ES2880402T3 - Conjugados de anticuerpo-fármaco basados en eribulina y métodos de uso - Google Patents
Conjugados de anticuerpo-fármaco basados en eribulina y métodos de uso Download PDFInfo
- Publication number
- ES2880402T3 ES2880402T3 ES17711475T ES17711475T ES2880402T3 ES 2880402 T3 ES2880402 T3 ES 2880402T3 ES 17711475 T ES17711475 T ES 17711475T ES 17711475 T ES17711475 T ES 17711475T ES 2880402 T3 ES2880402 T3 ES 2880402T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- seq
- linker
- cancer
- moiety
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC1[C@](CC[C@@](C[C@]2C)*[C@](C)C2=C)*C(CCC(CC2)(*C(C3)[C@@]4(C)OC(CC(CC(C*[C@@]5CC(C*#C)OC)[C@@]5OC)=O)CC5)OC2C3OC45I=C)C1 Chemical compound CC1[C@](CC[C@@](C[C@]2C)*[C@](C)C2=C)*C(CCC(CC2)(*C(C3)[C@@]4(C)OC(CC(CC(C*[C@@]5CC(C*#C)OC)[C@@]5OC)=O)CC5)OC2C3OC45I=C)C1 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68033—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a maytansine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/10—Immunoglobulins specific features characterized by their source of isolation or production
- C07K2317/14—Specific host cells or culture conditions, e.g. components, pH or temperature
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Cell Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un conjugado de anticuerpo-fármaco de Fórmula (I): Ab-(L-D)p (I) en donde (i) Ab es un anticuerpo anti-receptor alfa de folato internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ ID NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT. (ii) D es eribulina; (iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y (iv) p es un número entero de 1 a 8.
Description
DESCRIPCIÓN
Conjugados de anticuerpo-fármaco basados en eribulina y métodos de uso
CAMPO DE LA INVENCIÓN
La presente descripción se relaciona con conjugados de anticuerpo fármaco (ADC, por sus siglas en inglés) que se unen a dianas antigénicas oncológicas humanas tales como el receptor alfa de folato y/o proporcionan actividad de fármaco antitubulina. La descripción, además, se relaciona con métodos y composiciones útiles en el tratamiento y el diagnóstico de cánceres que expresan el receptor alfa de folato y/o se pueden someter a tratamiento al destruir la tubulina.
ANTECEDENTES DE LA INVENCIÓN
El cáncer está entre las principales causas de morbilidad y mortalidad en todo el mundo, con aproximadamente 14 millones de casos nuevos y 8,2 millones de muertes relacionadas con el cáncer en 2012. Las casusas más comunes de muerte por cáncer son cánceres de: pulmón (1,59 millones de muertes); hígado (745.000 muertes); estómago (723.000 muertes); colorrectal (694.000 muertes); mama (521.000 muertes); y esófago (400.000 muertes). Se espera que la cantidad de casos nuevos de cáncer aumente aproximadamente 70 % en las próximas dos décadas, hasta aproximadamente 22 millones de casos de cáncer nuevos por año (Informe de World Cancer 2014).
Los microtúbulos son proteínas citoesqueléticas filamentosas dinámicas que participan en una variedad de funciones celulares, incluida la migración y el transporte intracelular, la señalización celular y el mantenimiento de la forma de la célula. Los microtúbulos también tienen una función esencial en la división celular mitótica al formar el huso mitótico necesario para segregar los cromosomas en dos células secundarias. Las funciones biológicas de microtúbulos en todas las células se regulan en gran parte mediante su dinámica de polimerización, que se produce por la adición no covalente, reversible de dímeros de tubulina a y p en ambos extremos del microtúbulo. Este comportamiento dinámico y control resultante sobre la longitud del microtúbulo es vital para el funcionamiento adecuado del huso mitótico. Incluso una alteración menor de la dinámica del microtúbulo puede comprometer el punto de control del huso, interrumpir la progresión del ciclo celular en la mitosis y, posteriormente, conducir a la muerte celular (Mukhtar et al. (2014) Mol. Cancer Ther. 13:275-84). Debido a su rápida división celular, las células cancerosas son generalmente más sensibles a compuestos que se unen a tubulina y alteran su función normal, en comparación con células normales. Por este motivo, los inhibidores de tubulina y otros agentes dirigidos a microtúbulos se han convertido en una clase de fármacos prometedora para el tratamiento del cáncer (Dumontet y Jordan (2010) Nat. Rev. Drug Discov. 9:790-803).
El receptor alfa de folato (FRA, por sus siglas en inglés) es una proteína de membrana enlazada a glicofosfatidilinositol (GPI) que se une a folato. Aunque no se comprende completamente la función del FRA en la biología de los tejidos normales y cancerosos, se encuentra muy sobreexpresado en un alto porcentaje de cánceres de ovario de origen epitelial (O'Shannessy et al. (2013) Int. J. Gynecol. Pathol. 32(3):258-68), así como en un porcentaje de carcinomas pulmonares no microcíticos (Christoph et al. (2014) Clin. Lung Cancer 15(5):320-30). El FRA también tiene expresión limitada en tejidos normales. Estas propiedades convierten al FRA en una diana atractiva para la inmunoterapia para el cáncer.
Diamantis et al. (2016) Brit. J. Cancer 114:362-367 describen una revisión de conjugados anticuerpo-fármaco, incluidos conjugados de anticuerpo anti-FRA.
COMPENDIO DE LA INVENCIÓN
En un primer aspecto, la presente invención proporciona un conjugado de anticuerpo-fármaco de la Fórmula (I):
Ab-(L-D)p (I)
en donde (i) Ab es un anticuerpo anti-receptor alfa de folato internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR, por sus siglas en inglés) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR, por sus siglas en inglés) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ ID NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT; (ii) D es eribulina; (iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y (iv) p es un número entero de 1 a 8. El anticuerpo o fragmento de unión a antígeno puede comprender una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24. p puede ser un número entero de 3 a 4. El anticuerpo o fragmento de unión a antígeno puede comprender un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig humana.
En un segundo aspecto, la presente invención proporciona una composición que comprende múltiples copias del conjugado de anticuerpo-fármaco del primer aspecto, en donde la p promedio de los conjugados de anticuerpofármaco en la composición es de aproximadamente 3,2 a aproximadamente 3,8 o de aproximadamente 3,6 a aproximadamente 4,4.
En un tercer aspecto, la presente invención proporciona el primer aspecto o el segundo aspecto que puede ser para uso en el tratamiento de un cáncer que expresa el receptor alfa de folato. El cáncer es un cáncer gástrico, un cáncer de ovario seroso, un cáncer de ovario de células claras, un cáncer pulmonar no microcítico, un cáncer colorrectal, un cáncer de mama triple negativo, un cáncer de endometrio, un carcinoma de endometrio seroso, un tumor carcinoide pulmonar o un osteosarcoma.
En un cuarto aspecto, la presente invención proporciona una composición farmacéutica que comprende el conjugado de anticuerpo-fármaco del primer aspecto o la composición del segundo aspecto, y un portador farmacéuticamente aceptable.
En un quinto aspecto, la presente invención proporciona un método para producir el conjugado de anticuerpo-fármaco del primer aspecto o la composición del segundo aspecto, que comprende hacer reaccionar un anticuerpo o fragmento de unión a antígeno con un enlazador escindible ligado a eribulina en condiciones que permiten la conjugación.
En un sexto aspecto, la presente invención proporciona un método para determinar si un paciente responderá al tratamiento con el conjugado de anticuerpo-fármaco de uno cualquiera del primer aspecto o la composición del segundo aspecto, que comprende proporcionar una muestra biológica del paciente y poner en contacto la muestra biológica con el conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 4 o la composición de la reivindicación 5. La muestra biológica puede ser una biopsia tumoral de un cáncer gástrico, un cáncer de ovario seroso, un cáncer de ovario de células claras, un cáncer pulmonar no microcítico, un cáncer colorrectal, un cáncer de mama triple negativo, un cáncer de endometrio, un carcinoma de endometrio seroso, un tumor carcinoide pulmonar o un osteosarcoma.
En un séptimo aspecto, la presente invención proporciona una composición que comprende múltiples copias de un conjugado de anticuerpo-fármaco de la Fórmula (I):
Ab-(L-D)p (I)
en donde (i) Ab es un anticuerpo anti-receptor alfa de folato internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ ID NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT; (ii) D es eribulina; (iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y (iv) p es el número promedio de restos -L-D por Ab, en donde la p promedio de los conjugados de anticuerpo-fármaco en la composición es de aproximadamente 3,6 a aproximadamente 4,4. El anticuerpo o fragmento de unión a antígeno puede comprender una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24.
La presente descripción describe, en parte, nuevos compuestos con actividad biológica contra células tumorales. Los compuestos pueden inhibir el crecimiento tumoral en mamíferos y pueden ser útiles para tratar a pacientes con cáncer humanos.
La presente descripción se relaciona, más específicamente, con compuestos de conjugado de anticuerpo-fármaco que son capaces de unirse, internalizarse y destruir células tumorales (p. ej., células tumorales que expresan FRA). Se describen compuestos de conjugado anticuerpo-fármaco que comprenden un enlazador que acopla un resto de fármaco a un resto de anticuerpo. Los compuestos de conjugado de anticuerpo-fármaco (ADC) se pueden representar mediante la Fórmula I:
Ab-(L-D)p (I)
en donde Ab es un anticuerpo internalizante o un fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral;
D es eribulina;
L es un enlazador escindible que acopla covalentemente el Ab a D; y
p es un número entero de 1 a 20.
Como se describe en la presente memoria, el enlazador es estable fuera de una célula, de modo que el ADC permanece intacto cuando está presente en condiciones extracelulares, pero es capaz de escindirse en la internalización en una célula, p. ej., una célula cancerosa. Como se describe en la presente memoria, el resto de fármaco eribulina se escinde del resto de anticuerpo cuando el ADC ingresa a una célula que expresa un antígeno específico para el resto de anticuerpo del ADC, y la escisión libera una forma no modificada de eribulina. Como se describe en la presente memoria, el enlazador comprende un resto escindible que se posiciona de modo que ninguna parte del enlazador ni del resto de anticuerpo permanezca unida al resto de fármaco eribulina después de la escisión.
Como se describe en la presente memoria, el resto escindible en el enlazador es un resto de péptido escindible. Como se describe en la presente memoria, un ADC que comprende un resto de péptido escindible demuestra niveles de aglomeración más bajos, mejor relación anticuerpo:fármaco, mayor destrucción específica de células cancerosas, menor destrucción inespecífica de células no cancerosas y/o carga de fármaco más alta (p) con respecto a un ADC que comprende un resto escindible alternativo. Como se describe en la presente memoria, agregar un resto escindible aumenta la citotoxicidad y/o la potencia con respecto a un enlazador no escindible. Como se describe en la presente memoria, la mayor potencia y/o citotoxicidad es en un cáncer que expresa niveles moderados del antígeno al que se dirige el resto de anticuerpo del ADC (p. ej., expresión moderada de FRA). Como se describe en la presente memoria, el resto de péptido escindible es escindible mediante una enzima, y el enlazador es un enlazador escindible por enzima. Como se describe en la presente memoria, la enzima es catepsina y el enlazador es un enlazador escindible por catepsina. Como se describe en la presente memoria, el enlazador escindible por enzima (p. ej., el enlazador escindible por catepsina) exhibe una o más de las propiedades mejoradas mencionadas anteriormente, en comparación con un mecanismo de escisión alternativo.
Como se describe en la presente memoria, el resto de péptido escindible en el enlazador comprende una unidad de aminoácido. Como se describe en la presente memoria, la unidad de aminoácido comprende valina-citrulina (Val-Cit). Como se describe en la presente memoria, un ADC que comprende Val-Cit demuestra mayor estabilidad, menor destrucción inespecífica de células, mayor destrucción específica de células, niveles de aglomeración más bajos y/o carga de fármaco más alta con respecto a un ADC que comprende una unidad de aminoácido alternativa o resto escindible alternativo.
Como se describe en la presente memoria, el enlazador comprende al menos una unidad espaciadora que liga el resto de anticuerpo al resto escindible. Como se describe en la presente memoria, la unidad espaciadora en el enlazador puede comprender al menos un resto de polietilenglicol (PEG). El resto de PEG puede comprender, por ejemplo, -(PEG)m-, en donde m es un número entero de 1 a 10. Como se describe en la presente memoria, la unidad espaciadora en el enlazador comprende (PEG)2. Como se describe en la presente memoria, un ADC que comprende una unidad espaciadora más corta (p. ej., (PEG)2) demuestra niveles de aglomeración más bajos y/o carga de fármaco más alta con respecto a un ADC que comprende una unidad espaciadora más larga (p. ej., (PEG)8) a pesar de la longitud más corta del enlazador.
Como se describe en la presente memoria, la unidad espaciadora en el enlazador se acopla el resto de anticuerpo del ADC a través de un resto de maleimida (Mal). Como se describe en la presente memoria, un ADC que comprende un enlazador acoplado al resto de anticuerpo a través de un Mal demuestra carga de fármaco más alta con respecto a un ADC que comprende un enlazador acoplado al resto de anticuerpo a través de un resto alternativo. Como se describe en la presente memoria, el Mal en el enlazador es reactivo con un residuo cisteína en el resto de anticuerpo. Como se describe en la presente memoria, el Mal en el enlazador se liga al resto de anticuerpo a través de un residuo cisteína. Como se describe en la presente memoria, la Mal-unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)m, p. ej., Mal-(PEG)2. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2. Como se describe en la presente memoria, la Mal-unidad espaciadora acopla el resto de anticuerpo al resto escindible en el enlazador. Como se describe en la presente memoria, el resto escindible en el enlazador es un resto de péptido escindible, p. ej. una unidad de aminoácido. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-Val-Cit.
Como se describe en la presente memoria, el resto escindible en el enlazador se liga directamente al resto de fármaco eribulina del ADC, y el resto escindible se conecta directamente al resto de anticuerpo o se conecta a través de una unidad espaciadora. Como se describe en la presente memoria, la unidad espaciadora también acopla el resto escindible en el enlazador al resto de fármaco eribulina. Como se describe en la presente memoria, la unidad espaciadora que acopla el resto escindible en el enlazador al resto de fármaco eribulina es autodestructiva. Como se describe en la presente memoria, el espaciador autodestructivo es capaz de liberar eribulina no modificada en una célula diana. Como se describe en la presente memoria, la unidad espaciadora autodestructiva comprende un alcohol p-aminobencílico. Como se describe en la presente memoria, la unidad espaciadora autodestructiva comprende paminobenciloxicarbonilo (pAB). El pAB en el enlazador, como se describe en la presente memoria, acopla el resto escindible al resto de fármaco eribulina. Como se describe en la presente memoria, el resto escindible es un resto de péptido escindible, p. ej. una unidad de aminoácido. Como se describe en la presente memoria, el enlazador comprende Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende Val-Cit-pAB y una unidad espaciadora de PEG que liga el enlazador al resto de anticuerpo a través de una Mal.
Como se describe en la presente memoria, p es un número entero de 1 a 6, de 2 a 5 o, preferiblemente, de 3 a 4. Como se describe en la presente memoria, p es 4. Como se describe en la presente memoria, se proporciona una
agrupación de ADC, y la p promedio en la agrupación es aproximadamente 4 (p. ej., 3,5-4,5, tales como aproximadamente 3,8). Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-Val-Cit-pAB y p es 4. Como se describe en la presente memoria, se proporciona una agrupación de ADC, en donde cada ADC comprende un enlazador Mal-(PEG)2-Val-Cit-pAB y la p promedio en la agrupación es aproximadamente 4 (p. ej., 3,5-4,5, tales como aproximadamente 3,8).
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante (Ab o resto Ab) del ADC es un anticuerpo anti-receptor alfa de folato (FRA) o fragmento de anticuerpo internalizante, y se puede unir a células tumorales que expresan FRA (es decir, el ADC se dirige a células que expresan FRA). Como se describe en la presente memoria, el ADC que comprende un resto Ab anti-FRA y un resto de péptido escindible demuestra niveles de aglomeración más bajos, mejor relación anticuerpo:fármaco, mayor destrucción específica de células cancerosas, menor destrucción inespecífica de células no cancerosas, carga de fármaco más alta (p), mayor citotoxicidad y/o potencia con respecto a un enlazador no escindible o un mecanismo de escisión alternativo. Como se describe en la presente memoria, la mayor potencia y/o citotoxicidad es en un cáncer que expresa niveles moderados del antígeno al que se dirige el resto de anticuerpo del ADC (p. ej., expresión moderada de FRA). Como se describe en la presente memoria, el resto de péptido escindible es escindible mediante una enzima, y el enlazador es un enlazador escindible por enzima. Como se describe en la presente memoria, la enzima es catepsina y el enlazador es un enlazador escindible por catepsina. Como se describe en la presente memoria, el enlazador escindible por enzima (p. ej., el enlazador escindible por catepsina) exhibe una o más de las propiedades mejoradas mencionadas anteriormente, en comparación con un mecanismo de escisión alternativo. Como se describe en la presente memoria, el enlazador es un Mal-(PEG)m-Val-Cit-pAB.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une al receptor alfa de folato (FRA) y se dirige a células tumorales que expresan FRA. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende tres regiones de determinación de la complementariedad de cadena pesada (CDR, por sus siglas en inglés) y tres CDR de cadena ligera, en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:2, la CDR2 de cadena pesada que consiste en la SEQ ID NO:3 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:4; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:7, la CDR2 de cadena ligera que consiste en la SEQ ID NO:8 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:9, según se definen mediante el sistema de numeración de Kabat; o en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO: 13, la CDR2 de cadena pesada que consiste en la SEQ ID NO:14 y la CDR3 de cadena pesada que consiste en la SEQ ID NO: 15; y las CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:16, la CDR2 de cadena ligera que consiste en la SEQ ID NO: 17 y la CDR3 de cadena ligera que consiste en la SEQ ID NO: 18, según se definen mediante el sistema de numeración de IMGT. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende secuencias de marco humanas. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio variable de cadena pesada de la SEQ ID NO:23 y un dominio variable de cadena ligera de la SEQ ID NO:24. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante compite por la unión y/o se une al mismo epítopo que un anticuerpo que comprende un dominio variable de cadena pesada de la SEQ ID NO:23 y un dominio variable de cadena ligera de la SEQ ID NO:24. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une a un epítopo que comprende alanina-histadinalisina-ácido aspártico (AHKD, por sus siglas en inglés) (SEQ ID NO:365) (O'Shannessy et al., (2011) Oncotarget 2:1227-43). Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une a un epítopo que comprende NTSQEAHKDv Sy L (SEQ ID NO:366).
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante es un anticuerpo anti-FRA internalizante o fragmento de unión a antígeno internalizante. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende tres CDR de cadena pesada y tres CDR de cadena ligera, en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:2, la CDR2 de cadena pesada que consiste en la SEQ ID NO:3 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:4; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:7, la CDR2 de cadena ligera que consiste en la SEQ ID NO:8 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:9, según se definen mediante el sistema de numeración de Kabat; o en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:13, la CDR2 de cadena pesada que consiste en la SEQ ID NO:14 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:15; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:16, la CDR2 de cadena ligera que consiste en la SEQ ID NO:17 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:18, según se definen mediante el sistema de numeración de IMGT; el enlazador comprende Mal-(PEG)2-Val-CitpAB; y p es 4. Como se describe en la presente memoria, se proporciona una agrupación de tales ADC y p es aproximadamente 4 (p. ej., aproximadamente 3,8). Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio variable de cadena pesada de la
SEQ ID NO:23 y un dominio variable de cadena ligera de la SEQ ID NO:24. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante compite por la unión y/o se une al mismo epítopo que un anticuerpo que comprende un dominio variable de cadena pesada de la SEQ ID NO:23 y un dominio variable de cadena ligera de la SEQ ID NO:24. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une a un epítopo que comprende la SEQ ID NO:365. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une a un epítopo que comprende la SEQ ID NO:366.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une al receptor de factor de crecimiento epidérmico humano 2 (her2, por sus siglas en inglés) y se dirige a células tumorales que expresan her2. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende tres regiones de determinación de la complementariedad de cadena pesada (CDR) y tres CDR de cadena ligera, en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:71, la CDR2 de cadena pesada que consiste en la SEQ ID NO:72 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:73; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:74, la CDR2 de cadena ligera que consiste en la SEQ ID NO:75 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:76, según se definen mediante el sistema de numeración de Kabat; o en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:191, la CDR2 de cadena pesada que consiste en la SEQ ID NO:192 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:193; y las CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:194, la CDR2 de cadena ligera que consiste en la SEQ ID NO: 195 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:196, según se definen mediante el sistema de numeración de IMGT. Como se describe en la presente memoria, el anticuerpo o fragmento de unión a antígeno internalizante comprende secuencias de marco humanas. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio variable de cadena pesada de la SEQ ID NO:27 y un dominio variable de cadena ligera de la SEQ ID NO:28. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante compite por la unión y/o se une al mismo epítopo que un anticuerpo que comprende un dominio variable de cadena pesada de la SEQ ID NO:27 y un dominio variable de cadena ligera de la SEQ ID NO:28.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante es un anticuerpo anti-her2 internalizante o fragmento de unión a antígeno internalizante. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende tres CDR de cadena pesada y tres CDR de cadena ligera, en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:71, la CDR2 de cadena pesada que consiste en la SEQ ID NO:72 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:73; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:74, la CDR2 de cadena ligera que consiste en la SEQ ID NO:75 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:76, según se definen mediante el sistema de numeración de Kabat; o en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:191, la CDR2 de cadena pesada que consiste en la SEQ ID NO:192 y la CDR3 de cadena pesada que consiste en la SEQ ID NO: 193; y las CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO: 194, la CDR2 de cadena ligera que consiste en la SEQ ID NO:195 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:196, según se definen mediante el sistema de numeración de IMGT; el enlazador comprende Mal-(PEG)2-Val-CitpAB; y p es 4. Como se describe en la presente memoria, se proporciona una agrupación de tales ADC y p es aproximadamente 4 (p. ej., aproximadamente 3,8). Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio variable de cadena pesada de la SEQ ID NO:27 y un dominio variable de cadena ligera de la SEQ ID NO:28. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante compite por la unión y/o se une al mismo epítopo que un anticuerpo que comprende un dominio variable de cadena pesada de la SEQ ID NO:27 y un dominio variable de cadena ligera de la SEQ ID NO:28.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante se une al mesotelina (MSLN, por sus siglas en inglés) y se dirige a células tumorales que expresan MSLN. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende tres regiones de determinación de la complementariedad de cadena pesada (CDR) y tres CDR de cadena ligera, en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:65, la CDR2 de cadena pesada que consiste en la SEQ ID NO:66 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:67; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:68, la CDR2 de cadena ligera que consiste en la SEQ ID NO:69 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:70, según se definen mediante el sistema de numeración de Kabat; o en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO: 185, la CDR2 de cadena pesada que consiste
en la SEQ ID NO:186 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:187; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:188, la CDR2 de cadena ligera que consiste en la SEQ ID NO:189 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:190, según se definen mediante el sistema de numeración de IMGT. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio variable de cadena pesada de la SEQ ID NO:25 y un dominio variable de cadena ligera de la SEQ ID NO:26. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante compite por la unión y/o se une al mismo epítopo que un anticuerpo que comprende un dominio variable de cadena pesada de la SEQ ID NO:25 y un dominio variable de cadena ligera de la SEQ ID NO:26.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante es un anticuerpo anti-MSLN internalizante o fragmento de unión a antígeno internalizante. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende tres CDR de cadena pesada y tres CDR de cadena ligera, en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:65, la CDR2 de cadena pesada que consiste en la SEQ ID NO:66 y la CDR3 de cadena pesada que consiste en la SEQ ID NO:67; y las tres CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO:68, la CDR2 de cadena ligera que consiste en la SEQ ID NO:69 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:70, según se definen mediante el sistema de numeración de Kabat; o en donde las CDR de cadena pesada comprenden la CDR1 de cadena pesada que consiste en la SEQ ID NO:185, la CDR2 de cadena pesada que consiste en la SEQ ID NO:186 y la CDR3 de cadena pesada que consiste en la SEQ ID NO: 187; y las CDR de cadena ligera comprenden la CDR1 de cadena ligera que consiste en la SEQ ID NO: 188, la CDR2 de cadena ligera que consiste en la SEQ ID NO:189 y la CDR3 de cadena ligera que consiste en la SEQ ID NO:190, según se definen mediante el sistema de numeración de IMGT; el enlazador comprende Mal-(PEG)2-Val-CitpAB; y p es 4. Como se describe en la presente memoria, se proporciona una agrupación de tales ADC y p es aproximadamente 4 (p. ej., aproximadamente 3,8). Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio variable de cadena pesada de la SEQ ID NO:25 y un dominio variable de cadena ligera de la SEQ ID NO:26. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno internalizante compite por la unión y/o se une al mismo epítopo que un anticuerpo que comprende un dominio variable de cadena pesada de la SEQ ID NO:25 y un dominio variable de cadena ligera de la SEQ ID NO:26.
También se describen en la presente memoria composiciones que comprenden múltiples copias de cualquiera de los ADC descritos, en donde la carga de fármaco promedio (p promedio) del ADC en la composición es entre aproximadamente 3 y 4, o aproximadamente 3,5 a aproximadamente 4,5, o aproximadamente 4. Como se describe en la presente memoria, la p promedio es entre aproximadamente 3,2 y 3,8. Como se describe en la presente memoria, la p promedio es entre aproximadamente 3,6 y 4,4.
También se describen en la presente memoria composiciones que comprenden -L-D, en donde D es eribulina; y L es un enlazador escindible que se acopla covalentemente a D. Como se describe en la presente memoria, el enlazador escindible se acopla covalentemente a la amina C-35 en eribulina. Como se describe en la presente memoria, el enlazador escindible comprende Val-Cit. Como se describe en la presente memoria, el enlazador escindible comprende una unidad espaciadora de PEG. Como se describe en la presente memoria, el enlazador escindible comprende Mal-(PEG)2-Val-Cit-pAB.
Además, en la presente memoria se describen composiciones farmacéuticas que comprenden un ADC y un diluyente, portador y/o excipiente farmacéuticamente aceptable.
Otro aspecto de la presente descripción incluye usos terapéuticos y diagnósticos para los compuestos y composiciones de ADC descritos, p. ej., en el tratamiento del cáncer. Otro aspecto incluye métodos para el tratamiento de un cáncer que expresan un antígeno al que se dirige el resto de anticuerpo del ADC, tal como FRA. Como se describe en la presente memoria, se describen métodos para destruir o inhibir la proliferación de células tumorales o células cancerosas al administrar una cantidad terapéuticamente eficaz y/o régimen de uno cualquiera de los ADC descritos. Otro aspecto incluye métodos para detectar células tumorales o células cancerosas que expresan FRA al usar los ADC descritos, y métodos para seleccionar pacientes con cáncer que responderán al tratamiento con los ADC descritos. Como se describe en la presente memoria, el cáncer es un cáncer gástrico, un cáncer de ovario seroso, un cáncer de ovario de células claras, un cáncer pulmonar no microcítico, un cáncer colorrectal, un cáncer de mama triple negativo, un cáncer de endometrio, un carcinoma de endometrio seroso, un tumor carcinoide pulmonar o un osteosarcoma. También se describen métodos para producir los ADC descritos.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La Figura 1 muestra una de las metodologías usadas para preparar los ADC MORAb-003. En esta estrategia, se generan cisteínas no apareadas a través de reducción parcial con equivalentes molares limitados del agente reductor
TCEP distinto de tiol. Esta estrategia reduce preferencialmente las uniones disulfuro intercatenarias que enlazan la cadena ligera y la cadena pesada (un para por emparejamiento H-L) y las dos cadenas pesadas en la región bisagra (dos pares por emparejamiento H-H en el caso de la IgG1 humana), mientras deja las uniones disulfuro intracatenarias intactas.
La Figura 2 muestra un método para sintetizar maleimida-(PEG)2-Val-Cit-pAB-eribulina (mal-(PEG)2-VCP-eribulina).
La Figura 3 muestra un análisis SDS-PAGE de condiciones de reducción para MORAb-003. Los carriles se indican a la derecha de la figura. El carril M corresponde al estándar de proteína; el carril 1 corresponde a MORAb-003 sin tratar; el carril 2 corresponde a 5,3 mg/mL reducido en 70,6 gM de TCEP; el carril 3 corresponda a MORAb-003 5,3 mg/mL reducido en 141,2 gM de TCEP; el carril 4 corresponda a MORAb-003 1,5 mg/mL reducido en 20 gM de TCEP; y el carril 5 corresponde a MORAb-003 1,5 mg/mL reducido en 40 gM de TCEP. Las identidades de cada banda se indican en el gel derecho inferior. "H" indica cadena pesada. "L" indica cadena ligera.
La Figura 4 muestra un análisis SDS-PAGE de condiciones de reducción para MORAb-003. El carril 1 corresponde al estándar de proteína; el carril 2 corresponde a MORAb-003 sin tratar; el carril 3 corresponde a MORAb-003 tratado en una relación entre MORAb-003:TCEP de 1:1; carril 4 corresponde a MORAb-003 tratado en una relación entre MORAb-003:TCEP de 1:2; carril 5 corresponde a MORAb-003 tratado en una relación entre MORAb-003:TCEP de 1:3; y el carril 6 corresponde a MORAb-003 tratado en una relación entre MORAb-003:TCEP de 1:4.
La Figura 5 muestra un análisis SDS-PAGE no reductor de ADC de MORAb-003 seleccionados, incluidos M-MMAE (carril 2), M-DM1 (carril 3), M-0026 (carril 4), M-0260 (carril 5), M-0267 (carril 6), M-0272 (carril 7), M-0285 (carril 8), M-0292 (carril 9), M-027-0381 (carril 10) y M-0284 (carril 11).
La Figura 6A muestra los resultados de un ensayo de citotoxicidad por vecindad de MORAb-003-maleimido-PEG2-Val-Cit-pAB-eribulina (M3-VCP-eribulina o "MORAb-202"). La Figura 6B muestra los resultados de un ensayo de citotoxicidad por vecindad de MORAb-003-maleimido-(CH2)5-Val-Cit-pAB-ER-001150828 (M3-ER-61318). La Figura 6C muestra los resultados de un ensayo de citotoxicidad por vecindad de MORAb-003-PEG-pAB-duostatina 3 (M3-027-0285). La información que se muestra en las respectivas leyendas de figura proporciona la línea celular:agente evaluado (línea celular/líneas celulares cultivadas, densidad de siembra de la 1a/2a línea celular).
Las Figuras 7A y 7B muestra la distribución de relación entre fármaco y anticuerpo (DAR, por sus siglas en inglés) para ADC MORAb-003-VCP-eribulina (Figura 7A) y MORAb-003-0285 (Figura 7B) con respecto a MORAb-003 no conjugado. Los números sobre cada pico indican la DAR de la especie individual.
La Figura 8 muestra los resultados de un análisis de citotoxicidad - competición entre MORAb-003-VCP-eribulina y MORAb-003 no conjugado (2 gM) en células IGROV1 o SJSA-1.
La Figura 9 muestra la cinética de peso corporal para cada grupo de ratones CD-1 (promedio del grupo y SEM) tratado con una dosis intravenosa individual de vehículo (PBS), o MORAb-202 a 10, 20, 40 o 80 mg/kg.
La Figura 10 muestra la cinética de peso corporal para cada grupo de ratones CD-1 (promedio de grupo y SEM) tratado intravenosamente con PBS, o con eribulina a 0,4, 0,8, 1,6 o 3,2 mg/kg, según un régimen de dosificación q4dx3 (dosis administradas una vez cada cuatro días para 3 dosis totales).
La Figura 11 muestra la cinética de crecimiento tumoral para cada grupo de ratones CB17-SCID con implante de células hNSCLC NCI-H2110 (promedio de grupo y SEM) y tratados con una dosis intravenosa individual de PBS, MORAb-003-VCP-eribulina (MORAb-202) a 1,2,5 o 5 mg/kg, o MORAb-003-0285 a 5 mg/kg.
La Figura 12 muestra los volúmenes tumorales de ratones CB17-SCID individuales con implante de células hNSCLC NCI-H2110, así como el promedio del grupo y SEM, el día 17. Los grupos se trataron con una dosis intravenosa individual de PBS, MORAb-003-VCP-eribulina (MORAb-202) a 1, 2,5 o 5 mg/kg, o MORAb-003-0285 a 5 mg/kg.
La Figura 13 muestra la cinética de peso corporal para cada grupo de ratones CB17-SCID con implante de NCI-H2110 (promedio de grupo y SEM) tratados con una dosis intravenosa individual de PBS, MORAb-003-VCP-eribulina (MORAb-202) a 1,2,5 o 5 mg/kg, o MORAb-003-0285 a 5 mg/kg.
La Figura 14 muestra la cinética de crecimiento tumoral para cada grupo de ratones CB17-SCID con implante de NCI-H2110 (promedio de grupo y SEM) tratado intravenosamente con vehículo (PBS) o con eribulina a 0,5, 0,2, 0,8 o 1,6 mg/kg, según un régimen de dosificación q4dx3.
La Figura 15 muestra los volúmenes tumorales de ratones CB17-SCID individuales con implante de NCI-H2110, así como el promedio del grupo y SEM, el día 24. Los grupos se trataron intravenosamente con vehículo (PBS) o con eribulina a 0,5, 0,2, 0,8 o 1,6 mg/kg, según un régimen de dosificación q4dx3.
La Figura 16 muestra la cinética de cambio de peso corporal para cada grupo de ratones CB17-SCID con implante de NCI-H2110 (promedio de grupo y SEM) tratado intravenosamente con vehículo (PBS) o con eribulina a 0,5, 0,2, 0,8 o 1,6 mg/kg, según un régimen de dosificación q4dx3.
La Figura 17 muestra la potencia de MORAb-003-VCP-eribulina (MORAb-202) sobre células IGROV1, OVCAR3, NCI-H2110, A431-A3, y SJSA-1, según se mide mediante el ensayo de citotoxicidad Crystal Violet.
La Figura 18 muestra la cinética de crecimiento tumoral para cada grupo de ratones CB17-SCID con implante de NCI-H2110 (promedio de grupo y SEM) tratados con una dosis intravenosa individual de PBS o MORAb-003-VCP-eribulina (MORAb-202) a 1,2,5 o 5 mg/kg.
Las Figuras 19A y 19B muestran la cinética de crecimiento tumoral (Figura 19A) la cinética de cambio de peso corporal (Figura 19B) para cada grupo de ratones NSCLC PDx (LXFA-737) que llevan tumor (promedio de grupo y SEM) tratados con una dosis intravenosa individual de vehículo (PBS), MORAb-003 at 5 mg/kg o MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg.
Las Figuras 20A y 20B muestran relaciones de volumen tumoral individuales (Figura 20A) y la cinética de cambio de peso corporal (Figura 20B) para cada grupo de ratones que llevan tumor de cáncer de endometrio PDx (Endo-12961) (promedio de grupo y SEM) tratados con una dosis intravenosa individual de PBS, eribulina a 0,1 o 3,2 mg/kg o MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg. Las Figuras 20C y 20D muestran la cinética de crecimiento tumoral (Figura 20C) y la cinética de cambio de peso corporal (Figura 20D) para cada grupo de ratones que llevan tumor de cáncer de endometrio PDx (Endo-10590) (promedio de grupo y SEM) tratados con una dosis intravenosa individual de PBS, eribulina a 0,1 o 3,2 mg/kg o MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg.
La Figura 21A muestra la tinción inmunohistoquímica (IHC) de tejido tumoral en ratones que llevan tumor TNBC PDx (OD-BRE-0631) con un anticuerpo anti-IgG humana. Los tejidos tumorales de ratones tratados con una dosis intravenosa individual de vehículo (derecha), o MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg (izquierda), se recogieron y tiñeron 5 días postratamiento. La Figura 21B muestra la tinción IHC de tejido tumoral en ratones que llevan tumor TNBC PDx (OD-BRE-0631) con un anticuerpo para a-actina de músculo liso (SMA, por sus siglas en inglés)-FITC. Los tejidos tumorales de ratones no tratados se recogieron 2 días antes del tratamiento (izquierda), mientras que los tejidos tumorales de ratones tratados con una dosis intravenosa individual de MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg se recogieron 5 días postratamiento (derecha). La Figura 21C muestra la cinética de crecimiento tumoral para cada grupo de ratones que llevan tumor TNBC PDx (OD-BRE-0631) (promedio de grupo y SEM) tratados con una dosis intravenosa individual de vehículo (PBS) o MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg.
La Figura 22 muestra la diferenciación de células madre mesenquimales de médula ósea humana (BM-MSC, por sus siglas en inglés) en cultivo con células MKN-74 después del tratamiento con vehículo (PBS o etanol), eribulina, MORAb-003 o MORAb-003-VCP-eribulina (MORAb-202), según se mide mediante análisis de citometría de flujo. Stro-1+/CD105+, CD34+/CD31- y NG2+ son marcadores de MSC, adipocitos y pericitos, respectivamente.
La Figura 23 muestra en análisis del transcurso del tiempo de tejidos tumorales de ratones CB17-SCID con implante de NCI-H2110 tratados con una dosis intravenosa individual de vehículo (PBS) o MORAb-003-VCP-eribulina (MORAb-202) a 5 mg/kg, teñidos con un anticuerpo para a-actina de músculo liso (SMA)-FITC. Los tejidos tumorales se recogieron y tiñeron el día 0 y en los días 3, 5, 7 y 9 postratamiento. Eje Y: % = [recuento de células teñidas / recuento de células totales] * 100. Eje X: día (recuento de células totales).
DESCRIPCIÓN DETALLADA DE DESCRIPCIONES ILUSTRATIVAS
Las composiciones y métodos descritos pueden comprenderse más fácilmente mediante la referencia a la siguiente descripción detallada tomada en conexión con las figuras adjuntas, que forman parte de la presente descripción. Se entenderá que las composiciones y métodos descritos no se limitan a las composiciones y métodos específicos descritos y/o que se muestran en la presente memoria. El alcance de la invención se define por medio de las reivindicaciones.
A lo largo de este texto, las descripciones se refieren a composiciones y métodos para usar dichas composiciones. Cuando la descripción describe o reivindica una característica o descripción asociada con una composición, dicha característica o descripción se aplica igualmente a los métodos para usar dicha composición. Asimismo, cuando la descripción describe o reivindica una característica o descripción asociada con un método para usar una composición, dicha característica o descripción se aplica igualmente a la composición.
Cuando se expresa un intervalo de valores, incluye descripciones que usan cualquier valor particular dentro del intervalo. Además, la referencia a los valores indicados en los intervalos incluye todos y cada uno de los valores dentro de dicho intervalo. Todos los intervalos son inclusivos de sus puntos extremos y combinables. Cuando se expresan valores como aproximaciones, mediante el uso del antecedente “aproximadamente”, se entenderá que el valor particular forma otra descripción. La referencia a un valor numérico particular incluye al menos dicho valor particular, a menos que el contexto claramente indique de cualquier otra manera. El uso de "o" significará "y/o" a menos que el contexto específico de su uso indique de cualquier otra manera. Cuando una referencia y la memoria descriptiva difieran, la memoria descriptiva prevalecerá.
Se comprenderá que ciertas características de las composiciones y métodos descritos que, para mayor claridad, se describen en la presente memoria en el contexto de descripciones independientes, también pueden proporcionarse
en combinación en una única descripción. En cambio, diversas características de las composiciones y métodos que, para mayor brevedad, se describen en el contexto de una única descripción, pueden describirse también por separado o en cualquier subcombinación.
Definiciones
A lo largo de la memoria descriptiva y las reivindicaciones se usan diversos términos en relación con aspectos de la descripción. A dichos términos se debe dar su significado común en la técnica, a menos que se indique de cualquier otra manera. Otros términos definidos específicamente deben interpretarse en consonancia con las definiciones descritas en la presente memoria.
Según se usan en la presente memoria, las formas singulares de “un”, “una”, “el/la” incluyen formas plurales, a menos que el contexto indique claramente de cualquier otra manera.
Los términos "aproximadamente" o "de forma aproximada" en el contexto de valores e intervalos numéricos se refieren a valores o intervalos que se aproximan o son cercanos a los valores o intervalos mencionados de modo que la descripción se pueda llevar a cabo como se pretende, tal como con una cantidad deseada de ácidos nucleicos o polipéptidos en una mezcla de reacción, como será evidente para el experto a partir de las enseñanzas contenidas en la presente memoria. Esto se debe, al menos en parte, a las propiedades variables de composiciones de ácido nucleico, edad, raza, género, variaciones anatómicas y fisiológicas y la inexactitud de los sistemas biológicos. Por lo tanto, estos términos abarcan valores más allá de los que resultan del error sistemático.
Los términos "conjugado de anticuerpo-fármaco", "conjugado de anticuerpo", "conjugado", "inmunoconjugado" y "ADC" se usan de manera intercambiable, y hacen referencia a un compuesto o derivado de este que está enlazado a un anticuerpo (p. ej., un anticuerpo anti-FRA) y se define mediante la fórmula genérica: Ab-(L-D)p (Fórmula I), en donde Ab = un resto de anticuerpo (es decir, anticuerpo o fragmento de unión a antígeno), L = un resto de enlazador, D = un resto de fármaco y p = el número de restos de fármaco por resto de anticuerpo.
El término "anticuerpo" se usa en el sentido más amplio para hacer referencia a una molécula de inmunoglobulina que reconoce y se une específicamente a una diana, tal como una proteína, polipéptido, carbohidrato, polinucleótido, lípido o combinaciones de los anteriores, a través de al menos un sitio de reconocimiento de antígeno dentro de la región variable de la molécula de inmunoglobulina. La cadena pesada de un anticuerpo está compuesta por un dominio variable de cadena pesada (Vh) y una región constante de cadena pesada (Ch). La cadena ligera está compuesta por un dominio variable de cadena ligera (Vl) y un dominio constante de cadena ligera (Cl). A los efectos de la presente solicitud, los dominios variables de cadena pesada y cadena ligera maduros comprenden cada uno tres regiones de determinación de la complementariedad (CDR1, CDR2 y CDR3) dentro de cuatro regiones marco (FR1, FR2, FR3 y FR4) dispuestas desde el extremo N al extremo C: FR1, CDR1, FR2, CDR2, FR3, CDR3 y FR4. Un "anticuerpo" puede ser de origen natural o hecho por el hombre, tal como anticuerpos monoclonales producidos por tecnología de hibridoma convencional. El término "anticuerpo" incluye anticuerpos monoclonales de longitud completa y anticuerpos policlonales de longitud completa, así como también fragmentos de anticuerpo tales como Fab, Fab', F(ab')2, Fv y anticuerpos de cadena simple. Un anticuerpo puede ser uno cualquiera de las cinco clases principales de inmunoglobulinas: IgA, IgD, IgE, IgG y IgM, o subclases de estas (p. ej., los isotipos IgG1, IgG2, IgG3, IgG4). El término abarca, además, anticuerpos humanos, anticuerpos quiméricos, anticuerpos humanizados y cualquier molécula de inmunoglobulina modificada que contiene un sitio de reconocimiento de antígeno siempre que demuestre la actividad biológica deseada.
El término "anticuerpo monoclonal", según se usa en la presente memoria, se refiere a un anticuerpo obtenido a partir de una población de anticuerpos sustancialmente homogéneos, es decir, los anticuerpos individuales que comprenden la población son idénticos excepto las posibles mutaciones de origen natural que pueden estar presentes en cantidades menores. Los anticuerpos monoclonales son altamente específicos, se dirigen contra un único epítopo antigénico. En cambio, las preparaciones de anticuerpos convencionales (policlonales) típicamente incluyen una multitud de anticuerpos dirigidos contra (o específicos para) diferentes epítopos. El modificador "monoclonal" indica el carácter del anticuerpo como obtenido a partir de una población de anticuerpos sustancialmente homogénea y no se debe interpretar que requiere la producción del anticuerpo a través de ningún método particular. Por ejemplo, los anticuerpos monoclonales que se usan según la presente descripción pueden elaborarse mediante el método de hibridoma descrito, en primera instancia, por Kohler et al., (1975) Nature 256:495 o pueden elaborarse mediante métodos de ADN recombinantes (ver, p. ej., la patente estadounidense núm. 4.816.567). Los anticuerpos monoclonales también pueden aislarse a partir de bibliotecas de anticuerpos en fagos mediante el uso de las técnicas descritas en Clackson et al. (1991) Nature 352:624-8 y Marks et al. (1991) J. Mol. Biol. 222:581-97, por ejemplo.
Los anticuerpos monoclonales descritos en la presente memoria específicamente incluyen anticuerpos "quiméricos", en los cuales una porción de la cadena pesada y/o ligera es idéntica u homóloga a secuencias correspondientes en anticuerpos derivados de una especie particular o que pertenecen a una clase o subclase de anticuerpo particular, mientras que el resto de la(s) cadena(s) es idéntico u homólogo a secuencias correspondientes en los anticuerpos derivados de otra especie o que pertenecen a otra clase o subclase de anticuerpo, así como fragmentos de dichos anticuerpos, siempre que se unan específicamente al antígeno diana y/o exhiban la actividad biológica deseada.
El término "anticuerpo humano", según se usa en la presente memoria, se refiere a un anticuerpo producido por un humano o un anticuerpo que tiene una secuencia de aminoácidos de un anticuerpo producido por un humano.
El término "anticuerpo quimérico", según se usa en la presente memoria, se refiere a anticuerpos en donde la secuencia de aminoácido de la molécula de inmunoglobulina deriva de dos o más especies. En algunas instancias, las regiones variables de ambas cadenas pesada y ligera corresponden a las regiones variables de anticuerpos derivados de una especie con la especificidad, afinidad y actividad deseadas, mientras que las regiones constantes son homólogas a anticuerpos derivados de otra especie (p. ej. humana) para minimizar una respuesta inmunitaria en la segunda especie.
Según se usa en la presente memoria, el término "anticuerpo humanizado" se refiere a formas de anticuerpos que contienen secuencias de anticuerpos no humanos (p. ej., murinos), así como anticuerpos humanos. Dichos anticuerpos son anticuerpos quiméricos que contienen una secuencia mínima derivada de inmunoglobulina no humana. En general, el anticuerpo humanizado comprenderá sustancialmente todos de al menos uno, y típicamente dos, dominios variables, donde todos o sustancialmente todos los bucles hipervariables corresponden a aquellos de una inmunoglobulina no humana, y todas o sustancialmente todas las regiones marco (FR) son aquellas de una secuencia de inmunoglobulina humana. El anticuerpo humanizado opcionalmente también comprenderá al menos una porción de una región constante de inmunoglobulina (Fc), típicamente la de una inmunoglobulina humana. El anticuerpo humanizado puede modificarse adicionalmente mediante la sustitución de residuos, ya sea en la región marco de Fv y/o dentro de los residuos no humanos reemplazados para refinar y optimizar la especificidad, afinidad y/o actividad del anticuerpo.
El término "fragmento de unión a antígeno" o "porción de unión a antígeno" de un anticuerpo, según se usa en la presente memoria, se refiere a uno o más fragmentos de un anticuerpo que conservan la capacidad de unirse específicamente a un antígeno (p. ej., FRA). Los fragmentos de unión a antígeno preferiblemente también conservan la capacidad de internalizarse en una célula que expresa antígeno. En algunas descripciones, los fragmentos de unión a antígeno también conservan la actividad efectora inmunitaria. Se ha demostrado que fragmentos de un anticuerpo de longitud completa pueden llevar a cabo la función de unión a antígeno de un anticuerpo de longitud completa. Los ejemplos de fragmentos de unión abarcados dentro del término "fragmento de unión a antígeno" o "porción de unión a antígeno" de un anticuerpo incluyen (i) un fragmento Fab, un fragmento monovalente que consiste en los dominios Vl, Vh, Cl y Ch1 ; (ii) un fragmento F(ab')2 , un fragmento bivalente que comprende dos fragmentos Fab enlazados por un puente disulfuro en la región de bisagra; (iii) un fragmento Fd que consiste en los dominios Vh y Cm; (iv) un fragmento Fv que consiste en los dominios Vl y Vh de un brazo simple de un anticuerpo; (v) un fragmento dAb, que comprende un dominio variable simple, p. ej., un dominio Vh (ver, p. ej., Ward et al. (1989) Nature 341:544-6; y Winter et al., WO 90/05144); y (vi) una región de determinación de la complementariedad (CDR) aislada. Además, si bien los dos dominios del fragmento Fv, Vl y Vh, están codificados por genes separados, se pueden ligar al usar métodos recombinantes, mediante un enlazador sintético que les permite formarse como una única cadena proteica en la que las regiones Vl y Vh se emparejan para formar moléculas monovalentes (conocidas como Fv de cadena simple (scFv, por su sigla en inglés)). Ver, p. e j, Bird et al. (1988) Science 242:423-6; y Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-83. También se pretende que el término "fragmento de unión a antígeno" o "porción de unión a antígeno" de un anticuerpo abarque dichos anticuerpos de cadena simple, y se conocen en la técnica como un tipo ilustrativo de fragmento de unión que puede internalizarse en células después de la unión. Ver, p. ej., Zhu et al. (2010) 9:2131-41; He et al. (2010) J. Nucl. Med. 51:427-32; y Fitting et al. (2015) MAbs 7:390-402. Como se describe en la presente memoria, las moléculas de scFv se pueden incorporar en una proteína de fusión. También se abarcan otras formas de anticuerpos de cadena simple, tales como diacuerpos. Los diacuerpos son anticuerpos bivalentes, biespecíficos en los que los dominios Vh y Vl se expresan en una cadena polipeptídica simple, pero con el uso de un enlazador que es demasiado corto para permitir que se emparejen los dos dominios en la misma cadena, forzando así a que los dominios se emparejen con dominios complementarios en otra cadena y creando dos sitios de unión a antígeno (ver, p. ej., Holliger et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-8; y Poljak et al. (1994) Structure 2:1121-3). Los fragmentos de unión a antígeno se obtienen al usar técnicas convencionales conocidas por los expertos en la técnica, y los fragmentos de unión se analizan para determinar su utilidad (p. ej., afinidad de unión, internalización) de la misma forma que los anticuerpos intactos. Los fragmentos de unión a antígeno pueden prepararse mediante la escisión de la proteína intacta, p. ej., mediante escisión por proteasa o química.
"Internalizarse", según se usa en la presente memoria en referencia a un anticuerpo o fragmento de unión a antígeno, se refiere a un anticuerpo o fragmento de unión a antígeno que es capaz es capaz de ser tomado a través de la membrana de bicapa lipídica de la célula hacia un compartimiento interno (es decir, "internalizarse") después de unirse a la célula, preferiblemente, en un compartimiento degradativo en la célula. Por ejemplo, un anticuerpo anti-FRA internalizante es uno que es capaz de ser tomado en la célula después de unirse a FRA en la membrana celular.
El término "receptor alfa de folato" o "FRA", según se usa en la presente memoria, se refiere a cualquier forma natural de FRA humano. El término abarca FRA de longitud completa (p. ej., la secuencia de referencia de NCBI: NP_000793; SEQ ID NO: 19), así como cualquier forma de FRA humano que resulta del procesamiento celular. El término también abarca variantes de origen natural de FRA que incluyen, pero no se limitan a, variantes de empalme, variantes alélicas e isoformas. El FRA se puede aislar a partir de un ser humano o se puede producir de manera recombinante o mediante métodos sintéticos.
El término "anticuerpo anti-FRA" o "anticuerpo que se une específicamente a FRA" se refiere a cualquier forma de anticuerpo o fragmento de este que se une específicamente a FRA, y abarca anticuerpos monoclonales (incluidos anticuerpos monoclonales de longitud completa), anticuerpos policlonales y fragmentos de anticuerpo biológicamente funcionales, siempre que se unan específicamente a FRA. Preferiblemente, el anticuerpo anti-FRA usado en los ADC descritos en la presente memoria es un anticuerpo internalizante o fragmento de anticuerpo internalizante. MORAb-003 es un anticuerpo anti-FRA humano internalizante ilustrativo. Según se usan en la presente memoria, los términos "específico", "se une específicamente" y "unirse específicamente" hacen referencia a la unión selectiva del anticuerpo al epítopo del antígeno diana. Se puede evaluar la especificidad de unión de los anticuerpos al comparar la unión a un antígeno apropiado con la unión a un antígeno irrelevante o mezcla de antígenos en un conjunto de condiciones dado. Si el anticuerpo se una al antígeno apropiado con al menos 2, 5, 7, y preferiblemente 10 veces más afinidad que al antígeno irrelevante o mezcla de antígeno, se considera entonces que es específico. Según se describe en la presente memoria, un anticuerpo específico es uno que solo se une al antígeno de FRA, pero no se une (o exhibe unión mínima) a otros antígenos.
El término "receptor de factor de crecimiento epidérmico 2 humano", "her2" o "her2/neu", según se usa en la presente memoria, se refiere a cualquier forma natural de her2 humano. El término abarca her2 de longitud completa (p. ej., la secuencia de referencia de NCBI: NP_004439.2; SEQ ID NO: 21), así como cualquier forma de her2 humano que resulta del procesamiento celular. El término también abarca variantes de origen natural de her2 que incluyen, pero no se limitan a, variantes de empalme, variantes alélicas e isoformas. El her2 se puede aislar a partir de un ser humano o se puede producir de manera recombinante o mediante métodos sintéticos.
El término "anticuerpo anti-her2" o "anticuerpo que se une específicamente a her2" se refiere a cualquier forma de anticuerpo o fragmento de este que se une específicamente a her2, y abarca anticuerpos monoclonales (incluidos anticuerpos monoclonales de longitud completa), anticuerpos policlonales y fragmentos de anticuerpo biológicamente funcionales, siempre que se unan específicamente a her2. La patente estadounidense núm. 5.821.337 proporciona secuencias de unión a her2 ilustrativas que incluyen secuencias de anticuerpo anti-her2 ilustrativas. Preferiblemente, el anticuerpo anti-her2 usado en los ADC descritos en la presente memoria es un anticuerpo internalizante o fragmento de anticuerpo internalizante. Trastuzumab es un anticuerpo anti-her2 humano internalizante ilustrativo.
El término "epítopo" se refiere a la porción de un antígeno que un anticuerpo puede reconocer y a la que puede unirse específicamente. Cuando el antígeno es un polipéptido, los epítopos pueden formarse a partir de aminoácidos contiguos o aminoácidos no contiguos yuxtapuestos mediante el plegado terciario del polipéptido. El epítopo al que se une un anticuerpo se puede identificar al usar cualquier técnica de mapeo de epítopos conocida en la técnica, incluida la cristalografía de rayos X para la identificación de epítopos mediante visualización directa del complejo antígenoanticuerpo, así como el monitoreo de la unión del anticuerpo a fragmentos o variaciones mutadas del antígeno, o la monitorización de la accesibilidad de disolvente a diferentes partes del anticuerpo y el antígeno. Las estrategias ilustrativas usadas para mapear epítopos para anticuerpo incluyen, pero no se limitan a, barrido de oligo-péptido basado en matriz, proteólisis limitada, mutagénesis dirigida a sitio, mapeo por mutagénesis de alto rendimiento, intercambio de hidrógeno-deuterio y espectrometría de masas (ver, p. ej., Gershoni et al. (2007) 21:145-56; y Hager-Braun and Tomer (2005) Expert Rev. Proteomics 2:745-56).
La unión competitiva y la preclasificación de epítopos también se pueden usar para determinar anticuerpos que comparten epítopos idénticos o superpuestos. La unión competitiva se puede evaluar al usar un ensayo de bloqueo cruzado, tal como el ensayo descrito en "Antibodies, A Laboratory Manual", Cold Spring Harbor Laboratory, Harlow and Lane (1a edición 1988, 2a edición 2014). Como se describe en la presente memoria, la unión competitiva se identifica cuando un anticuerpo o proteína de unión de prueba reduce la unión de un anticuerpo o proteína de unión de referencia a un antígeno diana tal como FRA o her2 (p. ej., una proteína de unión que comprende CDR y/o dominios variables seleccionados de los identificados en las Tablas 2, 4 y 6), en al menos aproximadamente 50 % en el ensayo de bloqueo cruzado (p. ej., 50 %, 60 %, 70 %, 80 %, 90 %, 95 %, 99 %, 99,5 % o más, o cualquier porcentaje intermedio) y/o viceversa. Como se describe en la presente memoria, la unión competitiva puede deberse a epítopos compartidos o similares (p. ej., parcialmente superpuestos) o puede deberse al impedimento estérico cuando los anticuerpos o proteínas de unión se unen a epítopos cercanos. Ver, p. ej., Tzartos, Methods in Molecular Biology (Morris, ed. (1998) tomo 66, págs. 55-66). Como se describe en la presente memoria, la unión competitiva se puede usar para clasificar grupos de proteínas de unión que comparten epítopos similares, p. ej., aquellos que compiten por la unión se pueden "preclasificar" como un grupo de proteínas de unión que tienen epítopos superpuestos o cercanos, mientras que aquellos que no compiten se colocan en un grupo separado de proteínas de unión que no tienen epítopos superpuestos ni cercanos.
El término "kon" o "ka" se refiere a la constante de velocidad para la asociación de un anticuerpo con el antígeno para formar el complejo anticuerpo/antígeno. La velocidad se puede determinar al usar ensayos estándares, tales como un ensayo Biacore o ELISA.
El término "koff" o "kd" se refiere a la constante de velocidad de disociación de un anticuerpo del complejo anticuerpo/antígeno. La velocidad se puede determinar al usar ensayos estándares, tales como un ensayo Biacore o ELISA.
El término “Kd” se refiere a la constante de disociación en equilibrio de una interacción anticuerpo-antígeno particular. La Kd se calcula mediante ka/kd. La velocidad se puede determinar al usar ensayos estándares, tales como un ensayo Biacore o ELISA.
El término "p" o "relación anticuerpo:fármaco" o "relación entre fármaco y anticuerpo" o "DAR" se refiere al número de restos de fármaco por resto de anticuerpo, es decir, la carga de fármaco, o el número de restos -L-D por anticuerpo o fragmento de unión a antígeno (Ab) en ADC de Fórmula I. En composiciones que comprenden múltiples copias de ADC de Fórmula I, "p" se refiere al número promedio de restos -L-D por anticuerpo o fragmento de unión a antígeno, también denominada carga de fármaco promedio.
Un "enlazador" o "resto enlazador" es cualquier resto químico que es capaz de ligar covalentemente un compuesto, normalmente un resto de fármaco tal como un agente quimioterapéutico, a otro resto tal como un resto de anticuerpo. Los enlazadores pueden ser susceptibles o sustancialmente resistentes a la escisión inducida por ácido, la escisión inducida por peptidasa, la escisión inducida por luz, la escisión inducida por esterasa y/o la escisión de la unión disulfuro, en condiciones en las que el compuesto o el anticuerpo permanece activo.
El término "agente" se usa en la presente memoria para hacer referencia a un compuesto químico, una mezcla de compuestos químicos, una macromolécula biológica, o un extracto hecho a partir de materiales biológicos. El término "agente terapéutico", "fármaco" o "resto de fármaco" se refiere a un agente que es capaz de modular un proceso biológico y/o tiene actividad biológica.
El término "agente quimioterapéutico" o "agente anticanceroso" se usa en la presente memoria para hacer referencia a todos los compuestos químicos que son eficaces para tratar el cáncer, independientemente del mecanismo de acción. La inhibición de la metástasis o angiogénesis es frecuentemente una propiedad de un agente quimioterapéutico. Los ejemplos no limitantes de agentes quimioterapéuticos incluyen agentes alquilantes, por ejemplo, mostazas de nitrógeno, compuestos de etilenimina y sulfonatos de alquilo; antimetabolitos, por ejemplo, antagonistas de ácido fólico, purina o pirimidina; agentes antimitóticos, por ejemplo, agentes antitubulina tales como eribulina o mesilato de eribulina (Halaven™) o derivados de estos, alcaloides de la vinca, y auristatinas; antibióticos citotóxicos; compuestos que dañan o interfieren en la expresión o replicación del ADN, por ejemplo, aglutinantes de surco menor de ADN; y antagonistas del receptor del factor de crecimiento. Además, los agentes quimioterapéuticos incluyen anticuerpos, moléculas biológicas y moléculas pequeñas. Un agente quimioterapéutico puede ser un agente citotóxico o citostático. El término "agente citostático" se refiere a un agente que inhibe o suprime el crecimiento celular y/o la multiplicación de las células.
El término "agente citotóxico" se refiere a una sustancia que causa la destrucción celular principalmente al interferir en la actividad de expresión y/o funcionamiento de la célula. Los ejemplos de agentes citotóxicos incluyen, pero no se limitan a, agentes antimitóticos, tales como eribulina, auristatinas (p. ej., monometil auristatina E (MMAE), monometil auristatina F (MMAF)), maitansinoides (p. ej., maitansina), dolastatinas, duostatinas, criptoficinas, alcaloides de la vinca (p. ej., vincristina, vinblastina), taxanos, taxoles y colquicinas; antraciclinas (p. ej., daunorrubicina, doxorrubicina, dihidroxiantracindiona); antibióticos citotóxicos (p. ej., mitomicinas, actinomicinas, duocarmicinas (p. ej., CC-1065), auromicinas, duomicinas, caliqueamicinas, endomicinas, fenomicinas); agentes alquilantes (p. ej., cisplatino); agentes intercalantes (p. ej., bromuro de etidio); inhibidores de topoisomerasa (p. ej., etopósido, tenopósido); radioisótopos, tales como At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212 o 213, P32, e isótopos radioactivos de lutecio (p. ej., Lu177); y toxinas de origen bacteriano, fúngico, vegetal o animal (p. ej., ricina (p. ej., cadena A de la ricina), toxina diftérica, exotoxina A de Pseudomonas (p. ej., PE40), endotoxina, mitogelina, combrestatina, restrictocina, gelonina, alfasarcina, abrina (p. ej., cadena A de abrina), modecina (p. ej., cadena A de modecina), curicina, crotina, inhibidor de Sapaonaria officinalis, glucocorticoide).
El término "eribulina", según se usa en la presente memoria, se refiere a un análogo sintético de halicondrina B, un compuesto macrocíclico que se aisló originalmente a partir de la esponja marina Halichondria okadais. El término "resto de fármaco eribulina" se refiere al componente de un ADC que tiene la estructura de eribulina, y se acopla al enlazador del ADC a través de su amina C-35. La eribulina es un inhibidor de la dinámica de los microtúbulos, que se cree que se une a tubulina e induce la interrupción del ciclo celular en la fase G2/M al inhibir el ensamblaje del huso mitótico. El término "mesilato de eribulina" se refiere a la sal mesilato de eribulina, que se comercializa con el nombre comercial Halaven™.
El término "homólogo" se refiere a una molécula que exhibe homología con respecto a otra molécula, por ejemplo, al tener secuencias de residuos químicos que son iguales o similares en las posiciones correspondientes.
El término "inhibir" o "inhibición de", según se usa en la presente memoria, significa reducir en una cantidad medible, y puede incluir, pero no obligatoriamente, la prevención o inhibición completa.
El término "negativa para la diana" o "negativa para el antígeno diana" se refiere a la ausencia de la expresión del antígeno diana por parte de una célula o tejido. El término "positiva para la diana" o "positiva para el antígeno diana" se refiere a la presencia de la expresión del antígeno diana. Por ejemplo, una célula o una línea celular que no expresa un antígeno diana puede describirse como negativa para la diana, mientras que una célula o línea celular que expresa un antígeno diana puede describirse como positiva para la diana.
El término "destrucción por vecindad" o "efecto por vecindad" se refiere a la destrucción de células negativas para la diana en presencia de células positivas para la diana, en donde la destrucción de células negativas para la diana no se observa en ausencia de células positivas para la diana. El contacto célula a célula, o al menos la proximidad entre células positivas para la diana y negativas para la diana, posibilita la destrucción por vecindad. Este tipo de destrucción se puede distinguir de la "destrucción inespecífica", que se refiere a la destrucción indiscriminada de células negativas para la diana. La "destrucción inespecífica" se puede observar en la ausencia de células positivas para la diana.
El término "cáncer" se refiere a la afección fisiológica en mamíferos en la que una población de células se caracteriza por tener un crecimiento celular no regulado. Los ejemplos de cánceres incluyen, pero no se limitan a, carcinoma, linfoma, blastoma, sarcoma y leucemia. Los ejemplos más particulares de dichos cánceres incluyen cáncer de células escamosas, cáncer microcítico de pulmón, cáncer no microcítico de pulmón, adenocarcinoma de pulmón, carcinoma de células escamosas de pulmón, cáncer del peritoneo, cáncer hepatocelular, cáncer gastrointestinal, cáncer pancreático, glioblastoma, cáncer de cuello uterino, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama (p. ej. cáncer de mama triple negativo), osteosarcoma, melanoma, cáncer de colon, cáncer colorrectal, cáncer de endometrio (p. ej., seroso) o uterino, carcinoma de las glándulas salivales, cáncer de riñón, cáncer de hígado, cáncer de próstata, cáncer vulvar, cáncer de tiroides, carcinoma hepático y varios tipos de cánceres de vías respiratorias y digestivas altas. El cáncer de mama triple negativo se refiere al cáncer de mama que es negativo para la expresión de los genes para el receptor de estrógeno (ER, por sus siglas en inglés), el receptor de progesterona (PR, por sus siglas en inglés) o Her2/neu.
Los términos "tumor" y "neoplasia" se refieren a cualquier masa de tejido que resulta del crecimiento celular o proliferación excesivos, ya sea benigno o maligno, incluidas lesiones precancerosas.
Los términos "célula cancerosa" y "célula tumoral" se refieren a células individuales o la población total de células derivadas de un tumor, incluidas células no oncógenas y células madre cancerosas. Según se usa en la presente memoria, el término "célula tumoral" se modificará mediante el término "no oncógena" cuando se haga referencia solamente a aquellas células tumorales que carecen de la capacidad de renovarse y diferenciarse para distinguir dichas células tumorales de las células madre cancerosas.
Los términos "sujeto" o "paciente" se usan de manera intercambiable en la presente memoria para hacer referencia a cualquier animal, tal como cualquier mamífero que incluye, pero no se limita a, seres humanos, primates no humanos, roedores y similares. Como se describe en la presente memoria, el mamífero es un ratón. Como se describe en la presente memoria, el mamífero es un ser humano.
El término "coadministración" o administración "en combinación con" uno o más agentes terapéuticos incluye la administración simultánea y consecutiva en cualquier orden.
Una "composición farmacéutica" se refiere a una preparación que está en una forma tal para permitir la administración y posteriormente proporcionar la actividad biológica prevista del ingrediente(s) activo(s) y/o para lograr un efecto terapéutico, y que no contiene componentes adicionales que son inaceptablemente tóxicos para un sujeto al cual se administraría la formulación. La composición farmacéutica puede ser estéril.
Un "excipiente farmacéutico" comprende un material tal como un adyuvante, un portador, agentes de ajuste del pH y tampón, agentes de ajuste de la tonicidad, agentes humectantes, conservante y similares.
"Farmacéuticamente aceptable" significa aprobado o que puede ser aprobado por una agencia reguladora del gobierno federal o estatal, o que se incluye en la Farmacopea de los EE. UU. u otra farmacopea generalmente reconocida, para uso en animales y, más particularmente, en seres humanos.
Una "cantidad eficaz" de un ADC como se describe en la presente memoria es una cantidad suficiente para llevar a cabo un propósito establecido específicamente, por ejemplo, para producir un efecto terapéutico después de la administración, tal como una reducción en la velocidad del crecimiento tumoral o el volumen tumoral, una reducción en un síntoma del cáncer, o algunos otros indicios de la eficacia del tratamiento. Una cantidad eficaz se puede determinar de manera rutinaria en relación con el propósito establecido. El término "cantidad terapéuticamente eficaz" se refiere a una cantidad de un ADC eficaz para tratar una enfermedad o un trastorno en un sujeto. En el caso del cáncer, una cantidad terapéuticamente eficaz del ADC puede reducir el número de células cancerosas, reducir el tamaño del tumor, inhibir (p. ej., enlentecer o detener) la metástasis tumoral; inhibir (p. ej., enlentecer o detener) el crecimiento tumoral y/o aliviar uno o más síntomas. Una "cantidad profilácticamente eficaz" se refiere a una cantidad eficaz, en dosificaciones y durante períodos de tiempo necesarios, para lograr el resultado profiláctico deseado. Típicamente, dado que se usa una dosis profiláctica en los sujetos con anterioridad o en una etapa temprana de la enfermedad, la cantidad profilácticamente eficaz será menor que la cantidad terapéuticamente eficaz.
Según se usa en la presente memoria, "tratar" o "terapéutico" y términos relacionados gramaticalmente, hacen referencia a cualquier mejora de cualquier consecuencia de la enfermedad, tal como supervivencia prolongada, menos morbilidad y/o una disminución de los efectos secundarios que son los subproductos de una modalidad terapéutica alternativa. Como se aprecia sin inconvenientes en la técnica, se prefiere la erradicación completa de la enfermedad, pero no es un requisito para un acto de tratamiento. "Tratamiento" o "tratar", según se usa en la presente memoria, se refiere a la administración de un ADC descrito a un sujeto, p. ej., un paciente. El tratamiento puede ser para curar,
sanar, aliviar, mitigar, alterar, remediar, mejorar, paliar, favorecer o afectar el trastorno, los síntomas del trastorno o la predisposición al trastorno, p. ej., un cáncer.
Como se describe en la presente memoria, se usa un ADC etiquetado. Las "etiquetas" adecuadas incluyen radionúclidos, enzimas, sustratos, cofactores, inhibidores, restos fluorescentes, restos quimioluminiscentes, partículas magnéticas y similares.
Por "proteína", según se usa en la presente memoria, se entiende al menos dos aminoácidos acoplados covalentemente. El término abarca polipéptidos, oligopéptidos y péptidos. Como se describe en la presente memoria, los dos o más aminoácidos acoplados covalentemente se acoplan mediante una unión peptídica. La proteína puede estar compuesta por aminoácidos y uniones peptídicas de origen natural, por ejemplo, cuando la proteína se produce recombinantemente al usar sistemas de expresión y células hospedantes. Alternativamente, la proteína puede incluir aminoácidos sintéticos (p. ej., homofenilalanina, citrulina, ornitina y norleucina), o estructuras peptidomiméticas, es decir, "análogos peptídicos o proteicos", tales como peptoides. Los peptoides son una clase ilustrativa de peptidomiméticos cuyas cadenas laterales se anexan al átomo de nitrógeno de la cadena principal del péptido, en lugar de a los a-carbonos (dado que están en aminoácidos), y tienen diferente unión de hidrógeno y características conformacionales en comparación con péptidos (ver, p. ej., Simon et al. (1992) Proc. Natl. Acad. Sci. USA 89:9367). Como tal, los peptoides pueden ser resistentes a la proteólisis u otras condiciones fisiológicas o de almacenamiento, y eficaces para permear la membrana celular. Dichos aminoácidos sintéticos se pueden incorporar, en particular, cuando el anticuerpo se sintetiza in vitro mediante métodos convencionales conocidos en la técnica. Además, se puede usar cualquier combinación de residuos/estructuras peptidomiméticos, sintéticos y de origen natural. "Aminoácido" también incluye residuos iminoacídicos, tales como prolina e hidroxiprolina. El "grupo R" o "cadena lateral" del aminoácido puede estar en la configuración (L) o (S). Como se describe en la presente memoria, los aminoácidos están en la configuración (L) o (S).
Una "proteína recombinante" es una proteína producida al usar técnicas recombinantes que usan cualesquiera técnicas y métodos conocidos en la técnica, es decir, a través de la expresión de un ácido nucleico recombinante. En la técnica se conocen métodos y técnicas para la producción de proteínas recombinantes.
Una proteína "aislada" no está acompañada por al menos algo del material con el cual se asocia normalmente en su estado natural, por ejemplo, que constituye al menos aproximadamente 5 %, o al menos aproximadamente 50 % en peso de la proteína total en una muestra dada. Se entiende que la proteína aislada puede constituir de 5 a 99,9 % en peso del contenido de proteína total dependiendo de las circunstancias. Por ejemplo, la proteína se puede producir en una concentración significativamente más alta a través del uso de un promotor inducible o promotor de alta expresión, de modo que la proteína se produzca a niveles de concentración mayores. La definición incluye la producción de un anticuerpo en una amplia variedad de organismos y/o células hospedantes conocidas en la técnica.
Para las secuencias de aminoácidos, la identidad y/o similitud de secuencia se puede determinar al usar técnicas estándares conocidas en la técnica, que incluyen, pero no se limitan a, el algoritmo de identidad secuencial local de Smith y Waterman, 1981, Adv. Appl. Math. 2:482, el algoritmo de alineación de identidad secuencial de Needleman and Wunsch (1970) J. Mol. Biol. 48:443, el método de búsqueda de similitud de Pearson y Lipman (1988) Proc. Nat. Acad. Sci. USA 85:2444, implementaciones informáticas de estos algoritmos (GAP, BEs T fiT, FAs Ta y TfASTA en el paquete de software de Wisconsin Genetics, Genetics Computer Group, 575 Science Drive, Madison, Wis.), el programa de secuencias Best Fit descrito por Devereux et al, (1984) Nucl. Acid Res. 12:387-95, preferiblemente, al usar las configuraciones predeterminadas o mediante inspección. Preferiblemente, el porcentaje de identidad se calcula mediante FastDB en función de los siguientes parámetros: penalización por malapareamiento de 1; penalización por espacio de 1; penalización por tamaño de espacio de 0,33; y penalización de ligadura de 30 ("Current Methods in Sequence Comparison and Analysis", Macromolecule Sequencing and Synthesis, Selected Methods and Applications, págs. 127-149 (1988), Alan R. Liss, Inc.).
Un ejemplo de un algoritmo útil es PILEUP. PILEUP crea una alineación de múltiples secuencias a partir de un grupo de secuencias relacionadas al usar alineaciones progresivas por pares. También puede trazarse un árbol que muestre las relaciones de agrupamiento usadas para crear la alineación. PILEUP usa una simplificación del método de alineación progresiva de Feng & Doolittle (1987) J. Mol. Evol. 35:351-60; el método es similar al descrito por Higgins y Sharp (1989) CABIOS 5:151-3. Los parámetros de PILEUP útiles incluyen una ponderación de espacio predeterminada de 3,00, una ponderación de longitud de espacio predeterminada de 0,10 y espacios finales ponderados.
Otro ejemplo de un algoritmo útil es el algoritmo BLAST, descrito en: Altschul et al. (1990) J. Mol. Biol. 215:403-10; Altschul et al. (1997) Nucleic Acids Res. 25:3389-402; y Karin et al. (1993) Proc. Natl. Acad. Sci. USA 90:5873-87. Un programa BLAST particularmente útil es al programa WU-BLAST-2 que se obtuvo de Altschul et al. (1996) Methods in Enzymology 266:460-80. El WU-BLAST-2 usa varios parámetros de búsqueda, la mayoría de los cuales se encuentran configurados según los valores predeterminados. Los parámetros ajustables se configuran con los siguientes valores: lapso de superposición=1, fracción de superposición=0,125, umbral de palabra (T)=II. Los parámetros HSP S y HSP S2 son valores dinámicos y establecidos por el propio programa dependiendo de la composición de la secuencia particular o composición de la base de datos particular con respecto a la cual se busca la secuencia de interés; sin embargo, los valores pueden ajustarse para aumentar la sensibilidad.
Un algoritmo útil adicional es BLAST con espacios según informan Altschul et al., (1993) Nucl. Acids Res. 25:3389-402. El BLAST con espacios usa puntuaciones de sustitución de BLOSUM-62; el parámetro T de umbral configurado en 9; el método de dos eventos para activar las extensiones sin espacios, carga las longitudes de los espacios de k una ponderación de 10+k; Xu configurado en 16 y Xg configurado en 40 para la etapa de búsqueda en bases de datos y en 67 para la etapa de resultado de los algoritmos. Las alineaciones con espacios se activan mediante una puntuación correspondiente a aproximadamente 22 bits.
Generalmente, la homología, similitud o identidad de aminoácidos entre las proteínas descritas en la presente memoria y variantes de estas, incluidas variantes de FRA, variantes de her2, variantes de secuencias de tubulina y variantes de dominios variables de anticuerpo (incluidas CDR variantes individuales) son al menos 80 % con respecto a las secuencias expuestas en la presente memoria y, más típicamente, con homologías o identidades preferiblemente en aumento de al menos 85 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % y casi 100 % o 100 %.
De manera similar, el "porcentaje (%) de identidad secuencial de ácido nucleico" con respecto a la secuencia de ácido nucleico de los anticuerpos y otras proteínas identificadas en la presente memoria se define como el porcentaje de residuos nucleotídicos en una secuencia candidata que son idénticos a los residuos nucleotídicos en la secuencia codificante de la proteína de unión a antígeno. Un método específico utiliza el módulo BLASTN de WU-BLAST-2 configurado con los parámetros predeterminados, con un lapso de superposición y una fracción de superposición configurados en 1 y 0,125, respectivamente.
Aunque el sitio o la región para introducir una variación de secuencia de aminoácidos está predeterminado, no es necesario que la mutación esté predeterminada per se. Por ejemplo, para optimizar el rendimiento de una mutación en un sitio dado, se puede llevar a cabo mutagénesis aleatoria en un codón o región diana, y analizar las variantes de CDR de proteína de unión a antígeno expresadas para determinar la combinación óptima de la actividad deseada. Se conocen técnicas para hacer mutaciones de sustitución en sitios predeterminados de ADN que tienen una secuencia conocida, por ejemplo, mutagénesis de cebador MI3 y mutagénesis por PCR.
Conjugados de anticuerpo-fármaco
Los compuestos de la presente descripción incluyen aquellos con actividad anticancerosa. En particular, los compuestos incluyen un resto de anticuerpo (incluido un fragmento de unión a antígeno de este) conjugado (es decir, acoplado covalentemente mediante un enlazador) con un resto de fármaco, en donde el resto de fármaco cuando no está conjugado con un resto de anticuerpo tiene un efecto citotóxico o citostático. Como se describe en la presente memoria, el resto de fármaco exhibe citotoxicidad reducida o nula cuando está unido en un conjugado, pero recobra la citotoxicidad después de la escisión del enlazador y el resto de anticuerpo. Como se describe en la presente memoria, el resto de fármaco exhibe destrucción por vecindad reducida o nula cuando está unido en un conjugado (p. ej., al usar un enlazador no escindible), pero exhibe mayor destrucción por vecindad después de la escisión de un conjugado (p. ej., un conjugado que tiene un resto escindible Val-Cit escindible).
El desarrollo y la producción de un ADC para uso como un agente terapéutico humano, p. ej., como un agente oncológico, puede requerir más que la identificación de un anticuerpo capaz de unirse a una diana o dianas deseadas y el acoplamiento a un fármaco usado por sí solo para tratar el cáncer. Enlazar el anticuerpo al fármaco puede tener efectos significativos e impredecibles sobre la actividad de uno o ambos del anticuerpo y el fármaco, efectos que variarán dependiendo del tipo de enlazador y/o fármaco elegido. Por lo tanto, como se describe en la presente memoria, los componentes del ADC se seleccionan para (i) retener una o más propiedades terapéuticas exhibidas por el anticuerpo y restos de fármaco en aislamiento, (ii) mantener las propiedades de unión específicas del resto de anticuerpo; (iii) optimizar la carga de fármaco y las relaciones entre fármaco y anticuerpo; (iv) permitir el suministro, p. ej., el suministro intracelular, del resto de fármaco a través del acoplamiento estable al resto de anticuerpo; (v) retener la estabilidad del ADC como un conjugado intacto hasta el transporte o suministro a un sitio diana; (vi) minimizar la aglomeración del ADC antes o después de la administración; (vii) permitir el efecto terapéutico, p. ej., efecto citotóxico, del resto de fármaco después de la escisión en el entorno celular; (viii) exhibir eficacia de tratamiento anticanceroso in vivo semejante o superior a la del anticuerpo y restos de fármaco en aislamiento; (ix) minimizar la destrucción inespecífica mediante el resto de fármaco; y/o (x) exhibir propiedades farmacocinéticas y farmacodinámicas, capacidad de formulación y perfiles toxicológicos/inmunológicos deseables. Puede ser necesario someter a cribado cada una de estas propiedades para identificar un ADC mejorado para uso terapéutico (Ab et al. (2015) Mol. Cancer Ther. 14:1605-13).
Como se describe en la presente memoria, los ADC descritos en la presente memoria exhiben propiedades inesperadamente favorables en algunas o cada una de las categorías indicadas anteriormente. Por ejemplo, como se describe en la presente memoria, las construcciones de ADC que comprenden un acoplamiento Mal a un anticuerpo, una unidad espaciadora de PEG (preferiblemente una unidad espaciadora de PEG corta) y/o enlazador escindible peptídico (p. ej., un enlazador Val-Cit) exhiben perfiles de carga de fármaco, aglomeración y/o estabilidad sorprendentemente favorables, y/o conservan la función de unión del anticuerpo, la actividad del fármaco y/o destrucción por vecindad mejorada, mientras reducen la destrucción inespecífica, en comparación con ADC que usan otras estructuras de enlazador escindible o no escindible.
Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a un anticuerpo (p. ej., un anticuerpo anti-FRA tal como MORAb-003) exhibe propiedades particularmente favorables en todas las categorías indicadas, en comparación con otros enlazadores escindibles o no escindibles que ligan eribulina a un resto de anticuerpo. Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a un anticuerpo (p. ej., un anticuerpo anti-FRA tal como MORAb-003) exhibe propiedades de destrucción por vecindad particularmente favorables en comparación con un ADC no escindible. Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a un anticuerpo (p. ej., un anticuerpo anti-FRA tal como MORAb-003) exhibe propiedades de destrucción por vecindad particularmente favorables en comparación con un ADC que usa estructuras de enlazador escindible alternativas.
Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a MORAb-003 exhibes una relación fármaco:anticuerpo más alta y más deseable (es decir, una relación de aproximadamente 3-4) con respecto a un ADC, p. ej., que comprende un enlazador acoplado al anticuerpo a través de un resto alternativo (p. ej., un resto de succinimida). Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a MORAb-003 exhibes una relación fármaco:anticuerpo más alta y más deseable, y/o niveles de aglomeración menores con respecto a un ADC, p. ej., que comprende una unidad espaciadora más larga (p. ej., (PEG)8). Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a MORAb-003 demuestra una relación fármaco:anticuerpo más alta y más deseable, niveles de aglomeración menores, mayor destrucción específica y/o menor destrucción inespecífica con respecto a un ADC, p. ej., que comprende un resto escindible alternativo (es decir, un resto escindible no peptídico, tal como un disulfuro o sulfonamida escindible). Como se describe en la presente memoria, un ADC que comprende un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a MORAb-003 demuestra mayor estabilidad, mayor destrucción específica, menor destrucción inespecífica, niveles de aglomeración menores y/o una relación fármaco:anticuerpo más alta y más deseable con respecto a un ADC, p. ej., que comprende una unidad de aminoácido alternativa (p. ej., Ala-Ala-Asn) o un resto escindible alternativo (p. ej., un disulfuro o sulfonamida escindible).
Como se describe en la presente memoria, algunas o todos los rasgos deseables descritos anteriormente para ADC que comprenden un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a MORAb-003 se pueden observar con ADC que comprenden el enlazador Mal-(PEG)2-Val-Cit-pAB-eribulina-toxina conjugado con un anticuerpo anti-her2 tal como trastuzumab, o un anticuerpo anti-mesotelina.
Los compuestos de ADC de la presente descripción pueden suministrar selectivamente una dosis eficaz de un agente citotóxico o citostático a células cancerosas o a tejido tumoral. Se ha descubierto que los ADC descritos tienen actividad citotóxica y/o citostática potente contra células que expresan el antígeno diana respectivo (p. ej., FRA o her2). Como se describe en la presente memoria, la actividad citotóxica y/o citostática del ADC depende del nivel de expresión del antígeno diana en una célula. Como se describe en la presente memoria, los ADC descritos son particularmente eficaces en la destrucción de células cancerosas que expresan un nivel alto de antígeno diana, en comparación con células cancerosas que expresan el mismo antígeno en un nivel bajo. Como se describe en la presente memoria, los ADC descritos son particularmente eficaces en la destrucción de células cancerosas que expresan el antígeno diana en un nivel moderado, en comparación con células cancerosas que expresan el mismo antígeno en un nivel bajo. Los cánceres que expresan FRA alto ilustrativos incluyen, pero no se limitan a, cáncer de ovario (p. ej., cáncer de ovario seroso, cáncer de ovario de células claras), carcinoide pulmonar, cáncer de mama triple negativo, cáncer de endometrio y cáncer de pulmón no microcítico (p. ej., adenocarcinoma). Los cánceres que expresan FRA moderado ilustrativos incluyen, pero no se limitan a, cáncer gástrico y cáncer colorrectal. Los cánceres que expresan FRA bajo ilustrativos incluyen, pero no se limitan a, melanoma y linfoma. Los cánceres que expresan her2 alto ilustrativos incluyen, pero no se limitan a, cáncer de mama, cáncer gástrico, cáncer esofágico, cáncer de ovario y cáncer de endometrio. Los cánceres que expresan her2 moderado ilustrativos incluyen, pero no se limitan a, cáncer de pulmón y cáncer de vejiga.
Como se describe en la presente memoria, la escisión de un ADC libera eribulina del resto de anticuerpo y el enlazador. Como se describe en la presente memoria, la escisión y liberación de la eribulina mejora la citotoxicidad del ADC. Como se describe en la presente memoria, un ADC que comprende un enlazador escindible es particularmente eficaz en la destrucción de células cancerosas, incluida destrucción por vecindad, en comparación con un tratamiento semejante con un ADC que comprende un enlazador no escindible. Como se describe en la presente memoria, un ADC que comprende un enlazador escindible (p. ej., un enlazador Val-Cit) demuestra mayor destrucción celular específica y/o menor destrucción celular inespecífica con respecto a un ADC que comprende un enlazador no escindible (p. ej., un enlazador de (PEG)2 o (PEG)4 no escindible), particularmente en donde las células y/o el cáncer tratados con el ADC no expresan niveles altos del antígeno diana.
Como se describe en la presente memoria, los ADC descritos también demuestran actividad de destrucción por vecindad, pero baja citotoxicidad inespecífica. Sin desear limitarse a la teoría, la actividad de destrucción por vecindad de un ADC puede ser particularmente beneficiosa cuando su penetración en un tumor sólido es limitada y/o la expresión del antígeno diana entre las células tumorales es heterogénea. Como se describe en la presente memoria, un ADC que comprende un enlazador escindible es particularmente eficaz en la destrucción por vecindad y/o demuestra actividad de destrucción por vecindad mejorada en comparación con un tratamiento semejante con un a Dc que comprende un enlazador no escindible.
En la presente memoria se proporcionan compuestos de ADC que comprenden un anticuerpo o fragmento de unión a antígeno de este (Ab) que se dirige a una célula tumoral, un resto de fármaco (D), y un resto de enlazador (L) que acopla covalentemente el Ab al D. En ciertos aspectos, el anticuerpo o fragmento de unión a antígeno es capaz de unirse a un antígeno asociado a un tumor (p. ej., FRA o her2) con alta especificidad y alta afinidad. Como se describe en la presente memoria, el anticuerpo o fragmento de unión a antígeno se internaliza en una célula diana después de la unión, p. ej., en un compartimiento degradativo en la célula. Los ADC preferidos son, por lo tanto, aquellos que se internalizan después de la unión a una célula diana, sufren degradación y liberan el resto de fármaco para destruir células cancerosas. El resto de fármaco se puede liberar desde el anticuerpo y/o el resto de enlazador del ADC mediante acción enzimática, hidrólisis, oxidación o cualquier otro mecanismo.
Un ADC ilustrativo tiene la Fórmula I:
Ab-(L-D)p (I)
en donde Ab = resto de anticuerpo (es decir, anticuerpo o fragmento de unión a antígeno), L = resto de enlazador, D = resto de fármaco y p = el número de restos de fármaco por resto de anticuerpo.
Anticuerpos
El resto de anticuerpo (Ab) de Fórmula I incluye dentro de su alcance cualquier anticuerpo o fragmento de unión a antígeno que se une específicamente a un antígeno diana en una célula cancerosa. El anticuerpo o fragmento de unión a antígeno se puede unir a un antígeno diana con una constante de disociación (Kd) de <1 mM, <100 nM o <10 nM, o cualquier cantidad intermedia, según se mide mediante, p. ej., el análisis BIAcore®. Como se describe en la presente memoria, la Kd es 1 pM a 500 pM. Como se describe en la presente memoria, la Kd es entre 500 pM a 1 pM, 1 pM a 100 nM, o 100 mM a 10 nM.
Como se describe en la presente memoria, el resto de anticuerpo es un anticuerpo de cuatro cadenas (también denominado una inmunoglobulina), que comprende dos cadenas pesadas y dos cadenas ligeras. Como se describe en la presente memoria, el resto de anticuerpo es un medio cuerpo de dos cadenas (una cadena ligera y una cadena pesada), o un fragmento de unión a antígeno de una inmunoglobulina.
Como se describe en la presente memoria, el resto de anticuerpo es un anticuerpo internalizante o fragmento de unión a antígeno de este internalizante. Como se describe en la presente memoria, el anticuerpo internalizante se une a un antígeno de cáncer diana expresado en la superficie de una célula e ingresa a la célula después de la unión. Como se describe en la presente memoria, el resto de fármaco del ADC se libera del resto de anticuerpo del ADC después de que el ADC ingresa y está presente en una célula que expresa el antígeno de cáncer diana (es decir, después de que el ADC se ha internalizado).
Las secuencias de aminoácidos y ácido nucleico de anticuerpos ilustrativos de la presente descripción se exponen en las Tablas 1-9.
Tabla 1. Anticuerpos

Tabla 2. Secuencias de aminoácidos de regiones variables de mAb

(continuación)

(continuación)

Tabla 3. Secuencias de ácido nucleico que codifican regiones variables de mAb

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Tabla 4. Secuencias de aminoácidos de CDR de mAb según Kabat
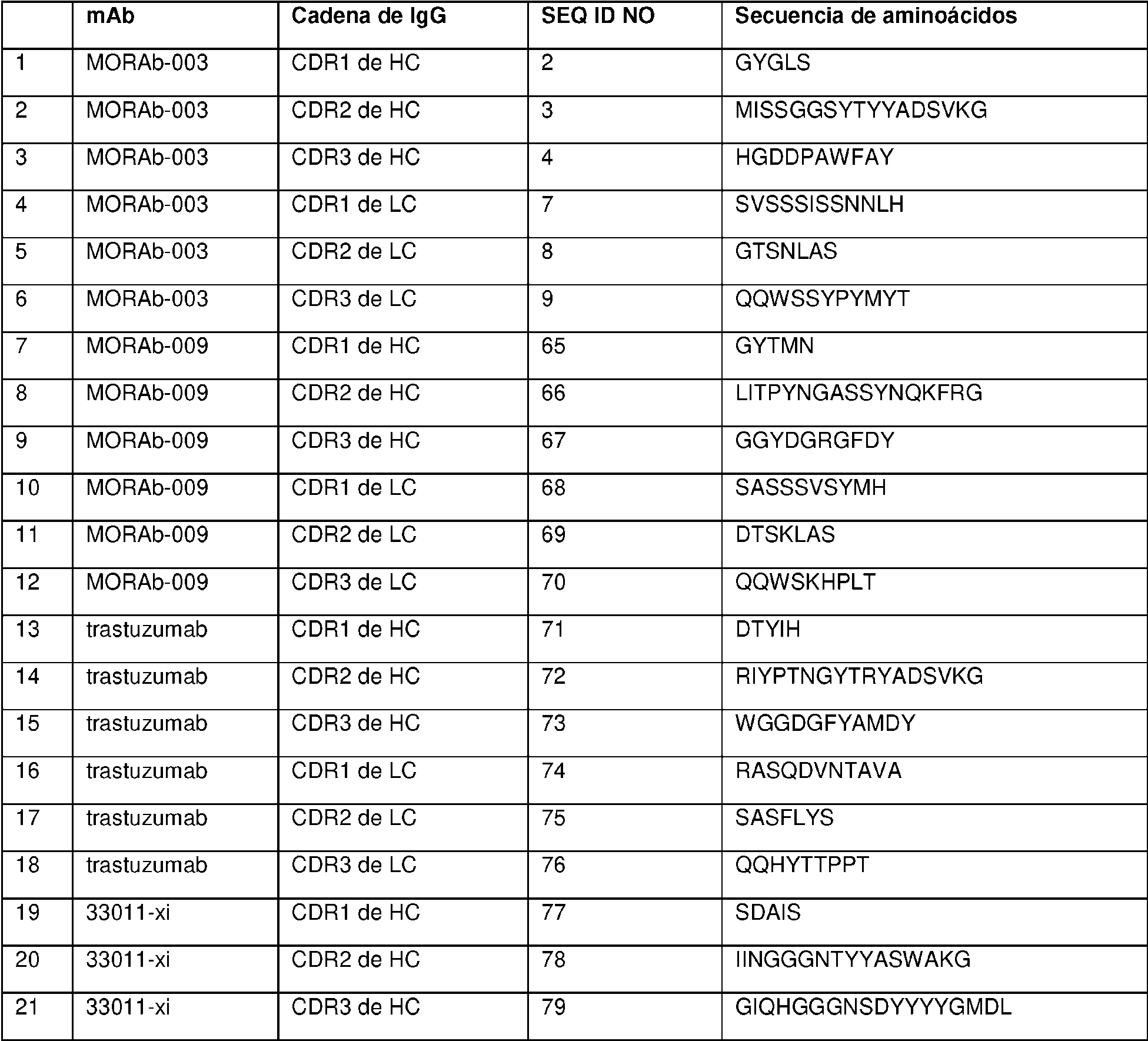
(continuación)

(continuación)

Tabla 5. Secuencias de ácido nucleico que codifican CDR de mAb según Kabat

(continuación)

(continuación)

Tabla 6. Secuencias de aminoácidos de CDR de mAb según IMGT
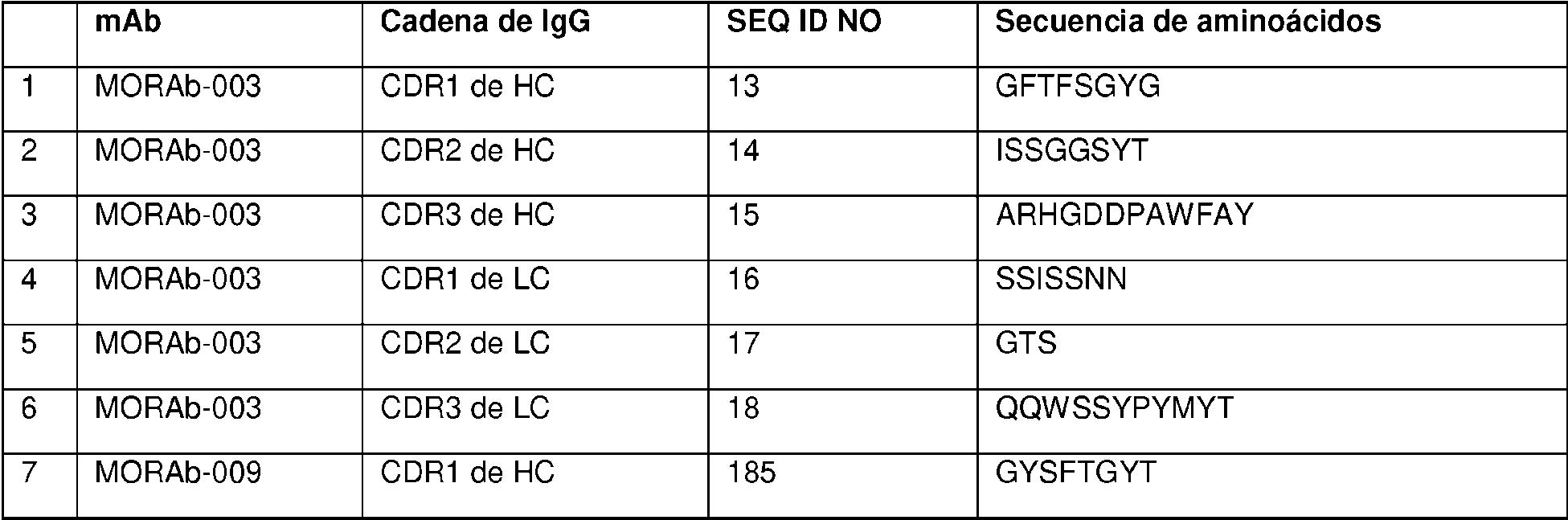
(continuación)

(continuación)
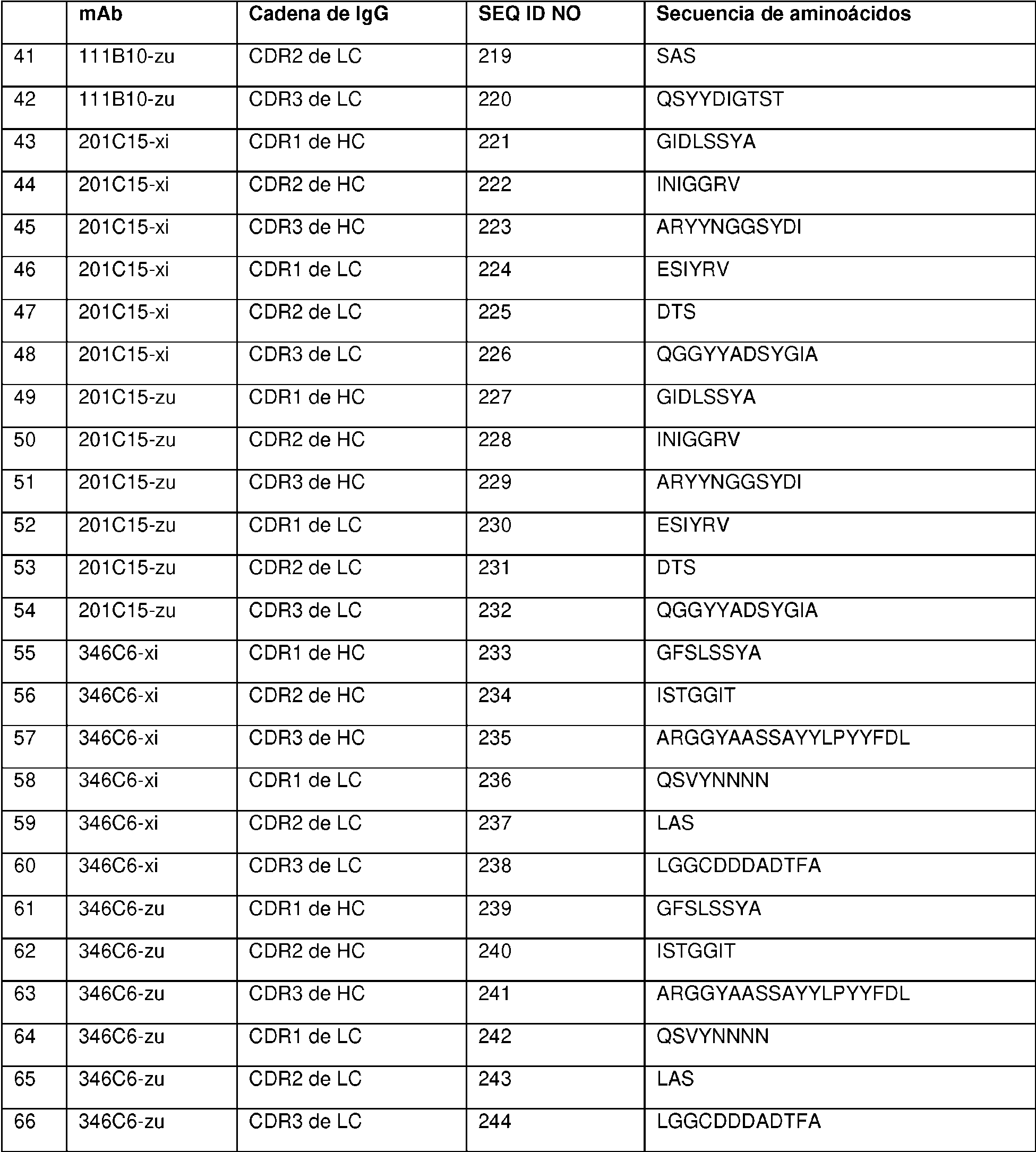
Tabla 7. Secuencias de ácido nucleico que codifican CDR de mAb según IMGT

(continuación)

(continuación)

Tabla 8. Secuencias de aminoácidos de cadenas de Ig de mAb de longitud completa

(continuación)

(continuación)

(continuación)

(continuación)
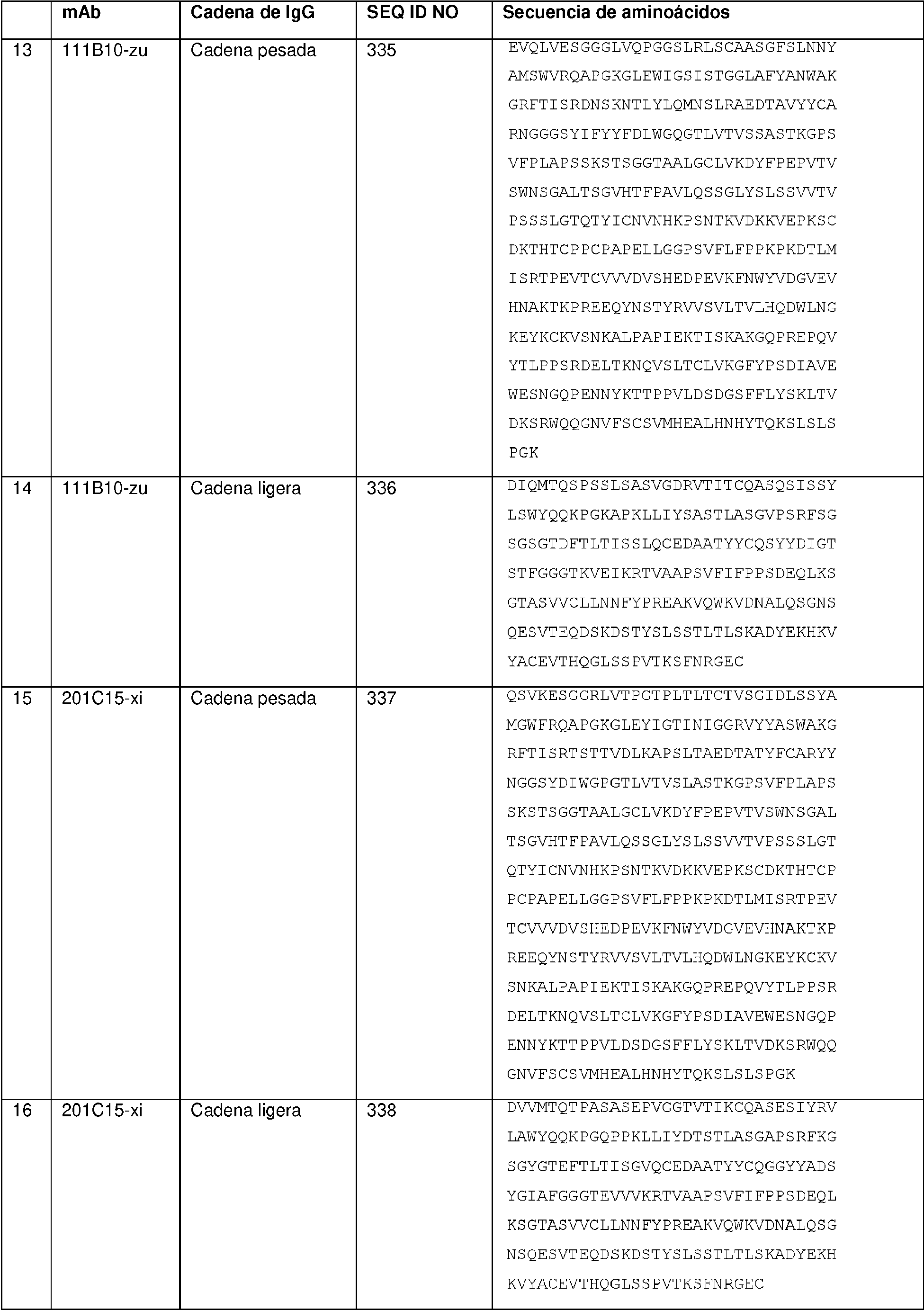
(continuación)

(continuación)
Tabla 9. Secuencias de ácido nucleico que codifican cadenas de Ig de mAb de longitud completa*

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Como se describe en la presente memoria, un ADC descrito en la presente memoria puede comprender cualquier conjunto de dominios variables de cadena pesada y ligera indicados en las tablas anteriores (p. ej., dominios variables de cadena pesada y ligera de MORAb-003 o dominios variables de cadena pesada y ligera de trastuzumab), o el conjunto de seis secuencias de CDR del conjunto de cadena pesada y ligera. Como se describe en la presente memoria, el ADC comprende, además, dominios constantes de cadena pesada y ligera humanos o fragmentos de estos. Por ejemplo, el ADC puede comprender un dominio constante de cadena pesada de IgG humana (tal como una IgG1) y un dominio constante de cadena ligera kappa o lambda humana. Como se describe en la presente memoria, el resto de anticuerpo de los ADC descritos comprende un dominio constante de cadena pesada del subtipo 1 de inmunoglobulina G humana (IgG1) con un dominio constante de cadena ligera kappa de Ig humana.
Como se describe en la presente memoria, el antígeno de cáncer diana para un ADC es el receptor alfa de folato ("FRA").
Como se describe en la presente memoria, el anticuerpo anti-FRA o fragmento de unión a antígeno de este comprende tres CDR de cadena pesada y tres CDR de cadena ligera como se indican a continuación: CDR1 de cadena pesada (HCDR1) que consiste en la SEQ ID NO:2, CDR2 de cadena pesada (HCDR2) que consiste en la SEQ ID NO:3, CDR3 de cadena pesada (HCDR3) que consiste en la SEQ ID NO:4; CDR1 de cadena ligera (LCDR1) que consiste en la SEQ ID NO:7, CDR2 de cadena ligera (LCDR2) que consiste en la SEQ ID NO:8 y CDR3 de cadena ligera (LCDR3) que consiste en la SEQ ID NO:9, según se definen mediante el sistema de numeración de Kabat (Kabat, Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, Md. (1987 y 1991))).
Como se describe en la presente memoria, el anticuerpo anti-FRA o fragmento de unión a antígeno de este comprende tres CDR de cadena pesada y tres CDR de cadena ligera como se indican a continuación: CDR1 de cadena pesada que consiste en la SEQ ID NO:13, CDR2 de cadena pesada que consiste en la SEQ ID NO: 14, CDR3 de cadena pesada que consiste en la SEQ ID NO: 15; CDR1 de cadena ligera que consiste en la SEQ ID NO: 16, CDR2 de cadena ligera que consiste en la SEQ ID NO: 17 y la CDR3 de cadena ligera que consiste en la SEQ ID NO: 18, según se definen mediante el sistema de numeración de IMGT (International ImMunoGeneTics Information System (IMGT®)).
Como se describe en la presente memoria, el anticuerpo anti-FRA o fragmento de unión a antígeno de este comprende una región variable de cadena pesada que comprende la secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de la SEQ ID NO:24. Como se describe en la
presente memoria, el anticuerpo anti-FRA o fragmento de unión a antígeno de este comprende la secuencia de aminoácidos de región variable de cadena pesada de la SEQ ID NO:23 y la secuencia de aminoácidos de la región variable de cadena ligera de la SEQ ID NO:24, o secuencias que son al menos 95 % idénticas a las secuencias mencionadas anteriormente. Como se describe en la presente memoria, el anticuerpo anti-FRA o fragmento de unión a antígeno de este tiene una secuencia de aminoácidos de región variable de cadena pesada que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:23 y una secuencia de aminoácidos de región variable de cadena ligera que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:24.
Como se describe en la presente memoria, el anticuerpo anti-FRA comprende un dominio constante de cadena pesada de IgG1 humana con un dominio constante de cadena ligera kappa de Ig humana.
Como se describe en la presente memoria, el anticuerpo anti-FRA comprende la secuencia de aminoácidos de cadena pesada de la SEQ ID NO:1 o una secuencia que es al menos 95 % idéntica a la SEQ ID NO:1, y la secuencia de aminoácidos de cadena ligera de la SEQ ID NO:6 o una secuencia que es al menos 95 % idéntica a la SEQ ID NO:6. Como se describe en la presente memoria, el anticuerpo comprende la secuencia de aminoácidos de cadena pesada de la SEQ ID NO:1 y la secuencia de aminoácidos de cadena ligera de la SEQ ID NO:6, o secuencias que son al menos 95 % idénticas a las secuencias mencionadas anteriormente. Como se describe en la presente memoria, el anticuerpo anti-FRA tiene una secuencia de aminoácidos de cadena pesada que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:1 y/o una secuencia de aminoácidos de cadena ligera que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:6. Como se describe en la presente memoria, el anticuerpo anti-FRA comprende una cadena pesada codificada por la secuencia de nucleótidos de la SEQ ID NO:11 (con los nucleótidos que codifican la secuencia líder), o la SEQ ID NO:345 (sin los nucleótidos que codifican la secuencia líder); y una cadena ligera codificada por la secuencia de nucleótidos de la SEQ ID NO:12 (con los nucleótidos que codifican la secuencia líder), o la SEQ ID NO:346 (sin los nucleótidos que codifican la secuencia líder). Como se describe en la presente memoria, la secuencia de aminoácidos de cadena pesada carece de la lisina del extremo C. Como se describe en la presente memoria, el anticuerpo anti-FRA tiene la secuencia de aminoácidos del anticuerpo producido mediante una línea celular depositada de conformidad con los términos del Tratado de Budapest en la Colección Estadounidense de Cultivos Tipo (ATCC, 10801 University Blvd., Manassas, Va. 20110-2209) el 24 de abril de 2006, con el núm. de acceso PTA-7552, o tales secuencias que carecen de la lisina del extremo C de cadena pesada. Como se describe en la presente memoria, el anticuerpo anti-FRA es MORAb-003 (nombre de USAN: farletuzumab) (Ebel et al. (2007) Cancer Immunity 7:6) o un fragmento de unión a antígeno de este.
Como se describe en la presente memoria, el antígeno de cáncer diana para un ADC es el receptor de factor de crecimiento epidérmico 2 humano ("her2").
Como se describe en la presente memoria, el anticuerpo anti-her2 o fragmento de unión a antígeno de este comprende tres CDR de cadena pesada y tres CDR de cadena ligera como se indican a continuación: CDR1 de cadena pesada (HCDR1) que consiste en la SEQ ID NO:71, CDR2 de cadena pesada (HCDR2) que consiste en la SEQ ID NO:72, CDR3 de cadena pesada (HCDR3) que consiste en la SEQ ID NO:73; CDR1 de cadena ligera (LCDR1) que consiste en la SEQ ID NO:74, CDR2 de cadena ligera (LCDR2) que consiste en la SEQ ID NO:75 y CDR3 de cadena ligera (LCDR3) que consiste en la SEQ ID NO:76, según se definen mediante el sistema de numeración de Kabat.
Como se describe en la presente memoria, el anticuerpo anti-her2 o fragmento de unión a antígeno de este comprende tres CDR de cadena pesada y tres CDR de cadena ligera como se indican a continuación: CDR1 de cadena pesada que consiste en la s Eq ID n O:191, CDR2 de cadena pesada que consiste en la SEQ ID NO: 192, CDR3 de cadena pesada que consiste en la SEQ ID NO: 193; CDR1 de cadena ligera que consiste en la SEQ ID NO: 194, CDR2 de cadena ligera que consiste en la SEQ ID NO: 195 y la CDR3 de cadena ligera que consiste en la SEQ ID NO: 196, según se definen mediante el sistema de numeración de IMGT.
Como se describe en la presente memoria, el anticuerpo anti-her2 o fragmento de unión a antígeno de este comprende una región variable de cadena pesada que comprende la secuencia de aminoácidos de la SEQ ID NO:27 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de la SEQ ID NO:28. Como se describe en la presente memoria, el anticuerpo anti-her2 o fragmento de unión a antígeno de este comprende la secuencia de aminoácidos de región variable de cadena pesada de la SEQ ID NO:27 y la secuencia de aminoácidos de la región variable de cadena ligera de la SEQ ID NO:28, o secuencias que son al menos 95 % idénticas a las secuencias mencionadas anteriormente. Como se describe en la presente memoria, el anticuerpo anti-her2 o fragmento de unión a antígeno de este tiene una secuencia de aminoácidos de región variable de cadena pesada que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:27 y/o una secuencia de aminoácidos de región variable de cadena ligera que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:28.
Como se describe en la presente memoria, el anticuerpo anti-her2 comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig humana.
Como se describe en la presente memoria, el anticuerpo anti-her2 comprende la secuencia de aminoácidos de cadena pesada de la SEQ ID NO:327 o una secuencia que es al menos 95 % idéntica a la SEQ ID NO:327, y la secuencia de aminoácidos de cadena ligera de la SEQ ID NO:328 o una secuencia que es al menos 95 % idéntica a la SEQ ID NO:328. Como se describe en la presente memoria, el anticuerpo comprende la secuencia de aminoácidos de cadena pesada de la SEQ ID NO:327 y la secuencia de aminoácidos de cadena ligera de la SEQ ID NO:328, o secuencias que son al menos 95 % idénticas a las secuencias mencionadas anteriormente. Como se describe en la presente memoria, el anticuerpo anti-her2 tiene una secuencia de aminoácidos de cadena pesada que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:327 y una secuencia de aminoácidos de cadena ligera que es al menos 96 %, al menos 97 %, al menos 98 % o al menos 99 % idéntica a la SEQ ID NO:328. Como se describe en la presente memoria, el anticuerpo anti-her2 es trastuzumab, o un fragmento de unión a antígeno de este.
Como se describe en la presente memoria, el anticuerpo anti-FRA o fragmento de unión a antígeno de este comprende las tres CDR de cadena pesada y las CDR de cadena ligera de MORAb-003 o en donde las CDR incluyen no más de una, dos, tres, cuatro, cinco o seis adiciones, eliminaciones o sustituciones aminoácidos de la HCDR1 (SEQ ID NO:2 según Kabat, o SEQ ID NO:13 según IMGT), HCDR2 (SEQ ID NO:3 según Kabat, o SEQ ID NO:14 según IMGT), HCDR3 (SEQ ID NO:4 según Kabat, o SEQ ID NO:15 según IMGT); LCDR1 (SEQ ID NO:7 según Kabat, o SEQ ID NO:16 según IMGT), LCDR2 (SEQ ID NO:8 según Kabat, o SEQ ID NO:17 según IMGT) y LCDR3 (SEQ ID NO:9 según Kabat, o SEQ ID NO:18 según IMGT).
Como se describe en la presente memoria, el anticuerpo anti-her2 o fragmento de unión a antígeno de este comprende las tres CDR de cadena pesada y las CDR de cadena ligera de trastuzumab o en donde las CDR incluyen no más de una, dos, tres, cuatro, cinco o seis adiciones, eliminaciones o sustituciones aminoácidos de la HCDR1 (SEQ ID NO:71 según Kabat, o SEQ ID NO:191 según IMGT), HCDR2 (SEQ ID NO:72 según Kabat, o SEQ ID NO:192 según IMGT), HCDR3 (SeQ ID NO:73 según Kabat, o SeQ ID NO:193 según IMGT); LCDR1 (SEQ ID NO:74 según Kabat, o SeQ ID NO:194 según IMGT), LCDR2 (SEQ ID NO:75 según Kabat, o SEQ ID NO:195 según IMGT) y LCDR3 (SEQ ID NO:76 según Kabat, o s Eq ID NO:196 según IMGT).
Como se describe en la presente memoria, las sustituciones de aminoácidos son de residuos simples. Las inserciones normalmente serán del orden de aproximadamente 1 a aproximadamente 20 residuos aminoacídicos, si bien se pueden tolerar inserciones considerablemente mayores, siempre que se conserve la función biológica (p. ej., unión a FRA o her2). Las eliminaciones normalmente varían de aproximadamente 1 a aproximadamente 20 residuos aminoacídicos, aunque en algunos casos las eliminaciones pueden ser mucho mayores. Las sustituciones, eliminaciones, inserciones o cualquier combinación de estas se pueden usar para llegar a un derivado o variante final. Generalmente estos cambios se hacen en unos pocos aminoácidos para minimizar la alteración de la molécula, particularmente, la inmunogenicidad y especificidad de la proteína de unión a antígeno. Sin embargo, se pueden tolerar cambios mayores en ciertas circunstancias. Las sustituciones conservadoras se hacen generalmente según el siguiente gráfico representado como la Tabla 10.
Tabla 10
Residuo original Sustituciones ilustrativas
Ala Ser
Arg Lys
Asn Gln, His
Asp Glu
Cys Ser
Gln Asn
Glu Asp
Gly Pro
His Asn, Gln
Ile Leu, Val
Leu Ile, Val
(continuación)
Residuo original Sustituciones ilustrativas
Lys Arg, Gln, Glu
Met Leu, Ile
Phe Met, Leu, Tyr
Ser Thr
Thr Ser
Trp Tyr
Tyr Trp, Phe
Val Ile, Leu
Se hacen cambios sustanciales en la función o identidad inmunológica al seleccionar sustituciones que son menos conservadoras que las que se muestran en la Tabla 10. Por ejemplo, se pueden hacer sustituciones que afectan más significativamente: la estructura de la cadena principal polipeptídica en el área de la alteración, por ejemplo, la estructura helicoidal alfa o de láminas beta; la carga o hidrofobicidad de la molécula en el sitio diana; o el volumen de la cadena lateral. Las sustituciones que, en general, se espera que produzcan los mayores cambios en las propiedades del polipéptido son aquellas en que (a) un residuo hidrófilo, p. ej., serilo o treonilo, se sustituye con (o por) un residuo hidrófobo, p. ej., leucilo, isoleucilo, fenilalanilo, valilo o alanilo; (b) una cisteína o prolina se sustituye con (o por) cualquier otro residuo; (c) un residuo que tiene una cadena lateral electropositiva, p. ej., lisilo, arginilo o histidilo, se sustituye con (o por) un residuo electronegativo, p. ej., glutamilo o aspartilo; o (d) un residuo que tiene una cadena lateral voluminosa, p. ej., fenilalanina, se sustituye con (o por) uno que no tiene una cadena lateral, p. ej., glicina.
Como se describe en la presente memoria, cuando se usan secuencias de anticuerpo variantes en un ADC, las variantes típicamente exhiben la misma actividad biológica cualitativa y desencadenarán la respuesta inmunitaria, si bien también se pueden seleccionar variantes para modificar las características de las proteínas de unión a antígeno según sea necesario. Alternativamente, la variante se puede diseñar de modo que se altere la actividad biológica de la proteína de unión a antígeno proteína. Por ejemplo, se pueden alterar o quitar sitios de glicosilación, como se describe en la presente memoria.
Se pueden usar varios anticuerpos con los ADC usados en la presente memoria para dirigirse a células cancerosas. Como se muestra más adelante, el enlazador-toxinas en los ADC descritos en la presente memoria son sorprendentemente eficaces con diferentes anticuerpos dirigidos a antígenos tumorales. Los antígenos adecuados expresados en células tumorales, pero no en células sanas, o expresados en células tumorales en un nivel más alto que en células sanas, se conocen en la técnica, así como los anticuerpos dirigidos contra ellos. Estos anticuerpos se pueden usar con los enlazadores y toxina (p. ej., eribulina) descritos en la presente memoria. Como se describe en la presente memoria, el resto de anticuerpo se dirige a FRA. Como se describe en la presente memoria, el resto de anticuerpo que se dirige a FRA es MORAb-003. Como se describe en la presente memoria, aunque los enlazadores y toxina (eribulina) descritos son sorprendentemente eficaces con varios anticuerpos dirigidos a tumores diferentes, los restos de anticuerpo dirigidos a FRA tales como MORAb-003 proporcionan una relación fármaco-anticuerpo, direccionamiento a tumor, destrucción por vecindad, eficacia de tratamiento particularmente mejores y destrucción inespecífica reducida. La eficacia de tratamiento mejorada se puede medir in vitro o in vivo, y puede incluir velocidad de crecimiento tumoral reducida y/o volumen tumoral reducido.
Como se describe en la presente memoria, se usan anticuerpos contra otras dianas antigénicas y proporcionan al menos algunas de las propiedades funcionales favorables de un ADC que comprende un resto de anticuerpo dirigido a FRA tal como MORAb-003 (p. ej., mejor relación fármaco:anticuerpo, mejor eficacia de tratamiento, destrucción inespecífica reducida, etc.). Como se describe en la presente memoria, algunas o todas estas propiedades funcionales favorables se observan cuando los enlazadores y toxina (eribulina) descritos se conjugan con un resto de anticuerpo dirigido a her2 tal como trastuzumab. Como se describe en la presente memoria, el resto de anticuerpo se dirige a her2. Como se describe en la presente memoria, el resto de anticuerpo que se dirige a her2 es trastuzumab. Como se describe en la presente memoria, algunas o todas estas propiedades funcionales favorables se observan cuando los enlazadores y toxina (eribulina) descritos se conjugan con un resto de anticuerpo dirigido a MSLN tal como MORAb-009. Como se describe en la presente memoria, el resto de anticuerpo se dirige a MSLN. Como se describe en la presente memoria, el resto de anticuerpo que se dirige a MSLN es MORAb-009.
Enlazadores
Como se describe en la presente memoria, el enlazador en un ADC es estable extracelularmente en un modo suficiente para que sea terapéuticamente eficaz. Como se describe en la presente memoria, el enlazador es estable fuera de una célula, de modo que el ADC permanece intacto cuando está presente en condiciones extracelulares (p. ej., antes del transporte o suministro a una célula). El término "intacto/a" usado en el contexto de un ADC, significa que el resto de anticuerpo permanece acoplado al resto de fármaco. Según se usa en la presente memoria, "estable" en el contexto de un enlazador o ADC que comprende un enlazador, significa que no más 20 %, no más de aproximadamente 15 %, no más de aproximadamente 10 %, no más de aproximadamente 5 %, no más de aproximadamente 3 % o no más de aproximadamente 1 % de los enlazadores (o cualquier porcentaje intermedio) en una muestra de ADC se escinden (o en el caso de un ADC entero está de cualquier otra manera no intacto) cuando el ADC está presente en condiciones extracelulares.
Puede determinarse si un enlazador es estable extracelularmente, por ejemplo, al incluir un ADC en plasma durante un período de tiempo predeterminado (p. ej., 2, 4, 6, 8, 16 o 24 horas) y después cuantificar de la cantidad de resto de fármaco libre presente en el plasma. La estabilidad puede dar tiempo al ADC para localizar células tumorales diana y prevenir la liberación prematura del fármaco, que podría bajar el índice terapéutico del ADC al dañar de manera indiscriminada tejidos normales y tumorales. Como se describe en la presente memoria, el enlazador es estable fuera de una célula diana y libera el resto de fármaco desde el ADC una vez dentro de la célula, de modo que el resto de fármaco puede unirse a su diana (p. ej., a microtúbulos). Por lo tanto, un enlazador eficaz: (i) mantendrá las propiedades de unión específicas del resto de anticuerpo; (ii) permitirá el suministro, p. ej., el suministro intracelular del resto de fármaco a través del acoplamiento estable al resto de anticuerpo; (iii) permanecerá estable e intacto hasta que el ADC se haya transportado o suministrado a su sitio diana; y (iv) permitirá el efecto terapéutico, p. ej., efecto citotóxico, del resto de fármaco después de la escisión.
Los enlazadores pueden tener un impacto sobre las propiedades físico-químicas de un ADC. Dado que muchos agentes citotóxicos son de naturaleza hidrófoba, enlazarlos al anticuerpo con un resto hidrófobo adicional puede conducir a la aglomeración. Los aglomerados de ADC son insolubles y a menudo limitan la carga de fármaco alcanzable en el anticuerpo, lo que puede afectar de manera negativa la potencia del ADC. Los aglomerados proteicos de sustancias biológicas, en general, también se han relacionado con mayor inmunogenicidad. Como se muestra más adelante, los enlazadores descritos en la presente memoria dan como resultado ADC con niveles de aglomeración más bajos y niveles deseables de carga de fármaco.
Un enlazador puede ser "escindible" o "no escindible" (Ducry and Stump, Bioconjugate Chem. (2010) 21:5-13). Los enlazadores escindibles están diseñados para liberar el fármaco cuando se someten a ciertos factores ambientales, p. ej., cuando se internalizan en la célula diana, mientras que los enlazadores no escindibles generalmente dependen de la degradación del propio resto de anticuerpo.
Como se describe en la presente memoria, el enlazador es un enlazador no escindible. Como se describe en la presente memoria, el resto de fármaco del ADC se libera mediante la degradación del resto de anticuerpo. Los enlazadores no escindibles tienden a permanecer asociados covalentemente con al menos un aminoácido del anticuerpo y el fármaco después de la internalización por parte de y la degradación dentro de la célula diana. Los enlazadores no escindibles comúnmente incluyen una ligadura tioéter, que se prepara mediante la conjugación de un grupo tiol en el fármaco o el anticuerpo con un grupo maleimida o haloacetamida en el anticuerpo o fármaco, respectivamente (Goldmacher et al., In Cancer Drug Discovery and Development: Antibody-Drug Conjugates and Immunotoxins (G. L. Phillips ed., Springer, 2013)). Un enlazador no escindible ilustrativo comprende tioéter, ciclohexilo, carboxilato de N-succinimidil 4-(N-maleimidometil) ciclohexano-1 (SMCC), N-hidroxisuccinimida (NHS), o uno o más restos de polietilenglicol (PEG), p. ej., 1, 2, 3, 4, 5 o 6 restos de PEG. Como se describe en la presente memoria, el enlazador no escindible comprende (PEG)2. Como se describe en la presente memoria, el enlazador no escindible comprende (PEG)4.
Como se describe en la presente memoria, el enlazador es un enlazador escindible. Un enlazador escindible se refiere a cualquier enlazador que comprende un resto escindible. Según se usa en la presente memoria, el término "resto escindible" se refiere a cualquier unión química que se puede escindir. Las uniones químicas escindibles adecuadas se conocen en la técnica e incluyen, pero no se limitan a, uniones lábiles a ácido, uniones lábiles a proteasa/peptidasa, uniones fotolábiles, uniones disulfuro y uniones lábiles a esterasa. Los enlazadores que comprenden un resto escindible pueden permitir la liberación del resto de fármaco desde el ADC a través de la escisión en un sitio particular en el enlazador. Como se describe en la presente memoria, la escisión del anticuerpo desde la toxina enlazada activa o aumenta la actividad de la toxina. Como se describe en la presente memoria, un ADC que comprende un enlazador escindible (p. ej., un enlazador Val-Cit) demuestra mayor destrucción celular específica y/o menor destrucción celular inespecífica, en comparación con un ADC que comprende un enlazador no escindible (p. ej., un enlazador de (PEG)2 o (PEG)4 no escindible). Como se describe en la presente memoria, un ADC que comprende un enlazador escindible exhibe mejor eficacia de tratamiento con respecto a un ADC que comprende un enlazador no escindible cuando las células y/o el cáncer tratados con el ADC no expresan niveles altos del antígeno diana (p. ej., FRA o her2). Como se describe en la presente memoria, la escisión del anticuerpo desde la toxina enlazada es necesaria para lograr una mejor eficacia de tratamiento de un ADC, según se mide in vitro e/o in vivo.
Como se describe en la presente memoria, el enlazador es escindible en condiciones intracelulares, de modo que la escisión del enlazador libera suficientemente el resto de fármaco del resto de anticuerpo en el entorno intracelular para
activar el fármaco y/o hacer que el fármaco sea terapéuticamente eficaz. Como se describe en la presente memoria, el resto de fármaco no se escinde del resto de anticuerpo hasta que el ADC ingresa a una célula que expresa un antígeno específico para el resto de anticuerpo del ADC, y el resto de fármaco se escinde del resto de anticuerpo después de ingresar a la célula. Como se describe en la presente memoria, el enlazador comprende un resto escindible que se posiciona de modo que ninguna parte del enlazador ni del resto de anticuerpo permanezca unida al resto de fármaco después de la escisión. Los enlazadores escindibles ilustrativos incluyen enlazadores lábiles a ácido, enlazadores sensibles a proteasa/peptidasa, enlazadores fotolábiles, enlazadores que contienen dimetil, disulfuro o sulfonamida.
Como se describe en la presente memoria, el enlazador es un enlazador sensible al pH y es sensible a la hidrólisis con ciertos valores de pH. Típicamente, el enlazador sensible al pH es escindible en condiciones ácidas. Esta estrategia de escisión generalmente aprovecha el pH más bajo en los compartimientos intracelulares endosómicos (pH ~5-6) y lisosómicos (pH ~4,8) en comparación con el citosol (pH ~7,4), para desencadenar la hidrólisis de un grupo lábil a ácido en el enlazador, tal como una hidrazona (Jain et al. (2015) Pharm Res 32:3526-40). Como se describe en la presente memoria, el enlazador es un enlazador lábil a ácido e/o hidrolizable. Por ejemplo, se puede usar un enlazador lábil a ácido que es hidrolizable en el lisosoma y contiene un grupo lábil a ácido (p. ej., una hidrazona, una semicarbazona, una tiosemicarbazona, una amida cis-aconítica, un ortoéster, un acetal, un cetal o similares). Ver, p. ej., las patentes estadounidenses núms. 5.122.368; 5.824.805; 5.622.929; Dubowchik and Walker (1999) Pharm. Therapeutics 83:67-123; Neville et al. (1989) Biol. Chem. 264:14653-61. Dichos enlazadores son relativamente estables en condiciones de pH neutro, tales como las de la sangre, pero son inestables en un pH menor que 5,5 o 5,0, el pH aproximado del lisosoma. Como se describe en la presente memoria, el enlazador hidrolizable es un enlazador tioéter (tal como, p. ej., un tioéter acoplado al agente terapéutico a través de una unión acilhidrazona). Ver, p. ej., la patente estadounidense núm. 5.622.929.
Como se describe en la presente memoria, el enlazador es escindible en condiciones reductoras. Como se describe en la presente memoria, el enlazador es escindible en presencia de un agente reductor, tal como glutatión o ditiotreitol. Como se describe en la presente memoria, el enlazador disulfuro escindible o un enlazador sulfonamida escindible.
Como se describe en la presente memoria, el enlazador es un enlazador disulfuro escindible. En la técnica se conoce una variedad de enlazadores disulfuro, que incluyen, por ejemplo, aquellos que pueden formarse al usar SATA (N-succinimidil-5-acetiltioacetato), SPDP (N-succinimidil-3-(2-piridilditio)propionato), SPDB (N-succinimidil-3-(2-piridilditiio)butirato) y SMPT (N-succinimidil-oxicarbonil-alfa-metil-alfa-(2-piridil-ditio)tolueno), SPDB y SMPT. Ver, p. ej., Thorpe et al. (1987) Cancer Res. 47:5924-31; Wawrzynczak et al., En Immunoconjugates: Antibody Conjugates in Radioimagery and Therapy of Cancer (C. W. Vogel ed., Oxford U. Press, 1987). Ver también la patente estadounidense núm. 4.880.935. Los enlazadores disulfuro típicamente se usan para explotar la abundancia de tioles intracelulares, que puede facilitar la escisión de sus uniones disulfuro. Las concentraciones intracelulares del tiol más abundante, glutatión reducido, están generalmente en el intervalo de 1-10 nM, que es aproximadamente 1000 veces más alta que la del tiol de bajo peso molecular más abundante en la sangre (es decir, cisteína) en aproximadamente 5 pM (Goldmacher et al., En Cancer Drug Discovery and Development: Antibody-Drug Conjugates and Immunotoxins (G. L. Phillips ed., Springer, 2013)). Las enzimas intracelulares de la familia de proteína disulfuro isomerasa también pueden contribuir a la escisión intracelular de un enlazador disulfuro. Según se usa en la presente memoria, un enlazador disulfuro escindible se refiere a cualquier enlazador que comprende un resto disulfuro escindible. El término "resto disulfuro escindible" se refiere a una unión disulfuro que se puede escindir y/o reducir, p. ej., mediante un tiol o enzima. Como se describe en la presente memoria, el resto disulfuro escindible es disulfidilo-dimetilo.
Como se describe en la presente memoria, el enlazador es un enlazador sulfonamida escindible. Según se usa en la presente memoria, un enlazador sulfonamida escindible se refiere a cualquier enlazador que comprende un resto sulfonamida escindible. El término "resto sulfonamida escindible" se refiere a un grupo sulfonamida, es decir, grupo sulfonilo conectado a un grupo amina, en donde la unión azufre-nitrógeno se puede escindir.
Como se describe en la presente memoria, el enlazado puede ser un enlazador de tipo dendrítico para el acoplamiento covalente de más de un resto de fármaco a un resto de anticuerpo a través de un resto enlazador multifuncional ramificado. Ver, p. ej., Sun et al. (2002) Bioorg. Med. Chem. Lett. 12:2213-5; Sun et al. (2003) Bioorg. Med. Chem.
11:1761 -8. Los enlazadores dendríticos pueden aumentar la relación molar entre el fármaco y el anticuerpo, es decir, la carga de fármaco, que está relacionada con la potencia del ADC. Por lo tanto, cuando un resto de anticuerpo tiene solo un grupo tiol de cisteína reactivo, por ejemplo, se pueden acoplar muchos restos de fármaco a través de un enlazador dendrítico. Como se describe en la presente memoria, el resto de enlazador o enlazador-resto de fármaco se puede acoplar al anticuerpo a través de química de puente disulfuro reducido o tecnología de utilización de lisina limitada. Ver, p. ej., las publicaciones internacionales núms. WO2013173391 y WO2013173393.
Como se describe en la presente memoria, el enlazador es escindible mediante un agente de escisión, p. ej., una enzima, que está presente en el entorno intracelular (p. ej., dentro de un lisosoma o endosoma o caveola). El enlazador puede ser, p. ej., un enlazador peptídico que se escinde mediante una enzima peptidasa o proteasa intracelular, incluida, pero sin limitarse a una proteasa lisosómica o endosómica. Como se describe en la presente memoria, el enlazador es un enlazador peptídico escindible. Según se usa en la presente memoria, un enlazador peptídico escindible se refiere a cualquier enlazador que comprende un resto peptídico escindible. El término "resto de péptido escindible" se refiere a cualquier unión química que enlaza aminoácidos (derivados aminoacídicos naturales o
sintéticos) que se puede escindir mediante un agente que está presente en el entorno intracelular. Por ejemplo, un enlazador puede comprender una secuencia de alanina-alanina-asparagina (Ala-Ala-Asn) o una secuencia de valinacitrulina (Val-Cit) que es escindible mediante una peptidasa tal como catepsina, p. ej., catepsina B.
Como se describe en la presente memoria, el enlazador es un enlazador escindible por enzima y un resto de péptido escindible en el enlazador es escindible mediante la enzima. Como se describe en la presente memoria, el resto de péptido escindible es escindible mediante una enzima lisosómica, p. ej., catepsina. Como se describe en la presente memoria, el enlazador es un enlazador escindible por catepsina. Como se describe en la presente memoria, el resto de péptido escindible en el enlazador es escindible mediante una cisteína catepsina lisosómica, tal como catepsina B, C, F, H, K, L, O, S, V, X o W. Como se describe en la presente memoria, el resto de péptido escindible es escindible mediante catepsina B. Un dipéptido ilustrativo que se puede escindir mediante catepsina B es valina-citrulina (Val-Cit) (Dubowchik et al. (2002) Bioconjugate Chem. 13:855-69). Como se describe en la presente memoria, un ADC que comprende un resto de péptido escindible demuestra niveles de aglomeración más bajos y/o carga de fármaco más alta (p) con respecto a un ADC que comprende un resto escindible alternativo (p. ej., un resto disulfuro escindible o un resto sulfonamida escindible).
Como se describe en la presente memoria, el enlazador o el resto de péptido escindible en el enlazador comprende una unidad de aminoácido. Como se describe en la presente memoria, la unidad de aminoácido permite la escisión del enlazador mediante una proteasa, facilitando así la liberación del resto de fármaco desde el ADC después de la exposición a una o más proteasas intracelulares, tales como una o más enzimas lisosómicas (Doronina et al. (2003) Nat. Biotechnol. 21:778-84; Dubowchik and Walker (1999) Pharm. Therapeutics 83:67-123). Las unidades de aminoácido ilustrativas incluyen, pero no se limitan a, dipéptidos, tripéptidos, tetrapéptidos y pentapéptidos. Los dipéptidos ilustrativos incluyen, pero no se limitan a, valina-citrulina (Val-Cit), alanina-asparagina (Ala-Asn), alaninafenilalanina (Ala-Phe), fenilalanina-lisina (Phe-Lys), alanina-lisina (Ala-Lys), alanina-valina (Ala-Val), valina-alanina (Val-Ala), valina-lisina (Val-Lys), lisina-lisina (Lys-Lys), fenilalanina-citrulina (Phe-Cit), leucina-citrulina (Leu-Cit), isoleucina-citrulina (Ile-Cit), triptófano-citrulina (Trp-Cit) y fenilalanina-alanina (Phe-Ala). Los tripéptidos ilustrativos incluyen, pero no se limitan a, alanina-alanina-asparagina (Ala-Ala-Asn), glicina-valina-citrulina (Gly-Val-Cit), glicinaglicina-glicina (Gly-Gly-Gly), fenilalanina-fenilalanina-lisina (Phe-Phe-Lys) y glicina-fenilalanina-lisina (Gly-Phe-Lys). Otras unidades de aminoácidos ilustrativas incluyen, pero no se limitan a, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg y Phe-N9-Nitro-Arg, como se describen en, p. ej., la patente estadounidense núm. 6.214.345. Como se describe en la presente memoria, la unidad de aminoácido en el enlazador comprende Val-Cit. Como se describe en la presente memoria, la unidad de aminoácido en el enlazador comprende Ala-Ala-Asn. Como se describe en la presente memoria, un ADC que comprende Val-Cit demuestra menor destrucción inespecífica de células, mayor destrucción específica de células, niveles de aglomeración más bajos y/o carga de fármaco más alta (p) con respecto a un ADC que comprende una unidad de aminoácido alternativa o resto escindible alternativo. Una unidad de aminoácido puede comprender residuos aminoacídicos de origen natural y/o aminoácidos menores y/o análogos de aminoácidos de origen no natural, tales como la citrulina. Las unidades de aminoácidos pueden diseñarse y optimizarse para la escisión enzimática mediante una enzima particular, por ejemplo, una proteasa asociada a un tumor, una proteasa lisosómica tal como catepsina B, C, D o S, o una proteasa plasmina.
Como se describe en la presente memoria, el enlazador en cualquiera de los ADC descritos en la presente memoria puede comprender al menos una unidad espaciadora que liga el resto de anticuerpo al resto de fármaco. Como se describe en la presente memoria, la unidad espaciadora liga un sitio de escisión (p. ej., un resto de péptido escindible) en el enlazador al resto de anticuerpo. Como se describe en la presente memoria, el enlazador y/o unidad espaciadora en el enlazador, es sustancialmente hidrófilo. Un enlazador hidrófilo se puede usar para reducir la extensión en la que el fármaco puede bombearse fuera de células cancerosas resistentes a través de multirresistencia a fármacos (MDr , por sus siglas en inglés) o transportadores funcionalmente similares. En algunos aspectos, el enlazador incluye uno o más restos de polietilenglicol (PEG), p. ej., 1,2, 3, 4, 5 o 6 restos de PEG. Como se describe en la presente memoria, el enlazador es un enlazador de PEG más corto y proporciona estabilidad mejorada y aglomeración reducida con respecto a enlazadores de PEG más largos.
Como se describe en la presente memoria, la unidad espaciadora en el enlazador comprende uno o más restos de PEG. Como se describe en la presente memoria, la unidad espaciadora comprende -(PEG)m-, y m es un número entero de 1 a 10. Como se describe en la presente memoria, m varía de 1 a 10; de 2 a 8; de 2 a 6; de 2 a 5; de 2 a 4; o de 2 a 3. Como se describe en la presente memoria, m es 8. Como se describe en la presente memoria, m es 4. Como se describe en la presente memoria, m es 3. Como se describe en la presente memoria, m es 2. Como se describe en la presente memoria, la unidad espaciadora comprende (PEG)2 , (PEG)4, (PEG)s, (PEG)9, (PEG)3-triazol-(PEG)3, (PEG)4-triazol-(PEG)3 o dibencilcicloocteno-triazol-(PEG)3. Como se describe en la presente memoria, la unidad espaciadora comprende (PEG)2. Como se describe en la presente memoria, un ADC que comprende una unidad espaciadora más corta (p. ej., (PEG)2) demuestra niveles de aglomeración más bajos y/o carga de fármaco más alta (p) con respecto a un ADC que comprende una unidad espaciadora más larga (p. ej., (PEG)s).
Como se describe en la presente memoria, la unidad espaciadora en el enlazador comprende un resto alquilo. Como se describe en la presente memoria, la unidad espaciadora comprende -(CH2)n-, y n es un número entero de 1 a 10 (es decir, n puede ser 1,2, 3, 4, 5, 6, 7, 8, 9 o 10). Como se describe en la presente memoria, n es 5. Como se describe en la presente memoria, un ADC que comprende una unidad espaciadora más corta (p. ej., (CH2)s) demuestra niveles
de aglomeración más bajos y/o carga de fármaco más alta (p) con respecto a un ADC que comprende una unidad espadadora más larga (p. ej., (PEG)8).
Una unidad espaciadora se puede usar, por ejemplo, para enlazar el resto de anticuerpo al resto de fármaco, ya sea directamente o indirectamente. Como se describe en la presente memoria, la unidad espaciadora enlaza el resto de anticuerpo al resto de fármaco directamente. Como se describe en la presente memoria, el resto de anticuerpo y el resto de fármaco se acoplan a través de una unidad espaciadora que comprende uno o más restos de PEG (p. ej., (PEG)2 o (PEG)4). Como se describe en la presente memoria, la unidad espaciadora enlaza el resto de anticuerpo al resto de fármaco indirectamente. Como se describe en la presente memoria, la unidad espaciadora enlaza el resto de anticuerpo al resto de fármaco indirectamente a través de un resto escindible (p. ej., un péptido escindible, un disulfuro escindible o una sulfonamida escindible) y/o un resto de acoplamiento para ligar la unidad espaciadora al resto de anticuerpo, p. ej., un resto de maleimida.
La unidad espaciadora, como se describe en la presente memoria, se acopla al resto de anticuerpo (es decir, el anticuerpo o fragmento de unión a antígeno) a través de un resto de maleimida (Mal). Como se describe en la presente memoria, un ADC que comprende un enlazador acoplado al resto de anticuerpo a través de un resto de maleimida demuestra carga de fármaco más alta (p) con respecto a un ADC que comprende un enlazador acoplado al resto de anticuerpo a través de un resto de acoplamiento alternativo tal como un resto de succinimida.
Una unidad espaciadora que se acopla al anticuerpo o fragmento de unión a antígeno a través de un Mal se denomina en la presente memoria una "Mal-unidad espaciadora". El término "resto de maleimida", según se usa en la presente memoria, significa un compuesto que contiene un grupo maleimida y que es reactivo con un grupo sulfhidrilo, p. ej., un grupo sulfhidrilo de un residuo cisteína en el resto de anticuerpo. Otros grupos funcionales que son reactivos con grupos sulfhidrilo (tioles) incluyen, pero no se limitan a, yodoacetamida, bromoacetamida, vinilpiridina, disulfuro, disulfuro de piridilo, isocianato e isotiocianato. Como se describe en la presente memoria, la Mal-unidad espaciadora es reactiva con un residuo cisteína en el anticuerpo o fragmento de unión a antígeno. Como se describe en la presente memoria, la Mal-unidad espaciadora se liga al anticuerpo o fragmento de unión a antígeno a través del residuo de cisteína. Como se describe en la presente memoria, la Mal-unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, la Mal-unidad espaciadora comprende un resto alquilo.
Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora y un resto de péptido escindible. Como se describe en la presente memoria, el resto de péptido escindible comprende una unidad de aminoácido. Como se describe en la presente memoria, la unidad de aminoácido comprende Val-Cit. Como se describe en la presente memoria, la unidad de aminoácido comprende Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora y Val-Cit. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2 y Val-Cit. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)m y Val-Cit, donde m es 2 a 8 o 2 a 5, o 2, 3, 4 o 5. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)8 y Val-Cit. Como se describe en la presente memoria, el enlazador comprende Mal-(CH2)5 y Val-Cit. Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora y Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2 y Ala-Ala-Asn.
Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora y un resto de disulfuro escindible. Como se describe en la presente memoria, el resto disulfuro escindible es disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora y disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3 y disulfidilo-dimetilo.
Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora y un resto de sulfonamida escindible. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3 y sulfonamida.
Como se describe en la presente memoria, la unidad espaciadora se acopla al anticuerpo o fragmento de unión a antígeno a través de un resto de succinimida (OSu). Una unidad espaciadora que se acopla al anticuerpo o fragmento de unión a antígeno a través de una OSu se denomina en la presente memoria una "OSu-unidad espaciadora". El término "resto de succinimida", según se usa en la presente memoria, significa un compuesto que contiene un compuesto de succinimida que es reactivo con un grupo amina, p. ej., un grupo amina de un residuo lisina en el resto de anticuerpo. Un resto de succinimida ilustrativo es N-hidroxisuccinimida (NHS). Como se describe en la presente memoria, la OSu-unidad espaciadora es reactiva con un residuo lisina en el anticuerpo o fragmento de unión a antígeno. Como se describe en la presente memoria, la OSu-unidad espaciadora se liga al anticuerpo o fragmento de unión a antígeno a través del residuo de lisina. Como se describe en la presente memoria, la OSu-unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, la OSu-unidad espaciadora comprende un resto alquilo.
Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora y un resto de péptido escindible. Como se describe en la presente memoria, el resto de péptido escindible comprende una unidad de aminoácido. Como se describe en la presente memoria, la unidad de aminoácido comprende Val-Cit. Como se describe en la presente memoria, la unidad de aminoácido comprende Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora y Val-Cit. Como se describe en la presente memoria,
el enlazador comprende OSu-(PEG)2 y Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)g y Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(CH2)5 y Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3 y Val-Cit. Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora y Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)2 y Ala-Ala-Asn.
Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora y un resto de disulfuro escindible. Como se describe en la presente memoria, el resto disulfuro escindible es disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora y disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3 y disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilcicloocteno-triazol-(PEG)3 y disulfidil-dimetilo.
Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora y un resto de sulfonamida escindible. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3 y sulfonamida. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilciclooctenotriazol-(PEG)3 y sulfonamida.
Como se describe en la presente memoria, la Mal-unidad espaciadora o la OSu-unidad espaciadora acopla el resto de anticuerpo (es decir, el anticuerpo o fragmento de unión a antígeno) al resto escindible en el enlazador. Como se describe en la presente memoria, la Mal-unidad espaciadora o la OSu-unidad espaciadora acopla el resto de anticuerpo o fragmento de unión a antígeno a un resto de péptido escindible. Como se describe en la presente memoria, el resto de péptido escindible comprende una unidad de aminoácido. Como se describe en la presente memoria, el enlazador comprende Mal-unidad espaciadora-unidad de aminoácido o OSu-unidad espaciadora-unidad de aminoácido. Como se describe en la presente memoria, la Mal-unidad espaciadora o la OSu-unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, la Mal-unidad espaciadora o la OSu-unidad espaciadora comprende un resto alquilo. Como se describe en la presente memoria, la unidad de aminoácido comprende Val-Cit. Como se describe en la presente memoria, la unidad de aminoácido comprende Ala-Ala-Asn.
Como se describe en la presente memoria, el enlazador comprende la estructura: Mal-unidad espaciadora-Val-Cit. Como se describe en la presente memoria, el enlazador comprende la estructura: Mal-(PEG)2-Val-Cit. Como se describe en la presente memoria, el enlazador comprende la estructura: Mal-(PEG)2-Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)s-Val-Cit. Como se describe en la presente memoria, el enlazador comprende Mal-(CH2)5-Val-Cit. Como se describe en la presente memoria, el enlazador comprende la Malunidad espaciadora-Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-Ala-Ala-Asn.
Como se describe en la presente memoria, el enlazador comprende OSu-unidad espaciadora-Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)2-Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)g-Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(CH2)5-Val-Cit. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-Val-Cit. Como se describe en la presente memoria, el enlazador comprende la OSu-unidad espaciadora-Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)2-Ala-Ala-Asn.
Como se describe en la presente memoria, la Mal-unidad espaciadora o la OSu-unidad espaciadora acopla el resto de anticuerpo o fragmento de unión a antígeno a un resto de disulfuro escindible. Como se describe en la presente memoria, el enlazador comprende la Mal-unidad espaciadora-disulfuro o la OSu-unidad espaciadora-disulfuro. Como se describe en la presente memoria, el disulfuro es disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende Mal-unidad espaciadora-disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3-disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende OSu-unidad espaciadora-disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-disulfidilo-dimetilo. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilcicloocteno-triazol-(PEG)3-disulfidilo-dimetilo.
Como se describe en la presente memoria, la Mal-unidad espaciadora o la OSu-unidad espaciadora acopla el resto de anticuerpo o fragmento de unión a antígeno a un resto de sulfonamida escindible. Como se describe en la presente memoria, el enlazador comprende Mal-unidad espaciadora-sulfonamida u OSu-unidad espaciadora-sulfonamida. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3-sulfonamida. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-sulfonamida. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilcicloocteno-triazol-(PEG)3-sulfonamida.
Como se describe en la presente memoria, el resto escindible en el enlazador se liga directamente al resto de fármaco. Como se describe en la presente memoria, otra unidad espaciadora se usa para acoplar el resto escindible en el enlazador al resto de fármaco. Como se describe en la presente memoria, el resto de fármaco es eribulina. Como se describe en la presente memoria, la eribulina se acopla al resto escindible en el enlazador mediante una unidad espaciadora. Como se describe en la presente memoria, la eribulina se acopla al resto escindible en el enlazador mediante una unidad espaciadora autodestructiva. Como se describe en la presente memoria, la eribulina se acopla
al resto escindible en el enlazador mediante una unidad espadadora autodestructiva, el resto escindible comprende Val-Cit, y una unidad espaciadora adicional que comprende PEG liga el resto enlazador al resto de anticuerpo. Como se describe en la presente memoria, la eribulina se liga a un anticuerpo anti-FRA a través de una Mal-unidad espaciadora en el enlazador ligada a un resto escindible Val-Cit y una unidad espaciadora autodestructiva pAB. Como se describe en la presente memoria, la eribulina se liga a un anticuerpo anti-her2 a través de una Mal-unidad espaciadora en el enlazador ligada a un resto escindible Val-Cit y una unidad espaciadora autodestructiva pAB.
Una unidad espaciadora puede ser "autodestructiva" o "no autodestructiva". Una unidad espaciadora "no autodestructiva" es una unidad en la cual parte o toda la unidad espaciadora permanece unida al resto de fármaco tras la escisión del enlazador. Los ejemplos de unidades espaciadoras no autodestructivas incluyen, pero no se limitan a, una unidad espaciadora de glicina y una unidad espaciadora de glicina-glicina. Las unidades espaciadoras no autodestructivas eventualmente pueden degradarse con el tiempo, pero no liberan fácilmente un fármaco natural enlazado por completo en condiciones celulares. Una unidad espaciadora "autodestructiva" permite la liberación del resto de fármaco natural en condiciones intracelulares. Un "fármaco natural" es uno donde ninguna parte de la unidad espaciadora u otra modificación química permanece después de la escisión/degradación de la unidad espaciadora.
La química de la autodestrucción se conoce en la técnica y podría seleccionarse sin inconvenientes para los ADC descritos. Como se describe en la presente memoria, la unidad espaciadora que acopla el resto escindible en el enlazador al resto de fármaco (p. ej., eribulina) es autodestructiva y sufre autodestrucción simultáneamente con o poco antes/después de la escisión del resto escindible en condiciones intracelulares.
Como se describe en la presente memoria, la unidad espaciadora autodestructiva en el enlazador comprende una unidad de p-aminobencilo. Como se describe en la presente memoria, un alcohol p-aminobencílico (pABOH) se acopla a una unidad de aminoácido u otro resto escindible en el enlazador a través de una unión amida, y se produce un carbamato, metilcarbamato o carbonato entre el pABOH y el resto de fármaco (Hamann et al. (2005) Expert Opin. Ther. Patents 15:1087-103). Como se describe en la presente memoria, la unidad espaciadora autodestructiva es o comprende p-aminobenciloxicarbonilo (pAB). Sin desear limitarse a la teoría, se cree que la autodestrucción de pAB implica una reacción de 1,6-eliminación espontánea (Jain et al. (2015) Pharm Res 32:3526-40).
Como se describe en la presente memoria, la estructura del p-aminobenciloxicarbonilo (pAB) usado en los ADC descritos se muestra a continuación:

Como se describe en la presente memoria, la unidad espaciadora autodestructiva acopla el resto escindible en el enlazador a la C-35 amina en la eribulina. Como se describe en la presente memoria, la unidad espaciadora autodestructiva es pAB. Como se describe en la presente memoria, el pAB acopla el resto escindible en el enlazador a la C-35 amina en la eribulina. Como se describe en la presente memoria, el pAB sufre autodestrucción después de la escisión del resto escindible, y la eribulina se libera desde el ADC en su forma activa, natural. Como se describe en la presente memoria, un anticuerpo anti-FRA (p. ej., MORAb-003) se liga a la C-35 amina de eribulina mediante un enlazador que comprende Mal-(PEG)2-Val-Cit-pAB. Como se describe en la presente memoria, un anticuerpo antiher2 (p. ej., trastuzumab) se liga a la C-35 amina de eribulina mediante un enlazador que comprende Mal-(PEG)2-Val-Cit-pAB.
Como se describe en la presente memoria, el pAb sufre autodestrucción después de la escisión de un resto de péptido escindible en el enlazador. Como se describe en la presente memoria, el resto de péptido escindible comprende una unidad de aminoácido. Como se describe en la presente memoria, el enlazador comprende una unidad de aminoácidopAB. Como se describe en la presente memoria, la unidad de aminoácido es Val-Cit. Como se describe en la presente memoria, el enlazador comprende Val-Cit-pAB (VCP). Como se describe en la presente memoria, la unidad de aminoácido es Ala-Ala-Asn. Como se describe en la presente memoria, el enlazador comprende Ala-Ala-Asn-pAB.
Como se describe en la presente memoria, el pAb sufre autodestrucción después de la escisión de un resto de disulfuro escindible en el enlazador. Como se describe en la presente memoria, el enlazador comprende disulfuro-pAB. Como se describe en la presente memoria, el enlazador comprende disulfidilo-dimetilo-pAB.
Como se describe en la presente memoria, el pAb sufre autodestrucción después de la escisión de un resto de sulfonamida escindible en el enlazador. Como se describe en la presente memoria, el enlazador comprende sulfonamida-pAB.
Como se describe en la presente memoria, el resto de anticuerpo del ADC se conjuga con el resto de fármaco a través de un enlazador, en donde el enlazador comprende una Mal-unidad espaciadora, una unidad de aminoácido escindible y un pAB. Como se describe en la presente memoria, la unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, la unidad espaciadora comprende un resto alquilo. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)2-Ala-Ala-Asn-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)8-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)8-Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(CH2)5-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(CH2)5-Val-Cit-pAB.
Como se describe en la presente memoria, el resto de anticuerpo del ADC se conjuga con el resto de fármaco a través de un enlazador, en donde el enlazador comprende una Mal-unidad espaciadora-disulfuro-pAB. Como se describe en la presente memoria, la unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3-disulfuro-pAB. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3-disulfidilo-dimetilo-pAB.
Como se describe en la presente memoria, el resto de anticuerpo del ADC se conjuga con el resto de fármaco a través de un enlazador, en donde el enlazador comprende una Mal-unidad espaciadora-sulfonamida-pAB. Como se describe en la presente memoria, la unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, el enlazador comprende Mal-(PEG)4-triazol-(PEG)3-sulfonamida-pAB.
Como se describe en la presente memoria, el resto de anticuerpo del ADC se conjuga con el resto de fármaco a través de un enlazador, en donde el enlazador comprende una OSu-unidad espaciadora-unidad de aminoácido-pAB. Como se describe en la presente memoria, la unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, la unidad espaciadora comprende un resto alquilo. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)2-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)2-Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)2-Ala-Ala-Asn-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)9-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)9-Val-CitpAB. Como se describe en la presente memoria, el enlazador comprende OSu-(CH2)5-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(CH2)5-Val-Cit-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-unidad de aminoácido-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-Val-Cit-pAB.
Como se describe en la presente memoria, el resto de anticuerpo del ADC se conjuga con el resto de fármaco a través de un enlazador, en donde el enlazador comprende una OSu-unidad espaciadora-disulfuro-pAB. Como se describe en la presente memoria, la unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-disulfuro-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-disulfidilo-dimetilo-pAB-. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilcicloocteno-triazol-(PEG)3-disulfuro-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilcicloocteno-triazol-(PEG)3-disulfidilo-dimetilo-pAB.
Como se describe en la presente memoria, el resto de anticuerpo del ADC se conjuga con el resto de fármaco a través de un enlazador, en donde el enlazador comprende una OSu-unidad espaciadora-sulfonamida-pAB. Como se describe en la presente memoria, la unidad espaciadora comprende un resto de PEG. Como se describe en la presente memoria, el enlazador comprende OSu-(PEG)3-triazol-(PEG)3-sulfonamida-pAB. Como se describe en la presente memoria, el enlazador comprende OSu-dibencilcicloocteno-triazol-(PEG)3-sulfonamida-pAB.
Como se describe en la presente memoria, el enlazador se diseña para facilitar la destrucción por vecindad (la destrucción de células vecinas) a través de la escisión después de la internalización celular y la difusión del enlazadorresto de fármaco y/o el resto de fármaco solo a las células vecinas. Como se describe en la presente memoria, el enlazador promueve la internalización celular. Como se describe en la presente memoria, el enlazador se diseña para minimizar la escisión en el entorno extracelular y reducir así la toxicidad para el tejido inespecífico (p. ej., tejido no canceroso), mientras se conserva la unión del ADC al tejido diana y la destrucción por vecindad de tejido canceroso que no expresa un antígeno diana del resto de anticuerpo de un ADC, pero que rodea al tejido cancerosos diana que expresa dicho antígeno. Como se describe en la presente memoria, un enlazador que comprende un resto de maleimida (Mal), un resto de polietilenglicol (PEG), valina-citrulina (Val-Cit o "vc") y un pAB proporciona estas características funcionales. Como se describe en la presente memoria, un enlazador que comprende Mal-(PEG)2-Val-Cit-pAB es particularmente eficaz en proporcionar estas características funcionales, p. ej., cuando se liga a un resto de anticuerpo anti-FRA tal como MORAb-003 y un resto de fármaco tal como eribulina. Como se describe en la presente memoria, al menos algunas de estas características funcionales también se pueden observar sin un resto de anticuerpo anti-FRA y/o sin MORAb-003. Por ejemplo, como se describe en la presente memoria, un enlazador que comprende Mal-(PEG)2-Val-Cit-pAB es eficaz en proporcionar algunas o todas estas características funcionales, p. ej., cuando se liga a un resto de anticuerpo anti-her2 tal como trastuzumab y un resto de fármaco tal como eribulina.
Como se describe en la presente memoria, el resto de anticuerpo se conjuga con el resto de fármaco a través de un enlazador que comprende un resto de maleimida (Mal), un resto de polietilenglicol (PEG), valina-citrulina (Val-Cit o "vc") y un pAB. Como se describe en la presente memoria, el resto de maleimida acopla covalentemente el enlazadorresto de fármaco al resto de anticuerpo y el pAB actúa como una unidad espaciadora autodestructiva. Dicho enlazador puede denominarse el enlazador "m-vc-pAB", el enlazador "Mal-VCP", el enlazador "Mal-(PEG)2-VCP'' o el enlazador "Mal-(PEG)2-Val-Cit-pAB". Como se describe en la presente memoria, el resto de fármaco es eribulina. La estructura de Mal-(PEG)2-Val-Cit-pAB-eribulina se proporciona en la Tabla 46. El pAB del enlazador Mal-(PEG)2-Val-Cit-pAB se acopla a la C-35 amina en la eribulina.
Se descubrió que los ADC que comprenden Mal-(PEG)2-Val-Cit-pAB-eribulina demuestran una combinación particular de propiedades deseables, particularmente cuando se emparejan con un anticuerpo anti-FRA tal como MORAb-003 o un fragmento de unión a antígeno de este. Estas propiedades incluyen, pero no se limitan a, niveles eficaces de carga de fármaco (p ~4), bajos niveles de aglomeración, estabilidad condiciones de almacenamiento o cuando está en circulación en el cuerpo (p. ej., estabilidad sérica), afinidad conservada por células que expresan la diana semejante a la del anticuerpo no conjugado, citotoxicidad potente contra células que expresan la diana, bajos niveles de destrucción celular inespecífica, elevados niveles de destrucción por vecindad y/o actividad anticancerosa eficaz in vivo, todas en comparación con ADC que usan otro enlazador-toxina y/o restos de anticuerpo. Aunque se conocen numerosas opciones de enlazador y combinaciones de espaciadores y sitios de escisión en la técnica y pueden proporcionar ciertos beneficios en una o más de estas categorías funcionales, la combinación particular del enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a un resto de anticuerpo tal como un anticuerpo anti-FRA (p. ej., MORAb-003) puede proporcionar propiedades buenas o superiores en todo el espectro de propiedades funcionales deseables para un ADC terapéutico. Como se describe en la presente memoria, las propiedades funcionales buenas o superiores proporcionadas mediante la combinación particular de un enlazador Mal-(PEG)2-Val-Cit-pAB que liga eribulina a un resto de anticuerpo se pueden observar con este enlazador-toxina conjugado con, p. ej., un anticuerpo anti-her 2 tal como trastuzumab.
Como se describe en la presente memoria, el ADC comprende Mal-(PEG)2-Val-Cit-pAB-eribulina y un resto de anticuerpo que comprende un anticuerpo internalizante o un fragmento de unión a antígeno de este que conserva la capacidad de dirigirse a e internalizarse en una célula tumoral. Como se describe en la presente memoria, el ADC comprende Mal-(PEG)2 -Val-Cit-pAB-eribulina y un anticuerpo internalizante o un fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa FRA. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa FRA comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ ID NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa FRA comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa FRA comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig.
Como se describe en la presente memoria, el ADC tiene la Fórmula I:
Ab-(L-D)P (I)
en donde:
(i) Ab es un anticuerpo anti-receptor alfa de folato (FRA) internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ ID NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT;
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es un número entero de 1 a 20.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24. Como se describe en la presente memoria, el anticuerpo internalizante es MORAb-003. Como se describe en la presente memoria, p es de 1 a 8, o 1 a 6. Como se describe en la presente memoria, p es de 2 a 8, o 2 a 5. Como se describe en la presente memoria, p es de 3 a 4. Como se describe en la presente memoria, p es 4.
Como se describe en la presente memoria, el ADC comprende Mal-(PEG)2-Val-Cit-pAB-eribulina y un anticuerpo internalizante o un fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa her2. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa her2 comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:71 (HCDR1), SEQ ID NO:72 (HCDR2) y SEQ ID NO:73 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:74 (LCDR1), SEQ ID NO:75 (LCDR2) y SEQ ID n O:76 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:191 (HCDR1), SEQ ID NO:192 (HCDR2) y SEQ ID No :193 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:194 (LCDR1), SEQ ID NO:195 (LCDR2) y SEQ ID NO:196 (LCDR3), según se definen mediante el sistema de numeración de IMGT. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa her2 comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:27 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:28. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa her2 comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig.
Como se describe en la presente memoria, el ADC tiene la Fórmula I:
Ab-(L-D)p (I)
en donde:
(i) Ab es un anticuerpo anti-receptor de factor de crecimiento epidérmico humano 2 (her2) internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:71 (HCDR1), SEQ ID NO:72 (HCDR2) y SEQ ID NO:73 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (Lc DR) que comprenden las secuencias de aminoácidos de SEQ ID NO:74 (LCDR1), SEQ ID NO:75 (LCDR2) y SEQ ID NO:76 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:191 (HCDR1), SEQ ID NO:192 (HCDR2) y s Eq ID NO:193 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:194 (LCDR1), SEQ ID NO:195 (LCDR2) y SEQ iD NO:196 (LCDR3), según se definen mediante el sistema de numeración de IMGT;
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es un número entero de 1 a 20.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:27 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:28. Como se describe en la presente memoria, el anticuerpo internalizante es trastuzumab. Como se describe en la presente memoria, p es de 1 a 8, o 1 a 6. Como se describe en la presente memoria, p es de 2 a 8, o 2 a 5. Como se describe en la presente memoria, p es de 3 a 4. Como se describe en la presente memoria, p es 4.
Como se describe en la presente memoria, el ADC comprende Mal-(PEG)2-Val-Cit-pAB-eribulina y un anticuerpo internalizante o un fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa mesotelina (MSLN). Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa MSLN comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:65 (HCDR1 ), SEQ ID NO:66 (HCDR2) y SEQ ID NO:67 (HCDR3); y tres regiones de determinación de
la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:68 (LCDR1), SEQ ID NO:69 (LCDR2) y SeQ ID NO:70 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:185 (HCDR1), SEQ ID NO:186 (HCDR2) y SEQ ID No :187 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:188 (LCDR1), SEQ ID NO:189 (LCDR2) y SEQ ID NO:190 (LCDR3), según se definen mediante el sistema de numeración de IMGT. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa MSLN comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:25 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:26. Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante que se dirige a una célula tumoral que expresa MSLN comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig.
Como se describe en la presente memoria, el ADC tiene la Fórmula I:
Ab-(L-D)p (I)
en donde:
(i) Ab es un anticuerpo anti-mesotelina internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:65 (HCDR1), SEQ ID NO:66 (HCDR2) y SEQ ID NO:67 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:68 (LCDR1), SEQ ID NO:69 (LCDR2) y SeQ iD No :70 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:185 (HCDR1), SEQ ID NO:186 (HCDR2) y SEQ ID NO:187 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:188 (LCDR1), SEQ ID NO:189 (LCDR2) y SEQ ID NO:190 (LCDR3), según se definen mediante el sistema de numeración de IMGT;
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es un número entero de 1 a 20.
Como se describe en la presente memoria, el anticuerpo internalizante o fragmento de unión a antígeno de este internalizante comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:25 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:26. Como se describe en la presente memoria, el anticuerpo internalizante es MORAb-003, MORAb-009 o trastuzumab. Como se describe en la presente memoria, p es de 1 a 8, o 1 a 6. Como se describe en la presente memoria, p es de 2 a 8, o 2 a 5. Como se describe en la presente memoria, p es de 3 a 4. Como se describe en la presente memoria, p es 4.
Restos de fármaco
El resto de fármaco (D) de los ADC descritos en la presente memoria puede ser cualquier agente quimioterapéutico. Las clases útiles de agentes quimioterapéuticos incluyen, por ejemplo, agentes antitubulina. Como se describe en la presente memoria, el resto de fármaco es un agente antitubulina. Los ejemplos de agentes antitubulina incluyen criptoficina y eribulina. El resto de fármaco preferido para uso en los ADC descritos es eribulina.
Como se describe en la presente memoria, el resto de fármaco es eribulina. Como se describe en la presente memoria, el enlazador del ADC se acopla a través de la C-35 amina en la eribulina.
Como se describe en la presente memoria, la forma natural de eribulina usada para ligarla al enlazador y el resto de anticuerpo se muestra a continuación:

Como se describe en la presente memoria, un intermediario, el cual es el precursor del enlazador, se hace reaccionar con el resto de fármaco en condiciones apropiadas. Como se describe en la presente memoria, los grupos reactivos se usan en el fármaco y/o el intermediario o enlazador. El producto de la reacción entre el fármaco y el intermediario, o el fármaco derivado, se hace reaccionar posteriormente con el anticuerpo o fragmento de unión a antígeno en condiciones apropiadas. Alternativamente, el enlazador o intermediario primero se puede hacer reaccionar con el anticuerpo o un anticuerpo derivado, y después hacerse reaccionar con el fármaco o fármaco derivado.
Una cantidad de reacciones diferentes están disponibles para el acoplamiento covalente de fármacos y/o enlazadores con el resto de anticuerpo. Esto se logra a menudo mediante la reacción de uno o más residuos aminoacídicos de la molécula de anticuerpo, incluidos los grupos amina de la lisina, los grupos de ácido carboxílico libre del ácido glutámico y el ácido aspártico, los grupos sulfhidrilo de la cisteína y los diversos restos de los aminoácidos aromáticos. Por ejemplo, se puede producir un acoplamiento covalente no específico al usar una reacción de carbodiimida para enlazar un grupo carboxi (o amino) de un compuesto a un grupo amino (o carboxi) de un resto de anticuerpo. Además, también se pueden usar agentes bifuncionales tales como dialdehídos o imidoésters para enlazar el grupo amino en un compuesto a un grupo amino de un resto de anticuerpo. La reacción de base Schiff también se encuentra disponible para el acoplamiento de fármacos a agentes de unión. Este método implica la oxidación por peryodato de un fármaco que contiene grupos glicol o hidroxi y, por lo tanto, forma un aldehído el cual después se hace reaccionar con el agente de unión. El acoplamiento se produce a través de la formación de una base Schiff con grupos amino del agente de unión. Los isotiocianatos también pueden usarse como agentes acopladores para enlazar covalentemente fármacos a agentes de unión. Otras técnicas son conocidas para el experto en la técnica y se encuentran dentro del alcance de la presente descripción.
Carga de fármaco
La carga de fármaco está representada por byp y también se denomina en la presente memoria relación entre fármacoy-anticuerpo (DAR). La carga de fármaco puede variar de 1 a 20 restos de fármaco por resto de anticuerpo. Como se describe en la presente memoria, p es un número entero de 1 a 20. Como se describe en la presente memoria, p es un número entero de 1 a 10, 1 a 9, 1 a 8, 1 a 7, 1 a 6, 1 a 5, 1 a 4, 1 a 3 o 1 a 2. Como se describe en la presente memoria, p es un número entero de 2 a 10, 2 a 9, 2 a 8, 2 a 7, 2 a 6, 2 a 5, 2 a 4 o 2 a 3. Como se describe en la presente memoria, p es un número entero de 3 a 4. Como se describe en la presente memoria, p es 1, 2, 3, 4, 5 o 6, preferiblemente, 3 o 4.
La carga de fármaco puede estar limitada por la cantidad de sitios de acoplamiento en el resto de anticuerpo. Como se describe en la presente memoria, el resto de enlazador (L) del ADC se acopla al resto de anticuerpo a través de un grupo químicamente activo en uno o más residuos aminoacídicos en el resto de anticuerpo. Por ejemplo, el enlazador se puede acoplar al resto de anticuerpo a través de un grupo amino, imino, hidroxilo, tiol o carboxilo libre (p. ej., al extremo N o C, al grupo épsilon amino de uno o más residuos lisina, al grupo ácido carboxílico libre de uno o más residuos ácido glutámico o ácido aspártico o al grupo sulfhidrilo de uno o más residuos cisteína). El sitio al cual se acopla el enlazador puede ser un residuo natural en la secuencia de aminoácidos del resto de anticuerpo, o se puede introducir en el resto de anticuerpo, p. ej., mediante tecnología de ADN recombinante (p. ej., al introducir un residuo cisteína en la secuencia de aminoácidos) o mediante bioquímica proteica (p. ej., mediante reducción, ajuste del pH o hidrólisis).
Como se describe en la presente memoria, la cantidad de restos de fármaco que se pueden conjugar con un resto de anticuerpo está limitada por la cantidad de residuos cisteína libres. Por ejemplo, cuando el acoplamiento es un grupo tiol de cisteína, un anticuerpo puede tener solo uno o uno pocos grupos tiol de cisteína, o puede tener solo uno o uno pocos grupos tiol suficientemente reactivos a través de los cuales puede acoplarse un enlazador. Generalmente, los anticuerpos no contienen muchos grupos tiol de cisteína libres y reactivos que puedan enlazarse a un resto de fármaco. De hecho, la mayoría de los residuos tiol de cisteína en anticuerpos existen como puentes disulfuro. El sobreacoplamiento del enlazador-toxina a un anticuerpo puede desestabilizar al anticuerpo al reducir los residuos cisteína disponibles para formar puentes disulfuro. Por lo tanto, una relación óptima fármaco:anticuerpo debería aumentar la potencia del ADC (al aumentar la cantidad de restos de fármaco acoplados por anticuerpo) sin
desestabilizar el resto de anticuerpo. Como se describe en la presente memoria, una relación óptima puede ser de aproximadamente 3-4.
Como se describe en la presente memoria, un enlazador acoplado a un resto de anticuerpo a través de un resto de Mal proporciona una relación de aproximadamente 3-4. Como se describe en la presente memoria, un enlazador acoplado a un resto de anticuerpo a través de un resto alternativo (p. ej., un resto de OSu) puede proporcionar una relación menos óptima (p. ej., una relación más baja, tal como aproximadamente 0-3). Como se describe en la presente memoria, un enlazador que comprende una unidad espaciadora corta (p. ej., una unidad espaciadora de PEG corta tal como (PEG)2 o (PEG)4, o una unidad espaciadora de alquilo corta tal como (CH2)5) proporciona a relación de aproximadamente 3-4. Como se describe en la presente memoria, un enlazador que comprende una unidad espaciadora más larga (p. ej., (PEG)8) puede proporcionar una relación menos óptima (p. ej., una relación más baja, tal como aproximadamente 0-3). Como se describe en la presente memoria, un enlazador que comprende un resto de péptido escindible proporciona una relación de aproximadamente 3-4. Como se describe en la presente memoria, un enlazador que comprende un resto escindible alternativo (p. ej., un disulfuro escindible o una sulfonamida escindible) puede proporcionar una relación menos óptima (p. ej., una relación más baja, tal como aproximadamente 0-3). Como se describe en la presente memoria, un ADC que comprende Mal-(PEG)2-Val-Cit-pAB-eribulina ligado a un anticuerpo tal como un anticuerpo anti-FRA (p. ej., MORAb-003) tiene una relación de aproximadamente 3-4. Como se describe en la presente memoria, una relación de aproximadamente 3-4 se observa con un ADC que comprende Mal-(PEG)2-Val-Cit-pAB-eribulina ligado a un anticuerpo diferente, tal como un anticuerpo anti-her2 (p. ej., trastuzumab). Como se describe en la presente memoria, la relación óptima observada con ADC que comprenden el Mal-(PEG)2-Val-Cit-pAB-eribulina enlazador-toxina es independiente del anticuerpo.
Como se describe en la presente memoria, un resto de anticuerpo se expone a condiciones reductoras antes de la conjugación para generar uno o más residuos cisteína libres. Un anticuerpo, como se describe en la presente memoria, se puede reducir con un agente reductor tal como ditiotreitol (DTT) o tris(2-carboxietil)fosfina (TCEP), en condiciones reductoras parciales o totales, para generar grupos tiol de cisteína reactivos. Las cisteínas no emparejadas se pueden generar a través de la reducción parcial con equivalentes molares limitados de TCEP, lo que reduce preferencialmente las uniones disulfuro intercatenarias que enlazan la cadena ligera y la cadena pesada (un para por emparejamiento H-L) y las dos cadenas pesadas en la región bisagra (dos pares por emparejamiento H-H en el caso de la IgG1 humana), mientras deja las uniones disulfuro intracatenarias intactas (Stefano et al. (2013) Methods Mol. Biol.
1045:145-71). Como se describe en la presente memoria, las uniones disulfuro dentro de los anticuerpos se reducen electroquímicamente, p. ej., al emplear un electrodo de trabajo que aplica un voltaje reductor y oxidante alternante. Esta estrategia puede permitir el acoplamiento en línea de la reducción de unión disulfuro con un dispositivo analítico (p. ej., un dispositivo de detección electroquímica, un espectrómetro de NMR o un espectrómetro de masas) o un dispositivo de separación química (p. ej., un cromatógrafo de líquidos (p. ej., un HPLC) o un dispositivo de electroforesis (ver, p. ej., la publicación estadounidense núm. 20140069822)). Como se describe en la presente memoria, un anticuerpo se somete a condiciones de desnaturalización para revelar grupos nucleófilos reactivos en residuos aminoacídicos, tales como lisina o cisteína.
La carga de fármaco de un ADC se puede controlar de diferentes modos, p. ej., al: (i) limitar el exceso molar del intermediario fármaco-enlazador o reactivo de enlazador con respecto al anticuerpo; (ii) limitar el tiempo o la temperatura de reacción de conjugación; (iii) condiciones reductoras parciales o limitantes de la modificación del tiol de cisteína; y/o (iv) genomanipular mediante técnicas recombinantes la secuencia de aminoácidos del anticuerpo de modo que la cantidad y posición de residuos cisteína se modifique para controlar la cantidad y/o posición de los acoplamientos enlazador-fármaco.
Como se describe en la presente memoria, los residuos cisteína libres se introducen en la secuencia de aminoácidos del resto de anticuerpo. Por ejemplo, se pueden preparar anticuerpos genomanipulados con cisteína, en donde uno o más aminoácidos de un anticuerpo original se reemplazan con un aminoácido de cisteína. Cualquier forma de anticuerpo se puede genomanipular de estar manera, es decir, mutar. Por ejemplo, un fragmento de anticuerpo Fab original se puede genomanipular para formar un Fab genomanipulado con cisteína que se denomina 'TioFab''. De manera similar, un anticuerpo monoclonal original se puede genomanipular para formar un 'TioMab''. Una mutación de sitio único produce un residuo cisteína genomanipulado único en un TioFab, mientras que una mutación de sitio único produce dos residuos cisteína genomanipulados en un TioMab, debido a la naturaleza dimérica del anticuerpo de IgG. El ADN que codifica una variante de secuencia de aminoácidos del polipéptido original se puede preparar mediante una variedad de métodos conocidos en la técnica (ver, p. ej., los métodos descritos en WO2006/034488). Estos métodos incluyen, pero no se limitan a, la preparación mediante mutagénesis dirigida al sitio (o mediada por oligonucleótido), mutagénesis por PCR y mutagénesis de casete de un ADN preparado anteriormente que codifica el polipéptido. También se pueden construir variantes de anticuerpos recombinantes también mediante manipulación del fragmento de restricción o mediante PCR de extensión de superposición con oligonucleótidos sintéticos. Los ADC de Fórmula I incluyen, pero no se limitan a, anticuerpos que tienen 1,2, 3 o 4 aminoácidos de cisteína genomanipulados (Lyon, et al. (2012) Methods Enzymol. 502:123-38). Como se describe en la presente memoria, uno o más residuos cisteína libres ya están presentes en un resto de anticuerpo, sin el uso de genomanipulación, en tal caso se pueden usar los residuos cisteína libres existentes para conjugar el resto de anticuerpo a un resto de fármaco.
Como se describe en la presente memoria, una carga de fármaco más alta (p. ej., p>5) puede provocar aglomeración, insolubilidad, toxicidad o pérdida de la permeabilidad celular de ciertos conjugados de anticuerpo-fármaco. La carga
de fármaco más alta también puede afectar de manera negativa la farmacocinética (p. ej., el aclaramiento) de ciertos ADC. Como se describe en la presente memoria, una carga de fármaco más baja (p. ej., p <3) puede reducir la potencia de ciertos ADC contra células que expresan la diana y/o células vecinas. Como se describe en la presente memoria, la carga de fármaco para un a Dc de la presente descripción varía de 1 a aproximadamente 8; de aproximadamente 2 a aproximadamente 6; de aproximadamente 2 a aproximadamente 5; de aproximadamente 3 a aproximadamente 5; o de aproximadamente 3 a aproximadamente 4.
Cuando más de un grupo nucleófilo hace reacción con un intermediario de fármaco-enlazador o un reactivo de resto enlazador y posteriormente un reactivo de resto de fármaco, en una mezcla de reacción que comprende múltiples copias del resto de anticuerpo y el resto enlazador, el producto resultante puede ser una mezcla de compuestos de ADC con una distribución de uno o más restos de fármaco acoplados a cada copia del resto de anticuerpo en la mezcla. Como se describe en la presente memoria, la carga de fármaco de una mezcla de ADC resultante de una reacción de conjugación varía de 1 a 20 restos de fármaco acoplados por resto de anticuerpo. La cantidad promedio de restos de fármaco por resto de anticuerpo (es decir, la carga de fármaco promedio, o p promedio) se puede calcular mediante cualquier método convencional conocido en la técnica, p. ej., mediante espectrometría de masas (p. ej., LC-MS de fase inversa) y/o cromatografía de líquidos de alto rendimiento (p. ej., HIC-HPLC). Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo se determina mediante cromatografía de interacción hidrófoba-cromatografía de líquidos de alto rendimiento (HIC-HPLC, por sus siglas en inglés). Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo se determina mediante cromatografía de líquidos de fase inversa-espectrometría de masas (LC-MS, por sus siglas en inglés). Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de aproximadamente 3 a aproximadamente 4; de aproximadamente 3,1 a aproximadamente 3,9; de aproximadamente 3,2 a aproximadamente 3,8; de aproximadamente 3,2 a aproximadamente 3,7; de aproximadamente 3,2 a aproximadamente 3,6; de aproximadamente 3,3 a aproximadamente 3,8; o de aproximadamente 3,3 a aproximadamente 3,7. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de aproximadamente 3,2 a aproximadamente 3,8. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de aproximadamente 3,8. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de 3 a 4; de 3,1 a 3,9; de 3,2 a 3,8; de 3,2 a 3,7; de 3,2 a 3,6; de 3,3 a 3,8; o de 3,3 a 3,7. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de 3,2 a 3,8. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de 3,8.
Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de aproximadamente 3,5 a aproximadamente 4,5; de aproximadamente 3,6 a aproximadamente 4,4; de aproximadamente 3,7 a aproximadamente 4,3; de aproximadamente 3,7 a aproximadamente 4,2; o de aproximadamente 3,8 a aproximadamente 4,2. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de aproximadamente 3,6 a aproximadamente 4,4.Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de aproximadamente 4,0. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de 3,5 a 4,5; de 3,6 a 4,4; de 3,7 a 4,3; de 3,7 a 4,2; o de 3,8 a 4,2. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de 3,6 a 4,4. Como se describe en la presente memoria, la cantidad promedio de restos de fármaco por resto de anticuerpo es de 4,0.
Como se describe en la presente memoria, el término "aproximadamente" según se usa con respecto a la cantidad promedio de restos de fármaco por resto de anticuerpo significa /- 10 %.
Los compuestos de ADC individuales, o "especies", se pueden identificar en la mezcla mediante espectroscopía de masas y separarse mediante UPLC o HPLC, p. ej., cromatografía de interacción hidrófoba (HIC-HPLC). Como se describe en la presente memoria, un ADC homogéneo o casi homogéneo con un valor de carga único se puede aislar de la mezcla de conjugación, p. ej., mediante electroforesis o cromatografía.
Como se describe en la presente memoria, una carga de fármaco y/o una carga de fármaco promedio de aproximadamente 4 proporciona propiedades beneficiosas. Como se describe en la presente memoria, una carga de fármaco y/o una carga de fármaco promedio menor que aproximadamente 4 puede dar como resultado un nivel inaceptablemente elevado de especies de anticuerpo no conjugadas, que pueden competir con el ADC por la unión a un antígeno diana y/o proporcionar una eficacia de tratamiento reducida. Como se describe en la presente memoria, una carga de fármaco y/o una carga de fármaco promedio mayor que aproximadamente 4 puede dar como resultado un nivel inaceptablemente elevado de heterogeneidad de producto y/o aglomeración de ADC. Una carga de fármaco y/o carga de fármaco promedio de más de aproximadamente 4 también puede afectar la estabilidad del ADC, debido a la pérdida de una o más uniones químicas necesarias para estabilizar el resto de anticuerpo.
Como se describe en la presente memoria, un ADC tiene la Fórmula I:
Ab-(L-D)p (I)
en donde:
(i) Ab es un anticuerpo anti-receptor alfa de folato internalizante o fragmento de unión a antígeno de este que comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24;
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es un número entero de 3 a 4.
Como se describe en la presente memoria, un ADC tiene la Fórmula I:
Ab-(L-D)p (I)
en donde:
(i) Ab es un anticuerpo anti-receptor de factor de crecimiento epidérmico humano 2 internalizante o fragmento de unión a antígeno de este que comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:27 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:28;
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es un número entero de 3 a 4.
Como se describe en la presente memoria, p es 4.
La presente descripción incluye métodos para producir los ADC descritos. A grandes rasgos, los ADC comprenden un anticuerpo o fragmento de unión a antígeno como el resto de anticuerpo, un resto de fármaco y un enlazador que liga el resto de fármaco y el resto de anticuerpo. Como se describe en la presente memoria, los a Dc se pueden preparar al usar un enlazador que tiene funcionalidades reactivas para acoplarse covalentemente al resto de fármaco y al resto de anticuerpo. Por ejemplo, como se describe en la presente memoria, un tiol de cisteína de un resto de anticuerpo puede formar una unión con un grupo funcional reactivo de un enlazador o un intermediario de fármaco-enlazador (p. ej., un resto de maleimida) para producir un ADC. La generación de los ADC puede lograrse mediante cualquier técnica conocida para el experto en la técnica.
Como se describe en la presente memoria, un ADC se produce al poner en contacto un resto de anticuerpo con un enlazador y un resto de fármaco de manera secuencial, de modo que el resto de anticuerpo se enlace covalentemente con el enlazador primero, y después el intermediario anticuerpo-enlazador preformado hace reacción con el resto de fármaco. El intermediario de anticuerpo-enlazador puede someterse o no a una etapa de purificación antes de ponerse en contacto con el resto de fármaco. Como se describe en la presente memoria, un ADC se produce al poner en contacto un resto de anticuerpo con un compuesto de enlazador fármaco preformado al hacer reaccionar un enlazador con un resto de fármaco. El compuesto de enlazador-fármaco preformado puede someterse o no a una etapa de purificación antes de ponerse en contacto con el resto de anticuerpo. Como se describe en la presente memoria, el resto de anticuerpo hace contacto con el enlazador y el resto de fármaco en una mezcla de reacción, lo que permite la formación simultánea de las uniones covalentes entre el resto de anticuerpo y el enlazador, y entre el enlazador y el resto de fármaco. Este método de producir ADC puede incluir una reacción, en donde el resto de anticuerpo hace contacto con el resto de anticuerpo antes de la adición del enlazador a la mezcla de reacción, y viceversa. Como se describe en la presente memoria, un ADC se produce al hace reaccionar un resto de anticuerpo con un enlazador ligado a un resto de fármaco, tal como Mal-(PEG)2-Val-Cit-pAB-eribulina, en condiciones que permiten la conjugación.
Los ADC preparados de acuerdo con los métodos descritos anteriormente pueden someterse a una etapa de purificación. La etapa de purificación puede implicar cualesquiera métodos bioquímicos conocidos en la técnica para purificar proteínas, o cualquier combinación de métodos de estos. Estos incluyen, pero no se limitan a, filtración de flujo tangencial (TFF, por sus siglas en inglés), cromatografía de afinidad, cromatografía de intercambio iónico, cualquier cromatografía basada en carga o punto isoeléctrico, cromatografía de modo mixto, p. ej., CHT (hidroxiapatita cerámica), cromatografía de interacción hidrófoba, cromatografía de exclusión por tamaño, diálisis, filtración, precipitación selectiva o cualquier combinación de estas.
Usos terapéuticos y composiciones
En la presente memoria se describen métodos para usar los ADC descritos en el tratamiento de un sujeto para un trastorno, p. ej., un trastorno oncológico. Los ADC se pueden administrar solos o en combinación con un segundo agente terapéutico, y se pueden administrar en cualquier formulación, dosificación y régimen de dosificación farmacéuticamente aceptable. La eficacia del tratamiento con ADC se puede evaluar para determinar la toxicidad, así como indicadores de eficacia y ajustarse en consecuencia. Las mediciones de la eficacia incluyen, pero no se limitan
a, un efecto citostático y/o citotóxico observado in vitro o in vivo, volumen tumoral reducido, inhibición del crecimiento tumoral y/o supervivencia prolongada.
Los métodos para determinar si un ADC ejerce un efecto citostático y/o citotóxico sobre una célula se conocen. Por ejemplo, la actividad citotóxica o citostática de un ADC puede medirse al: exponer células de mamífero que expresen una proteína diana del ADC en un medio de cultivo celular; cultivar las células durante un período de aproximadamente 6 horas a aproximadamente 5 días; y medir la viabilidad celular. Los ensayos in vitro basados en células también pueden usarse para medir la viabilidad (proliferación), citotoxicidad y la inducción de apoptosis (activación de la caspasa) del ADC.
Para determinar si un conjugado de anticuerpo-fármaco ejerce un efecto citostático, se puede usar un ensayo de incorporación de timidina. Por ejemplo, las células cancerosas que expresan un antígeno diana en una densidad de 5000 células/pocillo de una placa de 96 pocillos pueden cultivarse durante un período de 72 horas y exponerse a 0,5 |jC¡ de 3H-timidina durante las 8 horas finales del período de 72 horas. La incorporación de 3H-timidina en células del cultivo se mide en la presencia y ausencia del ADC.
Para determinar la citotoxicidad, se puede medir la necrosis o apoptosis (la muerte celular programada). Típicamente, la necrosis se encuentra acompañada por una permeabilidad en aumento de la membrana plasmática; la hinchazón de la célula y la ruptura de la membrana plasmática. La apoptosis típicamente se caracteriza por la incisión de la membrana, la condensación del citoplasma, y la activación de endonucleasas endógenas. La determinación de cualquiera de estos efectos en células cancerosas indica que un ADC es útil en el tratamiento de cánceres.
La viabilidad celular puede medirse, p. ej., mediante la determinación en una célula de la captación de un tinte tal como rojo neutro, azul tripán, Crystal Violet o azul ALAMAR™ (ver, p. ej. Page et al., (1993) Intl. J. Oncology 3:473-6). En dicho ensayo, las células se incuban en medios que contienen el tinte, las células se lavan y el tinte restante, que refleja la captación celular del tinte, se mide espectrofotométricamente. Como se describe en la presente memoria, la potencia in vitro de los ADC preparados se evalúa en un ensayo que usa Crystal Violet. El Crystal Violet es un tinte de triarilmetano que se acumula en el núcleo de células viables. En este ensayo, las células se exponen a los ADC o agentes testigo durante un período de tiempo definido, después del cual las células se tiñen con Crystal Violet, se lavan exhaustivamente con agua, después se solubilizan con SDS al 1 % y se leen espectrofotométricamente. El tinte de unión a proteína sulforrodamina B (SRB) puede usar también para medir la citotoxicidad (Skehan et al. (1990) J. Natl. Cancer Inst. 82:1107-12).
La apoptosis puede cuantificarse mediante, por ejemplo, la medición de la fragmentación de ADN. Se encuentran disponibles métodos fotométricos comerciales para la determinación in vitro cuantitativa de la fragmentación de ADN Los ejemplos de dichos ensayos, incluido TUNEL (que detecta la incorporación de nucleótidos etiquetados en ADN fragmentado) y ensayos basados en ELISA, se describen en Biochemica (1999), núm. 2, págs. 34-37 (Roche Molecular Biochemicals).
La apoptosis también puede determinarse mediante la medición de cambios morfológicos en una célula. Por ejemplo, como con la necrosis, la pérdida de la integridad de la membrana plasmática puede determinarse mediante la medición de la captación de ciertos tintes (p. ej., un tinte fluorescente tal como, por ejemplo, naranja de acridina o bromuro de etidio). Un método para medir la cantidad de células apoptóticas ha sido descrito por Duke y Cohen, Current Protocols in Immunology (Coligan et al. eds., (1992) págs. 3.17.1-3.17.16). También se pueden teñir las células con tinte de ADN (p. ej., naranja de acridina, bromuro de etidio o yoduro de propidio) y observar las células para determinar la condensación de cromatina y la marginación a lo largo de la membrana nuclear interna. Otros cambios morfológicos que se pueden medir para determinar la apoptosis incluyen, p. ej., condensación citoplasmática, incisión creciente de la membrana y encogimiento celular.
Los ADC descritos también puede evaluarse para determinar la actividad de destrucción por vecindad. La destrucción por vecindad actividad se puede determinar, p. ej., mediante un ensayo que emplea dos líneas celulares, una positiva para el antígeno diana y una negativa para el antígeno diana. Las líneas celulares preferiblemente se etiquetan para diferenciarlas. Por ejemplo, se pueden cocultivar células IGROV1 (FRA+) con verde Nuclight™ (NLG) y h L-60 (FRA-) con rojo Nuclight™ (NLR), tratarse con un ADC anti-FRA con posterior monitoreo de la citotoxicidad. La destrucción de las células negativas para el antígeno diana cuando se mezclan con células positivas para el antígeno diana es indicativa de la destrucción por vecindad, mientras que la destrucción de células negativas para el antígeno diana en ausencia de células positivas para el antígeno diana es indicativa de destrucción inespecífica.
En algunos aspectos, la presente descripción presenta un método para destruir, inhibir o modular el crecimiento de, o interferir en el metabolismo de, una célula o tejido canceroso al alterar la tubulina. El método puede usar con cualquier sujeto cuando la alteración de la tubulina proporciona un beneficio terapéutico. Los sujetos que se pueden beneficiar con la alteración de la tubulina incluyen, pero no se limitan a, aquellos que tienen o están en riesgo de tener un cáncer gástrico, cáncer de ovario (p. ej., cáncer de ovario seroso), cáncer de pulmón (p. ej., cáncer de pulmón no microcítico), cáncer de mama (p. ej., cáncer de mama triple negativo), cáncer de endometrio (p. ej., carcinoma de endometrio seroso), osteosarcoma, sarcoma de Kaposi, cáncer de célula reproductora testicular, leucemia, linfoma (p. ej., enfermedad de Hodgkin, linfoma no hodgkiniano), mieloma, cáncer de vías respiratorias y digestivas altas, cáncer esofágico, cáncer pancreático, cáncer de próstata, cáncer cerebral (p. ej., glioblastoma), cáncer de tiroides, cáncer
colorrectal y/o cáncer de piel (p. ej., melanoma), o cualquier metástasis de estos (Dumontet and Jordán (2010) Nat. Rev. Drug Discov. 9:790-803). Como se describe en la presente memoria, los ADC descritos pueden administrarse en cualquier célula o tejido que expresa FRA, tal como una célula o tejido canceroso que expresa FRA. Una descripción ilustrativa incluye un método para inhibir la señalización celular mediada por FRA o un método para destruir una célula. El método puede usarse con cualquier célula o tejido que expresa FRA, tal como una célula cancerosa o una lesión metastásica. Los ejemplos no limitantes de cánceres que expresan FRA incluyen cáncer gástrico, cáncer de ovario seroso, cáncer de ovario de células claras, cáncer de pulmón no microcítico, cáncer colorrectal, cáncer de mama triple negativo, cáncer de endometrio, carcinoma de endometrio seroso, carcinoide pulmonar y osteosarcoma. Los ejemplos no limitantes de células que expresan FRA incluyen IGROV1 y células de carcinoma de ovario humanas OVCAR3, célula de carcinoma de pulmón no microcítico humanas NCI-H2110 y células que comprenden un ácido nucleico recombinante que codifica FRA o una porción de este.
Como se describe en la presente memoria, los ADC descritos pueden administrarse en cualquier célula o tejido que expresa her2, tal como una célula o tejido canceroso que expresa her2. Una descripción ilustrativa incluye un método para inhibir la señalización celular mediada por her2 o un método para destruir una célula. El método puede usarse con cualquier célula o tejido que expresa her2, tal como una célula cancerosa o una lesión metastásica. Los ejemplos no limitantes de cánceres que expresan her2 incluyen cáncer de mama, cáncer gástrico, cáncer de vejiga, cáncer de célula urotelial, cáncer esofágico, cáncer de pulmón, cáncer de cuello uterino, cáncer de endometrio y cáncer de ovario (English et al. (2013) Mol. Diagn. Ther. 17:85-99). Los ejemplos no limitantes de células que expresan her2 incluyen células de carcinoma gástrico humanas NCI-N87-luc, células de carcinoma ductal de mama humanas ZR75 y BT-474 y células que comprenden un ácido nucleico recombinante que codifica her2 o una porción de este.
Como se describe en la presente memoria, los ADC descritos pueden administrarse en cualquier célula o tejido que expresa mesotelina (MSLN), tal como una célula o tejido canceroso que expresa MSLN. Una descripción ilustrativa incluye un método para inhibir la señalización celular mediada por MSLN o un método para destruir una célula. El método puede usarse con cualquier célula o tejido que expresa MSLN, tal como una célula cancerosa o una lesión metastásica. Los ejemplos no limitantes de cánceres que expresan MSLN incluyen mesotelioma, cáncer pancreático (p. ej., adenocarcinoma pancreático), cáncer de ovario, y cáncer de pulmón (p. ej., adenocarcinoma pulmonar) (Wang, et al. (2012) PLoS ONE 7:e33214). Los ejemplos no limitantes de células que expresan MSLN incluyen células de carcinoma de ovarios humanas OVCAR3, células endometroides humanas HEC-251, células de mesotelioma de células escamosas de pulmón humanas H226 y células que comprenden un ácido nucleico recombinante que codifica MSLN o una porción de este.
Los métodos ilustrativos incluyen las etapas de poner en contacto la célula con un ADC, como se describe en la presente memoria, en una cantidad eficaz, es decir, cantidad suficiente para destruir la célula. El método puede usar en células en cultivo, p. ej., in vitro, in vivo, ex vivo o in situ. Por ejemplo, las células que expresan FRA, her2 y/o MSLN (p. ej., células recogidas mediante biopsia de un tumor o lesión metastásica; células de una línea celular de cáncer establecida; o células recombinantes), se pueden cultivar in vitro en medio de cultivo y la etapa de contacto puede producir al agregar el ADC al medio de cultivo. El método resultará en la destrucción de células que expresan FRA, her2 y/o MSLN, incluso, en particular, células tumorales que expresan FRA, her2 y/o MSLN. Alternativamente, el ADC se puede administrar a un sujeto mediante cualquier vía de administración adecuada (p. ej., intravenosa, subcutánea, o contacto directo con un tejido tumoral) para que tenga un efecto in vivo.
El efecto in vivo de una composición terapéutica de ADC descrita puede evaluarse en un modelo animal adecuado. Por ejemplo, se pueden usar modelos de cáncer xenógenos, en donde los explantes de cáncer o tejidos xenojertados traspasados se introducen en animales con inmunidad comprometida, tales como ratones lampiños o SCID (Klein et al., (1997) Nature Med. 3:402-8). La eficacia se puede predecir mediante el uso de ensayos que midan la inhibición de la formación tumoral, la remisión tumoral o la metástasis y similares.
También se pueden usar ensayos in vivo que evalúan la promoción de la apoptosis. Como se describe en la presente memoria, se pueden examinar xenoinjertos de ratones que llevan tumor tratados con la composición terapéutica para determinar la presencia de focos apoptóticos y compararlos con ratones que llevan xenoinjerto testigo no tratados. La extensión en que se encuentran los focos apoptóticos en los tumores de los ratones tratados proporciona una indicación de la eficacia terapéutica de la composición.
Además, se describen en la presente memoria métodos para tratar un cáncer. Los ADC descritos en la presente memoria se pueden administrar a un sujeto mamífero no humano o humano con fines terapéuticos. Los métodos terapéuticos implican administrar a un mamífero que tiene un tumor una cantidad biológicamente eficaz de un ADC que comprende un agente quimioterapéutico seleccionado (p. ej., eribulina) enlazado a un anticuerpo de direccionamiento que se une a un antígeno expresado, que es accesible para la unión, o que está localizado en una superficie de la célula cancerosa. Una descripción ilustrativa es un método para suministrar un agente quimioterapéutico a una célula que expresa FRA, que comprende conjugar el agente quimioterapéutico con un anticuerpo que se une inmunoespecíficamente a un epítopo de FRA y exponer la célula al ADC. Las células tumorales ilustrativas que expresan FRA para las cuales se indican los ADC de la presente descripción incluyen células de un cáncer gástrico, un cáncer de ovario seroso, un cáncer pulmonar no microcítico, un cáncer colorrectal, un cáncer de mama (p. ej., un cáncer de mama triple negativo), un carcinoide pulmonar, un osteosarcoma, un cáncer de endometrio y un carcinoma de endometrio con histología serosa.
Otra descripción ilustrativa es un método para suministrar un agente quimioterapéutico a una célula que expresa her2, que comprende conjugar el agente quimioterapéutico con un anticuerpo que se une inmunoespecíficamente a un epítopo de her2 y exponer la célula al ADC. Las células tumorales ilustrativas que expresan her2 para las cuales se indican los ADC de la presente descripción incluyen células de un cáncer de mama, un cáncer gástrico, un cáncer de vejiga, un carcinoma de células uroteliales, un cáncer esofágico, un cáncer de pulmón, un cáncer de cuello uterino, un cáncer de endometrio y un cáncer de ovario.
Otra descripción ilustrativa es un método para suministrar un agente quimioterapéutico a una célula que expresa MSLN, que comprende conjugar el agente quimioterapéutico con un anticuerpo que se une inmunoespecíficamente a un epítopo de MSLN y exponer la célula al ADC. Las células tumorales ilustrativas que expresan MSLN para las cuales se indican los ADC de la presente descripción incluyen células de un mesotelioma, un cáncer pancreático (p. ej., un adenocarcinoma pancreático), un cáncer de ovario y un cáncer de pulmón (p. ej., adenocarcinoma pulmonar).
Otra descripción ilustrativa es un método para tratar a un paciente que tiene o está en riesgo de tener un cáncer que expresan un antígeno diana para el resto de anticuerpo del ADC, tal como FRA, her2 o MSLN, que comprende administrar al paciente una cantidad terapéuticamente eficaz de un ADC de la presente descripción. Como se describe en la presente memoria, el paciente no responde o responde escasamente al tratamiento con un anticuerpo anti-FRA cuando se administra solo y/o al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo. Como se describe en la presente memoria, el paciente no responde o responde escasamente al tratamiento con un anticuerpo anti-her2 cuando se administra solo y/o al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo. Como se describe en la presente memoria, el paciente no responde o responde escasamente al tratamiento con un anticuerpo anti-MSLN cuando se administra solo y/o al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo. Como se describe en la presente memoria, el paciente es intolerante al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo. Por ejemplo, un paciente puede requerir dosis de eribulina para tratar un cáncer que conducen a toxicidad sistémica, que se supera mediante el suministro dirigido a un cáncer que expresa un antígeno diana para el resto de anticuerpo del ADC tal como FRA, her2, o MSLN y reduce así la destrucción inespecífica.
Otra descripción ilustrativa es un método para reducir o inhibir el crecimiento de un tumor que expresan un antígeno diana (p. ej., un tumor que expresa FRA, un tumor que expresa her2 o un tumor que expresa MSLN), que comprende administrar una cantidad terapéuticamente eficaz de un ADC. Como se describe en la presente memoria, el tratamiento es suficiente para reducir o inhibir el crecimiento del tumor de un paciente, reducir el número o tamaño de lesiones metastásicas, reducir la carga tumoral, reducir la carga tumoral primaria, reducir la invasividad, prolongar el tiempo de supervivencia y/o mantener o mejorar la calidad de vida. Como se describe en la presente memoria, el tumor es resistente o insensible al tratamiento con un anticuerpo anti-FRA cuando se administra solo y/o al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo. Como se describe en la presente memoria, el tumor es resistente o insensible al tratamiento con un anticuerpo anti-her2 cuando se administra solo y/o al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo. Como se describe en la presente memoria, el tumor es resistente o insensible al tratamiento con un anticuerpo anti-MSLN cuando se administra solo y/o al tratamiento con un resto de fármaco (p. ej., eribulina) cuando se administra solo.
Además, los anticuerpos de la presente descripción pueden administrarse a un mamífero no humano que expresa un antígeno al cual es ADC es capaz de unirse para fines veterinarios o como un modelo animal de la enfermedad humana. Con respecto a lo último, dichos modelos animales pueden ser útiles para evaluar la eficacia terapéutica de los ADC descritos (p. ej., evaluar las dosificaciones y ciclos de tiempo de administración).
Además, en la presente memoria se describen usos terapéuticos de los ADC descritos. Una descripción ilustrativa es el uso de un ADC en el tratamiento de un cáncer que expresa el antígeno diana (p. ej., un cáncer que expresa FRA, un cáncer que expresa her2 o un cáncer que expresa MSLN). También se describen ADC para uso en el tratamiento de un cáncer que expresa el antígeno diana (p. ej., un cáncer que expresa FRA, un cáncer que expresa her2 o un cáncer que expresa MSLN). Los métodos para identificar sujetos que tienen cánceres que expresan FRA, her2 y/o MSLN se conocen en la técnica y se pueden usar para identificar pacientes adecuadas para el tratamiento con un ADC descrito.
Otra descripción ilustrativa es el uso de un ADC en un método para fabricar un medicamento para el tratamiento de un cáncer que expresa el antígeno diana (p. ej., un cáncer que expresa FRA, un cáncer que expresa her2 o un cáncer que expresa MSLN).
Las composiciones terapéuticas usadas en la práctica de los métodos que anteceden pueden formularse en composiciones farmacéuticas que comprenden un portador farmacéuticamente aceptable adecuado para el método de suministro deseado. Una descripción ilustrativa es una composición farmacéutica que comprende un ADC de la presente descripción y un portador farmacéuticamente aceptable. Los portadores adecuados incluyen cualquier material que, cuando se combina con la composición terapéutica, retiene la función antitumoral de la composición terapéutica y que, generalmente, no es reactivo con el sistema inmunitario del paciente. Los portadores farmacéuticamente aceptables incluyen todos y cualquiera de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardantes de la absorción y similares que son fisiológicamente compatibles. Los ejemplos de portadores farmacéuticamente aceptables incluyen uno o más de agua,
disolución salina, disolución salina tamponada con fosfato, dextrosa, glicerol, etanol, sal de mesilato y similares, así como combinaciones de estos. En muchos casos, será preferible incluir agentes isotónicos, por ejemplo, azúcares, polialcoholes tales como manitol, sorbitol o cloruro de sodio en la composición. Los portadores farmacéuticamente aceptables pueden comprender, además, cantidades menores de sustancias auxiliares tales como agentes humectantes o emulsionantes, conservantes o tampones, que potencian el tiempo de conservación o la eficacia del ADC.
Las formulaciones terapéuticas se pueden solubilizar y administrar a través de cualquier vía de administración capaz de suministrar la composición terapéutica al sitio del tumor. Las vías de administración potencialmente eficaces incluyen, pero no se limitan a, intravenosa, parenteral, intraperitoneal, intramuscular, intratumoral, intradérmica, intraorgánica, ortotípica y similares. Las preparaciones proteicas terapéuticas pueden liofilizarse y almacenarse como polvos estériles, preferiblemente al vacío, y después reconstituirse en agua bacteriostática (que contiene, por ejemplo, el conservante alcohol bencílico) o en agua estéril antes de la inyección. Las formulaciones terapéuticas pueden comprender un ADC o una sal farmacéuticamente aceptable de este, p. ej., una sal de mesilato.
Los ADC descritos en la presente memoria pueden administrarse en una dosificación que varía de aproximadamente 0,2 mg/kg a aproximadamente 10 mg/kg a un paciente que lo necesita. Como se describe en la presente memoria, el ADC se administra al paciente a diario, bimensualmente o en cualquier período de tiempo intermedio. Las dosificaciones y protocolos de administración para el tratamiento de cánceres al usar los métodos que anteceden variarán con el método y el cáncer diana, y dependerán generalmente de varios otros factores apreciados en la técnica.
Se conocen varios sistemas de suministro y pueden usarse para administrar uno o más ADC de la presente descripción. Los métodos para administrar los ADC incluyen, pero no se limitan a, administración parenteral (p. ej., intradérmica, intramuscular, intraperitoneal, intravenosa y subcutánea), administración epidural, administración intratumoral y administración a través de la mucosa (p. ej., vías intranasal y oral). Además, se puede emplear la administración pulmonar, p. ej., mediante el uso de un inhalador o nebulizador y la formulación con un agente de aerosolización. Ver, p. ej., las composiciones y métodos para la administración pulmonar descritas en las patentes estadounidenses núms.
6.019.968, 5.985.320, 5.985.309, 5.934.272, 5.874.064, 5.855.913, 5.290.540 y 4.880.078; y las publicaciones PCT núms. WO 92/19244, WO 97/32572, WO 97/44013, WO 98/31346 y WO 99/66903. Los ADC se pueden administrar mediante cualquier vía conveniente, por ejemplo, mediante infusión o inyección en bolo, o mediante absorción a través de revestimientos epiteliales o mucocutáneos (p. ej., mucosa oral, rectal e intestinal, etc.). La administración puede ser sistémica o local.
Las composiciones terapéuticas descritas en la presente memoria pueden ser estériles y estables en las condiciones de fabricación y almacenamiento. Como se describe en la presente memoria, uno o más de los ADC o composiciones farmacéuticas, se suministra como un polvo liofilizado esterilizado seco o concentrado libre de agua en un contenedor sellado herméticamente y puede reconstituirse (p. ej., con agua o disolución salina) hasta la concentración apropiada para la administración a un sujeto. Preferiblemente, uno o más de los agentes profilácticos o terapéuticos o composiciones farmacéuticas se suministra como un polvo liofilizado estéril seco en un contenedor sellado herméticamente en una dosificación unitaria de al menos 5 mg, al menos 10 mg, al menos 15 mg, al menos 25 mg, al menos 35 mg, al menos 45 mg, al menos 50 mg, al menos 75 mg, o al menos 100 mg, o cualquier cantidad intermedia. Como se describe en la presente memoria, los ADC liofilizados o composiciones farmacéuticas se almacenan a entre 2 °C y 8 °C en el contenedor original. Como se describe en la presente memoria, uno o más de los ADC o composiciones farmacéuticas descritos en la presente memoria se suministra en forma líquida en un contenedor sellado herméticamente, p. ej., un contenedor que indica la cantidad y concentración del agente. Como se describe en la presente memoria, la forma líquida de la composición administrar se suministra en un contenedor sellado herméticamente de al menos 0,25 mg/ml, al menos 0,5 mg/ml, al menos 1 mg/ml, al menos 2,5 mg/ml, al menos 5 mg/ml, al menos 8 mg/ml, al menos 10 mg/ml, al menos 15 mg/ml, al menos 25 mg/ml, al menos 50 mg/ml, al menos 75 mg/ml o al menos 100 mg/ml de ADC. La forma líquida se puede almacenar a entre 2 °C y 8 °C en el contenedor original.
Como se describe en la presente memoria, los ADC descritos se pueden incorporar en una composición farmacéutica adecuada para administración parenteral. La disolución inyectable puede estar compuesta por una forma de dosificación líquida o liofilizada en un tubo de vidrio o vial ámbar, ampolla o jeringa precargada, u otro dispositivo de suministro o almacenamiento conocido.
Las composiciones descritas en la presente memoria pueden estar en una variedad de formas. Estas incluyen, por ejemplo, formas de dosificación líquidas, semisólidas y sólidas, tales como disoluciones líquidas (p. ej., disoluciones que se puedan suministrar como inyección o infusión), dispersiones o suspensiones, comprimidos, píldoras, polvos, liposomas y supositorios. La forma preferida depende del modo de administración y la aplicación terapéutica previstos.
Como se describe en la presente memoria, el tratamiento implica la administración de un bolo simple o repetida de la preparación de ADC a través de una vía de administración aceptable.
Los pacientes se pueden evaluar para determinar los niveles de antígeno diana en una muestra dada (p. ej., los niveles de células que expresan el antígeno diana) para auxiliar en la determinación del régimen de dosificación más eficaz, etc. Una descripción ilustrativa es un método para determinar si un paciente responderá al tratamiento con un ADC de la presente descripción, que comprende proporcionar una muestra biológica del paciente y poner en contacto la
muestra biológica con el ADC. Las muestras biológicas ilustrativas incluyen tejido o fluido corporal, tal como un exudado inflamatorio, sangre, suero, fluido intestinal, muestra de materia fecal o biopsia tumoral (p. ej., una biopsia tumoral derivada de un paciente que tiene o está en riesgo de un cáncer que expresan antígeno diana, p. ej., un cáncer que expresa FRA, un cáncer que expresa her2 o un cáncer que expresa MSLN). Como se describe en la presente memoria, se puede obtener una muestra (p. ej., un tejido y/o fluido corporal) de un sujeto, y se puede usar un método inmunológico adecuado para detectar y/o medir la expresión proteica del antígeno diana (p. ej., FRA, her2 o MSLN). Dichas evaluaciones también se usan para fines de monitoreo a lo largo de la terapia, y son útiles para medir el éxito terapéutico en combinación con la evaluación de otros parámetros.
Como se describe en la presente memoria, la eficacia de un ADC se puede evaluar al poner en contacto una muestra de tumor de un sujeto con el ADC y evaluar la velocidad del crecimiento tumoral o el volumen. Como se describe en la presente memoria, cuando se ha determinado que un ADC es eficaz, puede administrarse al sujeto.
Las estrategias terapéuticas indicadas anteriormente se pueden combinar con una cualquiera de una amplia variedad de regímenes quirúrgicos, de quimioterapia o radioterapia adicionales.
También se describen en la presente memoria usos de uno o más de los ADC descritos en la fabricación de un medicamento para el tratamiento del cáncer, p. ej., según los métodos descritos anteriormente. Como se describe en la presente memoria, los ADC descritos en la presente memoria se usan para el tratamiento del cáncer, p. ej., según los métodos descritos anteriormente.
Como se describe en la presente memoria, los kits para su uso en el laboratorio y aplicaciones terapéuticas descritos en la presente memoria están dentro del alcance de la presente descripción. Dichos kits pueden comprender un portador, envase o contenedor que se divide en compartimientos para recibir uno o más contenedores tales como viales, tubos y similares, donde cada uno del(de los) contenedor(es) comprende uno de los elementos separados que se usarán en un método descrito en la presente memoria, junto con una etiqueta o prospecto que comprende instrucciones de uso, tal como un uso descrito en la presente memoria. Los kits pueden comprender un contenedor que comprende un resto de fármaco. La presente descripción también describe uno o más de los ADC o composiciones farmacéuticas de estos, envasados en un contenedor sellado herméticamente, tal como una ampolla o un sobrecito, que indica la cantidad del agente.
Los kits pueden comprender el contenedor descrito anteriormente y uno o más contenedores distintos asociados con este que comprenden materiales deseables desde el punto de vista comercial y del usuario, incluidos tampones, diluyentes, filtros, agujas, jeringas; etiquetas de portador, envase, contenedor, vial y/o tubo que indican el contenido e/o instrucciones de uso, y prospectos del envase con instrucciones de uso.
Una etiqueta puede estar presente en o con el contenedor para indicar que la composición se usa para una terapia específica o aplicación no terapéutica, tal como una aplicación de pronóstico, profilaxis, diagnóstico o laboratorio. Una etiqueta también puede indicar orientaciones para el uso in vivo o in vitro, tales como las descritas en la presente memoria. Las orientaciones y otra información también se pueden incluir en un prospecto(s) o etiqueta(s), que se incluye con o en el kit. La etiqueta puede estar en el contenedor o asociada con este. Una etiqueta puede estar en un contenedor cuando se moldean o graban letras, números u otros caracteres que forman la etiqueta en el propio contenedor. Una etiqueta puede estar asociada con un contenedor cuando se encuentra presente dentro de un receptáculo o portador que también contiene el contenedor, p. ej., como un prospecto del envase. La etiqueta puede indicar que la composición se usa para diagnosticar o tratar una afección, tal como un cáncer descrito en la presente memoria.
EJEMPLO 1
1. Materiales y métodos
El MORAb-003 usado para la preparación de ADC fue del Lote núm. AA0312.
1.1 Citotoxinas
Las estructuras de las citotoxinas conjugables se muestran en la Tabla 11.





1.2 Conjugación de anticuerpo-fármaco
1.2.1 Reducción parcial con el uso de TCEP
Las condiciones de reducción parcial para MORAb-003 se establecieron mediante la concentración variable del agente reductor distinto de tiol tris(2-carboxietil)fosfina (TCEP), la concentración de anticuerpo y el tiempo de reducción. Se intercambió el tampón de MORAb-003 a la disolución salina tamponada con fosfato de Dulbecco (DPBS, por sus siglas en inglés) que contenía ácido etilendiaminatetraacético 1 mM (EDTA, por sus siglas en inglés), después se concentró hasta 10 mg/mL al usar concentración centrífuga con filtros centrífugos con corte de peso molecular (MWCO, por sus siglas en inglés) de 10 kD. Los anticuerpos se diluyeron hasta la concentración apropiada y se agregó TCEP en la concentración final indicada y se mezcló suavemente durante 1 hora a temperatura ambiente. El TCEP se retiró mediante desalación al usar columnas de desalación rotativa de 5 o 10 mL Zeba™ con DPBS/EDTA 1 mM como tampón (Thermo Fisher, 40 kD MWCO), según el protocolo del fabricante. Las muestras se analizaron para determinar el contenido de tiol libre al usar el kit de cuantificación fluorométrica de Tiol (Abeam), según el protocolo del fabricante. Se llevó a cabo un análisis SDS-PAGE en condiciones no reductoras para terminar la extensión y ubicación de la ruptura de la unión disulfuro, como se describe en la sección 1.3.3. En algunos casos, los MAb desalados se llevaron hasta 1-2 mg/mL mediante dilución en DPBS y se sometieron a biotinilación para determinar la conjugabilidad y la relación entre fármaco y anticuerpo (DAR). Se agregó maleimido-PEG2-biotina 10 mM (Thermo Fisher) en dimetilsulfóxido (DMSO) al anticuerpo (mAb) en una relación molar de 10:1 y se incubaron a temperatura ambiente durante 4 horas con agitación suave. Después de la conjugación, se retiró el compuesto sin reaccionar mediante desalación al usar columnas de desalación rotativa Zeba™ (Thermo Fisher). Después se analizaron las muestras mediante LC-MS para la determinación de la DAR, como se detalla en la sección 1.3.4.
1.2.2 Conjugación de citotoxina
El anticuerpo parcialmente reducido se llevó hasta 2,5 mg/mL en 0,5X DPBS, EDTA 0,5 mM y se mezcló exhaustivamente. Los codisolventes orgánicos, si se usaron, después se agregaron y mezclaron exhaustivamente. Los codisolventes examinados fueron propilenglicol (20 % y 50 % de concentración final), dimetilsulfóxido (DMSO) (10 %), N,N-Dimetilformamida (20 %), N,N-dimetilacetamida (20 %) y W,W-dimetilpropionamida (20 %). La citotoxina modificada con maleimido (disolución madre 6 mM en DMSO) se agregó a anticuerpos en una relación molar de 1:6 (mAb:compound) y se mezcló exhaustivamente. La conjugación procedió a temperatura ambiente durante 3,5 horas, con mezcla suave. Se eligió 50 % propilenglicol al 50 % como el modificador orgánico final y se usó en todas las reacciones de conjugación posteriores.
1.2.3 Purificación
El anticuerpo conjugado se purificó al usar la(s) columna(s) de desalación 26/10 HiTrap® (GE Healthcare) con cromatografía llevada a cabo en una cromatografía de líquidos de proteína rápida (FPLC, por sus siglas en inglés) (GE Healthcare), para retirar la maleimido-citotoxina y el propilenglicol sin reaccionar. Los ADC de MORAb-003, incluido MORAb-003-mal-VCP-eribulina (MORAb-202) se formularon en DPBS (se usó el tampón de formulación como tampón de ejecución durante la cromatografía FPLC).
1.3 Caracterización biofísica
1.3.1 Ensayo de BCA
El reactivo de ácido bicinconínico (BCA) preparado (200 pL) se agregó a 25 pL de ADC diluidos en serie o estándar de gamma globina bovina (Thermo Fisher) 2 mg/mL y las muestras se mezclaron exhaustivamente. Las muestras se incubaron a 37 °C durante 20 min. Las placas se leyeron a 595 nm en un lector de placa SpectraMax® M5 (Molecular Devices). Los datos se analizaron al usar SoftMax® Pro (ver 3.2) con un modelo de ajuste de 4 parámetros.
1.3.2 Análisis por SEC-HPLC
La aglomeración del anticuerpo se analizó mediante cromatografía de líquidos de alto rendimiento-exclusión por tamaño (SEC-HPLC) al usar un Agilent 1100. El mAb se diluyó hasta 1 mg/mL en DPBS. El anticuerpo (20 pL) se inyectó en una columna de guarda TSKgel® SuperSW (4,6 mm x 3,5 cm, tamaño de poro 4 pm, Tosoh Bioscience), posteriormente en una columna TSKgel® SuperSW3000 (4,6 mm x 30 cm, tamaño de poro 4 pm), se eluyó desde la columna con fosfato de sodio 0,1 M que contenía NaCl 0,15 M y NaN3 al 0,05 %, a pH 7,4, a una velocidad de flujo de 0,3 mL/min durante 20 min. Todos los datos se analizaron al usar el programa informático Agilent ChemStation. El porcentaje de aglomeración se calculó como [PAaglomerado/PAtotal]*100, donde PA = área de pico integrada.
1.3.3 Análisis por SDS-PAGE
Las muestras de proteína (0,1-10 pg) se llevaron hasta 1X con tampón de muestra de dodecilsulfato de litio (LDS, por sus siglas en inglés). Para las muestras no reducidas, la incubación se llevó a cabo a temperatura ambiente durante 10 min antes de la electroforesis. Para las muestras reducidas, se agregó ditiotreitol (DTT) hasta una concentración final de 20 mM y las muestras se calentaron hasta 95 °C durante 10 min y se colocaron sobre hielo antes de la electroforesis. Las muestras se cargaron sobre geles de Bis-Tris SDS-PAGE de 10, 12, o 15 pocillos (Thermo Fisher)
con 1X MOPS o 1X MES como tampón de ejecución. La electroforesis se llevó a cabo a 185 V (voltaje constante) durante 1 hora. Los geles se tiñeron con disolución de tinción InstantBlue (Expedeon) y se destiñeron en agua. La documentación se realizó en un sistema de documentación de gel UltraLum al usar filtros anaranjados de 600 nm.
1.3.4 Análisis por UPLC/ESI-MS de la relación entre fármaco y anticuerpo (DAR)
Los ADC se desglicosilaron al usar PNGasa F (New England BioLabs). Se agregaron tampón G7 (10 pL) y PNGasa F (2 pL) al mAb (90 pL, 1 mg/mL en DPBS). La reacción se incubó en un microondas Discover (CEM) durante 2 ciclos: (1) potencia de microondas 10 W, 37 °C, 10 min, con posterior pausa de 5 min; (2) potencia de microondas 2 W, 37 °C, 10 min. Una porción de la muestra se redujo mediante la adición de DTT hasta una concentración final de 20 mM, con posterior incubación a 60 °C durante 3 min. Las muestras después se analizaron al usar una cromatografía de líquidos de rendimiento ultra alto (UPLC, por sus siglas en inglés) Waters Acquity y espectrómetro de masas Premier cuadrupolo con tiempo de vuelo (Q-Tof, por sus siglas en inglés). Las muestras (0,5-2 pg cada una) se inyectaron en una columna de desalación MassPrep™ a 65 °C, se eluyeron desde la columna con un equilibrio de 5 min en 95 % de fase móvil A, un gradiente de 10 min (5-90 % B) y un reequilibrio de 10 min en 95 % de fase móvil A, a 0,05 mL/min. La fase móvil A fue ácido fórmico al 0,1 % en agua. La fase móvil B fue ácido fórmico al 0,1 % en acetonitrilo. El espectrómetro de masas Q-Tof se ejecutó en modo V de ion positivo con detección en el intervalo de 500-4000 m/z. Los parámetros de la fuente fueron los siguientes: voltaje capilar, 2,25 kV (anticuerpo intacto)-2,50 kV (anticuerpo reducido); voltaje de cono de muestreo, 65,0 V (anticuerpo intacto) o 50,0 V (anticuerpo reducido); temperatura de fuente, 100°C; temperatura de desolvatación 250 °C; flujo de gas de desolvatación, 550 L/h. El pico de proteína se deconvolucionó al usar la función MassLynx® MaxEnt 1. Las intensidades relativas de cada una de las masas de cadena pesada y ligera no conjugadas, con conjugación simple y con conjugación múltiple se combinaron para calcular la DAR global al usar la fórmula:
... n(lHC+n)]/£lHCtOt]
donde Ilc+1 es la intensidad de masa de la cadena ligera conjugada con una citotoxina, Ilc+2 es la intensidad de masa de la cadena ligera conjugada con dos citotoxinas, etc. Ihc son las intensidades de las cadenas pesadas conjugadas correspondientes, y XLctot y ^iHctot son las intensidades combinadas de todas las cadenas ligeras y cadenas pesadas no conjugadas y conjugadas, respectivamente.
1.3.5 Análisis de DAR por HIC-HPLC
Además del análisis de DAR mediante el análisis por UPLC/ionización por electronebulización (ESI, por sus siglas en inglés)-MS, la DAR de MORAb-003-vcp-eribulina y la DAR de MORAb-003-0285 también se analizaron al usar HPLC de interacción hidrófoba (HIC-HPLC, por sus siglas en inglés). Las muestras se inyectaron en una TSKgel® Éter-5 PW, 7,5 mm ID x 7,5cm, tamaño de poro 10 pM y se eluyeron desde la columna con un equilibrio de 3 min en 100 % de fase móvil A, un gradiente de 15 min (0-100 % de B), una retención de 5 min en 100 % de B, un cambio de 1 min a 100 % de A y un reequilibrio de 5 min en 100 % de fase móvil A, a 0,7 mL/min. La fase móvil A fue fosfato de sodio 25 mM, sulfato de amonio 1,5 M, pH 7,0. La fase móvil B fue fosfato de sodio 25 mM, isopropanol al 25 %, pH 7,0. La detección se realizó a 280 nm (referencia de 320 nm). La DAR se determinó mediante la fórmula:
[AUC+i 2(AUC+2) 3(AUC+3) ...n(AUC-n)]/XAUCtot]
donde AUC+1 es el área bajo la curva para el pico de mAb correspondiente al ADC conjugado con una citotoxina, AUC+2 es el área bajo la curva para el pico de mAb correspondiente al ADC conjugado con dos citotoxinas, etc. XAUCtot es el área bajo la curva combinada para todos los picos.
1.4 Análisis de citotoxicidad
1.4.1 Ensayo con Crystal Violet
Se subcultivaron y sembraron las células IGROV1 (FRhi) y SJSA-1 (FRneg) a 10.000 células/pocillo en medio de crecimiento completo en placas de cultivo tisular de 96 pocillos, se incubaron a 37 °C, con CO2 al 5 % durante la noche (16 horas). Típicamente, los reactivos de prueba se diluyeron en serie 1:4 en placas de dilución de pocillos de 2 mL profundidad, comenzando con 1 pM (10 diluciones en total). Se agregaron 100 pL de muestras diluidas a las placas con células (comenzando con una concentración de muestras de prueba de 500 nm). Las placas se incubaron 37 °C, con CO2 al 5 % durante 48 horas adicionales. El medio se descartó, las placas se lavaron una vez con 200 pL de DPBS, se tiñeron con 50 pL de disolución de Crystal Violet al 0,2 % a temperatura ambiente durante 15 min y después se lavaron exhaustivamente con agua del grifo. Las placas se secaron con aire y se disolvió el Crystal Violet con 200 pL de disolución de SDS al 1 %. Las placas se leyeron a 570 nm. Los datos se analizaron al usar GraphPad Prism 6. Los ensayos se llevaron a cabo con el uso de una densidad de siembra de 1000 células por pocillo y la exposición al compuesto fue durante un total de 5 días. Cuando se deseó una exposición durante un plazo más corto, se retiró el medio que contenía los agentes citotóxicos después 4 horas y se reemplazó con medio de crecimiento nuevo antes de la incubación de 5 días. Para OVCAR3, CaOV3 y NCI-H2110, las células se sembraron a 3000 células/pocillo y se
incubaron durante 5 días con ADC. Para los experimentos de competición, los ADC ajustados se preincubaron con 2 pM (final) de MORAb-003 no conjugado antes de la incubación con las células.
1.4.2 Ensayo de destrucción por vecindad
El día antes del comienzo del estudio, se sembraron células IGROV1 con verde Nuclight™ (NLG) a 5000 células/pocillo en placas de fondo redondo de 96 pocillos, con posterior centrifugación a 1000 rpm durante 3 min a temperatura ambiente para garantizar la formación de un sedimento celular. La placa se colocó en el recipiente de un Incucyte Zoom® (EssenBio science) y se incubó a 37 °C/CO2 al 5 % durante la noche. El programa se configuró para recoger imágenes del crecimiento celular y para determinar las cantidades totales de células con tinción nuclear verde y con tinción nuclear roja, así como la confluencia de fase de las células cada dos horas. El día del experimento, el ADC de MORAb-003 o el fármaco libre se diluyó en medio RPMI completo y se diluyó en serie, comenzando con 400 nm. Se agregaron 50 pl de disolución de citotoxina a las células NLG-IGROV1 y se incubaron durante 30 min. Durante el período de incubación, las células HL-60 (FRneg) con rojo Nuclight™ (NLR) se diluyeron hasta 2x105, 1x105 o 5x104 célula/mL con medios nuevos. Se agregaron 50 pL de la suspensión de células NLR-HL60 o medio solo a los pocillos con NLG-IGROV1, con posterior centrifugación a 1000 rpm durante 3 min a temperatura ambiente para garantizar la nueva formación del sedimento celular. La placa se colocó nuevamente en el recipiente de Incucyte Zoom (EssenBio science) y se incubó a 37 °C/CO2 al 5 % durante hasta 5 días. El crecimiento celular relativo de NLG-IGROV1 se determinó mediante comparación con muestras con ausencia de ADC o fármaco libre agregado al usar recuentos de células verdes. El crecimiento celular relativo de HL60 se realizó de manera similar, excepto que se determinó el recuento de células rojas. La determinación de los valores de CI50 para NLG-IGROV1 y NLR-HL-60 se determinó al usar Prism (GraphPad).
1.4.3 Ensayo de estabilidad en suero
Se mezclaron exhaustivamente 20 pL de ADC de MORAb-003 con 80 pL de DPBS, suero humano concentrado normal (Bioreclamation, Lote BRH552911), o suero de ratón concentrado normal (Bioreclamation, Lote MSE152591), y se incubaron a 37 °C durante 0, 4, 24 y 48 horas. Después de la incubación, las muestras se congelaron y se almacenaron a -20 °C hasta la evaluación en los ensayos de citotoxicidad y unión. Para los análisis de citotoxicidad, las muestras se evaluaron en células IGROV1 y SJSA-1, como se detalla en la sección 1.4.1. Para la evaluación de la unión, las muestras se evaluaron al usar un ensayo MSD ECL basado en disolución. Las muestras se incubaron con receptor alfa de folato biotinilado y anti-MORAb-003 con identificador sulfo antes de la captura en una placa de estreptavidina y se detectaron al usar electroquimioluminiscencia con un MSD Sector Imager 2400.
2. Resultados
2.1 Preparación de ADC de MORAb-003
Para seleccionar la mejor combinación de enlazador y citotoxina para conjugar con MORAb-003, se prepararon ADC al usar tres metodologías. Según la estrategia de conjugación que se muestra en la Figura 1, se generan cisteínas no apareadas a través de reducción parcial con equivalentes molares limitados del agente reductor TCEP distinto de tiol. Esta estrategia reduce preferencialmente las uniones disulfuro intercatenarias que enlazan la cadena ligera y la cadena pesada (un para por emparejamiento H-L) y las dos cadenas pesadas en la región bisagra (dos pares por emparejamiento H-H en el caso de la IgG 1 humana), mientras deja las uniones disulfuro intracatenarias intactas.
La segunda estrategia de conjugación para preparar ADC de MORAb-003 utilizó química de puente disulfuro reducido. La química de puente disulfuro reducido repuentea los tioles libres de los residuos cisteína liberados durante el proceso de reducción parcial, al imitar la función de la unión disulfuro y, por lo tanto, conserva la estabilidad y función del ADC.
La tercera estrategia de conjugación para preparar ADC de MORAb-003 empleó utilización de lisina limitada. La utilización de lisina limitada resulta en la conjugación de una cantidad muy limitada de las lisinas expuestas a disolvente 70+ estimadas disponibles en una molécula de IgG humana típica, y puede proporcionar potencialmente mezclas del producto de ADC con homogeneidad más baja con respecto a las estrategias que implican la modificación de cisteínas.
2.1.1 Preparación de VCP-eribulina para ADC de MORAb-003
Se disolvió eribulina (1) (10 mg, 14 pmol) (Figura 2) en W,W-dimetilformamida (DMF) (1mL) y se mezclaron bien. Se agregó W,W-diisopropiletilamina (base de Hunig o /Pr2NEt) (3,6 pL, 21 pmol) y Fmoc-Val-Cit-para-aminobencil-paranitrofenol (Fmoc-VCP-PNP) (2) (16 mg, 21 pmol, Concortis Biosystems, núm. de cat. VC1003). La mezcla de reacción se agitó a temperatura ambiente durante 4-16 horas, se monitorizó al usar un kit de prueba de ninhidrina (Anaspec, núm. de cat. 25241) hasta que la reacción se completó. Después se agregó dietilamina (Et2NH) (0,014 mL, 0,14 mmol) a la mezcla de reacción, se agitó durante 2 horas a 18-25 °C para retirar el grupo protector Fmoc. La reacción se monitorizó con el uso de un kit de prueba de ninhidrina. Después de completarse, se evaporó el disolvente al vacío para proporcionar VCP-eribulina (3) bruta (16 mg), se purificó al usar una columna ZOBAX SB-C18 (tamaño de poro 5 pm, 9,4 x 150 mm) en un sistema de HPLC Waters Alliance e2695 en la fase móvil de H2O-CH3CN que contenía ácido fórmico al 0,1 %, a través de un gradiente de 15-70 % de B. La VCP-eribulina (3) (16 mg) se disolvió en DMF (1 mL). Se agregaron la base de Hunig (7,2 pL, 41 pmol) y maleimido-PEG2-NHS (4) (9,7 mg, 27 pmol). La mezcla de reacción se agitó a 18-25 °C durante 3 horas. La mezcla de reacción se purificó mediante HPLC (H2O-CH3CN) que
contenía ácido fórmico al 0,1 %) a través de un gradiente de 15-70 % de B. El disolvente se retiró mediante liofilización para proporcionar mal-(PEG)2-Val-Cit-p-aminobenciloxicarbonil (pAB)-eribulina (mal-(PEG)2-VCP-eribulina) (5).
2.1.2 Optimización de condiciones de reducción
Se prepararon ADC de MORAb-003 al generar cisteínas no apareadas a través de reducción parcial con equivalentes molares limitados del agente reductor tris(2-carboxietil)fosfina (TCEP) distinto de tiol. Se llevó a cabo una investigación inicial con MORAb-003, donde se variaron la concentración de anticuerpo, la concentración de TCEP y el tiempo de incubación, con el objetivo de generar un promedio de 4 sitios conjugables por molécula de anticuerpo. El número de sitios de tiol libre se determinó al usar un ensayo de determinación cuantitativa de tiol fluorométrico. Los resultados de este análisis se muestran en la Tabla 12. También se analizó la extensión de la ruptura de la unión H-H y H-L después de una incubación de 10 min, 30 min, 60 min o 120 min mediante SDS-PAGE (Figura 3). Para este análisis, se cargaron muestras no reducidas y reducidas en un gel de SDS-PAGE y se llevó a cabo la electroforesis a 185 V durante 1 hora. En la Figura 3, el carril M corresponde al estándar de proteína. El carril 1 corresponde a MORAb-003 no reducido, sin tratar. El carril 2 corresponde a MORAb-003 (5,3 mg/mL) reducido en 70,6 pM de TCEP. El carril 3 corresponde a MORAb-003 (5,3 mg/mL reducido) en 141,2 pM de TCEP. El carril 4 corresponde a MORAb-003 (1,5 mg/mL) reducido en 20 pM de TCEP. El carril 5 corresponde a MORAb-003 (1,5 mg/mL) reducido en 40 pM de TCEP. Las identidades de cada banda se indican en el gel derecho inferior. "H" indica cadena pesada, mientras que "L" indica cadena ligera.

El análisis del SDS-PAGE y el contenido de tiol sugirió que la incubación de 60 min para 5,3 mg/mL de mAb en una relación molar de 4 veces entre TCEP y mAb proporcionó un punto de partida razonable, dado que la reducción limitada de los disulfuros intramoleculares pareció estar presente (según se determinó por el contenido de tiol libre) y permaneció muy poco mAb no reducido (el mAb no reducido actuaría como un inhibidor competitivo en estudios in vitro e in vivo al usar ADC preparados). Se llevaron a cabo estudios adicionales con MORAb-003 con concentraciones iniciales de 5,0 mg/mL para confirmar esta relación molar optimizada entre TCEP y mAb al usar análisis SDS-PAGE (Figura 4). En la Figura 4, el carril 1 corresponde al estándar de proteína. El carril 2 corresponde a MORAb-003 no reducido, sin tratar. El carril 3 corresponde a MORAb-003 tratado con una relación de MORAb-003:TCEP de 1:1. El carril 4 corresponde a MORAb-003 tratado con una relación de MORAb-003:TCEP de 1:2. El carril 5 corresponde a MORAb-003 tratado con una relación de MORAb-003:TCEP de 1:3. El carril 6 corresponde a MORAb-003 tratado con una relación de MORAb-003:TCEP de 1:4. La conjugación con el uso de maleimido-PEG2-biotina también se llevó a cabo de manera posterior a la reducción y extracción de TCEP, para simular la conjugación de la citotoxina para la preparación del ADC. El análisis de DAR se llevó a cabo al usar LC-MS. Los resultados de estos estudios se proporcionan en la Tabla 13.
Tabla 13. Optimización de condiciones de reducción de MORAb-003 - niveles de conjugación con maleimido-PEG2-biotina

LC, nivel de biotina de cadena ligera; HC, nivel de biotina de cadena pesada; DAR, biotina por mAb [DAR = 2(LC) 2(HC)].
Después de la conjugación de la biotina, el análisis de tiol libre indicó que no había tiol libre presente en MORAb-003-biotina. Esto indicó que, después de la reducción de las uniones disulfuro, la conjugación típicamente se produjo en ambos tioles generados, y que cualesquiera disulfuros reducidos, no conjugados sufrieron reoxidación para reformar las uniones disulfuro. Las condiciones finales elegidas para la reducción para la generación de ADC fueron concentración de anticuerpo de 5,0 mg/mL, concentración de TCEP de 110 pM y tiempo de incubación de 60 min. Esto conduce a un mAb con una DAR de 4 después de la conjugación.
2.1.3 Optimización de la conjugación de ADC
Dado que la primera citotoxina usada para la preparación de ADC fue criptoficina, que es un compuesto hidrófobo, se llevaron a cabo experimentos de optimización de conjugación iniciales con un anticuerpo anti-mesotelina humana "sustituto" que tiene dos cisteínas no apareadas disponibles para conjugación (una por cadena ligera) en ubicaciones específicas. Esto facilita en gran medida el análisis de la eficacia de la conjugación mediante espectrometría de masas, dado que solo debe analizar la cadena ligera. La valoración del propilenglicol durante la conjugación de maleimido-LL3-criptoficina con el anticuerpo sustituto se llevó a cabo con posterior análisis de la eficacia de conjugación de la cadena ligera mediante LC-MS (Tabla 14).
Tabla 14. Optimización de la concentración de propilenglicol en la reacción de conjugación

El propilenglicol al 50 % dio como resultado una ocupación total de los sitios disponibles y se eligió como la concentración final a usarse. No se observó pérdida en la unión del mAb después de la conjugación (no se muestran los datos), lo que indica que el propilenglicol no tuvo efectos nocivos para el anticuerpo. Por lo tanto, las condiciones de reacción de conjugación finales elegidas fueron 2,5 mg/mL de mAb (final), relación molar de 6:1 de maleimidoenlazador-citotoxina:mAb en 0,5X DPBS (concentración final después de la adición del propilenglicol), EDTA 0,5 mM,
propilenglicol al 50 %, pH 7,2 durante 3,5-4 horas a temperatura ambiente. En estas reacciones, se agrega propilenglicol antes de la adición de maleimido-enlazador-citotoxina.
2.1.4 Preparación de ADC y caracterización biofísica
Las condiciones de reducción y conjugación establecidas, descritas en la sección 2.1.2, se usaron para preparar los primeros 10 ADC de MORAb-003 indicados en la Tabla 15. Los ADC restantes se prepararon mediante puenteo disulfuro reducido o utilización de lisina limitada, con las excepciones de M-MMAE y M-DM1. M-MMAE y M-DM1 se prepararon por Concortis Biosystems, Inc., y se recibieron en forma conjugada.
La química de puente disulfuro reducido puentea los tioles libres producidos durante el proceso de reducción parcial y proporciona una citotoxina por disulfuro reducido. En teoría, un anticuerpo de DAR = 4 tendría ambos disulfuros de H-L y bisagra repuenteados, lo que proporciona un ADC con mayor estabilidad y homogeneidad con respecto a las estrategias de conjugación tradicionales. La utilización de lisina limitada resulta en la conjugación de una cantidad muy limitada de las lisinas expuestas a disolvente 70+ estimadas disponibles en una molécula de IgG humana típica. Los conjugados de MORAb-003 preparados al usar este método dieron como resultado una DAR de 2,0, lo que sugiere que se utilizó una única lisina por par de H-L.
Todos los ADC se purificaron mediante cromatografía de desalación HiPrep 26/10 y se formularon en DPBS. El análisis de DAR se llevó a cabo en todos los ADC preparados mediante LC-MS y se determinaron los niveles de aglomeración mediante SEC-HPLC. Los resultados de estos análisis de DAR y aglomeración se indican en la Tabla 15 junto al ADC respectivo.
Tabla 15. Análisis biofísicos de ADC de MORAb-003

(continuación)

Los valores de DAR para todos los ADC estuvieron en el intervalo predeterminado (DAR entre 3 y 4). Los niveles de aglomerados para los ADC basados en criptoficina fueron significativamente más altos que lo deseado (>10 %), mientras que las maleimido-enlazador-citotoxinas basadas en eribulina (VCP-eribulina) y basadas en maitansina (ER-001161318, ER-001161319 y M-MMAE) demostraron todas niveles de aglomerados aceptables. Se llevó a cabo investigación de otros codisolventes orgánicos en las reacciones de conjugación con MORAb-003 al usar VCP-criptoficina. Los codisolventes evaluados fueron DMSO (10 %), W,W-dimetilformamida (20 %), W,W-dimetilacetamida (20 %) y W,W-dimetilpropionamida (20 %). Los niveles de aglomerados después de la conjugación al usar estos codisolventes fueron todos iguales o mayores que el propilenglicol al 50 %.
Se llevó a cabo un análisis de SDS-PAGE no reductor en un subconjunto de los ADC (Figura 5). Dado que se determinó una DAR de 4 para todos estos ADC, se creyó que estos ADC deberían migrar como IgG intacta de ~ 160 kD, dado que ambos disulfuros de H-L y ambos de bisagra deberían estar repuenteados. Este subconjunto de ADC incluyó M-MMAE (carril 2), M-DM1 (carril 3), M-0026 (carril 4), M-0260 (carril 5), M-0267 (carril 6), M-0272 (carril 7), M-0285 (carril 8), M-292 (carril 9), M-027-0381 (carril 10) y M-0384 (carril 11) (Figura 5). En la Figura 5, el carril 1 corresponde al estándar de proteína.
Está claro a partir de este análisis que, para los ADC con química de puente disulfuro reducido (carriles 4-9, 11), hay una especie monovalente de H-L significativa (80 kD), además del ADC intacto. Esto indica que hay un puenteo disulfuro de bisagra intracatenario significativo además del punteo de bisagra intercatenario. El análisis por SEC-HPLC indica que los ADC migran como una IgG intacta única, lo que indica que aquellos ADC con puenteo H-H intracatenario, las cadenas pesadas están asociadas no covalentemente en el ADC final.
2.2 Análisis de potencia in v itro de ADC de MORAb-003
2.2.1 Citotoxicidad en células IGROV1 y SJSA-1
Se evaluó la potencia in vitro de los ADC preparados al usar un ensayo con Crystal Violet como se detalla en la sección 1.4.1.
Se llevó a cabo un cribado inicial de todos los ADC de MORAb-003 en células IGROV1 (FRhi(+++)) y SJSA-1 (FRneg(-)). Las células IGROV1 tienen como origen el carcinoma epitelial de ovario humano y expresan niveles altos del receptor alfa de folato (FR), el antígeno diana de MORAb-003. Las células SJSA-1 son una línea celular de tumor de osteosarcoma humano que es negativo para el receptor alfa de folato. El cribado de ADC seleccionados también se llevó a cabo en células CaOV3 (carcinoma de ovario humano, FRmed(++)), NCI-H2110 (carcinoma de pulmón no microcítico humano, FRmed(++)) y/o OVCAR3 (carcinoma de ovario humano, FRmed(++)). Los resultados de este cribado se proporcionan en la Tabla 16.
Tabla 16. Cribado de citotoxicidad (CI50) de ADC de MORAb-003 en varias líneas celulares tumorales

(continuación)

Todos los valores son CI50 en nM, y son valores medios de experimentos repetidos, cuando se llevaron a cabo.
El ADC de VCP-eribulina fue potente (54 pM) en células IGROV1 y tuvo escasa destrucción en células SJSA-1. Para estas líneas celulares, el ADC de VCP-eribulina demostró mayor potencia y especificidad con respecto a los ADC con valores de DAR equivalentes, tales como M-MMAE y M-DM1. El ADC de VCP-eribulina también demostró potente citotoxicidad en líneas celulares tumorales que expresan FR adicionales con origen en carcinoma de ovario (CaOV3 y OVCAR3) y pulmón no microcítico (NC-H2110).
Los ADC de VCP-eribulina, LL2-criptoficina, LL3-criptoficina, VCP-criptoficina, ER-001161318, ER-001161319, y ER-001159200 exhibieron citotoxicidad específica (>2 logaritmos de especificidad) en células CaOV3 (FRmed(++)). Varios de estos ADC exhibieron potencia subnanomolar. Los conjugados de criptoficina también demostraron niveles altos de potencia (23 pM - 86 pM) en células IGROV1, pero, con la excepción de VCP-criptoficina, también demostraron citotoxicidad medible en células SJSA-1. Los conjugados de maitansina escindibles ER-001161318 y ER-001161319 tuvieron una potencia intermediaria sobre IGROV1 (0,26 nM y 0,49 nM), y poca destrucción inespecífica de células SJSA-1.
Todos los conjugados de utilización de lisina limitada no demostraron especificidad y no se evaluaron adicionalmente. Los conjugados escindibles que usan tecnología de puente disulfuro reducido de duostatina-3 (M-0285), duostatina-5 (M-0115) y duostatina-14 (M-292 y M-0026) demostraron todos citotoxicidad específica en la línea celular IGROV1, con poca citotoxicidad sobre la línea celular SJSA-1. Los conjugados de duostatina-3 y duostatina-5, derivados de auristatina, tuvieron una potencia ligeramente más alta, después los conjugados de duostatina-14, que es un derivado de maitansina. Las potencies y especificidades fueron semejantes a las del conjugado M-MMAE testigo, que usa un enlazador Val-Cit-pAB acoplado a monometil E. Los conjugados de química de disulfuro reducido no escindibles carecieron todos de potencia o especificidad suficiente, y no se analizaron adicionalmente.
2.2.2 Citotoxicidad en línea celular de cáncer de ovario que expresa el receptor de folato humana CaOV3
También se determinó la potencia de ADC de MORAb-003 seleccionados en líneas celulares tumorales de ovario humanas OVCAR3 y CaOV3, así como en la línea celular de NSCLC humana NCI-H2110 (Tabla 16). En la línea celular de ovario humana CaOV3, los conjugados de criptoficina demostraron potencia más alta medible que el conjugado de VCP-eribulina, a diferencia de lo observado en las células IGROV1. Esto puede deberse al nivel de expresión más bajo del receptor alfa de folato células CaOV3 en comparación con IGROV1, o la potencia más alta de criptoficina en estas células, en comparación con eribulina. Los conjugados basados en maitansina ER-001161318, ER-001161319 y ER-001159200 tuvieron todos potencias similares a, o menores que, VCP-eribulina.
2.3 Destrucción por vecindad de VCP-eribulina, ER-001161318 y M-0285
Para evaluar la actividad de destrucción por vecindad, se creó un ensayo con el uso de dos líneas celulares etiquetadas. En este ensayo, las células IGROV1 (FRhi) etiquetadas con verde Nuclight™ y HL-60 (FRneg) etiquetadas con rojo Nuclight™ se cocultivaron en diferentes relaciones de número de células y se trataron con ajustes de ADC de MORAb-003 VCP-eribulina, ER-001161318 o M-0285. El VCP-eribulina es un a Dc basado en eribulina que comprende a un enlazador escindible maleimido-PEG2-Val-Cit-pAB, mientras que ER-001161318 es un ADC basado en maitansina que comprende un enlazador escindible maleimido-(CH2)5-Val-Cit-pAB y M-0285 es un ADC basado en duostatina que comprende un enlazador escindible PEG-pAB. La citotoxicidad se monitorizó mediante un generador de imágenes celulares Incucyte Zoom®. Los resultados de este ensayo de citotoxicidad por vecindad se muestran en la Tabla 17 y las Figuras 6A-C.
Tabla 17. Actividad de destrucción por vecindad de VCP-eribulina en el cocultivo de líneas celulares positivas para FR y negativas para FR

Cuando las células HL-60 (FRneg) se cultivaron en una relación 2:1 con las células IGROV1 (FRhi), el tratamiento con MORAb003-VCP-eribulina dio como resultado un aumento de 2-log en la destrucción de las células HL-60, en comparación con las células HL-60 solas (Tabla 17 y Figura 6A). Estos datos sugieren que las células negativas para
la diana receptor alfa de folato (FR) se destruyen de manera más eficaz mediante MORAb003-VCP-eribulina cuando se cocultivan con células positivas para la diana FR, lo que se denomina en la presente memoria destrucción por vecindad. La destrucción por vecindad se puede distinguir de la destrucción inespecífica, que se define como la destrucción de células negativas para la diana que están solas, en ausencia de e independientes del cocultivo con células positivas para la diana. El aumento observado en la destrucción por vecindad fue también casi idéntico al aumento observado después del tratamiento de las células HL-60 con eribulina libre, lo que indica un posible mecanismo para el efecto de vecindad. Sin desear limitarse a ninguna teoría, el MORAb003-VCP-eribulina puede escindirse en o cerca de células IGROV1 positivas para FR, que también sufren apoptosis y liberan eribulina libre en el cultivo. La citotoxina liberada puede destruir células HL-60 negativas para FR.
En cambio, se observó apenas un ligero desplazamiento para MORAb003-ER-001161318 (Figura 6B) y no se observó ningún desplazamiento con MORAb003-0285 (Figura 6C). Cuando se bajó la relación de HL-60:IGr OV1 de 2:1 a 1:2, se observó destrucción medible de las células HL-60, con respecto a las células HL-60 solas, para MORAb003-ER-001161318, mientras que el efecto de vecindad permaneció bajo, aunque detectable, para MORAb003-0285. Estos datos sugieren que, en relación con la destrucción por vecindad, los ADC de MORAb-003 evaluados se pueden clasificar VCP-eribulina > ER-001161318 > M-0285.
2.4 Análisis de estabilidad en suero
Dada la prolongada semivida en circulación in vivo de los ADC y el potencial de toxicidad si las citotoxinas se liberan en la circulación, los ADC deben demostrar estabilidad en suero. Se preincubaron los ADC de MORAb-003 VCP-eribulina, ER-001161319 y M-0285 en suero humano o de ratón a 37 °C durante hasta 48 horas, después se evaluaron en un ensayo de citotoxicidad con las células IGROV1 y SJSA-1. ER-001161319 es un ADC basado en maitansina que comprende el mismo enlazador escindible que VCP-eribulina, maleimido-PEG2-Val-Cit-pAB. Se incluyeron testigos de PBS y suero para corregir cualesquiera efectos del suero sobre el rendimiento del ensayo. Los resultados de este estudio se muestran en la Tabla 18.
Tabla 18. Estabilidad en suero de ADC de MORAb-003

Los recuadros sombreadas jmdicití disminución sisniíicatLva de la patencia
can respecto a la muestra de 7=0.
Aunque VCP-eribulina y M-0285 fueron estables durante al menos 48 horas en cualquier suero, ER-001161319 demostró una caída significativa en la potencia después de 48 horas. Esto puede deberse a la ligadura aziridinocarbamato con la maitansina, que no se ha descrito en la literatura anteriormente. La forma del compuesto liberado puede no ser muy potente, dado que no se observó aumento en la citotoxicidad en células SJSA-1.
2.5 Estudios in v itro con MORAb003-VCP-eribulina
2.5.1 Análisis HIC-HPLC de DAR y heterogeneidad del producto
Se analizaron MORAb003-VCP-eribulina y MORAb003-0285 mediante HIC-HPLC para evaluar la DAR mediante un método alternativo y examinar la heterogeneidad del producto y el contenido de anticuerpo no conjugado (competidor). Se mostró que MORAb003-VCP-eribulina tiene especies de DAR de 0, 2, 4, y 6, lo que está en consonancia con el método usado para la reducción y conjugación (Figura 7A). Se observaron cantidades muy bajas de DAR = 0 especie. En general la DAR, en función de los cálculos de AUC, fue 3,80, lo que está en consonancia con los valores determinados mediante LC-MS. MORAb003-0285 migró como un pico único mediante HIC-HPLC, lo que indica una única especie de DAR (Figura 7B). Se le asignó una DAR de 4,0.
2.5.2 Especificidad mediante ensayos de competencia
Se demostró la especificidad para el antígeno de la citotoxicidad de MORAb-003-VCP-eribulina para el conjugado de VCP-eribulina al usar un formato de ensayo de competencia (Figura 8). En este experimento, se coincubaron ajustes de MORAb-003-VCP-eribulina (concentración inicial de 100 nM) con 2 |uM de MORAb-003 no conjugado. El MORAb-003 no conjugado proporcionó un desplazamiento de 2-log en la potencia en células IGROV1, similar a los resultados obtenidos con IMGN853,
el ADC de maitansina anti-receptor alfa de folato humano de Immunogen que ahora está en ensayos clínicos de Fase II, en células KB (Moore et al., 2015 American Society of Clinical Oncology (ASCO) Annual Meeting, Abstract 5518).
2.5.3 Citotoxicidad en células de NSCLC NCI-H2110
La citotoxicidad de MORAb003-VCP-eribulina y MORAb003-0285 sobre la línea celular de NSCLC humano NCI-H2110 se llevó a cabo al usar el ensayo con Crystal Violet. Los resultados de este ensayo se muestran en la Tabla 16. MORAb003-VCP-eribulina tuvo una CI50 de 0,73 nM, mientras que MORAb003-0285 tuvo una CI50 de 14 nM.
2.6 Estudios in v ivo
2.6.1 Dosis máxima tolerada (MTD) de MORAb-003-VCP-eribulina (MORAb-202) en cepa de ratón CD-1
Ratones CD-1 no tratados recibieron por inyección intravenosa 200 pL de MORAb-202 según el cronograma en la Tabla 19. Se midió el peso corporal antes de la dosis el día de la dosificación, 24 horas después de la dosis y tres veces por semana a continuación. Los animales se observaron para asegurar el bienestar clínico a lo largo de toda la duración del estudio. Dos semanas después de la dosificación, se midió el peso corporal terminal y se registró. Los ratones sacrificados al final del estudio (y si se sacrificó o encontró muerto a cualquier ratón durante el estudio) se procesaron para la necropsia. Los órganos se examinaron para detectar signos de daño tisular.
Tabla 19. Diseño del estudio

No se observó pérdida de peso corporal significativa en ninguno de los grupos de tratamiento en comparación con el grupo testigo tratado con PBS ni ningún hallazgo clínico que indicase toxicidad durante el tratamiento. El peso corporal de los ratones individuales se muestra en la Tabla 20, y el promedio grupal y el EEM se muestra en la Tabla 21. La cinética de cambio del peso corporal para cada grupo (promedio grupal y EEM) se muestran en la Figura 9. MORAb-202 en dosis de hasta 80 mg/kg a través de administración de bolo intravenoso no produjo no toxicidad. Por lo tanto, la DMT está por encima de 80 mg/kg.


2.6.2 Dosis máxima tolerada de eribulina en ratones CD-1
Ratones CD-1 no tratados recibieron por inyección intravenosa 200 pL de eribulina según el cronograma en la Tabla 22. Se midió el peso corporal tres veces por semana, incluso antes de la dosis en cada día de dosificación y 24 horas después de cada dosis. Los animales se observaron para asegurar el bienestar clínico a lo largo de toda la duración del estudio (dos semanas después de la última dosis). El peso corporal terminal se midió y se registró. Los ratones sacrificados al final del estudio (y si se sacrificó o encontró muerto a cualquier ratón durante el estudio) se procesaron para la necropsia. Los órganos se examinaron para detectar signos de daño tisular.
Tabla 22. Diseño del estudio

No se observó pérdida de peso corporal significativa ni se observaron hallazgos clínicos que indicasen toxicidad en los animales a los que se administró eribulina en dosis de hasta 1,6 mg/kg, al usar el régimen de dosificación q4dx3 (una vez cada cuatro días durante 3 dosis totales). La administración de 3,2 mg/kg con el mismo cronograma indujo piloerección en los tres ratones después de la segunda dosis. Se observó pérdida de peso extrema (23 % de pérdida en un ratón, núm. 552, después de la segunda dosis; 17 % y 8 % en el resto, núm. 551 y núm. 552, después de la tercera dosis), en comparación con el testigo tratado con PBS. No se observaron grandes cambios en los órganos de los ratones durante la necropsia. El peso corporal de los ratones individuales se muestra en la Tabla 23, y el promedio grupal y el EEM se muestra en la Tabla 24. La cinética de cambio del peso corporal para cada grupo (promedio grupal y EEM) se muestran en la Figura 10.
La eribulina en dosis de hasta 1,6 mg/kg, al usar un régimen de dosificación de q4dx3, no produjo toxicidad, mientras que 3,2 mg/kg indujo pérdida de peso extrema. Por lo tanto, la DMT de eribulina, en este estudio, es 1,6 mg/kg, q4dx3.


2.6.3 Evaluación de la dosis mínima eficaz de MORAb003-VCP-eribulina (MORAb-202) en modelo de hNSCLC NCI-H2110 en ratones CB17-SCID
Se implantaron células de NSCLC humano, NCI-H2110, traspaso 47, subcutáneamente en 30 ratones CB17 SCID (hembras, 5 a 6 semanas de vida, peso 20 gramos). Después de 14 días de la implantación, los ratones se asignaron aleatoriamente a cinco grupos. El volumen tumoral promedio en cada grupo el día de tratamiento (Día 0) varió entre 154-175 mm3 (Tabla 27). Los ratones participantes se trataron con MORAb003-VCP-eribulina (MORAb-202) (Núm. de lote NB2900-87E 10/07/15) con 1,2,5 o 5 mg/kg, con MORAb-003-0285 (Núm. de lote 042-150-002) como testigo con 5 mg/kg, o con PBS, según el diseño del estudio (Tabla 25). Cada grupo se retiró del estudio cuando el volumen tumoral en cualquier animal en el grupo fue >2000 mm3. El último grupo se terminó en el Día 61.
Tabla 25. Diseño del estudio

Los volúmenes tumorales de los ratones individuales se muestran en la Tabla 26, y el promedio grupal y el EEM se muestra en la Tabla 27. La cinética de crecimiento tumoral para cada grupo (promedio grupal y el error estándar de la media, EEM) se muestran en la Figura 11 y los volúmenes tumorales en ratones individuales, así como el promedio grupal y el EEM se muestran en la Figura 12. En función de los volúmenes tumorales del día 17 (cuando se observó el primer volumen tumoral >2000 mm3), MORAb-202 causó una inhibición del crecimiento tumoral (ICT) de 47 % con 1 mg/kg (P = 0,002 con respecto a la disolución salina), una ICT de 96 % con 2,5 mg/kg (p <0,0001 con respecto a la disolución salina). Sin embargo, los tumores remitidos volvieron a crecer una a dos semanas después del final del tratamiento. No se detectó ningún tumor en ratones tratados con 5 mg/kg de MORAb-202. Estos ratones permanecieron libres de tumor durante más de 60 días.
El peso corporal de los ratones individuales se muestra en la Tabla 28, y el promedio grupal y el EEM se muestra en la Tabla 29. La cinética de cambio del peso corporal para cada grupo (promedio grupal y EEM) se muestran en la Figura 13.
No se observó pérdida de peso corporal significativa en ninguno de los grupos de tratamiento en comparación con el testigo.
MORAb-202 mostró un efecto significativo sobre el crecimiento tumoral de NCI-H2110. La remisión tumoral se logró mediante un tratamiento por bolo con 2,5 mg/kg con una ICT de 94 % (con respecto a PBS). Por lo tanto, la dosis mínima eficaz de MORAb-202 es 2,5 mg/kg, evaluada en este modelo. La erradicación tumoral completa se logró con una dosis individual de 5 mg/kg. No se observó crecimiento tumoral durante más de 60 días.

ctJ







2.6.4 Evaluación de la dosis mínima eficaz de eribulina en modelo de hNSCLC NCI-H2110 en ratones CB17-SCID
Se implantaron células de NSCLC humano, H2110, traspaso 46, subcutáneamente en 30 ratones CB17 SCID (hembras, 5 a 6 semanas de vida, peso 20 gramos). Después de 11 días de la implantación, los ratones se asignaron aleatoriamente a cinco grupos. Los cinco animales con los volúmenes tumorales que se desviaban más del promedio se excluyeron. El volumen tumoral promedio en cada grupo el día de tratamiento (Día 0) varió entre 87,6-89,4 mm3 (Tabla 32). Los ratones participantes se trataron con eribulina (Núm. de lote N1201193) con 0,05, 0,2, 0,8 o 1,6 mg/kg, o con PBS, según el diseño del estudio (Tabla 30). Cada grupo se terminó, respectivamente, cuando se observó el primer volumen tumoral >2000 mm3 dentro del grupo. El estudio se terminó en el Día 38 (30 días después de la última dosis).
Tabla 30. Diseño del estudio

Los volúmenes tumorales de los ratones individuales se muestran en la Tabla 31, y el promedio grupal y el EEM se muestra en la Tabla 32. La cinética de crecimiento tumoral para cada grupo (promedio grupal y EEM) se muestran en la Figura 14 y los volúmenes tumorales en ratones individuales, así como el promedio grupal y el EEM el Día 24 (cuando se observó el volumen tumoral >2000mm3 en ratones tratados con PBS) se muestran en la Figura 15. La eribulina causó una ICT de 50,5 % (sin remisión tumoral observada) con 0,05 mg/kg (P = 0,0026 con respecto a la disolución salina); una ICT de ~ 99 % con 0,2, 0,8 o 1,6 mg/kg (los valores de la p fueron <0,0001 para los 3 grupos en comparación con la disolución salina). La dosis mínima eficaz que indujo remisión tumoral es 0,2 mg/kg. Sin embargo, la mayoría de los tumores que remitieron en estos ratones (3/5 en el grupo con 0,2 mg/kg, 4/5 en el grupo con 0,8 mg/kg y 2/5 en el grupo con 1,6 mg/kg) volvieron a crecer o permanecieron medibles a lo largo de toda la duración del estudio (30 días después de la última dosis).
El peso corporal de los ratones individuales se muestra en la Tabla 33, y el promedio grupal y el EEM se muestra en la Tabla 34. La cinética de cambio del peso corporal para cada grupo (promedio grupal y EEM) se muestran en la Figura 16.
No se observó pérdida de peso corporal significativa en ninguno de los grupos de tratamiento en comparación con el grupo testigo tratado con disolución salina. No se observaron hallazgos clínicos que indicasen toxicidad durante el tratamiento.
La eribulina, con 0,2 mg/kg y más alta, administrada q4dX3 i.v., mostró un efecto significativo sobre el crecimiento tumoral de H2110. Se logró la remisión tumoral. Cuando se administró una dosis más baja (de 0,05 mg/kg), no se logró una remisión tumoral. Por lo tanto, la dosis mínima eficaz de evaluada en este estudio es 0,2 mg/kg.





EJEMPLO 2
1. Materiales y métodos
Se sintetizó MORAb003-VCP-eribulina (MORAb-202) al conjugar MORAb-003 (anti-receptor alfa de folato humano humanizado) con el compuesto MAL-PEG2-Val-Cit-PAB-eribulina (ER-001159569) descrito en la sección 1.1 del Ejemplo 3. El método de conjugación se describe en la sección 1.4.1 del Ejemplo 4.
1.1 Modelos tumorales
Las líneas celulares tumorales humanas usadas en la evaluación in vitro adicional de MORAb-202 incluyen IGROV1 (carcinoma de ovario humano, FRhi (+++)), OVCAR3 (carcinoma de ovario humano, FRmed(++)), NCI-H2110 (carcinoma de pulmón no microcítico humano, FRmed(++)), A431-A3 (A431 línea celular original transfectada establemente con mesotelina humana, FRlo(+/-)), SJSA-1 (osteosarcoma humano, FRneg(-)) y HL-60 (leucemia humana, FRneg(-)). Todas estas líneas celulares se obtuvieron directamente de la Colección Estadounidense de Cultivos Tipo (ATCC, por sus silgas en inglés). Para estudios in vivo, se establecieron modelos de ratón por xenoinjerto derivados de pacientes con cáncer de pulmón no microcítico, cáncer de mama triple negativo y cáncer de endometrio y se mantuvieron en Oncotest GmbH (Freiburgo, Alemania), Oncodesign (Dijon, Francia) y EPO Berlin-Buch GmbH (Berlín, Alemania), respectivamente.
1.2 Análisis de citotoxicidad in v itro
1.2.1 Ensayo con Crystal Violet
Se subcultivaron y sembraron las células IGROV1 (FRhi(+++)), A431-A3 (FRlo(+/-)) y SJSA-1 (FRneg(-)) a 10.000 células/pocillo en medio de crecimiento completo en placas de cultivo tisular de 96 pocillos, se incubaron a 37 °C, con CO2 al 5 % durante la noche (16 horas). Típicamente, los reactivos de prueba se diluyeron en serie 1:4 en placas de dilución de pocillos de 2 mL profundidad, comenzando con 1 pM (10 diluciones en total). Se agregaron 100 pL de muestras diluidas a las placas con células (comenzando con una concentración de muestras de prueba de 100 nM). Las placas se incubaron 37 °C, con CO2 al 5 % durante 48 horas adicionales. El medio se descartó, las placas se lavaron una vez con 200 pL de DPBS, se tiñeron con 50 pL de disolución de Crystal Violet al 0,2 % a temperatura ambiente durante 15 min y después se lavaron exhaustivamente con agua del grifo. Las placas se secaron con aire y se disolvió el Crystal Violet con 200 pL de disolución de SDS al 1 %. Las placas se leyeron a 570 nm. Los datos se analizaron al usar GraphPad Prism 6. Para OVCAR3 (FRmed(++)) y NCI-H2110 (FRmed(++)), las células se sembraron a 3000 células/pocillo y se incubaron durante 5 días con MORAb-202.
1.3 Estudios in v ivo
1.3.1 Modelo de xenoinjerto de NCI-H2110
Preparación de los animales: Se alojaron ratones CB17 SCID (hembras, 6 semanas de vida) en 5 ratones por jaula ventilada. Se dejaron disponibles gránulos de alimento y botella de agua esterilizados, ad lib, para los animales. Los animales se aclimataron durante 5-7 días antes de la implantación del tumor.
Cultivo celular: Las células de NCI-H2110 humanas se descongelaron a partir de una disolución madre congelada (NB2813-65) y se cultivaron en medio RPMI-1640 complementado con suero fetal bovino (FBS, por sus siglas en inglés) al 10 % en CO2 al 5 % a 37 °C. Después de dos traspasos, tras alcanzar la confluencia en aproximadamente 70 %, las células se recogieron al usar la disolución de disociación celular, se lavaron dos veces con medio libre de suero y se contaron.
Implantación de tumor: La suspensión celular en medio libre de suero se mezcló con matrigel helado a 1:1 (v:v) hasta una concentración final de 1,0 x 108 células/mL. Cada ratón recibió una inyección subcutáneamente con 100 pL de la mezcla a 1,0 x 107 células/ratón. Se usó una aguja 27G para todas las inyecciones. Se monitorizó el bienestar clínico de los ratones y los tumores se midieron mediante un compás calibrador digital tres veces a la semana, comenzado el Día 3 después de la implantación. Se calculó el volumen tumoral (mm3) al usar la fórmula: A (mm) x L (mm) x D (mm) x n/6. Cuando los tumores alcanzaron ~100 mm3 (en un promedio de >70 hasta ~ 130 mm3), los animales se asignaron aleatoriamente a 4-5 por grupo. Los 5 animales con los volúmenes tumorales que se desviaban más del promedio se excluyeron.
Diseño del estudio: Los ratones experimentales que participaron recibieron una inyección intravenosa con 200 pL de vehículo o MORAb-202 con 1,0, 2,5 y 5 mg/kg, según el diseño del estudio (Tabla 35), el día de la aleatorización. Se midió el peso corporal antes de la dosis, y dos veces por semana durante el estudio. Al final del estudio, se midió el peso corporal terminal y se registró. Los animales se sacrificaron cuando el volumen tumoral individual superó los 2000 mm3. Los criterios para la terminación temprana antes de alcanzar el volumen tumoral máximo admitido incluyeron: (1) ulceración tumoral mayor que 50 % del tumor (v:v); (2) parálisis; (3) pérdida de peso corporal >20 %; y (4) 50 % de los animales dentro del grupo alcanzaron la terminación. Todo ratón sacrificado o encontrado muerto durante el estudio se procesó al seguir el procedimiento terminal descrito anteriormente.
Tabla 35. Diseño del estudio

1.3.2 Modelos de xenoinjerto derivado de paciente (PDx)
1.3.2.1 Modelo PDx de cáncer de pulmón no m icrocítico (NSCLC): LXFA-737 (Oncotest)
Implantación de tumor: Se obtuvieron fragmentos de tumor de NSCLC de xenoinjertos de tumor LXFA-737 traspasados en serie en ratones lampiños. Después de la extracción de los ratones donantes, los tumores se cortaron en fragmentos (3-4 mm de longitud de borde) y se colocaron en disolución salina tamponada con fosfato (PBS) que contenía penicilina/estreptomicina al 10 %. Los animales receptores se anestesiaron mediante inhalación de isoflurano y recibieron implantes de tumor unilaterales o bilaterales subcutáneamente en el flanco. Los xenoinjertos tumorales se implantaron con uno o dos tumores por ratón en un índice de prendimiento <65 %. En el caso de un prendimiento bilateral, uno de estos tumores se extrajo antes de la aleatorización. Los animales e implantes tumorales se monitorizaron a diario hasta que el crecimiento del tumor sólido fue detectable en una cantidad suficiente de animales. En la aleatorización, se determinó el volumen de los tumores en crecimiento. Los animales que cumplieron los criterios de aleatorización (es decir, tener tumores de 50-250 mm3, preferiblemente 80-200 mm3) se distribuyeron en grupos experimentales que consistían en 5-6 animales por grupo, con el objetivo de alcanzar una mediana y media de volúmenes tumorales por grupo semejantes de aproximadamente 100-120 mm3. Los animales no usados para los experimentos se sacrificaron. El día de la aleatorización se diseñó como el Día 0 del experimento.
Diseño del estudio: Los ratones experimentales que participaron recibieron una inyección intravenosa con vehículo, MORAb-003 con 5 mg/kg o MORAb-202 con 5 mg/kg, según el diseño del estudio (Tabla 36), el día de la aleatorización. Se midió el peso corporal antes de la dosis en cada día de dosificación, y dos veces por semana durante el estudio. Al final del estudio, se midió el peso corporal terminal y se registró. Los animales se sacrificaron cuando el volumen tumoral individual superó los 2000 mm3.
Tabla 36. Diseño del estudio

1.3.2.2 Modelo PDx de cáncer de mama trip le negativo (TNBC) OD-BRE-0631 (Oncodesign)
Implantación de tumor: Nueve ratones lampiños SWISS hembra recibieron inyecciones subcutáneamente en el flanco derecho con fragmentos tumorales de TNBC derivados de pacientes. Los ratones que tenían tumores se sacrificaron cuando el volumen tumoral alcanzó 500-1000 mm3 y los tumores se extirparon quirúrgicamente. Los fragmentos de tumor (30-50 mg) se implantaron ortotópicamente en la región del cuerpo adiposo mamario de 34 ratones lampiños SWISS hembra 24 a 72 horas después una irradiación a cuerpo entero con una fuente de gamma (2 Gy, 60Co, BioMEP, Francia). Cuando los tumores alcanzaron un volumen medio de 200-300 mm3, 24 de los 34 animales en total se asignaron aleatoriamente a dos grupos (n=12 animales) según su volumen tumoral individual al usar el programa informático Vivo Manager® (Biosystemes, Couternon, Francia). Se llevó a cabo una prueba estadística (análisis de varianza) para evaluar la homogeneidad entre los grupos. El día de la aleatorización se diseñó como el Día 0 del experimento.
Diseño del estudio: El Día 1 (un día después de la aleatorización y dos días antes del tratamiento), 3 ratones de cada uno de los dos grupos sin tratar se sacrificaron. Los ratones experimentales restantes recibieron una inyección intravenosa con vehículo o MORAb-202 con 5 mg/kg, según el diseño del estudio (Tabla 37), el Día 3. El Día 8 (cinco días después del tratamiento), 3 ratones de cada uno de los dos grupos tratados se sacrificaron. Inmediatamente después del sacrificio, se recogió el tejido tumoral y se fijó en formalina tamponada neutra al 4 % durante 24 a 48 horas, y después se embebió en parafina (Histosec®, Merck, Darmstadt, Alemania). La muestra embebida en parafina se almacenó a temperatura ambiente para el posterior análisis de inmunohistoquímica.
Tabla 37. Diseño del estudio

Análisis de inmunohistoquímica (IHC): Se llevó a cabo la tinción para IHC de los tejidos tumorales fijados en formalina, embebidos en parafina para evaluar la ocupación de MORAb-202 y la expresión de fibroblastos asociados con el cáncer. Antes de la tinción, se retiró la cera de los portaobjetos y se recuperó el antígeno en un Lab Vision™ PT Module (Thermo Scientific), en tampón de citrato (pH 6,0) precalentado hasta 85 °C, al usar el siguiente programa: calentar hasta 97 °C; incubar a 97 °C durante 30 min; y enfriar hasta 60 °C. Los portaobjetos después se transfirieron a agua doble destilada a temperatura ambiente durante 5 min. La tinción se llevó a cabo en un Lab Vision™ Autostainer 360 (Thermo Scientific). A grandes rasgos, los portaobjetos se lavaron dos veces en 1X disolución salina tamponada con Tris/Tween-20 (TBST) durante 6 min/lavado. Las secciones de tejido después se incubaron en tampón de bloqueo (300 pL) (suero de cabra al 10 %(Jackson Immunoresearch Laboratory Inc., núm. de cat. 005-000-121) diluido en seroalbúmina bovina (BSA, por sus siglas en inglés) al 3 % - disolución salina tamponada con fosfato (PBS)) durante 1 hora, se incubaron en anticuerpo conjugado (200 pL) (Tabla 38) durante 1 hora, y se lavaron cinco veces en 1X TBST durante 6 min/lavado. Los portaobjetos se contratiñeron con DAPI en medios de montaje y se dejó que las cubiertas de portaobjetos se endurecieran durante 30 min. Los portaobjetos se procesaron en un escáner Panoramic MIDI (3DHISTECH) y las imágenes de IHC se analizaron al usar el programa informático Halo (Indica Labs). Los anticuerpos usados en este análisis se dirigían a a-actina de músculo liso (SMA), que es un marcador específico para fibroblastos asociados al cáncer e IgG humana, que puede detectar la presencia de MORAb-202.
Tabla 38. Anticuerpos para IHC

1.3.2.3 Modelos PDx de cáncer de endometrio: Endo-12961 y Endo-10590 (EPO Berlin)
Implantación de tumor: Se obtuvieron fragmentos de tumor de cáncer de endometrio de xenoinjertos de tumor Endo-12961 y Endo-10590 traspasados en serie y se almacenaron como disolución madre en nitrógeno fluido. Los fragmentos de tumor se implantaron subcutáneamente en el flanco izquierdo de 40 ratones NMRI nu/nu hembra y se monitorizó el volumen tumoral. Los ratones con un volumen tumoral de 100-160 mm3 se asignaron aleatoriamente a cuatro grupos (Grupos A-D, Tabla 39). Se incluyeron ratones satélite para la aleatorización en un quinto grupo (Grupo E, Tabla 39). Cada grupo consistía en 8 animales. El día de la aleatorización se diseñó como el Día 0 del experimento.
Diseño del estudio: Los ratones experimentales que participaron recibieron una inyección intravenosa con PBS, eribulina con 3,2 mg/kg o 0,1 mg/kg o MORAb-202 con 5 mg/kg, según el diseño del estudio (Tabla 39), el día de la aleatorización. El crecimiento tumoral se evaluó mediante la medición de dos diámetros perpendiculares dos veces a la semana y se calcularon los valores del volumen tumoral (TV, por sus siglas en inglés), el volumen tumoral relativo (RTV, por sus siglas en inglés) y los valores de tratados/testigo (T/C, por sus siglas en inglés). También se evaluó el peso corporal dos veces a la semana como un parámetro para la toxicidad, con el cálculo del peso corporal por grupo y los cambios en el peso corporal (BWC, por sus siglas en inglés) con respecto al inicio del tratamiento. Los animales se sacrificaron cuando el volumen tumoral individual superó 1 cm3 o al final del estudio.
Tabla 39. Diseño del estudio

1.4 Mecanismo de acción
1.4.1 Sistema de cocultivo tridim ensional (3D) en zPredicta
Todos los experimentos de cocultivo en 3D que contenían células madre mesenquimales (MSC, por sus siglas en inglés) se llevaron a cabo en zPredicta, al usar sistemas de matriz extracelular 3D específicos para el órgano tales como rStomach™. Las células madre mesenquimales de médula ósea (BM-MSC, por sus siglas en inglés) en rStomach™ se cocultivaron con la línea celular de cáncer gástrico Nuc Red Light MKN-74 por cuadruplicado en formato de 48 pocillo durante 12 días. Las células MKN-74 anteriormente mostraron que expresaban suficiente receptor alfa de folato (FR) para que el tratamiento con MORAb-202 indujera apoptosis celular. Antes del cultivo, las BM-MSC se evaluaron para determinar la expresión del antígeno diana y los marcadores de diferenciación de MSC (Tabla 40) mediante citometría de flujo.
Tabla 40. Marcadores de diferenciación de MSC

Los cultivos de rStomach™ se trataron con MORAb-202, anticuerpo MORAb-003 no conjugado, eribulina o testigo, como se describe en la Tabla 41. Los testigos incluyeron cultivos sin tratar y tratados con vehículo (PBS y DMSO). La diferenciación de MSC se monitorizó mediante microscopía óptica. Una vez que se observó diferenciación visible, las muestras se recogieron para la tinción y el análisis de citometría de flujo.
Tabla 41. Tratamientos de cocultivo

1.4.2 Análisis de transcurso de tiempo del efecto de MORAb-202 sobre fibroblastos asociados con el cáncer Se prepararon ratones con tumor de xenoinjerto de H2110 subcutáneo como se describe en la sección 1.3.1. Las muestras de tumor se recogieron en los días 0, 3, 5, 7 y 9 después de la administración del vehículo o MORAb-202
con 5 mg/kg. Las muestras de tumor recogidas se procesaron en portaobjetos y se analizó la expresión de los fibroblastos asociados con el cáncer mediante IHC como se describe en la sección 1.3.2.2.
2. Resultados
2.1 Análisis de citotoxicidad in v itro
2.1.1 Citotoxicidad de MORAb-202
Se evaluó la potencia in vitro de MORAb-202 al usar un ensayo con Crystal Violet, como se detalla en la sección 1.2.1. Se llevó a cabo un cribado en las células IGROV1 (FRhi(+++)), OVCAR3 (FRmed(++)), NCI-H2110 (FRmed(++)), A431-A3 (FRlo(+/-)) y SJSA-1 (FRneg(-)). Los resultados de este cribado se proporcionan en la Figura 17 y la Tabla 42.
Tabla 42. Cribado de citotoxicidad (CE5 0 ) de MORAb-202 en varias líneas celulares tumorales

MORAb-202 exhibió citotoxicidad dependiente de la expresión del receptor alfa de folato contra líneas celulares tumorales y bajos niveles de destrucción inespecífica. MORAb-202 demostró del nivel de potencia más alto (0,01 nM) en células IGROV1, con poca citotoxicidad (>100 nM) en células SJSA-1 negativas para el receptor alfa de folato. Se observó potencia intermedia en células OVCAR3 y NCI-H2110 (0,16 nM y 0,74 nM).
2.2 Estudios in v ivo
2.2.1 Eficacia de MORAb-202 en el modelo de xenoinjerto de NC1-H2110
Los ratones que llevan tumor de H2110 subcutáneo recibieron una inyección intravenosa con vehículo o MORAb-202 con 1,2,5 y 5 mg/kg. Se observó remisión tumoral significativa después de una dosis individual de MORAb-202 con 5 mg/kg (Figura 18 y Tabla 43). Al usar este modelo de xenoinjerto con alta expresión de receptor alfa de folato y administraciones de dosis individuales, se mostró que la ventana terapéutica para MORAb-202 es 1 mg/kg para el retraso del crecimiento tumoral (con enfermedad estable) y >2,5 mg/kg para la remisión tumoral. En este estudio, MORAb-202 con una dosis de 2,5 mg/kg dio como resultado una respuesta parcial y MORAb-202 con una dosis de 5 mg/kg dio como resultado una respuesta completa.
Tabla 43. Actividad antitumoral de MORAb-202 en el modelo de xenoinjerto de NC1-H2110

2.2.2 Eficacia de MORAb-202 en el modelo PDx de NSCLC: LXFA-737
Los ratones con tumor PDx de NSCLC subcutáneo recibieron una inyección intravenosa con vehículo, MORAb-003 con 5 mg/kg o MORAb-202 con 5 mg/kg. Una dosis individual de MORAb-202 (5 mg/kg) dio como resultado una remisión tumoral significativa en este modelo, a diferencia de la dosis individual del anticuerpo MORAb-003 no conjugado (5 mg/kg), que no demostró actividad antitumoral significativa (Figura 19A). Cinco de los seis ratones en total tratados con MORAb-202 se consideraron libres de tumor el día 32 del estudio (Tabla 44), y cuatro permanecieron libres de tumor hasta el Día 74 (terminación del estudio). Además, no se observó pérdida de peso corporal significativa en el grupo de tratamiento en comparación con el grupo testigo tratado con vehículo, lo que indica que no hubo toxicidad durante el tratamiento (Figura 19B).
Tabla 44. Actividad antitumoral de MORAb-202 en el modelo PDx de NSCLC:

2.2.3 Eficacia relativa de MORAb-202 y eribulina en modelos PDx de cáncer de endometrio: Endo-12961 y Endo-10590
Los xenoinjertos Endo-12961 y Endo-10590 expresan niveles altos de receptor alfa de folato. Los ratones con tumor PDx de cáncer de endometrio subcutáneo recibieron una inyección intravenosa con PBS, eribulina con 3,2 mg/kg o 0,1 mg/kg o MORAb-202 con 5 mg/kg. La dosis máxima tolerada (DMT) de eribulina en este modelo es 3,2 mg/kg, mientras que 0,1 mg/kg es equivalente a la dosificación de eribulina proporcionada por MORAb-202 administrado con 5 mg/kg. A lo largo del comienzo del estudio, se observó actividad antitumoral significativa después del tratamiento con MORAb-202 (5 mg/kg) y la dosis DMT de eribulina (3,2 mg/kg) en ambos modelos animales, mientras que no se observó actividad antitumoral significativa después del tratamiento con eribulina con 0,1 mg/kg (Figuras 20A y 20C). Sin embargo, los tumores que remitieron en ratones tratados con eribulina con 3,2 mg/kg comenzaron a crecer nuevamente durante la duración del estudio, mientras que no se notó nuevo crecimiento tumoral significativo en ratones tratados con MORAb-202. En este estudio, se halló que MORAb-202 es significativamente más eficaz que eribulina. El tratamiento con eribulina también afectó temporalmente el peso corporal en la primera semana postratamiento (Figuras 20B y 20D). En cambio, no se observó pérdida de peso corporal en animales tratados con MORAb-202.
2.3 Mecanismo de acción de MORAb-202
2.3.1 IHC y eficacia de MORAb-202 en el modelo PDx de TNBC: OD-BRE-0631
Los ratones con tumor PDx de TNBC subcutáneo recibieron una inyección intravenosa con vehículo o MORAb-202 con 5 mg/kg. Se recogió el tejido tumoral de ratones en cada grupo antes del tratamiento (Día 1) y después del tratamiento (Día 8). Los análisis por IHC de los tejidos tumorales recogidos revelaron que MORAb-202 ocupa células tumorales que expresan receptor alfa de folato cinco días postratamiento (Día 8), después de la administración el Día 3 como una dosis individual (5 mg/kg). La ocupación celular se evaluó al usar un anticuerpo anti-IgG humana (Figura 21A). El tratamiento con MORAb-202 también mostró disminuir la estructura de fibroblastos asociados con el cáncer, como se muestra mediante tinción para IHC con un anticuerpo anti-a-actina de músculo liso (SMA)-FITC (Figura 21B). En relación con la eficacia, el tratamiento con MORAb-202 dio como resultado una remisión tumoral máxima a los 11 días postratamiento, medida por un volumen tumoral relativo (RTV) de 0,62 (Figura 21C).
2.3.2 Efecto de MORAb-202, MORAb-003 y eribulina en sistema de cocultivo 3D
Las células madre mesenquimales de médula ósea (BM-MSC) en rStomach™ (zPredicta) se cocultivaron con la línea celular de cáncer gástrico MKN-74 durante 12 días. Antes del cultivo, las BM-MSC se evaluaron para determinar la expresión del receptor alfa de folato y los marcadores de diferenciación de MSC mediante citometría de flujo. Los cultivos de rStomach™ después se trataron con MORAb-202, anticuerpo MORAb-003 no conjugado, eribulina o testigo. Una vez que se observó diferenciación de MSC visible mediante microscopía óptica, las muestras se recogieron para la tinción y el análisis de citometría de flujo. Los resultados de estos análisis se muestran en la Figura 22.
Una duración total de tratamiento de 7 días, con reposición del tratamiento durante este período, fue suficiente para producir un efecto medible sobre la diferenciación de BM-MSC humanas en cultivo con células MKN-74. Con respecto al testigo con vehículo, el tratamiento con MORAb-202 (10 nM) dio como resultado un aumento en las poblaciones de MSC y adipocitos, y una disminución en las poblaciones de pericitos (Tabla 45). Estos datos indican que MORAb-202 puede tener un efecto significativo sobre el microambiente tumoral.
Tabla 45. Efecto de MORAb-202, MORAb-003 y eribulina en sistema de cocultivo 3D

2.3.3 Análisis de transcurso de tiempo del efecto de MORAb-202 sobre fibroblastos asociados con el cáncer Se recogieron muestras de tumor de ratones con tumor de xenoinjerto de H2110 subcutáneo en los Días 0, 3, 5, 7 y 9 después de la administración del vehículo o MORAb-202 con 5 mg/kg. Las muestras de tumor recogidas se procesaron en portaobjetos y se analizó la expresión de los fibroblastos asociados con el cáncer (CAF) mediante IHC. La estructura de la red de CAF, según se evaluó y cuantificó mediante tinción con un anticuerpo anti-a-actina de músculo liso (SMA)-FITC fue prominente el Día 3 y Día 5, después de la administración de una dosis individual de MORAb-202 con 5 mg/kg (Figura 23). Sin embargo, al Día 7, gran parte de esta estructura había disminuido significativamente.
EJEMPLO 3
1. Materiales y métodos
Se sintetizaron compuestos de eribulina conjugables que tienen las estructuras que se muestran en la Tabla 46 según los siguientes procedimientos, y se usaron en la preparación de ADC (Ejemplo 4).
Todos disolventes usados en las reacciones de síntesis fueron de grado anhidro (EMD Millipore). Todos los disolventes usados para el procesamiento o purificación fueron de grado apto para cromatografía de líquidos de alto rendimiento (HPLC) (EMD Millipore). A menos que se indique de cualquier otra manera, todas las sustancias químicas están disponibles comercialmente. La cromatografía en columna se llevó a cabo al usar un Biotage® SP4. La extracción del disolvente se llevó a cabo al usar un evaporador rotatorio (Büchi Labortechik AG) o un evaporador centrífugo (Genevac, SP scientific). La cromatografía de líquidos preparativa-espectrometría de masas (LC/m S) se llevó a cabo al usar un sistema Waters AutoPurification y una columna XTerra MS C18 (5 pm, 19 mm x 100 mm) en condiciones de fase móvil ácida. Los espectros de resonancia magnética nuclear (NMR, por sus siglas en inglés) se registraron al usar cloroformo deuterado (CDCta) a menos que se indique de cualquier otra manera y se registraron a 400 o 500 MHz al usar un instrumento Varian (Agilent Technologies). Los espectros de masa se registraron al usar un LC/MS Waters Acquity Ultra Performance. Según se usa en la presente memoria, el término "inerte" se refiere al reemplazo del aire en un reactor (p. ej., un vaso de reacción, un matraz, un reactor de vidrio) con un gas inerte esencialmente libre de humedad, tal como nitrógeno o argón. Las multiplicidades se indican al usar las siguientes abreviaciones: s= singulete, d=doblete, t=triplete, q=cuarteto, quint=quinteto, sxt=sexteto, m=multiplete, dd=doblete de dobletes, ddd=doblete de dobletes de dobletes, dt=doblete de tripletes, br s=un singulete amplio.
Tabla 46. Compuestos de eribulina conjugables











(continuación)



Ċ
Ĩcontinuación)

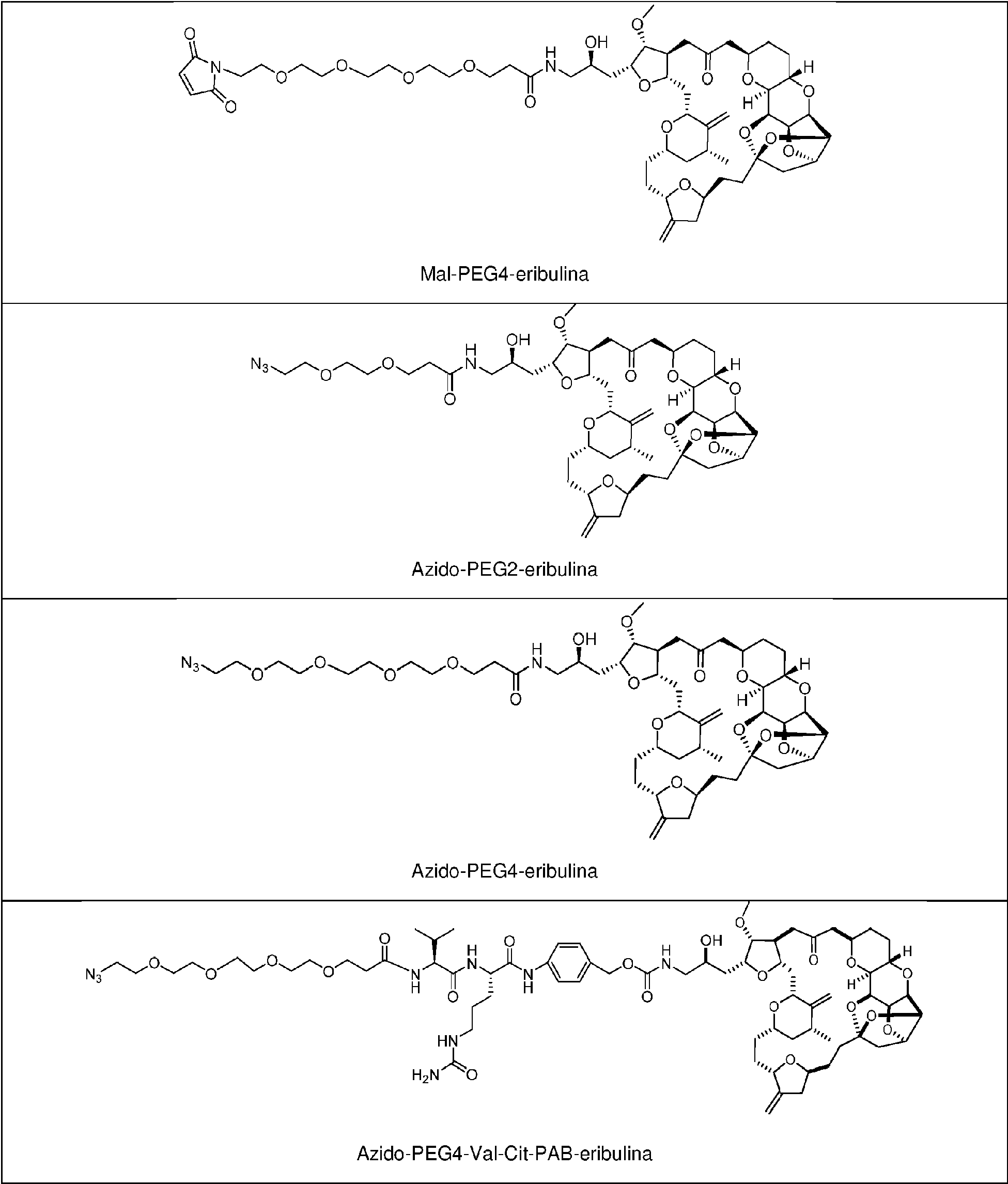

1.1 Preparación de MAL-PEG2-Val-Cit-PAB-eribulina (ER-001159569)

Se disolvió eribulina (ER-000086526) (61,5 mg, 0,074 mmol) en N,N-dimetilformamida (DMF) (6,0 mL) y después se mezcló con la base de Hunig (0,027 mL, 0,156 mmol) y Fmoc-Val-Cit-PAB-PNP (86 mg, 0,112 mmol). La reacción se agitó a temperatura ambiente durante 18 horas hasta que se completó el acoplamiento, según se determinó mediante análisis de cromatografía de líquidos de alto rendimiento (HPLC). Se agregó dietilamina (0,078 mL, 0,745 mmol) a la mezcla, y la mezcla se agitó durante 2 horas adicionales hasta que la reacción se completó. El disolvente se retiró mediante evaporación y el residuo se purificó mediante cromatografía rápida para obtener Val-Cit-PAB-eribulina (ER-001228950) como un sólido blanco (60 mg, 71 % de rendimiento). 1HRMN (400 MHz, CD3OD) 5 ppm 7,56 (d, J = 8,4 Hz, 2H), 7,32 (d, J = 8,4 Hz, 2H), 5,14 (s, 1H), 5,06 (d, J = 12,4 Hz, 1H), 5,03 (s, 1H), 5,01 (d, J= 12,4 Hz, 1H), 4,87 (s, 1H), 4,83 (s, 1H), 4,71 (t, J = 4,4 Hz, 1H), 4,62 (t, J = 4,4 Hz, 1H), 4,57 (dd, J = 4,8, 8,8 Hz, 1H), 4,47 (d, J = 10,8 Hz, 1H), 4,32-4,27 (m, 2H), 4,18 (dd, J = 4,8, 6,4 Hz, 1H), 4,13-4,07 (m, 2H), 3,98 (t, J = 10,4 Hz, 1H), 3,88-3,82 (m, 3H), 3,76-3,70 (m, 4H), 3,60 (d, J = 6,0 Hz, 1H), 3,38 (s, 3H), 3,26-3,10 (m, 3H), 2,93 (dd, J = 2,0, 11,2 Hz, 1H), 2,91-2,84 (m, 1H), 2,75-2,64 (m, 2H), 2,44-2,29 (m, 5H), 2,21-1,97 (m, 8H), 1,93-1,83 (m, 3H), 1,79-1,72 (m, 5H), 1,68-1,29 (m, 8H), 1,11 (d, J = 6,8 Hz, 3H), 1,07-1,01 (m, 1H), 1,06 (d, J = 7,2 Hz, 3H), 1,02 (d, J= 7,2 Hz, 3H). LCMS (M+H)=1135,7.
Se disolvió Val-Cit-PAB-eribulina (ER-001228950) (16 mg, 14 pmol) en DMF (1 mL). Después, se agregaron N,N-diisopropiletilamina (7,2 pL, 41 pmol) y Mal-PEG2-NHS (9,7 mg, 27 pmol) a esta disolución a temperatura ambiente y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Después de que se completó la reacción, la mezcla bruta se purificó mediante HPLC de fase inversa al usar una fase móvil de acetonitrilo-agua que contenía ácido fórmico al 0,1 %. Las fracciones recogidas se concentraron al vacío a temperatura ambiente en un baño de agua no calentado para proporcionar MAL-PEG2-Val-Cit-PAB-eribulina (ER-001159569) (7,1 mg, 5,2 pmol, 38 % de rendimiento). 1HRMN (400 MHz, CD3OD) 5 ppm 7,59 (d, J = 8,4 Hz, 2H), 7,31 (d, J = 8,4 Hz, 2H), 6,81 (s, 2H), 5,13 (s, 1H), 5,06 (d, J= 12,4 Hz, 1H), 5,02 (s, 1H), 5,01 (d, J= 12,4 Hz, 1H), 4,87 (s, 1H), 4,82 (s, 1H), 4,71 (t, J = 4,0 Hz, 1H), 4,61 (t, J = 4,4 Hz, 1H), 4,50 (dd, J = 5,2, 9,2 Hz, 1H), 4,47 (d, J= 10,8 Hz, 1H), 4,32-4,27 (m, 2H), 4,19 (dd, J= 6,8, 11,6 Hz, 1H), 4,13-4,07 (m, 2H), 3,98 (t, J= 10,4 Hz, 1H), 3,88-3,82 (m, 3H), 3,76-3,64 (m, 6H), 3,62-3,51 (m, 6H), 3,38 (s, 3H), 3,22-3,08 (m, 4H), 2,93 (dd, J = 2,4, 9,6 Hz, 1H), 2,92-2,84 (m, 1H), 2,76-2,63 (m, 2H), 2,52 (t, J = 6,0 Hz, 2H), 2,44-2,29 (m, 5H), 2,21 -1,97 (m, 8H), 1,93-1,83 (m, 3H), 1,80-1,66 (m, 5H), 1,66-1,28 (m, 10H), 1,11 (d, J = 6,4 Hz, 3H), 1,07-1,01 (m, 1H), 0,99 (d, J= 6,8 Hz, 3H), 0,97 (d, J= 6,4 Hz, 3H). LCMS (M+H)=1374,9.
1.2 Preparación de NHS-PEG2-Val-Cit-PAB-eribulina (ER-001236940)

Se mezclaron Val-Cit-PAB-eribulina (ER-001228950) (45 mg, 0,04 mmol) y bis(2,5-d ioxopi rrolidin-1 -il) 3,3'-(etano-1,2-diilbis(oxi))dipropanoato (79 mg, 0,198 mmol) en DMF (1,5 mL), y después se agregó Et3N (44,2 gl, 0,317 mmol). La mezcla se agitó durante 18 horas hasta que se completó la reacción, según se determinó mediante análisis de HPLC. El disolvente se evaporó y el residuo se purificó mediante cromatografía rápida para obtener NHS-PEG2-Val-Cit-PAB-eribulina (ER-001236940) como un sólido blanco (38 mg, 68 % de rendimiento). 1HRMN (400 MHz, CD3OD) 5 ppm 7,58 (d, J= 8,4 Hz, 2H), 7,33 (d, J= 8,4 Hz, 2H), 5,14 (s, 1H), 5,05 (d, J= 12,4 Hz, 1H), 5,03 (s, 1H), 5,01 (d, J = 12,4 Hz, 1H), 4,87 (s, 1H), 4,83 (s, 1H), 4,71 (t, J = 4,4 Hz, 1H), 4,62 (t, J = 4,4 Hz, 1H), 4,51 (dd, J = 4,8, 8,8 Hz, 1H), 4,50 4,47 (m, 1H), 4,32-4,27 (m, 2H), 4,21 (dd, J = 4,8, 6,4 Hz, 1H), 4,14-4,08 (m, 2H), 3,99 (t, J = 10,4 Hz, 1H), 3,88-3,82 (m, 3H), 3,78-3,70 (m, 4H), 3,62 (s, 2H), 3,62-3,58 (m, 1H), 3,50-3,46 (m, 2H), 3,39 (s, 4H), 3,36 (s, 3H), 3,22-3,08 (m, 3H), 2,93 (dd, J = 2,0, 11,2 Hz, 1H), 2,91-2,87 (m, 1H), 2,84 (s, 2H), 2,80 (s, 2H), 2,75-2,64 (m, 2H), 2,59-2,52 (m, 2H), 2,44-2,29 (m, 5H), 2,21-1,97 (m, 10H), 1,93-1,83 (m, 3H), 1,79-1,72 (m, 5H), 1,68-1,29 (m, 8H), 1,11 (d, J= 6,8 Hz, 3H), 1,08-0,98 (m, 1H), 1,00 (d, J = 7,2Hz, 3H), 0,98 (d, J = 7,2 Hz, 3H). LCMS (M+H)=1421,0.
1.3 Preparación de NHS-(CH2)5-Val-Cit-PAB-eribulina (ER-001236941)
Se disolvió ácido heptanodioico (1,6 g, 9,99 mmol) en tetrahidrofurano (THF) (100 mL) y después se agregó 1-hidroxipirrolidina-2,5-diona (2,299 g, 19,98 mmol), con la posterior adición de DCC (4,12 g, 19,98 mmol). La mezcla se agitó a temperatura ambiente durante 18 horas hasta que el análisis de HPLC indicó que la reacción se había completado. El sólido se retiró mediante filtración a través de una almohadilla de celite y se lavó con THF (3 x 2 mL). El filtrado combinado se concentró y se purificó mediante cromatografía rápida para proporcionar bis(2,5-dioxopirrolidin-1-il) heptanodioato (ER-001236140) como un sólido blanco (2,5 g, 71 % de rendimiento). 1HRMN (400 MHz) 5 ppm 2,83 (s, 8H), 2,64 (t, J = 7,6 Hz, 4H), 1,80 (dt, J= 7,6 Hz, 4H), 1,59-1,51 (m, 2H). LCMS (M+H)=355,2.
Se preparó NHS-(CH2)5-Val-Cit-PAB-eribulina (ER-001236941) (8,5 mg, 47 % de rendimiento) a partir de VCP-eribulina (ER-001228950) y bis(2,5-dioxopirrolidin-1 -il) heptanodioato (ER-001236140) al usar el mismo procedimiento según se describió anteriormente para la preparación de NHS-PEG2-Val-Cit-PAB-eribulina (ER-001236940). 1HRMN (400 MHz, CDaOD) 5 ppm 7,56 (d, J= 8,4 Hz, 2H), 7,30 (d, J= 8,4 Hz, 2H), 5,13 (s, 1H), 5,04 (d, J= 12,0 Hz, 1H), 5,01 (s, 1H), 5,00 (d, J= 12,4 Hz, 1H), 4,86 (s, 1H), 4,82 (s, 1H), 4,70 (t, J= 4,4 Hz, 1H), 4,60 (t, J= 4,4 Hz, 1H), 4,50 (dd, J= 4,8, 8,8 Hz, 1H), 4,46 (d, J = 10,8 Hz, 1H), 4,36-4,25 (m, 2H), 4,17 (dd, J = 4,8, 6,4 Hz, 1H), 4,13-4,06 (m, 2H), 3,97 (t, J= 10,4 Hz, 1H), 3,87-3,80 (m, 3H), 3,74-3,68 (m, 2H), 3,37 (s, 3H), 3,20-3,06 (m, 4H), 2,94 (dd, J = 2,0, 11,2 Hz, 1H), 2,90-2,82 (m, 1H), 2,82 (s, 4H), 2,74-2,65 (m, 2H), 2,61 (t, J= 8,0 Hz, 2H), 2,46-2,26 (m, 7H), 2,24-1,81 (m, 13H), 1,78 1,28 (m, 19H), 1,10 (d, J = 6,8 Hz, 3H), 1,06-0,96 (m, 1H), 0,97 (d, J = 7,2 Hz, 3H), 0,95 (d, J= 7,2 Hz, 3H). LCMS (M+H)=1375,1.
1.4 Preparación de Mal-(CH2)5 -Val-Cit-PAB-eribulina (ER-001235638)

Se disolvió eribulina (ER-000086526) (10 mg, 0,012 mmol) en DMF (1 mL) y se mezcló con MC-Val-Cit-PAB-PNP (9,02 mL, 0,012 mmol) y la base de Hunig (4,44 pL, 0,025 mmol). La mezcla después se agitó a temperatura ambiente durante 12 horas hasta que el análisis de HPLC indicó que la reacción se había completado. La mezcla de reacción se concentró y se purificó mediante cromatografía rápida para proporcionar Mal-(CH2)5-Val-Cit-PAB-eribulina (ER-001235638) como un sólido blanco (11,3 mg, 63 % de rendimiento). 1HRMN (400 MHz, CD3OD) 5 ppm 7,57 (d, J= 8,4 Hz, 2H), 7,31 (d, J= 8,4 Hz, 2H), 6,79 (s, 2H), 5,13 (s, 1H), 5,05 (d, J= 12,4 Hz, 1H), 5,02 (s, 1H), 5,00 (d, J= 12,4 Hz, 1H), 4,87 (s, 1H), 4,83 (s, 1H), 4,71 (t, J= 4,4 Hz, 1H), 4,61 (t, J= 4,4 Hz, 1H), 4,56-4,46 (m, 3H), 4,35-4,27 (m, 2H), 4.20- 4,07 (m, 4H), 3,98 (t, J= 10,8 Hz, 1H), 3,87-3,83 (m, 3H), 3,73-3,70 (m, 2H), 3,48 (t, J= 7,6 Hz, 2H), 3,38 (s, 3H), 3.20- 3,08 (m, 4H), 2,93 (dd, J = 1,6, 9,6 Hz, 1H), 2,89-2,85 (m, 1H), 2,69 (dt, J = 11,2, 16,8 Hz, 2H), 2,44-2,33 (m, 5H), 2,27-1,83 (m, 13H), 1,78-1,68 (m, 5H), 1,66-1,27 (m, 14H), 1,11 (d, J = 7,2 Hz, 3H), 1,07-0,98 (m, 1H), 0,98 (d, J = 7,2 Hz, 3H), 0,96 (d, J = 7,2 Hz, 3H). LCMS (M+H)=1328,9.
1.5 Preparación de Mal-PEG8-Val-Cit-PAB-eribulina (ER-001242287)

Se mezclaron VCP-eribulina (ER-001228950) (10 mg, 8,808 pmol) y 2,5-dioxopirrolidin-1-il 1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-3-oxo-7,10,13,16,19,22,25,28-octaoxa-4-azahentriacontan-31-oato (6,07 mg, 8,808 pmol) en DMF (1 mL), con la posterior adición de Et3N (9,82 pl, 0,07 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 18 horas hasta que el análisis de HPLC indicó que la reacción se había completado. El disolvente se retiró mediante evaporación y el residuo se purificó mediante cromatografía rápida para proporcionar Mal-PEG8-Val-Cit-PAB-eribulina (ER-001242287) como un sólido blanco (3,0 mg, 20 % de rendimiento). 1HRMN (400 MHz, CD3OD) 5 ppm 7,58 (d, J = 8,4 Hz, 2H), 7,29 (d, J = 8,4 Hz, 2H), 6,80 (s, 2H), 5,12 (s, 1H), 5,04 (d, J = 12,4 Hz, 1H), 5,01 (s, 1H), 4,99 (d, J = 12,4 Hz, 1H), 4,85 (s, 1H), 4,80 (s, 1H), 4,69 (t, J = 4,4 Hz, 1H), 4,59 (t, J = 4,4 Hz, 1H), 4,50-4,42 (m, 2H), 4,32-4,24 (m, 2H), 4,20-4,14 (m, 2H), 4,12-4,04 (m, 3H), 3,96 (t, J = 10,4 Hz, 1H), 3,86-3,80 (m, 3H), 3,76-3,57 (m, 4H), 3,48 (t, J = 6,0 Hz, 1H), 3,36 (s, 3H), 3,20-3,08 (m, 3H), 2,91 (dd, J = 2,0, 11,2 Hz, 1H), 2,90-2,82 (m, 1H), 2,74 2,60 (m, 2H), 2,44-2,29 (m, 5H), 2,21-1,97 (m, 10H), 1,93-1,83 (m, 3H), 1,79-1,20 (m, 19H), 1,09 (d, J= 6,8 Hz, 3H), 1,04-0,98 (m, 1H), 0,99 (d, J= 7,2 Hz, 3H), 0,97 (d, J= 7,2 Hz, 3H). LCMS (M+H)=1711,6.
1.6 Preparación de NHS-PEG9-Val-Cit-PAB-eribulina (ER-001242288)
Se preparó NHS-PEG9-Val-Cit-PAB-eribulina (ER-001242288) (13 mg, 85 % de rendimiento) a partir de VCP-eribulina (ER-001228950) y BisNHS-PEG9 al usar el mismo procedimiento según se describió anteriormente para la preparación de NHS-PEG2-Val-Cit-PAB-eribulina (ER-001236940). 1HRMN (400 MHz, CD3OD) 5 ppm 7,61 (d, J= 8,4 Hz, 2H), 7,32 (d, J = 8,4 Hz, 2H), 5,16 (s, 1H), 5,06 (d, J = 12,4 Hz, 1H), 5,01 (s, 1H), 5,00 (d, J= 12,4 Hz, 1H), 4,87 (s, 1H), 4,82 (s, 1H), 4,71 (t, J= 4,4 Hz, 1H), 4,61 (t, J= 4,4 Hz, 1H), 4,52-4,45 (m, 2H), 4,34-4,26 (m, 2H), 4,20-4,19 (m, 1 H), 4,14-4,06 (m, 2H), 3,98 (t, J= 10,4 Hz, 1H), 3,88-3,80 (m, 3H), 3,76-3,70 (m, 4H), 3,66-3,58 (m, 37H), 3,38 (s, 3H), 3,24-3,10 (m, 3H), 2,93 (dd, J = 2,0, 11,2 Hz, 1H), 2,91-2,84 (m, 1H), 2,84 (s, 4H), 2,76-2,64 (m, 2H), 2,58-2,50 (m, 4H), 2,46-2,28 (m, 5H), 2,22-1,96 (m, 8H), 1,91-1,82 (m, 3H), 1,79-1,68 (m, 5H), 1,64-1,24 (m, 8H), 1,11 (d, J = 6,8 Hz, 3H), 1,08-0,96 (m, 1H), 0,99 (d, J= 7,2 Hz, 3H), 0,97 (d, J= 7,2 Hz, 3H). LCMS (M+H)=1729,7.
1.7 Preparación de NHS-PEG3-triazol-PEG3-Val-Cit-PAB-eribulina (ER-001243700)

Se disolvió VCP-eribulina (ER-001228950) (25 mg, 0,022 mmol) en DMF (2,5 mL) y después se mezcló con Et3N (24,55 gl, 0,176 mmol) y Azida-PEG3-NHS (8,34 mg, 0,024 mmol). La mezcla se agitó a temperatura ambiente durante 18 horas hasta que el análisis de HPLC indicó que la reacción se había completado. La mezcla se concentró al vacío y el residuo se purificó mediante prep-HPLC (MeCN y agua con ácido fórmico al 0,1 %). Las fracciones que contenían azida-PEG3-Val-Cit-PAB-eribulina se extrajeron con diclorometano (CH2Cl2) (3 x 20 mL), y el CH2Cl2 se evaporó para obtener azida-PEG3-Val-Cit-PAB-eribulina (ER-001243116) como un sólido blanco (18,9 mg, 63 % de rendimiento).
1HRMN (400 MHz, CD3OD) 5 ppm 7,58 (d, J= 8,4 Hz, 2H), 7,30 (d, J= 8,4 Hz, 2H), 5,14 (s, 1H), 5,04 (d, J= 12,4 Hz, 1H), 5,03 (s, 1H), 5,01 (d, J= 12,4 Hz, 1H), 4,85 (s, 1H), 4,81 (s, 1H), 4,70 (t, J = 4,4 Hz, 1H), 4,61 (t, J = 4,4 Hz, 1H),
4,52-4,48 (m, 2H), 4,31-4,25 (m, 2H), 4,20-4,15 (m, 1H), 4,13-4,07 (m, 2H), 3,99 (t, J = 10,4 Hz, 1H), 3,84-3,79 (m, 3H), 3,77-3,65 (m, 4H), 3,64-3,56 (m, 13H), 3,38 (s, 3H), 3,20-3,05 (m, 3H), 2,95-2,80 (m, 2H), 2,75-2,60 (m, 2H), 2,55 2,50 (m, 2H), 2,43-2,25 (m, 5H), 2,21-1,97 (m, 8H), 1,93-1,83 (m, 3H), 1,79-1,72 (m, 5H), 1,68-1,29 (m, 10H), 1,08 (d, J= 6,8 Hz, 3H), 1,05-0,95 (m, 1H), 0,98 (d, J= 7,2 Hz, 3H), 0,95 (d, J = 7,2 Hz, 3H). LCMS (M+H)=1365,1.
Se mezclaron azida-PEG3-VCP-eribulina (ER-001243116) (9,6 mg, 7,035 pmol) y 3-(2-(2-(prop-2-in-1-iloxi)etoxi)etoxi)propanoato de 2,5-dioxopirrolidin-1-ilo (6,61 mg, 0,021 mmol) en agua (0,6 mL) y t-Butanol (1,8 mL). La mezcla se burbujeó con N2 durante 45 min. Se agregó yoduro de cobre sobre amberlist-21 (1,23 mmol/g, 10 mg) a la mezcla y se burbujeó con N2 a través de la mezcla durante otros 30 min. La mezcla de reacción después se agitó a temperatura ambiente durante 72 horas hasta que se consumió por completo el material de partida. No se observó el producto de éster de NHS deseado mediante el análisis por LCMS, solo el ácido carboxílico hidrolizado. La mezcla se filtró a través de una almohadilla de celite corta para retirar la resina de Cul. El filtrado se concentró al vacío, y el residuo resultante se purificó mediante cromatografía de capa delgada preparativa (prep-TLC) (20 % de MeOH/CH2Cl2) para obtener ácido-PEG3-triazol-PEG3-Val-Cit-PAB-eribulina (ER-001243701) como un sólido blanco (3,7 mg, 33 % de rendimiento). LCMS (ES) (M+H)=1581,2.
Se disolvió la ácido-PEG3-triazol-PEG3-Val-Cit-PAB-eribulina (ER-001243701) (3,0 mg, 1,898 pmol) en DMF (200 pL) y se agregó 1-hidroxipirrolidina-2,5-diona (0,437 mg, 3,796 pmol), con la posterior adición de 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDC) (0,728 mg, 3,796 pmol). La reacción se completó aproximadamente un 50 % después de la agitación a temperatura ambiente durante 18 horas. Se agregó EDC (1,46 mg, 7,8 pmol) y la mezcla se agitó durante otras 18 horas hasta que el análisis de HPLC indicó >95 % de conversión a NHS-PEG3-triazol-PEG3-Val-Cit-PAB-eribulina. La mezcla se concentró al vacío, y el residuo se purificó mediante prep-TLC (15 % de MeOH/CH2Cl2) para proporcionar NHS-PEG3-triazol-PEG3-Val-Cit-PAB-eribulina (ER-001243700) como un sólido blanco (2,2 mg, 69 % de rendimiento). 1HRMN (400 MHz, CD3OD) 5 ppm 8,00 (s, 1H), 7,59 (d, J= 8,0 Hz, 2H), 7,31 (d, J= 8,4 Hz, 2H), 5,13 (s, 1H), 5,04 (d, J= 12,4 Hz, 1H), 5,02 (s, 1H), 5,00 (d, J= 12,4 Hz, 1H), 4,87 (s, 1H), 4,83 (s, 1H), 4,71 (t, J= 4,0 Hz, 1H), 4,63 (s, 2H), 4,61 (t, J= 4,4 Hz, 1H), 4,57-4,55 (m, 2H), 4,51-4,45 (m, 1H), 4,32-4,28 (m, 2H), 4,21-4,17 (m, 2H), 4,13-4,10 (m, 2H), 3,98 (t, J = 10,8 Hz, 1H), 3,88-3,80 (m, 5H), 3,75-3,70 (m, 4H), 3,68-3,55 (m, 18H), 3,45-3,40 (m, 2H), 3,38 (s, 3H), 3,20-3,08 (m, 4H), 2,93-2,80 (m, 2H), 2,75-2,50 (m, 2H), 2,68 (s, 4H), 2,48 2,30 (m, 7H), 2,28-1,92 (m, 10H), 1,90-1,68 (m, 8H), 1,65-1,27 (m, 8H), 1,11 (d, J= 6,8 Hz, 3H), 1,05-0,95 (m, 1H), 0,99 (d, J= 7,2 Hz, 3H), 0,97 (d, J= 6,8 Hz, 3H). LCMS (M+H)=1678,3.
1.8 Preparación de Mal-PEG2-Ala-Ala-Asn-PAB-eribulina (ER-001231679) y Mal-PEG2-(Ala-Ala-Asn-PAB)2-eribulina (ER-001231690)

Se disolvió eribulina (ER-000086526) (10 mg, 0,014 mmol) en DMF (0,5 mL) y se mezcló con la base de Hunig (3,59 gL, 0,021 mmol). Después se agregó (9H-fluoren-9-il)metil ((S)-1-(((S)-1-(((S)-4-amino-1-((4-((((4-nitrofenoxi)carbonil)oxi)metil)fenil)amino)-1,4-dioxobutan-2-il)amino)-1-oxopropan-2-il)amino)-1-oxopropan-2-il)carbamato (15,76 mg, 0,021 mmol) y la disolución amarilla resultante se agitó a temperatura ambiente durante 3 días hasta que el análisis de HPLC indicó que el material de partida se había consumido por completo. Se agregó dietilamina (14,23 gl, 0,137 mmol) a la mezcla de reacción, que después se agitó a temperatura ambiente durante 2 horas adicionales hasta que hubo una escisión del 100 % de la protección de Fmoc. La mezcla de reacción se concentró para retirar la dietilamina, y el residuo se redisolvió en DMF (1,5 mL). Se agregó Et3N (0,015 mL, 0,11 mmol) a temperatura ambiente, con la posterior adición de 3-(2-(2-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1 -il)etoxi)etoxi)propanoato de 2,5-dioxopirrolidin-1-ilo (9,71 mg, 0,027 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 horas hasta que la reacción se completó, según se determinó mediante análisis de LCMS. La mezcla se concentró a vacío alto, y se purificó mediante cromatografía rápida para obtener Mal-PEG2-Ala-Ala-Asn-PAB-eribulina (ER-001231679) (9,2 mg, 49 % de rendimiento) y Mal-PEG2-(Ala-Ala-Asn-PAB)2-eribulina (ER-001231690) (6,0 mg, 18 % de rendimiento) como aceites incoloros.
Mal-PEG2-Ala-Ala-Asn-PAB-eribulina (ER-001231679): 1HRMN (400 MHz) 5 ppm 9,23 (s, 1H), 8,00 (d, J = 7,6 Hz, 1H), 7,61 (d, J = 8,4 Hz, 2H), 7,38 (d, J= 6,8 Hz, 1H), 7,24 (d, J= 8,4 Hz, 2H), 7,13 (d, J= 7,2 Hz, 1H), 6,68 (s, 2H), 6,30 (s a, 1H), 6,04-6,00 (m, 1H), 5,77 (s a, 1H), 5,42 (s a, 1H), 5,07 (s, 1H), 5,06-4,98 (m, 2H), 4,93 (s, 1H), 4,88 (s, 1H), 4,90-4,82 (m, 1H), 4,80 (s, 1H), 4,69 (t, J = 4,0 Hz, 1H), 4,60 (t, J = 4,0 Hz, 1H), 4,49-4,42 (m, 1H), 4,38-4,25 (m,
4H), 4,19 (t, J= 4,8 Hz, 1H), 4,15-4,08 (m, 1H), 4,03 (t, J= 4,8 Hz, 1H), 3,97-3,85 (m, 3H), 3,83-3,50 (m, 12H), 3,41 (s, 3H), 3,50-3,10 (m, 3H), 3,02-2,64 (m, 6H), 2,52-2,30 (m, 7H), 2,30-1,65 (m, 14H), 1,65-1,20 (m, 12H), 1,10 (d, J= 6,8 Hz, 3H), 1,13-1,05 (m, 1H). LCMS (M+Na)=1396,6.
Mal-PEG2-(Ala-Ala-Asn-PAB)2-eribulina (ER-001231690): 1HRMN (400 MHz, CDaOD) 5 ppm 7,65 (d, J= 8,4 Hz, 2H), 7,60 (d, J= 8,4 Hz, 2H), 7,28 (d, J= 8,8 Hz, 2H), 7,23 (d, J= 8,4 Hz, 2H), 6,79 (s, 2H), 5,13 (s, 1H), 5,02 (s, 1H), 5,06 4,98 (m, 4H), 4,87 (s, 1H), 4,82 (s, 1H), 4,85-4,72 (m, 2H), 4,71 (t, J= 4,8 Hz, 1H), 4,61 (t, J = 4,4 Hz, 1H), 4,47 (d, J= 11,2 Hz, 1H), 4,30-4,06 (m, 9H), 3,97 (t, J= 4,8 Hz, 1H), 3,89-3,80 (m, 3H), 3,75-3,48 (m, 12H), 3,38 (s, 3H), 3,17 (d, J= 6,8 Hz, 2H), 2,94-2,62 (m, 8H), 2,50-2,28 (m, 7H), 2,22-1,65 (m, 14H), 1,58-1,30 (m, 18H), 1,10 (d, J = 6,8 Hz, 3H), 1,06-0,97 (m, 1H). LCMS (M+Na)=1802,8.
1.9 Preparación de NHS-PEG2-Ala-Ala-Asn-PAB-eribulina (ER-001231691)

Se preparó Ala-Ala-Asn-PAB-eribulina (ER-001231678) (15 mg, rendimiento cuantitativo) a partir de eribulina (ER-000086526) y Fmoc-Ala-Ala-Asn-PAB-PNP al usar el mismo procedimiento según se describió anteriormente para la preparación de Val-Cit-PAB-eribulina (ER-001228950). LCMS (M+H)=1135.5.
Se preparó NHS-PEG2-Ala-Ala-Asn-PAB-eribulina (ER-001231691) (12,4 mg, 64 % de rendimiento) a partir de Ala-Ala-Asn-PAB-eribulina (ER-001231678) y BisNHS-PEG2 al usar el mismo procedimiento según se describió anteriormente para la preparación de NHS-PEG2-Val-Cit-PAB-eribulina (ER-001236940). 1HRMN (400 MHz) 5 ppm 9,21 (s, 1H), 7,95 (d, J= 8,0 Hz, 1H), 7,62 (d, J= 8,8 Hz, 2H), 7,58-7,52 (m, 1H), 7,28 (s a, 1H), 7,24 (d, J = 8,4 Hz, 2H), 7,10 (s a, 1H), 6,29 (d, J= 12,4 Hz, 1H), 5,83 (s a, 1H), 5,38 (s a, 1H), 5,07 (s, 1H), 5,05-4,95 (m, 2H), 4,93 (s, 1H), 4,88 (s, 1H), 4,90-4,83 (m, 1H), 4,81 (s, 1H), 4,69 (t, J= 4,4 Hz, 1H), 4,60 (t, J= 4,4 Hz, 1H), 4,46-4,41 (m, 1H), 4,36 4,25 (m, 4H), 4,19 (dd, J= 4,8, 6,0 Hz, 1H), 4,15-4,09 (m, 1H), 4,03 (dd, J= 4,8, 6,0 Hz, 1H), 3,99-3,89 (m, 3H), 3,853,50 (m, 10H), 3,41 (s, 3H), 3,40-3,10 (m, 3H), 3,01-2,60 (m, 10H), 2,60-2,35 (m, 7H), 2,35-1,65 (m, 14H), 1,65-1,20 (m, 14H), 1,10 (d, J = 6 , 8 Hz, 3H), 1,15-1,03 (m, 1H). LCMS (ES) (M+H)=1442,7.
1.10 Preparación de azida-PEG3-disulfuro-PAB-eribulina (ER-001237508)

Se disolvió ácido 4-(((terc-butildimetilsilM)oxi)metil)benzoico (1,0 g, 3,754 mmol) en diclorometano (DCM) (25 mL) y se enfrió hasta 0 °C. Después se agregó trietilamina (0,549 mL, 3,941 mmol), posteriormente difenil fosforazidato (1,085 mg, 3,941 mmol). La mezcla de reacción se calentó lentamente hasta temperatura ambiente y se agitó durante 14 horas. La mezcla bruta se diluyó con acetato de etilo (EtOAc)/Hep (1:1, 100 mL) y se pasó a través de un tapón de sílice corto con elución con EtOAc/Hep (50 %). El disolvente se retiró al vacío para proporcionar 1 , 10 g de azida de 4 (((tercbutildimetilsilil)oxi)metil)benzoilo (ER-001131970). 1H RMN (400 MHz) 5 ppm 7,98 (d, 2 H, J = 8,0 Hz), 7,40 (d, 2 H, J = 8,0 Hz), 4,79 (s, 2 H), 0,94 (s, 9 H), 0,10 (s, 6 H).
La azida de 4-(((terc-butildimetilsilil)oxi)metil)benzoilo (ER-001131970) (1,1 g, 3,775 mmol), disuelta en tolueno (20 mL), se calentó a 110°C durante 3 horas. Si bien el producto no se mostró como una única mancha, el análisis de cromatografía de capa delgada (TLC) indicó que el material de partida se había consumido. La mezcla de reacción después se enfrió hasta temperatura ambiente, y se transfirió a un vial sellado en nitrógeno y se almacenó como una disolución en tolueno (1 mL = 32,6 mg) a -20 °C.
Se agregó trietilamina (0,099 mL, 0,709 mmol) a una disolución de terc-butil((4-isocianatobencil)oxi)dimetilsilano (165 mg, 0,626 mmol) en tolueno (5 mL), posteriormente alcohol (90,0 mg, 0,591 mmol), y la mezcla de reacción se agitó durante 6 horas a 36 °C. Se monitoreó el progreso de la reacción mediante UPLC/MS. Después, se agregó una disolución saturada de carbonato de sodio hidrógeno (NaHCO3) (10 mL) y se extrajo con EtOAc/Hep (1:1,60 mL), se lavó con salmuera, se secó sobre sulfato de sodio y se concentró. El material bruto se purificó mediante cromatografía rápida (EtOAc/Hep 10 % a 40 %) para obtener 215 mg de (4-(((tercbutildimetilsilil) oxi)metil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001131973). 1H RMN (400 MHz) 5 ppm 7,34 (d, 2 H, J= 8,4 Hz), 7,26 (d, 2 H, J= 7,6 Hz), 6,63 (s a, 1 H), 4,69 (s, 2 H), 4,17 (s, 2 H), 2,42 (s, 3 H), 1,35 (s, 6 H), 0,93 (s, 9 H), 0,08 (s, 6 H).
Se disolvieron (4-(((tercbutildimetilsilil)oxi)metil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001131973) (198 mg, 0,476 mmol) y 4-metilbencenosulfonato de 2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etilo (325 mg, 0,87 mmol) en DMF (6 , 6 mL). Después, se agregó carbonato de cesio (621 mg, 1,905 mmol), posteriormente yoduro de tetrabutilamonio (45 mg, 0,122 mmol) y la mezcla de reacción se agitó durante 15 horas a 36 °C. Se monitoreó el progreso de la reacción mediante UPLC/MS. Después, se agregó una disolución saturada de NH4Cl (30 mL) y se extrajo con EtOAc/Hep (2:1, 150 mL), se lavó con salmuera (10 mL), se secó sobre sulfato de sodio y se concentró al vacío. El material bruto se purificó mediante cromatografía rápida (EtOAc/Hep 20 % a 50 %) para obtener 248 mg de (2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)(4-(((tercbutildimetilsilil)oxi)metil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001140141). 1H RMN (400 MHz) 5 ppm 7,28 (d, 2 H, J= 8,4 Hz), 7,20 (d, 2 H, J= 8,0 Hz), 4,73 (s, 2 H), 4,06 (s a, 2 H), 3,83 (dd, 2 H, J = 6,4, 5,6 Hz), 3,68- 3,56 (m, 12 H), 3,37 (dd, 2 H, J = 5,6, 5,2 Hz), 2,33 (s, 3 H), 1,14 (s a, 6 H), 0,93 (s, 9 H), 0,09 (s, 6 H).
Se disolvió (2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)(4-(((tercbutildimetilsilil)oxi)metil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001140141) (81 mg, 0,131 mmol) en una mezcla de metanol (5 mL) y agua (0,5 mL). Después, se agregó ácido acético (0,5 mL, 8,734 mmol) a la mezcla de reacción y se agitó durante 14 horas a 38 °C. La mezcla de reacción se enfrió hasta temperatura ambiente y el disolvente se retiró al vacío. El residuo se diluyó con EtOAc (30 mL), se lavó con agua (2 x 5 mL), NaHCO3 y salmuera (3 mL), se secó en sulfato de sodio y se concentró al vacío. El material bruto se purificó mediante cromatografía rápida (EtOAc/Hep 30 % a 90 %) para obtener 61,0 mg de (2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)(4-(hidroximetil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001140549). 1H RMN (400 MHz) 5 ppm 7,34 (d, 2 H, J = 8 , 8 Hz), 7,26 (d, 2 H, J = 8,0 Hz), 4,69 (d, 2 H, J= 4,4 Hz), 4,06 (s a, 2 H), 3,84 (dd, 2 H, J = 6,2, 6,2 Hz), 3,66-3,56 (m, 12 H), 3,37 (dd, 2 H, J = 5,2, 5,2 Hz), 2,33 (s, 3 H), 1,74 (s a, 1 H), 1,14 (s a, 6 H).
Se disolvió (2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)(4-(hidroximetil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001140549) (60 mg, 0,119 mmol) en Dc M (2 mL) y Py (0,019 mL, 0,239 mmol) y se enfrió hasta 0 °C. Después, se agregaron carbonocloridato de 4-nitrofenilo (38,5 mg, 0,191 mmol) en DCM (2 mL) y dimetilaminopiridina (DMAP) (2,9 mg, 0,024 mmol) y la mezcla de reacción se agitó durante 30 min a 0 °C. La mezcla de reacción se calentó lentamente hasta temperatura ambiente y se agitó hasta que se consumió el material de partida (aproximadamente 2,5 horas). Después, se retiró el disolvente al vacío y el residuo se purificó mediante cromatografía rápida (EtOAc/Hep 10 % a 35 %) para obtener 78 mg de (2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)(4-((((4-nitrofenoxi)carbonil)oxi)metil)fenil)carbamato de 2 -metil-2 -(metildisulfanil)propilo (ER-001M0550). 1H Rm N (400 MHz) 5 ppm 8,27 (dd, 2 H, J= 6 ,8 , 2,4 Hz), 7,41 (d, 2 H, J= 8 , 8 Hz), 7,37 (dd, 2 H, J= 7,2, 2,4 Hz), 7,33(d, 2 H, J= 8 , 8 Hz), 5,27 (s, 2 H), 4,08 (s a, 2 H), 3,85 (dd, 2 H, J = 5,8, 5,8 Hz), 3,66- 3,57 (m, 12 H), 3,36 (dd, 2 H, J = 5,2, 5,2 Hz), 2,33 (s a, 3 H), 1,19 (s a, 6 H).
Se colocó (2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)(4-((((4-nitrofenoxi)carbonil)oxi)metil)fenil)carbamato de 2-metil-2-(metildisulfanil)propilo (ER-001140550) (30 mg, 0,045 mmol) en DCM (3 mL, 46,625 mmol) en un matraz de 25 ml con nitrógeno y se enfrió hasta 0 °C. Se agregaron amina (40,8 mg, 0,049 mmol) en DCM (2 mL) y la base de Hunig (0,024 mL, 0,135 mmol), posteriormente DMAP (1,4 mg, 0,011 mmol). La mezcla de reacción después se calentó lentamente hasta temperatura ambiente, se agitó durante 3 horas, se concentró al vacío, y se purificó mediante cromatografía rápida (EtOAc/Hep 50 % a 100 %, posteriormente MeOH/EtOAc 3 % a 8 %) para obtener 45,0 mg de azida-PEG3-disulfuro-PAB-eribulina pura (ER-001237508). 1H RMN (400 MHz) 5 ppm 7,32 (d, 2 H, J= 8,0 Hz), 7,25 (d, 2 H, J = 7,2 Hz), 5,28 (dd, 1 H, J = 5,6, 5,6 Hz), 5,11-5,04 (m, 3 H), 4,93 (s, 1 H), 4,88 (s, 1 H), 4,81 (s, 1 H), 4,69 (dd, 1 H, J= 4,4, 4,4 Hz), 4,60 (dd, 1 H, J = 4,2, 4,2 Hz), 4,36 (s a, 1 H), 4,33(dd, 1 H, J = 4,0, 2,0), 4,29 (ddd, 1 H, J = 9,6, 4,4, 4,4 Hz), 4,18 (dd, 1 H, J = 6,4, 4,4 Hz), 4,14-4,04 (m, 3 H), 4,03 (dd, 1 H, J = 6,4, 4,4 Hz), 3,97-3,89 (m, 3 H), 3,84-3,78 (m, 3 H), 3,67-3,56 (m, 14 H), 3,42 (s, 3 H), 3,40-3,35 (m, 1 H), 3,37 (dd, 2 H, J = 5,2, 5,2 Hz), 3,27 (d, 1 H, J = 3,2 Hz), 3,20 (ddd, 1 H, J= 12,8, 6,0, 6,0 Hz), 2,91-2,83 (m, 2 H), 2,70 (dd, 1 H, J= 16,0, 10,0 Hz), 2,52-2,40 (m, 3 H), 2,35-2,13 (m, 9 H), 2,10-2,06 (m, 1 H), 2,01-1,89 (m, 4 H), 1,78-1,64 (m, 4 H), 1,60-1,52 (m, 4 H), 1,49-1,28 (m, 5 H), 1,22-1,07 (m, 6 H), 1,09 (d, 3 H, J= 6,0 Hz).
1.11 Preparación de Mal-PEG4-triazol-PEG3-disulfuro-PAB-eribulina (ER-001237504)

Una mezcla de azida (9,0 mg, 7,151 gmol) y 3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-N-(3,6,9,12-tetraoxapentadec-14-in-1-il)propanamida (6 , 8 mg, 0,018 mmol) en terc-butanol (1,5 mL) y agua (0,5 mL) se desgasificó durante 45 min. Después, se agregó yoduro de cobre sobre amberlist-21 (1,23 mmol/g, 10 mg) y se desgasificó durante 30 min adicionales. La mezcla de reacción se agitó a temperatura ambiente durante 18 horas y se monitorizó mediante UPLC/MS. El material de partida se consumió en gran medida y el producto deseado se mostró como un pico principal. La mezcla después se separó de la resina, y se purificó en HPLC (acetonitrilo/agua con ácido fórmico al 0,05 %) para obtener 1,5 mg de Mal-PEG4-triazol-PEG3-disulfuro-PAB-eribulina (ER-001237504). 1H RMN (400 MHz) 5 ppm 7,74 (s, 1 H), 7,32 (d, 2 H, J= 8,4 Hz), 7,27-7,25 (m, 2 H), 6,69 (s a, 2 H), 5,43 (dd, 1 H, J = 5,6, 5,6 Hz), 5,14-5,06 (m, 3 H), 4,95 (s, 1 H), 4,89 (s, 1 H), 4,82 (s, 1 H), 4,70 (dd, 1 H, J = 4,4, 4,4 Hz), 4,66 (s, 2 H), 4,62 (dd, 1 H, J = 4,4, 4,4 Hz), 4,52(dd, 1 H, J = 5,2, 5,2 Hz), 4,38-4,31 (m, 2 H), 4,30 (ddd, 1 H, J = 10,4, 4,0, 4,0 Hz), 4,20 (dd, 1 H, J = 6,4, 4,4 Hz), 4,16-4,05 (m, 3 H), 4,04 (dd, 1 H, J = 6,4, 4,4 Hz), 3,99-3,91 (m, 3 H), 3,87-3,80 (m, 6 H), 3,70-3,59 (m, 22 H), 3,53 (dd, 2 H, J= 5,2, 5,2 Hz), 3,44 (s, 3 H), 3,43-3,36 (m, 3 H), 3,29 (d, 1 H, J = 2,8 Hz), 3,18 (ddd, 1 H, J = 12,9, 6,2, 6,2 Hz), 2,92-2,84 (m, 2 H), 2,72 (dd, 1 H, J = 16,0, 10,0 Hz), 2,54-2,42 (m, 5 H), 2,37-1,90 (m, 19 H), 178-1,52 (m, 3 H), 1,50-1,14 (m, 16 H), 1,10 (d, 3 H, J= 6,0 Hz). LCMS (M+H)=1642,1.
1.12 Preparación de NHS-PEG3-triazol-PEG3-disulfuro-PAB-eribulina (ER-001244129)

Una mezcla de azida (9 mg, 7,151 gmol) y 3-(2-(2-(prop-2-in-1-iloxi)etoxi)etoxi)propanoato de 2,5-dioxopirrolidin-1-ilo (4,5 mg, 14,30 gmol) en terc-butanol (1 mL) y agua (0,5 mL) se desgasificó durante 45 min. Después, se agregó yoduro de cobre sobre amberlist-21 (1,23 mmol/g, 10 mg, 7,151 gmol) y se desgasificó durante 30 min adicionales. La mezcla de reacción se agitó a temperatura ambiente durante 18 horas y se monitorizó mediante UPLC/MS. El material de partida se consumió en gran medida y el producto deseado se mostró como un pico principal. La mezcla después se separó de la resina por filtración, se extrajo con DCM (15 mL), se lavó con salmuera (3 x 3 mL), se secó sobre sulfato de sodio y se concentró al vacío. El residuo (5 mg, 3,39 gmol) se azeotropeó con tolueno, se disolvió en THF (1 mL) y se enfrió hasta 0 °C. Se agregó DCC (4,2 mg, 0,02 mmol), posteriormente 1-hidroxipirrolidina-2,5-diona (2,2 mg, 0,019 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 18 horas. El material de partida se consumió en gran medida y el producto deseado se mostró como un pico principal, según se determinó mediante UPLC/MS. La mezcla de reacción después se concentró y se purificó en TLC preparativa (DCM//-propanol, 8 %) para proporcionar 2,5 mg de NHS-PEG3-triazol-PEG3-disulfuro-PAB-eribulina (ER-001244129) como un aceite incoloro. 1H RMN (400 MHz, CD2Cl2) 5 ppm 7,72 (s, 1 H), 7,32 (d, 2 H, J= 8 , 8 Hz), 7,25 (d, 2 H, J= 8 , 8 Hz), 5,08-5,04 (m, 3 H), 4,93 (s, 1 H), 4,85 (s, 1 H), 4,78 (s, 1 H), 4,64 (dd, 1 H, J= 4,4, 4,4 Hz), 4,58 (s, 2 H), 4,55 (dd, 1 H, J = 4,4, 4,4 Hz), 4,48 (dd, 2 H, J = 5,0, 5,0 Hz), 4,32 (d, 1 H, J = 6 , 6 Hz), 4,27-4,22 (m, 2 H), 4,14 (dd, 1 H, J = 6 ,6 , 4,8 Hz), 4,10-4,01 (m, 3 H), 4,00 (dd, 1 H, J = 6 ,8 , 4,4 Hz), 3,92-3,78 (m, 9 H), 3,65-3,53 (m, 19 H), 3,44-3,39 (m, 4 H), 3,37 (s, 3 H), 3,26 (d, 1 H, J = 3,2 Hz), 3,13 (ddd, 1 H, J = 12,4, 6,0, 6,0 Hz), 2,91 -2,73 (m, 11 H), 2,70-2,64 (m, 2 H), 2,54-2,41 (m, 3 H), 2,38-1,80 (m, 16 H), 1,74-1,52 (m, 3 H), 1,41-1,13 (m, 10 H), 1,07 (d, 3 H, J= 6,4 Hz). LCMS (M+H)=1572,3.
1.13 Preparación de azida-PEG3-sulfonamida-PAB-eribulina (ER-001138856)

Se disolvió 4-(((terc-butildimetilsilil)oxi)metil)anilina (315 mg, 1,327 mmol) en DCM (10 mL) y se enfrió hasta 0 °C. Después, se agregó piridina (0,268 mg, 3,317 mmol), posteriormente cloruro de 5-cianopiridina-2-sulfonilo (365 mg, 1,801 mmol) en DCM (10 mL) durante 15 min. La mezcla de reacción se calentó lentamente hasta temperatura ambiente durante 1 hora y se agitó durante 2 horas. La mezcla de reacción se diluyó con EtOAc (50 mL), se lavó con salmuera, se secó sobre sulfato de sodio y se concentró al vacío para obtener 610 mg (103 %) de N-(4-(((tercbutildimetilsilil)oxi)metil)fenil)-5-cianopiridina-2-sulfonamida (ER-001137670). El producto bruto fue razonablemente puro, aunque coloreado. 1H RMN (400 MHz) 5 ppm 8,94 (dd, 1 H, J = 1,8, 0,6 Hz), 8,10 (dd, 1 H, J = 8,4, 2,0 Hz), 7,99 (dd, 1 H, J = 8,0, 0,8 Hz), 7,18 (d, 2 H, J = 8,2 Hz), 7,15 (s a, 1 H), 7,11 (dd, 2 H, J= 6 ,8 , 0,8 Hz), 4,64 (s, 2 H), 0,90 (s, 9 H), 0,05 (s, 6 H).
Se disolvieron N-(4-(((terc-butildimetilsilil)oxi)metil)fenil)-5-cianopiridina-2-sulfonamida (ER-001137670) (105,0 mg, 0,26 mmol) y 4-metilbencenosulfonato de 2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etilo (143 mg, 0,383 mmol) en DMF (4 mL). Después, se agregó carbonato de potasio (K2CO3) (144 mg, 1,041 mmol), posteriormente yoduro de tetrabutilamonio
(19,2 mg, 0,052 mmol) y la mezcla de reacción se agitó durante 36 horas a 50 °C. Se monitoreó el progreso de la reacción mediante UPLC/MS. Después, se agregó una disolución saturada de NH4Cl (10 mL), se extrajo con EtOAc/Hep (2:1, 30 mL), se lavó con salmuera, se secó sobre sulfato de sodio y se concentró. El material bruto se purificó mediante cromatografía rápida (EtOAc/Hep 25 % a 80 %) para obtener 118,0 mg de N-(2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)-N-(4-(((terc-butildimetilsilil)oxi)metil)fenil)-5-cianopiridina-2-sulfonamida (ER-001138452) (75 %). 1H RMN (400 MHz) 5 ppm 8,99 (dd, 1 H, J = 1,8, 0,6 Hz), 8,08 (dd, 1 H, J = 8,2, 2,2 Hz), 7,86 (dd, 1 H, J = 8,0, 0,8 Hz), 7,24 (d, 2 H, J = 10 Hz), 7,09 (d, 2 H, J = 8 , 8 Hz), 4,69 (s, 2 H), 4,06 (dd, 2 H, J = 6,0, 6,0 Hz), 3,67 (dd, 2 H, J = 5,2, 5,2 Hz), 3,65 - 3,62 (m, 4 H), 3,58 (dd, 2 H, J = 6,2, 6,2 Hz), 3,56 - 3,53 (m, 4 H), 3,38 (dd, 2 H, J = 5,2, 5,2 Hz), 0,93 (s, 9 H), 0,08 (s, 6 H).
Se disolvió N-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etil)-N-(4-(((tercbutildimetilsilil)oxi)metil)fenil)-5-cianopiridina-2-sulfonamida (ER-001138452) (150 mg, 0,248 mmol) en metanol ( 6 mL). Después, se agregó agua (0,60 mL), posteriormente ácido acético (AcOH) (0,60 mL, 10,481 mmol). La mezcla de reacción se calentó lentamente hasta 38 °C, y se agitó durante 14 horas. Se retiró gran parte del solvente al vacío. El residuo se diluyó con EtOAc (30 mL), se lavó con agua (2 x 5 mL), NaHCO3 y salmuera, se secó en sulfato de sodio y se concentró al vacío. El material bruto se purificó mediante cromatografía rápida (EtOAc/Hep 35 % a 90 %) para obtener 105,0 mg de N-(2-(2-(2-(2-azidoetoxi)etoxi) etoxi)etil)-5-ciano-N-(4-(hidroximetil)fenil)piridina-2-sulfonamida (ER-001138455) (84 %). 1H r Mn (400 MHz) 5 ppm 8,99 (d, 1 H, J = 1,2 Hz), 8,09 (dd, 1 H, J = 8,4, 2,0 Hz), 7,88 (dd, 1 H, J = 8,4, 0,8 Hz), 7,30 (d, 2 H, J = 8 , 8 Hz), 7,15 (d, 2 H, J = 8,4 Hz), 4,67 (s, 2 H), 4,06 (dd, 2 H, J = 6,2, 6,2 Hz), 3,66 (dd, 2 H, J = 5,0, 5,0 Hz), 3,65 - 3,58 (m, 6 H), 3,55 - 3,51 (m, 4 H), 3,38 (dd, 2 H, J = 5,2, 5,2 Hz.
Se disolvió N-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etil)-5-ciano-N-(4-(hidroximetil)fenil)piridina-2-sulfonamida (ER-001138455) (45 mg, 0,092 mmol) en DCM (3 mL) y se enfrió hasta 0 °C después de la adición de piridina (0,015 mL, 0,183 mmol). Después, se agregó carbonocloridato de 4-nitrofenilo (20,3 mg, 0,101 mmol) en DCM (2 mL) y DMAP (2,3 mg, 0,018 mmol). La mezcla de reacción se calentó lentamente hasta temperatura ambiente y se agitó durante 2 horas. La UPLC/MS indicó que permanecía parte del material de partida. La mezcla de reacción después se concentró al vacío y se purificó mediante cromatografía rápida (EtOAc/Hep 12 % a 40 %) para obtener 35 mg de (4-nitrofenil) carbonato de 4-((N-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etil)-5-cianopiridina)-2-sulfonamido)bencilo (ER-001235286) (58 %) y 20 mg de material de partida. 1H RMN (400 MHz) 5 ppm 8,99 (d, 1 H, J = 0,8 Hz), 8,27 (dd, 2 H, J = 9,2, 2,0 Hz), 8,12 (dd, 1 H, J = 7,6, 2,0 Hz), 7,92 (d, 1 H, J = 8,4 Hz), 7,38 (d, 4 H, J = 9,6 Hz), 7,26 (d, 2 H, J = 8 , 8 Hz), 5,45 (s, 2 H), 4,06 (dd, 2 H, J = 5,8, 5,8 Hz), 3,67 - 3,58 (m, 8 H), 3,58 - 3,50 (m, 4 H), 3,38 (dd, 2 H, J = 6,1, 6,1 Hz).
Se colocó (4-nitrofenil) carbonato de 4-(N-(2-(2-(2-(2-azidoetoxi)etoxi)etoxi)etil)-5-cianopiridina-2-sulfonamido)bencilo (ER-001235286) (35,0 mg, 0,053 mmol) en un matraz de 25 mL con nitrógeno y se enfrió hasta 0 °C. Después, se agregaron amina (48,5 mg, 0,059 mmol) en DCM (3 mL, 46,625 mmol) y la base de Hunig (0,037 mL, 0,214 mmol), posteriormente DMAP (2,61 mg, 0,021 mmol). La mezcla de reacción se agitó durante 30 min a 0 °C y después durante 6 horas adicionales a temperatura ambiente. La mezcla de reacción se concentró al vacío y se purificó mediante cromatografía rápida (EtoAc/Hep 50 % a 10 0 %, posteriormente MeOH/EtOAc 3 % a 8 %) para obtener 61,0 mg de azida-PEG3-sulfonamida-PAB-eribulina pura (e R-001138856). 1H RMN (400 MHz) 5 ppm 8,98 (d, 1 H, J = 1,2 Hz), 8,10 (dd, 1 H, J = 8,2, 1, 8 Hz), 7,87 (d, 1 H, J = 8,0 Hz), 7,26 (d, 2 H, J = 6 , 8 Hz), 7,13 (d, 2 H, J = 8,4 Hz), 5,29 (dd, 1 H, J = 5,6, 5,6 Hz), 5,08-5,00 (m, 3 H), 4,92 (s, 1 H), 4,87 (s, 1 H), 4,80 (s, 1 H), 4,68 (dd, 1 H, J = 4,6, 4,6 Hz), 4,59 (dd, 1 H, J = 4,6, 4,6 Hz), 4,38-4,30 (m, 2 H), 4,28 (ddd, 1 H, J= 10,4, 4,0, 4,0, Hz), 4,17 (dd, 1 H, J= 6,2, 4,6 Hz), 4,13 4,01 (m, 4 H), 3,97-3,88 (m, 3 H), 3,82-3,78 (m, 1 H), 3,67-3,50 (m, 15 H), 3,41 (s, 3 H), 3,40-3,33 (m, 1 H), 3,37 (dd, 2 H, J = 4,8, 4,8 Hz), 3,27 (d, 1 H, J = 3,2 Hz), 3,15 (ddd, 1 H, J = 12,8, 6,4, 6,4 Hz), 2,90-2,82 (m, 2 H), 2,70 (dd, 1 H, J = 16,0, 10,0 Hz), 2,51-2,40 (m, 3 H), 2,34-2,13 (m, 7 H), 2,10-2,05 (m, 1 H), 1,99-1,88 (m, 4 H), 1,78-1,64 (m, 5 H), I , 62-1,52 (m, 2 H), 1,50-1,29 (m, 4 H), 1,08 (d, 3 H, J= 6 , 8 Hz).
1.14 Preparación de Mal-PEG4-triazol-PEG3-sulfonamida-PAB-eribulina (ER-001237505)

Una mezcla de azida (10 mg, 8,023 gmol) y 3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-N-(3,6,9,12-tetraoxapentadec-14-in-1-il)propanamida (9,20 mg, 0,024 mmol) en terc-butanol (2,1 mL) y agua (0,7 mL) se desgasificó durante 45 min.
Después, se agregó yoduro de cobre sobre amberlist-21 (1,23 mmol/g, 15 mg) y se desgasificó durante 30 min adicionales. La mezcla de reacción se agitó a temperatura ambiente durante 18 horas y se monitorizó mediante UPLC/MS. El material de partida se consumió en gran medida y el producto deseado se mostró como un pico principal.
La mezcla de reacción después se separó de la resina y se purificó en TLC preparativa (DCM/metanol, 7 %) para proporcionar 5,5 mg de Mal-PEG4-triazol-PEG3-sulfonamida-PAB-eribulina (ER-001237505). 1H RMN (400 MHz, CD2Cl2) 5 ppm 9,01 (s, 1 H), 8,15 (dd, 1 H, J = 8,0, 1,8 Hz), 7,87 (d, 1 H, J = 8,0 Hz), 7,75 (s, 1 H), 7,28 (d, 2 H, J = 8,0 Hz), 7,14 (d, 2 H, J = 8,4 Hz), 6,68 (s, 2 H), 6,47 (s a, 1 H), 5,44 (s a, 1 H), 5,10-5,02 (m, 3 H), 4,94 (s, 1 H), 4,86 (s, 1 H), 4,80 (s, 1 H), 4,68 (dd, 1 H, J = 4,4, 4,4 Hz), 4,59 (s, 2 H), 4,56 (dd, 1 H, J = 4,4, 4,4 Hz), 4,51 (dd, 2 H, J = 5,2, 5,2, Hz), 4,34(d, 1 H, J = 7,6, Hz), 4,30-4,23 (m, 2 H), 4,19 - 4,14 (m, 2 H), 4,08 (dd, 1 H, J = 4,0, 4,0 Hz), 4,03 -3,98 (m, 2 H), 3,94 - 3,72 (m, 8 H), 3,68 - 3,46 (m, 28 H), 3,38 (s, 3 H), 3,38 - 3,33 (m, 3 H), 3,27 (d, 1 H, J = 3,2 Hz), 3,16 - 3,02 (m, 2 H), 2,90 - 2,81 (m, 2 H), 2,68 (dd, 1 H, J= 16,2, 9,8 Hz), 2,54-2,40 (m, 7H), 2,40-1,8 (m, 11 H), 1,80 1,50 (m, 3 H), 1,48-1,25 (m, 3 H), 1,09 (d, 3 H, J = 6,4 Hz). LCMS (M+H)=1630,0.
1.15 Preparación de NHS-PEG3-triazol-PEG3-sulfonamida-PAB-eribulina (ER-001244623)

Una mezcla de azida (14 mg, 0,011 mmol) y 3-(2-(2-(prop-2-in-1-iloxi)etoxi)etoxi)propanoato de 2,5-dioxopirrolidin-1-ilo (8,80 mg, 0,028 mmol) en terc-butanol (2 mL) y agua (1 mL) se desgasificó durante 45 min. Después, se agregó yoduro de cobre sobre amberlist-21 (1,23 mmol/g, 20 mg) y se desgasificó durante 30 min adicionales. La mezcla de reacción se agitó a temperatura ambiente durante 18 horas y se monitorizó mediante UPLC/MS. El material de partida se consumió en gran medida y el producto deseado se mostró como un pico principal. La mezcla de reacción después se separó de la resina mediante extracción con DCM (2 x 10 mL). La capa de DCM se lavó con salmuera (4 x 5 mL), se secó en sulfato de sodio y se concentró para proporcionar el producto deseado (que se usó en la siguiente etapa sin purificación adicional).
El ácido bruto (15,0 mg, 10,255 pmol) se disolvió en THF (1,5 mL) y se enfrió hasta 0 °C. Después se agregó DCC (15,2 mg, 0,074 mmol), posteriormente 1-hidroxipirrolidina-2,5-diona (8,3 mg, 0,072 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 18 horas. La UPLC/MS indicó que el material de partida se consumió en gran medida y el producto deseado se mostró como un pico principal. La mezcla de reacción se concentró y se purificó en TLC preparativa (DCM//'-propanol, 8 %) para proporcionar 2,5 mg de NHS-PEG3-triazol-PEG3-sulfonamida-PAB-eribulina (ER-001244623). 1H RMN (400 MHz, CD2Ch) 5 ppm 9,00 (s, 1 H), 8,12 (d, 1 H, J= 8,4 Hz), 8,00 (d, 1 H, J = 8,0 Hz), 7,72 (s, 1 H), 7,26 (d, 2 H, J = 8,0 Hz), 7,12 (d, 2 H, J = 8,0 Hz), 5,37 (s a, 1 H), 5,08-5,02 (m, 3 H), 4,93 (s, 1 H), 4,85 (s, 1 H), 4,78 (s, 1 H), 4,66-4,62 (m, 1 H), 4,58-4,56 (m, 4 H), 4,33 (d, 1 H, J = 10,8 Hz), 4,29-4,21 (m, 2 H), 4,10-3,96 (m, 4 H), 3,93-3,76 (m, 6 H), 3,74-3,44 (m, 27 H), 3,36 (s, 3 H), 3,34-3,24 (m, 2 H), 3,15-3,06 (m, 1 H), 2,97 (s a, 1 H), 2,90-2,78 (m, 8 H), 2,74-2,08 (m, 13 H), 2,05 -1,78 (m, 5 H), 1,73-1,50 (m, 2 H), 1,41 -1,25 (m, 4 H), 1,07 (d, 3 H, J = 6,0 Hz). LCMS (M+H)=1560,0.
1.16 Preparación de Mal-PEG2-eribulina
Se disolvió eribulina (5 mg, 7 pmol) en DMF (0,5 mL) y se mezcló con maleimido-PEG2-NHS (5 mg, 14 pmol; Broadpharm, núm. de cat. BP-21680) y la base de Hunig (2,4 pL, 14 pmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción después se purificó mediante HPLC (gradiente de aguaacetonitrilo 30-70 % que contenía ácido fórmico al 0,1 %). El eluyente se recogió por masa y se liofilizó hasta secarse. El rendimiento final fue 3,7 mg (3,8 pmol, 54 %). La masa exacta previsto fue 968,5 Da. La masa medida fue 969,6 Da [M+H].
1.17 Preparación de Mal-PEG4-eribulina
Se disolvió eribulina (5 mg, 7 pmol) en DMF (0,5 mL) y se mezcló con maleimido-PEG4-NHS (6,2 mg, 14 pmol; Broadpharm, núm. de cat. BP-20554) y la base de Hunig (2,4 pL, 14 pmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción después se purificó mediante HPLC (gradiente de aguaacetonitrilo 30-70 % que contenía ácido fórmico al 0,1 %). El eluyente se recogió por masa y se liofilizó hasta secarse. El rendimiento final fue 3,7 mg (3,5 pmol, 50 %). La masa exacta previsto fue 1056,5 Da. La masa medida fue 1057,7 Da [M+H].
1.18 Preparación de azido-PEG2-eribulina
Se disolvió eribulina (5 mg, 7 gmol) en DMF (0,5 mL) y se mezcló con azido-PEG2-NHS (4,2 mg, 14 gmol; Broadpharm, núm. de cat. BP-20524) y la base de Hunig (2,4 gL, 14 gmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción después se purificó mediante HPLC (gradiente de agua-acetonitrilo 30-70 % que contenía ácido fórmico al 0,1 %). El eluyente se recogió por masa y se liofilizó hasta secarse. El rendimiento final fue 2,2 mg (2,4 gmol, 34 %). La masa exacta previsto fue 914,5 Da. La masa medida fue 915,7 Da [M+H].
1.19 Preparación de azido-PEG4-eribulina
Se disolvió eribulina (5 mg, 7 gmol) en DMF (0,5 mL) y se mezcló con azido-PEG4-NHS (5,5 mg, 14 gmol; Broadpharm, núm. de cat. BP-20518) y la base de Hunig (2,4 gL, 14 gmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción después se purificó mediante HPLC (gradiente de agua-acetonitrilo 30-70 % que contenía ácido fórmico al 0,1 %). El eluyente se recogió por masa y se liofilizó hasta secarse. El rendimiento final fue 3,0 mg (3,0 gmol, 43 %). La masa exacta previsto fue 1002,5 Da. La masa medida fue 1003,7 Da [M+H].
1.20 Preparación de azido-PEG4-Val-Cit-PAB-eribulina
Se disolvió eribulina (15 mg, 21 gmol) en DMF (1,5 mL) y se mezcló bien. Después, se agregaron la base de Hunig (5,5 gL, 32 gmol) y Fmoc-VCP-PNP (24 mg, 22 gmol; Levena Biopharma, núm. de cat. VC1003). La mezcla de reacción se agitó a temperatura ambiente durante la noche (16 horas). Después de que se completó la reacción, se agregó dietilamina (20 gL, 0,21 mmol) a la mezcla de reacción y se agitó durante 2 horas a temperatura ambiente para retirar el grupo protector Fmoc. La reacción de desprotección se monitoreó con el uso de un espectrómetro de masas Waters SQD. Después de que se completó la reacción, la mezcla de reacción se transfirió a un tubo de microcentrífuga de 1,5 mL prepesado. El disolvente se evaporó al vacío al usar un concentrador Centrivap refrigerado con la temperatura ajustada a 30 °C. El rendimiento fue 16 mg (14 gmol) de NH2-Val-Cit-pAB-eribulina bruta (masa exacta 1134,6 Da, 67 % de rendimiento).
Se disolvió NH2-Val-Cit-pAB-eribulina (16 mg, 14,1 gmol) en DMF (1,5 mL). Después, se agregaron la base de Hunig (7,2 gL, 41 gmol) y azido-PEG4-NHS (11 mg, 28,2 gmol). La mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción después se purificó mediante HPLC (gradiente de agua-acetonitrilo 48-72 % que contenía ácido fórmico al 0,1 %). El eluyente se recogió a m/z 1409, y se liofilizó para proporcionar azido-PEG4-Val-Cit-PAB-eribulina (masa exacta 1407,7 Da). Se obtuvo 13 mg (9,2 gmol) de azido-PEG4-Val-Cit-PAB-eribulina (65 % de rendimiento de la etapa, 44 % del global).
EJEMPLO 4
1. Materiales y métodos
Todos los reactivos usados se obtuvieron de proveedores comerciales con grado apto para investigación o superior, a menos que se indique de cualquier otra manera.
1.1 Anticuerpos
El MORAb-003 (anti-receptor alfa de folato humano humanizado, 25 mg/mL) y el MORAb-009 (anti-mesotelina humana quimérico de ratón-humano, 25 mg/mL) usados en los siguientes estudios fueron del lote núm. NB02962-19 y el lote núm. 030A14, respectivamente. El trastuzumab se obtuvo comercialmente (Clingen) y fue del lote núm. 503345.
Los anticuerpos anti-mesotelina humana quimérico de conejo-humano y humanizado que tienen una cisteína no apareada en LCcys80 (Tabla 1) se expresaron en células 293F transitoriamente o como concentrados seleccionados establemente. El medio acondicionado se purificó y descisteiniló como se describe en la sección 1.4.1.2.1.
1.2 Citotoxinas
Los compuestos de eribulina conjugables se sintetizaron como se describe en el Ejemplo 3 (Tabla 46). Las disoluciones madre (10 mM) se prepararon en DMSO y se almacenaron a -20 °C hasta el uso.
1.3 Líneas celulares tumorales
Las líneas celulares tumorales humanas usadas en los análisis de ADC de MORAb-003, MORAb-009 y trastuzumab preparados con compuestos de maleimido/succinimida (OSu)/azido-enlazador-eribulina (Tabla 46) incluyen IGROV1 (carcinoma de ovario humano, FRhi, MSLNneg), NCI-H2110 (carcinoma de pulmón no microcítico humano, FRmed, MSLNmed), A431 (FRneg, MSLNneg), NCI-N87-luc (carcinoma gástrico humano, FRo MSLNmed, her2hi), NUGC3 (adenocarcinoma gástrico humano, FRneg, MSLNneg, her2neg), ZR75 (carcinoma ductal de mama humano, FRneg, MSLNneg, her2med) y BT-474 (carcinoma ductal de mama humano, FRneg, MSLNneg, her2hi). Las líneas celulares tumorales humanas usadas en los análisis de anticuerpos anti-mesotelina humana LCcys80 quimérico de conejohumano y humanizado conjugados con MAL-PEG2-Val-Cit-PAB-eribulina (ER-001159569) fueron A3 (A431 transfectada establemente con mesotelina humana, MSLNhi), OVCAR3 (carcinoma de ovario humano, MSLNhi), HEC-251 (endometroide humano, MSLNmed), H226 (mesotelioma de células escamosas de pulmón humano, MSLNto) y A431 original (MSLNneg). Todas las líneas celulares usadas se obtuvieron directamente de la Colección
Estadounidense de Cultivos Tipo (ATCC), con las excepciones de IGROV1 (obtenida del Instituto Nacional del Cáncer, con permiso) y A3 (generada en Morphotek a partir de A431 original).
1.4 Conjugación de anticuerpo-fármaco
1.4.1 Conjugación basada en cisteína al usar maleimidas
1.4.1.1 Conjugación con disulfuros intercatenarios
1.4.1.1.1 Reducción parcial
Se intercambió el tampón de MORAb-003 y MORAb-009 a disolución salina tamponada con fosfato de Dulbecco (DPBS), y después se concentró hasta 20 mg/mL al usar concentración centrífuga. Se agregó un volumen igual de 270 pM tris(2-carboxietil)fosfina (TCEP) en 1X DPBS con EDTA 2 mM y la reducción se llevó a cabo mediante mezcla suave durante 80 min a temperatura ambiente. El trastuzumab se redujo parcialmente de manera similar, excepto que la reducción se llevó a cabo mediante mezcla suave durante 40 min a temperatura ambiente.
1.4.1.1.2 Conjugación
El compuesto de maleimido-enlazador-eribulina (en DMSO) se conjugó con los anticuerpos parcialmente reducidos en una relación molar de 1:6 (mAb:compuesto). El compuesto se agregó a propilenglicol al 50 % en DPBS y se mezcló bien. Después, se agregó un volumen igual de anticuerpo parcialmente reducido y se mezcló suavemente (concentración final de propilenglicol de 25 %). La conjugación procedió durante 3,5 a 4 horas a temperatura ambiente.
1.4.1.2 Conjugación con LCcys80
1.4.1.2.1 Descisteinilación
Al usar una ÁKTA Explorer (GE Healthcare), se equilibró una columna de proteína A (GE Healthcare) con 10 volúmenes de columna (CV, por sus siglas en inglés) de fosfato de sodio 20 mM, EDTA 10 mM, pH 7,2 (tampón de equilibrio). Después se cargó el medio acondicionado con posterior lavado del material no unido con 10 CV de tampón de equilibrio. La columna se lavó con 16 CV de fosfato de sodio 20 mM, EDTA 10 mM, cisteína 5 mM, pH 7,2 a 0,5 mL/min durante 16 horas para retirar el grupo de protección. La columna después se lavó con 60 CV de Tris 20 mM, pH 7,5 a 0,5 mL/min durante 60 horas. El anticuerpo descisteinilado se eluyó al usar 5 CV de glicina 0,1 M, pH 2,9 y se neutralizó inmediatamente al usar un volumen al 5 % de Tris 2 M, pH 9,0. Las fracciones que contenían los anticuerpos se agruparon y dializaron en DPBS al usar un MWCO 20K Slide-A-Lyzer (Thermo Fisher).
1.4.1.2.2 Conjugación
El anticuerpo descisteinilado se llevó hasta 5,0 mg/mL en DPBS, EDTA 1mM y se preparó propilenglicol al 50 % en DSBS, EDTA 1 mM. Se agregó MAL-PEG2-Val-Cit-PAB-eribulina (ER-001159569) (12 mM en DMSO) al propilenglicol al 50 % y se mezclaron exhaustivamente. Después, se agregó un volumen igual de anticuerpo descisteinilado en una relación molar de 1:4 (mAb:compuesto) y se mezcló suavemente. La conjugación procedió durante 3,5 a 4 horas a temperatura ambiente.
1.4.2 Conjugación basada en amina al usar succinimidas
1.4.2.1 Conjugación
El anticuerpo (MORAb-003 o MORAb-009, no reducido) se llevó hasta 10,0 mg/mL en bicarbonato de sodio 0,1 M, pH 8.3, se preparó propilenglicol al 50 % en bicarbonato de sodio 0,1 M, pH 8,3. Se agregó succinimida (OSu)-enlazadoreribulina (en DMSO) al propilenglicol al 50 % y se mezclaron exhaustivamente. Después, se agregó un volumen igual de anticuerpo en una relación molar de 1:4 (mAb:compuesto) y se mezcló exhaustivamente. La conjugación procedió durante 1 hora a temperatura ambiente. La reacción de conjugación se inactivó con la adición de un volumen 1:20 de Tris 1 M, pH 8,0, y el ADC se purificó como se describe en la sección 1.4.4.
1.4.3 Conjugación basada en amina de dos etapas al usar química alquina-azida promovida por cepa (SPAAC)
1.4.3.1 Derivación de dibencilciclooctina (DBCO)
El anticuerpo (MORAb-003 o MORAb-009, no reducido) se llevó hasta 10,0 mg/mL en bicarbonato de sodio 0,1 M, pH 8.3, se preparó propilenglicol al 50 % en bicarbonato de sodio 0,1 M, pH 8,3. Se agregó NHS-PEG4-DBCO (Click Chemistry Tools, 50 mM en DMSO) al propilenglicol al 50 % y se mezclaron exhaustivamente. Después, se agregó un volumen igual de anticuerpo en una relación molar de 1:4 (mAb:compuesto) y se mezcló exhaustivamente. La conjugación procedió durante 1 hora a temperatura ambiente. El NHS-PEG4-DBCO sin reaccionar se retiró, como se describe en la sección 1.4.4.
1.4.3.2 Conjugación
Se preparó propilenglicol al 50 % en DPBS. Se agregaron los compuestos de azido-enlazador-eribulina al propilenglicol al 50 % y se mezclaron exhaustivamente. Después, se agregó un volumen igual de MORAb-003 o MORAb-009 modificado con DBCO a la mezcla en una relación molar de 1:4 (mAb:compuesto) y se mezcló exhaustivamente. Se dejó que la conjugación de SPAAC procediera durante la noche a temperatura ambiente. El NHS-PEG4-DBCO sin reaccionar se retiró, como se describe en la sección 1.4.4.
1.4.4 Purificación
El anticuerpo conjugado se purificó al usar columna(s) de desalación HiTrap (GE Healthcare). Se llevó a cabo cromatografía en una cromatografía de líquidos de proteína rápida (FPLC) (GE Healthcare), al usar 1X DPBS como tampón de ejecución, para retirar la maleimido/OSu/azido-enlazador-eribulina y el propilenglicol. El contenido de proteína final se determinó mediante un ensayo de BCA como se describe en la sección 1.3.1 del Ejemplo 1.
1.5 Caracterización biofísica
1.5.1 Análisis por SEC-HPLC
La aglomeración de los ADC se analizó mediante cromatografía de líquidos de alto rendimiento-exclusión por tamaño (SEC-HPLC) al usar un HPLC Agilent 1260. El ADC se diluyó hasta 1 mg/mL en DPBS. El ADC (10 pL) después se inyectó en una columna de guarda Advanced SEC 300A (4,6 mm x 3,5 cm, tamaño de poro 2,7 pm, Agilent), posteriormente una columna AdvancedBio 300A (4,6 mm x 30 cm, tamaño de poro 2,7 pm). El ADC se eluyó desde la columna con fosfato de sodio 0,1 M que contenía NaCl 0,15 M e IPA al 5 %, pH 7,4, a una velocidad de flujo de 0,25 mL/min durante 28 min. Todos los datos se analizaron al usar el programa informático Agilent ChemStation. El porcentaje de aglomeración se calculó como [PAaglomerado/PAtotal]*100, donde PA = área de pico integrada.
1.5.2 Análisis por HIC-HPLC de la relación entre fármaco y anticuerpo (DAR)
Se analizó la DAR al usar HPLC de interacción hidrófoba (HIC-HPLC). Las muestras se inyectaron en una TSKgel® Butil-NP5, 4,6 mm ID x 3,5cm, columna no porosa de tamaño 2,5 pM (Tosol Bioscience) y se eluyeron con un equilibrio de 3 min en 100 % de fase móvil A, un gradiente de 15 min (0-100 % de B), una retención de 5 min en 100 % de B, un cambio de 1 min a 100 % de A y un reequilibrio de 5 min en 100 % de fase móvil A, a 0,7 mL/min. La fase móvil A fue fosfato de sodio 25 mM, sulfato de amonio 1,5 M, pH 7,0. La fase móvil B fue fosfato de sodio 25 mM, isopropanol al 25 %, pH 7,0. La detección se llevó a cabo a 280 nm (referencia de 320 nm). La DAR se determinó mediante la fórmula:
[AUC+i 2(AUC+2) 3(AUC+3) ... n(AUC+n)]/2AUCtot]
donde AUC+1 es el área bajo la curva para el pico de anticuerpo correspondiente al ADC conjugado con una citotoxina, AUC+2 es el área bajo la curva para el pico de anticuerpo correspondiente al ADC conjugado con dos citotoxinas, etc. XAUCtot es el área bajo la curva combinada para todos los picos.
1.5.3 Análisis de DAR por HIC-HPLC
La DAR también se analizó al usar un método de LC-MS con un Waters Alliance HPLC con detección SQD/PDA. Las muestras se inyectaron en una columna Proteomix RP-1000 (5 pM, 1000A, 4,6 mm x 15 cm, Sepax) a 65 °C, y se eluyeron con un equilibrio de 3 min en 25 % de B, un gradiente lineal de 27 min de 25 %-55 % de B, una retención de 5 min a 55 % de B, un cambio de 1 min a 90 % de B, una retención de 5 min a 90 % de B, un cambio de 1 min nuevamente a 25 % de B, y un reequilibrio de 5 min a 25 % de B. La fase móvil A fue TFA al 0,1 % en agua y la fase móvil B fue TFA al 0,1 % en acetonitrilo. La elución después se dividió (10:1) en los detectores PDA y SQD. El detector SQD se configuró con ES positiva, voltaje de capilar a 3,2 kV, voltaje de cono a 40 V, extractor a 3 V y lente de RF a 0,2 V, temperatura de fuente a 150 °C y temperatura de desolvatación a 250 °C. Los datos de masa se adquirieron a 200-2000m/z durante 40 min, en modo continuo, tiempo barrido 1 segundo. Los datos se analizaron y deconvolucionar fuera de línea al usar MassLynx y MaxEnt1. La DAR se calculó al usar la fórmula:
2[[AUC lc+i 2(AUClc+2) 3(AUClc+3) ... n(AUCLC+n)]/2lLctot]+
2[[AU C hc+i 2(AUC hc+2) 3(AUChc+3) ... n(AUCHC+n)]/2AUCHctot]
donde AUClc+1 es el área bajo la curva del pico de cadena ligera conjugada con una citotoxina, AUClc+2 es el área bajo la curva del pico de cadena ligera conjugada con dos citotoxinas, etc. AUChc es el área bajo la curva de las cadenas pesadas correspondientes, y XAUCLctot y XAUCHctot son el área bajo la curva combinada de todas las cadenas ligeras y cadenas pesadas no conjugadas y conjugadas, respectivamente.
1.5.4 Análisis de DAR por UPLC/ESI-MS de ADC de LCcys80
Se redujo el ADC (1 mg/mL) mediante la adición de DTT hasta una concentración final de 20 mM, con posterior incubación a 60 °C durante 3 min. Las muestras después se analizaron al usar una cromatografía de líquidos de rendimiento ultra alto Waters Acquity y espectrómetro de masas Premier Q-Tof. Las muestras (0,5-2 pg cada una) se
inyectaron en una columna de micro desalación MassPrep a 65 °C, se eluyeron desde la columna con un equilibrio de 5 min en 95 % de fase móvil A, un gradiente de 10 min (5-90 % B) y un reequilibrio de 10 min en 95 % de fase móvil A, a 0,05 mL/min. La fase móvil A fue ácido fórmico al 0,1 % en agua. La fase móvil B fue ácido fórmico al 0,1 % en acetonitrilo. El espectrómetro de masas Q-Tof se ejecutó en modo V de ion positivo con detección en el intervalo de 500-4000 m/z. Los parámetros de la fuente fueron los siguientes: voltaje capilar, 2,25 kV (anticuerpo intacto)-2,50 kV (anticuerpo reducido); voltaje de cono de muestreo, 65,0 V (anticuerpo intacto) o 50,0 V (anticuerpo reducido); temperatura de fuente, 105 °C; temperatura de desolvatación 250 °C; flujo de gas de desolvatación, 550 L/h. El pico de proteína de cadena ligera se deconvolucionó al usar la función MassLynx MaxEnt 1. Las intensidades relativas de las masas de cadena ligera no conjugadas y con conjugación simple se usaron para calcular la DAR global al usar la fórmula:
2 [ [ . C - i / X [ . C , o t ]
donde LC+1 es la intensidad de masa de la cadena ligera conjugada con una citotoxina y £LCtot es las intensidades combinadas de cadena ligera no conjugada y conjugada.
1.6 Caracterización de unión
1.6.1 BIAcore
Las concentraciones de anticuerpo se ajustaron hasta 2 qg/mL en tampón HBS-P+ (GE Healthcare). Los anticuerpos no modificados o ADC, se inyectaron en un sensor anti-IgG humana en un BIAcore T100 (GE Healthcare) durante 1 min a una velocidad de flujo de 10 qL/min. Para registrar la asociación del antígeno con al anticuerpo capturado, se inyectó una serie de concentraciones crecientes de antígeno durante 300 seg a una velocidad de flujo de 30 qL/min. Para los anticuerpos anti-mesotelina, el intervalo de concentraciones fue 10 nM - 0,041 nM. Para los ADC MORAb-003 y MORAb-009, el intervalo de concentraciones fue 100 nM - 0,41 nM. La disociación del antígeno se monitorizó durante 30 min a la misma velocidad de flujo. La superficie del sensor se regeneró al inyectar MgCl23 M durante 2 x 30 seg a una velocidad de flujo de 30 qL/min. Los sensogramas se analizaron con el programa informático Biacore T100 Evaluation al usar un modelo de unión de Langumuir a 1:1.
1.6.2 ELISA - Receptor alfa de folato
El receptor alfa de folato humano recombinante se diluyó hasta 115 ng/mL en tampón de recubrimiento (tampón de carbonato-bicarbonato 50 mM, pH 9,6) y se recubrió sobre placas negras de 96 pocillos Maxisorp (Thermo, núm. de cat. 43711, 100 qL/pocillo) a 4 °C, durante la noche. La disolución de recubrimiento se descartó y las placas se lavaron tres veces al usar 1X PBS con tampón Tween-20 al 0.05 % (PBST). Las placas se bloquear en 300 qL de tampón de bloqueo (BSA al 1 % en PBST) a temperatura ambiente durante 2 horas en un agitador orbital. MORAb-003 y los ADC de MORAb-003 se diluyeron hasta 1000 ng/mL en tampón de bloqueo, después se diluyeron en serie 2 veces para obtener un intervalo de 1000 ng/mL a 0,98 ng/mL. El tampón de bloqueo se descartó y se agregó 100 qL/pocillo de anticuerpo diluido a las placas. Las placas se incubaron a temperatura ambiente durante 2 horas en un agitador orbital. La disolución de anticuerpo se descartó y las placas se lavaron tres veces al usar PBST. Se agregó 100 qL/pocillo de disolución de anti-IgG humana de cabra (H+L)-HRP (dilución 1:10.000 en tampón de bloqueo) a las placas y las placas se incubaron a temperatura ambiente durante 1 hora en un agitador orbital. La disolución de anticuerpo secundario se descartó y las placas se lavaron tres veces al usar PBST. Se agregó 100 qL/pocillo de disolución de trabajo de sustrato de peroxidasa fluorógena QuantaBlu (Thermo, núm. de cat. 15169) a las placas y las placas se incubaron a temperatura ambiente durante 30 min. Se leyó la fluorescencia con excitación 325 nm/emisión 420 nm al usar un SpectraMax M5 (Molecular Devices). Los datos se analizaron al usar el programa informático SoftMaxPro 5.4.2 con ajuste de 4 parámetros.
1.6.3 ELISA - Mesotelina
La mesotelina humana recombinante se diluyó hasta 1 qg/mL en tampón de recubrimiento (tampón de carbonatobicarbonato 50 mM, pH 9,6) y se recubrió sobre placas negras de 96 pocillos Maxisorp (Thermo, núm. de cat. 43711, 100 qL/pocillo) a 4 °C, durante la noche. La disolución de recubrimiento se descartó y las placas se lavaron tres veces al usar 1X PBS con tampón Tween-20 al 0.05 % (PBST). Las placas se bloquear en 300 qL de tampón de bloqueo (BSA al 1 % en PBST) a temperatura ambiente durante 2 horas en un agitador orbital. MORAb009 y los ADC de MORAb-009 se diluyeron hasta 1000 ng/mL en tampón de bloqueo, después se diluyeron en serie 2,5 veces para obtener un intervalo de 1000 ng/mL a 0,105 ng/mL. El tampón de bloqueo se descartó y se agregó 100 qL/pocillo de anticuerpo diluido a las placas. Las placas se incubaron a temperatura ambiente durante 2 horas en un agitador orbital. La disolución de anticuerpo se descartó y las placas se lavaron tres veces al usar PBST. Se agregó 100 qL/pocillo de disolución de anti-IgG humana de cabra (H+L)-HRP (dilución 1:10.000 en tampón de bloqueo) a las placas y las placas se incubaron a temperatura ambiente durante 1 hora en un agitador orbital. La disolución de anticuerpo secundario se descartó y las placas se lavaron tres veces al usar PBST. Se agregó 100 qL/pocillo de disolución de trabajo de sustrato de peroxidasa fluorógena QuantaBlu (Thermo, núm. de cat. 15169) a las placas y las placas se incubaron a temperatura ambiente durante 30 min. Se leyó la fluorescencia con excitación 325 nm/emisión 420 nm al usar un SpectraMax M5 (Molecular Devices). Los datos se analizaron al usar el programa informático SoftMaxPro 5.4.2 con ajuste de 4 parámetros.
1.7 Análisis de citotoxicidad
1.7.1 Ensayo con Crystal Violet
Se subcultivaron y sembraron las células IGROV1 (FRh¡, MSLNneg), NCI-H2110 (FRmed, MSLNmed) y A431 (FRneg, MSLNneg) a 5000 células/pocillo en medio de crecimiento completo en placas de cultivo tisular de 96 pocillos, se incubaron a 37 °C, con CO2 al 5 % durante la noche (16 horas). Los reactivos de prueba se diluyeron en serie 1:3 en placas de dilución de pocillos de 2 mL profundidad, comenzando con 200 nM (10 diluciones en total). Las muestras diluidas (100 pL) se agregaron a las placas con células (comenzando con una concentración de muestras de prueba de 100 nM). Las placas se incubaron 37 °C, con CO2 al 5 % durante 5 días adicionales. Después, se descartó el medio. Las placas se lavaron una vez con 200 pL de DPBS, se tiñeron con 50 pL de disolución de Crystal Violet al 0,2 % a temperatura ambiente durante 15 min y después se lavaron exhaustivamente con agua del grifo. Las placas se secaron con aire y se disolvió el Crystal Violet con 200 pL de disolución de SDS al 1 %. Las placas se leyeron a 570 nm. Los datos se analizaron al usar GraphPad Prism 6.
2. Resultados
2.1 Caracterización biofísica de ADC de MORAb-003, MORAb-009 y trastuzumab
Se prepararon ADC de MORAb-003 (anti-receptor alfa de folato humano humanizado), MORAb-009 (anti-mesotelina humana quimérico de ratón-humano), y trastuzumab (anti-her2 humano humanizado) al usar los compuestos de eribulina conjugables indicados en la Tabla 46 según uno de los tres métodos de conjugación, incluidos: (1) reducción parcial de disulfuros intercatenarios del anticuerpo al usar el reductor distinto de tiol TCEP, con posterior conjugación al usar construcciones de maleimido-espaciador-enlazador-eribulina reactivas con tiol; (2) conjugación directa con residuos lisina del anticuerpo al usar construcciones de succinimida (OSu)-espaciador-enlazador-eribulina; y (3) conjugación con residuos lisina del anticuerpo al usar una estrategia de dos etapas, por la cual primero se conjugó OSu-PEG4-dibencilciclooctina con residuos lisina y después se llevó a cabo la conjugación ortogonal de las construcciones de azido-espaciador-enlazador-eribulina al usar SPAAC.
Después de la purificación, se determinaron los niveles de aglomeración para todos los ADC de MORAb-003, MORAb-009, y trastuzumab mediante SEG-HPLC y se analizó la relación entre fármaco y anticuerpo (DAR) al usar LC-MS de fase inversa y/o HIC-HPLC. La DAR para todos los ADC basados en maleimida se analizó al usar LC-MS de fase inversa e HlC-HPLC. Se observó típicamente una diferencia en los valores de DAR menor que 0,3 entre los dos métodos. En cambio, la DAR para todos los ADC preparados a través de la conjugación a través de residuos lisina se analizó solo mediante LC-MS, dado que el alto grado de heterogeneidad de estos ADC impide la resolución de especies de DAR individuales mediante HIC-HPLC. La unión al antígeno diana también se analizó al usar ELISA, para los ADC de MORAb-003 y MORAb-009. Los resultados de los análisis de DAR y aglomeración se muestran en la Tabla 47 junto al ADC respectivo.




2.1.1 ADC de MORAb-003, MORAb-009 y trastuzumab
No se observaron diferencias significativas entre MORAb-003, MORAb-009 y trastuzumab en relación con la eficacia de conjugación y los parámetros biofísicos. Todos los ADC demostraron valores de DAR y niveles de formación de aglomerados similares.
2.1.2 ADC basados en maleimida
Para los ADC basados en maleimida, los espaciadores pentilo y PEG2 apareados con un sitio de escisión val-cit-pAB escisión y un espaciador PEG2 apareado con un sitio de escisión ala-ala-asn-pAB proporcionaron valores de DAR entre 3,5 y 4,0 mediante LC-MS de fase inversa e HIC-HPLC, además de bajos niveles de aglomerado (<5 %). Sin embargo, cuando el espaciador se alargó a PEG (apareados con un sitio de escisión val-cit-pAB), los niveles de aglomerado aumentaron (11-18 %) y la eficacia de conjugación disminuyó, con resultados de valores de DAR entre 1.1 y 2,3. Ver, p. ej., los valores de porcentaje de aglomeración y DAR de MORAb003/MORAb009-ER-001159569 (enlazador PEG corto) y MORAb003/MORAb009-1242287 (enlazador PEG largo) en la Tabla 47.
Para los ADC preparados con un sitio de escisión de disulfidilo-pAB, se observaron valores de DAR bajos (1,0-1,6), junto con niveles de aglomerado relativamente altos (10-14 %). Se observaron valores de DAR significativamente más bajos cuando estos ADC se analizaron mediante LC-MS en lugar de HIC-HPLC (ver, p. ej., los valores de DAR por LC-MS/HIC-HPLC para MORAb003/MORAb009-ER1237504 y MORAb003/MORAb009-ER1237505 en la Tabla 47) Este resultado sugiere que el sitio de escisión del enlazador exhibe inestabilidad según el pH, dado que la fase móvil del análisis de LC-MS es aproximadamente 3,0, mientras que la fase móvil del análisis de HIC-HPLC es neutra.
Para los ADC preparados con un sitio de escisión de sulfonamida, se observaron niveles de aglomerado bajos (<5 %). De manera similar a los ADC de disulfidilo-pAB, se observaron valores de DAR más bajos cuando se analizaron mediante LC-MS (1,8-2,3) en lugar de HIC-HPLC (3,9), lo que indica que el sitio de escisión del enlazador exhibe inestabilidad según el pH.
Para los enlazadores no escindibles de PEG2 y PEG4 , se observó conjugación eficaz que dio como resultado valores de DAR entre 4,0 y 4,7. Los ADC de MORAb-009 con estos enlazadores no escindibles también demostraron bajos niveles de aglomeración (<2 %), mientras que se observaron niveles de aglomeración ligeramente más altos para los ADC de MORAb-003 correspondientes (4 % y 10 % para PEG2 y PEG4 , respectivamente).
2.1.3 ADC basados en succinimida
Todos los ADC preparados al usar succinimida acoplada con espaciador-enlazador-eribulina dieron como resultado valores de DAR <1,0. Para confirmar que esta eficacia de conjugación más baja (con respecto a las maleimidas) no fue una consecuencia del propio procedimiento de conjugación, estos ADC se volvieron a elaborar al usar una relación más alta de compuesto:anticuerpo y se volvieron a analizar al usar los mismos métodos de análisis de DAR. Se obtuvieron resultados similares, lo que sugiere que, sin desear limitarse a la teoría, los valores de DAR más bajos son una propiedad inherente de la combinación de succinimida y eribulina, y que las maleimidas se pueden conjugar de manera más eficaz. La eficacia de la conjugación de succinimida se aumentó a través d el uso de un método de dos etapas, por el cual primero se agregó DBCO al anticuerpo al usar NHS-DBCO, con la posterior adición de los compuestos azido. Esta estrategia da como resultado valores de DAR más altos, según se miden mediante análisis por HPLC de fase inversa, en comparación con conjugación directamente con los residuos lisina del anticuerpo. Para los ADC basados en succinimida que tienen enlazadores de sulfonamida (escindible), val-cit-pAB (escindible) o PEG2/PEG4 (no escindible), los valores de DAR que resultan de la conjugación de dos etapas fueron similares a los determinados para los ADC basados en maleimida que tienen un sitio de escisión de sulfonamida. Sin desear limitarse a la teoría, este resultado sugiere nuevamente que los valores de DAR más bajos para las reacciones de conjugación con succinimida-espaciador-enlazador-eribulina son una propiedad inherente de la combinación de succinimida y eribulina.
2.2 Caracterización de unión de ADC de MORAb-003 y MORAb-009
Para los ADC de MORAb-003, no se observaron diferencias significativas entre ADC de eribulina con enlazador basado en maleimida no escindible y el MORAb-003 original en relación con la unión al antígeno diana. Para otros ADC de MORAb-003 de eribulina con enlazador basado en maleimida, se observó típicamente una pérdida de 2 a 3 veces en la unión al antígeno diana con respecto al MORAb-003 original mediante análisis ELISA. Sin embargo, no hubo una correlación evidente entre la longitud del enlazador o composición del enlazador y los valores de CE50 más bajos. De manera similar, para los ADC de MORAb-003 de eribulina con enlazador basado en succinimida, se observó generalmente una pérdida de 0 a 3 veces en la unión al antígeno diana con respecto al MORAb-003 no conjugado. Nuevamente, no hubo una correlación evidente entre la longitud del enlazador o composición del enlazador y los valores de CE50 más bajos. Para los ADC de MORAb-009, todos los ADC tuvieron una disminución menor que 2 veces en los valores de CE50, con respecto al MORAb-009 original.
2.3 Análisis de citotoxicidad in v itro de ADC de MORAb-003, MORAb-009 y trastuzumab
Se evaluó la potencia in vitro de ADC de MORAb-003, MORAb-009 y trastuzumab preparados al usar un ensayo de citotoxicidad basado en células con Crystal Violet. Las líneas celulares seleccionadas para el cribado con ADC de MORAb-003 y MORAb-009 fueron IGROV1, NCI-H2110 y A431. Las células IGROV1 tienen como origen el carcinoma epitelial de ovario humano y expresan niveles altos del receptor alfa de folato, pero no mesotelina (es decir, reactivas a MORAb-003). Las células NCI-H2110 tienen como origen el carcinoma de pulmón no microcítico humano y expresan niveles moderados del receptor alfa de folato y de mesotelina (es decir, reactivas a MORAb-003 y MORAb-009). Las células testigo A431 son células que tienen como origen el carcinoma epidérmico humano y no expresan ningún antígeno diana. Los resultados de este cribado se muestran en la Tabla 48. Los ADC de MORAb-003, MORAb-009 y trastuzumab ADC que comprenden el enlazador-toxina maleimido-PEG2-val-cit-pAB-eribulina (VCP-eribulina) también se evaluaron en líneas celulares de cáncer gástrico y de mama adicionales, incluidas NCI-N87 (FRo MSLNmed, her2hi), BT-474 (FRneg, MSLNneg, her2hi), ZR-75 (FRneg, MSLNneg, her2med) y NUGC3 (FRneg, MSLNneg, her2neg). Los resultados de este cribado se muestran en la Tabla 49.





2.3.1 Citotoxicidad de ADC basados en maleimida
Todos los ADC de MORAb-003 y MORAb-009 basados en maleimida exhibieron citotoxicidad específica en células IGROV1, con una diferencia de 2-3 órdenes de magnitud en la potencia observada entre anticuerpos. Los ADC de MORAb-003 con val-cit-pAB-eribulina demostraron potencia más alta en la línea celular IGROV1 que los ADC de MORAb-003 con PEG2 o PEG4 no escindible, pero el múltiplo de especificidad permaneció sin modificación. Se observaron tendencias similares para los ADC de MORAb-009, donde los ADC de MORAb-009 no escindibles demostraron una citotoxicidad más baja en células IGROV1 que los ADC de MORAb-009 con val-cit-pAB-eribulina.
Los ADC de MORAb-009 basados en maleimida con enlazadores basados en disulfidilo y sulfonamida demostraron potencia más alta en la línea celular NCI-H2110 que en la línea celular IGROV1. Esto puede deberse a la potencial inestabilidad de los enlazadores en el cultivo, como se describe más adelante. También se observó una citotoxicidad potente con los ADC de MORAb-003 correspondientes. En cambio, los ADC de MORAb-003 y MORAb-009 basados en maleimida con enlazadores no escindibles demostraron una potencia relativamente baja en células NCI-H2110. Sin desear limitarse a la teoría, este resultado sugiere que, con una expresión de la diana más baja, la escisión eficaz y la liberación de la carga eficaz pueden mejorar la citotoxicidad.
Los ADC con un enlazador val-cit-pAB escindible por enzima o un enlazador no escindible demostraron niveles bajos de destrucción inespecífica en las células testigo A431 (CI50 >100 nM), mientras que ADC con un enlazador ala-alaasn-pAB escindible por enzima exhibió una destrucción débil pero detectable de estas células testigo. Esto indica que los enlazadores val-cit-pAB escindibles por enzima pueden ser más estables en cultivo que los enlazadores ala-alaasn-pAB escindibles por enzima. Además, los ADC de MORAb-009 con un espaciador de PEG2 más corto demostraron citotoxicidad más alta en células IGROV1 que los ADC correspondientes con un espaciador de PEG8 más largo. Esta misma tendencia se observó en células NCI-H2110 para los ADC de MORAb-003 y MORAb-009, donde las longitudes de espaciador más cortas dieron como resultado una citotoxicidad más alta.
Los ADC con enlazadores basados en sulfonamida generalmente demostraron valores de DAR más altos y niveles de aglomerado más bajos que los ADC correspondientes con enlazadores basados en disulfidilo. Sin embargo, se observó una destrucción a nivel de nM de las células testigo A431 en ambas categorías de estos ADC, lo que sugiere que los enlazadores basados en disulfidilo y sulfonamida fueron menos estables en cultivo que los enlazadores escindibles por enzima en las condiciones de ensayo examinadas.
El enlazador-toxina maleimido-PEG2-val-cit-pAB-eribulina (VCP-eribulina) específico se examinó adicionalmente para determinar la especificidad y potencia en diferentes líneas celulares de cáncer gástrico y de mama. El VCP-eribulina se conjugó con MORAb-003 y MORAb-009, además del anticuerpo anti-her2 humano trastuzumab. El MORAb-003-VCP-eribulina demostró una destrucción débil pero específica en células NCI-N87, que expresan niveles bajos de receptor alfa de folato (FR), y poca destrucción en las tres líneas celulares negativas para FR restantes. El MORAb-009-VCP-eribulina también demostró citotoxicidad potente en células NCI-N87, que expresan niveles moderados de mesotelina. Trastuzumab-VCP-eribulina fue muy potente (3 - 6 pM, CI50) en células NCI-N87 y BT-474, las dos líneas celulares que expresan niveles altos de her2, y también fue potente en las células de cáncer de mama ZR-75, que solo expresan moderadamente her2. Todos los ADC de MORAb-003, MORAb-009 y trastuzumab VCP-eribulina demostraron baja citotoxicidad en células NUGC3, que no expresan FR, mesotelina ni her2, los respectivos antígenos diana.
2.3.2 Citotoxicidad de ADC basados en succinimida
Las tendencias en la citotoxicidad de los ADC basados en succinimida fueron similares a las de los ADC basados en maleimida para las células IGROV1, donde los ADC con espaciador PEG8 demostraron baja citotoxicidad además de valores de DAR bajos. Se observó generalmente una citotoxicidad más en baja células IGROV1 y NCI-H2110 para ADC basados en succinimida con enlazadores escindibles por enzima en comparación con los ADC basados en maleimida correspondientes, que se debió con mayor probabilidad a los valores de DAR más bajos. También se observó destrucción inespecífica de células A431 con enlazadores basados en disulfidilo y sulfonamida, de manera similar a los ADC basados en maleimida correspondientes. Esto señala la mayor inestabilidad que potencialmente surge del sitio de escisión, en lugar de la química de conjugación.
Cuando se llevó a cabo una conjugación de dos etapas, se observaron valores de DAR más altos con respecto a los obtenidos con la estrategia de conjugación con succinimida directa. Estos valores de DAR más altos se correlacionan con una potencia más alta. Para los ADC de VCP-eribulina MORAb-003, se observó una citotoxicidad potente en células IGROV1 y NCI-H2110. Aunque los ADC de MORAb-003 no escindibles demostraron potencia en células IGROV1 (1- 4 nM), todavía fueron menos potentes que el ADC de VCP-eribulina MORAb-003 preparados con este método (38 pM), aunque los valores de DAR fueron semejantes. Además, los ADC de MORAb-003 no escindibles preparados al usar el método de dos etapas fueron ligeramente menos potentes que los ADC basados en maleimida correspondientes en la línea celular IGROV1, lo que puede deberse a sus valores de DAR más bajos. De manera similar a sus contrapartes basados en maleimida, los ADC no escindibles preparados al usar el método de dos etapas también perdieron casi toda la citotoxicidad en células NCI-2110.
2.4 Caracterización biofísica de ADC anti-mesotelina humana (LCcys80)
Se conjugó MAL-PEG2-Val-Cit-PAB-eribulina (ER-001159569) con ocho anticuerpos anti-mesotelina humana diferentes (Tabla 1). Las afinidades de unión de los anticuerpos originales se determinaron mediante análisis BIAcore, como se describió anteriormente en la sección 1.6.1. Los niveles de aglomeración se determinaron para todos los ADC de anti-mesotelina humana mediante SEC-HPLC y se analizó la DAR al usar HIC-HPLC. La potencia in vitro se evaluó al usar el ensayo de citotoxicidad basado en células con Crystal Violet en A3 (A431 transfectada establemente con mesotelina humana (MSLN), MSLNhi), OVCAR3 (ovario humano, MSLNhi), HEC-251 (endometroide humano, MSLNmed), H226 (mesotelioma de células escamosas de pulmón humano, MSLNto) y A431 original (MSLNneg). Los resultados de los análisis de DAR, aglomeración y citotoxicidad se muestran en la Tabla 50.

Todos los ADC anti-mesotelina humana retuvieron niveles de aglomeración bajos (<10 % de aglomerado) y demostraron potencia alta en las líneas celulares diana. Se observó potencia alta en A3 y OVCAR3, mientras que las células HEC-251 y H226 fueron relativamente resistentes a la citotoxicidad del ADC.
Secuencias seleccionadas:
SEQ ID NO: 1 (Cadena pesada de MORAb-003 (HC))
1 EVQLVESGGG WQPGRSLRL SCSASGFTFS GYGLSWVRQA PGKGLEWVAM
51 ISSGGSYTYY ADSVKGRFAI SRDNAKNTLF LQMDSLRPEDTGVYFCARHG
101 DDPAWFAYWG QGTPVTVSSA STKGPSVFPL APSSKSTSGGTAALGCLVKD
151 YFPEPVTVSW NSGALTSGVH TFPAVLQSSG LYSLSSWTV PSSSLGTQTY
201 ICNWHKPSN TKVDKKVEPK SCDKTHTCPP CPAPELLGGP SVFLFPPKPK
251 DTLMISRTPE VTCVWDVSH EDPEVKFNWY VDGVEVHNAKTKPREEQYNS
301 TYRWSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISKAKGQPREPQV
351 YTLPPSRDEL TKNQVSLTCL VKGFYPSDIA VEWESNGQPENNYKTTPPVL
401 DSDGSFFLYS KLTVDKSRWQ QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
SEQ ID NO: 2 (CDR1 de HC de MORAb-003; Kabat): GYGLS
SEQ ID NO: 3 (CDR2 de HC de MORAb-003; Kabat): MISSGGSYTYYADSVKG
SEQ ID NO: 4 (CDR3 de HC de MORAb-003; Kabat): HGDDPAWFAY
SEQ ID NO: 5 (secuencia de aminoácidos de preproteína de longitud completa de cadena pesada de MORAb-003; secuencia líder subrayada)
i MGWSCIILFL VATATGVHSE VQLVESGGGV VQPGRSLRLS CSASGFTFSG
51 YGLSWVRQAP GKGLEWVAMI SSGGSYTYYA DSVKGRFAIS RDNAKNTLFL
101 QMDSLRPEDT GVYFCARHGD DPAWFAYWGQ GTPVTVSSAS TKGPSVFPLA
151 PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
201 YSLSSWTVP SSSLGTQTYI CNVNHKPSNT KVDKKVEPKS CDKTHTCPPC
251 PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVWDVSHE DPEVKFNWYV
301 DGVEVHNAKT KPREEQYNST YRWSVLTVL HQDWLNGKEY KCKVSNKALP
351 APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV
401 EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH
451 EALHNHYTQK SLSLSPGK
SEQ ID NO: 6 (Cadena ligera de MORAb-003 (LC))
1 DIQLTQSPSS LSASVGDRVT ITCSVSSSIS SNNLHWYQQK PGKAPKPWIY
51 GTSNLASGVP SRFSGSGSGT DYTFTISSLQ PEDIATYYCQ QWSSYPYMYT
101 FGQGTKVEIK RTVAAPSVFI FPPSDEQLKS GTASWCLLN NFYPREAKVQ
151 WKVDNALQSG NSQESVTEQD SKDSTYSLSS TLTLSKADYE KHKVYACEVT
201 HQGLSSPVTK SFNRGEC
SEQ ID NO: 7 (CDR1 de LC de MORAb-003; Kabat): SVSSSISSNNLH
SEQ ID NO: 8 (CDR2 de LC de MORAb-003; Kabat): Kabat): GTSNLAS
SEQ ID NO: 9 (CDR3 de LC de MORAb-003; Kabat): QQWSSYPYMYT
SEQ ID NO: 10 secuencia de aminoácidos de preproteína de longitud completa de cadena ligera de MORAb-003 (secuencia líder subrayada)
1 MGWSCIILFL VATATGVHSD IQLTQSPSSL SASVGDRVTI TCSVSSSISS
51 NNLHWYQQKP GKAPKPWIYG TSNLASGVPS RFSGSGSGTD YTFTISSLQP
101 EDIATYYCQQ WSSYPYMYTF GQGTKVEIKR TVAAPSVFIF PPSDEQLKSG
151 TASWCLLNN FYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST
201 LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGEC
S E Q ID NO : 11 (n t de HC de M O R A b -003 )
1 ATGGGATGGA GCTGTATCAT CCTCTTCTTG GTAGCAACAG CTACAGGTGT 51 CCACTCCGAG GTCCAACTGG TGGAGAGCGG TGGAGGTGTT GTGCAACCTG 101 GCCGGTCCCT GCGCCTGTCC TGCTCCGCAT CTGGCTTCAC CTTCAGCGGC 151 TATGGGTTGT CTTGGGTGAG ACAGGCACCT GGAAAAGGTC TTGAGTGGGT 201 TGCAATGATT AGTAGTGGTG GTAGTTATAC CTACTATGCA GACAGTGTGA 251 AGGGTAGATT TGCAATATCG CGAGACAACG CCAAGAACAC ATTGTTCCTG 301 CAAATGGACA GCCTGAGACC CGAAGACACC GGGGTCTATT TTTGTGCAAG 351 ACATGGGGAC GATCCCGCCT GGTTCGCTTA TTGGGGCCAA GGGACCCCGG 401 TCACCGTCTC CTCAGCCTCC ACCAAGGGCC CATCGGTCTT CCCCCTGGCA 451 CCCTCCTCCA AGAGCACCTC TGGGGGCACA GCGGCCCTGG GCTGCCTGGT 501 CAAGGACTAC TTCCCCGAAC CGGTGACGGT GTCGTGGAAC TCAGGCGCCC 551 TGACCAGCGG CGTGCACACC TTCCCGGCTG TCCTACAGTC CTCAGGACTC 601 TACTCCCTCA GCAGCGTGGT GACCGTGCCC TCCAGCAGCT TGGGCACCCA 651 GACCTACATC TGCAACGTGA ATCACAAGCC CAGCAACACC AAGGTGGACA 701 AGAAAGTTGA GCCCAAATCT TGTGACAAAA CTCACACATG CCCACCGTGC 751 CCAGCACCTG AACTCCTGGG GGGACCGTCA GTCTTCCTCT TCCCCCCAAA 801 ACCCAAGGAC ACCCTCATGA TCTCCCGGAC CCCTGAGGTC ACATGCGTGG 851 TGGTGGACGT GAGCCACGAA GACCCTGAGG TCAAGTTCAA CTGGTACGTG 901 GACGGCGTGG AGGTGCATAA TGCCAAGACA AAGCCGCGGG AGGAGCAGTA 951 CAACAGCACG TACCGTGTGG TCAGCGTCCT CACCGTCCTG CACCAGGACT 1001 GGCTGAATGG CAAGGAGTAC AAGTGCAAGG TCTCCAACAA AGCCCTCCCA 1051 GCCCCCATCG AGAAAACCAT CTCCAAAGCC AAAGGGCAGC CCCGAGAACC 1101 ACAGGTGTAC ACCCTGCCCC CATCCCGGGA TGAGCTGACC AAGAACCAGG 1151 TCAGCCTGAC CTGCCTGGTC AAAGGCTTCT ATCCCAGCGA CATCGCCGTG 1201 GAGTGGGAGA GCAATGGGCA GCCGGAGAAC AACTACAAGA CCACGCCTCC 1251 CGTGCTGGAC TCCGACGGCT CCTTCTTCTT ATATTCAAAG CTCACCGTGG 1301 ACAAGAGCAG GTGGCAGCAG GGGAACGTCT TCTCATGCTC CGTGATGCAT 1351 GAGGCTCTGC ACAACCACTA CACGCAGAAG AGCCTCTCCC TGTCTCCCGG 1401 GAAATGA
SEQ ID NO: 12 (nt de LC de MORAb-003)
1 ATGGGATGGA GCTGTATCAT CCTCTTCTTG GTAGCAACAG CTACAGGTGT
51 CCACTCCGAC ATCCAGCTGA CCCAGAGCCC AAGCAGCCTG AGCGCCAGCG
101 TGGGTGACAG AGTGACCATC ACCTGTAGTG TCAGCTCAAG TATAAGTTCC
151 AACAACTTGC ACTGGTACCA GCAGAAGCCA GGTAAGGCTC CAAAGCCATG
201 GATCTACGGC ACATCCAACC TGGCTTCTGG TGTGCCAAGC AGATTCAGCG
251 GTAGCGGTAG CGGTACCGAC TACACCTTCA CCATCAGCAG CCTCCAGCCA
301 GAGGACATCG CCACCTACTA CTGCCAACAG TGGAGTAGTT ACCCGTACAT
351 GTACACGTTC GGCCAAGGGA CCAAGGTGGA AATCAAACGA ACTGTGGCTG
401 CACCATCTGT CTTCATCTTC CCGCCATCTG ATGAGCAGTT GAAATCTGGA
451 ACTGCCTCTG TTGTGTGCCT GCTGAATAAC TTCTATCCCA GAGAGGCCAA
501 AGTACAGTGG AAGGTGGATA ACGCCCTCCA ATCGGGTAAC TCCCAGGAGA
551 GTGTCACAGA GCAGGACAGC AAGGACAGCA CCTACAGCCT CAGCAGCACC
601 CTGACGCTGA GCAAAGCAGA CTACGAGAAA CACAAAGTCT ACGCCTGCGA
651 AGTCACCCAT CAGGGCCTGA GCTCGCCCGT CACAAAGAGC TTCAACAGGG
701 GAGAGTGTTA A
SEQ ID NO: 13 (CDR1 de HC de MORAb-003; IMGT): GFTFSGYG
SEQ ID NO: 14 (CDR2 de HC de MORAb-003; IMGT): ISSGGSYT
SEQ ID NO: 15 (CDR3 de HC de MORAb-003; IMGT): ARHGDDPAWFAY
SEQ ID NO: 16 (CDR1 de LC de MORAb-003; IMGT): SSISSNN
SEQ ID NO: 17 (CDR2 de LC de MORAb-003; IMGT): GTS
SEQ ID NO: 18 (CDR3 de LC de MORAb-003; IMGT): QQWSSYPYMYT
SEQ ID NO: 19 (FRA humano)
1 maqrmttqll lllvwvavvg eaqtriawar tellnvcmna khhkekpgpe dklheqcrpw 61 rknaccstnt sqeahkdvsy lyrfnwnhcg emapackrhf iqdtclyecs pnlgpwiqqv 121 dqswrkervl nvplckedce qwwedcrtsy tcksnwhkgw nwtsgfnkca vgaacqpfhf 181 yfptptvlcn eiwthsykvs nysrgsgrci qmwfdpaqgn pneevarfya aamsgagpwa 241 awpfllslal mllwlls
S E Q ID N O : 20 (n u c le ó tid o de F R A h u m an o)
1 cattccttgg tgccactgac cacagctctt tcttcaggga cagacatggc tcagcggatg 61 acaacacagc tgctgctcct tctagtgtgg gtggctgtag taggggaggc tcagacaagg 121 attgcatggg ccaggactga gcttctcaat gtctgcatga acgccaagca ccacaaggaa 181 aagccaggcc ccgaggacaa gttgcatgag cagtgtcgac cctggaggaa gaatgcctgc 241 tgttctacca acaccagcca ggaagcccat aaggatgttt cctacctata tagattcaac 301 tggaaccact gtggagagat ggcacctgcc tgcaaacggc atttcatcca ggacacctgc 361 ctctacgagt gctcccccaa cttggggccc tggatccagc aggtggatca gagctggcgc 421 aaagagcggg tactgaacgt gcccctgtgc aaagaggact gtgagcaatg gtgggaagat 481 tgtcgcacct cctacacctg caagagcaac tggcacaagg gctggaactg gacttcaggg 541 tttaacaagt gcgcagtggg agctgcctgc caacctttcc atttctactt ccccacaccc 601 actgttctgt gcaatgaaat ctggactcac tcctacaagg tcagcaacta cagccgaggg 661 agtggccgct gcatccagat gtggttcgac ccagcccagg gcaaccccaa tgaggaggtg 721 gcgaggttct atgctgcagc catgagtggg gctgggccct gggcagcctg gcctttcctg 781 cttagcctgg ccctaatgct gctgtggctg ctcagctgac ctccttttac cttctgatac 841 ctggaaatcc ctgccctgtt cagccccaca gctcccaact atttggttcc tgctccatgg 901 tcgggcctct gacagccact ttgaataaac cagacaccgc acatgtgtct tgagaattat 961 ttggaaaaaa aaaaaaaaaa aa
SEQ ID NO: 21 (her2 humano)
1 melaalcrwg lllallppga astqvctgtd mklrlpaspe thldmlrhly qgcqvvqgnl 61 eltylptnas lsflqdiqev qgyvliahnq vrqvplqrlr ivrgtqlfed nyalavldng 121 dplnnttpvt gaspgglrel qlrslteilk ggvliqrnpq lcyqdtilwk difhknnqla 181 ltlidtnrs r achpcspmck gsrcwgesse dcqsltrtvc aggcarckgp lptdccheqc 241 aagctgpkhs dclaclhfnh sgicelhcpa lvtyntdtfe smpnpegryt fgascvtacp 301 ynylstdvgs ctlvcplhnq evtaedgtqr cekcskpcar vcyglgmehl revravtsan 361 iqefagckki fgslaflpes fdgdpasnta plqpeqlqvf etleeitgyl yisawpdslp 421 dlsvfqnlqv irgrilhnga ysltlqglgi swlglrslre lgsglalihh nthlcfvhtv 481 pwdqlfrnph qallhtanrp edecvgegla chqlcarghc wgpgptqcvn csqflrgqec 541 veecrvlqgl preyvnarhc lpchpecqpq ngsvtcfgpe adqcvacahy kdppfcvarc 601 psgvkpdlsy mpiwkfpdee gacqpcpinc thscvdlddk gcpaeqrasp ltsiisavvg 661 illvvvlgvv fgilikrrqq kirkytmrrl lqetelvepl tpsgampnqa qmrilketel 721 rkvkvlgsga fgtvykgiwi pdgenvkipv aikvlrents pkankeilde ayvmagvgsp 781 yvsrllgicl tstvqlvtql mpygclldhv renrgrlgsq dllnwcmqia kgmsyledvr 841 lvhrdlaarn vlvkspnhvk itdfglarll dideteyhad ggkvpikwma lesilrrrft 901 hqsdvwsygv tvwelmtfga kpydgipare ipdllekger lpqppictid vymimvkcwm 961 idsecrprfr elvsefsrma rdpqrfvviq nedlgpaspl dstfyrslle dddmgdlvda 1021 eeylvpqqgf fcpdpapgag gmvhhrhrss strsgggdlt lglepseeea prsplapseg 1081 agsdvfdgdl gmgaakglqs lpthdpsplq rysedptvpl psetdgyvap ltcspqpeyv 1141 nqpdvrpqpp spregplpaa rpagatlerp ktlspgkngv vkdvfafgga venpeyltpq 1201 ggaapqphpp pafspafdnl yywdqdpper gappstfkgt ptaenpeylg ldvpv
S E Q ID N O : 22 (n u c le ó tid o de he r2 hu m an o)
1 ATGGAGCTGG CGGCCTTGTG CCGCTGGGGG CTCCTCCTCG CCCTCTTGCC CCCCGGAGCC 61 GCGAGCACCC AAGTGTGCAC CGGCACAGAC ATGAAGCTGC GGCTCCCTGC CAGTCCCGAG 121 ACCCACCTGG ACATGCTCCG CCACCTCTAC CAGGGCTGCC AGGTGGTGCA GGGAAACCTG 181 GAACTCACCT ACCTGCCCAC CAATGCCAGC CTGTCCTTCC TGCAGGATAT CCAGGAGGTG 241 CAGGGCTACG TGCTCATCGC TCACAACCAA GTGAGGCAGG TCCCACTGCA GAGGCTGCGG 301 ATTGTGCGAG GCACCCAGCT CTTTGAGGAC AACTATGCCC TGGCCGTGCT AGACAATGGA 361 GACCCGCTGA ACAATACCAC CCCTGTCACA GGGGCCTCCC CAGGAGGCCT GCGGGAGCTG 421 CAGCTTCGAA GCCTCACAGA GATCTTGAAA GGAGGGGTCT TGATCCAGCG GAACCCCCAG 481 CTCTGCTACC AGGACACGAT TTTGTGGAAG GACATCTTCC ACAAGAACAA CCAGCTGGCT 541 CTCACACTGA TAGACACCAA CCGCTCTCGG GCCTGCCACC CCTGTTCTCC GATGTGTAAG 601 GGCTCCCGCT GCTGGGGAGA GAGTTCTGAG GATTGTCAGA GCCTGACGCG CACTGTCTGT 661 GCCGGTGGCT GTGCCCGCTG CAAGGGGCCA CTGCCCACTG ACTGCTGCCA TGAGCAGTGT 721 GCTGCCGGCT GCACGGGCCC CAAGCACTCT GACTGCCTGG CCTGCCTCCA CTTCAACCAC 781 AGTGGCATCT GTGAGCTGCA CTGCCCAGCC CTGGTCACCT ACAACACAGA CACGTTTGAG 841 TCCATGCCCA ATCCCGAGGG CCGGTATACA TTCGGCGCCA GCTGTGTGAC TGCCTGTCCC 901 TACAACTACC TTTCTACGGA CGTGGGATCC TGCACCCTCG TCTGCCCCCT GCACAACCAA 961 GAGGTGACAG CAGAGGATGG AACACAGCGG TGTGAGAAGT GCAGCAAGCC CTGTGCCCGA 1021 GTGTGCTATG GTCTGGGCAT GGAGCACTTG CGAGAGGTGA GGGCAGTTAC CAGTGCCAAT 1081 ATCCAGGAGT TTGCTGGCTG CAAGAAGATC TTTGGGAGCC TGGCATTTCT GCCGGAGAGC 1141 TTTGATGGGG ACCCAGCCTC CAACACTGCC CCGCTCCAGC CAGAGCAGCT CCAAGTGTTT 1201 GAGACTCTGG AAGAGATCAC AGGTTACCTA TACATCTCAG CATGGCCGGA CAGCCTGCCT 1261 GACCTCAGCG TCTTCCAGAA CCTGCAAGTA ATCCGGGGAC GAATTCTGCA CAATGGCGCC 1321 TACTCGCTGA CCCTGCAAGG GCTGGGCATC AGCTGGCTGG GGCTGCGCTC ACTGAGGGAA 1381 CTGGGCAGTG GACTGGCCCT CATCCACCAT AACACCCACC TCTGCTTCGT GCACACGGTG 1441 CCCTGGGACC AGCTCTTTCG GAACCCGCAC CAAGCTCTGC TCCACACTGC CAACCGGCCA 1501 GAGGACGAGT GTGTGGGCGA GGGCCTGGCC TGCCACCAGC TGTGCGCCCG AGGGCACTGC 1561 TGGGGTCCAG GGCCCACCCA GTGTGTCAAC TGCAGCCAGT TCCTTCGGGG CCAGGAGTGC 1621 GTGGAGGAAT GCCGAGTACT GCAGGGGCTC CCCAGGGAGT ATGTGAATGC CAGGCACTGT
1681 TTGCCGTGCC ACCCTGAGTG TCAGCCCCAG AATGGCTCAG TGACCTGTTT TGGACCGGAG 1741 GCTGACCAGT GTGTGGCCTG TGCCCACTAT AAGGACCCTC CCTTCTGCGT GGCCCGCTGC 1801 CCCAGCGGTG TGAAACCTGA CCTCTCCTAC ATGCCCATCT GGAAGTTTCC AGATGAGGAG 1861 GGCGCATGCC AGCCTTGCCC CATCAACTGC ACCCACTCCT GTGTGGACCT GGATGACAAG 1921 GGCTGCCCCG CCGAGCAGAG AGCCAGCCCT CTGACGTCCA TCATCTCTGC GGTGGTTGGC 1981 ATTCTGCTGG TCGTGGTCTT GGGGGTGGTC TTTGGGATCC TCATCAAGCG ACGGCAGCAG 2041 AAGATCCGGA AGTACACGAT GCGGAGACTG CTGCAGGAAA CGGAGCTGGT GGAGCCGCTG 2101 ACACCTAGCG GAGCGATGCC CAACCAGGCG CAGATGCGGA TCCTGAAAGA GACGGAGCTG 2161 AGGAAGGTGA AGGTGCTTGG ATCTGGCGCT TTTGGCACAG TCTACAAGGG CATCTGGATC 2221 CCTGATGGGG AGAATGTGAA AATTCCAGTG GCCATCAAAG TGTTGAGGGA AAACACATCC 2281 CCCAAAGCCA ACAAAGAAAT CTTAGACGAA GCATACGTGA TGGCTGGTGT GGGCTCCCCA 2341 TATGTCTCCC GCCTTCTGGG CATCTGCCTG ACATCCACGG TGCAGCTGGT GACACAGCTT 2401 ATGCCCTATG GCTGCCTCTT AGACCATGTC CGGGAAAACC GCGGACGCCT GGGCTCCCAG 2461 GACCTGCTGA ACTGGTGTAT GCAGATTGCC AAGGGGATGA GCTACCTGGA GGATGTGCGG 2521 CTCGTACACA GGGACTTGGC CGCTCGGAAC GTGCTGGTCA AGAGTCCCAA CCATGTCAAA 2581 ATTACAGACT TCGGGCTGGC TCGGCTGCTG GACATTGACG AGACAGAGTA CCATGCAGAT 2641 GGGGGCAAGG TGCCCATCAA GTGGATGGCG CTGGAGTCCA TTCTCCGCCG GCGGTTCACC 2701 CACCAGAGTG ATGTGTGGAG TTATGGTGTG ACTGTGTGGG AGCTGATGAC TTTTGGGGCC 2761 AAACCTTACG ATGGGATCCC AGCCCGGGAG ATCCCTGACC TGCTGGAAAA GGGGGAGCGG 2821 CTGCCCCAGC CCCCCATCTG CACCATTGAT GTCTACATGA TCATGGTCAA ATGTTGGATG 2881 ATTGACTCTG AATGTCGGCC AAGATTCCGG GAGTTGGTGT CTGAATTCTC CCGCATGGCC 2941 AGGGACCCCC AGCGCTTTGT GGTCATCCAG AATGAGGACT TGGGCCCAGC CAGTCCCTTG 3001 GACAGCACCT TCTACCGCTC ACTGCTGGAG GACGATGACA TGGGGGACCT GGTGGATGCT 3061 GAGGAGTATC TGGTACCCCA GCAGGGCTTC TTCTGTCCAG ACCCTGCCCC GGGCGCTGGG 3121 GGCATGGTCC ACCACAGGCA CCGCAGCTCA TCTACCAGGA GTGGCGGTGG GGACCTGACA 3181 CTAGGGCTGG AGCCCTCTGA AGAGGAGGCC CCCAGGTCTC CACTGGCACC CTCCGAAGGG 3241 GCTGGCTCCG ATGTATTTGA TGGTGACCTG GGAATGGGGG CAGCCAAGGG GCTGCAAAGC 3301 CTCCCCACAC ATGACCCCAG CCCTCTACAG CGGTACAGTG AGGACCCCAC AGTACCCCTG 3361 CCCTCTGAGA CTGATGGCTA CGTTGCCCCC CTGACCTGCA GCCCCCAGCC TGAATATGTG 3421 AACCAGCCAG ATGTTCGGCC CCAGCCCCCT TCGCCCCGAG AGGGCCCTCT GCCTGCTGCC 3481 CGACCTGCTG GTGCCACTCT GGAAAGGCCC AAGACTCTCT CCCCAGGGAA GAATGGGGTC 3541 GTCAAAGACG TTTTTGCCTT TGGGGGTGCC GTGGAGAACC CCGAGTACTT GACACCCCAG 3601 GGAGGAGCTG CCCCTCAGCC CCACCCTCCT CCTGCCTTCA GCCCAGCCTT CGACAACCTC 3661 TATTACTGGG ACCAGGACCC ACCAGAGCGG GGGGCTCCAC CCAGCACCTT CAAAGGGACA 3721 CCTACGGCAG AGAACCCAGA GTACCTGGGT CTGGACGTGC CAGTGTGA
Claims (13)
1. Un conjugado de anticuerpo-fármaco de Fórmula (I):
Ab-(L-D)p (I)
en donde
(i) Ab es un anticuerpo anti-receptor alfa de folato internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ iD NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT.
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es un número entero de 1 a 8.
2. El conjugado de anticuerpo-fármaco de la reivindicación 1, en donde el anticuerpo o fragmento de unión a antígeno comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24.
3. El conjugado de anticuerpo-fármaco de la reivindicación 1 o la reivindicación 2, en donde p es un número entero de 3 a 4.
4. El conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 3, en donde el anticuerpo o fragmento de unión a antígeno comprende un dominio constante de cadena pesada de IgG1 humana y un dominio constante de cadena ligera kappa de Ig humana.
5. Una composición que comprende múltiples copias del conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 4, en donde la p promedio de los conjugados de anticuerpo-fármaco en la composición es de aproximadamente 3,2 a aproximadamente 3,8 o de aproximadamente 3,6 a aproximadamente 4,4.
6. Un conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 4 o la composición de la reivindicación 5 para uso en el tratamiento de un cáncer que expresa receptor alfa de folato.
7. El conjugado de anticuerpo-fármaco para el uso de la reivindicación 6, en donde el cáncer es un cáncer gástrico, un cáncer de ovario seroso, un cáncer de ovario de células claras, un cáncer pulmonar no microcítico, un cáncer colorrectal, un cáncer de mama triple negativo, un cáncer de endometrio, un carcinoma de endometrio seroso, un tumor carcinoide pulmonar o un osteosarcoma.
8. Una composición farmacéutica que comprende el conjugado de anticuerpo-fármaco de cualquiera de las reivindicaciones 1 a 4 o la composición de la reivindicación 5 y un portador farmacéuticamente aceptable.
9. Un método para producir el conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 4 o la composición de la reivindicación 5, que comprende hacer reaccionar un anticuerpo o fragmento de unión a antígeno con un enlazador escindible ligado a eribulina en condiciones que permiten la conjugación.
10. Un método para determinar si un paciente responderá al tratamiento con el conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 4 o la composición de la reivindicación 5, que comprende proporcionar una muestra biológica del paciente y poner en contacto la muestra biológica con el conjugado de anticuerpo-fármaco de una cualquiera de las reivindicaciones 1 a 4 o la composición de la reivindicación 5.
11. El método de la reivindicación 10, en donde la muestra biológica es una biopsia tumoral de un cáncer gástrico, un cáncer de ovario seroso, un cáncer de ovario de células claras, un cáncer pulmonar no microcítico, un cáncer colorrectal, un cáncer de mama triple negativo, un cáncer de endometrio, un carcinoma de endometrio seroso, un tumor carcinoide pulmonar o un osteosarcoma.
12. Una composición que comprende múltiples copias de un conjugado de anticuerpo-fármaco de Fórmula (I):
Ab-(L-D)p (I)
en donde
(i) Ab es un anticuerpo anti-receptor alfa de folato internalizante o fragmento de unión a antígeno de este internalizante que comprende tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:2 (HCDR1), SEQ ID NO:3 (HCDR2) y SEQ ID NO:4 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:7 (LCDR1), SEQ ID NO:8 (LCDR2) y SEQ iD NO:9 (LCDR3), según se definen mediante el sistema de numeración de Kabat; o tres regiones de determinación de la complementariedad de cadena pesada (HCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:13 (HCDR1), SEQ ID NO:14 (HCDR2) y SEQ ID NO:15 (HCDR3); y tres regiones de determinación de la complementariedad de cadena ligera (LCDR) que comprenden las secuencias de aminoácidos de SEQ ID NO:16 (LCDR1), SEQ ID NO:17 (LCDR2) y SEQ ID NO:18 (LCDR3), según se definen mediante el sistema de numeración de IMGT.
(ii) D es eribulina;
(iii) L es un enlazador escindible que comprende Mal-(PEG)2-Val-Cit-pAB; y
(iv) p es la cantidad promedio de restos -L-D por Ab, en donde la p promedio de los conjugados de anticuerpo-fármaco en la composición es de aproximadamente 3,6 a aproximadamente 4,4.
13. La composición de la reivindicación 12, en donde el anticuerpo o fragmento de unión a antígeno comprende una región variable de cadena pesada que comprende una secuencia de aminoácidos de la SEQ ID NO:23 y una región variable de cadena ligera que comprende una secuencia de aminoácidos de la SEQ ID NO:24.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662302562P | 2016-03-02 | 2016-03-02 | |
| PCT/US2017/020529 WO2017151979A1 (en) | 2016-03-02 | 2017-03-02 | Eribulin-based antibody-drug conjugates and methods of use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2880402T3 true ES2880402T3 (es) | 2021-11-24 |
Family
ID=58347964
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17711475T Active ES2880402T3 (es) | 2016-03-02 | 2017-03-02 | Conjugados de anticuerpo-fármaco basados en eribulina y métodos de uso |
Country Status (35)
| Country | Link |
|---|---|
| US (4) | US10548986B2 (es) |
| EP (2) | EP3423105B1 (es) |
| JP (6) | JP6599019B2 (es) |
| KR (4) | KR102702620B1 (es) |
| CN (4) | CN114272389B (es) |
| AR (3) | AR107787A1 (es) |
| AU (3) | AU2017225982B2 (es) |
| BR (1) | BR112018067379A2 (es) |
| CA (2) | CA3297123A1 (es) |
| CL (3) | CL2018002456A1 (es) |
| CO (1) | CO2018008667A2 (es) |
| CY (1) | CY1124628T1 (es) |
| DK (1) | DK3423105T3 (es) |
| ES (1) | ES2880402T3 (es) |
| HR (1) | HRP20211125T1 (es) |
| HU (1) | HUE054726T2 (es) |
| IL (3) | IL292946B2 (es) |
| JO (3) | JOP20170053B1 (es) |
| LT (1) | LT3423105T (es) |
| MA (1) | MA45280B1 (es) |
| MD (1) | MD3423105T2 (es) |
| MX (4) | MX2018010562A (es) |
| MY (2) | MY199595A (es) |
| PE (3) | PE20231050A1 (es) |
| PH (1) | PH12018501847A1 (es) |
| PL (1) | PL3423105T3 (es) |
| PT (1) | PT3423105T (es) |
| RS (1) | RS62108B1 (es) |
| RU (1) | RU2754369C2 (es) |
| SG (2) | SG10202007520WA (es) |
| SI (1) | SI3423105T1 (es) |
| SM (1) | SMT202100416T1 (es) |
| TW (3) | TWI772288B (es) |
| UA (1) | UA125024C2 (es) |
| WO (1) | WO2017151979A1 (es) |
Families Citing this family (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2172487A1 (en) * | 2005-04-22 | 2010-04-07 | Morphotek Inc. | Antibodies with Immune Effector Activity that Internalize in Folate Receptor Alpha-Positive Cells |
| US11130815B2 (en) | 2015-06-24 | 2021-09-28 | Jcr Pharmaceuticals Co., Ltd. | Fusion proteins containing a BDNF and an anti-human transferrin receptor antibody |
| TWI769982B (zh) | 2015-06-24 | 2022-07-11 | 日商Jcr製藥股份有限公司 | 通過血腦障壁之抗人類運鐵蛋白受體抗體 |
| TWI761413B (zh) | 2016-12-26 | 2022-04-21 | 日商Jcr製藥股份有限公司 | 通過血腦障壁之新穎抗人類運鐵蛋白受體抗體 |
| AU2017385274B2 (en) | 2016-12-26 | 2024-02-22 | Jcr Pharmaceuticals Co., Ltd. | Fusion protein including BDNF |
| ES2931533T3 (es) | 2017-04-05 | 2022-12-30 | Harvard College | Compuesto macrocíclico y usos del mismo |
| US11498892B2 (en) | 2017-07-06 | 2022-11-15 | President And Fellows Of Harvard College | Fe/Cu-mediated ketone synthesis |
| HUE061306T2 (hu) | 2017-07-06 | 2023-06-28 | Harvard College | Halikondrinok szintézise |
| CA3081539A1 (en) * | 2017-11-08 | 2019-05-16 | Yafei Shanghai Biolog Medicine Science & Technology Co., Ltd. | Conjugates of biomolecule and use thereof |
| CN111566113B (zh) | 2017-11-15 | 2024-01-09 | 哈佛大学的校长及成员们 | 大环化合物及其用途 |
| MX2020012940A (es) | 2018-06-01 | 2021-03-25 | Eisai R&D Man Co Ltd | Metodos de uso de moduladores de empalme. |
| JOP20200268A1 (ar) | 2018-06-01 | 2020-10-26 | Eisai R&D Man Co Ltd | تضفير مترافقات عقار جسم مضاد معدل وطرق استخدامها |
| TW202019487A (zh) * | 2018-08-06 | 2020-06-01 | 日商第一三共股份有限公司 | 抗體-藥物結合物及微管蛋白抑制劑之組合 |
| PE20211473A1 (es) | 2018-12-13 | 2021-08-05 | Eisai Randd Man Co Ltd | Conjugados de anticuerpo-farmaco de herboxidieno y metodos de uso |
| JP7607564B2 (ja) * | 2018-12-21 | 2024-12-27 | ノバルティス アーゲー | Pmel17に対する抗体及びその結合体 |
| CN110568121A (zh) * | 2019-07-11 | 2019-12-13 | 山东省药学科学院 | 一种艾日布林及含艾日布林的制剂中有关物质的检测方法 |
| JP7657213B2 (ja) * | 2019-11-07 | 2025-04-04 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 抗メソテリンエリブリン抗体-薬物コンジュゲート及び使用方法 |
| US20230074385A1 (en) * | 2019-12-23 | 2023-03-09 | Eisai R&D Management Co., Ltd. | Method for producing eribulin-based antibody-drug conjugate |
| CN118949063A (zh) * | 2020-01-22 | 2024-11-15 | 上海森辉医药有限公司 | 艾日布林衍生物的药物偶联物、其制备方法及其在医药上的应用 |
| CN113274507B (zh) * | 2020-02-20 | 2025-02-28 | 亚飞(上海)生物医药科技有限公司 | 靶向递送和激活的免疫刺激性偶联复合物的制备和用途 |
| CN113509557A (zh) * | 2020-04-09 | 2021-10-19 | 嘉兴优博生物技术有限公司 | 靶向蛋白酶降解平台(ted) |
| AR122546A1 (es) | 2020-06-05 | 2022-09-21 | Eisai R&D Man Co Ltd | Conjugados de anticuerpo anti bcma-fármaco y métodos de uso |
| EP4178624A2 (en) * | 2020-07-07 | 2023-05-17 | Bionecure Therapeutics, Inc. | Maytansinoids as adc payloads and their use for the treatment of cancer |
| KR102810379B1 (ko) * | 2020-08-07 | 2025-05-21 | 주식회사 피노바이오 | 데옥시사이티딘계 항암제 및 실릴 에테르 함유 링커를 포함하는 접합체 및 이의 용도 |
| US12590977B2 (en) * | 2020-09-11 | 2026-03-31 | Alexion Pharmaceuticals, Inc. | Anti-ceruloplasmin antibodies and uses thereof |
| US20230398232A1 (en) * | 2020-09-28 | 2023-12-14 | Navrogen, Inc. | Composition and Use of Alternatively Formatted Anti-Mesothelin Antibodies for the Treatment of Cancer |
| WO2022082068A1 (en) * | 2020-10-18 | 2022-04-21 | Ardeagen Corporation | Anti-msln binding agents, conjugates thereof and methods of using the same |
| CN114560940B (zh) * | 2020-11-27 | 2023-07-14 | 缔码生物科技(武汉)有限公司 | 一种抗SIRPα兔重组单克隆抗体及其制备方法和应用 |
| US20240100177A1 (en) * | 2020-12-04 | 2024-03-28 | Dyne Therapeutics, Inc. | Antibody-oligonucleotide complexes and uses thereof |
| EP4241789A4 (en) * | 2020-12-08 | 2025-04-02 | Nona Biosciences (Shanghai) Co., Ltd. | Protein-drug conjugate and site-specific conjugation method |
| US20230390411A1 (en) * | 2021-01-28 | 2023-12-07 | Nanjing Chempion Biotechnology Co., Ltd. | Conjugate and use thereof |
| CN116568707A (zh) * | 2021-01-29 | 2023-08-08 | 上海翰森生物医药科技有限公司 | 一种抗体药物偶联物及其医药用途 |
| JP7717832B2 (ja) * | 2021-04-10 | 2025-08-04 | ジェンマブ エー/エス | Folr1結合剤、そのコンジュゲートおよびこれを使用する方法 |
| EP4349371A4 (en) * | 2021-06-02 | 2025-06-25 | Bio-Thera Solutions, Ltd. | DRUG CONJUGATE AND USE THEREOF |
| AU2022316425A1 (en) * | 2021-07-22 | 2024-02-01 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Drug conjugate of eribulin derivative |
| EP4385522A1 (en) * | 2021-08-13 | 2024-06-19 | Kunshan Xinyunda Biotech Co., Ltd. | Microtubule inhibitor-based antibody-drug conjugate |
| CN117881431A (zh) * | 2021-08-24 | 2024-04-12 | 昆山新蕴达生物科技有限公司 | 一种由可断裂连接子偶联的抗体偶联药物 |
| CN117677400A (zh) * | 2021-09-16 | 2024-03-08 | 正大天晴药业集团股份有限公司 | 抗her3抗体药物偶联物及其组合物和用途 |
| WO2023055376A1 (en) * | 2021-09-30 | 2023-04-06 | Virtuoso Binco, Inc. | Multispecific antibodies for targeting cd47 and icam1 and methods of use thereof |
| WO2023061466A1 (zh) * | 2021-10-14 | 2023-04-20 | 江苏恒瑞医药股份有限公司 | 一种艾日布林衍生物的制备方法 |
| CN117980327A (zh) | 2021-11-03 | 2024-05-03 | 杭州多禧生物科技有限公司 | 抗体的特异性偶联 |
| CN116199740A (zh) * | 2021-12-01 | 2023-06-02 | 上海生物制品研究所有限责任公司 | 抗体药物偶联物及其用途 |
| CN118401258A (zh) * | 2021-12-09 | 2024-07-26 | 昆山新蕴达生物科技有限公司 | 一种亲和力改善的抗体-药物偶联物、其制备方法及应用 |
| WO2023124963A1 (zh) * | 2021-12-27 | 2023-07-06 | 昆山新蕴达生物科技有限公司 | 一种可逆反应减少的抗体-药物偶联物,其制备方法及应用 |
| WO2023143320A1 (zh) * | 2022-01-28 | 2023-08-03 | 博瑞生物医药(苏州)股份有限公司 | 用于制备抗体药物偶联物的连接子、化合物及用途 |
| CN118591394A (zh) * | 2022-01-28 | 2024-09-03 | 启德医药科技(苏州)有限公司 | 抗体药物偶联物、药物组合物及用途 |
| CN118647408A (zh) * | 2022-02-16 | 2024-09-13 | 苏州宜联生物医药有限公司 | 抗体-艾日布林或其衍生物的偶联物、其中间体、制备方法、药物组合物和用途 |
| CR20240375A (es) * | 2022-03-11 | 2025-01-06 | Mablink Bioscience | Conjugados anticuerpo-fármaco y sus usos |
| JP2025512327A (ja) | 2022-04-12 | 2025-04-17 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | エリブリンベースの抗体-薬物コンジュゲート及び使用方法 |
| US20250249114A1 (en) * | 2022-04-12 | 2025-08-07 | Bio-Thera Solutions, Ltd. | Method for treating her2-positive solid tumor |
| KR20250084935A (ko) * | 2022-10-14 | 2025-06-11 | 진콴텀 헬스케어 (쑤저우) 씨오., 엘티디. | 링커-페이로드 화합물, 접합체 및 이의 응용 분야 |
| CN117917248A (zh) * | 2022-10-20 | 2024-04-23 | 英诺湖医药(杭州)有限公司 | 酶裂解连接子及包含其的配体-艾瑞布林偶联物 |
| EP4619038A2 (en) * | 2022-11-14 | 2025-09-24 | Cornell University | Anionically functionalized polypeptides, and methods of making same and uses thereof |
| KR20250133788A (ko) | 2023-01-17 | 2025-09-08 | 시스트이뮨, 인코포레이티드 | 에리불린 약물 접합체 |
| TW202444422A (zh) | 2023-01-19 | 2024-11-16 | 大陸商江蘇恆瑞醫藥股份有限公司 | 一種含艾日布林衍生物藥物偶聯物的醫藥組成物 |
| JP2026505404A (ja) * | 2023-02-13 | 2026-02-13 | ブリス バイオファーマシューティカル(ハンゾウ)カンパニー,リミテッド | がんの治療に使用される抗体-薬物複合体 |
| CN120957757A (zh) * | 2023-05-12 | 2025-11-14 | 四川科伦博泰生物医药股份有限公司 | 大环类药物偶联物及其制备方法和用途 |
| EP4710946A1 (en) * | 2023-05-12 | 2026-03-18 | Medilink Therapeutics (Suzhou) Co., Ltd. | Multi-projectile antibody-drug conjugate, and preparation method therefor and use thereof |
| CN120957758A (zh) * | 2023-05-12 | 2025-11-14 | 四川科伦博泰生物医药股份有限公司 | 大环化合物及其制备方法和用途 |
| JP2024165695A (ja) | 2023-05-18 | 2024-11-28 | セイコーエプソン株式会社 | 印刷方法、印刷ヘッドユニットおよびロボットシステム |
| EP4725969A1 (en) | 2023-06-09 | 2026-04-15 | Latticon (Suzhou) Biopharmaceuticals Co., Ltd. | Anti-her2 complementary bispecific antibody-drug conjugate, preparation method therefor and use thereof |
| US12319747B2 (en) | 2023-07-03 | 2025-06-03 | Medicovestor, Inc. | Methods of using anti-SP17 immunotherapeutics |
| WO2025027529A1 (en) | 2023-07-31 | 2025-02-06 | Advesya | Anti-il-1rap antibody drug conjugates and methods of use thereof |
| WO2025078841A2 (en) | 2023-10-11 | 2025-04-17 | Antikor Biopharma Limited | Antibodies, conjugates, and uses thereof |
| TW202530697A (zh) | 2023-10-13 | 2025-08-01 | 日商衛材R&D企管股份有限公司 | 用於基於艾日布林之抗體-藥物結合物之生物標記及使用方法 |
| TW202518024A (zh) | 2023-10-13 | 2025-05-01 | 日商衛材R&D企管股份有限公司 | 抗體-藥物軛合物代謝物 |
| US12364777B2 (en) | 2023-10-20 | 2025-07-22 | Medicovestor, Inc. | Homodimeric antibodies for use in treating cancers and methods of use |
| WO2025106586A1 (en) * | 2023-11-13 | 2025-05-22 | Luxvitae Therapeutics Inc. | Macrocyclic ketone compounds and applications thereof |
| CN120131991A (zh) * | 2023-12-13 | 2025-06-13 | 成都百利多特生物药业有限责任公司 | 抗人Claudin18.2抗体-喜树碱类药物偶联物及其医药用途 |
| TW202530255A (zh) | 2023-12-15 | 2025-08-01 | 法商亞維西亞有限公司 | 抗il-1rap結合結構域及其抗體-藥物偶聯物 |
| US12116410B1 (en) | 2023-12-26 | 2024-10-15 | Medicovestor, Inc. | Methods of manufacturing dimeric antibodies |
| US12600795B2 (en) | 2023-12-26 | 2026-04-14 | Medicovestor, Inc. | Oligomeric IgG for immunotherapeutics and diagnostics |
| WO2025162492A1 (zh) * | 2024-01-29 | 2025-08-07 | 甘李药业股份有限公司 | 一种连接子、配体-药物偶联物及其医药用途 |
| US12240900B1 (en) | 2024-02-02 | 2025-03-04 | Medicovestor, Inc. | Nucleic acids, vectors, and cells that encode antibodies and other proteins that bind folate receptor alpha |
| US12258396B1 (en) | 2024-02-02 | 2025-03-25 | Medicovestor, Inc. | Methods of using immunotherapeutics that bind folate receptor alpha |
| US12378314B1 (en) | 2024-02-02 | 2025-08-05 | Medicovestor, Inc. | Proteins that bind folate receptor alpha including fully-human antibodies |
| WO2025199464A1 (en) | 2024-03-22 | 2025-09-25 | Eisai R&D Management Co., Ltd. | Anti-trop2 antibody-drug conjugates and methods of use |
| WO2025228172A1 (zh) * | 2024-04-28 | 2025-11-06 | 上海拓济医药有限责任公司 | 艾日布林衍生物及其抗体药物偶联物和医药用途 |
| WO2026037417A1 (zh) * | 2024-08-15 | 2026-02-19 | 上海睿智医药技术有限公司 | 艾日布林相关的新型活性分子、含其的连接子-药物毒素分子化合物和它们在抗体偶联物中的应用 |
Family Cites Families (227)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4880935A (en) | 1986-07-11 | 1989-11-14 | Icrf (Patents) Limited | Heterobifunctional linking agents derived from N-succinimido-dithio-alpha methyl-methylene-benzoates |
| US4880078A (en) | 1987-06-29 | 1989-11-14 | Honda Giken Kogyo Kabushiki Kaisha | Exhaust muffler |
| IL106992A (en) | 1988-02-11 | 1994-06-24 | Bristol Myers Squibb Co | Acylhydrazone derivatives of anthracycline and methods for their preparation |
| EP0368684B2 (en) | 1988-11-11 | 2004-09-29 | Medical Research Council | Cloning immunoglobulin variable domain sequences. |
| WO1992019244A2 (en) | 1991-05-01 | 1992-11-12 | Henry M. Jackson Foundation For The Advancement Of Military Medicine | A method for treating infectious respiratory diseases |
| EP1400536A1 (en) | 1991-06-14 | 2004-03-24 | Genentech Inc. | Method for making humanized antibodies |
| US5622929A (en) * | 1992-01-23 | 1997-04-22 | Bristol-Myers Squibb Company | Thioether conjugates |
| US5934272A (en) | 1993-01-29 | 1999-08-10 | Aradigm Corporation | Device and method of creating aerosolized mist of respiratory drug |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US6019968A (en) | 1995-04-14 | 2000-02-01 | Inhale Therapeutic Systems, Inc. | Dispersible antibody compositions and methods for their preparation and use |
| ATE234635T1 (de) | 1995-12-22 | 2003-04-15 | Bristol Myers Squibb Co | Verzweigte hydrazongruppen enthaltende kuppler |
| DK0885002T3 (da) | 1996-03-04 | 2011-08-22 | Penn State Res Found | Materialer og fremgangsmåder til forøgelse af cellulær internalisering |
| US5874064A (en) | 1996-05-24 | 1999-02-23 | Massachusetts Institute Of Technology | Aerodynamically light particles for pulmonary drug delivery |
| US5985309A (en) | 1996-05-24 | 1999-11-16 | Massachusetts Institute Of Technology | Preparation of particles for inhalation |
| US5855913A (en) | 1997-01-16 | 1999-01-05 | Massachusetts Instite Of Technology | Particles incorporating surfactants for pulmonary drug delivery |
| DE69732306T2 (de) | 1997-01-16 | 2006-01-12 | Massachusetts Institute Of Technology, Cambridge | Zubereitung von partikelhaltigen arzneimitteln zur inhalation |
| CA2336139C (en) | 1998-06-24 | 2008-10-14 | Advanced Inhalation Research, Inc. | Large porous particles emitted from an inhaler |
| US6019963A (en) | 1998-11-20 | 2000-02-01 | D.S.C. Products, Inc. | Deodorizing composition containing tea tree and eucalyptus oils |
| US7109019B2 (en) | 1999-01-06 | 2006-09-19 | The Regents Of The University Of California | Gene cluster for production of the enediyne antitumor antibiotic C-1027 |
| CA2445826C (en) | 2001-05-02 | 2014-03-25 | Purdue Research Foundation | Treatment and diagnosis of macrophage mediated disease |
| US7214660B2 (en) | 2001-10-10 | 2007-05-08 | Neose Technologies, Inc. | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| US7265085B2 (en) | 2001-10-10 | 2007-09-04 | Neose Technologies, Inc. | Glycoconjugation methods and proteins/peptides produced by the methods |
| US7265084B2 (en) | 2001-10-10 | 2007-09-04 | Neose Technologies, Inc. | Glycopegylation methods and proteins/peptides produced by the methods |
| US8008252B2 (en) | 2001-10-10 | 2011-08-30 | Novo Nordisk A/S | Factor VII: remodeling and glycoconjugation of Factor VII |
| US7173003B2 (en) | 2001-10-10 | 2007-02-06 | Neose Technologies, Inc. | Granulocyte colony stimulating factor: remodeling and glycoconjugation of G-CSF |
| US7179617B2 (en) | 2001-10-10 | 2007-02-20 | Neose Technologies, Inc. | Factor IX: remolding and glycoconjugation of Factor IX |
| US7795210B2 (en) | 2001-10-10 | 2010-09-14 | Novo Nordisk A/S | Protein remodeling methods and proteins/peptides produced by the methods |
| US7696163B2 (en) | 2001-10-10 | 2010-04-13 | Novo Nordisk A/S | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| US7297511B2 (en) | 2001-10-10 | 2007-11-20 | Neose Technologies, Inc. | Interferon alpha: remodeling and glycoconjugation of interferon alpha |
| US7125843B2 (en) | 2001-10-19 | 2006-10-24 | Neose Technologies, Inc. | Glycoconjugates including more than one peptide |
| US7399613B2 (en) | 2001-10-10 | 2008-07-15 | Neose Technologies, Inc. | Sialic acid nucleotide sugars |
| US7439043B2 (en) | 2001-10-10 | 2008-10-21 | Neose Technologies, Inc. | Galactosyl nucleotide sugars |
| US7226903B2 (en) | 2001-10-10 | 2007-06-05 | Neose Technologies, Inc. | Interferon beta: remodeling and glycoconjugation of interferon beta |
| US7157277B2 (en) | 2001-11-28 | 2007-01-02 | Neose Technologies, Inc. | Factor VIII remodeling and glycoconjugation of Factor VIII |
| EP1531846A4 (en) | 2002-02-27 | 2006-04-19 | Us Gov Health & Human Serv | Conjugates of ligand, linker and cytotoxic agent and related compositions and methods of use |
| SI1545613T1 (sl) * | 2002-07-31 | 2011-11-30 | Seattle Genetics Inc | Avristatinski konjugati in njihova uporaba za zdravljenje raka avtoimunske bolezni ali infekcijskebolezni |
| JP2004119755A (ja) | 2002-09-27 | 2004-04-15 | Alps Electric Co Ltd | 磁気検出素子及びその製造方法 |
| US20040071540A1 (en) | 2002-10-15 | 2004-04-15 | Lucas Philip J. | Disposable/recyclable pallet system and method |
| US20080025989A1 (en) | 2003-02-20 | 2008-01-31 | Seattle Genetics, Inc. | Anti-cd70 antibody-drug conjugates and their use for the treatment of cancer and immune disorders |
| EP2055189A1 (en) | 2003-04-09 | 2009-05-06 | Neose Technologies, Inc. | Glycopegylation methods and proteins/peptides produced by the methods |
| JP2007505914A (ja) | 2003-09-18 | 2007-03-15 | コンビナトアールエックス インコーポレーティッド | 新生物の治療のための薬物の併用方法 |
| US6997024B2 (en) | 2003-10-01 | 2006-02-14 | Truth Hardware Corporation | Pull door lock |
| US20050148535A1 (en) | 2003-10-30 | 2005-07-07 | Lacasse Eric | IAP nucleobase oligomers and oligomeric complexes and uses thereof |
| US8012944B2 (en) | 2003-10-30 | 2011-09-06 | Pharmascience Inc. | Method for treating cancer using IAP antisense oligomer and chemotherapeutic agent |
| CA2556027C (en) | 2004-02-12 | 2015-11-24 | Morphotek, Inc. | Monoclonal antibodies that specifically block biological activity of a tumor antigen |
| US7117807B2 (en) | 2004-02-17 | 2006-10-10 | University Of Florida Research Foundation, Inc. | Dynamically modifiable polymer coatings and devices |
| US20080253960A1 (en) | 2004-04-01 | 2008-10-16 | The Trustees Of The University Of Pennsylvania Center For Technology Transfer | Lipoprotein-Based Nanoplatforms |
| WO2005115454A2 (en) | 2004-05-07 | 2005-12-08 | Massachusetts Institute Of Technology | Methods and compositions for cancer treatment relating to brca1 brct domain recognition of phosphorylated bach1 |
| EP2522663B1 (en) | 2004-06-03 | 2015-04-01 | Eisai R&D Management Co., Ltd. | Intermediates for the preparation of halichondrin B |
| KR101270829B1 (ko) | 2004-09-23 | 2013-06-07 | 제넨테크, 인크. | 시스테인 유전자조작 항체 및 접합체 |
| JO3000B1 (ar) * | 2004-10-20 | 2016-09-05 | Genentech Inc | مركبات أجسام مضادة . |
| US20060148014A1 (en) | 2004-12-09 | 2006-07-06 | Sergei Agoulnik | Tubulin isotype screening in cancer therapy using hemiasterlin analogs |
| EP1831697A4 (en) | 2004-12-09 | 2011-01-26 | Eisai R&D Man Co Ltd | SEARCH FOR TUBULINISOTYPES IN CANCER THERAPY USING HALICHONDRIN B ANALOG |
| JP5171621B2 (ja) | 2005-07-07 | 2013-03-27 | シアトル ジェネティックス, インコーポレイテッド | フェニルアラニン側鎖修飾をc末端に有するモノメチルバリン化合物 |
| US8871720B2 (en) | 2005-07-07 | 2014-10-28 | Seattle Genetics, Inc. | Monomethylvaline compounds having phenylalanine carboxy modifications at the C-terminus |
| GB0515025D0 (en) | 2005-07-21 | 2005-08-31 | Novartis Ag | Organic compounds |
| AU2006279304A1 (en) | 2005-08-19 | 2007-02-22 | Endocyte, Inc. | Multi-drug ligand conjugates |
| WO2007031734A1 (en) | 2005-09-14 | 2007-03-22 | Ucb Pharma S.A. | Comb polymers |
| CA2632903C (en) | 2005-12-02 | 2015-11-24 | Vianova Labs, Inc. | Treatment of cancer and other diseases |
| EP2021502A4 (en) | 2006-05-09 | 2010-08-25 | Mas Metabolic Analytical Servi | GENES AND ATYPICAL MARKERS IN TYPE 2 DIABETES AND OBESITY |
| US20140037539A1 (en) | 2012-07-27 | 2014-02-06 | Rutgers, The State University Of New Jersey | Antibody cocktails for breast cancer radioimmunotherapy |
| US20150030534A1 (en) | 2013-07-26 | 2015-01-29 | Rutgers, The State University Of New Jersey | Antibody cocktails for breast cancer radioimmunotherapy |
| US8398956B2 (en) | 2007-01-11 | 2013-03-19 | Immunomedics, Inc. | In vivo copper-free click chemistry for delivery of therapeutic and/or diagnostic agents |
| WO2008101231A2 (en) | 2007-02-16 | 2008-08-21 | Endocyte, Inc. | Methods and compositions for treating and diagnosing kidney disease |
| CN104127878A (zh) | 2007-03-14 | 2014-11-05 | 恩多塞特公司 | 结合配体连接的微管溶素递药缀合物 |
| EP1977765A1 (en) | 2007-04-03 | 2008-10-08 | Diatos | Peptide prodrugs |
| RU2489437C2 (ru) | 2007-10-03 | 2013-08-10 | Эйсай Ар Энд Ди Менеджмент Ко., Лтд. | Промежуточные соединения и способы синтеза аналогов галихондрина в |
| MX2010003718A (es) * | 2007-10-19 | 2010-04-21 | Genentech Inc | Anticuerpos anti-tenb2 producidos por ingenieria de cisteina y conjugados de farmaco y anticuerpo. |
| JP5065228B2 (ja) | 2007-10-30 | 2012-10-31 | 富士通株式会社 | 携帯端末装置 |
| EP2265283B1 (en) | 2008-03-18 | 2014-09-03 | Seattle Genetics, Inc. | Auristatin drug linker conjugates |
| WO2009121031A1 (en) | 2008-03-27 | 2009-10-01 | Vascular Biosciences, Inc. | Methods of novel therapeutic candidate identification through gene expression analysis in vascular-related diseases |
| US20100260668A1 (en) | 2008-04-29 | 2010-10-14 | Abbott Laboratories | Dual Variable Domain Immunoglobulins and Uses Thereof |
| BRPI0910482A2 (pt) | 2008-04-29 | 2019-09-24 | Abbott Lab | imunoglobinas de domínio variável duplo e usos das mesmas |
| EP2276506A4 (en) * | 2008-04-30 | 2014-05-07 | Immunogen Inc | EFFICIENT CONJUGATES AND HYDROPHILIC BINDER |
| BRPI0913366A8 (pt) | 2008-06-03 | 2017-07-11 | Abbott Lab | Imunoglobulinas de domínio variável duplo e seus usos |
| PE20100092A1 (es) | 2008-06-03 | 2010-03-12 | Abbott Lab | Inmunoglobulina con dominio variable dual y usos de la misma |
| NZ603698A (en) | 2008-07-08 | 2014-03-28 | Abbvie Inc | Prostaglandin e2 dual variable domain immunoglobulins and uses thereof |
| WO2010029986A1 (ja) | 2008-09-12 | 2010-03-18 | 参天製薬株式会社 | スルホン酸エステル構造を導入したフェニル基を置換基として有する新規1,2,3,4-テトラヒドロキノキサリン誘導体からなるグルココルチコイド受容体アゴニスト |
| MX2011005953A (es) | 2008-12-04 | 2011-08-17 | Abbott Lab | Inmunoglobulinas de dominio variable dual y usos de las mismas. |
| EP2403531A4 (en) | 2009-03-05 | 2013-02-27 | Abbott Lab | IL-17 BINDING PROTEINS |
| US20140339088A1 (en) | 2009-03-09 | 2014-11-20 | Virginia Tech Intellectual Properties, Inc. | Dielectrophoresis methods for determining a property of a plurality of cancer cells |
| NZ596711A (en) | 2009-05-01 | 2013-11-29 | Dual variable domain immunoglobulins and uses thereof | |
| US8686520B2 (en) * | 2009-05-29 | 2014-04-01 | International Business Machines Corporation | Spin-torque magnetoresistive structures |
| UY32808A (es) | 2009-07-29 | 2011-02-28 | Abbott Lab | Inmunoglobulinas como dominio variable dual y usos de las mismas |
| BR112012003346A2 (pt) * | 2009-08-15 | 2016-11-16 | Genentech Inc | metodo de tratamento de um paciente diagnostico com cancer de mama metastico previamente tratado kit para tratamento de cancer de mama metastatico previamente tratado em um paciente humano metodo para instruir um paciente humano com cancer metodo promocional e metodo comercial |
| KR20120060877A (ko) * | 2009-09-01 | 2012-06-12 | 아보트 러보러터리즈 | 이원 가변 도메인 면역글로불린 및 이의 용도 |
| GB0917044D0 (en) | 2009-09-29 | 2009-11-18 | Cytoguide As | Agents, uses and methods |
| AU2010306677B2 (en) | 2009-10-15 | 2013-05-23 | Abbvie Inc. | Dual variable domain immunoglobulins and uses thereof |
| UY32979A (es) | 2009-10-28 | 2011-02-28 | Abbott Lab | Inmunoglobulinas con dominio variable dual y usos de las mismas |
| NZ599866A (en) | 2009-11-06 | 2014-09-26 | Plexxikon Inc | Compounds and methods for kinase modulation, and indications therefor |
| WO2011094339A1 (en) | 2010-01-26 | 2011-08-04 | Eisai R&D Management Co., Ltd. | Furo [3, 2 -b] pyrane derivatives useful in the synthesis of halichondrin b analogs |
| US8710035B2 (en) | 2010-03-24 | 2014-04-29 | The University Of Chicago | Methods and compositions related to glucocorticoid receptor antagonists and breast cancer |
| CN105837411B (zh) | 2010-03-24 | 2019-06-18 | 俄亥俄州立大学 | 用于葡萄糖转运抑制的组合物和方法 |
| KR101539684B1 (ko) | 2010-05-14 | 2015-07-27 | 애브비 인코포레이티드 | Il-1 결합 단백질 |
| JP6055404B2 (ja) | 2010-06-15 | 2016-12-27 | ゲンマブ エー/エス | 組織因子に対するヒト抗体薬物結合体 |
| UY33492A (es) | 2010-07-09 | 2012-01-31 | Abbott Lab | Inmunoglobulinas con dominio variable dual y usos de las mismas |
| US8735546B2 (en) | 2010-08-03 | 2014-05-27 | Abbvie Inc. | Dual variable domain immunoglobulins and uses thereof |
| PE20140229A1 (es) | 2010-08-26 | 2014-03-27 | Abbvie Inc | Inmunoglobulinas con dominio variable dual y usos de las mismas |
| US9089570B2 (en) | 2010-09-03 | 2015-07-28 | Tactical Therapeutics Inc | Compositions for treating cancers having acquired resitance to prior chemotherapeutic and targeted drugs using carboxyamidotriazole orotate |
| US9192677B2 (en) | 2010-09-07 | 2015-11-24 | Johannes Kepler Universität Linz | Biodegradable, water soluble and pH responsive poly(organo)phosphazenes |
| WO2012065019A2 (en) | 2010-11-12 | 2012-05-18 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of p13k alpha |
| WO2012065057A2 (en) | 2010-11-12 | 2012-05-18 | Exelixis, Inc. | Phosphatidylinositol 3-kinase inhibitors and methods of their use |
| AU2011336396B2 (en) | 2010-12-02 | 2016-02-04 | Immunomedics, Inc. | In vivo copper-free click chemistry for delivery of therapeutic and/or diagnostic agents |
| CN103517921B (zh) | 2010-12-21 | 2017-07-18 | Abbvie 公司 | IL‑1‑α和‑β双特异性双重可变结构域免疫球蛋白及其用途 |
| WO2012088290A2 (en) | 2010-12-22 | 2012-06-28 | Abbott Laboratories | Tri-variable domain binding proteins and uses thereof |
| WO2012088302A2 (en) | 2010-12-22 | 2012-06-28 | Abbott Laboratories | Half immunoglobulin binding proteins and uses thereof |
| US20140024539A1 (en) | 2011-02-02 | 2014-01-23 | Translational Genomics Research Institute | Biomarkers and methods of use thereof |
| ES2720659T3 (es) | 2011-02-15 | 2019-07-23 | Vaxiion Therapeutics Llc | Composiciones terapéuticas y métodos para la administración dirigida, mediante minicélulas bacterianas, de moléculas bioactivas basadas en moléculas dirigidas que contienen Fc y anticuerpos |
| US9597340B2 (en) | 2011-02-25 | 2017-03-21 | Arena Pharmaceuticals, Inc. | Cannabinoid receptor modulators |
| WO2012119077A1 (en) | 2011-03-02 | 2012-09-07 | Morphotek Inc. | Co -administration of eribulin and farletuzumab for the treatment of breast cancer |
| WO2012118978A1 (en) | 2011-03-03 | 2012-09-07 | The Regents Of The University Of Colorado, A Body Corporate | Methods for treating oncovirus positive cancers |
| CN103502218B (zh) | 2011-03-04 | 2016-08-17 | 生命科技公司 | 用于缀合生物分子的化合物和方法 |
| AU2012229123B2 (en) | 2011-03-15 | 2017-02-02 | British Columbia Cancer Agency Branch | Methods of treating breast cancer with anthracycline therapy |
| EP2686441B1 (en) | 2011-03-18 | 2019-05-08 | Eisai R&D Management Co., Ltd. | Methods and uses for predicting response to eribulin |
| EP2508525A1 (en) | 2011-04-05 | 2012-10-10 | Bayer Pharma Aktiengesellschaft | Substituted 2,3-dihydroimidazo[1,2-c]quinazoline salts |
| US9132131B2 (en) | 2011-04-21 | 2015-09-15 | Saint Louis University | Use of adenosine A3 receptor agonists for treatment of neuropathic pain |
| KR102356286B1 (ko) | 2011-05-27 | 2022-02-08 | 암브룩스, 인코포레이티드 | 비-천연 아미노산 연결된 돌라스타틴 유도체를 함유하는 조성물, 이를 수반하는 방법, 및 용도 |
| KR20140037105A (ko) | 2011-05-27 | 2014-03-26 | 암브룩스, 인코포레이티드 | 비-천연 아미노산 연결된 돌라스타틴 유도체를 함유하는 조성물, 이를 수반하는 방법, 및 용도 |
| WO2012170640A1 (en) | 2011-06-07 | 2012-12-13 | The Trustees Of Columbia University In The City Of New York | Methods and compositions for trail-drug combination therapy |
| WO2012171020A1 (en) | 2011-06-10 | 2012-12-13 | Mersana Therapeutics, Inc. | Protein-polymer-drug conjugates |
| US8815226B2 (en) | 2011-06-10 | 2014-08-26 | Mersana Therapeutics, Inc. | Protein-polymer-drug conjugates |
| CA2842492A1 (en) | 2011-08-05 | 2013-02-14 | Bioasis Technologies, Inc. | P97 fragments with transfer activity |
| EP2785687B1 (en) | 2011-11-30 | 2019-02-20 | Sandoz AG | Process for preparation of (3r)-2,4-di-leaving group-3-methylbut-1-ene |
| CN104024237B (zh) | 2011-12-16 | 2016-02-24 | 阿方拉研究股份有限公司 | 用于制备3-((2s,5s)-4-亚甲基-5-(3-氧代丙基)四氢呋喃-2-基)丙醇衍生物的方法及其有用的中间体 |
| US9764038B2 (en) | 2011-12-23 | 2017-09-19 | Innate Pharma | Enzymatic conjugation of antibodies |
| JP2015502397A (ja) | 2011-12-23 | 2015-01-22 | ファイザー・インク | 部位特異的コンジュゲーションのための操作された抗体定常領域、ならびにそのための方法および使用 |
| CN104080793B (zh) | 2011-12-29 | 2017-09-19 | 阿方拉研究股份有限公司 | 2‑((2s,3s,4r,5r)‑5‑((s)‑3‑氨基‑2‑羟基丙‑1‑基)‑4‑甲氧基‑3‑(苯磺酰甲基)四氢呋喃‑2‑基)乙醛衍生物以及它们的制备方法 |
| AR092790A1 (es) | 2012-02-01 | 2015-05-06 | Euro Celtique Sa | Derivados bencimidazolicos del acido hidroxamico |
| WO2013130093A1 (en) * | 2012-03-02 | 2013-09-06 | Genentech, Inc. | Biomarkers for treatment with anti-tubulin chemotherapeutic compounds |
| AU2013201584A1 (en) | 2012-03-12 | 2013-09-26 | Merrimack Pharmaceuticals, Inc. | Methods for treating pancreatic cancer using combination therapies comprising an anti-ErbB3 antibody |
| US9358235B2 (en) | 2012-03-19 | 2016-06-07 | Plexxikon Inc. | Kinase modulation, and indications therefor |
| CN104470535A (zh) | 2012-03-29 | 2015-03-25 | 阿尔托生物科学有限公司 | 用于治疗肿瘤的方法 |
| WO2013142999A1 (en) | 2012-03-30 | 2013-10-03 | Alphora Research Inc. | Synthetic process for preparation of macrocyclic c1-keto analogs of halichondrin b and intermediates useful therein |
| BR112014024494A2 (pt) | 2012-04-02 | 2017-08-08 | Merrimack Pharmaceuticals Inc | dosagem e administração de anticorpos anti-igf-1r e anti-erbb3 monoespecíficos e biespecíficos |
| HK1208727A1 (en) | 2012-05-15 | 2016-03-11 | Diatech Oncology Llc | Tumor cell isolation/purification process and methods for use thereof |
| US9981046B2 (en) | 2012-05-15 | 2018-05-29 | Concortis Biosystems, Corp., a wholly owned Subsidiary of Sorrento Therapeutics, Inc. | Drug-conjugates, conjugation methods, and uses thereof |
| US10800856B2 (en) | 2012-06-07 | 2020-10-13 | Ambrx, Inc. | Prostate-specific membrane antigen antibody drug conjugates |
| WO2013182668A1 (en) | 2012-06-08 | 2013-12-12 | F. Hoffmann-La Roche Ag | Mutant selectivity and combinations of a phosphoinositide 3 kinase inhibitor compound and chemotherapeutic agents for the treatment of cancer |
| SG11201408494UA (en) | 2012-06-19 | 2015-02-27 | Ambrx Inc | Anti-cd70 antibody drug conjugates |
| WO2014005010A2 (en) | 2012-06-29 | 2014-01-03 | Nanostring Technologies, Inc. | Methods of treating breast cancer with gemcitabine therapy |
| WO2014005089A2 (en) | 2012-06-29 | 2014-01-03 | The Research Foundation Of State University Of New York | Polyenolic zinc-binding agents (pezbins) actively promote inactivation of cancer stem cells and potentiate cytotoxic anti-tumor drug substances |
| PL2872157T3 (pl) | 2012-07-12 | 2020-07-13 | Hangzhou Dac Biotech Co., Ltd | Koniugaty wiążących komórkę cząsteczek ze środkami cytotoksycznymi |
| US20140069822A1 (en) | 2012-09-10 | 2014-03-13 | Antec Leyden B.V. | Electrochemical reduction of disulfide bonds in proteinaceous substances and electrochemical cell for carrying out such reduction |
| WO2014047199A1 (en) | 2012-09-19 | 2014-03-27 | The Research Foundation For The State University Of New York | Novel prodrugs for selective anticancer therapy |
| DK2903616T3 (da) | 2012-10-04 | 2018-01-29 | Ab Science | Anvendelse af masitinib i kombination med gemcitabin til behandling af en undergruppe af patienter, der lider af pankreascancer |
| WO2014058317A1 (en) | 2012-10-10 | 2014-04-17 | Stichting Het Nederlands Kanker Instituut-Antoni van Leeuwenhoek Ziekenhuis | Methods and means for predicting resistance to anti-cancer treatment |
| WO2014063443A1 (zh) | 2012-10-22 | 2014-05-01 | Zhang Yuliang | 自冷式热力做功方法 |
| FI2911699T4 (fi) | 2012-10-23 | 2025-12-09 | Synaffix Bv | Muokattu vasta-aine, vasta-ainekonjugaatti ja menetelmä niiden valmistamiseksi |
| MX340090B (es) | 2012-11-05 | 2016-06-23 | Pfizer | Analogos de spliceostatina. |
| US10131682B2 (en) | 2012-11-24 | 2018-11-20 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to a cell binding molecules |
| BR112015012536A2 (pt) | 2012-12-03 | 2017-07-11 | Calithera Biosciences Inc | tratamento de câncer com inibidores heterocíclicos da glutaminase |
| US9243058B2 (en) | 2012-12-07 | 2016-01-26 | Amgen, Inc. | BCMA antigen binding proteins |
| TW201425336A (zh) * | 2012-12-07 | 2014-07-01 | Amgen Inc | Bcma抗原結合蛋白質 |
| US10226535B2 (en) | 2012-12-10 | 2019-03-12 | Mersana Therapeutics, Inc. | Auristatin compounds and conjugates thereof |
| CA2892863C (en) | 2012-12-10 | 2022-03-15 | Mersana Therapeutics, Inc. | Polymeric scaffold based on phf for targeted drug delivery |
| WO2014093640A1 (en) | 2012-12-12 | 2014-06-19 | Mersana Therapeutics,Inc. | Hydroxy-polmer-drug-protein conjugates |
| US20150314007A1 (en) | 2012-12-21 | 2015-11-05 | Glykos Finland Oy | Linker-payload molecule conjugates |
| US20150182634A1 (en) | 2012-12-28 | 2015-07-02 | Cody P. Coyne | Molecular Design and Chemical Synthesis of Pharmaceutical-Ligands and Pharmaceutical-Pharmaceutical Analogs with Multiple Mechanisms of Action |
| WO2014113792A1 (en) | 2013-01-19 | 2014-07-24 | New York University | Hydrogen-bond surrogate peptides and peptidomimetics for p53 reactivation |
| EP2945631A4 (en) | 2013-01-19 | 2016-06-29 | Univ New York | OLIGOOXOPIPERAZINE FOR THE REACTIVATION OF P53 |
| US20140220112A1 (en) | 2013-02-01 | 2014-08-07 | Zoneone Pharma, Inc. | Transformation of drug cyclodextrin complex compositions into compositions of mixtures of lipid vesicle encapsulated drug and cyclodextrin drug complexes |
| EP4309657A3 (en) | 2013-02-01 | 2024-02-28 | Celator Pharmaceuticals, Inc. | Remote loading of sparingly water-soluble drugs into liposomes |
| US9849193B2 (en) | 2013-02-08 | 2017-12-26 | University Of Louisville Research Foundation, Inc. | Nanoparticles for drug delivery |
| EP2958568B1 (en) | 2013-02-22 | 2020-03-04 | University Of Houston | Phosphaplatins as neuroprotective agents |
| US9376437B2 (en) | 2013-03-13 | 2016-06-28 | Oncoceutics, Inc | 7-benzyl-4-(2-methylbenzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one, salts thereof and methods of using the same in combination therapy |
| WO2015073072A1 (en) | 2013-11-15 | 2015-05-21 | Oncoceutics, Inc. | 7-benzyl-4-(2-methylbenzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1h)-one |
| CA2905181C (en) * | 2013-03-13 | 2020-06-02 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof for providing targeted therapy |
| WO2014160130A1 (en) | 2013-03-13 | 2014-10-02 | Oncoceutics, Inc. | Combination therapy with 7-benzyl-10-(2-methylbenzyl)-2,6,7,8,9,10-hexahydroimidazo[1,2-a]pyrido[4,3-d]pyrimidin-5(3h)-one |
| US20140271923A1 (en) | 2013-03-14 | 2014-09-18 | Christopher Brian Reid | Compositions & formulations for preventing and treating chronic diseases that cluster in patients such as cardiovascular disease, diabetes, obesity, polycystic ovary syndrome, hyperlipidemia and hypertension, as well as for preventing and treating other diseases and conditions |
| US20160022829A1 (en) | 2013-03-14 | 2016-01-28 | Mersana Therapeutics, Inc. | Tubulysin compounds and conjugates thereof |
| US20160038456A1 (en) | 2013-03-14 | 2016-02-11 | University Of Florida Research Foundation, Incorporated | Regulation of cancer using natural compounds and/or diet |
| PL2968440T3 (pl) | 2013-03-15 | 2019-12-31 | Zymeworks Inc. | Związki cytotoksyczne i antymitotyczne oraz sposoby ich stosowania |
| US9295731B2 (en) | 2013-04-01 | 2016-03-29 | Mark Quang Nguyen | Cleavable drug conjugates, compositions thereof and methods of use |
| WO2014168721A2 (en) | 2013-04-12 | 2014-10-16 | Tufts Medical Center | Methods and systems for designing and/or characterizing soluble lipidated ligand agents |
| WO2014176284A1 (en) | 2013-04-22 | 2014-10-30 | Avelas Biosciences, Inc. | Selective drug delivery compositions and methods of use |
| GB2513405A (en) | 2013-04-26 | 2014-10-29 | Adc Biotechnology Ltd | Method of synthesising ADCs using affinity resins |
| PL2991683T3 (pl) | 2013-05-02 | 2020-03-31 | Glykos Finland Oy | Koniugaty glikoproteiny lub glikanu z toksycznym ładunkiem |
| AR096238A1 (es) | 2013-05-15 | 2015-12-16 | Alphora Res Inc | Proceso para preparar derivados de 3-((2s,5s)-4-metilen-5-(3-oxopropil)tetrahidrofuran-2-il)propanol e intermediarios útiles para el mismo |
| GB201309807D0 (en) | 2013-05-31 | 2013-07-17 | Pharma Mar Sau | Antibody drug conjugates |
| WO2014197871A2 (en) | 2013-06-06 | 2014-12-11 | Igenica Biotherapeutics, Inc. | Antibody-drug conjugates, compositions and methods of use |
| US11229711B2 (en) | 2013-06-06 | 2022-01-25 | Magenta Therapeutics, Inc. | Linkers for antibody-drug conjugates and related compounds, compositions, and methods of use |
| EP3010547B1 (en) | 2013-06-20 | 2021-04-21 | Innate Pharma | Enzymatic conjugation of polypeptides |
| JP6744212B2 (ja) | 2013-06-21 | 2020-08-19 | イナート・ファルマ・ソシエテ・アノニムInnate Pharma Pharma S.A. | ポリペプチドの酵素的結合 |
| US20160299147A1 (en) | 2013-06-26 | 2016-10-13 | Afg Technologies S.À.R.L. | Screening, diagnosis, prognostication and treatment of ovarian cancer |
| CA2916537C (en) | 2013-07-03 | 2021-07-27 | Alphora Research Inc. | Synthetic process for preparation of macrocyclic c1-keto analogs of halichondrin b and intermediates useful therein including intermediates containing -so2-(p-tolyl) groups |
| WO2015017729A1 (en) | 2013-07-31 | 2015-02-05 | Virginia Tech Intellectual Properties, Inc. | Dielectrophoresis methods for determining a property of a plurality of cancer cells |
| WO2015017034A1 (en) | 2013-08-02 | 2015-02-05 | Gourdie Robert G | Methods of treating a cancer through targeted disruption of alpha connexin 43-zonula occludens-1 (zo-1) interaction |
| AU2014312310A1 (en) | 2013-08-28 | 2016-04-07 | Abbvie Stemcentrx Llc | Novel SEZ6 modulators and methods of use |
| AU2014312215B2 (en) | 2013-08-28 | 2020-02-27 | Abbvie Stemcentrx Llc | Site-specific antibody conjugation methods and compositions |
| US20150093399A1 (en) | 2013-08-28 | 2015-04-02 | Bioasis Technologies, Inc. | Cns-targeted conjugates having modified fc regions and methods of use thereof |
| EP3044332A1 (en) | 2013-09-09 | 2016-07-20 | British Columbia Cancer Agency Branch | Methods and kits for predicting outcome and methods and kits for treating breast cancer with radiation therapy |
| WO2015038426A1 (en) | 2013-09-13 | 2015-03-19 | Asana Biosciences, Llc | Self-immolative linkers containing mandelic acid derivatives, drug-ligand conjugates for targeted therapies and uses thereof |
| US20150093331A1 (en) | 2013-10-01 | 2015-04-02 | OncoLock Co., Ltd. | Biomarkers for breast cancer |
| US9878049B2 (en) | 2013-10-09 | 2018-01-30 | The University Of Akron | High drug loading system to co-deliver anticancer drugs and nucleic acids for cancer therapy |
| CN105813654B (zh) | 2013-10-11 | 2019-05-31 | 美国政府(由卫生和人类服务部的部长所代表) | Tem8抗体及其用途 |
| EA032231B1 (ru) | 2013-10-11 | 2019-04-30 | Мерсана Терапьютикс, Инк. | Конъюгаты белок-полимер-лекарственное средство |
| CA2927022C (en) | 2013-10-11 | 2018-08-21 | Asana Biosciences, Llc | Protein-polymer-drug conjugates |
| US9617521B2 (en) | 2013-10-21 | 2017-04-11 | Hemoshear, Llc | In vitro model for a tumor microenvironment |
| EP3689881B1 (en) | 2013-11-04 | 2022-01-12 | Eisai R&D Management Co., Ltd. | Macrocyclization reactions and intermediates useful in the synthesis of analogs of halichondrin b |
| US9457019B2 (en) | 2013-11-07 | 2016-10-04 | Deciphera Pharmaceuticals, Llc | Methods for inhibiting tie-2 kinase useful in the treatment of cancer |
| WO2015069266A1 (en) | 2013-11-07 | 2015-05-14 | Flynn Daniel L | Methods for inhibiting tie2 kinase useful in the treatment of cancer |
| IL311202A (en) | 2013-11-15 | 2024-05-01 | Oncoceutics Inc | 7-Benzyl-4-(2-methylbenzyl)-2, 4, 6, 7, 8, 9-hexahydroimidazo [1,2-A] pyrido[3,4-E] pyrimidine-5(1H)one, its salts and methods Use |
| WO2015081282A1 (en) | 2013-11-27 | 2015-06-04 | Redwood Bioscience, Inc. | Hydrazinyl-pyrrolo compounds and methods for producing a conjugate |
| MX384292B (es) | 2013-12-06 | 2025-03-14 | Eisai R&D Man Co Ltd | Metodos utiles en la sintesis de analogos de halicondrina b. |
| WO2015095784A1 (en) | 2013-12-19 | 2015-06-25 | Luminus Biosciences, Inc. | Solid nanoparticle formulation of microtubule inhibitors with reduced ostwald ripening for oral administration |
| US10669296B2 (en) | 2014-01-10 | 2020-06-02 | Rgenix, Inc. | LXR agonists and uses thereof |
| CN105899539B (zh) | 2014-01-10 | 2021-11-09 | 博笛生物科技有限公司 | 用于免疫疗法的化合物和组合物 |
| EP3092027A4 (en) | 2014-01-10 | 2017-09-06 | Atossa Genetics Inc. | Transpapillary methods and compositions for diagnosing and treating breast conditions |
| CN104784699B (zh) | 2014-01-20 | 2019-05-03 | 博瑞生物医药(苏州)股份有限公司 | 叶酸受体结合配体-药物偶联物 |
| CA2937034A1 (en) * | 2014-02-03 | 2015-08-06 | Riken | Anti-tissue factor monoclonal antibody |
| AU2015212733B2 (en) | 2014-02-03 | 2018-06-07 | Eidgenoessische Technische Hochschule Zurich | Small molecule drug conjugates |
| EP2913064A1 (en) | 2014-02-26 | 2015-09-02 | celares GmbH | Branched drug-linker conjugates for the coupling to biological targeting molecules |
| TW201617326A (zh) | 2014-03-06 | 2016-05-16 | Alphora研發股份有限公司 | (s)-1-((2r,3r,4s,5s)-5-烯丙-3-甲氧-4-(對甲苯磺醯甲基)四氫呋喃-2-基)-3-氨基丙-2-醇之結晶衍生物 |
| CA2941161A1 (en) | 2014-03-13 | 2015-09-17 | F.Hoffmann-La Roche Ag | Methods and compositions for modulating estrogen receptor mutants |
| US9260478B2 (en) * | 2014-04-04 | 2016-02-16 | Shanghui Hu | Potent and efficient cytotoxic peptides and antibody-drug conjugates thereof and their synthesis |
| CN106572988B (zh) | 2014-04-08 | 2022-04-08 | 卫理公会医院 | Inos抑制性组合物及其作为乳腺癌治疗剂的用途 |
| EP3131587A4 (en) | 2014-04-14 | 2017-11-01 | Endocyte, Inc. | Drug delivery conjugates for treating resistant cancer and for use in combination therapy |
| WO2015161247A1 (en) * | 2014-04-17 | 2015-10-22 | Igenica Biotherapeutics, Inc. | Humanized anti-c16orf54 antibodies and methods of use thereof |
| BR112016024363A2 (pt) * | 2014-04-25 | 2017-10-10 | Pf Medicament | conjugado de anticorpo-fármaco e seu uso para tratamento de câncer |
| KR101628872B1 (ko) * | 2014-05-28 | 2016-06-09 | 주식회사 레고켐 바이오사이언스 | 자가-희생 기를 포함하는 화합물 |
| TWI695011B (zh) * | 2014-06-18 | 2020-06-01 | 美商梅爾莎納醫療公司 | 抗her2表位之單株抗體及其使用之方法 |
| WO2015195925A1 (en) * | 2014-06-18 | 2015-12-23 | Mersana Therapeutics, Inc. | Protein-polymer-drug conjugates and methods of using same |
| WO2016008112A1 (en) * | 2014-07-16 | 2016-01-21 | Medshine Discovery Inc. | Linkers and application towards adc thereof |
| EP3069734A1 (en) * | 2015-03-17 | 2016-09-21 | Exiris S.r.l. | Cryptophycin-based antibody-drug conjugates with novel self-immolative linkers |
| US10975112B2 (en) * | 2015-06-16 | 2021-04-13 | Hangzhou Dac Biotech Co., Ltd. | Linkers for conjugation of cell-binding molecules |
| WO2015151079A2 (en) | 2015-06-20 | 2015-10-08 | Hangzhou Dac Biotech Co, Ltd | Auristatin analogues and their conjugates with cell-binding molecules |
-
2017
- 2017-03-02 EP EP17711475.8A patent/EP3423105B1/en active Active
- 2017-03-02 KR KR1020247000482A patent/KR102702620B1/ko active Active
- 2017-03-02 CA CA3297123A patent/CA3297123A1/en active Pending
- 2017-03-02 SM SM20210416T patent/SMT202100416T1/it unknown
- 2017-03-02 KR KR1020187028407A patent/KR102445255B1/ko active Active
- 2017-03-02 CN CN202111360563.8A patent/CN114272389B/zh active Active
- 2017-03-02 CN CN202111360555.3A patent/CN114191428B/zh active Active
- 2017-03-02 MY MYPI2021003997A patent/MY199595A/en unknown
- 2017-03-02 WO PCT/US2017/020529 patent/WO2017151979A1/en not_active Ceased
- 2017-03-02 HU HUE17711475A patent/HUE054726T2/hu unknown
- 2017-03-02 MA MA45280A patent/MA45280B1/fr unknown
- 2017-03-02 RU RU2018134331A patent/RU2754369C2/ru active
- 2017-03-02 AU AU2017225982A patent/AU2017225982B2/en active Active
- 2017-03-02 IL IL292946A patent/IL292946B2/en unknown
- 2017-03-02 PE PE2023001172A patent/PE20231050A1/es unknown
- 2017-03-02 SG SG10202007520WA patent/SG10202007520WA/en unknown
- 2017-03-02 DK DK17711475.8T patent/DK3423105T3/da active
- 2017-03-02 TW TW106106901A patent/TWI772288B/zh active
- 2017-03-02 TW TW112144655A patent/TWI871094B/zh active
- 2017-03-02 BR BR112018067379-0A patent/BR112018067379A2/pt active Search and Examination
- 2017-03-02 AR ARP170100524A patent/AR107787A1/es unknown
- 2017-03-02 IL IL320940A patent/IL320940A/en unknown
- 2017-03-02 JP JP2018545210A patent/JP6599019B2/ja active Active
- 2017-03-02 UA UAA201809739A patent/UA125024C2/uk unknown
- 2017-03-02 MY MYPI2018001424A patent/MY189113A/en unknown
- 2017-03-02 HR HRP20211125TT patent/HRP20211125T1/hr unknown
- 2017-03-02 SI SI201730837T patent/SI3423105T1/sl unknown
- 2017-03-02 PT PT177114758T patent/PT3423105T/pt unknown
- 2017-03-02 IL IL261428A patent/IL261428B2/en unknown
- 2017-03-02 MX MX2018010562A patent/MX2018010562A/es unknown
- 2017-03-02 JO JOP/2017/0053A patent/JOP20170053B1/ar active
- 2017-03-02 CN CN201780022381.5A patent/CN108883198B/zh active Active
- 2017-03-02 TW TW111125421A patent/TWI825834B/zh active
- 2017-03-02 KR KR1020227022690A patent/KR102456433B1/ko active Active
- 2017-03-02 PE PE2018001578A patent/PE20181953A1/es unknown
- 2017-03-02 US US15/448,497 patent/US10548986B2/en active Active
- 2017-03-02 MD MDE20190061T patent/MD3423105T2/ro unknown
- 2017-03-02 PL PL17711475T patent/PL3423105T3/pl unknown
- 2017-03-02 ES ES17711475T patent/ES2880402T3/es active Active
- 2017-03-02 RS RS20210889A patent/RS62108B1/sr unknown
- 2017-03-02 CA CA3013791A patent/CA3013791A1/en active Pending
- 2017-03-02 LT LTEP17711475.8T patent/LT3423105T/lt unknown
- 2017-03-02 CN CN202111360093.5A patent/CN114191563A/zh active Pending
- 2017-03-02 EP EP20204172.9A patent/EP3824909A1/en active Pending
- 2017-03-02 SG SG11201806515RA patent/SG11201806515RA/en unknown
- 2017-03-02 KR KR1020227022691A patent/KR20220101204A/ko not_active Ceased
- 2017-03-02 PE PE2023001171A patent/PE20231049A1/es unknown
-
2018
- 2018-02-09 US US15/892,921 patent/US10322192B2/en active Active
- 2018-08-16 CO CONC2018/0008667A patent/CO2018008667A2/es unknown
- 2018-08-28 CL CL2018002456A patent/CL2018002456A1/es unknown
- 2018-08-30 PH PH12018501847A patent/PH12018501847A1/en unknown
- 2018-08-31 MX MX2023003808A patent/MX2023003808A/es unknown
- 2018-08-31 MX MX2023003806A patent/MX2023003806A/es unknown
- 2018-08-31 MX MX2023003809A patent/MX2023003809A/es unknown
-
2019
- 2019-10-01 JP JP2019181469A patent/JP6870051B2/ja active Active
- 2019-12-04 US US16/702,720 patent/US20200297860A1/en not_active Abandoned
-
2020
- 2020-05-13 JP JP2020084451A patent/JP2020128413A/ja active Pending
-
2021
- 2021-01-07 CL CL2021000049A patent/CL2021000049A1/es unknown
- 2021-01-07 CL CL2021000048A patent/CL2021000048A1/es unknown
- 2021-02-10 AR ARP210100338A patent/AR121301A2/es unknown
- 2021-02-10 AR ARP210100339A patent/AR121302A2/es unknown
- 2021-04-13 JO JOP/2021/0073A patent/JOP20210073A1/ar unknown
- 2021-04-13 JO JOP/2021/0074A patent/JOP20210074A1/ar unknown
- 2021-08-02 CY CY20211100686T patent/CY1124628T1/el unknown
- 2021-08-25 JP JP2021137504A patent/JP7254861B2/ja active Active
-
2022
- 2022-12-07 US US18/062,996 patent/US20230398228A1/en not_active Abandoned
-
2023
- 2023-03-29 JP JP2023053129A patent/JP7556086B2/ja active Active
- 2023-12-20 AU AU2023285804A patent/AU2023285804B2/en active Active
-
2024
- 2024-09-11 JP JP2024157430A patent/JP2024174991A/ja active Pending
-
2026
- 2026-02-24 AU AU2026201384A patent/AU2026201384A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2023285804B2 (en) | Eribulin-based antibody-drug conjugates and methods of use | |
| ES2991607T3 (es) | Anticuerpo anti-CDH6 y conjugado de anticuerpo anti-CDH6 y fármaco | |
| ES2814227T3 (es) | Anticuerpos anti-CD123 y conjugados de los mismos | |
| ES2796560T3 (es) | Moléculas de unión específicas para ASCT2 y usos de las mismas | |
| ES2930255T3 (es) | Anticuerpos anti-Ror2, fragmentos de anticuerpos, sus inmunoconjugados y usos de los mismos | |
| KR20200131840A (ko) | 항-her2 2파라토프 항체-약물 접합체 및 이의 사용 방법 | |
| BR112021010936A2 (pt) | Conjugados anticorpo-fármaco de herboxidieno e métodos de uso | |
| AU2020414996A1 (en) | Method for producing eribulin-based antibody-drug conjugate | |
| KR20230106606A (ko) | 항체 약물 접합체 | |
| HK40053532A (en) | Eribulin-based antibody-drug conjugates and methods of use | |
| HK1261001B (en) | Eribulin-based antibody-drug conjugates and methods of use |