FR2840302A1 - Derives d'isoindolones, procede de preparation et intermediaire de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant - Google Patents
Derives d'isoindolones, procede de preparation et intermediaire de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant Download PDFInfo
- Publication number
- FR2840302A1 FR2840302A1 FR0206783A FR0206783A FR2840302A1 FR 2840302 A1 FR2840302 A1 FR 2840302A1 FR 0206783 A FR0206783 A FR 0206783A FR 0206783 A FR0206783 A FR 0206783A FR 2840302 A1 FR2840302 A1 FR 2840302A1
- Authority
- FR
- France
- Prior art keywords
- isoindol
- dihydro
- oxo
- guanidine
- acetyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
- C07D209/46—Iso-indoles; Hydrogenated iso-indoles with an oxygen atom in position 1
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Anesthesiology (AREA)
- Transplantation (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Indole Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
La présente invention concerne les nouveaux dérivés d'isoindolones de formule générale (I)dans laquelle R1 et R2 sont indépendamment : hydrogène, alkyle (C1 -C4), alkényle, alkynyle, aryle, hétéroaryle, halogène, nitro, amino, alkylamino (C1 -C4), NRaRb, alkoylamino (C1-C4), hydroxy, alkoxy (C1-C4), S(O)nR7 (n=0,1, 2), carboxy, alkoxycarbonyle (C1-C4), alkylcarbonyle (C1-C4), carboxamide, cyano, polyfluoroalkyle (C1 -C4), polyfluoroalkoxy (C1 -C3), SO3H, où R3 représente un atome d'hydrogène, un groupe aryle, hétéroaryle ou une chaîne du type Alk-R8, R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8), polyfluoroalkyle (C1-C4), aryle, hétéroaryle, hydroxy, alkoxy (C1 -C4), carboxy, carboxamide, amino, alkylamino (C1-C4), NRaRb, où R4, R5 et R6 représentent indépendamment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4), où R7 est un alkyle linéaire ou ramifié (C1-C4), où Ra et Rb représentent R7, ou bien Ra et Rb forment ensemble avec l'atome d'azote auquel ils sont rattachés un hétérocycle à 5 ou 6 chaînons pouvant contenir éventuellement un autre hétéroatome choisi parmi O, S, ou N
Description
<Desc/Clms Page number 1>
DERIVES D'ISOINDOLONES, PROCEDE DE PREPARATION ET
INTERMEDIAIRES DE CE PROCEDE A TITRE DE MEDICAMENTS ET
COMPOSITIONS PHARMACEUTIQUES LES RENFERMANT La présente invention concerne les composés nouveaux isoindolones de formule générale (I)

dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), alkényle, alkynyle, aryle, hétéroaryle, halogène, nitro, amino, alkylamino (C1-C4), NRaRb, alkoylamino (C1-C4), hydroxy, alkoxy (CI- C4), S(O)nR7 (n=0,l,2), carboxy, alkoxycarbonyle (C1-C4), alkylcarbonyle (C1-C4), carboxamide, cyano, polyfluoroalkyle (C1-C4), polyfluoroalkoxy (C1-C3), S03H, RI et R2 étant eux-même éventuellement substitués par un groupement alkyle linéaire ou ramifié (C1-C4) où R3 représente un atome d'hydrogène, un groupe aryle, hétéroaryle ou une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8), polyfluoroalkyle (CI-C4), aryle, hétéroaryle, hydroxy, alkoxy (C1-C4), carboxy, carboxamide, amino, alkylamino (C1-C4), NRaRb, où R4, R5 et R6 représentent indépendamment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4) où R7 est un alkyle linéaire ou ramifié (C1-C4)
INTERMEDIAIRES DE CE PROCEDE A TITRE DE MEDICAMENTS ET
COMPOSITIONS PHARMACEUTIQUES LES RENFERMANT La présente invention concerne les composés nouveaux isoindolones de formule générale (I)
dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), alkényle, alkynyle, aryle, hétéroaryle, halogène, nitro, amino, alkylamino (C1-C4), NRaRb, alkoylamino (C1-C4), hydroxy, alkoxy (CI- C4), S(O)nR7 (n=0,l,2), carboxy, alkoxycarbonyle (C1-C4), alkylcarbonyle (C1-C4), carboxamide, cyano, polyfluoroalkyle (C1-C4), polyfluoroalkoxy (C1-C3), S03H, RI et R2 étant eux-même éventuellement substitués par un groupement alkyle linéaire ou ramifié (C1-C4) où R3 représente un atome d'hydrogène, un groupe aryle, hétéroaryle ou une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8), polyfluoroalkyle (CI-C4), aryle, hétéroaryle, hydroxy, alkoxy (C1-C4), carboxy, carboxamide, amino, alkylamino (C1-C4), NRaRb, où R4, R5 et R6 représentent indépendamment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4) où R7 est un alkyle linéaire ou ramifié (C1-C4)
<Desc/Clms Page number 2>
où Ra et Rb représentent R7, ou bien Ra et Rb forment ensemble avec l'atome d'azote auquel ils sont rattachés un hétérocycle à 5 ou 6 chaînons pouvant contenir éventuellement un autre hétéroatome choisi parmi 0, S, ou N et leurs sels pharmaceutiquement acceptables.
Dans les définitions précédentes et celles qui suivent, les radicaux cycloalkyles représentent des alkyles cycliques comprenant de 3 à 8 carbones ; les radicaux halogènes sont soit chlore, brome, fluor ou iode ; les radicaux polyfluoroalkyles (CI - C4) sont des alkyles de 1 à 4 carbones substitués par 1 à 9 halogènes identiques ou non identiques en particulier, difluorométhyle, trifluorométhyle, pentafluoroéthyl, 1,1,1-trifluoroéthyle; 1,1,1-trifluoropropyle; 1,1,1-trifluorobutyle ; les radicaux polyfluoroalkoxy (C1-C3) sont des alkoxy de 1 à 3 carbones substitués par 1 à 7 halogènes identiques ou non identiques en particulier le trifluorométhoxy ; les radicaux alkényles contiennent 2 à 6 atomes de carbone et une à 3 doubles liaisons conjuguées ou non en chaîne droite ou ramifiée ; les radicaux alkynyles contiennent 2 à 6 atomes de carbone et 1 à 3 triples liaisons conjuguées ou non en chaîne droite ou ramifiée ; les radicaux aryles sont choisis parmi phényle, naphtyle ou indényle ; radicaux hétéroaryles contiennent 3 à 10 chaînons, contenant éventuellement un ou plusieurs hétéroatomes choisis parmi oxygène, soufre et azote en particulier, thiazolyle, thiényle, pyrrolyle, pyridazinyle, pyridinyle, pyrimidinyle, furyle, imidazolyle, isoxazolyle, oxazolyle, pyrazinyle, tetrazolyle, triazolyle.
L'invention a pour objet l'utilisation des dérivés d'isoindolones de formule (I) et leurs sels pharmaceutiquement acceptables pour la préparation de médicaments, compositions pharmaceutiques en tant qu'inhibiteurs de l'échangeur Na+/H+ (NHE).
Selon, l'invention les composés d'isoindolones possèdent des propriétés antiarythmiques, telles que celles qui sont importantes par exemple pour le traitement des maladies qui surviennent dans le cas de phénomènes de manque d'oxygène. Par suite de leurs propriétés pharmacologiques, les composés conviennent
<Desc/Clms Page number 3>
remarquablement comme médicaments antiarythmiques à composante cardioprotectrice pour la prophylaxie des infarctus et le traitement des infarctus ainsi que pour le traitement de l'angine de poitrine, et ils peuvent aussi inhiber ou fortement réduire de manière préventive les processus pathophysiologiques lors de la survenue de lésions induites par une ischémie, en particulier lors du déclenchement d'arythmies cardiaques induites par une ischémie. Du fait de leurs effets protecteurs contre les situations hypoxiques et ischémiques pathologiques, les composés de formule I selon l'invention peuvent être utilisés comme médicament pour le traitement des les lésions aiguës ou chroniques induites par une ischémie ou des maladies induites de ce fait de manière primaire ou secondaire. Ceci concerne leur utilisation comme médicaments pour des opérations chirurgicales, par exemple dans le cas de transplantations d'organes, où les composés peuvent être utilisés aussi bien pour la protection des organes du donneur avant et pendant le prélèvement, pour la protection des organes prélevés, par exemple lors du traitement avec des liquides de bain physiologiques ou lors de leur conservation dans des liquides de bain physiologiques, qu'au cours du transfert dans l'organisme du receveur. Les composés sont aussi de précieux médicaments à effet protecteur lors de la réalisation d'opérations chirurgicales d'angioplastie, par exemple sur le c#ur et aussi sur les vaisseaux périphériques. Du fait de leur effet protecteur contre les lésions induites par une ischémie, les composés conviennent aussi comme médicaments pour le traitement des ischémies du système nerveux, en particulier du système nerveux central, et ils conviennent par exemple pour le traitement de l'attaque ou de l'oedème cérébral. En outre, les composés de formule I selon l'invention conviennent aussi pour les traitements de formes du choc, comme par exemple du choc allergique, du choc cardiogène, du choc hypovolémique et du choc bactérien.
En outre, les composés de formule I selon l'invention se caractérisent par un effet inhibiteur sur la prolifération des cellules, par exemple sur la prolifération cellulaire des fibroblastes et sur la prolifération des cellules lisses des muscles des vaisseaux. C'est pourquoi, les composés de formule I peuvent être utilisés comme agents thérapeutiques précieux pour les maladies dans lesquelles la prolifération
<Desc/Clms Page number 4>
cellulaire constitue une cause primaire ou secondaire, et ils peuvent donc être utilisés comme agents antiathérosclérotiques, agents contre les complications diabétiques tardives, les maladies cancéreuses, les maladies fibreuses comme la fibrose pulmonaire, la fibrose hépatique ou la fibrose rénale, les hypertrophies et hyperplasies d'organes, en particulier l'hyperplasie de la prostate ou l'hypertrophie de la prostate.
Les composés selon l'invention sont des inhibiteurs actifs de NHE qui est augmenté dans de nombreuses maladies (hypertonie essentielle, athérosclérose, diabète, etc. ), même dans les cellules qui sont d'accès facile pour les mesures, par exemple dans les érythrocytes, les thrombocytes ou les leucocytes. C'est pourquoi, les composés selon l'invention conviennent comme outils scientifiques simples, par exemple dans leur utilisation comme agents de diagnostic pour la détermination et la distinction de formes déterminées de l'hypertonie, mais aussi de l'athérosclérose, du diabète, des maladies prolifératives, etc. En outre, les composés de formule I conviennent pour la thérapie préventive pour éviter la genèse de l'hypertension sanguine, par exemple de l'hypertonie essentielle.
On a constaté en outre que les composés de formule I présentent une influence favorable sur les lipoprotéines sériques. On sait d'une manière générale que des valeurs trop élevées des graisses du sang, ce que l'on appelle les hyperlipoprotéinémies, constituent un facteur de risque essentiel pour la survenue de modifications artériosclérotiques des vaisseaux, en particulier de la maladie coronarienne. C'est pourquoi l'abaissement des lipoprotéines sériques augmentées a une importance pour la prophylaxie et la régression des modifications athérosclérotiques. Une importance particulière revient non seulement à la réduction du cholestérol sérique total mais aussi à l'abaissement de la proportion des fractions lipidiques athérogènes spécifiques de ce cholestérol total, en particulier des lipoprotéines de base densité (LDL) et des lipoprotéines de très basse densité (VLDL), car ces fractions lipidiques représentent un facteur de risque athérogène. Par contre, une fonction protectrice contre la maladie coronarienne est attribuée au lipoprotéines de haute densité. Ainsi, les agents hypolipidémiques doivent être en mesure d'abaisser non seulement le cholestérol total mais aussi en particulier les
<Desc/Clms Page number 5>
fractions VLDL et LDL du cholestérol sérique. On a maintenant trouvé que les composés de formule I présentent de précieuses propriétés thérapeutiquement valorisables concernant l'influence sur le niveau des lipides sériques. C'est ainsi qu'ils peuvent abaisser significativement la concentration sérique augmentée des LDL et des VLDL, telle qu'elle peut être observée par exemple lors de l'absorption alimentaire augmentée d'une nourriture riche en cholestérol et en lipides ou lors de modifications pathologiques du métabolisme, par exemple lors d'hyperlipidémies d'origine génétique. C'est pourquoi ils peuvent être utilisés pour la prophylaxie et la régression des modifications athérosclérotiques du fait qu'ils suppriment un facteur de risque causal. En font partie non seulement les hyperlipidémies primaires, mais aussi certaines hyperlipidémies secondaires, telles que celles qui apparaissent par exemple dans le diabète. Par ailleurs, les composés de formule I peuvent conduire à une nette réduction des infarctus induits par des anomalies du métabolisme et en particulier à une diminution significative de la taille des infarctus induits et de leur degré de gravité. Par ailleurs, les composés de formule I peuvent protéger efficacement contre les lésions endothéliales induites par des anomalies du métabolisme. Du fait de cette protection des vaisseaux contre le syndrome du disfonctionnement endothélial, les composés de formule I sont des médicaments utiles pour la prévention et pour le traitement des spasmes des artères coronaires, de l'athérogenèse et de l'athérosclérose, de l'hypertrophie du ventricule gauche et de la cardiomyopathie dilatée, et des maladies thrombotiques.
Les composés cités peuvent donc être utilisés avantageusement pour la préparation d'un médicament pour le traitement de l'hypercholestérolémie, pour la préparation d'un médicament pour la prévention de l'athérogenèse, pour la préparation d'un médicament pour la prévention et le traitement de l'athérosclérose, pour la préparation d'un médicament pour la prévention et le traitement des maladies qui sont déclenchées par un niveau de cholestérol augmenté, pour la préparation d'un médicament pour la prévention et le traitement des maladies qui sont déclenchées par un disfonctionnement endothélial, pour la préparation d'un médicament pour la prévention et le traitement de l'hypertonie induite par l'athérosclérose, pour la
<Desc/Clms Page number 6>
préparation d'un médicament pour la prévention et le traitement des thromboses induites par l'athérosclérose, pour la préparation d'un médicament pour la prévention et le traitement des lésions ischémiques induites par l'hypercholestérolémie et un disfonctionnement endothélial et des lésions de reperfusion post-ischémiques, pour la préparation d'un médicament pour la prévention et le traitement des hypertrophies cardiaques et des cardiomyopathies induites par l'hypercholestérolémie et un disfonctionnement endothélial, pour la préparation d'un médicament pour la prévention et le traitement des spasmes des artères coronaires et des infarctus du myocarde induits par l'hypercholestérolémie et un disfonctionnement endothélial, pour la préparation d'un médicament pour le traitement des affections citées en combinaison avec des substances abaissant la tension sanguine, de préférence avec des inhibiteurs de l'enzyme de conversion de l'angiotensine (ACE) et des antagonistes des récepteurs de l'angiotensine, une combinaison d'un inhibiteur de NHE de formule I avec une substance active abaissant le niveau des graisses sanguines, de préférence avec un inhibiteur de la HMG-CoA-réductase, (par exemple Lovastatine ou Pravastatine), ce dernier entraînant un effet hypolipidémique et augmentant ainsi les propriétés hypolipidémiques de l'inhibiteur de NHE de formule I, se révèle être une combinaison favorable à effet renforcé et utilisation de substances actives réduite.
Les composés de formule-1peuvent être utilisés comme nouveaux médicaments pour abaisser un niveau accru de lipides sanguins, ainsi qu'en combinaison avec des médicaments dont l'action abaisse la tension sanguine et/ou hypolipidémique.
Les composés de formule-1peuvent être utilisés comme nouveaux médicaments pour leurs applications thérapeutiques dans le traitement des maladies comme inhibiteurs de NHE et en particulier de NHE-1.
La présente invention concerne également les procédés de synthèse de dérivés isoindolones de formule (I)
<Desc/Clms Page number 7>

Par ailleurs les composés de formule (I) peuvent se présenter sous la forme de tautomères, racémiques, énantiomères et diastéréoisomères. Ces derniers font également partie de l'invention.
Parmi les composés de formule (I) utiles selon l'invention on peut citer les composés suivants : N-[(2-Méthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Ethyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine

N-[(3-Oxo-2-propyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[2-(3-Oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine N-[(2-Isopropyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[2-(2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-propionyl]-guanidine N-[(2-Isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-2-méthyl-propionyl]- guanidine N-[(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine
N-[(3-Oxo-2-propyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[2-(3-Oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine N-[(2-Isopropyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[2-(2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-propionyl]-guanidine N-[(2-Isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-2-méthyl-propionyl]- guanidine N-[(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine
<Desc/Clms Page number 8>

N-[2-(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-2-méthylpropionyl]-guanidine N-[(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 1 H-isoindol-1 -yl)-acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-4-méthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(2-Isobutyl-5-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-6-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-7-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol-1 -yl)-acétyl]-guanidine N-[(6-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(4-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-amino-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl] -guanidine N-[(4-hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-l-yl)-acétyl]-guanidine N-[(5- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétyl]-guanidine N-[(6- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine
<Desc/Clms Page number 9>

N-[(7- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-5-isopropoxy-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]guanidine N-[(6-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine N-[(5-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine

N-[(6-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(6-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]- guanidine N-[(5-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine
N-[(6-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]- guanidine N-[(5,6-Dichloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acetyl]-guanidine
N-[(4,7-Dichloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-l-yl)-acetyl]-guanidine N-[(4-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-l H-isoindol-1-yl)-acétyl]-guanidine
<Desc/Clms Page number 10>

N-[(5-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl] -guanidine N-[(4,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(4,5-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(5-Bromo-2-isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(6-Bromo-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-3-oxo-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine N-[(2-Isobutyl-3-oxo-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(2-Cyclopropylméthyl-3-oxo-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(2-Cyclopropylméthyl-3-oxo-6-trifluorométhyl-2,3-dihydro-1 1 H-isoindol- 1 -yl)- acétyl] -guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine
N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(3-Oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine
<Desc/Clms Page number 11>
N-[(3-Oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1H-isoindol-1-yl)- acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine
N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(4-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine

N-[(5-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-I-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine Acide [l-(2-guanidino-l-méthyl-2-oxo-éthyl)-3-oxo-l,3-dihydro-isoindol-2-yl]- acétique N-{2-[3-Oxo-2-(2-pyrrolidin-l-yl-éthyl)-2,3-dihydro-lH-isoindol-l-yl]-propionyl}- guanidine N-[2-(2-Hydroxy-éthyl)-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine leurs racémiques, énantiomères, diastéréoisomères, tautomères ainsi que leurs sels pharmaceutiquement acceptables, et plus particulièrement les composés suivants :
N-[(2-Méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Ethyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-0xo-2-propyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine
<Desc/Clms Page number 12>
N-[2-(3-Oxo-2-propyl-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine N-[(2-Isopropyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine
N-[2-(2-Butyl-3-oxo-2,3-dihydro- 1 H-isoindol- 1 -yl)-propionyl]-guanidine N-[(2-Isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine

N-[(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-4-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-5-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-6-méthyl-3-oxo-2,3-dihydro- 1H-isoindol-1-yl)-acétyl]-guanidine
N-[(5-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Isobutyl-5-isopropoxy-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(5-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine
N-[(6-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidineN- [(5-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine
<Desc/Clms Page number 13>

N-[(6-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]guanidine N-[(6-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 lH-isoindol- 1 -yl)-acétyl]guanidine N-[(6-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine N-[(6-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro- 1H-isoindol-1-yl)-acétyl]guanidine

N-[(5,6-Dichloro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acetyl]-guanidine N-[(7-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(4,7-Difluoro-2-isobutyl-3 -oxo-2,3 -dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N- [(5-Bromo-2-isobutyl-3-oxo-2,3-dihydro- 1H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Bromo-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Isobutyl-3-oxo-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine
N-[(2-Isobutyl-3-oxo-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]- guanidine
<Desc/Clms Page number 14>
N-[(2-Cyclopropylméthyl-3-oxo-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(2-Cyclopropylméthyl-3-oxo-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl] -guanidine

N-[(3-Oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine Acide [1-(2-guanidino-1-méthyl-2-oxo-éthyl)-3-oxo-1,3-dihydro-isoindol-2-yl]acétique N- {2-[3-Oxo-2-(2-pyrrolidin-1-yl-éthyl)-2,3-dihydro-1 H-isoindol-1-yl]-propionyl} guanidine N-[2-(2-Hydroxy-éthyl)-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine leurs racémiques, énantiomères, diastéréoisomères, tautomères ainsi que leurs sels pharmaceutiquement acceptables.
N-[(3-Oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)acétyl]-guanidine Acide [1-(2-guanidino-1-méthyl-2-oxo-éthyl)-3-oxo-1,3-dihydro-isoindol-2-yl]acétique N- {2-[3-Oxo-2-(2-pyrrolidin-1-yl-éthyl)-2,3-dihydro-1 H-isoindol-1-yl]-propionyl} guanidine N-[2-(2-Hydroxy-éthyl)-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine leurs racémiques, énantiomères, diastéréoisomères, tautomères ainsi que leurs sels pharmaceutiquement acceptables.
Les composés de formule (I) pour lesquels R4 et R6 représentent hydrogène peuvent être préparés à partir des phthalimides de formule (II) selon le schéma général de synthèse suivant:
<Desc/Clms Page number 15>

Le schéma de synthèse général est le suivant : a) on fait réagir un hydrure avec un phtalimide (de formule II) dans un alcool aliphatique b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec un trialkylphosphonoacétate et une base c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4) La réaction de réduction a s'effectue de préférence au moyen d'un hydrure tel que le borohydrure de potassium ou de sodium, au sein d'un alcool aliphatique (1-4C), de préférence le méthanol, ou du tétrahydrofuranne, à une température comprise entre 0 C et la température d'ébullition du milieu réactionnel.
La réaction b s'effectue généralement en présence d'un alkoxycarbonylméthylène triphénylphosphorane approprié au sein d'un solvant tel que le toluène, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel,
<Desc/Clms Page number 16>
ou bien en présence d'un trialkylphosphonoacétate approprié et d'une base telle que l'hydrure de sodium au sein d'un solvant tel que le 1,2-diméthoxyéthane, à une température comprise entre 0 C et la température d'ébullition du milieu réactionnel.
La réaction c s'effectue généralement en présence de chlorhydrate de guanidinium et d'une base telle que le tert-butylate de potassium au sein d'un solvant inerte tel que le diméthylformamide, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel, ou bien en présence de guanidine au sein d'un solvant tel qu'un alcool (C1-C4), de préférence l'isopropanol à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel.
Alternativement, certains composés de formule (I) pour lesquels R4 et R6 représentent hydrogène peuvent être préparés à partir des aldéhydes de formule (III) selon le schéma général de synthèse suivant:

Le schéma de synthèse général est le suivant :
Le schéma de synthèse général est le suivant :
<Desc/Clms Page number 17>
a) on fait réagir un composé de formule III avec alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec un trialkylphosphonoacétate et une base b) on met le produit obtenu en présence d'une amine de type R3NH2 (R3 ayant la même signification que dans la formule I) et d'un carbodiimide c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool C1-C4.
La réaction a s'effectue généralement en présence d'un alkoxycarbonylméthylène triphénylphosphorane approprié au sein d'un solvant inerte tel que le toluène, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel, ou bien en présence d'un trialkylphosphonoacétate approprié et d'une base telle que l'hydrure de sodium au sein d'un solvant tel que le 1,2-diméthoxyéthane, à une température comprise entre 0 C et la température d'ébullition du milieu réactionnel.
La réaction b s'effectue en présence de l'amine R3NH2 appropriée. On opère généralement en présence d'un agent de condensation utilisé en chimie peptidique tel qu'un carbodiimide (par exemple le N,N'-dicyclohexylcarbodiimide) ou le N,N'-diimidazole carbonyle, dans un solvant inerte tel qu'un éther (tétrahydrofuranne, dioxanne par exemple), un amide (diméthylformamide) ou un solvant inerte chloré (chlorure de méthylène, dichloro-1,2-éthane, chloroforme par exemple) à une température comprise entre 0 C et la température d'ébullition du milieu réactionnel.
La réaction c s'effectue généralement en présence de chlorhydrate de guanidinium et d'une base telle que le tert-butylate de potassium au sein d'un solvant inerte tel que le diméthylformamide, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel, ou bien en présence de guanidine au sein d'un solvant tel qu'un alcool (CI-C4), de préférence l'isopropanol à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel.
<Desc/Clms Page number 18>
Les composés de formule (I) pour lesquels R4 représente un groupe alkyle et R6 représente hydrogène peuvent être préparés à partir des phthalimides de formule (II) selon le schéma général de synthèse suivant:
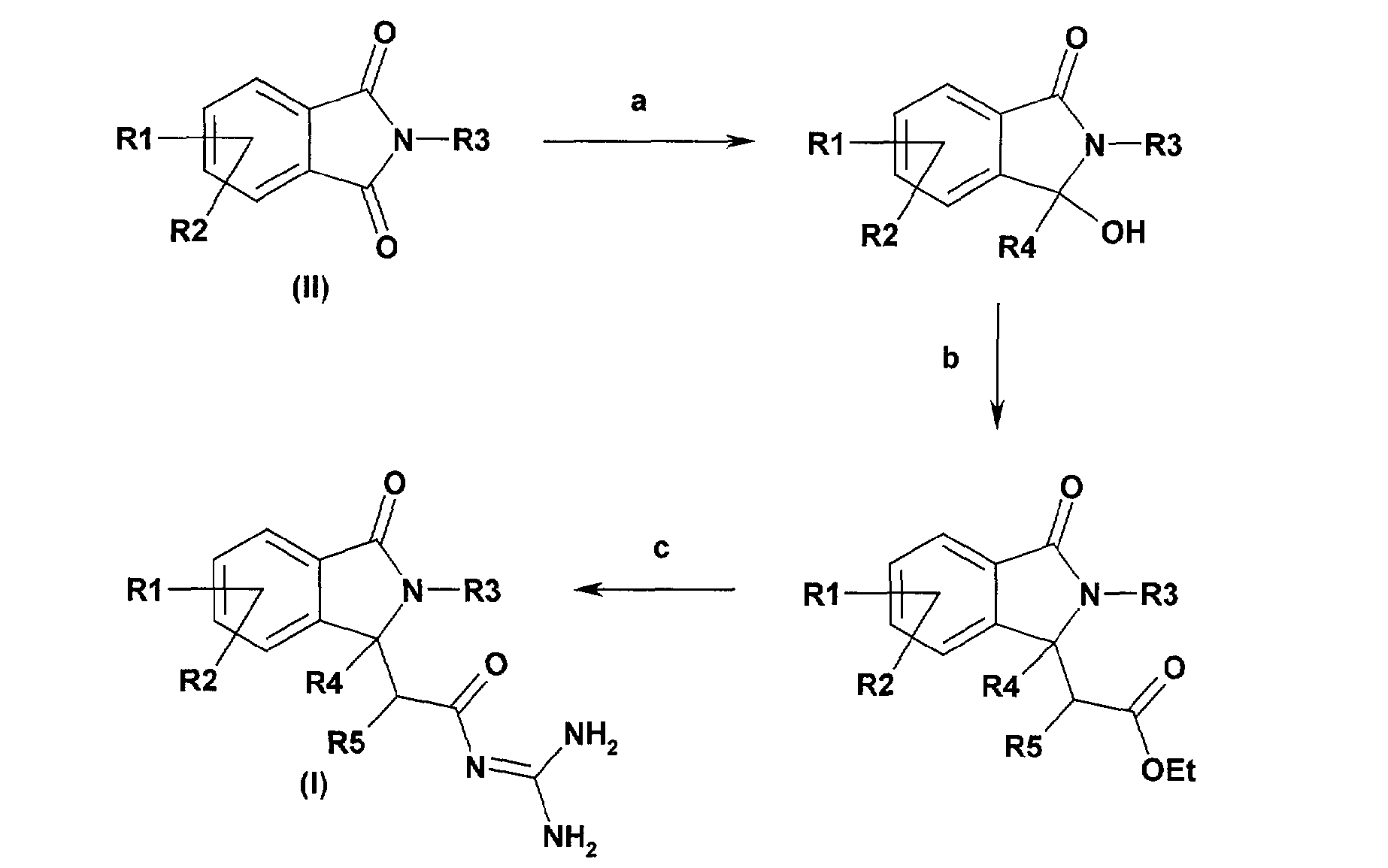
Le schéma de synthèse général est le suivant : a) on fait réagir un phtalimide (de formule II) avec un halogénure d'alkylmagnésium ou avec un alkyllithien dans un éther b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec le 1-éthoxy-1-triméthylsiloxyéthylène et un acide de Lewis c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4) La réaction a s'effectue de préférence au moyen d'un halogénure d'alkylmagnésium ou d'un alkyllithien, au sein d'un solvant tel qu'un éther, de préférence le
Le schéma de synthèse général est le suivant : a) on fait réagir un phtalimide (de formule II) avec un halogénure d'alkylmagnésium ou avec un alkyllithien dans un éther b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec le 1-éthoxy-1-triméthylsiloxyéthylène et un acide de Lewis c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4) La réaction a s'effectue de préférence au moyen d'un halogénure d'alkylmagnésium ou d'un alkyllithien, au sein d'un solvant tel qu'un éther, de préférence le
<Desc/Clms Page number 19>
tétrahydrofuranne, à une température comprise entre 0 C et la température d'ébullition du milieu réactionnel.
La réaction b peut s'effectuer en présence d'un alkoxycarbonylméthylène triphénylphosphorane approprié au sein d'un solvant tel que le toluène, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel, ou bien en présence de 1-éthoxy-1-triméthylsiloxyéthylène et d'un acide de Lewis tel que le chlorure de titane (IV) ou le triméthylsilyltriflate au sein d'un solvant inerte tel que le dichlorométhane, à une température comprise entre -78 C et 20 C. La préparation de dérivés tels que le 1-éthoxy-1-triméthylsiloxyéthène est décrite dans Synth. Commun. 1987, 17, 1.
La réaction c s'effectue généralement en présence de chlorhydrate de guanidinium et d'une base telle que le tert-butylate de potassium au sein d'un solvant inerte tel que le diméthylformamide, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel, ou bien en présence de guanidine au sein d'un solvant tel qu'un alcool (C1-C4), de préférence l'isopropanol à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel.
Les composés de formule (I) pour lesquels R6 représente un groupe alkyle peuvent être préparés à partir des esters de formule (IV) selon le schéma général de synthèse suivant:
<Desc/Clms Page number 20>

Le schéma de synthèse général est le suivant : a) on fait réagir un phtalimide (de formule II) avec un halogénure d'alkylmagnésium ou d'un alkyllithien dans un éther b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec le 1-éthoxy-1-triméthylsiloxyéthylène et un acide de Lewis c) puis on fait réagir le produit obtenu en présence de diisopropylamidure de lithium avec un R6-Hal d) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4) La réaction a peut s'effectuer en présence de diisopropylamidure de lithium dans un solvant inerte tel qu'un éther (le tétrahydrofuranne de préférence) et en présence d'un halogénure d'alkyle R6-Hal approprié à une température comprise entre -78 C et 0 C.
<Desc/Clms Page number 21>
La réaction b s'effectue généralement en présence de chlorhydrate de guanidinium et d'une base telle que le tert-butylate de potassium au sein d'un solvant inerte tel que le diméthylformamide, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel, ou bien en présence de guanidine au sein d'un solvant tel qu'un alcool (CI-C4), de préférence l'isopropanol à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel.
Les composés de formule (II) lorsqu'ils ne sont pas commerciaux peuvent être par exemple préparés (voie a) à partir des anhydrides (V) correspondants en présence de l'amine R3NH2 appropriée et d'un acide tel que l'acide para-toluènesulfonique, au sein d'un solvant tel que le toluène, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel ; ou bien (voie b) par la méthode dite de Gabriel, à partir des phthalimidures de potassium (VI) correspondants en présence de l'halogénure d'alkyle R3X approprié et au sein d'un solvant tel que le diméthylformamide à une température comprise entre 0 C et la température d'ébullition du milieu réactionnel par application ou adaptation de la méthode décrite dans Tetrahedron 1998, 54, 14437.

Les composés de formule (V) lorsqu'ils ne sont pas commerciaux peuvent être par exemple préparés à partir des acides phthaliques correspondants au sein de
Les composés de formule (V) lorsqu'ils ne sont pas commerciaux peuvent être par exemple préparés à partir des acides phthaliques correspondants au sein de
<Desc/Clms Page number 22>
l'anhydride acétique, à une température comprise entre 20 C et la température d'ébullition du milieu réactionnel.
Lorsque cela est nécessaire, on utilise un groupe protecteur de la fonction amine, alcool ou acide et des méthodes de déprotection tel que ceux décrits par T.W. GREENE, Protective groups in Organic Synthesis, J. Wiley-Interscience Publication (1991).
Les composés de formule (I) sont isolés et peuvent être purifiés par les méthodes connues habituelles, par exemple par cristallisation, chromatographie ou extraction.
Les composés de formule (I) peuvent être éventuellement transformés en sels d'addition avec un acide minéral ou organique par action d'un tel acide au sein d'un solvant organique tel qu'un alcool, une cétone, un éther ou un solvant chloré. Ces sels font également partie de l'invention.
Comme exemples de sels pharmaceutiquement acceptables, peuvent être cités les sels suivants : benzènesulfonate, bromhydrate, chlorhydrate, citrate, éthanesulfonate, fumarate, gluconate, iodate, maléate, iséthionate, méthanesulfonate, méthylène-bis-boxynaphtoate, nitrate, oxalate, pamoate, phosphate, salicylate, succinate, sulfate, tartrate, théophyllinacétate et p-toluènesulfonate.
Leurs CI50 ont été calculées par un test FLIPR.
Le test est réalisé dans le FLIPR (Fluorescent imaging plate reader) doté de plaques Microtitre de 96 puits à fond clair et à parois noires. Les lignées cellulaires transfectées qui expriment les différents sous-types de NHE en fonction de celui que l'on veut tester et n'ont aucune activité NHE endogène en raison de la mutagenèse et de la sélection subséquente), ont atteint le jour précédent une densité d'environ 25 000 cellules par puits.
Le milieu de croissance des cellules transfectées (Iscove +10% de sérum f#tal de veau) contient en plus du G418 comme antibiotique de sélection pour garantir la présence des séquences transfectées.
<Desc/Clms Page number 23>
Le test proprement dit commence par l'élimination du milieu de croissance et par l'ajout de 100 III de tampon de chargement par puits (5 M de BCECF-AM [2',7'-bis-(2-carboxyéthyl)-5-(6)-carboxyfluorescéine, acétoxyméthyl ester] dans 20 mM de NH4Cl, 115mM de chlorure de choline, 1 mM de CaCl2, 5 mM de KCl, 20 mM de HEPES, 5 mM de glucose ; pH 7,4 [ajusté avec KOH]. Les cellules sont ensuite incubées pendant 20 minutes à 37 C. Cette incubation entraîne le chargement dans les cellules du colorant fluorescent, dont l'intensité de fluorescence dépend du pH,, et du NH4C1, ce qui entraîne une légère alcalinisation des cellules.
Le progéniteur BCECF-AM, colorant non fluorescent, est en tant qu'ester apte à traverser la membrane. Le colorant proprement dit, qui n'est pas apte à traverser la membrane, est libéré à l'intérieur de la cellule par des estérases.
Après cette incubation de 20 minutes, le tampon de chargement, qui contient du NH4CI et du BCECF-AM libre est éliminé en effectuant trois lavages dans le dispositif de lavage cellulaires (Tecan Columbus) avec à chaque lavage 400 III de tampon de lavage (133,8 mM de chlorure de choline, 4,7 mM de KCI, 1,25 mM de MgCl2, 1,25 mM de CaCl2, 0,97 mM de K2HP04, 0,23 mM de KH2P04, 5 mM de HEPES, 5 mM de glucose ; pH de 7,4 [ajusté avec KOH]. Le volume résiduel restant dans les puits est de 90 III (éventuellement entre 50 et 125 l). Cette étape de lavage élimine le BCECF-AM libre et entraîne une acidification intracellulaire (pHi de 6,3- 6,4) due à l'élimination des ions ammonium externes.
Comme l'équilibre de l'ammonium intracellulaire avec l'ammoniaque et des protons par l'élimination de l'ammonium extracellulaire et par la traversée immédiate subséquente de l'ammoniaque à travers la membrane des cellules est perturbé, le processus de lavage entraîne qu'il reste des protons intracellulaires, ce qui est à l'origine de l'acidification intracellulaire. Cette acidification peut entraîner finalement la mort des cellules lorsqu'elle dure suffisamment longtemps. Il est important ici que le tampon de lavage soit dépourvu de sodium (<1 mM), sinon les ions sodium extracellaires entraîneraient une augmentation immédiate du pH, en raison de l'activité des isoformes de NHE clonées. Il est également important que tous les
<Desc/Clms Page number 24>
<Desc/Clms Page number 25>
Les composés de formule (I) présentent une activité très intéressante et en particulier certains composés ont une CI50 inférieure à 10 M.
Les exemples suivants illustrent l'invention.
Exemple 1 :

N-r (2-Méthyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyl]-guanidine A une suspension de 5,2 g de tert-butylate de potassium dans 100 cm3 de diméthylformamide, on ajoute 4,3 g de chlorure de guanidinium. Le mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 1 heure puis on ajoute une solution de 2 g de (2-méthyl-3-oxo-2,3-dihydro- lH-isoindol-l-yl)-acétate d'éthyle dans 20 cm3de diméthylformamide. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis 100 cm3d'eau sont ajoutés. Le pH est ajusté à 8 par ajout de 50 cm3 d'acide chlorhydrique IN et le mélange est concentré sous pression réduite (0,6 kPa) à une température voisine de 30 C. Le résidu d'évaporation est repris dans l'eau puis filtré.

On obtient ainsi 0,35 g de N-[(2-méthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine, sous forme d'un solide écru fondant à 214 C. Spectre de masse El : m/z 246 (M+), m/z 159 (pic de base), m/z 146.

N-r (2-Méthyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyl]-guanidine A une suspension de 5,2 g de tert-butylate de potassium dans 100 cm3 de diméthylformamide, on ajoute 4,3 g de chlorure de guanidinium. Le mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 1 heure puis on ajoute une solution de 2 g de (2-méthyl-3-oxo-2,3-dihydro- lH-isoindol-l-yl)-acétate d'éthyle dans 20 cm3de diméthylformamide. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis 100 cm3d'eau sont ajoutés. Le pH est ajusté à 8 par ajout de 50 cm3 d'acide chlorhydrique IN et le mélange est concentré sous pression réduite (0,6 kPa) à une température voisine de 30 C. Le résidu d'évaporation est repris dans l'eau puis filtré.
On obtient ainsi 0,35 g de N-[(2-méthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine, sous forme d'un solide écru fondant à 214 C. Spectre de masse El : m/z 246 (M+), m/z 159 (pic de base), m/z 146.
(2-Méthyl-3 -oxo-2,3 -dihydro- 1 H-isoindol- 1 -vl)-acétate d'éthyle A une suspension de 4,5 g de 3-hydroxy-2-méthyl-2,3-dihydro-isoindol-l-one dans 110 cm3 de toluène, on ajoute 11,5 g d'éthoxycarbonylméthylène triphénylphosporane. Le mélange réactionnel est chauffé au reflux sous agitation pendant 16 heures, puis il est refroidi à une température voisine de 20 C. Le mélange est ensuite concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. L'huile résiduelle est reprise dans 50 cm3 d'éther diéthylique. Le précipité formé est filtré puis lavé 2 fois avec 10 cm3d'éther diéthylique. Le filtrat est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C pour donner une huile orange qui est purifiée par chromatographie sous pression d'argon
<Desc/Clms Page number 26>
(60 kPa), sur une colonne de gel de silice (granulométrie 20-45 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (70/30,65/35, 60/40 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées sous pression réduite (2 kPa) à une température voisine de 30 C. On obtient ainsi 4,1 g de

(2-méthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune. (Rf = 0,25, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).

3- Hvdroxy-2-méthyl-2J-dihydro-isoindol-I-one A une suspension de 10 g de N-méthyl-phthalimide dans 220 cm3de méthanol sous atmosphère inerte, on ajoute lentement 3,4 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute au goutte à goutte 200 cm3 d'eau distillée. Le solvant est ensuite évaporé en partie (environ 120 cm3) sous pression réduite (2 kPa) à une température voisine de 35 C et le résidu est dilué avec 400 cm3d'eau distillée . Le mélange est extrait avec 400 cm3 d'acétate d'éthyle. La phase organique est séchée sur sulfate de magnésium, filtrée puis concentrée à sec sous pression réduite (2 kPa) à une température voisine de 30 C. On obtient ainsi 4,5 g de 3-hydroxy-2-méthyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre blanche fondant à 130 C.
(2-méthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune. (Rf = 0,25, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
3- Hvdroxy-2-méthyl-2J-dihydro-isoindol-I-one A une suspension de 10 g de N-méthyl-phthalimide dans 220 cm3de méthanol sous atmosphère inerte, on ajoute lentement 3,4 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute au goutte à goutte 200 cm3 d'eau distillée. Le solvant est ensuite évaporé en partie (environ 120 cm3) sous pression réduite (2 kPa) à une température voisine de 35 C et le résidu est dilué avec 400 cm3d'eau distillée . Le mélange est extrait avec 400 cm3 d'acétate d'éthyle. La phase organique est séchée sur sulfate de magnésium, filtrée puis concentrée à sec sous pression réduite (2 kPa) à une température voisine de 30 C. On obtient ainsi 4,5 g de 3-hydroxy-2-méthyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre blanche fondant à 130 C.
Exemple 2 :

N -[ (2-Isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyll-guanidine La N-[(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 5 g de tert-butylate de potassium, de 5,2 g de chlorure de guanidinium et de 2,5 g de (2-isobutyl-3-oxo-2,3-dihydro-IH- isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 24 heures, puis il est filtré. Le filtrat est repris avec 150 cm3 d'eau et 200 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite avec 2 fois 200 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés et concentrés à sec
N -[ (2-Isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyll-guanidine La N-[(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 5 g de tert-butylate de potassium, de 5,2 g de chlorure de guanidinium et de 2,5 g de (2-isobutyl-3-oxo-2,3-dihydro-IH- isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 24 heures, puis il est filtré. Le filtrat est repris avec 150 cm3 d'eau et 200 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite avec 2 fois 200 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés et concentrés à sec
<Desc/Clms Page number 27>
sous pression réduite (0,6 kPa) à une température voisine de 45 C. Le résidu d'évaporation est repris dans l'éther diéthylique et le précipité formé est filtré puis lavé plusieurs fois à l'éther diéthylique. Le solide est séché sous pression réduite (10 Pa) à une température voisine de 45 C. On obtient ainsi 1,5 g de N-[(2-isobutyl-3- oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine, sous forme d'un solide blanc fondant à 250 C. Spectre de masse El : m/z 288 (M+), m/z 201 (pic de base).

(2-Isobutyl-3 -oxo-2,3 -dihydro-lH-isoindol-l-vD-acétated'éthyle A une suspension de 1,6 g d'hydrure de sodium à 60% dans 60 cm3de 1,2diméthoxyéthane sous atmosphère inerte et refroidie à 0 C sous agitation, on ajoute au goutte à goutte 7,7 cm de triéthylphosphonoacétate en maintenant la température inférieure à 10 C. Le mélange réactionnel est laissé se réchauffer à une température voisine de 20 C puis il est agité pendant 45 minutes. On ajoute alors 5,3 g de 3- hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et le mélange est chauffé à reflux pendant 3,5 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 40 cm3d'eau distillée puis 100 cm3 d'éther diéthylique.
Après décantation, la phase aqueuse est extraite 2 fois avec 100 cm d'éther diéthylique. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 18 C pour donner une huile jaune pâle qui est purifiée par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (60/40 puis 50/50 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 30 C.
On obtient ainsi 6,3 g de (2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle, sous forme d'une huile jaune pâle. (Rf = 0,56, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
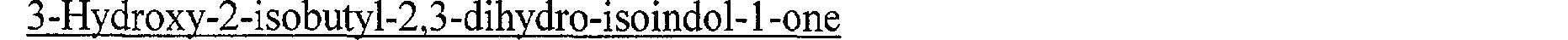
3-Hydroxy-2-isobpUI-2,3-dihydro-isoindol-1 -one La 3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 6,5 g de N-isobutyl-phthalimide dans 60 cm3de méthanol et
3-Hydroxy-2-isobpUI-2,3-dihydro-isoindol-1 -one La 3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 6,5 g de N-isobutyl-phthalimide dans 60 cm3de méthanol et
<Desc/Clms Page number 28>
de 1,7 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte 50 cm3 d'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 35 C et le résidu est extrait 3 fois avec 60 cm3de dichlorométhane. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 25 C pour donner une huile jaune pâle qui est purifiée par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 40-63 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (60/40 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 5,8 g de 3-hydroxy-2-isobutyl-2,3dihydro-isoindol-1-one, sous forme d'un solide blanc fondant à 82 C.
N-Isobutyl-phthalimide A une suspension de 5,2 g d'anhydride phthalique dans 50 cm3de toluène sous agitation, on ajoute une solution de 3,2 cm3d'isobutylamine dans 3 cm3 de toluène.
Le mélange réactionnel est chauffé à une température voisine de 60 C pendant 1 heure, puis à une température voisine de 100 C pendant 2 heures. Un Dean-Stark est alors installé sur le réacteur et le mélange réactionnel est chauffé à une température voisine de 130 C pendant 2 heures, puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans 50 cm3 d'une solution saturée de bicarbonate de sodium et extrait 2 fois avec 75 cm3de dichlorométhane. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 6,5 g de N-Isobutyl-phthalimide, sous forme d'un solide blanc fondant à 92 C.
Exemple 3 :

(- )-N-[(2- Isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyl)-guanidine
(- )-N-[(2- Isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyl)-guanidine
<Desc/Clms Page number 29>
La (-)-N-[(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 2,6 g de tert-butylate de potassium, de 2,6 g de chlorure de guanidinium et de 1,25 g de (-)-(2-isobutyl-3-oxo- 2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 40 heures, puis il est filtré. Le filtrat est repris avec 80 cm3 d'eau et 120 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite avec 2 fois 120 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés et concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 40 C.
Le résidu d'évaporation est repris dans 30 cm3d'éther diéthylique et le précipité formé est filtré puis lavé trois fois avec 5 cm3 d'éther diéthylique. Le solide est séché sous pression réduite (10 Pa) à une température voisine de 45 C. On obtient ainsi

0,75 g de (-)-N-[(2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'un solide blanc cassé fondant à 264 C. [alpha]D20= -10,2 +/-0,6 dans le méthanol à 0,5%). Spectre de masse El : m/z 288 (M+), m/z 245, m/z 201, m/z 132.
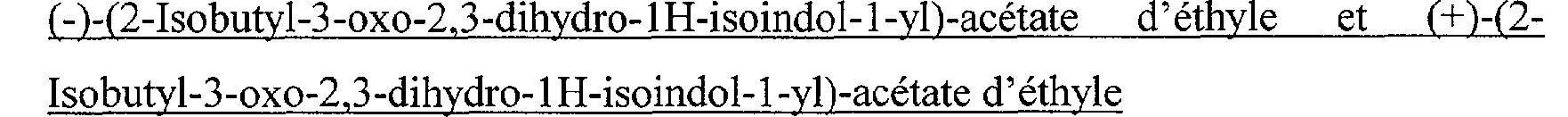
(- )-(2-Isobutyl-3-oxo-2.3-dihydro-IH-isoindol-I-vl)-acétate d'éthyle et (+)-(2Isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol-1 -vl>acétate d'éthyle Le (-)-(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et le (+)-(2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sont obtenus par dédoublement de 3,0 g de (2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle par chromatographie CLHP sur une colonne chirale de type WHELK-01SS 10 m en éluant successivement avec des mélanges heptane/ isopropanol (90/10 en volumes) puis heptane/ éthanol (90/10 puis 50/50 en volumes). Les fractions contenant le premier énantiomère sont réunies et concentrées sous pression réduite (1 kPa) à une température voisine de 40 C. Le résidu est séché sous pression réduite (3 kPa) à une température voisine de 40 C . On obtient ainsi 1,3 g de (-)-(2-isobutyl-3-
oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle sous forme d'une huile ocre pâle visqueuse, ([alpha]D20= -16,2 +/-0,6 dans le DMSO à 0,5%). Les fractions contenant le deuxième énantiomère sont réunies et concentrées sous pression réduite (1kPa) à une température voisine de 40 C. Le résidu est séché sous pression réduite (3 kPa) à une
0,75 g de (-)-N-[(2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'un solide blanc cassé fondant à 264 C. [alpha]D20= -10,2 +/-0,6 dans le méthanol à 0,5%). Spectre de masse El : m/z 288 (M+), m/z 245, m/z 201, m/z 132.
(- )-(2-Isobutyl-3-oxo-2.3-dihydro-IH-isoindol-I-vl)-acétate d'éthyle et (+)-(2Isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol-1 -vl>acétate d'éthyle Le (-)-(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et le (+)-(2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sont obtenus par dédoublement de 3,0 g de (2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle par chromatographie CLHP sur une colonne chirale de type WHELK-01SS 10 m en éluant successivement avec des mélanges heptane/ isopropanol (90/10 en volumes) puis heptane/ éthanol (90/10 puis 50/50 en volumes). Les fractions contenant le premier énantiomère sont réunies et concentrées sous pression réduite (1 kPa) à une température voisine de 40 C. Le résidu est séché sous pression réduite (3 kPa) à une température voisine de 40 C . On obtient ainsi 1,3 g de (-)-(2-isobutyl-3-
oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle sous forme d'une huile ocre pâle visqueuse, ([alpha]D20= -16,2 +/-0,6 dans le DMSO à 0,5%). Les fractions contenant le deuxième énantiomère sont réunies et concentrées sous pression réduite (1kPa) à une température voisine de 40 C. Le résidu est séché sous pression réduite (3 kPa) à une
<Desc/Clms Page number 30>
température voisine de 40 C . On obtient ainsi 1,0 g de (+)-(2-isobutyl-3-oxo-2,3dihydro-1H-isoindol-1-yl)-acétate d'éthyle sous forme d'une huile jaune pâle visqueuse, (aD20= -15,1 +/-0,7 dans le DMSO à 0,5%). Le (2-isobutyl-3-oxo-2,3dihydro-1H-isoindol-1-yl)-acétate d'éthyle est décrit à l'exemple 2.
Exemple 4 :

(+)-N-[(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acét]nanidine La (+)-N-[(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 2,0 g de tert-butylate de potassium, de 2,1 g de chlorure de guanidinium et de 1,0 g de (+)-(2-isobutyl-3-oxo- 2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 40 heures, puis il est filtré. Le filtrat est repris avec 70 cm3 d'eau et 100 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite avec 2 fois 100 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés et concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 40 C.
(+)-N-[(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acét]nanidine La (+)-N-[(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 2,0 g de tert-butylate de potassium, de 2,1 g de chlorure de guanidinium et de 1,0 g de (+)-(2-isobutyl-3-oxo- 2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 40 heures, puis il est filtré. Le filtrat est repris avec 70 cm3 d'eau et 100 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite avec 2 fois 100 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés et concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 40 C.
Le résidu d'évaporation est repris dans 30 cm3d'éther diéthylique et le précipité formé est filtré puis lavé trois fois avec 5 cm3 d'éther diéthylique. Le solide est séché sous pression réduite (10 Pa) à une température voisine de 45 C. On obtient ainsi 0,56 g de (+)-N-[(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine, sous forme d'un solide jaune orangé fondant à 264 C. (aD20= +13,9 +/-0,6 dans le méthanol à 0,5%). Le (+)-(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle est décrit à l'exemple 3. Spectre de masse DCI : m/z 289 (M+H)+.
Exemple 5 :
N-[3-Oxo-2-propyl-2,3-dihydro-1H-isoindol-1-1 -acéiyl]-uanidine La N-[(3-oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 3,9 g de tert-butylate de potassium, de 3,3 g de chlorure de guanidinium et de 1,8 g de (3-oxo-2-propyl-2,3-dihydro-lH-
N-[3-Oxo-2-propyl-2,3-dihydro-1H-isoindol-1-1 -acéiyl]-uanidine La N-[(3-oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 3,9 g de tert-butylate de potassium, de 3,3 g de chlorure de guanidinium et de 1,8 g de (3-oxo-2-propyl-2,3-dihydro-lH-
<Desc/Clms Page number 31>
isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 1 heure, puis on ajoute 60 cm3 d'eau. La phase aqueuse est extraite avec 3 fois 50 cm3d'acétate d'éthyle, puis elle est concentrée à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C. Le résidu est repris dans l'eau, trituré et filtré. Le solide est repris dans le méthanol puis le solvant est évaporé à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C. On obtient ainsi 0,6 g de N-[(3-oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine, sous forme d'un solide cotonneux jaune pâle fondant à 229 C. Spectre de masse El : m/z 274 (M+), m/z 187, m/z 86 (pic de base).
(3-Oxo-2-Propyl-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle Le (3-oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,8 g d'hydrure de sodium à 60% dans 20 cm3de 1,2-diméthoxyéthane, 4,0 cm3 de triéthylphosphonoacétate et 1,9 g de 3- hydroxy-2-propyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (50/50 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 30 C.

