ES2956471T3 - Tratamientos del angioedema hereditario - Google Patents
Tratamientos del angioedema hereditario Download PDFInfo
- Publication number
- ES2956471T3 ES2956471T3 ES20734283T ES20734283T ES2956471T3 ES 2956471 T3 ES2956471 T3 ES 2956471T3 ES 20734283 T ES20734283 T ES 20734283T ES 20734283 T ES20734283 T ES 20734283T ES 2956471 T3 ES2956471 T3 ES 2956471T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- solvate
- pharmaceutically acceptable
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010019860 Hereditary angioedema Diseases 0.000 title claims abstract description 333
- 238000011282 treatment Methods 0.000 title claims abstract description 193
- 150000001875 compounds Chemical class 0.000 claims description 368
- 230000001154 acute effect Effects 0.000 claims description 127
- 108090000113 Plasma Kallikrein Proteins 0.000 claims description 94
- 150000003839 salts Chemical class 0.000 claims description 69
- 239000012453 solvate Substances 0.000 claims description 61
- 238000003776 cleavage reaction Methods 0.000 claims description 43
- 230000007017 scission Effects 0.000 claims description 43
- 208000024891 symptom Diseases 0.000 claims description 40
- 238000000034 method Methods 0.000 claims description 31
- 230000004913 activation Effects 0.000 claims description 18
- 102100034869 Plasma kallikrein Human genes 0.000 claims description 15
- 108010080865 Factor XII Proteins 0.000 claims description 13
- 102000000429 Factor XII Human genes 0.000 claims description 13
- 230000008961 swelling Effects 0.000 claims description 10
- 108010071241 Factor XIIa Proteins 0.000 claims description 8
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 claims description 8
- 208000004998 Abdominal Pain Diseases 0.000 claims description 7
- 239000006186 oral dosage form Substances 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- 206010061218 Inflammation Diseases 0.000 claims description 6
- 230000003466 anti-cipated effect Effects 0.000 claims description 6
- 230000004054 inflammatory process Effects 0.000 claims description 6
- 229920000168 Microcrystalline cellulose Polymers 0.000 claims description 5
- 239000000314 lubricant Substances 0.000 claims description 5
- 229940016286 microcrystalline cellulose Drugs 0.000 claims description 5
- 235000019813 microcrystalline cellulose Nutrition 0.000 claims description 5
- 239000008108 microcrystalline cellulose Substances 0.000 claims description 5
- 210000001519 tissue Anatomy 0.000 claims description 5
- 229920002785 Croscarmellose sodium Polymers 0.000 claims description 4
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 claims description 4
- 239000011230 binding agent Substances 0.000 claims description 4
- 229960001681 croscarmellose sodium Drugs 0.000 claims description 4
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims description 4
- 239000007884 disintegrant Substances 0.000 claims description 4
- 235000019359 magnesium stearate Nutrition 0.000 claims description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 4
- 206010015150 Erythema Diseases 0.000 claims description 3
- 206010015216 Erythema marginatum Diseases 0.000 claims description 3
- 208000014674 injury Diseases 0.000 claims description 3
- 230000003340 mental effect Effects 0.000 claims description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 3
- 230000002250 progressing effect Effects 0.000 claims description 3
- 230000008733 trauma Effects 0.000 claims description 3
- 206010012735 Diarrhoea Diseases 0.000 claims description 2
- 206010013952 Dysphonia Diseases 0.000 claims description 2
- 206010013975 Dyspnoeas Diseases 0.000 claims description 2
- 206010019233 Headaches Diseases 0.000 claims description 2
- 208000010473 Hoarseness Diseases 0.000 claims description 2
- 206010027940 Mood altered Diseases 0.000 claims description 2
- 208000000112 Myalgia Diseases 0.000 claims description 2
- 206010028813 Nausea Diseases 0.000 claims description 2
- 206010047700 Vomiting Diseases 0.000 claims description 2
- 231100000869 headache Toxicity 0.000 claims description 2
- 230000007510 mood change Effects 0.000 claims description 2
- 208000013465 muscle pain Diseases 0.000 claims description 2
- 230000008693 nausea Effects 0.000 claims description 2
- 230000008673 vomiting Effects 0.000 claims description 2
- 206010042674 Swelling Diseases 0.000 claims 1
- 229940126155 plasma kallikrein inhibitor Drugs 0.000 abstract description 6
- 102000003827 Plasma Kallikrein Human genes 0.000 description 77
- 102100035792 Kininogen-1 Human genes 0.000 description 64
- 108010000487 High-Molecular-Weight Kininogen Proteins 0.000 description 57
- 239000003826 tablet Substances 0.000 description 55
- 230000000694 effects Effects 0.000 description 47
- 229960000633 dextran sulfate Drugs 0.000 description 43
- 239000003814 drug Substances 0.000 description 29
- 229940079593 drug Drugs 0.000 description 28
- 229940068196 placebo Drugs 0.000 description 27
- 239000000902 placebo Substances 0.000 description 27
- 239000003112 inhibitor Substances 0.000 description 23
- 230000005764 inhibitory process Effects 0.000 description 20
- 238000012360 testing method Methods 0.000 description 20
- 238000003556 assay Methods 0.000 description 19
- 108700040183 Complement C1 Inhibitor Proteins 0.000 description 18
- 102000055157 Complement C1 Inhibitor Human genes 0.000 description 18
- 238000011321 prophylaxis Methods 0.000 description 18
- 238000001994 activation Methods 0.000 description 17
- 238000011156 evaluation Methods 0.000 description 16
- 239000000523 sample Substances 0.000 description 16
- 238000004458 analytical method Methods 0.000 description 13
- 229940009550 c1 esterase inhibitor Drugs 0.000 description 13
- 230000003285 pharmacodynamic effect Effects 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 239000000243 solution Substances 0.000 description 13
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 230000003197 catalytic effect Effects 0.000 description 12
- 230000036470 plasma concentration Effects 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 230000004064 dysfunction Effects 0.000 description 11
- 239000007941 film coated tablet Substances 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 10
- 239000008280 blood Substances 0.000 description 10
- 230000003389 potentiating effect Effects 0.000 description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 239000000499 gel Substances 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 208000028185 Angioedema Diseases 0.000 description 8
- 229940127379 Kallikrein Inhibitors Drugs 0.000 description 8
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 238000003119 immunoblot Methods 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 230000035772 mutation Effects 0.000 description 8
- 230000001960 triggered effect Effects 0.000 description 8
- 101800004538 Bradykinin Proteins 0.000 description 7
- QXZGBUJJYSLZLT-UHFFFAOYSA-N H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH Natural products NC(N)=NCCCC(N)C(=O)N1CCCC1C(=O)N1C(C(=O)NCC(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CO)C(=O)N2C(CCC2)C(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CCCN=C(N)N)C(O)=O)CCC1 QXZGBUJJYSLZLT-UHFFFAOYSA-N 0.000 description 7
- 206010020751 Hypersensitivity Diseases 0.000 description 7
- 102100027637 Plasma protease C1 inhibitor Human genes 0.000 description 7
- 208000026935 allergic disease Diseases 0.000 description 7
- QXZGBUJJYSLZLT-FDISYFBBSA-N bradykinin Chemical compound NC(=N)NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)CCC1 QXZGBUJJYSLZLT-FDISYFBBSA-N 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 239000007850 fluorescent dye Substances 0.000 description 7
- 238000003018 immunoassay Methods 0.000 description 7
- 229950005287 lanadelumab Drugs 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- UPSFMJHZUCSEHU-JYGUBCOQSA-N n-[(2s,3r,4r,5s,6r)-2-[(2r,3s,4r,5r,6s)-5-acetamido-4-hydroxy-2-(hydroxymethyl)-6-(4-methyl-2-oxochromen-7-yl)oxyoxan-3-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)oxan-3-yl]acetamide Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@H]1[C@H](O)[C@@H](NC(C)=O)[C@H](OC=2C=C3OC(=O)C=C(C)C3=CC=2)O[C@@H]1CO UPSFMJHZUCSEHU-JYGUBCOQSA-N 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 102000002397 Kinins Human genes 0.000 description 6
- 108010093008 Kinins Proteins 0.000 description 6
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- 108050007539 Plasma protease C1 inhibitor Proteins 0.000 description 6
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 6
- 230000002411 adverse Effects 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 235000019439 ethyl acetate Nutrition 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 230000001174 ascending effect Effects 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 230000001055 chewing effect Effects 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 108010011867 ecallantide Proteins 0.000 description 5
- 230000002255 enzymatic effect Effects 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 230000009246 food effect Effects 0.000 description 5
- -1 for example Proteins 0.000 description 5
- VBGWSQKGUZHFPS-VGMMZINCSA-N kalbitor Chemical compound C([C@H]1C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]2C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=3C=CC=CC=3)C(=O)N[C@H](C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)NCC(=O)NCC(=O)N[C@H]3CSSC[C@H](NC(=O)[C@@H]4CCCN4C(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CO)NC(=O)[C@H](CC=4NC=NC=4)NC(=O)[C@H](CCSC)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCC(O)=O)CSSC[C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC3=O)CSSC2)C(=O)N[C@@H]([C@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=2NC=NC=2)C(=O)N2CCC[C@H]2C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N1)[C@@H](C)CC)[C@H](C)O)=O)[C@@H](C)CC)C1=CC=CC=C1 VBGWSQKGUZHFPS-VGMMZINCSA-N 0.000 description 5
- 108090000765 processed proteins & peptides Proteins 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 206010010904 Convulsion Diseases 0.000 description 4
- 208000005139 Hereditary Angioedema Types I and II Diseases 0.000 description 4
- 101001081555 Homo sapiens Plasma protease C1 inhibitor Proteins 0.000 description 4
- 102000001399 Kallikrein Human genes 0.000 description 4
- 108060005987 Kallikrein Proteins 0.000 description 4
- 230000007815 allergy Effects 0.000 description 4
- 229940088949 cinryze Drugs 0.000 description 4
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 4
- 231100000673 dose–response relationship Toxicity 0.000 description 4
- 230000002068 genetic effect Effects 0.000 description 4
- 102000044507 human SERPING1 Human genes 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- 230000001568 sexual effect Effects 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000007916 tablet composition Substances 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- OINHGEBQNPOQFY-UHFFFAOYSA-N 1-[[4-(hydroxymethyl)phenyl]methyl]pyridin-2-one Chemical compound C1=CC(CO)=CC=C1CN1C(=O)C=CC=C1 OINHGEBQNPOQFY-UHFFFAOYSA-N 0.000 description 3
- UXNXMBYCBRBRFD-MUUNZHRXSA-N 2-[3-(aminomethyl)phenyl]-N-[5-[(R)-(3-cyanophenyl)-(cyclopropylmethylamino)methyl]-2-fluorophenyl]-5-(trifluoromethyl)pyrazole-3-carboxamide Chemical compound NCc1cccc(c1)-n1nc(cc1C(=O)Nc1cc(ccc1F)[C@H](NCC1CC1)c1cccc(c1)C#N)C(F)(F)F UXNXMBYCBRBRFD-MUUNZHRXSA-N 0.000 description 3
- MXLCESBWIHSGLC-UHFFFAOYSA-N 3-fluoro-4-methoxypyridine-2-carbonitrile Chemical compound COC1=CC=NC(C#N)=C1F MXLCESBWIHSGLC-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 208000002193 Pain Diseases 0.000 description 3
- 208000037048 Prodromal Symptoms Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 230000003187 abdominal effect Effects 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000008186 active pharmaceutical agent Substances 0.000 description 3
- 235000015111 chews Nutrition 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 230000035487 diastolic blood pressure Effects 0.000 description 3
- 238000001647 drug administration Methods 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 235000021471 food effect Nutrition 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 239000001087 glyceryl triacetate Substances 0.000 description 3
- 235000013773 glyceryl triacetate Nutrition 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 230000009610 hypersensitivity Effects 0.000 description 3
- 229960003943 hypromellose Drugs 0.000 description 3
- QURWXBZNHXJZBE-SKXRKSCCSA-N icatibant Chemical compound NC(N)=NCCC[C@@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2SC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@H](CC3=CC=CC=C3C2)C(=O)N2[C@@H](C[C@@H]3CCCC[C@@H]32)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)C[C@@H](O)C1 QURWXBZNHXJZBE-SKXRKSCCSA-N 0.000 description 3
- 108700023918 icatibant Proteins 0.000 description 3
- 229960001062 icatibant Drugs 0.000 description 3
- 230000002109 interictal effect Effects 0.000 description 3
- 229960001021 lactose monohydrate Drugs 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- DISUFGSPBQZCJO-UHFFFAOYSA-N methyl 3-(methoxymethyl)-1-[[4-[(2-oxopyridin-1-yl)methyl]phenyl]methyl]pyrazole-4-carboxylate Chemical compound COCC1=NN(C=C1C(=O)OC)CC1=CC=C(C=C1)CN1C(C=CC=C1)=O DISUFGSPBQZCJO-UHFFFAOYSA-N 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 230000036407 pain Effects 0.000 description 3
- 230000002093 peripheral effect Effects 0.000 description 3
- 229940012957 plasmin Drugs 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000000634 powder X-ray diffraction Methods 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 230000036387 respiratory rate Effects 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 239000003001 serine protease inhibitor Substances 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- WWWHABZPALNMBR-UHFFFAOYSA-N tert-butyl N-[(3-fluoro-4-methoxypyridin-2-yl)methyl]carbamate Chemical compound C(C)(C)(C)OC(NCC1=NC=CC(=C1F)OC)=O WWWHABZPALNMBR-UHFFFAOYSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000036962 time dependent Effects 0.000 description 3
- 239000004408 titanium dioxide Substances 0.000 description 3
- 229960005196 titanium dioxide Drugs 0.000 description 3
- 229960002622 triacetin Drugs 0.000 description 3
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 2
- GBICOISGPUROBJ-UHFFFAOYSA-N 1-[[4-(chloromethyl)phenyl]methyl]pyridin-2-one Chemical compound ClCC1=CC=C(CN2C=CC=CC2=O)C=C1 GBICOISGPUROBJ-UHFFFAOYSA-N 0.000 description 2
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 2
- VRMNRFURXHSQPQ-UHFFFAOYSA-N 3-(methoxymethyl)-1-[[4-[(2-oxopyridin-1-yl)methyl]phenyl]methyl]pyrazole-4-carboxylic acid Chemical compound COCC1=NN(C=C1C(=O)O)CC1=CC=C(C=C1)CN1C(C=CC=C1)=O VRMNRFURXHSQPQ-UHFFFAOYSA-N 0.000 description 2
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 2
- 108010082126 Alanine transaminase Proteins 0.000 description 2
- 208000019901 Anxiety disease Diseases 0.000 description 2
- 108010003415 Aspartate Aminotransferases Proteins 0.000 description 2
- 102000004625 Aspartate Aminotransferases Human genes 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 102000010631 Kininogens Human genes 0.000 description 2
- 108010077861 Kininogens Proteins 0.000 description 2
- KGMPDQIYDKKXRD-UHFFFAOYSA-N N-[(3-fluoro-4-methoxypyridin-2-yl)methyl]-3-(methoxymethyl)-1-[[4-[(2-oxopyridin-1-yl)methyl]phenyl]methyl]pyrazole-4-carboxamide Chemical compound FC=1C(=NC=CC=1OC)CNC(=O)C=1C(=NN(C=1)CC1=CC=C(C=C1)CN1C(C=CC=C1)=O)COC KGMPDQIYDKKXRD-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 239000002033 PVDF binder Substances 0.000 description 2
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 2
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 2
- 101150097162 SERPING1 gene Proteins 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- 208000024780 Urticaria Diseases 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 150000001409 amidines Chemical class 0.000 description 2
- 239000003098 androgen Substances 0.000 description 2
- 229940030486 androgens Drugs 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 230000036506 anxiety Effects 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical class NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 229940075791 berinert Drugs 0.000 description 2
- 229940121533 berotralstat Drugs 0.000 description 2
- 230000002146 bilateral effect Effects 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 230000036760 body temperature Effects 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 150000001793 charged compounds Chemical class 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 239000003433 contraceptive agent Substances 0.000 description 2
- 229940109239 creatinine Drugs 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 229940000406 drug candidate Drugs 0.000 description 2
- 229960001174 ecallantide Drugs 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 238000001952 enzyme assay Methods 0.000 description 2
- 239000002532 enzyme inhibitor Substances 0.000 description 2
- 229940125532 enzyme inhibitor Drugs 0.000 description 2
- 229940011871 estrogen Drugs 0.000 description 2
- 239000000262 estrogen Substances 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 230000001815 facial effect Effects 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 238000001502 gel electrophoresis Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000003054 hormonal effect Effects 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 238000010191 image analysis Methods 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 2
- 229940018902 kalbitor Drugs 0.000 description 2
- 238000009533 lab test Methods 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000002547 new drug Substances 0.000 description 2
- 231100001079 no serious adverse effect Toxicity 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 230000002028 premature Effects 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- 230000000306 recurrent effect Effects 0.000 description 2
- 229940127558 rescue medication Drugs 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 210000002345 respiratory system Anatomy 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 208000011117 substance-related disease Diseases 0.000 description 2
- 150000003890 succinate salts Chemical class 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 108010036927 trypsin-like serine protease Proteins 0.000 description 2
- 230000008728 vascular permeability Effects 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical class CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- YUHZIUAREWNXJT-UHFFFAOYSA-N (2-fluoropyridin-3-yl)boronic acid Chemical class OB(O)C1=CC=CN=C1F YUHZIUAREWNXJT-UHFFFAOYSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- PGOHTUIFYSHAQG-LJSDBVFPSA-N (2S)-6-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-4-methylsulfanylbutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-methylpentanoyl]amino]-3-sulfanylpropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-oxopentanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-5-oxopentanoyl]amino]hexanoic acid Chemical compound CSCC[C@H](N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(O)=O PGOHTUIFYSHAQG-LJSDBVFPSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- ABEXEQSGABRUHS-UHFFFAOYSA-N 16-methylheptadecyl 16-methylheptadecanoate Chemical compound CC(C)CCCCCCCCCCCCCCCOC(=O)CCCCCCCCCCCCCCC(C)C ABEXEQSGABRUHS-UHFFFAOYSA-N 0.000 description 1
- ZVXKYWHJBYIYNI-UHFFFAOYSA-N 1h-pyrazole-4-carboxamide Chemical compound NC(=O)C=1C=NNC=1 ZVXKYWHJBYIYNI-UHFFFAOYSA-N 0.000 description 1
- GLDQAMYCGOIJDV-UHFFFAOYSA-N 2,3-dihydroxybenzoic acid Chemical class OC(=O)C1=CC=CC(O)=C1O GLDQAMYCGOIJDV-UHFFFAOYSA-N 0.000 description 1
- VHVPQPYKVGDNFY-DFMJLFEVSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2r,4s)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-DFMJLFEVSA-N 0.000 description 1
- AAHXVRZINNDWAF-UHFFFAOYSA-N 2-bromo-3-fluoro-4-methoxypyridine Chemical compound COC1=CC=NC(Br)=C1F AAHXVRZINNDWAF-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical class C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 125000004070 6 membered heterocyclic group Chemical group 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- 102000004411 Antithrombin III Human genes 0.000 description 1
- 108090000935 Antithrombin III Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 229940122155 Bradykinin receptor antagonist Drugs 0.000 description 1
- 201000007120 C1 inhibitor deficiency Diseases 0.000 description 1
- QAGYKUNXZHXKMR-UHFFFAOYSA-N CPD000469186 Natural products CC1=C(O)C=CC=C1C(=O)NC(C(O)CN1C(CC2CCCCC2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 208000004652 Cardiovascular Abnormalities Diseases 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 208000005487 Cytochrome P-450 CYP2C9 Inhibitors Diseases 0.000 description 1
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 1
- 208000009011 Cytochrome P-450 CYP3A Inhibitors Diseases 0.000 description 1
- 102100039205 Cytochrome P450 3A4 Human genes 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000028782 Hereditary disease Diseases 0.000 description 1
- 101000975003 Homo sapiens Kallistatin Proteins 0.000 description 1
- 101001091365 Homo sapiens Plasma kallikrein Proteins 0.000 description 1
- 101001077723 Homo sapiens Serine protease inhibitor Kazal-type 6 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 235000017309 Hypericum perforatum Nutrition 0.000 description 1
- 244000141009 Hypericum perforatum Species 0.000 description 1
- 206010073257 Idiopathic angioedema Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010022052 Injection site bruising Diseases 0.000 description 1
- 206010022061 Injection site erythema Diseases 0.000 description 1
- 206010022086 Injection site pain Diseases 0.000 description 1
- 241000764238 Isis Species 0.000 description 1
- 229940122920 Kallikrein inhibitor Drugs 0.000 description 1
- 102100038297 Kallikrein-1 Human genes 0.000 description 1
- 101710176219 Kallikrein-1 Proteins 0.000 description 1
- 102100023012 Kallistatin Human genes 0.000 description 1
- 150000000994 L-ascorbates Chemical class 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000007177 Left Ventricular Hypertrophy Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000024556 Mendelian disease Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 206010028116 Mucosal inflammation Diseases 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 101100028920 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) cfp gene Proteins 0.000 description 1
- QSLJIVKCVHQPLV-PEMPUTJUSA-N Oxandrin Chemical compound C([C@@H]1CC2)C(=O)OC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@](C)(O)[C@@]2(C)CC1 QSLJIVKCVHQPLV-PEMPUTJUSA-N 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 206010033647 Pancreatitis acute Diseases 0.000 description 1
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 241000233805 Phoenix Species 0.000 description 1
- 102100038124 Plasminogen Human genes 0.000 description 1
- 108010051456 Plasminogen Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 206010053262 Skin swelling Diseases 0.000 description 1
- LKAJKIOFIWVMDJ-IYRCEVNGSA-N Stanazolol Chemical compound C([C@@H]1CC[C@H]2[C@@H]3CC[C@@]([C@]3(CC[C@@H]2[C@@]1(C)C1)C)(O)C)C2=C1C=NN2 LKAJKIOFIWVMDJ-IYRCEVNGSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 102000002262 Thromboplastin Human genes 0.000 description 1
- 108010000499 Thromboplastin Proteins 0.000 description 1
- 238000008050 Total Bilirubin Reagent Methods 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- OGALXJIOJZXBBP-UHFFFAOYSA-N [4-(chloromethyl)phenyl]methanol Chemical compound OCC1=CC=C(CCl)C=C1 OGALXJIOJZXBBP-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 208000005707 acquired angioedema Diseases 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 201000003229 acute pancreatitis Diseases 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical class OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 230000003024 amidolytic effect Effects 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000001567 anti-fibrinolytic effect Effects 0.000 description 1
- 239000000504 antifibrinolytic agent Substances 0.000 description 1
- 229940082620 antifibrinolytics Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 125000005002 aryl methyl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- 238000013475 authorization Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 238000009811 bilateral tubal ligation Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000023555 blood coagulation Effects 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 238000010241 blood sampling Methods 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 229960000517 boceprevir Drugs 0.000 description 1
- LHHCSNFAOIFYRV-DOVBMPENSA-N boceprevir Chemical compound O=C([C@@H]1[C@@H]2[C@@H](C2(C)C)CN1C(=O)[C@@H](NC(=O)NC(C)(C)C)C(C)(C)C)NC(C(=O)C(N)=O)CC1CCC1 LHHCSNFAOIFYRV-DOVBMPENSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 1
- 229960000623 carbamazepine Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000013132 cardiothoracic surgery Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- ZCIGNRJZKPOIKD-CQXVEOKZSA-N cobicistat Chemical compound S1C(C(C)C)=NC(CN(C)C(=O)N[C@@H](CCN2CCOCC2)C(=O)N[C@H](CC[C@H](CC=2C=CC=CC=2)NC(=O)OCC=2SC=NC=2)CC=2C=CC=CC=2)=C1 ZCIGNRJZKPOIKD-CQXVEOKZSA-N 0.000 description 1
- 229960002402 cobicistat Drugs 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 108700005721 conestat alfa Proteins 0.000 description 1
- 229960000562 conivaptan Drugs 0.000 description 1
- JGBBVDFNZSRLIF-UHFFFAOYSA-N conivaptan Chemical compound C12=CC=CC=C2C=2[N]C(C)=NC=2CCN1C(=O)C(C=C1)=CC=C1NC(=O)C1=CC=CC=C1C1=CC=CC=C1 JGBBVDFNZSRLIF-UHFFFAOYSA-N 0.000 description 1
- 229940124558 contraceptive agent Drugs 0.000 description 1
- 230000002254 contraceptive effect Effects 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- POZRVZJJTULAOH-LHZXLZLDSA-N danazol Chemical compound C1[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=CC2=C1C=NO2 POZRVZJJTULAOH-LHZXLZLDSA-N 0.000 description 1
- 229960000766 danazol Drugs 0.000 description 1
- 238000013498 data listing Methods 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229960002086 dextran Drugs 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 1
- 229960004671 enzalutamide Drugs 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000007888 film coating Substances 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 150000002357 guanidines Chemical class 0.000 description 1
- 210000004247 hand Anatomy 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 208000030407 hereditary angioedema with normal C1Inh Diseases 0.000 description 1
- 125000000623 heterocyclic group Chemical class 0.000 description 1
- 230000000423 heterosexual effect Effects 0.000 description 1
- 238000002657 hormone replacement therapy Methods 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 238000009802 hysterectomy Methods 0.000 description 1
- YKLIKGKUANLGSB-HNNXBMFYSA-N idelalisib Chemical compound C1([C@@H](NC=2[C]3N=CN=C3N=CN=2)CC)=NC2=CC=CC(F)=C2C(=O)N1C1=CC=CC=C1 YKLIKGKUANLGSB-HNNXBMFYSA-N 0.000 description 1
- 229960003445 idelalisib Drugs 0.000 description 1
- 238000005417 image-selected in vivo spectroscopy Methods 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 238000011337 individualized treatment Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 231100000546 inhibition of ovulation Toxicity 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 238000012739 integrated shape imaging system Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- 229960004130 itraconazole Drugs 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 229940039088 kininogenase Drugs 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- HDKYHCGUBVWJHC-UHFFFAOYSA-N methyl 5-(methoxymethyl)-1h-pyrazole-4-carboxylate Chemical compound COCC1=NNC=C1C(=O)OC HDKYHCGUBVWJHC-UHFFFAOYSA-N 0.000 description 1
- 150000002746 methyltestosterones Chemical class 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 229960001800 nefazodone Drugs 0.000 description 1
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 description 1
- 230000018791 negative regulation of catalytic activity Effects 0.000 description 1
- 229960000884 nelfinavir Drugs 0.000 description 1
- QAGYKUNXZHXKMR-HKWSIXNMSA-N nelfinavir Chemical compound CC1=C(O)C=CC=C1C(=O)N[C@H]([C@H](O)CN1[C@@H](C[C@@H]2CCCC[C@@H]2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-HKWSIXNMSA-N 0.000 description 1
- LAIZPRYFQUWUBN-UHFFFAOYSA-L nickel chloride hexahydrate Chemical compound O.O.O.O.O.O.[Cl-].[Cl-].[Ni+2] LAIZPRYFQUWUBN-UHFFFAOYSA-L 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 238000009806 oophorectomy Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 229940127234 oral contraceptive Drugs 0.000 description 1
- 239000003539 oral contraceptive agent Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 229960000464 oxandrolone Drugs 0.000 description 1
- 229960002296 paroxetine Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 210000003800 pharynx Anatomy 0.000 description 1
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 1
- 229960002695 phenobarbital Drugs 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 238000000678 plasma activation Methods 0.000 description 1
- 229960001589 posaconazole Drugs 0.000 description 1
- RAGOYPUPXAKGKH-XAKZXMRKSA-N posaconazole Chemical compound O=C1N([C@H]([C@H](C)O)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@H]3C[C@@](CN4N=CN=C4)(OC3)C=3C(=CC(F)=CC=3)F)=CC=2)C=C1 RAGOYPUPXAKGKH-XAKZXMRKSA-N 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229940124606 potential therapeutic agent Drugs 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 238000009597 pregnancy test Methods 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 238000009117 preventive therapy Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- 229940095055 progestogen systemic hormonal contraceptives Drugs 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 229940043437 protein kinase A inhibitor Drugs 0.000 description 1
- 239000012656 protein kinase A inhibitor Substances 0.000 description 1
- 108010065251 protein kinase modulator Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 229960000311 ritonavir Drugs 0.000 description 1
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 1
- 238000009490 roller compaction Methods 0.000 description 1
- 229940009560 ruconest Drugs 0.000 description 1
- 231100000279 safety data Toxicity 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000012430 stability testing Methods 0.000 description 1
- 229960000912 stanozolol Drugs 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 201000009032 substance abuse Diseases 0.000 description 1
- 231100000736 substance abuse Toxicity 0.000 description 1
- 201000006152 substance dependence Diseases 0.000 description 1
- 235000021151 substantial meals Nutrition 0.000 description 1
- 229960005404 sulfamethoxazole Drugs 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- JLKIGFTWXXRPMT-UHFFFAOYSA-N sulphamethoxazole Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 JLKIGFTWXXRPMT-UHFFFAOYSA-N 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 230000035488 systolic blood pressure Effects 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- BBAWEDCPNXPBQM-GDEBMMAJSA-N telaprevir Chemical compound N([C@H](C(=O)N[C@H](C(=O)N1C[C@@H]2CCC[C@@H]2[C@H]1C(=O)N[C@@H](CCC)C(=O)C(=O)NC1CC1)C(C)(C)C)C1CCCCC1)C(=O)C1=CN=CC=N1 BBAWEDCPNXPBQM-GDEBMMAJSA-N 0.000 description 1
- 229960002935 telaprevir Drugs 0.000 description 1
- 108010017101 telaprevir Proteins 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- GYDJEQRTZSCIOI-LJGSYFOKSA-N tranexamic acid Chemical compound NC[C@H]1CC[C@H](C(O)=O)CC1 GYDJEQRTZSCIOI-LJGSYFOKSA-N 0.000 description 1
- 229960000401 tranexamic acid Drugs 0.000 description 1
- LQCLVBQBTUVCEQ-QTFUVMRISA-N troleandomycin Chemical compound O1[C@@H](C)[C@H](OC(C)=O)[C@@H](OC)C[C@@H]1O[C@@H]1[C@@H](C)C(=O)O[C@H](C)[C@H](C)[C@H](OC(C)=O)[C@@H](C)C(=O)[C@@]2(OC2)C[C@H](C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)OC(C)=O)[C@H]1C LQCLVBQBTUVCEQ-QTFUVMRISA-N 0.000 description 1
- 229960005041 troleandomycin Drugs 0.000 description 1
- 238000002562 urinalysis Methods 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- BCEHBSKCWLPMDN-MGPLVRAMSA-N voriconazole Chemical compound C1([C@H](C)[C@](O)(CN2N=CN=C2)C=2C(=CC(F)=CC=2)F)=NC=NC=C1F BCEHBSKCWLPMDN-MGPLVRAMSA-N 0.000 description 1
- 229960004740 voriconazole Drugs 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
Landscapes
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Dermatology (AREA)
- Diabetes (AREA)
- Pain & Pain Management (AREA)
- Otolaryngology (AREA)
- Pulmonology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicinal Preparation (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
La presente invención se refiere a tratamientos del angioedema hereditario (AEH). En particular, la presente invención proporciona tratamientos a demanda del angioedema hereditario (AEH) mediante la administración oral de un inhibidor de calicreína plasmática a un paciente que lo necesita a demanda. También se proporcionan tratamientos regulares (o continuos) de AEH. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Tratamientos del angioedema hereditario
[0001] La presente invención se refiere a tratamientos del angioedema hereditario (HAE). En particular, la presente invención proporciona un compuesto de Fórmula A para su uso en tratamientos a demanda del angioedema hereditario (HAE) mediante la administración oral de un inhibidor de la calicreína plasmática a un paciente que lo necesite a demanda.
Antecedentes de la invención
[0002] Los inhibidores de la calicreína plasmática tienen varias aplicaciones terapéuticas, en particular en el tratamiento del angioedema hereditario.
[0003] La calicreína plasmática es una serina proteasa similar a la tripsina que puede liberar cininas a partir de cininógenos (véase K. D. Bhoola et al., "Kallikrein-Kinin Cascade", Encyclopedia of Respiratory Medicine, p483-493; J. W. Bryant et al., "Human plasma kallikrein-kinin system: physiological and biochemical parameters" Cardiovascular and haematological agents in medicinal chemistry, 7, p234-250, 2009; K. D. Bhoola et al., Pharmacological Rev., 1992, 44, 1; y D. J. Campbell, "Towards understanding the kallikrein-kinin system: insights from the measurement of kinin peptides", Brazilian Journal of Medical and Biological Research 2000, 33, 665-677). Es un miembro esencial de la cascada intrínseca de la coagulación sanguínea, aunque su papel en esta cascada no implica la liberación de bradicinina ni la escisión enzimática. La precalicreína plasmática está codificada por un único gen y puede sintetizarse en el hígado, así como en otros tejidos. Es secretada por los hepatocitos como una precalicreína plasmática inactiva que circula en el plasma como un complejo heterodímero unido a un cininógeno de alto peso molecular (HK) que se activa para dar lugar a la calicreína plasmática activa. Este sistema de activación por contacto (o sistema de contacto) puede ser activado por superficies cargadas negativamente que activan el Factor XII (FXII) a Factor XIIa (FXlla), por ciertas proteasas, por ejemplo, plasmina (Hofman et al Clin Rev Allergy Immunol 2016), que pueden no requerir superficies negativas, o por proteínas mal plegadas (Maas et al J Clinical Invest 2008). La FXlla interviene en la conversión de la precalicreína plasmática en calicreína plasmática y en la posterior escisión del cininógeno de alto peso molecular (HK) para generar bradicinina, una potente hormona inflamatoria. Las cininas son potentes mediadores de la inflamación que actúan a través de receptores acoplados a proteínas G y los antagonistas de las cininas (como los antagonistas de los receptores de bradicinina) se han investigado anteriormente como posibles agentes terapéuticos para el tratamiento de diversos trastornos (F. Marceau y D. Regoli, Nature Rev., Drug Discovery, 2004, 3, 845-852).
[0004] Se cree que la calicreína plasmática desempeña un papel en varios trastornos inflamatorios. El principal inhibidor de la calicreína plasmática es el inhibidor de la serpina C1 esterasa. Los pacientes que presentan una deficiencia genética del inhibidor de la C1 esterasa padecen angioedema hereditario (HAE), que provoca hinchazón intermitente de cara, manos, garganta, tracto gastrointestinal y genitales. Las ampollas que se forman durante los episodios agudos contienen altos niveles de calicreína plasmática, que escinde el cininógeno de alto peso molecular (HK) liberando bradicinina, lo que provoca un aumento de la permeabilidad vascular. Se ha demostrado que el tratamiento con un inhibidor de la calicreína plasmática de grandes proteínas es eficaz para tratar el HAE al impedir la liberación de bradicinina, que provoca un aumento de la permeabilidad vascular (A. Lehmann "Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery" Expert Opin. Biol. Ther. 8, p1187-99).
[0005] El angioedema hereditario es un trastorno hereditario poco frecuente que se caracteriza por ataques agudos recurrentes en los que los fluidos se acumulan fuera de los vasos sanguíneos, bloqueando el flujo normal de sangre o líquido linfático y provocando una rápida hinchazón de los tejidos, como en las manos, los pies, las extremidades, la cara, el tracto intestinal o las vías respiratorias. Así pues, el "angioedema hereditario" puede definirse como cualquier trastorno caracterizado por episodios recurrentes de angioedema mediado por bradicinina (por ejemplo, hinchazón grave) causado por una disfunción/fallo/mutación hereditaria. Actualmente se conocen tres categorías de HAE: (i) HAE de tipo 1, (ii) HAE de tipo 2 y (iii) HAE normal con inhibidor de C1 (HAE normal C1 -Inh). Sin embargo, el campo del HAE se está desarrollando rápidamente, por lo que es de esperar que en el futuro se definan otros tipos de HAE.
[0006] Sin querer atarse a la teoría, se cree que el HAE de tipo 1 está causado por mutaciones en el gen SERPING1 que conducen a niveles reducidos de inhibidor de C1 en la sangre. Sin querer ceñirnos a la teoría, se cree que el HAE de tipo 2 está causado por mutaciones en el gen SERPING1 que provocan una disfunción del inhibidor C1 en la sangre. Sin querer ceñirnos a la teoría, la causa del HAE C1-Inh normal está menos definida y la disfunción/fallo/mutación genética subyacente puede resultar a veces desconocida. Lo que sí se sabe es que la causa del HAE normal C1-Inh no está relacionada con la reducción de los niveles o la disfunción del inhibidor de C1 (a diferencia de los tipos 1 y 2 de HAE). El HAE C1-Inh normal puede diagnosticarse revisando los antecedentes familiares y observando que el angioedema se ha heredado de una generación anterior (por lo que se trata de un angioedema hereditario). El HAE C1 -Inh normal también puede diagnosticarse determinando que existe una disfunción/fallo/mutación en un gen distinto de los relacionados con el inhibidor de C1. Por ejemplo, se ha informado de que la disfunción/fallo/mutación del plasminógeno puede causar HAE C1-Inh normal (véase, por ejemplo, Veronez et al., Front Med (Lausana). 2019 Feb 21;6:28. doi: 10.3389/fmed.2019.00028; o Recke et al., Clin Transl Allergy. 2019 Feb 14;9:9. doi: 10.1186/s13601-019-0247-x.). También se ha informado de que la disfunción/fallo/mutación del Factor XII puede causar HAE C1-Inh normal (véase, por
ejemplo, Mansi et al. 2014 The Association for the Publication of the Journal of Internal Medicine Journal of interna! Medicine, 2015, 277; 585-593; o Maat et al. J Thromb Haemost. 2019 Jan;17(1):183-194. doi: 10.1111/jth.14325).
[0007] Los ataques agudos de HAE suelen progresar a través de tres etapas clínicamente diferenciadas: una etapa prodrómica inicial (que suele durar hasta 12 horas), seguida de una etapa de inflamación y, por último, una etapa de absorción. La mayoría de los ataques de HAE se anuncian con síntomas prodrómicos. Dos tercios de los pródromos aparecieron menos de 6 horas antes de un ataque de HAE y ningún pródromo aparece más de 24 horas antes de un ataque de HAE (Magerl et al. Clinical and Experimental Dermatology (2014) 39, pp298-303). Por ejemplo, pueden empezar a observarse los siguientes síntomas prodrómicos: una ligera hinchazón (que afecta sobre todo a la cara y el cuello), un tipo típico de dolor abdominal, un enrojecimiento típico de la piel denominado "eritema marginado". Un ataque está plenamente desarrollado cuando ha alcanzado la máxima inflamación y la máxima expresión de dolor (por ejemplo, ataque abdominal), molestias (por ejemplo, ataque periférico) o amenaza para la vida (por ejemplo, ataque laríngeo). Una vez que el ataque ha alcanzado su punto álgido, el periodo posterior hasta la normalización viene determinado por el tiempo que tarda en desaparecer la inflamación y en reabsorberse el líquido que ha penetrado en los tejidos.
[0008] Anteriormente se han descrito inhibidores de la calicreína plasmática sintéticos y de moléculas pequeñas, por ejemplo por Garrett et al. ("Peptide aldehyde...." J. Peptide Res. 52, p62-71 (1998)), T. Griesbacher et al. ("Involvement of tissue kallikrein but not plasma kallikrein in the development of symptoms mediated by endogenous kinins in acute pancreatitis in rats" British Journal of Pharmacology 137, p692-700 (2002)), Evans ("Selective dipeptide inhibitors of kallikrein" WO03/076458 ), Szelke et al. ("Kininogenase inhibitors" WO92/04371 ), D. M. Evans et al. (Immunolpharmacology, 32, p115-116 (1996)), Szelke et al. ("Kininogen inhibitors" WO95/07921 ), Antonsson et al. ("New peptides derivatives" WO94/29335 ), J. Corte et al. ("Six membered heterocycles useful as serine protease inhibitors" WO2005/123680 ), J. Stürzbecher et al. (Brazilian J. Med. Biol. Res 27, p1929-34 (1994)), Kettner et al. (US 5.187.157 ), N. Teno et al. (Chem. Pharm. Bull. 41, p1079-1090 (1993)), W. B. Young et al. ("Small molecule inhibitors of plasma kallikrein" Bioorg. Med. Chem. Letts. 16, p2034-2036 (2006)), Okada et al. ("Development of potent and selective plasmin and plasma kallikrein inhibitors and studies on the structure-activity relationship" Chem. Pharm. Bull. 48, p1964-72 (2000)), Steinmetzer et al. ("Trypsin-like serine protease inhibitors and their preparation and use" WO08/049595 ), Zhang et al. ("Discovery of highly potent small molecule kallikrein inhibitors" Medicinal Chemistry 2, p545-553 (2006)), Sinha et al. ("Inhibitors of plasma kallikrein" WO08/016883 ), Shigenaga et al. ("Plasma Kallikrein Inhibitors" WO2011/118672 ), y Kolte et al. ("Biochemical characterization of a novel high-affinity and specific kallikrein inhibitor", British Journal of Pharmacology (2011), 162(7), 1639-1649). Asimismo, Steinmetzer et al. ("Serine protease inhibitors" WO2012/004678) describen análogos peptídicos ciclizados que son inhibidores de la plasmina humana y la calicreína plasmática.
[0009] Como ya se ha explicado, el HAE puede manifestarse en pacientes que presentan una deficiencia o disfunción genética del inhibidor de la C1 esterasa. Así, algunos de los tratamientos actuales del HAE consisten en administrar un inhibidor de la C1 esterasa para normalizar la deficiencia o la disfunción del inhibidor de la C1 esterasa. Dichos tratamientos pueden ser profilácticosfes decir, administrados en ausencia de síntomas de ataque agudo de HAE para prevenir/reducir la probabilidad de un ataque agudo de HAE) y/o tratamientos agudosfes decir, administrados cuando se notan síntomas de ataque agudo de HAE para intentar detener o reducir la gravedad del ataque agudo de HAE).
[0010] Cinryze® y Haegarda® contienen un inhibidor de la C1 esterasa y están indicados para prevenir los ataques agudos de HAEfes decir, tratamiento profiláctico). El tratamiento con Cinryze® requiere la preparación de una solución a partir de un polvo, que se inyecta cada 3 ó 4 días. Del mismo modo, el tratamiento con Haegarda® requiere la preparación de una solución a partir de un polvo, que se inyecta dos veces por semana. No siempre es posible que un paciente se autoadministre estos tratamientos y, en tal caso, es necesario que acuda a una clínica para recibir el tratamiento. Así pues, ambos tratamientos profilácticos suponen una elevada carga para el paciente. Además, el prospecto de la FDA para Haegarda® indica que "no debe utilizarse para tratar un ataque agudo de HAE", por lo que el paciente puede necesitar un tratamiento adicional si sufre un ataque de HAE.
[0011] Berinert® y Ruconest® contienen un inhibidor de la C1 esterasa y están indicados para tratar los ataques agudos de HAE. Ambos tratamientos también implican la preparación de una solución inyectable seguida de una inyección. Este proceso puede resultar pesado para el paciente, sobre todo cuando sufre un ataque agudo de HAE. La autoadministración de la dosis no siempre es posible, y si no lo es, la administración del fármaco puede retrasarse considerablemente, lo que aumenta la gravedad del ataque agudo de HAE para el paciente.
[0012] Hasta la fecha, los únicos inhibidores selectivos de la calicreína plasmática aprobados para uso médico en el tratamiento del HAE son Kalbitor® (principio activo ecalantida) y Takhzyro® (principio activo lanadelumab). Ambos tratamientos se formulan como soluciones inyectables. La ecallantida es un inhibidor de la calicreína plasmática de gran tamaño proteico que presenta riesgo de reacciones anafilácticas. De hecho, recientemente se ha retirado la solicitud de autorización de comercialización de Kalbitor® en la UE porque se considera que sus beneficios no compensan sus riesgos. Lanadelumab es un anticuerpo monoclonal recombinante de cadena ligera IgG1 kappa totalmente humano. Las reacciones adversas notificadas del tratamiento con lanadelumab incluyen hipersensibilidad, dolor en el punto de inyección, eritema en el punto de inyección y hematomas en el punto de inyección. En la etiqueta autorizada por la EMA de Takhzyro® (principio activo lanadelumab) se indica que "no está destinado al tratamiento de los ataques agudos de HAE" y que "en caso de ataque de HAE, debe iniciarse un tratamiento individualizado con una medicación de rescate aprobada". Además, al tratarse de inyecciones, ambos tratamientos implican una elevada carga para el paciente.
[0013] Berotralstat (BCX7353) se está investigando como tratamiento oral una vez al día para la prevención de los ataques de HAE. Hwang et al. (Immunotherapy (2019) 11(17), 1439-1444) afirma que las dosis más altas se asociaron con más efectos adversos gastrointestinales, lo que indica una mayor toxicidad a niveles más altos.
[0014] Otros inhibidores de la calicreína plasmática conocidos en la técnica son generalmente moléculas pequeñas, algunas de las cuales incluyen grupos funcionales altamente polares e ionizables, como guanidinas o amidinas. Recientemente se han descrito inhibidores de la calicreína plasmática que no presentan funcionalidades de guanidina o amidina. Por ejemplo Brandl et al. ("N-((6-amino-piridin-3-yl)methyl)-heteroaryl-carboxamides as inhibitors of plasma kallikrein" WO2012/017020 ), Evans et al. ("Benzylamine derivatives as inhibitors of plasma kallikrein" WO2013/005045 ), Allan et al. ("Benzylamine derivatives" WO2014/108679 ), Davie et al. ("Heterocyclic derivatives" WO2014/188211), y Davie et al. ("N-((het)arylmethyl)-heteroaryl-carboxamides compounds as plasma kallikrein inhibitors" WO2016/083820).
[0015] El solicitante ha desarrollado una serie novedosa de compuestos que son inhibidores de la calicreína plasmática, que se divulgan en el documento WO2016/083820 ( PCT/GB2015/053615 ). Estos compuestos demuestran una buena selectividad para la calicreína plasmática. Uno de estos compuestos es la N-[(3-fluoro-4-metoxipiridin-2-il)metil]-3-(metoximetil)-1-({4-[(2-oxopiridin-1-il)metil]fenil}metil)pirazol-4-carboxamida. La denominación N-[(3-fluoro-4-metoxipiridin-2-il)metil]-3-(metoximetil)-1-({4-[(2-oxopiridin-1-il)metil]fenil}metil)pirazol-4-carboxamida denota la estructura representada en la Fórmula A.

[0016] El documento WO2019/106361 se refiere a formas farmacéuticas sólidas orales que comprenden un inhibidor de la calicreína plasmática, en particular una forma sólida (Forma 1) del compuesto de Fórmula A.
[0017] Por lo tanto, existe la necesidad de un tratamiento del HAE que sea menos oneroso para el paciente a fin de mejorar su cumplimiento. En particular, existe la necesidad de un tratamiento del HAE que pueda administrarse por vía oral. También existe la necesidad de un tratamiento oral de los ataques agudos de HAE a demanda, por ejemplo, tras el reconocimiento de los síntomas de un ataque agudo de HAE. También es necesario un tratamiento profiláctico del HAE para reducir la probabilidad de que se produzca un ataque agudo de HAE. También existe la necesidad de un tratamiento de los ataques agudos de HAE que pueda ser utilizado a demanda por el paciente y que no requiera una dosificación regular (o continua), por ejemplo, un tratamiento que no requiera inyecciones dos veces por semana.
Descripción de la invención
[0018] La invención proporciona un compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para su uso en el tratamiento del angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) a un paciente que lo necesite a demanda,

[0019] Hasta hoy, no existen tratamientos orales a demanda autorizados para el HAE, y todos los tratamientos autorizados son inyectables. Los ataques de HAE se resuelven más rápido y son más breves tras un tratamiento precoz (Maurer M et al. PLoS ONE 2013;8(2): e53773. doi:10.1371/journal.pone.0053773) y, por tanto, la intervención precoz cuando se espera un ataque, o está en curso, es esencial para gestionar de forma deseable la enfermedad. Los tratamientos inyectables adolecen de retraso en la dosificación porque el paciente puede tener que preparar la forma farmacéutica o incluso desplazarse al hospital para recibir el tratamiento. Por lo tanto, el tratamiento del HAE se ve a menudo perjudicado por la dosificación tardía causada por la elevada carga que supone para el paciente. De hecho, Maurer M et al. explican que más del 60% de los pacientes se administran el inyectable contra el HAE más de una hora después del inicio del ataque. Sin querer ceñirnos a la teoría, se cree que los tratamientos inyectables contra el HAE adolecen de una dosificación tardía por razones como la incomodidad (la autoadministración no siempre es posible), el dolor (tanto durante como después de la inyección) y la esperanza (en lugar de tratar, los pacientes suelen limitarse a esperar un ataque menos grave). La presente invención pretende resolver este problema.
[0020] La presente invención proporciona un compuesto de Fórmula A para su uso en el tratamiento del HAE que mejora en comparación con cualquier tratamiento del HAE actualmente disponible. La presente invención proporciona un compuesto de Fórmula A para su uso en el tratamiento oral del HAE que es particularmente útil como tratamiento a demanda de los ataques agudos de HAE, y/o como tratamiento a demanda para reducir la probabilidad de un ataque agudo de HAE. Específicamente, como se describe en el presente documento, el compuesto de Fórmula A para su uso en tratamientos según la invención (i) tienen un rápido inicio de acción, (ii) son potentes, (iii) tienen un buen perfil de seguridad, y (iv) tienen efectos farmacodinámicos prolongados.
[0021] Se proporciona el compuesto de Fórmula A (o una sal y/o solvato farmacéuticamente aceptable del mismo) para su uso en el tratamiento del angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A (o una sal y/o solvato farmacéuticamente aceptable del mismo) a un paciente que lo necesite a demanda.
[0022] En cualquiera de los tratamientos de la invención aquí descritos, el término "compuesto de Fórmula A" es una abreviatura de "compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o solvato del mismo)". El término "solvato" se utiliza aquí para describir un complejo molecular que comprende el compuesto de la invención y una o más moléculas disolventes farmacéuticamente aceptables, por ejemplo, etanol o agua. El término "hidrato" se emplea cuando el disolvente es agua y, para evitar cualquier duda, el término "hidrato" queda englobado por el término "solvato".
[0023] En cualquiera de los tratamientos de la invención aquí descritos, el término "sal farmacéuticamente aceptable" significa una sal fisiológica o toxicológicamente tolerable e incluye, cuando sea apropiado, sales de adición de bases farmacéuticamente aceptables y sales de adición de ácidos farmacéuticamente aceptables. Por ejemplo (i) cuando un compuesto para uso según la invención contiene uno o más grupos ácidos, por ejemplo grupos carboxi, las sales de adición de base farmacéuticamente aceptables que pueden formarse incluyen sales de sodio, potasio, calcio, magnesio y amonio, o sales con aminas orgánicas, tales como, dietilamina, N metilglucamina, dietanolamina o aminoácidos (por ej. lisina) y similares; (ii) cuando un compuesto para uso según la invención contiene un grupo básico, tal como un grupo amino, las sales de adición ácida farmacéuticamente aceptables que pueden formarse incluyen hidrocloruros, hidrobromuros, sulfatos, fosfatos, acetatos, citratos, lactatos, tartratos, mesilatos, succinatos, oxalatos, fosfatos, esilatos, tosilatos, bencenosulfonatos, naftalendisulfonatos, maleatos, adipatos, fumaratos, hipuratos, alcanforatos, xinafoatos, pacetamidobenzoatos, dihidroxibenzoatos, hidroxinaftoatos, succinatos, ascorbatos, oleatos, bisulfatos y similares.
[0024] También pueden formarse hemisales de ácidos y bases, por ejemplo, hemisulfato y sales hemicálcicas.
[0025] Para una revisión de las sales adecuadas, véase "Handbook of Pharmaceutical Salts: Properties, Selection and Use" de Stahl y Wermuth (Wiley-VCH, Weinheim, Alemania, 2002).
[0026] En el contexto del HAE, la persona experta entenderá por tratamiento "a demanda" que el compuesto de Fórmula A se administra cuando se necesita terapia en relación con un ataque agudo específico de HAE. Como se describe en el presente documento, este ataque específico de HAE puede estar en curso (por ejemplo, el tratamiento se inicia al reconocer un síntoma de un ataque agudo de HAE) o ser probable que se produzca (por ejemplo, cuando el paciente prevé que podría inducirse o desencadenarse un ataque agudo de HAE). Pueden administrarse múltiples dosis del compuesto de Fórmula A como parte del tratamiento a demanda, pero estas dosis múltiples se administrarán en relación con el mismo ataque agudo de HAE. En otras palabras, "a demanda" no requiere la administración del compuesto de Fórmula A de forma continua a intervalos regulares (por ejemplo, una vez a la semana, dos veces a la semana, etc.) independientemente de que se produzca un ataque agudo de HAE. Esto contrasta con algunos otros tratamientos conocidos del HAE (por ejemplo, los tratamientos con Cinryze® y Haegarda®, descritos anteriormente) que requieren una dosificación continua y regular para la terapia. En cambio, en los tratamientos de la invención, el compuesto de Fórmula A se toma cuando el paciente requiere efectos terapéuticos de acción rápida. Entre los tratamientos "a demanda" particulares de la invención se incluyen: (i) tratar un ataque agudo de HAE a demanda, cuando el compuesto de Fórmula A se administra al reconocer un síntoma de un ataque agudo de HAE, y (ii) reducir profilácticamente la probabilidad de un
ataque de HAE a demanda, por ejemplo, cuando se prevé que podría inducirse (o desencadenarse) un ataque agudo de HAE. A continuación se analizan con más detalle.
[0027] En cualquiera de los tratamientos de la invención aquí descritos, el paciente es preferiblemente un ser humano. El HAE es una enfermedad hereditaria y los pacientes de todas las edades pueden sufrir ataques de HAE. Por consiguiente, el paciente humano puede ser un niño (de 0 a 18 años) o un adulto (de 18 años o más). Concretamente, el paciente puede tener 12 años o más. El paciente también puede tener 2 años o más.
[0028] Como se demuestra en los ejemplos, el compuesto de Fórmula A es un potente inhibidor de la calicreína plasmática. Como ya se ha explicado, la inhibición de la calicreína plasmática inhibe la escisión del kininógeno de alto peso molecular que contribuye a un ataque de HAE. Además, y como se demuestra en el Ejemplo 4, el compuesto de Fórmula A también es capaz de reducir la escisión de la precalicreína plasmática y la generación del Factor XIIa (FXlla) tras la activación del sistema de contacto. Estos ventajosos efectos adicionales apoyan que el tratamiento de la invención sea altamente eficaz y se demuestran en particular cuando la concentración del compuesto de Fórmula A es de al menos 500 ng/mL de plasma. Puede observarse una concentración plasmática de al menos 500 ng/mL tras la administración de una dosis de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 o aproximadamente 80 mg) del compuesto de Fórmula A.
[0029] En consecuencia, en cualquiera de los tratamientos de la invención aquí divulgados, particularmente siguiendo una dosis del compuesto de Fórmula A de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg), además de inhibir la calicreína plasmática, los tratamientos también pueden reducir la escisión de la precalicreína plasmática para generar calicreína plasmática y/o reducir la generación de Factor XIIa (FXlla) tras la administración. Así, en algunas realizaciones, particularmente después de una dosis del compuesto de Fórmula A de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg), los tratamientos pueden bloquear la escisión de precalicreína plasmática para generar calicreína plasmática y/o bloquear la escisión de FXII para generar FXlla.
[0030] Los términos "ataque agudo de HAE" y "ataque agudo de HAE" se utilizan indistintamente en el presente documento. El término "angioedema hereditario" se refiere a cualquier angioedema mediado por bradicinina causado por una disfunción, fallo o mutación genética heredada. En consecuencia, el término "HAE" incluye al menos el HAE de tipo 1, el HAE de tipo 2 y el HAE inhibidor de C1 normal (HAE C1-Inh normal).
El compuesto de Fórmula A para su uso en tratamientos de ataques agudos de HAE a demanda
[0031] Un aspecto de la invención proporciona el compuesto de Fórmula A para su uso en el tratamiento de un ataque agudo de angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A a un paciente que lo necesite, en el que el compuesto de Fórmula A se administra oralmente a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE.
[0032] Aunque cada ataque de HAE puede ser diferente en gravedad y en cuanto a la zona afectada, los pacientes que padecen HAE, los profesionales médicos con conocimientos de HAE y los cuidadores de pacientes con HAE son (y de hecho la persona experta lo sería) astutos a la hora de identificar los síntomas de un ataque agudo de HAE. Estos síntomas incluyen, entre otros: hinchazón de tejidos como manos, pies, extremidades, cara, tracto intestinal y/o vías respiratorias; fatiga; dolor de cabeza; dolores musculares; hormigueo en la piel; dolor abdominal; náuseas; vómitos; diarrea; dificultad para tragar; ronquera; dificultad para respirar; y/o cambios de humor. Así, en algunas realizaciones, la administración del compuesto de Fórmula A puede tener lugar tras el reconocimiento de al menos uno de los síntomas anteriores.
[0033] La persona experta también entenderá que "administrado tras el reconocimiento de un síntoma de un ataque de HAE" significa que la administración se produce tan pronto como sea factible tras el reconocimiento del síntoma de un ataque agudo de HAE. Por ejemplo, se espera que los pacientes tengan el compuesto de Fórmula A fácil y rápidamente disponible en todo momento (muy probablemente en forma de una composición farmacéuticamente aceptable) para garantizar que el tratamiento pueda tener lugar en cuanto se reconozca un síntoma de un ataque de HAE. En otras palabras, el tratamiento se produce a demanda. Por ejemplo, en algunas realizaciones, el compuesto de Fórmula A para uso según la invención puede administrarse en el plazo de 1 hora desde que se reconoce el síntoma de un ataque agudo de HAE, preferiblemente en el plazo de 30 minutos, en el plazo de 20 minutos, en el plazo de 10 minutos o en el plazo de 5 minutos desde que se reconoce el síntoma de un ataque agudo de HAE.
[0034] Si el síntoma de un ataque agudo de HAE se reconoce en la fase prodrómica, una realización de la invención consiste en que el compuesto de Fórmula A para uso según la invención puede administrarse en la fase prodrómica de un ataque agudo de HAE. En estas circunstancias, el síntoma reconocido puede ser una ligera hinchazón, en particular,
una ligera hinchazón que afecta a la cara y el cuello. Además, o como alternativa, el síntoma puede ser dolor abdominal, en particular, se considera que el dolor abdominal es característico de un ataque de HAE. Además, o como alternativa, el síntoma puede ser un enrojecimiento de la piel, como el eritema marginado.
[0035] El compuesto de Fórmula A para su uso de acuerdo con la invención puede evitar que un ataque agudo de HAE aumente en gravedad. En algunas circunstancias, el tratamiento puede acortar la duración del ataque, y a veces incluso detenerlo en su totalidad. Por ejemplo, el tratamiento puede detener la progresión de un ataque de HAE periférico o de un ataque de HAE abdominal. En algunas realizaciones, el compuesto de Fórmula A para uso según la invención puede suprimir la aparición posterior de hinchazón, a veces completamente, y en particular cuando el tratamiento se inicia en la fase prodrómica. En particular, en algunas realizaciones, se puede evitar que el ataque agudo de HAE progrese a la fase de inflamación cuando el tratamiento se inicia en la fase prodrómica.
[0036] El compuesto de Fórmula A puede ser suficiente para tratar el ataque agudo de HAE por sí solo, es decir, sin que se administre al paciente ningún principio activo farmacéutico distinto del compuesto de Fórmula A.
[0037] Así, en algunas realizaciones de la invención, no se administra al paciente ningún principio activo farmacéutico distinto del compuesto de Fórmula A para tratar el ataque agudo de HAE. En particular, en algunas realizaciones, el compuesto de Fórmula A para su uso en los tratamientos de la invención no requiere la administración de ningún ingrediente farmacéutico activo para tratar un ataque de HAE (por ejemplo, un medicamento de rescate como pdC1INH, rhC1INH o icatibant) distinto del compuesto de Fórmula A. Más concretamente, en algunas realizaciones, no se administra al paciente ningún principio activo farmacéutico para tratar un ataque de HAE (por ejemplo, un medicamento de rescate como pdC1 INH, rhC1INH o icatibant) distinto del compuesto de Fórmula A.
[0038] Alternativamente, en algunas realizaciones, el compuesto de Fórmula A para su uso en los tratamientos de la invención puede utilizarse en combinación con otros tratamientos del HAE. Por ejemplo, en algunas realizaciones, el compuesto de Fórmula A para uso en la terapia aguda a demanda descrita en el presente documento puede utilizarse como "complemento" de otro tratamiento del HAE. En algunas realizaciones, el paciente puede estar tomando otro tratamiento profiláctico del HAE y podría utilizar el compuesto de Fórmula A para su uso en los tratamientos a demanda aquí descritos para tratar un ataque agudo de HAE que no fue prevenido por el otro tratamiento profiláctico del HAE.
[0039] Por ejemplo, en algunas realizaciones, el compuesto de Fórmula A para su uso en un método para tratar el HAE en un paciente que ya está tomando un inhibidor de C1 (como Cinryze®, Haegarda®, Berinert®) para profilaxis se proporciona comprendiendo: administrar oralmente el compuesto de Fórmula A al paciente a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE. En otra realización, el compuesto de Fórmula A para su uso en un método para tratar el HAE en un paciente que ya está tomando lanadelumab como profilaxis se proporciona comprendiendo: administrar oralmente el compuesto de Fórmula A al paciente a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE. En otra realización, el compuesto de Fórmula A para su uso en un método para tratar el HAE en un paciente que ya está tomando berotralstat como profilaxis se proporciona comprendiendo: administrar oralmente el compuesto de Fórmula A al paciente a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE.
[0040] En cualquiera de los tratamientos anteriores, el síntoma puede ser reconocido por el paciente. En cualquiera de los tratamientos anteriores, el síntoma puede ser reconocido por un profesional médico con conocimientos sobre el HAE. En cualquiera de los tratamientos anteriores, el síntoma puede ser reconocido por un cuidador del paciente.
[0041] El compuesto de Fórmula A para uso en tratamientos según la invención puede reducir la proporción de ataques de HAE que progresan en un nivel o más en una escala Likert de 5 puntos (5LS). El compuesto de Fórmula A para su uso en tratamientos según la invención puede reducir la proporción de ataques de HAE que progresan en un nivel o más en un 5LS en las 12 horas siguientes a la administración del compuesto. El compuesto de Fórmula A para uso en tratamientos según la invención puede mejorar el tiempo de resolución de un ataque de HAE a "ninguno" en un 5LS. 5LS es una escala conocida en la técnica (véase, por ejemplo, Allergy Asthma Proc. 2018 Jan 1;39(1):74-80. doi: 10.2500/aap.2018.39.4095) que pueden utilizarse para informar de la gravedad de los ataques de HAE y, por ejemplo, pueden utilizarse para informar de los ataques como "ninguno", "leve", "moderado", "grave" o "muy grave".
[0042] El compuesto de Fórmula A para uso en tratamientos según la invención puede reducir la proporción de ataques de HAE que se califican como "peor" o "mucho peor" en una pregunta de transición de 7 puntos (7TQ). El compuesto de Fórmula A para su uso en tratamientos según la invención puede aumentar la proporción de ataques de HAE que se califican como "mejores" o "mucho mejores". El 7TQ es un índice conocido en la técnica que puede utilizarse para puntuar la progresión de un ataque de HAE e informar de los ataques como "mucho mejor", "mejor", "un poco mejor", "sin cambios", "un poco peor", "peor" o "mucho peor".
[0043] En algunas realizaciones de cualquiera de los tratamientos a demanda de un ataque agudo de HAE de la invención, se puede administrar al paciente una única dosis del compuesto de Fórmula A para tratar el ataque agudo de HAE. En algunas otras realizaciones de cualquiera de los tratamientos a demanda de los ataques agudos de HAE de la invención, se pueden administrar al paciente múltiples dosis del compuesto de Fórmula A para tratar el ataque agudo de HAE. Por ejemplo, el tratamiento a demanda puede comprender la administración de dos dosis del compuesto de Fórmula A en un
período de 24 horas a partir del momento de la toma de la primera dosis. Alternativamente, el tratamiento a demanda puede comprender la administración de tres dosis del compuesto de Fórmula A en un período de 24 horas a partir del momento de la toma de la primera dosis. Alternativamente, el tratamiento a demanda puede comprender la administración de cuatro dosis del compuesto de Fórmula A en un período de 24 horas a partir del momento de la toma de la primera dosis. Cuando se toman varias dosis, éstas pueden espaciarse uniformemente, de modo que transcurra un periodo de tiempo aproximadamente igual entre cada dosis, por ejemplo, tomando la dosis siguiente a las 8 horas, 16 horas y 24 horas de la primera dosis.
[0044] En algunas realizaciones de cualquiera de los tratamientos a demanda de un ataque agudo de HAE de la invención, se puede administrar al paciente la dosis diaria en dos dosis al día. Estas dos dosis pueden administrarse simultánea, separada o secuencialmente. En algunas realizaciones, las dos dosis pueden administrarse en cualquier momento del día, y el intervalo entre las dos dosis depende del paciente y de la gravedad del ataque agudo de HAE. En algunas realizaciones, la segunda dosis puede administrarse dentro de las 2 horas siguientes a la primera (más específicamente, entre 1 y 2 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse entre aproximadamente 1 y aproximadamente 4 horas después de la primera (más específicamente, entre aproximadamente 1 y 3 horas, aproximadamente 2 y 3 horas, o entre 3 horas y aproximadamente 4 horas, después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse entre unas 4 y unas 12 horas después de la primera (más específicamente, entre unas 4 y unas 8 horas, o a unas 6 horas, después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse entre aproximadamente 2 y aproximadamente 6 horas después de la primera (más específicamente, entre aproximadamente 3 y aproximadamente 6 horas, después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 8 horas siguientes a la primera (más específicamente, entre 4 y 8 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 12 horas siguientes a la primera (más específicamente, entre 8 y 12 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 16 horas siguientes a la primera (más específicamente, entre 12 y 16 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 20 horas siguientes a la primera (más específicamente, entre las 16 y las 20 horas siguientes a la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 24 horas siguientes a la primera (más específicamente, entre las 20 y las 24 horas siguientes a la primera dosis). En estas realizaciones, cada una de las dos dosis puede ser de 600 mg del compuesto de Fórmula A.
[0045] En cualquiera de los tratamientos a demanda de un ataque agudo de HAE de la invención, se puede administrar al paciente la dosis diaria en dos dosis al día, en las que la segunda dosis se puede administrar al menos unas 6 horas después de la primera dosis. El paciente puede recibir la dosis diaria en dos dosis al día, en las que la segunda dosis puede administrarse entre 5 y 7 horas después de la primera. Más concretamente, se puede administrar al paciente la dosis diaria en dos dosis al día, en las que la segunda dosis se administra unas 6 horas después de la primera. En estas realizaciones, cada una de las dos dosis puede ser de 600 mg del compuesto de Fórmula A. Cada una de estas dosis de 600 mg puede ser dos comprimidos que comprenden 300 mg del compuesto de Fórmula A.
[0046] En algunas realizaciones de cualquiera de los tratamientos a demanda de un ataque agudo de HAE de la invención, se puede administrar al paciente la dosis diaria en tres dosis al día. Estas tres dosis pueden administrarse simultánea, separada o secuencialmente. En algunas realizaciones, las tres dosis pueden administrarse en cualquier momento del día, y el intervalo entre las tres dosis depende del paciente y de la gravedad del ataque agudo de HAE. En algunas realizaciones, la segunda y la tercera dosis pueden administrarse dentro de las 4 horas siguientes a la primera. Más específicamente, la segunda dosis puede administrarse entre 1 y 3 horas después de la primera dosis y la tercera dosis puede administrarse entre 3 y 4 horas después de la primera dosis. La segunda dosis puede administrarse entre aproximadamente 4 y 12 horas después de la primera (más específicamente, entre aproximadamente 4 y 8 horas, o aproximadamente 6 horas después de la primera dosis), y la tercera dosis puede administrarse entre aproximadamente 4 y 12 horas después de la segunda (más específicamente, entre aproximadamente 4 y 8 horas, o aproximadamente 6 horas después de la segunda dosis). Más concretamente, la segunda dosis puede administrarse aproximadamente 2 horas después de la primera dosis y la tercera dosis puede administrarse aproximadamente 4 horas después de la primera dosis. En algunas realizaciones, la segunda y la tercera dosis pueden administrarse dentro de las 8 horas siguientes a la primera. Más específicamente, la segunda dosis puede administrarse entre 3 y 5 horas después de la primera dosis y la tercera dosis puede administrarse entre 7 y 8 horas después de la primera dosis. Más concretamente, la segunda dosis puede administrarse unas 4 horas después de la primera dosis y la tercera dosis puede administrarse unas 8 horas después de la primera dosis. En algunas realizaciones, la segunda y la tercera dosis pueden administrarse dentro de las 16 horas siguientes a la primera. Más específicamente, la segunda dosis puede administrarse entre 7 y 9 horas después de la primera dosis y la tercera dosis puede administrarse entre 15 y 16 horas después de la primera dosis. Más concretamente, la segunda dosis puede administrarse unas 8 horas después de la primera dosis y la tercera dosis puede administrarse unas 16 horas después de la primera dosis. En estas realizaciones, cada una de las tres dosis puede ser de 600 mg del compuesto de Fórmula A.
[0047] En cualquiera de los tratamientos a demanda de un ataque agudo de HAE de la invención, se puede administrar al paciente la dosis diaria en tres dosis al día, en las que la segunda y tercera dosis se pueden administrar al menos unas 6 horas después de la dosis precedente. El paciente puede recibir la dosis diaria en tres dosis al día, en las que la segunda dosis puede administrarse entre 5 y 7 horas después de la primera dosis, y la tercera dosis puede administrarse entre 11 y 13 horas después de la primera dosis. Más específicamente, el paciente puede recibir la dosis diaria en tres dosis al
día, en las que la segunda dosis puede administrarse unas 6 horas después de la primera dosis y la tercera dosis puede administrarse unas 12 horas después de la primera dosis. En estas realizaciones, cada una de las tres dosis puede ser 600 mg del compuesto de Fórmula A. Cada una de estas dosis de 600 mg puede ser dos comprimidos que comprenden 300 mg del compuesto de Fórmula A.
[0048] Pueden administrarse múltiples dosis si, por ejemplo, persiste un ataque de HAE tras la administración de la primera dosis. Cuando se utiliza en este contexto, "persiste" puede significar que, por ejemplo, la primera dosis no impide que un ataque agudo de HAE aumente en gravedad, o que la primera dosis no detiene el ataque de HAE en su totalidad, o que la primera dosis no disminuye la gravedad del ataque de HAE. En consecuencia, el compuesto de Fórmula A para su uso en tratamientos a demanda de un ataque de HAE de la invención puede comprender la administración de una primera dosis y, a continuación, la administración de una segunda dosis si el ataque de HAE persiste tras la administración de la primera dosis. El compuesto de Fórmula A para uso en tratamientos a demanda de un ataque de HAE de la invención también puede comprender administrar una primera dosis, y luego administrar una segunda dosis si el ataque de HAE persiste después de administrar la primera dosis, y luego administrar una tercera dosis si el ataque de hAe persiste después de administrar la segunda dosis. En cada caso, cada dosis posterior puede administrarse simultánea, separada o secuencialmente. En cada caso, cada dosis posterior puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la dosis precedente. En cada caso, cada dosis puede comprender 600 mg del compuesto, por ejemplo, administrado como dos comprimidos que comprenden 300 mg.
[0049] Específicamente, el compuesto de Fórmula A para uso en los tratamientos a demanda de un ataque agudo de HAE de la invención puede comprender la administración de una primera dosis que comprende 600 mg del compuesto (por ejemplo, como dos comprimidos que comprenden 300 mg del compuesto), y luego administrar una segunda dosis que comprende 600 mg del compuesto (por ejemplo, como dos comprimidos que comprenden 300 mg del compuesto) si el ataque de HAE persiste después de administrar la primera dosis. La segunda dosis puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la primera dosis. Si el ataque de hA e persiste después de la segunda dosis, los tratamientos a demanda de un ataque agudo de HAE de la invención pueden comprender la administración de una tercera dosis que contenga 600 mg del compuesto (por ejemplo, dos comprimidos que contengan 300 mg del compuesto). La tercera dosis puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la segunda dosis.
[0050] Pueden administrarse múltiples dosis incluso si la gravedad del ataque de HAE parece haberse reducido (o incluso detenido en su totalidad) tras la administración de la primera dosis para evitar que el ataque de HAE vuelva a aumentar en gravedad. Por ejemplo, se pueden utilizar dosis múltiples para la tranquilidad del paciente, por ejemplo, para calmar su ansiedad. En consecuencia, el compuesto de Fórmula A para su uso en tratamientos a demanda de un ataque de HAE de la invención puede comprender la administración de una primera dosis, y luego administrar una segunda dosis incluso si la gravedad del ataque de HAE parece haberse reducido (o incluso detenido en su totalidad) después de la administración de la primera dosis para evitar que el ataque de HAE vuelva a aumentar en gravedad. Incluso si la gravedad del ataque de HAE parece haberse reducido (o incluso detenido en su totalidad) tras la administración de la primera y/o segunda dosis, el compuesto de Fórmula A para su uso en tratamientos a demanda de un ataque de HAE de la invención también puede comprender la administración de una tercera dosis para evitar que el ataque de HAE vuelva a aumentar en gravedad. En cada caso, cada dosis posterior puede administrarse simultánea, separada o secuencialmente. En cada caso, cada dosis posterior puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la dosis precedente. En cada caso, cada dosis puede comprender 600 mg del compuesto, por ejemplo, administrado como dos comprimidos que comprenden 300 mg del compuesto.
[0051] Específicamente, el compuesto de Fórmula A para su uso en los tratamientos a demanda de un ataque agudo de HAE de la invención puede comprender la administración de una primera dosis que comprende 600 mg del compuesto (por ejemplo, como dos comprimidos que comprenden 300 mg del compuesto), y luego la administración de una segunda dosis que comprende 600 mg del compuesto (por ejemplo, como dos comprimidos que comprenden 300 mg del compuesto), incluso si la gravedad del ataque de HAE parece haberse reducido (o incluso detenido en su totalidad) después de la administración de la primera dosis, para evitar que el ataque de HAE vuelva a aumentar en gravedad. La segunda dosis puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la primera dosis. Incluso si la gravedad del ataque de HAE parece haberse reducido (o incluso detenido en su totalidad) tras la administración de la primera y/o segunda dosis, el compuesto de Fórmula A para su uso en los tratamientos a demanda de un ataque agudo de HAE de la invención puede comprender la administración de una tercera dosis que contenga 600 mg del compuesto (por ejemplo, dos comprimidos que contengan 300 mg del compuesto) para evitar que el ataque de HAE vuelva a aumentar en gravedad. La tercera dosis puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la segunda dosis.
[0052] El compuesto de Fórmula A para su uso en los tratamientos a demanda de un ataque agudo de HAE de la invención puede comprender no administrar más de tres dosis en un periodo de 24 horas (por ejemplo, tres dosis que comprendan 600 mg del compuesto, opcionalmente como 6 comprimidos cada uno de los cuales comprenda 300 mg del compuesto).
El compuesto de Fórmula A para su uso en el tratamiento profiláctico a demanda de los ataques agudos de HAE.
[0053] Un aspecto de la invención proporciona el compuesto de Fórmula A para su uso en el tratamiento del angioedema
hereditario (HAE) que comprende: administrar oralmente el compuesto de Fórmula A a un paciente que lo necesite, en el que el compuesto de Fórmula A se administra oralmente a demanda para reducir profilácticamente la probabilidad de un ataque agudo de HAE.
[0054] En algunas realizaciones, el compuesto de Fórmula A puede administrarse para prevenir un ataque agudo de HAE.
[0055] Como se discutió anteriormente, el compuesto de Fórmula A para uso de acuerdo con la invención no requiere dosificación del compuesto de Fórmula A a intervalos regulares para proporcionar terapia profiláctica. En efecto, el compuesto de Fórmula A se administra a demanda. Por ejemplo, el compuesto de Fórmula A puede administrarse a demanda para reducir la probabilidad de un ataque agudo de hA e (por ejemplo, para prevenir un ataque agudo de HAE) cuando se prevé que se inducirá (o desencadenará) un ataque agudo de HAE, es decir, cuando se prevé que el paciente sufrirá un ataque agudo de HAE. En algunas realizaciones, el paciente puede anticipar que se inducirá (o desencadenará) un ataque agudo de HAE. En algunas realizaciones, un profesional médico, como un profesional médico con conocimientos de HAE, puede anticipar que se inducirá (o desencadenará) un ataque agudo de HAE. En algunas realizaciones, un cuidador del paciente puede anticipar que se inducirá (o desencadenará) un ataque agudo de HAE. Por ejemplo, un ataque agudo de HAE puede ser inducido (o desencadenado) por diversos estímulos, como traumas físicos (por ejemplo, procedimientos médicos, dentales o quirúrgicos) y/o estrés (por ejemplo, situaciones de mucho estrés, como el estrés mental, que en algunos casos puede estar asociado a la realización de exámenes o al estrés mental asociado a un procedimiento médico, dental o quirúrgico). Por ejemplo, un ataque agudo de HAE puede ser inducido (o desencadenado) por los elevados niveles de estrés/ansiedad del paciente cuando éste podría esperar tener un ataque de HAE. Además, la frecuencia de los casos de ataques agudos de HAE puede variar con el tiempo en el mismo paciente. A menudo, los pacientes pueden sufrir periodos en los que la frecuencia de los ataques agudos de HAE es mayor de lo normal. Así pues, se puede anticipar un ataque agudo de HAE durante los periodos en los que el paciente sufre ataques agudos de HAE con mayor frecuencia de lo normal. Quienes estén familiarizados con el HAE sabrán que los ataques agudos de HAE pueden inducirse (o desencadenarse) de este modo. Los pacientes, los profesionales médicos con conocimientos sobre el HAE y los cuidadores de los pacientes también pueden ser astutos a la hora de anticiparse a dichos desencadenantes. Así, de acuerdo con la invención, el compuesto de Fórmula A puede administrarse a demanda cuando se prevé que el paciente será sometido a uno o más de estos estímulos o circunstancias.
[0056] Como se ha comentado anteriormente, se puede administrar al paciente el compuesto de Fórmula A como parte de un tratamiento profiláctico a demanda de un ataque agudo de HAE. Como ya se ha comentado, este tratamiento reduce la probabilidad de que se produzca un ataque agudo de HAE. Sin embargo, en algunas circunstancias, un paciente puede sufrir un ataque agudo de HAE. Por lo tanto, una realización de la invención es que al paciente se le puede administrar el compuesto de Fórmula A como parte de un tratamiento profiláctico a demanda de un ataque agudo de HAE, como se ha comentado anteriormente, que comprende además tomar una dosis a demanda del compuesto de Fórmula A al reconocer un síntoma de un ataque agudo de HAE para tratar un ataque agudo de HAE en caso de que surja. Estos tratamientos a demanda de los ataques agudos de HAE se han comentado anteriormente.
[0057] Así, en algunas realizaciones, se proporciona el compuesto de Fórmula A para su uso en un método para tratar el angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A a un paciente que lo necesite, en el que el compuesto de Fórmula A se administra oralmente a demanda para reducir profilácticamente la probabilidad de un ataque agudo de HAE, comprendiendo además administrar oralmente el compuesto de Fórmula A a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE.
[0058] En algunas realizaciones de cualquiera de los tratamientos a demanda de un ataque agudo de HAE de la invención, se puede administrar al paciente una única dosis del compuesto de Fórmula A para tratar el ataque agudo de HAE. En algunas otras realizaciones de cualquiera de los tratamientos a demanda de los ataques agudos de HAE de la invención, se pueden administrar al paciente múltiples dosis del compuesto de Fórmula A para tratar el ataque agudo de HAE. Por ejemplo, el tratamiento a demanda puede comprender la administración de dos dosis del compuesto de Fórmula A en un período de 24 horas a partir del momento de la toma de la primera dosis. Alternativamente, el tratamiento a demanda puede comprender la administración de tres dosis del compuesto de Fórmula A en un período de 24 horas a partir del momento de la toma de la primera dosis. Alternativamente, el tratamiento a demanda puede comprender la administración de cuatro dosis del compuesto de Fórmula A en un período de 24 horas a partir del momento de la toma de la primera dosis. Cuando se toman varias dosis, éstas pueden espaciarse uniformemente, de modo que haya un periodo de tiempo aproximadamente igual entre cada dosis, por ejemplo, tomando la dosis siguiente a las 8 horas, 16 horas y 24 horas de la dosis inicial.
[0059] En algunas realizaciones de cualquiera de los tratamientos profilácticos a demanda de los ataques agudos de HAE aquí descritos, se pueden administrar al paciente dos dosis al día. Estas dos dosis pueden administrarse simultánea, separada o secuencialmente. En algunas realizaciones, las dos dosis pueden administrarse en cualquier momento del día, siendo el intervalo entre las dos dosis específico para el paciente. En algunas realizaciones, la segunda dosis puede administrarse dentro de las 2 horas siguientes a la primera (más específicamente, entre 1 y 2 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse entre aproximadamente 1 y aproximadamente 4 horas después de la primera (más específicamente, entre aproximadamente 1 y 3 horas, aproximadamente 2 y 3 horas, o entre 3 horas y aproximadamente 4 horas, después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse entre unas 4 y unas 12 horas después de la primera (más específicamente, entre unas 4 y unas 8
horas, o a unas 6 horas, después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse entre aproximadamente 2 y aproximadamente 6 horas después de la primera (más específicamente, entre aproximadamente 3 y aproximadamente 6 horas, después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 8 horas siguientes a la primera (más específicamente, entre 4 y 8 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 12 horas siguientes a la primera (más específicamente, entre 8 y 12 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 16 horas siguientes a la primera (más específicamente, entre 12 y 16 horas después de la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 20 horas siguientes a la primera (más específicamente, entre las 16 y las 20 horas siguientes a la primera dosis). En algunas realizaciones, la segunda dosis puede administrarse dentro de las 24 horas siguientes a la primera (más específicamente, entre las 20 y las 24 horas siguientes a la primera dosis). En estas realizaciones, cada una de las dos dosis puede ser de 600 mg del compuesto de Fórmula A.
[0060] En cualquiera de los tratamientos profilácticos a demanda de ataques agudos de HAE descritos en el presente documento, se puede administrar al paciente la dosis diaria en dos dosis al día, en las que la segunda dosis se puede administrar al menos unas 6 horas después de la primera dosis. El paciente puede recibir la dosis diaria en dos dosis al día, en las que la segunda dosis puede administrarse entre 5 y 7 horas después de la primera. Más concretamente, se puede administrar al paciente la dosis diaria en dos dosis al día, en las que la segunda dosis se administra unas 6 horas después de la primera. En estas realizaciones, cada una de las dos dosis puede ser de 600 mg del compuesto de Fórmula A. Cada una de estas dosis de 600 mg puede ser dos comprimidos que comprenden 300 mg del compuesto de Fórmula A.
[0061] En algunas realizaciones de cualquiera de los tratamientos profilácticos a demanda de los ataques agudos de HAE aquí descritos, se puede administrar al paciente la dosis diaria en tres dosis al día. Estas tres dosis pueden administrarse simultánea, separada o secuencialmente. En algunas realizaciones, las tres dosis pueden administrarse en cualquier momento del día, siendo el intervalo entre las tres dosis específico para el paciente. En algunas realizaciones, la segunda y la tercera dosis pueden administrarse dentro de las 4 horas siguientes a la primera. Más específicamente, la segunda dosis puede administrarse entre 1 y 3 horas después de la primera dosis y la tercera dosis puede administrarse entre 3 y 4 horas después de la primera dosis. La segunda dosis puede administrarse entre aproximadamente 4 y 12 horas después de la primera (más específicamente, entre aproximadamente 4 y 8 horas, o aproximadamente 6 horas después de la primera dosis), y la tercera dosis puede administrarse entre aproximadamente 4 y 12 horas después de la segunda (más específicamente, entre aproximadamente 4 y 8 horas, o aproximadamente 6 horas después de la segunda dosis). Más concretamente, la segunda dosis puede administrarse aproximadamente 2 horas después de la primera dosis y la tercera dosis puede administrarse aproximadamente 4 horas después de la primera dosis. En algunas realizaciones, la segunda y la tercera dosis pueden administrarse dentro de las 8 horas siguientes a la primera. Más específicamente, la segunda dosis puede administrarse entre 3 y 5 horas después de la primera dosis y la tercera dosis puede administrarse entre 7 y 8 horas después de la primera dosis. Más concretamente, la segunda dosis puede administrarse unas 4 horas después de la primera dosis y la tercera dosis puede administrarse unas 8 horas después de la primera dosis. En algunas realizaciones, la segunda y la tercera dosis pueden administrarse dentro de las 16 horas siguientes a la primera. Más específicamente, la segunda dosis puede administrarse entre 7 y 9 horas después de la primera dosis y la tercera dosis puede administrarse entre 15 y 16 horas después de la primera dosis. Más concretamente, la segunda dosis puede administrarse unas 8 horas después de la primera dosis y la tercera dosis puede administrarse unas 16 horas después de la primera dosis. En estas realizaciones, cada una de las tres dosis puede ser de 600 mg del compuesto de Fórmula A.
[0062] En cualquiera de los tratamientos profilácticos a demanda de ataques agudos de HAE descritos en el presente documento, se puede administrar al paciente la dosis diaria en tres dosis al día, en las que la segunda y tercera dosis se pueden administrar al menos unas 6 horas después de la dosis precedente. El paciente puede recibir la dosis diaria en tres dosis al día, en las que la segunda dosis puede administrarse entre 5 y 7 horas después de la primera dosis, y la tercera dosis puede administrarse entre 11 y 13 horas después de la primera dosis. Más específicamente, el paciente puede recibir la dosis diaria en tres dosis al día, en las que la segunda dosis puede administrarse unas 6 horas después de la primera dosis y la tercera dosis puede administrarse unas 12 horas después de la primera dosis. En estas realizaciones, cada una de las tres dosis puede ser 600 mg del compuesto de Fórmula A. Cada una de estas dosis de 600 mg puede ser dos comprimidos que comprenden 300 mg del compuesto de Fórmula A.
[0063] Pueden administrarse múltiples dosis si, por ejemplo, existe una necesidad continuada de reducir profilácticamente la probabilidad de un ataque agudo de HAE (por ejemplo, si el paciente sigue previendo que podría inducirse un ataque de HAE, como se ha comentado anteriormente). En consecuencia, el compuesto de Fórmula A para su uso en tratamientos a demanda de un ataque de HAE de la invención puede comprender la administración de una primera dosis, y luego administrar una segunda dosis si existe una necesidad continuada de reducir profilácticamente la probabilidad de un ataque agudo de HAE después de administrar la primera dosis. El compuesto de Fórmula A para uso en tratamientos a demanda de un ataque de HAE de la invención también puede comprender la administración de una primera dosis, y luego la administración de una segunda dosis si existe una necesidad continuada de reducir profilácticamente la probabilidad de un ataque agudo de HAE después de administrar la primera dosis, y luego la administración de una tercera dosis si existe una necesidad continuada de reducir profilácticamente la probabilidad de un ataque agudo de HAE después de administrar la segunda dosis. En cada caso, cada dosis posterior puede administrarse simultánea, separada o
secuencialmente. En cada caso, cada dosis posterior puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la dosis precedente. En cada caso, cada dosis puede comprender 600 mg del compuesto, por ejemplo, administrado como dos comprimidos que comprenden 300 mg del compuesto.
[0064] Específicamente, los tratamientos profilácticos a demanda de ataques agudos de HAE descritos en el presente documento pueden comprender la administración de una primera dosis que comprenda 600 mg del compuesto (por ejemplo, como dos comprimidos cada uno de los cuales comprenda 300 mg del compuesto), y luego administrar una segunda dosis que comprenda 600 mg del compuesto (por ejemplo, como dos comprimidos cada uno de los cuales comprenda 300 mg del compuesto) si existe una necesidad continua de reducir profilácticamente la probabilidad de un ataque agudo de HAE después de administrar la primera dosis. La segunda dosis puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la primera dosis. Si existe una necesidad continuada de reducir profilácticamente la probabilidad de un ataque agudo de HAE después de la segunda dosis, el compuesto de Fórmula A para su uso en los tratamientos a demanda de un ataque agudo de HAE de la invención puede comprender la administración de una tercera dosis que contenga 600 mg del compuesto (por ejemplo, dos comprimidos de 300 mg cada uno). La tercera dosis puede administrarse al menos unas 6 horas (por ejemplo, unas 6 horas) después de la segunda dosis.
[0065] Los tratamientos profilácticos a demanda de ataques agudos de HAE descritos en el presente documento pueden comprender no administrar más de tres dosis en un periodo de 24 horas (por ejemplo, tres dosis que comprendan 600 mg del compuesto, opcionalmente como 6 comprimidos cada uno de los cuales comprenda 300 mg del compuesto).
[0066] Como se ha descrito anteriormente, la invención proporciona un compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para su uso en el tratamiento del angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) a un paciente que lo necesite a demanda.

[0067] En algunas realizaciones, la invención proporciona un compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para su uso en el tratamiento del angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A a un paciente que lo necesite, en el que el compuesto de Fórmula A se administra oralmente para reducir profilácticamente la probabilidad de un ataque agudo de HAE, en el que el compuesto de Fórmula A se administra regularmente al paciente, comprendiendo además administrar oralmente el compuesto de Fórmula A a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE.
[0068] El término "administrado regularmente" significa administrar el compuesto de Fórmula A continuamente a intervalos regulares (por ejemplo, una vez a la semana, dos veces a la semana, etc.) para proporcionar un tratamiento eficaz. El profesional sanitario comprendería fácilmente lo que se entiende por administración regular (o continua).
Dosificación
[0069] En cualquiera de los tratamientos de la invención aquí descritos, el compuesto de Fórmula A se administra por vía oral en una cantidad terapéuticamente eficaz.
[0070] En algunas realizaciones, el compuesto de Fórmula A puede administrarse en una dosis diaria de entre aproximadamente 5 mg y aproximadamente 2000 mg por día. "Dosis diaria": la cantidad total administrada en un día. Más específicamente, el compuesto de Fórmula A puede administrarse en una dosis diaria de entre aproximadamente 100 mg
y aproximadamente 1500 mg, aproximadamente 300 mg y aproximadamente 1800 mg, aproximadamente 100 mg y aproximadamente 1400 mg, aproximadamente 200 mg y aproximadamente 1200 mg, aproximadamente 300 mg y aproximadamente 1200 mg, aproximadamente 600 mg y aproximadamente 1200 mg, aproximadamente 450 mg y aproximadamente 900 mg, aproximadamente 500 mg y aproximadamente 1000 mg, aproximadamente 450 mg y aproximadamente 600 mg, aproximadamente 500 mg y aproximadamente 700 mg (más específicamente, 600 mg), aproximadamente 800 mg y aproximadamente 1000 mg al día, aproximadamente 900 mg y aproximadamente 1400 mg (más específicamente 1200 mg), o aproximadamente 900 mg y aproximadamente 1200 mg. En una realización específica, la dosis diaria es de 300 mg. En otra realización específica, la dosis diaria es de 600 mg. En otra realización específica, la dosis diaria es de 900 mg. En otra realización específica, la dosis diaria es de 1200 mg. En otra realización específica, la dosis diaria es de 1800 mg.
[0071] La dosis diaria puede administrarse como una sola dosis, o subdividirse en múltiples dosis para su administración periódica durante el día. A su vez, cada dosis puede administrarse como una única forma farmacéutica, o subdividirse en múltiples formas farmacéuticas. Por ejemplo, una dosis diaria de 1200 mg puede administrarse como dos dosis subdivididas de 600 mg, donde cada una de estas dosis subdivididas puede administrarse como dos dosis subdivididas de 300 mg. Cuando se utilicen múltiples dosis y múltiples formas farmacéuticas, éstas pueden administrarse simultáneamente, por separado o secuencialmente.
[0072] En algunas realizaciones, cada forma farmacéutica unitaria única que comprende el compuesto de Fórmula A comprende entre aproximadamente 5 mg y aproximadamente 1000 mg, aproximadamente 50 mg y aproximadamente 800 mg, aproximadamente 100 mg y aproximadamente 700 mg, aproximadamente 200 mg y aproximadamente 700 mg, aproximadamente 300 mg y aproximadamente 700 mg, o aproximadamente 500 mg y aproximadamente 700 mg del compuesto de Fórmula A. En algunas realizaciones, cada forma farmacéutica unitaria que comprende el compuesto de Fórmula A comprende: aproximadamente 5 mg, aproximadamente 10 mg, aproximadamente 20 mg, aproximadamente 40, aproximadamente 80 mg, aproximadamente 160 mg, aproximadamente 300 mg, aproximadamente 400 mg, aproximadamente 450 mg, aproximadamente 500 mg o aproximadamente 600 mg.
[0073] Cada dosis administrada al paciente puede comprender 600 mg del compuesto que puede subdividirse en dos comprimidos que comprenden 300 mg del compuesto.
[0074] Alternativamente, cada dosis puede comprender 300 mg del compuesto que puede ser un comprimido que comprende 300 mg del compuesto.
[0075] En una realización específica, se administra al paciente una dosis diaria de 600 mg, que se administra como una dosis.
[0076] En otra realización específica, se administra al paciente una dosis diaria de 1200 mg, que se administra como dos dosis, y en particular cuando la segunda dosis se administra entre 2 y 6 horas de la primera, preferentemente entre aproximadamente 3 y 6 horas de la primera dosis.
[0077] En otra realización específica, se administra al paciente una dosis diaria de 1800 mg, que se administra como tres dosis, y en particular cuando la segunda dosis se administra entre 2 y 8 horas de la primera (por ejemplo, a unas 2 horas, unas 4 horas, unas 6 horas o unas 8 horas), y la tercera dosis se administra entre unas 4 y 16 horas de la primera dosis (por ejemplo, a unas 4 horas, unas 6 horas, unas 8 horas, unas 12 horas o unas 16 horas).
[0078] Los tratamientos de la invención implican la administración oral. En cualquiera de los tratamientos de la invención, el compuesto de Fórmula A puede administrarse como una forma farmacéutica oral que comprende el compuesto de Fórmula A y excipientes farmacéuticamente aceptables. La forma farmacéutica oral puede ser un comprimido o una cápsula. En una realización, la forma farmacéutica oral es un comprimido. En otra realización, la forma farmacéutica oral es una cápsula.
[0079] El compuesto de Fórmula A para uso en los tratamientos de la invención puede comprender no administrar más de tres dosis en un período de 24 horas. Específicamente, si cada dosis comprende 600 mg del compuesto, esto significa que el compuesto de Fórmula A para su uso en los tratamientos de las invenciones puede comprender no administrar más de 1800 mg del compuesto en un período de 24 horas. Si cada dosis que comprende 600 mg del compuesto se subdivide en dos dosis (por ejemplo, comprimidos) que comprenden 300 mg del compuesto, el compuesto de Fórmula A para su uso en los tratamientos de la invención puede comprender la administración de no más de seis dosis, cada una de las cuales comprende 300 mg del compuesto, en un período de 24 horas, en el que cada dosis puede ser un comprimido.
[0080] La forma farmacéutica puede ser un comprimido que comprenda celulosa microcristalina como diluyente, croscarmelosa sódica como desintegrante, polivinilpirrolidona como aglutinante y, opcionalmente, estearato de magnesio como lubricante. En un comprimido preferido, el compuesto de Fórmula A comprende: (i) al menos aproximadamente un 40 % en peso del comprimido (más específicamente aproximadamente un 40 % en peso a aproximadamente un 60 % en peso), en comparación con la masa total del comprimido; (ii) aproximadamente un 25 % en peso a aproximadamente un 60 % en peso del diluyente (más específicamente aproximadamente un 25 % en peso a aproximadamente un 40 % en
peso, en comparación con la masa total del comprimido; (iii) entre 1 % y 15 % en peso del desintegrante (más específicamente entre 2 % y 6 % en peso), comparado con la masa total del comprimido; (iv) entre 1 % y 20 % en peso del aglutinante (más específicamente entre 2 % y 5 % en peso), comparado con la masa total del comprimido; y cuando esté presente, (v) entre 0,1 % y 5 % de lubricante.1 a aproximadamente 5 % en peso de lubricante (más específicamente, aproximadamente 0,1 % en peso a aproximadamente 1,5 % en peso), en comparación con la masa total del comprimido. La forma farmacéutica puede ser un comprimido que contenga 300 mg del compuesto.
[0081] El comprimido puede comprender además excipientes extragranulares que comprenden: celulosa microcristalina como diluyente extragranular, croscarmelosa sódica como desintegrante extragranular, polivinilpirrolidona como aglutinante extragranular y/o estearato de magnesio como lubricante extragranular.
[0082] Las formas farmacéuticas descritas en el presente documento (por ejemplo, los comprimidos) pueden estar recubiertas con película, en la que el recubrimiento de la película puede comprender uno o más de hipromelosa, lactosa monohidrato, dióxido de titanio y triacetina.
Otras particularidades del compuesto de Fórmula A para su uso en los tratamientos de la invención
[0083] Como se muestra en el presente documento, el compuesto de Fórmula A tiene un rápido inicio de acción. Específicamente, el compuesto de Fórmula A es un potente inhibidor de la actividad de la calicreína plasmática y es altamente eficaz para interrumpir el bucle de retroalimentación positiva del sistema de activación por contacto entre la calicreína plasmática, la precalicreína, el Factor XII (FXII) y el Factor XIIa (FXlla). Los datos farmacocinéticos y farmacodinámicos aquí proporcionados demuestran que estos efectos se muestran rápidamente tras la administración oral del compuesto de Fórmula A. En consecuencia, el compuesto de Fórmula A para su uso en los tratamientos de la invención son de acción rápida y, por tanto, son particularmente adecuados para tratar el HAE a demanda.
[0084] Como se discutió anteriormente, el compuesto de Fórmula A para uso en los tratamientos de la invención son particularmente ventajosos cuando la concentración del compuesto de Fórmula A es de al menos 500 ng/mL en plasma. Puede observarse una concentración plasmática de al menos 500 ng/mL tras la administración de una dosis de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg) del compuesto de Fórmula A.
[0085] El compuesto de Fórmula A para su uso en los tratamientos según la invención proporciona una protección rápida frente a la escisión del HK (cininógeno de alto peso molecular) que es particularmente adecuada para reducir profilácticamente las posibilidades de un ataque agudo de HAE y/o para acortar la gravedad (o incluso detener) un ataque agudo de HAE en curso. Como se describe aquí, el compuesto de Fórmula A para su uso en los tratamientos según la invención también tienen un efecto farmacodinámico prolongado. Los efectos farmacodinámicos del compuesto de Fórmula A que están relacionados con el tratamiento del HAE incluyen proporcionar protección frente a la escisión de HK, que como se ha comentado anteriormente, puede causar un ataque agudo de HAE. Por ejemplo, el compuesto de Fórmula A puede proporcionar protección contra la escisión de HK al menos (i) inhibiendo la calicreína plasmática, (ii) reduciendo la escisión de la precalicreína plasmática, y/o (iii) reduciendo la generación del Factor XIIa a partir del Factor XII.
