TW202128704A - 雙環亞磷醯胺之製造方法 - Google Patents
雙環亞磷醯胺之製造方法 Download PDFInfo
- Publication number
- TW202128704A TW202128704A TW109135908A TW109135908A TW202128704A TW 202128704 A TW202128704 A TW 202128704A TW 109135908 A TW109135908 A TW 109135908A TW 109135908 A TW109135908 A TW 109135908A TW 202128704 A TW202128704 A TW 202128704A
- Authority
- TW
- Taiwan
- Prior art keywords
- group
- compound
- formula
- salt
- general formula
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 87
- -1 bicyclic phosphoramidite Chemical class 0.000 title claims description 303
- 239000000178 monomer Substances 0.000 claims abstract description 18
- 150000001875 compounds Chemical class 0.000 claims description 383
- 238000000034 method Methods 0.000 claims description 310
- 238000006243 chemical reaction Methods 0.000 claims description 187
- 239000002904 solvent Substances 0.000 claims description 127
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 122
- 239000003153 chemical reaction reagent Substances 0.000 claims description 109
- 150000003839 salts Chemical class 0.000 claims description 109
- 125000003118 aryl group Chemical group 0.000 claims description 102
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 95
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 87
- 125000002252 acyl group Chemical group 0.000 claims description 75
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 70
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical group CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 69
- 239000003795 chemical substances by application Substances 0.000 claims description 67
- 239000003054 catalyst Substances 0.000 claims description 62
- 239000002585 base Substances 0.000 claims description 60
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 56
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical group [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 54
- 125000003545 alkoxy group Chemical group 0.000 claims description 52
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical group [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 51
- 239000012190 activator Substances 0.000 claims description 50
- 125000001931 aliphatic group Chemical group 0.000 claims description 48
- 125000000217 alkyl group Chemical group 0.000 claims description 46
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 37
- 125000003277 amino group Chemical group 0.000 claims description 35
- 229910052799 carbon Inorganic materials 0.000 claims description 35
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 34
- 229910052751 metal Inorganic materials 0.000 claims description 33
- 239000002184 metal Substances 0.000 claims description 33
- 125000006239 protecting group Chemical group 0.000 claims description 33
- 230000001681 protective effect Effects 0.000 claims description 33
- 238000010511 deprotection reaction Methods 0.000 claims description 32
- 229910052736 halogen Inorganic materials 0.000 claims description 32
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 32
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid group Chemical group S(O)(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 32
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical group N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 29
- 239000002777 nucleoside Substances 0.000 claims description 29
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 28
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 28
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 28
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 28
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 claims description 28
- 125000005843 halogen group Chemical group 0.000 claims description 27
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 27
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 claims description 26
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 claims description 26
- 239000003638 chemical reducing agent Substances 0.000 claims description 26
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 claims description 25
- 239000002808 molecular sieve Substances 0.000 claims description 24
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 24
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical group [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 claims description 24
- 108091034117 Oligonucleotide Proteins 0.000 claims description 23
- 239000002253 acid Substances 0.000 claims description 23
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 22
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 22
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical group C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 claims description 21
- 150000003833 nucleoside derivatives Chemical class 0.000 claims description 21
- 229910052763 palladium Inorganic materials 0.000 claims description 21
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical group CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 claims description 18
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 claims description 18
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 claims description 18
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims description 18
- 239000001257 hydrogen Substances 0.000 claims description 18
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 18
- 239000002773 nucleotide Substances 0.000 claims description 18
- 125000003729 nucleotide group Chemical group 0.000 claims description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 17
- 235000011114 ammonium hydroxide Nutrition 0.000 claims description 17
- 239000005977 Ethylene Substances 0.000 claims description 16
- DRUIESSIVFYOMK-UHFFFAOYSA-N Trichloroacetonitrile Chemical compound ClC(Cl)(Cl)C#N DRUIESSIVFYOMK-UHFFFAOYSA-N 0.000 claims description 16
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 claims description 16
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 claims description 15
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 claims description 15
- 239000003377 acid catalyst Substances 0.000 claims description 15
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 15
- 150000002367 halogens Chemical group 0.000 claims description 15
- 239000003446 ligand Substances 0.000 claims description 15
- 229910052698 phosphorus Inorganic materials 0.000 claims description 15
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 claims description 14
- 125000005604 azodicarboxylate group Chemical group 0.000 claims description 14
- 230000002140 halogenating effect Effects 0.000 claims description 14
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 14
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical group C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 14
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical group N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 claims description 13
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical group ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 claims description 13
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 13
- XGDRLCRGKUCBQL-UHFFFAOYSA-N 1h-imidazole-4,5-dicarbonitrile Chemical compound N#CC=1N=CNC=1C#N XGDRLCRGKUCBQL-UHFFFAOYSA-N 0.000 claims description 12
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 claims description 12
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 claims description 12
- 239000011574 phosphorus Substances 0.000 claims description 12
- 229910052697 platinum Inorganic materials 0.000 claims description 12
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 claims description 11
- 239000005695 Ammonium acetate Substances 0.000 claims description 11
- BHIIGRBMZRSDRI-UHFFFAOYSA-N [chloro(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(Cl)OC1=CC=CC=C1 BHIIGRBMZRSDRI-UHFFFAOYSA-N 0.000 claims description 11
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 claims description 11
- 235000019257 ammonium acetate Nutrition 0.000 claims description 11
- 229940043376 ammonium acetate Drugs 0.000 claims description 11
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 11
- 229910052801 chlorine Inorganic materials 0.000 claims description 11
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical group CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 claims description 11
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 claims description 10
- 241000208340 Araliaceae Species 0.000 claims description 10
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 10
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 claims description 10
- 235000003140 Panax quinquefolius Nutrition 0.000 claims description 10
- 229940073608 benzyl chloride Drugs 0.000 claims description 10
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 claims description 10
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 10
- 239000002274 desiccant Substances 0.000 claims description 10
- 235000008434 ginseng Nutrition 0.000 claims description 10
- 108020004707 nucleic acids Proteins 0.000 claims description 10
- 102000039446 nucleic acids Human genes 0.000 claims description 10
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 9
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 claims description 9
- 239000001099 ammonium carbonate Substances 0.000 claims description 9
- 235000012501 ammonium carbonate Nutrition 0.000 claims description 9
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 claims description 9
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 9
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 claims description 9
- 235000019253 formic acid Nutrition 0.000 claims description 9
- 229910000073 phosphorus hydride Inorganic materials 0.000 claims description 9
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 claims description 9
- 229940104230 thymidine Drugs 0.000 claims description 9
- JAPYIBBSTJFDAK-UHFFFAOYSA-N 2,4,6-tri(propan-2-yl)benzenesulfonyl chloride Chemical compound CC(C)C1=CC(C(C)C)=C(S(Cl)(=O)=O)C(C(C)C)=C1 JAPYIBBSTJFDAK-UHFFFAOYSA-N 0.000 claims description 8
- RKVHNYJPIXOHRW-UHFFFAOYSA-N 3-bis[di(propan-2-yl)amino]phosphanyloxypropanenitrile Chemical group CC(C)N(C(C)C)P(N(C(C)C)C(C)C)OCCC#N RKVHNYJPIXOHRW-UHFFFAOYSA-N 0.000 claims description 8
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 8
- 239000012445 acidic reagent Substances 0.000 claims description 8
- 229960005305 adenosine Drugs 0.000 claims description 8
- 239000004305 biphenyl Substances 0.000 claims description 8
- 235000010290 biphenyl Nutrition 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 150000002243 furanoses Chemical class 0.000 claims description 8
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 claims description 8
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 8
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 claims description 7
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 7
