JP2020007366A - ジアザビシクロオクタン誘導体の結晶とその製造法 - Google Patents
ジアザビシクロオクタン誘導体の結晶とその製造法 Download PDFInfo
- Publication number
- JP2020007366A JP2020007366A JP2019171587A JP2019171587A JP2020007366A JP 2020007366 A JP2020007366 A JP 2020007366A JP 2019171587 A JP2019171587 A JP 2019171587A JP 2019171587 A JP2019171587 A JP 2019171587A JP 2020007366 A JP2020007366 A JP 2020007366A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- vii
- compound represented
- tert
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 *OC([C@@](CC[C@](C1)N2OCc3ccccc3)N1C2=O)=O Chemical compound *OC([C@@](CC[C@](C1)N2OCc3ccccc3)N1C2=O)=O 0.000 description 17
- ZNDPBDWRYDUUNY-UEKLYDJUSA-N CC(C)(C)OC(N(C[C@@H](CC1)NOCc2ccccc2)[C@@H]1C(ON(C(C1C2CC=CC1)=O)C2=O)=O)=O Chemical compound CC(C)(C)OC(N(C[C@@H](CC1)NOCc2ccccc2)[C@@H]1C(ON(C(C1C2CC=CC1)=O)C2=O)=O)=O ZNDPBDWRYDUUNY-UEKLYDJUSA-N 0.000 description 1
- LKDHLZVWSJUXEB-ONIZESMZSA-N CC(C)(C)OC(N(C[C@@H](CC1)NOCc2ccccc2)[C@@H]1C(ON(C(C1C2CCCC1)O)C2=O)=O)=O Chemical compound CC(C)(C)OC(N(C[C@@H](CC1)NOCc2ccccc2)[C@@H]1C(ON(C(C1C2CCCC1)O)C2=O)=O)=O LKDHLZVWSJUXEB-ONIZESMZSA-N 0.000 description 1
- ALHJASVQSVZADW-SFHVURJKSA-N CC(C)(C)OC(NCCONC([C@H](CCCCC1)N1C(NOCc1ccccc1)=O)=O)=O Chemical compound CC(C)(C)OC(NCCONC([C@H](CCCCC1)N1C(NOCc1ccccc1)=O)=O)=O ALHJASVQSVZADW-SFHVURJKSA-N 0.000 description 1
- NMSZPDNMVGIKCY-WSFNDRFOSA-N C[C@H](C1)C=C[C@H]1C(CC(N1OC([C@H](CC[C@@H](C)C2)N2C(NOCc2ccccc2)=O)=O)=O)C1=O Chemical compound C[C@H](C1)C=C[C@H]1C(CC(N1OC([C@H](CC[C@@H](C)C2)N2C(NOCc2ccccc2)=O)=O)=O)C1=O NMSZPDNMVGIKCY-WSFNDRFOSA-N 0.000 description 1
Images

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/429—Thiazoles condensed with heterocyclic ring systems
- A61K31/43—Compounds containing 4-thia-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula, e.g. penicillins, penems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/542—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame ortho- or peri-condensed with heterocyclic ring systems
- A61K31/545—Compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins, cefaclor, or cephalexine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【解決手段】下記式(VII)、特に式(VII−1)で示されるジアザビシクロオクタン誘導体の結晶とその製造法を提供する。

【選択図】なし
Description
(上記式中、R3は後述するものと同様である)
(上記式中、R3は後述するものと同様、OBnはベンジルオキシ、Bocはtert−ブトキシカルボニルである)
で示される化合物の製造法であって、下記式(III):
で示される化合物を、化合物:R3ONH2と反応させて下記式(IV):
で示される化合物とし、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムで処理し、下記式(VI):
で示される化合物とした後、側鎖R3ONHC(=O)−が保護基を有する場合、酸により当該保護基を除去、続いて反応液に貧溶媒を加えて粗生成物を沈殿化し、下記式(VII−CR):
(上記各式中、OBnはベンジルオキシ、R1は2,5−ジオキソピロリジン−1−イル、1,3−ジオキソ−3a,4,7,7a‐テトラヒドロ−1H−イソインドール−2(3H)−イル、1,3−ジオキソヘキサヒドロ−1H−イソインドール−2(3H)−イル、または3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル、R3はC1−6アルキルまたはヘテロシクリルを示す。R3は0から5個のR4で修飾されていても良く、R4は連続して置換されてもよい。ここでR4はC1−6アルキル、ヘテロシクリル、R5(R6)N−、または保護基である。また、R5とR6は各々独立して水素またはC1−6アルキルを示すか、あるいは一緒になってヘテロシクリルを形成する。さらに、R3、R5及びR6は任意の位置で閉環することができる。)
で示される粗の化合物とした後、式(VII−CR)で示される粗の化合物と氷冷した緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、貧溶媒を加えて、結晶化することを特徴とする、製造法に関する。
で示される化合物の製造法であって下記式(III):
(上記各式中、R1、R3およびOBnは、上記のとおりである)
で示される化合物を、化合物:R3ONH2と反応させることを特徴とする、製造法に関する。
で示される化合物の製造法であって、下記式(IV):
(上記各式中、R3およびOBnは、上記のとおりである)
で示される化合物を、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムで処理することを特徴とする、製造法に関する。
で示される粗の化合物の製造法であって、下記式(VI):
(上記各式中、R3は、上記のとおりである)
で示される化合物を、側鎖R3ONHC(=O)−が保護基を有する場合、酸により当該保護基を除去、続いて反応液にエステル系貧溶媒を加えて粗生成物を沈殿化することを特徴とする、製造法に関する。
で示される化合物の製造法であって、下記式(VII−CR):
(上記各式中、R3は、上記のとおりである)
で示される粗の化合物と氷冷した緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、アルコール系貧溶媒を加えて、結晶化することを特徴とする、製造法に関する。
2−(tert−ブトキシカルボニルアミノ)エチル、
2−アミノエチル、
2−((tert−ブトキシカルボニル)(メチル)アミノ)エチル、
2−(メチルアミノ)エチル、
2−((tert−ブトキシカルボニル)(イソプロピル)アミノ)エチル、
2−(イソプロピルアミノ)エチル、
2−(ジメチルアミノ)エチル、
(2S)−2−((tert−ブトキシカルボニル)アミノ)プロピル、
(2S)−2−(アミノ)プロピル、
(2R)−2−((tert−ブトキシカルボニル)アミノ)プロピル、
(2R)−2−(アミノ)プロピル、
3−((tert−ブトキシカルボニル)アミノ)プロピル、
3−(アミノ)プロピル、
(2S)−tert−ブトキシカルボニルアゼチジン−2−イルメチル、
(2S)−アゼチジン−2−イルメチル、
(2R)−tert−ブトキシカルボニルピロリジン−2−イルメチル
(2R)−ピロリジン−2−イルメチル
(3R)−tert−ブトキシカルボニルピペリジン−3−イルメチル、
(3R)−ピペリジン−3−イルメチル、
(3S)−tert−ブトキシカルボニルピロリジン−3−イル、
(3S)−ピロリジン−3−イル、
1−(tert−ブトキシカルボニル)アゼチジン−3−イル、
アゼチジン−3−イル、
から選ばれる化合物の製造法であって、(1)〜(5)いずれか記載の方法であることを特徴とする、製造法に関する。
で示される化合物の製造法であって、下記式(III):
で示される化合物を、塩基存在下tert−ブチル 2−(アミノオキシ)エチルカーバメートと反応させて下記式(IV−1):
で示される化合物とし、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムと処理し、下記式(VI−1):
で示される化合物とした後、トリフルオロ酢酸によりtert−ブトキシカルボニル(Boc)基を除去、続いて反応液に酢酸エチルを滴下することにより粗生成物を沈殿化し、下記式(VII−1−CR):
(上記各式中、R1およびOBnは、上記のとおりである)
で示される粗の化合物とした後、式(VII−1−CR)で示される粗の化合物と氷冷した燐酸緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、イソプロパノールを加えて結晶化することを特徴とする、製造法に関する。
で示される化合物の製造法であって、下記式(III):
(上記各式中、R1およびOBnは、上記のとおりである)
で示される化合物を、塩基存在下tert−ブチル 2−(アミノオキシ)エチルカーバメートと反応させることを特徴とする、製造法に関する。
で示される化合物の製造法であって、下記式(IV−1):
(上記各式中、OBnは、上記のとおりである)
で示される化合物を、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムと処理することを特徴とする、製造法に関する。
で示される粗の化合物の製造法であって、下記式(VI−1):
で示される化合物を、トリフルオロ酢酸によりtert−ブトキシカルボニル(Boc)基を除去、続いて反応液に酢酸エチルを滴下することにより粗生成物を沈殿化することを特徴とする、製造法に関する。
で示される化合物の製造法であって、下記式(VII−1−CR):
で示される粗の化合物と氷冷した燐酸緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、イソプロパノールを加えて結晶化することを特徴とする、製造法に関する。
で示される化合物であって、粉末X線回析図形において面間隔(d)7.34、5.66、5.53、5.30、5.02、4.66、4.37、4.28、4.06、3.68、3.62、3.47、3.36、3.30、3.16、3.11、3.03、2.99、及び2.50Åに特徴的なピークを有するI形結晶に関する。
で示される化合物であって、粉末X線回析図形において面間隔(d)9.46、5.62、5.23、5.10、5.00、4.91、4.67、4.45、4.29、3.96、3.78、3.71、3.52、3.24、3.18、3.10、3.02、2.88、2.81、2.77、2.67、2.50、及び2.45Åに特徴的なピークを有するII形結晶に関する。
で示される化合物であって、粉末X線回析図形において面間隔(d)8.32、6.10、5.98、5.51、5.16、5.07、4.85、4.70、4.61、4.35、4.20、4.06、4.00、3.95、3.77、3.73、3.65、3.42、3.39、3.36、3.26、3.23、3.13、3.09、2.99、2.81、及び2.52Åに特徴的なピークを有するIII形結晶に関する。
で示される化合物であって、粉末X線回析図形において面間隔(d)7.88、6.41、5.20、4.67、4.50、4.02、3.81、3.75、3.70、3.62、3.38、3.23、3.20、及び2.74Åに特徴的なピークを有するIV形結晶に関する。
(上記式(VII)中、R3はC1−6アルキルまたはヘテロシクリルを示す。R3は0から5個のR4で修飾されていても良く、R4は連続して置換されてもよい。ここでR4はC1−6アルキル、ヘテロシクリル、R5(R6)N−、または保護基である。また、R5とR6は各々独立して水素またはC1−6アルキルを示すか、あるいは一緒になってヘテロシクリルを形成する。さらに、R3、R5及びR6は任意の位置で閉環することができる。)
「R3、R5及びR6は任意の位置で閉環することができる」とは、R3がC1−6アルキルを示し、かつR3を修飾するR4、すなわちR5(R6)N−に含まれるR5またはR6がC1−6アルキルを示す場合に、R3およびR5またはR6が一緒になって、3〜7員の飽和環を形成することができることを意味する。
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート、
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート、
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(メチル)カーバメート、
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(メチル)カーバメート、
(2S,5R)−N−[2−(メチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(プロパン−2−イル)カーバメート、
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(プロパン−2−イル)カーバメート、
(2S,5R)−7−オキソ−N−[2−(プロパン−2−イルアミノ)エトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
(2S,5R)−6−ベンジルオキシ−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
テトラブチルアンモニウム (2S,5R)−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
(2S,5R)−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル {(2S)−1−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート、
テトラブチルアンモニウム tert−ブチル {(2S)−1−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート、
(2S,5R)−N−{[(2S)−2−アミノプロピル]オキシ}−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル {(2R)−1−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート、
テトラブチルアンモニウム tert−ブチル {(2R)−1−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート、
(2S,5R)−N−{[(2R)−2−アミノプロピル]オキシ}−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル {3−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロピル}カーバメート、
テトラブチルアンモニウム tert−ブチル {3−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロピル}カーバメート、
(2S,5R)−N−(3−アミノプロポキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル (2S)−2−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート、
テトラブチルアンモニウム tert−ブチル (2S)−2−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート、
(2S,5R)−N−[(2S)−アゼチジン−2−イルメトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル (2R)−2−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピロリジン−1−カルボキシレート、
テトラブチルアンモニウム tert−ブチル (2R)−2−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピロリジン−1−カルボキシレート、
(2S,5R)−7−オキソ−N−[(2R)−ピロリジン−2−イルメトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル (3R)−3−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピペリジン−1−カルボキシレート、
テトラブチルアンモニウム tert−ブチル (3R)−3−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピペリジン−1−カルボキシレート、
(2S,5R)−7−オキソ−N−[(3R)−ピペリジン−3−イルメトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル (3S)−3−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]ピロリジン−1−カルボキシレート、
テトラブチルアンモニウム tert−ブチル (3S)−3−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]ピロリジン−1−カルボキシレート、
(2S,5R)−7−オキソ−N−[(3S)−ピロリジン−3−イルオキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
tert−ブチル 3−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート、
テトラブチルアンモニウム tert−ブチル 3−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート、
(2S,5R)−N−(アゼチジン−3−イルメトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
から選ばれる化合物であり、下記化合物群が挙げられる。
(上記式中、P2はtert−ブトキシカルボニル(Boc)などの保護基または水素、P3はベンジルオキシ(OBn)、テトラブチルアンモニウムスルホオキシ、またはスルホオキシを示す。)
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート、
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド、
であり、上記式(IV−1)、(VI−1)、(VII−1―CR)、(VII−1)で示される。
本発明により提供される医薬は、式(VII)で表される化合物および医学的に許容されるその塩、並びにそれらの水和物またはその溶媒和物からなる群から選ばれる物質を有効成分とし含有することを特徴としており、経口的または非経口的に投与されるが、好ましくは非経口的に投与される。本発明の化合物とβ−ラクタム系抗生物質は、用事個別に調製した各々の薬剤を併用して、同時または別々に投与する方法、あるいは予め両者の薬剤を混合した一般的には1または2以上の製剤用添加物(担体)を用いて、医薬組成物を製造し、投与することができる。
(上記式(III)、(IV)、(VI)、(VII−CR)、(VII)中、OBn、R1及びR3は上述されたものと同様である。)
(上記式(IV)中、OBn、R1及びR3は上述されたものと同様である。)
で示される化合物から式(IV)で示される化合物とする工程は以下のように実施される。
で示される化合物を上記式(VI)で示される化合物とする工程は以下のように実施される。
水素化分解に用いられる水素の供給源は水素ガスであり、水素圧は大気圧〜1MPaの範囲で選ばれ、好ましくは大気圧〜0.5MPaである。水素の供給量は少なくとも化学量論量以上の量が用いられる。
で示される化合物を上記式(VII−CR)で示される化合物とする工程は以下のように実施される。
で示される化合物を上記式(VII)で示される化合物とする工程は以下のように実施される。
I、II、及びIII形結晶は、DSC、含水イソプロパノールへの溶解度、粉末X線回析図形の面間隔パターンで区別される。含水イソプロパノールへの溶解度はI形結晶が最も低く、IIとIII形結晶は同程度である。
IV形結晶に関しては、水溶液とせずにI,II、またはIII形結晶をメタノール、エタノール、またはイソプロパノール中で懸濁撹拌することにより得ることもできる。
撹拌時間は析出速度に依存するが、1〜24時間、好ましくは1〜15時間撹拌する。
析出した結晶は通常の濾過、洗浄、通気乾燥または真空乾燥することで、結晶形態の式(VII)で示される化合物を得ることができる。溶媒和した結晶の場合は、品温、乾燥減量、加湿限定真空乾燥、加湿通気乾燥等の管理手段により過乾燥を回避する。
メチル (2S,5R)−5−(ベンジルオキシアミノ)ピペリジン−2−カルボキシレート の二塩酸塩
工程1
メチル (2S,5S)−5−ヒドロキシピペリジン−2−カルボキシレート
2M 塩化水素−メタノール溶液(12.8L)に市販の(2S,5S)−5−ヒドロキシピペリジン−2−カルボン酸(プレラベルHPLC含量84%、正味912.22g、洗い込み2M 塩化水素−メタノール 3.1L)を加え、3時間還流した(内温63〜67℃)。反応液を冷却後、1,4−ジオキサン(12.8L)を加えて溶媒を減圧留去した。残渣(4.1kg)に酢酸エチル(18.3L)と氷冷44%炭酸カリウム水溶液(23.7L)を加えて有機層を分層し、水層をさらに酢酸エチル(3x18.3L)で抽出した。50%炭酸カリウム水溶液(7.3L)を各々の有機層にて分液、有機層を併せて無水炭酸カリウム(2.37kg)で乾燥、ろ過、溶媒を減圧留去した。残渣をトルエン(9.1L)に溶解し、活性炭9.2gを加えて30分間攪拌、ろ過、溶媒を減圧留去した。残渣を酢酸エチル(9.1L)にて置換濃縮して淡黄色油状の標題化合物1130gを得た(プレラベルHPLC含量78.9%、正味891.57g、収率89%)。
メチル (2S,5S)−5−ヒドロキシ−1−(2,2,2−トリフルオロアセチル)ピペリジン−2−カルボキシレート
メチル (2S,5S)−5−ヒドロキシピペリジン−2−カルボキシレート(プレラベルHPLC含量78.8%、正味459.48g)の脱水酢酸エチル溶液(7.4L)を−40℃に冷却し、トリエチルアミン(1300g)を加え、次いでトリフルオロ酢酸無水物(1349g、洗い込み脱水酢酸エチル100mL)を−40〜−12℃にて30分間で滴下した。滴下終了後15分で−2℃に昇温し75分間撹拌、更に混合物に水(1277mL)を加え25℃にて1時間攪拌した。混合物を水(8.4L)に投入(酢酸エチル4.5Lで洗い込み)、更に酢酸エチル(2x9.8L)にて抽出、合併有機層を1M 塩酸(8.5L)、飽和重曹水(8.5L)、飽和食塩水(8.5L)にて順次洗浄し、無水硫酸ナトリウム(1.8kg)で乾燥、濾過した。有機層の溶媒を減圧留去した後、残渣に酢酸エチル(3.6L)を加えて置換濃縮し、残渣を真空乾燥し標題化合物793.4gを得た(HPLC含量81.5%、正味648.66g、収率88%)。
メチル (2S,5R)−5−(ベンジルオキシアミノ)−1−(2,2,2−トリフルオロアセチル)ピペリジン−2−カルボキシレート
メチル (2S,5S)−5−ヒドロキシ−1−(2,2,2−トリフルオロアセチル)ピペリジン−2−カルボキシレート(HPLC含量81.5%、正味556.23g)の脱水アセトニトリル溶液4.0Lを−40℃に冷却し、2,6−ルチジン(259.24g)を加え(アセトニトリル 100mLで洗浄)、トリフルオロメタンスルホン酸無水物(645.72g)を−43〜−37℃にて1時間10分かけて滴下した(アセトニトリル 100mLで洗浄)。反応液を−35℃にて50分攪拌後、ベンジルオキシアミン(550.27g)を−35℃以下で滴下し、アセトニトリル(500mL)で洗い込んだ。反応液を徐々に−5℃まで上昇させた後、2,6−ルチジン(259.24g)を加えて、5℃で40時間攪拌した。混合物を1.8Lまで濃縮後、酢酸エチル(12.4L)で希釈し、水(12.4L)、10%クエン酸水溶液(4x8L+4.7L)、飽和重曹水(6.3L)、飽和食塩水(7.2L)で順次洗浄した。有機層を無水硫酸ナトリウムにて乾燥、濾過後、減圧濃縮した。残渣を真空乾燥し、標題化合物867.73gを得た(HPLC含量71.56%、収率79%)。
メチル (2S,5R)−5−(ベンジルオキシアミノ)−1−(2,2,2−トリフルオロアセチル)ピペリジン−2−カルボキシレート 塩酸塩
メチル (2S,5R)−5−(ベンジルオキシアミノ)−1−(2,2,2−トリフルオロアセチル)ピペリジン−2−カルボキシレート(HPLC含量70.13%,正味673.20g)を酢酸エチル(4.8L)で希釈、活性炭48gを投入し、1時間撹拌、混合物を濾過し酢酸エチル2Lにて洗浄した。濾液を酢酸エチル4.7Lで希釈、室温で1M 塩化水素−酢酸エチル溶液(2.7L)を添加して15分撹拌、次いでヘキサン28.6Lを投入し、0℃に冷却した。3時間撹拌した後、結晶を濾過し、ヘキサン/酢酸エチル=4/1(3L)にて洗浄後、真空乾燥して標題化合物724.0gを得た(HPLC含量91.72%、収率90%)。
メチル (2S,5R)−5−(ベンジルオキシアミノ)ピペリジン−2−カルボキシレート の二塩酸塩
メチル (2S,5R)−5−(ベンジルオキシアミノ)−1−(2,2,2−トリフルオロアセチル)ピペリジン−2−カルボキシレート 塩酸塩(HPLC含量92.01%、正味732.25g)を2M 塩化水素−メタノール溶液(15L)に溶解、27時間加熱還流した。混合物を室温まで冷却、3Lまで減圧濃縮した。混合物をメタノール2.7Lで希釈し、次いで酢酸エチル16.3Lを加え1時間撹拌した。析出した結晶を濾過し、酢酸エチル(3x1.1L)にて洗浄、真空乾燥して標題化合物572.0gを得た(HPLC含量98.06%、収率92%)。
1H NMR (400 MHz, D2O) δ 1.40-1.51 (m, 1H), 1.61-1.72 (m, 1H), 1.90-1.94 (m, 1H), 2.25-2.30 (m, 1H), 2.80 (t, J = 11.2 Hz, 1H), 3.19-3.27 (m, 1H), 3.51-3.55 (m, 1H), 3.66 (s, 3H), 3.87-3.91 (m, 1H), 4.68 (s, 2H), 7.27 (s, 5H); MS m/z 265 [M-2HCl+H]+.
(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸
工程1
メチル (2S,5R)−5−(ベンジルオキシアミノ)ピペリジン−2−カルボキシレート
メチル (2S,5R)−5−(ベンジルオキシアミノ)ピペリジン−2−カルボキシレート, 二塩酸塩(参考例1、1.319g)に酢酸エチル(20mL)、50%炭酸カリウム水溶液(20mL)を加えて分液し、水層を酢酸エチル(15mL)にて3回抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムにて乾燥、濾過後、減圧濃縮、終夜真空乾燥し、標題化合物を975mg得た(収率94%)。
1H NMR (400 MHz, CDCl3) δ 1.25-1.35 (m, 1H), 1.49-1.59 (m, 1H), 1.89-2.11 (m, 2H), 2.45 (t, J = 11.7 Hz, 1H), 2.96-3.03 (m, 1H), 3.28-3.39 (m, 2H), 3.72 (s, 3H), 4.68 (s, 2H), 7.26-7.35 (m, 5H); MS m/z 265 [M+H]+.
メチル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート
メチル (2S,5R)−5−(ベンジルオキシアミノ)ピペリジン−2−カルボキシレート(1.154g、4.37mmol)に脱水アセトニトリル(198mL)を加え、氷冷した。5℃以下でトリエチルアミン(1.60mL)、ジホスゲン(0.389mL)を順次滴下し、2℃で20分攪拌した。次いで、反応液に4−ジメチルアミノピリジン(70.0mg)を加え、室温にて10時間攪拌した。反応液を減圧濃縮し、酢酸エチルによる置換濃縮を3回行った後、溶液を30mLまで濃縮した。ここに酢酸エチル(20mL)、水(40mL)を加え、分液し、分離した水層を酢酸エチル(30mL)にて2回抽出した。併せた有機層を5%クエン酸水溶液(40mL)、6.5% 重曹水(30mL)、5%食塩水(30mL)で順次洗浄し、無水硫酸ナトリウムにて乾燥、濾過後、減圧濃縮した。得られた残渣1.16gを酢酸エチル(5.5mL)で希釈し、n−ヘキサン(11mL)を加え、種晶を接種し結晶化させた。更にn−ヘキサン(49mL)を加え、0℃にて1時間攪拌後、結晶を濾過し、n−ヘキサン(60mL)にて洗浄後、真空乾燥し無色結晶性粉末の標題化合物を882.3mg得た(収率71%)。
1H NMR (400 MHz, CDCl3) δ 1.65-1.70 (m, 1H), 2.03-2.12 (m, 3H), 2.90 (d, J = 12.0 Hz, 1H), 3.07 (m, 1H), 3.32 (m, 1H), 4.12 (dd, J = 4.6&4.4 Hz, 1H), 4.91 (d, J = 11.2 Hz, 1H), 5.06 (d, J = 11.2 Hz, 1H), 7.35-7.44 (m, 5H); MS m/z 291 [M+H]+.
(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸
メチル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(809.0mg、2.79mmol)にテトラヒドロフラン(8mL)、水(3.6mL)を加え、0.5M 水酸化リチウム水溶液(6.41mL)を4.9℃以下で10分かけて滴下した。反応液を2℃で2時間攪拌後、水(30mL)を加え、酢酸エチル(25mL)にて洗浄した。分離した水層に酢酸エチル(15mL)を加え、1M 塩酸水溶液にて水層をpH4.0に調整し、酢酸エチルで2回抽出した(酢酸エチル:トータル65mL)。分離した水層を1M 塩酸水溶液でpH3.4に調整し、酢酸エチルで1回抽出後、水層をpH2.4に調整し、酢酸エチル抽出を2回行った。計5回の酢酸エチル抽出液(175mL)を飽和食塩水(40mL)で洗浄し、無水硫酸ナトリウムにて乾燥、濾過後、減圧濃縮した。得られた残渣759.1mgを酢酸エチル(5mL)で希釈し、n−ヘキサン(3mL)を加え、種晶を接種し結晶化させた。更に酢酸エチル/n−ヘキサン(5/3)溶液(8mL)を加えて攪拌後、n−ヘキサン(20mL)を加え、4℃で14時間攪拌した。結晶を濾過し、n−ヘキサン(55mL)にて洗浄後、真空乾燥し無色結晶性粉末の標題化合物を633.6mg得た(収率82%)。
1H NMR (400 MHz, CDCl3) δ 1.67 (m, 1H), 2.04-2.26 (m, 3H), 2.85 (d, J = 12.0 Hz, 1H), 3.13 (m, 1H), 3.35 (m, 1H), 4.12 (m, 1H), 4.91 (d, J = 11.3 Hz, 1H), 5.06 (d, J = 11.3 Hz, 1H), 7.37-7.44 (m, 5H); MS m/z 277 [M+H]+.
2,5−ジオキソピロリジン−1−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート
工程1
(2S,5R)−5−((ベンジルオキシ)アミノ)−1−(tert−ブトキシカルボニル)ピペリジン−2−カルボン酸
メチル (2S,5R)−5−(ベンジルオキシアミノ)ピペリジン−2−カルボキシレート, 二塩酸塩(参考例1、65.4g、200mmol)を水(400mL)、1,4−ジオキサン(270mL)に溶解し、氷冷して5M 水酸化ナトリウム水溶液(132mL)を加えて1時間撹拌した。反応液に5M 塩酸(12mL)、炭酸カリウム(27.6g)、ジ−tert−ブチルジカーボネート(48g)を加え、室温に昇温し終夜攪拌した。反応液を濃縮した水溶液を酢酸エチルで洗浄し、クエン酸・一水和物にてpH3.3に調整、酢酸エチル(500mL)で2回抽出、飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥、ろ過、溶媒を減圧濃縮、さらに酢酸エチルで置換濃縮し、標題化合物68.7gを得た(定量的)。本化合物は精製することなく次工程に用いられた。一部を酢酸エチル/ヘキサンより結晶化しその構造を確認した。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9H), 1.50-1.72 (m, 2H), 1.98-2.10 (m, 2H), 3.12-3.19 (m, 2H), 4.13-4.20 (m, 1H), 4.76 (d, J = 11.5 Hz), 4.70 (d, J = 11.5 Hz), 4.85-4.92 (m, 1H), 7.26-7.35 (m, 5H); MS m/z 351 [M+H]+.
2,5−ジオキソピロリジン−1−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート
(2S,5R)−5−((ベンジルオキシ)アミノ)−1−(tert−ブトキシカルボニル)ピペリジン−2−カルボン酸(参考例3の工程1、700mg、2mmol)を脱水テトラヒドロフラン(10mL)に溶解し、−20℃に冷却した。混合物にクロロギ酸イソブチル(300mg)とトリエチルアミン(444mg)を順次滴下し15分間撹拌した。反応液に1−ヒドロキシピロリジン−2,5−ジオン(253mg)を加え30分間撹拌し、室温でさらに30分間撹拌した。反応液を酢酸エチル(35mL)で希釈し、10%クエン酸(10mL)、飽和重曹水(10mL)、飽和食塩水(10mL)で順次洗浄、無水硫酸マグネシウムで乾燥、濾過、溶媒を減圧留去して残渣985mgを得た。本残渣全量を脱水クロロホルム(10mL)に溶解し、トリエチルアミン(303mg)を加えて氷冷した。混合物にトリホスゲン(237mg)を加え30分間撹拌し、メタノール(0.1mL)を加えて30分間撹拌した。次いでメタンスルホン酸(1.3mL)のジクロロメタン(4.0mL)溶液を滴下しさらに30分間撹拌した。混合物を氷冷1M 炭酸水素カリウム水溶液(2.4g/20mL)に滴下し30分間撹拌、クロロホルム(10mL)を加えて分層、有機層を1M 塩酸(10mL)、飽和重曹水(10mL)、飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥、濾過、減圧濃縮した。残渣に接種し、固体にヘキサン/酢酸エチル(1/2、3mL)を加え撹拌し、濾過、ヘキサン/酢酸エチル(1/1、3mL)、ヘキサン(3mL)で順次洗浄し、結晶の標題化合物556mgを得た(収率75%)。
1H NMR (400 MHz, CDCl3) δ 1.70-1.77 (m, 1H), 2.04-2.27 (m, 3H), 2.80-2.90 (m, 4H), 3.09-3.19 (m, 2H), 3.35 (br.s., 1H), 4.48 (d, J = 6.9 Hz, 1H), 4.92 (d, J = 11.3 Hz, 1H), 5.07 (d, J = 11.3 Hz, 1H), 7.35-7.45 (m, 5H); MS m/z 374 [M+H]+.
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート
(2S,5R)−5−((ベンジルオキシ)アミノ)−1−(tert−ブトキシカルボニル)ピペリジン−2−カルボン酸(参考例3の工程1、14.0g、41.09mmol)を脱水テトラヒドロフラン(200mL)に溶解し、−20℃付近に冷却した。混合物にクロロギ酸イソブチル(6.11g)、次いでトリエチルアミン(8.86g)を滴下し同温で15分間撹拌した。次に反応液に(1R,2S,6R,7S)−4−ヒドロキシ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−3,5−ジオン(7.87g)を加え同温で30分間、さらに室温で30分間撹拌した。反応液を酢酸エチル(700mL)で希釈し、氷冷10%クエン酸(200mL)、飽和重曹水(200mL)、飽和食塩水(200mL)で順次洗浄、無水硫酸マグネシウムで乾燥、濾過した。溶媒を減圧留去し再度酢酸エチルにて置換濃縮して得られた残渣25.1g(正味収率92%)全量を脱水クロロホルム(180mL)に溶解し、トリエチルアミン(5.5g)を加えて氷冷した。混合物にトリホスゲン(4.29g)を加え30分間撹拌し、次いでメタノール(1mL)を加えて30分間撹拌した。反応液にメタンスルホン酸(23.5mL)のジクロロメタン(30mL)溶液を滴下し更に30分間撹拌した。混合物を氷冷1M 炭酸水素カリウム水溶液(43.5g/200mL)に滴下し30分間撹拌、クロロホルム(100mL)を加えて分層、有機層を1M 塩酸(200mL)、飽和重曹水(200mL)、飽和食塩水(200mL)で順次洗浄した。各水層はクロロホルム(100mL)で順次逆抽出した。有機層を併せて無水硫酸マグネシウムで乾燥、濾過した。溶媒を減圧濃縮して得られた残渣をクロロホルム(70mL)に溶解しヘキサン(100mL)を加えて30分間撹拌晶析、更にヘキサン(100mL)を加えて1時間撹拌した。結晶をろ取乾燥して、標題化合物15.4gを得た(含量100%、収率88%)。
HPLC:COSMOSIL 5C18 MS-II 4.6X150mm, 35℃, 0.02M TFA/CH3CN=50/50, 1.0mL/min, UV210nm, RT 7.1min; 鏡像異性体過剰率99.9%ee以上: CHIRALPAK AD-H、4.6x150mm、40℃, Hexane/EtOH=1/1、UV210nm、1mL/min、RT 37.3min (cf. enantiomer 16.5min); Mp 196℃; [α]26 D+12.686 (c 0.885, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.52 (d, J = 9.1 Hz, 1H), 1.70 (m, 1H), 1.78 (d, J = 9.1 Hz, 1H), 2.01-2.26 (m, 3H), 3.04-3.17 (m, 2H), 3.32 (m, 3H), 3.45 (br.s., 2H), 4.41 (d, J = 6.7 Hz, 1H), 4.91 (d, J = 11.4 Hz, 1H), 5.06 (d, J = 11.4 Hz, 1H), 6.19 (br.s., 2H), 7.33-7.46 (m, 5H); MS m/z 438 [M+H]+.
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)
工程1
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (IV−1)
(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(4.30g、15.56mmol)の脱水酢酸エチル(47mL)溶液を−30℃に冷却し、クロロギ酸イソブチル(2.17g、洗い込み脱水酢酸エチル1mL)、トリエチルアミン(1.61g、洗い込み脱水酢酸エチル1mL)、を順次滴下し、−30℃にて1時間撹拌した。反応液にtert−ブチル 2−(アミノオキシ)エチルカーバメート(3.21g)の脱水酢酸エチル(4mL)溶液を加え(洗い込み脱水酢酸エチル1mL)、0℃まで1.5時間かけて昇温、さらに終夜撹拌した。混合物を8% クエン酸水溶液(56mL)、飽和重曹水(40mL)、飽和食塩水(40mL)で順次洗浄し、無水硫酸マグネシウムで乾燥後、濾過、5mLまで濃縮、さらにエタノール(10mL)で6mLまで置換濃縮した。得られた溶液にエタノール(3mL)、へキサン(8mL)を加え氷冷、接種し15分間撹拌した。混合物にヘキサン(75mL)を2時間かけて滴下し終夜撹拌した。析出結晶をろ取、ヘキサンで洗浄、真空乾燥して標題化合物5.49gを得た(正味4.98g、収率74%)。
HPLC:COSMOSIL 5C18 MS-II 4.6X150mm, 33.3mM phosphate buffer/MeCN = 50/50, 1.0mL/min, UV210nm, RT 4.4 min; 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m, 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br.d., J = 11.6 Hz, 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br.s., 1H), 7.34-7.48 (m, 5H), 9.37 (br.s., 1H); MS m/z 435 [M+H]+.
tert−ブチル {2−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (V−1)
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート(3.91g、9.01mmol)のメタノール溶液(80mL)に、10%パラジウム炭素触媒(50%含水、803mg)を加え、水素雰囲気下、45分間撹拌した。反応液をセライト濾過し、減圧濃縮後、標題化合物を3.11g得た(定量的)。
HPLC:COSMOSIL 5C18 MS-II 4.6X150mm, 33.3mM phosphate buffer/MeCN =75/25, 1.0mL/min, UV210nm, RT 3.9 min; 1H NMR (400 MHz, CD3OD) δ 1.44 (s, 9H), 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J = 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS m/z 345 [M+H]+.
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (VI−1)
tert−ブチル {2−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート(3.09g、8.97mmol)のジクロロメタン(80mL)溶液に、2,6−ルチジン(3.20mL)、三酸化イオウ−ピリジン錯体(3.58g)を加え、室温で終夜撹拌した。反応液を半飽和重曹水にあけ、水層をクロロホルムにて洗浄後、水層に硫酸水素テトラブチルアンモニウム(3.47g)とクロロホルム(30mL)を加え、10分間撹拌した。水層をクロロホルムで抽出後、得られた有機層を無水硫酸ナトリウムにて乾燥、濾過後、減圧濃縮し標題化合物5.46gを得た(収率91%)。
HPLC:COSMOSIL 5C18 MS-II 4.6X150mm, 33.3mM phosphate buffer/MeCN = 80/20, 1.0mL/min, UV210nm, RT 2.0 min; 1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.4 Hz, 12H), 1.37-1.54 (m, 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30-2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s, 1H), 9.44 (br.s., 1H); MS m/z 425 [M-Bu4N+2H]+.
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート(5.20g、7.82mmol)のジクロロメタン(25mL)溶液に、氷冷下トリフルオロ酢酸(25mL)を加え、0℃にて1時間撹拌した。反応液を減圧濃縮し、得られた残渣をジエチルエーテルにて洗浄後、重曹水にてpH7に調整し、オクタデシルシリカゲルカラムクロマトグラフィー精製(水)を行い、凍結乾燥後、標題化合物を1.44g得た(収率57%)。
HPLC:COSMOSIL 5C18 MS-II 4.6X150mm, 33.3mM phosphate buffer/MeCN = 99/1, 1.0mL/min, UV210nm, RT 3.1 min; 1H NMR (400 MHz, D2O) δ 1.66-1.76 (m, 1H), 1.76-1.88 (m, 1H), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02(d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz, 2H), 3.18 (br d, J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz,1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4&3.2 Hz, 1H); MS m/z 325 [M+H]+.
(2S,5R)−N−[2−(メチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−2)
工程1
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(メチル)カーバメート (IV−2)
参考例5と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(390mg、1.41mmol)とtert−ブチル (2−(アミノオキシ)エチル)(メチル)カーバメート(436mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物347.8mgを得た(収率55%)。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9H), 1.58-1.70 (m, 1H), 1.88-2.07 (m, 2H), 2.25-2.36 (m, 1H), 2.70-3.08 (m, 2H), 2.88 (s, 3H), 3.23-3.41 (m, 2H), 3.51-3.68 (m, 1H), 3.83-4.10 (m, 3H), 4.90 (d, J = 11.4 Hz, 1H), 5.06 (d, J = 11.4 Hz, 1H), 7.32-7.47 (m, 5H), 10.11 (br s, 1H); MS m/z 449 [M+H]+.
tert−ブチル {2−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(メチル)カーバメート (V−2)
参考例5と同様の手法にて、上記工程1の化合物全量より標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.46 (s, 9H), 1.73-1.83 (m, 1H), 1.86-2.00 (m, 1H), 2.01-2.13 (m, 1H), 2.14-2.28 (m, 1H), 2.93 (s, 3H), 3.04 (d, J = 10.8 Hz, 1H), 3.08-3.18 (m, 1H), 3.43-3.55 (m, 2H), 3.65-3.72 (m, 1H), 3.79-3.88 (m, 1H), 3.92-4.05 (m, 2H); MS m/z 359 [M+H]+.
(2S,5R)−N−[2−(メチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−2)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(メチル)カーバメートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.2 Hz, 12H), 1.36-1.53 (m, 8H), 1.47 (s, 9H), 1.57-1.77 (m, 9H), 1.83-1.98(m, 1H), 2.13-2.25 (m, 1H), 2.28-2.40 (m, 1H), 2.82-2.96 (m, 4H), 3.22-3.42 (m, 11H), 3.60-4.08 (m, 3H), 4.34 (br.s., 1H), 10.15 (br.s., 1H); MS m/z 437 [M-Bu4N]-.
1H NMR (500 MHz, D2O) δ 1.73-1.97 (m, 2H), 1.98-2.07 (m, 1H), 2.08-2.18 (m, 1H), 2.74 (s, 3H), 3.09 (d, J = 12.0 Hz, 1H), 3.21-3.32 (m, 3H), 4.04 (dd, J = 7.5, 2.0 Hz, 1H), 4.10-4.23 (m, 3H); MS m/z 337 [M-H]-.
(2S,5R)−7−オキソ−N−[2−(プロパン−2−イルアミノ)エトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−3)
工程1
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(プロパン−2−イル)カーバメート (IV−3)
(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(414mg、1.50mmol)の脱水ジクロロメタン(14.1mL)溶液をアルゴン雰囲気下0℃に冷却し、クロロギ酸イソブチル(245.9mg)、次いでトリエチルアミン(197mg)を順次加え30分攪拌した。この反応混合物にtert−ブチル (2−(アミノオキシ)エチル)(イソプロピル)カーバメート(596mg)を滴下し、投入終了後に室温へと昇温し一時間攪拌した。この反応混合物を0.5M 塩酸、飽和食塩水で順次洗浄し、有機層を硫酸マグネシウムにて乾燥後、減圧濃縮し得られた残渣をシリカゲルカラムクロマトグラフィ−に付し、標題化合物578.4mgを得た(収率81%)。
1H NMR (400 MHz, CDCl3) δ 1.15 (d, J = 6.8 Hz, 6H), 1.46 (s, 9H), 1.55-1.70 (m, 1H), 1.89-2.07 (m, 2H), 2.25-2.37 (m, 1H), 2.73-2.90 (m, 1H), 2.98-3.08 (m, 1H), 3.22-3.38 (m, 2H), 3.40-3.60 (m, 1H), 3.83-4.06 (m, 4H), 4.90 (d, J = 11.2 Hz, 1H), 5.06 (d, J = 11.2 Hz, 1H), 7.35-7.46 (m, 5H), 10.29 (br.s., 1H); MS m/z 477 [M+H]+.
tert−ブチル {2−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(プロパン−2−イル)カーバメート (V−3)
参考例5と同様の手法にて、上記工程1の化合物全量より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.09-1.23 (m, 6H), 1.46 (s, 9H), 1.73-2.27 (m, 4H), 3.06 (d, J = 11.6 Hz, 1H), 3.08-3.50 (m, 4H), 3.64-3.73 (m, 1H), 3.79-3.98 (m, 3H); MS m/z 387 [M+H]+.
(2S,5R)−7−オキソ−N−[2−(プロパン−2−イルアミノ)エトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−3)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(プロパン−2−イル)カーバメートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (d, J = 7.4 Hz, 12H), 1.10-1.20 (m, 6H), 1.33-1.77 (m, 17H), 1.46 (s, 9H), 1.84-1.97 (m, 1H), 2.12-2.25 (m, 1H), 2.28-2.40 (m, 1H), 2.79-2.95 (m, 1H), 3.17-3.45 (m, 9H), 3.50-3.67 (m, 1H), 3.80-4.07 (m, 5H), 4.34 (br.s., 1H), 10.36 (br.s., 1H); MS m/z 465 [M-Bu4N]-.
1H NMR (500 MHz, D2O) δ 1.28 (d, J = 6.5 Hz, 6H),1.74-1.83 (m, 1H), 1.85-1.96 (m, 1H), 1.98-2.14 (m, 2H), 3.11 (d, J = 12.5 Hz, 1H), 3.22-3.30 (m, 3H), 3.40 (quint, J = 6.5 Hz, 1H), 4.01 (br d, J = 5.5 Hz, 1H), 4.09-4.18 (m, 3H); MS m/z 367 [M+H]+.
(2S,5R)−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−4)
工程1
(2S,5R)−6−ベンジルオキシ−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (IV−4)
(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(553mg、2.00mmol)の脱水ジクロロメタン(10mL)溶液をアルゴン雰囲気下0℃に冷却し、クロロギ酸イソブチル(289μL、2.20mmol)を滴下した。次いで、トリエチルアミン(293μL)を加え30分攪拌することで、反応系内に混合酸無水物を調製した。この反応混合物に2−(アミノオキシ)−N,N−ジメチルエタナミン 2塩酸塩(591mg)とトリエチルアミン(930μL)を脱水ジクロロメタン(7.0mL)で洗いこみながらゆっくりと加え、そのままの温度で一時間攪拌した。この反応混合物を濾過後、残渣をメタノールで洗浄し、濾液を減圧濃縮した。得られた残渣をジクロロメタンと水に溶解し、ジクロロメタンで抽出した有機層を硫酸マグネシウムにて乾燥後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィ−(アミノシリカ、クロロホルム/メタノール=10/1)に付し,無色油状の標題化合物291.1mgを得た。(収率40%)
1H NMR (400 MHz, CDCl3) δ 1.45-1.85 (m, 4H), 2.29 (s, 6H), 2.60 (t, J = 5.2 Hz, 2H), 2.81 (d, J = 11.6 Hz, 1H), 2.97 (br.d., J = 11.6 Hz, 1H), 3.28-3.34 (m, 1H), 3.92-4.07 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 7.35-7.48 (m, 5H); MS m/z 363 [M+H]+.
(2S,5R)−N−[2−(ジメチルアミノ)エトキシ]−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (V−4)
参考例5と同様の手法にて、上記工程1の化合物全量より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.74-1.84 (m, 1H), 1.87-1.98 (m, 1H), 2.03-2.12 (m, 1H), 2.15-2.24 (m, 1H), 2.36 (s, 6H), 2.67-2.74 (m, 2H), 3.07 (br.d., J = 11.6 Hz, 1H), 3.12 (br.d., J = 11.6 Hz, 1H), 3.67-3.72 (m, 1H), 3.83 (br.d., J = 6.4 Hz, 1H), 3.96-4.06 (m, 2H); MS m/z 273 [M+H]+.
(2S,5R)−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−4)
参考例5と同様の手法にて得られた反応混合物をクロロホルムで希釈、水洗してピリジニウム (2S,5R)−N−[2−(ジメチルアミノ)エトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミドを得、飽和重曹水にて中和した後にオクタデシルシリカゲルカラムクロマトグラフィー精製後、標題化合物130.7mgを得た(2工程収率43%)。
1H NMR (400 MHz, D2O) δ 1.68-1.84 (m, 2H), 1.86-2.04 (m, 2H), 2.80 (s, 6H), 3.09-3.17 (m, 2H), 3.17-3.29 (m, 2H), 3.80-3.90 (m, 1H), 4.02-4.13 (m, 3H); MS m/z 353 [M+H]+.
(2S,5R)−N−{[(2S)−2−アミノプロピル]オキシ}−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−5)
工程1
tert−ブチル {(2S)−1−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート (IV−5)
参考例7と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(414mg、1.50mmol)と、(S)−tert−ブチル (1−(アミノオキシ)プロパン−2−イル)カーバメート(550mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物585.6mgを得た(収率87%)。
1H NMR (400 MHz, CDCl3) δ 1.17 (d, J = 6.4 Hz, 3H), 1.44 (s, 9H), 1.55-1.70 (m, 1H), 1.90-2.10 (m, 2H), 2.26-2.34 (m, 1H), 2.80 (d, J = 12.0 Hz, 1H), 3.06 (br.d., J = 12.0 Hz, 1H), 3.27-3.34 (m, 1H), 3.64-3.74 (m, 1H), 3.86-3.98 (m, 3H), 4.81 (br.d., J = 7.6 Hz, 1H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 7.34-7.45 (m, 5H), 9.68 (br.s., 1H); MS m/z 449 [M+H]+.
tert−ブチル {(2S)−1−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート (V−5)
参考例5と同様の手法にて、上記工程1の化合物全量より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.16 (d, J = 6.4 Hz, 3H), 1.44 (s, 9H), 1.74-1.84 (m, 1H), 1.86-1.98 (m, 1H), 2.03-2.12 (m, 1H), 2.21 (br.dd., J = 15.2, 6.8 Hz, 1H), 3.06 (d, J = 12.0 Hz, 1H), 3.14 (br.d., J = 12.0 Hz, 1H), 3.68-3.72 (m, 1H), 3.74-3.87 (m, 4H); MS m/z 359 [M+H]+.
(2S,5R)−N−{[(2S)−2−アミノプロピル]オキシ}−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−5)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル {(2S)−1−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメートを得た(定量的)。MS m/z 437[M−Bu4N]−.
上記テトラブチルアンモニウム塩全量をトリフルオロ酢酸により脱保護し、オクタデシルシリカゲルカラムクロマトグラフィー精製後、標題化合物117.1mgを得た(3工程収率26%)。
1H NMR (400 MHz, D2O) δ 1.17 (d, J = 6.8 Hz, 3H), 1.66-1.89 (m, 2H), 1.91-2.08 (m, 2H), 3.02 (d, J = 12.0 Hz, 1H), 3.18 (br.d., J = 12.0 Hz, 1H), 3.47-3.58 (m, 1H), 3.82 (dd, J = 11.8, 9.4 Hz, 1H), 3.92-4.02 (m, 2H), 4.05-4.10 (m, 1H); MS m/z 339 [M+H]+.
(2S,5R)−N−{[(2R)−2−アミノプロピル]オキシ}−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−6)
工程1
tert−ブチル {(2R)−1−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート (IV−6)
参考例7と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(414mg、1.50mmol)と(R)−tert−ブチル (1−(アミノオキシ)プロパン−2−イル)カーバメート(569mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物625mgを得た(収率93%)。
1H NMR (400 MHz, CDCl3) δ 1.14 (d, J = 6.4 Hz, 3H), 1.43 (s, 9H), 1.53-1.70 (m, 1H), 1.90-2.06 (m, 2H), 2.28-2.36 (m, 1H), 2.79 (d, J = 12.0 Hz, 1H), 3.02 (br.d., J = 12.0 Hz, 1H), 3.28-3.33 (m, 1H), 3.56-3.68 (m, 1H), 3.84 (dd, J = 11.2, 3.6 Hz, 1H), 3.92-4.04 (m, 2H), 4.66 (br d, J = 8.0 Hz, 1H), 4.91 (d, J = 11.2 Hz, 1H), 5.06 (d, J = 11.2 Hz, 1H), 7.35-7.45 (m, 5H), 9.94 (br.s., 1H); MS m/z 449 [M+H]+.
tert−ブチル {(2R)−1−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメート (V−6)
参考例5と同様の手法にて、上記工程1の化合物全量より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.15 (d, J = 6.4 Hz, 3H), 1.44 (s, 9H), 1.73-1.84 (m, 1H), 1.86-2.00 (m, 1H), 2.01-2.12 (m, 1H), 2.19-2.29 (m, 1H), 3.06 (d, J = 11.6 Hz, 1H), 3.10-3.20 (m, 1H), 3.67-3.72 (m, 1H), 3.73-3.92 (m, 4H); MS m/z 359 [M+H]+.
(2S,5R)−N−{[(2R)−2−アミノプロピル]オキシ}−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−6)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル {(2R)−1−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロパン−2−イル}カーバメートを得た(定量的)。MS m/z 437[M−Bu4N]−.
1H NMR (400 MHz, D2O) δ 1.17 (d, J = 6.8 Hz, 3H), 1.66-1.78 (m, 1H), 1.78-1.88 (m, 1H), 1.90-2.06 (m, 2H), 3.02 (d, J = 12.0 Hz, 1H), 3.18 (br.d., J = 12.0 Hz, 1H), 3.48-3.58 (m, 1H), 3.83 (dd, J = 11.8, 9.0 Hz, 1H), 3.94 (br.d., J = 7.2 Hz, 1H), 3.98 (dd, J = 11.8, 3.4 Hz, 1H), 4.06-4.10 (m, 1H); MS m/z 339 [M+H]+.
(2S,5R)−N−(3−アミノプロポキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−7)
工程1
tert−ブチル {3−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロピル}カーバメート (IV−7)
参考例5と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(390mg、1.41mmol)とtert−ブチル (3−(アミノオキシ)プロピル)カーバメート(730mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物398.1mgを得た(収率63%)。
1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 1.50-1.67 (m, 1H), 1.75-1.86 (m, 2H), 1.88-2.07 (m, 2H), 2.28-2.37 (m, 2H), 2.77 (d, J = 11.0 Hz, 1H), 3.01 (br.d., J = 11.0 Hz, 1H), 3.20-3.38 (m, 3H), 3.89-4.04 (m, 3H), 4.90 (d, J = 11.4 Hz, 1H), 5.05 (d, J = 11.4 Hz, 1H), 5.17 (br.s., 1H), 7.36-7.45 (m, 5H), 9.21 (br.s., 1H); MS m/z 449 [M+H]+.
tert−ブチル {3−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロピル}カーバメート (V−7)
参考例5と同様の手法にて、上記工程1の化合物(392.8mg、876μmol)より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.43 (s, 9H), 1.73-1.99 (m, 4H), 2.01-2.12 (m, 1H), 2.13-2.24 (m, 1H), 3.07 (d, J = 11.6 Hz, 1H), 3.09-3.21 (m, 3H), 3.69 (br.s., 1H), 3.80-3.96 (m, 3H); MS m/z 359 [M+H]+.
(2S,5R)−N−(3−アミノプロポキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−7)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル {3−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロピル}カーバメートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.4 Hz, 12H), 1.33-1.53 (m, 8H), 1.47 (s, 9H), 1.55-1.96 (m, 12H), 2.14-2.23 (m, 1H), 2.31-2.41 (m, 1H), 2.85 (br.d., J= 11.2 Hz, 1H), 3.15-3.42 (m, 11H), 3.88-4.07 (m, 3H), 4.35 (br.s., 1H), 5.27 (br s, 1H), 9.26 (br.s., 1H); MS m/z 437 [M-Bu4N]-.
1H NMR (400 MHz, D2O) δ 1.67-2.05 (m, 6H), 3.00-3.19 (m, 4H), 3.82-3.94 (m, 3H), 4.05-4.10 (m, 1H); MS m/z 337 [M-H]-.
(2S,5R)−N−[(2S)−アゼチジン−2−イルメトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−8)
工程1
tert−ブチル (2S)−2−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート (IV−8)
参考例8と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(553mg、2.00mmol)と、(S)−tert−ブチル 2−((アミノオキシ)メチル)アゼチジン−1−カルボキシレート(578mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物760.1mgを得た(収率83%)。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9H), 1.56-1.70 (m, 1H), 1.88-2.07 (m, 3H), 2.23-2.34 (m, 2H), 2.84 (d, J = 11.6 Hz, 1H), 3.02 (d, J = 11.6 Hz, 1H), 3.28 (br s, 1H), 3.77-4.03 (m, 4H), 4.06-4.15 (m, 1H), 4.37-4.48 (m, 1H), 4.89 (d, J =11.6 Hz, 1H), 5.04 (d, J = 11.6 Hz, 1H), 7.34-7.44 (m, 5H), 10.63 (br.s., 1H); MS m/z 461 [M+H]+.
tert−ブチル (2S)−2−{[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート (V−8)
参考例5と同様の手法にて、上記工程1の化合物(699mg、1.52mmol)より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.44 (s, 9H), 1.74-1.85 (m, 1H), 1.86-1.99 (m, 1H), 2.02-2.14 (m, 1H), 2.16-2.40 (m, 3H), 3.06 (d, J = 11.6 Hz, 1H), 3.10-3.17 (m, 1H), 3.67-3.74 (m, 1H), 3.75-3.93 (m, 3H), 4.01 (dd, J = 10.6, 10.6 Hz, 1H), 4.14 (dd, J = 10.6, 10.6 Hz, 1H), 4.37-4.47 (m, 1H); MS m/z 371 [M+H]+.
(2S,5R)−N−[(2S)−アゼチジン−2−イルメトキシ]−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−8)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル (2S)−2−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.2 Hz, 12H), 1.30-2.10 (m, 19H), 1.46 (s, 9H), 2.12 -2.39 (m, 3H), 2.89 (br.d., J = 12.0 Hz, 1H), 3.23-3.39 (m, 9H), 3.76-3.93 (m, 3H), 3.95-4.06 (m, 1H), 4.08-4.18 (m, 1H), 4.33 (br.s., 1H), 4.37-4.50 (m, 1H); MS m/z 449 [M-Bu4N]-.
1H NMR (500 MHz, D2O) δ 1.71-1.83 (m, 1H), 1.84-1.97 (m, 1H), 1.98-2.16 (m, 2H), 2.36-2.49 (m, 1H), 2.50-2.61 (m, 1H), 3.10 (d, J = 12.0 Hz, 1H), 3.22-3.30 (m, 1H), 3.92-4.12 (m, 5H), 4.25-4.36 (m, 1H), 4.68-4.77 (m, 1H); MS m/z 351 [M+H]+.
(2S,5R)−7−オキソ−N−[(2R)−ピロリジン−2−イルメトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−9)
工程1
tert−ブチル (2R)−2−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピロリジン−1−カルボキシレート (IV−9)
参考例5と同様の手法にて(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(390mg、1.41mmol)と、(R)−tert−ブチル 2−((アミノオキシ)メチル)ピロリジン−1−カルボキシレート(796mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物336mgを得た(収率50%)。
1H NMR (400 MHz, CDCl3) δ 1.45 (s, 9H), 1.52-1.72 (m, 1H), 1.80-2.09 (m, 6H), 2.27-2.39 (m, 1H), 2.84 (br.d., J = 12.4 Hz, 1H), 2.96-3.08 (m, 1H), 3.28-3.44 (m, 3H), 3.60-3.86 (m, 2H), 3.89-4.06 (m, 1H), 4.14-4.29 (m, 1H), 4.90 (d, J = 11.2 Hz, 1H), 5.06 (d, J = 11.2 Hz, 1H), 7.32-7.47 (m, 5H), 10.56 (s, 1H); MS m/z 475 [M+H]+.
tert−ブチル (2R)−2−{[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピロリジン−1−カルボキシレート (V−9)
参考例5と同様の手法にて、上記工程1の化合物全量より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.46 (s, 9H), 1.73-2.27 (m, 8H), 3.06 (d, J = 11.6 Hz, 1H), 3.09-3.18 (m, 1H), 3.24-3.40 (m, 2H), 3.67-3.71 (m, 1H), 3.73-4.12 (m, 4H); MS m/z 385 [M+H]+.
(2S,5R)−7−オキソ−N−[(2R)−ピロリジン−2−イルメトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−9)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル (2R)−2−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピロリジン−1−カルボキシレートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.4 Hz, 12H), 1.34-1.51 (m, 8H), 1.46 (s, 9H), 1.55-1.78 (m, 10H), 1.80-2.01 (m, 4H), 2.11-2.23 (m, 1H), 2.29-2.42 (m, 1H), 2.88 (br.d., J = 11.2 Hz, 1H), 3.21-3.43 (m, 10H), 3.60-3.86 (m, 2H), 3.88-4.07 (m, 2H), 4.16-4.28 (m, 1H), 4.34 (br.s., 1H), 10.62 (br s, 1H); MS m/z 463 [M-Bu4N+2H]+.
1H NMR (500 MHz, D2O) δ 1.66-2.18 (m, 8H), 3.14 (d, J = 12.8 Hz, 1H), 3.23 (br.d., J = 12.8 Hz, 1H), 3.30 (t, J = 7.3 Hz, 2H), 3.89 (ddd, J = 8.2, 8.2, 3.4 Hz, 1H), 3.92-4.01 (m, 2H), 4.09-4.18 (m, 2H); MS m/z 365 [M+H]+.
(2S,5R)−7−オキソ−N−[(3R)−ピペリジン−3−イルメトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−10)
工程1
tert−ブチル (3R)−3−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピペリジン−1−カルボキシレート (IV−10)
参考例8と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(390mg、1.41mmol)と、(R)−tert−ブチル 3−((アミノオキシ)メチル)ピペリジン−1−カルボキシレート(527mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物333mgを得た(収率48%)。
1H NMR (400 MHz, CDCl3) δ 1.15-2.10 (m, 8H), 1.45 (s, 9H), 2.25-2.40 (m, 1H), 2.70-3.08 (m, 4H), 3.27-3.37 (m, 1H), 3.65-4.00 (m, 5H), 4.90 (d, J = 11.2 Hz, 1H), 5.05 (d, J = 11.2 Hz, 1H), 7.34-7.46 (m, 5H), 9.22 (br.s., 1H); MS m/z 489 [M+H]+.
tert−ブチル (3R)−3−{[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピペリジン−1−カルボキシレート (V−10)
参考例5と同様の手法にて、上記工程1の化合物全量より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.24-1.37 (m, 1H), 1.40-1.56 (m, 1H), 1.45 (s, 9H), 1.64-1.73 (m, 1H), 1.75-2.00 (m, 4H), 2.03-2.13 (m, 1H), 2.15-2.26 (m, 1H), 2.65-2.95 (m, 2H), 3.06 (d, J = 12.0 Hz, 1H), 3.13 (br.d., J = 12.0 Hz, 1H), 3.67-3.91 (m, 5H), 4.01-4.08 (m, 1H); MS m/z 399 [M+H]+.
(2S,5R)−7−オキソ−N−[(3R)−ピペリジン−3−イルメトキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−10)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル (3R)−2−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}ピペリジン−1−カルボキシレートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (dd, J = 7.6&6.8 Hz, 12H), 1.11-1.99 (m, 23H), 1.46 (s, 9H), 2.12-2.24 (m, 1H), 2.30-2.42 (m, 1H), 2.67-2.96 (m, 3H), 3.19-3.38 (m, 9H), 3.70-3.99 (m, 5H), 4.35 (br.s., 1H), 9.16 (br.s., 1H); MS m/z 477 [M-Bu4N]-.
1H NMR (400 MHz, D2O) δ 1.16-1.28 (m, 1H), 1.54-1.88 (m, 5H), 1.92-2.16 (m, 3H), 2.72 (t, J = 12.2 Hz, 1H), 2.81 (ddd, J = 12.8&12.8&3.5 Hz, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15-3.28 (m, 2H), 3.37-3.44 (m, 1H), 3.70 (dd, J = 10.3&7.6 Hz, 1H), 3.79 (dd, J = 10.3&5.0 Hz, 1H), 3.88-3.94 (m, 1H), 4.06-4.10 (m, 1H); MS m/z 377 [M-H]-.
(2S,5R)−7−オキソ−N−[(3S)−ピロリジン−3−イルオキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−11)
工程1
tert−ブチル (3S)−3−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]ピロリジン−1−カルボキシレート (IV−11)
参考例5と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(553mg、2.00mmol)と、(S)−tert−ブチル 3−(アミノオキシ)ピロリジン−1−カルボキシレート(606mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物920.4mgを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9H), 1.61-1.68 (m, 1H), 1.89-2.09 (m, 3H), 2.15-2.19 (m, 1H), 2.28-2.34 (m, 1H), 2.75 (d, J = 11.6 Hz, 1H), 2.95-3.06 (m, 1H), 3.31 (br s, 1H), 3.35-3.68 (m, 4H), 3.97 (d, J = 7.6 Hz, 1H), 4.60 (br.d., J= 23.2 Hz, 1H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 7.26-7.43 (m, 5H), 9.08 (br.d., J = 23.2 Hz, 1H); MS m/z 461 [M+H]+.
tert−ブチル (3S)−3−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]ピロリジン−1−カルボキシレート (V−11)
参考例5と同様の手法にて、上記工程1の化合物(869mg、1.89mmol)より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.47 (s, 9H), 1.75-2.12 (m, 4H), 2.13-2.25 (m, 2H), 3.05 (d, J = 12.0 Hz, 1H), 3.13 (br.d., J = 12.0 Hz, 1H), 3.25-3.50 (m, 2H), 3.61 (br.d., J = 13.2 Hz, 1H), 3.70 (br.s., 1H), 3.86 (br d, J = 7.2 Hz, 1H), 4.32-4.38 (m, 1H), 4.54-4.62 (m, 1H); MS m/z 371 [M+H]+.
(2S,5R)−7−オキソ−N−[(3S)−ピロリジン−3−イルオキシ]−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−11)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル (3S)−3−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]ピロリジン−1−カルボキシレートを得た(定量的)。MS m/z 449[M−Bu4N]−.
上記テトラブチルアンモニウム塩全量をトリフルオロ酢酸により脱保護し、オクタデシルシリカゲルカラムクロマトグラフィー精製後、標題化合物170.7mgを得た(3工程収率26%)。
1H NMR (400 MHz, D2O) δ 1.71-1.92 (m, 2H), 1.95-2.18 (m, 3H), 2.21-2.30 (m, 1H), 3.07 (d, J = 12.2 Hz, 1H), 3.24 (br.d., J = 12.2 Hz, 1H), 3.31-3.45 (m, 3H), 3.51 (d, J = 13.6 Hz, 1H), 3.99 (br.d., J = 6.0 Hz, 1H), 4.10-4.14 (m, 1H), 4.72-4.77 (m, 1H); MS m/z 349 [M-H]-.
(2S,5R)−N−(アゼチジン−3−イルメトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−12)
工程1
tert−ブチル 3−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート (IV−12)
参考例8と同様の手法にて、(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(553mg、2.00mmol)と、tert−ブチル 3−((アミノオキシ)メチル)アゼチジン−1−カルボキシレート(564mg)より得られた粗生成物をシリカゲルカラムクロマトグラフィーに付し、標題化合物699.7mgを得た(収率76%)。
1H NMR (400 MHz, CDCl3) δ 1.43 (s, 9H), 1.54-1.70 (m, 1H), 1.87-2.06 (m, 2H), 2.27-2.35 (m, 1H), 2.75 (d, J = 11.6 Hz, 1H), 2.80-2.90 (m, 1H), 3.01 (br.d., J = 11.6 Hz, 1H), 3.32 (br.s., 1H), 3.68-3.76 (m, 2H), 3.94 (br.d., J = 7.6 Hz, 1H), 4.00-4.15 (m, 4H), 4.90 (d, J = 11.8 Hz, 1H), 5.05 (d, J = 11.8 Hz, 1H), 7.35-7.44 (m, 5H), 9.08 (br s, 1H); MS m/z 461 [M+H]+.
tert−ブチル 3−{[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート (V−12)
参考例5と同様の手法にて、上記工程1の化合物(642mg、1.39mmol)より、標題化合物を得た(定量的)。
1H NMR (400 MHz, CD3OD) δ 1.43 (s, 9H), 1.74-1.85 (m, 1H), 1.86-1.97 (m, 1H), 2.04-2.13 (m, 1H), 2.16-2.24 (m, 1H), 2.84-2.94 (m, 1H), 3.05 (d, J = 11.6 Hz, 1H), 3.13 (br.d., J = 11.6 Hz, 1H), 3.68-3.82 (m, 3H), 3.83 (br.d., J = 6.8 Hz, 1H), 3.97-4.06 (m, 4H); MS m/z 371 [M+H]+.
(2S,5R)−N−(アゼチジン−3−イルメトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−12)
参考例5と同様の手法にて、上記工程2の化合物全量より、テトラブチルアンモニウム tert−ブチル 3−{[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレートを得た(定量的)。
1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.2 Hz, 12H), 1.37-1.51 (m, 8H), 1.46 (s, 9H), 1.54-1.75 (m, 9H), 1.82-1.97 (m, 1H), 2.13-2.25 (m, 1H), 2.29-2.40 (m, 1H), 2.77-2.95 (m, 2H), 3.24-3.40 (m, 9H), 3.64-4.16 (m, 7H), 4.36 (br.s., 1H), 9.16 (br.s., 1H); MS m/z 449 [M-Bu4N]-.
1H NMR (400 MHz, D2O) δ 1.65-1.89 (m, 2H), 1.92-2.06 (m, 2H), 3.06 (d, J = 12.4 Hz, 1H), 3.10-3.22 (m, 2H), 3.90-4.00 (m, 5H), 4.07-4.14 (m, 3H); MS m/z 351 [M+H]+.
(3aR,7aS)−2−ヒドロキシ−3a,4,7,7a‐テトラヒドロ−1H−イソインドール−1,3(2H)−ジオン
1H NMR (400 MHz, CDCl3) δ 2.20-2.31 (m, 2H), 2.56-2.65 (m, 2H), 3.08-3.14 (m, 2H), 5.91 (dt, J = 0.9, 2.7 Hz, 2H); MS m/z 166 [M-H]-.
(3aR,7aS)−1,3−ジオキソ−3a、4,7,7a‐テトラヒドロ−1H−イソインドール−2(3H)−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート
1−tert−ブチル 2−((3aR,7aS)−1,3−ジオキソ−3a,4,7,7a‐テトラヒドロ−1H−イソインドール−2(3H)−イル) (2S,5R)−5−((ベンジルオキシ)アミノ)ピペリジン−1,2−ジカルボキシレート
1H NMR (400 MHz, CDCl3) δ 1.47 (bs, 9H), 1.59-1.75 (m, 2H), 2.04-2.32 (m, 2H), 2.16-2.35 (m, 2H), 2.61 (d, J = 15.2 Hz, 2H), 3.14-3.24 (m, 4H), 4.15-4.22 (m, 1H), 4.71 (q, J=11.6 Hz, 2H), 5.03 (bs, 1H), 5.97 (bs, 2H), 7.26-7.38 (m, 5H); MS m/z 500 [M+H]+.
上記工程1の化合物(4.689g、9.386mmol)を脱水クロロホルム(50mL)に溶解し、トリエチルアミン(1.40g)を加えて氷冷した。混合物にトリホスゲン(1.09g)を加え0.5時間撹拌し、TLCにて標題化合物への収束を確認した。混合物に氷冷下メタノール(0.255mL)を加え30分間撹拌した後、メタンスルホン酸(8.89g)を加えて30分間撹拌し、TLCにて標題化合物への収束を確認した。混合物を氷冷1M 炭酸水素カリウム水溶液(11.1g/100mL)に滴下し0.5時間撹拌、クロロホルム(30mL)を加えて分層、有機層を1M 塩酸(70mL)、飽和重曹水(70mL)、飽和食塩水(70mL)で順次洗浄、無水硫酸マグネシウムで乾燥、濾過、減圧濃縮した。残渣をクロロホルム(16mL)に溶解しヘキサン(24mL)を加えて15分間撹拌、さらにヘキサン(8mL)を加えて15分間撹拌熟成した。析出した固体をろ取、クロロホルム/ヘキサン(2/3)の混液で洗浄、減圧乾燥し、無色結晶性粉末の標題化合物3.51gを得た(収率88%)。
1H NMR (400 MHz, CDCl3) δ 1.67-1.77 (m, 1H), 2.08 (d, J = 14.2 Hz, 1H), 2.14-2.26 (m, 2H), 2.30 (d, J = 13.8 Hz, 2H), 2.55-2.66 (m, 2H), 3.10-3.24 (m, 4H), 3.34 (bs, 1H), 4.45 (d, J = 6.4 Hz, 1H), 4.91 (d, J = 11.2 Hz, 1H), 5.06 (d, J = 11.4 Hz, 1H), 5.97 (bs, 2H), 7.34-7.45 (m, 5H); MS m/z 426 [M+H]+.
(3aR,7aS)−2−ヒドロキシヘキサヒドロ−1H−イソインドール−1,3(2H)−ジオン
1H NMR (400 MHz, CDCl3) δ 1.45 (dt, J = 3.0, 5.9 Hz, 4H), 1.71-1.90 (m, 4H), 2.84-2.92 (m, 2H), 6.01 (brs, 1H); MS m/z 168 [M-H]-.
(3aR,7aS)−1,3−ジオキソヘキサヒドロ−1H−イソインドール−2(3H)−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート
1−tert−ブチル 2−((3aR,7aS)−1,3−ジオキソヘキサヒドロ−1H−イソインドール−2(3H)−イル) (2S,5R)−5−((ベンジルオキシ)アミノ)ピペリジン−1,2−ジカルボキシレート
1H NMR (400 MHz, CDCl3) δ 1.35-1.58 (m, 13H), 1.62 (bs, 1H), 1.76 (bs, 2H), 1.90 (bs, 4H), 1.95-2.15 (m, 2H), 3.00 (bs, 2H), 3.15-3.30 (m, 2H), 4.16-4.25 (m, 1H), 4.72 (q, J = 11.6 Hz, 2H), 5.30-5.53 (m, 1H), 7.26-7.38 (m, 5H); MS m/z 502 [M+H]+.
上記工程1の化合物(4.521g、9.01mmol)を脱水クロロホルム(50mL)に溶解し、トリエチルアミン(1.350g)を加えて氷冷した。混合物にトリホスゲン(1.043g)を加え0.5時間撹拌し、TLCにて標題化合物への収束を確認した。混合物に氷冷下メタノール(0.245mL)を加え30分間撹拌した後、メタンスルホン酸(8.53g)を加えて30分間撹拌し、TLCにて標題化合物への収束を確認した。混合物を氷冷1M 炭酸水素カリウム水溶液(10.668g/90mL)に滴下し0.5時間撹拌、クロロホルム(33mL)を加えて分層、有機層を1M 塩酸(70mL)、飽和重曹水(70mL)、飽和食塩水(70mL)で順次洗浄、無水硫酸マグネシウムで乾燥、濾過、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/酢酸エチル=6/1)に付し、無色固体の標題化合物3.106gを得た(収率81%)。
1H NMR (400 MHz, CDCl3) δ 1.50 (bs, 4H), 1.62 (bs, 1H), 1.68-1.84 (m, 1H), 1.91 (bs, 4H), 2.04-2.27 (m, 2H), 3.02 (bs, 2H), 3.15 (s, 2H), 3.35 (bs, 1H), 4.47 (d, J = 6.6 Hz, 1H), 4.92 (d, J = 11.2 Hz, 1H), 5.07 (d, J = 11.4 Hz, 1H), 7.34-7.45 (m, 5H); MS m/z 428 [M+H]+.
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (IV−1)
実施例1a
(2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボン酸(4.80kg、17.373mol)の脱水酢酸エチル(62L)溶液を−30℃に冷却し、クロロギ酸イソブチル(2.52kg)、トリエチルアミン(1.85kg)、を順次滴下し、−30℃にて15分間撹拌した。反応液にtert−ブチル 2−(アミノオキシ)エチルカーバメートの脱水酢酸エチル溶液(15wt%、23.45kg)を30分で加え(洗い込み脱水酢酸エチル2L)、0℃まで1時間かけて昇温した。混合物を8%クエン酸水溶液(65L)、5%重曹水(60L)、水(60L)で順次洗浄し、24Lまで濃縮した。濃縮液に酢酸エチル(24L)を加え24Lまで置換濃縮する操作を2回行い、得られた濃縮液に酢酸エチル(29L)、へキサン(72L)を加え、終夜撹拌した。混合物にヘキサン(82L)を滴下し2時間撹拌した。析出結晶をろ取、ヘキサンで洗浄、真空乾燥して標題化合物5.51kgを得た(収率76%)。このものの機器データは参考例5の工程1のものと一致した。
2,5−ジオキソピロリジン−1−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(参考例3、373mg、1mmol)を脱水ジクロロメタン(5mL)に溶解し、氷冷下にtert−ブチル 2−(アミノオキシ)エチルカーバメート(194mg)の脱水ジクロロメタン(2mL、洗い込み1mL)溶液を加え、1時間撹拌した。反応液を酢酸エチル(65mL)で希釈、10%クエン酸(20mL)、飽和重曹水(20mL)、飽和食塩水(20mL)で順次洗浄、無水硫酸マグネシウムで乾燥、濾過、減圧濃縮して標題化合物362mgを得た(収率83%)。このものの機器データは参考例5の工程1のものと一致した。
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(参考例4、49.7g、113.6mmol)を脱水酢酸エチル(650mL)に懸濁し、室温でtert−ブチル 2−(アミノオキシ)エチルカーバメート(24.2g)の脱水酢酸エチル(134mL)溶液とトリエチルアミン(13.8g)を加え、2.5時間撹拌した。反応液を酢酸エチル(0.8L)で希釈、氷冷0.25M 塩酸(1L)、飽和重曹水(1L)、水(1L)で順次洗浄、減圧濃縮して、標題化合物48.08gを得た(収率98%、HPLCエリア面積比99%以上)。このものの機器データは参考例5の工程1のものと一致した。
tert−ブチル {2−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (V−1)
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (IV−1、5.52kg、12.705mol)のメタノール溶液(85L)に、10%パラジウム炭素触媒(50%含水、0.55kg)を加え、水素加圧(0.1MPa)下、1時間撹拌した。触媒を濾過し、固体をメタノール(25L)で洗浄した。ろ液を併せて、液温10℃以下で39Lまで減圧濃縮した。濃縮液にアセトニトリル(44L)を加えて液温10℃以下で39Lまで置換濃縮する操作を2回行い、混合物を0℃に冷却して終夜撹拌した。析出結晶をろ取、アセトニトリル(24L)で洗浄、真空乾燥して標題化合物を3.63kg得た(収率83%)。このものの機器データは参考例5の工程2のものと一致した。
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (VI−1)
実施例3a
アセトニトリル(51L)に、水(51mL)、tert−ブチル {2−[({[(2S,5R)−6−ヒドロキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート(V−1、3.53kg、10.251mol)、三酸化イオウ−ピリジン錯体(3.95kg)、2,6−ルチジン(2.21kg)を順次加え、35〜45℃で終夜撹拌した。混合物をろ過して不溶物を除き、固体をアセトニトリル(11L)で洗浄、ろ液を併せて17Lまで濃縮した。濃縮液を10℃以下に冷却し、9%燐酸二水素ナトリウム水溶液(60L)、酢酸エチル(113L)で分層し、有機層を再度9%燐酸二水素ナトリウム水溶液(11L)で抽出した。得られた水層に酢酸エチル(113L)、30%硫酸水素テトラブチルアンモニウムの水溶液(12.87kg)、37%燐酸二水素ナトリウム水溶液(56.5kg)を加え、15分間撹拌した。有機層を分層し、20%燐酸2水素ナトリウム水溶液(60L)で洗浄、無水硫酸マグネシウム(2.5kg)にて乾燥、濾過後、減圧濃縮した。濃縮液中に析出した標題化合物の結晶は酢酸エチルで溶解して全液量を20Lに調整し、標題化合物の酢酸エチル溶液32.55kgを得た(正味6.25kg、収率92%)。本溶液はさらに精製することなく次工程に付した。
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (IV−1、515mg、1.16mmol)のイソプロパノール(7mL)溶液に、水(5mL)、三酸化硫黄−トリメチルアミン錯体(196mg)、トリエチルアミン(0.0407mL)、10%パラジウム炭素触媒(53.3%含水、95.0mg)を加え、水素雰囲気下、室温にて2時間撹拌した。10%パラジウム炭素触媒(53.3%含水、63.5mg)を追加し、水素雰囲気下、室温にて3時間撹拌後、アルゴン置換し、室温にて1時間撹拌した。反応液の触媒をセライト濾過し、イソプロパノール/水(1/1、40mL)で洗浄後、ミリポア濾過、イソプロパノール/水(1/1、15mL)で洗浄、イソプロパノールを減圧留去した。得られた水溶液に燐酸二水素ナトリウム(5.29g)、酢酸エチル(20mL)、硫酸水素テトラブチルアンモニウム(476mg)を加え、室温にて10分間撹拌後、酢酸エチルにて2回抽出した。有機層を硫酸ナトリウムにて乾燥、濾過、濃縮後、標題化合物702mgを得た(収率91%)。
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート (IV−1、5.0g、11.51mmol)のイソプロパノール(80mL)溶液に、10%パラジウム炭素触媒(50%含水、0.5g)を加え、水素雰囲気化2時間撹拌した。反応液の触媒をセライト濾過し、固体をイソプロパノール(15mL)で洗浄、ろ液を併せ水(47.5mL)、三酸化硫黄−トリメチルアミン錯体(1.8g)、トリエチルアミン(0.237g)を加え、25〜30℃で24時間撹拌した。混合物を47mLまで減圧濃縮し、燐酸二水素ナトリウム(11.87g)、酢酸エチル(200mL)、硫酸水素テトラブチルアンモニウム(4.688g)を加えて10分間撹拌した。有機層を分層し、水層をさらに酢酸エチル(2x100mL)で抽出、有機相を併せて硫酸マグネシウムで乾燥、濾過した。ろ液の有機溶媒を減圧濃縮し、標題化合物の酢酸エチル溶液を得た(正味6.522g、収率85%)
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR)
実施例4a
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート(VI−1、788g、正味467.1g、0.701mol)のジクロロメタン(934mL)溶液を窒素気流下にて−20℃に冷却し、トリフルオロ酢酸(934mL)を15分間で滴下、0℃に昇温して1時間撹拌した。反応液を−20℃に冷却しジイソプロピルエーテル(4.17L)を滴下し、混合物を−6℃に昇温して1時間撹拌した。沈殿をろ過、ジイソプロピルエーテル(2x1L)にて懸濁洗浄、湿固体を真空乾燥して標題化合物342.08gを得た(正味222.35g、収率98%、HPLCエリア面積比96.1%、CE/TFA27mol%)。
テトラブチルアンモニウム tert−ブチル {2−[({[(2S,5R)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}カーバメート(VI−1、酢酸エチル溶液15.60kg、正味2.98kg)を冷却し液温0℃以下で9Lまで濃縮した。濃縮液にジクロロメタン(9L)を加え、窒素気流下で混合物を−20℃に冷却した。混合物にトリフルオロ酢酸(16.5L)を−5℃以下にて約1時間で滴下、−5〜0℃にて1時間撹拌した。反応液に0℃に冷却した酢酸エチルを7℃以下で分割添加し(4x8.3L、24.6L、合計57.8L)、0℃で終夜撹拌した。沈殿をろ過、酢酸エチル(13.5L、9L)にて懸濁洗浄、終夜真空乾燥して標題化合物1.74kgを得た(正味1.43kg、収率99%、HPLCエリア面積比99.1%、CE/TFA10mol%、GC/EtOAc14%)。
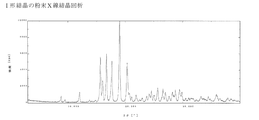
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のI形結晶
実施例5a
0.5M 酢酸緩衝液(pH5.5、35mL)を氷冷し、(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR、36g)と冷却した5M 水酸化ナトリウム水溶液を交互に加えてpHを5.5に調整し、オクタデシルシリカゲルカラムクロマトグラフィー(3.6L)に付し、水で溶出した。活性フラクションを集め、水浴温度35℃にて減圧濃縮し、析出した結晶を終夜真空乾燥した。得られた結晶2.10gを粉砕した後、氷冷下にてイソプロパノール/水(19/1、13mL)を加え、0℃にて1時間撹拌した。懸濁液を濾過し、冷イソプロパノール/水(19/1、80mL)にて洗浄し、得られた結晶を真空ポンプして乾燥後、標題化合物1.68gを得た(収率80%)。DSC吸熱ピーク 111℃。60%イソプロパノール水溶液への溶解度;0.44%(10℃)、0.48%(20℃)。標題化合物は粉末X線回析図形において下記表4と図1に示すような特徴的なピークパターンを示した。なお、測定に際し粉末X線回析装置は株式会社リガクのRINT2100を用い、X線源としてはCuKα1、管電圧40kV、管電流40mA、スキャンスピード4°/min、走査範囲は2θ=3から40°で測定した。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR、正味4.253g)を0.2M 燐酸緩衝液(pH6.5、73mL)に溶解しpH5.5とし、水(20mL)で希釈した。混合物を130mLまで濃縮、レジン精製(SP207、260mL)に付し、水(238mL)、10%イソプロパノール水溶液(780mL)にて溶出した。活性フラクションを集め、30mLまで減圧濃縮し、活性炭(精製白鷺、87mg)を投入し室温にて30分撹拌した。活性炭をメンブランフィルターでろ過、ろ液を凍結乾燥に付し、非晶質形態の(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)を4.07g得た(収率95.7%)。本非晶質形態の化合物(0.2g)を水(0.8mL)に溶解し、室温でイソプロパノール(1.2mL)を加え、I形結晶(実施例5a、1mg)を接種して撹拌子で3時間撹拌、析出結晶を濾過、乾燥し標題化合物0.1gを得た(収率50%)。本結晶は粉末X線回析図形において実施例5aの結晶と同様のピークパターンを示した。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR、正味2.113g)と0.2M 燐酸緩衝液(pH6.5、73mL)を交互に加えpH4.6に調整し、水(27mL)で希釈した。混合物を80mLまで減圧濃縮後、0.2M燐酸緩衝液(pH6.5、16mL)でpH5.4とし、水(48mL)で希釈した。本混合物をレジン精製(SP207、240mL)に付し、水(276mL)、10%イソプロパノール水溶液(720mL)にて溶出した。活性フラクションを集め、12mLまで減圧濃縮し、活性炭(精製白鷺、40mg)を投入し室温にて30分撹拌した。活性炭をメンブランフィルターでろ過、水で14mLに希釈した。水溶液にI形結晶(実施例5b、6mg)を接種し、室温で撹拌子にて撹拌して得られた懸濁液にイソプロパノール(84mL)を1時間かけて滴下した。滴下終了後、3時間撹拌、析出結晶を濾過、乾燥し標題化合物1.834gを得た(収率86.8%)。水分:5.37%,脱水物換算含量95.3%,HPLCエリア面積比99.3%。本結晶は粉末X線回析図形において実施例5aの結晶と同様のピークパターンを示した。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のII形結晶
実施例6a
0.2M 燐酸緩衝液(pH6.5、0.8L)を10℃以下に冷却し、撹拌しながら(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR、正味49.96g)と冷却した0.2M 燐酸緩衝液(pH6.5)を交互に少量ずつ加えてpHが4.2〜5.5の間でpHを調整、最終的にpH5.5に調整した。混合物を水で希釈(全量1.8L)、液温18℃以下で1.6Lまで減圧濃縮した。濃縮液を水で1.8Lに希釈(HPLCエリア面積比96.7%)、レジン(セパビーズSP207、3L)精製に付し、水(0.83L)と10%イソプロパノール水溶液で溶出して活性フラクションを集めた。活性フラクションを併せ(1.5L)、液温15℃以下で0.5Lまで減圧濃縮し、活性炭(0.88g)を加えて30分間撹拌した。活性炭をメンブランフィルターでろ過、水(0.05Lx2)で洗浄した。ろ液を併せ液温15℃以下で0.2Lまで減圧濃縮し、液温を10〜15℃に調整、混合物にイソプロパノール(0.25L)を10分間で滴下し、1時間撹拌後さらにイソプロパノール(0.6L)を15分間で滴下した。混合物を1時間撹拌し析出した結晶をろ取、イソプロパノール(0.2L)で洗浄、湿結晶の品温が20℃になるまで真空乾燥し標題化合物44.69gを得た(収率85%、水分5.9%、HPLCエリア面積比100%)。DSC吸熱ピーク 92℃。60%イソプロパノール水溶液への溶解度;0.67%(10℃)、0.74%(20℃)。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のII形結晶の転移実験
実施例6aの結晶懸濁液を少量取り、撹拌子により室温1日撹拌し、析出結晶を集め、粉末X線結晶回析に付したところ、他の結晶形への転移は観察されなかった。
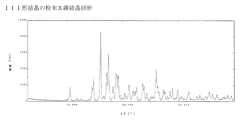
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のIII形結晶
実施例7a
0.2M 燐酸緩衝液(pH6.5、3.0L)を10℃以下に冷却し、撹拌しながら(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR、正味133.334g)と冷却した0.2M 燐酸緩衝液(pH6.5、1.8L)を交互に少量ずつ加えてpHが5.1〜5.5の間でpHを調整、最終的にpH5.3に調整した。混合物を液温18℃以下で3.6Lまで減圧濃縮した。濃縮液のpHを0.2M 燐酸緩衝液(pH6.5)でpH5.5に調整し、水で4.8Lに希釈、レジン(セパビーズSP207、7L)精製に付し、水(7.2L)と10%イソプロパノール水溶液で溶出して活性フラクションを集めた。活性フラクションを併せ(3.1L)、液温15℃以下で1.8Lまで減圧濃縮し、活性炭(2.66g)を加えて30分間撹拌した。活性炭をメンブランフィルターでろ過、水(0.39L)で洗浄した。ろ液を併せ液温18℃以下で0.6Lまで減圧濃縮した。濃縮液の液温を20〜25℃に調整、イソプロパノール(0.77L)を滴下後に、II形結晶(実施例6a、0.63g)を接種し1時間撹拌した。混合物にさらにイソプロパノール(1.93L)を1.5時間かけて滴下し、30分間撹拌した。析出した結晶をろ取、イソプロパノール(1L)で洗浄、湿結晶の品温が20℃になるまで真空乾燥しII形結晶を少量含む標題化合物127.3gを得た(収率90%、水分5.3%、HPLCエリア面積比99.9%)。本工程で得られた結晶を種結晶とし同様の工程を実施、さらに得られた結晶を次回の種結晶とすることで粉末X線結晶回析で単一のIII形結晶の標題化合物を得た。DSC吸熱ピーク 102℃。60%イソプロパノール水溶液への溶解度;0.76%(10℃)、0.80%(20℃)。
標題化合物は粉末X線回析図形において下記表6と図3に示すような特徴的なピークパターンを示した。なお、測定に際し粉末X線回析装置は株式会社リガクのRINT2100を用い、X線源としてはCuKα1、管電圧40kV、管電流40mA、スキャンスピード4°/min、走査範囲は2θ=3から40°で測定した。
0.2M燐酸緩衝液(pH6.5、7.2L)を10℃以下に冷却し、撹拌しながら(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1―CR、正味1.2kg)と氷冷した0.2M 燐酸緩衝液(pH6.5、3.5L)を交互に少量ずつ加えてpHが4.2〜4.8の間でpHを調整、最終的にpH4.6に調整した。混合物を水(19.3L)で希釈(全量30L)、液温18℃以下で24Lまで減圧濃縮した。濃縮液のpHを0.2M 燐酸緩衝液(pH6.5、2.4L)でpH5.4に調整し(HPLCエリア面積比98.5%)、水で43.2Lに希釈、レジン(セパビーズSP207、75L)精製に付し、水(83L)と10%イソプロパノール水溶液で溶出して活性フラクションを集めた。活性フラクションを併せ(33L)、液温15℃以下で7.2Lまで濃縮し、活性炭(24g)を加えて30分間撹拌した。活性炭をメンブランフィルターでろ過、水(0.4Lx2)で洗浄した。ろ液を併せ、液温を20〜25℃に調整、III形結晶(実施例7a、3.6g)を接種した。混合物にイソプロパノール(50.4L)を1時間かけて滴下し、終夜撹拌した。析出した結晶をろ取、イソプロパノール(4.8L)で洗浄、湿結晶の品温が20℃になるまで真空乾燥し標題化合物1.17kgを得た(収率90%、水分5.3%、HPLCエリア面積比100%)。本結晶は粉末X線回析図形において実施例7aの結晶と同様のピークパターンを示した。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のIII形結晶の転移実験
実施例7aの工程にて、接種直後とイソプロパノール滴下後の懸濁液を少量取り、それぞれ撹拌子により室温1日、4日撹拌し、析出結晶を集め、粉末X線結晶回析に付したところ、他の結晶形への転移は観察されなかった。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のIII形結晶のXRD−DSC実験
実施例7aのIII形結晶の加熱による結晶相の変化をXRD−DSCにて観察した。加熱、冷却条件は、60%RH一定、昇降温速度2℃/minにて室温から160℃まで昇温し、63℃まで冷却し、継時的に試料のDSCとXRDを測定した。なお、測定に際し粉末X線回析装置は株式会社リガクのSmartLabとXRD−DSCを用い、X線源としてはCuKα1、管電圧45kV、管電流200mA、スキャンスピード80°/min、走査範囲は2θ=5から35°で測定した。
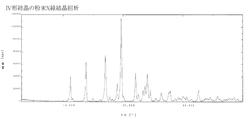
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のIV形結晶
実施例8a
実施例7aのIII形結晶0.20gを水2mLに溶解させ、撹拌しながらメタノール(30mL)を滴下し、20−25℃で終夜静置した。混合物をろ過し、メタノール(2x2mL)で洗浄、室温で終夜真空乾燥して標題化合物0.13gを得た(収率68%)。
標題化合物は粉末X線回析図形において下記表7と図4に示すような特徴的なピークパターンを示した。なお、測定に際し粉末X線回析装置は株式会社リガクのRINT2100を用い、X線源としてはCuKα1、管電圧40kV、管電流40mA、スキャンスピード4°/min、走査範囲は2θ=3から40°で測定した。
実施例7aのIII形結晶25gにメタノール(200mL)を加え、20−25℃で3.5時間撹拌した。混合物をろ過し、メタノール(2x20mL)で洗浄、室温で終夜真空乾燥して標題化合物23gを得た(収率99%)。本結晶は粉末X線回析図形において実施例8aの結晶と同様のピークパターンを示した。
実施例7aのIII形結晶25gにエタノール(200mL)を加え、20−25℃で3.5時間撹拌した。混合物をろ過し、エタノール(2x20mL)で洗浄、室温で終夜真空乾燥して標題化合物23gを得た(収率99%)。本結晶は粉末X線回析図形において実施例8aの結晶と同様のピークパターンを示した。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のIV形結晶の転移実験
実施例8a〜cのIV形結晶を取り、イソプロパノール/水(6/1)を加え25℃または40℃で1週間まで懸濁撹拌した。12時間後、24時間後(1日)、48時間後(2日)、72時間後(3日)、96時間後(4日)及び168時間後(1週間)にサンプルを取り、通気乾燥後に粉末X線回折に付したところ、いずれの撹拌時間においても他の結晶形への転移は観察されなかった。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のI〜IV形結晶の安定性評価
実施例7aのIII形結晶を水に溶解し凍結乾燥に付し、非晶体の(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)を得た。また、本非晶体の化合物と実施例5〜8のI〜IV形結晶をそれぞれスクリュウ管に量り、各温湿度条件下安定性試験を実施した。類縁物質、含量、水分測定法は以下のとおりである。結果を表9に示す。
被験物質を水に溶かして試料溶液とする。試料溶液5μLにつき,次の条件でJP16液体クロマトグラフィー<2.01>により試験を行い、個々の類縁物質の量(%)と類縁物質総量(%)及び含量を求める。
カラム:Waters Atlantis dc18、5μm,4.6×250mm
カラム温度:35℃付近の一定温度
注入量:5μL
検出器:紫外吸光光度計(測定波長:210nm)
移動相A:リン酸水素二アンモニウム1.32gを水900mLに溶かし,リン酸を加えてpH3.0に調整した後,水を加えて1000mLとする。
移動相B:液体クロマトグラフィー用アセトニトリル
勾配プログラム:移動相A及び移動相Bの混合比を次のように変えて制御する。
流量:1.0mL/min
保持時間:約6.5min
測定時間:30min
本品約20mgを精密に量り,JP16 水分測定法<2.48> 電量滴定法により試験を行う。
(2S,5R)−N−(2−アミノエトキシ)−7−オキソ−6−(スルホオキシ)−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキサミド (VII−1)のIII形結晶の包装容器での安定性評価
III形結晶を下記条件にて包装し、実施例9aの分析条件にて、各温湿度条件下安定性試験を実施した。結果を表10〜12に示す。
包装容器
内装:一重低密度ポリエチレン・ナイロン製結束バンド
外装:一重アルミラミネート袋・ヒートシール
tert−ブチル {2−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]エチル}(メチル)カーバメート (IV−2)
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(参考例4、144mg、0.329mmol)を脱水ジクロロメタン(2.5mL)に溶解し、tert−ブチル (2−(アミノオキシ)エチル)(メチル)カーバメート(88.8mg)の脱水ジクロロメタン(0.5mL)溶液を加え、室温で18時間撹拌した。反応液を酢酸エチル(10mL)で希釈、0.25M 塩酸、飽和重曹水、飽和食塩水で順次洗浄、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮して、標題化合物132mgを得た(収率89%)。このものの機器データは参考例6の工程1のものと一致した。
tert−ブチル {3−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]プロピル}カーバメート (IV−7)
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(参考例4、148mg、0.339mmol)を脱水ジクロロメタン(2.5mL)に溶解し、tert−ブチル 3−(アミノオキシ)プロピルカーバメート(90.9mg)の脱水ジクロロメタン(0.5mL)溶液を加え、室温で18時間撹拌した。反応液を酢酸エチル(10mL)で希釈、0.25M 塩酸、飽和重曹水、飽和食塩水で順次洗浄、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮して、標題化合物134mgを得た(収率88%)。このものの機器データは参考例11の工程1のものと一致した。
tert−ブチル (2S)−2−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート (IV−8)
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(参考例4、145mg、0.331mmol)を脱水ジクロロメタン(2.5mL)に溶解し、(S)−tert−ブチル 2−((アミノオキシ)メチル)アゼチジン−1−カルボキシレート(93.2mg)の脱水ジクロロメタン(0.5mL)溶液を加え、室温で21時間撹拌した。反応液を酢酸エチル(10mL)で希釈、0.25M 塩酸、飽和重曹水、飽和食塩水で順次洗浄、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮して、標題化合物127mgを得た(収率83%)。このものの機器データは参考例12の工程1のものと一致した。
tert−ブチル (3S)−3−[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]ピロリジン−1−カルボキシレート (IV−11)
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(参考例4、145mg、0.332mmol)を脱水ジクロロメタン(2.5mL)に溶解し、(S)−tert−ブチル 3−(アミノオキシ)ピロリジン−1−カルボキシレート(91.6mg)の脱水ジクロロメタン(0.5mL)溶液を加え、室温で19時間撹拌した。反応液を酢酸エチル(10mL)で希釈、0.25M 塩酸、飽和重曹水、飽和食塩水で順次洗浄、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮して、標題化合物145mgを得た(収率95%)。このものの機器データは参考例15の工程1のものと一致した。
tert−ブチル 3−{[({[(2S,5R)−6−ベンジルオキシ−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクト−2−イル]カルボニル}アミノ)オキシ]メチル}アゼチジン−1−カルボキシレート (IV−12)
(1R,2S,6R,7S)−3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル (2S,5R)−6−(ベンジルオキシ)−7−オキソ−1,6−ジアザビシクロ[3.2.1]オクタン−2−カルボキシレート(実施例4、140mg、0.320mmol)を脱水ジクロロメタン(2.5mL)に溶解し、tert−ブチル 3−((アミノオキシ)メチル)アゼチジン−1−カルボキシレート(91.5mg)の脱水ジクロロメタン(0.5mL)溶液を加え、室温で20時間撹拌した。反応液を酢酸エチル(10mL)で希釈、0.25M 塩酸、飽和重曹水、飽和食塩水で順次洗浄、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮して、標題化合物132mgを得た(収率90%)。このものの機器データは参考例16の工程1のものと一致した。
Claims (34)
- 下記式(III):
で示される化合物を、化合物:R3ONH2と反応させて下記式(IV):
で示される化合物とし、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムで処理し、下記式(VI):
で示される化合物とした後、側鎖R3ONHC(=O)−が保護基を有する場合、酸により当該保護基を除去、続いて反応液に貧溶媒を加えて粗生成物を沈殿化し、下記式(VII−CR):
で示される粗の化合物とした後、式(VII−CR)で示される粗の化合物と氷冷した緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、貧溶媒を加えて、結晶化することによる下記式(VII):
(上記各式中、OBnはベンジルオキシ、R1は2,5−ジオキソピロリジン−1−イル、1,3−ジオキソ−3a,4,7,7a‐テトラヒドロ−1H−イソインドール−2(3H)−イル、1,3−ジオキソヘキサヒドロ−1H−イソインドール−2(3H)−イル、または3,5−ジオキソ−4−アザトリシクロ[5.2.1.02,6]デカ−8−エン−4−イル、R3はC1−6アルキルまたはヘテロシクリルを示す。R3は0から5個のR4で修飾されていても良く、R4は連続して置換されてもよい。ここでR4はC1−6アルキル、ヘテロシクリル、R5(R6)N−、または保護基である。また、R5とR6は各々独立して水素またはC1−6アルキルを示すか、あるいは一緒になってヘテロシクリルを形成する。さらに、R3、R5及びR6は任意の位置で閉環することができる。)
で示される化合物の製造法。 - 下記式(III):
で示される化合物を、化合物:R3ONH2と反応させることによる下記式(IV):
(上記各式中、R1、R3およびOBnは、請求項1に記載のとおりである)
で示される化合物の製造法。 - 下記式(IV):
で示される化合物を、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムで処理することによる、下記式(VI):
(上記各式中、R3およびOBnは、請求項1に記載のとおりである)
で示される化合物の製造法。 - 下記式(VI):
で示される化合物を、側鎖R3ONHC(=O)−が保護基を有する場合、酸により当該保護基を除去、続いて反応液にエステル系貧溶媒を加えて粗生成物を沈殿化することによる、下記式(VII−CR):
(上記各式中、R3は、請求項1に記載のとおりである)
で示される粗の化合物の製造法。 - 下記式(VII−CR):
で示される粗の化合物と氷冷した緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、アルコール系貧溶媒を加えて、結晶化することによる下記式(VII):
(上記各式中、R3は、請求項1に記載のとおりである)
で示される化合物の製造法。 - 式(IV)、(VI)、(VII−CR)、(VII)にてR3が、
2−(tert−ブトキシカルボニルアミノ)エチル、
2−アミノエチル、
2−((tert−ブトキシカルボニル)(メチル)アミノ)エチル、
2−(メチルアミノ)エチル、
2−((tert−ブトキシカルボニル)(イソプロピル)アミノ)エチル、
2−(イソプロピルアミノ)エチル、
2−(ジメチルアミノ)エチル、
(2S)−2−((tert−ブトキシカルボニル)アミノ)プロピル、
(2S)−2−(アミノ)プロピル、
(2R)−2−((tert−ブトキシカルボニル)アミノ)プロピル、
(2R)−2−(アミノ)プロピル、
3−((tert−ブトキシカルボニル)アミノ)プロピル、
3−(アミノ)プロピル、
(2S)−tert−ブトキシカルボニルアゼチジン−2−イルメチル、
(2S)−アゼチジン−2−イルメチル、
(2R)−tert−ブトキシカルボニルピロリジン−2−イルメチル
(2R)−ピロリジン−2−イルメチル
(3R)−tert−ブトキシカルボニルピペリジン−3−イルメチル、
(3R)−ピペリジン−3−イルメチル、
(3S)−tert−ブトキシカルボニルピロリジン−3−イル、
(3S)−ピロリジン−3−イル、
1−(tert−ブトキシカルボニル)アゼチジン−3−イル、
アゼチジン−3−イル、
から選ばれる化合物の請求項1〜5いずれか記載の製造法。 - 下記式(III):
で示される化合物を、塩基存在下tert−ブチル 2−(アミノオキシ)エチルカーバメートと反応させて下記式(IV−1):
で示される化合物とし、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムと処理し、下記式(VI−1):
で示される化合物とした後、トリフルオロ酢酸によりtert−ブトキシカルボニル(Boc)基を除去、続いて反応液に酢酸エチルを滴下することにより粗生成物を沈殿化し、下記式(VII−1−CR):
で示される粗の化合物とした後、式(VII−1−CR)で示される粗の化合物と氷冷した燐酸緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、イソプロパノールを加えて結晶化することによる下記式(VII−1):
(上記各式中、R1およびOBnは、請求項1に記載のとおりである)
で示される化合物の製造法。 - 下記式(III):
で示される化合物を、塩基存在下tert−ブチル 2−(アミノオキシ)エチルカーバメートと反応させることによる下記式(IV−1):
(上記各式中、R1およびOBnは、請求項1に記載のとおりである)
で示される化合物の製造法。 - 下記式(IV−1):
で示される化合物を、水素雰囲気化にパラジウム炭素触媒と処理し、同時または連続して含水溶媒中で触媒量の塩基存在下に三酸化硫黄−トリメチルアミン錯体により硫酸化反応に付し、硫酸水素テトラブチルアンモニウムと処理することによる、下記式(VI−1):
(上記各式中、OBnは、請求項1に記載のとおりである)
で示される化合物の製造法。 - 下記式(VI−1):
で示される化合物を、トリフルオロ酢酸によりtert−ブトキシカルボニル(Boc)基を除去、続いて反応液に酢酸エチルを滴下することにより粗生成物を沈殿化することによる、下記式(VII−1−CR):
で示される粗の化合物の製造法。 - 下記式(VII−1−CR):
で示される粗の化合物と氷冷した燐酸緩衝液を交互に加えてpH4〜5.5の溶液とし、必要に応じて合成吸着剤による脱塩後に濃縮し、温調後に必要に応じて接種、イソプロパノールを加えて結晶化することによる下記式(VII−1):
で示される化合物の製造法。 - 粉末X線回析図形において面間隔(d)7.34、5.66、5.53、5.30、5.02、4.66、4.37、4.28、4.06、3.68、3.62、3.47、3.36、3.30、3.16、3.11、3.03、2.99、及び2.50Åに特徴的なピークを有する式(VII−1):
で示される化合物のI形結晶。 - 上記式請求項12記載のI形結晶を製造する、請求項1から11いずれか記載の製造法。
- 式(VII−1)で示される化合物の溶液を20〜25℃に温調し、I形結晶を接種、撹拌後にさらにイソプロパノールを加えることによる、請求項12記載のI形結晶の製造法。
- 場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項12記載のI形結晶の使用。
- アンピシリン、アモキシシリン、ピペラシリン、チカルシリン、フロモキセフ、セフォタキシム、セフトリアキソン、セフタジジム、セフェピム、セフタロリン、セフトロザン、イミペネム、メロペネム、ビアペネム、ドリペネム、エルタペネム、アズトレオナムからなる群より選択されるβ−ラクタム系抗生物質と、場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項12記載のI形結晶の使用。
- 粉末X線回析図形において面間隔(d)9.46、5.62、5.23、5.10、5.00、4.91、4.67、4.45、4.29、3.96、3.78、3.71、3.52、3.24、3.18、3.10、3.02、2.88、2.81、2.77、2.67、2.50、及び2.45Åに特徴的なピークを有する式(VII−1):
- 請求項17記載のII形結晶を製造する、請求項1から11いずれか記載の製造法。
- 式(VII−1)で示される化合物の溶液を10〜15℃に温調し、イソプロパノールを加えて撹拌することによる、請求項17記載のII形結晶の製造法。
- 場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項17記載のII形結晶の使用。
- アンピシリン、アモキシシリン、ピペラシリン、チカルシリン、フロモキセフ、セフォタキシム、セフトリアキソン、セフタジジム、セフェピム、セフタロリン、セフトロザン、イミペネム、メロペネム、ビアペネム、ドリペネム、エルタペネム、アズトレオナムからなる群より選択されるβ−ラクタム系抗生物質と、場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項17記載のII形結晶の使用。
- 粉末X線回析図形において面間隔(d)8.32、6.10、5.98、5.51、5.16、5.07、4.85、4.70、4.61、4.35、4.20、4.06、4.00、3.95、3.77、3.73、3.65、3.42、3.39、3.36、3.26、3.23、3.13、3.09、2.99、2.81、及び2.52Åに特徴的なピークを有する式(VII−1):
で示される化合物のIII形結晶。 - 請求項22記載のIII形結晶を製造する、請求項1から11いずれか記載の製造法。
- 式(VII−1)で示される化合物の溶液を20〜25℃に温調し、III形結晶を接種、イソプロパノールを加えて撹拌することによる、請求項22記載のIII形結晶の製造法。
- 場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項22記載のIII形結晶の使用。
- アンピシリン、アモキシシリン、ピペラシリン、チカルシリン、フロモキセフ、セフォタキシム、セフトリアキソン、セフタジジム、セフェピム、セフタロリン、セフトロザン、イミペネム、メロペネム、ビアペネム、ドリペネム、エルタペネム、アズトレオナムからなる群より選択されるβ−ラクタム系抗生物質と、場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項22記載のIII形結晶の使用。
- 粉末X線回析図形において面間隔(d)7.88、6.41、5.20、4.67、4.50、4.02、3.81、3.75、3.70、3.62、3.38、3.23、3.20、及び2.74Åに特徴的なピークを有する式(VII−1):
で示される化合物のIV形結晶。 - 請求項27記載のIV形結晶を製造する、請求項1から11いずれか記載の製造法。
- 式(VII−1)で示される化合物の溶液を20〜25℃に温調し、メタノールを加えて撹拌することによる、請求項27記載のIV形結晶の製造法。
- 請求項12、17、または22記載のI、II、またはIII形結晶を、メタノール、エタノールまたはイソプロパノール中で撹拌することによる、請求項27記載のIV形結晶の製造法。
- 場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項27記載のIV形結晶の使用。
- アンピシリン、アモキシシリン、ピペラシリン、チカルシリン、フロモキセフ、セフォタキシム、セフトリアキソン、セフタジジム、セフェピム、セフタロリン、セフトロザン、イミペネム、メロペネム、ビアペネム、ドリペネム、エルタペネム、アズトレオナムからなる群より選択されるβ−ラクタム系抗生物質と、場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項27記載のIV形結晶の使用。
- 場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項12、17、22、または27記載のI、II、III、またはIV形結晶の混合物の使用。
- アンピシリン、アモキシシリン、ピペラシリン、チカルシリン、フロモキセフ、セフォタキシム、セフトリアキソン、セフタジジム、セフェピム、セフタロリン、セフトロザン、イミペネム、メロペネム、ビアペネム、ドリペネム、エルタペネム、アズトレオナムからなる群より選択されるβ−ラクタム系抗生物質と、場合によっては医学的に許容される担体を含んでなる医薬組成物製造のための、請求項12、17、22、または27記載のI、II、III、またはIV形結晶の混合物の使用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021145422A JP7182677B2 (ja) | 2013-10-08 | 2021-09-07 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013211242 | 2013-10-08 | ||
| JP2013211242 | 2013-10-08 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015541603A Division JP6617029B2 (ja) | 2013-10-08 | 2014-10-08 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021145422A Division JP7182677B2 (ja) | 2013-10-08 | 2021-09-07 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020007366A true JP2020007366A (ja) | 2020-01-16 |
| JP6995810B2 JP6995810B2 (ja) | 2022-01-17 |
Family
ID=52813113
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015541603A Active JP6617029B2 (ja) | 2013-10-08 | 2014-10-08 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
| JP2019171587A Active JP6995810B2 (ja) | 2013-10-08 | 2019-09-20 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
| JP2021145422A Active JP7182677B2 (ja) | 2013-10-08 | 2021-09-07 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015541603A Active JP6617029B2 (ja) | 2013-10-08 | 2014-10-08 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021145422A Active JP7182677B2 (ja) | 2013-10-08 | 2021-09-07 | ジアザビシクロオクタン誘導体の結晶とその製造法 |
Country Status (22)
| Country | Link |
|---|---|
| US (5) | US10131665B2 (ja) |
| EP (4) | EP3613740A1 (ja) |
| JP (3) | JP6617029B2 (ja) |
| KR (2) | KR102487297B1 (ja) |
| CN (2) | CN111153903A (ja) |
| AR (4) | AR097971A1 (ja) |
| AU (1) | AU2014332960B2 (ja) |
| BR (2) | BR122022016622B1 (ja) |
| CA (2) | CA2926071C (ja) |
| DK (1) | DK3067355T3 (ja) |
| ES (2) | ES2842225T3 (ja) |
| HK (1) | HK1219479A1 (ja) |
| HU (1) | HUE051925T2 (ja) |
| IL (2) | IL244969B (ja) |
| MX (2) | MX383661B (ja) |
| MY (1) | MY199066A (ja) |
| NZ (5) | NZ719568A (ja) |
| RU (1) | RU2695219C2 (ja) |
| SG (1) | SG11201602723RA (ja) |
| TW (1) | TWI673269B (ja) |
| WO (1) | WO2015053297A1 (ja) |
| ZA (1) | ZA201602054B (ja) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2693898C2 (ru) | 2012-05-30 | 2019-07-05 | Мейдзи Сейка Фарма Ко., Лтд. | НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ |
| DK3050883T3 (da) | 2013-09-24 | 2020-05-25 | Meiji Seika Pharma Co Ltd | Fremgangsmåde til fremstilling af diazabicyclooctanderivater og mellemprodukter |
| EP3613740A1 (en) | 2013-10-08 | 2020-02-26 | Meiji Seika Pharma Co., Ltd. | Preparation of a diazabicyclooctane derivative |
| RU2732129C2 (ru) * | 2014-12-05 | 2020-09-11 | Мейдзи Сейка Фарма Ко., Лтд. | Способ производства кристаллов производного диазабициклооктана и стабильного лиофилизированного препарата |
| US10208041B2 (en) * | 2016-10-07 | 2019-02-19 | Hoffman-La Roche Inc. | Diazabicyclooctane compounds |
| ES2946917T3 (es) | 2017-07-21 | 2023-07-27 | Antabio Sas | Compuestos químicos |
| US10584123B2 (en) | 2017-09-27 | 2020-03-10 | Fedora Pharmaceuticals Inc. | Pharmaceutical forms of diazabicyclooctane derivatives and manufacturing method thereof |
| WO2019064065A1 (en) | 2017-09-27 | 2019-04-04 | F. Hoffmann-La Roche Ag | PHARMACEUTICAL FORMS OF DIAZABICYCLOOCTANE DERIVATIVES AND METHOD FOR THE PRODUCTION THEREOF |
| KR20200091395A (ko) | 2017-09-27 | 2020-07-30 | 페도라 파마슈티칼스 인코포레이티드 | 디아자비시클로옥탄 유도체의 결정형 및 그의 제조 방법 |
| CN107941944B (zh) * | 2017-11-23 | 2020-09-15 | 中山奕安泰医药科技有限公司 | 一种(2s,5r)-苄氧胺基哌啶-2-甲酸乙酯草酸盐的检测方法 |
| CN107991404B (zh) * | 2017-11-23 | 2021-01-08 | 中山奕安泰医药科技有限公司 | 一种(2s,5r)-苄氧胺基哌啶-2-甲酰胺的检测方法 |
| EP3572411A1 (en) | 2018-05-21 | 2019-11-27 | Antabio SAS | Thiazole derivatives as metallo-beta-lactamase inhibitors |
| CN119707784A (zh) * | 2018-09-21 | 2025-03-28 | 株式会社Api | 氨基酸衍生物的制备方法 |
| EP4180105A4 (en) * | 2020-07-13 | 2024-08-28 | NGK Insulators, Ltd. | REFINING PROCESS |
| CN113552250A (zh) * | 2021-06-30 | 2021-10-26 | 海南海灵化学制药有限公司 | 一种盐酸头孢唑兰的杂质检测方法 |
| WO2024263708A2 (en) * | 2023-06-23 | 2024-12-26 | Board Of Trustees Of Michigan State University | Ornithine decarboxylase (odc) inhibitors and uses thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130225554A1 (en) * | 2011-12-02 | 2013-08-29 | Naeja Pharmaceutical Inc. | New bicyclic compounds and their use as antibacterial agents and beta-lactamase inhibitors |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5038509B1 (ja) | 1970-10-21 | 1975-12-10 | ||
| JPS6019759B2 (ja) | 1977-11-24 | 1985-05-17 | イ−ライ・リリ−・アンド・カンパニ− | 非経口投予用11115152139199東京都中央区京橋2丁目3番13号 |
| GB1589317A (en) | 1977-11-24 | 1981-05-13 | Lilly Co Eli | Freeze drying cephalothin sodium |
| JPS60255723A (ja) | 1984-05-31 | 1985-12-17 | Ono Pharmaceut Co Ltd | メシル酸ガベキサートの結晶性凍結乾燥製剤及びその製造方法 |
| JPH07117533B2 (ja) | 1985-08-28 | 1995-12-18 | 株式会社生体科学研究所 | トランスフエリンおよびその用途 |
| US20030220521A1 (en) | 1989-07-27 | 2003-11-27 | G.D. Searle & Co. | Renal-selective prodrugs for control of renal sympathetic nerve activity in the treatment of hypertension |
| TW264475B (ja) | 1991-09-20 | 1995-12-01 | Takeda Pharm Industry Co Ltd | |
| WO1995029913A1 (en) | 1994-05-02 | 1995-11-09 | Shionogi & Co., Ltd. | Crystal of pyrrolidylthiocarbapenem derivative, lyophilized preparation containing said crystal, and process for producing the same |
| JP2843444B2 (ja) | 1994-05-02 | 1999-01-06 | 塩野義製薬株式会社 | ピロリジルチオカルバペネム誘導体の結晶,該結晶を含む凍結乾燥製剤,およびその製造方法 |
| DE19531874C1 (de) | 1995-08-30 | 1996-10-02 | Daimler Benz Ag | Seitenwandbaugruppe für eine Kraftfahrzeugkarosserie |
| FR2812635B1 (fr) | 2000-08-01 | 2002-10-11 | Aventis Pharma Sa | Nouveaux composes heterocycliques, preparation et utilisation comme medicaments notamment comme anti- bacteriens |
| FR2825705B1 (fr) | 2001-06-08 | 2005-05-20 | Aventis Pharma Sa | Nouveaux composes heterocycliques, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens |
| FR2835186B1 (fr) | 2002-01-28 | 2006-10-20 | Aventis Pharma Sa | Nouveaux composes heterocycliques, actifs comme inhibiteurs de beta-lactamases |
| FR2921060B1 (fr) | 2007-09-14 | 2012-06-15 | Novexel | Nouveau procede de preparation d'une piperidine disubsituee et nouveaux intermediaires |
| ES2533826T3 (es) | 2008-01-18 | 2015-04-15 | Merck Sharp & Dohme Corp. | Inhibidores de beta-lactamasa |
| FR2930553B1 (fr) | 2008-04-29 | 2010-05-21 | Novexel | Composes azabicycliques, leur preparation et leur utilisation comme medicaments, notamment inhibiteurs de beta-lactamases |
| FR2936798B1 (fr) | 2008-10-03 | 2012-09-28 | Novexel | Nouveaux composes heterocycliques azotes, leur preparation et leur utilisation comme medicaments antibacteriens. |
| US20120053350A1 (en) | 2009-04-30 | 2012-03-01 | Ian Mangion | Preparation of alkyl esters of n-protected oxo-azacycloalkylcarboxylic acids |
| FR2951171A1 (fr) | 2009-10-09 | 2011-04-15 | Novexel | Nouveau sel de sodium d'un compose azabicyclique sous forme enantiomere cristallisee et nouvelles formes polymorphes et pseudopolymorphes ainsi que leur preparation |
| JP2012122603A (ja) * | 2010-11-15 | 2012-06-28 | Nifco Inc | コネクタ装置 |
| SI2657234T1 (sl) | 2010-12-22 | 2017-06-30 | Meiji Seika Pharma Co., Ltd. | Derivat optično-aktivnega diazabiciklooktana in metoda za njegovo izdelavo |
| US8772490B2 (en) | 2010-12-22 | 2014-07-08 | Meiji Seika Pharma Co., Ltd. | Optically active diazabicyclooctane derivatives and process for preparing the same |
| KR102143660B1 (ko) * | 2011-06-17 | 2020-08-11 | 화이자 안티-인펙티브스 에이비 | 트랜스-7-옥소-6-(술포옥시)-1,6-디아자비시클로[3,2,1]옥탄-2-카르복스아미드 및 그의 염을 포함하는 헤테로시클릭 화합물의 제조 방법 |
| RU2578370C2 (ru) | 2011-08-27 | 2016-03-27 | Вокхардт Лимитед | Производные 1,6- диазабицикло [3,2,1] октан-7-она и их применение при лечении бактериальных инфекционных болезней |
| MX340533B (es) | 2011-08-30 | 2016-07-13 | Wockhardt Ltd | Derivados de 1,6- diazabiciclo [3,2,1] octan-7-ona y su uso en el tratamiento de infecciones bacterianas. |
| EP3052497B1 (en) | 2011-09-13 | 2017-06-28 | Wockhardt Limited | Nitrogen containing compounds and their use |
| US9505761B2 (en) | 2011-12-02 | 2016-11-29 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and beta-lactamase inhibitors |
| RU2693898C2 (ru) | 2012-05-30 | 2019-07-05 | Мейдзи Сейка Фарма Ко., Лтд. | НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ |
| BR112015003592B1 (pt) * | 2012-08-25 | 2020-04-14 | Wockhardt Ltd | derivados de 1,6-diazabiciclo[3,2,1]octan-7-ona e seu uso no tratamento de infecções bacterianas |
| DK3050883T3 (da) | 2013-09-24 | 2020-05-25 | Meiji Seika Pharma Co Ltd | Fremgangsmåde til fremstilling af diazabicyclooctanderivater og mellemprodukter |
| EP3613740A1 (en) | 2013-10-08 | 2020-02-26 | Meiji Seika Pharma Co., Ltd. | Preparation of a diazabicyclooctane derivative |
| IN2014MU01192A (ja) | 2014-03-29 | 2015-10-02 | Wockhardt Ltd | |
| RU2732129C2 (ru) | 2014-12-05 | 2020-09-11 | Мейдзи Сейка Фарма Ко., Лтд. | Способ производства кристаллов производного диазабициклооктана и стабильного лиофилизированного препарата |
-
2014
- 2014-10-08 EP EP19202412.3A patent/EP3613740A1/en not_active Withdrawn
- 2014-10-08 BR BR122022016622-9A patent/BR122022016622B1/pt active IP Right Grant
- 2014-10-08 KR KR1020227012598A patent/KR102487297B1/ko active Active
- 2014-10-08 MX MX2018015407A patent/MX383661B/es unknown
- 2014-10-08 EP EP19202413.1A patent/EP3613741A1/en not_active Withdrawn
- 2014-10-08 BR BR112016006266-3A patent/BR112016006266B1/pt active IP Right Grant
- 2014-10-08 US US15/027,956 patent/US10131665B2/en active Active
- 2014-10-08 CN CN202010091853.6A patent/CN111153903A/zh active Pending
- 2014-10-08 NZ NZ719568A patent/NZ719568A/en unknown
- 2014-10-08 SG SG11201602723RA patent/SG11201602723RA/en unknown
- 2014-10-08 CA CA2926071A patent/CA2926071C/en active Active
- 2014-10-08 KR KR1020167009770A patent/KR102388529B1/ko active Active
- 2014-10-08 NZ NZ743374A patent/NZ743374A/en unknown
- 2014-10-08 MY MYPI2019007794A patent/MY199066A/en unknown
- 2014-10-08 NZ NZ757222A patent/NZ757222A/en unknown
- 2014-10-08 NZ NZ757220A patent/NZ757220A/en not_active IP Right Cessation
- 2014-10-08 HK HK16107473.3A patent/HK1219479A1/zh unknown
- 2014-10-08 AR ARP140103759A patent/AR097971A1/es not_active Application Discontinuation
- 2014-10-08 DK DK14852849.0T patent/DK3067355T3/da active
- 2014-10-08 ES ES14852849T patent/ES2842225T3/es active Active
- 2014-10-08 EP EP14852849.0A patent/EP3067355B1/en active Active
- 2014-10-08 RU RU2016116501A patent/RU2695219C2/ru active
- 2014-10-08 NZ NZ757221A patent/NZ757221A/en unknown
- 2014-10-08 TW TW103134993A patent/TWI673269B/zh active
- 2014-10-08 ES ES17195437T patent/ES2901198T3/es active Active
- 2014-10-08 AU AU2014332960A patent/AU2014332960B2/en not_active Ceased
- 2014-10-08 JP JP2015541603A patent/JP6617029B2/ja active Active
- 2014-10-08 WO PCT/JP2014/076875 patent/WO2015053297A1/ja not_active Ceased
- 2014-10-08 CN CN201480055342.1A patent/CN105612159A/zh active Pending
- 2014-10-08 CA CA3110803A patent/CA3110803C/en active Active
- 2014-10-08 MX MX2016004506A patent/MX361659B/es active IP Right Grant
- 2014-10-08 HU HUE14852849A patent/HUE051925T2/hu unknown
- 2014-10-08 EP EP17195437.3A patent/EP3299370B1/en active Active
-
2016
- 2016-03-29 ZA ZA2016/02054A patent/ZA201602054B/en unknown
- 2016-04-06 IL IL244969A patent/IL244969B/en unknown
-
2018
- 2018-10-01 US US16/148,774 patent/US10604522B2/en active Active
-
2019
- 2019-09-20 JP JP2019171587A patent/JP6995810B2/ja active Active
-
2020
- 2020-02-18 US US16/793,843 patent/US11414417B2/en active Active
-
2021
- 2021-09-07 JP JP2021145422A patent/JP7182677B2/ja active Active
- 2021-12-22 IL IL289275A patent/IL289275B2/en unknown
-
2022
- 2022-07-07 US US17/859,015 patent/US12221443B2/en active Active
-
2024
- 2024-03-01 AR ARP240100517A patent/AR132030A2/es unknown
- 2024-03-01 AR ARP240100515A patent/AR132028A2/es unknown
- 2024-03-01 AR ARP240100516A patent/AR132029A2/es unknown
- 2024-10-22 US US18/922,948 patent/US20250059191A1/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130225554A1 (en) * | 2011-12-02 | 2013-08-29 | Naeja Pharmaceutical Inc. | New bicyclic compounds and their use as antibacterial agents and beta-lactamase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| 平山令明編, 有機化合物結晶作製ハンドブック −原理とノウハウ−, JPN6011053065, 25 July 2008 (2008-07-25), pages 57 - 84, ISSN: 0004524175 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7182677B2 (ja) | ジアザビシクロオクタン誘導体の結晶とその製造法 | |
| ES2758507T3 (es) | Nuevo inhibidor de B-lactamasa y método para producir el mismo | |
| CA2924953C (en) | Process for producing diazabicyclooctane derivative and intermediate thereof | |
| RU2838718C2 (ru) | Кристаллические формы производного диазабициклооктана и способ их получения | |
| RU2840014C2 (ru) | Кристаллические формы производного диазабициклооктана и способ их получения | |
| RU2847390C2 (ru) | Кристаллические формы производного диазабициклооктана и способ их получения | |
| RU2801220C2 (ru) | Кристаллические формы производного диазабициклооктана и способ их получения | |
| HK40023145A (en) | Preparation of a diazabicyclooctane derivative | |
| HK40023146A (en) | Preparation of a diazabicyclooctane derivative | |
| HK1226731A1 (en) | Crystals of diazabicyclooctane derivative and production method for crystals of diazabicyclooctane derivative | |
| HK40027891A (zh) | 二氮杂二环辛烷衍生物的结晶及其制备方法和应用以及医药组合物 | |
| HK1226731B (en) | Crystals of diazabicyclooctane derivative and production method for crystals of diazabicyclooctane derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20191018 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191018 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200923 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200917 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20201120 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210119 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20210615 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210907 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20210907 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20210917 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20210921 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20211207 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20211215 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6995810 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |