ES2934789T3 - Agonistas del receptor glp-1 y usos del mismo - Google Patents
Agonistas del receptor glp-1 y usos del mismoInfo
- Publication number
- ES2934789T3 ES2934789T3 ES17817174T ES17817174T ES2934789T3 ES 2934789 T3 ES2934789 T3 ES 2934789T3 ES 17817174 T ES17817174 T ES 17817174T ES 17817174 T ES17817174 T ES 17817174T ES 2934789 T3 ES2934789 T3 ES 2934789T3
- Authority
- ES
- Spain
- Prior art keywords
- methyl
- oxy
- acid
- carboxylic
- pyridin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Gynecology & Obstetrics (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Vascular Medicine (AREA)
- Psychology (AREA)
Abstract
En el presente documento se proporcionan ácidos 6-carboxílicos de benzimidazoles y 4-aza-, 5-aza-, 7-aza- y 4,7-diaza-benzimidazoles como agonistas de GLP-1R, procesos para fabricar dichos compuestos y métodos que comprenden la administración de dichos compuestos. a un mamífero que lo necesite. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Agonistas del receptor glp-1 y usos del mismo
Campo de la invención
En la presente memoria se proporcionan ácidos 6-carboxílicos de benzimidazoles y 4-aza-, 5-aza-, 7-aza-, y 4,7-diaza-benzimidazoles como agonistas del GLP-1R, procesos para hacer dichos compuestos, y procedimientos que comprenden la administración de dichos compuestos a un mamífero que necesite los mismos.
Antecedentes de la invención
La diabetes es un problema importante de salud pública debido a su creciente prevalencia y a los riesgos de salud asociados. La enfermedad se caracteriza por unos niveles elevados de glucosa en la sangre como consecuencia de defectos en la producción de insulina, en la acción de la insulina o en ambas. Se reconocen dos formas principales de diabetes, el tipo 1 y el tipo 2. La diabetes de tipo 1 (T1D) se desarrolla cuando el sistema inmunitario del organismo destruye las células beta del páncreas, las únicas células del cuerpo que producen la hormona insulina que regula la glucosa en sangre. Para sobrevivir, las personas con diabetes de tipo 1 se deben administrar insulina por medio de una inyección o una bomba. La diabetes mellitus de tipo 2 (denominada generalmente como DMT2) suele comenzar con una resistencia a la insulina o cuando la producción de insulina es insuficiente para mantener un nivel de glucosa aceptable.
En la actualidad, se dispone de varios enfoques farmacológicos para tratar la hiperglucemia y, posteriormente, la DMT2 (Hampp, C. et al. Use of Antidiabetic Drugs in the U.S., 2003 a 2012, Diabetes Care 2014, 37, 1367 a 1374). Se pueden agrupar en seis clases principales, cada una de las cuales actúa a través de un mecanismo primario diferente: (A) Secretoras de insulina, que incluyen las sulfonilureas (por ejemplo, glipizida, glimepirida, gliburida), meglitinidas (por ejemplo, nateglidina, repaglinida), inhibidores de la dipeptidil peptidasa IV (DPP-IV) (por ejemplo, sitagliptina, vildagliptina, alogliptina, dutogliptina, linagliptina, saxogliptina), y agonistas del receptor del péptido similar al glucagón-1 (GLP-1R) (por ejemplo, liraglutida, albiglutida, exenatida, lixisenatida, dulaglutida, semaglutida), que aumentan la secreción de insulina al actuar sobre las células beta pancreáticas. Las sulfonilureas y las meglitinidas tienen una eficacia y una tolerabilidad limitadas, provocan un aumento de peso y suelen inducir hipoglucemias. Los inhibidores de la DPP-IV tienen una eficacia limitada. Los agonistas del GLP-1R comercializados son péptidos que se administran por inyección subcutánea. La liraglutida está aprobada además para el tratamiento de la obesidad. (B) Se cree que las biguanidas (por ejemplo, la metformina) actúan principalmente por medio de la disminución de la producción hepática de glucosa. Las biguanidas suelen provocar trastornos gastrointestinales y acidosis láctica, lo que limita aún más su uso. (C) Los inhibidores de la alfa-glucosidasa (por ejemplo, la acarbosa) disminuyen la absorción intestinal de la glucosa. Estos agentes suelen provocar alteraciones gastrointestinales. (D) Las tiazolidinedionas (por ejemplo, la pioglitazona y la rosiglitazona) actúan sobre un receptor específico (el receptor gamma activado por el proliferador de peroxisomas) en los tejidos hepático, muscular y graso. Regulan el metabolismo de los lípidos mejorando posteriormente la respuesta de estos tejidos a las acciones de la insulina. El uso frecuente de estos fármacos puede provocar un aumento de peso y puede inducir edema y anemia. (E) La insulina se utiliza en los casos más graves, sola o en combinación con los agentes anteriores, y su uso frecuente también puede provocar un aumento de peso y conlleva un riesgo de hipoglucemia. (F) Los inhibidores del cotransportador 2 ligado a la sodio-glucosa (SGLT2) (por ejemplo, dapagliflozina, empagliflozina, canagliflozina, ertugliflozina) inhiben la reabsorción de glucosa en los riñones y, por lo tanto, reducen los niveles de glucosa en la sangre. Esta clase emergente de fármacos puede estar asociada a la cetoacidosis y a las infecciones del tracto urinario.
Sin embargo, con la excepción de los agonistas del GLP-1R y los inhibidores del SGLT2, los fármacos tienen una eficacia limitada y no abordan los problemas más importantes, la disminución de la función de las células p y la obesidad asociada.
La obesidad es una enfermedad crónica de gran prevalencia en la sociedad moderna y está asociada a numerosos problemas médicos, que incluyen la hipertensión, la hipercolesterolemia y las enfermedades coronarias. Además, está altamente correlacionada con la DMT2 y la resistencia a la insulina, esta última generalmente acompañada de hiperinsulinemia o hiperglucemia, o ambas. Además, la DMT2 se asocia a un riesgo entre dos y cuatro veces mayor de sufrir una enfermedad coronaria. Actualmente, el único tratamiento que elimina la obesidad con alta eficacia es la cirugía bariátrica, pero este tratamiento es costoso y arriesgado. La intervención farmacológica suele ser menos eficaz y estar asociada a efectos secundarios. Por lo tanto, es evidente la necesidad de una intervención farmacológica más eficaz, con menos efectos secundarios y una administración cómoda.
Aunque la DMT2 se asocia más comúnmente con la hiperglucemia y la resistencia a la insulina, otras enfermedades asociadas a la DMT2 incluyen la resistencia hepática a la insulina, la intolerancia a la glucosa, la neuropatía diabética, la nefropatía diabética, la retinopatía diabética, la obesidad, la dislipidemia, la hipertensión, la hiperinsulinemia y la enfermedad del hígado graso no alcohólico (HGNA).
La HGNA es la manifestación hepática del síndrome metabólico y es un espectro de afecciones hepáticas que abarca la esteatosis, la esteatohepatitis no alcohólica (EHNA), la fibrosis, la cirrosis y, en última instancia, el
carcinoma hepatocelular. La HGNA y la EHNA se consideran las principales enfermedades del hígado graso, dado que representan la mayor proporción de individuos con lípidos hepáticos elevados. La gravedad de la NAFLD/NASH se basa en la presencia de lípidos, el infiltrado de células inflamatorias, el abombamiento de los hepatocitos y el grado de fibrosis. Aunque no todos los individuos con esteatosis evolucionan a EHNA, una porción importante lo hace.
El GLP-1 es una hormona incretina de 30 aminoácidos de longitud segregada por las células L del intestino en respuesta a la ingestión de alimentos. Se ha demostrado que el GLP-1 estimula la secreción de insulina de forma fisiológica y dependiente de la glucosa, disminuye la secreción de glucagón, inhibe el vaciado gástrico, disminuye el apetito y estimula la proliferación de las células beta. En experimentos no clínicos, el GLP-1 promueve la competencia continua de las células beta al estimular la transcripción de genes importantes para la secreción de insulina dependiente de la glucosa y al promover la neogénesis de las células beta (Meier, et al. Biodrugs. 2003; 17 (2): 93 a 102).
En un individuo sano, el GLP-1 desempeña un papel importante en la regulación de los niveles de glucosa en sangre posprandiales al estimular la secreción de insulina dependiente de la glucosa por parte del páncreas, lo que provoca un aumento de la absorción de glucosa en la periferia. El GLP-1 también suprime la secreción de glucagón, lo que conduce a una reducción de la producción hepática de glucosa. Además, el GLP-1 retrasa el vaciado gástrico y ralentiza la motilidad del intestino delgado retrasando la absorción de los alimentos. En las personas con DMT, el aumento postprandial normal de GLP-1 está ausente o reducido (Vilsboll T, et al. Diabetes. 2001. 50: 609 a 613).
Holst (Physiol. Rev. 2007, 87, 1409) y Meier (Nat. Rev. Endocrinol. 2012, 8, 728) describen que los agonistas de los receptores de GLP-1, tales como el GLP-1, la liraglutida y el exendin-4, tienen 3 actividades farmacológicas principales para mejorar el control glucémico en pacientes con DMT2 por medio de la reducción de la glucosa en ayunas y posprandial (FPG y PPG): (i) aumento de la secreción de insulina dependiente de la glucosa (mejora de la primera y la segunda fase), (ii) actividad supresora del glucagón en condiciones de hiperglucemia, (iii) retraso de la velocidad de vaciado gástrico que da lugar a una absorción retardada de la glucosa derivada de las comidas. El documento WO 2011/143365 también desvela compuestos heterocíclicos útiles para tratar la diabetes y la obesidad.
Sigue existiendo la necesidad de una prevención y/o tratamiento de fácil administración para las enfermedades cardiometabólicas y asociadas.
Descripción detallada de la invención
La presente invención se refiere a compuestos de la Fórmula I

o a una sal aceptable para uso farmacéutico de los mismos, en la que
cada R1 es independientemente halógeno, -CN, alquilo C1-3, o -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 está sustituido con 0 a 3 átomos de F;
m es 0, 1, 2 o 3.
cada R2 es independientemente F, Cl, o-CN;
p es 0, 1 o 2;
cada R3 es independientemente F, -OH, -CN, alquilo C1-3, -Oalquilo C1-3, o -cicloalquilo C3-4, o 2 R3s se pueden ciclizar juntos para formar -espirocicloalquilo C3-4, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3, cicloalquilo, o espirocicloalquilo puede ser sustituido como la valencia lo permita con 0 a 3 átomos de F y con 0 a 1 -OH; q es 0, 1 o 2;
Y es CH o N;
R4 es alquilo C1-3, alquileno Cü.3-cicloalquilo C3-6, alquileno C0-3-R5, o alquileno C1-3-R6, en el que dicho alquilo puede estar sustituido como la valencia lo permita con 0 a 3 sustituyentes seleccionados independientemente de 0 a 3 átomos de F y 0 a 1 sustituyente seleccionado de alquileno C0-1-CN alquileno Cü.1-ORo, y -N(RN)2, y en el que dicho alquileno y cicloalquilo pueden estar sustituidos independientemente como la valencia lo permita con 0 a 2 sustituyentes seleccionados independientemente de 0 a 2 átomos de F y 0 a 1 sustituyente seleccionado de alquileno C0-1-CN, alquileno Cü.1-ORo, y -N(RN)2;
R5 es un heterocicloalquilo de 4 a 6 miembros, en el que dicho heterocicloalquilo puede estar sustituido con 0 a 2
sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 1 oxo (=O),
0 a 1 -CN,
0 a 2 átomos de F, y
0 a 2 sustituyentes seleccionados independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede ser sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F,
0 a 1 -CN, y
0 a 1 -ORO;
R6 es un heteroarilo de 5 a 6 miembros, en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 halógenos,
0 a 1 sustituyente seleccionado entre -ORO y -N(RN)2, y
0 a 2 alquilo C1-3, en el que el alquilo puede estar sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F, y
0 a 1 -ORO;
cada RO es independientemente H, o alquilo C1-3, en el que alquilo C1-3 puede estar sustituido con 0 a 3 átomos de F;
cada RN es independientemente H o alquilo C1-3;
Z1 es CH o N;
Z2 y Z3 son cada uno independientemente -CRZ o N, a condición de que cuando Z1 o Z3 sea N, Z2 sea -CRZ; y cada RZ es independientemente H, F, Cl, o -CH3.
Otra realización se refiere a los compuestos de la Fórmula II

m es 0 o 1;
R2 es F;
p es 0 o 1; y
q es 0 o 1.
Otra realización se refiere a los compuestos de las Fórmulas I o II, en los que
m es 0 o 1;
q es 0 o 1; y
R3 es -F, -CH3, -CH2CH3, -CH2OH, -CF3, isopropilo o ciclopropilo, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de la Fórmula III

o a una sal aceptable para uso farmacéutico de los mismos, en los que
m es 0 o 1;
R2 es F;
p es 0 o 1;
R3 es alquilo C1-2, en el que alquilo C1-2 puede estar sustituido, como la valencia lo permita, con 0 a 3 átomos de F; y
q es 0 o 1.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que cada R1 es independientemente F, Cl, -CN, -CH3 o -CF3, o una sal aceptable para uso farmacéutico de los mismos. Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que
R3 es -CH3;
q es 0 o 1; y
R4 es -CH2CH2OCH3, alquileno C1-3-R5, o alquileno C1-3-R6, o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que
R4 es -CH2-R5, en el que R5 es el heterocicloalquilo de 4 a 5 miembros, en el que dicho heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 átomos de F, y
0 a 1 sustituyente seleccionado entre -OCH3 y -CH2OCH3;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heterocicloalquilo es

en el que el heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionados independientemente entre:
0 a 1 oxo (O=),
0 a 1 -CN,
0 a 2 átomos de F, y
0 a 2 sustituyentes seleccionados independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede estar sustituido independientemente con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F,
0 a 1 -CN, y
0 a 1 -ORO,
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heterocicloalquilo es

en el que el heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionados independientemente entre:
0 a 1 -CN,
0 a 2 átomos de F, y
0 a 2 sustituyentes seleccionados independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede estar sustituido independientemente con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F,
0 a 1 -CN, y
0 a 1 -ORO, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heterocicloalquilo es

en el que el heterocicloalquilo puede estar sustituido con 0 a 1 sustituyente como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionado entre:
-CN,
un átomo de F, y
0 a 1 sustituyente seleccionado independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede ser sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F,
0 a 1 -CN, y
0 a 1 -ORO, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heterocicloalquilo es

y en los que el heterocicloalquilo puede estar sustituido con 0 a 1 sustituyente como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionado entre:
-CN,
un átomo de F, y
0 a 1 sustituyente seleccionado independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede ser sustituido con 0 a 3 sustituyentes como la valencia lo permita:
0 a 3 átomos de F,
0 a 1 -CN, o
0 a 1 -ORO, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heterocicloalquilo es

y en los que el heterocicloalquilo puede estar sustituido como la valencia lo permita con 0 a 1 metilo, en el que dicho metilo puede estar sustituido con 0 a 3 átomos de F, o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a compuestos seleccionados independientemente de uno o cualquier combinación de los siguientes:
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2R)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[4-(6-{[(4-ciano-2-fluorofenil)(metil-d2)]oxi}piridin-2-il)piperidin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(3R)-tetrahidrofurano-3-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(3R)-tetrahidrofurano-3-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(3S)-tetrahidrofurano-3-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2R)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2R)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(2,4-difluorobencil)oxi]-5-fluoropiridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico; o
ácido 2-{[(2S)-4-{6-[(4-cianobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1Hbenzimidazol-6-carboxílico;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a un compuesto que es el ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a un compuesto que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a un compuesto que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
Otra realización es la sal tris del ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico.
Otra realización es el ácido libre de 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico.
Otra realización se refiere a un compuesto que es el ácido 2-{[4-(6-{[(4-ciano-2-fluorofenil)(metil-d2)]oxi}piridin-2-il)piperidin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a un compuesto que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que
R4 es -CH2-R6, en los que R6 es el heteroarilo de 5 miembros, en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 halógenos, en los que el halógeno se selecciona independientemente entre F y Cl,
0 a 1 -OCH3, y
0 a 1 -CH3, -CH2CH3, -CF3, o -CH2CH2OCH3;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heteroarilo es

en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionados independientemente entre:
0 a 2 halógenos, en los que el halógeno se selecciona independientemente entre F y Cl,
0 a 1 sustituyente seleccionado entre -ORO y -N(RN)2, o
0 a 2 alquilo C1-3, en el que el alquilo puede estar sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F, y
0 a 1 -ORO;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heteroarilo es

en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionados independientemente entre:
0 a 2 halógenos, en los que el halógeno se selecciona independientemente entre F y Cl,
0 a 1 sustituyente seleccionado entre -ORO y -N(RN)2, o
0 a 2 alquilo C1-3, en el que el alquilo puede estar sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F, y
0 a 1 -ORO;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de las Fórmulas I, II o III, en los que el heteroarilo es
m í N
[.IM -a lqu ilo C1-3 ^ N-alquilo C1-3


en el que el alquilo C1-3 de dicho heteroarilo puede estar sustituido con 0 a 3 sustituyentes como la valencia lo permita, por ejemplo, sustituyendo al hidrógeno, seleccionados independientemente entre:
0 a 3 átomos de F, y
0 a 1 -ORO;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a compuestos seleccionados independientemente de uno o cualquier combinación de los siguientes:
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metil-1H-1,2,3-triazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H
benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-doro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-4-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-doro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-etil-1H-1,2,3-triazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,2-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,2-oxazol-3-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1H-1,2,3-triazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-(1,3-oxazol-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxílico; o
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-(1,3-oxazol-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxílico;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a compuestos seleccionados independientemente de uno o cualquier combinación de los siguientes:
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico; o
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a compuestos seleccionados independientemente de uno o cualquier combinación de los siguientes:
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-7-fluoro-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-7-fluoro-1-(2-metoxietil)-1 H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico; ácido 2-{[(2S)-4-{6-[(4-cianobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]-5-fluoropiridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico; o
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metoxiciclobutil)metil]-1H-benzimidazol-6-carboxílico;
o una sal aceptable para uso farmacéutico del mismo.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que Z1, Z2 y Z3 son cada uno CRZ, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que RZ es H, o una sal aceptable para uso farmacéutico de los mismos. Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que Z1, Z2 y Z3 son cada uno CH, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que p es 0 o 1; yR2 es F.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que R3 es -CH3, o -CF3; y q es 1, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que cada R1 es independientemente F, Cl o -CN, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que R4 es -CH2-R5, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de las Fórmulas I, II o III, en los que R4 es -CH2-R5, o una sal aceptable para uso farmacéutico de los mismos.
Otra realización se refiere a los compuestos de otras realizaciones de la presente memoria, por ejemplo, los compuestos de la Fórmula I, II o III, en los que el compuesto es el ácido libre.
En otra realización, la invención proporciona una composición farmacéutica que comprende un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, en mezcla con al menos un excipiente aceptable para uso farmacéutico.
La invención también incluye las siguientes realizaciones:
un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, para su uso como medicamento;
un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, para su uso en la prevención y/o el tratamiento de las enfermedades cardiometabólicas y asociadas que se tratan en la presente memoria, que incluyen la DMT2, la prediabetes, la EHNA y las enfermedades cardiovasculares;
un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, para su uso en un procedimiento de tratamiento de una enfermedad para la que está indicado un agonista del GLP-1R, en un sujeto que necesita dicha prevención y/o tratamiento, que comprende administrar al sujeto una cantidad eficaz para uso terapéutico de un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria;
el uso de un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, para la fabricación de un medicamento para el tratamiento de una enfermedad o afección para la que está indicado un agonista del GLP-1R;
un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, para su uso en el tratamiento de una enfermedad o afección para la que está indicado un agonista del GLP-1R; o
una composición farmacéutica para el tratamiento de una enfermedad o afección para la que está indicado un agonista del GLP-1R, que comprende un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria.
Cada Ejemplo o sal aceptable para uso farmacéutico del mismo se puede reivindicar individualmente o agruparse en cualquier combinación con cualquier número de todas y cada una de las realizaciones descritas en la presente memoria.
La invención también se refiere a una composición farmacéutica que comprende un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, para su uso en el tratamiento y/o prevención de enfermedades cardiometabólicas y asociadas que se tratan en la presente memoria, que incluyen la DMT2, la prediabetes, la EHNA y la enfermedad cardiovascular.
Otra realización de la invención se refiere a un compuesto de las Fórmulas I, II o III, o una sal aceptable para uso farmacéutico del mismo, como se define en cualquiera de las realizaciones descritas en la presente memoria, para su uso en el tratamiento y/o el tratamiento de enfermedades cardiometabólicas y asociadas, que incluyen la diabetes (T1D y/o T2DM, que incluyen la prediabetes), la T1D idiopática (Tipo 1b) diabetes autoinmune latente en adultos (LADA), T2DM de inicio temprano (EOD), diabetes atípica de inicio en la juventud (YOAD), diabetes de inicio en la madurez de los jóvenes (MODY), diabetes relacionada con la malnutrición, diabetes gestacional, hiperglucemia, resistencia a la insulina, resistencia hepática a la insulina, alteración de la tolerancia a la glucosa, neuropatía diabética, nefropatía diabética, enfermedad renal (por ejemplo, trastorno renal agudo, disfunción tubular, cambios proinflamatorios en los túbulos proximales), retinopatía diabética, disfunción adipocitaria, depósito adiposo visceral, apnea del sueño, obesidad (incluida la obesidad hipotalámica y la obesidad monogénica) y comorbilidades relacionadas (por ejemplo, osteoartritis e incontinencia urinaria), trastornos alimentarios (que incluyen el síndrome de atracones, la bulimia nerviosa y la obesidad sindrómica tal como los síndromes de Prader-Willi y Bardet-Biedl), aumento de peso por el uso de otros agentes (por ejemplo, por el uso de esteroides y antipsicóticos), deseo excesivo de azúcar, dislipidemia (que incluyen hiperlipidemia, hipertrigliceridemia, aumento del colesterol total, colesterol LDL alto y colesterol HDL bajo), hiperinsulinemia, NAFLD (que incluyen enfermedades relacionadas tales como esteatosis, NASH, fibrosis, cirrosis y carcinoma hepatocelular), enfermedades cardiovasculares, aterosclerosis (incluida la enfermedad de las arterias coronarias), enfermedad vascular periférica, hipertensión, disfunción endotelial, alteración de la distensibilidad vascular, insuficiencia cardíaca congestiva, infarto de miocardio (por ejemplo, necrosis y apoptosis), accidente cerebrovascular, accidente cerebrovascular hemorrágico, accidente cerebrovascular isquémico, lesión cerebral traumática, hipertensión pulmonar, reestenosis tras angioplastia, claudicación intermitente, lipemia postprandial, acidosis metabólica, cetosis, artritis, osteoporosis, Enfermedad de Parkinson, hipertrofia ventricular izquierda, enfermedad arterial periférica, degeneración macular, cataratas, glomeruloesclerosis, insuficiencia renal crónica, síndrome metabólico, síndrome X, síndrome premenstrual, angina de pecho, trombosis, aterosclerosis, ataques isquémicos transitorios, reestenosis vascular, alteración del metabolismo de la glucosa, condiciones de alteración de la glucosa plasmática en ayunas, hiperuricemia, gota, disfunción eréctil, trastornos de la piel y del tejido conectivo, psoriasis, ulceraciones en los pies, colitis ulcerosa, lipoproteinemia hiper apo B, enfermedad de Alzheimer, esquizofrenia, deterioro de la cognición, enfermedad inflamatoria intestinal, síndrome del intestino corto, enfermedad de Crohn, colitis, síndrome del intestino irritable, prevención o tratamiento del síndrome del ovario poliquístico y tratamiento de la adicción (por ejemplo, alcoholismo y/o drogadicción).
Las abreviaturas utilizadas en la presente memoria son las siguientes:
El término “alquilo”, como se utiliza en la presente memoria, significa un grupo hidrocarburo monovalente de cadena recta o ramificada de fórmula -CnH(2n+1). Los ejemplos no limitantes incluyen el metilo, el etilo, el propilo, el butilo, el 2-metil-propilo, el 1,1 -dimetiletilo, el pentilo y el hexilo.
El término “alquileno”, como se utiliza en la presente memoria, significa un grupo hidrocarburo divalente de cadena recta o ramificada de fórmula -CnH2n-. Los ejemplos no limitantes son el etileno y el propileno.
El término “cicloalquilo”, como se utiliza en la presente memoria, significa un grupo hidrocarburo cíclico y monovalente de fórmula -CnH(2n-1) que contiene al menos tres átomos de carbono. Los ejemplos no limitantes incluyen el ciclopropilo, el ciclobutilo, el ciclopentilo y el ciclohexilo.
El término “halógeno”, como se utiliza en la presente memoria, se refiere al fluoruro, cloruro, bromuro o yoduro. El término “heterocicloalquilo”, como se utiliza en la presente memoria, se refiere a un grupo cicloalquilo en el que uno o más de los grupos metileno del anillo (-CH2-) ha sido sustituido por un grupo seleccionado entre -O-, -S- o nitrógeno, en el que el nitrógeno puede proporcionar un punto de unión o puede estar sustituido como se proporciona en cada realización. Cuando el nitrógeno proporciona un punto de unión, un dibujo estructural de un heterocicloalquilo tendría un hidrógeno en dicho nitrógeno. Generalmente, el heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de oxo, -CN, halógeno, alquilo y -Oalquilo y el alquilo puede estar más sustituido. Se observará que cuando hay una sustitución 0, el heterocicloalquilo no está sustituido.
El término “heteroarilo”, como se utiliza en la presente memoria, se refiere a un hidrocarburo aromático monocíclico que contiene de 5 a 6 átomos de carbono en el que al menos uno de los átomos de carbono del anillo ha sido sustituido por un heteroátomo seleccionado entre el oxígeno, el nitrógeno y el azufre. Dicho grupo heteroarilo puede estar unido a través de un átomo de carbono del anillo o, cuando la valencia lo permita, a través de un átomo de nitrógeno del anillo. Generalmente, el heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de halógeno, OH, alquilo, O-alquilo, y amino (por ejemplo, NH2,
NHalquilo, N(alquilo)2), y el alquilo puede ser sustituido adicionalmente. Se observará que cuando hay 0 sustituciones, el heteroarilo no está sustituido.
Temperatura ambiente: TA.
Metanol: MeOH.
Etanol: EtOH.
Isopropanol: iPrOH.
Acetato de etilo: EtOAc.
Tetrahidrofurano: THF.
Tolueno: PhCH3.
Carbonato de cesio: Cs2CO3.
Bis(trimetilsilil)amida de litio: LiHMDS.
Butóxido de sodio: NaOtBu.
T-butóxido de potasio: KOtBu.
Diisopropilamida de litio: LDA.
Trietilamina: Et3N.
N,N-diisopropiletil amina: DIPEA.
Carbonato de potasio: K2CO3.
Dimetil formamida: DMF.
Acetamida de dimetilo: DMAc.
Sulfóxido de dimetilo: DMSO.
N-metil-2-pirrolidinona: NMP.
Hidruro de sodio: NaH.
Ácido trifluoroacético: TFA.
Anhídrido trifluoroacético: TFAA.
Anhídrido acético: Ac2O.
Diclorometano: DCM
1.2- dicloroetano: DCE.
Ácido clorhídrico: HCI.
1,8-Diazabiciclo[5.4.0]undec-7-eno: DBU.
Complejo borano-dimetilsulfuro: BH3-DMS.
Complejo borano-tetrahidrofurano: BH3-THF.
Hidruro de litio y aluminio: LAH.
Ácido acético: AcOH.
Acetonitrilo: MeCN.
ácido p-Toluenosulfónico: pTSA.
Acetona de dibencilidina: DBA.
2,2'-Bis(difenilfosfino)-1,1'-binaftaleno: BINAP.
1,1'-Ferrocenediil-bis(difenilfosfina): dppf.
1.3- Bis(difenilfosfino)propano: Dp Pp .
ácido 3-cloroperbenzoico: m-CPBA.
Éter terc-butílico: MTBE.
Metanosulfonilo: Ms.
N-Metilpirrolidinona: NMP.
Cromatografía en capa fina: TLC.
Cromatografía de fluidos supercríticos: SFC.
4-(Dimetilamino)piridina: DMAP.
Terc-Butiloxicarbonilo: Boc.
1- [Bis(dimetilamino)metileno]-1H-1,2,3-triazol[4,5-b]piridinio 3-oxid hexafluorofosfato: HATU.
Éter de petróleo: PE.
2- (1H-Benzotriazol-1-il)-1,1,3,3-tetrametiluronio hexafluorofosfato: HBTU.
2-Amino-2-(hidroximetil)propano-1,3-diol: tris.
tris(dibencilideneacetona)dipaladio: Pd2(dba)3
los espectros de resonancia magnética nuclear (RMN) de1H fueron en todos los casos consistentes con las estructuras propuestas. Los desplazamientos químicos característicos (8) se indican en partes por millón en relación con la señal de protones residuales en el disolvente deuterado (CHCh a 7,27 ppm; CD2H0 D a 3,31 ppm; MeCN a 1,94 ppm; DMSo a 2,50 ppm) y se indican mediante el uso de las abreviaturas convencionales para designar los picos principales: por ejemplo, s, singlete; d, doblete; t, triplete; q, cuarteto; m, multiplete; br, amplio. Los espectros de RMN de 1H se obtuvieron con intensidades de campo de 400 o 600 MHz si no se indicaba.
Como se utiliza en la presente memoria, una línea ondulada, denota un punto de unión de un sustituyente a otro grupo.
denota un punto de unión de un sustituyente a otro grupo.
Los compuestos e intermedios descritos a continuación se nombraron mediante el uso de la convención de
nomenclatura proporcionada con ChemBioDraw Ultra, Versión 13.0 (CambridgeSoft Corp., Cambridge, Massachusetts) o ACD/Labs, Versión 12 (Advanced Chemistry Development, Inc., Toronto, Ontario). Las convenciones de nomenclatura proporcionadas con ChemBioDraw Ultra, Versión 13.0 y ACD/Labs, Versión 12 son muy conocidas por los expertos en la técnica y se considera que las convenciones de nomenclatura proporcionadas con ChemBioDraw Ultra, Versión 13.0 y ACD/Labs, Versión 12 se ajustan en general a las recomendaciones de la IUPAC (Unión Internacional de Química Pura y Aplicada) sobre Nomenclatura de la Química Orgánica y a las normas del Índice CAS. Se observará que los nombres químicos pueden tener sólo paréntesis o pueden tener paréntesis y corchetes. Los descriptores estereoquímicos también se pueden colocar en diferentes lugares dentro del propio nombre, dependiendo de la convención de nomenclatura. Una persona con conocimientos ordinarios en la técnica reconocerá estas variaciones de formato y entenderá que proporcionan la misma estructura química.
Las sales aceptables para uso farmacéutico de los compuestos de la invención incluyen las sales de adición de ácido y de base de los mismos.
Las sales de adición de ácido adecuadas se forman a partir de ácidos que forman sales no tóxicas. Los ejemplos incluyen el acetato, el aspartato, el benzoato, el besilato, el bicarbonato/carbonato, el bisulfato/sulfato, el borato, el camsilato, el ciclamato, el citrato, el edisilato, el esilato, el formiato, el fumarato, el glucepato, el gluconato, el glucuronato, el hexafluorofosfato, el hibenzato, el clorhidrato/cloruro, el bromhidrato/bromuro, el yodidrato/yoduro, el isetionato, el lactato, el malato, el maleato, el malonato, el mesilato, el metilsulfato, el naftilato, el 2-napsilato, el nicotinato, el nitrato, el orotato, el oxalato, el palmitato, el pamoato, el fosfato/fosfato de hidrógeno/fosfato de dihidrógeno, el piroglutamato, el sacarato, el estearato, el succinato, el tannato, el tartrato, el tosilato, el trifluoroacetato, el ácido 1,5-naftalendisulfónico y las sales de trifluoroacetato.
Las sales básicas adecuadas se forman a partir de bases que forman sales no tóxicas. Los ejemplos incluyen las sales de aluminio, arginina, benzatina, calcio, colina, dietilamina, bis(2-hidroxietil)amina (diolamina), glicina, lisina, magnesio, meglumina, 2-aminoetanol (olamina), potasio, sodio, 2-Amino-2-(hidroximetil)propano-1,3-diol (tris o trometamina) y cinc.
También se pueden formar hemisales de ácidos y bases, por ejemplo, sales de hemisulfato y hemicálcicas. Para una revisión de las sales adecuadas, véase “Handbook of Pharmaceutical Salts: Properties, Selection, and Use por Stahl y Wermuth (Wiley-VCH, 2002).
Las sales aceptables para uso farmacéutico de los compuestos de la invención, se pueden preparar, respectivamente, por uno o más de tres procedimientos:
(i) por medio de la reacción del compuesto de la Fórmula I con el ácido o la base deseados;
(ii) por medio de la eliminación de un grupo protector lábil al ácido o a la base de un precursor adecuado del compuesto de la Fórmula I o la apertura en anillo de un precursor cíclico adecuado, por ejemplo, una lactona o una lactama, mediante el uso del ácido o la base deseados; o
(iii) por medio de la conversión de una sal del compuesto de la Fórmula I en otra por reacción con un ácido o una base apropiados o por medio de una columna de intercambio iónico adecuada.
Las tres reacciones se suelen llevar a cabo en solución. La sal resultante puede precipitar de la solución y recolectarse por filtración o se puede recuperar por evaporación del disolvente. El grado de ionización de la sal resultante puede variar desde completamente ionizada hasta casi no ionizada.
Los compuestos de la Fórmula I, y las sales aceptables para uso farmacéutico de los mismos, pueden existir en formas solvatadas y no solvatadas. El término “solvato” se utiliza en la presente memoria para describir un complejo molecular que comprende el compuesto de la Fórmula I, o una sal aceptable para uso farmacéutico del mismo, y una o más moléculas de disolvente aceptables para uso farmacéutico, por ejemplo, el etanol. El término “hidrato” se emplea cuando dicho disolvente es el agua.
Un sistema de clasificación actualmente aceptado para los hidratos orgánicos es el que define los hidratos de sitio aislado, de canal o coordinados por iones metálicos - véase Polymorphism in Pharmaceutical Solids por K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995). Los hidratos de sitio aislados son aquellos en los que las moléculas de agua están aisladas del contacto directo entre sí por moléculas orgánicas intermedias. En los hidratos de canal, las moléculas de agua se encuentran en los canales de la red, junto a otras moléculas de agua. En los hidratos coordinados con iones metálicos, las moléculas de agua están unidas al ion metálico.
Cuando el disolvente o el agua están fuertemente unidos, el complejo puede tener una estequiometría bien definida e independiente de la humedad. Si, sin embargo, el disolvente o el agua está unido débilmente, como en solvatos de canal y compuestos higroscópicos, el contenido de agua/disolvente dependerá de la humedad y las condiciones de secado. En estos casos, la no estequiometría será la norma.
También se incluyen en el ámbito de la invención los complejos multicomponentes (diferentes de las sales y los solvatos) en los que el fármaco y al menos otro componente están presentes en cantidades estequiométricas o no estequiométricas. Los complejos de este tipo incluyen los clatratos (complejos de inclusión de fármacos) y los cocristales. Estos últimos se definen normalmente como complejos cristalinos de componentes moleculares neutros
que se unen por medio de interacciones no covalentes, pero también podrían ser complejos de una molécula neutra con una sal. Los cocristales se pueden preparar por cristalización en fusión, por recristalización a partir de disolventes o por molienda física de los componentes - véase Chem Commun, 17, 1889 a 1896, por O. Almarsson y M. J. Zaworotko (2004). Para una revisión general de complejos de múltiples componentes, véase J Pharm. Sci., 64 (8), 1269 a 1288 por Haleblian (Agosto 1975).
Los compuestos de la invención pueden existir en un continuo de estados sólidos que van desde totalmente amorfos a totalmente cristalinos. El término “amorfo” se refiere a un estado en el que el material carece de orden de largo alcance a nivel molecular y, dependiendo de la temperatura, puede presentar las propiedades físicas de un sólido o un líquido. Normalmente, estos materiales no presentan patrones de difracción de rayos X característicos y, aunque presentan las propiedades de un sólido, se describen más formalmente como un líquido. Al calentarse, se produce un cambio de propiedades de sólido a líquido que se caracteriza por un cambio de estado, normalmente de segundo orden (“transición vítrea”). El término “cristalino” se refiere a una fase sólida en la que el material tiene una estructura interna regular y ordenada a nivel molecular y da un patrón de difracción de rayos X distintivo con picos definidos. Estos materiales, cuando se calientan lo suficiente, también presentan las propiedades de un líquido, pero el paso de sólido a líquido se caracteriza por un cambio de fase, normalmente de primer orden (“punto de fusión”).
Los compuestos de la Fórmula I también pueden existir en un estado mesomórfico (mesofase o cristal líquido) cuando se someten a condiciones adecuadas. El estado mesomórfico es intermedio entre el estado cristalino verdadero y el estado líquido verdadero (ya sea fundido o en solución). El mesomorfismo que surge como resultado de un cambio de temperatura se describe como “termotrópico” y el que resulta de la adición de un segundo componente, tal como el agua u otro disolvente, se describe como “lipotrópico”. Los compuestos que tienen el potencial de formar mesofases liotrópicas se describen como “anfifílicos” y consisten en moléculas que poseen un grupo de cabeza polar iónico (tal como -COO'Na+, -COO'K+, o -SO3-Na') o no iónico (tal como -N'N+(c H3)3). Para más información, véase Crystals and the Polarizing Microscope por N. H. Hartshorne y A. Stuart, 4ta edición (Edward Arnold, 1970).
Los compuestos de la Fórmula I pueden presentar polimorfismo y/o uno o más tipos de isomería (por ejemplo, isomería óptica, geométrica o tautomérica). Los compuestos de la Fórmula I también pueden ser etiquetados isotópicamente. Dicha variación está implícita en los compuestos de la Fórmula I definidos como tales por referencia a sus características estructurales y, por lo tanto, dentro del alcance de la invención.
Los compuestos de la Fórmula I que contienen uno o más átomos de carbono asimétricos pueden existir como dos o más estereoisómeros. Cuando un compuesto de la Fórmula I contiene un grupo alquenilo o alquenileno, son posibles los isómeros geométricos cis/trans (o Z/E). Cuando los isómeros estructurales son interconvertibles a través de una barrera de baja energía, se puede producir isomerismo tautomérico (“tautomerismo”). Esto puede adoptar la forma de tautomerismo de protones en compuestos de la Fórmula I que contengan, por ejemplo, un grupo imino, ceto u oxima, o el denominado tautomerismo de valencia en compuestos que contengan una fracción aromática. A partir de ello se deduce que un mismo compuesto puede presentar más de un tipo de isomería.
Las sales aceptables para uso farmacéutico de los compuestos de la Fórmula I también pueden contener un contraión ópticamente activo (por ejemplo, d-lactato o l-lisina) o racémico (por ejemplo, dl-tartrato o dl-arginina).
Los isómeros cis/trans se pueden separar por medio de técnicas convencionales muy conocidas por los expertos en la técnica, por ejemplo, cromatografía y cristalización fraccionada.
Las técnicas convencionales para la preparación/aislamiento de enantiómeros individuales incluyen la síntesis quiral a partir de un precursor ópticamente puro adecuado o la resolución del racemato (o el racemato de una sal o derivado) mediante el uso de, por ejemplo, cromatografía líquida de alta presión (HPLC) quiral. Alternativamente, el racemato (o un precursor racémico) puede reaccionar con un compuesto ópticamente activo adecuado, por ejemplo, un alcohol, o, en el caso de que el compuesto de la Fórmula I contenga una fracción ácida o básica, una base o un ácido tal como la 1 -feniletilamina o el ácido tartárico. La mezcla diastereomérica resultante se puede separar por cromatografía y/o cristalización fraccionada y uno o ambos diastereoisómeros se convierten en el(los) correspondiente(s) enantiómero(s) puro(s) por medios muy conocidos por los expertos en la técnica. Los compuestos quirales de la Fórmula I (y sus precursores quirales) se pueden obtener en forma enriquecida enantioméricamente por medio de cromatografía, típicamente HPLC, en una resina asimétrica con una fase móvil que consiste en un hidrocarburo, típicamente heptano o hexano, que contiene de 0 a 50% en volumen de isopropanol, típicamente de 2% a 20%, y de 0 a 5% en volumen de una alquilamina, típicamente 0,1% de dietilamina. La concentración del eluido permite obtener la mezcla enriquecida. Se puede emplear la cromatografía quiral mediante el uso de fluidos sub y supercríticos. Los procedimientos para la cromatografía quiral útiles en algunas realizaciones de la presente invención son conocidos en la técnica (véase, por ejemplo, Smith, Roger M., Loughborough University, Loughborough, UK; Chromatographic Science Series (1998), 75 (Supercritical Fluid Chromatography with Packed Columns), págs. 223 a 249 y las referencias citadas en el mismo). En algunos ejemplos relevantes de la presente memoria, las columnas se obtuvieron de Chiral Technologies, Inc, West Chester, Pennsylvania, EE.UU., una filial de Daicel® Chemical Industries, Ltd., Tokio, Japón.
Cuando cualquier racemato cristaliza, son posibles cristales de dos tipos diferentes. El primer tipo es el compuesto
racémico (verdadero racemato) mencionado anteriormente, en el que se produce una forma homogénea de cristal que contiene ambos enantiómeros en cantidades equimolares. El segundo tipo es la mezcla racémica o conglomerado, en el que se producen dos formas de cristal en cantidades equimolares que comprenden cada una un único enantiómero. Aunque las dos formas cristalinas presentes en una mezcla racémica tienen propiedades físicas idénticas, pueden tener propiedades físicas diferentes en comparación con el verdadero racemato. Las mezclas racémicas se pueden separar por medio de técnicas convencionales conocidas por los expertos en la técnica; véase, por ejemplo, "Stereochemistry of Organic Compounds" por E. L. Eliel y S. H. Wilen (Wiley, 1994). Hay que destacar que los compuestos de la Fórmula I se han dibujado en la presente memoria en una única forma tautomérica, todas las formas tautoméricas posibles están incluidas en el ámbito de la invención.
La presente invención incluye todos los compuestos de la Fórmula I etiquetados isotópicamente aceptables para uso farmacéutico, en los que uno o más átomos se sustituyen por átomos que tiene el mismo número atómico, pero una masa atómica o número de masa diferente de la masa atómica o número de masa que predomina en la naturaleza. Los ejemplos de isótopos adecuados para su inclusión en los compuestos de la invención incluyen isótopos de hidrógeno, tales como 2H y 3H, carbono, tales como 11C, 13C y 14C, cloro, tal como 36Cl, flúor, tal como 18F, yodo, tal como 123I y 125I, nitrógeno, tal como 13N y 15N, oxígeno, tal como 15O, 17O y 18O, fósforo, tal como 32P, y azufre, tal como 35S.
Ciertos compuestos de la Fórmula I marcados isotópicamente, por ejemplo, los que incorporan un isótopo radiactivo, son útiles en los estudios de distribución tisular de fármacos y/o sustratos. Los isótopos radiactivos tritio, es decir, 3H, y carbono-14, es decir, 14C, son particularmente útiles para este propósito en vista de su facilidad de incorporación y medios de detección.
La sustitución con isótopos más pesados, tales como deuterio, es decir, 2H, puede ofrecer ciertas ventajas terapéuticas derivadas de una mayor estabilidad metabólica, por ejemplo, un aumento de la vida media in vivo o una reducción de las necesidades de dosificación.
La sustitución con isótopos emisores de positrones, tales como 11C, 18F, 15O y 13N, puede ser útil en los Estudios de Topografía por Emisión de Positrones (PET) para examinar la ocupación de los receptores del sustrato.
Los compuestos de la Fórmula I marcados isotópicamente se pueden preparar generalmente por medio de técnicas convencionales conocidas por los expertos en la técnica o por medio de procesos análogos a los descritos en los Ejemplos y Preparaciones adjuntos, mediante el uso de un reactivo apropiado marcado isotópicamente en lugar del reactivo no marcado empleado previamente.
Los disolventes aceptables para uso farmacéutico de acuerdo con la invención incluyen aquellos en los que el disolvente de cristalización puede estar isotópicamente sustituido, por ejemplo, D2O, d6-acetona, d6-DMSO.
Un compuesto de la Fórmula I se puede administrar en forma de profármaco. Así, ciertos derivados de un compuesto de la Fórmula I que pueden tener poca o ninguna actividad farmacológica por sí mismos pueden, cuando se administran en o sobre el cuerpo, ser convertidos en un compuesto de la Fórmula I que tenga la actividad deseada, por ejemplo, por escisión hidrolítica, particularmente la escisión hidrolítica promovida por una enzima esterasas o peptidasas. Estos derivados se denominan "profármacos”. Se puede encontrar más información sobre el uso de profármacos en 'Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) y 'Bioreversible Carriers in Drug Desigrí, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association). También se puede hacer referencia a Nature Reviews/Drug Discovery, 2008, 7, 355 y Current Opinion in Drug Discovery and Development, 2007, 10, 550.
Los profármacos se pueden producir, por ejemplo, por medio de la sustitución de las funcionalidades apropiadas presentes en los compuestos de la Fórmula I por ciertas fracciones conocidas por los expertos en la técnica como "pro-fracciones”, como se describe, por ejemplo, en 'Design of Prodrugs' de H. Bundgaard (Elsevier, 1985) y Y. M. Choi-Sledeski y C. G. Wermuth, 'Designing Prodrugs and Bioprecursors' en Practice of Medicinal Chemistry, (Cuarta Edición), Capítulo 28, 657 a 696 (Elsevier, 2015).
Así, un profármaco es (a) un derivado éster o amida de un ácido carboxílico en un compuesto de la Fórmula I; (b) un derivado éster, carbonato, carbamato, fosfato o éter de un grupo hidroxilo en un compuesto de la Fórmula I (c) un derivado amida, imina, carbamato o amina de un grupo amino en un compuesto de la Fórmula I; (d) un derivado oxima o imina de un grupo carbonilo en un compuesto de la Fórmula I; o (e) un grupo metilo, alcohol primario o aldehído que pueda ser oxidado metabólicamente a un ácido carboxílico en un compuesto de la Fórmula I.
Algunos ejemplos específicos de profármacos incluyen:
(i) cuando el compuesto de la Fórmula I contiene una funcionalidad de ácido carboxílico (-COOH), un éster del mismo, tal como un compuesto en el que el hidrógeno de la funcionalidad de ácido carboxílico del compuesto de la Fórmula I se sustituye por alquilo C1-C8 (por ejemplo, etilo) o (alquilo C-i-C8)C(=O)OCH2-(por ejemplo, ‘BuC(=O)OCH2-);
(ii) cuando el compuesto de la Fórmula I contiene una funcionalidad de alcohol (-OH), un éster de la misma, tal como un compuesto en el que el hidrógeno de la funcionalidad de alcohol del compuesto de la Fórmula I se sustituye por -CO(alquilo C1-C8) (por ejemplo, metilcarbonilo) o el alcohol se esterifica con un aminoácido;
(iii) cuando el compuesto de la Fórmula I contiene una funcionalidad de alcohol (-OH), un éter del mismo, tal como un compuesto en el que el hidrógeno de la funcionalidad de alcohol del compuesto de la Fórmula I se sustituye por (alquilo C1-C8 )C(=O)OCH2- o -CH2OP(=O)(OH)2;
(iv) cuando el compuesto de la Fórmula I contenga una funcionalidad de alcohol (-OH), un fosfato del mismo, tal como un compuesto en el que el hidrógeno de la funcionalidad de alcohol del compuesto de la Fórmula I se sustituya por -P(=O)(OH)2 o -P(=O)(ONa)2 o -P(=O)(O')2Ca2+;
(v) cuando el compuesto de la Fórmula I contenga una funcionalidad amino primaria o secundaria (-NH2 o -NHR en el que R t H), una amida del mismo, por ejemplo, un compuesto en el que, de acuerdo con el caso, uno o ambos hidrógenos de la funcionalidad amino del compuesto de la Fórmula I se sustituyan por alcanoilo (C1-C10), -COCH2NH2 o el grupo amino se derive con un aminoácido;
(vi) cuando el compuesto de la Fórmula I contenga una funcionalidad amino primaria o secundaria (-NH2 o -NHR en el que R t H), una amina de la misma, por ejemplo, un compuesto en el que, de acuerdo con el caso, uno o ambos hidrógenos de la funcionalidad amino del compuesto de la Fórmula I sean sustituidos por -CH2OP(=O)(OH)2;
(vii) en el que el grupo ácido carboxílico del compuesto de la Fórmula I se sustituye por un grupo metilo, un grupo -CH2OH o un grupo aldehido.
Ciertos compuestos de la Fórmula I pueden actuar por sí mismos como profármacos de otros compuestos de la Fórmula I. También es posible que dos compuestos de la Fórmula I se unan en forma de profármaco. En determinadas circunstancias, se puede crear un profármaco de un compuesto de la Fórmula I por medio del enlace interno de dos grupos funcionales en un compuesto de la Fórmula I, por ejemplo, por medio de la formación de una lactona.
Administración y dosificación
Típicamente, un compuesto de la invención se administra en una cantidad efectiva para tratar una condición como se describe en la presente memoria. Los compuestos de la invención se pueden administrar como compuesto per se, o alternativamente, como una sal aceptable para uso farmacéutico. A efectos de administración y dosificación, el compuesto per se o la sal aceptable para uso farmacéutico del mismo se denominarán simplemente compuestos de la invención.
Los compuestos de la invención se administran por cualquier vía adecuada en forma de una composición farmacéutica adaptada a dicha vía, y en una dosis eficaz para el tratamiento previsto. Los compuestos de la invención se pueden administrar por vía oral, rectal, vaginal, parenteral o tópica.
Los compuestos de la invención se pueden administrar por vía oral. La administración oral puede implicar la deglución, de forma que el compuesto entra en el tracto gastrointestinal, o se puede emplear la administración bucal o sublingual, por la que el compuesto entra en el torrente sanguíneo directamente desde la boca.
En otra realización, los compuestos de la invención también se pueden administrar directamente en el torrente sanguíneo, en el músculo o en un órgano interno. Los medios adecuados para la administración parenteral incluyen por vía intravenosa, intraarterial, intraperitoneal, intratecal, intraventricular, intrauretral, intraesternal, intracraneal, intramuscular y subcutánea. Los dispositivos adecuados para la administración parenteral incluyen inyectores con aguja (que incluyen microagujas), inyectores sin aguja y técnicas de infusión.
En otra realización, los compuestos de la invención también se pueden administrar por vía tópica sobre la piel o las mucosas, esto es, por vía dérmica o transdérmica. En otra realización, los compuestos de la invención también se pueden administrar por vía intranasal o por inhalación. En otra realización, los compuestos de la invención se pueden administrar por vía rectal o vaginal. En otra realización, los compuestos de la invención también se pueden administrar directamente al ojo o al oído.
El régimen de dosificación para los compuestos de la invención y/o las composiciones que contienen dichos compuestos se basa en una variedad de factores, que incluyen el tipo, la edad, el peso, el sexo y la condición médica del paciente; la gravedad de la afección; la vía de administración; y la actividad del compuesto particular empleado. Por lo tanto, el régimen de dosificación puede variar mucho. En una realización, la dosis diaria total de un compuesto de la invención es típicamente de aproximadamente 0,001 a aproximadamente 100 mg/kg (es decir, mg de compuesto de la invención por kg de peso corporal) para el tratamiento de las condiciones indicadas discutidas en la presente memoria. En otra realización, la dosis diaria total del compuesto de la invención es de aproximadamente 0,01 a aproximadamente 30 mg/kg, y en otra realización, de aproximadamente 0,03 a aproximadamente 10 mg/kg, y en otra realización, de aproximadamente 0,1 a aproximadamente 3. No es raro que la administración de los compuestos de la invención se repita una pluralidad de veces en un día (típicamente no más de 4 veces). Por lo general, se pueden utilizar múltiples dosis al día para aumentar la dosis diaria total, si se desea. Para la administración oral, las composiciones se pueden proporcionar en forma de comprimidos que contengan 0,1,
0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 30,050,0, 75,0, 100, 125, 150, 175, 200, 250 y 500 miligramos del principio activo para el ajuste sintomático de la dosis al paciente. Un medicamento suele contener entre 0,01 mg y 500 mg del principio activo, o en otra realización, entre 1 mg y 100 mg del principio activo. Por vía intravenosa, las dosis pueden oscilar entre aproximadamente 0,01 y aproximadamente 10 mg/kg/minuto durante una infusión a velocidad constante.
Los sujetos adecuados de acuerdo con la invención incluyen sujetos mamíferos. En una realización, los humanos son sujetos adecuados. Los sujetos humanos pueden ser de cualquier sexo y estar en cualquier etapa de desarrollo.
Composiciones Farmacéuticas
En otra realización, la invención comprende composiciones farmacéuticas. Tales composiciones farmacéuticas comprenden un compuesto de la invención presentado con un portador aceptable para uso farmacéutico. También pueden estar presentes otras sustancias farmacológicamente activas. Como se utiliza en la presente memoria, el “portador aceptable para uso farmacéutico” incluye todos y cada uno de los disolventes, medios de dispersión, revestimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardadores de la absorción, y similares que sean fisiológicamente compatibles. Los ejemplos de portadores aceptables para uso farmacéutico incluyen uno o más de agua, solución salina, solución salina tamponada con fosfato, dextrosa, glicerol, etanol y similares, así como combinaciones de los mismos, y pueden incluir agentes isotónicos, por ejemplo, azúcares, cloruro de sodio o polialcoholes tales como manitol o sorbitol en la composición. Sustancias aceptables para uso farmacéutico, tales como agentes humectantes o pequeñas cantidades de sustancias auxiliares, tales como agentes humectantes o emulsionantes, conservantes o tampones, que mejoran la vida útil o la eficacia del anticuerpo o de la porción de anticuerpo.
Las composiciones de esta invención pueden estar en una variedad de formas. Entre ellas se encuentran, por ejemplo, formas de dosificación líquidas, semisólidas y sólidas, tales como soluciones líquidas (por ejemplo, soluciones inyectables e infusibles), dispersiones o suspensiones, comprimidos, píldoras, polvos, liposomas y supositorios. La forma depende del modo de administración previsto y de la aplicación terapéutica.
Las composiciones típicas se presentan en forma de soluciones inyectables o infusibles, tales como las composiciones similares a las utilizadas para la inmunización pasiva de seres humanos con anticuerpos en general. Un modo de administración es el parenteral (por ejemplo, intravenoso, subcutáneo, intraperitoneal, intramuscular). En otra realización, el anticuerpo se administra por infusión o inyección intravenosa. En otra realización, el anticuerpo se administra por inyección intramuscular o subcutánea.
La administración oral de una forma de dosis sólida puede ser, por ejemplo, presentada en unidades discretas, tales como cápsulas duras o blandas, píldoras, cachets, pastillas o tabletas, cada una de las cuales contiene una cantidad predeterminada de al menos un compuesto de la invención. En otra realización, la administración oral puede ser en forma de polvo o gránulos. En otra realización, la forma de la dosis oral es sub-lingual, tal como, por ejemplo, una pastilla. En dichas formas farmacéuticas sólidas, los compuestos de la Fórmula I se combinan normalmente con uno o más adyuvantes. Dichas cápsulas o comprimidos pueden contener una formulación de liberación controlada. En el caso de las cápsulas, los comprimidos y las píldoras, las formas farmacéuticas también pueden comprender agentes amortiguadores o se pueden preparar con revestimientos entéricos.
En otra realización, la administración oral puede ser en forma de dosis líquida. Las formas de dosificación líquida para la administración oral incluyen, por ejemplo, emulsiones, soluciones, suspensiones, jarabes y elixires aceptables para uso farmacéutico que contienen diluyentes inertes comúnmente utilizados en la técnica (por ejemplo, agua). Dichas composiciones también pueden comprender adyuvantes, tales como agentes humectantes, emulsionantes, suspensores, aromatizantes (por ejemplo, edulcorantes) y/o perfumantes.
En otra realización, la invención comprende una forma de dosis parenteral. “La administración parenteral” incluye, por ejemplo, las inyecciones subcutáneas, las inyecciones intravenosas, las inyecciones intraperitoneales, las inyecciones intramusculares, las inyecciones intraesternales y la infusión. Las preparaciones inyectables (es decir, las suspensiones acuosas u oleaginosas inyectables) se pueden formular de acuerdo con la técnica conocida mediante el uso de agentes dispersantes, humectantes y/o suspensores adecuados.
En otra realización, la invención comprende una forma de dosis tópica. “La administración tópica” incluye, por ejemplo, la administración transdérmica, tal como a través de parches transdérmicos o dispositivos de iontoforesis, la administración intraocular o la administración intranasal o por inhalación. Las composiciones para la administración tópica también incluyen, por ejemplo, geles tópicos, aerosoles, ungüentos y cremas. Una formulación tópica puede incluir un compuesto que mejore la absorción o la penetración del ingrediente activo a través de la piel u otras zonas afectadas. Cuando los compuestos de la presente invención se administran por medio de un dispositivo transdérmico, la administración se llevará a cabo mediante el uso de un parche del tipo de depósito y membrana porosa o de una variedad de matriz sólida. Las formulaciones típicas para este fin incluyen geles, hidrogeles, lociones, soluciones, cremas, ungüentos, polvos, apósitos, espumas, películas, parches cutáneos, obleas, implantes, esponjas, fibras, vendajes y microemulsiones. También se pueden utilizar liposomas. Los portadores típicos son alcohol, agua, aceite mineral, vaselina líquida, vaselina blanca, glicerina, polietilenglicol y
propilenglicol. Se pueden incorporar potenciadores de la penetración - véase, por ejemplo, B. C. Finnin y T. M. Morgan, J. Pharm. Sci., vol. 88, págs. 955 a 958, 1999.
Las formulaciones adecuadas para la administración tópica al ojo incluyen, por ejemplo, gotas oculares en las que el compuesto de esta invención se disuelve o suspende en un portador adecuado. Una formulación típica adecuada para la administración ocular o auditiva se puede presentar en forma de gotas de una suspensión o solución micronizada en una solución salina estéril, isotónica y de pH ajustado. Otras formulaciones adecuadas para la administración ocular y auditiva incluyen pomadas, implantes biodegradables (por ejemplo, esponjas de gel absorbibles, colágeno) y no biodegradables (por ejemplo, silicona), obleas, lentes y sistemas particulados o vesiculares, tales como niosomas o liposomas. Un polímero tal como ácido poliacrílico reticulado, polivinilalcohol, ácido hialurónico, un polímero celulósico, por ejemplo, hidroxipropilmetilcelulosa, hidroxietilcelulosa o metilcelulosa, o un polímero heteropolisacárido, por ejemplo, goma gelana, se puede incorporar junto con un conservante, tal como cloruro de benzalconio. Dichas formulaciones también se pueden administrar por iontoforesis.
Para la administración intranasal o por inhalación, los compuestos de la invención se suministran convenientemente en forma de solución o suspensión desde un recipiente de pulverización de bomba que es exprimido o bombeado por el paciente o como una presentación en aerosol desde un recipiente presurizado o un nebulizador, con el uso de un propulsor adecuado. Las formulaciones adecuadas para administración intranasal se administran típicamente en forma de polvo seco (ya sea solo, como una mezcla, por ejemplo, en una mezcla seca con lactosa, o como una partícula de componente mixto, por ejemplo, mezclada con fosfolípidos, tal como fosfatidilcolina) a partir de un inhalador de polvo seco o como un aerosol de un recipiente presurizado, bomba, pulverizador, atomizador (preferentemente un atomizador que utiliza electrohidrodinámica para producir una niebla fina) o nebulizador, con o sin el uso de un propulsor adecuado, tal como 1,1,1,2-tetrafluoroetano o 1,1,1,2,3,3,3-heptafluoropropano. Para el uso intranasal, el polvo puede comprender un agente bioadhesivo, por ejemplo, quitosano o ciclodextrina.
En otra realización, la invención comprende una forma de dosis rectal. Dicha forma de dosificación rectal puede ser en forma de, por ejemplo, un supositorio. La manteca de cacao es la base tradicional de los supositorios, pero se pueden utilizar diversas alternativas de acuerdo con lo que convenga.
También se pueden usar otros materiales portadores y modos de administración conocidos en la técnica farmacéutica. Las composiciones farmacéuticas de la invención se pueden preparar por medio de cualquiera de las técnicas farmacéuticas conocidas, tales como los procedimientos de formulación y administración eficaces. Las consideraciones anteriores respecto a las formulaciones eficaces y los procedimientos de administración son muy conocidas en la técnica y se describen en los libros de texto estándar. La formulación de medicamentos se discute, por ejemplo, en Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Nueva York, N.Y., 1980 y Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3ra Ed.), American Pharmaceutical Association, Washington, 1999.
Co-administración
Los compuestos de la invención se pueden usar solos o en combinación con otros agentes terapéuticos. La invención proporciona cualquiera de los usos, procedimientos o composiciones definidos en la presente memoria, en los que el compuesto de cualquier realización de la Fórmula I de la presente memoria, o una sal aceptable para uso farmacéutico del mismo, o un solvato aceptable para uso farmacéutico de dicho compuesto o sal, se utiliza en combinación con uno o más agentes terapéuticos mencionados en la presente memoria.
La administración de dos o más compuestos “en combinación” significa que todos los compuestos se administran lo suficientemente cerca en el tiempo como para que cada uno pueda generar un efecto biológico en el mismo marco temporal. La presencia de un agente puede alterar los efectos biológicos del otro u otros compuestos. Los dos o más compuestos se pueden administrar de forma simultánea, concurrente o secuencial. Además, la administración simultánea se puede llevar a cabo por medio de la mezcla de los compuestos antes de la administración o administrando los compuestos en el mismo momento pero como formas de dosificación separadas en el mismo o diferente lugar de administración.
Las frases “administración concurrente”, “coadministración”, “administración simultánea” y “administrado simultáneamente” significan que los compuestos se administran en combinación.
En otra realización, la invención proporciona compuestos para su uso en procedimientos de tratamiento que incluyen la administración de compuestos de la presente invención en combinación con uno o más agentes farmacéuticos, en los que el otro u otros agentes farmacéuticos se pueden seleccionar de entre los agentes discutidos en la presente memoria.
En una realización, los compuestos de esta invención se administran con un agente antidiabético que incluye, pero no se limita a, una biguanida (por ejemplo, metformina), una sulfonilurea (por ejemplo, tolbutamida, glibenclamida, gliclazida, clorpropamida, tolazamida, acetohexamida, glicopiramida, glimepirida o glipizida), una tiazolidinediona (por ejemplo, pioglitazona, rosiglitazona o lobeglitazona), un glitazar (por ejemplo, saroglitazar, aleglitazar, muraglitazar o tesaglitazar), una meglitinida (por ejemplo, nateglinida, repaglinida), un inhibidor de la dipeptidil
peptidasa 4 (DPP-4) (por ejemplo sitagliptina, vildagliptina, saxagliptina, linagliptina, gemigliptina, anagliptina, teneligliptina, alogliptina, trelagliptina, dutogliptina u omarigliptina), una glitazona (por ejemplo, pioglitazona, rosiglitazona, balaglitazona, rivoglitazona o lobeglitazona), un inhibidor del transportador 2 de sodio y glucosa (SGLT2) (por ejemplo, empagliflozina, canagliflozina, dapagliflozina, ipragliflozina, ipragliflozina, tofogliflozina, etabonato de sergliflozina, etabonato de remogliflozina o ertugliflozina), un inhibidor de SGLTL1, un agonista de GPR40 (agonista de FFAR1/FFA1, por ejemplo, fasiglifam), un péptido insulinotrópico dependiente de la glucosa (GIP) y análogos de los mismos, un inhibidor de la alfa glucosidasa (por ejemplopor ejemplo, voglibosa, acarbosa o miglitol), o una insulina o un análogo de la insulina, incluidas las sales aceptables para uso farmacéutico de los agentes específicamente mencionados y los solvatos aceptables para uso farmacéutico de dichos agentes y sales.
En otra realización, los compuestos de esta invención se administran con un agente antiobesidad que incluye pero no se limita al péptido YY o un análogo del mismo, un agonista del receptor de neuropéptidos Y tipo 2 (NPYR2), un antagonista de NPYR1 o NPYR5, un antagonista del receptor cannabinoide tipo 1 (CB1R), un inhibidor de la lipasa (por ejemplo, orlistat), un péptido proislet humano (HIP), un agonista del receptor 4 de la melanocortina (por ejemplo, setmelanotide), un antagonista del receptor 1 de la hormona concentradora de melanina, un agonista del receptor X farnesoide (FXR) (por ejemplo, ácido obetichólico), zonisamida, fentermina (sola o en combinación con topiramato), un inhibidor de la recaptación de norepinefrina/dopamina (por ejemplo, buproprión), un antagonista de los receptores opiáceos (por ejemplo, naltrexona), una combinación de inhibidor de la recaptación de norepinefrina/dopamina y antagonista de los receptores opiáceos (por ejemplo, una combinación de bupropión y naltrexona), un análogo del GDF-15, sibutramina, un agonista de la colecistoquinina, amilina y análogos de los mismos (por ejemplo, pramlintida), leptina y análogos de los mismos (por ejemplo, metroleptina), un agente serotoninérgico (por ejemplo, lorcaserina), un inhibidor de la metionina aminopeptidasa 2 (MetAP2) (por ejemplo, beloranib o ZGN-1061), fendimetrazina, dietilpropión, benzfetamina, un inhibidor de SGLT2 (por ejemplo, empagliflozina, canagliflozina, dapagliflozina, ipragliflozina, ipragliflozina, tofogliflozina, etabonato de sergliflozina, etabonato de remogliflozina o ertugliflozina), un inhibidor de SGLTL1, un inhibidor dual de SGLT2/SGLT1 un modulador del receptor del factor de crecimiento de fibroblastos (FGFR), un activador de la proteína quinasa activada por el AMP (AMPK), biotina, un modulador del receptor MAS o un agonista del receptor del glucagón (solo o en combinación con otro agonista del GLP-1R, por ejemplo, liraglutida, exenatida, dulaglutida, albiglutida, lixisenatida o semaglutida), incluidas las sales aceptables para uso farmacéutico de los agentes específicamente mencionados y los solvatos aceptables para uso farmacéutico de dichos agentes y sales.
En otra realización, los compuestos de esta invención se administran con un agente para tratar la EHNA, que incluyen, pero sin limitarse a, PF-05221304, un agonista de FXR (por ejemplo, ácido obetichólico), un agonista de PPAR a/8 (por ejemplo, elafibranor), un conjugado sintético de ácido graso y ácido biliar (por ejemplo, aramchol), un inhibidor de caspasas (por ejemplo, emricasan), un anticuerpo monoclonal anti-lisil oxidasa homóloga 2 (LOXL2) (por ejemplo, simtuzumab), un inhibidor de la galectina 3 (por ejemplo, GR-MD-02), un inhibidor de la MAPK5 (por ejemplo, GS-4997), un antagonista dual del receptor de quimioquinas 2 (CCR2) y CCR5 (por ejemplo, cenicriviroc), un agonista del factor de crecimiento de fibroblastos 21 (FGF2l) (por ejemplo, BMS-986036), un antagonista del receptor de leucotrienos D4 (LTD4) (por ejemplo, tipelukast), un análogo de la niacina (por ejemplo, ARI 3037MO), un inhibidor de la ASBT (por ejemplo, volixibat), un inhibidor de la acetil-CoA carboxilasa (ACC) (por ejemplo, NDI 010976), un inhibidor de la cetohexocinasa (KHK), un inhibidor de la diacilgliceril aciltransferasa 2 (DgAT2), un antagonista del receptor CB1, un anticuerpo anti-CB1R o un inhibidor de la quinasa reguladora de la señal de apoptosis 1 (ASK1), incluidas las sales aceptables para uso farmacéutico de los agentes específicamente mencionados y los solvatos aceptables para uso farmacéutico de dichos agentes y sales.
Estos agentes y compuestos de la invención se pueden combinar con vehículos aceptables para uso farmacéutico tales como solución salina, solución de Ringer, solución de dextrosa y similares. El régimen de dosificación concreto, es decir, la dosis, el momento y la repetición, dependerá de cada persona y de su historial médico.
Los portadores, excipientes o estabilizadores aceptables no son tóxicos para los receptores en las dosis y concentraciones empleadas, y pueden comprender tampones tales como fosfato, citrato y otros ácidos orgánicos; sales tales como cloruro de sodio; antioxidantes como ácido ascórbico y metionina; conservantes (tales como el cloruro de octadecildimetilbencilamonio; el cloruro de hexametonio; el cloruro de benzalconio, el cloruro de bencetonio; el fenol, el alcohol butílico o el bencílico; los alquilparabenos, tales como el metilo o el propilparabeno; el catecol; el resorcinol; ciclohexanol; 3-pentanol; y m-cresol); polipéptidos de bajo peso molecular (menos de aproximadamente 10 residuos); proteínas, tales como la albúmina de suero, la gelatina o las Igs; polímeros hidrófilos, tales como la polivinilpirrolidona; aminoácidos, tales como la glicina, la glutamina, la asparagina, la histidina, la arginina o la lisina; monosacáridos, disacáridos y otros hidratos de carbono tales como la glucosa, la manosa o las dextrinas; agentes quelantes tales como el EDTA; azúcares tales como la sacarosa, el manitol, la trehalosa o el sorbitol; contra-iones formadores de sal tales como el sodio; complejos metálicos (por ejemplo, complejos de Zn-proteína); y/o tensioactivos no iónicos como TWEEN™, PLURONICS™ o polietilenglicol (PEG).
Los liposomas que contienen estos agentes y/o compuestos de la invención se preparan por medio de procedimientos conocidos en la técnica, tales como los descritos en la Patente de los Estados Unidos Núms.
4.485.045 y 4.544.545. Los liposomas con mayor tiempo de circulación se desvelan en la Patente de los Estados Unidos Núm. 5.013.556. Se pueden generar liposomas particularmente útiles por el procedimiento de evaporación en fase inversa con una composición lipídica que comprende fosfatidilcolina, colesterol y fosfatidiletanolamina
derivada de PEG (PEG-PE). Los liposomas se extruyen a través de filtros de tamaño de poro definido para obtener liposomas con el diámetro deseado.
Estos agentes y/o los compuestos de la invención también pueden quedar atrapados en microcápsulas preparadas, por ejemplo, por técnicas de coacervación o por polimerización interfacial, por ejemplo, microcápsulas de hidroximetilcelulosa o gelatina y microcápsulas de poli(metilmetacrilato), respectivamente, en sistemas coloidales de administración de fármacos (por ejemplo, liposomas, microesferas de albúmina, microemulsiones, nanopartículas y nanocápsulas) o en macroemulsiones. Dichas técnicas se desvelan en Remington, The Science and Practice of Pharmacy, 20ma edición, Mack Publishing (2000).
Se pueden usar preparados de liberación sostenida. Los ejemplos adecuados de preparaciones de liberación sostenida incluyen matrices semipermeables de polímeros sólidos hidrofóbicos que contienen el compuesto de las Fórmulas I, II o III, cuyas matrices se presentan en forma de artículos moldeados, por ejemplo, películas o microcápsulas. Entre los ejemplos de matrices de liberación sostenida se encuentran los poliésteres, los hidrogeles (por ejemplo, el poli(2-hidroxietilmetacrilato) o el “poli(vinilalcohol)”), los polilactidos (la Patente de los Estados Unidos Núm. 3.773.919), copolímeros de ácido L-glutámico y 7 etil-L-glutamato, acetato de etileno-vinilo no degradable, copolímeros degradables de ácido láctico-ácido glicólico tales como los utilizados en LUPRON DEPOT™ (microesferas inyectables compuestas de copolímero de ácido láctico-ácido glicólico y acetato de leuprolida), isobutirato de acetato de sacarosa y ácido poli-D-(-)-3-hidroxibutírico.
Las formulaciones que se utilicen para la administración intravenosa deben ser estériles. Esto se consigue fácilmente, por ejemplo, por medio de la filtración a través de membranas de filtración estériles. Los compuestos de la invención se colocan generalmente en un recipiente que tiene un puerto de acceso estéril, por ejemplo, una bolsa de solución intravenosa o un vial que tiene un tapón perforable por una aguja de inyección hipodérmica.
Se pueden preparar emulsiones adecuadas mediante el uso de emulsiones de grasa disponibles en el mercado, tales como Intralipid™, Liposyn™, Infonutrol™, Lipofundin™ y Lipiphysan™. El ingrediente activo puede estar disuelto en una composición de emulsión premezclada o, alternativamente, puede estar disuelto en un aceite (por ejemplo, aceite de soja, aceite de cártamo, aceite de semilla de algodón, aceite de sésamo, aceite de maíz o aceite de almendras) y una emulsión formada al mezclarse con un fosfolípido (por ejemplo, fosfolípidos de huevo, fosfolípidos de soja o lecitina de soja) y agua. Se apreciará que se pueden añadir otros ingredientes, por ejemplo glicerol o glucosa, para ajustar la tonicidad de la emulsión. Las emulsiones adecuadas suelen contener hasta un 20% de aceite, por ejemplo, entre un 5 y un 20%. La emulsión grasa puede comprender gotas de grasa de entre 0,1 y 1,0 micrómetros, en particular de entre 0,1 y 0,5 micrómetros, y tener un pH en el intervalo de 5,5 a 8,0.
Las composiciones de emulsión pueden ser las que se preparan por medio de la mezcla de un compuesto de la invención con Intralipid™ o los componentes del mismo (aceite de soja, fosfolípidos de huevo, glicerol y agua). Las composiciones para inhalación o insuflación incluyen soluciones y suspensiones en disolventes acuosos u orgánicos aceptables para uso farmacéutico, o mezclas de los mismos, y polvos. Las composiciones líquidas o sólidas pueden contener excipientes aceptables para uso farmacéutico adecuados, como se ha indicado anteriormente. En algunas realizaciones, las composiciones se administran por vía oral o respiratoria nasal para un efecto local o sistémico. Las composiciones en disolventes preferentemente estériles y aceptables desde el punto de vista farmacéutico se pueden nebulizar mediante el uso de gases. Las soluciones nebulizadas se pueden respirar directamente desde el dispositivo de nebulización o el dispositivo de nebulización puede estar conectado a una máscara facial, una tienda de campaña o una máquina de respiración de presión positiva intermitente. Las composiciones en solución, en suspensión o en polvo se pueden administrar, preferentemente por vía oral o nasal, a partir de dispositivos que administren la formulación de forma adecuada.
Kits
Otro aspecto de la invención proporciona kits que comprenden el compuesto de las Fórmulas I, II o III o composiciones farmacéuticas que comprenden el compuesto de las Fórmulas I, II o III de la invención. Un kit puede incluir, además del compuesto de las Fórmulas I, II o III de la invención o de su composición farmacéutica, agentes diagnósticos o terapéuticos. Un kit también puede incluir instrucciones para su uso en un procedimiento diagnóstico o terapéutico. En algunas realizaciones, el kit incluye el compuesto de las Fórmulas I, II o III, o una composición farmacéutica del mismo y un agente de diagnóstico. En otras realizaciones, el kit incluye el compuesto de las Fórmulas I, II o III, o una composición farmacéutica del mismo.
En otra realización, la invención comprende kits que son adecuados para su uso en la realización de los procedimientos de tratamiento descritos en la presente memoria. En una realización, el kit contiene una primera forma de dosificación que comprende uno o más de los compuestos de la invención en cantidades suficientes para llevar a cabo los procedimientos de la invención. En otra realización, el kit comprende uno o más compuestos de la invención en cantidades suficientes para llevar a cabo los procedimientos de la invención y un recipiente para la dosificación y un recipiente para la dosificación.
Preparación
Los compuestos de las Fórmulas I, II o III se pueden preparar por los procedimientos generales y específicos que se describen a continuación, mediante el uso de los conocimientos generales comunes de los expertos en la técnica de la química orgánica sintética. Estos conocimientos generales comunes se pueden encontrar en libros de referencia estándar tales como Comprehensive Organic Chemistry, Ed. Barton y Ollis, Elsevier Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock, John Wiley and Sonsy Compendium of Organic Synthetic Methods, Vols. I a XII (publicado por Wiley-Interscience). Los materiales de partida utilizados en la presente memoria están disponibles comercialmente o se pueden preparar por medio de procedimientos rutinarios conocidos en la técnica.
En la preparación de los compuestos de las Fórmulas I, II o III, se observa que algunos de los procedimientos de preparación descritos en la presente memoria pueden requerir la protección de la funcionalidad remota (por ejemplo, amina primaria, amina secundaria, carboxilo en los precursores de la Fórmula I). La necesidad de dicha protección variará de acuerdo con la naturaleza de la funcionalidad a distancia y las condiciones de los procedimientos de preparación. La necesidad de dicha protección es fácilmente determinable por los expertos en la técnica. El uso de tales procedimientos de protección/desprotección también está dentro de la habilidad en la técnica. Para una descripción general de los grupos de protección y su uso, véase T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, Nueva York, 1991.
Por ejemplo, ciertos compuestos contienen aminas primarias o funcionalidades de ácido carboxílico que pueden interferir con las reacciones en otros sitios de la molécula si se dejan sin protección. En consecuencia, dichas funcionalidades pueden ser protegidas por un grupo protector apropiado que puede ser eliminado en una etapa posterior. Los grupos protectores adecuados para la protección de aminas y ácidos carboxílicos incluyen aquellos grupos protectores comúnmente utilizados en la síntesis de péptidos (tales como el N-t-butoxicarbonilo (Boc), el benciloxicarbonilo (Cbz) y el 9-fluorenilmetilenoxicarbonilo (Fmoc) para las aminas y los ésteres de alquilo inferior o de bencilo para los ácidos carboxílicos) que generalmente no son químicamente reactivos bajo las condiciones de reacción descritas y que típicamente se pueden eliminar sin alterar químicamente otra funcionalidad en los compuestos de la Fórmula I.
Los esquemas descritos a continuación pretenden proporcionar una descripción general de la metodología empleada en la preparación de los compuestos de la presente invención. Algunos de los compuestos de la presente invención pueden contener centros quirales simples o múltiples con la designación estereoquímica (R) o (S). Será evidente para los expertos en la técnica que todas las transformaciones sintéticas se pueden llevar a cabo de manera similar, tanto si los materiales son enantioenriquecidos como racémicos. Además, la resolución del material ópticamente activo deseado puede tener lugar en cualquier punto deseado de la secuencia mediante el uso de procedimientos muy conocidos tales como los descritos en la presente memoria y en la bibliografía química.
En los esquemas que siguen, las variables Y, Z1, Z2, Z3, R1,R2, R3, R4, m, p y q son como se describen en la presente memoria para los compuestos de las Fórmulas I, II o III, a menos que se indique lo contrario. Para los esquemas proporcionados a continuación, cada X1, X2, X3 y X4 puede ser independientemente un grupo de salida tal como cualquier alquil o aril sulfonato (por ejemplo, mesilato, tosilato o triflato), o un halógeno o cualquier otro grupo que pueda ser desplazado por una amina o utilizado en una reacción de acoplamiento mediada por metales. X4 también puede ser un ácido carboxílico protegido (es decir, un éster). Cuando el grupo protector se identifica como
Pg1, puede ser un grupo protector de alquil amina tal como el bencilo, el benceno o similares; un grupo protector de carbamato tal como Boc, Cbz o similares; o un grupo protector de amida tal como la trifluoroacetamida. Cuando el grupo protector se identifica como Pg2, puede ser un grupo protector ácido tal como el metilo, el etilo, el bencilo, el tbutilo o similares. R4a es alquilo C1-2, alquileno Cü.2-cicloalquilo C3-6, alquileno C0-2-R5, o alquileno C1-2-dicho alquilo, alquileno o cicloalquilo puede estar sustituido de forma independiente, como la valencia lo permita, con
0 a 3 átomos de F y 0 a 1 sustituyente seleccionado de forma independiente entre alquileno C0-1-ORO y -N(Rn)2.
La piridina sustituida 6 se puede preparar como se discute en el Esquema 1. Una 2,6-dihalopiridina (1, sintetizada o adquirida comercialmente) se puede hacer reaccionar con un ácido borónico sustituido o un éster de boronato (2) en presencia de un catalizador de paladio y un complejo de ligando a la manera de una reacción de Suzuki (Maluenda y Navarro, Molecules, 2015, 20, 7528 a 7557) para proporcionar compuestos de la fórmula general 3. Para obtener los mejores resultados en la reacción de Suzuki, el halógeno X2 es preferentemente Cl, Br o I. La reducción de la olefina para proporcionar compuestos de la estructura general 4 se llevaría a cabo bajo una atmósfera de hidrógeno (15 a
100 psi H2) en un disolvente alcohólico tal como MeOH o EtOH o alternativamente un disolvente orgánico aprótico tal como EtOAc o THF en presencia de un catalizador apropiado tal como paladio sobre carbono, Pd(OH)2 sobre carbono (catalizador de Pearlman) o PtO2 (catalizador de Adams). Alternativamente, la reducción se puede llevar a cabo por procedimientos alternativos conocidos por los expertos en la técnica, mediante el uso de reactivos tales como el trietil silano u otros silanos, bajo catálisis ácida o metálica, o reductores metálicos, tales como el magnesio o similares. Alternativamente, la olefina puede ser funcionalizada por procedimientos conocidos por los expertos en la técnica para introducir grupos R3. Por ejemplo, la olefina podría ser hidroborada para producir un alcohol que podría ser alquilado o convertido posteriormente en un grupo nitrilo, F o alquilo. La conversión a compuestos de la estructura general 5 se puede llevar a cabo por medio de un acoplamiento C-O Buchwald-Hartwig (Lundgren y Stradiotto, Aldrich Chimica Acta, 2012, 45, 59 a 65) entre los compuestos de la estructura general 4 y un alcohol bencílico adecuadamente sustituido en presencia de un catalizador de paladio o cobre y un complejo de ligando. Un halógeno X1 preferente es el Cl. Estas reacciones se llevan a cabo generalmente entre 0 y 110 °C en disolventes
orgánicos apróticos tales como, pero sin limitación, el 1,4-dioxano y el PhCH3 con adición de bases tales como CS2CO3, LiHMDS o NaOtBu. Alternativamente, la reacción de 4 con un alcohol bencílico adecuadamente sustituido en un disolvente aprótico tal como DMF o THF en presencia de una base fuerte tal como NaH, KOtBu o LiHMDS puede dar lugar a compuestos de la estructura general 5. Los sustituyentes X1 preferentes para esta reacción incluyen F y Cl o sulfonas (por ejemplo, SO2Me). La eliminación del Pg1 se podría llevar a cabo con numerosos procedimientos descritos en la bibliografía para proporcionar aminas 6.
Esquema 1

Alternativamente, como se muestra en el Esquema 2, los ésteres de piperidina apropiadamente sustituidos de la estructura general 7 se pueden hacer reaccionar con 1 en presencia de una base fuerte tal como LiHMDS o LDA u otra base adecuada en un disolvente orgánico aprótico tal como THF para obtener compuestos de la estructura general 8. Para obtener los mejores resultados en la preparación de compuestos tales como el 8 , X2 es preferentemente F o Cl. La eliminación del Pg2 por medio de hidrólisis de ésteres para dar lugar a los ácidos carboxílicos 9 se puede llevar a cabo de forma tradicional, tal como con hidróxido de litio, sodio o potasio acuoso en un disolvente miscible con agua, tal como MeOH, EtOH, THF o similares. Al someter los ácidos carboxílicos 9 al calor (60 a 120 °C) en un disolvente apropiado, tal como DCE o PhCH3, se producirá la descarboxilación para dar lugar a compuestos de fórmula general 4 que se utilizarán como se describe en el Esquema 1 para obtener las aminas 6.
Esquema 2

El Esquema 3 proporciona una preparación alternativa de los compuestos 5. La reacción de 1 con un alcohol bencílico adecuadamente sustituido en un disolvente aprótico tal como DMF o THF en presencia de una base fuerte tal como NaH, KOtBu o LiHMDS puede dar lugar a compuestos de la estructura general 10. Los sustituyentes X1 preferentes para esta reacción incluyen F y Cl, mientras que los sustituyentes X2 pueden incluir Cl, Br o I. Alternativamente, se pueden usar condiciones de acoplamiento C-O de Buchwald-Hartwig similares a la preparación de 5 para preparar 10 con los sustituyentes X1 preferentes Cl, Br o I. Se pueden usar condiciones de reacción Suzuki similares a la preparación de la estructura general 3 para preparar compuestos de estructura general 11 a partir de 10. Los sustituyentes X2 preferentes para su uso en el acoplamiento incluyen Cl, Br o I. La olefina se puede reducir por medio de los procedimientos descritos anteriormente en el Esquema 1 para obtener compuestos de estructura general 5 que posteriormente se utilizan para obtener aminas 6.
Esquema 3

Como se indica en el Esquema 4, la conversión de 10 en compuestos de la estructura general 12 se puede llevar a cabo por medio de un acoplamiento C-N Buchwald-Hartwig entre compuestos de la estructura general 10 y una piperazina adecuadamente sustituida y protegida en presencia de un catalizador de paladio o cobre y un complejo de ligando. Los sustituyentes X2 preferentes para el uso en el acoplamiento incluyen Cl, Br o I. Estas reacciones se llevan a cabo generalmente entre 0 y 110 °C en disolventes orgánicos apróticos tales como, pero no limitados a, 1,4-dioxano y PhCH3 con base añadida tal como Cs2CO3, LiHMDS o NaOtBu. La eliminación del Pg1 se podría llevar a cabo con numerosos procedimientos descritos en la bibliografía para proporcionar aminas 13.
Esquema 4
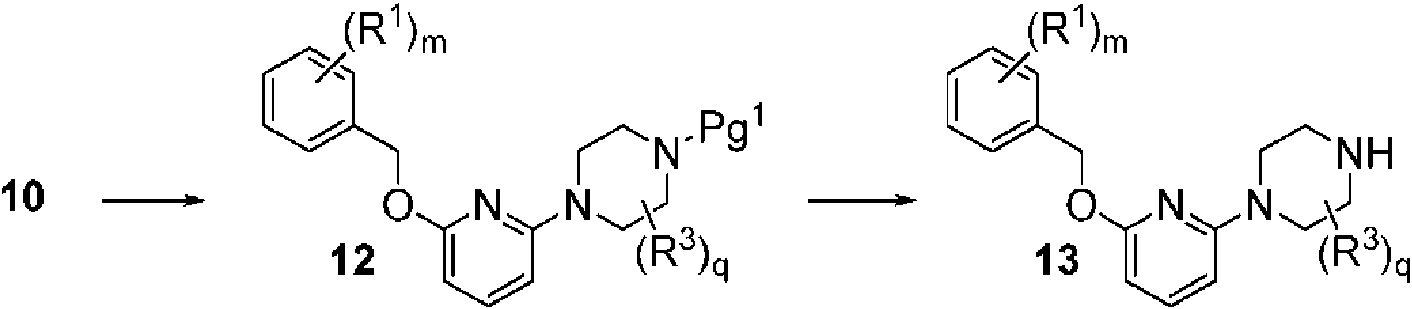
Los compuestos de la estructura 14 (Esquema 5) se pueden convertir en compuestos de la estructura general 15 por medio de los procedimientos descritos anteriormente en el Esquema 1 o el Esquema 2. Los sustituyentes X2 preferentes para su uso en el acoplamiento incluyen Cl, Br o I. La conversión de los intermedios 15 en sus respectivos N-óxidos 16 se puede llevar a cabo con oxidantes tales como el ácido 3-cloroperoxibenzoico, Oxone® u otro oxidante adecuado. El reordenamiento a compuestos de la estructura 17 puede ser afectado por el tratamiento con un anhídrido ácido orgánico tal como Ac2O o TFAA en solventes apróticos con una base amina orgánica apropiada tal como Et3N, DIPEA u otra base adecuada. La preparación de éteres de bencilo de la estructura general 18 se puede lograr por medio de procedimientos de alquilación estándar con bromuros de bencilo adecuadamente sustituidos o por medio de protocolos de alquilación estándar de Mitsunobu (Swamy et al., Chem. Rev. 2009, 109, 2551 a 2651) con alcoholes bencílicos convenientemente sustituidos. La eliminación del Pg1 se puede llevar a cabo por numerosos procedimientos descritos en la bibliografía para proporcionar aminas 19.
Esquema 5

El compuesto 20 (Esquema 6 ) se puede hacer reaccionar con una piperazina adecuadamente sustituida y protegida en presencia de una base adecuada tal como Cs2CO3, K2CO3, NaH o LiHMDS o una base orgánica tal como Et3N, DIPEA o DBU en un disolvente aprótico polar tal como, pero no limitado a, DMF, DMAc, DMSO o NMP para obtener compuestos de la estructura general 21. Los sustituyentes X1 y X2 preferentes para el uso en el acoplamiento incluyen F y Cl; el más preferente es F. Los éteres bencílicos 22 se pueden preparar de forma análoga a los compuestos 10 del Esquema 3. Alternativamente, por medio de la realización de las etapas anteriores en orden inverso, se pueden preparar compuestos de la estructura general 25 a partir del mismo material de partida 20. La eliminación del Pg1 se puede llevar a cabo por numerosos procedimientos descritos en la bibliografía para proporcionar las aminas 23 y 26.
Esquema 6

Los compuestos de amina preparados por medio de los procedimientos descritos en los Esquemas 1 a 6 , designados colectivamente como aminas 27, se pueden alquilar con un 2-bromoacetato protegido en presencia de una base adecuada tal como K2CO3, Et3N, NaH o LiHMDS en un disolvente aprótico polar tal como, pero no limitado a, DMF, DMAc, DMSO o NMP para obtener compuestos de la estructura general 28. Se puede llevar a cabo una hidrólisis estándar de ésteres para obtener los ácidos 29. Si Pg2 es t-butilo, se pueden utilizar procedimientos de desprotección ácida estándar tales como TFA/DCM, HCl/1,4-dioxano, HCl/EtOAc u otras condiciones adecuadas para obtener los ácidos 29.
Esquema 7

Los compuestos de la estructura general 30 (Esquema 8) pueden reaccionar con aminas R4NH2 en presencia de bases tales como carbonato de sodio, potasio o cesio, -bicarbonato, hidróxido, acetato o una base amínica orgánica tal como Et3N, DIPEA, DBU y similares en un disolvente aprótico polar tal como, pero no limitado a, THF, DMF, DMAc, DMSO o NMP o un disolvente prótico tal como agua, MeOH, EtOH o iPrOH o una mezcla de los mismos para obtener compuestos de la estructura general 31. Se observará que si un ejemplo proporciona un R4 con un centro enantiomérico resuelto, el otro enantiómero o una mezcla racémica del mismo se podría obtener por medio de la
selección del material de partida apropiado. Los sustituyentes X3 preferentes incluyen F, Cl y Br, los grupos X4 preferentes incluyen Cl, Br, -CO2-Pg2. La reducción del grupo nitro se puede efectuar por hidrogenación a 1 a 6 atm H2 con un catalizador metálico tal como paladio sobre carbono o níquel Raney en un disolvente prótico tal como MeOH o EtOH o aprótico tal como DMF, THF o EtOAc. Alternativamente, el grupo nitro se puede reducir con hierro, cinc, SnCl2 u otro metal adecuado en un medio ácido tal como HCI 1N, AcOH o NH4Cl acuoso en THF para proporcionar compuestos de estructura general 32 (Esquema 8a). Los compuestos tales como el 33 pueden ser acilados por haluros de acilo de forma estándar o por carboxilatos por medio de protocolos estándar de acoplamiento de amidas para proporcionar los compuestos 34. La reducción a los compuestos 35 se puede llevar a cabo en condiciones estándar con agentes reductores tales como LAH o BH3-THF o BH3-DMS (Esquema 8b).
Esquema 8

Los compuestos de diamina 32 y 35 preparados a través de los procedimientos descritos en los Esquemas 8a y 8b, designados colectivamente como diamina 37 (Esquema 9), pueden ser acilados con ácidos de estructura general 29 bajo protocolos de acoplamiento de amida estándar para entregar aminas 38 que existirán como una mezcla de 100% 38a a 100% 38b. Esta mezcla de aminas 38 puede ser ciclada para obtener compuestos de estructura general 39 por diversos procedimientos. Las aminas 38 se pueden calentar con un agente deshidratante tal como el T3P® o un alcohol alquílico tal como el n-butanol en condiciones de microondas (10 a 60 min a 120 a 180 °C) para obtener los compuestos 39. Alternativamente, la mezcla de compuestos 38 se puede calentar en condiciones ácidas tales como AcOH de 60 a 100 °C o en condiciones básicas tales como NaOH acuoso o KOH en 1,4-dioxano de 60 a 100 °C para proporcionar 39. Los compuestos de la estructura general 39 (X4 = Cl, Br o I) se pueden convertir en ésteres de la estructura 40 por carbonilación catalizada por paladio bajo una atmósfera de monóxido de carbono de 15 a 100 psi a una temperatura de 20 a 100 °C con un alcohol apropiado tal como MeOH o EtOH u otro alcohol alquílico. La hidrólisis del éster 40 se puede llevar a cabo como se describe en el Esquema 7 para proporcionar los ácidos 41.
Para los compuestos 38 en los que X4 = CO2-Pg2, la conversión a éster 40 se lleva a cabo en condiciones similares a las descritas anteriormente, excepto por el uso del procedimiento de ciclación básica, en el que el compuesto 41 se puede aislar directamente de la mezcla de reacción. Para los compuestos 40 en los que X4 es CO2tBu, la desprotección al ácido 41 se puede llevar a cabo en las condiciones ácidas descritas en el Esquema 7.
Esquema 9
R4
HN Z ^ X 4
h2( A ' z2
37

Además, la diamina 37 se puede convertir en el 2-clorometil-benzimidazol 42 (Esquema 10) por diversos procedimientos. El tratamiento con cloruro de 2-cloroacetilo en un disolvente aprótico tal como el 1,4-dioxano, seguido por un calentamiento a 40 a 100 °C durante 2 a 18 hs, puede dar lugar al benzimidazol 42 deseado, en el que Z1, Z2 y Z3 son CH. En los casos en los que Z1, Z2 y Z3 no son todos CRz, tras el tratamiento con cloruro de 2-cloroacetilo en un disolvente aprótico tal como el 1,4-dioxano durante 30 min a 4 hs, se cambia el disolvente por un medio ácido tal como AcOH o TFA, seguido por un calentamiento a 40 a 100 °C durante 2 a 18 hs para proporcionar el compuesto deseado 42. La diamina 37 también se puede tratar con anhídrido cloroacético a una temperatura entre 0 y 80 °C en un disolvente aprótico tal como, pero no limitado a, 1,4-dioxano, THF o MeCN, seguido por un calentamiento de 2 a 18 hs a 60 a 100 °C para obtener el compuesto deseado 42. Además, la diamina 37 se puede tratar con 2-cloro-1,1,1-trimetoxietano en un disolvente aprótico tal como, pero no limitado a, 1,4-dioxano, THF o MeCN, o un disolvente prótico, por ejemplo, MeOH o EtOH, en presencia de un catalizador ácido, por ejemplo, pTSA, a 20 a 100 °C. Alternativamente, las diaminas 37 se pueden calentar a 100 a 180 °C con ácido 2 hidroxiacético en un disolvente aprótico, tal como el mesitileno pero sin limitarse a él, para proporcionar un intermedio hidroximetilado. La conversión del grupo hidroximetilo en el compuesto clorometilo 42 se puede llevar a cabo por procedimientos estándar, que incluyen el tratamiento con SOCh en un disolvente aprótico. Los compuestos de la estructura general 42 se pueden hacer reaccionar con los compuestos 27 en presencia de bases tales como carbonato de sodio, potasio o cesio, -bicarbonato, NaH o una base amina orgánica tal como Et3N, DIPEA, DBU y similares en un disolvente aprótico polar tal como, por ejemplo, pero sin limitarse a THF, MeCN, DMF, DMAc, DMSO o NMP, para obtener los compuestos 39 (X4 = CI, Br, I) o los compuestos 40 (X4 = CO2-Pg2) que posteriormente se utilizan para obtener los compuestos 41 por medio de los procedimientos descritos en el Esquema 9.
Esquema 10

Alternativamente, los compuestos de la estructura general 42 pueden reaccionar con piperazinas apropiadamente sustituidas y protegidas para proporcionar los compuestos 43 (Esquema 11). La eliminación del Pg1 se podría llevar a cabo con numerosos procedimientos descritos en la bibliografía para proporcionar aminas 44. La conversión a compuestos de la estructura general 39 (X4 = Cl, Br o I) o 40 (X4 = CO2-Pg2) se puede llevar a cabo por medio de un acoplamiento C-N Buchwald-Hartwig entre compuestos de las estructuras generales 10 y como se ha descrito anteriormente en el Esquema 4. Los compuestos de la estructura general 39 o 40 se pueden usar entonces para obtener compuestos de la estructura 41 por medio de los procedimientos descritos en el Esquema 9.
Esquema 11

Ejemplos

6-cloro-3',6'-dihidro-[2,4'-bipiridin]-1'(2'H)-carboxilato de terc-butilo
Se cargó un recipiente de reacción equipado con un condensador de reflujo con 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidropiridin-1(2H)-carboxilato de terc-butilo (1,5 g, 4,9 mmol), 2,6-dicloropiridina (1,4 g, 9,7 mmol), Pd(dppf)Cl2 (0,34 g, 0,49 mmol) y carbonato de cesio (3,5 g, 11 mmol). Se añadió una solución de 1,4-dioxano (15 ml) y agua (3 ml) y la mezcla se calentó a 90 °C bajo N2 (g). Después de 7 hs, la mezcla se dejó enfriar hasta TA y se filtró a través de una almohadilla de Celite® con EtOAc (50 ml). La mezcla se diluyó con agua (20 ml), la capa acuosa se extrajo con EtOAc (3 x 50 ml) y las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y el disolvente se eliminó a presión reducida. El material bruto se purificó por medio de cromatografía en columna eluyendo con 10% de EtOAc en heptano para obtener el Intermedio 1 como un aceite incoloro (1,1 g, 75%). RMN de 1H (CDCla) 8: 7,57 (t, 1H), 7,23 (d, 1H), 7,14 (d, 1H), 6,66 (br s, 1H), 4,11 (br s, 2H), 3,61 (br s, 2H), 2,57 (br s, 2H), 1,43 a 1,52 (m, 9H).

4-(6-cloropiridin-2-il)piperidin-1-carboxilato de terc-butilo
A una solución agitada del Intermedio 1 (0,55 g, 1,9 mmol) en MeOH (19 ml) se le añadió PtO2 (0,042 g, 0,19 mmol). La solución se sometió a una atmósfera de hidrógeno (30 PSI) a TA. Después de 3 hs, la solución se filtró a través de un tapón de Celite®, se lavó con MeOH (2 x 15 ml) y se concentró a presión reducida. El material bruto se purificó por medio de cromatografía en columna eluyendo con 30% de EtOAc en heptano para obtener el Intermedio 2 (0,22 g, 40%) como un aceite incoloro. RMN de 1H (CDCh) 8: 7,57 (t, 1H), 7,15 (d, 1H), 7,05 (d, 1H), 4,23 (br s, 2H), 2,80 (d, 3H), 1,89 (d, 2H), 1,60 a 1,73 (m, 2H), 1,45 (s, 9H).

Bis(4-metilbencenosulfonato) de 2-((4-doro-2-fluorobencM)oxi)-6-(piperidm-4-M)piridma
Etapa 1:
Un recipiente de reacción equipado con un condensador de reflujo se cargó con el Intermedio 2 (6,5 g, 22 mmol), (4-cloro-2-fluorofenil)metanol (3,5 g, 22 mmol), Pd2(dba)3 (1,0 g, 1,1 mmol), BINAP (1,4 g, 2,2 mmol) y carbonato de cesio (14 g, 44 mmol). Se añadió tolueno (73 ml) y la mezcla se calentó a 100 °C. Después de 16 hs, se dejó enfriar la mezcla hasta TA, se filtró a través de Celite® con EtOAc (100 ml) y se concentró a presión reducida. El material bruto se purificó por medio de cromatografía en columna eluyendo con 10% de EtOAc en PE para obtener 4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-carboxilato de terc-butilo como un aceite amarillo (7,6 g, 82%). RMN de 1H (CDCla) 8: 7,51 (dd, 1H), 7,39 a 7,47 (m, 1H), 7,06 a 7,18 (m, 2H), 6,73 (d, 1H), 6,62 (d, 1H), 5,40 (s, 2H), 4,22 (br s, 2H), 2,83 (m, 2H), 2,73 (tt, 1H), 1,81 a 1,94 (m, 2H), 1,64 a 1,79 (m, 2H), 1,50 (s, 9H).
Etapa 2:
A una solución agitada de 4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-carboxilato de terc-butilo (50 g, 120 mmol) en EtOAc (700 ml) se añadió pTSA-H2O (59 g, 310 mmol). La mezcla se calentó a 60 °C. Después de 30 min, la solución se dejó enfriar hasta TA. El precipitado sólido resultante se sometió a una suspensión durante 16 hs, se recolectó por filtración y se secó a presión reducida para obtener el Intermedio 3 como un sólido (81 g, quant). RMN de 1H (600 MHz, DMSO-d6) 8: 8,55 (br s, 1H), 8,28 (d, 1H), 7,68 (t, 1H), 7,60 (t, 1H), 7,48 (d, 4H), 7,32 (d, 1H), 7,12 (d, 4H), 6,89 (d, 1H), 6,74 (d, 1H), 5,38 (s, 2H), 3,37 (d, 2H), 2,98 a 3,09 (m, 2H), 2,87 a 2,96 (m, 1H), 2,29 (s, 6H), 1,96 a 2,01 (m, 2H), 1,80 a 1,94 (m, 2H).

Bis(4-metilbencenosulfonato) de 3-vluoro-4-((6-(piperidin-4-il)piridin-2-il)oxi)metil)benzonitrilo
Etapa 1:
A una solución de diisopropilamina (92 ml, 656 mmol) en THF (350 ml) a -26 °C se añadió n-butilitio en heptanos (2,6 M, 250 ml, 650 mmol) durante 15 min. La mezcla se enfrió a -30 °C y se añadió una solución de piperidin-1,4-dicarboxilato de 1-(ferc-butil) 4-metilo (156 g, 641 mmol) en THF (150 ml) durante 25 min. Después de 10 min, se añadió una solución de 2,6-dicloropiridina (94 g, 635 mmol) en THF (150 ml) durante 2 min. La mezcla se calentó a 25 °C durante 2,5 hs y después se enfrió a 8 °C y se trató con HCI 6 M (125 ml) durante 20 min para llevar el pH de la mezcla a ~7 a 8. La mezcla se diluyó con agua (100 ml) y MTBE (150 ml) y las capas se separaron. La capa acuosa se extrajo con MTBE (150 ml) y las capas orgánicas combinadas se lavaron con salmuera (150 ml), se secaron sobre MgSO4. El disolvente se eliminó a presión reducida para proporcionar 1 -(terc-butil) 4-(6-cloropiridin-2-il)piperidin-1,4-dicarboxilato bruto (241 g) como un aceite amarillo, que se utilizó en la siguiente etapa sin purificación. RMN de 1H de una muestra purificada (400 MHz, CDCh) 8: 7,62 (t, 1H), 7,21 (d, 2H), 3,83 (br s, 2H), 3,71 (s, 3H), 3,14 (br s, 2H), 2,41 (d, 2H), 2,08 (ddd, 2H), 1,45 (s, 9H).
Etapa 2:
El 1 -(terc-butil) 4-(6-cloropiridin-2-il)piperidin-1,4-dicarboxilato bruto (241 g, supuesto 635 mmol) se disolvió en MeOH (400 ml) a 43 °C y se trató con 4 M aq. NaOH (300 ml) durante 20 min. La mezcla se calentó a 50 °C y se agitó durante 35 min. A continuación, la mezcla se enfrió a 11 °C y el pH se ajustó a ~2 por medio de la adición de HCI 6 M (200 ml) durante 25 min mientras se seguía enfriando a 5 °C, tras lo cual se formó un precipitado sólido. La suspensión espesa se diluyó con agua (300 ml) y se agitó durante 40 min, tras lo cual el sólido se recolectó por filtración, se lavó con agua y se secó al vacío a 50 °C para proporcionar un sólido blanco (224 g). El sólido se trituró en heptano (750 ml) a 45 °C durante 45 min. La mezcla se enfrió a 16 °C y el sólido se recolectó por filtración, se lavó con heptano y se secó para proporcionar el ácido 1-(terc-butoxicarbonNo)-4-(6-doropiridin-2-N)piperidin-4-carboxílico (187 g, 549 mmol, 86% en dos etapas) como un sólido blanco.
Etapa 3:
Una solución de ácido 1-(terc-butoxicarbonilo)-4-(6-cloropiridin-2-il)piperidin-4-carboxílico (187 g, 549 mmol) en DCE (900 ml) se calentó a 82 °C durante la noche y posteriormente se enfrió a 20 °C. La mezcla se trató con Magnesol® (30 g) durante 40 min. La suspensión espesa se filtró a través de una almohadilla de Magnesol® (30 g) y los sólidos se lavaron con MTBE:heptano 1:1 (300 ml). El filtrado se concentró a presión reducida para dar un sólido amarillo pálido, que se trituró en heptano (250 ml) a 50 °C. La mezcla se enfrió a 12 °C y el sólido se recolectó por filtración, se lavó con heptano y se secó al vacío a 45 °C para proporcionar 4-(6-cloropiridin-2-il)piperidin-1-carboxilato de tercbutilo (143 g, 481 mmol, 88%) como un sólido. RMN de 1H (600 MHz, CDCh) 8: 7,58 (t, 1H), 7,17 (d, 1H), 7,06 (d, 1H), 4,25 (br s, 2H), 2,66 a 2,93 (m, 3H), 1,91 (d, 2H), 1,69 (qd, 2H), 1,47 (s, 9H).
Etapa 4:
Una mezcla de 4-(6-cloropiridin-2-il)piperidin-1 -carboxilato de terc-butilo (100 g, 337 mmol), 3-fluoro-4-(hidroximetil)benzonitrilo (53,9 g, 357 mmol) y Cs2CO3 (170 g, 522 mmol) en dioxano (900 ml) se desoxigenó con 5 ciclos de vacío/llenado de nitrógeno. Se añadieron JohnPhos ([1,1'-bifenil]-2-il-di-terc-butilfosfina, 2,02 g, 6,77 mmol) y Pd2(dba)3 (3,10 g, 3,39 mmol) y se aplicaron otros 2 ciclos de vacío/llenado de nitrógeno. A continuación, la mezcla se calentó a 95 °C durante 3 hs. Se añadieron más JohnPhos (660 mg, 2,21 mmol) y Pd2(dba)3 (990 mg, 1,08 mmol) y el calentamiento continuó durante la noche. La mezcla se enfrió a 20 °C y se filtró a través de una almohadilla de Celite®, lavando con MTBE (250 ml). El filtrado se concentró a presión reducida para dar un aceite rojo-anaranjado (174 g). Este material se disolvió en MTBE/hexano al 30% (600 ml), se agitó con Magnesol® (20 g) y Darco® G-60 (10 g) durante 70 min y posteriormente se filtró a través de una almohadilla de sílice (100 g), lavando con MTBE/hexano al 50% (600 ml). El filtrado se concentró a presión reducida y se azeotró con EtOAc (100 ml) para proporcionar 4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1-carboxilato de terc-butilo como un aceite (147 g), que se utilizó sin más purificación. RMN de 1H de una muestra purificada (600 MHz, CDCh) 8: 7,62 (t, 1H), 7,53 (t, 1H), 7,44 (d, 1H), 7,37 (d, 1H), 6,75 (d, 1H), 6,65 (d, 1H), 5,49 (s, 2H), 4,20 (br s, 2H), 2,81 (br s, 2H), 2,70 (tt, 1H), 1,82 (d, 2H), 1,67 (d, 2H), 1,49 (s, 9H).
Etapa 5:
A una solución agitada de 4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1-carboxilato de terc-butilo (147 g, supuesto 337 mmol) en EtOAc (1,8 L) a TA se añadió pTsA-H2O (161 g, 846 mmol). La mezcla se calentó a 60 °C, lo que dio lugar a la evolución del gas y a la formación de sólidos. La mezcla se agitó durante 1,5 hs, tras lo cual se añadió pTSA-H2O (12 g, 63 mmol) y se continuó agitando durante 45 min. La suspensión espesa se enfrió a 17 °C y los sólidos se recolectaron por filtración, se lavaron con EtOAc (200 ml) y se secaron para obtener 205 g de sólido. Este material se disolvió en MeOH (500 ml) a 55 °C y se diluyó con EtOAc (1 L). La suspensión espesa resultante se enfrió a 20 °C y los sólidos se recolectaron por filtración, se lavaron con EtOAc:MeOH 9:1 (100 ml) y EtOAc (250 ml) y se secaron para proporcionar el Intermedio 4 (176,6 g, 269 mmol, 80% en dos etapas) como un sólido blanco. RMN de 1H (600 MHz, DMSO-d6) 8: 8,53 (br s, 1H), 8,26 (br s, 1H), 7,89 (d, 1H), 7,67 a 7,78 (m, 3H), 7,48 (d, 4H), 7,11 (d, 4H), 6,90 (d, 1H), 6,79 (d, 1H), 5,48 (s, 2H), 3,35 (d, 2H), 2,96 a 3,09 (m, 2H), 2,79 a 2,96 (m, 1H), 2,29 (s, 6H), 1,93 a 2,03 (m, 2H), 1,77 a 1,90 (m, 2H).

Intermedio 5
Ácido 2-(4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)acético
Etapa 1:
A una mezcla de NaH (60% en aceite, 118 g, 863 mmol) en DMF (6 ml) se le añadió metanol (39,9 g, 236 mmol). La mezcla se agitó a 30 °C durante 1 hora. La mezcla se diluyó con agua (500 ml), y se extrajo con EtOAc (400 ml * 3). Las capas orgánicas se combinaron, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía flash (columna de gel de sílice, 10:1 PE/EtOAc) para obtener 74 g de 2-(4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1 -il)acetato de etilo (84%) como un aceite amarillo. RMN de 1H (400 MHz, CDCh) 87,51 (t, 1H), 7,45 (t, 1H), 7,09 a 7,17 (m, 2H), 6,75 (d, 1H), 6,61 (d, 1H), 5,41 (s, 2H), 4,22 (q, 2H), 3,27 (s, 2H), 3,07 (d, 2H), 2,54 a 2,65 (m, 1H), 2,32 (td, 2H), 1,93 a 2,07 (m, 2H), 1,85 a 1,92 (m, 2H), 1,30 (t, 3H).
Etapa 2:
A una solución de 2-(4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)acetato de etilo (73 g, 179 mmol) en EtOH (270 ml) se añadió NaOH 5 M (156 ml, 780 mmol). La solución se agitó a 25 °C durante 2 hs. La mezcla se acidificó hasta un pH ~3,5 con HCl 1 M. El precipitado resultante se recolectó por filtración. Los sólidos se lavaron con agua y se secaron al vacío para obtener 54 g del Intermedio 5 (78%) como un sólido amarillo pálido. RMN de 1H (400 MHz, DMSO-d6) 87,65 a 7,72 (m, 1H), 7,62 (t, 1H), 7,47 (dd, 1H), 7,32 (dd, 1H), 6,92 (d, 1H), 6,73 (d, 1H), 5,40 (s, 2H), 4,13 (s, 2H), 3,58 (d, 2H), 3,16 a 3,26 (m, 2H), 2,89 (br s, 1H), 2,00 a 2,19 (m, 4H); LC-MS = 378,8.

Intermedio 6
4-(5-fluoro-6-oxo-1,6-dihidropiridin-2-il)piperidin-1-carboxilato de terc-butilo
Etapa 1:
A una solución de 2-bromo-5-fluoropiridina (20 g, 110 mmol) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidropiridin-1(2H)-carboxilato de terc-butilo (35,1 g, 114 mmol) en THF (240 ml) se añadió Pd(PPh3)4 (13,1 g, 11,4 mmol), y Na2CO3 (24,1 g, 227 mmol). La mezcla de reacción amarilla resultante se agitó a 90 °C durante 48 hs. La reacción se enfrió a TA, se diluyó con agua (100 ml) y se extrajo con EtOAc (3 * 200 ml). Los extractos orgánicos combinados se lavaron con salmuera (100 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente 1 a 20% de EtOAc/PE) para obtener 5-fluoro-3',6'-dihidro-[2,4'-bipiridin]-1'(2'H)-carboxilato de ferc-butilo (31 g, 98%) como un aceite incoloro. RMN de 1H (CDCla) 88,41 (t, 1H), 7,37 (dd, 2H), 6,52 (br s, 1H), 4,13 (d, 2H), 3,65 (m, 2H), 2,57 a 2,70 (m, 2H), 1,49 (s, 9H).
Etapa 2:
A una solución incolora de 5-fluoro-3',6'-dihidro-[2,4'-bipiridin]-1'(2'H)-carboxilato de ferc-butilo (31 g, 110 mmol) en EtOAc (300 ml) se añadió Pd/C húmedo al 10% (1,2 g, 5,6 mmol). La mezcla negra se agitó a 25 °C bajo H2 (15 psi) durante 16 hs. La mezcla se filtró a través de una almohadilla Celite® y se concentró a presión reducida para obtener 4-(5-fluoropiridin-2-il)piperidin-1-carboxilato de ferc-butilo (31 g, 99%) como un aceite incoloro. RMN de 1H (CDCh) 8 8,39 (d, 1H), 7,34 (td, 1H), 7,15 (dd, 1H), 4,25 (br s, 2H), 2,74 a 2,93 (m, 3H), 1,89 (d, 2H), 1,69 (qd, 2H), 1,48 (s, 9H).
Etapa 3:
A una solución de 4-(5-fluoropiridin-2-il)piperidin-1-carboxilato de ferc-butilo (31 g, 110 mmol) en DCM (400 ml) se añadió m-CPBA (47,7 g, 276 mmol) a 0 °C. La mezcla de reacción resultante se agitó a TA durante 16 hs. La suspensión blanca se filtró y el filtrado se apagó con una solución ac. de Na2SO3 (200 ml). La capa ac. se separó y se extrajo con DCM (3 * 200). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente de 0,5 a 4% de MeOH/DCM) para obtener 1-óxido de 2-(1-(ferc-butoxicarbonil)piperidin-4-il)-5-fluoropiridina (20 g, 61%) como un sólido. r Mn de 1H (CDCh) 88,21 (dd, 1H), 7,11 a 7,18 (m, 1H), 7,02 a 7,09 (m, 1H), 4,26 (br s, 2H), 3,58 (m, 1H), 2,89 (br s, 2H), 2,02 (d, 2H), 1,43 a 1,52 (m, 11H).
Etapa 4:
A una solución de 1-óxido de 2-(1-(tere-butox¡carbon¡lo)p¡per¡d¡n-4-¡l)-5-fluorop¡r¡d¡na (10 g, 34 mmol) en THF (150 ml) a 0 °C se añadió Et3N (6,83 g, 67,5 mmol), y TFAA (70,9 g, 337 mmol), gota a gota. La mezcla se agitó a 0 °C durante 1 hora y a TA durante 16 hs. La solución amarilla clara se apagó con una solución ac. de NaHCO3 (400 ml). El pH se ajustó a ~4 con TFA y la mezcla se extrajo con EtOAc (3 * 200 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (4 a 80% de EtOAc en PE) para dar el Intermedio 6 (5,4 g, 54%) como un sólido. RMN de 1H (CDCla) 812,92 (br s, 1H), 7,17 (dd, 1H), 5,97 (dd, 1H), 4,25 (br s, 2H), 2,86 (br s, 2H), 2,72 (t, 1H), 1,95 (d, 2H), 1,56 (qd, 2H), 1,48 (s, 9H).

Ácido 2-(4-(6-((4-cianobencil)oxi)-5-fluoropiridin-2-il)piperidin-1-il)acético
Etapa 1:
A una solución del Intermedio 6 (2,0 g, 6,8 mmol), el alcohol 4-cianobencílico (1,35 g, 10,1 mmol) y la 1,1'-(azodicarbon¡l)d¡piper¡d¡na (2,55 g, 10,1 mmol ) en PhCH3 (30 ml) se añadió tri-n-butilfosfina (2,05 g, 10,1 mmol), gota a gota, bajo una atmósfera de N2. La solución amarilla clara resultante se agitó a 80 °C bajo atmósfera de N2 durante 48 hs. La mezcla se diluyó con EtOAc (100 ml) y se lavó con agua (100 ml). La fase orgánica se secó sobre Na2SO4 (25,00 g), se filtró y se concentró a presión reducida. El producto bruto se purificó por medio de cromatografía flash (0 a 15% de EtOAc/PE) para dar 4-(6-((4-c¡anobenc¡l)ox¡)-5-fluorop¡r¡d¡n-2-¡l)piper¡d¡n-1-carboxilato de tere-butilo (1,72 g, 62% de rendimiento) como un aceite incoloro. RMN de 1H (CDCh) 87,67 (d, 2H), 7,58 (d, 2H), 7,29 (dd, 1H), 6,71 (dd, 1H), 5,51 (s, 2H), 4,20 (br s, 2H), 2,81 (t, 2H), 2,69 (dt, 1H), 1,81 (d, 2H), 1,65 (br s, 2H), 1,49 (s, 9H).
Etapa 2:
A una solución de 4-(6-((4-c¡anobenc¡l)oxi)-5-fluorop¡r¡d¡n-2-¡l)p¡per¡d¡n-1-carbox¡lato de terc-butilo (1,72 g, 4,18 mmol) en DCM (10 ml) se añadió TFA (2 ml). La solución amarilla clara resultante se agitó a 25 °C durante 2 hs. La mezcla se concentró a presión reducida para dar trifluoroacetato de 4-(((3-fluoro-6-(piper¡d¡n-4-¡l)p¡rid¡n-2-¡l)oxi)met¡l)benzon¡tr¡lo (1,3 g, quant.) como un sólido amarillo claro. RMN de 1H (CD3OD) 87,74 (d, 2H), 7,63 (d, 2H), 7,46 (dd, 1H), 6,89 (dd, 1H), 5,56 (s, 2H), 3,42 a 3,53 (m, 2H), 3,11 (td, 2H), 2,96 (tt, 1H), 2,03 a 2,13 (m, 2H), 1,87 a 2,02 (m, 2H).
Etapa 3:
A una solución incolora de 4-((3-fluoro-6-(p¡per¡d¡n-4-il)p¡r¡d¡n-2-¡l)ox¡)met¡l)benzon¡tr¡lo trifluoroacetato (1,3 g, 4,2 mmol) y 2-bromoacetato de etilo (767 mg, 4,59 mmol) en MeCN (20 ml) se añadió K2CO3 (2,89 g, 20,9 mmol). La suspensión blanca resultante se agitó a 60 °C durante 3 hs y se dejó a TA durante 16 hs. La mezcla se filtró y se concentró a presión reducida. El producto bruto se purificó por cromatografía flash (0 a 33% de EtOAc en PE) para dar 2-(4-(6-((4-c¡anobenc¡l)oxi)-5-fluorop¡r¡d¡n-2-¡l)p¡per¡d¡n-1-¡l)acetato de etilo (1,07 g, 65%) como un sólido amarillo claro. RMN de 1H (CDCh) 87,67 (d, 2H), 7,58 (d, 2H), 7,28 (dd, 1H), 6,72 (dd, 1H), 5,52 (s, 2H), 4,21 (m, 2H), 3,26 (s, 2H), 3,06 (d, 2H), 2,55 (tt, 1H), 2,29 (dt, 2H), 1,94 (dq, 2H), 1,77 a 1,86 (m, 2H), 1,30 (m, 3H).
Etapa 4:
A una solución de 2-(6-((4-c¡anobenc¡l)oxi)-5-fluorop¡r¡d¡n-2-¡l)p¡per¡d¡n-1-¡l)acetato de etilo (1,07 g, 2,69 mmol) en MeOH (10 ml) se añadió, gota a gota, una solución de NaOH (162 mg, 4,04 mmol) en agua (2 ml). La solución incolora resultante se agitó a 25 °C durante 3 hs. La mezcla se diluyó con agua (30 ml), se extrajo con MTBE (30 ml). La fase orgánica se acidificó a pH ~7 con HCI 2 M y se liofilizó durante 16 hs. El producto bruto se purificó por cromatografía flash (gradiente de 0 a 5% de MeOH/DCM) para dar el Intermedio 7 (850 mg, rendimiento del 86%) como un sólido. RMN de 1H (400 MHz, CD3OD) 87,75 (d, 2H), 7,67 (d, 2H), 7,46 (dd, 1H), 6,92 (dd, 1H), 5,59 (s, 2H), 3,71 a 3,80 (m, 2H), 3,35 (s, 2H), 3,10 a 3,27 (m, 2H), 2,90 a 3,06 (m, 1H), 2,11 a 2,29 (m, 2H), 2,01 a 2,10 (m, 2H), LC-MS(ES+): 369,9 (M+H).

Intermedio 8
(3R,4R)-4-(6-((4-doro-2-fluorobencM)oxi)piridm-2-M)-3-hidroxipiperidm-1-carboxMato de rac-terc-butilo A una solución del Intermedio 1 (800 mg, 1,9 mmol) en THF (15 ml) a 0 °C bajo una atmósfera de nitrógeno se añadió el complejo borano-THF (1 M en THF, 2,1 ml, 2,1 mmol). La mezcla de reacción se agitó a 0 °C durante 10 min y posteriormente se calentó a 30 °C durante 30 min. A continuación, el recipiente de reacción se enfrió a 0 °C, se abrió al aire y se añadió lentamente una solución de NaOH (190 mg, 4,8 mmol) en agua (5 ml) y peróxido de hidrógeno (30% en peso en agua, 0,86 ml, 9,6 mmol). La mezcla se calentó a 26 °C y se agitó durante 16 hs. Se añadieron Na2SO3 acuoso (15 ml) y NaHCO3 (15 ml) a la suspensión blanca resultante y la mezcla se extrajo con DCM (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El material bruto se purificó por medio de cromatografía en columna eluyendo con EtOAc en PE (gradiente de 10% a 30% a 60%) para obtener el Intermedio 8 como un aceite incoloro (320 mg, 38%). LC-MS(ES+): 437 (M+H), 459 (M+Na).

Intermedio 9
Trifluoroacetato de rac-(3R,4R)-4-(6-((4-Cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-3-ol
A una solución del Intermedio 8 (60 mg, 0,14 mmol) en DCM (2 ml) se añadió TFA (0,5 ml) a TA, y la mezcla se agitó durante 1 hora. La mezcla de reacción se concentró a presión reducida para obtener el Intermedio 9 bruto como un aceite amarillo claro, que se utilizó sin purificación. LC-MS(ES+): 337 (M+H). FALTA

Intermedio 10
Clorhidrato de rac-2-((4-cloro-2-fluorobencil)-6-((3R,4R)-3-fluoropiperidin-4-il)piridina
A una solución del Intermedio 8 (60 mg, 0,14 mmol) en DCM (6 ml) a 0 °C bajo una atmósfera de nitrógeno se añadió DAST (trifluoruro de dietilaminosulfuro, 38 mg, 0,23 mmol). La mezcla resultante se agitó a 0 °C durante 10 min y después a TA durante 2 hs. A continuación se añadió agua a la solución y se extrajo la mezcla con EtOAc (3 x 100 ml). La solución orgánica combinada se lavó con salmuera y posteriormente se concentró a presión reducida. El producto bruto se purificó por medio de pre-TLC (PE:EtOAc = 10:1) para dar (3R,4R)-4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-M)-3-fluoropiperidin-1 -carboxilato de rac-ferc-butilo (80 mg), que se utilizó sin más purificación. El material se disolvió en DCM (2 ml) a TA y se añadió HCI 4 M en EtOAc (1 ml), gota a gota. La mezcla se agitó durante 1 hora antes de concentrarse a presión reducida para dar el Intermedio 10 como un sólido blanco. RMN de 1H (400 MHz, CD3OD) 67,65 a 7,77 (m, 1H), 7,51 (t, 1H), 7,18 a 7,32 (m, 2H), 7,00 (d, 1H), 6,81 (d,
1H), 5,45 (s, 2H), 5,09 a 5,33 (m, 1H), 3,72 (ddd, 1H), 3,40 a 3,49 (m, 1H), 3,40 a 3,49 (m, 1H), 3,32 a 3,38 (m, 1H), 3,13 a 3,25 (m, 1H), 2,08 a 2,39 (m, 2H). Nota: la estereoquímica de los sustituyentes de piperidina se asignó como trans por analogía con los precedentes publicados (véase, por ejemplo, el documento WO 2010/022055), pero no se confirmó experimentalmente.

Clorhidrato de rac-(3R,4S)-4-(6-((4-cloro-2-fluorobencN)oxi)piridm-2-N)piperidm-3-ol
Etapa 1:
A una solución del Intermedio 8 (170 mg, 0,39 mmol) en DCM (5 ) a 0 °C se añadió Et3N (0,16 ml, 1,2 mmol) y MsCI (58 mg, 0,51 mmol), y la mezcla se agitó durante 2 hs. La mezcla se diluyó con DCM (30 ml), se lavó con solución saturada de NH4Cl ac. saturado y salmuera, se secó sobre Na2SO4 y se concentró a presión reducida para obtener un aceite amarillo. Este material se disolvió en DMSO (1,5 ml) y se añadió a una suspensión de formiato de cesio (140 mg, 0,78 mmol) en DMSO (1 ml). La mezcla se agitó a 120 °C durante 4 hs y a 25 °C durante 14 hs. La mezcla se vertió en agua (15 ml) y se extrajo con EtOAc (3 * 15 ml). La solución orgánica combinada se lavó con salmuera, se secó sobre Na2SO4 y se concentró a presión reducida. El producto bruto se purificó por pre-TLC (PE:EtOAc = 4:1) para obtener un aceite incoloro (60 mg).
El aceite se disolvió en MeOH (2 ml) a TA, se añadió K2CO3 y la mezcla se agitó durante 1 hora. La mezcla se diluyó con EtOAc, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a presión reducida para dar un producto bruto. El producto se purificó por medio de SFC preparativa para obtener (3R,4S)-4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-3-hidroxipiperidin-1-carboxilato de rac-terc-butilo como una goma amarilla (20 mg, 12%). LC-MS(ES+): 437 (M+H), 459 (M+Na)
Procedimiento de SFC: Columna: OJ (250mm * 30mm, 5 micrómetros); Fase móvil: CO2 con 15% de iPrOH (0,1% de NH4OH); Caudal: 60 ml/min; Longitud de onda: 220 nm. Tiempo de retención = 3,65 min.
Etapa 2:
A una solución de (3R,4S)-4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-3-hidroxipiperidin-1-carboxilato de rac-tercbutilo (20 mg, 0,046 mmol) en EtOAc (4 ml) a 0 °C se añadió HCl 4 M en EtOAc (4 ml), y la mezcla se agitó durante 2 hs. La mezcla se concentró a presión reducida para obtener el ejemplo bruto del Intermedio 11 como un aceite amarillo claro, que se utilizó sin purificación. LC-MS(ES+): 337 (M+H). FALTA

Intermedio 12a
Trifluoroacetato de rac-2-((4-cloro-2-fluorobencM)oxi)-6-((3R,4S)-3-metNpiperidm-4-N)piridma
Intermedio 12b Trifluoroacetato de rac-2-((4-cloro-2-fluorobencN)oxi)-6-((3R,4R)-3-metMpiperidm-4-N)piridma Etapa 1:
El (3R,4S)-4-(6-cloropiridin-2-il)-3-metilpiperidin-1 -carboxilato de rac-terc-butilo y el (3R,4R)-4-(6-cloropiridin-2-il)-3-metilpiperidin-1 -carboxilato de rac-terc-butilo se prepararon por medio de una vía análoga a la descrita para los Intermedios 1 y 2 mediante el uso de 3-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidropiridin-1(2H)-carboxilato de terc-butilo en la reacción de Suzuki. La mezcla de isómeros cis y trans se separó por cromatografía en columna eluyendo con EtOAc en PE (gradiente 0 a 15%). El isómero trans (rac-3R,4S) eluyó primero.
(3R,4S)-4-(6-doropiridin-2-il)-3-metilpiperidin-1-carboxilato de rac-terc-butilo: RMN de 1H (400 MHz, CDCI3) 87,56 (t, 1H), 7,71 (d, 1H), 7,04 (d, 1H), 4,22 (br s, 2H), 2,76 (br s, 1H), 2,43 a 2,39 (m, 2H), 2,02 a 1,92 (m, 1H), 1,79 a 1,71 (m, 2H), 1,48 (s, 9H), 0,70 (d, 3H).
(3R,4R)-4-(6-doropiridin-2-il)-3-metilpiperidin-1-carboxilato de rac-terc-butilo: RMN de 1H (400 MHz, CDCI3) 87,59 (t, 1H), 7,16 (d, 1H), 6,99 (d, 1H), 4,36 (br s, 1H), 4,01 (br s, 1H), 3,05 (dt, 2H), 2,79 (br s, 1H), 2,33 (q, 1H), 2,07 a 2,01 (m, 1H), 1,71 (d, 1H), 1,46 (s, 9H), 0,66 (d, 3H).
Etapa 2:
Los intermedios 12a y 12b se prepararon a partir de los respectivos isómeros separados de cloropiridina por medio de eterificación de forma análoga al intermedio 3, etapa 1, y desprotección de forma análoga al intermedio 9, y se utilizaron sin purificación.

Bis clorhidrato de 3-fluoro-4-((6-(piperazin-1-il)piridin-2-il)oxi)metil)benzonitrilo
Etapa 1:
Esta reacción se llevó a cabo en dos partidas paralelas; el ejemplo de preparación por partidas es el siguiente: A una suspensión agitada de KOtBu (313 g, 2,79 mol) en THF (4,0 L) se añadió alcohol 4-ciano-2-fluorobencílico (281 g, 1,86 mol) en porciones entre 10 a 15 °C. La mezcla se agitó a 15 °C durante 45 min y se añadió 2,6-dicloropiridina (230 g, 1,55 mol) en varias porciones a la mezcla de reacción a 15 °C y la mezcla se agitó a 15 °C durante 18 hs. La mezcla se vertió en NH4O ac. sat. (10 L). Se añadió EtOAc (10 L) y la mezcla se agitó durante 15 min. La mezcla se filtró a través de una almohadilla de Celite®. La capa orgánica se separó y la capa ac. se extrajo con EtOAc (2 * 6,0 L). Las capas orgánicas combinadas se lavaron con salmuera (5,0 L), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía en columna sobre gel de sílice (gradiente PE/EtOAc 10 a 15%) para dar 4-(((6-cloropiridin-2-il)oxi)metil)-3-fluorobenzonitrilo como un sólido amarillo claro. Las partidas combinadas produjeron 550 g (67%). RMN de 1H (CDCh) 87,67 (t, 1H), 7,58 (t, 1H), 7,48 (dd, 1H), 7,40 (dd, 1H), 6,97 (d, 1H), 6,75 (d, 1H), 5,49 (s, 2H).
Etapa 2:
A una solución agitada de 4-((6-cloropiridin-2-il)oxi)metil)-3-fluorobenzonitrilo (180 g, 0,685 mol) y piperazin-1-carboxilato de terc-butilo (140 g, 0,754 mol) en phcH3 (2,0 L) se añadió Cs2CO3 (446 g, 1,37 mol), BINAP (42,6 g, 0,0685 mol) y Pd2(dba)3 (31,4 g, 0,0343 mol) bajo N2 a 15 °C. La mezcla se desgasificó y se rellenó con N2 tres veces. La mezcla resultante se calentó a 120 °C bajo N2 durante 18 hs. La mezcla de reacción se enfrió a 80 °C y se filtró a través de una almohadilla de Celite®. La torta del filtro se lavó con EtOAc (4 * 1,0 L) y las capas orgánicas combinadas se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía en columna sobre gel de sílice (gradiente PE/EtOAc 10 a 15%). El producto se trituró con PE (1,0 L) con agitación a 10 °C durante 2 hs. Los sólidos se recolectaron por filtración para obtener 4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperazin-1-carboxilato de terc-butilo (168 g, 76%) como un sólido blanquecino. RMN de 1H (CDCh) 87,62 (t, 1H), 7,41 a 7,49 (m, 2H), 7,38 (dd, 1H), 6,20 (dd, 2H), 5,45 (s, 2H), 3,37 a 3,57 (m, 8H), 1,49 (s, 9H).
Etapa 3:
A una solución de EtOH (2,8 ml, 48 mmol) en EtOAc (20 ml) se añadió cloruro de acetilo (2,0 ml, 28 mmol), gota a gota. Después de agitar durante 1 hora a 40 °C, se añadió en una porción 4-(6-((4-ciano-2-fluorobencil)oxi)piperazin-1-il) piperacilato de terc-butilo (1,75 g, 4,24 mmol) y la mezcla se agitó a 40 °C durante 2 hs. La reacción se dejó enfriar a TA y se agitó durante 1 hora. Se añadió EtOAc (10 ml) a la suspensión blanca y la suspensión espesa resultante se agitó vigorosamente a TA durante 1 hora. El sólido se recolectó por filtración para proporcionar la sal bis HCl del producto deseado Intermedio 13 (1,45 g, 89%) como un sólido. RMN de 1H (600 MHz, DMsO-d6 ) 89,45 (br s, 2H), 7,89 (d, 1H), 7,65 a 7,73 (m, 2H), 7,55 (m, 1H), 6,44 (d, 1H), 6,22 (d, 1H), 5,42 (s, 2H), 3,61 a 3,74 (m, 4H), 3,09 (br s, 4H).

Intermedio 14
Clorhidrato de (S)-1-(6-((4-cloro-2-fluorobencil)oxi)-5-fluoropiridin-2-il)-3-metilpiperazina
Etapa 1:
A una solución de 2,3,5-trifluoropiridina (1,5 g, 11 mmol) y alcohol 4-doro-2-fluorobencílico (1,81 g, 11,3 mmol) en NMP (20 ml) se añadió K2CO3 (4,67 g, 33,8 mmol) a 25 °C y la mezcla se agitó a 100 °C durante 16 hs. La mezcla se vertió en agua (30 ml) y posteriormente se extrajo con EtOAc (3 * 50 ml). Los extractos orgánicos combinados se lavaron con salmuera (3 * 40 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (gradiente de 0 a 5% de EtOAc/PE) para dar 2-((4-cloro-2-fluorobencil)oxi)-3,6-difluoropiridina (2,45 g, 80%) como un aceite incoloro. RMN de 1H (CDCh) 6 7,41 a 7,54 (m, 2H), 7,11 a 7,20 (m, 2H), 6,47 (ddd, 1H), 5,44 (s, 2H).
Etapa 2:
A una solución de 2-((4-cloro-2-fluorobencil)oxi)-3,6-difluoropiridina (200 mg, 0,731 mmol) y (S)-2-metilpiperazin-1-carboxilato de terc-butilo (161 mg, 0,804 mmol) en DMSO (3 ml) se añadió K2CO3 (303 mg, 2,19 mmol) a TA. La reacción se agitó a 120 °C durante 18 hs. La mezcla se vertió en agua (10 ml) y posteriormente se extrajo con EtOAc (3 * 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (3 * 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de Prep-TLC (15% de EtOAc/PE) para dar (S)-4-(6-((4-cloro-2-fluorobencil)oxi)-5-fluoropiridin-2-il)-2-metilpiperazin-1-carboxilato de tercbutilo (60 mg, 18%) como un aceite incoloro. LC-MS(eS+): 397,9 (M+H-tBu).
Etapa 3:
A una solución de (S)-4-(6-((4-cloro-2-fluorobencil)oxi)-5-fluoropiridin-2-il)-2-metilpiperazin-1-carboxilato (60 mg, 0,13 mmol) en DCM (3 ml) se añadió HCl/EtOAc (3 ml). La solución se agitó a 30 °C durante 0,5 hs. La suspensión se concentró a presión reducida para obtener el Intermedio 14 (50 mg, 89%) como un sólido. LC-MS(ES+): 353,9 (M+H).

Intermedio 15
Clorhidrato de (S)-1-(6-((4-cloro-2-fluorobencil)oxi)-3-fluoropiridin-2-il)-3-metilpiperazina
Etapa 1:
A una solución de 2,3,5-trifluoropiridina (500 mg, 3,76 mmol) y piperazin-1-carboxilato de terc-butilo (753 mg, 3,76 mmol) en MeCN (8 ml) se añadió Et3N (1,14 g, 11,3 mmol) a 30 °C y la reacción se calentó y se agitó a 70 °C durante 16 hs. La mezcla de reacción se vertió en agua (20 ml) y se extrajo con EtOAc (3 * 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (2 * 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (0 a 5% de EtOAc/PE) para dar (S)-4-(3,6-difluoropiridin-2-il)-2-metilpiperazin-1 -carboxilato de terc-butilo (870 mg, 74%) como un aceite marrón pálido. RMN de 1H (CDCh) 67,24 a 7,34 (m, 1H), 6,22 (ddd, 1H), 4,31 (br s, 1H), 4,09 (ddt, 1H), 3,92 (dt, 2H), 3,22 (td, 1H), 3,13 (dd, 1H), 2,93 (td, 1H), 1,49 (s, 9H), 1,23 (d, 3H).
Etapa 2:
A una solución de alcohol 4-cloro-2-fluorobencílico (102 mg, 0,638 mmol) en DMF (2 ml), se le añadió NaH (44,7 mg, 1,12 mmol, 60% en aceite mineral). La mezcla amarilla se agitó a 30 °C durante 15 min. A continuación se añadió una solución de (S)-4-(3,6-difluoropiridin-2-il)-2-metilpiperazin-1-carboxilato de terc-butilo (100 mg, 0,319 mmol) en DMF (2 ml) a TA. La mezcla de reacción se agitó a 90 °C durante 18 hs. La mezcla de reacción se vertió en agua (30 ml) y posteriormente se extrajo con EtOAc (3 * 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (3 * 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de Prep-TLC (EtOAc:PE 5:1) para dar (S)-4-(6-((4-cloro-2-fluorobencil)oxi)-3-fluoropiridin-2-il)-2-metilpiperazin-1-carboxilato de terc-butilo (136 mg, 47%) como un aceite incoloro. LC-MS(ES+): 397,9 (M+H - tBu).
Etapa 3:
A una solución de (S)-4-(6-((4-cloro-2-fluorobencil)oxi)-3-fluoropiridin-2-il)-2-metilpiperazin-1-carboxilato de terc-butilo (136 mg, 0,300 mmol) en Dc M (4 ml) se añadió HCl/EtOAc (4 ml). La solución se agitó a 30 °C durante 2 hs. La suspensión se concentró a presión reducida para dar el Intermedio 15 (132 mg, quant.) como un sólido amarillo pálido. LC-MS(ES+): 354,1 (M+H).

4-amino-3-(metilamino)benzoato de metilo
Etapa 1:
A una solución de 3-fluoro-4-nitrobenzoato de metilo (5,10 g, 25,6 mmol) en THF (60 ml) se añadió metilamina (38,4 ml, 76,8 mmol, 2 M en THF), gota a gota, durante 10 min. La solución de color amarillo pálido se volvió naranja intenso inmediatamente después de la adición y se agitó 2 hs a TA. La mezcla se diluyó con Et2O (100 ml) y la capa orgánica separada se lavó con agua (50 ml) y salmuera (50 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para obtener 5,26 g de 3-(metilamino)-4-nitrobenzoato de metilo (98%) como un sólido de color naranja intenso. RMN de 1H (400 MHz, CDCla) 88,21 (d, 1H), 7,99 (br s, 1H), 7,54 (d, 1H), 7,23 (dd, 1H), 3,94 (s, 3H), 3,08 (d, 3H); LC-MS(ES+): 211,1 (M+H).
Etapa 2:
Se disolvió 3-(metilamino)-4-nitrobenzoato de metilo (5,26 g, 25,0 mmol) en EtOH (150 ml). La solución se añadió a una botella Parr® de 500 ml previamente cargada con 1 g de Pd/C al 10% (50% de agua). La mezcla se agitó bajo una atmósfera de H2 de 50 psi durante 1 hora a TA. La mezcla se filtró y la torta del filtro se lavó con EtOH (100 ml). El filtrado incoloro se concentró a presión reducida para obtener 4,38 g del Intermedio 16 (97%) como un sólido blanquecino. RMN de 1H (400 MHz, CDCla) 87,47 (dd, 1H), 7,35 (d, 1H), 6,69 (d, 1H), 3,88 (s, 3H), 3,75 (br s, 2H), 3,22 (br s, 1H), 2,92 (s, 3H); MS(APCI+): 181,1 (M+H).

2-(Clorometil)-1-metil-1H-benzo[d]imidazol-6-carboxilato de metilo
El Intermedio 16 (206 mg, 1,14 mmol) se disolvió en dioxano (11,5 ml) y se trató con cloruro de cloroacetilo (109 pL, 1,37 mmol). La mezcla se agitó a 100 °C durante 3 hs y se enfrió a TA. Se añadió Et3N (0,8 ml, 7 mmol) y heptano (10 ml) y se filtró. El filtrado se concentró a presión reducida y el producto bruto se purificó por medio de cromatografía flash (columna de gel de sílice, 40% de EtOAc/heptanos) para obtener 120 mg del Intermedio 17 (44%). RMN de 1H (400 MHz, CDCh) 8 8,14 (s, 1H), 8,01 (d, 1H), 7,78 (d, 1H), 4,87 (s, 2H), 3,97 (s, 3H), 3,94 (s, 3H); LC-MS(ES+): 239,1 (M+H).

4-Amino-3-((2-metoxietil)amino)benzoato de metilo
Etapa 1:
A una solución incolora de 3-fluoro-4-nitrobenzoato de metilo (50 g, 250 mmol) en THF (400 ml) se añadió Et3N (40,7 g, 402 mmol, 55,8 ml) seguido por la adición de 2-metoxietilamina (30,2 g, 402 mmol) en THF (100 ml), gota a gota, a TA. La solución amarilla resultante se agitó a 55 °C durante 18 hs. La solución se enfrió a TA y se concentró a presión reducida para eliminar el THF. El sólido amarillo resultante se disolvió en EtOAc (800 ml) y se lavó con una solución ac. sat. de NH4O (250 ml). La fase ac. se separó y extrajo con EtOAc (200 ml). Las capas orgánicas combinadas se lavaron con salmuera (3 * 250 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para proporcionar 3-((2-metoxietil)amino)-4-nitrobenzoato de metilo (60,2 g, 94% de rendimiento) como un sólido amarillo. RMN de 1H (CDCh) 88,23 (d, 1H), 8,17 (br s, 1H), 7,58 (d, 1H), 7,25(dd, 1H), 3,95 (s, 3H), 3,69 a 3,73 (m, 2H), 3,56 (m, 2H), 3,45 (s, 3H); LC-MS(ES+): 255,4 (M+H).
Etapa 2:
A una solución de 3-((2-metoxietil)amino)-4-nitrobenzoato de metilo (30 g, 118 mmol) en MeOH (500 ml) se añadió Pd/C (10 g, 94 mmol). Esta reacción se agitó a TA bajo 15 psi de H2 durante 18 hs. La suspensión negra se filtró a través de Celite® y la torta del filtro se lavó con MeOH (500 ml). Los filtrados combinados se concentraron al vacío para dar el Intermedio 18 (26,5 g, quant.) como un aceite marrón que se solidificó al reposar. RMN de 1H (400 MHz, CDCla) 87,48 (dd, 1H), 7,36 (d, 1H), 6,69 (d, 1H), 3,87 (s, 3H), 3,77 (br.s, 2H), 3,68 (t, 2H), 3,41 (s, 3H), 3,32 (t, 2H); LC-MS(ES+): 224,7 (M+H).

Clorhidrato de 2-(clorometil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo
Una solución del Intermedio 18 (5,0 g, 24 mmol) en dioxano (100 ml) se calentó a 100 °C, se añadió una solución de anhídrido cloroacético (4,1 g, 24,5 mmol) en dioxano (60 ml) a través de un embudo de adición durante 10 hs y se agitó durante otras 12 hs a 100 °C. Al día siguiente, la reacción se enfrió a TA y el dioxano se eliminó a presión reducida. La mezcla de reacción cruda se disolvió en EtOAc y se lavó con una solución saturada de NaHCO3. La capa de EtOAc se separó, se secó sobre Na2SO4 y se filtró. Se añadió una solución de HCl 4 M en dioxano (1,1 equiv.) a la solución de EtOAc del producto con agitación constante. La sal HCl del producto deseado se precipitó como un sólido amarillo pálido. La suspensión se agitó durante 1 hora y el producto se recolectó por filtración para obtener el Intermedio 19 como un sólido amarillo (6,1 g 86%).RMN de 1H (600 MHz, CD3OD) 88,64 (s, 1H), 8,30 (d, 1H), 7,92 (d, 1H), 5,32 (s, 2H), 4,84 (m, 2H), 3,99 (s, 3H), 3,83 (t, 2H), 3,31 (s, 3H). LC-MS(ES+): 283,2 (M+H).

Intermedio 20
Metanosulfonato de (S)-oxetan-2-ilmetilo
Etapa 1:
A una solución de f-butóxido de potasio (670 g, 5,98 mol) en t-BuOH (5 L) se añadió yoduro de trimetilsulfoxonio (1,32 kg, 5,98 mol) a 25 °C. La mezcla se calentó a 60 °C y se agitó durante 30 min. A continuación se añadió (S)-2-((benzoxi)metil)oxirano (500 g, 2,99 mol). La solución se calentó a 80 °C durante 2 hs. La mezcla se enfrió a 25 °C y se filtró a través de Celite®. Los sólidos se lavaron con PE (3 * 2 L). El filtrado se trató con agua (10 L) y se extrajo con PE (2 * 5 L). La capa orgánica se lavó con salmuera, se secó, se filtró y se concentró in vacuo. El producto bruto se purificó por medio de cromatografía en columna (gradiente PE/EtoAc de 15:1 a 10:1) para obtener (S)-2-((benzoxi)metil)oxetano (280 g, 52,6%) como un aceite claro. RMN de 1H (400 MHz, CDCh) 6 7,15 a 7,34 (m, 5H), 4,90 (tdd, 1H), 4,44 a 4,67 (m, 4H), 3,49 a 3,63 (m, 2H), 2,44 a 2,66 (m, 2H).
Etapa 2:
Esta reacción se llevó a cabo en dos partidas paralelas; a continuación se presenta una partida de ejemplo: A una solución de (S)-2-((benzoxi)metil)oxetano (140 g, 780 mmol) en THF (1,4 L) se añadió Pd(OH)2 (14 g) bajo un manto de nitrógeno. La mezcla se calentó a 45 °C y se agitó bajo H2 (50 psi) durante 16 hs. La mezcla se enfrió a 25 °C y se filtró a través de Celite® para obtener el compuesto deseado (S)-oxetan-2-ilmetanol como una solución en THF. Se comprobó una pequeña alícuota por RMN de 1H y la solución restante se utilizó directamente en la siguiente etapa. RMN de 1H (400 MHz, DMSO-d6 ) 6 4,76 a 4,90 (m, 1H), 4,66 (tdd, 1H), 4,46 (ddd, 1H), 4,37 (td, 1H), 3,47 (dd, 2H), 2,32 a 2,58 (m, 2H).
Etapa 3:
Esta reacción se llevó a cabo en dos partidas paralelas; a continuación se presenta una partida de ejemplo: A una solución de (S)-oxetan-2-ilmetanol (de la Etapa 2, supuesta 69 g, 780 mmol) en THF (1,4 L) se añadió Et3N (197 g, 1,95 mol) a 0 °C. Se añadió anhídrido metanosulfónico (204 g, 1,17 mol), gota a gota, manteniendo la temperatura interna por debajo de 10 °C. La mezcla se agitó a 25 °C durante 2 hs. Se combinaron las dos partidas y la mezcla se trató con agua (1 L) y se separaron las capas. La fase ac. se extrajo con DCM (3 x 2 L). La solución orgánica combinada se secó, se filtró y se concentró a presión reducida. El producto bruto se purificó por medio de cromatografía en columna (gradiente EtOAc/PE 50 a 100%) para obtener el Intermedio 20 (250 g, 96% en dos etapas) como un aceite amarillo. RMN de 1H (400 MHz, CDCla) 64,98 a 5,09 (m, 1H), 4,69 (ddd, 1H), 4,59 (td, 1H), 4,37 (d, 2H), 3,11 (s, 3H), 2,72 a 2,82 (m, 1H), 2,64 (tdd, 1H).

(S)-4-Nitro-3-((oxetan-2-ilmetil)amino)benzoato de metilo
Etapa 1:
A una solución de metanosulfonato de (S)-oxetan-2-ilmetilo (180 g, 1,08 mol) en DMF (1,2 L), se añadió azida de sodio (105 g, 1,62 mol). La mezcla se calentó a 80 °C y se agitó durante 16 hs. La mezcla se enfrió a 0 °C y se trató con éter dietílico (1,5 L) y la suspensión resultante se agitó durante 30 min. Los sólidos se eliminaron por filtración y la torta del filtro se lavó con éter dietílico (2 * 200 ml). El éter dietílico se eliminó al vacío a 25 °C para obtener una solución de (S)-2-(azidometil)oxetano en DMF (~1,2 L), que se utilizó directamente en la siguiente etapa.
Etapa 2:
La reacción se llevó a cabo en tres partidas paralelas; a continuación se presenta una partida de ejemplo: A una solución de (S)-2-(azidometil)oxetano (supuesta 41 g, 360 mmol) en DMF (~400 ml) y THF (1 L) se añadió 10% de
Pd/C (50% en peso húmedo, 13 g) bajo un manto de nitrógeno. La mezcla se agitó a 25 °C bajo H2 (50 psi) durante 16 hs. La solución se filtró a través de Celite®, se añadió Pd/C al 10% (seco, 4,0 g) y la mezcla se agitó a 40 °C bajo H2 (50 psi) durante 3 hs, después de lo cual el análisis de TLC indicó la reacción completa. La mezcla se enfrió a 0 °C y se combinaron las tres partidas. La mezcla se filtró a través de Celite® para obtener una solución de (S)-2-(aminometil)oxetano en DMF (~1,4 L) y THF (~2,6 L), que se utilizó directamente en la siguiente etapa.
Etapa 3:
A una solución de (S)-2-(aminometil)oxetano (supuesta 94 g, 1,08 mol) en DMF (~1,4 L) y THF (~2,6 L) se añadió Et3N (327 g, 3,24 mol) y 3-fluoro-4-nitrobenzoato de metilo (200 g, 1,0 mol) a 25 °C. La mezcla se agitó a 25 °C durante 16 hs. La mezcla se concentró a presión reducida para eliminar el THF y la solución restante se diluyó con agua (1 L). La mezcla se extrajo con EtOAc (2 x 1,5 L). Los extractos orgánicos combinados se lavaron con salmuera (2 x), se secaron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía en columna (gradiente EtOAc/PE = 10 a 50%) para obtener el Intermedio 21 (158 g, 55%) como un sólido amarillo. RMN de 1H (600 MHz, CDCh) 88,38 (br s, 1H), 8,25 (d, 1H), 7,64 (s, 1H), 7,27 (d, 1H), 5,13 a 5,20 (m, 1H), 4,70 a 4,82 (m, 1H), 4,64 (td, 1H), 3,95 (s, 3H), 3,57 a 3,71 (m, 2H), 2,71 a 2,86 (m, 1H), 2,55 a 2,70 (m, 1H); MS(ES+) = 266,7.

(S)-4-Amino-3-((oxetan-2-ilmetil)amino)benzoato de metilo
El Intermedio 21 (15 g, 56 mmol) se disolvió en THF (100 ml) en un reactor Parr®. Se añadió Pd/C (10% p/p, 1,5 g) al reactor y la mezcla se agitó a TA bajo 50 psi de H2 durante 4 hs. La mezcla se filtró a través de Celite® y el filtrado se concentró a presión reducida para obtener el Intermedio 22 (12,3 g, 92%) como un sólido de color canela. RMN de 1H (600 MHz, CDCla) 87,49 (dd, 1H), 7,39 (d, 1H), 6,70 (d, 1H), 5,05 a 5,18 (m, 1H), 4,76 (ddd, 1H), 4,62 (dt, 1H), 3,87 (s, 3H), 3,42 a 3,50 (m, 1H), 3,34 a 3,40 (m, 1H), 2,71 a 2,82 (m, 1H), 2,60 (ddt, 1H).

(S)-2-(ClorometM)-1-(oxetan-2-NmetM)-1H-benzo[d]imidazol-6-carboxNato de metilo
A una solución del Intermedio 22 (127 g, 0,54 mol) en MeCN (500 ml) se añadió 2-cloro-1,1,1-trimetoxietano (76,2 ml, 0,57 mol) y pTSA-H2O (5,12 g, 26,9 mmol). La mezcla se calentó a 60 °C durante 1 hora. La reacción se enfrió a TA y se concentró a presión reducida.. El producto bruto resultante se trituró en EtOAc/heptano al 50%. Los sólidos se recolectaron por filtración para obtener el Intermedio 23 (79 g, 50%) como un sólido de color canela. RMN de 1H (600 MHz, CDCla) 88,12 (s, 1H), 8,00 (d, 1H), 7,79 (d, 1H), 5,16 a 5,26 (m, 1H), 5,03 (s, 2H), 4,57 a 4,66 (m, 2H), 4,48 a 4,56 (m, 1H), 4,33 (m, 1H), 3,95 (s, 3H), 2,71 a 2,81 (m, 1H), 2,36 a 2,47 (m, 1H).

6-Cloro-5-nitropicolinato de metilo
Etapa 1:
Se añadió lentamente 2-cloro-6-metil-3-nitropiridina (97 g, 560 mmol) a un matraz previamente cargado con 18 M H2SO4 (400 ml) con agitación. Se añadió trióxido de cromo (169 g, 1,69 mol) a la mezcla de reacción en pequeñas porciones manteniendo la temperatura por debajo de 50 °C. La mezcla de reacción se agitó a 15 °C durante 20 hs. La goma verde resultante se vertió en 2 kg de hielo y los sólidos resultantes se recolectaron por filtración y se secaron al vacío para producir 6-cloro-5-nitrógeno. La mezcla de reacción se agitó a 15 °C durante 20 hs. La goma verde resultante se vertió en 2 kg de hielo y los sólidos resultantes se recolectaron por filtración y se secaron al vacío para obtener ácido 6-cloro-5-nitropicolínico (103 g, 90%) como un sólido pálido. RMN de 1H (400 MHz, DMSO-d6 ) 88,70 (d, 1H), 8,24 (d, 1H).
Etapa 2:
A una suspensión de ácido 6-cloro-5-nitropicolínico (103 g, 508,51 mmol) en CH2Ch (1 L) se añadió cloruro de oxalilo (129 g, 1,02 mol) y DMF (6 ml) a 0 °C. La mezcla de reacción se agitó a 15 °C durante 1 hora. Se añadió MeOH (60 ml) a la mezcla de reacción a 15 °C. La solución se agitó a 15 °C durante 10 min más. La solución amarilla se concentró a presión reducida y el producto bruto resultante se purificó por cromatografía en columna (EtOAc/PE: 0 a 20%) para obtener el Intermedio 24 (106 g, 96%) como un sólido. RMN de 1H (400 MHz, CD3OD) 8 8,55 (d, 1H), 8,27 (d, 1H), 4,01 (s, 3H).

Intermedio 25
(S)-5-nitro-6-((oxetano-2-ilmetilo)amino)picolinato de metilo
Etapa 1:
Se preparó una solución de (S)-2-(aminometil)oxetano (supuesta 152 g, 1,7 mol) en DMF (3 L) y THF (3 L) a partir del Intermedio 20 como se describe para el Intermedio 21 (etapas 1 y 2). El Intermedio 24 (270 g, 1,25 mol) y Et3N (500 g, 5,1 mol) se añadieron a una solución del Intermedio 20 (152 g, 1,7 mol) en DMF (3 l ) y THF (3 L) a 25 °C. La mezcla se agitó a 25 °C durante 16 hs. La mezcla se agitó a 25 °C durante 16 hs. La mezcla se concentró a presión reducida para eliminar el THF y se añadió agua (5 L). La mezcla se extrajo con EtOAc (2 * 5 L) y las soluciones orgánicas combinadas se lavaron con salmuera (2 x), se secaron y se concentraron a presión reducida. El material bruto se combinó con un segundo partida de producto bruto de un experimento similar (70 g) y los sólidos se trituraron con PE:EtOAc (4:1, 500 ml) durante 2 hs. Los sólidos se recolectaron por filtración y se secaron para proporcionar el Intermedio 25 (304 g, 52%) como un sólido amarillo. RMN de 1H (400 MHz, CDCh) 88,58 (br s, 1H), 8,56 (d, 1H), 7,39 (d, 1H), 5,08 a 5,18 (m, 1H), 4,73 (ddd, 1H), 4,61 (td, 1H), 4,06 a 4,16 (m, 1H), 3,98 (s, 3H), 3,88 a 3,97 (m, 1H), 2,68 a 2,80 (m, 1H), 2,55 (tdd, 1H).

(S)-5-amino-6-((oxetan-2-ilmetil)amino)picolinato de metilo
El Intermedio 25 (10 g, 37 mmol) se suspendió en MeOH (150 ml) y se trató con Pd/C al 10% (1,0 g) y la mezcla se agitó a TA bajo 50 psi de H2 durante 4 hs. La mezcla se filtró a través de Celite® y el filtrado se concentró a presión reducida para producir el Intermedio 26 (8,4 g, 95%) como un aceite amarillo que se solidificó en reposo. RMN de 1H (600 MHz, CDCla) 87,49 (d, 1H), 6,86 (d, 1H), 5,06 a 5,15 (m, 1H), 4,68 a 4,77 (m, 1H), 4,53 a 4,63 (m, 2H), 3,91 (s, 3H), 3,80 a 3,86 (m, 2H), 3,72 (br s, 2H), 2,68 a 2,78 (m, 1H), 2,52 a 2,61 (m, 1H).

(S)-2-(Clorometil)-3-(oxetan-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxilato de metilo
En un matraz de 2L y 3 cuellos equipado con un agitador mecánico de cabeza se tomó el Intermedio 26 (43,0 g, 181 mmol) en THF (780 ml). La suspensión rosa pálida resultante se trató con una solución de anhídrido cloroacético (33,5 g, 190 mmol en 100 ml de THF) por medio de un embudo de adición durante 30 min. La solución ámbar clara resultante se agitó a TA durante 2 hs y posteriormente se calentó a 60 °C durante 7 hs. La mezcla de reacción se enfrió a TA. Se eliminaron aproximadamente 400 ml de disolvente de la reacción a presión reducida en un evaporador rotatorio. La solución resultante se diluyó con EtOAc, (500 ml) y se trató con una solución ac. sat. de NaHcO3 (200 ml). La mezcla bifásica se agitó a TA durante 30 min. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (500 ml). Las capas orgánicas combinadas se lavaron con salmuera (500 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para proporcionar el Intermedio 27 (52,5 g, 98% de rendimiento) como un sólido marrón amarillento. RMN de 1H (600 Mhz, CDCla) 88,14 (d, 2H), 5,19 a 5,28 (m, 1H), 4,99 a 5,16 (m, 2H), 4,70 a 4,88 (m, 2H), 4,55 a 4,67 (m, 1H), 4,24 a 4,44 (m, 1H), 4,01 (s, 3H), 2,70 a 2,88 (m, 1H), 2,37 a 2,53 (m, 1H); LC-MS(ES+): 296,4 (M+H).

cis
(+/-) 3-(((2-metociclopentilo)metilo)amino)-4-nitrobenzoato de metilo
Etapa 1:
A un matraz que contenía cis (+/-)-2-(aminometil)ciclopentan-1-ol (300 mg, 2,60 mmol), 3-fluoro-4-nitrobenzoato de metilo (571 mg, 2,87 mmol) y Et3N (1,1 ml, 7,8 mmol) se añadió DMF y la mezcla se agitó durante la noche. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. El extracto orgánico se concentró a presión reducida y el producto bruto se purificó por medio de cromatografía flash (EtOAc/heptanos) para obtener cis (+/-) 3-((-2-hidroxiciclopentilo)amino)-4-nitrobenzoato de metilo (493 mg, 64%). RMN de 1H (CDCh) 8 8,21 (br s, 1 H), 8,19 (d,
1H), 7,62 (s, 1H), 7,20 (d, 1H), 4,38 (br s, 1H), 3,94 (s, 3H), 3,60 (ddd, 1H), 3,38 a 3,50 (m, 1H), 2,13 a 2,25 (m, 1H), 1,84 a 2,01 (m, 3H), 1,57 a 1,78 (m, 4H).
Etapa 2:
A un matraz que contenía una solución de cis (+/-) 3-(((2-hidroxicidopentilo)metil)amino)-4-nitrobenzoato (0,48 g, 1,6 mmol) en DCM ( 50 ml) se añadió 1,8-bis(dimetilamino)naftaleno (0,35 g, 1,6 mmol). La solución se agitó durante 5 min y posteriormente se añadió tetrafluoroborato de trimetiloxonio (0,48 g, 3,3 mmol) en porciones durante 10 min. La mezcla de reacción se agitó 18 horas más a TA. Se añadió agua al matraz y la mezcla resultante se extrajo con DCM. Las capas orgánicas combinadas se filtraron y la solución se concentró a presión reducida. La mezcla cruda se purificó por cromatografía flash (EtOAc/heptanos) para obtener el Intermedio 28 (0,4 g, 80%). RMN de 1H (600 MHz, CDCla) 88,19 (d, 1H), 7,61 (s, 1H), 7,20 a 7,12 (m, 1H), 3,93 (s, 3H), 3,57 a 3,50 (m, 1H), 3,43 (dd, 6,3 Hz, 1H), 3,30 (s, 3H), 2,22 (d, 1H), 1,89 a 1,43 (m, 7H).

Intermedio 29
3-Fluoro-4-nitrobenzoato de tere-butilo
El ácido 3-fluoro-4-nitrobenzoico (2,60 g, 14,0 mmol) se disolvió en THF (30 ml), la mezcla se trató con anhídrido de Boc (6,13 g, 28,1 mmol) y DMAP (525 mg, 4,21 mmol), y se agitó a TA. Rápidamente se formó una suspensión espesa que se agitó durante 3 hs a 40 °C, tiempo durante el cual la suspensión espesa se convirtió en una solución de color canela. Tras concentrar la mezcla de reacción a presión reducida, el residuo se disolvió en EtOAc, se adsorbió sobre gel de sílice y se eluyó a través de una almohadilla corta de gel de sílice con EtOAc/Heptano al 50%. El filtrado se concentró a presión reducida para producir el Intermedio 29 (8,88 g, 68%) como un sólido amarillo. RMN de 1H (600 MHz, CDCla) 88,05 a 8,09 (m, 1H), 7,86 a 7,90 (m, 2H), 1,61 (s, 9H).

Intermedio 30
5-Bromo-N3-metilpiridin-2,3-diamina
Intermedio 315-Bromo-N3,6-dimetilpiridin-2,3-diamina
El Intermedio 30 se sintetizó de acuerdo con el procedimiento de la bibliografía (Choi, J. Y. et al. J. Med. Chem.
2012, 55, 852 a 870). El Intermedio 31 se sintetizó siguiendo el mismo procedimiento.

2-(Clorometil)-1-((1-metil-1H-imidazol-5-il)metil)-1 H-benzo[d]imidazol-6-carboxilato de metilo
Etapa 1:
A una solución incolora de 3-fluoro-4-nitrobenzoato de metilo (1,0 g, 5,0 mmol) en DMF (60 ml) se añadió lentamente (1-metil-1H-imidazol-5-il)metanamina (670 mg, 6,0 mmol) y Et3N (762 mg, 7,53 mmol). La solución se agitó a 60 °C durante 16 hs. La mezcla de reacción se vertió en H2O (30 ml) y se extrajo con DCM (3 * 30 ml). Los extractos
orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (20% de MeOH/DCM). El sólido amarillo obtenido se trituró con 30:1 PE/EtOAc para obtener 3-(((1-metil-1H-imidazol-5-il)metil)amino)-4-nitrobenzoato de metilo (1,2 g, 82%) como un sólido amarillo. RMN de 1H (CDCla) 88,26 (d, 1H), 7,96 (br s, 1H), 7,71 (d, 1H), 7,50 (s, 1H), 7,35 (dd, 1H), 7,13 (s, 1H), 4,55 (d, 2H), 3,97 (s, 3H), 3,68 (s, 3H).
Etapa 2:
A una suspensión amarilla de 3-((1-metil-1H-imidazol-5-il)metil)amino)-4-nitrobenzoato de metilo (5,46 g, 18,8 mmol) en MeOH (160 ml) se añadió Pd/C húmedo al 10% (1 g). La mezcla se agitó bajo 1 atm de H2 durante 36 hs a 20 °C. La mezcla de reacción se filtró y la torta del filtro se lavó con MeOH (200 ml). El filtrado se concentró a presión reducida para obtener 4-amino-3-((1-metil-1H-imidazol-5-il)amino)benzoato de metilo (4,8 g, 98%) como un sólido marrón. RMN de 1H (DMSO-d6 ) 8 7,56 (s, 1H), 7,18 (d, 1H), 7,13 (s, 1H), 6,87 (s, 1H), 6,55 (d, 1H), 5,50 (s, 2H), 4,84 (t, 1H), 4,23 (d, 2H), 3,73 (s, 3H), 3,63 (s, 3H).
Etapa 3:
Una mezcla roja de 4-amino-3-((1-metil-1H-imidazol-5-il)amino)benzoato de metilo (780 mg, 3,00 mmol) y ácido 2-hidroxiacético (342 mg, 4,49 mmol) en mesitileno (8 ml) se agitó a 140 °C bajo N2 durante 14 hs y a 25 °C durante 48 hs. La solución amarilla clara se decantó para dar un residuo marrón que se disolvió en MeOH (50 ml) y se concentró a presión reducida. El producto bruto se purificó por cromatografía flash (20% de MeOH/DCM) para dar 2-(hidroximetil)-1-((1-metil-1H-imidazol-5-il)metil)-1H-benzo[d]imidazol-6-carboxilato de metilo (318 mg, 35%) como una espuma amarilla. RMN de 1H (DMSO-d6 ) 88,13 (d, 1H), 7,83 (dd, 1H), 7,71 (d, 1H), 7,60 (s, 1H), 6,59 (s, 1H), 5,69 (s, 2H), 4,76 (s, 2H), 3,91 (s, 1H), 3,84 (s, 3H), 3,53 (s, 3H).
Etapa 4:
A una suspensión amarilla de 2-(hidroximetil)-1-((1-metil-1H-imidazol-5-il)metil)-1H-benzo[d]imidazol-6-carboxilato (500 mg, 1,66 mmol) en DCM (10 ml) y DMF (3 ml) se añadió SOCh (990 mg, 0,60 ml, 8,32 mmol), gota a gota, a TA. La mezcla de reacción se agitó a TA durante 1 hora, se concentró a presión reducida y el residuo marrón resultante se trituró con DCM (10 ml). Los sólidos se recolectaron por filtración, se lavaron con DCM (5 ml) y se secaron al vacío para dar el Intermedio 32 (431 mg, 73%) como un sólido blanquecino. RMN de 1H (400 MHz, DMSO-d6 ) 89,17 (s, 1H), 8,31 (s, 1H), 7,91 a 7,99 (m, 1H), 7,77 a 7,87 (m, 1H), 7,11 (s, 1H), 5,92 (s, 2H), 5,13 (s, 2H),) 3,87 (s, 3H), 3,86 (s, 3H); MS(ES+): 319,0 (M+H).
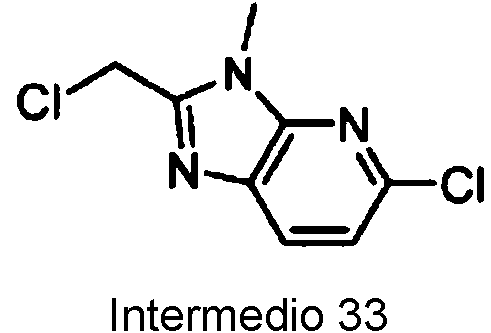
5-Cloro-2-(clorometil)-3-metil-3H-imidazo[4,5-b]piridina
Etapa 1:
A una suspensión de 2,6-dicloro-3-nitropiridina (200 g, 1,04 mol) y Na2CO3 (132 g, 1,24 mol) en EtOH (1 L) se añadió 2,0 M MeNH2 en THF (622 ml, 1,24 mol), gota a gota, a 0 °C por medio de una jeringa. Tras la adición, la mezcla de reacción se agitó a 18 °C durante 6 hs. La mezcla amarilla se filtró y el filtrado se concentró a presión reducida para dar un sólido amarillo. El producto bruto se purificó por medio de cromatografía flash (PE/EtOAc 0 a 5%) para obtener 6-cloro-N-metil-3-nitropiridin-2-amina (158 g, 81% de rendimiento) como un sólido amarillo. RMN de 1H (DMSO-d6 ) 88,72 (br s, 1H), 8,41 (d, 1H), 6,76 (d, 1H), 3,00 (d, 3H).
Etapa 2:
A una solución de 6-bromo-4-cloro-5-fluoro-3-metilanilina (-2) (15,8 g, 84,2 mmol) en AcOH (20 ml), se añadió NaNO2 (15,4 g, 276 mmol). La mezcla amarilla se agitó a 80 °C durante 3 hs. La reacción se enfrió a TA y se filtró. La torta del filtro se lavó con EtOAc (2 x 100). Las capas orgánicas combinadas se concentraron a presión reducida y el producto bruto se purificó por medio de cromatografía flash (120 g de gel de sílice, 50% de EtOAc/PE) para obtener 3-amino-6-cloro-2-metilaminopiridina (8,40 g, 63% de rendimiento) como un sólido marrón. RMN de 1H (CDCla) 86,80 (d, 1H), 6,50 (d, 1H), 3,39 (br s, 2H), 3,01 (s, 3H).
Etapa 3:
A una solución de 3-amino-6-cloro-2-metilaminopiridina (50,0 g, 317 mmol) en dioxano (1,2 L) se añadió cloruro de cloroacetilo (55,5 ml, 698 mmol) y la mezcla se agitó a 15 °C durante 50 min. La mezcla marrón se concentró a
presión reducida para dar un sólido marrón que se tomó en TFA (1,2 L) y se agitó a 80 °C durante 60 hs. La mezcla se concentró a presión reducida para dar un aceite marrón. El aceite se diluyó con EtOAc (1 L) y se neutralizó con una solución ac. sat. de NaHCO3. Cuando la evolución de CO2 disminuyó, las capas se separaron y la capa ac. se extrajo con EtOAc (200 ml). Los extractos orgánicos se combinaron, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente 10 a 25% de EtOAc/PE) para obtener el Intermedio 33 (61,0 g, 79% de rendimiento) como un sólido amarillo. RMN de 1H (400 MHz, DMSO-d6 ) 88,13 (d, 1H), 7,37 (d, 1H), 5,11 (s, 2H), 3,84 (s, 3H).

2-((4-(6-((4-Cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo
A una mezcla del Intermedio 3 (13,0 g, 23,8 mmol) y el Intermedio 19 (6,72 g, 23,8 mmol) y K2CO3 (16,4 g, 119 mmol) en MeCN (200 ml) se agitó a 50 °C durante 12 hs. La mezcla se enfrió a TA y se vertió en agua (200 ml). La mezcla se extrajo con EtOAc (5 * 500 ml), y las capas orgánicas combinadas se lavaron con salmuera (2 * 500 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo bruto se purificó por medio de cromatografía flash (120 g de gel de sílice, 0 a 2% de gradiente de MeOH/DCM) para obtener el Intermedio 34 (12,5 g, 93%) como un sólido amarillo pálido. RMN de 1H (400 MHz, CDCh) 88,16 (s, 1H), 7,97 (d, 1H), 7,75 (d, 1H), 7,50 (t, 1H), 7,44 (t, 1H), 7,11 (m, 2H), 6,73 (d, 1H), 6,61 (d, 1H), 5,41 (s, 2H), 4,64 (t, 2H), 3,96 (s, 3H), 3,92 (s, 2H), 3,79 (t, 2H), 3,31 (s, 3H), 2,99 (d, 2H), 2,58 a 2,67 (m, 1H), 2,29 (t, 2H), 1,78 a 1,91 (m, 4H).

1-(2-Metoxietil)-2-((4-(6-oxo-1,6-dihidropiridin-2-il)piperidin-1-il)metil)-1H-benzo[d]imidazol-6-carboxilato de metilo
A una suspensión agitada del Intermedio 34 (500 mg, 0,88 mmol) en MeOH (10 ml) se añadió HCl 4 M en dioxano (4,5 ml, 20 mmol). La reacción se calentó a 70 °C y se agitó durante 18 hs. A continuación, la mezcla se enfrió a TA y se concentró a presión reducida. El residuo se tomó en una solución ac. sat. de NaHCO3 y se extrajo con DCM (3 x). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se disolvió en un mínimo de DCM y se añadió lentamente PE hasta que se formó el precipitado. La mezcla se agitó para granular sólidos durante 2 hs. Los sólidos se aislaron por filtración y se lavaron con Pe para dar el Intermedio 35 (280 mg, 75%) como un sólido blanquecino. RMN de 1H (600 MHz, CDCh) 8 11,43 (br s, 1H), 8,13 (s, 1H), 7,96 (d, 1H), 7,73 (d, 1H), 7,36 (dd, 1H), 6,39 (d, 1H), 6,02 (d, 1H), 4,61 (t, 2H), 3,96 (s, 3H), 3,92 (s, 2H), 3,75 (t, 2H), 3,29 (s, 3H), 2,99 (d, 2H), 2,51 (t, 1H), 2,30 (t, 2H), 1,93 (d, 2H), 1,72 (qd, 2H).

2-((4-(6-Cloropiridin-2-il)piperidin-1-il)metil)-1-metil-1H-benzo[d]imidazol-6-carboxilato de metilo
Etapa 1:
A una solución incolora del Intermedio 2 (6,00 g, 20,2 mmol) en DCM (60 ml) se añadió HCl/EtOAc 4 M (60 ml) y la solución se volvió turbia. La suspensión se agitó a 20 °C durante 2 hs y, a continuación, se concentró a presión reducida para dar clorhidrato de 2-cloro-6-(piperidin-4-il)piridina (5,45 g, 99%) como un sólido. RMN de 1H (DMSO-d6 ) 89,32 (br s, 1H), 8,95 (br s, 1H), 7,83 (t, 1H), 7,37 (d, 1H), 7,31 (d, 1H), 3,31 (d, 2H), 2,89 a 3,06 (m, 3H), 1,85 a 2,04 (m, 4H).
Etapa 2:
A una mezcla de clorhidrato de 2-cloro-6-(piperidin-4-il)piridina (5,45 g, 20,2 mmol) y K2CO3 (8,38 g, 60,6 mmol) en DMF (50 ml) se añadió 2-bromoacetato de etilo (4,05 g, 24,3 mmol). La mezcla se agitó a 20 °C durante 2 hs y posteriormente se diluyó con EtOAc (300 ml) y se lavó con agua (100 ml). La capa orgánica se lavó con salmuera (200 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente de EtOAc/PE de 5 a 15%) para obtener 2-(4-(6-cloropiridin-2-il)piperidin-1-il)acetato de etilo (5,44 g, 95%) como un aceite amarillo. RMN de 1H (CDCh) 8 7,59 (t, 1H), 7,16 (d, 1H), 7,11 (d, 1H), 4,21 (q, 2H), 3,26 (s, 2H), 3,08 (d, 2H), 2,72 (tt, 1H), 2,31 (dt, 2H), 1,82 a 2,02 (m, 4H), 1,29 (t, 3H).
Etapa 3:
A una solución de 2-(4-(6-cloropiridin-2-il)piperidin-1-il)acetato de etilo (5,44 g, 19,2 mmol) en EtOH (50 ml) se añadió HCl 5 M (10,1 ml, 121 mmol). La solución se agitó a 25 °C durante 2 hs. La mezcla de reacción se apagó con HCl 1 M y se extrajo con DCM/MeOH (10:1, 5 * 80 ml). Los extractos orgánicos combinados se secaron sobre MgSO4, se filtraron y se concentraron a presión reducida para obtener ácido 2-(4-(6-cloropiridin-2-il)piperidin-1-il)acético (4,50 g, 92%) como un sólido amarillo. RMN de 1H (CD3OD) 87,71 (t, 1H), 7,24 (d, 2H), 3,20 (d, 2H), 3,13 (br s, 2H), 2,70 a 2,83 (m, 1H), 2,29 (br s, 2H), 1,83 a 2,06 (m, 4H).
Etapa 4:
A una solución amarilla de ácido 2-(4-(6-cloropiridin-2-il)piperidin-1-il)acético (4,50 g, 17,7 mmol) y el Intermedio 16 (3,50 g, 19,4 mmol) en DMF (50 ml) se añadió HATU (8,06 g, 21,2 mmol) a tA. La mezcla de reacción se agitó a 15 °C durante 20 min y posteriormente se añadió Et3N (3,58 g, 35,3 mmol). La mezcla amarilla se agitó a 50 °C durante 2 hs. La mezcla marrón resultante se vertió en agua (160 ml) y se extrajo con EtOAc (3 * 100 ml). Los extractos orgánicos combinados se lavaron con salmuera (3 * 100 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente de MeOH/DCM de 0 a 5%) para obtener 4-amino-3-(2-(4-(6-cloropiridin-2-il)piperidin-1-il)-N-metilacetamido)benzoato de metilo (7,37 g, cantidad) como un aceite amarillo. lC-MS(ES+): 417,1 (M+H).
Etapa 5:
Una mezcla de 4-amino-3-(2-(6-cloropiridin-2-il)piperidin-1-il)-N-metilacetamido)benzoato de metilo (7,37 g, 17,7 mmol) en AcOH (100 ml) se agitó a 60 °C durante 16 hs. La mezcla marrón se concentró a presión reducida para dar un aceite marrón que se tomó en EtOAc (300 ml) y se lavó con una solución ac. sat. de NaHCO3 (100 ml). La capa orgánica se lavó con salmuera (3 * 100 ml), se secó sobre MgSO4, se filtró y se concentró a presión reducida. El producto bruto se purificó por cromatografía flash (gradiente EtOAc/PE 0 a 50%) para obtener el Intermedio 36 (3,51 g, 50%) como un sólido amarillo. RMN de 1H (CDCla) 8 RMN de 1H (400 MHz, CDCla) 88,14 (s, 1H), 7,98 (d, 1H), 7,75 (d, 1H), 7,58 (t, 1H), 7,17 (d, 1H), 7,10 (d, 1H), 3,99 (s, 3H), 3,97 (s, 3H), 3,95 (br s, 2H), 3,09 (d, 2H), 2,77 (br s, 1H), 2,43 (br s, 2H), 1,83 a 2,04 (m, 4H); LC-MS(ES+): 399,1 (M+H).

Ejemplo 1A-01
Clorhidrato de ácido 2-((4-(6-((4-Cloro-2-fluorobencM)oxi)pmdm-2-N)piperidm-1-N)metM)-1-metiMH-benzo[d]imidazol-6-carboxílico
Etapa 1:
El Intermedio 17 (115 mg, 0,482 mmol), el Intermedio 3 (178 mg, 0,554 mmol) y el K2CO3 (133 mg, 0,96 mmol) se combinaron en MeCN (4,8 ml) y la mezcla se dejó agitar a 35 °C durante 3 hs. La reacción se enfrió a TA, se diluyó con EtOAc y se extrajo con agua. La capa orgánica se secó sobre MgSO4, se filtró y se concentró a presión reducida. El producto bruto resultante se purificó por medio de cromatografía flash (24 g de sílice, de 0 a 100% de EtOAc/heptano) para obtener 215 mg de 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-benzo[d]imidazol-6-carboxilato de metilo (85%) como una espuma blanca. RMN de 1H (400 MHz, CDCh) 88,10 (br s, 1H), 7,95 (d, 1H), 7,72 (d, 1H), 7,51 a 7,36 (m, 2H), 7,07 (br s, 2H), 6,71 (br s, 1H), 6,57 (d, 1H), 5,38 (br s, 2H), 3,95 (s, 3H), 3,93 (s, 3H), 3,84 (br s, 2H), 2,97 (br s, 2H), 2,59 (br s, 1H), 2,27 (br s, 2H), 1,75 a 1,93 (m, 4H); LC-MS(ES+): 523,3 (M+H).
Etapa 2:
El 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-benzo[d]imidazol-6-carboxilato de metilo (215 mg, 0,411 mmol) se suspendió en MeoH (4 ml) y se trató con NaOH 2 M (820 pL, 1,64 mmol). La reacción se dejó en agitación a 40 °C durante 3 hs y 14 hs a TA. La reacción se calentó de nuevo hasta 40 °C y se acidificó con HCl 1 M (2,50 ml, 2,50 mmol). La mezcla se dejó enfriar hasta TA y, cuando empezó a formarse un precipitado, se sopló vapor de N2 sobre la reacción para eliminar aproximadamente la mitad del MeOH. El sólido se recolectó por filtración, se lavó con H2O (2 * 2 ml) y se secó bajo N2 para obtener el Ejemplo 1A-01 (155 mg, 69%) como un sólido. RMN de 1H (600 MHz, DMSO-d6) 812,74 (br s, 1H), 8,15 (s, 1H), 7,79 (d, 1H), 7,58 a 7,65 (m, 2H), 7,54 (t, 1H), 7,43 (d, 1H), 7,27 (d, 1H), 6,85 (d, 1H), 6,65 (d, 1H), 5,34 (s, 2H), 3,94 (s, 3H), 3,82 (s, 2H), 2,93 (d, 2H), 2,57 (t, 1H), 2,19 (t, 2H), 1,73 a 1,80 (m, 2H), 1,64 a 1,73 (m, 2H); LC-MS(ES+): 509,2 (M+H).
Los compuestos enumerados en la Tabla 1 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis del Compuesto 1A-01 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron por medio de procedimientos muy conocidos por los expertos en la técnica y pueden incluir la cromatografía en gel de sílice, la HPLC o la cristalización a partir de la mezcla de reacción. Los compuestos finales se pueden haber aislado como neutros o como sales de ácidos o bases.
Tabla 1





Ejemplo 2A-01
Clorhidrato de ácido 2-((4-(6-((4-doro-2-fluorobencM)oxi)pmdm-2-N)piperidm-1-N)metM)-3-metN-3H-imidazo[4,5-b]piridin-5-carboxílico
Etapa 1:
Una mezcla amarilla del Intermedio 3 (92,3 g, 119 mmol, 4 eq de sal TFA), el Intermedio 33 (25,9 g, 120 mmol) y K2CO3 (98,5 g, 713 mmol) en MeCN (300 ml) se agitó a 50 °C durante 16 hs. La mezcla amarilla se vertió en agua (300 ml) y se extrajo con EtOAc (3 x 500 ml). Las capas orgánicas combinadas se lavaron con salmuera (500 ml), se secaron sobre MgSO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente de MeOH/DCM 0 a 5%) para obtener 5-cloro-2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-3-metil-3H-imidazo[4,5-b]piridina (59,0 g, 99%) como un sólido amarillo. RMN de 1H (400 MHz, CDCh) 87,92 (d, 1H), 7,49 (m, 1H), 7,43 (m, 1H), 7,21 (d, 1H), 7,10 a 7,13 (m, 1H), 7,09 (d, 1H), 6,72 (d, 1H), 6,60 (d, 1H), 5,40 (s, 2H), 3,98 (s, 3H), 3,84 (s, 2H), 2,97 (d, 2H), 2,51 a 2,73 (m, 1H), 2,29 (m, 2H), 1,73 a 1,97 (m, 4H).
Etapa 2:
Una solución amarilla de 5-cloro-2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-3-metil-3H-imidazo[4,5-b]piridina (59,0 g, 118 mmol), Dp Pp (6,80 g, 16,5 mmol), Pd(OAc)2 (3,65 g, 16,3 mmol) y Et3N (125 g, 1240 mmol) en MeOH (800 ml) y DMF (100 ml) se agitó a 80 °C bajo 50 psi de CO durante 16 hs. La solución naranja resultante se concentró a presión reducida hasta obtener un aceite marrón, que se diluyó con EtOAc (300 ml) y se lavó con agua (200 ml). La capa orgánica se lavó con salmuera (2 x 200 ml), se secó sobre MgSO4, se filtró y se concentró a presión reducida. El producto bruto se combinó con el producto de una reacción similar a escala de 11 g y se purificó por medio de cromatografía flash (gradiente 50 a 100% de EtOAc/PE) para obtener 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-3-metil-3H-imidazo[4,5-b]piridin-5-carboxilato de metilo (62,6 g, 85%) como un sólido amarillo pálido. RMN de 1H (400 MHz, CDCla) 88,13 (d, 1H), 8,07 (d, 1H), 7,49 (t, 1H), 7,43 (t, 1H), 7,12 (t, 1H), 7,08 a 7,11 (m, 1H), 6,73 (d, 1H), 6,60 (d, 1H), 5,40 (s, 2H), 4,09 (s, 3H), 4,03 (s, 3H), 3,90 (s, 2H), 2,93 a 3,05 (m, 2H), 2,55 a 2,69 (m, 1H), 2,31 (dt, 2H), 1,79 a 1,97 (m, 4H).
Etapa 3:
El 2-((6-((4-doro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-3-metil-3H-imidazo[4,5-b]piridin-5-carboxilato de metilo (57,0 g, 109 mmol) se suspendió en MeOH (1 L) y se trató con NaOH 2 M (218 ml). La suspensión espesa se agitó 5 min a TA y posteriormente se calentó a 85 °C durante 3 hs. La mezcla se filtró a través de Celite® y el filtrado claro se recalentó a 70 °C. La reacción se acidificó con HCl 2 M (272 ml) y se dejó enfriar a TA. Se formó un sólido y se dejó que la suspensión espesa se agitara durante 18 horas a TA. Los sólidos se recolectaron por filtración para entregar el Ejemplo 2A-01 (57,1 g, 96%) como un sólido blanco marfil. RMN de 1H (400 MHz, DMSO-d6 ) 813,20 (br s, 1H), 11,07 (br s, 1H), 8,27 (d, 1H), 8,07 (d, 1H), 7,57 a 7,78 (m, 2H), 7,47 (m, 1H), 7,32 (m, 1H), 6,92 (d, 1H), 6,74 (d, 1H), 5,40 (s, 2H), 4,84 (br s, 2H), 3,97 (s, 3H), 3,86 (br s, 2H), 3,37 (br s, 2H), 2,93 (br s, 1H), 1,85 a 2,36 (m, 4H); LC-MS(ES+): 510,2 (M+H).

Clorhidrato de ácido 2-((4-(6-((4-doro-2-fluorobencN)oxi)piridm-2-N)piperidm-1-N)metM)-1-(2-metoxietM)-1H-imidazo[4,5-b]piridin-6-carboxílico Ejemplo 2A-02
Etapa 1:
A un matraz que contiene una solución de ácido metoxiacético (1,00 g, 11,1 mmol) en DMF (30 ml) se añadió HATU (6,33 g, 16,7 mmol) y Et3N (3,37 g, 33,3 mmol). Tras agitar durante 20 min, se añadió en porciones 2,3-diamino-5-bromopiridina (2,3 g, 12 mmol) y la mezcla de reacción resultante se agitó durante la noche. Tras 15 hs, se añadió agua y la solución se extrajo con EtOAc. Las capas orgánicas combinadas se secaron y el disolvente se eliminó a presión reducida. El compuesto bruto se purificó por cromatografía flash (gradiente de 0 a 80% de EtOAc/heptano) para producir N-(2-amino-5-bromo-piridin-3-il)-2-metoxiacetamida (2,3 g, 80%). RMN de 1H (400 MHz, CDCh) 88,09 (s, 1H), 8,06 (d, 1H), 8,03 (s, 1H), 7,83 (d, 1H), 4,08 (s, 2H), 3,53 (s, 3H); LC-MS(ES+): 260,2 (M+H).
Etapa 2:
A una solución de N-(2-amino-5-bromopiridin-3-il)-2-metoxiacetamida (3,3 g, 13 mmol) en THF se añadió una solución 1 M de BH3 en THF (14 ml) durante 10 min, y se agitó a TA durante la noche, se añadió agua a la reacción lentamente para apagar el exceso de borano, y la mezcla se extrajo con EtOAc. La capa de EtOAc se secó y se concentró a presión reducida. El producto bruto se disolvió en MeOH y se añadió HCl en dioxano (1,0 equiv) y se agitó durante 2 hs. El exceso de metanol se eliminó a presión reducida para obtener el producto bruto. El compuesto se purificó por cromatografía flash con un gradiente de 0 a 70% de EtOAc en heptanos para obtener 5-bromo-W3-(2-metoxietil)piridin-2,3-diamina como un aceite marrón (1,1 g, 35%). RMN de 1H (600 MHz, CDCh) 87,83 (d, 1H), 6,95 (d, 1H), 5,56 (s, 2H), 3,77 (t, 1H), 3,66 (t, 2H), 3,42 (s, 3H), 3,22 (q, 2H); LC-MS(ES+): 246,1.
Etapa 3:
Se tomó 5-Bromo-W3-(2-metoxietil)piridin-2,3-diamina (400 mg, 1,63 mmol) en 8 ml de dioxano (8 ml) y se trató con cloruro de cloroacetilo (0,284 ml, 3,58 mmol). La mezcla se agitó a TA. El disolvente se eliminó a presión reducida y el residuo resultante se tomó en TFA (8 ml) y se agitó a 80 °C durante 18 hs. La reacción se enfrió a TA y se concentró a presión reducida. El aceite marrón resultante se recolectó en EtOAc (50 ml) y se neutralizó con una solución ac. sat. de NaHCO3. Después de que la evolución de CO2 hubiera disminuido, se separaron las capas y se extrajo la capa acuosa con EtOAc adicional (20 ml). Los extractos orgánicos se combinaron, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto resultante se purificó por medio de cromatografía flash (gradiente 0 a 80% de EtOAc/heptano) para obtener 6-bromo-2-(clorometil)-1-(2-metoxietil)-1H-imidazo[4,5-b]piridina (176 mg, 36%) como un sólido de color canela. RMN de 1H (600 MHz, CDCh) 88,59 (s, 1H), 7,90 (s, 1H), 4,93 (s, 2H), 4,45 (m, 2H), 3,72 (m, 2H), 3,29 (s, 3H); LC-MS(ES+): 306,1 (M+H).
Etapa 4:
Una mezcla del Intermedio 3 (294 mg, 0,97 mmol, base libre), 6-bromo-2-(clorometil)-1-(2-metoxietil)-1H-imidazo[4,5-b]piridina (341 mg, 1,06 mmol), KI (48 mg, 0,29 mmol) y W,W-diisopropiletilamina (0,51 ml, 0,97 mmol) en MeCN (8 ml) se agitó a 60 °C durante 16 hs. La mezcla se vertió en agua y se extrajo con EtOAc. La capa orgánica se secó, se filtró y se concentró a presión reducida. El producto bruto se purificó por medio de cromatografía flash (gradiente 0 a 100% de EtOAc/heptano) para obtener 6-bromo-2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(2-metoxietil)-1H-imidazo[4,5-b]piridina (406 mg, 71%) como un aceite de color canela. RMN de 1H (600
MHz, CDCla) 88,54 (s, 1H), 7,91 (s, 1H), 7,50 (m, 1H), 7,43 (m, 1H), 7,11 (m, 2H), 6,73 (d, 1H), 6,60 (d, 1H), 5,41 (s, 2H), 4,54 (m, 2H), 3,92 (s, 2H), 3,76 (m, 2H), 3,30 (s, 3H), 2,97 (d, 2H), 2,58 a 2,67 (m, 1H), 2,31 (m, 2H), 1,76 a 1,93 (m, 4H).
Etapa 5:
A una mezcla de 6-bromo-2-((4-(6-((4-doro-2-fluorobencN)oxi)piridin-2-N)pipendin-1-N)metN)-1-(2-metoxietN)-1H-imidazo[4,5-b]piridina (610 mg, 1,04 mmol), acetato de paladio(M) (47 mg, 0,21 mmol), y dppp (128 mg, 0,31 mmol) se añadió d Mf (4 ml), MeOH (16 ml) y trimetilamina (1,44 ml, 10,4 mmol). La reacción se calentó a 80 °C con agitación bajo una atmósfera de CO de 50 psi durante 20 hs. La mezcla de reacción se enfrió a TA y se repartió entre agua y EtOAc. La capa orgánica se separó y se secó sobre MgSO4, se filtró y se concentró a presión reducida. El material bruto se purificó por medio de cromatografía flash (gradiente de 0 a 5% de MeOH en DCM) para producir 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(2-metoxietil)-1H-imidazo[4,5-b]piridin-6-carboxilato de metilo (540 mg, 92%) como una goma color canela. RMN de 1H (600 MHz, CDCla) 89,18 (s, 1H), 8,39 (s, 1H), 7,49 (t, 1H), 7,43 (t, 1H), 7,10 (t, 2H), 6,73 (d, 1H), 6,60 (d, 1H), 5,40 (s, 2H), 4,64 (t, 2H), 4,00 a 3,90 (m, 5H), 3,78 (t, 2H), 3,29 (s, 3H), 2,99 (d, 2H), 2,62 (m, 1H), 2,27 a 2,40 (m, 2H), 1,79 a 1,91 (m, 4H).
Etapa 6:
A una solución de 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(2-metoxietil)-1H-imidazo[4,5-b]piridin-6-carboxilato de metilo (2,0 g de 3,5 mmol) en MeOH (60 ml) se añadió NaOH 2 M (8,9 ml) y la mezcla se calentó a 60 °C durante 1 hora,0 g, 3,5 mmol) en MeOH (60 ml) se añadió NaOH 2 M (8,9 ml) y la mezcla se calentó a 60 °C durante 1 hora. La reacción se enfrió a TA y se acidificó con HCl 1 M hasta pH ~4. La mezcla se concentró a presión reducida para eliminar el MeOH y el sólido se recolectó por filtración y se secó al vacío para obtener el Ejemplo 2A-02 (1,7 g 82%) como un sólido. RMN de 1H (400 MHz, DMSO-d6 ) 8 13,35 (br s, 1H), 10,90 (br s, 1H), 9,01 (d, 1H), 8,70 (d, 1H), 7,69 (t, 1H), 7,63 (t, 1H), 7,47 (dd, 1H), 7,32 (dd, 1H), 6,93 (d, 1H), 6,73 (d, 1H), 5,40 (s, 2H), 4,86 (br s, 2H), 4,70 (br s, 2H), 3,81 (br s, 2H), 3,65 (m, 2H), 3,20 (s, 3H), 2,94 (br s, 1H), 2,08 a 2,25 (m, 4H); LC-MS(ES+): 554,2 (M+H).

Ejemplo 2A-03
Ácido 2-((4-(6-((4-doro-2-fluorobencM)oxi)pmdm-2-N)piperídm-1-N)metM)-1-metiMH-imidazo[4,5-c]pmdm-6-carboxílico
Etapa 1:
A una solución agitada de 2,4-dibromo-5-nitropiridina (0,21 g, 0,72 mmol) en THF (4,1 ml) se añadió metil amina en THF (2 M, 1,2 ml, 2,5 mmol). Tras 0,5 hs, la solución se diluyó con agua (5 ml). La fase acuosa se extrajo con EtOAc (3 x 15 ml), las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SO4 anhidro, se filtraron y el disolvente se eliminó a presión reducida. El material bruto se purificó por medio de cromatografía en columna (50% de EtOAc/heptano) para obtener 2-bromo-N-metil-5-nitropiridin-4-amina como un sólido amarillo (0,15 g, 90%). RMN de 1H (CDCh) 88,99 (s, 1H), 6,95 (s, 1H), 3,08 (d, 3H).
Etapa 2:
A una solución agitada de 2-bromo-N-metil-5-nitropiridin-4-amina (0,22 g, 0,96 mmol) en AcOH (4,8 ml), se añadió Fe (0,053 g, 0,96 mmol). La solución se calentó a 75 °C. Después de 5 hs, la solución se filtró a través de un tapón de Celite®, se lavó con EtOAc (10 ml) y se apagó con una solución sat. de Na2CO3. La fase acuosa se extrajo con EtOAc (2 x 10 ml), las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron, se trataron con HCl en dixoano (4 M, 2,4 ml, 9,6 mmol) y el disolvente se eliminó a presión reducida. El material bruto se agitó en Et2O/PE durante 30 min, y el sólido resultante se recolectó por filtración, se lavó con PE y se secó a presión reducida para proporcionar clorhidrato de 6-bromo-N4-metilpiridin-3,4-diamina (0,20 g, 88%). RMN de 1H (CD3OD) 87,48 (s, 1H), 6,95 (s, 1H), 3,04 (s, 3H).
Etapa 3:
A una solución agitada de clorhidrato de 6-bromo-N4-metilpiridin-3,4-diamina (0,15 g, 0,52 mmol) en DMF (2,4 ml) se añadió el Intermedio 5 (0,18 g, 0,48 mmol) seguido por DIPEA (0,25 ml, 1,4 mmol) y HBTU (0,18 g, 0,57 mmol).
Después de 2 hs, la solución se concentró a presión reducida, se diluyó con EtOAc (20 ml) y se lavó con una solución sat. de Na2CO3. La capa orgánica se secó sobre Na2SO4 anhidro, se filtró y el disolvente se eliminó a presión reducida. La amida cruda N-(6-bromo-4-(metilamino)piridin-3-il)-2-(4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)acetamida se disolvió en 1,4-dioxano (5 ml), se trató con NaOH (2 M, 2,4 ml, 4,8 mmol) y se calentó a 100 °C. Tras 0,5 hs, la solución se diluyó con agua (10 ml). La fase acuosa se extrajo con CH2Ch (3 x 10 ml), las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y el disolvente se eliminó a presión reducida. El material bruto se purificó por medio de cromatografía en columna eluyendo con EtOAc para obtener 6-bromo-2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-imidazo[4,5-c]piridina como un aceite marrón (0,19 g, 72%). RMN de 1H (CDCh) 88,74 (s, 1H), 7,44 a 7,51 (m, 2H), 7,41 (t, 1H), 7,08 (t, 2H), 6,71 (d, 1H), 6,59 (d, 1H), 5,39 (s, 2H), 3,89 (s, 3H), 3,83 (s, 2H), 2,94 (d, 2H), 2,60 (ddd, 1H), 2,28 (t, 2H), 1,85 a 1,90 (m, 2H), 1,75 a 1,84 (m, 2H).
Etapa 4:
A un vial que contenía 6-bromo-2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-metiMH-imidazo[4,5-c]piridina (0,060 g, 0,11 mmol), Dp Pp (0,011 g, 0,028 mmol) y Pd(oAc)2 (0,035 g, 0,015 mmol) se añadió Dm F (0,4 ml) seguido por MeOH (2,6 ml) y Et3N (0,13 ml, 1,1 mmol). La solución se calentó a 80 °C bajo una atmósfera de CO (50 psi). Tras 16 hs, la solución se diluyó con salmuera (5 ml). La fase acuosa se extrajo con EtOAc (2 x 10 ml), las capas orgánicas combinadas se secaron sobre MgSO4 anhidro, se filtraron y el disolvente se eliminó a presión reducida. El material bruto se purificó por medio de cromatografía en columna eluyendo con MeOH al 5% en CH2Ch para obtener 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-metiMH-imidazo[4,5-c]piridin-6-carboxilato de metilo como un aceite amarillo (0,060 g, quant.). Rm N de 1H (600 MHz, CDCh) 8 9,11 (s, 1H), 8,29 (s, 1H), 7,45 a 7,54 (m, 1H), 7,35 a 7,45 (m, 1H), 7,10 (t, 2H), 6,73 (d, 1H), 6,61 (d, 1H), 5,40 (s, 2H), 4,05 (s, 3H), 4,02 (s, 3H), 3,91 (s, 2H), 2,98 (d, 2H), 2,58 a 2,68 (m, 1H), 2,32 (t, 2H), 1,74 a 1,95 (m, 4H).
Etapa 5:
A una solución agitada de 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-imidazo[4,5-c]piridin-6-carboxilato de metilo (0,041 g, 0,078 mmol) en MeOH (0,78 ml) se añadió una solución de NaOH en agua (2 M, 0,14 ml) bajo agitación a 35 °C. Después de 2 hs, la solución se acidificó a pH ~4 con HCl en agua (1 M), se enfrió a 0 °C, se diluyó con agua (0,5 ml) y se dejó reposar durante 2 hs. El precipitado sólido resultante se sometió a una suspensión durante 1 hora, se recolectó por filtración, se lavó con agua (2 x 1 ml) y se secó a presión reducida para proporcionar el Ejemplo 2A-03 como un sólido (21 mg, 48%). RMN de 1H (400MHz, CD3OD) 8 : 9,10 (br s, 1H), 8,57 (br s, 1H), 7,67 (br t, 1H), 7,52 (br t, 1H), 7,13 a 7,33 (m, 2H), 6,95 (d, 1H), 6,75 (d, 1H), 5,46 (s, 2H), 4,92 (s, 2H), 3,92 a 4,18 (m, 5H), 3,45 (br s, 2H), 3,08 (br s, 1H), 2,11 a 2,46 (m, 4H). LC-MS(ES+): 510,3 (M+H).
Los compuestos enumerados en la Tabla 2 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 2A-01, 2A-02 y 2A-03 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron por medio de procedimientos muy conocidos por los expertos en la técnica y pueden incluir la cromatografía en gel de sílice, la HPLc o la cristalización a partir de la mezcla de reacción. Los compuestos finales se pueden haber aislado como neutros o como sales de ácidos o bases.
Tabla 2
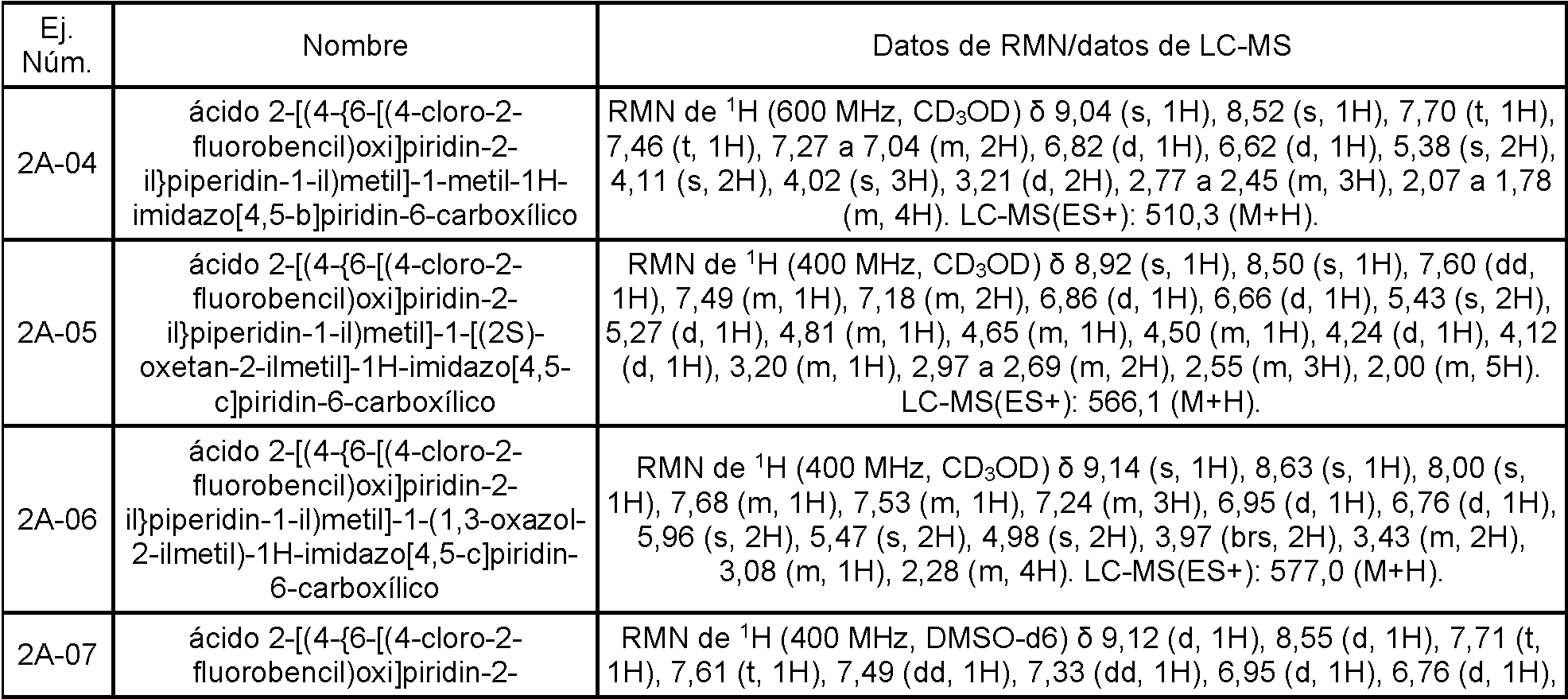



Ejemplo 3A-01
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico
Etapa 1:
A una solución agitada del Intermedio 22 (49,8 g, 211 mmol) en MeCN (300 ml) se añadió 2-cloro-1,1,1-trimetoxietano (30,0 ml, 223 mmol) seguido por pTSA-H2O (2,0 g, 10 mmol). Tras 1 hora a 60 °C, se añadieron MeCN (400 ml), K2CO3 (116 g, 841 mmol) y el Intermedio 3 (52,4 g, 90,2 mmol). Tras 2 hs, la solución se trató con agua (1,6 L), se dejó enfriar a TA y se agitó durante 2 hs. El precipitado sólido resultante se recolectó por filtración, se lavó con agua (2 x 300 ml) y se secó a presión reducida para proporcionar (S)-2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(oxetan-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo como un sólido (102 g, 84%). RMN de 1H (DMSO-d6 ) 88,30 (s, 1H), 7,82 (d, 1H), 7,67 (d, 1H), 7,62 (t, 1H), 7,55 (t, 1H), 7,45 (d, 1H), 7,29 (d, 1H), 6,87 (d, 1H), 6,67 (d, 1H), 5,37 (s, 2H), 5,04 a 5,16 (m, 1H), 4,82 (dd, 1H), 4,62 a 4,73 (m, 1H), 4,44 a 4,52 (m, 1H), 4,37 (dt, 1H), 3,96 (d, 1H), 3,87 (s, 3H), 3,78 (d, 1H), 3,00 (d, 1H), 2,85 (d, 1H), 2,66 a 2,76 (m, 1H), 2,54 a 2,64 (m, 1H), 2,38 a 2,49 (m, 1H), 2,24 (t, 2,11 a 2,21 (m, 1H), 1,60 a 1,88 (m, 4H).
Etapa 2:
A una solución agitada de (S)-2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(oxetan-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo (7,2 g, 12 mmol) en MeOH (50 ml) y THF (50 ml) se añadió NaOH 2 M (25 ml, 50 mmol). Después de 2 hs a 45 °C, se dejó enfriar la solución hasta TA, se diluyó con agua (100 ml) y se acidificó a pH ~6 con ácido cítrico en agua (1 M, 20 ml). El precipitado sólido resultante se sometió a una suspensión durante 1 hora, se recolectó por filtración, se lavó con agua (100 ml) y se secó a presión reducida para obtener el Ejemplo 3A-01 como un sólido (6,4 g, 91%). RMN de 1H (DMSO-d6 ) 8 12,84 (br s, 1H), 8,27 (s, 1H), 7,80 (d, 1H), 7,59 a 7,67 (m, 2H), 7,55 (t, 1H), 7,45 (dd, 1H), 7,29 (dd, 1H), 6,86 (d, 1H), 6,67 (d, 1H), 5,37 (s, 2H), 5,06 a 5,17 (m, 1H), 4,80 (dd, 1H), 4,66 (dd, 1H), 4,44 a 4,53 (m, 1H), 4,38 (dt, 1H), 3,95 (d, 1H), 3,78 (d, 1H), 3,00 (d, 1H), 2,85 (d,
1H), 2,64 a 2,77 (m, 1H), 2,54 a 2,64 (m, 1H), 2,40 a 2,48 (m, 1H), 2,20 a 2,29 (m, 1H), 2,17 (t, 1H), 1,61 a 1,85 (m, 4H). LC-MS(ES+): 565,4 (M+H).

Ejemplo 4A-01
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico
Etapa 1:
A una solución agitada del Intermedio 22 (33,6 g, 142 mmol) en MeCN (285 ml) se añadió 2-cloro-1,1,1-trimetoxietano (20,1 ml, 149 mmol) seguido por pTSA-H2O (1,35 g, 7,1 mmol). Tras 2 hs a 50 °C, se añadieron MeCN (280 ml), K2CO3 (79 g, 570 mmol) y el Intermedio 4 (93,2 g, 142 mmol). Tras 2 hs, la solución se trató con agua (800 ml), se dejó enfriar a TA y se agitó durante 2 hs. El precipitado resultante se recolectó por filtración, se lavó con MeCN al 10% en agua (150 ml) agua (2 x 200 ml) y se secó a presión reducida para proporcionar (S)-2-((4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(oxetan-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo como un sólido incoloro (77 g, 95%). RMN de 1H (600 MHz, DMSO-d6 ) 88,28 (s, 1H), 7,87 (d, 1H), 7,80 (d, 1H), 7,55 a 7,73 (m, 4H), 6,87 (d, 1H), 6,70 (d, 1H), 5,45 (s, 2H), 5,04 a 5,19 (m, 1H), 4,81 (dd, 1H), 4,66 (dd, 1H), 4.41 a 4,54 (m, 1H), 4,36 (dt, 1H), 3,94 (d, 1H), 3,86 (s, 3H), 3,76 (d, 1H), 2,97 (d, 1H), 2,82 (d, 1H), 2,63 a 2,77 (m, 1H), 2,49 a 2,63 (m, 1H), 2,37 a 2,46 (m, 1H), 2,18 a 2,29 (m, 1H), 2,05 a 2,18 (m, 1H), 1,47 a 1,82 (m, 4H).
Etapa 2:
A una solución agitada de (S)-2-((4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1 -il)metil)-1-(oxetan-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo (4 g, 7 mmol) en MeCN (70 ml) se añadió una solución de 1,5,7-triazabiciclo[4.4.0]dec-5-eno en agua (0,97 M, 14,7 ml). Después de 20 hs, la solución se acidificó a pH ~6 con ácido cítrico en agua (2 M, 7 ml) y se diluyó con agua (50 ml). La fase acuosa se extrajo con EtOAc (2 x 75 ml), las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y el disolvente se eliminó a presión reducida para dar un sólido blanquecino. El material bruto se purificó por medio de cromatografía en columna eluyendo con MeOH/DCM (0:100 a 8:92) para obtener el Ejemplo 4A-01 como un sólido (3,65 g, 90%). RMN de 1H (400 MHz, DMSO-d6 ) 812,75 (br s, 1H), 8,27 (s, 1H), 7,89 (d, 1H), 7,80 (d, 1H), 7,68 a 7,72 (m, 2H), 7,60 a 7,67 (m, 2H), 6,89 (d, 1H), 6,72 (d, 1H), 5,47 (s, 2H), 5,11 (d, 1H), 4,74 a 4,86 (m, 1H), 4,62 a 4,72 (m, 1H), 4,43 a 4,53 (m, 1H), 4,35 a 4.42 (m, 1H), 3,95 (d, 1H), 3,77 (d, 1H), 2,98 (d, 1H), 2,84 (d, 1H), 2,65 a 2,77 (m, 1H), 2,53 a 2,64 (m, 1H), 2,37 a 2,45 (m, 1H), 2,10 a 2,28 (m, 2H), 1,57 a 1,84 (m, 4H). LC-MS(ES+): 556,6 (M+H).
Sal tris del Ejemplo 4A-01 2-ácido [(4-{6-[(4-ciano-2-fluorobencM)oxi]piridm-2-M}piperidm-1-M)metM]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico
A una solución agitada del Ejemplo 4A-01 (6,5 g, 11,7 mmol) en 1-propanol (275 ml) a 70 °C se añadió una solución acuosa de tris (2,0 M, 6,1 ml, 12,2 mmol), gota a gota, durante la cual la solución permaneció homogénea. Tras agitar durante 5 min, se añadieron los cristales de semillas y se dejó enfriar la mezcla a TA durante 2 hs. Tras agitar durante toda la noche a TA, se había formado un sólido. El sólido se recolectó por filtración, se lavó con 1-propanol (2 x 30 ml) y se secó, primero bajo una corriente de nitrógeno y posteriormente en una estufa de vacío a 45 °C durante 15 hs, para dar la sal tris del Ejemplo 4A-01 (6,95 g, 88%) como un sólido cristalino. RMN de 1H (600 MHz, DMSO-d6 ) 8 : 8,20 (s, 1H), 7,89 (d, 1H), 7,79 (d, 1H), 7,70 (br s, 2H), 7,64 (t, 1H), 7,56 (d, 1H), 6,89 (d, 1H), 6,72 (d, 1H), 5,47 (s, 2H), 5,11 (qd, 1H), 4,77 (dd, 1H), 4,64 (dd, 1H), 4,44 a 4,53 (m, 1H), 4,38 (dt, 1H), 3,93 (d, 1H), 3,76 (d, 1H), 3,35 (br s, 9H), 2,98 (d, 1H), 2,85 (d, 1H), 2,64 a 2,75 (m, 1H), 2,54 a 2,64 (m, 1H), 2,40 a 2,49 (m, 1H), 2,08 a 2,26 (m, 2H), 1,56 a 1,83 (m, 4H). mp = 194 °C.

Ejemplo 5A-01
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico
Etapa 1:
Una solución del Intermedio 13 (5 g, 14,4 mmol) en MeOH:CH2Cl2 al 5% (60 ml) se trató con una solución ac. sat. de Na2CO3 (60 ml). La solución bifásica se agitó enérgicamente durante 30 min y se separó la capa orgánica. La capa orgánica se secó, se filtró y se concentró a presión reducida para entregar 4-(((6-(4-piperazin-1-il)piridin-2-il)oxi)metil)-3-fluorobenzonitrilo (4,4 g, quant.) como un semisólido.
Etapa 2:
A un matraz que contenía una solución de 4-((6-(4-piperazin-1 -il)piridin-2-il)oxi)metil)-3-fluorobenzonitrilo (1,58 gramos, 5,06 mmol) en MeCN (15 ml) se añadió el Intermedio 23 (1,40 g, 5,06 mmol) y K2CO3 (3,50 g, 25,3 mmol). La suspensión resultante se agitó durante 2 hs a 50 °C. Después de 2 hs, la mezcla se trató con agua (30 ml), se dejó enfriar a TA y se agitó durante 2 hs. El sólido se recolectó por filtración, se lavó con agua: MeCN (2:1) (2 x 30 ml) y se secó a presión reducida para proporcionar (S)-2-((4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperazin-1-il)metil)-1-(oxetan-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo (2,47 g, 86%) como un sólido. RMN de 1H (600 MHz, CDCla) 88,16 (s, 1H), 7,98 (d, 1H), 7,76 (d, 1H), 7,59 (t, 1H), 7,42 (dt, 2H), 7,34 (d, 1H), 6,17 (dd, 2H), 5,42 (s, 2H), 5,23 (dd, 1H), 4,77 a 4,58 (m, 3H), 4,38 (dt, 1H), 4,05 a 3,95 (m, 2H), 3,95 (s, 3H), 3,46 (d, 4H), 2,80 a 2,69 (m, 1H), 2,62 (t, 4H), 2,50 a 2,38 (m, 1H).
Etapa 3:
A un matraz que contenía una solución de (S)-2-((4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperazin-1-il)metil)-1-(oxetan-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo (2,5 g, 4,3 mmol) en una mezcla 1:1 de iPrOH y THF (140 ml) se añadió 1,4 equiv de LiOH (0,14 g, 6,1 mmol) y la solución resultante se calentó a 45 °C durante 15 hs. La solución se dejó enfriar a TA, se diluyó con agua (50 ml) y se acidificó a pH ~6 con ácido cítrico en agua. La solución resultante se extrajo con EtOAc. La capa de EtOAc se secó y el disolvente se eliminó a presión reducida para obtener el producto bruto. El producto bruto se purificó por cromatografía flash (10% de MeOH en CH2Ch) para obtener el Ejemplo 5A-01 (0,86 g, 35%) como un sólido. RMN de 1H (600 MHz, CDCla) 88,23 (s, 1H), 8,06 (d, 1H), 7,83 (d, 1H), 7,59 (t, 1H), 7,46 a 7,39 (m, 2H), 7,34 (d, 1H), 6,18 (dd, 2H), 5,43 (s, 2H), 5,28 a 5,20 (m, 1H), 4,81 a 4,58 (m, 3H), 4,44 a 4,33 (m, 1H), 4,04 (d, 2H), 3,48 (m, 4H), 2,82 a 2,71 (m, 1H), 2,65 (m, 4H), 2,46 (dd, 1H). LC-MS(ES+): 557,2 (M+H).

Ejemplo 6A-01
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico
Etapa 1:
A un matraz de 3 L equipado con un agitador mecánico, cargado con el Intermedio 4 (106 g, 161 mmol) se añadió
MeCN (886 ml), K2CO3 (89,0 g, 644 mmol) y el Intermedio 27 (52,4 g, 177 mmol). La mezcla se agitó a 60 °C durante 2 hs. La mezcla de reacción se vertió en un matraz Erlenmeyer de 4 L y se diluyó con 1,8 L de agua. La suspensión resultante se agitó a TA durante 4 horas para obtener una suspensión de color amarillo claro. Los sólidos se recolectaron por filtración y se secaron en una estufa de vacío a 45 °C durante la noche para producir el (S)-2-((4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1 -il)metil)-3-(oxetan-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxilato de metilo deseado (88,6 g, 96%) como un sólido amarillo claro. RMN de 1H (600 MHz, DMSO-d6 ) 88,16 (d, 1H), 8,01 (d, 1H), 7,87 (d, 1H), 7,61 a 7,74 (m, 3H), 6,88 (d, 1H), 6,71 (d, 1H), 5,46 (s, 2H), 5,11 a 5,26 (m, 1H), 4,85 (dd, 1H), 4,73 (dd, 1H), 4,43 a 4,60 (m, 1H), 4,37 (dt, 1H), 3,96 a 4,04 (m, 1H), 3,89 a 3,95 (m, 3H), 2,87 a 3,01 (m, 2H), 2,66 a 2,81 (m, 1H), 2,55 a 2,64 (m, 1H), 2,52 (br s, 3H), 2,24 (q, 2H), 1,64 a 1,81 (m, 3H); LC-MS(ES+): 571,5 (M+H).
Etapa 2:
En un matraz de 1L y 3 bocas, equipado con un agitador mecánico, se cargó (S)-2-((4-(6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-3-(oxetan-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxilato de metilo (35,5 g, 62,21 mmol). Se añadieron MeCN (350 ml) y agua (70 ml) al matraz. La mezcla resultante se agitó a TA durante 30 min para formar una suspensión espesa. Se añadió lentamente LiOH-H2O (2,92 g, 68,4 mmol) como un sólido. La suspensión resultante se agitó a 40 °C durante 1 hora. La mezcla de reacción se enfrió a TA y se trató, gota a gota, con ácido cítrico 1,0 M (15,5 ml) hasta que el pH de la suspensión alcanzó ~5. La suspensión resultante se agitó a TA durante 4 hs. Los sólidos resultantes se recolectaron por filtración, los sólidos se enjuagaron con ~20 ml de agua y posteriormente se secaron bajo una corriente de N2 durante 4 hs. Los sólidos se secaron durante 72 hs adicionales a 40 °C en una estufa de vacío para secar y producir el Ejemplo 6A-01 (31,2 g, 90%.) como un sólido. RMN de 1H (600 MHz, DMSO-d6 ) 8 13,03 (br s, 1H), 8,15 (d, 1H), 8,00 (d, 1H), 7,87 (d, 1H), 7,67 a 7,73 (m, 2H), 7,64 (t, 1H), 6,88 (d, 1H), 6,71 (d, 1H), 5,45 (s, 2H), 4,93 a 5,03 (m, 1H), 4,87 (s, 1H), 4,70 (d, 1H), 4,36 a 4,45 (m, 1H), 4,23 a 4,35 (m, 1H), 4,05 (d, 1H), 3,79 (d, 1H), 2,93 a 3,06 (m, 1H), 2,76 a 2,88 (m, 1H), 2,54 a 2,69 (m, 1H), 2,34 a 2,46 (m, 1H), 2,25 (d, 2H), 2,05 a 2,21 (m, 1H), 1,73 (d, 3H), 1,47 a 1,67 (m, 1H); LC-MS(ES+): 557,6 (M+H).

Ejemplo 7A-01
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico
Etapa 1:
A una suspensión de la sal HCl de oxazol-2-ilmetanamina (491 mg, 3,65 mmol) y el Intermedio 29 (800 mg, 3,32 mmol) en DMF (5 ml) se añadió K2CO3 (1,04 g, 6,63 mmol). La reacción se agitó a 60 °C durante 2 hs. Se añadió la sal HCl de oxazol-2-ilmetanamina (100 mg, 1,0 mmol) y la reacción se agitó durante 30 min más a 60 °C. La reacción se enfrió a TA y se diluyó con agua (30 ml) y se extrajo con EtOAc (60 ml). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo naranja se purificó por medio de cromatografía flash (12 g de gel de sílice, gradiente 0 a 50% de EtOAc/heptano) para obtener 4-nitro-3-((oxazol-2-ilmetil)benzoato de tere-butilo (764 mg, 75%) como un sólido naranja. RMN de 1H (CDCh) 88,48 (br s, 1H), 8,23 (d, 1H), 7,68 (d, 1H), 7,61 (d, 1H), 7,28 (dd, 1H), 7,15 (s, 1H), 4,72 (d, 2H), 1,60 (s, 9H).
Etapa 2:
A una solución de 4-nitro-3-((oxazol-2-ilmetil)benzoato de tere-butilo (15 g, 47 mmol) en THF (100 ml) se añadió 10% de paladio sobre carbono (1,5 g, 10% en peso), y la mezcla se agitó bajo 50 psi de H2 a TA durante 6 hs. La mezcla de reacción se filtró a través de Celite® para dar una solución oscura. El filtrado se filtró a través de una segunda almohadilla Celite® y el filtrado se concentró a presión reducida para obtener 4-amino-3-((oxazol-2-ilmetil)benzoato de terc-butilo (13,1 g, 92%) como una espuma oscura. RMN de 1H (CDCh) 8 7,62 (s, 1H), 7,43 (dd, 1H), 7,35 (d, 1H), 7,08 (s, 1H), 6,66 (d, 1H), 4,44 (s, 2H), 1,56 (s, 9H).
Etapa 3:
A una solución agitada de 4-amino-3-((oxazol-2-ilmetil)benzoato de terc-butilo (13 g, 45 mmol) en MeCN (100 ml) se añadió 2-cloro-1,1,1-trimetoxietano (9,0 ml, 65 mmol) y pTSA-H2O (400 mg, 2,1 mmol) y la mezcla se calentó a 60
°C durante 3 hs. A continuación, la reacción se enfrió a TA y se concentró a presión reducida. El producto bruto se purificó por medio de cromatografía flash (120 g de gel de sílice, gradiente 0 a 100% de EtOAc/heptano) para obtener 2-(clorometil)-1-(oxazol-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de ferc-butilo (11,6 g, 74%) como un sólido amarillo claro. RMN de 1H (CDCh) 88,19 (d, 1H), 7,98 (dd, 1H), 7,77 (d, 1H), 7,64 (d, 1H), 7,12 (d, 1H), 5,64 (s, 2H), 5,00 (s, 2H), 1,62 a 1,66 (m, 9H).
Etapa 4:
A una suspensión de 2-(clorometil)-1-(oxazol-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de ferc-butilo (10,1 g, 29 mmol) y el Intermedio 4 (11,2 g, 29,1 mmol) en MeCN (100 ml) se añadió K2CO3 (16,1 g, 116 mmol). La mezcla se agitó a 60 °C durante 2 hs y posteriormente se diluyó con agua (200 ml) y se agitó durante 4 hs más a TA. Los sólidos resultantes se recolectaron por filtración para obtener 2-((6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(oxazol-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de terc-butilo (16,23 g, 89%) como un sólido. RMN de 1H (DMSO-d6 ) 88,13 (s, 1H), 8,04 (s, 1H), 7,88 (d, 1H), 7,78 (dd, 1H), 7,70 (br s, 2H), 7,66 (d, 1H), 7,62 (t, 1H), 7,13 (s, 1H), 6,79 (d, 1H), 6,69 (d, 1H), 5,91 (s, 2H), 5,44 (s, 2H), 3,84 (s, 2H), 2,80 (d, 2H), 2,46 (d, 1H), 2,05 a 2,13 (m, 2H), 1,64 (d, 2H), 1,55 (s, 9H), 1,35 a 1,43 (m, 2H).
Etapa 5:
A una solución de 2-((6-((4-ciano-2-fluorobencil)oxi)piridin-2-il)piperidin-1 -il)metil)-1 -(oxazol-2-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de terc-butilo (31,1 g, 50,0 mmol) en DCE (300 ml) se añadió TfA (40 ml, 530 mmol). La mezcla se calentó a 70 °C durante 4 hs y posteriormente se enfrió lentamente hasta TA y se agitó durante la noche. La reacción se concentró a presión reducida y el residuo se disolvió en MeOH (100 ml) y agua (300 ml). . Se añadió NaHCO3 (85 ml), gota a gota, para llevar la solución a un pH ~7. Los sólidos resultantes se agitaron para granular durante 3 hs, y posteriormente se recolectaron por filtración para entregar el Ejemplo 7A-01 (27,3 g, 96%) como un sólido. RMN de 1H (600 MHz, DMSO-d6 ) 812,93 (br s, 1H), 8,19 (s, 1H), 8,03 (s, 1H), 7,88 (d, 1H), 7,82 (d, 1H), 7,70 (br s, 2H), 7,65 (d, 1H), 7,62 (t, 1H), 7,12 (s, 1H), 6,80 (d, 1H), 6,66 a 6,71 (m, 1H), 5,90 (s, 2H), 5,43 (s, 2H), 3,84 (s, 2H), 2,81 (d, 2H), 2,46 (m, 1H), 2,10 (t, 2H), 1,64 (d, 2H), 1,36 a 1,46 (m, 2H); LC-MS(ES+): 568,3 (M+H).

Ejemplo 8A-01
2-((6-((4-metilbencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de amonio
A un vial de 1 dram se añadió el Intermedio 35 (20 mg, 47 pmol) seguido por alcohol 4-metilbencílico (100 micrómetrosol). Se añadió THF (500 pl) seguido por el reactivo de Tsunoda (cianometileno tributario fosforano, 0,5 M en THF, 400 pl, 0,20 mmol) y la mezcla se calentó a 70 °C durante 3 hs. La reacción se enfrió a TA y se concentró a presión reducida. El residuo se disolvió en MeOH (1 mL). Se añadió NaOH 1 M (0,15 ml, 150 pmol) y la mezcla se calentó a 60 °C durante 3 hs y posteriormente se mantuvo a TA durante 48 hs. La mezcla se concentró a presión reducida y el producto bruto se purificó por SFC preparativa para obtener el Ejemplo 8A-01 (10,7 mg, 45%). Procedimiento SFC (Columna: Phenomenex Biphenyl 4,6x150 mm, 5 micrómetros; Fase móvil: CO2 (v/v); Fase móvil B: Metanol c/ 0,2% de NH4OH (v/v) 85% de CO2 / 15% de Metanol c/ 0,2% de NH4OH Lineal en 8 min, MANTENER a 70% de CO2 / 30% de Metanol c/ 0,2% de NH4OH a 10 min. Flujo: 75 mL/min. Presión trasera: 120 Bar; Tiempo de retención 2,56 min; LC-MS(ES+): 515,4 (M+H).
Los compuestos enumerados en la Tabla 3 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 8A-01 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron por medio de procedimientos muy conocidos por los expertos en la técnica y pueden incluir la cromatografía en gel de sílice, la HPLC o la cristalización a partir de la mezcla de reacción. Los compuestos finales se pueden haber aislado como neutros o como sales de ácidos o bases.
T a b la 3


ácido 2-((4-(6-(benciloxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-benzo[d]imidazol-6-carboxílico
Etapa 1:
Una mezcla del Intermedio 36 (100 mg, 0,251 mmol), alcohol bencílico (48,2 mg, 0,446 mmol), BINAP (23,2 mg, 0,0373 mmol), Pd2(dba)3 (15,2 mg, 0,0166 mmol) y Cs2CO3 (123 mg, 0,378 mmol) en PhMe (2 ml) se agitó a 100 °C durante 14 hs. La mezcla marrón se diluyó con DCM (50 ml) y se filtró. El filtrado se concentró a presión reducida para dar un aceite marrón que se purificó por medio de pre-TLC (DCM:MeOH = 20:1) para obtener 2-((4-(6-(benzoxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-benzo[d]imidazol-6-carboxilato de metilo (99,7 mg, 84%) como un sólido amarillo. RMN de 1H (CD3OD) 88,32 (s, 1H), 8,02 (dd, 1H), 7,79 (d, 1H), 7,59 a 7,70 (m, 1H), 7,40 a 7,48 (m, 2H), 7,35 (m, 2H), 7,23 a 7,32 (m, 1H), 6,90 (d, 1H), 6,73 (d, 1H), 5,40 (s, 2H), 4,79 (s, 2H), 3,96 (s, 6 H), 3,91 (d, 2H), 3,40 (m, 2H), 3,05 (br s, 1H), 2,14 a 2,38 (m, 4H).
Etapa 2:
A una solución de 2-((6-(benzoxi)piridin-2-il)piperidin-1-il)metil)-1-metil-1H-benzo[d]imidazol-6-carboxilato de metilo (90,0 mg, 0,191 mmol) en MeOH (3 ml) se añadió NaOH 3,0 M (2,0 ml, 6,0 mmol). La mezcla se agitó a 40 °C durante 4 hs. La mezcla de reacción se neutralizó con HCl 1 M y la suspensión espesa resultante se extrajo con (DCM:MeOH 10:1, 2 x 40 ml). Los extractos orgánicos combinados se secaron sobre MgSO4, se filtraron y concentraron a presión reducida. El sólido amarillo se purificó por HPLC preparativa (Columna: Waters Xbridge Prep OBD C18100 x 19 mm x 5 micrómetros; Fase móvil: de 5% de MeCN en agua [0,1% TFA] a 95% de MeCN en agua [0,1% TFA]; Longitud de onda: 220 nm; Caudal: 25 ml/min) para entregar el Ejemplo 9A-01 (33 mg, 28%) como un sólido. Debido al disolvente de purificación, el compuesto final era probablemente la sal de trifluoroacetato. RMN de 1H (400 MHz, CD3OD) 88,31 (s, 1H), 8,03 (dd, 1H), 7,78 (d, 1H), 7,65 (t, 1H), 7,40 a 7,46 (m, 2H), 7,35 (t, 2H), 7,25 a 7,31 (m, 1H), 6,90 (d, 1H), 6,73 (d, 1H), 5,40 (s, 2H), 4,79 (s, 2H), 3,96 (s, 3H), 3,90 (d, 2H), 3,40 (m, 2H), 3,05 (br s, 1H), 2,14 a 2,37 (m, 4H); LC-MS(ES+): 457,1 (M+H).
Los compuestos enumerados en la Tabla 4 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 9A-01 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron mediante el uso de HPLC. Debido al disolvente de purificación, los compuestos finales aislados por medio de los procedimientos PF-AB01 y PF-AB10 eran probablemente sales de trifluoroacetato, mientras que los compuestos aislados por medio del procedimiento PF-CD05 son probablemente sales de amonio.
Tabla 4



Ejemplo 10A-01
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico
Etapa 1:
A una solución incolora de 3-fluoro-4-nitrobenzoato de metilo (302 mg, 1,52 mmol) y oxazol-5-ilmetanamina (164 mg, 1,67 mmol) en DMF (5,0 ml) se añadió Et3N (460 mg, 4,55 mmol) lentamente a 20 °C. La solución marrón se agitó a 60 °C durante 36 hs. La mezcla se diluyó con EtOAc (50 ml) y se lavó con H2O (50 ml). La fase orgánica se separó y la fase ac. se extrajo con EtOAc (2 * 50 L). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (10 a 100% de EtOAc/PE) para dar 4-nitro-3-((oxazol-5-ilmetil)benzoato de metilo (320 mg, 76%) como un sólido naranja. RMN de 1H (CDCh) 88,26 (d, 2H), 7,89 (s, 1H), 7,66 (d, 1H), 7,35 (dd, 1H), 7,11 (s, 1H), 4,68 (d, 2H), 3,96 (s, 3H).
Etapa 2:
A una suspensión amarilla de 4-nitro-3-((oxazol-5-ilmetil)benzoato de metilo (67 mg, 0,24 mmol) en MeOH (8 ml) se añadió 10% de Pd/C (10,3 mg). La mezcla se agitó bajo 1 atm de H2 a TA durante 1 hora. Los sólidos se eliminaron por filtración y se lavaron con MeOH (20 ml). Las capas orgánicas combinadas se concentraron a presión reducida para dar 4-amino-3-((oxazol-5-ilmetil)benzoato de metilo (56 mg, 94%) como un sólido blanco. LC-MS(ES+): 247,9 (M+H).
Etapa 3:
A una solución amarilla del Intermedio 5 (85 mg, 0,22 mmol), 4-amino-3-((oxazol-5-ilmetil)amino)benzoato (55,5 mg, 0,224 mmol) y HATU (111 mg, 0,292 mmol) en DMF (2 ml) se añadió Et3N (114 mg, 1,12 mmol, 0,15 ml). La solución amarilla se agitó a 25 °C durante 16 hs. La mezcla se vertió en H2O (8 ml) y se extrajo con EtOAc (3 * 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por pre-TLC (EtOAc) para dar 4-(2-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)acetamido)-3-((oxazol-5-ilmetil)amino)benzoato de metilo (58 mg, 43%) como un aceite amarillo. LC-MS(ES+): 630,0 (M+Na).
Etapa 4:
Una solución amarilla de 4-(2-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)acetamido)-3-((oxazol-5-ilmetil)amino)benzoato de metilo (58 mg, 0,095 mmol) en AcOH (0,5 ml) se agitó a 60 °C durante 3 hs y posteriormente a TA durante 16 hs. El residuo amarillo se neutralizó con una solución ac. sat. de Na2CO3 y se extrajo con DCM (3 * 10 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(oxazol-5-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo (56 mg, 99%) como un aceite amarillo. LC-MS(ES+): 612,0 (M+Na).
Etapa 5:
A una solución de 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-(oxazol-5-ilmetil)-1H-benzo[d]imidazol-6-carboxilato de metilo (56 mg, 0,095 mmol) en THF (1 ml) y MeOH (0,2 ml) se añadió NaOH 2 M (0,0949 ml, 0,190 mmol). La solución amarilla se agitó a 25 °C durante 16 hs y posteriormente se mantuvo durante 48 hs a 25 °C. La solución amarilla se concentró a presión reducida y el residuo se disolvió en H2O (5 ml), se acidificó a pH ~5 con HCl 1 M y se extrajo con DCM (5 x 10 ml). Los extractos orgánicos combinados se concentraron a presión reducida y el producto bruto resultante se purificó por medio de HPLC preparativa (Columna: Waters Xbridge Prep OBD C18 150 * 30 mm * 5 micrómetros; Fase móvil: de 5% de MeCN en agua [0,1% TFA] a 95% de MeCN en agua [0,1% TFA]; Longitud de onda: 220 nm; Caudal: 25 ml/min) para entregar el Ejemplo 10A-01 (22 mg, 33%) como un sólido. Debido al disolvente de purificación, el compuesto final se aisló probablemente como la sal de trifluoroacetato. RMN de 1H (400 MHz, CD3OD) 88,41 (s, 1H), 8,18 (s, 1H), 8,03 (dd, 1H), 7,80 (d, 1H), 7,61 a 7,70 (m, 1H), 7,52 (t, 1H), 7,36 (s, 1H), 7,20 a 7,30 (m, 2H), 6,94 (d, 1H), 6,74 (d, 1H), 5,78 (s, 2H), 5,45 (s, 2H), 4,91 (br s, 2H), 3,97 (d, 2H), 3,42 (br s, 2H), 3,07 (br s, 1H), 2,17 a 2,33 (m, 4H); LC-MS(ES+): 576,1 (M+H).

Ejemplo 10A-02
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico
Etapa 1:
A una solución del Intermedio 29 (200 mg, 0,829 mmol) en DMF (8 ml) se añadió (1-etil-1H-imidazol-5-il)metanamina (104 mg, 0,829 mmol) y NaHCO3 (348 mg, 4,15 mmol). La mezcla de reacción se agitó a 60 °C durante 16 hs. La mezcla de reacción se vertió en agua (10 ml) y posteriormente se extrajo con EtOAc (2 * 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (2 * 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (0 a 5% de MeOH/DCM) para dar 3-((1-etil-1H-imidazol-5-il)amino)-4-nitrobenzoato de terc-butilo (105 mg, 37%) como un aceite rojo pálido. RMN de 1H (CDCh) 8 8,23 (d, 1H), 7,96 (br s, 1H), 7,66 (d, 1H), 7,57 (s, 1H), 7,28 (dd, 1H), 7,12 (s, 1H), 4,54 (d, 2H), 4,00 (q, 2H), 1,62 (s, 9H), 1,47 (t, 3H).
Etapa 2:
A una solución de 3-((etil-1H-imidazol-5-il)metil)amino)-4-nitrobenzoato de terc-butilo (105 mg, 0,303 mmol) en MeOH (3 ml) y H2O (1 ml) se añadió polvo de Fe (59,2 mg, 1,06 mmol) y NH4Cl (292 mg, 5,46 mmol). La mezcla de reacción se agitó a 80 °C durante 50 min. La mezcla de reacción se vertió en agua (10 ml) y se extrajo con EtOAc (3 x 15 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener 4-amino-3-((etil-1H-imidazol-5-il)amino)benzoato de terc-butilo (93 mg, 97%) como un sólido marrón pálido que se utilizó directamente en la siguiente etapa.
Etapa 3:
A una solución de color amarillo pálido del Intermedio 5 (55 mg, 0,15 mmol) y DMF (1 ml) se añadió HATU (66,2 mg, 0,174 mmol). La mezcla se agitó a 30 °C durante 10 min. Se añadió una solución de 4-amino-3-((etil-1H-imidazol-5-il)amino)benzoato de terc-butilo (45,9 mg, 0,145 mmol) y DIPEA (56,3 mg, 0,436 mmol) en DMF (1 ml) y la reacción se agitó a 30 °C durante 16 hs. La mezcla se vertió en agua (10 ml) y posteriormente se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se lavaron con NH4Cl ac. (3 x 20 ml), salmuera (2 x 20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por medio de Prep-TLC (5% de MeOH/DCM) para dar 4-amino-3-(2-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)-N-((1-etil-1H-imidazol-5-il)metil)acetamido)benzoato de terc-butilo (60 mg, 61%) como una goma marrón pálida. LC-MS(ES+): 699,4 (M+Na).
Etapa 4:
Una solución de color marrón pálido de dar 4-amino-3-(2-(4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)-N-((etil-1H-imidazol-5-il)metil)acetamido)benzoato de terc-butilo (60 mg, 0,089 mmol) en AcOH (2 ml) se agitó a 60 °C durante 16 hs. La mezcla de reacción se concentró al vacío para eliminar el AcOH y proporcionar 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-((etil-1H-imidazol-5-il)metil)-1H-benzo[d]imidazol-6-carboxilato de terc-butilo (56 mg, 96%) como una goma de color marrón pálido que se utilizó en la siguiente etapa sin más purificación. LC-MS(ES+): 681,3 (M+Na).
Etapa 5:
A una solución de color marrón pálido de 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)piperidin-1-il)metil)-1-((etil-1H-imidazol-5-il)metil)-1H-benzo[d]imidazol-6-carboxilato de terc-butilo (56 mg, 0,085 mmol) en DCM (2 ml) se añadió TFA (1 ml). La mezcla de reacción se agitó a TA (10 °C) durante 16 hs. La mezcla de reacción se concentró a presión reducida y el producto bruto se purificó por HPLC preparativa (Columna: Waters Xbridge Prep OBD C18150 x 30 mm x 5 micrómetros; Fase móvil: de 5% de MeCN en agua [0,1% TFA] a 95% de MeCN en agua [0,1% TFA]; Longitud de onda: 220 nm; Caudal: 25 ml/min) para entregar el Ejemplo 10A-02 (37 mg, 48%) como un sólido beige. Debido al disolvente de purificación, el compuesto se aisló probablemente como la sal de trifluoroacetato. RMN de 1H (400 MHz, CD3OD) 89,10 (d, 1H), 8,26 (s, 1H), 8,07 (dd, 1H), 7,88 (d, 1H), 7,66 (t, 1H), 7,52 (t, 1H), 7,19 a 7,28 (m, 2H), 7,08 (d, 1H), 6,93 (d, 1H), 6,73 (d, 1H), 5,88 (s, 2H), 5,45 (s, 2H), 4,84 (s, 2H), 4,36 (q, 2H), 3,98 (d, 2H), 3,41 (t, 2H), 3,06 (t, 1H), 2,14 a 2,40 (m, 4H), 1,58 (t, 3H); LC-MS(ES+): 603,1 (M+H).
Los compuestos enumerados en la Tabla 5 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 10A-01 o 10A-02 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron por medio de procedimientos muy conocidos por los expertos en la técnica y pueden incluir la cromatografía en gel de sílice, la HPLC o la cristalización a partir de la mezcla de reacción. Los compuestos finales se pueden haber aislado como neutros o como sales de ácidos o bases.
Tabla 5








Los compuestos enumerados en la Tabla 6 a continuación se prepararon por síntesis paralela mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 10A-01 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron mediante el uso de HPLC. Debido al disolvente de purificación, los compuestos finales aislados por medio de los procedimientos PF-AB01 y PF-AB10 eran probablemente sales de trifluoroacetato, mientras que los compuestos aislados por medio del procedimiento PF-CD05 son probablemente sales de amonio.
Tabla 6





Ejemplo 12A-01 Ácido 2-((6-((4-doro-2-fluorobencM)oxi)pmdm-2-M)-2-(trífluorometN)piperazm-1-N)metM)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxílico, enantiómero 1
Ejemplo 12A-02 Ácido 2-((6-((4-doro-2-fluorobencM)oxi)pmdm-2-M)-2-(trífluorometN)piperazm-1-N)metM)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxílico, enantiómero 2
Etapa 1:
A una solución de alcohol 4-cloro-2-fluorobencílico (15,0 g, 93,4 mmol) en DMF (250 ml) se añadió NaH (4,48 g, 112 mmol, 60% susp.) a 0 °C. Tras agitar a 15 °C durante 40 min, se añadió 2,6-dicloropiridina (16,6 g, 112 mmol). La mezcla resultante se agitó a 15 °C durante 3 hs. La mezcla se vertió en agua (1 L) y se extrajo con EtOAc (2 x 200 ml). Las capas orgánicas combinadas se lavaron con una solución sat. de NH4Cl (500 ml), salmuera (500 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto bruto se purificó por cromatografía flash (PE) para dar 2-cloro-6-((4-cloro-2-fluorobencil)oxi)piridina (19,2 g, 75%) como un sólido. RMN de 1H (CDCla) 87,55 (t, 1H), 7,47 (t, 1H), 7,15 (t, 2H), 6,94 (d, 1H), 6,71 (d, 1H), 5,40 (s, 2H).
Etapa 2:
A una solución de 3-(trifluorometil)piperazin-1-carboxilato de terc-butilo (100 mg, 0,393 mmol) en MeCN (2 ml) se añadió el Intermedio 19 (111 mg, 0,393 mmol), yoduro de tetrabutilamonio (145 mg, 0,39 mmol) y N,N-diisopropiletil amina (152 mg, 1,18 mmol). La mezcla de reacción se agitó a 150 °C durante 1 hora en condiciones de microondas. La mezcla de reacción se concentró a presión reducida y se purificó por medio de pre-TLC (33% de EtOAc/PE) para obtener 2-((4-(terc-butoxicarbonil)-2-(trifluorometil)piperazin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo (100 mg, 51%) como un aceite amarillo. MS(ES+): 501,1 (M+H).
Etapa 3:
A una solución de 2-((4-(terc-butoxicarbonil)-2-(trifluorometil)piperazin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo (100 mg, 0,2 mmol) en EtOAc (5 ml) se añadió HCl-EtOAc (5 ml). La mezcla de reacción se agitó a TA durante 2 hs. La mezcla de reacción se concentró a presión reducida. El producto bruto se disolvió en DCM (10 ml), se lavó con una solución ac. sat. de K2CO3, se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar 1-(2-metoxietil)-2-((2-(trifluorometil)piperazin-1-il)metil)-1H-benzo[d]imidazol-6-carboxilato de metilo (42 mg, 53%) como un aceite amarillo. MS(ES+): 401,0 (M+H).
Etapa 4:
A una solución de 1-(2-metoxietil)-2-((2-(trifluorometil)piperazin-1-il)metil)-1H-benzo[d]imidazol-6-carboxilato de metilo (90 mg, 0,22 mmol) en PhCH3 (2 ml) se añadió 2-cloro-6-((4-cloro-2-fluorobencil)oxi)piridina (61,2 mg, 0,225 mmol), Pd2(dba)3 (20,6 mg, 0,1 mmol), BInAp (28 mg, 0,045 mmol) y Cs2CO3 (220 mg, 0,674 mmol). La mezcla de reacción se agitó a 100 °C durante 16 hs. La mezcla de reacción se concentró a presión reducida y el producto bruto se purificó por medio de TLC preparativa (33% de EtOAc/PE) para dar el producto racémico como un aceite amarillo. La mezcla racemática se separó por medio de SFC quiral preparativa (Columna: Whelk-01 250 x 30 mm x 10 micrómetros; Fase móvil: 45% de isopropanol (1% de NH4OH)/CO2; Caudal: 50 ml/min) para obtener los enantiómeros separados del 2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-(trifluorometil)piperazin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo:
Enantiómero 1: (28 mg, 20%); tiempo de retención (15,98 min);
Enantiómero 2: (33 mg, 23%); tiempo de retención (20,92 min).
Etapa 5:
A una solución del Enantiómero 1 de la Etapa 4 (28 mg, 0,044 mmol) en MeOH (5 ml) se añadió 2 M NaOH (1 mL). La mezcla de reacción se agitó a 50 °C durante 2 hs. La mezcla de reacción se enfrió a 0 °C y se acidificó con HCl 1 M hasta alcanzar un pH ~4. La mezcla de reacción se extrajo con EtOAc (3 x 10 ml), se secó sobre Na2SO4 y se concentró a presión reducida. El producto bruto se purificó por HPLC preparativa (Columna: Waters Xbridge Prep OBD C18 150 x 30 mm x 5 micrómetros; Fase móvil: de 55% de MeOH en agua [0,1% TFA] a 75% de MeOH en agua [0,1% TFA]; gradiente de 10 min; Longitud de onda: 220 nm; Caudal: 25 ml/min) para entregar el Ejemplo 12A-01 (12,2 mg, 37%) como un sólido. Debido al disolvente de purificación, el compuesto se aisló probablemente como la sal de trifluoroacetato. Datos de LC-MS analíticos: Xtimate C18 5 x 30 mm, 3 micrómetros; Fase móvil: 1% de MeCN en agua (0,1% de TFA) a 5% de MeCN en agua (0,1% de TFA) en 1 min; posteriormente de 5% de MeCN en agua (0,1% de TFA) a 100% de MeCN (0,1% de TFA) en 5 min; mantener en 100% de MeCN (0,1% de TFA) durante 2 min; volver a 1,0% de MeCN en agua (0,1% de TFA) a 8,01 min, y mantener 2 min. Caudal: 1,2 ml/min; Tiempo de retención 4,465 min, MS(ES+): 622,2 (M+H).
El Ejemplo 12A-02 se preparó de forma similar a partir de la Etapa 4, Enantiómero 2 (33 mg, 0,052 mmol) y se purificó mediante el uso del mismo procedimiento de HPLC preparativa para obtener el Ejemplo 12A-02 (9,8 mg, 28%) como un sólido. Debido al disolvente de purificación, el compuesto se aisló probablemente como la sal de trifluoroacetato. Datos de LC-MS analíticos: Tiempo de retención 4,469 min, MS(ES+): 622,2 (M+H).
Los compuestos enumerados en la Tabla 7 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 12A-01 y 12A-02 mediante el uso de los materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de forma análoga a las vías descritas anteriormente para otros intermedios. Los compuestos se purificaron por medio de procedimientos muy conocidos por los expertos en la técnica y pueden incluir la cromatografía en gel de sílice, la HPLc o la cristalización a partir de la mezcla de reacción. Los compuestos finales se pueden haber aislado como neutros o como sales de ácidos o bases.
Tabla 7



Ejemplos 13A-01 y 13A-02 ácido
trans
2-{[4-[6-[(4-doro-2-fluorobencN)oxi]piridm-2-M}-2-metNpiperidm-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico, enantiómeros 1 y 2
Ejemplos 13A-03 y 13A-04 cis ácido 2-{[4-[6-[(4-doro-2-fluorobendl)oxi]piridm-2-M}-2-metNpiperidm-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico, enantiómeros 1 y 2
Etapa 1:
Una mezcla de 2-[(4-cloro-2-fluorobencil)oxi]-6-(2-metilpiperidin-4-il)piridina (350 mg, 0,86 mmol) [preparada como una mezcla de estereoisómeros por una vía similar a la utilizada para el Intermedio 3], el Intermedio 19 (220 mg, 0,78 mmol) y K2CO3 (540 mg, 3,9 mmol) en MeCN (6 ml) se agitó a 60 °C durante 16 hs. La mezcla se filtró y el filtrado se concentró a presión reducida. El residuo se purificó por medio de cromatografía en columna eluyendo con EtOAc/PE (1:1) para obtener 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1 -il)metil)-1 -(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo (250 mg, 55%, aceite amarillo) como una mezcla de cuatro estereoisómeros. La mezcla de estereoisómeros se separó por SFC en una columna quiral mediante el uso de la condición 1 siguiente para obtener los picos limpios 1, 3 y 4, junto con el pico 2 que no era puro. El pico 2 se repurificó por SFC mediante el uso de la condición 2. Los tiempos de retención indicados se refieren a la condición SFC 1. La estereoquímica relativa se asignó por medio de RMN 2D. No se asignó la configuración absoluta de cada isómero.
Pico 1 (tiempo de retención 5,6 min): trans 2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 1.
Pico 2 (tiempo de retención 5,8 min): trans 2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 2.
Pico 3 (tiempo de retención 6,4 min): cis 2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 1.
Pico 4 (tiempo de retención 6,9 min): cis 2-((4-(6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 2.
Condición SFC 1: Columna: AD (250 mm x 30 mm, 5 micrómetros); Fase móvil: CO2 c/ 35% de EtOH (0,1% de NH4OH); Caudal: 70 ml/min; Longitud de onda: 220 nm.
Condición SFC 2: Columna: AD (250 mm x 30 mm, 5 micrómetros); Fase móvil: CO2 con 40% de iPrOH (0,1% de NH4OH); Caudal: 60 ml/min; Longitud de onda: 220 nm
Etapa 2:
Los ésteres metílicos de la Etapa 1 se convirtieron en los ácidos libres por medio de tratamiento con NaOH en MeOH como se ha descrito anteriormente para obtener los cuatro ejemplos del título.
Ejemplo 13A-01 (a partir de trans 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 1): RMN de 1H (600MHz, CD3OD) 8: 8,32 (d, 1H), 8,01 (dd, 1H), 7,77 (d, 1H), 7,68 (dd, 1H), 7,49 (t, 1H), 7,17 a 7,31 (m, 2H), 6,97 (d, 1H), 6,76 (d, 1H), 5,37 a 5,50 (m, 2H), 4,75 a 4,86 (m, 2H), 4,62 (t, 2H), 4,19 (br s, 1H), 3,76 (t, 2H), 3,66 (d, 1H), 2,49 (ddd, 1H), 2,30 (m, 1H), 2,16 a 2,24 (m, 1H), 2,10 (dt, 1H), 1,62 (d, 2H); LC-MS(ES+): 567,1 (M+H).
Ejemplo 13A-02 (a partir de trans 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 2): RMN de 1H (400 MHz, CD3OD) 8 8,31 (d, 1H), 8,00 (dd, 1H), 7,76 (d, 1H), 7,67 (dd, 1H), 7,48 (t, 1H), 7,22 (m, 2H), 6,96 (d, 1H), 6,75 (d, 1H), 5,47 a 5,38 (m, 2H), 4,80 (m, 1H), 4,61 (m, 2H), 4,18 (m, 1H), 3,80 a 3,59 (m, 3H), 2,48 (m, 1H), 2,29 (m, 1H), 2,19 (m, 1H), 2,09 (m, 1H), 1,61 (d, 3H); LC-MS(ES+): 567,1 (M+H).
Ejemplo 13A-03 (a partir del cis 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 1): RMN de 1H (400 MHz, CD3OD) 8 8,34 (d, 1H), 8,05 (dd, 1H), 7,81 (d, 1H), 7,73 a 7,64 (m, 1H), 7,53 (m, 1H), 7,26 (m, 2H), 6,94 (d, 1H), 6,76 (d, 1H), 5,48 (s, 2H), 5,12 (d, 1H), 4,77 (d, 1H), 4,64 (m, 2H), 4,01 a 3,82 (m, 2H), 3,78 (m, 2H), 3,52 a 3,42 (m, 1H), 3,15 (m, 1H), 2,35 a 2,05 (m, 4H), 1,57 (d, 3H); LC-MS(ES+): 567,1 (M+H).
Ejemplo 13A-04 (a partir del cis 2-((6-((4-cloro-2-fluorobencil)oxi)piridin-2-il)-2-metilpiperidin-1-il)metil)-1-(2-metoxietil)-1H-benzo[d]imidazol-6-carboxilato de metilo enantiómero 2): RMN de 1H (400 MHz, CD3OD) 8 8,34 (d, 1H), 8,05 (dd, 1H), 7,81 (d, 1H), 7,70 a 7,64 (m, 1H), 7,53 (t, 1H), 7,26 (m, 2H), 6,94 (d, 1H), 6,76 (d, 1H), 5,48 (s, 2H), 5,12 (d, 1H), 4,77 (d, 1H), 4,64 (t, 2H), 3,99 a 3,83 (m, 2H), 3,78 (t, 2H), 3,52 a 3,42 (m, 1H), 3,19 a 3,09 (m, 1H), 2,31 a 2,05 (m, 4H), 1,57 (d, 3H); LC-MS(ES+): 567,1 (M+H).
Los ésteres metílicos de los compuestos enumerados en la Tabla 8 a continuación se prepararon mediante el uso de procedimientos análogos a los descritos anteriormente para la síntesis de los Ejemplos 10A-01 mediante el uso de 2-aminometiltetrahidrofurano racémico o 3-aminometiltetrahidrofurano y otros materiales de partida apropiados que están disponibles comercialmente, preparados mediante el uso de preparaciones muy conocidas por los expertos en la técnica, o preparados de manera análoga a las vías descritas anteriormente para otros intermedios. Los estereoisómeros del THF se separaron por SFC para dar los intermedios del éster como estereoisómeros simples. A continuación, los ésteres metílicos se hidrolizaron como se ha descrito para el ejemplo 10A-01 para proporcionar los compuestos enumerados en la Tabla 8. Los tiempos de retención y los procedimientos de cromatografía para los intermedios de ésteres metílicos se muestran en la tabla. No se asignó la estereoquímica del estereocentro del THF en cada compuesto.
Tabla 8



* Procedimientos de separación y tiempos de retención para los ésteres metílicos de los ejemplos:
Procedimiento A: Procedimiento de preparación: Columna: AD (250 mm x 30 mm, 10 micrómetros); Fase móvil: CO2 con 50% de MeOH (0,1% de NH4OH); Caudal: 80 ml/min; Longitud de onda: 220 nm. Procedimiento analítico: Columna: AD (50 mm x 4,6 mm, 3 micrómetros); Fase móvil: CO2 con 40% de EtOH (0,05% de NHEt2); Caudal: 4 ml/min; Longitud de onda: 220 nm
Procedimiento B: Columna: AD (250 mm x 30 mm, 10 micrómetros); Fase móvil: CO2 con 40% de MeOH (0,1% de NH4OH); Caudal: 80 ml/min; Longitud de onda: 220 nm
Procedimiento C: Columna: IC (250 mm x 30 mm, 10 micrómetros); Fase móvil: CO2 con 50% de MeOH (0,1% de NH4OH); Caudal: 80 ml/min; Longitud de onda: 220 nm
Procedimiento D: Columna: AD (250 mm x 30 mm, 10 micrómetros); Fase móvil: CO2 con 50% de MeOH (0,1% de NH4OH); Caudal: 80 ml/min; Longitud de onda: 220 nm
Procedimiento E: Columna: OD (250 mm x 30 mm, 10 micrómetros); Fase móvil: CO2 con 45% de EtOH (0,1% de NH4OH); Caudal: 70 ml/min; Longitud de onda: 220 nm
Procedimiento F: Columna: OJ (250 mm x 30 mm, 10 micrómetros); Fase móvil: CO2 c/ 30% de EtOH (0,1 NH4OH); Caudal: 80 ml/min; Longitud de onda: 220 nm
CHO GLP-1R Clon H6 - Ensayo 1
La actividad agonista mediada por el GLP-1R se determinó con un ensayo funcional basado en células mediante el uso de un kit de detección de AMPc HTRF (Fluorescencia Homogénea Resuelta en el Tiempo) (cAMP HI Range Assay Kit; CisBio cat. Núm. 62AM6PEJ) que mide los niveles de AMPc en la célula. El procedimiento es un inmunoensayo competitivo entre el AMPc nativo producido por las células y el AMPc exógeno marcado con el colorante d2. La unión del trazador se visualiza con un mAb anti-cAMP marcado con Cryptate. La señal específica (es decir, la transferencia de energía) es inversamente proporcional a la concentración de AMPc en el estándar o en la muestra experimental.
La secuencia de codificación del GLP-1R humano (Secuencia de Referencia del NCBI NP_002053,3, que incluyen la variante natural Gly168Ser) se subclonó en pcDNA3 (Invitrogen) y se aisló una línea celular que expresaba de forma estable el receptor (designada Clon H6 ). Los análisis de unión por saturación (procedimiento de ensayo de filtración) mediante el uso de 125I-GLP- 17-36 (Perkin Elmer) mostraron que las membranas plasmáticas derivadas de esta línea celular expresan una alta densidad de GLP-1R (Kd: 0,4 nM, Bmáx.: 1900 fmol/mg de proteína).
Las células se retiraron de la criopreservación, se resuspendieron en 40 ml de solución salina tamponada con fosfato de Dulbecco (DPBS - Lonza Cat. Núm. 17-512Q) y se centrifugaron a 800 x g durante 5 min a 22 °C. A continuación, el gránulo celular se resuspendió en 10 ml de medio de crecimiento [DMEM/F12 1:1 con HEPES, L-Gln, 500 ml (DMEM/F12 Lonza Cat. Núm. 12-719F), 10% de suero fetal bovino inactivado por calor (Gibco Cat. Núm. 16140-071), 5 ml de Pen-Strep 100X (Gibco Cat. Núm. 15140-122), 5 ml de L-Glutamina 100X (Gibco Cat. Núm. 25030-081) y 500 pg/ml de Geneticina (G418) (Invitrogen Núm. 10131035)]. Se contó una muestra de 1 ml de la suspensión celular en medio de crecimiento en un Becton Dickinson ViCell para determinar la viabilidad celular y el recuento de células por ml. A continuación, se ajustó la suspensión celular restante con medios de crecimiento para obtener 2.000 células viables por pocillo mediante el uso de un dispensador de reactivos Matrix Combi Multidrop, y las células se dispensaron en una placa de ensayo blanca de 384 pocillos tratada con cultivo de tejidos (Corning 3570). A continuación, la placa de ensayo se incubó durante 48 horas a 37 °C en un entorno humidificado con un 5% de dióxido de carbono.
Se diluyeron concentraciones variables de cada compuesto a ensayar (en DMSO) en un tampón de ensayo (HBSS con Calcio/Magnesio (Lonza/BioWhittaker cat. Núm. 10-527F) /0,1% de BSA (Sigma Aldrich cat. Núm. A7409-1L)/20 mM HEPES (Lonza/BioWhittaker cat. Núm. 17-737E) que contenía 100 micrómetros de 3-isobutil-1-metilxantina (IBMX; Sigma cat. Núm. I5879). La concentración final de DMSO es del 1%.
Después de 48 horas, se retiró el medio de crecimiento de los pozos de la placa de ensayo, y las células se trataron con 20 pl del compuesto diluido en serie en tampón de ensayo durante 30 minutos a 37 °C en un entorno
humidificado con 5% de dióxido de carbono. Tras la incubación de 30 minutos, se añadieron 10 |jl de AMPc d2 marcado y 10 j l de anticuerpo anti AMPc (ambos diluidos 1:20 en tampón de lisis celular; como se describe en el protocolo de ensayo del fabricante) a cada pocillo de la placa de ensayo. A continuación, las placas se incubaron a temperatura ambiente y, tras 60 minutos, se leyeron los cambios en la señal HTRF con un lector de placas multietiqueta Envision 2104, mediante el uso de una excitación de 330 nm y emisiones de 615 y 665 nm. Los datos brutos se convirtieron en nM de AMPc por interpolación de una curva estándar de AMPc (como se describe en el protocolo de ensayo del fabricante) y se determinó el porcentaje de efecto en relación con una concentración saturante del agonista completo GLP- 17-36 (1 micrómetros) incluida en cada placa. Las determinaciones de la CE50 se hicieron a partir de las curvas de dosis-respuesta del agonista analizadas con un programa de ajuste de curvas mediante el uso de una ecuación logística de dosis-respuesta de 4 parámetros.
CHO GLP-1 R Clon C6 - Ensayo 2
La actividad agonista mediada por el GLP-1R se determinó con un ensayo funcional basado en células mediante el uso de un kit de detección de AMPc HTRF (Fluorescencia Homogénea Resuelta en el Tiempo) (cAMP HI Range Assay Kit; Cis Bio Cat. Núm. 62AM6PEJ) que mide los niveles de AMPc en la célula. El procedimiento es un inmunoensayo competitivo entre el AMPc nativo producido por las células y el AMPc exógeno marcado con el colorante d2. La unión del trazador se visualiza con un mAb anti-cAMP marcado con Cryptate. La señal específica (es decir, la transferencia de energía) es inversamente proporcional a la concentración de AMPc en un estándar o en una muestra experimental.
La secuencia codificante del GLP-1R humano (Secuencia de Referencia NCBI NP_002053,3, que incluyen la variante natural Leu260Phe) se subclonó en pcDNA5-FRT-TO y se aisló una línea celular CHO clonal que expresaba de forma estable una baja densidad de receptores mediante el uso del sistema Flp-In™ T-Rex™, como lo describe el fabricante (ThermoFisher). Los análisis de unión por saturación (procedimiento de ensayo de filtración) mediante el uso de 125I-GLP-1 (Perkin Elmer) mostraron que las membranas plasmáticas derivadas de esta línea celular (designada como clon C6 ) expresan una baja densidad de GLP-1R (Kd 0,3 nM, Bmáx.: 240 fmol/mg de proteína), en relación con la línea celular clon H6.
Las células se retiraron de la criopreservación, se resuspendieron en 40 ml de solución salina tamponada con fosfato de Dulbecco (DPBS - Lonza Cat. Núm. 17-512Q) y se centrifugaron a 800 x g durante 5 min a 22 °C. Se aspiró el DPBS y se resuspendió el gránulo celular en 10 ml de medio de crecimiento completo (DMEM:F12 11Mezcla con HEPES, L-Gln, 500 ml de DMEM/F12 Lonza Cat. Núm. 12-719F), 10% de suero fetal bovino inactivado por calor (Gibco Cat. Núm. 16140-071), 5 ml de Pen-Strep 100X (Gibco Cat. Núm. 15140-122), 5 ml de L-Glutamina 100X (Gibco Cat. Núm. 25030-081), 700 jg/ml de Higromicina (Invitrogen Cat. Núm. 10687010) y 15 jg/ml de Blasticidina (Gibco Cat. Núm. R21001). Se contó una muestra de 1 ml de la suspensión celular en medio de crecimiento en un Becton Dickinson ViCell para determinar la viabilidad celular y el recuento de células por ml. A continuación, se ajustó la suspensión celular restante con medios de crecimiento para obtener 1600 células viables por pocillo mediante el uso de un dispensador de reactivos Matrix Combi Multidrop, y las células se dispensaron en una placa de ensayo blanca de 384 pocillos tratada con cultivo de tejidos (Corning 3570). A continuación, la placa de ensayo se incubó durante 48 hs a 37 °C en un entorno humidificado (95% de O2, 5% de CO2)
Se diluyeron concentraciones variables de cada compuesto a ensayar (en DMSO) en el tampón de ensayo [HBSS con calcio/magnesio (Lonza/BioWhittaker Cat. Núm. 10-527F) /0,1% de BsA (Sigma Aldrich Cat. Núm. A7409-1L)/20 mM HEPES (Lonza/BioWhittaker Cat. Núm. 17-737E)] que contenía 100 micrómetros de 3-isobutil-1-metilxantina (IBMX; Sigma Cat. Núm. I5879). La concentración final de DMSO en la mezcla de compuesto/tampón de ensayo es del 1 %.
Después de 48 horas, se retiró el medio de crecimiento de los pozos de la placa de ensayo y las células se trataron con 20 j l del compuesto diluido en serie en tampón de ensayo durante 30 min a 37 °C en un entorno humidificado (95% de O2, 5% de CO2). Tras la incubación de 30 min, se añadieron 10 j l de AMPc d2 marcado y 10 j l de anticuerpo anti AMPc (ambos diluidos 1:20 en tampón de lisis celular; como se describe en el protocolo de ensayo del fabricante) a cada pocillo de la placa de ensayo. A continuación, las placas se incubaron a temperatura ambiente y, tras 60 minutos, se leyeron los cambios en la señal HTRF con un lector de placas multietiqueta Envision 2104, mediante el uso de una excitación de 330 nm y emisiones de 615 y 665 nm. Los datos brutos se convirtieron en nM de AMPc por interpolación de una curva estándar de AMPc (como se describe en el protocolo de ensayo del fabricante) y se determinó el porcentaje de efecto en relación con una concentración saturante del agonista completo GLP-1 (1 micrómetros) incluida en cada placa. Las determinaciones de la CE50 se hicieron a partir de curvas de respuesta a la dosis de agonista analizadas con un programa de ajuste de curvas mediante el uso de una ecuación logística de respuesta a la dosis de 4 parámetros.
En la Tabla 9, los datos del ensayo se presentan con dos (2) cifras significativas como la media geométrica (CE50s) y la media aritmética (Emáx.) basada en el número de réplicas enumeradas (Número). Una celda en blanco significa que no hay datos para ese Ejemplo o que no se ha calculado el Emáx.
Tabla 9






Claims (27)
1. Un compuesto de la Fórmula I

o una sal aceptable para uso farmacéutico del mismo, en la que
cada R1 es independientemente halógeno, -CN, alquilo C1-3, o-Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 está sustituido con 0 a 3 átomos de F;
m es 0, 1, 2 o 3.
cada R2 es independientemente F, Cl, o -CN;
p es 0, 1 o 2 ;
cada R3 es independientemente F, -OH, -CN, alquilo C1-3, -Oalquilo C1-3, o -cicloalquilo C3-4, o 2 R3 se pueden ciclizar juntos para formar -espirocicloalquilo C3-4, en el que el alquilo de alquilo C1-3 y Oalquilo C1.3, el cicloalquilo, o el espirocicloalquilo pueden ser sustituidos como la valencia lo permita con 0 a 3 átomos de F y con 0 a 1 -OH;
q es 0, 1 o 2 ;
Y es CH o N;
R4 es alquilo C1-3, alquileno Co-3-cicloalquilo C3-6, alquileno C0.3-R5, o alquileno C1-3-R6, en el que dicho alquilo puede estar sustituido como la valencia lo permita con 0 a 3 sustituyentes seleccionados independientemente de 0 a 3 átomos de F y 0 a 1 sustituyente seleccionado de alquileno C0-1-CN alquileno C0-1-OR°, y -N(RN)2, y en el que dicho alquileno y cicloalquilo pueden estar sustituidos independientemente como la valencia lo permita con 0 a 2 sustituyentes seleccionados independientemente de 0 a 2 átomos de F y 0 a 1 sustituyente seleccionado de alquileno C0-1-CN, alquileno C0-1-OR°, y -N(RN)2;
R5 es un heterocicloalquilo de 4 a 6 miembros, en el que dicho heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 1 oxo (=O),
0 a 1 -CN,
0 a 2 átomos de F, y
0 a 2 sustituyentes seleccionados independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede ser sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F,
0 a 1 -CN, y
0 a 1 -OR°;
R6 es un heteroarilo de 5 a 6 miembros, en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 halógenos,
0 a 1 sustituyente seleccionado entre -OR° y -N(RN)2, y
0 a 2 alquilo C1-3, en el que el alquilo puede estar sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F, y
0 a 1 -OR°;
cada R° es independientemente H, o alquilo C1-3, en el que alquilo C1-3 puede estar sustituido con 0 a 3 átomos de F;
cada RN es independientemente H o alquilo C1-3;
Z1 es CH o N;
Z2 y Z3 son cada uno independientemente -CRZ o N, a condición de que cuando Z1 o Z3 sea N, Z2 sea -CRZ; y
cada RZ es independientemente H, F, Cl, o -CH3.
2. El compuesto de la reivindicación 1, en el que el compuesto es un compuesto de la Fórmula II

o una sal aceptable para uso farmacéutico del mismo, en el que
m es 0 o 1 ;
R2 es F;
p es 0 o 1 ; y
q es 0 o 1.
3. El compuesto de la reivindicación 1 o la reivindicación 2, en el que el compuesto es un compuesto de la Fórmula III

o una sal aceptable para uso farmacéutico del mismo, en el que
m es 0 o 1 ;
R2 es F;
p es 0 o 1 ;
R3 es alquilo C1-2, en el que alquilo C1-2 puede estar sustituido, como la valencia lo permita, con 0 a 3 átomos de F; y
q es 0 o 1.
4. El compuesto de cualquiera de las reivindicaciones 1 a 3, en el que cada R1 es independientemente F, Cl, -CN, -CH3, o -CF3, o una sal aceptable para uso farmacéutico del mismo.5
5. El compuesto de cualquiera de las reivindicaciones 1 a 4, en el que el heterocicloalquilo es

en el que el heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 1 oxo (O=),
0 a 1 -CN,
0 a 2 átomos de F, y
0 a 2 sustituyentes seleccionados independientemente de alquilo C1-3 y -Oalquilo C1-3, en el que el alquilo de alquilo C1-3 y Oalquilo C1-3 puede estar sustituido independientemente con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F,
0 a 1 -CN, y
0 a 1 -OR°,
o una sal aceptable para uso farmacéutico del mismo.
6. El compuesto de cualquiera de las reivindicaciones 1 a 5, en el que R4
es -CH2-R5, en el que R5 es el heterocicloalquilo de 4 a 5 miembros, en el que dicho heterocicloalquilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 átomos de F, y
0 a 1 sustituyente seleccionado entre -OCH3 y -CH2OCH3;
o una sal aceptable para uso farmacéutico del mismo.
7. El compuesto de cualquiera de las reivindicaciones 1 a 4, en el que dicho heteroarilo es

y en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 halógenos,
0 a 1 sustituyente seleccionado entre -OR° y -N(RN)2, y
0 a 2 alquilo C1-3, en el que el alquilo puede estar sustituido con 0 a 3 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 3 átomos de F, y
0 a 1 -OR°;
o una sal aceptable para uso farmacéutico del mismo.
8. El compuesto de cualquiera de las reivindicaciones 1 a 4, y 7, en el que R4 es -CH2-R6, en el que R6 es el heteroarilo de 5 miembros, en el que dicho heteroarilo puede estar sustituido con 0 a 2 sustituyentes como la valencia lo permita seleccionados independientemente de:
0 a 2 halógenos, en los que el halógeno se selecciona independientemente entre F y Cl, 0 a 1 -OCH3, y 0 a 1 -CH3, -CH2CH3, -CF3, o -CH2CH2OCH3;
o una sal aceptable para uso farmacéutico del mismo.
9. Un compuesto de acuerdo con la reivindicación 1, que es
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2R)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[4-(6-{[(4-ciano-2-fluorofenil)(metil-d2)]oxi}piridin-2-il)piperidin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H
imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-doro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1 H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(3R)-tetrahidrofurano-3-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(3R)-tetrahidrofurano-3-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(3S)-tetrahidrofurano-3-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2R)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2R)-tetrahidrofurano-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(28)-4-{6-[(2,4-difluorobencil)oxi]-5-fluoropiridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(28)-4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(28)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(28)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-3-[(2S)-oxetan-2-ilmetil]-3H-imidazo[4,5-b]piridin-5-carboxílico; o
ácido 2-{[(2S)-4-{6-[(4-cianobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1 H-benzimidazol-6-carboxílico;
o una sal aceptable para uso farmacéutico de la misma.
10. Un compuesto de acuerdo con la reivindicación 1, que es

o una sal aceptable para uso farmacéutico del mismo.
11. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
12. El compuesto de la reivindicación 11 que es un ácido libre.
13. El compuesto de la reivindicación 11 que es una sal aceptable para uso farmacéutico.
14. El compuesto de la reivindicación 13 que es una sal tris.
15. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
16. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]-5-fluoropiridin-2-il}piperidin-1-il)metil]-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
17. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-{[4-(6-{[(4-ciano-2-fluorofenil)(metild2)]oxi}piridin-2-il)piperidin-1-il]metil}-1-[(2S)-oxetan-2-ilmetil]-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
18. Un compuesto de acuerdo con la reivindicación 1, que es
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metil-1H-1,2,3-triazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(etil-1H-imidazol-5-il)metil]-1H
benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-doro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-4-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(2,4-difluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-[(1-etil-1H-1,2,3-triazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,2-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,2-oxazol-3-ilmetil)-1 H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1 H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1H-1,2,3-triazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-etil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metil-1H-imidazol-5-il)metil]-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-(1,3-oxazol-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxílico; o
ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-3-(1,3-oxazol-2-ilmetil)-3H-imidazo[4,5-b]piridin-5-carboxílico;
o una sal aceptable para uso farmacéutico del mismo.
19. Un compuesto de acuerdo con la reivindicación 1, que es

o una sal aceptable para uso farmacéutico del mismo.
20. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-5-ilmetil)-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
21. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso farmacéutico del mismo.
22. Un compuesto de acuerdo con la reivindicación 1 que es el ácido 2-[(4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(1,3-oxazol-2-ilmetil)-1H-benzimidazol-6-carboxílico, o una sal aceptable para uso
farmacéutico del mismo.
23. Un compuesto de acuerdo con la reivindicación 1, que es
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperidin-1-il)metil]-7-fluoro-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-7-fluoro-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cianobencil)oxi]piridin-2-il}piperazin-1-il)metil]-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cianobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-ciano-2-fluorobencil)oxi]piridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cloro-2-fluorobencil)oxi]-5-fluoropiridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-{[(2S)-4-{6-[(4-cianobencil)oxi]-5-fluoropiridin-2-il}-2-metilpiperazin-1-il]metil}-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico;
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-(2-metoxietil)-1H-benzimidazol-6-carboxílico; o
ácido 2-[(4-{6-[(4-cloro-2-fluorobencil)oxi]piridin-2-il}piperidin-1-il)metil]-1-[(1-metociclobutil)metil]-1 H-benzimidazol-6-carboxílico;
o una sal aceptable para uso farmacéutico del mismo.
24. Una composición farmacéutica que comprende el compuesto de la Fórmula I de acuerdo con cualquiera de las reivindicaciones 1 a 23, o una sal aceptable para uso farmacéutico del mismo. y un excipiente aceptable para uso farmacéutico.
25. Un compuesto de cualquiera de las reivindicaciones 1 a 23 o una sal aceptable para uso farmacéutico del mismo, para su uso en un procedimiento de tratamiento de enfermedades cardiometabólicas y asociadas que comprende la administración a un mamífero que necesita dicho tratamiento de una cantidad eficaz para uso terapéutico en la que la enfermedad se selecciona entre T1D, T2DM, prediabetes, T1D idiopática, LADa , EOD, YOAD, MODY, diabetes relacionada con la malnutrición, diabetes gestacional, hiperglucemia, resistencia a la insulina, resistencia a la insulina hepática, intolerancia a la glucosa, neuropatía diabética, nefropatía diabética, enfermedad renal, retinopatía diabética, disfunción adipocítica, depósito adiposo visceral, apnea del sueño, obesidad, trastornos alimentarios, aumento de peso por el uso de otros agentes, ansia excesiva de azúcar, dislipidemia, hiperinsulinemia, NAFLD, NASH, fibrosis, cirrosis, carcinoma hepatocelular, enfermedad cardiovascular, aterosclerosis, enfermedad arterial coronaria, enfermedad vascular periférica, hipertensión, disfunción endotelial, alteración de la distensibilidad vascular, insuficiencia cardíaca congestiva, infarto de miocardio, accidente cerebrovascular, accidente cerebrovascular hemorrágico, accidente cerebrovascular isquémico, lesión cerebral traumática, hipertensión pulmonar, reestenosis tras angioplastia, claudicación intermitente, lipemia postprandial, acidosis metabólica, cetosis, artritis, osteoporosis, Enfermedad de Parkinson, hipertrofia ventricular izquierda, enfermedad arterial periférica, degeneración macular, cataratas, glomeruloesclerosis, insuficiencia renal crónica, síndrome metabólico, síndrome X, síndrome premenstrual, angina de pecho, trombosis, aterosclerosis, ataques isquémicos transitorios, reestenosis vascular, alteración del metabolismo de la glucosa, condiciones de alteración de la glucosa plasmática en ayunas, hiperuricemia, gota, disfunción eréctil, trastornos de la piel y del tejido conectivo, psoriasis, ulceraciones en los pies, colitis ulcerosa, lipoproteinemia hiper apo B, enfermedad de Alzheimer, esquizofrenia, deterioro de la cognición, enfermedad inflamatoria intestinal, síndrome del intestino corto, enfermedad de Crohn, colitis, síndrome del intestino irritable, prevención o tratamiento del síndrome del ovario poliquístico y tratamiento de la adicción.
26. Un compuesto, o una sal aceptable para uso farmacéutico del mismo, para su uso en un procedimiento de tratamiento, como se reivindica en la reivindicación 25, en el que la enfermedad es T2DM, NASH, NAFLD u obesidad.
27. Un compuesto, o una sal aceptable para uso farmacéutico del mismo, para su uso en un procedimiento de tratamiento, de acuerdo con la reivindicación 25 o 26, en el que el mamífero es un ser humano.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662435533P | 2016-12-16 | 2016-12-16 | |
| PCT/IB2017/057577 WO2018109607A1 (en) | 2016-12-16 | 2017-12-01 | Glp-1 receptor agonists and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2934789T3 true ES2934789T3 (es) | 2023-02-27 |
| ES2934789T9 ES2934789T9 (es) | 2023-04-26 |
Family
ID=60702906
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17817174T Active ES2934789T3 (es) | 2016-12-16 | 2017-12-01 | Agonistas del receptor glp-1 y usos del mismo |
Country Status (40)
| Country | Link |
|---|---|
| US (7) | US10208019B2 (es) |
| EP (1) | EP3555064B9 (es) |
| JP (2) | JP6637641B1 (es) |
| KR (2) | KR102314286B1 (es) |
| CN (1) | CN110325530B (es) |
| AR (1) | AR110387A1 (es) |
| AU (1) | AU2017374860B2 (es) |
| CA (1) | CA2988721C (es) |
| CL (1) | CL2019001651A1 (es) |
| CO (1) | CO2019006046A2 (es) |
| CR (1) | CR20190289A (es) |
| CU (1) | CU24573B1 (es) |
| CY (1) | CY1125716T1 (es) |
| DK (1) | DK3555064T5 (es) |
| DO (1) | DOP2019000166A (es) |
| EA (1) | EA037318B1 (es) |
| EC (1) | ECSP19041071A (es) |
| ES (1) | ES2934789T3 (es) |
| FI (1) | FI3555064T3 (es) |
| GE (2) | GEP20217265B (es) |
| HR (1) | HRP20221437T1 (es) |
| HU (1) | HUE060533T2 (es) |
| IL (1) | IL267288B (es) |
| LT (1) | LT3555064T (es) |
| MA (1) | MA56480B1 (es) |
| MD (1) | MD3555064T2 (es) |
| MX (1) | MX387259B (es) |
| MY (1) | MY195385A (es) |
| PE (1) | PE20191501A1 (es) |
| PH (1) | PH12019501360B1 (es) |
| PL (1) | PL3555064T3 (es) |
| PT (1) | PT3555064T (es) |
| RS (1) | RS63849B9 (es) |
| RU (1) | RU2740135C1 (es) |
| SI (1) | SI3555064T1 (es) |
| TW (1) | TWI713809B (es) |
| UA (1) | UA122035C2 (es) |
| UY (1) | UY37514A (es) |
| WO (1) | WO2018109607A1 (es) |
| ZA (1) | ZA201904616B (es) |
Families Citing this family (178)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI735681B (zh) | 2016-10-24 | 2021-08-11 | 瑞典商阿斯特捷利康公司 | 化合物 |
| EP3555064B9 (en) | 2016-12-16 | 2023-03-01 | Pfizer Inc. | Glp-1 receptor agonists and uses thereof |
| TWI831738B (zh) | 2016-12-16 | 2024-02-11 | 美商瑞塔醫藥有限責任公司 | 用於抑制RORγ及其他用途的嘧啶三環烯酮衍生物 |
| JOP20190144A1 (ar) | 2016-12-16 | 2019-06-16 | Janssen Pharmaceutica Nv | إيميدازو بيرولو بيريدين كمثبطات لعائلة jak الخاصة بإنزيمات الكيناز |
| LT3555097T (lt) | 2016-12-16 | 2022-08-25 | Janssen Pharmaceutica Nv | Imidazo[4,5-d]pirolo[2,3-b]piridino junginiai kaip janus kinazės inhibitoriai |
| PT3494116T (pt) | 2017-01-30 | 2020-01-28 | Astrazeneca Ab | Moduladores de recetores de estrogénio |
| US12372530B2 (en) * | 2017-11-15 | 2025-07-29 | Beth Israel Deaconess Medical Center, Inc. | Markers for the diagnosis and treatment of non-alcoholic steatohepatitis (NASH) and advanced liver fibrosis |
| WO2019222677A1 (en) | 2018-05-18 | 2019-11-21 | Incyte Corporation | Fused pyrimidine derivatives as a2a / a2b inhibitors |
| CA3045644C (en) * | 2018-06-13 | 2024-01-16 | Pfizer Inc. | Glp-1 receptor agonists and uses thereof |
| IL279348B2 (en) | 2018-06-15 | 2026-01-01 | Reata Pharmaceuticals Inc | Decahydro-2h-naphth[1,2-d]imidazole compounds for inhibition of IL-17 and ROR-gamma |
| TW202016110A (zh) | 2018-06-15 | 2020-05-01 | 比利時商健生藥品公司 | Jak激酶家族之小分子抑制劑 |
| ES2943510T3 (es) | 2018-06-15 | 2023-06-13 | Pfizer | Agonistas del receptor GLP-1 y usos del mismo |
| WO2019246461A1 (en) | 2018-06-20 | 2019-12-26 | Reata Pharmaceuticals, Inc. | Cysteine-dependent inverse agonists of nuclear receptors ror-gamma/ror-gamma-t and methods of treating diseases or disorders therewith |
| GEP20237548B (en) | 2018-07-05 | 2023-10-10 | Incyte Corp | Fused pyrazine derivatives as a2a /a2b inhibitors |
| CR20210110A (es) | 2018-08-31 | 2021-05-13 | Pfizer | Combinaciones para tratamiento de ehna/ehgna y enfermedades relacionadas |
| CR20210341A (es) | 2018-11-22 | 2021-11-25 | Qilu Regor Therapeutics Inc | Agonistas de glp-ir y usos de los mismos |
| CN111320609A (zh) | 2018-12-13 | 2020-06-23 | 拓臻股份有限公司 | 一种THRβ受体激动剂化合物及其制备方法和用途 |
| TWI829857B (zh) | 2019-01-29 | 2024-01-21 | 美商英塞特公司 | 作為a2a / a2b抑制劑之吡唑并吡啶及三唑并吡啶 |
| AU2020256647B2 (en) | 2019-04-12 | 2025-07-31 | Qilu Regor Therapeutics Inc. | GLP-1R agonists and uses thereof |
| US10954221B2 (en) | 2019-04-12 | 2021-03-23 | Qilu Regor Therapeutics Inc. | GLP-1R agonists and uses thereof |
| CA3138642A1 (en) | 2019-04-29 | 2020-11-05 | Confo Therapeutics N.V. | Screening methods and assays for use with transmembrane proteins, in particular with gpcrs |
| WO2020234726A1 (en) * | 2019-05-20 | 2020-11-26 | Pfizer Inc. | Combinations comprising benzodioxol as glp-1r agonists for use in the treatment of nash/nafld and related diseases |
| WO2020261144A1 (en) | 2019-06-28 | 2020-12-30 | Pfizer Inc. | 5-(thiophen-2-yl)-1h-tetrazole derivatives as bckdk inhibitors useful for treating various diseases |
| TW202115086A (zh) | 2019-06-28 | 2021-04-16 | 美商輝瑞大藥廠 | Bckdk抑制劑 |
| TWI751585B (zh) * | 2019-06-28 | 2022-01-01 | 美商美國禮來大藥廠 | 類升糖素肽1受體促效劑 |
| WO2021018023A1 (zh) * | 2019-08-01 | 2021-02-04 | 济南泰达领创医药技术有限公司 | 小分子glp-1受体调节剂 |
| CN114945366B (zh) | 2019-09-13 | 2025-01-07 | 林伯士萨顿公司 | Hpk1拮抗剂和其用途 |
| TWI771766B (zh) | 2019-10-04 | 2022-07-21 | 美商輝瑞股份有限公司 | 二醯基甘油醯基轉移酶2 抑制劑 |
| CA3157525A1 (en) * | 2019-10-25 | 2021-04-29 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| KR20210059584A (ko) * | 2019-11-15 | 2021-05-25 | 일동제약(주) | Glp-1 수용체 작용제 및 이의 용도 |
| JP7386997B2 (ja) * | 2019-11-15 | 2023-11-27 | イルドン ファーマシューティカル カンパニー リミテッド | Glp-1受容体アゴニストおよびその使用 |
| KR20210069000A (ko) * | 2019-12-02 | 2021-06-10 | 현대약품 주식회사 | Glp-1 수용체 작용제 |
| MX2022007105A (es) * | 2019-12-10 | 2022-07-11 | Pfizer | Formas solidas de sal de 1,3-dihidroxi-2-(hidroximetil)propan-2-am ina de acido 2-((4-((s)-2-(5-cloropiridin-2-il)-2-metilbenzo[d][1, 3] dioxol-4-il)piperidin-1-il)metil)-1-(((s)-oxetan-2-il) metil)-1h- benzo[d]imidazol-6-carboxilico. |
| FI4073051T3 (fi) | 2019-12-10 | 2024-03-28 | Lilly Co Eli | Menetelmä ja välituote oksetan-2-yylimetanamiinin valmistamiseksi |
| WO2021154796A1 (en) | 2020-01-29 | 2021-08-05 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| CN119841865A (zh) | 2020-02-07 | 2025-04-18 | 加舒布鲁姆生物公司 | 杂环glp-1激动剂 |
| WO2021160127A1 (en) * | 2020-02-13 | 2021-08-19 | Gasherbrum Bio, Inc. | Heterocyclic glp-1 agonists |
| JP2022058085A (ja) | 2020-02-24 | 2022-04-11 | ファイザー・インク | ジアシルグリセロールアシルトランスフェラーゼ2阻害剤とアセチル-CoAカルボキシラーゼ阻害剤との組合せ |
| JP2021134211A (ja) | 2020-02-24 | 2021-09-13 | ファイザー・インク | Nafld/nashおよび関連疾患の処置のための組合せ |
| MY207021A (en) * | 2020-03-18 | 2025-01-24 | Lg Chemical Ltd | Glp-1 receptor agonist, pharmaceutical composition comprising same, and method for preparing same |
| CN115279750B (zh) * | 2020-03-18 | 2024-06-21 | 株式会社Lg化学 | Glp-1受体激动剂、包含该激动剂的药物组合物及其制备方法 |
| EP4125896B1 (en) * | 2020-03-27 | 2025-05-07 | Pfizer Inc. | Treatment of type 2 diabetes with 2-[(4-{6-[(4-cyano-2-fluorobenzyl)oxy]pyridin-2-yl} piperidin-1-yl)methyl]-1-[(2s)-oxetan-2-ylmethyl]-1h-benzimidazole-6-carboxylic acid or a pharmaceutically salt thereof |
| TW202144340A (zh) * | 2020-04-03 | 2021-12-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 稠合咪唑類衍生物、其製備方法及其在醫藥上的應用 |
| CN113493447B (zh) * | 2020-04-03 | 2024-06-11 | 轩竹(北京)医药科技有限公司 | Glp-1受体激动剂 |
| PE20221838A1 (es) | 2020-04-24 | 2022-11-29 | Astrazeneca Ab | Formulaciones farmaceuticas |
| WO2021214253A1 (en) | 2020-04-24 | 2021-10-28 | Astrazeneca Ab | Dosage regimen for the treatment of cancer |
| FI4143183T3 (fi) * | 2020-04-29 | 2026-01-07 | Gasherbrum Bio Inc | Heterosyklisiä glp-1-agonisteja |
| FI4157832T3 (fi) * | 2020-05-27 | 2024-08-29 | Qilu Regor Therapeutics Inc | Glp-1r-agonistien suola- ja kidemuotoja ja niiden käyttötarkoituksia |
| PH12022553313A1 (en) * | 2020-06-04 | 2023-04-12 | Hangzhou Sciwind Biosciences Co Ltd | Five-membered heteroaromatic imidazole compound and use thereof |
| MX2022015706A (es) | 2020-06-09 | 2023-01-24 | Pfizer | Antagonistas del receptor de melanocortina 4 y usos de estos. |
| CN117645601A (zh) * | 2020-06-10 | 2024-03-05 | 重庆康丁医药技术有限公司 | 一种具有心血管益处的glp-1小分子 |
| US20230234968A1 (en) * | 2020-06-10 | 2023-07-27 | Medshine Discovery Inc. | Methyl-substituted benzobisoxazole compound and use thereof |
| CN113816948B (zh) * | 2020-06-19 | 2023-08-11 | 江苏恒瑞医药股份有限公司 | 稠合咪唑类衍生物、其制备方法及其在医药上的应用 |
| WO2021254470A1 (zh) * | 2020-06-19 | 2021-12-23 | 江苏恒瑞医药股份有限公司 | 6-氧代-3,6-二氢吡啶类衍生物、其制备方法及其在医药上的应用 |
| WO2021259309A1 (zh) * | 2020-06-24 | 2021-12-30 | 广州市恒诺康医药科技有限公司 | Glp-1受体激动剂及其药物组合物和用途 |
| PE20231181A1 (es) | 2020-08-06 | 2023-08-11 | Gasherbrum Bio Inc | Agonistas del glp-1 heterociclicos |
| TW202214622A (zh) | 2020-08-06 | 2022-04-16 | 大陸商上海齊魯銳格醫藥研發有限公司 | Glp-1r促效劑及其用途 |
| PE20231206A1 (es) * | 2020-08-21 | 2023-08-17 | Terns Pharmaceuticals Inc | Compuestos como agonistas de glp-1r |
| WO2022042691A1 (en) * | 2020-08-28 | 2022-03-03 | Gasherbrum Bio, Inc. | Heterocyclic glp-1 agonists |
| AU2021306414B2 (en) | 2020-09-01 | 2026-01-29 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Fused imidazole derivative, preparation method therefor, and medical use thereof |
| JP2023541601A (ja) | 2020-09-10 | 2023-10-03 | プレシリックス・ナームローゼ・ベンノートシヤープ | Fapに対する抗体フラグメント |
| EP4211139A4 (en) | 2020-09-10 | 2024-12-18 | Gasherbrum Bio, Inc. | HETEROCYCLIC GLP-1 AGONISTS |
| WO2022068772A1 (zh) * | 2020-09-29 | 2022-04-07 | 深圳信立泰药业股份有限公司 | 一种苯并咪唑类衍生物及其制备方法和医药用途 |
| EP4227299A4 (en) * | 2020-10-12 | 2025-04-09 | Hangzhou Zhongmeihuadong Pharmaceutical Co., Ltd. | GLP-1 RECEPTOR AGONIST, BENZIMIDAZOLONE AND ITS USE |
| JP2023546054A (ja) * | 2020-10-13 | 2023-11-01 | ガシャーブラム・バイオ・インコーポレイテッド | ヘテロ環glp-1アゴニスト |
| EP4229050A4 (en) * | 2020-10-13 | 2024-12-11 | Gasherbrum Bio, Inc. | Heterocyclic glp-1 agonists |
| TWI912388B (zh) * | 2020-10-14 | 2026-01-21 | 大陸商上海齊魯銳格醫藥研發有限公司 | Glp-1r促效劑的晶型及其用途 |
| CN114478497B (zh) * | 2020-11-12 | 2023-10-20 | 杭州中美华东制药有限公司 | 芳基烷基酸类glp-1受体激动剂及其用途 |
| EP4247804A1 (en) | 2020-11-20 | 2023-09-27 | Gilead Sciences, Inc. | Polyheterocyclic glp-1 r modulating compounds |
| US20240246959A1 (en) * | 2020-11-27 | 2024-07-25 | Shenzhen Salubris Pharmaceuticals Co., Ltd. | Benzimidazole derivative and preparation method therefor and medical use thereof |
| CN114591308B (zh) | 2020-12-03 | 2024-03-08 | 苏州闻泰医药科技有限公司 | 一类glp-1r受体激动剂化合物及其用途 |
| US20220193063A1 (en) * | 2020-12-15 | 2022-06-23 | Pfizer Inc. | Metabolites of glp1r agonists |
| US12065434B2 (en) * | 2020-12-15 | 2024-08-20 | Pfizer Inc. | Metabolites of GLP1R agonists |
| WO2022135572A1 (zh) * | 2020-12-25 | 2022-06-30 | 四川海思科制药有限公司 | 一种五并五元环衍生物及其在医药上的应用 |
| CN114805336B (zh) * | 2021-01-20 | 2026-04-14 | 江苏恒瑞医药股份有限公司 | 稠合咪唑类化合物、其制备方法及其在医药上的应用 |
| CN117043154A (zh) * | 2021-01-28 | 2023-11-10 | 卡莫特医疗有限公司 | Gpcr受体激动剂、包含其的药物组合物以及它们的使用方法 |
| CA3209593A1 (en) * | 2021-01-28 | 2022-08-04 | Carmot Therapeutics, Inc. | Gpcr receptor agonists, pharmaceutical compositions comprising the same, and methods for their use |
| CN117120090A (zh) | 2021-02-12 | 2023-11-24 | 林伯士萨顿公司 | Hpk1拮抗剂和其用途 |
| WO2022184849A1 (en) | 2021-03-04 | 2022-09-09 | Les Laboratoires Servier | Glp-1r agonists, uses and pharmaceutical compositions thereof |
| EP4301756A4 (en) | 2021-03-05 | 2025-02-26 | Nimbus Saturn, Inc. | Hpk1 antagonists and uses thereof |
| EP4304711A1 (en) | 2021-03-11 | 2024-01-17 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| WO2022192430A1 (en) * | 2021-03-11 | 2022-09-15 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| BR112023018286A2 (pt) | 2021-03-11 | 2023-10-31 | Janssen Pharmaceutica Nv | Lorpucitinib para uso no tratamento de distúrbios mediados por jak |
| CN116940561A (zh) | 2021-03-22 | 2023-10-24 | 杭州中美华东制药有限公司 | 噻吩类glp-1受体激动剂及其用途 |
| WO2022202864A1 (ja) | 2021-03-24 | 2022-09-29 | 塩野義製薬株式会社 | 縮合環を有するglp-1受容体作動薬を含有する医薬組成物 |
| WO2022213062A1 (en) | 2021-03-29 | 2022-10-06 | Nimbus Saturn, Inc. | Hpk1 antagonists and uses thereof |
| PH12023552860A1 (en) * | 2021-04-21 | 2024-05-20 | Gilead Sciences Inc | Carboxy-benzimidazole glp-1r modulating compounds |
| WO2022235717A1 (en) * | 2021-05-03 | 2022-11-10 | Carmot Therapeutics, Inc. | Benzimidazoyl glp-1 receptor agonists, pharmaceutical compositions comprising the same, and methods for their use |
| TWI843104B (zh) | 2021-05-20 | 2024-05-21 | 美商美國禮來大藥廠 | 類升糖素肽1受體促效劑 |
| WO2022268152A1 (zh) | 2021-06-24 | 2022-12-29 | 杭州中美华东制药有限公司 | Glp-1受体激动剂及其组合物和用途 |
| WO2023001237A1 (en) * | 2021-07-21 | 2023-01-26 | Hepagene Therapeutics (HK) Limited | Glucagon-like peptide-1 receptor modulators and uses thereof |
| CN113480534B (zh) * | 2021-07-23 | 2022-05-13 | 广州必贝特医药股份有限公司 | 苯并咪唑或氮杂苯并咪唑-6-羧酸类化合物及其应用 |
| TW202321240A (zh) * | 2021-08-02 | 2023-06-01 | 大陸商深圳信立泰藥業股份有限公司 | Glp-1r促效劑化合物的鹽、包括其的藥物組合物及其用途 |
| EP4382525A4 (en) * | 2021-08-04 | 2025-07-16 | Shanghai Hansoh Biomedical Co Ltd | CYCLOALKENE DERIVATIVE REGULATOR, PREPARATION METHOD AND USE THEREOF |
| US20250066337A1 (en) | 2021-08-26 | 2025-02-27 | Pfizer Inc. | Amorphous Form of (S)-2-(5-((3-Ethoxypyridin-2-YL)Oxy)Pyridin-3-YL)-N-(Tetrahydrofuran-3-YL)Pyrimidine-5-Carboxamide |
| IL310377A (en) | 2021-08-30 | 2024-03-01 | Mindrank Ai Ltd | Novel aryl ether substituted heterocyclic compound as glp1r agonist |
| CN117940422A (zh) * | 2021-08-31 | 2024-04-26 | 辉瑞大药厂 | 2-[(4-{6-[(4-氰基-2-氟芐基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2s)-氧杂环丁烷-2-基甲基]-1h-苯并咪唑-6-羧酸,1,3-二羟基-2-(羟甲基)丙-2-胺盐的固体形式 |
| CN113548982B (zh) * | 2021-09-03 | 2022-05-06 | 上海三牧化工技术有限公司 | 一种4-氰基-2-氟苄醇的制备方法 |
| WO2023038039A1 (ja) | 2021-09-08 | 2023-03-16 | 塩野義製薬株式会社 | 抗肥満作用の関与する疾患の予防及び治療用医薬 |
| KR20240068737A (ko) * | 2021-09-27 | 2024-05-17 | 테른스 파마슈티칼스, 인크. | Glp-1r 효능제로서의 벤즈이미다졸 카복실산 |
| WO2023057414A1 (en) | 2021-10-05 | 2023-04-13 | Astrazeneca Ab | Certain octahydrofuro[3,4- b]pyrazines as glp-1 receptor modulators |
| WO2023057429A1 (en) | 2021-10-05 | 2023-04-13 | Astrazeneca Ab | Certain 2,5-diazabicyclo[4.2.0]octanes and octahydrofuro[3,4- b]pyrazines as glp-1 receptor modulators |
| AU2022358915A1 (en) | 2021-10-05 | 2024-05-09 | Astrazeneca Ab | Certain 2,5-diazabicyclo[4.2.0]octanes as glp-1 receptor modulators |
| TWI843243B (zh) * | 2021-10-22 | 2024-05-21 | 大陸商盛世泰科生物醫藥技術(蘇州)股份有限公司 | 作為glp-1受體激動劑的化合物、包含其的藥物組成物及其用途 |
| AU2022375634A1 (en) | 2021-10-25 | 2024-06-06 | Terns Pharmaceuticals, Inc. | Compounds as glp-1r agonists |
| CR20240183A (es) | 2021-12-01 | 2024-06-07 | Pfizer | Inhibidores y/o degradadores de bckdk |
| CN115850296B (zh) * | 2021-12-03 | 2025-03-11 | 杭州先为达生物科技股份有限公司 | 一种噻吩并咪唑类化合物的晶型及其制备方法 |
| ES3039638T3 (en) | 2021-12-06 | 2025-10-23 | Pfizer | Melanocortin 4 receptor antagonists and uses thereof |
| JPWO2023106310A1 (es) * | 2021-12-07 | 2023-06-15 | ||
| US20250066338A1 (en) | 2021-12-16 | 2025-02-27 | Astrazeneca Ab | Certain 3-azabicyclo[3.1.0]hexanes as glp-1 receptor modulators |
| WO2023111145A1 (en) | 2021-12-16 | 2023-06-22 | Astrazeneca Ab | Certain 3-azabicyclo[3.1.0]hexanes as glp-1 receptor modulators |
| WO2023124824A1 (zh) * | 2021-12-29 | 2023-07-06 | 海思科医药集团股份有限公司 | 一种glp-1激动剂的盐及其晶型和在医药上的应用 |
| CN118613476A (zh) * | 2022-01-10 | 2024-09-06 | 西藏海思科制药有限公司 | 一种glp-1激动剂盐及其晶型及在医药上的应用 |
| CN119343343A (zh) * | 2022-02-09 | 2025-01-21 | 加舒布鲁姆生物公司 | 杂环glp-1激动剂 |
| WO2023151575A1 (en) * | 2022-02-09 | 2023-08-17 | Gasherbrum Bio Inc. | Heterocyclic glp-1 agonists |
| KR20240150488A (ko) | 2022-02-23 | 2024-10-15 | 테른스 파마슈티칼스, 인크. | Glp-1r 작용제로서의 화합물 |
| WO2023169436A1 (zh) | 2022-03-08 | 2023-09-14 | 广州市联瑞制药有限公司 | 苯并双环类化合物及其制备方法和应用 |
| CN119137124A (zh) * | 2022-03-09 | 2024-12-13 | 加舒布鲁姆生物公司 | 杂环glp-1激动剂 |
| WO2023179542A1 (en) | 2022-03-21 | 2023-09-28 | Gasherbrum Bio , Inc. | 5,8-dihydro-1,7-naphthyridine derivatives as glp-1 agonists for the treatment of diabetes |
| WO2023182869A1 (en) * | 2022-03-25 | 2023-09-28 | Ildong Pharmaceutical Co., Ltd. | Novel salt of glp-1 receptor agonist compound, preparation method thereof and pharmaceutical composition comprising thereof |
| WO2023198140A1 (en) | 2022-04-14 | 2023-10-19 | Gasherbrum Bio, Inc. | Heterocyclic glp-1 agonists |
| WO2023203135A1 (en) | 2022-04-22 | 2023-10-26 | Precirix N.V. | Improved radiolabelled antibody |
| CA3251753A1 (en) | 2022-05-02 | 2023-11-09 | Precirix N.V. | PRE-TARGETTING |
| CN117003744B (zh) * | 2022-07-18 | 2025-08-15 | 厦门市博瑞来医药科技有限公司 | 五元并六元化合物、其中间体、制备方法、组合物和应用 |
| JP2025523949A (ja) * | 2022-07-22 | 2025-07-25 | ファイザー・インク | 2-[(4-{6-[(4-シアノ-2-フルオロベンジル)オキシ]ピリジン-2-イル}ピペリジン-1-イル)メチル]-1-[(2s)-オキセタン-2-イルメチル]-1h-ベンズイミダゾール-6-カルボン酸、1,3-ジヒドロキシ-2-(ヒドロキシメチル)プロパン-2-アミン塩を調製するための方法および中間体 |
| CN115536638B (zh) * | 2022-08-15 | 2023-10-13 | 上海交通大学 | 一种三氮唑类化合物及其应用 |
| WO2024051700A1 (zh) * | 2022-09-05 | 2024-03-14 | 德睿智药(苏州)新药研发有限公司 | 作为glp1r激动剂的新型芳基氘代苄醚取代杂环类化合物 |
| IL319453A (en) * | 2022-09-06 | 2025-05-01 | Mindrank Therapeutics Suzhou New Drug Res And Development Co Ltd | Polymorph of a GLP-1R agonist compound, method of preparation thereof and use thereof |
| US20260097037A1 (en) | 2022-09-22 | 2026-04-09 | Shionogi & Co., Ltd. | Fused ring compound having glp-1 receptor agonist effect |
| IL319486A (en) | 2022-10-07 | 2025-05-01 | Pfizer | HSD17B13 Inhibitors and/or Retarders |
| AU2023364628A1 (en) | 2022-10-18 | 2025-04-03 | Pfizer Inc. | Patatin-like phospholipase domain-containing protein 3 (pnpla3) modifiers |
| WO2024102625A1 (en) | 2022-11-11 | 2024-05-16 | Eli Lilly And Company | Glucagon-like peptide 1 receptor agonists |
| IL320810A (en) | 2022-11-16 | 2025-07-01 | Lilly Co Eli | Glucagon-like peptide 1 receptor agonists |
| TW202440556A (zh) * | 2022-12-13 | 2024-10-16 | 中國大陸商杭州中美華東製藥有限公司 | 鹽型的glp-1受體激動劑、其晶型和製備方法及其藥物組合物和用途 |
| KR20250117453A (ko) | 2022-12-16 | 2025-08-04 | 화이자 인코포레이티드 | 3-플루오로-4-하이드록시벤즈아미드-함유 억제제 및/또는 분해제 및 이의 용도 |
| JP2026500865A (ja) | 2023-01-13 | 2026-01-08 | 中国科学院上海薬物研究所 | Glp-1受容体アゴニストとして使用される4-アルコキシベンゾイミダゾール-6-カルボン酸誘導体 |
| EP4660194A1 (en) * | 2023-02-02 | 2025-12-10 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Salt and crystal form of cycloalkene compound, and preparation method therefor and use thereof |
| CN118496200A (zh) * | 2023-02-14 | 2024-08-16 | 成都康弘药业集团股份有限公司 | 吲唑啉酮类化合物及其制备方法和用途 |
| AU2024222719A1 (en) | 2023-02-16 | 2025-08-21 | Gasherbrum Bio, Inc. | Heterocyclic glp-1 agonists |
| WO2024206647A1 (en) * | 2023-03-29 | 2024-10-03 | Terns Pharmaceuticals, Inc. | Polymorphic forms and salts of a glp-1r agonist |
| US20240398794A1 (en) | 2023-04-07 | 2024-12-05 | Terns Pharmaceuticals, Inc. | COMBINATIONS OF GLP-1R AND THRß AGONISTS AND METHODS OF USE THEREOF |
| WO2024212742A1 (zh) * | 2023-04-10 | 2024-10-17 | 上海研健新药研发有限公司 | 一种glp-1r激动剂,其制备方法和应用 |
| EP4695226A1 (en) | 2023-04-14 | 2026-02-18 | Pfizer Inc. | Glucose-dependent insulinotropic polypeptide receptor antagonists and uses thereof |
| CN118812522A (zh) * | 2023-04-21 | 2024-10-22 | 苏州闻泰医药科技有限公司 | 一种glp-1r受体激动剂化合物盐、其晶型、其制备方法和应用 |
| WO2025011664A1 (zh) | 2023-07-12 | 2025-01-16 | 歌礼制药(中国)有限公司 | Glp-1r激动剂的制剂及其制备方法 |
| WO2025015268A1 (en) | 2023-07-13 | 2025-01-16 | Aconcagua Bio, Inc. | Modulators of calcitonin receptor and/or amylin receptor activity |
| WO2025015269A1 (en) | 2023-07-13 | 2025-01-16 | Aconcagua Bio, Inc. | Compounds, compositions, and methods |
| CN119330949A (zh) | 2023-07-21 | 2025-01-21 | 石药集团中奇制药技术(石家庄)有限公司 | 一类多环化合物及其用途 |
| CN116675680B (zh) * | 2023-08-02 | 2023-10-20 | 药康众拓(北京)医药科技有限公司 | 一种氘代化合物及其制备方法、药物和应用 |
| TW202521533A (zh) | 2023-09-14 | 2025-06-01 | 香港商歌禮製藥(中國)有限公司 | Glp-1r 激動劑及其治療方法 |
| CN121889388A (zh) * | 2023-09-21 | 2026-04-17 | 尹诺卫医药有限公司 | 制备取代的稠合咪唑衍生物的方法 |
| WO2025069009A1 (en) | 2023-09-29 | 2025-04-03 | Graviton Bioscience Bv | Rock2 inhibitors in the treatment of obesity |
| WO2025083727A1 (en) * | 2023-10-20 | 2025-04-24 | Dr. Reddy's Laboratories Limited | Amorpous solid dispersons of danuglipron free acid and process thereof |
| WO2025099561A1 (en) | 2023-11-07 | 2025-05-15 | Pfizer Inc. | Oral controlled-release matrix formulations of 2-[(4-{6-[(4-cyano-2-fluorobenzyl)oxy]pyridin-2-yl}piperidin-1-yl)methyl]-1-[(2s)-oxetan-2-ylmethyl]-1h-benzimidazole-6-carboxylic acid or a pharmaceutically salt thereof |
| WO2025104668A1 (en) | 2023-11-17 | 2025-05-22 | Pfizer Inc. | Anti-gastric inhibitory polypeptide receptor (gipr) antibodies and antibody conjugates for the treatment of metabolic disorders |
| US12291530B1 (en) | 2023-11-24 | 2025-05-06 | Ascletis Pharma (China) Co., Limited | GLP-1R agonist and therapeutic method thereof |
| TW202543613A (zh) * | 2023-12-27 | 2025-11-16 | 香港商英矽智能科技知識產權有限公司 | 作為glp-1r激動劑的新型化合物及其用途 |
| TW202542163A (zh) * | 2023-12-27 | 2025-11-01 | 香港商英矽智能科技知識產權有限公司 | 作為glp-1r激動劑的新型化合物及其用途 |
| WO2025158275A1 (en) * | 2024-01-24 | 2025-07-31 | Pfizer Inc. | Combination therapy using glucose-dependent insulinotropic polypeptide receptor antagonist compounds and glp-1 receptor agonist compounds |
| TW202539633A (zh) | 2024-02-01 | 2025-10-16 | 美商輝瑞股份有限公司 | 葡萄糖依賴性促胰島素多肽受體拮抗劑及其用途 |
| WO2025171341A2 (en) | 2024-02-08 | 2025-08-14 | Aconcagua Bio, Inc. | Compounds and compositions for treating conditions associated with calcitonin receptor and/or amylin receptor activity |
| WO2025171340A1 (en) | 2024-02-08 | 2025-08-14 | Aconcagua Bio, Inc. | The treatment of calcitonin- and/or amylin-receptor associated conditions |
| KR20250125490A (ko) | 2024-02-14 | 2025-08-22 | 주식회사 종근당 | Glp-1 수용체 작용제 및 이의 용도 |
| WO2025171806A1 (en) * | 2024-02-18 | 2025-08-21 | Rezubio Pharmaceuticals Co., Ltd | Methods for weight management and mitigation of vesceral malaise |
| WO2025189141A1 (en) | 2024-03-08 | 2025-09-12 | Annapurna Bio, Inc. | Methods for treating obesity and increasing weight loss |
| KR20250152464A (ko) * | 2024-04-16 | 2025-10-23 | 한미약품 주식회사 | 흡입제 조성물 |
| US20250326741A1 (en) | 2024-04-22 | 2025-10-23 | Pfizer Inc. | Glucose-dependent insulinotropic polypeptide receptor antagonists and uses thereof |
| WO2025252098A1 (zh) * | 2024-06-05 | 2025-12-11 | 杭州中美华东制药有限公司 | Glp-1受体激动剂的口服药物组合物及其制备方法 |
| WO2026013531A1 (en) | 2024-07-10 | 2026-01-15 | Pfizer Inc. | Spiro[2.5]octane compounds |
| WO2026033382A1 (en) | 2024-08-07 | 2026-02-12 | Pfizer Inc. | Heterocyclic glp-1r modulators |
| WO2026042018A1 (en) | 2024-08-21 | 2026-02-26 | Pfizer Inc. | 4-{4-[(1-{[4-(propan-2-yl)phenyl]carbamoyl)-d-prolyl)amino]cyclohexyl}benzoic acid derivatives as gipr antagonists for the treatment of diabetes |
| WO2026051998A1 (en) | 2024-09-04 | 2026-03-12 | Aconcagua Bio, Inc. | Compounds, compositions, and methods |
| WO2026078617A1 (en) | 2024-10-11 | 2026-04-16 | Pfizer Inc. | Glucose-dependent insulinotropic polypeptide receptor antagonists and uses thereof |
| WO2026078672A1 (en) | 2024-10-13 | 2026-04-16 | Pfizer Inc. | Glucose-dependent insulinotropic polypeptide receptor antagonists and uses thereof |
| WO2026078670A1 (en) | 2024-10-13 | 2026-04-16 | Pfizer Inc. | Heterocyclic derivatives as glucose-dependent insulinotropic polypeptide receptor antagonists |
| WO2026078673A1 (en) | 2024-10-13 | 2026-04-16 | Pfizer Inc. | Glucose-dependent insulinotropic polypeptide receptor antagonists and uses thereof |
| WO2026078674A1 (en) | 2024-10-13 | 2026-04-16 | Pfizer Inc. | Heterocyclic derivatives as glucose-dependent insulinotropic polypeptide receptor antagonists and uses thereof |
| WO2026078671A1 (en) | 2024-10-13 | 2026-04-16 | Pfizer Inc. | Heterocyclic compounds as glucose-dependent insulinotropic polypeptide receptor antagonists |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| US4485045A (en) | 1981-07-06 | 1984-11-27 | Research Corporation | Synthetic phosphatidyl cholines useful in forming liposomes |
| US4544545A (en) | 1983-06-20 | 1985-10-01 | Trustees University Of Massachusetts | Liposomes containing modified cholesterol for organ targeting |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| GB9305623D0 (en) | 1993-03-18 | 1993-05-05 | Merck Sharp & Dohme | Therapeutic agents |
| CA2156836A1 (en) | 1993-03-18 | 1994-09-29 | Janusz Jozef Kulagowski | Benzimidazole derivatives |
| US6960589B2 (en) * | 2001-03-09 | 2005-11-01 | Abbott Laboratories | Benzimidazoles that are useful in treating sexual dysfunction |
| US20020169166A1 (en) * | 2001-03-09 | 2002-11-14 | Cowart Marlon D. | Benzimidazoles that are useful in treating sexual dysfunction |
| SE0100903D0 (sv) | 2001-03-15 | 2001-03-15 | Astrazeneca Ab | Compounds |
| US20040127504A1 (en) | 2002-09-06 | 2004-07-01 | Cowart Marlon D. | Benzimidazoles that are useful in treating sexual dysfunction |
| WO2007029847A1 (ja) * | 2005-09-07 | 2007-03-15 | Banyu Pharmaceutical Co., Ltd. | 二環性芳香族置換ピリドン誘導体 |
| WO2008012623A1 (en) | 2006-07-25 | 2008-01-31 | Pfizer Products Inc. | Benzimidazolyl compounds as potentiators of mglur2 subtype of glutamate receptor |
| WO2010022055A2 (en) | 2008-08-20 | 2010-02-25 | Amgen Inc. | Inhibitors of voltage-gated sodium channels |
| WO2010115736A2 (en) | 2009-04-02 | 2010-10-14 | Merck Serono S.A. | Dihydroorotate dehydrogenase inhibitors |
| MA34300B1 (fr) * | 2010-05-13 | 2013-06-01 | Amgen Inc | Composés azotés hétérocycliques convenant comme inhibiteurs de la pde10 |
| BR112013021236B1 (pt) | 2011-02-25 | 2021-05-25 | Merck Sharp & Dohme Corp | composto derivado de benzimidazol, e, composição |
| EP3555064B9 (en) | 2016-12-16 | 2023-03-01 | Pfizer Inc. | Glp-1 receptor agonists and uses thereof |
-
2017
- 2017-12-01 EP EP17817174.0A patent/EP3555064B9/en active Active
- 2017-12-01 AU AU2017374860A patent/AU2017374860B2/en active Active
- 2017-12-01 JP JP2019531391A patent/JP6637641B1/ja active Active
- 2017-12-01 KR KR1020197020714A patent/KR102314286B1/ko active Active
- 2017-12-01 HR HRP20221437TT patent/HRP20221437T1/hr unknown
- 2017-12-01 GE GEAP201715110A patent/GEP20217265B/en unknown
- 2017-12-01 GE GEAP202115110A patent/GEAP202115110A/en unknown
- 2017-12-01 FI FIEP17817174.0T patent/FI3555064T3/fi active
- 2017-12-01 RU RU2019118485A patent/RU2740135C1/ru active
- 2017-12-01 LT LTEPPCT/IB2017/057577T patent/LT3555064T/lt unknown
- 2017-12-01 MX MX2019007077A patent/MX387259B/es unknown
- 2017-12-01 SI SI201731287T patent/SI3555064T1/sl unknown
- 2017-12-01 MD MDE20191184T patent/MD3555064T2/ro unknown
- 2017-12-01 DK DK17817174.0T patent/DK3555064T5/da active
- 2017-12-01 UA UAA201906738A patent/UA122035C2/uk unknown
- 2017-12-01 HU HUE17817174A patent/HUE060533T2/hu unknown
- 2017-12-01 ES ES17817174T patent/ES2934789T3/es active Active
- 2017-12-01 CU CU2019000056A patent/CU24573B1/es unknown
- 2017-12-01 WO PCT/IB2017/057577 patent/WO2018109607A1/en not_active Ceased
- 2017-12-01 MA MA56480A patent/MA56480B1/fr unknown
- 2017-12-01 PE PE2019001229A patent/PE20191501A1/es unknown
- 2017-12-01 PT PT178171740T patent/PT3555064T/pt unknown
- 2017-12-01 EA EA201991193A patent/EA037318B1/ru unknown
- 2017-12-01 KR KR1020217032471A patent/KR102466418B1/ko active Active
- 2017-12-01 MY MYPI2019003381A patent/MY195385A/en unknown
- 2017-12-01 RS RS20221178A patent/RS63849B9/sr unknown
- 2017-12-01 CN CN201780086550.1A patent/CN110325530B/zh active Active
- 2017-12-01 PH PH1/2019/501360A patent/PH12019501360B1/en unknown
- 2017-12-01 CR CR20190289A patent/CR20190289A/es unknown
- 2017-12-01 PL PL17817174.0T patent/PL3555064T3/pl unknown
- 2017-12-13 US US15/839,901 patent/US10208019B2/en active Active
- 2017-12-13 CA CA2988721A patent/CA2988721C/en active Active
- 2017-12-13 TW TW106143701A patent/TWI713809B/zh active
- 2017-12-14 UY UY0001037514A patent/UY37514A/es not_active Application Discontinuation
- 2017-12-15 AR ARP170103545A patent/AR110387A1/es unknown
-
2018
- 2018-12-14 US US16/220,184 patent/US10669259B2/en active Active
-
2019
- 2019-06-07 EC ECSENADI201941071A patent/ECSP19041071A/es unknown
- 2019-06-11 CO CONC2019/0006046A patent/CO2019006046A2/es unknown
- 2019-06-12 IL IL267288A patent/IL267288B/en active IP Right Grant
- 2019-06-14 DO DO2019000166A patent/DOP2019000166A/es unknown
- 2019-06-14 CL CL2019001651A patent/CL2019001651A1/es unknown
- 2019-07-15 ZA ZA2019/04616A patent/ZA201904616B/en unknown
- 2019-12-20 JP JP2019230341A patent/JP6982054B2/ja active Active
-
2020
- 2020-04-29 US US16/861,646 patent/US10851081B2/en active Active
- 2020-10-30 US US17/085,534 patent/US11512070B2/en active Active
-
2022
- 2022-10-24 US US18/049,052 patent/US11802121B2/en active Active
- 2022-12-22 CY CY20221100805T patent/CY1125716T1/el unknown
-
2023
- 2023-09-28 US US18/477,050 patent/US12319669B2/en active Active
-
2025
- 2025-05-02 US US19/196,906 patent/US20250257052A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2934789T3 (es) | Agonistas del receptor glp-1 y usos del mismo | |
| ES2943510T3 (es) | Agonistas del receptor GLP-1 y usos del mismo | |
| ES3012643T3 (en) | Glp-1 receptor agonists and uses thereof | |
| BR112019012211B1 (pt) | Agonistas de receptor de glp-1, composição farmacêutica que compreende os mesmos e usos dos mesmos | |
| OA19245A (en) | GLP-1 receptor agonists and uses thereof. | |
| HK40008766B (zh) | Glp-1受体激动剂及其用途 | |
| HK40008766A (en) | Glp-1 receptor agonists and uses thereof |