[0086] En algunas realizaciones, el compuesto de Fórmula A para su uso en los tratamientos según la invención puede proporcionar protección frente a la escisión de HK (cininógeno de alto peso molecular) dentro de una hora posdosificación, y en particular cuando la dosis del compuesto de Fórmula A es de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg). En algunas realizaciones, el compuesto de Fórmula A para su uso en los tratamientos según la invención puede proporcionar protección contra la escisión del HK (cininógeno de alto peso molecular) dentro de los 45 minutos posteriores a la dosis, o dentro de los 30 minutos posteriores a la dosis. En estas realizaciones, la protección frente a la escisión del HK (cininógeno de alto peso molecular) se determina comparando los niveles de HK en plasma no tratado con los niveles de HK en plasma tratado , es decir, plasma de sujetos que han recibido una dosis del compuesto de Fórmula A, y activando después el plasma con sulfato de dextrano para activar el sistema de contacto e inducir la escisión del HK. Si el nivel de HK en el plasma tratado es superior al nivel de HK en el plasma no tratado, entonces el HK ha sido protegido de la escisión del HK en el plasma activado.
[0087] En algunas realizaciones de la invención, el tratamiento puede inhibir al menos el 80% de la actividad de la calicreína plasmática dentro de los 30 minutos posteriores a la dosis y en particular cuando la dosis del compuesto de Fórmula A es de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg). En algunas realizaciones de la invención, el tratamiento puede inhibir al menos el 90% de la actividad de la calicreína plasmática dentro de los 30 minutos posteriores a la dosis, y en particular cuando la dosis del compuesto de Fórmula A es de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente
800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg). En algunas realizaciones de la invención, el tratamiento puede inhibir al menos el 95% de la actividad de la calicreína plasmática dentro de los 30 minutos posteriores a la dosis, y en particular cuando la dosis del compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o solvato del mismo es de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg). En las realizaciones en las que se menciona la inhibición de la actividad de la calicreína plasmática, la inhibición de la actividad de la calicreína plasmática se determina por hidrólisis dependiente del tiempo de un sustrato fluorogénico (por ejemplo. (H-D-Pro-Phe-Arg-AFC; Peptide Protein Research) según procedimientos conocidos en la técnica. En estas realizaciones, la inhibición de la actividad de la calicreína plasmática se determina en plasma obtenido de sujetos que han tomado una dosis del compuesto de Fórmula A que posteriormente se ha activado con sulfato de dextrano para emular una situación de HAE.
[0088] En algunas realizaciones de la invención, puede alcanzarse una concentración terapéuticamente eficaz del compuesto de Fórmula A dentro de los 20 minutos posteriores a la dosis.
[0089] En algunas realizaciones de la invención, la Tmáx del compuesto de Fórmula A puede estar entre 30 minutos y 3 horas posdosificación, preferentemente entre 30 minutos y 2 horas posdosificación.
[0090] En algunas realizaciones de la invención, el tratamiento puede inhibir al menos el 90% de la actividad de la calicreína plasmática durante al menos el periodo de tiempo comprendido entre 45 minutos y 2 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 100 mg y 200 mg (preferiblemente 160 mg). En algunas realizaciones, el tratamiento puede inhibir al menos el 90% de la actividad de la calicreína plasmática durante al menos el periodo de tiempo comprendido entre 20 minutos y 4 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 100 mg y 200 mg (preferiblemente 160 mg). En algunas realizaciones, el tratamiento puede inhibir al menos el 90% de la actividad de la calicreína plasmática durante al menos el periodo de tiempo comprendido entre 30 minutos y 10 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 300 mg y 800 mg (preferiblemente 600 mg). En algunas realizaciones, el tratamiento puede inhibir al menos el 95% de la actividad de la calicreína plasmática durante al menos el periodo de tiempo comprendido entre 20 minutos y 6 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 300 mg y 800 mg (preferiblemente 600 mg). En algunas realizaciones, el tratamiento puede inhibir al menos el 99% de la actividad de la calicreína plasmática durante al menos el periodo de tiempo comprendido entre 20 minutos y 6 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 300 mg y 800 mg (preferiblemente 600 mg). De nuevo, en estas realizaciones, la inhibición de la actividad de la calicreína plasmática se determina en plasma obtenido de sujetos que han tomado una dosis del compuesto de Fórmula A que posteriormente se ha activado con sulfato de dextrano para emular una situación de HAE.
[0091] En algunas realizaciones, los efectos farmacodinámicos del compuesto de Fórmula A que están relacionados con el tratamiento del HAE pueden mantenerse durante al menos 12 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 300 mg y 800 mg (preferiblemente 600 mg). En algunas realizaciones, el tratamiento puede inhibir al menos el 50% de la actividad de la calicreína plasmática durante al menos 10 horas posdosificación, y en particular cuando la dosis del compuesto de Fórmula A está comprendida entre 100 mg y 200 mg (preferiblemente 160 mg). En estas realizaciones, los efectos farmacodinámicos significan al menos (i) inhibición de la calicreína plasmática, (ii) protección frente a la escisión de HK / reducción de la escisión de HK, (iii) protección frente a (o reducción de) la escisión del Factor XII para generar Factor XIIa, y/o (iv) protección frente a (o reducción de) la escisión de la precalicreína plasmática para generar calicreína plasmática. Por lo tanto, los compuestos de Fórmula A utilizados en los tratamientos según la invención son candidatos adecuados para ser tratamientos ventajosamente eficaces de los ataques agudos de HAE, ya que son de acción rápida y potentes (por ejemplo, inhibidores) durante un periodo de tiempo suficientemente largo.
[0092] Como se discutió anteriormente, en cualquiera de los tratamientos de la invención, el compuesto de Fórmula A puede inhibir la calicreína plasmática.
[0093] En cualquiera de los tratamientos de la invención, particularmente siguiendo una dosis del compuesto de Fórmula A de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg), el compuesto de Fórmula A puede inhibir la escisión del Factor XII para generar Factor XIIa. En cualquiera de los tratamientos de la invención, particularmente siguiendo una dosis del compuesto de Fórmula A de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg), el compuesto de Fórmula A puede inhibir la escisión de precalicreína
plasmática en calicreína plasmática. En cualquiera de los tratamientos de la invención, particularmente siguiendo una dosis del compuesto de Fórmula A de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg), el compuesto de Fórmula A puede dar lugar a la inhibición (por ejemplo, bloqueo) de la activación del sistema de contacto durante hasta 6 horas posdosificación. En algunas realizaciones, cuando se administra una dosis de al menos aproximadamente 60 mg (más específicamente, al menos aproximadamente 70 mg o aproximadamente 80 mg, tales como aproximadamente 80 mg a aproximadamente 900 mg, aproximadamente 100 mg a aproximadamente 800 mg, aproximadamente 200 mg a aproximadamente 700 mg, aproximadamente 300 mg a aproximadamente 600 mg, o aproximadamente 400 mg a aproximadamente 600 mg, específicamente 600 mg), la activación del sistema de contacto puede inhibirse (por ejemplo, bloquearse) durante al menos 6 horas, por ejemplo, durante entre 6 horas y 12 o 18 horas posdosificación.
Figuras
[0094] En las Figuras, el término "Compuesto" significa el compuesto de Fórmula A.
Figura 1 : Patrón de difracción de rayos X del compuesto de Fórmula A generado en el Ejemplo 1.
Figura 2A : Resultados del ensayo que muestran la actividad de inhibición de la calicreína plasmática del compuesto de Fórmula A y un inhibidor de C1 C1-INH en plasma diluido activado con sulfato de dextrano (DXS). Figura 2B . Resultados del ensayo que muestran la actividad de inhibición de la calicreína plasmática del compuesto de Fórmula A y un inhibidor de C1 (C1-INH) en plasma no diluido activado por DXS.
Figura 3A . Resultados del ensayo comparando la actividad de inhibición de la calicreína plasmática del compuesto de Fórmula A y C1-INH en plasma diluido activado por DXS.
Figura 3B : Resultados del ensayo comparando la actividad de inhibición del compuesto de Fórmula A y C1 -INH tras la adición a plasma humano preactivado sin diluir. Los datos se expresan como fluorescencia total a lo largo del tiempo (unidades de fluorescencia) media ± SEM de n=3 experimentos.
Figura 4 A : Resultados de ensayos (bioanalíticos) que muestran las concentraciones plasmáticas del compuesto de Fórmula A entre 0 y 24 horas posdosis, en sujetos en ayunas de ocho (8) cohortes de dosis única ascendente. Figura 4B . Tabla de valores Cmáx determinados a partir de los resultados del ensayo (bioanalítico) mostrados en la Figura 4A.
Figura 5A : Resultados de ensayos que muestran la actividad de la calicreína plasmática en plasma no diluido activado con DXS para las cohortes 6 a 8 (160 mg, 300 mg y 600 mg).
Figura 5 B : Resultados del ensayo que muestran la actividad plasmática media de la calicreína y la concentración plasmática media del compuesto de Fórmula A en plasma no diluido en sujetos de la cohorte 8 (dosis de 600 mg). Figura 6 A : Resultados del ensayo que muestran las mediciones cinéticas fluorescentes medias que indican un tiempo de retraso en la actividad catalítica durante la activación del sistema de contacto en plasma no diluido activado por DXS de un sujeto que ha recibido una dosis de 600 mg del compuesto de Fórmula A.
Figura 6 B : Ampliación de la Figura 6A entre 0 y 5 minutos tras la activación catalítica.
Figura 7 : Resultados del ensayo que muestran el porcentaje medio de protección HK en puntos temporales seleccionados posdosificación en plasma no diluido activado con DXS para las cohortes 6 a 8 (160 mg, 300 mg y 600 mg), y una imagen de gel WES de representación de los datos de inmunoblot.
Figura 8 : Resultados del ensayo que muestran el efecto del compuesto de Fórmula A sobre la escisión de HK activada por DXS en puntos temporales seleccionados post-dosis en la cohorte 8 (600mg), y una representación en gel de WES de los datos de inmunoblot.
Figura 9 : Resultados del ensayo que muestran el efecto del compuesto de Fórmula A en la escisión de precalicreína plasmática activada por DXS (PPK), en puntos temporales seleccionados posdosificación en la cohorte 8 (600 mg), y una imagen de gel de WES de representación de los datos de inmunoblot.
Figura 10 : Resultados del ensayo que muestran el efecto del compuesto de Fórmula A sobre la generación de FXlla activada por DXS, en puntos temporales seleccionados posdosificación en la cohorte 8 (600mg), y una imagen de gel WES de representación de los datos de inmunoblot.
Figura 11 : Resultados de ensayo (bioanalíticos) que muestran el efecto de la concentración plasmática del compuesto de Fórmula A en varias etapas posdosis en la cohorte 8 (600mg) en los puntos temporales seleccionados para el análisis HK, FXlla, PPK.
Figura 12 . Los resultados del ensayo no muestran ningún efecto significativo de los alimentos sobre la actividad inhibidora de la calicreína plasmática del compuesto de Fórmula A en plasma no diluido activado por DXS.
Figuras 13A y 13B : Resultados del ensayo que muestran el curso temporal de la escisión de HK activada por dextranosulfato en plasma entero no diluido de HAE determinado mediante inmunoelectrotransferencia, y una imagen representativa de blot.
Figuras 14a y 14B : Resultados del ensayo que muestran la respuesta a la dosis del compuesto de Fórmula A sobre los niveles de HK de longitud completa en plasma de control sano activado con dextrano sulfato y plasma de HAE, e imágenes representativas de gel del sistema WES.
Figura 15 : Datos farmacocinéticos preliminares del estudio de fase 2 actualmente en curso.
Figura 16A : Concentraciones plasmáticas medias a lo largo del tiempo de 4 cohortes en el estudio de dosis múltiples de fase 1.
Figura 16B : Concentraciones plasmáticas medias a lo largo del tiempo (escala semilogarítmica) de 4 cohortes en
el estudio de dosis múltiples de fase 1.
[0095] Las realizaciones proporcionadas en el presente documento pueden comprenderse mejor haciendo referencia a los siguientes ejemplos. Estos ejemplos pretenden ser ilustrativos de los tratamientos proporcionados en el presente documento, pero no son en modo alguno limitativos. De hecho, el alcance de la invención viene definido por las reivindicaciones.
[0096] Aunque en el presente documento se proporcionan ejemplos de ciertas realizaciones particulares, será evidente para los expertos en la materia que pueden realizarse diversos cambios y modificaciones. Dichas modificaciones también se incluyen en el ámbito de aplicación de las reivindicaciones adjuntas.
Detalles Experimentales Generales
[0097] En los siguientes ejemplos, se utilizan las siguientes abreviaturas y definiciones:

[0098] Todas las reacciones se llevaron a cabo bajo una atmósfera de nitrógeno a menos que se especifique lo contrario.
[0099] Los espectros 1H NMR se registraron en un espectrómetro Bruker (400MHz) o en un espectrómetro JEOL (400MHz) con referencia al disolvente de deuterio y a rt.
[0100] Los iones moleculares se obtuvieron mediante LCMS que se llevó a cabo utilizando una columna Chromolith
Speedrod RP-18e, 50 * 4,6 mm, con un gradiente lineal del 10% al 90% de 0,1% de HCO2H/MeCN en 0,1% de HCO2H/H2O durante 13 min, caudal de 1,5 mL/min, o utilizando Agilent, X-Select, ácido, 5-95% de MeCN/agua durante 4 min. Los datos se recogieron utilizando un espectrómetro de masas Thermofinnigan Surveyor MSQ con ionización por electospray junto con un sistema LC Thermofinnigan Surveyor.
[0101] Alternativamente, los iones moleculares se obtuvieron usando LCMS que se llevó a cabo usando una columna Agilent Poroshell 120 EC-C18 (2.7μm, 3.0 * 50mm) con 0.1% v/v de ácido fórmico en agua [eluyente A]; MeCN [eluyente B]; Tasa de flujo 0.8mL/min y 1.5 minutos de tiempo de equilibrio entre muestras, gradiente mostrado a continuación. La detección de masas se realizó con el espectrómetro de masas API 2000 (electrospray).
Gradiente:
0102

[0103] Cuando los productos se purificaron mediante cromatografía flash, "sílice" se refiere a gel de sílice para cromatografía, de 0,035 a 0,070 mm (malla 220 a 440) (por ejemplo, gel de sílice 60 de Merck), y una presión aplicada de nitrógeno de hasta 10 p.s.i aceleró la elución en columna. Las purificaciones por HPLC preparativa de fase inversa se realizaron con un sistema de bombeo de gradiente binario Waters 2525 a caudales típicos de 20 mL/min utilizando un detector de matriz de fotodiodos Waters 2996.
[0104] Todos los disolventes y reactivos comerciales se utilizaron tal como se recibieron.
[0105] Los nombres químicos se generaron utilizando software automatizado como el software Autonom proporcionado como parte del paquete ISIS Draw de MDL Information Systems o el software Chemaxon proporcionado como componente de MarvinSketch o como componente del IDBS E-WorkBook.
[0106] Los patrones de difracción de polvo de rayos X se recogieron en un difractómetro Philips X-Pert MPD y se analizaron utilizando las siguientes condiciones experimentales (Método A), a menos que se especifique lo contrario:
Ánodo del tubo: Cu
Tensión del generador: 40 kV
Corriente del tubo: 40 mA
Longitud de onda alfa1: 1.5406 A
Longitud de onda alfa2: 1.5444 A
Ángulo inicial [20]: 4
Ángulo final [20]: 40
Escaneado continuo
[0107] Aproximadamente 2 mg de la muestra analizada se comprimieron suavemente en el portamuestras de sílice de corte oblicuo simple con fondo cero para XRPD. A continuación, la muestra se cargó en el difractómetro para su análisis.
Ejemplo 1 - Preparación del compuesto de Fórmula A
A. 1 -(4-hidroximetil-bencil)-1H-piridin-2-ona
[0108] Se disolvió 4-(Clorometil)bencilalcohol (5,0 g, 31,93 mmol) en acetona (150 mL). Se añadieron 2-hidroxipiridina (3,64 g, 38,3 mmol) y carbonato potásico (13,24 g, 95,78 mmol) y la mezcla de reacción se agitó a 50 °C durante 3 h, tras lo cual el disolvente se eliminó in vacuo y el residuo se recogió en cloroformo (100 mL). Esta solución se lavó con agua (30 mL), salmuera (30 mL), se secó (Na2SO4) y se evaporó in vacuo. El residuo se purificó mediante cromatografía flash (sílice), eluyente 3% MeOH / 97% CHCb, para dar un sólido blanco identificado como 1 -(4-hidroximetil-bencil)-1H-piridin-2-ona (5,30 g, 24,62 mmol, 77% de rendimiento).
[M+Na]+ = 238
B. 1-(4-clorometil-bencil)-1H-piridin-2-ona
[0109] 1-(4-hidroximetil-bencil)-1H-piridin-2-ona (8,45 g, 39,3 mmol), DCM seco (80 mL) y trietilamina (7,66 ml, 55,0 mmol) se enfriaron en un baño de hielo. Se añadió cloruro de metanosulfonilo (3,95 ml, 51,0 mmol) y se agitó en baño de hielo
durante 15 min. Se retiró el baño de hielo y se continuó agitando a temperatura rt durante toda la noche. La mezcla de reacción se repartió entre DCM (100 mL) y solución acuosa saturada de NH4Cl (100 mL). La capa acuosa se extrajo con más DCM (2 * 50 mL) y los orgánicos combinados se lavaron con salmuera (50 mL), se secaron sobre Na2SÜ4, se filtraron y se concentraron para dar 1-(4-clorometil-bencil)-1H-piridin-2-ona (8,65 g, 36,6 mmol, 93 % de rendimiento) como sólido amarillo pálido.
[MH]+ = 234,1
C. 3-(metoximetil)-1-(4-((2-oxopiridin-1(2H)-il)metil)bencilo)-1H-pirazol-4-carboxilato de metilo
[0110] Se añadió carbonato de potasio (519 mg, 3,76 mmol) a una solución de 3-(metoximetil)-1H-pirazol-4-carboxilato de metilo (320 mg, 1,88 mmol; CAS no. 318496-66-1 (sintetizado según el método descrito en WO 2012/009009)) y 1-(4-(clorometil)bencil)piridin-2(1 H)-ona (527 mg, 2,26 mmol) en DMF (5 mL) y se calentó a 60 °C durante la noche. La mezcla de reacción se diluyó con EtOAc (50 mL) y se lavó con salmuera (2 * 100 mL), se secó sobre sulfato de magnesio, se filtró y se redujo in vacuo. El producto bruto se purificó mediante cromatografía flash (columna de 40 g, 0-100% EtOAc en isohexanos) para obtener dos regioisómeros. El segundo isómero de la columna se recogió para obtener 3-(metoximetil)-1-(4-((2-oxopiridin-1(2H)-il)metil)bencilo)-1H-pirazol-4-carboxilato de metilo (378 mg, 1,01 mmol, 53,7 % de rendimiento) como una goma incolora.
[MH]+ = 368,2
D. Ácido 3-(metoximetil)-1-(4-((2-oxopiridin-1(2H)-il)metil)bencilo)-1H-pirazol-4-carboxílico
[0111] Al 3-(metoximetil)-1-(4-((2-oxopiridin-1(2H)-il)metil)bencilo)-1H-pirazol-4-carboxilato de metilo (3,77 g, 10,26 mmol) en THF (5 mL) y MeOH (5 mL) se añadió solución de NaOH 2M (15,39 ml, 30,8 mmol) y se agitó a rt durante la noche. Se añadió HCl 1M (50 mL) y se extrajo con EtOAc (50 mL). La capa orgánica se lavó con salmuera (50 mL), se secó sobre sulfato de magnesio, se filtró y se redujo in vacuo para dar ácido 3-(metoximetil)-1-(4-((2-oxopiridin-1(2H)-il)metil)bencilo)-1H-pirazol-4-carboxílico (1,22 g, 3,45 mmol, 33,6 % de rendimiento) como polvo blanco.
[MH]+ = 354,2
E. 3-Fluoro-4-metoxi-piridina-2-carbonitrilo
[0112] En un gran vial de microondas, se añadió cianuro de cobre (I) (1,304 g, 14,56 mmol) a una solución de 2-bromo-3-fluoro-4-metoxipiridina (1 g, 4,85 mmol) en DMF (5 mL). El vial de reacción se cerró herméticamente y se calentó a 100 °C durante 16 h. La mezcla de reacción se diluyó con agua (20 mL) y EtOAc (20 mL). La suspensión espesa se sonicó y requirió agua adicional (40 mL) y EtOAc (2 * 50 mL) con sonicación para romper el sólido precipitado. Las capas combinadas se filtraron a través de un tapón de celita y se aisló la capa orgánica, se lavó con salmuera (50 mL), se secó sobre sulfato de magnesio, se filtró y el disolvente se eliminó a presión reducida para dar un sólido verde pálido identificado como el compuesto deseado 3-fluoro-4-metoxi-piridina-2-carbonitrilo (100 mg, 0,578 mmol, rendimiento del 12 %).
F. Éster terc-butílico del ácido (3-fluoro-4-metoxi-piridin-2-ilmetil)-carbámico
[0113] Se disolvió 3-fluoro-4-metoxi-piridina-2-carbonitrilo (100 mg, 0,578 mmol) en metanol anhidro (10 mL, 247 mmol) y se añadió hexahidrato de cloruro de níquel (14 mg, 0,058 mmol) seguido de dicarbonato de di-terc-butilo (255 mg, 1,157 mmol). La solución verde pálida resultante se enfrió en un baño de hielo-sal hasta -5 °C y a continuación se añadió borohidruro sódico (153 mg, 4,05 mmol) por porciones manteniendo la temperatura de reacción ~0 °C. La solución de color marrón oscuro se dejó agitar a 0 °C y lentamente se dejó calentar hasta rt y luego se dejó agitar a rt durante 3 h. La mezcla de reacción se evaporó a sequedad a 40°C para obtener un residuo negro que se diluyó con DCM (10 mL) y se lavó con hidrogenocarbonato sódico (10 mL). Se formó una emulsión, por lo que los orgánicos se separaron via un cartucho separador de fases y se concentraron. El líquido crudo se purificó por cromatografía eluyendo con EtOAc / iso-Hexano para obtener el compuesto del título, éster terc-butílico del ácido (3-fluoro-4-metoxi-piridin-2-ilmetil)-carbámico como un aceite amarillo claro (108 mg, 62 % de rendimiento).
[MH]+ = 257
G. Sal clorhidrato de C-(3-Fluoro-4-metoxi-piridin-2-il)-metilamina
[0114] El éster terc-butílico del ácido (3-fluoro-4-metoxi-piridin-2-ilmetil)-carbámico (108 mg, 0,358 mmol) se tomó en alcohol isopropílico (1 mL) y luego se añadió HCl (6N en alcohol isopropílico) (1 mL, 0,578 mmol) a rt y se dejó agitar a 40°C durante 2 horas. La mezcla de reacción se concentró a presión reducida y después se trituró con éter, se sonicó y se decantó para dar un sólido de color crema (75 mg, 55% de rendimiento) identificado como sal de clorhidrato de C-(3-fluoro-4-metoxi-piridin-2-il)-metilamina.
[MH]+ = 157
Ejemplo________1a_______ -________N-[(3-Fluoro-4-metox¡p¡r¡d¡n-2-¡l)met¡l1-3-(metox¡met¡l)-1 -({4-[(2-oxopiridin-1-¡l)met¡l1fen¡l}met¡l)p¡razol-4-carboxam¡da (Compuesto de Fórmula A)
[0115] El ácido 3-(metoximetil)-1-(4-((2-oxopiridin-1(2H)-il)metil)bencil)-1H-pirazol-4-carboxílico (825 mg, 2,34 mmol) y la sal de clorhidrato de C-(3-fluoro-4-metoxi-piridin-2-il)-metilamina (450 mg, 2,34 mmol) se disolvieron en DCM mientras se
enfriaba a 0°C. Se añadió clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (627,0 mg, 3,27 mmol), HOBt (378,8 mg, 2,80 mmol) y trietilamina (1,63 mL, 1182 mmol) mientras se agitaba, se dejó que la mezcla se calentara hasta rt y se continuó agitando durante 20 horas. Se añadió cloroformo (50 mL), se lavó la mezcla con NaHCO3(aq) saturado y se redujo in vacuo. El material bruto se purificó por cromatografía eluyendo con metanol/DCM. El disolvente se eliminó in vacuo y el sólido resultante se trituró con éter dietílico. Los sólidos resultantes se recogieron por filtración para obtener el compuesto de Fórmula A.
[MH]+ = 492,0
[0116] NMR (CD3OD) ó: 3.41 (3H, s), 4.03 (3H, s), 4.65 (2H, s), 4.72 (2H, d, J=2.3Hz), 5.24 (2H, s), 5.37 (2H, s), 6.44 (1H, td, J = 1.4, 6.8Hz), 6.62 (1H, d, J = 9.0Hz), 7.18-7.22 (1H, m), 7.31-7.38 (4H, m), 7.56-7.60 (1H, m), 7.75 (1H, dd, J = 1.9, 7.1Hz), 8.18 (1H, s), 8.27 (1H, d, J = 5.6Hz) ppm.
[0117] En la Figura 1 se muestra un difractograma XRPD del compuesto de Fórmula A resultante del procedimiento anterior.

Ejemplo 2 - Preparación de una forma farmacéutica que comprende el compuesto de Fórmula A Mezcla y Compactación con Rodillo
[0119] Equipamiento: Micro Compactador de rodillos y granulador Freund Vector TFC Lab (el compactador de rodillos y el granulador son entidades independientes). A continuación se indican los parámetros del equipo:

Método
[0120] Se prepararon dos formulaciones de comprimidos (Comprimidos A y B) de acuerdo con el siguiente método a escala de mezcla de 30 g para producir comprimidos con componentes en las cantidades que se muestran a continuación.
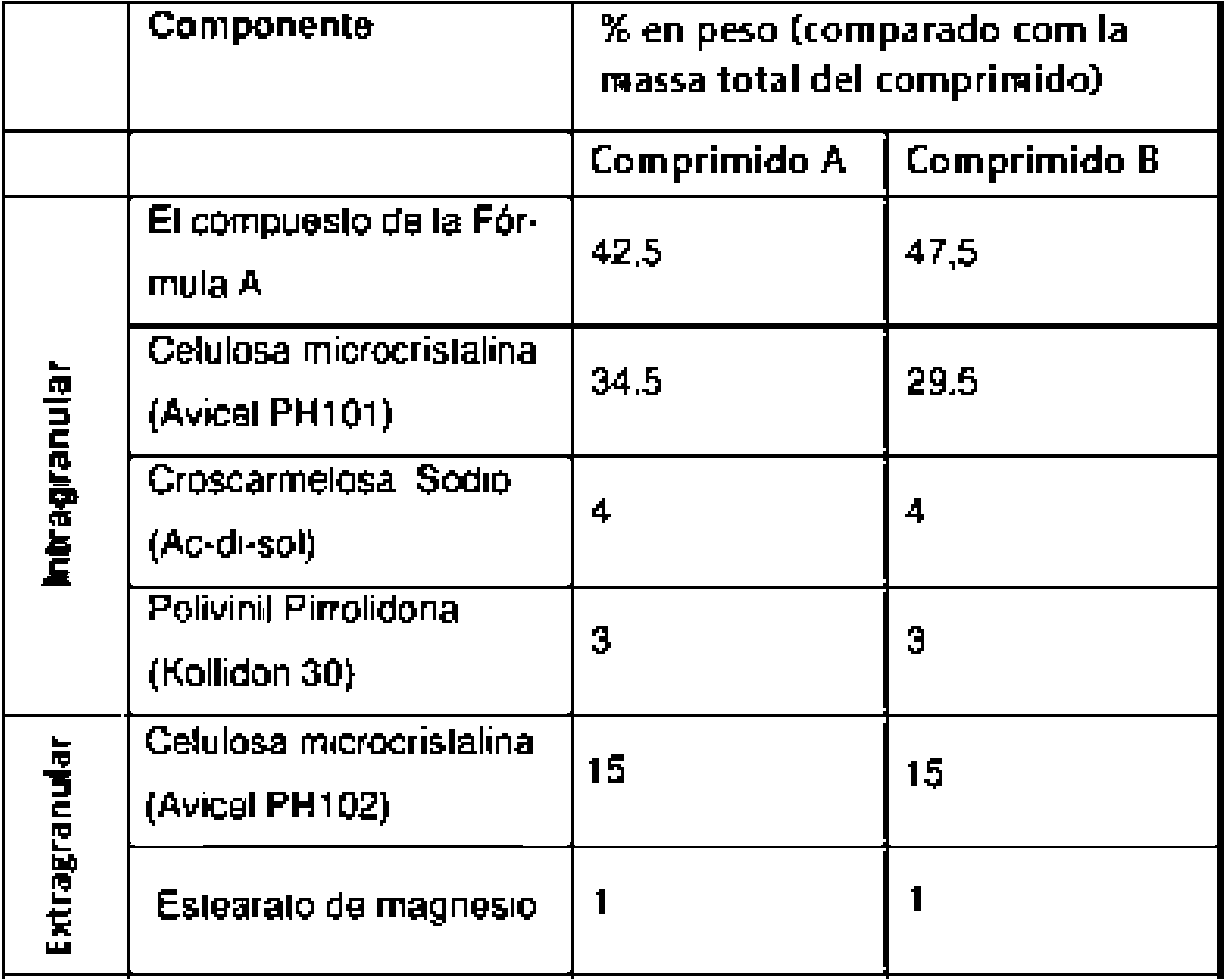
[0121] Para cada uno de los comprimidos, se prepararon mezclas haciendo pasar los componentes intragranulares a través de un tamiz de 355 μm a una escala adecuada para el alcance del compactador de rodillos en un recipiente de vidrio utilizando una Mezcladora Turbula a 34 rpm. A continuación, la mezcla se hizo pasar por el compactador de rodillos utilizando los parámetros descritos anteriormente. Las cintas producidas se recogieron en un recipiente de tamaño adecuado. A continuación, las cintas recogidas se sometieron al granulador fijado con un tamiz de 1 mm y los gránulos resultantes se recogieron para su posterior procesamiento.
Tableteado
[0122] Equipamiento: Prensa de comprimidos de estación única RIVA Mini. A continuación, se muestran los parámetros del equipo:

[0123] Los gránulos se mezclaron posteriormente con sus excipientes extragranulares, respectivamente. Los excipientes extragranulares se prepararon tamizándolos a través de un tamiz de 355 μm en un recipiente de vidrio utilizando una Mezcladora Turbula a 34 rpm. A continuación, se dispensó el peso objetivo del comprimido y se comprimió manualmente
en comprimidos. El comprimido A se comprimió con una fuerza de compresión de 7,2 a 8,8 kN. El comprimido B se comprimió con una fuerza de compresión de 6,9 a 7,7 kN.
[0124] Los comprimidos resultaron ser robustos. Los comprimidos A y B se sometieron posteriormente a pruebas de estabilidad a largo plazo.
[0125] La producción de comprimidos según el método descrito anteriormente se ha escalado a 180 g con un tiempo de compactación con rodillo de aproximadamente 60 minutos.
Ejemplo 3 - Comparación del compuesto de Fórmula A con un inhibidor C1 (C1 -INH)
[0126] Objetivo: Identificar las propiedades bioquímicas y biofísicas del compuesto de Fórmula A que contribuyen a su eficacia óptima en el control del Sistema Calicreína Cinina en plasma. A continuación, estas propiedades se comparan con las del C1-INH como referencia terapéutica para el HAE.
Métodos:
[0127] La actividad inhibidora de la calicreína plasmática in vitro se determinó utilizando métodos estándar publicados (véase, por ejemplo, Johansen et al., Int. J. Tiss. Reac. 1986, 8, 185; Shori et al., Biochem. Pharmacol., 1992, 43, 1209; Stürzebecher et al., Biol. Chem. Hoppe-Seyler, 1992, 373, 1025). La calicreína plasmática humana (Protogen) se incubó a 25 °C con el sustrato fluorogénico H-DPro-Phe-Arg-AFC y diversas concentraciones del compuesto de ensayo. La actividad enzimática residual (velocidad inicial de reacción) se determinó midiendo el cambio en la absorbancia óptica a 410 nm y se determinó el valor IC50 para el compuesto de ensayo.
[0128] La velocidad de formación del complejo enzima-inhibidor (Kon) se determinó utilizando PKa purificada mezclada rápidamente con una solución que contenía sustrato fluorogénico y un rango de concentración de inhibidor. A continuación, se utilizó el establecimiento de la inhibición en función del tiempo para calcular la velocidad de formación del complejo enzima-inhibidor para cada concentración de inhibidor. El Kon se calculó trazando la tasa de inhibición frente a la concentración de inhibidor. Los datos de la Tabla 1 se presentan en μM’1 sec'1.
[0129] La actividad catalítica de la PKa en plasma activado con sulfato de dextrano (DXS, Sigma; 10 μg/ml) (1:4 diluido o sin diluir, plasma de control VisuCon-F, Affinity Biologicals Inc) se determinó mediante la hidrólisis dependiente del tiempo de sustrato fluorogénico. Para determinar el 1C50 y la eficacia, se añadió el compuesto de Fórmula A o C1-INH (Sigma Cat #E0518) antes (Figuras 2A y 2B) o después (Figura 3A) de la adición de DXS al plasma.
[0130] La escisión de HK activada por DXS en plasma no diluido se realizó en ausencia o presencia de 300 nM de inhibidor de PKa y se cuantificó mediante electroforesis en gel SDS-PAGE, utilizando geles Criterion TGX Precast al 7,5% (Biorad). La transferencia se realizó a una membrana Immunobilon-FL PVDF. El análisis de imágenes se realizó con el sistema de imágenes LICOR. Para la inmunotransferencia tradicional se utilizó el anticuerpo monoclonal de ratón anti-HK (MAB15692, R&D systems). Los datos se presentan como % de HK restante tras 20 min de incubación con DXS en comparación con los niveles de HK en plasma no activado (Tabla 1).
[0131] La fracción libre de plasma se determinó utilizando el sistema "Rapid Equilibrium Dialysis" (Thermo Scientific), los compuestos de prueba se prepararon a 5 μM en plasma humano puro y se dializaron contra tampón fosfato durante 5 horas a 37°C. La cuantificación del compuesto particionado en dos cámaras del dispositivo de diálisis se realizó mediante LCMS/ MS. Fracción de compuesto no unido a proteínas plasmáticas presentada como % del total.
[0132] La capacidad del compuesto para inhibir la actividad enzimática del plasma preactivado se evaluó mediante la adición del compuesto después de la estimulación DXS. Se mezclaron alícuotas de plasma (20 μL) con una solución de 2,5 μL que contenía 1.300 mM de sustrato fluorogénico (H-DPro-Phe-Arg-AFC) y una solución de 2,5 μL de sulfato de dextrano (DXS; 100 μg/mL) que actuaba como activador de la vía plasmática calicreína-cinina. La actividad enzimática se midió inmediatamente mediante la monitorización de la acumulación de fluorescencia liberada del sustrato por escisión del sustrato durante 16 minutos. A los 3,5 minutos de la adición de DXS, se añadieron 5 μl de inhibidores o de control de agua en cada pocillo. El compuesto se probó a concentraciones de 300, 1000 y 3000 nM. También se incluyeron C1-INH a una concentración de 3000 nM y controles con vehículo. Los datos se presentan en la Figura 3B.
Resultados:
[0133] Como se muestra en la Figura 2, en ensayos que utilizan el sustrato fluorogénico, el compuesto de Fórmula A parece ser un inhibidor altamente potente de PKa con una potencia de 17 veces y 20 veces frente a C1-INH añadido exógenamente en plasma diluido (Figura 2A) y plasma sin diluir (Figura 2B), respectivamente. La Tabla 1 muestra el perfil bioquímico de las terapias probadas en este ejemplo.
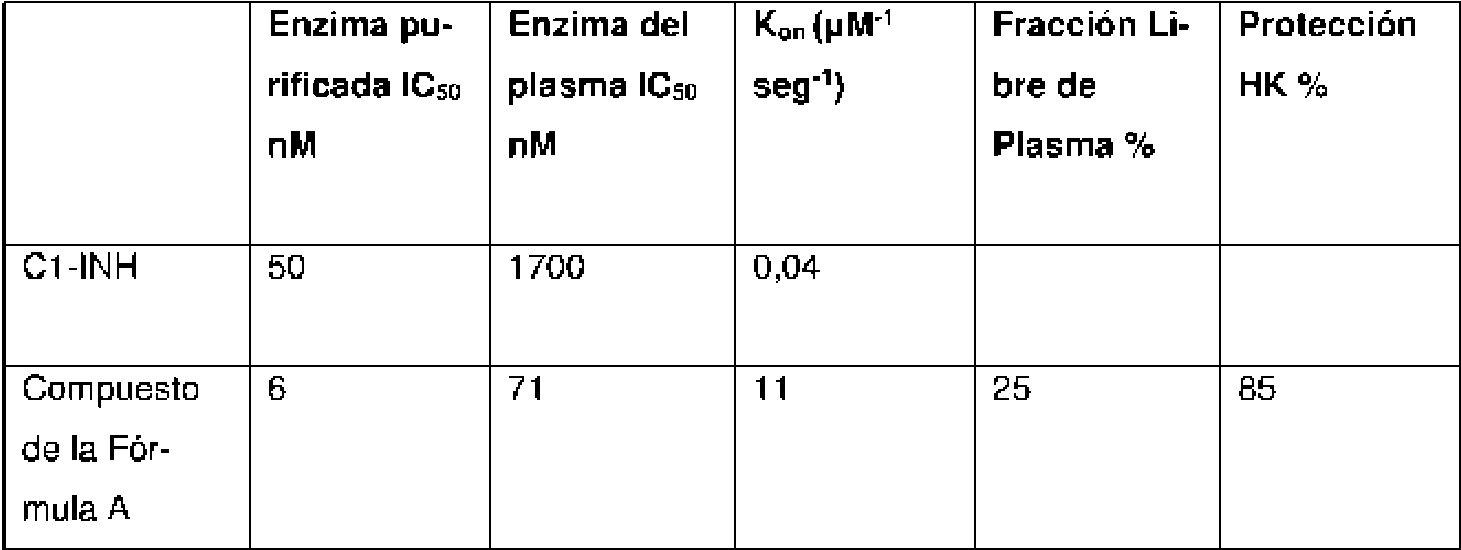
[0134] La Figura 3A muestra una comparación de los efectos de los dos inhibidores: compuesto de Fórmula A y C1 -INH, sobre la actividad de la calicreína plasmática en plasma (diluido 1:4) activado con DXS. Ambos inhibidores se añadieron a concentraciones diez veces superiores a su IC50 al plasma aproximadamente 100 segundos después de la adición del DXS.
[0135] La Figura 3B muestra que la adición del compuesto de Fórmula A después de la activación del plasma causa una inhibición rápida y dependiente de la dosis de la actividad enzimática en comparación con la acción más lenta de C1-INH. La Tabla 2 muestra la potencia bioquímica y la selectividad del compuesto de Fórmula A frente a enzimas humanas aisladas utilizando métodos bibliográficos como para el ensayo in vitro de calicreína plasmática descrito anteriormente.

Ejemplo 4 - Estudio de fase I de dosis única ascendente en varones sanos y efecto alimentario del compuesto [0136] Objetivo: Evaluar los efectos farmacodinámicos (PD) del compuesto de Fórmula A cuando se administra por vía oral utilizando ensayos ex vivo de plasma entero para la actividad catalítica de la calicreína plasmática y la escisión de HK, en muestras de un Estudio de Fase 1 de Dosis Única Ascendente en varones adultos sanos. Asimismo, se pretendía investigar la seguridad, tolerabilidad y efectos farmacocinéticos (PK) del compuesto de Fórmula A cuando se administra por vía oral.
Métodos:
[0137] Este estudio fue aleatorizado, doble ciego, controlado con placebo, de dosis única ascendente (SAD) y estudios cruzados para efecto alimentario y formulaciones en cápsula/comprimido.
[0138] A 64 participantes masculinos sanos (n=6 activos, 2 placebo por cohorte, 8 cohortes SAD) se les administraron dosis únicas ascendentes del compuesto de Fórmula A: 5, 10, 20, 40, 80, 160, 300 o 600 mg en una cápsula.
[0139] Se administró a 8 participantes 100 mg del compuesto de Fórmula A en un estudio cruzado de la cápsula y una formulación en comprimido.
[0140] Se administró a 12 participantes 600 mg del compuesto de Fórmula A en un estudio cruzado de efecto alimentario.
[0141] Se tomaron muestras para la evaluación farmacocinética (PK) y PD a intervalos repetidos durante 48 horas.
[0142] Las muestras de plasma utilizadas para la evaluación de la PK se analizaron mediante un método validado de espectrometría de masas en tándem por cromatografía líquida (LC MS/MS).
[0143] Las mediciones PD se determinaron en plasma no diluido estimulado con sulfato de dextrano (DXS) utilizando un ensayo enzimático fluorogénico y un inmunoensayo de clivaje HK basado en capilares.
[0144] La actividad catalítica de PKa en muestras de plasma estimuladas con DXS (Sigma; 10 μg/mL) del estudio de fase 1 del compuesto de Fórmula A se determinó mediante la hidrólisis dependiente del tiempo de sustrato fluorogénico en todas las muestras de todas las partes del estudio.
[0145] El tiempo hasta la aparición de actividad enzimática amidolítica detectable en plasma estimulado con DXS (tiempo de retraso) se calculó a partir del ensayo de actividad catalítica. La sensibilidad de detección de la tasa de actividad catalítica en plasma basada en el uso de un fluorímetro Spark (Tecan) es un aumento de fluorescencia para alcanzar 1AF unidad/seg.
[0146] La escisión de HK estimulada por DXS, en plasma no diluido se cuantificó mediante inmunoensayo capilar en el Sistema Wes (ProteinSimple) utilizando anticuerpo monoclonal anti-HK y detección basada en quimioluminiscencia. La escisión de HK mediada por calicreína plasmática en plasma humano citratado sin diluir se indujo mediante la activación del sistema de contacto con DXS (6,25 μg/ml) a 4°C en muestras seleccionadas de la fase DUA.
[0147] La escisión estimulada por DXS de la precalicreína plasmática y el Factor XII (FXII) se cuantificaron por inmunoensayo capilar en el Sistema Wes (ProteinSimple) de forma análoga.
Resultados:
[0148] La Figura 4A muestra las concentraciones plasmáticas del compuesto de Fórmula A de 0 a 24 horas posdosis. Como puede observarse, cuando se administra por vía oral, el compuesto de Fórmula A alcanzó una exposición plasmática rápida y dependiente de la dosis en el intervalo de dosis ensayadas de 5 mg a 600 mg. La Figura 4A muestra las curvas de concentración y la Figura 4B muestra la Cmáx para cada cohorte de TAS. El compuesto de Fórmula A se administró en forma de cápsula y el sujeto estaba en ayunas.
[0149] La Figura 5A muestra ensayos enzimáticos en plasma activado sin diluir realizados en muestras de las cohortes 6, 7 y 8. Las dosis de 160 mg y superiores demostraron una inhibición media >90% de la actividad catalítica de la calicreína plasmática entre 45 min y 2 h para la cohorte 6, entre 20 min y 4 h para la cohorte 7. Una dosis de 600 mg (cohorte 8) proporcionó una inhibición >90% de la actividad catalítica de la calicreína plasmática entre 30 min y 6 h posdosis y una inhibición >50% durante 10 h (Figura 5B).
[0150] Las mediciones fluorescentes cinéticas del ensayo enzimático en plasma sin diluir pueden trazarse como curvas de progresión del ensayo (Figuras 6A y 6B). Estas curvas ponen de manifiesto que el compuesto de Fórmula A no sólo tiene un efecto inhibidor sobre la actividad enzimática, sino que también aumenta el tiempo hasta la aparición de la actividad catalítica durante la activación del sistema de contacto (tiempo de retardo). En los primeros momentos tras la administración de la dosis, las muestras de plasma no mostraban una actividad catalítica detectable incluso tras una activación prolongada con el potente activador DXS. En esta prueba, se administró al sujeto una dosis de 600 mg en una formulación de comprimidos.
[0151] La Figura 7 muestra el porcentaje medio de protección HK en plasma no diluido activado por DXS (cohorte DUA 6 (160 mg), 7 (300 mg) y 8 (600 mg)). Como se muestra, las tres dosis del compuesto de Fórmula A fueron capaces de inhibir la actividad catalítica de la calicreína plasmática por encima del 90% durante un periodo de tiempo. La duración de estos efectos PD fue proporcional a la dosis. Se ha demostrado que el compuesto de Fórmula A protege al HK de la escisión activada por Dx S en plasma no diluido durante al menos 10 horas tras una dosis única de 600 mg.
[0152] En la Figura 7, se generó la imagen representativa de gel del sistema WES en muestras duplicadas de plasma sin diluir /- activación DXS de un solo sujeto de la cohorte 8 que recibió 600 mg del compuesto de Fórmula A en comparación con la dosis previa (P-D).
[0153] En la Figura 7, se evaluó la escisión de HK tras la activación DXS de muestras de plasma sin diluir en puntos temporales seleccionados de las cohortes 6 a 8. Los datos se expresan como media /- SEM, n=6.
[0154] Para evaluar si el compuesto de Fórmula A también reducía la generación de calicreína plasmática y Factor XIIa, se utilizaron inmunoensayos para cuantificar los niveles de proteínas del sistema de contacto en plasma activado por DXS en predosis y hasta 12 horas posdosis de 600 mg administrados oralmente en cápsulas. Los resultados de estos ensayos se muestran en las Figuras 8 a 11 y muestran que el compuesto de Fórmula A no sólo reduce la escisión de HK sino que también reduce la escisión de PPK y reduce la generación de FXlla. Estos resultados sugieren que el compuesto de Fórmula A inhibe el sistema de activación por contacto via interrupción del bucle de retroalimentación positiva mediado por la activación de FXII estimulada por PKa.
[0155] La Figura 12 muestra que no se observó ningún efecto significativo de la comida sobre el perfil farmacodinámico (PD) de un comprimido de 600 mg suministrado en estado alimentado y en ayunas. Como puede verse, los efectos de la DP se observan rápidamente en los estados de alimentación y ayuno, con una inhibición de la calicreína plasmática >90% alcanzada a los 30 minutos en ambos estados.
[0156] No se notificaron acontecimientos adversos graves en el ensayo de fase I. Tampoco hubo indicios de tolerabilidad. Ningún sujeto se retiró del ensayo.
[0157] Estos datos demuestran que el compuesto de Fórmula A tiene un efecto inhibidor sobre los sistemas de activación por bradicinina y por contacto. Como ya se ha comentado, estos efectos farmacodinámicos están implicados en trastornos como el HAE. Estos datos también muestran que el compuesto de Fórmula A tiene un perfil farmacocinético adecuado para la administración oral.
Ejemplo 5 - Inmunoensayos que investigan el compuesto de fórmula A en la protección del cininógeno de alto peso molecular (HK) frente a la escisión mediada por PKa en plasma de HAE y de control.
Método:
[0158] La escisión de cininógeno de alto peso molecular (HK) en plasma humano citratado sin diluir se indujo mediante la activación del sistema de contacto con sulfato de dextrano (DXS, Sigma #31395-10G; 6,25 μg/ml) sobre hielo húmedo. Se adquirió un pool de plasma humano normal (CONTROL) (VisuCon-F Frozen Normal Control plasma) a Affinity Biologicals Inc. Se preparó un stock de trabajo de 10mM del compuesto de fórmula A ("el compuesto") en DMSO y se diluyó en 1X PBS hasta las respectivas concentraciones finales descritas. Se obtuvo plasma de sujetos con HAE (n=6) y se confirmó la deficiencia de inhibidor de C1 mediante inmunoelectrotransferencia. A continuación, se determinó la protección de HK frente a la escisión mediada por PKa en plasma entero no diluido estimulado con DXS mediante dos métodos: inmunoelectrotransferencia tradicional e inmunoensayo capilar semiautomatizado.
[0159] Inmunoelectrotransferencia: La electroforesis en gel SDS-PAGE se realizó utilizando geles Criterion TGX Precast al 7,5% (Bio-rad). La transferencia se realizó a una membrana de PVDF Immobilon-FL. El análisis de imágenes se realizó con el sistema de imágenes LICOR. Para la inmunotransferencia tradicional se utilizó un anticuerpo monoclonal de ratón anti-HK humana (MAB15692, R&D systems).
Inmunoensayo capilar en el sistema WES (ProteinSimple):
[0160] Preparación de las muestras: Combine una parte de la mezcla maestra fluorescente 5* con cuatro partes de la muestra de plasma 1:200. Mezclar en vórtex. Calentar las muestras la mezcla maestra fluorescente y la escalera biotinilada a 95°C durante 5 minutos, agitar en vórtex y cargar en la placa WES. Para este método de detección basado en la quimioluminiscencia se utilizó el anticuerpo monoclonal anti-HK humana mediante el sistema Wes (ProteinSimple).
[0161] Análisis: Recoger la medida del área del pico obtenida en el software Compass (archivo cbz) para el peso molecular HK de longitud completa de la muestra del punto temporal respectivo con activación inducida por DXS. El área del pico se define como el área calculada para el perfil del pico espectral para HK. Para medir la inhibición de la calicreína plasmática por el compuesto, se calculó el porcentaje de Hk de longitud completa detectado.
Resultados:
[0162] Las Figuras 13A y 13B muestran el curso temporal de la escisión de HK activada por sulfato de dextrano en plasma entero no diluido de HAE determinado mediante inmunoelectrotransferencia, y un blot representativo.
[0163] Las Figuras 14A y 14B muestran una imagen de gel representativa del sistema WES y que el compuesto de Fórmula A proporciona protección dependiente de la dosis contra la escisión de HK tanto en plasma de HAE como en plasma de control sano estimulado con sulfato de dextrano determinado mediante inmunoensayo basado en capilares utilizando el sistema WES.
Ejemplo 6 - Estudio de fase 2 del compuesto de Fórmula A
[0164] Objetivo: Evaluar la eficacia y seguridad del compuesto de Fórmula A en el tratamiento a demanda de los ataques de angioedema en sujetos adultos con angioedema hereditario de tipo I o II.
Métodos:
[0165] El estudio es un ensayo clínico aleatorizado, doble ciego, controlado con placebo, de fase 2, cruzado, que evalúa la eficacia y la seguridad del compuesto de fórmula A ("el compuesto"), un inhibidor oral de la calicreína plasmática, en el tratamiento a demanda de los ataques de angioedema en sujetos adultos con angioedema hereditario de tipo I o II (número EudraCT: 2018-004489-32).
Objetivos:
Objetivo Principal:
[0166]
Investigar la eficacia del compuesto comparado con placebo para detener la progresión de un ataque periférico o abdominal de angioedema hereditario (HAE).
Objetivos Secundarios:
[0167]
Investigar la seguridad y tolerabilidad del compuesto.
Investigar el perfil farmacocinético (PK) del compuesto cuando se toma durante el periodo intercrítico entre ataques de HAE.
Investigar el perfil farmacodinámico (PD) del compuesto en la reducción de la concentración de cininógeno de alto peso molecular (HK) escindido residual durante el periodo intercrítico entre ataques de HAE.
Investigar el perfil de PD del compuesto en la reducción de la actividad enzimática plasmática activada durante el periodo intercrítico entre los ataques de HAE.
Configuración:
[0168] Se trata de un ensayo clínico cruzado de fase 2, de dos partes, dos secuencias y dos periodos (2x2). Los sujetos con HAE de tipo I o II serán reclutados a través de centros de tratamiento de HAE de Europa y EE.UU.
[0169] En la Parte 1, los sujetos recibirán una dosis oral única de 600 mg del compuesto para investigar la seguridad, PK y PD del compuesto durante el período intercrítico entre ataques de HAE.
[0170] Los sujetos adultos elegibles >18 años de edad se someterán a una evaluación de cribado para la inclusión en el estudio, recibirán el fármaco del estudio, seguido de una evaluación de 4 horas, en la clínica, de seguridad y PK / PD.
[0171] En la Parte 2, los sujetos serán aleatorizados 1:1 a 2 secuencias de tratamiento. Esta parte del estudio se realizará fuera de la clínica o el hospital. En la Secuencia 1 (brazo de estudio 1) los sujetos recibirán una dosis única de 600 mg del compuesto para tratar el primer ataque de HAE elegible. Tras la resolución de este ataque, los sujetos recibirán una segunda dosis única de placebo para tratar el segundo ataque de HAE elegible.
[0172] En la Secuencia 2 (brazo de estudio 2), los sujetos recibirán una dosis única de placebo para tratar el primer ataque de HAE elegible. Tras la resolución de este ataque, los sujetos recibirán una segunda dosis única de 600 mg del compuesto para tratar el segundo ataque de HAE elegible.
[0173] Se requiere un período mínimo de lavado de 48 horas entre cada dosis del fármaco en estudio.
[0174] Los ataques laríngeos o faciales no son susceptibles de tratamiento. Los ataques de HAE deben tratarse en la primera hora de aparición y antes de alcanzar la gravedad en la escala global de gravedad de los ataques. Los sujetos también deben ser capaces de identificar el inicio de un ataque de HAE. En el momento en que se produzca el ataque de HAE elegible, los sujetos lo notificarán al médico dedicado al estudio o a la persona cualificada designada con una descripción del ataque de HAE. El médico dedicado al estudio o la persona cualificada designada confirmará la elegibilidad del ataque de HAE y aceptará que se administre el fármaco del estudio. Los ataques de HAE requieren la documentación, en el Diario del Sujeto, de la localización del ataque, los síntomas del ataque, la hora de inicio, la gravedad del ataque y la hora de la última comida sustancial antes de la dosis. Los sujetos tomarán el fármaco del estudio, según las instrucciones, y completarán evaluaciones cronometradas de sus síntomas de ataque de HAE durante un periodo de 48 horas, tal y como se documenta a continuación en la Tabla 3. El médico dedicado al estudio o la persona cualificada designada se pondrá en contacto con el sujeto en las 24h siguientes al ataque de HAE elegible para confirmar la seguridad y el bienestar del sujeto. Se indicará a los sujetos que se pongan en contacto con el médico dedicado al estudio o con la persona cualificada designada en caso de que surjan problemas de seguridad. En caso de hipersensibilidad, los sujetos deben ponerse en contacto con el médico dedicado al estudio o con la persona cualificada designada, o con el servicio de urgencias más cercano. El médico dedicado al estudio o su designado cualificado estará disponible 24 horas al día y 7 días a la semana para recibir las llamadas de los sujetos.
Tabla 3: Frecuencia de la Evaluación del Sujeto

*en caso de que tratamiento convencional de ataque, el sujeto deberá realizar evaluaciones cada 30 min durante las 4 h siguientes a la primera administración del tratamiento de ataque convencional. Después de esto, el sujeto debe volver a la frecuencia original de las evaluaciones basadas en el momento de la administración del fármaco del estudio.
[0175] Los sujetos volverán a la clínica después del primer ataque de HAE, antes del segundo ataque de HAE, para someterse a controles de seguridad, incluida la notificación de acontecimientos adversos (AA), el registro de constantes vitales y la revisión del Diario del Sujeto.
[0176] Una vez que se hayan tratado dos ataques de HAE en la Parte 2, el sujeto regresará a la clínica para someterse a los últimos controles de seguridad, incluidos el informe de EA, el registro de signos vitales y la toma de muestras de sangre para mediciones de seguridad en el laboratorio.
[0177] Se permite el tratamiento convencional del ataque después de 4 horas, o antes si se justifica, tras la toma del fármaco del estudio, siempre que el sujeto considere que los síntomas del ataque de HAE son lo suficientemente graves como para requerir tratamiento según el régimen terapéutico habitual del sujeto, o se considere que no se puede administrar el tratamiento con el fármaco del estudio, o se asocien con síntomas laríngeos o faciales. Antes de utilizar un tratamiento convencional contra los ataques, los sujetos lo notificarán al médico dedicado al estudio o a la persona cualificada designada, quien confirmará que el tratamiento convencional es adecuado según el protocolo y el informe del sujeto sobre la gravedad de los síntomas. Los sujetos pueden tratar sus crisis de HAE con su tratamiento convencional para las crisis (pdC1INH o rhC1INH intravenoso [iv] o icatibant).
Medicamento en investigación:
[0178] El compuesto de fórmula A - comprimido recubierto con película de 100 mg. Contienen los siguientes excipientes: celulosa microcristalina, croscarmelosa sódica, povidona, estearato de magnesio; el recubrimiento estético contiene hipromelosa, lactosa monohidrato, dióxido de titanio y triacetina.
[0179] Placebo al compuesto 100 mg comprimido recubierto con película. Contienen celulosa microcristalina, dióxido de silicio coloidal, almidón glicolato sódico y estearil fumarato sódico y están recubiertos con una película; el recubrimiento estético contiene hipromelosa, lactosa monohidrato, dióxido de titanio y triacetina.
[0180] En este estudio no se permiten modificaciones de la dosis del fármaco en estudio.
Número de Sujetos:
[0181] Aproximadamente 60 sujetos serán inscritos en el estudio para asegurar que 50 sujetos completen el estudio. Población:
[0182] La población del estudio incluirá sujetos masculinos y femeninos de 18 años o más con HAE de tipo I o II.
Criterios de Inclusión:
[0183]
1. Hombres o mujeres adultos mayores de 18 años.
2. Diagnóstico confirmado de HAE tipo I o II en cualquier momento de la historia clínica:
a. Historial clínico documentado compatible con HAE (episodios de inflamación subcutánea o mucosa no pruriginosa sin urticaria acompañante) Y
b. Antígeno del inhibidor de la C1-esterasa (C1-INH) o nivel funcional <40% del nivel normal. Los sujetos con un nivel de antígeno o de C1-INH funcional entre el 40% y el 50% del nivel normal pueden ser incluidos si también
tienen un nivel de C4 por debajo del rango normal y antecedentes familiares compatibles con HAE de tipo I o II.
3. Al menos 3 ataques de HAE documentados en los últimos 93 días, según la historia clínica.
4. Acceso al tratamiento convencional de los ataques de HAE y capacidad para utilizarlo.
5. Funciones orgánicas adecuadas, tal como se definen a continuación:
a. Hemoglobina dentro del rango normal;
b. Cociente internacional normalizado (INR)< 1,2;
c. Tiempo de tromboplastina parcial activada (aPTT) ≤ límite superior de la normalidad (ULN);
d. Creatinina < 1x ULN;
e. Aclaramiento de creatinina (CrCI) > 60 mL/min;
f. Alanina aminotransferasa (ALT) ≤ 2x ULN;
g. Aspartato aminotransferasa (AST) ≤ 2x ULN;
h. Bilirrubina total ≤ 1,5x ULN;
i. Leucocitos ≤ 1,5x ULN;
j. Trombocitos ≤ 1,5x ULN.
6. Las mujeres en edad fértil deben comprometerse a utilizar métodos anticonceptivos altamente eficaces desde la visita de cribado hasta el final de los procedimientos de seguimiento del ensayo.
[0184] Los métodos anticonceptivos altamente efectivos incluyen:
a. Anticoncepción hormonal sólo con progestágenos asociada a la inhibición de la ovulación: oral / inyectable / implantable.
[0185] (Los anticonceptivos hormonales que contienen estrógenos están excluidos según el criterio de exclusión 3). b. Dispositivo intrauterino (DIU).
c. Sistema intrauterino liberador de hormonas (SIH).
d. Oclusión tubárica bilateral.
e. Pareja vasectomizada (siempre que la pareja sea la única pareja sexual de la mujer en edad fértil y que la pareja vasectomizada haya recibido una evaluación médica del éxito de la cirugía).
f. Abstinencia sexual (este método no es aceptable en Suiza).
Nota: La abstinencia sexual sólo se considerará un método altamente eficaz si se define como la abstinencia de relaciones heterosexuales. La fiabilidad de la abstinencia sexual debe evaluarse en relación con la duración del ensayo clínico y el estilo de vida preferido y habitual del sujeto.
7. Las mujeres en edad no fértil, definidas como estériles quirúrgicamente (estado posterior a histerectomía, ooforectomía bilateral o ligadura de trompas bilateral) o posmenopáusicas durante al menos 12 meses, no necesitan anticonceptivos durante el estudio.
8. Los varones con parejas femeninas en edad fértil deben aceptar la abstinencia o utilizar un método anticonceptivo altamente eficaz, tal como se define en el criterio de inclusión 6, desde la visita de cribado hasta el final de los procedimientos de seguimiento del ensayo.
9. Proporcionar el consentimiento informado firmado y estar dispuestos y ser capaces de cumplir los requisitos y procedimientos del estudio.
Criterios de exclusión:
[0186]
1. Cualquier diagnóstico concomitante de otra forma de angioedema crónico, como deficiencia adquirida del inhibidor C1, HAE con C1-INH normal (también conocido como HAE de tipo III), angioedema idiopático o angioedema asociado a urticaria.
2. Uso actual de C1INH, andrógenos, lanadelumab o ácido tranexámico para la profilaxis del HAE.
3. Uso de inhibidores de la enzima convertidora de angiotensina (ACE) o cualquier medicamento que contenga estrógenos con absorción sistémica (como anticonceptivos orales o terapia hormonal sustitutiva) en los 93 días previos al tratamiento inicial del estudio.
4. Uso de andrógenos (por ejemplo, estanozolol, danazol, oxandrolona, metiltestosteronas, testosterona) o antifibrinolíticos en los 30 días previos al tratamiento inicial del estudio.
5. Uso de lanadelumab en las 10 semanas previas al tratamiento inicial del estudio.
6. Uso de inhibidores e inductores potentes de CYP3A4/CYP2C9 durante la participación en el ensayo.
Nota: Estos medicamentos incluyen, entre otros, los siguientes cobicistat, conivaptán, itraconazol, ketoconazol, posaconazol, voriconazol, ritonavir, boceprevir, telaprevir, troleandomicina, claritromicina, carbamazepina, enzalutamida, mitotano, fenitαna, fenobarbital, fluconazol, isoniazida, metronidazol, paroxetina, sulfametoxazol, rifampicina, St. John's Wort, diltiazem, idelalisib, nefazodona y nelfinavir.
7. Electrocardiograma (ECG) anormal clínicamente significativo en la Visita 1 y antes de la dosis en la Visita 2. Esto incluye, pero no se limita a, un QTcF > 470 mseg (para mujeres) o > 450 mseg (para hombres), un PR > 220 mseg o contracciones prematuras ventriculares y/o auriculares que sean más frecuentes que ocasionales y/o se produzcan como pares o más agrupadas.
8. Cualquier antecedente clínicamente significativo de angina de pecho, infarto de miocardio, síncope, arritmias cardiacas clínicamente significativas, hipertrofia ventricular izquierda, cardiomiopatía o cualquier otra anomalía cardiovascular. 9. Cualquier otra disfunción sistémica (por ejemplo, gastrointestinal, renal, respiratoria, cardiovascular) o enfermedad o trastorno significativo que, en opinión del investigador, pudiera poner en peligro la seguridad del sujeto al participar en el
ensayo.
10. Antecedentes de abuso o dependencia de sustancias que pudieran interferir con la realización del estudio, según determine el investigador.
11. Alergia o intolerancia conocida a la lactosa.
12. Hipersensibilidad conocida al compuesto o al placebo o a cualquiera de los excipientes.
13. Participación en un estudio clínico de investigación intervencionista en un plazo de 93 días o en un plazo de 5 semividas desde la última dosis del fármaco en investigación (lo que sea más largo) antes del tratamiento inicial del estudio.
14. Cualquier sujeto embarazada o en periodo de lactancia.
Evaluaciones:
[0187] Parte 1: Se recogerán muestras de sangre para las mediciones de PK y PD en los siguientes momentos: Predosis (0h), 15 min, 30 min, 45 min, 1h, 1,5h, 2h, 3h y 4h posdosis. Los signos vitales (presión arterial sistólica [PAS], presión arterial diastólica [PAD], frecuencia del pulso [PR], frecuencia respiratoria [FR] y temperatura corporal) se medirán predosis (0h), 1h y 4h posdosis. Las muestras para las evaluaciones de seguridad post-tratamiento en laboratorio se tomarán con las muestras de 4h PK / PD.
[0188] Parte 2: Tras la ingesta del fármaco del estudio, se realizarán evaluaciones de la gravedad general de los ataques de HAE y de los cambios en la gravedad de los ataques de HAE durante un periodo de 48 horas, tal y como se documenta en la Tabla 3 anterior.
Variables de Eficacia:
[0189] Se evaluará el tiempo hasta el uso del tratamiento de ataque convencional. El diario del sujeto recogerá los criterios de valoración de la eficacia, incluido el tiempo transcurrido hasta el uso del tratamiento convencional de la crisis y la gravedad de la crisis de HAE.
[0190] La gravedad general de los ataques de HAE se evaluará en una escala de Likert de 5 puntos (5LS) y se puntuará como nula, leve, moderada, grave y muy grave.
[0191] El cambio en la gravedad de los ataques de HAE se evaluará mediante una pregunta de transición de 7 puntos (7TQ), puntuada como Mucho mejor / Mejor / Un poco mejor / Sin cambios / Un poco peor / Peor / Mucho peor.
[0192] El tipo de síntomas de ataque de HAE (dolor abdominal, dolor cutáneo e hinchazón cutánea) se evaluará cada uno en una escala analógica visual (VAS) de 100 mm anclada en 0 (ninguno) y 100 (muy grave).
Variables de Seguridad:
[0193]
AE, incluidos los efectos adversos graves (SAE).
Resultados de pruebas de laboratorio (química clínica, hematología, coagulación y análisis de orina).
Signos vitales (PAS, PAD, PR, RR, temperatura corporal).
Hallazgos del examen físico.
Resultados del ECG.
Prueba de embarazo (mujeres en edad fértil).
Criterios de Evaluación de la Eficacia
Criterios de Evaluación Primarios de Eficacia:
[0194]
Tiempo hasta el uso del tratamiento de ataque convencional.
Criterios de Evaluación Secundarios de Eficacia:
[0195]
Proporción de ataques de HAE que progresan un nivel o más en el 5LS o que requieren tratamiento convencional del ataque en las 12h siguientes al fármaco del estudio.
Tiempo transcurrido entre el tratamiento y (1) progresión de la gravedad global del ataque en el 5LS en un nivel o más, o (2) uso de tratamiento convencional del ataque, lo que ocurra primero en las 12h siguientes.
Criterios de Evaluación Exploratorios:
[0196]
Gravedad global acumulada del ataque en el 5LS tras el fármaco del estudio expresada como área bajo la curva (AUC) para el compuesto 600 mg frente a placebo.
Proporción de ataques de HAE que requieren tratamiento convencional.
Proporción de ataques de HAE calificados como "peor" o "mucho peor" en el TQ.
Proporción de ataques de HAE calificados como "mejores" o "mucho mejores" en el TQ.
Tiempo transcurrido desde la administración del fármaco del estudio hasta la resolución completa del ataque de HAE (calificación de ninguno) en la escala global de gravedad del ataque (5LS).
Es hora de que el ataque del HAE se califique peor o mucho peor en el TQ.
Es hora de que el ataque de HAE se califique mejor o mucho mejor en el TQ.
Métodos Estadísticos Generales y Tipos de Análisis
Conjuntos de Análisis:
[0197]
Conjunto de seguridad (SAF): Sujetos que hayan tomado al menos una dosis del fármaco en estudio (incluida la dosis del fármaco en estudio de la Parte 1).
Conjunto de análisis completo (para eficacia) (FAS): Todos los sujetos aleatorizados que recibieron ambas dosis del fármaco del estudio en la Parte 2.
Por conjunto de protocolos (de eficacia) (PPS): Sujetos aleatorizados en la Parte 2 que recibieron el compuesto ambas dosis del fármaco del estudio en la Parte 2 y no tienen desviaciones importantes del protocolo.
Conjunto de análisis PK / PD: Todos los sujetos para los que se tomaron muestras PK / PD en la Parte 1.
Tamaño de la muestra:
[0198] Se propone un tamaño de muestra de 50 sujetos (25 por secuencia) para proporcionar una potencia del 90% para las pruebas al nivel alfa del 5% (2 caras) para el criterio de valoración primario del tiempo hasta el uso del tratamiento de ataque convencional. El tamaño de la muestra se ha calculado partiendo de la base de que el 40% de los sujetos utilizarán el tratamiento de ataque convencional en la rama de control y el 10% en la rama experimental, y de que los datos entre sujetos tienen una correlación mínima. La hipótesis de correlación mínima debe ser conservadora con respecto al tamaño de la muestra. Se inscribirán aproximadamente 60 sujetos para garantizar que 50 completen el estudio.
[0199] Se propone un sobremuestreo del 20% (10 sujetos) para tener en cuenta a los sujetos que no completen ambos periodos de tratamiento debido a ataques de HAE infrecuentes o no elegibles, o a los sujetos que interrumpan el ensayo antes de tiempo, por cualquier motivo. Así pues, el número de participantes en el estudio se considerará suficiente para abordar la hipótesis de eficacia primaria una vez que 50 sujetos hayan completado ambos periodos de tratamiento. Dado que no se requiere más exposición y podría considerarse innecesaria, se pedirá a los sujetos en curso que no hayan completado ambos periodos que regresen al centro del estudio y completen la Visita 4 (visita de interrupción precoz). Los datos de todos los sujetos, completos e incompletos, se analizarán en el conjunto de seguridad.
Consideraciones Generales:
[0200] Los datos individuales de los sujetos se presentarán en listados de datos de sujetos. Se calcularán estadísticas descriptivas adecuadas para los datos continuos y categóricos y se resumirán en formato tabular.
Análisis de Muestras:
[0201] Los AE se codificarán utilizando el diccionario Medical Dictionary for Regulatory Activities (MedDRA) (v21.0 o superior) y se clasificarán por término preferido y clase de órgano del sistema (SOC). Las listas de acontecimientos adversos emergentes del tratamiento (TEAE), TEAE graves y TEAE que causan interrupción prematura se proporcionarán por grupo de secuencia, y se clasificarán además por gravedad del EATT y relación con el fármaco del estudio.
Análisis de Eficacia:
Criterio de Evaluación Principal
[0202] El criterio de valoración primario, el tiempo transcurrido hasta el uso del tratamiento de ataque convencional, se analizará mediante una generalización de la prueba de Gehan propuesta por Feingold y Gillespie (1996) (Ensayos cruzados con datos censurados. Statistics in Medicine 1996; 15(10): 953-967) para reflejar las medidas repetidas en cada tema. Los sujetos serán tratados como censurados si no se produce empeoramiento en las 12h siguientes al fármaco del estudio.
Criterios de Evaluación Secundarios
[0203] La proporción de ataques de HAE que empeoran en un nivel o más en el 5LS o que requieren tratamiento convencional del ataque en las 12h siguientes al fármaco del estudio se analizará mediante la prueba de Prescott (1981) (La comparación de las tasas de éxito en ensayos cruzados en presencia de un efecto de orden. Applied Statistics 1981; 30: 9-15) para comparar los brazos de tratamiento.
[0204] Se seguirá un enfoque similar al utilizado para el criterio de valoración primario para el análisis del tiempo transcurrido entre el fármaco del estudio y el empeoramiento del ataque de HAE en un nivel o más en el 5LS o el uso de un tratamiento convencional del ataque, lo que ocurra primero dentro de las 12h. Además de las pruebas descritas anteriormente, se presentarán estadísticas descriptivas para los criterios de valoración primarios, secundarios y exploratorios, en cada caso comparando el compuesto con el placebo, tales como:
Gravedad global acumulada del ataque en el 5LS tras el fármaco del estudio expresada como AUC para el compuesto 600 mg frente a placebo.
Proporción de ataques de HAE que requieren tratamiento convencional.
Proporción de ataques de HAE calificados como "peor" o "mucho peor" en el TQ.
Proporción de ataques de HAE calificados como "mejores" o "mucho mejores" en el TQ.
Tiempo transcurrido desde la administración del fármaco del estudio hasta la resolución completa del ataque de HAE (calificación de ninguno) en la escala global de gravedad del ataque (5LS).
Es hora de que el ataque del HAE se califique peor o mucho peor en el TQ.
Es hora de que el ataque de HAE se califique mejor o mucho mejor en el TQ.
Análisis PK:
[0205] Los parámetros PK no compartimentales incluirán la concentración máxima en plasma (Cmáx), el tiempo para alcanzar la Cmáx en plasma (tmáx) y el área bajo la curva desde el tiempo 0 hasta la última muestra (AUC0-t). La modelización de la PK por compartimentos describirá la PK del compuesto y generará la Cmáx, la tmáx, el AUC, el aclaramiento aparente (CL/F), el volumen aparente de distribución (Vd/F) y la semivida de eliminación terminal estimada (t1^ ) subyacentes.
[0206] Los parámetros PK del compuesto se determinarán a partir de los datos individuales de concentración frente al tiempo utilizando Phoenix WinNonlin. En caso de desviación del tiempo teórico, se utilizará el tiempo real de la muestra de sangre en el cálculo de los parámetros PK derivados. Se enumerarán y resumirán las concentraciones individuales y los parámetros PK derivados del compuesto en plasma para cada tratamiento. Los datos de concentración-tiempo individuales y medios geométricos se trazarán en escalas lineales y semilogarítmicas.
Análisis PD:
[0207] El efecto del compuesto sobre la actividad de la calicreína plasmática (PKa) se analizará utilizando dos medidas exploratorias de la actividad de la enzima PKa en plasma:
Un ensayo para determinar la inhibición de la actividad de la enzima calicreína plasmática activada exógenamente a partir de muestras de plasma obtenidas antes y después de recibir el compuesto.
Un ensayo para medir el nivel de protección de la escisión del sustrato de cininógeno de alto peso molecular (HK) (contenido en plasma entero) frente a la actividad enzimática de la calicreína plasmática.
[0208] La PD se resumirá para cada tratamiento. Los datos individuales y medios se facilitarán en un apéndice del informe final del estudio clínico.
Datos PK preliminares de la Parte 1 del estudio:
[0209] En el momento de presentar esta solicitud, se han cotejado y analizado los datos PK preliminares de 27 pacientes con HAE, que se muestran en la Tabla 4 y la Figura 15.
Tabla 4

[0210] Así, estos resultados preliminares muestran que el compuesto de Fórmula A demuestra un perfil farmacocinético adecuado para la administración oral a demanda en pacientes con HAE. El estudio está en curso en el momento de su presentación.
Ejemplo 7 - Estudio de fase 1 de dosis múltiples en adultos sanos
[0211] Objetivo: Evaluar la seguridad, la tolerabilidad, la farmacocinética y la variación del QTc con respecto al valor basal tras la administración del compuesto formulado en comprimidos recubiertos con película de 100 mg en sujetos adultos sanos.
Objetivo Principal:
[0212]
Investigar la seguridad y tolerabilidad de dosis múltiples del compuesto.
Objetivos Secundarios:
[0213]
Investigar la farmacocinética (PK) de dosis múltiples del compuesto.
Evaluar los efectos del compuesto sobre los parámetros ECG, incluida la relación concentración-QTc, tras la administración del compuesto 100 mg Comprimidos recubiertos con película (KalVista Pharmaceuticals) a sujetos adultos sanos.
Objetivos Exploratorios:
[0214]
Investigar la farmacodinámica (PD) de dosis múltiples del compuesto.
Métodos:
[0215] Se trata de un estudio de fase 1, doble ciego, controlado con placebo, de múltiples dosis y múltiples cohortes para evaluar la seguridad y tolerabilidad del compuesto, así como los efectos sobre el ECG del compuesto formulado como comprimidos recubiertos con película de 100 mg en sujetos adultos sanos de ambos sexos.
[0216] Se prevé evaluar cuatro (4) cohortes. Las cohortes 1, 2 y 3 incluirán 8 sujetos cada una. La cohorte 4 incluirá 18 sujetos. Se intentará incluir el mismo número de hombres y mujeres en cada cohorte.
[0217] Durante el estudio, se administrarán dosis orales de 600 mg del compuesto como comprimidos recubiertos con película (seis comprimidos de 100 mg) o 6 comprimidos de placebo iguales una vez cada 8 horas (Cohorte 1) cada 4 horas (Cohorte 2), o cada 2 horas (Cohortes 3 y 4) a sujetos adultos sanos de ambos sexos hasta una dosis total de 1800 mg. En las cohortes 1, 2 y 3, 6 sujetos recibirán el compuesto en forma de comprimidos recubiertos con película de 100 mg y 2 sujetos recibirán el placebo para un total de 8 sujetos por cohorte. En la Cohorte 4, 12 sujetos recibirán el compuesto en forma de comprimidos recubiertos con película de 100 mg y 6 sujetos recibirán el placebo para un total de 18 sujetos.
[0218] La progresión de la Cohorte 1 a la Cohorte 2 y de la Cohorte 2 a la Cohorte 3 se producirá tras la revisión de los datos de seguridad (análisis de laboratorio, constantes vitales, ECG de seguridad y acontecimientos adversos) capturados durante la realización de la Cohorte 1 y la Cohorte 2. La progresión a la Cohorte 4 se producirá tras la revisión de los datos de seguridad y farmacocinéticos de la Cohorte 3. Se revisarán los datos farmacocinéticos de la Cohorte 3 para garantizar que la Cmáx de la 3a dosis sea lo suficientemente alta como para respaldar la evaluación del cambio en el intervalo QTc con respecto al valor basal.
[0219] Se conectará un monitor Holter a cada sujeto para registrar continuamente ECG. El monitor se colocará 1 hora antes de la primera dosis y permanecerá colocado hasta después de la última extracción de sangre. Los electrodos para el monitor Holter serán comprobados por un miembro del personal de la clínica a intervalos apropiados para asegurarse de que están fijados.
[0220] Se recogerán muestras de sangre antes de la dosis, a intervalos después de la primera dosis y a intervalos de más de 24 horas después de la dosis final (tercera dosis) (40 horas desde la dosis inicial en la Cohorte 1, 32 horas desde la dosis inicial en la Cohorte 2, 28 horas desde la dosis inicial en las Cohortes 3 y 4) en cada cohorte. Los sujetos permanecerán confinados en el centro clínico desde al menos 10 horas antes de la dosis hasta después de la última extracción de muestras de sangre en cada cohorte del estudio y volverán a la clínica entre 5 y 7 días después de la última dosis para las evaluaciones de seguridad.
[0221] La farmacocinética del compuesto se medirá mediante un procedimiento analítico plenamente validado y el efecto farmacodinámico sobre la actividad enzimática de inhibición de la calicreína plasmática se evaluará mediante una evaluación farmacodinámica exploratoria.
[0222] Se realizará un análisis estadístico para evaluar la relación entre las concentraciones plasmáticas del fármaco y el cambio desde la línea de base en los efectos ECG de la formulación de prueba.
Administración del tratamiento
Cohort 1
[0223] Los sujetos recibirán el tratamiento de prueba o placebo cada 8 horas durante un período de 16 horas (3 administraciones de: 6 * 100 mg del compuesto como comprimidos recubiertos con película de 100 mg o administraciones de dosis placebo a las 0, 8 y 16 horas, dosis total de 1800 mg del compuesto o placebo) según un programa de aleatorización de dos tratamientos bajo observación directa. Cada dosis se administrará con 240 mL de agua a temperatura ambiente. Se indicará a los sujetos que traguen los comprimidos enteros, sin masticarlos ni morderlos. Cualquier sujeto que muerda o mastique los comprimidos será eliminado del estudio. Inmediatamente después de la dosificación se realizará una revisión bucal
Cohort 2
[0224] Los sujetos recibirán el tratamiento de prueba o placebo cada 4 horas durante un período de 8 horas (3 administraciones de: 6 * 100 mg del compuesto como comprimidos recubiertos con película de 100 mg o administraciones de dosis placebo a las 0, 4 y 8 horas, dosis total de 1800 mg del compuesto o placebo) según un programa de aleatorización de dos tratamientos bajo observación directa. Cada dosis se administrará con 240 mL de agua a temperatura ambiente. Se indicará a los sujetos que traguen los comprimidos enteros, sin masticarlos ni morderlos. Cualquier sujeto que muerda o mastique los comprimidos será eliminado del estudio. Inmediatamente después de la dosificación se realizará una comprobación bucal para asegurarse de que los comprimidos se han tragado enteros sin masticarlos ni morderlos.
Cohortes 3 y 4
[0225] Los sujetos recibirán el tratamiento de prueba o placebo cada 2 horas durante un período de 4 horas (3 administraciones de: 6 * 100 mg del compuesto como comprimidos recubiertos con película de 100 mg o administraciones de dosis placebo a las 0, 2 y 4 horas, dosis total de 1800 mg del compuesto o placebo) según un programa de aleatorización de dos tratamientos bajo observación directa. Cada dosis se administrará con 240 mL de agua a temperatura ambiente. Se indicará a los sujetos que traguen los comprimidos enteros, sin masticarlos ni morderlos. Cualquier sujeto que muerda o mastique los comprimidos será eliminado del estudio. Inmediatamente después de la dosificación se realizará una comprobación bucal para asegurarse de que los comprimidos se han tragado enteros sin masticarlos ni morderlos.
[0226] Todos los sujetos ayunarán (excepto agua) durante al menos 8 horas antes de la primera dosis. Tras la dosis inicial, los sujetos continuarán en ayunas hasta al menos 6 horas después de la primera dosis.
Método de Asignación de los Sujetos a los Grupos de Tratamiento:
Cohortes 1, 2 y 3
[0227] Los sujetos serán aleatorizados de tal manera que 6 sujetos recibirán el producto de prueba y 2 sujetos recibirán el placebo. Como medida de seguridad, se incorporará un esquema de dosificación centinela para cada cohorte, en el que un sujeto recibirá el producto de prueba y otro el producto placebo, seguidos por el resto de la cohorte.
Cohorte 4
[0228] Los sujetos serán aleatorizados de tal manera que 12 sujetos reciban el producto de prueba y 6 sujetos reciban el placebo.
[0229] El programa de aleatorización se generará antes de la primera cohorte de dosificación utilizando SAS®, Versión 9.4 o superior.
Resultados:
[0230] No se reportaron eventos adversos serios durante el estudio y ningún sujeto fue discontinuado debido a un AE. Todos los acontecimientos adversos notificados se consideraron de gravedad "leve" y tuvieron un desenlace de "recuperado/resuelto" al final del estudio.
[0231] No se identificaron efectos clínicamente relevantes sobre los parámetros ECG estudiados.
[0232] La Figura 16A muestra las concentraciones plasmáticas medias del compuesto de Fórmula A después de la dosis inicial para cada cohorte.
[0233] La Figura 16B muestra las concentraciones plasmáticas medias (escala semilogarítmica) del compuesto de fórmula A para cada cohorte.
[0234] Estos datos demuestran que el compuesto de Fórmula A tiene un perfil farmacocinético adecuado para la administración oral cuando se administra en dosis múltiples. Los resultados sugieren además que el compuesto de
Fórmula A puede dosificarse con seguridad a intervalos regulares.
Claims (32)
1. Un compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para su uso en el tratamiento del angioedema hereditario (HAE) a demanda que comprende: administrar oralmente el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) a un paciente que lo necesite a demanda,

2. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 1,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) es para uso en el tratamiento de un ataque agudo de angioedema hereditario (HAE) a demanda y se administra oralmente a demanda tras el reconocimiento de un síntoma de un ataque agudo de HAE.
3. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 2,
en el que el síntoma de un ataque agudo de HAE reconocido es al menos uno de los siguientes: hinchazón de los tejidos; fatiga; dolor de cabeza; dolores musculares; hormigueo en la piel; dolor abdominal; náuseas; vómitos; diarrea; dificultad para tragar; ronquera; falta de aliento; y/o cambios de humor.
4. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 2 o 3,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra oralmente a demanda en el plazo de 1 hora desde que se reconoce el síntoma de un ataque agudo de HAE.
5. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 2 a 4,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra oralmente a demanda en los 30 minutos, 20 minutos, 10 minutos o 5 minutos siguientes al reconocimiento del síntoma de un ataque agudo de HAE.
6. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 2 a 5,
en el que el compuesto de Fórmula A (o una sal o solvato farmacéuticamente aceptable del mismo) se administra por vía oral a demanda en la fase prodrómica de un ataque agudo de HAE.
7. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 6,
en el que el síntoma reconocido es al menos uno de los siguientes: una ligera hinchazón, dolor abdominal o enrojecimiento de la piel.
8. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 7,
en el que el síntoma reconocido es el eritema marginado.
9. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 1 a 8,
en el que el tratamiento acorta la duración del ataque agudo de HAE.
10. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 6,
en el que el tratamiento evita que el ataque agudo de HAE progrese a la fase de inflamación de un ataque agudo de HAE.
11. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la
reivindicación 1,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra oralmente a demanda para reducir profilácticamente la probabilidad de un ataque agudo de HAE.
12. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 11,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra oralmente a demanda cuando se prevé que se inducirá un ataque agudo de HAE.
13. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 12,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra oralmente a demanda cuando se prevé que un ataque agudo de HAE será inducido por un traumatismo físico y/o estrés.
14. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 13,
en el que se prevé que un ataque agudo de HAE será inducido por los traumas físicos de un procedimiento dental y/o el estrés mental asociado a un procedimiento dental.
15. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 11 a 14,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra por vía oral a demanda para prevenir un ataque agudo de HAE.
16. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquier reivindicación precedente,
donde el compuesto (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra como una forma farmacéutica oral que comprende: (i) el compuesto (o una sal farmacéuticamente aceptable y/o un solvato del mismo), y (ii) excipientes farmacéuticamente aceptables.
17. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 16,
donde la forma farmacéutica oral es un comprimido que comprende celulosa microcristalina como diluyente, croscarmelosa sódica como desintegrante, polivinilpirrolidona como aglutinante y, opcionalmente, estearato de magnesio como lubricante.
18. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquier reivindicación precedente,
en el que el compuesto (o una sal farmacéuticamente aceptable y/o un solvato del mismo) (i) inhibe la calicreína plasmática, (ii) reduce la escisión de la precalicreína plasmática, y/o (iii) reduce la generación del Factor XIIa a partir del Factor XII.
19. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 18,
en el que se administra al paciente una dosis del compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) tal que el plasma del paciente tiene una concentración del compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) de al menos 500 ng/mL.
20. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 19,
en el que se administra al paciente al menos 60 mg del compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo).
21. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquier reivindicación precedente,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) bloquea la activación del sistema de contacto durante un máximo de seis horas.
22. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquier reivindicación precedente,
en el que el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) se administra a una dosis diaria de entre 5 mg y 2000 mg.
23. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquier reivindicación precedente,
en el que el compuesto de Fórmula A se administra en una dosis diaria de entre 100 mg y 1500 mg, entre 300 mg y 1800
mg, entre 100 mg y 1400 mg al día, entre 200 mg y 1200 mg, entre 300 mg y 1200 mg, entre 600 mg y 1200 mg, entre 450 mg y 900 mg, entre 500 mg y 1000 mg, entre 450 mg y 600 mg, entre 500 mg y 700 mg, entre 800 mg y 1000 mg al día, entre 900 mg y 1400 mg, o entre 900 mg y 1200 mg.
24. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquier reivindicación precedente,
en el que se administra al paciente la dosis diaria en dos dosis en un periodo de 24 horas a partir de la toma de la primera dosis.
25. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 24,
en el que las dos dosis se administran simultánea, separada o secuencialmente.
26. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 24 o 25,
en la que la segunda dosis se administra entre 2 y 6 horas después de la primera, preferiblemente entre 3 y 6 horas después de la primera dosis.
27. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 24 o 25,
en la que la segunda dosis puede administrarse al menos unas 6 horas después de la primera dosis.
28. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 1 a 23,
en el que se administra al paciente el compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) en tres dosis al día.
29. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 28,
en el que las tres dosis se administran simultánea, separada o secuencialmente.
30. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 28 o 29,
donde la segunda y tercera dosis pueden administrarse al menos unas 6 horas después de la dosis anterior.
31. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según cualquiera de las reivindicaciones 21 a 30,
en el que cada dosis comprende aproximadamente 600 mg del compuesto de fórmula A.
32. El compuesto de Fórmula A (o una sal farmacéuticamente aceptable y/o un solvato del mismo) para uso según la reivindicación 31,
en el que cada dosis se administra en forma de dos comprimidos, cada uno de los cuales contiene aproximadamente 300 mg del compuesto de fórmula A.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962861725P | 2019-06-14 | 2019-06-14 | |
| GBGB1910116.1A GB201910116D0 (en) | 2019-07-15 | 2019-07-15 | Treatments of hereditary angioedema |
| PCT/GB2020/051439 WO2020249977A1 (en) | 2019-06-14 | 2020-06-15 | Treatments of hereditary angioedema |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2956471T3 true ES2956471T3 (es) | 2023-12-21 |
Family
ID=67700145
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20734283T Active ES2956471T3 (es) | 2019-06-14 | 2020-06-15 | Tratamientos del angioedema hereditario |
Country Status (34)
| Country | Link |
|---|---|
| US (1) | US20220218680A1 (es) |
| EP (2) | EP4282474A3 (es) |
| JP (3) | JP7356518B2 (es) |
| KR (1) | KR102748728B1 (es) |
| CN (1) | CN114126612A (es) |
| AR (1) | AR119158A1 (es) |
| AU (1) | AU2020293614B2 (es) |
| BR (1) | BR112021024664A2 (es) |
| CA (1) | CA3142218A1 (es) |
| CL (2) | CL2021003244A1 (es) |
| DK (1) | DK3982960T3 (es) |
| EA (1) | EA202193019A1 (es) |
| ES (1) | ES2956471T3 (es) |
| FI (1) | FI3982960T3 (es) |
| GB (1) | GB201910116D0 (es) |
| HR (1) | HRP20230696T1 (es) |
| HU (1) | HUE063163T2 (es) |
| IL (1) | IL288615A (es) |
| LT (1) | LT3982960T (es) |
| MA (1) | MA56187B1 (es) |
| MD (1) | MD3982960T2 (es) |
| MX (1) | MX2021014557A (es) |
| MY (1) | MY205687A (es) |
| PH (1) | PH12021552966A1 (es) |
| PL (1) | PL3982960T3 (es) |
| PT (1) | PT3982960T (es) |
| RS (1) | RS64412B1 (es) |
| SG (1) | SG11202113304YA (es) |
| SI (1) | SI3982960T1 (es) |
| SM (1) | SMT202300261T1 (es) |
| TW (1) | TW202112370A (es) |
| UA (1) | UA129869C2 (es) |
| WO (1) | WO2020249977A1 (es) |
| ZA (1) | ZA202110685B (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201609607D0 (en) | 2016-06-01 | 2016-07-13 | Kalvista Pharmaceuticals Ltd | Polymorphs of N-(3-Fluoro-4-methoxypyridin-2-yl)methyl)-3-(methoxymethyl)-1-({4-((2-oxopy ridin-1-yl)methyl)phenyl}methyl)pyrazole-4-carboxamide and salts |
| GB201719881D0 (en) | 2017-11-29 | 2018-01-10 | Kalvista Pharmaceuticals Ltd | Solid forms of plasma kallikrein inhibitor and salts thereof |
| GB201910116D0 (en) * | 2019-07-15 | 2019-08-28 | Kalvista Pharmaceuticals Ltd | Treatments of hereditary angioedema |
| EP4010333A1 (en) | 2019-08-09 | 2022-06-15 | Kalvista Pharmaceuticals Limited | Plasma kallikrein inhibitors |
| EP4232031A1 (en) * | 2020-10-23 | 2023-08-30 | Kalvista Pharmaceuticals Limited | Treatments of angioedema |
| JP2024505596A (ja) * | 2021-02-09 | 2024-02-06 | カルビスタ・ファーマシューティカルズ・リミテッド | 遺伝性血管性浮腫の治療 |
| EP4480545A3 (en) | 2022-04-27 | 2025-02-26 | Kalvista Pharmaceuticals Limited | Formulations of a plasma kallikrein inhibitor |
| CN116003386B (zh) * | 2022-11-20 | 2024-03-26 | 药康众拓(北京)医药科技有限公司 | 一种氘代n-苄基吡啶酮吡唑甲酰胺类化合物、药物组合物和用途 |
| WO2025172692A1 (en) | 2024-02-13 | 2025-08-21 | Kalvista Pharmaceuticals Limited | Oral sebetralstat for the treatment of an attack of hereditary angioedema |
| WO2025172693A1 (en) | 2024-02-13 | 2025-08-21 | Kalvista Pharmaceuticals Limited | Oral sebetralstat for the treatment of an attack of hereditary angioedema |
| WO2026003532A1 (en) | 2024-06-28 | 2026-01-02 | Kalvista Pharmaceuticals Limited | Sebetralstat for the treatment of anxiety associated with hereditary angioedema |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5187157A (en) | 1987-06-05 | 1993-02-16 | Du Pont Merck Pharmaceutical Company | Peptide boronic acid inhibitors of trypsin-like proteases |
| GB9019558D0 (en) | 1990-09-07 | 1990-10-24 | Szelke Michael | Enzyme inhibitors |
| SE9301911D0 (sv) | 1993-06-03 | 1993-06-03 | Ab Astra | New peptide derivatives |
| US5589467A (en) | 1993-09-17 | 1996-12-31 | Novo Nordisk A/S | 2,5',N6-trisubstituted adenosine derivatives |
| GB0205527D0 (en) | 2002-03-08 | 2002-04-24 | Ferring Bv | Inhibitors |
| US7429604B2 (en) | 2004-06-15 | 2008-09-30 | Bristol Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| WO2008016883A2 (en) | 2006-07-31 | 2008-02-07 | Activesite Pharmaceuticals, Inc. | Inhibitors of plasma kallikrein |
| DE102006050672A1 (de) | 2006-10-24 | 2008-04-30 | Curacyte Discovery Gmbh | Hemmstoffe des Plasmins und des Plasmakallikreins |
| JP2013121919A (ja) | 2010-03-25 | 2013-06-20 | Astellas Pharma Inc | 血漿カリクレイン阻害剤 |
| WO2012004678A2 (en) | 2010-07-07 | 2012-01-12 | The Medicines Company (Leipzig) Gmbh | Serine protease inhibitors |
| EP2595986A2 (en) | 2010-07-14 | 2013-05-29 | Addex Pharma SA | Novel 2-amino-4-pyrazolyl-thiazole derivatives and their use as allosteric modulators of metabotropic glutamate receptors |
| US9290485B2 (en) | 2010-08-04 | 2016-03-22 | Novartis Ag | N-((6-amino-pyridin-3-yl)methyl)-heteroaryl-carboxamides |
| GB2494851A (en) | 2011-07-07 | 2013-03-27 | Kalvista Pharmaceuticals Ltd | Plasma kallikrein inhibitors |
| GB201300304D0 (en) | 2013-01-08 | 2013-02-20 | Kalvista Pharmaceuticals Ltd | Benzylamine derivatives |
| EP2999697B1 (en) | 2013-05-23 | 2017-04-19 | Kalvista Pharmaceuticals Limited | Heterocyclic derivates |
| EP2815749A1 (en) * | 2013-06-20 | 2014-12-24 | IP Gesellschaft für Management mbH | Solid form of 4-amino-2-(2,6-dioxopiperidine-3-yl)isoindoline-1,3-dione having specified X-ray diffraction pattern |
| EP2886107A1 (en) * | 2013-12-17 | 2015-06-24 | ObsEva S.A. | Oral formulations of pyrrolydine derivatives |
| GB201421083D0 (en) | 2014-11-27 | 2015-01-14 | Kalvista Pharmaceuticals Ltd | Enzyme inhibitors |
| GB201721515D0 (en) * | 2017-12-21 | 2018-02-07 | Kalvista Pharmaceuticals Ltd | Dosage forms comprising a plasma kallikrein inhibtor |
| GB201910116D0 (en) | 2019-07-15 | 2019-08-28 | Kalvista Pharmaceuticals Ltd | Treatments of hereditary angioedema |
-
2019
- 2019-07-15 GB GBGB1910116.1A patent/GB201910116D0/en not_active Ceased
-
2020
- 2020-06-15 CN CN202080043365.6A patent/CN114126612A/zh active Pending
- 2020-06-15 RS RS20230620A patent/RS64412B1/sr unknown
- 2020-06-15 AU AU2020293614A patent/AU2020293614B2/en active Active
- 2020-06-15 HR HRP20230696TT patent/HRP20230696T1/hr unknown
- 2020-06-15 EP EP23181261.1A patent/EP4282474A3/en active Pending
- 2020-06-15 PL PL20734283.3T patent/PL3982960T3/pl unknown
- 2020-06-15 SG SG11202113304YA patent/SG11202113304YA/en unknown
- 2020-06-15 CA CA3142218A patent/CA3142218A1/en active Pending
- 2020-06-15 BR BR112021024664A patent/BR112021024664A2/pt not_active Application Discontinuation
- 2020-06-15 UA UAA202106869A patent/UA129869C2/uk unknown
- 2020-06-15 LT LTEPPCT/GB2020/051439T patent/LT3982960T/lt unknown
- 2020-06-15 MY MYPI2021007320A patent/MY205687A/en unknown
- 2020-06-15 WO PCT/GB2020/051439 patent/WO2020249977A1/en not_active Ceased
- 2020-06-15 FI FIEP20734283.3T patent/FI3982960T3/fi active
- 2020-06-15 KR KR1020217043374A patent/KR102748728B1/ko active Active
- 2020-06-15 ES ES20734283T patent/ES2956471T3/es active Active
- 2020-06-15 SI SI202030247T patent/SI3982960T1/sl unknown
- 2020-06-15 EA EA202193019A patent/EA202193019A1/ru unknown
- 2020-06-15 DK DK20734283.3T patent/DK3982960T3/da active
- 2020-06-15 HU HUE20734283A patent/HUE063163T2/hu unknown
- 2020-06-15 MD MDE20220440T patent/MD3982960T2/ro unknown
- 2020-06-15 PH PH1/2021/552966A patent/PH12021552966A1/en unknown
- 2020-06-15 US US17/617,439 patent/US20220218680A1/en active Pending
- 2020-06-15 TW TW109120108A patent/TW202112370A/zh unknown
- 2020-06-15 EP EP20734283.3A patent/EP3982960B1/en active Active
- 2020-06-15 PT PT207342833T patent/PT3982960T/pt unknown
- 2020-06-15 JP JP2021571931A patent/JP7356518B2/ja active Active
- 2020-06-15 MA MA56187A patent/MA56187B1/fr unknown
- 2020-06-15 MX MX2021014557A patent/MX2021014557A/es unknown
- 2020-06-15 SM SM20230261T patent/SMT202300261T1/it unknown
- 2020-06-16 AR ARP200101681A patent/AR119158A1/es unknown
-
2021
- 2021-12-02 IL IL288615A patent/IL288615A/en unknown
- 2021-12-06 CL CL2021003244A patent/CL2021003244A1/es unknown
- 2021-12-20 ZA ZA2021/10685A patent/ZA202110685B/en unknown
-
2023
- 2023-03-06 CL CL2023000639A patent/CL2023000639A1/es unknown
- 2023-08-14 JP JP2023131860A patent/JP7688078B2/ja active Active
-
2025
- 2025-05-21 JP JP2025084646A patent/JP2025124716A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2956471T3 (es) | Tratamientos del angioedema hereditario | |
| JP7640474B2 (ja) | 血管性浮腫の治療 | |
| US20240122909A1 (en) | Treatments of hereditary angioedema | |
| US20230381162A1 (en) | Treatments of angioedema | |
| US20250281397A1 (en) | Formulations of a plasma kallikrein inhibitor | |
| HK40101443A (en) | Treatments of hereditary angioedema | |
| HK40070596B (en) | Treatments of hereditary angioedema | |
| HK40070596A (en) | Treatments of hereditary angioedema | |
| EA048676B1 (ru) | Лечение наследственного ангионевротического отека | |
| EP4673437A1 (en) | New solid form of a plasma kallikrein inhibitor | |
| JP2022069377A (ja) | 血管性浮腫の処置 |