ES2908655T3 - Composiciones farmacéuticas sólidas y procedimientos para su producción - Google Patents
Composiciones farmacéuticas sólidas y procedimientos para su producción Download PDFInfo
- Publication number
- ES2908655T3 ES2908655T3 ES10723816T ES10723816T ES2908655T3 ES 2908655 T3 ES2908655 T3 ES 2908655T3 ES 10723816 T ES10723816 T ES 10723816T ES 10723816 T ES10723816 T ES 10723816T ES 2908655 T3 ES2908655 T3 ES 2908655T3
- Authority
- ES
- Spain
- Prior art keywords
- sodium
- group
- optionally substituted
- pharmaceutical composition
- buffer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/145—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2095—Tabletting processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2893—Tablet coating processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4833—Encapsulating processes; Filling of capsules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Obesity (AREA)
- Dermatology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Ophthalmology & Optometry (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Urology & Nephrology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Inorganic Chemistry (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un procedimiento para preparar una composición farmacéutica que comprende las etapas de: (a-1) granular en húmedo al menos un ingrediente activo que es un compuesto de fórmula (I): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, donde: Ra se selecciona de entre el grupo que consiste en C1-3 alifático, C1-3 fluoroalifático, -R1, -T-R1, -R2, y -T-R2; T es una cadena de alquileno C1-3 opcionalmente sustituida con flúoro; R1' es un grupo arilo, heteroarilo o heterociclilo opcionalmente sustituido; R2 se selecciona de entre el grupo que consiste en halo, -C≡C-R3, -CH=CH-R3, -N(R4)2, y -OR5; R3 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; Cada R4 es independientemente hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; o dos R4 en el mismo átomo de nitrógeno, tomados junto con el átomo de nitrógeno forman un anillo heteroarilo de 5 a 6 miembros o heterociclilo de 4 a 8 miembros opcionalmente sustituido que tiene, además del átomo de nitrógeno, 0-2 heteroátomos en el anillo seleccionado de entre N, O y S; R5 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; Rb se selecciona de entre el grupo que consiste en fluoro, cloro, -CH3, -CF3, -OH, -OCH3, -OCF3, -OCH2CH3, y - OCH2CF3; y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes y desintegrantes en presencia de un disolvente adecuado para formar una mezcla húmeda; (a-2) secar la mezcla húmeda de la etapa (a-1), para formar gránulos secos; (a-3) moler los gránulos secos de la etapa (a-2), para formar gránulos molidos; y (a-4) mezclar los gránulos molidos de la etapa (a-3) con un tampón y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes, desintegrantes, lubricantes y deslizantes; donde se añade un relleno durante la etapa (a-1), durante la etapa (a-4), o durante ambas etapas (a-1) y (a-4).
Description
DESCRIPCIÓN
Composiciones farmacéuticas sólidas y procedimientos para su producción
ANTECEDENTES DE LA INVENCIÓN
Campo de la invención
Esta invención se refiere a nuevas composiciones farmacéuticas sólidas y procedimientos para la producción a granel de dichas composiciones. Esta invención también proporciona las composiciones farmacéuticas para uso en el tratamiento del cáncer.
Antecedentes de la invención
En general, las composiciones farmacéuticas sólidas comprenden un ingrediente farmacéuticamente activo, como una molécula pequeña, mezclado con excipientes farmacéuticamente aceptables en cantidades apropiadas para mantener la actividad original del ingrediente activo. Estas composiciones normalmente se administran a los pacientes en forma de comprimidos o cápsulas.
Se pueden encontrar ejemplos de ingredientes activos particulares en Patente EE. UU. No. 7.572.784, Publicación EE. UU. No. 2008/0045501, Publicación EE. UU. No. 2008/0167292, y Solicitud EE. UU. No. 61/306,047, depositada el 19 de febrero de 2010 que divulga compuestos que inhiben las enzimas quinasa Aurora. Estas solicitudes también describen procedimientos para la preparación de estos compuestos, composiciones farmacéuticas que contienen estos compuestos y procedimientos para la profilaxis y terapia de enfermedades, trastornos o afecciones asociadas con la sobreexpresión. y/o amplificación de quinasas Aurora, incluyendo, pero sin limitarse a, trastornos proliferativos de células tales como cáncer.
Consideraciones importantes durante la fabricación de composiciones farmacéuticas sólidas incluyen la conservación de la forma cristalina del ingrediente activo y el mantenimiento de la estabilidad química y física del ingrediente activo. Los fabricantes generalmente tienen como diana a una vida útil de 2 a 3 años para las composiciones farmacéuticas.
Por lo tanto, existe la necesidad de desarrollar composiciones farmacéuticas sólidas que sean estables y tengan una biodisponibilidad favorable. En particular, existe la necesidad de composiciones farmacéuticas sólidas que comprendan inhibidores de la quinasa Aurora.
El documento US 2007/104785 A1 divulga en su título "comprimidos de linezolid forma III y procedimientos para su preparación". Faure y col., European Journal of Pharmaceutics and Biopharmaceutics, vol. 52, núm. 3, páginas 269 -277 se titula "control de procedimientos y escalado de procedimientos de granulación húmeda farmacéutica: una revisión".
DESCRIPCIÓN DE LA INVENCIÓN
La presente invención proporciona procedimientos para preparar las composiciones farmacéuticas sólidas descritas en esta invención. Estos procedimientos ofrecen una estrategia alternativa para procesar formulaciones basadas en granulación húmeda que requieren un tampón para la biodisponibilidad en comparación con los procedimientos de la técnica anterior. Se ha determinado que el orden de adición de los excipientes durante el procedimiento de granulación afecta la disolución del producto farmacéutico cuando se almacena para estudios de estabilidad.
En otro aspecto, la invención proporciona composiciones farmacéuticas que pueden prepararse mediante tales procedimientos, que comprenden los ingredientes activos, tampones y excipientes adicionales como se describe en esta invención.
En otro aspecto más, la invención proporciona composiciones farmacéuticas como se describe en esta invención para su uso en procedimientos para tratar trastornos.
La patente y/o la bibliografía científica a la que se hace referencia en esta invención establece el conocimiento que está disponible para los expertos en la materia. A menos que se defina de otro modo, todos los términos técnicos y científicos utilizados en esta invención tienen el mismo significado que entienden comúnmente los expertos en la técnica a la que se refiere esta invención. Aunque se pueden usar procedimientos y materiales similares o equivalentes a los descritos en esta invención en la práctica o ensayo de la presente invención, a continuación se describen procedimientos y materiales adecuados. En el caso de inconsistencias, prevalecerá la presente divulgación, incluidas las definiciones. Además, los materiales, procedimientos y ejemplos son sólo ilustrativos y no pretenden ser limitativos.
Definiciones:
El término "ingrediente activo" se usa en esta invención para referirse a un componente de una composición farmacéutica que es farmacéutica o fisiológicamente activo.
El término "excipiente farmacéuticamente aceptable" se usa en esta invención para referirse a un material que es compatible con un sujeto receptor, preferiblemente un mamífero, más preferiblemente un ser humano, y es adecuado para administrar un agente activo al sitio diana sin terminar la actividad. del agente La toxicidad o los efectos adversos, si los hubiere, asociados con el excipiente preferiblemente son proporcionales a una relación riesgo/beneficio para el uso previsto del ingrediente activo. Clases de excipientes farmacéuticamente aceptables incluyen, entre otros, tensioactivos, aglutinantes, desintegrantes, lubricantes, deslizantes, rellenos y tampones.
El término "aproximadamente" se usa en esta invención para significar alrededor de, en la región de, más o menos o alrededor. Cuando el término "aproximadamente" se usa junto con un intervalo numérico, modifica ese intervalo al extender los límites por encima y por debajo de los valores numéricos establecidos. En general, el término "aproximadamente" se utiliza en esta invención para modificar un valor numérico por encima y por debajo del valor establecido por una variación de 10 %.
Como se usa en esta invención, "% p/p" se usa para referirse al peso como un porcentaje de un peso total que se usa como base para calcular el porcentaje en peso de un componente individual. A modo de ejemplo, para una composición a granel, el % p/p de un componente individual puede calcularse como un porcentaje del peso total de todos los componentes de la composición a granel. A modo de otro ejemplo, para una sola forma de dosificación oral, el % p/p de un componente individual puede calcularse como un porcentaje del peso total de todos los componentes de la forma de dosificación oral única. Por ejemplo, cuando la forma de dosificación oral única es un comprimido recubierto, el peso total puede ser el peso total de todos los componentes del comprimido recubierto, incluidos los recubrimientos. Alternativamente, el peso total puede ser el peso total de todos los componentes del comprimido sin incluir los revestimientos.
Como se usa en esta invención, el término "comprende" significa "incluye, pero no se limita a".
Como se usa en esta invención, un "sujeto" es preferiblemente un ave o un mamífero, como un ser humano, pero también puede ser un animal que necesita tratamiento veterinario, por ejemplo, animales domésticos (por ejemplo, perros, gatos y similares), animales de granja (por ejemplo, vacas, ovejas, aves, cerdos, caballos y similares) y animales de laboratorio (por ejemplo, ratas, ratones, cobayas y similares).
Como se usa en esta invención, "tratar" o "tratamiento" significa prevención, alivio parcial o cura de una enfermedad, trastorno o afección.
Como se usa en esta invención, "cantidad terapéuticamente efectiva" pretende describir una cantidad de un compuesto, composición, medicamento u otro ingrediente activo eficaz para producir el efecto terapéutico deseado.
Como se usa en esta invención, el término "quinasa Aurora" se refiere a cualquiera de una familia de quinasas serina/treonina implicadas en la progresión mitótica. Una variedad de proteínas celulares que desempeñan un papel en la división celular son sustratos para la fosforilación por las enzimas quinasas Aurora, incluidas, entre otras, histona H3, p 53, CENP-A, cadena ligera reguladora de miosina II, proteína fosfatasa-1, TPX-2 , INCENP, survivina, topoisomerasa II alfa, vimentina, MBD-3, MgcRacGAP, desmina, Ajuba, XIEg5 (en Xenopus), Ndc10p (en levadura en ciernes) y D-TACC (en Drosophila). Las enzimas quinasa Aurora también son sustratos para la autofosforilación, por ejemplo, en Thr288. A menos que el contexto indique lo contrario, el término "quinasa Aurora" se refiere a cualquier proteína quinasa Aurora de cualquier especie, incluidas, entre otras, Aurora A, Aurora B y Aurora C, preferiblemente Aurora A o B. Preferiblemente, la proteína quinasa Aurora es una quinasa Aurora humana.
El término "inhibidor de quinasa Aurora" o "inhibidor de la quinasa Aurora" se utiliza para referirse a un compuesto que tiene una estructura como se define en esta invención, que es capaz de interactuar con una quinasa Aurora e inhibir su actividad enzimática. Inhibir la actividad enzimática de la quinasa Aurora significa reducir la capacidad de una quinasa Aurora para fosforilar un péptido o proteína sustrato. En varias realizaciones, dicha reducción de la actividad de la quinasa Aurora es al menos aproximadamente 50 %, al menos aproximadamente 75 %, al menos aproximadamente 90 %, al menos aproximadamente 95 %, o al menos aproximadamente 99 %. En diversas realizaciones, la concentración de inhibidor de quinasa Aurora necesaria para reducir la actividad enzimática de una quinasa Aurora es inferior a 1 pM, inferior a 500 nM, inferior a 100 nM o inferior a 50 nM.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
En un aspecto, la presente invención proporciona procedimientos para preparar una composición farmacéutica que comprende las etapas de:
(a-1) granular en húmedo al menos un ingrediente activo que es un compuesto de fórmula (/):

o una sal farmacéuticamente aceptable del mismo, donde:
Ra se selecciona de entre el grupo que consiste en C1-3 alifático, C1-3 fluoroalifático, -R1, -TR1, -R2, y -TR2;
T es una cadena de alquileno C1-3 opcionalmente sustituida con flúoro;
R1’ es un grupo arilo, heteroarilo o heterociclilo opcionalmente sustituido;
R2 se selecciona de entre el grupo que consiste en halo, -C=C-R3, -CH=CH-R3, -N(R4)2 , y -OR5;
R3 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Cada R4 es independientemente hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; o dos R4 en el mismo átomo de nitrógeno, tomados junto con el átomo de nitrógeno forman un anillo heteroarilo de 5 a 6 miembros o heterociclilo de 4 a 8 miembros opcionalmente sustituido que tiene, además del átomo de nitrógeno, 0-2 heteroátomos en el anillo seleccionado de entre N, O y S;
R5 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Rb se selecciona de entre el grupo que consiste en fluoro, cloro, -CH3 , -CF3 , -OH, -OCH3 , -OCF3 , -OCH2CH3y -OCH2CF3 , y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes y desintegrantes en presencia de un disolvente adecuado para formar una mezcla húmeda;
(a-2) secar la mezcla húmeda de la etapa (a-1), para formar gránulos secos;
(a-3) moler los gránulos secos de la etapa (a-2), para formar gránulos molidos; y
(a-4) mezclar los gránulos molidos de la etapa (a-3) con un tampón y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes, desintegrantes, lubricantes y deslizantes;
donde se añade un relleno durante la etapa (a-1), durante la etapa (a-4), o durante ambas etapas (a-1) y (a-4). En otra realización, los procedimientos de la invención también comprenden la etapa de (b-1) cargar la mezcla resultante de la etapa (a-4) en una cápsula.
En otra realización adicional, los procedimientos de la invención comprenden la etapa de (c-1) formar comprimidos con la mezcla resultante de la etapa (a-4) para formar un comprimido. En algunas realizaciones, los procedimientos de la invención comprenden agregar un lubricante durante la etapa (a-4) y a continuación (c-1) formar comprimidos con la mezcla resultante de la etapa (a-4) para formar un comprimido.
En aún otra realización, los procedimientos de la invención también comprenden la etapa de (c-2) recubrir el comprimido resultante de la etapa (c-1). En algunas realizaciones, los comprimidos están recubiertos con película, o con recubrimiento entérico, o ambos. En algunas otras realizaciones, los comprimidos están recubiertos con película y entéricamente.
En una realización adicional, la etapa de granulación en húmedo (a-1) de los procedimientos de la invención está precedida por la etapa (a-0) que mezcla en seco al menos un ingrediente activo y, opcionalmente, uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes, desintegrantes y rellenos.
En algunas realizaciones, la etapa (a-4) se puede realizar como una sola etapa de mezcla durante la cual todos los excipientes farmacéuticamente aceptables se agregan al mismo tiempo. En otras realizaciones, la etapa (a-4) se puede realizar como etapas de mezcla consecutivas durante las cuales se agrega un excipiente farmacéuticamente aceptable a la vez. En aún otras realizaciones, durante la etapa (a-4), se pueden agregar uno o más lubricantes después de que se hayan agregado todos los demás excipientes farmacéuticamente aceptables.
La etapa de granulación en húmedo (a-1) descrita en esta invención puede tener lugar en cualquier sistema o aparato de granulación convencional. Ejemplos de tales equipos de granulación incluyen, entre otros, granuladores de alto cizallamiento, granuladores de lecho fluido, granuladores de fusión en caliente, granuladores basados en un recipiente, granuladores basados en extrusión, granuladores basados en esferonización y granuladores basados en secado por pulverización. Un ejemplo de un granulador de alto cizallamiento es el granulador de alto cizallamiento Diosna P1-6, fabricado por DIOSNA Dierks & Sohne GmbH, Alemania. Un ejemplo de un granulador de lecho fluido es el granulador de lecho fluido por lotes GPCG-1 de Glatt Air Techniques, Inc., e E . UU.
