JP2004337172A - プロ−ウロキナーゼ変異種 - Google Patents
プロ−ウロキナーゼ変異種 Download PDFInfo
- Publication number
- JP2004337172A JP2004337172A JP2004200871A JP2004200871A JP2004337172A JP 2004337172 A JP2004337172 A JP 2004337172A JP 2004200871 A JP2004200871 A JP 2004200871A JP 2004200871 A JP2004200871 A JP 2004200871A JP 2004337172 A JP2004337172 A JP 2004337172A
- Authority
- JP
- Japan
- Prior art keywords
- pro
- variant
- activity
- lys
- native
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 108010073863 saruplase Proteins 0.000 title claims abstract description 7
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims abstract description 71
- 230000033885 plasminogen activation Effects 0.000 claims abstract description 16
- 230000002537 thrombolytic effect Effects 0.000 claims abstract description 15
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims abstract description 13
- 108010049003 Fibrinogen Proteins 0.000 claims abstract description 12
- 102000008946 Fibrinogen Human genes 0.000 claims abstract description 12
- 229940012952 fibrinogen Drugs 0.000 claims abstract description 12
- 230000009089 cytolysis Effects 0.000 claims abstract description 10
- 239000004472 Lysine Substances 0.000 claims abstract description 7
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 claims abstract description 6
- 125000003275 alpha amino acid group Chemical group 0.000 claims abstract 3
- 108020004414 DNA Proteins 0.000 claims description 26
- 102000053602 DNA Human genes 0.000 claims description 13
- 208000032843 Hemorrhage Diseases 0.000 claims description 5
- 108020004511 Recombinant DNA Proteins 0.000 claims description 5
- 208000034158 bleeding Diseases 0.000 claims description 5
- 230000000740 bleeding effect Effects 0.000 claims description 5
- 230000009885 systemic effect Effects 0.000 abstract description 6
- 238000011287 therapeutic dose Methods 0.000 abstract description 5
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 231
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 223
- 229960005356 urokinase Drugs 0.000 description 223
- 230000000694 effects Effects 0.000 description 80
- 150000001413 amino acids Chemical group 0.000 description 44
- 235000001014 amino acid Nutrition 0.000 description 36
- 125000003412 L-alanyl group Chemical class [H]N([H])[C@@](C([H])([H])[H])(C(=O)[*])[H] 0.000 description 32
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 30
- 108010073385 Fibrin Proteins 0.000 description 28
- 102000009123 Fibrin Human genes 0.000 description 28
- 229950003499 fibrin Drugs 0.000 description 28
- 210000004027 cell Anatomy 0.000 description 26
- 230000035772 mutation Effects 0.000 description 26
- 102000013566 Plasminogen Human genes 0.000 description 23
- 108010051456 Plasminogen Proteins 0.000 description 23
- 102000010911 Enzyme Precursors Human genes 0.000 description 22
- 108010062466 Enzyme Precursors Proteins 0.000 description 22
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical group O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 17
- 230000004913 activation Effects 0.000 description 17
- 108090000623 proteins and genes Proteins 0.000 description 16
- 108010088842 Fibrinolysin Proteins 0.000 description 15
- 229940012957 plasmin Drugs 0.000 description 15
- 108010038061 Chymotrypsinogen Proteins 0.000 description 13
- 125000002066 L-histidyl group Chemical group [H]N1C([H])=NC(C([H])([H])[C@](C(=O)[*])([H])N([H])[H])=C1[H] 0.000 description 12
- 239000012634 fragment Substances 0.000 description 12
- 230000014509 gene expression Effects 0.000 description 12
- 238000000034 method Methods 0.000 description 12
- 238000006467 substitution reaction Methods 0.000 description 12
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 11
- 230000003993 interaction Effects 0.000 description 11
- 238000002741 site-directed mutagenesis Methods 0.000 description 11
- 108090000317 Chymotrypsin Proteins 0.000 description 10
- 241000588724 Escherichia coli Species 0.000 description 10
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 10
- 229960002376 chymotrypsin Drugs 0.000 description 10
- 239000013598 vector Substances 0.000 description 10
- 108091034117 Oligonucleotide Proteins 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- 108050006955 Tissue-type plasminogen activator Proteins 0.000 description 9
- 108010027252 Trypsinogen Proteins 0.000 description 9
- 102000018690 Trypsinogen Human genes 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 235000018102 proteins Nutrition 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- 230000027455 binding Effects 0.000 description 8
- 229940035893 uracil Drugs 0.000 description 8
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 7
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 7
- 235000004279 alanine Nutrition 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 229960000485 methotrexate Drugs 0.000 description 7
- 238000003199 nucleic acid amplification method Methods 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 230000015556 catabolic process Effects 0.000 description 6
- 238000006731 degradation reaction Methods 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 108091005804 Peptidases Proteins 0.000 description 5
- 102000035195 Peptidases Human genes 0.000 description 5
- 239000004365 Protease Substances 0.000 description 5
- 108090000631 Trypsin Proteins 0.000 description 5
- 102000004142 Trypsin Human genes 0.000 description 5
- 230000003321 amplification Effects 0.000 description 5
- 230000002255 enzymatic effect Effects 0.000 description 5
- 239000003527 fibrinolytic agent Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 230000000087 stabilizing effect Effects 0.000 description 5
- 229960000103 thrombolytic agent Drugs 0.000 description 5
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 4
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- 108091028043 Nucleic acid sequence Proteins 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- 108010022999 Serine Proteases Proteins 0.000 description 4
- 102000012479 Serine Proteases Human genes 0.000 description 4
- 108010022394 Threonine synthase Proteins 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 108020001096 dihydrofolate reductase Proteins 0.000 description 4
- MUCZHBLJLSDCSD-UHFFFAOYSA-N diisopropyl fluorophosphate Chemical compound CC(C)OP(F)(=O)OC(C)C MUCZHBLJLSDCSD-UHFFFAOYSA-N 0.000 description 4
- 108010068611 fibrin fragment E-2 Proteins 0.000 description 4
- 229960005051 fluostigmine Drugs 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- 239000013615 primer Substances 0.000 description 4
- 230000001737 promoting effect Effects 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 239000012588 trypsin Substances 0.000 description 4
- 210000005253 yeast cell Anatomy 0.000 description 4
- 101150074155 DHFR gene Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000003146 anticoagulant agent Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 102000004419 dihydrofolate reductase Human genes 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 230000001939 inductive effect Effects 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000000717 retained effect Effects 0.000 description 3
- 239000001488 sodium phosphate Substances 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 3
- 241000024188 Andala Species 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 241000699802 Cricetulus griseus Species 0.000 description 2
- 230000006820 DNA synthesis Effects 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 208000005189 Embolism Diseases 0.000 description 2
- 108010013369 Enteropeptidase Proteins 0.000 description 2
- 102100029727 Enteropeptidase Human genes 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- 208000001435 Thromboembolism Diseases 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 238000005094 computer simulation Methods 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000208 fibrin degradation product Substances 0.000 description 2
- 230000003480 fibrinolytic effect Effects 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 230000008863 intramolecular interaction Effects 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000003239 susceptibility assay Methods 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 230000001131 transforming effect Effects 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- MXHRCPNRJAMMIM-SHYZEUOFSA-N 2'-deoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-SHYZEUOFSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 108010039627 Aprotinin Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXXGVUVQWQGGIG-YUMQZZPRSA-N Glu-Gly-Arg Chemical compound OC(=O)CC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCCN=C(N)N PXXGVUVQWQGGIG-YUMQZZPRSA-N 0.000 description 1
- LLEUXCDZPQOJMY-AAEUAGOBSA-N Glu-Trp Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@H](CCC(O)=O)N)C(O)=O)=CNC2=C1 LLEUXCDZPQOJMY-AAEUAGOBSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 206010062713 Haemorrhagic diathesis Diseases 0.000 description 1
- 108091027305 Heteroduplex Proteins 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 240000002853 Nelumbo nucifera Species 0.000 description 1
- 235000006508 Nelumbo nucifera Nutrition 0.000 description 1
- 235000006510 Nelumbo pentapetala Nutrition 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 239000012506 Sephacryl® Substances 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 1
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 1
- 102000006943 Uracil-DNA Glycosidase Human genes 0.000 description 1
- 108010072685 Uracil-DNA Glycosidase Proteins 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229960004405 aprotinin Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N aspartic acid group Chemical group N[C@@H](CC(=O)O)C(=O)O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 150000001510 aspartic acids Chemical class 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- PXXJHWLDUBFPOL-UHFFFAOYSA-N benzamidine Chemical compound NC(=N)C1=CC=CC=C1 PXXJHWLDUBFPOL-UHFFFAOYSA-N 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 108091092356 cellular DNA Proteins 0.000 description 1
- 239000013000 chemical inhibitor Substances 0.000 description 1
- BULLHNJGPPOUOX-UHFFFAOYSA-N chloroacetone Chemical compound CC(=O)CCl BULLHNJGPPOUOX-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- MXHRCPNRJAMMIM-UHFFFAOYSA-N desoxyuridine Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 238000001641 gel filtration chromatography Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 208000031169 hemorrhagic disease Diseases 0.000 description 1
- 235000014304 histidine Nutrition 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- -1 hydrogen ions Chemical class 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 1
- 238000012933 kinetic analysis Methods 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 108010064755 lys-plasmin Proteins 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000033607 mismatch repair Effects 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000000302 molecular modelling Methods 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 150000003839 salts Chemical group 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000006152 selective media Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 230000002047 thymogen Effects 0.000 description 1
- 108010014252 thymogen Proteins 0.000 description 1
- 229960000187 tissue plasminogen activator Drugs 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 230000003462 zymogenic effect Effects 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21073—Serine endopeptidases (3.4.21) u-Plasminogen activator (3.4.21.73), i.e. urokinase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6456—Plasminogen activators
- C12N9/6459—Plasminogen activators t-plasminogen activator (3.4.21.68), i.e. tPA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6456—Plasminogen activators
- C12N9/6462—Plasminogen activators u-Plasminogen activator (3.4.21.73), i.e. urokinase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21069—Protein C activated (3.4.21.69)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Veterinary Medicine (AREA)
- Enzymes And Modification Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Peptides Or Proteins (AREA)
- Compounds Of Unknown Constitution (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
【課題】治療的用量で投与された場合に、望ましくない上記の非特異的かつ全身的なプラスミノーゲン活性化を開始させにくい、あるいはさせない、pro−UK変異種を開発する。
【解決手段】ネイティブなプロ−ウロキナーゼ(pro−UK)のアミノ酸配列を必須としてなる血栓融解活性のあるpro−UK変異種であって、位置300のリジンがヒスチジンになっており、患者に投与された場合にネイティブなpro−UKよりもフィブリノーゲン溶解および非特異的プラスミノーゲン活性化を誘導しないものであるpro−UK変異種
【選択図】なし
【解決手段】ネイティブなプロ−ウロキナーゼ(pro−UK)のアミノ酸配列を必須としてなる血栓融解活性のあるpro−UK変異種であって、位置300のリジンがヒスチジンになっており、患者に投与された場合にネイティブなpro−UKよりもフィブリノーゲン溶解および非特異的プラスミノーゲン活性化を誘導しないものであるpro−UK変異種
【選択図】なし
Description
本発明は、血栓融解治療に有用な新たな形態のプロウロキナーゼに関する。
プロ−ウロキナーゼ(pro-UK)は1本鎖形態のセリンプロテアーゼ前駆体であり、プラスミンにより活性化されて2本鎖ウロキナーゼ(UK)を形成する。pro−UKおよびUKはともにチモーゲンであるプラスミノーゲンを活性化し、あるいは、フィブリンのクロットに包含される一連の血漿蛋白を分解する活性酵素であるプラスミンに変換する。したがって、pro−UKおよびUKはともに血栓閉栓症の治療に使用されている。
かかる治療により引き起こされうるある種の望ましくない副作用が存在する。pro−UKおよびUKはともにフィブリン分解、フィブリノーゲン溶解(フィブリノーゲノリシス)および血漿板ならびに血管壁のある部分の分解を導く非特異的プラスミノーゲン活性化、そして出血性素質を引き起こしうる。pro−UKはより選択的、すなわち、UKよりも低用量でもプラスミノゲン活性化に特異的である。なぜなら、pro−UKはフィブリン結合プラスミノゲンのみを活性化し、一方、UKはすべてのプラスミノーゲンを活性化するからである。しかしながら、低用量におけるpro−UKの特異性は、血栓融解効果に必要とされる高用量で投与されている場合には失われる可能性がある。
pro−UKは、「不活性」なチモーゲンおよび「活性のある」酵素の双方に似たある種の特性を有する。一方、pro−UKのチモーゲン特性は、血漿中での不活性な挙動、ドデシル硫酸ナトリウムに対して安定な阻害剤複合体を形成しないこと、およびジイソプロピルフルオロホスフェート(DFP)またはGlu−Gly−Arg、あるいはUKに対する有効な化学阻害剤クロロメチルケトンによる阻害に対する抵抗性を包含する。pro−UKはまた、UKよりも200倍低いプラスミノーゲン活性化活性を有する。
他方、pro−UKの酵素特性は、合成基質およびプラスミノーゲンに対する測定可能な本来的活性であり、それは、トリプシノーゲンまたはキモトリプシンのごとき他のセリンプロテアーゼチモーゲンよりも104.0〜4.3倍高い。pro−UKはまた、プラスミノーゲンに対してUKよりも低いミカエリス定数(KM)を有し、フィブリンフラグメントE2の存在下においては、pro−UKは、UK形態へと活性化されなくても、プラスミノーゲンに対して十分な活性があることが示されている。
血栓融解剤として使用可能であるにもかかわらず、治療的用量で投与された場合には、pro−UKの高い本来的な酵素活性は、望ましくない上記の非特異的かつ全身的なプラスミノーゲン活性化を開始させる。結果として、pro−UKでの血栓融解治療は、やはり有害な副作用に関連しているといえる。
治療的用量で投与された場合に、望ましくない上記の非特異的かつ全身的なプラスミノーゲン活性化を開始させにくい、あるいはさせない、pro−UK変異種を開発することが本発明の解決課題であった。
本発明者らは上記課題を解決せんと鋭意研究を重ね、ネイティブなプロ−ウロキナーゼ(pro−UK)のアミノ酸配列を必須としてなるpro−UK変異種であって、位置300のリジンがヒスチジンになっているpro−UK変異種が上記課題を解決するものであることを見出し、本発明を完成するに至った。
すなわち、本発明は、
(1)ネイティブなプロ−ウロキナーゼ(pro−UK)のアミノ酸配列を必須としてなる血栓融解活性のあるpro−UK変異種であって、位置300のリジンがヒスチジンになっており、患者に投与された場合にネイティブなpro−UKよりもフィブリノーゲン溶解および非特異的プラスミノーゲン活性化を誘導しないものであるpro−UK変異種;
(2)(1)記載のpro−UK変異種をコードしているDNA分子;
(3)(2)記載の組み換え型DNA分子で形質転換された細胞;
(4)ネイティブなpro−UKよりも出血性合併症を引き起こさないものである(1)記載のpro−UK変異種;
(5)ネイティブなpro−UKのアミノ酸配列からなるpro−UK変異種であって、位置300のリジンがヒスチジンになっている(1)記載のpro−UK変異種;
(6)(5)記載のpro−UK変異種をコードしているDNA分子;
(7)(6)記載の組み換え型DNA分子で形質転換された細胞
を提供するものである。
(1)ネイティブなプロ−ウロキナーゼ(pro−UK)のアミノ酸配列を必須としてなる血栓融解活性のあるpro−UK変異種であって、位置300のリジンがヒスチジンになっており、患者に投与された場合にネイティブなpro−UKよりもフィブリノーゲン溶解および非特異的プラスミノーゲン活性化を誘導しないものであるpro−UK変異種;
(2)(1)記載のpro−UK変異種をコードしているDNA分子;
(3)(2)記載の組み換え型DNA分子で形質転換された細胞;
(4)ネイティブなpro−UKよりも出血性合併症を引き起こさないものである(1)記載のpro−UK変異種;
(5)ネイティブなpro−UKのアミノ酸配列からなるpro−UK変異種であって、位置300のリジンがヒスチジンになっている(1)記載のpro−UK変異種;
(6)(5)記載のpro−UK変異種をコードしているDNA分子;
(7)(6)記載の組み換え型DNA分子で形質転換された細胞
を提供するものである。
治療的用量で投与された場合に、pro−UKの高い本来的な酵素活性を保持しつつも、望ましくない非特異的かつ全身的なプラスミノーゲン活性化を開始させにくい、あるいはさせないpro−UK変異種が提供される。
本発明は、天然に存在するネイティブなpro−UKの望ましくない本来的かつ非特異的な酵素活性の低下を示すが、フィブリン(フラグメントYまたはE2)により促進される(活性化される)能力においてネイティブなpro−UKと少なくとも同程度に有効で、プラスミノーゲンをプラスミンに変換し、2本鎖UKへの変換後も血栓融解活性を保持する変異種形態のpro−UKを設計できることを見いだしたことに基づく。結果として、これらのpro−UK変異種は、患者に投与した場合、非特異的プラスミノーゲン活性化および出血性合併症がネイテイブなpro−UKよりも抑えられる。
これらのpro−UK変異種は、フィブリンによってネイティブなpro−UKと同程度に促進されるため、ネイティブなpro−UKと比較すると血栓融解剤として優れており、それゆえ、同じフィブリン溶解効率を有するが、それらは血漿中では真に不活性であるためネイテイブなpro−UKよりもずっと高いフィブリン結合プラスミノーゲンに対する特異性を有する。この不活性な挙動の結果、これらのpro−UK変異種を、ネイティブなpro−UKより多量、より有効な用量で患者に投与することができる。
したがって、本発明は、pro−UK変異種がネイティブなpro−UKよりもフィブリノーゲン溶解および非特異的プラスミノーゲン活性化を誘導しないようにする変異を包含するネイティブなpro−UKのアミノ酸配列を有する血栓融解活性のあるpro−UK変異種を特徴とする。好ましくは、変異は、通常には、ネイティブなpro−UKのアミノ酸配列におけるネイティブなアミノ酸の新しいアミノ酸への置換であり、その変異により、pro−UK変異種は、ネイティブなpro−UKよりも少なくとも10倍低い本来の活性を有し、患者に投与した場合、ネイティブなpro−UKと比較して、プラスミン活性化後に実質的に同じフィブリンでの促進活性および血栓融解活性を有するのである。
本明細書の用語「ネイティブ」は、天然に存在する形態もしくは野生型の蛋白、あるいは天然に存在する蛋白におけるアミノ酸を意味する。「新しい」アミノ酸は、ネィテイブな蛋白中においてはその位置に通常は存在しないアミノ酸である。本明細書の用語「変異」は、1個のネイティブなアミノ酸のただ1個の新しいアミノ酸への置換、または2個またはそれ以上のネイティブなアミノ酸の2個またはそれ以上の新しいアミノ酸への置換を包含する。
詳細には、変異は、通常には、ネイティブなpro−UKの柔軟性のあるループ(アミノ酸297〜313)における1個のネイティブなアミノ酸の1個の新しいアミノ酸への置換であってもよい。例えば、新しいアミノ酸は、Lys300、Gly299、および/またはGlu301の、例えばアラニンまたはヒスチジンへの置換であってもよい。さらに、新しいアミノ酸は、Lys313の、例えばアラニンまたはヒスチジンへの置換、あるいはTyr306の、例えばグリシンへの置換であってもよい。
さらに、変異は、Ala175の新しいアミノ酸への置換、およびTyr187の第2の新しいアミノ酸への置換であって、pro−UK変異種においてチモーゲン3回転軸(zymogen triad)を形成するものであってもよい。例えば、新しいアミノ酸はセリンであってもよく、第2の新しいアミノ酸はヒスチジンであってもよい。本発明はさらに、チモーゲン3回転軸を形成する2つの変異の誘導のごとき変異の組み合わせを特徴とし、さらにLys300の第3の新しいアミノ酸への置換およびGlu301の第4の新しいアミノ酸への置換を包含する。かかつ変異種は、例えば、新しいアミノ酸としてセリン、第2の新しいアミノ酸としてヒスチジン、第3の新しいアミノ酸としてアラニンまたはヒスチジン、そして第4の新しいアミノ酸としてアラニンを有するであろう。
さらに本発明は、DNA分子、例えば、本明細書記載のpro−UK変異種のいずれかをコードする組み換えDNA分子を特徴とする。かかる変異種は、単一、2個、または多数の置換を含むことができる。また本発明は、かかるDNA分子で形質転換された細胞、例えば、哺乳動物、細菌または酵母細胞を包含する。また本発明は、pro−UK変異種をコードしているDNA分子で細胞を形質転換し、その細胞を培養して変異種を発現させ、次いで、その変異種を単離することによる、本明細書記載のpro−UK変異種の製造方法を特徴とする。
さらに、本発明は、血栓融解量の本明細書記載のpro−UK変異種のいずれかを患者に投与することを特徴とする患者における血栓閉栓症の治療方法を特徴とする。本明細書の用語「血栓融解量」は、全身的な出血を誘導することなく、患者におけるフィブリンクロットを溶解するpro−UK変異種の量である。
pro−UK変異体の開発
UKおよびpro−UKの3次元(「3−D」)構造はまだ決定されていない。しかしながら、pro−UK/UKのB−鎖(プロテアーゼのドメインである)は、X線結晶回折により3−D構造がよく知られているトリプシンならびにキモトリプシン、およびそれらの各前駆体であるトリプシノーゲンならびにキモトリプシノーゲンと有意に相同的である。これらの相同性は30%より高いので、これらの分子と並べた場合のpro−UK/UKの骨格は、コア残基に関して約1.0Åまたはそれより小さい。それゆえ、pro−UK/UKの予想構造は信頼できる。
UKおよびpro−UKの3次元(「3−D」)構造はまだ決定されていない。しかしながら、pro−UK/UKのB−鎖(プロテアーゼのドメインである)は、X線結晶回折により3−D構造がよく知られているトリプシンならびにキモトリプシン、およびそれらの各前駆体であるトリプシノーゲンならびにキモトリプシノーゲンと有意に相同的である。これらの相同性は30%より高いので、これらの分子と並べた場合のpro−UK/UKの骨格は、コア残基に関して約1.0Åまたはそれより小さい。それゆえ、pro−UK/UKの予想構造は信頼できる。
アミノ酸配列に関する「相同性」は、2個またはそれ以上のアミノ酸配列間の類似性または同一性のパーセントをいう。ある2個の蛋白の相同性を、例えば、配列分析ソフトウェア(例えば、ウィスコンシン州マディソン(Madison)のウィスコンシン大学のザ・シークエンス・アナリシス・ソフトウェア・パッケージ・オブ・ザ・ジェネティクス・コンピューター・グループ(the Sequence Analysis Software Package of the Genetics Computer Group))により決定することができる。相同性のパーセント値は、正確な合致ならびに保存的アミノ酸置換を伴う配列に割り当てられる。ここに、保存的アミノ酸置換とは、例えば、1のアミノ酸の別の同様なアミノ酸での置換であり、得られる蛋白の特性および活性にわずかしか変化を与えないかまたは変化を与えない。
トリプシン(トリプシノーゲン)およびキモトリプシン(キモトリプシノーゲン)に対するpro−UK/UKの構造上の相同性に基づいてpro−UK変異種が設計され、pro−UKのフィブリン特異性が、Glu−プラスミノーゲンにおける特別なコンホーメーション変化を誘導するフィブリンフラグメントE2による選択的促進によるものであることが見いだされた(リウ(Liu)およびグレビッチ(Gurewich),バイオケミストリー(Biochemistry),第31巻:6311〜6317頁(1992年))。pro−UK変異種を設計するために、出願人は、キモトリプシノーゲンおよびキモトリプシンそれぞれのポリペプチド骨格に関する空間座標に基づいて、さらにエラスターゼおよびトリプシン(トリプシノーゲン)の構造を加味して、pro−UK/UKのプロテアーゼドメイン(B鎖)の3−D構造のコンピューターモデルを開発した。さらに出願人は、2種の分子モデリングプログラム、CHARMM(高分子のエネルギーの最小化および力学の計算用のプログラムであり、ハーバード大学化学科により開発され、ブルックス(Brooks)ら,ジャーナル・オブ・コンピュ・ケミ(J.Comp.Chem.),第4巻:187〜217頁(1983年)に記載されている)、ならびに米国カリフォルニア州サンフランシスコのシリコン・グラフィックス・インコーポレイテッド(Sillicon Graphics,Inc.)により開発されたQUANTARを使用した。
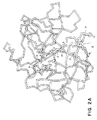
図1、2a、および2bに示すように、このモデルは、pro−UKのB鎖(プロテアーゼドメイン)の3次元構造を明確に示す。当該モデルは、キモトリプシン(キモトリプシノーゲン)の構造と類似のβ−バーレル構造(β-barrel structure)である2個のサブドメインにより構築されており、以下の特徴を有する。このB鎖における各原子の空間座標が十分に決定されている。分子表面上のIle159は、A鎖のペプチド130〜158と共有結合している。pro−UKの「活性化ドメイン」は、3つの配列(位置297〜313、344〜353、および376〜383のアミノ酸)からなる。配列297〜313は、キモトリプシノーゲンまたはトリプシノーゲンの同様のループよりも柔軟性が小さいがやはり「ぐらついて」いる「柔軟性のあるループ」(図2aおよび2bの青い部分)を形成する。柔軟性のあるループがあるコンホーメーションに存在する場合、ループ中のLys300のε−位の正に荷電したアミノ基がAsp355の負に荷電したカルボキシル基と相互作用する。この相互作用は、活性部位であるSer356を、他の2つの主要活性部位の残基を含んでいる電荷中継システム内での相互作用にとり最も好ましい位置にSer356のヒドロキシル基が存在するという理想的な位置のごく近傍に引き付ける。すなわち、Lys313も別方向からAsp355と相互作用するのである。それゆえ、この間にpro−UKドメインの活性部位および基質結合ポケットが、2本鎖形態への変換を伴わずに優先的に形成される。しかしながら、この「活性コンホーメーション」になる確率はわずか0.5%であるため、開裂されていない1本鎖のpro−UKは、活性のある2本鎖形態の活性のうちの約0.5%しか有していない。
図2cおよび3に示すように、2本鎖形態においては、開裂されたIle159の新しいアミノ基はAsp355とともに安定な「塩ブリッジ」を形成し、これにより活性化ドメインが安定化され、分子が十分に活性化される。これとは別に、フラグメントE2結合プラスミノーゲンからの相互作用のごとき外力が「柔軟性のあるループ」(図2cの黄)の活性コンホーメーションを安定化させる場合には、開裂されていない1本鎖形態のpro−UKも十分に活性となるであろう。
さらに、pro−UKのモデルは、キモトリプシノーゲンのいわゆる「チモーゲン3回転軸」("zymogen triad")(Asp194、His40およびSer32により形成される水素結合ネットワーク)を含んでいない。なぜなら、この3回転軸はネイティブなpro−UKには存在しないからである。キモトリプシン3回転軸のHis40およびSer32は、pro−UKにおいてはそれぞれTyr187およびAla175に置換されている。「チモーゲン3回転軸」は、キモトリプシノーゲンの活性部位(酸素ホール)の領域の不活性コンホーメーションを安定化するのに重要である。
このモデルを用いて、出願人は、pro−UKおよびUKのプロテアーゼドメインにおける特異的アミノ酸置換を選択、モデリングして特別なpro−UK変異種を設計し、次いで、これらの変異種のうちのあるものを構築し、発現させ、特徴づけた。非特異的プラスミノーゲン活性化の低下および十分に保持された血栓融解活性という所望の目的を実現する変異種の4つのクラスを以下に記載する。
pro−UKのLys300の部位特異的突然変異
X線結晶回折を用いる研究は、なぜ大部分のプロテアーゼの1本鎖チモーゲン形態が「活性なし」であるのか、そして、いかにしてただ1つの結合の開裂により十分な活性のある酵素の形成が誘導されるのかについての説明の助けとなった。しかしながら、pro−UKは、他の大部分のチモーゲンよりも大きい本来的活性を有しているが、1本鎖組織プラスミノーゲンアクチベーター(SC−t−PA)よりは活性が小さい。とりわけ、トリプシノーゲンおよびキモトリプシノーゲンはそれぞれの活性化された2本鎖酵素形態の活性のわずか約10−6倍の活性しか有していない。SC−t−PAは2本鎖t−PAの活性の約10−1.5倍の活性を有する。pro−UKはその2本鎖誘導体、すなわちUKの活性の約10−2.3倍の活性を有する。
X線結晶回折を用いる研究は、なぜ大部分のプロテアーゼの1本鎖チモーゲン形態が「活性なし」であるのか、そして、いかにしてただ1つの結合の開裂により十分な活性のある酵素の形成が誘導されるのかについての説明の助けとなった。しかしながら、pro−UKは、他の大部分のチモーゲンよりも大きい本来的活性を有しているが、1本鎖組織プラスミノーゲンアクチベーター(SC−t−PA)よりは活性が小さい。とりわけ、トリプシノーゲンおよびキモトリプシノーゲンはそれぞれの活性化された2本鎖酵素形態の活性のわずか約10−6倍の活性しか有していない。SC−t−PAは2本鎖t−PAの活性の約10−1.5倍の活性を有する。pro−UKはその2本鎖誘導体、すなわちUKの活性の約10−2.3倍の活性を有する。
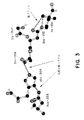
セリンプロテアーゼチモーゲンの活性酵素への活性化の主要な態様は、チモーゲンの新しいアミノ末端α−アミノ基のチモーゲンの表面ポケット中への移動であり、そこで当該アミノ基が、蛋白の活性部位のセリンに配列において隣接しているアスパラギン酸とともにいわゆる「塩ブリッジ」を形成しうる。この場合の配列を図式的に図3に示す。塩ブリッジは、2本鎖UKの新しいアミノ末端(Ile159)またはpro−UKにおける代用物とアスパラギン酸残基(Asp355)の側鎖の負に荷電したカルボキシル基との間に形成されるイオン結合または静電気的相互作用である。
この塩ブリッジ形成により、触媒の鍵となるものであり、His、AspおよびSer(図式2a〜2c(赤)、および図3)の3つの主要な活性部位残基により形成される荷電遅延またはプロトン輸送システム内での相互作用にとり最も好ましい新たな位置へのセリンのヒドロキシル基の回転を生じる。さらに塩ブリッジの形成は、3つの配列(297〜313、344〜353、および376〜383)からなる、pro−UKのいわゆる「活性化ドメイン」を安定化する。
不活性チモーゲンであるトリプシノーゲンおよびキモトリプシノーゲンにおいては、これらの活性化ドメインはX線結晶回折ではイメージ化されず、そのことはそれらが空間内に固定されていないのではなく、むしろぐらついていることが原因であるとされている。これらのドメインは基質結合ポケットを取り囲んであり、そのことは、基質に対してチモーゲンが結合しないことの説明となりうる。活性化された酵素であるトリプシンおよびキモトリプシンにおいては、これらのドメインは、ヒューバー(Huber)ら,アカウント・オブ・ケミカル・リサーチ(Acct.Chem.Res.),第11巻:114〜122頁(978年)に記載のごとくX線結晶回折において明らかに可視化される。
ヘッケル(Heckel)ら,ジャーナル・コンピュ・エイディド・モレ・デス(J.Comp.Aided.Mol.Des.),第2巻:7〜14頁(1988年)およびペーターソン(Peterson)ら,バイオケミストリー(Biochem.),第29巻:3451〜4357頁(1990年)に記載されているように、SC−t−PAにおいては、Lys416(t−PAにおける番号づけによる)のε−アミノ基の正電荷がAsp477(t−PAの番号づけによる)との代用塩ブリッジ効果を提供して、活性部位のSer478(t−PAにおける番号づけによる)を理想的な位置のごく近傍へと引き付けると思われる。pro−UKにおいては、SC−t−PAにおけるLys416と同じ位置にLys300(pro−UKにおける番号づけによる)が存在する。pro−UKのLys300の正に荷電したε−アミノ基は、UKの新しい末端α−アミノ基(Ile159の)の代用物となることが要求される。しかしながら、その影響を弱めると思われる隣接したGlu301が存在する。さらにAsn302残基におけるグリコシレーション部位ならびに近傍のSer303残基におけるリン酸化部位も存在する。この配置は、pro−UKの高い本質的活性(キモトリプシノーゲンと比べて)の部分的な提供(SC−t−PAと比べて)の説明となりうる。
これらの考慮に基づいて、出願人は、Lys300および/またはその周囲の残基がいくつかの異なるタイプのアミノ酸に変異して、この位置とAsp355(pro−UKにおける番号づけ)との間の電気的相互作用を調節している変異種であって、本来的活性ができるかぎり低下しているがフィブリンでの促進能およびネイティブなUKの2本鎖形態の活性を保持している一群の変異種を分類した。この変異株のグループを、以下の1から5までの番号を付したpro−UK変異種により説明する。部位特異的突然変異法および下記の遺伝子発現によりこれらの変異種を作り出した。
以下に実施例を示して本発明をさらに詳細かつ具体的に説明するが、実施例は本発明を限定するものと解すべきではない。
変異種1:Lys300→Ala
中性アミノ酸であるアラニン(Ala)をpro−UKのLys300と置換したが、その置換は、分子の折り畳みには影響しないが位置300とAsp355との間の塩ブリッジを除去して本来的活性を減少させるように設計された。コンピューターモデル上で設計されるとすぐに、この変異種を作成し、下記のごとく試験した。結果は、この変異種は測定可能な本来的活性をほとんど有しておらず、それゆえ、血漿中においてきわめて安定であり、10mg/mlよりも高濃度で24時間にわたりインキュベーションしてもフィブリノーゲン分解を誘導しなかった。この安定性は、ネイテイブなpro−UKがインビトロにおいて血漿フィブリノーゲンの50%の分解を誘導した濃度よりも少なくとも10倍高かった。この変異種の2本鎖形態の活性(プラスミン活性化後)は、フィブリンフラグメントE2によるその促進(pro−UKの促進の40%)と同様に低下した(UKの活性の33%)。
中性アミノ酸であるアラニン(Ala)をpro−UKのLys300と置換したが、その置換は、分子の折り畳みには影響しないが位置300とAsp355との間の塩ブリッジを除去して本来的活性を減少させるように設計された。コンピューターモデル上で設計されるとすぐに、この変異種を作成し、下記のごとく試験した。結果は、この変異種は測定可能な本来的活性をほとんど有しておらず、それゆえ、血漿中においてきわめて安定であり、10mg/mlよりも高濃度で24時間にわたりインキュベーションしてもフィブリノーゲン分解を誘導しなかった。この安定性は、ネイテイブなpro−UKがインビトロにおいて血漿フィブリノーゲンの50%の分解を誘導した濃度よりも少なくとも10倍高かった。この変異種の2本鎖形態の活性(プラスミン活性化後)は、フィブリンフラグメントE2によるその促進(pro−UKの促進の40%)と同様に低下した(UKの活性の33%)。
変異種2:Lys300→His
第2の変更として、pro−UKモデルのLys300をヒスチジン(His)に置換した。ヒスチジンは水素イオンを獲得することができ、6.5より低いpHにおいては正に荷電するが、リジンのε−アミノ基の電荷ほど強くない。pro−UKモデルにおいて、His300のイミダゾール基は、外部よりも低いエネルギー状態において、分子内部に包み込まれる。なぜなら、イミダゾール基はやや疎水的であるからである。このことにより、イミダゾール基はAsp355に向いているために正電荷を有することができ、局部的に環境が酸性となる。それゆえ、弱い塩ブリッジがAsp355とHis300との間に存在し、His300は弱いLys300のように挙動することとなる。Hisの静電気的荷電状況は局部的環境に依存し、さらに上記のごとくHis300が柔軟性のあるループ領域(297〜313)中に存在するので、モジュレーター(フラグメントE2)はやはりpro−UKの本来的活性を調節する。Lys300→AlaのデータおよびLysと比較した場合のHisの特性に基づくと、変異種2の本来的活性もまた、ネイティブなpro−UKの本来的活性よりも10倍低いはずであるが、変異種1の本来的活性よりは高いはずである。しかしながら、2本鎖形態の活性およびフィブリンでの促進能はともに保持されるはずであり、それらは変異種1よりは高いはずであるが、ネイテイブなpro−UKよりは低いはずである。
第2の変更として、pro−UKモデルのLys300をヒスチジン(His)に置換した。ヒスチジンは水素イオンを獲得することができ、6.5より低いpHにおいては正に荷電するが、リジンのε−アミノ基の電荷ほど強くない。pro−UKモデルにおいて、His300のイミダゾール基は、外部よりも低いエネルギー状態において、分子内部に包み込まれる。なぜなら、イミダゾール基はやや疎水的であるからである。このことにより、イミダゾール基はAsp355に向いているために正電荷を有することができ、局部的に環境が酸性となる。それゆえ、弱い塩ブリッジがAsp355とHis300との間に存在し、His300は弱いLys300のように挙動することとなる。Hisの静電気的荷電状況は局部的環境に依存し、さらに上記のごとくHis300が柔軟性のあるループ領域(297〜313)中に存在するので、モジュレーター(フラグメントE2)はやはりpro−UKの本来的活性を調節する。Lys300→AlaのデータおよびLysと比較した場合のHisの特性に基づくと、変異種2の本来的活性もまた、ネイティブなpro−UKの本来的活性よりも10倍低いはずであるが、変異種1の本来的活性よりは高いはずである。しかしながら、2本鎖形態の活性およびフィブリンでの促進能はともに保持されるはずであり、それらは変異種1よりは高いはずであるが、ネイテイブなpro−UKよりは低いはずである。
変異種3:Gly299→His/Lys300→Ala
pro−UKモデルにおいて、pro−UK主鎖のGly299のカルボニル基の酸素がAsp355と水素結合を形成する。かかる結合の形成はキモトリプシノーゲンおよびトリプシノーゲンにも存在する。モデルにおいてGly299をHisに置換した場合、His299とAsp355との間に強い塩ブリッジが形成され、それはHis300とAsp355との間のブリッジよりも強力であり、それゆえ、このブリッジの形成が防止される。さらに、His299の局部的環境は、活性部位、基質結合ポケット、または他の内部領域の変化によってよりも、pro−UKの柔軟性のあるループによって影響を受けにくい。対照的に、His300変異種(変異種2)は逆の挙動をするはずであり、すなわち、ループ領域のコンホーメーション変化によって、より影響を受けるはずである。Glyと比較した場合のHisの特性に基づくと、変異種3の本来的活性および2本鎖形態の活性は変異種2の2倍よりも高いはずである。しかしながら、そのフィブリンでの促進能は変異種2よりも20%以上低いはずであり、変異種1よりも高いはずである。
pro−UKモデルにおいて、pro−UK主鎖のGly299のカルボニル基の酸素がAsp355と水素結合を形成する。かかる結合の形成はキモトリプシノーゲンおよびトリプシノーゲンにも存在する。モデルにおいてGly299をHisに置換した場合、His299とAsp355との間に強い塩ブリッジが形成され、それはHis300とAsp355との間のブリッジよりも強力であり、それゆえ、このブリッジの形成が防止される。さらに、His299の局部的環境は、活性部位、基質結合ポケット、または他の内部領域の変化によってよりも、pro−UKの柔軟性のあるループによって影響を受けにくい。対照的に、His300変異種(変異種2)は逆の挙動をするはずであり、すなわち、ループ領域のコンホーメーション変化によって、より影響を受けるはずである。Glyと比較した場合のHisの特性に基づくと、変異種3の本来的活性および2本鎖形態の活性は変異種2の2倍よりも高いはずである。しかしながら、そのフィブリンでの促進能は変異種2よりも20%以上低いはずであり、変異種1よりも高いはずである。
変異種4:Lys300→Ala/Glu301→Ala
正に荷電したLys300を中性のアラニンに置換した場合、Glu301の負電荷は強くなり過ぎてIle159のα−アミノ基をそのAsp355との相互作用から引き離してしまう。このことは、変異種1の活性化された2本鎖形態(UK)の血栓融解活性の低下を引き起こす。それゆえ、この問題を解決するために、Lys300およびGlu301を中性のアラニンに置換することにより二重変異種を設計した。当該モデルおよびLysおよびGluと比較した場合の中性のAlaの特性に基づくと、変異種4は変異種1とほとんど同じ特性を有するはずであるが、変異種1の2本鎖形態の活性よりも2倍高い2本鎖形態の活性を有するはずである。
正に荷電したLys300を中性のアラニンに置換した場合、Glu301の負電荷は強くなり過ぎてIle159のα−アミノ基をそのAsp355との相互作用から引き離してしまう。このことは、変異種1の活性化された2本鎖形態(UK)の血栓融解活性の低下を引き起こす。それゆえ、この問題を解決するために、Lys300およびGlu301を中性のアラニンに置換することにより二重変異種を設計した。当該モデルおよびLysおよびGluと比較した場合の中性のAlaの特性に基づくと、変異種4は変異種1とほとんど同じ特性を有するはずであるが、変異種1の2本鎖形態の活性よりも2倍高い2本鎖形態の活性を有するはずである。
変異種5:Lys300→His/Glu301→Ala
この変異種は変異種2と変異種4の組み合わせである。pro−UKの変異種1のAla300の特性に基づくと、正に荷電した残基(300)は1本鎖の本来的活性および2本鎖形態の活性の双方を促進するのに重要である。それゆえ、この二重変異種である変異種5は、本来的活性およびフィブリンでの促進能においては変異種2と同様であるはずであるが、より高い2本鎖形態の活性を有するはずである。
この変異種は変異種2と変異種4の組み合わせである。pro−UKの変異種1のAla300の特性に基づくと、正に荷電した残基(300)は1本鎖の本来的活性および2本鎖形態の活性の双方を促進するのに重要である。それゆえ、この二重変異種である変異種5は、本来的活性およびフィブリンでの促進能においては変異種2と同様であるはずであるが、より高い2本鎖形態の活性を有するはずである。
変異種1から5までは、いかにしてネイティブなpro−UKと比較した場合の本来的活性、2本鎖形態の活性およびフィブリンでの促進能を調節するかを示すものである。これらの調節は、部位特異的突然変異法および構造モデリングを組み合わせることにより、実際のpro−UK変異種において遂行される。本質的にはこれらの変異種は、(1)フィブリノーゲン分解の減少(低い本来的活性)、(2)フィブリンでの促進が不変(フィブリンでの促進能が保持される)、(3)ネイティブなpro−UKと比較した場合のプラスミン活性化後の活性(2本鎖形態の活性)が同じ、からなる3つの目的を達成するものである。それゆえ、これらの変異種は治療上非常に有用であろう。
pro−UKの柔軟性のあるループの部位特異的突然変異
本来的活性に対して重要なpro−UKの別の構造は、アミノ酸297〜313により形成されるいわゆる「柔軟性のあるループ」であり(図1、図2aおよび2b(青))、それは、すべてのセリンプロテアーゼならびにそれらのチモーゲンにおける、最も歪んでいるかまたはぐらついている領域である。この領域は、酵素/チモーゲン機能に対するある種の変調に関する機構を提供しうる。
本来的活性に対して重要なpro−UKの別の構造は、アミノ酸297〜313により形成されるいわゆる「柔軟性のあるループ」であり(図1、図2aおよび2b(青))、それは、すべてのセリンプロテアーゼならびにそれらのチモーゲンにおける、最も歪んでいるかまたはぐらついている領域である。この領域は、酵素/チモーゲン機能に対するある種の変調に関する機構を提供しうる。
このループはキモトリプシンにおいては開裂されるが、t−PAおよびpro−UKにおいてはLys416/300変異種は本来的活性を有していた。pro−UK/UKモデルに基づいて、このループの明らかな「活性」コンホーメーションを同定した(図2bの青)。それは、ネイティブなpro−UKにおいて、他の種々の「不活性」コンホーメーションと動的平衡にある。「活性」コンホーメーションにおいては、ループの唯一の正に荷電した残基であるLys300は分子内部に対面する位置にあってAsp355と塩ブリッジを形成し、十分に活性のある触媒部位を生じる。対照的に、「不活性」コンホーメーションにおいては、ループ(図2a中、青)は、Lys300の正電荷がAsp355との一直線からはずれるように移動させられており、そのことによりチモーゲンが不活性となる。
pro−UKについて観察された活性は、平衡は不活性コンホーメーション寄りであり、活性コンホーメーションはわずか0.5%であることを示す。該0.5%は、その活性のある2本鎖形態(UK)に対するpro−UKの本来的活性のパーセントに対応する。
初めの研究は、柔軟性のあるループにおいて、3つの負に荷電した残基(Glu301、Asp305、およびGlu310)の側鎖はいつもLys300とは反対側のループの面に存在することを示した。不活性に荷電した残基の側鎖が分子の外側に保持される場合、Lys300のε−アミノ基は高い確率でAsp355と塩ブリッジを形成するであろう。換言すれば、活性コンホーメーションは、3つの負に荷電した残基が分子の外側に保持される場合に好ましいものである。
pro−UKがフラグメントE2結合Glu−プラスミノーゲンと相互作用する場合に、フラグメントE2−Glu−プラスミノーゲン複合体からの外的な静電気力は、これら3つの負に荷電した側鎖(Glu301、Asp305、およびGlu310)を分子から引きつけることにより、活性コンホーメーションを安定化させる。それゆえ、Lys300は分子の内側に対面するように配置され、Asp355と塩ブリッジを形成する。結果として、2本鎖形態へ変換されなくてもpro−UKは十分に活性となる。
ループの柔軟性をさらに増加させうる柔軟性のあるループの部位特異的突然変異は、「活性」コンホーメーションとなる確率を0.5%よりも低下させるであろう。ネイテイブなpro−UKと比較した場合に、この変異種が2本鎖形態への変換後に同様の2本鎖形態の活性を保持し、同様のフィブリンでの促進能を保持するならば、この変異種は、前記の3つの目的を達成する優れた活性剤であろう。かかる変異種は血漿において低い本来的活性を有するが、フィブリン(フラグメントE2)存在下および2本鎖形態においては十分に活性があるであろう。
これらの考えに基づき、出願人は、「活性」コンホーメーションの確率は低いが、フラグメントE2−Glu−プラスミノーゲン複合体による「活性」コンホーメーションへの誘導可能性ならびに2本鎖形態の活性を保持している第2のグループの変異種を分類した。このグループの変異種を、下記の6番のpro−UK変異種により説明する。
変異種6:Tyr306→Gly
pro−UKの本来的活性は、「活性」および[不活性」コンホーメーション間の平衡の機能であるという考えに基づけば、ループの柔軟性の増加は本来的活性を低下させるはずである。それゆえ、ループのβ−ターン構造が破壊されてその柔軟性が増加したTyr306→Gly変異種を設計した。結果として、この変異種は本来的活性が低下しているはずであるが、フラグメントE2による促進、2本鎖形態の活性、およびプラスミン活性化は保存されるはずである。それゆえ、変異種6は良好な治療上の有用性を有するであろう。
pro−UKの本来的活性は、「活性」および[不活性」コンホーメーション間の平衡の機能であるという考えに基づけば、ループの柔軟性の増加は本来的活性を低下させるはずである。それゆえ、ループのβ−ターン構造が破壊されてその柔軟性が増加したTyr306→Gly変異種を設計した。結果として、この変異種は本来的活性が低下しているはずであるが、フラグメントE2による促進、2本鎖形態の活性、およびプラスミン活性化は保存されるはずである。それゆえ、変異種6は良好な治療上の有用性を有するであろう。
pro−UKのLys313の部位特異的突然変異
pro−UKのLys313はUKの新しい末端α−アミノ基のもう1つの代用物である。図1および2bに示すように、Lys313はLys300と同様に挙動して、さらにpro−UKの本来的活性の増加を引き起こすはずであるAsp355との塩ブリッジを形成しうる。Lys313とAsp355との相互作用は、柔軟性のあるループの動きにより調節される。それゆえ、この相互作用の除去は、さらにpro−UKの本来的活性を低下させるであろう。これらの考えに基づき、出願人は、以下の変異種7および8を設計した。
pro−UKのLys313はUKの新しい末端α−アミノ基のもう1つの代用物である。図1および2bに示すように、Lys313はLys300と同様に挙動して、さらにpro−UKの本来的活性の増加を引き起こすはずであるAsp355との塩ブリッジを形成しうる。Lys313とAsp355との相互作用は、柔軟性のあるループの動きにより調節される。それゆえ、この相互作用の除去は、さらにpro−UKの本来的活性を低下させるであろう。これらの考えに基づき、出願人は、以下の変異種7および8を設計した。
変異種7:Lys313→Ala
図1および2bに示すように、Lys313(図2b中、黄)は柔軟性のあるループ(図1の297〜313、図2bにおいて青)の末端に存在し、Asp355に近接している。Lys300と同様に、そのε−アミノ基の正電荷は、UKの新しいα−アミノ基の代用物として作用してLys300とは反対の方向からAsp355と塩ブリッジを形成する。有利なことに、新しい末端アミノ基(Ile159)が生成して2本鎖UKにおいてAsp355とのもう1つの塩ブリッジを競争的に形成する際にこの塩ブリッジは破壊されるので、この変異種は2本鎖UKの活性に影響しない。
図1および2bに示すように、Lys313(図2b中、黄)は柔軟性のあるループ(図1の297〜313、図2bにおいて青)の末端に存在し、Asp355に近接している。Lys300と同様に、そのε−アミノ基の正電荷は、UKの新しいα−アミノ基の代用物として作用してLys300とは反対の方向からAsp355と塩ブリッジを形成する。有利なことに、新しい末端アミノ基(Ile159)が生成して2本鎖UKにおいてAsp355とのもう1つの塩ブリッジを競争的に形成する際にこの塩ブリッジは破壊されるので、この変異種は2本鎖UKの活性に影響しない。
対照的に、Lys300とAsp355との間の電荷の相互作用は2本鎖形態において存在し、2本鎖形態の活性に貢献するので、Lys300は2本鎖形態の活性に影響する。同様に、さらにSC−t−PAの同等のアミノ酸位置にLys残基が存在し、その高い本来的活性に関与していることが示されている。
変異種8:Lys313→His
この変異種においては、Lysよりも弱い正電荷を有するHisを用いてLys313を置換した。上記変異種2に関して記載したのと同様の理論に基づき、この変異種を変異種7の調節体として設計した。
この変異種においては、Lysよりも弱い正電荷を有するHisを用いてLys313を置換した。上記変異種2に関して記載したのと同様の理論に基づき、この変異種を変異種7の調節体として設計した。
変異種7および8はLys313との塩ブリッジの形成を妨げるので、それらは両方とも、ネイテイブなpro−UKよりも低い本来的活性を有するが、変異種1または2よりも本来的活性は高いはずである。それらの2本鎖形態の活性は、本質的にネイティブなUK(2本鎖形態)と同じはずである。それらのフィブリンでの促進能も保持されるはずであり、ネイティブなUKとほぼ同じはずである。それゆえ、これらの変異種も、臨床的に有用であろう。
pro−UKにおいて「チモーゲン3回転軸」を形成するための部位特異的突然変異
キモトリプシンおよびトリプシンのX線構造は、「チモーゲン3回転軸」("zymogen triad")(Asp194、His40およびSer32からなる)による水素結合のネットワークの形成は、キモトリプシノーゲンの活性部位(酸素ホール(oxygen hole))の領域の不活性コンホーメーションの安定化にとり重要であることを示す。しかしながら、出願人は、pro−UKまたはt−PAにおいてはこのチモーゲン3回転軸が生じないことを見いだした。キモトリプシノーゲン3回転軸のHis40およびSer32は、pro−UKにおいてはそれぞれTyr187およびAla175により表される。この失われた3回転軸も、pro−UKまたはt−PAの高い本来的活性に貢献しているかもしれない。所望の特性を有するpro−UK変異種を設計するためのもう1つのアプローチとして、「チモーゲン3回転軸」をネイティブなpro−UK中に構築して、フィブリン分解活性に必要な他の機能に影響することなく、その本来的活性を低下させることができる。
キモトリプシンおよびトリプシンのX線構造は、「チモーゲン3回転軸」("zymogen triad")(Asp194、His40およびSer32からなる)による水素結合のネットワークの形成は、キモトリプシノーゲンの活性部位(酸素ホール(oxygen hole))の領域の不活性コンホーメーションの安定化にとり重要であることを示す。しかしながら、出願人は、pro−UKまたはt−PAにおいてはこのチモーゲン3回転軸が生じないことを見いだした。キモトリプシノーゲン3回転軸のHis40およびSer32は、pro−UKにおいてはそれぞれTyr187およびAla175により表される。この失われた3回転軸も、pro−UKまたはt−PAの高い本来的活性に貢献しているかもしれない。所望の特性を有するpro−UK変異種を設計するためのもう1つのアプローチとして、「チモーゲン3回転軸」をネイティブなpro−UK中に構築して、フィブリン分解活性に必要な他の機能に影響することなく、その本来的活性を低下させることができる。
変異種9:Ala175→Ser/Tyr187→His
キモトリプシノーゲンにおいて見られるのと同様の「チモーゲン3回転軸」をpro−UK中に導入するために、この二重変異種を設計した。この変異はpro−UKの本来的活性を低下させるはすであるが、フラグメントE2による促進および2本鎖形態の活性には影響しないはずである。それゆえ、この変異種もまた、上記3つの目的を達成するはずであり、血栓融解剤として有用であるはずである。
キモトリプシノーゲンにおいて見られるのと同様の「チモーゲン3回転軸」をpro−UK中に導入するために、この二重変異種を設計した。この変異はpro−UKの本来的活性を低下させるはすであるが、フラグメントE2による促進および2本鎖形態の活性には影響しないはずである。それゆえ、この変異種もまた、上記3つの目的を達成するはずであり、血栓融解剤として有用であるはずである。
混合変異
出願人は、pro−UKの本来的活性が、いくつかの異なる分子内相互作用に依存することを見いだした。該相互作用は、1)1本鎖形態におけるUKの新たな末端α−アミノ基の代用物としてのLys300またはLys313;2)「チモーゲン3回転軸」の欠如;および3)柔軟性のあるループ(297〜313)の低い柔軟性ならびに活性コンホーメーションの高い確率を包含する。したがって、ただ1つの態様に基づく変異によっては、本来的活性は最大限に除去されないであろうし、かかる変異は損傷を与えるものであり、血栓融解活性に必要な2本鎖形態の活性およびフィブリンでの促進活性の低下を伴うであろう。しかしながら、これらの分子内相互作用のうち2つまたはそれ以上に影響する多重変異により、最適なpro−UK変異種を作ることができる。これらの考えに基づき、出願人は、以下の変異種10から17までにより説明される一群の組み合わせ変異種を設計した。
出願人は、pro−UKの本来的活性が、いくつかの異なる分子内相互作用に依存することを見いだした。該相互作用は、1)1本鎖形態におけるUKの新たな末端α−アミノ基の代用物としてのLys300またはLys313;2)「チモーゲン3回転軸」の欠如;および3)柔軟性のあるループ(297〜313)の低い柔軟性ならびに活性コンホーメーションの高い確率を包含する。したがって、ただ1つの態様に基づく変異によっては、本来的活性は最大限に除去されないであろうし、かかる変異は損傷を与えるものであり、血栓融解活性に必要な2本鎖形態の活性およびフィブリンでの促進活性の低下を伴うであろう。しかしながら、これらの分子内相互作用のうち2つまたはそれ以上に影響する多重変異により、最適なpro−UK変異種を作ることができる。これらの考えに基づき、出願人は、以下の変異種10から17までにより説明される一群の組み合わせ変異種を設計した。
変異種10から17のすべてが、本来的活性を低下させる人工的なチモーゲン3回転軸を有しており、大部分は、中和されているかまたは正電荷をより少なくされているLys313を有しており、塩ブリッジの形成を防止、または塩ブリッジを弱体化するようになっていて、さらに本来的活性を低下させるようになっている。同様に、これらの組み合わせ変異種のうちのいくつかは、中和されているかまたは正電荷をより少なくされているLys300を有している。Tyr306→Gly変異は、さらに柔軟性を提供することにより、柔軟性のあるループが活性コンホーメーションとなる確率を減じ、Glu301→Ala変異は、Ile159とAsp355との間の相互作用を妨げ、それゆえ、2本鎖形態の活性を保持する。
変異種10:Ala175→Ser/Tyr187→His/Lys313→Ala
変異種11:Ala175→Ser/Tyr187→His/Tyr306→Gly
変異種12:Ala175→Ser/Tyr187→His/Lys313→His
変異種13:Ala175→Ser/Tyr187→His/Tyr306→Gly/Lys313→Ala
変異種14:Ala175→Ser/Tyr187→His/Lys300→Ala/Glu301→Ala/Lys313→Ala
変異種15:Ala175→Ser/Tyr187→His/Lys300→His/Glu301→Ala/Lys313→Ala
変異種16:Ala175→Ser/Tyr187→His/Lys300→Ala/Glu301→Ala/Lys313→His
変異種17:Ala175→Ser/Tyr187→His/Lys300→His/Glu301→Ala/Lys313→His
変異種11:Ala175→Ser/Tyr187→His/Tyr306→Gly
変異種12:Ala175→Ser/Tyr187→His/Lys313→His
変異種13:Ala175→Ser/Tyr187→His/Tyr306→Gly/Lys313→Ala
変異種14:Ala175→Ser/Tyr187→His/Lys300→Ala/Glu301→Ala/Lys313→Ala
変異種15:Ala175→Ser/Tyr187→His/Lys300→His/Glu301→Ala/Lys313→Ala
変異種16:Ala175→Ser/Tyr187→His/Lys300→Ala/Glu301→Ala/Lys313→His
変異種17:Ala175→Ser/Tyr187→His/Lys300→His/Glu301→Ala/Lys313→His
これらすべての変異種は、提案された3つの目的:(1)低い本来的活性および血漿中での高い安定性、(2)フィブリンでの促進能が不変であることおよび著しく高いフィブリン特異性、および(3)ネイティブなpro−UKと比較した場合の、プラスミン活性化後の同等な2本鎖形態の活性、を最適に達成するはずである。それゆえ、これらの変異種は、出血性合併症を誘発することなく、ネイティブなpro−UKよりも高用量で効率的に血栓クロットを溶解するはずである。そのうえ、それらは、フィブリンがないと血漿中において非常に不活性であり、それゆえ、極めて安全であるため、それらは、閉塞性フィブリン血栓の形成を予防する薬物として有用なはずである。したがって、これらの変異種は高い治療上の有用性を有する。
pro−UK変異種の製造
オリゴヌクレオチド部位特異的突然変異
本明細書記載のpro−UK変異種の製造に適した1のタイプの部位特異的突然変異は、オリゴヌクレオチド部位特異的突然変異であり、これは存在するDNA配列、例えば、ネイティブなpro−UKのDNA配列の特異的変化を可能にする。ネイティブなpro−UKをコードしている遺伝子は十分に特徴づけられており、例えば、デイビッド・ディチェク(David Dichek)博士(NIH)またはパオロ・サルミエントス(Paolo Sarmientos)博士(イタリアのミラノのプリム(Primm))から入手することができる。ATCC番号はDNA57329または細菌/ファージ57238である。また、配列は、UKHUなる名称のプロテイン・アイデンテイティー・リソース(Protein Identity Resource)なるNIHコンピューターデータベースから利用できる。pro−UKのアミノ酸配列を図1に示す。
オリゴヌクレオチド部位特異的突然変異
本明細書記載のpro−UK変異種の製造に適した1のタイプの部位特異的突然変異は、オリゴヌクレオチド部位特異的突然変異であり、これは存在するDNA配列、例えば、ネイティブなpro−UKのDNA配列の特異的変化を可能にする。ネイティブなpro−UKをコードしている遺伝子は十分に特徴づけられており、例えば、デイビッド・ディチェク(David Dichek)博士(NIH)またはパオロ・サルミエントス(Paolo Sarmientos)博士(イタリアのミラノのプリム(Primm))から入手することができる。ATCC番号はDNA57329または細菌/ファージ57238である。また、配列は、UKHUなる名称のプロテイン・アイデンテイティー・リソース(Protein Identity Resource)なるNIHコンピューターデータベースから利用できる。pro−UKのアミノ酸配列を図1に示す。
配列が対象の変異を含んでいるオリゴヌクレオチドプライマーを合成し、プライマーをネイティブな配列を含んでいる鋳型とハイブリダイズさせ、次いで、それをT4DNAポリメラーゼにより伸長させることにより、オリゴヌクレオチド部位特異的突然変異を行う。得られた生成物は、オリゴヌクレオチドにおける変異によるミスマッチを含むヘテロ2本鎖分子である。例えば、イー・コリ(E.coli)細胞中で、ミスマッチの修復の際に当該変異を「固定」する。この方法の詳細は、例えば、アウスベル(Ausbel)ら(編),カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology),第8.1章(グリーン・パブリッシング・アソシエーツ(Greene Publishing Associates)1989年,Supp.13)に記載されており、参照により本明細書に取り入れる。
この部位特異的突然変異法は、チミジンのかわりに少量のウラシル残基を含むDNA鋳型の使用に基づく(図4)。ウラシル含有DNAは、イー・コリdut−ung−株において生産される。このdut−ung−を組み合わせた変異株において、チミジンのかわりにデオキシウリジンがDNAに取り込まれ、それは除去されない。かくして、変更されるべき配列を含むベクターをdut−ung−宿主中で増殖させて部位特異的突然変異用のウラシル含有DNA鋳型調製することができる。図4に示すように、変異したオリゴヌクレオチドプライマーをDNA鋳型にアニーリングし、T4DNAポリメラーゼおよびT4DNAリガーゼで処理して2本鎖環状分子を得る。
オリゴヌクレオチドプライマーは高品質、すなわち、不完全なDNA合成を引き起こす低分子混入物質がないように精製されているべきである。いくつかの場合、特に40ヌクレオチドより大きいオリゴヌクレオチドに関しては、ポリアクリルアミドゲル電気泳動による精製が必要であるかもしれない。
部位特異的突然変異プロトコールの典型的なインビトロ反応において、ウラシル含有DNA鋳型は通常の鋳型と区別できない。鋳型中のdUMPはTMPと同じコーディング能力を有するので、ウラシルは変異原性ではない。そのうえ、鋳型におけるウラシルの存在はインビトロDNA合成に対して阻害的でない。かくして、所望のDNA配列の変化を有するが、TMP残基のみを含みdUMP残基を含まない相補鎖の生成のために、このDNAを鋳型として使用できる。
インビトロ反応を完了した後、例えば、ウラシルN−グリコシラーゼの作用により、あるいはインビトロ反応の分画されていない生成物を野生型(dut+ung+)イー・コリ細胞に導入することにより、ウラシルを鋳型鎖から除去する。グリコシラーゼでの処理はウラシルを遊離させ、鋳型鎖に致命的なアピリミジニック部位(AP部位)(apyrimidinic site)を生じさせる。かくして、鋳型鎖は生物学的に不活性にされ、大部分の子孫は、所望の変化を有する伝染性の相補鎖から生じる。このことは非常に効率的な変異DNAの生成(典型的には50%)を引き起こし、DNA配列分析による変異DNAのスクリーニングを可能にする。
スミス(Smith),アニュ・レビュ・ジェネティ(Ann.Rev.Genet.),第19巻:423〜463頁(1986年)に記載のごとく、高い効率で変異種が得られるプライマー伸長によるインビトロ突然変異のいくつかの変法が開発されている。本明細書記載の方法は、迅速かつ効率的に変異DNAを回収する、特別なウラシル含有鋳型に適用される単純な部位特異的突然変異プロトコールであり、もともと、クンケル(Kunkel),プロシーディングス・オブ・ナショナル・アカデミー・オブ・サイエンシズ・ユーエスエイ(Proc.Natl.Acad.Sci.U.S.A.),第82巻:488〜492頁(1985年)、およびクンケルら,メソッズ・イン・エンザイモロジー(Meth.Enzymol.),第154巻:367〜382頁(1987年)に記載されているものであり、これらを参照により本明細書に取り入れる。原理的には、この同じ鋳型を大部分の他のプロトコールに適用することができる。
pro−UK遺伝子をプラスミドM13mp18のHindIII/XbaI部位中に連結し、このプラスミドをイー・コリ中に形質転換し、ssDNAを単離し、次いで、上記のごとくオリゴヌクレオチド部位特異的突然変異を行うことにより、変異種1を作り出した。
pro−UK変異種DNAの発現
所望の変異を有するpro−UK DNAが得られたならば、それを適当な発現ベクター中に組み込まなければならない。次いで、このベクターをpro−UK変異種を発現する細胞系、例えば、細菌、哺乳動物、または酵母に導入し、該変異種を培地から集め、あるいは酵母細胞から集め、次いで、精製しなければならない。これらの方法は分子生物学の分野の当業者によく知られており、例えば、上記アウスベル(Ausbel)ら(編),カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology),第9章および第16章;サムブルック(Sambrook)、フリッチュ(Fritsch)およびマニアティス(Maniatis),モレキュラー・クローニング(Molecular Cloning)(第2版),第16章(コールド・スプリング・ハーバー・ラボラトリー・プレス(Cold Spring Harbor Laboratory Press),1989年)に詳細に記載されており、これらを参照により本明細書に取り入れる。
所望の変異を有するpro−UK DNAが得られたならば、それを適当な発現ベクター中に組み込まなければならない。次いで、このベクターをpro−UK変異種を発現する細胞系、例えば、細菌、哺乳動物、または酵母に導入し、該変異種を培地から集め、あるいは酵母細胞から集め、次いで、精製しなければならない。これらの方法は分子生物学の分野の当業者によく知られており、例えば、上記アウスベル(Ausbel)ら(編),カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology),第9章および第16章;サムブルック(Sambrook)、フリッチュ(Fritsch)およびマニアティス(Maniatis),モレキュラー・クローニング(Molecular Cloning)(第2版),第16章(コールド・スプリング・ハーバー・ラボラトリー・プレス(Cold Spring Harbor Laboratory Press),1989年)に詳細に記載されており、これらを参照により本明細書に取り入れる。
変異pro−UK遺伝子を哺乳動物細胞系に導入できるいくつかの方法がある。1の方法は、チャイニーズ・ハムスターの卵巣(「CHO」)細胞中へのベクターのトランスフェクションを包含する。この方法において、変異pro−UK遺伝子は選択可能なマーカーとともに同時トランスフェクションされ、安定に宿主細胞染色体中に組み込まれ、次いで、増幅される。CHO細胞系は長時間にわたり大量の変異pro−UKを産生するという理由で、CHO細胞系が好ましい。CHO DXB11またはCHO DG44のごとき細胞系がコロンビア大学のローレンス・チェイシン(Lawrence Chasin,Columbia University)から入手できる。細胞系CHO GRAはジェネティクス・インステイチュートのランドール・コウフマン(Randall Koufman,Genetics Institute)から入手できる。
別の発現方法は、変異DNAを含むベクターをPET−19Bイー・コリ発現系(ウィスコンシン州マジソンのノバーゲン(Novagen))中に導入することを包含する。例えば、部位特異的突然変異を行った後、変異種1(Lys300→Ala)をコードしているDNAを配列分析して変異が起こったことを確認し、次いで、これを非常に効率的なイー・コリ遺伝子発現系であるPET−19B(ウィスコンシン州マジソンのノバーゲン(Novagen)社製)のNdeI/XhaI部位中に連結し、イー・コリ中に形質転換した。形質転換されたイー・コリを培養し、インデューサーIPTGを対数増殖期に添加することによりpro−UK変異種の発現を誘導した。
ベクターを哺乳動物細胞系に導入したならば、宿主染色体内のトランスフェクションされたDNAの増加したコピー数に関して選択することにより、所望蛋白、例えば、変異pro−UKの発現を増加させることが望ましい。トランスフェクションされたDNA同時増幅することは、トランスフェクションされたDNAによりコードされる蛋白の発現の100ないし1000倍の増加をもたらす。文献に記載された20種上の選択可能で増幅可能な遺伝子があるが、大部分の実験例および成功例は、メトトレキサート選択およびトランスフェクションされたジヒドロ葉酸レダクターゼ遺伝子を用いるものであった。例えば、ジヒドロ葉酸レダクターゼ欠損CHO細胞を用いて、メトトレキサート耐性に関する選択による同時増幅によって高レベルの変異pro−UK遺伝子の発現を行うことができる。
CHO細胞発現ベクターを用いる増幅
pEDシリーズのジシストロニックベクター(dicistronic vectors)用いて、安定にトランスフェクションされた細胞において標的遺伝子の高レベル発現を行うことができる(図5)。これらのベクターは、標的遺伝子、例えば、変異pro−UK遺伝子の挿入用に、選択可能で増幅可能なマーカー遺伝子であるジヒドロ葉酸レダクターゼ(「DHFR」)遺伝子に続くクローニング配列を有している。適当なベクターで形質転換されたジヒドロ葉酸レダクターゼ欠損CHO細胞を、ヌクレオシド不含培地で増殖する能力により選択する。有効なDHFR機能の阻害剤であるメトトレキサートの存在濃度を順次増加させていく選択サイクルにより、組み込まれたDNAの増幅および所望遺伝子産物の増加した発現を行う。図5に示すpEDベクターは、カウフマン(Kaufman)ら,ヌクレイック・アシッズ・リサーチ(Nucl.Acids.Res.),第19巻:4485〜4490頁(1991年)に記載されており、これを参照により本明細書に取り入れる。
pEDシリーズのジシストロニックベクター(dicistronic vectors)用いて、安定にトランスフェクションされた細胞において標的遺伝子の高レベル発現を行うことができる(図5)。これらのベクターは、標的遺伝子、例えば、変異pro−UK遺伝子の挿入用に、選択可能で増幅可能なマーカー遺伝子であるジヒドロ葉酸レダクターゼ(「DHFR」)遺伝子に続くクローニング配列を有している。適当なベクターで形質転換されたジヒドロ葉酸レダクターゼ欠損CHO細胞を、ヌクレオシド不含培地で増殖する能力により選択する。有効なDHFR機能の阻害剤であるメトトレキサートの存在濃度を順次増加させていく選択サイクルにより、組み込まれたDNAの増幅および所望遺伝子産物の増加した発現を行う。図5に示すpEDベクターは、カウフマン(Kaufman)ら,ヌクレイック・アシッズ・リサーチ(Nucl.Acids.Res.),第19巻:4485〜4490頁(1991年)に記載されており、これを参照により本明細書に取り入れる。
エレクトロポーレーション、リン酸カルシウム、またはリポソームにより媒介される方法のいずれかを用いて、適当なベクター、例えば、pED中の変異pro−UKおよびDHFR遺伝子でCHO細胞をトランスフェクションしてもよく、そのことは、カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology)に詳細に記載されている。リン酸カルシウム処理、次いで、グリセロールによるショックを与えることは、CHO DXB11細胞について好ましい。次いで、トランスフェクションされた細胞を選択培地に置き、数日間増殖させた後、約500個の細胞を含む大型の健康的なコロニーを増幅に使用する。増幅の前に、選択されたコロニーを別々のプレート上で増殖させる。
安定なトランスフェクション体を増幅する前に、細胞DNAをサザン分析により分析するか、あるいは変異pro−UKに関する機能的アッセイを用いることによりRNAを分析して、細胞に所望pro−UK遺伝子が組み込まれたことを確認すべきである。次いで、選択された細胞を、メトトレキサート溶液中に入れ、その選択レベルを増加させていく。なぜなら、メトトレキサートはDHFRの有効な阻害剤であるからである。細胞をこの培地中に分配することにより、DHFRレベルを上昇させている細胞について選択する。一般的には、このことは、トランスフェクションされた遺伝子のコピー数を増加させることにより行われる。同時に、DHFR遺伝子とともに同時トランスフェクションされた変異pro−UK遺伝子もコピー数を増加させる。用いるメトトレキサートのレベルが高くなればなるほど、DHFR遺伝子のコピー数が最も大きい細胞が選び出され、それゆえ、所望の変異pro−UK遺伝子が得られる。細胞が20ないし80μMのメトトレキサート中で増殖する場合、細胞は500ないし2000コピーのトランスフェクションされた変異pro−UK遺伝子を有する。
pro−UK変異種の抽出および精製
例えば、哺乳動物または細菌細胞系により変異pro−UKが発現された後、それを培地から抽出し、精製しなければならない。酵母細胞の培養の場合には、まず、例えば、ガラスビーズを用いる機械的破砕により酵母細胞を破砕して、変異pro−UKを含有する細胞抽出液を得なければならない。培地または細胞抽出液からの活性のある変異pro−UKの精製は、一般的には、1)液相/液相抽出または最初の濾過工程、2)疎水的親和性クロマトグラフィー、3)抗体親和性クロマトグラフィー、そして4)ゲル濾過クロマトグラフィーからなる工程を包含する。これらの段階は分子生物学の分野の当業者によく知られており、カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology)第10章に詳細に記載されている。
例えば、哺乳動物または細菌細胞系により変異pro−UKが発現された後、それを培地から抽出し、精製しなければならない。酵母細胞の培養の場合には、まず、例えば、ガラスビーズを用いる機械的破砕により酵母細胞を破砕して、変異pro−UKを含有する細胞抽出液を得なければならない。培地または細胞抽出液からの活性のある変異pro−UKの精製は、一般的には、1)液相/液相抽出または最初の濾過工程、2)疎水的親和性クロマトグラフィー、3)抗体親和性クロマトグラフィー、そして4)ゲル濾過クロマトグラフィーからなる工程を包含する。これらの段階は分子生物学の分野の当業者によく知られており、カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current Protocols in Molecular Biology)第10章に詳細に記載されている。
pro−UK変異種1を含んでいるイー・コリ細胞を超音波処理により溶解し、エンテロキナーゼ開裂部位(DDDDK)を伴うpro−UKのN末端の前にポリ−Hisを挿入してあるため、溶解物をニッケル親和性クロマトグラフィーカラムに適用した。精製されたpro−UK変異種を固定化エンテロキナーゼとともにインキュベーションして、ニッケル親和性カラムでの再クロマトグラフィーを用いてN−末端ポリ−Hisを除去した。S−セファロース、ヒドロキシアパタイト、セファクリルS−200、次いで、ウィンクラー(Winkler)およびブレイダー(Blader),バイオケミストリー(Biochemistry),第25巻:4041〜4045頁(1986年)、およびオルシニ(Orsini)ら,ヨーロピアン・ジャーナル・オブ・バイオケミストリー(Eur.J.Biochem.),第195巻:691〜697頁(1991年)に記載のごとく、再生させた後ベンズアミジンカラムによるクロマトグラフィーにより、さらに試料を精製した。ジイソプロピルフルオロホスフェート(DFP)処理によりすべての少量の混入UKを除去した。
pro−UK変異種の分析
変異種1について行ったように、pro−UK変異種の特性を、イー・コリにより産生された組み換え型pro−UKと比較することができる。本来的活性、早い時点におけるGlu−プラスミノーゲンのフラグメントE2による促進、および血漿環境におけるプラスミン活性化ならびにクロット溶解に対する感受性を試験して少量のpro−UK変異種について得られ、いかにして変異種がインビトロにおいて血栓融解剤として作用するのかを予想するアッセイプロフィールを決定した。
変異種1について行ったように、pro−UK変異種の特性を、イー・コリにより産生された組み換え型pro−UKと比較することができる。本来的活性、早い時点におけるGlu−プラスミノーゲンのフラグメントE2による促進、および血漿環境におけるプラスミン活性化ならびにクロット溶解に対する感受性を試験して少量のpro−UK変異種について得られ、いかにして変異種がインビトロにおいて血栓融解剤として作用するのかを予想するアッセイプロフィールを決定した。
本来的活性のアッセイ
これらのアッセイは、UKの合成基質であるS2444の加水分解の動的分析およびGlu−プラスミノーゲンの活性化に基づく。
これらのアッセイは、UKの合成基質であるS2444の加水分解の動的分析およびGlu−プラスミノーゲンの活性化に基づく。
S2444の加水分解:4.0nMのUKまたは1.0μMのpro−UK変異種1を、0.05Mリン酸ナトリウム、0.15M NaCl、0.2%BSAおよび0.01%ツイン80(pH7.8)中の0.03、0.06、0.12、0.18、0.24、0.3、0.6、1.2、1.8および2.4mMの濃度範囲のS2444とともに室温でインキュベーションした。マイクロタイタープレートリーダー上で、対照波長490nmに対する410nmにおけるODの直線的増加(410/490nm)により反応速度を測定した。変異種1はS2444加水分解活性をほとんど示さなかった。
Glu−プラスミノーゲンの活性化:マイクロタイタープレートリーダー(ダイナテック(Dynatech)MR5000)上で、対照波長490nmおよび選択波長410nm(410/490nm)における経時的な反応混合物のOD増加を測定することにより、Glu−プラスミノーゲン活性化の時間−吸光度曲線を得た。反応混合物は、S2251(1.5mM)、Glu−プラスミノーゲン(1.0、1.5、2.5、3.5、4.5、5.5、7.5、10.0μM)およびUK(0.2nM)またはpro−UK変異種1(5.0nM)を含んでいた。0.05Mリン酸ナトリウム、0.15M NaCl、0.2%BSAおよび0.01%ツイン80(pH7.8)中に反応物質を調製し、室温でインキュベーションした。変異種1は、約4IU/mgのプラスミノーゲン活性化活性を示し、これは、約400IU/mgの活性を有するネイティブなpro−UKのわずか1%の活性であった。
フィブリンフラグメントE−2によるプラスミノーゲン活性化に関するアッセイ
フラグメントE−2存在下でのpro−UK変異種1によるGlu−プラスミノーゲン活性化を、マイクロタイタープレートリーダー上で、対照波長490nmおよび選択波長410nm(410/490nm)における経時的な反応混合物のOD増加を測定することにより測定した。反応混合物は、0.05Mリン酸ナトリウム、0.15M NaCl、0.2%BSAおよび0.01%ツイン80(室温でpH7.8)中、1.5mM S2251(プラスミンに対する合成基質)、Glu−プラスミノーゲン(1.0、2.0、4.0および8.0μM)、5.0μMのフラグメントE−2、および1.0nMのpro−UK変異種1を含有していた。フィブリンフラグメントE2存在下での変異種1によるプラスミノーゲン活性化は、ネイティブなpro−UKのわずか約40%であった。ネイティブなpro−UKのプラスミノーゲン活性化は、フィブリン促進のない場合と比べてフィブリンフラグメントE−2存在下では250倍であった。
フラグメントE−2存在下でのpro−UK変異種1によるGlu−プラスミノーゲン活性化を、マイクロタイタープレートリーダー上で、対照波長490nmおよび選択波長410nm(410/490nm)における経時的な反応混合物のOD増加を測定することにより測定した。反応混合物は、0.05Mリン酸ナトリウム、0.15M NaCl、0.2%BSAおよび0.01%ツイン80(室温でpH7.8)中、1.5mM S2251(プラスミンに対する合成基質)、Glu−プラスミノーゲン(1.0、2.0、4.0および8.0μM)、5.0μMのフラグメントE−2、および1.0nMのpro−UK変異種1を含有していた。フィブリンフラグメントE2存在下での変異種1によるプラスミノーゲン活性化は、ネイティブなpro−UKのわずか約40%であった。ネイティブなpro−UKのプラスミノーゲン活性化は、フィブリン促進のない場合と比べてフィブリンフラグメントE−2存在下では250倍であった。
プラスミン感受性のアッセイ
プラスミン感受性のアッセイはLys−プラスミンによるpro−UK変異種の活性化の動的研究である。0、0.1、0.2、0.4、0.6、0.8、1.0、1.2、1.4、2.5、3.5および5.0μMの濃度範囲のpro−UK変異種1を、0.05M Tris−HCl、0.15M NaCl,0.01M CaCl2および0.01%ツイン80,pH7.4中、1.2mMのS2444の存在下で、室温において一定時間インキュベーションした。プラスミンが存在しない場合の同じ濃度範囲のpro−UK変異種を、対照としてS2444とともにインキュベーションした。0.1nMのプラスミンは、対照条件下においてはS2444の加水分解に対して直接的な影響を及ぼさなかった。pro−UK変異種1から生じたUKの量を、マイクロタイタープレートリーダー(MR5000型;バージニア州アレクサンドリアのダイナテック・ラボラトリーズ・インコーポレイテッド(Dynatech Laboratories Inc.))上で、対照波長490nmおよび選択波長410nm(410/490nm)における経時的な反応混合物のOD増加により測定した。変異種1は、ネイティブなpro−UKと同様のプラスミン感受性を有し、KM2.44μM、kcat3.04nM/分であった。
プラスミン感受性のアッセイはLys−プラスミンによるpro−UK変異種の活性化の動的研究である。0、0.1、0.2、0.4、0.6、0.8、1.0、1.2、1.4、2.5、3.5および5.0μMの濃度範囲のpro−UK変異種1を、0.05M Tris−HCl、0.15M NaCl,0.01M CaCl2および0.01%ツイン80,pH7.4中、1.2mMのS2444の存在下で、室温において一定時間インキュベーションした。プラスミンが存在しない場合の同じ濃度範囲のpro−UK変異種を、対照としてS2444とともにインキュベーションした。0.1nMのプラスミンは、対照条件下においてはS2444の加水分解に対して直接的な影響を及ぼさなかった。pro−UK変異種1から生じたUKの量を、マイクロタイタープレートリーダー(MR5000型;バージニア州アレクサンドリアのダイナテック・ラボラトリーズ・インコーポレイテッド(Dynatech Laboratories Inc.))上で、対照波長490nmおよび選択波長410nm(410/490nm)における経時的な反応混合物のOD増加により測定した。変異種1は、ネイティブなpro−UKと同様のプラスミン感受性を有し、KM2.44μM、kcat3.04nM/分であった。
血漿フィブリノーゲン分解活性のアッセイ
pro−UK変異種1(0〜100μg/ml)またはpro−UK(0〜10μg/ml)を、1.0mlの貯留しておいた血漿中で、6、16、または24時間インキュベーションし、次いで、0.2mlのアプロチニン(10000KIU/ml)を添加し、一定時間後に残存する血漿フィブリノーゲンを、スロンビンクロット法により測定した。変異種1は、ネイティブなpro−UKよりも約100倍低い血漿フィブリノーゲン分解活性を有していた。すなわち、1.0μg/mlのネイティブなpro−UKは9.0μg/mlのフィブリノーゲンの50%を6時間以内に分解するが、同じ効果を得るには100μg/mlの変異種1が必要である。
pro−UK変異種1(0〜100μg/ml)またはpro−UK(0〜10μg/ml)を、1.0mlの貯留しておいた血漿中で、6、16、または24時間インキュベーションし、次いで、0.2mlのアプロチニン(10000KIU/ml)を添加し、一定時間後に残存する血漿フィブリノーゲンを、スロンビンクロット法により測定した。変異種1は、ネイティブなpro−UKよりも約100倍低い血漿フィブリノーゲン分解活性を有していた。すなわち、1.0μg/mlのネイティブなpro−UKは9.0μg/mlのフィブリノーゲンの50%を6時間以内に分解するが、同じ効果を得るには100μg/mlの変異種1が必要である。
フィブリンクロット溶解アッセイ
0.25mlの血漿から調製された125I−標識クロットを、グレビッチ(Gurewich)ら,クリニカル・インベスティゲーションズ(Clin.Invest.),第73巻:1731ページ(1980年)に記載のごとく調製した。クロット溶解実験を、10、20、30、40、70、および80μg/mlの濃度範囲のpro−UK変異種1または5、10、30、50、75、100および150ng/mlのt−PA、およびt−PAとpro−UKの一定の組み合わせととのみ、3mlの血漿中で行った。放射活性の遊離から溶解を定量化し、時間に対する完全な溶解における値のパーセントとして表現した。変異種1のフィブリンクロット溶解活性はネイティブなpro−UKの50%あった。すなわち、200U/mlのネイティブなpro−UKはクロット(1cm3)を6時間以内に溶解するが、同じ効果を得るためには、同量の変異種1の場合12時間かかる。
0.25mlの血漿から調製された125I−標識クロットを、グレビッチ(Gurewich)ら,クリニカル・インベスティゲーションズ(Clin.Invest.),第73巻:1731ページ(1980年)に記載のごとく調製した。クロット溶解実験を、10、20、30、40、70、および80μg/mlの濃度範囲のpro−UK変異種1または5、10、30、50、75、100および150ng/mlのt−PA、およびt−PAとpro−UKの一定の組み合わせととのみ、3mlの血漿中で行った。放射活性の遊離から溶解を定量化し、時間に対する完全な溶解における値のパーセントとして表現した。変異種1のフィブリンクロット溶解活性はネイティブなpro−UKの50%あった。すなわち、200U/mlのネイティブなpro−UKはクロット(1cm3)を6時間以内に溶解するが、同じ効果を得るためには、同量の変異種1の場合12時間かかる。
pro−UK変異種の治療的使用
pro−UKおよびUKと同様に、血栓融解剤としてpro−UK変異種を使用し投与する。pro−UK変異種を医薬上許容される担体、例えば、セイラインと混合し、血管内投与、例えば、静脈内もしくは動脈内投与、または皮下注射により投与する。pro−UK変異種を、20ないし60mgの丸薬として注入するか、または40〜80mg/時の割合で静脈から輸液してもよい。pro−UK変異種は、ネイティブないしpro−UKよりもずっと高い血漿での安定性を有し、非特異的プラスミノーゲン活性化を誘導する可能性がずっと低いので、高用量、例えば、200mg/時までの輸液を使用することもできる。
pro−UKおよびUKと同様に、血栓融解剤としてpro−UK変異種を使用し投与する。pro−UK変異種を医薬上許容される担体、例えば、セイラインと混合し、血管内投与、例えば、静脈内もしくは動脈内投与、または皮下注射により投与する。pro−UK変異種を、20ないし60mgの丸薬として注入するか、または40〜80mg/時の割合で静脈から輸液してもよい。pro−UK変異種は、ネイティブないしpro−UKよりもずっと高い血漿での安定性を有し、非特異的プラスミノーゲン活性化を誘導する可能性がずっと低いので、高用量、例えば、200mg/時までの輸液を使用することもできる。
本発明は、治療的用量で投与された場合に、pro−UKの高い本来的な酵素活性を保持しつつも、望ましくない非特異的かつ全身的なプラスミノーゲン活性化を開始させにくい、あるいはさせないpro−UK変異種を提供するものなので、製薬産業等において利用可能である。
配列表

Claims (7)
- ネイティブなプロ−ウロキナーゼ(pro−UK)のアミノ酸配列(配列番号:1)を必須としてなる血栓融解活性のあるpro−UK変異種であって、位置300のリジンがヒスチジンになっており、患者に投与された場合にネイティブなpro−UKよりもフィブリノーゲン溶解および非特異的プラスミノーゲン活性化を誘導しないものであるpro−UK変異種。
- 請求項1記載のpro−UK変異種をコードしているDNA分子。
- 請求項2記載の組み換え型DNA分子で形質転換された細胞。
- ネイティブなpro−UKよりも出血性合併症を引き起こさないものである請求項1記載のpro−UK変異種。
- ネイティブなpro−UKのアミノ酸配列(配列番号:1)からなるpro−UK変異種であって、位置300のリジンがヒスチジンになっている請求項1記載のpro−UK変異種。
- 請求項5記載のpro−UK変異種をコードしているDNA分子。
- 請求項6記載の組み換え型DNA分子で形質転換された細胞。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/087,163 US5472692A (en) | 1993-07-02 | 1993-07-02 | Pro-urokinase mutants |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP7503598A Division JPH08511953A (ja) | 1993-07-02 | 1994-06-28 | プロウロキナーゼ変異種 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004337172A true JP2004337172A (ja) | 2004-12-02 |
Family
ID=22203481
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP7503598A Pending JPH08511953A (ja) | 1993-07-02 | 1994-06-28 | プロウロキナーゼ変異種 |
| JP2004200871A Pending JP2004337172A (ja) | 1993-07-02 | 2004-07-07 | プロ−ウロキナーゼ変異種 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP7503598A Pending JPH08511953A (ja) | 1993-07-02 | 1994-06-28 | プロウロキナーゼ変異種 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5472692A (ja) |
| EP (2) | EP0714435B1 (ja) |
| JP (2) | JPH08511953A (ja) |
| CN (1) | CN1115412C (ja) |
| AT (1) | ATE239084T1 (ja) |
| CA (1) | CA2165977A1 (ja) |
| DE (1) | DE69432592T2 (ja) |
| ES (1) | ES2197165T3 (ja) |
| NZ (1) | NZ269061A (ja) |
| PT (1) | PT714435E (ja) |
| WO (1) | WO1995001427A1 (ja) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7544500B2 (en) * | 1999-11-13 | 2009-06-09 | Talecris Biotherapeutics, Inc. | Process for the production of a reversibly inactive acidified plasmin composition |
| US6964764B2 (en) * | 1999-11-13 | 2005-11-15 | Talecris Biotherapeutics, Inc. | Method of thrombolysis by local delivery of reversibly inactivated acidified plasmin |
| US6355243B1 (en) * | 1999-11-13 | 2002-03-12 | Bayer Corporation | Method of thrombolysis by local delivery of active plasmin |
| US6969515B2 (en) | 1999-11-13 | 2005-11-29 | Talecris Biotherapeutics, Inc. | Method of thrombolysis by local delivery of reversibly inactivated acidified plasmin |
| WO2002061064A2 (de) * | 2001-02-01 | 2002-08-08 | Roche Diagnostics Gmbh | Verfahren zur herstellung von rekombinantem trypsin |
| CN1311076C (zh) * | 2003-04-16 | 2007-04-18 | 普罗特奥姆技术公司 | 生产重组尿激酶的方法 |
| US7070958B2 (en) * | 2003-04-18 | 2006-07-04 | Thrombolytic Science, Inc. | Methods of making pro-urokinase mutants |
| CA2426115A1 (en) * | 2003-04-18 | 2004-10-18 | Victor Gurewich | Methods, devices, and compositions for lysis of occlusive blood clots while sparing wound sealing clots |
| US20070218535A1 (en) * | 2005-11-28 | 2007-09-20 | Xinli Lin | Methods for production of recombinant alpha1-antitrypsin |
| CN101024827B (zh) * | 2006-02-22 | 2012-05-30 | 中国科学院福建物质结构研究所 | 尿激酶催化结构域突变体的表达、纯化以及结晶 |
| CN100441683C (zh) * | 2006-03-24 | 2008-12-10 | 中国人民解放军军事医学科学院生物工程研究所 | 低分子量尿激酶突变体及其表达载体 |
| US7837992B2 (en) * | 2006-06-22 | 2010-11-23 | Beth Israel Deaconess Medical Center | C-1 inhibitor prevents non-specific plasminogen activation by a prourokinase mutant without impeding fibrin-specific fibrinolysis |
| US20070298024A1 (en) * | 2006-06-22 | 2007-12-27 | Thrombolytic Science, Inc. | C-1 inactivator inhibits two-chain urokinase mutant and limits hemostatic bleeding during thrombolysis |
| SG175602A1 (en) | 2006-07-05 | 2011-11-28 | Catalyst Biosciences Inc | Protease screening methods and proteases identified thereby |
| EP1936531A1 (en) * | 2006-12-20 | 2008-06-25 | Thomson Licensing | Methods and device for secure software installation |
| WO2008103146A2 (en) * | 2007-02-23 | 2008-08-28 | Landing Biotech, Inc. | Treating atherosclerosis |
| WO2009149199A2 (en) * | 2008-06-04 | 2009-12-10 | Talecris Biotherapeutics, Inc. | Composition, method and kit for preparing plasmin |
| US20100143325A1 (en) * | 2008-12-09 | 2010-06-10 | Vascular Laboratory, Inc. | Composition And Methods Involving Thrombolytic Agents |
| PT2403865E (pt) | 2009-03-03 | 2015-11-18 | Grifols Therapeutics Inc | Métodos de preparação de plasminogénio |
| CA2953789A1 (en) | 2013-07-12 | 2015-01-15 | Emory University | Diagnostic assay to predict cardiovascular risk |
| RU2553533C2 (ru) * | 2013-10-23 | 2015-06-20 | Общество с ограниченной ответственностью "Международный биотехнологический центр "Генериум" ("МБЦ "Генериум") | Способ выделения и очистки рекомбинантной человеческой проурокиназы м5 |
| EP3708226A1 (en) | 2014-11-03 | 2020-09-16 | Thrombolytic Science, LLC | Methods and compositions for safe and effective thrombolysis |
| WO2018232305A1 (en) | 2017-06-16 | 2018-12-20 | Thrombolytic Science, Llc | Methods and compositions for thrombolysis |
| CA3095080A1 (en) | 2018-03-27 | 2019-10-03 | Coen MAAS | Targeted thrombolysis for treatment of microvascular thrombosis |
| CA3123872A1 (en) * | 2018-12-28 | 2020-07-02 | Catalyst Biosciences, Inc. | Modified urokinase-type plasminogen activator polypeptides and methods of use |
| US11613744B2 (en) | 2018-12-28 | 2023-03-28 | Vertex Pharmaceuticals Incorporated | Modified urokinase-type plasminogen activator polypeptides and methods of use |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1989007641A1 (en) * | 1988-02-10 | 1989-08-24 | Genzyme Corporation | Enhancement of the therapeutic properties of glycoprotein |
| US5108901A (en) * | 1988-09-02 | 1992-04-28 | Genentech, Inc. | Tissue plasminogen activator having zymogenic or fibrin specific properties |
| EP0387380A1 (en) * | 1989-03-17 | 1990-09-19 | BEHRINGWERKE Aktiengesellschaft | Mutants of urinary plasminogen activator, their production and use |
-
1993
- 1993-07-02 US US08/087,163 patent/US5472692A/en not_active Expired - Fee Related
-
1994
- 1994-06-28 WO PCT/US1994/007278 patent/WO1995001427A1/en not_active Ceased
- 1994-06-28 EP EP94922037A patent/EP0714435B1/en not_active Expired - Lifetime
- 1994-06-28 CN CN94193246A patent/CN1115412C/zh not_active Expired - Lifetime
- 1994-06-28 EP EP03009709A patent/EP1361276A1/en not_active Withdrawn
- 1994-06-28 CA CA002165977A patent/CA2165977A1/en not_active Abandoned
- 1994-06-28 ES ES94922037T patent/ES2197165T3/es not_active Expired - Lifetime
- 1994-06-28 AT AT94922037T patent/ATE239084T1/de not_active IP Right Cessation
- 1994-06-28 DE DE69432592T patent/DE69432592T2/de not_active Expired - Fee Related
- 1994-06-28 NZ NZ269061A patent/NZ269061A/en unknown
- 1994-06-28 JP JP7503598A patent/JPH08511953A/ja active Pending
- 1994-06-28 PT PT94922037T patent/PT714435E/pt unknown
-
2004
- 2004-07-07 JP JP2004200871A patent/JP2004337172A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP0714435A4 (en) | 1997-08-20 |
| NZ269061A (en) | 1997-03-24 |
| DE69432592D1 (de) | 2003-06-05 |
| US5472692A (en) | 1995-12-05 |
| CN1115412C (zh) | 2003-07-23 |
| ES2197165T3 (es) | 2004-01-01 |
| PT714435E (pt) | 2003-08-29 |
| EP0714435A1 (en) | 1996-06-05 |
| EP0714435B1 (en) | 2003-05-02 |
| ATE239084T1 (de) | 2003-05-15 |
| JPH08511953A (ja) | 1996-12-17 |
| EP1361276A1 (en) | 2003-11-12 |
| DE69432592T2 (de) | 2004-04-01 |
| CA2165977A1 (en) | 1995-01-12 |
| WO1995001427A1 (en) | 1995-01-12 |
| CN1130402A (zh) | 1996-09-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5472692A (en) | Pro-urokinase mutants | |
| JP2794028B2 (ja) | チモーゲン的またはフィブリン特異的特性を有する組織プラスミノーゲン活性化因子 | |
| JPH07186A (ja) | ヒトウロキナーゼを発現する組換え発現ベクター及び形質転換細胞 | |
| US5338546A (en) | Tissue plasminogen activator variants with decreased clearance | |
| AU630879B2 (en) | A tissue plasminogen activator analogue | |
| CA2271697C (en) | Tissue type plasminogen activator (t-pa) variants: compositions and methods of use | |
| WO1998021320A9 (en) | TISSUE TYPE PLASMINOGEN ACTIVATOR (t-PA) VARIANTS: COMPOSITIONS AND METHODS OF USE | |
| US6706504B1 (en) | Tissue type plasminogen activator (t-PA) variants: compositions and methods of use | |
| EP0236040A2 (en) | Amino acid modified prourokinase and method of preparation | |
| FLOHL | Single-chain urokinase-type plasminogen activators: new hopes for clot-specific lysis | |
| JP3126381B2 (ja) | 減少されたクリアランスを有する組織プラスミノーゲンアクチベーター変異体 | |
| US5688664A (en) | Thrombin activatable plasminogen analogues | |
| JPH03500125A (ja) | プロウロキナーゼの低分子量誘導体及び該誘導体を含有する薬剤組成物 | |
| EP0316068A1 (en) | Modified low molecular weight plasminogen activator and method of preparation | |
| Sun et al. | Mutagenesis at Pro309 of single‐chain urokinase‐type plasminogen activator alters its catalytic properties | |
| Cederholm‐Williams | Molecular biology of plasminogen activators and recombinant DNA progress | |
| Jeon et al. | Cloning, expression, and characterization of a cDNA encoding snake venom metalloprotease | |
| Yan et al. | Construction, expression, and characterization of a recombinant annexin B1-low molecular weight urokinase chimera in Escherichia coli | |
| JP2532193B2 (ja) | 組織プラスミノ―ゲン活性化因子置換変異体 | |
| EP0400054A1 (en) | Modified gene sequences encoding modified tpa and products therefrom | |
| JPH07501946A (ja) | フィブリン特異性が改善されたt‐PA置換変異体 | |
| JP2008301813A (ja) | 新規アグレカナーゼ分子 | |
| Wallén et al. | Effects of structural modifications on the properties of tissue plasminogen activator (tPA) | |
| Peng et al. | Mutation of Arg154 to Gly154 in urokinase augments its fibrin‐specificity | |
| Hernandez-Barrantes et al. | 1-Matrix Metalloproteinase (MTl-MMP) and. Matrix |