JP2010077205A - ポリアミド樹脂組成物 - Google Patents
ポリアミド樹脂組成物 Download PDFInfo
- Publication number
- JP2010077205A JP2010077205A JP2008244716A JP2008244716A JP2010077205A JP 2010077205 A JP2010077205 A JP 2010077205A JP 2008244716 A JP2008244716 A JP 2008244716A JP 2008244716 A JP2008244716 A JP 2008244716A JP 2010077205 A JP2010077205 A JP 2010077205A
- Authority
- JP
- Japan
- Prior art keywords
- polyamide resin
- diamine
- resin composition
- temperature
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Compositions Of Macromolecular Compounds (AREA)
Abstract
【解決手段】カルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂に、無機充填材を含むことを特徴とするポリアミド樹脂組成物。
【選択図】なし
Description
しかしながら、このポリアミド樹脂は、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンの3種のジアミンを特定の比率で用いたポリオキサミド樹脂ではない。
S. W. Shalaby., J. Polym. Sci., 11, 1(1973) L. Franco et al., Macromolecules., 31, 3912(1988)
本発明で用いるポリアミドは、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂に、無機充填材を含むことを特徴とするポリアミド樹脂組成物(PA92/62T)である。
本発明に用いるポリアミド樹脂PA92/62Tは、ポリアミドを製造する方法として知られている任意の方法を用いて製造することができる。本発明者らの研究によれば、ジアミン及び蓚酸ジエステルをバッチ式又は連続式で重縮合反応させることにより得ることができる。具体的には、以下の操作で示されるような、(i)前重縮合工程、(ii)後重縮合工程の順で行うのが好ましい。
まず原料の蓚酸ジエステルを容器内に仕込む。容器は、後に行う重縮合反応の温度および圧力に耐え得るものであれば、特に制限されない。その後、容器を原料のジアミンと混合する温度まで昇温させ、次いでジアミンを注入し重縮合反応を開始させる。原料を混合する温度は、原料の蓚酸ジエステルおよびジアミンの融点以上、沸点未満の温度であり、かつ蓚酸ジエステルとジアミンの重縮合反応によって生じるポリオキサミドが熱分解しない温度であれば特に制限されない。例えば、1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン、1,6−ヘキサンジアミンの混合物からなり、かつC9ジアミン(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比が1:99〜99:1であるジアミンと蓚酸ジブチルを原料とするポリオキサミド樹脂の場合、上記混合温度は15℃から300℃が好ましい。また、C9ジアミン(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比は、約5:95〜90:10の場合、常温で液状か又は50℃程度に加温するだけで液化するので取り扱いやすいためより好ましい。混合温度が縮合反応によって生成するアルコールの沸点以上の場合、アルコールを留去、凝縮する装置を備えた容器を用いるのが望ましい。また、縮合反応によって生成するアルコール存在下で加圧重合する場合には、耐圧容器を用いる。蓚酸ジエステルとジアミンの仕込み比は、蓚酸ジエステル/上記ジアミンで、0.8〜1.2(モル比)、好ましくは0.91〜1.09(モル比)、更に好ましくは0.98〜1.02(モル比)である。
本発明に用いるポリアミド樹脂PA92Cの分子量に特別の制限はないが、96%濃硫酸を溶媒とし、ポリアミド樹脂濃度が1.0g/dlの溶液を用い、25℃で測定した相対粘度ηrが1.8〜6.0の範囲内である。好ましくは2.0〜5.5であり、2.5〜4.5が特に好ましい。ηrが1.8より低いと成形物が脆くなり物性が低下する。一方、ηrが6.0より高いと溶融粘度が高くなり、成形加工性が悪くなる。
本発明は、上記のポリアミド樹脂に補強繊維を配合することを特徴とするが、その補強繊維としては、格別に限定されず、ガラス繊維、炭素繊維、金属繊維、鉱物繊維などの無機繊維、ポリアミド樹脂より強靭なアラミド繊維などの有機繊維を挙げることができる。補強繊維を配合することで、組成物の強度、耐クリープ性などの物性が改良される効果が顕著である。特にガラス繊維と炭素繊維が好ましい。
本発明で使用できる無機粒子としては、金属、金属酸化物、無機化合物などの粒子が挙げられ、用途に応じて適宜選択できる。具体的には、例えばタングステン、鉄、亜鉛、錫、鉛、銅などの金属、タングステン銅、タングステン銀などの金属合金、酸化鉄、酸化亜鉛などの金属酸化物、更に、硫化モリブデン等の硫化物、などの粒子が挙げられる。
本発明に用いるポリアミド樹脂PA92/62Tには、本発明の効果を損なわない範囲で他のジカルボン酸成分を混合する事が出来る。蓚酸以外の他のジカルボン酸成分としては、マロン酸、ジメチルマロン酸、コハク酸、グルタル酸、アジピン酸、2−メチルアジピン酸、トリメチルアジピン酸、ピメリン酸、2,2−ジメチルグルタル酸、3,3−ジエチルコハク酸、アゼライン酸、セバシン酸、スベリン酸などの脂肪族ジカルボン酸、また、1,3−シクロペンタンジカルボン酸、1,4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、さらにテレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、2,7−ナフタレンジカルボン酸、1,4−ナフタレンジカルボン酸、1,4−フェニレンジオキシジ酢酸、1,3−フェニレンジオキシジ酢酸、ジ安息香酸、4,4’−オキシジ安息香酸、ジフェニルメタン−4,4’−ジカルボン酸、ジフェニルスルホン−4,4’−ジカルボン酸、4,4’−ビフェニルジカルボン酸などの芳香族ジカルボン酸などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。さらに、トリメリット酸、トリメシン酸、ピロメリット酸などの多価カルボン酸を溶融成形が可能な範囲内で用いることもできる。蓚酸以外の他のジカルボン酸成分の配合量は、一般的には全ジカルボン酸成分を基準に5モル%以下が好ましい。
本発明のポリアミド樹脂組成物の成形方法としては、ポリアミド樹脂と補強繊維を予めブレンドし、あるいは成形機の途中で補強繊維を投入する、射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミド樹脂組成物に適用できる公知の成形加工法はすべて可能であり、これらの成形法によってフィルム、シート、成形品、繊維などに加工することができる。
本発明の繊維補強ポリアミド樹脂組成物を用いた成形物は、その優れた特性のゆえに様々な用途において有用であり、従来ポリアミド樹脂組成物の成形物が用いられてきた各種成形品、シート、フィルム、パイプ、チューブ、モノフィラメント、繊維、容器等として自動車部材、コンピューター及び関連機器、光学機器部材、電気・電子機器、情報・通信機器、精密機器、土木・建築用品、医療用品、家庭用品など広範な用途に使用できる。とりわけ、補強されているので自動車、電気・電子機器などの用途に有用である。
以下、実施例を挙げて本発明を具体的に説明するが、本発明はこれらにより何ら制限されるものではない。
なお、実施例中の測定は以下の方法により行った。
ηrはポリアミドの96%硫酸溶液(濃度:1.0g/dl)を使用してオストワルド型粘度計を用いて25℃で測定した。
Tm及びTcは、PerkinELmer社製PYRIS Diamond DSC用いて窒素雰囲気下で測定した。30℃から300℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、300℃で3分保持したのち、−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、次に300℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。可塑剤含有試料では得られたDSCチャートから昇温ファーストランの吸熱ピーク温度を、可塑剤を含まない試料では昇温セカンドランの吸熱ピーク温度をそれぞれ降温ファーストランの発熱ピーク温度を各試料の結晶化温度とした。
Tdは島津製作所社製THERMOGRAVIMETRIC ANALYZER TGA−50を用い、熱重量分析(TGA)により測定した。20ml/分の窒素気流下室温から500℃まで10℃/分の昇温速度で昇温し、Tdを測定した。
溶融粘度はティー・エイ・インスツルメント・ジャパン社製溶融粘弾性測定装置ARESに25mmのコーン・プレートを装着して、窒素中、290℃、せん断速度0.1s-1の条件で測定した。
東邦マシナリー社製真空プレス機TMB−10を用いてフィルム成形を行った。500〜700Paの減圧雰囲気下290℃(ナイロン6を用いた場合は260℃、ナイロン66を用いた場合は290℃、ナイロン12を用いた場合は230℃)で5分間加熱溶融させた後、5MPaで1分間プレスを行いフィルム成形した。次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却結晶化させてフィルムを得た。
ポリアミド樹脂を(5)の条件で成形したフィルム(寸法:20mm×10mm、厚さ0.25mm;重量約0.05g)を23℃のイオン交換水に浸漬し、所定時間ごとにフィルムを取り出し、フィルムの重量を測定した。フィルム重量の増加率が0.2%以内の範囲で3回続いた場合にポリアミド樹脂フィルムへの水分の吸収が飽和に達したと判断して、水に浸漬する前のフィルムの重量(Xg)と飽和に達した時のフィルムの重量(Yg)から式(1)により飽和吸水率(%)を算出した。
飽和吸水率(%)=100(Y−X)/X (1)
なお、上記フィルムの成形直後の重量(Xg)と上記フィルムを成形後に湿度65%、温度23℃で平衡に達したときの重量(Yg)から式(1)により算出した吸水率(平衡吸水率)をウェットでの吸水率として表中に記載した。
本発明によって得られるポリアミドの熱プレスフィルムを以下に列挙する薬品中に7日間浸漬した後に、フィルムの重量残存率(%)及び外観の変化を観測した。濃塩酸、64%硫酸、氷酢酸のそれぞれの溶液においては23℃において浸漬した試料について試験を行った。
本発明によって得られるポリアミドの熱プレスフィルムをオートクレーブに入れ、水(pH=7)、0.5mol/l硫酸(pH=1)、1mol/l水酸化ナトリウム水溶液(pH=14)中でそれぞれ121℃、60分間処理した後の重量残存率(%)、及び外観変化を調べた。
以下に示す〔1〕〜〔4〕の測定は、下記の試験片を樹脂温度290℃(ナイロン6を用いた場合は260℃、ナイロン66を用いた場合は290℃、ナイロン12を用いた場合は230℃)、金型温度80℃の射出成形により成形し、これを用いて行った。成形後直ちに調湿せずに23℃で評価したものをドライ、成形後に湿度65%、温度23℃で調湿した後に23℃で評価したものをウェットとして表中に記載した。
〔1〕 引張試験(引張降伏点強度):ASTM D638に記載のTypeIの試験片を用いてASTM D638に準拠して測定した。
〔2〕 曲げ試験(曲げ弾性率):試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D790に準拠し、23℃で測定した。
〔3〕 アイゾット衝撃強度:試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D256に準拠し、23℃で測定した。
〔4〕 荷重たわみ温度:試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D648に準拠し、荷重1.82MPaで測定した。
(5)の条件で成形したフィルムを、23℃の飽和塩化カルシウム水溶液に浸漬した。一日後、フィルムの外観を目視で観察し、クラックの有無を評価した。
600mlのトルエンと600mlのイソオクタンの混合溶液に0.1gのターシャルブチルパーオキサイドと0.01gのステアリン酸銅を加えた酸化ガソリン溶液を60℃とし、その中に30本の引張試験用試験片を浸漬し、一週間毎に溶液を替え、30日間の物性(引張強度)変化を測定した。
攪拌機、温度計、トルクメーター、圧力計、ダイアフラムポンプを直結した原料投入口、窒素ガス導入口、放圧口、圧力調節装置及びポリマー抜出し口を備えた内容積が約150リットルの圧力容器に蓚酸ジブチル28.230kg(139.56モル)を仕込み、圧力容器の内部を純度が99.9999%の窒素ガスで0.5MPaに加圧した後、次に常圧まで窒素ガスを放出する操作を5回繰り返し、窒素置換を行った後、封圧下、攪拌しながら系内を昇温した。約30分間かけてシュウ酸ジブチルの温度を100℃にした後、1,9−ノナンジアミン1.241kg(7.84モル)と2−メチル−1,8−オクタンジアミン19.639kg(124.04モル)と1,6−ヘキサンジアミン0.893kg(7.68モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が5.62:88.88:5.50)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.13であった。
蓚酸ジブチル28.462kg(140.71モル)を仕込み、1,9−ノナンジアミン16.448kg(103.88モル)と2−メチル−1,8−オクタンジアミン2.903kg(18.34モル)と1,6−ヘキサンジアミン2.150kg(18.50モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が73.83:13.03:13.14)を仕込んだほかは、製造例1と同様に反応を行ってポリアミドを得た。得られたポリアミドは白色の強靭なポリマーで、ηr=2.97であった。
蓚酸ジブチル30.238kg(149.49モル)を仕込み、1,9−ノナンジアミン4.486kg(28.33モル)と2−メチル−1,8−オクタンジアミン4.486kg(28.33モル)と1,6−ヘキサンジアミン10.79kg(92.85モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が18.95:18.95:62.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1.5時間かけて温度を270℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が270℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を285℃にし、285℃において1.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.88であった。
蓚酸ジブチル29.864kg(147.64モル)を仕込み、1,9−ノナンジアミン5.598kg(35.36モル)と2−メチル−1,8−オクタンジアミン5.598kg(35.36モル)と1,6−ヘキサンジアミン8.941kg(76.92モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が23.95:23.95:52.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が250℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を270℃にし、270℃において2時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.83であった。
蓚酸ジブチル29.107kg(143.89モル)を仕込み、1,9−ノナンジアミン5.641kg(35.63モル)と2−メチル−1,8−オクタンジアミン10.028kg(63.34モル)と1,6−ヘキサンジアミン5.223kg(44.93モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が24.76:44.02:31.22)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が240℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を265℃にし、265℃において3時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.11であった。
蓚酸ジブチル28.40kg(140.4モル)を仕込み、1,9−ノナンジアミン11.11kg(70.2モル)と2−メチル−1,8−オクタンジアミン11.11kg(70.2モル)の混合物をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.35であった。
ジアミン原料として1,9−ノナンジアミン22.25kg(140.4モル)だけを用いて、製造例1と同様に反応を行ってポリアミドを得た。得られた重合物は黄白色のポリマーであり、ηr=2.78であった。
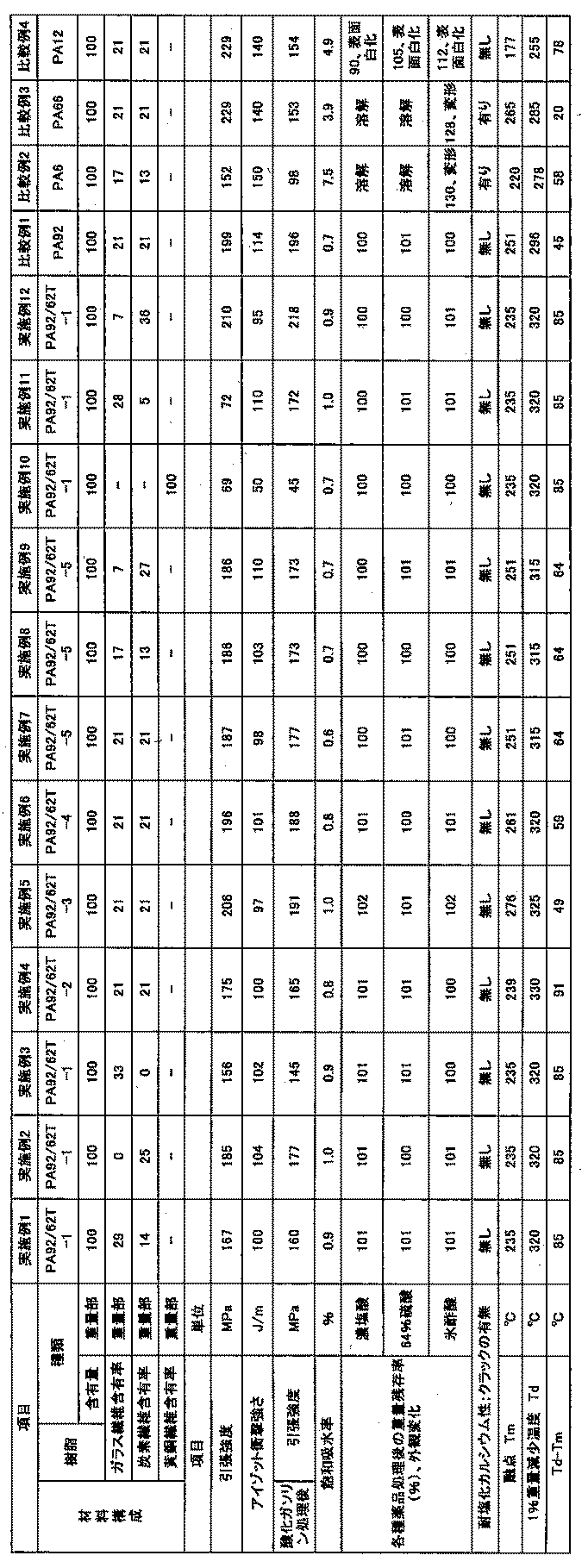
製造例1〜5、参考製造例1及び比較製造例1で製造したポリアミドPA92/62T−1〜PA92/62T−5、PA92C、PA−92、並びにナイロン6(宇部興産製、UBEナイロン1015B)、ナイロン66(宇部興産製、UBEナイロン2020B)及びナイロン12(UBESTA3020U)と、ガラス繊維(日本電気硝子製ECST-289(繊維径13μm)、炭素繊維(東邦テナックス(株)ベスファイトHTA-C6NR(繊維径7μm)、黄銅繊維(繊維径80μm)を用いて、表2に示した割合の混合物を作成した。
Claims (8)
- カルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂に、無機充填材を含むことを特徴とするポリアミド樹脂組成物。
- 前記無機充填材が、補強繊維及び/又は無機粒子である、請求項1記載のポリアミド樹脂組成物。
- 前記ポリアミド樹脂は、96%硫酸を溶媒とし、濃度が1.0g/dlのポリアミド樹脂溶液を用いて25℃で測定した相対粘度(ηr)が1.8〜6.0である、請求項1または2に記載のポリアミド樹脂組成物。
- 前記ポリアミド樹脂は、窒素雰囲気下、10℃/分の昇温速度で測定した熱重量分析における1%重量減少温度と窒素雰囲気下、10℃/分の昇温速度で測定した示差走査熱量法により測定した融点との温度差が50℃以上である請求項1〜3のいずれか1項に記載のポリアミド樹脂組成物。
- 前記ポリアミド樹脂は、C9ジアミン中の1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミンのモル比が5:95〜95:5であるジアミン成分とからなる、請求項1〜4のいずれか1項に記載のポリアミド樹脂組成物。
- 前記補強繊維が、ガラス繊維及び/又は炭素繊維である、請求項2〜5のいずれか1項に記載のポリアミド樹脂組成物。
- 前記無機粒子が、タングステン粒子である、請求項2〜5のいずれかに記載のポリアミド樹脂組成物。
- 前記無機粒子が、磁性粒子である、請求項2〜5のいずれかに記載のポリアミド樹脂組成物。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008244716A JP5584963B2 (ja) | 2008-09-24 | 2008-09-24 | ポリアミド樹脂組成物 |
| PCT/JP2009/060979 WO2009151145A1 (ja) | 2008-06-10 | 2009-06-10 | 新規なポリアミド樹脂組成物及びポリアミド樹脂含有製品 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008244716A JP5584963B2 (ja) | 2008-09-24 | 2008-09-24 | ポリアミド樹脂組成物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010077205A true JP2010077205A (ja) | 2010-04-08 |
| JP5584963B2 JP5584963B2 (ja) | 2014-09-10 |
Family
ID=42208022
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008244716A Expired - Fee Related JP5584963B2 (ja) | 2008-06-10 | 2008-09-24 | ポリアミド樹脂組成物 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5584963B2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013126830A (ja) * | 2011-12-19 | 2013-06-27 | Nsk Ltd | 電動パワーステアリング装置 |
| CN109517375A (zh) * | 2018-10-14 | 2019-03-26 | 金旸(厦门)新材料科技有限公司 | 一种导电尼龙材料及其制备方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05506466A (ja) * | 1990-02-20 | 1993-09-22 | エクソン・ケミカル・パテンツ・インク | 酸素バリヤー |
| JPH07228689A (ja) * | 1994-02-16 | 1995-08-29 | Kuraray Co Ltd | ポリアミド樹脂 |
| JP2000191771A (ja) * | 1998-12-25 | 2000-07-11 | Kuraray Co Ltd | ポリアミドおよびその組成物 |
| JP2003213126A (ja) * | 2002-01-25 | 2003-07-30 | Kanebo Ltd | 高比重樹脂組成物 |
| JP2004083817A (ja) * | 2002-08-29 | 2004-03-18 | Kuraray Co Ltd | ポリアミド |
| JP2004269550A (ja) * | 2003-03-05 | 2004-09-30 | Toray Ind Inc | 遮音性および流動性に優れたポリアミド樹脂組成物およびそれからなる成形品 |
| JP2004352890A (ja) * | 2003-05-29 | 2004-12-16 | Daisee Kogyo Kk | 樹脂組成物 |
| JP2006057033A (ja) * | 2004-08-23 | 2006-03-02 | Ube Ind Ltd | 低吸水性部材 |
-
2008
- 2008-09-24 JP JP2008244716A patent/JP5584963B2/ja not_active Expired - Fee Related
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05506466A (ja) * | 1990-02-20 | 1993-09-22 | エクソン・ケミカル・パテンツ・インク | 酸素バリヤー |
| JPH07228689A (ja) * | 1994-02-16 | 1995-08-29 | Kuraray Co Ltd | ポリアミド樹脂 |
| JP2000191771A (ja) * | 1998-12-25 | 2000-07-11 | Kuraray Co Ltd | ポリアミドおよびその組成物 |
| JP2003213126A (ja) * | 2002-01-25 | 2003-07-30 | Kanebo Ltd | 高比重樹脂組成物 |
| JP2004083817A (ja) * | 2002-08-29 | 2004-03-18 | Kuraray Co Ltd | ポリアミド |
| JP2004269550A (ja) * | 2003-03-05 | 2004-09-30 | Toray Ind Inc | 遮音性および流動性に優れたポリアミド樹脂組成物およびそれからなる成形品 |
| JP2004352890A (ja) * | 2003-05-29 | 2004-12-16 | Daisee Kogyo Kk | 樹脂組成物 |
| JP2006057033A (ja) * | 2004-08-23 | 2006-03-02 | Ube Ind Ltd | 低吸水性部材 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013126830A (ja) * | 2011-12-19 | 2013-06-27 | Nsk Ltd | 電動パワーステアリング装置 |
| CN109517375A (zh) * | 2018-10-14 | 2019-03-26 | 金旸(厦门)新材料科技有限公司 | 一种导电尼龙材料及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5584963B2 (ja) | 2014-09-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5056763B2 (ja) | ポリアミド樹脂 | |
| JP5796573B2 (ja) | ポリアミド樹脂 | |
| JP2009235225A (ja) | ポリアミド樹脂 | |
| JPWO2013062089A1 (ja) | ポリアミド樹脂及びそれからなる成形品 | |
| JPWO2012036303A1 (ja) | 耐衝撃性に優れるポリオキサミド樹脂及び耐衝撃性部品 | |
| JP5321434B2 (ja) | Smtコネクタ用ポリアミド樹脂組成物 | |
| JP5572922B2 (ja) | エンジン冷却水系部品用ポリアミド樹脂組成物、及び当該組成物から成形させたエンジン冷却水系部品 | |
| JP2009298853A (ja) | ポリアミド樹脂組成物 | |
| JP5584968B2 (ja) | 可塑剤含有ポリアミド樹脂組成物及び成形物 | |
| JP5584963B2 (ja) | ポリアミド樹脂組成物 | |
| JP2009298868A (ja) | ポリアミド樹脂組成物及び成形物 | |
| JP2009235223A (ja) | 自動車部材用ポリアミド樹脂 | |
| JP5621220B2 (ja) | 導電性ポリアミド樹脂組成物及びケーブルハウジング | |
| JP5347930B2 (ja) | 電子写真用部材 | |
| JP2010018794A (ja) | 導電性ポリアミド樹脂組成物 | |
| JP5446795B2 (ja) | Icトレイ用ポリアミド樹脂組成物及びicトレイ | |
| JP5584966B2 (ja) | 耐熱剤含有樹脂組成物及び該耐熱剤含有樹脂組成物から形成された成形物 | |
| JP2009298856A (ja) | 耐熱剤含有樹脂組成物及び該耐熱剤含有樹脂組成物から形成された成形物 | |
| JP2009298857A (ja) | ポリアミド樹脂組成物及び該ポリアミド樹脂組成物から形成された成形物 | |
| JP2013095792A (ja) | 充填材含有ポリアミド樹脂組成物 | |
| JP2009298852A (ja) | 可塑剤含有ポリアミド樹脂組成物及び成形物 | |
| JP5458845B2 (ja) | 電子写真用部材 | |
| JP5446796B2 (ja) | 新規なポリアミド樹脂を含む電子写真用部材 | |
| JP2009298854A (ja) | ポリアミド樹脂及び層状珪酸塩を含む複合材料 | |
| JP5584967B2 (ja) | ポリアミド樹脂組成物及び該ポリアミド樹脂組成物から形成された成形物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110830 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130820 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131017 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140624 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140707 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5584963 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |