a cabo en placas de gel de sílice 60 F254 (Merck) utilizando disolventes de grado reactivo. La cromatografía en columna abierta se realizó en gel de sílice, tamaño de partículas de 60 Á, malla = 230-400 (Merck) utilizando técnicas estándar. La cromatografía en columna ultrarrápida automatizada se realizó utilizando cartuchos listos para conectar de Merck, en gel de sílice irregular, tamaño de partícula de 15-40 gm (columnas ultrarrápidas desechables de fase normal) en un sistema SPOT o LAFLASH de Armen Instrument.
La configuración estereoquímica absoluta de algunos de los compuestos se determinó utilizando dicroísmo circular vibracional (VCD). Se midieron en un Equinox 55 de Bruker equipado con un PMA 37, en una celda líquida de KBr utilizando CD2Cl2 como disolvente (PEM: 1350 cm-1, LIA: 1 mV, resolución: 4 cm-1). Se puede consultar una descripción del uso de VCD para la determinación de la configuración absoluta en Dyatkin A.B. et. al, Chirality, 14:215-219 (2002).
Siempre que se indique en la presente la notación “RS”, esta indica que el compuesto es una mezcla racémica a menos que se indique lo contrario. La configuración estereoquímica de algunos compuestos se ha designado “R ’ o “S” cuando se ha separado la mezcla. El exceso enantiomérico de los compuestos mostrados en la presente se determinó mediante el análisis de la mezcla racémica mediante cromatografía de fluidos supercríticos (SFC) seguida de comparación por SFC del (de los) enantiómero(s) separado(s).
Preparación de intermedios
Descripción 1 - Intermedio 1
Se disolvió ácido ciclopropilacético ([CAS 5239-82-7], 50 g, 500 mmol) en CH2CL (300 mL) y a continuación, se añadió SOCl2 (100 mL). La mezcla de reacción se agitó a 60 °C durante 2 h y a continuación se evaporó el disolvente para producir el intermedio 1 (53 g, 90%), que se utilizó sin purificación adicional.
Descripción 2 - intermedio 2

A una solución de 2,4-dicloro-3-yodopiridina ([CAS 343781-36-2], 290 g, 1058 mmol) en DMF (1.7 L) se añadió 2,2-difluoro-2-(fluorosulfonil)acetato de metilo ([CAS 680-15-9], 403 g, 2098 mmol) y CuI (403 g, 2.13 mol), a continuación, la reacción se calentó a 100 °C durante 5 h.
La reacción se enfrió y se filtró. El filtrado se diluyó con H2O y se extrajo con Et2O y se lavó con una solución de NH3. La capa orgánica se secó (Na2SO4), se filtró y se concentró al vacío para reducir el intermedio 2 (160 g), que se utilizó sin purificación adicional.
Descripción 3 - intermedio 3

A una solución de NaH (60% en aceite, 24 g, 600 mmol) en DMF (2 L) a 0 °C se añadió alcohol bencílico (35 g, 325 mmol) y a continuación la reacción se agitó durante 2 min. Se añadió el intermedio 2 (160 mg, 741 mmol) en una porción y se agitó a 0 °C durante 1 h. La reacción se diluyó mediante la adición de H2O y se extrajo con Et2O. La capa orgánica se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó mediante columna cromatográfica en gel de sílice (eluyente: éter de petróleo/EtOAc = 20/1). Se recogieron las fracciones puras y se evaporó el disolvente para producir el intermedio 3 (100 g, 38%).
Descripción 4 - intermedio 4

A una solución del intermedio 3 (100 g, 277 mmol) en 1,4-dioxano (1.5 L) se añadió hidrato de NH2NH2 (solución al 85% en agua,300 g, 9.11 mol) y a continuación la reacción se calentó en un tubo sellado a 160° durante 2 h. La mezcla se concentró al vacío, se disolvió en DCM y se lavó con NaHCO3. La capa orgánica se secó (Na2SO4), se filtró y se concentró al vacío para producir el intermedio 4 (90 g, 90%), que se utilizó sin purificación adicional.
Descripción 5 - intermedio 5

A una solución del intermedio 4 (90 g, 318 mmol) en CH2Cl2 (1.5 L) se añadió trietilamina (64.3 g, 636 mmol), la mezcla se enfrió hasta 0 °C y a continuación se añadió una solución del intermedio 1 (53 g, 449 mmol) en CH2G 2. La solución se agitó a TA durante 1 H. La mezcla de reacción se lavó con una sol. ac. sat. de NaHCO3 y se extrajo con CH2G 2. La capa orgánica se secó (Na2SO4), se filtró y se concentró al vacío para producir el intermedio 5 (104.4 g, 90%).
Descripción 6
intermedio 9

A una solución del intermedio 5 (101 g, 277 mmol) en CH3CN (1.2 L) se añadieron oxicloruro de fósforo (V) (84.7 g, 553 mmol) y W,W-diisopropiletilamina (71.3 g, 553 mmol). La mezcla de reacción se agitó a 90 °C durante 38 h. A continuación, la reacción se diluyó con DCM y se lavó con una solución de Na2CO3. La capa orgánica se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en columna en gel de sílice (eluyente: éter de petróleo/EtOAc = 4/1). Se recogieron las fracciones puras y se evaporó el disolvente para producir el intermedio 9 (31.39 g, 41%).
Descripción 7 - intermedio 13

Se añadió (Ph3P)4Pd (2.096 g, 1.81 mmol) a una solución agitada del intermedio 9 (10 g, 36.28 mmol) y 4,4,5,5-tetrametil-2-vinil-1,3,2-dioxoborolano ([CAS 75927-49-0], 7.77 mL, 43.53 mmol) en dioxano desoxigenado (30 mL) y una solución saturada de NaHCO3 desoxigenada (30 mL) en nitrógeno. La mezcla se agitó a 100 °C durante 18 h. La mezcla se diluyó con EtOAc/agua y se filtró a través de un lecho de tierras de diatomeas. El filtrado se trató con salmuera y se extrajo con EtOAc. La capa orgánica se separó, se secó (Na2SO4), se filtró y los disolventes se evaporaron al vacío. El producto crudo se purificó mediante cromatografía en columna ultrarrápida (sílice; EtOAc en CH2G 2 de 0/100 a 5/95). Las fracciones deseadas se recogieron y se concentraron al vacío para producir el intermedio 13 (6.08, 63%) como un sólido amarillo.
Descripción 8
intermedio 17

Se añadieron tetróxido de osmio (2.5% in t-BuOH, 10.103 mL, 0.781 mmol) y a continuación peryodato de sodio 12.53 g, 58.58 mmol) en agua (48.5 mL) a una suspensión del Intermedio 13 (6.08 g, 20.02 mmol) en dioxano (192 mL). La mezcla se agitó a temperatura ambiente durante 2 h.
La mezcla se trató con agua y EtOAc y se filtró a través de un techo de tierra de diatomeas. El filtrado se extrajo con EtOAc. La capa orgánica se separó, se secó (Na2SO4), se filtró y se evaporaron los disolventes al vacío. El producto crudo se lavó con Et2O y se filtró y se secó para producir intermedio 17 (4.25 g, 79%) como un sólido marrón.
Descripción 9
(a) intermedios 21 a, 21b y 21c

intermedio 21a intermedio 21b intermedio 21c
Se añadió bromuro de metilmagnesio (1.4 M en THF, 12.40 mL, 17.37 mmol) gota a gota a una suspensión agitada del intermedio 17 (4.25 g, 15.79 mmol) en THF (281.07 mL) a -20 °C en una atmósfera de N2. La mezcla se agitó a -20 °C durante 45 minutos. El crudo se trató con una sol. sat. de NH4G y se extrajo con EtOAc. La capa orgánica se separó, se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (sílice: MeOH en DCM de 0/100 a 4/96). Se recogieron las fracciones deseadas y se concentraron al vacío para producir intermedio 21a (mezcla racémica) (2.96 g, 66%). El intermedio 21a (1.82 g) se purificó mediante SFC:
[fase estacionaria: CHIRALPAK AD-H (5pm 250 x20 mm), fase móvil: 80% de CO2, 20% de EtOH] para producir 21b (enantiómero R) (0.453 g, 10%) como un sólido gris pálido y el intermedio 21c (enantiómero S) (0.439 g, 10%).
Preparación del compuesto final
Ejemplo 1
(a) Síntesis de los compuestos 4-b, 6-b y 5-b

Compuesto 4-B Compuesto 6-B Compuesto 5-B
Se añadió DIAD (2.07 mL, 10.52 mmol) gota a gota a una solución agitada del intermedio 21a (2 g, 7.01 mmol), 2,4-difluorofenol (1.00 mL, 10.52 mmol) y trifenilfosfina (2.76 g, 10.52 mmol) en THF (74.18 mL) a 0 °C y en una atmósfera de nitrógeno. La mezcla se agitó a 100 °C durante 10 minutos con irradiación de microondas. La mezcla se diluyó con EtOAc y se lavó con una sol. sat. de NaHCO3. La capa orgánica se separó, se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (sílice; MeOH en DCM de 0/100 a 97/3). Las fracciones deseadas se recogieron y concentraron al vacío. El residuo se purificó con DIPE para obtener el compuesto 4-b (1.46 g, 52%) como un sólido blanco, que se purificó mediante SFC quiral [fase estacionaria: Chiralpak AD (5pm 250*30 mm, fase móvil: 85% de CO2, 15% de iPrOH)], lo que produjo el compuesto 6-b (0.659 g, 24%) y el compuesto 5-b (0.693 g, 25%).
(b) Síntesis alternativa del compuesto 6-b

Comp. N° 6-b
Se añadió DIAD (31.06 pL, 0.16 mmol) a una solución agitada del intermedio 21 b (30 mg, 0.11 mmol), 2,4-difluorofenol (15.07 pL, 0.16 mmol) y trifenilfosfina (41.38 mg, 0.16 mmol) en THF (1.11 mL) a 0 °C y en una atmósfera de nitrógeno. La mezcla se agitó a 100 °C durante 10 minutos con irradiación de microondas. La mezcla se diluyó con EtOAc y se lavó con una sol. sal. de NaHCO3. La capa orgánica se separó, se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (sílice; MeOH en d Cm de 0/100 a 97/3). Las fracciones deseadas se recogieron y se concentraron al vacío. El residuo se purificó con DIPE para obtener el compuesto 6-b (40 mg, 96%) como un sólido blanco.
(c) Síntesis de la sal de clorhidrato del compuesto 6-b (HCl)
Se añadió DIAD (207.06 pL, 1.05 mmol) gota a gota a una solución agitada del intermedio 21 b (200 mg, 0.70 mmol), 2,4-difluorofenol (100.45 pL, 1.05 mmol) y trifenilfosfina (275.84 mg, 1.0516 mmol) en THF (4 mL) a 0 °C y en una atmósfera de nitrógeno. La mezcla se agitó a 100 °C durante 15 minutos con irradiación de microondas. La mezcla se diluyó con EtOAc y se lavó con una sol. sat. de NaHCO3. la capa orgánica se separó, se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó mediante RP HPLC (fase estacionaria: C18 XBridge 30 x 100 mm 5 pm, fase móvil: gradiente de un 60% de una solución al 0.1% de NH4CO3H/NH4OH en agua, pH 9, y un 40% de CH3CN a un
43% de una solución al 0.1% de NH4CO3H/NH4OH en agua, pH 9, y un 57% de CH3CN), para generar un residuo sólido blanco que se disolvió en Et2O (8 mL) y 1,4-dioxano (0.5 mL). A la solución obtenida de esa manera se añadió HCl (4M en dioxano, 200 pL) gota a gota. El precipitado sólido blanco se filtró, se lavó con Et2O, se secó (Na2SO4) y se evaporó al vacío. El residuo blanco obtenido de esta manera se purificó con Et2O para obtener el compuesto 6-b-HCl (110 mg, 36%) como un sólido blanco.
Parte analítica
Rotaciones ópticas
Las rotaciones ópticas se midieron en un polarímetro 341 de Perkin-Elmer con una lámpara de sodio y se presentaron de la siguiente manera: [a]° (A, c g/100mL, disolvente, T°C).
[a]AT = (100a) / (l x c): donde l es la longitud de paso en dm y c es la concentración en g/100 mL para una muestra a una temperatura T (°C) y una longitud de onda A (en nm). Si la longitud de onda de la luz utilizada es 589 nm (la línea D de sodio), entonces el símbolo D se puede utilizar en su lugar. Siempre se debe proporcionar el signo de la rotación (+ o -). Cuando se utiliza esta ecuación la concentración y el disolvente siempre se proporcionan en paréntesis después de la rotación. La rotación se indica utilizando grados y no se proporcionan unidades de concentración (se asume que es g/100 mL).
lc m s
Para la caracterización por (LC)MS, se utilizaron los siguientes métodos.
Procedimiento general
La medida por cromatografía líquida de alta resolución (HPLC) se realiza utilizando una bomba LC, un haz de diodos (DAD) o un detector UV y una columna como se especifica en los métodos respectivos. Si fue necesario, se incluyeron detectores adicionales (véase la tabla de los métodos más adelante).
Desde la columna se llevó el flujo al espectrómetro de masas (MS) que se configuró con una fuente de iones a presión atmosférica. La configuración de los parámetros de ajuste está comprendida en las competencias del experto (por ejemplo, intervalo de barrido, tiempo de permanencia...) con el fin de obtener iones que permitan la identificación del peso molecular monoisotópico nominal del compuesto (PM). La adquisición de datos se realizó con el software apropiado. Los compuestos se describen según sus tiempos de retención experimentales (tR) e iones. Si no se especifica de manera diferente en la tabla de los datos, el ion molecular mencionado corresponde a [M+H]+ (molécula protonada) y/o [M-H]- (molécula desprotonada). En caso de que el compuesto no fuera directamente ionizable se especifica el tipo de aducto (es decir, [M+NH4]+, [M+HCOO]-, e tc .) . Para las moléculas con múltiples patrones isotópicos (Br, Cl...), el valor indicado es el obtenido para la masa isotópica más baja. Todos los resultados se obtuvieron con incertidumbres experimentales que son las que se asocian habitualmente con los métodos utilizados. A continuación, “SQD” significa detector de cuadrupolo único, “TA” temperatura ambiente, “BEH” híbrido de sílice/puente de etilsiloxano, “HSS” sílice de resistencia elevada, “DAD” detector de haz de diodos.
Tabla B: Códigos del método de LCMS (flujo expresado en mL/min;
temperatura de la columna (T) en aC: tiempo de ejecución en minutos).
Puntos de fusión
Los valores son valores máximos y se obtienen con las incertidumbres experimentales que están asociadas habitualmente con este método analítico.
Aparato FP 81HT/FP90 de Mettler
Se determinaron los puntos de fusión para varios compuestos en tubos capilares abiertos en un aparato FP81HT/FP90 (Mettler-Toledo). Los puntos de fusión se midieron con un gradiente de temperatura de 1, 3, 5 o 10 °C/minuto. La temperatura máxima fue de 300 °C. El punto de fusión se leyó en una pantalla digital.
Tabla C: Patos fisicoquímicos. tiempo de retención (tR) en min. pico [M+H1+ (molécula protonada), método LCMS v pf (punto de fusión en aC), (n.d. = no determinado).

s fc -ms
Procedimiento general
La medida de SFC se realizó utilizando un sistema analítico de Berger instruments que comprendía un módulo de control del fluido con bomba dual FCM-1200 para suministrar dióxido de carbono (CO2) y un modificador, un mostrador de líquidos automático de CTC Analytics, un módulo de control térmico TCM-20000 para calentar la columna desde la temperatura ambiente hasta 80 °C. Se utilizó un detectorde matriz de fotodiodos 1100 UV de Agilent equipado con una celda de flujo a presión elevada que soporta hasta 400 bar. Se dividió el flujo desde la columna hacia un espectrómetro de MS. Se configuró el detector de MS con una fuente de ionización a presión atmosférica. Los siguientes parámetros de ionización para el espectrómetro de masas ZQ de Waters son: corona: 9 pa, temp de la fuente: 140 °C, cono: 30 V, temp de la sonda 450 °C, extractor 3 V, gas de desolvatación 400 L/h, gas el cono 70 L/h. Se utilizó nitrógeno como el gas nebulizador. La adquisición de datos se realizó con un sistema de datos Micromass MassLynx-Openlynx de Waters.
Método 1: Además del procedimiento general: La separación quiral analítica en SFC-MS se llevó a cabo en una columna CHIRALPAK AD DAICEL (10 pm, 4.6 x 250 mm) a 35 °C con una tasa de flujo de 3.0 mL/min. La fase móvil es un 85% de CO2, 15% de iPrOH (+0.3% de iPrNH2) que se mantiene 7 min en modo isocrático.
Tabla P: Patos de SFC analítica - tR significa tiempo de retención (en minutos), [M+H]+ significa una masa protonada del compuesto, el método se refiere al método utilizado para el análisis de SFC/MS de los compuestos enantioméricamente puros. La medida se comparó con la mezcla.

Resonancia magnética nuclear (RMN)
Se registraron los espectros de 1H RMN en un espectrómetro DPX-400 de Bruker con secuencias de pulso estándar, que funcionaba 400 MHz y 500 MHz, respectivamente. Los desplazamientos químicos (5) se indican en partes por millón (ppm) en campo bajo respecto al tetrametilsilano (TMS), que se utilizó como patrón interno.
Comp, n,26-b: 1H RMN (400 MHz, CDCI3) 5 ppm 0.30 - 0.38 (m, 2 H), 0.59 - 0.68 (m, 2 H), 1.14 - 1.22 (m, 1 H), 1.72 (d, J=6.5 Hz, 3 H), 3.02 - 3.14 (m, 2 H), 5.84 (c, J=6.3 Hz, 1 H), 6.67 - 6.73 (m, 1 H), 6.80 - 6.89 (m, 2 H), 7.30 (d, J=7.4 Hz, 1 H), 8.11 (d, J=7.4 Hz, 1 H)
Pruebas in vitro
Los compuestos proporcionados en la presente invención son moduladores alostéricos positivos de mGluR2. Parece que estos compuestos potencian las respuestas ante glutamato al unirse a un sitio alostérico que no es el sitio de unión del glutamato. La respuesta de mGluR2 a una concentración de glutamato aumenta cuando los compuestos están presentes. Se espera que el compuesto 6-b tenga su efecto sustancialmente en mGluR2 en virtud de su capacidad para potenciar la función del receptor. Los efectos de los moduladores alostéricos positivos estudiados en mGluR2 utilizando el método del ensayo de unión de [35S]GTPy S descrito más adelante y que es adecuado para la identificación de tales compuestos y más especialmente el compuesto 6-b, se muestran en la Tabla E.
Ensayo de unión de [35S1GTPy S
El ensayo de unión de [35S]GTPy S es un ensayo basado en membrana funcional utilizado para estudiar la función del receptor acoplado a la proteína G (GPCR) en el cual se mide la incorporación de una forma no hidrolizable de GTP, [35S]GTPy S (guanosina 5’-trifosfato, marcado con 35S que emite radiación gamma). La subunidad a de la proteína G cataliza el intercambio de guanosina 5’-difosfato (GDP) por guanosina trifosfato (GTP) y tras la activación de GPCR por un agonista, [35S]GTPy S, se incorpora y no se puede escindir para continuar el ciclo de intercambio (Harper (1998) Current Protocols in Pharmacology 2.6.1-10, John Wiley & Sons, Inc.). La cantidad de incorporación de [35S]GTPy S radioactivo es una medida directa de la actividad de la proteína G y, por lo tanto, se puede determinar la actividad del agonista. Se muestra que los receptores de mGlu2 se acoplan preferencialmente a la proteína Gai, un acoplamiento preferencial para este método, y por lo tanto se utilizan ampliamente para estudiar la activación del receptor de los receptores de mGlu2 tanto en líneas celulares recombinantes es como en tejidos. En este documento describimos el uso del ensayo de unión de [35S]GTPy S utilizando membranas de células transfectadas con el receptor mGlu2 y adaptadas de Schaffhauser et al. (Molecular Pharmacology, 2003, 4:798-810) para la detección de las propiedades de modulación alostérica positiva (PAM) de los compuestos de esta invención.
Preparación de la membrana
Las células CHO se cultivaron hasta preconfluencia y se estimularon con butirato 5 mM durante 24 h. A continuación, se recogieron las células raspando en PBS y la suspensión celular se centrifugó (10 min a 4000 RPM en una centrífuga de sobremesa). El sobrenadante se desechó y el sedimento se resuspendió con cuidado en Tris-HCl 50 mM, pH 7.4 mezclando en un vórtex y pipeteando arriba y abajo. La suspensión se centrifugó a 16,000 RPM (rotor Sorvall RC-5C plus SS-34) durante 10 minutos y se desechó el sobrenadante. El sedimento se homogeneizó en Tris-HCl 5 mM, pH 7.4 utilizando un homogeneizador ultra-turrax y se centrifugó de nuevo (18,000 RPM, 20 min, 4 °C). El sedimento final se resuspendió en Tris-HCl 50 mM, pH 7.4 y se almacenó a -80 °C en alícuotas apropiadas antes de su uso. Se determinó la concentración de proteínas mediante el método de Bradford (Bio-Rad, EE. UU.) con albúmina de suero bovino como patrón.
Ensayo de unión de f35S1GTPvS
La medida de la actividad moduladora alostérica positiva de mGluR2 de los compuestos de prueba se realizó de la siguiente manera. Los compuestos de prueba y el glutamato se diluyeron en tampón de ensayo que contenía ácido HEPES 10 mM, sal HEPES 10 mM, pH 7.4, NaCl 100 mM, MgCl2 3 mM y GDP 10 pM. Se descongelaron las membranas que contenían el receptor de mGlu2 humano en hielo y se diluyeron en tampón de ensayo suplementado con 14 pg/mL de saponina. Las membranas se preincubaron con el compuesto solo o junto con una concentación predefinida (~CE2ü) de glutamato (ensayo PAM) durante 30 min a 30 °C. Después de la adición de [35S]GTPy S (c.f.
0.1 nM), las mezclas ensayo se agitaron brevemente y se incubaron más tiempo para permitir la incorporación de [35S]GTPy S tras la activación (30 minutos, 30 °C). Las mezclas del ensayo finales contuvieron 7 pg de la proteína de la membrana en ácido HEPES 10 mM, sal HEPES 10 mM, pH 7.4, NaCl 100 mM, MgCl23 mM, GDP 10 pM y 2 pg/mL de saponina. El volumen de reacción total fue de 200 pL. Las reacciones se hicieron terminar mediante filtración rápida a través de placas Unifilter-96 GF/B (Perkin Elmer, Massachusetts, EE. UU.) utilizando un recolectador Filtermate Universal de 96 pocillos. Los filtros se lavaron 6 veces con_NaH2PO4/10 mM/Na2HPO410 mM, pH 7.4. A continuación, los filtros se secaron con aire, y se añadieron 40 pL de cóctel de centelleo líquido (Microscint-O) a cada pocillo. La radiactividad unida a la membrana se contó en un contador de centelleo y luminiscencia de microplacas de Perkin Elmer.
Análisis de los datos
Las curvas de concentración-respuesta de los compuestos representativos de la presente invención (obtenidos en presencia de CE20 de glutamato agonista de mGluR2 para determinar la modulación alostérica positiva (PAM) se generaron utilizando la interfaz del software Lexis (desarrollado en J&J). Los datos se calcularon como % de la respuesta de glutamato de control, definida como la respuesta máxima que se genera tras la adición de gluamato solo. Las curvas sigmoideas de concentración-respuesta que representaron gráficamente estos porcentajes frente al log de
concentración del compuesto de prueba se analizaron utilizando análisis de regresión no lineales. A continuación, se calculó la concentración que produce la mitad del efecto máximo como CE50.
Los siguientes valores de pCE50 se calcularon como el -log CE50, cuando la CE50 se expresa en M. El Emáx se define como el efecto máximo relativo (es decir, el % de efecto máximo respecto a la respuesta de glutamato de control). La siguiente Tabla E muestra los datos farmacológicos obtenidos para el compuesto 6-b y los datos farmacológicos reales obtenidos para los compuestos de referencia.
Tabla E: Datos farmacológicos para Ios compuestos de acuerdo con la invención.

Todos los compuestos se estudiaron en presencia de glutamato agonistas de mGluR2 con una concentración CE20 predeterminada, para determinar la modulación alostérica positiva. Se calcularon los valores de pCE50 a partir de un experimento de concentración-respuesta de al menos 8 concentraciones.
B) e s tu d io s ANTICONVULSIVOS y e s tu d io s DE r e f e r e n c ia co n c o m p u e s t o s DE mGluR2 (a g o n is ta o r t o s t é r ic o y PAM)
Generalidades
Preparación de compuestos y disoluciones de prueba
Los compuestos de prueba se administraron usando una razón óptima de volumen de líquido con respecto a líquido corporal. Se administraron los compuestos de prueba a ratones a un volumen de 0.01 mL/g de peso corporal (White, H.S., et al., General principles: Experimental selection, quantification, and evaluation of antiepileptic drugs, in Antiepileptic Drugs, cuarta edición, R. H. Levy, R.H. Mattson, and B. S. Meldrum, Editors. 1995, Raven Press, Ltd.: Nueva York. págs. 99-110). Para la administración subcutánea (s.c.), se administraron los compuestos de prueba en un pliegue de piel laxo a lo largo del dorso del animal excepto el compuesto 6-b, que se administró por vía oral (p.o). Para cada uno de los ensayos realizados con los compuestos de prueba (excepto para el compuesto 6-b), se administraron concentraciones de compuesto finales como disolución acuosa en Hp-p-CD al 20%. Para el compuesto 6-b, primero se preparó una disolución madre de Hp-p-CD al 40% y se usó para formular el compuesto 6-b a las concentraciones deseadas para los ensayos mediante la vía oral; se administraron concentraciones de compuesto finales como disolución acuosa en Hp-p-CD al 20%. Para los grupos de vehículo, se usó una disolución de Hp-p-CD al 20%.
Para el compuesto de referencia LY-404039, se administraron concentraciones de compuesto finales como una solución salina s.c.
Para el compuesto de referencia CAS 1092453-15-0, se administraron concentraciones de compuesto finales en vehículo de Hp-p-CD al 10% (+NaCl) tras la disolución.
Las concentraciones finales de levetiracetam se administraron en una disolución acuosa de metilcelulosa (MC) al 0.5%, que se administró mediante una inyección intraperitoneal (i.p.).
Reactivos críticos
a) Dioluciones de vehículo
Metilcelulosa (MC) al 0.5%
Disolución madre de hidroxipropil-p-ciclodextrina (Hp-p-CD) al 40%
b) Dioluciones diversas
Se añadió tetracaína (disolución al 0.5% p/v) gota a gota a partir de una botella de plástico con cuentagotas a los ojos de todos los animales que recibirían posteriormente una estimulación eléctrica mediante electrodos corneales.
Animales y cría de animales
Se obtuvieron ratones albinos CF n.° 1 macho adultos (26-35 g) de Charles River, Portage, Michigan. Los animales siguieron una dieta adecuada (Prolab RMH 3000) y se les permitió acceso libre a comida y agua, excepto en el tiempo breve durante el que se retiraron de su jaula para las pruebas. Se permitió que los animales recién recibidos en el laboratorio se tomaran el tiempo suficiente para corregir posibles restricciones de comida y agua que hubieran tenido lugar durante el tránsito antes de emplearse en los ensayos. Todos los ratones se acomodaron en jaulas de plástico en salas especialmente construidas con humedad controlada, intercambio de aire e iluminación controlada (12 horas encendida - 12 horas apagada). Los animales se acomodaron, alimentaron y manipularon según las recomendaciones de la Publicación del Consejo Nacional "Guide for the Care and Use of Laboratory animals".
Alteración motora mínima (MMI, por sus siglas en inglés):
Se evaluó la MMI aguda mediante una combinación de observaciones directas del animal en busca de síntomas manifiestos de la función muscular o neurológica del animal. En ratones, se usó el procedimiento Rotarod para describir la deficiencia muscular o neurológica mínima. Cuando se coloca un ratón en un rodillo que rota a una velocidad de 6 rpm, el animal puede mantener su equilibrio durante periodos de tiempo prolongados. Se consideró que el animal estaba intoxicado si se caía de su rodillo giratorio tres veces durante un periodo de 1 min.
Determinación de la mediana de la dosis eficaz y tóxica (DEsn y DTsn)
En la determinación de una DE50 o DT50 para cada compuesto de prueba, la primera dosis administrada es habitualmente la misma dosis que la que se usó en una determinación de TPE exitosa. Cuando la dosis inicial empleada era eficaz o tóxica en más del 50% de los animales, la siguiente dosis sería la mitad de la dosis inicial; si la dosis inicial empleada era eficaz o tóxica en menos del 50% de los animales, la siguiente dosis sería el doble de la dosis inicial. Se seleccionaron terceras y cuartas dosis para producir una línea de respuesta a la dosis uniformemente espaciada. Debería haber un mínimo de 4 puntos o bien incluyendo o bien que se encuentran entre el 0 y el 100%.
Determinación de TPE:
Se administraron compuestos de prueba a grupos generalmente cada uno de cuatro animales y cada grupo se sometió a prueba en uno de cinco puntos de tiempo: 0.25, 0.5, 1,2, o 4 h tras el tratamiento (White et al. 1995). Se determinó el TPE usando el ensayo de 6 Hz (32 mA). Se consideró que el tiempo (0.25, 0.5, 1,2 o 4 h tras el tratamiento) en el que se observó una protección máxima era el tiempo del efecto máximo (TPE, por sus siglas en inglés).
Se sometieron a prueba los compuestos en el ensayo de 6 Hz (32 y/o 44 mA) en el TPE determinado para este estudio, o determinado previamente, para varias dosis, que comprendían dosis que provocaban desde una protección escasa o nula hasta una protección total.
Se calcularon la DE50 y el intervalo de confianza del 95% (IC) usando el análisis Probit en un programa informático proporcionado en el laboratorio (Finney “Probit Analysis” 34d DE 1971, Londres: Cambridge University Press).
Recogida de suero para el análisis pK/pD
En diversas pruebas, se sacrificaron los animales tras los ensayos, y se recogió sangre del torso y/o tejido cerebral (cerebros enteros) para la cuantificación de los niveles de fármaco. Inmediatamente después de los ensayos, los animales se decapitaron y se recogió sangre del torso en un tubo BD Vacutainer® que contenía K2EDTA y se enfrió sobre hielo hasta la centrifugación. Tras la centrifugación (13000-18000 rpm, 5-7 min), se retiró el plasma, se transfirió a un tubo para microcentrífuga etiquetado y se conservó a -80 °C. Para la recogida del tejido cerebral, se retiraron los cerebros inmediatamente tras la decapitación y se congelaron de forma instantánea. Se colocó la muestra congelada en un tubo para centrífuga etiquetado y se conservó a -80 °C.
Prueba de convulsión psieomotora de 6 Hz en ratones
Se usa la prueba de convulsión de 6 Hz como modelo de convulsiones límbicas farmacorresistentes. La convulsión de 6 Hz presenta resistencia a fenitoína, carbamazepina, lamotrigina, y topiramato (Barton et al. "Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy" Epilepsy Research 2001, vol.47, págs. 217-222).
Método para la prueba de convulsión psicomotora de 6 Hz
Se indujeron convulsiones focales en ratones mediante estimulación corneal (6 Hz, pulso rectangular de 0.2 ms, duración de 3 s; Barton et al. 2001). Se sometieron a prueba los ratones o bien a 32 mA o bien a 44 mA. Antes de la estimulación, se aplicaron gotas con tetracaína al 0.5% a cada ojo. Las convulsiones provocadas por la estimulación corneal en este ensayo se caracterizan por una fase clónica mínima seguida de comportamientos automatísticos estereotipados, que incluyen aturdimiento, espasmos de las extremidades delanteras, contracción de las vibrisas y cola de Straub. Se consideró que los animales que no presentaban estos comportamientos estaban protegidos.
e je m p lo i - e s tu d io s co n lo s c o m p u e s t o s de r e fe r e n c ia 1 y 2
1.1. Estudio de combinación con el Comp. de referencia n.21, Comp. de referencia n.22 y levetiracetam
En primer lugar, se sometió a prueba cada compuesto individualmente a una dosis que presentó actividad mínima en la prueba de 6 Hz, 44 mA en cada TPE del compuesto. Cuando se administraron los compuestos de PAM de mGluR2 y levetiracetam en combinación (mismas dosis y puntos de tiempo que en los ensayos indviduales) se observó protección casi completa en la prueba de 6 Hz, 44 mA (tabla 2). Además de registrar los datos de eficacia y toxicidad para estos compuestos solos o en combinación, se recogieron tanto muestras de plasma como de cerebro de cada uno de los groupos para análisis farmacocinéticos/farmacocinética. No se observó interacción farmacocinética basándose en los niveles de compuesto en las muestras de plasma y cerebro (datos no mostrados). En resumen, los compuestos de referencia 1 y 2 presentaron interacción farmacocinética positiva con levetiracetam en el modelo de 6 Hz que no parece deberse a la interacción farmacocinética, y sin alteración motora creciente (tablas 2, 2a, 2b). También se sometió a prueba el efecto de 1 dosis de compuesto de referencia 2 en la respuesta a la dosis de LEV. Tal como se muestra en la tabla 3, hubo un desplazamiento de ~200 veces en la DE50 de LEV en comparación con cuando se sometió a prueba LEV solo. Parecía que LEV aumentaba la potencia del Comp. de referencia n.° 2 ligeramente (tabla 3).
1.2. Análisis isobolográfico de las interacciones entre el Comp. de referencia n.2 1 y levetiracetam en el modelo de convulsiones de 6 Hz
Se llevaron a cabo estudios isobolográficos para la administración combinada del Comp. de referencia n.° 1 con LEV en el ensayo de 6 Hz (44 mA). Se llevaron a cabo estudios según los métodos previamente descritos (Madsen et al. 2011). se determinaron los valores de DE50 iniciales tanto para el Comp. de referencia n.° 1 como para LEV y se usaron para calcular los valores de DE50 (+ error estándar de la media, EEM) para tres combinaciones de razón de dosis fija (LEV: Comp. de referencia n.° 1): 1:3, 1:1, y 3:1. Las dosis usadas eran proporcionales a los valores de DE50 calculados. Por ejemplo, la razón de dosis usada para el paradigma 1:1 se basó en 0.5 x DE50 para LEV y 0.5 x DE50 para el Comp. de referencia n.° 1. De manera similar, el paradigma 1:3 usó 0.25 x DE50 para LEV y 0.75 x DE50 para el Comp. de referencia n.° 1. La razón de dosis 3:1 usó 0.75 x DE50 de LEV y 0.25 x DE50 para el Comp. de referencia n.° 1. Las dosis de tratamiento experimentales (véase la tabla 4) se basaron en los valores teóricos y se ajustaron según los efectos observados. Los valores de DE50 determinados experimentalmente (+ EEM) para cada combinación de razón de dosis fija se compararon con los valores teóricos (prueba de la t) para fines estadísticos. Se determinó que la razón de dosis era supra-aditiva (sinérgica) si el valor de DE50 determinado experimentalmente era significantivamente más bajo que la DE50 teórica. Posteriormente, se determinaron las dosis combinadas experimentales para los mismos paradigmas en la prueba de convulsión de 6 Hz (tabla 4 a continuación). El estudio isobolográfico con el compuesto 1 y levetiracetam en el modelo de 6 Hz demostró una interacción farmacodinámica sinérgica significativa en todas las razones de dosis evaluadas y se corresponde estrechamente con los niveles en plasma del compuesto de referencia 1. Además, no se observó alteración motora en ninguna de las razones de dosis evaluadas sugiriendo que la interacción de farmacodinámica sinérgica no produce una toxicidad motora aumentada.
1.3. Modelo de estimulación corneal de ratón y estudios con el compuesto de referencia 1
Se estimularon electricamente ratones con un estímulo de 3 segundos, 3 mA, 60 Hz, dos veces al día usando electrodos corneales hasta un criterio de 5 convulsiones consecutivas de estadio 5 tal como se definió por Racine (Racine "Modification of seizure activity by electrical stimulation" II. motor seizure" Electroenceph Clin Neurophysiol 1972, 32, págs. 281-294). Después de que los ratones alcanzasen un estado estimulado estable, se administró el compuesto o vehículo de prueba y, en el TPE determinado previamente, se aplicó a cada animal el estímulo eléctrico indicado anteriormente. Tras la estimulación, se observaron los animals para determinar la presencia o ausencia de la actividad de convulsión alcanzada en la escala de Racine (0 - 5) representando 5 el estado más alto irguiéndose y cayendo. Se sometieron a prueba una dosis de LEV y dos dosis del Comp. de referencia n.° 1 individualmente y en combinación contra convulsiones corneales estimuladas. La combinación del compuesto de referencia 1 con levetiracetam en este modelo sugirió una interacción de farmacodinámica positiva (tabla 5 a continuación).
Se presenta un resumen de los datos para los compuestos sometidos a prueba solos en la tabla 1 y se enumeran resultados adicionales de estudios realizados según el ejemplo 1 en las tablas 2-5 a continuación.
Tabla 1: Resumen de Ios datos anticonvulsivos agudos en el modelo de 6 Hz a 32 mA y 44 mA para Ios compuestos de PAM de mGluR21 (referencia), 2 (referencia), 11 (referencia), 2-a (referencia), 25-a (referencia), 6 b y compuesto de referencia LY-404039 tras la administración s.c. (excepto el compuesto 6-b, que se sometió a prueba por vía p.o.). TPE significa tiempo del efecto máximo, IC significa intervalo de confianza, s.c. significa subcutáneo, p.o. significa por vía oral, n.t. significa no sometido a prueba. Se determinó el TPE en la prueba de 6 Hz, 32 mA. Generalmente se observan los efectos a dosis que no producen alteración en la prueba de Rotarod. Para los compuestos 11 y 2-a se proporcionan los valores individuales de experimentos de repetición. Para el compuesto de referencia 25a, se usaron ambos puntos de tiempo de 0.25 y de 1 h para los estudios de 6 Hz (44 mA).

Tabla 1a: Resumen de la determinación del TPE a 6 Hz, 32 mA para el Comp. de referencia n.21
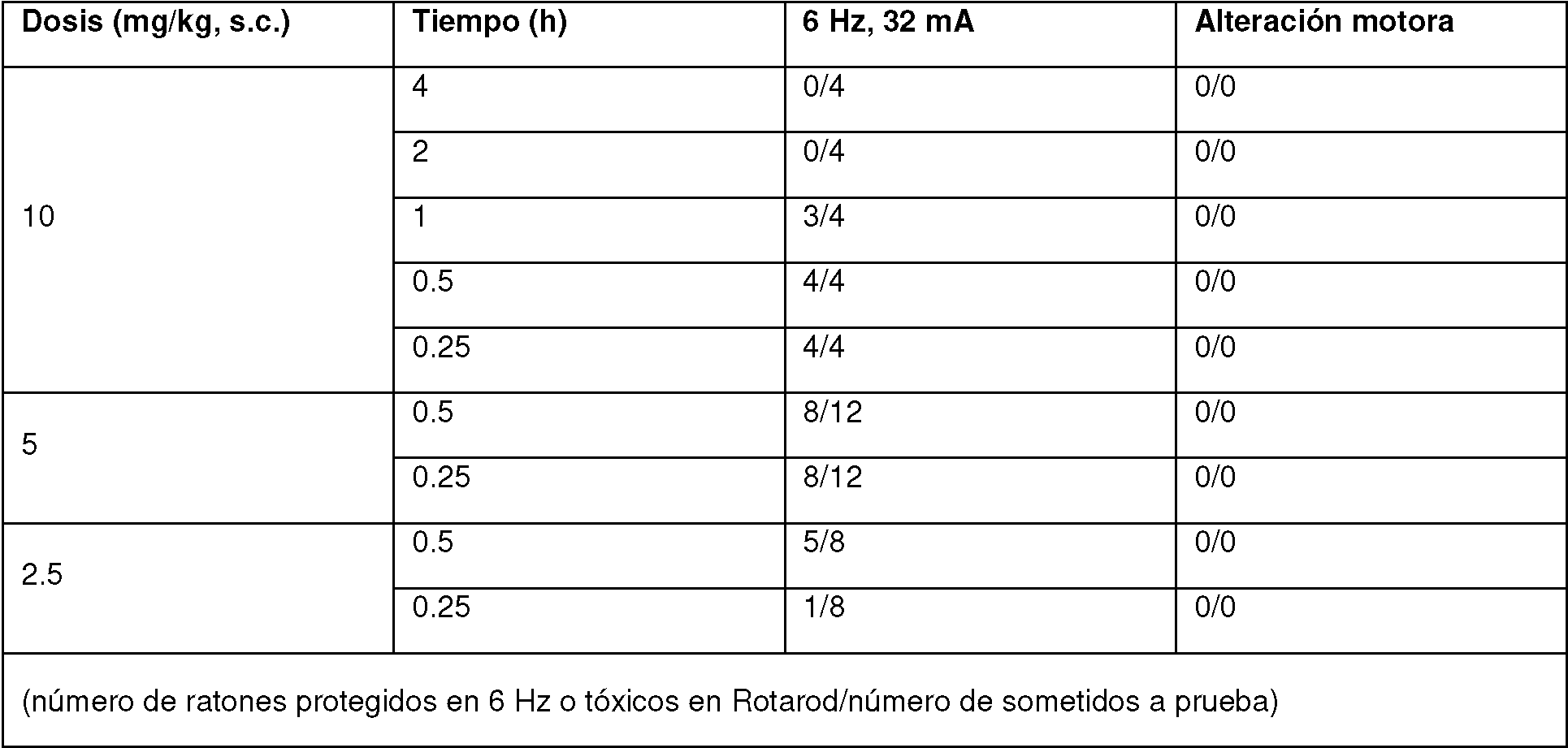
Tabla 1b: Estudios de dosis-respuesta para el Comp. de referencia n.21. Se determinó previamente que el TPE para el Comp. de referencia n.° 1 era de 0.5 h (resultados mostrados anteriormente en la tabla 1a). Se administraron varias dosis del Comp. de referencia n.° 1 en este TPE y se sometieron a prueba en el ensayo de 6 Hz, usando intensidades de estímulos tanto de 32 como de 44 mA.

