A.定 義
アミノ酸:この文章の中で定義されているすべてのアミノ酸は天然のL−型の立体配置にある。標準的なポリペプチドの命名法(J.Biol, Chem., 243, 3557-59 (1969)) に従い、アミノ酸残基の略号は以下の対応表に示した通りとする。
注目すべきことは、この文章の中ではアミノ酸残基の配列はすべて、式の左から右へ向かう方向を従来のアミノ末端からカルボキシル末端へ向かう方向とする式で表わしてあることである。
塩基対(bp) : DNA二重らせん中の、アデニン(A)とチミジン(T)との、またはシトシン(C)とグアニン(G)との定まった組合わせ。
クローン:一つの祖先細胞または分子をもつ多類の同等な細胞または分子をいう。
相補的塩基: DNAまたは RNAが二本鎖構造をとる際に自然に対を作るヌクレオチド。
相補的ヌクレオチド配列: DNAまたは RNAの一本鎖分子のヌクレオチド配列がもう一つの一本鎖に、結果として生じる水素結合によって特異的にハイブリダイズするほど十分に、その塩基配列に対して相補的である配列。
保存された:ある、あらかじめ選択した(対照)配列について、もし、その配列が、選択した配列の正確な相補体に、非ランダムにハイブリダイズするなら、その配列は保存される。
下流:発現の進行方向をさらに進んだ配列を定義する。たとえば、ある遺伝子のペプチドをコードしている部位は、開始コドンより下流にある。
発現:構造遺伝子によってポリペプチドを産生するために行なわれる工程。転写と翻訳とを組合わせたもの。
遺伝子(シストロン):核酸であってそのヌクレオチド配列がRNAまたはポリペプチドをコードしているもの。遺伝子は RNAでも DNAでもよい。情報をコードしている個々の部分(エキソン)の間に介在する配列(イントロン)の他に、情報をコードしている領域に先行する、および後に続く領域(リーダーおよびトレーラー)も含まれる。
ハイブリッド形成:十分に相補的なヌクレオチド配列(核酸の鎖)が対合し、相補的な塩基対の間の水素結合の確立によって二本鎖またはヘテロ二本鎖を形成すること。2つの相補的なポリヌクレオチドの間の特異的、すなわちランダムではない相互作用であり、拮抗的に阻害することができる。
リガンド:リガンドは、特定の受容体タンパク質との特異的な相互作用によって結合される構造部分を含んでいる分子に属する。
ヌクレオチド:糖部分(ペントース)、リン酸基、および含窒素複素環式塩基を含んでいる、 DNAまたは RNAのモノマー単位。塩基はグリコシドの炭素(ペントースの1′炭素)を経て糖部分に結合しており、この塩基と糖との化合物がヌクレオシドである。ヌクレオシドがペントースの3′または5′位にリン酸基を含んでいる時それをヌクレオチドという。操作によって結合させたヌクレオチドの配列をこの文章の中では代表的には「塩基配列」または「ヌクレオチド配列」、およびそれらの文法的に同等なものとよび、この文章の中では式の左から右へ向かう方向を従来の5′末端から3′末端へ向かう方向とする式で表わしている。
オリゴヌクレオチドまたはポリヌクレオチド:ヌクレオチドの単鎖または二本鎖のポリマー。この文章の中で使われているように、「オリゴヌクレオチド」およびその文法的に同等であるものは、あらゆる範囲の核酸を含むであろう。オリゴヌクレオチドは典型的には、直鎖のリボヌクレオチドを含む核酸分子に属するものであろう。正確な大きさは多くの因子に依存するが、本技術分野において周知のように使用の最終的な条件に依存している。
読み取り枠:タンパク質に翻訳され得る(可能性のある)ヌクレオチド配列で、通常は DNA配列。
ポリペプチドおよびペプチド:隣接したα−アミノ基とカルボキシル基との間のペプチド結合により互いに結合している一連のアミノ酸残基を示すために、この文章の中で互換性をもって使用されている用語。
受容体:受容体および受容体タンパク質は、他の分子に(又は分子と)特異的に結合する、生物学的に活性のあるタンパク質を含んでいる分子を示すために、この文章の中で用いた用語である。
組換えDNA (rDNA)分子:遺伝子のような核酸の配列を、本発明の DNA分子配列に対して操作して結合させることにより産生されるDNA分子。従って組換え DNA分子は、普通自然にはいっしょに見つかることのない少なくとも2つのヌクレオチド配列を含んでいる、ハイブリッド DNA分子である。共通の生物学的起源をもたない、すなわち進化的に異なるrDNAを「非相同」であるという。
事実上相同:ある特定の試料としての配列または分子、たとえば突然変異した配列が、一つまたはそれ以上の置換、欠失、または付加によって対照配列から変化していても、その正味の効果は、対照と試料との間で、逆に機能するような相違性を生じる結果には終わらないことを意味する。本発明の目的のためには、90パーセント以上のアミノ酸配列の類似性を有し、同等の生物学的活性を有し、かつ同等の発現の特徴を有するアミノ酸配列を、事実上相同とみなす。40パーセント以上の類似性を有するアミノ酸配列は、事実上類似しているとみなす。相同性かまたは類似性かを決定する目的のためには、対照となる配列の切端または内部の欠失は、続いて起こる分子の修飾、たとえば糖鎖形成のように、無視すべきである。より低い相同性を有し、かつ匹敵する生物活性を有する配列は、対等とみなす。
トランスフェクション:真核細胞において、加えた DNAの取りこみにより新たな遺伝マーカーを獲得することである。
ベクター:核酸であり、好ましくは DNA分子であって、細胞内で自律複製ができ、そこに、核酸の一部分、たとえば遺伝子またはポリヌクレオチド(好ましくはDNA)を効果的に結合させて、結合した部分の複製をもたらすことができるもの。
一つ以上のタンパク質をコードしている核酸(好ましくはDNA)部分(遺伝子)の発現を導くことができるベクターを、この文章の中では「発現ベクター」という。他の種類の重要なベクターは、逆転写酵素を用いて作られたmRNA由来のcDNA (相補的DNA)のクローニングを可能にする。
クローニングベクター:外来性ヌクレオチド配列(好ましくはDNA)をその中に挿入させてクローン化させることのできるプラスミドまたはウィルス。
発現ベクター:外来性ヌクレオチド配列(好ましくはDNA)をその中に挿入させて発現させることのできるプラスミドまたはウィルス。
B.詳細な説明
1.ヒトのクラス1MHC 分子
本発明のヒトのクラスIMHC 分子は、HLA-A, HLA-B, HLA-C, HLA-E, HLA-F 、および HLA-Gを含むグループから選択し、より好ましくは、HLA-A, HLA-B、および HLA-Cを含むグループから選択した。この分子は可溶性または不溶性のいずれの形状でも使用することができる。可溶型(「 sol」)においては、トランスメンブランドメインに先行している、選択した HLA分子をコードしているヌクレオチド配列中に停止コドンが入るように操作する。
ヒトのクラスIMHC 分子をコードしているヌクレオチド配列は、たとえばJY, BM92, WIN, NOC、およびMGのような形質転換した細胞株のように、適当な変異体をもっている既知の確立された細胞株から単離することができるが、適当なプライマーを用いたポリメラーゼ連鎖反応(PCR) によって、その遺伝子部分からヌクレオチドを合成する方がより実用的である。この方法を用いて完全な長さの HLAのcDNAをクローン化することに成功しており、たとえば HLA-A25,HLA-A2, HLA-B7, HLA-B57, HLA-B51、および HLA-B37の配列は各々、受託番号M32321, M32322, M32317, M32318, M32319、およびM32320により、ジェンバンクデータベースに寄託されている。
共通配列を含めて、既知の、部分的で推定上の HLAのアミノ酸配列およびヌクレオチド配列が公表されており(たとえば、 Zemmour, Parham共著、Immunogenetics 33, 310-320 (1991) 参照) 、 HLAの変異体を発現している細胞株も知られており、広く一般に、多くはアメリカン・タイプ・カルチャー・コレクション(「ATCC」) から入手することもできる。従って、 PCRを用いてヒトのクラスIMHC をコードしているヌクレオチド配列を合成することが可能であり、次にそれをベクターに効果的に結合させ、適当な細胞を形質転換させ、そこで発現させることができる。
本発明の、ヒトのクラスIMHC 分子およびヒトのβ2ミクログロブリンを産生するための特に好ましい方法は、この文章の中に記した PCR反応産物を作るためのポリメラーゼ連鎖反応(PCR) における、プライマーとしてあらかじめ選択したオリゴヌクレオチドの使用に依存している。 MHCおよびβ2ミクログロブリン遺伝子の調製は、典型的にはプライマーの伸長によって、好ましくはポリメラーゼ連鎖反応(PCR) の一つの型におけるプライマーの伸長により成就される。
もし遺伝子が (PCR)増幅により産生されるためのものであれば、情報をコードしている核酸のそれぞれの鎖が増幅されるように2つのプライマー、すなわち PCRプライマーのペアーを用いなければならない。(簡単のために、典型的な HLAの変異体の配列が論議されるだろうが、記載されている PCR増幅法は、現在完全な配列が知られていないものを含めて、β2ミクログロブリンおよびすべてのHLA変異体の合成に対して同様に適用することができる。第一のプライマーはナンセンス(マイナスまたは相補的な)鎖の一部となり、HLA(プラスまたは情報をコードしている)鎖の中に保存されているヌクレオチド配列にハイブリダイズする。
情報をコードしている DNAの同族体を作るためには、従って第一のプライマーは、 MHC遺伝子内の保存されている領域、好ましくは各 HLAグループ内の共通配列部分又は同様に保存されている領域、すなわちHLA-A, HLA-B, HLA-C 、および多型性の低いグループであるHLA-E, HLA-F、およびHLA-G内の保存領域にハイブリダイズする(すなわち相補的となる)よう選択する。第二のプライマーは情報をコードしている(プラス)鎖の一部となり、マイナス鎖に保存されているヌクレオチド鎖とハイブリダイズする。 HLAをコードしている DNAの同族体を作るためには、第二のプライマーは従って、リーダー領域または最初の枠組み領域をコードしている範囲内にあるような、 HLAをコードしている遺伝子の5′末端にある、保存されているヌクレオチド配列とハイブリダイズするように選択する。
注目すべきことは、情報をコードしている DNA同族体の増幅においては、第二のプライマーの保存された5′のヌクレオチド配列が、 Lohらによって記載されているように(Science 243, 217-220 (1989))、ターミナルデオキシヌクレオチジルトランスフェラーゼを用いて、外から加えた配列に対して相補的になり得ることである。第一プライマーと第二プライマーの片方または両方は、制限酵素認識部位を限定するヌクレオチド配列を含むことができる。その部位は増幅された免疫グロブリン遺伝子に対して非相同であることが可能であり、典型的にはプライマーの5′末端またはその付近にある。
PCR プライマーペアーの第一プライマーは、情報をコードしているかまたは意味のある核酸の鎖にハイブリダイズするという理由から、この文章の中ではしばしば「センスプライマー」とよぶ。さらに PCRプライマーペアーの第二プライマーは、情報をコードしていないかまたはアンチセンスである核酸の鎖、すなわち情報をコードしている鎖に相補的な鎖とハイブリダイズするという理由から、この文章の中ではしばしば「アンチセンプライマー」とよぶ。多数の第一プライマーおよび(または)多数の第二プライマーは、たとえば一つの種類の第一プライマーが多数の異なる第二プライマーと対合して多数の異なるプライマーペアーが形成されるように、各々の増幅ごとに利用することができる。そのかわりとして、第一および第二プライマーの個々のペアーを用いることもできる。いずれにせよ、第一および第二プライマーの、同じかまたは異なる組合わせを用いた増幅による増幅産物は、遺伝子ライブラリーの多様性の増加に結びつく。
制限部位を限定する部分が存在する場合には、それは典型的にはプライマーの5′末端にある、非プライミング部位に局在している。第一プライマーに限定される制限部位は、典型的には第二プライマーに限定される制限部位を認識しない制限酵素によって認識される部位であるが、その目的は互いに非相補的な付着末端を有し、ベクター内への方向性のある挿入ができる DNA分子の産生を可能にすることである。
一つの態様において、本発明はプライマーの3′末端に局在するプライミング領域を有するプライマーを形成する、ポリヌクレオチドのセットを利用している。プライミング領域は、典型的には最も3′よりの(3′末端)15から30ヌクレオチド塩基である。各プライマーの3′末端のプライミング部位は、核酸の合成を触媒するプライマーとして作用すること、すなわち3′末端からプライマー伸長反応を開始することができる。プライマーの片方または両方は5′末端(最も5′より)の非プライミング部位、すなわち鋳型 HLAとハイブリダイズしない領域を、付加的に含むことができる。
PCR においては、各プライマーは第二プライマーと組合って作用し、標的となる核酸の配列を増幅する。 PCRに用いる PCRプライマーの選択は、この文章の中でヒトのクラスIMHC 分子およびβ2ミクログロブリンの産生に関して論議したような考慮により決定する。すなわちプライマーは、選択した遺伝子に保存されている配列に相補的なヌクレオチド配列を有する。有用なプライミング配列を以下に開示する。
HLA 遺伝子のクローニングに用いる戦術は、本技術分野において周知のように、種々の HLA遺伝子を構成している核酸のタイプ、複雑さ、および純度に依存するであろう。他の因子は、遺伝子が増幅および(または)突然変異するためのものであるかどうかを含む。
一般に HLA遺伝子は、mRNAおよび(または)ゲノム DNAのセンスストランドのように、ポリペプチドをコードしている鎖に含まれる。もし HLAが二重鎖のゲノム DNAの形をとっていれば、通常ではまず融解により変性して単鎖とする。 HLA鎖はその配列を、ペアーの各々の構成員があらかじめ選択したヌクレオチド配列を有しているPCRプライマーペアーで処理( 接触) することにより PCR反応を受ける。PCR プライマーペアーは、 HLA配列内に保持されている、好ましくは少なくとも約10ヌクレオチドの長さの、さらに好ましくは少なくとも約20ヌクレオチドの長さのヌクレオチド配列にハイブリダイズすることにより、プライマー伸長反応を開始することができる。
PCR 反応は、 PCR緩衝液中で、 PCRプライマーペアー、好ましくはあらかじめ決められた量の PCRプライマーペアーを、 HLA遺伝子の核酸、好ましくはあらかじめ決められた量の HLA遺伝子の核酸と混合し、 PCR反応混合物を作ることにより行う。反応混合物を、ポリヌクレオチドを合成する条件の下に、あらかじめ代表的に決めた、PCR 反応産物の形成に十分な時間をかけて維持し、それにより多数の異なる HLAをコードしている DNA同族体を産生する。
もう一つの戦術においては、その目的はゲノム二重鎖 DNAのアンチセンス鎖、またはmRNAが逆転写酵素反応を受けることによって産生するポリヌクレオチドのような、 HLAのポリヌクレオチド相補体を供することにより、 HLA変異体から、 HLA変異体をコードしている遺伝子をクローン化することである。そのような相補体を産生するための方法は、本技術分野において周知である。
PCR 反応は、いかなる適切な方法を用いても行うことができる。一般には、それは緩衝水溶液、すなわち PCR緩衝液中において、好ましくはpH7〜9において、最も好ましくは約8において行う。好ましくはモル過剰(ゲノム核酸では一般にプライマー:鋳型は106:1)のプライマーを、鋳型鎖を含んでいる緩衝液に混ぜ合わせる。この方法の効率を高めるため、大きなモル過剰が好ましい。
PCR 緩衝液は好ましくはまたデオキシリボヌクレオチド三リン酸であるdATP, dCTP, dGTP、およびdTTPと、典型的には耐熱性のポリメラーゼを、すべてプライマー伸長 (ポリヌクレオチド合成)反応に適した量で含んでいる。結果として得られる溶液(PCR混合物) を約90℃〜 100℃で、約1〜10分間、好ましくは1〜4分間加熱する。この加熱期間の後、溶液を冷却してプライマーハイブリダイゼーションに好ましい温度である54℃にする。合成反応は室温から、それ以上ではポリメラーゼ(誘導剤)がもはや十分に機能しない温度になるまで行うことができる。従って、たとえばもし誘導剤としてDNAポリメラーゼを用いるとすると、温度は一般に約40℃より高くはない。代表的な PCR緩衝液は以下のものを含む:50mM KCl, 10mM Tris-HCl, pH8.3, 1.5mM MgCl2, 0.001% (wt/vol)ゼラチン、 200μMdATP, 200μM dTTP, 200μM dCTP, 200μM dGTP 、および緩衝液 100μlに対し 2.5単位のサーマス・アクアティクス (Thermusaquaticus) DNAポリメラーゼI(米国特許No.4,889,818) 。
誘導剤は、酵素を含めて、プライマー伸長産物の合成を成就すべく機能するいかなる化合物または系でもよい。この目的に適した酵素はたとえば、大腸菌 DNAポリメラーゼI、大腸菌 DNAポリメラーゼIのクレグノウフラグメント、T4 DNAポリメラーゼ、他の利用可能な DNAポリメラーゼ、逆転写酵素、および熱安定性酵素を含めて、適切な方法でのヌクレオチドの組合せを促進させて、各々の核酸鎖に相補的な、プライマー伸長産物を形成することのできる他の酵素を含む。一般的に、合成は各々のプライマーの3′末端で始まり、鋳型鎖に沿って5′方向に、合成が終結するまで進み、異なる長さの分子を産生する。しかしながら上記の方法と同じ方法を用いて、5′末端から合成を開始し、上流方向へ進ませる誘導剤もあり得る。
誘導剤は、酵素を含めて、 RNAプライマー伸長産物の合成を成就すべく機能する化合物または系でもよい。好ましい態様においては、誘導剤はT7 RNAポリメラーゼ、T3 RNAポリメラーゼ、またはSP6 RNAポリメラーゼのような DNA依存 RNAポリメラーゼを含んでいる。これらのポリメラーゼは相補的な RNAポリヌクレオチドを産生する。RNA ポリメラーゼの高い代謝回転速度は、Chamberlinらによって記載されているように、出発物質であるポリヌクレオチドを増幅させる(酵素 (The Enzymes)、 P.Boyer編、P.87-108、アカデミック・プレス, ニューヨーク (1982))。
T7 RNAポリメラーゼのもう一つの利点は、cDNAの一部を、一つあるいはそれ以上の変異原性オリゴデオキシヌクレオチド(ポリヌクレオチド)で置き換え、この部分的にミス対合した鋳型を Joyceら(Nuc.Acid.Res. 17, 711-712 (1989))によって記載されているように、直接転写することにより、ポリヌクレオチド合成に突然変異を導入することができることである。転写に基づく増幅システムは、Gingerasらにより記載されている(PCRプロトコル・方法および応用への入門書 (PCR Protocols, A Guideto Methods and Applications)、p245-252、アカデミックプレス社,サンディエゴ, カリフォルニア, (1990)) 。
もし誘導剤が DNA依存 RNAポリメラーゼであって、リボヌクレオチド三リン酸を取りこむならば、プライマー伸長反応混合物には十分な量の ATP, CTP, GTP、および UTPを加え、結果として生じる反応液を上述のように処理する。
新たに合成された鎖、およびその相補的核酸鎖は、二重鎖分子を形成し、それを、この工程の次に続く段階で使用することができる。
HLA 系の一つの遺伝子または、多数の異なる遺伝子についてのHLA変異体をコードしている DNA同族体の産生の後は、 DNA分子は典型的にさらに増幅している。 DNA分子は、自律的に複製するベクターへのとり込みのような古典的技法によって増殖させることができるが、ベクターへの挿入に先立ち、まずポリメラーゼ連鎖反応(PCR)を行うことによって増幅させる方が好まれる。 PCRは典型的にはサーモサイクリングにより、すなわち、 PCR反応混合物の温度を、約10℃〜約40℃を下限とし、上限を約90℃〜約 100℃とする温度範囲の間で、くり返し上げ下げすることにより行う。温度の上げ下げは連続的であってもよいが、好ましくは、ポリペプチド合成、変性、およびハイブリダイゼーションに有利な各々の温度において、相補的な温度安定性のある区切りをもった段階的なものである。
PCR 増幅法は米国特許No.4,683,192、 4,683,202、 4,800,159、および 4,965,188ならびに、「 PCRテクノロジー・ DNA増幅の原理と応用」 (PCR Technology:Principles and Applications for DNAAmplification)、H.Erlich編、ストックトン出版、ニューヨーク(1989)、および「 PCRプロトコル、方法および応用への入門書」(PCR Protocols:A Guide to Methods and Applications) Innis共著、アカデミックプレス, サンディエゴ, カリフォルニア, (1990)を含めて、少なくともいくつのテキストに記載されている。
この文章の中で用いた、種々の好ましい方法およびプライマーを以下に記載しているが、たとえばNilsonら、Cell 58, 707 (1989), Ennisら、PNAS USA 87, 2833-7 (1990)、および Zemmourら、Immunogenetics33, 310-20 (1991) にも記載されている。特にプライマーの設計は、保存配列の選択と共に、 HLA対立遺伝子 (たとえば−A,−B,−C,−E,−F、または−G対立遺伝子)の5′および3′の非翻訳領域の比較から設計することが好ましい。5′および3′プライマーに制限部位を取り込ませて、増殖産物を配列決定ベクターまたは発現ベクターにサブクローンすることもできる。4塩基のスペーサー配列を制限部位の近くに置いて、増幅産物の酵素による切断の効率を高めることも役に立つ。
以下のプライマーは、好ましくは別々の反応における、 HLA−A,−B,−C,−E,−F、および−G cDNA の増幅に好ましい。結果として得られるcDNAは、この文章の中に記載したように、クローン化して配列を決定することができる。これらのプライマーは、すべての既知の、および現在は未知の、すべてのタイプの HLAの増幅への使用に適している。
HLA A
5′プライマー:5′CC ACC ATG GCC GTC ATG GCG CCC 3′
(配列番号1)
3′プライマー:5′GG TCA CAC TTT ACA AGC TCT GAG 3′
(配列番号2)
HLA B
5′プライマー:5′CC ACC ATG CTG GTC ATG GCG CCC 3′
(配列番号3)
3′プライマー:5′GG ACT CGA TGT GAG AGA CAC ATC 3′
(配列番号4)
HLA C
5′プライマー:5′CC ACC ATG CGG GTC ATG GCG CCC 3′
(配列番号5)
3′プライマー:5′GG TCA GGC TTT ACA AGC GAT GAG 3′
(配列番号6)
HLA E
5′プライマー:5′CC ACC ATG CGG GTA GAT GCC CTC C 3′
(配列番号7)
3′プライマー:5′GG TTA CAA GCT GTG AGA CTC AGA 3′
(配列番号8)
HLA F
5′プライマー:5′CC ACC ATG GCG CCC CGA AGC CTC 3′
(配列番号9)
3′プライマー:5′GG TCA CAC TTT ATT AGC TGT GAG A 3′
(配列番号10)
HLA G
5′プライマー:5′CC ACC ATG GCG CCC CGA ACC CTC 3′
(配列番号11)
3′プライマー:5′GG TCA CAA TTT ACA AGC CGA GAG 3′
(配列番号12)
好ましい態様においては、第一プライマーと第二プライマーとのただ一つのペアーを、増幅反応のたびに使用する。各々が、多数の異なるプライマーペアーを用いている、多数の異なる増幅から得た増幅反応産物はそこで組合わせる。しかしながら、本発明はまた、共増幅(2組のプライマーを使用)、および複合増幅(およそ8,9、または10組までのプライマーを使用)を経た DNA同族体の産生も意図している。
好ましい態様においては、 PCR法は、単にヒトのクラスIをコードしている DNA分子の一種を産生するためだけでなく、高度に多型である HLA座にみられるものを模倣することができる突然変異を誘導するため、または、単一の親クローンから多様性を作り出し、それによってクラスIMHC 分子をコードしている DNAの「ライブラリー」に、より大きな異質性をもたらすために使用される。第一に、PCR 法自体が、本技術分野において周知の因子の一種のために、固有の特性として変異原性であるということに注目すべきである。
第二に、先に引用した米国特許No.4,683,195に記載されているように、突然変異を誘導する変形に加えて、他の、突然変異を誘導する PCRの変形も用いることができる。たとえば、 PCR反応混合物は、伸長産物に取り込まれるべき一つ以上のヌクレオチドの量を変えて作ることができる。そのような条件下では PCR反応の進行は、特定の塩基の不足の結果として、伸長産物内にヌクレオチドの置換をもたらしめる。
同様に、ほぼ等しいモル量のヌクレオチドを、サイクルをX回能率的に回転させる量だけ、最初の PCR反応混合物に取り込ませることができ、そこで反応混合物をX回より多く、たとえば2X回だけ回転させる。そのかわりとして、反応混合物の中にイノシンのような、通常では増幅された HLA変異体の核酸には見られないヌクレオチド誘導体を取り込ませることにより、 PCR反応の間に突然変異を誘導することができる。次に続く in vivoでの増幅の間に、ヌクレオチド誘導体はかわりのヌクレオチドに置き換わり、それにより点突然変異が誘導される。
ヒトのクラスIMHC をコードしている発現可能なヌクレオチド鎖をクローン化するために利用できる好ましい方法は以下の通りである。選択した遺伝子(または複数の遺伝子)を含んでいる細胞は、必要に応じて追加した(たとえば、牛胎児血清および(または)抗生物質)、適当な培地(たとえばRPMI 1640)中で生長させることができる。選択した培養物から全細胞 RNAを調製し、次いでcDNAの最初の鎖を作る。cDNAは、たとえば、オリゴ(dT) およびトリのミエロブラストシスウィルスの逆転写酵素を用いて合成することができる。クローン化した産物の単離および回収の助けとして放射線標識物を利用することもできる。
次に産物を、好ましくは抽出し、沈殿させ、この文章の中に示したように、適当な PCRプライマーペアーを用いて PCRの標的として使用する。増幅は、有効な方法としキット、たとえばジーンアンプ(Gene Amp) キットおよび DNAサーマルサイクラー(パーキン・エルマー/シータス)を用いて行うこともできる。種々のプロトコルが有効であり、一つの増幅プロトコルは30サイクル回転し、各々のサイクルは94℃で60秒、65℃で60秒、および72℃で90秒から成り、さらに72℃で10分が続く。もう一つのプロトコルは20サイクル回転し、94℃で60分、65℃で1秒、さらに72℃でいろいろな長さの時間(すなわち出発時には50秒であり、1サイクルごとに1秒ずつ増えていく)から成る。さらに72℃で10分後に増幅は終了する。
典型的には、次に PCR産物をサブクローンし、既知の方法で配列を決定する。たとえば、産物を抽出し、さらに逆抽出し(たとえばフェノール/クロロフォルムで)、沈殿させ、適当な酵素で、たとえばHindIII で、適当な時間をかけて消化する。2本鎖切断した産物を単離し、精製し(たとえば、ガラスビーズで)、同様に切断したベクターに連結させる。 MHC分子の発現のために選択した細胞株の特徴に基づき、次いで適したベクターを選択する。たとえば、 MHCの配列分析のための回収を目的として、大腸菌の中で MHCを発現させることができる。そのかわりとして、CD8細胞の活性化を目的として、本発明の形質転換細胞の中で MHCを発現させることができる。適したベクターの選択については、本章の他の場所に、さらに詳しく論じてある。
2. DNA発現ベクター
本発明のベクターは細胞内で自律的に複製することの可能な核酸(好ましくはDNA)分子であり、そこに DNAセグメント、たとえば遺伝子またはポリヌクレオチドを効果的に結合させて、結合した部分の複製をもたらすようにすることができる。本発明においては、ベクターの配列に効果的に結合されるためのヌクレオチドセグメントは、哺乳類のクラスIMHC 分子の少なくとも一部をコードしている。好ましくは、 MHC遺伝子の、ペプチドをコードしている配列全体を、ベクターに挿入して発現させるが、しかし MHCの非コード配列のいくつかも同様に含んでいるベクターを構築することもあり得る。
好ましくは、 MHCの非コード配列は排除する。かわりに、可溶(「 sol」) 型のクラスIMHC 分子のヌクレオチド配列を使用することができる。「 sol」型は、α3ドメインの末端か、またはトランスメンブランドメインの前に「停止」コドンを含む点において不溶型と異なる。もう一つの好ましいベクターは、発現のためベクターに効果的に結合している哺乳類のβ2ミクログロブリン分子の少なくとも一部をコードしているヌクレオチド配列を含む。クラスIMHC 分子およびβ2ミクログロブリンの両方をコードしているヌクレオチド配列を含んでいるベクターを構築することも可能である。
好ましいベクターは、方向性のある連結に適したヌクレオチド配列によって、発現のため効果的に結合している一つ以上の翻訳可能な DNA配列を含むカセットを含んでいる。カセットは好ましくは、翻訳可能な DNA配列が、方向性のある連結に適したヌクレオチド配列により、直接カセット内に挿入された時に産生されるペプチドまたはタンパク質を発現するための、 DNA発現調節配列を含んでいる。カセットはまた、好ましくは翻訳可能な DNA配列から上流にプロモーター配列を含み、さらに哺乳類の MHC配列より下流に、ポリアデニル化配列を含んでいる。カセットは選択マーカーを含むこともできるが、しかしそのようなマーカーは、別の発現ベクターの配列に効果的に結合しているヌクレオチド配列にコードする方が好ましい。
発現ベクターは、適合する宿主内において、哺乳類のクラスIMHCポリペプチド、又はβ2ミクログロブリン、またはその両方のような構造遺伝子の産物を発現することができるという特徴を有する。特に、この文章の中で開示した発現ベクターは、ヒトのクラスIMHC分子および(または)ヒトβ2ミクログロブリンを発現することができる。
この文章の中で用いているように、「ベクター」という用語は、異なる遺伝環境の間で、すでに効果的に結合されている他の核酸を輸送することのできる核酸分子に属する。好ましいベクターは、自律的複製が可能であって、ベクターに効果的に結合している核酸(DNA)セグメントに存在している構造遺伝子産物の発現が可能である。
DNA 配列又はセグメントについてこの文章の中で用いているように、「効果的に結合している」という句は、配列またはセグメントが一片の DNAに、単鎖状でも二重鎖状ででも、共有結合によって結合していることを意味する。
本発明のカセットが効果的に結合するベクターの選択は、本技術分野において周知のように、たとえばベクターの複製とタンパク質の発現、および形質転換されるべき宿主細胞のような、希望する機能特性に直接的に依存しており、これらのことが組換え DNAを構築するという本技術分野における固有の限界となっている。
種々の態様においては、 MHC変異体および抗原ペプチドを含めて、本発明において有用なポリペプチドを産生するためにベクターを利用している。そのようなベクターは、好ましくは、有用な量のタンパク質またはポリペプチドの産生に適した「宿主」細菌細胞と共に利用される。そのようなベクターは、原核細胞のレプリコン、すなわち、それによって形質転換した宿主細菌細胞のような原核生物の宿主細胞内において、組換え DNA分子の染色体外における自律的複製および維持を指示する能力を有するヌクレオチド配列を含むことができる。
そのようなレプリコンは、本技術分野において周知である。さらに、原核生物のレプリコンを含むそれらの態様はまた、その発現が、それによって形質転換する宿主細菌細胞に対して薬剤耐性のような選択的利益を与える遺伝子も含むことができる。細菌の典型的な薬剤耐性遺伝子は、アンピシリンまたはテトラサイクリンに対する耐性を与える遺伝子である。ベクターはまた典型的には、翻訳可能なヌクレオチド配列の挿入に便利な制限部位を含む。
典型的なベクターは、バイオラッド・ラボラトリース(カリフォルニア州,リッチモンド)から入手されるプラスミド pUC8, pUC9, pUC18,pBR322、およびpBR329、ファルマシア (ニュージャージー州, ピスカタウェイ) から入手される pPLおよびpKK223、および pBSとM13mp19(ストラタジーン, カリフォルニア州, ラジョラ) を含む。他の典型的なベクターはpCMU (Nilsonら、Cell, 58, 707 (1989))を含む。他の適切なベクターも既知の方法により合成することができ、たとえばこの文章の中で種々の適用に用いたベクターであるpCMU/Kb およびpCMUIIは、pCMUIV (Nilsonら、上記) の変形である。
方向性のある連結のために採用したヌクレオチド配列、すなわちポリリンカーは、発現ベクターの領域であり、(1)複製およびヌクレオチド配列の上流および下流への輸送のために効果的に結合しており、(2)あるヌクレオチド配列の、ベクターへの方向性のある連結のための場所または手段を供する。典型的には、方向性ポリリンカーは二つ以上の制限酵素認識配列または制限部位を限定するヌクレオチド配列である。制限切断時には、二つの部位は、翻訳可能なヌクレオチド配列が発現ベクターに結合できる、付着末端を生じる。
好ましくは、それら二つの制限部位は、制限切断に際し、非相補的であり、翻訳可能なヌクレオチド配列の、カセットへの方向性のある挿入がそれによって可能になる付着末端を供する。ある態様においては、方向性のある連結法は、上流のヌクレオチド配列か下流のヌクレオチド配列、またはその両方に存在するヌクレオチドにより供される。もう一つの態様においては、方向性のある連結に適応したヌクレオチド配列は、多数の方向性のあるクローニング法を限定するヌクレオチド配列を含む。方向性のある連結に適したヌクレオチド配列が、多数の制限部位を限定しているところを、多クローニング部位とよぶ。
翻訳可能なヌクレオチド配列は、直線的に連続したヌクレオチドであり、一つの読みとり枠内に一つのポリペプチドをコードしている、中断のない連続した少なくとも8コドンを備えたものである。好ましくは、ヌクレオチド配列は DNA配列である。さらに好ましくは、翻訳可能なヌクレオチド鎖の上流に、プロモーター配列がある。好ましくは、プロモーターは条件プロモーター(たとえば、誘導可能な)である。この文章の中で用いられる好ましい条件プロモーターは、メタロチオネインプロモーターまたは、熱ショックプロモーターである。
ベクターは、いかなる周知のベクター構築技術を利用しても構築することができる。しかしながらこれらの技術は、宿主細胞のゲノムに挿入されるべき翻訳可能なヌクレオチド配列が、その上流を適当なプロモーターと接しておりさらに、本発明のいくつかの変形においては、翻訳可能なヌクレオチド配列はその下流をポリアデニル化部位に接しているという程度には変形してある。このことは、「宿主」細胞が昆虫細胞であり、ヌクレオチド配列がトランスフェクションにより伝達される場合には特に好ましい。
トランスフェクションは、カルシウムリン酸法、DEAEデキストラン法、安定伝達法、電気穿孔法、またはリポソーム仲介法を含む多くの方法により行うことができる。周知のトランスフェクション法、および、細胞内にヌクレオチドを導入するための他の方法を述べている多くのテキストを利用することができる(「分子生物学における現在のプロトコル」(Current Protocols in Molecular Biology)、John Wiley,Sons, ニューヨーク (1991) などを参照) 。
ベクター自体は、ウィルスベクター(RNAまたはDNA)、むきだしの直鎖または環状 DNA、核酸物質と、細胞内に挿入されるべき何らかのポリペプチドとを含んでいる小胞またはエンベロープのような、いかなる適したタイプのものでもよい。小胞については、リポソームのような脂質小胞を構築するための技術が周知である。そのようなリポソームは、リポソームの外面に、抗体または他の特異的に結合する分子を供給するような、他の従来通りの技術を用いて、特定の細胞を標的とすることができる。 A.Huangら、J.Biol.Chem. 255,8015-8018 (1980)などを参照のこと。
最も有用なベクターは、「宿主」細胞、すなわち形質転換されるべき細胞の性質により、次にあげるもののうち1つ以上を含めた複数の要素をもつ:(1)高いコピー数への増幅のための、SV40の複製起点、(2)高レベルの転写開始のための効果的なプロモーター要素、(3)mRNA切断およびポリアデニル化配列のような、mRNAをプロセシングするシグナル(さらに、しばしば介在配も同様)、(4)「外来」 DNAの挿入のための、複数の制限酵素部位を含んでいるポリリンカー、(5)プラスミド DNAを安定に取りこんだ細胞の選択に用いることのできる選択マーカー、(6)細菌細胞における増殖を可能にする、プラスミド複製調節配列。
上記のものに加えて、多くのベクターも、外部刺激により調節されている誘導可能な発現システムを含んでいる。誘導された転写に必要な、多数のプロモーターからの配列が同定され、誘導可能な発現を得るための発現ベクター内に操作により挿入されている。いくつかの有用なベクターは、β−インターフェロン、熱ショック、重金属イオン、およびステロイド(たとえばグルココルチコイド)による誘導に基づくものである。(Kaufman, Meth.Enzymol. 185, 487-511 (1990) などを参照。)
好ましい態様においては、ベクターはまた選択マーカーを含んでいる。発現の後、翻訳可能なヌクレオチド配列の産物は、次にその配列に対する抗体を用いて精製することができる。選択マーカーの一例は、ネオマイシン耐性である。phshsneo, phsneo、又はpcopneoのような、ネオマイシン耐性をコードしているプラスミドは、トランスフェクトしたものを選択培地中で増殖させることにより、選択した遺伝子(または複数の遺伝子)を発現している細胞集団を確かめることができるように、各々のトランスフェクションに含ませていくことが可能である。
好ましい態様においては、翻訳可能なヌクレオチド配列は、調節可能な適当な転写プロモーター、翻訳調節配列、および翻訳可能なヌクレオチド配列の、正しい方向への挿入を平易にするためのポリリンカーと共にプラスミド中に取り込ませることができ、ドロソフィラのような真核細胞、または大腸菌のような原核細胞の中で、従来通りの技法を用いて発現させることができる。好ましくは、転写開始のためのプロモーターを限定する5′の調節配列、転写可能な DNA配列の上流の5′末端に効果的に結合している、リボソーム結合部位がある。
たとえば大腸菌などのような、形質転換した、またはトランスフェクトした細胞において、高いレベルの遺伝子発現を行うためには、大量のmRNAを生じる強力なプロモーターだけでなく、mRNAの効率のよい翻訳を確実にするための、リボソーム結合部位も用いることが必要である。たとえば大腸菌においては、リボソーム結合部位は開始コドン(AVG) 、および開始コドンの3〜11ヌクレオチド上流に局在する3〜9ヌクレオチドの長さの配列を含む(Shineら、Nature, 254, 34 (1975)) 。シャイン・ダルガーノ(SD) 配列と呼ばれる、 AGGAGGU配列は、大腸菌の 16SmRNAの3′末端に相補的である。
リボソームの、mRNAおよびmRNAの3′末端の配列への結合は、(1)SD配列と16stRNA の3′末端との間の相補性の度合い、および(2)SD配列と AUGとの間におかれている間隔およびおそらくは DNA配列、を含んでいるいくつかの因子により影響をうけることが可能である。 (Robertら、PNAS 76, 760 (1979a), Robersら、PNAS 76, 5596 (1979b) 、Guarenteら、Science, 209, 1428(1980),およびGuarenteら、Cell, 20, 543 (1980)などを参照のこと。) この間隔を系統だてて変えたプラスミドにおける、遺伝子発現のレベルを測定することにより、一般的には最適化を行うことができる。
異なるmRNAの比較は、統計学的に、−20から+13の位置(AUGのAを0位とする、Goldら、Ann.Rev.Microbiol. 35, 365 (1918) 参照)に、好ましい配列があることを示した。リーダー配列も、翻訳に対して劇的な影響をもつことが示されてきた(Robertsら、1979、上記)。リボソームの結合も、リボソームの結合に影響を与える AUGに続く、ヌクレオチド配列によって影響を受けることがある(Taniguchiら、J.Mol.Biol. 118, 533 (1978) 参照) 。
本発明に従って用いることのできるベクターは、熱ショックプロモーターを含んでいる。このようなプロモーターは、本技術分野において周知である(Stellarら、EMBO J. 4, 167-171 (1985) 参照) 。もしこのプロモーターを使うのであれば、ポリアデニル化部位を加えることも好ましい。
本発明に従って用いるための好ましいベクターは、プラスミドであり、さらに好ましくは、コピー数の高いプラスミドである。ベクターが誘導可能なプロモーター配列を含んでいることもまた好ましいが、それは、そのようなベクター(しばしば生来のものではない、またはキメラ状のヌクレオチド鎖を運ぶように構築されている)が導入される細胞に対して、選択圧を制限する傾向があるからである。選択したベクターが、選択した宿主における発現に最も適していることも好ましい。
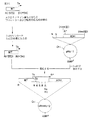
もし宿主細胞集団がドロソフィラ細胞の培養物であれば、適合するベクターは、 p25−lacZ (Bello, Couble 共著Nature 346, 480 (1990)参照) 、または pRmHa−1,−2、又は−3(Bunchら、Nucl.Acid Res. 16, 1043-1061 (1988) 参照) のようなベクターと機能的に等しいベクターを含む。好ましい態様においては、ベクターは pRmHa−3であり、図1に示した。このベクターは、好ましくは MHC配列が挿入されている部位の上流にある、メタロチオネインプロモーターを含み、ポリアデニル化部位は、好ましくは前記 MHC配列の下流にある。ドロソフィラ細胞は、本発明によれば好ましい宿主であり、特にシュナイダー2細胞のようなドロソフィラ細胞は、プロモーターの活性化に必要とされる、必須なトランス作用因子を有し、従ってさらになお好ましい。
発現ベクター pRmHa−3は、細菌のプラスミド pRmHa−1に基づくものであり、後者はプラスミド pUC18に基づくものである。そのプラスミド pUC18はアメリカン・タイプ・カルチャー・コレクション (ATCC, ロックビル,メリーランド州) に、受託番号 37253を以って寄託されている。
pRmHa−3ベクターは、プロモーターである、メタロチオネイン遺伝子(配列1〜421, SEQ ID NO 13)の、R1および Stu部位を除去した、翻訳されない5′リーダー配列を含んでいる(図1C参照)。それはまた、ポリアデニル化部位を含んでいるドロソフィラの ADH遺伝子 (配列6435〜7270, SEQ ID NO 14) の3′部分も含んでいる。従って、クローン化された DNAは、メタロチオネインプロモーターにより転写の制御を受け、さらにポリアデニル化されるであろう。プラスミド pRmHa−1の構築は、 Bunchらにより記載されている(Nucl.Acid Res. 16, 1043-1061 (1988)) 。
プラスミド pRmHa−3および pRmHa−2の構築(後者はEcoRIフラグメントとして除去することのできる、メタロチオネインプロモーター配列を有する)を、図1A,B、およびCに示した。本発明に従って使用するための好ましいプラスミドである pRmHa−3に関しては、 PstI, SphI、およびHindIII がプロモーターフラグメント内にあり、従って単一ではない。 Xbaは ADHフラグメント(3′末端から4塩基)にあり、単一ではない。しかし、次の制限部位は、 pRmHa−3においては単一である:EcoRI, SacI, KpnI, SmaI, BamHI, SalI,Hinc2 、および AccI。
本発明の DNA発現ベクターにおけるカセットは、翻訳可能な DNA配列の挿入に際し、適当な宿主において本発明の融合タンパク質を発現することのできるヌクレオチド配列を形成している、ベクターの領域である。発現に適したヌクレオチド配列をシストロンと呼ぶ。従って、カセットは、好ましくは一つ以上の翻訳可能な DNA配列に効果的に結合している、 DNA発現調節要素を含んでいる。シストロンは、翻訳可能な DNA配列が、調節要素の間に目的に合ったヌクレオチド配列を介し、方向性をもって挿入(方向性をもって連結)される時に形成される。結果として得られる DNA配列、すなわち挿入された DNAは、好ましくは、適当な読み取り枠の中に効果的に結合している。
DNA発現調節配列は、構造遺伝子産物を発現させるための、 DNA発現シグナルのセットを含んで成り、周知のように、シストロンが構造遺伝子産物を発現することができるように、シストロンに効果的に結合している5′および3′の要素を含む。
5′調節配列は、転写開始のためのプロモーター、および翻訳可能な DNA配列の上流の5′末端に効果的に結合した、リボソーム結合部位を定義する。
従って、本発明の DNA発現ベクターは、転写可能な DNA配列のベクターのカセット部分にクローニングして、本発明の融合タンパク質の発現が可能なシストロンを生じるためのシステムを供する。
3.細胞株
本発明の好ましい細胞系は、培養において連続的に増殖することができ、その細胞表面上に哺乳類クラスIMHC 分子を発現することの可能な細胞株である。細菌、酵母、昆虫、および哺乳類の細胞株を含めて、いかなる種類の形質転換および非形質転換細胞または細胞株もこの目的にかなっている。(培養法および、大腸菌および酵母菌などのような各種細胞株の使用法についての要約および方法については、「分子生物学における今日のプロトコル」(Current protocols in Molecular Biology) John Wiley, Sons, ニューヨーク、を参照のこと。)
好ましくは、細胞株は真核細胞の株である。さらに好ましくは、細胞株は変温性(すなわち哺乳類の細胞株よりも温度変化に対する感受性が低い)であり、より好ましくは昆虫細胞の株である。ガ(ATCC CCL 80)、ヨトウムシ (ATCC CRL 1711)、カの幼虫(ATCC 行CCL 125, CCL 126, CRL 1660, CRL 1591, CRL 6585, CRL 6586) およびカイコ(ATCC CRL 8851) を含めて、種々の昆虫の細胞株が、本発明による使用に役立つ。好ましい態様においては、細胞株はシュナイダー細胞株(J.Embryol.Exp.Morph. 27, 353〜365 (1972)) のようなドロソフィラ細胞株であり、好ましくは細胞株はM3培地での増殖に適したシュナイダー2細胞株(S2/M3) (Lindguistら、Drosophila Information Service 58, 163 (1982) 参照) である。
シュナイダー細胞は、本質的には以下のようにして調製することができる。ドロソフィラ・メラノガスター (Drosophilla melanoga-ster)(オレゴン−R) の卵を、およそ4時間ごとに集め、 2.5%次亜塩素酸ナトリウム水溶液中で卵膜を除去し、70%エタノールに20分間、さらに0.05% HgCl2を含む70%エタノールに20分間浸して表面を殺菌する。滅菌した蒸留水中で完全にすすいだ後、あらかじめ培養液で湿らせておいた、ミリポアフィルターで裏打ちした滅菌ずみのメトリセル黒色フィルターを含んでいるペトリ皿に、卵を移した。22℃のインキュベータ内に卵を一晩静置し、20〜24時間令の時点で培養に移した。胚を各々二分の一または三分の一に切り、次いで 0.2%トリプシン(1:250 、ディフコ) を含むリナルディニ塩類溶液(Rinaldini, Nature(ロンドン) 173, 1134〜1135 (1954))中に、室温で、20〜45分間静置した。 100〜300 の胚を用いて各々の培養を開始した。
ウシ胎児血清(FBS) を加えた後、断片を 100×gで2〜3分間遠心し、培養液1.25mlに再び浮遊させ、T−9ガラスフラスコに播種した。培養物は、周囲の空気を気相として、約22〜27℃± 0.5℃に維持した。好ましくは、シュナイダーの培養液(Schneider, J.Exp.Zool. 156, 91-104 (1964)、Schneider, J.Embryol.Exp.Morph. 15, 271〜279 (1966)) に、培養液 100mlあたり 500mgの細菌学用ペプトンおよび不活性化した FBSを15%加えたものを用いた。pH (好ましくは 6.7〜6.8)は、0.01%フェノール赤を用いて検査した。
細胞株は、好ましくは3〜7日ごとに継代培養して維持した。細胞は容易にガラスに接着するが、トリプシン処理を必要とする程には強固に接着しておらず、典型的には単なるピペッティングによりほとんどの細胞をフラスコの底から流し出すことができる。細胞の形態学的な外観は、 Schneider (J.Embryol.Exp.Morph. 27, 353〜365(1972)) により記載されている。それらは基本的には外観上は上皮性であり、直径では5〜11μm、長さでは11〜35μmの変動がある。小さなくぼみのある円形細胞は、他の細胞全体にランダムに分散させることができる。
好ましくは、シュナイダー2細胞は、ペニシリン(100単位/ml)およびストレプトマイシン(100mg/ml) を含めて、10%の FBSを加えたシュナイダーのドロソフィラ培地中で維持する。細胞は 0.5×105 /ml以上の密度に維持し、24〜30℃の温度範囲で増殖させることが好ましい。細胞は24時間より早く2倍になる傾向があり、高い細胞濃度、すなわち約2×107 /mlまたはそれより高い濃度になるまで増殖する。細胞は、後で使用または分析するため、90% FBSおよび10%DMSO中に凍結することができる。細胞を−70℃に置き、次いで液体窒素中に貯蔵することができる。
シュナイダー2細胞と同定される、本発明による好ましい細胞株は、ベダペスト条約の要求に従い、1992年2月18日、アメリカン・タイプ・カルチャー・コレクション(ATCC)に寄託し、受託番号 CRL−10974 を得た。
好ましい態様においては、細胞は哺乳類クラスIMHC 遺伝子を発現することのできる形質転換した細胞株であり、すなわち好ましくは、ヒトのクラスIMHC 遺伝子は、この細胞株により発現が可能である。細胞系が哺乳類β2ミクログロブリンを発現できることも好ましく、さらに好ましくはβ2ミクログロブリンはヒトβ2である。さらになお好ましい細胞株は、安定な、または一時的な発現が可能である。
ベクターは、本発明による細胞株を形質転換またはトランスフェクトするために用いることができる。細胞株の形質転換またはトランスフェクションに使用できる多くのベクターが入手でき、これらのベクターについては前文に、さらに詳しく述べられている。しかしながら簡単に言えば、好ましい態様においては、本発明の細胞は、発現ベクターに各々挿入されている(すなわち効果的に結合している)(ヒト)MHCのH鎖およびβ2ミクログロブリンをコードしているcDNAでトランスフェクトしている。
さらに好ましい態様においては、ベクターは、ヒトのクラスIMHC 分子またはβ2ミクログロブリンをコードしている発現可能なヌクレオチド配列が、この文章の中に開示した技術を用いて挿入されている、ドロソフィラの発現プラスミド pRmHa−3を含む。好ましくは MHCをコードしているcDNA、およびβ2ミクログロブリンをコードしているcDNAは、別々の発現プラスミドに効果的に結合しており、さらに培養細胞内へ同時にトランスフェクトする。
そのかわりに、 MHCおよびβ2ミクログロブリンをコードしているcDNAは同一の発現ベクターに効果的に結合しており、同じプラスミドによって同時にトランスフェクトする。もう一つの変形においては、 MHC,β2ミクログロブリン、およびIL2のようなサイトカインをコードしているcDNAは、発現プラスミドに効果的に結合しており、本発明の細胞株内へ同時にトランスフェクトする。 HLA遺伝子の選択、適当なベクターの構築、およびプライマーの選択については前文のB・1およびB・2章においてさらに詳しく記載した。
形質転換に成功した細胞、すなわち、本発明によって、発現可能なヒトのヌクレオチド配列を含んでいる細胞は、周知の技法により同定される。たとえば、本発明のcDNAおよびrDNAの導入の結果得られた細胞は、クローン化してモノクローナルコロニーを産生することができる。これらのコロニーから細胞を集め、溶解し、それらの DNA含量を、rDNAの存在に関して、Southern (J.Mol.Biol. 98, 503(1975)) により記載されているような方法を用いて測定することができる。
rDNAの存在についての直接的な分析に加え、rDNAが、主題であるキメラ状ポリペプチドの発現を指示することができるなら、周知の免疫学的方法により、形質転換またはトランスフェクトの成功を確認することができる。たとえば、ある発現ベクターで形質転換に成功した細胞は、適当な抗体を用いて容易に測定される特定の抗原性を示すタンパク質を産生することができる。さらに、形質転換またはトランスフェクションの成功は、上文に記載したように、ネオマイシン耐性のようなマーカー配列を運んでいる付加的なベクターを用いて確認することができる。
温度を下げても増殖を続けることのできる培養物もまた好ましい。たとえば、培養物はおよその室温、たとえば約24〜27℃に維持することが好ましい。他の態様においては、特にCD8細胞の活性化の工程においては、培養物をより高い温度に維持する。従って、本発明による培養物は、約30℃から約37℃の温度の挑戦に耐え得ることが好ましい。培養物へのβ2ミクログロブリンの添加は、少なくとも30℃の挑戦に対して安定化させ、β2ミクログロブリンおよびペプチドの添加は、より高い温度、すなわち37℃における、さらに大きな熱安定性をもたらす。
従って、本発明の細胞株は、以下のことが可能な細胞系、すなわち(1)低温でも予定した期間は増殖を続けること、(2)発現ベクターによる形質転換、(3)β2ミクログロブリンの発現、(4)一つ以上の型のクラスIMHC 分子の発現、(5)クラスI分子上へのペプチドの充填、および(6)空の、またはペプチドを充填したクラスIMHC 分子の、細胞表面上での発現、が可能であることがさらになお好ましい。
本発明の好ましい態様においては、ドロソフィラ細胞の培養を確立し、(1)ヒトのクラスIMHC 分子をコードしている一つ以上のヌクレオチド配列および、(2)ヒトβ2ミクログロブリンをコードしている少なくとも一つのヌクレオチド配列、に効果的に結合しているベクターでトランスフェクトする。これらの配列は同一のベクターに、効果的に結合することもできるが、好ましくはβ2ミクログロブリンのための配列と、ヒト MHCのための配列は、別々のベクター内にある。選択を目的とするためには、たとえばネオマイシン耐性のような選択マーカーを効果的に結合しているベクターで、細胞をトランスフェクトすることも有利である。その後、ヒトのクラスIMHC 分子およびβ2ミクログロブリンを発現している細胞を選び出し、培養中に維持する。
空の MHC、またはペプチドを充填した MHCの発現のための培養物を用意するためにはまず、たとえば CuSO4誘導のように、あらかじめ決めた時間の誘導が必要である。適当な培養時間の後、たとえば、およそ12〜48時間後、あらかじめ決めた濃度(たとえば約 100μg/ml) でペプチドを加える。ペプチドは、下文B・5章において論議したように調製することができる。さらにインキュベートした後(たとえば、27℃で約12時間)の培養物は、CD8細胞の活性化に使用する用意ができている。
この追加した培養時間は、短縮してもよいし、さもなければ省略されてもよいのだが、もしそれが休止期のCD8細胞かまたはCD8の先駆細胞の添加に先立ってインキュベートするためのものであれば、培養物は温度の挑戦に対して次第に安定していく傾向があるということが、本発明者らの観察である。たとえば、本発明による培養物でペプチドを加えてあった培養物は、たとえ37℃での培養時間を延長したとしても、有意な量の、ペプチドを充填したクラスIMHC 分子を発現することができる。
形質転換した宿主細胞の培養に有用な栄養培地は、本技術分野において周知であり、多くの販売元から入手できる。宿主細胞が哺乳類細胞である態様においては、好ましくは無血清培地を使用する。
4.ヒトβ2ミクログロブリン
治療上実用的な量の、表面に発現されたヒトのクラスIMHC 分子の産生が可能な細胞株を確立するためには、本発明の細胞株を、β2ミクログロブリンをコードしているヌクレオチド配列に操作しやすいように結合させたベクターと、同時にトランスフェクトさせて、細胞株の中でヒトのクラスIMHC 分子の発現が適切なレベルで達成されるようにすることが好ましい。マウスβ2ミクログロブリンのような哺乳類のβ2ミクログロブリンをコードしているヌクレオチド配列は、本発明の細胞株に発現される、ヒトのクラスIMHC分子の安定性を高めるが、その細胞株を、ヒトβ2ミクログロブリンをコードしている発現可能なヌクレオチド配列を、操作しやすいように結合したベクターを用いて同時にトランスフェクトすることが好ましい。
前文において、B・2章に論議したように、本発明による好ましいベクターは、発現のため効果的にベクターに結合している、哺乳類β2ミクログロブリン分子の少なくとも一部をコードしているヌクレオチド配列を含む。クラスIMHC 分子とβ2ミクログロブリンの両方をコードしているヌクレオチド配列を含んでいるベクターを構築することも可能である。
ヒトβ2ミクログロブリンのcDNAの配列は公表されており(Suggsら、PNAS 78, 6613-17, 1981参照) 、その配列を、以下のプライマーを用いたポリメラーゼ連鎖反応(PCR) に鋳型として使用することができる。
primers:
5′プライマー
5′GCTTGGATCCAGATCTACCATGTCTCGCTCCGTGGCCTTAGCTGTGCTCGCGCTACTCTC 3′
(配列番号15)
3′プライマー
5′GGATCCGGATGGTTACATGTCGCGATCCCACTTAAC 3′
(配列番号16)
プライマーを、標準的な PCR反応(前文のB・1章、および、そこに引用した文献を参照)に使用した。反応産物をフェノールで抽出し、ジーンクリーン・キット (バイオ 101, サンディエゴ, カリフォルニア州) で精製し、BamHIで消化し、pBS(ストラタジーン,ラジョラ, カリフォルニア州) のBamHI部位にクローン化する。配列を確認した後、このBamHIフラグメントを適当な発現ベクターのBamHI部位にクローン化する。好ましい態様においては、ヒトβ2ミクログロブリンcDNAを合成し、発現ベクター pRmHa−3に操作しやすいように結合する。
4.ペプチド
実質的には、ウィルス抗原に加えて、すべての細胞タンパク質を用いて、クラスIMHC の強力なリガンドとして働く、関連したペプチドフラグメントを生じることができる。ほとんどの哺乳類細胞においては、従っていかなる特定の MHCペプチド複合体も、細胞表面上に見られる MHCをコードしている分子のごく一部を表わしているにすぎない。従って、CD8細胞を特異的に活性化するための増加した能力をもつ、表面に発現されたヒトのクラスIMHC 分子を産生するためには、適当な大きさと抗原性のあるペプチドフラグメントを単離し、クラスI分子の上に充填することが好ましい。
本発明のペプチドはクラスIMHC 分子に結合する。結合は、in vitroと同様に in vivoでも作り出すことのできる生物学的条件下に起こる。ペプチドの結合の厳密な性質は、本発明の実行にとって知る必要はない。
好ましい態様においては、クラスIMHC 分子上に充填されるべきペプチドは抗原性がある。ペプチドは均一の大きさで、好ましくはオクタマーまたはノナマーから成り、最も好ましくはオクタマーである。 MHC分子上に充填するため調製したペプチドが一種類のものであること、すなわち、 MHC上に充填されるすべてのペプチドが、大きさおよび配列において同一であることも好ましい。この方法により、単一抗原性のある、ペプチドを充填した MHC分子を産生することができる。
ペプチドは種々の手段により細胞に提示することができる。好ましくは、ペプチドを、細胞内のペプチドのプールにそれらを取り込ませることのできる方法により提示することができる。たとえば、浸透圧的充填によりペプチドを提示することができる。典型的には、ペプチドは培養液に加える。培養物に加えるペプチドはそのままのペプチドまたはタンパク質の形で加えることができるが、その後細胞内の作用、たとえば酵素的分解により分解される。そのかわりとして、細胞培養物に加える前に、化学的分解(たとえば臭化シアン分解)またはプロテアーゼ(たとえばキモトリプシン)のような、他の何らかの方法で、そのままのポリペプチドまたはタンパク質を分解することもできる。他の態様においては、ペプチドを、エピトープとなるアミノ酸配列を含んでもよいし含まなくてもよい、より小さなセグメントとして提示する。
好ましい態様においては、十分な量のタンパク質(または複数のタンパク質)またはペプチド(または複数のペプチド)を細胞の培養物に加えて、クラスIMHC 分子に結合させ、続いて高濃度のペプチド(好ましくは各々の MHCには同じ種類のペプチドが結合する)を、本発明のヒトのクラスIMHC を発現している細胞の表面上に提示する。細胞内でペプチドを MHC分子に提示する前に、ヒトのクラスIMHC とヒトβ2ミクログロブリンを結合させて、すなわちヘテロダイマーを形成することも好ましい。
本発明の他の態様においては、本発明の形質転換細胞にペプチドを加えて、細胞によって発現されている MHC分子の熱安定性を高めるようにした。上述のように、ペプチドは好ましくは培養液に加える。クラスI分子に結合する抗原性ペプチドは、 MHC分子の熱安定性を保つために役立ち、また細胞表面での発現を増加させる。 MHC分子に結合するペプチドを加えた培養物は、従って、ペプチドを添加しない培養物より、温度の挑戦に対して有意に低い感受性を示した。
本発明の一つの態様においては、抗原ペプチドを、形質転換した、またはトランスフェクトした細胞株に対し、種々の形で提示する。たとえば、完全なタンパク質または他の抗原ポリペプチドを、たとえば化学的または酵素的に分解し、この形で細胞に加えることができる。たとえば、興味の対象となるタンパク質をキモトリプシンで分解し、結果として得られるペプチドの「フラグメント」の混合物を、形質転換した、またはトランスフェクトした細胞の培養物に加える。
これらの細胞は、そこで、適当なペプチド(しばしば、より小さなペプチドであり、好ましくは、オクタマーまたはノナマーから成る)を「選び」、クラスIMHC 分子の上に充填することができる。そのかわりとして、タンパク質またはポリペプチドの全配列を適当なベクターにクローン化して原核細胞に挿入し、それにより、細胞は有意な量の抗原ポリペプチドを産生し、それを集め、精製し、ペプチドに消化し、そこで形質転換したまたはトランスフェクトした原核細胞の培養物に加えることができ、細胞は発現された MHCの上に充填すべきペプチドを、ここでもまた「選ぶ」ことができる。
6.休止しているCD8細胞または、CD8先駆細胞
休止しているCD8細胞(またはCD8先駆細胞)すなわち、標的となる特異抗原に対してまだ活性化されていないT細胞は、好ましくはCD8細胞の、本発明の形質転換した培養物とのインキュベートに先立ち、患者から抽出する。患者からのCD8先駆細胞の採集は、CD8細胞の特異的に活性化されるべき能力を妨げる可能性のある他の処置または治療の開始に先立って行うことが好ましい。たとえば、もし新生物または腫瘍のある個体を治療しようとするのであれば、化学療法および放射線処理の開始に先立って細胞試料を入手し、それを培養することが好ましい。
リンパ球の抽出法および培養法は周知である。たとえば、Rosen-bergの、米国特許No.4,690,915は、リンホサイトフェレシス (lyn-phocyto pheresis) により多数のリンパ球を得る方法を記載している。使用された適切な培養条件は哺乳類細胞のためのものであり、典型的には37℃で行なった。
CD8先駆細胞の分離および(または)その培養物の濃縮にはまた、種々な方法を用いることができる。細胞の分離のための一般的な方法の例は、特異的にコートした表面への、細胞の直接的な結合を含む。他の例では、CD8細胞を含んでいる、ヒト末梢血リンパ球(PBL)を、フィコール・ハイパック(Ficoll-Hypaque) 勾配遠心法により単離した。 PBLリンパ芽球は、その後ただちに使用してもよいが、 FBSを含む10% DMSO(シグマ・ケミカル社 モンタナ州, セントルイス) 中で凍結した後、液体窒素中に保存することもでき、それにより細胞の生存率およびリンパ球としての機能を保つことができる。
先駆細胞の分離および (または)培養物の濃縮についてのその他の方法は、以下の例を含む。全血からリンパ球に富む PBL集団を調製した後、そこからCD8リンパ球の亜集団を、CD8受容体抗原の存在に向けた、親和性(アフィニティー)に基づく分離技術により単離する。これらのアフィニティーに基づく技法は、蛍光で活性化した細胞選別法 (FACS) を含む、フローサイトメトリー、細胞接着および類似の方法を含む。(Scher, Mage共著、「基礎免疫学」(Funda-mental Immunology)W.E.Paul編、P.767-780, リバープレス, ニューヨーク (1984) などを参照のこと。)
アフィニティー法は、アフィニティー試薬の源として、抗CD8受容体抗体を使用することができる。そのかわりとして、CD8受容体の天然のリガンドまたはリガンド類似体を、アフィニティー試薬として用いることもできる。これらの方法に用いるための様々な抗T細胞モノクローナル抗体、および抗CD8モノクローナル抗体は、一般的にアメリカン・タイプ・カルチャー・コレクション(ロックビル,メリーランド州)、およびファルマシア(サンディエゴ,カリフォルニア州)を含めた種々の販売元から入手することができる。抗原の選択により、適する抗体が異なることがある。(T細胞を含めたヒト白血球の命名法、抗原の名称、および、あてがわれる抗体についての議論および総説は、Knapp らの「現代免疫学」(Immunology Today 10, 253-258 (1989)を参照のこと。)
たとえば、モノクローナル抗体OKT4 (抗−CD4,ATCC No.CRL 8002) 、OKT5 (ATCC No.CRL 8013および8016) 、OKT8 (抗CD8, ATCC No.CRL 8014) 、およびOKT9 (ATCC No.CRL 8021)は、「細胞株およびハイブリドーマのATCCカタログ」(ATCC Catalo-gue of Cell Lines and Hybridomas (ATCC, ロックビル, メリーランド州) には、ヒトTリンパ球、ヒトT細胞サブセット、および活性化T細胞のそれぞれに反応性があると定義されている。T細胞種の同定および単離のための、他の様々な抗体を入手することができる。
好ましくは、 PBLを次いで精製する。たとえば、フィコール勾配をこの目的のために利用することができる。精製した PBLを次いで、あらかじめ適当な抗原ペプチドとインキュベートしておいた同系のドロソフィラ細胞と混合する。
7.CD8細胞のin vitroでの活性化
特異的な細胞障害性T細胞の産生のためのin vitroでの条件を最適化するために、刺激細胞の培養を、ある適した培地に維持した。好ましい態様においては、刺激細胞はドロソフィラ細胞であり、それらは無血清培地(たとえば、エックスセ(Excell) 400) に維持することが好ましい。
刺激細胞を、活性化されるべき細胞、たとえばCD8先駆細胞と共にインキュベートするに先立ち、ヒトのクラスIMHC 分子上に充填し、刺激細胞の表面上に発現するに十分な量の抗原ペプチドを、刺激細胞の培養物に加える。本発明によれば、ペプチドの十分な量とは、約 200、および好ましくは 200以上のヒトのクラスIMHC 分子に、ペプチドを充填して、個々の刺激細胞の表面上に発現させることのできる量である。好ましくは、刺激細胞を、20μg/mlより高濃度のペプチドとインキュベートする。
休止しているCD8細胞、またはCD8先駆細胞は、次いで適当な量の刺激細胞と共に、CD8細胞を活性化するに十分な期間、培養によりインキュベートする。好ましくは、CD8細胞は、抗原特異的方法で活性化する。刺激細胞に対する休止しているCD細胞、またはCD8先駆細胞(エフェクター)の割合は、個々に異なり、さらにはある個体のもつリンパ球の、培養条件に対する順応性、および、本文中に記載した治療様式が使用される、疾病の状態またはその他の状態の、性質およびきびしさのような変数に依存している。しかしながら、好ましくは、リンパ球対刺激細胞(たとえばドロソフィラ細胞)の比は、およそ30:1〜 300:1の範囲である。たとえば、一つの態様においては、3×107 個のヒト PBLと1×106 個のドロソフィラ細胞を混合し、 RPMI 1640培地20ml中に維持した。
エフェクター/刺激細胞培養は、治療上実用的な、または効果的な数のCD8細胞を刺激するために必要な期間維持することができる。一般的な条件下においては、最適期間はおよそ1日と5日の間であり、「プラトー」、すなわち特異的CD8活性化レベルの「最大値」は、一般的には培養5日後に見られる。本発明の一つの態様においては、in vitroでのCD8細胞の活性化は、細胞株の感染の後、短い期間内に検出される。一つの態様においては、CD8細胞の活性化が可能なトランスフェクトした細胞においての、一時的な発現を、トランスフェクト後48時間以内に検出できた。このことは、ヒトのクラスIMHC 分子を発現している形質転換細胞の安定な、または一時的な培養が、CD8細胞の活性化に効果的であることを明らかに示している。
8.CD8細胞の、ドロソフィラ細胞からの分離
活性化したCD細胞は、刺激を与える(たとえばドロソフィラ)細胞から、周知の方法の一種を用いて効果的に分離することができる。たとえば、刺激細胞に対して、または刺激細胞上に充填されているペプチドに対して、またはCD8細胞(またはそれらの断片)に対して特異的なモノクローナル抗体を用いて、これらの適切な相補性のあるリガンドを結合することができる。抗体をつけた分子は、適当な方法、たとえば周知の免疫沈降または、免疫検定法により、刺激細胞−エフェクター細胞混合物から抽出することができる。
9.活性化したCD8細胞の投与
活性化したCD8細胞の、効果的な細胞障害性のある量は、これらのキラー細胞の最終的な標的である細胞の量およびタイプによって変わるように、in vitroと in vivoでの使用によっても変わり得る。その量は、患者の状態によっても変わり、従事者により、すべての適切な因子の考慮を経て決定すべきである。しかしながら、マウスでは約5×106 〜5×107 個の細胞を用いるのに対し、成人では好ましくは約1×106 〜約1×1012個を、さらに好ましくは約1×108 〜約1×1011個を、さらになお好ましくは、約1×109 〜1×1010個の活性化したCD8細胞を利用する。
前文に論議したように、好ましくは治療を受ける個体への投与に先立ち、活性化したCD8細胞を、ドロソフィラ細胞の培養物から回収する。しかしながら、今日存在する、および提案されている他の方法と異なり、本発明の方法が腫瘍原性のない細胞培養系(すなわちドロソフィラ細胞)を用いていることに注目することは重要である。従って、もしドロソフィラ細胞と、活性化したCD8細胞との完全な分離が行われないとしても、少数のドロソフィラ細胞の投与に伴う固有の危険は、哺乳類の、発がんを促進する細胞の投与が極めて危険であるのに対し、何も知られていない。
細胞成分の再導入の方法は、本技術分野では周知であり、Honsikらによる米国特許No.4,844,893ならびに Rosenbergによる米国特許No.4,690,915に例示されている方法を含む。たとえば、CD8細胞の静脈内注入による投与が適している。
10. HLAのタイピング
先に述べたように、 HLAのハプロタイプ/アロタイプは個体ごとに異なり、本発明の実施に対しては本質的なことではないが、個体の HLAを決定することは、しばしば有用である。 HLAタイプは、標準的なタイピング方により、フィコール勾配で精製した PBLを用いて決定することができる。精製した PBLを、適当な抗原ペプチド(たとえば、ウィルス感染、がん、または悪性腫瘍に関する治療用の適用には、ウィルスに特異的または、がんに特異的なタンパク質に由来するペプチド)と共にあらかじめインキュベートしておいた同系のドロソフィラ細胞と混ぜる。
特定のウィルスまたはがんに特異的な抗原の性質が明らかになっているような場合には、例のようなウィルスの、または悪性な条件を使い続けるには、これらのエピトープをコードしている合成したペプチドを使うことが好ましい。好ましい抗原ペプチドが正確に決定されていない場合には、ウィルスまたはがんに特異的なタンパク質のプロテアーゼ分解物を使うことができる。それらの抗原の源として、ウィルスまたはがんに特異的なタンパク質をコードしているcDNAを、たとえばこの文章の中に開示した方法により、細菌の発現ベクターにクローン化し、細菌を形質転換することができる。
HLA のタイピングの後、もし好ましい HLAを発現しているドロソフィラ細胞が得られない場合には、ポリメラーゼ連鎖反応により、好ましい HLAをコードしているcDNAをクローン化することができる。前文において、B・1章に開示したプライマー(SEQ ID NO 1からSEQ ID NO 12まで) を用いて適切な HLA−A,−B,−C,−E,−F、またはGのcDNAを、別々の反応において増幅することができ、下文のA2・1において、 HLAについて開示した方法で、クローン化し、配列を決定することができる。クローン化した HLAを発現している安定な細胞株をここで、ドロソフィラ細胞において確立することができる。かわりの方法として、 PCR反応からのクローン化した組換え体の大部分の集団を一時的に発現している昆虫細胞の集団を、invitro でのCD8の活性化に用いることができる。
11.ドロソフィラの分裂促進因子
本発明に従って培養した細胞の上清が、特異的な活性化されたCD8細胞を産生するという、細胞株の能力を増進または回復させることが今わかった。約10%のドロソフィラの培養物上清を、リンパ球の培養物、または固定した細胞に加えるとCD8を効率よく活性化する。同系クラスI抗原を発現しており、初代培養では、CD8を活性化することができなかった培養細胞は、ドロソフィラ細胞の培養物上清を加えた後、特異的なCD8を産生することができた。
ドロソフィラの培養物から単離される因子であり、ドロソフィラの細胞培養において、CD8の活性化に重要なインパクトを持つと思われる因子は、ドロソフィラの分裂促進因子である。分裂促進因子は、安定性の高い、可溶性な分泌物質として特徴づけられ、4℃で4か月保存した後でもその活性を維持している。分裂促進因子は比較的大きな分子量、すなわちスペロース6(ファルマシア,ピツカタウェイ,ニュージャージー州)ゲルろ過法によれば500kDaを有する。それはまたモノQセファロース(ファルマシア,ピツカタウェイ,ニュージャージー州)に強く結合する。ドロソフィラ細胞の分裂促進因子は、明らかにB細胞の増殖を誘導し、マクロファージを活性化する。
ドロソフィラ中に発見された分裂促進因子は、他の昆虫の分裂促進因子の代表的なものであって、効果的な治療用アジュバントを供する可能性がある。たとえば、感染した動物から細胞をとり、それを用いてドロソフィラ細胞を刺激し、続いて血中リンパ球の初代培養物、およびドロソフィラの分裂促進因子と混合する。次いで、この混合物をその動物の治療に用いることができる。
下記の実施例によって、本発明をさらに説明する。これらの実施例は本発明を限定しない。
実施例1.ヒトクラスI MHC 分子
A. pRmHa−3発現ベクターの調製
SphI線状化 pRmHa−1DNA 発現ベクターを後述するように pRmHa−2発現ベクターの SphI制限消化から生ずるDNA 断片と結合することによって、本発明において記載するドロソフィラ・シュネイダー(Drosophila Schneider)2細胞中でMHC タンパク質を発現するとき使用する pRmHa−3発現ベクターを構成した。この方法における pRmHa−1と pRmHa−2断片との結合を実施して、 pRmHa−1の中に存在する2つのEcoRI制限エンドヌクレアーゼクローニング部位の1つを除去した。こうして、生ずる pRmHa−3発現ベクターは、種々のMHC をコードするDNA 断片が実施例に記載するように挿入された多数のクローニング部位(ポリリンカー)の中にただ1つのEcoRI制限部位を含有した。
1. pRmHa−1発現ベクターの調製
pRmHa−1発現ベクターは、メタロチオネインプロモーター、金属応答コンセンサス配列(MTと表示する)およびドロソフィラ・メラノガスター(Drosophila melanogaster)から単離されたポリアデニル化シグナルを含有するアルコールデヒドロゲナーゼ(ADH)を含有し、Bunch ら、Nucleic Acids Res.16:1043-61 (1988)に記載されているようにして構成した。最終の pRmHa−1構成体の概略を第1B図に示す。ATCC受け入れ番号 37253を有するプラスミド発現ベクターpUC18 を源ベクターとして使用し、これから引き続いてここに記載するベクターを誘導した。
pUC18 プラスミドは多数のクローニング部位の中に5′から3′に次の制限部位を含有し、それらのすべては第1図にpUC18 誘導ベクターの概略的表示の中に示されていない:EcoRI; SacI; KpnI;同一位置に位置する SmaIおよび SmaI;BamHI; XbaI; SalI;同一位置に位置する AccIおよびHincII; PstI; SphIおよびHindIII 。 pUC18ベクターをまずHindIII で消化して線状化pUC18 を形成した。次いで、Maniatisら、Molecular Cloning :A Laboratory Manual 、編、Cold Spring Harbor Laboratory 、ニューヨーク(1082)に記載するように、HindIII 末端にDNA ポリメラーゼIの大きい断片をフィルインすることによって、平滑末端をつくった。
生ずる線状化平滑末端pUC18 ベクターを、ポリアデニル化シグナルを含有するドロソフィラ・メラノガスター(Drosophila melanogaster)ADH 遺伝子からの 740塩基対(bp)のHinfI断片と結合した。プラスミドpSACIから、まず、Goldbergら、PNAS USA 77:5794-5798 (1980)に記載されているように、HinfI断片で消化し、次いでクレノーで平滑末端として配列識別番号14に記載するヌクレオチド配列を生成することによって、結合したADH アレレを単離した。ランダムの高分子量の(15kbより大きい)を含有するバクテリオファージラムダのライブラリーから選択したドロソフィラ(Drosophila)DNA の 4.7キロ塩基(kb)のEcoRI断片を、pBR322(ATCC受け入れ番号31344)の中にサブクローニングすることによって、ADH アレレを含有するpSACIベクターを構成した。
Kreitman,Nature 304:412-417 (1983)に記載されているように、5′HinfI制限部位はADH 遺伝子の中の1770位置に自然に見出された。3′HinfI部位は、ADH 遺伝子がクローニングされているpUC18 ベクターから誘導された。この位置はADH 遺伝子バンクの位置2500における XhoI部位に対して4塩基3′であった。ADH 断片は、ADH mRNAの3′非翻訳部分中のポリアデニル化/切断配列の35bp上流からポリアデニル化シグナルの 700bp下流まで延長した。ADH 遺伝子断片を含有する生ずるpUC18 誘導ベクターを、第1A図に示すように pHA−1と表示する。
421bp のEcoRI/ StuIMT遺伝子断片は、ドロソフィラ・メラノガスター(Drosophila melanogaster )ゲノムDNA ライブラリーの中においてほぼ15.3kbのDNA を含有するクローンから得られた。成虫DNA の MboI部分的消化で調製されたライブラリーをラムダ誘導EMBL4の中でクローニングした。この断片はMTプロモーターおよびドロソフィラ(Drosophila)MT遺伝子の金属応答コンセンサス要素を含有した(Maroniら、Genetics 112:493-504 (1986))。この領域は、プロモーターおよびヌクレオチド1+に転写開始部位を含有し、MT遺伝子の位置− 370からヌクレオチド位置+54に相当した(配列識別番号13)。
次いで、生ずる断片を、前以てEcoRIおよび SmaIで線状化した上で調製した pHA−1の中に結合した。 StuI消化によりつくられたMT中の3′平滑末端は、 SmaI消化によりつくられたpHA −1中の平滑末端と適合した。5′ドロソフィラ(Drosophila)MT遺伝子および3′ADH 遺伝子断片を含有する生ずるpUC18 誘導ベクターを pRmHa−1と表示した。 pRmHa−1発現ベクターは、第1B図に示されており、複製起点(ori)およびpUC18からのアンピシリンに対する耐性(Ampr ) を与えるβ−ラクタマーゼ遺伝子を第1A図に示すように pHa−1ベクター上に含有した。 pRmHa−1の線図は、また、MT遺伝子断片の5′−3′隣接位置、多数のクローニング部位およびADH 遺伝子断片を示す。 pRmHa−1ベクターは、下のC.に記載するように、 pRmHa−3発現ベクターの構成において使用した。
2. pRmHa−2発現ベクターの調製
pRmHa−2の構成を第1A図に示す。 pRmHa−2発現ベクターを構成するために、 pRmHa−1の構成について前述したようにわずかの変更を加えて、上で調製したMT断片をpUC18 誘導ベクター pHA−1の中に挿入した。上で調製したEcoRI。 StuI単離MT遺伝子断片の StuI部位にEcoRIリンカーを加えて、両末端にEcoRI制限部位を有するメタロチオネイン断片を形成した。次いで前以てEcoRIで線状化したADH 断片含有pUC18 発現ベクターの中に、生ずる断片を結合した。
生ずるpUC18 誘導ベクターは、5′ドロソフィラ(Drosophila)MT遺伝子断片および多数のクローニング部位に対して5′に2つのEcoRI制限部位を有する3′ADH 遺伝子断片を含有し、 pRmHa−2と表示する。 pRmHa−2発現ベクターは、第1A図に示されており、複製起点(ori)およびpUC18 からのアンピシリンに対する耐性(Ampr )を与えるβ−ラクタマーゼ遺伝子を含有した。 pRmHa−2の線図は、また、MT遺伝子断片の5′−3′隣接位置、多数のクローニング部位およびADH 遺伝子断片を示す。 pRmHa−2ベクターは、下のC.に記載するように、 pRmHa−3発現ベクターの構成において pRmHa−1と一緒に使用した。
3. pRmHa−3発現ベクターの調製
ただ1つのEcoRI制限部位を有する pRmHa−3発現ベクターを調製するために、 pRmHa−2からの断片を pRmHa−1の中に結合した。この構成のために、上のb.において調製した pRmHa−2をまず SphIで消化した。生ずる SphI断片はMT遺伝子の中央において開始しそして多数のクローニング部位中の SphI部位に延長し、これをまず pRmHa−2ベクターから単離し、次いで上のA.1.において調製した pRmHa−1の中に結合した。 pRmHa−1ベクターを前以て修飾してMT遺伝子断片に対して5′のEcoRI制限部位を除去し、次いで SphIで線状化した。この方法は第1B図に概略的に示されている。 pRmHa−1中のEcoRI部位を除去するために、このベクターをまずEcoRIで消化して線状化ベクターを形成し、次いでヤエナリヌクレアーゼで平滑末端とし、そして再結合した。
次いでEcoRI部位を欠如する pRmHa−1ベクターを SphIで消化して、 pRmHa−2から SphI断片のインサートに相当する領域を除去し、そして線状化 pRmHa−1ベクターを形成する。次いで pRmHa−2からの SphI断片を SphI線状化 pRmHa−1の中に結合して、 pRmHa−3発現ベクターを形成した。 pRmHa−3ベクターの概略は第1C図に示されている。 pRmHa−3を誘導したpUC18 ベクターからの種々の制限部位の相対位置は、この図面上に示されている。
さらに、問題のMHC 遺伝子をその中にクローニングした多数のクローニング部位(ポリリンカー)により分離されたMTおよびADH 遺伝子断片の相対位置および長さは、この図面上に示されている。pUC18から誘導された pRmHa−3ベクターは、pUC18 複製起点およびアンピシリン耐性を与えるβ−ラクタマーゼ遺伝子を含有する。こうして、本発明において調製されそして pRmHa−3の多数のクローニング部位の中にクローニングされたMHC をコードするDNA 断片は、MTプロモーターにより転写的に調節され、そしてこれをADH 遺伝子を介してポリアデニル化した。
B.cDNAの合成
HLA A2.2 を合成するために、完全なA2.2 をコードするcDNA(参照、Holmesら、J.Immunol.139 :936-41(1987)、発表された配列について)を、M13mp19 プラスミド、すなわち、商業的に入手可能なバクテリオファージのベクター(ストラタジーン(Stratagene)、 ラジョラ、カリフォルニア州)の中にクローニングする。A2の発表された配列から誘導されたプライマーを使用するPCR により、cDNAを合成する。cDNAをM13mp19 クローンから NotI(クレノーでフィリングされたオーバーハング)/EcoRI断片として解放する。(クレノー断片は、大腸菌(E.coli)DNA polIをスブチリシンで処理して生産された、大腸菌(E.coli)DNA ポリメラーゼI分子の一部分である。
それらを使用して、制限ヌクレアーゼにより生産されたDNA 分子の末端において5′または3′のオーバーハングを「フィル・アウト(fill out)」する。)BgIII (末端がクレノーでフィリングされている)およびEcoRIで消化されたpSP64Tの中に、 NotI/EcoRI断片を挿入する。pSP64Tは、それ自身の開始コドンを含有するcDNAに効率よく翻訳されるmRNA(β−ブロビン)から5′および3′フランキング領域を提供するようにデザインされたSP6クローニングベクターである。この翻訳SP6 ベクターは、 pSP64−X βm を BalIおよびBstEIIで消化し、互い違い末端をT4DNA ポリメラーゼでフィルインし、そして結合により BglIIリンカーを加えることによって構成された。 BalIはβ−グロビンcDNAをATG (開始コドン)の2塩基上流で切断し、そしてBstEIIはTAA (停止コドン)の8塩基上流で切断する。
pSP64Tの中にただ1つの BglIIが存在するので、ポリリンカー断片において、 PatIからEcoRIまでを切断する制限酵素をなお使用して転写のためにプラスミドを線状化することができる。(参照、Kreig およびMelton,Nucleic Acids Res.12:7057-7070,(1984)、これは、また、プラスミドpSP64 −X βm の構成を記載している)。生ずるプラスミドをEcoRI(クレノーで末端をフィリングした)およびHindIII で切断し、pCMUIIの中にHindIII (5′)と StuI(3′)との間においてクローニングする。(参照、Paabo ら、EMBO J.5:1921-1927 (1986)。)全体のcDNAをHindIII (クレノーで末端をフィリングした)BamHI断片として除去し、 SmaIおよびBamHIで切断した pRmHa−3の中にクローニングする。
トランスメンブレンドメインの直前に前述のA2.2 cDNAの中に停止コドンを操作することによって、HLA A2.2 を調製した。真核生物の発現ベクターpCMUIIの中のHindIII 5′と StuI3′(上を参照)との間にクローニングしたA2.2 cDNAを MboIIおよびBamHIで切断し、次のオリゴヌクレオチドを挿入することによって突然変異を達成する:
5′プライマー: 5′GGAGCCGTGACTGACTGAG 3 ′
(配列識別番号17)
3′プライマー: 5′CCCTCGGCACTGACTGACTCCTAG 3′
(配列識別番号18)
生ずる組換えプラスミドをHindIII で切断し、オーバーハング末端をクレノーでフィリングし、次いでBamHIで切断して制限断片を解放し、これをA2.2 全長と同一方法で pRmHa−3の中にクローニングする。
HLA A2.1 cDNAを次のようにして調製する。A2.1 の発表された配列(KollerおよびOrr ,J.Immunol.134 :2727-2733 (1985)) から誘導された次のプライマーおよびNilsson ら、Cell 58 :707-718(1989)に記載された反応条件を使用するPCR により、切頭HLAA2.1 cDNA配列を合成する。
5′プライマー: 5′GCGGATCCATGGCCGTCATGGCGCCC 3′
(配列識別番号19)
3′プライマー: 5′CGGAATTCTCATCAGGGCTTCGGCAGCCC 3 ′
(配列識別番号20)
生ずるPCR 断片をpBS (ストラタジーン(Stratagene)、ラジョラ、カリフォルニア州)の中にクローニングし、そして配列をジデオキシ配列決定により評価する。HLA A2.1 のコーディング配列の大部分をコードする 800bpの断片をこのプラスミドから AvaIおよび StuIで切除し、そして前以て pRmHa−3の中にクローニングされたHLA A2.2 トランスメンブレン配列において同一断片と置換するために使用する(上を参照)。
HLA B7をB7の発表された配列(参照、例えば、Zemmour およびParham,Immunogenetics 33 :310-320 (1991)) から誘導されたプライマーを使用するPCR により合成し、BamHI部位によりフランキングし、 pRmHa−3のBamHI部位の中に直接クローニングする。(参照、Soodら、Immunogenetics 22 :101-121 (1988))。
B27の発表された配列(参照、例えば、Zemmour およびParham、Immunogenetics 33 :310-320 (1991)) から誘導されたプライマーを使用するPCR により、HLA B27 cDNAを合成する。それ以上の詳細はすぐ下に記載されている。
HLA B27 sol cDNAを上のB27 cDNAのように調製するが、クローンpB1 のアルファ3ドメインの末端に停止コドンを組み込む(参照、Szotz ら、PNAS 83 :1428(1986))。切頭および全長の両者のB27cDNAは、5′末端を修飾したベクターpDS5において得られる(参照、Stueber ら、EMBO J.3:3143-3148) (1986))。部位特異的突然変異誘発を実施してcDNAの5′末端を伸長する。次の配列(大文字)を発表されたB27 cDNAクローンに付加する;スラッシュマーク(/)後の配列は発表された配列の開始である:
GGATCCTCTCAGACGCCGAGATGCGGGTC /acggcgccc...
(配列識別番号21)
完全なB27 およびB27 sol cDNAをpD55から、まずApaLI(クレノーで末端をフィリングした)およびBamHIで切断することによって切り出す。生ずる断片を SalI(末端をクレノーでフィリングした)およびBamHIで切断した pRmHa−3の中に方向的にクローニングする。
任意の好ましいHLA をコードするcDNAをポリメラーゼ連鎖反応によりクローニングすることができる。上の節B.1.の開示プライマー(配列識別番号1〜配列識別番号12)を使用して適当なHLA−A,−B,−C,−E,−F、または−G cDNAを別々の反応において増幅することができ、次いでこれらを上にHLA A2.1 について開示した方法に記載されているようにクローニングし、そして配列決定することができる。ヒト細胞からのcDNAの調製は、Ennis ら、PNAS USA 87 :2833-2837 (1990)に開示されているように実施する。簡単に述べると、血液試料を個体から獲得し、そして細胞を遠心後に集め、そして全体のRNA の調製に使用する。第1鎖cDNAをオリゴ(dT)および鳥類骨髄芽球ウイルス逆転写酵素使用することによって合成する。
生ずるcDNAを、上の節B.1.において記載するように1または2以上の適当なプライマーおよびジーンアンプ(GeneAmp)キットおよびサーマルサイクラー(パーキン−エルマー/セツス(Perkin−Elmer /Cetus ))を使用するPCR 増幅反応において使用する。反応条件は好ましくは次の通りである。100ng のcDNA鋳型および50ピコモルの各オリゴヌクレオチドのプライマーを使用する。30サイクルを次のようにして実施する:(a)94℃において1分;(b)60℃において1分;および(c)72℃において1分30秒。次いでPCR 反応を100 ℃に10分間加熱してTaq ポリメラーゼを殺し、そしてDNA の末端をT4ポリメラーゼ(ストラタジーン(Straragene)、 カリフォルニア州サンディエゴ)により平滑末端とする。
ヒトβ2ミクログロブリンcDNAを発表された部分的cDNA水性(参照、Suggs ら、PNAS 78 :6613-17, 1981)を使用して調製し、次のプライマーを使用するポリメラーゼ連鎖反応(PCR)のための鋳型として使用する:
5′プライマー:
5′GCTTGGATCCAGATCTACCATGTCTCGCTCCGTGGCCTTAGCTGTGCTCGCGCTACTCTC 3 ′
(配列識別番号15)
3′プライマー:
5′GGATCCGGATGGTTACATGTCGCGATCCCACTTAAC 3′
(配列識別番号16)
プライマーを標準のPCR 反応において使用する(参照、Nilssonら、Cell 58 :707 (1989)) 。反応生成物をフェノールで抽出し、ジーンクリーン(Geneclean)キット(Bio 101 、カリフォルニア州サンディエゴ)で精製し、BamHIで消化し、そしてpBS のBamHI部位(ストラタジーン(Stratagene)、カリフォルニア州ラジョラ)の中にクローニングする。配列の評価後、このBamHI断片をpRmHa−3のBamHI部位の中にクローニングする。
実施例に記載するように、ネズミクラスIcDNAを種々の場合において使用した。ネズミクラスIcDNAを次のようにして調製した。
H−2Kb :完全なKb 分子をコードするcDNAを次のようにして構成した発現プラスミドpCMU/Kb から得る。リーダー配列およびアルファIドメインの大部分を欠如する部分的H−2Kb cDNAを、Reyes ら、PNAS 79 :3270-74(1982)の方法に従い調製し、pH202 を生成する。このcDNAを使用して全長の分子を発生する。Not I部位によりフランキングされた5′プライマーを使用するPCR反応において鋳型としてH−2Kb をコードするゲノムのクローン(Caligan ら、Nature 291:35-39, 1981)を使用して、次いでリーダー配列の最後の7アミノ酸をコードする21ヌクレオチドおよびアルファIドメインの開始に対して相補的な18ヌクレオチドおよび StyI部位を含む領域に対して相補的な3′プライマーを使用して、前記欠如した配列を得る。生ずる断片をpH202 と StyI部位において結合する。シグナル配列の残部をコードする5′配列を、Db cDNA(下を参照)からBamHI/ NotI断片として得られる。全体のコーディング配列を発現プラスミドからBamHI断片として切断し、そしてBamHIで切断したpRmHa-3 の中にクローニングする。
H−2Ld :完全なLd 分子をコードするcDNAを発現プラスミドpCMUIV/Ld から得る(参照、JolyおよびOldstone, Gene 97:213,1991)。完全なcDNAを真核生物の発現ベクターpCMUIV/Ld からBamHI断片として切断し、そしてKb として pRmHa−3の中にクローニングする。
H−2Db :完全なDb 分子をコードするcDNAをpCMUIV/Db から得る(参照、JolyおよびOldstone, Science 253 :1283-85,1991) 。完全なcDNAを真核生物の発現ベクターpCMUIV/Db からBamHI断片として切断し、そしてKb として pRmHa−3の中にクローニングする。
ネズミβ2ミクログロブリン:全長のネズミβ2ミクログロブリンcDNAをHindIII (5′)(クレノーでフィリングした)/ BglII(3′)断片を pSV2neo (ATCC No.37149)ネズミβ2ミクログロブリンcDNAから形成し、そして SmaIおよびBamHIで切断した pRmHa−3の中にクローニングする。
前述したように、pCMUベクター(pCMUIV) を真核生物の発現ベクターpC8IG から Nilssonら、前掲、に記載されているように誘導する。引き続いて、ベクターpC81G をpA81G (Paabo ら、Cell 33 :445-453 (1983))から、Paabo ら、EMBO J.5:1921-7(1986)に開示されている方法に従い誘導する。簡単に述べると、これらのベクターを次のようにして構成した。
ベクターpA81G の220bp の長さのリーダー配列を、 DdeI断片に対して80bpのHindIII を欠失することによって短縮する(Herisse ら、NAR 8 :2173-2192 (1980)により発表された配列のヌクレオチド1286-1366);この構成体をpB81G と呼ぶ。転写開始の部位に対してそれぞれヌクレオチド-112から+19までの領域にわたる、アルファーグロブリン遺伝子のHinfI断片を、 pUC9の PstIの部位の中にサブクローニングし、SV40の72bpの反復エンハンサーをプロモーターから上流に挿入した。
このプロモーター−エンハンサー要素をまず SV40T−抗原のコーディング配列の前でクローニングすることによって試験し、そして生ずる構成体をHeLa細胞(ATCC CCL 185)の中にトランスフェクションした。細胞を2日後に固定し、そしてT−抗原について間接的免疫蛍光により染色する。多数の染色された核が見出され、アルファ−グロビン断片の強いプロモーター活性を示す。次いでベクターpB81G 中のSV40プロモーターをこの効率よいプロモーター要素で置換して構成体pB81G を得た。
pB81G をHindIII およびBamHIで消化し、そしてキセノプス・ラエビス(Xnenpus laevis)β−グロビンcDNAの5′非翻訳配列をコードするオリゴヌクレオチドを挿入することによってpCMUIVを構成し、pSP64Tの発生に使用した(Krieg ら、NAR 12:7057-7070(1984)) 。しかしながら、BamHI部位をpSP64Tにおいて使用した BglII部位の代わりに選択した。pC81G のHindIII とBamHIとの間に挿入された配列は、次の通りであった:
5′AGCTTGAGCACTTGTTCTTTTTGCAGAAGCTCAGAATAAACGCTCAACTTTG 3′
(配列識別番号22)
ベクターphshsneoはネオマイシン(G418 )耐性を与え、そして追加の熱ショックプロモーター(hs)配列をもつphsneoの誘導体(pUCChsneo)であり、これはSteller ら、EMBO J.4:167(1985)に記載されているように商業的に入手可能な pUC8から合成できる。これらのベクターの中に含有される熱ショックプロモーターはhsp70プロモーターである。ネオマイシン耐性(G418 耐性)を与える他の有用なベクターは、コスミドsmart 2(ATCC 37588)(これはドロソフィラ(Drosophila)hsp70 プロモーターのコントロール下に発現される)、およびプラスミドベクターpcopneo (ATCC 37409)を包含する。
C.発現ベクターの中への遺伝子の挿入
制限生成物を1%アガロースゲルの電気泳動にかける(Maniatisら、Molecular Cloning :A Laboratory Manual, Cold SpringHarbor Laboratory (1982))。cDNAをコードする制限断片をゲルから切り出し、そして製造業者(バイオ(Bio) 101、カリフォルニア州サンディエゴ)の指示に従い「ジーンクリーン(Geneclean)」を使用してアガロースから精製する。ドロソフィラ(Drosophila)発現プラスミド pRmHa−3(参照、Bunch ら、Nucleic Acids Res.16:1043-61, 1988)を製造業者(ファーマシア(Pharmacia)、ニュージャージイ州ピスカタェイ)に指示に従いOne Phor All緩衝液中で適当な制限酵素で切断し、そして製造業者の文献(ベーリンガー・マンヘイム(Boehringer Mannheim)、インジアナ州インディアナポリス)に記載されているようにアルカリ性ホスファターゼで処理する。
100ng の切断しかつホスファターゼ処理した pRmHa−3ベクターを300ng のアガロースゲル精製したクラスIMHC 重鎖cDNAまたはβ2ミクログロブリンcDNAと混合し、そして製造業者の文献に記載されているようにT4DNA リガーゼおよびOne Phor all緩衝液を使用して結合する。16℃において5時間インキュベーションした後、結合混合物を使用して能力大腸菌(E.coli)JM83(Maniatisら、前掲(1982))を形質転換する。
Maniatisら、前掲に開示されている方法を使用して必要なcDNAを調製する;簡単に述べると、方法は次の通りである。アンピシリンを含有する寒天プレート上で大腸菌(E.coli)をプレートすることによって、形質転換体を選択する。アンピシリン耐性コロニーを個々に液体培養において増殖させ、そしてDNA をアルカリ性溶解ミニプレプ法を使用して調製する。ベクター中のMHC 重鎖cDNAの存在およびその向きを制限マッピングにより決定する。メタロチオネインプロモーターに関して正しい向きでcDNAcDNAをもつベクターを含有する細菌を、アルカリ性溶解法および塩化セシウム勾配精製を使用するDNA の大規模調製のために使用する。得られるDNA の量を分光光度測定的に決定する。
D.シュネイダー(Schneider)細胞のトランスフェクションおよび標識化
シュネイダー(Schneider)II細胞を、10%胎児仔ウシ血清(55℃において1時間加熱処理した)、 100単位/mlのペニシリン、 100mg/mlのストレプトマイシン、および1mM グルタミン)を補充したシュネイダー培地(ギブコ(Gibco)/BRL 、ニューヨーク州グランドアイランド)中で増殖する。(便宜上、この補充した培地を以後シュネイダー培地と呼ぶ。)細胞を27℃において増殖し、そして典型的には新しい培地中で1:17に希釈することによって7日毎に継代する。
50%シュネイダー/50%エクスセル(Excell) 401 で最初に希釈することによって、細胞を血清不含培地(100 単位/mlペニシリン、 100mg/ストレプトマイシン、および1mM グルタミン、および 500μg /ml G418 を補充したエクスセル 400または 401(JRH バイオサイエンス(Bioscience)、カンサス州レネクサ)中の増殖に変換する。1週後、細胞を10%シュネイダー培地/90%エクスセル 401の中に継代し、そして1週後 100%エクスセル 401の中に継代することができる。細胞をこの培地中で維持し、そして新しい培地中で2:17に希釈することによって7日毎に継代する。
15×106 シュネイダー細胞を106 細胞/mlの濃度で85mmのペトリ皿中でプレートアウトする。12時間後、リン酸カルシウム/DNA 沈澱物を、後述するように調製し(1ml)、細胞に滴々添加する。48時間後、上澄み液を注意して除去し、そして細胞を175cm2のフラスコに 500μg /mlジェネチシン(Geneticin)(G418)(ギブコ/BRl、ニューヨーク州グランドアイランド)を含有するシュネイダー培地中の50mlの合計の体積で移す。21日後、20mlの培養物を500 μg /mlのG418 を含有する30mlのシュネイダー培地を含有する新しいフラスコに取り出す。
10日後、フラスコに弱く付着しかつほぼ24時間の倍加時間で増殖した細胞の安定な集団が得られ、引き続いてこれらの細胞を培養し、そして前述したように選択した培地中で継代する。5〜20×106 細胞を遠心により集め、そしてそれらを1mlの細胞凍結培地(93%胎児仔ウシ血清/7%ジメチルスルホキシド)中に再懸濁させることによって、これらの細胞の凍結したアリコートを調製する。次いでアリコートを−70℃において1週間配置し、引き続いて液体窒素貯蔵に移す。
リン酸カルシウム沈澱物をPaaoら、EMBO J.5:1921-27(1986)に記載されているように調製し、ただし25μg の DNA/トランスフェクションを使用する。次のDNA の組み合わせを使用して示したトランスフェクション体を調製する:
(a)MHC クラスI重鎖単独:23μg の重鎖発現ベクター DNA+2μg のphshsneoDNA 。
(b)MHC クラスI重鎖単独+β2ミクログロブリン:11.5μg の重鎖発現ベクターDNA +11.2μg のβ2ミクログロブリン(ヒトまたはマウス)発現ベクター DNA+2μg のphshsneoDNA 。
代謝的標識化の24時間前に、細胞を1mMのCuSO4 を含有するシュネイダー培地中で3〜5×106 細胞/mlの細胞密度(10ml/85mmペトリ皿)でプレートアウトする。標識化の30分前に、培地を皿から吸引し、そして細胞を2×10mlのPBS で洗浄し、次いでメチオニンおよびシステインを含有しないグレイセズ(Craces)昆虫培地(ギブコ/BRl 、ニューヨーク州グランドアイランド、からの特別の注文)中で20分間インキュベーションし、次いで0.1mCiの 35Sトランス(Trans)標識(ニュー・イングランド・ニュークリアー(NewEngland Nuclear);デュポン(duPon)、マサチュセッツ州ボストン)を含有する1mlのこの培地中でインキュベーションする。
標識化期間後、標識化溶液を吸引し、そして細胞を直ちに氷上で氷冷 PBS/1%トリトン× 100(1ml)で溶解するか、あるいはメチオニンを含有するシュネイダー培地またはエクスセル 400培地(JRH バイオサイエンス)の存在下にチェイス期間後に溶解する。可溶性クラスI MHC分子を分析している場合、チェイス培地を集める。
次の操作のすべてはリゼイトを冷く(8℃より低く)保持して実施する。リゼイトをエッペンドルフ(Eppendorf)管の中に集め、マイクロフージ(microfuge)管中で13,000×g において15分間遠心し、 100mlのプロテインAセファローズの10%のスラリーを含有する新鮮な管に移し、そして転倒型回転装置上に2時間配置する。マイクロフージ中で15分間さらに遠心後、細胞リゼイトは分析することができる。
ネズミMHC を利用する実験において、シュネイダー2細胞を前述のネズミMHC 組換え体でCaPO4 沈澱方法に従いトランスフェクションする;各重鎖を単独で、あるいはβ2ミクログロブリンをコードするベクターとの50:50との混合物としてトランスフェクションする。ネオマイシン耐性をコードするプラスミドのphshsneoDNA を各トランスフェクションにおいて含め、こうして MHCクラス1を安定に発現した細胞の集団を選択培地(Geneticin G418−サルフェート、ギブコ/BR1、ニューヨーク州グランドアイランド)中でトランスフェクション体を増殖することによって得ることができるようにする。
E.ペプチドの発生
本発明による抗原性ペプチドは自然に見出される源から得ることができるか、あるいは既知の方法により合成することができる。ここに開示する種々の実施例において、ペプチドはアプライド・バイオシステムス(Applied Biosystems)合成装置、ABI431A(カリフォルニア州フォスターシティー)で合成し、引き続いてHPLCにより精製する。ここに開示する種々の実験において使用する抗原性ペプチドは、下に記載するものを包含する。
オバルミン(8)1 1−文字コード: SIINFEKL;
3−文字コード: serIleIleAsnPheGluLysLeu
(配列番号 23)(残基 5−12)
オバルミン(24)1 1−文字コード: EQLESIINFEKLTEWTSSNVMEER;
3−文字コード: GluGlnLeuGluSerIleIleAsnPheGluLysLeuThrGluTrpThrSerSerAsnValMetGluGluArg
(配列番号 23)
VSV NP2 1−文字コード: RGYVYQGL
3−文字コード: ArgGlyTyrValTyrGlnGlyLeu
(配列番号 24)
インフルエンザ NP2 1−文字コード: ASNENMETM
3−文字コード: AlaSerAsnGluAsnMetGluThrMet
(配列番号 25)
LCMV NP3 1−文字コード: SERPQASGVYMGNL
3−文字コード: SerGluArgProGlnAlaSerGlyValTyrMetGlyAsnLeu
(配列番号 26)
LCMV GP24 1−文字コード: DSSGVENPGGYCTK
3−文字コード: AspSerSerGlyValGluAsnProGlyGlyTyrCysThrLys
(配列番号 27)
HIV GAG A (9)5 1−文字コード: QMKDCTERQ
3−文字コード: GlnMetLysAspCysThrGluArgGln
(配列番号 28)
HIV GAG B (9)5 1−文字コード: KRWIILGLN
3−文字コード: LysArgTrpIleIleLeuGlyLeuAsn
(配列番号 29)
HIV Vpr (9)5 1−文字コード: FRIGCRHSR
3−文字コード: PheArgIleGlyCysArgHisSerArg
(配列番号 30)
HIV POL (9)5 1−文字コード: ILKEPVHGV
3−文字コード: IleLeuLysGluProValHisGlyVal
(配列番号 31)
INF マトリックス(10)6 1−文字コード: ILGFVFTLTV
3−文字コード: IleLeuGlyPheValPheThrLeuThrVal
(配列番号 32)
1 Carbone ら、J. Exp. Med. 169:603-12(1989)
2 Van Bleek ら、Nature 348:213-216 (1990)
3 Whitton ら、J.Virol. 63 :4303-10 (1989)
4 Oldstoneら、J. Exp. Med. 168:559-570 (1988).
5 Nixon およびMcMichael, AIDS 5:1049(1991); 参照
また、Nixon ら、AIDS 4:841-5 (1990).
6 Gotch ら、J. Exp. Med. 168:2045-57 (1988).
「ランダム」ペプチドの単離または合成は、また、とくに前駆体CD8細胞を刺激する可能性が最も強いペプチドを空のMHC 分子を負荷するために特定のエピトープを確認しようとするとき、適当であることがある。プロテアソーム(proteasomes)を使用する(参照、例えば、実施例2. B. 6)か、あるいはタンパク質またはポリペプチドを分解する−−例えば、キモトリプシンにより消化する−−ことによって、「ランダム」ペプチドの混合物を生成することができるか、あるいはペプチドを合成することができる。本発明の細胞系をタンパク質およびポリペプチドをヒトクラス1 MHC分子上に負荷できるより小さいペプチドに分解することができることが観察されたが、より小さいペプチド−−例えば、8マーまたは9マー−−を細胞培養物の中に直接導入していっそう急速なおよび発現プロセスを促進することが好ましい。
ペプチド、例えば、8,9および18マーのアミノ酸ペプチドを合成する場合、すべての種々のアミノ酸は合成の各サイクルの間に組み込むことが好ましい。しかしながら、種々のパラメーター、例えば、ある種のアミノ酸の溶媒不混和性−−はある種のアミノ酸を欠如するペプチドを含有する混合物を生ずることがあることに注意すべきである。こうして、この方法を必要に応じて−−すなわち、溶媒および反応条件を変更することによって−−調節して最大の種々のペプチドを生成すべきである。
MHC 分子によるペプチドの選択における「好ましさ」を決定するために、異なる長さの1系列のペプチドを、したがって、表3に示すように合成した。異なる長さのペプチドおよび異なるβ2ミクログロブリンにより付与される熱安定性の量を定量するために、細胞リゼイトにおけるこれらの種々の分子の安定性を検査した。放射性細胞リゼイトを、前述したようにネズミまたはヒトをもつKb /β2,Ld /β2およびDb /β2を発現するドロソフィラ(Drosophila)細胞から調製した。次いでリゼイトのアリコートを取り、そして記載するように添加を行った。
各処理のために、5本の管を調製した;次いでこれらを4℃〜47℃の異なる温度において1時間インキュベーションし、その後クラス1を免疫沈澱させ、そして SDS−PAGEにより分析した。次いでオートラジオグラムをレーザーデンシトメーターを使用して走査し、そして試料をインキュベーションした温度に対してシグナルの量をプロットした。分子の50%が安定である温度をグラフから計算し、そして異なる種の相対的熱安定性の測度として使用した。前述したように、ヒトβ2ミクログロブリンと複合化したネズミ重鎖は、ネズミβ2と複合化する場合よりも、ほぼ6〜8°高い温度において安定であった。
また、ペプチドおよび異種β2ミクログロブリンにより付与される安定性は加法的であることが観察された。12〜25マーに比較して、8〜9マーを使用する場合、クラス1分子の熱安定性の大きい増加が起こる;事実、より大きいペプチドと比較して8〜9マーにより付与された安定化の間の差は従来観察されたものよりなお大きいことがあるが、ペプチドがHPLCにより精製されていてさえ、より大きいペプチドの8〜9マーの多少の汚染が存在するようである。
今回、クラス1分子の熱安定性は、明らかに、(1)β2ミクログロブリンの由来、(2)ペプチドの存在、および(3)このペプチドの長さおよび配列に依存することが示された。
F.熱安定性表面発現MHC の発生
1.ネズミMHC を使用するパラメーターの洗練
ヒトクラス1 MHC分子を発現する細胞を使用する実験の前に、ネズミMHC を使用して種々のパラメーターの最適化を実施した。第1に、ドロソフィラ(Drosophila)細胞中で発現されたネズミMHC クラス1分子はペプチド不含または「空の」クラス1分子の特性を有することが評価された。
例えば、第2A図および第2B図は、ネズミまたはヒトβ2ミクログロブリンをもつKb /β2,Ld /β2およびDb /β2を発現するドロソフィラ(Drosophila)細胞から調製したアッセイの結果を示す。Kb /β2,Ld /β2およびDb /β2を発現するドロソフィラ(Drosophila)細胞を35Sメチオニンで45分間標識化し、そして前述したようにリゼイトを調製した。各リゼイトを4つのアリコートに分割し、これらにOVA, NP またはGP2を添加したか、あるいはペプチドをまったく添加しなかった。4℃において1時間インキュベーションした。次いでクラス1分子をリゼイトから免疫沈澱させ、そして SDS−PAGEにより分析した。
OVA またはNPペプチドをリゼイトに添加したとき、温度の対抗に対するKb /β2の劇的な安定化が起こった;同様に、Ld /β2はNPペプチドの添加により安定化された。Db /β2は他の2つの分子のように温度不安定性であるように思われなかった;それにもかかわらず、GP2 ペプチドをリゼイトに添加したとき、分子の安定性の増加が見られるが、OVA またはNPペプチドの添加はこれを達成しない。2つのバンドを各クラス1について免疫沈澱したことを見ることができ、処理された(下)または処理されない形態のゴルジを表し、両者の分子はリゼイトへのペプチドの添加により等しくよく安定化されるように思われる。未処理Kb /β2またはGP2リゼイトにおいて観察された少量の「30℃耐性」タンパク質はバックグラウンドである。なぜなら、標識化Ld /β2細胞のリゼイトへのY3の添加はこのバンドの単離を生じたからである(示されていない)。
次に、ネズミ重鎖を熱安定化するとき、ヒトβ2ミクログロブリンがネズミβ2ミクログロブリンより優れる可能性を研究した。エクスセル400 培地中で増殖したドロソフィラ(Drosophila)細胞へのヒトβ2の添加はすべての3つのネズミクラス1分子の定常状態のレベルを増加するばかりでなく、かつまた分子を37℃において少なくとも3時間安定性とすることが発見された。ヒトβ2を添加したときのネズミクラス1の安定性の増加は、培地中のβ2濃度を増加する結果であった;この可能性は除外した。ヒトβ2ミクログロブリンをコードするcDNAをドロソフィラ(Drosophila)発現ベクター pRmHa−3の中にクローニングし、そして前述したように種々のネズミクラス1重鎖cDNAおよび耐性マーカープラスミドを含有する発現プラスミドと共トランスフェクションした。
G418 選択後、細胞を適当な抗クラス1抗体で表面発現について染色し、強く発現する細胞をFACSを使用して選択した。細胞の安定な集団が得られた(データは示されていない)。次いで細胞をエクスセル400 中で増殖するように条件づけ、そして37℃において異なる長さの時間の間インキュベーションした。ヒトβ2ミクログロブリンを使用して合成したマウスクラス1分子は、エクスセル培地に外因性β2を添加しないで37℃における3時間のインキュベーションに対して安定であった(データは示されていない)。われわれの結果が示すように、ネズミクラス1分子の熱安定性はペプチドの存在およびβ2ミクログロブリンの由来に依存する。(第3A図および第3B図参照。)
さらに、今回、細胞表面で発現されたクラス1分子は適当なサイズのペプチドの添加によりさらに安定化されることが決定された。(第5A図〜第5C図参照。)1系列の異なる長さのペプチドを、表3に示すように、合成した(実施例5.2.参照)。これらは長さが8アミノ酸であるペプチドを包含する;RGYVYQGL(配列識別番号24)は小胞性口内炎ウイルス(VSV)Gタンパク質中のエピトープとして特徴づけられた。
同様に、OVA の8マーペプチドSIINFEKL(配列識別番号23、残基5−12)はKb /β2により提示されるオバルブミン中のペプチドとして同定され、そして9アミノ酸長さのペプチドASNENMETM(配列識別番号25)はDb /β2により提示されるインフルエンザウイルスの核タンパク質のエピトープとして特徴づけられた。これらの「親」ペプチドをエクスセル中で培養しそしてネズミクラス1を発現するドロソフィラ(Drosophila)細胞に添加すると、OVA ペプチドによるKb /β2およびNPペプチドによるDb /β2の37℃における熱安定性を生じた。
異なる長さのペプチドおよび異なるβ2ミクログロブリンにより付与される熱安定性の量を定量するために、細胞リゼイト中のこれらの種々の分子の安定性を検査した。前述したようにヒトまたはネズミβ2ミクログロブリンをもつKb /β2,Ld /β2およびDb /β2を発現するドロソフィラ(Drosophila)細胞から、放射性細胞リゼイトを調製した。次いで、実施例1.D.に記載するようにリゼイトのアリコートを取り、そして添加を実施した。各処理のために、5本の管を調製した;次いでこれらを4℃〜47℃の異なる温度において1時間インキュベーションし、その後クラス1を免疫沈澱させ、そして SDS−PAGEにより分析した。
次いでオートラジオグラムをレーザーデンシトメーターを使用して走査し、そして試料をインキュベーションした温度に対してシグナルの量をプロットした。分子の50%が安定である温度をグラフから計算し、そして異なる種の相対的熱安定性の測度として使用した。前述したように、ヒトβ2ミクログロブリンと複合化したネズミ重鎖は、ネズミβ2と複合化する場合よりも、ほぼ6〜8°高い温度において安定であった。
興味あることには、ペプチドおよび異種β2ミクログロブリンにより付与される安定性は加法的である。12〜25マーに比較して、8〜9マーを使用する場合、クラス1分子の熱安定性の大きい増加が起こる;事実、より大きいペプチドと比較して8〜9マーにより付与された安定化の間の差は従来観察されたものよりなお大きいことがあるが、ペプチドがHPLCにより精製されていてさえ、より大きいペプチドの8〜9マーの多少の汚染が存在するようである。
ペプチドの不存在下または存在下にネズミまたはヒトβ2と共発現されたとき、クラス1 MHC重鎖(示した)の50%がTX100 リゼイト中で安定である温度(℃)を次の表に示す。
ペプチドがβ2ミクログロブリンの不存在下に空のクラス1分子に結合することができるかどうかという問題を取り扱うために、35Sメチオニンで1時間標識化したKb またはDb クラス1重鎖を発現するドロソフィラ(Drosophila)細胞から洗浄剤リゼイトを調製し、そして処理した。Kb 分子をY3抗体で、そしてDb をB22. 249 抗体で免疫沈澱させた。低いレベルの免疫反応性Kb 重鎖のみを未処理リゼイト中で検出することができた。クラス1またはヒトβ2ミクログロブリンに結合するペプチドの添加は、免疫反応性物質の量を増加する;両者の添加は非常に大きい量の免疫反応性重鎖を生じた。同様な結果がDb について得られた。
こうして、重鎖はβ2ミクログロブリンの不存在下にペプチドに結合することができるように思われる;しかしながら、この結合は非常に安定なコンフォメーションを生じないか、あるいは使用する抗体はこのコンフォメーションによく結合しない。β2ミクログロブリンの添加は、また、多分同じ理由で免疫反応性分子の数を増加する。これらの2つの試薬の作用は加法的であり、3成分の複合体が分子の最も安定な形態であることおよび/またはこの分子が抗体により最もよく認識されることを示唆する。
これらの結果は、30℃の温度処理後、リゼイトから重鎖/β2複合体を「再構成」することができるかどうかという問題を発生する。Kb /β2を発現するドロソフィラ(Drosophila)細胞からのリゼイトを30℃において1時間処理し、その後NPペプチドおよびヒトβ2ミクログロブリンを添加した。4℃において4時間インキュベーションした後、クラス1重鎖をY3または抗細胞質テイル抗体K193 で免疫沈澱させた。抗血清の調製は実施例2.1 に記載されている。
前に報告されているように、トリトンX−100 リゼイト中の空のクラス1分子の温度処理はそれらの分解を生じず(抗細胞質テイル抗体K193 で30℃において処理した後、Kb 分子を等しく免疫沈澱させることができる)、むしろそれはそれらの変性を生ずることが発見された。さらに、これらの変性された分子が取るコンフォメーションは、それらがもはやβ2ミクログロブリンと結合しないか、あるいはY3によりまたはペプチドおよびヒトβ2ミクログロブリンの添加により認識される分子に回復することができるようなものである。こうして、クラス1重鎖は単独であるいはそれらをβ2ミクログロブリンと複合化したとき、ペプチドと結合することができると結論することは合理的である。
しかしながら、ペプチド負荷したヒトクラス1 MHCの表面の発現は、重鎖がβ2ミクログロブリンと複合化した後、分子のペプチドの負荷を最もよく促進するように思われる。
2.ヒトMHC の発現
クラス1分子の熱安定性がβ2ミクログロブリンの起源、ペプチドの存在、およびこのペプチドの長さおよび配列に依存すると、いったんわれわれが決定したとき、ペプチド負荷ヒトクラス1 MHC分子の発現を介してCD8細胞を特別に活性化することができる細胞系をつくるとき、われわれはこの情報を利用した。
熱安定性はクラス1分子の固有の性質であるように思われる;それは多分進化して、ペプチドを含有しないか、あるいは劣った結合性質のペプチド(ほとんど熱安定性を付与しない)を含有するクラス1分子が自己破壊することを保証する。このようにして、細胞はその表面上の空のクラス1分子の数を最小にする。なぜなら、外因的に誘導されたペプチドは結合しそして提示されうることにおいて、このような場合は危険であるからである。
ヒトβ2をもつ昆虫細胞中で発現されるヒトクラス1分子(B27およびA2.1)は37℃において延長したインキュベーションに対して安定ではない;クラス1分子上のペプチドの負荷に欠如することが示された突然変異の細胞系T2の中でヒトクラス1分子は発現されない(HoskenおよびBevan,Science 248 ; 367-70 (1990); Cerundolo ら、Nature 345;449-452 (1990)) 。こうして、重鎖とβ2ミクログロブリンとの間の親和性は分子の共進化を通して注意して保存され、こうして空のクラス1分子、あるいは結合性に劣るペプチドを有する分子は「宿主」生物の体温において自己破壊するように思われる。
ヒトクラス1 MHC分子はシュネイダー細胞の中で発現された。ヒトβ2ミクログロブリンおよびHLA A2.2 Y,HLA A2.1 ,HLAB7、またはHLA B27共発現する細胞系は、防止記載された方法により確立された。簡単に述べると、上のタンパク質をコードするcDNAをドロソフィラ(Drosophila)発現ベクター pRmHa−3の中にクローニングし、そしてヒトβ2ミクログロブリン含有プラスミドおよびphshsneoプラスミドとここに開示する方法によりシュネイダー細胞の中に共トランスフェクションした。3〜4週後、G418耐性細胞の集団を新鮮な選択培地で1:5に希釈した。
いったん細胞の健康に増殖する集団が得られると、CuSO4 を細胞のアリコートに添加しそして24時間後、β2ミクログロブリンとアソシエーションしたとき、ヒトクラス1重鎖の単形性決定基を認識するモノクローナル抗体W6/32(ATCC HB95 、ベセスダ、マリイランド州)を使用するフローサイトメトリーにより細胞を分析した。(参照、Barnstableら、Cell 14 : 9 (1978))。ヒトクラス1分子の各々の高いレベルの表面発現はCuSO4 の添加により誘発された(データは示されていない)。これらの安定な集団を、後述するようにサイトフルオロメトリーによる細胞の高い発現のために貯蔵した。すべての引き続く実験のために使用したのは、これらの選別した細胞の集団である。
FACS分析の24時間前に、CuSO4 を安定にトランスフェクションした細胞(3〜4×106 細胞/ml)に1mMの最終濃度に添加し、これによりトランスフェクションした遺伝子からの発現を「スイッチオン」する。細胞を24ウェルのクラスター皿(2ml/ウェル)中でプレートアウトする。FACS分析の8時間前に、CuSO4 培地を50μg/mlの濃度のペプチドを含むか、あるいは含まない新鮮な培地(1ml)と置換する。皿を37℃に室内で平の表面上に種々の時間間隔で移して37℃の温度の対抗を実施した後、分析のために細胞を収獲する。
シュネイダー細胞上のクラス1MHC の表面発現を分析するために、細胞のアリコート(5×105 )を氷上の管の中に移し、遠心(1,000×g、4分間)により集め、3mlの PBS/1%BSA 、0.02%アジ化ナトリウムの中に再懸濁させ、遠心により集め、そしてPCR 増幅反応一次抗体(腹水Y3,28:14:8S,30. 5.7 ,W6/32、希釈1:200)を含有する PBS/BSA (0.5ml)の中に再懸濁させる。ウサギ抗血清を1:500 に希釈し、そしてB22. 293 ハイブリドーマ上澄み液を直接使用する。
氷上で1時間インキュベーションした後、細胞を3mlの PBS/BSA 中で2回洗浄し、そしてFITC標識化二次抗体(Cappell, Durham、ノースカロライナ州)および1mg/mlのヨウ化プロピジウムを含有する 0.5mlの PBS/BSA の中に再懸濁させる。氷上で30分間インキュベーションした後、細胞を PBS/BSA 中で1回洗浄し、そしてこの緩衝液中に1×106 /mlの濃度で再懸濁させる。次いで試料をFACS440 (Becton Dickinson)により分析する。ヨウ化プロピジウムで染色した死亡した細胞を、分析においてライブ・ ゲート(live gate)を含めることによって排除する。
細胞の選別のために、上に概説した同一手順を使用するが、ただしすべての染色操作を無菌のフードの中で実施する。抗体を含む溶液を濾過滅菌し、そしてシュネイダー培地またはエクスセル400 を PBS/BSA の代わりに使用する。一次抗体を特別に結合する細胞を、ベクトン・ディキンソン(Becton Dickinson)細胞ソーターにより選別する。選別した細胞(8×105 )を培地中で1回洗浄した後、2×105 細胞/mlの濃度でプレートアウトする。
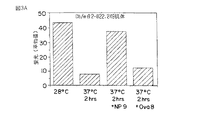
ドロソフィラ(Drosophila)細胞の表面上で発現されたヒトクラス1分子が空であることを証明するために、細胞を37℃において2時間インキュベーションし、そして細胞表面の発現をサイトフルオロメトリーにより分析した。細胞を37℃において2時間インキュベーションする場合、HLA B27およびA2.1 の両者の表面発現は大きく減少する;しかしながら、クラス1分子に結合することが知られているHIV ペプチド中で細胞を前インキュベーションすると、クラス1に対する有意な熱安定性が得られるが、結合しないペプチドはほとんど効果をもたない(第5A図参照)。(HIVのPOL タンパク質からの9アミノ酸のペプチドILKEPVHGV(配列識別番号31)は結合しそしてHLA A2.1 を安定化する。HIV のVpr タンパク質からの9アミノ酸のペプチドは結合しそしてB27 (FRIGCRHSR ; 配列識別番号30)を安定化する。これらのデータが示すように、ドロソフィラ(Drosophila)細胞の表面上でヒトクラス1分子は空であり、そして特定のHIV ペプチドを結合することによって安定化することができる。
第5A図は、ドロソフィラ(Drosophila)細胞の表面上に発現されたHLA B27およびHLA A2.1 のHIV ペプチドによるペプチド誘発熱安定性を示す。HLA B27またはA2.1 を発現するドロソフィラ(Drosophila)細胞を示すペプチドとインキュベーションし、次いで28℃に維持するか、あるいは37℃において2時間インキュベーションした後、抗体W6/32(ATCC HB95 から)およびサイトフルオロメトリーの使用によりクラス1分子の表面発現を分析する。各細胞の集団の平均蛍光をインキュベーション条件に対してプロットして示す。HIV POL ペプチド(ILKEPVHGV、配列識別番号31)はA2.1を安定化し、B27を安定化しないが、HIV Vpr ペプチド(FRIGCRHSR、配列識別番号30)はB27を安定化し、A2.1 を安定化しない。
第5B図が示すように、ドロソフィラ(Drosophila)細胞中で発現されたHLA A2.2 YはトリトンX−100 リゼイト中の空のクラス1分子の特性を有する。A2.2 Yを発現するドロソフィラ(Drosophila)細胞から調製した放射性トリトンX−100 リゼイトのアリコートを取り、そして次のようにペプチドを添加した:AおよびB:添加なし;C:HIV GAG(B)ペプチドKRWIILGLN(配列識別番号29);D:インフルエンザペプチドILGFVFTLTV(配列識別番号32);E:ランダム8マーペプチド;F:ランダム9マーペプチド。アリコートB〜Fを37℃において1時間インキュベーションした後、W6/32で免疫沈澱させそしてSDS PAGEにより分析する。
第5C図が示すように、ドロソフィラ(Drosophila)細胞中で発現されたHLA B7はトリトンX−100 リゼイト中の空のクラス1分子の特性を有する。リゼイトを第5B図におけるように、HLA B7を発現するドロソフィラ(Drosophila)細胞中で調製した。示すように、アリコートにペプチドを与えない(−P)か、あるいはランダム18マーまたは9マーのペプチドを与えた。アリコートの半分を、W6/32を使用する免疫沈澱およびSDS PAGEの前に、39℃において1時間インキュベーションした。
ドロソフィラ(Drosophila)細胞中で発現されたHLA B7およびA2.2 Yは、また、トリトンTX100 細胞リゼイト中で熱安定性であることが発見されたことにおいて、空のであることが示された(第5B図および第5C図参照);しかしながら、それらは温度対抗前にリゼイトにペプチドを添加することによって安定化することができなかった。とくに、インフルエンザマトリックスから誘導された10マー、ILGFVFTLTV(配列識別番号32)(Gotchら、J. Exp. Med.168 : 2045-57)は、HLA A2.2 を安定化することを見ることができ、同様にランダム9マーはHLA A2.2 を安定化し、また、HLAB7を安定化する。
ネズミクラス1について前述したように、トリトンTX100 細胞リゼイトの温度対抗アッセイを実施した。これらの研究からの結論は、ドロソフィラ(Drosophila)細胞中で発現されたヒトクラス1分子が突然変異のヒト細胞系T2中で発現された空の分子のすべての特性を有することである(Salterら、前掲(1985) ; HoskenおよびBevan 、前掲(1990))。
実施例2.クラス1分子について最適な結合特性をもつペプチドの発生
A.論考および外観
最近のデータが証明しているように、クラス1分子はコンセンサス配列のモチーフを含有するペプチドに結合する。(参照、Falkら、Nature 351: 290-6(1991);Van Bleek およびNathenson, Nature348 : 213-6 (1990)。)。こうして、クラス1分子の所定のアレレ形態に結合したペプチドのアミノ−およびカルボキシル−末端の残基はわずかの型のアミノ酸に限定されるように思われ、そして1または2以上の内部の残基は特定の位置を占有しなくてはならないことがある。さらに、クラス1結合ペプチドの全体の長さは8または9アミノ酸残基に制限されるように思われる。Falkら、前掲(1991);Van Bleek およびNathenson 、前掲(1990)。
ネズミKb 分子へのペプチドの結合の検査において、オバルブミンのペプチド、すなわち、配列SIINFEKL(配列識別番号23、残基5〜12)をもつ OVA−8は、生体内でKb 分子に結合し、最適な結合のモチーフを含有し、そして生体外でKb 分子に強く結合する。オバルブミン分子中のこのペプチドの配列は配列… QLEおよびTEW …によりフランキングされているので、いくつかのプロテアーゼ、非常に広い基質特異性をもつプロテアーゼ、あるいは多数のタンパク質分解活性をもつ酵素複合体は完全なオバルブミンからの OVA−8ペプチドの発生に関係するに違いないことが明らかである。
単離されたプロテアソームがKb 分子に結合する最適なペプチドを発生するかどうかを決定するために、「空の」クラス1Kb 分子、すなわち、結合したペプチドをもたないものが非常に熱安定性であるという発見を使用することは有用であった(Ljunggrenら、前掲(1990))。「空の」Kb 分子を洗浄剤中で30℃における1時間インキュベーションは、Kb 分子と反応することができないKb 特異的抗体Y3をβ2M依存性とする。なぜなら、β2Mは重鎖から急速に解離するからである。
適当なペプチド、例えば、 OVA−8の結合はサブユニットの解離を防止して、Y3をクラス1複合体に結合させる。生体内でKb 分子により提示されることができない OVA−8と同一長さのペプチド(例えば、Db に結合するペプチドSNENMETM(SerAsnGluAsnMetGluThrMet)(配列識別番号25、残基2−9)、Townsendら、Cell 44 : 959-968 (1986))は、Kb クラス1分子の加熱誘発解離を防止することができなかった(第3図参照)。配列EQLESIINFEKLTEWTSSNVMEER(配列識別番号23)(これは OVA−8の配列を包含する、下線で示す)はKb 分子に弱く結合する。しかしながら、 OVA−24の弱い結合はKb −β2M複合体の30℃における解離を防止するために不十分であった。
使用した種々の抗血清を次のようにして調製した。H−2Kb (LPDCKVMVHDPSLAまたはLeuProAspCysLysValMetValHisAspProSerLeuAla)(配列識別番号33)のCOOH末端の配列に相当する合成ペプチドに接合されたキーホールリンペットリンホシアニン(カルバイオケム(Calbiochem)、カリフォルニア州ラジョラ)でウサギを免疫化することによって、抗血清K193 を発生させる。ハイブリドーマHB176 (ATCC 、マリイランド州ロックビレ)を使用してY3腹水を調製する。(参照、また、Hammerlingら、PNAS USA 79 :4737−4741(1982)。)
腹水28−14−8SをハイブリドーマHB27(ATCC、マリイランド州ロックビレ)から調製する。(参照、Ozato およびSachs,J. Immunol. 126 :317-321 (1980); Myers ら、J. Immunol. 142 : 2751-58 (1989).) 。腹水30−5−7をハイブリドーマB22. 249 からの培養上澄み液から調製する(参照、Hammerlingら、Immunogenetics 8: 433-445 (1979); Allenら、Nature 309: 279−81 (1984)。)精製したネズミMHC に対するポリクローナル抗体をKvist ら、Scand. J. Immunol. 7: 265-76 (1978)。FITC標識化二次抗体(親和精製したヤギ抗マウスおよびヤギ抗ウサギIgG)はカッペル(Cappell)(ノースカロライナ州ダーハム)から入手した。
このアッセイ系を使用して、クラス1分子上にペプチドを負荷できない RMA−S細胞からの生合成的に標識化された「空の」Kb 分子を、β2M依存性Kb 特異的モノクローナル抗体Y3とインキュベーションした。抗体結合分子を SDS−PAGEにより分離し、そしてオートラジオグラフィーにより可視化する。 SDS−PAGEは次のように実施する。プロテインAセファローズビーズにより集めた免疫沈澱物を1mlの 0.1% TX100/PBS で5回洗浄し、そしてSDS PAGE試料緩衝液中で直ちに沸騰させるか、あるいは示す場合試料の半分をエンドグリコシダーゼHで消化した後沸騰させた(参照、Jacksonら、EMBO J. 9 : 3135-3162 (1990))。次いで試料を10〜15%の勾配SDS PAGEゲル上で分析し、アンプリファイ (Amplify)(アマーシャム(Amersham、イリノイ州アーリントンハイツ) で処理した後、コダック(Kodak)(ニューヨーク州ロチェスター)XOmat ARフィルムを使用して−70℃においてオートラジオグラフィーにかける。
Kb 鎖に相当するバンドをデンシトメトリーにより定量した。データ(示されていない)を発生させるために、 RMA−S細胞の「空の」クラス1Kb 分子含有リゼイトをペプチドおよび/または精製したプロテアソームとインキュベーションした後、4℃または30℃において1時間インキュベーションした。Kb 分子をモノクローナル抗体Y3で免疫沈澱させ、そして SDS−PAGEにより分析した。「空の」Kb 分子を OVA−24;プロテアソーム単独; OVA−24およびプロテアソーム; OVA−24、プロテアソーム、およびp−ヒドロキシ水銀ベンゾエート;および OVA−8とインキュベーションした。「空の」Kb 含有リゼイト単独を分析した(データは示されていない)。免疫沈澱の前に、試料を30℃において1時間加熱対抗するか、あるいは4℃に同一時間の間保持した(データは示されていない)。OVA −8ペプチドを75μMの最終濃度で使用し、そして OVA−24を75μMの最終濃度で使用した。
4℃において一夜インキュベーションしたKb 分子は、抗体との反応性の欠如により判定して、ほとんど完全に解離したが、 OVA−8ペプチドを含有するインキュベーション混合物から定量的に回収された。
偶数のレーンのデンシトメトリーの走査(示されていない)は、加熱対抗後に回収されたKb の量の決定を可能とした。データは加熱対抗後に回収Kb / OVA−8複合体の量の百分率として表した。OVA −8とインキュベーションしたKb 分子の大部分は、30℃の処理後に無傷で残った。より長いペプチドの OVA−24はKb 分子の安定化において OVA−8と同じようには効率よくなく、そして OVA−24の弱い結合についての前のデータに従い、熱処理後にKb 分子の約30%が残った。インキュベーション混合物が OVA−24および精製したプロテアソームを含有したとき、増加量のKb 分子を免疫沈澱することができ、そしてKb 分子のほぼ80%は30℃への暴露後に生き残った。
Kb 分子の安定性の増加は OVA−24から誘導された1種または2種以上のペプチドのためであることは、プロテアソーム単独がリゼイト中でKb −β2M複合体の解離を防止できるペプチドを発生することができなかったという事実から明らかであった。この結論は、また、開始剤p−ヒドロキシ水銀ベンゾエート(Rivett,Arch. Biochem. Biophys. 268 : 1-8 (1989))で処理したプロテアソームが、 OVA−24から、30℃においてKb 分子を安定化することができないペプチドを発生することができないという事実により支持される。
これらのデータが証明するように、単離されたプロテアソームは弱くKb 結合性の前駆体ペプチドから1種または2種以上のペプチド発生することができ、こうしてクラス1分子は内因性ペプチドによるのと同一程度に安定化された。結局、 OVA−24から誘導された1種または2種以上のペプチドは約8アミノ酸長さであり、そしてOVA −8ペプチドの配列を包含しなくてはならない。さらに、変性した、還元した、およびアルキル化したオバルブミンをプロテアソームとインキュベーションした同様な実験は、また、熱安定性Kb 分子の回収を生じた(データは示されていない)。
B.手順
1.細胞の培養
8%胎児仔ウシ血清、2mMグルタミン、ペニシリン(100μg/ml)、およびストレプトマイシン(100μg/ml)を補充したダルベッコ変性イーグル培地(DMEM、ギブコ(Gibco)、ニューヨーク州グランドアイライド)中でHLA 細胞(ATCC No. CCL 185)。4種類の異なるH−2ハプロタイプの脾細胞(H−2k ,s ,d 、およびq ) 、RMA −S,T1,T2およびネズミ星状細胞をRPMI 1640(ギブコ(Gibco) 、ニューヨーク州グランドアイランド)中で増殖させた。RMA −S、ネズミ突然変異H−2b リンパ腫細胞系(Ljungrenおよび Karre, J. Exp. Med. 162: 1745-59(1985);Karre ら、Nature319 : 675-8 (1986))は、内因性抗原を提示することができない(Townsendら、Nature 340: 443-8 (1989) ; TownsendおよびBodmer, Ann. Rev. Immunol. 7: 601-24 (1989)) 。
T1およびT2はT細胞リンパ腫CEM に融合されたヒト突然変異Bリンパ芽球様細胞系LBL 721. 174 (0.174)の誘導体である(Salterら、Immunogenetics 21 : 235-7 (1985))。T1, 0.174−CEM ハイブリッドは、 0.174から誘導されたクラス1 MHCを高いレベルで発現した。ヒト染色体6の両者のコピーの損失のために選択したT2は、0.174 の低いクラス1発現の表現型を有する(HoskenおよびBevan,Science 248 : 367-70 (1990)) 。
2.代謝的放射線標識化、免疫沈澱、およびデンシトメトリー
細胞の代謝的標識化を、Jackson ら、EMBO J. 9 : 3153-3162 (1990) に記載されているように、次の変更を加えて実施した。特記しない限り、代謝的標識化の前に細胞をインターフェロンガンマ(IFN−ガンマ、ベーリンガー・マンヘイム(Boehringer Mannheim)、インジアナ州インディアナポリス)(2500U/ml)で96時間処理した。パルス培地は 0.15mCi/mlのL [35S] メチオニン欠如およびシステイン欠如DMEMを含有した。細胞を日常的に4時間標識化し、次いで通常の培地の存在下に異なる時間(36時間まで)のチェイス期間を置いた。免疫沈澱、 SDS−PAGE (BlobelおよびDobberstein,J. Cell Biol. 67: 835-51 (1975)) およびフルオログラフィー(BonnerおよびLaskey, Eur. J. Biochem. 46 : 83-88 (1974))を記載されているように実施した(Jacksonら、EMBO J. 9 :3135−3162 (1990))。
第1次元の非平衡化pH勾配のゲル電気泳動(アンフォライン pH3.5−10)を、Jones, Selected Methods in Cellular Immunology, Mishell およびShigii編、Freeman 、サンフランシスコ、1980, pp. 398-400 、に記載されているように実施した。タンパク質の中に組み込まれた放射能を定量するために、LKB ウルトロスキャン(Ultroscan)XL(ブロウマ(Brouma)、スウェーデン国)を使用する走査デンシトメトリーにより、フルオログラフ上のバンドの強度を決定した。放射性マーカーのタンパク質、ウシ血清アルブミン(67kD)、オバルブミン(46kD)、炭酸脱水素酵素(30kD)およびラクトグロブリンA(18.4kD)をニュー・イングランド・ニュークリアー(New England Nuclear)(デュポン、ボストン、マサチュセッツ州)から購入した。
パルスチェイスの分析のために、輸送の分析に使用する抗体の数に依存して種々の時点からのリゼイトのアリコートを管の中に取った。抗体(1μlの腹水、Y3,30. 5. 7,28:14:8S;W6/32)、または抗血清(K193,K270)、 100mlのハイブリドーマ培養上澄み液をリゼイトに添加する;次いでこの混合物を2〜12時間放置した後、プロテインAセファローズとの1時間のインキュベーションおよびマイクロフージ中の5秒の回転により抗体を集める。
3.抗血清、モノクローナル抗体およびペプチド
ウサギ抗ヒトおよび抗ラットプロテアソーム血清は、A. Ichihara博士により提供された(Tanakaら、J. Cell. Pysiol. 139: 34-41 (1986))。Y3腹水、マウスH2−Kb に対するモノクローナル抗体を、ATCCからのハイブリドーマno. HB176 を使用して調製した(Hammerlingら、PNAS USA 79 : 4737-4741 (1982))。ヒトTリンパ球からの IFN−ガンマをベーリンガー・マンヘイム(Boehringer Mannheim)(インジアナ州インディアナポリス)から入手した。マウス IFN−ガンマをジェネテク(Genetech)(カリフォルニア州サウスサンフランシスコ)およびアムゲン・バイオロジカルス(Amgen Biologicals)(カリフォルニア州サウザンドオークス)から入手した。ペプチドをアプライド・バイオシステムス(Applied Biosystems)430 Aペプチド合成装置で固相技術により合成し、そしてさらにC18逆相カラムクロマトグラフィーのカラム(VyDac)で精製した。
4.HLA 細胞ホモジネートの硫酸アンモニウムの分画
未処理のおよび IFN−ガンマ処理した均質化HeLa細胞を遠心して核および細胞破片を除去し、そして生ずる上澄み液を記載されているように硫酸アンモニウムの分画にかけた(Rivett, J. Biol. Chem. 260 : 12600-12612 (1985))。上澄み液の中に存在する26Sプロテアソーム複合体を38%の飽和において硫酸アンモニウムで選択的に沈澱させたが、上澄み液の中に存在する19Sプロテアソーム複合体は38%の分画工程後に60%の飽和において沈澱させた(Waxmanら、J. Biol. Chem. 262: 2451-57 (1987))。
5.ミクロソームおよび細胞質ゾルの分画
未処理のおよび IFN−ガンマ処理したHeLa細胞を4時間標識化し、均質化し、そして分画遠心分離にかけた。ホモジネートをまず15,000rpm において30分間回転して核および細胞破片を除去した。生ずる上澄み液を次いで 100,000×gで30分間回転してミクロソームを沈澱させた。上澄み液画分(細胞質ゾルの画分)およびペレット(粗製のミクロソームno画分)中のプロテアソームを別々に免疫沈澱させ、そして2次元のゲル電気泳動により分析した。
6.プロテアソームの精製
Skilton ら(1991)に記載されている手順の変更を使用して、新鮮なウシ肝臓からプロテアソームを精製した。簡単に述べると、新鮮なウシ肝臓を 10mM Tris−HCl (pH7.2), 50mM KCl, 0.1mM EDTA,0.1mM DTT 、および20%グリセロール(v/v)(「緩衝液A」)中で均質化した。ホモジネートを 105,000×gで1時間遠心した。上澄み液をまず硫酸アンモニウムで38%の飽和に処理して、大きいタンパク質凝集物ならびにプロテアソームの26Sの形態を除去した(Waxmanら、J. Biol. Chem. 262: 2451-57 (1987))。次いで生ずる上澄み液を硫酸アンモニウムで60%の飽和に処理して、プロテアソームの16Sの形態を選択的に沈澱させた。ペレットを小さい体積の緩衝液Aの中に再懸濁させた。
再懸濁させた60%の硫酸アンモニウム画分を3つの順次のクロマトグラフィー工程にかけた:DEAE−セファローズ(Sepharose)、モノ(Mono)Q10/10のアニオン交換クロマトグラフィー、そして最後にスペロース(Superose)6カラムのゲル濾過。各クロマトグラフィー後、プロテアソーム含有画分を SDS−PAGEによりおよびHough ら、J. Biol. Chem. 262:8303−8313 (1987) に記載されているようにペプチダーゼ活性の分析により同定した。使用したペプチド基質は次の通りであった:N−スクシニル−ala −ala −phe −MCA, N−スクシニル−leu −leu −val −tyr −MCA 、およびt−ブトキシカルボニル−phe −ser −arg −MCA 。
7.ペプチドの結合アッセイ
RMA−S細胞の「空の」クラス1Kb 分子含有リゼイトを変更された手順(Ljunggrenら、Nature 346: 476-80 (1990)) 。簡単に述べると、 RMA−S細胞をRPMI 1640 中で26℃において8時間培養した。次いで細胞をPBS 中で1回すすぎ、そして再び10:1の比のメチオニン欠如およびシステイン欠如RPMI 1640 および通常のRPMI1640の中に 0.25mCi/mlのL− [35S] メチオニンおよび 0.25mCi/mlのL− [35S] システインを含有する培地中で26℃において16時間インキュベーションした。細胞をPBS 中ですすぎそして前述したように溶解した。次いで「空の」クラス1Kb 分子含有リゼイトをペプチドおよび/または精製したプロテアソームと26℃において1時間インキュベーションした後、30℃においてさらに1時間インキュベーションした。次いでこの混合物をモノクローナル抗体Y3で免疫沈澱させ、そして SDS−PAGEにより分析した。
実施例3.生体内の細胞障害性エフェクターの誘発
A.浸透圧的負荷
SC2および 3T3細胞へのオバルブミンタンパク質の浸透圧的負荷を、Moore ら、Cell 54 : 777-785 (1988) に記載されているように実施した。アッセイの手順は次の通りである。96ウェルの皿において、1×105 のドロソフィラ(Drosophila)細胞(負荷したペプチド/タンパク質を含むか、あるいは含まない)または 3T3細胞を、10%胎児ウシ血清を補充した 200μlのRPMI培地中で、1×105 B3/CD8T細胞ハイブリドーマ細胞と同時培養した。
24時間のインキュベーション後、これらの培養からの 100μlの上澄み液を5,000CTLL 細胞を含有する 100μlのRPMIに添加した。1μCiの 3H−チミジン(アマーシャム(Amersham))を添加したとき、細胞を37℃において24時間同時培養した。37℃においてさらに15時間インキュベーションした後、CTLL細胞の中への放射線標識の組み込みをシンチレーションカウンティングにより決定した。
ネズミ MHCを使用して実施したアッセイは、また、昆虫細胞がクラス1分子上にペプチドを負荷できることを評価した。特定の抗原を含有する MHC細胞を 200〜500 程度に少ない数で発現する細胞を、T細胞により検出することができる。ドロソフィラ(Drosophila)細胞はクロムを蓄積しないので、B3/CD8,T細胞ハイブリドーマに基づく抗原提示アッセイを使用した。B3/CD8は、B3,H−2Kb クラス1分子により提示されるオバルブミンのペプチド253−276 に対して特異的な細胞障害性T細胞、およびCD8を有するIL−2分泌細胞系の間のハイブリドーマである(参照。Carbone ら、前掲、1989)。抗原性刺激したとき、B3/CD8は、IL−2依存性細胞系CTLL中の 3H−チミジンの組み込みにより測定して、IL−2を生産する(Gillisら、J. Immunol. 120 : 202791978))。こうして、IL−2の生産量を測定することによって、T細胞の認識によりアッセイすることができる。
OVA ペプチドを誘導できるオバルブミンタンパク質の細胞内プールを得るために、オバルブミン(シグマ・ケミカル・カンパニー(Sigma Chemical Co.)、ミゾリー州)を、Moore ら、前掲(1988)に記載されているように、細胞の中に浸透圧的に負荷した。負荷直後に、細胞をT細胞ハイブリドーマと混合した。2日のインキュベーション後、培地を除去し、そしてIL−2についてアッセイした。IL−2依存性細胞系CTLLの増殖を支持する培地の能力によりIL−2の量を決定し(Gillisら、前掲、1978)、そして細胞の中に組み込まれた放射性チミジンの量により増殖を定量した。
第4図において見ることができるように、オバルブミンペプチドを培地に添加した場合、T細胞はドロソフィラ(Drosophila)細胞に対してよく応答したが、細胞にオバルブミンタンパク質を負荷した場合、認識は起こらなかった。昆虫細胞の細胞表面上で発現されたMHC クラス1分子は、ペプチドを培地に添加した場合ペプチドに結合することができ、そしてT細胞により認識されるように正しい関係でペプチドを提示することができることにおいて、完全に機能的である。
B.生体外条件の最適化
特定の細胞障害性T細胞の発生のための生体外条件を最適化するために、ドロソフィラ(Drosophila)細胞刺激因子の培養は好ましくは血清不含培地(例えば、エクスセル400)中で維持される。ドロソフィラ(Drosophila)細胞の刺激因子の細胞を好ましくは>20μg/mlのペプチドとインキュベーションする。有効なエフェクター:刺激因子の比(リンパ球:ドロソフィラ(Drosophila)細胞の比)は、好ましくは約30, :1〜 300:1の範囲である。一般に5日の培養後に、最大の特異性のCD8は観察される。死滅アッセイについての標的細胞の培養は、好ましくは、血清不含培地中で維持する。
C. 51 Cr解放細胞障害アッセイ
細胞仲介細胞溶解活性をCr51または解放アッセイで検出した。200 μl中の105 の51Cr標識化標的細胞の比溶解の百分率を種々のリンパ球/標的細胞の比について決定し、そして比細胞障害性/生存能力のあるエフェクターの数をプロットすることによって、各リンパ球の集団について線量−応答曲線を決定した。自発的51Cr解放値は合計の組み込まれた標識の5%〜15%であった。比51Cr解放の百分率を次式に従い計算した:(ER−MR)(MR−SR)×100 、ここでERは観測された実験的51Cr解放であり、SRは培地単独中でT細胞系をインキュベーションすることによって決定された自発的解放であり、そしてMRはアッセイの間にHCl 中の 200μl中でインキュベーション後に標的細胞から解放された最大放射能である。
D.ネズミクラス1およびペプチドを発現するドロソフィラ(Drosophila)細胞を使用する一次生体外刺激による細胞障害性エフェクターの誘発
ペプチドOVA, NP(インフルエンザ)およびNP)LcMV)をペプチド源として使用して、一次生体外CD8応答を発生するそれらの能力を評価した。(第6A図および第6B図参照。)それぞれ、H2−Kb +抗原およびH2−Ld +抗原を発現するドロソフィラ(Drosophila)細胞で免疫化した C57BL/6およびBalb/Cマウスからの脾細胞の5日間のインキュベーションは、ペプチドの存在下に腫瘍EL4およびP815 を効果的に溶解するエフェクターを発生した。
P815(ATCC RIB 64)(H−2d )は DBA/2マウスからの肥満細胞種である。EL4(ATCC TIB 39)は C57BL/6(H−2b ) から胸腺腫である。EL4をヒトβアクチンプロモーターをもつプラスミド構成体中でOVA cDNAでトランスフェクションして、OVA 生産細胞系EL4−OVA を誘導する。これらの腫瘍は10%胎児ウシ血清FBS を補充したRPMI 1640 中で静止懸濁培養物として維持する(参照、Mooreら、Cell 54 : 777-785 (1988))。Jurkat (ATCC CRL 8163)は HLAB7を発現するヒト細胞系である。JurkatA2は、Jurkat細胞をHLAA2−1で、例えば、ここに開示する方法によりトランスフェクションすることによって生産されたヒト細胞系である。(参照、また、Irwin ら、J. Exp. Med. 170: 1091-1101 (1989))。
合成ペプチドはCD8の認識の間に内因的に合成されたウイルスのタンパク質と置換することができる。発生した特異的CD8は、EL4(オバルブミンcDNAでトランスフェクションした)の表面においてクラス1により提示された内因的に合成されたペプチドを認識することができた。(参照、Moore ら、Cell 54 : 777-785 (1988)。)同様に、インフルエンザNPペプチドを負荷したDb に対して生体外で発生したCD8は、インフルエンザウイルス株/A/PR/8で感染したEL4標的細胞を認識した(第7図参照)。(参照、Carbone ら、J. Exp. Med. 167: 1767-1779 (1988) ;株A/PR/8/34は、また、アメリカン・タイプ・カルチャー・コレクション(AmericanType Culture Collection)から入手可能であり、そして受け入れ番号 no. ATCC VR−95)。
E.抗ペプチド細胞障害性細胞の特性決定
ペプチド特異的認識がT細胞活性のためであるかどうか決定するために、これらの系統を抗CD8(OKT8,ATCC No. CRL 8001)または抗CD4(OKT4,ATCC No. CRL 8002)および/または相補体で処理した後、それらをアッセイ培養物に添加した。抗CD8処理のみが細胞障害性を壊滅した。
これらの特異的CD8を、それぞれ、Db またはKb でBALB 3T3 (ATCC, CCL 163)細胞をトランスフェクションすることによって生産された 3T3/Db および 3T3/Kb 細胞上で試験した。(他の容易に入手可能な 3T3培養物は、また、使用のために入手可能であり、ATCC CCL 163.2を包含する。)好ましくは、安定なトランスフェクション体は 3T3をpCMU/Kb またはpCMU/Db およびpSV2neo により共トランスフェクションして調製され、そしてG418 で選択する。(参照、また、JolyおよびOldstone、前掲(1991))。抗OVA CD8の応答はDb またはKb 制限され、これらの活性が古典的クラス1 MHC制限を示すことを確証する。
ペプチドの刺激によりCD8を誘発する能力は、クラス1制限された認識を研究する、比較的簡単な技術を提供する。T細胞は自然の形態または外来抗原よりむしろ変性された形態に対して特異的であるので、ペプチドは有効なワクチン接種の潜在的手段を表す。
F.特異的CD8細胞の発生
ドロソフィラ(Drosophila)細胞のペプチドクラス1複合体をグルタルアルデヒド 0.001%で固定し、次いで洗浄しそしてリンパ球同時培養において刺激因子として使用した。非常に弱い細胞障害活性は5日の同時培養後に検出され、生きている(すなわち、固定されない)ドロソフィラ(Drosophila)細胞が特異的CD8の発生において優れることを確証する。固定された細胞に10%のドロソフィラ(Drosophila)培養物を添加すると、特異的CD8の発生が回復する。(第8A図および第8B図参照。)
シンジェネイッククラス1抗原を発現する培養した細胞は、通常一次培養においてCD8を活性化することができず、ドロソフィラ(Drosophila)細胞の培養上澄み液の添加後、特異的CD8を発生することができた。
応答体の集団からCD4+およびクラスII+細胞を除去すると、ドロソフィラ(Drosophila)細胞上に負荷されたペプチドで発生するCD8活性を減少する。こうして、ペプチド負荷ドロソフィラ(Drosophila)細胞により誘発された一次応答はCD4+細胞およびクラスII依存性であるように思われる。
実施例4.処理の生体内効能
A.腫瘍の拒絶
この中に開示する方法を使用して、今回、本発明の特異的、活性化CD8細胞を使用してヒトにおいて標的細胞を特異的に殺すことが可能である。本質的に、処理方法は次の工程を包含する:(1)処置すべき個体からT細胞を含有する試料を得る;(2)空のクラス1 MHC細胞に抗原性ペプチドの少なくとも1つの種を負荷し、ここでペプチドは標的細胞から誘導されたペプチドの少なくとも一部分に対して実質的に相同性である;(3)T細胞を生体外で活性化CD8細胞を生産するために十分な量のペプチド負荷クラス1 NHC細胞と混合する;(4)活性化CD8を培養物から収獲する;そして(5)処置すべき個体に活性化CD8細胞を投与する。
この方法はマウスのモデルにおいて有望な結果を既に示している。例えば、1つの研究において、 C57BL/6雌マウス(約6〜8週齢)のちょうど耳の背後に、3×106 (3MM)EL4/OVA 腫瘍細胞を皮下注射した。腫瘍細胞を C57BL/6マウスの中で単一の継代により腫瘍発生性とした。EL4/OVA 腫瘍細胞を皮下接種する日に、マウスにドロソフィラ(Drosophila)/Kb /OVA 刺激CD8細胞を腫瘍細胞注射部位の隣に(近位に)注射するか、あるいは静脈内に注射した。
第8日に、腫瘍の寸法を測定した。3MM腫瘍細胞のみを与えた5匹の対照マウスは12mm×12mmの大きさの腫瘍を有した。腫瘍細胞および 24MM CD8細胞注射VIを受けた3匹のマウスを検査した;腫瘍の大きさはそれらのマウスにおいて(a)2mm×2mm,(b)5mm×5mm、および(c)12mm×12mmであった。腫瘍細胞および腫瘍注射部位の隣に 24MM CD8細胞を受けた5匹のマウスを検査した。結果は次の通りであった:(i)3引きのマウスは第8日に検出可能な腫瘍を有した;(ii)1匹のマウスは1mm×1mmの大きさの腫瘍を有した;そして(iii )1匹のマウスは5mm×5mmの大きさの腫瘍を有した。
第11日に、腫瘍の大きさは次のように決定された:
引き続く検査において、種々の養生法を適用した後、マウスにおける腫瘍の体積を測定した。前述したように、マウスに3×106 (3MM)EL4/OVA 腫瘍細胞を注射した。EL4/OVA 腫瘍細胞を皮下接種した日に、下表2に記載するように、マウスをドロソフィラ(Drosophila)/Kb /OVA 刺激CD8で腹腔内、皮下または静脈内に注射した。第6,8,10, 12および20日に腫瘍体積を測定した。5匹のマウスを各実験群および対照群に含めた。結果を下表4に記載する。
B.インフルエンザの処置
例えば、インフルエンザ特異的CD8集団はこの中に開示する技術に従い発生することができ、そして致死的インフルエンザ感染に対して個体を保護するとき有効であることを証明することができる。生体外のペプチドのプライミングから誘導されたウィルス反応性CD8は生体内の保護を提供することができ、そして主として生体外のCD8の刺激は広範な種類の抗ウィルスおよび抗レトロウィルスのワクチンのデザインにおいて有用であることを証明することができる。
C.クラス1でトランスフェクションしたドロソフィラ (Droso-phila)細胞を使用するマウスのプライミング
50×106 ドロソフィラ (Drosophila) 細胞の腹腔内 (IP) 注射は、特異的CD8の誘発に導いた。このプライミングの検出は制限希釈分析を使用して実施した。 OVAに対して特異的なCD8の頻度は自然のマウスにおいて約106 であった。ペプチドで被覆したドロソフィラ (Drosophila) 細胞を注射した後、頻度は約10-3である(データは示されていない)。
第8A図および第8B図はトランスフェクションしたドロソフィラ (Drosophila) 細胞を使用する生体内のプライミング図解しており、ここで応答体は C57BL/6マウス脾細胞である。第8A図において、細胞の一部分を固定する;第8B図において、ドロソフィラ (Drosophila) 細胞のすべては固定されていない。
二次的CD8応答のために、 C57BL/6マウスをドロソフィラ (Drosophila) 細胞(5×107)の1回の腹腔内注射によりプライミングし、そして免疫化後5〜15日に試験した。(第8B図参照。)活性化CD8の生体内誘発は腫瘍の拒絶を明らかに刺激する。
他の実験において、 C57BL/6マウスを3MM EL4 OVAで皮下注射した。5日後、群IのマウスにKb /OVA を発現する50MM(生きている)ドロソフィラ (Drosophila) 細胞を皮下注射した;群IIマウスにKb /OVA をもつ50MM固定ドロソフィラ (Drosophila) 細胞を皮下注射した;群III マウスは対照群であり、そして第5日に追加の注射を与えなかった。この実験の結果を第9図に図解されており、これは群Iのマウスが最も長い期間生存しかつより大きい数で生存したことを示す。
一次CD8をB6アンプライムドマウスにおいてドロソフィラ (Drosophila) 細胞中のペプチド負荷クラス1により誘発した。CD8細胞を生体外でドロソフィラ (Drosophila) 細胞で1回刺激し、22℃において36時間培養し、異なる濃度の OVAペプチドとインキュベーションした。ペプチドの最終の最小量は約20μg/mlであった。エフェクターを収獲し、そして RMA−S細胞上で50μgペプチドで試験した。CD8細胞の集団は RMA−S OVA−8を認識することができた。(第6A図および第6B図参照)。
他の実験において、オバルブミンタンパク質をエンドプロテイナーゼ Glu−c(黄色ブドウ球菌(Staph.aureus) から、(ベーリンガー・マンヘイム(Boehringer Mannheim)、インジアナ州インディアナポリス)で分解した。この混合物をネズミ MHCをもつドロソフィラ (Drosophila) 細胞培養物に添加した。約5日後、CD8細胞をエフェクターに暴露した;ある数のCD8細胞は RMA−S OVA−8を認識することができた。
D.標的細胞レベルにおけるペプチドの滴定
最近の研究は、9アミノ酸の長さのペプチドが合成ペプチドの調製物の中に少量の種として存在することを証明した。9アミノ酸より長いものは低い親和性でH−2Kb クラス 1 MHC分子に結合する。ペプチドの滴定実験において、16マーのペプチドより50倍低い濃度の9マーのペプチドはT細胞の応答の誘発のために十分であることが示された。同一ペプチドの滴定を標的細胞の感作について実施した。結果は、標的細胞の感作が生体外の応答の誘発に要求されるより約1000倍低いペプチド濃度で達成されることを示す。
E.トランスジェニックマウスにおけるCD8の発生
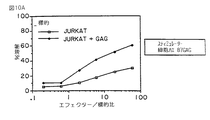
この研究において、HLA A2.1, HLA B7、および HLA B27を発現するトランスジェニックマウスを使用した。マウスの脾細胞をシンジェネイックHLA(それぞれ、A2.1,B7 、およびB27)を発現するドロソフィラ (Drosophila) 細胞と培養した。特異的CD8の応答は HIVの3つのペプチドに対して発生された。(第10A図、第10B図および第10C図参照。)
同系交配マウス C57BL/6y(H2b ) およびBalb/c(H2d )マウス、ならびに種々のトランスジェニックマウスをこれらの研究において使用する。トランスジェニックマウスは次のものを包含した。簡単に述べると、ゲノム HLA−B7クローン(5′および3′フランキング領域の数 100塩基対を含む)をB6× SJLマウス胚の中にマイクロインジェクションする(スクリップス・リサーチ・インスチチュート・トランスジェニック・ファシリティ(Scripps Res-earch Institute Transgenic Facility)、カリフォルニア州ラジョラ) 。B7トランスジェニック系統は C57BL/6(H2b ) に戻し交雑されている(スクリップス・リサーチ・インスチチュート・トランスジェニック・ファシリティ(Scripps Research InstituteTransgenic Facility)、カリフォルニア州ラジョラ)。
B7トランスジェニックマウスは既知の方法に従い生産することができる;われわれのものはゲノムクローンをB6× SJL胚の中に注入し、そしてそれらを C57BL/6に戻し交雑することによって生産された(スクリップス・リサーチ・インスチチュート・トランスジェニック・ファシリティ(Scripps Research Institute Transgenic Facility)、カリフォルニア州ラジョラ)。(参照、また、 Krimpenfortら、EMBO J.6(8) :1673(1987) 、B27トランスジェニックマウスを発生する他の方法について。) 。β2ミクログロブリントランスジェニックマウスおよびトランスジェニックA2−1マウスは、既知の技術を使用して同様な方法で生産する(参照、例えば、 Irwinら、J.Exp.Med.170 :1091-1101(1989)) 。
F.ヒトにおける特異的HIV CD8の発生
血清反応陰性のドナーからの PBLを、2つの HIVペプチドに対して特異的なCD8を発生するそれらの能力について試験した。これらのドナーはHLA A2, HLA B7または HLA B27である。5日間の培養後、非常に強いCD8活性が培養物中に両者 HIVペプチドに対して検出することができた。(第11A図、第11B図、および第11C図参照。)
ヒトPBL をフィコール−ハイパーク(Ficoll-Hypaque) 勾配遠心(ファーマシア(Pharmacia)、ニュージャージイ州ピスカタェイ)により単離する。 PBLリンパ球を10%DMSOを含有する PBS中で凍結後、液体窒素の中に貯蔵する。細胞の生存能力およびリンパ球の機能は保存される。
ペプチドに対して特異的なCD8細胞を、シンジェネイックペプチドクラス1複合体を発現するドロソフィラ (Drosophila) 細胞とのシンジェネイック一次培養において発生させる。これらの培養は、7% CO2中で37℃において直立25cm2 フラスコ中の5×10-5M β−メルカプトエタノールを補充した20mlの RPMI1640培地中においてヒトマウスまたはヒトPBL の3×107 応答性脾細胞および生きている106 ドロソフィラ (Drosophila) 細胞を使用して確立される。5日間の培養後、CD8細胞を収獲する。
他の実験において、ヒトPBL を収獲し、そして5日の期間にわたってヒトHLA A2.1を発現するドロソフィラ (Drosophila) 細胞で刺激し、そして HIV POLペプチドを負荷した。次いで刺激した PBLを HIV感染した Jurkat A2.1細胞上で試験した。この実験の結果を第12図に示す。生体外で刺激したヒトPBL は明らかに感染し細胞上のA2.1 POLを認識し、そして前記細胞を殺すことができた。
実施例5.生体外の可溶性、空のクラス 1 MHC分子の直接結合
A.手 順
H−2Kb :実施例1.Bにおいて前述したように調製した。
H−2Kb sol:Kb sol cDNAはKb の誘導体であり、そして既知の方法、例えば Ennisら、PNAS USA 87 :2833-7(1990) および Zemmourら、Immunogenetics 33 : 310-20(1991)に記載されている方法に従う PCRにより突然変異させることができる。詳しくは、アミノ酸位置+275 におけるアルファドメインの末端に停止コドンが挿入された切頭Kb 分子をコードするcDNAを、pCMU発現プラスミドからBamHI断片として切除し、そしてKb cDNAとして pRmHa−3の中にクローニングする。
Kb sol cDNAは完全なKb cDNA(上を参照)の誘導体であり、 StyIに示すを包含する5′オリゴヌクレオチド、および次の3′オリゴヌクレオチドを使用する PCR反応において鋳型として使用される:
5′ ATATGGATCCTCACCATCTCAGGGTGAGGGGC 3′
(配列識別番号34)
生ずる PCR断片を平滑末端とし、pBS(ストラタジーン(Stratagene)、カリフォルニア州ラジョラ)の SmaI部位の中にクローニングし、配列決定し、そしてKb の残りの5′配列を StyI部位の中にクローニングする。完全なKb sol タンパク質をコードするcDNAは、BamHI制限断片として得ることができた。
H−2Db およびH−2Ld は上の実施例1.B.に記載するように調製する。
Kb α1α2α3ドメイン(274残基) およびネズミβ2ミクログロブリン(99残基)をコードするcDNAを、それぞれ、メタロチオネインプロモーター pRmHa−3を収容する発現ベクターの独特BamHI部位の中にクローニングした(Bunchら、Nucleic Acids Res.16:1043-1061(1988))。ドロソフィラ (Drosophila) S2/M3細胞を、従来記載されたリン酸カルシウム沈澱法により、プラスミドphshsneo(ネオマイシン耐性遺伝子を含有する)に加えてこれらの組換えプラスミドで形質転換した。
ネオマイシン類似体の抗生物質G418に対して選択された形質転換した細胞を血清不含培地中の27℃において増殖させ、そして可溶性重鎖Kb β2ミクログロブリンwo0.7mM CuSO4 の添加により共発現した。Kb の可溶性のアセンブリングされたヘテロダイマーを、培養上澄み液から、アフィニティクロマトグラフィーにより抗Kb モノクローナル抗体Y3を使用して、次いでファーマシア(Pharmacia)モノ(Mono) QFPLCカラムのイオン交換クロマトグラフィーにより製造業者(ファーマシア(Pharmacia) 、ニュージャージイ州ピスカタェイ)の指示に従い精製した。
Kb 調製の SDS−ポリアクリルアミドゲルの電気泳動(SDS−PAGE) および引き続くクーマッシーブルーによる染色は、約32,000における相対的分子量(Mr)の1つのバンドおよび検出可能な不純物を含まない約12,000におけるMrの1つのバンドのみを示した。高度に精製したKb をリン酸塩緩衝液(PBS)に対して透析し、濾過滅菌し、そしてそれ以上の研究のために使用した。可溶性Kb (「Kb sol 」)タンパク質(43.2kDa)の吸光係数は、 280nmにおいて69,200M-1cm-1である。
1%TX−100 を含むか、あるいは含まない PBS中の精製したKb 溶液(0.3μM)を変化する温度(すなわち、4℃、23℃、32℃、37℃、42℃、および47℃)に1時間暴露した。次いでタンパク質をモノクローナル抗体Y3およびプロテインAセファローズビーズ(ファーマシア(Pharmacia)、ニュージャージイ州ピスカタェイ) と4℃において、それぞれ、インキュベーションすることによって免疫沈澱した。試料を12.5% SDS−PAGEにより分析し、次いでクーマッシーブルーで染色した。ゲル上の2つの厚いバンドは抗体Y3の重鎖および文字コードである。
他の手順において、Kb sol (0.3μM)を PBS中で23℃において50μMのペプチドと2時間インキュベーションして、Kb sol −ペプチド複合体を形成させた。1%TX−100の添加後、試料を12℃、37℃、または47℃の温度に1時間暴露した。前述したように、複合体を免疫沈澱させ、そして SDS−PAGEにより分析した。第3手順において、Kb sol (2.7μM)を、それぞれ、50μMの OVA−8、 VSV−8または SEV−8ペプチドと23℃において2時間インキュベーションした。試料を5%ポリアクリルアミドIEF ゲル上の適用した。 IEFをpH5〜7から展開し、そしてゲルを銀で染色した。
次に、 VSV−8ペプチドをクロラミン−T法(Hunterら、Nature 194 : 495-6(1962))を使用して放射線標識化しそして遊離 125IをC18カラム(OPCカートリッジ、アプライド・バイオシステムス(Applied Biosystems) 、カリフォルニア州フォスターシティー)により除去した。標識化ペプチドをC18逆相HPLCによりさらに精製した。溶離後、標識化ペプチドを凍結乾燥し、そして PBSの中に再懸濁させた。 274nmにおいてチロシンの吸光係数(1420M-1cm-1)を使用して、[125I]VSV−8(約 250Ci/mmol)の比活性を分光光度測定的に決定した。第1に、Kb sol (0.5μM) を[125I]VSV−8(1.5nM) および非標識化 VSV−8(50nM) と23℃において16時間混合して、複合体を形成した。
試料の一部分をゲル濾過(スペロース(Superose) 12、ファーマシア(Pharmacia)、ニュージャージイ州ピスカタェイ) より PBS中で分析した。溶離後、各画分 (0.05ml)の中に含有される放射能を測定した。タンパク質を 280nmにおける吸収によりモニターした。第2手順において、[125I]VSV−8(0.39nM) を1%ウシ血清アルブミン(BSA) を含有する PBS中のKb solの種々の濃度と混合した。23℃において2〜16時間インキュベーションした後、Kb sol −ペプチド複合体を遊離ペプチドから小さいゲル濾過(Bio−Gel P30 、バイオラド (BioRad) 、カリフォルニア州リッチモンド)により PBS中で分離した。
P30ゲル濾過は、5分以内で、結合したペプチドおよび遊離のペプチド95%を越える分離を可能とした。結合したペプチドおよび遊離のペプチドの放射能を測定し、そしてデータを線形回帰により分析した。提供されたKb sol の最大レベルにおいて、合計の標識化ペプチドの約65%が結合した。Kb sol への標識化ペプチドのこの最大の結合能力は、多分 VSV−8に結合した 125Iにより放射能のために、経時的に劣化した。
第3手順において、各試料は0.39nMの[125I]VSV−8(約18,000cpm)、示した濃度の非標識化ペプチド、および非標識化ペプチドの不存在下に約50%の[125I]VSV−8結合性を与える30nMのKb solを72μlの最終体積で含有した。すべての成分は溶解し、そして1%BSA を含有する PBS中で希釈した。23℃において2〜16時間インキュベーションした後、50μlの試料をP30ゲル濾過により前述したように分析した。非標識化ペプチドについての解離定数を、記載されているように[125I]VSV−8およびKb sol に対する[125I] VSV−8の結合の50%阻止を与える非標識化ペプチドのモル濃度から決定した。(参照、Mullerら、Meth.Enzymol.92 :589-601(1983)。)
次いでKb sol (0.3μM) および[125I]VSV−8(0.39nm)を4℃、23℃、および37℃においてインキュベーションし、そしてアソシエーションを種々の時間においてP30ゲル濾過により決定した。必要なとき、示した濃度におけるインキュベーションの前に、ネズミβ2ミクログロブリンを添加した。ネズミβ2ミクログロブリンは、組換えドロソフィラ (Drosophila) 細胞の培養上澄み液からの抗β2ミクログロブリンポリクローナル抗体K355を使用して、アフィニティクロマトグラフィーにより調製した。(参照、また、Logdbergら、Molec.Immnol.14 : 577-587(1979) 。)
他の実験において、Kb sol (0.3μMまたは 1.8μM) および[125I]VSV−8(2.4nM)を23℃において2時間インキュベーションし、そしてペプチド−Kb sol 複合体をP30ゲル濾過により単離した。試料は非常に少量の[125I]VSV−8およびKb sol 複合体(最大において、 2.4nM)および約50〜 300nmの最終濃度で空のKb sol を含有した。いくつかの試料に、3μMのβ2ミクログロブリン、3μMのβ2ミクログロブリン+20μMの非標識化 VSV−8、20μMの非標識化 VSV−8、または1%TX−100 を添加した。試料を37℃において種々の時間インキュベーションし、そして解離の程度をP30カラムの通過により決定した。
B.論 考
クラス 1 MHC分子は細胞障害性Tリンパ球に対して抗原性ペプチド提示する。クラス 1 MHC分子への生体外のペプチドの直接結合は、結合部位における前以て結合したペプチドの存在 (ChenおよびPerhm,Nature 337:743-5(1989))または結合特異性の欠如により妨害された。(参照、例えば、 Frelingerら、J.Exp.Med.172 :827-34(1990);Choppin ら、J.Exp.Med.172 : 889-99(1990);Chenら、J.Exp.Med.172 : 931-6(1990) 。)
切頭H−2Kb sol およびネズミβ2ミクログロブリン遺伝子で形質転換したドロソフィラ (Drosophila) 細胞から精製した、可溶性、空のクラス 1 MHC分子へのペプチドの結合の生体外分析をここに開示する。結果が証明するように、ペプチドの結合は非常に急速であり、そして自然にプロセシングされたペプチド(オクタペプチド;参照、例えば、 Van Bleekら、Nature348 : 213-6(1990) ; Falkら、Nature 351: 290-6(1991)) はナノモル範囲のKb sol に対して最高の親和性を有し、そしてオクタペプチドと複合化したKb sol は安定であるが、わずかに短いか、あるいは長いペプチドと複合化したものは寿命が短いことを示す。
遊離重鎖とβ2ミクログロブリンとの間の相互作用は洗浄剤の不存在下において基本的には可逆的である。ペプチドは、β2ミクログロブリンを解離しないで、空のクラス 1 MHC分子に自発的に結合する。しかしながら、過剰のβ2ミクログロブリンは、ヘテロダイマーが不安定である条件下にβ2ミクログロブリンとの遊離重鎖の再アソシエーションの結果として、空のクラス1へのペプチドの結合を明らかに促進する。
可溶性H−2Kb 分子(重鎖のα1α2α3ドメインから構成されている)およびネズミβ2ミクログロブリンを、切頭重鎖およびβ2ミクログロブリン遺伝子で同時に形質転換したドロソフィラ (Drosophila) 細胞の培養上澄み液から調製した。予備的実験が示唆するように、ドロソフィラ (Drosophila) 細胞は細胞表面上の内因性ペプチドを欠くクラス 1 MHC分子を発現する。空のクラス 1 MHC分子の性質のいくつかは、それらが37℃において安定性に劣り、そしてそれらの構造はペプチドの結合により安定化されるという観察を包含する。(参照、例えば、Schumacherら、Cell 62 :563-7(1990); Ljunggren ら、Naure 346 : 476-80(1990)。)
精製した可溶性Kb がまた空であることを確証するために、洗浄剤不含溶液中のそれらの安定性を検査した(データは示されていない)。驚くべきことには、47℃に1時間加熱したタンパク質はコンフォメーションナル抗体Y3を使用する免疫沈澱によりよく回収された。この予期せざる結果により、われわれは1%トリトンX−100(ポリオキシエチレン(9)オクチルフェニルエーテル)をタンパク質溶液に添加した。なぜなら、クラス1細胞の安定性を試験する同様な実験を洗浄剤リゼイト中で常に実施されたからである(参照、Schumacherら、前掲)。洗浄剤の存在下に得られた結果は、精製したKb solが今回37℃において不安定であることを示す(データは示されていない)。
この証拠および他の証拠が示唆するように、Kb sol ヘテロダイマーは高温において重鎖およびβ2ミクログロブリンに解離し、そして洗浄剤はβ2ミクログロブリンが解離した遊離重鎖と再アソシエーションすることを防止することができる(下を参照)。第2に、精製したKb sol をペプチドで可溶化する可能性を研究した。最初に記載した実験の結果が証明したように、自然にプロセシングされたペプチドであることが示された(参照、 Van Bleekら、前掲)オクタペプチド(小胞性口内炎ウィルスのヌクレオキャプシドタンパク質[VSV−8] 、下表3参照) と混合したときのみ、タンパク質を安定化することができる。これらの観察は、前述の空のクラス1分子の特性と一致する。
精製したKb sol 分子が空であるという独立の支持は、自然条件下の等電点電気泳動(IEF)により提供される(データは示されていない)。ドロソフィラ (Drosophila) 細胞から精製された可溶性Kb は、ヒトリンパ芽球様細胞系から精製した HLA−A2分子より非常に簡単なパターンを示した。(参照、Silberら、Nature 350:619-22(1991)) 。 IEF上の HLA−a2の複雑なパターンは、多分分子に結合した不均質ペプチドの存在の結果である。精製したKb solの簡単なバンドは、内因性ペプチドの不存在を示す。
さらに、抗原性ペプチドとのKb sol のインキュベーションは IEFゲル上のバンドの明確なシフトを引き起こし、ペプチドの結合のためのKb solの等電点の変化を反映した。前に結合した内因性ペプチドの除去後、 HLA−A2をペプチドと「再構成条件」においてインキュベーションしないかぎり、 HLA−A2分子をペプチドと単に混合したとき、このようなバンドのシフトは HLA−A2分子において観察されなかったことに注意すべきである。一緒にすると、自然 IEFについてのこれらの観察は、また、ドロソフィラ (Drosophila) 細胞から精製した可溶性Kb が空であることを示す。
125I標識化 VSV−8とKb sol とのアソシエーションはゲル濾過により証明された(示されていない)。高分子量物質の放射能はペプチド−Kb sol 複合体に相当するが、低分子量物質のそれは遊離ペプチドを表す。非標識化VSV およびオバルブミン(OVA) ペプチドは標識化 VSV−8と競争することができ(下を参照)、[125I]VSV −8がKb sol 分子に特異的に結合することを示す。逆相HPLCは、Kb 結合[125I]VSV−8がインプットペプチドと同一の保持時間を有することを明らかにした。標識化 VSV−8のKb sol への結合は飽和可能であり、約33nMの解離定数(KD ) を示す。スキャッチャードプロットのx軸から、標識化 VSV−8の約65%はKb に結合することができることが認められた。
Kb に対する種々のペプチドの親和性を決定するために[125I] VSV−8を使用する競合ラジオイムノアッセイ(RIA) を実施した(データは示されていない)。 RIAのために使用した阻害性ペプチドを表5に記載する。各ペプチドについてのKD を同様に要約する。
自然にプロセシングされた大きさのペプチド(VSVおよび OVAについて8マー、およびセンダイウィルスのヌクレオタンパク質[SEV]について9マー)は、 2.7〜4.7nM の範囲で最高のかつ顕著に類似する親和性を有した。自然のペプチドのこの過度に高い親和性は最近の観察と一致する。(参照、例えば、Schumacherら、Naurte 350:703-6(1991); Christnickら、Nature 352:60-70(1991) 。) しかしながら、1または2程度に少ない残基だけ短いか、あるいは長いペプチドは親和性を2〜 100ファルターだけ低下した。
親和性のこの減少は非常に長いペプチドについてなおいっそう激烈である;すなわち、24マーのペプチド(OVA−24)の親和性は OVA−8のそれより10,000倍低い。これらの結果は、より長いペプチドを使用する初期の報告がマイクロモルの範囲の親和性を主張している理由を説明することを助ける。(参照、例えば、 FrelingerらおよびChoppinら、両者共前掲。) カルボキシル末端におけるペプチドの延長はアミノ末端における延長より親和性に対する崩壊性が非常に低いことは、とくに興味あることである。
HLA−A2の3次元の構造に従い、ペプチド結合性みぞは逆平行β鎖上の2つの長いαらせんにより形成され、そしてくぼみは約25オングストロームの長さであり、これは約8残基の延長したペプチド鎖を収容することが提案された(参照、例えば、Bjorkmanら、Nature 329:506-12(1987)) 。くぼみの1末端において、α1およびα2らせんは一緒に緊密に閉じるようになるが、他方の末端において、くぼみはかなり開いている。ここで、 VSVおよび OVAの両者のペプチドはくぼみに同一向きで結合し、そしてペプチドのカルボキシル末端はくぼみの比較的開いた末端と相互作用することができるので、カルボキシル末端におけるペプチドの延長は厳しい立体障害を引き起こさないと推測される。
次いで実験を実施して、それぞれ、4℃および23℃におけるKb へのペプチドの結合速度を検査した。結合はことに23℃において非常に急速であり、標識化ペプチドの極めて低い濃度(約0.4nM)においてさえ、約5分の半減期である。これは約2時間のアソシエーションの半減期を示す従来の観察と対照的である。(参照、例えば、 Choppinら、前掲。)再び、合計の標識化ペプチドの65%のみが結合することができた。過剰のβ2ミクログロブリンの添加は、Kb ヘテロダイマーが安定である(集合体で止まる)ような低い温度においてペプチドの結合の反応速度論に影響を与えなかった。
これが意味するように、β2ミクログロブリンの交換はペプチドの結合のための前提条件ではない;すなわち、ペプチドはβ2ミクログロブリンの解離なしに空のクラス1分子に自発的に結合することができる。対照的に、過剰の遊離β2ミクログロブリンは37℃においてペプチドの結合を明らかに促進する(データは示されていない)。添加するβ2ミクログロブリンの濃度を増加したとき、より多くのペプチドがKb 分子に結合した。空のKb は37℃において不安定であるので、ヘテロダイマーの小部分は重鎖およびβ2ミクログロブリンに解離するに違いなく、これにより、ヘテロダイマーは遊離重鎖および遊離β2ミクログロブリンと平衡にあるに違いない。
次いで、β2ミクログロブリンの添加はペプチドに結合できるヘテロダイマーの形成に向かって平衡をシフトするであろう。この見解は次の最近の観察により支持される。すなわち、通常の細胞表面上の実質的な数のクラス1遊離重鎖が存在し、そして外因的に添加されたβ2ミクログロブリンは、β2ミクログロブリンと遊離重鎖との再アソシエーションの結果として、空のクラス1分子へのペプチドの結合を促進する。(参照、例えば、Rockら、Cell 65 : 611-620(1991); Kozlowskiら、Nature 349:74-77(1991) ; Vitielloら、Science 250 : 1423-6(1990)。)
37℃においてペプチドの解離の反応速度論を次いで観察した。[125I]VSV−8およびKb 複合体をゲル濾過により単離した直後に、50または 300nMのKb を含有する試料を37℃の温度に暴露した。いくつかの試料に3μMのβ2ミクログロブリンおよび/または20μMの非標識化 VSV−8、または1%のTX−100 を補充した。Kb からの標識化ペプチドの解離を種々の時間において測定した(示されていない)。
大過剰の非標識化ペプチドの存在において、ペプチドの解離速度は一次反応速度論に従い、解離の半減期は約36分であった(3.2×10-4/秒の解離速度定数)。標識化ペプチドのこの予期せざる、比較的に急速な解離は安定なペプチド−クラス1複合体のいくつかの現在の見解と一致しない。事実、確認された結果(示されていない)は、Kb および VSV−8複合体が安定であることを証明した。この解釈は非標識化 VSV−8のそれ(3.7nM) に比較して放射線標識化 VSV−8の10倍低い親和性(33nM) から発生するに違いない。
洗浄剤を非標識化ペプチドの代わりに添加したとき、一次反応速度論がまた観察され、洗浄剤はペプチドの解離プロセスを可逆的とすることを示した。対照的に、ペプチドの解離のプロフィルは非標識化ペプチドまたは洗浄剤の不存在下に一次反応速度論に従わなかった。これが示唆するように、ペプチドのアソシエーション/解離は可逆的であり、そしてペプチドの結合はヘテロダイマーの濃度に依存する(Kb の50nMと 300nMとの間の反応速度論を比較する) 。過剰のβ2ミクログロブリンを添加したとき、これはいっそう明らかとなった。
これらの結果は、完全でないにしても、洗浄剤の不存在下に、重鎖、β2ミクログロブリンおよびペプチドの間の相互作用は37℃において基本的に可逆的であるという従来の議論を支持する。洗浄剤、例えば、TX−100 はβ2ミクログロブリンが37℃において遊離重鎖と再アソシエーションするのを防止することができるであろう。これは、Kb が洗浄剤の不存在下に高温にいったん加熱されると、コンフォメーションナル抗体により効率よく免疫沈澱することができる理由を合理的に説明することができるであろう(示されていない)。
興味あることは、β2ミクログロブリンの添加は過剰の非標識化ペプチドの存在下のペプチドの解離を抑制せず、標識化ペプチドがβ2ミクログロブリンの解離なしに複合体から解放されることを示す。しかしながら[125I]VSV−8の親和性は自然ペプチドのそれより約10倍低いことを推奨すべきである。したがって、これは必ずしも自然ペプチドの場合ではない。
生体外のペプチド結合アッセイを使用して研究において、クラス1分子へのペプチドの結合は簡単な質量作用およびリガンド−レセプターの相互作用であることが示唆される。ここにおいて使用したアプローチは、クラス1分子へのペプチドの結合の特異性およびペプチド−クラス1分子の複合体とT細胞レセプターとの相互作用の特性決定を可能とする。
実施例6.治療的応用
A.クラス1分子のバンク
ドロソフィラ (Drosophila) 細胞系の溜または「バンク」を確立しそして維持することができ、各細胞系は50〜100 の最も共通のクラス1分子の1つを発現する。これらのタンパク質をコードするタンパク質はそれらを含有する細胞系から−−例えば、ポリメラーゼ連鎖反応(参照、 Ennisら、PNAS USA 87 :2833-7(1990))により得られた HLA変異型に基づいてクローニングし、そして適当なベクター、例えば、昆虫発現ベクターの中に挿入して、各 HLA変異型を発現する細胞系を発生することができる。
例えば、次のプロトコールに従う試験を使用して、選択のウィルスから誘導されたどのペプチドが異なるクラス1分子に最もよく結合するかを決定することができる。種々の培養物を適当に標識化するか、あるいはカタログ化して、特定のペプチドとともにどのクラス1分子が最もよく使用されるかを示すことができる。あるいは、必要に応じて一時的培養物を確立することができる。ここにおいて論じたように、昆虫細胞の培養物をベクターとほぼ48時間インキュベーションした後、培養物は明らかに空の MHC分子を発現することができ、これらの分子はCD8細胞を活性化する目的で選択の1または2以上のペプチドを負荷することができる。
B.「特別の」細胞系の調製
HLAの型別後、好ましい HLAを発現するドロソフィラ(Drosophila)細胞が入手可能でない場合、好ましい HLAをコードするcDNAをポリメラーゼ連鎖反応の使用によりクローニングすることができる。上の節B.1.に開示されているプライマー(配列識別番号1〜配列識別番号12)を使用して適当な HLA−A,−B,−C,−E,−F、または−GcDNA を別々の反応において増幅し、次いでこれらを上の実施例1、節1に開示されている方法に記載されているようにクローニングしそして配列決定することができる。次いで DNAを PCR反応からジーン・クリーン(Gene Clean) キット(Bio 101 、カリフォルニア州サンディエゴ) で精製し、そして pRmHa−3の SmaI部位に直接結合する。
個々のクローンを単離し、配列を評価し、そして HLAを発現する安定なドロソフィラ (Drosophila) 細胞を確立する。あるいは、組換えプラスミドの大量の集団を大規模に増殖し、そして DNAを塩化セシウム勾配により精製することができる。次いで精製した DNAを使用してリン酸カルシウム沈澱技術によりシュネイダー細胞をトランスフェクションする。24時間後、沈澱を洗浄して細胞を除去し、そして1mM CuSO4を含有する新鮮なシュネイダー培地と置換する。48時間後、一時的にトランスフェクションされた細胞の大量の集団を、シンジェネイックペプチドまたは特定のタンパク質のプロテアーゼ消化物とのインキュベーション後、CD8の生体外活性のために使用する。
次いで、クローニングした HLAを発現する安定な細胞系をドロソフィラ (Drosophila) 細胞中で確立する。あるいは、 PCR反応からのクローニングされた組換え分子の大量の集団を一時的に発現する昆虫細胞の集団を生体内のCD8活性化に使用することができる。
また、細胞系が発現した MHCが生体内で発現された要素でない場合、ペプチドとインキュベーションしたクラス1MHC を発現する昆虫細胞を使用して、ハプロタイプ特異的CD8を生体外で活性化することができる。これは、アレレの制限のために生体内で不可能である、特定の MHCとアソシエーションした特定の抗原を認識するCD8細胞を増殖する独特の機会を提供する。例えば、インフルエンザウィルスの核タンパク質からのペプチド(NP) は通常Db 分子に限定される;しかしながら、このようなペプチドはKb に結合することができ(Db よりいっそう弱くであるが) そしてKb に対するある程度の熱安定性を発生することができる(第3図参照)。
さらに、NPペプチドと前インキュベーションし、そしてB6マウスからの脾細胞と同時培養したKb 発現ドロソフィラ (Drosophila) 細胞は、NPペプチドとアソシエーションしたKb 分子を特異的に認識するCD8の生体外活性化を生ずる。さらに、オバルブミンから誘導されたKb 制限ペプチド(OVA) およびDb 発現ドロソフィラ (Drosophila)細胞は、 OVAペプチドを含有するDb を特異的に認識するCD8の増殖を生ずる。このようなCD8はオバルブミンタンパク質コードするcDNAでトランスフェクションした細胞(EL4 OVA)を殺すことができ、生体内で多少のDb 分子が OVAペプチドを負荷されることを示す。
したがって、この系は、生体内ではペプチドのための制限要素ではない、クラス1分子により提示される特異的抗原に対してCD8を増殖する機会を提供する。CD8により認識されかつ殺されるべき細胞について十分な抗原が生体内で前記クラス1により圧力されるが、抗原は生体内でこのようなCD8を増殖するために十分ではない。ドロソフィラ (Drosophila) 細胞により発現される空のクラス1分子にペプチドを負荷することによって、過剰の抗原性ペプチドをクラス1分子に非競合的環境において提供し、こうしてこの複合体を認識する特異的CD8を活性化するために十分な抗原をクラス1により圧力されるようにすることによって、生体内の制限を無視することが多分可能である。
C.エイズの処置
生体外活性化された細胞を生体内治療のために患者に投与することができる。好ましくは、個体のクラス1遺伝子型(ハプロタイプ)をまず決定する。普通の組織の型別はこの目的に適当であり、そして処置センターにおいてあるいはいくつかの適当な商業的操作により実施することができる。個体の1または2以上の HLA型がいったん決定されると、個々の患者に適当なペプチドとクラス1分子との最良の組み合わせが確認され、そして前述したように調製され、そして適当なドロソフィラ (Drosophila) 細胞およびペプチドが患者の処置のために提供される。
次いで、患者の血液からの休止または前駆体のCD8(T)細胞を、ドロソフィラ (Drosophila) 細胞の培養により生産された適当なペプチド負荷MHC で刺激する。活性化後、CD8細胞を患者の血流の中に再導入し、そして患者の疾患のプロセスをモニターし続ける。細胞成分を取り出しそして再導入する方法はこの分野において知られており、そして米国特許第 4,844,893号(Honsikら)および米国特許第 4,690,915号(Rosenberg) の中に例示されるような手順を包含する。
疾患が十分に軽減されるまで、追加の処置を必要に応じて投与することができる。同様な処置のプロトコールは、他の免疫抑制された個体、例えば、移植の患者、中年過ぎの患者などとともに使用するために適当である。
D.癌の処置
癌の患者において、前述のものに類似する処置の手順を利用する。しかしながら、このような患者において、腫瘍の塊を減少する普通の治療をここに記載する免疫治療に先行するであろうことが予測される。したがって、免疫細胞を破壊する傾向がある普通の治療、例えば、放射線または化学療法の開始前に、推定上の患者からの血液試料を獲得しそして貯蔵(例えば、凍結により)することが好ましい。存在する場合、わずかの形態の癌はウィルスの感染に直接応答して発生するので、免疫処置のための標的ペプチドが容易に観察されることが少ない。
しかしながら、最近の研究が示すように、腫瘍遺伝子ras, neu、およびp53中の突然変異はすべての癌の場合の50%程度に多さで癌に寄与する。こうして、分子のこれらの突然変異した領域から誘導されたペプチドは、現在の治療のための標的として主要な候補である。ここに開示するプロトコールに従い、個々の患者のために最良のペプチドとクラス1分子との組み合わせを決定し、そして投与することができる。
例えば、多数の腫瘍は影響を受けた個体から誘導されたCD8細胞により生体外で認識される抗原を発現する。こうして、正常細胞中で発現されないこのような抗原、ならびにCD8細胞にそれらを提示する HLA型を、正確にターゲッティングされた免疫治療のために、本発明の方法により同定することができる。例えば、van der Bru-ggenらは、その発現が特定の遺伝子により指令されそしてHLA A1により提示されるように思われる抗原を記載した(Science 254 :1643-1647(1991))。種々のヒト腫瘍抗原が単離されそして記載されているので、それらはここに記載するような免疫治療の応用のためのすぐれた候補となる。
E.組み合わせた治療/接合体
望ましくない活性化されたCD8細胞−−すなわち、移植片の拒絶を刺激することができるものの処置または除去のために、「空の」クラス1分子をさらに毒性のタンパク質(例えば、シュードモナス毒素)と接合することができる。接合体を構成し、次いでクラス1分子の部分に適当なペプチドを負荷し、次いでこれはクラス1分子−ペプチドの組み合わせのためのレセプターをもつT細胞を「誘引し」または「捜し出す」ので、抗原特異的CD8細胞を排除することができる。このアプローチは、また、移植片拒絶の原因となるT細胞を排除することができる。
例えば、1つのこのような手順は次の工程を包含する:(1)可溶性、空の、ヒトクラス 1 MHC分子をドロソフィラ (Drosophila) 細胞の培養物から単離する;(2)空のヒトクラス 1 MHC分子を毒素に生体外で結合する;(3)生体外で空の結合されたクラス 1 MHC分子を抗原性ペプチド(例えば、移植された組織から誘導されたペプチド)と、 MHC分子にペプチドを負荷させるために十分な時間の間接触させる;(4)ペプチドを負荷された、結合した分子を単離する;そして(5)処置を必要とする個体、例えば、移植を行った個体に前記ペプチド負荷分子を投与する。
同様に、ある種の明確なT細胞の集団は糖尿病、慢性関節リウマチおよび他の推定上の自己免疫疾患の原因として関係づけられている。このような細胞は、また、前述の手順により選択的に排除することができる。また、このプロトコールをアレルギー性患者の適用が考えられるであろう。
他の別の治療のモードにおいて、本発明の生体外で活性化されたCD8細胞を他の免疫原と組み合わせて投与することができる。例えば、米国特許第 5,045,320号に開示されている大きい多価の免疫原を活性化されたCD8細胞と組み合わせて投与することができる。
また、T細胞の分化および活性化を仲介するサイトカイン、例えば、IL2およびIL4を同様によく投与することができることが考えられる。なぜなら、サイトカインは生体内で腫瘍に対するT細胞の応答を刺激することができるからである。IL2は、CTL(CD8)前駆体の増殖および分化においておよびCD8の増殖において、主要な役割を演ずると信じられる。癌患者へのIL2の投与は、腫瘍特異的T細胞の誘発に関係するように思われる、改良された抗腫瘍の応答にしばしば関連する。しかしながら、IL2の最良の治療効果はIL2の全身的投与よりむしろ連続的局所的投与により得ることができる。こうして、IL2遺伝子構成体で腫瘍細胞をトランスフェクションすることによって、局所的送り出しを達成することができることがここにおいて示唆される。
KarasuyamaおよびMelcher,Eur.J.Immunol.18:97-104(1988)に記載されているように、IL2 cDNAを構成する。IL2の完全なcDNA配列をプラスミドpBMGneo IL2から XhoI断片として獲得し(参照、KarasuyamaおよびMelcher 、前掲) そして pRmHa−3の SalI部位の中に直接結合する。正しい向きでインサートをもつ組換え pRmHa−3プラスミド(HindIII を使用する制限マッピングにより決定する)をセシウム勾配により精製し、そしてリン酸カルシウム技術によりシュネイダー2細胞を共トランスフェクションするために使用する。
(プラスミドDNA の混合物をこの目的のために調製した:10μgのIL2 cDNA含有 pRmHa−3、6μgの MHCクラス1重鎖またはβ2ミクログロブリンを含有する pRmHa−3の各々および2μgのphshsneoDNA 。) 重鎖、β2ミクログロブリンおよびIL2を発現するように CuSO4で誘発可能な、安定な細胞系をG418培地中でトランスフェクション体を増殖することによって得た。
これらの安定な細胞系をペプチドで被覆し、そして前述したように生体外アッセイにおいて使用した。IL2でトランスフェクションした腫瘍細胞は親の腫瘍細胞に対するCTL(CD8)活性を増強し、そして生体内の抗腫瘍または細胞障害性の応答の誘発におけるCD4およびTヘルパーの機能をバイパスする。したがって、IL2遺伝子との共トランスフェクションによりドロソフィラ (Drosophila) 系の可能性を増加することが、ここにおいて示唆される。
以上の説明は本発明の例示を意図し、本発明を限定しない。本発明の精神および範囲から逸脱しないで多数の変化および変更を実施することができる。