JP3848663B2 - 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 - Google Patents
非a非b非c非d非e型肝炎試薬及びそれらの使用方法 Download PDFInfo
- Publication number
- JP3848663B2 JP3848663B2 JP2004176507A JP2004176507A JP3848663B2 JP 3848663 B2 JP3848663 B2 JP 3848663B2 JP 2004176507 A JP2004176507 A JP 2004176507A JP 2004176507 A JP2004176507 A JP 2004176507A JP 3848663 B2 JP3848663 B2 JP 3848663B2
- Authority
- JP
- Japan
- Prior art keywords
- hgbv
- seq
- antibody
- sequence
- clone
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images














































Landscapes
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Saccharide Compounds (AREA)
Description
本発明は、ヒトにおいて肝炎を惹起する一群の感染性ウイルス性物質、特にかかるウイルス群から誘導されるポリヌクレオチド、それらにコードされるポリペプチド、かかるポリペプチドに特異的に結合する抗体、並びに、かかる材料を使用する診断薬及びワクチンに係わる。
肝炎は、血液製剤の輸血、臓器移植及び血液透析によってドナーからレシピエントに感染される最も重要な疾患の1つである。また肝炎は、汚染された食物及び水の摂取や、人同士の接触によっても感染し得る。ウイルス性肝炎は固有のウイルス遺伝子及び複製モードを有する一群のウイルス性物質を含み、種々の感染経路で肝損傷の程度は様々であるが、肝炎を惹起することが知られている。急性ウイルス性肝炎は、黄疸、肝圧痛及び、アスパラギン酸トランスアミナーゼ(AST)、アラニントランスアミナーゼ(ALT)及びイソクエン酸デヒドロゲナーゼ(ISD)のごとき肝トランスアミナーゼレベルの上昇を含む特定の患者症状によって臨床診断される場合もあるが、臨床的に顕在化しないこともあり得る。ウイルス性肝炎物質としては、肝炎A型ウイルス(HAV)、肝炎B型ウイルス(HBV)、肝炎C型ウイルス(HCV)、肝炎δ型ウイルス(HDV)、肝炎E型ウイルス(HEV)、エプスタイン−バールウイルス(EBV)、及びサイトメガロウイルス(CMV)が挙げられる。
1960年代後期に使用可能となった、献血血液をHBV表面抗原(HBsAg)の存在についてスクリーニングする特異的血清アッセイは、血液レシピエントの輸血後肝炎(PTH)の発生を低減させることにおいて良い結果を与えてはいるが、PTHは相変わらず有意な率で発生している。H.J.Alterら,Ann.Int.Med.77:691−699(1972);H.J.Alterら,Lancet ii:838−841(1975)。研究者らは、これまでヒトにおいて肝炎を惹起することが知られているウイルス(HAV、HBV、CMV及びEBV)への暴露と関係なくウイルス性肝炎を惹起する、「非A非B型肝炎」(NANBH)と名付けられた新規の物質を探し始めた。例えばS.M.Feinstoneら,New Engl.J.Med.292:767−770(1975);編者無記名,Lancet ii:64−65(1975);B.N.FieldsのF.B.Hollinger及びD.M.Knipeら,Virology,Raven Press,New York,pp2239−2273(1990)参照。
静脈内薬物使用者における急性NANBHの多発性発症;輸血後NANBHを獲得した患者の固有の感染後潜伏期間;チンパンジー交差攻撃(cross−challenge)実験の結果;感染チンパンジーの超微細構造肝病理分析;及び疑似患者のクロロホルムに対する抵抗性の差など、幾つかの疫学的及び実験的な証拠により1種以上の非経口感染NANB物質の存在が示されている。J.L.Dienstag,Gastroenterology 85:439−462(1983);J.L.Dienstag,Gastroenterology 85,743−768(1983);F.B.Hollingerら,J.Infect.Dis.142:400−407(1980);F.ChisariのD.W.Bradley編,Advances in Hepatitis Research,Masson,New York,pp.268−280(1984);及び、D.W.Bradleyら,J.Infect.Dis.148:254−265(1983)。
急性肝炎を発症した外科医から得た血清試料は、タマリン(Saguinus species)に接種すると肝炎を誘発することが示された。4匹のタマリンのうち4匹ともが接種後数週間以内に肝酵素の上昇を生じ、これから、該外科医の血清中の物質がタマリンに肝炎を誘発した可能性があることが判る。種々の非ヒト霊長目動物において順次継代されることにより、この肝炎は感染性物質によって惹起されることが判り、濾過実験からはこの物質がウイルス性であることが示された。上記外科医及びタマリンにおいて肝炎の原因となった感染性物質は「GB物質(GB agent)」と名付けられた。F.Deinhardtら,J.Exper.Med.125:673−688(1967);F.Dienhardtら,J.Exper.Med.,前出;E.Taborら,J.Med.Virol.5:103−108(1980);R.O.Whittingtonら,Viral and Immunological Diseases in Nonhuman Primates,Alan R.Liss,Inc.,New York,pp.221−224(1983)。
GB物質はヒトにおいてNANBHを惹起する物質であり得ること、及びGB物質は種々の実験室で研究された既知のNANBH物質とは関連のないことが示されているが、GB物質に関する決定的な、また最終的な研究は知られておらず、ウイルス性物質は発見されていないし、分子学的に特性分析されてもいない。F.Deinhardtら,Am.J.Med.Sci.270:73−80(1975);及びJ.L.Dienstagら,Nature 264:260−261(1976)。更にE.Taborら,J.Med.Virol.,前出;E.Taborら,J.Infect.Dis.140:794−797(1979);R.O.Whittingtonら,前出;及びP.Karayiannisら,Hepatology 9:186−192(1989)参照。
初期の研究では、GB物質はいかなる既知のヒト肝炎ウイルスとも無関係であるとされていた。S.M.Feinstoneら,Science 182:1026−1028(1973);P.J.Provostら,Proc.Soc.Exp.Biol.Med.148;532−539(1975);J.L.Melnick,Intervirology 18:105−106(1982);A.W.Holmesら,Nature 243:419−420(1973);及び、F.Deinhardtら,Am.J.Med.Sci.,前出。しかしながら、GB物質がヒトにおいて肝炎感染を誘発するウイルスであるのか、またはGB血清によって活性化され、一旦活性化されると他のタマリンに容易に継代してそれらに肝炎を誘発する潜在的タマリンウイルスであるのかが問題とされた。また、GB陽性血清を接種されたマーモセットの極一部は臨床上肝炎を発症せず(52匹のうち4匹,7.6%)、かかる動物は自然免疫を有していた可能性があり、従ってGB物質はマーモセットウイルスであり得ることも示された。W.P.Parksら,J.Infect.Dis.120:539−547(1969);W.P.Parksら,J.Infect.Dis.120:548−559(1969)。形態的研究は不明瞭であり、ある報告書の免疫電子顕微鏡調査では、GB物質は、パルボウイルスの球構造と類似の、寸法分布が20〜22nmの免疫複合体を形成していると示されているが、他の研究では、GB陽性タマリンの肝ホモジネートから得られた免疫電子顕微鏡データから正二十面体対称構造の34〜36nmの凝集体(aggregares)が検出され、GB物質はカリチ様ウイルスであると報告されている。例えばJ.D.Almeidaら,Nature 261:608−609(1976);J.L.Dienstagら,Nature,前出参照。
主に非経口感染によって生じるHCVと、腸内感染するHEVとの2種類の肝炎を惹起するウイルスが最近になって発見され報告された。例えば、Q.L.Chooら,Science 244:359−362(1989),G.Kuoら,Science 244:362−364(1989),欧州特許出願公開第0318216号明細書(1989年3月31日公開),G.R.Reyesら,Science 247:1335−1339(1990)参照。HCVは、NANBH物質が原因とみられるPTHの大部分及び輸血によるものでない急性NANBHの多くに関与している。編者無記名,Lancet 335:1431−1432(1990);J.L.Dienstag,Gastroenterology 99:1177−1180(1990);及び、M.J.Alterら,JAMA 264:2231−2235(1990)。
ドナー試料中のHCV抗体を検出することにより、血液供給系におけるNANBH感染血液の70〜80%が除外されるが、HCVの発見及び検出によって肝炎感染が完全に予防されているわけではない。H.Alterら,New Eng.J.Med.321:1494−1500(1989)。最近の文献で、別の肝炎物質がPTH、並びにPTHと関連のない集団獲得急性及び/または慢性肝炎の原因であり得るかどうかが問題とされた。例えば、1988〜1990年にフランスで行われた予測臨床標本調査においてモニターとなった181人の患者のうち、調査人は全部で18のPTH症例を記載している。これら18人の患者のうちの13人は、抗HCV抗体、HBsAg、HBV及びHCV核酸について陰性であった。筆者らにはPTHを惹起する非A非B非C型物質の潜在的な重要性が推量された。V.Thiersら,J.Hepatology 18:34−39(1993)。1985〜1988年にドイツで行われた別の調査でモニターとなった1,476人の患者のうち、記録された22のPTH症例はHBVまたはHCV感染とは関係なかった。T.Petersら,J.Med.Virol.39:139−145(1993)。
肝炎を惹起する新規の固有のウイルス群から誘導される物質、例えばポリヌクレオチド、そこにコードされている組換え及び合成ポリペプチド、かかるポリペプチドに特異的に結合する抗体、並びにかかる物質を使用する診断薬及びワクチンを同定及び提供することは有利となろう。このような物質により、急性及び/または慢性ウイルス性肝炎をより正確に診断する医療機関の能力が著しく向上し、提供された血液及び臓器において非A、非B及び非C型肝炎を検出することにより、より安全な血液及び臓器を供給し得るであろう。
(発明の要約)
本発明は、肝炎GBウイルス(HGBV)のゲノムまたはその相補体に選択的にハイブリダイズし得るHGBVから誘導された精製ポリヌクレオチドまたはそのフラグメントであって、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率(identity)、より好ましくは40%の一致率、更に好ましくは60%の一致率、最も好ましくは80%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とする精製ポリヌクレオチドまたはそのフラグメントを提供する。更に、肝炎GBウイルス(HGBV)のゲノムまたはその相補体に選択的にハイブリダイズし得るHGBVから誘導される組換えポリヌクレオチドまたはそのフラグメントも提供されるが、ここで、前記ヌクレオチドはHGBVの少なくとも1つのエピトープをコードする配列を含んでおり、前記組換えヌクレオチドは、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とする。かかる組換えポリヌクレオチドは組換えベクター内に含まれ、更に前記ベクターを用いて形質転換された宿主細胞を構成する。
本発明は、肝炎GBウイルス(HGBV)のゲノムまたはその相補体に選択的にハイブリダイズし得るHGBVから誘導された精製ポリヌクレオチドまたはそのフラグメントであって、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率(identity)、より好ましくは40%の一致率、更に好ましくは60%の一致率、最も好ましくは80%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とする精製ポリヌクレオチドまたはそのフラグメントを提供する。更に、肝炎GBウイルス(HGBV)のゲノムまたはその相補体に選択的にハイブリダイズし得るHGBVから誘導される組換えポリヌクレオチドまたはそのフラグメントも提供されるが、ここで、前記ヌクレオチドはHGBVの少なくとも1つのエピトープをコードする配列を含んでおり、前記組換えヌクレオチドは、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とする。かかる組換えポリヌクレオチドは組換えベクター内に含まれ、更に前記ベクターを用いて形質転換された宿主細胞を構成する。
本発明は更に、肝炎GBウイルス(HGBV)ゲノムから誘導されるヌクレオチド配列を含むHGBV組換えポリヌクレオチドまたはそのフラグメントであって、組換えベクター内に含まれ、更に前記ベクターを用いて形質転換された宿主細胞を構成し、前記配列がHGBVのエピトープをコードするHGBV組換えポリヌクレオチドまたはそのフラグメントを提供する。HGBV組換えポリヌクレオチドは、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とする。本発明は、肝炎GBウイルス(HGBV)から誘導されるDNAまたはRNAの読取り枠を含む組換え発現系であって、前記読取り枠がHGBVゲノムまたはcDNAの配列を含んでおり、且つ前記読取り枠が所望の宿主と適合性のある制御配列に操作可能に結合されている組換え発現系を提供し、更に、前記組換え発現系を用いて形質転換された細胞及び、前記細胞によって産生される長さが少なくとも約8個のアミノ酸のポリペプチドを提供する。
本発明は更に、肝炎GBウイルス(HGBV)ポリペプチドまたはそのフラグメントの調製物、即ち、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率、より好ましくは40%の一致率、更に好ましくは60%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とするアミノ酸配列またはそのフラグメントを含む組換えポリペプチドを含む精製HGBVを提供する。ポリクローナル抗体及びモノクローナル抗体、並びに、少なくとも1種の肝炎GBウイルス(HGBV)ポリペプチドまたはそのフラグメントを含む融合ポリペプチド、真核性または原核性宿主内で産生されたときに粒子を形成し得るアミノ酸配列を有する非HGBVポリペプチドと、少なくとも1つのHGBVエピトープとを含む、肝炎GB(HGBV)感染に対して免疫原性である粒子、及び、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とする、肝炎GBウイルス(HGBV)に対するポリヌクレオチドプローブが提供される。
少なくとも1つの肝炎GBウイルス(HGBV)エピトープを含むポリペプチドを製造するためのアッセイキット及び方法であって、HGBV−A、HGBV−B及びHGBV−Cからなる群から選択されるアミノ酸配列と少なくとも35%の一致率を有するアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムを特徴とするポリペプチドをコードする配列を含む発現ベクターを用いて形質転換された宿主細胞をインキュベートすることからなるアッセイキット及び方法も提供される。更には、固相、組換えもしくは合成ペプチド、またはプローブを使用する方法を含む、検査試料中のHGBV核酸、抗原及び抗体を検出する方法も提供される。更に、ワクチン、肝炎GBウイルス(HGBV)に感染させた組織培養増殖細胞、免疫応答を生じるのに十分な量の少なくとも1つのHGBVエピトープを含む単離免疫原性ポリペプチドまたはそのフラグメントを個体に投与することからなる、肝炎GBウイルス(HGBV)に対する抗体を製造する方法も提供される。更に、ポリヌクレオチドもしくはポリペプチドまたはこれらのフラグメントを含む診断試薬も提供される。
(発明の詳細)
本発明は、新たに確認された病因物質である非A、非B、非C、非D、及び非E型肝炎を惹起する物質、総称して「肝炎GBウイルス」または「HGBV」の特性分析法を提供する。本発明は、HGBV病因物質の存在を判定する方法、HGBVに感染したヒトまたはタマリンの個体由来の感染血清、血漿または肝ホモジネートから生成された病因物質の核酸を得、これまでに単離されていないウイルス性物質のゲノムから新たに合成される抗原を検出し、非感染個体と比較して感染個体のみに認められる生成物を産生したクローンを選択する方法を提供する。
本発明は、新たに確認された病因物質である非A、非B、非C、非D、及び非E型肝炎を惹起する物質、総称して「肝炎GBウイルス」または「HGBV」の特性分析法を提供する。本発明は、HGBV病因物質の存在を判定する方法、HGBVに感染したヒトまたはタマリンの個体由来の感染血清、血漿または肝ホモジネートから生成された病因物質の核酸を得、これまでに単離されていないウイルス性物質のゲノムから新たに合成される抗原を検出し、非感染個体と比較して感染個体のみに認められる生成物を産生したクローンを選択する方法を提供する。
HGBVから誘導される核酸配列の部分は、検査試料中のHGBVの存在を判定するため、及び天然変種を単離するためのプローブとして有用である。かかる配列により、HGBVゲノム内にコードされているHGBV抗原のポリペプチド配列を得ることもできるし、診断試験の標準または試薬として及び/またはワクチンの成分として有用なポリペプチドを製造することもできる。かかるポリペプチド配列内に含まれる少なくとも1つのエピトープに対するモノクローナル抗体及びポリクローナル抗体もまた、診断試験及び治療薬に、抗ウイルス性物質のスクリーニングに、かかる核酸配列が誘導されるHGBV物質の単離に有用である。HGBVゲノムのその他の部分の単離及び配列決定も、かかる核酸配列から誘導されるプローブまたはPCRプライマーを使用することにより行うことができ、従って、HGBVの診断及び/または治療において予防薬及び治療薬として有用となろうHGBVの別のプローブ及びポリペプチドを製造することができる。
本発明の1つの態様によれば、精製HGBVポリヌクレオチド、組換えHGBVポリヌクレオチド、HGBVゲノムから誘導された配列を含む組換えポリヌクレオチド、HGBVのエピトープをコードする組換えポリペプチド、HGBVのエピトープをコードする合成ペプチド、上記組換えポリペプチドのいずれかを含む組換えベクター、及び、かかるベクターを用いて形質転換された宿主細胞が提供される。これらの組換えポリペプチド及び合成ペプチドは単独でも組合せても使用することができるし、或いは、HGBVのエピトープを与える他の物質と一緒に使用することもできる。
本発明の別の態様においては、精製HGBV、精製HGBV由来のポリペプチドの調製物、精製HGBVポリペプチド、HGBVに含まれるエピトープと免疫学的に同一であるエピトープを含む精製ポリペプチドが提供される。
本発明の更に別の態様においては、所望の宿主と適合性である制御配列に操作可能に連結されている、HGBVゲノムまたはHGBV cDNAから誘導されたDNAの読取り枠(ORF)を含む組換え発現系、該組換え発現系を用いて形質転換された細胞、及び、該形質転換細胞によって産生されたポリペプチドが提供される。
本発明の別の態様は、少なくとも1つの組換えHGBVポリペプチド、HGBVゲノムまたはHGBV cDNAから誘導された配列を含む少なくとも1つの組換えポリペプチド、HGBVエピトープを含む少なくとも1つの組換えポリペプチド、HGBVポリペプチドを含む少なくとも1つの融合ポリペプチドを含む。
更に本発明は、HGBVの少なくとも1つのエピトープに特異的に結合するモノクローナル抗体を製造する方法、少なくとも1つのHGBVエピトープに特異的に結合するポリクローナル抗体の精製調製物、並びに、診断、予防及び治療用途を含む、かかる抗体を使用する方法を提供する。
本発明の更に別の態様においては、真核性宿主中で産生されたときに粒子を形成し得るアミノ酸配列を有する非HGBVポリペプチドと、HGBVエピトープとを含む、HGBV感染に対して免疫原性の粒子が提供される。
更に、HGBVに対するポリヌクレオチドプローブも提供される。
本発明は、適当な容器に入った、HGBVから誘導されたポリヌクレオチドの存在及び/または量を検出するために使用し得る試薬であって、約8個またはそれ以上のヌクレオチドからなるHGBV由来のヌクレオチド配列を含むポリヌクレオチドプローブを含む試薬;適当な容器に入った、HGBV抗原の存在及び/または量を検出するための試薬であって、検出すべきHGBV抗原に対する抗体を含む試薬;及び、適当な容器に入った、HGBV抗原に対する抗体の存在及び/または量を検出するための試薬であって、HGBV抗原中に存在するHGBVエピトープを含むポリペプチドを含む試薬を含むキットを提供する。種々のアッセイ形式のための他のキットも本明細書に記載の本発明によって提供される。
本発明の他の態様として、固相に付着された少なくとも1種のHGBVエピトープを含むポリペプチドと、固相に付着されたHGBVエピトープに対する抗体とが挙げられる。更に、HGBVエピトープを含むポリペプチドをコードする配列を含む発現ベクターを用いて形質転換された宿主細胞を、該ポリペプチドの発現が可能となる条件下でインキュベートすることからなる、HGBVエピトープを含むポリペプチドを製造する方法と、該方法によって製造されたHGBVエピトープを含むポリペプチドも本発明に含まれる。
本発明は更に、本発明の組換えまたは合成ポリペプチド及び本発明の抗体を、いずれもシグナル生成化合物を使用し得る種々の形式で使用するアッセイを提供する。検出手段を与えるためのシグナル生成化合物を使用しないアッセイも提供される。記載の全てのアッセイは、通常は抗原もしくは抗体のいずれかまたは両方を検出するものであり、検査試料を少なくとも1種の本発明の試薬と接触させて少なくとも1種の抗原/抗体複合体を形成させ、該複合体の存在を検出することを含む。かかるアッセイについては詳述する。
HGBVエピトープを含む免疫原性ペプチド、もしくはHGBVの不活化調製物、もしくはHGBVの弱毒化調製物を含むHGBV感染治療用ワクチン、または、HGBVエピトープを発現する組換えワクチンの使用、並びに/または合成ペプチドの使用も本発明に含まれる。有効なワクチンにはこれらの免疫原性ペプチドの組合せが使用され得る(例えば、組換え抗原、合成ペプチド及び天然ウイルス性抗原のカクテルが同時にまたは異なる時点で投与される)。これらのなかには、単独で使用し得るものもあるし、後で免疫原性エピトープを別に与えて補い得るものもある。更に本発明には、HGBVに対する抗体を産生させる方法であって、個体に、接種個体において免疫応答を生起するのに十分な量のHGBVエピトープを含む単離免疫原性ポリペプチドを投与することからなる方法も含まれる。
更に、HGBVに感染した組織培養増殖細胞も本発明によって提供される。
本発明の更に別の態様においては、同定されていない感染性物質のゲノムから誘導されたDNAまたはcDNAを単離する方法が提供されるが、該方法は代表相違分析(representational difference analysis,RDA)の他に類のない改良方法であって、これについては後述する。
(定義)
本明細書において使用される「肝炎GBウイルス」または「HGBV」なる用語は、ヒトにおいて非A、非B、非C、非D、非E型肝炎を惹起するウイルス種、及びそれらから誘導される弱毒化株または欠陥干渉粒子を総称的に示す。これには更に、汚染された食物、飲料水などによって感染された急性ウイルス性肝炎;(性行為、呼吸及び非経口経路伝染を含む)人同士の接触または静脈内薬物使用によって感染されるHGBVに起因する肝炎も含まれる。本発明方法により、HGBVを獲得した個体の同定が可能となる。個々には、HGBV単離体を特に「HGBV−A」、「HGBV−B」及び「HGBV−C」と表記する。本明細書においてHGBVゲノムはRNAからなる。HGBVのヌクレオチド配列及び推定アミノ酸配列の分析から、このウイルス群はFlaviridaeファミリーと類似のゲノム構成を有することが明らかである。絶対的ではないが、基本的には、ゲノム構成の類似性に基づき、the International Committee on the Taxonomy of Virusesは、このファミリーがFlavivirus、Pestivirus及び肝炎C型グループの3つの属からなると勧告している。アミノ酸レベルでの類似性を探すと、肝炎GBウイルスサブクローンの配列は、少しではあるが一部が肝炎C型ウイルスのものと類似であることが判る。ここで与えられる情報は、HGBVの他の株を分類するのに十分である。
本明細書において使用される「肝炎GBウイルス」または「HGBV」なる用語は、ヒトにおいて非A、非B、非C、非D、非E型肝炎を惹起するウイルス種、及びそれらから誘導される弱毒化株または欠陥干渉粒子を総称的に示す。これには更に、汚染された食物、飲料水などによって感染された急性ウイルス性肝炎;(性行為、呼吸及び非経口経路伝染を含む)人同士の接触または静脈内薬物使用によって感染されるHGBVに起因する肝炎も含まれる。本発明方法により、HGBVを獲得した個体の同定が可能となる。個々には、HGBV単離体を特に「HGBV−A」、「HGBV−B」及び「HGBV−C」と表記する。本明細書においてHGBVゲノムはRNAからなる。HGBVのヌクレオチド配列及び推定アミノ酸配列の分析から、このウイルス群はFlaviridaeファミリーと類似のゲノム構成を有することが明らかである。絶対的ではないが、基本的には、ゲノム構成の類似性に基づき、the International Committee on the Taxonomy of Virusesは、このファミリーがFlavivirus、Pestivirus及び肝炎C型グループの3つの属からなると勧告している。アミノ酸レベルでの類似性を探すと、肝炎GBウイルスサブクローンの配列は、少しではあるが一部が肝炎C型ウイルスのものと類似であることが判る。ここで与えられる情報は、HGBVの他の株を分類するのに十分である。
HGBV−CはHCVの一遺伝子型ではないことを示す幾つかの証拠が挙がっている。第1に、HGB−C配列を含む血清においてHCV抗体の存在を試験した。HCVに暴露されたかまたは感染した個体の常套検出方法は、HCV−1由来の3つ以上の領域から誘導される抗原を使用する抗体試験に依存する。かかる試験により、HCVの既知の遺伝子型に対する抗体を検出し得る(例えばSakamotoら,J.Gen.Virol.75:1761−1768(1994)及びStuyverら,J.Gen.Virol.74:1093−1102(1993)参照)。HCV特異的ELISAでは8つのうち6つのケースでGB−C配列を含む血清を検出できなかった。第2に、HCV抗体に対して血清反応陰性の幾つかのヒト血清は、高感度RT−PCRアッセイによるとHCVゲノムRNAに対して陽性であることが判った(Sugitani,Lancet 339:1018−1019(1992))。このアッセイでは、HGB−C配列を含む8つの血清のうち7つでHCV RNAを検出できなかった(表A)。即ち、血清学的アッセイ及び分子学的アッセイのいずれによってもHGBV−CはHCVの一遺伝子型ではない。
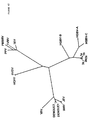
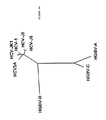
ヘリカーゼ領域内のHGB−Cの推定翻訳産物の一部をHGBV−A、HGBV−B、HCV−1、及びFlaviviridaeの別のメンバーの相同領域と並べ、一致した配列を系統発生学的に分析すると、HGBV−CはHCVグループの他のメンバーよりHGBV−Aに密接に関係することが判る。進化距離(evolutionary distance)0.42を示すHGBV−CとHGBV−Aの配列は、進化距離0.92を示すHGBV−CとHGBV−Bほどは離れていない(表33、後記)。即ち、HGBV−AとHGBV−CはGBウイルスの1つのサブグループのメンバーであり、HGBV−Bはそれ自体で1つのサブグループのメンバーであると考えられる。種々のHCV単離体由来のヘリカーゼ配列を系統発生学的に分析すると、それらは、最大進化距離が0.20である分岐の小さいグループを形成していることが判る(表32、後記)。HCVグループとHGBVグループとを比較すると、各グル−プからの任意の2つの配列の最小進化距離は0.69であることが判る。上記の距離の値を使用し、図42に表す系統樹を作成した。これらのウイルスの分岐度が比較的高いことから、GBウイルスは単に肝炎C型グループ内のタイプまたはサブタイプをなすのではなく、独自の系統発生学的グループを構成することが判る。HCVウイルスゲノムの小部分から誘導される配列情報を使用した系統発生学的分析は、新規の単離体を遺伝子型グループに割当てる容認可能な方法であることが示されている(Simmondsら,Hepatology 19:1321−1324(1994))。最近の分析では、代表的なHCV単離体由来のヘリカーゼ遺伝子内の110個のアミノ酸からなる配列を使用すれば、それらは、それぞれの遺伝子型に適当に分類される(Simmondsら,J.Gen.Virol.75:1053−1061(1994))。従って、示された進化距離は、多分、GBウイルスと肝炎C型ウイルスとの高度の分岐を正しく反映している。
先行特許出願において、「HGBV株はポリペプチドレベルで同定可能であり、HGBV株はポリペプチドレベルで40%以上が相同、好ましくは約60%以上が相同、更に好ましくは約80%以上が相同である」と述べられていた。「相同」なる用語は、使用されてはいるが、2つのポリヌクレオチドまたはポリペプチド配列の関連の度合いを示す場合に曖昧であり、実際には進化上の関連性を示唆するものである。当分野の最近の慣例によれば、「相同性」なる用語はもはや使用されず、代わりに、「類似性(similarity)」及び/または「一致性(identity)」なる用語を使用して2つのポリヌクレオチドまたはポリペプチド配列の関連の度合いを示している。アミノ酸配列の「類似性」及び/または「一致性」を決定する方法は当分野において公知であるが、例えば、アミノ酸配列を直接決定し、それを本明細書に与えられた配列と比較したり、推定HGBVのゲノム材料のヌクレオチド配列を(通常はcDNA中間体を介して)決定し、そこにコードされているアミノ酸配列を決定し、対応領域を比較するなどが挙げられる。通常、「一致性」とは、各ゲノム上の適当な場所においてHGBVのヌクレオチド配列と別の株のヌクレオチド配列、またはHGBVのアミノ酸配列と別の株のアミノ酸配列が正確に一致することを意味する。通常、「類似性」とは、適当な場所においてHGBVのアミノ酸配列と別の株のアミノ酸配列が正確に一致することを意味するが、その場合、アミノ酸は同一または類似の化学的及び/または物理的特性、例えば電荷または疎水性を有する。(the Genetics Computer Group,Madison,Wisconsin,53711から入手可能な)Wisconsin Sequence Analysis Package,バージョン8において使用可能なプログラム、例えばGAPプログラムにより、2つのポリヌクレオチドまたは2つのポリペプチドの配列間の一致率及び類似率を計算し得る。2つの配列間の一致率及び類似率を計算する他のプログラムも当分野において公知である。
更に、HGBV−A、HGBV−BまたはHGBV−Cの株を同定することにおいて、以下のパラメーターを単独または組合せて使用し得る。HGBV株は好ましくは約60%以上、より好ましくは約80%以上の遺伝的関連性があり得ると考えられることから、HGBV−A、HGBV−BまたはHGBV−Cとこれらの肝炎GBウイルスの1つの株との間のヌクレオチド配列全体の一致率は約45%以上であると推定される。
更に、HGBV株は好ましくは約40%以上、より好ましくは約60%以上、更に好ましくは約80%以上の遺伝的関連性があり得ると考えられることから、HGBV−AのゲノムとHGBV−Aのある株のゲノムとのアミノ酸レベルでの配列全体の一致率は約35%以上であろうと推定される。更に、2つ以上の連続配列の組合せで与えられているであろう、少なくとも約13ヌクレオチドからなる対応連続配列がある。また、HGBV株は好ましくは約40%以上、より好ましくは約60%以上、更に好ましくは約80%以上の遺伝的関連性があり得ると考えられることから、HGBV−BのゲノムとHGBV−Bのある株のゲノムとのアミノ酸レベルでの配列全体の一致率は約35%以上であると推定される。更に、2つ以上の連続配列の組合せで与えられているであろう、少なくとも13ヌクレオチドからなる対応連続配列がある。また、HGBV株は好ましくは約40%以上、より好ましくは約60%以上、更に好ましくは約80%以上の遺伝的関連性があり得ると考えられることから、HGBV−CのゲノムとHGBV−Cのある株のゲノムとのアミノ酸レベルでの配列全体の一致率は約35%以上であろうと推定される。更に、2つ以上の連続配列の組合せで与えられているであろう、少なくとも約13ヌクレオチドからなる対応連続配列がある。
本発明の組成物及び方法により、HGBV及びその存在し得る株の増殖、同定、検出及び単離が可能となる。更に本発明の組成物及び方法により、HGBVの存在し得る種々の株に対する診断薬及びワクチンの製造が可能となり、抗ウイルス性物質のスクリーニング処理に有用となる。情報は、ウイルス分類学者が該種に属する他の株を同定するのに十分となろう。本発明者らは、HGBVが本明細書に含まれる配列をコードすると考えている。かかる配列の存在をアッセイする方法は当分野において公知であり、例えばリガーゼ連鎖反応(LCR)、ポリメラーゼ連鎖反応(PCR)及びハイブリダイゼーションといった増幅方法が挙げられる。更に、かかる配列は、免疫原性ウイルスエピトープを見い出し得る読取り枠を含む。このエピトープは、他の既知の肝炎誘発ウイルスと比較し、HGBVに固有である。エピトープの固有性は、HGBVに対する免疫学的反応性と、肝炎A、B、C、D及びE型ウイルスに対する免疫学的反応性の欠如とによって判定し得る。免疫学的反応性を判定する方法は当分野において公知であり、例えばラジオイムノアッセイ(RIA)、酵素結合免疫吸着アッセイ(ELISA)、血球凝集反応(HA)、蛍光偏光イムノアッセイ(FPIA)が挙げられるが、適当な方法の幾つかの例を本明細書に記載する。
特定の配列、例えばHGBV cDNAまたはHGBVゲノム「から誘導された」ポリヌクレオチドとは、特定のヌクレオチド配列の領域に対応する、即ち類似であるかまたは相補的である、おおよそ少なくとも約6個のヌクレオチド、好ましくは少なくとも約8個のヌクレオチド、より好ましくは少なくとも約10〜12個のヌクレオチド、更に好ましくは少なくとも約15〜20個のヌクレオチドの配列からなるポリヌクレオチド配列を指す。ポリヌクレオチドを誘導する領域の配列は、HGBVゲノムに固有の配列に類似または相補的であるのが好ましい。配列がHGBVゲノムに固有の配列に相補的または類似であるか否かは、当業者には公知の方法で判断し得る。特定の配列の固有性を決定する方法として、例えばデータバンクにある配列との比較を行い得る。配列を誘導し得る領域としては、限定的ではないが、特異的エプトープをコードする領域、並びに非翻訳及び/または非転写領域が挙げられる。
誘導ポリヌクレオチドは必ずしもHGBVのヌクレオチド配列から物理的に誘導される必要はなく、限定的ではないが、ポリヌクレオチドを誘導する領域の塩基配列により与えられる情報に基づいた化学的合成、複製、または逆転写もしくは転写を含む任意の方法で生成し得る。更に、特定の配列に対応する領域の組合せは、当分野において公知の方法で所期の用途に応じて変更し得る。
特定の核酸配列またはHGBVゲノムから誘導される「ポリペプチド」または「アミノ酸配列」とは、配列内にコードされているポリペプチドと同一のアミノ酸配列を有するポリペプチド、または、少なくとも3〜5個のアミノ酸、より好ましくは少なくとも8〜10個のアミノ酸、更に好ましくは15〜20個のアミノ酸からなり、配列内にコードされているポリペプチドと免疫学的に一致し得る前記ポリペプチドの一部を指す。
本明細書において使用される「組換えポリペプチド」とは少なくとも、起源または操作によって、天然もしくはライブラリーの形態で関連するポリペプチドの全部または一部と関連せず、及び/またはそれが天然で結合しているもの以外のポリヌクレオチドに結合している、ゲノム、半合成または合成のポリペプチドを意味する。組換えまたは誘導ポリペプチドは必ずしもHGBVの特定の核酸配列またはHGBVゲノムから翻訳される必要はない。組換えまたは誘導ポリペプチドは、化学的合成もしくは組換え発現系の発現、または突然変異HGBVからの単離を含む任意の方法で生成することもできる。
本明細書において使用される「合成ペプチド」なる用語は、当業者には公知の方法で化学的に合成し得る、任意の長さの重合形態のアミノ酸を意味する。かかる合成ペプチドは種々の用途に有用である。
本明細書において使用される「ポリヌクレオチド」なる用語は、リボヌクレオチドまたはデオキシリボヌクレオチドいずれかの任意の長さの重合形態のヌクレオチドを意味する。この用語は分子の一次構造のみを指す。従って該用語には、二本鎖及び一本鎖DNA並びに二本鎖及び一本鎖RNAが含まれる。更に、メチル化及び/またはキャップ付加による修飾形態のポリヌクレオチド、並びに未修飾形態のポリヌクレオチドも含まれる。
「cDNAに対応する配列を含むHGBV」とは、HGBVが、特定のDNA内の配列と類似または相補的なポリヌクレオチド配列を含むことを意味する。該cDNAに対する類似または相補の程度は約50%以上、好ましくは少なくとも約70%、より好ましくは少なくとも約90%である。対応する配列の長さは少なくとも約70ヌクレオチド、好ましくは少なくとも約80ヌクレオチド、更に好ましくは少なくとも約90ヌクレオチドである。HGBVとcDNAの対応は当分野において公知の方法によって決定し得るが、例えば、配列決定した物質を記載のcDNAと直接比較したり、ハイブリダイズし一本鎖ヌクレアーゼにより消化し、消化されたフラグメントのサイズを決定することなどが挙げられる。
「精製ウイルスポリヌクレオチド」とは、ウイルスポリヌクレオチドが天然で関連するポリペプチドを実質的に含まない、即ちその約50%未満、好ましくは約70%未満、より好ましくは約90%未満を含まないHGBVゲノムまたはそのフラグメントを指す。ウイルスポリヌクレオチドを精製する方法は当分野において公知であり、例えば、カオトロピック(chaotropic)剤を用いて粒子を破壊し、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー、密度沈降によってポリヌクレオチド及びポリペプチドを分離することが挙げられる。「精製ウイルスポリペプチド」とは、ウイルスポリペプチドが天然で関連する細胞成分を実質的に含まない、即ちその約50%未満、好ましくは約70%未満、より好ましくは約90%未満を含まないHGBVポリペプチドまたはそのフラグメントを意味する。精製方法は当業者には公知である。
本明細書において使用される「ポリペプチド」とはアミノ酸の分子鎖を指し、特定の長さ産物を指すものではない。従って、ペプチド、オリゴペプチド及びタンパク質がポリペプチドの定義に含まれる。しかしながらこの用語には、ポリペプチドの発現後の修飾、例えばグリコシル化、アセチル化、リン酸化などは含まれないものとする。
「組換え宿主細胞」、「宿主細胞」、「細胞」、「細胞系」、「細胞培養物」、及び単細胞物質として培養された微生物または高等真核細胞系を示す他の同様の用語は、組換えベクターまたは他の移入DNAのレシピエントとして使用され得る、または使用された細胞を指し、トランスフェクトされた元の細胞の本来の子孫をも含む。
本明細書において使用される「レプリコン」とは、細胞内でポリヌクレオチド複製の自立単位として挙動する、プラスミド、染色体またはウイルスといった任意の遺伝子エレメントを意味する。即ちレプリコンはそれ自体の制御下で複製し得る。
「ベクター」は、別のポリヌクレオチドセグメントが複製及び/または発現し得るように付加されたレプリコンである。
「制御配列」なる用語は、それらが結合しているコーディング配列の発現を行うのに必要なポリヌクレオチド配列を指す。制御配列の特性は宿主生物に応じて異なる。原核細胞においては制御配列は一般にプロモーター、リボソーム結合部位及びターミネーターを含み、真核細胞においては制御配列は一般にプロモーター、ターミネーター、及び場合によってはエンハンサーを含む。従って「制御配列」なる用語は、少なくともその存在が発現に必要な全ての成分を含んでいるものとし、更には、存在すれば有利な追加成分、例えばリーダー配列を含んでもよい。
「操作可能に結合された」とは、上記成分が予定通りに機能し得る関係にある状況を指す。従って例えば、コーディング配列に「操作可能に結合された」制御配列は、制御配列に適合する条件下でコーディング配列の発現が行われるように連結されている。
「読取り枠」または「ORF」なる用語は、ポリペプチドをコードするポリヌクレオチド配列の領域を指す。この領域はコーディング配列の一部または全コーディング配列を与え得る。
「コーディング配列」は、適当な調節配列の制御下に置かれたときにmRNAに転写される、及び/またはポリペプチドに翻訳されるポリヌクレオチド配列である。コーディング配列の境界は、5’末端にある翻訳開始コドンと3’末端にある翻訳終結コドンとによって決定される。コーディング配列としては、限定的ではないが、mRNA、cDNA、及び組換えポリヌクレオチド配列が挙げられる。
「〜を用いて/として免疫学的に同定可能な」なる用語は、特定のポリペプチド、通常はHGBVタンパク質中に存在し、これに固有であるエピトープ及びポリペプチドが存在することを指す。免疫学的な一致(identity)は、抗体結合及び/または結合の競合によって判定し得る。かかる方法は当業者には公知であるし、本明細書にも記載される。エピトープの固有性は、公知のデータバンク、例えばGenBankにおいてエピトープをコードするポリヌクレオチド配列をコンピューター検索するか、または他の既知のタンパク質とアミノ酸配列を比較することにより判定し得る。
本明細書において使用される「エピトープ」とはポリペプチドの抗原決定基を意味する。恐らく、エピトープは、3個のアミノ酸をそのエピトープに固有の空間的配置で含み得る。通常エピトープは少なくとも5個のこのようなアミノ酸からなり、より一般的には少なくとも8〜10個のアミノ酸からなる。空間的配置を調査する方法は当分野において公知であり、例えばX線結晶分析及び2次元核磁気共鳴が挙げられる。
ポリペプチド内に含まれる特定のエピトープを抗体が認識したことから抗体に結合するとき、該ポリペプチドは該抗体に対して「免疫学的に反応性である」。免疫学的反応性は、抗体結合、特に抗体結合速度、及び/または、該抗体に対するエピトープを含む既知のポリペプチドを競合物質として使用する結合の競合によって判定し得る。ポリペプチドが抗体に対して免疫学的に反応性であるかどうかを決定する方法は当分野において公知である。
本明細書において使用される「HGBVエピトープを含む免疫原性ポリペプチド」なる用語は、天然HGBVポリペプチドまたはそのフラグメント、並びに、他の手段、例えば化学的合成または組換え微生物中でのポリペプチドの発現によって製造されたポリペプチドを意味する。
「形質転換」なる用語は、挿入方法に関係なく、外来ポリヌクレオチドを宿主細胞中に挿入することを指す。例えば、直接取込み、形質導入またはf−交配(f−mating)が含まれる。外来ポリヌクレオチドは、非組込みベクター、例えばプラスミドとして維持することもできるし、或いは、宿主ゲノム中に組込むこともできる。
「治療」とは予防及び/または加療を指す。
本明細書において使用される「個体」なる用語は、動物、特に哺乳動物種のものを指し、限定的ではないが、家畜、競技用動物、霊長目及びヒトを含むが、特に該用語はタマリン及びヒトを指す。
本明細書において使用される「正鎖」(または「+」)は、ポリペプチドをコードする配列を含む核酸を示す。「負鎖」(または「−」)は、「正」鎖の配列と相補的な配列を含む核酸を示す。
ウイルスの「陽性鎖ゲノム」は、RNAであろうとDNAであろうと、ゲノムが一本鎖であり、ウイルスポリペプチドをコードすることを示す。
「検査試料」とは、被分析物(例えば問題の抗体または問題の抗原)のソースとなる個体の成分を指す。かかる成分は当分野において公知である。検査試料には本発明の方法によって試験し得る生物試料が含まれるが、ヒト及び動物の体液、例えば全血、血清、血漿、脳脊髄液、尿、リンパ液、並びに、呼吸管、胃腸管及び尿生殖管の種々の外分泌物、涙、唾液、乳、白血球、ミエローマなど、並びに、生物学的液体、例えば細胞培養液上清、固定組織試料、固定細胞試料が挙げられる。
「精製HGBV」とは、ウイルスが通常関連する細胞構成物質、及び感染組織中に存在し得る他のタイプのウイルスから単離されたHGBVの調製物を指す。ウイルスを単離する方法は当業者には公知であり、例えば遠心及びアフィニティークロマトグラフィーが挙げられる。
「PNA」は、ある処理、例えばターゲットの存在を判定するようなアッセイに使用し得る「ペプチド核酸類縁物」を指す。PNAは、RNAターゲットまたはDNAに対する電気的に中性の物質である。例えばDNAプローブの代わりにアッセイに使用されるPNAプローブは、DNAプローブを使用したときには得られない利点を与える。かかる利点としては、製造可能性、大規模標識、再現性、安定性、イオン強度の変化に対する低感度性、及び、DNAまたはRNAを使用する方法に存在する酵素分解に対する抵抗性が挙げられる。PNAは、フルオレセイン、放射性核種、化学発光化合物などのシグナル生成化合物で標識することができる。このようにPNAは、DNAまたはRNAの代わりに方法に使用し得る。本明細書にはDNAを使用するアッセイを記載するが、必要であればアッセイ試薬を適当に変更し、PNAをRNAまたはDNAの代わりに使用することも常法の範囲内である。
(一般用途)
本発明の特定の組換えタンパク質、合成ペポチド、または精製ウイルスポリペプチドを製造したならば、その組換えまたは合成ペプチドを使用し、HGBVに対する抗原または抗体の存在を検出する本明細書に記載のごとき固有のアッセイを開発することができる。またかかる組成物を使用し、所望するHGBVの免疫学的エピトープに特異的に結合する特異的組換えタンパク質または合成ペプチドを用い、モノクローナル及び/またはポリクローナル抗体を開発することができる。少なくとも1種の本発明のポリヌクレオチドを使用して、当分野において公知の方法に従ってワクチンを開発することも考えられる。
本発明の特定の組換えタンパク質、合成ペポチド、または精製ウイルスポリペプチドを製造したならば、その組換えまたは合成ペプチドを使用し、HGBVに対する抗原または抗体の存在を検出する本明細書に記載のごとき固有のアッセイを開発することができる。またかかる組成物を使用し、所望するHGBVの免疫学的エピトープに特異的に結合する特異的組換えタンパク質または合成ペプチドを用い、モノクローナル及び/またはポリクローナル抗体を開発することができる。少なくとも1種の本発明のポリヌクレオチドを使用して、当分野において公知の方法に従ってワクチンを開発することも考えられる。
アッセイに使用される試薬は、アッセイに使用されるモノクローナル抗体もしくはモノクローナル抗体の混合物または(組換えもしくは合成)ポリペプチドといった試薬を各々が含む1つ以上の容器、例えばバイアルやボトルを包含する試験キットの形態で提供され得ると考えられる。当業者には公知の緩衝液、対照などの他の構成成分をかかる試験キットに含めてもよい。
「固相」(「固体支持体」)は当業者には公知であり、反応トレーのウェルの壁、試験管、ポリスチレンビーズ、磁性ビーズ、ニトロセルロースストリップ、膜、微粒子(例えばラテックス粒子)、ヒツジ(または他の動物)の赤血球、Duracytesなどが挙げられる。「固相」は限定的なものではなく、当業者によって選択され得る。即ち、ラテックス粒子、微粒子、磁性または非磁性ビーズ、膜、プラスチックチューブ、マイクロタイターウェルの壁、ガラスまたはシリコンチップ、ヒツジ(または他の適当な動物)の赤血球、及びDuracytesは全てが適当な例である。ペプチドを固相上に固定する適当な方法としては、イオン性、疎水性、共有結合性の相互作用などが挙げられる。本明細書において使用される「固相」とは、不溶性であるかまたは後続反応によって不溶性にし得る任意の材料を指す。固相は、捕獲剤を引き付けて固定する固有の能力において選択され得る。或いは固相は、捕獲剤を引き付けて固定する能力を有する補助的レセプターを保持し得る。補助的レセプターは、捕獲剤自体または捕獲剤に結合している帯電物質とは反対の電荷を有する帯電物質を含み得る。或いはまたレセプター分子は、固相上に固定(付着)されており、特異的結合反応によって捕獲剤を固定する能力を有する任意の特異的結合メンバーとすることもできる。アッセイ実施前またはアッセイ実施中に、レセプター分子によって捕獲剤を固相材料に間接的に結合することができる。固相は、プラスチック、誘導体化プラスチック、磁性または非磁性金属、ガラスまたはシリコン表面を備えた試験管、マイクロタイターウェル、シート、ビーズ、微粒子、チップ、ヒツジ(または他の適当な動物)の赤血球、Duracytes、並びに、当業者には公知の他の構成とし得る。
固相が、検出抗体がアクセスし得るに十分な多孔性及び抗原を結合するのに適当な表面親和性を有する任意の適当な多孔質材料からなることも考えられ、本発明の範囲内である。一般には微孔質構造が好ましいが、水和状態でゲル構造を有する材料も使用し得る。かかる有用な固体支持体としては、天然ポリマー炭水化物及びそれらの合成変性、架橋または置換誘導体、例えば寒天、アガロース、架橋アルギン酸、置換架橋グアゴム、特に硝酸及びカルボン酸とのセルロースエステル、混合セルロースエステル、セルロースエーテル;窒素を含む天然ポリマー、例えば架橋または変性ゼラチンを含むタンパク質及び誘導体;天然炭化水素ポリマー、例えばラテックス及びゴム;適当な多孔質構造を有するよう製造され得る合成ポリマー、例えばポリエチレン、ポリプロピレン、ポリスチレン、ポリ塩化ビニル、ポリ酢酸ビニルを含むビニルポリマー及びその部分加水分解誘導体、ポリアクリルアミド、ポリメタクリレート、上記ポリ縮合物のコポリマー及びターポリマー、例えばポリエステル、ポリアミド、及び他のポリマー、例えばポリウレタンまたはポリエポキシド;多孔質無機材料、例えばアルカリ土類金属及びマグネシウムの硫酸塩または炭酸塩(例えば硫酸バリウム、硫酸カルシウム、炭酸カルシウム)、アルカリ及びアルカリ土類金属、アルミニウム並びにマグネシウムのケイ酸塩;並びに、アルミニウムまたはケイ素の酸化物または水和物、例えばクレー、アルミナ、タルク、カオリン、ゼオライト、シリカゲルまたはガラス(これらの材料は上記ポリマー材料と一緒に充填剤として使用し得る);並びに、これらの混合物またはコポリマー、例えば既存の天然ポリマー上で合成ポリマーの重合を開始することにより得られるグラフトコポリマーが挙げられる。これらの材料は全てが、フィルム、シート、またはプレートといった適当な形状で使用することもできるし、紙、ガラス、プラスチックフィルム、またはファイバーといった適当な不活性担体に被覆、接着または積層することもできる。
ニトロセルロースの多孔質構造は、モノクローナル抗体を含む広範囲の試薬に対して優れた吸収性及び吸着性を示す。ナイロンも同様の特性を有しており、やはり適当である。上述のごとき多孔質固体支持体は厚さが約0.01〜0.05mm、好ましくは約0.1mmのシートの形態であると考えられる。孔径は広範囲で変えることができるが、約0.025〜15μ、特に約0.15〜15μであるのが好ましい。かかる支持体の表面は、抗原または抗体と支持体との共有結合を惹起する化学処理によって活性化し得る。一般には、あまり理解されていない疎水性の力によって多孔質材料上に吸着することにより、抗原または抗体は不可逆的に結合される。適当な固体支持体は米国特許出願第227,272号明細書にも記載されている。
「指示薬」は、HGBVの特異的結合メンバーに結合(付着)した、外部手段によって検出し得る測定可能シグナルを生成し得るまたは生成する「シグナル生成化合物」(標識)を含む。本明細書において使用される「特異的結合メンバー」とは特異的結合対、即ち一方の分子が化学的または物理的手段を介して第2の分子に特異的に結合する2つの分子のメンバーを意味する。HGBVに対する特異的結合対の抗体メンバーであるほか、指示薬は、ハプテン−抗ハプテン系、例えばビオチンまたは抗ビオチン、アビジンまたはビオチン、炭水化物またはレクチン、相補的ヌクレオチド配列、エフェクター分子またはレセプター分子、酵素補因子及び酵素、酵素阻害物質または酵素などとし得る。免疫反応性特異的結合メンバーは、サンドイッチアッセイにおいてはHGBVに、競合アッセイにおいては捕獲剤に、また間接アッセイにおいては任意の特異的結合メンバーに結合し得る、抗体、抗原、または抗体/抗原複合体であり得る。
考えられる種々の「シグナル生成化合物」(標識)としては、色原体、酵素のごとき触媒、フルオレセイン及びローダミンのごとき発光化合物、ジオキセタン、アクリジニウム、フェナントリジニウム及びルミノールのごとき化学発光化合物、放射性元素、直接可視標識が挙げられる。酵素の例としてはアルカリ性ホスファターゼ、西洋ワサビペルオキシダーゼ、β−ガラクトシダーゼなどが挙げられる。特定の標識の選択は限定的ではなく、標識はそれ自体でまたは1種以上の他の物質と一緒になってシグナルを生成することができる。
本発明は、特異的結合メンバーを使用するアッセイを提供する。本明細書において使用される「特異的結合メンバー」は特異的結合対、即ち一方の分子が化学的または物理的手段を介して第2の分子に特異的に結合する2種類の異なる分子のメンバーである。従って、一般的なイムノアッセイの抗原及び抗体特異的結合対のほかに、他の特異的結合対として、ビオチンとアビジン、炭水化物とレクチン、相補的ヌクレオチド配列、エフェクター分子とレポーター分子、補因子と酵素、酵素阻害物質と酵素などを挙げ得る。特異的結合対には更に、元の特異的結合対の類似物、例えば被分析物質類似物であるメンバーも含まれ得る。免疫反応性特異的結合メンバーとしては、組換えDNA分子によって形成されるものを含み、抗原、抗原フラグメント、モノクローナル及びポリクローナル抗体及び抗体フラグメント、これらの複合体が挙げられる。本明細書において使用される「ハプテン」なる用語は、抗体に結合することはできるが、担体タンパク質に結合していなければ抗体形成を誘起し得ない部分抗原または非タンパク質結合メンバーを指す。
本明細書において使用される「被分析物質」は、検査試料中に存在し得る検出すべき物質である。被分析物質は、それに対して天然特異的結合メンバー(例えば抗体)が存在するかまたはそれに対して特異的結合メンバーを調製し得る物質であり得る。即ち、被分析物質は、アッセイにおいて1つ以上の特異的結合メンバーに結合し得る物質である。「被分析物質」としては、任意の抗原性物質、ハプテン、抗体、及びこれらの組合せが挙げられる。特異的結合対のメンバーとして、被分析物質は天然特異的結合パートナー(対)によって検出することができ、例えば、ビタミンB12の測定には特異的結合対のメンバーとして固有因子タンパク質が使用され、葉酸を測定するには葉酸塩結合タンパク質が使用され、炭水化物の測定には特異的結合対のメンバーとしてレクチンが使用される。被分析物質としてはタンパク質、ペプチド、アミノ酸、ヌクレオチドターゲットなどが挙げられる。
種々の他の固相を使用する他の実施態様も考えられ、それらも本発明の範囲内である。例えば、欧州特許出願公開第0326100号明細書に対応する同時係属米国特許出願第150,278号明細書及び米国特許出願第375,029号明細書(欧州特許出願公開第0406473号明細書)に記載の、負電荷を有するポリマーを用いて固定可能な反応複合体を固定するイオン捕獲法を本発明に従って使用し、高速液相(fast solution−phase)免疫化学反応を行うことができる。固定可能な免疫複合体は、負電荷を有するポリアニオン/免疫複合体と予め処理して正電荷をもたせた多孔質マトリックスとのイオン相互反応によって反応混合物の残りの部分から分離し、EPO特許出願公開第0,273,115号明細書に対応する同時係属米国特許出願第921,979号明細書に記載のごとき化学発光シグナル測定に記載のものを含む、文献明記の種々のシグナル生成系を使用して検出し得る。
また本発明方法は、固相が(磁性または非磁性)微粒子からなる自動化及び半自動化系を含む、微粒子技術を使用する系での使用にも適合し得る。このような系として、EPO特許出願公開第0 425 633号明細書及び同第0 424 634号明細書にそれぞれ対応する係属米国特許出願第425,651号明細書及び同第425,643号明細書に記載のものが挙げられる。
イムノアッセイ用の走査型プローブ顕微鏡(SPM)を使用すると、本発明のモノクローナル抗体を容易に適合し得る。走査型プローブ顕微鏡、特に原子力(atomic force)顕微鏡においては、捕獲相、例えば少なくとも1種の本発明のモノクローナル抗体を固相に付着させ、走査型プローブ顕微鏡を使用し、固相の表面に存在し得る抗原/抗体複合体を検出する。走査型トンネル顕微鏡を使用すると、抗原/抗体複合体を検出するために多くのイムノアアッセイ系に通常は使用せねばならない標識が必要でなくなる。このような系は係属米国特許出願第662,147号明細書に記載されている。特異的結合反応をモニターするためにSPMは多くの態様で使用され得る。1つの実施態様においては、特異的結合パートナーの1つのメンバー(本発明のモノクローナル抗体である被分析物質特異的物質)を走査に適した表面に付着させる。被分析物質特異的物質の付着は、当業者には公知の方法に従って、プラスチックまたは金属表面の固相からなる試験片に吸着させることにより行い得る。或いは、誘導体化プラスチック、金属、シリコン、またはガラスの固相からなる試験片に特異的結合パートナー(被分析物質特異的物質)を共有結合させることもできる。共有結合方法は当業者には公知であり、特異的結合パートナーを試験片に不可逆的に結合する種々の手段を含む。試験片がシリコンまたはガラスである場合、特異的結合パートナーを付着させる前に表面を活性化する必要がある。トリエトキシアミノプロピルシラン(Sigma Chemical Co.,St.Louis,MOから入手可能)、トリエトキシビニルシラン(Aldrich Chemical Co.,Milwaukee,WI)及び(3−メルカプト−プロピル)−トリメトキシシラン(Sigma Chemical Co.,St.Louis,MO)といった活性化シラン化合物を使用し、それぞれアミノ、ビニル及びチオールといった反応性の基を導入することができる。このような活性化表面を使用して結合パートナーを(アミノまたはチオールの場合には)直接に結合することもできるし、活性化表面を更に、グルタルアルデヒド、ビス(スクシンイミジル)スベレート、SPPD9(スクシンイミジル 3−[2−ピリジルジチオ]プロピオネート)、SMCC(スクシンイミジル−4−[N−マレイミドメチル]シクロヘキサン−1−カルボキシレート)、SIAB(スクシンイミジル[4−ヨードアセチル]アミノベンゾエート)及びSMPB(スクシンイミジル4−[1−マレイミドフェニル]ブチレート)といったリンカーと反応させ、結合パートナーを表面から離すこともできる。ビニル基は、酸化して共有結合手段を与えることもできるし、特異的結合パートナーに対して複数の付着点を与え得るポリアクリル酸のごとき種々のポリマーを重合するためのアンカーとして使用することもできる。アミノ表面を種々の分子量の酸化デキストランと反応させ、種々のサイズ及び結合能の親水性リンカーを与えることもできる。酸化可能なデキストランの例として、Dextran T−40(分子量40,000ダルトン)、Dextran T−110(分子量110,000ダルトン)、Dextran T−500(分子量500,000ダルトン)、Dextran T−2M(分子量2,000,000ダルトン)(これら全てはPharmaciaから入手可能)、またはFicoll(分子量70,000ダルトン)(Sigma Chemical Co.,St Louis,MOから入手可能)が挙げられる。また、1988年1月29日出願係属米国特許出願第150,278号明細書及び1989年7月7日出願同第375,029号明細書に記載の方法及び化学物質を使用し、高分子電解質相互作用を使用して特定的結合パートナーを試験片の表面に固定することもできる。好ましい付着方法は共有結合によるものである。特異的結合メンバーを付着させたあと、非特異的結合を最少限に抑えるために表面を更に、血清、タンパク質、または他のブロッキング剤で処理してもよい。該表面がアッセイに適しているか検証するため、それを製造場所または使用時点で走査してもよい。走査方法は、試験片の特異的結合特性を変化させないものとする。
「サンドイッチ」イムノアッセイ及びプローブアッセイを含む種々の他のアッセイ形式を使用し得る。例えば、本発明のモノクローナル抗体を種々のアッセイ系に使用して、検査試料中のHGBVタンパク質の存在を、もしあるとするならば判定することができる。与えられるかかるモノクローナル抗体のフラグメントを使用してもよい。例えば高速アッセイ形式においては、固相に被覆したポリクローナルもしくはモノクローナル抗HGBV抗体またはそのフラグメントまたはこれらの抗体の組合せを、HGBVタンパク質を含み得る検査試料と接触させ、混合物を形成する。この混合物を、抗原/抗体複合体を形成するのに十分な時間及び条件下でインキュベートする。シグナル生成化合物を付着させた、HGBV領域に特異的に結合するモノクローナルもしくはポリクローナル抗体またはそのフラグメントまたはこれらの抗体の組合せを含む指示薬を、抗原/抗体複合体と接触させて第2の混合物を形成する。この第2の混合物を、抗体/抗原/抗体複合体を形成するのに十分な時間及び条件下でインキュベートする。検査試料中に存在しており固相上に捕獲されたHGBV抗原の存在は、もしあるとすれば、シグナル生成化合物によって生成される測定可能なシグナルを検出することにより判定される。検査試料中に存在するHGBV抗原の量は生成されたシグナルに比例する。
或いは、固体支持体に結合させたポリクローナルもしくはモノクローナル抗HGBV抗体またはそのフラグメントまたはこれらの抗体の組合せと、検査試料と、シグナル生成化合物を付着させた、HGBV抗原に特異的に結合するモノクローナルもしくはポリクローナル抗体またはそのフラグメントまたはこれらの抗体の組合せを含む指示薬とを接触させて混合物を形成する。この混合物を、抗体/抗原/抗体複合体を形成するのに十分な時間及び条件下でインキュベートする。検査試料中に存在しており固相上に捕獲されたHGBVタンパク質の存在は、もしあるとすれば、シグナル生成化合物によって生成される測定可能なシグナルを検出することにより判定される。検査試料中に存在するHGBVタンパク質の量は生成されたシグナルに比例する。
更に別のアッセイ形式においては、1種または2種以上組合せた本発明のモノクローナル抗体を、HGBVタンパク質に対する抗体を検出するための競合プローブとして使用し得る。例えばHGBVタンパク質を単独でまたは組合せて固相に被覆することができる。次いで、HGBV抗原に対する抗体を含む疑いのある検査試料を、シグナル生成化合物と少なくとも1種の本発明モノクローナル抗体とを含む指示薬と一緒に、検査試料及び指示薬と固相、または指示薬と固相とで、抗原/抗体複合体を形成するのに十分な時間及び条件下でインキュベートする。モノクローナル抗体の固相への結合の低下を定量的に測定し得る。非A、非B、非C、非D、非E型肝炎陰性が確認されている検査試料から生成されるシグナルと比較してシグナル低下が測定され得るならば、検査試料中に抗HGBV抗体が存在することが判る。
更に別の検出方法においては、本発明のモノクローナル抗体またはポリクローナル抗体の各々を、免疫組織化学分析による固定組織片及び固定細胞におけるHGBV抗原の検出に使用し得る。かかる抗体を直接標識する(フルオレセイン、コロイド金、西洋ワサビペルオキシダーゼ、アルカリ性ホスファターゼなど)か、または(本明細書に例示した種々の標識を用いた)第2標識抗種抗体を使用して標識して疾患の履歴を追跡する細胞化学分析も本発明の範囲内である。
更に、モノクローナル抗体をCNBr活性化セファロースと類似のマトリックスに結合し、細胞培養物、または血液及び肝といった生物組織由来の特異的HGBVタンパク質のアフィニティー精製に使用し、組換えまたは天然ウイルスHGBV抗原及びタンパク質を精製することもできる。
本発明のモノクローナル抗体は、治療を目的とするキメラ抗体の生成や、他の類似の用途に使用することもできる。
モノクローナル抗体またはそのフラグメントは、HGBV抗原を検出するために個々に提供され得る。本明細書において与えられるモノクローナル抗体(及びそのフラグメント)の組合せは、少なくとも1種の本発明の抗HGBV抗体と、他のHGBV領域に対する抗体との混合物または「カクテル」中の、各々が異なる結合特異性を有する成分として一緒に使用され得る。即ちこのカクテルは、HGBVタンパク質に対する本発明のモノクローナル抗体と、HGBVゲノムの他の抗原決定基に対する他のモノクローナル抗体とを含み得る。
かかるアッセイ形式に使用し得るポリクローナル抗体またはそのフラグメントは、アッセイに使用される特異的HGBV領域または他のHGBVタンパク質に特異的に結合すべきである。使用されるポリクローナル抗体は哺乳動物由来のものであるのが好ましく、ヒト、ヤギ、ウサギまたはヒツジの抗HGBVポリクローナル抗体を使用し得る。最も好ましくは、ポリクローナル抗体はウサギポリクローナル抗HGBV抗体である。アッセイに使用されるポリクローナル抗体は単独でもポリクローナル抗体のカクテルとしても使用し得る。アッセイ形式に使用されるカクテルは異なるHGBV特異性を有するモノクローナル抗体またはポリクローナル抗体いずれかからなるので、それらは、HGBV感染の診断、評価及び予防に、並びにHGBVタンパク質の分化及び特異性の研究に有用となろう。
全てのHGBVウイルスに共通な合成、組換えまたは天然ペプチドを使用することにより、HGBV群のウイルスがアッセイにおいて検出可能となり得ることが考えられ、本発明の範囲内である。HGBV−A、HGBV−B、HGBV−C、更には他のHGBVウイルス由来の種々のエピトープを同定する種々の合成、組換えまたは天然ペプチドをアッセイ形式に使用し得ることも本発明の範囲内である。後者の場合、それらを1つの固相に被覆してもよいし、各ペプチドをそれぞれ別個の固相、例えば微粒子上に被覆し、それらを合わせて、あとでアッセイに使用し得るペプチドの混合物を形成してもよい。このようなアッセイ形式の変形は当業者には公知であり、後述する。
別のアッセイ形式においては、HGBVに対する抗体及び/または抗原の存在を以下のように同時アッセイにおいて検出し得る。検査試料を、固相に付着された第1被分析物質に特異的な第1結合メンバーを含む第1被分析物質用捕獲剤と、第2固相に付着された第2被分析物質に対する第1結合メンバーを含む第2被分析物質用捕獲剤とに同時に接触させ、それによって混合物を形成する。この混合物を、捕獲剤/第1被分析物質複合体及び捕獲剤/第2被分析物質複合体を形成するのに十分な時間及び条件下でインキュベートする。このように形成された複合体を、シグナル生成化合物で標識された第1被分析物質に特異的な結合対のメンバーを含む指示薬と、シグナル生成化合物で標識された第2被分析物質に特異的な結合対のメンバーを含む指示薬とに接触させ、第2混合物を形成する。この第2混合物を、捕獲剤/第1被分析物質/指示薬複合体及び捕獲剤/第2被分析物質/指示薬複合体を形成するのに十分な時間及び条件下でインキュベートする。1種以上の被分析物質の存在は、検査試料中の1種以上の被分析物質の存在の指標として、いずれか一方または両方の固相上に形成された複合体に関連して生成されたシグナルを検出することにより判定される。このアッセイ形式においては、ヒト発現系から誘導されたタンパク質を、本明細書に記載のごとき哺乳動物発現系から誘導されたタンパク質から生成されるモノクローナル抗体と同様に使用し得る。かかるアッセイ系は、欧州特許出願公開第0473065号明細書に対応する係属米国特許出願第07/574,821号明細書、発明の名称“Simultaneous Assay for Detecting One Or More Analytes”に詳述されている。
更に別のアッセイ形式においては、組換えタンパク質及び/または合成ペプチドを使用して検査試料中の抗HGBVの存在を検出し得る。例えば、検査試料を、少なくとも1種の組換えタンパク質または合成ペプチドを付着させた固相と一緒にインキュベートする。これらを、抗原/抗体複合体を形成するのに十分な時間及び条件下で反応させる。インキュベーション後、抗原/抗体複合体を検出する。選択したアッセイ系に従い、指示薬を使用して検出を容易にし得る。別のアッセイ形式においては、検査試料を、本明細書に記載のごとく製造された組換えタンパク質または合成ペプチドを付着させた固相と接触させ、更に、好ましくは指示薬で標識された、該タンパク質に特異的なモノクローナルまたはポリクローナル抗体と接触させる。抗体/抗原複合体が形成されるのに十分な時間及び条件下でインキュベートした後、固相を遊離相から分離し、HGBV抗体の存在の指標としての標識を固相または遊離相において検出する。本発明のタンパク質を使用する他のアッセイ形式も考えられる。それらには、検査試料を、第1供給源由来の少なくとも1種の抗原を付着させた固相と接触させ、固相及び検査試料を、抗原/抗体複合体を形成するのに十分な時間及び条件下でインキュベートし、次いで固相を、第1供給源とは異なる第2供給源から誘導された標識抗原と接触させることを含む。例えば、E.coliのごとき第1供給源から誘導された組換えタンパク質を固相上の捕獲抗原として使用し、検査試料をこのように調製された固相に添加し、異なる供給源(即ちE.coli以外)から誘導された組換えタンパク質を指示薬の一部として使用する。同様に、固相上の組換え抗原と指示薬相中の合成ペプチドとを組合せることもできる。第1供給源由来のHGBVに特異的な抗原を捕獲抗原として使用し且つ異なる第2供給源由来のHGBVに特異的な抗原を使用するアッセイ形式も考えられる。組換え抗原の種々の組合せ、並びに合成ペプチド、精製ウイルスタンパク質の使用などが本発明の範囲に含まれる。このようなアッセイその他が本出願人名義の米国特許第5,254,458号明細書に記載されており、その内容は参照により本明細書に包含されるものとする。
他のアッセイ系では、ウイルスゲノム(またはそのフラグメント)を含むHGBVウイルス粒子またはサブウイルス粒子に、特異的抗体とウイルスタンパク質(ペプチドなど)の接触によって特異的に結合する(ポリクローナル、モノクローナルまたは天然)抗体が使用される。次いでこの捕獲された粒子をLCRまたはPCRなどの方法によって分析し、ウイルスゲノムが検査試料中に存在するかどうかを判定することができる。該方法に従って分析し得る検査試料としては、血液、肝、痰、尿、便、唾液などが挙げられる。かかる抗原捕獲増幅法を使用する利点は、特異的抗体を使用して検査試料中のウイルスゲノムを他の分子から分離し得ることである。このような方法は係属米国特許出願第08/141,429号明細書に記載されている。
本発明を固相を使用する場合について記載したが、本発明の抗体、タンパク質及びペプチドといった試薬は非固相アッセイ系においても使用し得ると考えられる。このようなアッセイ系は当業者には公知であり、本発明の範囲に含まれるものとする。
(材料及び方法)
(一般方法)
特に記載のない限り、本発明の実施には、分子生物学、微生物学、組換えDNA及び免疫学の分野における慣用的な公知の技術及び方法が使用される。このような技術は文献に詳述されている。例えば、J.Sambrookら,Molecular Cloning:A Laboratory Manual,第2版,Cold Spring Harbor Press,Cold Spring Harbor,N.Y.(1989);D.N.Glover編,DNA Cloning,Volumes I and II(1985);M.J.Gait編,Oligonucleotide Synthesis,(1984);B.D.Hamesら編,Nucleic Acid Hybridization,(1984);B.D.Hamesら編,Transcription and Translation,(1984);R.I.Freshney編,Animal Cell Culture,(1986);Immobilized Cells and Enzymes,IRL Press(1986);B.Perbal,A Practical Guide to Molecular Cloning,(1984);シリーズ,Methods in Enzymology,Academic Press,Inc.,Orlando,Florida;J.H.Millerら編,Gene Transfer Vectors For Mammalian Cells,Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y.(1987);Wuら編,Methods in Enzymology,Vol.154及び155;Mayerら編,Immunological Methods In Cell and Molecular Biology,Academic Press,London(1987);Scopes,Protein Purification:Principles and Practice,第2版,Springer−Verlag,N.Y.;及び、D.Weirら編,Handbook Of Experimental Immunology,Vol.I−IV(1986);N.Lisitisynら,Science 259:946−951(1993)参照。
(一般方法)
特に記載のない限り、本発明の実施には、分子生物学、微生物学、組換えDNA及び免疫学の分野における慣用的な公知の技術及び方法が使用される。このような技術は文献に詳述されている。例えば、J.Sambrookら,Molecular Cloning:A Laboratory Manual,第2版,Cold Spring Harbor Press,Cold Spring Harbor,N.Y.(1989);D.N.Glover編,DNA Cloning,Volumes I and II(1985);M.J.Gait編,Oligonucleotide Synthesis,(1984);B.D.Hamesら編,Nucleic Acid Hybridization,(1984);B.D.Hamesら編,Transcription and Translation,(1984);R.I.Freshney編,Animal Cell Culture,(1986);Immobilized Cells and Enzymes,IRL Press(1986);B.Perbal,A Practical Guide to Molecular Cloning,(1984);シリーズ,Methods in Enzymology,Academic Press,Inc.,Orlando,Florida;J.H.Millerら編,Gene Transfer Vectors For Mammalian Cells,Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y.(1987);Wuら編,Methods in Enzymology,Vol.154及び155;Mayerら編,Immunological Methods In Cell and Molecular Biology,Academic Press,London(1987);Scopes,Protein Purification:Principles and Practice,第2版,Springer−Verlag,N.Y.;及び、D.Weirら編,Handbook Of Experimental Immunology,Vol.I−IV(1986);N.Lisitisynら,Science 259:946−951(1993)参照。
本発明の試薬及び方法は、本明細書に記載のごとく改良された代表相違分析によって単離された、タマリンまたはヒトであるHGBV感染個体の血漿、血清または肝ホモジネート中に存在する極めて関連性のあるヌクレオチド配列群を与えることにより実現可能となる。このヌクレオチド配列群は、未感染個体由来のヒトまたはタマリンゲノムDNAにはハイブリダイズしないこと、HGBV感染個体の肝(または肝ホモジネート)、血漿または血清中にしか存在しないこと、及びGenBankに存在しないことから、ヒトまたはタマリンに由来するものではない。更に、該配列群は、HAV、HBV、HCV、HDV及びHEVゲノム内に含まれる配列と核酸レベルで有意な一致を示さず、翻訳産物でも一致レベルは有意とは考えられないほど低い。HGBV感染ヒト由来の感染性血清、血漿または肝ホモジネートはかかるポリヌクレオチド配列を含むが、未感染ヒト由来の血清、血漿または肝ホモジネートはかかる配列を含まない。これらのポリヌクレオチド配列の幾つかを含む感染肝をノーザンブロット分析すると、それらがウイルスゲノムと同様のサイズの大きなRNA転写物から誘導されていることが判る。HGBV感染ヒト由来の血清、血漿または肝ホモジネートはこのポリペプチドに結合する抗体を含むが、未感染ヒト由来の血清、血漿または肝ホモジネートはこのポリペプチドに対する抗体を含まず、かかる抗体は、急性非A、非B、非C、非D及び非E感染後の個体において誘起されている。これらを基準にすると該配列は、非A、非B、非C、非D及び非E型肝炎を惹起する、またはこれに関連のあるウイルスの配列であると考えられる。
この核酸配列群を使用し得ることにより、HGBV感染に起因する非A、非B、非C、非D、非E型肝炎を診断したり、血液ドナー、献血血液、血液製剤及び個体を感染についてスクリーニングすることにおいて有用なDNAプローブ及びポリペプチドを構築し得る。例えば、該配列から、例えばウイルスを保有する疑いのある被験者の血清中のウイルスゲノムの存在を検出するためのハイブリダイゼーションプローブまたはPCRプライマーとして有用であったり、また献血血液中のウイルスの存在をスクリーニングするために有用な、約8〜10またはそれ以上のヌクレオチドのDNAオリゴマーを合成し得る。当該核酸配列群により、HGBVに感染した際に生成される抗体の存在に対する診断試薬として有用な、HGBV特異的ポリペプチドを設計及び製造し得る。該核酸配列から誘導される精製ポリペプチドに対する抗体を使用し、感染個体及び血液中のウイルス性抗原を検出することもできる。かかる核酸配列により、HGBVに対するワクチンとして、及び抗体を製造するために使用し得るポリペプチドを設計及び製造することが可能となり、そうすればそれを疾患の予防及び/またはHGBV感染個体の治療に使用し得る。
かかる核酸配列群により、HGBVゲノムを更に特性分析することも可能となる。かかる配列から誘導されるポリヌクレオチドプローブを使用し、ゲノムまたはcDNAライブラリーを別の重複核酸配列についてスクリーニングし、更にそれを使用して重複した配列を得ることができる。ゲノムがセグメント化されていてもセグメントが共通配列を欠いていることがなければ、この方法を使用して全ゲノムの配列を得ることができる。しかしながら、ゲノムがセグメント化されている場合、記載のごときRDAクローニング処理を繰り返して後述のごとく修飾するか、後述のごとくλ−gt11血清学的スクリーニング処理を繰り返して後述のクローンを単離するか、或いは、精製HGBV粒子からゲノムを単離することにより、ゲノムの他のセグメントを得ることができる。
上記配列から誘導されるcDNA配列及びポリペプチド、並びにかかるポリペプチドに対する抗体も、HGBV原因物質の単離及び同定に有用である。例えば、かかる核酸配列から誘導されるポリペプチド中に含まれるHGBVエピトープに対する抗体をアフィニティークロマトグラフィーに基づく方法に使用し、ウイルスを単離することができる。或いは、抗体を使用し、他の方法で単離されたウイルス粒子を同定することもできる。単離されたウイルス粒子中のウイルス性抗原及びゲノム物質を更に特性分析することもできる。
HGBVゲノムを更に配列決定すること、HGBV抗原を更に特性分析すること、及びゲノムを特性分析することにより得られる情報により、HGBVに誘発される非A、非B、非C、非D、非E型肝炎の診断、予防及び治療に、並びに感染血液及び血液関連製剤のスクリーニングに使用し得る別のプローブ及びポリペプチド並びに抗体を設計及び合成することができる。
かかる核酸配列群が誘導されるゲノムから誘導される抗原、抗体及びポリヌクレオチドを含む、HGBVに対するプローブが得られれば、HGBVの生物学的特性を明らかにすることに特に使用される組織培養系の開発が可能となる。一旦これが既知となれば、HGBVの複製またはその感染を優先的に阻害する抗ウイルス性化合物をベースにして新規の治療方法を開発し得ると考えられる。
HGBVの病因物質を同定及び単離するために使用した1方法では、N.Lisitsynら,Science.259:946−951(1993)により報告されているような再現相違分析(RDA)として知られる公知方法の変法によりGB物質のクローニング/単離を行った。この方法は控除ハイブリダイゼーションの原理に基づいて2つの複合哺乳動物ゲノム間のDNA相違をクローニングする。要約すると、この方法では2つの被験ゲノムを(該当ターゲット配列を含む)「テスター」及び(正常DNAに相当する)「ドライバー」と属的に区別する。RDAに関するLisitsynらの記述は類似する複合DNAバックグラウンド間のDNA相違の同定及びクローニングに限られている。これらの相違は特定の細胞系、血液、血漿又は組織サンプル中に存在し且つ非感染細胞系、血液、血漿又は組織サンプル中に不在の任意の大きいDNAウイルス(例えば≧25,000DNA塩基対)を含み得る。HGBVは≦10,000塩基のDNA又はRNAゲノムを含む小さいウイルスであるらしいと先行文献に示されているので、小さいウイルスを検出できるようにRDAプロトコルを修正した。方法の主要段階を以下に記載し、図13に示す。
要約すると、段階1では市販キットを使用して全核酸(DNA及びRNA)を単離する。RDAではサンプルが高度に整合していることが必要である。理想的には、テスター及びドライバー核酸サンプルを同一源(動物、ヒト等)から得るべきである。高度に関連するものであれば、非同一材料をテスター及びドライバー核酸源に使用してもよい。RNAのランダムに開始される逆転写とそれに続いてランダムに開始されるDNA合成により全核酸から2本鎖DNAを生成する。この処理は、1本鎖RNAウイルス及び1本鎖DNAウイルスをRDAに使用可能な2本鎖DNA分子に変換するものである。未知ウイルスがDNA又はRNAゲノムの一方を有すると仮定するならば、文献に記載されているようにDNA単独又はRNA単独抽出手順を使用して2本鎖DNAを生成することができる。
段階2ではテスター及びドライバー核酸を増幅し、予備接種及び感染性血漿源から抽出された全核酸に相当する大量の材料(即ちテスターアンプリコン及びドライバーアンプリコン)を生成する。これは、4bp認識部位を有する制限エンドヌクレアーゼ(例えばSau3AI)で上記のように調製した2本鎖DNAを開裂することにより達せられる。DNAフラグメントをオリゴヌクレオチドアダプター(セット#1)に連結する。DNAフラグメントを末端充填し、PCR増幅する。PCR増幅後、オリゴヌクレオチドアダプター(セット#1)を制限エンドヌクレアーゼ消化(例えばSau3A I)により除去し、その後の控除ハイブリダイゼーション法で使用する大量のテスター及びドライバー核酸を遊離させる。
段階3の実験の目的はテスターゲノムに固有のDNAを集積することである。これは控除ハイブリダイゼーション及び動的集積を単一段階として組み合わせることにより達せられる。要約すると、オリゴヌクレオチドアダプターセット(#2又は#3)をテスターアンプリコンの5’末端に連結する。テスターアンプリコンと過剰のドライバーアンプリコンを混合し、変性させ、20時間ハイブリダイズさせる。テスター及びドライバーDNA間で共通して保存される大量の配列がこの時間中にアニールする。更に、テスターアプリコンに特有な配列が再アニールする。しかし、ハイブリダイゼーション時間が制限されているため、多少の1本鎖テスター及びドライバーDNAが残存する。
段階4では、再アニールしたテスター及びドライバーDNAの3’末端を文献に記載されているように高温で熱安定DNAポリメラーゼで充填する。テスターに固有の再アニールした配列はアニールした配列の両鎖上に連結されたアダプターを含む。こうしてこれらの分子の3’末端充填により2つのDNA鎖上にPCRプライマーに相補的な配列が生成される。従って、これらのDNA種はPCRにかけると指数的に増幅される。これとは対照的にテスター及びドライバーアンプリコン間で共通に保存された配列を含む比較的大量のハイブリッド分子(即ち一方の鎖はテスターアンプリコンに由来し、一方の鎖はドライバーアンプリコンに由来する)はPCRにより直線的に増幅される。これは、(テスターアンプリコンに由来する)鎖のみが連結されたアダプター配列を含み、3’末端充填がドライバーアダプターに由来する鎖上にPCRプライマーに相補的な配列のみを生起するためである。
段階5では、PCRを10増幅サイクル使用して該当2本鎖DNAを定量的に増量する。段階4で記載したように、再アニールしたテスター配列は指数的に増幅され、テスター及びドライバーアニール間で共通して保存された配列は直線的に増幅される。
段階6では、文献に記載されているようにヤエナリヌクレアーゼを使用して1本鎖DNAヌクレアーゼ消化により残存する1本鎖DNAを除去する。
段階7では、ヌクレアーゼ消化後に残存する2本鎖DNAを更に15〜25サイクルPCR増幅する。
最後に段階8ではこれらのDNA産物を制限エンドヌクレアーゼで開裂し、オリゴヌクレオチドアダプターを除去する。その後、これらのDNA産物を(前サイクルのRDAで使用されなかったオリゴヌクレオチドアダプターを使用して段階#3から開始する)次回の増幅にかけるか、又は適切なプラスミドベクターにクローニングし、更に分析する。
上記RDA法は当業者に公知の再現相違分析の変法である。予備接種及び感染性血清源からウイルスクローンを単離するように方法を変形した。これらの変形をテスター及びドライバーDNAのアンプリコンの調製に関して以下に説明する。まず第1に、出発材料は従来報告されているように哺乳動物細胞のゲノムDNAから得た2本鎖DNAではなく、タマリンから得た感染性予備接種生物血液サンプルから抽出した全核酸であった。他の生物サンプル(例えば臓器、組織、胆汁、糞便又は尿)を核酸源として使用してテスター及びドライバーアンプリコンを生成してもよい。第2に、出発核酸の量は文献に記載されているよりも実質的に少量である。第3に、6bpでなく4bp認識部位を有する制限エンドヌクレアーゼを使用した。これは従来技術と実質的に相違する。Lisitsynらの教示によると、RDAはアンプリコン(即ち再現物)の生成がハイブリダイズ(即ち控除)されるDNAの複合度を低下させるという理由で有効である。
従来技術では、6bp認識部位を有する制限酵素を使用してゲノムを分画した。これらの制限エンドヌクレアーゼは約4000bp毎に開裂する。他方、従来技術に記載されているPCR条件では配列の増幅寸法は≦1500bpである。従って、6bp配列を認識する制限酵素で分画されたDNAの複合種(例えばゲノム)のその後のPCR増幅の結果、制限エンドヌクレアーゼ消化後に寸法≦1500bpのDNAのフラクションを含むアンプリコンが生成される。RDAのハイブリダイゼーション段階が有効に行われるためにはDNA複合度のこの低下(10〜50分の1と推定される)が必要であると報告されている。複合度が低下しないならば、控除段階中にテスター中の固有配列は有効にハイブリダイズすることができず、従って、これらの固有配列はRDAの後続PCR段階中に指数的に増幅されない。
RDAを受ける核酸配列の複合度が低下すると、比較的小さいウイルスを単離するためにRDAを使用し難くなる。相互に1.5kb以内に2つの6bp認識部位が存在する確率はかなり低いので、小さい(≦10kb)ウイルスを単離できない恐れがある(表1)。

これに対して本発明者らは、より切断頻度の高い制限エンドヌクレアーゼを使用してRDA被験DNAを分画するならば、小さいウイルスのクローニングにRDAが有用であることを知見した。表1に示す通り、4bp認識部位酵素をベースとするアンプリコンは、約250bp毎に4bp認識部位フラグメントDNAを有する制限エンドヌクレアーゼのように、如何なる小さいウイルスからも数個のフラグメントをほぼ確実に含む。他方、DNA種のほぼ全部は4bp認識制限エンドヌクレアーゼによる消化後に≦1500bpで即ちPCR増幅されるので、アンプリコンはその生成起源である核酸源と同程度の複合度であると思われる。相対ウイルス配列コピー数は任意の特定又は内在配列コピー数よりも多いと予想されるので、テスターアンプリコン中に存在する固有ウイルス配列はハイブリダイゼーション段階(上記段階3)中に2本鎖分子を形成できるはずである。従って、これらの配列は上記のように指数的に増幅される。相対ウイルス配列コピー数がバックグラウンド又は内在核酸配列コピー数に近づくので、重複6bp配列を認識し(例えばBstYI又はHincII)且つ約1000bp毎に開裂する制限エンドヌクレアーゼを使用するか、又は6bp配列を認識する数個の制限エンドヌクレアーゼを同時に使用して、PCRによる増幅前にDNAを分画することができると推論される。こうして、テスターアンプリコンからウイルス配列を排除する危険を最小限にしながら、RDAを受けるアンプリコンの複合度を適度に低下させることができる。この方法の有用性は、本明細書中に記載する感染性タマリン血漿からのHGBV配列のクローニングにより立証される。
HGBV免疫反応性エピトープを同定するための免疫スクリーニング
以下に記載するような免疫スクリーニングも、HGBV配列を同定する付加的手段であった。急性又は慢性HGBV感染を有する下記パラメーター内で基準に合致する個体からの個々の血清、血漿又は肝臓ホモジネート又はそのプールを使用してウイルス粒子を単離する。これらの粒子から単離した核酸をゲノムライブラリー及び/又はウイルスゲノムに対するcDNAライブラリーの構築で鋳型として使用する。推定HGBV粒子を単離し、当業者に公知のλ−gt11又は類似のシステムでゲノム及び/又はcDNAライブラリーを構築するために使用する手順を以下に説明する。λ−gt11はβ−ガラクトシダーゼとの融合ポリペプチドとして挿入cDNAを特異的に発現させ、規定抗原に対する特異抗血清で以って多数の組換えファージをスクリーニングするために開発されたベクターである。cDNAを含むcDNAプールから生成されるλ−gt11 cDNAライブラリーを、非A、非B、非C、非D及び非E肝炎既往個体からの血清と特異的に結合することが可能なコード化エピトープについてスクリーニングする。V.Hunyhら,D.Glover編,DNA Cloning Techniques; A Practical Approach, IRL Press,Oxford,England,pp.49−78(1985)を参照されたい。約106〜107個のファージをスクリーニングして陽性ファージを同定、精製後、HGBV物質に先に感染した各個体からの血清と結合する特異性を試験する。下記基準に合致する患者に由来し且つこれらの基準に合致しない患者には存在しない血清又は血漿と選択的に結合するファージがその後の試験に好適である。オーバーラップする核酸配列を単離する方法を使用することにより、付加的な上流及び下流HGBV配列を含むクローンが得られる。単離クローン内でコードされるHGBV核酸配列のヌクレオチド配列を分析し、複合配列が1つの長い連続ORFを含むか否かを決定する。
以下に記載するような免疫スクリーニングも、HGBV配列を同定する付加的手段であった。急性又は慢性HGBV感染を有する下記パラメーター内で基準に合致する個体からの個々の血清、血漿又は肝臓ホモジネート又はそのプールを使用してウイルス粒子を単離する。これらの粒子から単離した核酸をゲノムライブラリー及び/又はウイルスゲノムに対するcDNAライブラリーの構築で鋳型として使用する。推定HGBV粒子を単離し、当業者に公知のλ−gt11又は類似のシステムでゲノム及び/又はcDNAライブラリーを構築するために使用する手順を以下に説明する。λ−gt11はβ−ガラクトシダーゼとの融合ポリペプチドとして挿入cDNAを特異的に発現させ、規定抗原に対する特異抗血清で以って多数の組換えファージをスクリーニングするために開発されたベクターである。cDNAを含むcDNAプールから生成されるλ−gt11 cDNAライブラリーを、非A、非B、非C、非D及び非E肝炎既往個体からの血清と特異的に結合することが可能なコード化エピトープについてスクリーニングする。V.Hunyhら,D.Glover編,DNA Cloning Techniques; A Practical Approach, IRL Press,Oxford,England,pp.49−78(1985)を参照されたい。約106〜107個のファージをスクリーニングして陽性ファージを同定、精製後、HGBV物質に先に感染した各個体からの血清と結合する特異性を試験する。下記基準に合致する患者に由来し且つこれらの基準に合致しない患者には存在しない血清又は血漿と選択的に結合するファージがその後の試験に好適である。オーバーラップする核酸配列を単離する方法を使用することにより、付加的な上流及び下流HGBV配列を含むクローンが得られる。単離クローン内でコードされるHGBV核酸配列のヌクレオチド配列を分析し、複合配列が1つの長い連続ORFを含むか否かを決定する。
本明細書に記載するようなHGBV配列から回収される配列(及びその相補体)及びその配列又は任意部分は、合成法を使用して調製してもよいし、本明細書に記載すると同様の方法を使用して合成法を部分配列の回収と組み合わせることにより調製してもよい。この記載は、完全HGBVゲノムに対応するゲノム又はcDNA配列を単離することが可能な方法に関する。しかしながら、これらの配列を単離するための他の方法も当業者に自明であり、本発明の範囲内であるとみなされる。
菌株寄託
HGBV核酸配列ライブラリーからの複製株(クローン2、4、10、16、18、23及び50)はブダペスト条約に基づき1994年2月10日付けで12301 Parklawn Drive,Rockville,Maryland 20852に所在のAmerican Type Culture Collectionに寄託し、寄託日から30年間、最新の試料分譲請求から5年間、又は米国特許の存続期間のうちのいずれか最長の期間保管される。上記寄託株及び本明細書に記載する他の全寄託材料は便宜の目的で呈示するに過ぎず、本明細書中の教示に鑑みて本発明を実施するために必要ではない。全寄託材料中のHGBV cDNA配列は参考資料として本明細書の一部とする。プラスミドは以下のA.T.C.C.寄託番号を付託された。クローン2はA.T.C.C.寄託番号69556、クローン4はA.T.C.C.寄託番号69557、クローン10はA.T.C.C.寄託番号69558、クローン16はA.T.C.C.寄託番号69559、クローン18はA.T.C.C.寄託番号69560、クローン23はA.T.C.C.寄託番号69561、クローン50はA.T.C.C.寄託番号69562を付与された。
HGBV核酸配列ライブラリーからの複製株(クローン2、4、10、16、18、23及び50)はブダペスト条約に基づき1994年2月10日付けで12301 Parklawn Drive,Rockville,Maryland 20852に所在のAmerican Type Culture Collectionに寄託し、寄託日から30年間、最新の試料分譲請求から5年間、又は米国特許の存続期間のうちのいずれか最長の期間保管される。上記寄託株及び本明細書に記載する他の全寄託材料は便宜の目的で呈示するに過ぎず、本明細書中の教示に鑑みて本発明を実施するために必要ではない。全寄託材料中のHGBV cDNA配列は参考資料として本明細書の一部とする。プラスミドは以下のA.T.C.C.寄託番号を付託された。クローン2はA.T.C.C.寄託番号69556、クローン4はA.T.C.C.寄託番号69557、クローン10はA.T.C.C.寄託番号69558、クローン16はA.T.C.C.寄託番号69559、クローン18はA.T.C.C.寄託番号69560、クローン23はA.T.C.C.寄託番号69561、クローン50はA.T.C.C.寄託番号69562を付与された。
HGBV核酸配列ライブラリーからの複製株(クローン11、13、48及び119)はブダペスト条約に基づき1994年4月29日付けで12301 Parklawn Drive,Rockville,Maryland 20852に所在のAmerican Type Culture Collectionに寄託し、寄託日から30年間、最新の試料分譲請求から5年間、又は米国特許の存続期間のうちのいずれか最長の期間保管される。上記寄託株及び本明細書に記載する他の全寄託材料は便宜の目的で呈示するに過ぎず、本明細書中の教示に鑑みて本発明を実施するために必要ではない。全寄託材料中のHGBV cDNA配列は参考資料として本明細書の一部とする。プラスミドは以下のA.T.C.C.寄託番号を付託された。クローン11はA.T.C.C.寄託番号69613、クローン13はA.T.C.C.寄託番号69611、クローン48はA.T.C.C.寄託番号69610、クローン119はA.T.C.C.寄託番号69612を付与された。
HGBV核酸配列ライブラリーからの付加的株(クローン4−B1.1、66−3A1.49、70−3A1.37及び78−1C1.17)はブダペスト条約に基づき1994年7月28日付けで12301 Parklawn Drive,Rockville,Maryland 20852に所在のAmerican Type Culture Collectionに寄託し、寄託日から30年間、最新の試料分譲請求から5年間、又は米国特許の存続期間のうちのいずれか最長の期間保管される。上記寄託株及び本明細書に記載する他の全寄託材料は便宜の目的で呈示するに過ぎず、明細書中の教示に鑑みて本発明を実施するために必要ではない。全寄託材料中のHGBV cDNA配列は参考資料として本明細書の一部とする。プラスミドは以下のA.T.C.C.寄託番号を付与された。クローン4−B1.1はA.T.C.C.寄託番号69666、クローン66−3A1.49はA.T.C.C.寄託番号69665、クローン70−3A1.37はA.T.C.C.寄託番号69664、クローン78−1C1.17はA.T.C.C.寄託番号69663を付与された。
クローンpHGBV−Cクローン#1はブダペスト条約に基づき1994年11月8日付けで12301 Parklawn Drive,Rockville,Maryland 20852に所在のAmerican Type Culture Collectionに寄託し、寄託日から30年間、最新の試料分譲請求から5年間、又は米国特許の存続期間のうちのいずれか最長の期間保管される。上記寄託株及び本明細書に記載する他の全寄託材料は便宜の目的で呈示するに過ぎず、本明細書上の教示に鑑みて本発明を実施するために必要ではない。pHGBV−Cクローン#1はA.T.C.C.寄託番号69711を付与された。全寄託材料中のHGBV cDNA配列は参考資料として本明細書の一部とする。
ウイルスポリペプチド及びフラグメントの調製
核酸配列を入手できることにより、いずれかの鎖でコードされるポリペプチドの抗原活性領域をコードする発現ベクターを構築することができる。これらの抗原活性領域は、例えばポリヌクレオチド結合タンパク質、ポリヌクレオチドポリメラーゼ、及びウイルス粒子の複製及び/又はアセンブリに必要な他のウイルスタンパク質を含むウイルスの非構造領域以外に、例えばエンベロープ(コート)又はコア抗原を含むウイルスの構造領域から誘導され得る。所望のポリペプチドをコードするフラグメントは慣用制限消化又は合成法によりゲノム又はcDNAクローンから得られ、例えばβ−ガラクトシダーゼ(β−gal)、スーパーオキシドジスムターゼ(SOD)又はCMP−KDOシンテターゼ(CKS)等の融合配列の部分を含み得るベクターに連結される。SODの融合配列を含むポリペプチドの製造に有用な方法及びベクターは1986年10月1日付け公開EPO第0196056号に記載されており、CKSの融合配列を含むポリペプチドの製造に有用な方法及びベクターは1989年9月13日付け公開EPO第0331961号に記載されている。いずれかの方向の鎖においてオープンリーディングフレームを含む核酸配列の任意の所望部分は成熟又は融合タンパク質等の組換えタンパクとして得られ、あるいはHGBVゲノム又はcDNAでコードされるポリペプチドを化学的合成により提供することもできる。
核酸配列を入手できることにより、いずれかの鎖でコードされるポリペプチドの抗原活性領域をコードする発現ベクターを構築することができる。これらの抗原活性領域は、例えばポリヌクレオチド結合タンパク質、ポリヌクレオチドポリメラーゼ、及びウイルス粒子の複製及び/又はアセンブリに必要な他のウイルスタンパク質を含むウイルスの非構造領域以外に、例えばエンベロープ(コート)又はコア抗原を含むウイルスの構造領域から誘導され得る。所望のポリペプチドをコードするフラグメントは慣用制限消化又は合成法によりゲノム又はcDNAクローンから得られ、例えばβ−ガラクトシダーゼ(β−gal)、スーパーオキシドジスムターゼ(SOD)又はCMP−KDOシンテターゼ(CKS)等の融合配列の部分を含み得るベクターに連結される。SODの融合配列を含むポリペプチドの製造に有用な方法及びベクターは1986年10月1日付け公開EPO第0196056号に記載されており、CKSの融合配列を含むポリペプチドの製造に有用な方法及びベクターは1989年9月13日付け公開EPO第0331961号に記載されている。いずれかの方向の鎖においてオープンリーディングフレームを含む核酸配列の任意の所望部分は成熟又は融合タンパク質等の組換えタンパクとして得られ、あるいはHGBVゲノム又はcDNAでコードされるポリペプチドを化学的合成により提供することもできる。
融合又は成熟形態のいずれかに関係なく、また分泌を可能にするシグナル配列を含むか否かに関係なく、所望のポリペプチドをコードする核酸配列を任意の適当な宿主に適切な発現ベクターに連結することができる。当該技術分野では真核及び原核両者の宿主系を使用して組換えタンパク質を形成することができ、本明細書にはこれらの宿主のいくつかを挙げる。次に溶解細胞又は培地からポリペプチドを単離し、所期用途に必要な程度まで精製する。精製は当業者に公知の方法により実施することができ、特に塩析、イオン交換樹脂クロマトグラフィー、アフィニティークロマトグラフィー、遠心分離等を使用することができる。このようなポリペプチドは診断薬として又は受動的免疫療法に使用することができる。更に、これらのポリペプチドに対する抗体は、HGBV粒子を単離及び同定するために有用である。HGBV抗原はHGBV粒子からも単離することができる。これらウイルス粒子は組織培養物又は感染個体中のHGBV感染細胞中で増殖させることができる。
抗原性ポリペプチドの調製及び固相との結合
ポリペプチドの抗原性領域又はフラグメントは比較的小さく、通常約8〜10アミノ酸長又はそれ以下である。5アミノ酸程度のフラグメントでも抗原性領域を特徴付けることができる。これらのセグメントはHGBV抗原の領域に対応し得る。HGBVゲノム又はcDNA配列を基本として使用することにより、HGBVポリペプチドの短いセグメントをコードする核酸配列を融合タンパク質又は単離ポリペプチドとして組換えにより発現させることができる。これらの短いアミノ酸配列は、化学的合成によっても得られる。化学的に合成された小さいポリペプチドは、得られた合成ポリペプチドが適正なエピトープを提供するように適正に配置されており且つ抗原性となるには小さ過ぎる場合に適切なキャリヤー分子に結合され得る。結合方法は当業者に公知であり、非限定的な例としては、N−スクシンイミジル−3−(2−ピリジルチオ)プロピオネート(SPDP)及びスクシンイミジル−4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート(SMCC)を使用する。システイン残基を加えることにより、スルフヒドリル基をもたないポリペプチドを修飾することができる。これらの物質は、該物質と一方のタンパク質上のペプチドシステイン残基の間にジスルフィド結合を生成し、他方のタンパク質中のリジン上のε−アミノ又は他の遊離アミノ基を介してアミド結合を生成する。このような種々のジスルフィド/アミド形成物質は公知である。他の2官能カップリング剤はジスルフィド結合でなくチオエステルを形成する。これらのチオエーテル形成剤の多くは市販されており、当業者に公知である。そのカルボキシ基は、スクシンイミド又は1−ヒドロキシ−2−ニトロ−4−スルホン酸ナトリウム塩と結合させることにより活性化することができる。宿主に有害な抗体の産生をそれ自体誘導しないものであれば、任意のキャリヤーを使用することができる。適切なキャリヤーとしては、特にタンパク質、多糖類(例えばラテックス官能化セファロース、アガロース、セルロース、セルロースビーズ)、ポリマーアミノ酸(例えばポリグルタミン酸、ポリリシン、アミノ酸コポリマー)及び不活性ウイルス粒子を挙げることができる。タンパク質基質の例としては、血清アルブミン、アオガイヘモシアニン、イムノグロブリン分子、チログロブリン、オボアルブミン、破傷風毒素及び当業者に公知の他のタンパク質を挙げることができる。
ポリペプチドの抗原性領域又はフラグメントは比較的小さく、通常約8〜10アミノ酸長又はそれ以下である。5アミノ酸程度のフラグメントでも抗原性領域を特徴付けることができる。これらのセグメントはHGBV抗原の領域に対応し得る。HGBVゲノム又はcDNA配列を基本として使用することにより、HGBVポリペプチドの短いセグメントをコードする核酸配列を融合タンパク質又は単離ポリペプチドとして組換えにより発現させることができる。これらの短いアミノ酸配列は、化学的合成によっても得られる。化学的に合成された小さいポリペプチドは、得られた合成ポリペプチドが適正なエピトープを提供するように適正に配置されており且つ抗原性となるには小さ過ぎる場合に適切なキャリヤー分子に結合され得る。結合方法は当業者に公知であり、非限定的な例としては、N−スクシンイミジル−3−(2−ピリジルチオ)プロピオネート(SPDP)及びスクシンイミジル−4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート(SMCC)を使用する。システイン残基を加えることにより、スルフヒドリル基をもたないポリペプチドを修飾することができる。これらの物質は、該物質と一方のタンパク質上のペプチドシステイン残基の間にジスルフィド結合を生成し、他方のタンパク質中のリジン上のε−アミノ又は他の遊離アミノ基を介してアミド結合を生成する。このような種々のジスルフィド/アミド形成物質は公知である。他の2官能カップリング剤はジスルフィド結合でなくチオエステルを形成する。これらのチオエーテル形成剤の多くは市販されており、当業者に公知である。そのカルボキシ基は、スクシンイミド又は1−ヒドロキシ−2−ニトロ−4−スルホン酸ナトリウム塩と結合させることにより活性化することができる。宿主に有害な抗体の産生をそれ自体誘導しないものであれば、任意のキャリヤーを使用することができる。適切なキャリヤーとしては、特にタンパク質、多糖類(例えばラテックス官能化セファロース、アガロース、セルロース、セルロースビーズ)、ポリマーアミノ酸(例えばポリグルタミン酸、ポリリシン、アミノ酸コポリマー)及び不活性ウイルス粒子を挙げることができる。タンパク質基質の例としては、血清アルブミン、アオガイヘモシアニン、イムノグロブリン分子、チログロブリン、オボアルブミン、破傷風毒素及び当業者に公知の他のタンパク質を挙げることができる。
HGBVエピトープを含むハイブリッド粒子免疫原の調製
HGBVエピトープの免疫原性は、粒子形成タンパク質(例えばHBV表面抗原にあるような)融合又は結合した哺乳動物又は酵母系で調製することにより強化することもできる。粒子形成タンパク質をコードする配列にHGBVエピトープを直接結合した構築物は、HGBVエピトープに対して免疫原性のハイブリッドを生成する。更に、調製されるHGBVに対して特異的なエピトープを含む全てのベクターは、異なる程度の免疫原性を有する。HGBV配列を含む粒子形成タンパク質から構築される粒子はHGBV及びHBVに対して免疫原性がある。
HGBVエピトープの免疫原性は、粒子形成タンパク質(例えばHBV表面抗原にあるような)融合又は結合した哺乳動物又は酵母系で調製することにより強化することもできる。粒子形成タンパク質をコードする配列にHGBVエピトープを直接結合した構築物は、HGBVエピトープに対して免疫原性のハイブリッドを生成する。更に、調製されるHGBVに対して特異的なエピトープを含む全てのベクターは、異なる程度の免疫原性を有する。HGBV配列を含む粒子形成タンパク質から構築される粒子はHGBV及びHBVに対して免疫原性がある。
B型肝炎表面抗原はS.cerevisiae及び哺乳動物細胞中で形成されて粒子に結合することが確認され、これらの粒子の形成はモノマーサブユニットの免疫原性を強化することが報告されている。P.Valenzuelaら,Nature 298:334(1982); P.Valenzuelaら,I.Millmanら編,Hepatitis B,Plenum Press,pp.225−236(1984)。構築物はHBsAgの免疫優性エピトープを含み得る。このような構築物は酵母で発現可能であることが報告されており、酵母発現のための異種ウイルス配列を含むハイブリッドが開示されている。例えばEPO第174,444号及びEPO第174,261号を参照されたい。これらの構築物はチャイニーズハムスター卵巣(CHO)細胞等の哺乳動物細胞で発現され得ることも報告されている。Michelleら,International Symposium on Viral Hepatitis,1984を参照されたい。HGBVにおいては粒子形成タンパク質をコードする配列の部分を、HGBVエピトープをコードするコドンで置換することができる。この置換では、酵母又は哺乳動物中で免疫原性粒子を形成するための単位の凝集を媒介するために不要な領域を削除し、こうしてHGBVエピトープとの競合から付加的なHGBV抗原部位を排除することができる。
ワクチン製造
ワクチンは、HGBV核酸配列又は該核酸配列に対応するHGBVゲノムに由来する1種以上の免疫原性ポリペプチド又は核酸から製造することができる。ワクチンはHGBVのエピトープを含む組換えポリペプチドを含み得る。これらのポリペプチドは細菌、酵母又は哺乳動物細胞中で発現させてもよいし、あるいはウイルス調製物から単離してもよい。種々の構造タンパク質が防御抗HGBV抗体を誘導するHGBVのエピトープを含み得ることも予想される。こうして、これらのワクチンの製造時には合成ペプチドも使用することができる。従って、HGBVの少なくとも1個のエピトープを含むポリペプチドをHGBVワクチン中で単独又は組み合わせて使用することができる。更に、非構造タンパク質及び構造タンパク質は、中和抗体を産生しないとしても、ウイルス病原性に対する防御を提供し得ると予想される。
ワクチンは、HGBV核酸配列又は該核酸配列に対応するHGBVゲノムに由来する1種以上の免疫原性ポリペプチド又は核酸から製造することができる。ワクチンはHGBVのエピトープを含む組換えポリペプチドを含み得る。これらのポリペプチドは細菌、酵母又は哺乳動物細胞中で発現させてもよいし、あるいはウイルス調製物から単離してもよい。種々の構造タンパク質が防御抗HGBV抗体を誘導するHGBVのエピトープを含み得ることも予想される。こうして、これらのワクチンの製造時には合成ペプチドも使用することができる。従って、HGBVの少なくとも1個のエピトープを含むポリペプチドをHGBVワクチン中で単独又は組み合わせて使用することができる。更に、非構造タンパク質及び構造タンパク質は、中和抗体を産生しないとしても、ウイルス病原性に対する防御を提供し得ると予想される。
上記に鑑み、HGBVに対する多価抗体は1種以上の構造タンパク質及び/又は1種以上の非構造タンパク質を含み得る。これらのワクチンは例えば組換えHGBVポリペプチド及び/又はウイルス粒子から単離されたポリペプチド及び/又は合成ペプチドから構成され得る。これらの免疫原エピトープは組み合わせて使用することができ、即ち組換えタンパク質、合成ペプチド及び/又はウイルス粒子から単離されたポリペプチドの混合物として使用することができ、これらのワクチンは同一又は異なる時刻に投与することができる。更に、ワクチンに不活化HGBVを使用することも可能であり得る。このような不活化はウイルス溶解物を調製してもよいし、肝炎様ウイルスの不活化を生じることが当業者に知られている他の手段、例えば有機溶媒もしくは界面活性剤による処理、又はホルマリン処理により実施してもよい。本発明では弱毒HGBV株調製物も開示する。HGBVにおけるタンパク質には他の公知ウイルスと交差反応できるものもあり、従って、HGBVと他のウイルスとの間に共有エピトープが存在し、これらの病原物質により生じる疾患の1種以上に対する防御抗体をもたらすと予想される。この確信に基づいて多重目的ワクチンを設計できることが予想される。
少なくとも1種の免疫原性ペプチドを活性成分として含むワクチンの調製は当業者に公知である。典型的には、このようなワクチンは溶液又は懸濁液としての注射剤として調製される。注射前に液体に溶解又は懸濁するのに適切な固体形態も製造可能である。調製物を乳化してもよいし、タンパク質をリポソームに封入してもよい。活性免疫原性成分は多くの場合には活性成分と適合可能な医薬的に許容可能な賦形剤と混合する。適切な賦形剤の非限定的な例としては、水、食塩水、デキストロース、グリセロール、エタノール等が挙げられ、種々の量のこれらの賦形剤を組み合わせて使用してもよい。ワクチンは更に、湿潤剤、乳化剤、pH緩衝剤及び/又はワクチンの効力を強化するアジュバント等の少量の補助物質も含有し得る。例えば、このようなアジュバントとしては、水酸化アルミニウム、N−アセチル−ムラミル−L−トレオニル−D−イソグルタミン(thr−DMP)、N−アセチル−ノルムラミル−L−アラニル−D−イソグルタミン(CGP 11687、別称nor−MDP)、N−アセチルムラミル−L−アラニル−D−イソグルタミニル−L−アラニン−2−(1’,2’ジパルミトイル−sn−グリセロ−3−ヒドロキシホスホリルオキシ)エチルアミン(CGP 19835A、別称MTP−PE)及びRIBI(MPL+TDM+CWS)の2%スクアレン/Tween−80(登録商標)エマルジョンが挙げられる。アジュバントの効力は、同様に種々のアジュバントから構成されるワクチン中のポリペプチドを投与した結果生じたHGBV抗原配列を含む免疫原性ポリペプチドに対する抗体の量を測定することにより決定することができる。
ワクチンは通常、静脈内又は筋肉内注射により投与される。他の投与方法に適切な別の製剤には座薬があり、場合によっては経口製剤でもよい。座薬の場合、慣用バインダー及びキャリヤーの非限定的な例としてはポリアルキレングリコール又はトリグリセリドが挙げられる。このような座薬は約0.5%〜約10%、好ましくは約1%〜約2%の活性成分を含有する混合物から形成することができる。経口製剤は例えば医薬グレードのマンニトール、ラクトース、澱粉、ステアリン酸マグネシウム、ナトリウムサッカリン、セルロース、炭酸マグネシウム等のような一般に使用される賦形剤を含有する。これらの組成物は溶液、懸濁液、タブレット、ピル、カプセル、徐放製剤又は散剤の形態をとることでがき、約10%〜約95%、好ましくは約25%〜約70%の活性成分を含有し得る。
ワクチン中で使用するタンパク質は中性又は塩形態としてワクチンに配合され得る。医薬的に許容可能な塩としては、(ペプチドの遊離アミノ基と共に形成され且つ)無機酸(例えば塩酸又はリン酸)又は有機酸(酢酸、蓚酸、酒石酸、マレイン酸)と共に形成される酸付加塩や、当業者に公知の他の塩を使用することができる。遊離カルボキシル基と共に形成される塩は無機塩基(例えば水酸化ナトリウム、水酸化カリウム、水酸化アンモニウム、水酸化カルシウム、水酸化鉄(III)等)及び有機塩基(例えばイソプロピルアミン、トリメチルアミン、2−エチルアミノエタノール、ヒスチジン、プロカイン)及び当業者に公知の他の塩基から誘導され得る。
ワクチンは剤形に適合可能な方法で且つ予防及び/又は治療上有効な量を投与される。投与量は一般に1回に抗原約5μg〜約250μgであり、被投与患者、患者の免疫系の抗体合成能力及び所望の防御度に依存する。投与するために必要な活性成分の厳密な量は処方医の判断にも依存し、患者毎に異なる。ワクチンは単一投与スケジュールで投与してもよいし、多重投与スケジュールで投与してもよい。多重投与では、1〜10回に分けて一次ワクチン接種過程を実施した後、免疫応答を維持及び/又は強化するために必要な期間後、例えば1〜4カ月後に二次投与を実施し、個体が必要とする場合には更に数カ月後にも投与する。投与計画は少なくとも一部には個体の必要により決定され、処方医の判断による。免疫原性HGBV抗原を含有するワクチンは他の免疫調節剤、例えば免疫グロブリンと併用投与し得ることが予想される。
HGBVエピトープに対する抗体の製造
本明細書中に記載するように調製した免疫原性ペプチドを使用してポリクローナル又はモノクローナル抗体を製造する。ポリクローナル抗体を製造する際には、選択した哺乳動物(例えばマウス、ウサギ、ヤギ、ウマ等)を少なくとも1種のHGBVエピトープを有する免疫原性ポリペプチドで免疫感作する。免疫感作した動物からの血清を適当なインキュベーション時間後に収集し、公知手順に従って処理する。HGBVエピトープに対するポリクローナル抗体を含有する血清が他の抗原に対する抗体を含有する場合には、例えばイムノアフィニティークロマトグラフィーによりポリクローナル抗体を精製することができる。ポリクローナル抗体を製造及び処理するための方法は当業者に公知であり、特にMayerとWalker編,Immunochemical Methods In Cell and Molecular Biology,Academic Press,London (1987)に記載されている。ポリクローナル抗体は先にHGBVに感染した哺乳動物からも得られる。アフィニティークロマトグラフィーを使用してHGBVに感染した個体の血清からHGBVエピトープに対する抗体を精製するための方法の1例をここに記載する。
本明細書中に記載するように調製した免疫原性ペプチドを使用してポリクローナル又はモノクローナル抗体を製造する。ポリクローナル抗体を製造する際には、選択した哺乳動物(例えばマウス、ウサギ、ヤギ、ウマ等)を少なくとも1種のHGBVエピトープを有する免疫原性ポリペプチドで免疫感作する。免疫感作した動物からの血清を適当なインキュベーション時間後に収集し、公知手順に従って処理する。HGBVエピトープに対するポリクローナル抗体を含有する血清が他の抗原に対する抗体を含有する場合には、例えばイムノアフィニティークロマトグラフィーによりポリクローナル抗体を精製することができる。ポリクローナル抗体を製造及び処理するための方法は当業者に公知であり、特にMayerとWalker編,Immunochemical Methods In Cell and Molecular Biology,Academic Press,London (1987)に記載されている。ポリクローナル抗体は先にHGBVに感染した哺乳動物からも得られる。アフィニティークロマトグラフィーを使用してHGBVに感染した個体の血清からHGBVエピトープに対する抗体を精製するための方法の1例をここに記載する。
HGBVエピトープに対するモノクローナル抗体も当業者により製造することができる。このような抗体を製造するための一般方法は周知であり、例えばKohlerとMilstein,Nature 256:494(1975)に記載されており、J.G.R.Hurrel編,Monoclonal Hybridoma Antibodies: Techniques and Applications,CRC Press Inc.,Boco Raton,FL(1982)に考察されており、L.T.Mimmsら,Virology 176:604−619(1990)により教示されている。不死化抗体産生細胞系は細胞融合により作成することができ、オンコジーンDNAによるBリンパ球の直接形質転換又はエプスタイン−バールウイルスによるトランスフェクション等の他の方法により作成することもできる。同様にM.Schreierら,Hybridoma Techniques,Scopes (1980) Protein Purification,Principles and Practice,第2版,Springer−Verlag,New York(1984); Hammerlingら,Monoclonal Antibodies and T−Cell Hybridomas (1981); Kennetら,Monoclonal Antibodies (1980)を参照されたい。モノクローナル抗体の使用及び技術の例は米国特許出願第748,292号、748,563号、610,175号、648,473号、648,477号及び648,475号に開示されている。
こうして開発されたHGBVエピトープに対するモノクローナル及びポリクローナル抗体は診断及び予後用途に有用であり、更に中和抗体は受動的免疫療法で有用である。モノクローナル抗体は特に抗イディオタイプ抗体を製造するために使用することができる。これらの抗イディオタイプ抗体は防御が所望される感染物質の抗原の「内部像」を有するイムノグロブリンである。例えば、A.Nisonoffら,Clin.Immunol.Immunopath.21:397−406(1981)及びDreesmanら,J.Infect.Dis.151:761(1985)を参照されたい。このようなイディオタイプ抗体を誘導するための方法は当業者に公知であり、例えばGrychら,Nature 316:74(1985); MacNamaraら,Science 226:1325(1984);及びUytdehaagら,J.Immunol.134:1225(1985)に例示されている。これらの抗イディオタイプ抗体はHGBV感染の治療及びHGBV抗原の免疫原性領域の解明にも有用であり得る。
診断オリゴヌクレオチドブローブ及びキット
単離したHGBV核酸配列の所定部分を基材として用いて、HGBVゲノムとハイブリダイズし、ウイルス剤の同定、ウイルスゲノムの更なる特性分析、及び羅患した個体でのウイルス検出に有用である約8ヌクレオチド以上のオリゴマーを切り出し又は合成により産生することができる。HGBVポリヌクレオチド用の天然又は誘導プローブは、ハイブリダイゼーションによる単一ウイルス配列の検出を可能とする長さである。6〜8ヌクレオチドが処理可能な長さであり得るが、10〜12ヌクレオチドの配列が好ましく、約20ヌクレオチドの配列が最も好ましいものであり得る。これらの配列が異質性に欠けた領域から誘導されることが好ましい。これらのプローブは、自動化オリゴヌクレオチド合成方法を含む常用の標準的な方法を用いて製造することができる。HGBVゲノムの任意の単一部分の相補体は満足すべきものであろう。完全相補性は、断片長さが増せば不要であり得るが、プローブとして使用するには望ましい。
単離したHGBV核酸配列の所定部分を基材として用いて、HGBVゲノムとハイブリダイズし、ウイルス剤の同定、ウイルスゲノムの更なる特性分析、及び羅患した個体でのウイルス検出に有用である約8ヌクレオチド以上のオリゴマーを切り出し又は合成により産生することができる。HGBVポリヌクレオチド用の天然又は誘導プローブは、ハイブリダイゼーションによる単一ウイルス配列の検出を可能とする長さである。6〜8ヌクレオチドが処理可能な長さであり得るが、10〜12ヌクレオチドの配列が好ましく、約20ヌクレオチドの配列が最も好ましいものであり得る。これらの配列が異質性に欠けた領域から誘導されることが好ましい。これらのプローブは、自動化オリゴヌクレオチド合成方法を含む常用の標準的な方法を用いて製造することができる。HGBVゲノムの任意の単一部分の相補体は満足すべきものであろう。完全相補性は、断片長さが増せば不要であり得るが、プローブとして使用するには望ましい。
診断試薬として使用するときには、血液又は血清のような分析すべき試験試料を処理してもよく、例えば試料内に含まれる核酸を抽出する。試料から得られた核酸をゲル電気泳動又は他のサイズ分離技術に付してもよいし、核酸試料をサイズ分離せずにドットブロットしてもよい。次いで、プローブを標識する。ラベルをプローブに付着させるための適切なラベル及び方法は当業界では公知であり、ニックトランスレーション又はキナーゼ反応(kinasing)により取り込まれる放射性ラベル、ビオチン、蛍光及び化学発光プローブが含まれるが、これらに限定はされない。これらのラベルの例は多数本明細書に開示されている。次いで、試料から抽出した核酸を、適切な厳密度のハイブリダイゼーション条件下にて標識プローブで処理する。
プローブをHGBVゲノムに対して完全に相補的にすることができる。従って、通常は偽陽性を回避するために厳密度の高い条件が望ましい。しかしながら、厳密度の高い条件は、プローブが異質性に欠けたHGBVゲノムの領域に相補的である場合にのみ使用すべきである。ハイブリダイゼーションの厳密度は、洗浄手順中の幾つかの要素(例えば温度、イオン強度、時間及びホルムアミド濃度)により決定される。例えばJ.Sambrook(上掲)を参照されたい。ハイブリダイゼーションは幾つかの様々な技術により実施することができる。増幅は、例えばリガーゼ連鎖反応(LCR)、ポリメラーゼ連鎖反応(PCR)、Q−βレプリカーゼ、NASBA等により実施することができる。
HGBVゲノム配列は、例えば約102〜103個の配列/mlの比較的低レベルで、感染した個体の血清中に存在し得ると考えられる。このレベルでは、ハイブリダイゼーションアッセイで、リガーゼ連鎖反応又はポリメラーゼ連鎖反応のような増幅技術を使用する必要があり得る。このような技術は当業界では公知である。例えば、“バイオブリッジ”系は末端デオキシヌクレオチドトランスフェラーゼを用いて、非改変3’−ポリ−dT−テールを核酸プローブに付加する(Enzo Biochem.Corp.)。ポリ−dT−テールを有するプローブを標的ヌクレオチド配列にハイブリダイズし、次いでビオチン改変ポリ−Aにハイブリダイズする。更には、被分析物質を、酵素標識オリゴヌクレオチドと相補的な一本鎖DNAプローブにアニーリングし、得られたテール付き二本鎖を酵素標識オリゴヌクレオチドにハイブリダイズするDNAハイブリダイゼーションアッセイがヨーロッパ特許第124221号に記載されている。ヨーロッパ特許第204510号は、被分析物質DNAを、テール(例えばポリ−dT−テール)を有するプローブ、及びプローブのテールにハイブリダイズする配列(例えばポリ−A配列)を有し、複数の標識鎖と結合し得るアンプリファイア鎖と接触させるDNAハイブリダイゼーションアッセイを記載している。この技術はまず、血清中の標的HGBV配列を約106個の配列/mlに増幅することからなり得る。これは、Saiki等、Nature 324:163(1986)に記載の方法に従って実施され得る。次いで、増幅した配列を、当業界では公知のようなハイブリダイゼーションアッセイを用いて検出することができる。標識され得るプローブ核酸配列を含む診断キットにプローブをパッケージすることができる。あるいは、プローブを標識せずに、標識用成分をキットに含ませてもよい。キットは更に、特定のハイブリダイゼーションプロトコルに必要な又は望ましい他の適切にパッケージされた試薬や材料、例えばアッセイ実施のための標準物や指示書を含み得る。
本発明で使用できる他の公知の増幅方法には、PNAS USA 87:1874−1878(1990)に教示され、Nature:350(No.6313):91−92(1991)でも議論されているいわゆる“NASBA”又は“3SR”技術や、Q−βレプリカーゼが含まれるが、これらに限定はされない。
本明細書に記載する試薬を用いて、蛍光インシトウハイブリダイゼーション(“FISH”)を実施することもできる。インシトウハイブリダイゼーションは、核酸ハイブリダイゼーション法により形態の損なわれていない組織、細胞又は染色体を採取して、特殊な遺伝情報の存在や、個体細胞内での前記情報の特定位置を実証することからなる。これは細胞の均質化及び標的配列の抽出を必要としないので、細胞集団中での配列の正確な位置や分布が判明する。インシトウハイブリダイゼーションは、細胞中に集中して含まれる問題の配列を同定し得、更には該配列を含む異質細胞集団中の細胞のタイプやフラクションを同定し得る。DNA及びRNAを同一のアッセイ試薬で検出することができる。PNAをFISH法で使用して、増幅せずに標的を検出することができる。シグナルの増加が望ましければ、多数の発蛍光団を使用してシグナルを増し、方法の感度を高めることができる。多数のオリゴヌクレオチドを用いる一工程法又は従来の多工程法を含む様々なFISH法が知られている。これらのタイプの方法を様々な手段(例えばフローサイトメトリー及び画像分析)により自動化できることは本発明の範囲内である。
イムノアッセイ及び診断キット
HGBV抗体及びその複合体を含む血清と免疫反応するポリペプチド、及びこれらのポリペプチド中のHGBV特異エピトープに対する抗体は共に、生物試験試料中でのHGBV抗体の存在又はウイルス及び/もしくはウイルス抗原の存在を検出するイムノアッセイで有用である。これらのイムノアッセイの設計は変形を受けることが多く、これらの変形は当業界では公知である。これらの変形は本明細書に記載する。イムノアッセイは、1個のウイルス抗原(例えばHGBV核酸配列を含むクローン、又はこれらのクローン中のHGBV核酸配列に由来する複合核酸配列、又はこれらのクローン中の核酸配列のソースであるHGBVゲノムから誘導されるポリペプチド)を使用し得る。あるいは、このイムノアッセイは、これらのソースから誘導されるウイルス抗原を組み合わせて使用してもよい。イムノアッセイは例えば、同一ウイルス抗原に対するモノクローナル抗体、又は異なるウイルス抗原に対するポリクローナル抗体を使用し得る。アッセイには、競合アッセイ、直接反応アッセイ又はサンドイッチ型アッセイに基づくものが含まれるが、これらに限定はされない。アッセイは、固相を使用してもよいし、免疫沈殿又は固相を使用しない他の任意の方法で実施してもよい。ラベルをシグナル生成化合物として使用するアッセイや、これらのラベルの例は本明細書に記載する。シグナルは更に、係属中の米国特許出願第608,849号、第070,647号、第418,981号、及び第687,785号に記載のビオチン及びアビジン、酵素ラベル又はビオチン抗ビオチン系を使用して増幅してもよい。HGBVの免疫優性領域に由来するエピトープを含む組換えポリペプチドは、感染個体の生物試験試料でウイルス抗体を検出するのに有用であり得る。抗体は、急性感染と非急性感染とを識別するのに有用であり得るとも考えられる。適切な材料(例えばHGBVエピトープ又はHGBVエピトープに対する抗体を含む本発明のポリペプチド)を、アッセイの実施に必要な他の試薬や材料、及び適切なアッセイ指示書と一緒に適切な容器にパッケージングすることにより、適切な試薬を含んだ免疫診断に適したキットを製造する。
HGBV抗体及びその複合体を含む血清と免疫反応するポリペプチド、及びこれらのポリペプチド中のHGBV特異エピトープに対する抗体は共に、生物試験試料中でのHGBV抗体の存在又はウイルス及び/もしくはウイルス抗原の存在を検出するイムノアッセイで有用である。これらのイムノアッセイの設計は変形を受けることが多く、これらの変形は当業界では公知である。これらの変形は本明細書に記載する。イムノアッセイは、1個のウイルス抗原(例えばHGBV核酸配列を含むクローン、又はこれらのクローン中のHGBV核酸配列に由来する複合核酸配列、又はこれらのクローン中の核酸配列のソースであるHGBVゲノムから誘導されるポリペプチド)を使用し得る。あるいは、このイムノアッセイは、これらのソースから誘導されるウイルス抗原を組み合わせて使用してもよい。イムノアッセイは例えば、同一ウイルス抗原に対するモノクローナル抗体、又は異なるウイルス抗原に対するポリクローナル抗体を使用し得る。アッセイには、競合アッセイ、直接反応アッセイ又はサンドイッチ型アッセイに基づくものが含まれるが、これらに限定はされない。アッセイは、固相を使用してもよいし、免疫沈殿又は固相を使用しない他の任意の方法で実施してもよい。ラベルをシグナル生成化合物として使用するアッセイや、これらのラベルの例は本明細書に記載する。シグナルは更に、係属中の米国特許出願第608,849号、第070,647号、第418,981号、及び第687,785号に記載のビオチン及びアビジン、酵素ラベル又はビオチン抗ビオチン系を使用して増幅してもよい。HGBVの免疫優性領域に由来するエピトープを含む組換えポリペプチドは、感染個体の生物試験試料でウイルス抗体を検出するのに有用であり得る。抗体は、急性感染と非急性感染とを識別するのに有用であり得るとも考えられる。適切な材料(例えばHGBVエピトープ又はHGBVエピトープに対する抗体を含む本発明のポリペプチド)を、アッセイの実施に必要な他の試薬や材料、及び適切なアッセイ指示書と一緒に適切な容器にパッケージングすることにより、適切な試薬を含んだ免疫診断に適したキットを製造する。
本明細書に詳述する組換えタンパク質を使用するアッセイ方式を設計することができる。本発明者らはCKSタンパク質を記載しているが、本発明でスーパーオキシドジスムターゼ(SOD)等のような他の発現系を使用して、様々な方法で使用できる融合タンパク質、例えばイムノアッセイの抗原、抗体産生用免疫原等を産生できるとも考えられる。ヒト試験試料中の特異被分析物質(例えばウイルスのような感染物質)に対する抗体の存在を検出するためのアッセイ方式では、ヒト試験試料を、少なくとも1種の組換えタンパク質(ポリペプチド)でコーティングした固相と接触させてインキュベートする。試験試料中に抗体が存在すれば、この抗体は抗原ポリペプチドと複合体を形成して、固相に固定化する。複合体の形成後、固相を洗浄して未結合の物質や試薬を除去する。複合体を指示試薬と反応させて、第2の複合体形成のための時間及び条件下でインキュベートする。生成したシグナルを検出して、試験試料中でのCKS組換えポリペプチドに対する抗体の存在を調べる。カットオフ値より高いシグナル生成は、試験試料中に被分析物質に対する抗体が存在することを示す。酵素のような多数の指示試薬を用いると、存在する抗体の量は生成するシグナルに比例する。試験試料の種類によっては、試験試料を適切な緩衝試薬で希釈してもよいし、濃縮してもよいし、操作せずに(“ニート”)固相と接触させてもよい。例えば通常は、予め希釈した血清もしくは血漿試料を試験するか又は尿のような試料を濃縮して、抗体の存在及び/又は存在する抗体の量を調べることが好ましい。
更には、研究中の感染物質のウイルスゲノムの様々な抗原エピトープに対するCKS融合タンパク質を使用して特異感染物質に対する抗体の存在を試験する前述のアッセイ方式では、2種以上の組換えタンパク質を使用することができる。従って、特異ウイルス抗原領域内のエピトープと、ウイルスゲノム由来の他の抗原領域に由来するエピトープとを含んでいる組換えポリペプチドを使用して、1種のエピトープに由来するポリペプチドを用いるよりも感度が増し、恐らくは特異性の高いアッセイを提供することが好ましいであろう。このようなアッセイは、確認(confirmatory)アッセイとして使用することができる。この特定のアッセイ方式では、既知量の試験試料を、組換えタンパク質/抗体複合体を形成するのに十分な時間及び条件下で少なくとも1種の組換えタンパク質でコーティングされた既知量の少なくとも1種の固相と接触させる。反応を生起するための時間、適切な条件下で、複合体を既知量の適切な指示試薬と接触させる。生成したシグナルを陰性試験試料と比較して、試験試料中での被分析物質に対する抗体の存在を調べる。微粒子のようなある種の固相を使用するときには、アッセイで使用される各組換えタンパク質が、個々の微粒子や、種々の被覆微粒子を合わせて製造される前記微粒子の混合物に付着して、各アッセイのために最適化され得る。 前述のアッセイ方式の変形には、各被分析物質に対する抗体の存在を検出するための同一又は異なる固相に付着した種々の被分析物質のCKS組換えタンパク質(例えば同一又は異なる固相上にコーティングされた1種の感染物質のある抗原領域に特異的なCKS組換えタンパク質)を、異なる感染物質のある抗原領域に特異的なCKS組換えタンパク質と共に取り込んで、一方(又は両方)の感染物質の存在を検出することが挙げられる。
他のアッセイ方式としては、抗原エピトープを含むCKS組換えタンパク質が、中和アッセイのような競合アッセイで有用である。中和アッセイを実施するには、ウイルスのような感染物質の抗原領域のエピトープを示す組換えポリペプチドを可溶化し、試料希釈液と混合して、0.5〜50.0μg/mlの最終濃度にする。所要により希釈した既知量の試験試料(好ましくは10μl)を反応ウェルに添加し、次いで組換えポリペプチドを含む試験希釈液400μlを添加する。所望とあれば、混合物を約15分〜2時間プレインキュベートしてもよい。次いで、本明細書に記載のCKS組換えタンパク質でコーティングした固相を反応ウェルに加え、約40℃で1時間インキュベートする。洗浄後、既知量の指示試薬、例えば結合希釈液中のペルオキシダーゼ標識ヤギ抗ヒトIgG 200μlを添加し、40℃で1時間インキュベートする。洗浄後、前述のような酵素結合体を使用するときには、酵素基質(例えばOPD基質)を添加して、室温で30分間インキュベートする。1N硫酸のような停止剤を反応ウェルに添加して、反応を停止させる。492nmで吸光度を読み取る。特異ポリペプチドに対する抗体を含む試験試料では、溶液中でのペプチドと前記抗体との競合結合によるシグナル生成は減少する。競合結合のパーセンテージは、組換えポリペプチドの存在下での試料の吸光度値を、同一希釈度の組換えポリペプチドの不在下でアッセイした試料の吸光度値と比較して計算することができる。従って、組換えタンパク質の存在下の試料と、組換えタンパク質の不在下の試料との間で生じるシグナルの差は、抗体の存在又は不在を調べるために使用する尺度となる。
他のアッセイ方式としは、イムノドットブロットアッセイ系で組換えタンパク質を使用することができる。イムノドットブロットアッセイ系は、ニトロセルロース固相担体上に並置された精製組換えポリペプチドのパネルを使用する。製造した固相担体を試料と接触させ、組換えタンパク質(他の特異結合要素)に対する特異抗体(特異結合要素)を捕捉して、特異結合要素対を形成する。捕捉した抗体は、指示試薬と反応させて検出する。1988年8月2日出願の米国特許出願第07/227,408号に記載されている機器内でレフレクタンスオプチックスアセンブリーを用いて、結合体特異反応を定量することが好ましい。関連米国特許出願第07/227,586号及び第07/227,590号(共に1988年8月2日出願)は更に、本出願人によるものであり、参考として本明細書に組み入れる米国特許第5,075,077号(1988年8月2日出願の米国特許出願第07/227,272号)と同様に、イムノドットアッセイを実施するのに有用な特定方法や装置を記載している。簡潔に言えば、ニトロセルロースベースの試験カートリッジを多数の抗原ポリペプチドで処理する。各ポリペプチドは、試験カートリッジ上の特異反応ゾーン内に含まれている。全ての抗原ポリペプチドがニトロセルロース上に置かれた後に、ニトロセルロース上の過剰結合部位を阻止する。次いで、試験カートリッジを試験試料と接触させて、試験試料が適切な抗体を含めば各反応ゾーンの各抗原ポリペプチドが反応するようにする。反応後、試験カートリッジを洗浄し、よく知られた適切な試薬を用いて、任意の抗原抗体反応を同定する。本明細書に引用した特許及び特許出願に記載されているように、全プロセスは自動化可能である。イムノドットブロットアッセイを実施するための方法及び装置に関連する前記出願明細書は参考として本明細書に組み入れる。
試験試料中に被分析物質の特異抗原に対する抗体を検出するために後述する第1及び第2の固相担体を使用するアッセイでCKS融合タンパク質を使用することができる。このアッセイ方式では、試験試料の第1のアリコートを、組換えタンパク質/被分析物質抗体複合体を形成するのに十分な時間及び条件下で、被分析物質に特異的なCKS組換えタンパク質でコーティングされた第1の固相担体と接触させる。次いで、複合体を、組換え抗原に特異的な指示試薬と接触させる。指示試薬を検出して、試験試料中での組換えタンパク質に対する抗体の存在を調べる。その後、試験試料の第2のアリコートを、組換えタンパク質/第2の抗体の複合体を形成するのに十分な時間及び条件下で、第2の抗体に特異的なCKS組換えタンパク質でコーティングされた第2の固相担体と接触させて、同一被分析物質の異なる抗原決定基の存在を調べる。複合体を、複合体の抗体に特異的な第2の指示試薬と接触させる。シグナルを検出して、試験試料中での抗体の存在を調べる。一方又は両方の被分析物質組換えタンパク質に対する抗体の存在は、試験試料中に抗被分析物質が存在することを示している。固相担体を同時に試験できるとも考えられる。
ハプテンの使用は当業界では公知である。CKS融合タンパク質を用いるアッセイでハプテンを使用して、アッセイの性能を高めることもできると考えられる。
プローブを用いたHGBVゲノム、ビリオン及びウイルス抗原の更なる特性分析
HGBV核酸配列を使用して、HGBVゲノムの配列、並びにHGBV剤の同定及び単離について更なる情報を得ることができる。従って、この知識は、HGBVの特性(例えばHGBVゲノムの種類、ウイルス粒子の構造、及びHGBVを構成する抗原の種類)の分析に役立つと考えられる。この情報により、付加的なポリヌクレオチドプローブ、HGBVゲノムに由来するポリペプチド、及びHGBVエピトープに対する抗体を得ることができる。これらは、HGBVにより発病する非A型、非B型、非C型、非D型及び非E型肝炎の診断及び/又は治療に有益である。
HGBV核酸配列を使用して、HGBVゲノムの配列、並びにHGBV剤の同定及び単離について更なる情報を得ることができる。従って、この知識は、HGBVの特性(例えばHGBVゲノムの種類、ウイルス粒子の構造、及びHGBVを構成する抗原の種類)の分析に役立つと考えられる。この情報により、付加的なポリヌクレオチドプローブ、HGBVゲノムに由来するポリペプチド、及びHGBVエピトープに対する抗体を得ることができる。これらは、HGBVにより発病する非A型、非B型、非C型、非D型及び非E型肝炎の診断及び/又は治療に有益である。
核酸配列情報は、HGBVゲノムの非確定領域に由来する更なる核酸配列を単離するためのプローブ又はPCRプライマーの設計に有用である。例えば、8個以上、好ましくは20個以上のヌクレオチドの配列を含み、HGBV核酸配列ファミリーの5’末端又は3’末端に近い領域に由来するPCRプライマー又は標識プローブを使用して、重複核酸配列をHGBVゲノム又はcDNAライブラリーから単離してもよいし、ウイルス核酸から直接単離してもよい。次いで、HGBV核酸配列と重複するが、該HGBV核酸配列のソースではないゲノムの領域に由来する配列を更に含んでいる前記配列を使用して、必ずしもクローン中の核酸配列とは重複しない他の重複断片を同定するためのプローブを合成してもよい。HGBVゲノムがセグメント化されていても共通配列が欠けていれば、ウイルスゲノムに由来する重複核酸配列の単離技術を使用して、全ウイルスゲノムの配列を決定することが可能である。あるいは、精製したHGBV粒子から単離したウイルスゲノムから、ゲノムセグメントの特性分析を行うことができる。HGBV粒子を精製し、精製手順中に該粒子を検出する方法は本明細書に記載する。ウイルス粒子からポリヌクレオチドゲノムを単離する手順は当業界ではよく知られている。次いで、単離したゲノムセグメントをクローニングして配列決定することができる。従って、HGBVゲノムをその種類とは関係なくクローニングして配列決定することが可能である。
HGBVゲノム又はcDNAライブラリーの構築方法は当業界では公知であり、このために有用なベクターも当業界では公知である。これらのベクターには、λ−gt11、λ−gt10等が含まれる。単離したポリヌクレオチドを適切な制限酵素で消化して、HGBVゲノム又はcDNA由来のプローブにより検出されるHGBV由来の核酸配列をクローンから単離し、配列決定してもよい。
これらの重複HGBV核酸配列に由来する配列情報は、ウイルスゲノム内の相同性エリア及び異質性エリアを決定するのに有用である。これは、種々のゲノム株及び/又は欠陥粒子集団の存在を示し得る。これは、本明細書に記載の技術を使用して、HGBVの単離中に、生物試料中のHGBV又はHGBV抗原又はHGBV核酸を検出するためのハイブリダイゼーションプローブの設計にとっても有用である。重複核酸配列を使用して、HGBVゲノム由来のポリペプチドに対する発現ベクターを産生してもよい。HGBVに対してユニークであると考えられるエピトープを含む抗原が核酸配列ファミリー内にコードされている。即ち、これらの抗原に対する抗体は、HAV、HBV、HCV及びHEVに感染した個体には存在せず、GenBankのゲノム配列と、これらの核酸配列及び前記ソースのポリヌクレオチド配列との間の相同性は小さいと考えられる。従って、HGBV核酸配列でコードされた抗原に対する抗体を使用して、感染個体から単離した非A型、非B型、非C型、非D型及び非E型粒子を同定してもよい。更には、これらはHGBV物質の単離にも有用である。
HGBV粒子は、例えばサイズ識別をベースとする技術(例えば沈降もしくは排除法)、又は密度をベースとする技術(例えば密度勾配超遠心分離、もしくはポリエチレングリコール(PEG)のような物質による沈殿、もしくはアニオンもしくはカチオン交換材料や、疎水性相互反応により結合する材料、及び親和性カラムのような様々な材料でのクロマトグラフィー)を含む当業界で公知の任意の方法により、感染個体の血清又は細胞培養物から単離することができる。単離手順中に、HGBV核酸配列に由来するプローブを用いて抽出ゲノムのハイブリダイゼーション分析を行って、又はHGBV核酸配列ファミリー内にコードされたHGBV抗原に対する抗体をプローブとして使用するイムノアッセイによりHGBVの存在を検出することができる。抗体はポリクローナルであっても、モノクローナルであってもよく、抗体をイムノアッセイで使用する前に精製することが所望され得る。固相に固定化したHGBV抗原に対する前記抗体は、イムノアフィニティークロマトグラフィーによるHGBVの単離に有用である。イムノアフィニティークロマトグラフィー法は当業界では公知であり、免疫選択活性を保持するように抗体を固相に固定化する方法を含んでいる。これらの方法には、吸着及び共有結合が含まれる。抗体の抗原結合部位が接近可能なままであるように、二官能価カップリング剤にスペーサー基を含ませてもよい。
精製手順中に、HGBVゲノム又はcDNA配列ファミリー、及び重複HGBV核酸配列に由来するポリヌクレオチドをプローブ又はプライマーとして使用して、HGBVの存在を核酸ハイブリダイゼーション又はPCRにより検出及び/又は検証してもよい。ウイルス粒子の破壊を引き起こす条件下で、例えばキレート化剤の存在下で洗剤を使用して画分を処理し、ウイルス核酸の存在をハイブリダイゼーション技術又はPCRにより調べる。個体(好ましくはタマリン)に単離したウイルス粒子を感染させ、次いで本明細書に記載の非A型、非B型、非C型、非D型及び非E型肝炎の症状が感染によるものかどうかを調べることにより、単離した粒子がHGBV感染誘発物質である別の確証が得られ得る。
次いで、精製した調製物から得た前述のウイルス粒子の特性を更に分析することができる。ゲノム核酸を精製すれば、これを試験して、RNAse又はDNAseIに対する感度を調べることができる。これらの試験に基づいて、HGBVをRNAゲノム又はDNAゲノムとして調べてもよい。当業界で公知の方法により、例えば電子鏡検法による可視化、密度勾配による移動及び沈降特性により、鎖の数及び環状性又は非環状性を調べることができる。HGBVゲノムのハイブリダイゼーションから、精製核酸の陰性又は陽性鎖数(strandedness)を調べることができる。更には、ゲノム材料がRNAならば、公知の技術(例えば逆転写酵素)により精製核酸をクローニングして配列決定することができる。次いで、ウイルス粒子に由来する核酸を用いて、全ゲノムをセグメント化の如何を問わず配列決定することが可能である。
保存された配列を含むポリペプチドの決定は、HGBVゲノムと結合するプローブ選択に有用であり、それはプローブを単離することにつながる。更には、保存された配列を、HGBV核酸配列に由来する配列と一緒に使用して、ゲノム配列を増幅する系で使用するプライマーを設計してもよい。更には、HGBVの構造を調べて、その成分を単離してもよい。例えば電子鏡検法により形態及び寸法を調べることができる。抗原が主要ウイルス成分中に存在するのか、少量ウイルス成分中に存在するのかを確かめ、単離した核酸配列内にコードされた特異抗原に対する抗体をプローブとして使用して、特異ウイルスポリペプチド抗原[例えばエンベロープ(コート)抗原又は内部抗原(例えば核酸結合タンパク質もしくはコア抗原)]や、ポリヌクレオチドポリメラーゼを同定して位置を調べることもできる。この情報は、診断及び治療用途に有用であり得る。例えば、ワクチン調製物中に外部抗原を含めることが好ましく、又は恐らく多価ワクチンは、構造タンパク質をコードするゲノムに由来するポリペプチド、及びゲノムの他の部分に由来するポリペプチド(例えば非構造ポリペプチド)からなり得る。
HGBV複製用の細胞培養系及び動物モデル系
一般に、HGBVの培養に適した細胞又は細胞系は、サル腎臓細胞(例えばMK2及びVERO)、ブタ腎臓細胞系(例えばPS)、ハムスター乳児腎臓細胞系(例えばBHK)、マウスマクロファージ細胞系(例えばP388D1、MK1及びMm1)、ヒトマクロファージ細胞系(例えばU−937)、ヒト末梢血白血球、ヒト付着単球、肝細胞又は肝細胞系(例えばHUH7及びHepG2)、胚もしくは胚細胞(例えばニワトリ胚線維芽細胞)、又は無脊椎動物、好ましくは昆虫に由来する細胞系(例えばショウジョウバエ細胞系)、更に好ましくは節足動物に由来する細胞系(例えば蚊細胞系もしくはマダニ細胞系)を含み得る。一次肝細胞を培養し、次いでこれにHGBVを感染させることも可能である。あるいは、肝細胞培養物を、感染個体(ヒト又はタマリン)の肝臓から誘導することができる。後者の場合は、in vivo感染させてin vitro継代させた細胞系の例である。更には、種々の不死化方法を使用して、肝細胞培養物に由来する細胞系を得ることができる。例えば、(肝細胞集団の増菌前後の)一次肝臓培養物を種々の細胞と融合して、安定性を維持することができる。更には、培養物に形質転換ウイルスを感染させるか、形質転換遺伝子をトランスフェクトして、永続的な又は半永続的な細胞系を生成してもよい。更には、肝臓培養物中の細胞を融合して、PehG2のような確定した細胞系にしてもよい。細胞融合方法は当業者にはよく知られており、とりわけPEG及びセンダイウイルスのような融合剤の使用を含んでいる。
一般に、HGBVの培養に適した細胞又は細胞系は、サル腎臓細胞(例えばMK2及びVERO)、ブタ腎臓細胞系(例えばPS)、ハムスター乳児腎臓細胞系(例えばBHK)、マウスマクロファージ細胞系(例えばP388D1、MK1及びMm1)、ヒトマクロファージ細胞系(例えばU−937)、ヒト末梢血白血球、ヒト付着単球、肝細胞又は肝細胞系(例えばHUH7及びHepG2)、胚もしくは胚細胞(例えばニワトリ胚線維芽細胞)、又は無脊椎動物、好ましくは昆虫に由来する細胞系(例えばショウジョウバエ細胞系)、更に好ましくは節足動物に由来する細胞系(例えば蚊細胞系もしくはマダニ細胞系)を含み得る。一次肝細胞を培養し、次いでこれにHGBVを感染させることも可能である。あるいは、肝細胞培養物を、感染個体(ヒト又はタマリン)の肝臓から誘導することができる。後者の場合は、in vivo感染させてin vitro継代させた細胞系の例である。更には、種々の不死化方法を使用して、肝細胞培養物に由来する細胞系を得ることができる。例えば、(肝細胞集団の増菌前後の)一次肝臓培養物を種々の細胞と融合して、安定性を維持することができる。更には、培養物に形質転換ウイルスを感染させるか、形質転換遺伝子をトランスフェクトして、永続的な又は半永続的な細胞系を生成してもよい。更には、肝臓培養物中の細胞を融合して、PehG2のような確定した細胞系にしてもよい。細胞融合方法は当業者にはよく知られており、とりわけPEG及びセンダイウイルスのような融合剤の使用を含んでいる。
細胞系のHGBV感染は、細胞内へのウイルス侵入を可能とする条件下で、細胞をウイルス調製物と共にインキュベートするような技術により達成され得ると考えられる。細胞に、単離したウイルスポリヌクレオチドをトランスフェクトさせてウイルス調製物を得ることも可能であり得る。組織培養細胞をトランスフェクトする方法は当業界では公知であり、エレクトロポレーション及びDEAE−デキストラン又はリン酸カルシウムによる沈殿を使用する技術が含まれるが、これに限定はされない。クローニングしたHGBVゲノム又はcDNAによるトラスフェクションでウイルスが複製され、in vitro増殖する。培養した細胞の他に、ウイルス複製に動物モデル系を使用してもよい。従って、HGBV複製はチンパンジー、更には例えばマーモセット及び乳児マウスで生起し得る。
HGBV抗ウイルス剤のスクリーニング
HGBV用の細胞培養系及び動物モデル系が利用できることから、HGBV複製を阻害する抗ウイルス剤、特に好ましくはウイルス複製を阻害しつつ細胞増殖を可能とする物質のスクリーニングも可能となる。これらのスクリーニング方法は当業界では公知である。一般に、ウイルス複製を支える細胞培養系で抗ウイルス剤がウイルス複製の阻止に及ぼす作用、次いで感染性又はウイルス病原性の阻害、また低レベルの毒性について、抗ウイルス剤を様々な濃度で動物モデル系にて検査する。本明細書に記載するHGBV抗原及びHGBVポリヌクレオチドの検出のための方法及び組成物は、抗ウイルス剤のスクリーニングに有用である。何故ならば、これらは、抗ウイルス剤のウイルス複製への作用の検出については、細胞プラークアッセイ又はID50アッセイよりも恐らく高感度の代替手段となるからである。例えば、本明細書に記載するHGBVポリヌクレオチドプローブを使用して、細胞培養物中に産生されるウイルス核酸の量を定量してもよい。これは、感染細胞核酸を標識したHGBVポリヌクレオチドプローブでハイブリダイズ又は競合ハイブリダイズすることにより実施され得る。更には、抗HGBV抗体を使用して、本明細書に記載するイムノアッセイにより、細胞培養物中のHGBV抗原を同定、定量してもよい。更には、競合アッセイにより感染細胞培養物中のHGBV抗原を定量することが所望され得るので、本明細書に記載するHGBV核酸配列内にコードされるポリペプチドはこれらのアッセイで有用である。一般に、HGBVゲノム又はcDNAに由来する組換えHGBVポリペプチドを標識し、細胞培養系で産生された抗原による、前記標識ポリペプチドとHGBVポリペプチドとの結合の阻害を監視する。これらの方法は、HGBVが細胞死を起こさずに細胞系で複製し得る場合に特に有用である。
HGBV用の細胞培養系及び動物モデル系が利用できることから、HGBV複製を阻害する抗ウイルス剤、特に好ましくはウイルス複製を阻害しつつ細胞増殖を可能とする物質のスクリーニングも可能となる。これらのスクリーニング方法は当業界では公知である。一般に、ウイルス複製を支える細胞培養系で抗ウイルス剤がウイルス複製の阻止に及ぼす作用、次いで感染性又はウイルス病原性の阻害、また低レベルの毒性について、抗ウイルス剤を様々な濃度で動物モデル系にて検査する。本明細書に記載するHGBV抗原及びHGBVポリヌクレオチドの検出のための方法及び組成物は、抗ウイルス剤のスクリーニングに有用である。何故ならば、これらは、抗ウイルス剤のウイルス複製への作用の検出については、細胞プラークアッセイ又はID50アッセイよりも恐らく高感度の代替手段となるからである。例えば、本明細書に記載するHGBVポリヌクレオチドプローブを使用して、細胞培養物中に産生されるウイルス核酸の量を定量してもよい。これは、感染細胞核酸を標識したHGBVポリヌクレオチドプローブでハイブリダイズ又は競合ハイブリダイズすることにより実施され得る。更には、抗HGBV抗体を使用して、本明細書に記載するイムノアッセイにより、細胞培養物中のHGBV抗原を同定、定量してもよい。更には、競合アッセイにより感染細胞培養物中のHGBV抗原を定量することが所望され得るので、本明細書に記載するHGBV核酸配列内にコードされるポリペプチドはこれらのアッセイで有用である。一般に、HGBVゲノム又はcDNAに由来する組換えHGBVポリペプチドを標識し、細胞培養系で産生された抗原による、前記標識ポリペプチドとHGBVポリペプチドとの結合の阻害を監視する。これらの方法は、HGBVが細胞死を起こさずに細胞系で複製し得る場合に特に有用である。
HGBV弱毒化株の調製
本明細書に記載する組織培養系及び/又は動物モデル系を用いて、HGBV弱毒化株を単離することが可能であり得る。これらの弱毒化株は、ワクチンとして又はウイルス抗原の単離に有用である。弱毒化株は、細胞培養物及び/又は動物モデルに何度も継代させた後に単離することができる。感染した細胞又は個体での弱毒化株の検出は、当業界で公知の以下の方法に従って達成可能であり、検出には、HGBVにコードされた1種以上のエピトープに対する抗体のプローブとしての使用、又は長さが少なくとも約8ヌクレオチドのHGBV配列を含むポリヌクレオチドのプローブとしての使用が含まれ得る。これに加え又はこれに代わって、本明細書に記載するHGBVのゲノム情報を使用し、また組換え技術を使用して弱毒化株を構築してもよい。通常、ウイルス複製ではなく、病原性に関連するポリペプチドをコードするゲノムの領域を欠失させる。ゲノムを構築すれば、HGBVに対する中和抗体を産生するエピトープを発現させることができる。次いで、変性ゲノムを使用して、HGBV複製を可能とする細胞を形質転換して、細胞をウイルス複製を可能とする条件下で増殖させることができる。弱毒化HGBV株はワクチン用としてだけでなく、ウイルス抗原の商業的産生のソースとしても有用である。何故ならば、これらのウイルスの処理は、ウイルス産生及び/又はウイルス製品の製造に係わる従業員に対してそれほど厳密な保護措置を必要としないからである。
本明細書に記載する組織培養系及び/又は動物モデル系を用いて、HGBV弱毒化株を単離することが可能であり得る。これらの弱毒化株は、ワクチンとして又はウイルス抗原の単離に有用である。弱毒化株は、細胞培養物及び/又は動物モデルに何度も継代させた後に単離することができる。感染した細胞又は個体での弱毒化株の検出は、当業界で公知の以下の方法に従って達成可能であり、検出には、HGBVにコードされた1種以上のエピトープに対する抗体のプローブとしての使用、又は長さが少なくとも約8ヌクレオチドのHGBV配列を含むポリヌクレオチドのプローブとしての使用が含まれ得る。これに加え又はこれに代わって、本明細書に記載するHGBVのゲノム情報を使用し、また組換え技術を使用して弱毒化株を構築してもよい。通常、ウイルス複製ではなく、病原性に関連するポリペプチドをコードするゲノムの領域を欠失させる。ゲノムを構築すれば、HGBVに対する中和抗体を産生するエピトープを発現させることができる。次いで、変性ゲノムを使用して、HGBV複製を可能とする細胞を形質転換して、細胞をウイルス複製を可能とする条件下で増殖させることができる。弱毒化HGBV株はワクチン用としてだけでなく、ウイルス抗原の商業的産生のソースとしても有用である。何故ならば、これらのウイルスの処理は、ウイルス産生及び/又はウイルス製品の製造に係わる従業員に対してそれほど厳密な保護措置を必要としないからである。
宿主及び発現制御配列
ゲノムをウイルスから抽出し、ゲノムライブラリーを作成、プロービングし、クローンの配列を決定し、発現ベクターを構築し、細胞を形質転換し、免疫アッセイを実施し、培養物中で細胞を増殖させるために使用する一般的な技術は当業界では公知であり、本発明に包含する。
ゲノムをウイルスから抽出し、ゲノムライブラリーを作成、プロービングし、クローンの配列を決定し、発現ベクターを構築し、細胞を形質転換し、免疫アッセイを実施し、培養物中で細胞を増殖させるために使用する一般的な技術は当業界では公知であり、本発明に包含する。
指定の宿主と適合する適切な制御配列を使用するときには、原核宿主細胞及び真核宿主細胞の両方を、所望のコード化配列の発現のために使用することができる。原核宿主としては、E.coliが最も頻繁に使用される。原核生物の発現制御配列には、場合によってオペレーター部分を含むプロモーターや、リボソーム結合部位が含まれる。原核宿主と適合するトランスファーベクターは通常、アンピシリン耐性及びテトラサイクリン耐性を付与するオペロンを含むプラスミドpBR322や、更に抗生物質耐性マーカーを付与する配列を含む種々のpUCベクターから誘導される。これらのマーカーを使用して、選択により良好な形質転換細胞を得ることができる。通常使用される原核生物制御配列には、β−ラクタマーゼ(ペニシリナーゼ)、ラクトースプロモーター系(Chang等,Nature 198:1056[1977])、(Goeddel等,Nucleic Acid Res 8:4057[1980]により報告されている)トリプトファンプロモーター系、λ誘導P1プロモーター及びN遺伝子リボソーム結合部位(Shimatake等,Nature 292:128[1981])、並びにtrp及びlacUV5プロモーターの配列に由来するハイブリッドTacプロモーター(De Boer等,Proc.Natl.Acad.Sci.USA 292:128[1983])が含まれる。前述の系は、特にE.coliに適合し得るが、所望とあれば、Bacillus株又はPseudomonas株のような他の原核宿主を対応する制御配列と共に使用してもよい。
真核宿主は、培養系の酵母及び哺乳動物細胞を含んでいる。Saccharomyces cerevisiae及びSaccharomyces carlsbergensisが最も一般的に使用されている酵母宿主であり、好都合な真菌宿主である。酵母適合性ベクターは、栄養要求突然変異株を原栄養性とするか又は野生型株に重金属耐性を付与して良好な形質転換細胞の選択を可能とするマーカーを保有する。酵母適合性ベクターは、2ミクロン複製オリジン(Broach等,Meth.Enz.101;307[1983])、CEN3とARS1の組み合わせ、又は複製を行うための他の手段(例えば宿主細胞ゲノム内への適切な断片の取り込みをもたらす配列)を使用し得る。酵母ベクターの制御配列は当業界では公知であり、3−ホスホフィセレートキナーゼ(phosphophycerate kinase)用プロモーターを含む解糖酵素合成用プロモーターを含んでいる。例えば、Hess等,J.Adv.Enzyme Reg.7:149(1968)、Holland等,Biochemistry 17:4900(1978)及びHitzeman J.Biol.Chem.255:2073(1980)を参照されたい。Holland,J.Biol.Chem.256:1385(1981)に報告されているエノラーゼ遺伝子に由来するようなターミネーターも含まれ得る。特に有用な制御系は、グリセルアルデヒド−3−ホスフェートデヒドロゲナーゼ(GAPDH)プロモーター又はアルコールデヒドロゲネーゼ(ADH)調節可能プロモーター、更にGAPDHから誘導されるターミネーター、また分泌が望ましければ、酵母αファクターに由来するリーダー配列を含む系であると考えられる。更には、操作可能に連結した転写調節領域及び転写開始領域は、野生型生物で先天的に関連しないようなものであり得る。
発現用宿主として利用可能な哺乳動物細胞系は当業界では公知であり、American Type Culture Collectionから入手可能な多数の不滅化細胞系が含まれる。これらには、HeLa細胞、チャイニーズハムスター卵巣(CHO)細胞、ハムスター乳児腎臓(BHK)細胞等が含まれる。哺乳動物細胞に適したプロモーターも当業界では公知であり、Simianウイルス40(SV40)、ラウス肉腫ウイルス(RSV)、アデノウイルス(ADV)、ウシ乳頭腫ウイルス(BPV)、サイトメガロウイルス(CMV)に由来するようなウイルスプロモーターが含まれる。哺乳動物細胞は更に、ターミネーター配列及びポリA付加配列を必要とし得る。発現を増加させるエンハンサー配列を含めてもよいし、遺伝子の増幅を誘発する配列も所望され得る。これらの配列は当業界では公知である。哺乳動物細胞の複製に適したベクターは、ウイルスレプリコン、又は非A型、非B型、非C型、非D型、非E型エピトープをコードする適切な配列の宿主ゲノム内への取り込みを確実にする配列を含み得る。HCVの哺乳動物発現系の一例は、1992年1月31日出願の米国特許出願第07/830,024号に記載されている。
形質転換
形質転換は、ポリヌクレオチドを宿主細胞内に導入するための公知の方法によって可能であり、ポリヌクレオチドをウイルス内に組み込んだウイルスによる宿主細胞の形質導入、及びポリヌクレオチドの直接取り込みを含む。選択する形質転換手順は、形質転換すべき宿主に依存する。直接の取り込みによる細菌の形質転換は一般に、塩化カルシウム又は塩化ルビジウムによる処理を使用する。Cohen,Proc.Natl.Acad.Sci.USA 69:2110(1972)。Graham等,Virology 52:526(1978)のリン酸カルシウム沈殿法又はその変形を用いて、直接の取り込みによる酵母形質転換を実施してもよい。
形質転換は、ポリヌクレオチドを宿主細胞内に導入するための公知の方法によって可能であり、ポリヌクレオチドをウイルス内に組み込んだウイルスによる宿主細胞の形質導入、及びポリヌクレオチドの直接取り込みを含む。選択する形質転換手順は、形質転換すべき宿主に依存する。直接の取り込みによる細菌の形質転換は一般に、塩化カルシウム又は塩化ルビジウムによる処理を使用する。Cohen,Proc.Natl.Acad.Sci.USA 69:2110(1972)。Graham等,Virology 52:526(1978)のリン酸カルシウム沈殿法又はその変形を用いて、直接の取り込みによる酵母形質転換を実施してもよい。
ベクター構築
ベクター構築は当業界で公知の方法を使用する。一般には、市販酵素の製造業者により一般に規定されている条件下にて、適切な制限酵素で処理して、部位特異的DNA開裂を実施する。通常、約20μlの緩衝溶液中の1〜10単位の酵素を用いて37℃で1〜2時間インキュベートすることにより、約1マイクログラム(μg)のプラスミド又はDNA配列を開裂する。制限酵素と共にインキュベートした後に、フェノール/クロロホルム抽出によりタンパク質を除去し、エタノールと共に沈殿させてDNAを回収する。開裂断片は、当業者に公知の方法により、ポリアクリルアミド又はアガロースゲル電気泳動法を用いて分離してもよい。
ベクター構築は当業界で公知の方法を使用する。一般には、市販酵素の製造業者により一般に規定されている条件下にて、適切な制限酵素で処理して、部位特異的DNA開裂を実施する。通常、約20μlの緩衝溶液中の1〜10単位の酵素を用いて37℃で1〜2時間インキュベートすることにより、約1マイクログラム(μg)のプラスミド又はDNA配列を開裂する。制限酵素と共にインキュベートした後に、フェノール/クロロホルム抽出によりタンパク質を除去し、エタノールと共に沈殿させてDNAを回収する。開裂断片は、当業者に公知の方法により、ポリアクリルアミド又はアガロースゲル電気泳動法を用いて分離してもよい。
混合物中に存在する適切なデオキシヌクレオチドトリホスフェート(dNTP)の存在下で,E.coli DNAポリメラーゼ1(クレノウ)を用いて付着端開裂断片をブラント末端にしてもよい。S1ヌクレアーゼによる処理を使用して、一本鎖DNA部分を加水分解してもよい。
T4 DNAリガーゼ及びATPを用い、標準的な緩衝液及び温度条件下で結合する。付着端結合は、ブラント末端結合ほどATPやリガーゼを必要としない。ベクター断片を結合混合物の一部として使用するときは、ベクター断片をしばしば細菌アルカリ性ホスファターゼ(BAP)又は仔ウシ腸アルカリ性ホスファターゼで処理して、5’−ホスフェートを除去し、ベクターの再結合を妨げる。あるいは、所望しない断片の制限酵素消化を使用して、結合を妨げることができる。結合混合物を、E.coliや、抗生物質耐性を含む方法により選択された良好な形質転換細胞のような適切なクローニング宿主内に形質転換し、次いで正しい構築についてスクリーニングする。
望ましいDNA配列の構築
Warner,DNA 3:401(1984)が記載するような自動化オリゴヌクレオチド合成機を用いて、合成オリゴヌクレオチドを産生してもよい。所望とあれば、標準的な反応条件を用いて、32P−ATPの存在下にてポリヌクレオチドキナーゼで処理して、合成鎖を32Pで標識することができる。DNA配列(例えばゲノム又はcDNAライブラリーから単離したDNA配列)を、公知の方法[例えばZoller,Nucleic Acid Res.10:6487(1982)に記載の特定部位の突然変異誘発]により改変してもよい。簡潔に言えば、改変すべきDNAを一本鎖配列としてファージ内にパッケージングし、改変すべきDNA部分に相補的であり、自身の配列に所望の改変を含む合成オリゴヌクレオチドをプライマーとして用いてDNAポリメラーゼで二本鎖DNAに変換する。ファージの各鎖の複製域を含む形質転換細菌の培養物を寒天平板培養して、プラークを得る。理論的には新しいプラークの50%が、突然変異配列を有するファージを含み、残りの50%は元の配列を有する。非改変配列ではなく、正しい鎖とのハイブリダイゼーションに適した温度及び条件下で、プラーク複製物を標識した合成プローブにハイブリダイズする。ハイブリダイゼーションにより同定された配列を回収して、クローニングする。
Warner,DNA 3:401(1984)が記載するような自動化オリゴヌクレオチド合成機を用いて、合成オリゴヌクレオチドを産生してもよい。所望とあれば、標準的な反応条件を用いて、32P−ATPの存在下にてポリヌクレオチドキナーゼで処理して、合成鎖を32Pで標識することができる。DNA配列(例えばゲノム又はcDNAライブラリーから単離したDNA配列)を、公知の方法[例えばZoller,Nucleic Acid Res.10:6487(1982)に記載の特定部位の突然変異誘発]により改変してもよい。簡潔に言えば、改変すべきDNAを一本鎖配列としてファージ内にパッケージングし、改変すべきDNA部分に相補的であり、自身の配列に所望の改変を含む合成オリゴヌクレオチドをプライマーとして用いてDNAポリメラーゼで二本鎖DNAに変換する。ファージの各鎖の複製域を含む形質転換細菌の培養物を寒天平板培養して、プラークを得る。理論的には新しいプラークの50%が、突然変異配列を有するファージを含み、残りの50%は元の配列を有する。非改変配列ではなく、正しい鎖とのハイブリダイゼーションに適した温度及び条件下で、プラーク複製物を標識した合成プローブにハイブリダイズする。ハイブリダイゼーションにより同定された配列を回収して、クローニングする。
プローブとのハイブリダイゼーション
Grunstein及びHogness,Proc.Natl.Acad.Sci.USA 73:3961(1975)に記載の手順を用いて、HGBVゲノム又はDNAライブラリーをプロービングしてもよい。簡潔に言えば、プロービングすべきDNAをニトロセルロースフィルター上に固定化し、変性し、0〜50%ホルムアミド、0.75M NaCl、75mMクエン酸ナトリウム、それぞれ0.02%(w/v)のウシ血清アルブミン(BSA)、ポリビニルピロリドン及びFicoll、50mMリン酸ナトリウム(pH6.5)、0.1%SDS並びに100μg/mlキャリヤー変性DNAを含む緩衝液でプレハイブリダイズする。緩衝液中のホルムアミドのパーセンテージ、及びプレハイブリダイゼーションやその後のハイブリダイゼーション工程の時間/温度条件は、必要とされる厳密度に依存する。厳密度条件が低くてもよいオリゴマープローブは一般に、ホルムアイドを低率に、温度をより低く、ハイブリダイゼーション時間をより長くして使用する。cDNA又はゲノム配列から誘導されるような30又は40個以上のヌクレオチドを含むプローブは一般に、例えば約40〜42℃のより高温と、例えば50%と高率のホルムアミドを使用する。プレハイブリダイゼーションの後に、32P標識オリゴヌクレオチドプローブを緩衝液に添加し、ハイブリダイゼーション条件下でフィルターをこの混合物中でインキュベートする。洗浄後、被処理フィルターをオートラジオグラフィーにかけて、ハイブリダイズしたプローブの位置を示す。元の寒天プレート上の対応位置にあるDNAを所望のDNAのソースとして使用する。
Grunstein及びHogness,Proc.Natl.Acad.Sci.USA 73:3961(1975)に記載の手順を用いて、HGBVゲノム又はDNAライブラリーをプロービングしてもよい。簡潔に言えば、プロービングすべきDNAをニトロセルロースフィルター上に固定化し、変性し、0〜50%ホルムアミド、0.75M NaCl、75mMクエン酸ナトリウム、それぞれ0.02%(w/v)のウシ血清アルブミン(BSA)、ポリビニルピロリドン及びFicoll、50mMリン酸ナトリウム(pH6.5)、0.1%SDS並びに100μg/mlキャリヤー変性DNAを含む緩衝液でプレハイブリダイズする。緩衝液中のホルムアミドのパーセンテージ、及びプレハイブリダイゼーションやその後のハイブリダイゼーション工程の時間/温度条件は、必要とされる厳密度に依存する。厳密度条件が低くてもよいオリゴマープローブは一般に、ホルムアイドを低率に、温度をより低く、ハイブリダイゼーション時間をより長くして使用する。cDNA又はゲノム配列から誘導されるような30又は40個以上のヌクレオチドを含むプローブは一般に、例えば約40〜42℃のより高温と、例えば50%と高率のホルムアミドを使用する。プレハイブリダイゼーションの後に、32P標識オリゴヌクレオチドプローブを緩衝液に添加し、ハイブリダイゼーション条件下でフィルターをこの混合物中でインキュベートする。洗浄後、被処理フィルターをオートラジオグラフィーにかけて、ハイブリダイズしたプローブの位置を示す。元の寒天プレート上の対応位置にあるDNAを所望のDNAのソースとして使用する。
構築の検証及び配列決定
標準的なベクター構築では、結合混合物をE.coli株XL−1 Blue又は他の適切な宿主内に形質転換し、良好な形質転換細胞を抗生物質耐性又は他のマーカーにより選択する。次いで、通常はClewell等,J.Bacteriol.110:667(1972)に記載のようなクロラムフェニコール増幅の後に、Clewell等,Proc.Natl.Acad.Sci.USA 62:1159(1969)の方法で、形質転換細胞由来のプラスミドを産生する。DNAを単離し、通常は制限酵素分析及び/又は配列決定により分析する。Messing等,Nucleic Acid Res.9:309(1981)にも記載されているSanger等,Proc.Natl.Acad.Sci.USA 74:5463(1977)のよく知られたジデオキシ法により、又はMaxam等,Methods in Enzymology 65:499(1980)に記載の方法により、配列決定してもよい。GCに富む領域で時々観察されるバンド圧縮の問題は、Barr等,Biotechniques 4:428(1986)に記載の方法により、T−デアゾグアノシンを用いて克服される。
標準的なベクター構築では、結合混合物をE.coli株XL−1 Blue又は他の適切な宿主内に形質転換し、良好な形質転換細胞を抗生物質耐性又は他のマーカーにより選択する。次いで、通常はClewell等,J.Bacteriol.110:667(1972)に記載のようなクロラムフェニコール増幅の後に、Clewell等,Proc.Natl.Acad.Sci.USA 62:1159(1969)の方法で、形質転換細胞由来のプラスミドを産生する。DNAを単離し、通常は制限酵素分析及び/又は配列決定により分析する。Messing等,Nucleic Acid Res.9:309(1981)にも記載されているSanger等,Proc.Natl.Acad.Sci.USA 74:5463(1977)のよく知られたジデオキシ法により、又はMaxam等,Methods in Enzymology 65:499(1980)に記載の方法により、配列決定してもよい。GCに富む領域で時々観察されるバンド圧縮の問題は、Barr等,Biotechniques 4:428(1986)に記載の方法により、T−デアゾグアノシンを用いて克服される。
エンザイムリンクドイムノソーベントアッセイ
エンザイムリンクドイムノソーベントアッセイ(ELISA)を使用して、抗原濃度又は抗体濃度を測定することができる。この方法は、酵素ラベルと抗原又は抗体との結合に依存し、結合酵素活性(生成シグナル)を定量ラベル(測定可能な生成シグナル)として使用している。酵素をラベルとして使用する方法、及びこのような酵素ラベルの例は本明細書に記載する。
エンザイムリンクドイムノソーベントアッセイ(ELISA)を使用して、抗原濃度又は抗体濃度を測定することができる。この方法は、酵素ラベルと抗原又は抗体との結合に依存し、結合酵素活性(生成シグナル)を定量ラベル(測定可能な生成シグナル)として使用している。酵素をラベルとして使用する方法、及びこのような酵素ラベルの例は本明細書に記載する。
HGBV核酸配列の作成
非A型、非B型、非C型、非D型、非E型物質のソースは、本明細書に記載する臨床的及び実験的基準に合致するHGBVウイルスに感染したヒト又はタマリンからの個々の血漿、血清もしくは肝臓ホモジネート又はそのプールである。あるいは、以下に記載する基準に合致する非A型、非B型、非C型、非E型肝炎に羅患した他の個体の血液をタマリンに実験的に感染させることができる。高レベルのアラニントランスフェラーゼ活性を含む個々の血漿、血清又は肝臓ホモジネート試料を多数合わせてプールを調製することができる。この活性は、HGBV感染による肝炎性損傷によるものである。本発明者らの実験のひとつで計算したウイルスのTID(タマリン感染価)は4×105個/ml以上であった(以下の実施例2を参照)。
非A型、非B型、非C型、非D型、非E型物質のソースは、本明細書に記載する臨床的及び実験的基準に合致するHGBVウイルスに感染したヒト又はタマリンからの個々の血漿、血清もしくは肝臓ホモジネート又はそのプールである。あるいは、以下に記載する基準に合致する非A型、非B型、非C型、非E型肝炎に羅患した他の個体の血液をタマリンに実験的に感染させることができる。高レベルのアラニントランスフェラーゼ活性を含む個々の血漿、血清又は肝臓ホモジネート試料を多数合わせてプールを調製することができる。この活性は、HGBV感染による肝炎性損傷によるものである。本発明者らの実験のひとつで計算したウイルスのTID(タマリン感染価)は4×105個/ml以上であった(以下の実施例2を参照)。
例えば、高力価であることが好ましいが、必ずしもそうでなくてもよい血漿、血清又は肝臓ホモジネートに由来する核酸ライブラリーを以下の方法で作成する。まず血漿、血清又は肝臓ホモジネートからウイルス粒子を単離する。次いで、アリコートを緩衝溶液(例えば50mMトリス−HCl(pH8.0)、1mM EDTA、100mM NaClを含む緩衝溶液)中に希釈する。例えば15,000×g、20℃で20分間の遠心分離により破片を除去する。次いで、当業者により慣用的に決定され得る適切な条件下で遠心分離を行って、得られた上清中のウイルス粒子をペレット化する。ウイルスゲノムを放出するために、ペレットをSDS懸濁液[例えば1%SDS、120mM EDTA、10mMトリス−HCl(pH7.5)を含み、更に2mg/mlのプロテイナーゼKを含んでいる懸濁液]のアリコート中に懸濁させ、次いで適切な条件下で、例えば45℃で90分間インキュベートして粒子を破壊する。例えば0.8μg MS2バクテリオファージRNAをキャリヤーとして添加し、この混合物をフェノール:クロロホルム[0.5Mトリス−HCl(pH7.5)、0.1%(v/v)β−メルカプトエタノール、0.1%(w/v)ヒドロキシキノロンで飽和させたフェノール]の1:1混合物で4回抽出し、次いでクロロホルムで2回抽出して核酸を単離する。水性相を例えば1−ブタノールで濃縮した後に、2.5容量の無水エタノールと共に−20℃で一晩沈殿させる。例えばBeckman SW41ローターにて、40,000rpm、4℃で90分間遠心分離にかけて核酸を回収し、これを水に溶解し、0.05%(v/v)ジエチルピロカーボネートで処理して、オートクレーブ処理する。
前記手順により得られた核酸を、例えば17.5mM CH3HgOHで変性し、次いでこの変性核酸を鋳型として用いてcDNAを合成し、例えばHuynh(1985、上掲)に記載の方法を用いてファージλ−gt11のEcoRI部位内にクローニングする。但し、逆転写酵素により第1核酸鎖の合成中にランダムプライマーをオリゴ(dT)12−18に代える(Taylor等,[1976]参照)。得られた二本鎖核酸配列を、例えばセファロースCL−4Bカラム上でのサイジングにより分別する。平均寸法が約400、300、200及び100塩基対の溶離材料をゲノムプール内にプールする。少なくとも1つのプール内のcDNAから、λ−gt11 cDNAライブラリーを作成する。あるいは、病因物質がDNAウイルスの場合、ゲノムDNAのクローニング方法が有用であり、これは当業者には公知である。
このようにして作成されたλ−gt11ゲノムライブラリーを、非A型、非B型、非C型、非E型肝炎既往個体(以下で説明する基準に合致する)からの血清、血漿又は肝臓ホモジネートと特異結合し得るエピトープについてスクリーニングする。Huyng等(上掲)の方法を用いて、約104〜107個のファージを血清、血漿又は肝臓ホモジネートを用いてスクリーニングする。125I又は他の適切なリポーター分子(例えばHRPO、アルカリ性ホスファターゼ等)で放射性標識したヒツジ抗ヒトIg抗血清で結合ヒト抗体を検出することができる。陽性ファージを同定して、精製する。次いで、同一方法を用い、先にHGBV物質に感染した所定人数の各ヒトの血清との結合の特異性について、前記ファージを試験する。理想的には、ファージは、試験する血清、血漿又は肝臓ホモジネートの全て又は大半と反応するポリペプチドをコードし、HGBV物質やA型、B型、C型、D型、E型肝炎について本明細書に記載した基準により“陰性”であることが確定した個体からの血清、血漿又は肝臓ホモジネートとは反応しない。これらの手順に従って、非A型、非B型、非C型、非D型、非E型であると同定された患者からの血清、血漿又は肝臓ホモジネートにより免疫学的に特異認識されるポリペプチドをコードするクローンを単離することができる。
以下で本発明を実施例により説明する。これらの実施例は例示的であって、本発明の範囲を限定するものではない。
(実施例)
本明細書に記載されている実施例は、HGBV群のウイルスの発見方法を詳細に記載するものである。該実施例は、HGBV群のHGBV−A、HGBV−B及びHGBV−Cウイルスの発見をたどり得るように年代順に記載されている。一般的には、先ず伝播性及び感染性の研究が行われた。本明細書に記載されているこれらの研究及びその後の研究により、HGBVとしてGB−A及びGB−Bの2種のHCV様ウイルスの存在が実証された。さらに、これも本明細書に詳細に記載されている退化性(degenerative)プライマーを用いたその後の実験によりHGBV−Cが発見された。血清研究により実証された該ウイルス群のヒトにおける罹患率、該ウイルス群のウイルス特性分析、その特定の属における他のウイルスとの関連性及びHGBV−A、HGBV−B及びHGBV−Cの相互関連性も教示する。
本明細書に記載されている実施例は、HGBV群のウイルスの発見方法を詳細に記載するものである。該実施例は、HGBV群のHGBV−A、HGBV−B及びHGBV−Cウイルスの発見をたどり得るように年代順に記載されている。一般的には、先ず伝播性及び感染性の研究が行われた。本明細書に記載されているこれらの研究及びその後の研究により、HGBVとしてGB−A及びGB−Bの2種のHCV様ウイルスの存在が実証された。さらに、これも本明細書に詳細に記載されている退化性(degenerative)プライマーを用いたその後の実験によりHGBV−Cが発見された。血清研究により実証された該ウイルス群のヒトにおける罹患率、該ウイルス群のウイルス特性分析、その特定の属における他のウイルスとの関連性及びHGBV−A、HGBV−B及びHGBV−Cの相互関連性も教示する。
(実施例1)
HGBVの伝播性
A.実験プロトコル。伝播性及び感染性研究用に、LEMSIP(Laboratory for Experimental Medicine and Surgery in Primates, Tuxedo, New York)から16匹のタマリン(Saguinus labiatsu)を入手した。動物全てをLEMSIPにより承認されたプロトコルに従ってLEMSIPで維持且つ監視した(注:動物1匹が自然死し、もう1匹の病弱な動物は感染力実験の開始前に安楽死させた)。血清肝酵素アラニントランスアミナーゼ(ALT)、γ−グルタミルトランスフェラーゼ(GGT)及びイソクエン酸デヒドロゲナーゼ(ICD)について、2〜3週間の間、1週間に一度又は2週間に一度得た血清試料に基づいて基準血清肝酵素値を決定した。接種に先立って各動物につき最低8つの血清肝酵素値を得た。平均肝酵素値+標準偏差の3.75倍に基づき、各動物についてカットオフ値(CO)を決定した。カットオフ値を超える肝臓酵素値は異常であり、肝臓が損傷を受けているとみなした。数匹のタマリンに以下に記載のように接種し、ALT、GGT及びICD血清レベルにおける変化を監視した。その後、監視過程の間の特定時期に、その後の実験用の血清及び組織を得るべく動物数匹を殺した。
HGBVの伝播性
A.実験プロトコル。伝播性及び感染性研究用に、LEMSIP(Laboratory for Experimental Medicine and Surgery in Primates, Tuxedo, New York)から16匹のタマリン(Saguinus labiatsu)を入手した。動物全てをLEMSIPにより承認されたプロトコルに従ってLEMSIPで維持且つ監視した(注:動物1匹が自然死し、もう1匹の病弱な動物は感染力実験の開始前に安楽死させた)。血清肝酵素アラニントランスアミナーゼ(ALT)、γ−グルタミルトランスフェラーゼ(GGT)及びイソクエン酸デヒドロゲナーゼ(ICD)について、2〜3週間の間、1週間に一度又は2週間に一度得た血清試料に基づいて基準血清肝酵素値を決定した。接種に先立って各動物につき最低8つの血清肝酵素値を得た。平均肝酵素値+標準偏差の3.75倍に基づき、各動物についてカットオフ値(CO)を決定した。カットオフ値を超える肝臓酵素値は異常であり、肝臓が損傷を受けているとみなした。数匹のタマリンに以下に記載のように接種し、ALT、GGT及びICD血清レベルにおける変化を監視した。その後、監視過程の間の特定時期に、その後の実験用の血清及び組織を得るべく動物数匹を殺した。
B.動物の接種(初期実験)。HGBVを接種した9匹のタマリンから肝炎の初期急性期(接種後19〜24日)の間に採取した血清から、既知の感染性タマリンGB血清プール(H205 GBパス11として示される11回継代)を調製した。該プールは、既に記載(J.L. Dienstagら, Nature 264,前出; E.Taborら,J. Med. Virol. 5, 前出)されており、関連病因物質を決定するために研究された。該プールのアリコートは、この実験研究に用いられるまでAbbott Laboratories(North Chicago, IL60064)において液体窒素貯蔵条件下に維持されていた。HGBVの他のアリコートはAmerican Type Culture Collection(A.T.C.C.),12301 Parklawn Drive, Rockville, MD20852から、A.T.C.C.受託番号第VR-806号の下に入手し得る。
第1日目に、初期群の残りの14匹中4匹ののタマリン、認識番号T−1053、T−1048、T−1057及びT−1061に、前以て1:50に希釈しておいたプールH205、11回継代0.25mlを静脈内接種した。これらの動物を肝酵素ALT、GGT及びICDの変化について毎週監視した。表2は、これら4匹のタマリン(T−1053、T−1048、T−1057及びT−1061)に関する接種前及び接種後の肝酵素データを示す。図1〜4は、接種前及び接種後のこれら4匹のタマリンのALTレベル及びICDレベルを示している。該データが示しているように、プールH205の1:50の希釈物を接種した4匹のタマリンのALT、GGT及びICDにCOを超える有意な上昇が得られた。
同日(第1日目)、1匹のタマリン(T−1047)に、プールされた正常タマリン血清0.25mlを静脈内接種して陰性対照として用い、もう1匹のタマリン(T−1042)には、プールされた正常ヒト血清を静脈内接種して追加の陰性対照とした。図5及び図6並びに表3は、2匹の対照タマリン(T−1047及びT−1042)の接種前及び接種後のALT及びICDレベルを示す。データが示しているように、該2匹の対照タマリンにおいては8週の間にALT又はICDに全く上昇が見られなかった。
同日(第1日目)、1匹のタマリン(T−1044)に、急性肝炎の発生から約3週間後の前記外科医(GB起源)から得た回復期血清0.2mlを静脈内接種した。この標本は−20℃で貯蔵されていた〔F. Deinhardtら, J. Exper. Med. 125:673-688 (1967)〕。もう1匹のタマリン(T−1034)に、該回復期血清0.1mlを接種した。図7及び図8並びに表4に示されているように、接種後11週の間には、これらのタマリンの血清肝酵素に上昇が全く見られなかった。従って、これらのデータは、元の患者から採取し、−20℃で貯蔵されていた回復期血清には感染性HGBVが検出されなかったことを示しており、それは、患者が感染から回復しており、且つ、ウイルスが患者の血清から除去されているか、又はウイルスタイターが−20℃での貯蔵により検出不能なレベルにまで減少していたことを示し得る。
C.その後の実験。タマリンT−1053は、接種後1週間で血清肝酵素に有意な上昇を示し、接種後11日目に肝酵素に関して再テストを行った。12日目に、血清肝酵素に有意な上昇が存在することが測定され、該動物をその日に殺した。その後の実験用に、血漿、肝臓及び脾臓の組織試料を得た。T−1053由来の血漿を以下の実施例3に記載のRDA手順用の出発物質とし、肝組織を以下の実施例8に用いた。
タマリンT−1048、T−1057及びT−1061を血清肝臓酵素について監視した。該動物は全て、接種後2週間以内に肝臓酵素レベルが上昇したことが認められた。これらの上昇値は接種後6週間以降に見られた。3匹のタマリンは全て、接種後84日目までに血清肝酵素レベルがCOを下回るレベルに減少したことが認められた。接種後97日目に、これら3匹のタマリン(T−1048、T−1057及びT−1061)を、タマリンT−1053由来のニート血漿0.10ml(感染性であると判明した、実施例2参照)を用いて再チャレンジし、血清肝酵素の上昇として示される肝炎が再誘発され得たかどうかを測定した。そのデータを表2、図1、図3及び図4に示す。データが示しているように、2匹のタマリン(T−1057及びT−1061)の血清肝酵素レベルは、接種後3週間ではCOを下回るレベルに留まった。1匹のタマリン(T−1048)は、再接種直後の2週間に血清肝酵素レベルに緩やかな上昇を示した。T−1048における緩やかな上昇は、HGBVによる肝臓組織の再感染によるものか又はH205の初期接種から完全に回復していないことによるものと推定された。
(実施例2)
感染性実験
A.実験プロトコル。実施例1(A)に記載のようにして4匹のタマリンに関する基準読み取り値を得た。接種前に各動物について簡単に基準血清肝臓酵素(ALT、GGT及びICD)の値を決定した。平均肝酵素値+標準偏差の3.75倍に基づいて各動物についてカットオフ値(CO)を決定した。カットオフ値を超える肝酵素は異常であり、肝臓に損傷があるとみなされた。
感染性実験
A.実験プロトコル。実施例1(A)に記載のようにして4匹のタマリンに関する基準読み取り値を得た。接種前に各動物について簡単に基準血清肝臓酵素(ALT、GGT及びICD)の値を決定した。平均肝酵素値+標準偏差の3.75倍に基づいて各動物についてカットオフ値(CO)を決定した。カットオフ値を超える肝酵素は異常であり、肝臓に損傷があるとみなされた。
B.タマリンの接種。接種後12日目に殺したタマリンT−1053由来の血漿(実施例1〔C〕参照)をその後の実験用の接種材料として用いた。1日目に、1匹のタマリン(T−1055)に、T−1053のニート血漿0.25mlを静脈内接種した。同日、2匹のタマリン(T−1038及びT−1051)に、プールされた正常タマリン血漿中に10−4(T−1038)又は10−5(T−1051)に順次希釈しておいたT−1053血漿0.25mlを静脈内接種した。同日、タマリンT−1049に、減少孔径(0.8μm、0.45μm、0.22μm及び0.10μm)の一連のフィルターを介して濾過し、プールされた正常タマリン血漿に10−4に希釈しておいたT−1053血漿0.25mlを静脈内接種した。
血清肝酵素ALT、GGT及びICDの変化について実施例1に記載のようにして毎週全タマリン(T−1055、T−1038、T−1051及びT−1049)を監視した。表5は、これら4匹のタマリンに関する接種前及び接種後の血清肝酵素データを示す。図9は、T−1055の接種前及び接種後のALT及びICD値を示す。図9を参照すると、血清肝酵素ALT及びICDにCDを超える上昇が生じたことがわかる。該タマリンを接種後12日目に殺した。図10及び図11は、それぞれタマリンT−1051及びT−1038のALT及びICDにおける接種前及び接種後の血清レベルを示す。図10及び図11を参照すると、接種後11日目まで両動物の血清肝酵素ALT及びICDに上昇が生じたことをわかる。T−1038を接種後14日目に殺した。表5及び図12は、T−1049に関して得られたデータを示す。表5及び図12からわかるように、接種後11日以内にT−1049にCOを超える血清肝酵素の上昇が認められた。
T−1049に関して行われた濾過実験は、HGBVが0.10μmのフィルターを通過し得ることを示し、それは、HGBVがウイルス性であり且つ直径が0.1μm未満であり得ることを示唆している。さらに、T−1038に関して行われた感染性滴定実験は、T−1053血清が1ml当たり少なくとも4×105のタマリン感染量を含んでいることを示している。
単一のHGBV病原体の伝播性を示すために、タマリンT−1044に、H205接種7日後に採取し、正常なタマリン血清に1:500希釈しておいたT−1057血清からなる接種材料0.25mlを接種した。接種後14〜63日からALTレベルにカットオフを超える緩やかな上昇(即ち、82〜106の範囲の上昇)が認められた。
接種後14日目に採取し、正常なタマリン血清に1:2希釈しておいたT−1044の血清0.25mlをタマリンT−1047及びT−1056に順次接種した。先ず、接種後42日目にT−1047及びT−1056にカットオフを超えるALTレベルの上昇が認められたが、それぞれ接種後64日目及び91日目に正常レベルに戻った。タマリンT−1058に、T−1053血清を用いてチャレンジ後22日目に採取したT−1057のニート血清0.25mlを接種した。接種後112日の間にはALTレベルに上昇は認められなかった。
(実施例3)
代表相違分析(控除ハイブリダイゼーション)
A.アンプリコン用二本鎖DNAの産生
本明細書の上記材料及び方法に記載され、図13に参照されている手順を用い、H205血清(実施例1C及び2B参照)の接種後12日目に採取したタマリンT−1053感染性血漿(実施例1A参照)由来の総核酸からテスターアンプリコンを合成した。接種前17〜30日の間にプールされたタマリンT−1053の接種前血漿からドライバーアンプリコンを合成した。簡単に言えば、両血漿を実施例2Bに記載のように0.1μmのフィルターを介して濾過した。次いで、市販のキット〔United States Biochemical (USB), Cleveland, OH, cat.#73750〕及び担体として10μgの酵母tRNAを用い、50μlの濾過血漿を抽出した。この核酸をランダムに開始して逆転写し、次いで市販のキットを用いランダム開始してDNA合成した。簡単に言えば、ランダムヘキサマーを用いて、製造業者に指示されたPerkin Elmer’s(Norwalk, CT)RNA PCRキット(cat.#N808-0017)を用いて80μlの逆転写反応を行い、20℃で10分間インキュベートし、次いで42℃で2時間インキュベートした。次いで反応を停止し、cDNA/RNA重複部(duplexes)を99℃で2分間インキュベートして変性させた。反応材料に、総量20μlの10μlの10×RP緩衝液〔100mMのNaCl、420mMのTris(pH8.0)、50mMのDTT、100μg/mlのBSA〕、250pmolのランダムヘキサマー及び13単位のSequenase(登録商標)バージョン2.0ポリメラーゼ(USB,cat.#70775)を補った。反応材料を20℃で10分間、次いで37℃で2時間インキュベートした。フェノール:クロロホルムで抽出し、エタノール沈殿させた後、製造業者により指示されたように、これらの反応の二本鎖DNA産物を30μlの反応材料容量中4単位の制限エンドヌクレアーゼSau3AI(New England Biolabs [NEB], cat.#169L)で30分間消化した。
代表相違分析(控除ハイブリダイゼーション)
A.アンプリコン用二本鎖DNAの産生
本明細書の上記材料及び方法に記載され、図13に参照されている手順を用い、H205血清(実施例1C及び2B参照)の接種後12日目に採取したタマリンT−1053感染性血漿(実施例1A参照)由来の総核酸からテスターアンプリコンを合成した。接種前17〜30日の間にプールされたタマリンT−1053の接種前血漿からドライバーアンプリコンを合成した。簡単に言えば、両血漿を実施例2Bに記載のように0.1μmのフィルターを介して濾過した。次いで、市販のキット〔United States Biochemical (USB), Cleveland, OH, cat.#73750〕及び担体として10μgの酵母tRNAを用い、50μlの濾過血漿を抽出した。この核酸をランダムに開始して逆転写し、次いで市販のキットを用いランダム開始してDNA合成した。簡単に言えば、ランダムヘキサマーを用いて、製造業者に指示されたPerkin Elmer’s(Norwalk, CT)RNA PCRキット(cat.#N808-0017)を用いて80μlの逆転写反応を行い、20℃で10分間インキュベートし、次いで42℃で2時間インキュベートした。次いで反応を停止し、cDNA/RNA重複部(duplexes)を99℃で2分間インキュベートして変性させた。反応材料に、総量20μlの10μlの10×RP緩衝液〔100mMのNaCl、420mMのTris(pH8.0)、50mMのDTT、100μg/mlのBSA〕、250pmolのランダムヘキサマー及び13単位のSequenase(登録商標)バージョン2.0ポリメラーゼ(USB,cat.#70775)を補った。反応材料を20℃で10分間、次いで37℃で2時間インキュベートした。フェノール:クロロホルムで抽出し、エタノール沈殿させた後、製造業者により指示されたように、これらの反応の二本鎖DNA産物を30μlの反応材料容量中4単位の制限エンドヌクレアーゼSau3AI(New England Biolabs [NEB], cat.#169L)で30分間消化した。
B.アンプリコンの合成
Sau3AI消化DNAを上記のように抽出、沈殿させた。反応材料を50〜55℃の乾燥加熱ブロックに入れ、4℃で1時間インキュベートすることにより、Sau3AI消化産物全体を、1×T4 DNAリガーゼ緩衝液(NEB)中で緩衝した30μlの反応材料容量中465pmolのR Bgl24(配列番号1)及び465pmolのR Bgl 12(配列番号2)にアニーリングした。400単位のT4 DNAリガーゼ(NEB, cat.#202S)を添加してアニーリングされた産物を連結した。16℃で14時間インキュベートした後、小規模のPCRを行った。10μlの連結反応材料を、60μlのH2O、20μlの5×PCR緩衝液(335mMのTris(pH8.8)、80mMの〔NH4〕2SO4、20mMのMgCl2、0.5μg/mlのウシ血清アルブミン及び50mMの2−メルカプトエタノール)、8μlの4mMdNTP株、2μl(124pmol)R Bgl 24(配列番号3)及び3.75単位のAmpli Taq(登録商標)DNAポリメラーゼ(Perkin Elmer, cat.#N808-1012)に加えた。GeneAmp(登録商標)9600サーモサイクラー(Perkin Elmer)中でPCR増幅を行った。試料を72℃で5分間インキュベートし、連結されたアダプターの5’突出末端を充填した。試料を25〜30サイクル(95℃で1分及び72℃で3分)で増幅し、次いで72℃で10分間延長増幅した。アガロースゲルにより首尾よくアンプリコンが合成されたこと(即ち、約100bp〜1500bpを超える範囲のPCR産物の塗抹標本)が確認された後で、テスター及びドライバーアンプリコンの大規模な増幅を行った。それぞれドライバー及びテスターの合成について先に記載されたようにして、40の100μlPCR及び8つの100μlPCRを設定した。100μlの反応材料当たり2μlの小規模PCR産物は、大規模アンプリコン産生用の鋳型としての役割を果たした。追加の15〜20サイクルの増幅を行うために、上記のようなサーモサイクリングを行った。次いでドライバー及びテスターDNA用のPCR反応材料をフェノール/クロロホルムで2回抽出し、イソプロパノールで沈殿させ、70%エタノールで洗浄、Sau3AIで消化してアダプターを開裂した。テスターアンプリコンを低融点アガロースゲル上でさらに精製した。簡単に言えば、テスターアンプリコンDNA10μgを2%SeaPlaque(登録商標)ゲル(FMC Bioproducts, Rockland, ME)上で移動させた。150〜1500塩基対のフラグメントをゲルから切り出し、ゲルのスライスを、3mlのH2O、400μlの0.5M MOPS及び400μlのNaClを用い、72℃で20分間融解させた。製造業者により指示されたように、Qiagen−tip20(Qiagen, Inc., Chatsworth, CA)を用いて融解したゲルスライスからDNAを回収した。
Sau3AI消化DNAを上記のように抽出、沈殿させた。反応材料を50〜55℃の乾燥加熱ブロックに入れ、4℃で1時間インキュベートすることにより、Sau3AI消化産物全体を、1×T4 DNAリガーゼ緩衝液(NEB)中で緩衝した30μlの反応材料容量中465pmolのR Bgl24(配列番号1)及び465pmolのR Bgl 12(配列番号2)にアニーリングした。400単位のT4 DNAリガーゼ(NEB, cat.#202S)を添加してアニーリングされた産物を連結した。16℃で14時間インキュベートした後、小規模のPCRを行った。10μlの連結反応材料を、60μlのH2O、20μlの5×PCR緩衝液(335mMのTris(pH8.8)、80mMの〔NH4〕2SO4、20mMのMgCl2、0.5μg/mlのウシ血清アルブミン及び50mMの2−メルカプトエタノール)、8μlの4mMdNTP株、2μl(124pmol)R Bgl 24(配列番号3)及び3.75単位のAmpli Taq(登録商標)DNAポリメラーゼ(Perkin Elmer, cat.#N808-1012)に加えた。GeneAmp(登録商標)9600サーモサイクラー(Perkin Elmer)中でPCR増幅を行った。試料を72℃で5分間インキュベートし、連結されたアダプターの5’突出末端を充填した。試料を25〜30サイクル(95℃で1分及び72℃で3分)で増幅し、次いで72℃で10分間延長増幅した。アガロースゲルにより首尾よくアンプリコンが合成されたこと(即ち、約100bp〜1500bpを超える範囲のPCR産物の塗抹標本)が確認された後で、テスター及びドライバーアンプリコンの大規模な増幅を行った。それぞれドライバー及びテスターの合成について先に記載されたようにして、40の100μlPCR及び8つの100μlPCRを設定した。100μlの反応材料当たり2μlの小規模PCR産物は、大規模アンプリコン産生用の鋳型としての役割を果たした。追加の15〜20サイクルの増幅を行うために、上記のようなサーモサイクリングを行った。次いでドライバー及びテスターDNA用のPCR反応材料をフェノール/クロロホルムで2回抽出し、イソプロパノールで沈殿させ、70%エタノールで洗浄、Sau3AIで消化してアダプターを開裂した。テスターアンプリコンを低融点アガロースゲル上でさらに精製した。簡単に言えば、テスターアンプリコンDNA10μgを2%SeaPlaque(登録商標)ゲル(FMC Bioproducts, Rockland, ME)上で移動させた。150〜1500塩基対のフラグメントをゲルから切り出し、ゲルのスライスを、3mlのH2O、400μlの0.5M MOPS及び400μlのNaClを用い、72℃で20分間融解させた。製造業者により指示されたように、Qiagen−tip20(Qiagen, Inc., Chatsworth, CA)を用いて融解したゲルスライスからDNAを回収した。
C.アンプリコンのハイブリッド形成及び選択的増幅
精製されたテスターDNAアンプリコン約2μlを上記のようにN Bgl 24(配列番号3)及びN Bgl 12(配列番号4)に連結した。最初の控除ハイブリッド形成のために、N Bglプライマーセットに連結されたテスターアンプリコン(0.5μg)とドライバーアンプリコン(20μg)とを混合し、フェノール/クロロホルムで抽出し、エタノールで沈殿させた。4μlのEE×3緩衝液(30mMのEPPS、20℃におけるpH8.0〔Sigma, St. Louis, MO〕、3mMのEDTA)にDNAを再懸濁し、35μlの鉱油を重層した。加熱変性(99℃で3分)させた後、変性したDNAに5MのNaCl1μlを加え、DNAを67℃で20時間ハイブリダイズした。水相を新しい管に移し、該試料に8μlのtRNA(5mg/ml)、次いで390μlのTE〔10mMのTris(pH8.0)及び1mMのEDTA〕を加えた。ハイブリダイズしたDNA溶液80μlを、H2O480μl、5×PCR緩衝液(上記)160μl、4mMのdNTP64μl及びAmpliTaq(登録商標)ポリメラーゼ6μl(30単位)に加えた。該溶液を72℃で5分間インキュベートして、連結されたN Bgl 24プライマーによって形成された5’オーバーハングを充填した。N Bgl 24(配列番号3、20μlのH2O中1.24nmol)を加え、反応材料をアリコート(100μl/管)に分け、上記のように10サイクルの増幅を行った。反応材料をプールし、フェノール/クロロホルムで2回抽出し、イソプロパノールで沈殿させ、70%エタノールで洗浄して、40μlのH2Oに再懸濁した。一本鎖DNAをヤエナリヌクレアーゼ(MBN)により除去した。簡単に言えば、増幅されたDNA20μlを、販売業者により記載のように、40μlの反応材料中20単位のMBN(NEB)で消化した。MBN消化物に50mMのTris(pH8.8)160μlを加えた。酵素を99℃で5分間熱不活化した。80μlのMBN消化DNAを上記のようにしてさらに15サイクルPCR増幅した。再び反応材料をプールし、フェノール/クロロホルムで2回抽出、イソプロパノールで沈殿させ、70%エタノールで洗浄、H2Oに再懸濁した。次いで、増幅されたDNA(3〜5μl)をSau3AIで消化し、上記のように抽出、沈殿させた。最終DNAペレットを100μlのTEに再懸濁した。
精製されたテスターDNAアンプリコン約2μlを上記のようにN Bgl 24(配列番号3)及びN Bgl 12(配列番号4)に連結した。最初の控除ハイブリッド形成のために、N Bglプライマーセットに連結されたテスターアンプリコン(0.5μg)とドライバーアンプリコン(20μg)とを混合し、フェノール/クロロホルムで抽出し、エタノールで沈殿させた。4μlのEE×3緩衝液(30mMのEPPS、20℃におけるpH8.0〔Sigma, St. Louis, MO〕、3mMのEDTA)にDNAを再懸濁し、35μlの鉱油を重層した。加熱変性(99℃で3分)させた後、変性したDNAに5MのNaCl1μlを加え、DNAを67℃で20時間ハイブリダイズした。水相を新しい管に移し、該試料に8μlのtRNA(5mg/ml)、次いで390μlのTE〔10mMのTris(pH8.0)及び1mMのEDTA〕を加えた。ハイブリダイズしたDNA溶液80μlを、H2O480μl、5×PCR緩衝液(上記)160μl、4mMのdNTP64μl及びAmpliTaq(登録商標)ポリメラーゼ6μl(30単位)に加えた。該溶液を72℃で5分間インキュベートして、連結されたN Bgl 24プライマーによって形成された5’オーバーハングを充填した。N Bgl 24(配列番号3、20μlのH2O中1.24nmol)を加え、反応材料をアリコート(100μl/管)に分け、上記のように10サイクルの増幅を行った。反応材料をプールし、フェノール/クロロホルムで2回抽出し、イソプロパノールで沈殿させ、70%エタノールで洗浄して、40μlのH2Oに再懸濁した。一本鎖DNAをヤエナリヌクレアーゼ(MBN)により除去した。簡単に言えば、増幅されたDNA20μlを、販売業者により記載のように、40μlの反応材料中20単位のMBN(NEB)で消化した。MBN消化物に50mMのTris(pH8.8)160μlを加えた。酵素を99℃で5分間熱不活化した。80μlのMBN消化DNAを上記のようにしてさらに15サイクルPCR増幅した。再び反応材料をプールし、フェノール/クロロホルムで2回抽出、イソプロパノールで沈殿させ、70%エタノールで洗浄、H2Oに再懸濁した。次いで、増幅されたDNA(3〜5μl)をSau3AIで消化し、上記のように抽出、沈殿させた。最終DNAペレットを100μlのTEに再懸濁した。
D.引き続いてのハイブリッド形成/増幅段階
上記のようにして、先のハイブリッド形成/選択的増幅材料由来のDNA100ngをJ Bglプライマーセット(配列番号5及び配列番号6)に連結した。このDNA(50ng)を20μgのドライバーアンプリコンと混合し、上記のようにしてハイブリッド形成及び増幅手順を繰り返したが、但し、サーモサイクリングの間の延長温度は70℃であって、N Bglプライマーセット(配列番号3及び配列番号4)の場合の72℃ではなく、最終増幅段階(NBN消化後の)は25サイクルであった。次いで2回目のハイブリッド形成−増幅産物100ngをN Bglプライマーセット(配列番号3及び配列番号4)に連結し、この材料200pgと20μgのドライバーアンプリコンとを混合し、上記のようにして3回目のハイブリッド形成/増幅を行ったが、最終増幅は25サイクルであった。
上記のようにして、先のハイブリッド形成/選択的増幅材料由来のDNA100ngをJ Bglプライマーセット(配列番号5及び配列番号6)に連結した。このDNA(50ng)を20μgのドライバーアンプリコンと混合し、上記のようにしてハイブリッド形成及び増幅手順を繰り返したが、但し、サーモサイクリングの間の延長温度は70℃であって、N Bglプライマーセット(配列番号3及び配列番号4)の場合の72℃ではなく、最終増幅段階(NBN消化後の)は25サイクルであった。次いで2回目のハイブリッド形成−増幅産物100ngをN Bglプライマーセット(配列番号3及び配列番号4)に連結し、この材料200pgと20μgのドライバーアンプリコンとを混合し、上記のようにして3回目のハイブリッド形成/増幅を行ったが、最終増幅は25サイクルであった。
HGBV接種前及び急性期T−1053血漿に体して行われた代表相違分析(RDA)から得られた産物の2%アガロースゲルを図14に示す。図14を参照すると、レーン1は、適切なサイズ(bp)のDNAフラグメントを有する150ngのHaeIII消化Phi−X174DNAマーカー(NEB)を含んでいる。ドライバーアンプリコン(レーン2)及びテスターアンプリコン(レーン3)の複合性は、試料中に見られるDNA産物の塗抹標本によって実証されている。この複合性は、テスター配列が1回目(レーン4)、2回目(レーン5)又は3回目(レーン6)のハイブリッド形成/選択的増幅を受けると劇的に低下する。
E.相違産物のクローニング
相違産物を、以下のようにpBluescriptIIKS+(Stratagene, La Jolla, CA, cat.#212207)のBamHI部位にクローニングした。0.5μgのpBluescriptIIをBamHI(10単位、NEB)で消化し、販売業者により指示されたように子牛腸管ホスファターゼ(10単位、NEB)を用いて5’脱リン酸化した。プラスミドをフェノール:クロロホルムで抽出し、エタノールで沈殿させ、70%エタノールで洗浄、10μlのH2Oに再懸濁した(最終濃度:1μl当たり約50ngのpBluescriptII)。2回目のハイブリッド形成/増幅産物由来の4つの最大バンドを上記のようにして2%低融点アガロースゲルから切り出した。TakaraDNA連結反応キット(Takara Biochemical, Berkeley, CA)を用い、融解した(72℃、5分)ゲルスライス4μlを、50μlの反応材料中50ngのBamHI−カット、脱リン酸化pBluescriptIIに連結した。16℃で3.5時間インキュベートした後、8μlの連結反応材料を用いて、販売業者に指示されたように、 E.coliコンピテントXL−1Blue細胞(Stratagene)を形質転換した。アンピシリン(150μg/ml)を補ったLBプレート上に形質転換混合物をのせ、37℃で一晩インキュベートした。得られたコロニーを液体培養中で増殖させ、ミニプレプ(miniprep)プラスミドDNAを当該分野に記載のようにして分析し、クローニング産物の存在を確認した。
相違産物を、以下のようにpBluescriptIIKS+(Stratagene, La Jolla, CA, cat.#212207)のBamHI部位にクローニングした。0.5μgのpBluescriptIIをBamHI(10単位、NEB)で消化し、販売業者により指示されたように子牛腸管ホスファターゼ(10単位、NEB)を用いて5’脱リン酸化した。プラスミドをフェノール:クロロホルムで抽出し、エタノールで沈殿させ、70%エタノールで洗浄、10μlのH2Oに再懸濁した(最終濃度:1μl当たり約50ngのpBluescriptII)。2回目のハイブリッド形成/増幅産物由来の4つの最大バンドを上記のようにして2%低融点アガロースゲルから切り出した。TakaraDNA連結反応キット(Takara Biochemical, Berkeley, CA)を用い、融解した(72℃、5分)ゲルスライス4μlを、50μlの反応材料中50ngのBamHI−カット、脱リン酸化pBluescriptIIに連結した。16℃で3.5時間インキュベートした後、8μlの連結反応材料を用いて、販売業者に指示されたように、 E.coliコンピテントXL−1Blue細胞(Stratagene)を形質転換した。アンピシリン(150μg/ml)を補ったLBプレート上に形質転換混合物をのせ、37℃で一晩インキュベートした。得られたコロニーを液体培養中で増殖させ、ミニプレプ(miniprep)プラスミドDNAを当該分野に記載のようにして分析し、クローニング産物の存在を確認した。
2回目のハイブリッド形成/増殖段階から得られた4つの最大産物のクローニングに加えて、3回目のハイブリッド形成/増殖段階から得られた産物の全集団をpBluescriptIIにクローニングした。50ngのpBluescriptIIベクター(上記のように合成した)を、上記のようにして、50μlの反応材料中10ngの3回目のハイブリッド形成/増殖産物に連結した。16℃で2時間インキュベートした後、10μlの連結反応産物を用いて、E.coliコンピテントXL−1Blue細胞を形質転換した。得られた形質転換産物由来の60のコロニーを増殖させ、ミニプレプDNAを合成し、上記のような当該分野において公知の方法で分析した。制限エンドヌクレアーゼ消化及びドットブロットハイブリッド形成法をユニーククローンを特定するために用いた。
(実施例4)
HGBVゲノムの抗原領域をコードするcDNAクローンの免疫分離
A.クローニング材料源としての濃縮ウイルスの調製
実施例3に示されている方法に加えて、以下の分離手順を用いて、HGBBVゲノムを分離した。3匹のタマリン(T−1055、T−1038及びT−1049)に、実施例2に記載のタマリンT−1053から調製した血清を接種した。表5を参照すると、接種後11日目までに3匹全てのタマリンに肝酵素値の上昇が見られた。タマリンT−1055を接種後12日目に殺し、タマリンT−1038及びT−1049を接種後14日目に殺した。これら3匹のタマリンそれぞれから由来の血清約3〜4mlをプールし、全量約11.3mlを得た。プールされた血清を15℃で15分間10,000×gで遠心して清澄化した。次いでこれを連続的に0.8、0.45、0.2及び0.1μmの注射器フィルターに通した。次いで該濾過された材料を、SW41−Tiローター中、4℃で68分間、41,000rpmで、0.3mlのCsClクッション〔密度:10mMのTris、150mMのNaCl、1mMのEDTA(pH8.0)中1.6g/ml〕を介して遠心して濃縮した。約0.6mlのCsCl層を遠心後に取り出し、3つの0.2mlアリコートに分け、−70℃で貯蔵した。
HGBVゲノムの抗原領域をコードするcDNAクローンの免疫分離
A.クローニング材料源としての濃縮ウイルスの調製
実施例3に示されている方法に加えて、以下の分離手順を用いて、HGBBVゲノムを分離した。3匹のタマリン(T−1055、T−1038及びT−1049)に、実施例2に記載のタマリンT−1053から調製した血清を接種した。表5を参照すると、接種後11日目までに3匹全てのタマリンに肝酵素値の上昇が見られた。タマリンT−1055を接種後12日目に殺し、タマリンT−1038及びT−1049を接種後14日目に殺した。これら3匹のタマリンそれぞれから由来の血清約3〜4mlをプールし、全量約11.3mlを得た。プールされた血清を15℃で15分間10,000×gで遠心して清澄化した。次いでこれを連続的に0.8、0.45、0.2及び0.1μmの注射器フィルターに通した。次いで該濾過された材料を、SW41−Tiローター中、4℃で68分間、41,000rpmで、0.3mlのCsClクッション〔密度:10mMのTris、150mMのNaCl、1mMのEDTA(pH8.0)中1.6g/ml〕を介して遠心して濃縮した。約0.6mlのCsCl層を遠心後に取り出し、3つの0.2mlアリコートに分け、−70℃で貯蔵した。
次いで、タマリンT−1034にこのペレット化材料(正常なタマリン血清中で調製した)の10−6希釈物0.25mlを接種した。先ず、接種後2週間でT−1034にALT肝酵素値の上昇が認められ、該値はその後7週間上昇を継続し、最終的に接種後10週までに正常化した(図30、実施例14を参照されたい)。この実験は、プールされたタマリン血清から濃縮した材料の感染性を示した。該材料は比較的高いタイターを有することが示されたので、このウイルス濃縮源を、以下に記載のような、cDNAライブラリー構築用の核酸源として用いた。
B.cDNAライブラリーの構築
販売業者の指示により市販のRNA抽出キット(Stratagene, La Jolla, CA)を用いて、濃縮ウイルス(上記)のアリコート(0.2ml)からRNAを抽出した。最終沈殿段階の前に、試料を4つの等量のアリコートに分け、次いで5μg/mlの酵母tRNAの存在下に沈殿させた。これらのアリコートの中一つだけをcDNA合成用に用いた。他のものは−80℃で貯蔵した。製造業者に指示されたように、市販のキット(Stratagene, La Jolla, CA)を用いて、RNAから、リン酸化され、ブラント末端化二本鎖cDNAを合成した。次いで、製造業者に指示されたように、T4DNAリガーゼキット(Stratagene, La, Jolla, CA)を用いて、10μlの反応材料容量中で二本鎖リンカー/プライマーをcDNA末端(センスストランド、配列番号7;アンチセンスストランド、配列番号8)に連結した。これによって、全てのcDNAを、NotI及びEcoRI制限酵素認識部位を含む同一の5’及び3’末端を有する混合物として得た〔G. Reyes及びJ. Kim, Mol. Cell. Probes 5:473-481 (1991); A. Akowitz及びL. Manuelidis, Gene 81:295-306 (1989); 並びにG. Inchauspeら, Viral Hepatitis and Liver Disease, F.B. Hollingerら, Eds., 382-387ページ(1991)〕。次いで、全cDNAがそれらの配列とは無関係に増幅されるように、リンカー/プライマーのセンスストランドオリゴヌクレオチドをPCR反応におけるプライマーとして用いた。この手順により、全cDNA集団内に存在する微量cDNAが、効率的にクローニングされるレベルにまで増幅され、それによって出発材料内の配列を表すcDNAライブラリーが形成された。
販売業者の指示により市販のRNA抽出キット(Stratagene, La Jolla, CA)を用いて、濃縮ウイルス(上記)のアリコート(0.2ml)からRNAを抽出した。最終沈殿段階の前に、試料を4つの等量のアリコートに分け、次いで5μg/mlの酵母tRNAの存在下に沈殿させた。これらのアリコートの中一つだけをcDNA合成用に用いた。他のものは−80℃で貯蔵した。製造業者に指示されたように、市販のキット(Stratagene, La Jolla, CA)を用いて、RNAから、リン酸化され、ブラント末端化二本鎖cDNAを合成した。次いで、製造業者に指示されたように、T4DNAリガーゼキット(Stratagene, La, Jolla, CA)を用いて、10μlの反応材料容量中で二本鎖リンカー/プライマーをcDNA末端(センスストランド、配列番号7;アンチセンスストランド、配列番号8)に連結した。これによって、全てのcDNAを、NotI及びEcoRI制限酵素認識部位を含む同一の5’及び3’末端を有する混合物として得た〔G. Reyes及びJ. Kim, Mol. Cell. Probes 5:473-481 (1991); A. Akowitz及びL. Manuelidis, Gene 81:295-306 (1989); 並びにG. Inchauspeら, Viral Hepatitis and Liver Disease, F.B. Hollingerら, Eds., 382-387ページ(1991)〕。次いで、全cDNAがそれらの配列とは無関係に増幅されるように、リンカー/プライマーのセンスストランドオリゴヌクレオチドをPCR反応におけるプライマーとして用いた。この手順により、全cDNA集団内に存在する微量cDNAが、効率的にクローニングされるレベルにまで増幅され、それによって出発材料内の配列を表すcDNAライブラリーが形成された。
製造業者により指示されたGeneAmp PCRキット(Perkin-Elmer)を用いて、PE−9600サーモサイクラー中で、センスストランドオリゴヌクレオチドプライマーの存在下に上記連結材料の1μlアリコートに対してPCRを行った(最終濃度:1μM;反応材料容量:50μl)。以下のようにして30サイクルのPCRを行った:94℃で0.5分間の変性、55℃で0.5分間のアニーリング及び72℃で1.5分間の延長。次いで、得られた産物の1μlアリコートを上記のようにして再増幅した。次いで、最終PCR反応産物を等量のフェノール−クロロホルム(1:1、v/v)で1回、等量のクロロホルムで1回抽出し、次いで、酢酸ナトリウム(最終濃度、0.3M)及び2.5容量の無水エタノールを加えて、ドライアイス上で10分間沈殿させた。得られたDNAペレットを水に再懸濁し、製造業者に指示されたように、制限酵素EcoRI(New England Biolabs)で消化した。次いで、製造業者に指示されたようにDNA結合樹脂(Prep-a-Gene, BioRad Laboratories)を用いて、消化されたcDNAを反応混合物から精製し、20μlの蒸留水中で溶離した。
cDNA(8μl)を、30μlの反応材料容量中3μgのラムダ gt11ベクターDNAアーム(Stratagen, La Jolla, CA)に4℃で1〜5日の間に連結した。製造業者に指示されたようにGigaPackIIIGoldパッケージ抽出物(Stratagene, 前出)を用いて、連結材料11μlをファージヘッドにパッケージした。得られたライブラリーは、組換え頻度が89.3%で、平均インサートサイズが約350塩基対の合計約173万メンバー(PFU)を含んでいた。
C.組換えGB cDNAライブラリーの免疫スクリーニング
cDNAライブラリーの免疫スクリーニングに用いた抗血清は、接種後に血清肝酵素レベルに上昇を示したタマリンから採取した。免疫スクリーニングには、2つの別個の抗血清プールを用いた。第1のプールは、2匹の動物(T−1048及びT−1051;それぞれ実施例1、表2と、実施例2、表5を参照されたい)由来の血清を含み、第2のプールは、1匹の動物(T−1034;図30、実施例14を参照されたい)由来の血清を含むものであった。用いられた特定の血清を表6に示す。
cDNAライブラリーの免疫スクリーニングに用いた抗血清は、接種後に血清肝酵素レベルに上昇を示したタマリンから採取した。免疫スクリーニングには、2つの別個の抗血清プールを用いた。第1のプールは、2匹の動物(T−1048及びT−1051;それぞれ実施例1、表2と、実施例2、表5を参照されたい)由来の血清を含み、第2のプールは、1匹の動物(T−1034;図30、実施例14を参照されたい)由来の血清を含むものであった。用いられた特定の血清を表6に示す。
cDNAライブラリーの免疫スクリーニングに用いるためにこれらの試料を選択した時点では、これらの試料は、1.4又は1.7組換えCKSタンパク質(実施例13)とのそれらの免疫反応性について試験されていなかった。従って、本明細書に示されている結果は、用いられた抗血清中のこれらの組換えタンパク質に対するHGBV抗体の有無に関するいずれの情報とも関係なく得られたものである。
組換えファージの免疫分離に用いた手順は、Young及びDavisにより記載された方法〔R.A. Young及びR.W. Davis, PNAS 80:1194-1198 (1983)〕に基づき、以下のように変更を加えたものであった。一方は、T−1048及びT−1051からプールした抗血清を用い、他方はT−1034由来の抗血清を用いた2回の免疫スクリーニング実験を行った。いずれに場合にも、抗体とE.coliタンパク質との非特異的相互作用を低減させるために、使用前に一次抗血清をE.coli抽出物に予備吸着させた。最初の実験では、T−1048/T−1051抗血清プールを用いて129万個の組換えファージを免疫スクリーニングした。2回目の実験では、T−1034抗血清を用いて30万個の組換えファージを免疫スクリーニングした。組換えファージライブラリーをE.coliストランドY1090r−のローン上に置き、37℃で3.5時間増殖させた。次いで、プレートをIPTG(10mM)で飽和したナイロンフィルターで覆い、42℃で3.5時間インキュベートした。次いでフィルターを、1%のBSA、1%のゼラチン及び3%のTween−20(「ブロッキング緩衝液」)を含むTris−塩水緩衝液中、22℃で1時間ブロックした。次いでフィルターを一次抗血清(ブロッキング緩衝液中の1:100希釈物)中、4℃で16時間インキュベートした。次いで一次抗血清を取り出して次回のプラーク精製用に取って置き、0.1%Tween−20を含むTris−塩水中でフィルターを4回洗浄した。次いで、フィルターを125−I−標識(又はアルカリ−ホスファターゼ結合)ヤギ抗ヒトIgG(Jackson ImmunoResearch, West Grove, PAから入手可能)を含むブロッキング緩衝液中22℃で60分間インキュベートし、上記のように洗浄し、次いでX線フィルムに暴露(又はJ. Sambrookら, Molecular Cloning: A Laboratory Manual, 第2版, Cold Spring Harbor Press, Cold Spring Harbor, N.Y., 1989におけるような確定した手順に従って発色)した。このライブラリーから5種の免疫陽性ファージ(4−3BI、48−1A1、66−3A1、70−3A1、78−1C1)を分離し、次いで、上記の方法を用いて3匹の感染タマリン(T−1048、T−1051、T−1034)由来の抗血清に対する結合特異性についてテストした。これらの組換え体は、3匹の感染タマリンそれぞれに由来する回復期血清と反応したが予備接種した血清とは反応しなかったポリペプチドをコードした(データは示さず)。
組換えファージによりコードされたポリペプチドの免疫学的反応性の特異性を証明するために、EcoRIクローニング部位に対して5’位に位置するプライマー(配列番号9)及び3’位に位置するプライマー(配列番号10)を用いて、PCRにより、各cDNAをラムダファージゲノムから回収した。次いで、PCR産物をEcoRIで消化し、実施例13に記載のようにして、E.coli発現プラスミドpJO201に次いで連結した。該cDNAをpJO201のEcoRI部位に挿入することにより、このcDNAの翻訳読み取り枠をラムダファージクローン中にあるがままに維持した。pJO201発現ベクター中のサブクローンを、4−3B1.1、48−1A1.1、66−3A1.49、70−3A1.37及び78−1C1.17とした。タマリンT−1034、T−1048及びT−1051由来の回復期血清(1:100希釈物)を含むこれらのcDNAを発現する培養物から調製されたE.coli溶解物の免疫ブロット分析(実施例13におけるような)により、各CKS融合タンパク質について予測されたサイズのタンパク質との特異的免疫学的反応性が示された(データは示していない)。該cDNAそれぞれのDNA配列を決定したが、これらのクローンはHGBV−Bウイルス(配列番号11)の配列とほぼ100%の配列同一性を有していることが見いだされた。4−3B1.1インサート(配列番号12及び13)の配列は、その全体は決定されなかったが、配列決定部分は、塩基対6834−7458由来のHGBV−B(配列番号11)中の配列部分との99.5%の配列同一性を示す。クローン4−3B1.1から得られた配列との同一性を示すHGBV−B(配列番号11)配列のこの領域は、+1読み取り枠に翻訳され、配列番号14として配列リストに示されている。48−1A1.1インサート(配列番号15)の配列は、塩基対4523−4752由来のHGBV−B(配列番号11、実施例9を参照されたい)の配列の部分との100%配列同一性を示す。配列番号15に対応するDNA配列は+1読み取り枠に翻訳され、配列番号16として配列リストに示されている。66−3A1.49インサート(配列番号17)の配列は、クローン48−1A1.1の配列と実質的に100%の配列同一性を示し、従って、配列リストにはタンパク質翻訳は示さない。70−3A1.37インサート(配列番号18)の配列は、塩基対6450−6732由来のHGBV−B(配列番号11)の配列の部分と100%の配列同一性を示すが、但し、HGBV−B配列(配列番号11)の塩基6630−6632に対応する3つの塩基対がかけている。配列番号18に対応するDNA配列は+2読み取り枠に翻訳され、配列番号19として配列リストに示されている。78−1C1.17インサート(配列番号20)の配列は、クローン70−3A1.37の配列と100%の配列同一性を示し、従って、配列リストにはタンパク質の翻訳は示さない。これらのデータは、ラムダgt11cDNAライブラリーから分離されたcDNAクローンがHGBV病原体のゲノムから誘導され、該ライブラリーがGB感染タマリン由来の血清によって免疫学的に特異的に認識されたポリペプチドをコードすることを示している。クローン48−1A1.1(「クローン48」)4−3B1.1、66−3A1.49、70−3A1.37及び78−1C1.17は、前記のAmerican Type Culture Collectionに寄託されている。
(実施例5)
HGBVクローンのDNA配列分析
実施例3で得られたユニーククローンをキット形態〔Sequenase(登録商標)バージョン2.0, USB〕のジデオキシヌクレオチド鎖停止法(Sangerら, 前出)を用いて配列形態した。これらの配列はオーバーラップしておらず、クローン4(配列番号21)、クローン2(配列番号22)、クローン10(配列番号23)、クローン11(配列番号24)、クローン13(配列番号25)、クローン16(配列番号26)、クローン18(配列番号27)、クローン23(配列番号28)、クローン50(配列番号29)及びクローン119(配列番号30)として配列リストに示されている。クローン4、2、10、11、13、16、18、23、50及び119はA.T.C.C.に寄託されている。クローン2はA.T.C.C.受託番号69556、クローン4はA.T.C.C.受託番号69557、クローン10はA.T.C.C.受託番号69558、クローン16はA.T.C.C.受託番号69559、クローン18はA.T.C.C.受託番号69560、クローン23はA.T.C.C.受託番号69561、クローン50はA.T.C.C.受託番号69562、クローン11はA.T.C.C.受託番号69613、クローン13はA.T.C.C.受託番号69611、及びクローン119はA.T.C.C.受託番号69612が付与された。
HGBVクローンのDNA配列分析
実施例3で得られたユニーククローンをキット形態〔Sequenase(登録商標)バージョン2.0, USB〕のジデオキシヌクレオチド鎖停止法(Sangerら, 前出)を用いて配列形態した。これらの配列はオーバーラップしておらず、クローン4(配列番号21)、クローン2(配列番号22)、クローン10(配列番号23)、クローン11(配列番号24)、クローン13(配列番号25)、クローン16(配列番号26)、クローン18(配列番号27)、クローン23(配列番号28)、クローン50(配列番号29)及びクローン119(配列番号30)として配列リストに示されている。クローン4、2、10、11、13、16、18、23、50及び119はA.T.C.C.に寄託されている。クローン2はA.T.C.C.受託番号69556、クローン4はA.T.C.C.受託番号69557、クローン10はA.T.C.C.受託番号69558、クローン16はA.T.C.C.受託番号69559、クローン18はA.T.C.C.受託番号69560、クローン23はA.T.C.C.受託番号69561、クローン50はA.T.C.C.受託番号69562、クローン11はA.T.C.C.受託番号69613、クローン13はA.T.C.C.受託番号69611、及びクローン119はA.T.C.C.受託番号69612が付与された。
配列は、BLASTNアルゴリズム(Altschulら, J. Mol. Biol. 215:403-410[1990])を用いて、GenBankデータベースで検索した。これらの配列はいずれもGenBankでは見つからなかったが、これは、これらの配列がまだ文献において特性分析されていないことを示している。該DNA配列は6つの可能な読み取り枠に翻訳され、配列リスト(配列番号21は配列番号31〜36に、配列番号22は配列番号37〜42に、配列番号23は配列番号43〜48に、配列番号26は配列番号49〜54に、配列番号27は配列番号55〜60に、配列番号28は配列番号61〜66に、配列番号29は配列番号67〜72に翻訳する)に示されている。配列番号24は配列番号73(実施例9に記載)に含まれ、配列番号74〜79に翻訳されている。配列番号25〜30は配列番号80(実施例9に記載)に含まれ、配列番号81〜86に翻訳されている。BLASTXアルゴリズム(Gishら, Nature Genetics 3:266-272 [1993])を用い、翻訳された配列をSWISS−PROTデータベースの検索に用いた。ここでも、これらの配列はSWISS−PROTでは見つからず、それは、これらの配列がまだ文献において特性分析されていないことを示している。
BLASTN、BLASTX及びFASTdbアルゴリズムを用いて相同体を検索すると、肝炎のCウイルスと低くはあっても幾分かの配列類似性が示される(以下の表7)。特に、クローン4(配列番号35)、10(配列番号44)、11(GB−4の残基1〜166、枠3〔配列番号76〕)、16(配列番号50)、23(配列番号65)、50(配列番号70及び72)並びに119(GB−Aの残基912〜988、枠3〔配列番号83〕)の翻訳は、アミノ酸レベルでの種々のHCV分離体に対する24.1〜45.1%の範囲の相同性がある。特に重要なのは、クローン10(配列番号44)の翻訳体が、HCVの推定RNA依存性RNAポリメラーゼに対してわずかな相同性を示したことである。他の正鎖ウイルスの推定RNA依存性RNAポリメラーゼ中に存在する保存アミノ酸(Jiangら, PNAS 90:10539-10543 [1993])とクローン10(配列番号44)の推定アミノ酸翻訳体との比較により、他のRNA依存性RNAポリメラーゼの保存アミノ酸残基もクローン10(配列番号44)中に保存されていることが明らかになった。これには、RNA依存性RNAポリメラーゼの正準(canonical)GDD(Gly−Asp−Asp)シグネチャー配列が含まれている。従って、クローン10(配列番号44)は、ウイルス性RNA依存性RNAポリメラーゼをコードするものと思われる。驚くべきことには、BLASTNアルゴリズムを用いた場合、クローン10(配列番号44)のみが、ヌクレオチドレベルでHCVとの配列相同性を示した。アミノ酸レベルで低いHCV相同性を有するクローン4(配列番号21)、16(配列番号26)、23(配列番号28)及び50(配列番号29)並びに119(配列番号30)は、GenBankの検索において、BLASTNでは検出されなかった。さらに、クローン2(配列番号37〜42)、13(配列番号25及び37〜42)並びに18(配列番号27及び55〜60)は、上記のGenBank又はSWISS−PROTOで検索した場合に、HCVに対する有意なヌクレオチド又はアミノ酸相同性は、下記のように示さなかった。
(実施例6)
HGBVクローンの外因性
HGBVクローンは、正常又はHGBV感染タマリン肝臓DNA、正常なヒトリンパ球DNA、酵母DNA又はE.coliDNAには検出されなかった。これは、サザン法によりHGBVクローン2(配列番号22)及び16(配列番号26)について示された。さらに、全HGBVクローンをゲノムPCRで分析して、タマリン、ヒト、酵母及びE.coliゲノムに関するHGBV配列の外因性起源を確認した。これらのデータは実施例5に記載のHGBV配列のウイルス性性質と一致している。
HGBVクローンの外因性
HGBVクローンは、正常又はHGBV感染タマリン肝臓DNA、正常なヒトリンパ球DNA、酵母DNA又はE.coliDNAには検出されなかった。これは、サザン法によりHGBVクローン2(配列番号22)及び16(配列番号26)について示された。さらに、全HGBVクローンをゲノムPCRで分析して、タマリン、ヒト、酵母及びE.coliゲノムに関するHGBV配列の外因性起源を確認した。これらのデータは実施例5に記載のHGBV配列のウイルス性性質と一致している。
A.サザンブロット法分析
HGBV感染タマリン及び正常なタマリンの肝臓ホモジネートを低速ペレット化してタマリンの肝臓の核を得た(以下に記載)。販売業者に指示されたように市販のキット(USB cat.#73750)を用いて核からDNAを抽出した。タマリンDNAを抽出過程の間にRNaseで処理した。ヒト胎盤DNA(Clontech, Palo Alto, CA)、酵母DNA(Saccharomyces cerevisiae, Clontech)及びE.coliDNA(Sigma)を市販源から得た。
HGBV感染タマリン及び正常なタマリンの肝臓ホモジネートを低速ペレット化してタマリンの肝臓の核を得た(以下に記載)。販売業者に指示されたように市販のキット(USB cat.#73750)を用いて核からDNAを抽出した。タマリンDNAを抽出過程の間にRNaseで処理した。ヒト胎盤DNA(Clontech, Palo Alto, CA)、酵母DNA(Saccharomyces cerevisiae, Clontech)及びE.coliDNA(Sigma)を市販源から得た。
販売業者の指示に従って各DNA試料をBamHI(NEB)で消化した。消化されたDNA(10μg)及びRNA産物(実施例3Bからそれぞれ0.5μgずつ)を1%アガロースゲル上で電気泳動させ、Sambrookらにより記載(pp.9.34ff)のようにHybond−N+ナイロンメンブラン(Amersham, Arlington Heights, IL)にキャピラリーでブロットした。DNAをメンブラン販売業者により指示されたようにアルカリ処理してメンブランに固定した。メンブランをRapid Hyb溶液(Amersham)中、65℃で30分間プレハイブリダイズした。
HGBV配列の放射線標識プローブをPCRにより調製した。1×PCR緩衝液II(Perkin Elmer)、2mMのMgCl2、20μMのdNTP、それぞれ1μMのクローン特異性センス及びアンチセンスプライマー(クローン2については、配列番号87及び88;クローン4については、配列番号89及び90;クローン10については、配列番号91及び92;クローン16については、配列番号93及び94;クローン18については、配列番号95及び96;クローン23については、配列番号97及び98;クローン50については、配列番号99及び100)、1ngのHGBVクローンプラスミド(実施例3〔E〕に記載)、60μCiα−32P−dATP(3000Ci/mmol)及び1.25単位のAmpliTaq(登録商標)ポリメラーゼ(Perkin Elmer)を用いて50μlのPCRを形成した。反応材料を、94℃で30秒、55℃で30秒及び72℃で30秒インキュベートして、合計30サイクルの増幅を行い、次いで72℃で3分間最終延長した。組み込まれなかった標識を、販売業者に指示されたようにQuick−Spin(登録商標)G−50スピンカラム(Boehringer Mannheim, Indianapolis, IN)により除去した。プレハイブリダイズしたメンブランに添加する前にプローブを変性させた(99℃、2分)。
プレハイブリダイズしたメンブラン(2×106dpm/ml)に、放射線標識したプローブを加え、Rapid Hyb(登録商標)販売業者に指示されたように、フィルターを65℃で2.5時間ハイブリダイズした。ハイブリダイズしたメンブランを、緩やかな厳格さ(1×SSC、65℃で0.1%SDS)で洗浄し、次いで増感板を用い、−80℃で72時間オートラジオグラフフィルムに暴露した。これらの条件は、類似の放射線標識プローブを用いて単一コピーの遺伝子を検出するために考案された。
結果は、クローン2(配列番号22)及びクローン16(配列番号26)配列が、正常又はHGBV感染タマリンの肝臓由来のDNA(それぞれ図15及び図16、レーン1B及び3B)、ヒトDNA(図15及び図16、レーン1A)、酵母DNA(図15及び図16、レーン2A)又はE.coliDNA(図15及び図16、レーン3A)にハイブリダイズしなかったことを示している。さらに、ドライバーアンプリコンDNA(図15及び図16、レーン4A、実施例2.B.に記載の予備HGBV接種されたタマリン血漿から誘導された)の場合にもハイブリッド形成は全く検出されなかった。対比的に、テスターアンプリコン(図15及び図16、レーン6A、実施例2.Bに記載の総核酸抽出及び逆転写ステップを用いた感染性HGBVタマリン血漿から誘導された)及び3回の控除/選択的増幅の産物(それぞれ1回目、2回目及び3回目の控除/選択的増幅からの産物に関する図15及び図16、レーン7A、8A及び4B)の場合には強力なハイブリッド形成シグナルが認められた。これらのデータは、HGBVクローン2(配列番号22)及び16(配列番号26)が感染源から増幅された核酸配列中に検出され、HGBVクローン2(配列番号22)及び16(配列番号26)が、タマリン、ヒト、酵母又はE.coliゲノムDNA配列からは誘導されないことを示している。
B.ゲノムのPCR分析
さらにHGBV配列の外因性を示し且つそれらのウイルス起源を支持するために、タマリン、ヒト、酵母及びE.coli由来のゲノムDNAに対してPCRを行った。前出のJ. Sambrookらによる記載のようにして、 正常なタマリンの腎臓及び肝臓組織由来のDNAを合成した。酵母、アカゲザルの腎臓及びヒト胎盤DNAをClonotechから得た。E.coliDNAはSigmaから得た。
さらにHGBV配列の外因性を示し且つそれらのウイルス起源を支持するために、タマリン、ヒト、酵母及びE.coli由来のゲノムDNAに対してPCRを行った。前出のJ. Sambrookらによる記載のようにして、 正常なタマリンの腎臓及び肝臓組織由来のDNAを合成した。酵母、アカゲザルの腎臓及びヒト胎盤DNAをClonotechから得た。E.coliDNAはSigmaから得た。
販売業者に指示されたように主としてPerkin-Elmer-Cetus製のGeneAmp(登録商標)試薬を用いてPCRを行った。各100μlの反応材料に対して300ngのゲノムDNAを用いた。HGBVクローン化配列(クローン2については、配列番号87及び88;クローン4については、配列番号89及び90;クローン10については、配列番号91及び92;クローン16については、配列番号93及び94;クローン18については、配列番号95及び96;クローン23については、配列番号97及び98;クローン50については、配列番号99及び100)由来のPCRプライマーを0.5μMの最終濃度で用いた。PCRを35サイクル(94℃で1分;55℃で1分;72℃で1分)行い、次いで、72℃で7分の伸長サイクルを行った。PCR産物をアガロースゲル電気泳動により分離し、臭化エチジウムを用いて核酸を直接染色した後で紫外線照射するか及び/又はサザン法によりニトロセルロースフィルターへトランスファーした後で放射線標識プローブにハイブリダイズして視覚化した。実施例6Aに記載のようにしてプローブを形成した。Digene(Belstville, MD)製のFast−Pair Hybridization Solution中でフィルターを3〜5時間プレハイブリダイズし、次いで100−200cpm/cm2を用い、42℃で15〜25時間Fast−Pair Hybridization Solution中でハイブリダイズした。G.G. Schlauderら, J. Virol. Methods 37:189-200 (1992)に記載のようにしてフィルターを洗浄し、 増感板を用い、−70℃で15〜72時間、Kodak X−Omat−ARフィルムに暴露した。
図17は、臭化エチジウム染色した1.5%アガロースゲルを示している。図18は、クローン16(配列番号26)由来の放射線標識プローブにハイブリダイズした後の同一ゲルからのサザンブロット法によるオートラジオグラムを示す。正常なタマリン肝臓及び腎臓DNAに0.05ゲノム当量(レーン17及び18)でスパイクされたクローン16(配列番号26)配列を検出することは可能であるにも拘わらず、クローン16(配列番号26)配列は、その外因性と一致して、ゲノムPCR分析により、タマリン(図17及び図18、レーン9及び10)、アカゲザル(レーン11)又はヒトゲノムDNA(レーン12)中にも、酵母又はE.coliDNA(データは示さず)中にも検出されなかった。さらに、ヒトドーパミンD1レセプター遺伝子由来のプライマー、1000−1019塩基対(センスプライマー)及び1533−1552塩基対(アンチセンスプライマー)(GenBankアクセス番号:X55760、R.K. Sunaharaら, Nature 347:80-83[1990])は、霊長類ゲノムDNA(タマリン腎臓、タマリン肝臓、アカゲザル及びヒトのDNAに対応する図17、レーン2、3、4及び5、)由来のドーパミンD1レセプターDNAを首尾よく増幅し、それは、低コピー数(即ち、単一コピー)配列を検出するこの方法の有用性を示している。レーン1及び8は、それぞれドーパミンD1レセプター及びクローンの16プライマー(配列番号93及び94)に対するH2O対照である。レーン6は、ドーパミンレセプタープライマーで増幅した100fgのクローン16(配列番号26)プラスミドDNAを含む。レーン14、15、16及び20は、それぞれ1、3、10及び100fgのクローン16(配列番号26)プラスミドDNAを含む。レーン7及び19はマーカーである。上記のクローン2、4、10、18、23及び50に特異的なPCRプライマーを用いて類似の結果を得た(データは示さず)。クローン2(配列番号22)、4(配列番号21)、10(配列番号23)、18(配列番号27)、23(配列番号28)及び50(配列番号29)はこの時点では結果が出ていない。しかし、クローン4(配列番号21)、10(配列番号23)、18(配列番号27)及び50(配列番号29)配列は、タマリン、ヒト、酵母及びE.coliDNA(アカゲザルは試験しなかった)には検出されず、これは、これらの配列が試験したゲノムDNA源に対して外因性であり、これらの配列のウイルス起源を支持するものであることを示している。
(実施例7)
タマリン血清におけるHGBV配列の存在
接種前及び急性期T−1053血漿のHGBVクローン配列の存在をPCRにより調べた。HGBVゲノムはDNA又はRNAであり得るので、PCR及びRT−PCRを行った。特に、全核酸を実施例3(A)に記載のようにして血漿から抽出した。実施例6(B)に記載のようにして当量の5μlの血漿核酸に対してPCRを行い、主として製造者の指示に従いPerkin-Elmer-Cetus製のGeneAmp(登録商標)RNA PCRキットを用いて、PCR中1μM濃度のプライマー(クローン2については、配列番号87及び88;クローン4については、配列番号89及び90;クローン10については、配列番号91及び92;クローン16については、配列番号93及び94;クローン18については、配列番号95及び96;クローン23については、配列番号97及び98;クローン50については、配列番号99及び100)を用いてRT−PCRを行った。ランダムヘキサマーを用いてcDNA合成を開始した。
タマリン血清におけるHGBV配列の存在
接種前及び急性期T−1053血漿のHGBVクローン配列の存在をPCRにより調べた。HGBVゲノムはDNA又はRNAであり得るので、PCR及びRT−PCRを行った。特に、全核酸を実施例3(A)に記載のようにして血漿から抽出した。実施例6(B)に記載のようにして当量の5μlの血漿核酸に対してPCRを行い、主として製造者の指示に従いPerkin-Elmer-Cetus製のGeneAmp(登録商標)RNA PCRキットを用いて、PCR中1μM濃度のプライマー(クローン2については、配列番号87及び88;クローン4については、配列番号89及び90;クローン10については、配列番号91及び92;クローン16については、配列番号93及び94;クローン18については、配列番号95及び96;クローン23については、配列番号97及び98;クローン50については、配列番号99及び100)を用いてRT−PCRを行った。ランダムヘキサマーを用いてcDNA合成を開始した。
PCR産物の臭化エチジウムによる染色及びハイブリッド形成により、急性期T−1053血漿には、HGBVクローン配列2(配列番号22)、4(配列番号2)、10(配列番号23)、16(配列番号26)、18(配列番号27)、23(配列番号28)及び50(配列番号29)が存在したが、予備接種T−1053血漿には存在しないことが示された(データは示さず)。さらに、HGBVクローン2(配列番号22)、4(配列番号21)、10(配列番号23)、18(配列番号27)、23(配列番号28)及び50(配列番号29)配列は、タマリンT−1053に注入されたHGBV接種材料であるH205中に検出することができた(実施例IB参照)。これらの結果を表8に要約する。HGBVクローン配列は急性期血漿においてRT−PCRによってのみ検出されたことに留意されたい。HGBVクローン配列が、RNAをcDNAに変換するための逆転写段階後に初めてPCRにより急性期血漿中に検出されたという事実は、これらのクローンのあるものとHCV分離体との低い相同性、及びHGBVクローン10(配列番号23;実施例5)のRNA依存性RNAポリメラーゼ中に認められた保存アミノ酸をコードする配列の存在と共に、HGBVがRNAウイルスであることを示唆している。
HGBVクローン16配列(配列番号26)を有するタマリン血漿パネルのRT−PCR分析を行って、実験的にHGBVに感染させた他の個体中のHGBVクローン16(配列番号26)の存在を確認した。先に記載(G.G. Schlauderら, J. Virological Methods 37:189-200 [1992])のようにして、H205の接種材料で実験的に感染させる前と感染させた後で得られたタマリン由来の血漿25μlから核酸を分離した。エタノールで沈殿させた核酸を3μlのDEPC処理H2Oに再懸濁した。主として製造業者の指示に従って、Perkin-Elmer-Cetus製のGeneAmpRNA PCRキットを用いて、cDNA合成及びPCRを行った。ランダムヘキサマーを用いてcDNA合成を開始した。得られたcDNAをクローン16プライマー(配列番号93及び94)を用い、0.5μMの最終濃度でPCRにかけた。PCRを35サイクル(94℃で1分;55℃で1分;72℃で1分)行い、次いで72℃で7分間延長サイクルを行った。PCR産物をアガロースゲル電気泳動により分離し、核酸を臭化エチジウムで直接染色した後で紫外線照射するか、及び/又は実施例6Bに記載のようにサザンブロット法によりニトロセルロースフィルターにトランスファーした後で放射線標識したプローブにハイブリダイズして視覚化した。
図19は、臭化エチジウム染色した1.5%アガロースゲルを示す。図20は、クローン16(配列番号26)由来の放射線標識プローブにハイブリダイズした後の同一ゲルからのサザンブロット法によるオートラジオグラムを示す。H2O及びヒトの正常血清がレーン1及び2に示されている。レーン3、19及び20はマーカーである。レーン4、8、12及び16は非感染タマリン血清由来であり、レーン6、10、14及び18は感染タマリン血清由来である。これらの結果は、HGBVクローン16配列(配列番号26)が、タマリンT−1053の外に、HGBVに感染した他の個体にも検出されたが、非感染個体には検出されなかったことを示している。5匹のH205感染動物由来の急性期血清をテストした。クローン16配列(配列番号26)は、これらの動物の中の3匹〔レーン10、T−1049、接種後14日目(dpi);レーン14、T−1051,28dpi;レーン18、T−1055、16dpi〕由来の血清中に検出された。クローン16配列(配列番号26)は、5匹の動物(レーン4、T−1048;レーン8、T−1049;レーン12、T−1051;レーン16、T−1055;T−1057は示さず)のいずれの接種前血清からも検出されなかった。これらの結果は、クローン16配列(配列番号26)が感染性HGBV病原体由来であり得ることを示唆している。5種の急性期血漿中の2種(レーン6、T−1048、28dpi;T−1057、14dpi、図示せず)に、クローン16配列(配列番号26)が不在であることは、タマリンにおけるHGBV感染の急性分解性質とあいまったクローン16RT−PCRの比較的低い感受性(クローン16配列(配列番号26)の約≧1000コピーを検出し得ると想定される)から説明し得る。従って、2匹の陰性動物由来の急性期血漿は、用いられたRT−PCRアッセイの検出レベルを下回るHGBVのタイターを含み得る。これら2匹の動物がクローン4(配列番号21)に対して陽性であったというRT−PCRによる観察結果(実施例14)は、1つのウイルス(クローン4を含む)のRNA配列の存在及び第2のウイルス(クローン16を含む)由来の検出可能なRNA配列の不在を反映している可能性がある。
(実施例8)
感染タマリンの肝臓におけるHGBV配列のノーザンブロット分析
HGBVクローン配列が、急性期タマリン血漿中でのRT−PCRとH205接種とにおいて検出できたことにより、これらの配列がHGBVゲノムを起源とする可能性が高まった。さらに別のRT−PCR検査によって、H205感染タマリンT−1053から抽出した肝臓RNA にHGBV配列が存在することが実証された(データは示されていない)。従って、HGBVゲノムのサイズを知るために、H205感染及び非感染タマリンの肝臓RNAとのノーザンブロット分析を行った。RNA単離キット(Stratagene, La Jolla, CA)をその推奨使用法に従って用い、H205感染タマリンT−1053の肝臓 1.25gと対照非感染タマリンT−1040の肝臓1.0gとから全細胞RNAを抽出した。全RNA(30μg)を、0.6Mホルムアルデヒドを含む1%アガロースゲルを通して電気泳動し(R.M. Fourney らFocus 10:5−7,[1988]) 、更に、上記の通りに20×SCC(pH7.0)中で毛管作用によってHybond−N ナイロン膜(Amersham)に移した。J.Sambrookら,Molecular Cloning−A Laboratory Manual, 第2版(1989)。そのRNA を上記ナイロン膜に対してUV架橋し、上記ナイロン膜を真空炉内で80℃で60分間焼き付けた。PIPES 0.05M とリン酸ナトリウム50mMとNaCl 100 mM とEDTA 1mMとSDS 5%とを含む溶液25ml中で、60℃で2時間、このブロットに前ハイブリッド形成させた。G.D. Vircaら,Biotechniques8:370 −371 (1990)。放射能標識化DNAプローブとのハイブリッド形成の前に、上記溶液を取り除き、新鮮な溶液 10 mlを加えた。ハイブリッド形成に使用したプローブは、クローン4(配列番号21; 221 bp) とクローン50(配列番号29; 337 bp) とヒトβ−アクチンに対応する2000 bp cDNAだった。P.Gunning, らMol. and Cell. Biol. 3:787−795 (1983)。ランダムプライマー標識化キット(Stratagene, La Jolla, CA)を推奨使用法に従って用い、 [α−32P]dATPの存在下でプローブ(50ng)を放射能標識化した。各プローブの比放射能は約109cpm/μg だった。ブロットを60℃で16時間ハイブリッド形成させ、上記の通りに洗浄し(G.D. Virca ら、前出)、−80℃でKodak X−Omat−ARフィルムに対して露出した。得られたオートラジオグラフの写真を図21A に示した。レーン1 とレーン3 とレーン5 は、T−1040からの肝臓RNA を含み、レーン2 とレーン4 とレーン6は、T−1053からの肝臓RNA を含む。レーン1 とレーン2 は、ヒトβ−アクチンcDNAプローブとハイブリッド形成し、レーン3 とレーン4 はクローン4 プローブ (配列番号21) とハイブリッド形成し、レーン5 とレーン6 はクローン50プローブ (配列番号29) とハイブリッド形成した。露出時間は、レーン1 とレーン2 とが−80℃で5 時間であり、レーン3 からレーン6までが−80℃で56時間であった。28S リボゾームRNA の位置と18S リボゾームRNA の位置は矢印で示されている。これらのリボゾームRNA の相対サイズは、各々6333ヌクレオチドと2366ヌクレオチドである。J. Sambrook ら、前出。
感染タマリンの肝臓におけるHGBV配列のノーザンブロット分析
HGBVクローン配列が、急性期タマリン血漿中でのRT−PCRとH205接種とにおいて検出できたことにより、これらの配列がHGBVゲノムを起源とする可能性が高まった。さらに別のRT−PCR検査によって、H205感染タマリンT−1053から抽出した肝臓RNA にHGBV配列が存在することが実証された(データは示されていない)。従って、HGBVゲノムのサイズを知るために、H205感染及び非感染タマリンの肝臓RNAとのノーザンブロット分析を行った。RNA単離キット(Stratagene, La Jolla, CA)をその推奨使用法に従って用い、H205感染タマリンT−1053の肝臓 1.25gと対照非感染タマリンT−1040の肝臓1.0gとから全細胞RNAを抽出した。全RNA(30μg)を、0.6Mホルムアルデヒドを含む1%アガロースゲルを通して電気泳動し(R.M. Fourney らFocus 10:5−7,[1988]) 、更に、上記の通りに20×SCC(pH7.0)中で毛管作用によってHybond−N ナイロン膜(Amersham)に移した。J.Sambrookら,Molecular Cloning−A Laboratory Manual, 第2版(1989)。そのRNA を上記ナイロン膜に対してUV架橋し、上記ナイロン膜を真空炉内で80℃で60分間焼き付けた。PIPES 0.05M とリン酸ナトリウム50mMとNaCl 100 mM とEDTA 1mMとSDS 5%とを含む溶液25ml中で、60℃で2時間、このブロットに前ハイブリッド形成させた。G.D. Vircaら,Biotechniques8:370 −371 (1990)。放射能標識化DNAプローブとのハイブリッド形成の前に、上記溶液を取り除き、新鮮な溶液 10 mlを加えた。ハイブリッド形成に使用したプローブは、クローン4(配列番号21; 221 bp) とクローン50(配列番号29; 337 bp) とヒトβ−アクチンに対応する2000 bp cDNAだった。P.Gunning, らMol. and Cell. Biol. 3:787−795 (1983)。ランダムプライマー標識化キット(Stratagene, La Jolla, CA)を推奨使用法に従って用い、 [α−32P]dATPの存在下でプローブ(50ng)を放射能標識化した。各プローブの比放射能は約109cpm/μg だった。ブロットを60℃で16時間ハイブリッド形成させ、上記の通りに洗浄し(G.D. Virca ら、前出)、−80℃でKodak X−Omat−ARフィルムに対して露出した。得られたオートラジオグラフの写真を図21A に示した。レーン1 とレーン3 とレーン5 は、T−1040からの肝臓RNA を含み、レーン2 とレーン4 とレーン6は、T−1053からの肝臓RNA を含む。レーン1 とレーン2 は、ヒトβ−アクチンcDNAプローブとハイブリッド形成し、レーン3 とレーン4 はクローン4 プローブ (配列番号21) とハイブリッド形成し、レーン5 とレーン6 はクローン50プローブ (配列番号29) とハイブリッド形成した。露出時間は、レーン1 とレーン2 とが−80℃で5 時間であり、レーン3 からレーン6までが−80℃で56時間であった。28S リボゾームRNA の位置と18S リボゾームRNA の位置は矢印で示されている。これらのリボゾームRNA の相対サイズは、各々6333ヌクレオチドと2366ヌクレオチドである。J. Sambrook ら、前出。
クローン4 プローブ (配列番号21) とクローン50プローブ(配列番号29) は、感染タマリン(T−1053) の肝臓から抽出したRNA 中に含まれるRNA 種とハイブリッド形成した (図21A 、レーン4 とレーン6)。このハイブリッド形成可能なRNA 種のサイズは、28S リボゾームRNA と18S リボゾームRNA と比べての相対移動度に基づいて約8300ヌクレオチドと計算された。上記両プローブは同一のRNA 種とハイブリッド形成したと考えられる。上記両プローブは非感染タマリン(T−1040) の肝臓から抽出したRNAとはハイブリッド形成しなかった (図21A、レーン3とレーン5)。これらの結果は、クローン4(配列番号21) 配列とクローン50 (配列番号29) 配列とが同じ8.3Kb 転写物内に存在していることを示している。
HGBV RNAゲノムの鎖形成度(strandedness)を知るために、鎖特異性(Strand-specific) 放射能標識化DNA プローブを、Perkin−Elmer からの GeneAmp(登録商標) PCRキットを推奨使用法に従って用い、非対称PCR によって調製した。精製クローン50 DNA (配列番号29) を、クローン50に陰性な鎖特異性プライマー(配列番号99) 又はクローン50に陽性な鎖特異性プライマー(配列番号100)のどちらかを含む別々の反応の中で、 1μM の最終濃度となるよう鋳型として使用した。この反応混合物は、こうした反応混合物中に通常含まれるdATPの代わりに、 {α32P −dATP](Amersham; 3000Ci/mmol)を含んでいた。上記鋳型を30回線形増幅した後に、非取込み [α32P −dATP] を Quick−Spin(登録商標) Sephadex G50 スピンカラム(Boehringer −Mannheim,Indianapolis, IN) を推奨使用法に従って用い、取り除いた。二重鎖クローン50 DNA (配列番号29) の10倍ずつ希釈した液を含むDNA ドットブロットに放射能標識化プローブをハイブリッド形成させ、上記両プローブが概ね同等の感度を有することを認めた(データは示していない)。上記の通りに非感染タマリン T−1040から抽出した肝臓RNA と感染タマリン T−1053から抽出した肝臓RNA とを合計で30μg 含むRNA ブロットと共に、放射能標識化プローブをハイブリッド形成させた。得られたオートラジオグラフの写真を図21B に示す。レーン1 とレーン3 は T−1040からの肝臓RNA を含み、レーン2 とレーン4 は T−1053からの肝臓RNA を含む。レーン1 とレーン2 はクローン50に陽性な鎖プローブと共にハイブリッド形成し (即ち、陽性鎖が放射能標識化され、陰性鎖を検出した:配列番号100)、レーン3 とレーン4 はクローン50に陰性な鎖プローブと共にハイブリッド形成した(即ち、陰性鎖が放射能標識化され、陽性鎖を検出した:配列番号99)。ブロットを−80℃で18時間露出した。28S リボゾームRNA の位置と18S リボゾームRNA の位置とが矢印で示されている。
図21B に示すように、クローン50に陽性及び陰性な鎖プローブ(各々配列番号100 と99)は、感染タマリン T−1053の肝臓から抽出した約8.3 キロベースのRNA 種とはハイブリッド形成した(図21B 、レーン2とレーン4)が、非感染タマリン T−1040の肝臓から抽出したRNA とはハイブリッド形成しなかった(図21B 、レーン1とレーン3 )。これは、上記で示したクローン4(配列番号21) 及びクローン50 (配列番号29) 二重鎖プローブを使用して得られたノーザンブロット結果と一致していた。より強いシグナルがクローン50に陰性な鎖プローブ (配列番号99) を用いて得られた(図21B 、レーン4 対レーン2 )ことは、感染タマリンの肝臓中に存在する優勢なRNA 種が陽性(即ち、コード化)鎖であることを示唆していた。
(実施例9)
HGBVクローン配列の伸長
A.HGBV配列の生成
既述の実施例3 の通りに得られ且つ実施例5の通りに配列決定されたクローンの各々は、HGBVゲノムの別々の領域から得られたと考えられた。従って、既に同定したクローンの間に残るHGBVゲノムの別の領域から配列を得るために、及び、そのRNA クローンの配列を確認するために、幾つかのPCR 移動実験(PCR walking experiment)を行った。
HGBVクローン配列の伸長
A.HGBV配列の生成
既述の実施例3 の通りに得られ且つ実施例5の通りに配列決定されたクローンの各々は、HGBVゲノムの別々の領域から得られたと考えられた。従って、既に同定したクローンの間に残るHGBVゲノムの別の領域から配列を得るために、及び、そのRNA クローンの配列を確認するために、幾つかのPCR 移動実験(PCR walking experiment)を行った。
全核酸を実施例3(A)で説明した通りに感染 T−1053血漿の50μl アリコートから抽出した。簡単に言えば、沈殿した核酸をDEPC処理した H2O 10μl 中に再懸濁させた。GeneAmp(登録商標) RNA PCRキット(Perkin −Elmer)で推奨使用法に従って標準RT−PCR を行った。簡潔に言えば、2mM MgCl2と 1μM プライマーによる抽出核酸のランダム感作逆転写反応のcDNA生成物に対して、PCR を行った。反応物に対して35サイクルの変性−アニーリング−伸長(94℃、30秒;55℃、30秒;72℃、 2分)を行い、その後で72℃で3分間の伸長を行った。アガロースゲル分析の前に反応物を 4℃に維持した。これらの生成物を慣用手法によりpT7 Blue T−ベクタープラスミド(Novagen)の中にクローン化した。表9は、上記反応を行った時に得られた結果を表している。
Sorensenら(J. Virol, 67:7118−7124[1993]) によって説明されているPCR 移動手法を変形した方法を、追加HGBV配列を得るために使用した。簡単に述べると、全核酸を感染タマリン T−1053血漿から抽出し、逆転写した。得られたcDNAを、MgCl2 2mMを使用したことを除いてはSorensenら(前出)の手順と同様に、50μl PCR 反応物(PCR 1) 中で増幅した。これらの反応物に対して35回の変性−アニーリング−伸長(94℃、30秒;55℃、30秒;72℃、 2分)を行い、その後72℃で3分間の伸長を行った。Sorensenら(前出)が説明している通りに、ストレプタビジンで被覆した常磁性ビーズ(Promega) を使用してビオチニル化生成物を単離した。Sorensenらが説明している通りに、合計で20〜35回の変性−アニーリング−伸長し、ストレプタビジンで精製した生成物に対して「入れ子(nested)」PCR(PCR 2)を行った。得られた生成物と、それらを生成するために使用したPCR プライマーとを表10に示す。
これらの生成物を、慣用手法により低融点アガロースゲルから単離し、pT7 Blue T−ベクタープラスミド(Novagen) の中にクローン化した。
上記の通りに、ウイルスゲノムRNA から5’末端配列を得るために、RNA リガーゼ仲介5’ RACE(cDNA末端の高速増幅:R apidA mplification of c DNA E nds)を使用した。簡単に述べると、5’ AmpliFINDER(登録商標)RACE キット(Clontech, Palo Alto, CA) を推奨使用法に従って使用した。ウイルスRNA の供給源は、上記の通りに抽出した急性期 T−1053血漿であった。逆転写(RT)のために使用した個々のウイルス種に固有のオリゴヌクレオチドと、第1のPCR 増幅(PCR 1) と第2のPCR 増幅(PCR 2) とを、表11に示す。結合アンカープライマーとその相補的PCR プライマーは上記製造者から提供を受けた。GeneAmp RNA PCR キット(Perkin Elmer)を使用して推奨使用法に従ってPCR を行った。
RNA リガーゼ仲介5’ RACE によって生成された生成物を、上記の通りに低融点アガロースゲルから単離し、pT7 Blue T−ベクタープラスミド(Novagen) の中にクローン化した。
HGBV−B 配列の5’末端と3’末端とにおける別の配列を得るために(下記の「HGBV内の2種類のHCV 様フラビウイルスの存在の証拠」を参照されたい)、RNA 環化実験(RNA circularization experiment)を行った。(この方法は、C.W. Mandlら(1991) Biotechniques, Vol 10 (4): 485 −486 に説明されている方法に基づいている。)(沈殿中にtRNAをグリコーゲン 1μg で置き換えたことを除いてH205接種後14日目の) T−1057血漿50μl から全核酸を精製した。核酸ペレットをDEPC処理した水16.3μl の中に再溶解し、 2×TAP 緩衝液(1×=50 mM NaOAc、pH 5.0、1 mM EDTA 、10mM 2−メルカプトエタノール、2m MATP)25 μl と、タバコ酸ピロホスファターゼ(20単位;Sigma) 8.7μl とを加えた。混合物を37℃で60分間温置した。(水で飽和させた)フェノールを使用し更にクロロホルムを使用して試料を抽出し、グリコーゲン(1μg)の存在下でNaOAc/EtOHを用いて沈殿させた。そのペレットをDEPC水83μl 中に溶解し、10×RNA リガーゼ緩衝液(New England Biolabs, NEB)10μl とRNase インヒビター(Perkin Elmer) 2μl とT4 RNAリガーゼ(NEB)5μl とを加えた。混合物を 4℃で16時間温置した。上記と同様にフェノールを使用し更にクロロホルムを使用して試料を抽出し、NaOAc/EtOHを用いて沈殿させた。
Superscript RT (GIBCO/BRL)をその推奨使用法に従って使用し且つ配列番号146 をプライマーとして用いる逆転写酵素(RT)反応で、結合RNA の1/10を使用した。RT反応混合物の半分を、ビオチニル化オリゴヌクレオチドプライマー (配列番号146)と第2のオリゴヌクレオチドプライマー (配列番号133)との存在下でPCR1のために上記の通り使用した。Sorensenら(前出)の通りにストレプタビジン被覆磁性ビーズを使用して、PCR1生成物を反応混合物から精製した。精製したPCR1生成物(30μl の中から 2μl )をPCR2のための鋳型として使用した。オリゴヌクレオチドプライマー (配列番号147,154)を使用するPCR2によって、1200 bp 生成物を得、この生成物をpT7 Blue T−ベクタープラスミドの中にクローン化し、下記の通りに配列決定した。この実験からの2つの独立したクローンの配列分析によって、(一方のクローンが18個のT 残基の配列を有し、他方のクローンが27個のT 残基の配列を有したが)既知の配列と重複する領域では100%の同一性を示し、さらに追加の塩基270 個の新たな配列とが明らかになった。
上記環化実験は、標準的な3’−RACE手法又は5’−RACE手法では得られなかったHGBV−B ウイルスゲノムの5’末端と3’末端の両方からの配列を与えた。しかし、新たに得られた配列から設計したプライマーを使用して行った別のPCR 実験の後でさえ、正確な5’−3’結合部を発見することは困難だった。従って、HGBV−B RNA ゲノムの5’末端をより適切に定性するために、 T−1053の肝臓から単離したRNA を使用してプライマー伸長実験を行った。
実施例7で説明した通り、 T−1053の肝臓と対照( 即ち、非感染)動物(T−1040) の肝臓とから全細胞RNA を単離した。アンチセンスオリゴヌクレオチド (配列番号155)を、T 4ポリヌクレオチドキナーゼ(NEB) を使用して約9.39×107CPM/μg の比放射能にγ−32P −ATP で末端標識化した(Sambrook ら)。プライマーを別々の反応において T−1053肝臓RNA と T−1040肝臓RNA との30μl にアニーリングし、上記(Sambrook ら)の通りにMMLV逆転写酵素(Perkin −Elmer)を使用して伸長した。生成物を6%シークエンシングゲル上で分析した。プライマー伸長のために使用したプライマーと同一のプライマーを使用したHGBV−B 環化クローンの1つから生成したシーケンスラダー(sequence ladder) が、サイズ標準としての役割を果たした。
176bp のプライマー伸長生成物を T−1053から得た。非感染動物(T−1040) から抽出した肝臓RNA を使用したプライマー伸長では、こうした生成物は得られず、従って、HGBV−B ゲノムから得られた生成物を示した。得られたこれらの生成物の長さは、感染動物の肝臓内に存在するそれのような上記ゲノムの5’末端が、AUG 開始コドンから442 ヌクレオチド長だけ上流に位置することを示した。
上記環化実験で得られた配列の3’位置を確認するために、予測される3’末端に対応するプライマーを使用してRT−PCR を行った(表2の反応1.25を参照されたい)。配列番号156 と配列番号157 とを使用した(上記の通りの)感染 T−1053血漿のRT−PCR では450bp の生成物が得られた。これとは対照的に、配列番号157 の相補と配列番号147 とを使用するRT−PCR では、検出可能なPCR 生成物が得られなかった(データは示していない)。これらのデータは上記ゲノムの3’末端がポリ T系の50ヌクレオチド長下流に位置することを示唆している。
表9 、表10、表11と上記RNA 環化実験とからのクローン化生成物を、実施例5で既に説明した通りに配列決定した。興味深いことに、反応1.4 、1.6 、1.9 、1.10、1.11のクローン化生成物がその末端において上記2つのプライマー配列の一方だけしか含まないことが発見され、このことは、これらの生成物が、誤ったランダムな合成開始事象(priming event) の結果として生じたことを示唆していた。PCR /配列決定実験によって、生成物1.4 、1.6 、1.9 、1.10、1.11中で検出した配列が、クローン4 (配列番号21) 及び/又はクローン50 (配列番号29) とに関連付けられた。加えて、反応の各々から得られた配列は、HCV との同一性が限定されていた。従って、これらの生成物は、PCR 生成物の一方の末端における誤った合成開始(priming) の結果ではあっても、真正のHGBV配列を含むと考えられた。反応1.14から得られた生成物も誤った合成開始の結果であると考えられた。この場合に、配列番号160 の相補が、反応1.14から得た生成物の5’末端に発見された (図22、GB−B)。GB−A 内にある配列番号161 から配列番号160 が得られているので、このことは予想外であった。しかし、反応1.14から得た生成物と反応2.8 から得た生成物との間の配列の同一性が、別のPCR /配列決定実験(データは示していない)と共に、反応1.14が真正 HGBV配列を含むことを実証した。配列番号160 の相補が配列番号162 の上流にあって、GB−B 配列に対してPCR プライマーとして働くのに十分な同一性を有することが明らかである。
上記の表9 と表10と表11とに示した生成物から得られた配列と、上記RNA 環化実験から得られた配列とを、GCG Package(Version 7) プログラムを使用してコンティグ(contig)の形にアセンブルした。このアセンブルしたコンティグの概要を図22に示す。GBコンティグA(GB−A)は長さ9493bpであり、その全てが配列決定されており、配列番号163 に示す。GB−A は、クローン2 (配列番号22) とクローン16 (配列番号26) とクローン23 (配列番号28) とクローン18 (配列番号27) とクローン11(配列番号24) とクローン10 (配列番号23) とを含む。配列番号163 は3つの読み可能枠に翻訳され、配列番号164 −392 としての配列表の形で示されている。GBコンティグB(GB−B)は 9143 bp であり、配列番号393 に示す。GB−B(配列番号393)は、クローン4 (配列番号21) とクローン50 (配列番号29) とクローン119(配列番号30) とクローン13 (配列番号25) とを含んでいる。配列番号393 は1つの開放読み枠に翻訳され、配列番号396 、397として配列表の形で示されている。5’末端と3’末端とからのUTR の各々を6つの読み枠に翻訳することが可能である。
B.HGBV内の2種類のHCV 様ウイルスの存在に関する証拠
1.2つの互いに異なったRNA 種を表すGB−A とGB−B との証拠
GB−A(配列番号163)及びGB−B(配列番号393)とHCV −1(GenBank accession#M67463)との比較によって、GB−A(配列番号163)及びGB−B(配列番号393)と HCV−1 とが全て異なった配列であることを示す。GB−A(配列番号163)及びGB−B(配列番号393)とHCV −1 との核酸配列のドットプロット分析を、GCGPackage(Version 7)を使用して行った。21のウィンドウサイズと14のストリンジェンシー(stringency)とを使用して、GB−A(配列番号163)及びGB−B(配列番号393)と HCV−1 とが互いに明らかに異なったヌクレオチド配列を含むことが発見された(図23)。従って、GB−A(配列番号163)及びGB−B(配列番号393)は、HCV の異なる株又は遺伝子型を表すものではなく、互いに異なる株又は遺伝子型を表すものでもない。この分析によって、限定されたヌクレオチド同一性を有する短い領域が、GB−A(配列番号163)とGB−B(配列番号393)のNS3 様配列とNS5b様配列との中に、並びにHCV のNS3 配列とNS5b配列の中に発見された。しかし、推定上のNTP 結合へリカーゼとRNA 依存性RNA ポリメラーゼの各々がNS3 とNS5bとに対応し、これが全てのフラビウイルス中に保存されているので、上記領域内のヌクレオチド同一性は驚くには値しない(下記参照)。GB−A(配列番号163)とGB−B(配列番号393)が別々のRNA 分子を表し、これらが同じRNA 分子の別々の領域でもないということが、5’ RACE 実験(上記)によって実証され、(実施例8で説明した)ノーザンブロットデータによって立証された。最初に、5’ RACE 実験によって、GB−A(配列番号163)とGB−B(配列番号393)との5’末端がそれぞれ異なることが示された。RNA 分子の5’末端は1種だけだから、GB−A(配列番号163)とGB−B(配列番号393)とは別々のRNA 分子を表す。第2に、クローン4とクローン50(両方ともGB−B[配列番号393]から得られた配列番号21、29、実施例8を参照されたい)を用いてノーザンブロット法によって感染タマリンの肝臓RNA 中に検出された8300塩基のRNA 分子は、GB−B(配列番号393 、9143 bp)のサイズに概ね一致した。GB−A とGB−B とが同一のRNA 分子のそれぞれ一部であるとすれば、少なくとも17,000個の塩基を有するノーザンブロット生成物がなければならない。これらのデータは、GB−A(配列番号163)とGB−B(配列番号393)とが、HCV の変形でもなく又は互いの変形でもない、2つの互いに異なったRNA 分子のヌクレオチド配列であることを示す。
1.2つの互いに異なったRNA 種を表すGB−A とGB−B との証拠
GB−A(配列番号163)及びGB−B(配列番号393)とHCV −1(GenBank accession#M67463)との比較によって、GB−A(配列番号163)及びGB−B(配列番号393)と HCV−1 とが全て異なった配列であることを示す。GB−A(配列番号163)及びGB−B(配列番号393)とHCV −1 との核酸配列のドットプロット分析を、GCGPackage(Version 7)を使用して行った。21のウィンドウサイズと14のストリンジェンシー(stringency)とを使用して、GB−A(配列番号163)及びGB−B(配列番号393)と HCV−1 とが互いに明らかに異なったヌクレオチド配列を含むことが発見された(図23)。従って、GB−A(配列番号163)及びGB−B(配列番号393)は、HCV の異なる株又は遺伝子型を表すものではなく、互いに異なる株又は遺伝子型を表すものでもない。この分析によって、限定されたヌクレオチド同一性を有する短い領域が、GB−A(配列番号163)とGB−B(配列番号393)のNS3 様配列とNS5b様配列との中に、並びにHCV のNS3 配列とNS5b配列の中に発見された。しかし、推定上のNTP 結合へリカーゼとRNA 依存性RNA ポリメラーゼの各々がNS3 とNS5bとに対応し、これが全てのフラビウイルス中に保存されているので、上記領域内のヌクレオチド同一性は驚くには値しない(下記参照)。GB−A(配列番号163)とGB−B(配列番号393)が別々のRNA 分子を表し、これらが同じRNA 分子の別々の領域でもないということが、5’ RACE 実験(上記)によって実証され、(実施例8で説明した)ノーザンブロットデータによって立証された。最初に、5’ RACE 実験によって、GB−A(配列番号163)とGB−B(配列番号393)との5’末端がそれぞれ異なることが示された。RNA 分子の5’末端は1種だけだから、GB−A(配列番号163)とGB−B(配列番号393)とは別々のRNA 分子を表す。第2に、クローン4とクローン50(両方ともGB−B[配列番号393]から得られた配列番号21、29、実施例8を参照されたい)を用いてノーザンブロット法によって感染タマリンの肝臓RNA 中に検出された8300塩基のRNA 分子は、GB−B(配列番号393 、9143 bp)のサイズに概ね一致した。GB−A とGB−B とが同一のRNA 分子のそれぞれ一部であるとすれば、少なくとも17,000個の塩基を有するノーザンブロット生成物がなければならない。これらのデータは、GB−A(配列番号163)とGB−B(配列番号393)とが、HCV の変形でもなく又は互いの変形でもない、2つの互いに異なったRNA 分子のヌクレオチド配列であることを示す。
T−1053のノーザンブロット分析とPCR 調査によれば、GB−A(配列番号163)とGB−B(配列番号393)とにそれぞれ対応する2つのRNA 種は、 T−1053の肝臓中に相等しい含量で存在するわけではないことが判明した。上記のように、いずれもGB−B(配列番号393)から得られたクローン4とクローン50(各々配列番号21、29) が、(実施例8 で説明した通り) T−1053の感染肝臓中に存在する8.3 kb RNA種とハイブリッド形成した。これとは対照的に、いずれもGB−A(配列番号163)から得られたクローン2 (配列番号22) とクローン10 (配列番号23) とクローン16 (配列番号26) とクローン23 (配列番号28) は、同一の実験において T−1053肝臓RNA とのハイブリッド形成を全く示さなかった(データは示していない)。加えて、クローン4 PCR とクローン16 PCRとが同等の感度を有することがプラズミド鋳型を使用して実証された(データは示していない)にもかかわらず、クローン16 PCRは、エチジウム染色によって T−1053肝臓RNA から生成されたcDNA上において、クローン4PCR に比べて著しく少ない生成物しか生成しなかった。これは、動物を犠牲にした時点で T−1053血漿中において発見された状況とは対照的だった。 T−1053血漿RNA から生成されたcDNA上におけるクローン4(GB −B(配列番号393 ) に特異的)PCR とクローン6(GB −A(配列番号163)に特異的)PCR に関するPCR 滴定実験は、相等しい量のGB−A(配列番号163)RNA とGB−B(配列番号393)RNA とが T−1053血漿中に存在することを示唆した (実施例4 、E.2)。従って、GB−A(配列番号163)RNA とGB−B(配列番号393) RNA とが T−1053血漿中に相等しい含量で存在していたにもかかわらず、動物を犠牲にした時点の T−1053肝臓中には、GB−A(配列番号163)RNA よりも多い量のGB−B(配列番号393)RNA が存在すると考えられた。これらの結果はさらに、 T−1053血漿中にGB−A(配列番号163)とGB−B(配列番号393)とに対応する2つの互いに異なったRNA 分子が存在することの更に別の証拠であり、これらのRNA が必ずしも感染タマリン肝臓RNA 中に相等しい含量で存在するわけではないということを示唆した。従って、GB−A(配列番号163)とGB−B(配列番号393)とが単一のウイルスゲノムの別々のセグメントを形成するとは考え難い。
2.GB−A(配列番号163)とGB−B(配列番号393)とが2つの互いに異なるウイルスのゲノムを表すということの証拠
感染力(infectivity) とPCR の検討により、GB−A(配列番号163)とGB−B(配列番号393)のウイルス性質に関する証拠が得られる。具体的には、濾過(0.1μm)されて10−4に希釈された又は濾過されずに10−5に希釈された T−1053血漿を接種されたタマリン T−1049とタマリン T−1051は、いずれもクローン4(GB −B[配列番号393])配列とクローン16(GB −A[配列番号163])配列の両方に関して陽性だった。接種の前は、これらの動物は二匹ともクローン4とクローン16とに対して陰性だった(実施例4、E.4 、E.5 )。従って、GB−A とGB−B とに対応する急性相 T−1053血漿中に含まれる2つのRNA 種を、濾過し、希釈して、他の動物に当該のGB−A(配列番号163)とGB−B(配列番号393)とのウイルスに予期される性質を維持するよう継代接種することが可能だった。GB−A とGB−B とが別個のウイルス粒子からのRNA 分子を表すことは、H205接種タマリンのPCR 調査によって明らかになった。具体的には、H205接種後にRT−PCR によって4匹のタマリンが全てクローン4(GB−B[配列番号393])に対して陽性になった。これとは対照的に、4匹のH205接種タマリンの内の1匹(T−1053) だけがクローン 16(GB−B[配列番号163])に対して陽性になったにすぎなかった(実施例4 、E.2 )。従って、GB−A(配列番号163)配列は T−1048と T−1057と T−1061には本当に存在しないと仮定し、且つ、クローン16 PCRの結果が陰性であることが感染力の低さに起因するわけではないと仮定すると、GB−B(配列番号393)配列に対応するウイルス(即ち、肝炎GBウイルスB[HGBV−B])が、GB−A(配列番号163)配列とは無関係に単独で継代接種されうると考えられた。T−1057からのHGBV−B のみの試料を、更に2回継代接種した(実施例4)。これらの動物では、GB−A(配列番号163)配列はRT−PCR によって検出されなかった。加えて、これらの動物において肝臓酵素のレベルが有意に上昇するとされており(実施例4 )、このことは、HGBV−B だけでもタマリンに肝炎を発症せしめることを示した。GB−B(配列番号393)配列が検出限界未満のタマリンにおいて、GB−A(配列番号163)配列が確認された。具体的には、GB−B のみの動物(T−1048、 T−1057、 T−1061)に T−1053血漿で誘導すると、クローン16に特異的なRT−PCR によって検出されるような、GB−A(配列番号163)のみのウイルス血症が発症した。 T−1057からのGB−A のみの血漿を更に1回継代接種した(実施例4)。従って、GB−A(配列番号163)配列に対応するウイルス(肝炎GBウイルス A[HGBV −A])はHGBV−B とは無関係に単独で複製するものと考えられた。HGBV−B の不在下でHGBV−A の継代接種を更に続けている。現在までのところ、HGBV−A がタマリンの肝炎を引き起こすか否かは判明していない。しかし、 T−1053誘導タマリンにHGBV−A ウイルス血症を伴う肝臓酵素のレベル上昇がないことや、 T−1057からのHGBV−A のみの血清を継代接種しても肝臓酵素のレベル上昇のないことは、タマリンにおけるHGBV−B の肝好性とは対照的である。
感染力(infectivity) とPCR の検討により、GB−A(配列番号163)とGB−B(配列番号393)のウイルス性質に関する証拠が得られる。具体的には、濾過(0.1μm)されて10−4に希釈された又は濾過されずに10−5に希釈された T−1053血漿を接種されたタマリン T−1049とタマリン T−1051は、いずれもクローン4(GB −B[配列番号393])配列とクローン16(GB −A[配列番号163])配列の両方に関して陽性だった。接種の前は、これらの動物は二匹ともクローン4とクローン16とに対して陰性だった(実施例4、E.4 、E.5 )。従って、GB−A とGB−B とに対応する急性相 T−1053血漿中に含まれる2つのRNA 種を、濾過し、希釈して、他の動物に当該のGB−A(配列番号163)とGB−B(配列番号393)とのウイルスに予期される性質を維持するよう継代接種することが可能だった。GB−A とGB−B とが別個のウイルス粒子からのRNA 分子を表すことは、H205接種タマリンのPCR 調査によって明らかになった。具体的には、H205接種後にRT−PCR によって4匹のタマリンが全てクローン4(GB−B[配列番号393])に対して陽性になった。これとは対照的に、4匹のH205接種タマリンの内の1匹(T−1053) だけがクローン 16(GB−B[配列番号163])に対して陽性になったにすぎなかった(実施例4 、E.2 )。従って、GB−A(配列番号163)配列は T−1048と T−1057と T−1061には本当に存在しないと仮定し、且つ、クローン16 PCRの結果が陰性であることが感染力の低さに起因するわけではないと仮定すると、GB−B(配列番号393)配列に対応するウイルス(即ち、肝炎GBウイルスB[HGBV−B])が、GB−A(配列番号163)配列とは無関係に単独で継代接種されうると考えられた。T−1057からのHGBV−B のみの試料を、更に2回継代接種した(実施例4)。これらの動物では、GB−A(配列番号163)配列はRT−PCR によって検出されなかった。加えて、これらの動物において肝臓酵素のレベルが有意に上昇するとされており(実施例4 )、このことは、HGBV−B だけでもタマリンに肝炎を発症せしめることを示した。GB−B(配列番号393)配列が検出限界未満のタマリンにおいて、GB−A(配列番号163)配列が確認された。具体的には、GB−B のみの動物(T−1048、 T−1057、 T−1061)に T−1053血漿で誘導すると、クローン16に特異的なRT−PCR によって検出されるような、GB−A(配列番号163)のみのウイルス血症が発症した。 T−1057からのGB−A のみの血漿を更に1回継代接種した(実施例4)。従って、GB−A(配列番号163)配列に対応するウイルス(肝炎GBウイルス A[HGBV −A])はHGBV−B とは無関係に単独で複製するものと考えられた。HGBV−B の不在下でHGBV−A の継代接種を更に続けている。現在までのところ、HGBV−A がタマリンの肝炎を引き起こすか否かは判明していない。しかし、 T−1053誘導タマリンにHGBV−A ウイルス血症を伴う肝臓酵素のレベル上昇がないことや、 T−1057からのHGBV−A のみの血清を継代接種しても肝臓酵素のレベル上昇のないことは、タマリンにおけるHGBV−B の肝好性とは対照的である。
急性相 T−1053血漿中の2種類のウイルスの存在は、H205接種物に基づくと考えられる。具体的には、実施例7のデータは、クローン16(配列番号26、GB−A[配列番号163]からのもの)が7匹の検査対象タマリンからの接種前の血漿中にはいずれにも存在しなかったことを示した。加えて、クローン2 、10、18、23(各々配列番号22、23、27、28、全てGB−A[配列番号163]からのもの)も、検査したHGBV接種タマリン血漿の何れにおいても検出されなかった(実施例7)。接種前のタマリンの血漿をクローン4 とクローン50(各々配列番号21、29、全てGB−B[配列番号393]からのもの)に関して検査した時にも、同様の陰性の結果が得られた。従って、HGBV−A とHGBV−B はいずれも接種前のタマリンの血漿中に存在しなかった。これに対し、上記クローンの全て(即ち、GB−A[配列番号163]からのクローン2 、10、16、18、23と、GB−B[配列番号393]からのクローン4 、50)が、H205接種物中に検出された(表7)。興味深いことに、T −1053肝臓から作られたcDNA(上記)に見られるように、H205からランダムに合成開始された同一のcDNAを使用した場合に、GB−A(配列番号163)中の幾つかの別々のPCR 標的は全て、GB−B(配列番号393)中の同様のPCR 標的の場合よりも生成物が少なかった(データは示していない)。従って、当初のGB接種物であるH205中には、HGBV−A 及びHGBV−B が存在すると結論付けられた。しかし、H205中にはHGBV−A よりも多くのHGBV−B が含まれると考えられる。H205接種物中のHGBV−A が相対的に少ないことが、H205接種後に4匹のタマリンの中の1匹だけが HGBV−A に対して陽性であったこと (実施例4、E.2)の理由と考えられる。
3.HGBV−A とHGBV−B とがフラビウイルス科(Flaviviridae)に属することの証拠
実施例5で説明したGB−A(配列番号165 −268 、270 −384 ,386 −392)の3つの枠翻訳生成物とGB−B(配列番号397)とをSWISS −PROTデータベースで検索すると、HCV の様々な菌株との、限定されてはいるが顕著なアミノ酸配列の相同性を示した。GB−A(配列番号164)とGB−B(配列番号393)とからの翻訳生成物は、様々なHCV 単離物(即ち、NS2 、NS3 、NS4 、NS5 )の非構造タンパク領域に対して最も近いホモロジーを示した。例えば、図24に示されるように、フラビウイルスの推定上のNTP 結合ヘリカーゼドメインにおける保存残基(*で表す)(図24A )と、全てのウイルス性RNA 依存性RNA ポリメラーゼのRNA 依存性RNA ポリメラーゼドメインにおける保存残基(*で表す)(図24B )は、同様に HCV−1 NS3及びNS5b(SWISS −PROT accession number p26664) と、GB−A(配列番号390)及びGB−B(配列番号397)の予期される翻訳生成物のいずれにも保持されていた。(Chooら, PNAS 88:2451−2455 [1991] とDomierら, Virology 158:20 −27 [1987] を参照されたい。)従って、GB−A ウイルスとGB−B ウイルスとはいずれも機能NTP 結合ヘリカーゼ(functional NTP −binding helicase) とRNA 依存性 RNA ポリメラーゼとをコードすると考えられた。しかし、GB−A(配列番号390)とGB−B(配列番号397)は、その間に完全なアミノ酸同一性があるわけではなく、及び/又は、HCV NS3 及びNS5bの他の領域内においてHCV とも完全なアミノ酸同一性がない。具体的には、図24A に示されるNS3 の残基200 個に亙って、GB−A(配列番号390 、残基1252−1449) ウイルスとHCV −1 (配列番号398)は47%同一であり、GB−B(配列番号397 、残基1212−1408) ウイルスとHCV −1 (配列番号398)は55%同一であり、GB−A(配列番号390 、残基1252−1449) ウイルスとGB−B(配列番号397 、残基1212−1408) ウイルスは43.5%同一だった。加えて、図24B に示されるNS3 の残基 100個に亙って、GB−A(配列番号390 、残基2644−2739) ウイルスと HCV−1 (配列番 号398)は36%同一であり、GB−B(配列番号397 、残基2513−2612) ウイルスとHCV−1 (配列番号398)は41%同一であり、GB−A(配列番号390 、残基2644−2739) ウイルスとGB−B(配列番号397 、残基2599−2698) ウイルスは44%同一だった。HCV と比較した場合に、GB−A(配列番号390)とGB−B(配列番号397)との他の推定上の非構造遺伝子にも、ホモロジーは比較的低かった。Genbank に登録された種々のHCV 配列と比較しても、GB−A ウイルスとGB−B ウイルスとの推定上の非構造タンパク質のホモロジーの全体的レベルは、GB−A(配列番号164)とGB−B(配列番号393)の両方はフラビウイルス科の2つの互いに異なったメンバーから得られることを示唆している。フラビウイルス類は、1つのNTP 結合ヘリカーゼドメインと1つのRNA 依存性RNA ポリメラーゼドメインとに対応する単一のゲノムRNA 分子を含む。推定上のRNA ヘリカーゼドメインと推定上のRNA 依存性RNA ポリメラーゼドメインとを各々に含む2つのコンティグの存在は、急性相 T−1053血漿中の2つのHCV 様フラビウイルスの存在と一致するものである。
実施例5で説明したGB−A(配列番号165 −268 、270 −384 ,386 −392)の3つの枠翻訳生成物とGB−B(配列番号397)とをSWISS −PROTデータベースで検索すると、HCV の様々な菌株との、限定されてはいるが顕著なアミノ酸配列の相同性を示した。GB−A(配列番号164)とGB−B(配列番号393)とからの翻訳生成物は、様々なHCV 単離物(即ち、NS2 、NS3 、NS4 、NS5 )の非構造タンパク領域に対して最も近いホモロジーを示した。例えば、図24に示されるように、フラビウイルスの推定上のNTP 結合ヘリカーゼドメインにおける保存残基(*で表す)(図24A )と、全てのウイルス性RNA 依存性RNA ポリメラーゼのRNA 依存性RNA ポリメラーゼドメインにおける保存残基(*で表す)(図24B )は、同様に HCV−1 NS3及びNS5b(SWISS −PROT accession number p26664) と、GB−A(配列番号390)及びGB−B(配列番号397)の予期される翻訳生成物のいずれにも保持されていた。(Chooら, PNAS 88:2451−2455 [1991] とDomierら, Virology 158:20 −27 [1987] を参照されたい。)従って、GB−A ウイルスとGB−B ウイルスとはいずれも機能NTP 結合ヘリカーゼ(functional NTP −binding helicase) とRNA 依存性 RNA ポリメラーゼとをコードすると考えられた。しかし、GB−A(配列番号390)とGB−B(配列番号397)は、その間に完全なアミノ酸同一性があるわけではなく、及び/又は、HCV NS3 及びNS5bの他の領域内においてHCV とも完全なアミノ酸同一性がない。具体的には、図24A に示されるNS3 の残基200 個に亙って、GB−A(配列番号390 、残基1252−1449) ウイルスとHCV −1 (配列番号398)は47%同一であり、GB−B(配列番号397 、残基1212−1408) ウイルスとHCV −1 (配列番号398)は55%同一であり、GB−A(配列番号390 、残基1252−1449) ウイルスとGB−B(配列番号397 、残基1212−1408) ウイルスは43.5%同一だった。加えて、図24B に示されるNS3 の残基 100個に亙って、GB−A(配列番号390 、残基2644−2739) ウイルスと HCV−1 (配列番 号398)は36%同一であり、GB−B(配列番号397 、残基2513−2612) ウイルスとHCV−1 (配列番号398)は41%同一であり、GB−A(配列番号390 、残基2644−2739) ウイルスとGB−B(配列番号397 、残基2599−2698) ウイルスは44%同一だった。HCV と比較した場合に、GB−A(配列番号390)とGB−B(配列番号397)との他の推定上の非構造遺伝子にも、ホモロジーは比較的低かった。Genbank に登録された種々のHCV 配列と比較しても、GB−A ウイルスとGB−B ウイルスとの推定上の非構造タンパク質のホモロジーの全体的レベルは、GB−A(配列番号164)とGB−B(配列番号393)の両方はフラビウイルス科の2つの互いに異なったメンバーから得られることを示唆している。フラビウイルス類は、1つのNTP 結合ヘリカーゼドメインと1つのRNA 依存性RNA ポリメラーゼドメインとに対応する単一のゲノムRNA 分子を含む。推定上のRNA ヘリカーゼドメインと推定上のRNA 依存性RNA ポリメラーゼドメインとを各々に含む2つのコンティグの存在は、急性相 T−1053血漿中の2つのHCV 様フラビウイルスの存在と一致するものである。
(実施例10)
PCR
HGBVとC 型肝炎ウイルスとの配列相関性を検出するために、PCR に基づく実験を次のように行った。様々な位置的起源からのHCV 単離体(Cha, T.−A.ら, J. Clin. Microbiol. 29:2528 −2534[1991]) において高度に保存されている、HCV ゲノムの5’−非翻訳領域(UTR) 配列に基づくPCR プライマー(J.H. Han, PNAS 88:1711 −1715 [1991])を、H205感染タマリン T−1053肝臓RNA における類似の配列を検出するために使用した。実施例8Aで説明した通りに、全細胞RNA を感染タマリン T−1053 の肝臓と非感染タマリン(T−1040) の肝臓とから抽出した。Perkin−Elmer から入手可能なキットを使用し、その推奨使用法に概ね従って、各RNA の試料30μg を逆転写しPCR 増幅した。HCV 5’−UTR の塩基 249−268 を含むアンチセンスプライマー(プライマー1)を上記逆転写酵素反応に使用した。プライマー1と、HCV 5’−UTR の塩基13−46を含むプライマー(プライマー2)とを、介在配列のPCR 増幅のために使用した。サーモサイクリング(thermocycling) の条件はCha ら(前出)による。 H205感染タマリン T−1053のHCV 5’−UTR 配列の検出感度を増大させるために、上記PCR 反応に対して、「入れ子」PCR プライマーを使用した第2の増幅反応を生じさせた。そのためのプライマーは、HCV 5’−UTR 内における上記プライマー1の配列と上記プライマー2の配列との間にある配列から得た。プライマー3はHCV 5’−UTR の塩基47−69からの配列を含み、アンチセンスプライマーであるプライマー4はHCV 5’−UTR の塩基 188−210 を含んでいた。この「入れ子」PCR 反応では、第1のPCR 反応からのPCR 生成物(反応体積合計100 μl から2 μl )をDNA 鋳型源として使用した。アニーリング温度が60℃でなく55℃であったことを除いてサーモサイクリング条件は上記と概ね同一だった。第2のPCR 反応で得られたPCR 生成物を、アガロースゲル電気泳動と臭化エチジウム染色とを使用して所期のDNA 生成物に関して分析した。HCV 5’−UTR 配列に基づくこの所期のDNA フラグメントのサイズ(Han ら、前出)は、第1のPCR 反応の生成物に関しては 253bpであり、入れ子PCR 反応の生成物に関しては 163bpであった。所期サイズのPCR 生成物が、実施例8Aで説明したHCV 感染チンパンジーの肝臓から抽出した全細胞RNA 30μg を使用して行った対照実験において得られ(データは示していない)、これは、この実験手順でHCV の5’−UTR を検出できることを示す。しかし、 T−1053肝臓 RNA 又は T−1040肝臓RNA の一方を使用した場合には、どちらの所期生成物も、生じた臭化エチジウム染色アガロースゲル上に発見できなかった(データは示していない)。このように所期の結果が得られなかったのは、(i) 上記試薬の5’−UTR の配列がHCV のそれと大きく異なっていたために、使用したオリゴヌクレオチドプライマーが効率的にアニールされず、従って、PCR 増幅が不可能になったということ、又は、(ii)上記試薬に5’−UTR が欠如していたということを示唆する。どちらの場合にも、上記試薬のヌクレオチド配列がHCV のそれと大きく異なっていることが、この結果から推定される。
PCR
HGBVとC 型肝炎ウイルスとの配列相関性を検出するために、PCR に基づく実験を次のように行った。様々な位置的起源からのHCV 単離体(Cha, T.−A.ら, J. Clin. Microbiol. 29:2528 −2534[1991]) において高度に保存されている、HCV ゲノムの5’−非翻訳領域(UTR) 配列に基づくPCR プライマー(J.H. Han, PNAS 88:1711 −1715 [1991])を、H205感染タマリン T−1053肝臓RNA における類似の配列を検出するために使用した。実施例8Aで説明した通りに、全細胞RNA を感染タマリン T−1053 の肝臓と非感染タマリン(T−1040) の肝臓とから抽出した。Perkin−Elmer から入手可能なキットを使用し、その推奨使用法に概ね従って、各RNA の試料30μg を逆転写しPCR 増幅した。HCV 5’−UTR の塩基 249−268 を含むアンチセンスプライマー(プライマー1)を上記逆転写酵素反応に使用した。プライマー1と、HCV 5’−UTR の塩基13−46を含むプライマー(プライマー2)とを、介在配列のPCR 増幅のために使用した。サーモサイクリング(thermocycling) の条件はCha ら(前出)による。 H205感染タマリン T−1053のHCV 5’−UTR 配列の検出感度を増大させるために、上記PCR 反応に対して、「入れ子」PCR プライマーを使用した第2の増幅反応を生じさせた。そのためのプライマーは、HCV 5’−UTR 内における上記プライマー1の配列と上記プライマー2の配列との間にある配列から得た。プライマー3はHCV 5’−UTR の塩基47−69からの配列を含み、アンチセンスプライマーであるプライマー4はHCV 5’−UTR の塩基 188−210 を含んでいた。この「入れ子」PCR 反応では、第1のPCR 反応からのPCR 生成物(反応体積合計100 μl から2 μl )をDNA 鋳型源として使用した。アニーリング温度が60℃でなく55℃であったことを除いてサーモサイクリング条件は上記と概ね同一だった。第2のPCR 反応で得られたPCR 生成物を、アガロースゲル電気泳動と臭化エチジウム染色とを使用して所期のDNA 生成物に関して分析した。HCV 5’−UTR 配列に基づくこの所期のDNA フラグメントのサイズ(Han ら、前出)は、第1のPCR 反応の生成物に関しては 253bpであり、入れ子PCR 反応の生成物に関しては 163bpであった。所期サイズのPCR 生成物が、実施例8Aで説明したHCV 感染チンパンジーの肝臓から抽出した全細胞RNA 30μg を使用して行った対照実験において得られ(データは示していない)、これは、この実験手順でHCV の5’−UTR を検出できることを示す。しかし、 T−1053肝臓 RNA 又は T−1040肝臓RNA の一方を使用した場合には、どちらの所期生成物も、生じた臭化エチジウム染色アガロースゲル上に発見できなかった(データは示していない)。このように所期の結果が得られなかったのは、(i) 上記試薬の5’−UTR の配列がHCV のそれと大きく異なっていたために、使用したオリゴヌクレオチドプライマーが効率的にアニールされず、従って、PCR 増幅が不可能になったということ、又は、(ii)上記試薬に5’−UTR が欠如していたということを示唆する。どちらの場合にも、上記試薬のヌクレオチド配列がHCV のそれと大きく異なっていることが、この結果から推定される。
HCV の実験的感染の急性相中に得られたチンパンジー血漿プールから核酸を実施例7で説明した通り単離した(G.Schlauderら,J. Clin. Microbiology 29:2175−2179[1991]) 。クローン16プライマー (配列番号93、94) を使用してRT−PCR を実施例7の通り行った。これらのプライマーに関する所期サイズのバンドは、臭化エチジウム染色によっても検出されず、クローン16に特異的なプローブに対するハイブリッド形成後にも検出されなかった(データは示していない)。こうした結果は、HCV に対するクローン16配列 (配列番号26) の無関係性を立証する。
(実施例11)
他の肝炎ウイルスに対するHGBV感染血清の反応性
H205接種物(T−1048、 T−1057、 T−1061) 又は T−1053接種物(T−1051) を使用したHGBV接種の前と後に血清標本を得、既知の肝炎ウイルスに接触した後に検出されることが多い抗体に関する検査を行った。A 型肝炎ウイルスに対する抗体(本出願人から入手可能なHAVAB 検定を使用)と、B 型肝炎コアの核タンパク質(本出願人から入手可能な Corzyme(登録商標)検査を使用)と、E 型肝炎ウイルス(HEV) (本出願人から入手可能なHEV EIA を使用)と、C 型肝炎ウイルス(HCV) (本出願人から入手可能なHCV 第2世代検査を使用)とに関して、上記標本を検査した。これらの検査は、各検査キットのパッケージに同封された説明書に従って行った。
他の肝炎ウイルスに対するHGBV感染血清の反応性
H205接種物(T−1048、 T−1057、 T−1061) 又は T−1053接種物(T−1051) を使用したHGBV接種の前と後に血清標本を得、既知の肝炎ウイルスに接触した後に検出されることが多い抗体に関する検査を行った。A 型肝炎ウイルスに対する抗体(本出願人から入手可能なHAVAB 検定を使用)と、B 型肝炎コアの核タンパク質(本出願人から入手可能な Corzyme(登録商標)検査を使用)と、E 型肝炎ウイルス(HEV) (本出願人から入手可能なHEV EIA を使用)と、C 型肝炎ウイルス(HCV) (本出願人から入手可能なHCV 第2世代検査を使用)とに関して、上記標本を検査した。これらの検査は、各検査キットのパッケージに同封された説明書に従って行った。
HGBV接種の前と後のどちらにおいても、検査したタマリンは何れも、HCV又はHEVに対する抗体に関して陽性ではなかった(表12を参照)。従って、HGBV感染は、HCV又はHEVに対する検出可能な抗血清をもたらさなかった。
上記タマリンの中の1匹(T−1061) は、HGBV接種の前と後のどちらにおいてもHAV 抗体に対して陽性であり、このことは、HAV に事前に罹患していたことを示唆した(表9 、 T−1061)。しかし、他の3匹のタマリン(T−1048、 T−1057、 T−1051) は、HGBV接種後において、HAV に特異的な抗体を全く示さなかった。従って、HGBV感染は抗HAV 反応を生じさせない。上記タマリンの中の1匹(T−1048) は、HGBV接種の前と後のどちらにおいてもHBV 抗体に対して陰性だった。上記タマリンの中の2匹(T−1061、 T−1057) は、HGBV接種の前に陽性だった。上記タマリンの中の1匹(T−1051) は、HGBV接種の前にはHBV 抗体に対して境界近くの陽性であったが、接種後には陰性だった。こうしたデータに基づく限り、HGBV試薬による感染がHBV 核に対する免疫反応を誘導するという証明は得られない。総論的に言えば、これらのデータは、HGBV試薬が独特(unique)なウイルス剤であり、ヒト肝炎に関連した一般のウイルス性試薬の何れにも関係しないことを実証した。
(実施例12)
HGBV感染肝臓のウェスタンブロット分析
上記の実施例1 と実施例2とに示したように、HGBVを接種したタマリンには肝臓酵素値の上昇が認められた。HGBVが実際に肝臓向性ウイルスである場合には、ウイルスタンパク質が感染肝臓細胞内で生成され、こうしたタンパク質に対する免疫反応が生じることが推測される。この実施例では、HGBV感染タマリンから得られた肝臓に特有(unique)のタンパク質が現れ、このタンパク質がHGBV感染後の回復期段階に得られたタマリン血清を使用するウェスタンブロットによって特異的に認識されるとすべき根拠を示す。
HGBV感染肝臓のウェスタンブロット分析
上記の実施例1 と実施例2とに示したように、HGBVを接種したタマリンには肝臓酵素値の上昇が認められた。HGBVが実際に肝臓向性ウイルスである場合には、ウイルスタンパク質が感染肝臓細胞内で生成され、こうしたタンパク質に対する免疫反応が生じることが推測される。この実施例では、HGBV感染タマリンから得られた肝臓に特有(unique)のタンパク質が現れ、このタンパク質がHGBV感染後の回復期段階に得られたタマリン血清を使用するウェスタンブロットによって特異的に認識されるとすべき根拠を示す。
HGBV感染タマリン肝臓と様々な対照用のタマリン肝臓とチンパンジー肝臓とを賽の目に切り、Omni−mixer ホモジナイザーを使用してPBS 中にホモジナイズした(5mlに対して肝臓約1g) 。得られた懸濁液を、遠心分離(10,000×g 、1時間、4 ℃)と、 5μm フィルタと 0.8μm フィルタと0.45μm フィルタとを通す限界濾過とによって清澄化した。この清澄化したホモジネートを、100S以上の全成分のペレット化を生じさせる条件下で遠心分離した。ペレット(100S肝臓フラクション)を少量の緩衝液中に溶解し、−70℃で貯蔵した。
標準的な方法と試薬(Laemmli 不連続ゲル)を使用してSDS ポリアクリルアミドゲル電気泳動(PAGE)を行った。100S肝臓フラクションを、SDS と 2−メルカプトエタノールとを含む試料緩衝液中に1:20に希釈し、 5分間95℃に加熱した。30〜45分間200 ボルトで12% アクリルアミド又は 4→15% アクリルアミド線形勾配ゲル(7cm×8cm)のいずれかによって上記タンパク質を電気泳動した。標準の方法と試薬を使用して、タンパク質をニトロセルロース膜に電気転写(electro−transfer) した。
標準的な方法を使用してウェスタンブロットを生じさせた。概略的に言えば、ニトロセルロース膜をTBS/Tween 中で簡単にすすぎ、 4℃のTBS/CS(100mM Tris、150mM NaCl、10mM EDTA 、0.18% Tween −20、4.0%仔ウシ血清、pH 8.0)中で一晩封鎖した。上記ニトロセルロース膜をMulti −screen装置の中に入れ、 600μg の血清を上記装置の溝の中に入れ、その後2 時間室温に維持し、一晩4 ℃で温置した。 Multi−screen装置から上記ニトロセルロース膜を取り除き、TBS/Twee (50mM Tris 、150mM NaCl、0.05% Tween −20、pH8.0 )中で5分間ずつ3回上記ニトロセルロース膜を洗浄した。そのニトロセルロース膜をヤギ抗ヒト:HRPO抱合体(0.2μg/ml TBS/CS)15ml中で室温で1時間温置した。上記の通りに洗浄した後に、ニトロセルロース膜をTMB 酵素基質溶液中に温置し、水ですすぎ、乾燥した。
犠牲時(GB接種12日後)に T−1053肝臓から単離して上記の通りに吸収転写したタンパク質は、GB接種後5週目、6週目、7週目、9週目、又は11週目から、 T−1057血清との反応時に約50〜80kDa の見掛け分子量を有する独特な抗原性タンパク質を示した。GB接種前又は接種3週後の T−1057血清と反応させた時には、上記バンドは存在しなかった。上記 T−1057血清の何れかと反応させた時に、上記バンドは、非接種タマリン(T−1040) から得られた肝臓タンパク質を含むレーン中には現れなかった。加えて、他のGB接種後の血清(GB接種後11週目の T−1048と、GB接種後 8週目の T−1051)に T−1053肝臓タンパク質を反応させた時には、同一サイズ (50〜80kDa)のタンパク質が現れたが、同じ上記動物から得た接種前血清と T−1053肝臓タンパク質を反応させた時には、この同一サイズのタンパク質は現れなかった。
他のGB接種タマリンからの肝臓組織中で、又は、HCV もしくはHBV のいずれかに感染させたチンパンジーの肝臓組織の中で、上記免疫反応性バンドが検出されるかを調べるために、更に別のウェスタンブロット実験を行った。各々の場合、肝臓タンパク質を含むニトロセルロースストリップを T−1048(GB接種後5 、8 、16週目)と T−1051(GB接種後 8、12週目)とから得た血清プールと反応させた。上記プール中では、5つの血清を同等の割合で混合した。50〜80kDa の反応性タンパク質バンドを、GB接種タマリン(GB接種後14日目に得た T−1038、 T−1049、 T−1055と、GB接種後12日目に得た T−1053)から得た全てのタマリン肝臓標本において検出した。この免疫反応性バンドは、 T−1040(非接種)から得た肝臓標本と、チンパンジー肝臓標本 (CHAS−457(HCV 接種前) 、CHAS−457(HCV +) 、 CRAIG−454(HCV +) 、MUNA−376(HBV +))とにおいては検出できなかった。
以上総合して、これらのデータは、HGBV感染タマリン肝臓に特異的に結合する、約50〜80kDa の見掛け分子量を有する免疫原性及び抗原性タンパク質の存在を実証するものだった。このHGBV関連タンパク質の性質(即ち、ウイルスをコードするか又は宿主起源か)は現在調査中である。HGBV関連タンパク質の起源に関係なく、これらの結果は、HGBV感染がHGBV感染タマリン肝臓中に存在する抗原に対する抗体反応を誘導する事実と整合している。
(実施例13)
CKS に基づく免疫原性HGBV−A 及びHGBV−B ポリペプチドの発現と検出
A.HGBV−A 及びHGBV−B 配列のクローン化
クローン化ベクターpJO200、pJO201、pJO202により、組換えタンパク質を CMP−KDO シンセターゼ (CKS)タンパク質に融合させた。これらのプラスミドの各々はプラスミドpBR322由来であるが、(E.coli CKSタンパク質を構成する全体で 248個のアミノ酸の中の最初の 239個をコードする)kdsB遺伝子フラグメントに融合した修飾ラックプロモーターと、kdsB遺伝子フラグメントの末端に融合した合成リンカーとを有する。この合成リンカーは、遺伝子挿入に要する複数の制限部位と、翻訳停止シグナルと、trpA rho依存性転写ターミネーターとを含んでいた。このリンカー領域内の特有の制限部位は、5’から3’方向に、 EcoRI 、SacI、KpnI、SmaI、BamHI 、XbaI、PstI、SphI、HindIIIを含んでいた。各々のプラスミドは、上記複数のクローン化部位内のいずれの読み枠の中へも挿入を可能にする。タンパク質合成のCKS 方法とCKS ベクターは、本明細書に引例として組み入れられている本出願人の米国特許第 5,124,255号に開示されており、一方、検定フォーマットと検査キットとにおけるCKS 融合タンパク質の使用は、本明細書に引例として組み入れられている本出願人の米国出願番号No. 07/903,043に開示されている。
CKS に基づく免疫原性HGBV−A 及びHGBV−B ポリペプチドの発現と検出
A.HGBV−A 及びHGBV−B 配列のクローン化
クローン化ベクターpJO200、pJO201、pJO202により、組換えタンパク質を CMP−KDO シンセターゼ (CKS)タンパク質に融合させた。これらのプラスミドの各々はプラスミドpBR322由来であるが、(E.coli CKSタンパク質を構成する全体で 248個のアミノ酸の中の最初の 239個をコードする)kdsB遺伝子フラグメントに融合した修飾ラックプロモーターと、kdsB遺伝子フラグメントの末端に融合した合成リンカーとを有する。この合成リンカーは、遺伝子挿入に要する複数の制限部位と、翻訳停止シグナルと、trpA rho依存性転写ターミネーターとを含んでいた。このリンカー領域内の特有の制限部位は、5’から3’方向に、 EcoRI 、SacI、KpnI、SmaI、BamHI 、XbaI、PstI、SphI、HindIIIを含んでいた。各々のプラスミドは、上記複数のクローン化部位内のいずれの読み枠の中へも挿入を可能にする。タンパク質合成のCKS 方法とCKS ベクターは、本明細書に引例として組み入れられている本出願人の米国特許第 5,124,255号に開示されており、一方、検定フォーマットと検査キットとにおけるCKS 融合タンパク質の使用は、本明細書に引例として組み入れられている本出願人の米国出願番号No. 07/903,043に開示されている。
表9と表10(実施例9)で説明した移動実験(walking experiment) から得られたHGBV−A 配列とHGBV−B 配列を、表13と表14に示した制限酵素(10単位、NEB )を使用して適切な pT7Blue T−ベクタークローンから切り離し、実施例3Bで説明した通りに1%低融点アガロースゲルから精製した。プラスミドpJO200、pJO201、pJO202を同一の制限酵素(10単位、NEB )によって消化し、細菌性アルカリフォスフォターゼ(GIBCO BRL, Grand Island, NY) で脱リン酸した。精製したHGBVフラグメントの各々を、消化し脱リン酸したpJO200、pJO201、pJO202の中に結合させ、実施例3Bで説明した通りにE. coli XL1 Blueに形質転換した。標準的なミニプレプ(miniprep)分析によって、CKS/HGBV発現ベクターの形成の成功を確認した。
別の2つのPCR 生成物を、特に発現のために準備した。これら2つの生成物は、4.1 、4.2 と表され、各々にHGBV−B コア領域とHGBV−A コア領域とをコードすると予想された(図22参照)。実施例9で説明したように、PCR 生成物4.1 を、プライマーコア B−s とプライマーコア B−a1 (配列番号708 、709)を使用して作成し、PCR 生成物4.2 を、プライマーコア A−s とプライマーコア2.2.1’ (配列番号710 、138)を使用して生成した。この4.1 センス及びアンチセンスプライマーは、末端となるように意図したEcoRI 及びBamHI 制限部位を各々に有した。上記のように、4.1PCR生成物を消化し、ゲル単離し、pJO200、pJO201、pJO202に結合させた。4.2PCR 生成物に関するセンスプライマーは、末端となるように意図したEcoRI 制限部位を有するが、そのアンチセンスプライマーは制限部位を持たなかった。従って、この生成物を、EcoRI によって切断し、ゲル単離し、既にBamHI で消化したpJO200、pJO201、pJO202に結合させ、DNA ポリメラーゼのクレノウフラグメントとdNTPとで末端修飾し、EcoRI で消化し、当業界で公知のように細菌性アルカリフォスフォターゼで脱リン酸した。
B.HGBV−A 及びHGBV−B 配列の発現
トリプトン32gm/L、酵母抽出物20gm/L、NaCl 5gm/L、pH7.4 、アンピシリン100mg/L 、グルコース3mM を含む媒質中で振とうしながら、CKS/HGBV発現ベクターを含むE. coli XL1 Blue培養物を37℃で生長させた。この培養物が 1.0〜2.0 のOD600 に達した時に、修飾ラックプロモーターからの発現を誘導するためIPTGを最終濃度1mM になるように加えた。更に3時間振とうしながら37℃で培養物を生長させ、集菌した。細胞ペレットを SDS/PAGE充填緩衝液(Tris pH6.8 62.5mM 、SDS 2%、グリセロール 10%、 2−メルカプトエタノール5%、ブロモフェノールブルー0.1mg/ml)中に10のOD600 となるよう再懸濁させ、5分間沸騰させた。調製した全細胞ライゼートのアリコートを10% SDS −ポリアクリルアミドゲル上に施し、40% メタノール/10% 酢酸中にクーマシーブルー染料0.2%を含む溶液で着色し、更に、バックグラウンドが透明になるまで16.5% メタノール/5%酢酸で脱色した。
トリプトン32gm/L、酵母抽出物20gm/L、NaCl 5gm/L、pH7.4 、アンピシリン100mg/L 、グルコース3mM を含む媒質中で振とうしながら、CKS/HGBV発現ベクターを含むE. coli XL1 Blue培養物を37℃で生長させた。この培養物が 1.0〜2.0 のOD600 に達した時に、修飾ラックプロモーターからの発現を誘導するためIPTGを最終濃度1mM になるように加えた。更に3時間振とうしながら37℃で培養物を生長させ、集菌した。細胞ペレットを SDS/PAGE充填緩衝液(Tris pH6.8 62.5mM 、SDS 2%、グリセロール 10%、 2−メルカプトエタノール5%、ブロモフェノールブルー0.1mg/ml)中に10のOD600 となるよう再懸濁させ、5分間沸騰させた。調製した全細胞ライゼートのアリコートを10% SDS −ポリアクリルアミドゲル上に施し、40% メタノール/10% 酢酸中にクーマシーブルー染料0.2%を含む溶液で着色し、更に、バックグラウンドが透明になるまで16.5% メタノール/5%酢酸で脱色した。
全細胞ライゼートを第2の10% SDS −ポリアクリルアミドゲル上に施し、イムノブロット検定のために電気泳動によってニトロセルロースに転写した。転写されたタンパク質を含むニトロセルロースのシートを室温で30分間封鎖溶液(Tris緩衝塩 類液中の5%Carnation 無脂肪ドライミルク)中に温置し、E.coli 細胞ライゼートに対して予め封鎖されたヤギ抗CKS 血清中で室温で1時間温置し、封鎖溶液中に1:1000に希釈した。このニトロセルロースシートをTris緩衝塩類液(TBS) で2回洗浄し、その後アルカリホスファターゼ抱合ウサギ抗ヤギIgG と共に室温で1時間温置し、封鎖溶液中で 1:1000に希釈した。このニトロセルロースシートをTBS で2回洗浄し、ニトロブルーテトラゾリウム(nitroblue tetrazolium) と5 −ブロモ−4 −クロロ−3 −インドリルホスフェートとを含むTBS 中で発色させた。各フラグメントに対する適切な読み枠を、適正な予想サイズの免疫反応性CKS 融合タンパク質の発現に基づいて定め、ベクター挿入結合部分についてはDNA 配列決定によって確認した。
上記フラグメント各々の適切な読み枠を決定した後に、適切な構成要素を含む培養物からの試料を、SDS −ポリアクリルアミドゲル電気泳動とウェスタンブロットとによって分析した。図25A は、上記CKS 融合タンパク質全細胞ライゼートを含む2つのクーマシー染色10%SDS−ポリアクリルアミドゲルを示す。レーン1とレーン16は、左に示されるキロダルトン単位の分子量標準を含む。ゲル1(HGBV−A 試料)上の展開順序は次の通りだった:レーン2 、クローン1.17誘導前:レーン3 −15、クローン4.2 、クローン1.17、クローン1.8 、クローン1.2 、クローン1.18 (配列番号390)、クローン1.19、クローン1.20、クローン1.21、クローン1.22 (配列番号390)、クローン2.12、クローン1.5 、クローン1.23、クローン2.18、各々全て誘導3時間後。ゲル2 (HGBV−B 試料)上の展開順序は次の通りだった:レーン17、クローン4.1 誘導前:レーン18−29、クローン4.1 、クローン1.15、クローン1.14、クローン2.8 、クローン1.13、クローン1.12、クローン2.1 、クローン1.7 、クローン1.3 、クローン1.4 、クローン1.16、クローン2.12、各々全て誘導3時間後。これらのタンパク質を上記と同じ順序で2つの別の10%ゲル上に施し、上記の通りにニトロセルロースに転写した。試料を、2匹の回復期の T−1048、 T−1051からの血清のプールを使用したウェスタンブロットで次のように分析した。上記試料を含むニトロセルロースシートを封鎖溶液中に30分間温置し、E.coliライゼート 10%と、XL1 −Blue/CKSライゼート6mg/mlと、表6(実施例4)で説明した回復期タマリン血清プールの1:100 希釈液とを含む封鎖溶液に移した。室温で一晩温置した後、上記ニトロセルロースシートをTBS 中で2回洗浄し、HRPO−抱合ヤギ抗ヒトIgG 中に室温で1時間温置し、封鎖溶液中に1:500 に希釈した。ニトロセルロースシートをTBS 中で2回洗浄し、4 −クロロ−1 −ナフトール 2 mg/mlと過酸化水素 0.02%とメタノール 17%とを含むTBS 中で発色させた。図25B に示されているように、3つのHGBV−B タンパク質、クローン1.4 、1.7 、4.1 のCKS 融合が、タマリン血清プールとの免疫反応を示した。クローン1.7 は、HGBV−B 免疫原性領域をコードする配列 (配列番号610)を含み、クローン1.4 は、同じ回復期タマリン血清プールを使用したcDNAライブラリー(実施例4)のイムノスクリーニングによって発見された、2つのHGBV−B 免疫原性領域をコードする配列 (配列番号12、13、18) を含んでいた。
上記のパラグラフで説明した試料も、軍医GB(the surgeon GB)からの急性肝炎の発病後約3週目に得られた回復期血清の1:100 希釈液を使用して、上記の通りにウェスタンブロットによって分析した。HGBV−A とHGBV−B とからの融合タンパク質の上記血清に対する反応性を表13、14に示す。HGBV−B タンパク質では1つ(2.1) だけがこの血清に対する反応性を示したが、この反応性は非常に低く、一方、2つのHGBV−A タンパク質 (1.22[配列番号390]、2.17)はこの血清に対する高い反応性を示した。これら2つのHGBV−A タンパク質は40個のアミノ酸について共通であり、このことは、1個以上のエピトープに対する反応性を反映している可能性がある。これら2つのHGBV−A タンパク質を、実施例16で説明した通りのELISA 検定で使用するために選択した。第11代継代接種GB材料(H205 GB pass 11) に感染したタマリンは、幾つかのHGBV−B エピトープに対して免疫反応を示し、HGBV−A エピトープに対しては免疫反応を示さなかったが、最初のGB源からの血清は、少なくとも1つの GBV −A エピトープに対して著しい反応性を示したということは興味深い。このことは、HGBV−A が軍医GBにおける肝炎の原因であった可能性を示唆した。
1.4 、1.7 、又は2.17 ELISAS(実施例15、16を参照されたい)によってCKS/HGBV−A 又はCKS/HGBV−B 融合タンパク質の1つ以上に対する抗体の存在を示した4つの別のヒト血清を、ウェスタンブロット分析のために選択した。これらの血清の3つ(G1−41、G1−14、G1−31)は西アフリカ「危険」集団からのものであり、4番目(341C)は、非 A−E 肝炎(エジプト)標本(これらの集団の詳細な説明については実施例15を参照されたい)からのものだった。上記のCKS 融合タンパク質の中の幾つかからの全細胞ライゼートを別の10% SDS −ポリアクリルアミドゲルに施し、上記の通りニトロセルロースに転写した。これらのブロットの各々を上記の通りに予め封鎖し、E. coli ライゼート 10%と XL1−Blue/CKSライゼート 6 mg/mlとを含む封鎖緩衝液中に1:100 に希釈したヒト血清標本の1つと共に一晩温置した。ブロットをTBS 中で2回洗浄し、HRPO抱合ヤギ抗ヒトIgG と反応させ、上記の通りに発色させた。
CKS/HGBV−B タンパク質を上記血清の2つ(G1−41、G1−14)を使用して分析し、その反応性を表13に示した。タマリン血清に対して反応性を示した上記3つのタンパク質に加えて、更に別の2つのタンパク質 (1.16、2.1)が2つのヒト血清のどちらか一方に対して反応性を示した。CKS/HGBV−A タンパク質を、上記4つのヒト血清全てと分析し、これらの反応性を表14に示す。GB血清に対して反応性を示した上記2つのタンパク質に加えて、更に別の3つのタンパク質(1.5、1.18、1.19) がヒト血清の1つ以上に対して反応性を示した。これらのタンパク質の内の2つ(1.5 、1.18)を、実施例16で説明する通りのELISA 検定に使用するために選択した。ウェスタンブロットによって及び/又は2つのHGBV−A タンパク質(1.18、2.17)と1つのHGBV−B タンパク質(1.7) とに対するELISA(実施例15、16)によって反応性を示すG1−31血清が、GB−C 配列 (配列番号673 、残基2274−2640) がそれから単離された血清である(実施例17)ということが特に興味深い。
b制限消化は、 pT7Blue T−ベクターからPCR フラグメントを遊離させるために、又は、4.1PCR生成物の直接消化のために使用した。
(実施例14)
免疫反応性HGBV−A 及びHGBV−B タンパク質のエピトープマッピング
A.HGBV−B タンパク質 1.7のエピトープマッピング
免疫原性領域の位置を確認するために、HGBV−B 免疫原性タンパク質1.7 内の重複サブクローンを、実施例7で説明した通り T−1053血清からのRT−PCR によって実施した。各PCR プライマーは、制限酵素消化を容易にするために5’末端側に余分の6つの塩基を有しており、その後にEcoRI 部位(センスプライマー)又はHindIII部位(アンチセンスプライマー)のいずれかが続いた。加えて、各アンチセンスプライマーはコーディング領域の直後に終止コドンを含んでいた。消化後に、実施例13で説明したように、各フラグメントをEcoRI/HindIII−消化pJO201の中にクローン化した。実施例13で説明した通り、CKS 融合タンパク質を発現させ、タマリン T−1048/T−1051血清を使用したウェスタンブロットによって分析した。 1.7−1 から 1.7−5 と称する5つの重複クローンを生成した。これらのクローンは、サイズがアミノ酸104〜110 個分の範囲にある1.7 タンパク質の領域をコードしている。各クローンを生成するために使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.7 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。cDNAライブラリー(実施例4)のイムノスクリーニングによって発見された免疫原性領域(配列番号678 )にまたがる、更に別の2つの重複クローンを生成した。 1.7−6 と 1.7−7 と称されるこれらのクローンの各々は、アミノ酸75個のポリペプチドをコードする。使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.7 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。アミノ酸 507個分の長さの1.7 タンパク質の中に、2つの免疫原性領域を確認した。即ち、 N末端付近の免疫原性領域は残基 1−105 の範囲内であり、上記タンパク質の中央付近の免疫原性領域は残基 185−410 の範囲内であった。これらの領域の各々の中に単一のエピトープ又は複数のエピトープがあるかどうかは、更に検討を要する。
免疫反応性HGBV−A 及びHGBV−B タンパク質のエピトープマッピング
A.HGBV−B タンパク質 1.7のエピトープマッピング
免疫原性領域の位置を確認するために、HGBV−B 免疫原性タンパク質1.7 内の重複サブクローンを、実施例7で説明した通り T−1053血清からのRT−PCR によって実施した。各PCR プライマーは、制限酵素消化を容易にするために5’末端側に余分の6つの塩基を有しており、その後にEcoRI 部位(センスプライマー)又はHindIII部位(アンチセンスプライマー)のいずれかが続いた。加えて、各アンチセンスプライマーはコーディング領域の直後に終止コドンを含んでいた。消化後に、実施例13で説明したように、各フラグメントをEcoRI/HindIII−消化pJO201の中にクローン化した。実施例13で説明した通り、CKS 融合タンパク質を発現させ、タマリン T−1048/T−1051血清を使用したウェスタンブロットによって分析した。 1.7−1 から 1.7−5 と称する5つの重複クローンを生成した。これらのクローンは、サイズがアミノ酸104〜110 個分の範囲にある1.7 タンパク質の領域をコードしている。各クローンを生成するために使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.7 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。cDNAライブラリー(実施例4)のイムノスクリーニングによって発見された免疫原性領域(配列番号678 )にまたがる、更に別の2つの重複クローンを生成した。 1.7−6 と 1.7−7 と称されるこれらのクローンの各々は、アミノ酸75個のポリペプチドをコードする。使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.7 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。アミノ酸 507個分の長さの1.7 タンパク質の中に、2つの免疫原性領域を確認した。即ち、 N末端付近の免疫原性領域は残基 1−105 の範囲内であり、上記タンパク質の中央付近の免疫原性領域は残基 185−410 の範囲内であった。これらの領域の各々の中に単一のエピトープ又は複数のエピトープがあるかどうかは、更に検討を要する。
B.HGBV−B タンパク質1.4 のエピトープマッピング
免疫原性領域の位置を確認するために、HGBV−B 免疫原性タンパク質1.4 内の重複サブクローンを、 T−1053血清からのRT−PCR によって実施した。各PCR プライマーは、制限酵素消化を容易にするために5’末端側に余分の6つの塩基を有しており、その後にEcoRI 部位(センスプライマー)又はBamHI 部位(アンチセンスプライマー)のいずれかが続いた。加えて、各アンチセンスプライマーはコーディング領域の直後に終止コドンを含んでいた。消化後に、実施例13で説明したように、各フラグメントを EcoRI/BamHI−消化pJO201の中にクローン化した。実施例13で説明した通り、CKS 融合タンパク質を発現させ、タマリン T−1048/T−1051血清を使用したウェスタンブロットによって分析した。1.4−1 から 1.4−4 と称する4つの重複クローンを生成した。これらのクローンは、サイズがアミノ酸 137〜138 個の範囲にある1.4 タンパク質の領域をコードしている。各クローンを生成するために使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.4 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。cDNAライブラリー(実施例4)のイムノスクリーニングによって発見された免疫原性領域にまたがる、更に別の2つの重複クローンを生成した。1.4−5 と 1.4−6 とで称されるこれらのクローンの各々は、アミノ酸75個の長さのポリペプチドをコードする。使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.4 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。残基 129−393 を含むアミノ酸 265個から構成される配列が、アミノ酸 522個分の長さの1.4 タンパク質の中の免疫原性領域であると確認した。ライブラリーのイムノスクリーニング(実施例4)によって上記配列内に2つの非隣接免疫原性クローンを確認したので、少なくとも2つのエピトープが上記領域内に存在する可能性がある。
免疫原性領域の位置を確認するために、HGBV−B 免疫原性タンパク質1.4 内の重複サブクローンを、 T−1053血清からのRT−PCR によって実施した。各PCR プライマーは、制限酵素消化を容易にするために5’末端側に余分の6つの塩基を有しており、その後にEcoRI 部位(センスプライマー)又はBamHI 部位(アンチセンスプライマー)のいずれかが続いた。加えて、各アンチセンスプライマーはコーディング領域の直後に終止コドンを含んでいた。消化後に、実施例13で説明したように、各フラグメントを EcoRI/BamHI−消化pJO201の中にクローン化した。実施例13で説明した通り、CKS 融合タンパク質を発現させ、タマリン T−1048/T−1051血清を使用したウェスタンブロットによって分析した。1.4−1 から 1.4−4 と称する4つの重複クローンを生成した。これらのクローンは、サイズがアミノ酸 137〜138 個の範囲にある1.4 タンパク質の領域をコードしている。各クローンを生成するために使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.4 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。cDNAライブラリー(実施例4)のイムノスクリーニングによって発見された免疫原性領域にまたがる、更に別の2つの重複クローンを生成した。1.4−5 と 1.4−6 とで称されるこれらのクローンの各々は、アミノ酸75個の長さのポリペプチドをコードする。使用したPCR プライマーと、コードされたポリペプチドのサイズと、1.4 配列内の位置と、タマリン T−1048/T−1051血清に対する反応性とを、表15に示す。残基 129−393 を含むアミノ酸 265個から構成される配列が、アミノ酸 522個分の長さの1.4 タンパク質の中の免疫原性領域であると確認した。ライブラリーのイムノスクリーニング(実施例4)によって上記配列内に2つの非隣接免疫原性クローンを確認したので、少なくとも2つのエピトープが上記領域内に存在する可能性がある。
C.HGBV−A タンパク質1.22 (配列番号390)及び2.17のエピトープマッピング
HGBV−A タンパク質1.22(配列番号390 )と2.17 (配列番号613)は、いずれもウェスタンブロットによってGB血清との免疫反応を示した(実施例13)。これらの2つのタンパク質はアミノ酸40個分だけ重なっているので、観察された免疫反応は、1つのエピトープによるか、又は2つ以上のエピトープの存在の結果として生じた可能性がある。完全な1.22/2.17配列はアミノ酸 641個分の長さだった。免疫原性領域を発見するために、この領域内の重複サブクローンを T−1053からRT−PCR によって実施した。各PCR プライマーは、制限酵素消化を容易にするために5’末端側に余分の6つの塩基を有しており、その後に、EcoRI 部位(センスプライマー)、又は、1.22/2.17−2 から1.22/2.17−6 に関してBamHI 部位(アンチセンスプライマー)のいずれかが続いた。しかし、クローン1.22/2.17−1 は内部EcoRI 部位を有するので、BamHI 部位がセンスプライマーとして使用され、HindIII部位がアンチセンスプライマーとして使用された。加えて、各アンチセンスプライマーはコーディング領域の直後に終止コドンを含んでいた。消化後に、実施例13で説明したように、各フラグメントをEcoRI/BamHI −消化(又は、1.22/2.17−1 に関してはBamHI/HindIII−消化)pJO201の中にクローン化した。実施例13で説明した通り、CKS 融合タンパク質を発現させ、GB血清を使用してウェスタンブロットによって分析した。このクローンはアミノ酸 115−116 個分のサイズ範囲の1.22/2.17 の領域をコードした。各クローンを生成するために使用したPCR プライマーと、コードされたポリペプチドのサイズと、HGBV−A ポリペプチド配列内の位置と、GB血清に対する反応性とを、表15に示す。1.22/2.17 タンパク質の中央では、免疫原性領域はアミノ酸 220個分の長さの領域に狭まっていた。これは、1.22と2.17の間のアミノ酸40個分の長さの重複領域を含み、従って、2つのタンパク質に共通する免疫反応は、共有している1つのエピトープによるか、又は複数のエピトープに起因していた可能性がある。
HGBV−A タンパク質1.22(配列番号390 )と2.17 (配列番号613)は、いずれもウェスタンブロットによってGB血清との免疫反応を示した(実施例13)。これらの2つのタンパク質はアミノ酸40個分だけ重なっているので、観察された免疫反応は、1つのエピトープによるか、又は2つ以上のエピトープの存在の結果として生じた可能性がある。完全な1.22/2.17配列はアミノ酸 641個分の長さだった。免疫原性領域を発見するために、この領域内の重複サブクローンを T−1053からRT−PCR によって実施した。各PCR プライマーは、制限酵素消化を容易にするために5’末端側に余分の6つの塩基を有しており、その後に、EcoRI 部位(センスプライマー)、又は、1.22/2.17−2 から1.22/2.17−6 に関してBamHI 部位(アンチセンスプライマー)のいずれかが続いた。しかし、クローン1.22/2.17−1 は内部EcoRI 部位を有するので、BamHI 部位がセンスプライマーとして使用され、HindIII部位がアンチセンスプライマーとして使用された。加えて、各アンチセンスプライマーはコーディング領域の直後に終止コドンを含んでいた。消化後に、実施例13で説明したように、各フラグメントをEcoRI/BamHI −消化(又は、1.22/2.17−1 に関してはBamHI/HindIII−消化)pJO201の中にクローン化した。実施例13で説明した通り、CKS 融合タンパク質を発現させ、GB血清を使用してウェスタンブロットによって分析した。このクローンはアミノ酸 115−116 個分のサイズ範囲の1.22/2.17 の領域をコードした。各クローンを生成するために使用したPCR プライマーと、コードされたポリペプチドのサイズと、HGBV−A ポリペプチド配列内の位置と、GB血清に対する反応性とを、表15に示す。1.22/2.17 タンパク質の中央では、免疫原性領域はアミノ酸 220個分の長さの領域に狭まっていた。これは、1.22と2.17の間のアミノ酸40個分の長さの重複領域を含み、従って、2つのタンパク質に共通する免疫反応は、共有している1つのエピトープによるか、又は複数のエピトープに起因していた可能性がある。
(実施例15)
HGBV−Bの血清学的研究
A.組換えタンパク精製手順
CKS融合タンパクを発現する細菌細胞培養物を凍結し、−70℃で保存した。三つの構築物それぞれからの細菌細胞を解凍し、リゾチームとDNアーゼを用いて粉砕処理し、次いでフッ化フェニルメタンスルホニルおよびその他のプロテアーゼ阻害剤の存在下、超音波処理して個々の組換え抗原と大腸菌タンパクの混合物を得た。三つの培養物それぞれについて、不溶性の組換え抗原を遠心分離により濃縮し、一連の洗浄工程に付して非組換え大腸菌タンパクの大部分を除去した。本プロトコールで使用した洗浄液の構成は、蒸留水、5%TritonX−100、および50mMのTris(8.5)であった。得られた沈殿物をドデシル硫酸ナトリウム(SDS)の存在下、溶解した。タンパク濃度を測定したあと、2−メルカプトエタノールを加え、混合物をセファクリルS300樹脂を用いたゲル濾過カラムクロマトグラフィーに付し、種々のタンパクをそのサイズによって分別、分離した。各分画を集め、SDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE)で分析した。この電気泳動で分離されたタンパクは、次いでクーマシーブリリアントブルーR250で染色し、分子量が約75kD(CKS−1.7/配列番号610)、80kD(CKS−1.4/配列番号611)、42kD(CKS−4.1/配列番号612)に相当するタンパクの有無を調べた。該当するタンパクを含有する分画をプールし、SDS−PAGEによって再度調査した。
HGBV−Bの血清学的研究
A.組換えタンパク精製手順
CKS融合タンパクを発現する細菌細胞培養物を凍結し、−70℃で保存した。三つの構築物それぞれからの細菌細胞を解凍し、リゾチームとDNアーゼを用いて粉砕処理し、次いでフッ化フェニルメタンスルホニルおよびその他のプロテアーゼ阻害剤の存在下、超音波処理して個々の組換え抗原と大腸菌タンパクの混合物を得た。三つの培養物それぞれについて、不溶性の組換え抗原を遠心分離により濃縮し、一連の洗浄工程に付して非組換え大腸菌タンパクの大部分を除去した。本プロトコールで使用した洗浄液の構成は、蒸留水、5%TritonX−100、および50mMのTris(8.5)であった。得られた沈殿物をドデシル硫酸ナトリウム(SDS)の存在下、溶解した。タンパク濃度を測定したあと、2−メルカプトエタノールを加え、混合物をセファクリルS300樹脂を用いたゲル濾過カラムクロマトグラフィーに付し、種々のタンパクをそのサイズによって分別、分離した。各分画を集め、SDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE)で分析した。この電気泳動で分離されたタンパクは、次いでクーマシーブリリアントブルーR250で染色し、分子量が約75kD(CKS−1.7/配列番号610)、80kD(CKS−1.4/配列番号611)、42kD(CKS−4.1/配列番号612)に相当するタンパクの有無を調べた。該当するタンパクを含有する分画をプールし、SDS−PAGEによって再度調査した。
精製抗原を含有するプール分画の免疫原性および構造の完全性を、実施例13に記載の方法に従ってニトロセルロースに対する電子移動およびイムノブロット法によって決定した。適当な陽性コントロールがなかったため、組換えタンパクは、各融合タンパクのCKS部分に対するモノクローナル抗体との反 応性によって同定した。CKS−1.7タンパク(配列番号 610)について、組換え抗原を検出すべく抗−CKSモノ クローナル抗体を用いてウェスタンブロット法で調べると、約75kDのところにシングルバンドが認められた。この結果はCKS−1.7タンパク(配列番号610)の予想サイズに一致している。CKS−1.4タンパク(配列番号611)については、抗−CKSモノクローナル抗体によって60kDと 70kDの間に四重のバンドパターンが検出された。観察されたこれらのバンドは完全長タンパクについて予想された長さより短く、恐らく欠失されたものであろうと思われる。CKS−4.1タンパク(配列番号52)をウェスタンブロット法で調べると、抗−CKSモノクローナル抗体によって組換え抗体が約42kDにおいてシングルバンドとして検出された。この結果はCKS−4.1タンパク(配列番号612)について予想されたサイズと一致している。
B.ポリスチレンビーズの被覆手順
上記タンパクを透析し、以下に記載のように、これらのタンパクのポリスチレンビーズ上での抗原性を評価した。これとは別に、三種の精製HGBV組換えタンパク(CKS−1.7/配列番号610、CKS−1.4/配列番号611、およびCKS−4.1/配列番号612)のそれぞれを用いてHGBVに対する抗体を検出すべく、酵素結合免疫吸収アッセイ(ELISA)を行った。これらのタンパクで行ったELISAをそれぞれ、1.7ELISA(CKS−1.7(配列番号610)組換えタンパク使用)、1.4ELISA(CKS−1.4(配列番号611)組換えタンパク使用)、および4.1ELISA(CKS−4.1(配列番号612)組換えタンパク使用)とする。最初の研究においては、1/4インチのポリスチレンビーズを種々の濃度の精製タンパクで被覆(約60ビーズ/ロット)し、無接種のタマリンからの血清を陰性コントロール、接種したタマリンからの回復期血清を陽性コントロールとして用いたELISA試験(下記)により評価した。その他のコントールとして、種々の肝炎ウイルスに対して陰性と判定された個体から得たヒト血清のプールを含めた。追加の陽性コントロールは、ビーズの被覆効率をモニターするためのCKSタンパクに対するモノクローナル抗体であった。陰性コントロールシグナルに対する陽性コントロールシグナルの比が最も高くなるようにビーズの被覆条件を選択し、ビーズ被覆プロセスのスケールアッセイに用いた。四つのELISA試験のそれぞれについて、1000ビーズからなるロットを少なくとも2ロット作成し、血清学的研究に用いた。
上記タンパクを透析し、以下に記載のように、これらのタンパクのポリスチレンビーズ上での抗原性を評価した。これとは別に、三種の精製HGBV組換えタンパク(CKS−1.7/配列番号610、CKS−1.4/配列番号611、およびCKS−4.1/配列番号612)のそれぞれを用いてHGBVに対する抗体を検出すべく、酵素結合免疫吸収アッセイ(ELISA)を行った。これらのタンパクで行ったELISAをそれぞれ、1.7ELISA(CKS−1.7(配列番号610)組換えタンパク使用)、1.4ELISA(CKS−1.4(配列番号611)組換えタンパク使用)、および4.1ELISA(CKS−4.1(配列番号612)組換えタンパク使用)とする。最初の研究においては、1/4インチのポリスチレンビーズを種々の濃度の精製タンパクで被覆(約60ビーズ/ロット)し、無接種のタマリンからの血清を陰性コントロール、接種したタマリンからの回復期血清を陽性コントロールとして用いたELISA試験(下記)により評価した。その他のコントールとして、種々の肝炎ウイルスに対して陰性と判定された個体から得たヒト血清のプールを含めた。追加の陽性コントロールは、ビーズの被覆効率をモニターするためのCKSタンパクに対するモノクローナル抗体であった。陰性コントロールシグナルに対する陽性コントロールシグナルの比が最も高くなるようにビーズの被覆条件を選択し、ビーズ被覆プロセスのスケールアッセイに用いた。四つのELISA試験のそれぞれについて、1000ビーズからなるロットを少なくとも2ロット作成し、血清学的研究に用いた。
簡単に述べると、ポリスチレンビーズを精製タンパクで被覆するにあたり、洗浄したビーズをシンチレーションバイアルに加え、ビーズを組換え抗原含有緩衝液に浸漬した(約0.233ml/ビーズ)。各組換え抗原につき種々の異なる濃度を用意し、これらをpH5.0〜9.5の範囲に調整した種々の異なる緩衝液とともに用いて評価した。次いでバイアルを40℃のインキュベーター中の回転装置上に2時間載置し、その後液体を吸引し、ビーズをpH6.8のリン酸緩衝食塩水(PBS)で3回洗浄した。次いでビーズを0.1%TritonX−100を用いて1時間、40℃で処理し、PBSで3回洗浄した。次に、ビーズを5%ウシ血清アルブミンでオーバーコートし、攪拌しながら40℃、1時間でインキュベートした。PBSでさらに洗浄を繰り返した後、ビーズを5%ショ糖で20分間室温でオーバーコートし、液体は吸引除去した。最後にビーズを空気乾燥し、HGBVに対する抗体を検出するためのELISA試験に使用した。
C.HGBVに対する抗体を検出するためのELISA手順
ELISAは間接アッセイ法で行った。即ち、血清または血漿を検体希釈剤で希釈し、固相に被覆した抗原と反応させた。洗浄工程の後、固相と結合したタマリンまたはヒト抗体を検出するために、ビーズをヒト免疫グロブリンを指向する西洋ワサビペルオキシダーゼ(HRPO)標識抗体と反応させた。切 捨て値を越えるシグナルを生じた検体は反応性ありとした。 ELISAに関する付加的な詳細を以下に記す。
ELISAは間接アッセイ法で行った。即ち、血清または血漿を検体希釈剤で希釈し、固相に被覆した抗原と反応させた。洗浄工程の後、固相と結合したタマリンまたはヒト抗体を検出するために、ビーズをヒト免疫グロブリンを指向する西洋ワサビペルオキシダーゼ(HRPO)標識抗体と反応させた。切 捨て値を越えるシグナルを生じた検体は反応性ありとした。 ELISAに関する付加的な詳細を以下に記す。
ELISAの様式は、抗原で被覆した固相を予め検体希釈剤(動物血清と非イオン活性剤を含有する緩衝溶液)で希釈したタマリン血清と接触させるものである。この検体希釈剤の調合に当たっては、特異的抗体が抗原で被覆された固相と結合する割合を高める一方、固相に対する非特異的な免疫グロブリンの結合に起因するバックグラウンドシグナルを減少させるように調合した。具体的には、10μlのタマリン血清を150μlの検体希釈剤で希釈し、回転攪拌した。この予備希釈された検体10μlを反応トレイ中のウェルに添加した後、200μlの検体希釈剤と抗原被覆ポリスチレンビーズを加えた。この反応トレイをダイナミックインキュベーター(アボットラボラトリーズ製)中でインキュベートした。インキュベーターは、室温で常に攪拌し続けた。1時間のインキュベーションの後、液体を吸引除去し、ビーズを含有するウェルを蒸留水で3回洗浄した(各回5ml)。次にコンジュゲート希釈剤(動物血清と非イオン活性剤を含有する緩衝溶液)で希釈したHRPO標識ヤギ抗ヒト免疫グロブリンを200μl各ウェルに添加し、反応トレイを上記のようにして再び1時間インキュベートした。液体を吸引除去し、ビーズを含有するウェルを蒸留水で上記のようにして3回洗浄した。抗原及び結合免疫グロブリンを有するビーズをウェルから除去し、各ビーズを試験管に入れ、0.3%o−フェニレンジアミン二塩酸塩の0.02%H2O2含有0.1Mクエン酸塩緩衝液(pH5.5)溶液300μlと反応させた。室温で30分保持後、1NのH2SO4を加えて反応を停止させた。492nmでの吸収を分光光度計で読み取った。生じた色は、試験サンプル中に存在する抗体の量と正比例する関係にあった。
HGBVエピトープに対する抗体を含まないと思われる検体とHGBVエピトープに対する抗体を含むと思われる検体とを分別するために、検体の各グループに対して予備的な切捨て値を設定した。
D.感染個体の血清におけるHGBV由来RNAの検出
タマリン血清またはヒト血清/血漿中における、HGBV−RNA存在下の1.7ELISAおよび1.4ELISAの血清反応データを相互に関連付けるために、RT−PCRを既に参照して本明細書に包含した米国特許出願第08/283,314号の実施例7と同様に実施した。実施にあたってはHGBVのクローン化された配列に由来するオリゴヌクレオチドを使用した。オリゴヌクレオチドのHGBV−Bゲノムに由来するクローン4(実施例7参照)およびHGBV−Aゲノムに由来するクローン16に対する最終濃度は0.5μMであった。
タマリン血清またはヒト血清/血漿中における、HGBV−RNA存在下の1.7ELISAおよび1.4ELISAの血清反応データを相互に関連付けるために、RT−PCRを既に参照して本明細書に包含した米国特許出願第08/283,314号の実施例7と同様に実施した。実施にあたってはHGBVのクローン化された配列に由来するオリゴヌクレオチドを使用した。オリゴヌクレオチドのHGBV−Bゲノムに由来するクローン4(実施例7参照)およびHGBV−Aゲノムに由来するクローン16に対する最終濃度は0.5μMであった。
E.タマリンの血清像
LEMSIPに収容されたタマリンから週単位で採血して血清を得、肝臓の酵素レベルを検査した。検体血清の残量はさらなる調査のためにアボットラボラトリーズに送った。
LEMSIPに収容されたタマリンから週単位で採血して血清を得、肝臓の酵素レベルを検査した。検体血清の残量はさらなる調査のためにアボットラボラトリーズに送った。
1.タマリンについてのELISAの結果(初期感染性の研究)
4匹のタマリン(T−1053、T−1048、T−1057、およびT−1061)にGB血清(H205GB継代数 11と称する)を接種した。接種後(PI)の最初1週間の間、タマリンT−1053では肝酵素の上昇が認められた。このタマリンは、PI12日で安楽死させた。タマリンT−1048、T−1057、およびT−1061は、接種後2週間までの間に肝酵素の上昇を示した。この上昇はPI8〜9週まで続いた(図2〜4)後、接種前のレベルに戻った。PI14週において、これら3匹のタマリンに対し、タマリンT−1053(この個体は感染力を有することが判明している−実施例2)から得た希釈していない血清0.10mlでの再接種を行った。
4匹のタマリン(T−1053、T−1048、T−1057、およびT−1061)にGB血清(H205GB継代数 11と称する)を接種した。接種後(PI)の最初1週間の間、タマリンT−1053では肝酵素の上昇が認められた。このタマリンは、PI12日で安楽死させた。タマリンT−1048、T−1057、およびT−1061は、接種後2週間までの間に肝酵素の上昇を示した。この上昇はPI8〜9週まで続いた(図2〜4)後、接種前のレベルに戻った。PI14週において、これら3匹のタマリンに対し、タマリンT−1053(この個体は感染力を有することが判明している−実施例2)から得た希釈していない血清0.10mlでの再接種を行った。
この3匹のタマリン(タマリンT−1048、T−1057、およびT−1061)の回復期の血清がCKS−1.7(配 列番号610)組換えタンパク、CKS−1.4(配列番号 611)組換えタンパク、CKS−4.1(配列番号612)組換えタンパクに対する抗体を有するか否かについて、ELISA(図3、4、5)を個々に行って検討した。1.7(配列番号610)、1.4(配列番号611)、4.1(配列番号612)、あるいは1.5(配列番号614)の各組換えタンパクに対する特異抗体は、どの接種前の検体からも検出されなかった。
図26に示すように、T−1048の血清においては、1.7および1.4ELISAの試験によってPI56〜84日で特異抗体が検出されたが、PI97および137日では検出されなかった。また、T−1048の血清は、4.1ELISAの試験では特異抗体を検出しなかった。図27に示すよ うに1.7タンパク(配列番号610)に対する抗体がT− 1057の血清においてPI56および63日に検出されたが、PI63日よりも後は検出されなかった。4.1タンパク(配列番号612)に対する抗体がPI28〜63日に検出されたが、PI84〜97では検出されなかった。上記のように、各タマリンに対し、PI97日に接種剤H205で再接種を行った。4.1タンパク(配列番号612)に対する特異抗体が PI112日およびPI126日に検出されたが、これは接種に対する既往反応を示唆するものである。1.4組換えタンパク(配列番号611)については抗体反応は認められなかった。
組換え1.4タンパク(配列番号611)に対する特異抗体がタマリンT−1061の血清においてPI84日からPI 112日の間で検出されたが、PI126日以降では検出されなかった。図28に示すように、タマリンT−1061の血清は1.7タンパク(配列番号610)および4.1タンパク(配列番号612)に対する抗体に関してPI350日で陰性であった。
2.タマリンについてのPCRの結果(初期感染性の研究)
タマリンT−1048およびT−1057の血清を選択し、実施例7に記載のクローン4から得たプライマーと実施例7に記載のクローン16から得たプライマーを用い、RT−PCRによってHGBV−RNAを調べた。
タマリンT−1048およびT−1057の血清を選択し、実施例7に記載のクローン4から得たプライマーと実施例7に記載のクローン16から得たプライマーを用い、RT−PCRによってHGBV−RNAを調べた。
HGBV−RNAは、RT−PCRによっては接種前10日および接種前17日の血清中のプライマーのいずれにおいても(T−1048)(図26)、あるいは接種前17、37、59日の血清中のプライマーのいずれにおいても(T−1057)(図27)検出されなかった。T−1048については、HGBV−RNAが、PI7〜137日の間に得た17の異なった血清のうちの15に関してクローン4からのプライマーを用いて検出された。HGBV−RNAは、PI7〜97日の期間に得た10種血清のいずれかにあっても、クローン 16から得たプライマーを用いるRT−PCRによっては検出されなかった。T−1053の血漿を接種すると、接種後8日および40日の期間に採取した血清5検体のうち4検体がクローン16に対して陽性であった。T−1057については、RT−PCRの結果が陽性であったのは、図27に示すようにPI7日から28日に採取した4検体で、クローン4からのプライマーを用いたものであった。各検体に対して行ったRT−PCRの結果は、PI28日後においては弱いハイブリダイゼーションシグナルを示した287日目を除いては、クローン4に対して陰性であった。PI7日〜97日に得たT−1057の6つの検体のいずれもが、クローン16からのプライマーを用いたRT−PCRが陽性であった。しかしながら、T−1053の接種後8日〜85日の間の血清は、クローン16からのプライマーの使用で陽性であった。
3.タマリンについてのELISAの結果(力価検定/伝播性の研究)
実施例2に記載のように、タマリンT−1053の血清を4匹のタマリンに接種した。これら4匹のタマリンのうち3匹を疾病の急性期(PI12日から14日の間)に安楽死させた。これら3匹のタマリンについて得たRT−PCRの結果を以下に記す。生き残ったタマリン(T−1051)は最初PI14日までに肝酵素値が上昇し、この上昇値は少なくともPI8週まで持続した。タマリンT−1051の検体を1.7ELISAおよび1.4ELISAで試験した。結果を図29に示す。特異抗体は接種後の最初の41日間に採取された血清においても接種前の血清においても検出されなかった。しかしながら、1.4タンパク(配列番号611)および1.7タンパク(配列番号610)に対する抗体反応がPI49日とPI113日の間に、また4.1タンパク(配列番号612)に対する抗体反応がPI28日とPI105日の間に認められた。タマリンはPI113日目に安楽死させた。
実施例2に記載のように、タマリンT−1053の血清を4匹のタマリンに接種した。これら4匹のタマリンのうち3匹を疾病の急性期(PI12日から14日の間)に安楽死させた。これら3匹のタマリンについて得たRT−PCRの結果を以下に記す。生き残ったタマリン(T−1051)は最初PI14日までに肝酵素値が上昇し、この上昇値は少なくともPI8週まで持続した。タマリンT−1051の検体を1.7ELISAおよび1.4ELISAで試験した。結果を図29に示す。特異抗体は接種後の最初の41日間に採取された血清においても接種前の血清においても検出されなかった。しかしながら、1.4タンパク(配列番号611)および1.7タンパク(配列番号610)に対する抗体反応がPI49日とPI113日の間に、また4.1タンパク(配列番号612)に対する抗体反応がPI28日とPI105日の間に認められた。タマリンはPI113日目に安楽死させた。
タマリン(T−1034)は、実施例1および表4に記載のように最近罹患した肝炎感染から回復しつつある患者(最初のGB源)の潜在的感染性血清0.1mlで前に接種したものである。肝酵素値の上昇はT−1034において接種後の約10週間認められなかった。この理由からタマリンT−1034は付加的研究において使用できるであろうと決定した。タマリンT−1034はHGBV調製物で接種した。このHGBV調製物は、タマリンT−1053の血清を予め接種した3匹のタマリン(T−1055、T−1038、T−1049)の血清プールから実施例4??の記載に従って調製したものである。
これらの3匹のタマリン(T−1055、T−1038、T−1049)に、実施例2の記載に従ってタマリンT−1053から調製した血清を接種した。PI11日までには3匹のタマリン全てで肝酵素の上昇が認められた。PI12日目にタマリンT−1055を殺した。タマリンT−1038とタマリンT−1049はPI14日目に殺した。これらのタマリンの血清をプールして、清澄化し、濾過した。この濾過したものの10−6希釈物(健常タマリン血清中に調製)0.25mlをタマリンT−1034に接種した。
T−1034に接種後2週間で肝酵素ALT値の上昇が最初に認められ、続く7週間の間上昇値を保ち、最後に接種後10週までに正常化した。図30に示したように1.4(配列番号22)組換えタンパクに対する特異抗体応答がPI49日に最初に検出され、PI56日〜118日の間検出され続けた。4.1(配列番号52)組換えタンパクに対する抗体応答は PI49日に最初に検出され、PI56日〜77日の間検出され続けた。但しPI84日から118日の間は検出されなかった。1.7(配列番号610)組換えタンパクに対する抗体応答はPI56日に最初に検出され、PI63日〜118日の間検出され続けた。タマリンはPI118日目に殺した。
実施例2に記載のように、タマリンT−1044は、H205で接種した7日後にT−1057からの血清で接種した。この接種物はクローン4プライマーで検出された配列に対してのみ陽性であった。クローン16プライマーを用いたRT−PCRでPI14日から63日の間、切捨て値を越えるゆるやかなALT値の上昇が見られた。前述のように、1.7(配列番号610)組換えタンパクに対する特異抗体応答は、PI63日〜84日の間、検出された。4.1(配列番号612)組換えタンパクに対する抗体応答や、1.4(配列番号611)組換えタンパクに対する抗体応答は検出されなかった。タマリンは、PI161日目に殺した。
4.タマリンについてのPCRの結果(力価検定/伝播性の研究)
接種に先立つ8週目の週にT−1049とT−1055から採取した血清および接種の日にT−1038から得た血清は、クローン16(配列番号26)およびクローン4(配列番号 21)に対し、RT−PCR陰性であった。タマリンT−1049とタマリンT−1055はクローン4の配列(配列番号21)に対して接種の1週間後、RT−PCR陽性であった(クローン16のPCRは実施しなかった)。殺す日に先立つ、T−1049(PI14日)並びにT−1055(PI11日)は、クローン4(配列番号21)およびクローン16の配列 (配列番号26)のいずれに対しても、RT−PCR陽性であった。タマリンT−1038は、殺す日(PI14日)には、両プライマーのいずれでも陽性であった。
接種に先立つ8週目の週にT−1049とT−1055から採取した血清および接種の日にT−1038から得た血清は、クローン16(配列番号26)およびクローン4(配列番号 21)に対し、RT−PCR陰性であった。タマリンT−1049とタマリンT−1055はクローン4の配列(配列番号21)に対して接種の1週間後、RT−PCR陽性であった(クローン16のPCRは実施しなかった)。殺す日に先立つ、T−1049(PI14日)並びにT−1055(PI11日)は、クローン4(配列番号21)およびクローン16の配列 (配列番号26)のいずれに対しても、RT−PCR陽性であった。タマリンT−1038は、殺す日(PI14日)には、両プライマーのいずれでも陽性であった。
図30から判るように、T−1034は、接種後に採取した(PI7日)最初の血清サンプルについてクローン4プライマーで検出された配列に対しRT−PCR陽性で、PI70日まで陽性でありつづけた。PI112日に採取したサンプルは陰性であった。これらのサンプル全てはクローン16プライマーによるRT−PCRで陰性であった。接種前の70日目および101日目に採取したサンプルは両プライマーのいずれでも陰性であった。
タマリンT−1051に関する図29から判るように、HGBV−RNAは、接種の8週間前に採取した血清サンプルにおいては、両プライマー(上記のクローン4とクローン16からのもの)のいずれによっても検出されなかった。HGBV−RNAは、PI7日〜69日の間に採取した6種の血清において、クローン4からのプライマーを用いたRT−PCRで検出されたが、PI77、84、91、あるいは105日に採取した血清では検出されなかった。接種後に採取した9つのサンプルにおいて、クローン16からのプライマーを用いたRT−PCRでHGBV−RNAが検出された。
図7に示すように、T−1044は、接種後(PI7日)に採取した最初の血清サンプルにおいては、クローン4プライマーで検出された配列に対し、RT−PCR陽性であり、PI63日まで陽性でありつづけた。PI77日〜119日の間に採取したサンプルは陰性であった。これらのすべてのサンプルは、クローン16プライマーに対してRT−PCR陰性であった。接種の42日前に採取したサンプルは、両プライマーのいずれに対しても陰性であった。
タマリンT−1047とT−1056に対し、PI14日に採取したT−1044血清を接種した。これらの両動物からPI7日〜64日の間に採取した9種のサンプルは、クローン4プライマー(配列番号8と9)によるRT−PCRは陽性であったが、クローン16プライマーでは陰性であった。
タマリンT−1058に、T−1053血清による誘発の22日後に採取した無希釈のT−1057血清を接種した。この接種物は、クローン16プライマーで検出された配列に対して陽性であったが、クローン4プライマーでのものには陰性であった。この動物から採取した血清サンプルを、GBV−配列[クローン16、クローン2、クローン10、クローン18]およびGB−B配列[クローン4およびクローン50]に由来するプライマーを用いて試験した。接種の9日前に採取したサンプルは、全てのプライマーでいずれも陰性であった。PI14日に採取したサンプルはクローン10と18のプライマーのみで陽性であった。PI21日に採取したサンプルはクローン16、10、および18のプライマーのみで陽性であった。PI28日に採取したサンプルはクローン18のプライマーのみで陽性であった。PI35日に採取したサンプルはクローン2、16、および18のプライマーのみで陽性であった。PI41日に採取したサンプルはクローン16および18のプライマーのみで陽性であった。クローン4およびクローン50からのプライマーでは、試験した全サンプルが陰性であった。
5.タマリンによる血清学的研究のまとめ
5匹のタマリンに対し、HGBVの種々の調製物を接種すると、接種後2週間までに肝酵素が上昇を始めた。この上昇値は続く6〜8週間の間持続した。一以上のHGBV組換え抗原、1.7、1.4、および4.1に対する特異抗体の応答が5匹全部のタマリンで観察された。いずれの場合も抗体はまず、PI6から10週で検出され、さらに2週間から7週間の間持続した。一般に、抗体レベルはピークに達し、次いで引き続く数週間の内に急速に下降した。抗体は、肝酵素の値が正常値に戻った後しばらくして検出可能となることがわかる。このことは、抗体の産生がウイルス感染の消散において何らかの作用をしていることを示唆するものである。
5匹のタマリンに対し、HGBVの種々の調製物を接種すると、接種後2週間までに肝酵素が上昇を始めた。この上昇値は続く6〜8週間の間持続した。一以上のHGBV組換え抗原、1.7、1.4、および4.1に対する特異抗体の応答が5匹全部のタマリンで観察された。いずれの場合も抗体はまず、PI6から10週で検出され、さらに2週間から7週間の間持続した。一般に、抗体レベルはピークに達し、次いで引き続く数週間の内に急速に下降した。抗体は、肝酵素の値が正常値に戻った後しばらくして検出可能となることがわかる。このことは、抗体の産生がウイルス感染の消散において何らかの作用をしていることを示唆するものである。
6.タマリンでのPCR研究のまとめ
ゲノムウォーキング実験の結果は、クローン4(配列番号21)とクローン16(配列番号26)が別々のRNA分子上にあることを示唆している。我々は従前、ウイルスゲノムには異なる2種類が存在し、その一つは一部にクローン4(配列番号21)を含有しもう一つは一部にクローン16(配列番号26)を含有する、という仮説を提起した。ある動物がクローン4からのプライマーで陽性でクローン16からのプライマーでは陰性であるという事実が観察されたことは、ウイルスゲノムに異なる2種類が存在することを支持するものである。しかしながら、クローン16(配列番号26)の配列が感染タマリンの内のあるものでは検出できなかったことは、クローン16のプライマーセットとクローン4のプライマーセットとの感度の下限の違いを反映している、という議論もできるかもしれない。もしこの後者の可能性が正しいとするならば、両方のプライマーともに陽性のタマリンは、これら二つのプライマーセットに関し、感度の差を示すはずである。上記の結果は二つのウイルス種の存在によって説明されるのであって二つのプライマーセットの感度の差によるものではない、という立場を補強するため、タマリンT−1057とT−1053のcDNAの一連の希釈列を用いてPCRを実施した。T−1057血清はクローン4プライマーの血清当量5×10−3μlで陽性、5×10−4μlで陰性であった。T−1057血清を20μlという多量で用いてRT−PCRをクローン16プライマーで行ったが、結果は陰性であった。もしこの差が二つのプライマーセット(クローン4対クローン16)の相対感度に帰するのであれば、他の検体もPCR試験を行ったとき4000倍もの高い終点希釈を示すであろうと予測される。しかしながら、T−1053血清に由来するcDNAは、クローン4(配列番号21)およびクローン16(配列番号26)の両クローンの配列に対し、いずれも血清当量2.5×10−4μlで陽性で、2.5×10−5μlで陰性であることが判った。よってこの結果はプライマーセットの感度の差では説明できず、contigB−クローン4(配列番号21)およびcontigA−クローン16(配列番号26)配列が、T−1057においては少なくとも4000倍異なる力価であるがT−1053では略等しい力価であるような別個のウイルスゲノムの上に存在する、という仮定と整合する。すなわちこのデータは、異なる検体において異なる相対終点力価を有する別個の二つのウイルスの存在と整合するものである。
ゲノムウォーキング実験の結果は、クローン4(配列番号21)とクローン16(配列番号26)が別々のRNA分子上にあることを示唆している。我々は従前、ウイルスゲノムには異なる2種類が存在し、その一つは一部にクローン4(配列番号21)を含有しもう一つは一部にクローン16(配列番号26)を含有する、という仮説を提起した。ある動物がクローン4からのプライマーで陽性でクローン16からのプライマーでは陰性であるという事実が観察されたことは、ウイルスゲノムに異なる2種類が存在することを支持するものである。しかしながら、クローン16(配列番号26)の配列が感染タマリンの内のあるものでは検出できなかったことは、クローン16のプライマーセットとクローン4のプライマーセットとの感度の下限の違いを反映している、という議論もできるかもしれない。もしこの後者の可能性が正しいとするならば、両方のプライマーともに陽性のタマリンは、これら二つのプライマーセットに関し、感度の差を示すはずである。上記の結果は二つのウイルス種の存在によって説明されるのであって二つのプライマーセットの感度の差によるものではない、という立場を補強するため、タマリンT−1057とT−1053のcDNAの一連の希釈列を用いてPCRを実施した。T−1057血清はクローン4プライマーの血清当量5×10−3μlで陽性、5×10−4μlで陰性であった。T−1057血清を20μlという多量で用いてRT−PCRをクローン16プライマーで行ったが、結果は陰性であった。もしこの差が二つのプライマーセット(クローン4対クローン16)の相対感度に帰するのであれば、他の検体もPCR試験を行ったとき4000倍もの高い終点希釈を示すであろうと予測される。しかしながら、T−1053血清に由来するcDNAは、クローン4(配列番号21)およびクローン16(配列番号26)の両クローンの配列に対し、いずれも血清当量2.5×10−4μlで陽性で、2.5×10−5μlで陰性であることが判った。よってこの結果はプライマーセットの感度の差では説明できず、contigB−クローン4(配列番号21)およびcontigA−クローン16(配列番号26)配列が、T−1057においては少なくとも4000倍異なる力価であるがT−1053では略等しい力価であるような別個のウイルスゲノムの上に存在する、という仮定と整合する。すなわちこのデータは、異なる検体において異なる相対終点力価を有する別個の二つのウイルスの存在と整合するものである。
HGBV−Bによるウイルス血症のみで肝酵素レベルの上昇を引き起こすのに十分であって、GBV−Aのみのウイルス血症段階では肝酵素レベル上昇は認められない、という観察結果から、HGBV−Bが恐らくこれらのタマリンにおける肝炎の原因物質であったことがわかった。HGBV−B抗原に対する免疫応答は、精々接種後150日という短期間出現した。これに対する説明としては、これらのELISAアッセイにおいて選定されたエピトープが、免疫応答が生成する主エピトープからのものではなかったということが考えられる。他の説明として、タマリンにおいては肝炎誘発は長期間の応答を必要とする重症ではない、ということも考えられる。これは、急性期に殺した、あるいは急性期の少し後で自然死した動物において、肝臓の炎症が軽症ないし非重症に止まった(結果は示さない)という組織学的証拠と整合する。
この研究で記載した6匹の動物の内の5匹において、PI112日までにHGBV−Bによるウイルス血症が散退した。一方、タマリンT−1048は136日間ウイルス血症から回復せず、死亡時(PI137日)にウイルス血症が認められた。GBV−A配列に陽性であった4匹の動物中で、3匹は、GBV−A配列が最初に出現してから77日後までにウイルス血症の解消をみた。これに対し、タマリンT−1061は、犠牲に供されるまでの245日間、ウイルス血症が認められた。加えて、タマリンT−1051は、犠牲に供されるまで(PI113日)ウイルス血症であったが、この持続するウイルス血症が最初にT−1053血漿で接種したことによるものか、あるいはその69日後にT−1053血漿で追加接種した結果によるものかは不明である。
HGBV−AおよびHGBV−Bの両方に陽性であった6匹の動物のピークALTの平均値は、HGBV−Bのみに陽性の4匹の動物の平均値より高かった。加えてピーク値に達するのは、平均してGBV−Bのみに陽性の動物よりもGBV−AおよびGBV−Bの両方に陽性の動物における方が早かった。これらの結果は、肝炎の強さは両剤が相当量で存在すること に関係するかもしれないことを示唆している。タマリンT− 1047とT−1056へのGBV−Bの追加継代接種において肝酵素値の上昇がGBV−Bウイルス血症でほとんどなかったことが観察されたが、この結果は、タマリンにおいては両剤がALTレベルの主たる上昇をもたらすのに必須ではないかという仮説を支持する。HGBV−B単独を用いた継代接種に加え、T−1058に接種物HGBV−Aを接種した時の初期の結果は、クローン4とクローン50のプライマーで検出可能なGB−B配列の不在によって示されるように、HGBV−Aは検出可能なHGBV−Bがなくとも独立に伝染しうることを示唆している。
F.ヒト集団でのHGBVへの暴露を示すための実験手順
種々のヒト集団から検体を得て、HGBV−Bに由来する組換えタンパクを用いた三種の別個のELISAによってHGBVに対する抗体の有無を調べた。実施例15Bのようにして1.7ELISAにおいてはCKS−1.7組換えタンパク(配列番号610)で固相を被覆し、1.4ELISAにおいてはCKS−1.4組換えタンパク(配列番号611)で固相を被覆し、また4.1ELISAでは4.1組換えタンパク(配列番号612)で固相を被覆して用いた。実施例15Eでも記したように、HGBVで接種したタマリンでは、これらのタンパクに対して特異的ではあるが短い抗体応答が認められた。この検出可能な免疫応答の短期的な性質からすると、結果が陰性のヒト集団においてHGBVに対して従前暴露された可能性がないとはいえない。
種々のヒト集団から検体を得て、HGBV−Bに由来する組換えタンパクを用いた三種の別個のELISAによってHGBVに対する抗体の有無を調べた。実施例15Bのようにして1.7ELISAにおいてはCKS−1.7組換えタンパク(配列番号610)で固相を被覆し、1.4ELISAにおいてはCKS−1.4組換えタンパク(配列番号611)で固相を被覆し、また4.1ELISAでは4.1組換えタンパク(配列番号612)で固相を被覆して用いた。実施例15Eでも記したように、HGBVで接種したタマリンでは、これらのタンパクに対して特異的ではあるが短い抗体応答が認められた。この検出可能な免疫応答の短期的な性質からすると、結果が陰性のヒト集団においてHGBVに対して従前暴露された可能性がないとはいえない。
ヒトの検体で行った血清学的研究は二重の目的を有するものであった。第一の目的は、種々のヒト集団における本発明の HGBV組換え抗原に対する抗体の血清学的存在を決定することであった。これらの研究は次の(1)〜(3)のテストを含む:(1)HGBVへの暴露に関し「低リスク」と考えられる集団(例えば合衆国における健康なボランティアの血液ドナーの人々);(2)HGBVへの暴露に関し「リスクあり」と考えられる集団(例えば経静脈麻薬使用者や血友病患者の検体は、非経口的に伝播した肝炎ウイルス(HBVおよびHCV)に対して血清反応がしばしば陽性となる;発展途上国に居住する個体からの検体は、経腸的に伝播したウイルス(HAVおよびHEV)に対して血清反応がしばしば陽性となる;(3)既知の肝炎ウイルス(HAV、HBV、HCV、HDV、またはHEV)への暴露や、サイトメガロウイルス(CMV)やエプスタイン−バーウイルス(EBV)などの肝炎関連のその他のウイルスへの暴露には関連のない、「非A−E−肝炎」を有する個体から得た検体グループ。非A−E−肝炎という一般的呼称に属するグループのメンバーの一部はHEVに対する抗体に関してはテストされていない場合がある。よって、1.7、1.4、あるいは4.1ELISAで反応性を有した非A−E群のすべての検体について、HEV−ELISAアッセイ(本出願人から入手可能)を行った。抗HEVは、三つの地域(パキスタン、アメリカ合衆国、およびニュージーランド)からのサンプルで以下に記載するように陽性の結果となった。
HGBVに対する暴露に関し「低リスク」と考えられる集団よりも、HGBVに対する暴露の「リスクあり」の集団や非A−E肝炎を有する個体における方が、より高い血清学的存在率がみられることが予想される。
本血清学的研究の第二の目的は、一以上のHGBVエピトープに対する抗体について陽性であることが判った検体RT−PCRで調べ、ウイルスが血清中に存在するかどうかを決定することであった。HBVおよびHCVは数カ月あるいは数年も持続するウイルス血症の疾病状態を、また、一般にHAVやHEVは一般に数週間持続する短期間のウイルス血症をもたらすことはよく知られている。急性で消退しつつある肝炎である、HBVやHCV感染の場合には、ウイルス血症の段階は数週間という短期間の持続の場合もあろう。このようにRT−PCRは、感染個体にウイルスが存在するという証拠を得るために用いることができる。しかしながら、ウイルス血症の状態は短期間でありうるので、ある剤に感染した個体でもその剤に関してRT−PCR陰性となることもある。
G.切捨て値の決定
間接方式を利用するその他のELISAアッセイにおける従前の経験から、予備的な切捨て値を、研究対象のタンパクに対する抗体に関して恐らく陰性であると思われる集団から得た吸収に基づいて算出すればよいことが判明している。予備的切捨て値は、集団の平均吸収値と、集団の平均値からの10の標準偏差との合計として計算した。切捨て値は一検査群の検査が行われるたびに用いられるものなので、切捨てを表現するより簡便な方法は、各アッセイのたびに5個重複して走らせる陰性コントロール(正常ヒト血漿プール−NFP)から決める方法であった。1.7、1.4、および4.1ELISAについては、陰性コントロールは、典型的には0.030と0.060の間の吸収を有していた。以下に記載のように、切捨て値は吸収値で約0.300から0.600であると計算されたが、これは陰性コントロールの値の10倍の吸収シグナルに匹敵するものであった。よって、検体が反応性であると考えられるのは、陰性対照(N)吸収値に対するサンプル(S)の吸収値(=S/N比)が10.0以上であることを要する。
間接方式を利用するその他のELISAアッセイにおける従前の経験から、予備的な切捨て値を、研究対象のタンパクに対する抗体に関して恐らく陰性であると思われる集団から得た吸収に基づいて算出すればよいことが判明している。予備的切捨て値は、集団の平均吸収値と、集団の平均値からの10の標準偏差との合計として計算した。切捨て値は一検査群の検査が行われるたびに用いられるものなので、切捨てを表現するより簡便な方法は、各アッセイのたびに5個重複して走らせる陰性コントロール(正常ヒト血漿プール−NFP)から決める方法であった。1.7、1.4、および4.1ELISAについては、陰性コントロールは、典型的には0.030と0.060の間の吸収を有していた。以下に記載のように、切捨て値は吸収値で約0.300から0.600であると計算されたが、これは陰性コントロールの値の10倍の吸収シグナルに匹敵するものであった。よって、検体が反応性であると考えられるのは、陰性対照(N)吸収値に対するサンプル(S)の吸収値(=S/N比)が10.0以上であることを要する。
H.追補的試験
最初の試験で反応性であるとされた検体は、原則として2個重複で再試験した。もし再試験時の吸収の一あるいは二者も切捨て値より高ければ、その検体は繰り返し反応性であると考えられた。繰り返し反応性であると考えられた検体については、さらにELISAのデータを補充すべく追補的アッセイを行った。十分量がある場合、繰り返し反応性検体はウェスタンブロットによって、抗体応答がCKS1.7(配列番号610)CKS1.4(配列番号611)またはCKS4.1(配列番号612)抗原を指向するものであって、問題とする主タンパクとともに固相に共に被覆されていたかもしれない大腸菌タンパクを指向するものではないことを確認した。ウェスタンブロットが陽性と考えられるためには、可視的なバンドが1.7タンパク(配列番号610)に対して80kD、1.4タンパク(配列番号611)に対して60から70kD、または4.1タンパク(配列番号612)に対して42kDにおいて検出される必要がある。ウェスタンブロットはELISAの感度に匹敵ないしそれを凌駕するほど最適化されていないので、陰性の結果が出ても直ちにELISAのデータを捨てることはしなかった。しかしながら、陽性の結果が出れば、それはELISAによって検出された反応性を補強するものである。
最初の試験で反応性であるとされた検体は、原則として2個重複で再試験した。もし再試験時の吸収の一あるいは二者も切捨て値より高ければ、その検体は繰り返し反応性であると考えられた。繰り返し反応性であると考えられた検体については、さらにELISAのデータを補充すべく追補的アッセイを行った。十分量がある場合、繰り返し反応性検体はウェスタンブロットによって、抗体応答がCKS1.7(配列番号610)CKS1.4(配列番号611)またはCKS4.1(配列番号612)抗原を指向するものであって、問題とする主タンパクとともに固相に共に被覆されていたかもしれない大腸菌タンパクを指向するものではないことを確認した。ウェスタンブロットが陽性と考えられるためには、可視的なバンドが1.7タンパク(配列番号610)に対して80kD、1.4タンパク(配列番号611)に対して60から70kD、または4.1タンパク(配列番号612)に対して42kDにおいて検出される必要がある。ウェスタンブロットはELISAの感度に匹敵ないしそれを凌駕するほど最適化されていないので、陰性の結果が出ても直ちにELISAのデータを捨てることはしなかった。しかしながら、陽性の結果が出れば、それはELISAによって検出された反応性を補強するものである。
繰り返し反応性のある検体で十分量あるものの対しては、クローン4プライマーを使用するRT−PCR(実施例15Dに記載に従って実施)を実施し、血清中のHGBV特異性ヌクレオチド配列を同定してもよい。結果が陽性であれば、その検体はウイルス血症であることがわかり、究極的にはヒト肝炎におけるHGBVの役割を確立する一助になるであろう。しかしながら、結果が陰性であってもそれはELISAの結果が誤りだったと解釈してはならない。タマリンによる研究において実施例15Eに記載したように、RT−PCRの結果は感染後の最初の数週間は陽性で、その後本発明のELISAで抗体がまさに検出されはじめた頃には陰性になっていた。この後の段階における検体はRT−PCR反応陰性であっても一方または両方のELISAにおいて陽性であるかもしれない。
I.低リスク検体における血清反応データ
ウィスコンシン州南東部の健康なボランティア血液供与者から100血清と100血漿から成る集団を得て、上記のELISAによって1.7(配列番号610)、1.4(配列番号611)、4.1(配列番号612)の各組換えタンパクに 対する抗体を検査した。血清および血漿について、1.7、1.4、および4.1ELISAで得られた吸収の値を別々にプロットした(図9−14)。
ウィスコンシン州南東部の健康なボランティア血液供与者から100血清と100血漿から成る集団を得て、上記のELISAによって1.7(配列番号610)、1.4(配列番号611)、4.1(配列番号612)の各組換えタンパクに 対する抗体を検査した。血清および血漿について、1.7、1.4、および4.1ELISAで得られた吸収の値を別々にプロットした(図9−14)。
1.7ELISAでは、血清検体と血漿検体に対する平均吸収値はそれぞれ0.072[標準偏差(SD)は0.061]および0.083(SD=0.055)であった。よって1.7ELISAでは血清と血漿に対する仮の切捨て値はそれぞれ0.499および0.468であった。上述のように陰性コントロールの吸収値をもとにして切捨て値を表すこともできる。S/N比が10.0を越える検体を反応性ありとした。この切捨て値を用いると、1.7(配列番号610)に対する抗体に対して試験した200検体の内の0検体が反応性であった。
1.4ELISAでは幾つかの検体(血清集団から3種、血漿集団から6種)が0.300を越える吸収値を示した(S/N比が6から12、予想される切捨て値に近いかそれ以上)。再試験すると、これらの検体の9種全部が10.0より低い S/N比を示した。血清検体と血漿検体に対する平均吸収値はそれぞれ0.072(SD=0.052)および0.108(SD=0.062)であった。1.4ELISAにおける切捨て値は、上記の式によって計算した。即ち、血清と血漿それぞれの集団に対する切捨て値はそれぞれ0.436および0.542であった。血清集団の中の一検体は、最初の試験で反応性とされたが、二重にして再試験したときには陰性であった。血漿集団の中の2検体も最初は試験で反応性とされたが、再試験では陰性であった。血漿100検体と血清100検体からなる第二の正常検体200の集団を試験した。先に提案した切捨てに基づき試験すると、血漿2検体および血清2検体が繰り返し反応性であった。
4.1ELISAでは血清検体と血漿検体に対する平均吸収値はそれぞれ0.070[標準偏差(SD)は0.037]および0.063(SD=0.040)であった。よって4.1ELISAにおいて、血清および血漿に対する仮の切捨て値はそれぞれ0.329および0.511であった。上述のように陰性コントロールの吸収値に基づいて切捨て値を決めることもできる:S/N比が10.0を越える検体を反応性ありとした。この切捨て値を用いると、4.1(配列番号612)に対する抗体について試験した100血漿検体の内の0検体、および 100血清検体の内の0検体が最初の試験で反応性を有しているとされた。
インターステート血液銀行(オハイオ州)から追加の760の血漿供与者の提供を受け、1.7ELISAおよび1.4ELISAの試験を行った。合計9検体が繰り返し反応性であった。両方のELISAで同時に反応性を有する検体はなく、全9検体が1.4ELISAで繰り返し反応性であった。
アメリカ合衆国に居住している血漿或いは血液提供者からの合計960検体について、1.7タンパクおよび1.4タンパクに対する抗体の有無を調査した。合計13検体が1.4ELISAにおいて繰り返し反応性であった。1.7ELISAで繰り返し反応性を有する検体はなかった。
以上まとめると、これらのデータは次のことを示している。すなわち、HGBV−Bからの組換え抗原を用いる一種以上のELISAアッセイにおいて、これまでのELISA結果によると、アメリカ合衆国の血液提供者から採取された960検体のうち13検体が抗体反応性であった。この結果からHGBVはアメリカ合衆国に固有のいわゆる風土病である可能性があると思われる。
これらのデータを表16に示す。
J.肝炎に関し「リスクあり」と考えられる検体
この研究でのデータを表16に示す。
この研究でのデータを表16に示す。
(i)西アフリカからの検体
西アフリカから入手した1300検体の内、合計181検体が一以上のELISAで繰り返し反応性であった。3種のELISA全部で繰り返し反応性の検体が1検体あった。合計43検体が1.7ELISAで繰り返し反応性を有し、91検体が1.4ELISAで繰り返し反応性を有し、51検体が4.1ELISAで繰り返し反応性を有した。
西アフリカから入手した1300検体の内、合計181検体が一以上のELISAで繰り返し反応性であった。3種のELISA全部で繰り返し反応性の検体が1検体あった。合計43検体が1.7ELISAで繰り返し反応性を有し、91検体が1.4ELISAで繰り返し反応性を有し、51検体が4.1ELISAで繰り返し反応性を有した。
1.7ELISAで繰り返し反応性であった6検体のうちの一つは、1.7タンパク(配列番号610)についてのウェスタンブロットで反応性であった。1.4ELISAで繰り返し反応性であった9検体のうちの9検体(100%)は、1.4タンパク(配列番号611)に対する抗体についてのウェスタンブロットで陽性であった。一検体が両方のタンパクについてのウェスタンブロットで陽性であった。4.1ELISAで繰り返し反応性であった12検体のうちの12検体(100%)は、4.1タンパク(配列番号612)に関するウェスタンブロットで陽性であった。
繰り返し反応性であった3つの検体(1.4ELISAで陽性であった一検体と、両ELISAおよびウェスタンブロットで陽性であった一検体を含む)について、クローン4からのプライマーを用いるRT−PCRによって上記のようにしてHGBV−RNAの有無を調べた。
得られたデータはHGBVが西アフリカの風土病である可能性を示唆するものであった。
(ii)経静脈麻薬使用者(IVDU)からの検体
セット1:112検体のうち3検体が1.4ELISAで陽性であった。5検体が4.1ELISAで、3検体が1.7ELISAで反応性であった。2サンプルが、2以上のELISAで陽性であった。
セット1:112検体のうち3検体が1.4ELISAで陽性であった。5検体が4.1ELISAで、3検体が1.7ELISAで反応性であった。2サンプルが、2以上のELISAで陽性であった。
セット2:経静脈麻薬使用者の集団から計99検体を採取した。この試験はイリノイ州シカゴのハインズ退役軍人管理病院(Hines Veteran’s Administration Hospital )で実施されたものの一部である。検体のいずれも1.7あるいは4.1 ELISAで反応性を有しなかった。一検体は1.4ELISAで繰り返し反応性が認められた。この繰り返し反応性の検体について、クローン4からのプライマーを用いるRT−PCRによって上記のようにしてHGBV−RNAの有無を調べた。この検体はRT−PCR陰性であった。
K.非A−E肝炎の個体から採取した検体
得られたデータを表16にまとめて示す。
得られたデータを表16にまとめて示す。
非A−E肝炎と診断された個体群から種々の検体集団を得て、実施例15Cに従って1.7、1.4、および4.1ELISAを実施した。使用した検体は次のものを含む:日本のクリニックからの180検体;ニュージーランドのクリニックからの56検体;ギリシャのクリニックからの73検体;エジプトのクリニックからの132検体;アメリカ合衆国テキサス州のクリニックからの64検体(セットT);ミネソタの研究センターからの72検体(セットM);米国からの62検体(セット#1);パキスタンのクリニックからの82検体;イタリーのクリニックからの10検体(幾つかの検体は血清量の不足のため実行可能なELISAアッセイすべては実施しなかった)。
(i)日本の検体
これらの180検体は85人の異なる患者からのものであった。全180検体のうち二つが1.7ELISAで繰り返し反応性であった。この二つの反応性を示す検体は2人の異なる患者からのものであった。上記2検体について、クローン4からのプライマーを用いるRT−PCRによって前記のように検査した。陽性の検体はなかった。
これらの180検体は85人の異なる患者からのものであった。全180検体のうち二つが1.7ELISAで繰り返し反応性であった。この二つの反応性を示す検体は2人の異なる患者からのものであった。上記2検体について、クローン4からのプライマーを用いるRT−PCRによって前記のように検査した。陽性の検体はなかった。
1.4ELISAで陽性の検体はなかった。
4.1ELISAについては、89検体中7検体が4.1アッセイで繰り返し反応性を示した(注:これら89検体は29人の異なる患者から得たものであった)。反応性の検体のうち5検体は一人の患者からのものであった。残る2検体は別の一人の患者からのものであった。
(ii)ニュージーランドの検体
56検体のうち計4検体が1.7、1.4、および4.1ELISAの一以上において繰り返し反応性であった。これらの内、二以上のELISAで反応性を示した検体はなかった。1検体が1.7ELISAで繰り返し反応性であり、2検体が1.4ELISAで繰り返し反応性であった。1検体が4.1ELISAで繰り返し反応性であった。二つの繰り返し反応性の検体についてPCRを実施した。その結果、両検体とも陰性であった。1.4ELISAで繰り返し反応性を示した1検体はHEVに対する抗体とも反応性を有した。
56検体のうち計4検体が1.7、1.4、および4.1ELISAの一以上において繰り返し反応性であった。これらの内、二以上のELISAで反応性を示した検体はなかった。1検体が1.7ELISAで繰り返し反応性であり、2検体が1.4ELISAで繰り返し反応性であった。1検体が4.1ELISAで繰り返し反応性であった。二つの繰り返し反応性の検体についてPCRを実施した。その結果、両検体とも陰性であった。1.4ELISAで繰り返し反応性を示した1検体はHEVに対する抗体とも反応性を有した。
(iii)ギリシャの検体
73検体中の計5検体が1.7および/または1.4ELISAで抗体反応性であった。これら73検体は計11人の患者から採取したものであった。繰り返し反応性の5検体中2検体は、両ELISAで繰り返し反応性を示したが、これらは別々の日に同一の患者から採取したものであった。二つの繰り返し反応性を示した検体をRT−PCRで調べたところ陰性であった。これらの検体のいずれも4.1ELISAでは抗体反応性を有しなかった。
73検体中の計5検体が1.7および/または1.4ELISAで抗体反応性であった。これら73検体は計11人の患者から採取したものであった。繰り返し反応性の5検体中2検体は、両ELISAで繰り返し反応性を示したが、これらは別々の日に同一の患者から採取したものであった。二つの繰り返し反応性を示した検体をRT−PCRで調べたところ陰性であった。これらの検体のいずれも4.1ELISAでは抗体反応性を有しなかった。
(iv)エジプトの検体
132検体中の計11検体が1.7、1.4、あるいは4.1ELISAで反応性を示した。8検体が1.7および1.4ELISAの両方で陽性であった。9検体が1.7ELISAで抗体反応性であり、9検体が1.4ELISAで反応性であった。1検体は4.1ELISAでは繰り返し反応性を示したが、1.7および1.4ELISAでは陰性であった。1.7ELISAで繰り返し反応性を示した1検体は、ウェスタンブロットで調べると1.7組換えタンパク(配列番号610)に対する抗体に関し陰性であった。1.4ELISAで繰り返し反応性を示した9検体中6検体は、ウェスタンブロットでは1.4組換えタンパク(配列番号611)に対する抗体に関し陽性であった。繰り返し反応性であったもののうち7検体についてRT−PCRで試験した。どの検体も反応性を示さなかった。これら全132検体は25人の異なる個体から別々の日に採取したものであった。繰り返し反応性を示した11検体は、異なる5個体からのものであった。これらの個体中の一個体(患者#101)においては、免疫応答は明白にタマリン(図31)で観察されたそれと相似であった。図31においてALTレベルが医者が症状を認めた時には上昇していたことに注意されたい。その後の検体ではALTレベルは低下し、1.4および1.7ELISAによって抗体が検出された。抗体応答は、タマリンで観察された血清反応状況と同じく、その後数週間にわたって低下した。追加の3人の患者(257、260、および340)は患者#101と同様の血清パターンを示した(図32−34参照)。これらのデータはHGBVがこれらの肝炎の症例において原因剤であるかもしれないという仮説を支持するものである。
132検体中の計11検体が1.7、1.4、あるいは4.1ELISAで反応性を示した。8検体が1.7および1.4ELISAの両方で陽性であった。9検体が1.7ELISAで抗体反応性であり、9検体が1.4ELISAで反応性であった。1検体は4.1ELISAでは繰り返し反応性を示したが、1.7および1.4ELISAでは陰性であった。1.7ELISAで繰り返し反応性を示した1検体は、ウェスタンブロットで調べると1.7組換えタンパク(配列番号610)に対する抗体に関し陰性であった。1.4ELISAで繰り返し反応性を示した9検体中6検体は、ウェスタンブロットでは1.4組換えタンパク(配列番号611)に対する抗体に関し陽性であった。繰り返し反応性であったもののうち7検体についてRT−PCRで試験した。どの検体も反応性を示さなかった。これら全132検体は25人の異なる個体から別々の日に採取したものであった。繰り返し反応性を示した11検体は、異なる5個体からのものであった。これらの個体中の一個体(患者#101)においては、免疫応答は明白にタマリン(図31)で観察されたそれと相似であった。図31においてALTレベルが医者が症状を認めた時には上昇していたことに注意されたい。その後の検体ではALTレベルは低下し、1.4および1.7ELISAによって抗体が検出された。抗体応答は、タマリンで観察された血清反応状況と同じく、その後数週間にわたって低下した。追加の3人の患者(257、260、および340)は患者#101と同様の血清パターンを示した(図32−34参照)。これらのデータはHGBVがこれらの肝炎の症例において原因剤であるかもしれないという仮説を支持するものである。
上の4人の患者からの7検体中、いずれの検体もRT−PCRではHGBV−RNA陽性ではなかった。この結果には幾つかの理由が考えられる。第一にウイルス血症の段階は非常に短期間であるかもしれず、その場合ウイルスは最初の採血の日までに血清から除去されてしまっていた可能性がある。第二にこれらの検体が船でエジプトから輸送されてきたことから、恐らく保存・輸送の過程で凍結、解凍を経るかその他の変化を受けたことが考えられ、その結果HGBV−RNAの検出能力が低下した可能性がある。
(v)アメリカ合衆国の検体(セットT)
アメリカ合衆国の64検体(セットT)で、1.7、1.4、4.1ELISAにおいて繰り返し反応性を有するものはなかった。
アメリカ合衆国の64検体(セットT)で、1.7、1.4、4.1ELISAにおいて繰り返し反応性を有するものはなかった。
(vi)アメリカ合衆国の検体(セットM)
アメリカ合衆国の72検体(セットM)のうち、計4検体が一以上のELISAで繰り返し反応性を示した。2検体は1.7および4.1ELISAで反応性を示した。1.7ELISAのみで反応性のものが1検体、4.1ELISAのみで反応性のものが1検体存在した。
アメリカ合衆国の72検体(セットM)のうち、計4検体が一以上のELISAで繰り返し反応性を示した。2検体は1.7および4.1ELISAで反応性を示した。1.7ELISAのみで反応性のものが1検体、4.1ELISAのみで反応性のものが1検体存在した。
(vii) アメリカ合衆国の検体(セット1)
非A−E肝炎のアメリカ合衆国検体(セット1)51検体中、3検体が一つまたは両ELISAで繰り返し反応性を示した。1検体は両ELISAで繰り返し反応性を示した。1検体は1.7ELISAで反応性を示し、3検体は1.4ELISAで繰り返し反応性を示した。両ELISAで陽性の検体は、1.4組換えタンパク(配列番号22)に対するウェスタンブロットで陽性であったが、1.7組換えタンパク(配列番号23)では陰性であった。追加の1検体は1.4ELISAで陽性、1.4組換えタンパク(配列番号611)に対してウェスタンブロット陽性であった。1.4ELISAで繰り返し反応性を示した1検体はHEVに対する抗体に反応性であった。
非A−E肝炎のアメリカ合衆国検体(セット1)51検体中、3検体が一つまたは両ELISAで繰り返し反応性を示した。1検体は両ELISAで繰り返し反応性を示した。1検体は1.7ELISAで反応性を示し、3検体は1.4ELISAで繰り返し反応性を示した。両ELISAで陽性の検体は、1.4組換えタンパク(配列番号22)に対するウェスタンブロットで陽性であったが、1.7組換えタンパク(配列番号23)では陰性であった。追加の1検体は1.4ELISAで陽性、1.4組換えタンパク(配列番号611)に対してウェスタンブロット陽性であった。1.4ELISAで繰り返し反応性を示した1検体はHEVに対する抗体に反応性であった。
(viii)パキスタンの検体
82検体中の計4検体が、1.4および/または1.7ELISAで繰り返し抗体反応性であった。両ELISAで反応性を示した検体はなかった。2検体が1.7ELISAで繰り返し反応性を示し、2検体が1.4ELISAで繰り返し反応性を示した。1.4ELISAで繰り返し反応性の2検体は、HEVに対する抗体にも反応性であった。82検体いずれも4.1ELISAでは陽性でなかった。
82検体中の計4検体が、1.4および/または1.7ELISAで繰り返し抗体反応性であった。両ELISAで反応性を示した検体はなかった。2検体が1.7ELISAで繰り返し反応性を示し、2検体が1.4ELISAで繰り返し反応性を示した。1.4ELISAで繰り返し反応性の2検体は、HEVに対する抗体にも反応性であった。82検体いずれも4.1ELISAでは陽性でなかった。
(ix)イタリーの検体
10検体いずれも、1.7、1.4、および4.1ELISAで繰り返し反応性を示さなかった。
10検体いずれも、1.7、1.4、および4.1ELISAで繰り返し反応性を示さなかった。
L.血清反応の結果の統計的意義
これらのデータは、HGBVタンパクに対する特異抗体(即ち1.7、1.4、または4.1ELISAで抗体に繰り返し反応性を示す検体)を、調査した集団の三つのカテゴリー全部で検出できることを示している。種々の検体カテゴリー(「低リスク」、「リスクあり」、および非A−E肝炎患者)から得た血清学上の結果を合わせ、χ二乗検定で統計的意義について分析した。データは、HGBVへの暴露に関し「リスクあり」と考えられる個体、あるいは分類未知の肝炎と診断された個体を含むボランティア血液提供者において、抗−HGBVの血清学的有病率を比較すると有意の差異が存在することを示した。
これらのデータは、HGBVタンパクに対する特異抗体(即ち1.7、1.4、または4.1ELISAで抗体に繰り返し反応性を示す検体)を、調査した集団の三つのカテゴリー全部で検出できることを示している。種々の検体カテゴリー(「低リスク」、「リスクあり」、および非A−E肝炎患者)から得た血清学上の結果を合わせ、χ二乗検定で統計的意義について分析した。データは、HGBVへの暴露に関し「リスクあり」と考えられる個体、あるいは分類未知の肝炎と診断された個体を含むボランティア血液提供者において、抗−HGBVの血清学的有病率を比較すると有意の差異が存在することを示した。
西アフリカ検体においては血清学的有病率は13.9%であって、これはベースライン上のグループ(表17)と比べ、p値0.000の下で有意に高かった。同様にIVDUにおいても、結果をボランティア提供者のデータと比較した時、統計的に有意の差が存在した(p値=0.000)。日本、ニュージーランド、アメリカ合衆国、エジプト、およびパキスタンを含む国々については、アメリカ合衆国のボランティア血液提供者に比べ、非A−E肝炎患者において抗体の存在に有意の差が存在した。
H.まとめ
これらのデータは、本明細書に記載の種々のELISAが、日本、ニュージーランド、アメリカ合衆国、エジプト、およびパキスタンを含む様々な地域におけるヒトの肝炎診断に有用である可能性を示唆している。これらのデータは、試験した検体の全カテゴリーにおいてHGBVに対する抗体の血清学的存在を過小に評価しているように思われる。さらなるHGBVエピトープが発見され、評価されるにつれ、現在は診断を下すことができない患者において肝炎を診断するにあたり、HGBVゲノム(群)に基づく試験の有用性がより重要なものとなることが期待される。注:これらの最初の研究ではRT−PCRの結果は陰性であったが、その後のデータから血清反応陽性の個体の血清でフラヴィ様ウイルス配列が見いだされた。
これらのデータは、本明細書に記載の種々のELISAが、日本、ニュージーランド、アメリカ合衆国、エジプト、およびパキスタンを含む様々な地域におけるヒトの肝炎診断に有用である可能性を示唆している。これらのデータは、試験した検体の全カテゴリーにおいてHGBVに対する抗体の血清学的存在を過小に評価しているように思われる。さらなるHGBVエピトープが発見され、評価されるにつれ、現在は診断を下すことができない患者において肝炎を診断するにあたり、HGBVゲノム(群)に基づく試験の有用性がより重要なものとなることが期待される。注:これらの最初の研究ではRT−PCRの結果は陰性であったが、その後のデータから血清反応陽性の個体の血清でフラヴィ様ウイルス配列が見いだされた。
上に記載したように、2種以上のHGBV株が存在する。これらは本発明の範囲内のものと考えられ、これを「肝炎GBウイルス(HGBV)」と呼ぶ。
(実施例16)
HGBV−Aの血清学的研究
A.組換えタンパク精製手順
CKS融合タンパクを発現する細菌細胞を凍結し、−70℃で保存した。各GBV−A構築物からの細菌細胞を解凍し、実施例15でGBV−B構築物について記載したようにして粉砕処理した。さらに、組換えタンパクを、実施例15でGBV−B組換えタンパクについて記載したようにして精製した。
HGBV−Aの血清学的研究
A.組換えタンパク精製手順
CKS融合タンパクを発現する細菌細胞を凍結し、−70℃で保存した。各GBV−A構築物からの細菌細胞を解凍し、実施例15でGBV−B構築物について記載したようにして粉砕処理した。さらに、組換えタンパクを、実施例15でGBV−B組換えタンパクについて記載したようにして精製した。
この精製手順で収集した分画を電気泳動で分離し、クーマシーブリリアントブルーR250で染色して、分子量が約60kD(CKS−1.5/配列番号614)、65kD(CKS−2.17/配列番号613)、55kD(CKS−1.18/配列番号390)、および66kD(CKS−1.22/配列番号390)に相当するタンパクの有無を調べた。該当するタンパクを含有する分画をプールし、SDS−PAGEによって再度調査した。
精製抗原を含有するプール分画の免疫原性および構造の完全性を、実施例13に記載の方法に従ってニトロセルロースに対する電子移動およびイムノブロット法によって決定した。適当な陽性コントロールがなかったため、組換えタンパクは、各融合タンパクのCKS部分に対するモノクローナル抗体との反応性によって同定した。CKS−1.5タンパク(配列番号614)について、組換え抗原を検出すべく抗−CKSモノクローナル抗体を用いてウェスタンブロット法で調べると、約60kDのところにシングルバンドが認められた。この結果はCKS−1.5タンパク(配列番号614)の予想サイズに 対応している。同様にCKS−2.17タンパク(配列番号613)、CKS−1.18タンパク(配列番号390)およびCKS−1.22タンパク(配列番号390)も、イムノブロット法で調べた結果予想されたサイズを有していた。
B.ポリスチレンビーズの被覆手順
上記タンパクを透析し、実施例15の記載に従って、これらのタンパクのポリスチレンビーズ上での抗原性を評価した。
上記タンパクを透析し、実施例15の記載に従って、これらのタンパクのポリスチレンビーズ上での抗原性を評価した。
C.HGBVに対する抗体を検出するためのELISAの手順
各種ELISAは実施例15の記載に従って実施した。
各種ELISAは実施例15の記載に従って実施した。
D.感染個体の血清におけるHGBV−RNAの検出
各ELISAで繰り返し反応性を示した検体について、実施例15のセクションDの記載に従ってHGBV−RNAの有無を調べた。
各ELISAで繰り返し反応性を示した検体について、実施例15のセクションDの記載に従ってHGBV−RNAの有無を調べた。
E.タマリンの血清像
すべてHGBV−Aゲノム由来のCKS1.5タンパク、CKS2.17タンパク、CKS1.18タンパク、あるいはCKS1.22タンパクを用いたELISAアッセイにおいて、タマリンからの血清はいずれも特異免疫反応を示さなかった。しかしながら、先の実施例に記載のように、感染タマリンの内のあるものではHGBV−AのRNAが検出された(実施例15中タマリンの血清像についてのまとめ参照)。
すべてHGBV−Aゲノム由来のCKS1.5タンパク、CKS2.17タンパク、CKS1.18タンパク、あるいはCKS1.22タンパクを用いたELISAアッセイにおいて、タマリンからの血清はいずれも特異免疫反応を示さなかった。しかしながら、先の実施例に記載のように、感染タマリンの内のあるものではHGBV−AのRNAが検出された(実施例15中タマリンの血清像についてのまとめ参照)。
F.ヒト集団についての血清学的研究のための実験手順
実施例15では、HGBV−Bに由来する組換え抗原を用いたELISAによって種々のヒトの集団におけるHGBV−Bに対する抗体の存在を評価した。その後、同じ検体の多くについて、CKS1.5組換えタンパク(配列番号614)を用いた1.5ELISA、CKS2.17組換えタンパク(配列番号613)を用いた2.17ELISA、CKS1.18組換えタンパク(配列番号390)を用いた1.18ELISA、およびCKS1.22組換えタンパク(配列番号390)をそれぞれ用いたELISAによって、HGBV−Aに対する抗体の有無を調べた。各タンパクは実施例15のように固相に被覆して用いた。実施例15に記載のように、HGBVを接種された回復期のタマリン5匹全部が、HGBV−B組換えタンパクに対して特異的抗体反応を示したが、短期間しか持続しなかった(1.7、1.4、および4.1ELISAで検出)。1.5、2.17、1.18、あるいは1.22ELISAにおいては、どのタマリンも検出可能な抗体反応を示さなかったが、西アフリカからのヒト検体の幾つかは、ウェスタンブロットでこれらの組換えタンパクの一以上のものについて特異的抗体反応を示し、ある外科医(GB剤の供給者)の提供になる肝炎発症22日目に採取した検体の一つは、ウェスタンブロットで試験したとき2.17組換えタンパクに対する特異的抗体反応を示した(実施例3参照)。本実施例では、様々なヒトの集団において抗体を検出するに際しての、1.5、2.17、1.18、および1.22ELISAの有用性を評価した。
実施例15では、HGBV−Bに由来する組換え抗原を用いたELISAによって種々のヒトの集団におけるHGBV−Bに対する抗体の存在を評価した。その後、同じ検体の多くについて、CKS1.5組換えタンパク(配列番号614)を用いた1.5ELISA、CKS2.17組換えタンパク(配列番号613)を用いた2.17ELISA、CKS1.18組換えタンパク(配列番号390)を用いた1.18ELISA、およびCKS1.22組換えタンパク(配列番号390)をそれぞれ用いたELISAによって、HGBV−Aに対する抗体の有無を調べた。各タンパクは実施例15のように固相に被覆して用いた。実施例15に記載のように、HGBVを接種された回復期のタマリン5匹全部が、HGBV−B組換えタンパクに対して特異的抗体反応を示したが、短期間しか持続しなかった(1.7、1.4、および4.1ELISAで検出)。1.5、2.17、1.18、あるいは1.22ELISAにおいては、どのタマリンも検出可能な抗体反応を示さなかったが、西アフリカからのヒト検体の幾つかは、ウェスタンブロットでこれらの組換えタンパクの一以上のものについて特異的抗体反応を示し、ある外科医(GB剤の供給者)の提供になる肝炎発症22日目に採取した検体の一つは、ウェスタンブロットで試験したとき2.17組換えタンパクに対する特異的抗体反応を示した(実施例3参照)。本実施例では、様々なヒトの集団において抗体を検出するに際しての、1.5、2.17、1.18、および1.22ELISAの有用性を評価した。
G.切捨て値の決定
1.5、2.17、1.18、および1.22ELISAにおける切捨て値を、実施例15に記載のようにして決定した。
1.5、2.17、1.18、および1.22ELISAにおける切捨て値を、実施例15に記載のようにして決定した。
H.追補的試験
実施例15に記載したように、最初反応性を示した検体については、原則として再試験を行った。ある検体が2回目も反応性を示した場合、ELISAによるデータをさらに補充すべく追加の試験(例えばウェスタンブロット)を実施した。ウェスタンブロットの結果が陽性と考えられるためには、観察可能ななバンドが1.5タンパク(配列番号614)に対して60kD、2.17タンパク(配列番号613)に対して65kD、1.18タンパク(配列番号390)に対して55kD、および1.22タンパク(配列番号390)に対して66kDにおいて検出される必要がある。ウェスタンブロットはELISAの感度に匹敵ないしはそれを凌駕するほど最適化されていないので、陰性の結果が出ても直ちにELISAのデータを捨てることはしなかった。しかしながら、陽性の結果が出れば、それはELISAによって検出された反応性を補強するものである。
実施例15に記載したように、最初反応性を示した検体については、原則として再試験を行った。ある検体が2回目も反応性を示した場合、ELISAによるデータをさらに補充すべく追加の試験(例えばウェスタンブロット)を実施した。ウェスタンブロットの結果が陽性と考えられるためには、観察可能ななバンドが1.5タンパク(配列番号614)に対して60kD、2.17タンパク(配列番号613)に対して65kD、1.18タンパク(配列番号390)に対して55kD、および1.22タンパク(配列番号390)に対して66kDにおいて検出される必要がある。ウェスタンブロットはELISAの感度に匹敵ないしはそれを凌駕するほど最適化されていないので、陰性の結果が出ても直ちにELISAのデータを捨てることはしなかった。しかしながら、陽性の結果が出れば、それはELISAによって検出された反応性を補強するものである。
実施例15で記載したように、繰り返し反応性のある検体で十分量あるものの対しては、プライマーを使用するRT−PCR(実施例15の記載に従って実施)を実施し、血清中のHGBV特異性ヌクレオチド配列を同定してもよい。
I.低リスク検体における血清学的データ
計252本の血漿検体をオハイオ州のインターステート血液銀行から入手し、1.5組換えタンパク(配列番号614)を用いた1.5ELISAで抗体の有無を検査した。この集団での平均吸収値は0.036(SD=0.022)であった。切捨て値は0.168と計算された。これはS/N比=10.0に対応する。760本の血漿検体(切捨て値決定のために用いた252検体を含む)について、1.5ELISAで抗体の有無を検査した。繰り返し反応性を示す検体はなかった。加えて、100本の血漿検体をウィスコンシン州南東部から入手し、1.5ELISAで抗体の有無を検査した。繰り返し反応性を示す検体はなかった。
計252本の血漿検体をオハイオ州のインターステート血液銀行から入手し、1.5組換えタンパク(配列番号614)を用いた1.5ELISAで抗体の有無を検査した。この集団での平均吸収値は0.036(SD=0.022)であった。切捨て値は0.168と計算された。これはS/N比=10.0に対応する。760本の血漿検体(切捨て値決定のために用いた252検体を含む)について、1.5ELISAで抗体の有無を検査した。繰り返し反応性を示す検体はなかった。加えて、100本の血漿検体をウィスコンシン州南東部から入手し、1.5ELISAで抗体の有無を検査した。繰り返し反応性を示す検体はなかった。
このように、1.5タンパクに対する抗体の存在を示す証拠はアメリカ合衆国の血液提供者においては見いだされなかった。
200本の検体をウィスコンシンの血液提供者から入手し、2.17組換えタンパク(配列番号60)を用いる2.17ELISAによって抗体の有無を検査した。この集団での平均吸収値は0.058(SD=0.025)であった。切捨て値は0.208と計算された。これはS/N比=10.0にほぼ対応する。検体の一つは繰り返し反応性を示した。このように、アメリカ合衆国の血液提供者(N=200)における血清学的有病率(seroprevalence)は比較的低かった。
上のパラグラフに記載のものと同じ200検体について、1.18および1.22ELISAで抗体の有無を検査した。どの検体も繰り返し反応性を示さなかった。このように、ボランティアの血液提供者からの検体については、1.5、2.17、1.18、および1.22ELISAから判断するかぎり、HGBV−Aタンパクに関して抗体陽性であるという証拠はなかった。
J.肝炎に関し「リスクあり」と考えられる検体
この研究でのデータを表18にまとめて示す。
この研究でのデータを表18にまとめて示す。
(i)西アフリカからの検体
1300検体の内、58検体が1.5ELISAで反応性を示した。繰り返し反応性であった18検体のうちの12検体は、1.5タンパク(配列番号614)に対する抗体についてのウェスタンブロットで陽性であった。817検体中43検体が2.17ELISAで反応性であった。これらの繰り返し反応性の検体については、2.17タンパク(配列番号613)に対する抗体に関するウェスタンブロットは行わなかった。
1300検体の内、58検体が1.5ELISAで反応性を示した。繰り返し反応性であった18検体のうちの12検体は、1.5タンパク(配列番号614)に対する抗体についてのウェスタンブロットで陽性であった。817検体中43検体が2.17ELISAで反応性であった。これらの繰り返し反応性の検体については、2.17タンパク(配列番号613)に対する抗体に関するウェスタンブロットは行わなかった。
817検体中6検体が1.22ELISAで反応性であった。353検体中9検体が1.18ELISAで反応性であった。2.17ELISAで反応性の21検体をウェスタンブロットで試験したところ、13検体が反応性であった。1.18ELISAで繰り返し反応性の8検体がウェスタンブロットで陽性であった。
これらのデータはHGBVが西アフリカの風土病である可能性を示唆するものである。
(ii)経静脈麻薬使用者からの検体
経静脈麻薬使用者の集団から全112検体を採取した。この試験はイリノイ州シカゴのハインズ退役軍人管理病院で実施されたものの一部である。1検体は2.17ELISAで繰り返し反応性が認められた。追加の1検体は1.18ELISAで反応性を示した。これらの検体のいずれも1.5あるいは1.22ELISAでは陽性ではなかった。
経静脈麻薬使用者の集団から全112検体を採取した。この試験はイリノイ州シカゴのハインズ退役軍人管理病院で実施されたものの一部である。1検体は2.17ELISAで繰り返し反応性が認められた。追加の1検体は1.18ELISAで反応性を示した。これらの検体のいずれも1.5あるいは1.22ELISAでは陽性ではなかった。
K.非A−E肝炎の個体から採取した検体
得られたデータを表18にまとめて示す。
得られたデータを表18にまとめて示す。
非A−E肝炎の個体群から種々の検体集団(実施例15Kに記載)を得て、1.5、2.17、1.18、および1.22ELISA(実施例15Cに記載)を実施した。幾つかの検体は血清量の不足のため全部のELISAアッセイは実施しなかった。
(i)日本の検体
89検体中の計4検体は、1.5ELISAで繰り返し反応性を示した。そのうちの3検体は1個人から、1検体は別の個人からのものであった。1.5ELISA、1.18ELISA、および1.22ELISAで陰性であった1検体は、2.17ELISAでは反応性を示した。1.18ELISAで反応性を示した検体はなかった。これらの検体は、1.22ELISAでは検査しなかった。
89検体中の計4検体は、1.5ELISAで繰り返し反応性を示した。そのうちの3検体は1個人から、1検体は別の個人からのものであった。1.5ELISA、1.18ELISA、および1.22ELISAで陰性であった1検体は、2.17ELISAでは反応性を示した。1.18ELISAで反応性を示した検体はなかった。これらの検体は、1.22ELISAでは検査しなかった。
(ii)ニュージーランドの検体
56検体のうち1.5ELISAで反応性を示したものはなかった。これらの検体は2.17ELISA、1.18ELISA、あるいは1.22ELISAでは検査しなかった。
56検体のうち1.5ELISAで反応性を示したものはなかった。これらの検体は2.17ELISA、1.18ELISA、あるいは1.22ELISAでは検査しなかった。
(iii)ギリシャの検体
67検体(10人の患者からのもの)のいずれもが1.5、2.17、あるいは1.22ELISAでは反応性を示さなかった。
67検体(10人の患者からのもの)のいずれもが1.5、2.17、あるいは1.22ELISAでは反応性を示さなかった。
(iv)エジプトの検体
132検体中に1.5ELISAに反応性の検体はなかった。検査可能であった132検体中、計7検体が2.17ELISAで反応性を有した。これらの検体は急性の非A−E肝炎患者25名からのものであった。25名の患者のうち3名が2.17ELISAで肝炎の発症のあと1日以上たったとき血清反応陽性であった。1.18、あるいは1.22ELISAで反応性の検体は存在しなかった。
132検体中に1.5ELISAに反応性の検体はなかった。検査可能であった132検体中、計7検体が2.17ELISAで反応性を有した。これらの検体は急性の非A−E肝炎患者25名からのものであった。25名の患者のうち3名が2.17ELISAで肝炎の発症のあと1日以上たったとき血清反応陽性であった。1.18、あるいは1.22ELISAで反応性の検体は存在しなかった。
(v)アメリカ合衆国の検体(セットM)
72検体のいずれも1.5ELISAで反応性を示さなかった。72検体中3検体は1.18ELISAで反応性を示した。2検体が2.17ELISAで、また4検体が1.22ELISAで反応性を示した。2サンプルは一以上のELISAで反応性を示した。
72検体のいずれも1.5ELISAで反応性を示さなかった。72検体中3検体は1.18ELISAで反応性を示した。2検体が2.17ELISAで、また4検体が1.22ELISAで反応性を示した。2サンプルは一以上のELISAで反応性を示した。
(vi)アメリカ合衆国の検体(セットT)
64検体のいずれも1.5、1.22、あるいは2.17ELISAで反応性を示さなかった。1検体は1.18で反応性を有した。
64検体のいずれも1.5、1.22、あるいは2.17ELISAで反応性を示さなかった。1検体は1.18で反応性を有した。
(vii)アメリカ合衆国の検体(セット1)
62検体中の計3検体が一以上のGBV−AのELISAで反応性を示した。2.17および1.22ELISAの両者で繰り返し反応性を示したものが1検体存在した。2.17ELISAのみに反応性の検体が1検体あり、別の1検体は1.22ELISAのみに反応性を示した。1.5あるいは1.18ELISAに反応性の検体は存在しなかった。
62検体中の計3検体が一以上のGBV−AのELISAで反応性を示した。2.17および1.22ELISAの両者で繰り返し反応性を示したものが1検体存在した。2.17ELISAのみに反応性の検体が1検体あり、別の1検体は1.22ELISAのみに反応性を示した。1.5あるいは1.18ELISAに反応性の検体は存在しなかった。
上に述べたように、2以上のHGBV株があり、あるいは、2以上の別種のウイルスを本明細書に記載の配列によって表すことができる。それらは本発明の範囲内のものと考えられ、これを「肝炎GBウイルス(HGBV)」と呼ぶ。
L.血清反応の結果の統計的意義
これらのデータによって、HGBV−Aタンパクに対する特異抗体(即ち1.5、2.17、1.18および1.22ELISAで抗体に繰り返し反応性を示す検体)は、HGBVへの暴露に関し「リスクあり」と考えられる個体および非A−E肝炎と診断された個体において検出されたが、アメリカ合衆国のボランティアからも売血者からもそれほど高頻度には検出されることがなかったことが示された。表19において、種々のカテゴリーの検体(表18の「低リスク」、「リスクあり」、および非A−E肝炎患者)からの血清反応の結果を合わせ、χ二乗検定で統計的意義について分析した。表18は検査した検体の総数におけるHGBVタンパクに対する抗体の血清学的存在をまとめたものであるが、これと異なり、表19のデータは、異なる個体(人)からの結果を示している。GBV−AのELISAについては、抗−HGBVの血清学的存在をボランティア血液提供者とHGBVへの暴露に関し「リスクあり」と考えられる個体(西アフリカ、但しIVDUではないもの)との間で比較すると、有意の差(p値=0.000)が存在することを示している。加えて、ボランティアの血液提供者と比較した場合、エジプトとアメリカ合衆国における非A−E肝炎患者におけるHGBV−Aに対する抗体の血清学的存在には、統計的に有意の差があった。このデータは、HGBV−Aに対する暴露と非A−E肝炎とに関連があることを示唆している。注:これらの最初の研究ではRT−PCRの結果は陰性であったが、その後のデータによって血清反応陽性の個体の血清においてフラヴィ様ウイルス配列が見いだされた(実施例19参照)。
これらのデータによって、HGBV−Aタンパクに対する特異抗体(即ち1.5、2.17、1.18および1.22ELISAで抗体に繰り返し反応性を示す検体)は、HGBVへの暴露に関し「リスクあり」と考えられる個体および非A−E肝炎と診断された個体において検出されたが、アメリカ合衆国のボランティアからも売血者からもそれほど高頻度には検出されることがなかったことが示された。表19において、種々のカテゴリーの検体(表18の「低リスク」、「リスクあり」、および非A−E肝炎患者)からの血清反応の結果を合わせ、χ二乗検定で統計的意義について分析した。表18は検査した検体の総数におけるHGBVタンパクに対する抗体の血清学的存在をまとめたものであるが、これと異なり、表19のデータは、異なる個体(人)からの結果を示している。GBV−AのELISAについては、抗−HGBVの血清学的存在をボランティア血液提供者とHGBVへの暴露に関し「リスクあり」と考えられる個体(西アフリカ、但しIVDUではないもの)との間で比較すると、有意の差(p値=0.000)が存在することを示している。加えて、ボランティアの血液提供者と比較した場合、エジプトとアメリカ合衆国における非A−E肝炎患者におけるHGBV−Aに対する抗体の血清学的存在には、統計的に有意の差があった。このデータは、HGBV−Aに対する暴露と非A−E肝炎とに関連があることを示唆している。注:これらの最初の研究ではRT−PCRの結果は陰性であったが、その後のデータによって血清反応陽性の個体の血清においてフラヴィ様ウイルス配列が見いだされた(実施例19参照)。
M.まとめ
これらのデータは、本発明に記載のELISAが西アフリカに居住している個体および非A−E肝炎の個体において抗体を検出するのに有用であることを示唆するものである。西アフリカの個体群においては肝炎に罹患するリスクは比較的高い。人々のほぼ85%近くが肝炎Bウイルスに対する抗体に血清反応陽性であり、約5%が肝炎Cウイルスに対する抗体に陽性である。これらのデータは、試験した検体の全カテゴリーにおいてHGBVに対する抗体の血清学的存在を過小に評価しているように思われる。さらなるHGBVエピトープが発見され、評価されるにつれ、現在は診断を下すことができない患者において肝炎を診断するにあたり、HGBVゲノム(群)に基づく試験の有用性がより重要なものとなることが期待される。
これらのデータは、本発明に記載のELISAが西アフリカに居住している個体および非A−E肝炎の個体において抗体を検出するのに有用であることを示唆するものである。西アフリカの個体群においては肝炎に罹患するリスクは比較的高い。人々のほぼ85%近くが肝炎Bウイルスに対する抗体に血清反応陽性であり、約5%が肝炎Cウイルスに対する抗体に陽性である。これらのデータは、試験した検体の全カテゴリーにおいてHGBVに対する抗体の血清学的存在を過小に評価しているように思われる。さらなるHGBVエピトープが発見され、評価されるにつれ、現在は診断を下すことができない患者において肝炎を診断するにあたり、HGBVゲノム(群)に基づく試験の有用性がより重要なものとなることが期待される。
(実施例18)
ヒトにおけるGB関連ウイルスの同定
A.理論
HGBV−AおよびHGBV−Bの両者のエピトープが同定された(実施例3)。これらを血清学的マーカーとして用いてヒトの血清および血漿サンプルをスクリーニングした(実施例5および6)。これらのマーカーのうちの幾つかについての血清学的活性と非A−E肝炎の発症頻度との間に顕著な相関があったが、これはHGBV−Bがヒトにおける非A−E肝炎の病因物質であることを示唆している(実施例5G)。しかしながら、GBヒト血清をウェスタンブロットで分析してみるとHGBV−Bエピトープに対する反応性は示されなかった(実施例3)。そのかわり、GBヒト血清に少なくとも一つのHGBV−Aエピトープが同定された。これはHGBV−AがGBにおける肝炎の病因物質であったことを示唆するものである。HGBV−A配列もHGBV−B配列も、RT−PCRでは非A−E肝炎の患者では同定されなかった(実施例5E)。そこで非A−E肝炎に罹患しているヒトにおけるHGBV−Aおよび/またはHGBV−B感染の証拠を定める必要がいまだ残っている。
ヒトにおけるGB関連ウイルスの同定
A.理論
HGBV−AおよびHGBV−Bの両者のエピトープが同定された(実施例3)。これらを血清学的マーカーとして用いてヒトの血清および血漿サンプルをスクリーニングした(実施例5および6)。これらのマーカーのうちの幾つかについての血清学的活性と非A−E肝炎の発症頻度との間に顕著な相関があったが、これはHGBV−Bがヒトにおける非A−E肝炎の病因物質であることを示唆している(実施例5G)。しかしながら、GBヒト血清をウェスタンブロットで分析してみるとHGBV−Bエピトープに対する反応性は示されなかった(実施例3)。そのかわり、GBヒト血清に少なくとも一つのHGBV−Aエピトープが同定された。これはHGBV−AがGBにおける肝炎の病因物質であったことを示唆するものである。HGBV−A配列もHGBV−B配列も、RT−PCRでは非A−E肝炎の患者では同定されなかった(実施例5E)。そこで非A−E肝炎に罹患しているヒトにおけるHGBV−Aおよび/またはHGBV−B感染の証拠を定める必要がいまだ残っている。
ヒト血清あるいは血漿源におけるHGBV−Aおよび/またはHGBV−B配列の同定が失敗に終わったことには幾つかの要因が考えられる。第一に、我々がRT−PCRによって検査したのは限られた数のHGBV−A血清陽性および/またはHGBV−B血清陽性サンプルであって、これらのサンプルの多くは保存歴が不明である。よってこれらのサンプル中に存在したウイルス性のRNAが不適当な保存の影響を受けた可能性がある。第二に、GB感染は自然に快癒するように見受けられる。そのため、GB配列が感染した個体の血清中に存在する期間幅は非常に狭い可能性がある。即ちGB配列を含有する血清サンプルを得る機会は極端に低いものとなろう。最後に、HGBV−Aおよび/またはHGBV−B配列を探すにあたって、限られた数のPCRプライマーセットを用いたことが挙げられる。HGBV−Aおよび/またはHGBV−BはRNAウイルスであるため、突然変異の率が高い傾向がある(ホランドら(1982年)サイエンス215:1577−1585)。即ち、検査したヒト血清中のHGBV−Aおよび/またはHGBV−B配列が我々のPCRプライマーの配列とは異なっていて、そのためHGBV−Aおよび/またはHGBV−Bが検出されない可能性がある。
HGBV−Aおよび/またはHGBV−Bのゲノムの変異性のために我々のPCR研究においてこれらのウイルスが妨害された可能性を示すため、HGBV−Aおよび/またはHGBV−Bの高度に保存されたNS3様の領域(図17参照)に合わせ、変性PCRプライマーを設計配置した。これらの高度に保存された領域がウイルスの複製サイクルにおいて必要な機能を果たしているからである。よってこれらの配列はHGBV−Aおよび/またはHGBV−B変異体においても保存されているはずである。この領域内に設計配置されたPCRプライマーは、RT−PCRによってHGBV−Aおよび/またはHGBV−BのゲノムRNAを検出できるはずである。加えて、HGBV−A、HGBV−BおよびHCV配列を特異的に増幅することのできる変性PCRプライマーを設計すれば、HGBV−A、HGBV−BおよびHCVに関連するウイルスから得た配列を増幅することができるかもしれないと我々は判断した。よって、もし別々のHGBV−A関連ウイルスまたはHGBV−B関連ウイルスの抗原と交差反応する抗体が存在するために、ヒトの血清および血漿サンプル(実施例5および6)で血清反応があまり検出されなかったのであるならば、我々はこれらのGB関連ウイルスから配列を得ることができるであろう。[これはニコルと彼の同僚が最近米国南西部における急性呼吸器疾患の大流行に関連する特殊なハンタウイルスを同定するために採用した実験手法に類似のものである。ニコルら、サイエンス262:914−917(1993)]
B.肝炎GBウイルスC(HGBV−C)のNS3様領域のクローニング
ウイルス感染のモデルでは、感染の初期段階でウイルス血症が起こり、これがウイルスタンパクに対するIgM抗体の検出と相関していることが多い。実施例5、6で記載したように、HGBV−A及びHGBV−Bに由来する組換えタンパクに対するIgG抗体が検出された検体は、ELISAにおいて免疫反応性を有していた。更に追加のELISAを実施し、IgM抗体がこれらのタンパクについて検出されるかどうかを調べた。西アフリカの個体(実施例5E−i)の血清反応陽性の検体の内のあるものは、組換えタンパクに対するIgM抗体に関して反応性であった(データ省略)。これらの検体はウイルスを含有する可能性が高いと考えられた。加えて、急性肝炎に罹患しているHGBV−A陽性およびHGBV−B陽性のエジプトの個体(実施例5F−vii )で、HGBV−AまたはHGBV−B組換えタンパクに対するIgM抗体が検出されない検体についても、急性肝臓疾患はウイルスの存在と関連している可能性があることから検査を実施した。これらのサンプルの核酸について、HGBV−A、HGBV−B、及びHCV−1の各配列を増幅する変性オリゴヌクレオチドプライマーを用いて“半入れ子”(hemi-nested) RT−PCRを実施した。使用機器は GeneAmp(登録商標)RNAPCRキット(パーキンエルマー製)を用い、製造業者の使用説明書に従って実施した。簡単に言えば、増幅の第一セットは、2mMのMgCl2および1μMのプライマーns3.1−sとns3.1−a(配列番号はそれぞれ671および672)で抽出した核酸のランダム−プライムド逆転写反応によるcDNA産物について行った。反応物に対し、変性−アニーリング−伸展[(94℃、30秒;37℃、30秒;2分かけて72℃;72℃、30秒)を3サイクル行い、次いで(94℃、30秒;55℃、30秒;72℃、30秒)を37サイクル行う]を40回行い、最後に72℃で10分間伸展させた。反応が終了したものは、4℃で保存した。増幅の第二セットは、最初のPCR産物の4%を鋳型として使用し、ns3.1−sおよびns3−a(配列番号はそれぞれ671および673)を“ヘミネスティッド”プライマーセットとして用いた他は上記と同様であった。PCRの第一及び第二のセットの産物をゲル濾過で分析した。
ウイルス感染のモデルでは、感染の初期段階でウイルス血症が起こり、これがウイルスタンパクに対するIgM抗体の検出と相関していることが多い。実施例5、6で記載したように、HGBV−A及びHGBV−Bに由来する組換えタンパクに対するIgG抗体が検出された検体は、ELISAにおいて免疫反応性を有していた。更に追加のELISAを実施し、IgM抗体がこれらのタンパクについて検出されるかどうかを調べた。西アフリカの個体(実施例5E−i)の血清反応陽性の検体の内のあるものは、組換えタンパクに対するIgM抗体に関して反応性であった(データ省略)。これらの検体はウイルスを含有する可能性が高いと考えられた。加えて、急性肝炎に罹患しているHGBV−A陽性およびHGBV−B陽性のエジプトの個体(実施例5F−vii )で、HGBV−AまたはHGBV−B組換えタンパクに対するIgM抗体が検出されない検体についても、急性肝臓疾患はウイルスの存在と関連している可能性があることから検査を実施した。これらのサンプルの核酸について、HGBV−A、HGBV−B、及びHCV−1の各配列を増幅する変性オリゴヌクレオチドプライマーを用いて“半入れ子”(hemi-nested) RT−PCRを実施した。使用機器は GeneAmp(登録商標)RNAPCRキット(パーキンエルマー製)を用い、製造業者の使用説明書に従って実施した。簡単に言えば、増幅の第一セットは、2mMのMgCl2および1μMのプライマーns3.1−sとns3.1−a(配列番号はそれぞれ671および672)で抽出した核酸のランダム−プライムド逆転写反応によるcDNA産物について行った。反応物に対し、変性−アニーリング−伸展[(94℃、30秒;37℃、30秒;2分かけて72℃;72℃、30秒)を3サイクル行い、次いで(94℃、30秒;55℃、30秒;72℃、30秒)を37サイクル行う]を40回行い、最後に72℃で10分間伸展させた。反応が終了したものは、4℃で保存した。増幅の第二セットは、最初のPCR産物の4%を鋳型として使用し、ns3.1−sおよびns3−a(配列番号はそれぞれ671および673)を“ヘミネスティッド”プライマーセットとして用いた他は上記と同様であった。PCRの第一及び第二のセットの産物をゲル濾過で分析した。
西アフリカの1サンプルは、約386bp(HGBV−A、HGBV−B、またはHCV産物の予想されるサイズ)にぼやけた半入れ子反応に由来するPCR産物を有していた。この産物を、本技術分野の書物に記載されているpT7ブルーT−ベクタープラスミド(ノヴァゲン製)にクローニングした。このクローンから得た配列(GBcontigC[GB−C]、配列番号673、残基2274−2640)をGBcontigA(GB−A、配列番号163、残基4438−4804)、GBcontigB(GB−B、配列番号393、残基4218−4587)、およびHCV−1(配列番号398)と比較した。図36は、これらの配列のヌクレオチド配列を示し、一方、表20はこれらの配列間の%相同性を示す。
図36と表20に示すように、GB−A、GB−B、およびHCV−1のヌクレオチドの比較の結果、これらの配列は 47.99〜61.66%互いに類似している。このことは、これらのウイルスのNTP結合性ヘリカーゼに存在する保存されたアミノ酸残基を考えると特に驚くべきことではない(実施例2B3、図17A)。西アフリカのサンプル(GB−C、配列番号673、残基2274−2640)から得たNS3PCR産物のヌクレオチドと他のウイルスのヌクレオチドを比較すると、GB−A(配列番号163、残基4438−4804)、GB−B(配列番号393、残基4218−4587)、およびHCV−1(配列番号398)からのNS3配列に関連しているが、明確に異なっていることが示唆されている。ウィスコンシン配列分析パッケージ(バージョン8)中の核酸および タンパクデータベースとGB−C(配列番号673、残基2274−2640)とのBLASTINおよびBLASTX解析の結果、HCVの株の幾つかは配列の同一性に限界があることが判明した。ヌクレオチドおよびアミノ酸に関するもっとも高いP値(すなわち偶然に配列が整合した確率)はそれぞれ1.9×10−20と5.3×10−31であった(データ省略)。以上を合わせると、これらのデータは、GB−C(配列番号673、残基2274−2640)がHGBV−A、HGBV−Bおよび我々がここでHGBV−Cと標記するHCVに関連するユニークなGB様のウイルスに由来するものであるかもしれないことを示唆している。
C.GB−Cは外因性である
GB−C配列に対するPCRプライマーを用い、上記のように、即ち例えば実施例6Bに記載の手順によって、この配列 がヒト、アカゲザル、S.cerevisiaeおよびE.coliのゲノム中に検出されるかどうかを決定した。PCRはパーキン−エルマー−シータスのGeneAmp(登録商標)を使用し、基本的に製造者の指示書に従って実施した。即ち、300ngのゲノムDNAを各100μlの反応物に対して使用した。PCRプライマー(配列番号675および676)を最終濃度1.0μMで使用した。PCRは40サイクル行い(94℃、30秒;55℃、30秒;72℃、30秒)、次いで72℃で10分間伸展させた。PCR産物をアガロースゲル電気泳動で分離し、臭化エチジウムで核酸を直接染色したのちUV照射で可視化し、さらにサザントランスファーでHybond−N+ナイロンフィルターに移してから放射標識プローブとハイブリダイズした。図37は、GB−C(配列番号673、残基2274−2640)から調製した放射線標識プローブとハイブリダイズした後、PCR産物をサザンブロットして得たPhosphoImage(モレキュラー・ダイナミクス社、カリフォルニア州サニーヴェイル)である。GB−C(配列番号673)は、300ngのヒトゲノムDNA中にGB−Cプラスミド鋳型の〜350fg(1ゲノムコピー当量、レーン5)および〜35fg(0.1ゲノムコピー当量、レーン6)が検出された以外、ヒト(図19、レーン1)、アカゲザル(レーン2)、S.cerevisiae(レーン3)、E.coli(レーン4)の各ゲノムDNAには検出されなかった。(レーン7は〜3.5fg[0.01ゲノムコピー当量]のGB−Cプラスミド鋳型からのPCR産物を300ngのヒトゲノムDNA中に含有する。)このように、0.1ゲノムコピー当量を検出できるゲノムPCRを使用してもGB−C(配列番号673)はヒト、アカゲザル、S.cerevisiaeおよびE.coliのゲノム中に検出できない。この結果は、GB−C(配列番号673)が外因性(即ちウイルス性)起源であるという予想と一致している。
GB−C配列に対するPCRプライマーを用い、上記のように、即ち例えば実施例6Bに記載の手順によって、この配列 がヒト、アカゲザル、S.cerevisiaeおよびE.coliのゲノム中に検出されるかどうかを決定した。PCRはパーキン−エルマー−シータスのGeneAmp(登録商標)を使用し、基本的に製造者の指示書に従って実施した。即ち、300ngのゲノムDNAを各100μlの反応物に対して使用した。PCRプライマー(配列番号675および676)を最終濃度1.0μMで使用した。PCRは40サイクル行い(94℃、30秒;55℃、30秒;72℃、30秒)、次いで72℃で10分間伸展させた。PCR産物をアガロースゲル電気泳動で分離し、臭化エチジウムで核酸を直接染色したのちUV照射で可視化し、さらにサザントランスファーでHybond−N+ナイロンフィルターに移してから放射標識プローブとハイブリダイズした。図37は、GB−C(配列番号673、残基2274−2640)から調製した放射線標識プローブとハイブリダイズした後、PCR産物をサザンブロットして得たPhosphoImage(モレキュラー・ダイナミクス社、カリフォルニア州サニーヴェイル)である。GB−C(配列番号673)は、300ngのヒトゲノムDNA中にGB−Cプラスミド鋳型の〜350fg(1ゲノムコピー当量、レーン5)および〜35fg(0.1ゲノムコピー当量、レーン6)が検出された以外、ヒト(図19、レーン1)、アカゲザル(レーン2)、S.cerevisiae(レーン3)、E.coli(レーン4)の各ゲノムDNAには検出されなかった。(レーン7は〜3.5fg[0.01ゲノムコピー当量]のGB−Cプラスミド鋳型からのPCR産物を300ngのヒトゲノムDNA中に含有する。)このように、0.1ゲノムコピー当量を検出できるゲノムPCRを使用してもGB−C(配列番号673)はヒト、アカゲザル、S.cerevisiaeおよびE.coliのゲノム中に検出できない。この結果は、GB−C(配列番号673)が外因性(即ちウイルス性)起源であるという予想と一致している。
D.GB−Cは追加のヒト血清サンプル中に検出できる
追加のHGBV−AおよびHGBV−B免疫反応性ヒト血清サンプルにRT−PCRを適用し、GB−C配列の存在について調べた。実施例7に記載のように、血清サンプルから抽出した核酸をランダムヘキサマーで逆転写し、cDNAに対して 35−40サイクルの増幅を行い(94℃、30秒;55℃、30秒;72℃、30−90秒)、次いで72℃で10分間伸展させた。GB−Cに特異的なPCRプライマー(g131−slとg131−al、配列番号675と676)を濃度1.0μMで使用した。PCR産物をアガロースゲル電気泳動で分離し、臭化エチジウムで核酸を直接染色したのちUV照射で可視化し、サザンブロットでHybond−N+ナイロンフィルターに移してから放射標識プローブとハイブリダイズした。西アフリカからの計48本のHGBV−免疫陽性サンプルを試験した。GB−Cが同定された最初のサンプルを含む西アフリカからの8サンプルは、GB−C配列に陽性であることがRT−PCRで判った。GB血清反応陰性の西アフリカ血清サンプルを計10本試験したが、いずれも検出可能なGB−C配列は有していなかった。陽性サンプル4本からのPCR産物をクローニングし、前記のように配列決定した。156にわたるヌクレオチドを調べた所、試験した4クローンの内の2つがGB−C配列(配列番号673、残基2274−2640)と同一であり、2クローン(配列番号677および678)がGB−C(配列番号673、残基2274−2640)と88.4%および83.6%相同の配列を含有していた(図38)。しかしながら、ヌクレオチドレベルにおけるダイバージェンスにもかかわらず、各クローンは非常に類似しており、配列番号678の予想された翻訳中にはただ一つのアミノ酸の相違が見られただけであることが、予想された翻訳で示された。
追加のHGBV−AおよびHGBV−B免疫反応性ヒト血清サンプルにRT−PCRを適用し、GB−C配列の存在について調べた。実施例7に記載のように、血清サンプルから抽出した核酸をランダムヘキサマーで逆転写し、cDNAに対して 35−40サイクルの増幅を行い(94℃、30秒;55℃、30秒;72℃、30−90秒)、次いで72℃で10分間伸展させた。GB−Cに特異的なPCRプライマー(g131−slとg131−al、配列番号675と676)を濃度1.0μMで使用した。PCR産物をアガロースゲル電気泳動で分離し、臭化エチジウムで核酸を直接染色したのちUV照射で可視化し、サザンブロットでHybond−N+ナイロンフィルターに移してから放射標識プローブとハイブリダイズした。西アフリカからの計48本のHGBV−免疫陽性サンプルを試験した。GB−Cが同定された最初のサンプルを含む西アフリカからの8サンプルは、GB−C配列に陽性であることがRT−PCRで判った。GB血清反応陰性の西アフリカ血清サンプルを計10本試験したが、いずれも検出可能なGB−C配列は有していなかった。陽性サンプル4本からのPCR産物をクローニングし、前記のように配列決定した。156にわたるヌクレオチドを調べた所、試験した4クローンの内の2つがGB−C配列(配列番号673、残基2274−2640)と同一であり、2クローン(配列番号677および678)がGB−C(配列番号673、残基2274−2640)と88.4%および83.6%相同の配列を含有していた(図38)。しかしながら、ヌクレオチドレベルにおけるダイバージェンスにもかかわらず、各クローンは非常に類似しており、配列番号678の予想された翻訳中にはただ一つのアミノ酸の相違が見られただけであることが、予想された翻訳で示された。
ギリシャ、エジプトおよびアメリカ合衆国の非A−E肝炎の個体から追加の血清サンプルを得て、前記のようにしてGB−C配列について検査した。これらのサンプルはいずれもGB−C配列は含有していなかった。サンプル中にGB−C配列が検出されなかったことには幾つかの理由が考えられる(上記、理論の項参照)。しかしながら、GB−C(配列番号673、残基2274−2640)と二つのGB−C変異体(配列番号678と677)との間における上述の配列変化は、もし密接に関連している西アフリカからのHGBV−Cがヌクレオチドレベルで15.1%異なることがあるならば、GB−Cに特異的なPCRプライマー(g131−sl、g131−al、配列番号675と676)が地理的に別個の隔離体であるGB−Cウイルスと、検出可能なPCR産物を産生するほどには十分ハイブリダイズしないかもしれないことを示唆している。この場合、ゲノムのより高度に保存された領域(5’UTR)に設計されたPCRプライマーによって、GB−C配列を西アフリカ血清サンプル中に検出できるかもしれない。
E.HGBV−C配列の伸展
実施例2Aに記載のPCRウォーキング法によって追加のGB−C配列を得た。即ち、当初GB−C(配列番号673、残基2274−2640)を同定するために用いた西アフリカのヒト血清から全核酸を抽出した。この核酸を前記のようにして逆転写した。その結果得られたcDNAを、2mMのMgCl2を用いたこと以外はソレンセンらによって記載された方法に従って50μlのPCR反応物(PCR1)中で増幅した。反応物を35サイクルの変性−アニーリング−伸展工程(94℃、30秒;55℃、30秒;72℃、90秒)に付し、次いで72℃で10分間伸展させた。ソレンセンらによって記載された方法に従ってストレプトアビジン被覆常磁性ビーズ(プロメガ社)によってビオチン化産物を単離した。次いでソレンセンらの記載する方法に従ってこのストレプトアビジンで精製した産物に上記と同じ全35サイクルの変性−アニーリング−伸展によってネスティッドPCR(PCR2)を行った。得られた産物およびこれを精製するために用いたPCRプライマーを表21に示す。
実施例2Aに記載のPCRウォーキング法によって追加のGB−C配列を得た。即ち、当初GB−C(配列番号673、残基2274−2640)を同定するために用いた西アフリカのヒト血清から全核酸を抽出した。この核酸を前記のようにして逆転写した。その結果得られたcDNAを、2mMのMgCl2を用いたこと以外はソレンセンらによって記載された方法に従って50μlのPCR反応物(PCR1)中で増幅した。反応物を35サイクルの変性−アニーリング−伸展工程(94℃、30秒;55℃、30秒;72℃、90秒)に付し、次いで72℃で10分間伸展させた。ソレンセンらによって記載された方法に従ってストレプトアビジン被覆常磁性ビーズ(プロメガ社)によってビオチン化産物を単離した。次いでソレンセンらの記載する方法に従ってこのストレプトアビジンで精製した産物に上記と同じ全35サイクルの変性−アニーリング−伸展によってネスティッドPCR(PCR2)を行った。得られた産物およびこれを精製するために用いたPCRプライマーを表21に示す。
さらに、上記のPCR1の条件下でオリゴヌクレオチドプライマー(配列番号669および670)を用いて1.3kb産物(C.16)を生成した。この産物を表21に記載のものと共にアガロースゲルから単離し、本技術分野で周知の方法によってpT7ブルーT−ベクタープラスミド(ノヴァゲン社)中にクローニングした。
クローニングした産物は、実施例5に記載のようにして配列決定した。GCGパッケージ(バージョン7)のプログラムによって配列を組み立てた。構築されたcontigの概略を図39に示す。GB−Cは長さが9034bpで、その全長が配列決定された。これを配列番号400−606に示す。これらの配列番号は三つの前進翻訳フレームと対応している。
(実施例19)
CKSに基づいた発現と免疫原性HGBV−Cポリペプチドの検出
実施例17(表13)に記載のウォーキング試験で得たHGBV−C配列を、実施例13の記載にしたがって表22(10ユニット、NEB)に掲載の制限酵素を用いてCKS発現ベクターpJO200、pJO201およびpJO202にクローニングした。追加のPCRクローン2種、C3/2およびC8/12、も発現した(図39)。配列番号681のプライマーによってPCR産物C3/2を生成し、PCR産物C8/12と配列番号685の補体を、実施例9の記載に従ってプライマー(配列番号693とその補体)を用いて生成した。このPCR産物を前記のようにしてpT7ブルー中にクローニングし、次いで表22に掲載した制限酵素で遊離させ、上記のpJO200、pJO201およびpJO202中にクローニングした。
CKSに基づいた発現と免疫原性HGBV−Cポリペプチドの検出
実施例17(表13)に記載のウォーキング試験で得たHGBV−C配列を、実施例13の記載にしたがって表22(10ユニット、NEB)に掲載の制限酵素を用いてCKS発現ベクターpJO200、pJO201およびpJO202にクローニングした。追加のPCRクローン2種、C3/2およびC8/12、も発現した(図39)。配列番号681のプライマーによってPCR産物C3/2を生成し、PCR産物C8/12と配列番号685の補体を、実施例9の記載に従ってプライマー(配列番号693とその補体)を用いて生成した。このPCR産物を前記のようにしてpT7ブルー中にクローニングし、次いで表22に掲載した制限酵素で遊離させ、上記のpJO200、pJO201およびpJO202中にクローニングした。
一以上のCKS/HGBV−AあるいはCKS/HGBV−B融合タンパクに対する抗体の存在が1.7、4.1、または2.17ELISA(実施例15、16参照)によって示唆された二つのヒト血清を選択し、ウェスタンブロットによって分析した。これらの血清のうちの一つ(240D)は非A−E肝炎(エジプト)の個体からのものであり、他方(G8−81)は西アフリカの個体のものでHGBVへの暴露について「リスクあり」(実施例15参照)のものであった。CKS/HGBV−C融合タンパクを発現し、これを前記のようにしてニトロセルロースシートに移した。記載されたようにブロットをプレブロックし、10%のE.coli溶解物と6mg/mlの XL1−ブルー/CKS溶解物を含有するブロッキングバッファーで1:100に希釈されたヒト血清サンプルの一つで一夜インキュベートした。ブロットはTBSで2回洗浄し、HRPO−接合ヤギ抗−ヒトIgGと反応させ、上記のように展開させた。結果を表22に示す。
HGBV−Cタンパクの内この二つの血清の一方あるいは他方と反応性を示したものがいくつかあり、その三つ(C.1、C.6、C.7)を選んでELISAアッセイ(実施例20参照)に付した。このようにして、HGBV−Aおよび/またはHGBV−Bタンパクと反応性を示すことが既に判っているサンプルは、HGBV−Cタンパクとも反応することが判明した。三つのHGBVウイルスからの複数のタンパクとの反応性は、ウイルス間でエピトープが共有された結果の交差反応によるものであるかもしれない。あるいは、これは複数種のウイルスの感染によるものかもしれず、さらにはその他の同定されていない要因によるものである可能性もある。
b:制限分解はpT7ブルーT−ベクターからPCRフラグメントを遊離させるために行った。ND=実施せず。
(実施例20)
GBV−Cの血清学的研究
A.組換えタンパク精製手順
CKS融合タンパクを発現する細菌細胞を凍結し、−70℃で保存した。各GBV−C構築物からの細菌細胞を解凍し、実施例15でGBV−B構築物について記載したようにして破砕処理した。さらに、組換えタンパクを、実施例15でGBV−B組換えタンパクについて記載したようにして精製した。
GBV−Cの血清学的研究
A.組換えタンパク精製手順
CKS融合タンパクを発現する細菌細胞を凍結し、−70℃で保存した。各GBV−C構築物からの細菌細胞を解凍し、実施例15でGBV−B構築物について記載したようにして破砕処理した。さらに、組換えタンパクを、実施例15でGBV−B組換えタンパクについて記載したようにして精製した。
この精製手順で収集した分画を電気泳動で分離し、クーマシーブリリアントブルーR250で染色して、分子量が約75kD(CKS−C.1/配列番号404)、71kD(CKS−C.6/配列番号404)、および49kD(CKS−C.7/配列番号404)であるタンパクの存否を調べた。予想された分子量のタンパクを示すバンドが、CKS−C.6およびCKSC.7組換えタンパクについて観察された。CKS−C.1タンパクについては、予想された分子量75kDの所ではなく、分子量62kDに対応する箇所にバンドが認められた。なぜ予想されたバンドと観察されたバンドが異なるのかは不明である。興味の対象のタンパクを含有する分画をプールし、SDS−PAGEによって再度調査した。
精製抗原を含有するプール分画の免疫原性および構造の完全性を、実施例13に記載の方法に従ってニトロセルロースに対する電子移動およびイムノブロット法によって決定した。適切な陽性対照がなかったため、組換えタンパクは、各融合タンパクのCKS部分に対するモノクローナル抗体との反応性によって同定した。CKS−C.1タンパク(配列番号404)の組換え抗原を検出すべく抗−CKSモノクローナル抗体を用いてウェスタンブロット法で調べると、約65kDのところにシングルバンドが認められた。この結果はCKS−C.1タンパク(配列番号404)の予想サイズの75kDとは異なっている。なお、CKS−C.6タンパク(配列番号404)およびCKS−C.7タンパク(配列番号404)については、イムノブロット法で調べた結果予想されたサイズを有していた。
B.ポリスチレンビーズの被覆手続き
上記タンパクを透析し、実施例15の記載に従って、これらのタンパクのポリスチレンビーズ上での免疫原性を評価した。
上記タンパクを透析し、実施例15の記載に従って、これらのタンパクのポリスチレンビーズ上での免疫原性を評価した。
C.HGBVに対する抗体を検出するためのELISAの手順
各種ELISAは実施例15の記載に従って実施した。
各種ELISAは実施例15の記載に従って実施した。
D.感染個体の血清におけるHGBV−RNAの検出
各ELISAで繰り返し反応性を示した検体について、実施例15のセクションDの記載に従ってHGBV−RNAを調べた。
各ELISAで繰り返し反応性を示した検体について、実施例15のセクションDの記載に従ってHGBV−RNAを調べた。
E.タマリンの血清像
HGBV−Cゲノム由来のCKSC.1タンパク、CKSC.6タンパク、あるいはCKSC.7タンパクを用いたELISAアッセイにおいて、タマリンからの血清はいずれも特異的免疫反応を示さなかった。実施例15中のタマリンの血清像についての記載参照。
HGBV−Cゲノム由来のCKSC.1タンパク、CKSC.6タンパク、あるいはCKSC.7タンパクを用いたELISAアッセイにおいて、タマリンからの血清はいずれも特異的免疫反応を示さなかった。実施例15中のタマリンの血清像についての記載参照。
F.追補的試験
実施例15に記載したように、最初反応性を示した検体については、典型的には再試験を行った。ある検体が繰り返し反応性を示した場合、ELISAによるデータをさらに補充すべく追加の試験(例えばウェスタンブロット)を実施することができる。ウェスタンブロットの結果が陽性と考えられるためには、観察可能なバンドがC.1タンパク(配列番号404)に対して65kD、C.6タンパク(配列番号404)に対して71kD、またはC.7タンパク(配列番号404)に対して49kDにおいて検出される必要がある。ウェスタンブロットはELISAの感度に匹敵しあるいはそれを越えるほど最適化されていないので、陰性の結果が出ても直ちにELISAのデータを捨てることはしなかった。しかしながら、陽性の結果が出ればそれはELISAによって検出された反応性を補強するものである。
実施例15に記載したように、最初反応性を示した検体については、典型的には再試験を行った。ある検体が繰り返し反応性を示した場合、ELISAによるデータをさらに補充すべく追加の試験(例えばウェスタンブロット)を実施することができる。ウェスタンブロットの結果が陽性と考えられるためには、観察可能なバンドがC.1タンパク(配列番号404)に対して65kD、C.6タンパク(配列番号404)に対して71kD、またはC.7タンパク(配列番号404)に対して49kDにおいて検出される必要がある。ウェスタンブロットはELISAの感度に匹敵しあるいはそれを越えるほど最適化されていないので、陰性の結果が出ても直ちにELISAのデータを捨てることはしなかった。しかしながら、陽性の結果が出ればそれはELISAによって検出された反応性を補強するものである。
実施例15で記載したように、繰り返し反応性のある検体で十分量あるものに対しては、RT−PCR(配列番号8と9に対応するプライマーを用い、実施例10の記載に従って実施)によって血清中のHGBV−C特異性ヌクレオチド配列を同定することができる。
G.実験手順
実施例15では、種々のヒト個体群におけるHGBV−BとHGBV−Aに対する抗体の存在を評価するために、HGBV−Bからの組換え抗原を使用したELISAを利用した。実施例14に記載したように、固相の上に被覆したCKS−C.1組換えタンパク(配列番号404)を用いたC.1ELISA、CKS−C.6組換えタンパク(配列番号404)を用いたC.6ELISA、およびCKS−C.7組換えタンパク(配列番号404)を用いたC.7ELISAを利用し、HGBV−Cに対する抗体についての試験を同じ検体の多くについて行なった。実施例15に述べたように、HGBVを接種した回復期にあるタマリンの5匹全てが、(1.7、1.4および4.1ELISAを用いて検出されたように)HGBV−B組換えタンパクに応答する特異的ではあるが短命の抗体を産生した。C.1、C.6およびC.7ELISAに応答する検出可能な抗体を産生したタマリンはなかったが、ヒトの検体の幾つかは、ウエスタンブロット法(実施例13参照)によって試験したとき、C.1、C.6およびC.7組換えタンパクに反応する特異的な抗体を産生した。本実施例では、種々のヒト個体群における抗体を検出する上でのC.1、C.6およびC.7ELISAの有用性を評価した。
実施例15では、種々のヒト個体群におけるHGBV−BとHGBV−Aに対する抗体の存在を評価するために、HGBV−Bからの組換え抗原を使用したELISAを利用した。実施例14に記載したように、固相の上に被覆したCKS−C.1組換えタンパク(配列番号404)を用いたC.1ELISA、CKS−C.6組換えタンパク(配列番号404)を用いたC.6ELISA、およびCKS−C.7組換えタンパク(配列番号404)を用いたC.7ELISAを利用し、HGBV−Cに対する抗体についての試験を同じ検体の多くについて行なった。実施例15に述べたように、HGBVを接種した回復期にあるタマリンの5匹全てが、(1.7、1.4および4.1ELISAを用いて検出されたように)HGBV−B組換えタンパクに応答する特異的ではあるが短命の抗体を産生した。C.1、C.6およびC.7ELISAに応答する検出可能な抗体を産生したタマリンはなかったが、ヒトの検体の幾つかは、ウエスタンブロット法(実施例13参照)によって試験したとき、C.1、C.6およびC.7組換えタンパクに反応する特異的な抗体を産生した。本実施例では、種々のヒト個体群における抗体を検出する上でのC.1、C.6およびC.7ELISAの有用性を評価した。
H.切捨て値の決定
C.1、C.6およびC.7ELISAの切捨て値を実施例15に記載したようにして決定した。
C.1、C.6およびC.7ELISAの切捨て値を実施例15に記載したようにして決定した。
I.低リスク検体を用いて得た血清学的データ
ウイスコンシン州南西部に住む健康なボランティアのドナーから100本の血清と100本の血漿を含む集団を得るとともに、C.1ELISAにおけるCKS−C.1(配列番号404)タンパク、C.6ELISAにおけるCKS−C.6(配列番号404)タンパク、およびC.7ELISAにおけるCKS−C.7(配列番号404)タンパクを含むHGBV−Cからの三種の組換えタンパクに対する抗体について試験を行なった。
ウイスコンシン州南西部に住む健康なボランティアのドナーから100本の血清と100本の血漿を含む集団を得るとともに、C.1ELISAにおけるCKS−C.1(配列番号404)タンパク、C.6ELISAにおけるCKS−C.6(配列番号404)タンパク、およびC.7ELISAにおけるCKS−C.7(配列番号404)タンパクを含むHGBV−Cからの三種の組換えタンパクに対する抗体について試験を行なった。
C.1ELISAについては、血清および血漿の検体の平均吸収値が、それぞれ0.049(標準偏差(SD)=0.040)と0.038(SD=0.029)であった。血清と血漿についての切捨て値はそれぞれ0.214と0.286と計算された。上記のように、切捨て値は、陰性対照の吸収値の関数として表すこともでき、10.0を越えるS/N値を有する検体は反応性であると考えられた。この切捨て値を用いたところ、100の血漿検体にはC.1タンパク(配列番号404)に対する抗体に初期反応性および繰り返し反応性がみとめられたものはなく、100の血清検体の内の一つでは、C.1タンパク(配列番号404)に対する抗体に初期反応性および繰り返し反応性が認められた。
C.6ELISAについては、血清および血漿の検体の平均吸収値が、それぞれ0.102(標準偏差(SD)=0.046)と0.105(SD=0.047)であった。10以上のS/N値を有する検体が反応性であると判断されるように切捨て値を設定した。この切捨て値を用いたところ、3個の検体(2個は血清の集団からで、1個は血漿の集団から)ではC.6タンパク(配列番号404)に対する抗体に対して繰り返し反応性がみとめられた。
C.7ELISAについては、血清および血漿の検体の平均吸収値が、それぞれ0.061(標準偏差(SD)=0.040)と0.050(SD=0.055)であった。10以上のS/N値を有する検体が反応性であると判断されるように切捨て値を設定した。この切捨て値を用いたところ、C.7タンパク(配列番号404)に対する抗体に対して繰り返し反応性であると考えられる検体は無かった。
したがって、米国人の血液提供者(N=200)の約1%にC1、C6またはC.7タンパクに対する抗体が存在することが証明された。
J.肝炎に対し「リスクあり」と考えられる検体
この研究のデータを表23にまとめて示す。
この研究のデータを表23にまとめて示す。
(i)西アフリカからの検体
137個の検体の内の計20個が、GBV−Cタンパクを用いたELISAの一種またはそれ以上において反応性であった。97個の内の合計12個がC.1ELISAにおいて繰り返し反応性であることが認められ、52個の内の3個がC.6ELISAにおいて繰り返し反応性であることが認められ、137個の検体の内の5個がC.7ELISAにおいて繰り返し反応性であることが認められた。C.1において反応性であった検体の内の3個をウエスタンブロット法で試験し、反応性であることが分かった。
137個の検体の内の計20個が、GBV−Cタンパクを用いたELISAの一種またはそれ以上において反応性であった。97個の内の合計12個がC.1ELISAにおいて繰り返し反応性であることが認められ、52個の内の3個がC.6ELISAにおいて繰り返し反応性であることが認められ、137個の検体の内の5個がC.7ELISAにおいて繰り返し反応性であることが認められた。C.1において反応性であった検体の内の3個をウエスタンブロット法で試験し、反応性であることが分かった。
これらのデータは、HGBVが西アフリカの風土病であることを示唆している。
(ii)経静脈麻薬使用者の検体
イリノイ州シカゴのハインズ退役軍人管理病院において行なわれた研究の一環として、経静脈麻薬使用者の集団から合計112個の検体を得た。112個の検体の内の計2個が一種またはそれ以上のタンパクに対して繰り返し反応性を有していた。1個の検体がC.1ELISAにおいて繰り返し反応性であることがみとめられ、1個の検体がC.7ELISAにおいて繰り返し反応性であることがみとめられた.C.6ELISAにおいて陽性であった検体は無かった。
イリノイ州シカゴのハインズ退役軍人管理病院において行なわれた研究の一環として、経静脈麻薬使用者の集団から合計112個の検体を得た。112個の検体の内の計2個が一種またはそれ以上のタンパクに対して繰り返し反応性を有していた。1個の検体がC.1ELISAにおいて繰り返し反応性であることがみとめられ、1個の検体がC.7ELISAにおいて繰り返し反応性であることがみとめられた.C.6ELISAにおいて陽性であった検体は無かった。
K.非A−E肝炎の個体から得た検体
この研究のデータを表23にまとめて示す。
この研究のデータを表23にまとめて示す。
非A−E肝炎を有する個体から種々の検体の集団(実施例15.Kに記載)を得、1.5、2.17、1.18、および1.22ELISA(実施例15.Cに記載)によって試験した。検体の量が不十分であったため、検体の全てを全てのELISAにおいて試験できなかった。
(i)日本の検体
合計89個の検体の内、C.1ELISAにおいて繰り返し反応性であるとみとめられたものは無かった。検体の量が少なかったため、C6またはC.7ELISAでの抗体についての試験は行なわなかった。
合計89個の検体の内、C.1ELISAにおいて繰り返し反応性であるとみとめられたものは無かった。検体の量が少なかったため、C6またはC.7ELISAでの抗体についての試験は行なわなかった。
(ii)ギリシャの検体
合計67個の検体をC.1およびC.7ELISAを用いて試験した。反応性のみとめられた検体は無かった。
合計67個の検体をC.1およびC.7ELISAを用いて試験した。反応性のみとめられた検体は無かった。
(iii)エジプトの検体
132個の検体の内の計18個が一つ以上のELISAにおいて反応性であるとみとめられた。C.1ELISAにおいて反応性であるとみとめられたものは無かった。計15個の検体がC.6ELISAにおいて反応性であるとみとめられ、3個の検体がC.7ELISAにおいて反応性であるとみとめられた。
132個の検体の内の計18個が一つ以上のELISAにおいて反応性であるとみとめられた。C.1ELISAにおいて反応性であるとみとめられたものは無かった。計15個の検体がC.6ELISAにおいて反応性であるとみとめられ、3個の検体がC.7ELISAにおいて反応性であるとみとめられた。
(iv)米国の検体(Mセット)
合計6個の検体が一以上のELISAにおいて反応性であると認められた。2個の検体がC.1ELISAにおいて繰り返し反応性であると認められた。4個の検体がC.6ELISAにおいて繰り返し反応性であると認められた。C.7ELISAにおいて繰り返し反応性の検体は無かった。
合計6個の検体が一以上のELISAにおいて反応性であると認められた。2個の検体がC.1ELISAにおいて繰り返し反応性であると認められた。4個の検体がC.6ELISAにおいて繰り返し反応性であると認められた。C.7ELISAにおいて繰り返し反応性の検体は無かった。
(v)米国からの検体(Tセット)
64個の検体の内で、C.1ELISAまたはC.6ELISAにおいて反応性であると認められたものは無かった。1個の検体がC.7ELISAにおいて繰り返し反応性であった。
64個の検体の内で、C.1ELISAまたはC.6ELISAにおいて反応性であると認められたものは無かった。1個の検体がC.7ELISAにおいて繰り返し反応性であった。
(vi)米国の種々の臨床現場からの検体(セット1)
総計すると、62個の検体の内の3個が一以上のELISAで反応性であると認められた。1個の検体がC.1ELISAおよびC.6ELISAの両方において繰り返し反応性であると認められた。2個の検体がC.7ELISAにおいて繰り返し反応性であると認められた。
総計すると、62個の検体の内の3個が一以上のELISAで反応性であると認められた。1個の検体がC.1ELISAおよびC.6ELISAの両方において繰り返し反応性であると認められた。2個の検体がC.7ELISAにおいて繰り返し反応性であると認められた。
上記のように、HGBVには二以上の株が存在する可能性があり、即ち二以上の別個のウイルスを本明細書に開示した配列によって表すことができる。これらは本発明の範囲内であると考えられ“肝炎GBウイルス(“HGBV”)”と称す。
L.血清反応の結果の統計的意義
これらのデータは、HGBVに晒されるリスクがあると考えられる個人、および非A−E肝炎であると診断された個人からは、HGBV−Cタンパク(即ち、C.1、C.6およびC.7ELISAの抗体に対して繰り返し反応性である検体)に特異的な抗体が検出され、米国からのボランティアまたは売血血液提供者からはこれらの抗体が低い割合で検出された。表24に、種々のカテゴリーの検体(表23に示す“低リスク”、“リスク有り”、および非A−E肝炎患者)について得られた血清反応の結果をグループ分けして示し、χ二乗検定によって統計的意義を分析した。試験した検体の総数におけるHGBVタンパクに対する抗体の血清学的にみた存在をまとめた表23のデータと異なり、表24のデータは、異なる個体(人)について得られた結果を反映している。GBV−CのELISAについては、そのデータは、ボランティアの血液提供者における抗HGBVの血清学的にみた存在と、IVDUではなくHGBVに晒される“リスクあり”と考えられる個体(西アフリカ)との比較においては有意差(p値=0.000)があることを示している。また、エジプトと米国の非A−E肝炎に罹患している個体におけるHGBV−Cに対する血清学的にみた抗体の存在は、ボランティアの血液提供者と比較した場合、統計的有意差があった。これらのデータによれば、HGBV−Cに晒されることは非A−E肝炎と関連していることが示唆される。注:これらの初期の研究ではRT−PCRの結果は陰性であったが、その後に得られたデータによって、血清学的に陽性の個体(実施例19参照)の血清におけるフラヴィ様ウイルス配列が判明した。
これらのデータは、HGBVに晒されるリスクがあると考えられる個人、および非A−E肝炎であると診断された個人からは、HGBV−Cタンパク(即ち、C.1、C.6およびC.7ELISAの抗体に対して繰り返し反応性である検体)に特異的な抗体が検出され、米国からのボランティアまたは売血血液提供者からはこれらの抗体が低い割合で検出された。表24に、種々のカテゴリーの検体(表23に示す“低リスク”、“リスク有り”、および非A−E肝炎患者)について得られた血清反応の結果をグループ分けして示し、χ二乗検定によって統計的意義を分析した。試験した検体の総数におけるHGBVタンパクに対する抗体の血清学的にみた存在をまとめた表23のデータと異なり、表24のデータは、異なる個体(人)について得られた結果を反映している。GBV−CのELISAについては、そのデータは、ボランティアの血液提供者における抗HGBVの血清学的にみた存在と、IVDUではなくHGBVに晒される“リスクあり”と考えられる個体(西アフリカ)との比較においては有意差(p値=0.000)があることを示している。また、エジプトと米国の非A−E肝炎に罹患している個体におけるHGBV−Cに対する血清学的にみた抗体の存在は、ボランティアの血液提供者と比較した場合、統計的有意差があった。これらのデータによれば、HGBV−Cに晒されることは非A−E肝炎と関連していることが示唆される。注:これらの初期の研究ではRT−PCRの結果は陰性であったが、その後に得られたデータによって、血清学的に陽性の個体(実施例19参照)の血清におけるフラヴィ様ウイルス配列が判明した。
(実施例21)
非A−E肝炎のヒトにおけるHGBV−Cの存在
HGBV−Cに特異的なELISAの生成により、非A−E肝炎に罹患している患者の免疫的に陽性の血清(HGBV−C血清学の実施例)を同定できるようになった。この血清を、非A−E肝炎であると立証された個体(表25)から得た幾つかの免疫的に陽性のHGBV−AとHGBV−Bと共に、HGBV−C配列についてRT−PCRによって試験した。HGBV−Cの異型もなるべく検出できるよう、第1回目の増幅において変性NS3オリゴヌクレオチドプライマーを使用してRT−PCRを行ない、これに続き、ネスト化されたGB−C特異性プライマーを用いて第2回目の増幅を行なった。具体的には、第1回目の増幅は、2mMのMgCl2、および1μMのプライマーns3.2−s1、ns3.2−a1(配列番号はそれぞれ711と712)を使用し、実施例6で述べたようにして調製された血清cDNA製品を用いて行なった。反応物に対して変性−アニーリング−伸展を40回繰り返し〔(94℃で 30秒、37℃で30秒、2分間かけて72℃に温度上昇、72℃で30秒)というサイクルを3回繰り返し、その後(94℃で30秒、50℃で30秒、72℃で30秒)のサイクルを37回〕、これに続き72℃で10分間伸展を行なった。得られた反応物を4℃に維持した。第2回目の増幅は、2mMのMgCl2、1μMのGB−C特異性のプライマー(配列番号675と676)、および鋳型としての4%の第1のPCR製品を使用して行なった。第2回目の増幅では、増幅すべき鋳型との塩基対の不適合を含んでいるようなオリゴヌクレオチドプライマーを有する特定の生成物をも増幅するために設計された温度循環計画を採用した〔Roux、Bio/Techniques、16:812〜814(1994年)〕。即ち、反応は、温度循環(94℃で20秒、55℃(各サイクル毎に0.3℃低下)で30秒、72℃で1分)を43回行なった後、(94℃で20秒、40℃で30秒、72℃で1分)で10回行い、最終伸展を72℃で10分行なった。PCR産物をアガロースゲル電気泳動法により単離し、臭化エチジウムを用いて核酸を直接染色した後、紫外線照射によって可視化し、サザントランスファーでHybond−N+ナイロンフィルターに移した後、GB−Cに対する放射線標識化プローブにハイブリダイゼーションを行なった。PCR産物を、当分野において述べられたようにクローン化するとともに配列決定した。
非A−E肝炎のヒトにおけるHGBV−Cの存在
HGBV−Cに特異的なELISAの生成により、非A−E肝炎に罹患している患者の免疫的に陽性の血清(HGBV−C血清学の実施例)を同定できるようになった。この血清を、非A−E肝炎であると立証された個体(表25)から得た幾つかの免疫的に陽性のHGBV−AとHGBV−Bと共に、HGBV−C配列についてRT−PCRによって試験した。HGBV−Cの異型もなるべく検出できるよう、第1回目の増幅において変性NS3オリゴヌクレオチドプライマーを使用してRT−PCRを行ない、これに続き、ネスト化されたGB−C特異性プライマーを用いて第2回目の増幅を行なった。具体的には、第1回目の増幅は、2mMのMgCl2、および1μMのプライマーns3.2−s1、ns3.2−a1(配列番号はそれぞれ711と712)を使用し、実施例6で述べたようにして調製された血清cDNA製品を用いて行なった。反応物に対して変性−アニーリング−伸展を40回繰り返し〔(94℃で 30秒、37℃で30秒、2分間かけて72℃に温度上昇、72℃で30秒)というサイクルを3回繰り返し、その後(94℃で30秒、50℃で30秒、72℃で30秒)のサイクルを37回〕、これに続き72℃で10分間伸展を行なった。得られた反応物を4℃に維持した。第2回目の増幅は、2mMのMgCl2、1μMのGB−C特異性のプライマー(配列番号675と676)、および鋳型としての4%の第1のPCR製品を使用して行なった。第2回目の増幅では、増幅すべき鋳型との塩基対の不適合を含んでいるようなオリゴヌクレオチドプライマーを有する特定の生成物をも増幅するために設計された温度循環計画を採用した〔Roux、Bio/Techniques、16:812〜814(1994年)〕。即ち、反応は、温度循環(94℃で20秒、55℃(各サイクル毎に0.3℃低下)で30秒、72℃で1分)を43回行なった後、(94℃で20秒、40℃で30秒、72℃で1分)で10回行い、最終伸展を72℃で10分行なった。PCR産物をアガロースゲル電気泳動法により単離し、臭化エチジウムを用いて核酸を直接染色した後、紫外線照射によって可視化し、サザントランスファーでHybond−N+ナイロンフィルターに移した後、GB−Cに対する放射線標識化プローブにハイブリダイゼーションを行なった。PCR産物を、当分野において述べられたようにクローン化するとともに配列決定した。
上記の方法を用い、GB−C.4、GB−C.5、GB−C.6、およびGB−C.7を得た。これらの配列は、82.1〜86.6%の割合でGB−C(配列番号400、塩基4167〜4365)と相同である。図40は、予期されたコドントリプレットにおける、GB−Cと相同な領域に整合したGB−C.4、GB−C.5、GB−C.6、およびGB−C.7の配列の相違を示す。この図に示すように、ヌクレオチドの相違の殆どは、GB−Cからのアミノ酸の変化を引き起こさない。このアミノ酸レベルでの全体的な配列の維持は、GB−C.4、GB−C.5、GB−C.6、およびGB−C.7が同じウイルスの異なった株であるHGBV−Cから得られたものであることを示唆している。さらに、ヌクレオチドレベルでの配列の相違の程度は、これらのPCR産物が以前に同定されたGB−C配列の何れかによる汚染の結果生じたものではないことを示している。
これらの個体の内の三つ(GB−C.4、GB−C.5、GB−C.6、およびGB−C.7源)については、A型肝炎、B型肝炎、またはC型肝炎に感染している証拠はなかった。病因が未知の肝炎に罹患している個体におけるGB−C配列の存在は、ヒト肝炎の原因物質の一つがHGBV−Cであることを示唆している。個体の内の2つ(GB−C.4とGB−C.5を含んでいる)については経時的試料が利用可能であった。これらの試料中のHGBV−Cの配列を追跡するために、クローン特異性のRT−PCRを開発した。即ち、血清から抽出した核酸を、実施例7において行なったようにランダムヘキサマーによって逆転写した。得られたcDNAを、94℃で30秒、55℃で30秒、72℃で30秒の条件での増幅を40回行い、その後、伸展を72℃で10分行なった。GB−C.4とGB−C.5に特異的なPCRプライマー(GB−C.4−s1とGB−C.4−a1、またはGB−C.5−s1とGB−C.5−a1)を1.0μMの濃度で使用した。PCR産物をアガロースゲル電気泳動法により単離し、臭化エチジウムを用いて核酸を直接染色した後、紫外線照射によって可視化し、サザントランスファーでHybond−N+ナイロンフィルターに移した後、放射線標識プローブにハイブリダイズした。
急性非A−E肝炎に罹患しているエジプト人の患者の血清においてGB−C.4が見いだされた。この患者はHGBV−Aタンパクについ血清反応陽性であった(HGBV−A ELISAの実施例参照)。このエジプト人の患者から得た5個の一連の試料のRT−PCRはウイルス血症を示し、これは血清ALT値が正常になった後少なくも20日の間持続した(表26)。血清ALTが正常になった後におけるGB−C配列の存在は、人によっては慢性の感染につながることを示唆した。しかし、この患者の試料はそれ以上は得られなかったため、HGBV−Cの慢性化について結論を出すことはできない。この問題を解決するため追加の試料を求めている段階である。
GB−C.5は、肝炎に関連する再生不良貧血に罹患していたカナダ人の患者から得た。この患者の各試料は、C.7ELISAにおいて血清学的に陽性であった(実施例20)。カナダ人の患者から再生不良貧血の間(症状の顕出後13日目)および死亡時(14日目、図41)において得た試料から、GB−C.5特異性のプライマー(GB−C.5−s1とGB−C.5−a1)を用いてGB−C.5を検出した。しかし、GB−C.5特異性のPCRは、症状の発生時(0日目、急性肝炎)および3日目(肝不全)においてGB−C.5配列を検出できなかった。したがって、最初の試料中にGB−C.5が検出限界以下のレベルで存在していたか否かは不明である。もし、存在していたとしたら、HGBV−Cは、その患者の再生不良貧血の原因物質であったであろう。しかし、GB−C.5は、再生不良の危期においてのみRT−PCRによって検出されたため、GB−C.5は、貧血の対処のために投与された血液製品から混入したものかもしれない。この場合、再生不良貧血へのHGBV−Cの関連はHCVと類似したものであろう〔ヒブス(Hibbs )ら、JAMA、267、2051〜2054(1992年)〕。
HGBV−CとHCVとの関係が遠いことに鑑み、HCV感染を検出するための現在の方法により、HGBV−Cを含むヒトの試料を認識できるか否かは興味の対象であった。HCVに暴露または感染した個体の通常の検査法は、HCV−1の3つまたはそれ以上の領域から得た抗原を利用する抗体試験によるものである。この試験により、殆どの個体におけるHCVの既知の遺伝子型の全てに対する抗体を検出できる〔サカモトら、J.Gen.Virol.75、1761〜1768(1994年)、スチュイバー(Stuyver)ら、J.Gen.Virol.74、1093〜1102(1993年)〕。HCVのための第2世代のELISAを、実施例10(表25)に記載されたHGBV−Cを含む試料に対して実施した。HGBV−Cを含む4つの試料の内の一つがHCV抗原に対して血清反応陽性であった。HCVに対して血清反応陰性のヒト血清中の限られたものだけが、非常に感度の高いRT−PCRアッセイにより、HCVゲノムRNAに対して陽性であることが示された〔スギタニ、1992年、#65〕。類似のRT−PCRアッセイ(実施例9に記載されたような)は、血清反応陽性の試料中のHCVウイルス血症の存在を確認した。しかし、血清反応HCV陰性の試料にはHCVウイルス血症であるものは存在しなかった。したがって、HGBV−C配列を含む4つの個体の内の一つはHCV感染の証拠を有しているものの、HCVの存在に関する本アッセイは、HGBV−Cの存在を正確に予測できなかった。この一人のHCV陽性の患者はHGBV−Cにも感染していたようである。この患者の肝炎がHCV、HGBV−C、または両方のウイルスの存在によるものであったかは不明確である。同じ患者においてHCVとHGBV−Cが発見されることは、これらの感染を受ける共通のリスクが存在することを示唆するものであろう。
上記のPCR手順を使用し、1994年に2人のボランティアの米国人提供者から得た血液の“正常(normal)”ユニット中に、GB−C配列(図41に示した先のGB−C分離株と〜85%相同、データは示していない)を同定した。これらのユニットは、試験の結果HBV、HCVについて陰性であるとされ、正常な血清ALT値を有していた。しかし、これらのユニットは1.4ELISAでの試験の結果、陽性とされた。免疫的にスクリーニングされた〜1000ユニットの内の少なくとも2つの“正常”血液ユニットにHGBV−Cが発見されたことは、このウイルスが現在、米国の血液供給に存在していることを示唆している。しかし、我々はHGBVタンパクから開発されたELISAおよびHGBV配列からのヌクレオチドプローブを使用して、これらの血液ユニットを同定できることを示す。
種々のHGBV配列(図41)には、多数の配列のバリエーションがあることに留意しなければならない。ウイルス性の核酸に対するPCRに基づくアッセイは、非常に敏感ではあるものの、オリゴヌクレオチドプライマーとウイルス鋳型との間の配列の一致に依存する。したがって、本研究で利用したPCRプライマーは、種々の分離物において十分保存されていないHGBV−Cゲノムの領域に位置していたため、試験したHGBV−Cウイルス血症の試料の全てを、ここで採用したRT−PCRによって検出できなかったのであろう。HCV5’の翻訳されていない領域中に発見されたように〔チャ(Cha ) ら、J.Clin.Microbiol.、29、2528〜2534(1991年)〕、HGBV−Cゲノムの良く保存された領域からのPCRプライマーを利用することにより、HGBV−Cウイルス血症の試料をより正確に検出できるようになろう。
2 切捨て値と報告された値との対比。値が1以上のもの(下線で示す)は陽性と考えられる。
3 肝炎に関連した再生不良貧血。
2 正常と報告された値との対比。値が10以上のものは陽性と考えられる。
(実施例22)
配列の比較と系統的分析
ヌクレオチドと予測したアミノ酸配列の比較、即ちアライメントを行なうことによってウイルスの近親性の程度についての情報を得ることができる。HGBV配列と他のウイルスの配列とのアライメントを行なうことにより、配列間の類似性と相同性の程度を量的に評価することができる。この情報は、HGBVウイルスの分類のための原理を開発するのに使用できる。一般に、二つのアミノ酸配列の間の類似性の計算は、アライメント中のアミノ酸対の側鎖の間の類似度程度に基づいて行なわれる。類似度は、アミノ酸の側鎖の物理化学的特徴、即ち、寸法、形状、電荷、水素結合の能力、および化学的反応性に基づいている。即ち類似したアミノ酸は類似の物理化学的特徴を備えた側鎖を有している。例えば、フェニルアラニンとチロシンは共に芳香性の側鎖を含んでおり、したがって化学的に類似であると見做すことができる。アミノ酸の化学についての議論は基礎的な生化学の教科書、例えば、Biochemistry、第3版、ルバート・スタイヤー(Lubert Stier)編、W.H.Freeman and Company、ニューヨーク、1988年に見ることができる。整列した2つのアミノ酸配列の間の相同性の計算は、一般的にアライメント中の同一のアミノ酸対の数を計数し、この数をアライメント中の配列の長さで割るという算術計算である。アミノ酸配列のアライメントに使用する方法と同様に、アライメントした2つのヌクレオチド配列の間の相同性の程度の決定は、アライメント中の同一のヌクレオチド塩基対の数を計数し、この数をアライメント中の配列の長さで割るという算術計算である。アラインした2つのヌクレオチド配列の間の類似性の計算は、アライメント中の種々の位置における対になった(即ち、整合した)対の間における遷移および転換に対して時として異なる値を用いる。しかし、一対のヌクレオチド配列間の類似性と相同性の点数は通常非常に接近しており、即ち1〜2%以内である。
配列の比較と系統的分析
ヌクレオチドと予測したアミノ酸配列の比較、即ちアライメントを行なうことによってウイルスの近親性の程度についての情報を得ることができる。HGBV配列と他のウイルスの配列とのアライメントを行なうことにより、配列間の類似性と相同性の程度を量的に評価することができる。この情報は、HGBVウイルスの分類のための原理を開発するのに使用できる。一般に、二つのアミノ酸配列の間の類似性の計算は、アライメント中のアミノ酸対の側鎖の間の類似度程度に基づいて行なわれる。類似度は、アミノ酸の側鎖の物理化学的特徴、即ち、寸法、形状、電荷、水素結合の能力、および化学的反応性に基づいている。即ち類似したアミノ酸は類似の物理化学的特徴を備えた側鎖を有している。例えば、フェニルアラニンとチロシンは共に芳香性の側鎖を含んでおり、したがって化学的に類似であると見做すことができる。アミノ酸の化学についての議論は基礎的な生化学の教科書、例えば、Biochemistry、第3版、ルバート・スタイヤー(Lubert Stier)編、W.H.Freeman and Company、ニューヨーク、1988年に見ることができる。整列した2つのアミノ酸配列の間の相同性の計算は、一般的にアライメント中の同一のアミノ酸対の数を計数し、この数をアライメント中の配列の長さで割るという算術計算である。アミノ酸配列のアライメントに使用する方法と同様に、アライメントした2つのヌクレオチド配列の間の相同性の程度の決定は、アライメント中の同一のヌクレオチド塩基対の数を計数し、この数をアライメント中の配列の長さで割るという算術計算である。アラインした2つのヌクレオチド配列の間の類似性の計算は、アライメント中の種々の位置における対になった(即ち、整合した)対の間における遷移および転換に対して時として異なる値を用いる。しかし、一対のヌクレオチド配列間の類似性と相同性の点数は通常非常に接近しており、即ち1〜2%以内である。
前に述べたように、HGBV作用物質とC型肝炎遺伝子型のアミノ酸配列の間の相同性は限られたものである。HGBV作用物質とHCVとの間の近親性の程度をより精度良く決定するために、HGBV−A、BおよびCの全体的な大きな開放読み枠(ORF)の配列、および幾つかの代表的なHCV分離物の大きなORFのアミノ酸配列を用いてアミノ酸配列アライメントを行った。さらに、ヌクレオチドレベルにおけるHGBV作用物質とHCVの間の近親性の程度は、HGBV−A、BおよびCの全体的なゲノムヌクレオチド配列、および幾つかの代表的なHCV分離物の全体的なゲノムヌクレオチド配列を用いて決定した。アミノ酸配列とヌクレオチド配列の間のアライメントは、53711、ウイスコンシン州マジゾン、サイエンスドライブ575にあるジェネティック・コンピュータ・グループ社から入手可能なウイスコンシン配列分析パッケージ(バージョン8)のプログラムGAPを用いて行なった。ギャップ作成とギャップ延長のペナルティーは、核酸配列のアライメントに対してはそれぞれ5.0と0.3であり、アミノ酸配列の比較に対してはそれぞれ3.0と0.1であった。GAPプログラムは、アラインされている2つの配列の間の百分率で表した類似性と相同性の程度を計算するために、ニードルマン(Needleman)とワンシュ(Wunsch)のアルゴリズム(J.Mol.Biol.48、443〜453、1970年)を使用している。
選択された数の主要なC型肝炎のウイルス(HCV)遺伝子型のヌクレオチド配列とアミノ酸配列をGenBankから入手した。これをそれらの登録番号とともに下記に示す。
大きな開放読み枠(即ち推定の前駆体ポリタンパク)の予想されるアミノ酸配列と、上記HCV遺伝子型の各々とHGBV分離物との間のヌクレオチド配列との間の対毎の比較結果を表28と29にそれぞれ示す。各HCV分離物の遺伝子型の指定を一番上の行に示す。この遺伝子型の指定は、シモンズ・ピー(Simmonds, P)ら(1994年)Hepatology、19、1321〜1324に記載されたHCV分離物のための命名法に基づく。
表28に示すデータは、HCV遺伝子型の間におけるアミノ酸配列の相同性の下限が69%であることを示している。この値は、シモンズら〔シモンズ・ピー(Simmonds, P )ら、Hepatology、19、1321〜1324(1994年)〕が示した値に非常に近い。シモンズらは、2つの主要な群からの8個の完全なHCVゲノムのコード化領域を比較して、67.1%〜68.6%のアミノ酸配列の類似度を示した。しかし、この著者は、類似度を計算した方法を記載しなかった。この値(69%)は、HCV−J8と他の主要なHCV分離物との比較についてオコモト(Okomoto )ら〔Virology、188、331〜341、1992〕が報告した値にも非常に近い。しかし、これらの研究者も、相同性を計算した方法を述べなかった。HGBVポリタンパク配列と各HCV遺伝子型との比較は、HGBVによってコードされたポリタンパクの配列は、HCVポリタンパク(表28)の何れのものに対しても33%以下の相同性しか示さないことを明らかにしている。ヌクレオチド配列(表29)の比較により、任意のHGBVと任意のHCV分離物との間では最大で44.2%の相同性があり、これに対し、HCVの分離物の間では最小でもヌクレオチド配列の相同性が64.9%である。したがって、HGBV−A、BおよびCとHCVとの間では、そのヌクレオチドと予想されるアミノ酸配列の相同性が、既知のHCV遺伝子型について確立された相同性の範囲から大きく外れたものであるため、HGBVウイルスはC型肝炎ウイルスの遺伝子型であると考えることはできない。
C型肝炎ウイルスとGB型肝炎のウイルスとの間の関係は、それらのアラインされたヌクレオチド、推定されたアミノ酸配列(即ち、大きな開放読み枠)、またはこれらの配列の一部について系統的分析を行なうことによって調べることができる。この方法をC型肝炎ウイルスに適用したところ、HCV分離物の可変性により配列の6つの均等に発散した主グループを描写することが示された〔シモンズ・ピー(Simmonds, P )ら、J.Gen.Virol.(1993年)74、2391〜2399およびシモンズ・ピー(Simmonds, P) ら、J.Gen.Virol.(1994年)75、1053〜1061〕。この分析の結果、C型肝炎ウイルスのための命名法が確立された〔シモンズ・ピー(Simmonds, P )ら、Hepatology、19、1321〜1324(1994年)〕、この命名法では、分離物を配列間の進化論的隔たりに基づいて遺伝子型に分類している。
GB型肝炎ウイルスとC型肝炎ウイルスとの間の系統的関係を決定するために、NS3の推定ヘリカーゼ遺伝子およびNS5Bの推定RNA−依存RNA−ポリメラーゼ(RdRp)内のアミノ酸配列のアライメントを行なった。また、Flaviviridae内の他のウイルスからの関連する配列、およびヘリカーゼ遺伝子およびポリメラーゼ遺伝子内においてFlaviviridaeのメンバーに対する進化論的近親性を有することが示されているウイルスもアライメントに含めた〔クーニン・イー・ヴイ(Koonin, E. V.) およびドルジャ・ブイ・ヴイ(Dolja, V. V )(1993年)、Crit.Rev.Biochem.Mol.Biol.28、375〜430、およびクーニン・イー・ヴイ(Koonin, E. V.) (1991年)、J.Gen.Virol.72、2179〜2206〕。
アミノ酸配列のアライメントは、ウイスコンシン配列分析パッケージ(バージョン8)のプログラムPILEUPを用いて行なった。アラインされた一対の配列の間の系統的な距離は、ジェイ・フェルセンスタイン(J. Felsenstein)〔フェルセンスタイン・ジェイ(1989年)、Cladistics 5、164〜166〕のご好意により提供を受けたPHYLIPパッケージ(バージョン3.5c、1993年)のPROTDISTプログラムを用いて決定した。この計算した距離は、プログラムNEIGHBOR(隣接結合設定)を使用して系統図 (phylogenetic trees)を組み立てるのに使用した。プログラムDRAWTREEを用いて系統図をプロットした。使用したウイルスの配列とそれらに対応するゲンバンク登録番号を表30に示す。ヘリカーゼ、RdRp、または完全な大きな開放読み枠内で行なったアライメントのための各HCV遺伝子型と各HGBVウイルスとの間の進化距離を、表31、32および33にそれぞれ示す。HCV遺伝子型と、HGBVウイルスおよび表30に列挙した他のウイルスとの間で計算した距離は示していない。ヘリカーゼ、RdRp、または完全な大きな開放読み枠配列のアミノ酸アライメントについて作られた系統図を図42、43および44にそれぞれ示す。
HGBV−A、BおよびCの、NS5B領域内でコードされた、推定RdRpと、幾つかのHCV遺伝子型のRdRp、伝染性ウイルス二種、幾つかの代表的なフラヴィウイルス、および幾つかのポジティプストランドのRNA植物ウイルスとの間でのアミノ酸配列のアライメントは、それらがポジティプストランドRNAウイルスのRdRpに関連する保存された配列の特色を有することを示した(データは示していない)。類似の分析に基づき、HGBV−AとHGBV−Bでコードされたヘリカーゼは、これらのポジティプストランドのRNAウイルスのヘリカーゼと著しい相同性を示す(データは示していない)。例外は、ヘリカーゼ遺伝子を保有していないと推定されるCARMV、TCV、およびMNSVである〔グイレー・エッチ(Guilley, H)ら、(1985年)Nucleic Acids Res.13、6663〜6677〕。これらの結果は、以前の系統的分析によって示された、これらのウイルスのヘリカーゼとRdRp遺伝子のスーパーグループへの関連に鑑みると予期できない結果ではなかった〔クーニン・イー・ヴイ(Koonin, E. V. )およびドルジャ・ブイ・ヴイ(Dolja, V. V )(1993年)、Crit.Rev.Biochem.Mol.Biol.28、375〜430〕。しかし、ヘリカーゼまたはRdRp配列(表31および33)の配列に基づくHGBV分離物とHCV分離物の間の系統的距離の調査は、これら2群の間には相当な隔たりがあることを示している。計算された隔たりは、HCV遺伝子型の間における近い関係を示し、2つの遺伝子型の間での最大距離は0.3696(RdRp距離)である。しかし、HGBV−A、−B、または−CとHCV群の任意のメンバーとの間のRdRpから計算された距離は、0.96042〜1.46261である。同様に、任意の2つのHCV遺伝子型についてのヘリカーゼアライメントから計算された値は0.044555〜0.19706の範囲であるのに対し、HCV群の任意のメンバーとHGBV−A、−B、または−Cとの間の距離は0.69130〜0.87120の範囲である。また、HCV遺伝子型とGBウイルスの完全な大きな開放読み枠の予想されるアミノ酸配列のアライメントは、HCV分離物の進化論的隔たりの範囲は狭く(0.17918〜0.39646)、一方、任意のGBウイルスと任意のHCV分離物の間の距離は最小でも1.68650であることを示す。したがって、GB肝炎ウイルスは、C型肝炎ウイルスの遺伝子型が示す範囲から外れた進化論的隔たりを示す。
HGBVおよびHCVの配列の系統的分析は、“HCV配列からのHGBV配列のダイバージェンスをHCV配列の間でのダイバージェンスとどの様にして比較できるか?特に、HCV配列相互のダイバージェンスよりもHCV配列からのHGBV配列のダイバージェンスの方が少ないのであろうか?”という疑問に答えようとするものである。もしHCV配列相互のダイバージェンスよりもHGBV配列のHCV配列からのダイバージェンスが小さいとしたら、満足しなければならない条件は、HGBV−A、HGBV−B、および/またはHGBV−Cの配列が、HCV配列の最も距離的に関連する対と同じ程度に、HCV配列の少なくとも一つと接近していることである(即ち、任意のHGBV配列から任意にHCV配列までの最小距離は、HCV配列の間で観察される最大値と等しいか小さい)。現在の配列データはこの条件を満たしていない。表31(RbRpアライメント)においては、最小HCV−HGBV距離は、最大HCV−HCVの距離の2.83倍であり、表32においては、最小HCV−HGBV距離は、最大HCV−HCVの距離の3.51倍である。したがって、データは、HGBV配列は、前に特徴づけしたHCV群のメンバーによってダイバージェンスが制限された群のメンバーであるとする着想を支持するものではない。
これらの相対距離は、ブートストラップに基づく試験を用いて調べることができる〔エフロン・ビー(Efron, B.)(1982年)、“ジャックナイフ、ブートストラップ、および他の再サンプリング計画”、Society Industrial and Applied Mathematics、フィラデルフィヤ、エルトン・ビー(Elton, B.) およびゴング・ジー(Gong, G.)、1983年)“ジャックナイフ、ブートストラップ、およびクロスバリデーションの楽しい研究”Am.Stat.37、36〜48〕。ブートストラップサンプリングから得られた結果を表32に示す。この表は、HCV−HGBVのダイバージェンス(全てのHCV−HGBV距離の内の最小値)のHCVダイバージェンス(全てのHCV−HCV距離の内の最大値)に対する比較を示すもので、PROTDISTプログラムを用いて計算したPAM距離に基づいている。配列アライメントにおけるカラムの1000のブートストラップ再サンプリングにおいて、HCV間の最大のダイバージェンスは、HCV配列からのHGBV配列のダイバージェンスの最小値を越えることは無かった(表34)。したがって、2つの分離した遺伝子からのコード領域のアライメントに基づく個々の測定においては、HCV遺伝子型の遺伝子配列のダイバージェンスの範囲内にHGBVの配列が入るという例は一つもない。慎重気味に推測するとしても、HGBVのためのデータを、HCV配列と同じ配列プールから引き出せる可能性は 100,000に一つ未満である。
本研究で利用したHCV配列が、HCV遺伝子型の最も大きくダイバージェンスしたものを代表していると仮定すると、これらの結果は、HGBV−A、B、およびCがHCVの遺伝子型でないことを示している。また、HGBV−AとHGBV−Cの間の関係は、それらのHGBV−Bに対する関係に比べより近いようであり、これは、HGBV−AとHGBV−Cが異なったウイルスの系列を代表するものであることを示唆している。同様にHGBV−Bは、その独自のウイルス系列を代表する唯一のものである。分析されたウイルス配列の間の相対旋回距離は、図45および46に示した根のない系統図を調べることによって容易に明らかになる。系統図において枝の長さは進化論的距離に比例している。HCVウイルスが近い進化論的関係を持つことは明らかであり、分析を行なうにあたりコード化されたゲノム配列の一部を使用するか、ゲノムの全体を使用するかを選択することと一致している(図44)。HGBV−A、HGBV−B、およびHGBV−Cと、Flaviviridaeの他のメンバーとの間のダイバージェンスの程度が大きいことは、C型肝炎ウイルスに最も緊密に関係しているものの、GB作用物質はHCVの遺伝子型とは考えることはできず、実際にはフラヴィウイルス科内の新しいグループを代表するものであろうことを示している。
本発明は、試験試料中におけるHGBV−A、HGBV−B、およびHGBV−Cの存在を決定するための試薬と方法を提供するものである。本明細書に記載したHGBV−A、HGBV−B、およびHGBV−Cに特異的なポリヌクレオチドまたはポリペプチド(またはその断片)、またはこれらのポリペプチドおよびポリヌクレオチドから産生される抗体は、一般に使用されているアッセイ用試薬と組み合わせることができ、また上記のウイルスに対する抗体を検出するための本アッセイ手順に組み込むことができ、これは本発明の範囲内と考えられるものである。あるいは、本明細書に記載したHGBV−A、HGBV−B、およびHGBV−Cに特異的なポリヌクレオチドまたはポリペプチド(またはその断片)、またはこれらのポリペプチドおよびポリヌクレオチド(またはその断片)から産生される抗体は、HGBV−A、HGBV−B、およびHGBV−Cウイルスを検出するためにそれぞれ別個に使用できる。
本発明の他の用途または変種は、本明細書の開示を検討すれば当業者にとって明らかなもである。したがって、本発明は添付の請求の範囲のみによって限定されることを意図するものである。
Claims (14)
- 配列番号397のアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムによってコードされるアミノ酸配列からなる肝炎GBウイルス(HGBV)のポリペプチド。
- 前記ポリペプチドが精製ポリペプチド、組換えポリペプチド又は合成ポリペプチドである、請求項1に記載のHGBVポリペプチド。
- 配列番号397のアミノ酸配列を含むポリプロテインをコードする読取り枠(ORF)を含む正鎖RNAゲノムによってコードされる肝炎GBウイルスのポリペプチドからなる診断用試薬。
- 肝炎GBウイルス(HGBV)抗原の少なくとも1つのエピトープに特異的に結合する精製抗体であって、前記エピトープが少なくとも5つのアミノ酸からなり、前記HGBV抗原が、(a)配列番号397を含むポリプロテインをコードする正鎖RNAウイルスゲノムによってコードされており、且つ(b)HGBV−AまたはHGBV−Cに特異的に結合する抗体とは免疫反応性でないことを特徴とする、前記精製抗体。
- 前記抗体がポリクローナル抗体またはモノクローナル抗体である、請求項4に記載の抗体。
- シグナル生成化合物に結合している、請求項4または5に記載の抗体。
- 検査試料中、肝炎GBウイルス(HGBV)の抗体の存在を検出するためのアッセイキットであって、少なくとも1つのHGBV抗原を有するポリペプチドの容器を含み、前記HGBV抗原が、(a)配列番号397を含むポリプロテインをコードする正鎖RNAウイルスゲノムによってコードされ、且つ(b)HGBV−AまたはHGBV−Cに特異的に結合する抗体とは免疫反応性でないことを特徴とする、前記アッセイキット。
- 肝炎GBウイルス(HGBV)の抗体を含む疑いのある検査試料中の同抗体を検出する方法であって、
(a)抗原/抗体複合体を形成するのに十分な時間及び条件下に、前記検査試料を、(i)少なくとも5つのアミノ酸からなる少なくとも1つのHGBVエピトープを含み、(ii)配列番号397を含むポリプロテインをコードする正鎖RNAウイルスゲノムによってコードされ、且つ(iii)HGBV−AまたはHGBV−Cに特異的に結合する抗体とは免疫反応性でないポリペプチドに接触させ、
(b)HGBVの抗体を含む前記複合体を検出する
ことからなる、前記抗体の検出方法。 - 前記ポリペプチドが組換え技術または合成手段により製造される、請求項8に記載の方法。
- ステップ(b)において、前記複合体を検出する前に、前記複合体を指示薬に接触させることを特徴とする、請求項8に記載の方法。
- 前記指示薬がシグナル生成化合物である、請求項10に記載の方法。
- 肝炎GBウイルス(HGBV)を含む疑いのある検査試料中のHGBV抗原を検出する方法であって、
(a)抗原/抗体複合体を形成するのに十分な時間及び条件下に、前記検査試料を、HGBV抗原の少なくとも5つのアミノ酸からなる少なくとも1つのエピトープに特異的に結合する抗体またはそのフラッグメントに接触させ、
(b)(i)配列番号397を含むポリプロテインをコードする正鎖RNAウイルスゲノムによってコードされ、且つ(ii)HGBV−AまたはHGBV−Cに特異的に結合する抗体とは免疫反応性でない前記HGBV抗原を含む前記複合体を検査試料中に検出する
ことからなる、前記抗原の検出方法。 - ステップ(b)において、前記複合体を検出する前に、前記複合体を指示薬に接触させてインキュベートすることを特徴とする、請求項12に記載の方法。
- 前記指示薬がシグナル生成化合物である、請求項13に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US19603094A | 1994-02-14 | 1994-02-14 | |
| US24265494A | 1994-05-13 | 1994-05-13 | |
| US28331494A | 1994-07-29 | 1994-07-29 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP52144195A Division JP3580822B2 (ja) | 1994-02-14 | 1995-02-14 | 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005041862A JP2005041862A (ja) | 2005-02-17 |
| JP3848663B2 true JP3848663B2 (ja) | 2006-11-22 |
Family
ID=27393563
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP52144195A Expired - Fee Related JP3580822B2 (ja) | 1994-02-14 | 1995-02-14 | 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 |
| JP10111629A Pending JPH10337193A (ja) | 1994-02-14 | 1998-03-31 | 肝炎gbウイルスエピトープに対する抗体及びその使用 |
| JP2004176507A Expired - Fee Related JP3848663B2 (ja) | 1994-02-14 | 2004-06-15 | 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP52144195A Expired - Fee Related JP3580822B2 (ja) | 1994-02-14 | 1995-02-14 | 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 |
| JP10111629A Pending JPH10337193A (ja) | 1994-02-14 | 1998-03-31 | 肝炎gbウイルスエピトープに対する抗体及びその使用 |
Country Status (1)
| Country | Link |
|---|---|
| JP (3) | JP3580822B2 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001278894A (ja) * | 2000-03-29 | 2001-10-10 | Osamu Ozawa | 血液循環障害予防および/または治療用医薬用組成物並びにその有効成分であるペプチド |
| EP1337849B1 (en) * | 2000-11-30 | 2009-11-18 | Boston Probes, Inc. | Methods and compositions for sorting and/or determining microorganisms |
| GB0806801D0 (en) * | 2008-04-15 | 2008-05-14 | Immunodiagnostic Systems Ltd | Substrate |
| CN111190001B (zh) * | 2020-01-10 | 2024-03-26 | 苏州睿瀛生物技术有限公司 | 免疫组化抗体专用稀释液及其制备方法 |
-
1995
- 1995-02-14 JP JP52144195A patent/JP3580822B2/ja not_active Expired - Fee Related
-
1998
- 1998-03-31 JP JP10111629A patent/JPH10337193A/ja active Pending
-
2004
- 2004-06-15 JP JP2004176507A patent/JP3848663B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005041862A (ja) | 2005-02-17 |
| JP3580822B2 (ja) | 2004-10-27 |
| JPH10337193A (ja) | 1998-12-22 |
| JPH09511137A (ja) | 1997-11-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2809956B2 (ja) | Nanbvの診断用薬 | |
| JP3813168B2 (ja) | B型肝炎ウイルス突然変異体、試薬及び検出方法 | |
| JPH10503642A (ja) | G型肝炎ウイルスおよびその分子クローニング | |
| WO1994018217A1 (en) | Non-a, non-b, non-c, non-d, non-e hepatitis reagents and methods for their use | |
| WO1995021922A2 (en) | Non-a, non-b, non-c, non-d, non-e hepatitis reagents and methods for their use | |
| JP3061258B2 (ja) | 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 | |
| EP0747482A2 (en) | Hepatitis GB virus recombinant proteins and uses thereof | |
| US5843450A (en) | Hepatitis GB Virus synthetic peptides and uses thereof | |
| US6451578B1 (en) | Non-A, non-B, non-C, non-D, non-E hepatitis reagents and methods for their use | |
| US6720166B2 (en) | Non-a, non-b, non-c, non-c, non-d, non-e hepatitis reagents and methods for their use | |
| JP3848663B2 (ja) | 非a非b非c非d非e型肝炎試薬及びそれらの使用方法 | |
| DK175975B1 (da) | NANBV-diagnostika og -vacciner | |
| US6586568B1 (en) | Non-A, non-B, non-C, non-D, non-E hepatitis reagents and methods for their use | |
| US6558898B1 (en) | Non-A, non-B, non-C, non-D, non-E hepatitis reagents and methods for their use | |
| EP0745129B1 (en) | Non-a, non-b, non-c, non-d, non-e hepatitis reagents and methods for their use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20060418 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060815 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060825 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |