JP5382000B2 - オキセタニル基を有するケイ素化合物の製造方法 - Google Patents
オキセタニル基を有するケイ素化合物の製造方法 Download PDFInfo
- Publication number
- JP5382000B2 JP5382000B2 JP2010544012A JP2010544012A JP5382000B2 JP 5382000 B2 JP5382000 B2 JP 5382000B2 JP 2010544012 A JP2010544012 A JP 2010544012A JP 2010544012 A JP2010544012 A JP 2010544012A JP 5382000 B2 JP5382000 B2 JP 5382000B2
- Authority
- JP
- Japan
- Prior art keywords
- silicon compound
- group
- reaction
- compound
- mol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
- C07F7/0838—Compounds with one or more Si-O-Si sequences
- C07F7/0872—Preparation and treatment thereof
- C07F7/0874—Reactions involving a bond of the Si-O-Si linkage
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Silicon Polymers (AREA)
Description
本発明は、オキセタニル基を有し、表面硬度の高い硬化物を与えることのできるケイ素化合物を、反応途中でゲル化の問題を生じることなく、高濃度で製造できる方法を提供することを課題とする。
一方、高い濃度で反応を行うことは、効率が上がり経済的であるものの、反応性シラン同士の接触確率が上がり、分子間反応が進んでゲルが生じやすくなる恐れがある。
本発明のケイ素化合物(C)の製造方法は、上記一般式(1)で表されるケイ素化合物(A)と上記一般式(2)で表されるケイ素化合物(B)を、1−プロパノール中でアルコール交換反応させる第一工程と、上記反応させて得られた組成物をアルカリ性条件下で加水分解共重縮合させる第二工程とを備えることを特徴とする。
本発明で得られるケイ素化合物(C)は、ケイ素化合物(A)、第1工程によりケイ素化合物(A)から生じるケイ素化合物(APと呼ぶ)、ケイ素化合物(B)及び第1工程によりケイ素化合物(B)から生じるケイ素化合物(BPと呼ぶ)との加水分解共重縮合によって生成するものである。以下に説明する第二工程により、A,B,AP,BPに含まれるシロキサン結合生成基、加水分解性基及びn−プロポキシ基の大部分はシロキサン結合に転化されるので、ケイ素化合物(C)は、シロキサン結合生成基、加水分解性基及びn−プロポキシ基が加水分解して形成された三次元のシロキサン結合(Si−O−Si)からなるポリシロキサンであるということができる。
上記の場合、ケイ素化合物(C)は、部分的にラダー(はしご)状、かご状又はランダム状の構造をとることができる。
さらに、残存するアルコキシ基の種類としては、残存アルコキシの中で少なくともプロポキシ基などの炭素数3以上のアルコキシ基が50モル%以上含まれることが好ましく、より好ましくは70モル%以上であり、さらに好ましくは90モル%以上含まれることが好ましい。炭素数2以下のメトキシ基やエトキシ基は反応性が高く、保存中に架橋反応を起こして保存安定性が低下する恐れがあるからである。
本発明におけるケイ素化合物(A)は、下記一般式(1)で表される化合物である。
SiX4 (1)
〔一般式(1)において、Xはシロキサン結合生成基であり、Xは同一であっても異なっても良い。〕
ケイ素化合物(A)はシロキサン結合生成基であるXを4個有するもの(Qモノマーとも呼ばれる。)であり、このシロキサン結合生成基は、ケイ素化合物(B)の加水分解性基との反応により、シロキサン結合を生成する。その結果得られるケイ素化合物(C)は、1つ以上の有機基を有するケイ素化合物を縮合させた場合にくらべて、SiやOといった無機成分の質量組成が大きくなり、結果的に耐熱性や硬度といった無機的な特徴をより強く示すようになるため、Qモノマーは得られる縮合物の無機部分を大きくする原料成分であるということができる。
上記シクロアルコキシ基としては、例えばシクロペンチルオキシ基及びシクロヘキシルオキシ基等が挙げられる。アラルキルオキシ基の例としては、ベンジルオキシ基、2−フェニルエチルオキシ基等が挙げられる。アリールオキシ基としては、例えばフェニルオキシ基、o−トルイルオキシ基、m−トルイルオキシ基、p−トルイルオキシ基及びナフチルオキシ基等が挙げられる。これらのうち、加水分解性が良好であることからアルコキシ基が好ましく、炭素数1〜3のアルコキシ基がより好ましく、原料入手が容易であり安価であること、並びに加水分解反応が制御しやすいことから、メトキシ基が更に好ましい。これらは1種のみ用いてもよく、2種以上を混合して用いてもよい。上記ケイ素化合物(A)として好ましい化合物は、テトラメトキシシラン、テトラエトキシシランおよびテトラn−プロポキシシランである。更に、入手が容易であることから、より好ましくはテトラメトキシシランである。
尚、シロキサン結合生成基の少なくとも一部は、後述のアルコール交換反応により、n−プロポキシ基に交換される。
本発明におけるケイ素化合物(B)は、オキセタニル基を有する有機基を有するものであり、本発明の製造方法により得られるケイ素化合物(C)にカチオン硬化性を付与するための成分であり、下記一般式(2)で表される化合物である。
〔式(2)において、R0はオキセタニル基を有する有機基であり、R0は同一であっても異なっても良く、Rは炭素数1〜6のアルキル基、炭素数7〜10のアラルキル基、炭素数6〜10のアリール基、またはオキセタニル基を有する有機基であり、Rは同一であっても異なっても良く、Yは加水分解性基であり、Yは同一であっても異なってもよく、nは0または1である。〕
更に、無機部分の割合量と溶剤への溶解性とのバランスから、nが0のケイ素化合物とnが1のケイ素化合物とを併用することができる。併用する場合、nが0のケイ素化合物とnが1のケイ素化合物との配合割合は、得られるケイ素化合物(C)を用いる用途により適宜選択される。本発明において好ましいのは、平均値でn=0〜0.5、さらに好ましくはn=0〜0.3の範囲となるものである。
本発明の第一工程は、上記一般式(1)で表されるケイ素化合物(A)と上記一般式(2)で表されるケイ素化合物(B)を、1−プロパノール中でアルコール交換反応させる反応工程である。第一工程において用いる有機ケイ素化合物(A)は1種のみでもよく、2種以上であってもよい。同様に、ケイ素化合物(B)は1種のみでもよく、2種以上であってもよい。
また、上記ケイ素化合物(B)が有する加水分解性基と1−プロパノールとのアルコール交換反応により、ケイ素化合物(B)は、上記1−プロパノール由来のn−プロポキシ基を含むものとなる。
この第一工程によりn−プロポキシ基を有する有機ケイ素化合物(A)の反応物(以下、「ケイ素化合物(AP)」という)と、n−プロポキシ基を有する有機ケイ素化合物(B)の反応物(以下、「ケイ素化合物(BP)」という)が製造される。
上記ケイ素化合物(AP)は、上記ケイ素化合物(A)が有する4個のシロキサン結合生成基のうち、少なくとも1個がn−プロポキシ基に交換されているものである。
また、上記ケイ素化合物(BP)は、上記ケイ素化合物(B)が有する2個または3個の加水分解性基のうち、少なくとも1個がn−プロポキシ基に交換されているものである。
また、第一工程のアルコールとしてメタノールやエタノールを用いた場合、得られる反応物は、1−プロパノールを用いた場合に得られる反応物よりも反応性が比較的高いメトキシ基やエトキシ基を有することとなり、得られた化合物(AP)と(BP)との加水分解共重縮合反応が均一に進行せずゲルが生じたり、安定性の低いものが得られるたりする恐れがある。
また、反応時間は5分〜30時間が好ましく、さらに好ましくは10分〜24時間、より好ましくは15分〜24時間である。
上記第一工程がアルカリ性条件下である場合、反応液のpHは7を超える値である。その場合、反応液のpHは、好ましくは8以上であり13以下である。さらに好ましくはpHが9以上である。
上記アルカリ剤としては、例えば、アンモニア、有機アミン類、水酸化テトラメチルアンモニウム、水酸化テトラエチルアンモニウム、水酸化テトラプロピルアンモニウム、水酸化テトラブチルアンモニウム、コリン、水酸化ナトリウム、水酸化カリウム及び水酸化カルシウム等が挙げられる。これらのうち、触媒活性の良好な第4級窒素原子を有するアンモニウム化合物が好ましく、更に入手が容易な水酸化テトラメチルアンモニウムがより好ましい。これらは1種のみ用いてもよく、2種以上を混合して用いてもよい。
上記第二工程は、上記第一工程により得られた組成物に、アルカリ性条件下で水を加え、加水分解共重縮合させる工程である。
上記第一工程により得られた組成物は、第一工程における反応後の反応溶液をそのまま用いることができ、上記組成物には、アルコール交換反応により得られたn−プロポキシ基を有するケイ素化合物(AP)及びケイ素化合物(BP)が含有される。
上記アルカリ剤としては、例えば、アンモニア、有機アミン類、水酸化テトラメチルアンモニウム、水酸化テトラエチルアンモニウム、水酸化テトラプロピルアンモニウム、水酸化テトラブチルアンモニウム、コリン、水酸化ナトリウム、水酸化カリウム及び水酸化カルシウム等が挙げられる。これらのうち、触媒活性の良好な第4級窒素原子を有するアンモニウム化合物が好ましく、更に入手が容易な水酸化テトラメチルアンモニウムがより好ましい。これらは1種のみ用いてもよく、2種以上を混合して用いてもよい。
また、中性条件下(pH7付近)では、加水分解共重縮合反応が進行し難い。
すなわち、アルカリ性条件下(pH7超)で有機ケイ素化合物(AP)と有機ケイ素化合物(BP)との加水分解共重縮合を進行させることによって保存安定性の高いケイ素化合物を得ることができる。
上記の適当な有機溶媒としては、メチルアルコール、エチルアルコール、イソプロピルアルコール等のアルコール類、アセトン、メチルエチルケトン等のケトン類、テトラヒドロフラン、1,4−ジオキサン等のエーテル類等が挙げられる。有機溶剤は2種類以上が併用されてもよい。アルコール類は、原料および生成物の溶解性が良好であり好ましい有機溶媒である。さらに、反応溶媒として使用している溶媒を用いて希釈することが好ましく、1−プロパノールが最も好ましい。
また、第二工程における反応温度は高いほど反応が速く進むが、低い方が副反応を抑えることができるので好ましい。0〜120℃が好ましく、さらに好ましくは10〜100℃であり、より好ましくは40〜100℃、特に好ましくは40〜80℃である。第二工程の反応時間としては30分〜30時間が好ましく、さらに好ましくは30分〜10時間、より好ましくは1〜8時間である。
また、本発明のケイ素化合物の製造方法において、上記第二工程の後、更に、中和工程、揮発成分除去工程(1)、溶解工程、洗浄工程及び揮発成分除去工程(2)を備えるのが好ましい。
反応溶媒として使用された有機溶剤が水と混和しないものであり、中間処理物の水による洗浄に適した有機溶剤である場合、またはアルコール等の水と混和する溶媒であったとしても、中間処理物の水による洗浄に適した有機溶剤を多量に追加することで洗浄工程を行うことが可能な場合には、この揮発成分除去工程(1)及び後述の溶解工程は省略することができるが、それでも揮発成分除去工程(1)を行うことは経済的に好ましい。
洗浄工程における、水と中間処理物とを混合し、水と中間処理物とを接触させる工程、及び水層と有機層(中間処理物)とを分離させる工程での温度は特に限定されないが、0〜70℃が好ましく、10〜60℃がより好ましく、分離時間が短縮される40〜60℃が更に好ましい。
ケイ素化合物(C)を単離することなく、洗浄用有機溶剤がそのままケイ素化合物(C)の溶剤として使用される場合には、揮発成分除去工程(2)は省略することができる。
(実施例1:ケイ素化合物C1の製造)
攪拌機および温度計を備えた反応器に、下記式(4)で表される3−エチル−3−((3−(トリメトキシシリル)プロポキシ)メチル)オキセタン(生成するケイ素化合物(C)において、T構造単位を与えるモノマーの一種であり、以下、「TMSOX」という)1.1kg(4.01mol)、テトラメトキシシラン(Q構造単位を与えるモノマーの一種であり、以下、「TMOS」という)1.1kg(7.24mol)と1−プロパノール1.1kgを仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液0.29kg(メタノール6.8mol、水酸化テトラメチルアンモニウム0.8mol)を徐々に加えた。60℃で1時間反応させた後、反応液を攪拌しながら水750g(41mol)と1−プロパノール750gの混合液を0.5時間かけて滴下した。滴下時間も含めて60℃で6時間反応させたあと、反応液に硝酸を加えて中和した。減圧下で有機溶剤と水を留去して、得られた残さをPGMEAに溶解させ、水洗を行うことで塩類や過剰の酸を除去した。得られたPGMEA溶液から減圧下で溶剤を留去し、無色の固体(化合物C1)を得た。収量1.2kg。仕込み原料の量から算出される質量収率は91%であった。
1H NMR分析は、ケイ素化合物C1の1gおよび内部標準物質であるヘキサメチルジシロキサン(以下、「HMDSO」という)の100mgを、それぞれ精秤して混合し、HMDSOのプロトンのシグナル強度を基準として定量計算を行った。この1H NMR分析により、ケイ素化合物(AP)、即ち、TMSOXに由来する構造単位(Tモノマー単位)の含有量および化合物C1のアルコキシ基の含有量を求め、これらを基にしてケイ素化合物(BP)、即ち、TMOSに由来する構造単位(Qモノマー単位)の含有量を計算した。その結果、得られた有機ケイ素化合物C1は、ケイ素化合物(AP)およびケイ素化合物(BP)が化学量論的に反応して得られた共重縮合物であることが確認された。
また、得られたケイ素化合物のMnは3,200、Mwは350,000であり、保持時間6分から10分付近の高分子領域にピークは見られなかった。Mw/Mnは108と算出され、上記ケイ素化合物C1において、無機部分の割合は50%であった。
実施例1で得られた化合物C1を等量のPGMEAに溶解させ60℃のオイルバスに浸し、経時的に外観を観察したところ5日経過してもゲル化せず均一溶液のままであった。
攪拌機および温度計を備えた反応器に、式(4)で表されるTMSOXを27.82g(0.1mol)、TMOSを28.19g(0.185mol)と1−プロパノール111.5gを仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液7.29g(メタノール0.17mol、水酸化テトラメチルアンモニウム20mmol)を徐々に加えた。60℃で1時間反応させた後、水18.72g(1.04mmol)と1−プロパノール20.0gの混合液を0.5時間かけて滴下した。滴下時間も含めて60℃で6時間反応させたあと、硝酸を加えて中和した。減圧下で有機溶剤と水を留去して、得られた残さをPGMEAに溶解させ、水洗を行うことで塩類や過剰の酸を除去した。得られたPGMEA溶液から減圧下で溶剤を留去し、無色の固体(化合物C2)を得た。収量28.8g。質量収率90%。
また、この有機ケイ素化合物C2についても、実施例1と同様にした1H NMR分析により、TMSOXとTMOSとが化学量論的に反応して得られた共重縮合物であることが確認された。
ケイ素化合物C2の1H NMRチャートから算出したアルコキシ基(ケイ素原子に結合したメトキシ基)の含有量は、仕込み原料に含まれていたアルコキシ基の全体に対して0.9%に相当する量であった。
得られたケイ素化合物のMnは3,500、Mwは400,000であり、保持時間6分から10分付近の高分子領域にピークは見られなかった。MW/Mnは114と算出された。
実施例2で得られた化合物C2を等量のPGMEAに溶解させ60℃のオイルバスに浸し、経時的に外観を観察したところ5日経過後には、ゲル化せず均一溶液のままであった。
攪拌機および温度計を備えた反応器に、式(4)で表されるTMSOXを27.82g(0.1mol)、TMOSを28.19g(0.185mol)と1−プロパノール111.5gを仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液7.29g(メタノール0.17mol、水酸化テトラメチルアンモニウム20mmol)を徐々に加えた。20℃で1時間反応させた後、水18.72g(1.04mmol)と1−プロパノール20.0gの混合液を0.5時間かけて滴下した。滴下時間も含めて20℃で6時間反応させたあと、硝酸を加えて中和した。減圧下で有機溶剤と水を留去して、得られた残さをPGMEAに溶解させ、水洗を行うことで塩類や過剰の酸を除去した。得られたPGMEA溶液から減圧下で溶剤を留去し、無色の固体(化合物C2)を得た。収量27.2g。質量収率85%。
また、この有機ケイ素化合物C2についても、実施例1と同様にした1H NMR分析により、TMSOXとTMOSとが化学量論的に反応して得られた共重縮合物であることが確認された。
ケイ素化合物C3の1H NMRチャートから算出したアルコキシ基(ケイ素原子に結合したメトキシ基)の含有量は、仕込み原料に含まれていたアルコキシ基の全体に対して2.0%に相当する量であった。
得られたケイ素化合物のGPC分析では、3,800、Mwは500,000で、保持時間6分から10分付近の高分子領域にピークは見られなかった。MW/Mnは132と算出された。
実施例3で得られた化合物C3を等量のPGMEAに溶解させ60℃のオイルバスに浸し、経時的に外観を観察したところ5日経過後には、わずかに粘度上昇があったがゲル化せず均一溶液のままであった。
攪拌機および温度計を備えた反応器に、TMSOX39.2g(0.14mol)、TMOS39.6g(0.26mol)とメタノール39.3gを仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液10.4g(メタノール0.24mol、水酸化テトラメチルアンモニウム28mmol)を徐々に加えた。21℃で1時間反応させた後、水26.3g(1.46mol)と1−プロパノール26.6gの混合液を滴下したところ、滴下中にゲル化してしまった。
攪拌機および温度計を備えた反応器に、TMSOX27.73g(0.1mol)、TMOS 28.12g(0.18mol)とメタノール 27.57gを仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液7.41g(メタノール0.18mol、水酸化テトラメチルアンモニウム 20mmol)を徐々に加えた。60℃で1時間反応させた後、水 18.9g(1mol)とメタノール 18.91gを滴下し、60℃で6時間反応させた。硝酸を加えて中和した。減圧下で有機溶剤と水を留去した。得られた残さをPGMEAに溶解させ、水洗を行うことで塩類や過剰の酸を除去しようとした。
水洗途中の有機層のGPCを測定したところ、保持時間6分から10分付近の高分子領域に大きなピークが見られ、分子量40万を超える高分子量成分が多量に存在することがわかり、安定性の悪いことが推測された。表1にはこの測定結果を載せた。また、水洗中水層と有機層の分離が非常に悪く、明確に分離することはできなかったために目的物を単離することができなかった。
攪拌機および温度計を備えた反応器に、TMSOX 27.70g(0.1mol)、TMOS 28.5g(0.185mol)とエタノール27.54を仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液7.3g(メタノール0.18mol、水酸化テトラメチルアンモニウム20mmol)を徐々に加えた。60℃で1時間反応させた後、水 18.74g(1mol)とエタノール 18.7gを滴下し、60℃で6時間反応させた。硝酸を加えて中和した。減圧下で有機溶剤と水を留去して、得られた残渣をPGMEAに溶解させ、水洗を行うことで塩類や過剰の酸を除去した。PGMEA溶液から溶剤を減圧下で留去し、淡黄色の固体を得た。収量29.08g。収率91%。
生成物を1H NMR分析およびIR(赤外吸収スペクトル)分析し、オキセタニル基が存在することを確認した。
また、この有機ケイ素化合物C4についても、実施例1と同様にした1H NMR分析により、ケイ素化合物(A)およびケイ素化合物(B)が化学量論的に反応して得られた共重縮合物であることが確認された。
ケイ素化合物C4の1H NMRチャートから算出したアルコキシ基(ケイ素原子に結合したメトキシ基)の含有量は、仕込み原料に含まれていたアルコキシ基の全体に対して2.6%に相当する量であった。
また、得られたケイ素化合物のMnは4,300、Mwは3,000,000と算出されたが、保持時間6分から10分付近の高分子領域に大きなピークが見られ、Mwは40万を超えており定量性の乏しい領域なので、表1にはMw>100万より大(>100万)と表記し、Mw/Mnも200より大(>200)と表記した。
比較例3で得られた化合物を等量のPGMEAに溶解させ60℃のオイルバスに浸し、経時的に外観を観察したところ翌日にはゲル化していた。残存アルコキシ基がエトキシ基のみのものは安定性が低いことがわかった。
攪拌機および温度計を備えた反応器に、TMSOX 27.74g(0.1mol)、TMOS 28.20g(0.185mol)と2−プロパノール27.54gを仕込んだ後、25%水酸化テトラメチルアンモニウムメタノール溶液7.3g(メタノール0.18mol、水酸化テトラメチルアンモニウム20mmol)を徐々に加えた。60℃で1時間反応させた後、水 18.87g(1mol)と2−プロパノール 18.88gを滴下し、60℃で6時間反応させた。硝酸を加えて中和した。減圧下で有機溶剤と水を留去して、得られた残渣をPGMEAに溶解させようとしたが、水洗を行うことで塩類や過剰の酸を除去した。PGMEA溶液から溶剤を減圧下で留去し、淡黄色の固体を得た。収量は28.73gで収率90%。
反応を終え、硝酸で中和した直後の反応液のガスクロ分析を行ったところ、シランモノマー由来のピークが残存していることがわかった。一方、反応終了後の溶液のGPC分析を行ったところ、また、得られたケイ素化合物のMnは3,300、Mwは11,000,000と算出されたが、保持時間6分から10分付近の高分子領域に大きなピークが見られ、Mwは40万を超えており定量性の乏しい領域なので、表1にはMw>100万より大(>100万)と表記し、Mw/Mnも200より大(>200)と表記した。モノマーが残る一方で、ポリマーの高分子量化が進んでいることから原料モノマーの加水分解・共重縮合反応が均一に進行していないことがわかった。
比較例4で得られたケイ素化合物を等量のPGMEAに溶解させ60℃のオイルバスに浸し、経時的に外観を観察したところ4日目にはゲル化していた。
攪拌機および温度計を備えた反応器に、TMSOX27.89g(0.1mol)、TMOS28.84g(0.185mol)と1−プロパノール27.69gを仕込んだ後、反応溶液を60℃へ昇温した。25%水酸化テトラメチルアンモニウム水溶液7.3g(水酸化テトラメチルアンモニウムとして20mmol)と水18.53g(1mol)と1−プロパノール 17.75gの混合液を滴下した。滴下を始めてすぐにゲル化した。すなわち、本発明の第一工程と第二工程を分離しない製造方法ではゲル化を抑えることはできなかった。
第一工程における「溶媒」とは、第一工程においてアルコール交換に用いた溶媒を示し、比較例5では独立した第一工程を有しないので「−」とした。第2工程における「C濃度」とは、原料モノマー(A),(B)の量から算出される、第二工程において完全に加水分解縮合したときのケイ素化合物(C)の生成質量の計算値を、第二工程における全仕込み質量で割ったときの%で表される。尚、モノマーが完全に加水分解縮合したときとは、加水分解縮合により4つのシロキサン結合生成基を持ったケイ素化合物(Qモノマー)からは、SiO2が、3つの加水分解性基を持ったケイ素化合物(Tモノマー)からはSiO1.5、2つの加水分解性基を持ったケイ素化合物(Dモノマー)からはSiO1が得られたことをいう。
上記ケイ素化合物の製造において、ゲル化することなく製造できたケイ素化合物(C)に関して、そのケイ素化合物の収率(%)及び残留アルコキシ基の含有量(%)を下記要領で測定及び算出した。
収率(%)は、(化合物Cの単離収量)/(QモノマーのアルコキシシランがすべてSiO2に変わり、TモノマーのアルコキシシランがすべてSiO1.5に、DモノマーのアルコキシシランがすべてSiO1に変わったと仮定したときの理論収量)×100によって算出した。残留アルコキシ基の含有量(%)は、1H−NMR(核磁気共鳴スペクトル)チャートから算出した。実施例1〜3の中でも、第二工程の反応温度が高かった実施例1および2では残留アルコキシ基の含有量が1%を下回っており、特に優れていた。
上記ケイ素化合物の安定性評価を行った。実施例で得られた化合物Cと比較例3、4で得られた化合物Cとをそれぞれを等重量のPGMEAに溶解させ、この溶液を5ml容量のガラス製サンプル管に2ml入れて密封し、60℃の空気恒温槽内で保管して経時的に外観を観察した。サンプル管をさかさまにしても液体が流れない状態をゲル化したと判断した。比較例3で得られた生成物は1日後、比較例4は4日後にはゲル化していたのに対し、実施例で得られた生成物は5日経ってもゲル化はしていなかった。
Mw/Mnは通常、ポリマーの多分散度と呼ばれる数字であり、全ての分子が同一である時に1になり、分子量分布が広がるほど大きくなる指標として理解されている。実施例1〜3ではこの値が200より小さかったが、比較例の内、反応中にゲル化しなかった比較例2〜4では200より大きい値になった。実施例1〜3では、第一工程を経ることにより、モノマーの反応性がバランスよく均一化したために、化合物Cの分子量も均一化したからであると考えられる。また実施例1のMw/Mnが108と、最も小さかった理由は第二工程がC濃度が25%となる仕込み濃度で、反応温度が60℃と、高温かつ高濃度の条件で反応させたために、縮合反応と同時にアルコール交換反応や加アルコール分解反応(アルコリシス反応)が活発に起き、結果的に分子量分布がより均一化された結果であると考えられる。
Claims (8)
- 下記一般式(1)で表されるケイ素化合物(A)および下記一般式(2)で表されるケイ素化合物(B)を、1−プロパノール中でアルコール交換反応させる第一工程と、ケイ素化合物(B)1モルに対して、ケイ素化合物(A)が0.3〜2.8モルの割合で、アルカリ性条件下で加水分解共重縮合させる第二工程とを含む、ケイ素化合物(C)の製造方法。
SiX4 (1)
(式(1)において、Xはシロキサン結合生成基であり、Xは同一であっても異なっていても良い。)
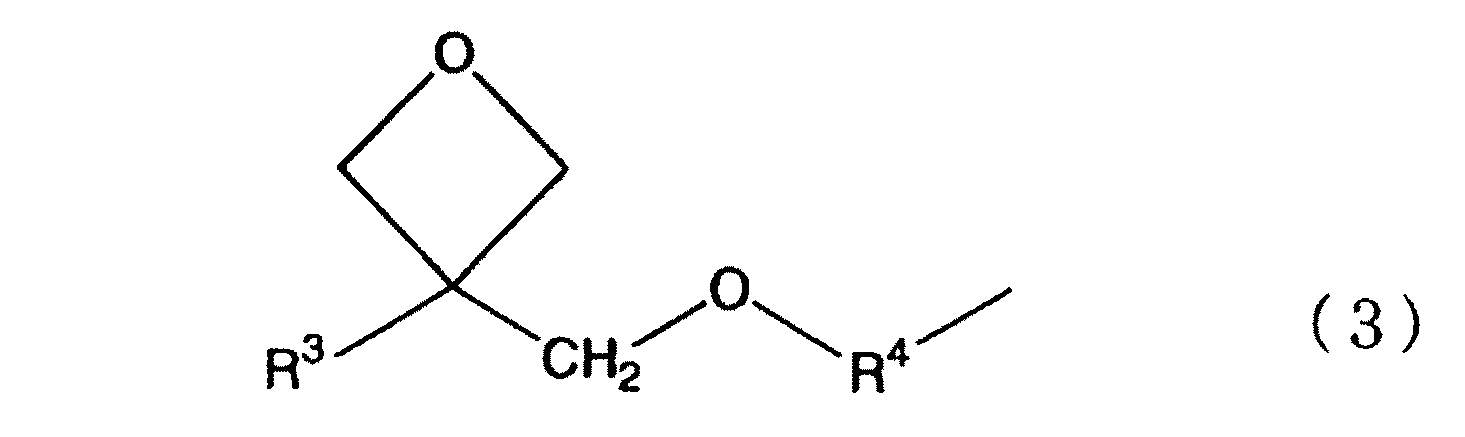
- 式(2)におけるR0が下記式(3)で表される有機基である、請求項1に記載のケイ素化合物(C)の製造方法。
- 式(2)におけるYがアルコキシ基、シクロアルコキシ基またはアリールオキシ基である、請求項1または2に記載のケイ素化合物(C)の製造方法。
- アルカリ性条件とするためのアルカリ剤として第4級窒素原子を有するアンモニウム化合物が使用される、請求項1〜3のいずれかに記載のケイ素化合物(C)の製造方法。
- アルカリ性条件とするためのアルカリ剤として水酸化テトラアルキルアンモニウムが使用される、請求項1〜4のいずれかに記載のケイ素化合物(C)の製造方法。
- ケイ素化合物(A)およびケイ素化合物(B)の合計モル数を100モルとした場合に、第二工程をアルカリ性条件とするためのアルカリ剤の使用量が1〜20モルである、請求項1〜5のいずれかに記載のケイ素化合物(C)の製造方法。
- 第二工程における反応温度が40〜100℃である、請求項1〜6のいずれかに記載のケイ素化合物(C)の製造方法。
- 第二工程におけるケイ素モノマーの仕込み濃度が、生成するケイ素化合物(C)の質量濃度に換算して、10%〜40質量%である、請求項1〜7のいずれかに記載のケイ素化合物(C)の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010544012A JP5382000B2 (ja) | 2008-12-26 | 2009-12-14 | オキセタニル基を有するケイ素化合物の製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008332075 | 2008-12-26 | ||
| JP2008332075 | 2008-12-26 | ||
| JP2010544012A JP5382000B2 (ja) | 2008-12-26 | 2009-12-14 | オキセタニル基を有するケイ素化合物の製造方法 |
| PCT/JP2009/070845 WO2010073933A1 (ja) | 2008-12-26 | 2009-12-14 | オキセタニル基を有するケイ素化合物の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2010073933A1 JPWO2010073933A1 (ja) | 2012-06-14 |
| JP5382000B2 true JP5382000B2 (ja) | 2014-01-08 |
Family
ID=42287549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010544012A Active JP5382000B2 (ja) | 2008-12-26 | 2009-12-14 | オキセタニル基を有するケイ素化合物の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8110613B2 (ja) |
| EP (1) | EP2371879B1 (ja) |
| JP (1) | JP5382000B2 (ja) |
| KR (1) | KR101621576B1 (ja) |
| CN (1) | CN102264801B (ja) |
| WO (1) | WO2010073933A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI519560B (zh) | 2014-11-24 | 2016-02-01 | 財團法人工業技術研究院 | 含氧雜環丁烷基與環氧基之樹脂與樹脂組成物 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11199673A (ja) * | 1998-01-13 | 1999-07-27 | Toagosei Co Ltd | 光カチオン硬化性樹脂組成物 |
| WO2004076534A1 (ja) * | 2003-02-27 | 2004-09-10 | Toagosei Co., Ltd | カチオン硬化性含ケイ素化合物の製造方法 |
| JP2008133204A (ja) * | 2006-11-27 | 2008-06-12 | Toagosei Co Ltd | 多官能オキセタンの製造方法 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5463084A (en) * | 1992-02-18 | 1995-10-31 | Rensselaer Polytechnic Institute | Photocurable silicone oxetanes |
| US6096903A (en) * | 1997-03-25 | 2000-08-01 | Ivoclar Ag | Hydrolysable and polymerizable oxetane silanes |
| JP3598749B2 (ja) | 1997-07-07 | 2004-12-08 | 東亞合成株式会社 | 光カチオン硬化性組成物の製造方法及び光カチオン硬化性ハードコート剤組成物 |
| US6121342A (en) * | 1998-01-13 | 2000-09-19 | Toagosei Co., Ltd. | Photocationically curable compositions and process for producing the same |
| DE19860361A1 (de) * | 1998-12-24 | 2000-06-29 | Espe Dental Ag | Vernetzbare Monomere auf Cyclosiloxanbasis, deren Herstellung und deren Verwendung in polymerisierbaren Massen |
| DE19934407A1 (de) * | 1999-07-22 | 2001-01-25 | Espe Dental Ag | Hydrolysierbare und polymerisierbare Silane mit geringer Viskosität und deren Verwendung |
| TWI300516B (ja) * | 2001-07-24 | 2008-09-01 | Jsr Corp | |
| US6743510B2 (en) * | 2001-11-13 | 2004-06-01 | Sumitomo Chemical Company, Limited | Composition comprising a cationic polymerization compound and coating obtained from the same |
| EP1403953A3 (en) * | 2002-09-26 | 2009-07-29 | FUJIFILM Corporation | Organic-inorganic hybrid material, organic-inorganic hybrid proton-conductive material and fuel cell |
| AU2003304346A1 (en) * | 2003-07-22 | 2005-02-04 | Canon Kabushiki Kaisha | Ink jet head and its manufacture method |
| US7524553B2 (en) * | 2004-05-31 | 2009-04-28 | Fujifilm Corporation | Optical film, polarizing plate and image display using the same |
| WO2006013863A1 (ja) * | 2004-08-04 | 2006-02-09 | Toagosei Co., Ltd. | ポリオルガノシロキサン及びそれを含む硬化性組成物 |
| JP3976778B2 (ja) * | 2005-02-08 | 2007-09-19 | 横浜ゴム株式会社 | オキセタン化合物およびそれを含む硬化性組成物 |
| JP4872549B2 (ja) * | 2005-11-15 | 2012-02-08 | セントラル硝子株式会社 | 熱線遮蔽膜形成基材の製法 |
| KR101587297B1 (ko) * | 2008-01-15 | 2016-01-20 | 도아고세이가부시키가이샤 | 옥세타닐기를 갖는 유기 규소 화합물 및 그의 제조 방법 및 경화성 조성물 |
| JP5454762B2 (ja) * | 2008-02-06 | 2014-03-26 | 東亞合成株式会社 | カチオン硬化性組成物 |
| CN102746467B (zh) * | 2008-04-22 | 2015-01-14 | 东亚合成株式会社 | 固化性组合物以及有机硅化合物的制备方法 |
-
2009
- 2009-12-14 JP JP2010544012A patent/JP5382000B2/ja active Active
- 2009-12-14 CN CN2009801526581A patent/CN102264801B/zh not_active Expired - Fee Related
- 2009-12-14 WO PCT/JP2009/070845 patent/WO2010073933A1/ja not_active Ceased
- 2009-12-14 EP EP09834736.2A patent/EP2371879B1/en not_active Not-in-force
- 2009-12-14 US US13/128,308 patent/US8110613B2/en active Active
- 2009-12-14 KR KR1020117011861A patent/KR101621576B1/ko not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11199673A (ja) * | 1998-01-13 | 1999-07-27 | Toagosei Co Ltd | 光カチオン硬化性樹脂組成物 |
| WO2004076534A1 (ja) * | 2003-02-27 | 2004-09-10 | Toagosei Co., Ltd | カチオン硬化性含ケイ素化合物の製造方法 |
| JP2008133204A (ja) * | 2006-11-27 | 2008-06-12 | Toagosei Co Ltd | 多官能オキセタンの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2371879A1 (en) | 2011-10-05 |
| CN102264801B (zh) | 2013-05-01 |
| JPWO2010073933A1 (ja) | 2012-06-14 |
| US8110613B2 (en) | 2012-02-07 |
| EP2371879A4 (en) | 2012-08-01 |
| KR20110099223A (ko) | 2011-09-07 |
| KR101621576B1 (ko) | 2016-05-16 |
| US20110245448A1 (en) | 2011-10-06 |
| WO2010073933A1 (ja) | 2010-07-01 |
| EP2371879B1 (en) | 2013-10-02 |
| CN102264801A (zh) | 2011-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114929784B (zh) | 低硅氧烷环含量的官能化q-t-硅氧烷基聚合物材料和制备方法 | |
| CN102741326A (zh) | 新型的具有伯胺官能团的有机改性硅氧烷和新型的具有季铵官能团的有机改性硅氧烷及二者的制备方法 | |
| JPH0718077A (ja) | カルビノール・官能シロキサンの製造法 | |
| JP5990909B2 (ja) | 片末端官能基含有オルガノポリシロキサン及びその製造方法 | |
| JP3598749B2 (ja) | 光カチオン硬化性組成物の製造方法及び光カチオン硬化性ハードコート剤組成物 | |
| JP3652151B2 (ja) | 硬化性ポリメチルシルセスキオキサンの製造方法 | |
| KR100492279B1 (ko) | 경화성폴리메틸실세스키옥산조성물 | |
| JP2009167325A (ja) | シラノール基含有硬化性籠型シルセスキオキサン化合物およびこれを用いた共重合体並びにそれらの製造方法 | |
| US20070055034A1 (en) | Process for producing cation-curable silicon compound | |
| WO2005000857A1 (ja) | 有機ケイ素化合物とその製造方法、およびポリシロキサンとその製造方法 | |
| JP5382000B2 (ja) | オキセタニル基を有するケイ素化合物の製造方法 | |
| KR101877599B1 (ko) | 폴리실록산의 제조 방법 | |
| RU2428438C2 (ru) | Способ получения полиорганосилоксанов на основе органоалкоксисиланов | |
| JPH07173220A (ja) | 無機アルコキシドもしくはハロシランを有する有機ポリマーの反応生成物 | |
| JP2006274082A (ja) | ラジカル重合性基を有する有機ケイ素化合物の製造方法 | |
| JP3199598B2 (ja) | (メタ)アクリロイルオキシル基含有オルガノポリシロキサンの製造方法 | |
| JP5454761B2 (ja) | オキセタニル基を有するケイ素化合物の製造方法 | |
| US8742051B2 (en) | Method of preparing a bodied siloxane resin including M, Q, and T-propyl units and capped with additional M units | |
| TWI468444B (zh) | An organopolysiloxane compound containing a? -keto ester group | |
| TW202440741A (zh) | 有機聚矽氧烷化合物、其衍生物及該些的製造方法 | |
| JP2006299121A (ja) | (ポリ)シロキサン化合物およびその製法 | |
| JPH11106511A (ja) | 有機ケイ素化合物及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130618 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130809 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130903 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130916 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5382000 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |