JP2004530657A - 上皮組織にわたるおよび上皮組織への薬物送達を増強するための組成物および方法 - Google Patents
上皮組織にわたるおよび上皮組織への薬物送達を増強するための組成物および方法 Download PDFInfo
- Publication number
- JP2004530657A JP2004530657A JP2002569108A JP2002569108A JP2004530657A JP 2004530657 A JP2004530657 A JP 2004530657A JP 2002569108 A JP2002569108 A JP 2002569108A JP 2002569108 A JP2002569108 A JP 2002569108A JP 2004530657 A JP2004530657 A JP 2004530657A
- Authority
- JP
- Japan
- Prior art keywords
- delivery
- conjugate
- arginine
- transporter
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 *C(C*CC(N)=O)C(*)=O Chemical compound *C(C*CC(N)=O)C(*)=O 0.000 description 3
- KQMAREZWIRETAM-UHFFFAOYSA-N CC(CSCC(C(N)=O)NC)=O Chemical compound CC(CSCC(C(N)=O)NC)=O KQMAREZWIRETAM-UHFFFAOYSA-N 0.000 description 1
- JGSACAANCVVBKM-UHFFFAOYSA-N CCC(OCCOC[n]1c(N=C(N)NC2=O)c2nc1)=O Chemical compound CCC(OCCOC[n]1c(N=C(N)NC2=O)c2nc1)=O JGSACAANCVVBKM-UHFFFAOYSA-N 0.000 description 1
Images























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/155—Amidines (), e.g. guanidine (H2N—C(=NH)—NH2), isourea (N=C(OH)—NH2), isothiourea (—N=C(SH)—NH2)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
- A61K38/13—Cyclosporins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/555—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound pre-targeting systems involving an organic compound, other than a peptide, protein or antibody, for targeting specific cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/555—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound pre-targeting systems involving an organic compound, other than a peptide, protein or antibody, for targeting specific cells
- A61K47/557—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound pre-targeting systems involving an organic compound, other than a peptide, protein or antibody, for targeting specific cells the modifying agent being biotin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/645—Polycationic or polyanionic oligopeptides, polypeptides or polyamino acids, e.g. polylysine, polyarginine, polyglutamic acid or peptide TAT
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/66—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid the modifying agent being a pre-targeting system involving a peptide or protein for targeting specific cells
- A61K47/665—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid the modifying agent being a pre-targeting system involving a peptide or protein for targeting specific cells the pre-targeting system, clearing therapy or rescue therapy involving biotin-(strept) avidin systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/0019—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules
- A61K49/0021—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules the fluorescent group being a small organic molecule
- A61K49/0041—Xanthene dyes, used in vivo, e.g. administered to a mice, e.g. rhodamines, rose Bengal
- A61K49/0043—Fluorescein, used in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0056—Peptides, proteins, polyamino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/085—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier conjugated systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/08—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by the carrier
- A61K49/10—Organic compounds
- A61K49/14—Peptides, e.g. proteins
- A61K49/146—Peptides, e.g. proteins the peptide being a polyamino acid, e.g. poly-lysine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Biomedical Technology (AREA)
- Ophthalmology & Optometry (AREA)
- Biochemistry (AREA)
- Nanotechnology (AREA)
- Genetics & Genomics (AREA)
- Radiology & Medical Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Gastroenterology & Hepatology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Medical Informatics (AREA)
- General Engineering & Computer Science (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Cardiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
Abstract
Description
【0001】
(発明の分野)
本発明は、皮膚膜および上皮膜(例えば、皮膚、胃腸上皮および気管支上皮を含む)にわたる薬物および他の化合物の送達を増強する組成物および方法の分野に関する。
【背景技術】
【0002】
(背景)
経皮薬物送達または経粘膜薬物送達は、様々な理由のために、魅力的な薬物送達経路である。胃腸薬分解および肝臓の最初の通過効果が回避される。さらに、経皮薬物送達および経粘膜薬物送達は、制御された持続性送達に十分適している(例えば、Elias、Percutaneous Absorption:Mechanisms−Methodology−Drug Delivery,Bronaugh & Maibach Eds.1〜12頁、Marcel Dekker,New York,1989を参照のこと)。多くの適用について、薬物を投与する従来の方法は、薬物の非常に高い初濃度に起因して、最適でない。経皮送達は、より高い均一性、より遅い薬物の送達速度を可能にし得る。さらに、患者コンプライアンスは、このような送達方法は、使用が容易で、快適、簡便かつ非侵襲性なので、奨励される。
【0003】
経皮送達および経粘膜送達のこれらの利点は、上被膜、皮膚の低浸透性(特に、薬物に対する)に起因して、多くの臨床的適用につながっていなかった。皮膚にわたる薬物の送達における困難は、皮膚の障壁特性に起因する。皮膚は、外部の不適環境に対する身体の境界を示す、構造的に複雑な薄膜である。皮膚は、表皮、真皮、皮下組織および付属(adenexal)構造(表皮の付属物)から構成される。表皮(皮膚の最外上皮組織)は、それ自身が、いくつかの層−−角質層、顆粒層、有棘層、および基底層から構成される。
【0004】
環境から無傷の皮膚に移動し、そして、無傷の皮膚を進む化合物は、最初に、角質層(皮膚の最外層)に浸透しなければならなく、この角質層は、緻密で、そして、非常に角質化されている。角質層は、コレステロールおよび脂肪酸から構成される「膠(glue)」によって、一緒に緊密に結合される、ケラチン充填皮膚細胞のいくつかの層から構成される。角質層の厚さは、身体部位に依存して変化する。これは、薬学的薬剤に対する皮膚の不浸透性を生じる角質層の存在である。この角質層は、基底層から皮膚表面(ここで、細胞は、最終的に剥がれる)への細胞移動によって自然に形成される。表面への細胞の進行に伴い、この細胞は、次第に、より乾燥され、そして角質化される。角質層にわたる浸透は、一般に、皮膚にわたる薬物浸透の律速段階である。例えば、Flynn,G.L.,Percutaneous Absorption:Mechanisms−Methodology−Drug Delivery,前述、27〜53頁を参照のこと。
【0005】
角質層を介した浸透の後、次いで、全身作用性薬物分子は、表皮、真皮に移動し、そしてこれらを通過し、最終的には、血流の毛細管壁を通過しなければならない。角質層の下に位置する表皮は、3つの層から構成される。これらの層の最外部は、顆粒層で、この顆粒層は、角質層に隣接して位置し、基底細胞およびケラチノサイトとは区別される細胞から構成され、これらの基底細胞およびケラチノサイトは、根底の層を形成し、獲得されたさらなるケラチンおよびより平坦な形状を有する。表皮のこの層の細胞は、顆粒を含み、この顆粒は、大部分がタンパク質フィラグリンから構成される。このタンパク質は、ケラチン複合体を形成するために、ケラチンフィラメントに結合すると思われている。細胞はまた、細胞を結合させるための「セメント」として機能する脂質を合成する。表皮(特に、顆粒層)は、アミノペプチダーゼのような酵素を含む。
【0006】
表皮の次の外層は有棘層で、この主細胞はケラチノサイトであり、このケラチノサイトは、基底細胞層を含む基底細胞由来である。ランゲルハンス細胞(有棘層でもまた見出される)は、抗原提示細胞であり、従って、皮膚に移動する抗原に対する免疫応答の増大に関する。この層の細胞は、一般に、接触感受性皮膚炎に関する。
【0007】
最内部の表皮層は、基底層すなわし基底細胞層であり、これらは、根底の真皮から基底細胞層を分離する薄基底膜に、半接着斑により結合される立方細胞の1つの細胞層からなる。基底層の細胞は、表皮の外部層の前駆物質として機能する、比較的未分化の増殖細胞である。基底細胞層としては、さらに、基底細胞、メラニン細胞が挙げられる。
【0008】
真皮は、表皮の下で見出され、この表皮は、連結乳頭間隆起および真皮乳頭からなる基底膜によって、真皮から分離される。真皮自身は、2つの層(乳頭真皮および網状真皮)から構成される。この真皮は、以下を構成する:線維芽細胞、組織球、内皮細胞、脈管周囲マクロファージ、ならびに、樹状細胞、肥胖細胞、平滑筋細胞、ならびに、末梢神経の細胞およびそれらの器官内レセプター。真皮としては、コラーゲンおよびレチクリンのような線維物質、ならびに細胞間質(主に、グリコサミノグリカン(ヒアルロン酸、コンドロイチン硫酸およびデルマタン硫酸))が挙げられる。
【0009】
いくつかの方法は、薬物の経皮輸送を増強することが提唱されている。例えば、化学エンハンサー(Burnette,R.R.Developmental Issues and Research Initiatives;Hadgraft J.,Ed.,Marcel Dekker:1989 247〜288頁)、イオン導入、および他のものが、使用されている。しかし、特に、皮膚にわたる薬物の送達を調査している、30余りの長い研究にも関わらず、12にも満たない薬物が、現在、例えば、皮膚用パッチ剤における経皮投与に利用可能であるにすぎない。
【0010】
血液脳関門にわたる薬物および他の分子の輸送はまた、問題がある。血液脳関門を構成Sする脳毛細管は、内皮細胞自身の間で接着結合を形成する内皮細胞から構成される(Goldsteinら、Scientific American 255:74〜83(1986);Pardridge,W.M.,Endocrin.Rev.7:314〜330(1986))。内皮細胞、および、細胞を連結する緊密な細胞間連結は、血液から脳への多くの分子の受動的な移動に対する障壁を形成する。血液脳関門の内皮細胞は、飲小胞をほとんど有さず、この飲小胞は、他の組織においては、毛細管壁にわたる無差別な輸送をいくらか可能にし得る。血液脳関門はまた、細胞を貫通し、従って、薬物および他の分子の無制限の通過を可能にする連続的な裂孔またはチャネルによっても妨害されない。
【0011】
従って皮膚、胃腸管、眼、および血液脳関門のような、上皮組織および内皮組織にわたる化合物(薬物を含む)の送達を増大する、改良された試薬および方法が必要である。本発明は、この必要性および他の必要性を満たす。
【発明の開示】
【課題を解決するための手段】
【0012】
(発明の要旨)
本発明は、化合物を動物の胃腸上皮に標的化する方法を提供する。本方法は、化合物および送達増強トランスポーターを含む結合体を、胃腸上皮に投与する工程を包含する。送達増強トランスポーター(これはまた、本発明によって提供される)は、十分なグアジニノ部分またはアミジノ部分を有し、送達増強トランスポーターの非存在下での化合物の送達と比較して、胃腸上皮への結合体の送達を増大する。いくつかの実施形態において、胃腸上皮への結合体の送達は、送達増強トランスポーターの非存在下での化合物の送達と比較して、少なくとも2倍増大される。より好ましい実施形態において、結合体の胃腸上皮への送達は、送達増強トランスポーターの非存在下での化合物の送達と比較して、少なくとも10倍増大される。送達増大トランスポーターおよび化合物は、代表的には、リンカーを介して結合される。さらに、この結合体は、化合物に連結された2つ以上の送達増強トランスポーターを含み得る。
【0013】
代表的には、送達増強トランスポーターは、50未満のサブユニットを含み、そして、少なくとも6つのグアニジノ部分およびアミジノ部分を含む。いくつかの実施形態において、このサブユニットはアミノ酸である。いくつかの実施形態において、送達増強トランスポーターは、6〜25のグアニジノ部分またはアミジノ部分、ならびに、より好ましくは7と15との間のグアニジノ部分、ならびになおさらに好ましくは、少なくとも6つの連続したグアニジノ部分および/またはアミジノ部分を有する。いくつかの実施形態において、送達増強トランスポーターは、本質的に5〜50のサブユニットからなり、その少なくとも50%は、グアニジノ残基またはアミジノ残基を含む。これらの実施形態のいくつかにおいて、このサブユニットは、天然アミノ酸または非天然アミノ酸である。例えば、いくつかの実施形態において、送達増強トランスポーターは、5〜25のアルギニン残基またはそのアナログを含む。例えば、トランスポーターは、7個の連続したD−アルギニンを含み得る。
【0014】
いくつかの実施形態において、送達増強トランスポーターは、7〜15のアルギニン残基またはアルギニンアナログを含む。送達増強トランスポーターは、少なくとも1つのアルギニン(これは、D−アルギニンである)を有し得、そして、いくつかの実施形態では、全てのアルギニンがD−アルギニンである。いくつかの実施形態において、アミノ酸の少なくとも70%がアルギニンまたはアルギニンアナログである。いくつかの実施形態において、送達増強トランスポーターは、少なくとも5つの連続したアルギニンまたはアルギニンアナログを含む。送達増強トランスポーターは、非ペプチド骨格を含み得る。さらに、いくつかの局面において、トランスポーターは、送達増強分子が、天然に存在するタンパク質に結合されるアミノ酸配列に結合されない。
【0015】
本発明の送達増強トランスポーターおよび方法は、薬物、診断剤、および目的の他の化合物を胃腸上皮に送達するのに有用である。本発明の方法および組成物は、化合物を特定の投与部位に送達するだけでなく、全身送達を提供するために使用される。いくつかの実施形態において、結合体は、頬に(bucally)または坐薬として投与される。この結合体の化合物は、疾患(例えば、炎症性腸疾患、結腸癌、潰瘍性大腸炎、胃腸潰瘍、便秘、ならびに、塩および水吸収の失調)に対する治療薬であり得る。従って、この化合物としては、免疫抑制剤、コルチコステロイド、緩下薬、抗生物質または抗腫瘍剤が挙げられ得る。本発明のいくつかの局面において、化合物は、回腸(iliem)および/または結腸に標的化される。
【0016】
上記で討論したように、送達される化合物は、リンカーによって送達増強トランスポーターに結合され得る。いくつかの実施形態において、このリンカーは、化合物が上皮組織および/もしくは内皮皮組織の1つ以上の層に移り、そして/または通過した後、生物学的活性形態で、送達増強トランスポーターからこの化合物を放出する、放出可能なリンカーである。いくつかの実施形態において、この化合物は、溶媒媒介切断によって、リンカーから放出される。結合体は、いくつかの実施形態において、酸性pHで実質的に安定であるが、化合物は、実質的に、生理学的pHで、送達増強トランスポーターから放出される。いくつかの実施形態において、この結合体の半減期は、皮膚または他の上皮組織もしくは内皮組織と接触するとき、5分と24時間の間である。例えば、半減期は、皮膚または他の上皮組織もしくは内皮組織と接触するとき、30分と2時間の間であり得る。
【0017】
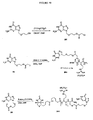
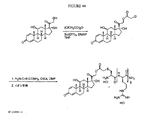
本発明の結合体構造の例としては、以下:
【0018】
【化4】

【0019】
好ましくは、Xは、以下の:−C(O)O−、−C(O)NH−、−OC(O)NH−、−S−S−、−C(S)O−、−C(S)NH−、−NHC(O)NH−、−SO2NH−、−SONH−、ホスフェート、ホスホネートホスフィネート、およびCR7R8からなる群より選択され、ここで、R7およびR8は、それぞれ独立してHおよびアルキルからなる群より選択される。いくつかの実施形態において、R4は、Sであり;R5は、NHR6であり;そしてR6は、水素、メチル、アリル、ブチルまたはフェニルである。いくつかの実施形態において、R2は、ベンジルであり;k、m、およびnは、おのおの1であり、そしてXはOである。いくつかの実施形態において、構造3を含む結合体で、Yは、Nであり、およびR2は、メチル、エチル、プロピル、ブチル、アリル、ベンジル、またはフェニルである。いくつかの実施形態において、R2は、ベンジルであり;k、m、およびnは、おのおの1であり、そしてXは、−OC(O)−である。構造4を含む結合体のいくつかの実施形態において;R4は、Sであり;R5は、NHR6であり;そしてR6は、水素、メチル、アリル、ブチルまたはフェニルである。構造4を含む結合体のいくつかの実施形態において;R5は、NHR6であり;R6は、水素、メチル、アリル、ブチルまたはフェニルであり;そしてkおよびmは、各々1である。
【0020】
本発明はまた、結合体を提供し、この結合体における生物学的薬剤からのリンカーの放出は、第1の、律速の分子内反応、続いて起こるリンカーの放出を起こすより速い分子内反応を含む。律速反応は、リンカーの置換基の適切な選択により、生理的pHよりも高いかまたは低いpHにおいても安定であるようにされ得る。しかし、一旦、結合体が、上皮組織または内皮組織の1つ以上の層を横切って通過すると、リンカーは、薬剤から切断される。この型のリンカーを有する化合物の例は以下のような構造6である:
【0021】
【化5】
【0022】
いくつかの実施形態において、Xは、−C(O)O−、−C(O)NH−、−OC(O)NH−、−S−S−、−C(S)O−、−C(S)NH−、−NHC(O)NH−、−SO2NH−、−SONH−、ホスフェート、ホスホネートホスフィネート、およびCR7R8からなる群より選択され、ここで、R7およびR8は、各々独立して、Hおよびアルキルからなる群より選択される。いくつかの実施形態において、R4はSであり;R5は、NHR6であり;R6は、水素、メチル、アリル、ブチルまたはフェニルである。
【0023】
好ましい実施形態において、本発明の組成物は、溶媒媒介型切断を受けやすいリンカーを含む。例えば、好ましいリンカーは、酸性のpHでは実質的に安定であるが、生理的pHでは実質的に切断される。
【0024】
(詳細な説明)
(定義)
「上皮組織」は、身体の表面、空間および腔の表面領域を覆う基本的な組織である。上皮組織は、主に、お互いに結合し、細胞によって代表的に生成される細胞外マトリックス(基底膜)にある上皮細胞で構成される。上皮組織は、細胞の形状に基づく3つの一般的な型を含む:扁平上皮、立方上皮および円柱上皮。扁平上皮(これは、肺および血管の輪郭となる)は、平らな細胞で作製される。立方上皮は、腎臓細管に並び、立方形状の細胞で構成されるが、円柱上皮細胞は、消化管に並び、円柱状の外見を有する。上皮組織はまた、その組織における細胞層の数に基づいて分類され得る。例えば、単純な上皮組織は、細胞の単一の層で構成され、これらの各々は基底膜上にある。「層状の」上皮組織は、お互いに積み重なったいくつかの組織で構成され;すべての細胞が基底膜と接触しているわけではない。「偽層状の」上皮組織は、すべて基底膜と接触するが、層状に見える細胞を有する。なぜなら、核が種々のレベルにあるからである。
【0025】
用語「経上皮」送達または「経上皮」投与は、局所投与による、身体表面または組織(例えば、無傷な皮膚または粘膜)の1以上の層を介した浸透による薬剤の送達または投与をいう。従って、この用語は、経皮投与(例えば、経皮吸収)および経粘膜投与の両方を含むことが意図される。送達は、例えば、組織のより深い層までであり得、そして/または血流への送達であり得る。
【0026】
本明細書中で使用される場合、「送達増強」、「透過増強」または「浸透増強」は、上皮組織または内皮組織の1以上の層へ、そして上皮組織または内皮組織の1以上の層を横切って送達される化合物の送達量および/または送達速度における増加に関する。送達の増強は、動物またはヒトの皮膚または他の組織の1以上の層を通過する化合物の速度および/または量を測定することによって、観察され得る。送達増強はまた、化合物が送達される組織への深さにおける増加、および/または上皮組織もしくは他の組織の1つ以上の細胞型への送達の程度における増加(例えば、皮膚または他の組織の線維芽細胞、免疫細胞、および内皮細胞への増加した送達)を含み得る。このような測定は、例えば、米国特許第5,891,462号に記載されるような拡散細胞装置を用いることによって、容易に得られる。
【0027】
皮膚または他の上皮膜もしくは内皮膜を通過する薬剤の送達および/または皮膚または他の上皮膜もしくは内皮膜中への薬剤の送達の量または速度は、時折、予め決定された領域の皮膚または他の組織を通過する化合物の量に関して定量され、これは、インタクトな破壊されていない成体の皮膚または粘膜組織の領域と規定される。この領域は一般に、約5cm2〜約100cm2の範囲、より一般には約10cm2〜約100cm2の範囲、なおより一般には約20cm2〜約60cm2の範囲である。
【0028】
用語「グアニジル」、「グアニジニル」および「グアニジノ」は、式−HN=C(NH2)NH(非プロトン化型)を有する部分をいうために相互変換可能に使用される。例として、アルギニンは、グアニジル(グアニジノ)部分を含み、そしてまた2−アミノ−5−グアニジノ吉草酸またはα−アミノ−δ−グアニジノ吉草酸という。「グアニジウム」は、陽性に荷電した共役酸型をいう。用語「グアニジノ部分」としては、例えば、グアニジン、グアニジニウム、グアニジン誘導体(例えば、(RNHC(NH)NHR’)、一置換グアニジン、モノグアニド、モノグアニド、ビグアニド、ビグアニド誘導体(例えば、(RNHC(NH)NHC(NH)NHR’)などが挙げられる。さらに、用語「グアニジノ部分」は、任意の1つ以上のグアニド単独または異なるグアニドの組合せを包含する。
「アミジニル」および「アミジノ」は、式C(=NH)(NH2)を有する部分をいう。「アミジニウム」は、陽性に荷電した共役酸形態をいう。
【0029】
用語「トランス−障壁濃度(trans−barrier)」または「トランス−組織濃度」は、特定の組成物が付加されている組織側の反対、または「トランス」である、上皮障壁組織または内皮障壁組織の1つ以上の層の側面に存在する化合物の濃度をいう。例えば、化合物が、皮膚に塗布される場合、皮膚の1つ以上の層を実質的に横切って測定された化合物の量は、化合物のトランス−障壁濃度である。
【0030】
「生物学的に活性な薬剤」または「生物学的に活性な基質」は、細胞による取込みの際、細胞の構造、機能、または組成物における観察可能な変化を生じる化学基質(例えば、低分子、高分子、または金属イオン)をいう。観察可能な変化としては、1つ以上のmRNAの発現の増加または減少、1つ以上のタンパク質の発現の増加または減少、タンパク質または他の細胞成分リン酸化、酵素の阻害または活性化、結合対のメンバー間での結合の阻害または活性化、代謝物の合成増加または減少した、速度増加または減少した細胞増殖などが挙げられる。
【0031】
用語「治療剤」、「治療組成物」および「治療基質」は、限定はないが、哺乳動物種の利益のために使用され得る任意の組成物をいう。このような薬剤は、例えば、イオン、有機低分子、ペプチド、タンパク質またはポリペプチド、オリゴヌクレオチド、およびオリゴ糖、の形態をとり得る。
【0032】
本明細書中で使用される場合、用語「高分子」は、以下によって例示されるがそれらに限定されない大分子(1000ダルトンよりも大きなMW)をいう:生物学的起源または合成起源のペプチド、タンパク質、オリゴヌクレオチドおよびポリヌクレオチドをいう。
【0033】
「有機低分子」は、1000ダルトン未満または1000ダルトンの分子量(MW)を有する炭素含有薬剤をいう。
【0034】
用語「非ポリペプチド剤」および「非ポリペプチド治療剤」は、送達増強トランスポーターを含まず、そしてポリペプチド以外の生物学的活性剤である、結合体の部分をいう。非ポリペプチド剤の例は、アンチセンスオリゴヌクレオチドであり、これはポリアルギニンペプチドに結合して、上皮組織または内皮組織の1つ以上の層中への送達および上皮組織または内皮組織の1つ以上の層を通過する送達を増強するための結合体を形成し得る。
【0035】
本明細書中で使用される「サブユニット」は、より大きなポリマー化合物を形成するために結合されるモノマー単位である。アミノ酸のセットは、サブユニットの例である。各アミノ酸は、共通の骨格(−C−C−N−)、およびその側鎖において異なる種々のアミノ酸を共有する。この骨格は、ポリペプチド中で繰り返される。サブユニットは、ポリマー骨格におけるエレメントの最短の繰り返しパターンを表す。例えば、2つのアミノ酸のペプチドは、ペプチドとみなされない。なぜならば、2つのアミノ酸は、ポリマー骨格における最短の繰り返しパターンのエレメントを有さないからである。
【0036】
用語「ポリマー」は、共有結合によって結合された2つ以上の同一または非同一のサブユニットの直鎖をいう。ペプチドは、ポリマーの例であり;ペプチドは、ペプチド結合(linkage)(アミド結合(bond))によって結合される同一または非同一のアミノ酸サブユニットからなり得る。
【0037】
本明細書中で使用される場合、用語「ペプチド」は、ペプチド結合によって結合された、D−アミノ酸もしくはL−アミノ酸、またはD−アミノ酸およびL−アミノ酸の混合物の単鎖からなる化合物をいう。一般に、ペプチドは、少なくとも2つのアミノ酸残基を含み、そして約50アミノ酸長未満である。D−アミノ酸は、本明細書中で小文字の1文字アミノ酸記号(例えば、rはD−アルギニン)によって、L−アミノ酸は、大文字の1文字アミノ酸記号(例えば、RはL−アルギニン)によって表される。ホモポリマーペプチドは、1文字アミノ酸記号、続いてそのペプチド中に連続して存在するアミノ酸の数によって表される(例えば、R7は、L−アルギニン残基からなるヘプタマーを表す)。
【0038】
本明細書中で使用される場合、用語「タンパク質」は、ペプチド結合によって連結された直状に整列されたアミノ酸からなる化合物をいうが、ペプチドとは対照的に、十分に規定された構造を有する。タンパク質は、ペプチドとは対照的に、一般に50以上のアミノ酸の全員からなる。
【0039】
本明細書中で使用される場合、「ポリペプチド」は、少なくとも2つのアミノ酸残基のポリマーをいい、そしてこれは1つ以上のペプチド結合を含む。「ポリペプチド」は、ポリペプチドが、十分に規定された構造を有するか否かに関係なく、ペプチドおよびタンパク質を包含する。
【0040】
(好ましい実施形態の説明)
本発明は、動物の上皮組織または内皮組織の1つ以上の層中ヘ、および動物の上皮組織または内皮組織の1つ以上の層を通過する、化合物(薬物、および他の生物学的に活性な化合物を含む)の移動を増強する組成物および方法を提供する。この方法は、組織を、送達増強トランスポーターと連結した目的の化合物を含む結合体と接触させる工程を包含する。本発明によって提供される送達増強トランスポーターは、1つ以上のインタクトな上皮組織の層および内皮組織の層中ならびに1つ以上のインタクトな上皮組織の層および内皮組織の層を通過する、結合体の送達を増加させるために十分なグアニジノ部分またはアミジノ部分を含む分子である。この方法および組成物は、薬物ならびに他の生物学的に活性な分子のトランス上皮送達およびトランス内皮送達に有用であり、そしてまた、画像化分子および診断分子の送達に有用である。本発明の方法および組成物は、それらの生物学的効果を示すトランス上皮輸送またはトランス内皮輸送を必要とし、かつ(送達増強トランスポーターまたはいくつかの他の改変体への結合なしで)それ自体では、このような組織を通過し、従って、生物学的活性を示すことができないか、またはほとんどできない化合物の送達に特に有用である。
【0041】
本発明の送達増強トランスポーターおよび方法は、目的の化合物のトランス上皮組織送達およびトランス内皮組織送達を得るための既に利用可能な方法について重大な利点を提供する。このトランスポーターは、予め薬物を通さない組織を通過する薬物および他の薬剤の送達を可能にする。例えば、薬物の皮膚への送達は、以前2、3の化合物を除いて全てについてほぼ不可能であったのに対し、本発明の方法は皮膚のような上皮組織の第1の層の細胞だけでなく、皮膚の1つ以上の層を通過して化合物を送達し得る。血液脳関門はまた、薬物の輸送ならびに他の診断試薬および治療試薬の輸送に耐性であり;本発明の方法およびトランスポーターは、このような輸送を得るための手段を提供する。
【0042】
送達増強トランスポーターは、送達増強トランスポーターの非存在下での化合物の送達と比較して、1つ以上のインタクトな上皮組織層および内皮組織の層中へ、および通過する結合体の送達を増加させる。いくつかの実施形態において、この送達増強トランスポーターは、HIV−1のtatタンパク質を用いて得られるトランスポーターにおいて結合体の送達を有意に増加させる(Frankelら(1991)PCT公開番号WO 91/09958)。送達はまた、tat基本領域(配列RKKRRQRRRを有する残基49〜57)を含むtatタンパク質のより短いフラグメントの使用において、有意に増加される(Barsoumら(1994) WO 94/04686およびFawellら(1994)Proc.Nat’l.Acad.Sci.USA 91:664−668)。好ましくは、本発明のトランスポーターを用いて得られる送達は、tat残基49〜57で得られる送達よりも、2倍、なおより好ましくは6倍、なおより好ましくは10倍、そしてなおより好ましくは20倍より増加される。いくつかの実施形態において、本発明の組成物は、tat残基49〜57を含まない。
【0043】
同様に、本発明の送達増強トランスポーターは、培養されたニューロンによって迅速に内在化されるAntennapediaのホメオドメイン由来の16アミノ酸ペプチド−コレステロール結合体と比較して、増加した送達を提供し得る(Brugidouら(1995) Biochem.Biophys.Res.Commun.214:685−93)。この領域(最小で43〜58残基)は、アミノ酸配列RQIKIWFQNRRMKWKKを有する。Herpes simplexタンパク質VP22(tatおよびAntennapediaドメインのような)は、細胞中への輸送を増強することが以前から公知であるが、内皮膜および上皮膜中への輸送および内皮膜および上皮膜を通過する輸送を増強することは知られていない(ElliotおよびO’Hare(1997)Cell 88 :223−33;Dilberら(1999)Gene Ther.6:12−21;Phelanら(1998)Nature Biotechnol.16:440−3).現在好ましい実施形態において、送達増強トランスポーターは、AntennapediaホメオドメインおよびVP22タンパク質と比較して有意に増加した送達を提供する。いくつかの実施形態において、本発明の組成物は、Antennapediaホメオドメイン、VP22タンパク質または8つ連続したアルギニンを含まない。
【0044】
(送達増強トランスポーターの構造)
本発明の送達増強トランスポーターは、化合物の送達を増加させるために十分なグアニジノ部分および/またはアミジノ部分を有する分子であり、ここで送達増強トランスポーターは、上皮組織(例えば、皮膚または粘膜)あるいは内皮組織(例えば、血液脳関門)の1つ以上の層中およびこれらを横切って付着される。この送達増強トランスポーターは、一般にグアニジノおよび/またはアミジノの側鎖部分に付着する骨格構造を含む。いくつかの実施形態において、この骨格は、サブユニット(例えば、繰り返しのモノマー単位)からなるポリマーであり、この少なくともいくつかのサブユニットは、グアニジノ部分またはアミジノ部分を含む。
【0045】
(A.グアニジノ部分および/またはアミジノ部分)
送達増強トランスポーターは、代表的に少なくとも5つのグアニジノ部分および/またはアミジノ部分、そしてより好ましくは7つ以上のこのような部分を示す。好ましくは、送達増強トランスポーターは、25またはそれよりも少ないグアニジノ部分および/またはアミジノ部分を有し、そしてしばしば、15またはそれよりも少ないこのような部分を有する。いくつかの実施形態において、この送達増強トランスポーターは、本質的に50またはそれよりも少ないサブユニット、からなり、そして本質的に25以下、20以下または15以下のサブユニットからなり得る。送達増強トランスポーターは、5つのサブユニット程度に短く、この場合、全てのサブユニットは、グアニジノ側鎖部分またはアミジノ側鎖部分を含む。送達増強トランスポーターは、例えば、少なくとも6つのサブユニットを有し得、そしていくつかの実施形態において、少なくとも7つまたは10のサブユニットを有し得る。一般に、少なくとも50%のサブユニットは、グアニジノ側鎖部分またはアミジノ側鎖部分を含む。送達増強トランスポーターにおいて、より好ましくは、少なくとも70%のサブユニット、およびいくつかの場合少なくとも90%のサブユニットが、グアニジノ側鎖部分またはアミジノ側鎖部分を含む。
【0046】
送達増強トランスポーター中のいくつかまたは全てのグアニジノ部分および/またはアミジノ部分は、連続し得る。例えば、送達増強トランスポーターは、6〜25の連続した、グアニジノおよび/またはアミジノ含有サブユニットを含み得る。7つ以上の連続したグアニジノおよび/またはアミジノ含有サブユニットが、いくつかの実施形態において存在する。いくつかの実施形態において、グアニジノ部分を含む各サブユニットは、少なくとも6つ連続したアルギニン残基を含むポリマーによって例示されるように、連続である。
【0047】
送達増強トランスポーターは、ペプチドによって例示される。アルギニン残基またはアルギニンのアナログは、グアニジノ部分を有するサブユニットを構築し得る。このようなアルギニン含有ペプチドは、全てのD−アミノ酸、全てのL−アミノ酸または混合されたD−アミノ酸およびL−アミノ酸のいずれかからなり得、そしてさらなるアミノ酸、アミノ酸アナログ、またはアルギニン残基間の他の分子を含み得る。必要に応じて、送達増強トランスポーターはまた、非アルギニン残基を含み得、送達されるべき化合物は直接またはリンカーを介してのいずれかが付着される。送達増強トランスポーターにおける少なくとも1つのD−アルギニンの使用は、結合体をその生物学的標的へと輸送する間、トランスポーターの生物学的安定性を増強し得る。いくつかの場合、送達増強トランスポーターは、少なくとも約50%のD−アルギニン残基であり、(ついでにこのサブユニットの全ては、D−アルギニン残基である)さらにより安定性のトランスポーターが使用される。送達増強トランスポーター分子が、ペプチドである場合、このトランスポーターは、送達増強トランスポーター分子を作製するアミノ酸が、天然に存在するタンパク質に付着するためのアミノ酸配列に付着しない。
【0048】
好ましくは、この送達増強トランスポーターは、線状である。好ましい実施形態において、上皮組織の1つ以上の層中および上皮組織の1つ以上の層を横切って送達される薬剤は、送達増強トランスポーターの末端に付着する。いくつかの実施形態において、薬剤は、結合体を形成するために単一の輸送ポリマーに連結される。他の実施形態において、薬剤に連結した1つより多い送達増強トランスポーターまたは単一の送達増強トランスポーターに連結した複数の薬剤を含み得る。
【0049】
より一般的には、各サブユニットが、以下のような高度に塩基性の側鎖部分を含むことが好ましい;(i)11よりも大きいpKa、より好ましくは12.5以上のpKaを有し、そして(ii)プロトン化状態で、共鳴安定化正荷電を共有する少なくとも2つのジャミナルなアミノ基(NH2)を含み、これらはこの部分に二座特性を与える。
【0050】
グアニジノ部分またはアミジノ部分は、側鎖のリンカーによって骨格に連結されることによって骨格から離れて伸びる。この側鎖原子は、好ましくは、メチレン炭素原子として提供されるが、いつ以上の他の原子(例えば、酸素、硫黄または窒素)もまた存在し得る。例えば、骨格に対するグアニジノ部分を結合するリンカーは、以下のように示され得る:
【0051】
【化6】
【0052】
いくつかの実施形態において、送達増強トランスポーターは、連結したサブユニットからなり、少なくともいくつかは、グアニジノ部分および/またはアミジノ部分を含む。グアニジノ部分および/またはアミジノ部分を有する適切なサブユニットの例は、以下に記載される。
【0053】
(アミノ酸)
いくつかの実施形態において、送達増強トランスポーターは、D−アミノ酸残基またはL−アミノ酸残基からなる。これらのアミノ酸は、天然に存在するアミノ酸でもあり得るし、天然に存在しないアミノ酸でもあり得る。アルギニン(α−アミノ−δ−グアニジノ吉草酸)およびα−アミノ−ε−アミジノ−ヘキサン酸(等配電子のアミジノアナログ)は、適切なグアニジノ含有アミノ酸サブユニット、およびアミジノ含有アミノ酸サブユニットの例である。アルギニン中のグアニジニウム基は、約12.5のpKaを有する。いくつかの好ましい実施形態において、トランスポーターは、少なくとも6つの連続したアルギニン残基からなる。
【0054】
他のアミノ酸(例えば、α−アミノ−β−グアニジノ−プロピオン酸、α−アミノ−γ−グアニジノ−酪酸、またはα−アミノ−ε−グアニジノ−カプロン酸(骨格鎖とグアニジウニウムの中心炭素との間に、それぞれ2、3または5の側鎖リンカー原子を含む)もまた、使用され得る。
【0055】
D−アミノ酸もまた、送達増強トランスポーターにおいて使用され得る。排他的にD−アミノ酸を含む組成物は、減少した酵素的分解の利点を有する。しかし、これらはまた、標的細胞内で大部分インタクトなまま維持され得る。このような安定性は、ポリマーがさらに結合される場合に、薬剤が生物学的に活性であるとすると、一般に問題ではない。結合体形態で不活性な薬剤について、作用部位で切断可能(例えば、細胞内の酵素媒介性切断または溶媒媒介性切断によって)なリンカーは、細胞またはオルガネラ中の薬剤の放出を促進するために結合体内に含まれるべきである。
【0056】
さらに、輸送部分は、以下の式のアミノ酸オリゴマーである:(ZYZ)nZ、(ZY)nZ、(ZYY)nZおよび(ZYYY)nZ。2001年2月7日に出願された米国特許出願番号09/779,693および2000年2月14日に出願された米国特許出願番号60/182166を参照のこと。式中の「Z」は、D−アルギニンまたはL−アルギニンである。「Y」は、グアニジル部分またはアミジニル部分を含まないアミノ酸である。添え字「n」は、2〜25の範囲の整数である。
【0057】
上記の輸送部分の式において、文字「Y」は、天然のアミノ酸または非天然のアミノ酸を表す。このアミノ酸は、本質的に、アミノ基(NH2またはNH−アルキル)およびカルボン酸基(CO2H)を(輸送部分への組込みの前に)有し、そしてグアニジル部分もアミジニル部分も含まない任意の化合物であり得る。このような化合物の例としては、D−アラニンおよびL−アラニン、D−システインおよびL−システイン、D−アスパラギン酸およびL−アスパラギン酸、D−グルタミン酸およびL−グルタミン酸、D−フェニルアラニンおよびL−フェニルアラニン、グリシン、D−ヒスチジンおよびL−ヒスチジン、D−イソロイシンおよびL−イソロイシン、D−リジンおよびL−リジン、D−ロイシンおよびL−ロイシン、D−メチオニンおよびL−メチオニン、D−アスパラギンおよびL−アスパラギン、D−プロリンおよびL−プロリン、D−グルタミンおよびL−グルタミン、D−セリンおよびL−セリン、D−スレオニンおよびL−スレオニン、D−バリンおよびL−バリン、D−トリプトファンおよびL−トリプトファン、D−ヒドロキシプロリンおよびL−ヒドロキシプロリン、D−チロシンおよびL−チロシン、サルコシン、β−アラニン、γ−アミノ酪酸ならびにε−アミノカプロン酸が挙げられる。上記のそれぞれの式において、各Yは、輸送部分に存在する任意の他のYから独立しているが、いくつかの実施形態において、全てのY基は、同じであり得る。
【0058】
好ましい実施形態の1つの群において、輸送部分は、式(ZYZ)nZを有し、ここで、各「Y」は、グリシン、β−アラニン、γ−アミノ酪酸およびε−アミノカプロン酸から独立して選択され、「Z」は、好ましくはL−アルギニンであり、そしてnは、好ましくは、2〜5の範囲の整数である。より好ましくは、各「Y」は、グリシンまたはε−アミノカプロン酸であり、そしてnは3である。実施形態のこの群において、グリシンの使用は、輸送部分が、組成物全体が、組換え方法によって調製され得るような、ポリペプチド生物学的薬剤に直接融合されるか、または共有結合される組成物にとって好ましい。例えば、固相方法を用いて輸送部分が収集される実施形態において、ε−アミノカプロン酸が好ましい。
【0059】
別の好ましい実施形態において、輸送部分は式(ZY)nZを有し、ここで各「Y」は、好ましくはグリシン、β−アラニン、γ−アミノ酪酸およびε−アミノカプロン酸から選択され、「Z」は、好ましくはL−アルギニンであり、そしてnは、好ましくは、4〜10の範囲の整数である。より好ましくは、各「Y」は、グリシンまたはε−アミノカプロン酸であり、そしてnは6である。特定の実施形態の上記の群と同様に、グリシンの使用は、輸送部分が、組成物全体が組換え方法によって調製され得るような、ポリペプチド生物学的薬剤に直接融合されるか、または共有結合される組成物にとって好ましい。輸送部分の溶液または固相構築物について、ε−アミノカプロン酸が好ましい。
【0060】
好ましい実施形態のなお別の群において、輸送部分は、式(ZYY)nZを有し、ここで、各「Y」は、好ましくは、グリシン、β−アラニン、γ−アミノ酪酸およびε−アミノカプロン酸から選択され、「Z」は、好ましくは、L−アルギニンであり、そしてnは、好ましくは、4〜10の範囲の整数である。より好ましくは、各「Y」は、グリシンまたはε−アミノカプロン酸であり、そしてnは6である。
【0061】
好ましい実施形態のなお別の群において、輸送部分は、式(ZYYY)nZを有し、ここで、各「Y」は、好ましくは、グリシン、β−アラニン、γ−アミノ酪酸およびε−アミノカプロン酸から選択され、「Z」は、好ましくはL−アルギニンであり、そしてnは、好ましくは、4〜10の範囲の整数である。より好ましくは、「Y」はグリシンであり、そしてnは6である。
【0062】
他の実施形態において、それぞれのY基は、輸送部分の特定の所望の特性を増強するように選択される。例えば、より疎水性の特徴を有する輸送部分が所望される場合、各Yは、代表的に疎水性アミノ酸(例えば、フェニルアラニン、フェニルグリシン、バリン、ロイシン、イソロイシン)として一緒にグループ化される、天然に存在するアミノ酸から選択され得る。同様に、いくつかのまたは全てのY基が親水性アミノ酸(例えば、リジン、セリン、スレオニン、グルタミン酸など)である場合、より親水性の特徴を有する輸送部分が、調製され得る。
【0063】
当業者は、輸送部分が、より大きいポリペプチド内のポリペプチドフラグメントであり得ることを理解する。例えば、輸送部分は、この部分に隣接するさらなるアミノ酸をなお有する式(ZYY)nZ(例えば、Xm(ZYY)nZ−Xp、ここで、添え字mおよびpは、0〜約10の整数を表し、そして各Xは、独立して天然アミノ酸または非天然アミノ酸である)であり得る。
【0064】
(他のサブユニット)
アミノ酸以外のサブユニットはまた、輸送ポリマー形成における使用のために選択され得る。このようなサブユニットとしては、ヒドロキシアミノ酸、N−メチル−アミノ酸アミノアルデヒドなどが挙げられ得るが、これらに限定されず、これらは還元されたペプチド結合を有するポリマーを生じる。他のサブユニット型が、次の節に議論されるような、選択された骨格の性質に依存して使用され得る。
【0065】
(B.骨格)
送達増強トランスポーターに含まれるグアニジノ部分および/またはアミジノ部分は、一般に線状の骨格に結合される。この骨格は、炭素、窒素、酸素、硫黄およびこのリン骨格鎖原子の大部分は代表的に炭素からなる種々の原子型を含み得る。末端グアニジノ基またはアミジノ基を含む複数の側鎖部分が、骨格に結合される。隣接の側鎖部分間の間隔は、代表的に一定であるが、本発明において使用される送達増強トランスポーターはまた、骨格に沿って側鎖部分間の種々の間隔を含み得る。
【0066】
より詳細な骨格リストとしては、N−置換アミド(CONRは、CONH結合を置換する)、エステル(CO2)、還元されたケト−メチレン(COCH2)またはメチレンアミノ(CH2NH)、チオアミン(CSNH)、ホスフィネート(PO2RCH2)ホスホアミデートおよびホスホアミデートエステル(PO2RNH)、レトロペプチド(NHCO)トランス−アルケン(CR=CH)、フルオロアルケン(CF=CH)、ジメチレン(CH2CH2)、チオエステル(CH2S)ヒドロキシエチレン(CH(OH)CH2)、メチレンオキシ(CH2O)、テトラゾール(CN4)、レトロチオアミド(NHCS)、レトロ還元された(retroreduced)(NHCH2)、スルホンアミド(SO2NH)、メチレンスルホンアミド(CHRSO2NH)、レロトスルホンアミド(NHSO2)、およびペプトイド(N−置換アミド)、およびマロネートサブユニットおよび/またはgem−ジアミノ−アルキルサブユニットを有する骨格(例えば、Fletcherら、((1998)Chem.Rev.98:763)により概説され、そして本明細書中で引用される参考文献によって詳述される。前述の置換基の多くは、ほとんどα−アミノ酸から形成される骨格に対して、ほぼ等配電子が挙げられる。ポリマー骨格を生じる。
【0067】
上記で意図されるように、ペプトイド骨格において、側鎖は、炭素原子よりもむしろ窒素原子の骨格に結合される。(例えば、Kessler(1993)Angew.Chem.Int.Ed.Engl.32:543;Zuckermanら(1992)Chemtracts−Macromol.Chem.4:80;およびSimonら(1992)Proc.Nat’l.Acad.Sci.USA 89:9367を参照のこと)。適切なペプトイド骨格の例は、ポリ−(N−置換)グリシン(ポリ−NSG)である。ペプトイドの合成は、例えば、米国特許第5,877,278号に記載される。用語が本明細書中で使用される場合、ペプトイド骨格を有するトランスポーターは、「非ペプチド」トランスポーターと考えられる。なぜならば、このトランスポーターは、天然に存在する側鎖位置を有するアミノ酸から構成されるからである。ペプトイド骨格を含む非ペプチド骨格は、増強された生物学的安定性(例えば、インビボでの酵素的分解に対する耐性)を提供する。
【0068】
C.送達増強トランスポーターの合成
送達増強トランスポーターは、当該分野で公知の任意の技術により構築される。例示のペプチドポリマーは、合成的に、好ましくはペプチド合成機(例えば、Applied Biosystems Model 433)を使用して作製され得るか、または当該分野で周知の方法により組み換え的に合成され得る。組み換え合成は一般的に、この送達増強トランスポーターが目的のポリペプチドまたはタンパク質と融合されるペプチドである場合に使用される。
【0069】
N−メチルアミノ酸およびN−ヒドロキシアミノ酸は、固相ペプチド合成において、従来のアミノ酸に置き換えられ得る。しかし、還元性のペプチド結合を有する送達増強トランスポーターの産生は、還元性のペプチド結合を含む二量体アミノ酸の合成を必要とする。このような二量体は、標準的な固相合成手法を使用してポリマー内に組み込まれる。他の合成手法は周知であり、そして、例えば、Fletcherら(1998)Chem.Rev.98:763、Simonら(1992)Proc.Nat’l.Acad.Sci.USA 89:9367、およびこれらの中に記載される引用文献の中に見出され得る。
【0070】
本発明の送達増強トランスポーターは、1つ以上の(例えば、グリシン、アラニン、およびシステインのような)非グアニジノ/非アミジノサブユニット、またはリンカー(例えば、アミノカプロン酸基)の末端を有し得、それらはその対応する送達増強トランスポーターを含む結合体の、組織縦断層の輸送の度合に、有意には影響しない。また、任意の遊離アミノ末端基は、インビボのユビキチン化を避けるために、保護基(例えば、アセチル基またはベンジル基)でキャップされ得る。
【0071】
このトランスポーターがペプトイドポリマーである場合、1つの合成方法は以下の工程を包含する:1)ペプトイドポリアミンを塩基およびピラゾール−1−カルボキシアミジンで処理して、混合物を提供する;2)この混合物を加熱し、次いで放置して冷ます;3)この冷ました混合物を酸性化する;そして4)この酸性化した混合物を精製する。好ましくは、工程1にて使用される塩基は炭酸塩(例えば、炭酸ナトリウム)であり、そして加熱工程2は、混合物を約24時間と約48時間の間にわたり約50℃に加熱する工程を包含する。精製工程は、好ましくは、クロマトグラフィー(例えば、逆相HPLC)を含む。
【0072】
D.生物学的に活性な薬剤との輸送ポリマーの結合
輸送される薬剤は、多くの実施形態に従って、送達増強トランスポーターと連結され得る。1つの実施形態において、この薬剤は、送達増強トランスポーターの末端との連結を介するか、または適切な連結基を介してその試薬の中の内部サブユニットとの連結を介するかのいずれかで、単一の送達増強トランスポーターと連結される。
【0073】
第2の実施形態において、この薬剤は上記と同様の様式で、1つより多くの送達増強トランスポーターと結合される。この実施形他は、隣接細胞の架橋を導き得るため、余り好ましくない。
【0074】
第3の実施形態において、この結合体は、送達増強トランスポーターの各々の末端と結合される2つの薬剤部分を含む。この実施形態に関して、その薬剤が10kDa未満の分子量を有することが、現在好まれる。
【0075】
先に述べた第1および第3の実施形態に関して、この薬剤は、一般的に、グアニジノ側鎖またはアミジノ側鎖が標的の膜と自由に相互作用するように、任意の一方のグアニジノ側鎖またはアミジノ側鎖とは結合されない。
【0076】
本発明の結合体は、直接的な合成スキームにより調製され得る。さらに、これら結合体生成物は、通常は、長さおよび組成が実質的に均一であり、そのためこれら生成物は不均一な混合物よりも強力な効果の一貫性および再現性を提供する。
【0077】
本発明の重要な局面に従って、任意の種々の型の生物学的に活性な薬剤との単一の送達増強トランスポーターの結合が、結合体中に巨大な疎水性部分の存在を必要とすらせずに、1層以上の上皮組織および内皮組織の中への、およびそれらを透過した、薬剤の取り込み割合を実質的に増強するのに十分であることが、本出願人らにより見出されている。実際に、巨大な疎水性部分を結合させることは、上皮組織または内皮組織を構成する細胞の脂質二重層への疎水性部分の接着に起因した、層横断輸送を有意に妨害し得るか、または阻害し得る。従って、本発明は、実質的な疎水性部分(例えば、脂質分子および脂肪酸分子)を含まない結合体を包含する。
【0078】
本発明の送達増強トランスポーターは、化学的方法または組み換え方法により、生物学的に活性な薬剤と共有結合で結合され得る。
【0079】
(1.化学的連結)
小有機分子および高分子のような生物学的に活性な薬剤は、当該分野で公知である多くの方法(例えば、Wong,S.S.編、Chemistry of Protein Conjugation and Cross−linking,CRC Press,Inc.,Boca Raton,FL(1991)参照のこと)を介して、(例えば、カルボジイミドを用いて)直接的に、または連結部分を介してのいずれかで、本発明の送達増強トランスポーターと連結され得る。特に、カルバメート結合、エステル結合、チオエーテル結合、ジスルフィド結合、およびヒドラゾン結合は、一般的に、形成が容易であり、そして大部分の適用に適している。エステル結合およびジスルフィド結合は、細胞膜を横断する物質の輸送の後、この結合が細胞質で直ちに分解されるべき場合に好まれる。
【0080】
種々の官能基(ヒドロキシル、アミノ、ハロゲン、等)は、輸送ポリマーと生物学的に活性な薬剤を結合させるために使用され得る。生物学的に活性な薬剤の活性部位の一部であることが公知でない基は、特にポリペプチドまたはその任意の部分が送達後にこの物質と結合したままであるべき場合に、好ましい。
【0081】
ポリマー(例えば、PCT出願US98/10571(公開番号 WO9852614)に記載されるように生成されたペプチド)は、一般的に、アミノ末端保護基(例えば、FMOC)を用いて作製される。このポリペプチドを合成樹脂から開裂させ、そしてその側鎖を脱保護するために使用される条件に残存し得る生物学的に活性な薬剤のために、このFMOCは、その薬剤が遊離のN末端アミンと連結され得るように、完成して樹脂に結合したポリペプチドのN末端から開裂され得る。このような場合に、結合される薬剤は、典型的に当該分野で周知の方法によって活性化されて、ポリマーのアミノ基とアミド結合またはカルバメート結合を効率的に生成する、活性エステル部分または活性カルボネート部分をそれぞれ生成する。無論、他の連結化学反応もまた使用され得る。
【0082】
副反応を最小限にするよう役立てるため、グアニジノ部分およびアミジノ部分は、従来の保護基(例えば、カルボベンジルオキシ基(CBZ)、ジ−t−BOC、PMC、Pbf、N−NO2など)を使用して保護され得る。
【0083】
カップリング反応は、任意の多数の溶媒(例えば、N,N−ジメチルホルムアミド(DMF)、N−メチルピロリジノン、ジクロロメタン、水など)の中での公知のカップリング法により実施される。例示的なカップリング試薬として、例えば、O−ベンゾトリアゾリルオキシテトラメチルウロニウム、ヘキサフルオロリン酸塩(HATU)、ジシクロヘキシルカルボジイミド、ブロモ−トリス(ピロリジノ)ホスホニウムブロミド(PyBroP)、などが挙げられる。他の試薬としては、例えば、N,N−ジメチルアミノピリジン(DMAP)、4−ピロリジノピリジン、N−ヒドロキシスクシンイミド、N−ヒドロキシベンゾトリアゾール、などが含まれ得る。
【0084】
(2.融合ポリペプチド)
本発明の送達増強トランスポーターは、当該分野で周知の方法に従って、目的のポリペプチドおよびその送達増強トランスポーターを含む融合タンパク質のベクターを構築することにより、組み換え方法により生物学的に活性なポリペプチド薬剤と結合され得る。一般的に、その送達増強トランスポーターの成分は、目的のポリペプチドのC末端またはN末端に、必要に応じて短いペプチドリンカーを介して、結合される。
【0085】
(3.放出可能なリンカー)
現在の好ましい実施形態において、生物学的に活性な薬剤は、特異的に開裂可能であるか、または放出可能である結合を使用して、送達増強トランスポーターと結合される。そのような結合の使用は、結合された送達増強トランスポーターが放出されるまで不活性である生物学的に活性な薬剤にとって、特に重要である。いくつかの場合において、送達増強トランスポーターと結合された薬物分子からなる結合体は、プロドラッグといわれ得、この場合、薬物からの送達増強トランスポーターの放出の結果、不活性型から活性型への薬物の転換を生じる。本明細書中で使用される場合、結合体の「開裂した」もしくは「開裂」、またはリンカーの「開裂した」もしくは「開裂」とは、トランスポーター分子からの生物学的薬剤の放出、それによる活性な生物学的薬剤の放出をいう。「特異的に開裂可能な」または「特異的に放出可能な」とは、トランスポーターが分解されること(例えば、タンパク質消化性の分解)よりもむしろ、開裂するトランスポーターと薬剤との間の結合をいう。
【0086】
いくつかの実施形態において、この結合は容易に開裂可能な結合であり、インビボに見出される条件下で開裂することが可能であることを意味する。従って、上皮組織および/または内皮組織の1つ以上の層の中への、そしてその層を介した透過の際、薬剤は、送達増強トランスポーターから放出される。容易に開裂可能な結合とは、例えば、特異的な活性(例えば、エステラーゼ、プロテアーゼ、ホスファターゼ、ペプチダーゼ、など)を有する酵素により、または加水分解により開裂される結合である。この目的のために、カルボン酸エステルおよびジスルフィド結合を含むリンカーが時折好ましく、そこでは前者の基は酵素的にまたは化学的に加水分解され、そして後者の基は、例えばグルタチオンの存在下において、ジスルフィド交換により供給される。この結合は、結合が上皮組織または内皮組織の1つ以上の層中に存在することが公知である酵素的活性により開裂可能であるように、選択され得る。例えば、皮膚の顆粒層は、比較的高い濃度のN−ペプチダーゼ活性を有する。
【0087】
特異的に開裂可能なリンカーは、トランスポーター分子上に設計され得る。例えば、プロテアーゼ認識部位、または他のそのような特異的に認識される酵素開裂部位を構成するアミノ酸は、トランスポーターを薬剤と連結するために使用され得る。あるいは、例えば、光または他の刺激への曝露により開裂可能である化学的リンカーまたは他の種類のリンカーは、目的の薬剤とトランスポーターを連結するために使用され得る。
【0088】
送達される薬剤および送達増強トランスポーターが、特異的に開裂可能なリンカーまたは特異的に放出可能なリンカーにより連結される結合体は、半減期を有する。本文脈においてこの用語「半減期」とは、上皮膜または内皮膜にこの結合体を投与した後、結合体の半量が分離されて遊離の薬剤を放出するのに要する時間量をいう。いくつかの実施形態についての半減期は、代表的に5分と24時間の間であり、そしてより好ましくは、30分と2時間の間である。結合体の半減期は、本発明に従って、以下に記載されるように「調整」され得るか、または改変され得る。
【0089】
いくつかの実施形態において、リンカーの開裂度合はpH依存性である。例えは、リンカーは酸性pH(例えば、pH6.5以下、より好ましくは約6以下、そしてさらにより好ましくは約5.5以下)において、薬剤と送達増強トランスポーターとの間の安定な連結を生成し得る。しかし、この結合体が生理学的なpH(例えば、pH7以上、好ましくは約pH7.4)に置かれた場合、このリンカーは開裂を受けて薬剤を放出する。このようなpH感受性は、例えば、プロトン化された場合(すなわち酸性pHにて)、求核剤として作用しない官能基を含むことにより獲得され得る。より高い(例えば、生理学的な)pHにて、この官能基はもはやプロトン化されず、従って求核剤として作用し得る。適切な官能基の例として、例えば、NおよびSが挙げられる。当業者は、自己開裂が起こるpHをうまく調整するために、このような官能基を使用し得る。
【0090】
別の実施形態において、この連結部分は自己犠牲を介して開裂される。輸送部分−生物学的に活性な化合物の結合体中のそのような連結部分は、生物学的に活性な化合物から遠位の求核剤(例えば、酸素、窒素および硫黄)および生物学的に活性な化合物に近位の開裂基(例えば、エステル、炭酸塩、カルバメート、およびチオカルバメート)を含む。開裂可能な基への求核剤の分子内攻撃の結果、共有結合の切断が生じ、それによって生物学的に活性な化合物から連結部分を放出する。
【0091】
自己犠牲の連結部分を含む結合体の例(例えば、生物学的に活性な薬剤−L−輸送部分の結合体)は、構造3、4および5により表される:
【0092】
【化7】
【0093】
構造3、実施例および好ましい実施形態により例示される連結基の第一の分岐は、式3aに例示される:
【0094】
【化8】
【0095】
従って、構造3について、以下の置換基が好ましい:AはNであり;R2はベンジルであり;k、mおよびnは1であり;Xは−OC(O)−であり、そしてYは−C(O)NH−である。
【0096】
構造4の結合は、式4aにより例示される:
【0097】
【化9】
【0098】
【化10】
【0099】
従って、好ましい実施形態の一群において、この結合体は式5により表され、ここで、Xは−OC(O)−であり;Yは−C(O)NH−であり;R4はSであり;R5はNHR6であり;そして添え字kおよびmは各々1である。好ましい実施形態の別の群において、この結合体は式2により表され、ここで、Xは−OC(O)−であり;Yは−NHC(O)−であり;R4はSであり;R5はCONH2であり;そして添え字kおよびmは各々1である。特に好ましい結合体は、R6が水素、メチル、アリル、ブチルまたはフェニルである結合体である。
【0100】
適切に保護されたジカルボン酸またはジアミンもまた、特定の生物学的薬剤と共に有用であるが、式6中に示される結合体により表される連結基は、一般的にヘテロな二機能性の型(例えば、ε−アミノカプロン酸、セリン、ホモセリン、−ブチル酪酸など)である。
【0101】
構造6に関して、以下の置換基が好ましい:R5はNHR6であり(ここで、R6は水素、メチル、アリル、ブチルまたはフェニルである);kは2であり;Xは−C(O)O−であり;そしてYは−C(O)NH−である。
【0102】
自己犠牲のリンカーは、代表的に、37℃の、pH約7.4の水中で、約10分と約24時間の間の半減期を有する分子内開裂を受ける。好ましくは、この開裂の半減期は、37℃で約7.4のpHでの水中で、約20分と約4時間の間である。より好ましくは、この開裂の半減期は、37℃で約7.4のpHでの水中で、約30分と約2時間の間である。
【0103】
構造3を有する結合体に関して、当業者は、種々のR2置換基により開裂の半減期を調節し得る。増加または減少した大きさのR2を使用して、当業者は、それぞれより長い半減期またはより短い半減期を有する結合体を獲得し得る。構造3中のR2は、好ましくはメチル、エチル、プロピル、ブチル、アリル、ベンジルまたはフェニルである。
【0104】
自己犠牲のリンカー中に塩基性基または酸性基がある場合、当業者はしばしば結合体の溶液のpHに従って、開裂の半減期を調節し得る。例えば、構造3の骨格のアミン基は、酸性pH(例えば、pH5.5)でプロトン化される。このアミンは、プロトン化される場合、分子内開裂を誘導する求核剤としては利用不可能である。しかし、生理学的pH(7.4)の媒質中への結合体の導入の際、このアミンは、半減期の有意な一部の時間に非プロトン化される。この開裂の半減期は、それに相応して減少する。
【0105】
1つの実施形態において、自己犠牲のリンカーの開裂は2段階で生じる:連結部分の一部の開裂を生じる、求核基の分子内反応;および、連結部分の残存部分の脱離である。この開裂の第1段階は、速度制限的であり、そしてpH感受性および半減期についてうまく調整され得る。
【0106】
構造6は、輸送部分−生物学的に活性な化合物の結合体中に組み込まれる、2段階の自己犠牲の部分の例である:
【0107】
【化11】
【0108】
式6の結合体を生成するための適切な連結基の例は:
【0109】
【化12】
【0110】
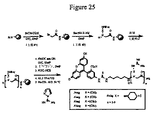
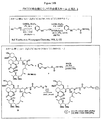
式6aの連結基を含む結合体の構造は、実施例23(図41もまた参照のこと)中に提供される。この実施例(および図)において、2,4−ジメチル−4−ヒドロキシメチルフェノール(41i)のα−クロロ酢酸エステルは、ジシクロヘキシルカルボジイミド(DCC)および4−ジメチルアミノピリジン(DMAP)を使用してレチノイン酸(41ii)とカップリングされ、中間体41iiiを生じる。アルギニン七量体の輸送部分のN末端にあるシステイン残基との、41iiiの続くカップリングにより、標的の結合体41ivを生じる。
【0111】
好ましくは、式6の結合体中に使用される連結基は、Arが置換または非置換のフェニレン基であり;R4がSであり;R5がNHR6であり(ここでR6は水素、メチル、アリル、ブチル、アセチルまたはフェニルである);kおよびmは1であり;Xは−C(O)O−であり;そしてYは−C(O)O−または−C(O)NH−である、基である。より好ましくは、R6は水素またはアセチルである。
【0112】
上記の連結基はアルギニン七量体を含む結合体に関して記載されているが、当業者はこの技術が本発明の「間隙の」アルギニン輸送部分との結合体に容易に適用されることを理解する。
【0113】
本発明の使用に対して、なお他に有用な連結基は、同時係属PCT出願中に記載されている。例えば、同様の組成物(例えば、生物学的に活性な薬剤および輸送オリゴマー)に対する連結基を記載する、PCT出願US98/10571(公開番号 WO9852614)およびUS00/23440(公開番号 WO01/13957)を参照のこと。これら出願中に記載される連結技術は、本発明の組成物において、同様の様式で使用され得る。
【0114】
従って、実施形態の1群において、連結部分は、生物学的に活性な化合物から遠位の第一の開裂可能な基、および生物学的に活性な化合物に近位の第二の開裂可能な基を含む。第一の開裂可能な基の開裂は、第二の開裂可能な基と分子内で反応し得る求核剤を生成して、それによって生物学的に活性な化合物から連結部分を開裂させる。第一の基が開裂される方法の例として、光照射および酵素に媒介される加水分解が挙げられる。この方法論は、PCT出願US98/10571(公開番号 WO9852614)中に論じられる、種々の関連低分子の結合体に関して例示されている。
【0115】
1つのアプローチにおいて、結合体は、PCT出願US00/23440(公開番号 WO01/13957)(PCT出願US98/10571(公開番号 WO9852614)もまた参照のこと)の図5A中に図示されるように、ジスルフィド結合を含み得、この出願は、リンカーとして役立つN−アセチル保護されたシステイン基により、細胞傷害性薬剤の6−メルカプトプリンと連結された輸送ポリマーTを包含する、結合体(I)を示す。従って、この細胞傷害性薬剤はジスルフィド結合により6−メルカプト基と結合されて、そしてこの輸送ポリマーはアミド結合を介してシステインカルボニル部分と結合される。還元またはジスルフィド交換によるジスルフィド結合の開裂は、遊離した細胞傷害性薬剤の放出を生じる。ジスルフィド含有結合体を合成する方法は、PCT出願US98/10571の実施例9A中に提供される。そこで記載される生成物は、N−アセチル−Cys−Ala−Alaリンカーにより6−メルカプトプリンと連結されるArg残基の七量体を含み、ここでこのAla残基は、そのジスルフィドをチオールにより接近させるための追加のスペーサー、そして細胞内の開裂のための還元剤として含まれる。この例におけるリンカーもまた、アミド結合の使用を例示し、そしてこの結合は細胞内で酵素的に開裂され得る。
【0116】
別のアプローチにおいて、この結合体は、電磁照射への曝露で開裂する、光開裂可能なリンカーを含む。この方法論の適用は、メタ−ニトロベンゾエートの連結部分を介して6−メルカプトプリンに連結される輸送ポリマーTを含む結合体(II)を示す、PCT出願US00/23440(公開番号 WO01/13957)の図5B中で、関連システムに対して提供される。ポリマーTはベンゾエートカルボニル基とのアミド結合によりニトロベンゾエート部分と連結され、そして細胞傷害性薬剤はその6−メルカプト基を介してp−メチレン基と結合される。この化合物は、NaOCH3/メタノールの存在下で加熱しながら、6−メルカプトプリンをp−ブロモメチル−m−ニトロ安息香酸と反応させ、続いてそのベンゾエートカルボン酸の輸送ポリマー(例えば、ポリマーと結合したγ−アミノ酪酸リンカーのアミノ基(例えば、PCT出願US98/10571の実施例9Bもまた参照のこと))とのカップリングにより、生成され得る。結合体の光照射は、メルカプトメチル部分にオルトであるニトロ基により、6−メルカプトプリンの放出を生じる。このアプローチは、特に生体の選択した領域への局所的な薬物活性化のため、当該分野で公知の光療法における利用を見出す。
【0117】
好ましい実施形態の一群では、以下のアプローチによって例示されるように、切断可能なリンカーは、生物学的に活性な薬剤からオリゴマーを切断するように協同し得る第1および第2の切断可能な基を含む。すなわち、切断可能なリンカーは、この薬剤に対して遠位にある第1の切断可能な基、およびこの薬剤に対して近位にある第2の切断可能な基を含み、その結果、第1の切断可能な基の切断は、第2の切断可能な基を切断するために分子内で反応し得る求核性部分を含むリンカー−薬剤結合体を生じ、それによってリンカーおよびポリマーから薬剤を放出する。
【0118】
再度、共有に係る同時係属中のPCT出願US00/23440(公開番号WO01/13957)を参照する。このPCT出願US00/23440において、図5Cは、抗癌剤である5−フルオロウラシル(5FU)に連結したトランスポーターポリマーTを含む結合体(III)を示す。この図において、連結は、改変されたリジル残基によって提供される。トランスポーターポリマーは、α−アミノ基に連結され、そして5−フルオロウラシルは、α−カルボニルを介して連結される。リジルε−アミノ基は、o−ヒドロキシメチルニトロベンゼンのカルバメートエステルに修飾されており、これは、結合体内に第1の光不安定で切断可能な基を含む。光照射は、結合体からニトロベンゼン部分を切断し、また迅速に分解して遊離α−アミノ基(有効な求核剤)を与えるカルバメートを残す。α−アミノ基の、5−フルオロウラシル基に対するアミド連結との分子内反応は、5−フルオロウラシル基の放出と共に環化を導く。
【0119】
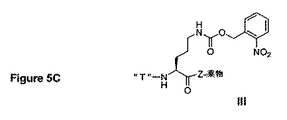
本発明において有用なさらに他のリンカーが、PCT出願US00/23440(公開番号WO01/13957)において提供されている。特に、US00/23440の図5Dは、抗癌剤であるパクリタキセルの2’−酸素に連結した送達増強トランスポーターTを含む結合体(IV)を例示する。この連結は、以下を含む連結部分によって提供される:(i)送達増強トランスポーターに結合した窒素原子、(ii)窒素原子に対してパラ位に位置するリン酸モノエステル、および(iii)窒素原子に対してメタ位のカルボキシメチル基であり、これは、カルボキシレートエステル結合によってパクリタキセルの2’−酸素に結合される。結合体からのリン酸基の酵素的切断は、遊離したフェノールヒドロキシル基を与える。次いで、この求核基は、カルボキシレートエステルと分子内反応し、その生物学的標的に完全に結合し得る遊離パクリタキセルを放出する。PCT出願US98/10571の実施例9Cは、この型の結合体を調製するための合成プロトコールを記載している。
【0120】
さらに他の適切なリンカーが、PCT出願US00/23440(公開番号WO01/13957)の図5Eにおいて例示されている。ここで提供されるアプローチにおいて、送達増強トランスポーターは、生物学的に活性な薬剤(例えば、パクリタキセル)に、アミノアルキルカルボン酸によって連結される。好ましくは、リンカーアミノ基は、3〜5個の鎖の原子(n=3〜5)、好ましくは、3個または4個のいずれかの鎖の原子(これらは、好ましくは、メチレン炭素として提供される)によって、リンカーカルボキシル炭素に連結される。図5Eに見られるように、リンカーアミノ基は、アミド結合によって送達増強トランスポーターに結合され、エステル結合によってパクリタキセル部分に結合される。アミド結合の酵素的切断は、送達増強トランスポーターを放出して、そして遊離した求核アミノ基を生成する。次いで、この遊離したアミノ基は、エステル基と分子内反応して、パクリタキセルからリンカーを放出し得る。
【0121】
別のアプローチにおいて、結合体は、あるpHで不安定であるが、別のpHで安定であるリンカーを含む。例えば、PCT出願US00/23440(公開番号WO01/13957)の図6は、生理学的pHで切断されるが、酸性pHで安定であるリンカーを有する結合体を合成する方法を例示する。好ましくは、このリンカーは、約6.6〜約7.6のpHで水中において、切断される。好ましくは、このリンカーは、約4.5〜約6.5のpHで水中において安定である。
【0122】
(送達増強トランスポーターの使用)
送達増強トランスポーターは、治療的適用、予防的適用および診断的適用における使用を見出す。この送達増強トランスポーターは、1層以上の皮膚または他の上皮組織(例えば、胃腸、肺、眼など)内に、およびそれらを横切って、あるいは、血液脳障壁のような内皮組織を横切って、診断的または生物学的に活性な試薬を運搬し得る。この特性により、この試薬は、それらの生物学的効果を発揮するために1層以上の組織層を横切って浸透しなければならない薬剤を送達することによって状態を処置するために有用となる。
【0123】
さらに、本発明のトランスポーターはまた、フリン(furin)インヒビターとして、単独でかまたは別の治療剤もしくは他の化合物と組み合わせて使用され得る。例えば、種々のポリアルギニントランスポーターに加えて、本明細書中に記載される合成トランスポーター(ペプトイド、および天然には存在しないアミノ酸を含有する合成トランスポーターを含む)が、フリンを阻害するために使用され得る。例えば、Cameronら、J.Biol.Chem.275(47):36741−9を参照のこと。フリンは、種々のプロタンパク質をそれらの活性成分へと変換するプロテアーゼである。フリンの阻害は、例えば、ビルレンスまたは複製をフリン活性に依存するウイルスによる感染を処置するために有用である。例えば、Molloyら、T.Cell Biol.9:28−35(1999)を参照のこと。
【0124】
同様に、本発明のトランスポーターは、カプテシンC(capthesin C)の有用なインヒビターである。例えば、特定のポリアルギニン化合物は、カプテシンCのインヒビターである。例えば、Hornら、Eur.J.Biochem.267(11):3330−3336(2000)を参照のこと。同様に、本発明のトランスポーター(合成アミノ酸を含有するものを含む)は、カプテシンCを阻害するために有用である。
【0125】
本発明の1つの局面では、フリンインヒビターアッセイを使用して、さらなるトランスポーターについてスクリーニングし得る。例えば、候補のトランスポーター化合物を、標準的な競合アッセイを使用して、フリンまたはカプテシンCに結合するその能力についてポリアルギニンと競合するその能力を試験し得る。あるいは、候補のトランスポーターは、Cameronら(前出)およびHornら(前出)において考察されたように、フリンプロテアーゼ活性を阻害するその能力についてスクリーニングされ得る。次いで、特に活性な候補が、組織(例えば、上皮)中へのおよび/または組織(例えば、上皮)を横切るトランスポーターとして作用するその能力についてさらに試験され得る。
【0126】
本発明の組成物および方法は、ヒトのおよび獣医学的な治療の分野において、特に有用性を有する。一般的に、投与される用量は、エフェクター部位にpM〜μMの濃度の治療用組成物を送達するのに有効である。適切な用量および濃度は、治療用の組成物または薬物、意図される送達の部位、および投与の経路のような因子に依存し、これらの全ては、当該分野で周知の方法に従って経験的に導き出され得る。さらなる指針は、当該分野で公知であるように、用量を評価するための実験動物モデルを使用する研究から得られ得る。
【0127】
必要に応じて適切な薬学的賦形剤を伴う本発明の化合物の投与は、任意の受容された投与様式を介して実行され得る。従って、投与は、例えば、静脈内投与、局所的投与、皮下投与、経皮投与、筋肉内投与、経口投与、関節内投与、非経口投与、腹膜投与、鼻腔内投与または吸入であり得る。従って、投与の適切な部位として、皮膚、気管支、胃腸、肛門、膣、眼、および耳が挙げられるが、これらに限定されない。これらの処方物は、好ましくは、正確な用量の単回投与に適切な単位用量形態において、例えば、錠剤、丸剤、カプセル、散剤、溶液、懸濁液、エマルジョン、坐剤、保持浣腸(retention enema)、クリーム、軟膏、ローション、エアロゾル、点眼剤などのような、固体、半固体、凍結乾燥した散剤、または液体投与形態の形態をとり得る。
【0128】
組成物は、代表的には、従来の薬学的キャリアまたは賦形剤を含み、さらに、他の医薬剤、キャリア、アジュバントなどを含み得る。好ましくは、この組成物は、本発明の化合物(単数または複数)の重量を基準に、約5%〜75%であり、残りは適切な薬学的賦形剤からなる。適切な賦形剤は、当該分野で周知の方法(例えば、REMINGTON’S PHARMACEUTICAL SCIENCES、第18編、Mack Publishing Co.,Easton,PA(1990))によって、特定の組成物および投与の経路に合わせられ得る。
【0129】
経口投与のためには、このような賦形剤としては、薬学的等級のマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、ナトリウムサッカリン、タルク(talcum)、セルロース、グルコース、ゼラチン、スクロース、炭酸マグネシウムなどが挙げられる。この組成物は、溶液、懸濁液、錠剤、丸剤、カプセル、散剤、持続性放出処方物などの形態をとり得る。
【0130】
いくつかの実施形態において、薬学的組成物は、丸剤、錠剤またはカプセルの形態をとり、このようにして、この組成物は、生物学的に活性な結合体と共に、以下のいずれかを含み得る:ラクトース、スクロース、リン酸二カルシウムなどのような希釈剤;デンプンまたはその誘導体のような崩壊剤;ステアリン酸マグネシウムなどのような潤滑剤;およびデンプン、アラビアゴム、ポリビニルピロリドン、ゼラチン、セルロースおよびそれらの誘導体のような結合剤。
【0131】
処方物の活性化合物は、例えば、ポリエチレングリコール(PEG)キャリア(例えば、PEG1000[96%]およびPEG4000[4%])中に配置される、約0.5%〜約50%の本発明の化合物を含む坐剤中に処方され得る。
【0132】
液体組成物は、キャリア(例えば、生理食塩水(例えば、0.9%w/v塩化ナトリウム)、水性デキストロース、グリセロール、エタノールなど)中に化合物(約0.5%〜約20%)、および任意の薬学的アジュバントを溶解または分散することによって調製され、例えば、静脈内投与のための溶液または懸濁液を形成し得る。この活性化合物はまた、保持浣腸に処方され得る。
【0133】
所望される場合、投与される組成物はまた、湿潤剤または乳化剤、pH緩衝剤(例えば、酢酸ナトリウム、ソルビタンモノラウレート、またはトリエタノールアミンオレアート)のような少量の非毒性補助物質を含み得る。
【0134】
局所投与の場合、組成物は、ローションまたは経皮パッチのような任意の適切な形式で投与される。吸入による送達の場合、この組成物は、乾燥散剤(例えば、吸入治療剤)として、または噴霧器を介する液体形態で、送達され得る。
【0135】
このような投与形態を調製するための方法は、公知であるか、または当業者に明らかである;例えば、Remington’s Pharmaceutical Scinces(前出)および同様の刊行物を参照のこと。投与される組成物は、どのような場合でも、本発明の教示に従って投与される際に処置される状態の軽減のために薬学的に有効な量の一定量のプロドラッグおよび/または活性化合物を含む。
【0136】
一般に、本発明の化合物は、治療的に有効な量、すなわち、処置をもたらすのに十分な用量で投与され、この用量は、処置される個体および状態に依存して変更する。代表的には、治療的に有効な1日の用量は、1日あたり、体重1kgあたり0.1〜100mgの薬物である。ほとんどの状態が、1日あたり体重1kgあたり約1〜約30mg、すなわち70kgのヒトの場合、1日あたり、約70mg〜2100mgの総用量の投与に応答する。
【0137】
結合体の安定性は、組成および送達増強トランスポーターの骨格および側鎖の立体化学によってさらに制御され得る。ポリペプチド送達増強トランスポーターの場合、D−異性体は、一般に内在性プロテアーゼに対して耐性があり、それ故に、血清および細胞内のより長い半減期を有する。それ故に、D−ポリペプチドポリマーは、より長い期間の作用が所望される場合に適切である。L−ポリペプチドポリマーは、プロテアーゼに対するそれらの感受性に起因して、より短い半減期を有し、それ故に、より短い作用効果を付与するために選択される。このことによって、副作用が観察されるとすぐに、治療を中止するこよによって、より容易に副作用が回避され得る。D−残基およびL−残基の混合物を含むポリペプチドは、中間安定性を有する。ホモ−D−ポリマーが、一般的に好ましい。
【0138】
(A.皮膚への適用)
本発明の送達増強トランスポーターは、生物学的に活性な薬剤および診断的な薬剤の皮膚を横切る送達を可能にする。驚くべきことに、このトランスポーターは、以前には薬物送達に対してほぼ浸透不可能な障壁であった角質層を横切って薬剤を送達し得る。角質層、すなわち皮膚の最外層は、コレステロールおよび脂肪酸からなる「糊剤(glue)」によって一緒に、しっかり結合される、死んだケラチン充填皮膚細胞のいくつかの層から構成される。一旦、この薬剤が本発明のトランスポーターによって角質層を通して送達されると、この薬剤は、角質層とともに表皮を構成する顆粒層、淡明層および胚芽層からなる生存可能な表皮に侵入し得る。本発明のいくつかの実施形態における送達は、表皮を通り、そして真皮(乳頭真皮および網様真皮の1つまたは両方を含む)に入る。
【0139】
皮膚の1層以上の層の貫入を得るこの能力は、抗細菌剤、抗真菌剤、抗ウイルス剤、抗増殖剤、免疫抑制剤、ビタミン、鎮痛剤、ホルモンなどのような化合物の効力を非常に増強し得る。莫大なこのような化合物は、当業者に公知である(例えば、HardmanおよびLimbird,Goodman & Gilman’s The Pharmacological Basis of Therapeutics,McGraw−Hill、New York、1996を参照のこと)。
【0140】
いくつかの実施形態において、薬剤は、上皮組織内に存在する血管内に送達され、それにより、全身的にこの薬剤の送達のための手段を提供する。送達は、濾胞内もしくは濾胞間、またはその両方のいずれかであり得る。皮膚の前処置は、結合体の送達に必要とされない。
【0141】
他の実施形態において、送達増強トランスポーターは、皮膚状態を処置し得る化粧品および薬剤を送達するために有用である。目的の皮膚内の標的細胞として、例えば、線維芽細胞、上皮細胞および免疫細胞が挙げられる。例えば、トランスポーターは、真皮内に見られる免疫細胞に対する抗炎症剤のような化合物を送達する能力を提供する。
【0142】
糖質コルチコイド(アドレノコルチコイドステロイド)は、皮膚を横切る送達が本発明の送達増強トランスポーターによって増強され得る化合物の1つである。本発明の結合体化した糖質コルチコイドは、例えば、炎症性皮膚疾患を処置するのに有用である。例示的な糖質コルチコイドとして、例えば、ヒドロコルチゾン、プレニゾン(prenisone(デルタゾン(deltasone)))およびプレドリゾンロン(predrisonlone)(ハイデルタゾル(hydeltasol))が挙げられる。特定の状態の例として、湿疹(アトピー性皮膚炎、接触性皮膚炎、アレルギー性皮膚炎を含む)、水疱性疾患、コラーゲン脈管疾患、サルコイドーシス、スウィート病、壊疽性膿皮症、I型反応性らい病、毛細血管性血管腫、扁平苔癬、剥脱性皮膚炎、結節性紅斑、ホルモン異常(挫瘡および多毛症を含む)、ならびに毒性表皮壊死症、多形性紅斑、皮膚性T細胞リンパ腫、円板状エリテマトーデスなどが挙げられる。
【0143】
レチノイドは、1層以上の皮膚または他の上皮組織もしくは内皮組織内への送達およびそれらを横切る送達を増強するために、本発明の送達増強トランスポーターを使用し得る生物学的に活性な薬剤の別の例である。現在使用されるレチノイドとして、例えば、レチノール、トレチノイン、イソトレチノイン、エトレチナート、アチトレチン(acitretin)、およびアロチノイド(arotinoid)が挙げられる。本発明の送達増強トランスポーターに結合したレチノイドを使用して処置可能である状態として、挫瘡、角質化障害、皮膚癌、前癌状態、乾癬、皮膚老化、円板状エリテマトーデス、硬化性粘液水腫、いぼ状表皮神経、角層下膿疱性皮膚炎、ライター症候群、いぼ、扁平苔癬、黒色表皮肥厚症、サルコイドーシス、グロヴァー病、汗孔角化症などが挙げられるが、これらに限定されない。
【0144】
細胞傷害性薬物および免疫抑制薬物は、本発明の送達増強トランスポーターが有用であるさらなるクラスの薬物を構成する。これらの因子は、乾癬のような過剰増殖疾患、ならびに水疱性皮膚疾患および白血球破砕性血管炎のような免疫疾患を処置するために通常使用される。本発明の送達増強トランスポーターに結合し得るこのような化合物の例として、代謝拮抗剤(例えば、メトトレキサート、アザチオプリン、フルオロウラシル、ヒドロキシ尿素、6−チオクアニン(thioquanine)、ミコフェノレート(mycophenolate)、クロランブシル、ビニクリスチン(vinicristine)、ビンブラスチンおよびダクチノマイシン)が挙げられるが、これらに限定されない。他の例は、アルキル化剤(例えば、シクロホスファミド、メクロロエタミン塩酸塩(mechloroethamine hydrochloride)、カルムスチンである。タキソール、タクロリムス(tacrolimus)およびビンブラスチンは、ダプソンおよびスルファサラジンのように、有用な生物学的薬剤のさらなる例である。シクロスポリン、FK506(タクロリムス)、およびラパマイシン(rapamycin)(例えば、米国特許第5,912,253号)のようなアスコマイシン(ascomycin))、ならびにこのような化合物のアナログのような免疫抑制剤が、特に関心が高い(例えば、Mollinsonら、Current Pharm.Design 4(5):367−380(1998);米国特許第5,612,350号;同第5,599,927号;同第5,604,294号;同第5,990,131号;同第5,561,140号;同第5,859,031号;同第5,925,649号;同第5,994,299号;同第6,004,973号および同第5,508,397号)。シクロスポリンとして、例えば、シクロスポリンA、B、C、D、GおよびMが挙げられる。例えば、米国特許第6,007,840号;および同第6,004,973号を参照のこと。例えば、このような化合物は、乾癬、湿疹(アトピー性皮膚炎、接触性皮膚炎、アレルギー性皮膚炎を含む)および円形脱毛症の処置に有用である。
【0145】
送達増強トランスポーターは、紅斑性狼瘡(円板状および全身性の両方)、皮膚筋炎、晩発性皮膚ポルフィリン症および多形光線疹のような状態を処置するために有用な薬剤と結合体化され得る。このような状態を処置するために有用な薬剤としては、例えば、キニーネ、クロロキン、ヒドロキシクロロキンおよびキナクリンが挙げられる。
【0146】
本発明の送達増強トランスポーターはまた、抗感染剤の経皮送達のために有用である。例えば、抗細菌剤、抗真菌剤および抗ウイルス剤が、送達増強トランスポーターに結合体化され得る。抗細菌剤は、挫瘡、皮膚感染などのような状態を処置するために有用である。抗真菌薬剤を用いて、体部白癬、足部白癬、爪真菌症、カンジダ症、でん風などを処置し得る。この結合体の送達増強特性に起因して、これらの結合体は、局所化した感染および広範に広がった感染の両方を処置するために有用である。抗真菌薬剤はまた、爪真菌症を処置するために有用である。抗真菌薬剤の例としては、イトラコナゾール、ミコナゾール(myconazol)およびフルコナゾールのようなアゾール抗真菌剤が挙げられるがこれらに限定されない。抗ウイルス薬剤の例としては、アシクロビル、ファンシクロビル(famciclovir)およびバラシクロビル(valacyclovir)が挙げられるがこれらに限定されない。このような薬剤は、ウイルス性疾患(例えば、ヘルペス)を処置するために有用である。
【0147】
本発明の送達増強トランスポーターへの結合体化による送達の増強が望ましい、生物学的に活性な薬剤の別の例は、抗ヒスタミン剤である。これらの薬剤は、蕁麻疹、アトピー性皮膚炎、接触皮膚炎、乾癬などに起因するかゆみのような状態を処置するために有用である。このような試薬の例としては例えば、テルフェナジン、アステミゾール、ロロタジン(lorotadine)、セチリジン、アクリバスチン、テメラスチン(temelastine)、シメチジン、ラニチジン、ファモチジン、ニザチジンなどが挙げられる。三環系抗うつ剤もまた、本発明の送達増強トランスポーターを用いて送達され得る。
【0148】
表面抗乾癬薬物(topical antipsoriasis drug)もまた目的のものである。コルチコステロイド、カルシポトリエン(calcipotriene)およびアントラリンのような薬剤は、本発明の送達増強トランスポーターに結合体化され得、そして皮膚に塗布され得る。
【0149】
本発明の送達増強トランスポーターはまた、皮膚および他の上皮組織の1以上の層内への、ならびに皮膚および他の上皮組織の1以上の層を通しての光化学療法剤の送達を増強するために有用である。このような化合物としては例えば、ソラレンなどが挙げられる。遮光剤成分もまた目的のものである;これらとしては、p−アミノ安息香酸エステル、シナメートおよびサリシレート、ならびにベンゾフェノン、アントラニレート(anthranilates)およびアボベンゾンが挙げられる。
【0150】
疼痛軽減剤および局所麻酔剤は、本発明の送達増強トランスポーターへの結合体化が処置を増強し得る別のクラスの化合物を構成する。リドカイン、ブピバカイン、ノボカイン、プロカイン、テトラカイン、ベンゾカイン、コカインおよびオピエートはとりわけ、本発明の送達増強トランスポーターに結合体化され得る化合物である。関節または関節付近の皮膚への疼痛軽減剤の適用(例えば、慢性関節リウマチに罹患した患者における)もまた意図される。
【0151】
目的の他の生物学的薬剤としては例えば、ミノキシジル、角質溶解剤、破壊剤(例えば、ポドフィリン、ヒドロキノン、カプサイシン、マソプロコール、コルヒチンおよび金が挙げられる。
【0152】
慢性関節リウマチにおいて生じるような炎症性関節の処置もまた、本発明のトランスポーターに結合体化された、このような処置のために有用な化合物を用いて処置され得る。
【0153】
(B.胃腸投与)
本発明の送達増強トランスポーターはまた、胃腸投与による結合体化薬物の送達のために有用である。胃腸投与は、全身的に活性な薬物および胃腸上皮において作用する薬物の両方について用いられ得る。
【0154】
送達増強トランスポーターに結合体化した適切な試薬を用いて処置され得る胃腸状態の中でも、クローン病のような炎症性腸疾患がある(例えば、シクロスポリン、FK506およびアスコマイシン;アミノサリチル酸塩(例えば、スルファサラジンおよびメサラミン);コルチコステロイド、例えば、プレドニゾンおよびメチルプレドニゾロン;免疫改変剤、例えば、アザチオプリン、6MP、メトトレキサート;ならびに抗生物質、例えば、メトロニダゾル、アンピシリン、シプロフロキサシンなど)。他の処置可能な胃腸状態としては、、潰瘍性大腸炎、胃腸の潰瘍、消化性潰瘍疾患、塩分および水分の吸収の失調(imbalance of salt and water absorption)(便秘、下痢または栄養失調を引き起こし得る)、異常増殖疾患などが挙げられる。潰瘍の処置としては例えば、胃酸分泌を減少させる薬物(例えば、H2ヒスタミンインヒビター(例えば、シメチジンおよびラニチジン)およびプロトン−カリウムATPaseのインヒビター(例えば、ランソプラゾールおよびオメプラゾール))ならびにHelicobacter pyloriに対する抗生物質が挙げられる。
【0155】
便秘の処置のために有用な化合物もまた、本発明のトランスポーターとの結合体において使用され得る。便秘を処置するために有用な化合物としては、例えば、ドキュセートナトリウム、ポロキサマー(poloxamer)188、デヒドロコール酸およびリシノール酸のような、界面活性剤緩下薬が挙げられる。例示的な界面活性剤緩下薬としては、例えば、フェノールフタレイン、ビサコジル、およびアントラキノン緩下薬(例えば、ダントロン)が挙げられる。
【0156】
抗生物質(特に、侵襲性の細菌(例えば、Shigella、SalmonellaおよびYersinia)に対して作用する抗生物質)はとりわけ、本発明の送達増強トランスポーターに結合体化した場合に有用である生物学的に活性な薬剤である。このような化合物としては例えば、ノルフロキサシン、シプロフロキサシン、トリメトプリム、スルファメチロキサゾールなどが挙げられる。
【0157】
例えば、結腸癌の処置のために、抗新生物剤もまた本発明の送達増強トランスポーターに結合体化され得、そして胃腸経路によって投与され得る。これらとしては例えば、シスプラチン、メトトレキサート、タキソール、フルオロウラシル、メルカプトプリン、ドキソルビシン(donorubicin)、ブレオマイシン、ストレプトゾシン、マイトマイシンなどが挙げられる。
【0158】
経口投与されたトランスポーターまたは活性化合物の胃腸および結腸への送達のために、その化合物が胃腸(GI)管または結腸に送達されるまで、その化合物が放出されないように、その化合物をコーティングまたはカプセル化することが有利であり得る。GI管または結腸への送達のために有用な方法および組成物は、例えば、米国特許第6,183,466号および同第6,120,803号に記載されている。
【0159】
(C.気道投与)
本発明の送達増強トランスポーターはまた、気道を介した薬物の投与を増強するために使用され得る。鼻粘膜、下咽頭、ならびに大気道構造および小気道構造を含む、気道は、薬物吸収のための大きな粘膜表面を提供する。本発明の送達増強トランスポーターにより提供される上皮組織の1つ以上の層へおよび1つ以上の層を横切る、結合体化された薬剤の増強された浸透は、気道送達が他の送達方法よりも優れた利点の拡大をもたらす。例えば、低用量の薬剤は、しばしば、所望の効果を得るために必要とされ、局所的な治療効果が、より迅速に生じ得、そして薬剤の全身性の治療的血液レベルが、直ちに得られる。薬理学的活性の迅速な発生は、気道投与から生じ得る。さらに、気道投与は、一般的に、比較的副作用がわずかしかない。
【0160】
本発明のトランスポーターは、肺性状態の処置のために有用な生物学的薬剤を送達するために使用され得る。鼻投与によって処置可能な状態の例としては、例えば、喘息が挙げられる。これらの化合物としては、抗炎症剤(例えば、コルチコステロイド、クロモリン、およびネドクロミル)、気管支拡張薬(例えば、β2選択性アドレナリン作用性(adronergic)薬物およびテオフィリン)、および免疫抑制薬物(例えば、シクロスポリンおよびFK506)が挙げられる。他の状態としては、例えば、アレルギー性鼻炎(これは、グルココルチコイドで処置可能である)、および慢性閉塞性肺疾患(気腫)が挙げられる。肺性組織に作用し、そして本発明のトランスポーターを使用して送達され得る、他の薬物としては、βアゴニスト、肥満細胞安定剤、抗生物質、抗真菌剤および抗ウイルス剤、界面活性剤、血管作用薬、鎮痛薬ならびにホルモンが挙げられる。
【0161】
気道投与は、肺性状態の処置のためだけでなく、循環器系を介した離れた標的器官への薬物の送達のためにも有用である。多種多様なこのような薬物および診断剤は、本発明の送達増強トランスポーターへの結合体化の後、気道を通じて投与され得る。
【0162】
(D.血液脳関門を横切る薬剤の送達)
送達増強トランスポーターはまた、血液脳関門を横切った生物学的に活性な薬剤および診断剤の送達のために有用である。これらの薬剤は、虚血の処置(例えば、抗アポトーシス薬物の使用)のため、ならびに種々の状態(例えば、精神分裂病、パーキンソン病、痛み(例えば、モルヒネ、アヘン製剤))を処置するための神経伝達物質および他の薬剤を送達するために有用である。5−ヒドロキシトリプタミンレセプターアンタゴニストは、片頭痛および不安のような状態を処置するために有用である。
【0163】
(E.診断画像化剤および造影剤)
本発明の送達増強トランスポーターはまた、上皮組織および/もしくは内皮組織の1つ以上の層へか、または1つ以上の層を横切る、診断画像化剤ならびに造影剤の送達のために有用である。診断剤の例としては、放射能で標識化される物質(例えば、99mTcグルコヘプトネート)、または磁気共鳴画像化(MRI)法で使用される物質(例えば、ガドリニウムドープしたキレート化剤(例えば、Gd−DTPA))が挙げられる。診断剤の他の例としては、細胞内で発現された場合に容易に検出可能なタンパク質(例えば、β−ガラクトシダーゼ、グリーン蛍光タンパク質、ルシフェラーゼなどが挙げられるが、これらに限定されない)をコードするマーカー遺伝子が挙げられる。多種多様な標識(例えば、放射性核種、蛍光物質(fluor)、酵素、酵素基質、酵素補因子、酵素インヒビター、リガンド(特に、ハプテン)など)が、使用され得る。
【0164】
(送達増強トランスポーターと共に有用な生物学的に活性な分子および診断分子)
送達増強トランスポーターは、診断的に使用する多種多様な生物学的に活性な薬剤および分子に結合体化され得る。
【0165】
(A.低有機分子)
低有機分子治療剤は、本明細書中に記載されるような直鎖状ポリマー組成物に有利に連結されて、上皮組織もしくは内皮組織の1つ以上の層を横切った輸送を容易にし得るかまたは増強し得る。例えば、高度に荷電した薬剤(例えば、レボドパ(L−3,4−ジヒドロキシ−フェニルアラニン;L−DOPA))の送達は、本明細書中に記載されるような送達増強トランスポーターへの連結に役立ち得る。ペプトイド薬剤およびペプチド模倣薬剤はまた、企図される(例えば、Langston(1997)DDT 2:255;Giannisら、(1997)Advances Drug Res.29:1)。また、本発明は、水溶液(例えば、血清および生理食塩水)に対して乏しい可溶性を有する低有機分子を送達するために有利である。従って、治療的効力がそれらの低い可溶性によって制限される化合物は、本発明に従ってより多くの用量で投与され得、そして細胞によるより高い取り込みレベルに起因して、非結合体化形態と比較して、結合体化形態においてモルベースでより有効であり得る。
【0166】
低分子の位相幾何学的表面の有意な部分は、しばしば、関与され、従って、生物学的活性に対して必要とされるので、特定の場合における結合体の低分子部分は、標的上皮組織を横切った後、生物学的活性を発揮するための低分子薬剤について、連結された送達増強トランスポーターおよびリンカー部分(存在する場合)から切断されることが必要であり得る。このような状況について、この結合体は、好ましくは、上皮組織を介して通過した後、遊離の薬物を放出するための切断可能なリンカーを含有する。
【0167】
図5Dおよび図5Eは、本発明の別の局面の例証であり、対応する非結合体化形態に関連して増強された透過性上皮(trans−epithelial)組織輸送速度を有する、タキサン抗癌結合体およびタキソイド抗癌結合体を含有する。この結合体は、特に、癌細胞の増殖を阻害するために有用である。タキサンおよびタキソイドは、細胞機能に有害な程度まで微小管の重合化を促進し(そして解重合を阻害し)、細胞複製を阻害し、そして最終的には細胞死をもたらすことによって、それらの抗癌作用を発揮すると考えられる。
【0168】
用語「タキサン」とは、パクリタキセル(図5F、R’=アセチル、R’’=ベンジル、登録商標「TAXOL」としても知られる)、ならびに図5Gにおいて図示されるように、パクリタキセルのA環、B環、C環およびD環を含有する骨格コアを有する天然に存在するアナログ、合成アナログ、または生物工学アナログのことをいう。図5Fはまた、「TAXOTERETM」(R’=H、R’’=BOC)の構造を示し、これは、Rhone−Poulencにより販売されるパクリタキセルの幾分より可溶性の合成アナログである。「タキソイド」とは、図5Hに示されるように、パクリタキセルの基本的なA環、B環およびC環を含有するパクリタキセルの天然に存在するアナログ、合成アナログまたは生物工学アナログをいう。実質的な合成的および生物学的な情報は、Suffness(1995)Taxol:Science and Applications,CRC Press,New York,NY,pp.237−239、特に12〜14章、および引き続くパクリタキセルの文献において論評されるように、種々のタキサン化合物およびタキソイド化合物の合成および活性に関して利用可能である。さらに、宿主の細胞株は、例えば、Suffnessの8章および13章に記載されるような、特定の癌のタイプに対するこれらの化合物の抗癌活性を推定するために利用可能である。
【0169】
送達増強トランスポーターは、タキサンまたはタキソイドにおける任意の適切な連結部位を介してタキサン部分またはタキソイド部分に結合体化される。簡単には、この輸送ポリマーは、上記のような連結ストラテジーを用いて、C2’−酸素原子、C7−酸素原子を介して連結される。C7−酸素を介する輸送ポリマーの結合体化は、その位置に結合体化されるにも関わらず抗癌活性および抗腫瘍活性を有するタキサン結合体をもたらす。従って、このリンカーは、切断可能または非切断可能であり得る。C2’−酸素を介する結合体化は、抗癌活性を有意に減少させ、そのため、切断可能なリンカーが、この部位に対する結合体に関して好ましい。他の部位の連結(例えば、C10)がまた、使用され得る。
【0170】
本発明のタキサン結合体およびタキソイド結合体が、タキソール(約0.25μg/mL)およびタキソテール(6〜7μg/mL)と比較して改善された水溶性を有することが理解される。従って、大量の可溶化剤(例えば、「CREMOPHOR EL」(ポリオキシエチル化ヒマシ油)、ポリソルベート80(ポリオキシエチレンソルビタンモノオレエート、「TWEEN80」としても知られる)、およびエタノール)が、必要でなく、その結果、典型的に、これらの可溶化剤に関連した副作用(例えば、アナフィラキシー、呼吸困難、高血圧、および潮紅)が、低減され得る。
【0171】
(B.金属)
金属は、実施例において例証されるように、本発明の送達増強トランスポーターに結合体化されたキレート剤(例えば、テキサフィリンまたはジエチレントリアミン五酢酸(DTPA))を使用して、上皮組織および内皮組織の1つ以上の層へおよび1つ以上の層を横切って、輸送され得る。これらの結合体は、画像化または処置のための金属イオンを送達するために有用である。例示的な金属イオンとしては、Eu、Lu、Pr、Gd、Tc99m、Ga67、In111、Y90、Cu67、およびCo57が挙げられる。結合体候補物を用いる予備的な膜輸送研究は、以下の実施形態例の節において記載されるように、細胞ベースのアッセイを用いて実施され得る。例えば、ユーロピウムイオンを用いて、細胞取り込みは、時間分解蛍光測定によってモニターされ得る。細胞傷害性である金属イオンについて、取り込みは、細胞傷害性によってモニターされ得る。
【0172】
(C.高分子)
本発明の増強された輸送方法は、多数の高分子(例えば、タンパク質、核酸、多糖類、およびそれらのアナログが挙げられるが、これらに限定されない)について、上皮組織または内皮組織の1つ以上の層への輸送および1つ以上の層を横切った輸送を増強させるために特に適している。例示的な核酸としては、DNAおよびRNAならびにそれらのアナログから形成されたオリゴヌクレオチドならびにポリヌクレオチドが挙げられ、これらは、相補的な標的(例えば、一本鎖標的または二本鎖標的に対するアンチセンス配列)へのハイブリダイゼーションのためにか、またはこの配列によってコードされる核酸転写物もしくはタンパク質を発現するために、設計された選択された配列を有する。アナログとしては、荷電した骨格アナログ、および好ましくは無電荷の骨格アナログ、例えば、ホスホネート(好ましくは、メチルホスホネート)、ホスホラミデート(N3’またはN5’)、チオホスフェート、無電荷のモルホリノベースのポリマー、およびタンパク質核酸(PNA)が挙げられる。このような分子は、例えば、酵素置換治療、遺伝子治療、およびアンチセンス治療のような種々の治療的レジメンにおいて使用され得る。
【0173】
例示の目的で、タンパク質核酸(PNA)は、その骨格がデオキシリボース骨格と構造的に同形性であるDNAのアナログである。この骨格は、核酸塩基が結合されるN−(2−アミノエチル)グリシン単位から構成される。4つ全ての天然の核酸塩基を含有するPNAは、ワトソン−クリック塩基対則に従う相補的なオリゴヌクレオチドとハイブリダイズし、そして塩基対認識に関して忠実なDNA模倣物である(Egholmら(1993)Nature 365:566−568)。PNAの骨格は、リン酸エステルでなくペプチド結合によって形成され、それをアンチセンス適用に対して十分に適応させる。この骨格が無電荷であるので、形成するPNA/DNA二重鎖またはPNA/RNA二重鎖は、通常よりも大きな熱安定性を示す。PNAは、それらがヌクレアーゼまたはプロテアーゼによって認識されないさらなる利点を有する。さらに、PNAは、標準的なt−Boc化学を用いる自動化ペプチド合成機で合成され得る。その結果、PNAは、本発明のトランスポーターポリマーに容易に連結される。
【0174】
上皮組織および内皮組織への輸送、ならびに上皮組織および内皮組織を横切る輸送が、本発明の方法を用いて増強され得る、アンチセンスオリゴヌクレオチドの例は、例えば、米国特許第5,594,122号に記載される。このようなオリゴヌクレオチドは、ヒト不全ウイルス(HIV)を処置するために標的化される。アンチセンスオリゴヌクレオチドへのトランスポーターポリマーの結合体化は、例えば、非常に確立された方法に従って、スクシネートリンカーを介して、ペプチドとオリゴヌクレオチドの5’末端との間のアミド架橋を形成することによって達成され得る。PNA結合体の使用は、PCT出願PCT/US98/10571の実施例11においてさらに例証される。この出願の図7は、実施例11に詳述されるように、T細胞によってγ−インターフェロン(γ−IFN)の分泌を阻害するためにPNA配列を含有する本発明の結合体で得られた結果を示す。示され得るように、このアンチセンスPNA結合体は、結合体が、約10μMを越えるレベルで存在する場合、γ−IFNの分泌をブロックするのに有効であった。対照的に、センスPNA結合体または非結合体化アンチセンスPNA単独では、阻害が観察されなかった。
【0175】
上皮組織または内皮組織の1つ以上の層を横切って輸送され得る別の分類の高分子は、タンパク質、および特に、酵素によって、例証される。治療的タンパク質としては、置換酵素が挙げられるが、これらに限定されない。治療的酵素としては、以下が挙げられるが、これらに限定されない:アルグルセラーゼ(alglucerase)(リソソームグルコセレブロシダーゼ欠損(ゴシェ病)を処置する使用のため)、α−L−イズロニダーゼ(ムコ多糖症Iを処置する使用のため)、α−N−アセチルグルコサミダーゼ(サンフィリポB症候群を処置する使用のため)、リパーゼ(膵機能不全症を処置する使用のため)、アデノシンデアミナーゼ(重篤な混合性免疫不全症候群を処置する使用のため)、およびトリオースホスフェートイソメラーゼ(トリオースホスフェートイソメラーゼ欠損に関連する神経筋機能不全を処置する使用のため)。
【0176】
さらに、そして本発明の重要な局面に従って、タンパク質抗原は、これらがペプチド中で分解される場合、抗原提示細胞(APC)の細胞質ゾルの区画に送達され得る。次いで、このペプチドは、小胞体中に輸送され、ここで、これらは、発生期のHLAクラスI分子と結合し、そして細胞表面上に提示される。このような「活性化」APCは、クラスI限定抗原特異的細胞傷害性T−リンパ球(CTL)の誘導因子として作用し得、次いで特定の抗原を提示する細胞の認識および破壊を進行させる。このプロセスを実施し得るAPCとしては、特定のマクロファージ、B細胞および樹状細胞が挙げられるが、これらに限定されない。1つの実施形態において、タンパク質抗原は、腫瘍細胞に対して免疫応答を誘発するかまたは促進するための腫瘍抗原である。CTLの引き続く活性化を伴うAPCの細胞質ゾルへの単離タンパク質または可溶性タンパク質の輸送は、例外的である。なぜなら、わずかな例外を除いて、単離タンパク質または可溶性タンパク質の注入が、APCの活性化またはCTLの誘導のいずれももたらさないからである。従って、本発明のトランスポーター増強組成物に結合体化される抗原は、インビトロまたはインビボで細胞性免疫グロブリン応答を刺激するように作用し得る。
【0177】
別の実施形態において、本発明は、有害な生物学的過程(例えば、微生物感染)を妨げるために細胞質ゾルへの免疫特異的な抗体または抗体フラグメントを送達するために有用である。最近の実験は、細胞内抗体が、植物細胞および哺乳動物細胞において有効な抗ウイルス剤であり得ることを示してきた(例えば、Tavladorakiら、(1993)Nature 366:469;およびShaheenら、(1996)J.Virol.70:3392)。これらの方法は、典型的に、抗体の重鎖および軽鎖が、単一のポリペプチドとして合成される、一本鎖の可変領域フラグメント(scFv)を使用してきた。可変性の重鎖および軽鎖は、通常、可撓性のリンカーペプチド(例えば、15個のアミノ酸のペプチド)によって分離されて、高親和性リガンド結合部位を保持する28kDaの分子を生成する。この技術の広範な適用に対する主要な障害は、感染細胞中への取り込みの効率であった。しかし、scFvフラグメントへトランスポーターポリマーを連結することによって、細胞取り込みの程度は、増加され得、免疫特異的なフラグメントが結合して、重要な微生物成分(例えば、HIV Rev、HIV逆転写酵素、およびインテグラーゼタンパク質)を無能にし得る。
【0178】
(D.ペプチド)
本明細書中に記載される増強された輸送方法によって送達されるペプチドとしては、エフェクターポリペプチド、レセプターフラグメントなどが挙げられるが、これらに限定されるべきでない。例としては、細胞内シグナルを媒介するタンパク質により使用されるリン酸化部位を有するペプチドが挙げられる。このようなタンパク質の例としては、プロテインキナーゼC、RAF−1、p21Ras、NF−κB、C−JUN、および膜レセプター(例えば、IL−4レセプター)の細胞質テイル(tail)、CD28、CTLA−4、V7、ならびにMHCクラスI抗原およびMHCクラスII抗原が挙げられるがこれらに限定されない。
【0179】
送達増強トランスポーターがまたペプチドである場合、合成は、上記に記載されるように、自動化ペプチド合成機を使用してか、または融合ペプチドをコードするポリヌクレオチドが産生される組換え方法のいずれかによって合成され得る。
【実施例】
【0180】
(実施例)
以下の実施例は、例証するために提供されるが、本発明を限定しない。
【0181】
(実施例1)
(ヌードマウスの皮膚へのD−アルギニンのビオチン化ポリマーの浸透)
この実施例は、ポリアルギニン七量体が、濾胞ならびに濾胞間の両方で、皮膚の層へ、および皮膚の層を横切って、そして真皮内へ、結合体化ビオチンを送達し得ることを示す。
【0182】
(方法)
ビオチン化ペプチドを、固相技術、ならびにApplied Biosystems 433ペプチド合成機によって市販のFmocアミノ酸、樹脂、および試薬(PE Biosystems,Foster City CA、およびBachem Torrence,CA)を使用して合成した。Fastmocサイクルを、カップリング試薬としてHBTU/HOBtの代わりに、O−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート(HATU)で使用した。ペプチドのアミノ末端へのビオチンの付加の前に、アミノカプロン酸(aca)を、結合体化し、そしてスペーサーとして作用させた。このペプチドを、1時間〜12時間の間、96%トリフルオロ酢酸、2%トリイソプロピルシラン、および2%フェノールを用いて樹脂から切断した。より長い反応時間は、アルギニンのポリマーからPdf保護基を完全に除去するために必要であった。その後、ペプチドを、樹脂から濾過して、ジエチルエーテルを用いて沈殿させ、逆相カラムHPLC(Alltech Altima,Chicago,IL.)を使用して精製し、そしてエレクトロスプレー質量分析法またはマトリクス支援レーザー脱離質量分析法のいずれか(Perceptive Biosystems,Boston,MA)を使用して特徴付けた。
【0183】
リン酸緩衝化生理食塩水(PBS)中に溶解した、アミノカプロン酸スペーサー(bio r7)を用いてアミノ末端に共有結合的に連結したビオチンを有する、変動させた濃度(1mM〜10μM)のD−アルギニンの七量体を、麻酔したヌードマウスの背中に塗布した。サンプル(100μl)を、賦形剤なしで液体として塗布し、VaselineTM障壁による分散を防ぎ、そして15分間浸透させた。この期間の最後に、この動物を、屠殺して、皮膚の関連切片を、切除し、封入剤(OCT)中に包埋し、そして凍結した。凍結切片(5ミクロン)を、低温槽を用いて切除し、スライド上に収集し、そして蛍光標識化ストレプトアビジン(Vector Laboratories,Burlingame,CA)で染色した。このスライドを、4℃で10分間アセトン中で固定し、風乾し、5分間PBS中に浸し、5分間正常ヤギ血清でブロックし、そして5分間PBSで洗浄した。この切片を、30μg/mlの蛍光標識化ストレプトアビジンを用いた30分間のインキュベーションにより染色し、PBSで洗浄し、2分間ヨウ化プロピジウム(1μg/ml)で対比染色し、そしてこの切片を、VectashieldTM封入剤で封入した。スライドを、蛍光顕微鏡によって分析した。対応する研究を、フルオレセイン−ストレプトアビジンではなくストレプトアビジン−西洋ワサビペルオキシダーゼを用いて実施した。ビオチン化ペプチドを、西洋ワサビペルオキシダーゼ基質ジアミノベンザジンを用いた切片の処理、および光学顕微鏡を用いた可視化によって可視化した。
【0184】
(結果)
ビオチン化アルギニン七量体を、表皮へおよび表皮を横切って、ならびに真皮へ、通過させた。視野の全ての細胞の細胞質ゾルおよび核は、蛍光性であり、実質的に、切片中のヌードマウス皮膚の全ての細胞への浸透を示す。染色パターンは、濾胞および濾胞間の両方であり、予期しない輸送に一致した。さらに、ポジティブな細胞は、乳頭真皮および細網真皮で明らかであった。対照的に、染色は、ビオチン単独、またはリン酸緩衝化生理食塩水単独で処理したマウスにおいて明らかでなかった。
【0185】
(実施例2)
(正常Balb/Cマウスの皮膚へのD−アルギニンのビオチン化ポリマーの浸透)
PBS中に溶解した、アミノカプロン酸スペーサー(bio r7)を用いてアミノ末端に共有結合的に連結したビオチンを有する、変動させた濃度(1mM〜100μM)のD−アルギニンの七量体を、麻酔したBalb/Cマウスの鼡径部の皮膚に塗布した。サンプル(100μl)を、賦形剤中の液体として塗布し、VaselineTM障壁による分散を防ぎ、そして30分間浸透させた。この期間の最後に、この動物を、屠殺して、皮膚の関連切片を、切除し、封入剤(OCT)中に包埋し、そして凍結した。凍結切片を、実施例1に記載されるように、低温槽を用いて切除し、スライド上に収集し、そして蛍光標識化ストレプトアビジン(Vector Laboratories,Burlingame,CA)で染色した。スライドを、蛍光顕微鏡によって分析した。
【0186】
(結果)
ヌードマウス由来の皮膚と同様に、ビオチン化アルギニン七量体を、表皮へおよび表皮を横切って、ならびに真皮へ、通過させた。視野の全ての細胞の細胞質ゾルおよび核は、蛍光性であり、実質的に、切片中のヌードマウス皮膚の全ての細胞への浸透を示す。染色パターンは、濾胞および濾胞間の両方であり、予期しない輸送に一致した。さらに、ポジティブな細胞は、乳頭真皮および細網真皮で明らかであった。対照的に、染色は、ビオチン単独、またはリン酸緩衝化生理食塩水単独で処理したマウスにおいて明らかでなかった。
【0187】
(実施例3)
(ヌードマウス上へ移植した正常なヒト皮膚へのD−アルギニンのビオチン化ポリマーの浸透)
PBS中に溶解した、アミノカプロン酸スペーサー(bio r7)を用いてアミノ末端に共有結合的に連結したビオチンを有する、変動させた濃度(1mM〜100μM)のD−アルギニンの七量体を、SCIDマウスの背中上のヒト包皮移植片に塗布した(例えば、Dengら(1997)Nature Biotechnol.15:1388−1391;Khavariら(1997)Adv.Clin.Res.15:27−35;ChoateおよびKhavari(1997)Human Gene Therapy 8:895−901を参照のこと)。サンプル(100μl)を、賦形剤中の液体として塗布し、VaselineTM障壁による分散を防ぎ、そして15分間浸透させた。この期間の最後に、動物を、屠殺して、皮膚の関連切片を、切除し、封入剤(OCT)中に包埋し、そして凍結した。凍結切片を、実施例1に記載されるように、低温槽を用いて切除し、スライド上に収集し、そして蛍光標識化ストレプトアビジン(Vector Laboratories,Burlingame,CA)で染色した。スライドを、蛍光顕微鏡によって分析した。
【0188】
(結果)
ヌードマウスおよび正常マウス由来の皮膚と同様に、ビオチン化アルギニン七量体を、ヒトの皮膚の、表皮へおよび表皮を横切って、ならびに真皮へ、通過させた。視野の全ての細胞の細胞質ゾルおよび核は、蛍光性であり、表皮および真皮へ、ならびに表皮および真皮を通した浸透を示す。強力な染色が、20×および40×の倍率で観察された。染色パターンは、濾胞および濾胞間の両方であり、予期しない輸送に一致した。さらに、ポジティブな細胞は、乳頭真皮および細網真皮で明らかであった。対照的に、染色は、ビオチン単独、またはリン酸緩衝化生理食塩水単独で処理したマウスにおいて明らかでなく、そして非常にわずかな染色が、低倍率または高倍率のいずれかで、ビオチン化アルギニン五量体結合体とともに観察された。
【0189】
(実施例4)
(プラスチックラップまたはローション賦形剤を用いるヌードマウスの皮膚へのD−アルギニンのビオチン化ポリマーの増加された浸透)
アミノカプロン酸スペーサー(bio r7)を用いてアミノ末端に共有結合的に連結したビオチンを有する、変動させた濃度(1mM〜100μM)のD−アルギニンの七量体を、PBS中に溶解し、そして等容量のLubridermTMと混合した。次いで、このローション混合物を、ヌードマウスの背中に塗布し、そして30分間、60分間、および120分間浸透させた。代替的に、サンプル(100μl)を、賦形剤なしの液体として塗布し、VaselineTMでシールされたサンプルにわたってプラスチックラップを覆うことによって蒸発するのを防いだ。このサンプルを、30分間、60分間、および120分間浸透させた。この期間の最後に、動物を、屠殺して、皮膚の関連切片を、切除し、封入剤(OCT)中に包埋し、そして凍結した。凍結切片を、実施例1に記載されるように、低温槽を用いて切除し、スライド上に収集し、そして蛍光標識化ストレプトアビジン(Vector Laboratories,Burlingame,CA)で染色した。スライドを、蛍光顕微鏡によって分析した。
【0190】
(結果)
ローションおよびプラスチックラップの両方とも、賦形剤なしでの染色と比較して増加した取り込みをもたらした。ローションは、結合体の増強された取り込みにおいてプラスチックラップよりもより効果的であった。ビオチン化アルギニン五量体を、表皮細胞層の細胞質ゾルおよび核の両方、濾胞および濾胞間の両方に到達する、いくつかの皮膚層へおよび皮膚層を横切って、通過させた。さらに、ポジティブな細胞は、乳頭真皮および細網真皮で明らかであった。
【0191】
(実施例5)
(ヌードマウスの皮膚へのD−アルギニンのビオチン化五量体、ビオチン化七量体、およびビオチン化九量体へ結合体化されたシクロスポリンの浸透)
(方法)
(A.送達増強トランスポーターへのシクロスポリンの連結)
(1.α−クロロアセチルシクロスポリンA誘導体の調製)
α−アセチルシクロスポリンA誘導体を、図1に示されるように調製した。シクロスポリンA(152.7mg、127μmol)およびクロロ酢酸無水物(221.7mg;1300μmol)を、N2雰囲気下乾燥フラスコ中に配置した。ピリジン(1.0mL)を、添加し、そしてこの溶液を50℃(油浴)まで加熱した。16時間後、この反応を、室温まで冷却して、水(4.0mL)でクエンチした。得られた懸濁液を、ジエチルエーテル(Σ15mL)で抽出した。合わせた有機層を、MgSO4で乾燥した。減圧下での溶媒の濾過およびエバポレーションは、黄色オイルを与え、これを、シリカゲル上のフラッシュクロマトグラフィー(溶出剤:EtOAc/ヘキサン:40%〜80%)によって精製し、136mg(106.4μmol、83%)の所望の産物を得た。
【0192】
(2.トランスポーター分子へのカップリング)
システイン含有ペプチドのα−クロロアセチルシクロスポリンA誘導体へのカップリングについての一般的手順を図2に示す。
【0193】
(a.標識ペプチド)
シクロスポリンA誘導体および標識ペプチド(1当量)を、N2雰囲気下でDMFに溶解した(約10mmolのシクロスポリンA誘導体/1mL DMF)。ジイソプロピルエチルアミン(10当量)を添加し、そして室温での攪拌を、全ての出発物質が消費されるまで(通常16時間後)続けた(図3)。溶媒を真空で除去し、そして粗製反応生成物を水に溶解し、そして逆相高速液体クロマトグラフィー(RP−HPLC)(溶離液:水/MeCN*TFA)により精製した。生成物を以下の収率で得た
【0194】
【化13】
ペプチド(34.7mg、15.3μmol)およびシクロスポリンA誘導体(19.6mg、15.3μmol)を、N2雰囲気下でDMF(1.0mL)に溶解した(図4)。ジイソプロピルエチルアミン(19.7mg、153μmol)を添加し、そして室温での攪拌を続けた。12時間後、溶媒を真空で除去した。粗製物質を水に溶解し、そしてRP−HPLC(溶離液:水/MeCN*TFA)で精製し、純粋な生成物(24.1mg、6.8mmol、44%)を得た。
【0195】
(B.皮膚を横切る輸送の分析)
PBSに溶解した種々の濃度(1mM〜100μM)の、D−アルギニンのビオチン化した五量体、七量体または九量体(bio r5、bio r7、またはbio r9)のいずれかに結合体化されたシクロスポリンを、ヌードマウスの背部に塗布した。サンプル(100μl)を、賦形剤中の液体として塗布し、そしてVaselineTMバリアにより分散を防ぎ、そして30分、60分および120分間浸透させた。この期間の終わりに、動物を屠殺し、皮膚の関連切片を切除し、封入剤(OCT)中に包埋し、そして凍結した。凍結された切片を、クリオスタットを使用して切断し、スライド上に集め、そして蛍光標識ストレプトアビジン(Vector Laboratories,Burlingame,CA)で実施例1に記載のように染色した。スライドを蛍光顕微鏡により分析した。
【0196】
(結果)
D−アルギニンのビオチン化七量体および九量体とのシクロスポリンの結合体は、ヌードマウスの皮膚の表皮に有効に進入し、そして表皮を横切って真皮中に進入した。対照的に、アルギニンの五量体とシクロスポリンとの間の結合体を使用すると、ほとんど取り込みは見られず、PBSコントロールでは染色は全く見られなかった。視野中の全ての細胞の細胞質ゾルおよび核は、蛍光性であり、表皮および真皮への浸透および表皮および真皮を通過する浸透を示していた。染色パターンは、小胞および小胞間の両方の予期しない輸送と一致していた。さらに、ポジティブな細胞は、乳頭真皮および網状真皮において明白であった。これらの結果は、十分なグアニジニル基が送達増強トランスポーターに含まれる場合にのみ顕著な取り込みを実証する。
【0197】
(実施例6)
(D−アルギニン七量体がヒト皮膚に浸透し得ることの実証)
ヒト皮膚とマウス皮膚とは、多数の点で有意に異なり、ヒト表皮は、かなり厚い。D−アルギニン七量体/シクロスポリンA(r7 CsA)結合体もまたヒト皮膚に浸透し得るか否かを決定するために、ビオチンr7 CsAを、SCIDマウスの背部上に移植されたヒト全厚皮膚に塗布した。マウス皮膚のように、結合体シクロスポリンAは、ヒト表皮および真皮に浸透した。蛍光は、ビオチン化ペプチド単独に曝露された組織における細胞の細胞質ゾルおよび核の両方において観察されたが、ビオチンr7 CsAで染色された切片では、蛍光の大部分は細胞質ゾルであり、シクロスポリンAの公知の細胞質標的に対するr7 CsAの結合と一致した。
【0198】
(実施例7)
(シクロスポリンA−トランスポーター結合体が真皮のT細胞に進入することの実証)
(方法)
放出可能なR7−CsA結合体によるIL−2分泌の阻害。Jurkat細胞(5×104)を、種々の濃度の放出可能でないR7−CsA結合体または放出可能なR7−CsA結合体またはCsAと共に37℃でインキュベートして、PMAおよびイオノマイシンでの刺激の前にCsAの活性形態を放出させた。続いて、10ng/mgのPMA(Sigma、St.Louis,MO)および1μMのイオノマイシン(CAlBiochem,San Diego,CA)を添加することによりT細胞を刺激して、IL−2を産生させた。培養物を、37℃で一晩インキュベートし、そして上清を収集し、そして蛍光ELISAを使用してIL−2を測定した。手短には、プレートを4μg/mlの抗ヒトIL−2抗体(BD Pharmingen,San Diego,CA)でコーティングし、10%FBSを含むPBSで1時間室温にてブロックし、洗浄し、そして上清を添加し、そして1時間インキュベートした。培地を除去し、そしてビオチン化抗ヒトIL−2(1.6μg/ml)を、1時間で添加した、このプレートを洗浄し、次いでユーロピウム標識ストレプトアビジン(0.04ng/ml)を1時間で添加した。もう1度洗浄した後、増強溶液を添加し、そして得られる蛍光を、Wallacプレートリーダー(Wallac,Turku,Finland)を使用して測定した。
【0199】
(結果)
ビオチン化D−アルギニン七量体−シクロスポリン(r7 CsA)結合体がインビボで炎症性皮膚内の浸潤性T細胞に到達するか否かを決定するために、ビオチンr7 CsAを、実験的に誘導された接触皮膚炎を有するマウスの背部上の炎症部位に塗布した。炎症性皮膚を、ローダミン標識ヤギ抗マウスCD3で染色して、T細胞を局在化し、そしてフルオレセイン標識ストレプトアビジンで染色してビオチンr7 CsAを局在化した。ビオチンr7 CsAは、種々の他の細胞(おそらく他の炎症性細胞および内在する繊維芽細胞に相当する)に加えて、組織中の全てのCD3[+]T細胞において見出された。これらのデータは、ビオチンr7 CsAが、炎症した皮膚に浸透して重要な標的Tリンパ球に到達するということを示す。
【0200】
(実施例8)
(アルギニンの短いオリゴマーおよびCsAの放出可能な結合体の合成、インビトロでの活性およびインビボでの活性)
シクロスポリンAの2級アルコールの修飾は、その生物学的活性の有意な損失を生じる。例えば、R.E.Handschumacherら、Science 226,544−7(1984)を参照のこと。結果として、細胞への輸送後のその結合体からの遊離シクロスポリンAの放出を確実にするために、シクロスポリンAを、図10に示すように、pH感受性リンカーを介してオリゴアルギニントランスポーターに結合した。得られた結合体は、酸性pHでは安定であるが、pH>7では、シクロスポリンAに隣接するカルボニルへの遊離アミンの付加を含む分子内環化を受け(図6)、これは、修飾されていないシクロスポリンAの放出をもたらす。
【0201】
放出可能な結合体の設計における別の修飾は、L−アルギニン(R)の使用であり、トランスポーター中にD−アルギニン(r)は使用しなかった。この結合体の最大の安定性、従って蛍光によるその位置の決定における精度を確実にするために、オリゴ−D−アルギニントランスポーターを組織学的実験に用いた一方で、L−アルギニンのオリゴマーを、その生物学的半減期を最小にするために放出可能な結合体の設計に組み込んだ。その設計と一致して、得られる放出可能な結合体は、酸性pHでは安定であるが、血清の非存在下での生理学的pHでは不安定であることが示された。pH7.4のPBS中のこの放出可能なシクロスポリンA結合体の半減期は、90分であった。
【0202】
(結果)
シクロスポリンAの放出可能なグアニジノ−七量体結合体は、インビトロでPMAおよびイオノマイシンで刺激されたヒトT細胞株(Jurkat)によるIL−2分泌を阻害することにより、生物学的に活性であることが示された。R.Wiskocilら、J.Immunol 134,1599−603(1985)を参照のこと。この結合体は、PMA/イオノマイシンの添加の12時間前に加えられ、そして用量依存性阻害が、放出可能なR7 CsA結合体により観察された。t−Boc保護基(これは、環化および結果としての活性薬物の放出を防ぐ)を保持していることにより放出可能な結合体と異なる、放出可能でないアナログ(図6)では、この阻害は観察されなかった。放出可能なR7シクロスポリン結合体のEC50は、アルコールに溶解されて放出可能な結合体と同時に加えられたCsAより約2倍高かった。
【0203】
放出可能なR7 CsA結合体を、接触皮膚炎のマウスモデルを使用して機能的活性についてインビボでアッセイした。1%の放出可能なR7 CSA結合体での処理は、耳の炎症において73.9%±4.0の減少を生じた(図7)。炎症の減少は、未処置の耳では全く見られず、処置された耳において見られた効果が、局所的でありかつ全身的ではないということを示していた。0.1%および0.01%のR7−CsAで処置されたマウスの耳においてより低い阻害が観察され(それぞれ、64.8%±4.0および40.9%±3.3)、この効果が力価測定が可能であることを実証した。フッ素化コルチコステロイドポジティブコントロールでの処置は、耳腫大における減少(34.1%±6.3)を生じたが、0.1%の放出可能なR7 CsAについて観察された減少より有意に低かった(図7)。未修飾のシクロスポリンA、ビヒクル単独、R7、または放出可能でないR7 CsAで処置されたマウスのいずれにおいても炎症の減少は観察されなかった。
【0204】
(実施例9)
(銅−DTPA−r7複合体およびガドリニウム−DTPA−r7複合体のヌードマウスの皮膚への浸透)
(方法)
1.金属錯体の調製
工程1−銅−ジエチレントリアミン五酢酸錯体(Cu−DTPA)の調製
炭酸銅(10mmol)およびジエチレントリアミン五酢酸(10mmol)を、水(150mL)に溶解した(図8)。18時間後、この溶液を、遠心分離して固体を除去した。この青色溶液を、デカントし、そして凍結乾燥して青色粉末を得た(収率>90%)。
【0205】
工程2−DTPAトランスポーターの調製
Cu−DTPAを、PE Applied Biosystems Peptide Synthesizer(ABI 433A)を使用してアミノカプロン酸スペーサーを介してトランスポーターに連結した(図9)。この材料を、トリフルオロ酢酸(TFA)(40mL)、トリイソプロピルシラン(100μL)およびフェノール(100μL)で18時間処理することにより樹脂から切断した。この樹脂を、濾別し、そしてこのペプチドをジエチルエーテル(80mL)を加えることにより沈殿させた。この溶液を遠心分離し、そして溶媒をデカントして除いた。粗製固体を、水/アセトニトリル勾配を使用する逆相HPLCにより精製した。TFAでの処理は、Cu2+イオンの損失を生じ、これを再挿入する必要があった。
【0206】
DTPA−aca−R7−CO2H(10mg,0.0063mmol)および硫酸銅(1.6mg,0.0063mmol)を、水(1mL)に溶解した。18時間穏やかに攪拌させ、そして凍結乾燥して生成物を白色粉末(10mg)として得た。
【0207】
(2.皮膚を横切る輸送の分析)
金属ジエチレントリアミン五酢酸(DTPA)錯体を、等モル量の金属塩とDTPAとを水中で18時間混合することにより形成した。この時間の終わりに、この溶液を遠心分離し、凍結し、そして凍結乾燥した。乾燥した粉末を質量分析法で特徴付けし、そして固相ペプチド合成に使用した。この金属−DTPA錯体を、ペプチド合成に使用される固相樹脂にまだ結合されているD−アルギニンまたはL−アルギニンのポリマーに結合させた。この金属−DTPA錯体を、アミノカプロン酸スペーサーを使用して結合した。トリフルオロ酢酸中のペプチド−DTPA−金属複合体の切断が金属を放出したことを除いて、固相ペプチド合成技術は、実施例1に記載されている。HPLC精製およびペプチド−DTPA複合体の凍結乾燥後に金属を置換する。この金属の置換には、ペプチド−アミノカプロン酸−DTPA複合体と共に等モル量の金属塩をインキュベーションすること、およびその後の凍結乾燥が含まれる。
【0208】
種々の濃度(1μM〜1mM)のCu−DTPA−aca−r7複合体を、ヌードマウスの腹部領域に15分、30分および45分間塗布した。コントロールとして、等モル量のCu−DTPA錯体をこの腹部領域にスポッティングした。このインキュベーション時間の終わりに、これらのサンプルを単純に拭き取り、そしてCu−DTPA−aca−r7複合体をスポッティングした皮膚では強い青色が明らかであり、そしてCu−DTPA単独をスポッティングした皮膚では強い青色が見られなかった。1mMの塗布の場合、3日間青色の色素が見られ、時間と共に減少したが、この期間の間ずっと明白であった。
【0209】
種々の濃度(1μM〜1mM)のGd−DTPA−aca−r7複合体を、BALB/cマウスの尾静脈中に100μlで注射する。Gdの分布は、磁気共鳴画像化を使用して実時間で観察される。この色素の分布は、血流全体で明らかであり、肝臓、脾臓、腎臓および心臓に入る。ウサギの頸動脈に注射される場合、この色素は、血液脳関門を横切って見られる。
【0210】
(実施例10)
(D−アルギニンのビオチン化五量体、七量体、および九量体に結合体化されたヒドロコルチゾンの、ヌードマウスの皮膚への浸透)
(方法)
(A.送達増強トランスポーターへのヒドロコルチゾンの連結)
(工程1−無水クロロ酢酸を用いたヒドロコルチゾンのアシル化)
ヒドロコルチゾン(200mg、0.55mmol)および無水クロロ酢酸(113mg、0,66mmol)のピリジン(5mL)溶液を、室温で2時間攪拌した(図10)。溶媒を留去し、そして粗生成物を、溶離液として50%ヘキサン/酢酸エチルを使用してシリカでクロマトグラフした。白色固体として生成物を単離した(139mg、58%)。
【0211】
(工程2−トランスポーターへの連結)
ヒドロコルチゾンのクロロ酢酸エステル(0.0137mmol)、システイン残基を含むトランスポーター(0.0137)、およびジイソプロピルエチルアミン(DIEA)(0.0274mmol)のジメチルホルムアミド(DMF)(1mL)溶液を、18時間室温で撹拌した(図11)。この物質を水/アセトニトリル勾配を用いて、逆相HPLCにより精製し、凍結乾燥して白色粉末を得た。
【0212】
r5結合体−12mg獲得(29%単離収率)
r7結合体−22mg獲得(55%単離収率)
R7結合体−13mg獲得(33%単離収率)
(B.皮膚を横断する輸送の分析)
PBSに溶解された様々な濃度(1mM〜100μM)の、D−アルギニンのビオチン化五量体、七量体、または九量体(bio r5、bio r7、またはbio r9)のいずれかに結合体化したヒドロコルチゾンを、ヌードマウスの背中に塗布した。サンプル(100μl)を、賦形剤内の液体として塗布し、そして、VaselineTMバリアによって分散することを妨げ、30分、60分、および120分間浸透させた。この期間の終わりに、動物を屠殺し、皮膚の関連切片を切除し、封入剤(OCT)で包埋し、凍結させた。凍結切片を、クリオスタットを用いて切断し、スライド上に収集し、そして、実施例1に記載されるように、蛍光標識したストレプトアビジン(Vector Laboratories,Burlingame,CA)で染色した。スライドを、蛍光顕微鏡によって分析した。
【0213】
(結果)
D−アルギニンのビオチン化七量体とヒドロコルチゾンとの結合体は、ヌードマウスの皮膚の表皮の中へおよび表皮を横切って、そして、皮膚の真皮の中へ効率良く入った。対照的に、アルギニンの五量体とヒドロコルジゾンとの間の結合体では、ほとんど取り込みが見られず、そして、PBSコントロールでは、全く染色が見られなかった。視野における全ての細胞の細胞質および核は蛍光性であり、表皮および真皮中に浸透し、そして、表皮および真皮を通ることを示した。この染色パターンは、小胞および小胞間の両方であった未予知の輸送と一致していた。さらに、陽性細胞は、乳頭真皮および網状真皮において明らかであった。これらの結果は、十分なグアニジル基が、送達増強トランスポーターに含まれる場合のみ、目立った取り込みを示す。
【0214】
(実施例11)
(D−アルギニンのビオチン化五量体、七量体、および九量体に結合体化したタキソールのヌードマウスの皮膚への浸透)
(方法)
(1.C−2’活性化タキソール誘導体のビオチン標識ペプチドへの結合体化)
(C−2’誘導体の合成)
タキソール(48.7mg、57.1μmol)を、N2雰囲気下で、CH2Cl2(3.0mL)中に溶解した。溶液を0℃に冷却した。ベンジル−(p−ヒドロキシベンゾエート)のクロロホルメート(200mmol(2.0mLのCH2Cl2中)、ベンジル−(p−ヒドロキシベンゾエート)およびジホスゲンから新しく調製した)のストック溶液を、0℃で添加し、そして、その温度での撹拌を5時間続け、その後、溶液を室温まで温めた(図12)。撹拌をさらに10時間続けた。溶媒を真空中で除去し、そして、粗物質を、シリカゲル上でのフラッシュクロマトグラフィーによって精製し(溶離液:EtOAc/ヘキサン30%〜70%)、所望のタキソールC−2’炭酸塩を得た(36.3mg、32.8μmol、57.4%)。
【0215】
(ビオチン標識ペプチドへのカップリング)
ビオチン化標識ペプチドへのカップリング手順を図13に示す。タキソール誘導体およびビオチン標識ペプチド(1.2当量)を、N2雰囲気下で、DMF中で溶解した(約10μmol/mL DMF)。ジイソプロピルエチレンアミン(DMF中1.2当量)およびDMAP(DMF中0.3当量)のストック溶液を添加し、そして、全出発物質が消費されるまで室温での撹拌を続けた。16時間後、溶媒を真空下で除去した。粗反応物混合物を水に溶解し、そして、RP−HPLCによって精製し(溶離液:水/MeCN*TFA)、示される収率の結合体を生成した:
B−aca−r5−K−タキソール:3.6mg、1.32mmol、20%。
【0216】
B−aca−r7−K−タキソール:9.8mg、3.01mmol、44%。
【0217】
B−aca−r9−K−タキソール:19.4mg、5.1mmol、67%。
【0218】
(未標識C−2’カルバメート:)
タキソール誘導体(12.4mg、11.2μmol)および未標識ペプチド(27.1mg、13.4μmol)を、N2雰囲気下で、DMF(1.5mL)に溶解した(図14)。ジイソプロピルエチレンアミン(1.7mg、13.4μmol)をDMF中にストック溶液として添加して、次いで、DMF中にストック溶液としてDMAP(0.68mg、5.6μmol)を添加した。室温での撹拌を、全ての出発物質が消費されるまで続けた。16時間後、溶媒を真空下で除去した。粗物質を水中に溶解し、そして、RP−HPLCによって精製し(溶離液:水/MeCN*TFA)、所望の生成物を得た(16.5mg、5.9μmol、53%)。
【0219】
(他のC−2’結合体)
タキソール誘導体(8.7mg、7.85μmol)をEtOAc(2.0mL)に溶解した。Pd/C(10%、4.0mg)を添加し、反応フラスコをH2で5回パージした(図15A)。水素雰囲気下で撹拌を7時間続けた。Pd/Cをろ過し、そして、溶媒を真空下で除去した。この方法で獲得された粗物質(6.7mg、6.58μmol、84%)は、純粋であって、次の工程でさらなる精製なしで使用され得た。
【0220】
遊離酸タキソール誘導体(18.0mg、17.7μmol)をCH2Cl2(2.0mL)に溶解した。ジシクロヘキシルカルボジイミド(4.3mg、21.3μmol)を、CH2Cl2(0.1mL)中にストック溶液として添加した。N−ヒドロキシスクシンイミド(2.0mg、17.7μmol)をDMF(0.1mL)にストック溶液として添加した(図15B)。室温で撹拌することを14時間続けた。この溶媒を真空下で除去し、そして得られた粗物質をシリカゲルのフラッシュクロマトグラフィーによって精製し(溶離液:EtOAc/ヘキサン40%〜80%)、所望の生成物を得た(13.6mg、12.2μmol、69%)。
【0221】
活性化タキソール誘導体(14.0mg、12.6μmol)およびペプチド(30.6mg、15.1μmol)を、N2雰囲気下で、DMF(3.0mL)に溶解した(図15C)。DMF(0.1mL)中にストック溶液として、ジイソプロピルエチレンアミン(1.94mg、15.1μmol)を添加して、次いで、0.1mLのDMF中にストック溶液としてDMAP(0.76mg、6.3μmol)を添加した。室温での撹拌を、全ての出発物質が消費されるまで続けた。20時間後、溶媒を真空下で除去した。粗物質を水に溶解し、そして、RP−HPLCによって精製し(溶離液:水/MeCN*TFA)、1:6の比率(それぞれ、カーボネート対カルバメート)で2種類の示されるタキソール結合体を生成した。
【0222】
(2.皮膚を横断する輸送の分析)
PBSに溶解された様々な濃度(1mM〜100μM)の、D−アルギニンのビオチン化五量体、七量体、または九量体(bio r5、bio r7、またはbio r9)のいずれかに結合体化したタキソールを、ヌードマウスの背中に適用した。サンプル(100μl)を、賦形剤内の液体として適用し、そして、VaselineTM障壁によって分散することを妨げ、30分、60分、および120分間浸透させた。この期間の終わりに、動物を屠殺し、皮膚の関連切片を切除し、封入剤(OCT)で包埋し、凍結させた。凍結切片を、クリオスタットを用いて切り出し、スライド上に収集し、そして、実施例1に記載されるように、蛍光標識したストレプトアビジン(Vector Laboratories,Burlingame,CA)で染色した。スライドを、蛍光顕微鏡によって分析した。
【0223】
(結果)
D−アルギニンのビオチン化七量体および九量体とタキソールとの結合体は、ヌードマウスの皮膚の表皮の中へおよび皮膚の表皮を横断して真皮の中へ効率的に進入した。対照的に、アルギニンの五量体とタキソールとの間での結合体では、ほとんど取り込みが見られず、そして、PBSコントロールでは、全く染色が見られなかった。視野における全ての細胞の細胞質および核が蛍光性であり、表皮および真皮中への浸透、および表皮および真皮の通過を示す。この染色パターンは、小胞および小胞間の両方の予期しない輸送と一致していた。さらに、陽性細胞は、乳頭状および網状真皮において明らかであった。これらの結果は、十分なグアニジル基が、送達増強トランスポーターに含まれる場合のみ、目立った取り込みを示す。
【0224】
(実施例12)
(タキソールおよび送達増強トランスポーターのpH放出可能なリンカーとの結合体)
この実施例は、生理学的pHに曝される際、送達増強トランスポーターから薬物を放出するリンカーによって、薬物に連結された送達増強トランスポーターを有するプロドラッグを合成するための一般的なストラテジーの使用を実証する。一般的には、薬物上の適切な部位は、α−クロロアセチル残基を有するように誘導体化される。次に、塩素が、保護されていないアミンを保有するシステイン残基のチオールと置換される。このスキームを、図16に示す。
【0225】
(方法)
(タキソール−2’−クロロアセチルの合成)
タキソール(89.5mg、104.9μmol)を、CH2Cl2(3.5mL)に溶解した。N2雰囲気下で、溶液を0℃以下に冷却した。無水α−クロロ酢酸(19.7mg、115.4μmol)を添加し、DIEA(14.8mg、115.4μmol)を続いて添加した。溶液を室温に昇温させた。薄層クロマトグラフィー(tlc)分析が、出発物質の完全な消費を示した後、溶媒を真空下で除去し、そして、粗物質をシリカゲルでフラッシュクロマトグラフィーによって精製し(溶離液:EtOAC/Hex20%〜50%)、所望の物質を得た(99.8mg、定量的)(図17)。
【0226】
【化14】
ペプチド(47.6mg、22.4μmol)を、N2雰囲気下でDMF(1.0mL)に溶解した。DIEA(2.8mg、22.4μmol)を添加した。DMF(1.0mL)中のタキソール−2’−クロロ酢酸(20.8mg、22.4μmol)の溶液を添加した。室温での撹拌を6時間続けた。0.1%TFA(1.0mL)を含む水を添加し、サンプルを凍結し、溶媒を凍結乾燥した。粗物質をRP−HPLCによって精製した(溶離液:水/MeCN*0.1%TFA:85%〜15%)。この反応のスキームを図18に示す。
【0227】
(関連結合体の合成)
上記に概要を述べた結合体の条件を用いて、示される3つのさらなる結合体を合成した。
【0228】
(細胞傷害性アッセイ)
タキソール結合体を、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニル−テトラゾリウム−ブロミド(MTT)染色還元における細胞傷害性について試験した。結果(図20に示す)は、易pH放出可能なリンカーを有するr7に結合したタキソール(CG 1062;図19に示される構造においてR=Ac)は、タキソール単独、または難pH放出可能なリンカーを有するr7に結合されたタキソール(CG 1040;図19に示される構造においてR=H)のいずれかよりも、有意により細胞障害性であることを示す。
【0229】
(実施例13)
(Tat49〜57由来の蛍光標識ペプチドの構造−機能関係)
(方法)
(概要)Rinkアミド樹脂およびBoc2OをNovabiochemより購入した。ジイソプロピルカルボジイミド、ブロモ酢酸、フルオレセインイソチオシアネート(FITC−NCS)、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノブタン、1,6−ジアミノヘキサン、トランス−1,6−ジアミノシクロヘキサン、およびピラゾール−1−カルボキシアミジンを全てAldrich(登録商標)より購入した。全ての溶媒および他の試薬を、販売元より購入し、そして、さらなる精製なしで使用した。モノ−Bocアミンを、文献の手順(クロロホルム中で、10当量のジアミンのおよび1当量のBoc2O、次いで未反応のジアミンを除去するための水でのワークアップ)を用いて、市販ジアミンより合成した(34)。
【0230】
【化15】
【0231】
合成された全てのペプチドおよびペプトイドは、アミノヘキサン酸スペーサーのアミノ基に共有結合されるフルオレセイン部分(FI)を有するN末端アミノ基に付着されたアミノヘキサン(ahx)酸部分を含む。全てのペプチドおよびペプトイドのカルボキシル末端は、カルボキサアミドである。
【0232】
(細胞の取り込みアッセイ)アルギニンホモポリマーおよびグアニジン置換ペプトイドをそれぞれ、PBS緩衝液(pH 7.2)中で溶解し、そして、それらの濃度を490nm(ε=67,000)におけるフルオレセインの吸収によって決定した。濃度を決定するためのこの方法の精度は、選択されたサンプルの秤量およびPBS緩衝液の既知の量においてそれらを溶解することによって確立される。UV分光法によって決定された濃度は、手動で秤量された量に相関する。Jurkat細胞(ヒトT細胞株)、マウスB細胞(CH27)、またはヒトPBL細胞を、10%胎仔ウシ血清およびDMEM中で増殖し、そして、これらのそれぞれを細胞の取り込み実験について使用した。様々な量のアルギニンおよびグアニジン置換ペプトイドのオリゴマーを、2%FCS/PBS(組み合わせて総量200μL)中の約3×106細胞に添加し、そして、マイクロタイタープレート(96ウェル)に配置し、そして、23℃または4℃で、様々な時間でインキュベートした。マイクロタイタープレートを遠心分離し、そして、細胞を単離し、PBS(3×250μL)で洗浄し、5分間37℃で0.05%トリプシン/0.53mM EDTAでインキュベートし、冷PBSで洗浄し、そして、0.1%ヨウ化プロピジウムを含むPBS中で再懸濁した。この細胞を蛍光フローサイトメトリー(FACScan,Becton Dickinson)を用いて分析し、そして、ヨウ化プロピジウムを使用して染色した細胞を、分析から除外した。示されるデータは、収集された5000細胞についての平均蛍光シグナルである。
【0233】
(アジ化ナトリウムを使用した細胞の取り込みの阻害)使用された細胞を、蛍光性ペプチドの添加の前に2%FCS/PBS緩衝液中に0.5%アジ化ナトリウムを使用して30分間プレインキュベートし、そして、その細胞をPBS緩衝液中の0.5%アジ化ナトリウムを使用して洗浄することを除き、以前に記載されるようにアッセイを行なった。細胞の取り込みの全てのアッセイを、アジ化ナトリウムの存在下および非存在下で並行して行なった。
【0234】
(細胞の取り込み速度アッセイ)細胞を、マイクロタイタープレート(96ウェル)において2%FCS/PBS(50μl)中で3連で、4℃で、0.5、1、2、および4分間、インキュベートしたのを除き、アッセイを上記のように行なった。2%FCS/PBS(5mL)中にサンプルを希釈することによって、反応をクエンチした。次いで、このアッセイをワークアップし、そして、以前に記載されたように、蛍光フローサイトメトリーによって分析した。
【0235】
(結果)
短いアルギニンリッチペプチドの細胞の取り込みのための構造的必要を決定するために、一連のTat49〜57の蛍光標識短縮アナログを、標準的な固相化学用いて、合成した。例えば、Atherton、E.ら、SOLID−PHASE PEPTIDE SYNTHESIS(IRL:Oxford,Engl.1989)を参照のこと。フルオレセイン部分をアミノ末端のアミノヘキサン酸スペーサーを介して付着した。次いで、これらの蛍光標識ペプチドがJurkat細胞に入る能力を、蛍光活性化細胞仕分け(FACS)を用いて分析した。試験されたペプチド構築物は、
【0236】
【化16】
【0237】
アミノ末端(Tat50〜57)またはカルボシキル末端(Tat49〜56)のいずれか由来のアルギニン残基の欠失は、親配列(Tat49〜57)と比較して、細胞内蛍光の80%の喪失を生じた。1アミノ酸短縮アナログに由来する、カルボキシル末端からのR−56のさらなる欠失(Tat49〜55)は、細胞内蛍光のさらなる60%の喪失を生じたが、アミノ末端由来のK−50の欠失(Tat51〜57)は、内部移行の量をさらに減少しなかった。これらの結果は、Tat49〜57の短縮アナログが、Jurkat細胞へのフルオレセインの細胞外送達において有意に効果が低く、アルギニン残基が、リジン残基よりも細胞の取り込みにより貢献するようであることを示す。