Tabla 2: Resumen de la interacción del Comp. de referencia n.21 y el Comp. de referencia n.22 con levetiracetam (LEV) en el modelo de convulsiones de 6 Hz, 44 mA de ratón. Los resultados se enumeran como número de ratones que presentan protección completa/número total de ratones sometidos a prueba en cada grupo de dosificación (en los niveles de dosificación de combinación o compuesto de prueba especificados).

Tabla 2a: Niveles en plasma y cerebro del Comp. de referencia n.21. en el estudio de combinación con levetiracetam (LEV). BQL significa por debajo del límite cuantificable.
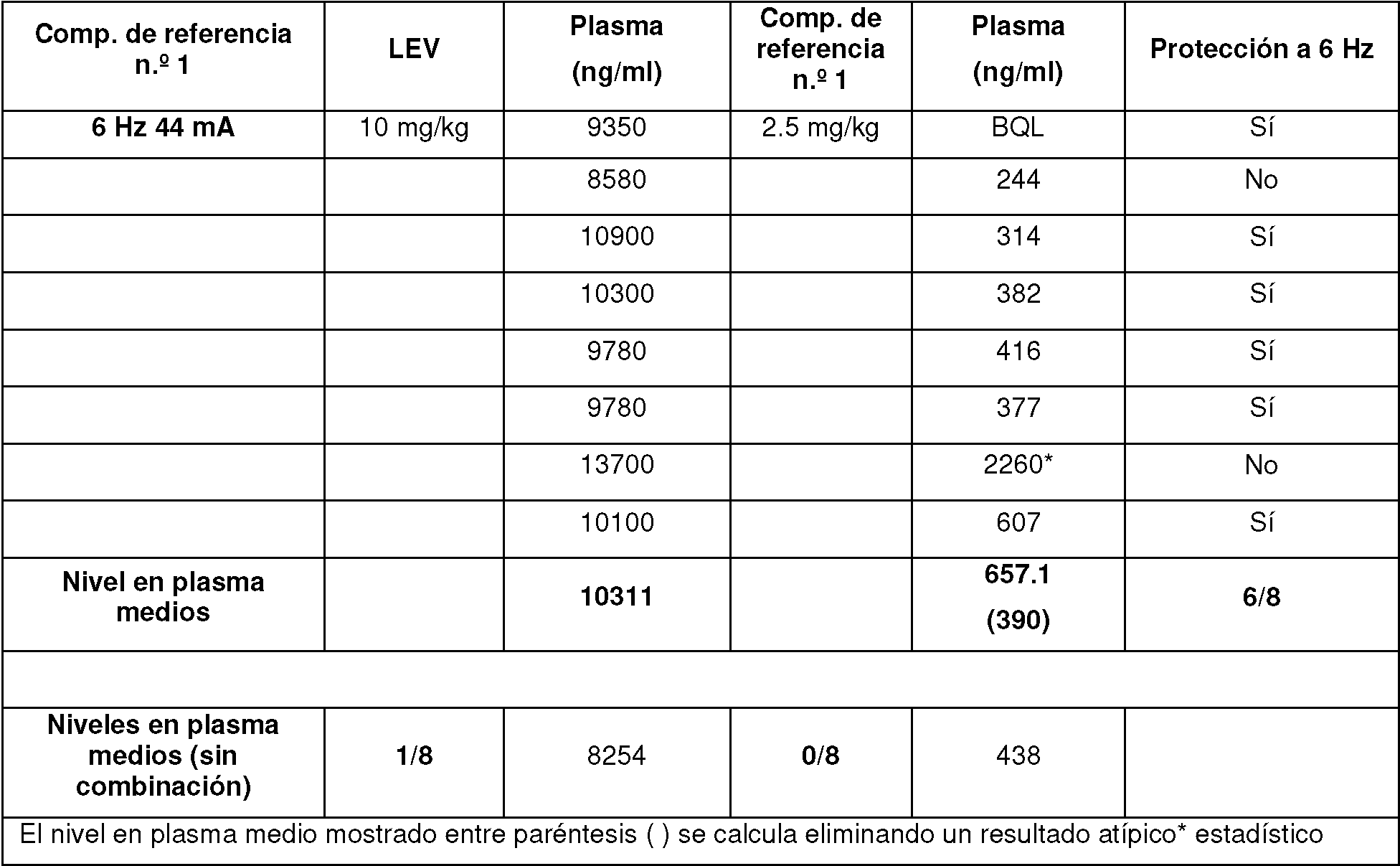
Tabla 2b: Niveles en plasma y cerebro del Comp. de referencia n.22 en el estudio de combinación con levetiracetam (LEV). AQL significa por encima del límite cuantificable.

Tabla 3: Determinaciones de DE50 en el modelo de convulsiones de 6 Hz (44 mA) para el Comp. de referencia n.22 y levetiracetam (LEV) solos y en combinación. LEV a una dosis de 10 mg/kg aumentó la potencia del Comp. de referencia n.° 2 (desplazamiento de ~5 veces en la DE50). Comp. de referencia n.° 2 a una dosis de 3 mg/kg aumentó tanto la eficacia (hasta el 100% de protección) como la potencia de LEV (desplazamiento de ~200 veces en la DE50). La figura 1 muestra la respuesta a la dosis para las determinaciones de DE50 a 6 Hz, 44 mA para el Comp. de referencia n.° 2 y LEV solos y en combinación.

Tabla 4: Resultados del Comp. de referencia n.21 y levetiracetam en el estudio isobolográfico.

El análisis isobolográfico (figura 2) demuestra que la combinación del Comp. de referencia n.° 1 y levetiracetam da como resultado un efecto sinérgico significativamente positivo.
Tabla 5: Resultados del estudio de combinación del Comp. de referencia n.21 y levetiracetam en el modelo de estimulación corneal en ratones.5*10

EJEMPLO 2 - ESTUDIOS CON LOS COMPUESTOS DE REFERENCIA 25-a Y 2-a
2.1. Estudio de combinación con el Comp. de referencia n.225-a y levetiracetam
Se realizaron estudios de respuesta a la dosis independientes en la prueba de 6 Hz, 44 mA para ambos compuestos para determinar los valores de DE50 en el TPE de 1 h i.p. para levetiracetam y de 1 h s.c. para el Comp. de referencia n.° 25-a. El valor de DE50 para el Comp. de referencia n.° 25-a era de 25.9 mg/kg y para levetiracetam se estimó que era de aproximadamente 345 mg/kg. Se repitió la respuesta a la dosis para levetiracetam con coadministración de 10 mg/kg del Comp. de referencia n.° 25-a (una dosis del Comp. de referencia n.° 25-a que sola no protegió en el modelo de 6 Hz, 44 mA). La coadministración de 10 mg/kg del Comp. de referencia n.° 25-a produjo una DE50 en la respuesta a la dosis de levetiracetam de 4.9 mg/kg (~70 veces inferior en comparación con levetiracetam solo) y proporcionó de manera importante una protección completa en el modelo de convulsiones de 6 Hz, 44 mA. Estos resultados sugieren que existe una interacción farmacodinámica positiva en el modelo de convulsiones de 6 Hz entre el Comp. de referencia n.° 25-a y levetiracetam.
Tabla 6: Determinación del tiempo para el efecto máximo para el Comp. de referencia n.225-a en el ensayo de 6 Hz (32 mA). Se usaron dos dosis en este estudio, de 10 y 20 mg/kg, a lo largo de varios puntos de tiempo (0.25 - 4 h). El compuesto mostró el mayor grado de protección en el ensayo de 6 Hz entre 0.25 y 1 h, que era más evidente a 20 mg/kg. Niveles en plasma del compuesto generalmente correspondian a la protección frente a la convulsión del comportamiento. Se usó un TPE de 0.25 h para los estudios de 6 Hz (32 mA) mientras que se usaron los puntos de tiempo tanto de 0.25 como de 1 h para los estudios de 6 Hz (44 mA).

Tabla 7: Estudios de respuesta a la dosis para el Comp. de referencia n.225-a en el ensayo de 6 Hz (32 mAa y 44 mAb)


Tabla 8: Estudios de combinación para el Comp. de referencia n.225-a con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA).5*10

2.2. Estudios de combinación con el Comp. de referencia n.22-a y levetiracetam
Se realizaron estudios de respuesta a la dosis en las pruebas de 6 Hz, 32 mA y 44 mA (tabla 9 a continuación) y en la prueba de combinación con levetiracetam (efecto del Comp. de referencia n.° 2-a sobre la respuesta a la dosis de LEV en la tabla 10a y el efecto de LEV sobre la respuesta a la dosis del Comp. de referencia n.° 2-a en la tabla 10b a continuación) de la misma manera que se describió para los estudios con el Comp. de referencia n.° 25-a y levetiracetam anteriormente.
Tabla 9: Estudios de respuesta a la dosis para el Comp. de referencia n.22-a en el ensayo de 6 Hz (32 mAa y 44 mA; 0.5 h de TPE) Se determinó un tiempo para el efecto máximo de 0.5 h en la prueba de 6 Hz, 32 mA (s.c.) y se usó para los estudios de 6 Hz (32 mA y 44 mA).

Tabla 10a: Estudios de combinación para el Comp. de referencia n.22-a con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA). Combinación de 10 mg/kg del Comp. de referencia n.° 2-a con dosis variables de levetiracetam.


Tabla 10b: Estudios de combinación para el Comp. de referencia n.22-a con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA). combinación de 350 mg/kg de levetiracetam con dosis variables del Comp. de referencia n.22-a.

A una dosis de 10 mg/kg s.c., el Comp. de referencia n.° 2-a aumenta la potencia de LEV, llevando a un desplazamiento de aproximadamente 35 veces en la DE50. Esto sugiere una relación farmacodinámica positiva (tabla 10a). A una dosis de 350 mg/kg i.p., LEV aumenta la potencia del Comp. de referencia n.° 2-a, conduciendo a un desplazamiento de aproximadamente 14 veces en la DE50. Esto sugiere una relación de farmacodinámica positiva (tabla 10b).
e je m p lo 3 - e s tu d io s CON EL c o m p u e s to 6-b
3.1. Estudio de combinación con el Comp. n.26-b y levetiracetam
Se realizaron estudios de respuesta a la dosis independientes en la prueba de 6 Hz, 44 mA para ambos compuestos para determinar los valores de DE50 en los TPE de 1 h i.p. para levetiracetam y de 0.5 h p.o. para el Comp. n.° 6-b. El valor de DE50 para el Comp. n.° 6-b era de 16.1 mg/kg y para levetiracetam se estimó que el valor era de aproximadamente 345 mg/kg. Se repitió la respuesta a la dosis para levetiracetam con coadministración de 10 mg/kg del Comp. n.° 6-b (una dosis del Comp. n.° 6-b que sola no protegió en el modelo de 6 Hz, 44 mA). La coadministración de 10 mg/kg del Comp. n.° 6-b produjo una DE50 en la respuesta a la dosis de levetiracetam de 2.4 mg/kg (~ 100 veces inferior en comparación con levetiracetam solo) y proporcionó de manera importante una protección completa en el modelo de convulsiones de 6 Hz, 44 mA. Estos resultados sugieren que existe una interacción farmacodinámica positiva en el modelo de convulsiones de 6 Hz entre el Comp. n.° 6-b y levetiracetam.
Los resultados de los estudios realizados con el compuesto 6-b se enumeran en las tablas 11-13 a continuación.
Tabla 11: Determinación del tiempo para el efecto máximo para el
Comp. n.26-b (p.o.) en el ensayo de 6 Hz (32 mA).

Tabla 12: Estudio de respuesta a la dosis para el Comp. n.26-b en el ensayo de 6 Hz (32 mA y 44 mA; 0.5 h de
TPE).


Tabla 13: Estudios de combinación para el Comp. n.26-b con LEV en el ensayo de 6 Hz (44 mA).

e je m p lo 4 - e s tu d io s co n el c o m p u e s to de r e fe r e n c ia LY404039
3.1. Estudio de combinación con LY404039 de referencia y levetiracetam
Se sometió a prueba el compuesto de referencia LY-404039 solo y en combinación con levetiracetam según los procedimientos ya descritos anteriormente en la presente. Los resultados de los estudios realizados con el compuesto de referencia LY-404039 se enumeran en las tablas 14-15.
Tabla 14: Estudios de respuesta a la dosis para el compuesto de referencia LY404039 en el ensayo de 6 Hz (32 mA y 44 mA) Se determinó un tiempo para el efecto máximo de 0.5 h en la prueba de 6 Hz, 32 mA (s.c.) y se usó para los estudios de 6 Hz (32 mA y 44 mA).

Tabla 15: Estudios de combinación para el compuesto de referencia LY404039
con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA).


A una dosis de 5 mg/kg del compuesto de referencia LY404039 aumenta la potencia de LEV, conduciendo a un desplazamiento de aproximadamente 27 veces en la DE50. Esto sugiere una relación farmacodinámica positiva.
e je m p lo 5 - e s tu d io s de r e fe r e n c ia co n el c o m p u e s t o de r e fe r e n c ia c a s 1092453-15-0
4.1. Estudio de combinación con el compuesto de referencia 2,3-dihidro-7-met¡l-5-[3-(1 -piperazinilmetil)-1,2,4-oxad¡azol-5-¡l]-2-[[4-(tr¡fluorometoxi)fen¡l]met¡l]-1H-¡so¡ndol-1-ona [CAS 1092453-15-0] (descrita en el documento WO 2008150233, WO 2011084098) y levetiracetam

Se sometió a prueba el compuesto de referencia CAS 1092453-15-0 solo y en combinación con levetiracetam según los procedimientos ya descritos anteriormente en la presente. Los resultados del ejemplo 5 se enumeran en las tablas 16-17.
Tabla 16: Estudios de respuesta a la dosis para el compuesto de referencia CAS 1092453-15-ü en el ensayo de 6 Hz (32 mAa)

Se observó baja actividad a las dosis y puntos de tiempo sometidos a prueba. La mayor actividad a 0.25-1 h en las dosis sometidas a prueba. Se realizaron estudios de combinación usando 20 mg/kg, s.c, 1 h de TPE en el ensayo de 6 Hz (44 mA).
Tabla 17: Estudios de combinación para el compuesto de referencia CAS 1092453-15-ü
con levetiracetam (LEV) en el ensayo de 6 Hz (44 mA).


E l conjunto d e d ato s actu al in d ica q u e la s m o lé c u la s d e PAM d e m G lu 2 o a g o n ista tien en a ctiv id ad a n tic o n v u ls iv a en el m odelo a n im a l d e 6 H z. L a s PA M d e m G lu 2 so m e tid a s a p ru e b a con p o te n cias d e C E 50 <d e 150 nM (tal co m o se d eterm inó en el e n s a y o d e [35S ] G T P gS ), p a rá m e tro s d e P K y pen etración en el cereb ro a p ro p ia d o s, m ostraron activ idad en el p a ra d ig m a d e 6 H z, tanto d e 32 co m o d e 44 m A. A d e m á s , to d a s la s m o lé c u la s so m e tid a s a p ru eb a m ostraron efe cto s s in é rg ic o s con L E V . En cam b io , la m o lé cu la d e re fe re n cia C A S 1092453 -15 -0 , q u e so lo e ra d éb ilm ente a ctiva ( C E 50 d e 562 nM) in vitro, no m ostró a ctiv id ad en n in g u n a d e la s p ru e b a s d e 6 H z, y tam p oco m ostró s in e rg ia con L E V . D e m a n e ra im portante, los d atos in d ican qu e, en c o n d ic io n e s d e c a ra c te ríst ic a s de P K c o m p a ra b le s y p en etración en el cereb ro a p ro p ia d a , la s PA M d e m G lu 2 m á s potentes, b a s á n d o s e en los v a lo re s de C E 50 in vitro, tam b ién p a re c ía n la s m á s potentes in vivo, su g irie n d o q u e la p o ten cia in v itro e in v ivo p u ed e e sta r v in c u la d a s . A d e m á s, s e o b serva ro n d e m a n e ra s iste m á tic a efecto s s in é rg ic o s con L E V co n d o s is d e PAM d e m G lu 2 s im ila re s a la D E 50 o b ten id a en el m odelo d e 32 m A o al m e n o s 2 v e c e s inferior q u e la D E 50 d e te rm in a d a en el p a ra d ig m a d e 44 m A (es d ecir, u n a d o sis in a ctiv a en la p ru e b a d e 44 m A cu a n d o s e so m etiero n a p ru e b a la s m o lé c u la s so la s).
T a m b ié n p a ra el co m p u e sto de re fe re n cia L Y 404039 , el a g o n ista d e m G lu 2 /3 , s e o b se rvó a ctiv id ad en a m b a s p ru e b a s d e 6 H z y s e o b se rvó s in e rg ia a u n a d o s is 3 v e c e s inferior a la D E 50 d e te rm in a d a en el m odelo d e 44 m A, q u e e ra in a ctiva cu a n d o s e so m e tía a p ru e b a so la .
B a s á n d o s e en los d ato s p re c lín ic o s d isp o n ib le s en el m odelo d e 6 H z, 44 m A, p a re c e q u e co m b in a r un lig an d o de S V 2 A potente y u n a PA M d e m G lu 2 potente, co n d u ce a u n a d ism u n u c ió n en la m e d ia n a d e la d o s is e fic a z o D E 50 del ligando d e SV2a , tal co m o L E V , entre 35 y 100 v e c e s .
P o r tanto, s in d e s e a r restrin g irse a la teo ría , s e s u g ie re qu e los co m p u e sto s d el m o d u lad o r a lo sté rico positivo del receptor g lu tam atérg ico m etabotrópico d e subtipo 2 (PA M d e m G lu R 2 ), en p a rticu la r co m p u e sto s d e PA M d e m G lu R 2 q u e tien en u n a p o ten cia d e C E 50 d e < 150 nM (tal co m o se d eterm inó en el e n s a y o d e [35S ] G T P gS ), en el q u e C E 50 e s la co n cen trac ió n qu e p rod u ce la m itad del efecto m áxim o en u n a c u rv a d e c o n c e n tra c ió n -re sp u e sta o b ten id a en p re se n c ia de C E 2üde glutam ato , y p a rá m e tro s d e P K y pen etración en el ce re b ro a p ro p ia d o s, d an co m o resultad o un a c o m b in a c ió n s in é rg ic a con un ligando de S V 2 A , en p a rticu la r levetiracetam , a d o s is no e f ic a c e s d e uno o a m b o s de co m p u e sto (a) y co m p u esto (b) d e la co m b in a ció n d e la in ven ción .
P o r tanto, en u n a re a liza c ió n a d ic io n a l, el co m p u e sto del m o d u lad o r a lo sté rico positivo d el receptor g lu tam atérg ico m etabotrópico d e subtipo 2 (PA M d e m G lu R 2 ) d e la co m b in a c ió n d e la in ven ció n tal co m o s e definió en la p resen te se s e le c c io n a de un co m p u esto d e PA M d e m G lu R 2 q u e tien e u n a p o ten cia d e C E 50 d e <150 nM (tal co m o s e determ inó en el e n s a y o d e [35S ] G T P gS ), en el q u e C E 50 e s la co n cen trac ió n q u e p rod u ce la mitad d el efecto m áxim o en u n a cu rv a d e c o n c e n tra c ió n -re sp u e sta o b ten id a en p re se n c ia d e C E 20d e glutam ato.
e je m p l o s te ó r ic o s
a ) r e l a c io n e s de c o m p o r t a m ie n t o d o m in a n t e -s u m is o (d s r , po r s u s s ig la s en in g lé s ) en en s a y o in v iv o en r a ta s
El ensayo de DSR se divide en dos modelos: Modelo de reducción del comportamiento dominante (RDBM, por sus siglas en inglés) de la manía y modelo de reducción del comportamiento sumiso (RSBM, por sus siglas en inglés) de la depresión. El RDBM, en el que se tratan los animales dominantes con el compuesto de prueba, predice la capacidad del compuesto de prueba para tratar la manía. El RSBM, en el que se tratan los animales sumisos con el compuesto de prueba, predice la capacidad del compuesto de prueba para tratar la depresión.
En este ensayo, se usan ratas Sprague Dawley macho (de 140 a 160 g) de Charles River Laboratories Wilmington, MA. Se reciben envíos de ratas en intervalos de dos semanas. Cada envío se someterá a una cuarentena de cinco días, un periodo de aclimatación de una semana y un proceso de selección de una semana, seguido de cinco semanas de tratamiento con vehículo o fármaco para los pares seleccionados.
Se acomodarán cuatro ratas en cada jaula. El acceso a la comida se restringirá a una hora por día después de los ensayos desde el lunes hasta el jueves. Después de los ensayos del viernes, las ratas tendrán acceso libre a la comida hasta someterlas nuevamente a ayuno el domingo. En ningún momento se privará a las ratas del acceso a agua. Los periodos de privación de alimentos utilizados tendrán un efecto menor en el aumento de peso, ya que el peso medio de la rata será de aproximadamente 300 g al final del estudio. Al concluir el experimento, las ratas se sacrificarán por decapitación, se recogerán la sangre del tronco y los cerebros para realizar experimentos in vitro y mediciones de la concentración de fármaco.
El aparato básico de los ensayos constaba de dos cámaras conectadas con un túnel sólo lo suficientemente largo como para permitir que pasara una rata a través cada vez. En el suelo, en el punto central del túnel, se colocará un envase con leche edulcorada. Este aparato básico se replicará, de manera que se pueda realizar un seguimiento por video de un total de cuatro pares de ratas de manera simultánea. La cámara puede distinguir ratas marcadas mediante diferentes colores. Por tanto, las cabezas de las ratas se colorearán con el fin de un seguimiento por video, de rojo en una jaula y amarillo en la otra jaula. Solo un animal puede tener acceso confortable al alimentador cada vez, pero ambos animales pueden beber leche durante la sesión diaria de cinco minutos. Durante las sesiones diarias de cinco minutos, se registrará el tiempo que pasa cada rata en la zona del alimentador con el software de seguimiento por video y se guardará en un archivo de texto.
La prueba empezará con una distribución aleatoria de las ratas por pares. Cada miembro de un par se colocará en una cámara opuesta del aparato de pruebas. Se registrará el tiempo que pasa cada animal en la zona del alimentador. Durante la primera semana (cinco días) de pruebas, los animales se habitúan al nuevo entorno. Se asignará la dominancia al animal con la puntuación más elevada durante la segunda semana de pruebas si se cumplen tres criterios. En primer lugar, debe existir una diferencia significativa (prueba de la t bilateral, P < 0.05) entre las puntuaciones de bebida diarias promedio de ambos animales. En segundo lugar, la puntuación del animal dominante debe ser al menos un 25% mayor que la puntuación del animal sumiso. Finalmente, no debe haber ninguna "inversión" durante la semana de selección del par, en la que la posible rata sumisa consiga una puntuación mejor que la de su compañero dominante en ocasiones aisladas. Idealmente, también habrá inversiones mínimas durante la semana de aclimatación. Solo pares de animales que cumplan estos criterios continuarán en el estudio.
Las diferencias significativas entre el tiempo que las ratas dominantes y sumisas pasan en el alimentador se determinarán mediante ANOVA usando el software GraphPad Prism (software GraphPad, Inc. San Diego, CA) seguido por una prueba de la t bilateral (P < 0.05). Se compararán los grupos de tratamiento usando valores del nivel de dominancia normalizados en los animales apareados. El nivel de dominancia es un valor que mide la relación social entre sujetos apareados. Nivel de dominancia (DL, por sus siglas en inglés) = FTD - FTS, donde FTD (por sus siglas en inglés) es el tiempo que pasan las ratas dominantes en el alimentador y FTS (por sus siglas en inglés) es el tiempo que pasan las ratas sumisas en el alimentador. La normalización se llevará a cabo según la fórmula:
Nivel de dominancia (semana n en %) =
(Nivel de dominancia (semana n)) / (Nivel de dominancia (semana 2)
La significación estadística de la diferencia en el nivel de dominancia entre el grupo control (pares de ratas en los que ambos animales tanto dominantes como sumisos se tratarán con vehículo) y el grupo de tratamiento (las ratas sumisas se tratarán con fármaco y las ratas dominantes con vehículo) se determinará mediante ANOVA, seguido por una prueba de la t. El valor del tiempo de comienzo de actividad al 50% de la respuesta (AOT-50) y la respuesta mínima y máxima al fármaco se calcularán basándose en la reducción del valor del nivel de dominancia usando un análisis de regresión no lineal (GraphPad Software, Inc., San Diego, CA). Para este cálculo se usán los valores normalizados de DL, donde los valores de DL para las semanas de tratamiento se normalizarán como un porcentaje del valor de la segunda semana (pretratamiento) para ese par según la fórmula anterior. En este contexto, el mínimo de la respuesta (DL) determina una actividad positiva del fármaco, que se corresponde con la eficacia, ya que los valores de DL se reducirán si la respuesta a un fármaco es positiva. En el caso de una respuesta negativa al fármaco (empeoramiento de los síntomas) los valores de DL aumentarán. Si el fármaco no presenta tal actividad el máximo de la respuesta no
excederá el 100%. Cualquier valor de DL máximo significativamente superior al valor de control (aproximadamente el 100%) indica una actividad negativa del fármaco.
Se evaluarán levetiracetam y compuesto de PAM de mGluR2 (p. ej. compuesto 6-b) en el RDBM con ratas según el procedimiento descrito en mayor detalle a continuación.
Se tratarán grupos de ratas dominantes p.o. QD con levetiracetam 10 mg/kg y compuesto de PAM de mGluR2 a varias concentraciones de desde aproximadamente 0.05 mg/kg (n > 3), a 0.5 mg/kg (n > 3), a 2.5 mg/kg (n > 3), a 5.0 mg/kg (n > 3) y a 50.0 mg/kg (n > 3). Un grupo control de ratas dominantes que se tratarán con 0.5% de metilcelulosa (n > 3) y un segundo grupo control de ratas dominantes se tratará i.p. QD con valproato de sodio a 30 mg/kg (n>6 de 2 estudios de n>3 cada uno).
Todos los tratamientos se administrarán aproximadamente 1 hora antes de los ensayos. Todos los tratamientos se iniciarán el sábado después de la segunda semana de ensayos (semana de selección). El levetiracetam y compuesto de PAM de mGluR2 se administrarán por vía oral (p.o.).
Cuando se tratan los animales dominantes con 10 mg/kg de levetiracetam y compuesto de PAM de mGluR2, la diferencia entre las ratas dominantes y sumisas desaparecerá después de la primera o segunda semana de tratamiento dependiendo de la dosis. De manera similar, cuando se tratan los animales dominantes con valproato de sodio, la diferencia entre las ratas dominantes y sumisas también desaparecerá después de la primera semana de tratamiento. Puede observarse un incremento de la permisividad de las ratas dominantes tratadas con levetiracetam y el compuesto de PAM de mGluR2 o valproato de sodio. Por tanto, las ratas dominantes tratadas permitirán que sus compañeras sumisas aumenten su tiempo en el alimentador.
Para comparar los diferentes efectos de fármacos y dosis, los datos se normalizarán respecto a los valores iniciales de la semana control. Se observará el efecto más fuerte de la combinación de levetiracetam y el compuesto PAM de mGluR2 cuando exista una diferencia significativa en los valores del nivel de dominancia (DL) entre las ratas tratadas con vehículo y con la combinación a partir de la segunda semana y que continua a lo largo de la duración del tratamiento de 5 semanas. En comparación, los animales que se tratarán con valproato de sodio (30 mg/kg) mostrarán de manera sistemática un nivel de dominancia disminuido después de la segunda semana de tratamiento incrementándose el efecto en las siguientes semanas.
Para estimar el tiempo de comienzo de actividad (AOT), los valores promedio diarios del tiempo de alimentador de los pares de animales dominantes y sumisos se representarán gráficamente y las diferencias significativas entre estos dos grupos se calcularán usando la prueba de la t bilateral.
Para comparar el tiempo de comienzo de actividad (AOT) entre diferentes tratamientos se estimará el tiempo de comienzo de actividad a partir del ajuste de regresión no lineal. El modelo de regresión no lineal se ajustará para los valores de DL diarios normalizados de cada fármaco, combinación y dosis.
Cabe esperar que los efectos de levetiracetam y el compuesto de PAM de mGluR2 en el RDBM sean dependientes de dosis.
En este ensayo, se espera que la combinación de levetiracetam y los compuestos de PAM de mGlu2 reduzcan el comportamiento dominante indicando que la combinación es activa como agente contra la manía.
b) c o m p r im id o s o r a le s
A modo de realización específica para una composición oral, se formulan 100 mg de un compuesto de PAM de mGluR2/agonista con una cantidad suficiente de lactosa finamente dividida para proporcionar una cantidad total de 580 a 590 mg para llenar una cápsula de gelatina dura de tamaño O.
Claims (18)
1. Una combinación que comprende
(a) un ligando de una proteína de la vesícula sinóptica 2A (“SV2A”) seleccionado del grupo que consiste en levetiracetam, brivaracetam y seletracetam; y
(b) un compuesto de modulador alostérico positivo (“PAM”) del receptor glutamatérgico metabotrópico de subtipo 2 (“mGluR2”) seleccionado de

2. La combinación de la reivindicación 1, en la que el modulador alostérico positivo del receptor glutamatérgico metabotrópico de subtipo 2 es

o una sal de clorhidrato del mismo.
3. La combinación según la reivindicación 1 o 2, en la que el ligando de SV2A es levetiracetam.
4. La combinación según la reivindicación 1 o 2, en la que el ligando de SV2A es brivaracetam.
5. Una composición farmacéutica que comprende una combinación según una cualquiera de las reivindicaciones 1 a 4, y un portador farmacéuticamente aceptable.
6. La composición farmacéutica según la reivindicación 5, formulada como una composición farmacéutica combinada.
7. La composición farmacéutica según la reivindicación 5, formulada en composiciones farmacéuticas separadas.
8. Un procedimiento para preparar la composición farmacéutica según la reivindicación 5 o 6, en el que la combinación según una cualquiera de las reivindicaciones 1 a 4 se mezcla de manera íntima con un portador farmacéuticamente aceptable.
9. Un producto que comprende la combinación tal como se define en una cualquiera de las reivindicaciones 1 a 4, como una preparación combinada para su uso simultáneo, secuencial o por separado en el tratamiento o la prevención de la epilepsia y trastornos relacionados; dolor neuropático; migraña o cefalea resistente; trastornos bipolares y trastornos relacionados.
10. Una combinación tal como se define en una cualquiera de las reivindicaciones 1 a 4, o una composición farmacéutica tal como se define en una cualquiera de las reivindicaciones 5 a 7, para su uso como medicamento.
11. Una combinación según una cualquiera de las reivindicaciones 1 a 4 para su uso en el tratamiento o la prevención de la epilepsia; dolor neuropático; migraña o cefalea resistente; y trastornos bipolares y trastornos relacionados.
12. La combinación para su uso según la reivindicación 11, en el tratamiento o la prevención de la epilepsia.
13. La combinación para su uso según la reivindicación 12, en la que la epilepsia es epilepsia resistente al tratamiento.
14. La combinación para su uso según la reivindicación 12, en la que la epilepsia es epilepsia con convulsiones de inicio parcial con o sin generalización.
15. La combinación para su uso según la reivindicación 12, en la que la epilepsia es epilepsia con convulsiones generalizadas.
16. La combinación para su uso según la reivindicación 12, donde la epilepsia es epilepsia con convulsiones tónicoclónicas generalizadas primarias.
17. Una combinación según una cualquiera de las reivindicaciones 1 a 4, o una composición farmacéutica tal como se define en una cualquiera de las reivindicaciones 5 a 7, para su uso en la prevención de la epileptogénesis.
18. Un producto farmacéutico o un envase comercial que comprende una combinación según una cualquiera de las reivindicaciones 1 a 4 junto con instrucciones para su uso simultáneo, secuencial o por separado en el tratamiento o la prevención de la epilepsia; dolor neuropático; migraña o cefalea resistente; y trastornos bipolares y trastornos relacionados.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461929795P | 2014-01-21 | 2014-01-21 | |
| EP14153887 | 2014-02-04 | ||
| EP14153880 | 2014-02-04 | ||
| EP14183324 | 2014-09-03 | ||
| EP14187429 | 2014-10-02 | ||
| US201462091668P | 2014-12-15 | 2014-12-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2860298T3 true ES2860298T3 (es) | 2021-10-04 |
Family
ID=65633001
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18182695T Active ES2860298T3 (es) | 2014-01-21 | 2015-01-20 | Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso |
| ES15700726T Active ES2748633T3 (es) | 2014-01-21 | 2015-01-20 | Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15700726T Active ES2748633T3 (es) | 2014-01-21 | 2015-01-20 | Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US11369606B2 (es) |
| JP (2) | JP6684936B2 (es) |
| KR (2) | KR20200036063A (es) |
| CL (3) | CL2016001815A1 (es) |
| DK (1) | DK3096790T3 (es) |
| ES (2) | ES2860298T3 (es) |
| HR (2) | HRP20191646T1 (es) |
| HU (2) | HUE053734T2 (es) |
| IL (4) | IL279202B2 (es) |
| LT (2) | LT3431106T (es) |
| ME (1) | ME03518B (es) |
| NZ (1) | NZ722385A (es) |
| PH (2) | PH12019500127B1 (es) |
| PT (1) | PT3096790T (es) |
| UA (1) | UA121965C2 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12396981B2 (en) | 2023-03-09 | 2025-08-26 | William Shulman | Methods of using DMT |
Family Cites Families (490)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2976146A (en) | 1958-11-28 | 1961-03-21 | Eastman Kodak Co | Novel cyan-forming couplers |
| GB1309692A (en) | 1970-02-13 | 1973-03-14 | Ucb Sa | N-substituted lactams |
| GB1039113A (en) | 1964-08-06 | 1966-08-17 | Ucb Sa | New n-substituted lactams |
| BE790440A (es) | 1971-10-23 | 1973-04-24 | Bayer Ag | |
| JPS524071B2 (es) | 1972-01-20 | 1977-02-01 | ||
| JPS538707B2 (es) | 1974-02-05 | 1978-03-31 | ||
| US3906953A (en) | 1974-05-23 | 1975-09-23 | American Optical Corp | Endoscopic surgical laser system |
| SU509578A1 (ru) | 1974-09-19 | 1976-04-05 | Стерлитамакский Химический Завод | Способ получени пропилендиаминов |
| GB1502312A (en) | 1975-03-20 | 1978-03-01 | Ici Ltd | Quinolone derivatives |
| IE43079B1 (en) | 1975-03-20 | 1980-12-17 | Ici Ltd | Quinolone derivatives |
| FR2311776A1 (fr) | 1975-05-23 | 1976-12-17 | Sogeras | Diamino-2,4 bromo-5 chloro-6 pyrimidines et procede pour leur preparation |
| GB1570494A (en) | 1975-11-28 | 1980-07-02 | Ici Ltd | Thienopyrimidine derivatives and their use as pesticides |
| JPS5382783A (en) | 1976-12-29 | 1978-07-21 | Yamanouchi Pharmaceut Co Ltd | Novel pyridone derivs and process for their preparation |
| ZA782648B (en) | 1977-05-23 | 1979-06-27 | Ici Australia Ltd | The prevention,control or eradication of infestations of ixodid ticks |
| DE2750288A1 (de) | 1977-11-10 | 1979-05-17 | Thomae Gmbh Dr K | Neue 9-(omega-heteroarylamino- alkylamino)-erythromycine, ihre salze, verfahren zu ihrer herstellung und diese enthaltende arzneimittel |
| JPS5676589A (en) | 1979-11-28 | 1981-06-24 | Tokyo Shibaura Electric Co | Wiring pattern |
| JPS5718381A (en) | 1980-07-08 | 1982-01-30 | Mitsubishi Electric Corp | Printed circuit board |
| JPS6057300B2 (ja) | 1980-09-11 | 1985-12-14 | 株式会社東芝 | 超電導コイル間のエネルギ転送装置 |
| US4432979A (en) | 1981-10-26 | 1984-02-21 | William H. Rorer, Inc. | Pyridone compounds |
| FI824314L (fi) | 1981-12-16 | 1983-06-17 | Sankyo Co | Thienopyrimidin-derivater, deras framstaellning och deras medicinska anvaendning |
| US4358453A (en) | 1982-01-08 | 1982-11-09 | Schering Corporation | 1,2,4-Triazolo[4,3-a]pyridines |
| US4520025A (en) | 1982-07-21 | 1985-05-28 | William H. Rorer, Inc. | Bicyclic nitrogen heterocyclic ethers and thioethers, and their pharmaceutical uses |
| JPS60159184A (ja) | 1984-01-27 | 1985-08-20 | Agency Of Ind Science & Technol | 水電解用陽極 |
| DE3406329A1 (de) | 1984-02-22 | 1985-08-22 | Merck Patent Gmbh, 6100 Darmstadt | Pyridone |
| GB8412358D0 (en) | 1984-05-15 | 1984-06-20 | Ucb Sa | Pharmaceutical composition |
| GB8412357D0 (en) | 1984-05-15 | 1984-06-20 | Ucb Sa | Pharmaceutical composition |
| US4550166A (en) | 1984-05-21 | 1985-10-29 | American Cyanamid Company | (Pyridinyl)-1,2,4-triazolo[4,3-a]pyridines |
| DE3717561A1 (de) | 1987-05-25 | 1988-12-08 | Thomae Gmbh Dr K | Indol-, isochinolin- und benzazepinderivate, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| US5175157A (en) | 1985-11-27 | 1992-12-29 | Boehringer Ingelheim Gmbh | Cyclic amine derivatives, pharmaceutical compositions containing these compounds and methods for preparing them |
| JPH081861Y2 (ja) | 1986-06-13 | 1996-01-24 | カシオ計算機株式会社 | 印刷装置のカ−トリツジ構造 |
| US4866074A (en) | 1987-05-08 | 1989-09-12 | Rorer Pharmaceutical Corporation | Naphtheridinone- and pyridooxazinone-pyridone compounds, cardiotonic compositions including same, and their uses |
| EP0308020A3 (en) | 1987-09-18 | 1990-12-05 | Merck & Co. Inc. | 5-(aryl and heteroaryl)-6-(aryl and heteroaryl)-1,2-dihydro-2-oxo 3-pyridinecarboxylic acids and derivatives thereof |
| US5260293A (en) | 1988-01-30 | 1993-11-09 | Merck Sharp & Dohme Limited | Pyrazines, pyrimidines and pyridazines useful in the treatment of senile dementia |
| GB8804448D0 (en) | 1988-02-25 | 1988-03-23 | Smithkline Beckman Intercredit | Compounds |
| JP2614081B2 (ja) | 1988-05-27 | 1997-05-28 | 大塚化学株式会社 | 光学活性β−ラクタム誘導体の製造法 |
| JPH02124871A (ja) | 1988-07-27 | 1990-05-14 | Dainippon Pharmaceut Co Ltd | 1位が置換された複素環式カルボン酸アミド誘導体 |
| DE68923527D1 (de) | 1988-10-20 | 1995-08-24 | Sandoz Ag | Faserreaktive Azofarbstoffe. |
| GB8827389D0 (en) | 1988-11-23 | 1988-12-29 | Ucb Sa | Process for preparation of(s)alpha-ethyl-2-oxo-1-pyrrolidineacetamide |
| US5032602A (en) | 1988-12-14 | 1991-07-16 | Bayer Aktiengesellschaft | Inhibiting HMG-CoA reductase with novel substituted 2-pyridones and pyrid-2-thiones |
| HU206337B (en) | 1988-12-29 | 1992-10-28 | Mitsui Petrochemical Ind | Process for producing pyrimidine derivatives and pharmaceutical compositions |
| JPH02277044A (ja) | 1989-04-19 | 1990-11-13 | Fuji Photo Film Co Ltd | ハロゲン化銀写真感光材料 |
| US5236917A (en) | 1989-05-04 | 1993-08-17 | Sterling Winthrop Inc. | Saccharin derivatives useful as proteolytic enzyme inhibitors and compositions and method of use thereof |
| US5280026A (en) | 1989-05-30 | 1994-01-18 | Smithkline Beecham Intercredit B.V. | Thienopyrimidines |
| AU622330B2 (en) | 1989-06-23 | 1992-04-02 | Takeda Chemical Industries Ltd. | Condensed heterocyclic compounds having a nitrogen atom in the bridgehead for use as fungicides |
| US4978663A (en) | 1989-08-16 | 1990-12-18 | Hoechst-Roussel Pharmaceuticals Inc. | 5-(1-aminocyclohexyl)-2(1H)-pyridinone compounds which have pharmaceutical utility |
| GB8926560D0 (en) | 1989-11-24 | 1990-01-17 | Zambeletti Spa L | Pharmaceuticals |
| IL96432A0 (en) | 1989-11-30 | 1991-08-16 | Schering Ag | Pesticidal compositions containing pyridine derivatives and novel pyridine derivatives |
| DE3940480A1 (de) | 1989-12-07 | 1991-06-13 | Bayer Ag | Chromogene enaminverbindungen, ihre herstellung und verwendung als farbbildner |
| WO1991009848A1 (en) | 1989-12-22 | 1991-07-11 | The Upjohn Company | Pyridinones useful as antiatherosclerotic agents |
| FR2657610A1 (fr) | 1990-01-29 | 1991-08-02 | Rhone Poulenc Agrochimie | Triazolopyridines herbicides. |
| GB9104238D0 (en) | 1990-03-16 | 1991-04-17 | Ici Pharma | 3-tetrazolylthiomethyl cephalosporin antibiotics |
| DE4008726A1 (de) | 1990-03-19 | 1991-09-26 | Basf Ag | Thieno(2,3-d)pyrimidinderivate |
| BR9101256A (pt) | 1990-03-30 | 1991-11-05 | Dowelanco | Composto,composicao fungicida,processo fungicida,composicao inseticida ou acaricida e processo inseticida ou acaricida |
| JP2503317Y2 (ja) | 1990-05-25 | 1996-06-26 | 日本電気株式会社 | 空気調節装置 |
| RU1796625C (ru) | 1990-06-27 | 1993-02-23 | Киевский Государственный Университет Им.Т.Г.Шевченко | 3-Амино-7-нитро-4(2,3,4-триметоксифенил)-2-фенил-1(2Н)изохинолон, обладающий аналептическим действием |
| ES2131506T3 (es) | 1990-09-21 | 1999-08-01 | Rohm & Haas | Dihidropiridacinonas y piridacinonas como fungicidas. |
| KR920008026A (ko) | 1990-10-24 | 1992-05-27 | 오노 화아마슈티칼 캄파니 리미팃드 | 이소퀴놀리논 유도체 또는 이의 무독성 산부가염 또는 이의 수화물, 이의 제조방법 및 이를 포함하는 약제 조성물 |
| AU1532492A (en) | 1991-04-18 | 1992-11-17 | Dr. Lo Zambeletti S.P.A. | Use of heterocyclic compounds for the treatment of inflammatory pain |
| DE4122240A1 (de) | 1991-07-05 | 1993-01-07 | Boehringer Mannheim Gmbh | Dibenz(b,e)azepinderivate und diese enthaltende arzneimittel |
| DE4129340A1 (de) | 1991-09-04 | 1993-03-11 | Merck Patent Gmbh | 1,2-dihydro-2-oxopyridine |
| US5332750A (en) | 1991-09-04 | 1994-07-26 | Merck Patent Gesellschaft Mit Beschrankter Haftung | 1,2-dihydro-2-oxopyridines |
| DE4131924A1 (de) | 1991-09-25 | 1993-07-08 | Hoechst Ag | Substituierte 4-alkoxypyrimidine, verfahren zu ihrer herstellung und ihre verwendung als schaedlingsbekaempfungsmittel |
| US5204198A (en) | 1991-10-28 | 1993-04-20 | Eastman Kodak Company | Photoelectrographic elements utilizing nonionic sulfonic acid photogenerators |
| US5416099A (en) | 1991-10-29 | 1995-05-16 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| DE4221583A1 (de) | 1991-11-12 | 1993-05-13 | Bayer Ag | Substituierte biphenylpyridone |
| JP2878531B2 (ja) | 1991-12-16 | 1999-04-05 | 富士写真フイルム株式会社 | ハロゲン化銀写真感光材料 |
| US5378720A (en) | 1991-12-19 | 1995-01-03 | Sterling Winthrop Inc. | Saccharin derivative proteolytic enzyme inhibitors |
| AU668694B2 (en) | 1991-12-19 | 1996-05-16 | Sanofi-Synthelabo | Saccharin derivative proteolytic enzyme inhibitors |
| TW219935B (es) | 1991-12-25 | 1994-02-01 | Mitsubishi Chemicals Co Ltd | |
| GB9200293D0 (en) | 1992-01-08 | 1992-02-26 | Wyeth John & Brother Ltd | Piperazine derivatives |
| GB9201694D0 (en) | 1992-01-27 | 1992-03-11 | Smithkline Beecham Intercredit | Compounds |
| JP3042156B2 (ja) | 1992-02-20 | 2000-05-15 | 田辺製薬株式会社 | ナフタレン誘導体、その製法及びその合成中間体 |
| DE4206045A1 (de) | 1992-02-27 | 1993-09-02 | Bayer Ag | Sulfonylbenzyl substituierte pyridone |
| US5922773A (en) | 1992-12-04 | 1999-07-13 | The Children's Medical Center Corp. | Glaucoma treatment |
| AU660132B2 (en) | 1992-12-21 | 1995-06-08 | Bayer Aktiengesellschaft | Substituted 4-phenyl-pyridones and 4-phenyl-2-alkoxypyridine |
| SE9300657D0 (sv) | 1993-02-26 | 1993-02-26 | Astra Ab | New compounds |
| US5814645A (en) | 1993-03-24 | 1998-09-29 | Bayer Aktiengesellschaft | Arylor hetaryl substituted nitrogen heterocycles and their use as pesticides |
| DE4316077A1 (de) | 1993-05-13 | 1994-11-17 | Bayer Ag | Substituierte Mono- und Bihydridylmethylpyridone |
| EP0702671A4 (en) | 1993-06-09 | 1996-06-19 | Smithkline Beecham Corp | BICYCLIC FIBRINOGEN ANTAGONISTS |
| DE4326758A1 (de) | 1993-08-10 | 1995-02-16 | Basf Ag | [1,3,4]Triazolo[1,5-a]pyridine |
| WO1995005383A1 (en) | 1993-08-19 | 1995-02-23 | Janssen Pharmaceutica N.V. | Vasocontrictive dihydrobenzopyran derivatives |
| RU2158126C2 (ru) | 1993-08-19 | 2000-10-27 | Жансен Фармасетика Н.В. | Замещенные арилоксиалкилдиамины, обладающие сосудосуживающим действием |
| WO1995006032A1 (fr) | 1993-08-20 | 1995-03-02 | Banyu Pharmaceutical Co., Ltd. | Inhibiteur de la tyrosine kinase |
| JP3701984B2 (ja) | 1993-08-31 | 2005-10-05 | サントリー株式会社 | ラベル化シクロプロパン誘導体およびその製造方法 |
| US5424435A (en) | 1993-10-18 | 1995-06-13 | Olin Corporation | 1-hydroxy-6-substituted-2-pyridones |
| US5500420A (en) | 1993-12-20 | 1996-03-19 | Cornell Research Foundation, Inc. | Metabotropic glutamate receptor agonists in the treatment of cerebral ischemia |
| US5679683A (en) | 1994-01-25 | 1997-10-21 | Warner-Lambert Company | Tricyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| WO1995024393A1 (en) | 1994-03-10 | 1995-09-14 | Fujisawa Pharmaceutical Co., Ltd. | Naphthalene derivatives as prostaglandin i2 agonists |
| ES2079323B1 (es) | 1994-06-21 | 1996-10-16 | Vita Invest Sa | Derivados de indol utiles para el tratamiento de la migraña, composicion y uso correspondientes. |
| GB9416554D0 (en) | 1994-08-19 | 1994-10-12 | Ciba Geigy Ag | Glutamate receptor |
| WO1996005828A1 (en) | 1994-08-24 | 1996-02-29 | Eli Lilly And Company | Pyrrolidinyl di-carboxylic acid derivatives as metabotropic glutamate receptor antagonists |
| US6017697A (en) | 1994-11-14 | 2000-01-25 | Eli Lilly And Company | Excitatory amino acid receptor protein and related nucleic acid compounds |
| US5473077A (en) | 1994-11-14 | 1995-12-05 | Eli Lilly And Company | Pyrrolidinyl di-carboxylic acid derivatives as metabotropic glutamate receptor agonists |
| US5512576A (en) | 1994-12-02 | 1996-04-30 | Sterling Winthrop Inc. | 2-substituted 1,2,5,-thiadiazolidin-3-one 1,1-dioxides and compositions and method of use thereof |
| US5789426A (en) | 1995-01-20 | 1998-08-04 | Cornell Research Foundation, Inc. | Method for the treatment of fibroproliferative disorders by application of inhibitors of protein hydroxylation |
| US5869486A (en) | 1995-02-24 | 1999-02-09 | Ono Pharmaceutical Co., Ltd. | Fused pyrimidines and pyriazines as pharmaceutical compounds |
| DE19507522C2 (de) | 1995-03-03 | 2003-05-28 | Basf Ag | Verfahren zur Herstellung von 3,4-Dihydroisochinolinverbindungen und 3,4-Dihydroisochinolinium-Salzen |
| US5869428A (en) | 1995-03-13 | 1999-02-09 | Ishihara Sangyo Kaisha Ltd. | Pyridonesulfonylurea compounds, process for their production and herbicides containing them |
| DE19510965A1 (de) | 1995-03-24 | 1996-09-26 | Asta Medica Ag | Neue Pyrido/3,2-e/pyrazinone mit antiasthmatischer Wirksamkeit und Verfahren zu deren Herstellung |
| US5948785A (en) | 1995-04-27 | 1999-09-07 | The Green Cross Corporation | Heterocyclic amide compounds and pharmaceutical use of the same |
| JPH08325248A (ja) | 1995-05-26 | 1996-12-10 | Chugoku Kayaku Kk | テトラゾール類の新規な合成試薬及びそれを用いたテトラゾール類の製造方法 |
| US5849587A (en) | 1995-06-09 | 1998-12-15 | Cornell Research Foundation, Inc. | Method of inhibiting viral replication in eukaryotic cells and of inducing apoptosis of virally-infected cells |
| US5659033A (en) | 1995-09-13 | 1997-08-19 | Neurogen Corporation | N-aminoalkylfluorenecarboxamides; a new class of dopamine receptor subtype specific ligands |
| SI0850235T1 (en) | 1995-09-15 | 2000-04-30 | Sanofi-Synthelabo | Quinolein-2(1h)-one derivatives as serotonin antagonists |
| US6130217A (en) | 1995-09-20 | 2000-10-10 | Pfizer Inc | Compounds enhancing antitumor activity of other cytotoxic agents |
| JPH1045750A (ja) | 1995-09-20 | 1998-02-17 | Takeda Chem Ind Ltd | アゾール化合物、その製造方法及び用途 |
| AR004010A1 (es) | 1995-10-11 | 1998-09-30 | Glaxo Group Ltd | Compuestos heterociclicos |
| CN1202167A (zh) | 1995-11-16 | 1998-12-16 | 伊莱利利公司 | 兴奋性的氨基酸衍生物 |
| DE69620445T2 (de) | 1995-12-08 | 2002-12-12 | Janssen Pharmaceutica N.V., Beerse | (imidazol-5-yl)methyl-2-chinolinoderivate als farnesyl protein transferase inhibitoren |
| US6313127B1 (en) | 1996-02-02 | 2001-11-06 | Zeneca Limited | Heterocyclic compounds useful as pharmaceutical agents |
| GB9602166D0 (en) | 1996-02-02 | 1996-04-03 | Zeneca Ltd | Aminoheterocyclic derivatives |
| GB9602294D0 (en) | 1996-02-05 | 1996-04-03 | Zeneca Ltd | Heterocyclic compounds |
| US6084084A (en) | 1996-02-21 | 2000-07-04 | Nps Pharmaceuticals, Inc. | Human metabotropic glutamate receptor |
| US5710274A (en) | 1996-02-28 | 1998-01-20 | Neurogen Corporation | N-aminoalkyldibenzofurancarboxamides; new dopamine receptor subtype specific ligands |
| US5756518A (en) | 1996-04-02 | 1998-05-26 | Kowa Co., Ltd. | Phenylene derivatives |
| JPH1029979A (ja) | 1996-04-12 | 1998-02-03 | Ajinomoto Co Inc | 新規ピリジン誘導体 |
| WO1997046532A1 (en) | 1996-06-03 | 1997-12-11 | Boehringer Ingelheim Pharmaceuticals, Inc. | 2-benzyl-4-sulfonyl-4h-isoquinolin-1,3-diones and their use as anti-inflammatory agents |
| WO1998006724A1 (en) | 1996-08-09 | 1998-02-19 | Yamanouchi Pharmaceutical Co., Ltd. | Metabotropic glutamate receptor agonists |
| DE19632423A1 (de) | 1996-08-12 | 1998-02-19 | Merck Patent Gmbh | Thienopyrimidine |
| CZ45699A3 (cs) | 1996-08-14 | 1999-05-12 | Zeneca Limited | Substituované deriváty pyrimidinu, způsob jejich přípravy a farmaceutický prostředek, který je obsahuje |
| US6159980A (en) | 1996-09-16 | 2000-12-12 | Dupont Pharmaceuticals Company | Pyrazinones and triazinones and their derivatives thereof |
| DE19638486A1 (de) | 1996-09-20 | 1998-03-26 | Basf Ag | Hetaroylderivate |
| DE19638484A1 (de) | 1996-09-20 | 1998-03-26 | Basf Ag | Hetaroylderivate |
| DE19644228A1 (de) | 1996-10-24 | 1998-04-30 | Merck Patent Gmbh | Thienopyrimidine |
| US6284794B1 (en) | 1996-11-05 | 2001-09-04 | Head Explorer Aps | Method for treating tension-type headache with inhibitors of nitric oxide and nitric oxide synthase |
| IL130181A0 (en) | 1996-12-05 | 2000-06-01 | Amgen Inc | Substituted pyrimidone and pyridone compounds and methods of use |
| ATE236188T1 (de) | 1997-01-24 | 2003-04-15 | Conpharma As | Gemcitabin-derivate |
| US5855654A (en) | 1997-01-30 | 1999-01-05 | Rohm And Haas Company | Pyridazinones as marine antifouling agents |
| FR2759366B1 (fr) | 1997-02-11 | 1999-04-16 | Centre Nat Rech Scient | Composes constituant notamment des effecteurs de recepteurs du systeme nerveux central sensibles aux amino acides neuroexcitateurs, leur preparation et leurs applications biologiques |
| US6262068B1 (en) | 1997-02-21 | 2001-07-17 | Bristol-Myers Squibb Company | Lactam derivatives as antiarrhythmic agents |
| WO1998038168A1 (en) | 1997-02-27 | 1998-09-03 | Tanabe Seiyaku Co., Ltd. | Isoquinolinone derivatives, process for preparing the same, and their use as phosphodiesterase inhibitors |
| ES2131463B1 (es) | 1997-04-08 | 2000-03-01 | Lilly Sa | Derivados de ciclopropilglicina con propiedades farmaceuticas. |
| GB9708945D0 (en) | 1997-05-01 | 1997-06-25 | Merck Sharp & Dohme | Therapeutic agents |
| DE19728996A1 (de) | 1997-07-07 | 1999-01-14 | Basf Ag | Triazolverbindungen und deren Verwendung |
| EP0891978B1 (en) | 1997-07-18 | 2002-03-20 | F. Hoffmann-La Roche Ag | 5H-Thiazolo (3,2-a) pyrimidine derivatives |
| JP2001515839A (ja) | 1997-07-18 | 2001-09-25 | ジョージタウン・ユニバーシティ | 二環式向代謝性グルタミン酸受容体リガンド |
| CA2295295A1 (en) | 1997-07-31 | 1999-02-11 | Celgene Corporation | Substituted alkanohydroxamic acids and method of reducing tnf.alpha. levels |
| PL192029B1 (pl) | 1997-08-14 | 2006-08-31 | Hoffmann La Roche | Zastosowanie heterocyklicznych eterów winylowych, nowe heterocykliczne etery winylowe, sposób ich wytwarzania i środek farmaceutyczny |
| US6358975B1 (en) | 1997-08-15 | 2002-03-19 | Johns Hopkins University | Method of using selective parp inhibitors to prevent or treat neurotoxicity |
| US20020028813A1 (en) | 1997-09-03 | 2002-03-07 | Paul F. Jackson | Thioalkyl compounds, methods, and compositions for inhibiting parp activity |
| US20020022636A1 (en) | 1997-09-03 | 2002-02-21 | Jia-He Li | Oxo-substituted compounds, process of making, and compositions and methods for inhibiting parp activity |
| US6121278A (en) | 1997-09-03 | 2000-09-19 | Guilford Pharmaceuticals, Inc. | Di-n-heterocyclic compounds, methods, and compositions for inhibiting parp activity |
| WO1999011622A1 (en) | 1997-09-03 | 1999-03-11 | Guilford Pharmaceuticals Inc. | Amino-substituted compounds, methods, and compositions for inhibiting parp activity |
| US6197785B1 (en) | 1997-09-03 | 2001-03-06 | Guilford Pharmaceuticals Inc. | Alkoxy-substituted compounds, methods, and compositions for inhibiting PARP activity |
| US6635642B1 (en) | 1997-09-03 | 2003-10-21 | Guilford Pharmaceuticals Inc. | PARP inhibitors, pharmaceutical compositions comprising same, and methods of using same |
| AU9740998A (en) | 1997-09-08 | 1999-03-29 | F. Hoffmann-La Roche Ag | Piperidine derivatives against malaria |
| EP0903343B1 (en) | 1997-09-19 | 2003-05-28 | SSP Co., Ltd. | Alpha-Substituted phenylpropionic acid derivative and medicine containing the same |
| US6465484B1 (en) | 1997-09-26 | 2002-10-15 | Merck & Co., Inc. | Angiogenesis inhibitors |
| WO1999016755A1 (en) | 1997-09-26 | 1999-04-08 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| WO2000012089A1 (en) | 1998-08-31 | 2000-03-09 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| US6162804A (en) | 1997-09-26 | 2000-12-19 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| WO1999018096A1 (en) | 1997-10-02 | 1999-04-15 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| US6455528B1 (en) | 1997-10-14 | 2002-09-24 | Mitsubishi Pharma Corporation | Piperazine compounds and medicinal use thereof |
| WO1999021992A2 (en) | 1997-10-23 | 1999-05-06 | Ganimed Pharmaceuticals Gmbh | Nucleic acid molecules encoding a glutamate receptor |
| AU1504599A (en) | 1997-12-17 | 1999-07-05 | Shionogi & Co., Ltd. | Novel pyridine compounds |
| ATE388139T1 (de) | 1997-12-18 | 2008-03-15 | Boehringer Ingelheim Pharma | Pyridone als hemmer der sh2-domäne der src- familie |
| US6013672A (en) | 1997-12-18 | 2000-01-11 | Uab Research Foundation | Agonists of metabotropic glutamate receptors and uses thereof |
| ES2304797T3 (es) | 1997-12-19 | 2008-10-16 | Amgen Inc. | Compuestos de piridina y piridazina sustituidos y su uso farmaceutico. |
| FR2772763B1 (fr) | 1997-12-24 | 2004-01-23 | Sod Conseils Rech Applic | Nouveaux analogues tetracycliques de camptothecines, leurs procedes de preparation, leur application comme medicaments et les compositions pharmaceutiques les contenant |
| IT1298155B1 (it) | 1998-01-19 | 1999-12-20 | Moreno Paolini | Composti pirimidin 3-ossido per il trattamento delle patologie muscolo-scheletriche, in particolare per il trattamento della |
| US6664250B2 (en) | 1998-01-20 | 2003-12-16 | Bristol-Myers Squibb Co. | Lactam derivatives as antiarrhythmic agents |
| ATE236118T1 (de) | 1998-01-28 | 2003-04-15 | Taisho Pharmaceutical Co Ltd | Fluorhaltige aminosäurederivate |
| BR9908004A (pt) | 1998-02-17 | 2001-12-18 | Tularik Inc | Composto, composição e método para prevençãoou supressão de uma infecção viral |
| JP3665567B2 (ja) | 1998-03-17 | 2005-06-29 | ファイザー・プロダクツ・インク | ビシクロ[2.2.1]ヘプタンおよび関連化合物 |
| KR20010042521A (ko) | 1998-04-08 | 2001-05-25 | 한스 루돌프 하우스, 헨리테 브룬너, 베아트리체 귄터 | N-피리도닐 제초제 |
| AU3170099A (en) | 1998-04-16 | 1999-11-08 | Yamanouchi Pharmaceutical Co., Ltd. | Remedies for obesity |
| EP0955301A3 (en) | 1998-04-27 | 2001-04-18 | Pfizer Products Inc. | 7-aza-bicyclo[2.2.1]-heptane derivatives, their preparation and use according to their affinity for neuronal nicotinic acetylcholine receptors |
| DE19822198C2 (de) | 1998-05-16 | 2003-02-13 | Wella Ag | Oxonolfarbstoffe enthaltende Mittel und Verfahren zur Erzeugung von semipermanenten Färbungen auf Haaren |
| CA2333770A1 (en) | 1998-06-04 | 1999-12-09 | Abbott Laboratories | Cell adhesion-inhibiting antinflammatory compounds |
| DE19826671A1 (de) | 1998-06-16 | 1999-12-23 | Hoechst Schering Agrevo Gmbh | 1,3-Oxazolin- und 1,3-Thiazolin-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Schädlingsbekämpfungsmittel |
| FR2781218B1 (fr) | 1998-07-15 | 2001-09-07 | Lafon Labor | Compositions pharmaceutiques comprenant des 2-quinolones |
| JP2000072751A (ja) | 1998-08-26 | 2000-03-07 | Tanabe Seiyaku Co Ltd | イソキノリノン誘導体 |
| JP2000072731A (ja) | 1998-08-31 | 2000-03-07 | Taisho Pharmaceut Co Ltd | 4−置換−2−アミノビシクロ[3.1.0]ヘキサン−2,6−ジカルボン酸誘導体及び製薬用組成物 |
| ATE269293T1 (de) | 1998-08-31 | 2004-07-15 | Taisho Pharmaceutical Co Ltd | 6-fluoro(3.1.0)hexan-derivate |
| CH694053A5 (de) | 1998-09-03 | 2004-06-30 | Hoffmann La Roche | Verfahren zur Herstellung von 2-Amino-bicyclo[3.1.0]hexan-2,6-dicarbonsäure-Derivaten. |
| US6284759B1 (en) | 1998-09-30 | 2001-09-04 | Neurogen Corporation | 2-piperazinoalkylaminobenzo-azole derivatives: dopamine receptor subtype specific ligands |
| SE9803518D0 (sv) | 1998-10-15 | 1998-10-15 | Astra Pharma Prod | Novel compounds |
| PE20001236A1 (es) | 1998-11-13 | 2000-11-10 | Lilly Co Eli | Moduladores del receptor de aminoacidos excitadores |
| US6133271A (en) | 1998-11-19 | 2000-10-17 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure thienopyrimidine derivatives |
| US5948911A (en) | 1998-11-20 | 1999-09-07 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells and related conditions by exposure to thienopyrimidine derivatives |
| EP1006112A1 (en) | 1998-12-01 | 2000-06-07 | Cerebrus Pharmaceuticals Limited | 3-Hydroxy-2(1H)-pyridinone or 3-hydroxy-4(1H)-pyridinone derivatives useful as reactive oxygen species (ROS) scavengers |
| EP1133474B1 (en) | 1998-12-04 | 2007-02-21 | Bristol-Myers Squibb Company | 3-substituted-4-arylquinolin-2-one derivatives as potassium channel modulators |
| US6245759B1 (en) | 1999-03-11 | 2001-06-12 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| TW564247B (en) | 1999-04-08 | 2003-12-01 | Akzo Nobel Nv | Bicyclic heteraromatic compound |
| GB9908175D0 (en) | 1999-04-09 | 1999-06-02 | Lilly Co Eli | Method of treating neurological disorders |
| US6723711B2 (en) | 1999-05-07 | 2004-04-20 | Texas Biotechnology Corporation | Propanoic acid derivatives that inhibit the binding of integrins to their receptors |
| US6972296B2 (en) | 1999-05-07 | 2005-12-06 | Encysive Pharmaceuticals Inc. | Carboxylic acid derivatives that inhibit the binding of integrins to their receptors |
| EP1183238A1 (en) | 1999-05-17 | 2002-03-06 | Eli Lilly And Company | Metabotropic glutamate receptor antagonists |
| CN1361768A (zh) | 1999-06-02 | 2002-07-31 | Nps药物有限公司 | 代谢移变的谷氨酸盐受体拮抗剂和它们治疗中枢神经系统疾病的用途 |
| EP1189873A1 (en) | 1999-06-03 | 2002-03-27 | Lilly, S.A. | Excitatory amino acid receptor modulators |
| JP2004509059A (ja) | 1999-06-03 | 2004-03-25 | アボット・ラボラトリーズ | 細胞接着抑制性抗炎症化合物 |
| JP4783967B2 (ja) | 1999-07-21 | 2011-09-28 | 大正製薬株式会社 | 含フッ素アミノ酸誘導体を有効成分とする医薬 |
| MXPA02001340A (es) | 1999-08-05 | 2004-07-16 | Prescient Neuropharma Inc | Compuestos novedosos 1,4-benzodiazepina y derivados de los mismos. |
| US6660753B2 (en) | 1999-08-19 | 2003-12-09 | Nps Pharmaceuticals, Inc. | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| US7040838B2 (en) | 1999-08-27 | 2006-05-09 | Kristar Enterprises, Inc. | High capacity catch basin filtration system with adjustable deflector ring |
| AU774451B2 (en) | 1999-10-15 | 2004-06-24 | F. Hoffmann-La Roche Ag | Benzodiazepine derivatives |
| CA2386980C (en) | 1999-10-15 | 2010-03-16 | Geo Adam | Benzodiazepine derivatives for use in acute or chronic neurological disorders |
| HRP20020349A2 (en) | 1999-10-19 | 2004-02-29 | Merck & Co Inc | Tyrosine kinase inhibitors |
| EP1230225A2 (en) | 1999-11-01 | 2002-08-14 | Eli Lilly And Company | Pharmaceutically active 4-substituted pyrimidine derivatives |
| GB2355982A (en) | 1999-11-03 | 2001-05-09 | Lilly Co Eli | Heterocyclic amino acids |
| CA2475026A1 (en) | 1999-12-01 | 2001-06-07 | Ucb, S.A. | A pyrrolidineacetamide derivative alone or in combination for treatment of cns disorders |
| AU2220801A (en) | 1999-12-22 | 2001-07-03 | Kyorin Pharmaceutical Co. Ltd. | Tricyclic compounds and addition salts thereof |
| IL150650A0 (en) | 2000-01-20 | 2003-02-12 | Eisai Co Ltd | Piperidine derivatives and pharmaceutical compositions containing the same |
| GB0002100D0 (en) | 2000-01-28 | 2000-03-22 | Novartis Ag | Organic compounds |
| WO2001056990A2 (en) | 2000-02-03 | 2001-08-09 | Eli Lilly And Company | Pyridine derivates as potentiators of glutamate receptors |
| GB0004297D0 (en) | 2000-02-23 | 2000-04-12 | Ucb Sa | 2-oxo-1 pyrrolidine derivatives process for preparing them and their uses |
| JP4064671B2 (ja) | 2000-02-25 | 2008-03-19 | エフ.ホフマン−ラ ロシュ アーゲー | アデノシン受容体モジュレーター |
| DE10012373A1 (de) | 2000-03-14 | 2001-09-20 | Dresden Arzneimittel | Verwendung von Pyrido[3,2-e]-pyrazinonen als Inhibitoren der Phosphodiesterase 5 zur Therapie von erektiler Dysfunktion |
| GB0007108D0 (en) | 2000-03-23 | 2000-05-17 | Novartis Ag | Organic compounds |
| WO2001072712A1 (en) | 2000-03-24 | 2001-10-04 | Cor Therapeutics, Inc. | ISOQUINOLONE INHIBITORS OF FACTOR Xa |
| GB0007193D0 (en) | 2000-03-25 | 2000-05-17 | Univ Manchester | Treatment of movrmrnt disorders |
| US6403588B1 (en) | 2000-04-27 | 2002-06-11 | Yamanouchi Pharmaceutical Co., Ltd. | Imidazopyridine derivatives |
| WO2001083481A1 (en) | 2000-04-27 | 2001-11-08 | Yamanouchi Pharmaceutical Co., Ltd. | Imidazopyridine derivatives |
| KR100551925B1 (ko) | 2000-04-28 | 2006-02-16 | 니혼노야쿠가부시키가이샤 | 2-할로벤조산의 제조 방법 |
| EP1288199A4 (en) | 2000-04-28 | 2005-10-12 | Shionogi & Co | INHIBITORS OF MMP-12 |
| AU2000258650A1 (en) | 2000-05-11 | 2001-11-20 | Eli Lilly And Co | 2-piperidone compounds for the treatment of cancer |
| NZ522674A (en) | 2000-05-11 | 2004-10-29 | Kenneth Curry | Novel spiro[2.4]heptane amino carboxy compounds and derivatives thereof |
| US20020009713A1 (en) | 2000-05-11 | 2002-01-24 | Miller Freda D. | Methods for identifying modulators of neuronal growth |
| US7081481B2 (en) | 2000-05-31 | 2006-07-25 | Eli Lilly And Company | Excitatory amino acid receptor modulators |
| IL152848A0 (en) | 2000-06-12 | 2003-06-24 | Eisai Co Ltd | 1,2-dihydropyridine derivatives, pharmaceutical compositions containing the same and methods for the production thereof |
| JP2002012533A (ja) | 2000-06-27 | 2002-01-15 | Kao Corp | 染毛剤組成物 |
| JP2002003401A (ja) | 2000-06-27 | 2002-01-09 | Japan Science & Technology Corp | 脳由来神経栄養因子誘導剤 |
| JP2004501627A (ja) | 2000-06-27 | 2004-01-22 | センター ナショナル デ ラ レシェルルシェ サイエンティフィック−シーエヌアールエス | 哺乳動物2pドメインメカノ感受性k+チャネル、そのクローニングおよび利用 |
| AU6785401A (en) | 2000-06-28 | 2002-01-08 | Taisho Pharmaceutical Co Ltd | Novel dicarboxylic acid derivatives |
| DE10031390A1 (de) | 2000-07-03 | 2002-01-17 | Knoll Ag | Pyrimidinderivate und ihre Verwendung zur Prophylaxe und Therapie der zerebralen Ischämie |
| US20020041880A1 (en) | 2000-07-05 | 2002-04-11 | Defeo-Jones Deborah | Method of treating cancer |
| US6861530B2 (en) | 2000-07-07 | 2005-03-01 | Kyowa Hakko Kogyo Co., Ltd. | Piperidine derivatives |
| JP2002040252A (ja) | 2000-07-27 | 2002-02-06 | Shiseido Co Ltd | コレステリック液晶層を含む光学シート、それを用いた情報記録体、情報記録方法並びに情報判別方法 |
| DE10038019A1 (de) | 2000-08-04 | 2002-02-14 | Bayer Ag | Substituierte Triazolopyrid(az)ine |
| JP4251868B2 (ja) | 2000-08-11 | 2009-04-08 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 2−アミノピリジン化合物および医薬用途 |
| CZ304344B6 (cs) | 2000-09-11 | 2014-03-19 | Novartis Vaccines & Diagnostics, Inc. | Chinolinonový derivát a jeho použití a farmaceutický prostředek s obsahem tohoto derivátu |
| WO2002022622A2 (en) | 2000-09-13 | 2002-03-21 | Georgetown University | Synthesis of 2-hydroxymethylglutamic acid and congeners thereof |
| JP2002105085A (ja) | 2000-09-28 | 2002-04-10 | Yamanouchi Pharmaceut Co Ltd | 新規イミダゾチアゾール誘導体 |
| CN1703403A (zh) | 2000-10-02 | 2005-11-30 | 詹森药业有限公司 | 趋代谢的谷氨酸盐受体拮抗剂 |
| DE10058663A1 (de) | 2000-11-25 | 2002-05-29 | Merck Patent Gmbh | Verwendung von Thienopyrimidinen |
| JPWO2002051849A1 (ja) | 2000-12-26 | 2004-04-22 | 第一製薬株式会社 | Cdk4活性阻害剤 |
| ES2330719T3 (es) | 2000-12-28 | 2009-12-15 | SHIONOGI & CO., LTD. | Derivados de 2-piridona con afinidad para el receptor cannabinoide de tipo 2. |
| JP2002308882A (ja) | 2001-02-08 | 2002-10-23 | Yamanouchi Pharmaceut Co Ltd | チエノピリミジン誘導体 |
| AU2002258400A1 (en) | 2001-02-16 | 2002-08-28 | Tularik Inc. | Methods of using pyrimidine-based antiviral agents |
| DE60239097D1 (de) | 2001-03-02 | 2011-03-17 | Gpc Biotech Ag | Drei-hybrid-assaysystem |
| DE10291003D2 (de) | 2001-03-08 | 2004-04-15 | Ilfa Industrieelektronik Und L | Mehrschichtige Leiterplatte |
| US6596731B2 (en) | 2001-03-27 | 2003-07-22 | Hoffmann-La Roche Inc. | Substituted imidazo[1,2-A] pyridine derivatives |
| CA2443144A1 (en) | 2001-04-02 | 2002-10-10 | Merck & Co., Inc. | In vivo methods of determining activity of receptor-type kinase inhibitors |
| EP1379522B1 (en) | 2001-04-12 | 2005-01-26 | F. Hoffmann-La Roche Ag | DIHYDRO-BENZO(b)(1,4)DIAZEPIN-2-ONE DERIVATIVES AS MGLUR2 ANTAGONISTS I |
| SK13682003A3 (sk) | 2001-04-12 | 2004-07-07 | F. Hoffmann-La Roche Ag | Dihydrobenzo[b][1,4]diazepin-2-ónové deriváty ako mGluR2 antagonisty II |
| SE0101579D0 (sv) | 2001-05-04 | 2001-05-04 | Astrazeneca Ab | New compounds |
| CA2446980A1 (en) | 2001-05-14 | 2002-11-21 | Argyrios G. Arvanitis | Substituted pyrazinones, pyridines and pyrimidines as corticotropin releasing factor ligands |
| US7144903B2 (en) | 2001-05-23 | 2006-12-05 | Amgen Inc. | CCR4 antagonists |
| CA2448306A1 (en) | 2001-05-30 | 2002-12-05 | Alteon Inc. | Method for treating fibrotic diseases or other indications |
| US6806268B2 (en) | 2001-05-30 | 2004-10-19 | Alteon, Inc. | Method for treating glaucoma V |
| AU2002310156A1 (en) | 2001-06-05 | 2002-12-16 | Boehringer Ingelheim Pharmaceuticals, Inc. | 1,4-disubstituted benzo-fused cycloalkyl urea compounds |
| CA2451057A1 (en) | 2001-06-14 | 2002-12-27 | Banyu Pharmaceutical Co., Ltd. | Novel isoxazolopyridone derivatives and use thereof |
| JP2005500294A (ja) | 2001-06-19 | 2005-01-06 | ブリストル−マイヤーズ スクイブ カンパニー | ホスホジエステラーゼ7に対するピリミジン阻害剤 |
| JP2003012653A (ja) | 2001-06-28 | 2003-01-15 | Yamanouchi Pharmaceut Co Ltd | キナゾリン誘導体 |
| WO2003011293A2 (en) | 2001-08-02 | 2003-02-13 | Neurocrine Biosciences, Inc. | Pyridinone and pyridazinone derivatives as gonadotropin-releasing hormone receptor antagonists |
| AU2002333524A1 (en) | 2001-09-11 | 2003-03-24 | Glaxosmithkline K.K. | Furo-and thienopyrimidine derivatives as angiogenesis inhibitors |
| ES2309206T3 (es) | 2001-10-02 | 2008-12-16 | Smithkline Beecham Corporation | Compuestos quimicos. |
| TWI330183B (es) | 2001-10-22 | 2010-09-11 | Eisai R&D Man Co Ltd | |
| TW200406466A (en) | 2001-11-13 | 2004-05-01 | Ciba Sc Holding Ag | Compositions comprising at least one oxonol dye and at least one metal complex |
| US6921762B2 (en) | 2001-11-16 | 2005-07-26 | Amgen Inc. | Substituted indolizine-like compounds and methods of use |
| GB0129260D0 (en) | 2001-12-06 | 2002-01-23 | Eisai London Res Lab Ltd | Pharmaceutical compositions and their uses |
| KR20040068240A (ko) | 2001-12-14 | 2004-07-30 | 노보 노르디스크 에이/에스 | 호르몬 민감성 리파아제의 활성을 감소시키기 위한 화합물및 그들의 사용 |
| WO2003059884A1 (en) | 2001-12-21 | 2003-07-24 | X-Ceptor Therapeutics, Inc. | Modulators of lxr |
| DE10164139A1 (de) | 2001-12-27 | 2003-07-10 | Bayer Ag | 2-Heteroarylcarbonsäureamide |
| CA2471642C (en) | 2001-12-27 | 2011-05-24 | Taisho Pharmaceutical Co., Ltd. | 6-fluorobicyclo[3.1.0]hexane derivatives |
| JP2005170790A (ja) | 2002-01-09 | 2005-06-30 | Ajinomoto Co Inc | N−アルキルスルフォニル置換アミド誘導体 |
| WO2003062192A1 (en) | 2002-01-17 | 2003-07-31 | Smithkline Beecham Corporation | Cycloalkyl ketoamides derivatives useful as cathepsin k inhibitors |
| EP1513522A2 (en) | 2002-01-18 | 2005-03-16 | Sri International | Methods of treating conditions associated with an edg receptor |
| US20050113283A1 (en) | 2002-01-18 | 2005-05-26 | David Solow-Cordero | Methods of treating conditions associated with an EDG-4 receptor |
| WO2003064428A1 (en) | 2002-01-29 | 2003-08-07 | H. Lundbeck A/S | Furano- and thienopyrimidines as neurokinase inhibitors |
| US6949542B2 (en) | 2002-02-06 | 2005-09-27 | Hoffman-La Roche Inc. | Dihydro-benzo[b][1,4]diazepin-2-one derivatives |
| AU2003210869A1 (en) | 2002-02-07 | 2003-09-02 | University Of Miami | Schwann cell and phosphodiesterase inhibitors based therapy |
| US20040116489A1 (en) | 2002-02-12 | 2004-06-17 | Massey Steven Marc | Synthetic excitatory amino acids |
| AU2003211931A1 (en) | 2002-02-13 | 2003-09-04 | Takeda Chemical Industries, Ltd. | Jnk inhibitor |
| RS52392B (sr) | 2002-02-14 | 2013-02-28 | Pharmacia Corporation | Supstituisani piridinoni kao modulatori p38 map kinaze |
| AU2003205558A1 (en) | 2002-02-19 | 2003-09-09 | H. Lundbeck A/S | Thioibotenic acid and derivatives thereof |
| US6833380B2 (en) | 2002-03-07 | 2004-12-21 | Warner-Lambert Company, Llc | Compounds that modulate PPAR activity and methods of preparation |
| MXPA04008822A (es) | 2002-03-14 | 2004-11-26 | Bayer Healthcare Ag | Aroilpiridinonas monociclicas. |
| WO2003082191A2 (en) | 2002-03-28 | 2003-10-09 | Merck & Co., Inc. | Substituted 2,3-diphenyl pyridines |
| CN1642580B (zh) | 2002-03-29 | 2010-05-26 | 詹森药业有限公司 | 放射性标记的喹啉和喹啉酮衍生物及其作为代谢性谷氨酸受体配体的制造用途 |
| WO2003084610A1 (en) | 2002-04-03 | 2003-10-16 | Eli Lilly And Company | Therapy for psychoses combining an atypical antipsychotic and an mglu2/3 receptor agonist |
| US6864261B2 (en) | 2002-05-02 | 2005-03-08 | Euro-Celtique S.A. | Therapeutic agents useful for treating pain |
| WO2003092595A2 (en) | 2002-05-02 | 2003-11-13 | Merck & Co., Inc | Tyrosine kinase inhibitors |
| CA2485166A1 (en) | 2002-05-21 | 2003-12-04 | Amgen Inc. | Substituted pyrimidinone and pyridinone compounds |
| EP1517915B1 (en) | 2002-06-11 | 2009-01-21 | Eli Lilly And Company | Prodrugs of excitatory amino acids |
| DE60309701T2 (de) | 2002-06-13 | 2007-09-06 | Vertex Pharmaceuticals Inc., Cambridge | 2-ureido-6-heteroaryl-3h-benzoimidazol-4-carbonsäurederivate und verwandte verbindungen als gyrase und/oder topoisomerase iv inhibitoren zur behandlung von bakteriellen infektionen |
| MY141867A (en) | 2002-06-20 | 2010-07-16 | Vertex Pharma | Substituted pyrimidines useful as protein kinase inhibitors |
| GB0214268D0 (en) | 2002-06-20 | 2002-07-31 | Celltech R&D Ltd | Chemical compounds |
| US20060063782A1 (en) | 2002-07-03 | 2006-03-23 | Murray Christopher W | 3-Hetero arylmethoxy ! pyridines and their analogues as p38 map kinase inhibitors |
| US7262194B2 (en) | 2002-07-26 | 2007-08-28 | Euro-Celtique S.A. | Therapeutic agents useful for treating pain |
| US20040138238A1 (en) | 2002-08-08 | 2004-07-15 | Dhanoa Dale S. | Substituted aminopyrimidine compounds as neurokinin antagonists |
| GB0218630D0 (en) | 2002-08-10 | 2002-09-18 | Tanabe Seiyaku Co | Novel compounds |
| GB0218800D0 (en) | 2002-08-13 | 2002-09-18 | Celltech R&D Ltd | Chemical compounds |
| WO2004017950A2 (en) | 2002-08-22 | 2004-03-04 | Piramed Limited | Phosphadidylinositol 3,5-biphosphate inhibitors as anti-viral agents |
| JP2006502143A (ja) | 2002-08-26 | 2006-01-19 | メルク エンド カムパニー インコーポレーテッド | 向代謝型グルタミン酸受容体のアセトフェノン増強因子 |
| EP1551369A4 (en) | 2002-08-28 | 2007-05-09 | Intermune Inc | COMBINATION THERAPY FOR THE TREATMENT OF FIBROTIC DISEASES |
| MY139563A (en) | 2002-09-04 | 2009-10-30 | Bristol Myers Squibb Co | Heterocyclic aromatic compounds useful as growth hormone secretagogues |
| BR0314192A (pt) | 2002-09-10 | 2005-07-26 | Novartis Ag | Antagonistas de receptores de mglu para tratamento de distúrbios associados com receptores de mglu incluindo vìcio e depressão |
| US20060147923A1 (en) | 2002-09-11 | 2006-07-06 | Daggett Lorrie P | Nucleic acid sequences encoding novel point mutations on mglur2 and mglur3 |
| AU2003269628A1 (en) | 2002-09-19 | 2004-04-08 | Boehringer Ingelheim (Canada) Ltd. | Non-nucleoside reverse transcriptase inhibitors |
| JP4391426B2 (ja) | 2002-09-19 | 2009-12-24 | ベーリンガー インゲルハイム (カナダ) リミテッド | 非ヌクレオシド逆転写酵素インヒビター |
| US20040058997A1 (en) | 2002-09-24 | 2004-03-25 | David Gordon Daniel | Method for treatment of disorders of personal attachment and deficient social interaction |
| TW200410975A (en) | 2002-09-26 | 2004-07-01 | Nihon Nohyaku Co Ltd | New pesticide and method for using it, new substituted thienopyrimidine derivative, its intermediate, and method for producing it |
| US7067658B2 (en) | 2002-09-30 | 2006-06-27 | Bristol-Myers Squibb Company | Pyridino and pyrimidino pyrazinones |
| US7998163B2 (en) | 2002-10-03 | 2011-08-16 | Boston Scientific Scimed, Inc. | Expandable retrieval device |
| DE60326259D1 (de) | 2002-10-23 | 2009-04-02 | Daiichi Pure Chemicals Co Ltd | Neue fructosylpeptidoxidase und deren nutzung |
| US20040138204A1 (en) | 2002-10-30 | 2004-07-15 | Harrington James Frederick | Compositions and methods for pain reduction |
| US7902203B2 (en) | 2002-11-01 | 2011-03-08 | Abbott Laboratories, Inc. | Anti-infective agents |
| EP2361913A1 (en) | 2002-11-01 | 2011-08-31 | Abbott Laboratories | Anti-infective agents |
| US6930117B2 (en) | 2002-11-09 | 2005-08-16 | The Procter & Gamble Company | N-alkyl-4-methyleneamino-3-hydroxy-2-pyridones |
| US7423148B2 (en) | 2002-11-21 | 2008-09-09 | Chiron Corporation | Small molecule PI 3-kinase inhibitors and methods of their use |
| AU2003289386A1 (en) | 2002-12-18 | 2004-07-09 | Takeda Pharmaceutical Company Limited | Jnk inhibitors |
| DE60330187D1 (de) | 2002-12-30 | 2009-12-31 | Celgene Corp | Fluoralkoxy-substituierte 1, 3-dihydro-isoindolyl-verbindungen und ihre pharmazeutischen verwendungen |
| MXPA05007485A (es) | 2003-01-14 | 2006-01-30 | Arena Pharm Inc | Derivados de arilo y heteroarilo 1,2,3-trisubstituidos como moduladores del metabolismo y la profilaxis y tratamiento de trastornos relacionados con ello tales como diabetes e hiperglicemia. |
| ITMI20030151A1 (it) | 2003-01-30 | 2004-07-31 | Recordati Ind Chimica E Farma Ceutica S P A | Uso di antagonisti selettivi del recettore mglu5 per il trattamento di disfunzioni neuromuscolari del tratto urinario inferiore. |
| EP1592684B1 (en) | 2003-02-04 | 2008-07-30 | F. Hoffmann-La Roche Ag | Malonamide derivatives as gamma-secretase inhibitors |
| DE10306250A1 (de) | 2003-02-14 | 2004-09-09 | Aventis Pharma Deutschland Gmbh | Substituierte N-Arylheterozyklen, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| ES2316964T3 (es) | 2003-02-24 | 2009-04-16 | Arena Pharmaceuticals, Inc. | Fenil-y piridilpipereidinia-derivados como moduladores del metabolismo de la glucosa. |
| PL378754A1 (pl) | 2003-03-03 | 2006-05-15 | F.Hoffmann-La Roche Ag | 2,5- i 2,6-Podstawione tetrahydroizochinoliny do stosowania jako modulatory 5-HT6 |
| ITFI20030058A1 (it) | 2003-03-06 | 2004-09-07 | Univ Firenze | Formulazioni farmaceutiche contenenti tiazolidinedioni |
| DE10311065A1 (de) | 2003-03-13 | 2004-09-23 | Abbott Gmbh & Co. Kg | Pyrimidin-2-on-Verbindungen und ihre therapeutische Verwendung |
| TW200508197A (en) | 2003-03-31 | 2005-03-01 | Ucb Sa | Indolone-acetamide derivatives, processes for preparing them and their uses |
| WO2004092123A2 (en) | 2003-04-10 | 2004-10-28 | Microbia, Inc. | Inhibitors of fungal invasion |
| JP2006523707A (ja) | 2003-04-15 | 2006-10-19 | アストラゼネカ アクツィエボラーグ | 治療化合物 |
| JP2004339080A (ja) | 2003-05-13 | 2004-12-02 | Tokyo Institute Of Technology | ピラゾ−ル誘導体を含有する高血圧治療剤 |
| JP4847868B2 (ja) | 2003-05-14 | 2011-12-28 | ニューロジェネティック ファーマシューティカルズ、 インコーポレイテッド | 化合物、及び、アミロイドベータの調節におけるその使用 |
| WO2005040337A2 (en) | 2003-05-20 | 2005-05-06 | The Regents Of The University Of California | METHODS FOR BINDING AGENTS TO β-AMYLOID PLAQUES |
| GB0315950D0 (en) | 2003-06-11 | 2003-08-13 | Xention Discovery Ltd | Compounds |
| WO2005002585A1 (en) | 2003-07-02 | 2005-01-13 | Warner-Lambert Company Llc | Combination of an allosteric inhibitor of matrix metalloproteinase-13 and a ligand to an alpha-2-delta receptor |
| WO2005007144A2 (en) | 2003-07-14 | 2005-01-27 | Decode Genetics Ehf | Method of diagnosis and treatment for asthma based on haplotype association |
| EA009920B1 (ru) | 2003-08-29 | 2008-04-28 | Вернэлис (Кембридж) Лимитед | Соединения пиримидотиофена |
| GB0320300D0 (en) | 2003-08-29 | 2003-10-01 | Cancer Rec Tech Ltd | Pyrimidothiophene compounds |
| GB0322016D0 (en) | 2003-09-19 | 2003-10-22 | Merck Sharp & Dohme | New compounds |
| EA010031B1 (ru) | 2003-12-02 | 2008-06-30 | Юсб, С.А. | Производные имидазола, способы их получения и применения |
| TW200533664A (en) | 2004-02-18 | 2005-10-16 | Astrazeneca Ab | Tetrazole compounds and their use as metabotropic glutamate receptor antagonists |
| DE102004009039A1 (de) | 2004-02-23 | 2005-09-08 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 8-[3-Amino-piperidin-1-yl]-xanthine, deren Herstellung und Verwendung als Arzneimittel |
| US7306631B2 (en) | 2004-03-30 | 2007-12-11 | The Procter & Gamble Company | Keratin dyeing compounds, keratin dyeing compositions containing them, and use thereof |
| AU2005233437A1 (en) | 2004-04-12 | 2005-10-27 | Sankyo Company, Limited | Thienopyridine derivatives |
| US7459562B2 (en) | 2004-04-23 | 2008-12-02 | Bristol-Myers Squibb Company | Monocyclic heterocycles as kinase inhibitors |
| WO2005121082A1 (en) | 2004-06-11 | 2005-12-22 | Ucb, S.A. | Process for preparing 2-oxo-1-pyrrolidine derivatives by intramolecular allylation |
| GB0413605D0 (en) | 2004-06-17 | 2004-07-21 | Addex Pharmaceuticals Sa | Novel compounds |
| US8063004B2 (en) | 2004-07-22 | 2011-11-22 | Malcera, L.L.C. | Chemical composition of matter for the liquefaction and dissolution of asphaltene and paraffin sludges into petroleum crude oils and refined products at ambient temperatures and method of use |
| EP1773792A1 (en) | 2004-07-30 | 2007-04-18 | Merck & Co., Inc. | Indanone potentiators of metabotropic glutamate receptors |
| EP1773774A4 (en) | 2004-07-30 | 2009-12-30 | Merck & Co Inc | HETEROCYCLIC ACETOPHENONE AMPLIFIERS OF METABOTROPIC GLUTAMATE RECEPTORS |
| JP2008508337A (ja) | 2004-08-02 | 2008-03-21 | シュバルツ ファルマ アクチェンゲゼルシャフト | インドリジンカルボキサミド並びにそのアザ及びジアザ誘導体 |
| WO2006024970A1 (en) | 2004-08-11 | 2006-03-09 | Koninklijke Philips Electronics, N.V. | Ultrasonic diagnosis of ischemic cardiodisease |
| TW200613272A (en) | 2004-08-13 | 2006-05-01 | Astrazeneca Ab | Isoindolone compounds and their use as metabotropic glutamate receptor potentiators |
| BRPI0514391A (pt) | 2004-08-18 | 2008-06-10 | Pharmacia & Upjohn Co Llc | compostos de triazolopiridina para o tratamento de inflamação |
| DE102004044884A1 (de) | 2004-09-14 | 2006-05-24 | Grünenthal GmbH | Substituierte bizyklische Imidazo-3-yl-amin-Verbindungen |
| GB0420722D0 (en) | 2004-09-17 | 2004-10-20 | Addex Pharmaceuticals Sa | Novel allosteric modulators |
| GB0420719D0 (en) | 2004-09-17 | 2004-10-20 | Addex Pharmaceuticals Sa | Novel allosteric modulators |
| EP1799273A2 (en) | 2004-10-07 | 2007-06-27 | Koninklijke Philips Electronics N.V. | Use of the staudinger ligation in imaging and therapy end kits for imaging and therapy |
| CN101048157A (zh) | 2004-10-25 | 2007-10-03 | 默克公司 | 代谢型谷氨酸受体的杂环二氢茚酮增强剂 |
| ES2333979T3 (es) | 2004-11-22 | 2010-03-03 | Eli Lilly And Company | Potenciadores de los receptores del glutamato. |
| US7434262B2 (en) | 2004-12-08 | 2008-10-07 | At&T Intellectual Property I, L.P. | Methods and systems that selectively resurrect blocked communications between devices |
| DE102004061288A1 (de) | 2004-12-14 | 2006-06-29 | Schering Ag | 3-Amino-Pyrazolo[3,4b]pyridine als Inhibitoren von Proteintyrosinkinasen, deren Herstellung und Verwendung als Arzneimittel |
| KR20070106690A (ko) | 2004-12-27 | 2007-11-05 | 아스트라제네카 아베 | 피라졸론 화합물 및 대사성 글루타메이트 수용체길항제로서 그들의 용도 |
| US7456289B2 (en) | 2004-12-31 | 2008-11-25 | National Health Research Institutes | Anti-tumor compounds |
| EP1855670A4 (en) | 2005-02-24 | 2010-05-05 | Merck Sharp & Dohme | BENZAZOLE AMPLIFIERS OF METABOTROPIC GLUTAMATE RECEPTORS |
| DE602006012815D1 (de) | 2005-03-23 | 2010-04-22 | Hoffmann La Roche | Acetylenylpyrazolopyrimidinderivate als mglur2-antagonsten |
| AU2006234627C1 (en) | 2005-04-08 | 2009-11-26 | Eisai R & D Management Co., Ltd. | Therapeutic agent for dyskinesia |
| EP2308870A3 (en) | 2005-06-01 | 2011-10-19 | UCB Pharma S.A. | 2-oxo-1-pyrrolidine deriatives, processes for preparing them and their uses |
| US7572807B2 (en) | 2005-06-09 | 2009-08-11 | Bristol-Myers Squibb Company | Heteroaryl 11-beta-hydroxysteroid dehydrogenase type I inhibitors |
| US7579360B2 (en) | 2005-06-09 | 2009-08-25 | Bristol-Myers Squibb Company | Triazolopyridine 11-beta hydroxysteroid dehydrogenase type I inhibitors |
| WO2006137350A1 (ja) | 2005-06-22 | 2006-12-28 | Kissei Pharmaceutical Co., Ltd. | 新規なフロピリジン誘導体、それを含有する医薬組成物およびそれらの用途 |
| KR20080035576A (ko) | 2005-08-05 | 2008-04-23 | 아스트라제네카 아베 | 트리사이클릭 벤즈이미다졸 및 대사체 글루타메이트 수용체조절제로서 이들의 용도 |
| CN101309905A (zh) | 2005-08-12 | 2008-11-19 | 阿斯利康(瑞典)有限公司 | 取代的异吲哚酮及其作为代谢型谷氨酸受体增效剂的用途 |
| WO2007027669A1 (en) | 2005-08-29 | 2007-03-08 | Cps Biofuels, Inc. | Improved biodiesel fuel, additives, and lubbricants |
| EP1764099A3 (en) | 2005-09-17 | 2007-05-09 | Speedel Experimenta AG | Diaminoalcohol derivatives for the treatment of Alzheimer, malaria, HIV |
| CN101268074B (zh) | 2005-09-17 | 2011-09-21 | 斯皮德尔实验股份公司 | 5-氨基-4-羟基-7-(咪唑并[1,2-a]吡啶-6-基甲基)-8-甲基-壬酰胺衍生物和相关用作肾素抑制剂治疗高血压的化合物 |
| US8492428B2 (en) | 2005-09-20 | 2013-07-23 | Mayo Foundation For Medical Education And Research | Small-molecule botulinum toxin inhibitors |
| AU2006298829B2 (en) | 2005-09-27 | 2011-03-03 | F. Hoffmann-La Roche Ag | Oxadiazolyl pyrazolo-pyrimidines as mGluR2 antagonists |
| ES2364901T3 (es) | 2005-11-15 | 2011-09-16 | Array Biopharma, Inc. | Procesos e intermedios para la preparación de derivados de n4-fenil-quinazolin-4-amina. |
| EP1963325A2 (en) | 2005-12-07 | 2008-09-03 | UCB Pharma, S.A. | Xanthine derivatives, processes for preparing them and their uses |
| TW200732313A (en) | 2005-12-15 | 2007-09-01 | Astrazeneca Ab | Oxazolidinone compounds and their use as metabotropic glutamate receptor potentiators |
| US8338621B2 (en) | 2005-12-21 | 2012-12-25 | Ucb S.A. | Process for the preparation of 2-oxo-1-pyrrolidine derivatives |
| TW200804281A (en) | 2006-02-16 | 2008-01-16 | Astrazeneca Ab | New metabotropic glutamate receptor-potentiating isoindolones |
| EP1993539A4 (en) | 2006-03-02 | 2010-05-19 | Glaxosmithkline Llc | THIAZOLONE AS A PI3 KINASE INHIBITOR |
| TWI417095B (zh) | 2006-03-15 | 2013-12-01 | Janssen Pharmaceuticals Inc | 1,4-二取代之3-氰基-吡啶酮衍生物及其作為mGluR2-受體之正向異位性調節劑之用途 |
| GB0606774D0 (en) | 2006-04-03 | 2006-05-10 | Novartis Ag | Organic compounds |
| GB0608263D0 (en) | 2006-04-26 | 2006-06-07 | Glaxo Group Ltd | Compounds |
| WO2007135527A2 (en) | 2006-05-23 | 2007-11-29 | Pfizer Products Inc. | Benzimidazolyl compounds |
| WO2007135529A2 (en) | 2006-05-23 | 2007-11-29 | Pfizer Products Inc. | Azabenzimidazolyl compounds as mglur2 potentiators |
| EA019757B1 (ru) | 2006-06-15 | 2014-06-30 | ЮСиБи ФАРМА ГМБХ | Фармацевтическая композиция с синергетическим противосудорожным эффектом |
| CA2655573C (en) | 2006-06-19 | 2014-04-29 | Toray Industries, Inc. | Therapeutic or prophylactic agent for multiple sclerosis |
| US20100035756A1 (en) | 2006-07-12 | 2010-02-11 | Syngenta Limited | Triazolophyridine derivatives as herbicides |
| US8198448B2 (en) | 2006-07-14 | 2012-06-12 | Amgen Inc. | Fused heterocyclic derivatives and methods of use |
| US8217177B2 (en) | 2006-07-14 | 2012-07-10 | Amgen Inc. | Fused heterocyclic derivatives and methods of use |
| PE20080403A1 (es) | 2006-07-14 | 2008-04-25 | Amgen Inc | Derivados heterociclicos fusionados y metodos de uso |
| WO2008012623A1 (en) | 2006-07-25 | 2008-01-31 | Pfizer Products Inc. | Benzimidazolyl compounds as potentiators of mglur2 subtype of glutamate receptor |
| WO2008012622A2 (en) | 2006-07-25 | 2008-01-31 | Pfizer Products Inc. | Azabenzimidazolyl compounds as potentiators of mglur2 subtype of glutamate receptor |
| WO2008032191A2 (en) | 2006-09-13 | 2008-03-20 | Astrazeneca Ab | Spiro-oxazolidinone compounds and their use as metabotropic glutamate receptor potentiators |
| EP2086973B1 (en) | 2006-10-11 | 2012-01-25 | Amgen Inc., | Imidazo- and triazolo-pyridine compounds and methods of use therof |
| US7994190B2 (en) | 2006-11-01 | 2011-08-09 | Bristol-Myers Squibb Company | Modulators of glucocorticoid receptor, AP-1, and/or NF-κB activity and use thereof |
| TW200831085A (en) | 2006-12-13 | 2008-08-01 | Merck & Co Inc | Non-nucleoside reverse transcriptase inhibitors |
| CN101679409B (zh) | 2006-12-22 | 2014-11-26 | Astex治疗学有限公司 | 双环杂环衍生化合物、其医药组合物和其用途 |
| WO2008078091A1 (en) | 2006-12-22 | 2008-07-03 | Astex Therapeutics Limited | Bicyclic heterocyclic compounds as fgfr inhibitors |
| CN101605792A (zh) | 2007-02-09 | 2009-12-16 | 阿斯利康(瑞典)有限公司 | 氮杂-异吲哚酮和它们作为亲代谢性谷氨酸受体增效剂-613的用途 |
| ES2320955B1 (es) | 2007-03-02 | 2010-03-16 | Laboratorios Almirall S.A. | Nuevos derivados de 3-((1,2,4)triazolo(4,3-a)piridin-7-il)benzamida. |
| GB0704407D0 (en) | 2007-03-07 | 2007-04-18 | Glaxo Group Ltd | Compounds |
| TW200900065A (en) | 2007-03-07 | 2009-01-01 | Janssen Pharmaceutica Nv | 3-cyano-4-(4-pyridinyloxy-phenyl)-pyridin-2-one derivatives |
| ATE537157T1 (de) | 2007-03-07 | 2011-12-15 | Janssen Pharmaceutica Nv | Substituierte phenoxy-n-alkylierte thiazoledindione als östrogenassoziierte rezeptor-alpha-modulatoren |
| CL2008000667A1 (es) | 2007-03-07 | 2008-11-03 | Janssen Pharmaceutica Nv | Compuestos derivados de fenoxi aminotiazolonas sustituidas, modulares de receptores alfa relacionados con estrogenos; composion farmaceutica que comprende a dichos compuestos; y sus uso para tratar cancer, sindrome metabolico, obesidad, diabetes. |
| TW200845978A (en) | 2007-03-07 | 2008-12-01 | Janssen Pharmaceutica Nv | 3-cyano-4-(4-tetrahydropyran-phenyl)-pyridin-2-one derivatives |
| TW200902508A (en) | 2007-03-07 | 2009-01-16 | Janssen Pharmaceutica Nv | Substituted phenoxy thiazolidinediones as estrogen related receptor-α modulators |
| TW200900391A (en) | 2007-03-07 | 2009-01-01 | Janssen Pharmaceutica Nv | 3-cyano-4-(4-phenyl-piperidin-1-yl)-pyridin-2-one derivatives |
| NZ579431A (en) | 2007-03-09 | 2012-04-27 | Sanofi Aventis | Substituted dihydro and tetrahydro oxazolopyrimidinones, preparation and use thereof |
| WO2008124085A2 (en) | 2007-04-03 | 2008-10-16 | Exelixis, Inc. | Methods of using combinations of mek and jak-2 inhibitors |
| WO2008130853A1 (en) | 2007-04-17 | 2008-10-30 | Astrazeneca Ab | Hydrazides and their use as metabotropic glutamate receptor potentiators - 681 |
| WO2008132142A2 (en) | 2007-04-27 | 2008-11-06 | Ucb Pharma S.A. | New heterocyclic derivatives useful for the treatment of cns disorders |
| TWI443090B (zh) | 2007-05-25 | 2014-07-01 | Abbvie Deutschland | 作為代謝性麩胺酸受體2(mglu2 受體)之正向調節劑之雜環化合物 |
| TWI417100B (zh) | 2007-06-07 | 2013-12-01 | Astrazeneca Ab | 二唑衍生物及其作為代謝型麩胺酸受體增效劑-842之用途 |
| TW200911255A (en) | 2007-06-07 | 2009-03-16 | Astrazeneca Ab | Metabotropic glutamate receptor oxadiazole ligands and their use as potentiators-841 |
| WO2009004430A1 (en) | 2007-06-29 | 2009-01-08 | Pfizer Inc. | N-benzyl oxazolidinones and related heterocycleic compounds as potentiators of glutamate receptors |
| PL2203439T3 (pl) | 2007-09-14 | 2011-06-30 | Addex Pharmaceuticals Sa | 1',3'-dipodstawione-4-fenylo-3,4,5,6-tetrahydro-2H, 1'H-[1, 4']bipirydynylo-2'-ketony |
| US8252937B2 (en) | 2007-09-14 | 2012-08-28 | Janssen Pharmaceuticals, Inc. | 1,3-disubstituted 4-(aryl-X-phenyl)-1H-pyridin-2-ones |
| US8722894B2 (en) | 2007-09-14 | 2014-05-13 | Janssen Pharmaceuticals, Inc. | 1,3-disubstituted-4-phenyl-1H-pyridin-2-ones |
| WO2009041567A1 (ja) | 2007-09-27 | 2009-04-02 | Banyu Pharmaceutical Co., Ltd. | メラニン凝集ホルモン受容体拮抗作用を有するジアリールケチミン誘導体 |
| US8119658B2 (en) | 2007-10-01 | 2012-02-21 | Bristol-Myers Squibb Company | Triazolopyridine 11-beta hydroxysteroid dehydrogenase type I inhibitors |
| RU2492170C9 (ru) | 2007-11-14 | 2013-12-27 | Орто-Макнейл-Янссен Фармасьютикалз, Инк. | Имидазо[1,2-а]пиридиновые производные и их применение в качестве положительных аллостерических модуляторов рецепторов mglur2 |
| AU2009206658A1 (en) | 2008-01-24 | 2009-07-30 | Merck Sharp & Dohme Corp. | 3,5-substituted-1,3-oxazolidin-2-one derivatives |
| JP5309162B2 (ja) | 2008-03-06 | 2013-10-09 | サノフイ | 置換されたジヒドロ、トリヒドロ及びテトラヒドロシクロアルキルオキサゾロピリミジノン、その製造及び使用 |
| DE102008001056A1 (de) | 2008-04-08 | 2009-10-15 | Robert Bosch Gmbh | Umlenkeinrichtung für einen Strahl einer elektromagnetischen Welle |
| US20110065669A1 (en) | 2008-05-15 | 2011-03-17 | Merck Sharp & Dohme Corp. | Oxazolobenzimidazole derivatives |
| AU2009246626A1 (en) | 2008-05-15 | 2009-11-19 | Merck Sharp & Dohme Corp. | Oxazolobenzimidazole derivatives |
| SA109300358B1 (ar) | 2008-06-06 | 2012-11-03 | استرازينيكا ايه بي | مقويات مستقبل جلوتامات ذي انتحاء أيضي من أيزو إندولون |
| TW201006801A (en) | 2008-07-18 | 2010-02-16 | Lilly Co Eli | Imidazole carboxamides |
| UY32049A (es) | 2008-08-14 | 2010-03-26 | Takeda Pharmaceutical | Inhibidores de cmet |
| TWI496779B (zh) | 2008-08-19 | 2015-08-21 | Array Biopharma Inc | 作為pim激酶抑制劑之三唑吡啶化合物 |
| WO2010022081A1 (en) | 2008-08-19 | 2010-02-25 | Array Biopharma Inc. | Triazolopyridine compounds as pim kinase inhibitors |
| JP5547194B2 (ja) | 2008-09-02 | 2014-07-09 | ジャンセン ファーマシューティカルズ, インコーポレイテッド. | 代謝型グルタミン酸受容体の調節因子としての3−アザビシクロ[3.1.0]ヘキシル誘導体 |
| RU2517181C2 (ru) | 2008-10-16 | 2014-05-27 | Орто-Макнейл-Янссен Фармасьютикалз, Инк. | Производные индола и бензоморфолина в качестве модулятора метаботропных глутаматных рецепторов |
| WO2010060589A1 (en) | 2008-11-28 | 2010-06-03 | Ortho-Mcneil-Janssen Pharmaceuticals, Inc. | Indole and benzoxazine derivatives as modulators of metabotropic glutamate receptors |
| AT507619B1 (de) | 2008-12-05 | 2011-11-15 | Oesterreichisches Forschungs Und Pruefzentrum Arsenal Ges M B H | Verfahren zur approximation des zeitlichen verlaufs von verkehrsdaten |
| CA2744946A1 (en) | 2009-02-04 | 2010-08-12 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11.beta.-hydroxysteroid dehydrogenase 1 |
| EP2393483B1 (en) | 2009-02-09 | 2017-06-28 | UCB Biopharma SPRL | Pharmaceutical compositions comprising brivaracetam |
| WO2010114726A1 (en) | 2009-03-31 | 2010-10-07 | Merck Sharp & Dohme Corp. | Aminobenzotriazole derivatives |
| EP2417135A1 (en) | 2009-04-07 | 2012-02-15 | Schering Corporation | Substituted triazolopyridines and analogs thereof |
| MY153913A (en) | 2009-05-12 | 2015-04-15 | Janssen Pharmaceuticals Inc | 7-aryl-1,2,4-triazolo[4,3-a]pyridine derivatives and their use as positive allosteric modulators of mglur2 receptors |
| CN102439008B (zh) * | 2009-05-12 | 2015-04-29 | 杨森制药有限公司 | 1,2,4-三唑并[4,3-a]吡啶衍生物及其用于治疗或预防神经和精神病症的用途 |
| MY153912A (en) | 2009-05-12 | 2015-04-15 | Janssen Pharmaceuticals Inc | 1, 2, 4,-triazolo[4,3-a[pyridine derivatives and their use as positive allosteric modulators of mglur2 receptors |
| WO2010141360A1 (en) | 2009-06-05 | 2010-12-09 | Merck Sharp & Dohme Corp. | Biaryl benzotriazole derivatives |
| US8952034B2 (en) | 2009-07-27 | 2015-02-10 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
| EP2470527A4 (en) | 2009-08-20 | 2013-02-27 | Merck Sharp & Dohme | ETHER-benzotriazole |
| CN102002040A (zh) | 2009-09-01 | 2011-04-06 | 上海药明康德新药开发有限公司 | 一种三唑并吡啶环化合物的合成方法 |
| AR078171A1 (es) | 2009-09-15 | 2011-10-19 | Sanofi Aventis | Dihidrobenzocicloalquiloximetil-oxazolopirimidinonas sustituidas, preparacion y uso de las mismas |
| WO2011034741A1 (en) | 2009-09-15 | 2011-03-24 | Merck Sharp & Dohme Corp. | Imidazopyridin-2-one derivatives |
| AR078173A1 (es) | 2009-09-15 | 2011-10-19 | Sanofi Aventis | Bifeniloximetil dihidro oxazolopirimidinonas sustituidas, su preparacion y su uso |
| AR078172A1 (es) | 2009-09-15 | 2011-10-19 | Sanofi Aventis | Fenoximetil dihidro oxazolopirimidinonas sustituidas y uso de las mismas como moduladores de receptores metabotropicos de mglur |
| JP5204071B2 (ja) | 2009-09-25 | 2013-06-05 | パナソニック株式会社 | 電気かみそり |
| CA2778194C (en) | 2009-10-23 | 2016-07-12 | Ucb Pharma, S.A. | 2-oxo-1-pyrrolidinyl imidazothiadiazole derivatives |
| EP2496569A2 (en) | 2009-11-02 | 2012-09-12 | MSD Oss B.V. | Heterocyclic derivatives |
| KR20120101551A (ko) | 2009-12-18 | 2012-09-13 | 얀센 파마슈티카 엔.브이. | Mglur5 수용체의 알로스테릭 조절자로서 바이사이클릭 티아졸 |
| AR079343A1 (es) | 2009-12-21 | 2012-01-18 | Lilly Co Eli | Compuesto de acido 2-amino-4-(4h-1,2,4-triazol-3-ilsulfanil)biciclo[3.1.0]hexano-2,6-dicarboxilico, composicion farmaceutica que lo comprende y su uso para preparar un medicamento util para tratar un trastorno psiquiatrico |
| WO2011109277A1 (en) | 2010-03-04 | 2011-09-09 | Merck Sharp & Dohme Corp. | Positive allosteric modulators of mglur2 |
| US8748632B2 (en) | 2010-03-19 | 2014-06-10 | Sanford-Burnham Medical Research Institute | Positive allosteric modulators of group II mGluRs |
| US8735397B2 (en) | 2010-03-29 | 2014-05-27 | Vanderbilt University | Method for treating schizophrenia and related diseases |
| US8314120B2 (en) | 2010-03-30 | 2012-11-20 | Abbott Gmbh & Co. Kg | Small molecule potentiators of metabotropic glutamate receptors |
| US8664214B2 (en) | 2010-03-30 | 2014-03-04 | AbbVie Deutschland GmbH & Co. KG | Small molecule potentiators of metabotropic glutamate receptors I |
| EP2563143A4 (en) | 2010-04-29 | 2013-09-25 | Merck Sharp & Dohme | SUBSTITUTED 1,3-BENZOTHIAZOL-2 (3H) -ONE AND [1,3-] THIAZOLO- [5,4-B] PYRIDINE-2 (-1H) -ONE AS POSITIVE ALLOSTERE MODULATORS OF MGLUR2 |
| KR20130094179A (ko) | 2010-04-30 | 2013-08-23 | 아스트라제네카 아베 | 대사형 글루탐산 수용체 양성 알로스테릭 조정자의 다형체 |
| EP2579717A4 (en) | 2010-06-09 | 2013-12-11 | Merck Sharp & Dohme | POSITIVE ALLOSTERIC MODULATORS OF MGLUR2 |
| CN101893589B (zh) | 2010-06-29 | 2012-10-17 | 中国人民解放军第三0二医院 | 一种无菌检查方法及其使用的全封闭集菌安瓿培养器 |
| AP2012006631A0 (en) | 2010-08-12 | 2012-12-31 | Boehringer Ingelheim Int | 6-Cycloalkyl-1, 5-dihydro-pyrazolo[3,4-D] pyrimidin-4-one derivatives and their use as PDE9A inhibitors |
| US8993779B2 (en) | 2010-08-12 | 2015-03-31 | Merck Sharp & Dohme Corp. | Positive allosteric modulators of MGLUR2 |
| US8785481B2 (en) | 2010-09-29 | 2014-07-22 | Merck Sharp & Dohme Corp. | Ether benzotriazole derivatives |
| CA2815120A1 (en) | 2010-11-08 | 2012-05-18 | Jose Ignacio Andres-Gil | Radiolabelled mglur2 pet ligands |
| EP2661435B1 (en) | 2010-11-08 | 2015-08-19 | Janssen Pharmaceuticals, Inc. | 1,2,4-TRIAZOLO[4,3-a]PYRIDINE DERIVATIVES AND THEIR USE AS POSITIVE ALLOSTERIC MODULATORS OF MGLUR2 RECEPTORS |
| PL2649069T3 (pl) | 2010-11-08 | 2016-01-29 | Janssen Pharmaceuticals Inc | Pochodne 1,2,4-triazolo[4,3-a]pirydyny i ich zastosowanie jako dodatnich allosterycznych modulatorów receptorów mGluR2 |
| CN103261195B (zh) | 2010-11-08 | 2015-09-02 | 杨森制药公司 | 1,2,4-三唑并[4,3-a]吡啶衍生物及其作为MGLUR2受体的正变构调节剂的用途 |
| BR112013020283A2 (pt) | 2011-02-09 | 2016-07-19 | Univ Johns Hopkins | "uso de inibidor de proteína de vesícula sináptica 2a (sv2a), valproato, levetiracetam, brivarecetam e seletracetam no tratamento de deficiência cognitiva, bem como composição farmacêutica compreendendo os mesmos" |
| WO2012143116A1 (en) | 2011-04-18 | 2012-10-26 | Ucb Pharma, S.A. | 4-oxo-1-imidazolidinyl imidazothiadiazole derivatives |
| US8957218B2 (en) | 2011-04-18 | 2015-02-17 | Ucb Pharma S.A. | 2-oxo-1-imidazolidinyl imidazothiadiazole derivatives |
| US9139576B2 (en) | 2011-05-03 | 2015-09-22 | Merck Sharp & Dohme Corp. | Aminomethyl biaryl benzotriazole derivatives |
| US20140088151A1 (en) | 2011-05-03 | 2014-03-27 | Douglas C. Beshore | Alhydroxymethyl biaryl benzotriazole derivatives |
| EP2705024B1 (en) | 2011-05-03 | 2015-12-16 | Merck Sharp & Dohme Corp. | Alkyne benzotriazole derivatives |
| US8765767B2 (en) | 2012-03-16 | 2014-07-01 | Bristol-Myers Squibb Company | Positive allosteric modulators of mGluR2 |
| US20130289019A1 (en) | 2012-04-26 | 2013-10-31 | Amazing Grace, Inc. | Methods of treating behaviorial and/or mental disorders |
| JP6211797B2 (ja) | 2012-05-14 | 2017-10-11 | パナソニック株式会社 | 除臭剤を備えた除臭装置、および除臭装置を備えた冷蔵庫 |
| US9682980B2 (en) | 2012-06-21 | 2017-06-20 | Bristol-Myers Squibb Company | Positive allosteric modulators of mGluR2 |
| US20140206667A1 (en) | 2012-11-14 | 2014-07-24 | Michela Gallagher | Methods and compositions for treating schizophrenia |
| WO2014078377A1 (en) | 2012-11-14 | 2014-05-22 | Agenebio, Inc. | Methods and compositions for treating schizophrenia |
| WO2014144663A1 (en) | 2013-03-15 | 2014-09-18 | The Johns Hopkins University | Methods and compositions for improving cognitive function |
| US9191739B2 (en) | 2013-03-25 | 2015-11-17 | Bose Corporation | Active reduction of harmonic noise from multiple rotating devices |
| JP6211798B2 (ja) | 2013-05-17 | 2017-10-11 | 富士機械製造株式会社 | 基板上の異物検査方法および異物検査装置 |
| JO3368B1 (ar) | 2013-06-04 | 2019-03-13 | Janssen Pharmaceutica Nv | مركبات 6، 7- ثاني هيدرو بيرازولو [5،1-a] بيرازين- 4 (5 يد)- اون واستخدامها بصفة منظمات تفارغية سلبية لمستقبلات ميجلور 2 |
| JO3367B1 (ar) | 2013-09-06 | 2019-03-13 | Janssen Pharmaceutica Nv | مركبات 2،1، 4- ثلاثي زولو [3،4-a] بيريدين واستخدامها بصفة منظمات تفارغية موجبة لمستقبلات ميجلور 2 |
| JOP20150179B1 (ar) | 2014-08-01 | 2021-08-17 | Janssen Pharmaceutica Nv | مركبات 6 ، 7 ثاني هيدرو بيرازولو [ 1، 5 الفا ] بيرازين – 4 (5 يد) – اون واستخدامها كمنظمات الوسترية سلبية لمستقبلات ملجور 2 |
| RU2695651C2 (ru) | 2014-12-11 | 2019-07-25 | Янссен Фармацевтика Нв | 1,2,4-ТРИАЗОЛО[4,3-a]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПОЛОЖИТЕЛЬНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ РЕЦЕПТОРОВ MGLUR2 |
| JP7070018B2 (ja) | 2018-04-19 | 2022-05-18 | コニカミノルタ株式会社 | 設定制御装置、設定制御方法およびプログラム |
-
2015
- 2015-01-20 HU HUE18182695A patent/HUE053734T2/hu unknown
- 2015-01-20 IL IL279202A patent/IL279202B2/en unknown
- 2015-01-20 ES ES18182695T patent/ES2860298T3/es active Active
- 2015-01-20 KR KR1020207009124A patent/KR20200036063A/ko not_active Ceased
- 2015-01-20 KR KR1020207031423A patent/KR20200126026A/ko not_active Ceased
- 2015-01-20 ES ES15700726T patent/ES2748633T3/es active Active
- 2015-01-20 NZ NZ72238515A patent/NZ722385A/en unknown
- 2015-01-20 LT LTEP18182695.9T patent/LT3431106T/lt unknown
- 2015-01-20 LT LTEP15700726.1T patent/LT3096790T/lt unknown
- 2015-01-20 ME MEP-2019-264A patent/ME03518B/me unknown
- 2015-01-20 DK DK15700726.1T patent/DK3096790T3/da active
- 2015-01-20 HR HRP20191646 patent/HRP20191646T1/hr unknown
- 2015-01-20 PT PT157007261T patent/PT3096790T/pt unknown
- 2015-01-20 UA UAA201608913A patent/UA121965C2/uk unknown
- 2015-01-20 HU HUE15700726A patent/HUE045610T2/hu unknown
- 2015-01-20 PH PH1/2019/500127A patent/PH12019500127B1/en unknown
-
2016
- 2016-07-15 CL CL2016001815A patent/CL2016001815A1/es unknown
- 2016-07-17 IL IL246802A patent/IL246802B/en active IP Right Grant
-
2019
- 2019-01-17 PH PH12019500128A patent/PH12019500128A1/en unknown
- 2019-03-07 JP JP2019041890A patent/JP6684936B2/ja not_active Expired - Fee Related
- 2019-03-07 JP JP2019041891A patent/JP6684374B2/ja active Active
- 2019-05-14 IL IL266624A patent/IL266624B2/en unknown
- 2019-05-14 IL IL266622A patent/IL266622B/en unknown
- 2019-12-19 US US16/720,750 patent/US11369606B2/en active Active
-
2021
- 2021-01-03 HR HRP20210001TT patent/HRP20210001T1/hr unknown
- 2021-04-20 CL CL2021001000A patent/CL2021001000A1/es unknown
- 2021-04-20 CL CL2021000999A patent/CL2021000999A1/es unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20250288584A1 (en) | Combinations comprising positive allosteric modulators or orthosteric agonists of metabotropic glutamatergic receptor subtype 2 and their use | |
| ES2860298T3 (es) | Combinaciones que comprenden moduladores alostéricos positivos del receptor glutamatérgico metabotrópico de subtipo 2 y su uso | |
| BR122020008941B1 (pt) | Combinações compreendendo moduladores alostéricos positivos ou agonistas ortostéricos do receptor glutamatérgico metabotrópico do subtipo 2, composição farmacêutica, e seus usos | |
| BR122021006155B1 (pt) | Produto compreendendo modulador alostérico positivo do receptor glutamatérgico metabotrópico do subtipo 2 ou ligante da proteína de vesícula sináptica 2a | |
| HK40053171A (en) | Combinations comprising positive allosteric modulators or orthosteric agonists of metabotropic glutamatergic receptor subtype 2 and their use | |
| EA045021B1 (ru) | Комбинации, содержащие положительные аллостерические модуляторы метаботропного глутаматергического рецептора 2 подтипа, и их применение |