On obtient ainsi 1,9 g de (3-oxo-2-propyl-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune. (Rf = 0,7, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (30/70 en volumes)).

3-H, droxrop,yl-2,3-dihydro-isoindol-1-one La 3-hydroxy-2-propyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 1,5 g de N-propyl-phthalimide dans 25 cm3de méthanol et de 0,48 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 35 C et le résidu est refroidi à 0 C. Le précipité obtenu est filtré puis lavé à l'eau
On obtient ainsi 1,9 g de (3-oxo-2-propyl-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune. (Rf = 0,7, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (30/70 en volumes)).
3-H, droxrop,yl-2,3-dihydro-isoindol-1-one La 3-hydroxy-2-propyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 1,5 g de N-propyl-phthalimide dans 25 cm3de méthanol et de 0,48 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 35 C et le résidu est refroidi à 0 C. Le précipité obtenu est filtré puis lavé à l'eau
<Desc/Clms Page number 32>
froide. Le solide est repris dans du dichlorométhane puis le solvant est évaporé à sec sous pression réduite (2 kPa) à une température voisine de 35 C. On obtient ainsi 1,0 g de 3-hydroxy-2-propyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre beige.
(Rf = 0,6, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (30/70 en volumes)).
Exemple 6 :
N-["f2-Ethvl-3-oxo-2,3-dihydro-lH-isoindol-l-vl)-acétyll-guanidine La N-[(2-éthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 4,3 g de tert-butylate de potassium, de 3,7 g de chlorure de guanidinium et de 1,9 g de (2-éthyl-3-oxo-2,3-dihydro-lH-isoindol- l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 60 cm3 d'eau. La phase aqueuse est extraite avec 3 fois 50 cm3d'acétate d'éthyle, puis elle est concentrée à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C. Le résidu est repris dans l'eau, trituré et filtré. Le solide est repris dans le méthanol puis le solvant est évaporé à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C. On obtient ainsi 0,56 g de N-[(2-éthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine, sous forme d'un solide écru fondant à 223 C. Spectre de masse El : m/z 260 (M+), m/z 173, m/z 160, m/z 132.

N-["f2-Ethvl-3-oxo-2,3-dihydro-lH-isoindol-l-vl)-acétyll-guanidine La N-[(2-éthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 4,3 g de tert-butylate de potassium, de 3,7 g de chlorure de guanidinium et de 1,9 g de (2-éthyl-3-oxo-2,3-dihydro-lH-isoindol- l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 60 cm3 d'eau. La phase aqueuse est extraite avec 3 fois 50 cm3d'acétate d'éthyle, puis elle est concentrée à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C. Le résidu est repris dans l'eau, trituré et filtré. Le solide est repris dans le méthanol puis le solvant est évaporé à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C. On obtient ainsi 0,56 g de N-[(2-éthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine, sous forme d'un solide écru fondant à 223 C. Spectre de masse El : m/z 260 (M+), m/z 173, m/z 160, m/z 132.
(2-Ethyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétate d'éthyle Le (2-éthyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,6 g d'hydrure de sodium à 60% dans 20 cm3 de 1,2-diméthoxyéthane, 3,2 cm de triéthylphosphonoacétate et 1,8 g de 3-hydroxy-2- éthyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (50/50 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 30 C. On obtient ainsi 2,0 g de
<Desc/Clms Page number 33>
(2-éthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle, sous forme d'une huile jaune pâle. (Rf = 0,7, chromatographie sur couche mince de gel de silice, éluant : dichlorométhane/méthanol (90/10 en volumes)).

3-H dy-2-éthyl-2,3-dihydro-isoindol-1-one La 3-hydroxy-2-éthyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 4,0 g de N-éthyl-phthalimide dans 20 cm3de méthanol et de 1,2 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le précipité obtenu est filtré puis lavé à l'eau froide. Le méthanol est ensuite évaporé en partie du filtrat sous pression réduite (2 kPa) à une température voisine de 35 C et le résidu est refroidi à 0 C. Le deuxième précipité ainsi obtenu est filtré puis lavé à l'eau froide. Les 2 fractions de solide sont séchées sous pression réduite (2 kPa) à une température voisine de 35 C. On obtient ainsi 1,9 g de 3-hydroxy-2-éthyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre blanche pailletée. (Rf = 0,5, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (30/70 en volumes)).
Exemple 7 :

N-r(2- Isopropyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyll-guanidine La N-[(2-isopropyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 1,5 g de tert-butylate de potassium, de 1,3 g de chlorure de guanidinium et de 0,7 g de (2-isopropyl-3-oxo-2,3dihydro-1AH-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 30 cm3d'eau. La phase aqueuse est extraite avec 3 fois 50 cm3d'acétate d'éthyle, puis elle est concentrée à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C.
N-r(2- Isopropyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyll-guanidine La N-[(2-isopropyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 1,5 g de tert-butylate de potassium, de 1,3 g de chlorure de guanidinium et de 0,7 g de (2-isopropyl-3-oxo-2,3dihydro-1AH-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 30 cm3d'eau. La phase aqueuse est extraite avec 3 fois 50 cm3d'acétate d'éthyle, puis elle est concentrée à sec sous pression réduite (0,6 kPa) à une température voisine de 45 C.
Le résidu est repris dans l'eau, trituré et filtré. Le solide est repris dans un mélange de dichlorométhane/ méthanol (90/10 en volumes), filtré et le filtrat est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice
<Desc/Clms Page number 34>
(granulométrie 15-40 m), en éluant par un mélange de dichlorométhane/ méthanol (90/10 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C.
On obtient ainsi 0,05 g de N-[(2-isopropyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine, sous forme d'un solide blanc. Spectre infra-rouge (KBr). Spectre de masse El : m/z 274 (M+), m/z 187, m/z 132. Spectre infra-rouge (KBr) : 3412; 1974 ; 1667 ; 1603 ; 1531; 1367 et 698 cm-1.

(2- Isopropyl- 3 -oxo- 23 -dihydro-l H - isoindol-l- yl)-acétate d'éthyle Le (2-isopropyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,63 g d'hydrure de sodium à 60% dans 20 cm3de 1,2-diméthoxyéthane, 3,1 cm3 de triéthylphosphonoacétate et 2,0 g de 3- hydroxy-2-isopropyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (60/40 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 30 C.

On obtient ainsi 0,84 g de (2-isopropyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune pâle. (Rf = 0,7, chromatographie sur couche mince de gel de silice, éluant : (90/10 en volumes)).

3-Hydroxv-2-isopropvl-2,3-dihydro-isoindol-l-one La 3-hydroxy-2-isopropyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 4,0 g de N-isopropyl-phthalimide dans 20 cm3de méthanol et de 1,1 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le solvant est ensuite évaporé à sec sous pression réduite (2 kPa) à une température voisine de 35 C. On obtient ainsi 6,1 g de 3-hydroxy-2-isopropyl-2,3-dihydro-isoindol-l-one, sous forme d'une cire blanche. (Rf = 0,65, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (30/70 en volumes)).
On obtient ainsi 0,84 g de (2-isopropyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune pâle. (Rf = 0,7, chromatographie sur couche mince de gel de silice, éluant : (90/10 en volumes)).
3-Hydroxv-2-isopropvl-2,3-dihydro-isoindol-l-one La 3-hydroxy-2-isopropyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 4,0 g de N-isopropyl-phthalimide dans 20 cm3de méthanol et de 1,1 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le solvant est ensuite évaporé à sec sous pression réduite (2 kPa) à une température voisine de 35 C. On obtient ainsi 6,1 g de 3-hydroxy-2-isopropyl-2,3-dihydro-isoindol-l-one, sous forme d'une cire blanche. (Rf = 0,65, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (30/70 en volumes)).
<Desc/Clms Page number 35>
Exemple 8 :

N-((2-Cyclopropvlméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl")-guanidine La N-[(2-cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 2,9 g de tert-butylate de potassium, de 2,5g de chlorure de guanidinium et de 1,5 g de (2-cyclopropylméthyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 20 cm3d'eau. La phase aqueuse est extraite avec 3 fois 100 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis puis concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 35 C. Le résidu est repris dans l'eau, trituré et filtré puis séché dans un dessiccateur. On obtient ainsi 1,2 g de N-[(2-cyclopropylméthyl-3-oxo-2,3dihydro-1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'une poudre écrue fondant à 229 C. Spectre de masse : DCI : m/z 287 (M+H)+.

N-((2-Cyclopropvlméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl")-guanidine La N-[(2-cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 2,9 g de tert-butylate de potassium, de 2,5g de chlorure de guanidinium et de 1,5 g de (2-cyclopropylméthyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 20 cm3d'eau. La phase aqueuse est extraite avec 3 fois 100 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis puis concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 35 C. Le résidu est repris dans l'eau, trituré et filtré puis séché dans un dessiccateur. On obtient ainsi 1,2 g de N-[(2-cyclopropylméthyl-3-oxo-2,3dihydro-1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'une poudre écrue fondant à 229 C. Spectre de masse : DCI : m/z 287 (M+H)+.
(2-Cyclopropylméthyl-3 -oxo-2,3 -dihydro- 1 H-isoindol- 1 -vD-acétate d'éthyle Le (2-cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,61 g d'hydrure de sodium à 60% dans 20 cm3de 1,2-diméthoxyéthane, 3,0 cm3 de triéthylphosphonoacétate et 1,55 g de 3-hydroxy-2-cyclopropylméthyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (70/30 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 30 C.
On obtient ainsi 1,45 g de (2-cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l- yl) -acétate d'éthyle, sous forme d'une huile incolore (Rf = 0,52, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).

3-Hvdroxy-2-cyclopropvlméthyl-2,3-dihydro-isoindol-l-one
<Desc/Clms Page number 36>
La 3-hydroxy-2-cyclopropylméthyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 4,3 g de N-cyclopropylméthyl-phthalimide dans 40 cm3de méthanol et de 1,2 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le précipité obtenu est filtré, puis le solide obtenu est repris dans le dichlorométhane et concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 4,1 g de 3-hydroxy-2-cyclopropylméthyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre blanche (Rf = 0,38, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (50/50 en volumes)).
N-Cyclopropylméthyl-phthalimide Le N-cyclopropylméthyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 4 g d'anhydride phthalique, de 2,3 cm3 de cyclopropylméthylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 40 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 2 heures, puis il est refroidi à une température voisine de 20 C et agité pendant 16 heures. Le mélange réactionnel est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans du dichlorométhane et lavé 2 fois avec une solution aqueuse saturée de bicarbonate de sodium. La phase organique est décantée, séchée sur sulfate de magnésium, filtrée puis concentrée à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 4,3 g de N-cyclopropylméthyl-phthalimide, sous forme d'un solide blanc cotoneux (Rf = 0,46, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (40/60 en volumes)).
Exemple 9 :
N-[(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yli-acét]-uanidine A 20 cm d'éthanol absolu sous atmosphère inerte on ajoute 0,22 g de sodium. Après disparition totale du sodium, on ajoute 0,94 g de chlorure de guanidinium. Le
N-[(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yli-acét]-uanidine A 20 cm d'éthanol absolu sous atmosphère inerte on ajoute 0,22 g de sodium. Après disparition totale du sodium, on ajoute 0,94 g de chlorure de guanidinium. Le
<Desc/Clms Page number 37>
mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 1 heure, puis on ajoute une solution de 2 g de (2-benzyl-3-oxo-2,3- dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 5 cm3d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 10 cm3d'eau et 30 cm3 d'éther diéthylique puis agité à une température voisine de 0 C pendant 15 minutes. Le précipité obtenu est filtré, lavé avec 2 fois 10 cm3d'eau glacée puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C . On obtient ainsi 1,2 g de

N-[(2-benzyl-3-oxo-2,3-dihydro- H-isoindol-I-yl)-acétyl]-guanidine, sous forme d'une poudre blanche fondant à 222-225 C. Spectre infra-rouge (KBr) 3488 ; 3344 ; 1621 ; 1382 et 704 cm-1.

N-[(2-benzyl-3-oxo-2,3-dihydro- H-isoindol-I-yl)-acétyl]-guanidine, sous forme d'une poudre blanche fondant à 222-225 C. Spectre infra-rouge (KBr) 3488 ; 3344 ; 1621 ; 1382 et 704 cm-1.
(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yD-acétated'éthyle Le (2-benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 3,2 g d'hydrure de sodium à 60% dans 200 cm3de 1,2-diméthoxyéthane, 16,4 cm3 de triéthylphosphonoacétate et 9,5 g de 3- hydroxy-2-benzyl-2,3-dihydro-isoindol-l-one. Le mélange est chauffé à reflux pendant 18 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 100 cm3d' eau puis 100 cm3 d'acétate d'éthyle. Après décantation, la phase aqueuse est extraite 2 fois avec 100 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm3de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (75/25 puis 67/33 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi

8,7 g de (2-benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune visqueuse. (Rf = 0,35, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (75/25 en volumes)).
8,7 g de (2-benzyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune visqueuse. (Rf = 0,35, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (75/25 en volumes)).
<Desc/Clms Page number 38>
3-Hydroxy-2-benzyl-2,3-dihydro-isoindol- 1 -one La 3-hydroxy-2-benzyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 10,2 g de N-benzyl-phthalimide dans 100 cm3de méthanol et de 2,6 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de 50 cm3 d'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C, puis on rajoute 50 cm3d'eau distillée. La phase aqueuse est extraite 3 fois avec 50 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm3 de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 9,5 g de 3-hydroxy-2-benzyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre blanche. (Rf = 0,20, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
N-Benzvl-phthalimide Le N-benzyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 10 g d'anhydride phthalique, de 7,3 cm3 de benzylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 100 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 3 heures, puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est repris dans 100 cm3 d'une solution aqueuse saturée de bicarbonate de sodium et la phase aqueuse est extraite 2 fois avec 100 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm3de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 10,3 g de N-benzyl-phthalimide, sous forme d'une poudre blanche. (Rf = 0,44, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
<Desc/Clms Page number 39>
Exemple 10 :

N-f (6-tet-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acét]-uanidine La N-[(6-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 9 à partir de 20 cm3d'éthanol absolu, de 0,37 g de sodium, de 1,56 g de chlorure de guanidinium et de 3,53 g de (6-tert-butyl- 2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 10 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 15 cm3d'eau et 45 cm d'éther diéthylique puis agité à une température voisine de 0 C pendant 2 heures. Le précipité obtenu est filtré, lavé avec 2 fois 10 cm3d'eau glacée puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C

. On obtient ainsi 2,5 g de N-[(6-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol- l-yl)-acétyl]-guanidine, sous forme d'une poudre brune fondant à 214-215 C. Spectre de masse: DCI : m/z 345 (M+H)+.

N-f (6-tet-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acét]-uanidine La N-[(6-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 9 à partir de 20 cm3d'éthanol absolu, de 0,37 g de sodium, de 1,56 g de chlorure de guanidinium et de 3,53 g de (6-tert-butyl- 2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 10 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 15 cm3d'eau et 45 cm d'éther diéthylique puis agité à une température voisine de 0 C pendant 2 heures. Le précipité obtenu est filtré, lavé avec 2 fois 10 cm3d'eau glacée puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C
. On obtient ainsi 2,5 g de N-[(6-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol- l-yl)-acétyl]-guanidine, sous forme d'une poudre brune fondant à 214-215 C. Spectre de masse: DCI : m/z 345 (M+H)+.
(5-fer-Butyl-2-isobutyl-3-oxo-2,3-dihvdro-lH-isoindol-l-yl)-acétate d'éthyle et (6tert-Butyl-2-isobutyl-3-oxo-2,3-dihvdro-1H-isoindol-1-yl)-acétate d'éthyle Le (5-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle et le (6-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétate d'éthyle sont préparés comme décrit dans l'exemple 2 à partir de 2,9 g d'hydrure de sodium à 60% dans 200 cm3de 1,2-diméthoxyéthane, 15,2 cm3 de triéthylphosphonoacétate et 9,6 g

d'un mélange de 5-tert-butyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one de 6tert-butyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one. Le mélange est chauffé à reflux pendant 18 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 110 cm3d' eau puis 100 cm3 d'acétate d'éthyle.
d'un mélange de 5-tert-butyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one de 6tert-butyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one. Le mélange est chauffé à reflux pendant 18 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 110 cm3d' eau puis 100 cm3 d'acétate d'éthyle.
Après décantation, la phase aqueuse est extraite 2 fois avec 100 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 100 cm3de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séparé par chromatographie CLHP sur
<Desc/Clms Page number 40>
une colonne chirale de type WHELK-01SS 10 [un en éluant successivement avec des mélanges heptane/ isopropanol (90/10 puis 50/50 en volumes). Les fractions contenant le premier régioisomère sont réunies et concentrées sous pression réduite (1 kPa) à une température voisine de 40 C. Le résidu est séché sous pression réduite (3 kPa) à une température voisine de 40 C . On obtient ainsi 1,81 g de (5-tert-butyl-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sous forme d'une huile visqueuse grise (Rf = 0,43, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)). Les fractions contenant le deuxième régioisomère sont réunies et concentrées sous pression réduite (1 kPa) à une température voisine de 40 C. Le résidu est séché sous pression réduite (3 kPa) à une température voisine de 40 C . On obtient ainsi 3,53 g de (6-tert-butyl-2-isobutyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sous forme d'une huile visqueuse grise. (Rf = 0,38, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
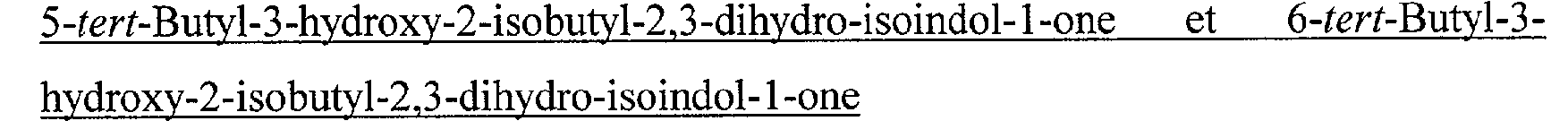
5-tert-Bptyl-3-~hydroxy-2-isobUyl-2,3-dihydro-isoindol-1 -one et 6-tet-Butyl-3h dy roxy-2-isobutyl-2,3-dihydro-isoindol-1-one La 5-tert-butyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one et la 6-tert-butyl-3- hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one sont préparées comme décrit dans l'exemple 1 à partir de 10,4 g de 4-tert-butyl-N-isobutyl-phthalimide dans 60 cm3 de méthanol et de 2,3 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 19 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de 50 cm3 d'eau distillée.
Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C, puis on rajoute 50 cm3d'eau distillée. La phase aqueuse est extraite 2 fois avec 100 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm3 de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 9,9 g d'un mélange de 5-tert-butyl-3-hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one et de 6-tert-butyl-3-hydroxy-2-isobutyl-2,3-
<Desc/Clms Page number 41>
dihydro-isoindol-1-one, sous forme d'une mousse jaune (Rf = 0,26 et 0,30 non attribués, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
4-tert-Butyl-N-isobutyl-phthalimide Le 4-tert-butyl-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 10 g d'anhydride 4-tert-butyl-phthalique et de 4,9 cm3 d'isobutylamine dans 100 cm de toluène. Le mélange réactionnel est chauffé à une température voisine de 75 C pendant 10 minutes puis on ajoute une quantité catalytique d'acide paratoluènesulfonique et le mélange est chauffé à une température voisine de 140 C pendant 3 heures. Après refroidissement à une température voisine de 60 C, le mélange réactionnel est concentré à sec sous pression réduite (2 kPa). Le résidu est repris dans un mélange de 50 cm3 d'eau et 30 cm3 d'une solution aqueuse saturée de bicarbonate de sodium et la phase aqueuse est extraite 2 fois avec 200 cm3 de dichlorométhane. Les extraits organiques sont réunis, lavés avec 50 cm de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 10,4 g 4- tert-butyl-N-isobutyl-phthalimide, sous forme d'une huile visqueuse jaune. (Rf = 0,75, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (75/25 en volumes)).
Exemple 11 :

N-[(5-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétvl]'-guanidine La N-[(5-tet-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 9 à partir de 10 cm3d'éthanol absolu, de 0,19 g de sodium, de 0,80 g de chlorure de guanidinium et de 1,81 g de (5-tert-butyl-
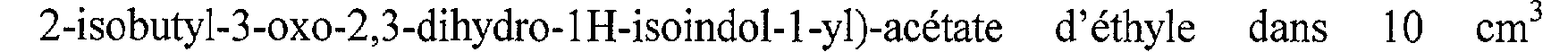
2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 10 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une
N-[(5-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétvl]'-guanidine La N-[(5-tet-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 9 à partir de 10 cm3d'éthanol absolu, de 0,19 g de sodium, de 0,80 g de chlorure de guanidinium et de 1,81 g de (5-tert-butyl-
2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 10 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une
<Desc/Clms Page number 42>
température voisine de 40 C. Le résidu est repris dans un mélange de 10 cm3d'eau et 30 cm3d'éther diéthylique puis agité à une température voisine de 0 C pendant 1 heures. Le précipité obtenu est filtré, lavé avec 2 fois 10 cm3 d'eau glacée puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C

. On obtient ainsi 1,1 g de N-[(5-tet-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindoll-yl)-acétyl]-guanidine, sous forme d'une poudre blanche fondant à 262-263 C. Le (5-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-IH-isoindol- 1-yl)-acétate d'éthyle est décrit dans l'exemple 10. (Analyse C19 H28 N4 02 % calculé C : 66,25,H : 8,19, N : 16,27, O : 9,29 % trouvé C : H : N : Exemple12 :
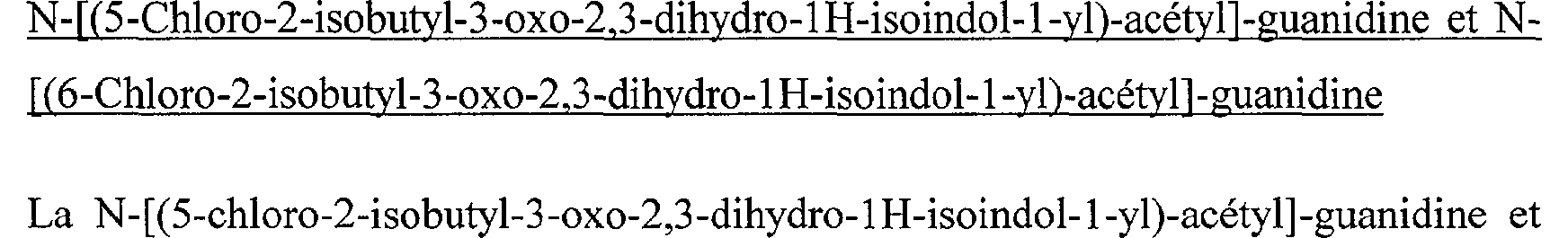
N- [(5 -Chloro-2-isobutyl-3-oxo-2,3 -dihvdro- 1 H-isoindol- 1 -vD-acétyli -guanidine et N- [(6-Chloro-2-isobutyl-3-oxo-2,3-dihvdro-lH-isoindol-l-vl)-acétyll-Ruanidine La N-[(5-chloro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine et la N-[(6-chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine sont préparées comme décrit dans l'exemple 9 à partir de 20 cm3d'éthanol absolu, de 0,35 g de sodium, de 1. 46 g de chlorure de guanidinium et de 3,1 g de (5-chloro-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et de (6-chloro-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 15 cm3d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 15 cm d'eau et 45 cm3 d'éther diéthylique puis agité à une température voisine de 0 C pendant 4 heures. Le précipité obtenu est filtré, lavé avec 2 fois 20 cm3d'eau glacée puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C . On obtient ainsi 1 g de N-[(5-chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-

acétyl]-guanidine et N-[(6-chloro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)- acétyl]-guanidine, sous forme d'une poudre blanche. (Analyse C15 H19 Cl N4 02 % calculé C : 55,81, H : 5,93,Cl : 10,98, N : 17,36,0 : 9,91 % trouvé C : 55,67,H : 6,18, Cl : 11,32, N : 17,05). Spectre de masse : m/z 323 (M+H)+.
. On obtient ainsi 1,1 g de N-[(5-tet-butyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindoll-yl)-acétyl]-guanidine, sous forme d'une poudre blanche fondant à 262-263 C. Le (5-tert-butyl-2-isobutyl-3-oxo-2,3-dihydro-IH-isoindol- 1-yl)-acétate d'éthyle est décrit dans l'exemple 10. (Analyse C19 H28 N4 02 % calculé C : 66,25,H : 8,19, N : 16,27, O : 9,29 % trouvé C : H : N : Exemple12 :
N- [(5 -Chloro-2-isobutyl-3-oxo-2,3 -dihvdro- 1 H-isoindol- 1 -vD-acétyli -guanidine et N- [(6-Chloro-2-isobutyl-3-oxo-2,3-dihvdro-lH-isoindol-l-vl)-acétyll-Ruanidine La N-[(5-chloro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine et la N-[(6-chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine sont préparées comme décrit dans l'exemple 9 à partir de 20 cm3d'éthanol absolu, de 0,35 g de sodium, de 1. 46 g de chlorure de guanidinium et de 3,1 g de (5-chloro-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et de (6-chloro-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 15 cm3d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 15 cm d'eau et 45 cm3 d'éther diéthylique puis agité à une température voisine de 0 C pendant 4 heures. Le précipité obtenu est filtré, lavé avec 2 fois 20 cm3d'eau glacée puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C . On obtient ainsi 1 g de N-[(5-chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-
acétyl]-guanidine et N-[(6-chloro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)- acétyl]-guanidine, sous forme d'une poudre blanche. (Analyse C15 H19 Cl N4 02 % calculé C : 55,81, H : 5,93,Cl : 10,98, N : 17,36,0 : 9,91 % trouvé C : 55,67,H : 6,18, Cl : 11,32, N : 17,05). Spectre de masse : m/z 323 (M+H)+.
<Desc/Clms Page number 43>

(5-CWoro-2-isobutyl-3-oxo-2J-dihvdro-IH-isoindol-l-yl)-acétate d'éthyle et (6chloro-2-isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétate d'éthyle Le (5-chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et le (6chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle sont préparés comme décrit dans l'exemple 2 à partir de 4 g d'hydrure de sodium à 60% dans 200 cm3de 1,2-diméthoxyéthane, 20,3 cm3 de triéthylphosphonoacétate et 12 g de 5- chloro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et de 6-chloro-3-hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one. Le mélange est chauffé à reflux pendant 18 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 200 cm3 d' eau puis 100 cm3 d'acétate d'éthyle. Après décantation, la phase aqueuse est extraite 2 fois avec 200 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 100 cm3de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est purifié par chromatographie sous pression d'argon (60 kPa), sur une cartouche de gel de silice (granulométrie 32-63 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (90/10 puis 80/20 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi
3,1 g de (5-chloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et de (6-chloro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile jaune visqueuse. (Rf = 0,34, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (75/25 en volumes)).
5-Chloro-3-hydroxv-2-isobutvl-2,3-dihydro-isoindol-l-one et 6-chloro-3-hydroxy-2- isobutyl-2,3-dihydro-isoindol-1-one La 5-chloro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et la 6-chloro-3hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one sont préparées comme décrit dans l'exemple 1 à partir de 12 g de 4-chloro-N-isobutyl-phthalimide dans 100 cm3 de méthanol et de 3 g de borohydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est refroidi à une
<Desc/Clms Page number 44>
température voisine de 0 C et on ajoute au goutte à goutte de 100 cm3d'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C. La phase aqueuse est extraite 2 fois avec 150 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm3de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 12 g de 5-chloro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et de 6-chloro-3-hydroxy- 2-isobutyl-2,3-dihydro-isoindol-l-one , sous forme d'une poudre blanche (Rf = 0,15, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
4-Chloro-N-isobutyl-phthalimide Le 4-chloro-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 10 g d'anhydride 4-chloro-phthalique, de 5,4 cm3d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 100 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 16 heures, puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est repris dans 100 cm3 d'une solution aqueuse saturée de bicarbonate de sodium et la phase aqueuse est extraite 2 fois avec 100 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 12 g de 4-chloroN-isobutyl-phthalimide, sous forme d'une poudre blanche. (Rf = 0,85, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
Exemple13 :

N-[(5-Bromo-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-)-acétyll-uanidine et Nl (6-bromo-2-isobutyl-3-oxo-2.3-dihydro-l 1 H-isoindol- 1 -yD-acétyl] -guanidine
N-[(5-Bromo-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-)-acétyll-uanidine et Nl (6-bromo-2-isobutyl-3-oxo-2.3-dihydro-l 1 H-isoindol- 1 -yD-acétyl] -guanidine
<Desc/Clms Page number 45>
La N-[(5-bromo-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine et la N-[(6-bromo-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine sont préparées comme décrit dans l'exemple 9 à partir de 30 cm3d'éthanol absolu, de 0,36 g de sodium, de 1. 5 g de chlorure de guanidinium et de 3,7 g de (5-bromo-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et de (6-bromo-2-
isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle dans 20 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 100 cm3d'eau et 200 cm3 d'acétate d' éthyle. La phase organique est lavée par 100 cm3d'eau puis par 150 cm3 d'une solution saturée de chlorure de sodium. Cette phase organique est séchée sur sulfate de magnésium et concentrée sous pression réduite (2 kPa) à une température voisine de 40 C . On obtient ainsi un solide blanc qui est ensuite repris et agité dans un mélange de 15 cm3d'eau et 50 cm3 de diéthyléther pendant 1. 5 heures à une température voisine de 20 C. Le solide blanc ainsi obtenu est filtré, lavé par 2 fois 50 cm3 de diéthyléther et séché dans un dessiccateur sous pression réduite (2 Pa) à une température voisine de 35 C. On obtient ainsi 0,92 g de N-[(5-bromo-2-isobutyl-3-
oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine et de N-[(6-bromo-2-isobutyl-3- oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine, sous forme d'une poudre blanche. (Analyse C15 H19 Br N4 02 % calculé C : H : 5,21, Br : 21,76, N : 15,26, O : 8,71 % trouvé C : H : 5,34, Cl : 14,72, N : Spectre de masse: El : m/z 366 (M+), m/z 279, m/z 86 (pic de base).

(5-Bromo-2-isobutyl-3-oxo-2J-dihydro-IH-isoindol-I-yl)-acétate d'éthyle et (6bromo-2-isobutyl-3-oxo-2 3 -dihydro-1 H-isoindol- 1-ylVacétated'éthyle : Le (5-bromo-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle et le (6-bromo-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle sont préparés comme décrit dans l'exemple 2 à partir de 5,0 g d'hydrure de sodium à 60% dans 250 cm3de 1,2-diméthoxyéthane, 24,8 cm3 de triéthylphosphonoacétate et 23,7 g de 5-bromo-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et de 6-bromo-3- hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one. Le mélange est chauffé à reflux
<Desc/Clms Page number 46>
pendant 4 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 300 cm3d'eau puis le mélange est extrait par 3 fois 250 cm3 de diéthyléther. Les extraits organiques sont réunis, lavés avec 300 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est purifié par chromatographie sous pression d'argon (80 kPa), sur une colonne de gel de silice (granulométrie 32-63 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (95/5 en volumes), de cyclohexane/méthanol (90/10) et de cyclohexane/acétate d'éthyle (80/20). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 3,72 g de (5-bromo-2-isobutyl-3-oxo-2,3-dihydro- lH-isoindol-l-yl)-acétate d'éthyle et de (6-bromo-2-isobutyl-3-oxo-2,3-dihydro-lH- isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile incolore. (Rf = 0,70, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).

5-Bromo-3-hydroxy-2-isobutvl-2,3-dihydro-isoindol-l-one et 6-bromo-3-hydroxy-2- isobutyl-2,3-=dihydro-isoindol-1-one La 5-bromo-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et la 6-bromo-3hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one sont préparées comme décrit dans l'exemple 1 à partir de 23,6 g de 4-bromo-N-isobutyl-phthalimide dans 200 cm3 de méthanol et de 4,5 g de borohydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de 175 cm3d'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C. La phase aqueuse est extraite 3 fois avec 200 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 23,7 g de 5-bromo-3-hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one et de 6-bromo-3-hydroxy-2-isobutyl-2,3- dihydro-isoindol-I-one , sous forme d'une poudre blanche (Rf = 0,79,
<Desc/Clms Page number 47>
chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
4-Bromo-N-isobutyl-phthalimide Le 4-bromo-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 20 g d'anhydride 4-bromo-phthalique, de 9,2 cm3 d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 200 cm3 de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 6 heures, puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C, puis le résidu est repris dans 350 cm3 d'acétate d'éthyle et 300 cm3 d'une solution saturée d'hydrogénocarbonate de sodium. La phase aqueuse est décantée puis extraite 2 fois avec 350 cm3d'acétate d'éthyle. Les extraits organiques réunis sont lavés avec 300 cm de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 23,6 g de 4-bromo-N-isobutyl-phthalimide, sous forme d'une poudre blanche (Rf = 0,91, chromatographie sur couche mince de gel de silice, éluant: cyclohexane/acétate d'éthyle (50/50 en volumes)).
Exemple 14 :
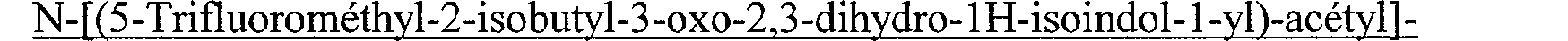
N-[(5-Trifluorométhyl-2-isobgtyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétylL- guanidine La N-[(5-trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine est préparée comme décrit dans l'exemple 9 à partir de 10 cm3d'éthanol absolu, de 0,19 g de sodium, de 0,78 g de chlorure de guanidinium et de 1,86 g de (5-
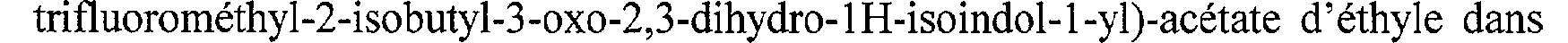
trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle dans 10 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est ensuite repris dans un mélange de 5 cm3 d'eau et 15 cm3 de diéthyléther puis reconcentré à sec dans les mêmes conditions. Le
N-[(5-Trifluorométhyl-2-isobgtyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétylL- guanidine La N-[(5-trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine est préparée comme décrit dans l'exemple 9 à partir de 10 cm3d'éthanol absolu, de 0,19 g de sodium, de 0,78 g de chlorure de guanidinium et de 1,86 g de (5-
trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle dans 10 cm3 d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est ensuite repris dans un mélange de 5 cm3 d'eau et 15 cm3 de diéthyléther puis reconcentré à sec dans les mêmes conditions. Le
<Desc/Clms Page number 48>
résidu est repris par 100 cm3d'acétate d'éthyle et la phase organique est lavée par 2 fois 60 cm3d'une solution aqueuse de soude IN puis par 2 fois 60 cm3 d'une solution d'acide chlorhydrique 4N. Les extraits aqueux acides sont réunis, traités par une solution aqueuse d'hydroxyde de sodium à 30% jusqu'à un pH de 14, puis extraits par 3 fois 50 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés par 100 cm d'une solution saturée de chlorure de sodium, séchés sur sulfate de magnésium et concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le solide jaune ainsi obtenu est repris dans 20 cm3de diéthyléther, agité à une température voisine de 20 C pendant 1 heure puis filtré. Le solide est lavé par 2 fois 20 cm3de diéthyléther puis séché dans un dessiccateur sous pression réduite (2 Pa) à une température voisine de 35 C. On obtient ainsi 0,34 g de N-[(5-trifluorométhyl-2-

isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine sous forme d'une poudre jaune fondant à 224 C. (Analyse C16 H19 F3 N4 02 % calculé C : 53,93, H : 5,37, F : N : 15,72, O : 8,98 % trouvé C : H : F : N : Exemple15 :

N-r(6- Trifluorométhyl-2-isobutyl-3-oxo-2.3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]- guanidine La N-[(6-trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl]- guanidine est préparée comme décrit dans l'exemple 9 à partir de 10 cm3d'éthanol absolu, de 0,13 g de sodium, de 0,52 g de chlorure de guanidinium et de 1,25 g de (6-

trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle dans 10 cm3d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est ensuite repris par un mélange de 5 cm3 d'eau et 15 cm3de diéthyléther puis reconcentré à sec dans les mêmes conditions. Le résidu est repris par 100 cm3d'acétate d'éthyle et la phase organique est lavée par 2 fois 50 cm3d'une solution aqueuse de soude IN puis par 30 cm3 d'une solution aqueuse d'acide chlorhydrique 4N. La phase aqueuse acide est traitée avec une solution aqueuse d'hydroxyde de sodium à 30% jusqu'à un pH de 14, puis extraite
isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine sous forme d'une poudre jaune fondant à 224 C. (Analyse C16 H19 F3 N4 02 % calculé C : 53,93, H : 5,37, F : N : 15,72, O : 8,98 % trouvé C : H : F : N : Exemple15 :
N-r(6- Trifluorométhyl-2-isobutyl-3-oxo-2.3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]- guanidine La N-[(6-trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl]- guanidine est préparée comme décrit dans l'exemple 9 à partir de 10 cm3d'éthanol absolu, de 0,13 g de sodium, de 0,52 g de chlorure de guanidinium et de 1,25 g de (6-
trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle dans 10 cm3d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est ensuite repris par un mélange de 5 cm3 d'eau et 15 cm3de diéthyléther puis reconcentré à sec dans les mêmes conditions. Le résidu est repris par 100 cm3d'acétate d'éthyle et la phase organique est lavée par 2 fois 50 cm3d'une solution aqueuse de soude IN puis par 30 cm3 d'une solution aqueuse d'acide chlorhydrique 4N. La phase aqueuse acide est traitée avec une solution aqueuse d'hydroxyde de sodium à 30% jusqu'à un pH de 14, puis extraite
<Desc/Clms Page number 49>
par 3 fois 50 cm3d'acétate d'éthyle. Les extraits organiques sont réunis et lavés par une solution saturée de chlorure de sodium, séchés sur sulfate de magnésium puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le solide blanc ainsi obtenu est repris dans 20 cm3 de diéthyléther et agité à une température voisine de 20 C, puis filtré. Le solide est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 0,099 g

de N-[(6-trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine sous forme d'un solide jaune pâle fondant à 154 C. Spectre de masse: El: m/z 356 (M+), m/z 269, m/z 200, m/z 86 (pic de base).

(5-Trifluométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle et (6-trifluométhyl-2-isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétate d'éthyle Le (5-trifluométhyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l -yl)-acétate d'éthyle et le (6-trifluométhyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sont préparés comme décrit dans l'exemple 2 à partir de 1,32 g d'hydrure de sodium à 60% dans 120 cm3de 1,2-diméthoxyéthane, 6,57 cm3 de triéthylphosphonoacétate et 12,07 g de 5-trifluométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et de 6- trifluométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one. Le mélange est chauffé à reflux pendant 24 heures puis refroidi à une température voisine de 20 C. Le mélange réactionnel est ensuite traité avec 150 cm3 d' eau puis le mélange est extrait par 3 fois 250 cm3de diéthyléther. Les extraits organiques sont réunis, lavés avec 150 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Un mélange de 2,3 g du résidu est injecté sur une colonne de 8 cm de diamètre contenant 1,2 kg de phase stationnaire chirale Whelk OI,SS. L'élution est réalisée avec une phase mobile composée d'un mélange de dichlorométhane, d'éthanol et d'heptane dans les proportion 14,5/ 0,5/ 85 v/v. Deux injections supplémentaires de 2,7 g et 3 g sont réalisées dans des conditions identiques. Les fractions contenant le premier régioisomère sont réunies et concentrées à sec sous pression réduite (1 kPa) à une température voisine de 40 C. On obtient ainsi 1,86 g de (5-trifluométhyl-2-isobutyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sous forme d'un solide blanc
de N-[(6-trifluorométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine sous forme d'un solide jaune pâle fondant à 154 C. Spectre de masse: El: m/z 356 (M+), m/z 269, m/z 200, m/z 86 (pic de base).
(5-Trifluométhyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle et (6-trifluométhyl-2-isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétate d'éthyle Le (5-trifluométhyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l -yl)-acétate d'éthyle et le (6-trifluométhyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sont préparés comme décrit dans l'exemple 2 à partir de 1,32 g d'hydrure de sodium à 60% dans 120 cm3de 1,2-diméthoxyéthane, 6,57 cm3 de triéthylphosphonoacétate et 12,07 g de 5-trifluométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et de 6- trifluométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one. Le mélange est chauffé à reflux pendant 24 heures puis refroidi à une température voisine de 20 C. Le mélange réactionnel est ensuite traité avec 150 cm3 d' eau puis le mélange est extrait par 3 fois 250 cm3de diéthyléther. Les extraits organiques sont réunis, lavés avec 150 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Un mélange de 2,3 g du résidu est injecté sur une colonne de 8 cm de diamètre contenant 1,2 kg de phase stationnaire chirale Whelk OI,SS. L'élution est réalisée avec une phase mobile composée d'un mélange de dichlorométhane, d'éthanol et d'heptane dans les proportion 14,5/ 0,5/ 85 v/v. Deux injections supplémentaires de 2,7 g et 3 g sont réalisées dans des conditions identiques. Les fractions contenant le premier régioisomère sont réunies et concentrées à sec sous pression réduite (1 kPa) à une température voisine de 40 C. On obtient ainsi 1,86 g de (5-trifluométhyl-2-isobutyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle sous forme d'un solide blanc
<Desc/Clms Page number 50>
(Rf = 0,64, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)). Les fractions contenant le deuxième régioisomère sont réunies et concentrées à sec sous pression réduite (1 kPa) à une température voisine de 40 C pour donner 2,79 g d'un solide blanc. Ce produit est repurifié par chromatographie sous pression d'argon (80 kPa), sur une cartouche de gel de silice (granulométrie 32-63 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (90/10 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 1,25 g de (6-trifluométhyl-2-isobutyl- 3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle, sous forme d'une huile incolore visqueuse (Rf = 0,46, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).

5-Trifluométhyl-3-h. d-2-isobutyl-2,3-dihydro-isoindol-1-one et 6-trifluométhyl- 3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one La 5-trifluométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et la 6trifluométhyl-3-hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one sont préparées comme décrit dans l'exemple 1 à partir de 14,9 g de 4-trifluorométhyl-N-isobutylphthalimide dans 300 cm3de méthanol et de 2,96 g de borohydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de 100 cm3d'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C. La phase aqueuse est extraite 3 fois avec 200 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés par 300 cm3 d'une solution saturée de chlorure de sodium, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 12,07 g de 5-trifluorométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol- 1-one et de 6-trifluorométhyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one , sous forme d'une poudre blanche. (Rf = 0,64, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (50/50 en volumes)).
<Desc/Clms Page number 51>
4-Trifluorométhyl-N-isobutyl-phthalimide Le 4-trifluorométhyl-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 14,8 g d'anhydride 4-trifluorométhyl-phthalique, de 7,2 cm3 d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 160 cm de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 5 heures, puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C et le résidu est repris dans 250 cm3d'acétate d'éthyle et 200 cm d'une solution aqueuse saturée d'hydrogénocarbonate de sodium. La phase aqueuse est décantée puis extraite 2 fois avec 250 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec de la saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 14,9 g de 4-trifluorométhyl-N-isobutylphthalimide, sous forme d'une poudre jaune (Rf = 0,59, chromatographie sur couche mince de gel de silice, éluant : d'éthyle (75/25 en volumes)).
L' anhydride 4-trifluorométhyl-phthalique peut être préparé par adaptation ou application de la méthode décrite par Cavalleri et coll., J. Med. Chem., 13(1), 148- 149, (1970).
Exemple 16 : N-[(5-Isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine

La N-[(5-isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétyll- guanidine est préparée comme décrit dans l'exemple 9 à partir de 20 cm3d'éthanol absolu, de 0,086g de sodium, de 0,36 g de chlorure de guanidinium et de 0,42 g de (5-

isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est ensuite repris par 50 cm3d'acétate d'éthyle et la phase organique est
La N-[(5-isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétyll- guanidine est préparée comme décrit dans l'exemple 9 à partir de 20 cm3d'éthanol absolu, de 0,086g de sodium, de 0,36 g de chlorure de guanidinium et de 0,42 g de (5-
isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est ensuite repris par 50 cm3d'acétate d'éthyle et la phase organique est
<Desc/Clms Page number 52>
lavée 2 fois avec 30 cm3 d'une solution aqueuse de soude IN puis 2 fois par 50 cm3 d'une solution aqueuse d'acide chlorhydrique IN. La phase aqueuse acide est traitée avec une solution aqueuse d'hydroxyde de sodium à 30% jusqu'à un pH de 14, puis extraite par 3 fois 30 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 50 cm de saumure saturée, séchés sur sulfate de magnésium et concentrés sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est trituré avec du diéthyléther puis filtré et séché dans un dessiccateur sous pression réduite (2 Pa) à une température voisine de 40 C pendant 2 heures. On obtient ainsi 0,0175 g de N- [(5-isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine sous forme d'une poudre blanche fondant à 208 C. R. M.N. 1H (300 MHz, (CD3)2S0 d6, 8 en ppm) : 0,75 (d, J = 6,5 Hz : 3H) ; 0,90 (d, J = 6,5 Hz : 3H) ; 1,30 (mt : 6H) ; 2,01 (mt : 1 H) ; 2,40 (dd, J = 15 et 6,5 Hz : 1 H) ; 2,65 (dd, J = 15 et 6,5 Hz : 1H) ; 3,00 (dd, J = 13,5 et 5,5 Hz : 1H) ; 3,53 (dd, J = 13,5 et 10 Hz : 1H) ; 4,68 (mt : 1H) ; 4,96 (t large, J = 6,5 Hz : 1H) ; de 6,40 à 7,10 (mf étalé : 2H) ; 6,98 (dd, J = 8,5 et 2 Hz : 1H) ; 7,09 (d, J = 2 Hz : 1H) ; 7,54 (d, J = 8,5 Hz : 1H) ; de 7,60 à 8,20 (mf étalé : 2H).

(5-Isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle Le (5-isopropyloxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,149 g d'hydrure de sodium à 60% dans 20 cm3de 1,2-diméthoxyéthane, 1,23 cm3 de triéthylphosphonoacétate et 1,08 g de 5- isopropyloxy-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one. Le mélange est chauffé à reflux pendant 5 heures puis refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 100 cm3d' eau puis le mélange est extrait par 3 fois 100 cm3de diéthyléther. Les extraits organiques sont réunis, lavés avec 100 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est purifié par chromatographie sous pression d'argon (80 kPa), sur une cartouche de gel de silice (granulométrie 32-63 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (90/10 en volumes) puis par un mélange de cyclohexane/acétate d'éthyle (80/20 en volumes). Les fractions contenant le produit
<Desc/Clms Page number 53>
attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 0,42 g de (5-isopropyloxy-2-isobutyl- 3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle sous forme d'une huile incolore. (Rf = 0,57, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).

5-Isoprop -xy-2-isobutyl-2,3-dihydro-isoindol-1-one La 5-isopropyloxy-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 1,16 g de N-isobutyl-4-isopropyloxyphthalimide dans 20 cm3de méthanol et de 0,24 g de borohydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 17 heures, puis refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de 30 cm3d'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C et 50 cm3d'eau sont additionnés. La phase aqueuse est extraite 3 fois avec 70 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés par 100 cm3d'une solution saturée de chlorure de sodium, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 1,08 g de 5-isopropyloxy-3- hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one sous forme d'une huile incolore visqueuse. (Rf = 0,50, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
N-Isobutyl-4-isopropyloxy-phthalimide Un mélange de 1 g de 4-hydroxy-N-isobutyl-phthalimide, 0,85 cm3de 2-bromopropane, 1,38 g de carbonate de potassium dans 5 cm3 de diméthylformamide est porté sous agitation à une température voisine de 60 C pendant 17 heures. Le mélange réactionnel est ensuite refroidi à une température voisine de 20 C puis concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris par 70 cm3d'eau, puis extrait 3 fois par 75 cm3 d'acétate d'éthyle.
Les phases organiques sont réunies, lavées par une solution saturée de chlorure de sodium, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression
<Desc/Clms Page number 54>
réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 1,16 g de Nisobutyl-4-isopropyloxy-phthalimide sous forme d'un solide blanc. (Rf = 0,50, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
4-Hydroxy-N-isobutyl-phthalimide Le 4-hydroxy-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 7,26 g d'anhydride 4-acétoxy-phthalique, de 7,35 cm3d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 75 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 4 heures, puis refroidi à une température voisine de 40 C et concentré à sec sous pression réduite (2 kPa). Le résidu est repris dans 175 cm3d'acétate d'éthyle et 150 cm3d'une solution saturée d'hydrogénocarbonate de sodium. La phase aqueuse est décantée puis extraite 2 fois avec 150 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 200 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris et agité dans 100 cm3 de cyclohexane à une température voisine de 20 C pendant 1,5 heures puis filtré. Le solide est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 4,0 g de 4-hydroxy-N-isobutyl-phthalimide sous forme d'un solide blanc (Rf = 0,25, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (75/25 en volumes)).
L' anhydride 4-acétoxy-phthalique peut être préparé par adaptation ou application de la méthode décrite par J. Sah, J. Med. Chem., 42 (16), 3014-3017, (1999) et N. J.
Hinde, J. Chem. Soc., Perkin Trans 2, 5, 1249-125, (1998).
Exemple 17 :

N -[ (7 - Fluoro-2-isobutyl- 3-oxo-2J-dihydro-1 H -isoindol-l-yl)-acétyll-guanidine
N -[ (7 - Fluoro-2-isobutyl- 3-oxo-2J-dihydro-1 H -isoindol-l-yl)-acétyll-guanidine
<Desc/Clms Page number 55>
La N-[(7-fluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 1,29 g de tert-butylate de potassium, de 1,32 g de chlorure de guanidinium et de 0,67 g de (7-fluoro-2-isobutyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est filtré. Le filtrat est repris avec 40 cm3d'eau et 60 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite avec 2 fois 60 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de sodium, filtrés et concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 55 C.
Le résidu d'évaporation est repris dans l'éther diéthylique et reconcentré à sec dans les mêmes conditions, puis repris dans l'eau et reconcentré à sec dans les mêmes conditions. Le résidu est purifié par chromatographie sous pression d'argon (50 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de dichlorométhane/méthanol (90/10 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 20 C. Le résidu est repris dans de l'éther diisopropylique, trituré, filtré puis séché. On obtient ainsi 0,15 g N-[(7-fluoro-2-isobutyl-3-oxo-2,3- dihydro-lH-isoindol-l-yl)-acétyl]-guanidine, sous forme d'un solide jaune pâle fondant à 205 C. R. M.N. 1H (300 MHz, (CD3)2S0 d6, 8 en ppm) : 0,74 (d, J = 6,5 Hz : 3H) ; 0,89 (d, J = 6,5 Hz : 2,06 (mt : 1H) ; 2,38 (dd, J = 15 et 7,5 Hz : 1H) ; 2,90 (dd, J = 15 et 4,5 Hz : 1H) ; 3,04 (dd, J = 13,5 et 4,5 Hz : 1H) ; 3,56 (dd, J = 13,5 et 10 Hz : 1H) ; 5,23 (dd, J = 7,5 et 4,5 Hz : 1H) ; de 6,20 à 7,00 (mf étalé : 2H) ; de 7,35 à 7,60 (mt : 3H) ; de 7,50 à 8,20 (mf étalé : 2H).

(7-Fluoro-2-isobutyl-3 -oxo-2.,3 -dihvdro-lH-isoindol-1 -y l)-acétate d'éthyle Le (7-fluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,2 g d'hydrure de sodium à 60% dans 15 cm3 de 1,2-diméthoxyéthane, 1,1 cm3de triéthylphosphonoacétate et 0,8 g de 4-fluoro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (50 kPa), sur une colonne de gel de silice (granulométrie 40-63 m), en éluant par un mélange de cyclohexane/acétate d'éthyle
<Desc/Clms Page number 56>
(80/20 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 20 C.
On obtient ainsi 0,44 g de (7-fluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)- acétate d'éthyle, sous forme d'une huile jaune. (Rf = 0,59, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).

4-Fluoro-3-hvdroxy-2-isobutyl-2,3-dihydro-isoindol-l-one La 4-fluoro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one est préparée comme décrit dans l'exemple 1 à partir de 3,9 g de 3-fluoro-N-isobutyl-phthalimide dans 20 cm3de méthanol et de 0,95 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 19 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le précipité obtenu est filtré, lavé à l'eau froide puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 3,5 g de 4-fluoro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one, sous forme d'une poudre blanche collante. (Rf = 0,36, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (60/40 en volumes)).
3-Fluoro-N-isobutyl-phthalimide Le 3-fluoro-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 3,4 g d'anhydride 3-fluoro-phthalique, de 2,0 cm3d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 20 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 3 heures. Après refroidissement à une température voisine de 20 C, le mélange réactionnel est concentré à sec sous pression réduite (2 kPa). Le résidu est repris dans du dichlorométhane et lavé avec une solution aqueuse saturée de bicarbonate de sodium. La phase organique est décantée, séchée sur sulfate de magnésium, filtrée puis concentrée à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 4,0 g de 3-fluoro-N-isobutyl-
<Desc/Clms Page number 57>
phthalimide, sous forme d'une poudre blanc cassé. (Rf = 0,73, chromatographie sur couche mince de gel de silice, éluant : dichlorométhane).
Exemple 18 :

N-[(5,6-Dichloro-2-isobutvl-3-oxo-2,3-dihydro-lH-isoindol-l-vl)-acétyl]-guanidine A 4,0 cm3 d'éthanol absolu sous atmosphère inerte on ajoute 0,04 g de sodium. Après disparition totale du sodium, on ajoute 0,16 g de chlorure de guanidinium. Le mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 45 minutes, puis il est filtré. Le filtrat est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est dilué dans 10 cm3 de tétrahydrofuranne, puis on ajoute une solution du chlorure de (5,6-dichloro-2-

isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyle préparé ci-dessous dans 5 cm de tétrahydrofuranne. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans 50 cm3 d'acétate d'éthyle puis lavé 2 fois avec 50 cm3 d'une solution d'acide chlorhydrique IN. Les extraits aqueux sont réunis puis le pH est ajusté à 14 par ajout d'une solution de soude à 30% et le mélange est extrait 3 fois avec 50 cm d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 100 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans l'acétate d'éthyle puis reconcentré à sec dans les mêmes conditions. Le résidu est repris dans 20 cm3d'éther diéthylique, trituré puis filtré. Le solide est séché dans un dessiccateur sous pression réduite (2 Pa) à une température voisine de 35 C . On obtient ainsi 0,04 g de N-[(5,6-dichloro-2-isobutyl-

3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'une poudre blanc cassé fondant à 154 C. Spectre de masse: DCI : m/z 357 (M+H)+.
N-[(5,6-Dichloro-2-isobutvl-3-oxo-2,3-dihydro-lH-isoindol-l-vl)-acétyl]-guanidine A 4,0 cm3 d'éthanol absolu sous atmosphère inerte on ajoute 0,04 g de sodium. Après disparition totale du sodium, on ajoute 0,16 g de chlorure de guanidinium. Le mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 45 minutes, puis il est filtré. Le filtrat est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est dilué dans 10 cm3 de tétrahydrofuranne, puis on ajoute une solution du chlorure de (5,6-dichloro-2-
isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyle préparé ci-dessous dans 5 cm de tétrahydrofuranne. Le milieu réactionnel est agité à une température voisine de 20 C pendant 16 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans 50 cm3 d'acétate d'éthyle puis lavé 2 fois avec 50 cm3 d'une solution d'acide chlorhydrique IN. Les extraits aqueux sont réunis puis le pH est ajusté à 14 par ajout d'une solution de soude à 30% et le mélange est extrait 3 fois avec 50 cm d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 100 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans l'acétate d'éthyle puis reconcentré à sec dans les mêmes conditions. Le résidu est repris dans 20 cm3d'éther diéthylique, trituré puis filtré. Le solide est séché dans un dessiccateur sous pression réduite (2 Pa) à une température voisine de 35 C . On obtient ainsi 0,04 g de N-[(5,6-dichloro-2-isobutyl-
3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'une poudre blanc cassé fondant à 154 C. Spectre de masse: DCI : m/z 357 (M+H)+.
Chlorure de (5.6-dichloro-2-isobutyl-3-oxo-2.3-dihydro-lH-isoindoI-l-yD-acétyle A une solution de 0,36 g d'acide (5,6-dichloro-2-isobutyl-3-oxo-2,3-dihydro-IH- isoindol-1-yl)-acétique dans 10 cm3 de dichlorométhane sous atmosphère inerte on
<Desc/Clms Page number 58>
ajoute au goutte à goutte 0,72 g de chlorure d'oxalyle. Le mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 3 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi le chlorure de (5,6-dichloro-2-isobutyl-3-oxo-2,3-dihydro-lH- isoindol-l-yl)-acétyle brut sous forme d'une huile verte visqueuse qui est directement utilisé ci-dessus.
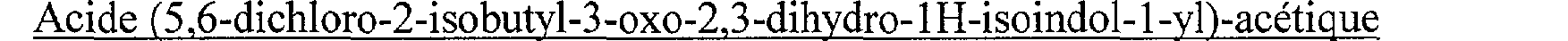
Acide (5,6-dichloro-2-isobntyl-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétique A 5 cm3 d'éthanol absolu sous atmosphère inerte on ajoute 0,09 g de sodium. Après disparition totale du sodium, on ajoute 0,04 g de chlorure de guanidinium. Le mélange réactionnel est agité sous atmosphère inerte à une température voisine de 20 C pendant 1,5 heures, puis on ajoute une solution de 0,93 g de (5,6-dichloro-2- isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle dans 10 cm3d'éthanol absolu. Le milieu réactionnel est agité à une température voisine de 20 C pendant 18 heures, puis il est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de 5 cm3d'eau et 15 cm3 d'éther diéthylique puis agité à une température voisine de 20 C pendant 1 heure. Le mélange est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans un mélange de dichlorométhane et d'acétate d'éthyle puis filtré. Le filtrat est purifié par chromatographie sous pression d'argon (80 kPa), sur une cartouche de gel de silice (granulométrie 32-63 m), en éluant par un mélange de dichlorométhane/méthanol (95/5 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans l'éther diéthylique, trituré puis filtré. Le solide est séché dans un dessiccateur sous pression réduite (2 Pa) à une température voisine de 35 C puis redissous dans 70 cm3d'acétate d'éthyle et traité avec 50 cm3d'une solution aqueuse de soude 1N. Après décantation, la phase organique est lavée avec 50 cm3d'une solution aqueuse de soude IN. Les extraits aqueux sont réunis puis traités avec une solution aqueuse d'acide chlorhydrique 5N jusqu'à pH 1. Après extraction avec 3 fois 50 cm3 d'acétate d'éthyle, les extraits organiques sont réunis, lavés avec 100 cm3 de saumure saturée, séchés sur sulfate de
<Desc/Clms Page number 59>
magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 0,36 g d'acide (5,6-dichloro-2-isobutyl- 3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétique, sous forme d'un solide jaune. (Rf = 0,33, chromatographie sur couche mince de gel de silice, éluant : dichlorométhane/méthanol (90/10 en volumes)).

(5,6-Dichloro-2-isobel-3-oxo-2,3-dihydro-IH-isoindol-1-yl)-acétate d'éthyle Le (5,6-dichloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,75 g d'hydrure de sodium à 60% dans 45 cm3de 1,2-diméthoxyéthane, 3,71 cm3 de triéthylphosphonoacétate et 3,42 g de 5,6-dichloro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one. Le mélange est chauffé à reflux pendant 5,5 heures puis il est refroidi à une température voisine de 20 C. Le mélange réactionnel est traité avec 75 cm3 d' eau puis 100 cm3 d'éther diéthylique. Après décantation, la phase aqueuse est extraite 2 fois avec 100 cm3 d'éther diéthylique. Les extraits organiques sont réunis, lavés avec 100 cm3de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est purifié par chromatographie sous pression d'argon (80 kPa), sur une cartouche de gel de silice (granulométrie 32-63 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (90/10 puis 80/20 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 0,93 g de (5,6-dichloro-2-isobutyl-3oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle sous forme d'un solide blanc (Rf
0,74, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).

0,74, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
5,6-Dichloro-3-hvdroxv-2-isobutvl-2,3-dihvdro-isoindol-l-one La 5,6-dichloro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 3,78 g de 4,5-dichloro-N-isobutylphthalimide dans 75 cm3de méthanol et de 0,75 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 16 heures,
<Desc/Clms Page number 60>
puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte de l'eau distillée. Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 40 C, puis on rajoute 100 cm3 d'acétate d'éthyle. Après décantation, la phase aqueuse est extraite 2 fois avec 75 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 150 cm3 de saumure, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 3,42 g de 5,6-dichloro-3-hydroxy-2- isobutyl-2,3-dihydro-isoindol-l-one sous forme d'un solide blanc cassé. (Rf = 0,69, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
4,5-Dichloro-N-isobutyl-phthalimide Le 4,5-dichloro-N-isobutyl-phthalimide peut être préparé comme décrit dans l'exemple 2 à partir de 10 g d'anhydride 4,5-dichloro-phthalique et de 4,6 cm3d' isobutylamine dans 100 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 10 minutes puis on ajoute une quantité catalytique d'acide para-toluènesulfonique et le mélange est chauffé à une température voisine de 140 C pendant 4 heures. Après refroidissement à une température voisine de 40 C, le mélange réactionnel est concentré à sec sous pression réduite (2 kPa). Le résidu est repris dans 250 cm3d'une solution aqueuse saturée de bicarbonate de sodium et le mélange est extrait 3 fois avec 200 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, lavés avec 200 cm3 de saumure saturée, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 2,8 g de 4,5-dichloro-N-isobutyl-phthalimide, sous forme d'un solide beige (Rf = 0,80, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
Exemple 19 :

N-[(4,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-)-acét]-uanidine
N-[(4,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-)-acét]-uanidine
<Desc/Clms Page number 61>
La N-[(4,7-difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-y l)-acétyl]- guanidine est préparée comme décrit dans l'exemple 1 à partir de 2,68 g de tertbutylate de potassium, de 2,74 g de chlorure de guanidinium et de 1,49 g de (4,7-
difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis il est filtré. Le filtrat est repris avec 80 cm3d'eau et 120 cm3 d'acétate d'éthyle. Après décantation, la phase organique est séparée et la phase aqueuse est extraite 2 fois avec 120 cm3d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés et concentrés à sec sous pression réduite (0,6 kPa) à une température voisine de 40 C. Le résidu d'évaporation est purifié par chromatographie sous pression d'argon (50 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par des mélanges successifs de dichlorométhane/méthanol (95/5 puis 90/10 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 35 C. Le résidu est repris dans de l'éther diéthylique, trituré, filtré puis séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On
obtient ainsi 0,16 g de N-[(4,7-difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l- yl) -acétyl]-guanidine, sous forme d'un solide jaune pâle fondant à 212 C. Spectre de masse : DCI : m/z 325 (M+H)+, m/z 263 (pic de base).
(4,7-Difluoro-2-isobutvl-3-oxo-2,3-dihydro-1H-isoindol-1-)-acétate d'éthyle Le (4,7-difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,6 g d'hydrure de sodium à 60% dans 35 cm3de 1,2-diméthoxyéthane, 3,1 cm3 de triéthylphosphonoacétate et 2,5 g de 4,7-difluoro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (60 kPa), sur une colonne de gel de silice (granulométrie 40-63 m), en éluant par des mélanges successifs de cyclohexane/acétate d'éthyle (80/20 puis 70/30 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 1,49 g (4,7-difluoro-2-isobutyl-3-oxo- 2,3-dihydro-lH-isoindol-l-yl)-acétate d'éthyle impur, sous forme d'une huile jaune
<Desc/Clms Page number 62>
foncé qui est utilisée directement à l'étape suivante. (Rf = 0,27, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (70/30 en volumes)).

4,7-Difluoro-3-hydroxv-2-isobutyl-2,3-dihydro-isoindol- 1 -one La 4,7-difluoro-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one est préparée comme décrit dans l'exemple 1 à partir de 6,41 g de 3,6-difluoro-N-isobutyl-phthalimide dans 70 cm3de méthanol et de 1,44 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 3 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte 45 cm3d'eau distillée.
Le méthanol est ensuite évaporé en partie sous pression réduite (2 kPa) à une température voisine de 30 C, puis on rajoute 80 cm3de dichlorométhane. Après décantation, la phase aqueuse est extraite 2 fois avec 80 cm3de dichlorométhane. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une température voisine de 30 C. Le résidu est séché dans un dessiccateur sous pression réduite (2 kPa) à une température voisine de 20 C. On obtient ainsi 5,68 g de 4,7-difluoro-3-hydroxy-2-isobutyl-2,3dihydro-isoindol-1-one, sous forme d'un solide jaune fondant à 129,5 C. (Rf = 0,48, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
3 , 6-Difluoro-N-i sobutyl-phthalimide Le 3,6-difluoro-N-isobutyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 5,0 g d'anhydride 3,6-difluoro-phthalique, de 2,7 cm3d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 50 cm3de toluène. Le mélange réactionnel est chauffé au reflux pendant 3 heures. Après refroidissement à une température voisine de 20 C, le mélange réactionnel est filtré puis concentré à sec sous pression réduite (2 kPa). Le résidu est repris dans 50 cm3d'une solution aqueuse saturée de bicarbonate de sodium et lavé 3 fois avec 80 cm3de dichlorométhane. Les extraits organiques sont réunis, séchés sur sulfate de magnésium, filtrés puis concentrés à sec sous pression réduite (2 kPa) à une
<Desc/Clms Page number 63>
température voisine de 40 C. On obtient ainsi 6,41 g de 3,6-difluoro-N-isobutylphthalimide, sous forme d'un solide jaune pâle. (Rf = 0,74, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
Exemple 20 :

N-[( 4-Méthyl-2-isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyll-guanidine La N-[(4-méthyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 0,68 g de tert-butylate de potassium, de 0,58 g de chlorure de guanidinium et de 0,36 g de (4-méthyl-2-

isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 20 cm3 d'eau. La phase aqueuse est extraite avec 3 fois 100 cm d'acétate d'éthyle. Les extraits organiques sont réunis puis concentrés à sec sous pression réduite (0,5 kPa) à une température voisine de 40 C. Le résidu est repris dans le dichlorométhane puis reconcentré dans les mêmes conditions. Le résidu est repris dans l'eau, trituré et filtré, puis repris dans le dichlorométhane, trituré et filtré. Le solide est ensuite séché dans un dessiccateur. On obtient ainsi 0,21 g N-[(4-méthyl-2-isobutyl-3-oxo-2,3-dihydro- 1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'une poudre blanche fondant à 270- 272 C (décomposition). R.M.N. 1H (300 MHz, (CD3)2SO d6, 8 en ppm) : 0,76 (d, J = 7 Hz : 3H) ; 0,91 (d, J = 7 Hz : 3H) ; 2,03 (mt : 1H) ;2,42 (dd, J = 15,5 et 6,5 Hz : 1H) ; 2,60 (dd, J = 15,5 et 6,5 Hz : 1H) ; 2,62 (s : 3H) ; 3,00 (dd, J = 14 et 5,5 Hz : 1H) ; 3,56 (dd, J = 14 et 10 Hz : 1H) ; 4,98 (t, J = 6,5 Hz : 1H) ;de 6,.40 à 7,10 (mf étalé : 2H) ; 7,21 (d large, J = 7,5 Hz : 1H) ; 7,36 (d large, J = 7,5 Hz : 1H) ; 7,42 (t, J = 7,5 Hz : 1H) ; de 7,50 à 8,30 (mf étalé : 2H).

N-[( 4-Méthyl-2-isobutyl-3-oxo-2J-dihydro-1 H-isoindol-I-yl)-acétyll-guanidine La N-[(4-méthyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 1 à partir de 0,68 g de tert-butylate de potassium, de 0,58 g de chlorure de guanidinium et de 0,36 g de (4-méthyl-2-
isobutyl-3-oxo-2,3-dihydro-lH-isoindol-1-yl)-acétate d'éthyle. Le milieu réactionnel est agité à une température voisine de 20 C pendant 20 heures, puis on ajoute 20 cm3 d'eau. La phase aqueuse est extraite avec 3 fois 100 cm d'acétate d'éthyle. Les extraits organiques sont réunis puis concentrés à sec sous pression réduite (0,5 kPa) à une température voisine de 40 C. Le résidu est repris dans le dichlorométhane puis reconcentré dans les mêmes conditions. Le résidu est repris dans l'eau, trituré et filtré, puis repris dans le dichlorométhane, trituré et filtré. Le solide est ensuite séché dans un dessiccateur. On obtient ainsi 0,21 g N-[(4-méthyl-2-isobutyl-3-oxo-2,3-dihydro- 1H-isoindol-1-yl)-acétyl]-guanidine, sous forme d'une poudre blanche fondant à 270- 272 C (décomposition). R.M.N. 1H (300 MHz, (CD3)2SO d6, 8 en ppm) : 0,76 (d, J = 7 Hz : 3H) ; 0,91 (d, J = 7 Hz : 3H) ; 2,03 (mt : 1H) ;2,42 (dd, J = 15,5 et 6,5 Hz : 1H) ; 2,60 (dd, J = 15,5 et 6,5 Hz : 1H) ; 2,62 (s : 3H) ; 3,00 (dd, J = 14 et 5,5 Hz : 1H) ; 3,56 (dd, J = 14 et 10 Hz : 1H) ; 4,98 (t, J = 6,5 Hz : 1H) ;de 6,.40 à 7,10 (mf étalé : 2H) ; 7,21 (d large, J = 7,5 Hz : 1H) ; 7,36 (d large, J = 7,5 Hz : 1H) ; 7,42 (t, J = 7,5 Hz : 1H) ; de 7,50 à 8,30 (mf étalé : 2H).
(4-Méthvl-2-isobutvl-3-oxo-2,3-dihvdro-lH-isoindol-l-vl)-acétated'éthyle Le (4-méthyl-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétate d'éthyle est préparé comme décrit dans l'exemple 2 à partir de 0,23 g d'hydrure de sodium à 60% dans 15 cm3 de 1,2-diméthoxyéthane, 1,1 cm3de triéthylphosphonoacétate et 0,63 g
<Desc/Clms Page number 64>
de 7-méthyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one. Le produit brut est purifié par chromatographie sous pression d'argon (50 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (70/30 en volumes). Les fractions contenant le produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C.

On obtient ainsi 0,36 g de (4-méthyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)acétate d'éthyle, sous forme d'une huile incolore.
On obtient ainsi 0,36 g de (4-méthyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)acétate d'éthyle, sous forme d'une huile incolore.
4-Méthyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-1-one et 7-méthyl-3-hydroxy-2- isobutvl-2,3-dihvdro-isoindol-l-one La 4-méthyl-3-hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one et la 7-méthyl-3- hydroxy-2-isobutyl-2,3-dihydro-isoindol-l-one sont préparées comme décrit dans l'exemple 1 à partir de 5,4 g de N-isobutyl-3-méthyl-phthalimide dans 20 cm3de méthanol et de 1,4 g de borhydrure de potassium. Le mélange réactionnel est agité à une température voisine de 20 C pendant 65 heures, puis il est refroidi à une température voisine de 0 C et on ajoute au goutte à goutte 10 cm3 d'eau distillée. Le mélange est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C et le résidu est repris successivement dans l'éther diéthylique puis dans le dichlorométhane. Le précipité obtenu est filtré, puis le filtrat est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est purifié par chromatographie sous pression d'argon (50 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (70/30 en volumes). Les fractions contenant chaque produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Les fractions contenant un mélange des deux produits attendus sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repurifié par chromatographie sous pression d'argon (50 kPa), sur une colonne de gel de silice (granulométrie 15-40 m), en éluant par un mélange de cyclohexane/acétate d'éthyle (80/20 en volumes). Les fractions contenant chaque produit attendu sont réunies et concentrées à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 0,74 g de 7-méthyl-3-hydroxy-2-
<Desc/Clms Page number 65>
isobutyl-2,3-dihydro-isoindol-l-one sous forme d'une poudre blanche (Rf = 0,54, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)) et 0,89 g de 4-méthyl-3-hydroxy-2-isobutyl-2,3dihydro-isoindol-1-one, sous forme d'un solide cotoneux blanc (Rf = 0,40, chromatographie sur couche mince de gel de silice, éluant : cyclohexane/acétate d'éthyle (50/50 en volumes)).
N-Isobutyl-3 -méthyl-phthalimide Le N-isobutyl-3-méthyl-phthalimide est préparé comme décrit dans l'exemple 2 à partir de 5,0 g d'anhydride 3-méthyl-phthalique, de 3,0 cm3d'isobutylamine et d'une quantité catalytique d'acide para-toluènesulfonique dans 50 cm3de toluène. Le mélange réactionnel est chauffé à une température voisine de 140 C pendant 2,5 heures, puis agité à une température voisine de 20 C pendant 16 heures. Le mélange réactionnel est concentré à sec sous pression réduite (2 kPa) à une température voisine de 40 C. Le résidu est repris dans du dichlorométhane et lavé 2 fois avec une solution aqueuse saturée de bicarbonate de sodium. La phase organique est décantée, séchée sur sulfate de magnésium, filtrée puis concentrée à sec sous pression réduite (2 kPa) à une température voisine de 40 C. On obtient ainsi 6,0 g de N-isobutyl-3méthyl-phthalimide, sous forme d'une huile couleur caramel, Rf = 0,76 ( chromatographie sur couche mince de gel de silice, éluant : dichlorométhane).
Exemple 21:

N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine La N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine est préparée comme décrit dans l'exemple 9. On obtient ainsi une poudre blanche amorphe, Rf = 0,10 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 315.
N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine La N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine est préparée comme décrit dans l'exemple 9. On obtient ainsi une poudre blanche amorphe, Rf = 0,10 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 315.
Exemple 22:
<Desc/Clms Page number 66>
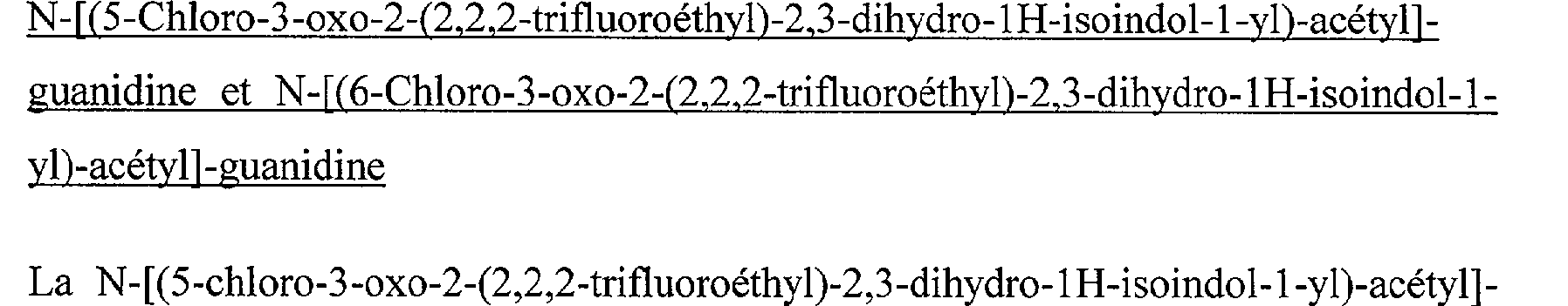
N- ff 5-Chloro-3 -oxo-2-(2,2.2-trifluoroéthyl)-23 -dihvdro- 1 H-isoindol- 1 -vD-acétyl] guanidine et N-r(6-Chloro-3-oxo-2-(2.2.2-trifluoroéthvl)-2,3-dihydro-lH-isoindol-lyl)-acétyll-guanidine La N-[(5-chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]- guanidine et la N-[(6-chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-lH-isoindol- 1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 9. On obtient ainsi une poudre blanche amorphe, Rf = 0,20 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 349.
Exemple 23:

N-[(3-Oxo-2- 2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-1H-isoindol-1-)- acétyl] -guanidine La N-[(3-oxo-2-(2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-lH-isoindol-l- yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 9. On obtient ainsi une poudre blanche amorphe, Rf = 0,23 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 3 84.
N-[(3-Oxo-2- 2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-1H-isoindol-1-)- acétyl] -guanidine La N-[(3-oxo-2-(2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-lH-isoindol-l- yl)-acétyl]-guanidine est préparée comme décrit dans l'exemple 9. On obtient ainsi une poudre blanche amorphe, Rf = 0,23 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 3 84.
Exemple 24 :

N-[(5-Chloro-2-cprop lyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyllguanidine et N-F(6-Chloro-2-çyclopropylméthyl-3-oxo-2,3-dihydro-IH-isoindol-1vl)-acétyl1-guanidine La N-[(5-chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1 H-isoindol-l-yl)-acétyl]- guanidine et la N-[(6-chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l- yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 8, Rf = 0,09 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 321.
N-[(5-Chloro-2-cprop lyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyllguanidine et N-F(6-Chloro-2-çyclopropylméthyl-3-oxo-2,3-dihydro-IH-isoindol-1vl)-acétyl1-guanidine La N-[(5-chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1 H-isoindol-l-yl)-acétyl]- guanidine et la N-[(6-chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-lH-isoindol-l- yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 8, Rf = 0,09 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 321.
<Desc/Clms Page number 67>
Exemple 25:

N-[(2-Cloprop lyl-3-oxo-5-trifluorométhyl-2,3-dihydro-1H-isoindol-1-yl)acétyll-guanidine et N-[(2-Cvclopropylméthyl-3-oxo-6-trifluorométhyl-2.3-dihydro- 1 H-isoindol- 1 -yl)-acétyl"|-guanidine La N-[(2-cyclopropylméthyl-3-oxo-5-trifluorométhyl-2,3-dihydro-l H-isoindol-1-yl)- acétyl]-guanidine et la N-[(2-cyclopropylméthyl-3-oxo-6-trifluorométhyl-2,3dihydro-1H-isoindol-1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 8, Rf = 0,11 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 355.
N-[(2-Cloprop lyl-3-oxo-5-trifluorométhyl-2,3-dihydro-1H-isoindol-1-yl)acétyll-guanidine et N-[(2-Cvclopropylméthyl-3-oxo-6-trifluorométhyl-2.3-dihydro- 1 H-isoindol- 1 -yl)-acétyl"|-guanidine La N-[(2-cyclopropylméthyl-3-oxo-5-trifluorométhyl-2,3-dihydro-l H-isoindol-1-yl)- acétyl]-guanidine et la N-[(2-cyclopropylméthyl-3-oxo-6-trifluorométhyl-2,3dihydro-1H-isoindol-1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 8, Rf = 0,11 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 355.
Exemple 26:

N-[(5-Chloro-3-oxo-2-(3,3,3-trifluoropr)-2,3-dihydro-1H-isoindol-1-yl -acétyllguanidine et N-f(6-Chloro-3-oxo-2-(3,3,3-trifluoroprop)-2,3-dihydro-1H-isoindol- 1 -vl)-acétyl]-guanidine La N-[(5-chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine et la N-[(6-chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-lH- isoindol-1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 1 à partir de 4-chloro-N-(3,3,3trifluoropropyl) -phthalimide, Rf = 0,12 (chromatographie sur couche mince de gel de silice, éluant : d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 363.
N-[(5-Chloro-3-oxo-2-(3,3,3-trifluoropr)-2,3-dihydro-1H-isoindol-1-yl -acétyllguanidine et N-f(6-Chloro-3-oxo-2-(3,3,3-trifluoroprop)-2,3-dihydro-1H-isoindol- 1 -vl)-acétyl]-guanidine La N-[(5-chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine et la N-[(6-chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-lH- isoindol-1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 1 à partir de 4-chloro-N-(3,3,3trifluoropropyl) -phthalimide, Rf = 0,12 (chromatographie sur couche mince de gel de silice, éluant : d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+ : m/z 363.
4-Chloro-N-(3,3,3-trifluoropropyl)-phthalimide Un mélange de 5,0 g d'acide 4-chloro-N-(3,3,3-trifluoropropyl)-phthalamique et de 30 mg d'acide para-toluènesulfonique dans 100 cm3de toluène est chauffé à reflux sous agitation pendant 7 heures. Le mélange réactionnel est ensuite concentré à sec sous pression réduite. On obtient ainsi 4,2 g de 5-chloro-N-(3,3,3-trifluoropropyl)-
<Desc/Clms Page number 68>
phthalimide, Rf = 0,5 (chromatographie sur couche mince de gel de silice, éluant : diisopropyl éther). Spectre de masse : m/z 278.

Acide 4-chloro-N-(3.3,3 -trifmoropropyD-phthalamique A un mélange de 7,5 g de 4-chloro-phthalimide et de 4,6 g de carbonate de potassium dans 40 cm3de diméthylformamide à une température voisine de 110 C et sous agitation, sont ajoutés au goutte à goutte 3,6 g de 1,1,1-trifluoro-3-iodo-propane.
Acide 4-chloro-N-(3.3,3 -trifmoropropyD-phthalamique A un mélange de 7,5 g de 4-chloro-phthalimide et de 4,6 g de carbonate de potassium dans 40 cm3de diméthylformamide à une température voisine de 110 C et sous agitation, sont ajoutés au goutte à goutte 3,6 g de 1,1,1-trifluoro-3-iodo-propane.
Après 11 heures d'agitation à une température voisine de 120 C, le mélange réactionnel est refroidi à une température voisine de 20 C puis versé dans 200 cm3 d'eau. Le mélange est acidifié avec une solution aqueuse diluée d'acide chlorhydrique jusqu'à un pH d'environ 3, puis il est extrait trois fois avec 100 cm3d'acétate d'éthyle.
La phase organique est séchée sur sulfate de magnésium puis concentrée à sec. On obtient ainsi 5,0 g d'acide 4-chloro-N-(3,3,3-trifluoropropyl)-phthalamique, Rf = 0,12 (chromatographie sur couche mince de gel de silice, éluant : d'éthyle).
4-Chloro-phthalimide Une solution de 10,0 g d'anhydride 4-chloro-phthalique dans 24,6 g de formamide est chauffée à une température voisine de 120 C sous agitation pendant 3 heures, puis elle est refroidie à une température voisine de 20 C et versée dans 100 cm3d'eau.
Après 30 minutes d'agitation, le mélange est filtré puis le précipité est séché sous vide à une température voisine de 60 C. On obtient ainsi 10,4 g de 4-chlorophthalimide sous forme d'un solide fondant à 171 C. Rf = 0,07 (chromatographie sur couche mince de gel de silice, éluant : dichlorométhane).
Exemple 27:

N-[3-Oxo-5-trifluorométhy3,3,3-trifluoroprop)-2,3-dihydro-1 H-isoindol-1-yl)acétyl] -guanidine et N-[(3-Oxo-6-trifluorométhvl-2-(3,3,3-trifluoropropyl dihydro-1 H-isoindol-1 -yl)-acétyl]-guanidine La N-[(3-oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1H-isoindol-1- yl)-acétyl]-guanidine et la N-[(3-oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-
N-[3-Oxo-5-trifluorométhy3,3,3-trifluoroprop)-2,3-dihydro-1 H-isoindol-1-yl)acétyl] -guanidine et N-[(3-Oxo-6-trifluorométhvl-2-(3,3,3-trifluoropropyl dihydro-1 H-isoindol-1 -yl)-acétyl]-guanidine La N-[(3-oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1H-isoindol-1- yl)-acétyl]-guanidine et la N-[(3-oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-
<Desc/Clms Page number 69>
dihydro-1H-isoindol-1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 1 à partir de 4-trifluorométhyl- N-(3,3,3-trifluoropropyl)-phthalimide, Rf = 0,12 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse : ES+ : m/z 397.
Le 4-trifluorométhyl-N- (3,3,3-trifluoropropyl)-phthalimide préparé de façon analogue au 4-chloro-N- (3,3,3-trifluoropropyl)-phthalimide dans l'exemple 26, à partir d'anhydride 4-trifluoroméhtyl-phthalique, Rf = 0,12, (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle).
L' anhydride 4-trifluorométhyl-phthalique peut être préparé par adaptation ou application de la méthode décrite par Cavalleri et coll., J. Med. Chem., 13(1), 148- 149,(1970).
Exemple 28:

N-[(5-Chloro-3-oxo-2-(4,4,4-trifluorobutyl -2,) 3idihydro-1H-isoindol-1-)-acétyl]guanidine et N-[(6-Chloro-3-oxo-2-(4,4,4-trifluorobutvn-2,3-dihydro-lH-isoindol-lvl)-acétvll-guanidine La N-[(5-chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-lH-isoindol-l-yl)-acétyl]guanidine et la N - [( 6-chloro- 3 -oxo- 2-( 4, 4,4- trifluoro butyl)- 2,3 -dihydro-l H - isoindol- 1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 8, Rf = 0,11(chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+: m/z 377.
N-[(5-Chloro-3-oxo-2-(4,4,4-trifluorobutyl -2,) 3idihydro-1H-isoindol-1-)-acétyl]guanidine et N-[(6-Chloro-3-oxo-2-(4,4,4-trifluorobutvn-2,3-dihydro-lH-isoindol-lvl)-acétvll-guanidine La N-[(5-chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-lH-isoindol-l-yl)-acétyl]guanidine et la N - [( 6-chloro- 3 -oxo- 2-( 4, 4,4- trifluoro butyl)- 2,3 -dihydro-l H - isoindol- 1-yl)-acétyl]-guanidine sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 8, Rf = 0,11(chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse: ES+: m/z 377.
Exemple 29 :

N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhl-2,3-dihydro-1 H-isoindol-1-yl acétyll-guanidine et N-F(3-Oxo-2-(4,4,4-trifluorobMtyl)-6-trifluoroméLhyl-2,3dihydro- 1 H-isoindol- 1 -vD-acétvl"|-guanidine
N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhl-2,3-dihydro-1 H-isoindol-1-yl acétyll-guanidine et N-F(3-Oxo-2-(4,4,4-trifluorobMtyl)-6-trifluoroméLhyl-2,3dihydro- 1 H-isoindol- 1 -vD-acétvl"|-guanidine
<Desc/Clms Page number 70>
La N-[(3-oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhyl-2,3-dihydro-1H-isoindol-1- yl)-acétyl]-guanidine et la N-[(3-oxo-2-(4,4,4-trifluorobutyl)-6-trifluorométhyl-2,3- dihydro-lH-isoindol-l-yl)-acétyl]-guanidine~sont préparées sous forme de mélange de deux régioisomères comme décrit dans l'exemple 27 à partir de 4-trifluorométhylN- (4,4,4-trifluorobutyl)-phthalimide, Rf = 0,12 (chromatographie sur couche mince de gel de silice, éluant : acétate d'éthyle/ méthanol (5: 1 en volumes)). Spectre de masse : ES+ : m/z411.
Exemple 30:

N-[2-(3-oxo-2-propyl-2J-dihydro-1 H -isoindoi-I-yl)-propionyll-guanidine On dissout 3,75 g (39,2 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et on ajoute 3,96 g (35,3 mmol) de tert-butylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,08 g (3,92 mmol) de 2-méthyl-3-(2-propylcarbamoylphényl) -acrylate d'éthyle dans 15 cm3de diméthylformamide. Après agitation à une température voisine de 20 C pendant environ 16 heures, le solvant est évaporé et le résidu est dissout dans une solution aqueuse d'acide chlorhydrique 2 N. Après extraction une seule fois avec du dichlorométhane, la phase aqueuse est basifiée avec de la potasse jusqu'à une valeur de pH égale à 12 et le précipité ainsi obtenu est filtré*. Ce dernier est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré, puis séché à froid ; obtient ainsi environ 400 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport d'environ 6:1 ( MS(ESI): M+H : 289,1/ Rt: 3,555 min).
N-[2-(3-oxo-2-propyl-2J-dihydro-1 H -isoindoi-I-yl)-propionyll-guanidine On dissout 3,75 g (39,2 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et on ajoute 3,96 g (35,3 mmol) de tert-butylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,08 g (3,92 mmol) de 2-méthyl-3-(2-propylcarbamoylphényl) -acrylate d'éthyle dans 15 cm3de diméthylformamide. Après agitation à une température voisine de 20 C pendant environ 16 heures, le solvant est évaporé et le résidu est dissout dans une solution aqueuse d'acide chlorhydrique 2 N. Après extraction une seule fois avec du dichlorométhane, la phase aqueuse est basifiée avec de la potasse jusqu'à une valeur de pH égale à 12 et le précipité ainsi obtenu est filtré*. Ce dernier est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré, puis séché à froid ; obtient ainsi environ 400 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport d'environ 6:1 ( MS(ESI): M+H : 289,1/ Rt: 3,555 min).
*Le premier filtrat obtenu est soumis à une double extraction avec du dichlorométhane. Les extraits organiques réunis sont séchés sur sulfate de sodium puis concentrés. Le résidu est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré, puis séché à froid. On isole ainsi 77 mg du deuxième diastéréoisomère (diastéréoisomère B) sous forme de chlorhydrate enrichi dans un rapport de 10:1 (MS(ESI): M+H : 289,1/ Rt: 3,544 min).
<Desc/Clms Page number 71>
Le diastéréoisomère A est dédoublé en deux énantiomères par chromatographie CLHP sur une colonne chirale (Chiralpak AD 250 x 4,6) en éluant par un mélange acétonitrile : iso-propanol : n-heptane (50: 3:4 en volumes); débit d'écoulement : ml/min. On obtient ainsi l'énantiomère Al: 3,106 min, sous forme de trifluoracétate ([alpha]D20= -0,073 dans le méthanol à 0,1%) et l'énantiomère A2: 3,522 min, sous forme de trifluoracétate , ([alpha]D20= +0,056 dans le méthanol à 0,1 %).
2-Méthvl-3 -(2-propylcarbamoyl-phénvl)-acrylate d'éthyle A une solution de 1,17 g (5,0 mmol) d'acide 2-(2-éthoxycarbonyl-propén-1-yl)- benzoïque dans 10 cm3de diméthylformamide, on ajoute à 0 C une solution de 0,69 cm3 (5,0 mmol) de triéthylamine et de 1,64 g (5,0 mmol) de 0- [(cyano(éthoxycarbonyl)méthylèn)amino]-1,1,3,3-tétraméthyluronium- tétrafluoroborate ("TOTU") dans 6 cm3de diméthylformamide. Après agitation pendant 30 min à 0 C, puis 30 min à une température voisine de 20 C, on introduit au goutte à goutte cette solution dans une deuxième solution constituée de 296 mg (5,0 mmol) de n-propylamine et de 0,69 cm3(5,0 mmol) de triéthylamine dans 10 cm3 de diméthylformamide, et on agite encore pendant 6 heures à une température voisine de 20 C. Après avoir laissé la solution reposer une nuit, on élimine le solvant sous vide et on dissout le résidu dans de l'acétate d'éthyle. Après lavage deux fois avec une solution de bicarbonate de sodium puis une fois avec une solution de chlorure de sodium, la phase organique est sèchée sur sulfate de sodium puis concentrée. On obtient ainsi 1,10 g de 2-méthyl-3-(2-propylcarbamoyl-phényl)-acrylate d'éthyle sous forme d'une huile jaunâtre, que l'on peut faire réagir dans l'étape suivante sans autre purification.
*Acide 2-(2-éthoxycarbonyl-propén-1-yl)-benzoïque Un mélange de 5,0 g (33,3 mmol) d'acide 2-formyl-benzoïque et 14,5 g (40,0 mmol) de 2- (triphénylphosphanylidène)-propionate dans 100 cm3 de diméthylformamide est agité à une température voisine de 20 C pendant 1 heure.
Après élimination du solvant, le résidu est dissout dans du dichlorométhane puis
<Desc/Clms Page number 72>
extrait deux fois avec une solution de bicarbonate de sodium. Les phases aqueuses sont réunies, lavées avec du dichlorométhane puis acidifiées avec une solution aqueuse d'acide chlorhydrique 6 N jusqu'à une valeur de pH comprise entre 1 et 2.
Après extraction deux fois avec du dichlorométhane, les phases organiques sont réunies, séchées sur sulfate de magnésium puis concentrées. On obtient ainsi l'acide 2-(2-éthoxycarbonyl-propén-l-yl)-benzoïque, sous forme d'une huile jaune, qui est utilisé directement dans l'étape suivante sans autre purification.
Exemple 31 :
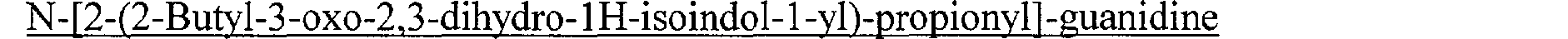
N-[2-(2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-)-pro ionyl]-uanidine La N-[2-(2-butyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine est préparée comme décrit dans l'exemple 30 à partir de 3,63 g (38,0 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et 3,84 g (34,2 mmol) de tertbutylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,10 g (3,80 mmol) de 3-(2butylcarbamoyl-phényl) -2-méthyl-acrylate d'éthyle dans 15 cm3 de diméthylformamide et on agite pendant 3 heures à une température voisine de 20 C.
N-[2-(2-Butyl-3-oxo-2,3-dihydro-1H-isoindol-1-)-pro ionyl]-uanidine La N-[2-(2-butyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine est préparée comme décrit dans l'exemple 30 à partir de 3,63 g (38,0 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et 3,84 g (34,2 mmol) de tertbutylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,10 g (3,80 mmol) de 3-(2butylcarbamoyl-phényl) -2-méthyl-acrylate d'éthyle dans 15 cm3 de diméthylformamide et on agite pendant 3 heures à une température voisine de 20 C.
Après avoir laissé la solution reposer une nuit, on élimine le solvant et on dissout le résidu dans une solution aqueuse d'acide chlorhydrique 2 N. Après extraction une seule fois avec du dichlorométhane, la phase organique est concentrée puis le résidu est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré puis séché à

froid. On obtient ainsi 1,07 g de N-[2-(2-butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)- propionyl]-guanidine sous forme d'un mélange des diastéréoisomères. Le pH de la phase aqueuse est ajusté à une valeur de 12 avec de la potasse et le précipité ainsi obtenu est filtré*. On reprend le solide ainsi obtenu dans une solution aqueuse d'acide chlorhydrique 2 N et après filtration et séchage à froid on obtient environ 106 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport de 5 :1 M+H : / Rt: 3,793 min).
froid. On obtient ainsi 1,07 g de N-[2-(2-butyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)- propionyl]-guanidine sous forme d'un mélange des diastéréoisomères. Le pH de la phase aqueuse est ajusté à une valeur de 12 avec de la potasse et le précipité ainsi obtenu est filtré*. On reprend le solide ainsi obtenu dans une solution aqueuse d'acide chlorhydrique 2 N et après filtration et séchage à froid on obtient environ 106 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport de 5 :1 M+H : / Rt: 3,793 min).
* Le premier filtrat obtenu est soumis à une double extraction avec du dichlorométhane. Les extraits organiques réunis sont séchés sur sulfate de sodium
<Desc/Clms Page number 73>
puis concentrés. Le résidu est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré puis séché à froid; on isole ainsi 38 mg du deuxième diastéréoisomère (diastéréoisomère B) sous forme de chlorhydrate enrichi dans un rapport supérieur à 10:1(MS(ESI): M+H : 303,1 / Rt: 3,754 min).
Le diastéréoisomère A est dédoublé en deux énantiomères par chromatographie CLHP sur une colonne chirale (Chiralpak AD 250 x 4,6) en éluant par un mélange acétonitrile : iso-propanol : n-heptane (50: 3:4 en volumes) contenant 0,3% de diéthylamine; débit d'écoulement : ml/min. On obtient ainsi l'énantiomère Al: 3,195 min, sous forme de trifluoracétate (aD20= -0,064 dans le méthanol à 0,2%) et l'énantiomère A2 : min, sous forme de trifluoracétate (aD20= +0,084 dans le méthanol à 0,2%).

3 -(2-Butylcarbamoyl-phényl)-2-méthyl-acrylate d'éthyle Le 3- (2-butylcarbamoyl-phényl)-2-méthyl-acrylate est préparé comme décrit dans l'exemple 30 à partir de 1,17 g (5,0 mmol) d'acide 2-(2-éthoxycarbonyl-propén- l-yl)-benzoïque dans 10 cm3de diméthylformamide, une solution de 0,69 cm3 (5,0 mmol) de triéthylamine, 1,64 g (5,0 mmol) de 0-

[(cyano(éthoxycarbonyl)méthylèn)amino]-1,1,3,3-tétraméthyluronium- tétrafluoroborate ("TOTU") dans 6 cm3de diméthylformamide et une deuxième solution constituée de 366 mg (5,0 mmol) de n-butylamine, 0,69 cm3(5,0 mmol) de triéthylamine dans 10 cm3de diméthylformamide. On obtient ainsi 1,13 g de 3-(2butylcarbamoyl-phényl) -2-méthyl-acrylate d'éthyle sous forme d'une huile jaunâtre, que l'on peut faire réagir dans l'étape suivante sans autre purification.
[(cyano(éthoxycarbonyl)méthylèn)amino]-1,1,3,3-tétraméthyluronium- tétrafluoroborate ("TOTU") dans 6 cm3de diméthylformamide et une deuxième solution constituée de 366 mg (5,0 mmol) de n-butylamine, 0,69 cm3(5,0 mmol) de triéthylamine dans 10 cm3de diméthylformamide. On obtient ainsi 1,13 g de 3-(2butylcarbamoyl-phényl) -2-méthyl-acrylate d'éthyle sous forme d'une huile jaunâtre, que l'on peut faire réagir dans l'étape suivante sans autre purification.
Exemple 32:
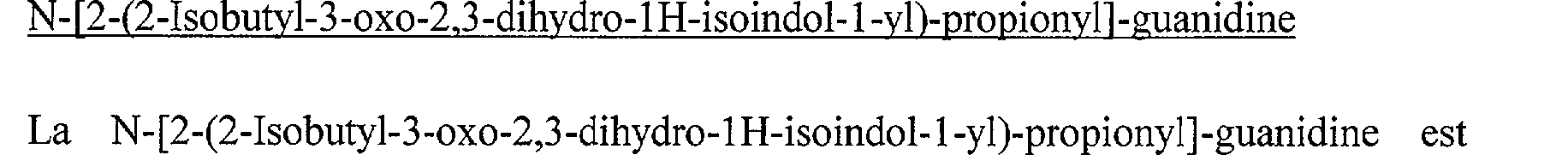
N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -vD-propionyl] -guanidine La N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-propionyl]-guanidine est préparée comme décrit dans l'exemple 30 à partir de 3,73 g (39,0 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et 3,94 g (35,1 mmol) de tert-
N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -vD-propionyl] -guanidine La N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-propionyl]-guanidine est préparée comme décrit dans l'exemple 30 à partir de 3,73 g (39,0 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et 3,94 g (35,1 mmol) de tert-
<Desc/Clms Page number 74>
butylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,13 g (3,90 mmol) de 3-(2isobutylcarbamoyl-phényl) -2-méthyl-acrylate d'éthyle dans 15 cm3 de diméthylformamide et on agite pendant 3 heures à une température voisine de 20 C.
Après avoir laissé la solution reposer une nuit, on élimine le solvant et on dissout le résidu dans une solution aqueuse d'acide chlorhydrique 2 N. Après extraction une seule fois avec du dichlorométhane, la phase organique est concentrée puis le résidu est repris dans une solution d'acide chlorhydrique 2 N, filtré puis séché à froid. On obtient ainsi 862 mg de N-[2-(2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)- propionyl]-guanidine sous forme d'un mélange des diastéréoisomères. Le pH de la phase aqueuse est ajusté à une valeur de 12 avec de la potasse et le précipité ainsi obtenu est filtré*. On reprend le solide ainsi obtenu dans une solution aqueuse d'acide chlorhydrique 2 N et après filtration et séchage à froid on obtient environ 255 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport de 9 :1 M+H : / Rt: 3,744 min).
* Le premier filtrat obtenu est soumis à une double extraction avec du dichlorométhane. Les extraits organiques réunis sont séchés sur sulfate de sodium puis concentrés. Le résidu est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré puis séché à froid ; isole ainsi 78 mg du deuxième diastéréoisomère (diastéréoisomère B) sous forme de chlorhydrate enrichi dans un rapport supérieur à 10:1(MS(ESI): M+H : 303,1 / Rt: 3,710 min).
Le diastéréoisomère A est dédoublé en deux énantiomères par chromatographie CLHP sur une colonne chirale (Chiralpak AD 250 x 4,6) en éluant par un mélange acétonitrile : iso-propanol : n-heptane (50: 3:4 en volumes) contenant 0,3% de diéthylamine; débit d'écoulement : ml/min. On obtient ainsi l'énantiomère Al: 3,895 min, sous forme de trifluoracétate ([alpha]D20= -0,086 dans le méthanol à 0,2%) et l'énantiomère A2 : min, sous forme de trifluoracétate (aD20= +0,114 dans le méthanol à 0,2%).

3 -(2- Isobutylcarbamoyl-phényl)- 2-méthyl-acrylate d'éthyle
<Desc/Clms Page number 75>
Le 3- (2-isobutylcarbamoyl-phényl)-2-méthyl-acrylate est préparé comme décrit dans l'exemple 30 à partir de 1,17 g (5,0 mmol) d'acide 2-(2-éthoxycarbonylpropén-1-yl)-benzoïque dans 10 cm3de diméthylformamide, une solution de 0,69 cm3 (5,0 mmol) de triéthylamine, 1,64 g (5,0 mmol) de 0- [(cyano(éthoxycarbonyl)méthylèn)amino] -1,1,3,3 -tétraméthyluronium- tétrafluoroborate ("TOTU") dans 6 cm3de diméthylformamide et une deuxième solution constituée de 366 mg (5,0 mmol) d' isobutylamine, 0,69 cm3 (5,0 mmol) de triéthylamine dans 10 cm3de diméthylformamide. On obtient ainsi 1,15 g de 3-(2isobutylcarbamoyl-phényl) -2-méthyl-acrylate d'éthyle sous forme d'une huile jaunâtre, que l'on peut faire réagir dans l'étape suivante sans autre purification.
Exemple 33:

N-[2-(2-Hvdroxv-éthvl)-3-oxo-2,3-dihydro-lH-isoindol-l-vl)-propionyl]-guanidine La N-[2-(2-hydroxy-éthyl)-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]- guanidine est préparée comme décrit dans l'exemple 30 à partir de 3,75 g (39,2 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et 3,96 g (35,3 mmol) de tert-butylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,09 g (3,92 mmol) de 3- [2-(2-hydroxy-éthylcarbamoyl)-phényl]-2-méthyl-acrylate dans 15 cm3 de diméthylformamide et on agite pendant 3 heures à une température voisine de 20 C. Après avoir laissé la solution reposer une nuit, on élimine le solvant et on dissout le résidu dans une solution aqueuse d'acide chlorhydrique 2 N. Après extraction une seule fois avec du dichlorométhane, le pH de la phase aqueuse est ajusté à une valeur de 12 avec de la potasse et le précipité ainsi obtenu est filtré*. On reprend le solide ainsi obtenu dans une solution aqueuse d'acide chlorhydrique 2 N et après filtration et séchage à froid on obtient environ 207 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport de 10:1(MS(ESI): M+H : 291,1 / Rt: 3,186 min).
N-[2-(2-Hvdroxv-éthvl)-3-oxo-2,3-dihydro-lH-isoindol-l-vl)-propionyl]-guanidine La N-[2-(2-hydroxy-éthyl)-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]- guanidine est préparée comme décrit dans l'exemple 30 à partir de 3,75 g (39,2 mmol) de chlorure de guanidinium dans 20 cm3de diméthylformamide et 3,96 g (35,3 mmol) de tert-butylate de potassium. On agite pendant 45 minutes à une température voisine de 20 C, puis on ajoute au goutte à goutte une solution de 1,09 g (3,92 mmol) de 3- [2-(2-hydroxy-éthylcarbamoyl)-phényl]-2-méthyl-acrylate dans 15 cm3 de diméthylformamide et on agite pendant 3 heures à une température voisine de 20 C. Après avoir laissé la solution reposer une nuit, on élimine le solvant et on dissout le résidu dans une solution aqueuse d'acide chlorhydrique 2 N. Après extraction une seule fois avec du dichlorométhane, le pH de la phase aqueuse est ajusté à une valeur de 12 avec de la potasse et le précipité ainsi obtenu est filtré*. On reprend le solide ainsi obtenu dans une solution aqueuse d'acide chlorhydrique 2 N et après filtration et séchage à froid on obtient environ 207 mg d'un des deux produits diastéréoisomères (diastéréoisomère A) sous forme de chlorhydrate enrichi dans un rapport de 10:1(MS(ESI): M+H : 291,1 / Rt: 3,186 min).
* Le premier filtrat obtenu est soumis à une double extraction avec du dichlorométhane. Les extraits organiques réunis sont séchés sur sulfate de sodium
<Desc/Clms Page number 76>
puis concentrés. Le résidu est repris dans une solution aqueuse d'acide chlorhydrique 2 N, filtré puis séché à froid ; isole ainsi 50 mg d'un mélange des deux diastéréoisomères (diastéréoisomères A et B) sous forme de chlorhydrate, dans la proportion 1:1(MS(ESI): M+H : 291,1 / Rt: 3,710 min).

3-[2-(2-Hvdroxy-éthvlcarbamoyl)-phényl]-2-méthyl-acrylate d'éthyle Le 3- [2-(2-hydroxy-éthylcarbamoyl)-phényl]-2-méthyl-acrylate est préparé comme décrit dans l'exemple 30 à partir de 1,17 g (5,0 mmol) d'acide 2-(2- éthoxycarbonyl-propén-l-yl)-benzoïque dans 10 cm3de diméthylformamide, une solution de 0,69 cm3(5,0 mmol) de triéthylamine, 1,64 g (5,0 mmol) de 0- [(cyano(éthoxycarbonyl)méthylèn)amino]-1,1,3,3-tétraméthyluronium- tétrafluoroborate ("TOTU") dans 6 cm3de diméthylformamide et une deuxième solution constituée de 306 mg (5,0 mmol) d' éthanolamine, 0,69 cm3(5,0 mmol) de triéthylamine dans 10 cm3de diméthylformamide. On obtient ainsi 1,14 g de 3-[2-(2hydroxy-éthylcarbamoyl) -phényl]-2-méthyl-acrylate d'éthyle sous forme d'une huile jaunâtre, que l'on peut faire réagir dans l'étape suivante sans autre purification.
Les compositions pharmaceutiques selon l'invention sont constitués par un composé de formule (I) ou un sel d'un tel composé, à l'état pur ou sous forme d'une composition dans laquelle il est associé à tout autre produit pharmaceutiquement compatible, pouvant être inerte ou physiologiquement actif. Les médicaments selon l'invention peuvent être employés par voie orale, parentérale, rectale ou topique.
Comme compositions solides pour administration orale, peuvent être utilisés des comprimés, des pilules, des poudres (capsules de gélatine, cachets) ou des granulés.
Dans ces compositions, le principe actif selon l'invention est mélangé à un ou plusieurs diluants inertes, tels que amidon, cellulose, saccharose, lactose ou silice, sous courant d'argon. Ces compositions peuvent également comprendre des substances autres que les diluants, par exemple un ou plusieurs lubrifiants tels que le stéarate de magnésium ou le talc, un colorant, un enrobage (dragées) ou un vernis.
<Desc/Clms Page number 77>
Comme compositions liquides pour administration orale, on peut utiliser des solutions, des suspensions, des émulsions, des sirops et élixirs pharmaceutiquement acceptables contenant des diluants inertes tels que l'eau, l'éthanol, le glycérol, les huiles végétales ou l'huile de paraffine. Ces compositions peuvent comprendre des substances autres que les diluants, par exemple des produits mouillants, édulcorants, épaississants, aromatisants ou stabilisants.
Les compositions stériles pour administration parentérale, peuvent être de préférence des solutions aqueuses ou non aqueuses, des suspensions ou des émulsions. Comme solvant ou véhicule, on peut employer l'eau, le propylèneglycol, un polyéthylèneglycol, des huiles végétales, en particulier l'huile d'olive, des esters organiques injectables, par exemple l'oléate d'éthyle ou d'autres solvants organiques convenables. Ces compositions peuvent également contenir des adjuvants, en particulier des agents mouillants, isotonisants, émulsifiants, dispersants et stabilisants. La stérilisation peut se faire de plusieurs façons, par exemple par filtration aseptisante, en incorporant à la composition des agents stérilisants, par irradiation ou par chauffage. Elles peuvent également être préparées sous forme de compositions solides stériles qui peuvent être dissoutes au moment de l'emploi dans de l'eau stérile ou tout autre milieu stérile injectable.
Les compositions pour administration rectale sont les suppositoires ou les capsules rectales qui contiennent, outre le produit actif, des excipients tels que le beurre de cacao, des glycérides semi-synthétiques ou des polyéthylèneglycols.
Les compositions pour administration topique peuvent être par exemple des crèmes, lotions, collyres, collutoires, gouttes nasales ou aérosols.
L'invention a pour objet les composés d'isoindolones de formule (I) et leurs sels pharmaceutiquement acceptables et leur utilisation pour la préparation de compositions pharmaceutiques destinées à prévenir et traiter les maladies tel que les infarctus, comme composés antiarythmiques à composantes cardioprotectrice, les angines de poitrine, les lésions induites par une ischémie, le traitement des ischémies du système nerveux, en particulier du système nerveux central, le traitement de
<Desc/Clms Page number 78>
l'attaque ou de l'oedème cérébral, les traitements de formes du choc comme le choc allergique, le choc cardiogène, le choc hypovolémique et le choc bactérien, l'hypertension sanguine, l'hypertonie essentielle, le traitement de la prolifération cellulaire des fibroblastes, les complications diabétiques tardives, les maladies cancéreuses, les maladies fibreuses comme la fibrose pulmonaire, la fibrose hépatique ou la fibrose rénale, les hypertrophies et hyperplasies d'organes, en particulier l'hyperplasie de la prostate ou l'hypertrophie de la prostate ; comme agents de diagnostic pour la détermination et la distinction de formes déterminées de l'hypertonie, mais aussi de l'athérosclérose, du diabète, des maladies prolifératives et dans le cadre des opérations chirurgicales dans le cas des transplantations d'organes.
Les composés selon l'invention sont particulièrement utiles pour le traitement et la prévention des maladies cardiovasculaires, des maladies du métabolisme, des maladies cancéreuses et des maladies fibreuses.
En thérapeutique humaine, les composés selon l'invention sont particulièrement utiles pour le traitement et/ou la prévention des infarctus, comme composés antiarythmiques à composante cardioprotectrice, des angines de poitrine, des lésions induites par une ischémie, des ischémies du système nerveux, en particulier du système nerveux central, l'attaque ou de l'oedème cérébral, des formes du choc comme le choc allergique, le choc cardiogène, le choc hypovolémique et le choc bactérien, l'hypertension sanguine, l'hypertonie essentielle, la prolifération cellulaire des fibroblastes, les complications diabétiques tardives, les maladies cancéreuses, les maladies fibreuses comme la fibrose pulmonaire, la fibrose hépatique ou la fibrose rénale, les hypertrophies et hyperplasies d'organes, en particulier l'hyperplasie de la prostate ou l'hypertrophie de la prostate.
En thérapeutique humaine, les composés selon l'invention sont également utiles comme agents de diagnostic pour la détermination et la distinction de formes déterminées de l'hypertonie, mais aussi de l'athérosclérose, du diabète, des maladies prolifératives.
<Desc/Clms Page number 79>
En thérapeutique humaine, les composés selon l'invention sont également utiles pour leurs applications aux opérations chirurgicales dans le cas de transplantations d'organes, des opérations chirurgicales d'angioplastie et en particulier pour la protection des organes donneurs et pour le transfert dans l' organisme receveur.
Les doses dépendent de l'effet recherché, de la durée du traitement et de la voie d'administration utilisée ; sont généralement comprises entre 0. 001 mg et 1000 mg par jour par voie orale pour un adulte .
D'une façon générale, le médecin déterminera la posologie appropriée en fonction de l'âge, du poids et de tous les autres facteurs propres au sujet à traiter.
Les exemples suivants illustrent des compositions selon l'invention : EXEMPLE A On prépare, selon la technique habituelle, des gélules dosées à 50 mg de produit actif ayant la composition suivante : - Composé de formule (I)..................................................... 50 mg - Cellulose........................................................................... 18 mg - Lactose............................................................................. 55 mg - Silice colloïdale................................................................. 1 mg - Carboxyméthylamidon sodique...................................... 10 mg - Talc................................................................................... 10 mg - Stéarate de magnésium.................................................. 1 mg EXEMPLE B On prépare selon la technique habituelle des comprimés dosés à 50 mg de produit actif ayant la composition suivante :
<Desc/Clms Page number 80>
- Composé de formule (I).................................................... 50 mg - Lactose............................................................................. 104 mg - Cellulose.......................................................................... 40 mg - Polyvidone....................................................................... 10 mg - Carboxyméthylamidon sodique........................................ 22 mg - Talc................................................................................... 10 mg - Stéarate de magnésium.................................................... 2 mg - Silice colloïdale................................................................. 2 mg - Mélange d'hydroxyméthylcellulose, glycérine, oxyde de titane (72-3,5-24,5) q. s.p. 1 comprimé pelliculé terminé à 245 mg EXEMPLE C On prépare une solution injectable contenant 10 mg de produit actif ayant la composition suivante :
- Composé de formule (I).................................................... 10 mg - Acide benzoïque.............................................................. 80 mg - Alcool benzylique.............................................................. 0,06 ml - Benzoate de sodium......................................................... 80 mg - Ethanol à 95 %.................................................................. 0,4 ml - Hydroxyde de sodium....................................................... 24 mg - Propylène glycol................................................................ 1,6 ml
<Desc/Clms Page number 81>
- Eau..........................................................................q.s.p. 4 ml La présente invention concerne également la méthode de prévention et de traitement des maladies comme les infarctus, des angines de poitrine, les lésions induites par une ischémie, le traitement des ischémies du système nerveux, en particulier du système nerveux central, le traitement de l'attaque ou de l'oedème cérébral, les traitements de formes du choc comme le choc allergique, le choc cardiogène, le choc hypovolémique et le choc bactérien, l'hypertension sanguine, l'hypertonie essentielle, la prolifération cellulaire des fibroblastes, les complications diabétiques tardives, les maladies cancéreuses, les maladies fibreuses comme la fibrose pulmonaire, la fibrose hépatique ou la fibrose rénale, les hypertrophies et hyperplasies d'organes, en particulier l'hyperplasie de la prostate ou l'hypertrophie de la prostate.
Claims (17)
- dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), alkényle, alkynyle, aryle, hétéroaryle, halogène, nitro, amino, alkylamino (C1-C4), NRaRb, alkoylamino (C1-C4), hydroxy, alkoxy (CI- C4), S(0)nR7 (n=0,1,2), carboxy, alkoxycarbonyle (C1-C4), alkylcarbonyle (C1-C4), carboxamide, cyano, polyfluoroalkyle (C1-C4), polyfluoroalkoxy (C1-C3), S03H, RI et R2 étant eux-même éventuellement substitués par un groupement alkyle linéaire ou ramifié (C1-C4) où R3 représente un atome d'hydrogène, un groupe aryle, hétéroaryle ou une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8), polyfluoroalkyle (CI-C4), aryle, hétéroaryle, hydroxy, alkoxy (C1-C4), carboxy, carboxamide, amino, alkylamino (C1-C4), NRaRb, où R4, R5 et R6 représentent indépendamment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4) où R7 est un alkyle linéaire ou ramifié (C1-C4)
 REVENDICATIONS 1. Composés de formule (I)<Desc/Clms Page number 83>où Ra et Rb représentent R7, ou bien Ra et Rb forment ensemble avec l'atome d'azote auquel ils sont rattachés un hétérocycle à 5 ou 6 chaînons pouvant contenir éventuellement un autre hétéroatome choisi parmi 0, S, ou N leurs racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables.
REVENDICATIONS 1. Composés de formule (I)<Desc/Clms Page number 83>où Ra et Rb représentent R7, ou bien Ra et Rb forment ensemble avec l'atome d'azote auquel ils sont rattachés un hétérocycle à 5 ou 6 chaînons pouvant contenir éventuellement un autre hétéroatome choisi parmi 0, S, ou N leurs racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables. - 2. Composés de formule (I) selon la revendication 1
 dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), halogène, amino, alkylamino (C1-C4), NRaRb, alkoylamino (C1-C4), hydroxy, alkoxy (C1-C4), carboxy, alkoxycarbonyle (C1-C4), polyfluoroalkyle (CI-C4), polyfluoroalkoxy (CI-C3), S03H, RI et R2 étant euxmême éventuellement substitués par un groupement alkyle linéaire ou ramifié (CIC4) où R3 représente une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8), polyfluoroalkyle (C1-C4), aryle, hétéroaryle, où R4, R5 et R6 représentent indépendemment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4)<Desc/Clms Page number 84>où Ra et Rb représentent un alkyle linéaire ou ramifié (C1-C4), ou bien Ra et Rb forment ensemble avec l'atome d'azote auquel ils sont ratachés un hétérocycle à 5 ou 6 chaînons pouvant contenir éventuellement un autre hétéroatome choisi parmi 0, S, ou N leurs racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables.
dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), halogène, amino, alkylamino (C1-C4), NRaRb, alkoylamino (C1-C4), hydroxy, alkoxy (C1-C4), carboxy, alkoxycarbonyle (C1-C4), polyfluoroalkyle (CI-C4), polyfluoroalkoxy (CI-C3), S03H, RI et R2 étant euxmême éventuellement substitués par un groupement alkyle linéaire ou ramifié (CIC4) où R3 représente une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8), polyfluoroalkyle (C1-C4), aryle, hétéroaryle, où R4, R5 et R6 représentent indépendemment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4)<Desc/Clms Page number 84>où Ra et Rb représentent un alkyle linéaire ou ramifié (C1-C4), ou bien Ra et Rb forment ensemble avec l'atome d'azote auquel ils sont ratachés un hétérocycle à 5 ou 6 chaînons pouvant contenir éventuellement un autre hétéroatome choisi parmi 0, S, ou N leurs racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables. - 3. Composés de formule (I) selon la revendication 1
 dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), halogène, hydroxy, alkoxy (CI-C4), polyfluoroalkyle (CI-C4), polyfluoroalkoxy (C1-C3), RI et R2 étant eux-même éventuellement substitués par un groupement alkyle linéaire ou ramifié (C1-C4) où R3 représente une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8) ou polyfluoroalkyle (C1-C4), où R4, R5 et R6 représentent indépendamment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4) leurs racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables.<Desc/Clms Page number 85>N-[(5-hydroxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(6- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[(7- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétylJ-guanidine N-[(4,7-Dichloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acetyl]-guanidine N-[(4-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine
dans laquelle RI et R2 sont indépendamment : hydrogène, alkyle (C1-C4), halogène, hydroxy, alkoxy (CI-C4), polyfluoroalkyle (CI-C4), polyfluoroalkoxy (C1-C3), RI et R2 étant eux-même éventuellement substitués par un groupement alkyle linéaire ou ramifié (C1-C4) où R3 représente une chaîne du type Alk-R8, Alk représentant une chaîne de 1 à 5 atomes de carbone en chaîne linéaire ou ramifiée et R8 représentant un atome d'hydrogène, un groupe cycloalkyle (C3-C8) ou polyfluoroalkyle (C1-C4), où R4, R5 et R6 représentent indépendamment un atome d'hydrogène ou un alkyle linéaire ou ramifié (C1-C4) leurs racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables.<Desc/Clms Page number 85>N-[(5-hydroxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(6- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[(7- hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétylJ-guanidine N-[(4,7-Dichloro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acetyl]-guanidine N-[(4-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(4-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(4-hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine
N-[(4-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-amino-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(4-hydroxy-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihy dro- IH-isoindol-1 -yl)-2-méthyl-propionyl]guanidine N-[2-(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-2-méthylpropionyl]-guanidine N-[(3-Oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-IH-isoindo1-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(2-Isobutyl-7-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine
N-[2-(2-Isobutyl-3-oxo-2,3-dihy dro- IH-isoindol-1 -yl)-2-méthyl-propionyl]guanidine N-[2-(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-2-méthylpropionyl]-guanidine N-[(3-Oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro-IH-isoindo1-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(2-Isobutyl-7-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine
- 4. Composé selon les revendications 1 à 3 caractérisé par le fait qu'il est choisi parmi :<Desc/Clms Page number 86>N-[(2-Isobutyl-I-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine leur racémiques, énantiomères, diastéréoisomères et leurs mélanges, leurs tautomères et leurs sels pharmaceutiquement acceptables.
 N-[(6-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[(4,5-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(6,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(4-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(6-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine
N-[(6-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acétyl]-guanidine N-[(4,5-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(6,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(4-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(6-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(7-Carboxy-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine - 5. Composé selon l'une quelconque des revendications 1 à 3 caractérisé par le fait qu'il est choisi parmi : N-[(2-Méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Ethyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-propyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[2-(3-0xo-2-propyl-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine
 N-[(2-Isopropyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[2-(2-Butyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine N-[(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine<Desc/Clms Page number 87>N-[(6-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl] -guanidine N-[(2-Isobutyl-5-isopropoxy-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(5-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(5-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]guanidine N-[(6-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]guanidine N-[(6-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl] - guanidine
N-[(2-Isopropyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[2-(2-Butyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine N-[(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine<Desc/Clms Page number 87>N-[(6-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl] -guanidine N-[(2-Isobutyl-5-isopropoxy-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(5-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Chloro-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(5-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]guanidine N-[(6-Chloro-2-cyclopropylméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1H-isoindol-1-yl)-acétyl]guanidine N-[(6-Chloro-3-oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine N-[(5-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl] - guanidine N-[(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-4-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-5-méthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Isobutyl-6-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine
N-[(2-Cyclopropylméthyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(2-Benzyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-4-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(2-Isobutyl-5-méthyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Isobutyl-6-méthyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(5-tert-Butyl-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine<Desc/Clms Page number 88>N-[(3-Oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1H-isoindol-1-yl)- acétyl]-guanidine
N-[2-(2-Isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-propionyl]-guanidine<Desc/Clms Page number 88>N-[(3-Oxo-5-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1H-isoindol-1-yl)- acétyl]-guanidine N-[(2-Cyclopropylméthyl-3-oxo-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(2-Cyclopropylméthyl-3-oxo-6-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine
N-[(2-Cyclopropylméthyl-3-oxo-5-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(2-Cyclopropylméthyl-3-oxo-6-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine N-[(3-Oxo-2-(2,2,2-trifluoroéthyl)-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine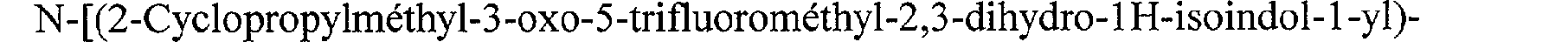 N-[(5,6-Dichloro-2-isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol- 1 -yl)-acetyl] -guanidine N-[(7-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(4,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(5-Bromo-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Bromo-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Isobutyl-3-oxo-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine N-[(2-Isobutyl-3-oxo-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]- guanidine
N-[(5,6-Dichloro-2-isobutyl-3-oxo-2,3-dihydro- 1 H-isoindol- 1 -yl)-acetyl] -guanidine N-[(7-Fluoro-2-isobutyl-3-oxo-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]-guanidine N-[(4,7-Difluoro-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(5-Bromo-2-isobutyl-3-oxo-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]-guanidine N-[(6-Bromo-2-isobutyl-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-acétyl]-guanidine N-[(2-Isobutyl-3-oxo-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)-acétyl]- guanidine N-[(2-Isobutyl-3-oxo-6-trifluorométhyl-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]- guanidine N-[(6-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl] guanidine N-[(5-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]- guanidine N-[(6-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine
N-[(6-Chloro-3-oxo-2-(3,3,3-trifluoropropyl)-2,3-dihydro- 1 H-isoindol- 1 -yl)-acétyl] guanidine N-[(5-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 1 H-isoindol- 1 -yl)-acétyl]- guanidine N-[(6-Chloro-3-oxo-2-(4,4,4-trifluorobutyl)-2,3-dihydro-1 H-isoindol-1-yl)-acétyl]guanidine <Desc/Clms Page number 89>N- 2-[3-Oxo-2-(2-pyrrolidin-1-yl-éthyl)-2,3-dihydro-1 H-isoindol-1-yl]-propionyl }- guanidine N-[2-(2-Hydroxy-éthyl)-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine leurs racémiques, énantiomères, diastéréoisomères, tautomères ainsi que leurs sels pharmaceutiquement acceptables.
<Desc/Clms Page number 89>N- 2-[3-Oxo-2-(2-pyrrolidin-1-yl-éthyl)-2,3-dihydro-1 H-isoindol-1-yl]-propionyl }- guanidine N-[2-(2-Hydroxy-éthyl)-3-oxo-2,3-dihydro-lH-isoindol-l-yl)-propionyl]-guanidine leurs racémiques, énantiomères, diastéréoisomères, tautomères ainsi que leurs sels pharmaceutiquement acceptables. N-[(3-Oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-6-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine Acide [l-(2-guanidino-l-méthyl-2-oxo-éthyl)-3-oxo-l,3-dihydro-isoindol-2-yl]- acétique
N-[(3-Oxo-6-trifluorométhyl-2-(3,3,3-trifluoropropyl)-2,3-dihydro-1 H-isoindol-1-yl)- acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-5-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine N-[(3-Oxo-2-(4,4,4-trifluorobutyl)-6-trifluorométhyl-2,3-dihydro-lH-isoindol-l-yl)- acétyl]-guanidine Acide [l-(2-guanidino-l-méthyl-2-oxo-éthyl)-3-oxo-l,3-dihydro-isoindol-2-yl]- acétique
- 6. Composé selon l'une quelconque des revendications 1 à 5 utilisé à titre de médicament.
- 7. Composition pharmaceutique caractérisée par le fait qu'elle comprend dans un milieu pharmaceutiquement acceptable, un composé défini selon l'une quelconque des revendications 1 à 5.
- 8. Médicament selon la revendication 6 caractérisé par le fait qu'il contient au moins un composé défini selon l'une quelconque des revendications 1 à 5 pour son application thérapeutique dans le traitement des maladies comme inhibiteur de NHE.
- 9 Médicament selon la revendication 6 caractérisé par le fait qu'il contient au moins un composé défini selon l'une quelconque des revendications 1 à 5 pour son application thérapeutique dans le traitement des maladies comme inhibiteur de NHE- 1.<Desc/Clms Page number 90>
- 10. Médicament caractérisé en ce qu'il contient au moins un composé défini selon l'une quelconque des revendications 1 à 6 pour son application thérapeutique dans le traitement et la prévention des maladies cardiovasculaires, des maladies du métabolisme, des maladies cancéreuses, des maladies fibreuses.
- 11. Médicament selon la revendication 10 caractérisé en ce que son application thérapeutique est utile dans le traitement et la prévention des infarctus, angine de poitrine, lésions induites par ischémie, ischémies du système nerveux, des attaque de l'oedème cérébral, des formes de choc, des complications diabétiques, des fibroses pulmonaires, fibroses hépatiques, fibroses rénales, des hypertrophies et hyperplasies d'organes, de l'hypertension sanguine, de l'hypertonie essentielle, de l'hypercholestérolémie, des lésions endothéliales, des spasmes des artères coronaires, de l'athérogenèse, de l'athérosclérose, de l'hypertrophie du ventricule gauche, de la cardiomyopathie dilatée, des maladies thrombotiques.
- 12. Médicament caractérisé en ce qu'il contient au moins un composé défini selon l'une quelconque des revendications 1 à 6 pour son application thérapeutique pour des opérations chirurgicales dans le cas de transplantations d'organes, des opérations chirurgicales d'angioplastie.
- 13. Médicament selon la revendication 12 pour son application aux transplantations d'organes pour la protection des organes donneurs et pour le transfert dans l'organisme receveur.
- 14. Procédé de préparation des composés de formule (I) tels que définis dans les revendications 1 à 3 pour lesquels R4 et R6 sont des hydrogènes caractérisé en ce que a) on fait réagir un hydrure avec un phtalimide (de formule II, RI, R2 et R3 ayant la même définition que dans la revendication 1) dans un alcool aliphatique<Desc/Clms Page number 91>b) on met le produit obtenu en présence d'une amine de type R3NH2 et d'un carbodiimide (R3 ayant la même signification que dans la revendication 1) c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4)
 b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec un trialkylphosphonoacétate et une base c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine un alcool (C1-C4)
b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec un trialkylphosphonoacétate et une base c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine un alcool (C1-C4) - 15. Procédé de préparation des composés de formule (I) tels que définis dans les revendications 1 à 3 pour lesquels R4 et R6 sont des hydrogènes caractérisé en ce que a) on fait réagir un composé de formule III (RI et R2 ayant la même définition que dans la revendcation 1) avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec un trialkylphosphonoacétate et une base
 <Desc/Clms Page number 92>
<Desc/Clms Page number 92> - 16. Procédé de préparation des composés de formule (I) tels que définis dans les revendications 1 à 3 pour lesquels R4 est un alkyle et R6 est un hydrogène caractérisé en ce que a) on fait réagir un phtalimide (de formule II telle que définie dans la revendication 14) avec un halogénure d'alkylmagnésium ou un alkyllithien dans un éther b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec du 1-éthoxy-1-triméthylsiloxyéthylène et un acide de Lewis c) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4)
- 17. Procédé de préparation des composés de formule (I) tels que définis dans les revendications 1 à 3 pour lesquels R6 est un alkyle caractérisé en ce que a) on fait réagir un phtalimide (de formule II telle que définie dans la revendication 14) avec un halogénure d'alkyl magnésium ou un alkyllithien dans un éther b) puis on fait réagir le produit obtenu avec un alkoxycarbonylméthylène triphénylphosphorane dans du toluène ou avec du 1-éthoxy-l-triméthylsiloxyéthylène et un acide de Lewis c) puis on fait réagir le produit obtenu en présence de diisopropylamidure de lithium avec un R6-Hal d) le produit obtenu est mis en présence de chlorure de guanidinium et d'une base ou avec de la guanidine dans un alcool (C1-C4)
Priority Applications (43)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0206783A FR2840302B1 (fr) | 2002-06-03 | 2002-06-03 | Derives d'isoindolones, procede de preparation et intermediaire de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant |
| JO200351A JO2357B1 (en) | 2002-06-03 | 2003-05-07 | Derivatives of isoandolone, method of preparation and intermediate compounds of this method, use as pharmaceutical compositions |
| YUP104804 RS51636B (sr) | 2002-06-03 | 2003-05-20 | Derivati n-((3-okso- 2,3-dihidro-1h-izoindol-1-il) acetil) gvanidina kao inhibitori nhe-1 za lečenje infarkta i angine pektoris |
| AT03730076T ATE324889T1 (de) | 2002-06-03 | 2003-05-20 | N-((3-oxo-2,3-dihydro-1h-isoindol-1- yl)acetyl)guanidin derivate als nhe1-inhibitoren zur behandlung von infarkten und angina pectoris |
| KR1020047019619A KR101149771B1 (ko) | 2002-06-03 | 2003-05-20 | 경색 및 협심증의 치료를 위한 nhe-1 억제제로서의n-((3-옥소-2,3-디하이드로-1h-이소인돌-1-일)아세틸)구아니딘 유도체 |
| PCT/EP2003/005279 WO2003101450A1 (fr) | 2002-06-03 | 2003-05-20 | Derives de guanidine n- ((3-oxo-2, 3-dihydro-1h-isoindole-1-yle) acetyle) comme inhibiteurs de nhe-1 pour traiter l'infarctus et l'angine pectorale |
| UA20041211001A UA81118C2 (en) | 2002-06-03 | 2003-05-20 | N-((3-oxo-2, 3-dihydro-1h-isoindol-1-yl) acetyl) guanidine derivatives as nhe-1 inhibitors for the treatment of infarction and angina pectoris |
| MXPA04011931A MXPA04011931A (es) | 2002-06-03 | 2003-05-20 | Derivados de n-((3-oxo-2, 3-dihidro-1h-isoindol-1-il)acetil)guanidina en calidad de inhibidores de nhe-1 para el tratamiento del infarto y la angina de pecho. |
| NZ536953A NZ536953A (en) | 2002-06-03 | 2003-05-20 | N-((3-oxo2,3-dihydro-1H-isoindol-1-yl) acetyl) guanidine derivatives as nhe-1 inhibitors for the treatment of infarction and angina pectoris |
| MEP27508 MEP27508A (en) | 2002-06-03 | 2003-05-20 | N- ((3-oxo2, 3-dihydro-1h-isoindol-1-yl) acetyl) guanidine derivatives as nhe-1 inhibitors for the treatment of infarction and angina pectoris |
| ES03730076T ES2261939T3 (es) | 2002-06-03 | 2003-05-20 | Derivados de n-((3-oxo-2,3-dihidro-1h-isoindol-1-il)acetil)guanidina como inhibidores de nhe1 para el tratamiento del infarto y la angina de pecho. |
| DK03730076T DK1528923T3 (da) | 2002-06-03 | 2003-05-20 | N-((3-oxo-2,3-dihydro-1H-isoindol-1-yl)-acetyl)guanidinderivater som NHE1-inhibitorer til behandling af infarkt og angina pectoris |
| EP20030730076 EP1528923B1 (fr) | 2002-06-03 | 2003-05-20 | Derives de guanidine n- ((3-oxo-2, 3-dihydro-1h-isoindole-1-yle) acetyle) comme inhibiteurs de nhe-1 pour traiter l'infarctus et l'angine pectorale |
| CNB038128500A CN1290833C (zh) | 2002-06-03 | 2003-05-20 | 用于治疗梗塞和心绞痛的nhe-1抑制剂n-((3-氧代-2,3-二氢-1h-异吲哚-1-基)乙酰基)胍衍生物 |
| HK05108389.7A HK1076383B (en) | 2002-06-03 | 2003-05-20 | N-((3-oxo2,3-dihydro-1h-isoindol-1-yl) acetyl) guanidine derivatives as nhe-1 inhibitors for the treatment of infarction and angina pectoris |
| BR0311556A BR0311556A (pt) | 2002-06-03 | 2003-05-20 | Derivados de n-((3-oxo-2,3-diidro-1h-isoindol-1-il)acetil)guanidina como inibidores de nhe-1 para o tratamento de enfarte e angina pectoris |
| OA1200400323A OA12864A (en) | 2002-06-03 | 2003-05-20 | N-(3-0X02,3-DIHYDRO-1H-ISOINDOL-1-YL)ACETYL) guanidine derivatives as NHE-1 inhibitors for the treatment of infarction and angina pectoris. |
| PL37218803A PL372188A1 (en) | 2002-06-03 | 2003-05-20 | N-((3-oxo-2,3-dihydro-1h-isoindol-1-yl)acetyl)guanidine derivatives as nhe1-inhibitors for the treatment of infarction and angina pectoris |
| PT03730076T PT1528923E (pt) | 2002-06-03 | 2003-05-20 | Derivados de n-((3-oxo-2,3-di-hidro-1h-isoindol-1-il)acetil)guanidina como inibidores de nhe-1 para o tratamento de enfarte e angina de peito |
| DE2003605042 DE60305042T2 (de) | 2002-06-03 | 2003-05-20 | N-((3-oxo-2,3-dihydro-1h-isoindol-1-yl)acetyl)guanidin derivate als nhe1-inhibitoren zur behandlung von infarkten und angina pectoris |
| HRP20041151AA HRP20041151B1 (hr) | 2002-06-03 | 2003-05-20 | Derivati n-((3-okso-2,3-dihidro-1h-izoindol-1-il)-acetil)gvanidina kao nhe-1 inhibitori za lijeäśenje infarkta i angine pektoris |
| RU2004138789A RU2318809C2 (ru) | 2002-06-03 | 2003-05-20 | Производные n-((3-оксо-2,3-дигидро-1h-изоиндол-1-ил)ацетил)гуанидина в качестве ингибиторов nhe-1 для лечения инфаркта и стенокардии |
| CA 2488373 CA2488373C (fr) | 2002-06-03 | 2003-05-20 | Derives de guanidine n- ((3-oxo-2, 3-dihydro-1h-isoindole-1-yle) acetyle) comme inhibiteurs de nhe-1 pour traiter l'infarctus et l'angine pectorale |
| SI200330302T SI1528923T1 (sl) | 2002-06-03 | 2003-05-20 | N-((3-okso-2,3-dihidro-1H-izoindol-1-il)acetil)gvanidinski derivati NHE1 za zdravljenje infarkta inangine pektoris |
| JP2004508808A JP4634793B2 (ja) | 2002-06-03 | 2003-05-20 | 梗塞および狭心症の治療のためのnhe−1抑制剤としてのn−((3−オキソ−2,3−ジヒドロ−1h−イソインドール−1−イル)アセチル)グアニジン誘導体 |
| AU2003240677A AU2003240677B2 (en) | 2002-06-03 | 2003-05-20 | N- ((3-oxo2, 3-dihydro-1H-isoindol-1-yl) acetyl) guanidine derivatives as NHE-1 inhibitors for the treatment of infarction and angina pectoris |
| US10/445,618 US7078428B2 (en) | 2002-06-03 | 2003-05-27 | Isoindolone derivatives, preparation process and intermediates of this process, their use as medicaments, and pharmaceutical compositions comprising them |
| PE2003000523A PE20040531A1 (es) | 2002-06-03 | 2003-05-29 | Derivados de isoindolona, proceso de preparacion, compuestos intermedios de este proceso y composiciones farmaceuticas que los contienen |
| TW92114642A TWI286545B (en) | 2002-06-03 | 2003-05-30 | Isoindolone derivatives, preparation process and intermediates of this process, their use as medicaments, and pharmaceutical compositions comprising them |
| HN2003000162A HN2003000162A (es) | 2002-06-03 | 2003-05-30 | Derivados de isoindolona, proceso de preparacion y compuestos intermedios de este proceso, su uso como medicamentos, y composiciones farmaceuticas que los contienen. |
| ARP030101923 AR039927A1 (es) | 2002-06-03 | 2003-05-30 | Derivados de isoindolona, proceso de preparacion y compuestos intermedios de este proceso, su uso como medicamentos, y composiciones farmaceuticas que los contienen |
| EG2003060521A EG24784A (en) | 2002-06-03 | 2003-06-01 | Isoindolone derivatives, preparation process and intermediates of this process, their use as medicaments and pharmaceutical comprising them. |
| MYPI20032036 MY142502A (en) | 2002-06-03 | 2003-06-02 | N-((3-oxo-2,3-dihydro-1h-isoindol-1-yl)acetyl)guanidine derivatives as nhe1-inhibitors for the treatment of infarction and angina pectoris |
| PA8574701A PA8574701A1 (es) | 2002-06-03 | 2003-06-02 | Derivados de isoindolona, proceso de preparacion y compuestos intermedios de este proceso, su uso como medicamentos, y composiciones farmaceuticas que los contienen |
| UY27835A UY27835A1 (es) | 2002-06-03 | 2003-06-03 | Derivados de isoindolona, proceso de preparacion y compuestos intermedios de este proceso, su uso como medicamentos, y composiciones farmaceuticas que los contienen. |
| ZA2004/09094A ZA200409094B (en) | 2002-06-03 | 2004-11-10 | N-((3-oxo2,3-dihydro-1h-isoindol-1-yl), acetyl) guanidine derivatives as nhe-1- inhibitor for the treatment of infarction and angina pectoris |
| MA27951A MA27308A1 (fr) | 2002-06-03 | 2004-11-16 | Derives de n-( (3-oxo-2, 3-dihydro- 1h-isoindol-1-yl) acetyl)guanidine en tant qu'inhibiteurs du nhe-1-destines au traitement de l'infractus et de l'angine de poitrine |
| TNP2004000237A TNSN04237A1 (en) | 2002-06-03 | 2004-11-30 | N- ((3-oxo2, 3-dihydro-1h-isoindol-1-yl) acetyl) guanidine derivatives as nhe-1 inhibitors for the treatment of infarction and angina pectoris |
| IL165520A IL165520A (en) | 2002-06-03 | 2004-12-02 | N-((3-oxo-2, 3-dihydro-1h-isoindol-1-yl), acetyl) guanidine derivatives as nhe - 1 - inhibitors, pharmaceutical compositions comprising them, process for their preparation and uses thereof |
| ECSP045472 ECSP045472A (es) | 2002-06-03 | 2004-12-02 | "derivados de n-((3-oxo-2,3-dihidro-1h-isoindol-1-il)acetil)guanidina en calidad de inhibidores de nhe-1 para el tratamiento del infarto y la angina de pecho" |
| NO20045660A NO329224B1 (no) | 2002-06-03 | 2004-12-27 | N-((3okso-2,3-dihydro-IH-isoindol-1-yl)acetyl)guanidinderivater, anvendelse derav, farmasoytisk sammensetning inneholdende en slik forbindelse samt fremgangsmater for fremstilling av forbindelsene |
| US11/365,319 US7589117B2 (en) | 2002-06-03 | 2006-03-01 | Isoindolone derivatives, preparation process and intermediates of this process, their use as medicaments, and pharmaceutical compositions comprising them |
| CY20061101057T CY1105115T1 (el) | 2002-06-03 | 2006-07-28 | Παραγωγα ν-((3-οξο-2,3-διυδρο-1η-ισοϊνδολ-1-υλ)ακετυλ)γουανιδινης ως nhe1-αναστολεις για την θepαπεια του εμφραγματος και της στηθαγχης |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0206783A FR2840302B1 (fr) | 2002-06-03 | 2002-06-03 | Derives d'isoindolones, procede de preparation et intermediaire de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2840302A1 true FR2840302A1 (fr) | 2003-12-05 |
| FR2840302B1 FR2840302B1 (fr) | 2004-07-16 |
Family
ID=29558914
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR0206783A Expired - Fee Related FR2840302B1 (fr) | 2002-06-03 | 2002-06-03 | Derives d'isoindolones, procede de preparation et intermediaire de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant |
Country Status (41)
| Country | Link |
|---|---|
| US (2) | US7078428B2 (fr) |
| EP (1) | EP1528923B1 (fr) |
| JP (1) | JP4634793B2 (fr) |
| KR (1) | KR101149771B1 (fr) |
| CN (1) | CN1290833C (fr) |
| AR (1) | AR039927A1 (fr) |
| AT (1) | ATE324889T1 (fr) |
| AU (1) | AU2003240677B2 (fr) |
| BR (1) | BR0311556A (fr) |
| CA (1) | CA2488373C (fr) |
| CY (1) | CY1105115T1 (fr) |
| DE (1) | DE60305042T2 (fr) |
| DK (1) | DK1528923T3 (fr) |
| EC (1) | ECSP045472A (fr) |
| EG (1) | EG24784A (fr) |
| ES (1) | ES2261939T3 (fr) |
| FR (1) | FR2840302B1 (fr) |
| HN (1) | HN2003000162A (fr) |
| HR (1) | HRP20041151B1 (fr) |
| IL (1) | IL165520A (fr) |
| JO (1) | JO2357B1 (fr) |
| MA (1) | MA27308A1 (fr) |
| ME (1) | MEP27508A (fr) |
| MX (1) | MXPA04011931A (fr) |
| MY (1) | MY142502A (fr) |
| NO (1) | NO329224B1 (fr) |
| NZ (1) | NZ536953A (fr) |
| OA (1) | OA12864A (fr) |
| PA (1) | PA8574701A1 (fr) |
| PE (1) | PE20040531A1 (fr) |
| PL (1) | PL372188A1 (fr) |
| PT (1) | PT1528923E (fr) |
| RS (1) | RS51636B (fr) |
| RU (1) | RU2318809C2 (fr) |
| SI (1) | SI1528923T1 (fr) |
| TN (1) | TNSN04237A1 (fr) |
| TW (1) | TWI286545B (fr) |
| UA (1) | UA81118C2 (fr) |
| UY (1) | UY27835A1 (fr) |
| WO (1) | WO2003101450A1 (fr) |
| ZA (1) | ZA200409094B (fr) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10344002B2 (en) | 2016-09-26 | 2019-07-09 | Nusirt Sciences, Inc. | Compositions and methods for treating metabolic disorders |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10356717A1 (de) * | 2003-12-02 | 2005-07-07 | Aventis Pharma Deutschland Gmbh | Verfahren zur Herstellung von (3-Oxo-2,3-dihydro-1H-isoindol-1-yl)-acetylguanidin-Derivaten |
| US7531569B2 (en) | 2003-12-02 | 2009-05-12 | Sanofi-Aventis Deutschland Gmbh | Process for preparing (3-oxo-2,3-dihydro-1H-isoindol-1-yl) acetylguanidine derivatives |
| KR100518184B1 (ko) * | 2003-12-27 | 2005-10-04 | 한국화학연구원 | 퓨란카보닐구아니딘 유도체, 그의 제조방법 및 그를포함하는 약학적 조성물 |
| TW200613272A (en) | 2004-08-13 | 2006-05-01 | Astrazeneca Ab | Isoindolone compounds and their use as metabotropic glutamate receptor potentiators |
| US7807706B2 (en) | 2005-08-12 | 2010-10-05 | Astrazeneca Ab | Metabotropic glutamate-receptor-potentiating isoindolones |
| US20070161648A1 (en) * | 2005-10-14 | 2007-07-12 | Hughes Terry V | Substituted dihydro-isoindolones useful in treating kinase disorders |
| TW200812962A (en) * | 2006-07-12 | 2008-03-16 | Astrazeneca Ab | New compounds I/418 |
| TWI417100B (zh) | 2007-06-07 | 2013-12-01 | Astrazeneca Ab | 二唑衍生物及其作為代謝型麩胺酸受體增效劑-842之用途 |
| SA109300358B1 (ar) | 2008-06-06 | 2012-11-03 | استرازينيكا ايه بي | مقويات مستقبل جلوتامات ذي انتحاء أيضي من أيزو إندولون |
| GB0811643D0 (en) | 2008-06-25 | 2008-07-30 | Cancer Rec Tech Ltd | New therapeutic agents |
| EP2588104B1 (fr) | 2010-07-01 | 2014-12-10 | Merck Sharp & Dohme Corp. | Modulateurs allostériques positifs du récepteur m1 de l'isoindolone |
| AR088320A1 (es) | 2011-10-14 | 2014-05-28 | Incyte Corp | Derivados de isoindolinona y pirrolopiridinona como inhibidores de akt |
| CN103193794B (zh) * | 2013-04-12 | 2016-04-06 | 中国药科大学 | 一种异吲哚酮并异噁唑类稠环化合物及其合成方法 |
| GB201517216D0 (en) | 2015-09-29 | 2015-11-11 | Cancer Res Technology Ltd And Astex Therapeutics Ltd | Pharmaceutical compounds |
| GB201517217D0 (en) | 2015-09-29 | 2015-11-11 | Astex Therapeutics Ltd And Cancer Res Technology Ltd | Pharmaceutical compounds |
| CN106316922B (zh) * | 2016-08-17 | 2019-02-01 | 华东师范大学 | 一种含c-3全取代氧化吲哚衍生物及其合成方法和应用 |
| GB201704965D0 (en) | 2017-03-28 | 2017-05-10 | Astex Therapeutics Ltd | Pharmaceutical compounds |
| GB201704966D0 (en) | 2017-03-28 | 2017-05-10 | Astex Therapeutics Ltd | Pharmaceutical compounds |
| CN110664803B (zh) * | 2019-09-19 | 2023-08-29 | 中山大学 | 异吲哚酮类化合物在制备防治溶骨性疾病药物中的应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001083470A1 (fr) * | 2000-04-28 | 2001-11-08 | Pfizer Products Inc. | Inhibiteurs de l'echangeur sodium-hydrogene de type 1 |
| WO2002024637A1 (fr) * | 2000-09-22 | 2002-03-28 | Aventis Pharma Deutschland Gmbh | Guanidides d'acide cinnamique substitues, procedes permettant de les produire, leur utilisation comme medicament et medicament les contenant |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2708605A1 (fr) * | 1993-07-30 | 1995-02-10 | Sanofi Sa | Dérivés du N-sulfonylindol-2-one, leur préparation, les compositions pharmaceutiques en contenant. |
| DK0612723T3 (da) * | 1993-02-20 | 1998-03-30 | Hoechst Ag | Substituerede benzoylguanidiner, fremgangsmåde til fremstilling, deres anvendelse som lægemiddel, som inhibitor af den cellulære Na+/H+-udbytning eller som diagnostikum samt lægemiddel med indhold deraf |
| FR2735166B1 (fr) * | 1995-06-08 | 1997-08-29 | Aerospatiale | Procede de fabrication d'un panneau ou analogue a proprietes structurale et acoustique et panneau ainsi obtenu |
| DE19737224A1 (de) * | 1997-08-27 | 1999-03-18 | Hoechst Marion Roussel De Gmbh | Pharmazeutisches Kombinationspräparat aus einem Inhibitor des Natrium-Wasserstoff-Austauschers und einem Arzneimittel zur Behandlung von Herz-Kreislauferkrankungen |
| DE19737463A1 (de) * | 1997-08-28 | 1999-03-04 | Hoechst Marion Roussel De Gmbh | Verwendung von Inhibitoren des Natrium-Wasserstoff-Austauschers zur Herstellung eines Arzneimittels zur Behandlung von Erkrankungen, die durch Protozoen verursacht werden |
| DE19738604A1 (de) * | 1997-09-04 | 1999-03-11 | Hoechst Marion Roussel De Gmbh | Die Verwendung von Hemmern des Natrium-Wasserstoff-Austauschers zur Herstellung eines Medikaments zur Verminderung der Giftigkeit cardiotoxischer Stoffe |
| PT1056729E (pt) * | 1998-02-27 | 2005-04-29 | Pfizer Prod Inc | Derivados de n-[ciclo di- ou tri-aza di-insaturado de cinco membros substituido) carbonil] guanidina para o tratamento de isquemia |
| DE19859727A1 (de) * | 1998-12-23 | 2000-06-29 | Aventis Pharma Gmbh | Die Verwendung von Hemmern des Natrium-Wasserstoff-Austauschers zur Herstellung eines Medikaments zur Verhinderung von altersbedingten Organ-Dysfunktionen, altersbedingten Erkrankungen zur Lebensverlängerung |
| US6534651B2 (en) * | 2000-04-06 | 2003-03-18 | Inotek Pharmaceuticals Corp. | 7-Substituted isoindolinone inhibitors of inflammation and reperfusion injury and methods of use thereof |
| ATE348097T1 (de) * | 2001-04-09 | 2007-01-15 | Chiron Corp | Guanidinoverbindungen als melanocortin-4-rezeptor (mc4-r) agonisten |
-
2002
- 2002-06-03 FR FR0206783A patent/FR2840302B1/fr not_active Expired - Fee Related
-
2003
- 2003-05-07 JO JO200351A patent/JO2357B1/en active
- 2003-05-20 ME MEP27508 patent/MEP27508A/xx unknown
- 2003-05-20 CN CNB038128500A patent/CN1290833C/zh not_active Expired - Fee Related
- 2003-05-20 SI SI200330302T patent/SI1528923T1/sl unknown
- 2003-05-20 CA CA 2488373 patent/CA2488373C/fr not_active Expired - Fee Related
- 2003-05-20 BR BR0311556A patent/BR0311556A/pt not_active IP Right Cessation
- 2003-05-20 DK DK03730076T patent/DK1528923T3/da active
- 2003-05-20 PT PT03730076T patent/PT1528923E/pt unknown
- 2003-05-20 EP EP20030730076 patent/EP1528923B1/fr not_active Expired - Lifetime
- 2003-05-20 NZ NZ536953A patent/NZ536953A/en not_active IP Right Cessation
- 2003-05-20 AT AT03730076T patent/ATE324889T1/de active
- 2003-05-20 PL PL37218803A patent/PL372188A1/xx unknown
- 2003-05-20 KR KR1020047019619A patent/KR101149771B1/ko not_active Expired - Fee Related
- 2003-05-20 MX MXPA04011931A patent/MXPA04011931A/es active IP Right Grant
- 2003-05-20 UA UA20041211001A patent/UA81118C2/uk unknown
- 2003-05-20 AU AU2003240677A patent/AU2003240677B2/en not_active Ceased
- 2003-05-20 RS YUP104804 patent/RS51636B/sr unknown
- 2003-05-20 ES ES03730076T patent/ES2261939T3/es not_active Expired - Lifetime
- 2003-05-20 DE DE2003605042 patent/DE60305042T2/de not_active Expired - Lifetime
- 2003-05-20 JP JP2004508808A patent/JP4634793B2/ja not_active Expired - Fee Related
- 2003-05-20 RU RU2004138789A patent/RU2318809C2/ru not_active IP Right Cessation
- 2003-05-20 WO PCT/EP2003/005279 patent/WO2003101450A1/fr not_active Ceased
- 2003-05-20 HR HRP20041151AA patent/HRP20041151B1/hr not_active IP Right Cessation
- 2003-05-20 OA OA1200400323A patent/OA12864A/en unknown
- 2003-05-27 US US10/445,618 patent/US7078428B2/en not_active Expired - Lifetime
- 2003-05-29 PE PE2003000523A patent/PE20040531A1/es not_active Application Discontinuation
- 2003-05-30 AR ARP030101923 patent/AR039927A1/es active IP Right Grant
- 2003-05-30 TW TW92114642A patent/TWI286545B/zh not_active IP Right Cessation
- 2003-05-30 HN HN2003000162A patent/HN2003000162A/es unknown
- 2003-06-01 EG EG2003060521A patent/EG24784A/xx active
- 2003-06-02 MY MYPI20032036 patent/MY142502A/en unknown
- 2003-06-02 PA PA8574701A patent/PA8574701A1/es unknown
- 2003-06-03 UY UY27835A patent/UY27835A1/es unknown
-
2004
- 2004-11-10 ZA ZA2004/09094A patent/ZA200409094B/en unknown
- 2004-11-16 MA MA27951A patent/MA27308A1/fr unknown
- 2004-11-30 TN TNP2004000237A patent/TNSN04237A1/en unknown
- 2004-12-02 EC ECSP045472 patent/ECSP045472A/es unknown
- 2004-12-02 IL IL165520A patent/IL165520A/en not_active IP Right Cessation
- 2004-12-27 NO NO20045660A patent/NO329224B1/no not_active IP Right Cessation
-
2006
- 2006-03-01 US US11/365,319 patent/US7589117B2/en not_active Expired - Fee Related
- 2006-07-28 CY CY20061101057T patent/CY1105115T1/el unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001083470A1 (fr) * | 2000-04-28 | 2001-11-08 | Pfizer Products Inc. | Inhibiteurs de l'echangeur sodium-hydrogene de type 1 |
| WO2002024637A1 (fr) * | 2000-09-22 | 2002-03-28 | Aventis Pharma Deutschland Gmbh | Guanidides d'acide cinnamique substitues, procedes permettant de les produire, leur utilisation comme medicament et medicament les contenant |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10344002B2 (en) | 2016-09-26 | 2019-07-09 | Nusirt Sciences, Inc. | Compositions and methods for treating metabolic disorders |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| FR2840302A1 (fr) | Derives d'isoindolones, procede de preparation et intermediaire de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant | |
| DE60221391T2 (de) | Substituierte indolsäurederivate als inhibitoren von plasminogen-aktivator-inhibitor-1 (pai-1) | |
| EP0501892B1 (fr) | Dérivés hétérocycliques diazotés N-substitués par un groupement biphénylméthyle, leur préparation, les compositions pharmaceutiques en contenant | |
| EP0636609A1 (fr) | Dérivés du 1-benzyl-1,3-dihydro-indol-2-one, leur préparation, les compositions pharmaceutiques en contenant | |
| FR2792314A1 (fr) | Nouveaux composes aminotriazoles, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| EP0580502A1 (fr) | 3-(hydroxybenzylidényl)-indoline-2-ones, et les compositions pharmaceutiques qui les contiennent | |
| EP0231139B1 (fr) | Nouveaux dérivés de dihydrobenzofuranne - et de chromane -carboxamides, leurs procédés de préparation et leur utilisation comme neuroleptiques | |
| EP1296976A1 (fr) | Derives de 1,3-dihydro-2h-indol-2-one et leur utilisation en tant que ligands des recepteurs v1b et v1a de l'arginine-vasopressine | |
| EP1720544A1 (fr) | Nouveaux derives azabicycliques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| EP0743312A1 (fr) | Nouveaux composés spiro hétérocycliques, leur procédé de préparation et les compositions pharmaceutiques les contenant | |
| FR2842525A1 (fr) | Derives de 3-guanidinocarbonyl-1-heteroaryl-indole, procede de preparation a titre de medicaments et compositions pharmaceutiques les renfermant | |
| FR2842524A1 (fr) | Compositions pharmaceutiques contenant un derive de 3-guanidinocarbonyl-1-heteroaryl-pyrrole, leur procede de preparation a titre de medicaments | |
| EP2419422B1 (fr) | Dérivés d'imidazolidine-2,4-dione et leur utilisation comme médicament | |
| EP0562936A1 (fr) | Imidazolines N-substituées par un groupement biphénylméthyle, leur préparation, les compositions pharmaceutiques en contenant | |
| EP1255757B1 (fr) | Derives d'aminoacides et leurs utilisation en tant qu'inhibiteurs de la nep, l' ace et l' ece | |
| EP0549407B1 (fr) | Nouveaux dérivés de pyrrolidine, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| EP0556119B1 (fr) | Dérivés de pyrrolidine portant en position 3 un groupe acide gras saturé ou non-saturé, procédé pour leur préparation et compositions pharmaceutiques ayant une activité d'inhibiteurs de thromboxane-A2 les contenant | |
| EP0698008A1 (fr) | Nouveaux derives du lupane, leur preparation et les compositions pharmaceutiques qui les contiennent | |
| EP1324998A1 (fr) | Nouveaux composes aminotriazolones, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| FR2842526A1 (fr) | Compositions pharmaceutiques contenant un derive de 3-guanidinocarbonyl-1-heteroaryl-indole, procede de preparation a titre de medicaments | |
| FR2758329A1 (fr) | Derives d'imidazole-4-butane boronique, leur preparation et leur utilisation en therapeutique | |
| EP0463944A1 (fr) | Acyl benzoxazolinones, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| EP0384843B1 (fr) | Dérivés de la 1-arylsulfonyl 2-pipéridinone, leur procédé et les intermédiaires de préparation, leur application comme médicaments et les compositions les renfermant | |
| FR2842527A1 (fr) | Derives d'acyloxypyrolidine, leur preparation et leur application en therapeutique | |
| FR2856062A1 (fr) | Derives 3-guanidinocarbonyl-heterocycle, procede de preparation et intermediaires de ce procede a titre de medicaments et compositions pharmaceutiques les renfermant |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PLFP | Fee payment |
Year of fee payment: 15 |
|
| PLFP | Fee payment |
Year of fee payment: 16 |
|
| PLFP | Fee payment |
Year of fee payment: 17 |
|
| ST | Notification of lapse |
Effective date: 20200205 |