【0238】
個々のアミノ酸残基の細胞の取り込みへの貢献を決定するために、Tat49〜57のそれぞれの部分でのアラニン置換を含むアナログを合成し、そして、FACS分析によって合成した(図22)。以下の構築物を試験した:
【0239】
【化17】
【0240】
トランスポーターペプチドのキラリティーが重要であるか否かを決定するために、対応するd−(d−Tat49〜57)、レトロ−l−(Tat57〜49)およびレトロ−逆異性体(d−Tat57〜49)を合成し、そして、FACSアッセイによって分析した(図23)。重要なことには、全ての3アナログは、Jurkat細胞に入る際、Tat49〜57よりもより効果的であった。これらの結果は、ペプチド骨格のキラリティーが細胞の取り込みが重要でないことを示した。興味深いことには、Tat49〜57において見出される1つのアルギニンおよび2つのリジンの代わりに、アミノ末端に位置する3つのアルギニン残基を有するレトロ−lアイソマー(Tat57〜49)は、取り込み増加したことを示した。従って、アミノ末端における残基は重要であるようであり、そして、アルギニンは、内部移行について、リジンよりもより効果的である。非天然のd−ペプチドの改善した細胞の取り込みは、アッセイで使用された2%FCS(胎仔ウシ血清)におけるタンパク質分解に対するそれらの増加した安定性に起因するようである。血清が除去される場合、d−ペプチドおよびl−ペプチドは、予想されるように等価であった。
【0241】
これらの最初の結果は、アルギニン含有量が、Tat49〜57の細胞の取り込みについて主な原因であることを示す。さらに、これらの結果は、アルギニンの短いオリゴマーが、細胞に入る際、リジン、オルニチン、およびヒスチジンの対応する短いオリゴマーよりもより効果的であることを実証した、本発明者らの以前の結果と一貫していた。確立されていなかったことは、アルギニンホモオリゴマーが、Tat49〜57よりもより効果的であるか否であった。この点に関して、Tat49〜57を、l−アルギニン(R5−R9)およびd−アルギニン(r5−r9)オリゴマーと比較した。Tat49〜57は、陽イオン性残基を含むが、その細胞内部移行は、R6およびR7との間の内部移行であり(図24)、このことは6つのアルギニン残基の存在が、細胞の取り込みについて最も重要な因子であることを示す。重要なことに、7−9アルギニン残基を含む結合体は、Tat49〜57よりもより優れた取り込みを示した。
【0242】
これらのアルギニンオリゴマーおよびTat49〜57の細胞に入る能力を定量的に比較するために、ミカエリス−メンテン速度分析を行なった。細胞の取り込みの速度は、30、60、120および240秒間、Jurkat細胞におけるペプチドのインキュベーション(3℃)後に決定した(表1)。結果のKm値は、r9およびR9が、Tat49 〜57よりもそれぞれ、約100倍および20倍速い速度で、細胞に入ることを明らかにした。比較のために、アンテナペンディア(Antennapedia)43〜59をまた分析し、そして、Tat49〜57より約2倍速く細胞に入ることを示したが、r9およびR9よりも有意に遅かった。
【0243】
【表1】
(Tat49−57のペプチド模倣アナログの設計および合成)
(方法)
ペプトイドポリアミン合成の一般的手順。ペプトイドを、Zuckermannによって開発された文献の手順に従って、樹脂を混合するために、フリッティングしたガラス装置および窒素の正圧を使用して、手動で合成した。例えば、Murphy,J.Eら、Proc.Natl.Acad.Sci.USA 95,1517−1522(1998);Simon,R.Jら、Proc.Natl.Acad.Sci.USA 89,9367−9371(1992);Zuckermann,R.Nら、J.Am.Chem.Soc.114,10646−10647(1992)を参照のこと。Fmoc置換Rinkアミド樹脂(0.2mmol)の、20%ピペリジン/DMF(5mL)での30分間(2×)の処理により、遊離樹脂結合アミンを得、これをDMF(3×5mL)で洗浄した。この樹脂を、DMF(5mL)中のブロモ酢酸(2.0mmol)の溶液で30分間処理した。この手順を繰り返した。次いで、この樹脂を洗浄し(3×5mL DMF)、そしてDMF(5mL)中のモノ−Bocジアミン(8.0mmol)の溶液で12時間処理した。これら2つの工程を、所望の長さのオリゴマーが得られるまで繰り返した(注記:DMF中のモノ−Bocジアミンの溶液を、収率を明らかに低下させることなく再利用し得た)。次いで、この樹脂を、DMF中のN−Fmoc−アミノヘキサン酸(2.0mmol)およびDIC(2.0mmol)で1時間処理し、そしてこれを繰り返した。次いで、Fmocを、20%ピペリジン/DMF(5mL)で30分間処理することによって、除去した。この工程を繰り返し、そしてこの樹脂をDMF(3×5mL)で洗浄した。次いで、遊離アミン樹脂を、DMF(5mL)中のフルオレセインイソチオシアネート(0.2mmol)およびDIEA(2.0mmol)で12時間処理した。次いで、この樹脂をDMF(3×5mL)およびジクロロメタン(5×5mL)で洗浄した。この樹脂からの切断を、95:5 TFA/トリイソプロピルシラン(8mL)を使用して達成した。減圧下での溶媒の除去により、粗製油を得、これを冷エーテル(20mL)で粉砕した。このように得た粗製混合物を遠心分離し、エーテルをデカンテーションによって除去し、そして得られる橙色固体を、逆相HPLC(0.1% TFA中H2O/CH3CN)により精製した。この生成物を、凍結乾燥によって単離し、そして電子スプレー質量分析によって、そして選択され場合には1H NMR分光法によって、特徴付けた。
【0244】
ペプトイドポリアミンの過グアニジニル化(Perguanidinylation)のための一般的手順。脱イオン水(5mL)に溶解したペプトイドアミン(0.1mmol)の溶液を、炭酸ナトリウム(1つのアミン残基あたり5当量)およびピラゾール−1−カルボキサミジン(1つのアミン残基あたり5当量)で処理し、そして24〜48時間50℃に加熱した。次いで、この粗製混合物を、TFA(0.5mL)で酸性化し、そして逆相HPLC(0.1%TFA中H2O/CH3CN)で直接精製した。この生成物を、電子スプレー質量分析によって特徴付け、そして凍結乾燥によって単離し、そして逆相HPLCによってさらに精製した。このグアニジン置換したペプトイドの純度は、分析逆相HPLC(0.1% TFA中H2O/CH3CN)によって決定した場合に、95%より高かった。
【0245】
(結果)
Tat47−59の細胞取り込みについて決定した構造−機能関係を利用して、本発明者らは、アルギニンオリゴマーの側鎖の1,4骨格間隔を保存するが、ステレオジェン中心が欠けているオリゴグリシン骨格を有するポリグアニジンペプトイド誘導体のセットを設計した。アルギニン様側鎖をアミド窒素に組み込むこれらのペプトイドを、これらのタンパク分解に対する予測される抵抗性、ならびに合成の潜在的な容易さおよび有意に低い費用に起因して、選択した。(Simonら、Proc.Natl.Acad.Sci.USA 89:9367−9371(1992);Zuckermannら、J.Am.Chem.Soc.114:10646−10647(1992))さらに、ラセミ化(頻繁に、ペプチド合成において遭遇する)は、ペプトイド合成においては問題ではない;そして「亜モノマー(sub−monomer)」ペプトイドアプローチは、側鎖スペーサーの容易な改変を可能にする。Tat49−57のオリゴウレア(oligurea)およびペプトイド−ペプチドハイブリッド(Hamyら、Proc.Natl.Acad.Sci.USA 94:3548−3553(1997))誘導体の調製は、以前に報告されているが、これらの細胞取り込みは、明白には研究されなかった。
【0246】
所望のペプトイドを、ペプトイドに対する「亜モノマー」アプローチ(Simonら;Zuckermannら)を使用して調製し、続いてアミノヘキサン酸スペーサーを介して、フルオレセイン部分をアミン末端に結合させた。固相樹脂からの切断の後に、このように得られた蛍光標識したポリアミンペプトイドを過剰のピラゾール−1−カルボキサミジン(Bernatowiczら、J.Org.Chem.57:2497−2502(1992)および炭酸ナトリウムで処理することによって、良好な収率(60〜70%)でポリグアニジンペプトイドに転換した(図25に示すように)。以前に報告された、単離されたN−Argユニットを含むペプトイドの合成は、ペプトイド合成の前のN−Argモノマーの合成(5〜7工程)および特殊化されかつ高価なグアニジン保護基(Pmc、Pbf)の使用に依存した(Kruijtzerら、Chem.Eur.J.4:1570−1580(1998);Heizmannら、Peptide Res.7:328−332(1994))。本明細書で報告される化合物は、過グアニジニル化工程を使用して調製される、ポリグアニジニル化ペプトイドの最初の例を提示する。この方法は、対応するポリアミンからのポリグアニジニル化化合物への容易なアクセスを提供し、そして過グアニジニル化ホモオリゴマーの合成のために、特に有用である。さらに、この方法は、高価な保護基(Pbf、Pmc)の使用を排除する。新規のトリフリル(triflyl)置換グアニル化剤を使用する、ペプチド基質の過グアニジニル化のさらなる例が、最近報告された(Feichtingerら、J.Org.Chem.63:8432−8439(1998))。
【0247】
蛍光標識されたポリグアニジンN−arg5ペプトイド、ポリグアニジンN−arg7ペプトイド、ポリグアニジンN−arg9ペプトイドの細胞取り込みを、Jurkat細胞およびFACS分析を使用して、対応するd−アルギニンペプチドr5、r7、r9(類似のタンパク分解性特性)と比較した。N−arg5、N−arg7、N−arg9を含む細胞の内側で測定した蛍光の量は、r5、r7、r9に関して見出される量に類似して、グアニジン残基の数に比例した:N−arg9>N−arg7>N−arg5(図26)。さらに、N−arg5ペプトイド、N−arg7ペプトイド、N−arg9ペプトイドは、対応するペプチドr5、r7、r9と比較して、わずかに低い量の細胞侵入のみを示した。これらの結果は、Tat49−57またはアルギニンオリゴマーのペプチド骨格に沿った水素結合が、細胞取り込みのために必要とされる構造的エレメントではなく、そしてオリゴマーのグアニジン置換ペプトイドが、アルギニンに富むペプチドの代わりに、分子トランスポーターとして利用され得ることを実証する。アジ化ナトリウムの添加は、インターナリゼーションを阻害し、このことは、ペプトイドの細胞取り込みもまたエネルギー依存性であることを示した。
【0248】
(実施例15)
(細胞取り込みに対する側鎖長の効果)
N−argペプトイドが効果的に細胞膜と交差することを確認した後に、細胞取り込みに対する側鎖長(メチレンの数)の効果を調査した。所定の数のグアニジン残基(5、7、9)については、細胞取り込みは、側鎖長に比例した。より長い側鎖を有するペプトイドは、より効率的な細胞取り込みを示した。グアニジンヘッド基と骨格との間に6つのメチレンスペーサーを有する9マーペプトイドアナログ(N−hxg9)は、対応するd−アルギニンオリゴマー(r9)より顕著により高い細胞取り込みを示した。取り込みの相対的順序は、N−hxg9(6メチレン)>N−btg9(4メチレン)>r9(3メチレン)>N−arg9(3メチレン)>N−etg9(2メチレン)であった(図27)。注記として、N−hxgペプトイドは、対応するd−アルギニンオリゴマーよりさらに大きな、顕著に高い細胞取り込みを示した。対応するヘプタマーおよびペンタマーの細胞取り込みもまた、同じ相対的傾向を示した。N−hxgペプトイドに含まれる側鎖がより長いほど、インターナリゼーションの量がさらに1つ多いグアニジン残基を含む対応するd−アルギニンオリゴマーに匹敵するような程度まで、細胞取り込みが改善された(図28)。例えば、N−hxg7ペプトイドは、r8に匹敵する細胞取り込みを示した。
【0249】
細胞取り込みの増加が、増加した長さの側鎖に起因したのか、またはそれらの疎水性の性質に起因したのかを確認するために、シクロヘキシル側鎖を含むペプトイドのセットを合成した。これらを、N−chg5ペプトイド、N−chg7ペプトイド、N−chg9ペプトイドと呼ぶ。これらは、N−hxgペプトイドと同じ数の側鎖炭素を含むが、異なる自由度を有する。興味深いことに、N−chgペプトイドは、以前にアッセイされたペプトイド(N−etgペプトイドを含む)の全てよりも、ずっと低い細胞取り込み活性を示した(図29)。従って、直鎖アルキルスペーサー基(spacing group)の、コンホメーションの可撓性および立体的に妨害されない性質が、効率的な細胞取り込みのために重要である。
【0250】
(考察)
ノナペプチドTat49−57は、以前に、原形質膜を通して効率的に転座することが示された。この研究の目的は、この効果のための構造的基礎を決定すること、およびこの情報を使用してより単純かつより効果的な分子トランスポーターを開発することであった。この目的のために、フルオレセイン標識に結合体化した、Tat49−57の短縮型誘導体およびアラニン置換誘導体を調製した。これらの誘導体は、Tat49−57と比較して非常に減少した細胞取り込みを示し、このことは、Tat49−57のカチオン性残基の全てが、効率的な細胞取り込みのために必要とされることを示す。カチオン性オリゴマーの短いオリゴマーに関する本発明者らの以前の研究と比較した場合に、これらの知見は、アルギニンのオリゴマーが、Tat49−57より優れており、そして確かにより容易かつ費用効率的に調製され得ることを示唆した。短いアルギニンオリゴマーとTat49−57との比較は、前者のメンバーが、実際に、より効率的に細胞に取り込まれることを示した。これは、初めて、Michaelis−Mentonの速度論分析によって、さらに定量され、この分析は、R9およびr9オリゴマーが、Tat49−57に関して見出される値より30倍および100倍大きなKm値を有することを示した。
【0251】
本発明者らのペプチドの研究において明らかとなった、グアニジノヘッド基の重要性およびオリゴマーキラリティーの明らかな不感受性を考慮して、本発明者らは、新規な一連のポリグアニジンペプトイドを設計し、そして合成した。アルギニン側鎖を組み込むペプトイドであるN−arg5、N−arg7、N−arg9は、対応するd−アルギニンペプチドr5、r7、r9に匹敵する細胞取り込みを示した。このことは、ペプチド骨格に沿った水素結合および骨格キラリティーが、細胞取り込みのために必須ではないことを示す。この観察は、これらのペプトイド、アルギニンオリゴマー、およびTat49−57(これらは全て、深く埋め込まれた骨格およびグアニジニウム優性表面を有する)の分子モデルと一致する。分子モデルは、これらの構造的特性が、ペプトイド骨格とグアニジノヘッド基との間に異なるアルキルスペーサーを有するオリゴマーにおいて、様々な程度で保持されることを、さらに明らかにする。従って、骨格と側鎖との間に2−(N−etg)、4−(N−btg)、および6−atom(N−hxg)のスペーサーを組み込む一連のペプトイドを調製し、そして細胞取り込みについて、N−argペプトイド(3原子スペーサー)およびd−アルギニンオリゴマーと比較した。側鎖の長さは、細胞侵入に対して、劇的な影響を有した。細胞取り込みの量は、N−hxg>N−btg>N−arg>N−etgの順で、側鎖の長さに比例した。細胞取り込みは、グアニジンヘッド基と骨格との間のアルキルスペーサーユニットの数が増加する場合に、改善された。重要なことに、N−hxg9は、r9より優れており、後者は、Tat49−57より100倍良好であった。この結果により、本発明者らは、グアニジノ基と骨格との間により長いオクチルスペーサー(N−ocg)を含むペプトイド誘導体を調製した。溶解度に関する問題点に起因して、本発明者らは、これらの化合物を試験しなかった。
【0252】
過グアニジニル化ペプチドと過グアニジニル化ペプトイドとの両方が、効率的に細胞に侵入するので、グアニジンヘッド基(骨格とは独立している)は、明らかに、細胞取り込みの重大な構造的決定因子である。しかし、いくつか(6より大きい)のグアニジン部分が分子の足場に存在することは、細胞内への能動的な移送のためには十分ではない。なぜなら、N−chgペプトイドは、細胞内に効率的に転座しなかったからである。従って、グアニジンヘッド基の重要性に加えて、細胞取り込みのために重要な構造/コンホメーション要件が存在する。
【0253】
要約すると、この研究は、Tat49−57が細胞に侵入する能力に影響を与える、一連の構造的特徴(配列の長さ、アミノ酸組成、およびキラリティーを含む)を同定した。これらの特徴は、一連の新規ペプトイドの設計のための設計図を提供し、これらのうちの17のメンバーが合成され、そして細胞取り込みに関してアッセイされた。重要なことには、N−hxg9トランスポーターは、細胞取り込みが、r9より優れており、これは、N−btg9に匹敵することが見出された。従って、これらのペプトイドトランスポーターは、Tat49−57より実質的に良好であることが示された。この研究は、ペプチド骨格およびその骨格に沿った水素結合が、細胞取り込みのために必要ではないこと、グアニジノヘッド基が、他のカチオン性サブユニットより優れていること、ならびに最も重要なことには、骨格とヘッド基との間のアルキル鎖の伸長が、優れたトランスポーターを提供することを、確認した。より良好な取り込み性能に加えて、これらの新規ペプトイドは、Tat49−57より優れたいくつかの利点(費用効果、アナログの合成の容易さ、およびプロテアーゼ安定性を含む)を与える。これらの特性は、それらの有意な水溶性(>100mg/mL)とともに、これらの新規ペプトイドが、薬物、薬物候補および他の薬剤の、細胞内への分子送達のための効果的なトランスポーターとして働き得ることを示す。
【0254】
(実施例16)
(イトラコナゾール−トランスポーター結合体の合成)
この実施例は、送達増強トランスポーターを、トリアゾール構造を含む化合物に結合させるための一般的なストラテジーの1つの適用を提供する。このスキーム(アルギニン(r7)送達増強トランスポーターへのイトラコナゾールの結合を例として使用する)を、図30に示す。このスキームにおいて、RはHまたはアルキルであり、nは1または2であり、そしてXはハロゲンである。
【0255】
この反応は、ハロゲン(例えば、Br、Fl、I)またはメチルエステルを有するアシル基を結合するために、トリアゾール環の窒素の四量化の使用を含む。化合物3を、HPLCによって単離した。D2O中でのプロトンNMRは、イトラコナゾールおよびトランスポーターのピークを明らかにした。
【0256】
メチルエステルは、70%以上の収率を提供したが、Br−プロピオン酸/エステル対を使用して得られた収率は、40〜50%であった。次いで、このアシル誘導体は、送達増強トランスポーターのアミンと反応して、結合体を形成する。あるいは、ハロゲン化アシル基が、アミド結合を介して最初にトランスポーター分子に結合し得、その後、薬物化合物との反応が実施される。
【0257】
(実施例17)
(FK506結合体の調製)
この実施例は、FK506が、送達増強トランスポーターに結合される結合体の調製を記載する。2つの異なるリンカーを使用し、これらの各々は、生理学的pH(pH5.5〜7.5)でFK506を放出したが、より酸性のpHでより長い半減期を有した。これらのスキームを、図31AおよびBに図示する。
【0258】
(リンカー1:6−マレイミドカプロンヒドラジドトリフルオロアセテート(スキームIおよびII))
無水メタノール(5mL)中のFK506(1)(0.1g、124.4μmol)、6−マレイミドカプロンヒドラジドトリフルオロアセテート(2)(0.126g、373.2μmol)およびトリフルオロ酢酸(触媒、1μL)の溶液を、室温で36時間攪拌した。この反応物を薄層クロマトグラフィーによってモニタリングし、出発物質のほぼ完全な消失を示した[TLC溶媒系−ジクロロメタン(95):メタノール(5)、Rf=0.3]。この反応混合物を乾燥するまで濃縮して、酢酸エチル(20mL)中に溶解させた。有機層を水および10%重炭酸ナトリウム溶液で洗浄し、次いで硫酸ナトリウムで乾燥させ、濾過し、そして濃縮した。残渣を溶出液としてジクロロメタン(96):メタノール(4)を使用するカラムクロマトグラフィーによって精製して、ヒドラゾン3(0.116g、92%)を得た。
【0259】
無水ジメチルホルムアミド(1mL)中の上記ヒドラゾン(3)(0.025g、24.7μmol)、トランスポーター(1×Bacar9CCONH2・9TFA、Bacar7CCONH2・7TFA、BacaCCONH2、NH2r7CCONH2・8TFA、NH2R7CCONH2・8TFA)およびジイソプロピルエチルアミン(1×)の溶液を、窒素下で36時間室温で攪拌し、この時点で、TLCは、出発ヒドラゾンの完全な消失を示した。溶媒を反応混合物からエバポレートし、そして残渣を、トリフルオロ酢酸緩衝化水およびアセトニトリルを使用する逆相HPLCによって精製した。
【0260】
種々のトランスポーターとの結合体の収率は、以下:
【0261】
【化18】
【0262】
全ての生成物の構造を、1H−NMRスペクトルおよびTOF MS分析によって確認した。
【0263】
(リンカー2:2−(2−ピリジニルジチオ)エチルヒドラジンカルボキシレート(スキームIIIおよびIV))
無水メタノール(5mL)中のFK506(1)(0.1g、124.4μmol)、2−(2−ピリジニルジチオ)エチルヒドラジンカルボキシレート(9)(0.091g、373.2μmol)およびトリフルオロ酢酸(触媒、1μL)の溶液を、室温で16時間攪拌した。この反応物を薄層クロマトグラフィーによってモニタリングし、出発物質のほぼ完全な消失を示した[TLC溶媒系−酢酸エチル、Rf=0.5]。この反応混合物を乾燥するまで濃縮して、酢酸エチル(20mL)中に溶解させた。有機層を水および10%重炭酸ナトリウム溶液で洗浄し、次いで硫酸ナトリウムで乾燥させ、濾過し、そして濃縮した。残渣を溶出液としてジクロロメタン(97):メタノール(3)を使用するカラムクロマトグラフィーによって精製して、ヒドラゾン10(0.091g、71%)を得た。
【0264】
(実施例18)
(胃腸管におけるトランスポーターの差示的取り込み)
(方法)
(胃腸吸収プロトコル)
Taconic(Germantown、NY)から購入した8週齢および10週齢の雌Swiss Websterマウスに対して実験を行った。マウスをNembutalで麻酔し、腹部に沿って正中切開を行った。腸を測定し、所望の区画の両端で縫合によって結紮し、そしてビオチン化ペプチドを管腔中に注入した(約100λ/インチ)。15分のインキュベーション後、この組織を取り出し、そして管腔をPBSで穏やかに洗浄した。
【0265】
CellGateトランスポーターが、口腔の扁平上皮に進入し得るか否かを決定するために、マウスをNembutalで麻酔し、その頭部を一方向に傾け、そしてその口にビオチン化ペプチドの溶液を配置した。15分後、液体をピペットによって除去した。
【0266】
(胃腸管領域の組織学的切片の調製)
ビオチン化ペプチドとのインキュベーションの直後に、麻酔したげっ歯類動物を頸部脱臼によって屠殺し、そして結紮した胃腸管の区画を取り出した。種々の区画の管腔を、プラスチック先端のシリンジを使用してOCTで満たし、OCTを満たしたボートに浸漬し、そして2−メチル−ブタン/ドライアイス溶液中ですばやく凍結させた。凍結切片を僅かに温め、そして2mmの厚さの矢状切断を、鋼鉄のかみそりの刃を使用して行った。切片をOCT型に配置し、そして2−メチル−ブタン/ドライアイス溶液中ですばやく凍結させた。切片(5μm)を、クリオスタット上で切断し、アセトン中で4℃で10分間固定し、そして風乾させた。PBS中で5分間の再水和後、切片を正常ウマ血清でブロッキングし、洗浄し、ストレプトアビジン−FITC(20μg/ml)と共に30分間インキュベートし、洗浄し、そしてヨウ化プロピジウム(PI)を含む取り付け媒体を用いて取り付けた。
【0267】
(組織培養)
Caco−2細胞をATCCから獲得し、解凍し、そしてペニシリン、ストレプトマイシン、グルタミンおよび10%ウシ胎仔血清を含むDMEM中で、コンフルエント(>250,000細胞/cm2)になるまで3〜5日間増殖させ、次いで数継代サイクルにわたって、60,000細胞/cm2で継代した。継代数25〜29を、Snapwell 12mm直径0.4ミクロン孔サイズのポリカーボネート膜(Corning Costar、Corning、N.Y.)上に、膜1枚当たり60,000細胞でプレートし、そして少なくとも21日間増殖させ、培地を一日おきに交換した。
【0268】
(Caco−2細胞における細胞取り込み)
懸濁物中の細胞へのD−アルギニンの蛍光オリゴマーの透過を分析するために、単層をトリプシン(0.05%溶液4.5ml、Gibco、Grand Rapids、MI)で処理し、個々の細胞懸濁物をスピンダウンした。細胞を再懸濁し、計数し、そして種々の濃度のFl aca r5、Fl aca r7、Fl aca r9、Fl aca k9(50〜0.8μM)で5分間処理し、洗浄し、400λ PBS、2%EBS、40ngPI/ml中に再懸濁し、フローサイトメトリーによって分析した。
【0269】
蛍光ペプチドが単層の一部のCaco−2細胞に進入する能力を分析するために、細胞を、lab−tek flaskette顕微鏡スライド(Nalge Nunc Int.、Naperville、IL)において、60,000細胞/mlの密度で4ml中に播種し、そして培地を一日おきに交換しながら21日間増殖させた。一旦単層が確立されると、これを100μM Fl aca r9 CONH2と共に5分間インキュベートした。この単層を、引き続いてPBS/2%FBSで2回洗浄して、標識ペプチドを除去し、蛍光顕微鏡を使用して分析した。
【0270】
(単層のCaco−2細胞を横切る輸送)
短いD−アルギニンのオリゴマーが、Caco−2細胞の単層を横切り得るか否かを分析する実験を、電位固定増幅器およびEasymount並行水平拡散チャンバ(Physiologic Instruments、San Diego、CA)を、再循環水浴に接続された95%酸素5%二酸化炭素ガスリフトシステムと共に使用して、実行した。
【0271】
電流測定値(I2−I1)を、試験化合物の添加の10分前を含めて、5分間隔で取り、単層がインタクトであることを保証した。ゼロ時間で、試験化合物を適切な濃度で添加し、そして測定値を記録した。測定値を60分間にわたって5分間隔でとり、経上皮電気抵抗(TEER)および短絡電流(Isc)を、これらの値から引き続いて計算した。
【0272】
(合成化学)
(シクロスポリンA)
この研究において使用したCsA結合体の詳細は、以前に記載されている(Rothbardら、Nature Medicine 6、1253 2000)。図6もまた参照のこと。簡潔には、CsAを、図6Aに示されるpH感受性リンカーを介して、D−アルギニン七量体に結合体化した。得られた結合体は、酸性pHで安定であるが、7より高いpHでは、CsAに隣接するカルボニルに対する遊離アミンの付加を含む分子内環化を受けて(図6B)、未修飾CsAの放出を生じる。
【0273】
(タキソール)
タキソールを、無水α−クロロ酢酸で処理し、C−2’クロロアセチル誘導体12を実質的に定量的な収率で生じた。
【0274】
【化19】
【化20】
【0276】
化合物13をC2’エステルカルボニルへのN末端窒素の求核攻撃を介してタキソールを放出するように設計した。この窒素のプロトン化状態は、この機構に重要である。なぜなら、遊離アミンのみがこの放出を行い得るからである。さらに、両方の結合体は、単純なエステル加水分解に対して感受性にする、共通のα−ヘテロ原子置換されたアセテート部分を共有する。これは、さらなる放出経路を提供する。
【0277】
(結腸内注入)
約200〜300gの雌のWisterラット(Simonsen、Gilroy、CA)を麻酔し、その腹部を剃毛し、正中切開を行い、そして各動物の上行結腸において1.5インチの切片を、縫合によって結紮した。タキソールおよびシクロスポリン(5mg/kg)を1:1v/vのCremophor EL:エタノール混合物中の溶液として注入したが、一方、等モル量のr7結合体を、PBS中で注入した。全ての場合において、注入される容積は、約500λであった。結腸を腔に戻し、そして切開を縫合を使用して閉じた。
【0278】
血液サンプルを、ゼロ時間(薬物注入前)および実験の持続期間の間30分毎に尾部静脈から採取し、そして90分後に終了した1匹の動物以外、実験的に決定した。凝固を、100λの0.5% EDTAを含むガラス管に血液を移すことによって阻害し、そしてこの血液を凍結させた。
【0279】
(全血からの薬物抽出およびHPLC MS MS分析)
タキソールまたはシクロスポリンのいずれかを、文献の手順の改変を使用して全血から抽出した。簡潔には、全血(100λ)を、5mlのジエチルエーテルを含むネジキャップ付のガラス管に移した。このサンプルを2分間にわたって激しくボルテックスし、遠心分離し、そしてドライアイス/メタノール中で凍結させた。エーテル層を別のガラス管に移し、そしてエーテルをエバポレートした。シクロスポリンの場合、残渣を1.5mlのメタノール、水、アセトニトリル(3:2:1)に再懸濁したが、タキソールについては、残渣を1.5mlのメタノール:アセトニトリル(1:1)に再懸濁した。サンプルをPerkin−Elmerシリーズ200自動サンプラー中に配置し、そしてShimazu HPLCシステムに接続されたC18逆カラム上に70℃で引き続いて注入し、70%メタノール、30%水性蟻酸アンモニウム緩衝液で溶出し、そして溶出液をPE Sciex API 3000タンデム質量分析器で分析した。既知の量のシクロスポリンA(10〜1000ng/ml)またはタキソール(1〜1000ng/ml)のいずれかを全血に添加し、そして以前に記載されたように抽出して、標準曲線を得た。シクロスポリンAを、2つの遷移(1220〜1203ダルトンおよび1220〜100ダルトンの両方)によってモニタリングした。1220種は、シクロスポリンA+アンモニアに対応し、1203はプロトン化シクロスポリンであるが、100は既知のフラグメントである。タキソールを2つの遷移(872〜855ダルトンおよび872〜110ダルトンの両方)によってモニタリングした。872種は、タキソールのアンモニウム付加物に対応し、855は、プロトン化親化合物であるが、110は、第二の四極子において優勢に見られるフラグメントである。
【0280】
サンプル中のシクロスポリンAおよびタキソールの総量を、標準曲線によって確立された値と適切なピークの積分面積を比較することによって決定した。
【0281】
(結果)
(胃腸管におけるトランスポーターの差示的取り込み)
短いD−アルギニンのオリゴマーは、懸濁物中で増殖した広範な種々の細胞株の原形質膜を迅速かつ効率的に通過することが示された。さらに、これらは、局所的に適用される場合に複数層の皮膚を透過し、静脈内に注入される場合に静脈および動脈の複数層の内皮細胞および平滑筋細胞を透過し、そして吸入される場合には複数層の肺組織を透過することが示されている。これらの化合物が、胃腸管の非角質化上皮に進入し得るか否かを決定するために、断食マウスの小腸および大腸の区画を結紮し、そしてD−アルギニンのビオチン化九量体(bio aca r9 CONH2)の溶液(100μM)を注入した。15分間の曝露後、組織の適切な区画を解剖し、凍結し、切片化し、そしてストレプトアビジンフルオレセインで染色して、ビオチン化ペプチドの位置を規定し、そしてヨウ化プロピジウムで切片中の全ての核を対比染色した。
【0282】
マウス十二指腸の管腔に注入された場合、バックグラウンドを超えて検出可能な染色は観察されなかった。乏しい染色はまた、ビオチン化ペプチドを空腸に注入した場合に見られた。複数の切片を走査した場合、検出可能な染色は、いくつかの絨毛の先端で見られた。均質な染色の第一の徴候は、回腸の切片を分析した場合に観察された。染色は、微絨毛の先端に局在し、そして陰窩の細胞には及ばなかった。回腸の全ての切片において見られたが、観察された蛍光は、皮膚、肺、または動脈もしくは静脈の内皮細胞において以前に観察された強度レベルには到達しなかった。
【0283】
小腸の比較的乏しい染色は、結腸領域が試験された場合に見られた染色とは、顕著に異なった。大腸の上行区画および横行区画の両方において、ビオチン化アルギニン九量体は、局所的に適用された場合、絨毛の全ての表面領域を染色し、いくつかの細胞層を透過し、表皮および真皮の強い染色を想起させた。さらに、陰窩の細胞は、結腸区画において強く染色され、そしてこの区画の全厚みの透過についての証拠が、いくつかの領域において観察された。
【0284】
組織学的分析は、胃腸管の非角質化上皮層へのbio r9の取り込みが、胃の幽門からの距離と共に増大する取り込みを伴って、有意に変化したことを実証した。回腸において観察された染色は、空腸において観察された染色よりも強く、空腸において観察された染色は、十二指腸において観察された染色よりも強かった。最も強い染色は、結腸の上行領域および横行領域において見られた。本発明を特定の理論または機構に限定することを意図しないが、上皮を裏打ちする粘膜の組成および量が、差示的染色における重要な因子であり得るというのが1つの理論である。
【0285】
(Caco−2細胞への細胞取り込み)
Caco−2細胞(市販のヒト結腸細胞)を、種々の量の蛍光標識したD−アルギニンの五量体、七量体および九量体(Fl aca r5、Fl aca r7、Fl aca r9)またはリジン九量体(Fl aca k7)と共に懸濁物中でインキュベートし、そしてフローサイトメトリーによって分析し、種々の他の懸濁細胞において以前に見られたパターン(Mitchellら、Peptide Research 56、318(2000))と類似のパターンが観察された(図32)。蛍光ペプチドの取り込みは、アルギニン含量と共に増大し、r9はr7より効率的であり、r7はr5よりも効率的であった。全てのアルギニンポリマーは、リジン九量体よりもより効率的に細胞に進入した。蛍光の速度、相対量、および延長した時間(数時間)にわたる内在化した蛍光ペプチドの見かけの流出の欠如の両方は、リンパ球を用いたより初期の実験を想起させた。
【0286】
細胞が懸濁物中に存在した場合にのみ見られたCaco−2細胞への短いアルギニンオリゴマーの迅速な透過が、人為的結果ではないことを確認するために、Fl aca r9 CONH2(50μM)を顕微鏡スライド上に単層として増殖したCaco−2細胞と共に5分間インキュベートし、洗浄し、そして蛍光顕微鏡によって分析した。懸濁細胞のフローサイトメトリーと一致して、単層中の実質的に全ての細胞は、5分後に蛍光であった。
【0287】
ペプチドがCaco−2細胞に迅速に進入する能力を、拡散チャンバ中の膜として単層のCaco−2細胞を配置し、そしてこれを、タキソールと異なる長さのアルギニンオリゴマーとの間の蛍光トランスポーター薬物結合体に曝露することによって、しっかりと確立した。種々の濃度のタキソール結合体(50〜0.08μM)を、頂端側に添加し、そして3分間、単層のCaco−2細胞に曝露した。この膜を装置から取り外し、細胞をトリプシン処理し、そして得られた細胞懸濁物をフローサイトメトリーによって分析した(図33)。ペプチド単独と同様に、薬物結合体は、迅速かつ効率的に単層を含む細胞に進入し、そしてより多いアルギニンを含む結合体がより効率的であった。
【0288】
本発明の理論を限定することを意図しないが、これらの実験は、フルオレセインまたは治療薬物(例えば、タキソール)のいずれかと結合体化した短いアルギニンオリゴマーが、懸濁物中に存在する場合および単層で増殖した場合の両方において、迅速にCaco−2細胞に進入するという仮説の強力な支持を提供する。
【0289】
(Caco−2単層を横切る輸送)
腸上皮の管腔膜を横切ることは、送達された(例えば、頬投与される)薬物の血中レベルを増大させるために必要である。このトランスポーターが腸上皮を横切り得るか否かを決定するために、単層のCaco−2細胞を培養物中で増殖させ、そして市販の拡散チャンバ中に膜として配置する。膜の完全性を、経上皮電気抵抗(TEER)が、100Ωcm2よりいつも大きいことを実証することによって、実証した(図34)。膜がインタクトであり、細胞間に有意な空間が存在しない場合にのみ、このような安定な抵抗パターンが観察される。
【0290】
膜が生存性かつ連続であることのさらなる証拠は、Lucifer Yellow(200μM)はこの単層を横切って運搬されなかったが、一方でヒドロコルチゾンが刊行された報告と一致する量で輸送されたことであった(図35)。
【0291】
4〜15サブユニットの範囲のD−アルギニンまたはL−アルギニンの種々の蛍光オリゴマーが、多数の膜を用いた複数の実験において頂部チャンバに配置された場合、基底外側のチャンバへは有意に輸送されなかった(図35)。
【0292】
一緒に考えて、本明細書中に示されるデータは、送達された化合物(例えば、フルオレセインまたはタキソールのいずれか)に結合体化した短いアルギニンオリゴマーが、Caco−2細胞単層に迅速に進入するが、Caco−2細胞単層を横切って輸送されないというモデルと一致する。このモデルはまた、本発明のトランスポーターが、エンドソームによってCaco−2細胞に進入せず、結腸上皮を横切って輸送され、十分理解された経路によって基底外側に排泄されることを意味する。CellGateトランスポーターは、細胞質ゾルに直接進入するようであり、そして基底外側での高い流出速度を有さないようである。
【0293】
(直腸内注入後の薬物血中レベルの測定)
単独、またはタキソールもしくはシクロスポリンのいずれかと結合した場合のいずれかの、フルオレセインで標識した短いアルギニンオリゴマーは、インビトロで単層のCaco−2細胞を横切ることができなかったが、この細胞株は、結腸のインビボの挙動を正確に模倣していないかもしれない。さらに、短いアルギニンオリゴマーとのインキュベーション後に結腸組織において見られた蛍光パターンは、このペプチドが、血管新生が生じることが公知の層に透過したことを実証した。
【0294】
本発明のトランスポーターが、経口投与された薬物の送達を増強し得るか否かを決定するために、シクロスポリンAおよびタキソールの放出可能な結合体を、直腸内に注入し、そして生じた、放出された薬物の血中レベルを、LC MS MSによって測定した。
【0295】
第一の実験を、リン酸緩衝化生理食塩水中に溶解された等モル量の放出可能なCsAのr7結合体と比較して、Cremophor EL:エタノール中のCsA5mg/kgの結腸内注入後のCsA血中レベルを測定するために設計した。親薬物の場合、血中レベルは30分後に約25ng/mlのCsAにまで上昇し、次いで基準レベルに近いレベルまで迅速に低下した(図36)。90分後には動物が死亡したので、さらなるデータは獲得しなかった。対照的に、等モル量のCsA−r7結合体を注入した場合、血中の検出可能なレベルのCsAは、3時間後にのみ観察された(図36)。短いアルギニンオリゴマーに結合体化された場合に変更されたCsAの薬物動態が再現性があるかどうかを決定するために、第三のラットに、10mg/kg相当の水溶性放出可能結合体を注入した。この動物の血中のCsA取り込み速度は、5mg/kgのこの結合体を注入した動物と類似した。より多くの結合体を投与すると、30分で小さい増大が見られたが、より多い量は、血中に2時間後にのみ現れ、血中レベルは3時間後に45ng/mlに達した。全体的パターンは、この結合体を注入した2匹の動物において類似であった。両方の場合において、循環中で測定されたCsAの全体量は、CsAを注入した場合よりも有意に多かった。
【0296】
CsA結合体の半減期は、約90分であり、これは、親化合物と比較した、血中CsAの出現の遅延と一致した。この事実を、結腸の円柱上皮における迅速かつ効率的な取り込み、およびCsAまたはタキソールの放出不能な結合体がインビトロで単層の結腸細胞を横切ることができないことを実証する組織学的データと合わせて、この現象を説明する理にかなった簡潔なモデルを導く。本発明を特定の機構または理論に限定することを意図しないが、この結合体は、Cremophor中で注入されたCsAよりもかなり効率的かつより迅速に結腸の円柱上皮に進入するが、結合体が加水分解されるまで上皮細胞中に保持され、そして毛細管を囲む内皮細胞を横切らないようである。一旦放出されると、CsAは、血中に自由に拡散するか、または能動的に輸送された。
【0297】
この仮説を試すために、タキソールおよび異なる2つのr7−タキソール結合体を結腸に注射し、そして薬物の血中レベルを30分間隔で測定した。2つのr7−タキソール結合体は、有意に異なる半減期(10分および5時間)を有し、そしてこの薬物結合体の加水分解が、循環での薬物の出現における律速段階であるという仮説を試すために使用された。本当であるならば、この前提は、10分の半減期を有するr7−タキソール結合体が、上皮においてタキソールを放出し、その結果、30分の時点で循環において検出され得ることを予測した。対照的に、タキソールは、より安定なr7結合体(t1/2=5時間)から放出されるべきではなく、そして実験の間に採取した血液サンプル中で検出されるべきではなかった。
【0298】
この前提は、血液中のタキソールの出現によって支持された(図37)。タキソールの検出可能レベルは、結腸中に注射された場合、2.5時間まで血液中に現われず、4時間および5.5時間に引き続いて波を有した。時間の関数として、血液中のタキソールの変動レベルは、そのパターンが、物質のいくらかを隔離し、かつ徐放性ビヒクルとして作用するCremophor能力を合理化するという公開された研究に一致する。対照的に、不安定で、水溶性であるタキソールのr7結合体(13)を投与した場合、200ng/mlより多いタキソールを、最も早い時点(30分)で観察し、1時間まで増加し続け、この時点でのレベルが5時間までゆっくり減少する。予測されるように、より安定で水溶性のタキソールr7結合体(14)の注射は、5時間内ではタキソールの有意な血中レベルを生じなかった。これらのデータは、本発明のトランスポーターが結腸上皮に侵入するが横切らないという仮説を支持する。これらは、水溶性を劇的に改善することによって、そして結腸における結合体の取り込み速度を増加することによって、薬物の経口バイオアベイラビリティーを改善し得る。血流への薬物の最終的な送達は、生体膜を介する薬物の固有の拡散能力、および結合体から放出の速度に基づくようである。
【0299】
(考察)
本明細書中に記載される実験における有意な発見は、ペプチドが十二指腸の上皮に入ることができなことである。これは、細胞型または組織に入らないトランスポーターの第1の例を表す。胃の幽門からさらに伸長した組織の取り込みの増加パターンは、興味深く、インヒビターの勾配(例えば、負に荷電した粘液)におそらく起因するようである。小腸の上部領域を染色するような不全は、結腸における激しい染色とは対照的に明確(in stark)であり、本発明のトランスポーターが、結腸への治療剤の選択的送達のために使用され得るという推測を導く。結腸の全ての内腔表面が強度の蛍光を発するのみでなく、染色パターンはまた、ビオチン化化合物が、複数の細胞層に浸透し得、そして組織の血管化領域に到達し得ることを示し、このことは、トランスポーターが、血流中への輸送を増強するはずであることを示唆する。
【0300】
しかし、結腸細胞株(Caco−2)の単層を用いる別の研究は、代替の機構を示唆する。Caco−2系において、トランスポーターは単層に迅速に侵入したが、単層を横断しなかった。細胞および組織への高速での輸送(しかし、細胞および組織からの制限された流出速度)を示すトランスポーターの例がいくつか存在する。これは、現在までに試験された全ての懸濁細胞株についての場合であり、最も研究された細胞株は、ヒトT細胞株(Jurket)である。短いオリゴマーのD−アルギニンは、これらの細胞に迅速に侵入し、そして非常に低い退出速度を示し、37℃で1時間のインキュベーション後、蛍光シグナルの5%未満を失う。この現象のなおより関連性のある例としては、放出可能なCsA r7結合体での複数の局所処置を受けるマウスの血流中で測定された、低レベルのCsAである。結腸についての場合のように、CsA結合体のビオチン化アナログで処理された皮膚の切片における染色パターンによって、薬物が真皮の高度に血管化した領域へ浸透したことを確立した。さらに、モノクローナル抗体を用いての染色は、高度に蛍光を発した真皮において顕著な細胞型の1つの毛細管の内皮細胞であったことを確立した。にもかかわらず、これらの動物の血液中のCsAの検出可能なレベルは、1日に2回適用される4%の軟膏での処理の10日後でもなお観察されなかった。この観察は、裏づけに乏しい(anecdotal)、この報告においてより研究された結果と一致する。
【0301】
皮膚、肺および心血管系中の複数の細胞層に侵入し、そして続いて抜け出すペプチドの能力は、これらのペプチドがリンパ球またはCaco−2細胞から抜け出す能力がないことに対して顕著に対称的である。特定の理論または機構に本発明を限定することを意図することなく、1つの差異は、タイトジャンクション、および組織構造において固有の他の膜構造によって結合されている細胞であって、これらの構造を介してペプチドが迅速に浸透する細胞であるが、懸濁液中、およびおそらく血流に接する側の内皮細胞の膜のの個々の細胞は、これらの特徴を欠損する。タイトジャンクションまたはギャップジャンクションのような構造が、トランスポーターの退出を可能にするために周囲の脂質を改変する場合、次いで、トランスポーターは、組織(皮膚または結腸)を通して迅速に拡散されるはずであるが、血流中へは輸送されない。この仮説との一致は、循環におけるタキソールおよびCsAの出現速度の変化を説明するため、この報告において構築されたモデルである。このモデルにて、アルギニンの短いオリゴマーは、結腸の円柱上皮または頬の扁平上皮への取り込みを非常に促進したが、血流中に薬物を輸送しなかった。観察された血中レベルは、結合体の加水分解後の、循環への上皮からの薬物の拡散、または能動輸送の結果であった。
【0302】
(実施例18)
(トランスポーター結合体の頬送達)
タキソールおよびCsAの頬送達は、濃縮した溶液(250λの5mg/kg)を(その側面に)横たわる麻酔されたラットの口腔へ添加する工程を包含する。血液サンプルを、0時間(薬物注射前)および実験の継続時間の間、30分毎に尾静脈から採取し、経験的に測定した。血液を100λの0.5%EDTAを含むガラス管に移すことによって、凝固を阻害し、そして血液を凍結した。
【0303】
口腔の扁平上皮層に侵入するトランスポーターの能力を、試験した。マウスを麻酔し、その頭を切り取り(tip)、その結果、ビオチン化r9溶液を投与し得た。この動物を、この位置で15分間維持し、その時点で屠殺し、そして舌および頬を解剖し、凍結し、切片化し、ストレプトアビジン−フルオレセインで染色し、そしてプロピジウムヨウ素で対比染色した。頬および舌の両方おいて、ビオチン化ペプチドは、複数の内皮層に迅速にかつ効率的に浸透し、そして組織の内部層へと深く浸透し皮膚の表皮および真皮の両方の染色を連想させる。
【0304】
第2の実施例において、ラットを麻酔し、そしてCremophor EL:エタノール1:1中のタキソールおよびCsA(5mg/kg)の両方の溶液、またはPBS中で等モルの、これらの薬物に対応するr7結合体を、実験の継続時間の間、動物の頬袋中で単純にインキュベートした。LC MSを使用して、タキソールおよび早放性r7結合体のみの血中レベルを測定した。口腔に投与した場合に血液中にタキソールが出現する量および動力学の両方(図38)は、結腸中に注射した場合と異なった。タキソール結合体の頬投与は、ほとんど効果がないようであり、結腸内注射と比較して8分の1未満の量のタキソールが循環中に観察さた。別の差異は、改変されていない薬物の出現速度である。口腔に投与される場合、タキソールは最初の時点よって血流中までに出現するが、結腸注射において、検出可能な量のタキソールは、数時間にわたって出現しない。頬投与での循環中の、タキソールの出現におけるタキソールとr7−タキソール結合体との間に差異は存在しないが、結合体が使用される場合、約2倍ほど多い物質が、循環に到達する。
【0305】
(実施例19)
この実施例は、pH感受性連結基を用いる、輸送部分にへのシイクロスポリンの結合体化を説明する(図6Aおよび図9Bを参照のこと)。
【0306】
この実施例において、シクロスポリンを、無水クロロ酢酸を用いてそのα−クロロ酢酸エステルに変換し、6iを得る(図6を参照のこと)。次いで、このエステル6iをベンジルアミンで処理し、6iiを得る。アミンとBoc−保護無水イミノニ酢酸との反応は、酸6iiiを提供し、次いで、これを、N−ヒドロキシスクシンイミドで活性化エステル(6iv)に変換する。L−アルギニン7量体との6ivのカップリングは、BOC−保護結合体6vを提供し、これを、確立された方法に従ってBOC保護基を除去することによって結合体6viに変換し得る。
【0307】
例えば、グリシン、ε−アミノカプロン酸またはγ−アミノブチル酪酸によって分離されたアルギニン基を有する輸送部分は、この実施例およびオリゴアルギニン輸送基を示す以下の実施例において、アルギニン7量体の代わりに使用され得る。
【0308】
(実施例20)
この実施例は、輸送部分へのアシクロビルの結合体化を説明する。
【0309】
a.r7CONH2へのアシクロビルの結合体化
この実施例は、連結基を介するr7CONH2へのアシクロビルの結合体化を説明する:
【0310】
【化21】
【0311】
【化22】
【0312】
【化23】
【0313】
【化24】
【0314】
【化25】
【0315】
【化26】
【0316】
【化27】
【0317】
i)ビオチン−アミノカプロン酸−r5−Cys(アシクロビル)−CONH2を、白色粉末として得た(36%)。
【0318】
【化28】
ii)ビオチン−アミノカプロン酸−r7−C(アシクロビル)−CONH2−を白色粉末として得た(33%)。
【0319】
【化29】
この実施例は、輸送部分へのヒドロコルチゾンの結合体化を説明する。
【0320】
a.r7CONH2へのヒドロコルチゾンの結合体化
i)ヒドロコルチゾンa−クロロエステルの調製:
【0321】
【化30】
【0322】
【化31】
【0323】
【化32】
【0324】
【化33】
【0325】
【化34】
【0326】
【化35】
【0327】
i)ビオチン−アミノカプロン酸−r5−C(ヒドロコルチゾン)−CONH2−
1.2モル当量よりもむしろ10モル当量のジイソプロピルエチルアミンを使用した。白色粉末を得た(65%)。
【0328】
【化36】
1.2モル当量よりもむしろ10モル当量のジイソプロピルエチルアミンを使用した。白色粉末を得た(36%)。
【0329】
【化37】
この実施例は、輸送部分へのタキソール結合体化を説明する。
【0330】
a.r7−CONH2へのタキソールの結合体化
この実施例は、タキソール結合体の調製への上記で概説した方法論の適用を説明する(図12を参照のこと)。
【0331】
i)タキソールa−クロロ酢酸エステルの調製
【0332】
【化38】
【0333】
ii)タキソール結合体の形成
【0334】
【化39】
【0335】
結合体(ここで、R=H)を、C2’エステルカルボニル上のN−末端窒素の求核攻撃を介して親薬物を放出するように設計した。遊離アミンのみがこの放出を可能にするので、この窒素のプロトン化状態はこの機構に対して重要である。さらに、両方の結合体は、単純なエステル加水分解に感受性である両方の結合体を作製する、共通のα−ヘテロ炭子を置換した酢酸部分を共有する。結合体は、さらなる放出経路を提供する。
【0336】
(実施例23)
この実施例は、輸送部分に活性試薬を連結する2つの方法を説明する。例証は、ポリ−D−Arg誘導体に連結されたレチノイン酸誘導体を提供するが、他の生物学的薬剤と本発明の輸送部分との間の連結に適用され得る。
【0337】
a.アルデヒド官能基を有する生物学的薬剤との間の連結
この実施例は、D−アルギニン(H2N−r9−CO2H・10TFA)のナノマーと全てのtrans−レチナールまたは13−cis−レチナールのいずれかとの間の結合体の調製を説明する。図40は、反応の略図を提供する。図40において見られるように、4Åのモレキュラーシーブの存在下、MeOH中での、いずれか一方のレチナールとH2N−r9−CO2H・10TFAとの、室温にて4時間の縮合は、レチナールアルデヒドとアミノ末端基との間でのSchiffベース型連結の形成を生じる。結合体の精製を、モレキュラーシーブを濾過し、そして減圧下でメタノールを除去することによって達成し得る。
【0338】
b.r7−CONH2へのレチノイン酸の結合体化
この実施例は、連結基を用いてレチノイン酸とr7−CONH2との間の結合体の調製を説明する。
【0339】
【化40】
【0340】
(実施例24)
この実施例は、輸送部分にアシクロビルのような活性な薬剤を連結する方法を説明する。図42を参照のこと。
【0341】
アシクロビル(1当量)を、窒素雰囲気下で、乾燥N,N−ジメチルホルムアミドに溶解した。無水クロロ酢酸(chloroacetic)(1当量)、ピリジン(1当量)およびDMAP(0.25当量)を、撹拌しながら引き続き反応に添加した。この反応を、室温にてさらに4時間撹拌させた。この反応を、減圧下、溶媒の除去によって停止した。この残渣を、塩化メチレン中に溶解し、そして飽和塩化アンモニウム水溶液、続いて飽和重炭酸アンモニウム水溶液およびブラインで洗浄した。有機層を、減圧下で濃縮し、そして残渣をシリカゲルクロマトグラフィーによって精製し、アシクロビルクロロアセチルエステルを得た。
【0342】
得られたクロロアセチルエステルを、窒素雰囲気下で、乾燥N,N−ジメチルホルムアミドに溶解した。この溶液を急速に撹拌しながらHunig’s塩基(1当量)およびAcHN−C−aca−R8−CONH2*8HClに添加した。この反応を、全ての開始物質が消費されたことをTLC分析が同定するまで進めさせる(ca 2時間)。この反応を、減圧下、溶媒の除去によって停止した。この残渣を、RP−HPLCによって精製し、所望のアシクロビル結合体を得た。
【0343】
(実施例25)
この実施例は、輸送部分にアシクロビルのような活性な薬剤を連結する方法を説明する。図43を参照のこと。
【0344】
ジスホスゲン(0.5当量)を、乾燥塩化メチレン中に溶解し、そして−10℃まで冷却した。この溶液に、塩化メチレン中の溶液としてトリエチルアミン(1当量)を添加した。この混合物を、15分間撹拌し、この時点でアシクロビルを塩化メチレン中の溶液としてこの反応物に添加した。この反応物を、室温にてさらに4時間、撹拌させた。この反応物を、飽和塩化アンモニウム水溶液でクエンチし、続いて飽和重炭酸アンモニウム水溶液およびブラインで洗浄した。有機層を、減圧下で濃縮し、そして残渣をシリカゲルを通す迅速な濾過によって精製し、アシクロビルクロロホルメートを得た。るえるブたしたンの接合体オチンがんゆう
クロロホルメートを、窒素雰囲気下で、乾燥塩化メチレンに溶解した。メルカプトエタノール(1当量)を、乾燥塩化メチレン溶液として、この反応物に添加した。この反応物を、窒素雰囲気下で10時間撹拌させた。この溶液を、減圧下で濃縮し、高真空下に24時間配置し、残ったメルカプトエタノールを除去した。この得られたメルカプトエチルカーボネートを、さらに精製することなく使用した。
【0345】
カーボネート(1当量)を、DMF/水に溶解した。急速に撹拌しながら、この溶液に活性化ペプチドNPYs−CR*−CONH2*8HCl(1当量)を添加した。明黄色が即時に発色し、そしてこの反応物を室温にてさらに5時間撹拌させた。この反応物を、RP−HPLCによって直接的に精製し、所望のアシクロビル結合体を得た。
【0346】
(実施例26)
この実施例は、輸送部分にコルチコイドステロイドのような活性な薬剤を連結する方法を説明する。図44を参照のこと。
【0347】
(プレドニゾロンα−クロロエステル−)
乾燥THF中のプレドニゾロン(1.38mmol)、スカンジウムトリフレート(0.83mmol)および無水クロロ酢酸(4.14mmol)の溶液に、ジメチルアミノピリジン(4.14mmol)を添加した。この溶液は、ジメチルアミノピリジンの添加で明黄色に変化した。30分後、この溶媒をエバポレートし、そして粗物質を酢酸エチル(100mL)へと取り上げた。この酢酸エチル層を、1.0N HClおよびブラインで洗浄した。この有機相を回収し、乾燥し(Na2SO4)、そしてエバポレートして生成物を白色固体として得た。
【0348】
H2N−C(プレドニゾロン)−r8−CONH2: ジメチルホルムアミド(1mL)中のプレドニゾロンα−クロロエステル(1当量)、H2N−C−r8−CONH2(1当量)およびジイソプロピルエチルアミン(1.2当量)の溶液を、18時間撹拌した。ジメチルホルムアミドを、エバポレートした。この粗生成物を、逆相HPLC(22mm×250mm C−18カラム、0.1%トリフルオロ酢酸での5〜10%CH3CN/H2O勾配、214nmおよび254nm UV検出)によって精製し、そして凍結乾燥し、9TFA塩を得た。次いで、この物質をイオン交換クロマトグラフィーに供し、プレドロニゾン結合体(9 HCl塩)を黄褐色固体として得た。
【0349】
本明細書中に記載される実施例および実施形態が、例示の目的のみのためであり、そしてその権利において、様々な改変または変化が、当業者に示唆されかつ本出願の精神および権限ならびに添付の特許請求の範囲内に含まれることは、理解される。本明細書中に引用されるすべての出版物、特許および特許出願は、全ての目的のための参考として本明細書中に援用される。
【図面の簡単な説明】
【0350】
【図1】図1は、α−クロロアセチルシクロスポリンA誘導体の調製のための反応スキームを示す。
【図2】図2は、システイン含有ペプチドのα−クロロアセチルシクロスポリンA誘導体とのカップリングのための一般的な手順を示す。
【図3】図3は、シクロスポリンA誘導体のビオチン標識ペプチドへのカップリングのための反応スキームを示す。
【図4】図4は、シクロスポリンA誘導体の標識していないペプチドとのカップリングのための反応スキームを示す。
【図5A】図5A〜Hは、切断可能なリンカーの種々の型を示し、これらのリンカーは、送達増強トランスポーターの生物学的に活性な薬剤または他の目的の分子との連結のために使用され得る。図5Aは、ジスルフィド結合の例を示す。
【図5B】図5Bは、光切断可能な(photocleavable)リンカーを示す。このリンカーは、電磁放射への曝露によって切断される。
【図5C】図5Cは、切断可能なリンカーとして使用される修飾されたリジル残基を示す。
【図5D】図5Dは、送達増強トランスポーターTが、抗癌剤パクリタキセルの2’酸素に連結された、結合体を示す。連結部分は、以下を含む(i)送達増強トランスポーターに付着した窒素原子、(ii)窒素原子に対しパラ位に位置するリン酸モノエステル、および(iii)窒素原子に対しメタ位のカルボキシメチル基(このカルボキシルメチル基は、カルボン酸エステル連結によってパクリタキセルの2’酸素に結合されている)。
【図5E】図5Eは、アミノアルキルカルボン酸による、送達増強トランスポーターの生物学的に活性な薬剤(例えば、パクリタキセル)への架橋であり;リンカーアミノ基は、アミド連結によって送達増強トランスポータに結合され、そして、エステル連結によってパクリタキセル部分に結合される。
【図5F】図5Fおよび図5Gは、パクリタキセルおよび「TAXOTERETM」(R’=H、R’’=BOC)についての構成骨格原子の化学構造および従来の番号付けを示す。
【図5G】図5Gは、タキソイド(taxoid)化合物についての一般的な化学構造および環原子番号付けを示す。
【図6】図6は、L−アルギニンの七量体とシクロスポリンAとの間の化学結合体についての合成スキーム(パネルA)、およびそのpH依存性化学放出(パネルB)を示す。α−クロロエステル(6i)を、置換をするためにヨウ化ナトリウムの存在下でベンジルアミンを用いて処理し、2級アミン(6ii)を得る。アミン(6ii)を、無水物(6)を用いて処理し、そして得られた粗酸(6iii)を、その対応するNHSエステル(6iv)に変換した。次いで、エステル(6iv)を、へプタ−L−アルギニンのアミノ末端とカップリングし、N−Boc保護されたCsA結合体(6v)を得る。最後に、ギ酸を用いるBoc保護基の除去によって、HPLC精製の後、そのオクタトリフルオロ酢酸塩として結合体(6vi)を得た。
【図7】図7は、放出可能なR7 CsAによる接触皮膚炎マウスにおける炎症の阻害を示す。Balb/cマウス(6〜7週)に、アセトンオリーブオイル中の0.7%DNFB(95:5)を100μl腹部に塗布した。3日後、動物の両耳を、アセトン中の0.5% DNFBで再刺激した。再刺激の1時間後、5時間後、および20時間後に、ビヒクルのみ、1% R7ペプチドのみ、1% CsA、1% 放出不可能なR7 CsA、0.01%/0.1%/1.0% 放出可能なR7 CsA、およびフッ化ステロイド陽性コントロール0.1% トリアムシノロンアセトニドのいずれかで、マウスを処置した。耳の炎症を、再刺激の24時間後に、バネを装着したキャリパーを用いて測定した。炎症の減少の百分率を、以下の式:(t−n)/(u−n)(ここで、t=処置した耳の厚さ、n=正常な未処置の耳の厚さ、およびu=いかなる処置もしていない炎症した耳の厚さである)を用いて計算した。各群において、N=20動物である。
【図8】図8は、銅−ジエチレントリアミンペンタ酢酸錯体(Cu−DTPA)の調製のための手順を示す。
【図9】図9は、Cu−DTPAをアミノカプロン酸を介してトランスポーターに連結するための手順を示す。
【図10】図10は、ヒドロコルチゾンの無水クロロ酢酸でのアシル化のための反応を示す。
【図11】図11は、アシル化ヒドロコルチゾンをトランスポーターに連結するための反応を示す。
【図12】図12は、タキソールのC−2’誘導体の調製のための反応を示す。
【図13】図13は、タキソール誘導体をビオチン標識ペプチドにカップリングするための反応の概略を示す。
【図14】図14は、標識していないペプチドをタキソールのC−2’誘導体にカップリングするための反応を示す。
【図15A】図15Aは、他のC−2’タキソール−ペプチド結合体の形成のための反応スキームを示す。
【図15B】図15Bは、他のC−2’タキソール−ペプチド結合体の形成のための反応スキームを示す。
【図15C】図15Cは、他のC−2’タキソール−ペプチド結合体の形成のための反応スキームを示す。
【図16】図16は、薬物または他の生物学的因子がpH−放出可能なリンカーによって送達増強トランスポーターに連結される結合体を合成するための、一般的なストラテジーを示す。
【図17】図17は、タキソール2’−クロロアセチル誘導体を合成するためのプロトコルの概略図を示す。
【図18】図18は、タキソール2’−クロロアセチル誘導体が、アルギニン七量体送達増強トランスポーターに連結されるストラテジーを示す。
【図19】図19は、図18に図示される反応条件を用いて作製され得る、3つのさらなるタキソール−r7結合体を示す。
【図20】図20は、タキソールおよび2つの異なるリンカーを用いる、3日間のMTT細胞傷害性アッセイの結果を示す。
【図21】図21は、以下のものの短縮型アナログのFACS細胞取り込みアッセイを示す:Tat49〜57(Fl−ahx−RKKRRQRRR)、Tat49〜56(Fl−ahx−RKKRRQRR)、Tat49〜55(Fl−ahx−RKKRRQR)、Tat50〜57(Fl−ahx−KKRRQRRR)、およびTat51〜57(Fl−ahx−KRRQRRR)。Jurkat細胞を、種々の濃度(12.5μMを示す)のペプチドとともに23℃で15分間インキュベートした。
【図22】図22は、以下のもののアラニン置換アナログのFACS細胞取り込みアッセイを示す:Tat49〜57:A−49(Fl−ahx−AKKRRQRRR)、A−50(Fl−ahx−RAKRRQRRR)、A−51(Fl−ahx−RKARRQRRR)、A−52(Fl−ahx−RKKARQRRR)、A−53(Fl−ahx−RKKRAQRRR)、A−54(Fl−ahx−RKKRRARRR)、A−55(Fl−ahx−RKKRRQARR)、A−56(Fl−ahx−RKKRRQRAR)、およびA−57(Fl−ahx−RKKRRQRRA)。Jurkat細胞を、種々の濃度(12.5μMを示す)のペプチドとともに23℃で12分間インキュベートした。
【図23】図23は、Tat49〜57のd−異性体およびretro−異性体:d−Tat49−57(Fl−ahx−rkkrrqrrr)、Tat57−49(Fl−ahx−RRRQRRKKR)、およびd−Tat57−49(Fl−ahx−rrrqrrkkr)のFACS細胞取り込みアッセイを示す。Jurkat細胞を、種々の濃度(12.5μMを示す)のペプチドとともに、23℃で15分間インキュベートした。
【図24】図24は、一連のアルギニンオリゴマーおよびTat49−57:R5(Fl−ahx−RRRRR)、R6(Fl−ahx−RRRRRR)、R7(Fl−ahx−RRRRRRR)、R8(Fl−ahx−RRRRRRRR)、R9(Fl−ahx−RRRRRRRRR)、r5(Fl−ahx−rrrrr)、r6(Fl−ahx−rrrrrr)、r7(Fl−ahx−rrrrrrr)、r8(Fl−ahx−rrrrrrrr)、r9(Fl−ahx−rrrrrrrrr)のFACS細胞取り込みを示す。Jurkat細胞を、種々の濃度(12.5μMを示す)のペプチドとともに23℃で4分間インキュベートした。
【図25】図25は、グアニジン置換ペプトイドの調製を示す。
【図26】図26は、ポリグアニジンペプトイドおよびd−アルギニンオリゴマーのFACS細胞取り込みを示す。Jurkat細胞を、種々の濃度(12.5μMを示す)のペプトイドおよびペプチドとともに23℃で4分間インキュベートした。
【図27】図27は、d−アルギニンオリゴマーおよびポリグアニジンペプトイドのFACS細胞取り込みを示す。Jurkat細胞を、種々の濃度(12.5μMを示す)の蛍光標識したペプトイドおよびペプチドとともに23℃で4分間インキュベートした。
【図28】図28は、d−アルギニンオリゴマーおよびN−hxgペプトイドのFACS細胞取り込みを示す。Jurkat細胞を、種々の濃度(6.3μMを示す)の蛍光標識したペプトイドおよびペプチドとともに23℃で4分間インキュベートした。
【図29】図29は、d−アルギニンオリゴマーおよびN−chgペプトイドのFACS細胞取り込みを示す。Jurkat細胞を、種々の濃度(12.5μMを示す)の蛍光標識したペプトイドおよびペプチドとともに23℃で4分間インキュベートした。
【図30】図30は、送達増強トランスポーターをトリアゾール環構造を含む薬物に結合するための一般的なストラテジーを示す。
【図31A】図31Aおよび図31Bは、FK506が送達増強トランスポーターに結合された結合体を作製するための合成スキームを示す。
【図31B】図31Aおよび図31Bは、FK506が送達増強トランスポーターに結合された結合体を作製するための合成スキームを示す。
【図32】図32は、Caco−2細胞に効果的に導入された、アルギニン(リジンではない)の短いオリゴマーを示す。Caco−2細胞を、示される各ペプチドと、異なる濃度でインキュベートし、洗浄し、そしてフローサイトメトリーによって分析した。取り込みは、アジ化ナトリウムを含むプレインキュベーションによって阻害され得、取り込みが、エネルギー依存性であったことを実証する。
【図33】図33は、蛍光性タキソールまたはD−アルギニンの七量体(r7)もしくはデカマー(r10)のいずれかとの放出不可能な蛍光性タキソール結合体に曝露されたCaco−2細胞の測定された蛍光を示す。
【図34】図34は、本報告において記載される実験の持続時間の間での100オームcm2より大きい安定な経上皮電気抵抗の測定により、Caco−2単層の完全性を実証する。
【図35】図35は、Caco−2細胞単層を介する輸送1時間後の拡散装置の基底外側チャンバーにおけるFl aca r5、Fl aca r9、ルシファーイエロー、またはヒドロコルチゾンのいずれかの蓄積を示す。
【図36】図36は、結腸内注射後、30分間隔のLC MS MSにより測定されたラットにおけるCsAの血中レベルを示す。CsAを、Cremophor El:エタノール 1:1の中において投与し、一方、CsA結合体を、PBSの中において投与した。CsA結合体(CGC1072)のPBS中、pH7.4、37℃での半減期は1.5時間であった。
【図37】図37は、結腸内注射後、30分間隔のLC MS MSにより測定されたラットにおけるタキソールの血中レベルを示す。タキソールを、Cremophor EL:エタノール 1:1の中において投与し、一方、2つのタキソール結合体を、PBSの中において投与した。遅延して放出する結合体(14)のPBS中、pH7.4、37℃での半減期は、5時間であったが、速放性結合体(13)の半減期は、10分であった。実施例18を参照のこと。
【図38】図38は、口腔送達後、30分間隔のLC MS MSにより測定されたラットにおけるタキソールの血中レベルを示す。タキソールを、Cremophor El:エタノール 1:1の中において投与し、一方、タキソール結合体を、PBSの中において投与した。結合体(13)のPBS中、pH7.4、37℃での半減期は、10分であった。
【図39】図39は、N末端システイン基を介すr7−アミドに対するアシクロビルの結合体を描く。ビオチン含有トランスポーターとの結合体もまた、示す。
【図40】図40は、レチナールとr9との間に形成される結合体を示す(スペーシングアミノ酸なしで示す)。
【図41】図41は、レチノール酸−r9結合体の調製における切断可能なリンカーの使用を描く。
【図42】図42は、アシクロビルのような活性薬剤のトランスポーター部分への連結方法を描く。
【図43】図43は、アシクロビルのような活性薬剤のトランスポーター部分への連結方法を描く。
【図44】図44は、コルチコイドステロイドのような活性な薬剤のトランスポーター部分への連結方法を描く。
Claims (31)
- 化合物を動物の胃腸上皮に標的化する方法であって、該方法は、該化合物および送達増強トランスポーターを含む結合体を、該胃腸上皮に投与する工程を包含し、ここで:
i.該化合物が、リンカーを介して該送達増強トランスポーターに結合され;そして、
ii.該送達増強トランスポーターが、50未満のサブユニットを含み、そして、少なくとも5つのグアニジノ部分またはアミジノ部分を含み、これによって、該送達増強トランスポーターの非存在下での該化合物の輸送と比較して、該胃腸上皮への該結合体の送達が増大される、
方法。 - 請求項1に記載の方法であって、ここで、前記結合体の前記胃腸上皮への送達が、前記送達増強トランスポーターの非存在下における前記化合物の送達と比較して、少なくとも2倍増大される、方法。
- 請求項1に記載の方法であって、ここで、前記結合体の前記胃腸上皮への送達が、前記送達増強トランスポーターの非存在下における前記化合物の送達と比較して、少なくとも10倍増大される、方法。
- 前記リンカーが、放出可能なリンカーである、請求項1に記載の方法。
- 前記サブユニットがアミノ酸である、請求項1に記載の方法。
- 請求項1に記載の方法であって、ここで、前記結合体が、以下:
R1は、前記化合物を含み;
Xは、生物学的に活性な化合物上の官能基と結合部分上の末端官能基との間で形成された結合であり;
Yは、輸送部分上の官能基および結合部分上の官能基から形成された結合であり;
Aは、NまたはCHであり;
R2は、水素、アルキル、アリール、アシルまたはアリルであり;
R3は、前記送達増強トランスポーターを含み、
R4は、S、O,NR6またはCR7R8であり;
R5は、H、OH、SHまたはNHR6であり;
R6は、水素、アルキル、アリール、アシルまたはアリルであり;
kおよびmは1および2から各々独立して選択され;ならびに
nは1〜10である、
方法。 - 請求項6に記載の方法であって、ここで、Xは、−C(O)O−、−C(O)NH−、−OC(O)NH−、−S−S−、−C(S)O−、−C(S)NH−、−NHC(O)NH−、−SO2NH−、−SONH−、ホスフェート、ホスホンネート、ホスフィンネート、およびCR7R8からなる群より選択され、ここで、R7およびR8は、各々、Hおよびアルキルからなる群より独立して選択される、方法。
- 前記結合体が構造3を含み、YはNであり、そして、R2はメチル、エチル、プロピル、ブチル、アリル、ベンジルまたはフェニルである、請求項6に記載の方法。
- R2はベンジルであり;k、mおよびnが各々1であり、そして、Xが−OC(O)−である、請求項6に記載の方法。
- 前記結合体は構造4を含み;R4はSであり;R5はNHR6であり;そして、R6は水素、メチル、アリル、ブチルまたはフェニルである、請求項6に記載の方法。
- 前記結合体は構造4を含み;R5はNHR6であり;R6は水素、メチル、アリル、ブチルまたはフェニルであり;そして、kおよびmは各々1である、請求項6に記載の方法。
- 請求項1に記載の方法であって、ここで、前記結合体が、以下:
R1は、前記化合物を含み;
Xは、生物学的に活性な化合物上の官能基と結合部分上の末端官能基との間で形成された結合であり;
Yは、輸送部分上の官能基および結合部分上の官能基から形成された結合であり;
Arは、オルト位またはパラ位に配置された結合化遊離基を有するアリール基であり、アリール基は置換されてもよいし、置換されなくてもよい;
R3は、前記送達増強トランスポーターを含み、
R4は、S、O,NR6またはCR7R8であり;
R5は、H、OH、SHまたはNHR6であり;
R6は、水素、アルキル、アリール、アリールアルキル、アシルまたはアリルであり;
R7およびR8は、水素またはアルキルから独立して選択され;そして
kおよびmは1および2から各々独立して選択される、
方法。 - 請求項12に記載の方法であって、ここで、Xは、−C(O)O−、−C(O)NH−、−OC(O)NH−、−S−S−、−C(S)O−、−C(S)NH−、−NHC(O)NH−、−SO2NH−、−SONH−、ホスフェート、ホスホンネート、ホスフィンネート、およびCR7R8からなる群より選択され、ここで、R7およびR8は、各々、Hおよびアルキルからなる群より独立して選択される、方法。
- R4はSであり;R5はNHR6であり;そして、R6は水素、メチル、アリル、ブチルまたはフェニルである、請求項12に記載の方法。
- 前記結合体が、少なくとも2つの送達増強トランスポーターを含む、請求項1記載の方法。
- 前記結合体が、頬に投与される、請求項1に記載の方法。
- 前記結合体が、坐薬として投与される、請求項1に記載の方法。
- 前記送達増強トランスポーターが、非ペプチド骨格を含む、請求項1に記載の方法。
- 請求項1に記載の方法であって、ここで、前記送達増強トランスポーターは、該送達増強トランスポーター分子が天然に存在するタンパク質中で結合されるアミノ酸配列に結合されない、方法。
- 前記送達増強トランスポーターが、2〜25のグアニジノ部分またはアミジノ部分を含む、請求項1に記載の方法。
- 前記送達増強トランスポーターが、7と15の間のグアニジノ部分を含む、請求項20に記載の方法。
- 前記送達増強トランスポーターが、少なくとも6つの連続したグアニジノ部分および/またはアミジノ部分を含む、請求項20に記載の方法。
- 請求項1に記載の方法であって、前記送達増強トランスポーターが、本質的に5〜50のアミノ酸からなり、これらのアミノ酸の少なくとも50%がアルギニンまたはそのアナログである、方法。
- 前記送達増強トランスポーターが、5〜25のアルギニン残基またはそのアナログを含む、請求項23に記載の方法。
- 少なくとも1つのアルギニンが、D−アルギニンである、請求項24に記載の方法。
- 前記アルギニンの全てがD−アルギニンである、請求項25に記載の方法。
- 前記送達増強トランスポーターを含む前記アミノ酸の少なくとも70%が、アルギニンまたはそのアナログである、請求項23に記載の方法。
- 前記送達増強トランスポーターが、7つの連続したD−アルギニンからなる、請求項23に記載の方法。
- 請求項1に記載の方法であって、前記化合物は、炎症性腸疾患、結腸癌、潰瘍性大腸炎、胃腸潰瘍、便秘、ならびに、塩および水吸収の失調からなる群より選択される疾患のための治療薬である、方法。
- 請求項1に記載の方法であって、前記化合物は、免疫抑制剤、コルチコステロイド、緩下薬、抗生物質または抗腫瘍剤からなる群より選択される、方法。
- 前記化合物が、回腸(iliem)および/または結腸に標的化される、請求項1に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/792,480 US6669951B2 (en) | 1999-08-24 | 2001-02-23 | Compositions and methods for enhancing drug delivery across and into epithelial tissues |
| PCT/US2002/005829 WO2002069930A1 (en) | 2001-02-23 | 2002-02-25 | Compositions and methods for enhancing drug delivery across and into epithelial tissues |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004530657A true JP2004530657A (ja) | 2004-10-07 |
| JP2004530657A5 JP2004530657A5 (ja) | 2005-12-22 |
Family
ID=25157022
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002567285A Pending JP2004533414A (ja) | 2001-02-23 | 2002-02-25 | 眼組織を横切る薬物送達および眼組織内への薬物送達を増強するための組成物および方法 |
| JP2002569108A Pending JP2004530657A (ja) | 2001-02-23 | 2002-02-25 | 上皮組織にわたるおよび上皮組織への薬物送達を増強するための組成物および方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002567285A Pending JP2004533414A (ja) | 2001-02-23 | 2002-02-25 | 眼組織を横切る薬物送達および眼組織内への薬物送達を増強するための組成物および方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (5) | US6669951B2 (ja) |
| EP (2) | EP1372626A4 (ja) |
| JP (2) | JP2004533414A (ja) |
| CA (2) | CA2438784A1 (ja) |
| MX (2) | MXPA03007590A (ja) |
| WO (2) | WO2002069930A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010514781A (ja) * | 2006-12-29 | 2010-05-06 | ルバンス セラピュティックス インク. | Hiv−tatに由来するポリペプチド断片を用いて安定化されるボツリヌス毒素を局所適用及び経皮送達する組成物及び方法 |
| JP2014522816A (ja) * | 2011-06-22 | 2014-09-08 | ビョーメ バイオサイエンシズ ピーブイティー.リミテッド | コンジュゲートベースの抗真菌性および抗菌性プロドラッグ |
| WO2019198316A1 (ja) * | 2018-04-09 | 2019-10-17 | 国立研究開発法人産業技術総合研究所 | 生理活性を持つ中分子の薬物動態を改善する方法、薬物動態の改善を利用した中分子ライブラリの製造法 |
| JP2020519622A (ja) * | 2017-05-11 | 2020-07-02 | ケアジェン カンパニー, リミテッドCaregen Co., Ltd. | イソトレチノインとペプチドの結合体 |
Families Citing this family (163)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7135191B2 (en) * | 1997-09-04 | 2006-11-14 | Zsolt Istvan Hertelendy | Urogenital or anorectal transmucosal vaccine delivery system |
| KR100606857B1 (ko) * | 1998-07-08 | 2006-08-01 | 키린-암젠 인코포레이티드 | 고분자 의약품 함유 분말상 경점막 투여 제제 |
| US20060025387A1 (en) * | 1998-12-23 | 2006-02-02 | Cytoscan Sciences Llc | Compositions and methods for the treatment of disorders of the central and peripheral nervous systems |
| US8722668B2 (en) | 1998-12-23 | 2014-05-13 | Daryl W. Hochman | Methods and compositions for the treatment of neuropathic pain and neuropsychiatric disorders |
| US7214711B2 (en) * | 1998-12-23 | 2007-05-08 | Neurotherapeutics Pharma Llc | Method of treating migraine headache without aura |
| US8008283B2 (en) * | 1998-12-23 | 2011-08-30 | Neurotherapeutics Pharma, Inc. | Methods and compositions for the treatment of neuropsychiatric disorders |
| US7229961B2 (en) * | 1999-08-24 | 2007-06-12 | Cellgate, Inc. | Compositions and methods for enhancing drug delivery across and into ocular tissues |
| US6669951B2 (en) * | 1999-08-24 | 2003-12-30 | Cellgate, Inc. | Compositions and methods for enhancing drug delivery across and into epithelial tissues |
| US8183339B1 (en) | 1999-10-12 | 2012-05-22 | Xigen S.A. | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US20040082509A1 (en) | 1999-10-12 | 2004-04-29 | Christophe Bonny | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| EP1301213B1 (en) * | 2000-07-21 | 2017-01-18 | ReVance Therapeutics, Inc. | Multi-component biological transport systems |
| US20040220100A1 (en) * | 2000-07-21 | 2004-11-04 | Essentia Biosystems, Inc. | Multi-component biological transport systems |
| ATE444756T1 (de) * | 2000-11-14 | 2009-10-15 | Vital Health Sciences Pty Ltd | Zusammensetzungen umfassend komplexe von tocopherolphosphatderivaten |
| AUPR549901A0 (en) * | 2001-06-06 | 2001-07-12 | Vital Health Sciences Pty Ltd | Topical formulation containing tocopheryl phosphates |
| MXPA03007358A (es) * | 2001-02-16 | 2004-12-13 | Cellgate Inc | Transportadores que contienen porciones de arginina separadas. |
| JP5052736B2 (ja) * | 2001-05-30 | 2012-10-17 | 中外製薬株式会社 | タンパク質製剤 |
| WO2003011303A1 (en) | 2001-07-27 | 2003-02-13 | Vital Health Sciences Pty Ltd | Dermal therapy using phosphate derivatives of electron transfer agents |
| AU2002359679B2 (en) * | 2001-12-11 | 2009-01-08 | Cellgate, Inc. | Guanidinium transport reagents and conjugates |
| CA2466536C (en) * | 2001-12-13 | 2012-02-07 | Vital Health Sciences Pty Ltd | Transdermal transport of compounds |
| JP2005514438A (ja) | 2001-12-21 | 2005-05-19 | ソーン, デイビッド エス. | 薬剤の可溶化、安定化、及び運搬のためのオリゴマー及びポリマーの使用 |
| FR2836474B1 (fr) * | 2002-02-22 | 2004-12-24 | Synt Em | Composes, compositions et methode pour le transport des molecules de cyclosporine a travers la barriere hemato-encephalique |
| WO2003090604A2 (en) * | 2002-04-24 | 2003-11-06 | University Of Florida | Method of endovascular brain mapping |
| AU2002950713A0 (en) | 2002-08-09 | 2002-09-12 | Vital Health Sciences Pty Ltd | Carrier |
| US20040157810A1 (en) * | 2002-08-23 | 2004-08-12 | Teicher Martin H. | Corticosteroid conjugates and uses thereof |
| US7458982B2 (en) * | 2002-10-04 | 2008-12-02 | Photokinetix, Inc. | Photokinetic delivery of biologically active substances using pulsed incoherent light |
| US6969514B2 (en) * | 2003-02-05 | 2005-11-29 | Soll David B | Method for treating elevated intraocular pressure, including glaucoma |
| EP1603512A2 (en) * | 2003-03-17 | 2005-12-14 | Albany Molecular Research, Inc. | Novel cyclosporins |
| US7332164B2 (en) * | 2003-03-21 | 2008-02-19 | Enzon Pharmaceuticals, Inc. | Heterobifunctional polymeric bioconjugates |
| CA2523672C (en) * | 2003-04-29 | 2012-07-17 | Avi Biopharma, Inc. | Compositions for enhancing transport of molecules into cells |
| JP4879020B2 (ja) | 2003-05-01 | 2012-02-15 | コーネル リサーチ ファウンデイション インコーポレイテッド | 細胞に分子を送達する方法及び担体複合体 |
| AU2004263124B2 (en) * | 2003-08-07 | 2009-01-15 | Avi Biopharma, Inc. | Sense antiviral compound and method for treating ssRNA viral infection |
| US20050136437A1 (en) * | 2003-08-25 | 2005-06-23 | Nastech Pharmaceutical Company Inc. | Nanoparticles for delivery of nucleic acids and stable double-stranded RNA |
| US20050148534A1 (en) * | 2003-09-22 | 2005-07-07 | Castellino Angelo J. | Small molecule compositions and methods for increasing drug efficiency using compositions thereof |
| US20050222068A1 (en) * | 2003-10-23 | 2005-10-06 | Mourich Dan V | Method and antisense composition for selective inhibition of HIV infection in hematopoietic cells |
| US20050203041A1 (en) * | 2003-09-23 | 2005-09-15 | Mourich Dan V. | Antisense compound and method for selectively killing activated T cells |
| US20050074506A1 (en) * | 2003-10-02 | 2005-04-07 | Brainsgate Ltd. | Targeted release of nitric oxide in the CNS circulation for modulating the BBB and treating disorders |
| DE10355559A1 (de) * | 2003-11-21 | 2005-06-23 | Orthogen Ag | Transskin |
| US20060182692A1 (en) | 2003-12-16 | 2006-08-17 | Fishburn C S | Chemically modified small molecules |
| AU2005218539A1 (en) * | 2004-03-01 | 2005-09-15 | Lumen Therapeutics, Llc | Compositions and methods for treating diseases |
| US9211248B2 (en) | 2004-03-03 | 2015-12-15 | Revance Therapeutics, Inc. | Compositions and methods for topical application and transdermal delivery of botulinum toxins |
| DK1720551T3 (da) * | 2004-03-03 | 2011-05-16 | Vital Health Sciences Pty Ltd | Alkaloidformuleringer |
| CN1946431B (zh) | 2004-03-03 | 2011-12-07 | 雷文斯治疗公司 | 用于肉毒毒素的局部施用和透皮递送的组合物和方法 |
| CA2558676C (en) | 2004-03-03 | 2019-04-16 | Essentia Biosystems, Inc. | Compositions and methods for topical diagnostic and therapeutic transport |
| CA2604243C (en) | 2004-04-14 | 2015-11-24 | Avirid, Inc. | Compositions with modified nucleases targeted to viral nucleic acids and methods of use for prevention and treatment of viral diseases |
| US20050288246A1 (en) | 2004-05-24 | 2005-12-29 | Iversen Patrick L | Peptide conjugated, inosine-substituted antisense oligomer compound and method |
| SI1768657T1 (sl) | 2004-06-23 | 2009-02-28 | Sirion Terapeutics Inc | Postopki in sestavki za zdravljenje oftalmičnih obolenj z derivati retinila |
| CA2575587C (en) * | 2004-08-03 | 2014-06-17 | Vital Health Sciences Pty Ltd | Carrier for enteral administration |
| US8129352B2 (en) | 2004-09-16 | 2012-03-06 | Avi Biopharma, Inc. | Antisense antiviral compound and method for treating ssRNA viral infection |
| US7378391B2 (en) * | 2004-09-29 | 2008-05-27 | Amr Technology, Inc. | Cyclosporin alkyne analogues and their pharmaceutical uses |
| WO2006039164A2 (en) * | 2004-09-29 | 2006-04-13 | Amr Technology, Inc. | Novel cyclosporin analogues and their pharmaceutical uses |
| WO2006041631A2 (en) * | 2004-10-06 | 2006-04-20 | Amr Technology, Inc. | Novel cyclosporin alkynes and their utility as pharmaceutical agents |
| US7442682B2 (en) * | 2004-10-19 | 2008-10-28 | Nitto Denko Corporation | Transepithelial delivery of peptides with incretin hormone activities |
| US8357664B2 (en) | 2004-10-26 | 2013-01-22 | Avi Biopharma, Inc. | Antisense antiviral compound and method for treating influenza viral infection |
| US7524829B2 (en) * | 2004-11-01 | 2009-04-28 | Avi Biopharma, Inc. | Antisense antiviral compounds and methods for treating a filovirus infection |
| JP2008519054A (ja) | 2004-11-04 | 2008-06-05 | シリオン セラピューティクス, インコーポレイテッド | レチノール−レチノール結合タンパク質(rbp)−トランスチレチン(ttr)複合体の形成のモジュレーター |
| EP1656951A1 (en) * | 2004-11-12 | 2006-05-17 | Xigen S.A. | Conjugates with enhanced cell uptake activity |
| US7603097B2 (en) | 2004-12-30 | 2009-10-13 | Valeo Radar Systems, Inc. | Vehicle radar sensor assembly |
| AU2006213686A1 (en) | 2005-02-09 | 2006-08-17 | Avi Bio Pharma, Inc. | Antisense composition and method for treating muscle atrophy |
| US20090239827A1 (en) * | 2005-03-03 | 2009-09-24 | Esra Ogru | Compounds having lipid lowering properties |
| SG160357A1 (en) | 2005-03-03 | 2010-04-29 | Revance Therapeutics Inc | Compositions and methods for topical application and transdermal delivery of botulinum toxins |
| CN101160318A (zh) | 2005-03-03 | 2008-04-09 | 雷文斯治疗公司 | 用于局部施用和经皮肤递送寡肽的组合物和方法 |
| US20060240032A1 (en) * | 2005-03-31 | 2006-10-26 | Hinrichs David J | Immunomodulating compositions and methods for use in the treatment of human autoimmune diseases |
| NZ565049A (en) | 2005-06-17 | 2012-02-24 | Vital Health Sciences Pty Ltd | A carrier comprising one or more DI and/or mono-(electron transfer agent) phosphate derivatives or complexes thereof |
| US8067571B2 (en) | 2005-07-13 | 2011-11-29 | Avi Biopharma, Inc. | Antibacterial antisense oligonucleotide and method |
| US8329668B2 (en) * | 2005-09-08 | 2012-12-11 | Avi Biopharma, Inc. | Antisense antiviral compound and method for treating picornavirus infection |
| US8080517B2 (en) | 2005-09-12 | 2011-12-20 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| WO2007031098A1 (en) | 2005-09-12 | 2007-03-22 | Xigen S.A. | Cell-permeable peptide inhibitors of the jnk signal transduction pathway |
| EP1924292A2 (en) * | 2005-09-16 | 2008-05-28 | Allergan, Inc. | Compositions and methods for the intraocular transport of therapeutic agents |
| WO2007038171A2 (en) | 2005-09-23 | 2007-04-05 | Nitto Denko Corporation | Peptide nucleic acid based guanidinium compounds |
| US7745392B2 (en) | 2005-09-23 | 2010-06-29 | Nitto Denko Corporation | Multi-valent guanidinium compounds for enhancing molecular translocation across cellular membranes and epithelial tissues |
| WO2007038172A2 (en) | 2005-09-23 | 2007-04-05 | Nitto Denko Corporation | Guanidinium carriers |
| CA2625918A1 (en) * | 2005-10-17 | 2007-04-26 | Neurotherapeutics Pharma, Inc. | Diuretic-like compound analogs useful for regulation of central nervous system disorders |
| US8501704B2 (en) | 2005-11-08 | 2013-08-06 | Sarepta Therapeutics, Inc. | Immunosuppression compound and treatment method |
| NZ568216A (en) | 2005-11-17 | 2012-09-28 | Revance Therapeutics Inc | Compositions and methods of topical application and transdermal delivery of botulinum toxins with reduced non-toxin proteins |
| US7740880B2 (en) * | 2006-03-03 | 2010-06-22 | University Of Utah Research Foundation | Polymeric carrier for delivery of small interfering RNA |
| US7696166B2 (en) * | 2006-03-28 | 2010-04-13 | Albany Molecular Research, Inc. | Use of cyclosporin alkyne/alkene analogues for preventing or treating viral-induced disorders |
| US7696165B2 (en) * | 2006-03-28 | 2010-04-13 | Albany Molecular Research, Inc. | Use of cyclosporin alkyne analogues for preventing or treating viral-induced disorders |
| US8785407B2 (en) * | 2006-05-10 | 2014-07-22 | Sarepta Therapeutics, Inc. | Antisense antiviral agent and method for treating ssRNA viral infection |
| WO2008066982A2 (en) * | 2006-09-25 | 2008-06-05 | Cmed Technologies Ltd. | A method of surface plasmon resonance (spr) technology to detect genomic aberrations in patients with chronic lymphocytic leukemia |
| EP2068936B1 (en) | 2006-09-27 | 2016-06-22 | Paolo Botti | Means and methods of enhancing delivery to biological systems |
| WO2008056207A1 (en) | 2006-11-08 | 2008-05-15 | Chongxi Yu | Transdermal delivery systems of peptides and related compounds |
| NZ598159A (en) * | 2006-12-29 | 2013-08-30 | Revance Therapeutics Inc | Transport molecules using reverse sequence hiv-tat polypeptides |
| EP2121929B1 (en) * | 2007-01-26 | 2011-11-23 | Industry-University Cooperation Foundation Hanyang University | Targeted delivery of sirna |
| US20080200441A1 (en) * | 2007-02-14 | 2008-08-21 | University Of Southern California | Estrogen receptor modulators associated pharmaceutical compositions and methods of use |
| EP3443976A1 (en) * | 2007-06-29 | 2019-02-20 | Sarepta Therapeutics, Inc. | Tissue specific peptide conjugates and methods |
| AU2015200600B2 (en) * | 2007-06-29 | 2017-04-20 | Sarepta Therapeutics, Inc. | Tissue specific peptide conjugates and methods |
| US20100016215A1 (en) | 2007-06-29 | 2010-01-21 | Avi Biopharma, Inc. | Compound and method for treating myotonic dystrophy |
| CN101842107B (zh) | 2007-07-26 | 2014-04-23 | 雷文斯治疗公司 | 抗微生物肽,组合物和使用方法 |
| AU2008295509A1 (en) * | 2007-08-29 | 2009-03-12 | Tufts University | Methods of making and using a cell penetrating peptide for enhanced delivery of nucleic acids, proteins, drugs, and adenovirus to tissues and cells, and compositions and kits |
| WO2009099636A1 (en) * | 2008-02-07 | 2009-08-13 | The Board Of Trustees Of The Leland Stanford Junior University | Conjugation of small molecules to octaarginine transporters for overcoming multi-drug resistance |
| EP2123262A1 (en) | 2008-05-20 | 2009-11-25 | Consorzio per il Centro di Biomedica Moleculare Scrl | Polyelectrolyte-encapsulated gold nanoparticles capable of crossing blood-brain barrier |
| WO2009143864A1 (en) | 2008-05-30 | 2009-12-03 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of chronic or non-chronic inflammatory digestive diseases |
| WO2009143865A1 (en) | 2008-05-30 | 2009-12-03 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of various diseases |
| CA2778604C (en) * | 2008-09-23 | 2021-10-05 | Laboratory Skin Care, Inc. | Active agent loaded uniform, rigid, spherical, nanoporous calcium phosphate particles and methods of making and using the same |
| AU2009335740B2 (en) * | 2008-12-17 | 2016-04-21 | Sarepta Therapeutics, Inc. | Antisense compositions and methods for modulating contact hypersensitivity or contact dermatitis |
| WO2010072228A1 (en) | 2008-12-22 | 2010-07-01 | Xigen S.A. | Novel transporter constructs and transporter cargo conjugate molecules |
| EP2427475B1 (en) | 2009-05-08 | 2020-11-04 | Techfields Biochem Co., Ltd. | High penetration prodrug compositions of peptides and peptide-related compounds |
| MX2011012875A (es) * | 2009-06-05 | 2012-02-28 | 13Therapeutics Inc | Peptidos inmunoreguladores y metodos de uso. |
| US20110269665A1 (en) | 2009-06-26 | 2011-11-03 | Avi Biopharma, Inc. | Compound and method for treating myotonic dystrophy |
| AU2010319314C1 (en) | 2009-11-13 | 2016-09-01 | Sarepta Therapeutics, Inc. | Antisense antiviral compound and method for treating influenza viral infection |
| AU2010321784B2 (en) | 2009-11-23 | 2014-04-24 | Research Development Foundation | Recombinant filaggrin polypeptides for cell importation |
| ES2999020T3 (en) | 2010-02-05 | 2025-02-24 | Phosphagenics Ltd | Carrier comprising non-neutralised tocopheryl phosphate |
| EP2552486B1 (en) | 2010-03-30 | 2020-08-12 | Phosphagenics Limited | Transdermal delivery patch |
| US8980253B2 (en) | 2010-04-26 | 2015-03-17 | Atyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of cysteinyl-tRNA synthetase |
| CA2797362C (en) | 2010-04-27 | 2020-12-08 | Atyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of isoleucyl trna synthetases |
| WO2011139853A2 (en) | 2010-04-28 | 2011-11-10 | Atyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of alanyl trna synthetases |
| CA2797393C (en) | 2010-04-29 | 2020-03-10 | Pangu Biopharma Limited | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of valyl trna synthetases |
| CA2797374C (en) | 2010-04-29 | 2021-02-16 | Pangu Biopharma Limited | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of asparaginyl trna synthetases |
| CN103096912A (zh) | 2010-05-03 | 2013-05-08 | Atyr医药公司 | 与苯丙氨酰-α-tRNA合成酶的蛋白片段相关的治疗、诊断和抗体组合物的创新发现 |
| US8961961B2 (en) | 2010-05-03 | 2015-02-24 | a Tyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related protein fragments of arginyl-tRNA synthetases |
| CN103140233B (zh) | 2010-05-03 | 2017-04-05 | Atyr 医药公司 | 与甲硫氨酰‑tRNA合成酶的蛋白片段相关的治疗、诊断和抗体组合物的发现 |
| JP6008844B2 (ja) | 2010-05-04 | 2016-10-19 | エータイアー ファーマ, インコーポレイテッド | p38MULTI−tRNA合成酵素複合体のタンパク質フラグメントに関連した治療用、診断用および抗体組成物の革新的発見 |
| EP2568996B1 (en) | 2010-05-14 | 2017-10-04 | aTyr Pharma, Inc. | Therapeutic, diagnostic, and antibody compositions related to protein fragments of phenylalanyl-beta-trna synthetases |
| CA2800375C (en) | 2010-05-27 | 2021-03-09 | Atyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of glutaminyl-trna synthetases |
| EP2575857B1 (en) | 2010-06-01 | 2018-01-24 | aTyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of lysyl-trna synthetases |
| WO2011160653A1 (en) | 2010-06-21 | 2011-12-29 | Xigen S.A. | Novel jnk inhibitor molecules |
| EP2593125B1 (en) | 2010-07-12 | 2017-11-01 | aTyr Pharma, Inc. | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of glycyl-trna synthetases |
| US8198429B2 (en) | 2010-08-09 | 2012-06-12 | Avi Biopharma, Inc. | Antisense antiviral compounds and methods for treating a filovirus infection |
| AU2011293294B2 (en) | 2010-08-25 | 2016-03-24 | Pangu Biopharma Limited | Innovative discovery of therapeutic, diagnostic, and antibody compositions related to protein fragments of Tyrosyl-tRNA synthetases |
| AU2010362444B2 (en) | 2010-10-14 | 2015-08-06 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of chronic or non-chronic inflammatory eye diseases |
| WO2012050673A1 (en) * | 2010-10-14 | 2012-04-19 | Wisconsin Alumni Research Foundation | Methods for the treatment of x-linked hypophosphatemia and related disorders |
| WO2012122586A1 (en) | 2011-03-15 | 2012-09-20 | Phosphagenics Limited | New composition |
| US8580748B2 (en) | 2011-04-06 | 2013-11-12 | 13Therapeutics, Inc. | Peptides for the treatment of hearing |
| US9161948B2 (en) | 2011-05-05 | 2015-10-20 | Sarepta Therapeutics, Inc. | Peptide oligonucleotide conjugates |
| US20130085139A1 (en) | 2011-10-04 | 2013-04-04 | Royal Holloway And Bedford New College | Oligomers |
| WO2013091670A1 (en) | 2011-12-21 | 2013-06-27 | Xigen S.A. | Novel jnk inhibitor molecules for treatment of various diseases |
| CN116898848A (zh) * | 2012-01-18 | 2023-10-20 | 苏州泰飞尔医药有限公司 | 治疗肺部疾病的高穿透力前药组合物和医药组合物 |
| WO2013162729A1 (en) | 2012-04-24 | 2013-10-31 | The Board Of Trustees Of The Leland Stanford Junior University | Method for delivery of small molecules and proteins across the cell wall of algae using molecular transporters |
| US9782491B2 (en) | 2012-06-04 | 2017-10-10 | Indiana University Research And Technology Corporation | Peptide conjugates for treating pain |
| US9089536B2 (en) | 2012-06-06 | 2015-07-28 | Brian J. Smith | Ophthalmic solution for absorbing ultraviolet radiation and method for absorbing ultraviolet radiation |
| EP2908818A4 (en) * | 2012-10-16 | 2016-07-13 | Endocyte Inc | CONJUGATES OF DRUG DELIVERY CONTAINING ARTIFICIAL AMINO ACIDS AND METHODS OF USE |
| CA2889833A1 (en) | 2012-10-28 | 2014-05-01 | Revance Therapeutics, Inc. | Compositions and methods for safe treatment of rhinitis |
| WO2014117035A1 (en) | 2013-01-24 | 2014-07-31 | Transderm, Inc. | COMPOSITIONS FOR TRANSDERMAL DELIVERY OF mTOR INHIBITORS |
| CA2915296A1 (en) * | 2013-06-11 | 2015-02-26 | Portage Pharmaceuticals Ltd. | Structure, manufacturing and uses of human-derived cell-permeable peptides conjugated with specific biologically active cargo peptides |
| ES2870085T3 (es) | 2013-06-26 | 2021-10-26 | Xigen Inflammation Ltd | Inhibidores peptídicos permeables a células de la ruta de transducción de señales de JNK para el tratamiento de la cistitis |
| WO2014206427A1 (en) | 2013-06-26 | 2014-12-31 | Xigen Inflammation Ltd. | New use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of various diseases |
| WO2015197097A1 (en) | 2014-06-26 | 2015-12-30 | Xigen Inflammation Ltd. | New use for jnk inhibitor molecules for treatment of various diseases |
| CA2925487C (en) | 2013-09-24 | 2023-11-07 | University Of Washington Through Its Center For Commercialization | Desmoglein 2 (dsg2) binding proteins and uses therefore in treating disorders associated with epithelial tissues |
| US10815276B2 (en) | 2014-05-21 | 2020-10-27 | Entrada Therapeutics, Inc. | Cell penetrating peptides and methods of making and using thereof |
| EP3613426A1 (en) | 2014-05-21 | 2020-02-26 | Entrada Therapeutics, Inc. | Cell penetrating peptides and methods of making and using thereof |
| US11484580B2 (en) | 2014-07-18 | 2022-11-01 | Revance Therapeutics, Inc. | Topical ocular preparation of botulinum toxin for use in ocular surface disease |
| EP3185880B1 (en) | 2014-08-27 | 2020-02-12 | Ohio State Innovation Foundation | Improved peptidyl calcineurin inhibitors |
| MA41795A (fr) | 2015-03-18 | 2018-01-23 | Sarepta Therapeutics Inc | Exclusion d'un exon induite par des composés antisens dans la myostatine |
| EP3302489A4 (en) | 2015-06-04 | 2019-02-06 | Sarepta Therapeutics, Inc. | METHOD AND COMPOUNDS FOR THE TREATMENT OF DISEASES AND SUFFERING IN CONNECTION WITH LYMPHOCYTES |
| CN114712308A (zh) | 2015-12-09 | 2022-07-08 | 磷肌酸有限公司 | 药物制剂 |
| CN115252807A (zh) | 2016-07-27 | 2022-11-01 | 利兰斯坦福初级大学董事会 | 用于核酸递送的解体型细胞穿透性复合物 |
| SMT202200366T1 (it) | 2016-12-19 | 2022-11-18 | Sarepta Therapeutics Inc | Coniugati di oligomeri per salto di esone per distrofia muscolare |
| EP3558903B1 (en) | 2016-12-21 | 2024-07-03 | Avecho Biotechnology Limited | Process for phosphorylating a complex alcohol |
| JP7108631B2 (ja) | 2017-01-06 | 2022-07-28 | パルヴェラ セラピューティクス、インク. | mTOR阻害剤の無水組成物およびその使用方法 |
| EP3681479B1 (en) * | 2017-09-15 | 2024-01-31 | Dyve Biosciences, Inc. | Sodium bicarbonate for use in the treatment of gout and related disorders |
| WO2019070962A1 (en) | 2017-10-04 | 2019-04-11 | Ohio State Innovation Foundation | BICYCLIC PEPTIDE INHIBITORS |
| EP3746105A4 (en) | 2018-01-29 | 2022-05-18 | Ohio State Innovation Foundation | CYCLIC PEPTIDYLIN HIBITORS OF THE CAL-PDZ-BINDING DOMAIN |
| EP3749365A4 (en) | 2018-01-29 | 2022-01-12 | Ohio State Innovation Foundation | PEPTIDY INHIBITORS OF THE CALCINEURIN-NFAT INTERACTION |
| TW201945014A (zh) | 2018-02-22 | 2019-12-01 | 美商安卓達治療股份有限公司 | 用於治療粒線體性神經胃腸腦病變之組合物及方法 |
| US11814341B2 (en) | 2018-05-04 | 2023-11-14 | Ohio State Innovation Foundation | Non-peptidic cell-penetrating motifs |
| EP3790890A4 (en) | 2018-05-09 | 2022-03-02 | Ohio State Innovation Foundation | CYCLIC, CELL-PENETRATING PEPTIDES WITH ONE OR MORE HYDROPHOBIC RAILS |
| KR102167755B1 (ko) | 2018-05-23 | 2020-10-19 | 주식회사 큐어바이오 | 단편화된 grs 폴리펩타이드, 이의 변이체 및 이들의 용도 |
| US11000513B2 (en) | 2018-07-02 | 2021-05-11 | Palvella Therapeutics, Inc. | Anhydrous compositions of mTOR inhibitors and methods of use |
| CN111601618A (zh) | 2018-09-14 | 2020-08-28 | 江阴贝瑞森制药有限公司 | 孟鲁司特与肽的新缀合物 |
| CN109010798A (zh) * | 2018-09-17 | 2018-12-18 | 中山大学中山眼科中心 | 一种多肽的新用途 |
| WO2020117429A1 (en) * | 2018-12-04 | 2020-06-11 | Purdue Research Foundation | Novel antibacterial cell-penetrating peptides |
| CN115768779A (zh) * | 2020-05-07 | 2023-03-07 | 上海偕怡医药科技有限公司 | 伊曲康唑前药及其用途 |
| US20240166691A1 (en) * | 2022-11-08 | 2024-05-23 | Alcon Inc. | Prodrugs and compositions for ophthalmology applications |
Family Cites Families (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE744988C (de) | 1941-02-01 | 1944-02-22 | Wilhelm Ulrich O | Handschraem- und Schlitzsaege |
| GB744988A (en) | 1952-01-31 | 1956-02-15 | Allen & Hanburys Ltd | Improvements in preparations of insulin |
| GB1541435A (en) | 1975-02-04 | 1979-02-28 | Searle & Co | Immunological materials |
| US4847240A (en) | 1978-01-16 | 1989-07-11 | The Trustees Of Boston University | Method of effecting cellular uptake of molecules |
| US4701521A (en) | 1978-07-17 | 1987-10-20 | The Trustees Of Boston University | Method of effecting cellular uptake of molecules |
| JPS55500053A (ja) | 1978-01-16 | 1980-01-31 | ||
| US4631190A (en) | 1981-06-26 | 1986-12-23 | Shen Wei C | Acidity-sensitive spacer molecule to control the release of pharmaceuticals from molecular carriers |
| US4880911A (en) | 1982-03-19 | 1989-11-14 | G. D. Searle & Co. | Fused polypeptides and methods for their detection |
| US4532207A (en) | 1982-03-19 | 1985-07-30 | G. D. Searle & Co. | Process for the preparation of polypeptides utilizing a charged amino acid polymer and exopeptidase |
| JPH0236570B2 (ja) | 1985-05-30 | 1990-08-17 | Toko Yakuhin Kogyo Kk | Shoenchintsunankozai |
| US5354844A (en) | 1989-03-16 | 1994-10-11 | Boehringer Ingelheim International Gmbh | Protein-polycation conjugates |
| US5162505A (en) | 1989-09-19 | 1992-11-10 | Centocor | Proteins modified with positively charged carriers and compositions prepared therefrom |
| US5028707A (en) | 1989-11-20 | 1991-07-02 | Board Of Regents, University Of Texas | 4-hydroxyquinaldic acid derivatives |
| ES2091916T3 (es) | 1989-12-21 | 1996-11-16 | Whitehead Biomedical Inst | Metodo para suministrar moleculas en el interior de celulas eucarioticas. |
| US5804604A (en) | 1989-12-21 | 1998-09-08 | Biogen, Inc. | Tat-derived transport polypeptides and fusion proteins |
| US5268164A (en) | 1990-04-23 | 1993-12-07 | Alkermes, Inc. | Increasing blood-brain barrier permeability with permeabilizer peptides |
| EP0554284B1 (en) | 1990-10-24 | 1996-12-18 | Allelix Biopharmaceuticals Inc. | Peptide-based inhibitors of hiv replication |
| US5646120A (en) | 1990-10-24 | 1997-07-08 | Allelix Biopharmaceuticals, Inc. | Peptide-based inhibitors of HIV replication |
| US5633230A (en) | 1990-10-24 | 1997-05-27 | Allelix Biopharmaceuticals, Inc. | Treatment of cytomegalovirus infection |
| US5674849A (en) | 1990-10-24 | 1997-10-07 | Allelix Biopharmaceuticals Inc. | Anti-viral compositions |
| US5831001A (en) | 1990-10-24 | 1998-11-03 | Allelix Biopharmaceuticals Inc. | Treatment of herpesvirus infection |
| WO1993004701A1 (en) | 1991-09-05 | 1993-03-18 | University Of Connecticut | Targeted delivery of poly- or oligonucleotides to cells |
| EP0637247B1 (en) | 1992-04-23 | 1998-08-19 | Allelix Biopharmaceuticals Inc. | Treatment of herpesvirus infection |
| CA2094658A1 (en) | 1992-04-23 | 1993-10-24 | Martin Sumner-Smith | Intracellular delivery of biochemical agents |
| GB9213077D0 (en) | 1992-06-19 | 1992-08-05 | Erba Carlo Spa | Polymerbound taxol derivatives |
| CA2135642C (en) | 1992-08-21 | 1999-12-14 | James G. Barsoum | Tat-derived transport polypeptides |
| EP0599303A3 (en) | 1992-11-27 | 1998-07-29 | Takeda Chemical Industries, Ltd. | Peptide conjugate |
| CA2152387C (en) | 1992-12-22 | 1998-10-27 | Michael Twist (Deceased) | Synergistic anti-viral compositions |
| CA2152373C (en) | 1993-10-22 | 1998-12-15 | Michael Twist (Deceased) | Treatment of cytomegalovirus infection |
| US6013628A (en) * | 1994-02-28 | 2000-01-11 | Regents Of The University Of Minnesota | Method for treating conditions of the eye using polypeptides |
| US6077835A (en) | 1994-03-23 | 2000-06-20 | Case Western Reserve University | Delivery of compacted nucleic acid to cells |
| US5716614A (en) | 1994-08-05 | 1998-02-10 | Molecular/Structural Biotechnologies, Inc. | Method for delivering active agents to mammalian brains in a complex with eicosapentaenoic acid or docosahexaenoic acid-conjugated polycationic carrier |
| US5620013A (en) | 1994-10-21 | 1997-04-15 | American Cyanamid Company | Method for destroying residual lens epithelial cells |
| US5783178A (en) | 1994-11-18 | 1998-07-21 | Supratek Pharma. Inc. | Polymer linked biological agents |
| AU4690596A (en) | 1994-12-30 | 1996-07-24 | Chiron Viagene, Inc. | Nucleic acid condensing agents with reduced immunogenicity |
| DE69735057T2 (de) | 1996-03-12 | 2006-08-31 | PG-TXL Co., L.P., Houston | Wasserlösliche paclitaxel-prodrogen |
| AU729643B2 (en) | 1996-05-01 | 2001-02-08 | Antivirals Inc. | Polypeptide conjugates for transporting substances across cell membranes |
| US5795909A (en) | 1996-05-22 | 1998-08-18 | Neuromedica, Inc. | DHA-pharmaceutical agent conjugates of taxanes |
| JP3770666B2 (ja) | 1996-09-20 | 2006-04-26 | 株式会社ティ・ティ・エス技術研究所 | 経粘膜吸収製剤用組成物 |
| JPH10204000A (ja) | 1997-01-22 | 1998-08-04 | Nof Corp | 経皮吸収促進剤 |
| US6228344B1 (en) | 1997-03-13 | 2001-05-08 | Mallinckrodt Inc. | Method of measuring physiological function |
| IL132941A0 (en) | 1997-05-21 | 2001-03-19 | Univ Leland Stanford Junior | Composition and method for enhancing transport across biological membranes |
| US6730293B1 (en) * | 1999-08-24 | 2004-05-04 | Cellgate, Inc. | Compositions and methods for treating inflammatory diseases of the skin |
| US7229961B2 (en) | 1999-08-24 | 2007-06-12 | Cellgate, Inc. | Compositions and methods for enhancing drug delivery across and into ocular tissues |
| CA2381425A1 (en) | 1999-08-24 | 2001-03-01 | Cellgate, Inc. | Enhancing drug delivery across and into epithelial tissues using oligo arginine moieties |
| US6669951B2 (en) | 1999-08-24 | 2003-12-30 | Cellgate, Inc. | Compositions and methods for enhancing drug delivery across and into epithelial tissues |
| US20020009491A1 (en) | 2000-02-14 | 2002-01-24 | Rothbard Jonathan B. | Compositions and methods for enhancing drug delivery across biological membranes and tissues |
| EP1301213B1 (en) | 2000-07-21 | 2017-01-18 | ReVance Therapeutics, Inc. | Multi-component biological transport systems |
| MXPA03007358A (es) * | 2001-02-16 | 2004-12-13 | Cellgate Inc | Transportadores que contienen porciones de arginina separadas. |
| US9810571B1 (en) | 2016-12-15 | 2017-11-07 | Chung-Hsiu Su | Hand truck with a loading weighing mechanism |
-
2001
- 2001-02-23 US US09/792,480 patent/US6669951B2/en not_active Expired - Fee Related
-
2002
- 2002-02-25 WO PCT/US2002/005829 patent/WO2002069930A1/en not_active Ceased
- 2002-02-25 JP JP2002567285A patent/JP2004533414A/ja active Pending
- 2002-02-25 WO PCT/US2002/005804 patent/WO2002067917A1/en not_active Ceased
- 2002-02-25 CA CA002438784A patent/CA2438784A1/en not_active Abandoned
- 2002-02-25 EP EP02713692A patent/EP1372626A4/en not_active Ceased
- 2002-02-25 JP JP2002569108A patent/JP2004530657A/ja active Pending
- 2002-02-25 MX MXPA03007590A patent/MXPA03007590A/es active IP Right Grant
- 2002-02-25 MX MXPA03007591A patent/MXPA03007591A/es not_active Application Discontinuation
- 2002-02-25 EP EP02731103A patent/EP1367995A4/en not_active Ceased
- 2002-02-25 CA CA002438326A patent/CA2438326A1/en not_active Abandoned
-
2003
- 2003-12-17 US US10/740,365 patent/US20040186045A1/en not_active Abandoned
-
2006
- 2006-10-02 US US11/542,278 patent/US20070173436A1/en not_active Abandoned
-
2010
- 2010-06-15 US US12/816,358 patent/US8623833B2/en not_active Expired - Fee Related
-
2013
- 2013-12-24 US US14/140,223 patent/US20140213532A1/en not_active Abandoned
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010514781A (ja) * | 2006-12-29 | 2010-05-06 | ルバンス セラピュティックス インク. | Hiv−tatに由来するポリペプチド断片を用いて安定化されるボツリヌス毒素を局所適用及び経皮送達する組成物及び方法 |
| JP2014522816A (ja) * | 2011-06-22 | 2014-09-08 | ビョーメ バイオサイエンシズ ピーブイティー.リミテッド | コンジュゲートベースの抗真菌性および抗菌性プロドラッグ |
| JP2020519622A (ja) * | 2017-05-11 | 2020-07-02 | ケアジェン カンパニー, リミテッドCaregen Co., Ltd. | イソトレチノインとペプチドの結合体 |
| JP7016886B2 (ja) | 2017-05-11 | 2022-02-07 | ケアジェン カンパニー,リミテッド | イソトレチノインとペプチドの結合体 |
| US11351266B2 (en) | 2017-05-11 | 2022-06-07 | Caregen Co., Ltd. | Conjugate of isotretinoin and peptide |
| US12005123B2 (en) | 2017-05-11 | 2024-06-11 | Caregen Co., Ltd. | Conjugate of isotretinoin and peptide |
| WO2019198316A1 (ja) * | 2018-04-09 | 2019-10-17 | 国立研究開発法人産業技術総合研究所 | 生理活性を持つ中分子の薬物動態を改善する方法、薬物動態の改善を利用した中分子ライブラリの製造法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1372626A4 (en) | 2007-08-01 |
| US20140213532A1 (en) | 2014-07-31 |
| US20020127198A1 (en) | 2002-09-12 |
| CA2438784A1 (en) | 2002-09-06 |
| US6669951B2 (en) | 2003-12-30 |
| US20070173436A1 (en) | 2007-07-26 |
| EP1367995A1 (en) | 2003-12-10 |
| EP1372626A1 (en) | 2004-01-02 |
| CA2438326A1 (en) | 2002-09-12 |
| EP1367995A4 (en) | 2007-08-01 |
| MXPA03007591A (es) | 2003-12-04 |
| US20040186045A1 (en) | 2004-09-23 |
| WO2002069930A1 (en) | 2002-09-12 |
| WO2002067917A1 (en) | 2002-09-06 |
| MXPA03007590A (es) | 2003-12-04 |
| US20110206610A1 (en) | 2011-08-25 |
| JP2004533414A (ja) | 2004-11-04 |
| US8623833B2 (en) | 2014-01-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6669951B2 (en) | Compositions and methods for enhancing drug delivery across and into epithelial tissues | |
| US6730293B1 (en) | Compositions and methods for treating inflammatory diseases of the skin | |
| US6593292B1 (en) | Compositions and methods for enhancing drug delivery across and into epithelial tissues | |
| US8729010B2 (en) | Compositions and methods for enhancing drug delivery across and into ocular tissues | |
| EP1401473B1 (en) | Transporters comprising spaced arginine moieties | |
| JP2003523982A (ja) | 生体膜および生体組織を横切る薬物送達を増強するための、組成物および方法 | |
| AU2002306500A1 (en) | Transporters comprising spaced arginine moieties | |
| AU2002303102A1 (en) | Compositions and methods for enhancing drug delivery across and into epithelial tissues | |
| HK1152869A (en) | Enhancing drug delivery across and into epithelial tissues using oligo arginine moieties | |
| AU2002245529A1 (en) | Compositions and methods for enhancing drug delivery across and into ocular tissues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050216 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050216 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081023 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090108 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090116 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090622 |