JP5480131B2 - ホスフィンオキシドビタミンd前駆物質の製造方法 - Google Patents
ホスフィンオキシドビタミンd前駆物質の製造方法 Download PDFInfo
- Publication number
- JP5480131B2 JP5480131B2 JP2010509907A JP2010509907A JP5480131B2 JP 5480131 B2 JP5480131 B2 JP 5480131B2 JP 2010509907 A JP2010509907 A JP 2010509907A JP 2010509907 A JP2010509907 A JP 2010509907A JP 5480131 B2 JP5480131 B2 JP 5480131B2
- Authority
- JP
- Japan
- Prior art keywords
- ether
- compound
- hydroxide
- group
- noh
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 phosphine oxide vitamin D Chemical class 0.000 title claims description 11
- 229930003316 Vitamin D Natural products 0.000 title description 8
- QYSXJUFSXHHAJI-XFEUOLMDSA-N Vitamin D3 Natural products C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C/C=C1\C[C@@H](O)CCC1=C QYSXJUFSXHHAJI-XFEUOLMDSA-N 0.000 title description 8
- 235000019166 vitamin D Nutrition 0.000 title description 8
- 239000011710 vitamin D Substances 0.000 title description 8
- 229940046008 vitamin d Drugs 0.000 title description 8
- 238000004519 manufacturing process Methods 0.000 title description 5
- 239000002243 precursor Substances 0.000 title description 3
- 238000000034 method Methods 0.000 claims description 39
- 150000001875 compounds Chemical class 0.000 claims description 19
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 18
- 125000006239 protecting group Chemical group 0.000 claims description 16
- 239000003444 phase transfer catalyst Substances 0.000 claims description 11
- YFPJFKYCVYXDJK-UHFFFAOYSA-N Diphenylphosphine oxide Chemical compound C=1C=CC=CC=1[P+](=O)C1=CC=CC=C1 YFPJFKYCVYXDJK-UHFFFAOYSA-N 0.000 claims description 10
- 239000003960 organic solvent Substances 0.000 claims description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical group CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 8
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 8
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 239000011737 fluorine Substances 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 6
- 239000007864 aqueous solution Substances 0.000 claims description 6
- 239000000203 mixture Substances 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 5
- 150000007529 inorganic bases Chemical class 0.000 claims description 5
- 150000007530 organic bases Chemical class 0.000 claims description 5
- 239000011541 reaction mixture Substances 0.000 claims description 5
- UZKWTJUDCOPSNM-UHFFFAOYSA-N 1-ethenoxybutane Chemical compound CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 claims description 4
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 claims description 4
- 230000008569 process Effects 0.000 claims description 4
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 claims description 4
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 150000003242 quaternary ammonium salts Chemical class 0.000 claims description 3
- FGTJJHCZWOVVNH-UHFFFAOYSA-N tert-butyl-[tert-butyl(dimethyl)silyl]oxy-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)O[Si](C)(C)C(C)(C)C FGTJJHCZWOVVNH-UHFFFAOYSA-N 0.000 claims description 3
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- SKYXLDSRLNRAPS-UHFFFAOYSA-N 1,2,4-trifluoro-5-methoxybenzene Chemical compound COC1=CC(F)=C(F)C=C1F SKYXLDSRLNRAPS-UHFFFAOYSA-N 0.000 claims description 2
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 claims description 2
- YFNONBGXNFCTMM-UHFFFAOYSA-N butoxybenzene Chemical compound CCCCOC1=CC=CC=C1 YFNONBGXNFCTMM-UHFFFAOYSA-N 0.000 claims description 2
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 claims description 2
- POLCUAVZOMRGSN-UHFFFAOYSA-N dipropyl ether Chemical compound CCCOCCC POLCUAVZOMRGSN-UHFFFAOYSA-N 0.000 claims description 2
- VNKYTQGIUYNRMY-UHFFFAOYSA-N methoxypropane Chemical compound CCCOC VNKYTQGIUYNRMY-UHFFFAOYSA-N 0.000 claims description 2
- DLRJIFUOBPOJNS-UHFFFAOYSA-N phenetole Chemical compound CCOC1=CC=CC=C1 DLRJIFUOBPOJNS-UHFFFAOYSA-N 0.000 claims description 2
- AGZRBJLATOQBCH-UHFFFAOYSA-N 1-methoxy-2-(2-methoxyphenoxy)benzene Chemical compound COC1=CC=CC=C1OC1=CC=CC=C1OC AGZRBJLATOQBCH-UHFFFAOYSA-N 0.000 claims 1
- MFGOFGRYDNHJTA-UHFFFAOYSA-N 2-amino-1-(2-fluorophenyl)ethanol Chemical compound NCC(O)C1=CC=CC=C1F MFGOFGRYDNHJTA-UHFFFAOYSA-N 0.000 claims 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 claims 1
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Inorganic materials [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 claims 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 claims 1
- 239000000920 calcium hydroxide Substances 0.000 claims 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 claims 1
- 150000002170 ethers Chemical class 0.000 claims 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 claims 1
- 239000000347 magnesium hydroxide Substances 0.000 claims 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 claims 1
- 229940125782 compound 2 Drugs 0.000 description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 229940125904 compound 1 Drugs 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 229940126214 compound 3 Drugs 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 150000003710 vitamin D derivatives Chemical class 0.000 description 5
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 239000011574 phosphorus Substances 0.000 description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 3
- 150000004714 phosphonium salts Chemical class 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 2
- 239000003872 25-hydroxy-cholecalciferol Substances 0.000 description 2
- OSDWBNJEKMUWAV-UHFFFAOYSA-N Allyl chloride Chemical compound ClCC=C OSDWBNJEKMUWAV-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229910052987 metal hydride Inorganic materials 0.000 description 2
- 150000004681 metal hydrides Chemical class 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 2
- AUONHKJOIZSQGR-UHFFFAOYSA-N oxophosphane Chemical compound P=O AUONHKJOIZSQGR-UHFFFAOYSA-N 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- WGTYBPLFGIVFAS-UHFFFAOYSA-M tetramethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C WGTYBPLFGIVFAS-UHFFFAOYSA-M 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 238000006257 total synthesis reaction Methods 0.000 description 2
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 2
- PKFBWEUIKKCWEW-WEZTXPJVSA-N (1r,3r)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]cyclohexane-1,3-diol Chemical class C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1C[C@@H](O)C[C@H](O)C1 PKFBWEUIKKCWEW-WEZTXPJVSA-N 0.000 description 1
- QYSXJUFSXHHAJI-YHJXBONMSA-N (1r,3z)-3-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-6-methylheptan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexan-1-ol Chemical class C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C\C=C1\C[C@H](O)CCC1=C QYSXJUFSXHHAJI-YHJXBONMSA-N 0.000 description 1
- IZNIJEWSKRQQLT-UHFFFAOYSA-N 1-(3-chloropentan-3-yl)-2-ethylbenzene Chemical compound CCC1=CC=CC=C1C(Cl)(CC)CC IZNIJEWSKRQQLT-UHFFFAOYSA-N 0.000 description 1
- ULDSHVAMTLXGCH-UHFFFAOYSA-N 1-cyclohexylideneethanol Chemical class CC(O)=C1CCCCC1 ULDSHVAMTLXGCH-UHFFFAOYSA-N 0.000 description 1
- CXBDYQVECUFKRK-UHFFFAOYSA-N 1-methoxybutane Chemical compound CCCCOC CXBDYQVECUFKRK-UHFFFAOYSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WDZOZGBMIGOESD-UHFFFAOYSA-N CC(C)(C)C=1C(=C(C=CC1)P(C1=CC=CC=C1)(CC=C1CCCCC1)=O)O[SiH](C)C Chemical compound CC(C)(C)C=1C(=C(C=CC1)P(C1=CC=CC=C1)(CC=C1CCCCC1)=O)O[SiH](C)C WDZOZGBMIGOESD-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical group OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 1
- LVRCYPYRKNAAMX-UHFFFAOYSA-M bis(triphenylphosphine)iminium chloride Chemical compound [Cl-].C1=CC=CC=C1[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)N=P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 LVRCYPYRKNAAMX-UHFFFAOYSA-M 0.000 description 1
- 210000002449 bone cell Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 229960005084 calcitriol Drugs 0.000 description 1
- GMRQFYUYWCNGIN-NKMMMXOESA-N calcitriol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-NKMMMXOESA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- XGRJZXREYAXTGV-UHFFFAOYSA-N chlorodiphenylphosphine Chemical compound C=1C=CC=CC=1P(Cl)C1=CC=CC=C1 XGRJZXREYAXTGV-UHFFFAOYSA-N 0.000 description 1
- 150000003983 crown ethers Chemical group 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- JQDCIBMGKCMHQV-UHFFFAOYSA-M diethyl(dimethyl)azanium;hydroxide Chemical compound [OH-].CC[N+](C)(C)CC JQDCIBMGKCMHQV-UHFFFAOYSA-M 0.000 description 1
- HPYNZHMRTTWQTB-UHFFFAOYSA-N dimethylpyridine Natural products CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229960002061 ergocalciferol Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- OFXSXYCSPVKZPF-UHFFFAOYSA-N methoxyperoxymethane Chemical compound COOOC OFXSXYCSPVKZPF-UHFFFAOYSA-N 0.000 description 1
- YULMNMJFAZWLLN-UHFFFAOYSA-N methylenecyclohexane Chemical compound C=C1CCCCC1 YULMNMJFAZWLLN-UHFFFAOYSA-N 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- MPQXHAGKBWFSNV-UHFFFAOYSA-N oxidophosphanium Chemical class [PH3]=O MPQXHAGKBWFSNV-UHFFFAOYSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000000849 parathyroid Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical class CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- RKHXQBLJXBGEKF-UHFFFAOYSA-M tetrabutylphosphanium;bromide Chemical compound [Br-].CCCC[P+](CCCC)(CCCC)CCCC RKHXQBLJXBGEKF-UHFFFAOYSA-M 0.000 description 1
- 229940073455 tetraethylammonium hydroxide Drugs 0.000 description 1
- LRGJRHZIDJQFCL-UHFFFAOYSA-M tetraethylazanium;hydroxide Chemical compound [OH-].CC[N+](CC)(CC)CC LRGJRHZIDJQFCL-UHFFFAOYSA-M 0.000 description 1
- BRKFQVAOMSWFDU-UHFFFAOYSA-M tetraphenylphosphanium;bromide Chemical compound [Br-].C1=CC=CC=C1[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 BRKFQVAOMSWFDU-UHFFFAOYSA-M 0.000 description 1
- AEFPPQGZJFTXDR-UHFFFAOYSA-M tetraphenylphosphanium;iodide Chemical compound [I-].C1=CC=CC=C1[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 AEFPPQGZJFTXDR-UHFFFAOYSA-M 0.000 description 1
- USFPINLPPFWTJW-UHFFFAOYSA-N tetraphenylphosphonium Chemical compound C1=CC=CC=C1[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 USFPINLPPFWTJW-UHFFFAOYSA-N 0.000 description 1
- WAGFXJQAIZNSEQ-UHFFFAOYSA-M tetraphenylphosphonium chloride Chemical compound [Cl-].C1=CC=CC=C1[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 WAGFXJQAIZNSEQ-UHFFFAOYSA-M 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- RYVBINGWVJJDPU-UHFFFAOYSA-M tributyl(hexadecyl)phosphanium;bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[P+](CCCC)(CCCC)CCCC RYVBINGWVJJDPU-UHFFFAOYSA-M 0.000 description 1
- LZOHWIXYBMRNAP-UHFFFAOYSA-M triphenyl(trityl)phosphanium;chloride Chemical compound [Cl-].C1=CC=CC=C1C([P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 LZOHWIXYBMRNAP-UHFFFAOYSA-M 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 239000011653 vitamin D2 Substances 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/50—Organo-phosphines
- C07F9/53—Organo-phosphine oxides; Organo-phosphine thioxides
- C07F9/5316—Unsaturated acyclic phosphine oxides or thioxides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
Description
2007年5月30日に出願の米国仮出願第60/940,866号の35 U.S.C. §119(e)による恩典をここに主張し、この全体の開示内容は本願明細書に含まれるものとする。
開示の分野
本開示は、一般的には、ホスフィンオキシドビタミンD前駆物質に関する。より詳しくは、本開示は、金属水素化物塩基を用いる代わりに、脱プロトンのための塩基水溶液と相間移動触媒を用いてホスフィンオキシドビタミンD前駆物質を調製する方法に関する。
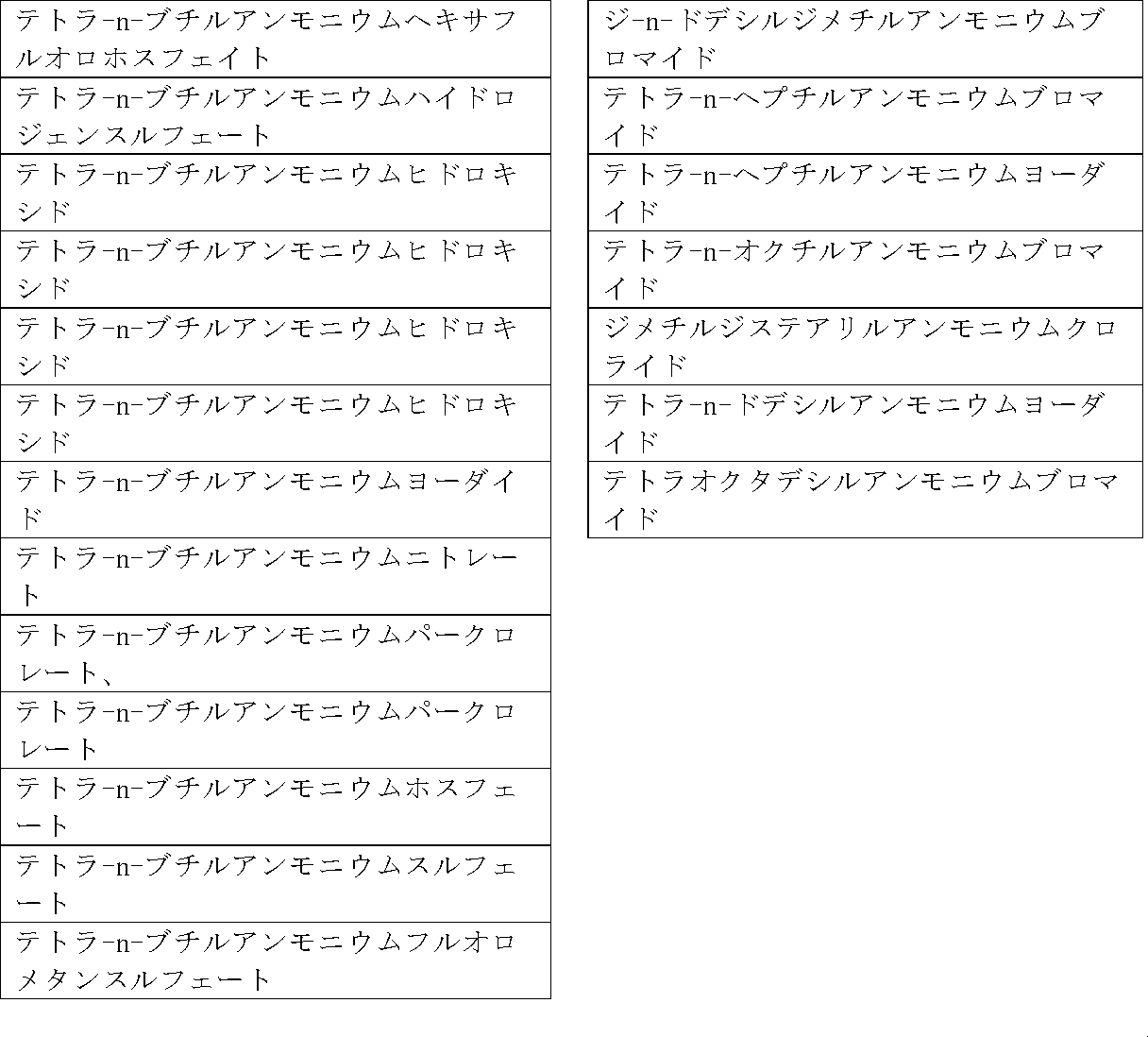
と、有機溶媒中ジフェニルホスフィンオキシド、塩基性水溶液及び相間移動触媒を含む二成分相反応混合物とを反応させて、式1の化合物を得ることを含む。
態様及び利点は、更に、以下の詳細な説明を検討することによって当業者に明らかになるであろう。本方法は実施態様が様々な形で可能であるが、以後の説明は、開示内容が説明的であるという了解の下で個々の実施態様を含み、本明細書に記載される個々の実施態様に本発明を限定するものではない。
化合物1は、下記式の化合物である:
明確にするために、波線は、以下の二つの構造の省略表現である:
化合物1と類似の化合物を調製する以前の方法は、米国特許第6,603,030号(“‘030特許”)に記載されている。‘030特許において、化合物2と類似の化合物をジフェニルホスフィンオキシドの塩と反応させて、化合物1を形成している。無水条件下、強塩基、例えば、金属水素化物がジフェニルホスフィンオキシドからプロトンを取り出して、無水溶媒中、例えば、ジメチルホルムアミド中で安定である、ジフェニルホスフィンオキシド金属塩を形成する。次に、金属塩と化合物2のハライドとをアニオン求核置換機序を経て反応させて、化合物1を形成する。
ここに開示された方法においては、水と有機溶媒の二相システムが使われている。このように、‘030特許プロセスにおいて形成されるタイプの塩は、それが水と反応することから存在させることができない。むしろ、化合物2のハロゲン化アルキルとジフェニルホスフィンオキシドとの間の求核置換反応が有機相で生じ、続いてHCl分子の脱離が塩基性部分と関連する相間移動触媒、例えば、水酸化テトラブチルアンモニウム(Bu4NOH)によって行われて、化合物2を形成する。この例において、塩基、Bu4NOHは、水相において水酸化カリウムとテトラブチルアンモニウム塩、例えば、Bu4NBrとの間の反応から形成することができ、これが有機相へ移動する。
本明細書に開示されるように、化合物1は、下記式の化合物2を、有機溶媒中のジフェニルホスフィンオキシド、塩基性水溶液及び相間移動触媒を含む二成分相反応混合物を用いて反応させて、式1の化合物を得ることによって製造される:
理論によって縛られることを望まないが、開示された方法において、化合物2の塩化アリルとジフェニルホスフィンオキシドが反応して、五価のリン中間体を形成することが仮定される。五価のリン化学の考察については、例えば、Moriarty, et al., Phosphorus, Sulfur, and Silicon, 109-100:237-240 (1996); Moriarty, et al., J. Am. Chem.. Soc., 112:8575-8577 (1990); and Moriarty, et al., J. Am. Chem. Soc., 113: 9374-9376 (1991)を参照のこと。次に、相間移動触媒は、水相から有機相まで水酸化イオンに付き添い、五価のリン中間体と反応するとともに化合物2を形成することができる。相間移動触媒の説明については、例えば、Rabinovitz, et al., Angew. Chem. Int. Ed. Engl., 25:960-970 (1986)を参照のこと。
任意の適切な反応温度を用いることができ、室温反応条件が適度な時間(例えば、約2時間)で結果を生じることが分かった。このように、好ましい範囲には、約0℃〜約40℃、約10℃〜約30℃、室温(例えば、約20℃〜約25℃)が含まれる。
水の溶解性が非常に低い相対的に無極性の溶媒は、好ましくは有機相、例えば、ジクロロメタン、トルエン、又はベンゼンに用いられる。二成分相反応混合物に好ましい有機溶媒は、環境にやさしいものである。適切な例としては、ジエチルエーテル、メトキシエーテル、エチルフェニルエーテル、メチルプロピルエーテル、エチルビニルエーテル、ハロエチルエーテル、ベンジルエーテル、ジブチルエーテル、ジプロピルエーテル、ブチルフェニルエーテル、ブチルビニルエーテル、シクロヘキシルビニルエーテル、及びt-ブチルメチルエーテルが挙げられる。環境にやさしい有機溶媒として、t-ブチルメチルエーテルが好ましい。
水相のための塩基性化合物は、有機塩基又は無機塩基であり得る。適切な無機塩基としては、LiOH、NaOH、Cs(OH)、Ca(OH)2、Mg(OH)2、Al(OH)3及び水酸化カリウムが挙げられるがこれらに限定されず、水酸化カリウムが好ましい。適切な有機塩基としては、水酸化ジエチルジメチルアンモニウム、水酸化テトラメチルアンモニウム(Me4NOH)、水酸化テトラエチルアンモニウム(Et4NOH)及び水酸化ベンジルトリメチルアンモニウム(BnMe3NOH)又は任意の水酸化テトラアルキルアンモニウム又は水酸化テトラアリールアンモニウムが挙げられるがこれらに限定されない。
反応は、相間移動触媒の存在下に行われる。四級塩やクラウンエーテルが企図される。塩化物、臭化物、硫酸水素塩、ヨウ化物、アンモニウム塩、ホスホニウム塩を含む四級塩が好ましい。四級アンモニウム塩やホスホニウム塩が好ましい。
四級アンモニウム塩としては、以下の表1に確認されるものが挙げられるがこれらに限定されない。トリエチルベンジルクロライドやテトラ-n-ブチルアンモニウムブロマイドが好ましい。
化合物2は、下記式の化合物を例えば、N-クロロスクシンイミド又はメシルクロライド/ルチジン/塩化リチウムの混合物又はトリホスゲンを用いて塩素化することによって製造することができる:
化合物2は、化合物3のアリルアルコールを化合物2のアリルクロライドに塩素化することによって化合物3から得られる。この塩素化は、ヘキサン、ジクロロメタン又はジメチルホルムアミドのような有機溶媒中で行われる。各モルの化合物3については、二分の一(1/2)モル以上のトリホスゲン、二(2)モル以上のN-クロロスクシンイミド又は二(2)モル以上のメシルクロライドと四(4)モル以上の塩化リチウムの混合物が塩素源として使用し得る。少なくとも2当量の有機塩基、好ましくはピリジンのような非プロトン性アミン塩基又は好ましくはトリエチルアミンが含まれるべきである。温度は、決定的なものではなく、-30℃〜50℃の範囲にあってもよい。約0℃の温度が好ましい。
多くの種類の化合物3が既知である。例えば、Perlman et al., Novel synthesis of 19-nor-vitamin D compounds, Tetrahedron Lett., 32(52):7663-6 (1991); Courtney et al., Asymmetric synthesis of a key ring A synthon for lα-hydroxy-19-nor vitamin D, Tetrahedron Lett., 39(21):3363-3366 (1998); Shiuey et al. Total synthesis of la-fluoro-25-hydroxycholecalciferol and -ergocalciferol., J. Org. Chem. 55(1):243-7 (1990); Reddy, Synthesis and activity of 3-epi vitamin D3 compounds for use in treatment of disorders involving aberrant activity of hyperproliferative skin, parathyroid, and bone cells, WIPO PCT Publication No. WO 98/51663; Sotojima, Preparation of cyclohexylideneethanol derivatives as intermediates for lα-hydroxy- and lα,25-dihydroxyvitamin D3; JP Kokai No. 05279283; Baggiolini et al., Stereoselective total synthesis of lα,25-dihydroxycholecalciferol, J. Am. Chem. Soc., 104(10):2945-8 (1982)を参照のこと。残りの種類の化合物3は、当該技術において既知の手順を用いてこれらの既知の化合物から製造することができる。このような製造は、当業者の技術の充分に範囲内である。
R2は、フッ素、水素、又は保護ヒドロキシル基であり得る。保護ヒドロキシ基は、酸素が環に結合し、保護基によって保護されている基である。上記のように、適切な保護基の選択は、当業者の範囲内である。例えば、適切な保護基は、Wuts et al., Greene's Protective Groups in Organic Synthesis, 4th ed., (Wiley Interscience: Hoboken, NJ) 2007に記載されている。好ましい保護ヒドロキシ基としては、シリル保護ヒドロキシ基、例えば、TBSによって保護されたヒドロキシが含まれる。TBS保護ヒドロキシ基の使用により、tert-ブチルジメチルシリルオキシド(“TBDMSO”)であるR2になる。本発明に用いられる任意の化合物について、R1及びR2は、同じか又は異なるヒドロキシ保護基を用いてもよい。好ましい方法において、R1はTBSであり、R2はフッ素又はTBDMSOである。
本発明の他の好ましい方法において、R1はTBSであり、R2はOR3であり、R3はTBSである。更に他の好ましい方法において、R1はTBSであり、R2はフッ素である。更に他の好ましい方法において、R1はTBSであり、R2は水素である。本発明において、化合物1、2及び3は、P(O)(Ph)2、Cl及びOHを、それぞれ、シス又はトランスの位置で有し得る。これらの化合物のいずれにおいても、R1及びR2は、これらが結合されているシクロヘキサン環の平面の上に
以下の実施例は、説明のために示され、本発明の範囲を制限するものではない。
3S-(3α,5β,Z)-2-2-2-メチレンビス(1,1-ジメチルエチル)ジメチルシリルオキシシクロヘキシリデンエチルジフェニルホスフィンオキシドの調製
薄層クロマトグラフィー(TLC)は、少量の出発物質(5-10%)のみを示した。反応生成物を、MTBE(50mL)と水(25mL)で希釈した。層を分離し、有機層を水(25mL)と食塩水(25mL)で洗浄し、無水Na2SO4で乾燥し、ろ過し、減圧下で濃縮した。粗製物(6.0g)をカラムクロマトグラフィ(EA:ヘキサン)で精製して、2.5g(〜50%)の所望のホスフィンオキシドを得た(HPLC純度が所望の94%)。この物質をカラムクロマトグラフィ(EA:ヘキサン)で再精製して、1.7gの純物質(1.7g、HPLC = 96.04%)を得た。1H NMRとHPLCによって同定と純度を証明した。残りの画分を集め、次のバッチに添加した。
塩基として、水酸化カリウムの代わりにそれぞれMe4NOH、Et4NOH、BnMe3NOHを用いても、肯定的な結果が得られた。
上記の説明は、理解を明らかにするためにだけ示され、本発明の範囲内の変更が当業者に明らかであり得るように、不必要な制限がそこから理解されるべきではない。
明細書全体に、方法が工程、成分、又は材料を含むように記載されている場合、特に記載されない限り、組成物が、本質的に詳述した工程、成分又は材料の任意の組み合わせからなり得るか又は詳述した工程、成分又は材料の任意の組み合わせからなり得ることも企図される。
本明細書に開示される方法、及びその個々の工程の実施は、手で及び/又は電子装置によって行われ得る。方法を具体的な実施態様によって記載してきたが、当業者は、方法と関連付けられた作用を行う他の方法が用いられてもよいことを容易に理解するであろう。例えば、特に記載されない限り、種々の工程の順序は、方法の範囲又は真意を逸脱せずに変更されてもよい。更に、個々の工程の一部が、追加の工程に合わせられ、省略され、又は更に細分され得る。
本明細書に引用したすべての特許、出版物及び参考文献の記載は、本願明細書に完全に含まれるものとする。本開示と組み込まれた特許、出版及び参考文献との間が不一致の場合には、本開示が支配すべきである。
Claims (19)
- 下記式:
の化合物の製造方法であって、下記式:
- R1が、シリル保護基である、請求項1に記載の方法。
- R1が、tert-ブチルジメチルシリル基である、請求項2に記載の方法。
- R2が、フッ素である、請求項1〜3のいずれか1項に記載の方法。
- R2が、tert-ブチルジメチルシリルオキシドである、請求項1〜3のいずれか1項に記載の方法。
- R2が、フッ素又はtert-ブチルジメチルシリルオキシドである、請求項1〜3のいずれか1項に記載の方法。
- X1とX2が一緒になってCH2である、請求項1〜6のいずれか1項に記載の方法。
- 有機溶媒が、ジエチルエーテル、メトキシフェニルエーテル、エチルフェニルエーテル、メチルプロピルエーテル、エチルビニルエーテル、ハロエチルエーテル、ベンジルエーテル、ジブチルエーテル、ジプロピルエーテル、ブチルフェニルエーテル、ブチルビニルエーテル、シクロヘキシルビニルエーテル、t-ブチルメチルエーテル、及びこれらの混合物からなる群より選ばれる、請求項1〜7のいずれか1項に記載の方法。
- 有機溶媒が、t-ブチルメチルエーテルを含む、請求項1〜8のいずれか1項に記載の方法。
- 塩基性水溶液が、無機塩基を含む、請求項1〜9のいずれか1項に記載の方法。
- 無機塩基が、水酸化リチウム、水酸化ナトリウム、水酸化セシウム、水酸化カルシウム、水酸化マグネシウム、水酸化アルミニウム、水酸化カリウム、及びこれらの混合物からなる群より選ばれる、請求項10に記載の方法。
- 無機塩基が、水酸化カリウムである、請求項10に記載の方法。
- 塩基性水溶液が、有機塩基を含む、請求項1〜9のいずれか1項に記載の方法。
- 有機塩基が、Me4NOH、Et4NOH、BnMe3NOH、及びこれらの混合物からなる群より選ばれる、請求項13に記載の方法。
- 相間移動触媒が、一つ以上の四級アンモニウム塩を含む、請求項1〜14に記載の方法。
- 相間移動触媒が、テトラ-n-ブチルアンモニウムブロマイドを含む、請求項15に記載の方法。
- 反応させる工程が、0℃〜40℃の範囲にある温度で行われる、請求項1〜16に記載の方法。
- 反応させる工程が、10℃〜30℃の範囲にある温度で行われる、請求項17に記載の方法。
- 反応させる工程が、室温で行われる、請求項18に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94086607P | 2007-05-30 | 2007-05-30 | |
| US60/940,866 | 2007-05-30 | ||
| PCT/IB2008/001321 WO2008146130A1 (en) | 2007-05-30 | 2008-05-23 | Process for producing phosphine oxide vitamin d precursors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010528099A JP2010528099A (ja) | 2010-08-19 |
| JP5480131B2 true JP5480131B2 (ja) | 2014-04-23 |
Family
ID=40074615
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010509907A Active JP5480131B2 (ja) | 2007-05-30 | 2008-05-23 | ホスフィンオキシドビタミンd前駆物質の製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8039676B2 (ja) |
| EP (1) | EP2167516B1 (ja) |
| JP (1) | JP5480131B2 (ja) |
| CN (1) | CN101754971A (ja) |
| CA (1) | CA2688083C (ja) |
| ES (1) | ES2544819T3 (ja) |
| WO (1) | WO2008146130A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2603519B1 (fr) * | 1986-07-29 | 1989-03-10 | Nergeco Sa | Procede de fabrication de rideaux souples de toute dimension et dispositif pour sa mise en oeuvre |
| CN111659471B (zh) * | 2020-06-18 | 2023-06-02 | 河北威远生物化工有限公司 | 一种用于合成甲氨基阿维菌素中间体亚胺化合物的催化剂及其应用 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3992432A (en) * | 1967-04-05 | 1976-11-16 | Continental Oil Company | Phase transfer catalysis of heterogeneous reactions by quaternary salts |
| FR2472575B1 (fr) * | 1979-12-28 | 1985-10-04 | Poudres & Explosifs Ste Nale | Procede de synthese d'oxydes de phosphines tertiaires et nouveaux oxydes de phosphines tertiaires |
| GB2198725B (en) * | 1986-11-05 | 1991-01-16 | Alter Sa | Use of a series of quaternary ammonium salts as phase-transfer catalysts |
| US5001250A (en) * | 1989-05-30 | 1991-03-19 | Occidental Chemical Corporation | Purification of bidentate organophosphorous extractants |
| AU650751B2 (en) * | 1991-05-28 | 1994-06-30 | Wisconsin Alumni Research Foundation | Novel synthesis of 19-nor vitamin D compounds |
| SG70009A1 (en) | 1996-05-23 | 2000-01-25 | Hoffmann La Roche | Vitamin d3 analogs |
| US6603030B1 (en) * | 1999-04-22 | 2003-08-05 | Hoffman-La Roche Inc. | Process for producing phosphineoxide vitamin D precursors |
| US6331642B1 (en) * | 1999-07-12 | 2001-12-18 | Hoffmann-La Roche Inc. | Vitamin D3 analogs |
| WO2006019169A1 (ja) * | 2004-08-17 | 2006-02-23 | Teijin Pharma Limited | 3-エピビタミンd3誘導体およびそれを用いる治療剤 |
| KR20080050420A (ko) * | 2005-08-18 | 2008-06-05 | 비옥셀 에스.피.에이. | 1α-플루오로-25-히드록시-16-23E-디엔-26,27-비스호모-20-에피-콜레칼시페롤의 합성 |
| US20100069339A1 (en) * | 2006-10-13 | 2010-03-18 | Bioxell S.P.A. | Novel method of treatment of male sub-fertility |
-
2008
- 2008-05-23 JP JP2010509907A patent/JP5480131B2/ja active Active
- 2008-05-23 WO PCT/IB2008/001321 patent/WO2008146130A1/en not_active Ceased
- 2008-05-23 EP EP08762721.2A patent/EP2167516B1/en active Active
- 2008-05-23 CA CA2688083A patent/CA2688083C/en active Active
- 2008-05-23 CN CN200880018041A patent/CN101754971A/zh active Pending
- 2008-05-23 ES ES08762721.2T patent/ES2544819T3/es active Active
- 2008-05-29 US US12/128,992 patent/US8039676B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CA2688083A1 (en) | 2008-12-04 |
| JP2010528099A (ja) | 2010-08-19 |
| CA2688083C (en) | 2015-07-07 |
| US20080300427A1 (en) | 2008-12-04 |
| US8039676B2 (en) | 2011-10-18 |
| WO2008146130A1 (en) | 2008-12-04 |
| EP2167516A1 (en) | 2010-03-31 |
| CN101754971A (zh) | 2010-06-23 |
| EP2167516A4 (en) | 2012-04-11 |
| EP2167516B1 (en) | 2015-07-22 |
| ES2544819T3 (es) | 2015-09-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA1221707A (en) | Process for the preparation of 1-hydroxylated vitamin d compounds | |
| JP5480131B2 (ja) | ホスフィンオキシドビタミンd前駆物質の製造方法 | |
| JP2014139151A (ja) | (e)−1−ハロ−エナミド誘導体又はその塩を製造する方法及び(e)−1−ハロ−エナミド誘導体又はその塩。 | |
| US6603030B1 (en) | Process for producing phosphineoxide vitamin D precursors | |
| JP2003335735A (ja) | パーフルオロイソプロピルアニリン類の製造方法 | |
| Li et al. | A convergent and stereoselective synthesis of a seco-precursor of macrolactin A | |
| JP4635251B2 (ja) | 有機ビスマス化合物およびその製法 | |
| CN104804039A (zh) | 有机磷系阻燃剂代谢物的合成方法 | |
| KR102376869B1 (ko) | 요오드 함유 규소 화합물의 제조 방법 | |
| JP5818073B2 (ja) | O−エチルS−n−プロピル(E)−[2−(シアノイミノ)−3−エチルイミダゾリジン−1−イル]ホスホノチオアートの製造方法 | |
| JP3806886B2 (ja) | (e)−エノールチオエーテル誘導体の立体選択的な製造方法 | |
| JP4508722B2 (ja) | 含フッ素リンイリド化合物及びその製造方法 | |
| JP3863956B2 (ja) | ホスホン酸ジエステルの新規な製造方法 | |
| JP2526810B2 (ja) | ヒドロキシシクロペンタノン誘導体及びその製造法 | |
| JP2026009455A (ja) | スフィンゴミエリン類の製造方法 | |
| JP2003113128A (ja) | イノラートアニオンの新規合成法 | |
| JP2003261555A (ja) | 2−トリフルオロメチル−4,5−ジヒドロオキセピン類およびその製造方法 | |
| JPH10109994A (ja) | アミドチオリン酸エステル誘導体の製造方法 | |
| PL216135B1 (pl) | Nowe syntony i ich zastosowanie do otrzymywania pochodnych 19-nor witamin D | |
| JPH01216965A (ja) | 2−アルコキシブロピオン酸アミド誘導体の製造方法 | |
| JPH09227544A (ja) | ジヒドロフラン誘導体の製造方法 | |
| JPH0770053A (ja) | フッ素化ビタミンd誘導体および製造方法 | |
| JPH09208566A (ja) | 光学活性オキサゾリン化合物及び不斉アリル酸化反応 | |
| KR20090121377A (ko) | 6-히드록시에틸페남 화합물의 제조 방법 | |
| JP2007063262A (ja) | α−グリコシド結合を有する3−フルオロシアル酸誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110520 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20120216 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130430 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130522 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130821 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140122 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140213 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5480131 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |