ES2924933T3 - Derivados de piridina y usos terapéuticos de los mismos como inhibidores de TRPC6 - Google Patents
Derivados de piridina y usos terapéuticos de los mismos como inhibidores de TRPC6 Download PDFInfo
- Publication number
- ES2924933T3 ES2924933T3 ES20197194T ES20197194T ES2924933T3 ES 2924933 T3 ES2924933 T3 ES 2924933T3 ES 20197194 T ES20197194 T ES 20197194T ES 20197194 T ES20197194 T ES 20197194T ES 2924933 T3 ES2924933 T3 ES 2924933T3
- Authority
- ES
- Spain
- Prior art keywords
- methoxy
- mmol
- pyridin
- carboxylic acid
- pyridine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102000003623 TRPC6 Human genes 0.000 title claims abstract 3
- 108050001421 Transient receptor potential channel, canonical 6 Proteins 0.000 title claims abstract 3
- 239000003112 inhibitor Substances 0.000 title description 8
- 230000001225 therapeutic effect Effects 0.000 title description 5
- 150000003222 pyridines Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 309
- 150000003839 salts Chemical class 0.000 claims abstract description 43
- 238000011282 treatment Methods 0.000 claims abstract description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 9
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 40
- 201000010099 disease Diseases 0.000 claims description 20
- 208000035475 disorder Diseases 0.000 claims description 20
- 206010028980 Neoplasm Diseases 0.000 claims description 19
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 claims description 19
- 208000004248 Familial Primary Pulmonary Hypertension Diseases 0.000 claims description 15
- 208000020875 Idiopathic pulmonary arterial hypertension Diseases 0.000 claims description 15
- 201000011510 cancer Diseases 0.000 claims description 14
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 11
- 206010020772 Hypertension Diseases 0.000 claims description 11
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 11
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 10
- 206010063837 Reperfusion injury Diseases 0.000 claims description 10
- 208000020832 chronic kidney disease Diseases 0.000 claims description 10
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 claims description 10
- 231100000854 focal segmental glomerulosclerosis Toxicity 0.000 claims description 10
- 230000000302 ischemic effect Effects 0.000 claims description 10
- 208000028867 ischemia Diseases 0.000 claims description 9
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 claims description 8
- 206010007572 Cardiac hypertrophy Diseases 0.000 claims description 8
- 208000006029 Cardiomegaly Diseases 0.000 claims description 8
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 8
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 claims description 8
- 201000000028 adult respiratory distress syndrome Diseases 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 8
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 7
- 208000014674 injury Diseases 0.000 claims description 7
- 206010013801 Duchenne Muscular Dystrophy Diseases 0.000 claims description 6
- 206010029164 Nephrotic syndrome Diseases 0.000 claims description 6
- 208000006673 asthma Diseases 0.000 claims description 6
- 206010012601 diabetes mellitus Diseases 0.000 claims description 6
- 201000006938 muscular dystrophy Diseases 0.000 claims description 6
- 201000011461 pre-eclampsia Diseases 0.000 claims description 6
- 208000024827 Alzheimer disease Diseases 0.000 claims description 5
- 206010018374 Glomerulonephritis minimal lesion Diseases 0.000 claims description 5
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 5
- 208000004883 Lipoid Nephrosis Diseases 0.000 claims description 5
- 208000037803 restenosis Diseases 0.000 claims description 5
- 208000014644 Brain disease Diseases 0.000 claims description 4
- XAOSVOZSYZXLDC-UHFFFAOYSA-N CCOC1=CC(N)=NN=C1C1CCN(CC1)C(=O)C1=NC=C(OC2=CC=C(C=C2)C(F)(F)F)C(OC)=C1 Chemical compound CCOC1=CC(N)=NN=C1C1CCN(CC1)C(=O)C1=NC=C(OC2=CC=C(C=C2)C(F)(F)F)C(OC)=C1 XAOSVOZSYZXLDC-UHFFFAOYSA-N 0.000 claims description 4
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 4
- 206010014561 Emphysema Diseases 0.000 claims description 4
- 208000023105 Huntington disease Diseases 0.000 claims description 4
- KDZRRQSRUXMZAE-UHFFFAOYSA-N N1=C(C=C(C(=N1)C1CCN(CC1)C(=O)C1=CC(=C(OC2=CC=C(C=C2)C(F)(F)F)C=N1)OC)C)N Chemical compound N1=C(C=C(C(=N1)C1CCN(CC1)C(=O)C1=CC(=C(OC2=CC=C(C=C2)C(F)(F)F)C=N1)OC)C)N KDZRRQSRUXMZAE-UHFFFAOYSA-N 0.000 claims description 4
- SMULRYIMTOWDEM-UHFFFAOYSA-N N1=NC(=CC(=C1C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=CC=C1)C)N Chemical compound N1=NC(=CC(=C1C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=CC=C1)C)N SMULRYIMTOWDEM-UHFFFAOYSA-N 0.000 claims description 4
- NGVFJNKJEASNHO-UHFFFAOYSA-N NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)OC)OC Chemical compound NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)OC)OC NGVFJNKJEASNHO-UHFFFAOYSA-N 0.000 claims description 4
- WUUUUIADANIROZ-UHFFFAOYSA-N NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=CC=C1)OCC Chemical compound NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=CC=C1)OCC WUUUUIADANIROZ-UHFFFAOYSA-N 0.000 claims description 4
- 208000018737 Parkinson disease Diseases 0.000 claims description 4
- 208000001647 Renal Insufficiency Diseases 0.000 claims description 4
- 208000028208 end stage renal disease Diseases 0.000 claims description 4
- 201000000523 end stage renal failure Diseases 0.000 claims description 4
- 201000006370 kidney failure Diseases 0.000 claims description 4
- 201000006417 multiple sclerosis Diseases 0.000 claims description 4
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 claims description 4
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 claims description 4
- 201000008482 osteoarthritis Diseases 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 4
- 230000008733 trauma Effects 0.000 claims description 4
- FYEHZCHYPSYCHO-UHFFFAOYSA-N C1(N)=NN=C(C(=C1)C)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(F)C=C1 Chemical compound C1(N)=NN=C(C(=C1)C)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(F)C=C1 FYEHZCHYPSYCHO-UHFFFAOYSA-N 0.000 claims description 3
- NBFDGEIKHLFJLY-UHFFFAOYSA-N NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)C(F)(F)F)OC Chemical compound NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)C(F)(F)F)OC NBFDGEIKHLFJLY-UHFFFAOYSA-N 0.000 claims description 3
- DTAHYUKCFJTZCI-UHFFFAOYSA-N NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)F)OC Chemical compound NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)F)OC DTAHYUKCFJTZCI-UHFFFAOYSA-N 0.000 claims description 3
- 206010063897 Renal ischaemia Diseases 0.000 claims description 3
- 230000035935 pregnancy Effects 0.000 claims description 3
- 229940079593 drug Drugs 0.000 claims description 2
- 238000000034 method Methods 0.000 abstract description 271
- 239000000543 intermediate Substances 0.000 abstract description 71
- 208000037765 diseases and disorders Diseases 0.000 abstract description 5
- 230000008569 process Effects 0.000 abstract description 4
- 239000011541 reaction mixture Substances 0.000 description 158
- 238000004128 high performance liquid chromatography Methods 0.000 description 130
- 230000002829 reductive effect Effects 0.000 description 102
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 95
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 92
- 230000015572 biosynthetic process Effects 0.000 description 92
- 238000003786 synthesis reaction Methods 0.000 description 91
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 89
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 85
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 50
- 239000000243 solution Substances 0.000 description 50
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 49
- 239000000203 mixture Substances 0.000 description 46
- 235000019439 ethyl acetate Nutrition 0.000 description 44
- 210000004027 cell Anatomy 0.000 description 43
- 238000010898 silica gel chromatography Methods 0.000 description 41
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 39
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 39
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 38
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 38
- 239000000047 product Substances 0.000 description 37
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 35
- 230000006870 function Effects 0.000 description 33
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 32
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 32
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 31
- IXCSERBJSXMMFS-UHFFFAOYSA-N hcl hcl Chemical compound Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 31
- -1 organic acid salts Chemical class 0.000 description 31
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 30
- 239000012267 brine Substances 0.000 description 30
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 30
- 239000012074 organic phase Substances 0.000 description 29
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 28
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 28
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 28
- 238000006243 chemical reaction Methods 0.000 description 25
- 238000003756 stirring Methods 0.000 description 24
- NIILYUCSOILJQE-UHFFFAOYSA-N methyl 5-hydroxy-4-methoxypyridine-2-carboxylate Chemical compound COC(=O)C1=CC(OC)=C(O)C=N1 NIILYUCSOILJQE-UHFFFAOYSA-N 0.000 description 23
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 22
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 22
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Chemical class OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 22
- HFWFHCQDNWAYBB-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC1=C(C=CC=C1)F Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC1=C(C=CC=C1)F HFWFHCQDNWAYBB-UHFFFAOYSA-N 0.000 description 21
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 21
- 239000007832 Na2SO4 Substances 0.000 description 21
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 21
- 230000014509 gene expression Effects 0.000 description 21
- 229910052938 sodium sulfate Inorganic materials 0.000 description 21
- 235000011152 sodium sulphate Nutrition 0.000 description 21
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 20
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 19
- 230000001404 mediated effect Effects 0.000 description 19
- 238000000746 purification Methods 0.000 description 19
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 18
- BJIMENNVCUGZFH-UHFFFAOYSA-N FC1=C(COC=2C(=CC(=NC=2)C(=O)O)OC)C=CC=C1 Chemical compound FC1=C(COC=2C(=CC(=NC=2)C(=O)O)OC)C=CC=C1 BJIMENNVCUGZFH-UHFFFAOYSA-N 0.000 description 18
- 239000011575 calcium Substances 0.000 description 18
- 239000013067 intermediate product Substances 0.000 description 18
- 229910052791 calcium Inorganic materials 0.000 description 17
- 238000004440 column chromatography Methods 0.000 description 17
- XRAAKYZYTBLTJK-UHFFFAOYSA-N 5-fluoro-4-methoxypyridine-2-carbonitrile Chemical compound COc1cc(ncc1F)C#N XRAAKYZYTBLTJK-UHFFFAOYSA-N 0.000 description 15
- 239000007821 HATU Substances 0.000 description 15
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 15
- 239000012298 atmosphere Substances 0.000 description 15
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- NCIVGAFVNZZUTJ-UHFFFAOYSA-N Cl.Cl.COc1cc(N)ncc1N1CCNCC1 Chemical compound Cl.Cl.COc1cc(N)ncc1N1CCNCC1 NCIVGAFVNZZUTJ-UHFFFAOYSA-N 0.000 description 14
- 239000002253 acid Substances 0.000 description 14
- 230000002378 acidificating effect Effects 0.000 description 14
- 229910052786 argon Inorganic materials 0.000 description 14
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 14
- 108091006146 Channels Proteins 0.000 description 13
- LCPQMGNKYCEZFZ-UHFFFAOYSA-N FC1=CC=C(OC=2C(=CC(=NC=2)C(=O)O)OC)C=C1 Chemical compound FC1=CC=C(OC=2C(=CC(=NC=2)C(=O)O)OC)C=C1 LCPQMGNKYCEZFZ-UHFFFAOYSA-N 0.000 description 13
- 239000012071 phase Substances 0.000 description 13
- XXMFJKNOJSDQBM-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;hydrate Chemical compound [OH3+].[O-]C(=O)C(F)(F)F XXMFJKNOJSDQBM-UHFFFAOYSA-N 0.000 description 12
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 12
- 238000004587 chromatography analysis Methods 0.000 description 12
- 239000002552 dosage form Substances 0.000 description 12
- 230000005764 inhibitory process Effects 0.000 description 12
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical class COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 11
- GMTIVFXNTFDTKA-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=CC=C1)C#N Chemical compound COC1=CC(=NC=C1OC1=CC=CC=C1)C#N GMTIVFXNTFDTKA-UHFFFAOYSA-N 0.000 description 11
- 235000019253 formic acid Nutrition 0.000 description 11
- 239000001257 hydrogen Substances 0.000 description 11
- 229910052739 hydrogen Inorganic materials 0.000 description 11
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 11
- 235000019341 magnesium sulphate Nutrition 0.000 description 11
- 239000002244 precipitate Substances 0.000 description 11
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- 239000012141 concentrate Substances 0.000 description 10
- 235000008504 concentrate Nutrition 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 150000002500 ions Chemical class 0.000 description 10
- 229910000027 potassium carbonate Inorganic materials 0.000 description 10
- 238000004007 reversed phase HPLC Methods 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 9
- SSCZVBVSLVFOFD-UHFFFAOYSA-N BrC=1C(=CC(=NC=1)N1C(=CC=C1C)C)OC Chemical compound BrC=1C(=CC(=NC=1)N1C(=CC=C1C)C)OC SSCZVBVSLVFOFD-UHFFFAOYSA-N 0.000 description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 9
- 108090000862 Ion Channels Proteins 0.000 description 9
- 102000004310 Ion Channels Human genes 0.000 description 9
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- 230000000747 cardiac effect Effects 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- 208000017169 kidney disease Diseases 0.000 description 9
- 239000012299 nitrogen atmosphere Substances 0.000 description 9
- 239000000377 silicon dioxide Substances 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 239000012528 membrane Substances 0.000 description 8
- 108020004999 messenger RNA Proteins 0.000 description 8
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 8
- FQFILJKFZCVHNH-UHFFFAOYSA-N tert-butyl n-[3-[(5-bromo-2-chloropyrimidin-4-yl)amino]propyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCNC1=NC(Cl)=NC=C1Br FQFILJKFZCVHNH-UHFFFAOYSA-N 0.000 description 8
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- 229910001424 calcium ion Inorganic materials 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 229910052708 sodium Inorganic materials 0.000 description 7
- TWXZYWWOMADXFJ-UHFFFAOYSA-N 5-bromo-4-methoxypyridin-2-amine Chemical compound COC1=CC(N)=NC=C1Br TWXZYWWOMADXFJ-UHFFFAOYSA-N 0.000 description 6
- PZOYLWXXFXENPV-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=C(C=C1)C(F)(F)F)C#N Chemical compound COC1=CC(=NC=C1OC1=CC=C(C=C1)C(F)(F)F)C#N PZOYLWXXFXENPV-UHFFFAOYSA-N 0.000 description 6
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 6
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 6
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 230000003834 intracellular effect Effects 0.000 description 6
- 238000012423 maintenance Methods 0.000 description 6
- 230000035772 mutation Effects 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 235000015320 potassium carbonate Nutrition 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 239000012453 solvate Substances 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 6
- BUVVJIGULSJYLF-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2h-pyridine-1-carboxylic acid Chemical compound O1C(C)(C)C(C)(C)OB1C1=CCN(C(O)=O)CC1 BUVVJIGULSJYLF-UHFFFAOYSA-N 0.000 description 5
- QBEYQHJZHIMZAZ-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=CC=C1)C(=O)O Chemical compound COC1=CC(=NC=C1OC1=CC=CC=C1)C(=O)O QBEYQHJZHIMZAZ-UHFFFAOYSA-N 0.000 description 5
- MLZMLRZRDCFLSO-UHFFFAOYSA-N Cl.Cl.COc1cc(N)ncc1C1CCNCC1 Chemical compound Cl.Cl.COc1cc(N)ncc1C1CCNCC1 MLZMLRZRDCFLSO-UHFFFAOYSA-N 0.000 description 5
- VELQYKUMZBYRCA-UHFFFAOYSA-N Cl.Cl.Cc1cc(N)nnc1C1CCNCC1 Chemical compound Cl.Cl.Cc1cc(N)nnc1C1CCNCC1 VELQYKUMZBYRCA-UHFFFAOYSA-N 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- 208000002193 Pain Diseases 0.000 description 5
- 229910002666 PdCl2 Inorganic materials 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 5
- 239000002671 adjuvant Substances 0.000 description 5
- 239000000556 agonist Substances 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 5
- 150000001768 cations Chemical class 0.000 description 5
- 230000002950 deficient Effects 0.000 description 5
- 210000002889 endothelial cell Anatomy 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 230000004907 flux Effects 0.000 description 5
- 210000002216 heart Anatomy 0.000 description 5
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 5
- 208000019423 liver disease Diseases 0.000 description 5
- 210000004072 lung Anatomy 0.000 description 5
- 239000012452 mother liquor Substances 0.000 description 5
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 5
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 5
- LETVJWLLIMJADE-UHFFFAOYSA-N pyridazin-3-amine Chemical compound NC1=CC=CN=N1 LETVJWLLIMJADE-UHFFFAOYSA-N 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- MXVLKZHAGVQJCH-UHFFFAOYSA-N tert-butyl 4-(6-aminopyridazin-3-yl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C1=CC=C(N)N=N1 MXVLKZHAGVQJCH-UHFFFAOYSA-N 0.000 description 5
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 5
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 5
- OJVAMHKKJGICOG-UHFFFAOYSA-N 2,5-hexanedione Chemical compound CC(=O)CCC(C)=O OJVAMHKKJGICOG-UHFFFAOYSA-N 0.000 description 4
- BAYGVMXZJBFEMB-UHFFFAOYSA-N 4-(trifluoromethyl)phenol Chemical compound OC1=CC=C(C(F)(F)F)C=C1 BAYGVMXZJBFEMB-UHFFFAOYSA-N 0.000 description 4
- MYSAEEDQBBBWAC-UHFFFAOYSA-N CC1=C(N=NC(N)=C1)C1=CCN(CC1)C(=O)OC(C)(C)C Chemical compound CC1=C(N=NC(N)=C1)C1=CCN(CC1)C(=O)OC(C)(C)C MYSAEEDQBBBWAC-UHFFFAOYSA-N 0.000 description 4
- FJLDYNGHCDYYLH-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC(C)C Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC(C)C FJLDYNGHCDYYLH-UHFFFAOYSA-N 0.000 description 4
- AVLQYHCLVCZBNH-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC1CC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC1CC1 AVLQYHCLVCZBNH-UHFFFAOYSA-N 0.000 description 4
- KBBQMQPMAVSOTF-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=C(C=C1)C(F)(F)F)C(=O)O Chemical compound COC1=CC(=NC=C1OC1=CC=C(C=C1)C(F)(F)F)C(=O)O KBBQMQPMAVSOTF-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- 108091005462 Cation channels Proteins 0.000 description 4
- IELAWEREGRKINC-UHFFFAOYSA-N Cl.Cl.COc1cc(N)nnc1C1CCNCC1 Chemical compound Cl.Cl.COc1cc(N)nnc1C1CCNCC1 IELAWEREGRKINC-UHFFFAOYSA-N 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- ZDEXYYDJRRYDED-UHFFFAOYSA-N FC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 Chemical compound FC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 ZDEXYYDJRRYDED-UHFFFAOYSA-N 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- IMKRBQQYKXHUMJ-UHFFFAOYSA-N NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)OC(C)(C)C)C Chemical compound NC1=CC(=C(N=N1)C1CCN(CC1)C(=O)OC(C)(C)C)C IMKRBQQYKXHUMJ-UHFFFAOYSA-N 0.000 description 4
- 101100426589 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) trp-3 gene Proteins 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 102000003629 TRPC3 Human genes 0.000 description 4
- 101150037542 Trpc3 gene Proteins 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 230000009460 calcium influx Effects 0.000 description 4
- 210000004413 cardiac myocyte Anatomy 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 230000008482 dysregulation Effects 0.000 description 4
- 230000003176 fibrotic effect Effects 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 4
- 235000011181 potassium carbonates Nutrition 0.000 description 4
- 210000001147 pulmonary artery Anatomy 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- VVDCRJGWILREQH-UHFFFAOYSA-N tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC(B2OC(C)(C)C(C)(C)O2)=C1 VVDCRJGWILREQH-UHFFFAOYSA-N 0.000 description 4
- MZQVKWDRBMUGCF-UHFFFAOYSA-N tert-butyl 4-(6-aminopyridazin-3-yl)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC(C=2N=NC(N)=CC=2)=C1 MZQVKWDRBMUGCF-UHFFFAOYSA-N 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- 230000001052 transient effect Effects 0.000 description 4
- SESOMTJUSDACSD-UHFFFAOYSA-N 2-chloro-5-fluoro-1h-pyridin-4-one Chemical compound FC1=CNC(Cl)=CC1=O SESOMTJUSDACSD-UHFFFAOYSA-N 0.000 description 3
- OYQMZELAVIVTOM-UHFFFAOYSA-N 2-chloro-5-fluoro-4-methoxypyridine Chemical compound COC1=CC(Cl)=NC=C1F OYQMZELAVIVTOM-UHFFFAOYSA-N 0.000 description 3
- RHMPLDJJXGPMEX-UHFFFAOYSA-N 4-fluorophenol Chemical compound OC1=CC=C(F)C=C1 RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 3
- ANGSKLSOBDQMPI-UHFFFAOYSA-N 5-bromo-2-(2,5-dimethylpyrrol-1-yl)-4-methylpyridine Chemical compound CC1=CC=C(C)N1C1=CC(C)=C(Br)C=N1 ANGSKLSOBDQMPI-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- MNJQDVVIWARPAX-UHFFFAOYSA-N C1(=CCN(CC1)C(=O)OC(C)(C)C)C1=C(OC)C=C(N)N=C1 Chemical compound C1(=CCN(CC1)C(=O)OC(C)(C)C)C1=C(OC)C=C(N)N=C1 MNJQDVVIWARPAX-UHFFFAOYSA-N 0.000 description 3
- FWAPTNTUKUVTDP-UHFFFAOYSA-N C1(CC1)COC=1C(=CC(=NC=1)C(=O)O)OC Chemical compound C1(CC1)COC=1C(=CC(=NC=1)C(=O)O)OC FWAPTNTUKUVTDP-UHFFFAOYSA-N 0.000 description 3
- LGFNMPQTUYRZPM-UHFFFAOYSA-N COC1=CC(=NC=C1OCC(C)C)C(=O)O Chemical compound COC1=CC(=NC=C1OCC(C)C)C(=O)O LGFNMPQTUYRZPM-UHFFFAOYSA-N 0.000 description 3
- 208000024172 Cardiovascular disease Diseases 0.000 description 3
- VTPGFFJOHYCBTN-UHFFFAOYSA-N Cl.Cl.CCOc1cc(N)ncc1C1CCNCC1 Chemical compound Cl.Cl.CCOc1cc(N)ncc1C1CCNCC1 VTPGFFJOHYCBTN-UHFFFAOYSA-N 0.000 description 3
- YWTNJSAEOGZMJF-UHFFFAOYSA-N Cl.Cl.Nc1ccc(nn1)C1CCNCC1 Chemical compound Cl.Cl.Nc1ccc(nn1)C1CCNCC1 YWTNJSAEOGZMJF-UHFFFAOYSA-N 0.000 description 3
- 206010070538 Gestational hypertension Diseases 0.000 description 3
- 201000005624 HELLP Syndrome Diseases 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 208000005347 Pregnancy-Induced Hypertension Diseases 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Chemical class OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 239000012317 TBTU Substances 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 208000027418 Wounds and injury Diseases 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 3
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 230000004094 calcium homeostasis Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000013375 chromatographic separation Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 239000013058 crude material Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 230000035876 healing Effects 0.000 description 3
- 230000013632 homeostatic process Effects 0.000 description 3
- 208000027866 inflammatory disease Diseases 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 230000010291 membrane polarization Effects 0.000 description 3
- 208000030159 metabolic disease Diseases 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 3
- 235000011007 phosphoric acid Nutrition 0.000 description 3
- 210000000557 podocyte Anatomy 0.000 description 3
- 208000036335 preeclampsia/eclampsia 1 Diseases 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 201000001474 proteinuria Diseases 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 230000000241 respiratory effect Effects 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 3
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 3
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- PWTCCMJTPHCGMS-YRBAHSOBSA-N 1-Oleoyl-2-acetyl-sn-glycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](CO)OC(C)=O PWTCCMJTPHCGMS-YRBAHSOBSA-N 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical class OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- HQNIMNQVKVPZES-UHFFFAOYSA-N 2-amino-1h-pyridin-4-one Chemical compound NC1=CC(O)=CC=N1 HQNIMNQVKVPZES-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- WRMNZCZEMHIOCP-UHFFFAOYSA-N 2-phenylethanol Chemical compound OCCC1=CC=CC=C1 WRMNZCZEMHIOCP-UHFFFAOYSA-N 0.000 description 2
- DTXVKPOKPFWSFF-UHFFFAOYSA-N 3(S)-hydroxy-13-cis-eicosenoyl-CoA Chemical compound NC1=CC=C(Cl)N=N1 DTXVKPOKPFWSFF-UHFFFAOYSA-N 0.000 description 2
- AXWRAIIIBRLXBP-UHFFFAOYSA-N 3-[1-(5,6,7,8-tetrahydro-4h-cyclohepta[b]thiophene-2-carbonyl)piperidin-4-yl]-1h-benzimidazol-2-one Chemical compound C1CCCCC(S2)=C1C=C2C(=O)N1CCC(N2C(NC3=CC=CC=C32)=O)CC1 AXWRAIIIBRLXBP-UHFFFAOYSA-N 0.000 description 2
- QMJYJTKXWDIUKV-UHFFFAOYSA-N 4-(6-amino-4-methylpyridin-3-yl)piperidine-1-carboxylic acid Chemical compound CC1=CC(N)=NC=C1C1CCN(C(O)=O)CC1 QMJYJTKXWDIUKV-UHFFFAOYSA-N 0.000 description 2
- IXVJBUVOOUSBJO-UHFFFAOYSA-N 4-cyclopropyloxyphenol Chemical compound C1=CC(O)=CC=C1OC1CC1 IXVJBUVOOUSBJO-UHFFFAOYSA-N 0.000 description 2
- JXPYTHDXGAGHBU-UHFFFAOYSA-N 5-[4-(trifluoromethyl)phenoxy]pyridine-2-carboxylic acid Chemical compound C1=NC(C(=O)O)=CC=C1OC1=CC=C(C(F)(F)F)C=C1 JXPYTHDXGAGHBU-UHFFFAOYSA-N 0.000 description 2
- ATXXLNCPVSUCNK-UHFFFAOYSA-N 5-bromo-2-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Br)C=N1 ATXXLNCPVSUCNK-UHFFFAOYSA-N 0.000 description 2
- XYKSKZHIGZFCEF-UHFFFAOYSA-N 5-hydroxy-4-methoxypyridine-2-carboxylic acid Chemical compound COc1cc(ncc1O)C(O)=O XYKSKZHIGZFCEF-UHFFFAOYSA-N 0.000 description 2
- CDGBAWNUZVFTOJ-UHFFFAOYSA-N 5-piperazin-1-ylpyridin-2-amine dihydrochloride Chemical compound Cl.Cl.Nc1ccc(cn1)N1CCNCC1 CDGBAWNUZVFTOJ-UHFFFAOYSA-N 0.000 description 2
- UWDLNRUFHRYMSE-UHFFFAOYSA-N 6-chloro-5-methylpyridazin-3-amine Chemical compound CC1=CC(N)=NN=C1Cl UWDLNRUFHRYMSE-UHFFFAOYSA-N 0.000 description 2
- 206010001580 Albuminuria Diseases 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 2
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- XMNZMNFBLSPKRM-UHFFFAOYSA-N C(=O)(N1CCC(CC1)C1=CC(OC)=C(N)N=N1)OC(C)(C)C Chemical compound C(=O)(N1CCC(CC1)C1=CC(OC)=C(N)N=N1)OC(C)(C)C XMNZMNFBLSPKRM-UHFFFAOYSA-N 0.000 description 2
- DRZKXVLQBPWCHG-MRXNPFEDSA-N C(C)(C)(C)OC(=O)N1[C@H](CN(CC1)C=1C=NC(=CC=1)[N+](=O)[O-])C(O[SiH2]C(C)(C)C)(C)C Chemical compound C(C)(C)(C)OC(=O)N1[C@H](CN(CC1)C=1C=NC(=CC=1)[N+](=O)[O-])C(O[SiH2]C(C)(C)C)(C)C DRZKXVLQBPWCHG-MRXNPFEDSA-N 0.000 description 2
- GAYJPTAWMBOQPN-UHFFFAOYSA-N C(C)(C)OC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 Chemical compound C(C)(C)OC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 GAYJPTAWMBOQPN-UHFFFAOYSA-N 0.000 description 2
- JLKQZHOMDUECEJ-UHFFFAOYSA-N C(C)OC1=CC(=NC=C1F)C#N Chemical compound C(C)OC1=CC(=NC=C1F)C#N JLKQZHOMDUECEJ-UHFFFAOYSA-N 0.000 description 2
- JZOXAHUIUCLUKO-UHFFFAOYSA-N C(C)OC1=CC(=NC=C1OC1=CC=C(C=C1)F)C#N Chemical compound C(C)OC1=CC(=NC=C1OC1=CC=C(C=C1)F)C#N JZOXAHUIUCLUKO-UHFFFAOYSA-N 0.000 description 2
- CYMBHJUPLKJIDP-UHFFFAOYSA-N C(C)OC1=CC(=NC=C1OC1=CC=CC=C1)C#N Chemical compound C(C)OC1=CC(=NC=C1OC1=CC=CC=C1)C#N CYMBHJUPLKJIDP-UHFFFAOYSA-N 0.000 description 2
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 2
- IJJBPDGMTZCNHA-UHFFFAOYSA-N C1(=CC(=CC=N1)OCCC)NC(=O)OC(C)(C)C Chemical compound C1(=CC(=CC=N1)OCCC)NC(=O)OC(C)(C)C IJJBPDGMTZCNHA-UHFFFAOYSA-N 0.000 description 2
- GNGREHVINDLYHJ-UHFFFAOYSA-N C1(=CN=C(C=C1OC)N)C1CCN(CC1)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1OC)N)C1CCN(CC1)C(=O)OC(C)(C)C GNGREHVINDLYHJ-UHFFFAOYSA-N 0.000 description 2
- VBHJBFQCXQTIJG-UHFFFAOYSA-N C1(CC1)OC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 Chemical compound C1(CC1)OC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 VBHJBFQCXQTIJG-UHFFFAOYSA-N 0.000 description 2
- QIQCXSCNPVIXAO-UHFFFAOYSA-N C1C(OC2=C(C=NC(N)=C2)C2CCN(CC2)C(=O)OC(C)(C)C)C1 Chemical compound C1C(OC2=C(C=NC(N)=C2)C2CCN(CC2)C(=O)OC(C)(C)C)C1 QIQCXSCNPVIXAO-UHFFFAOYSA-N 0.000 description 2
- YHOXFRPLXUOBRV-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OC(C)C Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OC(C)C YHOXFRPLXUOBRV-UHFFFAOYSA-N 0.000 description 2
- DBXDKGPPSVCZSV-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OC1CCCC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OC1CCCC1 DBXDKGPPSVCZSV-UHFFFAOYSA-N 0.000 description 2
- YSDYDQFUONKCMX-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OC1CCCCC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OC1CCCCC1 YSDYDQFUONKCMX-UHFFFAOYSA-N 0.000 description 2
- QDAGRTZRUZFUFL-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC QDAGRTZRUZFUFL-UHFFFAOYSA-N 0.000 description 2
- APJPJDAKELBSBA-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC(C)(C)C Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC(C)(C)C APJPJDAKELBSBA-UHFFFAOYSA-N 0.000 description 2
- JBYQCKXXXSYYNI-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC1(CC1)CF Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC1(CC1)CF JBYQCKXXXSYYNI-UHFFFAOYSA-N 0.000 description 2
- LBYKHZCNUNICMC-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC1=CC=C(C=C1)F Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC1=CC=C(C=C1)F LBYKHZCNUNICMC-UHFFFAOYSA-N 0.000 description 2
- JWHYNDMRGBNHSG-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC1CC(C1)(F)F Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC1CC(C1)(F)F JWHYNDMRGBNHSG-UHFFFAOYSA-N 0.000 description 2
- MLQRHZJHJZZZDY-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCC1CCC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCC1CCC1 MLQRHZJHJZZZDY-UHFFFAOYSA-N 0.000 description 2
- ZWNSXDNIWFULPA-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCCC Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCCC ZWNSXDNIWFULPA-UHFFFAOYSA-N 0.000 description 2
- WCJFISUTOWJJGP-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCCC1=CC=CC=C1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCCC1=CC=CC=C1 WCJFISUTOWJJGP-UHFFFAOYSA-N 0.000 description 2
- KDAQUQASOCUDDD-QMMMGPOBSA-N COC(=O)C1=NC=C(C(=C1)OC)O[C@@H](C)C1CC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)O[C@@H](C)C1CC1 KDAQUQASOCUDDD-QMMMGPOBSA-N 0.000 description 2
- KOPFOLOXMRLYRA-UHFFFAOYSA-N COC1=CC(=NC=C1N1CCNCC1)N Chemical compound COC1=CC(=NC=C1N1CCNCC1)N KOPFOLOXMRLYRA-UHFFFAOYSA-N 0.000 description 2
- SIMCHENCACNBMA-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=C(C=C1)OC(F)(F)F)C#N Chemical compound COC1=CC(=NC=C1OC1=CC=C(C=C1)OC(F)(F)F)C#N SIMCHENCACNBMA-UHFFFAOYSA-N 0.000 description 2
- SPQDXUCVYNOFFP-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=C(C=C1)OC)C#N Chemical compound COC1=CC(=NC=C1OC1=CC=C(C=C1)OC)C#N SPQDXUCVYNOFFP-UHFFFAOYSA-N 0.000 description 2
- LLUXWYSJIZKAJG-UHFFFAOYSA-N COC1=CC(=NC=C1OC1=CC=C(C=C1)OC)C(=O)O Chemical compound COC1=CC(=NC=C1OC1=CC=C(C=C1)OC)C(=O)O LLUXWYSJIZKAJG-UHFFFAOYSA-N 0.000 description 2
- FDLBJJIWFPXHDJ-UHFFFAOYSA-N COC1=CC(=NC=C1OCC(C(F)(F)F)C)C(=O)O Chemical compound COC1=CC(=NC=C1OCC(C(F)(F)F)C)C(=O)O FDLBJJIWFPXHDJ-UHFFFAOYSA-N 0.000 description 2
- WHWFJSNAJGQGGY-UHFFFAOYSA-N COC1=CC(=NC=C1OCC(C(F)(F)F)C)C(=O)OC Chemical compound COC1=CC(=NC=C1OCC(C(F)(F)F)C)C(=O)OC WHWFJSNAJGQGGY-UHFFFAOYSA-N 0.000 description 2
- GHOSNRCGJFBJIB-UHFFFAOYSA-N Candesartan cilexetil Chemical compound C=12N(CC=3C=CC(=CC=3)C=3C(=CC=CC=3)C3=NNN=N3)C(OCC)=NC2=CC=CC=1C(=O)OC(C)OC(=O)OC1CCCCC1 GHOSNRCGJFBJIB-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- TYCZBYSUMDSYBZ-UHFFFAOYSA-N Cl.Cl.CCCOc1cc(N)ncc1C1CCNCC1 Chemical compound Cl.Cl.CCCOc1cc(N)ncc1C1CCNCC1 TYCZBYSUMDSYBZ-UHFFFAOYSA-N 0.000 description 2
- FMUZJDUEGITZRA-DDWIOCJRSA-N Cl.OC[C@H]1CN(CCN1)c1ccc(nc1)[N+]([O-])=O Chemical compound Cl.OC[C@H]1CN(CCN1)c1ccc(nc1)[N+]([O-])=O FMUZJDUEGITZRA-DDWIOCJRSA-N 0.000 description 2
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 2
- 108010011459 Exenatide Proteins 0.000 description 2
- GVXWWTLNVVZLSC-UHFFFAOYSA-N FC(OC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1)F Chemical compound FC(OC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1)F GVXWWTLNVVZLSC-UHFFFAOYSA-N 0.000 description 2
- OUVXYXNWSVIOSJ-UHFFFAOYSA-N Fluo-4 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(F)C(=O)C=C3OC3=CC(O)=C(F)C=C32)N(CC(O)=O)CC(O)=O)=C1 OUVXYXNWSVIOSJ-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 206010020880 Hypertrophy Diseases 0.000 description 2
- 206010021143 Hypoxia Diseases 0.000 description 2
- 241000713666 Lentivirus Species 0.000 description 2
- YSDQQAXHVYUZIW-QCIJIYAXSA-N Liraglutide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC)C(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 YSDQQAXHVYUZIW-QCIJIYAXSA-N 0.000 description 2
- 108010019598 Liraglutide Proteins 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- BOUSXXWTDFIOTM-UHFFFAOYSA-N NC1=CC(=C(C=N1)C1CCN(CC1)C(=O)C1=CC(=C(C=N1)O)OC)OC Chemical compound NC1=CC(=C(C=N1)C1CCN(CC1)C(=O)C1=CC(=C(C=N1)O)OC)OC BOUSXXWTDFIOTM-UHFFFAOYSA-N 0.000 description 2
- DJHIRNDSJKKHMZ-UHFFFAOYSA-N Nc1ccc(nn1)C1CCNCC1 Chemical compound Nc1ccc(nn1)C1CCNCC1 DJHIRNDSJKKHMZ-UHFFFAOYSA-N 0.000 description 2
- SCWNAJXLGSGCJA-UHFFFAOYSA-N OC(=O)C(F)(F)F.COc1cc(ncc1OCC(C)C)C(=O)N1CCC(CC1)c1cnc(N)cc1OC Chemical compound OC(=O)C(F)(F)F.COc1cc(ncc1OCC(C)C)C(=O)N1CCC(CC1)c1cnc(N)cc1OC SCWNAJXLGSGCJA-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000004098 Tetracycline Substances 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 206010072810 Vascular wall hypertrophy Diseases 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000001099 ammonium carbonate Substances 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 229940125364 angiotensin receptor blocker Drugs 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 229960003065 bosentan Drugs 0.000 description 2
- SXTRWVVIEPWAKM-UHFFFAOYSA-N bosentan hydrate Chemical compound O.COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 SXTRWVVIEPWAKM-UHFFFAOYSA-N 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 229960000932 candesartan Drugs 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 230000010001 cellular homeostasis Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- GUDMZGLFZNLYEY-UHFFFAOYSA-N cyclopropylmethanol Chemical compound OCC1CC1 GUDMZGLFZNLYEY-UHFFFAOYSA-N 0.000 description 2
- 230000003436 cytoskeletal effect Effects 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 2
- 230000006806 disease prevention Effects 0.000 description 2
- KQTXIZHBFFWWFW-UHFFFAOYSA-L disilver;carbonate Chemical compound [Ag]OC(=O)O[Ag] KQTXIZHBFFWWFW-UHFFFAOYSA-L 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 229960001519 exenatide Drugs 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 150000004675 formic acid derivatives Chemical class 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 230000009368 gene silencing by RNA Effects 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 229960004580 glibenclamide Drugs 0.000 description 2
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 230000007954 hypoxia Effects 0.000 description 2
- 239000000367 immunologic factor Substances 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 238000005462 in vivo assay Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 229940039009 isoproterenol Drugs 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 229960002701 liraglutide Drugs 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- OVXMKONILMIYDX-UHFFFAOYSA-N methyl 4-methoxy-5-phenylmethoxypyridine-2-carboxylate Chemical compound C1=NC(C(=O)OC)=CC(OC)=C1OCC1=CC=CC=C1 OVXMKONILMIYDX-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- 210000000651 myofibroblast Anatomy 0.000 description 2
- 230000001114 myogenic effect Effects 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000004770 neurodegeneration Effects 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 210000002826 placenta Anatomy 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000008521 reorganization Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical class OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000007423 screening assay Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 230000035882 stress Effects 0.000 description 2
- 238000004808 supercritical fluid chromatography Methods 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- SHHHRQFHCPINIB-UHFFFAOYSA-N tert-butyl 3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC=CC1 SHHHRQFHCPINIB-UHFFFAOYSA-N 0.000 description 2
- CHZPLAKGFHZPMO-UHFFFAOYSA-N tert-butyl 4-(6-amino-4-methylpyridin-3-yl)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound CC1=CC(N)=NC=C1C1=CCN(C(=O)OC(C)(C)C)CC1 CHZPLAKGFHZPMO-UHFFFAOYSA-N 0.000 description 2
- HRWBTZONXATYQE-UHFFFAOYSA-N tert-butyl n-(6-chloro-5-methoxypyridazin-3-yl)carbamate Chemical compound COC1=CC(NC(=O)OC(C)(C)C)=NN=C1Cl HRWBTZONXATYQE-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- 235000019364 tetracycline Nutrition 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 150000003522 tetracyclines Chemical class 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Chemical class OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 102000038650 voltage-gated calcium channel activity Human genes 0.000 description 2
- 108091023044 voltage-gated calcium channel activity Proteins 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 1
- PIZQWRXTMGASCZ-UHFFFAOYSA-N (1-methylcyclopropyl)methanol Chemical compound OCC1(C)CC1 PIZQWRXTMGASCZ-UHFFFAOYSA-N 0.000 description 1
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- XFQNWPYGEGCIMF-HCUGAJCMSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].[Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 XFQNWPYGEGCIMF-HCUGAJCMSA-N 0.000 description 1
- DKKVKJZXOBFLRY-SCSAIBSYSA-N (1r)-1-cyclopropylethanol Chemical compound C[C@@H](O)C1CC1 DKKVKJZXOBFLRY-SCSAIBSYSA-N 0.000 description 1
- DKKVKJZXOBFLRY-BYPYZUCNSA-N (1s)-1-cyclopropylethanol Chemical compound C[C@H](O)C1CC1 DKKVKJZXOBFLRY-BYPYZUCNSA-N 0.000 description 1
- QEHXDOJPVIHUDO-UHFFFAOYSA-N (2-fluorophenyl)methanol Chemical compound OCC1=CC=CC=C1F QEHXDOJPVIHUDO-UHFFFAOYSA-N 0.000 description 1
- JILXQHYGCQCNBW-UHFFFAOYSA-N (2-phenylpiperazin-1-yl)-pyridin-2-ylmethanone Chemical class C=1C=CC=NC=1C(=O)N1CCNCC1C1=CC=CC=C1 JILXQHYGCQCNBW-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical class O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- GABDOIDBGZVFOB-SECBINFHSA-N (2R)-4-(6-amino-4-methylpyridin-3-yl)-2-(hydroxymethyl)piperazine-1-carboxylic acid Chemical compound NC1=CC(=C(C=N1)N1C[C@@H](N(CC1)C(=O)O)CO)C GABDOIDBGZVFOB-SECBINFHSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- MDZKTVVUZBIKGI-UHFFFAOYSA-N (3,3-difluorocyclobutyl)methanol Chemical compound OCC1CC(F)(F)C1 MDZKTVVUZBIKGI-UHFFFAOYSA-N 0.000 description 1
- GEZMEIHVFSWOCA-UHFFFAOYSA-N (4-fluorophenyl)methanol Chemical compound OCC1=CC=C(F)C=C1 GEZMEIHVFSWOCA-UHFFFAOYSA-N 0.000 description 1
- KZEDPVFJLQLDIZ-UHFFFAOYSA-N (5-diphenylphosphanyl-9,9-dimethylxanthen-4-yl)-diphenylphosphane Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1.C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZEDPVFJLQLDIZ-UHFFFAOYSA-N 0.000 description 1
- NDAUXUAQIAJITI-LBPRGKRZSA-N (R)-salbutamol Chemical compound CC(C)(C)NC[C@H](O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-LBPRGKRZSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical class OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- RYCNUMLMNKHWPZ-SNVBAGLBSA-N 1-acetyl-sn-glycero-3-phosphocholine Chemical compound CC(=O)OC[C@@H](O)COP([O-])(=O)OCC[N+](C)(C)C RYCNUMLMNKHWPZ-SNVBAGLBSA-N 0.000 description 1
- CYNYIHKIEHGYOZ-UHFFFAOYSA-N 1-bromopropane Chemical compound CCCBr CYNYIHKIEHGYOZ-UHFFFAOYSA-N 0.000 description 1
- RHDYQUZYHZWTCI-UHFFFAOYSA-N 1-methoxy-4-phenylbenzene Chemical compound C1=CC(OC)=CC=C1C1=CC=CC=C1 RHDYQUZYHZWTCI-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- OGSNAIXWIZRZLF-UHFFFAOYSA-N 2-(4-cyclopropyloxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C(C=C1)=CC=C1OC1CC1 OGSNAIXWIZRZLF-UHFFFAOYSA-N 0.000 description 1
- MXWFDXNJVQMDGA-UHFFFAOYSA-N 2-chloro-4-ethoxy-5-fluoropyridine Chemical compound CCOC1=CC(Cl)=NC=C1F MXWFDXNJVQMDGA-UHFFFAOYSA-N 0.000 description 1
- QOGXQLSFJCIDNY-UHFFFAOYSA-N 2-chloro-5-fluoropyridine Chemical compound FC1=CC=C(Cl)N=C1 QOGXQLSFJCIDNY-UHFFFAOYSA-N 0.000 description 1
- LUNMJRJMSXZSLC-UHFFFAOYSA-N 2-cyclopropylethanol Chemical compound OCCC1CC1 LUNMJRJMSXZSLC-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical class C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- GPEPNXRUEOKKDO-UHFFFAOYSA-N 3,3,3-trifluoro-2-methylpropan-1-ol Chemical compound OCC(C)C(F)(F)F GPEPNXRUEOKKDO-UHFFFAOYSA-N 0.000 description 1
- YWPMKTWUFVOFPL-UHFFFAOYSA-N 3,4-dihydro-2h-isoquinolin-1-one Chemical compound C1=CC=C2C(=O)NCCC2=C1 YWPMKTWUFVOFPL-UHFFFAOYSA-N 0.000 description 1
- OPXGIXNXHJHPLP-UHFFFAOYSA-N 3,8-diazabicyclo[3.2.1]octane-8-carboxylic acid Chemical compound C1NCC2CCC1N2C(=O)O OPXGIXNXHJHPLP-UHFFFAOYSA-N 0.000 description 1
- ZSULKAPJHICPKB-UHFFFAOYSA-N 3-(trifluoromethyl)cyclobutane-1-carboxylic acid Chemical compound OC(=O)C1CC(C(F)(F)F)C1 ZSULKAPJHICPKB-UHFFFAOYSA-N 0.000 description 1
- JTNUDWWUSCHHTF-UHFFFAOYSA-N 3-[6-(2,5-dimethylpyrrol-1-yl)-4-methoxypyridin-3-yl]-3,8-diazabicyclo[3.2.1]octane-8-carboxylic acid Chemical compound CC=1N(C(=CC=1)C)C1=CC(=C(C=N1)N1CC2CCC(C1)N2C(=O)O)OC JTNUDWWUSCHHTF-UHFFFAOYSA-N 0.000 description 1
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical class NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- YTEIHWVCQJZNEN-UHFFFAOYSA-N 3-pyridin-4-ylpyridine Chemical compound C1=CN=CC(C=2C=CN=CC=2)=C1 YTEIHWVCQJZNEN-UHFFFAOYSA-N 0.000 description 1
- OAKJRAGZSJBPTL-UHFFFAOYSA-N 4-(6-aminopyridin-3-yl)piperazine-1-carboxylic acid Chemical compound C1=NC(N)=CC=C1N1CCN(C(O)=O)CC1 OAKJRAGZSJBPTL-UHFFFAOYSA-N 0.000 description 1
- ZHHGGWNJDCRDHI-UHFFFAOYSA-N 4-(6-nitropyridin-3-yl)piperazine-1-carboxylic acid Chemical compound C1CN(C(=O)O)CCN1C1=CC=C([N+]([O-])=O)N=C1 ZHHGGWNJDCRDHI-UHFFFAOYSA-N 0.000 description 1
- BNHAYQSUBZKWAG-UHFFFAOYSA-N 4-(difluoromethoxy)phenol Chemical compound OC1=CC=C(OC(F)F)C=C1 BNHAYQSUBZKWAG-UHFFFAOYSA-N 0.000 description 1
- WDRJNKMAZMEYOF-UHFFFAOYSA-N 4-(trifluoromethoxy)phenol Chemical compound OC1=CC=C(OC(F)(F)F)C=C1 WDRJNKMAZMEYOF-UHFFFAOYSA-N 0.000 description 1
- ZZLCFHIKESPLTH-UHFFFAOYSA-N 4-Methylbiphenyl Chemical compound C1=CC(C)=CC=C1C1=CC=CC=C1 ZZLCFHIKESPLTH-UHFFFAOYSA-N 0.000 description 1
- LTXNDRMMTRTOJG-BZSJEYESSA-N 4-O-benzyl 1-O-tert-butyl (2R)-2-(1-hydroxyethyl)piperazine-1,4-dicarboxylate Chemical compound OC(C)[C@@H]1N(CCN(C1)C(=O)OCC1=CC=CC=C1)C(=O)OC(C)(C)C LTXNDRMMTRTOJG-BZSJEYESSA-N 0.000 description 1
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical class COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 1
- WXNZTHHGJRFXKQ-UHFFFAOYSA-N 4-chlorophenol Chemical compound OC1=CC=C(Cl)C=C1 WXNZTHHGJRFXKQ-UHFFFAOYSA-N 0.000 description 1
- MPJDLFXZHLPNKH-UHFFFAOYSA-N 4-methoxy-5-propoxypyridine-2-carboxylic acid Chemical compound CCCOc1cnc(cc1OC)C(O)=O MPJDLFXZHLPNKH-UHFFFAOYSA-N 0.000 description 1
- QEYQMWSESURNPP-UHFFFAOYSA-N 4-propan-2-yloxyphenol Chemical compound CC(C)OC1=CC=C(O)C=C1 QEYQMWSESURNPP-UHFFFAOYSA-N 0.000 description 1
- WJXSRLIHZNDTBQ-UHFFFAOYSA-N 5-[4-(trifluoromethyl)phenoxy]pyridine-2-carbonitrile Chemical compound FC(F)(F)c1ccc(Oc2ccc(nc2)C#N)cc1 WJXSRLIHZNDTBQ-UHFFFAOYSA-N 0.000 description 1
- MRBKHNNJMDNENM-UHFFFAOYSA-N 5-bromo-4-cyclopropyloxypyridin-2-amine Chemical compound C1=NC(N)=CC(OC2CC2)=C1Br MRBKHNNJMDNENM-UHFFFAOYSA-N 0.000 description 1
- JDNCMHOKWINDKI-UHFFFAOYSA-N 5-bromo-4-methylpyridin-2-amine Chemical compound CC1=CC(N)=NC=C1Br JDNCMHOKWINDKI-UHFFFAOYSA-N 0.000 description 1
- WGOLHUGPTDEKCF-UHFFFAOYSA-N 5-bromopyridin-2-amine Chemical compound NC1=CC=C(Br)C=N1 WGOLHUGPTDEKCF-UHFFFAOYSA-N 0.000 description 1
- BHXHRMVSUUPOLX-UHFFFAOYSA-N 5-fluoropyridine-2-carbonitrile Chemical compound FC1=CC=C(C#N)N=C1 BHXHRMVSUUPOLX-UHFFFAOYSA-N 0.000 description 1
- FXTDHDQFLZNYKW-UHFFFAOYSA-N 6-bromopyridazin-3-amine Chemical compound NC1=CC=C(Br)N=N1 FXTDHDQFLZNYKW-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 206010001497 Agitation Diseases 0.000 description 1
- 229940123338 Aldosterone synthase inhibitor Drugs 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- 102000005862 Angiotensin II Human genes 0.000 description 1
- 101800000733 Angiotensin-2 Proteins 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 206010003162 Arterial injury Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 239000005485 Azilsartan Substances 0.000 description 1
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 1
- 239000005711 Benzoic acid Chemical class 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- DGOHVOJMLZANBH-UHFFFAOYSA-N C(C)(C)(C)OC(=O)N1CCN(CC1)C=1C=NC(=CC=1OC)N Chemical compound C(C)(C)(C)OC(=O)N1CCN(CC1)C=1C=NC(=CC=1OC)N DGOHVOJMLZANBH-UHFFFAOYSA-N 0.000 description 1
- TZCQLPHIHIDITC-UHFFFAOYSA-N C(C)(C)(C)OC(=O)N1CCN(CC1)C=1C=NC(=CC=1OC)N1C(=CC=C1C)C Chemical compound C(C)(C)(C)OC(=O)N1CCN(CC1)C=1C=NC(=CC=1OC)N1C(=CC=C1C)C TZCQLPHIHIDITC-UHFFFAOYSA-N 0.000 description 1
- WMXJQORZUMDIEG-MRXNPFEDSA-N C(C)(C)(C)OC(=O)N1[C@H](CN(CC1)C=1C=NC(=CC=1)N)C(O[SiH2]C(C)(C)C)(C)C Chemical compound C(C)(C)(C)OC(=O)N1[C@H](CN(CC1)C=1C=NC(=CC=1)N)C(O[SiH2]C(C)(C)C)(C)C WMXJQORZUMDIEG-MRXNPFEDSA-N 0.000 description 1
- IWNFJXDDLCHHNO-UHFFFAOYSA-N C(C)(C)(C)OC(=O)NC1=CC(=C(C=N1)C1CCN(CC1)C(=O)OC(C)(C)C)OCC Chemical compound C(C)(C)(C)OC(=O)NC1=CC(=C(C=N1)C1CCN(CC1)C(=O)OC(C)(C)C)OCC IWNFJXDDLCHHNO-UHFFFAOYSA-N 0.000 description 1
- KEMXYUXDIDEAGE-UHFFFAOYSA-N C(C)OC1=CC(=NC=C1)NC(OC(C)(C)C)=O Chemical compound C(C)OC1=CC(=NC=C1)NC(OC(C)(C)C)=O KEMXYUXDIDEAGE-UHFFFAOYSA-N 0.000 description 1
- MJPAAUBEKGISJG-UHFFFAOYSA-N C(C)OC1=CC(=NC=C1OC1=CC=C(C=C1)F)C(=O)O Chemical compound C(C)OC1=CC(=NC=C1OC1=CC=C(C=C1)F)C(=O)O MJPAAUBEKGISJG-UHFFFAOYSA-N 0.000 description 1
- KBJSDGQCVLJBBG-UHFFFAOYSA-N C(C)OC1=CC(=NC=C1OC1=CC=CC=C1)C(=O)O Chemical compound C(C)OC1=CC(=NC=C1OC1=CC=CC=C1)C(=O)O KBJSDGQCVLJBBG-UHFFFAOYSA-N 0.000 description 1
- NNDFHMFXAFAWTJ-MRXNPFEDSA-N C(C1=CC=CC=C1)OC(=O)N1CCN([C@@H](C(=O)N(C)OC)C1)C(=O)OC(C)(C)C Chemical compound C(C1=CC=CC=C1)OC(=O)N1CCN([C@@H](C(=O)N(C)OC)C1)C(=O)OC(C)(C)C NNDFHMFXAFAWTJ-MRXNPFEDSA-N 0.000 description 1
- QPYMASZSKAUFJL-UHFFFAOYSA-N C(C1=CC=CC=C1)OC=1C(=CC(=NC=1)C(=O)O)OC Chemical compound C(C1=CC=CC=C1)OC=1C(=CC(=NC=1)C(=O)O)OC QPYMASZSKAUFJL-UHFFFAOYSA-N 0.000 description 1
- UQCNUBFAMLJXBP-UHFFFAOYSA-N C(OC(=O)NC1=NN=C(C(=C1)OC)C1=CCN(CC1)C(=O)OC(C)(C)C)(C)(C)C Chemical compound C(OC(=O)NC1=NN=C(C(=C1)OC)C1=CCN(CC1)C(=O)OC(C)(C)C)(C)(C)C UQCNUBFAMLJXBP-UHFFFAOYSA-N 0.000 description 1
- KHVWJJDWJJURSD-UHFFFAOYSA-N C(OC1=CC=C(OC2=C(OC)C=C(C(=O)O)N=C2)C=C1)(F)F Chemical compound C(OC1=CC=C(OC2=C(OC)C=C(C(=O)O)N=C2)C=C1)(F)F KHVWJJDWJJURSD-UHFFFAOYSA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- SLVVLHHREZYZQJ-GFCCVEGCSA-N C1(=CN=C(C=C1C)N)N1CCN([C@@H](CO)C1)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1C)N)N1CCN([C@@H](CO)C1)C(=O)OC(C)(C)C SLVVLHHREZYZQJ-GFCCVEGCSA-N 0.000 description 1
- RDDLNWPZYUSNFS-CYBMUJFWSA-N C1(=CN=C(C=C1C)N)N1CCN([C@H](C1)COC)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1C)N)N1CCN([C@H](C1)COC)C(=O)OC(C)(C)C RDDLNWPZYUSNFS-CYBMUJFWSA-N 0.000 description 1
- VNYLYYJVWOZBPK-UHFFFAOYSA-N C1(=CN=C(C=C1OC)N)N1CCN(C2(CC2)C1)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1OC)N)N1CCN(C2(CC2)C1)C(=O)OC(C)(C)C VNYLYYJVWOZBPK-UHFFFAOYSA-N 0.000 description 1
- VWHFOMCANVHLBQ-NSHDSACASA-N C1(=CN=C(C=C1OC)N)N1CCN([C@@H](C1)CO)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1OC)N)N1CCN([C@@H](C1)CO)C(=O)OC(C)(C)C VWHFOMCANVHLBQ-NSHDSACASA-N 0.000 description 1
- VWHFOMCANVHLBQ-LLVKDONJSA-N C1(=CN=C(C=C1OC)N)N1CCN([C@@H](CO)C1)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1OC)N)N1CCN([C@@H](CO)C1)C(=O)OC(C)(C)C VWHFOMCANVHLBQ-LLVKDONJSA-N 0.000 description 1
- BVMIPGCFSXGSGW-UHFFFAOYSA-N C1(=CN=C(C=C1OCCC)NC(=O)OC(C)(C)C)C1=CCN(CC1)C(=O)OC(C)(C)C Chemical compound C1(=CN=C(C=C1OCCC)NC(=O)OC(C)(C)C)C1=CCN(CC1)C(=O)OC(C)(C)C BVMIPGCFSXGSGW-UHFFFAOYSA-N 0.000 description 1
- FETNUIKXWCXHAW-UHFFFAOYSA-N C1=C(N)N=NC(C2=CC3N(C(CC3)C2)C(=O)OC(C)(C)C)=C1 Chemical compound C1=C(N)N=NC(C2=CC3N(C(CC3)C2)C(=O)OC(C)(C)C)=C1 FETNUIKXWCXHAW-UHFFFAOYSA-N 0.000 description 1
- YDPFDSXVYWMCMH-UHFFFAOYSA-N C1=C(N)N=NC(C2CC3N(C(CC3)C2)C(=O)OC(C)(C)C)=C1 Chemical compound C1=C(N)N=NC(C2CC3N(C(CC3)C2)C(=O)OC(C)(C)C)=C1 YDPFDSXVYWMCMH-UHFFFAOYSA-N 0.000 description 1
- GBLGOBIMJLXLSO-UHFFFAOYSA-N C1C(OC2=CC(N)=NC=C2C2=CCN(CC2)C(=O)OC(C)(C)C)C1 Chemical compound C1C(OC2=CC(N)=NC=C2C2=CCN(CC2)C(=O)OC(C)(C)C)C1 GBLGOBIMJLXLSO-UHFFFAOYSA-N 0.000 description 1
- IRHPKOVRFDGRJB-ZCFIWIBFSA-N CC(=O)[C@H]1CN(CCN1C(O)=O)C(O)=O Chemical compound CC(=O)[C@H]1CN(CCN1C(O)=O)C(O)=O IRHPKOVRFDGRJB-ZCFIWIBFSA-N 0.000 description 1
- SVKXBWFGLNIKRX-UHFFFAOYSA-N CC(COC=1C(=CC(=NC=1)C(=O)O)OC)(C)C Chemical compound CC(COC=1C(=CC(=NC=1)C(=O)O)OC)(C)C SVKXBWFGLNIKRX-UHFFFAOYSA-N 0.000 description 1
- CZCMKRWGAOQDHS-OAHLLOKOSA-N CC1=CC=C(N1C2=NC=C(C(=C2)OC)N3CCN([C@H](C3)COC)C(=O)O)C Chemical compound CC1=CC=C(N1C2=NC=C(C(=C2)OC)N3CCN([C@H](C3)COC)C(=O)O)C CZCMKRWGAOQDHS-OAHLLOKOSA-N 0.000 description 1
- ZLPHYBYUKWPHPU-UHFFFAOYSA-N COC(=O)C1=NC=C(C(=C1)OC)OCCC1CC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)OCCC1CC1 ZLPHYBYUKWPHPU-UHFFFAOYSA-N 0.000 description 1
- KDAQUQASOCUDDD-MRVPVSSYSA-N COC(=O)C1=NC=C(C(=C1)OC)O[C@H](C)C1CC1 Chemical compound COC(=O)C1=NC=C(C(=C1)OC)O[C@H](C)C1CC1 KDAQUQASOCUDDD-MRVPVSSYSA-N 0.000 description 1
- RNTHFSGYRVUSSX-UHFFFAOYSA-N COC(=O)C1=NC=C(OCC2(C)CC2)C(OC)=C1 Chemical compound COC(=O)C1=NC=C(OCC2(C)CC2)C(OC)=C1 RNTHFSGYRVUSSX-UHFFFAOYSA-N 0.000 description 1
- VJQFDEYQPMHXIZ-UHFFFAOYSA-N COC1=C(N)N=NC(=C1)C1=CCN(CC1)C(=O)OC(C)(C)C Chemical compound COC1=C(N)N=NC(=C1)C1=CCN(CC1)C(=O)OC(C)(C)C VJQFDEYQPMHXIZ-UHFFFAOYSA-N 0.000 description 1
- YLZGCQMTEFUWDV-UHFFFAOYSA-N COC1=CC(=NC=C1OC1CCCCC1)C(O)=O Chemical compound COC1=CC(=NC=C1OC1CCCCC1)C(O)=O YLZGCQMTEFUWDV-UHFFFAOYSA-N 0.000 description 1
- QXCYXNGRIXXMMV-UHFFFAOYSA-N COC1=CC(=NC=C1OCC1(CF)CC1)C(O)=O Chemical compound COC1=CC(=NC=C1OCC1(CF)CC1)C(O)=O QXCYXNGRIXXMMV-UHFFFAOYSA-N 0.000 description 1
- GKCQLRJDMQOMGP-UHFFFAOYSA-N COC1=CC(=NC=C1OCC1CC(C1)C(F)(F)F)C(=O)O Chemical compound COC1=CC(=NC=C1OCC1CC(C1)C(F)(F)F)C(=O)O GKCQLRJDMQOMGP-UHFFFAOYSA-N 0.000 description 1
- VFKZAFPHDGIVQV-UHFFFAOYSA-N COC1=CC(=NC=C1OCC1CC(C1)C(F)(F)F)C(=O)OC Chemical compound COC1=CC(=NC=C1OCC1CC(C1)C(F)(F)F)C(=O)OC VFKZAFPHDGIVQV-UHFFFAOYSA-N 0.000 description 1
- XSTLEOQQMKRZIW-UHFFFAOYSA-N COC1=CC(=NC=C1OCC1CCC1)C(O)=O Chemical compound COC1=CC(=NC=C1OCC1CCC1)C(O)=O XSTLEOQQMKRZIW-UHFFFAOYSA-N 0.000 description 1
- OQPWDWLSRYUIOS-UHFFFAOYSA-N COC1=CC(=NC=C1OCCC1=CC=CC=C1)C(=O)O Chemical compound COC1=CC(=NC=C1OCCC1=CC=CC=C1)C(=O)O OQPWDWLSRYUIOS-UHFFFAOYSA-N 0.000 description 1
- ZDLKPHSQRCQRMO-ZETCQYMHSA-N COC1=CC(=NC=C1O[C@@H](C)C1CC1)C(O)=O Chemical compound COC1=CC(=NC=C1O[C@@H](C)C1CC1)C(O)=O ZDLKPHSQRCQRMO-ZETCQYMHSA-N 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- WXQFIWOBRANJNL-DDWIOCJRSA-N Cl.COc1cc(N)ncc1N1CCN[C@@H](CO)C1 Chemical compound Cl.COc1cc(N)ncc1N1CCN[C@@H](CO)C1 WXQFIWOBRANJNL-DDWIOCJRSA-N 0.000 description 1
- MWCXXRLYXGAHGN-UHFFFAOYSA-N Cl.Cl.COc1cc(nnc1N)C1CCNCC1 Chemical compound Cl.Cl.COc1cc(nnc1N)C1CCNCC1 MWCXXRLYXGAHGN-UHFFFAOYSA-N 0.000 description 1
- WFGHRZMDIONHIU-UHFFFAOYSA-N Cl.Cl.Nc1cc(OC2CC2)c(cn1)C1CCNCC1 Chemical compound Cl.Cl.Nc1cc(OC2CC2)c(cn1)C1CCNCC1 WFGHRZMDIONHIU-UHFFFAOYSA-N 0.000 description 1
- ADQATROQODPNMO-UHFFFAOYSA-N ClC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 Chemical compound ClC1=CC=C(OC=2C(=CC(=NC=2)C#N)OC)C=C1 ADQATROQODPNMO-UHFFFAOYSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 208000016998 Conn syndrome Diseases 0.000 description 1
- OCUCCJIRFHNWBP-IYEMJOQQSA-L Copper gluconate Chemical class [Cu+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O OCUCCJIRFHNWBP-IYEMJOQQSA-L 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 208000014311 Cushing syndrome Diseases 0.000 description 1
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 229940124213 Dipeptidyl peptidase 4 (DPP IV) inhibitor Drugs 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical class OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- MARMYYDUFCXUBI-UHFFFAOYSA-N FC1(CC(C1)COC=1C(=CC(=NC=1)C(=O)O)OC)F Chemical compound FC1(CC(C1)COC=1C(=CC(=NC=1)C(=O)O)OC)F MARMYYDUFCXUBI-UHFFFAOYSA-N 0.000 description 1
- FZUGVIWNZUOPRL-UHFFFAOYSA-N FC1=CC=C(COC=2C(=CC(=NC=2)C(=O)O)OC)C=C1 Chemical compound FC1=CC=C(COC=2C(=CC(=NC=2)C(=O)O)OC)C=C1 FZUGVIWNZUOPRL-UHFFFAOYSA-N 0.000 description 1
- JFMUWYXOYUCWOO-UHFFFAOYSA-N FC=1C(=CC(=NC=1)C#N)OC(C)C Chemical compound FC=1C(=CC(=NC=1)C#N)OC(C)C JFMUWYXOYUCWOO-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical class OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- VPNYRYCIDCJBOM-UHFFFAOYSA-M Glycopyrronium bromide Chemical compound [Br-].C1[N+](C)(C)CCC1OC(=O)C(O)(C=1C=CC=CC=1)C1CCCC1 VPNYRYCIDCJBOM-UHFFFAOYSA-M 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000817629 Homo sapiens Dymeclin Proteins 0.000 description 1
- 101000601770 Homo sapiens Protein polybromo-1 Proteins 0.000 description 1
- 101000600434 Homo sapiens Putative uncharacterized protein encoded by MIR7-3HG Proteins 0.000 description 1
- 101000846110 Homo sapiens Short transient receptor potential channel 6 Proteins 0.000 description 1
- 101000894525 Homo sapiens Transforming growth factor-beta-induced protein ig-h3 Proteins 0.000 description 1
- 208000004454 Hyperalgesia Diseases 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 208000035154 Hyperesthesia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- CZGUSIXMZVURDU-JZXHSEFVSA-N Ile(5)-angiotensin II Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C([O-])=O)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=[NH2+])NC(=O)[C@@H]([NH3+])CC([O-])=O)C(C)C)C1=CC=C(O)C=C1 CZGUSIXMZVURDU-JZXHSEFVSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 1
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 1
- 208000000913 Kidney Calculi Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- LTXREWYXXSTFRX-QGZVFWFLSA-N Linagliptin Chemical compound N=1C=2N(C)C(=O)N(CC=3N=C4C=CC=CC4=C(C)N=3)C(=O)C=2N(CC#CC)C=1N1CCC[C@@H](N)C1 LTXREWYXXSTFRX-QGZVFWFLSA-N 0.000 description 1
- 206010024602 Lipiduria Diseases 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- XVVOERDUTLJJHN-UHFFFAOYSA-N Lixisenatide Chemical compound C=1NC2=CC=CC=C2C=1CC(C(=O)NC(CC(C)C)C(=O)NC(CCCCN)C(=O)NC(CC(N)=O)C(=O)NCC(=O)NCC(=O)N1C(CCC1)C(=O)NC(CO)C(=O)NC(CO)C(=O)NCC(=O)NC(C)C(=O)N1C(CCC1)C(=O)N1C(CCC1)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)CC)NC(=O)C(NC(=O)C(CC(C)C)NC(=O)C(CCCNC(N)=N)NC(=O)C(NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(CCC(O)=O)NC(=O)C(CCC(O)=O)NC(=O)C(CCSC)NC(=O)C(CCC(N)=O)NC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC=1C=CC=CC=1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)CNC(=O)C(N)CC=1NC=NC=1)C(C)O)C(C)O)C(C)C)CC1=CC=CC=C1 XVVOERDUTLJJHN-UHFFFAOYSA-N 0.000 description 1
- 208000004852 Lung Injury Diseases 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- UWWDHYUMIORJTA-HSQYWUDLSA-N Moexipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC(OC)=C(OC)C=C2C1)C(O)=O)CC1=CC=CC=C1 UWWDHYUMIORJTA-HSQYWUDLSA-N 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- ZKGNPQKYVKXMGJ-UHFFFAOYSA-N N,N-dimethylacetamide Chemical compound CN(C)C(C)=O.CN(C)C(C)=O ZKGNPQKYVKXMGJ-UHFFFAOYSA-N 0.000 description 1
- AVYVHIKSFXVDBG-UHFFFAOYSA-N N-benzyl-N-hydroxy-2,2-dimethylbutanamide Chemical compound C(C1=CC=CC=C1)N(C(C(CC)(C)C)=O)O AVYVHIKSFXVDBG-UHFFFAOYSA-N 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- DEAUKMQEDWSPFN-GOSISDBHSA-N N1(C(=CC=C1C)C)C1=CC(=C(C=N1)N1CCN([C@@H](CO)C1)C(=O)OC(C)(C)C)C Chemical compound N1(C(=CC=C1C)C)C1=CC(=C(C=N1)N1CCN([C@@H](CO)C1)C(=O)OC(C)(C)C)C DEAUKMQEDWSPFN-GOSISDBHSA-N 0.000 description 1
- DKMDRWWOLQJXCG-GFCCVEGCSA-N N1=C(C=C(C(=C1)N1C[C@@H](N(CC1)C(=O)OC(C)(C)C)COC)OC)N Chemical compound N1=C(C=C(C(=C1)N1C[C@@H](N(CC1)C(=O)OC(C)(C)C)COC)OC)N DKMDRWWOLQJXCG-GFCCVEGCSA-N 0.000 description 1
- WVGHZVITINFPIW-UHFFFAOYSA-N NC1=CC(=C(C=N1)C=1CCN(CC=1)C(=O)O)OC Chemical compound NC1=CC(=C(C=N1)C=1CCN(CC=1)C(=O)O)OC WVGHZVITINFPIW-UHFFFAOYSA-N 0.000 description 1
- JUALOUHZJJERQT-UHFFFAOYSA-N NC1=CC=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)F Chemical compound NC1=CC=C(N=N1)C1CCN(CC1)C(=O)C1=NC=C(C(=C1)OC)OC1=CC=C(C=C1)F JUALOUHZJJERQT-UHFFFAOYSA-N 0.000 description 1
- 206010029148 Nephrolithiasis Diseases 0.000 description 1
- 208000013901 Nephropathies and tubular disease Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- JMDHNBWGYBZBLK-UHFFFAOYSA-N O(C)C1=C(C=NC(C(=O)O)=C1)OC1=CC=C(C=C1)OC(C)C Chemical compound O(C)C1=C(C=NC(C(=O)O)=C1)OC1=CC=C(C=C1)OC(C)C JMDHNBWGYBZBLK-UHFFFAOYSA-N 0.000 description 1
- BMXFYYCHSFEFFH-UHFFFAOYSA-N O(C)C1=C(C=NC(C(=O)O)=C1)OC1=CC=C(C=C1)OC1CC1 Chemical compound O(C)C1=C(C=NC(C(=O)O)=C1)OC1=CC=C(C=C1)OC1CC1 BMXFYYCHSFEFFH-UHFFFAOYSA-N 0.000 description 1
- OSSCRRAWGUIUSI-UHFFFAOYSA-N O(C)C1=CC(C(=O)O)=NC=C1OC1=CC=C(Cl)C=C1 Chemical compound O(C)C1=CC(C(=O)O)=NC=C1OC1=CC=C(Cl)C=C1 OSSCRRAWGUIUSI-UHFFFAOYSA-N 0.000 description 1
- QOFIKMZUYHOPFD-UHFFFAOYSA-N O(C1=C(N2CC3N(C(CC3)C2)C(=O)OC(C)(C)C)C=NC(N)=C1)C Chemical compound O(C1=C(N2CC3N(C(CC3)C2)C(=O)OC(C)(C)C)C=NC(N)=C1)C QOFIKMZUYHOPFD-UHFFFAOYSA-N 0.000 description 1
- CHSFVCWRRBUSLU-UHFFFAOYSA-N O(C1=CN=C(C(=O)O)C=C1OC)C1CCCC1 Chemical compound O(C1=CN=C(C(=O)O)C=C1OC)C1CCCC1 CHSFVCWRRBUSLU-UHFFFAOYSA-N 0.000 description 1
- YQMUANDAAAAVDX-UHFFFAOYSA-N O(C1=CN=C(C=C1OC)C(=O)O)C(C)C Chemical compound O(C1=CN=C(C=C1OC)C(=O)O)C(C)C YQMUANDAAAAVDX-UHFFFAOYSA-N 0.000 description 1
- BJINIKFTLFHSGL-UHFFFAOYSA-N O(C1=CN=C(C=C1OC)C(=O)O)CC Chemical compound O(C1=CN=C(C=C1OC)C(=O)O)CC BJINIKFTLFHSGL-UHFFFAOYSA-N 0.000 description 1
- NQIJZXLCZRJBKL-UHFFFAOYSA-N O(C1=CN=C(C=C1OC)C(=O)O)CCC1CC1 Chemical compound O(C1=CN=C(C=C1OC)C(=O)O)CCC1CC1 NQIJZXLCZRJBKL-UHFFFAOYSA-N 0.000 description 1
- DWJMQURCJVTIHW-UHFFFAOYSA-N O=C(C1=NC=C(C(OC)=C1)OC1=CC=C(OC(F)(F)F)C=C1)O Chemical compound O=C(C1=NC=C(C(OC)=C1)OC1=CC=C(OC(F)(F)F)C=C1)O DWJMQURCJVTIHW-UHFFFAOYSA-N 0.000 description 1
- VAJOSFZFQDDYRT-UHFFFAOYSA-N OC(=O)C(F)(F)F.COc1cc(ncc1Oc1ccc(cc1)C(F)(F)F)C(=O)N1CCC(CC1)c1nnc(N)cc1C Chemical compound OC(=O)C(F)(F)F.COc1cc(ncc1Oc1ccc(cc1)C(F)(F)F)C(=O)N1CCC(CC1)c1nnc(N)cc1C VAJOSFZFQDDYRT-UHFFFAOYSA-N 0.000 description 1
- XKDPPEUBYOQKEJ-UHFFFAOYSA-N OC(=O)C(F)(F)F.COc1cc(ncc1Oc1ccccc1)C(=O)N1CCC(CC1)c1nnc(N)cc1C Chemical compound OC(=O)C(F)(F)F.COc1cc(ncc1Oc1ccccc1)C(=O)N1CCC(CC1)c1nnc(N)cc1C XKDPPEUBYOQKEJ-UHFFFAOYSA-N 0.000 description 1
- BJAKPBQLGWORBK-UHFFFAOYSA-N OC(=O)C(F)(F)F.OC(=O)C(F)(F)F.COc1cc(ncc1N1CCNCC1)-n1c(C)ccc1C Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F.COc1cc(ncc1N1CCNCC1)-n1c(C)ccc1C BJAKPBQLGWORBK-UHFFFAOYSA-N 0.000 description 1
- IRNOAQCJQYETTI-QGZVFWFLSA-N OC[C@@H]1N(CCN(C1)C=1C=NC(=CC=1)[N+](=O)[O-])C(=O)C1=NC=C(C(=C1)OC)OC1=CC=CC=C1 Chemical compound OC[C@@H]1N(CCN(C1)C=1C=NC(=CC=1)[N+](=O)[O-])C(=O)C1=NC=C(C(=C1)OC)OC1=CC=CC=C1 IRNOAQCJQYETTI-QGZVFWFLSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 239000005922 Phosphane Substances 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- SIOXPEMLGUPBBT-UHFFFAOYSA-N Picolinic acid Natural products OC(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 102100037516 Protein polybromo-1 Human genes 0.000 description 1
- 102100037401 Putative uncharacterized protein encoded by MIR7-3HG Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Chemical class OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- BPZSYCZIITTYBL-YJYMSZOUSA-N R-Formoterol Chemical compound C1=CC(OC)=CC=C1C[C@@H](C)NC[C@H](O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-YJYMSZOUSA-N 0.000 description 1
- 101100372319 Rattus norvegicus Utrn gene Proteins 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 206010061481 Renal injury Diseases 0.000 description 1
- 101100485158 Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) wzzE gene Proteins 0.000 description 1
- 201000004239 Secondary hypertension Diseases 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229940123518 Sodium/glucose cotransporter 2 inhibitor Drugs 0.000 description 1
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 1
- 208000036765 Squamous cell carcinoma of the esophagus Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Chemical class OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 102000003621 TRPC5 Human genes 0.000 description 1
- 101150042815 TRPC5 gene Proteins 0.000 description 1
- 102000003564 TRPC7 Human genes 0.000 description 1
- 101150095096 TRPM2 gene Proteins 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- 102100021398 Transforming growth factor-beta-induced protein ig-h3 Human genes 0.000 description 1
- 206010069363 Traumatic lung injury Diseases 0.000 description 1
- 101150090814 Trpc6 gene Proteins 0.000 description 1
- 101150033973 Trpc7 gene Proteins 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- IRFLMQHCAJMPCS-UHFFFAOYSA-N [1-(fluoromethyl)cyclopropyl]methanol Chemical compound OCC1(CF)CC1 IRFLMQHCAJMPCS-UHFFFAOYSA-N 0.000 description 1
- XBUQANULLSLVTC-UHFFFAOYSA-N [3-(trifluoromethyl)cyclobutyl]methanol Chemical compound FC(C1CC(C1)CO)(F)F XBUQANULLSLVTC-UHFFFAOYSA-N 0.000 description 1
- GELXYNKOMMJDNP-UHFFFAOYSA-N [3-[(2-chlorophenoxy)methyl]phenyl]-piperidin-1-ylmethanone Chemical compound ClC1=CC=CC=C1OCC1=CC=CC(C(=O)N2CCCCC2)=C1 GELXYNKOMMJDNP-UHFFFAOYSA-N 0.000 description 1
- GGKQWFQQUMWVOB-UHFFFAOYSA-N [5-chloro-2-[(6-fluoro-1,3-benzodioxol-5-yl)amino]-1,3-thiazol-4-yl]-(2,3-dimethylpiperidin-1-yl)methanone Chemical compound CC1C(C)CCCN1C(=O)C1=C(Cl)SC(NC=2C(=CC=3OCOC=3C=2)F)=N1 GGKQWFQQUMWVOB-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- ASMXXROZKSBQIH-VITNCHFBSA-N aclidinium Chemical compound C([C@@H](C(CC1)CC2)OC(=O)C(O)(C=3SC=CC=3)C=3SC=CC=3)[N+]21CCCOC1=CC=CC=C1 ASMXXROZKSBQIH-VITNCHFBSA-N 0.000 description 1
- 229940019903 aclidinium Drugs 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 206010069351 acute lung injury Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 229960004733 albiglutide Drugs 0.000 description 1
- OGWAVGNOAMXIIM-UHFFFAOYSA-N albiglutide Chemical compound O=C(O)C(NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)C(NC(=O)CNC(=O)C(NC(=O)CNC(=O)C(N)CC=1(N=CNC=1))CCC(=O)O)C(O)C)CC2(=CC=CC=C2))C(O)C)CO)CC(=O)O)C(C)C)CO)CO)CC3(=CC=C(O)C=C3))CC(C)C)CCC(=O)O)CCC(=O)N)C)C)CCCCN)CCC(=O)O)CC4(=CC=CC=C4))C(CC)C)C)CC=6(C5(=C(C=CC=C5)NC=6)))CC(C)C)C(C)C)CCCCN)CCCNC(=N)N OGWAVGNOAMXIIM-UHFFFAOYSA-N 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 229960001667 alogliptin Drugs 0.000 description 1
- ZSBOMTDTBDDKMP-OAHLLOKOSA-N alogliptin Chemical compound C=1C=CC=C(C#N)C=1CN1C(=O)N(C)C(=O)C=C1N1CCC[C@@H](N)C1 ZSBOMTDTBDDKMP-OAHLLOKOSA-N 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Chemical class OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 210000001132 alveolar macrophage Anatomy 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229950006323 angiotensin ii Drugs 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 229940065524 anticholinergics inhalants for obstructive airway diseases Drugs 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 229960001692 arformoterol Drugs 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 125000003289 ascorbyl group Chemical class [H]O[C@@]([H])(C([H])([H])O*)[C@@]1([H])OC(=O)C(O*)=C1O* 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 230000000778 atheroprotective effect Effects 0.000 description 1
- 230000003143 atherosclerotic effect Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- OVLHQUWQINALPR-UHFFFAOYSA-N azane;dichloromethane Chemical compound N.ClCCl OVLHQUWQINALPR-UHFFFAOYSA-N 0.000 description 1
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 1
- JPNZKPRONVOMLL-UHFFFAOYSA-N azane;octadecanoic acid Chemical class [NH4+].CCCCCCCCCCCCCCCCCC([O-])=O JPNZKPRONVOMLL-UHFFFAOYSA-N 0.000 description 1
- 229960002731 azilsartan Drugs 0.000 description 1
- KGSXMPPBFPAXLY-UHFFFAOYSA-N azilsartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NOC(=O)N1 KGSXMPPBFPAXLY-UHFFFAOYSA-N 0.000 description 1
- 229960004530 benazepril Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical class BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical class O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- XQPRBTXUXXVTKB-UHFFFAOYSA-M caesium iodide Chemical compound [I-].[Cs+] XQPRBTXUXXVTKB-UHFFFAOYSA-M 0.000 description 1
- 229960001713 canagliflozin Drugs 0.000 description 1
- VHOFTEAWFCUTOS-TUGBYPPCSA-N canagliflozin hydrate Chemical compound O.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1.CC1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(S1)=CC=C1C1=CC=C(F)C=C1 VHOFTEAWFCUTOS-TUGBYPPCSA-N 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 230000003727 cerebral blood flow Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- DRNAQRXLOSUHBQ-UHFFFAOYSA-N cphos Chemical group CN(C)C1=CC=CC(N(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 DRNAQRXLOSUHBQ-UHFFFAOYSA-N 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- WPOPOPFNZYPKAV-UHFFFAOYSA-N cyclobutylmethanol Chemical compound OCC1CCC1 WPOPOPFNZYPKAV-UHFFFAOYSA-N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- XCIXKGXIYUWCLL-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1 XCIXKGXIYUWCLL-UHFFFAOYSA-N 0.000 description 1
- 229960003834 dapagliflozin Drugs 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000002999 depolarising effect Effects 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 150000001982 diacylglycerols Chemical class 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 239000003603 dipeptidyl peptidase IV inhibitor Substances 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical class [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 229960005175 dulaglutide Drugs 0.000 description 1
- 108010005794 dulaglutide Proteins 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 229940048820 edetates Drugs 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 229960003345 empagliflozin Drugs 0.000 description 1
- OBWASQILIWPZMG-QZMOQZSNSA-N empagliflozin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(O[C@@H]3COCC3)=CC=2)=C1 OBWASQILIWPZMG-QZMOQZSNSA-N 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- 230000002121 endocytic effect Effects 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 230000008497 endothelial barrier function Effects 0.000 description 1
- 230000004528 endothelial cell apoptotic process Effects 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- 208000007276 esophageal squamous cell carcinoma Diseases 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethanedisulfonate group Chemical class C(CS(=O)(=O)[O-])S(=O)(=O)[O-] AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical class CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 150000002171 ethylene diamines Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000002795 fluorescence method Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229960002714 fluticasone Drugs 0.000 description 1
- MGNNYOODZCAHBA-GQKYHHCASA-N fluticasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(O)[C@@]2(C)C[C@@H]1O MGNNYOODZCAHBA-GQKYHHCASA-N 0.000 description 1
- 229960002848 formoterol Drugs 0.000 description 1
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 239000001530 fumaric acid Chemical class 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960005219 gentisic acid Drugs 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 150000002306 glutamic acid derivatives Chemical class 0.000 description 1
- 229940015042 glycopyrrolate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000004217 heart function Effects 0.000 description 1
- 210000004024 hepatic stellate cell Anatomy 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 238000012188 high-throughput screening assay Methods 0.000 description 1
- 102000043698 human TRPC6 Human genes 0.000 description 1
- DKAGJZJALZXOOV-UHFFFAOYSA-N hydrate;hydrochloride Chemical compound O.Cl DKAGJZJALZXOOV-UHFFFAOYSA-N 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000005286 illumination Methods 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- MGXWVYUBJRZYPE-YUGYIWNOSA-N incretin Chemical class C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=C(O)C=C1 MGXWVYUBJRZYPE-YUGYIWNOSA-N 0.000 description 1
- 239000000859 incretin Substances 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000010262 intracellular communication Effects 0.000 description 1
- 210000004020 intracellular membrane Anatomy 0.000 description 1
- 210000005061 intracellular organelle Anatomy 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 229940126181 ion channel inhibitor Drugs 0.000 description 1
- 230000005865 ionizing radiation Effects 0.000 description 1
- OEXHQOGQTVQTAT-JRNQLAHRSA-N ipratropium Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 OEXHQOGQTVQTAT-JRNQLAHRSA-N 0.000 description 1
- 229960001888 ipratropium Drugs 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 201000010901 lateral sclerosis Diseases 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229950008204 levosalbutamol Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229960002397 linagliptin Drugs 0.000 description 1
- 229920006008 lipopolysaccharide Polymers 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960001093 lixisenatide Drugs 0.000 description 1
- 108010004367 lixisenatide Proteins 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 229940125389 long-acting beta agonist Drugs 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 231100000515 lung injury Toxicity 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical class OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Chemical class 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 239000001630 malic acid Chemical class 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 150000004701 malic acid derivatives Chemical class 0.000 description 1
- 210000001349 mammary artery Anatomy 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- BCVXHSPFUWZLGQ-UHFFFAOYSA-N mecn acetonitrile Chemical compound CC#N.CC#N BCVXHSPFUWZLGQ-UHFFFAOYSA-N 0.000 description 1
- 229950004994 meglitinide Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000028161 membrane depolarization Effects 0.000 description 1
- 230000012241 membrane hyperpolarization Effects 0.000 description 1
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 101150089110 metN gene Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- PPTFIKIWCZHYRA-UHFFFAOYSA-N methyl 5-bromo-4-methoxypyridine-2-carboxylate Chemical compound COC(=O)C1=CC(OC)=C(Br)C=N1 PPTFIKIWCZHYRA-UHFFFAOYSA-N 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical class CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- 150000005451 methyl sulfates Chemical class 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 229960005170 moexipril Drugs 0.000 description 1
- 208000005264 motor neuron disease Diseases 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000754 myometrium Anatomy 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 1
- 238000013059 nephrectomy Methods 0.000 description 1
- 230000000955 neuroendocrine Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- VWBWQOUWDOULQN-UHFFFAOYSA-N nmp n-methylpyrrolidone Chemical compound CN1CCCC1=O.CN1CCCC1=O VWBWQOUWDOULQN-UHFFFAOYSA-N 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 108010071584 oxidized low density lipoprotein Proteins 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Chemical class OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical class OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229910000064 phosphane Inorganic materials 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 229920001992 poloxamer 407 Polymers 0.000 description 1
- 208000030761 polycystic kidney disease Diseases 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 229940124606 potential therapeutic agent Drugs 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960003611 pramlintide Drugs 0.000 description 1
- 108010029667 pramlintide Proteins 0.000 description 1
- NRKVKVQDUCJPIZ-MKAGXXMWSA-N pramlintide acetate Chemical compound C([C@@H](C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCCCN)[C@@H](C)O)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NRKVKVQDUCJPIZ-MKAGXXMWSA-N 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 208000013846 primary aldosteronism Diseases 0.000 description 1
- 201000009395 primary hyperaldosteronism Diseases 0.000 description 1
- 230000002206 pro-fibrotic effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 210000004129 prosencephalon Anatomy 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 230000036593 pulmonary vascular resistance Effects 0.000 description 1
- 108700027806 rGLP-1 Proteins 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 230000020874 response to hypoxia Effects 0.000 description 1
- 230000036390 resting membrane potential Effects 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 210000001908 sarcoplasmic reticulum Anatomy 0.000 description 1
- 229960004937 saxagliptin Drugs 0.000 description 1
- 108010033693 saxagliptin Proteins 0.000 description 1
- QGJUIPDUBHWZPV-SGTAVMJGSA-N saxagliptin Chemical compound C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QGJUIPDUBHWZPV-SGTAVMJGSA-N 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 229940125390 short-acting beta agonist Drugs 0.000 description 1
- 230000006807 siRNA silencing Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- LKZMBDSASOBTPN-UHFFFAOYSA-L silver carbonate Substances [Ag].[O-]C([O-])=O LKZMBDSASOBTPN-UHFFFAOYSA-L 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229960004034 sitagliptin Drugs 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000004055 small Interfering RNA Substances 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 208000019411 steroid-resistant nephrotic syndrome Diseases 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- WPLOVIFNBMNBPD-ATHMIXSHSA-N subtilin Chemical compound CC1SCC(NC2=O)C(=O)NC(CC(N)=O)C(=O)NC(C(=O)NC(CCCCN)C(=O)NC(C(C)CC)C(=O)NC(=C)C(=O)NC(CCCCN)C(O)=O)CSC(C)C2NC(=O)C(CC(C)C)NC(=O)C1NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C1NC(=O)C(=C/C)/NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C2NC(=O)CNC(=O)C3CCCN3C(=O)C(NC(=O)C3NC(=O)C(CC(C)C)NC(=O)C(=C)NC(=O)C(CCC(O)=O)NC(=O)C(NC(=O)C(CCCCN)NC(=O)C(N)CC=4C5=CC=CC=C5NC=4)CSC3)C(C)SC2)C(C)C)C(C)SC1)CC1=CC=CC=C1 WPLOVIFNBMNBPD-ATHMIXSHSA-N 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940066771 systemic antihistamines piperazine derivative Drugs 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- IKEGEFYKQOSMBB-GFCCVEGCSA-N tert-butyl (2R)-2-(2-tert-butylsilyloxypropan-2-yl)piperazine-1-carboxylate Chemical compound C(C)(C)(C)OC(=O)N1[C@H](CNCC1)C(O[SiH2]C(C)(C)C)(C)C IKEGEFYKQOSMBB-GFCCVEGCSA-N 0.000 description 1
- IKEGEFYKQOSMBB-LBPRGKRZSA-N tert-butyl (2S)-2-(2-tert-butylsilyloxypropan-2-yl)piperazine-1-carboxylate Chemical compound C(C)(C)(C)OC(=O)N1[C@@H](CNCC1)C(O[SiH2]C(C)(C)C)(C)C IKEGEFYKQOSMBB-LBPRGKRZSA-N 0.000 description 1
- BCPPNDHZUPIXJM-QMMMGPOBSA-N tert-butyl (2s)-2-(hydroxymethyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNC[C@H]1CO BCPPNDHZUPIXJM-QMMMGPOBSA-N 0.000 description 1
- JNHGCLCLMOCPCQ-UHFFFAOYSA-N tert-butyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-8-azabicyclo[3.2.1]oct-3-ene-8-carboxylate Chemical compound CC(C)(C)OC(=O)N1C(C2)CCC1C=C2B1OC(C)(C)C(C)(C)O1 JNHGCLCLMOCPCQ-UHFFFAOYSA-N 0.000 description 1
- GJKPPQXNOJOCQU-UHFFFAOYSA-N tert-butyl 4-(6-aminopyridin-3-yl)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC(C=2C=NC(N)=CC=2)=C1 GJKPPQXNOJOCQU-UHFFFAOYSA-N 0.000 description 1
- RMULRXHUNOVPEI-UHFFFAOYSA-N tert-butyl 4-(6-aminopyridin-3-yl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=CC=C(N)N=C1 RMULRXHUNOVPEI-UHFFFAOYSA-N 0.000 description 1
- UGJYTOMOCDGLRS-UHFFFAOYSA-N tert-butyl 4-(6-aminopyridin-3-yl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C1=CC=C(N)N=C1 UGJYTOMOCDGLRS-UHFFFAOYSA-N 0.000 description 1
- SUWKOEMQNOBJEQ-UHFFFAOYSA-N tert-butyl 4-(6-nitropyridin-3-yl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=CC=C([N+]([O-])=O)N=C1 SUWKOEMQNOBJEQ-UHFFFAOYSA-N 0.000 description 1
- ALEDJEMUMOYFAB-UHFFFAOYSA-N tert-butyl N-(5-bromo-4-ethoxypyridin-2-yl)carbamate Chemical compound BrC=1C(=CC(=NC=1)NC(OC(C)(C)C)=O)OCC ALEDJEMUMOYFAB-UHFFFAOYSA-N 0.000 description 1
- LOXPHLAWUQJODH-UHFFFAOYSA-N tert-butyl N-(5-bromo-4-propoxypyridin-2-yl)carbamate Chemical compound C1(=CC(=C(C=N1)Br)OCCC)NC(=O)OC(C)(C)C LOXPHLAWUQJODH-UHFFFAOYSA-N 0.000 description 1
- QKSQWQOAUQFORH-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonylimino]carbamate Chemical compound CC(C)(C)OC(=O)N=NC(=O)OC(C)(C)C QKSQWQOAUQFORH-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- LERNTVKEWCAPOY-DZZGSBJMSA-N tiotropium Chemical compound O([C@H]1C[C@@H]2[N+]([C@H](C1)[C@@H]1[C@H]2O1)(C)C)C(=O)C(O)(C=1SC=CC=1)C1=CC=CS1 LERNTVKEWCAPOY-DZZGSBJMSA-N 0.000 description 1
- 229940110309 tiotropium Drugs 0.000 description 1
- 229960002277 tolazamide Drugs 0.000 description 1
- OUDSBRTVNLOZBN-UHFFFAOYSA-N tolazamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 OUDSBRTVNLOZBN-UHFFFAOYSA-N 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 102000042565 transient receptor (TC 1.A.4) family Human genes 0.000 description 1
- 108091053409 transient receptor (TC 1.A.4) family Proteins 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- DNYWZCXLKNTFFI-UHFFFAOYSA-N uranium Chemical compound [U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U] DNYWZCXLKNTFFI-UHFFFAOYSA-N 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 1
- 235000008979 vitamin B4 Nutrition 0.000 description 1
- 235000021119 whey protein Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/45—Non condensed piperidines, e.g. piperocaine having oxo groups directly attached to the heterocyclic ring, e.g. cycloheximide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Pulmonology (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Cardiology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
- Pyridine Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
La invención se refiere a nuevos compuestos que tienen actividad inhibidora de TRPC6 y sus sales farmacéuticamente aceptables. La invención también se refiere a composiciones farmacéuticas que comprenden estos compuestos, usando estos compuestos en el tratamiento de varias enfermedades y trastornos, procesos para preparar estos compuestos e intermedios útiles en estos procesos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de piridina y usos terapéuticos de los mismos como inhibidores de TRPC6
Campo de la invención
La presente invención se refiere a compuestos y composiciones farmacéuticos, y a su uso en métodos para el tratamiento de estados cardiacos y respiratorios, enfermedad renal, enfermedad hepática, distrofia muscular, trastornos fibróticos, dolor, isquemia o lesión por reperfusión isquémica, y cáncer, así como para la inhibición del canal iónico receptor de potencial transitorio C6 (TRPC6).
Antecedentes
Existen una variedad de proteínas de canales iónicos para mediar en el flujo de iones a través de las membranas celulares. La expresión y la función apropiadas de las proteínas de canales iónicos son esenciales para el mantenimiento de la función celular, la comunicación intracelular, y similares. Un aspecto importante para lograr la homeostasis celular es el mantenimiento de concentraciones de iones apropiadas en diversos tipos de células durante el desarrollo y en respuesta a numerosos estímulos. Un gran número de diversos tipos de canales iónicos actúan para mantener la homeostasis celular moviendo los iones al interior y fuera de las células a través de la membrana plasmática, y moviendo, dentro de las células, los iones a través de las membranas de orgánulos intracelulares incluyendo, por ejemplo, el retículo endoplásmico, el retículo sarcoplásmico, las mitocondrias y los orgánulos endocíticos incluyendo los endosomas y los lisosomas. Numerosas enfermedades son el resultado de la desregulación del potencial de membrana o de la manipulación aberrante del calcio. Dada la importancia central de los canales iónicos en la modulación del potencial de membrana y del flujo de iones en las células, la identificación de agentes que puedan fomentar o inhibir canales iónicos particulares es de gran interés como herramientas de investigación y como posibles agentes terapéuticos.
Uno de tales canales es el canal receptor de potencial transitorio C6 (TRPC6). El TRPC6 pertenece a la familia más grande de canales iónicos TRP (véase, Desai et al., 2005 Eur J Physiol 451:11-18; Clapham et al., 2001 Nat Neurosci 2:387-396; Clapham, 2003 Nature 426: 517-524; Clapham et al., 2002 IUPHAR Compendium). El TRPC6 es un canal permeable al calcio, específicamente un canal catiónico permeable al calcio no selectivo. Además de los iones de calcio, los canales TRPC6 son permeables a otros cationes, por ejemplo, al sodio. Por tanto, los canales TRPC6 modulan no sólo la concentración de calcio intracelular, sino también el potencial de membrana, modulando el flujo de cationes incluyendo los iones de calcio y sodio. Aunque los canales catiónicos no selectivos tales como TRPC6 modulan, entre otras cosas, el flujo de iones de calcio, son mecanísticamente distintos de los canales de calcio dependientes del voltaje. Generalmente, los canales de calcio dependientes del voltaje responden a la despolarización de la diferencia de potencial a través de la membrana y pueden abrirse para permitir un flujo de entrada de calcio desde el medio extracelular y un rápido aumento en los niveles o las concentraciones de calcio intracelular. En cambio, los canales catiónicos no selectivos tales como TRPC6 dependen generalmente de la transducción de señales, son de larga duración y producen cambios menos rápidos en la concentración de iones. Muestran una actividad aumentada en respuesta a la producción del segundo mensajero, el diacilglicerol (Hofmann et al., 1999). Además, el TRPC6 puede responder a cambios en la presión. Estas diferencias mecanísticas están acompañadas por diferencias estructurales entre los canales dependientes del voltaje y los canales permeables a cationes. Por tanto, aunque muchos canales diversos actúan para regular el flujo de iones y el potencial de membrana en diversos tipos de células y en respuesta a numerosos estímulos, es importante reconocer las diferencias estructurales, funcionales y mecanísticas significativas entre las diferentes clases de canales iónicos.
La función de TRPC6 se ha implicado en, entre otras cosas, la modulación del tono miogénico. El TRPC6 presenta una alta expresión en células de músculo liso, células de músculo liso vascular, miocardiocitos, arterias pulmonares, la aorta, el corazón, el hígado, el cerebro y el riñón. La expresión de TRPC6, junto con experimentos llevados a cabo en ratones con genes inactivados y células en cultivo, sugieren que el TRPC6 puede proporcionar una diana útil para el tratamiento de hipertensión y otros estados cardiacos y vasculares, preeclampsia.
La mutación en el canal TRPC6 humano puede provocar glomeruloesclerosis focal y segmentaria (GEFS) (Winn et al., 2005, Reiser et al., 2005). Estas mutaciones, que se notifica que son de ganancia de función (Reiser et al., 2005), son suficientes para inducir la enfermedad. Además, la expresión elevada de TRPC6 se ha asociado con el síndrome nefrótico, la enfermedad de cambios mínimos y la nefropatía diabética (Moller et al., 2006, Ilatovskaya et al., 2013, Thilo et al., 2011)., u otros estados renales.
Basándose en su expresión y en el trabajo que lo implica en la señalización de TGF-B, también se piensa que el TRPC6 es importante en estados respiratorios, reestenosis, enfermedad hepática, distrofia muscular, trastornos fibróticos, dolor, isquemia y lesión por reperfusión isquémica, y determinadas formas de cáncer.
Yue et al. estudiaron los canales TRPC6 por su papel en la mediación de la proliferación de células de músculo liso de la arteria pulmonar, lo que puede conducir a hipertensión arterial pulmonar idiopática (HAPI). La hipertrofia medial vascular pulmonar provocada por un exceso de proliferación de células de músculo liso de la arteria pulmonar
(PASMC) es una causa principal de la resistencia vascular pulmonar elevada en pacientes con HAPI. Los autores hallaron que la expresión del TRPC6 era alta y que la expresión del TRPC3 era mínima en PASMC de tejido pulmonar sano. Sin embargo, en tejido pulmonar de pacientes con HAPI, la expresión de ARNm y proteínas de TRPC3 y TRPC6 se elevó significativamente en comparación con la de pacientes normotensos. Además, la proliferación de células PASMC derivadas de pacientes con HAPI se redujo notablemente tras la incubación con ARNip de TRPC6. Basándose en estos resultados, los autores concluyeron que el TRPC6 puede ser importante en la mediación de la proliferación de PASMC apropiada, y que la desregulación de TRPC6 puede conducir a una proliferación de PASMC aumentada y a hipertrofia medial vascular pulmonar observada en pacientes con HAPI (Yu et al., 2004 Proc Natl Acad Sci 101(38):13861-6). Se proporciona un respaldo adicional mediante la observación de que, en pacientes con HAPI, la frecuencia de un polimorfismo de un solo nucleótido en el promotor de TRPC6 que aumenta la expresión era significativamente mayor en comparación con sujetos normales (Yue, et al., 2009 Circulation 119: 2313-22).
Evidencias adicionales que implican la desregulación TRPC6 en HAPI provienen de estudios del bosentán, un bloqueador del receptor de endotelina dual que se ha usado clínicamente para tratar la HAPI. Este inhibidor disminuye la proliferación de PASMC, pero el mecanismo por el cual se produce no está claro. Curiosamente, el bosentán disminuye tanto la proliferación de PASMC como la expresión de TRPC6 en tejido pulmonar de pacientes con HAPI (Kunichika et al., 2004 Am J Respir Crit Care Med 170(10):1101-7).
La exposición prolongada al humo de cigarrillos (CS) en ratas dio como resultado un aumento en la expresión de ARNm y proteínas de TRPC6 en arterias pulmonares distales y se observaron efectos similares usando PASMC in vitro. El tratamiento con nicotina de PASMC de rata cultivadas reguló por incremento la expresión de TRPC6 y aumentó los niveles de calcio intracelular, ambos de los cuales se redujeron por el silenciamiento del ARNip de TRPC6 (Wang et al., 2014 Am J Physiol Cell Physiol 306:C364-73). Estos resultados sugieren un papel para el TRPC6 en la lesión pulmonar inducida por CS.
Las evidencias respaldan el papel del TRPC6 en trastornos pulmonares adicionales. En macrófagos alveolares de pacientes con enfermedad pulmonar obstructiva crónica (EPOC), se halló que la expresión de TRPC6 se elevaba en comparación con controles (Finney-Hayward et al., 2010 Am J Respir Cell Mol Biol 43:296-304). En células epiteliales con fibrosis quística humanas, el flujo de entrada de calcio mediado por TRPC6 aumenta de manera anómala y puede contribuir a la hipersecreción de moco. El ARNip-TRPC6 pudo reducir este flujo de entrada de calcio anómalo (Antigny et al. 2011 Am J Resp Cell Mol Biol, 44:83-90). En fibroblastos pulmonares de ratón, la actividad profibrótica de PDg F depende de la activación de TRPC6, lo que sugiere que la inhibición de TRPC6 reduciría la fibrosis pulmonar (Lei et al., 2014 Biomaterials 35:2868-77). Se demostró un papel del TRPC6 en la función de células endoteliales pulmonares en modelos de pulmón de ratón de edema inducido por isquemia-reperfusión e inflamación inducida por lipopolisacáridos en los que la deficiencia en TRPC6 pudo reducir la lesión pulmonar aguda conservando la función de barrera endotelial (Weissmann et al., 2011 Nat Comm, 3:649-58 y Tauseef et al., 2012 J Exp Med 209:1953-68).
Estudios recientes también implican el papel del TRPC6 en otros estados cardiacos, incluyendo hipertrofia cardiaca. Los corazones de pacientes con miocardiopatía dilatada tienen una expresión de ARNm de TRPC6 elevada en comparación con corazones normales (Kuwahara et al., 2006 J Clin Invest 116:3114-26). En modelos de ratón de hipertrofia cardiaca, los niveles de ARNm cardiaco de TRPC6 se elevan por sobrecarga de presión (Kuwahara et al., 2006 J Clin Invest 116:3114-26), tratamiento prolongado con isoproterenol (Xie et al., 2012 Nat Commun 3:1238) y miocardiopatía urémica inducida por nefrectomía parcial (Xie et al., 2015 J Am Soc Nephrol 26:1150-60). Además, la sobreexpresión específica del corazón de TRPC6 en los miocardiocitos de ratones transgénicos indujo hipertrofia cardiaca y muerte prematura (Kuwahara et al., 2006 J Clin Invest 116:3114-26).
Wu y colaboradores hallaron que los ratones transgénicos que expresan TRPC6 negativo dominante de manera específica del corazón tenían una respuesta hipertrófica cardiaca atenuada o bien tras una infusión de agonista neuroendocrino o bien tras una simulación de sobrecarga de presión, lo que indica que el TRPC6 es un componente de los complejos de canales que son mediadores esenciales de la hipertrofia (Wu et al., 2010 Proc Natl Acad Sci.
107:7000-05). Recientemente también han comenzado a mostrarse que los fármacos de molécula pequeña que seleccionan como diana TRPC6 son prometedores en el tratamiento de estados cardiacos. Por ejemplo, Seo y colaboradores demostraron que los antagonistas de TRPC6 y TRPC3 (GSK2332255B y GSK833503A) presentaban una inhibición dependiente de la dosis de la señalización de hipertrofia celular en miocardiocitos de neonatales y adultos (Seo et al., 2014 Proc Natl Acad Sci 111:1551-1556). De manera similar, los ratones deficientes en TRPC6 estaban protegidos frente a hipertrofia cardiaca inducida por isoproterenol (Xie et al., 2012 Nat Commun 3:1238).
La reducción de la actividad de TRPC6 puede ser beneficiosa para el tratamiento de enfermedad cardiovascular. In vitro, la tensión de cizalladura de la ateroprona induce niveles de ARNm de TRPC6 aumentados en células endoteliales (EC) vasculares humanas en comparación con condiciones de flujo ateroprotectoras (Thilo, et al., 2012 Hypertension 59:1232-40). La migración de EC es importante para la curación después de una lesión arterial, y se previno in vitro la inhibición mediada por lisofosfatidilcolina de la migración de EC en células de ratones deficientes en TRPC6. Además, una dieta de alto contenido en colesterol combinada con una lesión de la carótida no afectó a la curación en ratones deficientes en TRPC6 en comparación con controles de tipo natural (Rosembaum et al., 2015 J Vasc Surg 62:1040-47 y Chaudhuri et al., 2008 Mol Biol Cell 19: 3203-11). De manera similar, la lesión inducida por dilatación con globo de arterias mamarias internas humanas ex vivo dio como resultado niveles de ARNm de TRPC6 aumentados en
comparación con arterias no dilatadas (Bergdahl et al., 2005 Am J Physiol Cell Physiol 288:C872-80). La apoptosis de células endoteliales está implicada en el inicio y la progresión de lesiones ateroescleróticas, y se demostró que la apoptosis inducida por lipoproteínas de baja densidad oxidadas de EC aórticas humanas dependía de TRPC6 (Zhang et al., 2015 Sci Rep 5:9401-10). En un modelo de rata de isquemia prosencefálica, los niveles de ARNm de TRPC6 se aumentaron en SMC vasculares y se correlacionaron con un flujo sanguíneo cerebral reducido (Johannson et al., 2015 Acta Physiol 214:376-89).
Los estudios de Reiser, Winn y Schlondorff identificaron que las mutaciones en TRPC6 en pacientes son las causantes de la GEFS (Reiser et al., 2005 Nature Genet 37:739-744; Winn et al., 2005 Science 308:1801-1804; Schlondorff et al., 2009 Am J Physiol Cell Physiol 296:C558-69). Estudios posteriores identificaron mutaciones de TRPC6 adicionales asociadas con el síndrome nefrótico resistente a los esteroides (C. Sadowski et al., 2014 J Am Soc Nephrol 26:1279-89). Estudios adicionales demostraron que el TRPC6 es importante en la función normal de podocitos controlando el flujo de entrada de calcio y el factor nuclear de activación de células T activadas en el que la corriente elevada a través del canal está asociada con la lesión renal y la inducción de proteinuria (Moller et al., 2007 J Am Soc Nephrol 18:29-36 y Schlondorff et al., 2009 Am J Physiol Cell Physiol 296:C558-69). Además de las mutaciones de ganancia de función, se ha demostrado que la expresión de TRPC6 es elevada en enfermedades renales crónicas humanas incluyendo GEFS, enfermedad de cambios mínimos, glomerulonefritis membranosa y nefropatía diabética (Moller et al., 2007 J Am Soc Nephrol 18:29-36 y Thilo et al., 2011 Nephrol. Dial. Transplant 27:921-9), así como en modelos de ratón de lesión de podocitos (Moller et al., 2007 J Am Soc Nephrol 18:29-36). Se ha demostrado que los ratones deficientes en TRPC6 tienen albuminuria inducida por angiotensina II (Ang II) reducida (Eckel et al., 2011 J Am Soc Nephrol 22:526-35), mientras que la expresión específica de podocitos transgénicos de mutaciones de ganancia de función (GoF) humanas en ratones induce albuminuria y lesiones glomerulares (Krall et al., 2010 PLoS ONE e12859 y Canales et al., 2015 Brit J Medicine Med Res 5:1198-1212). En consecuencia, la inhibición de TRPC6 puede ser útil en el tratamiento de enfermedades renales crónicas. Estos hallazgos no sólo sugieren que el TRPC6 funciona normalmente para mantener una función renal apropiada, sino que también implica al TRPC6 como causa específica de al menos determinados casos de GEFS. Basándose en el posible papel del TRPC6 en la función renal, pueden usarse compuestos inhibidores de TRPC6 en el tratamiento o la mejora de enfermedades o estados renales crónicos provocados (en su totalidad o en parte) por la disfunción de TRPC6. Además, pueden usarse compuestos inhibidores de TRPC6 en el tratamiento o la mejora de los síntomas de enfermedades renales (por ejemplo, hipertensión, proteinuria, etc.), independientemente de la causa de la enfermedad.
El TRPC6 se expresa en el miometrio y en la placenta durante el embarazo (Ku et al., 2006 J Soc Gynecol Investig 13:217-225; Clarson et al., 2003 J Physiol 550:515-528). Como tal, el TRPc 6 puede contribuir al mantenimiento del tono miogénico apropiado en la placenta y/o al mantenimiento de la tensión arterial fetal y materna apropiada durante el embarazo.
Han surgido evidencias recientes que implican al TRPC6 en determinadas formas de cáncer. Varios grupos han establecido que la expresión de TRPC6 es elevada en células tomadas de pacientes con glioblastoma multiforme, el tipo más frecuente e incurable del cáncer de cerebro (Chigurupati, et al., 2010 Cancer Res, 70:418-427; Ding et al., 2010 J Natl Cancer Inst. 102:1052-1068). De manera similar, Ding et al. hallaron niveles elevados de TRPC6 en células de glioma humano, y la inhibición de TRPC6, farmacológicamente o con un mutante negativo dominante, suprimió el crecimiento celular in vitro. En dos modelos de xenoinjerto de gliomas humanos, la expresión mediada por lentivirus de TRPC6 negativo dominante en las células tumorales antes de la implantación subcutánea o intracraneal redujo el volumen tumoral en comparación con controles (Ding et al., J. Natl. Cancer Inst. 2010, 102, 1052-1068). También se halló que los niveles aumentados de TRPC6 estaban asociados con el cáncer de cuello uterino (Wan et al., 2012 Onco Targets Ther 5:171-176), el cáncer de mama (Dhennin-Duthille et al., 2011 Cell Physiol Biochem 28:813-822), el carcinoma de células renales (Song et al., 2013 Mol Biol Rep 40:5115-5122), el carcinoma epidermoide de cabeza y cuello (de Quiros, et al. 2013 BMC Cancer 13:116-127) y el carcinoma epidermoide de esófago (Zhang et al., 2013 Med Oncol 30:607), entre otros. En células de carcinoma hepatocelular, se demostró que la doxorrubicina, la hipoxia y la radiación ionizante aumentaron la expresión de ARNm de TRPC6, y que el TRPC6 se encuentra a niveles más altos en tejidos tumorales que en los tejidos no implicados. El TRPC6 elevado estaba asociado con la farmacorresistencia, que se disminuyó por el silenciamiento del ARN de TRPC6 in vitro. La administración mediante lentivirus de ARN de horquilla corta específico de TRPC6 a células tumorales Huh7 antes de la implantación en un modelo de xenoinjerto subcutáneo de ratón redujo el crecimiento tumoral y sensibilizó los tumores a la doxorrubicina (Wen et al., 2016 Sci Rep 6:23269). Estos hallazgos sugieren que el TRPC6 puede ser una diana terapéutica prometedora para el tratamiento del cáncer.
Las enfermedades hepáticas, incluyendo la esteatohepatitis no alcohólica, pueden tratarse reduciendo la actividad de TRPC6. La hipoxia aumentó la expresión de TRPC6 en una línea de células estrelladas hepáticas humanas en comparación con condiciones normóxicas. Usando estas células, el silenciamiento del ARN de TRPC6 reguló por disminución los transcritos para actina de músculo liso alfa y colágeno 1A1, ambos de los cuales están asociados con la fibrosis, en respuesta a la hipoxia (Iyer et al., 2015 Exp Cell Res 336:66-75).
La inhibición de TRPC6 puede proporcionar un beneficio a los pacientes con distrofia muscular de Duchenne (DMD). En el modelo mdx/utrn+/' de DMD usando miocardiocitos aislados, la deficiencia en TRPC6 restableció a la normalidad la fuerza de contractilidad estimulada por tensión y la respuesta transitoria al calcio en comparación con ratones que
poseen el gen TRPC6 de tipo natural, lo que sugiere que la inhibición de TRPC6 conservará la función cardiaca en pacientes con DMD (Seo et al., 2014 Circ Res 114:823-32).
Los trastornos fibróticos pueden tratarse con inhibidores de TRPC6. La sobreexpresión de TRPC6 indujo la activación de miofibroblastos, mientras que la deleción de TRPC6 redujo la transformación de miofibroblastos inducida por el factor de crecimiento transformante beta. Además, los ratones deficientes en TRPC6 demostraron una cicatrización dérmica y cardiaca reducida (Davis et al., 2012 Dev Cell 23:705-15).
Los inhibidores de TRPC6 pueden ser útiles para el tratamiento del dolor. La administración espinal de oligonucleótidos antisentido de TRPC6 redujo la hiperalgesia inducida por estímulos mecánicos, hipotónicos y térmicos en modelos de dolor preclínicos (Alessandri-Haber et al., 2009 J Neurosci 29:6217-28).
Modular una función de TRPC6 proporciona un medio para modular la homeostasis del calcio, la homeostasis del sodio, los niveles de calcio intracelular, la polarización de membrana (potencial de membrana en reposo) y/o los niveles catiónicos en una célula. Los compuestos que pueden modular una o más de las funciones de TRPC6 son útiles en muchos aspectos, incluyendo, pero sin limitarse a, el mantenimiento de la homeostasis del calcio; el mantenimiento de la homeostasis del sodio; la modulación de los niveles de calcio intracelular; la modulación de la polarización de membrana (potencial de membrana); la modulación de los niveles catiónicos; y/o el tratamiento o la prevención de enfermedades, trastornos o estados asociados con la homeostasis del calcio, la homeostasis del sodio, la dishomeostasis del calcio o del sodio, o la polarización/hiperpolarización de membrana (incluyendo hipoexcitabilidad e hiperexcitabilidad), y/o el tratamiento o la prevención de enfermedades, trastornos o estados asociados con la regulación o desregulación de la expresión o función de TRPC6.
El documento EP 1396487 A1 divulga derivados de fenilpiridincarbonilpiperazina como inhibidores de fosfodiesterasa de tipo 4 (PDE4) para su uso en el tratamiento de asma y EPOC.
El documento WO 2016/138114 divulga derivados de piperazina como inhibidores de BRG1 (gen 1 relacionado con Brahma), BRM (modificadores de la respuesta biológica) y/o PB1 (proteína polibromo-1) para su uso en el tratamiento del cáncer.
El documento WO 2014/191336 divulga derivados de 3,4-dihidro-2H-isoquinolin-1-ona y 2,3-dihidro-2H-isoindol-1-ona como inhibidores de aldosterona sintasa para su uso en el tratamiento de enfermedad renal crónica, insuficiencia cardiaca congestiva, hipertensión, aldosteronismo primario y síndrome de Cushing.
Existe la necesidad de antagonistas de TRPC6 altamente selectivos para tratar enfermedades o trastornos que pueden aliviarse mediante la modulación de TRPC6.
Breve sumario de la invención
La presente invención proporciona nuevos compuestos que modulan TRPC6 y, por tanto, son útiles para tratar una variedad de enfermedades y trastornos que pueden aliviarse mediante la modulación de TRPC6, incluyendo hipertensión, preeclampsia, reestenosis, un estado cardiaco o respiratorio, enfermedad renal, enfermedad hepática, distrofia muscular, trastornos fibróticos, dolor, isquemia o lesión por reperfusión isquémica, y cáncer. Esta invención también se refiere a composiciones farmacéuticas que comprenden estos compuestos, al uso de estos compuestos para el tratamiento de diversas enfermedades y trastornos, a procedimientos para preparar estos compuestos y a productos intermedios útiles en estos procedimientos.
La invención se refiere a las siguientes realizaciones:
Un compuesto seleccionado del grupo que consiste en:
[4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona,
[4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-(4-metoxi-5-fenoxi-piridin-2-il)-metanona,
[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona,
[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-il]-metanona,
[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-il]-metanona,
[4-(6-amino-4-etoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(fenoxi)-piridin-2-il]-metanona,
5- etoxi-6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)piridazin-3-amina, y
6- (1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)-5-metilpiridazin-3-amina.
Un compuesto, que es [4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-ilH5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona.
Un compuesto, que es [4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-(4-metoxi-5-fenoxi-piridin-2-il)-metanona. Un compuesto, que es [4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona.
Un compuesto, que es [4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-il]-metanona.
Un compuesto, que es [[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-il]-metanona.
Un compuesto, que es [4-(6-amino-4-etoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(fenoxi)-piridin-2-il]-metanona. Un compuesto, que es 5-etoxi-6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)piridazin-3-amina.
Un compuesto, que es 6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)-5-metilpiridazin-3-amina.
Una sal farmacéuticamente aceptable de cualquiera de los compuestos anteriores.
Una composición farmacéutica que comprende uno cualquiera de los compuestos anteriores, o una sal farmacéuticamente aceptable de los mismos, y opcionalmente un excipiente farmacéuticamente aceptable.
Cualquiera de los compuestos anteriores, o una sal farmacéuticamente aceptable de los mismos, para su uso en el tratamiento de una enfermedad o un trastorno que puede aliviarse mediante la inhibición de TRPC6, en el que la enfermedad o el trastorno se selecciona del grupo que consiste en hipertrofia cardiaca, isquemia, lesión por reperfusión isquémica, hipertensión, hipertensión arterial pulmonar, hipertensión arterial pulmonar idiopática, reestenosis, enfermedad pulmonar obstructiva crónica, fibrosis quística, enfermedad de Alzheimer, enfermedad de Parkinson, enfermedad de Huntington, esclerosis lateral amiotrófica (ELA), trastornos encefálicos inducidos por traumatismo, asma, enfermedad pulmonar obstructiva crónica, artritis reumatoide, artrosis, enfermedad intestinal inflamatoria, esclerosis múltiple, distrofia muscular, distrofia muscular de Duchenne, preeclampsia e hipertensión inducida por el embarazo, esteatohepatitis no alcohólica, enfermedad de cambios mínimos, glomeruloesclerosis focal y segmentaria (GEFS), síndrome nefrótico, nefropatía diabética o enfermedad renal diabética (ERD), enfermedad renal crónica, insuficiencia renal, enfermedad renal de fase terminal, isquemia o una lesión por reperfusión isquémica, cáncer, FPI (fibrosis pulmonar idiopática), SDRA (síndrome de dificultad respiratoria aguda), enfisema y diabetes.
Descripción detallada de la invención
La tabla 1 muestra los compuestos de la invención que pueden prepararse mediante los esquemas de síntesis y los ejemplos mostrados en la sección de ejemplos de síntesis a continuación, y métodos conocidos en la técnica.
Tabla 1.


En una realización, la invención se refiere a cualquiera de los compuestos representados en la tabla 1 anterior, y a las sales farmacéuticamente aceptables de los mismos.
DEFINICIONES GENERALES
A los términos no definidos específicamente en el presente documento deben dárseles los significados que les daría un experto en la técnica a la luz de la divulgación y del contexto. Sin embargo, tal como se usa en la memoria descriptiva, a menos que se especifique lo contrario, los siguientes términos tienen el significado indicado y se cumplen las siguientes convenciones.
En caso de que un compuesto de la presente invención se represente en forma de un nombre químico y como fórmula, en caso de cualquier discrepancia prevalecerá la fórmula.
A menos que se indique específicamente, a lo largo de la memoria descriptiva y de las reivindicaciones adjuntas, un nombre o una fórmula química dada debe abarcar los tautómeros y todos los esteroisómeros, isómeros ópticos e isómeros geométricos (por ejemplo, enantiómeros, diastereómeros, isómeros E/Z etc.) y racematos de los mismos, así como mezclas en diferentes proporciones de los enantiómeros independientes, mezclas de diastereómeros o mezclas de cualquiera de las formas anteriores en las que existen tales isómeros y enantiómeros, así como sales, incluyendo sales farmacéuticamente aceptables de los mismos y solvatos de los mismos tales como, por ejemplo, hidratos incluyendo solvatos de los compuestos libres o solvatos de una sal del compuesto.
Pueden prepararse compuestos enantioméricamente puros de esta invención o productos intermedios a través de síntesis asimétrica, por ejemplo, mediante la preparación y la posterior separación de compuestos o productos intermedios diastereoméricos apropiados que pueden separarse mediante métodos conocidos (por ejemplo, mediante
separación cromatográfica o cristalización) y/o mediante el uso de reactivos quirales, tales como materiales de partida quirales, catalizadores quirales o compuestos auxiliares quirales.
Además, el experto en la técnica sabe cómo preparar compuestos enantioméricamente puros a partir de las mezclas racémicas correspondientes, tales como mediante separación cromatográfica de las mezclas racémicas correspondientes sobre fases estacionarias quirales; o mediante resolución de una mezcla racémica usando un agente de resolución apropiado, por ejemplo, por medio de la formación de sales diastereoméricas del compuesto racémico con ácidos o bases ópticamente activos, la posterior resolución de las sales y la liberación del compuesto deseado a partir de la sal; o mediante la derivatización de los compuestos racémicos correspondientes con reactivos auxiliares quirales ópticamente activos, la posterior separación de diastereómeros y la retirada del grupo auxiliar quiral; o mediante la resolución cinética de un racemato (por ejemplo, mediante resolución enzimática); mediante la cristalización enantioselectiva a partir de un conglomerado de cristales enantiomorfos en condiciones adecuadas; o mediante la cristalización (fraccionada) a partir de un disolvente adecuado en presencia de un compuesto auxiliar quiral ópticamente activo.
La expresión “farmacéuticamente aceptable” se emplea en el presente documento para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que, dentro del alcance del juicio médico razonable, son adecuados para su uso en contacto con los tejidos de seres humanos y animales sin toxicidad, irritación, respuesta alérgica u otro problema o complicación excesivos, y compatibles con una relación beneficio/riesgo razonable. Tal como se usa en el presente documento, “sal farmacéuticamente aceptable” se refiere a derivados de los compuestos divulgados en los que el compuesto original se modifica preparando sales de ácido o de base del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales de ácidos orgánicos o minerales de residuos básicos tales como aminas; sales alcalinas u orgánicas de residuos ácidos tales como ácidos carboxílicos; y similares.
Por ejemplo, tales sales incluyen sales de ácido bencenosulfónico, ácido benzoico, ácido cítrico, ácido etanosulfónico, ácido fórmico, ácido fumárico, ácido gentísico, ácido bromhídrico, ácido clorhídrico, ácido maleico, ácido málico, ácido malónico, ácido mandélico, ácido metanosulfónico, ácido 4-metilbencenosulfónico, ácido fosfórico, ácido salicílico, ácido succínico, ácido sulfúrico, ácido tartárico y ácido trifluoroacético.
Pueden formarse sales farmacéuticamente aceptables adicionales con cationes de amoniaco, L-arginina, calcio, 2,2'-iminobisetanol, L-lisina, magnesio, A/-metil-D-glucamina, potasio, sodio y tris(hidroximetil)-aminometano.
Las sales farmacéuticamente aceptables de la presente invención pueden sintetizarse a partir del compuesto original que contiene un resto básico o ácido mediante métodos químicos convencionales. Generalmente, tales sales pueden prepararse haciendo reaccionar las formas de ácido o de base libre de estos compuestos con una cantidad suficiente de la base o el ácido apropiado en agua o en un diluyente orgánico tal como éter, acetato de etilo, etanol, isopropanol o acetonitrilo, o una mezcla de los mismos.
Sales de otros ácidos distintos de los mencionados anteriormente que, por ejemplo, son útiles para purificar o aislar los compuestos de la presente invención (por ejemplo, sales de trifluoroacetato, formiatos) también forman parte de la invención.
Muchos de los términos proporcionados anteriormente pueden usarse repetidamente en la definición de una fórmula o un grupo y, en cada caso, tienen uno de los significados proporcionados anteriormente, independientemente unos de otros.
La presente solicitud proporciona compuestos que pueden modular la función de TRPC6. También se proporcionan métodos que emplean estos compuestos. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en la modulación de una función de TRPC6 en una célula o un animal, en la que el compuesto inhibe un flujo de iones mediado por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en la modulación de una función de TRPC6 en una célula o un animal, en la que el compuesto inhibe un flujo de entrada de calcio mediado por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en la modulación de una función de TRPC6 en una célula o un animal, en la que el compuesto inhibe una reorganización citoesquelética mediada por TRPC6 o una alteración en la morfología celular. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de modulación de una función de TRPC6 en una célula, en la que el compuesto inhibe la corriente saliente mediada por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de modulación de una función de TRPC6 en una célula, en la que el compuesto inhibe la corriente entrante mediada por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de modulación de una función de TRPC6 en una célula, en la que el compuesto inhibe las corrientes tanto entrante como saliente mediadas por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de modulación de una función de TRPC6 en una célula, en la que el compuesto inhibe los aumentos mediados por TRPC6 en la concentración de calcio intracelular. Determinadas
realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de modulación de una función de TRPC6 en una célula, en la que el compuesto inhibe alteraciones en la morfología celular. Determinadas realizaciones también proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de prevención o tratamiento de una enfermedad o un estado relacionado con la función de TRPC6 en un sujeto, en la que el compuesto inhibe la corriente entrante mediada por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de prevención o tratamiento de una enfermedad o un estado relacionado con la función de TRPC6 en un sujeto, en la que el compuesto inhibe la corriente saliente mediada por TRPC6. Determinadas realizaciones también proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de prevención o tratamiento de una enfermedad o un estado relacionado con la función de TRPC6 en un sujeto, en la que el compuesto inhibe las corrientes tanto entrante como saliente mediadas por TRPC6. Determinadas realizaciones proporcionan una cantidad eficaz de un compuesto que inhibe una función de TRPC6 para su uso en un método de prevención o tratamiento de una enfermedad o un estado relacionado con la función de TRPC6 en un sujeto, en la que el compuesto inhibe el flujo de iones mediado por TRPC6. Obsérvese que la inhibición de una corriente particular se refiere a la capacidad de un compuesto para inhibir esa corriente (por ejemplo, entrante y/o saliente) en un ensayo o bien in vitro o bien in vivo. La inhibición de una corriente particular en un ensayo o bien in vivo o bien in vitro sirve como indicador para la actividad funcional particular del compuesto particular.
La presente invención proporciona una cantidad eficaz de un compuesto de la invención para su uso en métodos de tratamiento de un trastorno mediado por TRPC6 en un sujeto, en la que cada una de las variables anteriores se describen en el presente documento, por ejemplo, la descripción detallada a continuación.
La presente invención proporciona además una composición que comprende un compuesto de la invención y un excipiente, diluyente o portador farmacéuticamente aceptable para su uso en un método para tratar un trastorno mediado por TRPC6 en un sujeto.
La presente invención proporciona además una composición que comprende un compuesto de la invención y un excipiente, diluyente o portador farmacéuticamente aceptable para su uso en un método para tratar un trastorno mediado por TRPC6 en un sujeto, en la que el trastorno mediado por TRPC6 se selecciona del grupo que consiste en hipertrofia cardiaca, isquemia, lesión por reperfusión isquémica, hipertensión, hipertensión arterial pulmonar, hipertensión arterial pulmonar idiopática, reestenosis, enfermedad pulmonar obstructiva crónica, fibrosis quística, enfermedad de Alzheimer, enfermedad de Parkinson, enfermedad de Huntington, esclerosis lateral amiotrófica (ELA), trastornos encefálicos inducidos por traumatismo, asma, enfermedad pulmonar obstructiva crónica, artritis reumatoide, artrosis, enfermedad intestinal inflamatoria, esclerosis múltiple, distrofia muscular, preeclampsia e hipertensión inducida por el embarazo, esteatohepatitis no alcohólica, glomeruloesclerosis focal y segmentaria, síndrome nefrótico, nefropatía diabética o enfermedad renal diabética, insuficiencia renal, enfermedad renal de fase terminal, isquemia o una lesión por reperfusión isquémica, cáncer, FPI (fibrosis pulmonar idiopática), SDRA (síndrome de dificultad respiratoria aguda), enfisema y diabetes.
A menos que se indique específicamente, a lo largo de la memoria descriptiva y de las reivindicaciones adjuntas, un nombre o una fórmula química dada debe abarcar los tautómeros y todos los esteroisómeros, isómeros ópticos e isómeros geométricos (por ejemplo, enantiómeros, diastereómeros, isómeros E/Z etc.) y racematos de los mismos, así como mezclas en diferentes proporciones de los enantiómeros independientes, mezclas de diastereómeros o mezclas de cualquiera de las formas anteriores en las que existen tales isómeros y enantiómeros, así como sales, incluyendo sales farmacéuticamente aceptables de los mismos y solvatos de los mismos tales como, por ejemplo, hidratos incluyendo solvatos de los compuestos libres o solvatos de una sal del compuesto.
Algunos de los compuestos en la tabla 1 pueden existir en más de una forma tautomérica. La invención incluye métodos para usar todos de tales tautómeros.
La invención incluye derivados farmacéuticamente aceptables de los compuestos de la invención. Un “derivado farmacéuticamente aceptable” se refiere a cualquier éster o sal farmacéuticamente aceptable, o cualquier otro compuesto que, tras la administración a un paciente, es capaz de proporcionar (directa o indirectamente) un compuesto útil para la invención, o un metabolito farmacológicamente activo o residuo farmacológicamente activo del mismo. Debe entenderse que un metabolito farmacológicamente activo significa cualquier compuesto de la invención que puede ser metabolizado enzimática o químicamente. Este incluye, por ejemplo, compuestos de derivados hidroxilados u oxidados de la invención.
Tal como se usa en el presente documento, “sales farmacéuticamente aceptables” se refieren a derivados de los compuestos divulgados en los que el compuesto original se modifica preparando sales de ácido o de base del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales de ácidos orgánicos o minerales de residuos básicos tales como aminas; sales alcalinas u orgánicas de residuos ácidos tales como ácidos carboxílicos; y similares. Por ejemplo, tales sales incluyen acetatos, ascorbatos, bencenosulfonatos, benzoatos, besilatos, bicarbonatos, bitartratos, bromuros/bromhidratos, edetatos, camsilatos, carbonatos, cloruros/clorhidratos, citratos, edisilatos, etanodisulfonatos, estolatos, esilatos, formiatos, fumaratos, gluceptatos, gluconatos, glutamatos, glicolatos, glicolilarsenilatos, hexilresorcinatos, hidrabaminas, hidroximaleatos, hidroxinaftoatos, yoduros, isotionatos,
lactatos, lactobionatos, malatos, maleatos, mandelatos, metanosulfonatos, metilbromuros, metilnitratos, metilsulfatos, mucatos, napsilatos, nitratos, oxalatos, pamoatos, pantotenatos, fenilacetatos, fosfatos/difosfatos, poligalacturonatos, propionatos, salicilatos, estearatos, subacetatos, succinatos, sulfamidas, sulfatos, tanatos, tartratos, teoclatos, toluenosulfonatos, trietioduros, trifluoroacetatos, amonio, benzatinas, cloroprocaínas, colinas, dietanolaminas, etilendiaminas, megluminas y procaínas. Pueden formarse sales farmacéuticamente aceptables adicionales con cationes de metales tales como aluminio, calcio, litio, magnesio, potasio, sodio, zinc, y similares. (Véase también Pharmaceutical salts, Birge, S.M. et al., J. Pharm. Sci., (1977), 66, 1-19).
Las sales farmacéuticamente aceptables de la presente invención pueden sintetizarse a partir del compuesto original que contiene un resto básico o ácido mediante métodos químicos convencionales. Generalmente, tales sales pueden prepararse haciendo reaccionar las formas de ácido o de base libre de estos compuestos con una cantidad suficiente de la base o el ácido apropiado en agua o en un diluyente orgánico tal como éter, acetato de etilo, etanol, isopropanol o acetonitrilo, o una mezcla de los mismos.
Sales de otros ácidos distintos de los mencionados anteriormente que, por ejemplo, son útiles para purificar o aislar los compuestos de la presente invención (por ejemplo, sales de trifluoroacetato) también forman parte de la invención.
Los compuestos de la invención también incluyen sus formas isotópicamente marcadas. Una forma isotópicamente marcada de un agente activo de una combinación de la presente invención es idéntica a dicho agente activo salvo por el hecho de que uno o más átomos de dicho agente activo se ha reemplazado por un átomo o átomos que tienen una masa atómica o un número másico diferente de la masa atómica o el número másico de dicho átomo que habitualmente se encuentra en la naturaleza. Los ejemplos de isótopos que están fácilmente comercialmente disponibles y que pueden incorporarse en un agente activo de una combinación de la presente invención según procedimientos bien establecidos incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, por ejemplo, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente. Se contempla activo de una combinación de la presente invención, o una sal farmacéuticamente aceptable del mismo, que contiene uno o más de los isótopos anteriormente mencionados y/u otros isótopos de otros átomos, está dentro del alcance de la presente invención.
Los compuestos de la invención son sólo aquellos que se contempla que son “químicamente estables” tal como apreciarán los expertos en la técnica. Por ejemplo, un compuesto que tendría una “valencia colgante”, o un “carbanión”, no son compuestos contemplados por los métodos inventivos divulgados en el presente documento.
Para todos los compuestos divulgados anteriormente en el presente documento en esta solicitud, en el caso de que la nomenclatura entre en conflicto con la estructura, debe entenderse que el compuesto se define por la estructura.
Lista de abreviaturas
AA ácido acético
ACN/MeCN acetonitrilo
ac. acuoso
BEH columna híbrida con puentes de etileno
BOC terc-butiloxicarbonilo
°C grados centígrados
CDI di(imidazol-1-il)metanona
metanosulfonato de CPhos-3G-paladaciclo metanosulfonato de (2-diciclohexilfosfino-2',6'-bis(dimetilamino)-1,1'-bifenil)(2'-amino-1,1'-bifenil-2-il)paladio(II)
DCM diclorometano
DIPEA W,W-diisopropiletilamina
DMF N,N-dimetilformamida
DMA N,N-dimetilacetamida
DMSO dimetilsulfóxido
DTAD azodicarboxilato de di-terc-butilo
EE dietil éter
eq equivalente
ESI-EM espectrometría de masas de ionización por electropulverización EtOH etanol
EtOAc/ EE acetato de etilo
h hora
H2 hidrógeno
H3PO4 ácido fosfórico
HATU hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uranio
HCI ácido clorhídrico
HPLC cromatografía de líquidos de alta resolución
MeOH metanol
min minuto
Mel yodometano
ml mililitro
EM espectro de masas
NaH hidruro de sodio
NaOH hidróxido de sodio
NMP N-metil-2-pirrolidinona
Pd2(dba)3 tris(dibencilidenoacetona)dipaladio(0)
Pd/C paladio sobre carbono
PdCl2(dppf)CH2Cl2 [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(N)diclorometano Pd(OH)2 hidróxido de paladio
PE éter de petróleo
RP fase inversa
ta oTA temperatura ambiente (aproximadamente 25°C)
CFS cromatografía de fluidos supercríticos
TBTU tetrafluoroborato de benzotriazoliltetrametiuronio
TFA ácido trifluoroacético
THF tetrahidrofurano
CCF cromatografía en capa fina sobre SiO2
Xantphos 4,5-bis(difenilfosfino)-9,9-dimetilxanteno
Xphos de 2a gen. cloro(2-diciclohexilfosfino-2',4',6'-triisopropiM,1 '-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(II)
TPP trifenilfosfina
Métodos generales: a menos que se indique lo contrario, todas las reacciones tienen lugar a temperatura ambiente (aproximadamente 25°C), bajo una atmósfera inerte (por ejemplo, argón, N2) y en condiciones anhidras. Todos los compuestos se caracterizan mediante al menos uno de los siguientes métodos: 1H-RMN, HPLC, EM, HPLC-EM o punto de fusión.
Normalmente, el avance de la reacción se monitoriza mediante cromatografía en capa fina (CCF) o HPLC-EM. Los productos intermedios y los productos se purifican usando al menos uno de los siguientes métodos:
Cromatografía ultrarrápida sobre gel de sílice, recristalización, HPLC quiral de fluidos supercríticos (CFS) usando una columna RegisPack de 3,0 x 25,0 cm, eluyendo con una mezcla isocrática de MeOH, isopropilamina (IPA) y dióxido de carbono supercrítico a 125 bar; 80 ml/min, y/o HPLC de fase inversa usando una columna semipreparativa C18 eluyendo con un gradiente de:
• MeCN el 0,1% de TFA y H2O el 0,1% de TFA,
• MeCN el 0,1% de ácido fórmico y H2O el 0,1% de ácido fórmico, o
• MeCN y H2O que contiene NH4HCO32,5 mM
• MeCN y H2O el 0,1% de TFA,
• MeCN y H2O el 0,1% de NH3,
• MeCN y H2O y el 0,1% de TFA
• MeCN y H2O y el 0,1% de NH3
DATOS ANALÍTICOS
Los datos de espectrometría de masas (EM) notificados corresponden a la masa observada (por ejemplo, [M+H]+). El método de HPLC usado para caracterizar los compuestos de la invención y los compuestos de referencia adicionalmente divulgados en el presente documento se describe en la tabla 2.
Tabla 2. Métodos de HPLC

Este método se utiliza en todas las demás tablas en esta sección para los datos de ESI-EM y de tiempo de retención. Si se usa una HPLC-EM diferente, se indica en el texto.
Método 1
Modo de ionización ESI+/-. Columna: CSH C182,1x50 mm, 1,7 |im de diámetro de partícula. Gradiente: del 90% de A al 100% de B en 1,19 minutos, mantener al 100% de B hasta 1,70 minutos. Velocidad de flujo de 0,8 ml/min. A=(el 95% de agua el 5% de acetonitrilo el 0,05% de ácido fórmico), B=(acetonitrilo el 0,05% de ácido fórmico).
Método 2
Modo de ionización ESI+/-. Columna: BEH C182,1x50 mm, 1,7 |im de diámetro de partícula. Gradiente: del 90% de A al 100% de B en 4,45 minutos, mantener al 100% de B hasta 4,58 minutos. Velocidad de flujo de 0,8 ml/min. A=(el 95% de agua el 5% de acetonitrilo bicarbonato de amonio 2,5 mM), B=(acetonitrilo).
Método 3
Modo de ionización ESI+/-. Columna: BEH C182,1x50 mm, 1,7 |im de diámetro de partícula. Gradiente: del 90% de A al 95% de B en 1,19 minutos, mantener al 95% de B hasta 1,70 minutos. Velocidad de flujo de 0,8 ml/min. A=(el 95% de agua el 5% de acetonitrilo bicarbonato de amonio 2,5 mM), B=(acetonitrilo).
Método 4
Modo de ionización ESI+/-. Columna: HSS T32,1x100 mm, 1,8 |im de diámetro de partícula. Gradiente: el 100% de A, mantener durante 1,00 minuto, del 100% de A al 95% de B en 4,50 minutos, mantener al 100% de B hasta 4,91 minutos. Velocidad de flujo de 0,6 ml/min. A=(el 95% de agua el 5% de acetonitrilo el 0,05% de ácido fórmico) B=(acetonitrilo el 0,05% de ácido fórmico).
Método 5
Modo de ionización ESI+/-. Columna: CSH C182,1x50 mm, 1,7 |im de diámetro de partícula: Gradiente: del 90% de A al 100% de B en 4,45 minutos, mantener al 100% de B hasta 4,58 minutos. Velocidad de flujo de 0,8 ml/min A=(el 95% de agua el 5% de acetonitrilo el 0,05% de ácido fórmico), B=(acetonitrilo el 0,05% de ácido fórmico).
Método 6
Modo de ionización ESI+/-. Columna: HSS T32,1x100 mm, 1,8 |im de diámetro de partícula. Gradiente: del 95% de A al 100% de B en 3,65 minutos, mantener al 100% de B hasta 4,95 minutos. Velocidad de flujo de 0,6 ml/min. Temperatura de columna: 60°C. A=(el 95% de agua el 5% de acetonitrilo el 0,05% de ácido fórmico) B=(acetonitrilo el 0,05% de ácido fórmico).
Método 7 (temperatura de columna: 60°C)

Método 8 (temperatura de columna: 40°C)

Método 9


Método 10 (temperatura de columna: 60°C)

Método 11

Método 12

Método 13

Método 14

Ejemplos de síntesis
Los ejemplos a continuación son ilustrativos y, tal como reconoce un experto en la técnica, podrían modificarse los reactivos o las condiciones particulares según fuese necesario para los compuestos individuales sin experimentación indebida.
Los compuestos de la invención pueden prepararse mediante los métodos generales y los ejemplos presentados a continuación y métodos conocidos por los expertos habituales en la técnica. Las condiciones de reacción y los tiempos de reacción óptimos pueden variar dependiendo de los reactantes particulares usados. A menos que se especifique lo contrario, los disolventes, las temperaturas, las presiones y otras condiciones de reacción pueden seleccionarse fácilmente por un experto habitual en la técnica. Se proporcionan procedimientos específicos en la sección de ejemplos de síntesis. Los productos intermedios usados en las síntesis a continuación o bien están comercialmente disponibles o bien se preparan fácilmente mediante métodos conocidos por los expertos en la técnica. El avance de la reacción puede monitorizarse mediante métodos convencionales tales como cromatografía en capa fina (CCF) o cromatografía de líquidos de alta presión-espectrometría de masas (HPLC-EM). Los productos intermedios y los productos pueden purificarse mediante métodos conocidos en la técnica, incluyendo cromatografía en columna, Hp LC, CCF preparativa o recristalización.
Procedimiento de síntesis general
Los compuestos de la invención y los compuestos de referencia divulgados en el presente documento se preparan generalmente haciendo reaccionar un producto intermedio de ácido carboxílico con un producto intermedio de amina en condiciones apropiadas.

A éster terc-butílico del ácido piperazin-1-carboxílico (1,0 g, 5,37 mmol) y 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxipiridina (1,5 g, 5,37 mmol) en 1,4-dioxano (15 ml) se les añaden metanosulfonato de CPhos-G3-paladaciclo y terc-butóxido de sodio (216 mg, 16,1 mmol) y se desgasifica con nitrógeno durante 5 min Se agita la mezcla resultante a 100°C durante 10 h. Se filtra la mezcla de reacción a través de un lecho de sílice eluyendo con EtOAc y se concentra. Se purifica el producto en bruto mediante cromatografía en columna de gel de sílice para proporcionar el compuesto del título.
Rendimiento: 2,1 g (88%) Tr (HPLC): 1,15 min (método 1)

A éster terc-butílico del ácido 4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il]-piperazin-1-carboxílico (2,1 g, 4,73 mmol) en EtOH (10 ml) y agua (5 ml) se le añaden clorhidrato de hidroxilamina (1,64 g, 23,6 mmol) y trimetilamina (659 ^l, 4,73 mmol) y se agita a 80°C durante 18 h. Se concentra la mezcla de reacción a presión reducida. Se suspende el residuo en DCM y se filtra para retirar las sales. Se purifica el filtrado mediante cromatografía en columna de gel de sílice para proporcionar el compuesto del título.
Rendimiento: 1,07 g (73%)
Diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina

A éster ferc-butílico del ácido 4-(6-amino-4-metoxi-piridin-3-il)-piperazin-1-carboxílico (1,07 g, 3,47 mmol) en DCM (12 ml) se le añade HCI 4 M en 1,4-dioxano (4,34 ml, 17,35 mmol) y se agita a TA durante 2 h. Se concentra la mezcla de reacción a presión reducida.
Rendimiento: 976 mg (cuantitativo)
Éster terc-butílico del ácido 6-amino-4-metil-3’,6’-dihidro-2’H-[3,4’1bipiridinil-1’-carboxílico

A éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (1,24 g, 4,01 mmol) y 5-bromo-4-metil-piridin-2-ilamina (750 mg, 4,01 mmol) en 1,4-dioxano se les añaden disolución de Na2CO32 M (4,01 ml, 8,02 mmol) y PdCl2(dppf) (328 mg, 0,40 mmol). Se desgasifica la mezcla de reacción con nitrógeno durante 5 min y se agita en el microondas a 150°C durante 30 min Se diluye la reacción con EtOAc y agua y se separan las fases. Se extrae la fase ac. de nuevo con EtOAc. Se lavan las fases orgánicas combinadas con salmuera, se secan sobre MgSO4 y se concentran a vacío. Se purifica el residuo mediante cromatografía en gel de sílice para dar el compuesto del título.
Rendimiento: 1,1 g (95%) ESI-EM: m/z = 290 (M+H)+ Tr (HPLC): 1,82 min (método 2)

A éster terc-butílico del ácido 6-amino-4-metil-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico (1,10 g, 3,80 mmol) en MeOH (10 ml) se le añade Pd/C (405 mg, 0,38 mmol) bajo nitrógeno. Se desgasifica la mezcla de reacción y se somete a un globo de H2. Se filtra la reacción y se concentra a presión reducida. Se purifica el producto en bruto mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 511 mg (46%) ESI-EM: m/z = 292 (M+H)+ Tr (HPLC): 1,80 min (método 2)
Diclorhidrato de 4-metil-1’,2’,3’,4’,5’,6’-hexahidro-[3,4’1bipiridinil-6-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 6-amino-4-metil-3’,4’,5’,6’-tetrahidro-2’H-[3,4’]bipiridinil-1’-carboxílico (511 mg, 1,75 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 347 mg (75%) ESI-EM: m/z = 192 (M+H)+ Tr (HPLC): 0,36 min (método 2)
Éster terc-butílico del ácido 6-amino-3’,6’-dihidro-2’H-[3,4’1bipiridinil-1’-carboxílico

A éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (1,70 g, 5,50 mmol) y 5-bromo-piridin-2-ilamina (1,00 mg, 5,78 mmol) en 1,4-dioxano se les añaden disolución de Na2CO32 M (2 ml, 4,00 mmol) y PdCl2(dppf)CH2Cl2 (449 mg, 0,55 mmol). Se desgasifica la mezcla de reacción con nitrógeno durante 5 min y se agita a 120°C durante 16 h. Se evaporan todas las sustancias volátiles a presión reducida. Se purifica el material en bruto mediante cromatografía de fase normal para proporcionar el compuesto del título.
Rendimiento: 1,2 g (79%)
Éster terc-butílico del ácido 6-amino-3’,4’,5’,6’-tetrahidro-2’H-[3,4’1bipiridinil-1’-carboxílico

A éster terc-butílico del ácido 6-amino-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico (45,0 g, 163,4 mmol) en EtOH (1000 ml) se le añade Pd(OH)2 sobre carbono (4,5 g, 32,4 mmol) bajo nitrógeno. Se agita la mezcla de reacción a 30 PSI en un agitador de PARR durante 16 h. Se filtra la reacción a través de Celite®. Se evapora el filtrado a presión reducida y se purifica el residuo mediante cromatografía en columna de gel de sílice para obtener el compuesto del título.
Rendimiento: 23,7 g (79%)
Diclorhidrato de 1’,2’,3’,4’,5’,6’-hexahidro-[3,4’1bipiridinil-6-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 6-amino-3’,4’,5’,6’-tetrahidro-2’H-[3,4’]bipiridinil-1’-carboxílico (800 mg, 2,88 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.

A éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (10,0 g, 49,3 mmol) y 5-bromo-4-metoxi-piridin-2-ilamina (15,2 g, 49,3 mmol) en 1,4-dioxano (100 ml) se les añaden disolución de Na2CO32 M (2 ml, 148 mmol) y PdCl2(dppf)CH2Cl2 (3,93 g, 4,93 mmol). Se desgasifica la mezcla de reacción con nitrógeno durante 5 min y se agita a 120°C durante 16 h. Se evaporan todas las sustancias volátiles a presión reducida. Se diluye el residuo con agua y se extrae tres veces con EtOAc. Se lavan las fases orgánicas combinadas con salmuera, se secan sobre Na2SO4 y se concentran a presión reducida. Se purifica el material en bruto mediante cromatografía de fase normal para proporcionar el compuesto del título.

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 6-amino-4-metoxi-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico (750 mg, 2,46 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metil-3’,4’,5’,6’-tetrahidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 715 mg (95%) ESI-EM: m/z = 308 (M+H)+ Tr (HPLC): 0,88 min (método 5)
Diclorhidrato de 4-metoxi-1’,2’,3’,4’,5’,6’-hexahidro-[3,4’1bipiridinil-6-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 6-amino-4-metoxi-3’,4’,5’,6’-tetrahidro-2’H-[3,4’]bipiridinil-1’-carboxílico (715 mg, 2,33 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 745 mg (cuantitativo) ESI-EM: m/z = 208 (M+H)+ Tr (HPLC): 0,56 min (método 6)
Éster terc-butílico del ácido 4-(6-amino-piridazin-3-il)-3,6-dihidro-2H-piridin-1-carboxílico

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (977 mg, 3,16 mmol) y 6-cloro-piridazin-3-ilamina (500 mg, 2,87 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metoxi-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 590 mg (74,3%) ESI-EM: m/z = 276 (M+H)+ Tr (HPLC): 0,44 min (método 1)
Éster terc-butílico del ácido 4-(6-amino-piridazin-3-il)-piperidin-1-carboxílico

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(6-amino-piridazin-3-il)-3,6-dihidro-2H-piridin-1-carboxílico (5,40 g, 19,5 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metil-3’,4’,5’,6’-tetrahidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 3,93 g (72%) ESI-EM: m/z = 279 (M+H)+ Tr (HPLC): 0,38 min (método 1)
Diclorhidrato de 6-piperidin-4-il-piridazin-3-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(6-amino-piridazin-3-il)-piperidin-1-carboxílico (3,60 g, 12,9 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 2,30 g (cuantitativo) ESI-EM: m/z = 179 (M+H)+ Tr (HPLC): 0,32 min (método 1)

A éster terc-butílico del ácido (R)-2-hidrox¡met¡l-p¡perazin-1-carboxíl¡co (1,00 g, 4,62 mmol) en DMA (10 ml) se le añaden terc-butil-cloro-dimetil-silano (1,05 g, 6,94 mmol) e ¡midazol (944 mg, 13,9 mmol), y se agita la mezcla de reacción durante 14 h a ta. Se diluye la mezcla de reacción con EtOAc y se lava con agua y salmuera, se seca sobre Na2SO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 1,45 g (95%)
5-Bromo-2-(2,5-dimetil-pirrol-1-il)-4-metil-piridina

A 5-bromo-4-metil-p¡r¡din-2-¡lam¡na (2,00 g, 10,7 mmol) y hexano-2,5-diona (1,47 g, 12,8 mmol) en tolueno (50 ml) se les añade ácido para-toluenosulfónico (61,0 mg, 0,32 mmol), y se agita la mezcla de reacción durante 18 h a 140°C. Se vierte la mezcla de reacción en agua y se diluye en EtOAc. Se lava la fase orgánica separada con salmuera y se seca sobre MgSO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 2,68 g (95%)
Éster terc-butílico del ácido (R)-2-(terc-but¡l-d¡met¡l-s¡lan¡lox¡metN)-4-[6-(2,5-d¡met¡l-p¡rrol-1-¡l)-4-met¡l-p¡r¡d¡n-3-¡l1-piperazin-1 -carboxílico

A 5-bromo-2-(2,5-dimet¡l-p¡rrol-1-¡l)-4-met¡l-p¡r¡dina (1,00 g, 3,77 mmol) y éster terc-butílico del ácido (R)-2-(terc-butildimet¡l-s¡lan¡lox¡met¡l)-p¡peraz¡n-1-carboxíl¡co (1,25 g, 3,77 mmol) en 1,4-dioxano (13 ml) se les añaden terc-butóxido de sodio (1,09 g, 11,3 mmol) y metanosulfonato de CPhos-G3-paladaciclo (152 mg, 0,19 mmol). Se desgasifica la mezcla con nitrógeno durante 5 min, y se agita durante 18 h a 100°C. Se filtra la mezcla de reacción a través de un lecho de gel de sílice y se eluye con EtOAc. Se concentra el filtrado a presión reducida para proporcionar el compuesto del título.
Rendimiento: 1,67 g (86%) ESI-EM: m/z = 515 (M+H)+ Tr (HPLC): 1,56 min (método 1)
Éster terc-butílico del ácido (ft)-4-(6-amino-4-metil-piridin-3-il)-2-hidroximetil-piperazin-1-carboxílico r

Se agita una mezcla de éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metil-piridin-3-il]-piperazin-1-carboxílico (1,67 g, 3,24 mmol), clorhidrato de hidroxilamina (1,13 g, 16,2 mmol) y trimetilamina (452 |al, 3,24 mmol) en etanol (10 ml) y agua (5 ml) durante 18 h a 80°C. Se concentra la mezcla de reacción a presión reducida y se purifica el residuo mediante cromatografía de fase inversa para proporcionar el compuesto del título.
Rendimiento: 1,67 g (86%) Tr (HPLC): 0,66 min (método 3)
Diclorhidrato de [(ft)-4-(6-amino-4-metil-piridin-3-il)-piperazin-2-il1-metanol

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido (R)-4-(6-amino-4-metil-piridin-3-il)-2-hidroximetil-piperazin-1-carboxílico (450 mg, 1,40 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 412 mg (cuantitativo)
5-Bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina

Se sintetiza el compuesto del título a partir de 5-bromo-4-metoxi-piridin-2-ilamina (2,00 g, 9,85 mmol) según el procedimiento descrito para la síntesis del producto intermedio 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metil-piridina. Rendimiento: 2,48 g, (90%) ESI-EM: m/z = 283 (M+H)+ Tr (HPLC): 2,13 min (método 5)
Éster terc-butílico del ácido 7-[6-(2,5-dimetil-pirrol-1-N)-4-metoxi-piridin-3-il1-4,7-diaza-espiro[2,51octano-4-carboxílico

A 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina (1,25 g, 4,45 mmol) y éster terc-butílico del ácido 4,7-diazaespiro[2,5]octano-4-carboxílico (1,13 g, 5,34 mmol) en 1,4-dioxano (13 ml) se les añaden Cs2CO3(4,35 g, 13,3 mmol) y metanosulfonato de CPhos-G3-paladaciclo (359 mg, 0,45 mmol). Se desgasifica la mezcla con nitrógeno durante 5 min, y se agita durante 18 h a 100°C. Se extrae la mezcla de reacción con EtOAc, se lava con salmuera, se seca sobre MgsO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para proporcionar el compuesto del título.

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 7-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-¡l]-4,7-diaza-espiro[2,5]octano-4-carboxíl¡co (1,51 g, 3,66 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido (R)-4-(6-amino-4-metil-piridin-3-il)-2-hidroximetil-piperazin-1-carboxílico.
Rendimiento: 1,07 g (87%) ESI-EM: m/z = 335 (M+H)+ Tr (HPLC): 0,74 min (método 5)

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 7-(6-amino-4-metoxi-piridin-3-il)-4,7-diazaespiro[2,5]octano-4-carboxílico (1,07 g, 3,19 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 1,10 g (cuantitativo) ESI-EM: m/z = 235 (M+H)+ Tr (HPLC): 0,17 min (método 5)

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (1,74 g, 5,64 mmol) y 6-doro-4-metoxi-piridazin-3-ilamina (900 mg, 5,64 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metoxi-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 787 mg (46%) ESI-EM: m/z = 307 (M+H)+ Tr (HPLC): 0,59 min (método 5)
Éster terc-butílico del ácido 4-(6-amino-5-metoxi-piridazin-3-il)-piperidin-1-carboxílico

A éster terc-butílico del ácido 4-(6-amino-5-metoxi-piridazin-3-il)-3,6-dihidro-2H-piridin-1-carboxílico (785 mg, 2,56 mmol) en MeOH (10 ml) y ácido acético (1 ml) se le añade Pd/C (273 mg, 0,26 mmol) bajo nitrógeno. Se desgasifica la mezcla de reacción y se somete a un globo de H2. Se filtra la reacción y se concentra a presión reducida. Se purifica el producto en bruto mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 513 mg (65%) ESI-EM: m/z = 309 (M+H)+ Tr (HPLC): 0,54 min (método 5)
Diclorhidrato de 4-metoxi-6-piperidin-4-il-piridazin-3-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(6-amino-5-metoxi-piridazin-3-il)-piperidin-1-carboxílico (510 mg, 1,65 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 514 mg (cuantitativo) ESI-EM: m/z = 209 (M+H)+ Tr (HPLC): 0,14 min (método 5)
4-(6-{[(7erc-butoxi)carbonil1aminoM-metoxipiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de terc-butilo

Se sintetiza el compuesto del título a partir de 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de terc-butilo (4,76 g, 20 mmol) y N-(6-cloro-5-metoxipiridazin-3-il)carbamato de terc-butilo (4,00 g, 20 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metoxi-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 4,56 g (59%)

A 4-(6-{[(terc-butoxi)carbonil]amino}-4-metoxipiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de tere-butilo (1,50 g, 3,69 mmol) en MeOH (15 ml) se le añade Pd/C (1,18 g, 1,11 mmol) bajo una atmósfera de nitrógeno. Se desgasifica la mezcla de reacción y se somete a 30°C durante la noche a un globo de H2. Se trata la mezcla con Pd/C (0,3 g) y se agita a 30°C durante 3h. Se filtra la reacción y se concentra a presión reducida.
Rendimiento: 1,42 g (94%)
Diclorhidrato de 5-metoxi-6-(piperidin-4-il)piridazin-3-amina

Se sintetiza el compuesto del título a partir de 4-(6-{[(tere-butoxi)carbonil]amino}-4-metoxipiridazin-3-il)piperidin-1-carboxilato de tere-butilo (1,42 g, 3,48 mmol) según el procedimiento descrito para la síntesis de diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 0,99 g (cuantitativo)
Éster tere-butílico del ácido 4-(6-nitro-piridin-3-il)-piperazin-1-carboxílico

Se agitan 5-bromo-2-nitro-piridina (5,00 g, 24,63 mmol) y éster tere-butílico del ácido piperazin-1-carboxílico (13,7 g, 73,9 mmol) en NMP (50 ml) durante 3 h a 120°C. Se vierte la mezcla de reacción en agua. Se filtra el precipitado, se lava con agua y se seca para dar el compuesto del título.
Rendimiento: 6,80 g (90%)
Éster tere-butílico del ácido 4-(6-amino-piridin-3-il)-piperazin-1-carboxílico

Se agitan éster tere-butílico del ácido 4-(6-nitro-piridin-3-il)-piperazin-1-carboxílico (2,00 g, 65,9 mmol) y Pd/C (200 mg)
en etanol con un globo de H2 durante 3 h. Se filtra la mezcla de reacción y se concentra el filtrado a presión reducida. Rendimiento: 1,90 g (cuantitativo)
Diclorhidrato de 5-piperazin-1-il-piridin-2-ilamina

Se agitan éster terc-butílico del ácido 4-(6-amino-piridin-3-il)-piperazin-1-carboxílico (2,50 g, 8,98 mmol) en DCM (30 ml) y HCI 4 M en 1,4-dioxano (11,2 ml, 44,9 mmol) durante 16 h a ta. Se filtra la mezcla de reacción y se lava con éter para dar el compuesto del título.

A éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-piperazin-1-carboxílico (1,50 g, 4,54 mmol) y 5 bromo-2-nitropiridina (1,00 g, 4,93 mmol) en 1,4-dioxano (12 ml) se les añaden Cs2CO3 (4,44 g, 13,6 mmol), Pd2(dba)3 (208 mg, 0,23 mmol) y Xantphos (263 mg, 0,45 mmol). Se agita la mezcla de reacción a 100°C durante 24 h, se filtra a través de Celite® y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para proporcionar el compuesto del título.
Rendimiento: 1,35 g (66%) ESI-EM: m/z = 453 (M+H)+ Tr (HPLC): 1,31 min (método 1)
Éster terc-butílico del ácido (ft)-4-(6-amino-piridin-3-il)-2-(terc-butil-dimetil-silaniloximetil)-piperazin-1-carboxílico

Se agitan éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-(6-nitro-piridin-3-il)-piperazin-1-carboxílico (1,35 g, 2,98 mmol) y Pd/C (317 mg, 0,15 mmol) en metanol (20 ml) con un globo de H2 durante 24 h. Se filtra la mezcla de reacción a través de Celite®, se lava con metanol y se concentra el filtrado a presión reducida. Rendimiento: 1,26 g (cuantitativo)
Diclorhidrato de [(ft)-4-(6-amino-piridin-3-il)-piperazin-2-il1-metanol

Se agitan éster terc-butílico del ácido (R)-4-(6-amino-piridin-3-il)-2-(terc-butil-dimetil-silaniloximetil)-piperazin-1-carboxílico (1,26 g, 2,98 mmol) en DCM (10 ml) y HCI 4 M en 1,4-dioxano (7,5 ml, 30,0 mmol) durante 1 h a ta. Se concentra la mezcla de reacción a presión reducida, se suspende en éter, se filtra y se lava con éter para dar el compuesto del título.
Rendimiento: 838 mg (cuantitativo)
Éster terc-butílico del ácido (ft)-4-[6-(2,5-dimetil-pirrol-1-N)-4-metil-piridin-3-il1-2-hidroximetil-piperazin-1-carboxílico

A éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metil-piridin-3-il]-piperazin-1-carboxílico (8,56 g, 16,1 mmol) en THF (100 ml) se le añade fluoruro de tetrabutilamonio (16,1 ml, 16,1 mmol), y se agita la mezcla de reacción a TA durante 1,5 h. Se concentra la mezcla de reacción a presión reducida y se purifica el residuo mediante cromatografía en columna de gel de sílice para proporcionar el compuesto del título. Rendimiento: 6,10 g (91%) ESI-EM: m/z = 417 (M+H)+ Tr (HPLC): 0,98 min (método 1)
Éster terc-butílico del ácido (ft)-4-[6-(2,5-dimetilo 4-pirrol-1-il)-4-metil-piridin-3-il1-2-metoximetil-piperazin-1-carboxílico

A éster terc-butílico del ácido (R)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metil-piridin-3-il]-2-hidroximetil-piperazin-1-carboxílico (2,00 g, 4,80 mmol) y yoduro de metilo (915 mg, 7,20 mmol) en d Ma (15 ml) se les añade NaH al 60% (230 mg, 5,76 mmol). Se agita la mezcla de reacción durante 2 h a TA y se extingue con agua. Se extrae la mezcla tres veces con EtOAc, se lavan las fases orgánicas combinadas con salmuera, se secan sobre MgSO4, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 1,80 g (87%) ESI-EM: m/z = 431 (M+H)+ Tr (HPLC): 1,12 min (método 1)

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido (R)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metilpiridin-3-il]-2-metoximetil-piperazin-1-carboxílico (1,80 g, 4,18 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido (R)-4-(6-amino-4-metil-piridin-3-il)-2-hidroximetil-piperazin-1-carboxílico.
Rendimiento: 1,07 g (87%) ESI-EM: m/z = 353 (M+H)+ Tr (HPLC): 0,44 min (método 1)
Diclorhidrato de 5-((ft)-3-metoximetil-piperazin-1-il)-4-metil-piridin-2-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido (R)-4-(6-amino-4-metil-piridin-3-il)-2-metoximetil-piperazin-1-carboxílico (440 mg, 1,25 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 406 mg (cuantitativo)
Éster terc-butílico del ácido 4-(6-amino-4-metil-piridazin-3-il)-3,6-dihidro-2H-piridin-1 -carboxílico

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1 -carboxílico (538 mg, 1,74 mmol) y 6-cloro-5-metil-piridazin-3-ilamina (250 mg, 1,74 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metil-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 326 mg (65%) ESI-EM: m/z = 292 (M+H)+ Tr (HPLC): 0,51 min (método 5)
Éster terc-butílico del ácido 4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-carboxílico

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(6-amino-4-metil-piridazin-3-il)-3,6-dihidro-2H-piridin-1-carboxílico (326 mg, 1,12 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metil-3’,4’,5’,6’-tetrahidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 289 mg (88%) ESI-EM: m/z = 293 (M+H)+ Tr (HPLC): 0,60 min (método 5)
Diclorhidrato de 5-metil-6-piperidin-4-il-piridazin-3-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-carboxílico (175 mg, 0,60 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 154 mg (97%) ESI-EM: m/z = 193 (M+H)+ Tr (HPLC): 0,46 min (método 2)
5-Bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina

Se sintetiza el compuesto del título a partir de 5-bromo-4-metoxi-piridin-2-ilamina (10,6 g, 52,1 mmol) según el procedimiento descrito para la síntesis del producto intermedio 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metil-piridina. Rendimiento: 14,0 g (96%) ESI-EM: m/z = 283 (M+H)+ Tr (HPLC): 0,93 min (método 3)
Éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il1-piperazin-1 -carboxílico

A 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina (1,24 g, 4,41 mmol) y éster terc-butílico del ácido (R)-2-(terc-butildimetil-silaniloximetil)-piperazin-1-carboxílico (1,46 g, 4,41 mmol) en 1,4-dioxano (13 ml) se les añaden terc-butóxido
de sodio (1,27 g, 13,2 mmol) y metanosulfonato de CPhos-G3-paladaciclo (178 mg, 0,22 mmol). Se desgasifica la mezcla con nitrógeno durante 5 min, y se agita durante 4 h a 100°C. Se filtra la mezcla de reacción a través de un lecho de gel de sílice y se eluye con EtOAc. Se concentra el filtrado a presión reducida y se purifica el residuo mediante cromatografía en columna de fase inversa para dar el compuesto del título.

Se agitan éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxipiridin-3-il]-piperazin-1-carboxílico (1,68 g, 3,17 mmol), clorhidrato de hidroxilamina (l,10 g, 15,8 mmol) y trimetilamina (320 |al, 3,24 mmol) en etanol (6 ml) y agua (3 ml) durante 18 h a 80°C. Se añade clorhidrato de hidroxilamina (440 mg, 6,33 mmol) de nuevo y se agita a 80°C. Se concentra la mezcla de reacción a presión reducida y se purifica el residuo mediante cromatografía en columna de fase inversa (para proporcionar el compuesto del título.
Rendimiento: 620 mg (58%)
Clorhidrato de [(ft)-4-(6-amino-4-metoxi-piridin-3-il)-piperazin-2-il1-metanol

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido (R)-4-(6-amino-4-metoxi-piridin-3-il)-2-hidroximetil-piperazin-1-carboxílico (620 mg, 1,83 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 503 mg (cuantitativo)
Éster terc-butílico del ácido 3-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il1-3,8-diaza-biciclo[3.2.11octano-8-carboxílico

A 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina (1,00 g, 3,56 mmol) y éster terc-butílico del ácido 3,8-diazabiciclo[3.2.1]octano-8-carboxíl¡co (830 mg, 3,91 mmol) en 1,4-dioxano (13 ml) se les añaden terc-butóxido de sodio
(3,48 g, 10,7 mmol) y metanosulfonato de CPhos-G3-paladaciclo (287 mg, 0,36 mmol). Se desgasifica la mezcla con nitrógeno durante 5 min, y se agita durante 18 h a 80°C. Se extrae la mezcla de reacción con EtOAc, se lava con salmuera, se seca sobre MgSO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 760 mg (52%) ESI-EM: m/z = 412 (M+H)+ Tr (HPLC): 1,23 min (método 1)
Éster terc-butílico del ácido 3-(6-amino-4-metoxi-piridin-3-il)-3,8-diaza-biciclo[3.2.11octano-8-carboxílico

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 3-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-¡l]-3,8-diaza-b¡c¡clo[3.2.1]octano-8-carboxíl¡co (760 mg, 1,84 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 4-(6-amino-4-metoxi-piridin-3-il)-piperazin-1-carboxílico.
Rendimiento: 330 mg (54%) ESI-EM: m/z = 335 (M+H)+ Tr (HPLC): 1,75 min (método 6)
Diclorhidrato de 5-(3,8-diaza-biciclo[3.2.11oct-3-il)-4-metoxi-piridin-2-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 3-(6-amino-4-metoxi-piridin-3-il)-3,8-diazabiciclo[3.2.1]octano-8-carboxílico (330 mg, 0,99 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 330 mg (cuantitativo) ESI-EM: m/z = 235 (M+H)+ Tr (HPLC): 0,15 min (método 5)
(2^)-2-[Metoxi(metil)carbamoil1piperazin-1,4-dicarboxilato de 4-bencilo y 1-terc-butilo

Se agitan ácido (2R)-4-[(benciloxi)carbonil]-1-[(terc-butoxi)carbonil1piperazin-2-carboxílico (4,00 g, 11,0 mmol), DIPEA (5,1 ml, 27,4 mmol), Ha Tu (5,01 g, 13,2 mmol) y clorhidrato de N,O-dimetilhidroxilamina (1,29 g, 13,2 mmol) en DMA (40 ml) a TA durante 3 días. Se diluye la mezcla de reacción con EtOAc, se lava con agua y salmuera. Se seca la fase orgánica sobre MgSO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en
columna de gel de sílice para dar el compuesto del título.
Rendimiento: 4,44 g (99%) ESI-EM: m/z = 408 (M+H)+
(2ft)-2-Acetilpiperazin-1,4-dicarboxilato de 4-bencilo y 1-tere-butilo

A una mezcla enfriada hasta -20°C de (2R)-2-[metoxi(metil)carbamoil]-piperazin-1,4-dicarboxilato de 4-bencilo y 1 tere-butilo (4,40 g, 10,80 mmol) en THF (25 ml) se le añade gota a gota bromuro de metilmagnesio (5,40 ml, 16,20 mmol) y se agita a -20°C durante 30 min Se extingue la mezcla de reacción con disolución acuosa saturada de NH4Cl, se diluye con EtOAc y se lava con agua HCI 1 N y salmuera. Se seca la fase orgánica sobre MgSO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía para dar el producto deseado. Se realiza purificación adicional mediante separación por cromatografía quiral para dar el enantiómero R puro.
Rendimiento: 2,38 g (61%)
(2ft)-2-(1-hidroxietil)piperazin-1,4-dicarboxilato de 4-bencilo y 1-tere-butilo

Se añade borohidruro de sodio (0,36 g, 9,52 mmol) a (2R)-2-acetilpiperazin-1,4-dicarboxilato de 4-bencilo y 1-terebutilo (2,30 g, 6,35 mmol) en metanol (100 ml). Después de agitar la mezcla de reacción durante 30 min, se elimina el disolvente a presión reducida. Se purifica el residuo mediante cromatografía en sílice.
Rendimiento: 2,10 g (91%)

Se añade ferc-butil(cloro)dimetilsilano (1,30 g, 8,64 mmol) a (2R)-2-(1-hidroxietil)piperazin-1,4-dicarboxilato de 4-bencilo y 1-ferc-butilo (2,10 g, 5,76 mmol) e imidazol (1,18 g, 17,29 mmol) en diclorometano (15 ml). Se agita la mezcla de reacción durante la noche. Después de añadir agua (10 ml), se extrae la fase acuosa con diclorometano (2 x 25 ml). Se lavan las fases orgánicas combinadas con salmuera. Se seca la fase orgánica, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en sílice.
Rendimiento: 2,75 g (99,7%)
(2R)-2-{1 -f(ferc-butildimetilsiliDoxi1etil}piperazin-1 -carboxilato de ferc-butilo

Bajo una atmósfera (globo) de hidrógeno, se agitan (2R)-2-{1 -[(íerc-butildimetilsilil)-oxi]etil}piperazin-1,4-dicarboxilato de 4-bencilo y 1-ferc-butilo (2,75 g, 5,75 mmol) y Pd/C (0,20 g) a temperatura ambiente en etanol (50 ml) durante 2 h. Después de retirar el catalizador filtrando a través de Celite®, se elimina el disolvente a presión reducida. Se filtra el residuo a través de sílice eluyendo con MeOH al 10%/diclorometano.
Rendimiento: 1,89 g (96%)
(2R)-2-{1-[(ferc-butildimetilsili r )oxi1etil}-4-[6-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridin-3-illpiperazin-1-carboxilato de ferc-butilo

A (2R)-2-{1-[(tere-butildimetilsilil)oxi]etil]piperazin-1-carboxilato de tere-butilo (1,89 g, 5,49 mmol) y 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina (1,54 g, 5,49 mmol) en 1,4-dioxano (20 ml) se les añaden metanosulfonato de CPhos-G3-paladaciclo (0,22 g) y tere-butóxido de sodio (1,58 g, 16,5 mmol), y se rocía la mezcla de reacción con nitrógeno. Se agita la mezcla de reacción a 100°C durante 10 h. Se filtra la mezcla de reacción a través de un lecho de sílice eluyendo con EtOAc y se concentra. Se purifica el residuo dos veces mediante cromatografía en sílice para proporcionar los compuestos del título.
Rendimiento: (2R)-2-[(7S)-1-[(tere-butildimetilsilil)oxi]etil]-4-[6-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridin-3-il]piperazin-1-carboxilato de tere-butilo: 0,57 g (19%) y (2R)-2-[(7R)-1-[(tere-butildimetilsilil)oxi]etil]-4-[6-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridin-3-il]piperazin-1-carboxilato de tere-butilo: 0,78 g (26%)

Se calientan (2R)-2-[(7R)-1-[(tere-butildimetilsilil)oxi]etil]-4-[6-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridin-3-il]piperazin-1-carboxilato de tere-butilo (0,87 g, 1,60 mmol), clorhidrato de hidroxilamina (0,56 g, 7,99 mmol) y trimetilamina (0,22 ml, 1,60 mmol) en 8 ml de etanol y 4 ml de agua a 80°C durante 42 h. Se añade una cantidad adicional de clorhidrato de hidroxilamina (0,22 g, 3,19 mmol) y se agita la mezcla de reacción a 80°C durante la noche. Se concentra la mezcla de reacción a presión reducida, se lleva a diclorometano y se filtra. Se purifica el compuesto deseado mediante cromatografía en sílice.
Rendimiento: 0,20 g (36%)
Diclorhidrato de (7ft)-7-[(2ft)-4-(6-amino-4-metoxipiridin-3-il)piperazin-2-il]etan-1-ol

Se añade HCI 4 N en dioxano (0,71 ml, 2,84 mmol) a (2R)-4-(6-amino-4-metoxipiridin-3-il)-2-[(7R)-1-hidroxietil]piperazin-1-carboxilato de tere-butilo (0,20 g, 0,57 mmol) en 5 ml de diclorometano y se agita a TA durante 2 h. Se añade 1 ml adicional de HCI 4 N en dioxano y se agita durante 1 h a TA. Se concentra la mezcla de reacción a presión reducida. Se usa el residuo sin purificación adicional.
Rendimiento: 0,18 g (cuantitativo)
(2ft)-4-(6-amino-4-metoxipiridin-3-il)-2-[(7S)-1-hidroxietil]piperazin-1-carboxilato de terc-butilo

Se calientan (2R)-2-[(7S)-1-[(ferc-butildimetilsilil)oxi]etil]-4-[6-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridin-3-il]piperazin-1-carboxilato de terc-butilo (0,57 g, 1,04 mmol), clorhidrato de hidroxilamina (0,36 g, 5,21 mmol) y trimetilamina (0,15 ml, 1,04 mmol) en 4 ml de etanol y 2 ml de agua a 80°C durante 42 h. Se añade una cantidad adicional de clorhidrato de hidroxilamina (0,15 g, 2,09 mmol) y se agita la mezcla de reacción a 80°C durante la noche. Se concentra la mezcla de reacción a presión reducida, se lleva a diclorometano y se filtra. Se purifica el compuesto deseado mediante cromatografía en sílice y vuelve a purificarse mediante HPLC
Rendimiento: 0,12 g (33%),
Diclorhidrato de (7S)-1-[(2ft)-4-(6-amino-4-metoxipiridin-3-il)piperazin-2-il]etan-1-ol

Se añade HCl 4 N en dioxano (0,50 ml, 2,00 mmol) a (2R)-4-(6-amino-4-metoxipiridin-3-il)-2-[(7S)-1-hidroxietil]piperazin-1-carboxilato de ferc-butilo (0,12 g, 0,34 mmol) en 1 ml de diclorometano y se agita a TA durante 1 h. Se concentra la mezcla de reacción a presión reducida. Se usa el residuo sin purificación adicional.

Se sintetiza el compuesto del título a partir de 5-bromo-4-metoxi-piridin-2-ilamina (202 mg, 1,00 mmol) y éster terc-butílico del ácido 7-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3-oxa-9-aza-biciclo[3.3.1]non-6-eno-9-carboxílico (350 mg, 1,00 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster terc-butílico del ácido 6-amino-4-metil-3’,6’-dihidro-2’H-[3,4’]bipiridinil-1’-carboxílico.
Rendimiento: 220 mg (64%) ESI-EM: m/z = 348 (M+H)+ Tr (HPLC): 1,52 min (método 2)

A éster terc-butílico del ácido 7-(6-amino-4-metoxi-piridin-3-il)-3-oxa-9-aza-bicido[3.3.1]non-6-eno-9-carboxílico (220 mg, 0,63 mmol) en EtOAc (10 ml) se le añade Pd/C (67,0 mg, 0,06 mmol) bajo nitrógeno. Se desgasifica la mezcla de reacción, se coloca bajo un globo de H2 y se agita durante 18 h a 50°C. Se filtra la reacción a través de Celite®, se concentra a presión reducida y se purifica mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 145 mg (66%) ESI-EM: m/z = 350 (M+H)+ Tr (HPLC): 1,60 min (método 2)
Diclorhidrato de 4-metoxi-5-(3-oxa-9-aza-biciclo[3.3.11non-7-il)-piridin-2-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido 7-(6-amino-4-metoxi-piridin-3-il)-3-oxa-9-aza-biciclo[3.3.1]nonano-9-carboxílico (145 mg, 0,41 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 133 mg (cuantitativo) ESI-EM: m/z = 250 (M+H)+ Tr (HPLC): 0,15 min (método 5)
Éster terc-butílico del ácido (S)-2-(terc-butil-dimetil-silaniloximetil)-piperazin-1-carboxílico

A éster terc-butílico del ácido (S)-2-hidroximetil-piperazin-1-carboxílico (2,00 g, 9,25 mmol) en DMA (10 ml) se le añaden terc-butil-cloro-dimetil-silano (2,09 g, 13,9 mmol) e imidazol (1,89 g, 27,7 mmol), y se agita la mezcla de reacción durante 24 h a ta. Se diluye la mezcla de reacción con disolución de NH4Cl y se extrae con EtOAc. Se lava la fase orgánica con agua y salmuera, se seca sobre Na2SO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para dar el compuesto del título.
Rendimiento: 2,80 g (92%)
Éster ferc-butílico del ácido (S)-2-(ferc-butil-dimetil-silaniloximetN)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-ilpiperazin-1-carboxílico

Se sintetiza el compuesto del título a partir de 5-bromo-2-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridina (3,25 g, 11,6 mmol) y éster ferc-butílico del ácido (S)-2-(ferc-butil-dimetil-silaniloximetil)-piperazin-1-carboxílico (3,82 g, 11,6 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster ferc-butílico del ácido 3-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il]-3,8-diaza-biciclo[3.2.1]octano-8-carboxílico.

Se agitan éster ferc-butílico del ácido (S)-2-(ferc-butil-dimetil-silaniloximetil)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxipiridin-3-il]-piperazin-1-carboxílico (11,9 g, 22,4 mmol), clorhidrato de hidroxilamina (3,89 g, 56,0 mmol) y trimetilamina (7,8 ml, 56,0 mmol) en etanol (30 ml) y agua (15 ml) durante 18 h a 80°C. Se concentra la mezcla de reacción a presión reducida y se purifica el residuo mediante cromatografía en gel de sílice para dar el compuesto del título.

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido (S)-4-(6-amino-4-metoxi-piridin-3-il)-2-hidroximetil-piperazin-1-carboxílico (264 mg, 0,58 mmol) según el procedimiento descrito para la síntesis del producto intermedio diclorhidrato de 4-metoxi-5-piperazin-1-il-piridin-2-ilamina.
Rendimiento: 160 mg (cuantitativo)
Éster terc-butílico del ácido (ft)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il]-2-hidroximetil-piperazin-1-carboxílico

A éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il]-piperazin-1-carboxílico (8,56 g, 16,1 mmol) en THF (100 ml) se le añade TbA f (1 M en THF, 16,1 ml, 16,1 mmol). Se agita la mezcla de reacción durante 2,5 h a ta. Se concentra la mezcla de reacción a presión reducida y se purifica el residuo mediante cromatografía para obtener el producto del título.
Rendimiento: 180 mg (87%)
Éster terc-butílico del ácido (ft)-4-[6-(2,5-dimetil-pirrol-1-N)-4-metoxi-piridin-3-il]-2-metoximetil-piperazin-1-carboxílico

Se añade NaH (60%, 230 mg, 9,58 mmol) a éster terc-butílico del ácido (R)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il]-2-hidroximetil-piperazin-1-carboxílico (2,0 g, 4,80 mmol) y Mel (401 ^l, 7,20 mmol) en DMA (20 ml). Se agita la mezcla de reacción durante 2 h a ta. Se añade agua y se extrae la mezcla de reacción con EtOAc (3 veces). Se lavan las fases orgánicas combinadas con salmuera, se secan sobre MgSO4, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía de fase normal.
Rendimiento: 1,8 g (87%) ESI-EM: m/z = 431 (M+H)+ Tr (HPLC): 1,11 min (método 1)
Éster terc-butílico del ácido (ft)-4-(6-amino-4-metoxi-piridin-3-il)-2-metoximetil-piperazin-1-carboxílico

Se agitan éster terc-butílico del ácido (R)-4-[6-(2,5-dimetil-pirrol-1-il)-4-metoxi-piridin-3-il]-2-metoximetil-piperazin-1-carboxílico (1,8 g, 4,18 mmol), clorhidrato de hidroxilamina (1,45 g, 20,9 mmol) y trimetilamina (0,58 ml, 4,18 mmol) en etanol (10 ml) y agua (5 ml) a 80°C durante 18 h. Se concentra la mezcla de reacción a presión reducida, se suspende en DCM, se filtra para retirar las sales y se concentra de nuevo a presión reducida. Se purifica el residuo mediante cromatografía en columna de fase normal para dar el producto del título.
Rendimiento: 440 mg (30%) ESI-EM: m/z = 353 (M+H)+ Tr (HPLC): 0,44 min (método 1)
Diclorhidrato de 4-metoxi-5-((ft)-3-metoximetil-piperazin-1-il)-piridin-2-ilamina

Se sintetiza el compuesto del título a partir de éster terc-butílico del ácido (R)-4-(6-amino-4-metoxi-piridin-3-il)-2-metoximetil-piperazin-1-carboxílico (440 mg, 1,25 mmol) según el procedimiento descrito para la síntesis del producto intermedio clorhidrato de [(R)-4-(6-amino-4-metoxipiridin-3-il)-piperazin-2-il]-metanol.
Rendimiento: 406 mg (cuantitativo)
5-Fluoro-4-metoxi-piridin-2-carbonitrilo

Se lleva 2-cloro-5-fluoro-4-metoxi-piridina (1,00 g, 6,19 mmol) a un tubo sellado. Se añaden cianuro de zinc (799 mg, 6,81 mmol) y zinc (40,5 mg, 0,31 mmol) y se purga con argón. Luego se añaden PdCl2(dppf)CH2Cl2 (253 mg, 0,62 mmol) y NMP y se calienta la mezcla durante 45 min a 150°C en el microondas. Se añaden agua y EtOAc a la mezcla de reacción y se filtra a través de Celite®. Se lava la fase orgánica con disolución de bicarbonato de sodio, agua, salmuera y se seca sobre MgSO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna de gel de sílice para proporcionar el compuesto del título.
Rendimiento: 689 mg (73%) ESI-EM: m/z = 153 (M+H)+ Tr (HPLC): 0,61 min (método 1)
5-(4-Fluoro-fenoxi)-4-metoxi-piridin-2-carbonitrilo

Se calientan 5-fluoro-4-metoxi-piridin-2-carbonitrilo (6,00 g, 39,4 mmol), 4-fluorofenol (5,31 g, 47,3 mmol) y K2CO3 (12,0 g, 86,8 mmol) en NMP (12 ml) a 100°C durante 3 h en un tubo sellado. Se diluye la mezcla de reacción con agua y se extrae con EtOAc. Se lava la fase orgánica con salmuera y se seca sobre MgSO4, se filtra y se concentra a presión reducida. Se tritura el residuo mediante éter y heptano para dar el compuesto del título.
Rendimiento: 8,99 g (93%) ESI-EM: m/z = 245 (M+H)+ Tr (HPLC): 0,91 min (método 1)
Ácido 5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-carboxílico

Se agita 5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-carbonitrilo (8,50 g, 34,8 mmol) en disolución acuosa de NaOH 2 N (90 ml) a 100°C durante 6 h. Se enfría la mezcla de reacción hasta Ta y se ajusta el pH de la disolución a pH 4,5 con HCI 4 N. Se recoge el precipitado y se seca en un horno de secado para dar el compuesto del título.
Rendimiento: 8,80 g (96%) ESI-EM: m/z = 264 (M+H)+ Tr (HPLC): 1,58 min (método 4)
4-Metoxi-5-fenoxi-piridin-2-carbonitrilo

Se calientan 5-fluoro-4-metoxi-piridin-2-carbonitrilo (8,00 g, 52,6 mmol), fenol (5,94 g, 63,1 mmol) y K2CO3 (16,0 g, 115 mmol) en NMP (3 ml) a 100°C durante 3 h en un tubo sellado. Se diluye la mezcla de reacción con agua y se extrae con EtOAc. Se lava la fase orgánica con salmuera y se seca sobre MgSO4, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en gel de sílice para dar el compuesto del título.
Rendimiento: 11,5 g (93%) ESI-EM: m/z = 227 (M+H)+ Tr (HPLC): 0,92 min (método 1)
Ácido 4-metoxi-5-fenoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 4-metoxi-5-fenoxi-piridin-2-carbonitrilo (11,5 g, 50,8 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-carboxílico. Rendimiento: 9,57 g (77%) ESI-EM: m/z = 246 (M+H)+ Tr (HPLC): 2,64 min (método 4)
5-(4-Isopropoxi-fenoxi)-4-metoxi-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (500 mg, 3,29 mmol) y 4 isopropoxi-fenol (600 mg, 3,94 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5-fenoxi-piridin-2-carbonitrilo.
Rendimiento: 850 mg (91%) ESI-EM: m/z = 285 (M+H)+ Tr (HPLC): 1,02 min (método 1)
Ácido 5-(4-Isopropoxi-fenoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 5-(4-isopropoxi-fenoxi)-4-metoxi-piridin-2-carbonitrilo (200 mg, 0,70 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 190 mg (77%) Tr (HPLC): 0,73 min (método 1)
4-Metoxi-5-(4-metoxi-fenoxi)-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (500 mg, 3,29 mmol) y 4 metoxifenol (490 mg, 3,94 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5
fenoxi-piridin-2-carbonitrilo.
Rendimiento: 740 mg (88%)
Ácido 4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-carbonitrilo (740 mg, 2,89 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 610 mg (77%)
4-Metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (500 mg, 3,29 mmol) y 4 trifluorometil-fenol (639 mg, 3,94 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5-fenoxi-piridin-2-carbonitrilo.
Rendimiento: 320 mg (33%) ESI-EM: m/z = 294 (M+H)+ Tr (HPLC): 1,06 min (método 1)
Ácido 4-metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 4-metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-carbonitrilo (151 mg, 0,51 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 150 mg (93%)
5-(4-Cloro-fenoxi)-4-metoxi-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (500 mg, 3,29 mmol) y 4-clorofenol (507 mg, 3,94 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5-fenoxi-piridin-2-carbonitrilo.
Rendimiento: 695 mg (81%)
Ácido 5-(4-cloro-fenoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 5-(4-doro-fenoxi)-4-metoxi-piridin-2-carbonitrilo (645 mg, 2,47 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 622 mg (90%) ESI-EM: m/z = 280 (M+H)+
5-(4-Difluorometoxi-fenoxi)-4-metoxi-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (75,0 mg, 0,49 mmol) y 4-difluorometoxi-fenol (101 mg, 0,63 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5-fenoxi-piridin-2-carbonitrilo.
Rendimiento: 98,0 mg (68%) Tr (HPLC): 0,93 min (método 1)
Ácido 5-(4-difluorometoxi-fenoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 5-(4-difluorometoxi-fenoxi)-4-metoxi-piridin-2-carbonitrilo (98,0 mg, 0,34 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 94,0 mg (90%) Tr (HPLC): 0,60 min (método 1)
4-Ciclopropoxi-fenol

Se agitan 2-(4-ciclopropoxi-fenil)-4,4,5,5-tetrametil-[1,3,2]dioxaborolano (800 mg, 3,08 mmol) y 4-óxido de 4-metilmorfolina (1,03 g, 8,83 mmol) en THF (100 ml) a 75°C durante 1,5 h y luego durante 18 h a ta. Se concentra la mezcla de reacción a vacío y se purifica el residuo mediante cromatografía en gel de sílice para proporcionar el compuesto del título.
Rendimiento: 389 mg (84%)
5-(4-Ciclopropoxi-fenoxi)-4-metoxi-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (350 mg, 2,30 mmol) y 4-ciclopropoxi-fenol (389 mg, 2,59 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5-fenoxi-piridin-2-carbonitrilo.
Rendimiento: 342 mg (53%) Tr (HPLC): 1,00 min (método 1)
Ácido 5-(4-ciclopropoxi-fenoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 5-(4-ciclopropoxi-fenoxi)-4-metoxi-piridin-2-carbonitrilo (100 mg, 0,35 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 622 mg (90%) Tr (HPLC): 0,63 min (método 1)
4-Metoxi-5-(4-trifluorometoxi-fenoxi)-piridin-2-carbonitrilo

Se sintetiza el compuesto del título a partir de 5-fluoro-4-metoxi-piridin-2-carbonitrilo (115 mg, 0,76 mmol) y 4-trifluorometoxi-fenol (162 mg, 0,91 mmol) según el procedimiento descrito para la síntesis del producto intermedio 4-metoxi-5-fenoxi-piridin-2-carbonitrilo.
Rendimiento: 140 mg (60%)
Ácido 4-metoxi-5-(4-trifluorometoxi-fenoxi)-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de 4-metoxi-5-(4-trifluorometoxi-fenoxi)-piridin-2-carbonitrilo (150 mg, 0,48 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(4-fluoro-fenoxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 120 mg (75%)
Éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico

A éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol), trifenilfosfina (372 mg, 1,42 mmol) y alcohol 2-fluorobencílico (114 |al, 1,065 mmol) en THF (2 ml) se les añade azodicarboxilato de dietilo (646 |al, 1,42 mmol) a 0°C. Se permite que la mezcla de reacción se caliente hasta TA y se agita durante 16 h. Se concentra la mezcla resultante a vacío y se purifica el residuo mediante cromatografía en gel de sílice para dar el compuesto del título.
Rendimiento: 66,0 mg (32%) Tr (HPLC): 0,77 min (método 1)
Ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico

A éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico (66,0 mg, 0,23 mmol) en THF/agua/MeOH (3 ml/1 ml/1 ml) se le añade LiOH (38,0 mg, 0,91 mmol), y se agita la mezcla de reacción a ta. Se acidifica la mezcla de reacción hasta pH 4,5 con HCI 4 N y se concentra a vacío. Se disuelve el residuo en DCM y tolueno y se concentra de nuevo a presión reducida. Se usó el producto sin purificación adicional.
Rendimiento: 62,0 mg (99%) Tr (HPLC): 0,48 min (método 1)
Éster metílico del ácido 5-ciclobutilmetoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y ciclobutil-metanol (91,7 mg, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 132 mg (74%) Tr (HPLC): 0,80 min (método 1)
Ácido 5-ciclobutilmetoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-ciclobutilmetoxi-4-metoxi-piridin-2-carboxílico (132 mg, 0,53 mmol) según el procedimiento descrito para la síntesis del producto intermedio del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 124 mg (cuantitativo) Tr (HPLC): 0,53 min (método 1)
Éster metílico del ácido 4-metoxi-5-(1-metil-ciclopropilmetoxi)-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y (1-metil-ciclopropil)-metanol (103 mg, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 115 mg (65%) Tr (HPLC): 0,81 min (método 1)

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-cidobutilmetoxi-4-metoxi-piridin-2-carboxílico (115 mg, 0,46 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 108 mg (cuantitativo) Tr (HPLC): 0,52 min (método 1)
Éster metílico del ácido 5-ciclohexiloxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y ciclohexanol (111 |l, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 171 mg (91%) Tr (HPLC): 0,87 min (método 1)
Ácido 5-ciclohexiloxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-ciclohexiloxi-4-metoxi-piridin-2-carboxílico (131 mg, 0,49 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 124 mg (cuantitativo) Tr (HPLC): 0,57 min (método 1)
Éster metílico del ácido 5-(4-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y (4-fluoro-fenil)-metanol (115 |l, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 150 mg (62%) Tr (HPLC): 0,82 min (método 1)
Ácido 5-(4-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-(4-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico (150 mg, 0,44 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 177 mg (cuantitativo) Tr (HPLC): 0,82 min (método 1)
Éster metílico del ácido 5-ciclopentiloxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y ciclopentanol (96,7 |al, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 170 mg (95%) Tr (HPLC): 0,87 min (método 1)
Ácido 5-ciclopentiloxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-ciclopentiloxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,52 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 122 mg (99%) Tr (HPLC): 0,49 min (método 1)
Éster metílico del ácido 5-isobutoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (118 mg, 0,64 mmol) y alcohol isobutílico (71,6 mg, 0,97 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 141 mg (92%) Tr (HPLC): 0,78 min (método 1)
Ácido 5-isobutoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-isobutoxi-4-metoxi-piridin-2-carboxílico (141 mg, 0,59 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 133 mg (cuantitativo) Tr (HPLC): 0,51 min (método 1)
Éster metílico del ácido 5-ciclopropilmetoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y ciclopropilmetanol (84,2 |al, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 146 mg (87%) Tr (HPLC): 0,74 min (método 1)
Ácido 5-ciclopropilmetoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-ciclopropilmetoxi-4-metoxi-piridin-2-carboxílico (325 mg, 1,37 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 358 mg (cuantitativo) ESI-EM: m/z = 224 (M+H)+ Tr (HPLC): 0,40 min (método 5)
Éster metílico del ácido 5-benciloxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (118 mg, 0,64 mmol) y alcohol bencílico (100 |al, 0,97 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 140 mg (80%) Tr (HPLC): 0,79 min (método 1)
Ácido 5-benciloxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-benciloxi-4-metoxi-piridin-2-carboxílico (140 mg, 0,51 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 358 mg (99%) Tr (HPLC): 0,54 min (método 1)
Éster metílico del ácido 5-(3,3-difluoro-ciclobutilmetoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (118 mg, 0,64 mmol) y (3,3-difluoro-ciclobutil)-metanol (150 mg, 0,82 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 111 mg (47%) ESI-EM: m/z = 288 (M+H)+ Tr (HPLC): 1,20 min (método 5)
Ácido 5-(3,3-difluoro-ciclobutilmetoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-(3,3-difluoro-ciclobutilmetoxi)-4-metoxipiridin-2-carboxílico (110 mg, 0,38 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 73,4 mg (70%) ESI-EM: m/z = 274 (M+H)+ Tr (HPLC): 0,56 min (método 5)
Éster metílico del ácido 4-metoxi-5-propoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y 1-propanol (80,0 |al, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 114 mg (71%) Tr (HPLC): 0,69 min (método 1)
Ácido 4-metoxi-5-propoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 4-metoxi-5-propoxi-piridin-2-carboxílico (114 mg, 0,51 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y 2-ciclopropiletanol (91,7 mg, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 130 mg (73%) Tr (HPLC): 0,82 min (método 1)
Ácido 5-(2-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-(2-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico (130 mg, 0,52 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 122 mg (99%) Tr (HPLC): 0,53 min (método 1)
Éster metílico del ácido 4-metoxi-5-fenetiloxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y 2-feniletanol (128 |l, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 177 mg (87%) Tr (HPLC): 0,90 min (método 1)
Ácido 4-metoxi-5-fenetiloxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 4-metoxi-5-fenetiloxi-piridin-2-carboxílico (177 mg, 0,62 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 168 mg (cuantitativo) Tr (HPLC): 0,63 min (método 1)
Éster metílico del ácido 5-(2,2-dimetil-propoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y 2,2-dimetil-propan-1-ol (93,8 mg, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 160 mg (89%) Tr (HPLC): 0,92 min (método 1)
Ácido 5-(2,2-dimetil-propoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-(2,2-dimetil-propoxi)-4-metoxi-piridin-2-carboxílico (160 mg, 0,63 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 150 mg (99%) Tr (HPLC): 0,61 min (método 1)
Éster metílico del ácido 5-(1-fluorometil-ciclopropilmetoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (118 mg, 0,64 mmol) y (1-fluorometil-ciclopropil)-metanol (101 mg, 0,97 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 159 mg (92%) Tr (HPLC): 0,69 min (método 1)
Ácido 5-(1-fluorometil-ciclopropilmetoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-(1-fluorometil-ciclopropilmetoxi)-4-metoxipiridin-2-carboxílico (159 mg, 0,59 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 150 mg (cuantitativo) Tr (HPLC): 0,43 min (método 1)
Éster metílico del ácido 5-etoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y etanol (62,1 ^l, 1,07 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 151 mg (100%) Tr (HPLC): 0,92 min (método 1)
Ácido 5-etoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-etoxi-4-metoxi-piridin-2-carboxílico (151 mg, 0,71 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 140 mg (99%) Tr (HPLC): 0,83 min (método 1)
Éster metílico del ácido 5-((S)-1-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (118 mg, 0,64 mmol) y (R)-1-ciclopropil-etanol (83,2 mg, 0,97 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 102 mg (63%)
Ácido 5-((S)-1-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-((S)-1-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico (102 mg, 0,41 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 96,0 mg (100%) Tr (HPLC): 0,51 min (método 1)
Éster metílico del ácido 5-isopropoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (130 mg, 0,71 mmol) y propan-2-ol (81,5 |l, 0,97 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluoro-benciloxi)-4-metoxipiridin-2-carboxílico.
Rendimiento: 154 mg (96%) Tr (HPLC): 0,62 min (método 1)
Ácido 5-isopropoxi-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-isopropoxi-4-metoxi-piridin-2-carboxílico (154 mg, 0,68 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 144 mg (cuantitativo)
Éster metílico del ácido 5-((ft)-1-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico

Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-hidroxi-4-metoxi-piridin-2-carboxílico (118 mg, 0,64 mmol) y (S)-1-ciclopropil-etanol (83,2 mg, 0,97 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 101 mg (63%)
Ácido 5-((ft)-1-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico
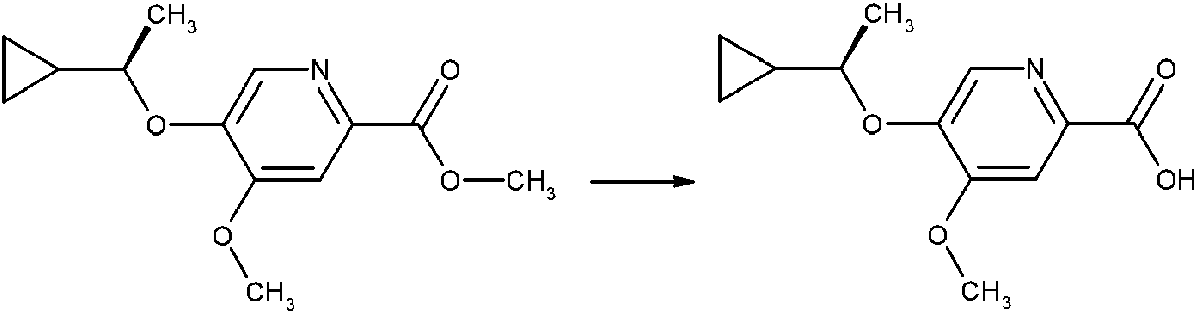
Se sintetiza el compuesto del título a partir de éster metílico del ácido 5-((R)-1-ciclopropil-etoxi)-4-metoxi-piridin-2-carboxílico (101 mg, 0,40 mmol) según el procedimiento descrito para la síntesis del producto intermedio ácido 5-(2-fluoro-benciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 94,0 mg (99%) Tr (HPLC): 0,51 min (método 1)
3-(Trifluorometil)ciclobutil1metanol
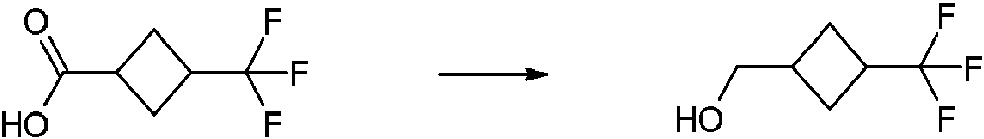
A ácido 3-(trifluorometil)ciclobutano-1-carboxílico (50 mg, 0,29 mmol) en THF (2 ml) se le añade CDI (57 mg, 0,36 mmol) y se agita a Ta durante 2 h. Se añade borohidruro de sodio (12 mg, 0,31 mmol) en agua (0,5 ml) y se agita la mezcla de reacción a TA durante 30 min Se acidifica la mezcla de reacción con HCI 1 M y se extrae con DCM. Se separan las fases orgánicas combinadas y se secan sobre Na2SO4, se filtran y se concentran.
Rendimiento: 45 mg (cuantitativo)
4-Metoxi-5-{[3-(trifluorometil)ciclobutil1metoxi}piridin-2-carboxilato de metilo

Se sintetiza el compuesto del título a partir de 5-hidroxi-4-metoxipiridin-2-carboxilato de metilo (53 mg, 0,29 mmol) y [3-(trifluorometil)ciclobutil]metanol (45 mg, 0,29 mmol) según el procedimiento descrito para la síntesis del producto intermedio éster metílico del ácido 5-(2-fluorobenciloxi)-4-metoxi-piridin-2-carboxílico.
Rendimiento: 90 mg (97%)
Ácido 4-metoxi-5-{[3-(trifluorometil)ciclobutil1metoxi}piridin-2-carboxílico
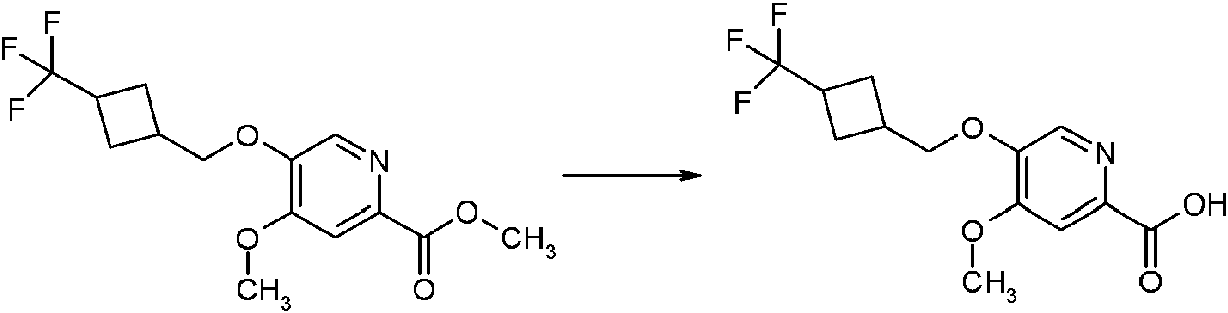
Se añade disolución acuosa de NaOH 4 M (0,55 ml, 2,2 mmol) a 4-metoxi-5-{[3-(trifluorometil)-ciclobutil]metoxi}-piridin-2-carboxilato de metilo (350 mg, 1,10 mmol) en 5 ml de metanol. Se agita la mezcla de reacción durante la noche a TA. Se añade disolución acuosa de HCl 4 M (0,5 ml) y se agita la mezcla de reacción durante 30 min Se evapora la mezcla de reacción a presión reducida. Se añade DMF al residuo y se purifica el compuesto deseado mediante HPLC. Rendimiento: 150 mg (45%)
4-Metoxi-5-(3,3,3-trifluoro-2-metilpropoxi)piridin-2-carboxilato de metilo

A 5-hidroxi-4-metoxipiridin-2-carboxilato de metilo (100 mg, 0,55 mmol) en THF se le añaden 3,3,3-trifluoro-2-metilpropan-1-ol (105 mg, 0,82 mmol) y trifenilfosfina (286 mg, 1,10 mmol) y seguido de azodicarboxilato de diisopropilo (221 mg, 1,10 mmol). Se agita la mezcla de reacción a TA durante 3 h, se evapora la mezcla de reacción a presión reducida y se purifica el residuo mediante HPLC. Se combinan las fracciones que contienen producto y se liofilizan.
Rendimiento: 160 mg (cuantitativo)
Ácido 4-metoxi-5-(3,3,3-trifluoro-2-metilpropoxi)piridin-2-carboxílico

Se añade disolución acuosa de NaOH 4 M (0,52 ml, 2,08 mmol) a 4-metoxi-5-(3,3,3-trifluoro-2-metilpropoxi)piridin-2-carboxilato de metilo (160 mg, 0,55 mmol) en metanol. Se agita la mezcla de reacción durante 2 h a Ta . Se neutraliza la mezcla de reacción con disolución acuosa de HCI 4 M y se evapora a presión reducida. Se usa el residuo sin purificación adicional.
Rendimiento: 150 mg (98%)
Procedimiento general:
En la tabla 3A se resumen los procedimientos para preparar los compuestos 29, 31,49, 56 y 66 de la invención, así como los compuestos de referencia 1-28, 30, 32-48, 50-55, 57-65 y 67-80. En la tabla 3B se resumen los análisis de los compuestos respectivos.
I: A ácido carboxílico (1 eq.) en DMA se le añade HATU (1,2 eq.) y se agita. Se añaden amina (1 eq.) y DIPEA (4,0 eq.) y se agitan durante 18 h a ta. Purificación mediante cromatografía en columna RP (ACN/agua, condiciones ácidas o básicas) o mediante cromatografía en gel de sílice.
II: Se agitan ácido carboxílico (1 eq.) y CDI (1,5 eq.) en DMA durante 30 min a ta. Se añaden amina (1 eq.) y DIPEA (2,0 eq.) y se agitan durante 3 h a ta. Purificación mediante cromatografía en columna RP (ACN/agua, condiciones ácidas o básicas) o mediante cromatografía en gel de sílice.
III: Se agitan amina (1,0 eq.), ácido carboxílico (0,9 eq.), TBTU (1,0 eq.) y DIPEA (4,0 eq.) en NMP durante 18 h a ta. Se purifica la mezcla de reacción filtrada mediante cromatografía en columna r P (ACN/agua, condiciones ácidas o básicas) o mediante cromatografía en gel de sílice.
Tabla 3A. Procedimientos generales para preparar los compuestos 29, 31,49, 56 y 66 de la invención, así como los compuestos de referencia 1-28, 30, 32-48, 50-55, 57-65 y 67-80.











Tabla 3B. Datos analíticos para los compuestos 29, 31, 49, 56 y 66 de la invención, así como los compuestos de referencia 1-28, 30, 32-48, 50-55, 57-65 y 67-80.



Síntesis de productos intermedios nitrogenados
Clorhidrato de [(ft)-4-(6-nitro-piridin-3-il)-piperazin-2-il1-metanol

Se agita éster terc-butílico del ácido (R)-2-(terc-butil-dimetil-silaniloximetil)-4-(6-nitro-piridin-3-il)-piperazin-1-carboxílico (1,73 g, 3,82 mmol) en DCM (10 ml) y HCI 4 M (9,55 ml, 38,2 mmol) a TA durante 2 h. Se concentra la
mezcla de reacción a presión reducida.

Se agitan clorhidrato de [(R)-4-(6-nitro-piridin-3-il)-piperazin-2-il]-metanol (60,0 mg, 0,21 mmol) y ácido 4-metoxi-5-fenoxi-piridin-2-carboxílico (42,8 mg, 0,18 mmol) en NMP (500 |al) con TBTU (70,1 mg, 0,22 mmol) y DIPEA (151 |al, 0,87 mmol) durante 18 h a ta. Se purifica la mezcla de reacción mediante cromatografía en columna RP (ACN/agua/NH4HCO3). Se purifica el residuo de nuevo mediante cromatografía en columna de fase normal (MeOH/DCM) para dar el compuesto del título.
Rendimiento: 95 mg (93%)

Se sintetiza el compuesto del título a partir de clorhidrato de [(R)-4-(6-nitro-piridin-3-il)-piperazin-2-il]-metanol (60,0 mg, 0,22 mmol) y ácido 4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-carboxílico (48,1 mg, 0,18 mmol) según el procedimiento descrito para la síntesis del producto intermedio [(R)-2-hidroximetil-4-(6-nitro-piridin-3-il)-piperazin-1-il]-(4-metoxi-5-fenoxi-piridin-2-il)-metanona.
Rendimiento: 102 mg (cuantitativo) ESI-EM: m/z = 496 (M+H)+ Tr (HPLC): 0,78 min (método 1) Procedimiento:
IV: Se agita producto intermedio nitrogenado (1 eq.) y Pd/C (10%) en MeOH durante 20 h a TA bajo una atmósfera de hidrógeno. Se concentra la mezcla de reacción a vacío y se purga con argón. Se filtra el residuo a través de Celite® y se lava con MeOH. Se concentra el filtrado a presión reducida y se purifica el producto en bruto mediante cromatografía en columna RP (ACN/agua, condiciones básicas o ácidas).
Tabla 4. Procedimientos para preparar los compuestos de referencia 81 y 82.

Los compuestos 85 y 87 de la invención, así como los compuestos de referencia 83, 84, 86, 88 y 89 se preparan
generalmente haciendo reaccionar un producto intermedio de ácido carboxílico con un producto intermedio de amina en condiciones similares a las descritas para el procedimiento general (I) en la tabla 3A. En la tabla 5B se resumen los análisis de los compuestos respectivos.
Síntesis de productos intermedios
4-Etoxi-5-fenoxipicolinonitrilo

A una disolución de 5-fluoro-4-isopropoxipicolinonitrilo (500 mg, 3,01 mmol) en DMF (10 ml) agitada a TA bajo una atmósfera de N2 se le añaden fenol (339,85 mg, 3,61 mmol) y K2CO3 (1,25 g, 9,03 mmol), se calienta la mezcla resultante hasta 100°C durante 3 h. Luego se diluye la mezcla de reacción con acetato de etilo (50 ml), se lava con agua y salmuera, se seca sobre Na2SO4 anhidro, se filtra y se concentra. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice.
Rendimiento: 530 mg (73%) m/z = 241 (M+H)+.
Ácido 4-etoxi-5-fenoxipicolínico

Se agita una mezcla de 4-etoxi-5-fenoxipicolinonitrilo (530 mg, 2,21 mmol) en disolución de hidróxido de sodio 2 N (10 ml) a 100°C durante la noche. Luego se acidifica la mezcla de reacción mediante HCI 1 N para ajustar a pH = 4 y se extrae con DCM (20 ml x 2). Se separan las fases orgánicas combinadas y se secan sobre Na2SO4, se filtran y se concentran para dar el producto deseado que puede usarse sin purificación adicional.

A una mezcla agitada de 5-bromo-4-ciclopropoxipiridin-2-amina (2,1 g, 9,17 mmol), éster terc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (4,25 g, 13,75 mmol) y Cs2CO3 (9,0 g, 27,50 mmol) en dioxano (60 ml) y agua (12 ml) a ta bajo una atmósfera de nitrógeno se le añade Pd(dppf)Ch (200 mg,
0,27 mmol). Se agita la mezcla resultante a 90°C durante 4 h. Luego se vierte la mezcla de reacción en agua helada y se extrae con DCM (50 ml x 3). Se lavan las fases orgánicas combinadas con salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 3 g (98%) m/z = 332 (M+H)+.
4-(6-Amino-4-ciclopropoxipiridin-3-il)piperidin-1-carboxilato de tere-butilo

A una disolución de 6-amino-4-ciclopropoxi-1’,2’,3’,6’-tetrahidro-[3,4’-bipiridina]-1’-carboxilato de tere-butilo (3 g, 9,05 mmol) en EtOH (40 ml) se le añade Pd(OH)2/C (2 g). Se agita la mezcla de reacción resultante a 25°C bajo una atmósfera de hidrógeno durante 16 h. Se retira el catalizador por filtración a través de Celite®, y se evapora el filtrado hasta sequedad a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 1,8 g (60%) m/z = 334 (M+H)+.
Diclorhidrato de 4-ciclopropoxi-5-(piperidin-4-il)piridin-2-amina

Se disuelve 4-(6-amino-4-ciclopropoxipiridin-3-il)piperidin-1-carboxilato de tere-butilo (1,6 g, 4,8 mmol) en una disolución de HCl (g) en EtOH (10 ml). Se agita la mezcla de reacción a TA durante 2 h. Después de completarse la reacción, se elimina el disolvente a presión reducida. Luego se tritura el producto en bruto con Et2O para dar el producto deseado que puede usarse sin purificación adicional.
Rendimiento: 1 g (90%) m/z = 234 (M+H)+.
(4-Propoxipiridin-2-il)carbamato de tere-butilo

A una disolución agitada de 2-aminopiridin-4-ol (1,25 g, 11,4 mmol) en N,N-dimetilacetamida (15 ml) se le añaden carbonato de cesio (7,42 g, 22,8 mmol), bromuro de propilo (1,24 ml, 13,6 mmol) y yoduro de cesio (2,95 g, 11,4 mmol). Se agita la mezcla resultante a 100°C durante 1 día. Se añade dicarbonato de di-tere-butilo (2,74 g, 12,6 mmol) a la mezcla de reacción y se agita a 100°C durante 16 h. Se diluye la mezcla de reacción con agua (20 ml) y se extrae con EtOAc (50 ml). Se separan las fases y se concentra la fase orgánica. Se purifica la mezcla en bruto mediante cromatografía en columna sobre gel de sílice para proporcionar el producto deseado.
Rendimiento: 787 mg (27%) m/z = 253 (M+H)+.
(5-Bromo-4-propoxipiridin-2-iDcarbamato de ferc-butilo

A una disolución agitada de (4-propoxipiridin-2-il)carbamato de ferc-butilo (0,79 g, 3,11 mmol) en ácido acético (5 ml) se le añade bromo (0,40 g, 2,49 mmol, en 1 ml de ácido acético) gota a gota a 0°C. Después de 0,5 h, se añade una cantidad adicional de ácido acético (8 ml) y se permite que la mezcla de reacción se caliente hasta temperatura ambiente. Después de 1 h, se concentra la mezcla y se purifica mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.

A una disolución de (5-bromo-4-propoxipiridin-2-il)carbamato de ferc-butilo (254 mg, 0,77 mmol) en dioxano (4 ml) se le añaden éster ferc-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico (596 mg, 1,93 mmol), carbonato de sodio (disolución acuosa 2 M, 0,77 ml) y [1,1’-bis(difenilfosfino)ferroceno]-dicloropaladio(II) (56 mg, 0,077 mmol). Se agita la mezcla de reacción a 100°C durante 24 h. Se diluye la mezcla de reacción con EtoAc (10 ml) y se filtra a través de un lecho de agente de filtración SuperCell. Se concentra el filtrado y se purifica mediante cromatografía en columna sobre gel de sílice para proporcionar el producto deseado.
Rendimiento: 333 mg (cuantitativo) m/z = 434 (M+H)+.
4-(6-((7erc-butoxicarbonil)amino)-4-propoxipiridin-3-il)piperidin-1-carboxilato de ferc-butilo

A 6-{[(ferc-butoxi)carbonil]amino}-4-propoxi-1’,2’,3’,6’-tetrahidro-[3,4’-bipiridina]-1’-carboxilato de ferc-butilo (333 mg, 0,77 mmol) en EtOH (18 ml) y EtoAc (3 ml) se le añade hidróxido de paladio sobre carbono (20% húmedo, 27 mg). Se agita la mezcla de reacción bajo una atmósfera de hidrógeno (43 psi) durante 3 días y se filtra a través de agente de filtración SuperCell. Se concentra el filtrado a presión reducida para proporcionar el producto deseado.
Rendimiento: 330 mg (98%) m/z = 436 (M+H)+.
Diclorhidrato de 5-(piperidin-4-il)-4-propoxipiridin-2-amina

A 4-(6-((tere-butoxicarbonil)amino)-4-propoxipiridin-3-il)piperidin-1-carboxilato de tere-butilo (330 mg, 0,76 mmol) en diclorometano (2 ml) se le añade una disolución de HCl en dioxano (2,00 ml, 4 M, 8,0 mmol). Se agita la mezcla de reacción durante 16 h y se concentra. Se tritura el residuo con DCM y se seca a vacío para proporcionar el producto deseado.
Rendimiento: 233 mg cuantitativo.
Diclorhidrato de 4-etoxi-5-(piperidin-4-il)piridin-2-amina

Puede sintetizarse diclorhidrato de 4-etoxi-5-(piperidin-4-il)piridin-2-amina de manera análoga al protocolo para la síntesis de diclorhidrato de 5-(piperidin-4-il)-4-propoxipiridin-2-amina. La alquilación de 2-aminopiridin-4-ol con bromuro de etilo y la posterior protección con Boc conduce a la formación de N-(4-etoxipiridin-2-il)carbamato de terebutilo. La bromación de N-(4-etoxipiridin-2-il)carbamato de tere-butilo conduce a la síntesis de (5-bromo-4-etoxipiridin-2- il)carbamato de tere-butilo. La posterior reacción con éster tere-butílico del ácido 4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1-carboxílico conduce a la formación de 6-{[(tercbutoxi)carbonil]amino}-4-etoxi-1’,2’,3’,6’-tetrahidro-[3,4’-bipiridina]-1’-carboxilato de tere-butilo. En la siguiente etapa, se obtiene 4-(6-((tere-butoxicarbonil)amino)-4-etoxipiridin-3-il)piperidin-1-carboxilato de tere-butilo a través de hidrogenación. La escisión del grupo protector Boc conduce a la síntesis de diclorhidrato de 4-etoxi-5-(piperidin-4-il)piridin-2-amina.
3- (6-Aminopiridazin-3-il)-8-azabiciclo[3.2.11oct-2-eno-8-carboxilato de tere-butilo

A 3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-8-azabiciclo[3.2.1]-oct-2-eno-8-carboxilato de tere-butilo (1,93 g, 5,75 mmol) y 6-bromopiridazin-3-amina (1,00 g, 5,75 mmol) en 1,4-dioxano (25 ml) se le añaden disolución ac. de Na2CO32 M (11,5 ml, 23,0 mmol) y catalizador Xphos de 2a generación (136 mg, 0,17 mmol). Se desgasifica la mezcla de reacción con argón y se agita a 100°C durante 2 h. Se evaporan todas las sustancias volátiles a presión reducida. Se purifica el material en bruto mediante cromatografía de fase normal para obtener el compuesto del título.
Rendimiento: 0,80 g (46%) ESI-EM: m/z = 303 (M+H)+
3-(6-Aminopiridazin-3-il)-8-azabiciclo[3.2.11octano-8-carboxilato de ferc-butilo

A 3-(6-aminopiridazin-3-il)-8-azabiciclo[3.2.1]oct-2-eno-8-carboxilato de ferc-butilo (0,80 g, 2,65 mmol) en MeOH (30 ml) se le añade Pd/C (250 mg) bajo nitrógeno. Se desgasifica la mezcla de reacción y se hidrogena a una atmósfera de hidrógeno de 3 bar a TA durante la noche. Se filtra la mezcla de reacción y se concentra a presión reducida. Rendimiento: 800 mg (cuantitativo) ESI-EM: m/z = 305 (M+H)+
Diclorhidrato de 6-{8-azabiciclo[3.2.11octan-3-il)piridazin-3-amina

A 3-(6-aminopiridazin-3-il)-8-azabiciclo[3.2.1]octano-8-carboxilato de ferc-butilo (800 mg, 2,63 mmol) en un volumen apropiado de DCM se le añade HCl 4 M en 1,4 dioxano y se agita a TA hasta que se completa la reacción. Se evaporan todas las sustancias volátiles a presión reducida.
Rendimiento: 700 mg (96%) ESI-EM: m/z = 205 (M+H)+
2-Cloro-5-fluoro-4-metoxipiridina

A 2-cloro-5-fluoropiridin-4-ol (1 g, 7,05 mmol) y K2CO3 (1,27 g, 9,16 mmol) en DMF (10 ml) se les añade yodometano (1,15 g, 8,13 mmol) a temperatura ambiente. Se agita la mezcla de reacción resultante a Ta durante 2 h. Se diluye la mezcla de reacción con agua (20 ml) y se extrae con EtOAc (30 ml x 2). Se lavan las fases orgánicas combinadas con agua y salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento^ g (91%) m/z = 162 (M+H)+
5-Fluoro-4-metoxipicolinonitrilo

A 2-cloro-5-fluoro-4-metoxipiridina (1,0 g, 6,2 mmol), cianuro de zinc (800 mg, 6,8 mmol) y dppf (34 mg, 0,62 mmol) en DMF (10 ml) agitados a TA bajo nitrógeno atmósfera, se les añade Pd2(dba)3 (56 mg, 0,62 mmol). Se agita la mezcla de reacción a 150°C bajo una atmósfera de nitrógeno durante 3 h. Luego se diluye la mezcla de reacción con agua (30 ml) y se extrae con EtOAc (30 ml x 2). Se lavan las fases orgánicas combinadas con agua y salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 700 mg (74 %) m/z = 153 (M+H)+.
4-Metoxi-5-(4-(trifluorometil)fenoxi)picolinonitrilo

A 5-fluoro-4-metoxipicolinonitrilo (700 mg, 4,6 mmol) en DMF (10 ml) se le añaden 4-(trifluorometil)fenol (746 mg, 4,6 mmol) y K2CO3 (636 mg, 4,6 mmol). Se agita la mezcla de reacción a 100°C durante 16 h. Se diluye la mezcla de reacción con agua (20 ml) y se extrae con EtOAc (20 ml x 2). Se combinan las fases orgánicas combinadas, se lavan con agua y salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 1 g (80 %) m/z = 295 (M+H)+.
Ácido 4-metoxi-5-(4-(trifluorometil)fenoxi)picolínico

A una disolución de NaOH (1,6 g, 40 mmol) en agua (20 ml) se le añade 4-metoxi-5-(4-(trifluorometil)fenoxi)picolinonitrilo (700 mg, 2,4 mmol). Se agita la mezcla de reacción a 100°C durante la noche. Se acidifica la mezcla de reacción mediante HCl 6 M para ajustar a pH = 2, se extrae con EtOAc (30 ml x 2). Se lavan las fases orgánicas combinadas con agua y salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida para dar el producto en bruto que puede usarse directamente sin purificación adicional.
Rendimiento: 700 mg (94%) m/z = 314 (M+H)+.
2-Cloro-5-fluoropiridin-4-ol

Bajo una atmósfera de nitrógeno a -78°C, a una disolución agitada de 2-cloro-5-fluoropiridina (5,0 g, 38 mmol) en tetrahidrofurano (50 ml), se le añade diisopropilamiduro de litio (24,7 ml, 49,4 mmol, 2 M en tetrahidrofurano) gota a gota a lo largo de 30 min. Se agita la mezcla de reacción a -78°C durante 2 h. Después de eso, se añade una disolución de borato de trimetilo (7,9 g, 76,03 mmol) en tetrahidrofurano (10 ml) gota a gota a lo largo de 20 min. Después de la adición, se agita la mezcla de reacción a TA durante otras 2 h. Se enfría la mezcla de reacción hasta 0°C y se añade ácido acético (6,5 ml). Se agita la mezcla de reacción a 0°C durante 30 min. Se añade peróxido de hidrógeno (11,5 ml, disolución al 30%) gota a gota a 0°C. Se agita la mezcla de reacción a TA durante la noche. Se extingue la mezcla de reacción con NaS2O4 acuoso saturado. Se añade HCl 5 N a la mezcla de reacción. Después de la extracción con EtOAc (50 ml x 3), se lavan las fases orgánicas combinadas con agua y salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en gel de sílice para dar el producto deseado.
Rendimiento: 3,8 g (68%). m/z = 149 (M+H)+.
2-Cloro-4-etoxi-5-fluoropiridina

A 2-cloro-5-fluoropiridin-4-ol (2, 3,0 g, 20,33 mmol) y carbonato de plata(I) (8,4 g, 30,50 mmol) en DMF (50 ml) se les añade yodoetano (9,51 g, 61,00 mmol) a 0°C bajo una atmósfera de nitrógeno. Se agita la mezcla de reacción a temperatura ambiente durante 3 h. Luego se diluye la mezcla de reacción con acetato de etilo (100 ml) y se lava con agua y salmuera, se seca sobre Na2SO4 anhidro, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 3,0 g (84%) m/z = 177 (M+H)+.
4-Etoxi-5-fluoropicolinonitrilo

A 2-cloro-4-etoxi-5-fluoropiridina (300 mg, 1,71 mmol) en DMF (10 ml) se le añaden dicianozinc (141 mg, 1,2 mmol), zinc (22,3 mg, 0,34 mmol) y Pd(dppf)Ch (50 mg) bajo una atmósfera de nitrógeno. Se agita la mezcla de reacción a 150°C durante 3 h. Se diluye la mezcla de reacción con acetato de etilo (50 ml), se lava con agua y salmuera, se seca sobre Na2SO4 anhidro, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 220 mg (78%) CL-EM: m/z 167 [M+H]+.
4-Etoxi-5-(4-fluorofenoxi)picolinonitrilo

A 4-fluorofenol (202 mg, 1,81 mmol) y K2CO3 (249 mg, 1,81 mmol) en DMF (5 ml) se les añade 4-etoxi-5-fluoropicolinonitrilo (200 mg, 1,2 mmol) en una porción. Se agita la mezcla de reacción a 100°C durante 3 h. Después de enfriar, se diluye la mezcla de reacción con acetato de etilo (20 ml), se lava con agua y salmuera, se seca sobre Na2SO4 anhidro, se filtra y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en columna sobre gel de sílice para dar el producto deseado.
Rendimiento: 220 mg (71%) m/z = 259 (M+H)+.
Ácido 4-etoxi-5-(4-fluorofenoxi)picolínico

Se agita una mezcla de 4-etoxi-5-(4-fluorofenoxi)picolinonitrilo (500 mg, 1,94 mmol) en una disolución acuosa de hidróxido de sodio 2 N (10 ml) a 100°C durante la noche. Después de enfriar, se acidifica la mezcla de reacción mediante HCl acuoso 1 N para ajustar a pH = 4 y se extrae con DCM (20 ml x 2). Se lavan las fases orgánicas combinadas con agua y salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida para dar el producto deseado en bruto.
Rendimiento: 490 mg (91%) m/z = 278 (M+H)+.
Tabla 5A. Procedimientos para preparar los compuestos 85 y 87 de la invención, así como los compuestos de referencia 83, 84, 86, 88 y 89.


Tabla 5B. Datos analíticos para los compuestos 85 y 87 de la invención, así como los compuestos de referencia 83, 84, 86, 88 y 89.

Procedimientos generales:
En la tabla 6A se resumen los procedimientos para preparar el compuesto 90 de la invención, así como el compuesto de referencia 91. En la tabla 6B se resumen los análisis de los compuestos respectivos.
V: A ácido carboxílico (1,0 eq.) (producto intermedio 2 en la siguiente tabla 6A) en DMF se le añaden DIPEA (3,0 eq.) y HATU (1,0 eq.) y se agita la mezcla de reacción durante 30 min a ta. Se añade amina (1,0 eq) (producto intermedio 1 en la siguiente tabla 6A) y se agita la mezcla de reacción durante la noche. Se purifica la mezcla de reacción filtrada mediante cromatografía en columna RP (ACN/agua TFA o condiciones básicas).
Tabla 6A. Procedimientos generales para preparar el compuesto 90 de la invención, así como el compuesto de referencia 91.

Tabla 6B. Datos analíticos para el compuesto 90 de la invención, así como el compuesto de referencia 91.

Compuestos de referencia 92 y 93:
Sal de TFA de 4-metoxi-5-[1-(4-metoxi-5-{[3-(trifluorometN)-ciclobutil1metoxi}-piridin-2-carbonil)piperidin-4-il1piridin-2-amina

Se agitan ácido 4-metoxi-5-(3-trifluorometil-ciclobutilmetoxi)-piridin-2-carboxílico (40 mg, 0,13 mmol), DIPEA (113 |al, 0,66 mmol), HATU (54 mg, 0,144 mmol) y diclorhidrato de 4-metoxi-5-(piperidin-4-il)piridin-2-amina (37 mg, 0,13 mmol) en DMF (2 ml) durante la noche a ta. Se purifica la mezcla de reacción mediante cromatografía en columna Rp (ACN/agua TFA) para obtener ambos estereoisómeros.
Rendimiento: compuesto 92 (isómero trans): 5 mg (6%) Tr en HPLC: 0,50 min (método 12) y compuesto 93 (isómero cis): 8 mg (10%) Tr en HPLC: 0,48 min (método 12), ESI-EM: m/z = 495 (M+H)+
Compuesto de referencia 94:
4-Metoxi-5-{1-[4-metoxi-5-(3,3,3-trifluoro-2-metilpropoxi)piridin-2-carbonil1piperidin-4-il}piridin-2-amina

Se agitan ácido 4-metoxi-5-(3,3,3-trifluoro-2-metilpropoxi)piridin-2-carboxílico (110 mg, 0,39 mmol), DIPEA (271 ^l, 1,58 mmol), HATU (150 mg, 0,39 mmol) y diclorhidrato de 4-metoxi-5-(piperidin-4-il)piridin-2-amina (121 mg, 0,43 mmol) en DMF (2 ml) durante 2 h a ta. Se purifica la mezcla de reacción mediante cromatografía en columna RP. Rendimiento: 110 mg (60%) ESI-EM: m/z = 469 (M+H)+Tr en HPLC: 0,71 min (método 13)
Enantiómeros de 4-metoxi-5-(1-{4-metoxi-5-[3,3,3-trifluoro-2-metilpropoxi1piridin-2-carbonil}piperidin-4-il)-piridin-2-amina (94):
4-Metoxi-5-(1-{4-metoxi-5-[(2S)-3,3,3-trifluoro-2-metilpropoxi1piridin-2-carbonil}piperidin-4-il)-piridin-2-amina y 4-metoxi-5-(1-{4-metoxi-5-[(2ft)-3,3,3-trifluoro-2-metilpropoxi1piridin-2-carboniDpiperidin-4-N)piridin-2-amina

Se separa adicionalmente 4-metoxi-5-{1-[4-metoxi-5-(3,3,3-trifluoro-2-metilpropoxi)piridin-2-carbonil1piperidin-4-il}piridin-2-amina (292 mg, 0,62 mmol) mediante cromatografía de fluidos supercríticos quiral (CFS, dióxido de carbono supercrítico/NH320 mM en EtOH, Chiral ART®, Amylose-SC 20x250 mm, 5 p,M) para obtener ambos enantiómeros 94a (primera fracción de elución) y 94b (segunda fracción de elución). La estereoquímica se asigna al azar.
Rendimiento: 70 mg (48%, compuesto 94a; Tr: 5,69 min) y 74 mg (50%, compuesto 94b; Tr: 6,23 min)
Ácido 5-hidroxi-4-metoxipiridin-2-carboxílico

Se añade hidróxido de potasio (6,28 g, 111,98 mmol) en 50 ml de agua a 5-bromo-4-metoxipiridin-2-carboxilato de metilo (5,00 g, 20,32 mmol) en 1,4-dioxano (50 ml). Se añaden di-ferc-butil-(2’,4’,6’-triisopropil-3,4,5,6-tetrametil-bifenil-2-il)-fosfano (1,57 g, 3,27 mmol) y tris(dibencilidenoacetona)dipaladio(0) (949 mg, 1,04 mmol) bajo argón. Se agita la mezcla de reacción a 100°C durante 2 h. Se filtra la mezcla de reacción y se concentra a presión reducida. Se acidifica el residuo con HCl 4 M y se filtra el sólido. Se concentra la fase líquida y se recoge el precipitado, se lava y se seca. Rendimiento: 2,61g (76%) ESI-EM: m/z = 170 (M+H)+
6-[4-(6-Amino-4-metoxipiridin-3-il)piperidin-1-carbonil]-4-metoxipiridin-3-ol

A ácido 5-hidroxi-4-metoxipiridin-2-carboxílico (100 mg, 0,59 mmol) en DMF (5 ml) se le añaden DIPEA (407 ^l, 2,36 mmol) y diclorhidrato de 4-metoxi-5-(piperidin-4-il)piridin-2-amina (331 mg, 1,18 mmol). Luego se añade HATU (225 mg, 0,59 mmol). Se agita la mezcla de reacción durante la noche a TA y se purifica mediante cromatografía en columna de fase inversa para proporcionar el compuesto del título.
Rendimiento: 140 mg (66%) ESI-EM: m/z = 359 (M+H)+ Tr (HPLC): 0,61 min (método 10)
Procedimientos generales:
En la tabla 7A se resumen un procedimiento para preparar el compuesto de referencia 95. En la tabla 7B se resume el análisis del compuesto de referencia 95 de la invención.
V[: A 6-[4-(6-amino-4-metoxipiridin-3-il)piperidin-1-carbonil]-4-metoxipiridin-3-ol (1,0 eq.) (producto intermedio 2 en la siguiente tabla 7A) en dioxano se le añaden alcohol (2,4 eq.) (producto intermedio 1 en la siguiente tabla 7A), TPP (2,7 eq.) y DTAD (2,5 eq.). Se agita la mezcla de reacción a 60°C durante 1 h. Si la reacción muestra una conversión completa, se purifica la mezcla de reacción mediante cromatografía en columna RP (ACN/agua TFA). Si la reacción no muestra completitud, se añaden TPP (2,7 eq.) y DTAD (2,5 eq.) adicionales hasta que se produzca la conversión. Después de cada adición, se agita la mezcla de reacción a 60°C durante 1 h. Se purifica la mezcla de reacción mediante cromatografía en columna RP (ACN/agua TFA).
Tabla 7A. Procedimientos generales para preparar el compuesto de referencia 95.

Tabla 7B. Datos analíticos para el compuesto de referencia 95.

Preparación alternativa del compuesto de referencia 15-{4-[5-(4-fluorofenoxi)-4-metoxipiridin-2-carbonil]piperazin-1-ilM-metoxipiridin-2-amina
5-Bromo-2-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridina

Se agitan 5-bromo-4-metoxi-piridin-2-ilamina (9,50 g, 46,79 mmol), hexano-2,5-diona (7,08 ml, 60,83 mmol) y ácido ptoluenosulfónico (0,81 g, 4,68 mmol) en tolueno (80 ml) durante la noche a 120°C usando un aparato de Dean-Stark. Se concentra la mezcla de reacción a presión reducida, se lleva a DCM y se purifica mediante cromatografía en gel de sílice (DCM).
Rendimiento: 7,60 g (58%) ESI-EM: m/z = 281 [M+H]+ Tr (HPLC): 1,13 min (método 7)

Se realiza la reacción bajo una atmósfera de argón. Se agitan 5-bromo-2-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridina (1,00 g, 3,56 mmol), piperazin-1-carboxilato de tere-butilo (0,73 g, 3,92 mmol), metanosulfonato de CPhos-3G (0,30 g, 0,36 mmol) y carbonato de cesio (3,48 g, 10,67 mmol) en 1,4-dioxano (15 ml) durante la noche a 80°C. Se filtra la mezcla de reacción y se concentra a presión reducida. Se lleva el residuo a DCM (20 ml) y se añade TFA (1,37 ml, 17,76 mmol). Se agita la mezcla de reacción durante 3 días a TA y, después de la adición de la misma cantidad de TFA, se agita la mezcla de reacción durante la noche a 40°C. Se evapora la mezcla de reacción hasta sequedad y se usa sin purificación adicional.
Rendimiento: 1,80 g (98%) ESI-EM: m/z = 287 [M+H]+ Tr (HPLC): 0,67 min (método 7)
4-Metoxi-5-(piperazin-1-il)piridin-2-amina

Se agitan bis(ácido trifluoroacético) de 1-[6-(2,5-dimetil-1H-pirrol-1-il)-4-metoxipiridin-3-il]piperazina (1,20 g, 2,33 mmol), clorhidrato de hidroxilamina (0,70 g, 10,03 mmol) y trietilamina (1,00 ml, 7,11 mmol) en EtOH/agua (1/1; 16 ml) durante la noche a 80°C. Se elimina el disolvente orgánico a presión reducida. Se purifica el residuo mediante RP-HPLC (ACN/agua NHs).
Rendimiento: 290 mg (60%) ESI-EM: m/z = 209 [M+H]+ Tr (HPLC): 0,35 min (método 11)
5-(4-Fluorofenoxi)-4-metoxipiridin-2-carbonitrilo

Se agitan 5-fluoro-4-metoxi-piridin-2-carbonitrilo (1,00 g; 6,57 mmol), 4-fluorofenol (0,88 g; 7,89 mmol) y carbonato de potasio (2,00 g; 14,46 mmol) en NMP a 105°C durante 1,5 horas. Se permite que la mezcla de reacción se enfríe hasta TA y se extrae con EtOAc. Se lava la fase orgánica con agua y salmuera, se separa, se seca sobre Na2SO4, se filtra y se concentra a presión reducida. Se somete a levigación el residuo con PE, se filtra y se seca en un horno de secado a 60°C.
Rendimiento: 1,54 g (96%) ESI-EM: m/z = 245 [M+H]+ Tr (HPLC): 1,03 min (método 7)
Ácido 5-(4-fluorofenoxi)-4-metoxipiridin-2-carboxílico

Se agitan 5-(4-fluorofenoxi)-4-metoxipiridin-2-carbonitrilo (1,54 g; 6,31 mmol) y NaOH (2 mol/l, disolución ac.; 15,40 ml, 30,80 mmol) a 105°C durante 10 horas. Se permite que la mezcla de reacción se enfríe hasta TA y se deja durante 3 días. Se filtra el precipitado resultante y se somete a levigación en agua. Se calienta la mezcla de reacción hasta 50°C y se ajuste el pH a pH 7 usando HCl (4 mol/l, disolución ac.). Se filtra el precipitado resultante, se lava con EE y se seca en un horno de secado a 60°C.
Rendimiento: 0,84 g (51%) ESI-EM: m/z = 264 [M+H]+ Tr (HPLC): 0,77 min (método 7)
5-{4-5-(4-Fluorofenoxi)-4-metoxipiridin-2-carbonil]piperazin-1-ilM-metoxipiridin-2-amina

Se agitan ácido 5-(4-fluorofenoxi)-4-metoxipiridin-2-carboxílico (0,40 g; 1,92 mmol), HATU (0,75 g; 1,97 mmol) y DIPEA (1,16 ml; 6,72 mmol) en DMF (10 ml) durante 30 minutos a TA. Se añade 4-metoxi-5-(piperazin-1-il)piridin-2-amina (0,52 g; 1,98 mmol) y se permite que la mezcla de reacción se agite a TA durante la noche. Se purifica la mezcla mediante RP-HPLC (ACN/agua NH3).
Rendimiento: 0,31 g (36%) ESI-EM: m/z = 454 [M+H]+Tr (HPLC): 0,88 min (método 11)
Preparación alternativa del compuesto de referencia 39 ácido trifluoroacético de 4-metoxi-5-{1-[4-metoxi-5-(2-metilpropoxi)piridin-2-carbonil]piperidin-4-il)-piridin-2-amina
4-Metoxi-5-(2-metilpropoxi)piridin-2-carboxilato de metilo

Se agitan 5-hidroxi-4-metoxipiridin-2-carboxilato de metilo (0,40 g, 2,18 mmol), 2-metilpropan-1-ol (0,40 ml, 4,37 mmol) y TPP (1,72 g, 6,55 mmol) en THF durante 10 minutos a TA. Se enfría la mezcla de reacción en un baño de hielo y se añade DTAD (1,51 g; 6,55 mmol). Después de 30 minutos, se purifica la mezcla de reacción mediante RP-HPLC (ACN/agua TFA).
Rendimiento: 0,30 g (57%) ESI-EM: m/z = 240 [M+H]+ Tr (HPLC): 0,85 min (método 7)
Ácido 4-metoxi-5-(2-metilpropoxi)piridin-2-carboxílico

Se agitan 4-metoxi-5-(2-metilpropoxi)piridin-2-carboxilato de metilo (0,30 g; 1,25 mmol) y NaOH (4 mol/l, disolución ac.; 0,47 ml; 1,88 mmol) en MeOH (8 ml) a TA durante 3 días. Se neutraliza el pH de la mezcla de reacción usando HCl (4 mol/l; disolución ac.) y se eliminan los disolventes a presión reducida. Se añaden DCM y una pequeña cantidad de MeOH al residuo. Se elimina el material insoluble por filtración y se eliminan las aguas madres a presión reducida. Se usa el residuo sin purificación adicional.
Rendimiento: 0,20 g (71%) ESI-EM: m/z = 226 [M+H]+ Tr (HPLC): 0,76 min (método 7)
6-Amino-4-metoxi-1’,2’,3’,6’-tetrahidro-[3,4’-bipiridina]-1’-carboxilato de ferc-butilo

Se realiza la reacción bajo una atmósfera de argón. Se purgan 5-bromo-4-metoxipiridin-2-amina (7,40 g; 32,80 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo (11,16 g; 36,08 mmol) y carbonato de sodio (2 mol/l, disolución ac.; 65,60 ml; 131,21 mmol) en 1,4-dioxano (300 ml) con argón. Después de 5 minutos, se añade Xphos de 2a gen. (0,77 g; 0,98 mmol) y se agita la mezcla de reacción durante la noche en un vial sellado a 100°C. Se concentra la mezcla de reacción a presión reducida. Se lleva el residuo a agua y se extrae
varias veces con EtOAc. Se secan las fases orgánicas combinadas sobre Na2SO4, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en gel de sílice (DCM/MeOH).
Rendimiento: 9,69 g (97%) ESI-EM: m/z = 306 [M+H]+ Tr (HPLC): 0,83 min (método 10)
4-(6-Amino-4-metoxipiridin-3-il)-piperidin-1-carboxilato de ferc-butilo

Bajo una atmósfera de hidrógeno (aparato de Parr; 50 psi), se agitan 6-amino-4-metoxi-1’,2’,3’,6’-tetrahidro-[3,4’-bipiridina]-1 ’-carboxilato de ferc-butilo (5,11 g; 16,73 mmol) y Pd/C (10%; 0,60 g) en MeOH (100 ml) a TA durante 41,5 horas. Se añade catalizador adicional dos veces y se hidrogena adicionalmente la mezcla de reacción. Después de retirar el catalizador por filtración, se concentran las aguas madres a presión reducida. Se usa el producto sin purificación adicional.
Rendimiento: 4,71 g (92%) ESI-EM: m/z = 308 [M+H]+ Tr (HPLC): 0,82 min (método 10)
Diclorhidrato de 4-metoxi-5-(piperidin-4-il)piridin-2-amina

Se agitan 4-(6-amino-4-metoxipiridin-3-il)-piperidin-1-carboxilato de ferc-butilo (6,90 g; 22,45 mmol) y HCl (4 mol/l; disolución en 1,4-dioxano; 69,00 ml; 224,47 mmol) en DCM (89,70 ml) a TA durante la noche. Se concentra la mezcla de reacción a presión reducida. Se somete a levigación el residuo en EE y se filtra. Se usa el producto sin purificación adicional.
Rendimiento: 5,30 g (84%) ESI-EM: m/z = 208 [M+H]+ Tr (HPLC): 0,66 min (método 11)
Ácido trifluoroacético de 4-metoxi-5-{1-[4-metoxi-5-(2-metilpropoxi)piridin-2-carbonil]piperidin-4-il}-piridin-2-amina

Se agitan ácido 4-metoxi-5-(2-metilpropoxi)piridin-2-carboxílico (80 mg; 0,36 mmol), diclorhidrato de 4-metoxi-5-(piperidin-4-il)piridin-2-amina (96 mg; 0,36 mmol), DIPEA (0,24 ml; 1,42 mmol) y HATU (149 mg; 0,39 mmol) en DMF (3 ml) a TA durante la noche. Se purifica la mezcla de reacción mediante RP-HPLC (ACN/agua TFA).
Rendimiento: 0,11 g (72%) ESI-EM: m/z = 415 [M+H]+ Tr (HPLC): 0,80 min (método 7)
Preparación alternativa del compuesto de referencia 176-{1-[5-(4-fluorofenoxi)-4-metoxipiridin-2-carbonil1piperidin-4-il)piridazin-3-amina
4-(6-Aminopiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo

Se realiza la reacción bajo una atmósfera de argón. Se purgan 6-cloropiridazin-3-amina (5,20 g; 40,14 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo (13,65 g; 44,15 mmol) y
carbonato de sodio (2 mol/l, disolución ac.; 80,28 ml; 160,56 mmol) en 1,4-dioxano (350 ml) con argón. Después de 5 minutos, se añade Xphos de 2a gen. (0,95 g; 1,20 mmol) y se agita la mezcla durante la noche en un vial sellado a 100°C. Se filtra la mezcla de reacción y se concentra a presión reducida. Se lleva el residuo a MeOH, se precipita con agua y se filtra. Se seca el precipitado resultante en un horno de secado a 50°C. Se usa el producto sin purificación adicional.
Rendimiento: cuantitativo ESI-EM: m/z = 277 [M+H]+ Tr (HPLC): 0,78 min (método 10)
4-(6-Aminopiridazin-3-il)-piperidin-1-carboxilato de ferc-butilo

Bajo una atmósfera de hidrógeno (aparato de Parr; 4 bar), se agitan 4-(6-aminopiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo (4,85 g; 17,55 mmol) y Pd/C (10%; 0,50 g) en MeOH (100 ml) a t A durante 3 horas. Después de retirar el catalizador por filtración, se concentran las aguas madres a presión reducida. Se usa el producto sin purificación adicional.
Rendimiento: cuantitativo ESI-EM: m/z = 279 [M+H]+ Tr (HPLC): 0,86 (método 11)
6-(Piperidin-4-il)piridazin-3-amina

Se agita 4-(6-aminopiridazin-3-il)-piperidin-1-carboxilato de ferc-butilo (4,89 g; 17,55 mmol) durante 1 hora en TFA (20 ml; 259,25 mmol). Se evapora el disolvente y se purifica el residuo mediante cromatografía en gel de sílice (DCM/MeOH NH3).
Rendimiento: cuantitativo ESI-EM: m/z = 179 [M+H]+ Tr (HPLC): pico de inyección (método 11) Alternativamente, se usa amina:
Diclorhidrato de 6-(piperidin-4-il)piridazin-3-amina

Se realiza la reacción usando una atmósfera de nitrógeno. Se agitan 4-(6-aminopiridazin-3-il)-piperidin-1-carboxilato de ferc-butilo (1,00 g; 3,59 mmol) y HCl (4 mol/l, disolución en 1,4-dioxano; 2,96 ml; 11,84 mmol) en ACN (6 ml) a 35-40°C durante 2 horas. Se enfría la mezcla de reacción hasta TA y se diluye con acetato de isopropilo. Después de 10 minutos de agitación, se elimina el precipitado resultante por filtración y se seca en un horno de secado a 45°C. Rendimiento: cuantitativo ESI-EM: m/z = 179 [M+H]+ Tr (HPLC): 0,94 min (método 14)
6-{1-[5-(4-Fluorofenoxi)-4-metoxipiridin-2-carbonil1piperidin-4-il)piridazin-3-amina

Se agitan ácido 5-(4-fluorofenoxi)-4-metoxipiridin-2-carboxílico (0,70 g; 2,66 mmol), HATU (1,52 g; 3,99 mmol) y DIPEA (1,83 ml; 10,64 mmol) en DMF (20 ml) durante 30 minutos. Se añade 6-(piperidin-4-il)piridazin-3-amina (0,71 g; 3,98 mmol) y se permite que la mezcla de reacción se agite a TA durante la noche. Se purifica la mezcla mediante RPHPLC (ACN/agua TFA). Para retirar la sal de trifluoroacetato, se lleva el producto a agua/EtOH (1,5/1) y se somete a levigación con bicarbonato unido a polímero. Después de 30 minutos de agitación, se filtra la mezcla y se concentra a presión reducida.
Rendimiento: 180 mg (16%) ESI-EM: m/z = 424 [M+H]+ Tr (HPLC): 0,77 min (método 7)
Alternativamente, puede obtenerse el compuesto del título de la siguiente manera:
6-{1-[5-(4-Fluorofenoxi)-4-metoxipiridin-2-carbonil]piperidin-4-il)piridazin-3-amina

Se agitan ácido 5-(4-fluorofenoxi)-4-metoxipiridin-2-carboxílico (0,50 g; 1,90 mmol) y CDI (0,46 g; 2,85 mmol) en NMP (1 ml) a TA durante 1 hora. Se añaden diclorhidrato de 6-(piperidin-4-il)piridazin-3-amina (0,52 g; 2,09 mmol) y DIPEA (0,99 ml; 5,70 mmol). Después de agitar durante 3 horas, se diluye la mezcla de reacción con agua y se extrae con EtOAc. Se separa la fase orgánica, se lava con agua y salmuera, se seca sobre MgSO4 y se filtra. Se concentran las aguas madres a presión reducida y se purifican mediante cromatografía en gel de sílice (DcM/MeOH). Se concentran las fracciones deseadas a presión reducida y se tratan con ACN/etil éter para proporcionar el producto del título en forma sólida.
Rendimiento: 0,27 g (34%) ESI-EM: m/z = 424 [M+H]+ Tr (HPLC): 0,49 min (método 1)
Preparación alternativa del compuesto de referencia 375-{1-[5-(ciclopropilmetoxi)-4-metoxipiridin-2-carbonil]piperidin-4-il)-4-metoxipiridin-2-amina
5-(Ciclopropilmetoxi)-4-metoxipiridin-2-carboxilato de metilo

Se enfrían 5-hidroxi-4-metoxipiridin-2-carboxilato de metilo (0,20 g; 1,09 mmol) y ciclopropilmetanol (88 |al; 1,09 mmol) en THF (3 ml) en un baño de hielo. Se añaden TPP (0,32 g; 1,20 mmol) y DtA d (0,28 g; 1,20 mmol). Se permite que la mezcla de reacción se caliente hasta TA durante la noche. Se concentra la mezcla de reacción a presión reducida y se purifica mediante RP-HPLC (ACN/agua TFA).
Rendimiento: 0,18 g (70%) ESI-EM: m/z = 238 [M+H]+ Tr (HPLC): 0,41 min (método 12)
Ácido 5-(ciclopropilmetoxi)-4-metoxipiridin-2-carboxílico

Se agitan 5-(ciclopropilmetoxi)-4-metoxipiridin-2-carboxilato de metilo (0,18 g; 0,76 mmol) y NaOH (4 mol/l, disolución ac.; 0,50 ml; 2,00 mmol) en MeOH (3 ml) a TA durante 1 hora. Se concentra la mezcla de reacción a presión reducida. Se lleva el residuo a agua y se lava con EtOAc. A la fase acuosa se le añade HCl (4 mol/l, disolución ac.; 0,5 ml) y se concentra a presión reducida. Se usa el producto sin purificación adicional.
Rendimiento: 0,13 g (74%) ESI-EM: m/z = 224 [M+H]+ Tr (HPLC): 0,30 min (método 12)
5-{1-[5-(Ciclopropilmetoxi)-4-metoxipiridin-2-carbonil]piperidin-4-ilM-metoxipiridin-2-amina

Se agitan ácido 5-(ciclopropilmetoxi)-4-metoxipiridin-2-carboxílico (50 mg; 0,22 mmol), diclorhidrato de 4-metoxi-5-(piperidin-4-il)piridin-2-amina (63 mg; 0,22 mmol), DIPEA (193 |l; 1,12 mmol) y HATU (94 mg; 0,25 mmol) en DMF (2 ml) a TA durante la noche. Se purifica la mezcla resultante mediante RP-h PLc (ACN/agua NH3).
Rendimiento: 45 mg (49%) ESI-EM: m/z = 413 [M+H]+ Tr (HPLC): 0,87 min (método 11)
Preparación alternativa del compuesto 90 ácido trifluoroacético de 6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)-5-metilpiridazin-3-amina
4-Metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonitrilo

Se agitan 5-fluoro-4-metoxi-piridin-2-carbonitrilo (4,69 g; 30,84 mmol), 4-trifluorometilfenol (5,00 g; 30,84 mmol) y carbonato de potasio (6,39 g; 46,27 mmol) en DMSO a 110°C durante 1 hora. Se permite que la mezcla de reacción se enfríe hasta TA y se diluye con agua. Se filtra el precipitado resultante, se lava con agua y se seca en un horno de secado a 50°C.
Rendimiento: 7,40 g (82%) ESI-EM: m/z = 295 [M+H]+ Tr (HPLC): 1,08 min (método 10)
Ácido 4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carboxílico

Se agitan 4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonitrilo (7,40 g; 25,51 mmol) y NaOH (4 mol/l, disolución ac.; 31,44 ml, 125,75 mmol) en MeOH (100 ml) a 70°C durante la noche. Se permite que la mezcla de reacción se enfríe hasta TAy se evapora el disolvente orgánico. Se diluye el disolvente restante con agua y se ajusta a pH 3 usando HCl (4 mol/l, disolución ac.). Se filtra el precipitado resultante y se seca en un horno de secado a 50°C.
Rendimiento: 6,80 g (51%) ESI-EM: m/z = 314 [M+H]+ Tr (HPLC): 0,87 min (método 10)
4-(6-Amino-4-metilpiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo

Se realiza la reacción bajo una atmósfera de argón. Se purgan 6-cloro-5-metilpiridazin-3-amina (3,00 g; 20,90 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo (7,11 g; 22,98 mmol) y carbonato de sodio (2 mol/l, disolución ac.; 41,79 ml; 83,58 mmol) en 1,4-dioxano (150 ml) con argón. Después de 5 minutos, se añade Xphos de 2a gen. (0,49 g; 0,63 mmol) y se agita la mezcla durante la noche en un vial sellado a 100°C. Se concentra la mezcla de reacción a presión reducida. Se lleva el residuo a agua y se extrae varias veces con EtOAc. Se lavan las fases orgánicas combinadas con salmuera, se secan sobre Na2SO4, se filtran y se concentran a presión reducida. Se purifica el residuo mediante cromatografía en gel de sílice (DCM/MeOH).
Rendimiento: 5,20 g (86%) ESI-EM: m/z = 291 [M+H]+ Tr (HPLC): 0,79 min (método 10)
4-(6-Amino-4-metilpiridazin-3-il)piperidin-1-carboxilato de ferc-butilo

Bajo una atmósfera de hidrógeno (aparato de Parr; 50 psi), se agitan 4-(6-amino-4-metilpiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de ferc-butilo (5,20 g; 17,91 mmol) y Pd/C (10%; 0,75 g) en MeOH (100 ml) a t A durante 17 horas. Después de retirar el catalizador por filtración, se concentran las aguas madres a presión reducida.
Rendimiento: 5,00 g (96%) ESI-EM: m/z = 293 [M+H]+ Tr (HPLC): 0,79 min (método 10)
Diclorhidrato de 5-metil-6-(piperidin-4-il)piridazin-3-amina

Se agitan 4-(6-amino-4-metilpiridazin-3-il)piperidin-1-carboxilato de ferc-butilo (4,91 g; 16,79 mmol) y HCl (4 mol/l; disolución en 1,4-dioxano; 73,65 ml; 251,90 mmol) en 1,4-dioxano (34,37 ml) a TA durante la noche. Se concentra la mezcla de reacción a presión reducida. Se somete a levigación el residuo en EtOAc y se filtra. Se usa el producto sin purificación adicional.
Rendimiento: cuantitativo ESI-EM: m/z = 193 [M+H]+ Tr (HPLC): 0,59 min (método 11)
Ácido trifluoroacético de 6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi1piridin-2-carbonil}piperidin-4-il)-5-metilpiridazin-3-amina

Se agitan ácido 4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carboxílico (0,12 g; 0,37 mmol), HATU (0,15 g; 0,39 mmol) y DIPEA (0,19 ml; 1,11 mmol) en DMF (3 ml) durante 30 minutos. Se añade diclorhidrato de 5-metil-6-(piperidin-4-il)piridazin-3-amina (0,10 g; 0,38 mmol) y se permite que la mezcla de reacción se agite a TA durante la noche. Se purifica la mezcla de reacción mediante Rp-HPLC (ACN/agua TFA).
Rendimiento: 0,12 g (55%) ESI-EM: m/z = 488 [M+H]+ Tr (HPLC): 0,86 min (método 7)
Preparación alternativa del compuesto de referencia 475-metoxi-6-[1-(4-metoxi-5-fenoxipiridin-2-carbonil)piperidin-4-il1piridazin-3-amina
4-Metoxi-5-fenoxipiridin-2-carbonitrilo

Se agitan 5-fluoro-4-metoxi-piridin-2-carbonitrilo (0,40 g; 2,63 mmol), fenol (0,25 g; 2,66 mmol) y carbonato de potasio
(0,54 g; 3,91 mmol) en DMSO (10 ml) a 110°C durante 2 horas. Se permite que la mezcla de reacción se enfríe hasta TA y se diluye con agua. Se extrae la fase acuosa varias veces con EtOAc. Se secan las fases orgánicas combinadas sobre Na2SO4, se filtran y se concentran a presión reducida.
Rendimiento: 0,55 g (92%) %) ESI-EM: m/z = 227 [M+H]+ Tr (HPLC): 1,01 min (método 7)
Ácido 4-metoxi-5-fenoxipiridin-2-carboxílico

Se agitan 4-metoxi-5-fenoxipiridin-2-carbonitrilo (0,54 g; 2,39 mmol) y NaOH (4 mol/l, disolución ac.; 3,00 ml, 12,00 mmol) en MeOH (10 ml) a 70°C durante la noche. Se permite que la mezcla de reacción se enfríe hasta TAy se evapora el disolvente orgánico. Se diluye el disolvente restante con agua y se acidifica hasta pH 3 usando HCl (4 mol/l, disolución ac.). Se filtra el precipitado resultante y se seca en un desecador.
Rendimiento: 0,30 g (51%) ESI-EM: m/z = 246 [M+H]+ Tr (HPLC): 0,72 min (método 10)

Se realiza la reacción bajo una atmósfera de argón. Se purgan éster terc-butílico del ácido (6-cloro-5-metoxi-piridazin-3-il)-carbámico (4,00 g; 15,40 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de terc-butilo (4,76 g; 15,40 mmol) y carbonato de sodio (2 mol/l, disolución ac.; 15,40 ml; 30,81 mmol) en 1,4-dioxano (80 ml) con argón. Después de 5 minutos, se añade Xphos de 2a gen. (1,26 g; 1,54 mmol) y se agita la mezcla durante la noche en un vial sellado a 90°C. Se concentra la mezcla de reacción a presión reducida. Se lleva el residuo a EtOAc y se lava con agua y salmuera. Se separa la fase orgánica y se concentra a presión reducida. Se purifica el residuo mediante cromatografía en gel de sílice (DCM/MeOH).
Rendimiento: 4,56 g (59%)
4-(6-{f(7erc-butoxi)carboni
n
amino
M
-metoxipiridazin-3-il)piperidin-1-carboxilato de terc-butilo

Bajo una atmósfera de hidrógeno (aparato de Parr; 50 psi), se agitan 4-(6-{[(terc-butoxi)carbonil]amino}-4-metoxipiridazin-3-il)-1,2,3,6-tetrahidropiridin-1-carboxilato de terc-butilo (4,55 g; 11,19 mmol) y Pd/C (10%; 3,57 g) en MeOH (45,5 ml) a 30°C durante la noche. Después de retirar el catalizador por filtración, se concentran las aguas madres a presión reducida.
Rendimiento: 3,67 g (80%)
Diclorhidrato de 5-metoxi-6-(piperidin-4-il)piridazin-3-amina

Se agitan 4-(6-{[(tere-butoxi)carbonil]amino}-4-metoxipiridazin-3-il)piperidin-1-carboxilato de tere-butilo (3,67 g; 8,98 mmol) y HCl (4 mol/l; disolución en 1,4-dioxano; 55,05 ml; 134,76 mmol) en 1,4-dioxano (26,69 ml) a TA durante la noche. Se concentra la mezcla de reacción a presión reducida. Se somete a levigación el residuo en EtOAc y se filtra. Se usa el producto sin purificación adicional.
Rendimiento: 2,07 g (82%) ESI-EM: m/z = 209 [M+H]+ Tr (HPLC): 0,60 min (método 11)
5-Metoxi-6-[1-(4-metoxi-5-fenoxipiridin-2-carbonil)piperidin-4-il1piridazin-3-amina

Se agitan ácido 4-metoxi-5-fenoxipiridin-2-carboxílico (0,10 g; 0,41 mmol), HATU (0,16 g; 0,419 mmol) y DIPEA (0,18 ml; 1,05 mmol) en DMF (3 ml) durante 30 minutos. Se añade diclorhidrato de 5-metoxi-6-(piperidin-4-il)piridazin-3-amina (0,12 g; 0,41 mmol) y se permite que la mezcla de reacción se agite a TA durante la noche. Se purifica la mezcla mediante RP-HPLC (ACN/agua n H3).
Rendimiento: 0,09 g (53%) ESI-EM: m/z = 436 [M+H]+ Tr (HPLC): 0,63 min (método 13)
Preparación alternativa del compuesto 29 ácido trifluoroacético de 6-{1-[5-(4-Fluorofenoxi)-4-metoxipiridin-2-carbonil1piperidin-4-il}-5-metilpiridazin-3-amina

Se agitan ácido 5-(4-fluorofenoxi)-4-metoxipiridin-2-carboxílico (60 mg; 0,23 mmol), diclorhidrato de 5-metil-6-(piperidin-4-il)piridazin-3-amina (60 mg; 0,23 mmol), HATU (95 mg; 0,25 mmol) y DIPEA (0,12 ml; 0,68 mmol) en DMF (3 ml) a TA durante 1 hora. Se purifica la mezcla mediante RP-HPLC (ACN/agua TFA).
Rendimiento: 73 mg (59%) ESI-EM: m/z = 438 [M+H]+ Tr (HPLC): 0,82 min (método 7)
Preparación alternativa del compuesto de referencia 91 ácido trifluoroacético de 5-metoxi-6-(1-{5-[4-(trifluorometil)fenoxi1piridin-2-carbonil}piperidin-4-il)piridazin-3-amina
5-[4-(Trifluorometil)fenoxi1piridin-2-carbonitrilo

Se agitan 2-ciano-5-fluoropiridina (3,54 g; 28,99 mmol), 4-trifluorometil-fenol (4,70 g; 28,99 mmol) y carbonato de potasio (6,01 g; 43,49 mmol) en d Ms O (150 ml) a 110°C durante 1 hora. Se diluye la mezcla de reacción con agua y se extrae con EtOAc. Se lava la fase orgánica con agua, se separa, se seca sobre Na2SO4, se filtra y se concentra a presión reducida.
Rendimiento: cuantitativo ESI-EM: m/z = 265 [M+H]+ Tr (HPLC): 1,03 min (método 10)
Ácido 5-[4-(trifluorometil)fenoxi]piridin-2-carboxílico

Se agitan 5-[4-(trifluorometil)fenoxi]piridin-2-carbonitrilo (3,87 g; 14,65 mmol) y NaOH (4 mol/l, disolución ac.; 18,31 ml, 73,24 mmol) en MeOH (50 ml) a 70°C durante la noche. Se concentra la mezcla de reacción a presión reducida. Se lleva el residuo a agua y se acidifica hasta pH 3 usando HCl (4 mol/l, disolución ac.). Se evapora por completo el disolvente orgánico y se filtra el precipitado resultante. Se lleva el residuo a DCM, se filtra y se seca en un horno de secado a 50°C.
Rendimiento: cuantitativo ESI-EM: m/z = 284 [M+H]+ Tr (HPLC): 0,68 min (método 11)
Ácido trifluoroacético de 5-metoxi-6-(1-{5-[4-(trifluorometiDfenoxi]piridin-2-carbonil}piperidin-4-il)piridazin-3-amina

Se agitan ácido 5-[4-(trifluorometil)fenoxi]piridin-2-carboxílico (0,10 g; 0,35 mmol), HATU (0,15 g; 0,39 mmol) y DIPEA (0,19 ml; 1,11 mmol) en DMF (3 ml) durante 30 minutos a TA. Se añade diclorhidrato de 5-metoxi-6-(piperidin-4-il)piridazin-3-amina (0,11 g; 0,37 mmol) y se permite que la mezcla de reacción se agite a TA durante la noche. Se purifica la mezcla mediante RP-HPLC (ACN/agua TFA).
Rendimiento: 0,06 g (31%) ESI-EM: m/z = 474 [M+H]+ Tr (HPLC): 0,87 min (método 7)
Preparación alternativa del compuesto 31 ácido trifluoroacético de 6-[1-(4-metoxi-5-fenoxipiridin-2-carbonil)piperidin-4-il]5-metilpiridazin-3-amina

Se agitan ácido 4-metoxi-5-fenoxipiridin-2-carboxílico (60 mg; 0,23 mmol), diclorhidrato de 5-metil-6-(piperidin-4-il)piridazin-3-amina (55 mg; 0,23 mmol), HATU (95 mg; 0,25 mmol) y DIp Ea (0,12 ml; 0,68 mmol) en DMf (3 ml) durante 1 hora a TA. Se purifica la mezcla mediante RP-HPLC (ACN/agua TFA).
Rendimiento: 69 mg (57%) ESI-EM: m/z = 420 [M+H]+ Tr (HPLC): 0,81 min (método 7)
EVALUACIÓN DE LA ACTIVIDAD BIOLÓGICA
Ensayo de cribado de alto rendimiento
Este ensayo de cribado mide la activación del canal iónico TRPC6 (canal catiónico receptor de potencial transitorio, subfamilia C, miembro 6) a través de la adición o bien del análogo del ligando DAG comercialmente disponible OAG (1 -oleoil-2-acetil-sn-glicerol) o bien del agonista de TRPC6 1-[1-(4,5,6,7,8-pentahidrociclohepta[2,1-d]tiofen-2-ilcarbonil)-4-piperidil]-3-hidrobencimidazol-2-ona (GSK1702934A). El ensayo utiliza un colorante de potencial de membrana (FMP) de éstertetrakis(acetoximetílico) del ácido 4-(6-acetoximetoxi-2,7-difluoro-3-oxo-9-xantenil)-4’-metil-2,2’-(etilendioxi)dianilina-N,N,N’,N’-tetraacético (Fluo4/AM) de sensor de calcio fluorescente FLIPR de Molecular Devices, que es un indicador sensible al voltaje con un extinguidor de fluorescencia. Los cambios (aumentos) en el potencial de concentración de calcio de membrana intracelular tal como se miden mediante el aumento en la señal de fluorescencia durante la despolarización de membrana proporcionan una medición de la actividad del canal.
Se transfectó de manera estable la línea HEK293/TREx comercialmente disponible (Invitrogen) con un constructo de TRPC6 y se cribó mediante obtención de imágenes de calcio convencional para hallar clones con expresión de TRPC6 tras la estimulación con tetraciclina 1 ^g/ml. Se mantuvieron estas células en el medio de crecimiento recomendado por el fabricante complementado con higromicina 100 ^g/ml para fomentar la retención del constructo de TRPC6. Después de crecer hasta casi confluencia, se sembraron en placas CellBind de 384 pocillos (Corning) las células a una densidad de ~35.000 células/pocillo en presencia de tetraciclina 1 ^g/ml, y se permitió que crecieran durante 20
30 h. Resultó una monocapa prácticamente confluente. Se retiró el medio de crecimiento de los pocilios y luego se cargaron 25 ml de Fluo4/AM diluido en solución de Ringer (6,5 g de NaCl, 0,42 g de KCI, 0,25 g de CaCh y 0,2 g de bicarbonato de sodio; pH 7,4) complementada con Pluronic F-127 al 1% en las células hasta una concentración final de 0,5 |iM y se incubaron durante 60 min, a temperatura ambiente. Luego se retiró la disolución de colorante de las células invirtiendo las placas con un movimiento brusco, y se reemplazó con 25 |il de solución de Ringer. Tras ~0,5 horas para la recuperación a partir de la carga, se sometieron a ensayo las células usando el sistema Hamamatsu
FDSS 6000, que permitía la iluminación a 485 nm. Se adquirieron fotogramas a una tasa de 0,2 Hz. Durante el ensayo, se agitaron continuamente con vórtex las placas, con mezclado con pipeta de los pocillos tras la adición de cada reactivo. Para el ensayo de cribado, se añadieron 26 |il de una reserva de compuesto diluida (a 50 |iM) a cada pocillo durante 2 minutos tras la recogida de un fondo pequeño (4 fotogramas). Luego se añadieron 13 |il de disolución de agonista que consistía en GSK1702934A 125 nM diluido en solución de Ringer con alto contenido en Ca2+ (que contenía Ca2+ 90 mM) a cada pocillo, logrando una concentración final de Ca2+ de 20 mM y compuesto de prueba de
10 |iM. Se recogieron datos durante ~3 minutos tras la adición de solución de Ringer con alto contenido en Ca2+. Se dividió la razón de fluorescencia para cada pocillo entre la intensidad de fluorescencia inicial para ese pocillo y se determinó la respuesta global promediando la razón de fluorescencia de los últimos 4 fotogramas adquiridos durante el experimento exceptuando el fotograma final. Se incluyeron controles negativos y positivos en cada placa. Los pocillos de control negativo consistían en células HEK293/TREx TRPC6 expuestas a tampón de ensayo y disolución de agonista, pero sin compuesto de prueba. Los pocillos de control positivo consistían en pocillos que consistían en células HEK293/TREx TRPC6 expuestas a 3-[(2-clorofenoxi)metil]fenil piperidil cetona 25 |iM (Chembridge) diluida en solución de Ringer y disolución de agonista. Estos controles definieron el 0% y el 100% de bloqueo, respectivamente, y se normalizó la intensidad de cada pocillo a estos valores.
Se determinaron las CI50 usando el método de fluorescencia anterior con la excepción de que, en lugar de someter a prueba los compuestos a 10 |iM, los compuestos se sometieron a prueba a concentraciones finales de 20 |iM, 6,667
|iM, 2,222 |iM, 0,741 |iM, 0,247 |iM, 0,082 |iM y 0,027 |iM. Los compuestos se sometieron a prueba todas las concentraciones. Se usó un software convencional para ajustar las curvas de CI50.
Tabla 8. Efectos antagonistas de los compuestos 29, 31, 49, 56, 66, 85, 87 y 90 de la invención y de los compuestos de referencia 1-28, 30, 32-48, 50-55, 57-65, 67-84, 86, 88, 89 y 91-95 contra TRPC6 (CI50)



La actividad biológica de los compuestos reivindicados también puede mostrarse usando un ensayo de pinzamiento zonal de TRPC6.
USO EN MÉTODOS DE TRATAMIENTO
La inhibición de TRPC6 es un medio atractivo para prevenir y tratar una variedad de enfermedades o estados que se agravan por la actividad de TRPC6. Los compuestos divulgados en el presente documento inhiben eficazmente la actividad de TRPC6. En particular, los compuestos de la invención son inhibidores de canales iónicos selectivos y tienen buena estabilidad metabólica en microsomas humanos. Más particularmente, los compuestos de la invención tienen una potencia y una selectividad muy buenas sobre el canal TRPC6 en comparación con otros canales TRP incluyendo TRPC3, TRPC5 y TRPC7. Por tanto, los compuestos de la invención son para su uso en métodos para el tratamiento de las enfermedades y los estados que se describen en las secciones de antecedentes y descripción detallada, incluyendo los siguientes estados y enfermedades:
estados cardiacos (por ejemplo, hipertrofia cardiaca), hipertensión (por ejemplo, primaria o secundaria), hipertensión arterial pulmonar (por ejemplo, HAPI), una enfermedad o un trastorno neurodegenerativo (por ejemplo, enfermedad de Alzheimer (EA), enfermedad de Parkinson, enfermedad de Huntington, esclerosis lateral amiotrófica (ELA) y otros trastornos encefálicos provocados por traumatismo u otras agresiones incluyendo el envejecimiento), enfermedades inflamatorias (por ejemplo, asma, enfermedad pulmonar obstructiva crónica, artritis reumatoide, artrosis, enfermedad intestinal inflamatoria, esclerosis múltiple y trastornos del sistema inmunitario), preeclampsia e hipertensión inducida por el embarazo, enfermedades renales (glomeruloesclerosis focal y segmentaria, síndrome nefrótico, nefropatía diabética, insuficiencia renal, enfermedad renal de fase terminal, enfermedad de cambios mínimos), isquemia o una lesión por reperfusión isquémica, cáncer, FPI (fibrosis pulmonar idiopática), SDRA (síndrome de dificultad respiratoria aguda) y trastornos metabólicos diabéticos tales como diabetes. Los métodos para prevenir o tratar cualquiera de las enfermedades y los estados anteriores o siguientes incluyen tratar cualquiera de los síntomas asociados con estas enfermedades o estados. Por ejemplo, los métodos para tratar una enfermedad renal contemplan tratar los síntomas incluyendo, pero sin limitarse a, hipertensión secundaria, proteinuria, lipiduria, hipercolesterolemia, hiperlipidemia y anomalías de la coagulación.
Debido al papel importante que desempeña la regulación del calcio en muchos procesos celulares incluyendo la activación celular, la reorganización citoesquelética, la expresión génica, el tráfico celular y la muerte celular apoptótica, la dishomeostasis del calcio está implicada en muchas enfermedades y trastornos. Estas enfermedades y
trastornos incluyen enfermedades y trastornos neurológicos y neurodegenerativos; enfermedades y trastornos inflamatorios tales como enfermedad intestinal inflamatoria y enfermedad de Crohn; enfermedad renal tal como hipercalcemia, cálculos renales y enfermedad renal poliquística; enfermedades y trastornos metabólicos incluyendo obesidad y diabetes; enfermedades y trastornos hepáticos y renales; enfermedad renal crónica, enfermedades y trastornos cardiovasculares incluyendo hipertensión; enfermedades respiratorias incluyendo EPOC, HAPI, asma y enfisema; y cánceres incluyendo cánceres de cerebro, mama, riñón, cuello uterino, próstata, tracto gastrointestinal, (por ejemplo, cáncer gástrico o cáncer de estómago), piel y epitelios.
Estos trastornos se han caracterizado bien en seres humanos, pero también existen con una etiología similar en otros mamíferos, y pueden tratarse mediante las composiciones farmacéuticas de la presente invención.
Por consiguiente, un compuesto de la invención, tal como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, puede usarse para la preparación de un medicamento para tratar una enfermedad o un trastorno mediado por TRPC6, incluyendo los mencionados anteriormente y en las secciones de antecedentes y descripción detallada.
Para uso terapéutico, los compuestos de la invención pueden administrarse a través de una composición farmacéutica en cualquier forma de dosificación farmacéutica convencional de cualquier manera convencional. Las formas de dosificación convencionales incluyen normalmente un portador farmacéuticamente aceptable adecuado para la forma de dosificación particular seleccionada. Las vías de administración incluyen, pero no se limitan a, intravenosa, intramuscular, subcutánea, intrasinovial, mediante infusión, sublingual, transdérmica, oral, tópica o mediante inhalación. Los modos de administración preferidos son por vía oral e intravenosa.
Los compuestos de esta invención pueden administrarse solos o en combinación con adyuvantes que potencian la estabilidad de los inhibidores, facilitan la administración de las composiciones farmacéuticas que los contienen en determinadas realizaciones, proporcionan una disolución o dispersión aumentada, aumentan la actividad inhibidora, proporcionan una terapia complementaria, y similares, incluyendo otros principios activos. En una realización, por ejemplo, pueden administrarse múltiples compuestos de la presente invención. Ventajosamente, tales terapias de combinación utilizan dosificaciones más bajas de los productos terapéuticos convencionales, evitando de ese modo la posible toxicidad y los efectos secundarios adversos que se producen cuando se usan esos agentes como monoterapias. Los compuestos de la invención pueden combinarse físicamente con los productos terapéuticos convencionales u otros adyuvantes para dar una única composición farmacéutica. Ventajosamente, los compuestos pueden administrarse entonces juntos en una única forma de dosificación. En algunas realizaciones, las composiciones farmacéuticas que comprenden tales combinaciones de compuestos contienen al menos aproximadamente el 5%, pero más preferiblemente al menos aproximadamente el 20%, de un compuesto de la invención (p/p) o una combinación de los mismos. El porcentaje óptimo (p/p) de un compuesto de la invención puede variar y se encuentra dentro del ámbito de los expertos en la técnica. Alternativamente, los compuestos de la presente invención y los productos terapéuticos convencionales u otros adyuvantes pueden administrarse por separado (ya sea en serie o en paralelo). La dosificación por separado permite una mayor flexibilidad en la pauta posológica.
Tal como se mencionó anteriormente, las formas de dosificación de los compuestos de esta invención pueden incluir portadores y adyuvantes farmacéuticamente aceptables conocidos por los expertos habituales en la técnica y adecuados para la forma de dosificación. Estos portadores y adyuvantes incluyen, por ejemplo, intercambiadores iónicos, alúmina, estearato de aluminio, lecitina, proteínas séricas, sustancias tamponantes, agua, sales o electrolitos y sustancias a base de celulosa. Las formas de dosificación preferidas incluyen comprimido, cápsula, comprimido oblongo, líquido, disolución, suspensión, emulsión, pastillas para chupar, jarabe, polvo reconstituible, gránulo, supositorio y parche transdérmico. Se conocen métodos para preparar tales formas de dosificación (véase, por ejemplo, H.C. Ansel y N.G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5a ed., Lea y Febiger (1990)). Los niveles de dosificación y los requisitos para los compuestos de la presente invención pueden seleccionarlos los expertos habituales en la técnica a partir de métodos y técnicas disponibles adecuados para un paciente particular. En algunas realizaciones, los niveles de dosificación oscilan desde aproximadamente 1 hasta 1000 mg/dosis para un paciente de 70 kg. Aunque una dosis al día puede ser suficiente, pueden administrarse hasta 5 dosis al día. Para las dosis orales, pueden requerirse hasta 2000 mg/día. Tal como apreciará el experto en la técnica, pueden requerirse dosis más bajas o más altas dependiendo de factores particulares. Por ejemplo, las pautas posológicas y los regímenes de tratamiento específicos dependerán de factores tales como el perfil de salud general del paciente, la gravedad y el avance del trastorno o de la disposición al mismo del paciente, y el criterio del médico responsable.
Los compuestos de la invención pueden usarse solos o en combinación de uno o más agentes terapéuticos adicionales. Los ejemplos no limitativos de agentes terapéuticos adicionales pueden incluir:
antagonistas del receptor de angiotensina II (bloqueadores del receptor de angiotensina (ARB)) tales como candesartán, eprosartán, candesartán, irbesartán, losartán, olmesartán, telmisartán, valsartán, azilsartán ymedoxomil;
inhibidores de enzimas convertidoras de angiotensina (por ejemplo, benazepril, captopril, enalapril, fosinopril, lisinopril, moexipril y perindopril);
antidiabéticos tales como inhibidores de alfa-glucosidasa (por ejemplo, miglitol y acarbosa), análogos de amilina (por ejemplo, pramlintida), inhibidores de dipeptidil peptidasa 4 (por ejemplo, alogliptina, sitagliptina, saxagliptina y linagliptina), miméticos de la incretina (por ejemplo, liraglutida, exenatida, liraglutida, exenatida, dulaglutida, albiglutida y lixisenatida), insulina, meglitinidas (por ejemplo, repaglinida y nateglinida), biguanidas (por ejemplo, metformina); inhibidores de SGLT-2 (por ejemplo, canagliflozina, empagliflozina y dapagliflozina), sulfonilureas (por ejemplo, clorpropamida, glimepirida, gliburida, glipizida, gliburida, tolazamida y tolbutamida) y tiazolidindionas (por ejemplo, rosiglitazona y pioglitazona);
broncodilatadores incluyendo beta-agonistas de acción corta y larga (por ejemplo, albuterol, levalbuterol, salmeterol, formoterol y arformoterol) y anticolinérgicos de acción corta y larga (ipratropio, tiotropio, umeclidino, glicopirrolato y aclidinio).
esteroides tales como fluticasona y budesonida.
Cuando se usan como tratamiento de combinación de una combinación farmacéutica, los compuestos de la invención y el uno o más agentes adicionales pueden administrarse en la misma forma de dosificación o en formas de dosificación diferentes. Los compuestos de la invención y el uno o más agentes adicionales pueden administrarse simultáneamente o por separado, como parte de una pauta.
Claims (12)
- REIVINDICACIONESi. Compuesto seleccionado del grupo que consiste en:[4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona,[4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-(4-metoxi-5-fenoxi-piridin-2-il)-metanona,[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona,[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-il]-metanona, [4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-il]-metanona,[4-(6-amino-4-etoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(fenoxi)-piridin-2-il]-metanona,5- etoxi-6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)piridazin-3-amina, y 6- (1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)-5-metilpiridazin-3-amina.
- 2. Compuesto según la reivindicación 1, en el que el compuesto es [4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona.
- 3. Compuesto según la reivindicación 1, en el que el compuesto es [4-(6-amino-4-metil-piridazin-3-il)-piperidin-1-il]-(4-metoxi-5-fenoxi-piridin-2-il)-metanona.
- 4. Compuesto según la reivindicación 1, en el que el compuesto es [4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[5-(4-fluoro-fenoxi)-4-metoxi-piridin-2-il]-metanona.
- 5. Compuesto según la reivindicación 1, en el que el compuesto es [4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-trifluorometil-fenoxi)-piridin-2-il]-metanona.
- 6. Compuesto según la reivindicación 1, en el que el compuesto es [[4-(6-amino-4-metoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(4-metoxi-fenoxi)-piridin-2-il]-metanona.
- 7. Compuesto según la reivindicación 1, en el que el compuesto es [4-(6-amino-4-etoxi-piridazin-3-il)-piperidin-1-il]-[4-metoxi-5-(fenoxi)-piridin-2-il]-metanona.
- 8. Compuesto según la reivindicación 1, en el que el compuesto es 5-etoxi-6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)piridazin-3-amina.
- 9. Compuesto según la reivindicación 1, en el que el compuesto es 6-(1-{4-metoxi-5-[4-(trifluorometil)fenoxi]piridin-2-carbonil}piperidin-4-il)-5-metilpiridazin-3-amina.
- 10. Sal farmacéuticamente aceptable de un compuesto según cualquiera de las reivindicaciones 1 a 9.
- 11. Composición farmacéutica que comprende uno cualquiera de los compuestos según las reivindicaciones 1 a 9, o sal farmacéuticamente aceptable de los mismos según la reivindicación 10, y opcionalmente un excipiente farmacéuticamente aceptable.
- 12. Compuesto según una cualquiera de las reivindicaciones 1 a 9, o sal farmacéuticamente aceptable de los mismos según la reivindicación 10, para su uso en el tratamiento de una enfermedad o un trastorno que puede aliviarse mediante la inhibición de TRPC6, en el que la enfermedad o el trastorno se selecciona del grupo que consiste en hipertrofia cardiaca, isquemia, lesión por reperfusión isquémica, hipertensión, hipertensión arterial pulmonar, hipertensión arterial pulmonar idiopática, reestenosis, enfermedad pulmonar obstructiva crónica, fibrosis quística, enfermedad de Alzheimer, enfermedad de Parkinson, enfermedad de Huntington, esclerosis lateral amiotrófica (ELA), trastornos encefálicos inducidos por traumatismo, asma, enfermedad pulmonar obstructiva crónica, artritis reumatoide, artrosis, enfermedad intestinal inflamatoria, esclerosis múltiple, distrofia muscular, distrofia muscular de Duchenne, preeclampsia e hipertensión inducida por el embarazo, esteatohepatitis no alcohólica, enfermedad de cambios mínimos, glomeruloesclerosis focal y segmentaria (GEFS), síndrome nefrótico, nefropatía diabética o enfermedad renal diabética (ERD), enfermedad renal crónica, insuficiencia renal, enfermedad renal de fase terminal, isquemia o una lesión por reperfusión isquémica, cáncer, FPI (fibrosis pulmonar idiopática), SDRA (síndrome de dificultad respiratoria aguda), enfisema y diabetes.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762577883P | 2017-10-27 | 2017-10-27 | |
| US201862628313P | 2018-02-09 | 2018-02-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2924933T3 true ES2924933T3 (es) | 2022-10-11 |
Family
ID=64270818
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20197194T Active ES2924933T3 (es) | 2017-10-27 | 2018-10-25 | Derivados de piridina y usos terapéuticos de los mismos como inhibidores de TRPC6 |
| ES18800513T Active ES2946274T3 (es) | 2017-10-27 | 2018-10-25 | Inhibidores de TRPC6 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18800513T Active ES2946274T3 (es) | 2017-10-27 | 2018-10-25 | Inhibidores de TRPC6 |
Country Status (30)
| Country | Link |
|---|---|
| US (4) | US10800757B2 (es) |
| EP (2) | EP3700902B8 (es) |
| JP (2) | JP7217273B6 (es) |
| KR (2) | KR20200125758A (es) |
| CN (1) | CN111527078B (es) |
| AU (2) | AU2018355743B2 (es) |
| CA (1) | CA3078769A1 (es) |
| CL (2) | CL2020001097A1 (es) |
| CY (2) | CY1125488T1 (es) |
| DK (2) | DK3786160T3 (es) |
| ES (2) | ES2924933T3 (es) |
| FI (1) | FI3700902T3 (es) |
| HR (2) | HRP20230502T1 (es) |
| HU (2) | HUE059450T2 (es) |
| IL (2) | IL274039B (es) |
| LT (2) | LT3700902T (es) |
| MX (1) | MX387232B (es) |
| MY (1) | MY202126A (es) |
| PE (1) | PE20210154A1 (es) |
| PH (1) | PH12020550503A1 (es) |
| PL (2) | PL3700902T3 (es) |
| PT (2) | PT3700902T (es) |
| RS (2) | RS64271B1 (es) |
| SA (1) | SA520411843B1 (es) |
| SG (2) | SG10202011632RA (es) |
| SI (2) | SI3700902T1 (es) |
| TW (2) | TWI780246B (es) |
| UA (1) | UA128474C2 (es) |
| WO (1) | WO2019081637A1 (es) |
| ZA (1) | ZA202002372B (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK3786160T3 (da) * | 2017-10-27 | 2022-08-22 | Boehringer Ingelheim Int | Pyridinderivater og terapeutiske anvendelser deraf som trpc6-inhibitorer |
| US11485740B2 (en) * | 2018-02-15 | 2022-11-01 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| US11370771B2 (en) * | 2018-02-16 | 2022-06-28 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| IL315313A (en) * | 2018-08-24 | 2024-10-01 | Xeniopro GmbH | Aromatic molecules for use in the treatment of pathological conditions |
| WO2020208002A1 (en) * | 2019-04-12 | 2020-10-15 | Boehringer Ingelheim International Gmbh | Inhibitors of trpc6 |
| JP7594274B2 (ja) | 2019-10-24 | 2024-12-04 | 国立大学法人大阪大学 | 難聴の予防および/または治療用医薬組成物 |
| WO2021160625A1 (en) | 2020-02-11 | 2021-08-19 | Klinikum Rechts Der Isar | Administration of calcium channel trpc6 inhibitors using balloons, stents or other medical devices |
| EP4135696A1 (en) * | 2020-04-16 | 2023-02-22 | Boehringer Ingelheim International GmbH | Inhibitors of trpc6 for treating respiratory conditions |
| AR121846A1 (es) | 2020-04-16 | 2022-07-13 | Teijin Pharma Ltd | Derivado de arilo o heteroarilo |
| CN113105318B (zh) * | 2021-02-23 | 2022-05-20 | 中山大学 | 一种2,2-二氟环丁烷-1-羧酸的制备方法及应用 |
| AU2022366192A1 (en) * | 2021-10-15 | 2024-02-29 | Boehringer Ingelheim International Gmbh | Trpc6 inhibitory compounds for treating sepsis |
| JP2025023360A (ja) * | 2021-12-06 | 2025-02-17 | 国立大学法人 熊本大学 | Trpc6の発現を抑制する方法 |
| AU2024328580A1 (en) | 2023-08-18 | 2026-02-05 | Boehringer Ingelheim International Gmbh | Inhibitors of trpc6 for treating focal segmental glomerulosclerosis |
Family Cites Families (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2071897A1 (en) | 1989-12-28 | 1991-06-29 | Richard A. Glennon | Sigma receptor ligands and the use thereof |
| AU676993B2 (en) | 1991-06-27 | 1997-04-10 | Virginia Commonwealth University | Sigma receptor ligands and the use thereof |
| NZ257955A (en) | 1992-12-02 | 1996-05-28 | Pfizer | Catechol diethers pharmaceutical compositions |
| CN1129581C (zh) | 1998-09-22 | 2003-12-03 | 山之内制药株式会社 | 氰基苯基衍生物 |
| WO2001064642A2 (en) | 2000-02-29 | 2001-09-07 | Cor Therapeutics, Inc. | Benzamides and related inhibitors of factor xa |
| EP1301484A2 (en) | 2000-07-20 | 2003-04-16 | Neurogen Corporation | Capsaicin receptor ligands |
| DE60230266D1 (de) | 2001-06-15 | 2009-01-22 | Astellas Pharma Inc | Phenylpyridincarbonyl piperazinderivate |
| JP4186518B2 (ja) * | 2001-06-15 | 2008-11-26 | アステラス製薬株式会社 | フェニルピリジン誘導体 |
| NI200300045A (es) | 2002-04-26 | 2005-07-08 | Pfizer Prod Inc | Inhibidores de triariloxiariloxipirimidin-2,4,6-triona de metaloproteinasa. |
| DE10222291A1 (de) | 2002-05-18 | 2003-11-27 | Schlafhorst & Co W | Faserbandzuführeinrichtung |
| CN1150176C (zh) | 2002-05-22 | 2004-05-19 | 上海医药工业研究院 | 芳烷酮哌嗪衍生物及其应用 |
| CA2581745A1 (en) | 2004-10-13 | 2006-04-27 | Neurogen Corporation | Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor |
| DE102005044814A1 (de) | 2005-05-19 | 2006-11-23 | Grünenthal GmbH | Substituierte Sprio-Verbindungen und deren Verwendung zur Herstellung von Arzneimitteln |
| PE20070795A1 (es) | 2005-10-28 | 2007-08-24 | Lilly Co Eli | Compuestos pirazol-urea-heterociclicos como inhibidores de cinasa |
| US7638531B2 (en) | 2005-12-21 | 2009-12-29 | Schering Corporation | Phenoxypiperidines and analogs thereof useful as histamine H3 antagonists |
| JP5019768B2 (ja) | 2006-03-23 | 2012-09-05 | 独立行政法人科学技術振興機構 | 新規低分子化合物およびその製造方法 |
| CA2681162C (en) | 2007-03-15 | 2015-11-24 | Novartis Ag | Benzyl and pyridine derivatives as modulators of hedgehog pathway |
| JP5082538B2 (ja) | 2007-03-28 | 2012-11-28 | Dic株式会社 | ピペラジン化合物 |
| JP2012512877A (ja) | 2008-12-18 | 2012-06-07 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | セロトニン5−ht2b受容体阻害剤 |
| US20100234603A1 (en) | 2009-03-13 | 2010-09-16 | Xin Linghu | Process for Making Substituted Aryl Sulfone Intermediates |
| US20110039850A1 (en) | 2009-08-12 | 2011-02-17 | Mhairi Copland | Leukemia Treatment |
| WO2011044001A1 (en) | 2009-10-09 | 2011-04-14 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| KR20120107962A (ko) | 2009-11-18 | 2012-10-04 | 노파르티스 아게 | 고형 종양 및 다른 악성종양의 치료를 위한 방법 및 조성물 |
| JP5879273B2 (ja) | 2010-03-01 | 2016-03-08 | ジーティーエックス・インコーポレイテッド | 癌を処置するための化合物 |
| ES2653966T3 (es) | 2010-04-27 | 2018-02-09 | Mitsubishi Tanabe Pharma Corporation | Nuevo derivado de amida y su uso como medicina |
| WO2011143365A1 (en) | 2010-05-13 | 2011-11-17 | Amgen Inc. | Nitrogen heterocyclic compounds useful as pde10 inhibitors |
| PE20130774A1 (es) | 2010-07-29 | 2013-06-26 | Rigel Pharmaceuticals Inc | Compuestos heterociclicos activadores de ampk y metodos para emplearlos |
| WO2012158784A2 (en) | 2011-05-16 | 2012-11-22 | Theodore Mark Kamenecka | Modulators of the nuclear hormone receptor ror |
| JP5800786B2 (ja) | 2011-10-26 | 2015-10-28 | 田辺三菱製薬株式会社 | 新規アミド誘導体を有効成分として含有する医薬組成物 |
| WO2013066714A1 (en) | 2011-10-31 | 2013-05-10 | Merck Sharp & Dohme Corp. | Inhibitors of the renal outer medullary potassium channel |
| CN103360343B (zh) | 2012-03-30 | 2017-04-19 | 凯惠药业(上海)有限公司 | 一种哌嗪酰胺类化合物的制备方法 |
| WO2014139144A1 (en) | 2013-03-15 | 2014-09-18 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| ES2804271T3 (es) * | 2013-05-27 | 2021-02-05 | Hoffmann La Roche | Nuevos compuestos de 3,4-dihidro-2H-isoquinolin-1-ona y 2,3-dihidro-isoindol-1-ona |
| US20150087673A1 (en) | 2013-09-26 | 2015-03-26 | Rigel Pharmaceuticals, Inc. | Methods for using and biomarkers for ampk-activating compounds |
| KR20160100408A (ko) | 2014-01-06 | 2016-08-23 | 리젠 파마슈티컬스 소시에떼 아노님 | 신규한 글루타미나제 저해제 |
| WO2015199206A1 (ja) | 2014-06-27 | 2015-12-30 | 塩野義製薬株式会社 | Trpv4阻害活性を有する6員環誘導体 |
| UY36391A (es) | 2014-11-05 | 2016-06-01 | Flexus Biosciences Inc | Compuestos moduladores de la enzima indolamina 2,3-dioxigenasa (ido1), sus métodos de síntesis y composiciones farmacèuticas que las contienen |
| MA41598A (fr) | 2015-02-25 | 2018-01-02 | Constellation Pharmaceuticals Inc | Composés thérapeutiques de pyridazine et leurs utilisations |
| WO2017066705A1 (en) | 2015-10-14 | 2017-04-20 | Aquinnah Pharmaceuticals, Inc. | Compounds, compositions and methods of use against stress granules |
| CN107176927B (zh) | 2016-03-12 | 2020-02-18 | 福建金乐医药科技有限公司 | 组蛋白去甲基化酶lsd1抑制剂 |
| PL3429591T3 (pl) | 2016-03-16 | 2023-07-17 | Kura Oncology, Inc. | Podstawione pochodne tieno[2,3-d]pirymidyny jako inhibitory meniny-mll i metody ich zastosowania |
| RU2019101420A (ru) | 2016-06-20 | 2020-07-21 | Ратгерс, Де Стейт Юниверсити Оф Нью Джерси | Терапевтические соединения |
| CN107540636A (zh) | 2016-06-29 | 2018-01-05 | 成都贝斯凯瑞生物科技有限公司 | 一种含氮杂环衍生物及其应用 |
| CN106317050B (zh) * | 2016-08-24 | 2018-11-23 | 烟台大学 | 一种苯基噻唑衍生物及其制备方法与应用 |
| JPWO2018159827A1 (ja) | 2017-03-03 | 2020-01-09 | 国立大学法人京都大学 | 植物ステロイドホルモン(ブラシノライド)様活性をもつ非ステロイド化合物の創製 |
| TW201902875A (zh) | 2017-03-15 | 2019-01-16 | 美商亞瑟尼克斯公司 | 聯芳基哌啶醯胺化合物及其使用方法 |
| DK3786160T3 (da) | 2017-10-27 | 2022-08-22 | Boehringer Ingelheim Int | Pyridinderivater og terapeutiske anvendelser deraf som trpc6-inhibitorer |
| EP3710458B1 (en) * | 2017-11-16 | 2026-03-25 | Principia Biopharma Inc. | Immunoproteasome inhibitors |
| US11485740B2 (en) * | 2018-02-15 | 2022-11-01 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| US11370771B2 (en) * | 2018-02-16 | 2022-06-28 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
-
2018
- 2018-10-25 DK DK20197194.2T patent/DK3786160T3/da active
- 2018-10-25 KR KR1020207031025A patent/KR20200125758A/ko not_active Withdrawn
- 2018-10-25 HU HUE20197194A patent/HUE059450T2/hu unknown
- 2018-10-25 LT LTEPPCT/EP2018/079276T patent/LT3700902T/lt unknown
- 2018-10-25 US US16/170,154 patent/US10800757B2/en not_active Ceased
- 2018-10-25 SI SI201830924T patent/SI3700902T1/sl unknown
- 2018-10-25 CN CN201880070122.4A patent/CN111527078B/zh active Active
- 2018-10-25 WO PCT/EP2018/079276 patent/WO2019081637A1/en not_active Ceased
- 2018-10-25 CA CA3078769A patent/CA3078769A1/en active Pending
- 2018-10-25 MY MYPI2020001894A patent/MY202126A/en unknown
- 2018-10-25 HU HUE18800513A patent/HUE062460T2/hu unknown
- 2018-10-25 PT PT188005136T patent/PT3700902T/pt unknown
- 2018-10-25 PL PL18800513.6T patent/PL3700902T3/pl unknown
- 2018-10-25 UA UAA202003145A patent/UA128474C2/uk unknown
- 2018-10-25 EP EP18800513.6A patent/EP3700902B8/en active Active
- 2018-10-25 SG SG10202011632RA patent/SG10202011632RA/en unknown
- 2018-10-25 DK DK18800513.6T patent/DK3700902T3/da active
- 2018-10-25 MX MX2020004283A patent/MX387232B/es unknown
- 2018-10-25 LT LTEP20197194.2T patent/LT3786160T/lt unknown
- 2018-10-25 FI FIEP18800513.6T patent/FI3700902T3/fi active
- 2018-10-25 AU AU2018355743A patent/AU2018355743B2/en active Active
- 2018-10-25 PL PL20197194.2T patent/PL3786160T3/pl unknown
- 2018-10-25 PE PE2020000424A patent/PE20210154A1/es unknown
- 2018-10-25 ES ES20197194T patent/ES2924933T3/es active Active
- 2018-10-25 HR HRP20230502TT patent/HRP20230502T1/hr unknown
- 2018-10-25 JP JP2020523445A patent/JP7217273B6/ja active Active
- 2018-10-25 HR HRP20220991TT patent/HRP20220991T1/hr unknown
- 2018-10-25 KR KR1020207014013A patent/KR102724066B1/ko active Active
- 2018-10-25 US US16/170,178 patent/US10889568B2/en active Active
- 2018-10-25 ES ES18800513T patent/ES2946274T3/es active Active
- 2018-10-25 PT PT201971942T patent/PT3786160T/pt unknown
- 2018-10-25 SG SG11202003367TA patent/SG11202003367TA/en unknown
- 2018-10-25 RS RS20230382A patent/RS64271B1/sr unknown
- 2018-10-25 TW TW107137733A patent/TWI780246B/zh active
- 2018-10-25 EP EP20197194.2A patent/EP3786160B1/en active Active
- 2018-10-25 SI SI201830732T patent/SI3786160T1/sl unknown
- 2018-10-25 TW TW109138850A patent/TWI780511B/zh active
- 2018-10-25 RS RS20220752A patent/RS63535B1/sr unknown
-
2020
- 2020-04-19 IL IL274039A patent/IL274039B/en unknown
- 2020-04-23 CL CL2020001097A patent/CL2020001097A1/es unknown
- 2020-04-26 SA SA520411843A patent/SA520411843B1/ar unknown
- 2020-04-27 PH PH12020550503A patent/PH12020550503A1/en unknown
- 2020-05-04 ZA ZA2020/02372A patent/ZA202002372B/en unknown
- 2020-09-22 IL IL277493A patent/IL277493B/en unknown
- 2020-10-02 CL CL2020002562A patent/CL2020002562A1/es unknown
- 2020-10-06 AU AU2020250185A patent/AU2020250185B2/en active Active
- 2020-10-08 JP JP2020170563A patent/JP6979502B2/ja active Active
- 2020-12-08 US US17/115,667 patent/US20210163449A1/en not_active Abandoned
-
2021
- 2021-05-28 US US17/334,097 patent/USRE49699E1/en active Active
-
2022
- 2022-08-18 CY CY20221100560T patent/CY1125488T1/el unknown
-
2023
- 2023-06-07 CY CY20231100270T patent/CY1126109T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2924933T3 (es) | Derivados de piridina y usos terapéuticos de los mismos como inhibidores de TRPC6 | |
| EP3752499B1 (en) | Inhibitors of trpc6 | |
| CN113677673B (zh) | Trpc6的抑制剂 | |
| HK40045092B (en) | Pyridine derivatives and therapeutic uses thereof as trpc6 inhibitors | |
| HK40045092A (en) | Pyridine derivatives and therapeutic uses thereof as trpc6 inhibitors | |
| BR112020007818B1 (pt) | Compostos derivados de piridina carbonila, sal farmaceuticamente aceitável dos mesmos, composição farmacêutica que os compreende e seu uso como inibidores de trpc6 | |
| EA042674B1 (ru) | Пиридинкарбонильные производные и их терапевтические применения в качестве ингибиторов trpc6 | |
| HK40035138B (en) | Inhibitors of trpc6 | |
| HK40035138A (en) | Inhibitors of trpc6 | |
| EA040737B1 (ru) | Пиридинкарбонильные производные и их терапевтические применения в качестве ингибиторов trpc6 |