En algunas realizaciones, la etapa de granulación en húmedo (a-1) dura entre aproximadamente 5 minutos y aproximadamente 60 minutos. En algunas realizaciones, al menos un ingrediente activo y, opcionalmente, uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes y desintegrantes se mezclan mientras se introduce un disolvente adecuado en el sistema de granulación para formar una mezcla húmeda. En algunas otras realizaciones, la etapa de granulación en húmedo incluye opcionalmente un tiempo de mezcla adicional después de que se haya introducido el disolvente adecuado en el sistema de granulación para lograr un punto final de granulación deseado. En algunas realizaciones, el tiempo de mezclado adicional se produce durante menos de 15 minutos, o menos de 10 minutos, o menos de 5 minutos. En algunas realizaciones, el tiempo de mezclado adicional se produce entre aproximadamente 1 minuto y aproximadamente 5 minutos, o entre aproximadamente 1 minuto y aproximadamente 4 minutos, o entre aproximadamente 1 minuto y aproximadamente 3 minutos, o entre aproximadamente 1 minuto y aproximadamente 2 minutos. En algunas realizaciones, el tiempo de mezclado adicional se produce durante aproximadamente 1 minuto, aproximadamente 2 minutos, aproximadamente 3 minutos, aproximadamente 4 minutos o aproximadamente 5 minutos.
En algunas realizaciones, el contenido de humedad durante la etapa de granulación en húmedo está entre aproximadamente el 15 % p/p a aproximadamente el 45 % p/p, donde el peso total es igual a la cantidad total de material en la etapa de granulación en húmedo. En algunas realizaciones, el contenido de humedad durante la etapa de granulación en húmedo está entre aproximadamente el 20 % p/p a aproximadamente el 40 % p/p, o entre aproximadamente el 25 % p/p a aproximadamente el 35 % p/p, o aproximadamente el 30 % p/p, donde el peso total es igual a la cantidad total de material en la etapa de granulación en húmedo. En algunas otras realizaciones, el contenido de humedad durante la etapa de granulación en húmedo es de aproximadamente el 15 % p/p, o aproximadamente el 20 % p/p, o aproximadamente el 25 % p/p, o aproximadamente el 30 % p/p, o aproximadamente el 35 % p/p, o aproximadamente el 40% p/p, donde el peso total es igual a la cantidad total de material en la etapa de granulación en húmedo.
La etapa de secado (a-2) descrita en esta invención puede tener lugar en cualquier sistema o aparato de secado convencional. Ejemplos de tales equipos de secado incluyen, pero no se limitan a, granuladores de lecho fluido y equipos para secado en bandeja, secado por microondas y secado al vacío. Un ejemplo de granulador de lecho fluido es GPCG-1, fabricado por Glatt Air Techniques, Ramsey, N j . En algunas realizaciones, la etapa de secado dura entre aproximadamente 5 minutos y aproximadamente 240 minutos. En algunas otras realizaciones, la etapa de secado dura entre aproximadamente 60 minutos y aproximadamente 240 minutos, o entre aproximadamente 180 minutos y aproximadamente 240 minutos. En algunas realizaciones, la temperatura del aire de entrada está entre aproximadamente 40 °C y aproximadamente 85 °C. En algunas otras realizaciones, la temperatura del aire de entrada está entre aproximadamente 50 °C y aproximadamente 80 °C, o entre aproximadamente 60 °C y aproximadamente 75 °C, o aproximadamente 70 °C.
La etapa de molienda (a-3) descrita en esta invención puede tener lugar en cualquier sistema o aparato de molienda convencional. Ejemplos de dichos equipos de molienda incluyen, pero no se limitan a, Comil® U3 (Quadro Engineering LP, Waterloo, ON, Canadá), FitzMill® (Fitzpatrick Co., Elmhurst, IL) y equipos para tamizar usando tamices. En algunas realizaciones, la etapa de molienda dura entre aproximadamente 2 minutos y aproximadamente 60 minutos.
La etapa de mezcla (a-4) descrita en esta invención puede tener lugar en cualquier mezcladora convencional, como mezcladoras en V, contenedores a granel intermedios (IBC), mezcladoras de tambor, mezcladoras de bolsa, mezcladoras de flujo cruzado y otras mezcladoras convencionales. Un ejemplo de una mezcladora convencional es una mezcladora PK, fabricada por Patterson-Kelley Co., East Stroudsburg, PA. En algunas realizaciones, la etapa de mezcla dura entre aproximadamente 5 minutos y aproximadamente 120 minutos. En algunas realizaciones, la velocidad de mezcla está entre aproximadamente 10 rpm y aproximadamente 60 rpm.
La etapa de mezcla en seco (a-0) descrita en esta invención puede tener lugar en cualquier mezcladora convencional, como mezcladoras en V, contenedores a granel intermedios (IBC), mezcladoras de tambor, mezcladoras de bolsa, mezcladoras de flujo cruzado y otras mezcladoras convencionales. Un ejemplo de una mezcladora convencional es una mezcladora PK, fabricada por Patterson-Kelley Co., East Stroudsburg, PA. En algunas realizaciones, la etapa de mezcla dura entre aproximadamente 5 minutos y aproximadamente 120 minutos. En algunas realizaciones, la velocidad de mezcla está entre aproximadamente 10 rpm y aproximadamente 60 rpm.
La etapa de carga de cápsulas (b-1) descrita en esta invención puede tener lugar en cualquier sistema o aparato convencional de llenado de cápsulas. En algunas realizaciones, el sistema de llenado de cápsulas está semiautomatizado y puede manejar lotes pequeños. Un ejemplo de un sistema de llenado de cápsulas de este tipo se
vende como In-Cap (Isopak Limited, Lincolnshire, Stamford, Reino Unido). En algunas realizaciones, el sistema de llenado de cápsulas es manual. Un ejemplo de un aparato de llenado de cápsulas de este tipo se vende como ProFill 100 (Torpac, Inc., Fairfield, NJ, EE. UU.). Un ejemplo de un encapsulador a escala comercial es un Zanasi 70C, un llenador de cápsulas de movimiento intermitente, fabricado por IMA Industria Macchine Automatiche SpA, Castenaso, Italia.
La etapa de formación de comprimidos (c-1) descrita en esta invención puede tener lugar en cualquier prensa de comprimidos convencional. Un ejemplo de equipo de formación de comprimidos es el mezclador PK de 8 estaciones PLC Piccola, fabricado por SMI Inc, Lebanon, NJ. En algunas realizaciones, la velocidad de formación de comprimidos está entre aproximadamente 10 rpm y aproximadamente 100 rpm.
La etapa de recubrimiento (c-2) descrita en esta invención puede tener lugar en cualquier sistema convencional de recubrimiento de comprimidos. Un ejemplo de equipo de recubrimiento de comprimidos convencional se vende como Labcoat I (O'Hara Technologies, Inc, Richmond Hill, ON, Canadá). En algunas realizaciones, la velocidad de recubrimiento está entre aproximadamente 10 rpm y aproximadamente 100 rpm. En algunas realizaciones, la velocidad de rociado de recubrimiento está entre aproximadamente 5 g/ minuto a aproximadamente 100 g/ minuto.
Rellenos adecuados incluyen, entre otros, lactosa, celulosa microcristalina, manitol, etilcelulosa, sorbitol, almidón, sacarosa, fosfato de calcio, celulosa en polvo, celulosa microcristalina silicificada, isomalta y mezclas de los mismos. En algunas realizaciones, la carga es celulosa microcristalina silicificada, celulosa microcristalina o mezclas de las mismas. En algunas otras realizaciones, el relleno es celulosa microcristalina.
Tensioactivos adecuados incluyen, entre otros, laurilsulfato de sodio, dodecilsulfato de sodio, polisorbatos (como Tween 20 y Tween 80), poloxámeros (como Poloxamer 335 y Poloxamer 407), monooleato de glicerilo y mezclas de los mismos. En algunas realizaciones, el tensioactivo es laurilsulfato de sodio, dodecilsulfato de sodio o mezclas de los mismos. En algunas realizaciones, el tensioactivo es laurilsulfato de sodio.
Aglutinantes adecuados incluyen, entre otros, polivinilpirrolidona, etilcelulosa, alginato de maltosa sódica, hidroxipropilmetilcelulosa (HPMC), ácido esteárico, almidón pregelatinizado y mezclas de los mismos. En algunas realizaciones, el aglutinante es HPMC, polivinilpirrolidona o mezclas de los mismos. En otras realizaciones, el aglutinante es polivinilpirrolidona.
Desintegrantes adecuados incluyen, entre otros, dióxido de silicio coloidal, celulosa en polvo, silicato de calcio, crospovidona, alginato de calcio, metilcelulosa, quitosano, carboximetilcelulosa, croscarmelosa de sodio, carboximetilalmidón, alginato de sodio, glicolato de almidón de sodio, pregelatinizado almidón y mezclas de los mismos. En algunas realizaciones, el desintegrante es croscarmelosa sódica, crospovidona o mezclas de las mismas. En otras realizaciones, el desintegrante es croscarmelosa sódica.
Lubricantes adecuados incluyen, entre otros, talco, estearato de magnesio, estearilfumarato de sodio, behenato de glicerilo, aceite vegetal hidrogenado, estearato de zinc, estearato de calcio, estearato de sacarosa, alcohol polivinílico, laurilsulfato de magnesio y mezclas de los mismos. En algunas realizaciones, el lubricante es estearato de magnesio, estearilfumarato de sodio o mezclas de los mismos. En otras realizaciones, el lubricante es estearilfumarato de sodio.
Deslizantes adecuados incluyen, entre otros, dióxido de silicio, dióxido de silicio coloidal, fosfato de calcio tribásico, estearato de magnesio, trisilicato de magnesio, celulosa en polvo, talco, almidón y mezclas de los mismos. En algunas realizaciones, el deslizante es talco, dióxido de silicio coloidal o mezclas de los mismos. En otras realizaciones, el deslizante es dióxido de silicio coloidal.
Disolventes adecuados para la etapa de granulación en húmedo de (a-1) incluyen, entre otros, agua, etanol, acetona y mezclas de los mismos.
Los procedimientos de la invención se pueden utilizar para la preparación de composiciones farmacéuticas sólidas que comprenden cualquier ingrediente activo adecuado para formular en forma sólida con un tampón. Personas expertas de manera ordinaria en la materia reconocerán que un ingrediente activo que tiene un resto básico se formularía mejor con un tampón ácido, y que un ingrediente activo que tuviera un resto ácido se formularía mejor con un tampón básico. Por lo tanto, tampones adecuados para uso en la presente invención incluyen tampones tanto ácidos como básicos. Por ejemplo, en algunas realizaciones, una solución acuosa del tampón tiene un pH de menos de aproximadamente 7,0. En otras realizaciones, una solución acuosa del tampón tiene un pH de al menos aproximadamente 7,0. Ejemplos de tales tampones son conocidos por los expertos en la materia y se pueden encontrar en el Handbook of Pharmaceutical Excipients (5a Edición), Publicaciones APhA.
En algunas realizaciones, una solución acuosa del tampón tiene un pH de menos de aproximadamente 7,0. En algunas otras realizaciones, una solución acuosa del tampón tiene un pH de entre aproximadamente 1,0 y aproximadamente 6,0, o entre aproximadamente 2,0 y aproximadamente 6,0, o entre aproximadamente 3,0 y aproximadamente 6,0, o entre aproximadamente 4,0 y aproximadamente 6,0, o entre aproximadamente 5,0 a aproximadamente 6,0. Tampones adecuados que tienen un pH inferior a aproximadamente 7,0 en solución acuosa incluyen, entre otros, citrato disódico,
citrato trisódico, acetato sódico, fosfato monopotásico, fosfato monosódico y mezclas de los mismos.
En algunas realizaciones, una solución acuosa del tampón tiene un pH de al menos de aproximadamente 7,0. En algunas otras realizaciones, una solución acuosa del tampón tiene un pH de entre aproximadamente 8,0 y aproximadamente 13,0, o entre aproximadamente 8,0 y aproximadamente 12,0, o entre aproximadamente 8,0 y aproximadamente 11,0, o entre aproximadamente 8,0 y aproximadamente 10,0, o entre aproximadamente 8,0 a aproximadamente 9,0.
Tampones adecuados que tienen un pH de al menos aproximadamente 7,0 en solución acuosa incluyen, entre otros, bicarbonato sódico, fosfato disódico, fosfato dipotásico, bicarbonato potásico, carbonato sódico, carbonato potásico y mezclas de los mismos. En algunas realizaciones, el tampón es bicarbonato de sodio, carbonato de sodio o mezclas de los mismos. En otras realizaciones, el tampón es bicarbonato de sodio.
Un ingrediente activo como se describe en esta invención es un compuesto de fórmula (A):

o una sal farmacéuticamente aceptable del mismo; donde:
Rf1 es hidrógeno, o Rf1 y Rf2 juntos forman un enlace;
Rf2 es hidrógeno, o Rf2 forma un enlace con Rf1 o Rx;
cada uno de Rx y Ry es independientemente hidrógeno, flúor o un alifático C 1-6 opcionalmente sustituido; o Rx y Ry, tomados junto con el átomo de carbono al que están unidos, forman un anillo cicloalifático de 3 a 6 miembros opcionalmente sustituido; o Rx y Rf2 juntos forman un enlace;
G es hidrógeno, un anillo alifático o B opcionalmente sustituido cuando Rf1 es hidrógeno; y G es hidrógeno, -OR5, -N(R4) 2 , -SR5, un anillo alifático opcionalmente sustituido, o B cuando Rf1 y Rf2 juntos forman un enlace;
El anillo A es un anillo arilo, heteroarilo, cicloalifático o heterociclilo de 5 ó 6 miembros sustituido o no sustituido; El anillo B es un anillo arilo, heteroarilo, heterociclilo o cicloalifático sustituido o no sustituido;
El anillo C es un anillo arilo, heteroarilo, cicloalifático o heterociclilo sustituido o no sustituido;
Ra es hidrógeno, -C(O)R1, -CO 2 R 1 , -SO 2 R 1 , o un alifático C 1-3 que tiene 0-2 sustituyentes seleccionados independientemente de entre R3 o R7;
Re es hidrógeno, -OR5, -N(R4) 2 , -SR5, -NR4C(O)R5, -NR4C(O)N(R4) 2 , -NR4CO 2 R6, -N(R4)SO 2 R6, -N(R4)SO 2 N(R4) 2 , o un alifático C 1-3 opcionalmente sustituido con R3 o R7;
R1 es alifático C 1-6 o un grupo arilo, heteroarilo o heterociclilo opcionalmente sustituido;
cada R3 se selecciona independientemente de entre el grupo que consiste en -halo, -OH, -O(C1-3 alquilo), -CN, -N(R4) 2 , -C(O)(C1-3 alquilo), -CO 2 H, -CO2(C1-3 alquilo), -C(O)NH 2 , y -C(O)NH(C1-3 alquilo);
Cada R4 es independientemente hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; o dos R4 en el mismo átomo de nitrógeno, tomados junto con el átomo de nitrógeno, forman un anillo heteroarilo de 5 a 6 miembros o heterociclilo de 4 a 8 miembros opcionalmente sustituido que tiene, además del átomo de nitrógeno, 0-2 heteroátomos en el anillo seleccionado de entre N, O y S;
cada R5 independientemente es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
cada R6 independientemente es un grupo alifático o arilo opcionalmente sustituido;
cada R7 independientemente es un grupo arilo, heterociclilo o heteroarilo opcionalmente sustituido.
De acuerdo con la presente invención, el ingrediente activo es un compuesto de fórmula (I):
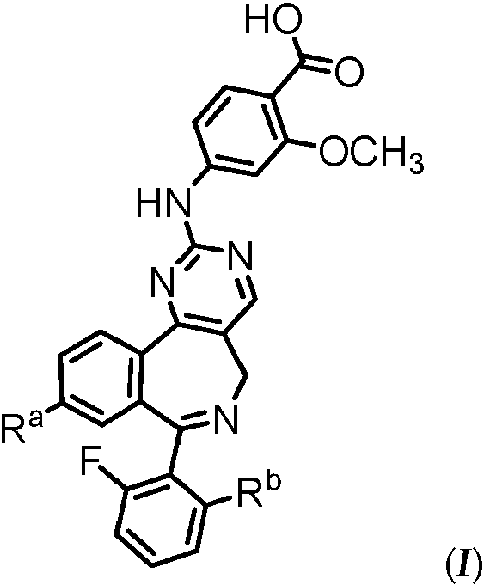
o una sal farmacéuticamente aceptable del mismo, donde:
Ra se selecciona delgrup° que consiste en C1-3 alifático, C1-3 fluoroalifático, -R1, -TR1, -R2y -TR2;
T es una cadena de alquileno C1-3 opcionalmente sustituida con flúoro;
R1’ es un grupo arilo, heteroarilo o heterociclilo opcionalmente sustituido;
R2 se selecciona de entre el grupo que consiste en halo, -C=C-R3, -CH=CH-R3, -N(R4)2 , y -OR5;
R3 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Cada R4 es independientemente hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; o dos R4 en el mismo átomo de nitrógeno, tomados junto con el átomo de nitrógeno forman un anillo heteroarilo de 5 a 6 miembros o heterociclilo de 4 a 8 miembros opcionalmente sustituido que tiene, además del átomo de nitrógeno, 0-2 heteroátomos en el anillo seleccionado de entre N, O y S;
R5 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Rb se selecciona de entre el grupo que consiste en fluoro, cloro, -CH3 , -CF3 , -OH, -OCH3 , -OCF3 , -OCH2CH3 , y -OCH2CF3.
Las definiciones de los grupos sustituyentes de los compuestos de fórmula (A) y fórmula (I) pueden encontrarse en Patente EE. UU. No. 7.572.784 y Publicación EE. UU. No. 2008/0167292, respectivamente. A menos que se indique lo contrario, las estructuras representadas en esta invención también incluyen formas solvatadas e hidratadas de los compuestos representados. También se incluyen dentro del alcance de la invención composiciones que comprenden sales farmacéuticamente aceptables de compuestos de fórmula (I), así como formas solvatadas e hidratadas de dichas sales.
Si se utilizan sales farmacéuticamente aceptables de los compuestos de fórmula (I) en las composiciones de la invención, las sales se derivan preferiblemente de ácidos y bases inorgánicos u orgánicos. Para revisiones de sales adecuadas, véase, por ejemplo, Berge y col., J. Pharm. Sci. 66:1-19 (1977) y Remington: The Science and Practice of Pharmacy, 20th Ed., ed. A. Gennaro, Lippincott Williams & Wilkins, 2000.
Ejemplos no limitativos de sales de adición de ácido adecuadas incluyen los siguientes: acetato, adipato, alginato, aspartato, benzoato, bencenosulfonato, bisulfato, butirato, citrato, canforato, canforsulfonato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, fumarato, lucoheptanoato, glicerofosfato, hemisulfato , heptanoato, hexanoato, clorhidrato, bromhidrato, yodhidrato, 2-hidroxietanosulfonato, lactato, maleato, metanosulfonato, 2-naftalenosulfonato, nicotinato, oxalato, pamoato, pectinato, persulfato, 3-fenil-propionato, picrato, pivalato, propionato, succinato, tartrato, tiocianato, tosilato y undecanoato.
Sales de adición de bases adecuadas incluyen, sin limitación, sales de amonio, sales de metales alcalinos, como sales de sodio y potasio, sales de metales alcalinotérreos, como sales de calcio y magnesio, sales con bases orgánicas, como diciclohexilamina, N-metil-D-glucamina, f-butilamina, etilendiamina, etanolamina y colina, y sales con aminoácidos tales como arginina, lisina, etc. Por ejemplo, los compuestos de fórmula (A), donde el anillo C está
sustituido con -CO2H pueden formularse como una sal de adición de base correspondiente, por ejemplo, una sal de sodio correspondiente.
Si se utilizan sales farmacéuticamente aceptables de los compuestos de fórmula (/) en las composiciones de la invención, las sales son preferiblemente sales de adición de base. Sales de adición de base adecuadas son las descritas anteriormente para los compuestos de fórmula (/). En algunas realizaciones, el ingrediente activo es un compuesto de fórmula (/), o una sal de sodio o potasio del mismo.
En algunas realizaciones, el ingrediente activo es una forma cristalina de un compuesto de fórmula (/). En algunas otras realizaciones, el ingrediente activo es una forma cristalina de una sal farmacéuticamente aceptable de un compuesto de fórmula (/). Algunos ejemplos de sales farmacéuticamente aceptables de los compuestos de fórmula (/) y formas cristalinas de los mismos se pueden encontrar en Patente EE. UU. No. 7.572.784, Publicación EE. UU.
2008/0167292, y Solicitud EE. UU. No. 61/306.047, depositada el 19 de febrero de 2010.
En aún una realización adicional, el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1), o una forma cristalina del mismo. En otra realización, el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1) monohidrato. En otra realización, el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1) polimorfo Forma 2, como se describe en Publicación EE. UU. No. 2008/0167292, y Solicitud EE. UU. No. 61/306.047, depositada el 19 de febrero de 2010.
Materiales adecuados que se pueden usar para recubrir con película los comprimidos en la etapa (c-2) incluyen, entre otros, Opadry® (Hidroxi propil metilcelulosa PEG) (Colorcon, West Point, PA), Opadry® II (Polivinil alcohol, PEG, talco, y dióxido de titanio), Opadry® fx, Opadry® amb, y mezclas de los mismos. En algunas realizaciones, el material de recubrimiento de película es Opadry®, Opadry® II o mezclas de los mismos. En otras realizaciones, el material de revestimiento de película es Opadry® II.
Materiales adecuados que se pueden usar para recubrir entéricamente los comprimidos en la etapa (c-2) incluyen, entre otros, Acryl- EZE ® (Copolímero de ácido metacrílico, talco, SLS, dióxido de titanio, bicarbonato de sodio, sílice, citrato de trietilo) (Colorcon, West Point, PA), ftalato de acetato de celulosa (CAP), copolímeros de acrilato de metilo y ácido metacrílico, succinato de acetato de celulosa, ftalato de hidroxipropilmetilcelulosa, succinato de acetato de hidroxipropilmetilcelulosa (succinato de acetato de hipromelosa), ftalato de polivinilacetato (PVAP), copolímeros de metacrilato de metilo-ácido metacrílico y mezclas de los mismos.
En algunas realizaciones, la composición farmacéutica producida después de la etapa (a-4) comprende aproximadamente del 1 % p/p a aproximadamente el 60 % p/p de ingrediente activo, aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de tampón, y aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de relleno En una realización adicional, la composición farmacéutica comprende desde aproximadamente 2 % p/p a aproximadamente el 22 % p/p del Compuesto 1, o una forma cristalina del mismo. En algunas realizaciones, la composición farmacéutica comprende desde aproximadamente 3 % p/p a aproximadamente el 15 % p/p del Compuesto 1, o una forma cristalina del mismo. En algunas otras realizaciones, la composición farmacéutica comprende aproximadamente 13,6 % p/p del Compuesto 1, o una forma cristalina del mismo.
En algunas realizaciones adicionales, la composición farmacéutica comprende un tampón de bicarbonato de sodio, donde el tampón de bicarbonato de sodio está presente en una cantidad de aproximadamente 10 % p/p a aproximadamente el 60 % p/p. En algunas realizaciones, el tampón de bicarbonato de sodio está presente en una cantidad de aproximadamente 20 % p/p a aproximadamente el 40 % p/p. En algunas otras realizaciones, el tampón de bicarbonato de sodio está presente en una cantidad de aproximadamente 30 % p/p.
En aún otras realizaciones, la composición farmacéutica comprende un relleno de celulosa microcristalina, donde el relleno de celulosa microcristalina está presente en una cantidad de aproximadamente 27 % p/p a aproximadamente el 53 % p/p.
En aún otras realizaciones, la composición a granel comprende un lubricante de estearilfumarato de sodio, donde el lubricante de estearilfumarato de sodio está presente en una cantidad de aproximadamente 0 % p/p a aproximadamente 3 % p/p.
En aún otras realizaciones, la composición farmacéutica producida después de la etapa (a-4) comprende, aproximadamente 0 % p/p a aproximadamente 5 % p/p de tensioactivo, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de aglutinante, y aproximadamente 0 % p/p a aproximadamente el 20 % p/p de desintegrante.
En aún otras realizaciones, se agrega aproximadamente del 30 % al 70 % de la cantidad total del desintegrante durante la etapa (a-1) y aproximadamente del 30 % al 70 % de la cantidad total del desintegrante se agrega durante la etapa (a- 4).
En otro aspecto, la presente invención proporciona composiciones farmacéuticas. En algunas realizaciones, las composiciones farmacéuticas de la invención pueden prepararse mediante los procedimientos descritos en esta invención. En algunas otras realizaciones, las composiciones farmacéuticas de la invención son composiciones a granel.
En una realización, la composición a granel comprende un ingrediente activo, un tampón y un relleno. En otra realización, la composición a granel comprende un ingrediente activo, un tampón, un relleno y, opcionalmente, uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en un lubricante, un tensioactivo, un aglutinante, un desintegrante y un deslizante. En otra realización más, la composición a granel comprende un ingrediente activo, un tampón, un relleno, un lubricante, un tensioactivo, un aglutinante y un desintegrante.
En una realización, la composición a granel comprende aproximadamente 1 % p/p a aproximadamente el 60 % p/p de ingrediente activo, aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de tampón, y aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de relleno En otra realización, la composición a granel comprende aproximadamente de 1 % p/p a aproximadamente el 60 % p/p de ingrediente activo, aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de tampón, aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de relleno, aproximadamente 0 % p/p a aproximadamente 5 % p/p de lubricante, aproximadamente 0 % p/p a aproximadamente 5 % p/p de tensioactivo, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de aglutinante, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de desintegrante, y aproximadamente 0 % p/p a aproximadamente 5 % p/p de un deslizante.
En otra realización, la composición a granel comprende aproximadamente de 1 % p/p a aproximadamente el 30 % p/p de ingrediente activo, aproximadamente del 30 % p/p a aproximadamente el 60 % p/p de tampón, aproximadamente del 20 % p/p a aproximadamente el 60 % p/p de relleno, aproximadamente 1 % p/p a aproximadamente 3 % p/p de lubricante, aproximadamente 0 % p/p a aproximadamente 3 % p/p de tensioactivo, aproximadamente 0 % p/p a aproximadamente el 10 % p/p de aglutinante, aproximadamente 0 % p/p a aproximadamente el 15 % p/p de desintegrante, y aproximadamente 0 % p/p a aproximadamente 2 % p/p de un deslizante.
En otra realización, la composición a granel comprende aproximadamente de 10 % p/p a aproximadamente el 16 % p/p de ingrediente activo, aproximadamente del 28 % p/p a aproximadamente el 40 % p/p de tampón, aproximadamente del 35 % p/p a aproximadamente el 45 % p/p de relleno, aproximadamente 1 % p/p a aproximadamente 2 % p/p de lubricante, aproximadamente 1 % p/p a aproximadamente 2 % p/p de tensioactivo, aproximadamente 3 % p/p a aproximadamente el 7 % p/p de aglutinante, aproximadamente 5 % p/p a aproximadamente el 10 % p/p de desintegrante, y aproximadamente 0 % p/p a aproximadamente 2 % p/p de un deslizante.
En otra realización, la composición a granel comprende aproximadamente el 13,6 % p/p de ingrediente activo, aproximadamente el 30,0 % p/p de tampón, y aproximadamente el 40,4 % p/p de relleno, aproximadamente 1,0 % p/p de lubricante, aproximadamente 2,0 % p/p de tensioactivo, aproximadamente el 5,0 % p/p de aglutinante, y aproximadamente el 8,0 % p/p de desintegrante.
Rellenos adecuados incluyen, entre otros, lactosa, celulosa microcristalina, manitol, etilcelulosa, sorbitol, almidón, sacarosa, fosfato de calcio, celulosa en polvo, celulosa microcristalina silicificada, isomalta y mezclas de los mismos. En algunas realizaciones, la carga es celulosa microcristalina silicificada, celulosa microcristalina o mezclas de las mismas. En algunas otras realizaciones, el relleno es celulosa microcristalina.
En algunas realizaciones, el relleno está presente en una cantidad de aproximadamente el 10 % p/p a aproximadamente el 80 % p/p. En otras realizaciones, el relleno está presente en una cantidad de aproximadamente 20 % p/p a aproximadamente el 60 % p/p, o aproximadamente el 25 % p/p a aproximadamente el 55 % p/p, o aproximadamente el 30 % p/p a aproximadamente el 50 % p/p, o aproximadamente el 35 % p/p a aproximadamente el 45 % p/p. En algunas realizaciones, el relleno está presente en una cantidad de aproximadamente el 20 % p/p, aproximadamente el 25 % p/p, alrededor de 30 % p/p, aproximadamente el 35 % p/p, aproximadamente el 40 % p/p, aproximadamente el 45 % p/p, aproximadamente 50 % p/p, aproximadamente el 55 % p/p, o aproximadamente el 65 % p/p. En algunas otras realizaciones, el relleno está presente en una cantidad de aproximadamente el 40,4 % p/p.
En algunas realizaciones, el relleno comprende un primer relleno, que se agrega durante la etapa (a-1), y un segundo relleno, que se agrega durante la etapa (a-4), que pueden ser iguales o diferentes. En algunas realizaciones, el primer relleno y el segundo relleno son iguales. En algunas otras realizaciones, solo está presente el primer relleno. Todavía en algunas otras realizaciones, solo está presente el segundo relleno.
En algunas realizaciones, el primer relleno y el segundo relleno se seleccionan cada uno independientemente de entre el grupo que consiste en lactosa, celulosa microcristalina, manitol, etilcelulosa, sorbitol, almidón, sacarosa, fosfato de calcio, celulosa en polvo, celulosa microcristalina silicificada, isomalta, y mezclas de los mismos. En algunas otras realizaciones, el primer relleno y el segundo relleno se seleccionan cada uno independientemente de entre el grupo que consiste en celulosa microcristalina silicificada, celulosa microcristalina o mezclas de las mismas. En ciertas
realizaciones particulares, el primer relleno y el segundo relleno son ambos celulosa microcristalina.
En algunas realizaciones, el primer relleno y el segundo relleno están presentes cada uno en la misma cantidad, siempre que la cantidad total de relleno no supere aproximadamente el 80 % p/p. En otras realizaciones, el primer relleno y el segundo relleno están presentes cada uno en la misma cantidad, siempre que la cantidad total de relleno no supere aproximadamente el 80 % p/p. En algunas otras realizaciones, el primer relleno y el segundo relleno están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente el 80 % p/p, siempre que la cantidad total de relleno no supere el 80 % p/p. En algunas otras realizaciones, el primer relleno y el segundo relleno están presentes cada uno independientemente en una cantidad de aproximadamente 5 % p/p a aproximadamente el 40 % p/p. En algunas otras realizaciones, el primer relleno y el segundo relleno están presentes cada uno independientemente en una cantidad de aproximadamente 10 % p/p a aproximadamente el 30 % p/p. En algunas otras realizaciones, el primer relleno y el segundo relleno están presentes cada uno independientemente en una cantidad de aproximadamente el 10 %. p/p, o aproximadamente el 15 % p/p, o aproximadamente el 20 % p/p, o aproximadamente el 25 % p/p, o aproximadamente 30 %, p/p. En algunas otras realizaciones, el primer relleno y el segundo relleno están presentes cada uno independientemente en una cantidad de aproximadamente 20 % p/p.
Tensioactivos adecuados incluyen, entre otros, laurilsulfato de sodio, dodecilsulfato de sodio, polisorbatos (como Tween 20 y Tween 80), poloxámeros (como Poloxamer 335 y Poloxamer 407), monooleato de glicerilo y mezclas de los mismos. En algunas realizaciones, el tensioactivo es laurilsulfato de sodio, dodecilsulfato de sodio o mezclas de los mismos. En algunas realizaciones, el tensioactivo es laurilsulfato de sodio.
En algunas realizaciones, el tensioactivo está presente en una cantidad de aproximadamente 0 % p/p a aproximadamente 5 % p/p. En algunas realizaciones, el tensioactivo está presente en una cantidad de aproximadamente el 0 % p/p a aproximadamente el 3 % p/p. En algunas realizaciones, el tensioactivo está presente en una cantidad de aproximadamente el 1 % p/p a aproximadamente el 2 % p/p. En algunas otras realizaciones, el tensioactivo está presente en una cantidad de aproximadamente 0,5 % p/p, o aproximadamente 1 % p/p, o aproximadamente el 1,5 % p/p, o aproximadamente 2 % p/p, o aproximadamente el 2,5 % p/p, o aproximadamente 3 % p/p. En algunas otras realizaciones, el tensioactivo está presente en una cantidad de aproximadamente 2 % p/p.
En algunas realizaciones, el tensioactivo comprende un primer tensioactivo, que se agrega durante la etapa (a-1), y un segundo tensioactivo que se agrega durante la etapa (a-4), que pueden ser iguales o diferentes. En algunas realizaciones, el primer tensioactivo y el segundo tensioactivo son iguales. En algunas otras realizaciones, solo está presente el primer tensioactivo. Todavía en algunas otras realizaciones, solo está presente el segundo tensioactivo.
En algunas realizaciones, el primer tensioactivo y el segundo tensioactivo se seleccionan cada uno independientemente de entre el grupo que consiste en laurilsulfato de sodio, dodecilsulfato de sodio, polisorbatos (como Tween 20 y Tween 80), poloxámeros (como Poloxamer 335 y Poloxamer 407), monooleato de glicerilo y mezclas de los mismos. En algunas realizaciones, el tensioactivo es laurilsulfato de sodio, dodecilsulfato de sodio o mezclas de los mismos. En algunas otras realizaciones, el primer tensioactivo y el segundo tensioactivo son ambos laurilsulfato de sodio.
En algunas realizaciones, el primer tensioactivo y el segundo tensioactivo están presentes cada uno en la misma cantidad, siempre que la cantidad total de tensioactivo no sea mayor que aproximadamente 5 % p/p. En otras realizaciones, el primer tensioactivo y el segundo tensioactivo están presentes cada uno cantidades diferentes, siempre que la cantidad total de tensioactivo no supere aproximadamente el 5 % p/p. En algunas otras realizaciones, el primer tensioactivo y el segundo tensioactivo están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente 5 % p/p, siempre que la cantidad total de tensioactivo no sea superior a aproximadamente 5 % p/p. En algunas otras realizaciones, el primer tensioactivo y el segundo tensioactivo están presentes cada uno independientemente en una cantidad de aproximadamente el 0,5 %. p/p, o aproximadamente 1 % p/p, o aproximadamente el 1,5 % p/p, o aproximadamente 2 % p/p, o aproximadamente el 2,5 % p/p, o aproximadamente 3 % p/p, siempre que la cantidad total de tensioactivo no sea superior a aproximadamente 5 % p/p. En algunas otras realizaciones, el primer tensioactivo y el segundo tensioactivo están presentes cada uno independientemente en una cantidad de aproximadamente 1 % p/p.
Aglutinantes adecuados incluyen, entre otros, polivinilpirrolidona, etilcelulosa, alginato de maltosa sódica, hidroxipropilmetilcelulosa (HPMC), ácido esteárico, almidón pregelatinizado y mezclas de los mismos. En algunas realizaciones, el aglutinante es HPMC, polivinilpirrolidona o mezclas de los mismos. En otras realizaciones, el aglutinante es polivinilpirrolidona.
En algunas realizaciones, el aglutinante está presente en una cantidad de aproximadamente 0 % p/p a aproximadamente 20 % p/p. En algunas realizaciones, el aglutinante está presente en una cantidad de aproximadamente el 0 % p/p a aproximadamente el 10 % p/p. En algunas otras realizaciones, el aglutinante está presente en una cantidad de aproximadamente 1 % p/p, o aproximadamente 2 % p/p, o aproximadamente 3 % p/p, o aproximadamente 4 % p/p, o aproximadamente 5 % p/p, o aproximadamente 6 % p/p, o aproximadamente 7 % p/p, o aproximadamente 8 % p/p, o aproximadamente 9 % p/p, o aproximadamente el 10 % p/p. En algunas otras realizaciones, el aglutinante está presente en una cantidad de aproximadamente 1 % p/p a aproximadamente 9 % p/p,
o aproximadamente 2 % p/p a aproximadamente 8 % p/p, o aproximadamente 3 % p/p a aproximadamente 7 % p/p, o aproximadamente 4 % p/p a aproximadamente 6 % p/p. En algunas otras realizaciones, el aglutinante está presente en una cantidad de aproximadamente 5 %.
En algunas realizaciones, el aglutinante comprende un primer aglutinante, que se agrega durante la etapa (a-1), y un segundo aglutinante que se agrega durante la etapa (a-4), que pueden ser iguales o diferentes. En algunas realizaciones, el primer aglutinante y el segundo aglutinante son iguales. En algunas otras realizaciones, solo está presente el primer aglutinante. Todavía en algunas otras realizaciones, solo está presente el segundo aglutinante.
En algunas realizaciones, el primer aglutinante y el segundo aglutinante se seleccionan cada uno independientemente de entre el grupo que consiste en polivinilpirrolidona, etilcelulosa, alginato de sodio y maltosa, hidroxipropilmetilcelulosa (HPMC), ácido esteárico, almidón pregelatinizado y mezclas de los mismos. En algunas otras realizaciones, el primer aglutinante y el segundo aglutinante se seleccionan cada uno independientemente de entre el grupo que consiste en HPMC, polivinilpirrolidona y mezclas de los mismos. En ciertas realizaciones particulares, el primer aglutinante y el segundo aglutinante son ambos polivinilpirrolidona.
En algunas realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno en la misma cantidad, siempre que la cantidad total de aglutinante no sea mayor que aproximadamente 20 % p/p. En otras realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno en la misma cantidad, siempre que la cantidad total de aglutinante no supere aproximadamente el 20 % p/p. En algunas otras realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente 20 % p/p, siempre que la cantidad total de aglutinante no sea superior a aproximadamente 20 % p/p. En algunas otras realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente el 10 % p/p. En algunas otras realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente el 5 % p/p. En algunas otras realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno independientemente en una cantidad de aproximadamente el 0,5 %. p/p, o aproximadamente 1 % p/p, o aproximadamente el 1,5 % p/p, o aproximadamente 2 % p/p, o aproximadamente el 2,5 % p/p, o aproximadamente 3 % p/p, o aproximadamente el 3,5 % p/p, o aproximadamente 4 % p/p, o aproximadamente el 4,5 % p/p, o aproximadamente 5 % p/p. En algunas otras realizaciones, el primer aglutinante y el segundo aglutinante están presentes cada uno independientemente en una cantidad de aproximadamente 2,5 % p/p.
Desintegrantes adecuados incluyen, entre otros, dióxido de silicio coloidal, celulosa en polvo, silicato de calcio, crospovidona, alginato de calcio, metilcelulosa, quitosano, carboximetilcelulosa, croscarmelosa de sodio, carboximetilalmidón, alginato de sodio, glicolato de almidón de sodio, pregelatinizado almidón y mezclas de los mismos. En algunas realizaciones, el desintegrante es croscarmelosa sódica, crospovidona o mezclas de las mismas. En otras realizaciones, el desintegrante es croscarmelosa sódica.
En algunas realizaciones, el desintegrante está presente en una cantidad de aproximadamente 0 % p/p a aproximadamente 20 % p/p. En algunas realizaciones, el desintegrante está presente en una cantidad de aproximadamente el 0 % p/p a aproximadamente el 15 % p/p. En algunas otras realizaciones, el desintegrante está presente en una cantidad de aproximadamente 1 % p/p a aproximadamente el 14 % p/p, o de aproximadamente 2 % p/p a aproximadamente el 13 % p/p, o de aproximadamente 3 % p/p a aproximadamente el 12 % p/p, o de aproximadamente 4 % p/p a aproximadamente el 11 % p/p, o de aproximadamente 5 % p/p a aproximadamente el 10 % p/p, o de aproximadamente 6 % p/p a aproximadamente 9 % p/p, o de aproximadamente 7 % p/p a aproximadamente 8 % p/p. En algunas otras realizaciones, el desintegrante está presente en una cantidad de aproximadamente 4 % p/p, o aproximadamente 5 % p/p, o aproximadamente 6 % p/p, o aproximadamente 7 % p/p, o aproximadamente 8 % p/p, o aproximadamente 9 % p/p, o aproximadamente 10 % p/p, o aproximadamente 11 % p/p, o aproximadamente el 12 % p/p. En algunas otras realizaciones, el desintegrante está presente en una cantidad de aproximadamente 8 % p/p.
En algunas realizaciones, el desintegrante comprende un primer desintegrante, que se agrega durante la etapa (a-1), y un segundo desintegrante, que se agrega durante la etapa (a-4), que pueden ser iguales o diferentes. En algunas realizaciones, el primer desintegrante y el segundo desintegrante son iguales. En algunas otras realizaciones, solo está presente el primer desintegrante. Todavía en algunas otras realizaciones, solo está presente el segundo desintegrante.
En algunas realizaciones, el primer desintegrante y el segundo desintegrante se seleccionan cada uno independientemente de entre el grupo que consiste en dióxido de silicio coloidal, celulosa en polvo, silicato de calcio, crospovidona, alginato de calcio, metilcelulosa, quitosano, carboximetilcelulosa, croscarmelosa de sodio, carboximetilalmidón, alginato de sodio, glicolato de almidón de sodio, almidón pregelatinizado y mezclas de los mismos. En algunas otras realizaciones, el primer desintegrante y el segundo desintegrante se seleccionan cada uno independientemente de entre el grupo que consiste en croscarmelosa sódica, crospovidona y mezclas de los mismos. En ciertas realizaciones particulares, el primer desintegrante y el segundo desintegrante son ambos croscarmelosa sódica.
En algunas realizaciones, el primer desintegrante y el segundo desintegrante están presentes cada uno en la misma cantidad, siempre que la cantidad total de desintegrante no supere aproximadamente el 20 %. p/p. En otras realizaciones, el primer desintegrante y el segundo desintegrante están presentes cada uno en la misma cantidad, siempre que la cantidad total de desintegrante no supere aproximadamente el 20 % p/p. En algunas otras realizaciones, el primer desintegrante y el segundo desintegrante están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente 20 % p/p, siempre que la cantidad total de desintegrante no sea superior a aproximadamente 20 % p/p. En algunas otras realizaciones, el primer desintegrante y el segundo desintegrante están presentes cada uno independientemente en una cantidad de aproximadamente 0 % p/p a aproximadamente 8 % p/p. En algunas otras realizaciones, el primer desintegrante y el segundo desintegrante están presentes cada uno independientemente en una cantidad de aproximadamente 1 % p/p, o aproximadamente 2 % p/p, o aproximadamente 3 % p/p, o aproximadamente 4 % p/p, o aproximadamente 5 % p/p, o aproximadamente 6 % p/p, o aproximadamente 7 % p/p, o aproximadamente 8 % p/p. En algunas otras realizaciones, el primer desintegrante y el segundo desintegrante están presentes cada uno independientemente en una cantidad de aproximadamente 4 % p/p.
En algunas realizaciones, el primer desintegrante y el segundo desintegrante comprenden cada uno independientemente de aproximadamente el 30 % a aproximadamente el 70 % de la cantidad total de desintegrante. En algunas realizaciones, el primer desintegrante y el segundo desintegrante comprenden cada uno independientemente de aproximadamente el 40 % a aproximadamente el 60 % de la cantidad total de desintegrante. En algunas realizaciones, el primer desintegrante y el segundo desintegrante comprenden cada uno independientemente de aproximadamente el 40 %, o aproximadamente el 50 %, o aproximadamente el 60 % de la cantidad total de desintegrante. En algunas realizaciones, el primer desintegrante y el segundo desintegrante comprenden cada uno independientemente aproximadamente el 50 % de la cantidad total de desintegrante.
Lubricantes adecuados incluyen, entre otros, talco, estearato de magnesio, estearilfumarato de sodio, behenato de glicerilo, aceite vegetal hidrogenado, estearato de zinc, estearato de calcio, estearato de sacarosa, alcohol polivinílico, laurilsulfato de magnesio y mezclas de los mismos. En algunas realizaciones, el lubricante es estearato de magnesio, estearilfumarato de sodio o mezclas de los mismos. En otras realizaciones, el lubricante es estearilfumarato de sodio.
En algunas realizaciones el lubricante está presente en una cantidad de aproximadamente 0 % p/p a aproximadamente 5 % p/p. En algunas realizaciones, el lubricante está presente en una cantidad de aproximadamente 1 % p/p a aproximadamente 3 % p/p de lubricante En algunas realizaciones, el lubricante está presente en una cantidad de aproximadamente 1 % p/p a aproximadamente 2 % p/p de lubricante En algunas otras realizaciones, el lubricante está presente en una cantidad de aproximadamente 0,5 % p/p a aproximadamente el 4,5 % p/p, o de aproximadamente 0.5 % p/p a aproximadamente 4 % p/p, o de aproximadamente 0,5 % p/p a aproximadamente el 3,5 % p/p, o de aproximadamente 0.5 % p/p a aproximadamente 3 % p/p. En algunas realizaciones, el lubricante está presente en una cantidad de aproximadamente el 0 % p/p a aproximadamente el 3 % p/p. En algunas otras realizaciones, el lubricante está presente en una cantidad de aproximadamente 1 % p/p.
Deslizantes adecuados incluyen, entre otros, dióxido de silicio, dióxido de silicio coloidal, fosfato de calcio tribásico, estearato de magnesio, trisilicato de magnesio, celulosa en polvo, talco, almidón y mezclas de los mismos. En algunas realizaciones, el deslizante es talco, dióxido de silicio coloidal o mezclas de los mismos. En otras realizaciones, el deslizante es dióxido de silicio coloidal.
En algunas realizaciones el deslizante está presente en una cantidad de aproximadamente 0 % p/p a aproximadamente 5 % p/p. En algunas realizaciones, el deslizante está presente en una cantidad de aproximadamente el 0 % p/p a aproximadamente el 2 % p/p. En algunas realizaciones, el deslizante está presente en una cantidad de aproximadamente el 0,3 % p/p a aproximadamente el 2 % p/p, o de aproximadamente el 0,8 % p/p a aproximadamente el 1,5 % p/p. En otras realizaciones, el deslizante está presente en una cantidad de aproximadamente el 0,5 % p/p, o aproximadamente el 0,7 % p/p, o aproximadamente 1 % p/p, o aproximadamente el 1,2 % p/p, o aproximadamente el 1,5 % p/p, o aproximadamente el 1,7 % p/p, o aproximadamente 2 % p/p. En algunas otras realizaciones, el deslizante está presente en una cantidad de aproximadamente 1 % p/p.
Disolventes adecuados para la etapa de granulación en húmedo de (a-1) incluyen, entre otros, agua, etanol, acetona y mezclas de los mismos.
En algunas realizaciones, la cantidad de solvente presente en la etapa de granulación en húmedo de (a-1) es de aproximadamente 10 % p/p a aproximadamente el 50 % p/p. En otras realizaciones, el solvente está presente en una cantidad de aproximadamente 15 % p/p a aproximadamente el 40 % p/p, o aproximadamente el 28 % p/p.
Como se ha descrito anteriormente, tampones adecuados para su uso en la presente invención incluyen tampones tanto ácidos como básicos. Por ejemplo, en algunas realizaciones, una solución acuosa del tampón tiene un pH de menos de aproximadamente 7,0. En otras realizaciones, una solución acuosa del tampón tiene un pH de al menos aproximadamente 7,0. Ejemplos de tales tampones son conocidos por los expertos en la materia y se pueden encontrar en el Handbook of Pharmaceutical Excipients (5a Edición), Publicaciones APhA.
En algunas realizaciones, una solución acuosa del tampón tiene un pH de menos de aproximadamente 7,0. En algunas otras realizaciones, una solución acuosa del tampón tiene un pH de entre aproximadamente 1,0 y aproximadamente 6,0, o entre aproximadamente 2,0 y aproximadamente 6,0, o entre aproximadamente 3,0 y aproximadamente 6,0, o entre aproximadamente 4,0 y aproximadamente 6,0, o entre aproximadamente 5,0 a aproximadamente 6,0. Tampones adecuados que tienen un pH inferior a aproximadamente 7,0 en solución acuosa incluyen, entre otros, citrato disódico, citrato trisódico, acetato sódico, fosfato monopotásico, fosfato monosódico y mezclas de los mismos.
En algunas realizaciones, una solución acuosa del tampón tiene un pH de al menos de aproximadamente 7,0. En algunas otras realizaciones, una solución acuosa del tampón tiene un pH de entre aproximadamente 8,0 y aproximadamente 13,0, o entre aproximadamente 8,0 y aproximadamente 12,0, o entre aproximadamente 8,0 y aproximadamente 11,0, o entre aproximadamente 8,0 y aproximadamente 10,0, o entre aproximadamente 8,0 a aproximadamente 9,0.
Tampones adecuados que tienen un pH de al menos aproximadamente 7,0 en solución acuosa incluyen, entre otros, bicarbonato sódico, fosfato disódico, fosfato dipotásico, bicarbonato potásico, carbonato sódico, carbonato potásico y mezclas de los mismos. En algunas realizaciones, el tampón es bicarbonato de sodio, carbonato de sodio o mezclas de los mismos. En otras realizaciones, el tampón es bicarbonato de sodio.
En algunas realizaciones el tampón está presente en una cantidad de aproximadamente 10 % p/p a aproximadamente 80 % p/p. En algunas realizaciones, el tampón está presente en una cantidad de aproximadamente el 15 % p/p a aproximadamente el 60 % p/p. En algunas otras realizaciones, el tampóm está presente en una cantidad de aproximadamente 20 % p/p a aproximadamente el 55 % p/p, o de aproximadamente 22 % p/p a aproximadamente 50 % p/p, o de aproximadamente 25 % p/p a aproximadamente el 45 % p/p, o de aproximadamente 28 % p/p a aproximadamente 40 % p/p. En algunas otras realizaciones, el tampón está presente en una cantidad de aproximadamente el 30 % p/p.
En una realización de la invención, la composición farmacéutica preparada por los procedimientos de la invención comprende aproximadamente el 30 % p/p de tampón de bicarbonato de sodio.
El ingrediente activo es un compuesto de fórmula (/), o una sal farmacéuticamente aceptable del mismo, como se describe anteriormente.
En algunas realizaciones, el ingrediente activo es una forma cristalina de un compuesto de fórmula (/). En algunas otras realizaciones, el ingrediente activo es una forma cristalina de una sal farmacéuticamente aceptable de un compuesto de fórmula (/). Algunos ejemplos de sales farmacéuticamente aceptables de los compuestos de fórmula (/) y formas cristalinas de los mismos se pueden encontrar en Publicación EE. UU. 2008/0167292, y Solicitud EE. UU. No. 61/306.047, depositada el 19 de febrero de 2010.
En aún una realización adicional, el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1), o una forma cristalina del mismo. En otra realización, el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1) monohidrato. En otra realización, el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1) polimorfo Forma 2, como se describe en Publicación EE. UU. No. 2008/0167292, y Solicitud EE. UU. No. 61/306.047, depositada el 19 de febrero de 2010.
En algunas realizaciones, el ingrediente activo está presente en una cantidad de aproximadamente 1 % p/p a aproximadamente el 60 % p/p. En algunas realizaciones, el ingrediente activo está presente en una cantidad de aproximadamente el 1 % p/p a aproximadamente el 30 % p/p. En algunas otras realizaciones, el ingrediente activo está presente en una cantidad de aproximadamente 5 % p/p a aproximadamente el 25 % p/p, o de aproximadamente el 10 % p/p a aproximadamente el 20 % p/p, o de aproximadamente el 11 % p/p a aproximadamente el 18 % p/p, o de aproximadamente el 12 % p/p a aproximadamente el 16 % p/p. En algunas otras realizaciones, el ingrediente activo está presente en una cantidad de aproximadamente 10 % p/p, o aproximadamente el 11 % p/p, o aproximadamente el 12 % p/p, o aproximadamente el 13 % p/p, o aproximadamente el 14 % p/p, o aproximadamente el 15 % p/p, o aproximadamente el 16 % p/p. En algunas otras realizaciones, el ingrediente activo está presente en una cantidad de aproximadamente 13,6 % p/p.
En otra realización, la composición a granel comprende aproximadamente 1 % p/p a aproximadamente el 60 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de bicarbonato de sodio, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de celulosa microcristalina, aproximadamente 0 % p/p a aproximadamente 5 % p/p de estearilfumarato de sodio, aproximadamente 0 % p/p a aproximadamente 5 % p/p de lauril sulfato de sodio, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de polivinilpirrolidona, y aproximadamente 0 % p/p a aproximadamente el 20 % p/p de croscarmelosa sódica.
En otra realización, la composición a granel comprende aproximadamente 1 % p/p a aproximadamente el 30 % p/p
del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 30 % p/p a aproximadamente el 60 % p/p de bicarbonato de sodio, aproximadamente el 20 % p/p a aproximadamente el 60 % p/p de celulosa microcristalina, aproximadamente 1 % p/p a aproximadamente 3 % p/p de estearilfumarato de sodio, aproximadamente 0 % p/p a aproximadamente 3 % p/p de lauril sulfato de sodio, aproximadamente 0 % p/p a aproximadamente el 10 % p/p de polivinilpirrolidona, y aproximadamente 0 % p/p a aproximadamente el 15 % p/p de croscarmelosa sódica.
En otra realización, la composición a granel comprende aproximadamente 10 % p/p a aproximadamente el 16 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 28 % p/p a aproximadamente el 40 % p/p de bicarbonato de sodio, aproximadamente el 35 % p/p a aproximadamente el 45 % p/p de celulosa microcristalina, aproximadamente 1 % p/p a aproximadamente 2 % p/p de estearilfumarato de sodio, aproximadamente 1 % p/p a aproximadamente 2 % p/p de lauril sulfato de sodio, aproximadamente 3 % p/p a aproximadamente el 7 % p/p de polivinilpirrolidona, y aproximadamente 5 % p/p a aproximadamente el 10 % p/p de croscarmelosa sódica.
En otra realización, la composición a granel comprende aproximadamente el 13,6 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 30,0 % p/p de bicarbonato de sodio, y aproximadamente el 40,4 % p/p de celulosa microcristalina, aproximadamente 1,0 % p/p de estearilfumarato de sodio, aproximadamente 2,0 % p/p de lauril sulfato de sodio, aproximadamente 5,0 % p/p de polivinilpirrolidona, y aproximadamente el 8,0 % p/p de croscarmelosa sódica.
En realizaciones adicionales, la invención proporciona composiciones farmacéuticas que pueden formularse en formas de dosificación unitaria. La expresión "forma de dosificación unitaria", tal como se usa en esta invención, se refiere a un conjunto físicamente discreto del agente adecuado para el paciente a tratar. En algunas realizaciones, la forma de dosificación unitaria es una forma de dosificación farmacéutica oral sólida. Ejemplos de formas sólidas de dosificación farmacéutica oral incluyen, entre otros, comprimidos, cápsulas, píldoras, polvos y gránulos. En algunas realizaciones, la forma de dosificación farmacéutica oral sólida es un comprimido. En algunas realizaciones, los comprimidos están recubiertos con película, o con recubrimiento entérico, o ambos. En algunas de tales realizaciones, el comprimido está recubierto entéricamente.
Los excipientes adecuados, que incluyen tampones, rellenos, lubricantes, tensioactivos, aglutinantes y desintegrantes que pueden usarse en las formas farmacéuticas orales sólidas de la invención se describen anteriormente.
En algunas realizaciones, el ingrediente activo de la forma de dosificación farmacéutica oral sólida contiene un resto ácido, como se describe anteriormente. El ingrediente activo es un compuesto de fórmula (/), o una sal farmacéuticamente aceptable de un compuesto de fórmula (/), como se describe anteriormente.
En algunas realizaciones, el ingrediente activo de la forma de dosificación farmacéutica oral sólida es una forma cristalina de un compuesto de fórmula (/). En algunas otras realizaciones, el ingrediente activo de la forma de dosificación farmacéutica oral sólida es una forma cristalina de una sal farmacéuticamente aceptable de un compuesto de fórmula (/). Algunos ejemplos de sales farmacéuticamente aceptables de los compuestos de fórmula (/) y formas cristalinas de los mismos se pueden encontrar en Publicación EE. UU. 2008/0167292, y Solicitud EE. UU. No.
61/306.047, depositada el 19 de febrero de 2010.
En aún una realización adicional, el ingrediente activo de la forma de dosificación farmacéutica oral sólida es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1), o una forma cristalina del mismo. En otra realización, el ingrediente activo de la forma de dosificación farmacéutica oral sólida es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1) monohidrato. En otra realización, el ingrediente activo de la forma de dosificación farmacéutica oral sólida es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1) polimorfo Forma 2, como se describe en Publicación EE. UU. No. 2008/0167292, y Solicitud EE. UU. No. 61/306.047, depositada el 19 de febrero de 2010.
Materiales adecuados que se pueden usar para recubrir con película los comprimidos en la incluyen, entre otros, Opadry® (Hidroxi propil metilcelulosa PEG) (Colorcon, West Point, PA), Opadry® II (Polivinil alcohol, PEG, talco, y dióxido de titanio), Opadry® fx, Opadry® amb, y mezclas de los mismos. En algunas realizaciones, el material de recubrimiento de película es Opadry®, Opadry® II o mezclas de los mismos. En otras realizaciones, el material de revestimiento de película es Opadry® II.
En algunas realizaciones, el material de recubrimiento de película está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente 0 % p/p a aproximadamente el 10 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos. En algunas realizaciones, el material de recubrimiento de película está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente 0 % p/p a aproximadamente el 8 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos. En otras realizaciones, el material de recubrimiento de película está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente 0 % p/p a aproximadamente 6 % p/p, o aproximadamente el 0,5 % p/p al 5,5 % p/p, o aproximadamente el 1,0 % p/p al 5,0 % p/p, o aproximadamente el 1,5 % p/p al 4,5 % p/p, o aproximadamente 2.0 %
p/p al 4,0 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos. En algunas realizaciones, el material de recubrimiento de película está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente el 3,6 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos.
Materiales adecuados que se pueden usar para recubrir entéricamente incluyen, entre otros, Acryl- EZE ® (Copolímero de ácido metacrílico, talco, SLS, dióxido de titanio, bicarbonato de sodio, sílice, citrato de trietilo) (Colorcon, West Point, PA), ftalato de acetato de celulosa (CAP), copolímeros de acrilato de metilo y ácido metacrílico, succinato de acetato de celulosa, ftalato de hidroxipropilmetilcelulosa, succinato de acetato de hidroxipropilmetilcelulosa (succinato de acetato de hipromelosa), ftalato de polivinilacetato (PVAP) ), copolímeros de metacrilato de metilo-ácido metacrílico y mezclas de los mismos.
En algunas realizaciones, el material de recubrimiento entérico está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente 0 % p/p a aproximadamente el 20 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos. En algunas realizaciones, el material de recubrimiento entérico está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente 0 % p/p a aproximadamente el 18 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos. En otras realizaciones, el material de recubrimiento entérico está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente 0 % p/p a aproximadamente el 15 % p/p, o aproximadamente el 5 % p/p al 13 % p/p, o aproximadamente el 7 % p/p a aproximadamente el 11 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos. En algunas realizaciones, el material de recubrimiento entérico está presente en la forma de dosificación farmacéutica oral sólida en una cantidad de aproximadamente el 9,4 % p/p, donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos.
En otra realización, la forma de dosificación farmacéutica oral sólida comprende aproximadamente 1 % p/p a aproximadamente el 60 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de bicarbonato de sodio, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de celulosa microcristalina, aproximadamente 0 % p/p a aproximadamente 5 % p/p de estearilfumarato de sodio, aproximadamente 0 % p/p a aproximadamente 5 % p/p de lauril sulfato de sodio, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de polivinilpirrolidona, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de croscarmelosa sódica, aproximadamente 0 % p/p a aproximadamente el 10 % p/p de recubrimiento de película, y aproximadamente 0 % p/p a aproximadamente el 20 % p/p de recubrimiento entérico.
En otra realización, la forma de dosificación farmacéutica oral sólida comprende aproximadamente 1 % p/p a aproximadamente el 30 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 30 % p/p a aproximadamente el 60 % p/p de bicarbonato de sodio, aproximadamente el 20 % p/p a aproximadamente el 60 % p/p de celulosa microcristalina, aproximadamente 1 % p/p a aproximadamente 3 % p/p de estearilfumarato de sodio, aproximadamente 0 % p/p a aproximadamente 3 % p/p de lauril sulfato de sodio, aproximadamente 0 % p/p a aproximadamente el 10 % p/p de polivinilpirrolidona,aproximadamente 0 % p/p a aproximadamente el 15 % p/p de croscarmelosa sódica, aproximadamente 0,5 % p/p a aproximadamente el 5,5 % p/p de recubrimiento de película, y aproximadamente 5 % p/p a aproximadamente el 13 % p/p de recubrimiento entérico.
En otra realización, la forma de dosificación farmacéutica oral sólida comprende aproximadamente 10 % p/p a aproximadamente el 16 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 28 % p/p a aproximadamente el 40 % p/p de bicarbonato de sodio, aproximadamente el 35 % p/p a aproximadamente el 45 % p/p de celulosa microcristalina, aproximadamente 1 % p/p a aproximadamente 2 % p/p de estearilfumarato de sodio, aproximadamente 1 % p/p a aproximadamente 2 % p/p de lauril sulfato de sodio, aproximadamente 3 % p/p a aproximadamente el 7 % p/p de polivinilpirrolidona, y aproximadamente 5 % p/p a aproximadamente el 10 % p/p de croscarmelosa sódica, aproximadamente 2 % p/p a aproximadamente el 4 % p/p de recubrimiento de película, y aproximadamente 7 % p/p a aproximadamente el 11 % p/p de recubrimiento entérico.
En otra realizacion, la forma de dosificacion farmaceutica oral solida comprende aproximadamente 11,9 % p/p del Compuesto 1, o una forma cristalina del mismo, aproximadamente el 26,1 % p/p de bicarbonato de sodio, y aproximadamente el 35,1 % p/p de celulosa microcristalina, aproximadamente el 0,9 % p/p de fumarato de estearilo de sodio, aproximadamente el 1,7 % p/p de lauril sulfato de sodio, aproximadamente el 4,4 % p/p de polivinilpirrolidona, aproximadamente el 7,0 % p/p de croscarmelosa sódica, aproximadamente 3,6 % p/p de recubrimiento transparente Opadry® , y aproximadamente el 9,4 % p/p de recubrimiento entérico blanco Acryl-EZE® donde el peso total incluye todos los componentes del comprimido recubierto, incluidos los recubrimientos.
Las composiciones farmacéuticas de la invención que comprenden compuestos de fórmula (/), o sales farmacéuticamente aceptables de los mismos, y el Compuesto 1, o formas cristalinas de los mismos, son particularmente útiles en aplicaciones terapéuticas relacionadas con enfermedades, trastornos o afecciones mediadas por la quinasa mitótica, particularmente enfermedades, trastornos o afecciones mediadas por la quinasa Aurora. La inhibición de la actividad de la quinasa mitótica puede servir para tratar una serie de enfermedades, que implican la supervivencia, proliferación y migración celular, incluido el cáncer, así como otras enfermedades de proliferación
celular.
Un aspecto de la invención, por lo tanto, proporciona una composición farmacéutica de la invención para uso en procedimientos para tratar trastornos mediados por la quinasa Aurora mediante la administración de una cantidad terapéuticamente efectiva de la composición farmacéutica de la invención. Como se usa en esta invención, el término "trastorno mediado por quinasa Aurora" incluye cualquier trastorno, enfermedad o afección que esté causada o caracterizada por un aumento en la expresión o actividad de la quinasa Aurora, o que requiera actividad de quinasa Aurora. El término "trastorno mediado por quinasa Aurora" también incluye cualquier trastorno, enfermedad o afección en la que la inhibición de la actividad de quinasa Aurora es beneficiosa. Los trastornos mediados por quinasa Aurora incluyen trastornos proliferativos. Ejemplos no limitativos de trastornos proliferativos incluyen trastornos proliferativos inflamatorios crónicos, por ejemplo, psoriasis y artritis reumatoide; trastornos oculares proliferativos, por ejemplo, retinopatía diabética; trastornos proliferativos benignos, por ejemplo, hemangiomas; y cáncer Ejemplos no limitativos de cáncer incluyen cáncer colorrectal, cáncer de ovario, cáncer de mama, cáncer gástrico, cáncer de próstata y cáncer de páncreas.
La estabilidad física y química de la forma de dosificación farmacéutica oral se puede probar de manera convencional, por ejemplo, la medición del tiempo de disolución o desintegración, o el contenido de humedad, o el ensayo del ingrediente activo o los productos de degradación después del almacenamiento a diferentes temperaturas para diferentes intervalos de tiempo.
Los compuestos y composiciones, según el procedimiento de la presente invención, pueden administrarse utilizando cualquier cantidad y cualquier vía de administración efectiva para el tratamiento de la enfermedad. La cantidad exacta requerida variará de un sujeto a otro, dependiendo de la especie, la edad y el estado general del sujeto, la gravedad de la infección, el ingrediente activo particular, su modo de administración y similares. Las composiciones farmacéuticas se formulan preferiblemente en una forma de dosificación unitaria farmacéutica oral para facilitar la administración y la uniformidad de la dosificación. La expresión "forma de dosificación unitaria", tal como se usa en esta invención, se refiere a un conjunto físicamente discreto del agente adecuado para el paciente a tratar. Sin embargo, se entenderá que el uso diario total de las composiciones de la presente invención será decidido por el médico responsable dentro del alcance del buen criterio médico. El nivel de dosis efectiva específica para cualquier sujeto en particular dependerá de una variedad de factores, incluyendo la enfermedad que se está tratando y la gravedad de la enfermedad; la actividad del compuesto específico empleado; la composición específica empleada; la edad, el peso corporal, el estado general de salud, el sexo y la dieta del paciente; el tiempo de administración, la vía de administración y la tasa de excreción del compuesto específico empleado; la duración del tratamiento; los fármacos usados en combinación con, o simultáneamente a, el compuesto específico empleado; y factores similares bien conocidos en la técnica médica.
En algunas realizaciones, la forma de dosificación unitaria comprende de aproximadamente 1 mg a aproximadamente 250 mg de ingrediente activo. En algunas otras realizaciones, la forma de dosificación unitaria comprende de aproximadamente 5 mg a aproximadamente 200 mg de ingrediente activo. En algunas otras realizaciones, la forma de dosificación unitaria comprende de aproximadamente 10 mg a aproximadamente 150 mg de ingrediente activo. En algunas otras realizaciones más, la forma de dosificación unitaria comprende de aproximadamente 10 mg a aproximadamente 100 mg de ingrediente activo.
Para que esta invención se entienda mejor, se exponen los siguientes ejemplos preparativos. Estos ejemplos ilustran cómo hacer o probar composiciones específicas, y no deben interpretarse como limitantes del alcance de la invención de ninguna manera.
EJEMPLOS
Sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d] [2]benzazepin-2-il]amino}-2-metoxibenzoato (Compuesto 1), y formas cristalinas del mismo se pueden preparar según los procedimientos sintéticos descritos en Publicación EE. UU. No. 2008/0167292 o Solicitud EE. UU. No. 61/306.047, depositada el 19 de febrero de 2010. Cuando se usa el Compuesto 1 en los ejemplos siguientes, se entenderá que el término se refiere al Compuesto 1, o a una forma cristalina del mismo.
Ejemplos de composiciones farmacéuticas que se pueden preparar utilizando los procedimientos de la presente invención se muestran en los ejemplos a continuación.
Ejemplo 1: La composición farmacéutica se muestra a continuación en la Tabla 1
Tabla 1: composición farmacéutica


Ejemplo 2: La composición farmacéutica se muestra a continuación en la Tabla 2
Tabla 2: Composición farmacéutica

Ejemplo 3: La composición farmacéutica se muestra a continuación en la Tabla 3
Tabla 3: Composición farmacéutica

Ejemplo 4: Se fabricó una granulación por lotes de 1,0 kg mediante el siguiente procedimiento. El compuesto 1 (0,06 kg) se tamizó y se mezcló con celulosa microcristalina (0,79 kg) en un granulador húmedo de alto cizallamiento Diosna P1-6. Se tamizaron polivinilpirrolidona (0,08 kg) y croscarmelosa sódica (0,08 kg) y se añadieron al granulador. El líquido de granulación (agua) se roció a una tasa de rociado predeterminada de 50 g/minuto utilizando una bomba peristáltica. Una vez que se alcanzó el punto final con un contenido de humedad aproximado del 38,5 % del material granulado húmedo, los gránulos húmedos resultantes se tamizaron y posteriormente se secaron utilizando un secador de lecho fluido GPCG-1. Los gránulos secos resultantes se tamizaron y pesaron. En base al peso obtenido, se realizó un cálculo para determinar la cantidad adecuada de componentes extragranulares. Bicarbonato de Sodio (30 % p/p), Croscarmelosa Sódica (3 % p/p) y estearilfumarato de sodio (1 % p/p) a continuación se mezclaron con los gránulos secos tamizados para dar un lote con la composición que se muestra en la Tabla 4.
Tabla 4: Composición farmacéutica

Ejemplo 5: Se fabricó una granulación por lotes de 0,65 kg mediante el siguiente procedimiento. El compuesto 1 (0,13 kg) se tamizó y se mezcló con celulosa microcristalina (0,40 kg) en un granulador húmedo de alto cizallamiento Diosna P1-6. Laurilsulfato de sodio (0,02 kg), polivinilpirrolidona (0,05 kg) y croscarmelosa sódica (0,05 kg) se tamizaron y se añadieron al granulador. El líquido de granulación (agua) se roció a una tasa de rociado predeterminada de 35 g/minuto utilizando una bomba peristáltica. Una vez que se alcanzó el punto final con un contenido de humedad aproximado del 37,5 % del material granulado húmedo, los gránulos húmedos resultantes se tamizaron y posteriormente se secaron utilizando un secador de lecho fluido GPCG-1. Los gránulos secos resultantes se tamizaron y pesaron. En base al peso obtenido, se realizó un cálculo para determinar la cantidad adecuada de componentes extragranulares. Bicarbonato de Sodio (30 % p/p), Croscarmelosa Sódica (3 % p/p) y estearilfumarato de sodio (1 % p/p) a continuación se mezclaron con los gránulos secos tamizados para dar un lote con la composición que se muestra en la Tabla 5.
Tabla 5: Composición farmacéutica

Ejemplo 6: Se fabricó una granulación por lotes de 4,8 kg mediante el siguiente procedimiento. El compuesto 1 (0,64 kg) se tamizó a través de un tamiz de malla 14 y se mezcló con celulosa microcristalina (1,95 kg), lauril sulfato de sodio (0,1 kg), polivinilpirrolidona (0,24 kg) y croscarmelosa sódica (0,24 kg) en un granulador húmedo de alto cizallamiento PMA25/65. Se roció agua purificada a una tasa de rociado predeterminada de 180-235 g/minuto utilizando una bomba peristáltica. En este procedimiento se roció un total de 1 kg de agua purificada. Una vez que se alcanzó el punto final con un contenido de humedad aproximado del 25 % del material granulado húmedo, los gránulos húmedos resultantes se tamizaron y posteriormente se secaron utilizando un secador de lecho fluido GPCG-1. Los gránulos secos resultantes se tamizaron y pesaron. En base al peso obtenido, se realizó un cálculo para determinar la cantidad adecuada de componentes extragranulares. Bicarbonato de Sodio (30 % p/p), Croscarmelosa Sódica (3 % p/p) y estearilfumarato de sodio (1 % p/p) a continuación se mezclaron con los gránulos secos tamizados para dar un lote con la composición que se muestra en la Tabla 5.
Ejemplo 7: Se fabricó una granulación por lotes de 4,8 kg mediante el siguiente procedimiento. El compuesto 1 (0,64 kg) se tamizó a través de un tamiz de malla 14 y se mezcló con celulosa microcristalina (1,95 kg), lauril sulfato de sodio (0,1 kg), polivinilpirrolidona (0,24 kg) y croscarmelosa sódica (0,24 kg) en un granulador húmedo de alto cizallamiento PMA25/65. Se roció agua purificada a una tasa de rociado predeterminada de 242 g/minuto utilizando una bomba peristáltica. En este procedimiento se roció un total de 0,8 kg de agua purificada. El amasado en húmedo se realizó durante 2 minutos después de rociar el agua en este ejemplo. Una vez que se alcanzó el punto final con un contenido de humedad aproximado del 20 % del material granulado húmedo, los gránulos húmedos resultantes se tamizaron y posteriormente se secaron utilizando un secador de lecho fluido GPCG-1. Los gránulos secos resultantes se tamizaron y pesaron. En base al peso obtenido, se realizó un cálculo para determinar la cantidad adecuada de componentes extragranulares. Bicarbonato de Sodio (30 % p/p), Croscarmelosa Sódica (3 % p/p) y estearilfumarato de sodio (1 % p/p) a continuación se mezclaron con los gránulos secos tamizados para dar un lote con la composición que se muestra en la Tabla 5.
Ejemplo 8: Se fabricó una granulación por lotes de 4,8 kg mediante el siguiente procedimiento. El compuesto 1 (0,64 kg) se tamizó a través de un tamiz de malla 14 y se mezcló con celulosa microcristalina (1,95 kg), lauril sulfato de sodio (0,1 kg), polivinilpirrolidona (0,24 kg) y croscarmelosa sódica (0,24 kg) en un granulador húmedo de alto cizallamiento PMA25/65. Se roció agua purificada a una tasa de rociado predeterminada de 200 g/minuto a 254 g/minuto utilizando una bomba peristáltica. En este procedimiento se roció un total de 0,8 kg de agua purificada. Una vez que se alcanzó el punto final con un contenido de humedad aproximado del 20 % del material granulado húmedo, los gránulos húmedos resultantes se tamizaron y posteriormente se secaron utilizando un secador de lecho fluido GPCG-1. Los gránulos secos resultantes se tamizaron y pesaron. En base al peso obtenido, se realizó un cálculo para determinar la cantidad adecuada de componentes extragranulares. Bicarbonato de Sodio (30 % p/p), Croscarmelosa Sódica (3 % p/p) y estearilfumarato de sodio (1 % p/p) a continuación se mezclaron con los gránulos secos tamizados para dar un lote con la composición que se muestra en la Tabla 5.
Ejemplo 9: Los gránulos mezclados finales resultantes del Ejemplo 5 se cargaron en una prensa de comprimidos Piccola de 10 estaciones con estampación cóncava redonda estándar de 7/32". Los gránulos se comprimieron para fabricar comprimidos con un peso total de 80 mg por comprimido (dosis de 10 mg del Compuesto 1). Los comprimidos resultantes se recubrieron con una primera capa de Opadry® Clear Coating seguida de una segunda capa de Acryl-eze® White Enteric Coating utilizando un recubridor de cubeta perforada Vector LDCS. La composición de los comprimidos recubiertos entéricamente resultantes se muestra a continuación en la Tabla 6. Los gránulos combinados finales resultantes del Ejemplo 5 también se pueden usar para formar las composiciones de comprimidos que se muestran a continuación en las Tablas 7 y 8.
Tabla 6: Composición del comprimido

Ejemplo 10: La composición del comprimido se muestra a continuación en la Tabla 7
Tabla 7: Composición del comprimido
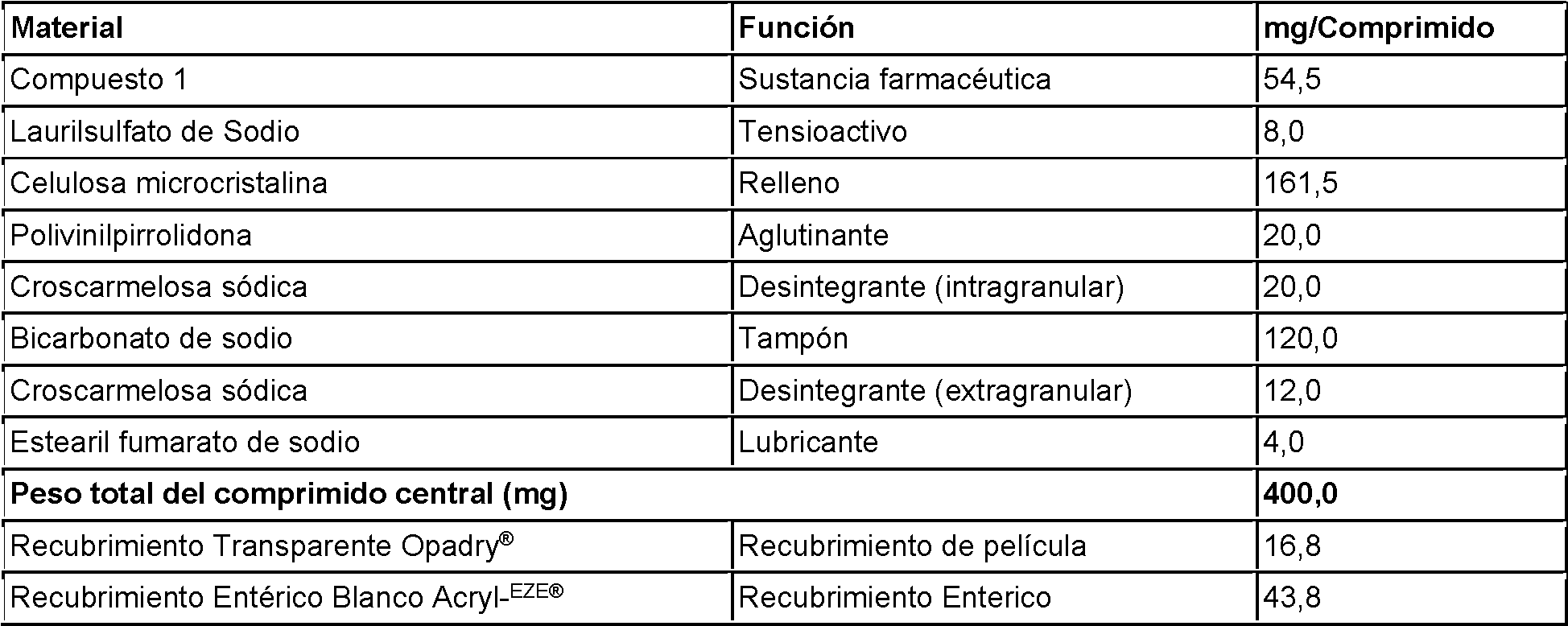

Ejemplo 11: La composición del comprimido se muestra a continuación en la Tabla 8
Tabla 8: Composición del comprimido

Ejemplo 12: Se fabricó una granulación por lotes de 1,0 kg mediante el siguiente procedimiento. El compuesto 1 (0,04 kg) se cribó y mezcló con celulosa microcristalina (0,55 kg), bicarbonato sódico (0,32 kg) y croscarmelosa sódica (0,05 kg) en un mezclador PK de 4 cuartos. A continuación, la mezcla bien mezclada se transfirió a un granulador de lecho fluido GPCG-1. Se mezcló polivinilpirrolidona (0,05 kg) con agua (0,3 kg) para preparar una solución aglutinante. La solución aglutinante se pulverizó a una velocidad de pulverización predeterminada de 21 g/minuto utilizando una bomba peristáltica. Una vez que toda la solución aglutinante se pulverizó en el granulador de lecho fluido GPCG-1, el procedimiento de secado continuó en el granulador de lecho fluido GPCG-1 hasta que se obtuvieron una temperatura de escape y un producto predeterminados. Los gránulos secos resultantes se tamizaron y pesaron. En base al peso obtenido, se realizó un cálculo para determinar la cantidad adecuada de componentes extragranulares. Croscarmelosa Sódica (3 % p/p) y estearilfumarato de sodio (1 % p/p) a continuación se mezclaron con los gránulos secos tamizados para dar un lote con la composición que se muestra en la Tabla 9.
Tabla 9: Composición farmacéutica

Si bien se ha descrito una cantidad de realizaciones de la presente invención, es evidente que los ejemplos básicos pueden alterarse para proporcionar otras realizaciones, los cuales utilicen los compuestos y procedimientos de la presente invención. Por lo tanto, se apreciará que el alcance de la presente invención debe definirse por las reivindicaciones adjuntas en lugar de por las realizaciones específicas, que han sido representadas a modo de ejemplo.
Claims (23)
1. Un procedimiento para preparar una composición farmacéutica que comprende las etapas de:
(a-1) granular en húmedo al menos un ingrediente activo que es un compuesto de fórmula (/):

o una sal farmacéuticamente aceptable del mismo, donde:
Ra se selecciona de entre el grupo que consiste en C1-3 alifático, C1-3 fluoroalifático, -R1, -T-R1, -R2,
y -T-R2;
T es una cadena de alquileno C1-3 opcionalmente sustituida con flúoro;
R1’ es un grupo arilo, heteroarilo o heterociclilo opcionalmente sustituido;
R2 se selecciona de entre el grupo que consiste en halo, -C=C-R3, -CH=CH-R3, -N(R4)2 , y -OR5;
R3 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Cada R4 es independientemente hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; o dos R4 en el mismo átomo de nitrógeno, tomados junto con el átomo de nitrógeno forman un anillo heteroarilo de 5 a 6 miembros o heterociclilo de 4 a 8 miembros opcionalmente sustituido que tiene, además del átomo de nitrógeno, 0-2 heteroátomos en el anillo seleccionado de entre N, O y S;
R5 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Rb se selecciona de entre el grupo que consiste en fluoro, cloro, -CH3 , -CF3 , -OH, -OCH3 , -OCF3 , -OCH2CH3 , y -OCH2CF3;
y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes y desintegrantes en presencia de un disolvente adecuado para formar una mezcla húmeda;
(a-2) secar la mezcla húmeda de la etapa (a-1), para formar gránulos secos;
(a-3) moler los gránulos secos de la etapa (a-2), para formar gránulos molidos; y
(a-4) mezclar los gránulos molidos de la etapa (a-3) con un tampón y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes, desintegrantes, lubricantes y deslizantes;
donde se añade un relleno durante la etapa (a-1), durante la etapa (a-4), o durante ambas etapas (a-1) y (a-4).
2. El procedimiento según la reivindicación 1, donde:
a) el procedimiento comprende además la etapa de (b-1) cargar la mezcla resultante de la etapa (a-4) en una cápsula; o
b) se agrega un lubricante durante la etapa (a-4), y donde el procedimiento comprende además la etapa de (c-1) formar comprimidos con la mezcla resultante de la etapa (a-4) para formar un comprimido, donde opcionalmente el procedimiento comprende además la etapa de (c-2) recubrir el comprimido resultante de la etapa (c-1), donde opcionalmente la etapa de recubrimiento (c-2) comprende recubrir con película y recubrir entéricamente el comprimido resultante de la etapa (c-1).
3. El procedimiento de la reivindicación 1, donde la etapa de granulación en húmedo está precedido por la etapa (a-0) que mezcla en seco el al menos un ingrediente activo y, opcionalmente, uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes, desintegrantes y rellenos.
4. El procedimiento de la reivindicación 1, donde el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato, o una forma cristalina del mismo.
5. El procedimiento de la reivindicación 1, donde el relleno se selecciona de entre el grupo que consiste en lactosa, celulosa microcristalina, manitol, etilcelulosa, sorbitol, almidón, sacarosa, fosfato de calcio, celulosa en polvo, celulosa microcristalina silicificada e isomalta, preferiblemente donde el relleno es celulosa microcristalina.
6. El procedimiento de la reivindicación 1, donde el tensioactivo de la etapa (a-1) o la etapa (a-4) se selecciona de entre el grupo que consiste en laurilsulfato de sodio, dodecilsulfato de sodio, polisorbatos, poloxámeros y monooleato de glicerilo, preferiblemente donde el tensioactivo es laurilsulfato de sodio.
7. El procedimiento de la reivindicación 1, donde el aglutinante de la etapa (a-1) o etapa (a-4) se selecciona de entre el grupo que consiste en polivinilpirrolidona, etilcelulosa, alginato de sodio maltosa, hidroxipropilmetilcelulosa, ácido esteárico y almidón pregelatinizado. , preferiblemente donde el aglutinante es polivinilpirrolidona.
8. El procedimiento de la reivindicación 1, donde el desintegrante de la etapa (a-1) y/o la etapa (a-4) se selecciona de entre el grupo que consiste en dióxido de silicio coloidal, celulosa en polvo, silicato de calcio, crospovidona, alginato de calcio, metilcelulosa, quitosano, carboximetilcelulosa, croscarmelosa de sodio, carboximetilalmidón, alginato de sodio, glicolato de almidón de sodio, y almidón pregelatinizado, preferiblemente donde el desintegrante es croscarmelosa sódica.
9. El procedimiento de la reivindicación 1, donde el lubricante se selecciona de entre el grupo que consiste en talco, estearato de magnesio, estearilfumarato de sodio, behenato de glicerilo, aceite vegetal hidrogenado, estearato de zinc, estearato de calcio, estearato de sacarosa, alcohol polivinílico y laurilsulfato de magnesio, preferiblemente donde el lubricante es estearilfumarato de sodio.
10. El procedimiento de la reivindicación 1, donde el tampón se selecciona de entre el grupo que consiste en bicarbonato de sodio, fosfato monosódico, fosfato disódico, fosfato monopotásico, fosfato dipotásico, bicarbonato de potasio, carbonato de sodio y carbonato de potasio, preferiblemente donde el tampón es bicarbonato de sodio .
11. El procedimiento de la reivindicación 1, donde la composición farmacéutica producida después de la etapa (a-4) comprende aproximadamente del 1 % p/p a aproximadamente el 60 % p/p de ingrediente activo, aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de tampón, y aproximadamente del 10 % p/p a aproximadamente el 80 % p/p de relleno.
12. El procedimiento de la reivindicación 11, donde:
a) la composición farmacéutica comprende aproximadamente 2 % p/p a aproximadamente el 22 % p/p del ingrediente activo, donde el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato, o una forma cristalina de los mismos; o
b) la composición farmacéutica comprende aproximadamente el 10 % p/p a aproximadamente el 60 % p/p del tampón, donde el tampón es bicarbonato de sodio; o
c) la composición farmacéutica comprende aproximadamente el 27 % p/p a aproximadamente el 53 % p/p del relleno, donde el relleno es celulosa microcristalina; o
d) la composición farmacéutica comprende además de aproximadamente 0 % p/p a aproximadamente 3 % p/p de lubricante, donde el lubricante es estearilfumarato de sodio.
13. El procedimiento de la reivindicación 1, donde el tampón es bicarbonato de sodio y está presente en una cantidad de aproximadamente 20 % p/p a aproximadamente 40 % p/p.
14. Una composición farmacéutica, donde la composición farmacéutica se prepara mediante el procedimiento de la reivindicación 1, que comprende:
(a) gránulos que comprenden un ingrediente activo de fórmula (I):

o una sal farmacéuticamente aceptable del mismo, donde:
Ra se selecciona de entre el grupo que consiste en C1-3 alifático, C1-3 fluoroalifático, -R1, -T-R1, -R2, y -T-R2;
T es una cadena de alquileno C1-3 opcionalmente sustituida con flúoro;
R1’ es un grupo arilo, heteroarilo o heterociclilo opcionalmente sustituido;
R2 se selecciona de entre el grupo que consiste en halo, -C=C-R3, -CH=CH-R3, -N(R4)2 , y -OR5;
R3 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Cada R4 es independientemente hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido; o dos R4 en el mismo átomo de nitrógeno, tomados junto con el átomo de nitrógeno forman un anillo heteroarilo de 5 a 6 miembros o heterociclilo de 4 a 8 miembros opcionalmente sustituido que tiene, además del átomo de nitrógeno, 0-2 heteroátomos en el anillo seleccionado de entre N, O y S;
R5 es hidrógeno o un grupo alifático, arilo, heteroarilo o heterociclilo opcionalmente sustituido;
Rb se selecciona de entre el grupo que consiste en fluoro, cloro, -CH3 , -CF3 , -OH, -OCH3 , -OCF3 , -OCH2CH3 , y -OCH2CF3;
y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes y desintegrantes; y
(b) un componente extragranular que comprende un tampón y opcionalmente uno o más excipientes farmacéuticamente aceptables seleccionados independientemente de entre el grupo que consiste en tensioactivos, aglutinantes, desintegrantes, lubricantes y deslizantes;
donde la composición comprende además un relleno dentro de los gránulos, dentro del componente extragranular, o dentro de los gránulos y el componente extragranular.
15. La composición farmacéutica de la reivindicación 14, donde el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato, o una forma cristalina del mismo.
16. La composición farmacéutica de la reivindicación 14, que comprende aproximadamente 1 % p/p a aproximadamente el 60 % p/p de ingrediente activo, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de tampón, y aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de relleno, opcionalmente donde:
el tampón se selecciona de entre el grupo que consiste en bicarbonato de sodio, fosfato monosódico, fosfato disódico, fosfato monopotásico, fosfato dipotásico, bicarbonato de potasio, carbonato de sodio y carbonato de potasio, preferiblemente el tampón es bicarbonato de sodio; y/o
el relleno se selecciona de entre el grupo que consiste en lactosa, celulosa microcristalina, manitol, etilcelulosa, sorbitol, almidón, sacarosa, fosfato de calcio, celulosa en polvo, celulosa microcristalina silicificada e isomalta, preferiblemente el relleno es celulosa microcristalina.
17. La composición agrícola de la reivindicación 15, que comprende:
a) aproximadamente 2 % p/p a aproximadamente el 22 % p/p del ingrediente activo, donde el ingrediente activo es sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato, o una forma cristalina de los mismos; o
b) aproximadamente el 10 % p/p a aproximadamente el 60 % p/p del tampón, donde el tampón es bicarbonato de sodio; o
c) aproximadamente el 27 % p/p a aproximadamente el 53 % p/p del relleno, donde el relleno es celulosa microcristalina; o
d) de aproximadamente 0 % p/p a aproximadamente 3 % p/p de lubricante, donde el lubricante es estearilfumarato de sodio.
18. La composición farmacéutica de la reivindicación 14, donde el tampón es bicarbonato de sodio y está presente en una cantidad de aproximadamente 20 % p/p a aproximadamente el 40 % p/p.
19. La composición farmacéutica de la reivindicación 14, que comprende aproximadamente 1 % p/p a aproximadamente el 60 % p/p de sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato, o una forma cristalina del mismo, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de bicarbonato de sodio, aproximadamente el 10 % p/p a aproximadamente el 80 % p/p de celulosa microcristalina, aproximadamente 0 % p/p a aproximadamente 5 % p/p de estearilfumarato de sodio, aproximadamente 0 % p/p a aproximadamente 5 % p/p de lauril sulfato de sodio, aproximadamente 0 % p/p a aproximadamente el 20 % p/p de polivinilpirrolidona, y aproximadamente 0 % p/p a aproximadamente el 20 % p/p de croscarmelosa de sodio.
20. La composición farmacéutica de la reivindicación 14, que comprende aproximadamente 13,6 % p/p de sodio 4-{[9-cloro-7-(2-fluoro-6-metoxifenil)-5H-pirimido[5,4-d][2]benzazepin-2-yl]amino}-2-metoxibenzoato, o una forma cristalina del mismo, aproximadamente 30,0 % p/p de bicarbonato de sodio, aproximadamente 40,4 % p/p de celulosa microcristalina, aproximadamente 1,0 % p/p de estearilfumarato de sodio, aproximadamente 2,0 % p/p de lauril sulfato de sodio, aproximadamente 5,0 % p/p de polivinilpirrolidona, y aproximadamente 8,0 % p/p de croscarmelosa sódica.
21. La composición farmacéutica de la reivindicación 20, donde la composición farmacéutica es una forma de dosificación farmacéutica oral sólida.
22. Una composición farmacéutica de cualquiera de las reivindicaciones 14--21 para su uso en el tratamiento del cáncer.
23. La composición farmacéutica para uso de la reivindicación 22, donde el cáncer es cáncer colorrectal, cáncer de ovario, cáncer de mama, cáncer gástrico, cáncer de próstata y cáncer de páncreas.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21649309P | 2009-05-18 | 2009-05-18 | |
| US26843809P | 2009-06-12 | 2009-06-12 | |
| PCT/US2010/001434 WO2010134965A1 (en) | 2009-05-18 | 2010-05-14 | Solid pharmaceutical compositions and processes for their production |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2908655T3 true ES2908655T3 (es) | 2022-05-03 |
Family
ID=42332398
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES10723816T Active ES2908655T3 (es) | 2009-05-18 | 2010-05-14 | Composiciones farmacéuticas sólidas y procedimientos para su producción |
Country Status (30)
| Country | Link |
|---|---|
| US (6) | US9173846B2 (es) |
| EP (1) | EP2432456B1 (es) |
| JP (2) | JP5802199B2 (es) |
| KR (1) | KR101809140B1 (es) |
| CN (2) | CN103860512A (es) |
| AR (1) | AR076879A1 (es) |
| AU (1) | AU2010250099B2 (es) |
| BR (1) | BRPI1012875B8 (es) |
| CA (1) | CA2762336C (es) |
| CL (1) | CL2011002918A1 (es) |
| CO (1) | CO6470892A2 (es) |
| CR (1) | CR20110626A (es) |
| DK (1) | DK2432456T3 (es) |
| DO (1) | DOP2011000358A (es) |
| EA (1) | EA024209B1 (es) |
| EC (1) | ECSP11011542A (es) |
| ES (1) | ES2908655T3 (es) |
| GE (1) | GEP20156328B (es) |
| IL (1) | IL216427A (es) |
| JO (1) | JO3635B1 (es) |
| MA (1) | MA33349B1 (es) |
| MX (1) | MX2011012368A (es) |
| MY (1) | MY178082A (es) |
| NZ (1) | NZ596883A (es) |
| PE (1) | PE20120316A1 (es) |
| SG (1) | SG176158A1 (es) |
| TW (1) | TWI597073B (es) |
| UY (1) | UY32647A (es) |
| WO (1) | WO2010134965A1 (es) |
| ZA (1) | ZA201108846B (es) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CL2007003244A1 (es) | 2006-11-16 | 2008-04-04 | Millennium Pharm Inc | Compuestos derivados de pirimido[5,4-d][2]benzazepina; composicion farmaceutica que comprende a dicho compuesto; y uso del compuesto para el tratamiento del cancer. |
| RU2535032C2 (ru) | 2008-12-22 | 2014-12-10 | Милленниум Фармасьютикалз, Инк. | Сочетание ингибиторов аврора киназы и анти-cd 20 антител |
| JO3635B1 (ar) | 2009-05-18 | 2020-08-27 | Millennium Pharm Inc | مركبات صيدلانية صلبة وطرق لانتاجها |
| JO3434B1 (ar) | 2009-07-31 | 2019-10-20 | Millennium Pharm Inc | مركبات صيدلانية لمعالجة السرطان وامراض واضطرابات اخري |
| BR112012020557A8 (pt) | 2010-02-19 | 2018-01-02 | Millennium Pharm Inc | formas cristalinas de 4-{[9-cloro-7-(2-flúor-6-metoifenil)-5h-pirimido[5,4-d][2]benzazepin-2-il]amino}-2-metoxibenzoato de sódio |
| AR086656A1 (es) | 2011-06-03 | 2014-01-15 | Millennium Pharm Inc | Combinacion de inhibidores de mek e inhibidores selectivos de la quinasa aurora a |
| ITBO20110461A1 (it) * | 2011-07-29 | 2013-01-30 | Alfa Wassermann Spa | Composizioni farmaceutiche comprendenti rifaximina, processi per la loro preparazione e loro uso nel trattamento di infezioni vaginali. |
| US20130303519A1 (en) | 2012-03-20 | 2013-11-14 | Millennium Pharmaceuticals, Inc. | Methods of treating cancer using aurora kinase inhibitors |
| WO2014153509A1 (en) | 2013-03-22 | 2014-09-25 | Millennium Pharmaceuticals, Inc. | Combination of catalytic mtorc 1/2 inhibitors and selective inhibitors of aurora a kinase |
| EP3076963A4 (en) | 2013-12-06 | 2017-09-13 | Millennium Pharmaceuticals, Inc. | Combination of aurora kinase inhibitors and anti-cd30 antibodies |
| EP3148513B1 (en) * | 2014-05-29 | 2020-02-26 | Novartis AG | Ceritinib formulation |
| BR112017012558A2 (pt) * | 2014-12-19 | 2018-01-02 | Merck Sharp & Dohme | Composições de (s)-n-(3-(6-isopropoxipiridin-3-il)-1h- indazol-5-il)-1-(2-(4-(4-(1-metil-1h-1,2,4-triazol-3-il) fenil)-3,6-diidropiridin-1(2h)-il)-2-oxoetil)-3-(metiltio) pirrolidina-3-carboxamida para preparações farmacêuticas |
| WO2017015316A1 (en) | 2015-07-21 | 2017-01-26 | Millennium Pharmaceuticals, Inc. | Administration of aurora kinase inhibitor and chemotherapeutic agents |
| SG10201609137PA (en) * | 2016-11-01 | 2018-06-28 | Xylonix Ip Holdings Pte Ltd | Gamma-polyglutamic acid and zinc compositions |
| SG10201609131YA (en) | 2016-11-01 | 2018-06-28 | Xylonix Ip Holdings Pte Ltd | Zinc-pga compositions and methods for treating cancer |
| CN110772423A (zh) * | 2019-09-20 | 2020-02-11 | 徐州立方机电设备制造有限公司 | 一种基于智能控制的胶囊生产设备 |
| KR20230041049A (ko) * | 2020-07-20 | 2023-03-23 | 세이지 테라퓨틱스, 인크. | 19-노르 c3,3-이치환된 c21-n-피라졸릴 스테로이드의 제형 및 이의 사용 방법 |
| US12133918B2 (en) | 2021-10-01 | 2024-11-05 | Griffin Gamma, Llc | Partially pre-gelatinized cassava starch as pharmaceutical excipient |
| KR102767479B1 (ko) * | 2022-06-13 | 2025-02-14 | 전라남도 | 홍화씨 타블렛 제조방법 및 이에 따라 제조된 홍화씨 타블렛 |
| CN120112284A (zh) * | 2022-08-19 | 2025-06-06 | 米拉蒂治疗股份有限公司 | 阿达格拉西布固体药物组合物 |
| CN121941495A (zh) | 2023-10-03 | 2026-04-28 | 武田药品工业株式会社 | 用于治疗小细胞肺癌的阿立塞替和紫杉醇 |
| WO2025245045A1 (en) | 2024-05-21 | 2025-11-27 | The Regents Of The University Of California | Methods of treating lung cancer |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4099012A (en) | 1975-08-28 | 1978-07-04 | Ciba-Geigy Corporation | 2-pyrazolyl-benzophenones |
| NZ192803A (en) | 1979-02-07 | 1984-08-24 | Hoffmann La Roche | Pyrimido-2-benzazepines and pharmaceutical compositions |
| US4481142A (en) | 1979-02-07 | 1984-11-06 | Hoffmann-La Roche Inc. | Pyrimido-2-benzazepines |
| US4469633A (en) | 1980-05-16 | 1984-09-04 | Hoffmann-La Roche Inc. | N-oxides of 5-oxo-1-phenyl-2-benzazepines |
| EP0273697A3 (en) | 1986-12-30 | 1989-11-29 | Merck & Co. Inc. | 2-benzazepines with 5- and 6- membered heterocyclic rings |
| US5166151A (en) | 1988-03-25 | 1992-11-24 | Merck & Co., Inc. | 2-Benzazepines with 5- and 6-membered heterocyclic rings, compositions and medical methods of use thereof |
| US5210082A (en) | 1991-05-16 | 1993-05-11 | Merck & Co., Inc. | 2-benzazepines with 5- and 6-membered heterocyclic rings to treat pain and anxiety disorders |
| US5747487A (en) | 1993-07-29 | 1998-05-05 | American Cyanamid Company | Tricyclic benzazepine vasopressin antagonists |
| GB9523675D0 (en) | 1995-11-20 | 1996-01-24 | Celltech Therapeutics Ltd | Chemical compounds |
| EP0888350A1 (en) | 1996-03-08 | 1999-01-07 | Zeneca Limited | Azolobenzazepine derivatives as neurologically active agents |
| EP0946523A1 (en) | 1996-12-23 | 1999-10-06 | Celltech Therapeutics Limited | Fused polycyclic 2-aminopyrimidine derivatives, their preparation and their use as protein tyrosine kinase inhibitors |
| GB9713087D0 (en) | 1997-06-20 | 1997-08-27 | Celltech Therapeutics Ltd | Chemical compounds |
| ES2191963T3 (es) | 1997-09-29 | 2003-09-16 | Meiji Seika Kaisha | Derivados triciclicos de triazolobenzazepina, proceso para la obtencion de los mismos y agentes antialergicos. |
| US6277844B1 (en) | 1998-09-14 | 2001-08-21 | Sydney Spector | Compound for selective treatment of malignant cells by inhibiting cell cycle progression, decreasing Bcl2, and increasing apoptosis |
| US6417207B1 (en) | 1999-05-12 | 2002-07-09 | Nitromed, Inc. | Nitrosated and nitrosylated potassium channel activators, compositions and methods of use |
| US7674480B2 (en) * | 2000-06-23 | 2010-03-09 | Teva Pharmaceutical Industries Ltd. | Rapidly expanding composition for gastric retention and controlled release of therapeutic agents, and dosage forms including the composition |
| US6989385B2 (en) | 2000-12-21 | 2006-01-24 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| SI20848A (sl) | 2001-03-14 | 2002-10-31 | Lek, Tovarna Farmacevtskih In Kemijskih Izdelkov, D.D. | Farmacevtska formulacija, ki vsebuje atorvastatin kalcij |
| US6686352B2 (en) | 2001-05-18 | 2004-02-03 | Hoffmann-La Roche Inc. | Substituted imidazo [1,5-a] pyrimido [5,4-d] [1] benzazepine derivatives |
| DE10135457A1 (de) | 2001-07-20 | 2003-02-06 | Adc Automotive Dist Control | Optische Sensoranordnung |
| JP4364637B2 (ja) | 2001-08-09 | 2009-11-18 | アクテリオン ファーマシューティカルズ リミテッド | 新規なベンゾ縮合へテロ環化合物 |
| CN1897950A (zh) | 2003-10-14 | 2007-01-17 | 惠氏公司 | 稠合芳基和杂芳基衍生物及其使用方法 |
| AU2004296877A1 (en) * | 2003-12-09 | 2005-06-23 | Incyte Corporation | Dosing methods for ss-D-2',3'-dideoxy-2',3'-didehydro-5-fluorocytidine antiviral therapy |
| WO2005076987A2 (en) | 2004-02-10 | 2005-08-25 | Santarus, Inc. | Combination of proton pump inhibitor, buffering agent, and nonsteroidal anti-inflammatory agent |
| AU2005243175B2 (en) | 2004-05-14 | 2011-12-01 | Millennium Pharmaceuticals, Inc. | Compounds and methods for inhibiting mitotic progression by inhibition of Aurora kinase |
| CN101084214A (zh) | 2004-11-17 | 2007-12-05 | 迈卡纳治疗股份有限公司 | 激酶抑制剂 |
| WO2006070198A1 (en) | 2004-12-30 | 2006-07-06 | Astex Therapeutics Limited | Pyrazole derivatives as that modulate the activity of cdk, gsk and aurora kinases |
| US20070104785A1 (en) * | 2005-07-29 | 2007-05-10 | Navale Suryakant V | Tablets of linezolid form iii and processes for their preparation |
| EA200870117A1 (ru) | 2005-12-23 | 2008-12-30 | Смитклайн Бичам Корпорейшн | Азаиндоловые ингибиторы аурора-киназ |
| JP2009523784A (ja) | 2006-01-16 | 2009-06-25 | ジュビラント・オルガノシス・リミテッド | 酸不安定化合物の安定な医薬製剤およびその製造方法 |
| TW200744583A (en) * | 2006-03-14 | 2007-12-16 | Ranbaxy Lab Ltd | Statin stabilizing dosage formulations |
| DE102006012301A1 (de) | 2006-03-15 | 2007-09-20 | Cemag-Anlagenbau-Dessau Gmbh | Herstellung von Zementklinker |
| US7718648B2 (en) | 2006-08-09 | 2010-05-18 | Millennium Pharmaceuticals, Inc. | Pyridobenzazepine compounds and methods for inhibiting mitotic progression |
| AU2007314282A1 (en) * | 2006-10-31 | 2008-05-08 | Achillion Pharmaceuticals, Inc. | Elvucitabine pharmaceutical compositions |
| CL2007003244A1 (es) * | 2006-11-16 | 2008-04-04 | Millennium Pharm Inc | Compuestos derivados de pirimido[5,4-d][2]benzazepina; composicion farmaceutica que comprende a dicho compuesto; y uso del compuesto para el tratamiento del cancer. |
| AR065802A1 (es) * | 2007-03-22 | 2009-07-01 | Schering Corp | Formulaciones de comprimidos que contienen sales de 8- [( 1- ( 3,5- bis- (trifluorometil) fenil) -etoxi ) - metil) -8- fenil -1, 7- diaza- spiro [ 4,5] decan -2- ona y comprimidos elaborados a partir de estas |
| US20090203671A1 (en) | 2007-11-27 | 2009-08-13 | Abbott Laboratories | Method of treating cancer |
| KR101848095B1 (ko) | 2008-06-26 | 2018-04-11 | 안테리오스, 인코퍼레이티드 | 경피 운반 |
| JO3635B1 (ar) | 2009-05-18 | 2020-08-27 | Millennium Pharm Inc | مركبات صيدلانية صلبة وطرق لانتاجها |
| JO3434B1 (ar) | 2009-07-31 | 2019-10-20 | Millennium Pharm Inc | مركبات صيدلانية لمعالجة السرطان وامراض واضطرابات اخري |
-
2010
- 2010-05-12 JO JOP/2010/0155A patent/JO3635B1/ar active
- 2010-05-14 EP EP10723816.4A patent/EP2432456B1/en active Active
- 2010-05-14 MA MA34433A patent/MA33349B1/fr unknown
- 2010-05-14 DK DK10723816.4T patent/DK2432456T3/da active
- 2010-05-14 ES ES10723816T patent/ES2908655T3/es active Active
- 2010-05-14 MY MYPI2011005572A patent/MY178082A/en unknown
- 2010-05-14 KR KR1020117029942A patent/KR101809140B1/ko active Active
- 2010-05-14 PE PE2011001987A patent/PE20120316A1/es active IP Right Grant
- 2010-05-14 EA EA201171435A patent/EA024209B1/ru unknown
- 2010-05-14 CN CN201410045241.8A patent/CN103860512A/zh active Pending
- 2010-05-14 GE GEAP201012505A patent/GEP20156328B/en unknown
- 2010-05-14 WO PCT/US2010/001434 patent/WO2010134965A1/en not_active Ceased
- 2010-05-14 AU AU2010250099A patent/AU2010250099B2/en active Active
- 2010-05-14 NZ NZ596883A patent/NZ596883A/en unknown
- 2010-05-14 CN CN2010800220567A patent/CN102427805A/zh active Pending
- 2010-05-14 BR BRPI1012875A patent/BRPI1012875B8/pt active IP Right Grant
- 2010-05-14 MX MX2011012368A patent/MX2011012368A/es active IP Right Grant
- 2010-05-14 CA CA2762336A patent/CA2762336C/en active Active
- 2010-05-14 JP JP2012511813A patent/JP5802199B2/ja active Active
- 2010-05-14 US US12/780,015 patent/US9173846B2/en active Active
- 2010-05-14 SG SG2011085289A patent/SG176158A1/en unknown
- 2010-05-18 TW TW099115844A patent/TWI597073B/zh active
- 2010-05-18 UY UY0001032647A patent/UY32647A/es not_active Application Discontinuation
- 2010-05-18 AR ARP100101730A patent/AR076879A1/es not_active Application Discontinuation
-
2011
- 2011-11-17 IL IL216427A patent/IL216427A/en active IP Right Grant
- 2011-11-18 CL CL2011002918A patent/CL2011002918A1/es unknown
- 2011-11-18 DO DO2011000358A patent/DOP2011000358A/es unknown
- 2011-11-24 CR CR20110626A patent/CR20110626A/es unknown
- 2011-12-01 ZA ZA2011/08846A patent/ZA201108846B/en unknown
- 2011-12-14 CO CO11172436A patent/CO6470892A2/es not_active Application Discontinuation
- 2011-12-16 EC EC2011011542A patent/ECSP11011542A/es unknown
-
2014
- 2014-09-19 JP JP2014191161A patent/JP6150777B2/ja active Active
-
2015
- 2015-09-22 US US14/860,829 patent/US9655856B2/en active Active
-
2017
- 2017-04-25 US US15/496,728 patent/US20170281551A1/en not_active Abandoned
-
2019
- 2019-02-27 US US16/287,419 patent/US10888523B2/en active Active
-
2021
- 2021-01-11 US US17/145,799 patent/US11786470B2/en active Active
-
2023
- 2023-09-06 US US18/462,342 patent/US20240091157A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2908655T3 (es) | Composiciones farmacéuticas sólidas y procedimientos para su producción | |
| ES2957912T3 (es) | Composiciones farmacéuticas que comprenden nilotinib | |
| CN109646404B (zh) | 葡萄糖激酶激活剂的口服制剂及其制备方法 | |
| KR101290925B1 (ko) | 코팅된 정제 제형 및 방법 | |
| RS66565B1 (sr) | Farmaceutska kombinacija, kompozicija i kombinovani preparat koji sadrži aktivator glukokinaze i inhibitor sglt-2 i postupci njihove pripreme i upotrebe | |
| TWI827535B (zh) | 乳酸鈣組成物及使用方法 | |
| US20170281642A1 (en) | Methods of treating acute myeloid leukemia or acute lymphoid leukemia using pharmaceutical compositions containing thienotriazolodiazepine compounds | |
| WO2014190898A1 (zh) | 一种异硫氰酸酯类化合物制剂 | |
| AU2016224503A1 (en) | Solid preparation | |
| ES2601503T3 (es) | Composición farmacéutica | |
| CA2999030A1 (en) | Pharmaceutical compositions comprising palmitoylethanolamide and naturalfatty acid amide hydrolase inhibitor(s) and use thereof in the treatmentof neuropathic pain | |
| ES2629739T3 (es) | Composición farmacéutica estable para el tratamiento de osteoporosis | |
| ES3033299T3 (en) | Crystalline forms of compounds for preventing or treating sensory hair cell death | |
| KR101379664B1 (ko) | 리세드론산 또는 그의 염 및 비타민 d를 포함하는 약학 조성물 | |
| JP2020075924A (ja) | エルロチニブを有効成分とする医薬錠剤 | |
| JP2020090456A (ja) | エルロチニブを有効成分とする医薬錠剤 | |
| KR20260062790A (ko) | 닌테다닙 또는 이의 약학적으로 허용가능한 염 및 메타크릴산 공중합체를 포함하는 약학 제제 | |
| HK40007318B (en) | Oral formulation of glucokinase activators and preparation method thereof | |
| KR20130031313A (ko) | 리세드론산 또는 그의 염 및 비타민 d를 포함하는 약학 조성물 |