ES2951817T3 - Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLR7/8 y usos de los mismos - Google Patents
Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLR7/8 y usos de los mismos Download PDFInfo
- Publication number
- ES2951817T3 ES2951817T3 ES18773000T ES18773000T ES2951817T3 ES 2951817 T3 ES2951817 T3 ES 2951817T3 ES 18773000 T ES18773000 T ES 18773000T ES 18773000 T ES18773000 T ES 18773000T ES 2951817 T3 ES2951817 T3 ES 2951817T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- methyl
- compounds
- alkyl
- antigen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Se describen compuestos de 1H-imidazoquinolina modificados con cadena alquílica de fórmula (J) o sales de los mismos como agonistas de los receptores tipo Toll 7 y 8 para mejorar las respuestas inmunitarias. También se proporcionan métodos para preparar composiciones farmacéuticas que contienen estos compuestos. La presente divulgación también describe métodos de uso de estos compuestos y composiciones farmacéuticas que contienen estos compuestos para el tratamiento de enfermedades en un sujeto tales como enfermedades infecciosas y cáncer. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLRH8 y usos de los mismos
Campo de la invención
La presente divulgación se refiere a compuestos agonistas de TLRH8 de imidazoquinolina de cadena alquílica modificada para potenciar las respuestas inmunitarias. La presente divulgación también se refiere a composiciones farmacéuticas que comprenden los compuestos de imidazoquinolina de cadena alquílica modificada, métodos de preparación de los mismos, métodos para estimular una respuesta inmunitaria y usos de las composiciones farmacéuticas en el tratamiento de enfermedades en un sujeto, por ejemplo, enfermedades infecciosas y cáncer.
Antecedentes de la invención
Los receptores de tipo Toll (TLR) son una familia de proteínas transmembrana que reconocen moléculas conservadas estructuralmente que se derivan y son exclusivas de patógenos, denominadas patrones moleculares asociados a patógenos. Como tales, los TLR funcionan en el sistema inmunitario de los mamíferos como sensores de primera línea de patrones moleculares asociados a patógenos, detectando la presencia de patógenos invasores (Takeuchi y Akira 2010 Cell 140:805-820). La participación de TLR en las células inmunitarias centinela provoca la biosíntesis de citocinas seleccionadas (por ejemplo, interferones de tipo I), la inducción de moléculas co-estimulantes, y el aumento de la capacidad de presentación de antígenos. Estos son importantes mecanismos moleculares que activan las respuestas inmunitarias innatas y adaptativas. Por consiguiente, los agonistas y antagonistas de los TLR encuentran uso en la modulación de las respuestas inmunitarias. Los agonistas de TLR se emplean normalmente para estimular respuestas inmunitarias, mientras que los antagonistas de TLR se emplean normalmente para inhibir respuestas inmunitarias (Gosu et al 2012 Molecules 17:13503-13529).
El genoma humano contiene 10 TLR conocidos, de estos TLR3, TLR7, TLR8 y TLR9 detectan ácidos nucleicos y sus productos de degradación. La distribución de TLR7, TLR8 y TLR9 está restringida a los compartimentos endolisosómicos de las células y se expresan preferentemente en células del sistema inmunitario. En la configuración de receptor dimérico activado TLR7 y TLR8 reconocen ARN monocatenario en un sitio de unión a ligando y los productos de degradación de ribonucleósidos guanosina y uridina, respectivamente, (así como ligandos de moléculas pequeñas con motivos estructurales relacionados) en un segundo sitio de unión a ligando (Zhang et al 2016 Immunity 45:737-748; Tanji et al 2015 Nat Struct Mol Biol 22:109-115). La participación de TLR7 en las células dendríticas plasmocitoides conduce a la inducción de interferón-a/p, que desempeña funciones esenciales en el control de la respuesta inmunitaria adaptativa (Bao y Liu 2013 Protein Cell 4:40-52). Compromiso de TLR8 en células dendríticas mieloides, monocitos y células dendríticas derivadas de monocitos induce un perfil prominente de citocinas proinflamatorias, caracterizado por una producción aumentada de factor de necrosis tumoral-a, interleucina-12 e IL-18 (Figenbrod et al 2015 J Immunol 195:1092-1099). Así pues, prácticamente todos los tipos principales de células monocíticas y dendríticas pueden ser activados por agonistas de TLR7 y TLR8 para convertirse en células presentadoras de antígenos altamente eficaces, promoviendo así una respuesta inmunitaria innata y adaptativa eficaz. La mayoría de los tipos de células presentadoras de antígeno expresan solo uno de estos dos receptores, por consiguiente, las moléculas pequeñas con bioactividad agonista potente frente a los receptores TLR7 y TLR8 son adyuvantes inmunitarios potencialmente más eficaces que los agonistas específicos para solo uno de estos TLR. Así pues, un agonista de molécula pequeña de TLRHTLR8 (TLRH8) con la bioactividad dual equilibrada provocaría respuestas inmunitarias innatas en una gama más amplia de células presentadoras de antígenos y otros tipos de células inmunitarias clave, incluyendo células dendríticas plasmocitoides y mieloides, monocitos, y linfocitos B (van Haren et al 2016 J Immunol 197:4413-4424; Ganapathi et al 2015 PLoS One 10:e0134640). Dichos agonistas duales potentes de TLRH8 también pueden ser eficaces para estimular respuestas inmunitarias antitumorales eficaces en el cáncer (Singh et al 2014 J Immunol 193:4722-4731; Sabado et al 2015 Cancer Immunol Res 3:278-287; Spinetti et al 2016 Oncoimmunol 5:e1230578; Patil et al 2016 Mini Rev Med Chem 16:309-322).
Se sabe que varias clases estructurales de moléculas pequeñas interactúan en el sitio de unión a ligando de guanosina/uridina y poseen niveles variables de bioactividad agonista de TLR 7 y/o TLR8 (véase, por ejemplo, Lu et al 2012 Clin Cancer Res 18:499-509; Patentes de EE. UU. N.° 5.446.153, 6.194.425, 6.110.929 y 7.199.131), incluidos los derivados de 1H-imidazo[4,5-c]quinolina que son agonistas de TLR7 o agonistas duales de TLRH8 (véase por ejemplo, Vasilakos y Tomai 2013 Expert Rev Vaccines 12:809-819; Patente de EE. UU. N.° 4.689.338). Una 1h -imidazo[4,5-c]quinolina de este tipo es 1-isobutil-1H-imidazo[4,5-c]quinolin-4-amina (imiquimod), un agonista específico de TLR7 que fue aprobado en 1997 para el tratamiento de la queratosis actínica, el carcinoma basocelular superficial y las verrugas genitales, y posteriormente fue aprobado para el tratamiento del carcinoma basocelular (véase por ejemplo, Hemmi et al 2002 Nat Immunol 3:196-200). Mientras que algunas 1H-imidazo[4,5-c]quinolinas muestran actividades agonistas de TLR7 o TLR8 selectivas, otras muestran actividades agonistas duales de TLRH8. Por ejemplo, se descubrió 1-bencil-2-butil-1H-imidazo[4,5-c]quinolin-4-amina era un agonista de TLR7 con bioactividad insignificante contra TLR8 (Shukla et al 2010 J Med Chem 53:4450-4465). Por el contrario, se descubrió que la 2-propil[1,3]tiazolo[4,5-c]quinolin-4-amina es un agonista de TLR8 con actividad insignificante contra TLR7 (Gorden et al 2005 J Immunol 174:1259-1268). Se descubrió que 1-(4-aminometilbencil)-2-butil-1H-imidazo[45-c]quinolin-4-amina (IMDQ) y 1-(3-aminometilbencil)-2-but es
de TLRH8 con una potente actividad agonista contra ambos receptores (véase por ejemplo, Shukla et al 2010 J Med Chem 53:4450-4465; Shukla et al 2010 Bioorg Med Chem Lett 10:6384-6386; Patente de EE. UU. N.° 8.728.486 (US 2012/294885); Patente de EE. UU. N.° 9.441.005).
Sin embargo, se ha demostrado que la rápida distribución sistémica de agonistas de TLRH8 basados en 1H-imidazo[4,5-c]quinolina solubles después de la administración subcutánea, intratumoral o intramuscular causa toxicidades significativas en los pacientes (véase por ejemplo, Vasilakos et al 2013 Expert Rev Vaccines 12:809-819; Savage et al 1996 Br J Cancer 74:1482-1486; Pockros et al 2007 J Hepatol 47:174-182). La activación del sistema inmunitario sistémico debido a la activación de los TLR en las células del bazo y el hígado causa un aumento en los niveles séricos de citocinas proinflamatorias, lo que a su vez causa síntomas similares a los de la gripe y otros acontecimientos adversos que limitan la utilidad de estos compuestos como agentes terapéuticos humanos a una vía de administración tópica. Así pues, sigue existiendo la necesidad de agentes terapéuticos de molécula pequeña con actividades agonistas de TLRH8 potentes y equilibradas que también posean propiedades fisicoquímicas que permitan composiciones farmacéuticas que promuevan la retención del compuesto en el sitio de inyección.
Breve sumario de la invención
La presente divulgación proporciona derivados de 1H-imidazo[4,5-c]quinolina de cadena alquílica modificada que son potentes agonistas de TLRH8 que presentan una bioactividad equilibrada frente a ambos receptores. En un aspecto, se proporciona un compuesto de fórmula (J):

o una sal
R0 es -(CH2)mRA o -(CH2)z(C(CH3)2)RA; m es 0, 1, 2 o 3; z es 1 o 2; y RA es cicloalquilo C3-C8 opcionalmente sustituido con de 1 a 4 grupos seleccionados independientemente del grupo que consiste en alquilo C1-C4 , alquileno C1-C4 y halógeno;
X es -NH-;
R1 es alquilo C3-C6 , -(CH2)pOR1a, -(CH2)pNHR1b o -(CH2)pR1c; donde R1a y R1b son independientemente alquilo C1-C3 ; R1c es cicloalquilo C3-C4 ; y p es 1 o 2;
R2 es NHR2 a ; donde R2a es H, Oh , NH2 o metilo;
cada R3 es independientemente halógeno, alquilo C1-C8 , -(alquilen C1-C7)-NH2 ,
o -CH2-fenilen-CH2NH2 ;
q es 0, 1, 2, 3 o 4; y
R4a y R4b son independientemente H o alquilo C1-C8.
En algunas realizaciones, R0 es -(CH2)mRA. En una variación, m es 1 o 2, y RA es ciclopropilo, ciclobutilo o ciclopentilo. En algunas realizaciones, R0 es -(CH2)z(C(CH3)2)RA. En una variación, z es 1 y RA es ciclopropilo, ciclobutilo o ciclopentilo.
En algunas realizaciones, RA es cicloalquilo C3-C8.
En algunas realizaciones, RA es cicloalquilo C3-C6 opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno. En una variación, m es 1 o 2.
En algunas realizaciones, RA es ciclopropilo opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno, y m es 1 o 2.
En algunas realizaciones, m es 0 o 1, y RA es ciclohexilo opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno.
En algunas realizaciones, R0 se selecciona entre el grupo que consiste en:

En algunas realizaciones, R1 es alquilo C3-C6 (por ejemplo, n-butilo). En algunas realizaciones, R1 es -(CH2)pO1a (por ejemplo, CH2OCH2CH3). En algunas realizaciones, R1 es -(CH2)pNHR1b (por ejemplo, CH2NHCH2CH3). En algunas realizaciones, R1 es (CH2)pR1c. En una variación, R1c es ciclopropilo.
En algunas realizaciones, R2 es NH2.
En algunas realizaciones, q es 0. En algunas realizaciones, q es 1 y R3 es alquilo C1-C8.
En algunas realizaciones, cada R4a y R4b es H.
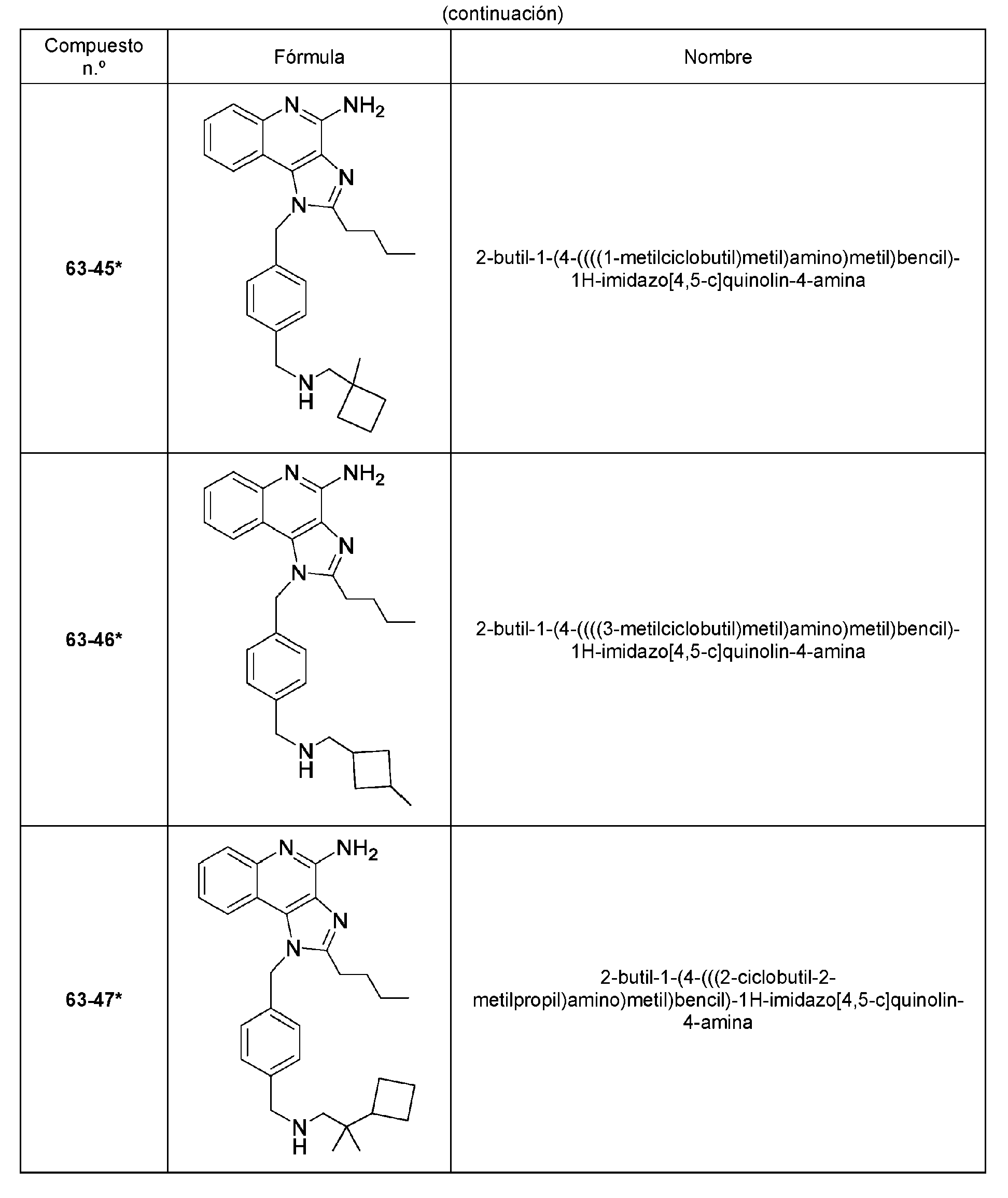
En algunas realizaciones, el compuesto se selecciona del grupo que consiste en los compuestos n.° 63-33, 63-35, 63 36 y 63-38 a 63-49 en la tabla 1, o una sal del mismo.
También se divulga un compuesto de fórmula (K):

o una sal del mismo, en donde:
n es un número entero de 4 a 21;
X es -NH- o -NH(C=O)-;
R1 es alquilo C3-C6 , -(CH2 )pOR1a, -(CH2 )pNHR1b o -(CH2 )pR1c; donde R1a y R1b son independientemente alquilo Ci -C3 ; R1c es cicloalquilo C3-C4 ; y p es 1 o 2;
R2 es NHR2 a ; donde R2a es H, Oh , NH2 o metilo;
cada R3 es independientemente halógeno, alquilo C1-C8 , -(alquilen C1-C7 )-NH2 o -CH2-fenilen-CH2NH2 ;
q es 0, 1, 2, 3 o 4; y
R4a y R4b son independientemente H o alquilo C1-C8 ,
siempre que el compuesto sea distinto de 2-butil-1-(4-((hexadecilamino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-32) o N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)palmitamida (Compuesto n.° 63-31).
Además se proporcionan composiciones farmacéuticas que comprenden (i) un compuesto de fórmula (J) y (ii) uno o más excipientes farmacéuticamente aceptables. En algunas realizaciones, las composiciones farmacéuticas comprenden además un antígeno. En algunas realizaciones, la composición farmacéutica se compone de excipientes farmacéuticamente aceptables que incluyen aceites de calidad USP y un modificador orgánico (por ejemplo, 95 % de aceite de sésamo/5 % de etanol). En algunas realizaciones, la composición farmacéutica se compone de excipientes farmacéuticamente aceptables que permiten una nanoemulsión de aceite en agua o una formulación liposómica, cuyos ejemplos son conocidos por los expertos en la materia. En algunas realizaciones, la composición farmacéutica puede incluir una mezcla de un antígeno o antígenos, incluyendo, pero sin limitación, antígenos o neoantígenos asociados a tumores.
La presente divulgación también proporciona una composición farmacéutica como se ha descrito anteriormente para su uso en un método para estimular una respuesta inmunitaria en un sujeto mamífero que lo necesite, que comprende administrar la composición al sujeto mamífero en una cantidad, a una frecuencia y durante un marco de tiempo suficiente para estimular una respuesta inmunitaria en el sujeto mamífero. En un aspecto, la respuesta inmunitaria es una respuesta inmunitaria local. En otro aspecto, la respuesta inmunitaria es una respuesta inmunitaria sistémica.
También se divulga una pluralidad de métodos para usar una composición farmacéutica descrita anteriormente en un sujeto mamífero, tal como un paciente humano. En un aspecto, se desvelan métodos para tratar el cáncer en un sujeto mamífero que lo necesite, que comprenden administrar al sujeto mamífero la composición farmacéutica en una cantidad suficiente para tratar el cáncer en el sujeto mamífero. En otro aspecto del método, la administración intratumoral comprende la inyección de la composición farmacéutica en al menos una lesión tumoral. En un aspecto del método, además se administra al sujeto una cantidad eficaz de un segundo agente terapéutico. En determinados aspectos, el agente terapéutico es un agente quimioterapéutico, un modulador epigenético, inductor de la muerte celular inmunogénica, o un antagonista de una molécula inhibidora del punto de control inmunitario. En otro aspecto, se describen métodos para inducir una respuesta de anticuerpos específica de antígeno en un sujeto mamífero que lo necesita, que comprenden administrar al sujeto mamífero la composición farmacéutica en una cantidad suficiente para inducir una respuesta de anticuerpos específica de antígeno y/o una respuesta de linfocitos T específica de antígeno en el sujeto mamífero. En un aspecto, se describen métodos para tratar o prevenir una enfermedad infecciosa en un sujeto mamífero que lo necesite, que comprenden administrar al sujeto mamífero la composición farmacéutica en una cantidad suficiente para tratar o prevenir una enfermedad infecciosa en el sujeto mamífero. En un aspecto, se describen métodos para tratar o prevenir un trastorno relacionado con IgE en un sujeto mamífero, que comprenden administrar la composición farmacéutica en una cantidad suficiente para tratar o prevenir un trastorno relacionado con IgE en el sujeto mamífero.
También se desvelan kits que comprenden composiciones farmacéuticas de la invención e instrucciones para su uso en el tratamiento de enfermedades infecciosas y/o cánceres. También se desvelan métodos para la fabricación de kits para su uso en el tratamiento de enfermedades infecciosas y/o cáncer.
Breve descripción de los dibujos
Las figuras 1A-B muestran el cambio en los niveles séricos de IL- 6 (figura 1A) e IL-12p40 (figura 1B) a lo largo del tiempo posterior a la inyección después de una sola administración subcutánea de los Compuestos n.° 63-00 (barras negras), 63-17 (barras blancas) y 63-10 (barras grises) a ratones de tipo silvestre como se describe en el ejemplo B3. Tamaño del grupo = 3, /- error típico de la media.
Las figuras 2A-C muestran la inhibición del crecimiento tumoral en ratones de tipo silvestre portadores de tumores CT26 singénicos después de la administración intratumoral repetida de los Compuestos n.° 63-18 (figura 2A), 63 33 (figura 2B) o 63-10 (figura 2C) como se describe en el ejemplo B4. A los animales se les administró la dosis descrita con control de vehículo (-•-), 20 μg de compuesto (-■-), 5 μg de compuesto (- -■- -), 0,5 μg de compuesto (--■ --), o 50 μg de un control positivo de agonista de TLR9 (-♦-). Tamaño del grupo = 5 para controles y 8 para condiciones experimentales, /- error típico de la media.
Las figuras 3A-B muestran la inhibición del crecimiento tumoral a lo largo del tiempo en los tumores inyectados (figura 3A) y distales (figura 3B) de ratones de tipo silvestre portadores de tumores CT26 después de la administración intratumoral repetida de composiciones farmacéuticas compuestas por un control de vehículo de nanoemulsión de aceite en agua a base de escualeno (-•-), una nanoemulsión de aceite en agua a base de escualeno con 50 ng del Compuesto n.° 63-10 (-■-) o una nanoemulsión de aceite en agua a base de escualeno con 50 ng del Compuesto n.° 63-10 más 5 lo
B5. Los animales recibieron la dosis descrita en los días experimentales 8, 12, 16 y 20. Tamaño del grupo = 8 para controles y todas las condiciones experimentales, los datos se expresan como volumen tumoral promedio (en mm3) /- error típico de la media. Las diferencias en los volúmenes tumorales entre los grupos el día 27 para el tumor inyectado o el día 23 para el tumor distal, se analizaron utilizando una prueba de Kruskall-Wallis seguida de una prueba posterior de Dunn para comparaciones de pares de grupos específicos, ns indica P > 0,050; * indica P ≤ 0,050.
Las figuras 4A-B muestran la inhibición del crecimiento tumoral a lo largo del tiempo en los tumores inyectados (figura 4A) y distales (figura 4B) de ratones de tipo silvestre portadores de tumores CT26 después de una única administración intratumoral en el día experimental 14 de composiciones farmacéuticas compuestas por control de vehículo de solución salina tamponada con fosfato (-•-), control de vehículo de solución salina tamponada con fosfato en combinación con 250 |jg de anticuerpo anti-PD-1 (-o-), 5.000 ng del Compuesto n.° 63-10 en 95 % de aceite de sésamo/5 % de etanol (v/v) en combinación con 250 jg de anticuerpo anti-PD-1 (-■-), 5.000 ng del Compuesto n.° 63-33 en 95 % de aceite de sésamo/5 % de etanol (v/v) en combinación con 250 jg de anticuerpo anti-PD-1 (-▲-), o 5.000 ng del Compuesto n.° 63-00 en solución salina tamponada con fosfato en combinación con 250 jg de anticuerpo anti-PD-1 (-▼-) como se describe en el ejemplo B6. Para todos los grupos de tratamiento con una combinación anti-PD-1, el tratamiento anti-PD-1 se administró por vía intraperitoneal los días experimentales 12, 15, 19, 22 y 26. Tamaño del grupo = 10 para controles y todas las condiciones experimentales, los datos se expresan como volumen tumoral promedio (en mm3) /- error típico de la media. Las diferencias en los volúmenes tumorales entre los grupos en el día experimental 29 se analizaron utilizando una prueba de Kruskall-Wallis seguida de una prueba posterior de Dunn para comparaciones de pares de grupos específicos. ns indica P > 0,050; * indica P ≤ 0,050; ** P ≤ 0,010.
Descripción detallada de la invención
La invención es como se define en la reivindicación 1. Los aspectos y realizaciones preferentes adicionales se definen en las reivindicaciones dependientes. Cualquier aspecto, realización y ejemplo de la presente divulgación que no esté dentro del alcance de las reivindicaciones adjuntas no forma parte de la invención y se proporciona meramente con fines ilustrativos.
La presente divulgación se refiere a derivados de 1H-imidazo[4,5-c]quinolina de cadena alquílica modificada que son potentes agonistas de TLRH8 que muestran una bioactividad equilibrada contra ambos receptores y poseen propiedades fisicoquímicas que permiten composiciones farmacéuticas que promueven la retención del compuesto en el sitio de inyección. La presente divulgación también se refiere a composiciones farmacéuticas que comprenden los compuestos de 1H-imidazo[4,5-c]quinolina de cadena alquílica modificada y métodos de preparación de los mismos, usos de las composiciones farmacéuticas para estimular una respuesta inmunitaria, y métodos para el tratamiento de enfermedades en un sujeto, por ejemplo, enfermedades infecciosas y cáncer.
I. Métodos generales y definiciones
La práctica de la presente divulgación empleará, a menos que se indique otra cosa, técnicas convencionales de química orgánica, química analítica, biología molecular, microbiología, biología celular, bioquímica e inmunología, que se encuentran entre las capacidades de la técnica. Tales técnicas se describen totalmente en la bibliografía, véase, por ejemplo: Fiesers' Reagents for Organic Synthesis, 25a edición (Ho, ed., Wiley, 2016); Comprehensive Organic Functional Groups Transformations, 2a edición (Katritsky y Taylor, eds., Elsevier, 2004); Comprehensive Organic Synthesis, versión 1-8 (Trost y Flemming, eds., Pergamon Press, 1991); Beilsteins Handbuch der Organischen Chemie, 4 (Auflage, ed., Springer-Verlag, 1934); Animal Cell Culture, sexta edición (Freshney, Wiley-Blackwell, 2010); Current Protocols in Cell Biology (Bonifacino et al., ed., John Wiley & Sons, Inc., 1996, incluyendo los suplementos hasta 2014); Current Protocols in Immunology (Coligan et al., eds., John Wiley & Sons, Inc., 1991 incluyendo los suplementos hasta 2014); Current Protocols in Molecular Biology (Ausubel et al., eds., John Wiley & Sons, Inc., 1987, incluyendo los suplementos hasta 2014); Molecular Cloning: A Laboratory Manual, tercera edición (Sambrook y Russell, Cold Spring Harbor Laboratory Press, 2001); y Molecular Cloning: A Laboratory Manual, cuarta edición (Green y Sambrook, Cold Spring Harbor Laboratory Press, 2012).
Los términos "individuo" y "sujeto" se refieren a mamíferos. "Mamíferos" incluyen, pero sin limitación, seres humanos, primates no humanos (por ejemplo, monos), animales de granja, animales de deporte (por ejemplo, caballos), roedores (por ejemplo, ratones y ratas) y mascotas (por ejemplo, perros y gatos).
El término "antígeno" se refiere a una sustancia que es reconocida y unida específicamente por un anticuerpo o por un receptor de antígeno de linfocitos T. Los antígenos pueden incluir péptidos, polipéptidos, proteínas, glucoproteínas, polisacáridos, hidratos de carbono complejos, azúcares, gangliósidos, lípidos y fosfolípidos; porciones de los mismos y combinaciones de los mismos. Los antígenos, cuando están presentes en las composiciones de la presente divulgación, pueden ser sintéticos o aislados de la naturaleza. Los antígenos adecuados para la administración en los métodos de la presente divulgación incluyen cualquier molécula capaz de provocar una respuesta de linfocitos B o linfocitos T específica de antígeno. Los haptenos están incluidos dentro del alcance de "antígeno". Un "hapteno" es un compuesto de bajo peso molecular que no es inmunogénico por sí mismo pero se vuelve inmunogénico cuando se conjuga con una molécula inmunogénica gen
Los "antígenos polipeptídicos" pueden incluir péptidos nativos purificados, péptidos sintéticos, péptidos modificados por ingeniería genética, péptidos recombinantes, extractos de péptidos crudos, o péptidos en un estado activo parcialmente purificado o sin purificar (tales como péptidos que forman parte de virus atenuados o inactivados, microorganismos o células), o fragmentos de dichos péptidos. Los antígenos polipeptídicos tienen preferentemente al menos seis residuos de aminoácidos de longitud, preferentemente de 8 a 1800 aminoácidos de longitud, más preferentemente de 9 a 1000 aminoácidos de longitud, o de 10 a 100 aminoácidos de longitud. De forma similar, en algunas realizaciones, el polipéptido es de aproximadamente 9 a aproximadamente 2000, de aproximadamente 9 a aproximadamente 1000, de aproximadamente 9 a aproximadamente 100 o de aproximadamente 9 a aproximadamente 60 aminoácidos de longitud. En algunas realizaciones, el polipéptido es de al menos (límite inferior) 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80 o 90 aminoácidos de longitud. En algunas realizaciones, el polipéptido es como máximo (límite superior) de 1000, 900, 800, 700, 600, 500, 400, 300, 250, 200, 150, 100, 50 o 25 aminoácidos de longitud. En algunas realizaciones, el antígeno polipeptídico es de 9 y 35 aminoácidos una longitud.
Como se usa en el presente documento, el término "inmunogénico" se refiere a un agente (por ejemplo, antígeno polipeptídico) que provoca una respuesta inmunitaria adaptativa tras la administración en condiciones adecuadas a un sujeto mamífero. La respuesta inmunitaria puede ser una respuesta mediada por linfocitos B (humoral) y/o linfocitos T (celular).
"Adyuvante" se refiere a una sustancia que, cuando se mezcla con un agente inmunogénico tal como un antígeno, mejora o potencia de forma no específica una respuesta inmunitaria al agente en el receptor tras la exposición a la mezcla.
El término "agonista" se usa en el sentido más amplio e incluye cualquier molécula que active la señalización a través de un receptor. Por ejemplo, un agonista de TLR7 se une a una proteína del receptor 7 de tipo toll y activa una vía de señalización de TLR7; un agonista de TLR8 se une a una proteína del receptor 8 de tipo toll y activa una vía de señalización de TLR8. Un agonista dual de TLRH8 se une a las proteínas del receptor de tipo toll 7 y del receptor de tipo toll 8 y activa las vías de señalización de TLR7 y TLR8.
La "estimulación" de una respuesta o parámetro incluye provocar y/o potenciar esa respuesta o parámetro en comparación con condiciones que, por lo demás, son las mismas excepto por el agente o la molécula, o como alternativa, en comparación con otra condición (por ejemplo, aumento en la señalización de TLR en presencia de un agonista de TLR en comparación con la ausencia del agonista de TLR). Por ejemplo, "estimulación" de una respuesta inmunitaria significa un aumento en la respuesta.
Una "cantidad eficaz" de un agente divulgado en el presente documento es una cantidad suficiente para llevar a cabo un fin específicamente indicado. Una "cantidad eficaz" puede determinarse empíricamente y de manera rutinaria, con respecto al fin indicado. Una "cantidad eficaz" o una "cantidad suficiente" de un agente es la cantidad adecuada para producir un efecto biológico deseado, tal como un resultado beneficioso, incluyendo un resultado clínico beneficioso. La expresión "cantidad terapéuticamente eficaz" se refiere a una cantidad de un agente (por ejemplo, modulador de TLR) eficaz para "tratar" una enfermedad o trastorno en un sujeto (por ejemplo, un mamífero tal como un ser humano).
Los términos "tratar" o "tratamiento" de una enfermedad se refieren a la ejecución de un protocolo, que puede incluir la administración de uno o más fármacos a un individuo (humano o no), en un esfuerzo por aliviar los signos o síntomas de la enfermedad. Así pues, "tratar" o el "tratamiento" no requiere el alivio completo de los signos o síntomas, no requiere cura y, de manera específica, incluye protocolos que solo tienen un efecto paliativo en el individuo. Como se usa en el presente documento, así como se entiende en la materia, "tratamiento" es una estrategia para obtener resultados beneficiosos o deseados, incluyendo resultados clínicos. Los resultados clínicos beneficiosos o deseados incluyen, pero sin limitación, el alivio o la mejora de uno o más síntomas, la disminución de la extensión de la enfermedad, la estabilización (es decir, no empeoramiento) de la patología, la prevención de la propagación de la enfermedad, el retraso o la ralentización de la progresión de la enfermedad, la mejora o el alivio de la patología, y la remisión (ya sea parcial o total), ya sea detectable o no detectable. "Tratamiento" también puede significar prolongar la supervivencia en comparación con la supervivencia esperada de un individuo que no recibe tratamiento.
"Atenuar" una enfermedad o trastorno significa que el alcance y/o las manifestaciones clínicas indeseables de la enfermedad o trastorno se reducen y/o la evolución temporal de la progresión de la enfermedad o trastorno se ralentiza, en comparación con el resultado esperado sin tratamiento. Especialmente en el contexto de la alergia, la atenuación puede ocurrir tras la estimulación de una respuesta inmunitaria Th1 contra uno o más alérgenos. Asimismo, la atenuación no ocurre necesariamente con la administración de una dosis, pero a menudo ocurre tras la administración de una serie de dosis. Así pues, se puede administrar una cantidad suficiente para atenuar una respuesta o trastorno en una o más dosis.
"Alquilo", como se usa en el presente documento, se refiere a una cadena de hidrocarburo univalente lineal (es decir, no ramificada) o ramificada saturada o una combinación de las mismas. Los grupos alquilo particulares son aquellos que tienen un número designado de átomos de carbono por ejemplo un grupo alquilo que tiene de 1 a 2 0 átomos de carbono (un "alquilo C1-C20"), que tiene de 1 a os
de carbono (un "alquilo C1-C8"), que tiene de 1 a 6 átomos de carbono (un "alquilo C1-C6"), que tiene de 2 a 6 átomos de carbono (un "alquilo C2-C6"), o que tiene de 1 a 4 átomos de carbono (un" alquilo C1-C4"). Los ejemplos de grupos alquilo incluyen, pero sin limitación, grupos tales como metilo, etilo, n-propilo, isopropilo, n-butilo, t-butilo, isobutilo, sec-butilo, homólogos e isómeros de, por ejemplo, n-pentilo, n-hexilo, n-heptilo, n-octilo y similares.
"Alquenilo", como se usa en el presente documento, se refiere a una cadena de hidrocarburo univalente lineal (es decir, no ramificada) o ramificada insaturada o una combinación de las mismas, que tiene al menos un sitio de instauración olefínica (es decir, que tiene al menos un resto de fórmula C=C). Los grupos alquenilo particulares son aquellos que tienen un número designado de átomos de carbono, por ejemplo, un grupo alquenilo que tiene de 2 a 20 átomos de carbono (un "alquenilo C2-C20"), que tiene de 2 a 10 átomos de carbono (un "alquenilo C2-C10"), que tienen de 2 a 8 átomos de carbono (un "alquenilo C2-C8"), que tiene de 2 a 6 átomos de carbono (un "alquenilo C2-C6"), o que tiene de 2 a 4 átomos de carbono (un "alquenilo C2-C4"). El grupo alquenilo puede estar en configuraciones "cis" o "trans", o como alternativa en configuraciones "E" o "Z". Los ejemplos de grupos alquenilo incluyen, pero sin limitación, grupos tales como etenilo (o vinilo), prop-1-enilo, prop-2-enilo (o alilo), 2-metilprop-1-enilo, but-1-enilo, but-2-enilo, but-3-enilo, buta-1,3-dienilo, 2-metilbuta-1,3-dienilo, homólogos e isómeros de los mismos y similares.
"Alquinilo", como se usa en el presente documento, se refiere a una cadena de hidrocarburo univalente lineal (es decir, no ramificada) o ramificada insaturada o una combinación de las mismas, que tiene al menos un sitio de insaturación acetilénica (es decir, que tiene al menos un resto de fórmula C=C). Los grupos alquinilo particulares son aquellos que tienen un número designado de átomos de carbono, por ejemplo, un grupo alquinilo que tiene de 2 a 20 átomos de carbono (un "alquinilo C2-C20"), que tiene de 2 a 10 átomos de carbono (un "alquinilo C2-C10"), que tiene de 2 a 8 átomos de carbono (un "alquinilo C2-C8"), que tiene de 2 a 6 átomos de carbono (un "alquinilo C2-C6"), o que tiene de 2 a 4 átomos de carbono (un" alquinilo C2-C4"). Los ejemplos de grupos alquinilo incluyen, pero sin limitación, grupos tales como etinilo (o acetilenilo), prop-1-inilo, prop-2-inilo (o propargilo), but-1-inilo, but-2-inilo, but-3-inilo, homólogos e isómeros de los mismos y similares.
"Alquileno", como se usa en el presente documento, se refiere a los mismos restos que alquilo, pero que tienen bivalencia. Los grupos alquileno particulares son aquellos que tienen de 1 a 6 átomos de carbono (un "alquileno C1-C6"), de 1 a 5 átomos de carbono (un "alquileno C1-C5"), de 1 a 4 átomos de carbono (un "alquileno C1-C4") o de 1 a 3 átomos de carbono (un" alquileno C1-C3"). Algunos ejemplos de grupos alquileno incluyen, pero sin limitación, grupos tales como metileno (-CH2- o =CH2), etileno (-CH2CH2- o =CHCH3), propileno (-CH2CH2CH2- o =CHCH2CH3), butileno (-CH2CH2CH2CH2- o =CHCH2CH2CH3), y similares.
"Cicloalquilo", como se usa en el presente documento, se refiere a estructuras de hidrocarburos univalentes cíclicos saturados o insaturados no aromáticos. Los grupos cicloalquilo particulares son aquellos que tienen un número designado de átomos de carbono anulares (es decir, anillos), por ejemplo, un grupo cicloalquilo que tiene de 3 a 12 átomos de carbono anulares (un cicloalquilo "C3-C12"). Un cicloalquilo preferido es un hidrocarburo cíclico que tiene de 3 a 8 átomos de carbono anulares (un "cicloalquilo C3-C8"), o que tiene de 3 a 6 átomos de carbono anulares (un "cicloalquilo C3-C6"). El cicloalquilo puede consistir en un anillo, tal como ciclohexilo o múltiples anillos, tal como adamantilo, pero excluye los grupos arilo. Un cicloalquilo que comprende más de un anillo puede ser condensado, espiro o puenteado o combinaciones de los mismos. Los ejemplos de grupos cicloalquilo incluyen, pero sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo, norbornilo, y similares.
"Cicloalquileno", como se usa en el presente documento, se refiere a los mismos restos que cicloalquilo, pero que tienen bivalencia. Los grupos cicloalquileno particulares son aquellos que tienen de 3 a 12 átomos de carbono (un "cicloalquileno C3-C12"), que tienen de 3 a 8 átomos de carbono anulares (un "cicloalquileno C3-C8"), o que tienen de 3 a 6 átomos de carbono anulares (un "cicloalquileno C3-C6"). Los ejemplos de grupos cicloalquileno incluyen, pero sin limitación, ciclopropileno, ciclobutileno, ciclopentileno, ciclohexileno, 1,2-ciclohexenileno, 1,3-ciclohexenileno, 1,4-ciclohexenileno, cicloheptilo, norbornilo, y similares. "Hidrocarbilo", como se usa en el presente documento, se refiere a e incluye un grupo univalente formado al eliminar un átomo de hidrógeno de un hidrocarburo no aromático, que pueden ser totalmente saturados, monoinsaturados o poliinsaturados, que tienen el número indicado de átomos de carbono (es decir, C1-C20 significa de uno a veinte átomos de carbono). Un grupo hidrocarbilo puede contener uno o más restos lineales, ramificados o cíclicos, o combinaciones de los mismos. Los grupos alquilo, alquenilo, alquinilo y cicloalquilo son subconjuntos particulares de grupos hidrocarbilo. Un grupo hidrocarbilo también puede contener un grupo alquilo, alquenilo o alquinilo sustituido además con uno o más grupos cicloalquilo; y/o un grupo cicloalquilo sustituido además con uno o más grupos alquilo, alquenilo y alquinilo. Un grupo hidrocarbilo puede estar sustituido, en una o más posiciones, con uno o más átomos de halógeno, tal como cloro o flúor. Los ejemplos de grupos hidrocarbilo incluyen, pero sin limitación, grupos como los siguientes:


y similares.
"Arilo", como se usa en el presente documento, se refiere a un grupo carbocíclico aromático insaturado que tiene un solo anillo (por ejemplo, fenilo) o múltiples anillos condensados (por ejemplo, naftilo o antrilo), donde uno o más de los anillos condensados pueden no ser aromáticos. Los grupos arilo particulares son aquellos que tienen de 6 a 14 átomos de carbono anulares (es decir, anulares) (un "arilo C6-C14"). Un grupo arilo que tiene más de un anillo en donde al menos un anillo es no aromático puede estar conectado a la estructura parental en una posición de anillo aromático o en una posición de anillo no aromático. En una variación, un grupo arilo que tiene más de un anillo en donde al menos un anillo no es aromático está conectado a la estructura parental en una posición de anillo aromático. Los ejemplos de arilo incluyen, pero sin limitación, grupos tales como fenilo, naftilo, 1-naftilo, 2-naftilo y similares.
"Arileno", como se usa en el presente documento, se refiere a los mismos restos que arilo, pero que tienen bivalencia. Los grupos arileno particulares son aquellos que tienen de 6 a 14 átomos de carbono anulares (un "arileno C6-C14"). Los ejemplos de arileno incluyen, pero sin limitación, grupos tales como fenileno, o-fenileno (es decir, 1,2-fenileno), m-fenileno (es decir, 1,3-fenileno), p-fenileno (es decir, 1,4-fenileno), naftileno, 1,2-naftileno, 1,2-naftileno, 1,4-naftileno y similares.
"Halo" o "halógeno" se refiere a elementos de la serie del Grupo 17 que tienen un número atómico de 9 a 85. Grupos halo preferidos incluyen flúor, cloro, bromo y yodo. Cuando un resto se sustituye por más de un halógeno, se puede hacer referencia a él mediante el uso de un prefijo correspondiente al número de residuos de halógeno fijados. Por ejemplo, dihaloarilo, dihaloalquilo y trihaloarilo, etc., se refieren a arilo y alquilo sustituidos con dos ("di") o tres ("tri") grupos halo, que pueden ser, pero no necesariamente, el mismo halo; por tanto, el 4-cloro-3-fluorofenilo está dentro del alcance de un dihaloarilo. Un grupo alquilo en el que cada hidrógeno se reemplaza con un grupo halo se denomina "perhaloalquilo". Un grupo perhaloalquilo preferido es trifluoroalquilo (-CF3). De forma similar, "perhaloalcoxi" se refiere a un grupo alcoxi en el que un halógeno ocupa el lugar de cada H en el hidrocarburo que forma el residuo alquilo del grupo alcoxi. Un ejemplo de un grupo perhaloalcoxi es trifluorometoxi (-OCF3).
"Amino" se refiere al grupo -NH2.
"Amino sustituido" se refiere al grupo -NR'R" d te
en hidrógeno, alquilo, alquilo sustituido, cicloalquilo, cicloalquilo sustituido, alquenilo, alquenilo sustituido, cicloalquenilo, cicloalquenilo sustituido, alquinilo, alquinilo sustituido, arilo, heteroarilo y heterociclilo, a condición de que al menos uno de R' y R" no sea hidrógeno.
"Opcionalmente sustituido" a menos que se especifique lo contrario significa que un grupo puede estar sin sustituir o sustituido con uno o más (por ejemplo, 1, 2, 3, 4 o 5) de los sustituyentes enumerados para ese grupo en el que los sustituyentes pueden ser iguales o diferentes. En una realización, un grupo opcionalmente sustituido tiene un sustituyente. En otra realización, un grupo opcionalmente sustituido tiene dos sustituyentes. En otra realización, un grupo opcionalmente sustituido tiene tres sustituyentes. En otra realización, un grupo opcionalmente sustituido tiene cuatro sustituyentes. En algunas realizaciones, un grupo opcionalmente sustituido tiene de 1 a 2, de 1 a 3, de 1 a 4 o de 1 a 5 sustituyentes.
"Modificador orgánico", a menos que se especifique lo contrario, significa uno de un grupo de disolventes normalmente usados para solubilizar compuestos químicos orgánicos. Este grupo puede incluir, pero sin limitación, ácido acético, acetona, anisol, 1-butanol, 2-butanol, acetato de butilo, ferc-butil metil éter, cumeno, dimetilsulfóxido, etanol, acetato de etilo, éter etílico, formiato de etilo, ácido fórmico, heptano, acetato de isobutilo, acetato de isopropilo, acetato de metilo, 3-metil-1-butanol, metiletilcetona, metilisobutilcetona, 2-metil-1-propanol, pentano, 1-pentanol, 1-propanol, 2-propanol (isopropanol), acetato de propilo y combinaciones de los mismos.
Además de la divulgación del presente documento, el término "sustituido", cuando se usa para modificar un grupo o radical especificado, también puede significar que uno o más átomos de hidrógeno del grupo o radical especificado son reemplazados cada uno, independientemente entre sí, por el mismo o diferentes grupos sustituyentes como se definen en el presente documento. En algunas realizaciones, un grupo que está sustituido tiene 1, 2, 3 o 4 sustituyentes, 1, 2 o 3 sustituyentes, 1 o 2 sustituyentes o 1 sustituyente.
A menos que se indique un isótopo específico de un elemento en una fórmula, la invención incluye todos los isotopólogos de los compuestos divulgados en el presente documento, tales como, por ejemplo, derivados deuterados de los compuestos (donde H puede ser 2H, es decir, D). Los isotopólogos pueden tener reemplazos isotópicos en cualquiera o en todas las ubicaciones de una estructura, o pueden tener átomos presentes en abundancia natural en cualquiera o todas las ubicaciones de una estructura.
Se aprecia que determinadas características de la invención, que están, por claridad, descritas en el contexto de realizaciones separadas, también pueden proporcionarse en combinación en una sola realización. Por el contrario, diversas características de la invención, que están, por brevedad, descritas en el contexto de una única realización, también pueden proporcionarse por separado o en cualquier subcombinación adecuada. Todas las combinaciones de las realizaciones que pertenecen a los grupos químicos representados por las variables están abarcadas específicamente por la presente invención y se desvelan en el presente documento igual que si todas y cada una de las combinaciones se hubiesen desvelado en el presente documento de manera individual y explícita, en la medida en que tales combinaciones abarquen compuestos que son compuestos estables (es decir, compuestos que pueden aislarse, caracterizarse y probarse para determinar su actividad biológica). Además, todas las subcombinaciones de los grupos químicos enumerados en las realizaciones que describen tales variables también están abarcadas específicamente por la presente invención y se desvelan en el presente documento igual que si todas y cada una de tales subcombinaciones de grupos químicos se hubiesen desvelado en el presente documento de manera individual y explícita.
Se entiende que los aspectos y realizaciones descritos en el presente documento como "que comprenden" incluyen realizaciones "que consisten en" y/o "que consisten esencialmente en".
Como se usan en el presente documento y en las reivindicaciones adjuntas, las formas singulares "un", "una" y "el/la" incluyen referentes plurales a menos que se indique lo contrario o quede claro por el contexto.
A menos que se indique claramente lo contrario, el término "aproximadamente" se usa para indicar que un valor incluye la desviación típica de error para el dispositivo o método que se emplea para determinar el valor. La referencia a "aproximadamente" un valor o parámetro en el presente documento incluye (y describe) realizaciones que se refieren a ese valor o parámetro en sí mismo. Por ejemplo, una descripción que hace referencia a "aproximadamente X" incluye la descripción de "X".
II. Compuestos
En un aspecto, se proporciona un compuesto de fórmula (J):

R0 es -(CH2)mRA o -(CH2)z(C(CH3)2)RA; m es 0, 1, 2 o 3; z es 1 o 2; y RA es cicloalquilo C3-C8 opcionalmente sustituido con de 1 a 4 grupos seleccionados independientemente del grupo que consiste en alquilo C1-C4 , alquileno C1-C4 y halógeno;
X es -NH-;
R1 es alquilo C3-C6 , -(CH2)pOR1a, -(CH2)pNHR1b o -(CH2)pR1c; donde R1a y R1b son independientemente alquilo C1-C3 ; R1c es cicloalquilo C3-C4 ; y p es 1 o 2;
R2 es NHR2 a ; donde R2a es H, Oh , NH2 o metilo;
cada R3 es independientemente halógeno, alquilo C1-C8 , -(alquilen C1-C7)-NH2 ,
o -CH2-fenilen-CH2NH2 ;
q es 0, 1, 2, 3 o 4; y
R4a y R4b son independientemente H o alquilo C1-C8.
En algunas realizaciones, R0 es -(CH2)mRA. En una variación, m es 1 o 2, y RA es ciclopropilo, ciclobutilo o ciclopentilo. En otra variación, m es 0 y RA es ciclobutilo, ciclopentilo o ciclohexilo.
En algunas realizaciones, RA es cicloalquilo C3-C8.
En algunas realizaciones, RA es cicloalquilo C3-C6 opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno. En una variación, m es 1 o 2.
En algunas realizaciones, RA es ciclopropilo opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno, y m es 1 o 2.
En algunas realizaciones, m es 0 o 1, y RA es ciclohexilo opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno.
En algunas realizaciones, RA es cicloalquilo C3-C6 opcionalmente sustituido con de 1 a 4 átomos de halógeno. En algunas realizaciones, RA es cicloalquilo C3-C6 opcionalmente sustituido con de 1 a 3 átomos de cloro o flúor. En algunas realizaciones, RA es cicloalquilo C3-C6 opcionalmente sustituido con de 1 a 2 átomos de cloro o flúor. En algunas realizaciones, RA es ciclobutilo opcionalmente sustituido con de 1 a 2 átomos de flúor. En una variación, m es 1.
En algunas realizaciones, R0 es -(CH2)z (C(CH3)2)RA. En una variación, z es 1 o 2, y RA es ciclopropilo, ciclobutilo o ciclopentilo. En una variación, z es 1 y RA es ciclopropilo, ciclobutilo o ciclopentilo.
En algunas realizaciones, R0 se selecciona entre el grupo que consiste en:


En algunas realizaciones, R0 se selecciona entre el grupo que consiste en:

En algunas realizaciones, R0 se selecciona entre el grupo que consiste en:


En algunas realizaciones, X es -NH-, R0 es -(CH2)mRA, m es 2 y RA es ciclopropilo, ciclobutilo o ciclopentilo.
En algunas realizaciones, X es -NH-, R0 es -(CH2)z(C(CH3)2)RA, z es 1 y RA es ciclopropilo, ciclobutilo o ciclopentilo. En algunas realizaciones, X es -NH-, R0 es -(CH2)mRA, m es 0 y RA es ciclobutilo, ciclopentilo o ciclohexilo.
En algunas realizaciones, R1 es alquilo C3-C6 (por ejemplo, n-butilo). En algunas realizaciones, R1 es propilo, butilo, pentilo o hexilo. En algunas realizaciones preferidas, R1 es n-butilo. En algunas realizaciones, R1 es n-pentilo. En algunas realizaciones, R1 es -(CH2)pO1a (por ejemplo, CH2OCH2CH3). En algunas realizaciones, R1 es -(CH2)pNHR1b (por ejemplo, CH2 NHCH2CH3). En algunas realizaciones, R1 es (CH2)pR1c. En una variación, R1c es ciclopropilo. En algunas realizaciones, R2 es NHR2 a , donde R2a es H, OH, NH2 o metilo. En algunas realizaciones, R2 es NH2. En algunas realizaciones, R2 es NHOH, NHNH2 o NHCH3.
En algunas realizaciones, R4a y R4b son independientemente H o alquilo C1-C8. En algunas realizaciones, cada R4a y R4b es H.
En algunas realizaciones, el resto fenilo del núcleo de 1H-imidazo[4,5-c]quinolina no está sustituido (es decir, q es 0). En algunas realizaciones, el resto fenilo del núcleo de 1H-imidazo[4,5-c]quinolina está sustituido con 1, 2, 3 o 4 sustituyentes seleccionados independientemente del grupo que consiste en halógeno, alquilo C1-C8 , -(alquilen C1-C7)-NH2 y -CH2-fenilen-CH2NH2. En algunas realizaciones, q es 1 y R3 es alquilo C1-C8.
Se pretende y se entiende que cuando estén presentes todas y cada una de las variaciones de X y R0 descritas para la fórmula (J) puede combinarse con todas y cada una de las variaciones de R1, p, R2 , q, R3 , R4a y R4b descritas para la fórmula (J) lo mismo que si todas y cada una de las combinaciones estuvieran descritas específica e individualmente. En algunas realizaciones, el compuesto de fórmula (J) es de fórmula (J-1):

o una sal del mismo, en donde R0 es hidrocarbilo C4-C21. En algunas realizaciones, R0 es -(CH2)mRA. En una variación, m es 1 o 2, y RA es ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo. En una variación, m es 2 y RA es ciclopropilo, ciclobutilo o ciclopentilo. En otra variación, m es 0 y RA es ciclobutilo, ciclopentilo o ciclohexilo. En algunas realizaciones, R0 es -(CH2)z(C(CH3)2)RA. En una variación, z es 1 y RA es ciclopropilo, ciclobutilo o ciclopentilo. En algunas realizaciones, RA está opcionalmente sustituido con 1-4 grupos seleccionados independientemente del grupo que consiste en metilo, metileno y halógeno.
También se divulga un compuesto de fórmula (K):

o una sal del mismo, en donde:
n es un número entero de 4 a 21;
X es -NH- o -NH(C=O)-;
R1 es alquilo C3-C6 , -(CH2)pOR1a, -(CH2)pNHR1b o -(CH2)pR1c; donde R1a y R1b son independientemente alquilo C1-C3 ; R1c es cicloalquilo C3-C4 ; y p es 1 o 2;
R2 es NHR2 a ; donde R2a es H, OH, NH2 o metilo;
cada R3 es independientemente halógeno, alquilo C1-C8 , -(alquilen C1-C7)-NH2 o -CH2-fenilen-CH2NH2 ;
q es 0, 1,2, 3 o 4; y
R4a y R4b son independientemente H o alquilo C1-C8 ,
siempre que el compuesto sea distinto de 2-butil-1-(4-((hexadecilamino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-32) o N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)palmitamida (Compuesto n.° 63-31).
Los compuestos representativos de la invención (marcados con un asterisco) se enumeran en la tabla 1.


(continuación)











ti ió
En algunas realizaciones, se proporciona un compuesto seleccionado del grupo que consiste en uno o más de los Compuestos n.° 63-33, 63-35, 63-36 y 63-38 a 63-49 en la tabla 1, o una sal del mismo.
Los compuestos de la presente divulgación son potentes agonistas de TLRH8 que muestran una bioactividad equilibrada frente a ambos receptores y poseen propiedades fisicoquímicas tales como una mayor hidrofobicidad. Los compuestos con bioactividad agonista potente frente a los receptores TLR7 y TLR8 son adyuvantes inmunitarios potencialmente más eficaces que los agonistas específicos solo para uno de estos TLR, y promoverían respuestas inmunitarias innatas en una gama más amplia de células presentadoras de antígenos y otros tipos de células inmunitarias clave, incluyendo células dendríticas plasmocitoides y mieloides, monocitos y linfocitos B (véase por ejemplo, Vasilakos et al 2013 Expert Rev Vaccines 12:809-819). Se sabe que los compuestos con propiedades fisicoquímicas, tales como una mayor hidrofobicidad, son compatibles con los enfoques de formulación a base de aceite y permiten composiciones farmacéuticas que promueven la retención del compuesto en el sitio de inyección.
En algunas realizaciones, los compuestos de fórmula (J) son capaces de activar tanto TLR7 como TLR8. En algunas realizaciones, el compuesto de fórmula (J) tiene una CE50 para TLR7 de aproximadamente 200 nM o menos y una CE50 para TLR8 de aproximadamente 2000 nM o menos, en donde los valores de CE50 son los descritos en el ejemplo B1. En algunas realizaciones, el compuesto de fórmula (J) tiene una CE50 para TLR7 de aproximadamente 50 nM o menos, y una CE50 para TLR8 de aproximadamente 1000 nM o menos. En algunas realizaciones, el compuesto de fórmula (J) tiene una CE50 para TLR7 de aproximadamente 200 nM aproximadamente 175 nM aproximadamente 150 nM, aproximadamente 125 nM, aproxima M,
aproximadamente 40 nM, aproximadamente 30 nM, aproximadamente 20 nM, aproximadamente 10 nM, aproximadamente 8 nM, aproximadamente 6 nM, aproximadamente 5 nM, aproximadamente 4 nM, aproximadamente 3 nM, aproximadamente 2 nM, aproximadamente 1 nM, o aproximadamente 0,5 nM; y una CE50 para TLR8 de aproximadamente 2000 nM, 1500 nM, 1000 nM, 900 nM, 800 nM, 700 nM, 600 nM, 500 nM, 400 nM, 300 nM, 200 nM, 100 nM, 50 nM, 25 nM, 10 nM, 5 nM, 1 nM o 0,5 nM.
Los compuestos de fórmula (J-1) son derivados N-alquílicos de 1-(4-(aminometil)bencil)-2-butil-1H-imidazo[4,5-c]quinolin-4-amina (IMDQ. IMDQ (Compuesto n.° 63-00) muestra potente actividades in vitro para ambos receptores TLR7 y TLR8 (véanse, por ejemplo, las Patentes de EE.UU. n.° 8.728.486 y 9.441.005). La modificación de la cadena alquílica de los compuestos de la presente invención en la amina bencílica aumenta la hidrofobicidad y, por lo tanto, permite que composiciones farmacéuticas que promueven la retención del compuesto en el sitio de inyección. Sin embargo, mientras que los derivados de cadenas alquílicas de longitud de cadena de carbono creciente pueden proporcionar aumentos significativos en la hidrofobicidad (por ejemplo, según lo estimado por el coeficiente de reparto calculado o cLogP), estos derivados también pueden tener una potencia agonista disminuida contra TLR7 y TLR8 , o tener bioactividad selectivamente disminuida contra uno de los dos receptores. Por ejemplo, el derivado de N-octadecanoílo (Compuesto n.° 63-13) es más de 40 veces menos potente contra TLR7 y 2 6 veces menos potente contra TLR8 que su congénere original IMDQ en los mismos ensayos de bioactividad de células inmunitarias humanas in vitro. Inesperadamente, el derivado de N-tetradecanoílo (Compuesto n.° 63-10) de IMDQ es solo 2 y 2,4 veces menos potente contra TLR7 y TLR8 , respectivamente, que IMDQ. Estos datos demuestran que una cadena alquílica de longitud óptima proporciona una potente bioactividad equilibrada de agonista de TLRH8, así como una mayor hidrofobicidad que permite la incorporación en composiciones farmacéuticas que promueven la retención del compuesto en el sitio de inyección.
Los compuestos de fórmula (J-1) son derivados N-alquílicos de 1-(4-(aminometil)bencil)-2-butil-1H-imidazo[4,5-c]quinolin-4-amina (IMDQ). Mientras que los derivados de cadenas alquílicas de longitud de cadena de carbono creciente pueden proporcionar aumentos en la hidrofobicidad (por ejemplo, según lo estimado por el coeficiente de reparto calculado o cLogP), estos derivados también pueden tener una potencia agonista disminuida contra TLR7 y t LR8 , o tener bioactividad selectivamente disminuida contra uno de los dos receptores. Por ejemplo, el derivado de 4-octadecilamino (Compuesto n.° 63-29) es 19 veces menos potente contra TLR7 y más de 12 veces menos potente contra TLR8 que su congénere original IMDQ en los mismos ensayos de bioactividad de células inmunitarias humanas in vitro (véase, por ejemplo, el ejemplo B1). De manera interesante, el derivado de 4-pentilamino (Compuesto n.° 63 17) es solo 2 veces menos potente contra TLR7 y es 2 veces más potente contra TLR8 que IMDQ, pero muestra una hidrofobicidad significativamente menor que el Compuesto n.° 63-29. Inesperadamente, el derivado de 4-(2-ciclopropiletil)amino (Compuesto n.° 63-33) es solo 4 veces menos potente contra TLR7 y 2,9 veces más potente contra TLR8 que IMDQ, y tiene un cLogP aumentado en comparación con la variante lineal de 5 carbonos (Compuesto n.° 63-17). Estos datos demuestran que una cadena alquílica de longitud óptima proporciona una potente bioactividad equilibrada de agonista de TLRH8, así como una mayor hidrofobicidad que puede permitir la incorporación en composiciones farmacéuticas que promuevan la retención del compuesto en el sitio de inyección.
Una molécula pequeña agonista de TLRH8 con potencia dual equilibrada también permitiría la síntesis y caracterización de un solo principio farmacéutico activo, facilitando así la fabricación NCF a costes más bajos y permitiendo una vía regulatoria más directa y predecible.
Los compuestos de fórmula (J) se pueden sintetizar de acuerdo con el esquema 1 y/o utilizando métodos conocidos en la técnica.
Esquema 1

en donde R1, R2, q y R3 son como se definen
Compuestos preferidos de fórmula (J) donde X es -NH-, R0 es -(CH2 )mRA, m es 1 o 2, y RA es ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo se pueden preparar utilizando los siguientes ácidos carboxílicos: ácido ciclopropanocarboxílico, ácido ciclopropilacético, ácido ciclobutanocarboxílico, ácido ciclobutilacético, ácido ciclopentanocarboxílico, ácido ciclopentilacético, ácido ciclohexanocarboxílico y ácido ciclohexanoacético. Compuestos preferidos de fórmula (J) donde X es -NH-, R0 es -(CH2 )mRA, m es 1, 2 o 3, y RA es ciclopropilo, 2-metilciclopropilo, 2,2,-dimetilciclopropilo, 1-metilciclopropilo, 1 -metilciclobutilo, 3-metilciclobutilo o 3-fluorociclobutilo se pueden preparar utilizando los siguientes ácidos carboxílicos: ácido ciclopropanocarboxílico, ácido 2-metilciclopropanocarboxílico, ácido 2,2-dimetilciclopropanocarboxílico, ácido 1-metilciclopropanocarboxílico, ácido 1-metilciclobutanocarboxílico, ácido 3-metilciclobutanocarboxílico o ácido 3-fluorociclobutanocarboxílico. Compuestos preferidos de fórmula (J) donde X es -NH-, R0 es -(CH2)(C(CH3)2)RAy RA es ciclopropilo o ciclobutilo se pueden preparar utilizando los siguientes ácidos carboxílicos: ácido ciclopropanocarboxílico o ácido ciclobutanocarboxílico. Se puede encontrar una descripción detallada del esquema de síntesis para el Compuesto representativo n.° 63-33 en el ejemplo S3.
En algunas realizaciones, donde R1 es alquilo C3-C6 (por ejemplo, n-butilo), R2 es NH2 , X es -NH-, R0 es -(CH2)mRA, m es 0 y RA es cicloalquilo, los compuestos se sintetizan de acuerdo con el esquema 1-2. Un compuesto de fórmula (J), en donde R1 es n-butilo, R2 es NH2 , X es -NH-, R0 es -(CH2 )mRA, m es 0 y RA es ciclohexilo (Compuesto n.° 63-49) de acuerdo con la síntesis descrita en el ejemplo S10.
Esquema 1-2

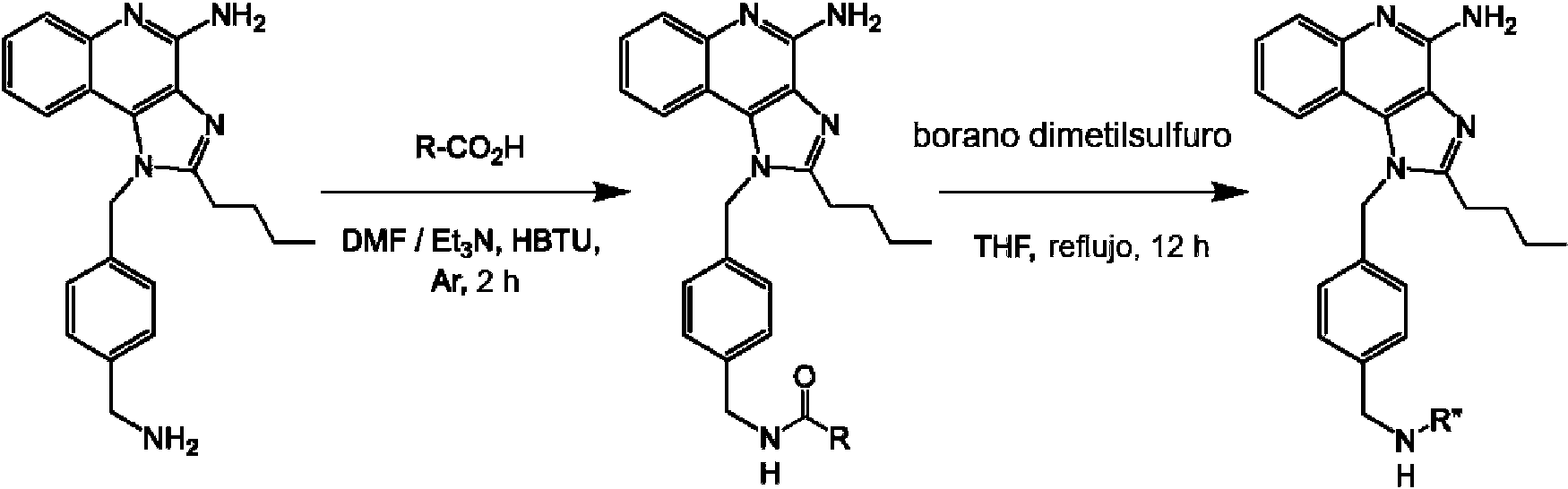
Los compuestos de fórmula (K) se pueden sintetizar de acuerdo con el esquema 2 y/o utilizando métodos conocidos en la técnica.
Esquema 2

en donde R1, R2, q y R3 son como se definen para la fórmula (K), R y R" son grupos alquilo lineales.
Compuestos preferidos de fórmula (K) donde X es -NH(C=O)- y n es un número entero de 4 a 21 siempre que el compuesto sea distinto de N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)palmitamida (Compuesto n.° 63-31) se puede preparar utilizando los siguientes ácidos carboxílicos; pentanoico, hexanoico, heptanoico, octanoico, nonanoico, decanoico, undecanoico, dodecanoico, tridecanoico, tetradecanoico, pentadecanoico, hexadecanoico, heptadecanoico, octadecanoico nonadecanoico icosanoico heneicosanoico y docosanoico Se puede encontrar una descripción detallada de y
63-17 en los ejemplos S1 y S2, respectivamente.
En algunos casos, donde R1 es alquilo C3-C6 (por ejemplo, n-butilo), R2 es NH2 , y q es 0, los compuestos se sintetizan de acuerdo con el esquema 3 o 4. Para una descripción más detallada de las etapas de reacción individuales útiles para preparar el Compuesto n.° 63-00, el compuesto de partida en los esquemas 3 y 4, véase, por ejemplo, las patentes de EE. UU. n.° 8.728.486 y 9.441.005.
Esquema 3

en donde R y R0 son grupos hidrocarbilo.
Esquema 4
en donde R y R" son grupos alquilo lineales.
Los expertos en la materia apreciarán que se pueden emplear otras rutas sintéticas para sintetizar la gama de compuestos descritos en la invención, incluidos diversos disolventes, catalizadores, agentes reductores, temperaturas, tiempos de reacción y condiciones atmosféricas.
Pueden usarse métodos y técnicas convencionales de separación y purificación para aislar los compuestos de la invención. Las técnicas pueden incluir cromatografía líquida de alta resolución (HPLC) con diferentes matrices (véase, por ejemplo, C18, C8, C4, etc.), cromatografía utilizando adsorbentes típicos (véase, por ejemplo, gel de sílice, carbón activado, alúmina, zeolitas y similares), recristalización y métodos de extracción diferencial (por ejemplo, líquidolíquido, fase sólida, y similares).
III. Composiciones farmacéuticas
También se proporcionan composiciones farmacéuticas que comprenden agonistas de TLRH8 de 1H-imidazo[4,5-c]quinolina de cadena alquílica modificada de la presente divulgación. Las composiciones farmacéuticas contienen rutinariamente uno o más excipientes farmacéuticamente aceptables. En algunas realizaciones, las composiciones farmacéuticas comprenden además un antígeno. Las composiciones farmacéuticas de la presente divulgación son preferentemente estériles y preferentemente esencialmente libres de endotoxinas.
Excipientes
Los excipientes farmacéuticamente aceptables de la presente divulgación incluyen, por ejemplo, aceites, lípidos, disolventes, agentes espesantes, tensioactivos agentes tamponantes agentes de ajuste de la tonicidad y conservantes (véase, por ejemplo, Pramanic as
composiciones farmacéuticas comprenden un excipiente que funciona como uno o más de un disolvente, un agente espesante, un agente tamponante y un agente de ajuste de la tonicidad (por ejemplo, el cloruro de sodio en solución salina puede servir tanto como vehículo acuoso como agente de ajuste de la tonicidad). Las composiciones farmacéuticas de la presente divulgación son adecuadas para las vías de administración parenteral y, en ciertos casos, preferentemente para la administración intratumoral. En determinadas realizaciones, las composiciones farmacéuticas de la presente divulgación no están destinadas a la administración enteral.
En algunas realizaciones, las composiciones farmacéuticas comprenden un excipiente a base de aceite para solubilizar el compuesto agonista de TLRH8 para permitir la administración parenteral, así como para promover la retención del compuesto en el sitio de inyección. Los ejemplos no limitantes de excipientes a base de aceite son conocidos por los expertos en la materia, incluyendo aceite de sésamo de calidad farmacéutica, aceite de soja, aceite de ricino, aceite de maíz, aceite de semilla de algodón, aceite de cacahuete, Miglyol®, aceite de escualeno, y similares. Estos aceites pueden ser purificados o refinados, mediante un proceso de cromatografía para reducir los niveles de impurezas polares, produciendo así productos de calidad USP-NHJP/Ph. Eur., que poseen propiedades y perfiles de impurezas consistentes.
En algunas realizaciones, el compuesto agonista de TLRH8 se solubiliza inicialmente en un excipiente de etanol al 100 % y a continuación se diluye en el aceite hasta una concentración final de entre 2 y 20 % de etanol para facilitar la solubilización del compuesto en el aceite. El etanol adecuado para su uso es aquel que no contiene agua o desnaturalizantes, véase, por ejemplo, etanol de 200 grados, alcohol deshidratado de calidad USP, etc.
En algunas realizaciones, las composiciones farmacéuticas comprenden un conservante. Los conservantes adecuados incluyen, por ejemplo, agentes antimicrobianos y antioxidantes. En realizaciones preferidas, la composición farmacéutica se prepara en condiciones estériles y está en un recipiente de un solo uso y, por tanto, no necesita la inclusión de un agente antimicrobiano. Un experto en la materia reconocerá que los antioxidantes de calidad farmacéutica, utilizados para evitar coloración, olores, o formación de peróxido, pueden añadirse a estas formulaciones a base de aceite, incluyendo pero sin limitarse a hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), butilhidroquinona terciaria (TBHQ), vitamina E, galato de propilo y similares. En determinadas realizaciones, la concentración de antioxidante añadido en la formulación es de al menos 10 ppm, 50 ppm, 100 ppm, 300ppm, 500 ppm y hasta 1000 ppm, para garantizar la estabilidad de la formulación a base de aceite durante períodos de hasta 1 año cuando se almacena a temperaturas de 5 °C a 40 °C.
En algunas realizaciones, las composiciones farmacéuticas comprenden una nanoemulsión a base de aceite en agua (véase, por ejemplo, Dowling et al 2017 JCI Insight 2:e91020) o una formulación a base de liposomas (véase, por ejemplo, VanHoeven et al 2017 Sci Rep 7:46426). En una realización que ejemplifica una composición farmacéutica basada en nanoemulsión de aceite en agua, los compuestos agonistas de TLRH8 de fórmula (J) o (K) se disuelven en una fase oleosa compuesta de fosfolípidos (por ejemplo, 1,2-dimiristoil-sn-glicero-3-fosfocolina; 1,2-dipalmitoleoilsn-glicero-3-fosfocolina; 1,2-diestearoil-sn-glicero-3-fosfocolina; 1,2-diestearoil-sn-glicero-3-fosfo-(1'-rac-glicerol); 1,2-dioleoil-sn-glicero-3-fosfocolina; L-a-fosfatidilcolina; y similares), aceites a base de triglicéridos (véase, por ejemplo, aceite de sésamo, aceite de soja, aceite de ricino, aceite de maíz, aceite de semilla de algodón, aceite de cacahuete, Miglyol®, aceite de escualeno y similares), y opcionalmente un modificador orgánico (por ejemplo, etanol). Posteriormente, una fase acuosa que contiene un tampón adecuado, agente isotónico, un agente emulsionante y, opcionalmente, un conservante se añade a la fase oleosa y se forma una emulsión en bruto mediante mezcla de alto cizallamiento (por ejemplo, Polytron®) durante 5-10 minutos. Finalmente, una nanoemulsión que contiene partículas con un diámetro medio en el intervalo de 100 - 150 nm (evaluado por dispersión de luz dinámica) y un índice de dispersidad en el intervalo de 0,1 - 0,2, se forma procesando la emulsión en bruto a través de un homogeneizador de alto cizallamiento (véase, por ejemplo, Microfluidizer M110P) durante 10 - 15 pases a 30.000 psi.
En otra realización que ejemplifica una composición farmacéutica basada en liposomas, los compuestos agonistas de TLRH8 de fórmula (J) se disuelven en modificadores orgánicos y una mezcla adecuada de fosfolípidos usando diversas proporciones de fosfolípidos con carga neutra, cargados positivamente, cargados negativamente y pegilados, con diferentes longitudes de cola de lípidos y colesterol (dependiendo de las propiedades fisicoquímicas deseadas del liposoma). A continuación, los disolventes orgánicos se eliminan usando un evaporador rotatorio y la película de lípido/compuesto se vuelve a disolver en un tampón acuoso hasta que la formulación es translúcida sin partículas visibles. Finalmente, los liposomas multilamelares resultantes se procesan mediante homogeneización de alto cizallamiento o una extrusora de membrana (por ejemplo, Lipex®) a liposomas unilamelares con un diámetro medio en el intervalo de 100 - 150 nm (evaluado por dispersión de luz dinámica) y un índice de dispersidad en el intervalo de 0,1 - 0,2. Estos ejemplos no son limitantes, y un experto en la materia reconocerá que las nanoemulsiones de aceite en agua y las composiciones farmacéuticas basadas en liposomas se pueden formar mediante cualquiera de varios métodos diferentes (véase, por ejemplo, Brito et al 2013 Seminar Immunol 25:130-145).
En algunas realizaciones, las composiciones farmacéuticas comprenden un agente espesante. Los agentes espesantes son particularmente útiles cuando la composición farmacéutica se va a liofilizar antes de la administración. En algunas realizaciones, el agente espesante es un lioprotector que ayuda en la estabilización y prevención de la degradación de los agentes activos durante la liofilización y/o durante el almacenamiento Los agentes espesantes adecuados son azúcares (mono-, di- y polisac sa
y rafinosa.
En algunas realizaciones, las composiciones farmacéuticas comprenden un agente tamponante. Los agentes tamponantes controlan el pH para inhibir la degradación del agente activo durante el procesamiento, almacenamiento y, opcionalmente, reconstitución. Los tampones adecuados incluyen, por ejemplo, sales que comprenden acetato, citrato, fosfato o sulfato. Otros tampones adecuados incluyen, por ejemplo, aminoácidos, tales como arginina, glicina, histidina y lisina. El agente tamponante puede comprender además ácido clorhídrico o hidróxido de sodio. En algunas realizaciones, el agente tamponante mantiene el pH de la composición dentro de un intervalo de 4 a 9. En algunas realizaciones, el pH es mayor que (límite inferior) 4, 5, 6, 7 u 8. En algunas realizaciones, el pH es menor que (límite superior) 9, 8, 7, 6 o 5. Esto es, el pH está en el intervalo de aproximadamente 4,0 a 9,0, en el que el límite inferior es menor que el límite superior.
En algunas realizaciones, las composiciones farmacéuticas comprenden un agente de ajuste de la tonicidad. Los agentes de ajuste de la tonicidad adecuados incluyen, por ejemplo, dextrosa, glicerol, cloruro de sodio, glicerina y manitol.
Antígenos
En un aspecto, la presente divulgación proporciona composiciones farmacéuticas que comprenden un antígeno. En algunas realizaciones, la composición farmacéutica comprende un agonista de TLRH8 de 1H-imidazo[4,5-c]quinolina de cadena alquílica modificada, uno o más excipientes y un antígeno. En algunas de estas realizaciones, el antígeno es un antígeno proteico. En algunas de estas realizaciones, el antígeno es un antígeno polisacarídico, que preferentemente está unido covalentemente a una proteína transportadora. En algunas realizaciones, el antígeno es un antígeno microbiano, un alérgeno o un antígeno asociado a un tumor. En algunas realizaciones, el antígeno es un antígeno vírico, un antígeno protozoario, un antígeno bacteriano o un antígeno fúngico. En algunas realizaciones, el antígeno tumoral es un autoantígeno o neoantígeno.
En algunas realizaciones, las composiciones farmacéuticas comprenden un antígeno microbiano seleccionado del grupo que consiste en un antígeno vírico, un antígeno bacteriano, un antígeno fúngico y un antígeno parasitario. En algunas realizaciones, el antígeno microbiano proviene de un microbio que causa una enfermedad infecciosa en un sujeto mamífero no humano. En algunas realizaciones, el antígeno microbiano procede de un microbio que causa una enfermedad infecciosa en un sujeto humano. En algunas realizaciones, la enfermedad infecciosa es causada por un virus, una bacteria, un hongo o un parásito protozoario. Los antígenos microbianos adecuados incluyen, por ejemplo, antígenos de adenovirus tipo 4, adenovirus tipo 7, Bacillus antracis (antrax), tuberculosis por Mycobacterium, Corynebacterium diphtheriae (por ejemplo, toxoide diftérico), Clostridium tetani (por ejemplo, toxoide tetánico), Bordetella pertussis, Haemophilus influenza tipo B, virus de la hepatitis A, virus de la hepatitis B (por ejemplo, HBsAg), virus del papiloma humano (tipos 6, 11, 16, 18, 31, 33, 45, 52 y 58) virus de la gripe tipo A y B (por ejemplo, hemaglutinina, neuraminadasa), virus de la gripe tipo B, virus paragripal, virus de la encefalitis japonesa, virus del sarampión, virus de las paperas, virus de la rubeola, Neisseria meningitidis (Grupos A, B, C, Y y W-135), Streptococcus pneumoniae (serotipos 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F y 23F), poliovirus, virus de la rabia, rotavirus, virus vaccinia, Salmonella typhi, virus de la varicela zoster y virus de la fiebre amarilla (véase, por ejemplo, Plotkin, S.A., Orenstein, W., Offit, P.A., Edwards K.M. (2017). Plotkin's Vaccines, 7a edición. Elsevier). En algunas realizaciones, el antígeno microbiano es un antígeno vírico del virus herpes simplex tipo 1 o 2, virus del herpes humano, virus de la inmunodeficiencia humana tipo 1 y virus respiratorio sincitial. En algunas realizaciones, el antígeno microbiano es un antígeno fúngico de Candida albicans, Aspergillus flavus, Cryptococcus neoformans, Histoplasma capsulatum, y Pneumocystis carinii. En algunas realizaciones, el antígeno microbiano es un antígeno parasitario de una especie de Leishmania, una especie de Plasmodium, una especie de Schistosoma, o una especie de Trypanosoma.
En algunas realizaciones, las composiciones farmacéuticas comprenden un alérgeno. En algunas realizaciones, el alérgeno es un antígeno ambiental tal como alérgenos de mamíferos, insectos, plantas y moho. En algunas realizaciones, el alérgeno de mamíferos incluye pelo y caspa. Los alérgenos de mamíferos adecuados incluyen, por ejemplo, Fel d 1 de gato, Bos d 2 de vaca, Can f I y Can f II de perro, Equ c1 de caballo y MUP de ratón. En algunas realizaciones, el alérgeno de insectos incluye heces y veneno de insectos. Ejemplos de alérgenos de insectos incluyen Sol i2 de hormiga, PLA y Hya de abeja, Bla g Bd9OK, Bla g4, GST, y Per a3 de cucaracha, Der p2, Der f2, Der p10 y Tyr p2 de ácaro del polvo, Dol m v de avispón, Aed a 1 de mosquito, y hialuronidasa y fosfolipasa de avispa amarilla. En algunas realizaciones, el alérgeno vegetal incluye alérgenos de hierba, maleza y árboles (por ejemplo, pólenes). Los alérgenos de hierba adecuados incluyen, por ejemplo, alérgenos del pasto azul de Kentucky, festuca de los prados, dáctilo, hierba roja, ballica perenne, césped vernal dulce, e hierba timothy. Ejemplos de alérgenos de plantas incluyen Hor v 9 de cebada, Bet v1 y v2 de abedul, Pru a 1 de cereza, Zml3 de maíz, Phl p 1, 2, 4, 5, 6, 7, 11 y 12, Hol 15, Cyn d 7 y d12 de hierba, Jun a 2, Cry j 1 y j2 de cedro, Jun o2 de enebro, Hev b7 látex, Sin a I de mostaza amarilla, Bra r 1 de colza, Amb a 1 de ambrosía, y Lol p1 de centeno. En algunas realizaciones, el alérgeno de moho es un alérgeno de Aspergillus fumigatus tal como Asp f 1, 2, 3, 4 y 6. En algunas realizaciones, el alérgeno es un alérgeno alimentario tal como un alérgeno de marisco, un alérgeno de legumbre, un alérgeno de frutos secos o un alérgeno de leche. Ejemplos de alérgenos alimentarios incluyen tropomiosina de camarones, Ara h 1, 2, 3, 8 y 9 de cacahuete, Jug r 1 y 3 de nogal, Cor a 1, 14 y 8 LTP de avellana, lactoalbúmina caseína y lactoferrina de leche de vaca
En algunas realizaciones, las composiciones farmacéuticas comprenden un antígeno tumoral. En algunas realizaciones, el antígeno tumoral comprende la secuencia de aminoácidos de una proteína de longitud completa o un fragmento de la misma (por ejemplo, un polipéptido de aproximadamente 10 a aproximadamente 100 aminoácidos de longitud). En algunas realizaciones, el antígeno tumoral comprende un fragmento de proteína o polipéptido de longitud completa de uno o más del grupo que consiste en WT1, MUC1, LMP2, HPV E6, HPV E7, EGFRvIII, Her-2/neu, idiotipo, Ma Ge A3, p53, NY-ESO-1 (CTAG1), PSMA, CEA, MelanAMart1, Ras, gp100, proteinasa 3, bcr-able, tirosinasa, survivina, PSA, hTERT, puntos de corte de translocación de sarcoma, EphA2, PAP, MP-IAP, AFP, EpCAM, ERG, NA17-A, PAX3, ALK, receptor de andrógenos, ciclina B1, MYCN, PhoC, TRP-2, mesotelina, PSCA, MAGE A1, CYP1B1, PLAC1, BORIS, ETV6-AML, NY-BR-1, RGS5, SART3, anhidrasa carbónica IX, PAX5, OY-TES1, proteína de esperma 17, LCK, HMWMAA, AKAP-4, SSX2, XAGE 1, B7-H3, legumaína, Tie 2, Page4, VEGFR2, MAD-CT-1, FAP, PAP, PDGFR beta, MAD-CT-2, CEA, TRP-1 (gp75), BAGE1, BAGE2, BAGE3, BAGE4, BAGE5, CAMEL, MAGE-A2, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12 y antígeno relacionado con Fos 1. En algunas realizaciones preferidas, el antígeno tumoral comprende una secuencia de aminoácidos o un fragmento de la misma de uno o más del grupo que consiste en gp100, hTERT, MAGE A1, MAGE A3, MAGE A10, MelanAMart1, NY-ESO-1, PSA, Ras, survivina, TRP1 (gp75), TRP2 y tirosinasa.
IV. Métodos de uso
Las composiciones farmacéuticas de la presente divulgación son adecuadas para una pluralidad de usos que implican la estimulación de una respuesta inmunitaria en un sujeto mamífero que lo necesite. Los sujetos mamíferos incluyen, pero sin limitación, seres humanos, primates no humanos, roedores, mascotas y animales de granja. En algunas realizaciones, las composiciones farmacéuticas son para administración al sujeto en una cantidad eficaz para lograr un resultado específico.
Posología y modo de administración
Como con todas las composiciones farmacéuticas, la cantidad eficaz y el modo de administración pueden variar en función de varios factores evidentes para un experto en la materia. Los factores a considerar incluyen la potencia del compuesto agonista de TLRH8 de 1H-imidazo[4,5-c]quinolina de cadena alquílica modificada, la capacidad del compuesto y la composición farmacéutica para promover la retención del compuesto agonista en el sitio de administración, la vía de administración y si la composición farmacéutica contiene un antígeno. Otros factores a considerar incluyen el resultado de modificación de la enfermedad que se logrará y el número/frecuencia de las dosis que se administrarán durante un régimen terapéutico.
Un intervalo de posología adecuado es aquel que proporciona el efecto clínico deseado. La posología puede determinarse por la cantidad del agonista de TLRH8 en la composición farmacéutica que necesita administrarse a un sujeto para producir una respuesta terapéutica deseada con acontecimientos adversos mínimos. Un intervalo de posología ilustrativo del compuesto agonista de TLRH8 administrado en una cantidad que se administrará según el peso del sujeto es de aproximadamente 1 a 5000 ng/kg, tal como aproximadamente entre 1 y 2.500 ng/kg, aproximadamente entre 1 y 1.000 ng/kg, aproximadamente entre 1 y 500 ng/kg, aproximadamente entre 1 y 250 ng/kg, aproximadamente entre 1 y 100 ng/kg, aproximadamente entre 1 y 50 ng/kg, aproximadamente entre 50 y 2.500 ng/kg, aproximadamente entre 50 y 1.000 ng/kg, aproximadamente entre 50 y 500 ng/kg, aproximadamente entre 100 y 5.000 ng/kg, aproximadamente entre 100 y 2.500 ng/kg, aproximadamente entre 100 y 1.000 ng/kg, aproximadamente entre 100 y 500 ng/kg, aproximadamente entre 500 y 5.000 ng/kg, aproximadamente entre 1.000 y 5.000 ng/kg, aproximadamente entre 2.000 y 5.000 ng/kg, aproximadamente entre 2.500 y 5.000 ng/kg, aproximadamente entre 3000 y 5000 ng/kg, o aproximadamente entre 4000 y 5000 ng/kg. En algunas realizaciones, la posología es superior a aproximadamente (límite inferior) 1, 5, 10, 50, 100, 150, 200, 250, 300, 350, 400, 450 o 500 ng/kg. En algunas realizaciones, la posología es inferior a aproximadamente (límite superior) 5000, 2000, 1000, 900, 800, 700, 600, 500, 450, 400, 350, 300, 250, 200, 150 o 100 ng/kg. Esto es, la posología está en el intervalo de aproximadamente 1 a 5000 ng/kg en el que el límite inferior es menor que el límite superior. Un intervalo de posología ilustrativo del agonista de TLRH8 administrado en una cantidad para administrar a un sujeto es aproximadamente entre 1 y 5000 ng. En algunas realizaciones, la posología puede ser incluso mayor, por ejemplo, aproximadamente entre 2.500 y 500.000 ng/kg, aproximadamente entre 5.000 y 500.000 ng/kg, aproximadamente entre 2.500 y 150.000 ng/kg, aproximadamente entre 2.500 y 100.000 ng/kg, aproximadamente entre 2.500 y 50.000 ng/kg, aproximadamente entre 2.500 y 25.000 ng/kg, aproximadamente entre 2.500 y 10.000 ng/kg, aproximadamente entre 10.000 y 500.000 ng/kg, aproximadamente entre 25.000 y 500.000 ng/kg, aproximadamente entre 50.000 y 500.000 ng/kg, aproximadamente entre 100000 y 500000 ng/kg, o aproximadamente entre 150000 y 500000 ng/kg.
En algunas realizaciones, cuando la composición farmacéutica comprende además un antígeno, el intervalo de posología de antígeno dado en una cantidad para administrar a un sujeto es de aproximadamente 1 |jg a 500 |jg. En algunas realizaciones, la posología de antígeno es de aproximadamente 1 jg a 50 jg . En algunas realizaciones, la posología de antígeno es superior a aproximadamente (límite inferior) 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 100, 200 o 400 jg . En algunas realizaciones, la posología de antígeno es inferior a aproximadamente (límite superior) 500, 400, 300, 200, 100, 50, 45, 40, 35, 30, 25, 20, 15 o 10 jg . Esto es, la posología de antígeno está en cualquier punto en el intervalo de aproximadamente 1 a 500 jg en el que el límite inferior es menor que el límite superior La posología óptima de antígeno se puede determinar por
Asimismo, una vía de administración adecuada es aquella que proporciona el efecto deseado. En general, las composiciones farmacéuticas de la presente divulgación están destinadas a la administración parenteral (por ejemplo, no a la administración oral ni rectal). Las vías de administración adecuadas incluyen inyección, tópica e inhalación. En particular, las composiciones farmacéuticas de la presente divulgación pueden administrarse por una vía tal como intratumoral, intramuscular, subcutánea, intravenosa, epidérmica (pistola de genes), transdérmica e inhalación. Los dispositivos adecuados para la administración por inhalación incluyen, por ejemplo, atomizadores, vaporizadores, nebulizadores y dispositivos de administración por inhalación de polvo seco. En algunas realizaciones, cuando las composiciones farmacéuticas estén destinadas a tratar un tumor sólido, las composiciones son para administración por vía intratumoral. En una realización, la administración intratumoral se realiza mediante inyección en al menos una lesión tumoral.
Una pauta posológica adecuada del agonista de TLRH8 formulado en la composición farmacéutica es aquella que proporciona el efecto deseado en un contexto profiláctico o terapéutico con acontecimientos adversos mínimos. El número de dosis administradas por una vía elegida puede ser una o más de una. La frecuencia posológica puede oscilar entre semanalmente, quincenalmente, mensualmente, cada dos meses, o de 3 a 12 meses entre dosis. Una frecuencia de dosis ilustrativa del agonista de TLRH8 es de aproximadamente una vez por semana a una vez cada 8 semanas. En algunas realizaciones, la frecuencia de la dosis es mayor que aproximadamente (límite superior) una vez cada 8, 6, 4, 2 o 1 semana. En algunas realizaciones, la frecuencia de la dosis es inferior a aproximadamente (límite inferior) una vez cada 7, 10 o 14 días. Un intervalo de frecuencia de dosis ilustrativo del agonista de TLRH8 que se administrará a un sujeto es de aproximadamente una vez cada semana a una vez cada 4 semanas. En algunos casos, se administran 2 dosis, administrándose la segunda dosis de uno a dos meses después de la primera dosis. En algunos casos, se administran 3 dosis, administrándose la segunda dosis de uno a dos meses después de la primera dosis y administrándose la tercera dosis de uno a cinco meses después de la segunda dosis. En otros casos, se puede administrar una serie de dosis durante un programa de tratamiento de 3 a 12 meses, donde la frecuencia de la dosis es una vez por semana, cada dos semanas, cada tres semanas, o mensualmente. En otras realizaciones, puede transcurrir un período de tiempo más corto o más largo entre las dosis. En determinadas realizaciones, el intervalo entre dosis sucesivas puede variar en términos de número de semanas o número de meses. En un caso, se puede administrar una serie de 2, 3, 4, 5 o 6 dosis semanales seguida de una segunda serie de dosis semanales en un momento posterior. Un experto en la materia podrá ajustar la pauta posológica midiendo resultados biológicos tales como respuestas de anticuerpos específicos de antígeno o regresión tumoral.
Estimulación de una respuesta inmunitaria
En un aspecto, la presente divulgación proporciona métodos para estimular una respuesta inmunitaria en un sujeto mamífero que lo necesite, que comprenden la administración a un sujeto mamífero de una composición farmacéutica en una cantidad y frecuencia suficientes para estimular una respuesta inmunitaria en dicho sujeto. "Estimular" una respuesta inmunitaria significa aumentar la respuesta inmunitaria, que puede surgir de provocar una respuesta inmunitaria de novo (por ejemplo, como consecuencia de un régimen de vacunación inicial) o potenciar una respuesta inmunitaria existente (por ejemplo, como consecuencia de un régimen de vacunación de refuerzo). En algunas realizaciones, estimular una respuesta inmunitaria comprende uno o más del grupo que consiste en: estimular la producción de IFNa, estimular la producción de interferones de tipo 1 y/o tipo 2, estimular la producción de IL-6, estimular la producción de TNFα, estimular la proliferación de linfocitos B, estimular la expresión génica asociada a la vía del interferón, estimular la expresión génica asociada a quimioatrayentes y estimular la maduración de células dendríticas plasmocitoides (pDC) o células dendríticas mieloides (mDC). Los métodos para medir la estimulación de una respuesta inmunitaria se conocen en la técnica y se describen en los ejemplos biológicos de la presente divulgación. En realizaciones en las que la composición farmacéutica comprende además un antígeno, estimular una respuesta inmunitaria comprende inducir una respuesta de anticuerpos específica de antígeno.
Por ejemplo, en algunas realizaciones en las que la composición farmacéutica comprende además un antígeno, la presente divulgación proporciona métodos para inducir una respuesta de anticuerpos y/o linfocitos T específicos de antígeno en un sujeto mamífero que lo necesite mediante la administración de la composición farmacéutica en una cantidad suficiente para inducir una respuesta de anticuerpos y/o linfocitos T específicos de antígeno en dicho sujeto. "Inducir" una respuesta de anticuerpos específicos de antígeno significa aumentar los títulos de anticuerpos específicos de antígeno por encima de un nivel umbral, tal como un título de referencia previo a la administración o un nivel de seroprotección. "Inducir" una respuesta de linfocitos T específicos de antígeno significa estimular los linfocitos T citotóxicos específicos de antígeno, generar linfocitos T específicos de antígeno que se alojan en sitios tumorales no inmunizados, generar linfocitos T que estén menos agotadas y/o potenciar la respuesta inmunitaria a antígenos tumorales adicionales mediante la propagación del epítopo.
El análisis (tanto cualitativo como cuantitativo) de la respuesta inmunitaria puede realizarse mediante cualquier método conocido en la técnica, incluyendo, pero sin limitación, medir la producción de anticuerpos específicos de antígeno (incluyendo la medición de subclases de anticuerpos específicos), activación de poblaciones específicas de linfocitos tales como linfocitos B y linfocitos T cooperadores, medir la expresión de un conjunto de genes específicos de un tipo de célula inmunitaria particular, producción de citocinas tales como IFNa IL-6 IL-12 IL-18 TNFα y/o liberación de histamina de basófilos o mastocitos. Los mét no
incluyen el ensayo de inmunoadsorción enzimática (ELISA). La producción de citocinas también puede medirse mediante ELISA. Análisis de expresión génica pueden ser realizados mediante ensayos de expresión génica TaqMan® o nCounter®. La activación de poblaciones específicas de linfocitos se puede medir mediante ensayos de proliferación y clasificación de células activadas por fluorescencia (FACS).
Preferentemente, se estimula (es decir, se provoca o se potencia) una respuesta inmunitaria de tipo Th1. Con referencia a la presente divulgación, se puede determinar la estimulación de una respuesta inmunitaria de tipo Th1 in vitro o ex-vivo midiendo la producción de citocinas a partir de células tratadas con un agente activo de la presente divulgación (derivados de 1H-imidazo[4,5-c]quinolinas de cadena alquílica que son agonistas de TLRH8) en comparación con las células de control no tratadas con el agente activo. Los ejemplos de "citocinas de tipo Th1" incluyen, pero sin limitación, IL-2, IL-12, IFNy e IFNa. Por el contrario, las "citocinas de tipo Th2" incluyen, pero sin limitación, IL-4, IL-5 y IL-13. Las células útiles para la determinación de la actividad inmunoestimuladora incluyen células del sistema inmunitario tales como células presentadoras de antígenos, linfocitos, y preferentemente macrófagos y linfocitos T. Las células inmunitarias adecuadas incluyen células primarias tales como células mononucleares de sangre periférica, incluyendo pDC, monocitos, mDC y linfocitos B o esplenocitos aislados de un sujeto mamífero.
También se puede determinar la estimulación de una respuesta inmunitaria de tipo Th1 en un sujeto mamífero tratado con un agente activo de la presente divulgación midiendo los niveles de IL-2, IL-12 e interferón antes y después de la administración, o en comparación con un sujeto de control no tratado con el agente activo. La estimulación de una respuesta inmunitaria de tipo Th1 también se puede determinar midiendo la proporción de títulos de anticuerpos de tipo Th1 a tipo Th2. Los anticuerpos de "tipo Th1" incluyen IgG1 e IgG3 humanas e IgG2a murina. Por el contrario, los anticuerpos de "tipo Th2" incluyen IgG2, IgG4 e IgE humanas e IgG1 e IgE murinas.
Tratamiento de una enfermedad
La presente divulgación proporciona además métodos para prevenir una enfermedad infecciosa en un sujeto mamífero que lo necesite, que comprenden la administración de una composición farmacéutica en una cantidad suficiente para prevenir una enfermedad infecciosa en dicho sujeto. Esto es, en algunas realizaciones, la presente divulgación proporciona vacunas profilácticas. En algunas realizaciones, el sujeto mamífero está en riesgo de exposición a un agente infeccioso. "Prevenir" una enfermedad infecciosa significa proteger a un sujeto de desarrollar una enfermedad infecciosa. En algunas realizaciones, prevenir una enfermedad infecciosa comprende además proteger a un sujeto de ser infectado con un agente infeccioso (por ejemplo, proteger a un sujeto de desarrollar una infección aguda o crónica). Adicionalmente, la presente divulgación proporciona métodos para mejorar un síntoma de una enfermedad infecciosa en un sujeto mamífero que lo necesite, que comprenden la administración de una composición farmacéutica en una cantidad suficiente para mejorar un síntoma de una enfermedad infecciosa en dicho sujeto. Esto es, en algunas realizaciones, la presente divulgación proporciona vacunas terapéuticas. En algunas realizaciones, el sujeto es infectado de forma aguda o crónica con un agente infeccioso. La enfermedad infecciosa puede ser una enfermedad vírica (por ejemplo, hepatitis, herpes o virus del papiloma humano), bacteriana, fúngica o parasitaria. En algunas realizaciones, la composición farmacéutica comprende además un antígeno vírico, bacteriano, fúngico o parasitario. "Mejorar" un síntoma de una enfermedad infecciosa significa mejorar un síntoma, preferentemente, disminuir el alcance de la enfermedad.
Por otra parte, la presente divulgación proporciona métodos para mejorar un síntoma de un trastorno relacionado con IgE en un sujeto mamífero que lo necesite, que comprenden la administración de una composición farmacéutica en una cantidad suficiente para mejorar un síntoma de un trastorno relacionado con IgE en dicho sujeto. En algunas realizaciones preferidas, el trastorno relacionado con IgE es una alergia. Las alergias incluyen, pero sin limitación, rinitis alérgica (fiebre del heno), sinusitis, eccema y urticaria. En algunas realizaciones, la composición farmacéutica comprende además un alérgeno. "Mejorar" un síntoma de un trastorno relacionado con IgE significa mejorar un síntoma, preferentemente, disminuir el alcance del trastorno. Por ejemplo, si el trastorno relacionado con IgE es rinitis alérgica, mejorar un síntoma significa reducir la hinchazón de la mucosa nasal, reducir la rinorrea (secreción nasal) y/o reducir los estornudos.
Además, la presente divulgación proporciona una pluralidad de métodos para tratar el cáncer en un sujeto mamífero que lo necesite, que comprenden la administración de una composición farmacéutica en una cantidad suficiente para tratar el cáncer en dicho sujeto. En determinadas realizaciones, la presente divulgación proporciona métodos para tratar el cáncer en un sujeto mamífero que lo necesite, que comprenden administrar una cantidad eficaz de una composición farmacéutica mediante administración intratumoral. En otro aspecto del método, la administración intratumoral comprende la inyección de la composición farmacéutica en al menos una lesión tumoral. En otros aspectos, tratar el cáncer comprende inducir la acumulación de linfocitos T específicos de antígeno tumoral en el tumor inyectado, por ejemplo, en mayor número que si la composición farmacéutica se hubiera administrado en un sitio extratumoral. En otros aspectos, tratar el cáncer comprende provocar una respuesta sistémica de linfocitos T específicos de antígeno tumoral, incluyendo, por ejemplo, una respuesta sistémica de linfocitos T específicos de antígeno tumoral de mayor magnitud que si se hubiera administrado la composición inmunogénica en un sitio extratumoral. En otros aspectos, tratar el cáncer comprende provocar una respuesta sistémica de linfocitos T específicos de antígeno tumoral. En otros a T
reguladores CD4+ FoxP3+ en el tumor inyectado. En otros aspectos, el sujeto tiene uno o más tumores no inyectados (lesiones primarias o metastásicas) además del tumor inyectado, y tratar el cáncer comprende uno o más de los siguientes: (a) reducir el número de tumores no inyectados; (b) reducir el volumen de tumores no inyectados; y (c) retardar el crecimiento de tumores no inyectados. En algunos aspectos, tratar el cáncer comprende uno o más de los siguientes: (d) aumentar el tiempo de supervivencia del sujeto; (e) reducir el volumen del tumor inyectado; y (f) retardar el crecimiento del tumor inyectado. En algunas realizaciones, cuando el cáncer es un tumor sólido, "tratar" el cáncer comprende reducir el tamaño del tumor sólido y cualquier lesión metastásica, o reducir de otro modo el número de células cancerosas viables. En otras realizaciones, cuando el cáncer es un tumor sólido, "tratar" el cáncer comprende retrasar el crecimiento del tumor sólido y cualquier lesión metastásica. En algunos aspectos, tratar el cáncer comprende aumentar la supervivencia sin progresión o aumentar el tiempo hasta la progresión. En otras realizaciones, el método comprende además administrar una cantidad eficaz de un segundo, o adicional, agentes terapéuticos al sujeto. "T ratar" el cáncer significa lograr un resultado clínico beneficioso, tal como provocar la remisión o prolongar de otro modo la supervivencia en comparación con la supervivencia esperada en ausencia de tratamiento. En algunas realizaciones preferidas, "tratar el cáncer" comprende evaluar la respuesta de un paciente a la composición inmunogénica de acuerdo con los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST versión 1.1) como se describe (véase, por ejemplo, Eisenhauer et al 2009 Eur J Cancer 45:228-247). Los criterios de respuesta para determinar respuestas antitumorales objetivas según RECIST incluyen: respuesta completa, respuesta parcial, enfermedad progresiva y enfermedad estable.
En algunas realizaciones, el tumor es un sarcoma, un carcinoma, o una queratosis actínica. En algunas realizaciones, el tumor es un linfoma. En algunas realizaciones, el cáncer se selecciona del grupo que consiste en cáncer de mama, cáncer de próstata, cáncer de pulmón, cáncer colorrectal, cáncer de útero, cáncer de vejiga, melanoma, cáncer de cabeza y cuello, linfoma no hodgkiniano, cáncer de riñón, cáncer de ovario, cáncer pancreático y cáncer de tiroides. En algunas realizaciones, el cáncer es un cáncer primario de un sitio seleccionado del grupo que consiste en la cavidad bucal, el sistema digestivo, el sistema respiratorio, la piel, la mama, el sistema genital, el sistema urinario, el sistema ocular, el sistema nervioso, el sistema endocrino y linfoma.
En algunas realizaciones, el método comprende además administrar al sujeto una cantidad eficaz de un segundo agente terapéutico. En algunas de estas realizaciones, el segundo agente terapéutico comprende un agente quimioterapéutico seleccionado del grupo que consiste en actinomicina, afatinib, alectinib, asparaginasa, azacitidina, azatioprina, bicalutamida, binimetinib, bleomicina, bortezomib, camptotecina, carboplatino, capecitabina, carmustina, certinib, cisplatino, clorambucilo, cobimetinib, crizotinib, ciclofosfamida, citarabina, dabrafenib, dacarbazina, daunorrubicina, docetaxel, doxifluridina, doxorrubicina, encorafenib, erlotinib, epirrubicina, epotilona, etopósido, fludarabina, flutamina, fluorouracilo, gefitinib, gemcitabina, hidroxiurea, idarrubicina, ifosfamida, imatinib, irinotecán, lapatinib, letrozol, mecloretamina, mercaptopurina, metotrexato, mitomicina, mitoxantrona, octreotida, oxaliplatino, paclitaxel, pemetrexed, raltitrexed, sorafenib, sunitinib, tamoxifeno, temozolomida, tenipósido, tioguanina, topotecán, trametinib, valrrubicina, vemurafenib, vinblastina, vincristina, vindesina, vinorelbina y combinaciones de los mismos. En algunas realizaciones, el segundo agente terapéutico comprende uno o ambos de un inhibidor de BRAF y un inhibidor de MEK. En algunas realizaciones, el segundo agente terapéutico comprende un modulador epigenético seleccionado del grupo que consiste en inhibidores de HDAC (véase, por ejemplo, voronistat [SAHA], romidepsina, entinostat, abexinostat, elinostat [CHR-3996], panobinostat, quisinostat [JNJ-26481585],4SC-202, resminostat [SB939], pracinostat [CI-9940] y valproato), Inhibidores de la ADN metiltransferasa (véase, por ejemplo, azacitidina, decitabina, zebularina, SGI-1027, RG-108 y sinfungina), y combinaciones de los mismos.
En algunas de estas realizaciones, el segundo agente terapéutico es un antagonista de una molécula inhibidora de punto de control inmunitario, por ejemplo, una molécula inhibidora de punto de control inmunitario seleccionada del grupo que consiste en PD-1, PD-L1, p D-L2, CTLA-4 (CD152), LAG-3, TIM-3, TIGIT, IL-10, indolamina 2,3-dioxigenasa (IDO), ligando-1 de glucoproteína P-selectina (PSGL-1) y TGF-beta. En algunas de estas realizaciones, el segundo agente terapéutico es un agonista de una molécula inmunoestimuladora. En algunas de estas realizaciones, la molécula inmunoestimuladora se selecciona del grupo que consiste en CD27, CD40, OX40 (CD134), GITR, 4-1BB (CD137), CD28 e ICOS (CD278). En algunas de estas realizaciones, el segundo agente terapéutico comprende un anticuerpo, fragmento o derivado del mismo. En algunas de estas realizaciones, el segundo agente terapéutico es un antagonista de una molécula inhibidora de punto de control inmunitario y el segundo agente terapéutico comprende un anticuerpo, fragmento o derivado del mismo. En algunas realizaciones, el método comprende además administrar radioterapia y/o administrar una cantidad eficaz de un segundo agente terapéutico al sujeto. En algunas de estas realizaciones, la cantidad eficaz de la composición inmunogénica y la cantidad eficaz del segundo agente terapéutico juntas dan como resultado un efecto aditivo o mejor contra el tumor. En algunas de estas realizaciones, la cantidad eficaz de la composición inmunogénica y la cantidad eficaz del segundo agente terapéutico juntas dan como resultado un efecto sinérgico contra el tumor.
En algunas realizaciones del método, el tratamiento de enfermedades infecciosas o cáncer no da como resultado el desarrollo de síntomas similares a los de la gripe de tal gravedad que esté contraindicada la administración repetida de la composición inmunogénica, en donde los síntomas similares a los de la gripe comprenden uno o más del grupo que consiste en fiebre, cefalea, escalofríos, mialgia y fatiga.
En algunas realizaciones, la presente divulg ca
(por ejemplo, un compuesto agonista de TLRH8 de fórmula (K), un excipiente o excipientes, y opcionalmente un antígeno) y un conjunto de instrucciones relacionadas con el uso de la composición para los métodos descritos en el presente documento. La composición farmacéutica de los kits se envasa apropiadamente. Si la composición farmacéutica es un líquido o una suspensión de nanopartículas, un vial de dióxido de silicio (por ejemplo, SCHOTT Type I plus®) con un tapón de caucho (por ejemplo, elastómero de halobutilo Exxpro) y una tapa de engarce de aluminio se suele utilizar como sistema de cierre del recipiente. En algunas realizaciones, los kits comprenden además un dispositivo (por ejemplo, una jeringa y una aguja) para la administración de la composición farmacéutica. En otras realizaciones, los kits comprenden además un sistema de jeringa/aguja precargada, autoinyectores o dispositivos sin aguja. Las instrucciones relacionadas con el uso de las composiciones farmacéuticas generalmente incluyen información sobre la dosis, el programa y la vía de administración para los métodos de uso previstos.
V. Ejemplos
Aunque la presente divulgación se ha descrito con cierto nivel de detalle a modo de ilustración y ejemplo con fines de claridad de entendimiento, para los expertos en la materia será evidente que se pueden poner en práctica determinados cambios y modificaciones. Por lo tanto, los siguientes ejemplos sintéticos y biológicos no deben interpretarse como limitantes del alcance de la presente divulgación, que se define por las reivindicaciones adjuntas.
Ejemplos de síntesis
Ejemplo S1: Síntesis de W-(4-((4-amino-2-butil-1H-imidazo[4,5-C]quinolin-1-il)metil)bencil)tetradecanamida (Compuesto n.° 63-10)
Parte A. Se añadió ácido nítrico (125 ml) a una suspensión espesa de quinolin a-2,4-dio l (50 g, 0,3 mol) en ácido acético (500 ml) y la mezcla de reacción se calentó a 65 °C durante tres horas. A continuación, la mezcla se enfrió a 5 - 10 °C y el material sólido se recogió por filtración, se lavó con agua fría y se secó al aire. A continuación, el sólido resultante se recristalizó en metanol y se secó al vacío para producir 58 g de 3-nitro-2,4-quinolinadiol como un sólido amarillo.
Parte B. Se añadió oxicloruro de fósforo (150 ml) a 3-nitro-2,4-quinolinadiol (58 g) en una atmósfera de argón y se calentó a 95 °C durante 4 horas. A continuación, la mezcla se enfrió a temperatura ambiente y se vertió sobre hielo picado con agitación constante. El producto precipitado se recogió por filtración, se lavó con agua, y se secó al vacío. El sólido en bruto se purificó mediante cromatografía ultrarrápida sobre gel de sílice usando hexano/acetato de etilo para producir 35 g de 2,4-dicloro-3-nitroquinolina.
Parte C. Se añadió (4-(aminometil)bencil)carbamato de tere-butilo (37,3 g, 0,16 mol, 1,1 equiv.) a una solución de 2,4-dicloro-3-nitroquinolina (35 g, 0,15 mol, 1,0 equiv.) en diclorometano anhidro (400 ml) y trimetilamina (16,0 g, 0,16 mol, 1,1 eq), y se agitó durante la noche a temperatura ambiente. Los disolventes se eliminaron a presión reducida y el producto en bruto se purificó mediante cromatografía ultrarrápida sobre gel de sílice usando hexano/acetato de etilo para producir 52 g de (4-(((2-cloro-3-nitroquinolin-4-il)amino)metil)bencil)carbamato de tere-butilo.
Parte D. Una solución de (4-(((2-cloro-3-nitroquinolin-4-il)amino)metil)bencil)carbamato de tere-butilo (52 g, 0,12 mol) en acetato de etilo (250 ml) se hidrogenó en presencia de platino al 5 % sobre carbono (2,0 g) y sulfato de sodio (52 g) usando un aparato de hidrogenación Parr a 60 psi durante 12 horas. El catalizador de platino y el sulfato de sodio se eliminaron filtrando a través de una capa de Celite® y el filtrado se concentró a presión reducida. El producto se purificó adicionalmente mediante cromatografía ultrarrápida sobre gel de sílice eluyendo con hexano/acetato de etilo para producir 32 g de (4-(((3-amino-2-cloroquinolin-4-il)amino)metil)bencil)carbamato de tere-butilo.
Parte E. Se añadió lentamente cloruro de pentanoílo (9,7 ml, 81,5 mmol, 1,05 eq) a una solución de (4-(((3-amino-2-cloroquinolin-4-il)amino)metil)bencil)carbamato de tere-butilo (32 g, 77,6 mmol, 1,0 eq) en tetrahidrofurano anhidro (350 ml) y piridina (30 ml) a 0 - 5 °C. A continuación, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 12 horas. Los disolventes se eliminaron a presión reducida y a continuación los sólidos se volvieron a disolver en acetato de etilo (400 ml), se lavaron sucesivamente con agua y bicarbonato de sodio saturado (150 ml) y finalmente se secaron sobre sulfato de magnesio anhidro. El producto se purificó mediante cromatografía ultrarrápida sobre gel de sílice, eluyendo con hexano/acetato de etilo para producir 22 g de (4-(((3-butiramido-2-cloroquinolin-4-il)amino)metil)bencil)carbamato de tere-butilo.
Parte F. Se añadió agua (80 ml) a una solución de (4-(((3-butiramido-2-cloroquinolin-4-il)amino)metil)bencil)carbamato de tere-butilo (22 g, 44,2 mmol, 1,0 eq) en etanol (320 ml), seguido de la adición de carbonato de potasio (12,2 g, 88,4 mmol, 2,0 eq), y la mezcla se calentó con agitación vigorosa a 55 °C durante 16 horas. Esta mezcla de reacción se concentró y el residuo se repartió entre acetato de etilo (500 ml) y agua (250 ml). A continuación, la capa de acetato de etilo se lavó con agua (100 ml), se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El producto se purificó mediante cromatografía ultrarrápida sobre gel de sílice, eluyendo con hexano/acetato de etilo para producir 15,4 g de (4-((2-butil-4-cloro-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)carbamato de tere-butilo.
Parte G. Se disolvió (4-((2-butil-4-cloro-1H-imidazo[45-C]quinolin-1-il)metil)bencil)carbamato de tere-butilo (154 g, 32,2 mmol, 1 eq) en dimetilformamida anhidra ol,
4 eq) a esta solución. La suspensión resultante se desgasificó y se agitó en atmósfera de argón a 110 - 115 °C, controlándose el progreso de la reacción mediante análisis por HPLC de fase inversa. Después de 18 horas, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en agua fría (500 ml) y se extrajo con acetato de etilo (3 x 100 ml). El extracto combinado se lavó con agua (2 x 75 ml), se secó sobre sulfato de magnesio, se filtró a través de Celite y se concentró a presión reducida para producir un sólido blanquecino. Este sólido se trató adicionalmente mediante recristalización con 1:1 de etilo/hexano para producir 12,5 g de (4-((4-azido-2-butil-1H-imidazo[4,5-C]quinolin-1-il)metil)bencil)carbamato de tere-butilo.
Parte H. Se añadió (4-((4-azido-2-butil-1H-imidazo[4,5-C]quinolin-1-il)metil)bencil)carbamato de tere-butilo (12,5 g, 25,7 mmol) a ácido clorhídrico concentrado (65 ml) y a esta suspensión se le añadió platino al 10 % sobre carbono (3,0 g). Esta mezcla de reacción se sometió a hidrogenación a 65 psi, controlándose el progreso de la reacción mediante análisis por HPLC de fase inversa. Después de 6 días, el catalizador se retiró por filtración y la torta filtrada se lavó con agua (2 x 25 ml). La torta se enfrió en un baño de hielo, se añadió hidróxido de sodio 1 N enfriado con hielo gota a gota mientras se agitaba vigorosamente hasta que el pH alcanzó 8,5, y el material se extrajo con diclorometano que contenía metanol al 5 % (4 x 75 ml). El extracto combinado se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó en una columna de gel de sílice utilizando un 8 % de metanol/diclorometano que contenía amoníaco acuoso al 1 % para producir 5,3 g de 1-(4-(aminometil)bencil)-2-butil-1H-imidazo[4,5-C]quinolin-4-amina.
Parte I. Se añadieron ácido mirístico (27,4 mg, 0,12 mmol, 1,2 eq) y trimetilamina (0,2 ml) a una solución de 1-(4-(aminometil)bencil)-2-butil-1H-imidazo[4,5-C]quinolin-4-amina (34 mg, 0,1 mmol, 1,0 eq) en dimetilformamida anhidra (2 ml), y la suspensión espesa se mezcló durante 5 minutos seguido de la adición de HBTU (47,4 mg, 0,125 mmol, 1,25 eq). Esta mezcla de reacción se agitó adicionalmente durante 2 horas en atmósfera de argón. El disolvente se retiró a presión reducida, el residuo se disolvió en acetato de etilo (30 ml) y se lavó con agua (2 x 10 ml), a continuación se secó con sulfato de magnesio y se concentró al vacío. Este producto se purificó mediante cromatografía en columna (6 % de metanol/diclorometano) para producir 35 mg de N-(4-((4-amino-2-butil-1H-imidazo[4,5-C]quinolin-1-il)metil)bencil)tetradecanamida (Compuesto n.° 63-10). La pureza del producto se evaluó en ~98 % mediante HPLC de fase inversa, la masa sintética prevista de 569,8 se confirmó mediante LC/MS, y la estructura sintética prevista se confirmó mediante RMN de protones de 300 MHz (CDCla): 87,98 (d, J = 8,1 Hz, 1H), 7,75 (d, J = 8,4 Hz, 1H), 7,67 (t, J = 8,4 Hz, 1H), 7,40 (t, J = 7,5 Hz, 1H), 7,30 (d, J = 8,1 Hz, 2H), 7,06 (d, J = 8,1 Hz, 2H), 5,95 (s, 2H), 4,33 (s, 2H), 3,74 (s, 2H), 3,01 (t, J = 7,8 Hz, 2H), 2,20 (t, J = 7,5 Hz, 2H), 1,82-1,9 (m, 2H), 1,42-1,70 (m,4H), 1,26-1,48 (m, 20 H), 0,85-1,05 (m, 6H).
Ejemplo S2: Síntesis de 2-butil-1-(4-((pentilamino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63 17)
Las Partes A - H fueron las mismas que en el ejemplo S1.
Parte I. Se añadieron ácido valérico (34 mg, 0,33 mmol, 1,2 eq) y trimetilamina (140 mg, 1,39 mmol, 5,0 eq) a una solución de 1-(4-(aminometil)bencil)-2-butil-1H-imidazo[4,5-c]quinolin-4-amina (100 mg, 0,28 mmol, 1,0 eq) en dimetilformamida anhidra (2 ml), y la suspensión espesa se mezcló durante 5 minutos seguido de la adición de HBTU (131 mg, 0,34 mmol, 1,25 equiv.). Esta mezcla de reacción se agitó adicionalmente durante 2 horas en una atmósfera de argón. La reacción se diluyó con acetato de etilo (100 ml) y se lavó con agua (3 x 30 ml), a continuación se secó con sulfato de magnesio y se concentró al vacío. El residuo en bruto se recogió en acetato de etilo y metanol y se purificó mediante cromatografía en columna (6 % de metanol/diclorometano) para producir 160 mg de N-(4-((4-amino-2-butil-1H-imidazo[4,5-C]quinolin-1-il)metil)bencil)pentanamida.
Parte J. Una solución de complejo de borano - dimetilsulfuro (2,0 M, 1,5 ml, exceso) se añadió a la solución de N-(4-((4-amino-2-butil-1H-imidazo[4,5-C]quinolin-1-il)metil)bencil)pentanamida (127 mg, 0,28 mmol, 1,0 eq) en tetrahidrofurano anhidro (5 ml) a temperatura ambiente y la mezcla de reacción se calentó a reflujo durante 12 horas. La mezcla se enfrió a temperatura ambiente, se inactivó con HCl 3 N (1 ml) y se agitó durante 4 horas. El pH de la mezcla de reacción se alcalinizó mediante la adición de hidróxido de sodio 2 N y el producto se extrajo con diclorometano (20 ml x 10). Las capas orgánicas combinadas se concentraron a presión reducida y el residuo se purificó mediante cromatografía ultrarrápida con 6 % de metanol/diclorometano como eluyente para producir 24 mg de 2-butil-1-(4-((pentilamino)metil)bencil)-1H-imidazo[4,5-C]quinolin-4-amina (Compuesto n.° 63-17). La pureza del producto se evaluó en ~96 % mediante HPLC de fase inversa, la masa sintética prevista de 429,6 se confirmó mediante LC/MS, y la estructura sintética prevista se confirmó mediante RMN de protones de 300 MHz (CDCl3): 87,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7,34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,80 (s, 2H), 2,89 (t, J = 7,6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,2-1,75 (m, 8H), 0,65-1,0 (m, 6H).
Ejemplo S3: Síntesis de 2-butil-1-(4-(((2-ciclopropiletil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-33)
Las Partes A - H fueron las mismas que en el ejemplo S1
Parte I. Se añadieron ácido cidopropilacético (33 mg, 0,33 mmol, 1,2 eq) y trimetilamina (140 mg, 1,39 mmol, 5,0 eq) a una solución de 1-(4-(aminometil)bencil)-2-butil-1H-imidazo[4,5-c]quinolin-4-amina (100 mg, 0,28 mmol, 1,0 eq) en dimetilformamida anhidra (2 ml), y la suspensión espesa se mezcló durante 5 minutos seguido de la adición de HBTU (131 mg, 0,34 mmol, 1,25 equiv.). Esta mezcla de reacción se agitó adicionalmente durante 2 horas en una atmósfera de argón. La reacción se diluyó con acetato de etilo (100 ml) y se lavó con agua (3 x 30 ml), a continuación se secó con sulfato de magnesio y se concentró al vacío. El residuo en bruto se recogió en acetato de etilo y metanol y se purificó mediante cromatografía en columna (6 % de metanol/diclorometano) para producir 140 mg de N-(4-((4-amino-2-butil-1 H-imidazo[4,5-c]quinolin-1 -il)metil)bencil)-2-ciclopropilacetamida (Compuesto n.° 63-34).
Parte J. Una solución de complejo de borano - dimetilsulfuro (2,0 M, 1,5 ml, exceso) se añadió a la solución de N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-2-ciclopropilacetamida (123 mg, 0,28 mmol, 1,0 eq) en tetrahidrofurano anhidro (5 ml) a temperatura ambiente y la mezcla de reacción se calentó a reflujo durante 12 horas. La mezcla se enfrió a temperatura ambiente, se inactivó con HCl 3 N (1 ml) y se agitó durante 4 horas. El pH de la mezcla de reacción se alcalinizó mediante la adición de hidróxido de sodio 2 N y el producto se extrajo con diclorometano (20 ml x 10). Las capas orgánicas combinadas se concentraron a presión reducida y el residuo se purificó mediante cromatografía ultrarrápida con 6 % de metanol/diclorometano como eluyente para producir 28 mg de 2-butil-1-(4-(((2-ciclopropiletil)amino)metil) bencilo)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-33). La pureza del producto se evaluó en un 97 % mediante HPLC de fase inversa, la masa sintética prevista de 427,6 se confirmó mediante LC/MS, y la estructura sintética prevista se confirmó mediante 1H RMN de 400 MHz (CDCb): 57,80 (dd, J = 8,5, 1,0 Hz, 1H), 7,70 (dd, J = 8,3, 1,1 Hz, 1H), 7,42 (m, J = 8,4. 7,0, 1,4 Hz, 1H), 7,29 (sap, 1H), 7,25 (sap, 1H), 7,10 (m, J = 8,2, 7,1, 1,3 Hz, 1H), 7,00 (d, J = 8,4 Hz, 2H), 5,92 (s a, 2H), 5,69 (s, 2H), 3,75 (s, 2H), 2,86 (dd, J = 8,0 Hz, 2H), 2,68 (dd, J = 8,0 Hz, 2H), 1,82-1,74 (m, 2H), 1,45-1,36 (m, 4H), 0,91 (t, J = 7,8 Hz, 3H), 0,91-0,85 (m, 1H), 0,68-0,60 (m, 1H), 0,42-0,37 (m, 2H), 0,04-0,00 (m, 2H).
Ejemplo S4: Síntesis de otros compuestos ilustrativos
Los compuestos de Nalquilo de fórmula (J-1) ilustrativos se sintetizaron usando procedimientos similares a los del Compuesto n.° 63-33. Usando técnicas conocidas por los expertos en la materia, el coeficiente de reparto calculado (cLogP) para los compuestos ilustrativos se determinó utilizando el algoritmo Molecular Descriptors en el software Molecular Operating Environment (véase, por ejemplo, Labute P, The Derivation and Applications of Molecular Descriptors Based Upon (Approximate) Surface Area; en Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery, J.Bajorath ed. 2003). Los datos 1HRMN y de espectrometría de masas se detallan a continuación para determinados compuestos de la presente invención, y la tabla 3 proporciona datos de pureza y valores de cLogP.

Compuesto n.° 63-33: 1H RMN (CDCls, 400 MHz): 57,80 (dd, J = 8,5, 1,0 Hz, 1H), 7,70 (dd, J = 8,3, 1,1 Hz, 1H), 7,42 (m, J = 8,4. 7,0, 1,4 Hz, 1H), 7,29 (sap, 1H), 7,25 (sap, 1H), 7,10 (m, J = 8,2, 7,1, 1,3 Hz, 1H), 7,00 (d, J = 8,4 Hz, 2H), 5,92 (s a, 2H), 5,69 (s, 2H), 3,75 (s, 2H), 2,86 (dd, J = 8,0 Hz, 2H), 2,68 (dd, J = 8,0 Hz, 2H), 1,82-1,74 (m, 2H), 1,45 1,36 (m, 4H), 0,91 (t, J = 7,8 Hz, 3H), 0,91-0,85 (m, 1H), 0,68-0,60 (m, 1H), 0,42-0,37 (m, 2H), 0,04-0,00 (m, 2H). Espectr. de masas: m/z 428,6 (M+1).
Compuesto n.° 63-35: 1H RMN (CDCb, 300 MHz): 87,8 (d, J= 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7,34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,72 (s, 2H), 3,85 (s, 2H), 2,89 (t, J = 7.6 Hz, 2H), 2,60 (t, J = 7,2 Hz, 2H), 2,20-2,35 (m, 1H), 1,8-2,1 (m, 2H), 1,65-1,8 (m, 6H), 1,4-1,55 (m, 4H), 0,94 (t, J = 7,3 Hz, 3H). Espectr. de masas: m/z 442,9 (M+1).
Compuesto n.° 63-36: 1H RMN (CDCla, 300 MHz): 87,8 (d, J= 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7,34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,72 (s, 2H), 3,80 (s a, 4H), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,62-1,84 (m, 7H), 1,0-1,20 (m, 1H), 0,94 (t, J = 7,3 Hz, 3H). Espectr. de masas: m/z 456,4 (M+1).
Compuesto n.° 63-39: 1H RMN (CD3OD, 300 MHz): 87,80 (d, J = 8,1 Hz, 1H), 7,67 (d, J = 8,4 Hz, 1H), 7,30-7,48 (m, 3H), 7,05-7,14 (m, 3H), 5,88 (s, 2H), 3,81 (s, 2H), 2,89 (t, J = 7,5, 15,6 Hz, 2H), 2,44-2,55 (m, 2H), 1,70-1,85 (m, 2H), 1,35-1,55 (m, 3H), 1,02 (s, J = 6 Hz, 3H), 0,94 (t, J = 7,5, 14,7 Hz, 3H), 0,50-0,75 (m, 2H), 0,20-0,35 (m, 2H). Espectr. de masas: m/z 428,4 [M+1],
Compuesto n.° 63-40: 1H RMN (CDCI3 , 300 MHz): 87,82 (d, J = 8,1 Hz, 1H), 7,73 (d, J = 8,4 Hz, 1H), 7,45 (t, J = 7,5, 15.0 Hz, 1H), 7,29 (d, J = 8,4 Hz, 2H), 7,15 (t, J = 7,5, 15,0 Hz, 1H), 7,03 (d, J = 8,4 Hz, 2H), 5,73 (s, 2H), 5,54 (s, 2H), 3,78 (s, 2H), 2,50-2,70 (m, 2H), 1,75-1,80 (m, 2H), 1,40-1,55 (m, 2H), 1,04 (s, 3H), 1,02 (s, 3H), 0,94 (t, J = 7,5, 14,7 Hz, 3H), 0,7-0,8 (m, 1H), 0,4-0,5 (m, 1H), 0,1-0,05 (m, 1H). Espectr. de masas: m/z 442,5 [M+1].
Compuesto n.° 63-42: 1H RMN (CDCI3 , 300 MHz): 87,80 (d, J = 8,1 Hz, 1H), 7,71 (d, J = 8,4 Hz, 1H), 7,43 (t, J = 7,5, 15.0 Hz, 1H), 7,31 (d, J = 8,4 Hz, 2H), 7,14 (t, J = 7,5, 15,0 Hz, 1H), 7,01 (d, J = 8,4 Hz, 2H), 5,71 (s, 2H), 3,76 (s, 2H), 2,89 (t, J = 7,5, 15,6 Hz, 2H), 2,70 (t, J = 7,2, 15,0 Hz, 2H), 1,77-1,92 (m, 2H), 1,70-1,84 (m, 4H), 0,98 (s, 3H), 0,94 (t, J = 7,5, 14,7 Hz, 3H), 0,18-0,35 (m, 4H). Espectr. de masas: m/z 442,5 [M+1].
Compuesto n.° 63-43: 1H RMN (CDCI3 , 300 MHz): 87,80 (d, J = 8,1 Hz, 1H), 7,71 (d, J = 8,4 Hz, 1H), 7,43 (t, J = 7,5, 15.0 Hz, 1H), 7,25-7,31 (m, 2H), 7,14 (t, J = 7,5, 15,0 Hz, 1H), 7,01 (d, J = 8,4 Hz, 2H), 5,71 (s, 2H), 5,59 (s, 2H), 3,75 (s, 2H), 2,88 (t a, 2H), 2,63 (t, J = 7,2, 14,1 Hz, 3H), 0,55-0,72 (m, 1H), 0,4 (d a, 2H), 0,01 (d, J = 4,5 Hz, 2H). Espectr. de masas: m/z 442,5 [M+1].
Compuesto n.° 63-46: 1H RMN (CDCI3 , 300 MHz): 87,81 (d, J = 8,1 Hz, 1H), 7,74 (d, J = 8,4 Hz, 1H), 7,45 (t, J = 7,5, 15.0 Hz, 1H), 7,29 (d, J = 8,4 Hz, 2H), 7,15 (t, J = 7,5, 15,0 Hz, 1H), 7,03 (d, J = 8,4 Hz, 2H), 5,73 (s, 2H), 5,51 (s, 2H), 3,74 (s, 2H), 2,86 (t, J = 7,5, 15,6 Hz, 2H), 2,60-2,7 (m, 2H), 2,1-2,4 (m, 3H), 1,6-1,85 (m, 4H), 1,40-1,55 (m, 2H), 0,95 1,15 (m, 5H), 0,94 (t, J = 7,5, 14,7 Hz, 3H). Espectr. de masas: m/z 442,4 [M+1].
Ejemplo S5: Síntesis de 2-butil-1-(4-(((cidopropilmetil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-38).
Parte A.Se añadió N,N-diisopropilcarbodiimida (272 mg, 2,15 mmol) a una solución de ácido ciclopropanocarboxílico (172 mg, 2,0 mmol) y pentafluorofenol (387 mg, 2,1 mmol) en diclorometano (4 ml), en presencia de cantidades catalíticas de N,N-dimetilaminopiridina (12 mg) y se agitó durante la noche a temperatura ambiente. A continuación, esta mezcla se diluyó con éter (20 ml), la urea precipitada se eliminó por filtración y el filtrado se concentró para obtener el producto en bruto. Este producto en bruto se suspendió en un 1 % de acetato de etilo/hexano y la urea precipitada residual se eliminó de nuevo por filtración. El filtrado resultante se concentró a presión reducida para producir 440 mg del producto carboxilato de (2,3,4,5,6-pentafluorofenil)-ciclopropano deseado.
Parte B. Se añadió el Compuesto n.° 63-00 (80 mg, 0,22 mmol) a una solución de carboxilato de (2,3,4,5,6-pentafluorofenil)-ciclopropano (61 mg, 0,24 mmol) en diclorometano (3 ml) en presencia de trietilamina (45 mg, 0,44 mmol) y se agitó durante 2 horas a temperatura ambiente. A continuación, la mezcla de reacción se concentró a presión reducida y se purificó por cromatografía ultrarrápida, eluyendo con 3-8 % de metanol/diclorometano que contiene 1 % de amoníaco acuoso, para obtener 78 mg de N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)ciclopropanocarboxamida como un sólido blanquecino.
Parte C. N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)ciclopropanocarboxamida (78 mg) se redujo con complejo de borano - dimetilsulfuro (3,5 eq) a 55 °C durante 12 horas. La reacción se enfrió a continuación a temperatura ambiente, se inactivó cuidadosamente con HCl 1 M (exceso) y se agitó durante 3 horas más a 55 °C. La reacción se enfrió a temperatura ambiente y se diluyó con agua (10 ml) y a continuación se extrajo con diclorometano (10 ml) para eliminar impurezas. El pH de la mezcla de reacción se ajustó a 8,0 mediante la adición de una solución de NaOH 1 M enfriada con hielo, se extrajo con diclorometano (3x10 ml), se secó sobre MgSO4y se concentró a presión reducida para producir un sólido blanquecino. Tras la recristalización con acetato de etilo/hexano 9:1, la reacción produjo 27 mg de 2-butil-1-(4-(((ciclopropilmetil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4 -amina (Compuesto n.° 63-38). Se evaluó que la pureza del producto era del 96 % puro mediante HPLc de fase inversa a 254 nm, la masa sintética prevista de 413,3 Daltons se confirmó mediante lC/MS, y la estructura sintética prevista se confirmó mediante 1H RMN de 300 MHz (CD3OD): 87,80 (d, J = 8,1 Hz, 1H), 7,67 (d, J = 8,4 Hz, 1H), 7,38-7,48 (m, 3H), 7,07-7,20 (m, 3H), 5,92 (s, 2H), 3,97 (s, 2H) 30 (t J = 78 153 Hz 2H) 266 (2 J = 72 Hz 2H) 178-190 (m, 2H), 1,40-1,60 (m, 2H), 1,0 (t, J = 7,5 Hz, 3H)
Ejemplo S6: Síntesis de 2-butil-1-(4-((((1-metilcidobutil)metil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-45).
Parte A.Se añadió N,N-diisopropilcarbodiimida (76 mg, 0,6 mmol) a una solución de ácido 1-metilcidobutanocarboxílico (57 mg, 0,5 mmol) y pentafluorofenol (94 mg, 0,52 mmol) en diclorometano (3 ml), en presencia de una cantidad catalítica de N,N-dimetilaminopiridina (6 mg) y se agitó durante la noche a temperatura ambiente. A continuación, esta mezcla se diluyó con éter (20 ml), la urea precipitada se eliminó por filtración y el filtrado se concentró para obtener el producto en bruto. Este producto en bruto se suspendió en un 1 % de acetato de etilo/hexano y la urea precipitada residual se eliminó de nuevo por filtración. El filtrado resultante se concentró a presión reducida para producir 126 mg del producto carboxilato de (2,3,4,5,6-pentafluorofenil)-1-metilciclobutano deseado.
Parte B. Se añadió el Compuesto n.° 63-00 (84 mg, 0,23 mmol) a una solución de carboxilato de (2,3,4,5,6-pentafluorofenil)-1-metilciclobutano (72 mg, 0,26 mmol) en diclorometano (3 ml) en presencia de trietilamina (48 mg, 0,47 mmol) y se agitó durante 2 horas a temperatura ambiente. A continuación, la mezcla de reacción se concentró a presión reducida y el residuo se lavó con un 5 % de acetato de etilo/hexano. El residuo se disolvió en diclorometano (15 ml), se lavó con HCl 1 M seguido de agua (20 ml), se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para producir 88 mg de N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-1-metilciclobutano-1-carboxamida como un sólido blanquecino.
Parte C. N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-1-metilciclobutano-1-carboxamida (88 mg) se redujo con complejo de borano - dimetilsulfuro (3,5 eq) a 55 °C durante 12 horas. La reacción se enfrió a continuación a temperatura ambiente, se inactivó cuidadosamente con HCl 2 M (exceso) y se agitó durante 3 horas más a 55 °C. La reacción se enfrió a temperatura ambiente y se diluyó con agua (10 ml), a continuación se extrajo con diclorometano (10 ml) para eliminar impurezas. El pH de la mezcla de reacción se ajustó a 8,0 mediante la adición de una solución de NaOH 1 M enfriada con hielo, se extrajo con diclorometano (3x10 ml), se secó sobre MgSO4y se concentró a presión reducida para producir un sólido blanquecino. Tras la recristalización con acetato de etilo/hexano 9:1, la reacción produjo 32 mg de 2-butil-1-(4-((((1-metilciclobutil)metil)amino)metil)bencil)-1H-imidazo[4, 5- c] quinolin-4-amina (Compuesto n.° 63-45). Se evaluó que la pureza del producto era del 96 % puro mediante HPLC de fase inversa a 254 nm, la masa sintética prevista de 441,3 Daltons se confirmó mediante lC/MS, y la estructura sintética prevista se confirmó mediante 1H RMN de 300 MHz (CDCla): 87,80 (d, J = 8,1 Hz, 1H), 7,71 (d, J = 8,4 Hz, 1H), 7,43 (t, J = 7,5, 15,0 Hz, 1H), 7,31 (d, J = 8,4 Hz, 2H), 7,14 (t, J = 7,5, 15,0 Hz, 1H), 7,01 (d, J = 8,4 Hz, 2H), 5,71 (s, 2H), 5,52 (s, 2H), 3,78 (s, 2H), 2,89 (t, J = 7,5, 15,6 Hz, 2H), 2,52 (s, 2H), 1,6-1,88 (m, 8H), 1,35-1,60 (m, 2H), 1,12 (s, 3H), 0,94 (t, J = 7,5, 14,7 Hz, 3H).
Ejemplo S7: Síntesis de 2-butil-1-(4-(((ciclobutilmetil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-44).
Parte A.Se añadió N,N-diisopropilcarbodiimida (127 mg, 1,0 mmol) a una solución de ácido ciclobutanocarboxílico (106 mg, 0,93 mmol) y pentafluorofenol (175 mg, 0,97 mmol) en diclorometano (3 ml), en presencia de una cantidad catalítica de N,N-dimetilaminopiridina (12 mg) y se agitó durante la noche a temperatura ambiente. A continuación, esta mezcla se diluyó con éter (20 ml), la urea precipitada se eliminó por filtración y el filtrado se concentró para obtener el producto en bruto. Este producto en bruto se suspendió en un 1 % de acetato de etilo/hexano y la urea precipitada residual se eliminó de nuevo por filtración. El filtrado resultante se concentró a presión reducida para producir 126 mg del producto carboxilato de (2,3,4,5,6-pentafluorofenil)ciclobutano deseado.
Parte B. Se añadió el Compuesto n.° 63-00 (62 mg, 0,17 mmol) a una solución de carboxilato de (2,3,4,5,6-pentafluorofenil)ciclobutano (50 mg, 0,18 mmol) en diclorometano (3 ml) en presencia de trietilamina (35 mg, 0,34 mmol) y se agitó durante 2 horas a temperatura ambiente. A continuación, la mezcla de reacción se concentró a presión reducida y el residuo se lavó con un 5 % de acetato de etilo/hexano. El residuo se disolvió en diclorometano (15 ml), se lavó con HCl 1 M seguido de agua (20 ml), se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para producir 85 mg de N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)ciclobutanocarboxamida como un sólido blanquecino.
Parte C. N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)ciclobutanocarboxamida (85 mg) se redujo con complejo de borano - dimetilsulfuro (3,5 eq) a 55 °C durante 12 horas. La reacción se enfrió a continuación a temperatura ambiente, se inactivó cuidadosamente con HCl 2 M (exceso) y se agitó durante 3 horas más a 55 °C. La reacción se enfrió a temperatura ambiente y se diluyó con agua (10 ml), a continuación se extrajo con diclorometano (10 ml) para eliminar impurezas. El pH de la mezcla de reacción se ajustó a 8,0 mediante la adición de una solución de NaOH 1 M enfriada con hielo, se extrajo con diclorometano (3x10 ml), se secó sobre MgSO4y se concentró a presión reducida para producir un sólido blanquecino. Tras la recristalización con acetato de etilo/hexano 9:1, la reacción produjo 21 mg de 2-butil-1-(4-(((ciclobutilmetil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4 -amina (Compuesto n.° 63-44). Se evaluó que la pureza del producto era del 95 % puro mediante HPLC de fase inversa a 254 nm, la masa sintética prevista de 427,3 Daltons se confirmó mediante lC/MS, y la estructura sintética prevista se confirmó mediante 1H RMN de 300 MHz (CDCla): 8780 (d J = 81 Hz 1H) 773 (d J = 84 Hz 1H) 745 (t J = 75, 15,0 Hz, 1H), 7,29 (d, J = 8,4 Hz, 2H), 7,15 (t H),
3,74 (s, 2H), 2,89 (t, J = 7,5, 15,6 Hz, 2H), 2,62 (d, J = 7,2 Hz, 2H), 2,40-2,58 (m, 1H), 1,75-2,15 (m, 7H), 1,60-1,70 (m, 2H), 1,35-1,50 (m, 2H), 0,94 (t, J = 7,5, 14,7 Hz, 3H).
Ejemplo S8: Síntesis de 2-butil-1-(4-(((2-cidobutil-2-metilpropil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63- 47).
Parte A.Se añadió N,N-diisopropilcarbodiimida (113 mg, 0,89 mmol) a una solución 3- cidobutil-3-metil-butan-2-ona (70 mg, 0,5 mmol) y pentafluorofenol (108 mg, 0,59 mmol) en diclorometano (3 ml), en presencia de una cantidad catalítica de N,N-dimetilaminopiridina (11 mg) y se agitó durante la noche a temperatura ambiente. A continuación, esta mezcla se diluyó con éter (20 ml), la urea precipitada se eliminó por filtración y el filtrado se concentró para obtener el producto en bruto. Este producto en bruto se suspendió en un 1 % de acetato de etilo/hexano y la urea precipitada residual se eliminó de nuevo por filtración. El filtrado resultante se concentró a presión reducida para producir 141 mg del producto (2,3,4,5,6-pentafluorofenil)-2-ciclobutil-2-metil-propanoato deseado.
Parte B. Se añadió el Compuesto n.° 63-00 (60 mg, 0,17 mmol) a una solución de (2,3,4,5,6-pentafluorofenil)-2-ciclobutil-2-metil-propanoato (54 mg, 0,18 mmol) en diclorometano (3 ml), en presencia de trietilamina (34 mg, 0,34 mmol) y se agitó a reflujo durante 3 días. A continuación, la mezcla de reacción se enfrió a temperatura ambiente, se concentró a presión reducida y el residuo se lavó con un 5 % de acetato de etilo/hexano. El residuo lavado se disolvió en diclorometano (15 ml), se lavó con HCl 1 M seguido de agua (20 ml), se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para producir 67 mg de N-(4-((4- amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-2-ciclobutil-2-metilpropanamida como un sólido blanquecino.
Parte C. N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-2-ciclobutil-2-metilpropanamida (67 mg) se redujo con complejo de borano - dimetilsulfuro (3,5 eq) a 55 °C durante 12 horas. La reacción se enfrió a continuación a temperatura ambiente, se inactivó cuidadosamente con HCl 2 M (exceso) y se agitó durante 3 horas más a 55 °C. La reacción se enfrió a temperatura ambiente y se diluyó con agua (10 ml) y a continuación se extrajo con diclorometano (10 ml) para eliminar impurezas. El pH de la mezcla de reacción se ajustó a 8,0 mediante la adición de una solución de NaOH 1 M enfriada con hielo, se extrajo con diclorometano (3x10 ml), se secó sobre MgSO4y se concentró a presión reducida para producir un sólido blanquecino. Tras la recristalización con acetato de etilo/hexano 9:1, la reacción produjo 13 mg de 2-butil-1-(4-(((2-ciclobutil-2-metilpropil)amino)metil)bencil)-1H-imidazo[4, 5 -c]quinolin-4-amina (Compuesto n.° 63-47). Se evaluó que la pureza del producto era del 92 % puro mediante HPLC de fase inversa a 254 nm, la masa sintética prevista de 469,3 Daltons se confirmó mediante LC/MS, y la estructura sintética prevista se confirmó mediante 1H r Mn de 300 MHz (CDCb): 87,80 (d, J = 8,1 Hz, 1H), 7,71 (d, J = 8,4 Hz, 1H), 7,43 (t, J = 7,5, 15,0 Hz, 1H), 7,31 (d, J = 8,4 Hz, 2H), 7,14 (t, J = 7,5, 15,0 Hz, 1H), 7,01 (d, J = 8,4 Hz, 2H), 6,60 (s a, 1H), 5,73 (s, 2H), 3,74 (s a, 4H), 2,90 (t, J = 7,5, 15,6 Hz, 2H), 2,35 (s, 2H), 1,20-1,8 (m, 10H), 0,94 (t, J = 7,5, 14,7 Hz, 3H), 0,90 (s, 6H).
Ejemplo S9: Síntesis de 2-butil-1-(4-(((2-ciclopropil-2-metilpropil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-41).
Parte A.Se añadió N,N-diisopropilcarbodiimida (127 mg, 1,0 mmol) a una solución de ácido 2-ciclopropil-2-metilpropanoico (106 mg, 0,93 mmol) y pentafluorofenol (175 mg, 0,97 mmol) en diclorometano (3 ml), en presencia de cantidades catalíticas de N,N-dimetilaminopiridina (12 mg) y se agitó durante la noche a temperatura ambiente. A continuación, esta mezcla se diluyó con éter (20 ml), la urea precipitada se eliminó por filtración y el filtrado se concentró para obtener el producto en bruto. Este producto en bruto se suspendió en un 1 % de acetato de etilo/hexano y la urea precipitada residual se eliminó de nuevo por filtración. El filtrado resultante se concentró a presión reducida para producir 130 mg del producto carboxilato de (2,3,4,5,6-pentafluorofenil)-2,2-dimetilciclopropano deseado.
Parte B. Se añadió el Compuesto n.° 63-00 (62 mg, 0,17 mmol) a una solución de carboxilato de (2,3,4,5,6-pentafluorofenil)-2,2-dimetilciclopropano (50 mg, 0,18 mmol) en diclorometano (3 ml), en presencia de trietilamina (35 mg, 0,34 mmol) y se agitó durante 2 horas a temperatura ambiente. A continuación, la mezcla de reacción se concentró a presión reducida y el residuo se lavó con un 5 % de acetato de etilo/hexano. El residuo lavado se disolvió en diclorometano (15 ml), se lavó con HCl 1 M seguido de agua (20 ml), se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para producir 85 mg de N-(4-((4- amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-2-ciclopropil-2-metilpropanamida como un sólido blanquecino.
Parte C. N-(4-((4-amino-2-butil-1H-imidazo[4,5-c]quinolin-1-il)metil)bencil)-2-ciclopropil-2-metilpropanamida (85 mg) se redujo con complejo de borano - dimetilsulfuro (3,5 eq) a 55 °C durante 12 horas. La reacción se enfrió a continuación a temperatura ambiente, se inactivó cuidadosamente con HCl 2 M (exceso) y se agitó durante 3 horas más a 55 °C. La reacción se enfrió a temperatura ambiente y se diluyó con agua (10 ml) y a continuación se extrajo con diclorometano (10 ml) para eliminar impurezas. El pH de la mezcla de reacción se ajustó a 8,0 mediante la adición de una solución de NaOH 1 M enfriada con hielo, se extrajo con diclorometano (3x10 ml), se secó sobre MgSO4y se concentró a presión reducida para producir un sólido blanquecino. Tras la recristalización con acetato de etilo/hexano 9:1, la reacción produjo 14 mg de 2-butil-1-(4-(((2-ciclopropil-2-metilpropil)amino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-41). Se evaluó que la pureza del producto era del 94 % puro mediante HPLC de fase inversa a 254 nm, la masa sintética prevista de 4553 Daltons se confirmó mediante LC/MS y la estructura sintética prevista se confirmó mediante 1H r M Hz,
1H), 7,45 (t, J = 7,5, 15,0 Hz, 1H), 7,29 (d, J = 8,4 Hz, 2H), 7,15 (t, J = 7,5, 15,0 Hz, 1H), 7,03 (d, J = 8,4 Hz, 2H), 5,72 (s, 2H), 5,62 (s, 2H), 3,79 (s, 2H), 2,90 (t, J = 7,5, 15,6 Hz, 2H), 2,87 (s, 2H), 1,75-1,90 (m, 2), 1,40-1,55 (m, 2H), 0,94 (t, J = 7,5, 14,7 Hz, 3H), 0,75 (s, 6H), 0,6-0,75 (m, 1H), 0,15-0,3 (m, 4H).
Ejemplo S10: Síntesis de 2-butil-1-(4-((cidohexilamino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-49).
Parte A. Se añadió cloruro de ferc-butildimetilsililo (3,31 g, 22 mmol) a una solución de alcohol 4-cianobencílico (2,66 g, 20 mmol) en N,N-dimetilformamida (20 ml) en presencia de imidazol (2,72 g, 40 mmol) a temperatura ambiente y se agitó durante 4 horas. La mezcla de reacción se vertió en agua (150 ml) y se extrajo con un 10 % de acetato de etilo/hexano (3 x 75 ml). El extracto orgánico combinado se lavó con agua, se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El producto resultante se purificó adicionalmente por cromatorgrafía ultrarrápida, eluyendo con hexano, para producir 4,84 g de 4-(((ferc-butildimetilsilil)oxi)metil)benzonitrilo.
Parte B. Una solución de 4-(((ferc-butildimetilsilil)oxi)metil)benzonitrilo (4,84 g) en metanol (200 ml) se hidrogenó en un aparato de hidrogenación Parr en presencia de níquel Raney (1,0 g de suspensión espesa en agua) bajo hidrógeno a 60 psi durante 4 horas. La reacción se filtró y el filtrado se concentró a presión reducida. El producto resultante, (4-(((ferc-butildimetilsilil)oxi)metil)fenil)metanamina (4,8 g), se usó sin purificación.
Parte C. Una solución de 4-cloro-3-nitroquinolina (4,16 g, 20 mmol) en diclorometano (100 ml) se añadió lentamente a una solución de 4-(ferc-butildimetilsiloximetil)bencilamina (4,8 g, 19 mmol) y diisopropiletilamina (DIPEA) (3,87 g, 30 mmol) en diclorometano (100 ml) y se agitó durante 12 horas. El disolvente se retiró a presión reducida, el residuo se disolvió en acetato de etilo (200 ml), se lavó con agua (2 x 100 ml) y se secó sobre sulfato de magnesio anhidro. El disolvente se retiró a presión reducida y el producto resultante se purificó por cromatografía ultrarrápida, eluyendo con acetato de etilo/hexano 1:1, para producir 4,67 g de (4-((4-(((ferc-butildimetilsilil)oxi)metil)bencil)amino)quinolin-3-il(oxo)-A4-azanol.
Parte D. Una solución de (4-((4-(((ferc-butildimetilsilil)oxi)metil)bencil)amino)-quinolin-3-il(oxo)-A4-azanol (4,67 g) en acetato de etilo (250 ml) se hidrogenó en presencia de paladio sobre carbono (10 %, 1,0 g) a 60 psi durante 4 horas. El catalizador se retiró por filtración, el disolvente se retiró del filtrado a presión reducida, y el producto, N4-(4-(((fercbutildimetilsilil)oxi)metil)bencil)quinolin-3,4-diamina (4,5 g), se usó sin purificación adicional.
Parte E. Se añadió gota a gota una solución de cloruro de valerilo (1,50 g, 12,46 mmol, 1,05 eq.) en diclorometano (30 ml) a una solución de N4-(4-(((ferc-butildimetilsilil)oxi)metil)-bencil)quinolina-3,4-diamina (4,5 g, 11,86 mmol) en piridina seca (20 ml) a 0 °C. Después de la adición, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 4 horas. El disolvente y la piridina se retiraron a presión reducida. El residuo se disolvió en diclorometano (300 ml) y a continuación se lavó sucesivamente con agua, solución saturada de bicarbonato de sodio (100 ml), solución saturada de sulfato de cobre (3 x 50 ml) y agua (100 ml), a continuación se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El producto, N-(4-((4-(((ferc-butildimetilsilil)oxi)metil)bencil)amino)quinolin-3-il)pentanamida (5,5 g), se usó sin purificación.
Parte F. Una solución de N-(4-((4-(((ferc-butildimetilsilil)oxi)metil)bencil)amino)-quinolin-3-il)pentanamida (5,5 g, 11,87 mmol) en una mezcla de etanol/agua (8:2 v/v, 150 ml) se calentó a 60 °C en presencia de carbonato de potasio (2,5 g, 18,11 mmol, 1,5 eq.) durante 18 horas. El disolvente se retiró a presión reducida. El residuo se repartió entre acetato de etilo (200 ml) y agua (100 ml), la capa de acetato de etilo se separó y se secó sobre sulfato de magnesio anhidro y el residuo se concentró a presión reducida. La purificación adicional se realizó mediante cromatografía ultrarrápida, eluyendo con un 20 % de acetato de etilo/hexano, para producir 3,43 g de 2-butil-1-(4-(((fercbutildimetilsilil)oxi)metil)bencil)-1H-imidazo[4, 5-c]quinolina.
Parte G. Se añadió ácido meta-cloroperbenzoico (60-70 %, 2,5 g) a una solución de 2-butil-1-(4-(((fercbutildimetilsilil)oxi)metil)bencil)-1H-imidazo[4,5-c]quinolina (3,43 g, 7,4 mmol) en diclorometano (200 ml) a temperatura ambiente y se agitó durante 6 horas. La reacción se inactivó añadiendo una solución saturada de sulfito de sodio (20 ml). La capa orgánica se separó, se lavó sucesivamente con una solución de solución saturada de bicarbonato de sodio (50 ml) y a continuación con agua (50 ml), se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. Se realizó una purificación adicional mediante cromatografía ultrarrápida con elución con un 25 % de acetato de etilo/hexano para producir 3,1 g de 2-butil-1-(4-(((ferc-butildimetilsilil)oxi)metil)-bencil)-1H-5A4-imidazo[4,5-c]quinolin-5-ol.
Parte H. Una solución de 2-butil-1-(4-(((ferc-butildimetilsilil)oxi)metil)bencil)-1H-5A4-imidazo[4,5-c]quinolin-5-ol (3,1 g, 6,5 mmol) en diclorometano (50 ml) se añadió a tri-n-butilamina (2,4 g, 13 mmol) y ftalimida (1,91 g, 13 mmol) y la mezcla de reacción se enfrió a 0 °C. Se añadió lentamente a la reacción una solución de cloruro de benzoílo (1,82 g, 13 mmol) en diclorometano (10 ml), y la mezcla se calentó a temperatura ambiente y se agitó durante 30 minutos. La reacción se diluyó con diclorometano (100 ml), se lavó sucesivamente con una solución acuosa saturada de cloruro de amonio (100 ml) y a continuación con agua (100 ml), se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El producto resultante se aisló por cromatografía ultrarrápida eluyendo con un 10 % de acetato de etilo/hexano, para producir 2,85 g de 2-(2-buti 4il)isoindolin-1,3-diona.
Parte I. Se añadió una solución 1 M de fluoruro de tetrabutilamonio (6 ml) a una solución de 2-(2-butil-1-(4-(((fercbutildimetilsilil)oxi)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-il)isoindolin-1,3-diona (2,85 g, 4,7 mmol) en tetrahidrofurano seco (1o ml) a 0 °C. Después de la adición, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 6 horas más. La reacción se inactivó mediante la adición de cloruro de amonio saturado (20 ml), se diluyó con acetato de etilo (100 ml), se lavó con agua, se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. Se realizó una purificación adicional mediante cromatografía ultrarrápida eluyendo con diclorometano/hexano (1:1) para producir 1,57 g de 2-(2-butil-1-(4-(hidroximetil)bencil)-1H-imidazo[4,5-c] quinolin-4-il)isoindolin-1,3-diona.
Parte J. Una solución de DMSO (2,5 g, 32 mmol) en CH2Cl2 (10 ml) se añadió a una solución de cloruro de oxalilo (2,0 g, 16 mmol) en diclorometano (10 ml) y tamices moleculares de 3 Å en CH2Cl2 (20 ml) a -78 °C en argón. Después de 15 minutos, se añadió lentamente gota a gota una solución de 2-(2-butil-1-(4-(hidroximetil)bencil)-1H-imidazo[4,5-c]quinolin-4-il)isoindolin-1,3-diona (1,57 g, 3,2 mmol ) en CH2Cl2 (3 ml). Después de 30 min, se añadió gota a gota Et3N (4,5 g, 45 mmol), la reacción se agitó durante 30 minutos a -78 °C y a continuación se dejó calentar lentamente a temperatura ambiente. Después de agitar durante otra hora a temperatura ambiente, la mezcla de reacción se inactivó mediante la adición de una solución saturada de cloruro de amonio. La capa orgánica se separó, se lavó con agua (25 ml), se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. Se realizó una purificación adicional mediante cromatografía ultrarrápida para producir 1,15 g de 4-((2-butil-4-(1,3-dioxoisoindolin-2-il)-1H-imidazo[4,5-c]quinolin-1-il) metil)benzaldehído.
Parte K. Una solución de 4-((2-butil-4-(1,3-dioxoisoindolin-2-il)-1H-imidazo[4,5-c]quinolin-1-il)metil)benzaldehído (2,33 mmol) y ciclohexilamina (7 mmol) en diclorometano se calienta a reflujo en presencia de una cantidad catalítica de ácido p-toluenosulfónico durante 12 horas. La mezcla de reacción se concentra a presión reducida. El producto en bruto, 2-(2-butil-1-(4-((ciclohexilimino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-il)isoindolin-1,3-diona, se usa sin purificación adicional.
Parte L. Se añade borohidruro de sodio (10 mmol) a una solución de 2-(2-butil-1-(4-((ciclohexilimino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4- il)isoindolin-1,3-diona en metanol a temperatura ambiente y se agitó durante 2 horas. La mezcla de reacción se inactiva con solución saturada de cloruro de amonio y se extrae con diclorometano (3 x 30 ml). La capa orgánica combinada se lava con agua, se seca sobre sulfato de magnesio anhidro y se concentra a presión reducida. El producto, 2-(2-butil-1-(4-((ciclohexilamino)metil)-bencil)-1 H-imidazo[4,5-c]quinolin-4-il)isoindolin-1,3-diona, se purifica por cromatografía ultrarrápida.
Parte M. Se añade hidrazina (100 mg) a una solución de 2-(2-butil-1-(4-((ciclohexilamino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-il)isoindolin-1,3-diona (0,38 mmol) en metanol y se agitó durante 12 horas. El disolvente se retira a presión reducida y el producto resultante, 2-butil-1-(4-((ciclohexilamino)metil)bencil)-1H-imidazo[4,5-c]quinolin-4-amina, se purifica por cromatografía ultrarrápida.
Ejemplo S11: Síntesis de compuestos de N-acilo de fórmula (J-2) ilustrativos
Los compuestos de N-acilo de fórmula (J-2) ilustrativos se sintetizaron usando procedimientos similares a los del Compuesto n.° 63-33, hasta la parte I. Los datos de 1HRMN y espectrometría de masas se detallan a continuación para determinados compuestos de la presente invención, y la tabla 4 proporciona datos de pureza y valores de cLogP.

Compuesto n.° 63-34: 1H RMN (CDCls, 400 MHz): 57,75 (dd, J = 8,3, 0,9 Hz, 1H), 7,67 (dd, J = 8,3, 1,1 Hz, 1H), 7,40 (m, J = 8,3, 7,1, 1,4 Hz, 1H), 7,22 (d, J = 8,2 Hz, 2H), 7,11 (m, J = 8,2, 7,1, 1,2 Hz, 1H), 6,98 (d, J = 8,2 Hz, 2H), 6,30 (tap, 1H), 5,68 (s, 2H), 4,42 (d, J = 6,0 Hz, 2H) 284 (ddap 2H) 216 (dd J = 72 Hz 2H) 181-175 (m 2H) 148 1,37 (m, 2H), 0,91 (t, J = 7,4 Hz, 3H), 0,57-0,5
Ejemplo S12: Síntesis de compuestos de fórmula (K) ilustrativos
Los compuestos de W-acilo de fórmula (K-2) ilustrativos se sintetizaron usando procedimientos similares a los del Compuesto n.° 63-10. Los datos de 1H RMN y espectrometría de masas se detallan a continuación para determinados compuestos de la presente invención, y la tabla 5 enumera los compuestos con datos de pureza así como valores de cLogP.
Los compuestos de Walquilo de fórmula (K-1) ilustrativos se sintetizaron usando procedimientos similares a los del Compuesto n.° 63-17. Los datos de 1H RMN y espectrometría de masas se detallan a continuación para determinados compuestos de la presente invención, y la tabla 6 proporciona datos de pureza y valores de cLogP.
Compuesto n.° 63-02: 1H RMN (CDCb, 400 MHz): 8 7,80 (dd, J = 8,4, 1,2 Hz, 1H), 7,64 (dd, J = 8,2, 1,0 Hz, 1H), 7,40 (m, J = 8,4, 7,0, 1,0 Hz, 1H), 7,20 (d, J = 8,2 Hz, 2H), 7,09 (m, J = 8,2, 7,1, 1,3 Hz, 1H), 6,96 (d, J = 8,2 Hz, 2H), 6,28 (s a, 2H), 6,02 (t a, J = 5,7 Hz, 1H), 5,65 (s, 2H), 4,37 (d, J = 5,8 Hz, 2H), 2,83 (dd, J = 7,8, 7,8 Hz, 2H), 2,15 (dd, J = 7,6, 7,6 Hz, 2H), 1,81-1,73 (m, 2H), 1,64-1,56 (m, 2H), 1,42 (dc, J = 15,0, 7,4 Hz, 2H), 1,32-1,21 (m, 4H), 0,91 (t, J = 7,3 Hz, 3H), 0,84 (t, J = 7,3 Hz, 3H). Espectr. de masas: m/z 468,2 (M+1).
Compuesto n.° 63-05: 1H RMN (CDCla, 300 MHz): 7,89 (d, J = 8,1 Hz, 1H), 7,86 (d, J = 8,1 Hz, 1H), 7,69 (t, 1H), 7,74 (t, J = 15,6, 8,4, 1H), 7,47 (t, J = 15,5, 7,5 Hz, 1H), 7,38 (m, 4H), 7,14 (s a, 1H) 7,08 (d, J=8,1 Hz) 5,85 (s, 2H), 4,53 (d, J=6 Hz, 2H), 2,99 (t, J = 7,8 Hz, 2H), 2,30 (t, J = 7,5 Hz, 2H), 1,90-1,97 (m, 4H), 1,52-1,75 (m, 4H), 1,26-1,48 (m, 14 H), 0,90-1,10 (m, 6 h ). Espectr. de masas: m/z 500,8 (M+1).
Compuesto n.° 63-06: 1H RMN (CDCb, 300 MHz): 8,05 (d, J = 8,1 Hz, 1H), 7,84 (d, J = 8,4 Hz, 1H), 7,74 (t, J = 8,4 Hz, 1H), 7,47 (t, J = 7,5 Hz, 1H), 7,38 (d, J = 8,1 Hz, 2H), 7,14 (d, J = 8,1 Hz, 2H), 6,04 (s, 2H), 4,42 (s, 2H), 3,74 (s, 2H), ), 3,10 (t, J = 7,8 Hz, 2H), 2,29 (t, J = 7,5 Hz, 2H), 1,90-1,97 (m, 2H), 1,52-1,75 (m,4H), 1,26-1,48 (m, 12 H), 0,90-1,10 (m, 6 H). Espectr. de masas: m/z 514,5 (M+1).
Compuesto n.° 63-07: 1H RMN (CDCls, 300 MHz): 7,88 (d, J = 8,1 Hz, 1H), 7,69 (d, J = 8,4 Hz, 1H), 7,50 (t, J=15,6, 8.4, 1H), 7,29 (d, J=8,1 Hz, 2H), 7,20 (t, J = 15,5, 7,5 Hz, 1H), 7,06 (d, J = 8,1 Hz, 2H), 5,90 (s, 2H), 4,33 (s, 2H), 3,74 (s, 2H), ), 3,0 (t, J = 15,0, 7,8 Hz, 2H), 2,21 (t, J = 14,7, 7,5 Hz, 2H), 1,90-1,97 (m, 2H), 1,42-1,70 (m,4H), 1,26-1,48 (m, 14 H), 0,85-1,05 (m, 6 H). Espectr. de masas: m/z 528,6 (M+1).
Compuesto n.° 63-08: 1H RMN (CDCb, 400 MHz): 8 7,74 (d, J = 8,2 Hz, 1H), 7,64 (d, J = 8,2 Hz, 1H), 7,37 (dd, J = 7.4, 7,4 Hz, 1H), 7,18 (d, J = 8,0 Hz, 2H), 7,07 (dd, J = 7,5, 7,5 Hz, 1H), 6,95 (d, J = 8,0 Hz, 2H), 5,98 (t a, J = 5,6 Hz, 1H), 5,89 (s a, 2H), 5,63 (s, 2H), 4,35 (d, J = 5,8 Hz, 2H), 2,81 (dd, J = 8,0, 8,0 Hz, 2H), 2,14 (dd, J = 7,6, 7,6 Hz, 2H), 1,80-1,71 (m, 2H), 1,61-1,56 (m, 2H), 1,39 (dc, J = 15,0, 7,4 Hz, 2H), 1,32-1,21 (m, 18H), 0,90 (t, J = 7,3 Hz, 3H), 0,86 (t, J = 7,3 Hz, 3H). Espectr. de masas: m/z 542,4 (M+1).
Compuesto n.° 63-09: 1H RMN (DMSO-d6 , 300 MHz): 8,18 (t, 1H), 7,8 (d, J = 8,4 Hz, 1H), 7,6 (d, J = 8,5 Hz, 1H), 7,34 (t, J = 8,4, 1H), 7,18 (d, J = 8,5 Hz, 2H), 6,9-7,12 (m, 3 H), 6,55 (s, 2H), 5,8 (s, 2H), 4,2 (d, 2H), 2,90 (t, J = 7,6 Hz, 2H), 2.2 (t, J = 7,2 Hz, 2H), 1,65-1,80 (m, 2H), 1,3-1,75 (m, 4H), 1,25-1,38 (m, 10 H), 0,65-1,0 (m, 6 H). Espectr. de masas: m/z 556,9 (M+1).
Compuesto n.° 63-10: 1H RMN (CDCb, 300 MHz): 7,98 (d, J = 8,1 Hz, 1H), 7,75 (d, J = 8,4 Hz, 1H), 7,67 (t, J = 8,4 Hz, 1H), 7,40 (t, J = 7,5 Hz, 1H), 7,30 (d, J = 8,1 Hz, 2H), 7,06 (d, J = 8,1 Hz, 2H), 5,95 (s, 2H), 4,33 (s, 2H), 3,74 (s, 2H), ), 3,01 (t, J = 7,8 Hz, 2H), 2,20 (t, J = 7,5 Hz, 2H), 1,82-1,9 (m, 2H), 1,42-1,70 (m,4H), 1,26-1,48 (m, 20 H), 0,85-1,05 (m, 6 H). Espectr. de masas: m/z 570,8 (M+1).
Compuesto n.° 63-11: 1H RMN (CDCb, 300 MHz): 7,98 (d, J = 8,1 Hz, 1H), 7,76 (d, J = 8,4 Hz, 1H), 7,65 (t, J = 8,4 Hz, 1H), 7,38 (t, J = 7,5 Hz, 1H), 7,30 (d, J = 8,1 Hz, 2H), 7,06 (d, J = 8,1 Hz, 2H), 5,95 (s, 2H), 4,33 (s, 2H), 3,74 (s, 2H), 3.03 (t, J = 7,8 Hz, 2H), 2,21 (t, J = 7,5 Hz, 2H), 1,82-1,9 (m, 2H), 1,42-1,70 (m,4H), 1,26-1,48 (m, 22 H), 0,85-1,05 (m, 6 H). Espectr. de masas: m/z 585,2 (M+1).
Compuesto n.° 63-31: 1H RMN (CD3OD, 300 MHz): 7,82 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,42 (t, J = 8,4, 1H), 7,34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,9 (s, 2H), 4,39 (s, 2H), ), 3,0 (t, J = 7,6 Hz, 2H), 2,25 (t, J = 7,2 Hz, 2H), 1,42-1,90 (m, 6 H), 1,2-1,4 (m, 13H), 0,65-1,0 (m, 6 H). Espectr. de masas: m/z 598,7 (M+1).
Compuesto n.° 63-13: 1H RMN (CDCb, 400 MHz): 8 7,77 (d, J = 8,2 Hz, 1H), 7,67 (d, J = 8,2 Hz, 1H), 7,41 (dd, J = 7.4, 7,4 Hz, 1H), 7,21 (d, J = 8,0 Hz, 2H), 7,11 (dd, J = 7,5, 7,5 Hz, 1H), 6,98 (d, J = 8,0 Hz, 2H), 5,87 (t a, J = 5,6 Hz, 1H), 5,87 (s a, 2H), 5,68 (s, 2H), 4,39 (d, J = 5,8 Hz, 2H), 2,85 (dd, J = 8,0, 8,0 Hz, 2H), 2,17 (dd, J = 7,6, 7,6 Hz, 2H), 1,83-1,75 (m, 2H), 1,65-1,58 (m, 2H), 1,43 (dc, J = 15,0, 7,4 Hz, 2H), 1,32-1,21 (m, 30H), 0,92 (t, J = 7,3 Hz, 3H), 0,88 (t, J = 7,3 Hz, 3H). Espectr. de masas: m/z 625,5 (M+1).
Compuesto n.° 63-00: 1H RMN (MeOHd4, 40 H),
7,36 (m, J = 8,4, 7,0, 1,4 Hz, 1H), 7,27 (d, J = 8,2 Hz, 2H), 7,03 (m, J = 8,3, 7,0, 1,3 Hz, 1H), 6,97 (d, J = 8,4 Hz, 2H), 5,72 (s, 2H), 3,72 (s, 2H), 2,88 (dd, J = 7,6, 7,6 Hz, 2H), 1,77-1,69 (m, 2H), 1,45-1,35 (m, 2H), 0,90 (t, J = 7,4 Hz, 3H). Espectr. de masas: m/z 360,2 (M+1).
Compuesto n.° 63-17: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,80 (s, 2H), ), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,2-1,75 (m, 8H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 430,3 (M+1).
Compuesto n.° 63-18: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,72 (s a, 4H), 3,76 (s, 2H), ), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,4-1,6 (m, 4H), 1,2-1,35 (s a, 4H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 444,6 (M+1).
Compuesto n.° 63-19: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,80 (s, 2H), ), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,4-1,6 (m, 4H), 1,2-1,35 (b, s, 8H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 458,4 (M+1).
Compuesto n.° 63-20: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,80 (s, 2H), ), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,4-1,6 (m, 4H), 1,2-1,35 (b, s, 10H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 472,4 (M+1).
Compuesto n.° 63-21: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,80 (s, 2H), ), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,4-1,6 (m, 4H), 1,2-1,35 (b, s, 12H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 486,5 (M+1).
Compuesto n.° 63-22: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,45 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,80 (s, 2H), ), 2,89 (t, J = 7.6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,84 (m, 2H), 1,4-1,6 (m, 4H), 1,2-1,35 (b, s, 14H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 500,5 (M+1).
Compuesto n.° 63-24: 1H RMN (CDCh, 300 MHz): 7,78 (d, J = 8,4 Hz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 7,43 (t, J = 8,4, 1H), 7,28 (d, J = 8,5 Hz, 2H), 7,10 (t, J = 8,4 Hz, 1H), 6,98 (d, J = 8,3 Hz, 2H), 5,70 (s a, 4H), 3,73 (s, 2H), ), 2,87 (t, J = 7,6 Hz, 2H), 2,58 (t, J = 7,2 Hz, 2H), 1,71-1,84 (m, 2H), 1,38-1,55 (m, 4H), 1,10-1,35 (m, 18H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 528,7 (M+1).
Compuesto n.° 63-32: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,73 (d, J = 8,5 Hz, 1H), 7,42 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,12 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,73 (s, 2H), 5,5 (s a, 2H) 3,75 (s, 2H), ), 2,90 (t, J = 7,6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,75-1,85 (m, 4H), 1,4-1,6 (m, 4H), 1,25-1,38 (m, 15 H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 584,9 (M+1).
Compuesto n.° 63-29: 1H RMN (CDCh, 300 MHz): 7,8 (d, J = 8,4 Hz, 1H), 7,73 (d, J = 8,5 Hz, 1H), 7,42 (t, J = 8,4, 1H), 7.34 (d, J = 8,5 Hz, 2H), 7,12 (t, J = 8,4 Hz, 1H), 7,02 (d, J = 8,3 Hz, 2H), 5,72 (s, 2H), 3,80 (s, 2H), 3,78 (s, 2H), 2,90 (t, J = 7,6 Hz, 2H), 2,62 (t, J = 7,2 Hz, 2H), 1,65-1,85 (m, 2H), 1,35-1,6 (m, 4H), 1,25-1,38 (m, 15 H), 0,65-1,0 (m, 6H). Espectr. de masas: m/z 612,8 (M+1).


Ejemplo S13: Síntesis de compuestos de N-alquilo de fórmula (K-1) ilustrativos adicionales
Los compuestos de N-alquilo n.° 63-16, 63-23 63-25 63-26 63-27 63-28 y 63-30 de fórmula (K-1) ilustrativos se sintetizan usando un procedimiento similar a se
usan los siguientes ácidos carboxílicos en lugar del ácido valérico usado para el Compuesto n.° 63-17: ácido nbutanoico (Compuesto n.° 63-16); ácido undecanoico (Compuesto n.° 63-23); ácido tridecanoico (Compuesto n.° 63 25); ácido tetradecanoico (Compuesto n.° 63-26); ácido pentadecanoico (Compuesto n.° 63-27); ácido heptadecanoico (Compuesto n.° 63-28); y ácido nonadecanoico (Compuesto n.° 63-30). Los derivados de N-acilo resultantes se reducen a continuación a los compuestos de N-alquilo finales de acuerdo con la Parte J descrita para el ejemplo S2.
Ejemplo S14: Síntesis de compuestos de N-acilo de fórmula (K-2) ilustrativos adicionales
Los compuestos de N-acilo n.° 63-01, 63-03, 63-04, 63-12, 63-14 y 63-15 de fórmula (K-2) ilustrativos se sintetizan usando un procedimiento similar al descrito para el Compuesto n.° 63-10 (Ejemplo S1) hasta la Parte H. En la Parte I, se usan los siguientes ácidos carboxílicos en lugar del ácido mirístico usado para el Compuesto n.° 63-10: ácido pentanoico (Compuesto n.° 63-01), ácido heptanoico (Compuesto n.° 63-03), ácido octanoico (Compuesto n.° 63-04), ácido heptadecanoico (Compuesto n.° 63-12), ácido nonadecanoico (Compuesto n.° 63-14), y ácido araquídico (Compuesto n.° 63-15), para formar los productos de N-acilo.
Ejemplos biológicos
Ejemplo B1. Ensayos biológicos in vitro y resultados
Métodos
Se prepararon células mononucleares de sangre periférica (PBMC) enriquecidas con células dendríticas plasmocitoides (pDC) a partir de la sangre de una serie de donantes humanos (3-5 donantes/experimento). Las PBMC se aislaron usando Ficoll-Paque Premium® (GE Healthcare, Chicago IL) usando métodos bien conocidos por los expertos en la materia. Las pDC se aislaron magnéticamente de la población total de PBMC recuperadas usando microperlas CD304 (BDCA-4/Neuropilin-1) (Miltenyi Biotec, San Diego CA), de acuerdo con las instrucciones del fabricante. A continuación, se añadieron pDC aisladas entre 1 y 2 x108 de las PBMC del donante correspondiente para el enriquecimiento relativo de este tipo celular. Cultivos duplicados de PBMC enriquecidas con pDC (2,5x106 células/ml en medio RPMI-1640 más suero fetal bovino al 10 %, cultivadas en placas de 96 pocillos) se incubaron durante 24 horas con los Compuestos n.° 63-02, 63-05 a 63-11,63-13, 63-31, 63-34, 63-38 a 63-47, y su congénere no modificado 1-(4-aminometilbencil)-2-butil-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-00) a 10 concentraciones diluidas en serie que cubren el intervalo de 0,1 nM a 400 nM. Se recogieron los sobrenadantes de cultivo y se midieron los niveles de proteína de interferón-alfa (IFNa) mediante ELISA (MabTech, Cincinnati OH), de acuerdo con las instrucciones del fabricante.
Los monocitos se aislaron magnéticamente a partir de PBMC, preparadas como se ha descrito anteriormente, después del etiquetado con microesferas CD14 (Miltenyi Biotec, San Diego CA), de acuerdo con las instrucciones del fabricante. Cultivos duplicados de monocitos (1x106 células/ml en RPMI-1640 más suero fetal bovino al 10 %, cultivadas en placas de 96 pocillos) se incubaron a continuación durante 24 horas con los Compuestos n.° 63-02, 63-05 a 63-11, 63-13, 63-31, 63-34, 63-38 a 63-47, y su congénere no modificado 1-(4-aminometilbencil)-2-butil-1H-imidazo[4,5-c]quinolin-4-amina (Compuesto n.° 63-00) a 10 concentraciones diluidas en serie que cubren el intervalo de 2 nM a 40 μM. Se recogieron los sobrenadantes de cultivo y se midieron los niveles de proteína del factor de necrosis tumoral alfa (TNFα) mediante ELISA (MabTech, Cincinnati OH), de acuerdo con las instrucciones del fabricante.
Resultados
El efecto de modificar ya sea i) el compuesto de fórmula (K-2), donde R es una cadena alquílica lineal de 5 (Compuesto n.° 63-02), 8 (Compuesto n.° 63-05), 9 (Compuesto n.° 63-06), 10 (Compuesto n.° 63-07), 11 (Compuesto n.° 63-08), 12 (Compuesto n.° 63-09), 13 (Compuesto n.° 63-10), 14 (Compuesto n.° 63-11), 15 (Compuesto n.° 63-31) o 17 (Compuesto n.° 63-13) carbonos añadidos al grupo amida en el resto bencilmetilo, o ii) el compuesto de fórmula (J-2), donde R0 es un resto ciclopropilmetilo con 4 (Compuesto n.° 63-34) carbonos añadidos al grupo amida en el resto bencilmetilo, sobre la bioactividad agonista in vitro de TLR7 (inducción de proteína IFNa en cultivos de PBMC enriquecidos con pDC) y TLR8 (inducción de proteína TNFα en cultivos de monocitos) se evaluó. La Tabla B1-1 resume las relaciones estructural, de cálculo de cLogP y de bioactividad del agonista TLRH8 para los Compuestos n.° 63-00, 63-02, 63-05 a 63-11, 63-13, 63-31 y 63-34. La potencia agonista de TLR7 y TLR8 se muestra en la tabla B1-1 y se notifica como la concentración eficaz al 50 % de la respuesta máxima, en nanomolar (CE50 en nM). La modificación del Compuesto n.° 63-00 con estas cadenas alquílicas generalmente dio como resultado una pérdida de 2 - 3x la potencia agonista de TLR7, a excepción de las variantes de carbono 5 (Compuesto n.° 63-02) y 17 (Compuesto n.° 63-13) que perdieron potencia 11x y 41x, respectivamente. Por el contrario, las variantes de la cadena de carbono 5 (Compuesto n.° 63-02), 8 (Compuesto n.° 63-05), 9 (Compuesto n.° 63-06), 10 (Compuesto n.° 63-07), 15 (Compuesto n.° 63-31) y 17 (Compuesto n.° 63-13) perdieron >10 - 26x de potencia agonista de TLR8 en comparación con el Compuesto n.° 63-00 no modificado. Inesperadamente, las variantes con longitudes de cadena de carbono de 11 (Compuesto n.° 63-08), 12 (Compuesto n.° 63-09), 13 (Compuesto n.° 63-10) y 14 (Compuesto n.° 63-11) no perdieron tanta actividad agonista, con el Compuesto n.° 63-10 demostrando solo una pérdida de 2,4x de potencia agonista de TLR8 en comparación con el Compuesto n.° 63-00 no modificado El Compuesto n° 63-34 donde R0 es un grupo ciclopropilmetilo, demostró una pérdida de 8 v ia
agonista de TLR8 en comparación con el Compuesto n.° 63-00 no modificado. Estos datos son comparables con la pérdida de potencia agonista de TLR7 y TLR8 observada para el compuesto n.° 63-02 modificado con cadena alquílica lineal de 5 carbonos.
Tabla B1-1. Relaciones estructural, de cálculo de cLogP y de bioactividad para compuestos seleccionados de

El efecto de ya sea modificar i) el compuesto de fórmula (K-1), donde R es una cadena alquílica lineal de 5 (Compuesto n.° 63-17), 6 (Compuesto n.° 63-18), 7 (Compuesto n.° 63-19), 8 (Compuesto n.° 63-20), 9 (Compuesto n.° 63-2l), 10 (Compuesto n.° 63-22), 12 (Compuesto n.° 63-24), 16 (Compuesto n.° 63-32), o 18 (Compuesto n.° 63-29) carbonos añadidos al grupo amino en el resto bencilmetilo, o ii) modificar el compuesto de fórmula (J-1), donde R0 es un resto ciclopropiletilo con 5 (Compuesto n.° 63-33), un resto ciclobutiletilo con 6 (Compuesto n.° 63-35), o un resto ciclopentiletilo con 7 (Compuesto n.° 63-36) carbonos añadidos al grupo amida sobre la bioactividad agonista in vitro de TLR7 (inducción de proteína IFNa en cultivos de PBMC enriquecidos con pDC) y TLR8 (inducción de proteína TNFα en cultivos de monocitos) se evaluó. La Tabla B1-2 resume las relaciones estructural, de cálculo de cLogP y de bioactividad del agonista TLRH8 para los Compuestos n.° 63-00, 63-17 hasta 63-22, 63-24, 63-29, 63-32, 63-33, 63 35 y 63-36. La potencia agonista de TLR7 y TLR8 se muestra en la tabla B1-2 y se notifica como la concentración eficaz al 50 % de la respuesta máxima, en nanomolar (CE50 en nM). La modificación del Compuesto n.° 63-00 con cadenas alquílicas lineales demostró una pérdida creciente de la potencia agonista de TLRH8 con un número creciente de carbonos, aunque las variantes 5 (Compuesto n.° 63-17) y 6 (Compuesto n.° 63-18) de carbonos demostraron una potencia agonista de TLR8 ligeramente mejorada. La modificación del Compuesto n.° 63-00 con tres grupos hidrocarbilo, ciclopropiletilo, ciclobutiletilo y ciclopentiletilo, causó una pérdida de 4 a 12 veces de la bioactividad agonista de TLR7, pero dio como resultado un aumento de 2-3 veces en la potencia agonista.
Tabla B1-2. Relaciones estructural, de cálculo de cLogP y de bioactividad para compuestos seleccionados de

fó l J 1 K 1

El efecto de modificar el compuesto de fórmula (J-1), donde R0 es un resto (ciclopropil)etilo (Compuesto n.° 63-33), resto (ciclobutil)etilo (Compuesto n.° 63-35), resto (ciclopentil)etilo (Compuesto n.° 63-36), resto (ciclopropil)metilo (Compuesto n.° 63-38), resto (2-metilciclopropil)metilo (Compuesto n.° 63-39), resto (2,2-dimetiiciclopropil)metilo (Compuesto n.° 63-40), resto (2-ciclopropN)-(2,2-dimetil)etilo (Compuesto n.° 63-41), resto (l-metilciclopropN)etilo (Compuesto n.° 63-42), resto (3-ciclopropil)propilo (Compuesto n.° 63-43), resto (ciclobutil)metilo (Compuesto n.° 63 44), resto (l-metilciclobutN)metilo (Compuesto n.° 63-45), resto (3-metilciclobutN)metilo (Compuesto n.° 63-46), o resto (2-cidobutN)-(2,2-dimetil)etilo (Compuesto n.° 63-47), sobre la bioactividad agonista in vitro de TLR7 (inducción de proteína IFNa en cultivos de PBMC humanos enriquecidos con pDC) y TLR8 (inducción de proteína TNFα en cultivos de monocitos humanos) se evaluó. La tabla B1-3 resume las relaciones de bioactividad de los agonistas de TLRH8 para los Compuestos n.° 63-00, 63-33, 63-35, 63-36 y 63-38 a 63-47. La potencia agonista de TLR7 y TLR8 que se muestra en la tabla B1-3 se notifica como un porcentaje de la concentración eficaz del compuesto al 50 % de la respuesta máxima determinada para el Compuesto n.° 63-00. La variación entre donantes de sangre humana en los niveles de citocinas secretadas por células inmunitarias purificadas usadas para evaluar la potencia de los compuestos agonistas de TLR7 y TLR8 da como resultado variaciones menores en los valores absolutos calculados para potencia de CE50 para un compuesto dado; para normalizar para este efecto, los datos en la tabla B1-3 se expresan como un porcentaje de la CE50 determinada para la estructura química basada en imidazoquinolina no modificada (Compuesto n.° 63-00).
Como se muestra en la Tabla B1-3, la modificación de la estructura química del Compuesto n.° 63-00 para producir compuestos de fórmula (J-1) con fracciones de cicloalquilo variables dio como resultado la atenuación de la potencia agonista de TLR7 hasta 11 veces. Las variantes de (ciclobutil)metilo (Compuesto n.° 63-44) y (l-metilciclobutil)metilo (Compuesto n.° 63-45) demostraron la menor cantidad de actividad agonista de TLR7 atenuada (1,2 y 1,9 veces menos de potencia, respectivamente), mientras que las variantes de (ciclopentil)etilo (Compuesto n.° 63-36) y (2-ciclobutil)-(2,2-dimetil)etilo (Compuesto n.° 63-47) demostraron la mayor cantidad de actividad agonista de TLR7 atenuada (10,9 y 9,0 veces menos de potencia, respectivamente). Inesperadamente, el mismo conjunto de modificaciones estructurales a la estructura química del Compuesto n.° 63-00 dio como resultado una potencia agonista de TLR8 comparable o mayor. Las variantes de (ciclobutil)metilo (Compuesto n.° 63-44) y (1-metilciclobutil)metilo (Compuesto n.° 63-45) demostraron el mayor aumento en la potencia agonista de TLR8 (5,9 y 7,7 veces mayor potencia, respectivamente). Por el contrario, las variantes de (1-metilciclopropil)etilo (Compuesto n.° 63-42) y (2-ciclobutil)-(2,2-dimetil)etilo (Compuesto n.° 63-47) demostraron una actividad agonista de TLR8 ligeramente mejorada a ligeramente inferior en comparación con el Compuesto n.° 63-00. Es más probable que una molécula pequeña agonista de TLRH8 con una potencia agonista de TLR7 y TLR8 más parecida produzca una activación comparable de los 2 sistemas de receptores tras la administración de una dosis terapéutica del compuesto en una composición farmacéutica dada, activando así una gama más amplia de tipos de células inmunitarias relevantes. Los compuestos con potencia dual equilibrada también permitirían la síntesis y caracterización de un solo principio farmacéutico activo, facilitando así la fabricación NCF a costes más bajos y permitiendo una vía regulatoria más directa y predecible.


Ejemplo B2. Preparación de composiciones farmacéuticas
Ejemplo B2-1. Preparación de composiciones farmacéuticas a base de aceite de sésamo. Los Compuestos n.° 63-17, 63-18, 63-10 y 63-33 se formularon para administración in vivo en 95 % de aceite de sésamo/5 % de etanol (v/v) como sigue. El aceite de sésamo Super Refined® se obtuvo de Croda Inc. (Edison, NJ) y el etanol (200 grados, calidad USP) se obtuvo de Pharmaco-AAPER (Brookfield, CT). Los compuestos se colocaron en un vial de vidrio y se añadió etanol al 100 % para preparar suspensiones de 2,75 mg/ml. Las soluciones se solubilizaron mediante agitación con vórtice durante 30 segundos y a continuación se mantuvieron en un baño de agua ultrasónico ajustado a 50 °C durante 30 minutos. A continuación, se transfirió un ml de estas soluciones a un vial de vidrio de 20 ml que contenía 16,0 g de aceite de sésamo, se mezcló en un mezclador de extremo a extremo durante 20 minutos a temperatura ambiente y a continuación se transfirió a un baño de agua a 90 °C durante 2 horas para garantizar la solubilización completa. Los compuestos formulados se enfriaron a 37 °C antes de la esterilización mediante filtración de 0,2 micras. Los compuestos formulados se almacenaron a 2 - 8 °C en viales de vidrio estériles con tapón de caucho. Las concentraciones de los componentes en la formulación final fueron las siguientes: 0,1 mg/ml (p/v) de compuesto en 95 % de aceite de sésamo y 5 % de etanol (v/v).
Ejemplo B2-2. Preparación de composiciones farmacéuticas basadas en nanoemulsiones de aceite en agua de escualeno. Los Compuestos n.° 63-17, 63-18, 63-10 y 63-33 se formularon para administración in vivo en nanoemulsiones a base de aceite en agua de escualeno como sigue. Escualeno (>98 %, líquido), Tween®80 (polisorbato 80), glicerol y dihidrato tribásico de citrato de sodio se obtuvieron de Sigma-Aldrich (St. Louis, MO). La 1,2-dioleoil-sn-glicero-3-fosfocolina (DOPC) se obtuvo de Avanti Polar Lipids (Alabaster, AL). El agua de calidad de cultivo celular (agua estéril para inyección) se obtuvo de Corning Life Sciences (Tewksbury, MA). Para formar la fase oleosa, se añadió DOPC (175,6 mg) a aceite de escualeno (1,4 ml) en un vial de vidrio de 4 ml. A continuación, la mezcla se incubó en un baño de agua con ultrasonidos a 70 °C durante 45 minutos, con breve agitación con vórtice cada 15 minutos, hasta que se disolvió el lípido. Se añadió el compuesto indicado (13,7 mg) a la solución de escualeno/DOPC y se agitó con vórtice vigorosamente durante un minuto. A continuación, esta solución se incubó en un baño de agua con ultrasonidos a 70 °C durante 30 minutos, con breve agitación con vórtice cada 10 minutos. Si es necesario para generar una solución clara, la mezcla se incubó adicionalmente en un baño de agua a 90 °C durante 2 horas con breve agitación con vórtice cada 15 minutos. Por separado, para formar la fase acuosa, se mezclaron Tween®80 (70 mg) y glicerol (315 mg) con una solución de citrato de sodio 100 mM pH 6,5 (3,5 ml) y agua (29,8 ml) en un tubo de polipropileno de 50 ml.
Se formó una emulsión de aceite en agua combinando la fase oleosa que contiene aceite de escualeno/DOPC/compuesto y la fase acuosa que contiene Tween®80/glicerol/citrato de sodio seguida de mezcla de alto cizallamiento con un mezclador Polytron® (Kinematica, Luzern CH) durante 5 minutos a 24.000 rpm. A continuación, la emulsión en bruto se sometió a homogeneización a alta presión usando un microfluidizador Microfluidics M-110P Microfluidizer® (Westwood, MA) durante 8 pases a aproximadamente 30.000 psi. El análisis por dispersión de luz dinámica (Malvern NanoS®, Malvern RU) indicó un diámetro medio de gota de aceite de 150 - 175 nm, con un índice de dispersidad de ≤0,15. A continuación, la formulación en nanoemulsión que contenía el compuesto se filtró de forma estéril con un filtro de jeringa estéril de 0,2 micras y se almacenó a 2 - 8 °C en viales de vidrio estériles con tapón de caucho.
Las concentraciones de diversos componentes en la formulación en nanoemulsión final fueron las siguientes: 0,4 mg/ml de compuesto (p/v), 4 % de aceite de escualeno (v/v), 0,5 % de DOPC (p/v), 0,2 % de Tween®80 (p/v), 0,9 % de glicerol (p/v) y acetato de sodio 10 mM. Preparaciones farmacéuticas para la administración in vivo de los compuestos en una formulación en nanoemulsión de aceite en agua de escualeno se preparó antes de su uso diluyendo 1:1 en solución salina tamponada con fosfato de Dulbecco, con mezcla por inversión suave.
Ejemplo B3. Ensayo de Activación Inmunitaria Sistémica in vivo
Se sabe que agonistas de TLR7 y TLRH8 de molécula pequeña derivados de la plantilla privilegiada de 1H-imidazo[4,5-c]quinolina (véase, por ejemplo, Imiquimod, Resiquimod) se distribuyen rápidamente al compartimento sistémico después de inyección intratumoral, subcutánea o intramuscular. La amplia distribución sistémica de estos compuestos agonistas en ratones de tipo silvestre induce respuestas de citocinas dependientes de TLR7, principalmente en las células del bazo y el hígado, que posteriormente se puede detectar en el suero dentro de 3 - 6 horas. El rápido aumento de los biomarcadores de citocinas séricas (por ejemplo, IL-6 e IL-12p40) se puede usar para evaluar la cinética de distribución sistémica de un agonista de TLR administrado localmente.
Se evaluó la cinética de distribución de las composiciones farmacéuticas compuestas por los Compuestos n.° 63-00, 63-17 o 63-10 formulados en 95 % de ace ón
subcutánea en ratones de tipo silvestre. Todos los procedimientos in vivo se llevaron a cabo de acuerdo con los protocolos aprobados por el Comité Institucional de Cuidado y Uso de Animales (IACUC). Los animales se alojaron en una instalación acreditada por la Asociación para la Acreditación y el Cuidado de Animales de Laboratorio (AALAC, Frederick MD). Se obtuvieron ratones BALB/c hembra de tipo silvestre (15 - 20 g) de Envigo (Hayward, CA) y se aclimataron durante 2 - 3 días antes de su uso.
Las composiciones farmacéuticas se realizaron con los tres compuestos, a una concentración final de 200 μg/ml, de manera similar a la descrita en el ejemplo B2-1. A T = 0, se anestesiaron grupos de 3 ratones con isoflurano al 1 % y se les inyectó por vía subcutánea en la almohadilla plantar derecha 5 pg de cada uno de los tres compuestos, o un vehículo de control, en 25 μl de 95 % de aceite de sésamo/5 % de etanol (v/v). A continuación, a T = 3, 6 y 24 horas, tres animales de cada grupo fueron anestesiados con isoflurano al 1 % para facilitar la extracción de sangre por punción cardíaca. Las muestras se procesaron hasta obtener suero y se almacenaron a -20 °C para su posterior análisis.
Se cuantificaron los niveles séricos de IL-6 e IL-12p40 mediante ELISA de cada animal individual para determinar si los compuestos derivatizados con cadenas alquílicas más largas mostraban una velocidad más lenta de distribución sistémica. El Compuesto n.° 63-00 de cadena alquílica no modificada indujo niveles séricos elevados de IL-6 e IL-12p40 a las 3 horas (IL-6 = 772 ± 141 pg/ml, IL-12p40 = 139.767 ± 31.024 pg/ml) y 6 horas (IL-6 = 160 ± 19 pg/ml, IL-12p40 = 142.359 ± 22.350 pg/ml) que volvió a la línea base a las 24 horas (IL-6 = 32 ± 2 pg/ml, IL-12p40 = 7.796 ± 1.545 pg/ml), como se muestra en la figura 1. La variante pentilamino, Compuesto n.° 63-17, mostró una magnitud y una cinética similares de producción inicial de citocinas en el compartimento sérico a las 3 horas (IL-6 = 853 ± 539 pg/ml, IL-12p40 = 84.731 ± 32.530 pg/ml) y a las 6 horas (IL-6 = 414 ± 105 pg/ml, IL-12p40 = 258.645 ± 19.982 μg/ml), así como un retorno a los niveles iniciales a las 24 horas (IL-6 = 33 ± 4 pg/ml, IL-12p40 = 19.546 ± 2.116 pg/ml). La variante tetradecanamida, Compuesto n.° 63-10, que poseía un valor de cLogP sustancialmente más alto, no demostró un aumento detectable en las citocinas séricas a las 3, 6 o 24 horas (3 horas: IL-6 = 49 ± 32 pg/ml, IL-12p40 = 1.012 ± 246 pg/ml; a las 6 horas: IL-6 = 33 ± 4 pg/ml, IL-12p40 = 2.179 ± 597 pg/ml; y a las 24 horas: IL-6 = 32 ± 2 μg/ml, IL-12p40 = 2.436 ± 237 pg/ml). Estos datos son consistentes con la interpretación de que, a pesar de que los tres compuestos demuestran la misma bioactividad agonista de TLR7 in vitro (véanse las tablas B1-1 y B1-2) la mayor hidrofobicidad del Compuesto n.° 63-10, así como su formulación en una composición farmacéutica de aceite de sésamo/etanol promueve la retención de la molécula en el sitio de inyección.
Ejemplo B4. Eficacia antitumoral de agonistas de TLRH8 de cadena alquílica modificados en ratones de tipo silvestre portadores de carcinoma de colon CT26
Se evaluó el efecto de dosis semanales repetidas de composiciones farmacéuticas administradas por vía intratumoral compuestas por el Compuesto n.° 63-10, 63-18 o 63-33 formuladas en 95 % de aceite de sésamo/5 % de etanol (v/v) sobre la inhibición del crecimiento tumoral en Ratones Balb/c portadores de carcinoma de colon CT26 singénico. Todos los procedimientos in vivo se llevaron a cabo de acuerdo con los protocolos aprobados por el Comité Institucional de Cuidado y Uso de Animales (IACUC). Los animales se alojaron en una instalación acreditada por la Asociación para la Acreditación y el Cuidado de Animales de Laboratorio (AALAC, Frederick, MD). Se obtuvieron ratones Balb/c hembra de tipo silvestre (15 - 20 g) de Envigo (Hayward, CA) y se aclimataron durante 2 - 3 días antes de su uso.
Las composiciones farmacéuticas se prepararon con los tres compuestos a concentraciones finales de 5, 50 y 200 μg/ml de manera similar a la descrita en el ejemplo B2-1. Un agonista de TLR9 CpG que previamente ha demostrado eficacia en este modelo de tumor murino se utilizó como control positivo (Wang et al. 2016 PNAS 113:E7240-E7249). El día 0, los ratones se anestesiaron con isoflurano al 1 % y se les inyectaron subcutáneamente en el flanco derecho 80.000 células tumorales CT26 en 200 μl de medio de cultivo RMPI-1640 más suero fetal bovino al 2,5 %. Se permitió que los tumores crecieran hasta que tuvieran ~35 mm3, momento en el cual los animales fueron asignados a grupos para comenzar el tratamiento. A los ratones se les inyectaron semanalmente durante 4 semanas por vía intratumoral 100 μl de una composición farmacéutica que comprendía 20, 5 o 0,5 μg del Compuesto n.° 63-10, o 20 o 5 μg de los Compuestos n.° 63-18 o 63-33, formulados en 95 % aceite de sésamo/5 % etanol (v/v), o un vehículo de control, dos veces por semana durante 3 semanas (días experimentales 9, 12, 16, 19, 23 y 26). El agonista de TLR9 CpG se inyectó por vía intratumoral con 100 μl de una composición farmacéutica compuesta por 50 μg de compuesto formulado en solución salina tamponada con fosfato en el mismo programa posológico. Los tamaños de los tumores se midieron dos veces por semana desde el día 8 hasta el día 30 con calibradores, con volúmenes tumorales calculados usando la fórmula: longitud, multiplicada por anchura, multiplicada por anchura, dividida por 2.
Los Compuestos n.° 63-10, 63-18 y 63-33 demostraron un fuerte control del crecimiento del tumor CT26 en el intervalo de dosis probadas en comparación con el control de vehículo (figura 2). El nivel de control del crecimiento tumoral observado para los Compuestos n.° 63-10, 63-18 y 63-33 fue comparable al del TLR9 CpG. Estos datos demuestran que los agonistas de TLRH8 de la presente invención poseen potentes efectos antitumorales en este sistema de modelo de crecimiento tumoral en ratón singénico que muestra un mayor control del crecimiento tumoral en comparación con el control de vehículo (y son comparables a la inhibición del crecimiento tumoral por TLR9 CpG). Estos datos son consistentes con la interpretación de que la inhibición del crecimiento tumoral para los Compuestos n.° 63-10, 63-18 y 63-33 se correlaciona con su bioactividad agonista de TLR7 in vitro (véanse por ejemplo las tablas B1-1 y B1-2) y es independiente de sus modi
Ejemplo B5. Eficacia antitumoral de agonistas de TLRH8 de cadena alquílica modificada coadministrados con antígenos asociados a tumores en ratones de tipo silvestre portadores de carcinoma de colon CT26 en dos flancos
El efecto de dosis semanales repetidas de composiciones farmacéuticas administradas por vía intratumoral compuestas por el Compuesto n.° 63-10 formulado en una nanoemulsión de aceite en agua a base de escualeno que se coformuló con el péptido AH-1 clase II asociado al tumor CT-26 (secuencia del epítopo inmunodominante del producto gp70 del gen retrovírico endógeno; véase, por ejemplo, Rice J, Buchan S y Stevenson F 2002 J Immunol 169:3908-3913) sobre la inhibición del crecimiento tumoral inyectado y distal se evaluó en ratones Balb/c portadores de carcinoma de colon CT26 portadores de tumores en 2 flancos. Todos los procedimientos in vivo se llevaron a cabo de acuerdo con protocolos aprobados por IACUC. Los animales fueron alojados en una instalación acreditada por la AALAC. Se obtuvieron ratones Balb/c hembra de tipo silvestre (15 - 20 g) de Envigo (Hayward, CA) y se aclimataron durante 2 - 3 días antes de su uso.
Las composiciones farmacéuticas compuestas por nanoemulsiones de aceite en agua a base de escualeno se prepararon generalmente como se describe en el ejemplo B2-2. Además de una nanoemulsión de aceite en agua a base de escualeno de control, se prepararon nanoemulsiones adicionales que incorporaron el Compuesto n.° 63-10 solo a una concentración final de 500 ng/ml, o el Compuesto n.° 63-10 a una concentración final de 500 ng/ml más el péptido AH-1 clase II a una concentración final de 500.000 ng/ml. Para esta última composición farmacéutica, el péptido AH-1 se disolvió inicialmente a una concentración de 2x en solución salina tamponada con fosfato, a continuación se formula en la nanoemulsión de aceite en agua a base de escualeno durante la etapa de mezcla final para producir una concentración final en la nanoemulsión de 500.000 ng/ml. El día 0, los ratones se anestesiaron con isoflurano al 1 % y se les inyectaron por vía subcutánea 80.000 células tumorales CT26 en 200 μl de medio de cultivo RMPI-1640 con suero fetal bovino al 2,5 % en el flanco derecho e izquierdo. Se permitió que los tumores crecieran hasta el día 8, cuando el tamaño promedio de los tumores había alcanzado aproximadamente 50 mm3, momento en el que los ratones se aleatorizaron en grupos y se les inyectaron por vía intratumoral en el tumor del flanco derecho con 100 μl del control de vehículo de nanoemulsión de aceite en agua a base de escualeno, nanoemulsión que contenía 50 ng del Compuesto n.° 63-10, o una nanoemulsión que contenía 50 ng del Compuesto n.° 63-10 más 50.000 ng de péptido antigénico tumoral AH-1. Estas tres composiciones farmacéuticas se inyectaron adicionalmente en el tumor del flanco derecho los días experimentales 12, 16 y 20. A continuación, se midieron los volúmenes tumorales derecho (inyectado) e izquierdo (distal) dos veces por semana desde el día 8 hasta el día 30 con calibradores, con volúmenes tumorales calculados usando la fórmula: longitud, multiplicada por anchura, multiplicada por anchura, dividida por 2.
La composición farmacéutica compuesta por una nanoemulsión de aceite en agua a base de escualeno que contenía 50 ng de los Compuestos n.° 63-10 demostró una tendencia hacia una mayor inhibición del crecimiento tumoral en el tumor inyectado (derecho) en comparación con el control de vehículo de nanoemulsión (figura 3A); sin embargo, esta inhibición del crecimiento tumoral no fue significativamente diferente del control de vehículo en el día 27. Por el contrario, la composición farmacéutica compuesta por una nanoemulsión de aceite en agua a base de escualeno que contenía 50 ng de los Compuestos n.° 63-10 más 50.000 ng del péptido asociado al tumor AH-1 demostró una inhibición del crecimiento tumoral significativamente mayor en el tumor inyectado (derecho) en comparación con el control del vehículo de nanoemulsión el día 27. Adicionalmente, la composición farmacéutica compuesta por una nanoemulsión de aceite en agua a base de escualeno que contenía 50 ng de los Compuestos n.° 63-10 demostró una tendencia hacia una mayor inhibición del crecimiento tumoral en el tumor distal (izquierdo) en comparación con el control de vehículo de nanoemulsión (figura 3B); sin embargo, esta inhibición del crecimiento tumoral no fue significativamente diferente del control de vehículo en el día 23. Por el contrario, la composición farmacéutica compuesta por una nanoemulsión de aceite en agua a base de escualeno que contenía 50 ng de los Compuestos n.° 63-10 más 50.000 ng del péptido asociado al tumor AH-1 demostró una inhibición del crecimiento tumoral significativamente mayor en el tumor distal (izquierdo) en comparación con el control del vehículo de nanoemulsión el día 23. Estos datos son consistentes con la interpretación de que la inhibición del crecimiento tumoral inyectado y distal por el Compuesto n.° 63-10 es superior cuando se administra conjuntamente a las células presentadoras de antígenos en el microentorno tumoral con un antígeno asociado al tumor CT26 añadido exógenamente.
Ejemplo B6. Eficacia antitumoral de los agonistas de TLRH8 de cadena alquílica modificada en combinación con la inhibición del punto de control inmunitario en ratones de tipo silvestre portadores de carcinoma de colon CT26 en doble flanco
El efecto de las composiciones farmacéuticas administradas por vía intratumoral compuestas por el Compuesto n.° 63 10 o 63-33 formulado en 95 % de aceite de sésamo/5 % de etanol (v/v), o el Compuesto n.° 63-00 formulado en solución salina tamponada con fosfato, que se dan en combinación con anticuerpo anti-ratón PD-1 (CD279) administrado por vía intraperitoneal (un inhibidor del punto de control inmunitario; Bio X Cell, Lebanon NH), sobre la inhibición del crecimiento tumoral se evaluó en ratones Balb/c con carcinoma de colon CT26 que portaban tumores en 2 flancos. Todos los procedimientos in vivo se llevaron a cabo de acuerdo con protocolos aprobados por IACUC. Los animales fueron alojados en una instalación acreditada por la AALAC. Se obtuvieron ratones Balb/c hembra de tipo silvestre (15 - 20 g) de Envigo (Hayward, CA) y se aclimataron durante 2 - 3 días antes de su uso
Las composiciones farmacéuticas se prepararon usando el Compuesto n.° 63-10 o 63-33 a concentraciones finales de 50.000 ng/ml de manera similar a la descrita para el ejemplo B2-1. La composición farmacéutica del Compuesto n.° 63 00 se preparó utilizando solución salina tamponada con fosfato a una concentración final de 50.000 ng/ml. El día 0, los ratones se anestesiaron con isoflurano al 1 % y se les inyectaron por vía subcutánea 80.000 células tumorales CT26 en 200 |jl de medio de cultivo RMPI-1640 más suero fetal bovino al 2,5 % en el flanco derecho e izquierdo. Se permitió que los tumores (izquierdo y derecho) crecieran hasta el día 8, cuando los tamaños de los tumores de los flancos derecho e izquierdo habían alcanzado aproximadamente 35 mm3, momento en el que se inyectó a los ratones por vía intraperitoneal 250 jg de anticuerpo anti-PD-1 formulado en solución salina tamponada con fosfato o un control de vehículo de solución salina tamponada con fosfato. Los tratamientos anti-PD-1 se repitieron los días experimentales 12, 15, 19, 22 y 26. En el día experimental 14, cuando los tumores del flanco derecho (inyectado) e izquierdo (distal) habían alcanzado aproximadamente 100 mm3, los ratones se distribuyeron aleatoriamente en grupos de tratamiento. A los grupos de tratamiento anti-PD-1 plus se les inyectó adicionalmente por vía intratumoral en el tumor del flanco derecho solo 100 j l de una composición farmacéutica que comprendía 5000 ng del Compuesto n.° 63-00 en solución salina tamponada con fosfato, 5000 ng del Compuesto n.° 63-10 en 95 % de aceite de sésamo/5 % de etanol (v/v), o 5000 ng del Compuesto No. 63-33 en 95 % de aceite de sésamo/5 % de etanol (v/v). Los tamaños de los tumores se midieron dos veces por semana desde el día 14 hasta el día 29 con calibradores, con volúmenes tumorales calculados usando la fórmula: longitud, multiplicada por anchura, multiplicada por anchura, dividida por 2.
Las composiciones farmacéuticas compuestas por 5000 ng del Compuesto n.° 63-00, 63-10 o 63-33 en combinación con el tratamiento anti-PD-1 demostraron una mayor inhibición del crecimiento tumoral en el tumor inyectado (flanco derecho) en comparación con el tratamiento anti-PD-1 solo (figura 4A). Esta inhibición del crecimiento tumoral fue significativamente diferente del control de tratamiento anti-PD-1 en el día 29 para los Compuestos n.° 63-33 y 63-00. Adicionalmente, las composiciones farmacéuticas compuestas por 5000 ng de los Compuestos n.° 63-00 o 60-33 en combinación con el tratamiento anti-PD-1 demostraron una tendencia hacia una mayor inhibición del crecimiento tumoral en el tumor distal (flanco izquierdo) en comparación con el tratamiento anti-PD-1 solo (figura 4B), aunque esta inhibición del crecimiento tumoral no alcanzó significación estadística a partir del control de tratamiento anti-PD-1 en el día 29. La composición farmacéutica compuesta por 5000 ng del Compuesto n.° 63-10 en combinación con el tratamiento anti-PD-1 no demostró mejora en la inhibición del crecimiento tumoral distal en comparación con el tratamiento anti-PD-1 solo. Estos datos son consistentes con la interpretación de que el Compuesto n.° 63-33, en combinación con el inhibidor del punto de control inmunitario anti-PD-1, es superior para controlar el crecimiento tumoral de CT26 inyectado y distal en comparación con el tratamiento con control de vehículo más anti-PD-1.
Claims (18)
1. Un compuesto de fórmula (J):

o una sal del mismo, en
R0 es -(CH2)mRA o -(CH2)z(C(CH3)2)RA; m es 0, 1, 2 o 3; z es 1 o 2; y RA es cicloalquilo C3-C8 opcionalmente sustituido con de 1 a 4 grupos seleccionados independientemente del grupo que consiste en alquilo C1-C4 , alquileno C1-C4 y halógeno;
X es -NH-;
R1 es alquilo C3-C6 , -(CH2)pOR1a, -(CH2)pNHR1b o -(CH2)pR1c; donde R1a y R1b son independientemente alquilo C1-C3 ; R1c es cicloalquilo C3-C4 ; y p es 1 o 2;
R2 es NHR2a; donde R2a es H, OH, NH2 o metilo;
cada R3 es independientemente halógeno, alquilo C1-C8 , -(alquilen C1-C7)-NH2 ,
o -CH2-fenilen-CH2NH2 ;
q es 0, 1, 2, 3 o 4; y
R4a y R4b son independientemente H o alquilo C1-C8.
2. El compuesto de la reivindicación 1, o una sal del mismo, en donde R0 es -(CH2)mRA.
3. El compuesto de la reivindicación 2, o una sal del mismo, en donde m es 2.
4. El compuesto de la reivindicación 1, o una sal del mismo, en donde R0 es

5. El compuesto de la reivindicación 4, o una sal del mismo, en donde z es 1.
6. El compuesto de una cualquiera de las reivindicaciones 2-5, o una sal del mismo, en donde RA es ciclopropilo, ciclobutilo o ciclopentilo.
7. El compuesto de una cualquiera de las reivindicaciones 2-5, o una sal del mismo, en donde RA es cicloalquilo C3-C6 opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo, metileno y halógeno.
8. El compuesto de la reivindicación 2, o una sal del mismo, en donde m es 1 o 2.
9. El compuesto de la reivindicación 8, o una sal del mismo, en donde RA es cicloalquilo C3-C8.
10. El compuesto de la reivindicación 8 o la reivindicación 9 o una sal del mismo, en donde RA es ciclopropilo opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno.
11. El compuesto de la reivindicación 2, o una sal del mismo, en donde m es 0 y RA es ciclohexilo opcionalmente sustituido con de 1 a 3 grupos seleccionados independientemente del grupo que consiste en metilo y metileno.
12. El compuesto de la reivindicación 1, o una sal del mismo, en donde R0 se selecciona entre el grupo que consiste en:


13. El compuesto de la reivindicación 1, en donde el compuesto se selecciona del grupo que consiste en los compuestos n.° 63-33, 63-35, 63-36 y 63-38 a 63-49 en la tabla 1, o una sal del mismo:




14. Una composición farmacéutica que comprende (i) el compuesto de acuerdo con una cualquiera de las reivindicaciones 1-13 o una sal del mismo; y (ii) un excipiente farmacéuticamente aceptable.
15. La composición farmacéutica de la reivindicación 14, que comprende además un antígeno.
16. La composición farmacéutica de la reivindicación 14 o la reivindicación 15 para su uso en un método de estimulación de una respuesta inmunitaria en un sujeto mamífero, que comprende administrar la composición al sujeto mamífero en una cantidad suficiente para estimular la respuesta inmunitaria en el sujeto mamífero.
17. La composición farmacéutica de la reivindicación 14 o la reivindicación 15 para su uso en un método de tratamiento o prevención de una enfermedad infecciosa en un sujeto mamífero, que comprende administrar la composición al sujeto mamífero en una cantidad suficiente para tratar o prevenir la enfermedad infecciosa en el sujeto mamífero.
18. La composición farmacéutica de la reivindicación 14 o la reivindicación 15 para su uso en un método de tratamiento del cáncer en un sujeto mamífero, que comprende administrar la composición al sujeto mamífero en una cantidad suficiente para tratar el cáncer en el sujeto mamífero.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762548848P | 2017-08-22 | 2017-08-22 | |
| PCT/US2018/047323 WO2019040491A1 (en) | 2017-08-22 | 2018-08-21 | MODIFIED ALKYL CHAIN IMIDAZOQUINOLINE TLR7 / 8 AGONIST COMPOUNDS AND USES THEREOF |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2951817T3 true ES2951817T3 (es) | 2023-10-25 |
Family
ID=63638354
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18773000T Active ES2951817T3 (es) | 2017-08-22 | 2018-08-21 | Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLR7/8 y usos de los mismos |
Country Status (5)
| Country | Link |
|---|---|
| US (4) | US10618896B2 (es) |
| EP (2) | EP4257198A3 (es) |
| JP (3) | JP7386536B2 (es) |
| ES (1) | ES2951817T3 (es) |
| WO (1) | WO2019040491A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2951817T3 (es) | 2017-08-22 | 2023-10-25 | Dynavax Tech Corp | Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLR7/8 y usos de los mismos |
| US10722591B2 (en) | 2017-11-14 | 2020-07-28 | Dynavax Technologies Corporation | Cleavable conjugates of TLR7/8 agonist compounds, methods for preparation, and uses thereof |
| WO2020162705A1 (ko) | 2019-02-08 | 2020-08-13 | 성균관대학교산학협력단 | 톨-유사 수용체 7 또는 8 작용자와 콜레스테롤의 결합체 및 그 용도 |
| BR112022008039A2 (pt) | 2019-10-29 | 2022-07-12 | Prime Reach Trading Ltd | Compostos de 4-amino-imidazoquinolina e usos relacionados |
| CA3170551A1 (en) | 2020-03-02 | 2021-09-10 | Yong Taik Lim | Live-pathogen-mimetic nanoparticles based on pathogen cell wall skeleton, and production method thereof |
| US12564630B2 (en) * | 2020-05-05 | 2026-03-03 | Virovax Llc | Vaccine adjuvants |
| US20230277525A1 (en) | 2020-08-04 | 2023-09-07 | Progeneer Inc | Conjugate of functional drug and toll-like receptor 7 or 8 agonist of which active site is temporarily inactivated and use thereof |
| JP7690221B2 (ja) | 2020-08-04 | 2025-06-10 | プロジェニア インコーポレイテッド | 動力学的に作用するアジュバントアンサンブル |
| US20230346924A1 (en) | 2020-08-04 | 2023-11-02 | Progeneer Inc. | Mrna vaccine comprising adjuvant capable of kinetic control |
| CN113967256B (zh) * | 2021-10-26 | 2022-09-30 | 山东大学 | 一种具有光热-化疗-免疫功能的纳米粒及其制备方法和应用 |
| WO2023081237A1 (en) * | 2021-11-03 | 2023-05-11 | Regents Of The University Of Minnesota | Toll-like receptor agonists and antagonists and uses thereof |
| CN119136795A (zh) * | 2021-11-10 | 2024-12-13 | 芝加哥大学 | 具有增强的疗效和降低的毒性的nfkb激活剂的作为佐剂的小分子免疫增强剂的缀合物 |
| CN116731015B (zh) * | 2023-05-18 | 2024-09-27 | 深圳威科森生物医药科技有限公司 | 咪唑并喹啉化合物及其制备方法、应用和组合物 |
| EP4731632A1 (en) * | 2023-06-21 | 2026-04-29 | The Trustees of The University of Pennsylvania | Toll-like receptor (tlr) agonist lipidoid compounds, lipid nanoparticles (lnps) comprising the same, and methods of use thereof |
| CN119320387B (zh) * | 2024-12-19 | 2025-06-27 | 杭州天龙药业有限公司 | 基于Toll样受体激动剂的疫苗佐剂脂质化合物及其应用 |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL73534A (en) | 1983-11-18 | 1990-12-23 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinoline-4-amines,their preparation and pharmaceutical compositions containing certain such compounds |
| JPH09500128A (ja) | 1993-07-15 | 1997-01-07 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | イミダゾ〔4,5−c〕ピリジン−4−アミン |
| JPH1180156A (ja) | 1997-09-04 | 1999-03-26 | Hokuriku Seiyaku Co Ltd | 1−(置換アリール)アルキル−1h−イミダゾピリジン−4−アミン誘導体 |
| UA67760C2 (uk) | 1997-12-11 | 2004-07-15 | Міннесота Майнінг Енд Мануфакчурінг Компані | Імідазонафтиридин та тетрагідроімідазонафтиридин, фармацевтична композиція, спосіб індукування біосинтезу цитокінів та спосіб лікування вірусної інфекції, проміжні сполуки |
| US6180095B1 (en) | 1997-12-17 | 2001-01-30 | Enzon, Inc. | Polymeric prodrugs of amino- and hydroxyl-containing bioactive agents |
| US6110929A (en) | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| US7238368B2 (en) | 1999-04-23 | 2007-07-03 | Alza Corporation | Releasable linkage and compositions containing same |
| ATE340592T1 (de) | 1999-04-23 | 2006-10-15 | Alza Corp | Konjugate enthaltend eine spaltbare bindung zur anwendung in einem liposom |
| US6677349B1 (en) | 2001-12-21 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| DK1545597T3 (da) | 2002-08-15 | 2011-01-31 | 3M Innovative Properties Co | Immunstimulerende sammensætninger og fremgangsmåde til stimulering af en immunrespons |
| JP2006503068A (ja) | 2002-09-26 | 2006-01-26 | スリーエム イノベイティブ プロパティズ カンパニー | 1h−イミダゾダイマー |
| CA2510375A1 (en) | 2002-12-20 | 2004-07-15 | 3M Innovative Properties Company | Aryl / hetaryl substituted imidazoquinolines |
| EP2572714A1 (en) | 2002-12-30 | 2013-03-27 | 3M Innovative Properties Company | Immunostimulatory Combinations |
| JP2006517974A (ja) | 2003-02-13 | 2006-08-03 | スリーエム イノベイティブ プロパティズ カンパニー | Irm化合物およびトル様受容体8に関する方法および組成物 |
| EP1615665A4 (en) | 2003-04-10 | 2010-10-06 | 3M Innovative Properties Co | DISTRIBUTION OF THE IMMUNE RESPONSE MODIFYING LINKS |
| US20040265351A1 (en) | 2003-04-10 | 2004-12-30 | Miller Richard L. | Methods and compositions for enhancing immune response |
| US7799800B2 (en) | 2003-08-14 | 2010-09-21 | 3M Innovative Properties Company | Lipid-modified immune response modifiers |
| PT1687305E (pt) | 2003-11-21 | 2008-10-17 | Novartis Ag | Derivados de 1h-imidazoquinolina como inibidores de proteína quinase |
| US7884207B2 (en) | 2004-06-18 | 2011-02-08 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| WO2007079086A1 (en) | 2005-12-28 | 2007-07-12 | Coley Pharmaceutical Group, Inc. | Pyrazoloalkyl substituted imidazo ring compounds and methods |
| EP1988896A4 (en) | 2006-02-22 | 2011-07-27 | 3M Innovative Properties Co | CONJUGATES TO MODIFY IMMUNE REACTIONS |
| JP2013512859A (ja) | 2009-12-03 | 2013-04-18 | 大日本住友製薬株式会社 | トール様受容体(tlr)を介して作用するイミダゾキノリン |
| US8728486B2 (en) | 2011-05-18 | 2014-05-20 | University Of Kansas | Toll-like receptor-7 and -8 modulatory 1H imidazoquinoline derived compounds |
| WO2013166110A1 (en) | 2012-05-02 | 2013-11-07 | Yale University | Tlr-agonist-conjugated antibody recruiting molecules (tlr_arms) |
| EP2674170B1 (en) | 2012-06-15 | 2014-11-19 | Invivogen | Novel compositions of TLR7 and/or TLR8 agonists conjugated to lipids |
| EP2732825B1 (en) | 2012-11-19 | 2015-07-01 | Invivogen | Conjugates of a TLR7 and/or TLR8 agonist and a TLR2 agonist |
| WO2014113634A1 (en) | 2013-01-17 | 2014-07-24 | University Of Kansas | Toll-like receptor 2-agonistic lipopeptides, and method of making the same |
| EP2769738B1 (en) | 2013-02-22 | 2016-07-20 | Invivogen | Conjugated TLR7 and/or TLR8 and TLR2 polycationic agonists |
| US10471139B2 (en) | 2013-08-15 | 2019-11-12 | The University Of Kansas | Toll-like receptor agonists |
| GB201321242D0 (en) | 2013-12-02 | 2014-01-15 | Immune Targeting Systems Its Ltd | Immunogenic compound |
| DK3092256T3 (da) | 2014-01-10 | 2022-06-20 | Birdie Biopharmaceuticals Inc | Forbindelser og sammensætninger til immunterapi |
| ES2908150T3 (es) | 2014-05-01 | 2022-04-27 | Novartis Ag | Compuestos y composiciones como agonistas del receptor de tipo Toll 7 |
| GB201418004D0 (en) | 2014-10-10 | 2014-11-26 | Isis Innovation | Polymer adjuvant |
| WO2017044803A1 (en) | 2015-09-09 | 2017-03-16 | The United States Of America, As Represented By The Secretary Department Of Health And Human Service | Expression vector delivery system and use thereof for inducing an immune response |
| EP3356371A4 (en) | 2015-09-29 | 2020-06-24 | The University of Chicago | POLYMER CONJUGATE VACCINE |
| EP3368092B9 (en) | 2015-10-29 | 2020-07-29 | Novartis AG | Antibody conjugates comprising toll-like receptor agonist |
| AU2017376460A1 (en) | 2016-12-13 | 2019-06-20 | Bolt Biotherapeutics, Inc. | Antibody adjuvant conjugates |
| US10328895B2 (en) | 2017-03-17 | 2019-06-25 | Autoliv Asp, Inc. | Seatbelt pretensioning retractor assembly including a pretensioner rod |
| AR111651A1 (es) | 2017-04-28 | 2019-08-07 | Novartis Ag | Conjugados de anticuerpos que comprenden agonistas del receptor de tipo toll y terapias de combinación |
| US20180311334A1 (en) | 2017-04-29 | 2018-11-01 | Yale University | Compositions and Methods for Inducing an Immune Response |
| ES2951817T3 (es) | 2017-08-22 | 2023-10-25 | Dynavax Tech Corp | Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLR7/8 y usos de los mismos |
| BR112020004505A2 (pt) | 2017-09-06 | 2020-09-15 | BioNTech SE | imidazoquinolinas substituídas como agonistas de tlr7 |
| US10722591B2 (en) | 2017-11-14 | 2020-07-28 | Dynavax Technologies Corporation | Cleavable conjugates of TLR7/8 agonist compounds, methods for preparation, and uses thereof |
-
2018
- 2018-08-21 ES ES18773000T patent/ES2951817T3/es active Active
- 2018-08-21 WO PCT/US2018/047323 patent/WO2019040491A1/en not_active Ceased
- 2018-08-21 EP EP23177052.0A patent/EP4257198A3/en active Pending
- 2018-08-21 EP EP18773000.7A patent/EP3672969B1/en active Active
- 2018-08-21 US US16/107,605 patent/US10618896B2/en active Active
- 2018-08-21 JP JP2020511364A patent/JP7386536B2/ja active Active
-
2020
- 2020-03-02 US US16/806,733 patent/US11124510B2/en active Active
-
2021
- 2021-08-16 US US17/403,312 patent/US11981670B2/en active Active
-
2023
- 2023-07-26 JP JP2023121658A patent/JP2023129623A/ja active Pending
-
2024
- 2024-04-08 US US18/629,809 patent/US20240368155A1/en active Pending
-
2025
- 2025-05-20 JP JP2025083816A patent/JP2025131620A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US11124510B2 (en) | 2021-09-21 |
| US20220033400A1 (en) | 2022-02-03 |
| US10618896B2 (en) | 2020-04-14 |
| US20190062329A1 (en) | 2019-02-28 |
| US20240368155A1 (en) | 2024-11-07 |
| EP3672969B1 (en) | 2023-07-05 |
| WO2019040491A1 (en) | 2019-02-28 |
| EP4257198A3 (en) | 2023-10-18 |
| EP3672969A1 (en) | 2020-07-01 |
| JP7386536B2 (ja) | 2023-11-27 |
| JP2025131620A (ja) | 2025-09-09 |
| US11981670B2 (en) | 2024-05-14 |
| JP2020531530A (ja) | 2020-11-05 |
| US20200199124A1 (en) | 2020-06-25 |
| EP4257198A2 (en) | 2023-10-11 |
| JP2023129623A (ja) | 2023-09-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2951817T3 (es) | Derivados de imidazoquinolina de cadena alquílica modificada como agonistas de TLR7/8 y usos de los mismos | |
| JP6710727B2 (ja) | 脂質付加された免疫反応調節化合物の組成物、製剤及び方法 | |
| CN108290845B (zh) | 嘧啶化合物 | |
| US11833247B2 (en) | Vaccine adjuvant formulation | |
| KR102283760B1 (ko) | 펩티드 접합된 입자 | |
| JP2021531266A (ja) | 免疫応答を調節可能な金属含有製剤のための組成物および方法 | |
| ES2389080T3 (es) | Fármaco que presenta un ligando de células reguladoras contenido en el liposoma | |
| CN114206378B (zh) | 基于tlr受体配体的疫苗佐剂 | |
| Verma et al. | CpG ODNs: Precision Immunotherapy and Recent Trends in Nano-Enabled Nucleotide Delivery | |
| EA046871B1 (ru) | Адъюванты вакцин на основе лигандов рецепторов tlr | |
| JP2014122165A (ja) | 免疫賦活剤 |