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 claims description 7
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 claims description 7
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 claims description 7
- 125000004849 alkoxymethyl group Chemical group 0.000 claims description 7
- 229910021529 ammonia Inorganic materials 0.000 claims description 7
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 claims description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 7
- 229940029575 guanosine Drugs 0.000 claims description 7
- 229910052740 iodine Inorganic materials 0.000 claims description 7
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 7
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical group C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 claims description 7
- MARUHZGHZWCEQU-UHFFFAOYSA-N 5-phenyl-2h-tetrazole Chemical compound C1=CC=CC=C1C1=NNN=N1 MARUHZGHZWCEQU-UHFFFAOYSA-N 0.000 claims description 6
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 claims description 6
- 229910052770 Uranium Inorganic materials 0.000 claims description 6
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 claims description 6
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 6
- 238000006317 isomerization reaction Methods 0.000 claims description 6
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 claims description 6
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 claims description 6
- 229940045145 uridine Drugs 0.000 claims description 6
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 claims description 5
- OVRNDRQMDRJTHS-KEWYIRBNSA-N N-acetyl-D-galactosamine Chemical group CC(=O)N[C@H]1C(O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-KEWYIRBNSA-N 0.000 claims description 5
- 235000019445 benzyl alcohol Nutrition 0.000 claims description 5
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 claims description 5
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical group CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 claims description 5
- 238000006460 hydrolysis reaction Methods 0.000 claims description 5
- 125000001424 substituent group Chemical group 0.000 claims description 5
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 claims description 5
- QFMZQPDHXULLKC-UHFFFAOYSA-N 1,2-bis(diphenylphosphino)ethane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCP(C=1C=CC=CC=1)C1=CC=CC=C1 QFMZQPDHXULLKC-UHFFFAOYSA-N 0.000 claims description 4
- WIKNOSQXZNVMEG-UHFFFAOYSA-N 1-methyl-1h-imidazol-1-ium;2,2,2-trifluoroacetate Chemical compound C[NH+]1C=CN=C1.[O-]C(=O)C(F)(F)F WIKNOSQXZNVMEG-UHFFFAOYSA-N 0.000 claims description 4
- QHYFBUJBVQBFPG-UHFFFAOYSA-N FC(C(=O)O)(F)F.C(C)(C)N1C=NC=C1 Chemical compound FC(C(=O)O)(F)F.C(C)(C)N1C=NC=C1 QHYFBUJBVQBFPG-UHFFFAOYSA-N 0.000 claims description 4
- FKTKIEPXXDOVLP-UHFFFAOYSA-N chloroform;1,5-diphenylpenta-1,4-dien-3-one Chemical compound ClC(Cl)Cl.C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 FKTKIEPXXDOVLP-UHFFFAOYSA-N 0.000 claims description 4
- 238000006880 cross-coupling reaction Methods 0.000 claims description 4
- 230000007062 hydrolysis Effects 0.000 claims description 4
- NRTYMEPCRDJMPZ-UHFFFAOYSA-N pyridine;2,2,2-trifluoroacetic acid Chemical group C1=CC=NC=C1.OC(=O)C(F)(F)F NRTYMEPCRDJMPZ-UHFFFAOYSA-N 0.000 claims description 4
- IBXMKLPFLZYRQZ-UHFFFAOYSA-N 1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 IBXMKLPFLZYRQZ-UHFFFAOYSA-N 0.000 claims description 3
- ZEMZPXWZVTUONV-UHFFFAOYSA-N 2-(2-dicyclohexylphosphanylphenyl)-n,n-dimethylaniline Chemical group CN(C)C1=CC=CC=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 ZEMZPXWZVTUONV-UHFFFAOYSA-N 0.000 claims description 3
- 125000004423 acyloxy group Chemical group 0.000 claims description 3
- 125000003282 alkyl amino group Chemical group 0.000 claims description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 3
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 150000002009 diols Chemical class 0.000 claims description 3
- 231100000219 mutagenic Toxicity 0.000 claims description 3
- 230000003505 mutagenic effect Effects 0.000 claims description 3
- 229910052760 oxygen Inorganic materials 0.000 claims description 3
- 239000001301 oxygen Substances 0.000 claims description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 125000004545 purin-9-yl group Chemical group N1=CN=C2N(C=NC2=C1)* 0.000 claims description 3
- 230000002194 synthesizing effect Effects 0.000 claims description 3
- 125000005233 alkylalcohol group Chemical group 0.000 claims description 2
- 229910052796 boron Inorganic materials 0.000 claims description 2
- 230000004048 modification Effects 0.000 claims description 2
- 238000012986 modification Methods 0.000 claims description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims 2
- 125000005278 alkyl sulfonyloxy group Chemical group 0.000 claims 2
- 229930182470 glycoside Natural products 0.000 claims 2
- 150000002338 glycosides Chemical class 0.000 claims 2
- 125000003172 aldehyde group Chemical group 0.000 claims 1
- HHSARRMUXPDGJD-UHFFFAOYSA-N butyl(dimethyl)silicon Chemical group CCCC[Si](C)C HHSARRMUXPDGJD-UHFFFAOYSA-N 0.000 claims 1
- 125000000101 thioether group Chemical group 0.000 claims 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims 1
- 230000000707 stereoselective effect Effects 0.000 abstract description 2
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 465
- 239000000243 solution Substances 0.000 description 222
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 204
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 180
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 171
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 105
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 90
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 88
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 81
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 73
- 238000003756 stirring Methods 0.000 description 64
- 239000010410 layer Substances 0.000 description 62
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 54
- 239000013078 crystal Substances 0.000 description 50
- 239000007788 liquid Substances 0.000 description 48
- 239000007864 aqueous solution Substances 0.000 description 47
- 238000000926 separation method Methods 0.000 description 46
- 239000007787 solid Substances 0.000 description 44
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 40
- 239000012044 organic layer Substances 0.000 description 40
- 238000010898 silica gel chromatography Methods 0.000 description 37
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 36
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 36
- 230000035484 reaction time Effects 0.000 description 36
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 36
- 206010013801 Duchenne Muscular Dystrophy Diseases 0.000 description 34
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 32
- 238000001953 recrystallisation Methods 0.000 description 32
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 30
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 27
- 239000012295 chemical reaction liquid Substances 0.000 description 27
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 27
- 229930195733 hydrocarbon Natural products 0.000 description 27
- 150000002430 hydrocarbons Chemical class 0.000 description 27
- 229940125782 compound 2 Drugs 0.000 description 26
- 238000006206 glycosylation reaction Methods 0.000 description 25
- 150000008282 halocarbons Chemical class 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 24
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- 150000002170 ethers Chemical class 0.000 description 24
- 239000000203 mixture Substances 0.000 description 24
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 23
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 21
- 239000000543 intermediate Substances 0.000 description 21
- 238000005481 NMR spectroscopy Methods 0.000 description 18
- 125000005129 aryl carbonyl group Chemical group 0.000 description 17
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 17
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 16
- 230000003472 neutralizing effect Effects 0.000 description 16
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 16
- 235000017557 sodium bicarbonate Nutrition 0.000 description 16
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 15
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 13
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 13
- 229940126657 Compound 17 Drugs 0.000 description 13
- 150000001721 carbon Chemical group 0.000 description 13
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 13
- MXZNUGFCDVAXLG-CHWSQXEVSA-N [(2S)-1-[(2R)-3-methyl-2-(pyridine-4-carbonylamino)butanoyl]pyrrolidin-2-yl]boronic acid Chemical compound CC(C)[C@@H](NC(=O)c1ccncc1)C(=O)N1CCC[C@@H]1B(O)O MXZNUGFCDVAXLG-CHWSQXEVSA-N 0.000 description 12
- 229940126208 compound 22 Drugs 0.000 description 12
- 238000001914 filtration Methods 0.000 description 12
- 229940035429 isobutyl alcohol Drugs 0.000 description 12
- 238000005406 washing Methods 0.000 description 12
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 11
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 11
- 150000001412 amines Chemical class 0.000 description 11
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 11
- 238000001816 cooling Methods 0.000 description 10
- 230000013595 glycosylation Effects 0.000 description 10
- 150000007530 organic bases Chemical class 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 10
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 10
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 9
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 9
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 9
- 239000005289 controlled pore glass Substances 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 9
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 8
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical group NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 8
- 150000007529 inorganic bases Chemical class 0.000 description 8
- LULXBAGMGMJJRW-UHFFFAOYSA-N n,2-bis(trimethylsilyl)acetamide Chemical compound C[Si](C)(C)CC(=O)N[Si](C)(C)C LULXBAGMGMJJRW-UHFFFAOYSA-N 0.000 description 8
- 150000007523 nucleic acids Chemical class 0.000 description 8
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 8
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 7
- 229940126086 compound 21 Drugs 0.000 description 7
- 238000012790 confirmation Methods 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 150000004678 hydrides Chemical class 0.000 description 7
- 150000002431 hydrogen Chemical group 0.000 description 7
- 239000011259 mixed solution Substances 0.000 description 7
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 7
- GVOISEJVFFIGQE-YCZSINBZSA-N n-[(1r,2s,5r)-5-[methyl(propan-2-yl)amino]-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 GVOISEJVFFIGQE-YCZSINBZSA-N 0.000 description 7
- 229940090181 propyl acetate Drugs 0.000 description 7
- 239000000377 silicon dioxide Substances 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- 229940113082 thymine Drugs 0.000 description 7
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 7
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 6
- 125000005103 alkyl silyl group Chemical group 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 229940126142 compound 16 Drugs 0.000 description 6
- 229940125810 compound 20 Drugs 0.000 description 6
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 6
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 6
- WFKAJVHLWXSISD-UHFFFAOYSA-N isobutyramide Chemical compound CC(C)C(N)=O WFKAJVHLWXSISD-UHFFFAOYSA-N 0.000 description 6
- 230000007935 neutral effect Effects 0.000 description 6
- 125000003835 nucleoside group Chemical group 0.000 description 6
- 229910052710 silicon Inorganic materials 0.000 description 6
- 239000010703 silicon Substances 0.000 description 6
- 239000002002 slurry Substances 0.000 description 6
- PHDIJLFSKNMCMI-ITGJKDDRSA-N (3R,4S,5R,6R)-6-(hydroxymethyl)-4-(8-quinolin-6-yloxyoctoxy)oxane-2,3,5-triol Chemical compound OC[C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)OCCCCCCCCOC=1C=C2C=CC=NC2=CC=1)O PHDIJLFSKNMCMI-ITGJKDDRSA-N 0.000 description 5
- 125000006283 4-chlorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Cl)C([H])([H])* 0.000 description 5
- GXGKKIPUFAHZIZ-UHFFFAOYSA-N 5-benzylsulfanyl-2h-tetrazole Chemical compound C=1C=CC=CC=1CSC=1N=NNN=1 GXGKKIPUFAHZIZ-UHFFFAOYSA-N 0.000 description 5
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 5
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 5
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 5
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 5
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 5
- 239000012141 concentrate Substances 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 125000006503 p-nitrobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1[N+]([O-])=O)C([H])([H])* 0.000 description 5
- 238000001556 precipitation Methods 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 235000010265 sodium sulphite Nutrition 0.000 description 5
- XHFLOLLMZOTPSM-UHFFFAOYSA-M sodium;hydrogen carbonate;hydrate Chemical compound [OH-].[Na+].OC(O)=O XHFLOLLMZOTPSM-UHFFFAOYSA-M 0.000 description 5
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 5
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 4
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 4
- QSSXJPIWXQTSIX-UHFFFAOYSA-N 1-bromo-2-methylbenzene Chemical compound CC1=CC=CC=C1Br QSSXJPIWXQTSIX-UHFFFAOYSA-N 0.000 description 4
- IHCCLXNEEPMSIO-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperidin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 IHCCLXNEEPMSIO-UHFFFAOYSA-N 0.000 description 4
- 125000004080 3-carboxypropanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C(O[H])=O 0.000 description 4
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- 239000005708 Sodium hypochlorite Substances 0.000 description 4
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 4
- 239000012346 acetyl chloride Substances 0.000 description 4
- 150000001298 alcohols Chemical class 0.000 description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- HRYZWHHZPQKTII-UHFFFAOYSA-N chloroethane Chemical compound CCCl HRYZWHHZPQKTII-UHFFFAOYSA-N 0.000 description 4
- 229940125758 compound 15 Drugs 0.000 description 4
- 229940125877 compound 31 Drugs 0.000 description 4
- 229940104302 cytosine Drugs 0.000 description 4
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 4
- 229960003750 ethyl chloride Drugs 0.000 description 4
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 4
- JIMAEIUQOBHHIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine;1-methylimidazole Chemical compound CN1C=CN=C1.CN(C)C1=CC=CC=N1 JIMAEIUQOBHHIA-UHFFFAOYSA-N 0.000 description 4
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 4
- 238000003419 tautomerization reaction Methods 0.000 description 4
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 4
- 229940035893 uracil Drugs 0.000 description 4
- 229910052727 yttrium Inorganic materials 0.000 description 4
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 3
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 3
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 3
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 3
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 3
- XDLJIIILKATPAQ-UHFFFAOYSA-N 2,8-dichloro-7h-purine Chemical compound ClC1=NC=C2NC(Cl)=NC2=N1 XDLJIIILKATPAQ-UHFFFAOYSA-N 0.000 description 3
- VWVRASTUFJRTHW-UHFFFAOYSA-N 2-[3-(azetidin-3-yloxy)-4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound O=C(CN1C=C(C(OC2CNC2)=N1)C1=CN=C(NC2CC3=C(C2)C=CC=C3)N=C1)N1CCC2=C(C1)N=NN2 VWVRASTUFJRTHW-UHFFFAOYSA-N 0.000 description 3
- JQMFQLVAJGZSQS-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-N-(2-oxo-3H-1,3-benzoxazol-6-yl)acetamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)NC1=CC2=C(NC(O2)=O)C=C1 JQMFQLVAJGZSQS-UHFFFAOYSA-N 0.000 description 3
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 3
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 3
- 125000006280 2-bromobenzyl group Chemical group [H]C1=C([H])C(Br)=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 3
- 125000006281 4-bromobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)C([H])([H])* 0.000 description 3
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 3
- 125000006181 4-methyl benzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])C([H])([H])* 0.000 description 3
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 3
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 3
- 208000007976 Ketosis Diseases 0.000 description 3
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 3
- NWBJYWHLCVSVIJ-UHFFFAOYSA-N N-benzyladenine Chemical compound N=1C=NC=2NC=NC=2C=1NCC1=CC=CC=C1 NWBJYWHLCVSVIJ-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 3
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 3
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 description 3
- HXYXTCJDWHHCBW-UHFFFAOYSA-N acetonitrile;toluene Chemical compound CC#N.CC1=CC=CC=C1 HXYXTCJDWHHCBW-UHFFFAOYSA-N 0.000 description 3
- 229960000643 adenine Drugs 0.000 description 3
- 125000004414 alkyl thio group Chemical group 0.000 description 3
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 3
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 229940126543 compound 14 Drugs 0.000 description 3
- 229940125833 compound 23 Drugs 0.000 description 3
- 229940125961 compound 24 Drugs 0.000 description 3
- 229940125846 compound 25 Drugs 0.000 description 3
- 229940125851 compound 27 Drugs 0.000 description 3
- 229940127204 compound 29 Drugs 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 239000008098 formaldehyde solution Substances 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 125000003843 furanosyl group Chemical group 0.000 description 3
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical class O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 3
- 238000007069 methylation reaction Methods 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 125000006505 p-cyanobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C#N)C([H])([H])* 0.000 description 3
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 3
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 235000015497 potassium bicarbonate Nutrition 0.000 description 3
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 3
- 239000011736 potassium bicarbonate Substances 0.000 description 3
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 3
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 3
- 150000003222 pyridines Chemical class 0.000 description 3
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- 125000002889 tridecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 3
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- OHVLMTFVQDZYHP-UHFFFAOYSA-N 1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-2-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound N1N=NC=2CN(CCC=21)C(CN1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)=O OHVLMTFVQDZYHP-UHFFFAOYSA-N 0.000 description 2
- KZEVSDGEBAJOTK-UHFFFAOYSA-N 1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-2-[5-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]ethanone Chemical compound N1N=NC=2CN(CCC=21)C(CC=1OC(=NN=1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)=O KZEVSDGEBAJOTK-UHFFFAOYSA-N 0.000 description 2
- HMUNWXXNJPVALC-UHFFFAOYSA-N 1-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C(CN1CC2=C(CC1)NN=N2)=O HMUNWXXNJPVALC-UHFFFAOYSA-N 0.000 description 2
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 2
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 2
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 2
- PUABNAWFNOFZPZ-UHFFFAOYSA-N 2,3,5,6,7,8,9,9a-octahydro-1h-benzo[7]annulene Chemical compound C1CCCCC2CCCC=C21 PUABNAWFNOFZPZ-UHFFFAOYSA-N 0.000 description 2
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 2
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 2
- SXAMGRAIZSSWIH-UHFFFAOYSA-N 2-[3-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,2,4-oxadiazol-5-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NOC(=N1)CC(=O)N1CC2=C(CC1)NN=N2 SXAMGRAIZSSWIH-UHFFFAOYSA-N 0.000 description 2
- WWSJZGAPAVMETJ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-3-ethoxypyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2)OCC WWSJZGAPAVMETJ-UHFFFAOYSA-N 0.000 description 2
- YJLUBHOZZTYQIP-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)NN=N2 YJLUBHOZZTYQIP-UHFFFAOYSA-N 0.000 description 2
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- MSHFRERJPWKJFX-UHFFFAOYSA-N 4-Methoxybenzyl alcohol Chemical compound COC1=CC=C(CO)C=C1 MSHFRERJPWKJFX-UHFFFAOYSA-N 0.000 description 2
- 125000006189 4-phenyl benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- ZAYHVCMSTBRABG-UHFFFAOYSA-N 5-Methylcytidine Natural products O=C1N=C(N)C(C)=CN1C1C(O)C(O)C(CO)O1 ZAYHVCMSTBRABG-UHFFFAOYSA-N 0.000 description 2
- ZAYHVCMSTBRABG-JXOAFFINSA-N 5-methylcytidine Chemical compound O=C1N=C(N)C(C)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 ZAYHVCMSTBRABG-JXOAFFINSA-N 0.000 description 2
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 2
- WTFUTSCZYYCBAY-SXBRIOAWSA-N 6-[(E)-C-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-N-hydroxycarbonimidoyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C/C(=N/O)/C1=CC2=C(NC(O2)=O)C=C1 WTFUTSCZYYCBAY-SXBRIOAWSA-N 0.000 description 2
- DFGKGUXTPFWHIX-UHFFFAOYSA-N 6-[2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]acetyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)C1=CC2=C(NC(O2)=O)C=C1 DFGKGUXTPFWHIX-UHFFFAOYSA-N 0.000 description 2
- RYYIULNRIVUMTQ-UHFFFAOYSA-N 6-chloroguanine Chemical group NC1=NC(Cl)=C2N=CNC2=N1 RYYIULNRIVUMTQ-UHFFFAOYSA-N 0.000 description 2
- 229930024421 Adenine Natural products 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N Glycolaldehyde Chemical compound OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 2
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 2
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- JGJBTOUVZMRNGR-UHFFFAOYSA-N [(4-cyano-2-methylbutan-2-yl)-propan-2-ylamino]phosphonous acid Chemical compound C(#N)CCC(C)(C)N(P(O)O)C(C)C JGJBTOUVZMRNGR-UHFFFAOYSA-N 0.000 description 2
- JAWMENYCRQKKJY-UHFFFAOYSA-N [3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-ylmethyl)-1-oxa-2,8-diazaspiro[4.5]dec-2-en-8-yl]-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]methanone Chemical compound N1N=NC=2CN(CCC=21)CC1=NOC2(C1)CCN(CC2)C(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F JAWMENYCRQKKJY-UHFFFAOYSA-N 0.000 description 2
- IRMNIXXVOOMKKP-UHFFFAOYSA-N [methoxy(diphenyl)methyl]benzene Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(OC)C1=CC=CC=C1 IRMNIXXVOOMKKP-UHFFFAOYSA-N 0.000 description 2
- AUALQMFGWLZREY-UHFFFAOYSA-N acetonitrile;methanol Chemical compound OC.CC#N AUALQMFGWLZREY-UHFFFAOYSA-N 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 125000001204 arachidyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000005098 aryl alkoxy carbonyl group Chemical group 0.000 description 2
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- JPYQFYIEOUVJDU-UHFFFAOYSA-N beclamide Chemical compound ClCCC(=O)NCC1=CC=CC=C1 JPYQFYIEOUVJDU-UHFFFAOYSA-N 0.000 description 2
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 2
- 239000003426 co-catalyst Substances 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- FPUGCISOLXNPPC-IOSLPCCCSA-N cordysinin B Chemical compound CO[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(N)=C2N=C1 FPUGCISOLXNPPC-IOSLPCCCSA-N 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000003074 decanoyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(*)=O 0.000 description 2
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 2
- WCGGWVOVFQNRRS-UHFFFAOYSA-N dichloroacetamide Chemical compound NC(=O)C(Cl)Cl WCGGWVOVFQNRRS-UHFFFAOYSA-N 0.000 description 2
- LIKFHECYJZWXFJ-UHFFFAOYSA-N dimethyldichlorosilane Chemical compound C[Si](C)(Cl)Cl LIKFHECYJZWXFJ-UHFFFAOYSA-N 0.000 description 2
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 125000002960 margaryl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 125000004043 oxo group Chemical group O=* 0.000 description 2
- BSCHIACBONPEOB-UHFFFAOYSA-N oxolane;hydrate Chemical compound O.C1CCOC1 BSCHIACBONPEOB-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 125000002958 pentadecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical class O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- 239000010452 phosphate Chemical class 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 2
- 125000005767 propoxymethyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])[#8]C([H])([H])* 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- RUPAXCPQAAOIPB-UHFFFAOYSA-N tert-butyl formate Chemical group CC(C)(C)OC=O RUPAXCPQAAOIPB-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229920001285 xanthan gum Polymers 0.000 description 2
- LODDFDHPSIYCTK-UHFFFAOYSA-N (2,4,6-trimethylphenyl)methanol Chemical compound CC1=CC(C)=C(CO)C(C)=C1 LODDFDHPSIYCTK-UHFFFAOYSA-N 0.000 description 1
- RUPONURESHSCBM-UHFFFAOYSA-N (3,4,5-trimethylphenyl)methanol Chemical compound CC1=CC(CO)=CC(C)=C1C RUPONURESHSCBM-UHFFFAOYSA-N 0.000 description 1
- PTHGDVCPCZKZKR-UHFFFAOYSA-N (4-chlorophenyl)methanol Chemical compound OCC1=CC=C(Cl)C=C1 PTHGDVCPCZKZKR-UHFFFAOYSA-N 0.000 description 1
- 239000001211 (E)-4-phenylbut-3-en-2-one Substances 0.000 description 1
- JUDOLRSMWHVKGX-UHFFFAOYSA-N 1,1-dioxo-1$l^{6},2-benzodithiol-3-one Chemical compound C1=CC=C2C(=O)SS(=O)(=O)C2=C1 JUDOLRSMWHVKGX-UHFFFAOYSA-N 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- YIWGJFPJRAEKMK-UHFFFAOYSA-N 1-(2H-benzotriazol-5-yl)-3-methyl-8-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carbonyl]-1,3,8-triazaspiro[4.5]decane-2,4-dione Chemical compound CN1C(=O)N(c2ccc3n[nH]nc3c2)C2(CCN(CC2)C(=O)c2cnc(NCc3cccc(OC(F)(F)F)c3)nc2)C1=O YIWGJFPJRAEKMK-UHFFFAOYSA-N 0.000 description 1
- LOSXTWDYAWERDB-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-2,3-dimethoxybenzene Chemical compound COC1=CC=CC(C(Cl)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1OC LOSXTWDYAWERDB-UHFFFAOYSA-N 0.000 description 1
- OBOHMJWDFPBPKD-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 OBOHMJWDFPBPKD-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical group CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- FPUGCISOLXNPPC-UHFFFAOYSA-N 2'-O-Methyladenosine Natural products COC1C(O)C(CO)OC1N1C2=NC=NC(N)=C2N=C1 FPUGCISOLXNPPC-UHFFFAOYSA-N 0.000 description 1
- RFCQJGFZUQFYRF-UHFFFAOYSA-N 2'-O-Methylcytidine Natural products COC1C(O)C(CO)OC1N1C(=O)N=C(N)C=C1 RFCQJGFZUQFYRF-UHFFFAOYSA-N 0.000 description 1
- OVYNGSFVYRPRCG-UHFFFAOYSA-N 2'-O-Methylguanosine Natural products COC1C(O)C(CO)OC1N1C(NC(N)=NC2=O)=C2N=C1 OVYNGSFVYRPRCG-UHFFFAOYSA-N 0.000 description 1
- SXUXMRMBWZCMEN-UHFFFAOYSA-N 2'-O-methyl uridine Natural products COC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 SXUXMRMBWZCMEN-UHFFFAOYSA-N 0.000 description 1
- RFCQJGFZUQFYRF-ZOQUXTDFSA-N 2'-O-methylcytidine Chemical compound CO[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N=C(N)C=C1 RFCQJGFZUQFYRF-ZOQUXTDFSA-N 0.000 description 1
- OVYNGSFVYRPRCG-KQYNXXCUSA-N 2'-O-methylguanosine Chemical compound CO[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=C(N)NC2=O)=C2N=C1 OVYNGSFVYRPRCG-KQYNXXCUSA-N 0.000 description 1
- UPQQXPKAYZYUKO-UHFFFAOYSA-N 2,2,2-trichloroacetamide Chemical compound OC(=N)C(Cl)(Cl)Cl UPQQXPKAYZYUKO-UHFFFAOYSA-N 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical group NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- SPSPIUSUWPLVKD-UHFFFAOYSA-N 2,3-dibutyl-6-methylphenol Chemical compound CCCCC1=CC=C(C)C(O)=C1CCCC SPSPIUSUWPLVKD-UHFFFAOYSA-N 0.000 description 1
- YHGKEORTCHVBQH-UHFFFAOYSA-M 2,4,6-tri(propan-2-yl)benzenesulfonate Chemical compound CC(C)C1=CC(C(C)C)=C(S([O-])(=O)=O)C(C(C)C)=C1 YHGKEORTCHVBQH-UHFFFAOYSA-M 0.000 description 1
- RMFWVOLULURGJI-UHFFFAOYSA-N 2,6-dichloro-7h-purine Chemical compound ClC1=NC(Cl)=C2NC=NC2=N1 RMFWVOLULURGJI-UHFFFAOYSA-N 0.000 description 1
- WJSVJNDMOQTICG-UHFFFAOYSA-N 2-amino-1-[(2-methyl-4-methylidene-5-oxooxolan-2-yl)methyl]-7h-purin-6-one Chemical compound NC1=NC=2N=CNC=2C(=O)N1CC1(C)CC(=C)C(=O)O1 WJSVJNDMOQTICG-UHFFFAOYSA-N 0.000 description 1
- OZVMOEUWPMYRQU-JUQFDLSGSA-N 2-amino-9-[(1r,4s,6r,7s)-7-hydroxy-4-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-6-yl]-3h-purin-6-one Chemical compound C1=NC(C(NC(N)=N2)=O)=C2N1[C@@H]1O[C@@]2(CO)CO[C@]1([H])[C@@H]2O OZVMOEUWPMYRQU-JUQFDLSGSA-N 0.000 description 1
- GZCIUTGUKIDCEK-BQIHAETKSA-N 2-amino-9-[(2R,3R,4S,5R)-5-ethyl-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-purin-6-one Chemical compound CC[C@]1(CO)O[C@H]([C@H](O)[C@@H]1O)N1C=NC2=C1N=C(N)NC2=O GZCIUTGUKIDCEK-BQIHAETKSA-N 0.000 description 1
- XKVATRYEPITAPH-UHFFFAOYSA-N 2-chloro-6-phenylmethoxy-7h-purine Chemical compound C=12NC=NC2=NC(Cl)=NC=1OCC1=CC=CC=C1 XKVATRYEPITAPH-UHFFFAOYSA-N 0.000 description 1
- HBJGQJWNMZDFKL-UHFFFAOYSA-N 2-chloro-7h-purin-6-amine Chemical compound NC1=NC(Cl)=NC2=C1NC=N2 HBJGQJWNMZDFKL-UHFFFAOYSA-N 0.000 description 1
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- JHYNEQNPKGIOQF-UHFFFAOYSA-N 3,4-dihydro-2h-phosphole Chemical class C1CC=PC1 JHYNEQNPKGIOQF-UHFFFAOYSA-N 0.000 description 1
- LJFFAOJTTGMNKP-UHFFFAOYSA-N 3-[bis(propan-2-ylamino)phosphoryl-chloroamino]propanenitrile Chemical compound CC(C)NP(=O)(NC(C)C)N(Cl)CCC#N LJFFAOJTTGMNKP-UHFFFAOYSA-N 0.000 description 1
- QWTBDIBOOIAZEF-UHFFFAOYSA-N 3-[chloro-[di(propan-2-yl)amino]phosphanyl]oxypropanenitrile Chemical compound CC(C)N(C(C)C)P(Cl)OCCC#N QWTBDIBOOIAZEF-UHFFFAOYSA-N 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- MYYYOBVDIRECDX-UHFFFAOYSA-N 4,5-dibromo-1h-imidazole Chemical compound BrC=1N=CNC=1Br MYYYOBVDIRECDX-UHFFFAOYSA-N 0.000 description 1
- BFEYPVSPIWUAEE-UHFFFAOYSA-N 4-amino-4-benzyl-5-methyl-1,3-dihydropyrimidin-2-one Chemical compound CC1=CNC(=O)NC1(N)CC1=CC=CC=C1 BFEYPVSPIWUAEE-UHFFFAOYSA-N 0.000 description 1
- XAASLEJRGFPHEV-UHFFFAOYSA-N 4-cyanobenzyl alcohol Chemical compound OCC1=CC=C(C#N)C=C1 XAASLEJRGFPHEV-UHFFFAOYSA-N 0.000 description 1
- KMTDMTZBNYGUNX-UHFFFAOYSA-N 4-methylbenzyl alcohol Chemical compound CC1=CC=C(CO)C=C1 KMTDMTZBNYGUNX-UHFFFAOYSA-N 0.000 description 1
- TXLINXBIWJYFNR-UHFFFAOYSA-N 4-phenylpyridine-2-carbonitrile Chemical compound C1=NC(C#N)=CC(C=2C=CC=CC=2)=C1 TXLINXBIWJYFNR-UHFFFAOYSA-N 0.000 description 1
- GAHCNYHAKKGGHF-UHFFFAOYSA-N 5,5-dimethylhexan-1-amine Chemical class CC(C)(C)CCCCN GAHCNYHAKKGGHF-UHFFFAOYSA-N 0.000 description 1
- CONKBQPVFMXDOV-QHCPKHFHSA-N 6-[(5S)-5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-2-oxo-1,3-oxazolidin-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C[C@H]1CN(C(O1)=O)C1=CC2=C(NC(O2)=O)C=C1 CONKBQPVFMXDOV-QHCPKHFHSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical group NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical class C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- NADTUDXSPFPHPK-UHFFFAOYSA-N B.[Na] Chemical compound B.[Na] NADTUDXSPFPHPK-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- AUUGOABOFBHQTL-UHFFFAOYSA-N C(C(C)C)(=O)OC(C(C)C)=O.C(C(C)C)Cl Chemical compound C(C(C)C)(=O)OC(C(C)C)=O.C(C(C)C)Cl AUUGOABOFBHQTL-UHFFFAOYSA-N 0.000 description 1
- TZQKSUVUQRSZQN-UHFFFAOYSA-N C(C)(=O)OCC.C.ClCCl Chemical compound C(C)(=O)OCC.C.ClCCl TZQKSUVUQRSZQN-UHFFFAOYSA-N 0.000 description 1
- LNMXAQDYIPHBOF-UHFFFAOYSA-N C1C(C2)C3CC2NC1C3 Chemical compound C1C(C2)C3CC2NC1C3 LNMXAQDYIPHBOF-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 125000000824 D-ribofuranosyl group Chemical group [H]OC([H])([H])[C@@]1([H])OC([H])(*)[C@]([H])(O[H])[C@]1([H])O[H] 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical class C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical class NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- VCUFZILGIRCDQQ-KRWDZBQOSA-N N-[[(5S)-2-oxo-3-(2-oxo-3H-1,3-benzoxazol-6-yl)-1,3-oxazolidin-5-yl]methyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C1O[C@H](CN1C1=CC2=C(NC(O2)=O)C=C1)CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F VCUFZILGIRCDQQ-KRWDZBQOSA-N 0.000 description 1
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical class [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical class [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Chemical group 0.000 description 1
- UATOFRZSCHRPBG-UHFFFAOYSA-N acetamide;hydrate Chemical compound O.CC(N)=O UATOFRZSCHRPBG-UHFFFAOYSA-N 0.000 description 1
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 1
- XEFCWBLINXJUIV-UHFFFAOYSA-N acetic acid;iodobenzene Chemical compound CC(O)=O.CC(O)=O.IC1=CC=CC=C1 XEFCWBLINXJUIV-UHFFFAOYSA-N 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000005426 adeninyl group Chemical group N1=C(N=C2N=CNC2=C1N)* 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 150000008431 aliphatic amides Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000005092 alkenyloxycarbonyl group Chemical group 0.000 description 1
- 238000007083 alkoxycarbonylation reaction Methods 0.000 description 1
- 125000004848 alkoxyethyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 1
- 125000001124 arachidoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000008430 aromatic amides Chemical class 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- UPABQMWFWCMOFV-UHFFFAOYSA-N benethamine Chemical class C=1C=CC=CC=1CNCCC1=CC=CC=C1 UPABQMWFWCMOFV-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical class C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- GSSJSUKUYXXPME-UHFFFAOYSA-N benzene;chloromethane Chemical group ClC.C1=CC=CC=C1 GSSJSUKUYXXPME-UHFFFAOYSA-N 0.000 description 1
- 150000008107 benzenesulfonic acids Chemical class 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229930008407 benzylideneacetone Natural products 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- GTXRYQGMNAPQDP-UHFFFAOYSA-N butyl-chloro-diphenylsilane Chemical group C=1C=CC=CC=1[Si](Cl)(CCCC)C1=CC=CC=C1 GTXRYQGMNAPQDP-UHFFFAOYSA-N 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- LHRSMZZXPVKEKR-UHFFFAOYSA-N chloromethane;ethyl acetate Chemical compound ClC.CCOC(C)=O LHRSMZZXPVKEKR-UHFFFAOYSA-N 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical class CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 150000001868 cobalt Chemical class 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 150000001879 copper Chemical class 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical class OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 150000005332 diethylamines Chemical class 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000005868 electrolysis reaction Methods 0.000 description 1
- 239000008393 encapsulating agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-M ethanimidate Chemical group CC([O-])=N DLFVBJFMPXGRIB-UHFFFAOYSA-M 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- PCIBVZXUNDZWRL-UHFFFAOYSA-N ethylene glycol monophosphate Chemical group OCCOP(O)(O)=O PCIBVZXUNDZWRL-UHFFFAOYSA-N 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 125000004705 ethylthio group Chemical group C(C)S* 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 1
- 150000002357 guanidines Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- YEMWINDXAKRPBK-UHFFFAOYSA-N heptane;2-methoxy-2-methylpropane Chemical compound COC(C)(C)C.CCCCCCC YEMWINDXAKRPBK-UHFFFAOYSA-N 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine Substances NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 125000000879 imine group Chemical group 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- XJTQJERLRPWUGL-UHFFFAOYSA-N iodomethylbenzene Chemical compound ICC1=CC=CC=C1 XJTQJERLRPWUGL-UHFFFAOYSA-N 0.000 description 1
- 150000002505 iron Chemical class 0.000 description 1
- 235000013847 iso-butane Nutrition 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000000400 lauroyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 125000000628 margaroyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- IZXGZAJMDLJLMF-UHFFFAOYSA-N methylaminomethanol Chemical class CNCO IZXGZAJMDLJLMF-UHFFFAOYSA-N 0.000 description 1
- 230000001035 methylating effect Effects 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- UIUXUFNYAYAMOE-UHFFFAOYSA-N methylsilane Chemical compound [SiH3]C UIUXUFNYAYAMOE-UHFFFAOYSA-N 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 150000002815 nickel Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 125000001402 nonanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940127073 nucleoside analogue Drugs 0.000 description 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 101150032584 oxy-4 gene Proteins 0.000 description 1
- 125000005740 oxycarbonyl group Chemical group [*:1]OC([*:2])=O 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- VEDDBHYQWFOITD-UHFFFAOYSA-N para-bromobenzyl alcohol Chemical compound OCC1=CC=C(Br)C=C1 VEDDBHYQWFOITD-UHFFFAOYSA-N 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004894 pentylamino group Chemical group C(CCCC)N* 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical class OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical class [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical class CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- KIDBBTHHMJOMAU-UHFFFAOYSA-N propan-1-ol;hydrate Chemical compound O.CCCO KIDBBTHHMJOMAU-UHFFFAOYSA-N 0.000 description 1
- XTUSEBKMEQERQV-UHFFFAOYSA-N propan-2-ol;hydrate Chemical compound O.CC(C)O XTUSEBKMEQERQV-UHFFFAOYSA-N 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 238000005475 siliconizing Methods 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000008399 tap water Substances 0.000 description 1
- 235000020679 tap water Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical group CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- BWHOZHOGCMHOBV-BQYQJAHWSA-N trans-benzylideneacetone Chemical compound CC(=O)\C=C\C1=CC=CC=C1 BWHOZHOGCMHOBV-BQYQJAHWSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- QZCCVLMMVYJZTD-UHFFFAOYSA-N trimethyl(trifluoromethylsulfonyl)silane Chemical compound C[Si](C)(C)S(=O)(=O)C(F)(F)F QZCCVLMMVYJZTD-UHFFFAOYSA-N 0.000 description 1
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- NNPPMTNAJDCUHE-UHFFFAOYSA-N trimethylmethane Natural products CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 1
- 125000000297 undecanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003021 water soluble solvent Substances 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
- C07H13/02—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids
- C07H13/04—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids having the esterifying carboxyl radicals attached to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H3/00—Compounds containing only hydrogen atoms and saccharide radicals having only carbon, hydrogen, and oxygen atoms
- C07H3/10—Anhydrosugars, e.g. epoxides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Microbiology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Compounds Of Unknown Constitution (AREA)
Abstract
根據本發明,可提供有用於製造複數之ENA單體之結晶性之2,4-交聯的共通中間體、該中間體之立體選擇性的製造方法、及使用該中間體的各ENA單體之有效率的製造方法。
Description
本發明係關於用以製造複數個2’-O,4’-C-伸乙基交聯核酸(ENA)單體的新穎結晶性之共通中間體、於使用該中間體之醣基化中立體選擇性地合成β-加成體的方法、該製造中間體之製法、及使用該製造中間體的ENA單體之製法,該共通中間體包含成為含有2’-O,4’-C-伸乙基交聯核酸(ENA)的寡核苷酸的原料的A、G、T、C等嘧啶鹼基或嘌呤鹼基。
ENA單體係於製造修飾核酸醫藥及診斷藥上重要的化合物。
ENA製造中的重要步驟為用以形成基本骨架之2,4-交聯化反應及醣基化反應。
已知一般而言,於醣基化反應,於2位有醯氧基存在的情形,藉由鄰基效應控制立體結構,獲得β體之核苷(參照專利文獻1),但於2,4-交聯體及2-去氧體等之2位不存有醯氧基的情形,優先獲得α體之核苷(參照專利文獻2)。
因此,於製造β體之ENA的情形,使用於進行2,4-伸乙基氧基化之前,於2位具有醯氧基的狀態下導入鹼基的方法(參照專利文獻3~4、非專利文獻1~2)。
然而,習用方法由於包含2,4-交聯前之醣基化前的共通中間體(2位有羥基或乙醯基氧基、5位有保護基-O-伸乙基的化合物)之許多中間體為油狀化合物,而必須進行管柱純化,由共通中間體至各ENA單體的步驟過程長,因而並非工業上可滿足的製造方法(參照非專利文獻2)。
[先前技術文獻]
[專利文獻]
[專利文獻1] WO98/39352
[專利文獻2] WO99/14226
[專利文獻3] WO00/47599
[專利文獻4] WO2013/191129
[非專利文獻]
[非專利文獻1] Bioorganic & Medicinal Chemistry Letters 12 (2002) 73-76
[非專利文獻2] Bioorganic & Medicinal Chemistry Letters 11 (2003) 2211-2226
[發明欲解決之課題]
本發明之目的係提供一種用以製造各ENA單體之有用的結晶性之2,4-交聯的共通中間體,提供一種該中間體之立體選擇性的製造方法,及提供一種使用該中間體的各ENA單體之有效率的製造方法。
[用以解決課題之手段]
本發明人等,針對ENA單體之有效率的製造方法深入鑽研的結果,發現新穎的結晶性之2,4-交聯的共通中間體、該共通中間體之製造方法、使用該共通中間體的ENA單體之製造方法,遂而完成本發明。該共通中間體為結晶,因能藉由晶析而純化,而適用於工業的製造。又,該共通中間體之製造方法由於利用取得容易的便宜原料,不需單離而實施複數步驟,故可藉由少數步驟且高產率獲得該共通中間體。又,藉由使用該共通中間體,可於將2,4-交聯的骨架進行醣基化步驟前構築,因而縮短鹼基導入後之步驟而改善產率,有效率地分別製作複數個ENA單體。再者,於使用該中間體的醣基化,藉由將1位之羥基以碘原子或溴原子取代,即使沒有2位之醯基之鄰基效應,亦可控制立體結構而選擇性地製造β體。於ENA單體製造中的最終步驟的AMIDITE化,藉由使用特定之活化劑及乾燥劑,可削減AMIDITE試藥之當量,有效率地製造ENA單體。
即,本發明包含以下之發明。
(1)一種通式(I)所表示的化合物,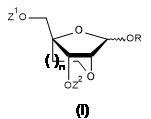 [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,R表示氫原子或脂肪族醯基,n表示0至4之整數];
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,R表示氫原子或脂肪族醯基,n表示0至4之整數];
(2)如(1)記載之化合物,其中R為氫原子或乙醯基;
(3)如(1)或(2)記載之化合物,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(4)如(1)或(2)記載之化合物,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(5)如(1)或(2)記載之化合物,其中Z1
及Z2
為苄基;
(6)如(1)至(5)中任一項記載之化合物,其中n為1;
(7)一種式(I’)所表示的化合物, ;
;
(8)一種式(I’’)所表示的化合物, ;
;
(9a)一種製造通式(III)所表示的化合物之方法,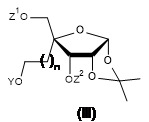 [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,Y表示經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基、低級烷氧基甲基、四氫哌喃基或矽基,n表示0至4之整數],
其特徵為包含:
(i)將通式(XXIX)所表示的化合物之1級羥基加以保護的步驟,
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,Y表示經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基、低級烷氧基甲基、四氫哌喃基或矽基,n表示0至4之整數],
其特徵為包含:
(i)將通式(XXIX)所表示的化合物之1級羥基加以保護的步驟, [式中,n表示0至4之整數],
(ii)將步驟(i)中所獲得的通式(XXX)所表示的化合物之羥基進行氧化的步驟,
[式中,n表示0至4之整數],
(ii)將步驟(i)中所獲得的通式(XXX)所表示的化合物之羥基進行氧化的步驟,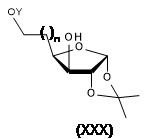 [式中,Y及n表示與前述同意義],
(iii)對步驟(ii)中所獲得的通式(XXXI)所表示的化合物之4位進行立體選擇性的羥基甲基化的步驟,
[式中,Y及n表示與前述同意義],
(iii)對步驟(ii)中所獲得的通式(XXXI)所表示的化合物之4位進行立體選擇性的羥基甲基化的步驟,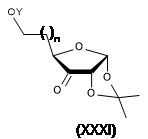 [式中,Y及n表示與前述同意義],
(iv)將步驟(iii)中所獲得的通式(XXXII)所表示的化合物之3位的羰基進行還原的步驟,
[式中,Y及n表示與前述同意義],
(iv)將步驟(iii)中所獲得的通式(XXXII)所表示的化合物之3位的羰基進行還原的步驟,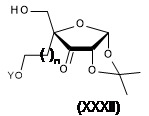 [式中,Y及n表示與前述同意義],
(v)將步驟(iv)中所獲得的通式(XXXIII)所表示的化合物之羥基加以保護的步驟,
[式中,Y及n表示與前述同意義],
(v)將步驟(iv)中所獲得的通式(XXXIII)所表示的化合物之羥基加以保護的步驟,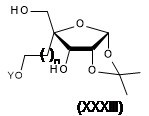 [式中,Y及n表示與前述同意義]。
[式中,Y及n表示與前述同意義]。
(10a)如(9a)記載之方法,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
(11a)如(9a)記載之方法,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
(12a)如(9a)記載之方法,其中Z1
及Z2
為苄基。
(13a)如(9a)至(12a)中任一項記載之方法,其中Y為三級丁基二苯基矽基、三級丁基二甲基矽基、四氫哌喃-2-基或三苯甲基。
(14a)如(9a)至(12a)中任一項記載之方法,其中Y為三苯甲基。
(15a)如(9a)至(14a)中任一項記載之方法,其中n為1。
(16a)如(9a)至(15a)中任一項記載之方法,其特徵為:於步驟(i)至(v)之各步驟中,將包含反應後之目的化合物的溶液之有機層進行水洗,將獲得的有機層直接使用於各後續步驟。
(9)一種製造通式(II)所表示的化合物之方法,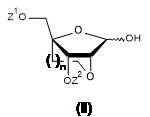 [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],
其特徵為包含:
(i)將通式(III)所表示的化合物之縮醛部位,
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],
其特徵為包含:
(i)將通式(III)所表示的化合物之縮醛部位,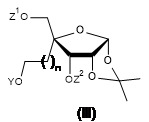 [式中,Z1
、Z2
及n表示與前述同意義,Y表示經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基、低級烷氧基甲基、四氫哌喃基或矽基],於酸觸媒存在下,低級烷基醇溶媒中,進行溶媒分解,而將Y去保護的步驟,
(ii)步驟(i)中所獲得的通式(IV)所表示的化合物之二醇部位進行環化的步驟,
[式中,Z1
、Z2
及n表示與前述同意義,Y表示經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基、低級烷氧基甲基、四氫哌喃基或矽基],於酸觸媒存在下,低級烷基醇溶媒中,進行溶媒分解,而將Y去保護的步驟,
(ii)步驟(i)中所獲得的通式(IV)所表示的化合物之二醇部位進行環化的步驟, [式中,Z1
、Z2
及n表示與前述同意義,A表示低級烷基],
(iii)進行步驟(ii)中所獲得的通式(V)所表示的化合物之變旋異構物(anomer)位之水解的步驟,
[式中,Z1
、Z2
及n表示與前述同意義,A表示低級烷基],
(iii)進行步驟(ii)中所獲得的通式(V)所表示的化合物之變旋異構物(anomer)位之水解的步驟, [式中,Z1
、Z2
、A及n表示與前述同意義];
[式中,Z1
、Z2
、A及n表示與前述同意義];
(10)如(9)記載之方法,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(11)如(9)記載之方法,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(12)如(9)記載之方法,其中Z1
及Z2
為苄基;
(13)如(9)至(12)中任一項記載之方法,其中A為甲基、乙基或丙基;
(14)如(9)至(12)中任一項記載之方法,其中A為甲基;
(15)如(9)至(14)中任一項記載之方法,其中Y為三級丁基二苯基矽基、三級丁基二甲基矽基、四氫哌喃-2-基或三苯甲基;
(16)如(9)至(14)中任一項記載之方法,其中Y為三苯甲基;
(17)如(9)至(16)中任一項記載之方法,其中n為1;
(18)如(9)至(17)中任一項記載之方法,其中酸觸媒為硫酸、對甲苯磺酸或甲磺酸;
(19)如(9)至(18)中任一項記載之方法,其中步驟(ii)使用三價磷試藥及偶氮二甲酸酯進行;
(20)如(19)記載之方法,其中三價磷試藥為三苯基膦或三(正丁基)膦;
(21)如(19)或(20)記載之方法,其中偶氮二甲酸酯為偶氮二甲酸二乙酯、偶氮二甲酸二異丙酯或偶氮二甲酸二(三級丁酯);
(22)如(9)至(21)中任一項記載之方法,其中步驟(iii)使用酸進行;
(23)如(22)記載之方法,其中酸為鹽酸、硫酸、三氟乙酸、甲磺酸或對甲苯磺酸;
(9b)如(9)記載之方法,其中Z1
及Z2
為苄基,
A為甲基,
Y為三苯甲基,
n為1;
(9c)如(9)記載之方法,其中酸觸媒為硫酸、對甲苯磺酸或甲磺酸,
步驟(ii)使用三價磷試藥及偶氮二甲酸酯進行,
步驟(iii)使用酸進行;
(9d)如(9)記載之方法,其中酸觸媒為硫酸、對甲苯磺酸或甲磺酸,
步驟(ii)使用三價磷試藥及偶氮二甲酸酯進行,
步驟(ii)所使用的三價磷試藥為三苯基膦或三(正丁基)膦,
步驟(ii)所使用的偶氮二甲酸酯為偶氮二甲酸二乙酯、偶氮二甲酸二異丙酯或偶氮二甲酸二(三級丁酯),
步驟(iii)使用酸進行,
步驟(iii)所使用的酸為鹽酸、硫酸、三氟乙酸、甲磺酸或對甲苯磺酸;
(9e)如(9)記載之方法,其中Z1
及Z2
為苄基,
A為甲基,
Y為三苯甲基,
n為1,
酸觸媒為硫酸、對甲苯磺酸或甲磺酸,
步驟(ii)使用三價磷試藥及偶氮二甲酸酯進行,
步驟(ii)所使用的三價磷試藥為三苯基膦或三(正丁基)膦,
步驟(ii)所使用的偶氮二甲酸酯為偶氮二甲酸二乙酯、偶氮二甲酸二異丙酯或偶氮二甲酸二(三級丁酯),
步驟(iii)使用酸進行,
步驟(iii)所使用的酸為鹽酸、硫酸、三氟乙酸、甲磺酸或對甲苯磺酸;
(24)一種製造通式(VI)所表示的化合物或其鹽之方法,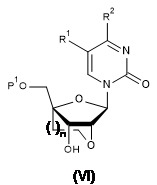 [式中,R1
表示低級烷基或氫原子,R2
表示羥基、胺基或經脂肪族醯基或芳香族醯基保護的胺基,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數],
其特徵為包含:
(i)使通式(II)所表示的化合物,
[式中,R1
表示低級烷基或氫原子,R2
表示羥基、胺基或經脂肪族醯基或芳香族醯基保護的胺基,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數],
其特徵為包含:
(i)使通式(II)所表示的化合物, [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],於溶媒中,與活化劑反應,將1位之羥基轉化為形成脫離基的基的步驟,及
(ii)使步驟(i)中所獲得的通式(VII)所表示的化合物,
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],於溶媒中,與活化劑反應,將1位之羥基轉化為形成脫離基的基的步驟,及
(ii)使步驟(i)中所獲得的通式(VII)所表示的化合物,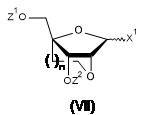 [式中,Z1
、Z2
及n表示與前述同意義,X1
表示形成脫離基的基],
與通式(VIII)所表示的化合物或其鹽,於溶媒中,鹵化劑存在下反應,
[式中,Z1
、Z2
及n表示與前述同意義,X1
表示形成脫離基的基],
與通式(VIII)所表示的化合物或其鹽,於溶媒中,鹵化劑存在下反應,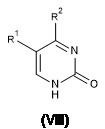 [式中,R1
及R2
表示與前述同意義],
而立體選擇性地獲得通式(IX)所表示的化合物或其鹽的步驟,
[式中,R1
及R2
表示與前述同意義],
而立體選擇性地獲得通式(IX)所表示的化合物或其鹽的步驟,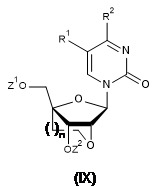 [式中,Z1
、Z2
、R1
、R2
及n表示與前述同意義];
[式中,Z1
、Z2
、R1
、R2
及n表示與前述同意義];
(25)如(24)記載之方法,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(26)如(24)記載之方法,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(27)如(24)記載之方法,其中Z1
及Z2
為苄基;
(28)如(24)至(27)中任一項記載之方法,其中P1
為4,4’-二甲氧基三苯甲基;
(29)如(24)至(28)中任一項記載之方法,其中X1
為鹵素原子、脂肪族醯氧基、鹵素取代的低級烷基亞胺氧基或鹵素取代的低級烷基磺醯氧基;
(30)如(24)至(28)中任一項記載之方法,其中X1
為碘原子、乙醯氧基或三氯乙醯亞胺氧基;
(31)如(24)至(30)中任一項記載之方法,其中n為1;
(32)如(24)至(31)中任一項記載之方法,其中R1
為甲基或氫原子;
(33)如(24)至(32)中任一項記載之方法,其中R2
為羥基或苄醯基胺基;
(34)如(24)至(31)中任一項記載之方法,其中R1
為甲基,R2
為羥基;
(35)如(24)至(31)中任一項記載之方法,其中R1
為甲基,R2
為苄醯基胺基;
(36)如(24)至(35)中任一項記載之方法,其包含:
(iii)使步驟(ii)中所獲得的通式(IX)所表示的化合物或其鹽,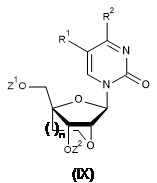 [式中,Z1
、Z2
、R1
、R2
及n表示與前述同意義],於溶媒中,與羥基之去保護試藥反應,而將Z1
及Z2
去保護的步驟、及
(iv)使步驟(iii)所獲得的二醇化合物或其鹽與1級羥基之保護試藥反應,而獲得通式(VI)所表示的化合物或其鹽的步驟,
[式中,Z1
、Z2
、R1
、R2
及n表示與前述同意義],於溶媒中,與羥基之去保護試藥反應,而將Z1
及Z2
去保護的步驟、及
(iv)使步驟(iii)所獲得的二醇化合物或其鹽與1級羥基之保護試藥反應,而獲得通式(VI)所表示的化合物或其鹽的步驟,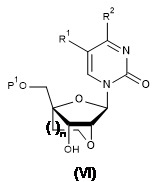 [式中,P1
、R1
、R2
及n表示與前述同意義];
[式中,P1
、R1
、R2
及n表示與前述同意義];
(37)如(24)至(36)中任一項記載之方法,其中活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯;
(38)如(24)至(37)中任一項記載之方法,其中鹵化劑為氯三甲基矽烷、溴三甲基矽烷或碘三甲基矽烷;
(24a)如(24)記載之方法,其中Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
n為1,
R1
為甲基,R2
為苄醯基胺基;
(24b)如(24)記載之方法,其中Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
X1
為碘原子、乙醯氧基或三氯乙醯亞胺氧基,
n為1,
R1
為甲基,R2
為苄醯基胺基;
(24c)如(24)記載之方法,其中活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯,
鹵化劑為氯三甲基矽烷、溴三甲基矽烷或碘三甲基矽烷;
(24d)如(24)記載之方法,其中Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
X1
為碘原子、乙醯氧基或三氯乙醯亞胺氧基,
n為1,
R1
為甲基,R2
為苄醯基胺基,
活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯,
鹵化劑為氯三甲基矽烷、溴三甲基矽烷或碘三甲基矽烷;
(39)一種製造通式(X)所表示的化合物或其鹽之方法,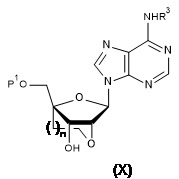 [式中,R3
表示脂肪族醯基或芳香族醯基,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數],
(i)使通式(II)所表示的化合物,
[式中,R3
表示脂肪族醯基或芳香族醯基,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數],
(i)使通式(II)所表示的化合物,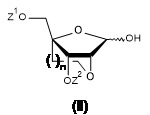 [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示與前述同意義],於溶媒中,與活化劑反應,而將1位之羥基轉化為形成脫離基的基的步驟,
(ii)步驟(i)中所獲得的通式(XI)所表示的化合物,
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示與前述同意義],於溶媒中,與活化劑反應,而將1位之羥基轉化為形成脫離基的基的步驟,
(ii)步驟(i)中所獲得的通式(XI)所表示的化合物,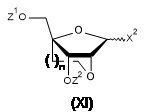 [式中,Z1
、Z2
及n表示與前述同意義,X2
表示形成脫離基的基],與通式(XII)所表示的化合物或其鹽,
[式中,Z1
、Z2
及n表示與前述同意義,X2
表示形成脫離基的基],與通式(XII)所表示的化合物或其鹽,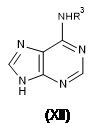 [式中,R3
表示與前述同意義],
於溶媒中,酸試藥存在下反應的步驟,
(iii)接著,使異構物化,立體選擇性地獲得通式(XIII)所表示的化合物或其鹽的步驟,
[式中,R3
表示與前述同意義],
於溶媒中,酸試藥存在下反應的步驟,
(iii)接著,使異構物化,立體選擇性地獲得通式(XIII)所表示的化合物或其鹽的步驟,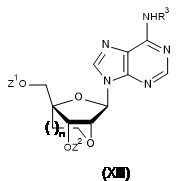 [式中,Z1
、Z2
、R3
及n表示與前述同意義];
[式中,Z1
、Z2
、R3
及n表示與前述同意義];
(39a)如(39)記載之方法,其中於步驟(iii),藉由加熱而使異構物化;
(40)如(39)記載之方法,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(41)如(39)記載之方法,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(42)如(39)記載之方法,其中Z1
及Z2
為苄基;
(43)如(39)至(42)中任一項記載之方法,其中P1
為4,4’-二甲氧基三苯甲基;
(44)如(39)至(43)中任一項記載之方法,其中X2
為鹵素原子、脂肪族醯氧基、鹵素取代的低級烷基亞胺氧基或鹵素取代的低級烷基磺醯氧基;
(45)如(39)至(43)中任一項記載之方法,其中X2
為乙醯氧基;
(46)如(39)至(45)中任一項記載之方法,其中n為1;
(47)如(39)至(46)中任一項記載之方法,其中R3
為乙醯基或苄醯基;
(48)如(39)至(46)中任一項記載之方法,其中R3
為苄醯基;
(49)如(39)至(48)中任一項記載之方法,其包含:
(iv)使步驟(iii)中所獲得的通式(XIII)所表示的化合物或其鹽, [式中,Z1
、Z2
、R3
及n表示與前述同意義],
於溶媒中,與羥基之去保護試藥反應,而將Z1
及Z2
去保護的步驟,及
(v)使步驟(iv)中所獲得的二醇化合物或其鹽、與1級羥基之保護試藥反應,選擇性保護1級羥基,藉此獲得通式(X)所表示的化合物或其鹽的步驟,
[式中,Z1
、Z2
、R3
及n表示與前述同意義],
於溶媒中,與羥基之去保護試藥反應,而將Z1
及Z2
去保護的步驟,及
(v)使步驟(iv)中所獲得的二醇化合物或其鹽、與1級羥基之保護試藥反應,選擇性保護1級羥基,藉此獲得通式(X)所表示的化合物或其鹽的步驟,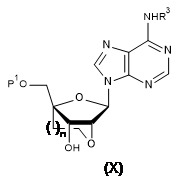 [式中,R3
、n及P1
表示與前述同意義];
[式中,R3
、n及P1
表示與前述同意義];
(50)如(39)至(49)中任一項記載之方法,其中活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯;
(51)如(39)至(50)中任一項記載之方法,其中酸試藥為三氟甲磺酸三甲基矽酯及三氟乙酸;
(39b)如(39)記載之方法,其中Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
n為1,
R3
為苄醯基;
(39c)如(39)記載之方法,其中Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
X2
為乙醯氧基,
n為1,
R3
為苄醯基;
(39d)如(39)記載之方法,其中活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯,
酸試藥為三氟甲磺酸三甲基矽酯及三氟乙酸;
(39e)如(39)記載之方法,其中Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
X2
為乙醯氧基,
n為1,
R3
為苄醯基,
活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯,
酸試藥為三氟甲磺酸三甲基矽酯及三氟乙酸;
(52)一種通式(XIV)所表示的化合物或其鹽, [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數];
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數];
(53)如(52)記載之化合物或其鹽,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(54)如(52)記載之化合物或其鹽,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(55)如(52)記載之化合物或其鹽,其中Z1
及Z2
為苄基;
(56)如(52)至(55)中任一項記載之化合物或其鹽,其中n為1;
(57)一種通式(XIV’)所表示的化合物或其鹽,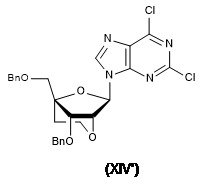 ;
;
(58)一種製造通式(X)所表示的化合物或其鹽之方法, [式中,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,R3
表示脂肪族醯基或芳香族醯基,n表示1至4之整數],
其特徵為包含:
(i)使通式(XIV)所表示的化合物或其鹽與胺基化劑反應,
[式中,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,R3
表示脂肪族醯基或芳香族醯基,n表示1至4之整數],
其特徵為包含:
(i)使通式(XIV)所表示的化合物或其鹽與胺基化劑反應,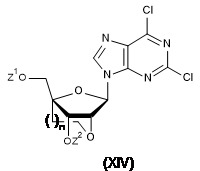 [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],
而將嘌呤環之6位之氯原子取代為胺基的步驟,
(ii)使步驟(i)中所獲得的通式(XV)所表示的化合物或其鹽,
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],
而將嘌呤環之6位之氯原子取代為胺基的步驟,
(ii)使步驟(i)中所獲得的通式(XV)所表示的化合物或其鹽,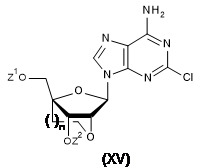 [式中,Z1
、Z2
及n表示與前述同意義],
藉由於溶媒中,金屬觸媒存在下,與還原劑反應,將嘌呤環之2位之氯原子取代為氫原子,而將Z1
及Z2
去保護,獲得通式(XVI)所表示的化合物或其鹽的步驟,
[式中,Z1
、Z2
及n表示與前述同意義],
藉由於溶媒中,金屬觸媒存在下,與還原劑反應,將嘌呤環之2位之氯原子取代為氫原子,而將Z1
及Z2
去保護,獲得通式(XVI)所表示的化合物或其鹽的步驟,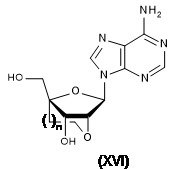 [式中,n表示與前述同意義];
[式中,n表示與前述同意義];
(58b)如(58)記載之方法,其中金屬觸媒為承載於碳上的金屬觸媒;
(59)如(58)記載之化合物或其鹽,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(60)如(58)記載之方法,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(61)如(58)記載之方法,其中Z1
及Z2
為苄基;
(62)如(58)至(61)中任一項記載之方法,其中P1
為4,4’-二甲氧基三苯甲基;
(63)如(58)至(62)中任一項記載之方法,其中n為1;
(64)如(58)至(63)中任一項記載之方法,其中R3
為乙醯基或苄醯基;
(65)如(58)至(63)中任一項記載之方法,其中R3
為苄醯基;
(66)如(58)至(65)中任一項記載之方法,其包含:
(iii)使步驟(ii)中所獲得的通式(XVI)所表示的化合物或其鹽與1級羥基之保護試藥反應, [式中,n表示與前述同意義],
而選擇性地保護1級羥基的步驟,
(iv)藉由使步驟(iii)中所獲得的通式(XVII)所表示的化合物或其鹽與醯化劑反應,
[式中,n表示與前述同意義],
而選擇性地保護1級羥基的步驟,
(iv)藉由使步驟(iii)中所獲得的通式(XVII)所表示的化合物或其鹽與醯化劑反應,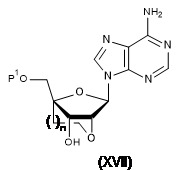 [式中,P1
及n表示與前述同意義],
而獲得通式(X)所表示的化合物或其鹽的步驟,
[式中,P1
及n表示與前述同意義],
而獲得通式(X)所表示的化合物或其鹽的步驟,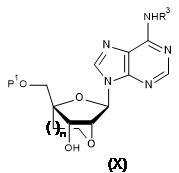 [式中,P1
、R3
及n表示與前述同意義];
[式中,P1
、R3
及n表示與前述同意義];
(67)如(58)至(66)中任一項記載之方法,其中胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨。
(68)如(58)至(67)中任一項記載之方法,其中金屬觸媒為鈀、氫氧化鈀或鉑;
(69)如(58)至(68)中任一項記載之方法,其中還原劑為氫、甲酸或甲酸銨;
(70)如(66)至(69)中任一項記載之方法,其中醯化劑為苯甲醯氯或苯甲酸酐;
(58a)如(58)記載之方法,其包含:
(iii-a)使(58)中的步驟(ii)中所獲得的通式(XVI)所表示的化合物或其鹽,
[式中,n表示與前述同意義],與1級羥基之保護試藥反應,而選擇性地保護1級羥基的步驟,
(iv-a)藉由使步驟(iii-a)中所獲得的通式(XVII)所表示的化合物或其鹽與醯化劑反應,
[式中,P1
及n表示與前述同意義],
而獲得通式(X)所表示的化合物或其鹽的步驟,
[式中,P1
、R3
及n表示與前述同意義],
Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
n為1,
R3
為苄醯基,
胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨,
金屬觸媒為鈀、氫氧化鈀或鉑,
還原劑為氫、甲酸或甲酸銨,
醯化劑為苯甲醯氯或苯甲酸酐;
(71)一種製造通式(XVIII)所表示的化合物或其鹽之方法,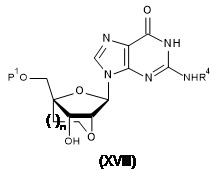 [式中,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,R4
表示脂肪族醯基或芳香族醯基,n表示1至4之整數],
其特徵為包含:
(i)使通式(XIV)所表示的化合物或其鹽,
[式中,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,R4
表示脂肪族醯基或芳香族醯基,n表示1至4之整數],
其特徵為包含:
(i)使通式(XIV)所表示的化合物或其鹽, [式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],
於溶媒中,鹼存在下,使與可經低級烷基、低級烷氧基、鹵素或氰基取代的芐醇反應,而將嘌呤環之6位之氯原子取代為低級烷基、低級烷氧基、可經鹵素或氰基取代的苄氧基的步驟,
(ii)使步驟(i)中所獲得的通式(XIX)所表示的化合物或其鹽,
[式中,Z1
及Z2
為相同或相異,表示羥基之保護基,n表示0至4之整數],
於溶媒中,鹼存在下,使與可經低級烷基、低級烷氧基、鹵素或氰基取代的芐醇反應,而將嘌呤環之6位之氯原子取代為低級烷基、低級烷氧基、可經鹵素或氰基取代的苄氧基的步驟,
(ii)使步驟(i)中所獲得的通式(XIX)所表示的化合物或其鹽,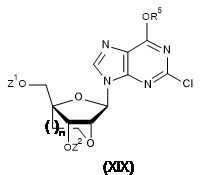 [式中,Z1
、Z2
及n表示與前述同意義,R5
表示可經低級烷基、低級烷氧基、鹵素或氰基取代的苄基],
於溶媒中,鈀觸媒、膦配位子存在下,與醯胺化劑交叉偶合,而獲得通式(XX)所表示的化合物或其鹽的步驟,
[式中,Z1
、Z2
及n表示與前述同意義,R5
表示可經低級烷基、低級烷氧基、鹵素或氰基取代的苄基],
於溶媒中,鈀觸媒、膦配位子存在下,與醯胺化劑交叉偶合,而獲得通式(XX)所表示的化合物或其鹽的步驟,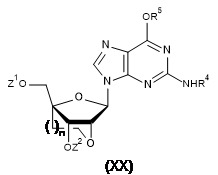 [式中,Z1
、Z2
、R4
、R5
及n表示與前述同意義];
[式中,Z1
、Z2
、R4
、R5
及n表示與前述同意義];
(72)如(71)記載之方法,其中Z1
及Z2
為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基;
(73)如(71)記載之方法,其中Z1
及Z2
為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基;
(74)如(71)記載之方法,其中Z1
及Z2
為苄基;
(75)如(71)至(74)中任一項記載之方法,其中P1
為4,4’-二甲氧基三苯甲基;
(76)如(71)至(75)中任一項記載之方法,其中n為1;
(77)如(71)至(76)中任一項記載之方法,其中R4
為異丁醯;
(78)如(71)至(77)中任一項記載之方法,其中R5
為苄基;
(79)如(71)至(78)中任一項記載之方法,其包含:
(iii)使步驟(ii)中所獲得的通式(XX)所表示的化合物或其鹽,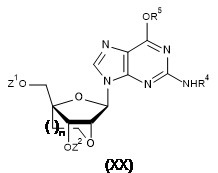 [式中,Z1
、Z2
、R4
、R5
及n表示與前述同意義]
於溶媒中,與羥基之去保護試藥反應,而將Z1
、Z2
及R5
去保護的步驟,
(iv)使步驟(iii)中所獲得的通式(XXI)所表示的化合物或其鹽,
[式中,Z1
、Z2
、R4
、R5
及n表示與前述同意義]
於溶媒中,與羥基之去保護試藥反應,而將Z1
、Z2
及R5
去保護的步驟,
(iv)使步驟(iii)中所獲得的通式(XXI)所表示的化合物或其鹽,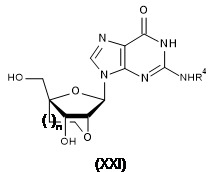 [式中,R4
及n表示與前述同意義],
與1級羥基之保護試藥反應,藉由選擇性保護1級羥基,而獲得通式(XVIII)所表示的化合物或其鹽的步驟,
[式中,R4
及n表示與前述同意義],
與1級羥基之保護試藥反應,藉由選擇性保護1級羥基,而獲得通式(XVIII)所表示的化合物或其鹽的步驟,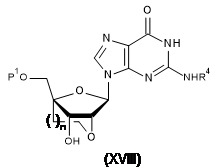 [式中,P1
、R4
及n表示與前述同意義];
[式中,P1
、R4
及n表示與前述同意義];
(80)如(71)至(79)中任一項記載之方法,其中鹼為氫氧化鈉、碳酸鈉、碳酸銫、三乙基胺、吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯;
(81)如(71)至(80)中任一項記載之方法,其中鈀觸媒為參(二亞苄基丙酮)(氯仿)二鈀、乙酸鈀(II)或參(二亞苄基丙酮)二鈀(0);
(82)如(71)至(81)中任一項記載之方法,其中膦配位子為4,5’-雙(二苯基膦基)-9,9’二甲基𠮿 、1,1’-雙(二苯基膦基)二茂鐵(1,1’-bis(diphenylphosphino) ferrocene)、1,2-雙(二苯基膦基)乙烷或2-二環己基膦基-2’-(N,N-二甲基胺基)聯苯;
、1,1’-雙(二苯基膦基)二茂鐵(1,1’-bis(diphenylphosphino) ferrocene)、1,2-雙(二苯基膦基)乙烷或2-二環己基膦基-2’-(N,N-二甲基胺基)聯苯;
(83)如(71)至(82)中任一項記載之方法,其中醯胺化劑為乙醯基醯胺、苄醯基醯胺或異丁基醯胺;
(84)如(79)至(83)中任一項記載之方法,其中羥基之去保護試藥為金屬觸媒及還原劑;
(84a)如(79)至(83)中任一項記載之方法,其中羥基之去保護試藥為承載於碳上的金屬觸媒及氫;
(85)如(84)記載之方法,其中金屬觸媒為鈀、氫氧化鈀或鉑;
(86)如(84)或(85)記載之方法,其中還原劑為氫、甲酸或甲酸銨;
(71a)如(71)記載之方法,其包含:
(iii-a)使(71)中的步驟(ii)中所獲得的通式(XX)所表示的化合物或其鹽,
[式中,Z1
、Z2
、R4
、R5
及n表示與前述同意義],
於溶媒中,與羥基之去保護試藥反應,而將Z1
、Z2
及R5
去保護的步驟,
(iv-a)使步驟(iii-a)中所獲得的通式(XXI)所表示的化合物或其鹽,
[式中,R4
及n表示與前述同意義],
與1級羥基之保護試藥反應,藉由選擇性保護1級羥基,而獲得通式(XVIII)所表示的化合物或其鹽的步驟,
[式中,P1
、R4
及n表示與前述同意義],
Z1
及Z2
為苄基,
P1
為4,4’-二甲氧基三苯甲基,
n為1,
R4
為異丁醯基,
鹼為氫氧化鈉、碳酸鈉、碳酸銫、三乙基胺、吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯,
鈀觸媒為參(二亞苄基丙酮)(氯仿)二鈀、乙酸鈀(II)或參(二亞苄基丙酮)二鈀(0),
膦配位子為4,5’-雙(二苯基膦基)-9,9’二甲基𠮿、1,1’-雙(二苯基膦基)二茂鐵、1,2-雙(二苯基膦基)乙烷或2-二環己基膦基-2’-(N,N-二甲基胺基)聯苯,
醯胺化劑為乙醯基醯胺、苄醯基醯胺或異丁基醯胺,
羥基之去保護試藥為承載於碳上的金屬觸媒及氫,
金屬觸媒為鈀、氫氧化鈀或鉑;
(87)一種製造通式(XXII)所表示的化合物或其鹽之方法,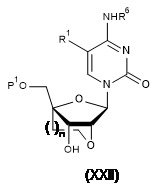 [式中,R1
表示低級烷基或氫原子,R6
表示脂肪族醯基或芳香族醯基,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數],
其特徵為包含:
(i)使通式(XXIII)所表示的化合物或其鹽,
[式中,R1
表示低級烷基或氫原子,R6
表示脂肪族醯基或芳香族醯基,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數],
其特徵為包含:
(i)使通式(XXIII)所表示的化合物或其鹽,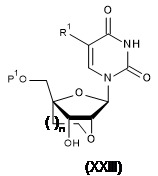 [式中,P1
、R1
及n表示與前述同意義],
於溶媒中,與羥基之保護試藥反應,將3’位之羥基保護的步驟,
(ii)使步驟(i)中所獲得的通式(XXIV)所表示的化合物或其鹽,
[式中,P1
、R1
及n表示與前述同意義],
於溶媒中,與羥基之保護試藥反應,將3’位之羥基保護的步驟,
(ii)使步驟(i)中所獲得的通式(XXIV)所表示的化合物或其鹽,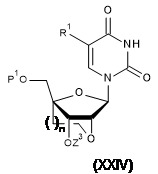 [式中,P1
、R1
及n表示與前述同意義,Z3
表示脂肪族醯基或芳香族醯基],
於溶媒中,鹼及觸媒存在下,與活化劑反應的步驟,
(iii)接著,藉由使與胺基化劑反應,而獲得通式(XXV)所表示的化合物或其鹽的步驟,
[式中,P1
、R1
及n表示與前述同意義,Z3
表示脂肪族醯基或芳香族醯基],
於溶媒中,鹼及觸媒存在下,與活化劑反應的步驟,
(iii)接著,藉由使與胺基化劑反應,而獲得通式(XXV)所表示的化合物或其鹽的步驟,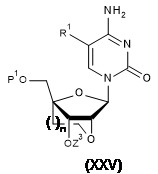 [式中,P1
、R1
、Z3
及n表示與前述同意義];
[式中,P1
、R1
、Z3
及n表示與前述同意義];
(88)如(87)記載之方法,其中P1
為三苯甲基;
(89)如(87)或(88)記載之方法,其中Z3
為乙醯基;
(90)如(87)至(89)中任一項記載之方法,其中n為1;
(91)如(87)至(90)中任一項記載之方法,其中R1
為甲基或氫原子;
(92)如(87)至(91)中任一項記載之方法,其中R6
為乙醯基或苄醯基;
(93)如(87)至(91)中任一項記載之方法,其中R6
為苄醯基;
(94)如(87)至(93)中任一項記載之方法,其包含:
(iv)使步驟(iii)中所獲得的通式(XXV)所表示的化合物或其鹽,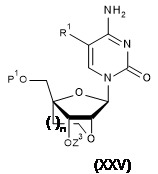 [式中,P1
、R1
、Z3
及n表示與前述同意義],
於溶媒中,與醯化劑反應的步驟,
(v)使步驟(iv)中所獲得的通式(XXVI)所表示的化合物或其鹽,
[式中,P1
、R1
、Z3
及n表示與前述同意義],
於溶媒中,與醯化劑反應的步驟,
(v)使步驟(iv)中所獲得的通式(XXVI)所表示的化合物或其鹽,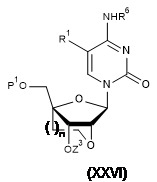 [式中,P1
、R1
、R6
、Z3
及n表示與前述同意義],與羥基之去保護試藥反應,藉由僅將Z3
去保護,而獲得通式(XXII)所表示的化合物或其鹽的步驟,
[式中,P1
、R1
、R6
、Z3
及n表示與前述同意義],與羥基之去保護試藥反應,藉由僅將Z3
去保護,而獲得通式(XXII)所表示的化合物或其鹽的步驟,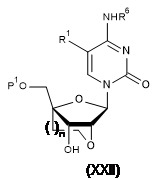 [式中,P1
、R1
、R6
及n表示與前述同意義];
[式中,P1
、R1
、R6
及n表示與前述同意義];
(95)如(87)至(94)中任一項記載之方法,其中觸媒為N,N-二甲基胺基吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯;
(96)如(87)至(95)中任一項記載之方法,其中活化劑為對甲苯磺醯氯或2,4,6-三異丙基苯磺醯氯;
(97)如(87)至(96)中任一項記載之方法,其中胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨;
(98)如(94)至(97)中任一項記載之方法,其中醯化劑為苯甲醯氯或苯甲酸酐;
(87a)如(87)記載之方法,其中P1
為三苯甲基,
Z3
為乙醯基,
n為1,
R1
為甲基或氫原子,
R6
為苄醯基;
(87b)如(87)記載之方法,其包含:
(iv-b)使(87)中的步驟(iii)中所獲得的通式(XXV)所表示的化合物或其鹽,
[式中,P1
、R1
、Z3
及n表示與前述同意義]
於溶媒中,與醯化劑反應的步驟,
(v-b)使步驟(iv-b)中所獲得的通式(XXVI)所表示的化合物或其鹽,
[式中,P1
、R1
、R6
、Z3
及n表示與前述同意義],
與羥基之去保護試藥反應,藉由僅將Z3
去保護,而獲得通式(XXII)所表示的化合物或其鹽的步驟,
[式中,P1
、R1
、R6
及n表示與前述同意義],
觸媒為N,N-二甲基胺基吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯,
活化劑為對甲苯磺醯氯或2,4,6-三異丙基苯磺醯氯,
胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨,
醯化劑為苯甲醯氯或苯甲酸酐;
(87c)如(87)記載之方法,其包含:
(iv-c)使(87)中的步驟(iii)中所獲得的通式(XXV)所表示的化合物或其鹽,
[式中,P1
、R1
、Z3
及n表示與前述同意義],
於溶媒中,與醯化劑反應的步驟,
(v-c)使步驟(iv-c)中所獲得的通式(XXVI)所表示的化合物或其鹽,
[式中,P1
、R1
、R6
、Z3
及n表示與前述同意義],
與羥基之去保護試藥反應,藉由僅將Z3
去保護,而獲得通式(XXII)所表示的化合物或其鹽的步驟,
[式中,P1
、R1
、R6
及n表示與前述同意義],
P1
為三苯甲基,
Z3
為乙醯基,
n為1,
R1
為甲基或氫原子,
R6
為苄醯基,
觸媒為N,N-二甲基胺基吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯,
活化劑為對甲苯磺醯氯或2,4,6-三異丙基苯磺醯氯,
胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨,
醯化劑為苯甲醯氯或苯甲酸酐;
(99)一種製造通式(XXVIII)所表示的化合物或其鹽之方法,
其使通式(XXVII)所表示的化合物或其鹽,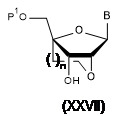 [式中,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,B表示可具有選自下述α群組的1或2個以上之取代基的2-側氧基-嘧啶-1-基或嘌呤-9-基,n表示0至4之整數],於溶媒中,活化劑及乾燥劑之存在下,與AMIDITE化試藥反應,
[式中,P1
表示可經1至3個之低級烷氧基取代的三苯甲基,B表示可具有選自下述α群組的1或2個以上之取代基的2-側氧基-嘧啶-1-基或嘌呤-9-基,n表示0至4之整數],於溶媒中,活化劑及乾燥劑之存在下,與AMIDITE化試藥反應,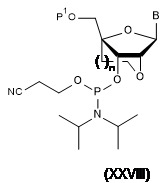 [式中,P1
、B及n表示與前述同意義];
(α群組)
羥基、
經保護的羥基、
低級烷氧基、
巰基、
經保護的巰基、
低級烷硫基、
胺基、
經保護的胺基、
低級烷基胺基、
低級烷基、及
鹵素原子;
[式中,P1
、B及n表示與前述同意義];
(α群組)
羥基、
經保護的羥基、
低級烷氧基、
巰基、
經保護的巰基、
低級烷硫基、
胺基、
經保護的胺基、
低級烷基胺基、
低級烷基、及
鹵素原子;
(100)如(99)記載之方法,其中P1
為4,4’-二甲氧基三苯甲基;
(101)如(99)或(100)記載之方法,其中B為2-側氧基-4-羥基-5-甲基嘧啶-1-基、胺基經保護的2-側氧基-4-胺基-嘧啶-1-基、胺基經保護的4-胺基-5-甲基-2-側氧基-嘧啶-1-基、胺基經保護的6-胺基嘌呤-9-基或胺基經保護的2-胺基-6-羥基嘌呤-9-基;
(102)如(99)或(100)記載之方法,其中B為2-側氧基-4-羥基-5-甲基嘧啶-1-基、2-側氧基-4-苄醯基胺基-嘧啶-1-基、4-苄醯基胺基-5-甲基-2-側氧基-嘧啶-1-基、6-苄醯基胺基嘌呤-9-基、或2-異丁醯基胺基-6-羥基嘌呤-9-基;
(103)如(99)至(102)中任一項記載之方法,其中n為1;
(104)如(99)至(103)中任一項記載之方法,其中活化劑為吡啶三氟乙酸鹽、N-甲基咪唑三氟乙酸鹽、N-異丙基咪唑三氟乙酸鹽、5-苄硫基四唑、5-苯基四唑、4,5-二氰基咪唑或2,4,5-四溴咪唑;
(105)如(99)至(103)中任一項記載之方法,其中活化劑為4,5-二氰基咪唑;
(106)如(99)至(105)中任一項記載之方法,其中AMIDITE化試藥為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(2-cyanoethyl N, N, N’, N’-tetraisopropyl phosphorodiamidite)或2-氰基乙基二異丙基氯亞磷醯胺(2-cyanoethyl diisopropyl chlorophosphoroamidide);
(107)如(99)至(105)中任一項記載之方法,其中AMIDITE化試藥為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺;
(108)如(99)至(107)中任一項記載之方法,其中乾燥劑為分子篩3A、分子篩4A、或分子篩5A;
(99a)如(99)記載之方法,其中P1
為4,4’-二甲氧基三苯甲基,
B為2-側氧基-4-羥基-5-甲基嘧啶-1-基、2-側氧基-4-苄醯基胺基-嘧啶-1-基、4-苄醯基胺基-5-甲基-2-側氧基-嘧啶-1-基、6-苄醯基胺基嘌呤-9-基、或2-異丁醯基胺基-6-羥基嘌呤-9-基,
n為1;
(99b)如(99)記載之方法,其中活化劑為吡啶三氟乙酸鹽、N-甲基咪唑三氟乙酸鹽、N-異丙基咪唑三氟乙酸鹽、5-苄硫基四唑、5-苯基四唑、4,5-二氰基咪唑或2,4,5-四溴咪唑,
AMIDITE化試藥為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺或2-氰基乙基二異丙基氯亞磷醯胺,
乾燥劑為分子篩3A、分子篩4A、或分子篩5A;
(99c)如(99)記載之方法,其中P1
為4,4’-二甲氧基三苯甲基,
B為2-側氧基-4-羥基-5-甲基嘧啶-1-基、2-側氧基-4-苄醯基胺基-嘧啶-1-基、4-苄醯基胺基-5-甲基-2-側氧基-嘧啶-1-基、6-苄醯基胺基嘌呤-9-基、或2-異丁醯基胺基-6-羥基嘌呤-9-基,
n為1,
活化劑為吡啶三氟乙酸鹽、N-甲基咪唑三氟乙酸鹽、N-異丙基咪唑三氟乙酸鹽、5-苄硫基四唑、5-苯基四唑、4,5-二氰基咪唑或2,4,5-四溴咪唑,
AMIDITE化試藥為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺或2-氰基乙基二異丙基氯亞磷醯胺,
乾燥劑為分子篩3A、分子篩4A、或分子篩5A;
(109)一種寡核苷酸之製造方法,其包含以下之步驟:
(A)藉由如(99)至(108)中任一項記載之方法而合成ENA單體的步驟,及
(B)使用步驟(A)中所獲得的ENA單體、其他之核酸之亞磷醯胺化合物及/或配位體之亞磷醯胺化合物,依據所期望的序列而使核苷酸鏈延長的步驟。
(110)如(109)記載之方法,其特徵為寡核苷酸係選自以下之DMD AO01~DMD AO15的任一者之式所表示的序列,
(DMD AO01)
HO-Ce2s
-Am1s
-Gm1s
-Te2s
-Te2s
-Um1s
-Gm1s
-Ce2s
-Ce2s
-Gm1s
-Ce2s
-Te2s
-Gm1s
-Ce2s
-Ce2s
-Ce2s
-Am1s
-Am1s
- CH2
CH2
OH(序列識別號1)
(DMD AO02)
HO-Te2s
-Gm1s
-Te2s
-Te2s
-Ce2s
-Te2s
-Gm1s
-Am1s
-Ce2s
-Am1s
-Am1s
-Ce2s
-Am1s
-Gm1s
-Te2s
-Te2s
-Te2s
-Gm1s
- CH2
CH2
OH(序列識別號2)
(DMD AO03)
HO-Ce2s
-Gm1s
-Ce2s
-Te2s
-Gm1s
-Cm1s
-Ce2s
-Ce2s
-Am1s
-Am1s
-Te2s
-Gm1s
-Ce2s
-Ce2s
-Am1s
-Um1s
-Ce2s
-Ce2s
- CH2
CH2
OH(序列識別號3)
(DMD AO04)
HO-Ce2s
-Am1s
-Te2s
-Am1s
-Am1s
-Te2s
-Gm1s
-Am1s
-Ae2s
-Am1s
-Am1s
-Ce2s
-Gm1s
-Cm1s
-Ce2s
-Gm1s
-Ce2s
-Ce2s
- CH2
CH2
OH(序列識別號4)
(DMD AO05)
HO-Te2s
-Um1s
-Ce2s
-Cm1s
-Ce2s
-Am1s
-Am1s
-Te2s
-Um1s
-Cm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Gm1s
-Ae2s
-Am1s
-Te2s
- CH2
CH2
OH(序列識別號5)
(DMD AO06)
HO-Ce2s
-Ce2s
-Am1s
-Um1s
-Te2s
-Um1s
-Gm1s
-Te2s
-Am1s
-Um1s
-Te2s
-Te2s
-Am1s
-Gm1s
-Ce2s
-Am1s
-Te2s
-Gm1s
- CH2
CH2
OH(序列識別號6)
(DMD AO07)
HO-Gm1s
-Gm1s
-Ce2s
-Te2s
-Gm1s
-Cm1s
-Te2s
-Te2s
-Um1s
-Gm1s
-Ce2s
-Cm1s
-Cm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Ce2s
-CH2
CH2
OH(序列識別號7)
(DMD AO08)
HO-Gm1s
-Ce2s
-Te2s
-Am1s
-Gm1s
-Gm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Gm1s
-Te2s
-Gm1s
-Cm1s
-Te2s
-Te2s
-Um1s
-CH2
CH2
OH(序列識別號8)
(DMD AO09)
HO-Am1s
-Ce2s
-Ce2s
-Gm1s
-Cm1s
-Ce2s
-Te2s
-Um1s
-Cm1s
-Ce2s
-Am1s
-Cm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Ae2s
-Gm1s
-CH2
CH2
OH; (序列識別號9)
(DMD AO10)
HO-Ge2s
-Ge2s
-Ce2s
-Ae2s
-Te2s
-Um1s
-Um1s
-Cm1s
-Um1s
-Am1s
-Gm1s
-Um1s
-Um1s
-Te2s
-Ge2s
-Ge2s
-Ae2s
-Ge2s
- CH2
CH2
OH(序列識別號10)
(DMD AO11)
HO-Gm1s
-Gm1s
-Ce2s
-Am1s
-Te2s
-Te2s
-Um1s
-Ce2s
-Te2s
-Am1s
-Gm1s
-Um1s
-Te2s
-Te2s
-Gm1s
-Gm1s
-Ae2s
-Gm1s
- CH2
CH2
OH(序列識別號11)
(DMD AO12)
HO-Ae2s
-Gm1s
-Te2s
-Um1s
-Te2s
-Gm1s
-Gm1s
-Ae2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Ae2s
-Gm1s
-Te2s
-Te2s
- CH2
CH2
OH(序列識別號12)
(DMD AO13)
HO-Ce2s
-Te2s
-Cm1s
-Ce2s
-Te2s
-Um1s
-Ce2s
-Ce2s
-Am1s
-Te2s
-Gm1s
-Am1s
-Ce2s
-Te2s
-Ce2s
-Am1s
-Am1s
-Gm1s
- CH2
CH2
OH(序列識別號13)
(DMD AO14)
HO-Ce2s
-Te2s
-Gm1s
-Am1s
-Am1s
-Gm1s
-Gm1s
-Te2s
-Gm1s
-Te2s
-Te2s
-Ce2s
-Te2s
-Te2s
-Gm1s
-Te2s
-Am1s
-Ce2s
- CH2
CH2
OH(序列識別號14)
(DMD AO15)
HO-Te2s
-Te2s
-Cm1s
-Ce2s
-Am1s
-Gm1s
-Ce2s
-Ce2s
-Am1s
-Te2s
-Te2s
-Gm1s
-Te2s
-Gm1s
-Te2s
-Te2s
-Gm1s
-Am1s
-CH2
CH2
OH(序列識別號15)
[於上述式中,左側表示5’末端,右側表示3’末端,A、G、C、U及T各自表示D-核呋喃糖經修飾且5’位之碳原子與左側所表示的結構單元進行硫代磷酸酯(phosphorothioate)鍵結而成的腺苷、鳥苷、胞苷、尿苷及胸苷;附於各核苷酸或核苷之e2s表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯,3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷的5’位碳原子鍵結,e2t表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯,3’位以-O-與3’末端之氫原子鍵結,m1s表示D-核呋喃糖經2’-O-甲基化,3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,m1t表示D-核呋喃糖經2’-O-甲基化,3’位以-O-與3’末端之氫原子鍵結]。
(111)如(109)記載之方法,其特徵為寡核苷酸為包含選自以下之GSD AO01~GSD AO16的任一式所表示的序列,配位體為下述式之X18
或X20
所表示者,
(GSDAO01)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號16)
(GSD AO02)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號17)
(GSD AO03)
X18
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號18)
(GSD AO04)
X18
-Am1s
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號19)
(GSD AO05)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號20)
(GSD AO06)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號21)
(GSD AO07)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號22)
(GSD AO08)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號23)
(GSD AO09)
X18
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號24)
(GSD AO10)
X18
-Am1s
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號25)
(GSD AO11)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H1(序列識別號26)
(GSD AO12)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號27)
(GSD AO13)
X20
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號28)
(GSD AO14)
X20
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號29)
(GSD AO15)
X20
-Am1s
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號30)
(GSD AO16)
X20
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號31)
[於上述式中,左側表示5’末端,右側表示3’末端,A、G、C、U及T各自表示D-核呋喃糖經修飾且5’位之碳原子與左側所表示的結構單元進行硫代磷酸酯鍵結而成的腺苷、鳥苷、胞苷、尿苷及胸苷;附於各核苷酸或核苷之e2s表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯,3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷的5’位碳原子鍵結,e2t表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯,3’位以-O-與3’末端之氫原子鍵結,m1s表示D-核呋喃糖經2’-O-甲基化,3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,m1t表示D-核呋喃糖經2’-O-甲基化,3’位以-O-與3’末端之氫原子鍵結。
於上述式中,X18
及X20
表示下述之式所表示的GalNAc單元。於下述之式中,與磷酸基鍵結的鍵結肢係表示與寡核苷酸之5’末端的碳原子鍵結,而形成磷酸二酯鍵。]
 [發明之效果]
[發明之效果]
依據本發明,能夠有效率地分別製作複數之ENA單體。
[用以實施發明的形態]
進一步詳細說明本發明。
於本發明,Z1
及Z2
之「羥基的保護基」,以及α群組之「經保護的羥基」之保護基,係指能夠藉由如氫解、水解、電分解及光分解的化學方法或於人體內藉由水解等之生物學的方法而裂解的保護基。就此種保護基而言,可列舉例如,如甲醯基、乙醯基、丙醯基、丁醯基、異丁醯基、戊醯基、三甲基乙醯基、纈草醯基、異纈草醯基、辛醯基、壬醯基、癸醯基、3-甲基壬醯基、8-甲基壬醯基、3-乙基辛醯基、3,7-二甲基辛醯基、十一醯基、十二醯基、十三醯基、十四醯基、十五醯基、十六醯基、1-甲基十五醯基、14-甲基十五醯基、13,13-二甲基十四醯基、十七醯基、15-甲基十六醯基、十八醯基、1-甲基十七醯基、十九醯基、二十醯基及二十一醯基的烷基羰基;如琥珀醯基、戊二醯基、己二醯基的羧基化烷基羰基、氯乙醯基、二氯乙醯基、三氯乙醯基、三氟乙醯基的鹵低級烷基羰基;如甲氧基乙醯基的低級烷氧基低級烷基羰基,如(E)-2-甲基-2-丁烯醯基的不飽和烷基羰基之「脂肪族醯基」;
如苄醯基、α-萘甲醯基、β-萘甲醯基的芳基羰基;如2-溴苄醯基、4-氯苄醯基的鹵芳基羰基;如2,4,6-三甲基苄醯基、4-甲苯甲醯基的低級烷基化芳基羰基;如4-大茴香醯基的低級烷氧基化芳基羰基、2-羧基苄醯基、3-羧基苄醯基、4-羧基苄醯基的羧基化芳基羰基;如4-硝基苄醯基、2-硝基苄醯基的硝基化芳基羰基;如2-(甲氧基羰基)苄醯基的低級烷氧基羰基化芳基羰基;如4-苯基苄醯基的芳基化芳基羰基的「芳香族醯基」;
如四氫哌喃-2-基、3-溴四氫哌喃-2-基、4-甲氧基四氫哌喃-4-基、四氫噻喃-2-基、4-甲氧基四氫噻喃-4-基的「四氫哌喃基或四氫噻喃基」;如四氫呋喃-2-基、四氫噻吩-2-基的「四氫呋喃基或四氫噻吩基」;
如三甲基矽基、三乙基矽基、異丙基二甲基矽基、三級丁基二甲基矽基、甲基二異丙基矽基、甲基二-三級丁基矽基、三異丙基矽基的三低級烷基矽基;如二苯基甲基矽基、二苯基丁基矽基、二苯基異丙基矽基、苯基二異丙基矽基的經1至2個芳基取代的三低級烷基矽基之類的「矽基」;
如甲氧基甲基、1,1-二甲基-1-甲氧基甲基、乙氧基甲基、丙氧基甲基、異丙氧基甲基、丁氧基甲基、三級丁氧基甲基的「低級烷氧基甲基」;
如2-甲氧基乙氧基甲基的「低級烷氧基化低級烷氧基甲基」;
如2,2,2-三氯乙氧基甲基、雙(2-氯乙氧基)甲基的「鹵低級烷氧基甲基」;
如1-乙氧基乙基、1-(異丙氧基)乙基的「低級烷氧基乙基」;
如2,2,2-三氯乙基的「鹵化乙基」;
如苄基、α-萘基甲基、β-萘基甲基、二苯基甲基、三苯基甲基、α-萘基二苯基甲基、9-蒽基甲基的「經1至3個之芳基取代的甲基」;
如4-甲基苄基、2,4,6-三甲基苄基、3,4,5-三甲基苄基、4-甲氧基苄基、4-甲氧基苯基二苯基甲基、4,4’-二甲氧基三苯基甲基、2-硝基苄基、4-硝基苄基、4-氯苄基、4-溴苄基、4-氰基苄基的「芳基環被低級烷基、低級烷氧基、鹵素、氰基取代的經1至3個之芳基取代的甲基」;
如甲氧基羰基、乙氧基羰基、三級丁氧基羰基、異丁氧基羰基的「低級烷氧基羰基」;
如2,2,2-三氯乙氧基羰基、2-三甲基矽基乙氧基羰基的「經鹵素或三低級烷基矽基取代的低級烷氧基羰基」;
如乙烯氧基羰基、芳氧基羰基的「烯氧基羰基」;
如苄氧基羰基、4-甲氧基苄氧基羰基、3,4-二甲氧基苄氧基羰基、2-硝基苄氧基羰基、4-硝基苄氧基羰基的1至2個之「芳基環可經低級烷氧基或硝基取代的芳烷基氧基羰基」。
於Z1
、Z2
之「羥基的保護基」,適合地為「脂肪族醯基」、「芳香族醯基」、「經1至3個之芳基取代的甲基」、「芳基環被低級烷基、低級烷氧基、鹵素、氰基取代之經1至3個之芳基取代的甲基」或「矽基」,更適合地為乙醯基、苄醯基、苄基、對甲氧基苄醯基、二甲氧基三苯甲基、單甲氧基三苯甲基或三級丁基二苯基矽基,進一步適合地為苄基。
於α群組之「經保護的羥基」,為「脂肪族醯基」或「芳香族醯基」,更適合地為苄醯基。
於本發明,A、R1
及α群組之「低級烷基」係表示碳數1~6之直鏈或分支鏈狀的烷基,可列舉例如,甲基、乙基、正丙基、異丙基、正丁基、異丁基、二級丁基、三級丁基、正戊基、異戊基、2-甲基丁基、新戊基、1-乙基丙基、正己基、異己基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基。
就A之「低級烷基」而言,適合地為甲基、乙基或丙基,更適合地為甲基。
就R1
之「低級烷基」而言,適合地為甲基。
於本發明,α群組之「低級烷氧基」係表示上述「低級烷基」與氧原子結合的基,可列舉例如,甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、異丁氧基、二級丁氧基或三級丁氧基,適合地為甲氧基或乙氧基。
於本發明,就α群組之「經保護的巰基」之保護基而言,可列舉例如,除上述羥基的保護基所列舉者之外,如甲硫基、乙硫基、三級丁硫基的烷硫基;如苄硫基的芳硫基等之「形成二硫化物(disulfide)的基」,適合地為「脂肪族醯基」或「芳香族醯基」,更適合地為苄醯基。
於本發明,α群組之「低級烷硫基」係表示上述「低級烷基」與硫原子鍵結的基,可列舉例如,甲硫基、乙硫基、丙硫基、異丙硫基、丁硫基、異丁硫基、二級丁硫基、三級丁硫基,適合地為甲硫基或乙硫基。
於本發明,就α群組之「經保護的胺基」之保護基而言,可列舉例如,如甲醯基、乙醯基、丙醯基、丁醯基、異丁醯基、戊醯基、三甲基乙醯基、纈草醯基、異纈草醯基、辛醯基、壬醯基、癸醯基、3-甲基壬醯基、8-甲基壬醯基、3-乙基辛醯基、3,7-二甲基辛醯基、十一醯基、十二醯基、十三醯基、十四醯基、十五醯基、十六醯基、1-甲基十五醯基、14-甲基十五醯基、13,13-二甲基十四醯基、十七醯基、15-甲基十六醯基、十八醯基、1-甲基十七醯基、十九醯基、二十醯基及二十一醯基的烷基羰基;如琥珀醯基、戊二醯基、己二醯基的羧基化烷基羰基;如氯乙醯基、二氯乙醯基、三氯乙醯基、三氟乙醯基的鹵低級烷基羰基;如甲氧基乙醯基的低級烷氧基低級烷基羰基;如(E)-2-甲基-2-丁烯醯基的不飽和烷基羰基等之「脂肪族醯基」;
如苄醯基、α-萘甲醯基、β-萘甲醯基的芳基羰基;如2-溴苄醯基、4-氯苄醯基的鹵芳基羰基;如2,4,6-三甲基苄醯基、4-甲苯甲醯基的低級烷基化芳基羰基;如4-大茴香醯基的低級烷氧基化芳基羰基;如2-羧基苄醯基、3-羧基苄醯基、4-羧基苄醯基的羧基化芳基羰基;如4-硝基苄醯基、2-硝基苄醯基的硝基化芳基羰基;如2-(甲氧基羰基)苄醯基的低級烷氧基羰基化芳基羰基;如4-苯基苄醯基的芳基化芳基羰基等之「芳香族醯基」;
如甲氧基羰基、乙氧基羰基、三級丁氧基羰基、異丁氧基羰基的「低級烷氧基羰基」;
如2,2,2-三氯乙氧基羰基、2-三甲基矽基乙氧基羰基的「經鹵素或三低級烷基矽基取代的低級烷氧基羰基」;
如乙烯氧基羰基、芳氧基羰基的「烯氧基羰基」;
如苄氧基羰基、4-甲氧基苄氧基羰基、3,4-二甲氧基苄氧基羰基、2-硝基苄氧基羰基、4-硝基苄氧基羰基的可經1至2個之「低級烷氧基或硝基取代芳基環的芳烷氧基羰基」,適合地為「脂肪族醯基」或「芳香族醯基」,更適合地為苄醯基。
於本發明,α群組之「低級烷基胺基」係表示胺基之1個或2個之氫原子經上述「低級烷基」取代的基,可列舉例如,甲基胺基、乙基胺基、丙基胺基、異丙基胺基、丁基胺基、異丁基胺基、二級丁基胺基、三級丁基胺基、二甲基胺基、二乙基胺基、二丙基胺基、二異丙基胺基、二丁基胺基、二異丁基胺基、二(二級丁基)胺基、二(三級丁基)胺基,適合地為甲基胺基、乙基胺基、二甲基胺基、二乙基胺基或二異丙基胺基。
於本發明,就X1
、X2
及α群組之「鹵素原子」而言,可列舉例如,氟原子、氯原子、溴原子或碘原子。
於本發明,就X1
及X2
之「形成脫離基的基」而言,可列舉例如,鹵素原子、脂肪族醯氧基、鹵素取代的低級烷基亞胺氧基或鹵素取代的低級烷基磺醯氧基。
於X1
之「形成脫離基的基」中,適合地為碘原子、乙醯氧基或三氯乙醯亞胺氧基。
於X2
之「形成脫離基的基」中,適合地為乙醯氧基。
於本發明,就Y之「經1至3個之芳基取代的甲基」而言,可列舉例如,苄基、α-萘基甲基、β-萘基甲基、二苯基甲基、三苯甲基、α-萘基二苯基甲基或9-蒽基甲基,適合地為三苯甲基。
於本發明,Y之「低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基」,可列舉例如,4-甲基苄基、2,4,6-三甲基苄基、3,4,5-三甲基苄基、4-甲氧基苄基、4-甲氧基苯基二苯基甲基、4、4’-二甲氧基三苯基甲基(4、4’-二甲氧基三苯甲基)、2-硝基苄基、4-硝基苄基、4-氯苄基、4-溴苄基或4-氰基苄基。
於本發明,Y之「低級烷氧基甲基」係指甲基與上述「低級烷氧基」結合的基。可列舉例如,甲氧基甲基、1,1-二甲基-1-甲氧基甲基、乙氧基甲基、丙氧基甲基、異丙氧基甲基、丁氧基甲基、三級丁氧基甲基,適合地為甲氧基甲基。
於本發明,就Y之「四氫哌喃基」而言,可列舉例如,四氫哌喃-2-基、3-溴四氫哌喃-2-基或4-甲氧基四氫哌喃-4-基,適合地為四氫哌喃-2-基。
於本發明,就Y之「矽基」而言,可列舉例如,如三甲基矽基、三乙基矽基、異丙基二甲基矽基、三級丁基二甲基矽基、甲基二異丙基矽基、甲基二-三級丁基矽基、三異丙基矽基的三低級烷基矽基;如二苯基甲基矽基、二苯基丁基矽基、二苯基異丙基矽基、苯基二異丙基矽基的經1至2個之芳基取代的三低級烷基矽基,適合地為三級丁基二苯基矽基或三級丁基二甲基矽基。
P1
之「可經1至3個之低級烷氧基取代的三苯甲基」係指三苯甲基中的苯基之1至3個氫原子經上述「低級烷氧基」取代的基,可列舉例如,三苯甲基、單甲氧基三苯甲基或二甲氧基三苯甲基,適合地為4,4’-二甲氧基三苯甲基。
於本發明,就R、R3
、R4
、R6
及Z3
之「脂肪族醯基」而言,可列舉例如,如甲醯基、乙醯基、丙醯基、丁醯基、異丁醯基、戊醯基、三甲基乙醯基、纈草醯基、異纈草醯基、辛醯基、壬醯基、癸醯基、3-甲基壬醯基、8-甲基壬醯基、3-乙基辛醯基、3,7-二甲基辛醯基、十一醯基、十二醯基、十三醯基、十四醯基、十五醯基、十六醯基、1-甲基十五醯基、14-甲基十五醯基、13,13-二甲基十四醯基、十七醯基、15-甲基十六醯基、十八醯基、1-甲基十七醯基、十九醯基、二十醯基及二十一醯基的烷基羰基;如琥珀醯基、戊二醯基、己二醯基的羧基化烷基羰基;如氯乙醯基、二氯乙醯基、三氯乙醯基、三氟乙醯基的鹵低級烷基羰基;如甲氧基乙醯基的低級烷氧基低級烷基羰基;如(E)-2-甲基-2-丁烯醯基的不飽和烷基羰基。
就R、R3
、R6
及Z3
之「脂肪族醯基」而言,適合地為乙醯基。
就R4
之「脂肪族醯基」而言,適合地為異丁醯基。
於本發明,就R3
、R4
、R6
及Z3
之「芳香族醯基」而言,可列舉例如,如苄醯基、α-萘甲醯基、β-萘甲醯基的芳基羰基;如2-溴苄醯基、4-氯苄醯基的鹵芳基羰基;如2,4,6-三甲基苄醯基、4-甲苯甲醯基的低級烷基化芳基羰基;如4-大茴香醯基的低級烷氧基化芳基羰基;如2-羧基苄醯基、3-羧基苄醯基、4-羧基苄醯基的羧基化芳基羰基;如4-硝基苄醯基、2-硝基苄醯基的硝基化芳基羰基;如2-(甲氧基羰基) 苄醯基的低級烷氧基羰基化芳基羰基;如4-苯基苄醯基的芳基化芳基羰基。
就R3
、R4
、R6
及Z3
之「芳香族醯基」而言,適合地為苄醯基。
於本發明,R2
之「經脂肪族醯基或芳香族醯基保護的胺基」係表示胺基經上述「脂肪族醯基」或「芳香族醯基」取代的基。就R2
之「經脂肪族醯基保護的胺基」而言,可列舉例如,甲醯基胺基、乙醯基胺基、丙醯基胺基、丁醯基胺基、異丁醯基胺基、戊醯基胺基、三甲基乙醯基胺基、纈草醯基胺基、氯乙醯基胺基、二氯乙醯基胺基、三氯乙醯基胺基、三氟乙醯基胺基、甲氧基乙醯基胺基或(E)-2-甲基-2-丁烯醯基胺基。就R2
之「經芳香族醯基保護的胺基」而言,可列舉例如,苄醯基胺基、α-萘甲醯基胺基、β-萘甲醯基胺基、2-溴苄醯基胺基、4-氯苄醯基胺基、2,4,6-三甲基苄醯基胺基、4-甲苯甲醯基胺基、4-大茴香醯基胺基、2-羧基苄醯基胺基、3-羧基苄醯基胺基、4-羧基苄醯基胺基、4-硝基苄醯基胺基、2-硝基苄醯基胺基、2-(甲氧基羰基)苄醯基胺基或4-苯基苄醯基胺基,適合地為苄醯基胺基。
於本發明,就R5
之「可經低級烷基、低級烷氧基、鹵素或氰基取代的苄基」而言,可列舉例如,苄基、4-甲基苄基、2,4,6-三甲基苄基、3,4,5-三甲基苄基、4-甲氧基苄基、4-氯苄基、4-溴苄基或4-氰基苄基,適合地為苄基。
上述通式(XXVII)及(XXVIII)中,B之「可具有選自α群組的1或2個以上之取代基的嘌呤-9-基」,適合地為6-胺基嘌呤-9-基(即,腺嘌呤基)、胺基經保護的6-胺基嘌呤-9-基、2,6-二胺基嘌呤-9-基、2-胺基-6-氯嘌呤-9-基、胺基經保護的2-胺基-6-氯嘌呤-9-基、2-胺基-6-氟嘌呤-9-基、胺基經保護的2-胺基-6-氟嘌呤-9-基、2-胺基-6-溴嘌呤-9-基、胺基經保護的2-胺基-6-溴嘌呤-9-基、2-胺基-6-羥基嘌呤-9-基(即,鳥嘌呤基)、胺基經保護的2-胺基-6-羥基嘌呤-9-基、胺基及羥基經保護的2-胺基-6-羥基嘌呤-9-基、6-胺基-2-甲氧基嘌呤-9-基、6-胺基-2-氯嘌呤-9-基、6-胺基-2-氟嘌呤-9-基、2,6-二甲氧基嘌呤-9-基、2,6-二氯嘌呤-9-基或6-巰基嘌呤-9-基,更適合地為胺基經保護的6-胺基嘌呤-9-基或胺基經保護的2-胺基-6-羥基嘌呤-9-基,進一步更適合地為6-苄醯基胺基嘌呤-9-基或2-異丁醯基胺基-6-羥基嘌呤-9-基。
上述通式(XXVII)及(XXVIII)中,B之「可具有選自α群組的1或2個以上之取代基的2-側氧基-嘧啶-1-基」,適合地為2-側氧基-4-胺基-嘧啶-1-基(即,胞嘧啶基)、胺基經保護的2-側氧基-4-胺基-嘧啶-1-基、2-側氧基-4-胺基-5-氟-嘧啶-1-基、胺基經保護的2-側氧基-4-胺基-5-氟-嘧啶-1-基、4-胺基-2-側氧基-5-氯-嘧啶-1-基、2-側氧基-4-甲氧基-嘧啶-1-基、2-側氧基-4-巰基-嘧啶-1-基、2-側氧基-4-羥基-嘧啶-1-基(即,尿嘧啶基)、2-側氧基-4-羥基-5-甲基嘧啶-1-基(即,胸腺嘧啶基)、4-胺基-5-甲基-2-側氧基-嘧啶-1-基(即,5-甲基胞嘧啶基)、胺基經保護的4-胺基-5-甲基-2-側氧基-嘧啶-1-基,更適合地為2-側氧基-4-羥基-5-甲基嘧啶-1-基、胺基經保護的2-側氧基-4-胺基-嘧啶-1-基或胺基經保護的4-胺基-5-甲基-2-側氧基-嘧啶-1-基,進一步更適合地為2-側氧基-4-羥基-5-甲基嘧啶-1-基、2-側氧基-4-苄醯基胺基-嘧啶-1-基或4-苄醯基胺基-5-甲基-2-側氧基-嘧啶-1-基。
於本發明,n為0至4之整數,適合地為0或1,更適合地為1。
「其鹽」係指因本發明之化合物可作成鹽,故稱其鹽,就如此的鹽而言,適合地可列舉如鈉鹽、鉀鹽、鋰鹽的鹼金屬鹽;如鈣鹽、鎂鹽的鹼土金屬鹽;如鋁鹽、鐵鹽、鋅鹽、銅鹽、鎳鹽、鈷鹽等之金屬鹽;如銨鹽的無機鹽;如三級辛基胺鹽、二苄基胺鹽、 啉鹽、葡萄糖胺鹽、苯基甘胺酸烷基酯鹽、乙二胺鹽、N-甲基還原葡萄糖胺鹽(N-methylglucamine salt)、胍鹽、二乙基胺鹽、三乙基胺鹽、二環己基胺鹽、N,N’-二苄基乙二胺鹽、氯普魯卡因鹽、普魯卡因鹽、二乙醇胺鹽、N-苄基-苯乙基胺鹽、哌
啉鹽、葡萄糖胺鹽、苯基甘胺酸烷基酯鹽、乙二胺鹽、N-甲基還原葡萄糖胺鹽(N-methylglucamine salt)、胍鹽、二乙基胺鹽、三乙基胺鹽、二環己基胺鹽、N,N’-二苄基乙二胺鹽、氯普魯卡因鹽、普魯卡因鹽、二乙醇胺鹽、N-苄基-苯乙基胺鹽、哌 鹽、四甲基銨鹽、參(羥基甲基)胺基甲烷鹽的有機鹽等之胺鹽;如氫氟酸鹽、鹽酸鹽、氫溴酸鹽、氫碘酸鹽的鹵素原子化氫酸鹽;硝酸鹽、過氯酸鹽、硫酸鹽、磷酸鹽等之無機酸鹽;如甲磺酸鹽、三氟甲磺酸鹽、乙磺酸鹽的低級烷磺酸鹽;如苯磺酸鹽、對甲苯磺酸鹽的芳基磺酸鹽;如乙酸鹽、蘋果酸鹽、反丁烯二酸鹽、琥珀酸鹽、檸檬酸鹽、酒石酸鹽、草酸鹽、順丁烯二酸鹽等之有機酸鹽;及如甘胺酸鹽、離胺酸鹽、精胺酸鹽、鳥胺酸鹽、麩胺酸鹽、天冬胺酸鹽的胺基酸鹽。
鹽、四甲基銨鹽、參(羥基甲基)胺基甲烷鹽的有機鹽等之胺鹽;如氫氟酸鹽、鹽酸鹽、氫溴酸鹽、氫碘酸鹽的鹵素原子化氫酸鹽;硝酸鹽、過氯酸鹽、硫酸鹽、磷酸鹽等之無機酸鹽;如甲磺酸鹽、三氟甲磺酸鹽、乙磺酸鹽的低級烷磺酸鹽;如苯磺酸鹽、對甲苯磺酸鹽的芳基磺酸鹽;如乙酸鹽、蘋果酸鹽、反丁烯二酸鹽、琥珀酸鹽、檸檬酸鹽、酒石酸鹽、草酸鹽、順丁烯二酸鹽等之有機酸鹽;及如甘胺酸鹽、離胺酸鹽、精胺酸鹽、鳥胺酸鹽、麩胺酸鹽、天冬胺酸鹽的胺基酸鹽。
於本發明,胸腺嘧碇衍生物、或鳥嘌呤衍生物之羰基可為「互變異構物」。互變異構物係指以平衡狀態存在的2種以上之結構異構物的1種,由某一異構物容易轉化成另一種異構物,且呈溶液中之互變異構物組的混合物而存在。於互變異構化為可能的條件下,達到互變異構物之化學平衡,但其正確比率受到包含溫度、溶媒及pH的數個要因左右。藉由互變異構物化而可相互轉換的互變異構物的概念,稱為互變異構性。
胸腺嘧碇衍生物及鳥嘌呤衍生物中的醯胺-醯亞胺酸互變異構之例,如以下所示。

於本發明,「羥基之保護試藥」係指對核苷類似物及其製造中間體所具有的羥基,用以導入前述「羥基之保護基」的試藥。例如,「羥基之保護基」為:
(1)苄基之情形:氯甲苯、溴甲苯、苄基碘、三氯乙醯亞胺苄酯(benzyl trichloroacetimidate)
(2)三苯甲基之情形:三苯氯甲烷(trityl chloride)、三苯甲基三氟磺酸酯、
(3)三級丁基二甲基矽基之情形:氯化三級丁基二甲基矽基、
(4)乙醯基之情形:乙酸酐、乙醯氯。
於本發明,「1級羥基之保護試藥」係指用於對核苷類似物之5位的糖羥基導入上述「可經1至3個之低級烷氧基取代的三苯甲基」的試藥,可列舉例如,三苯氯甲烷、三苯甲基三氟磺酸酯、4-單甲氧基三苯氯甲烷、4,4’-二甲氧基三苯氯甲烷、4,4’-二甲氧基三苯甲基三氟磺酸酯,適合地為4,4’-二甲氧基三苯氯甲烷。
於本發明,「羥基之去保護試藥」係指為了使上述「羥基之保護基」脫離而添加的反應助劑,可列舉例如,「羥基之保護基」為
(1)苄基之情形:鈀、氫氧化鈀、鉑等之金屬觸媒及氫、甲酸、甲酸銨等之還原劑、氯化鐵(III)、三氯化硼
(2)三苯甲基之情形:硫酸、對甲苯磺酸、甲磺酸等之酸觸媒
(3)乙醯基之情形:氫氧化鈉、氫氧化鉀、碳酸鈉等之無機鹼、氨、烷基胺等之有機鹼。
於本發明,金屬觸媒可為承載於載體(適合地為碳)或未承載於載體上的金屬觸媒,適合地為承載於碳上的金屬觸媒(適合地為承載於碳上的鈀、氫氧化鈀、或鉑,更適合地為承載於碳上的鈀)。
於本發明,「醯化劑」係指用以對核苷類似物之胺基導入上述「胺基之保護基」所使用的試藥,例如,
(1)乙醯基之情形:乙酸酐、乙醯氯
(2)苄醯基之情形:苯甲醯氯、苯甲酸酐、苯甲酸
(3)異丁醯基之情形:氯異丁烷異丁酸酐。
於本發明,「胺基化劑」係指用以將嘌呤環之6位之氯原子或嘧啶環之4位之羰基取代為胺基所使用的試藥,例如,氨、氨水溶液或如碳酸銨、乙酸銨的氨鹽類,適合地為氨水溶液。
於本發明,「醯胺化劑」係指於交叉偶合反應中用於將嘌呤環之6位之氯原子轉換為脂肪族醯胺或芳香族醯胺所使用的試藥,例如,乙醯基醯胺、苄醯基醯胺、異丁基醯胺,適合地為異丁基醯胺。
於本發明,醣基化反應中的「活化劑」係指用以將糖之1位之羥基轉換為上述「形成脫離基的基」所使用的試藥,例如,乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑、氯磷酸二苯酯,適合地為乙酸酐或三氯乙腈。
於本發明,醣基化反應中的「鹵化劑」係指為了使立體選擇性地進行醣基化反應、為了將上述「形成脫離基的基」鹵化而使用的試藥,可列舉例如氯三甲基矽烷(TMSCl)、溴三甲基矽烷(TMSBr)、碘三甲基矽烷(TMSI),適合地為TMSI。
於本發明,胺基化反應中的「活化劑」係指用以將羥基轉換為脫離基所使用的試藥,例如,對甲苯磺醯氯、2,4,6-三異丙基苯磺醯氯,適合地為2,4,6-三異丙基苯磺醯氯。
於本發明,AMIDITE化反應中的「活化劑」係用以形成AMIDITE試藥之活性中間體所使用的試藥,可列舉例如,5-苄硫基四唑、5-苯基四唑、二溴咪唑、二氰基咪唑、N-烷基咪唑三氟乙酸鹽等,適合地為4,5-二氰基咪唑。使用的活化劑之量,相對於通式(XXVII)之化合物,為0.01~1.0當量,較佳為0.1~0.5當量。
於本發明,AMIDITE化反應中的「AMIDITE化試藥」係指用以於核苷類似物之3位之糖羥基,導入含有用以形成核苷間鍵結的磷原子的基的試藥,可列舉例如,2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺、2-氰基乙基二異丙基氯亞磷醯胺,適合地為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺。使用的AMIDITE化試藥之量,相對於通式(XXVII)之化合物,為1.0~1.5當量,較佳為1.1~1.3當量。
於本發明,AMIDITE化反應中的「乾燥劑」係指用以吸收反應溶液中之水分所使用的試藥,例如,分子篩3A、分子篩4A、或分子篩5A,適合地為分子篩4A。
本發明之化合物(2A)可藉由以下所述的A法而製造。
(A法)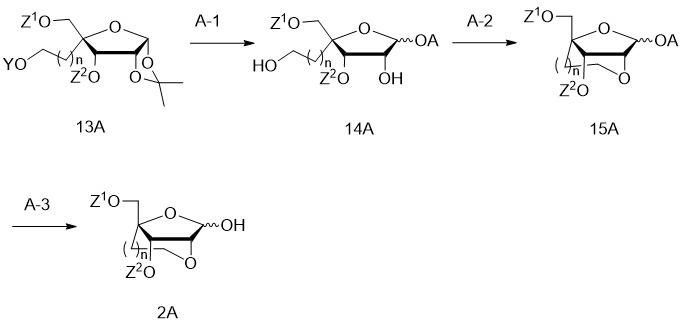
A法中,A、Y、Z1
及Z2
表示與前述同意義。Z1
、Z2
表示於Y之去保護條件下安定的羥基之保護基。
以下,對於A法之各步驟進行詳細說明。
(A-1步驟)
本步驟係於醇溶媒中、酸觸媒存在下,進行可藉由後述C法、D法而製造的化合物(13A)之1,2位羥基的保護基及Y的去保護,而製造化合物(14A)的步驟。又,n=0之情形,省略本步驟,使用藉由後述B法所製造的化合物(14A’)而進行A-2步驟即可。
就使用的醇溶媒而言,可列舉例如,甲醇、乙醇、丙醇等之醇類,但適合地為甲醇。
就使用的酸觸媒而言,可列舉硫酸、對甲苯磺酸、甲磺酸等,但適合地為硫酸。
反應溫度,通常為0℃至100℃,適合地為40至60℃。
反應時間雖依酸觸媒的種類及使用量而不同,但通常為1小時至48小時,適合地為10小時至24小時。
反應結束後,本反應之目的化合物(14A)的醇溶液,例如可將反應液以三乙基胺中和,添加水,藉此使為共生成物的甲氧基三苯基甲烷結晶化,藉由濾除而將含有目的化合物的含水醇溶液分離。再者,藉由以正庚烷進行分液洗淨而獲得的含水醇溶液中添加水、甲苯而分液,可藉由將獲得的有機層進行濃縮而獲得的甲苯溶液直接利用於下一步驟。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(A-2步驟)
本步驟係於溶媒中,使用三價磷試藥、偶氮二甲酸酯,進行A-1步驟所製造的化合物(14A)之二醇部位的環化反應,而製造化合物(15A)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類、苯、甲苯等之烴類、四氫呋喃、二甲基醚之類的醚類,但適合地為甲苯。
就使用的三價磷試藥而言,可列舉三苯基膦、三(正丁基)膦等,但適合地為三苯基膦。
就使用的偶氮二甲酸酯而言,可列舉偶氮二甲酸二乙酯、偶氮二甲酸二異丙酯、偶氮二甲酸二(三級丁酯)等,但適合地為偶氮二甲酸二異丙酯。
反應溫度通常為0℃至50℃,適合地為10至40℃。
反應時間雖依三價磷試藥的種類及使用量而不同,但通常為1小時至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(15A)之甲苯溶液,可藉由例如於反應液中添加氯化鎂,藉由攪拌使形成難溶性之膦錯體,而藉由過濾去除為共生成物的氧化膦。又,與為另一種之共生成物的肼酯有關,使用甲醇水而將甲苯層洗淨,亦可有效果地去除。藉由將獲得的有機層進行濃縮而獲得的甲苯溶液可直接利用於下一步驟。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(A-3步驟)
本步驟係於溶媒中,藉由使酸作用,進行A-2步驟所製造的化合物(15A)之變旋異構物位的水解反應,而製造化合物(2A)的步驟。
就使用的溶媒而言,可列舉例如,乙酸、水、醇等之水溶性溶媒、二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類、苯、甲苯等之烴類、四氫呋喃、二甲基醚之類的醚類,但適合地為乙酸。
就使用的酸而言,可列舉鹽酸、硫酸、三氟乙酸、甲磺酸、對甲苯磺酸等,但適合地為鹽酸。
反應溫度通常為0℃至50℃,適合地為20至30℃。
反應時間依酸的種類及使用量而不同,但通常為1小時至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(2A)係例如可於反應液中添加水,冷卻後藉由攪拌而使結晶化。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
前述之化合物(14A)(n=0之情形)係可利用以下所述的B法而製造。
(B法)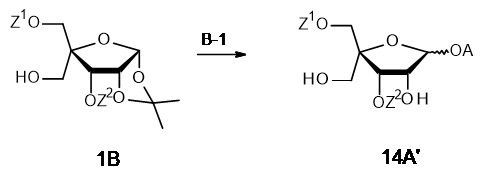 B法中,A、Z1
及Z2
表示與前述同意義。
B法中,A、Z1
及Z2
表示與前述同意義。
以下,對於B法之步驟進行詳細說明。
(B-1步驟)
本步驟係使用依據WO00/47599之化合物(7)之製造方法而製造的化合物(1B),而製造化合物(14A’)的步驟。化合物(14A’)係可藉由將依據A法A-1步驟而將化合物(1B)之1,2位羥基之保護基加以去保護而製造。
前述之化合物(13A)(n=1之情形)係可利用以下所述的C法而製造。
(C法)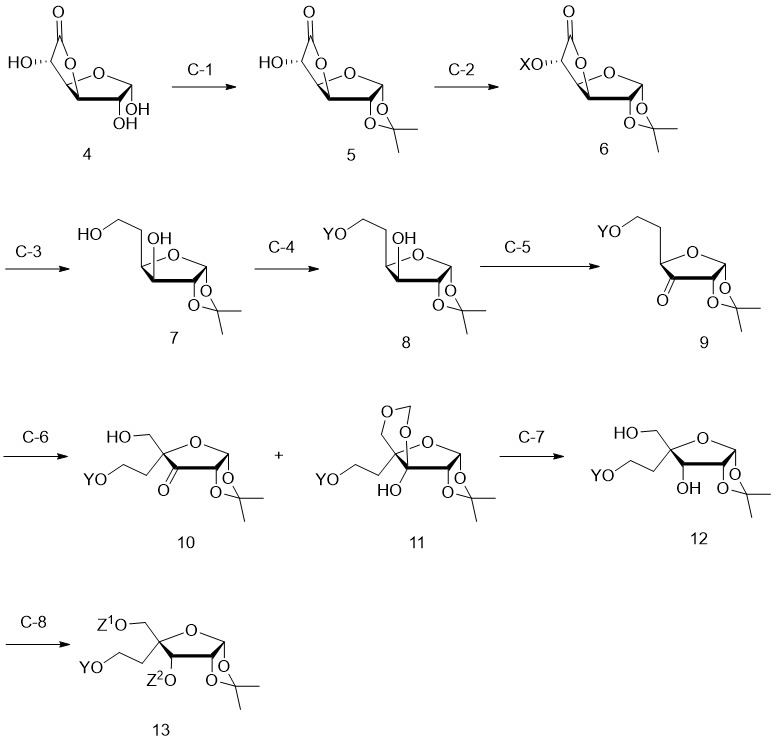
C法中,X係與氧原子一起形成脫離基的基。Y、Z1
及Z2
表示與前述同意義。
就X而言,可列舉例如,甲磺醯基、乙磺醯基之類的低級烷基磺醯基;三氟甲磺醯基之類的鹵素取代的低級烷基磺醯基;對甲苯磺醯基之類的芳基磺醯基;惟適合地為甲磺醯基或對甲苯磺醯基。
以下,對於C法之步驟進行詳細說明。
(C-1步驟)
本步驟係於酸觸媒存在下,使化合物(4)於丙酮溶媒中反應,而製造化合物(5)的步驟。
就使用的酸觸媒而言,可列舉硫酸、對甲苯磺酸、甲磺酸等,但適合地為硫酸。
反應溫度雖依使用的酸觸媒而不同,但通常為0℃至50℃,適合地為30至40℃。
反應時間雖依酸觸媒的種類及使用量而不同,但通常為10分鐘至24小時,適合地為10小時至20小時。
反應結束後,本反應之目的化合物(5)係藉由例如將反應液中和,將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-2步驟)
本步驟係於溶媒中,鹼存在下,使C-1步驟所製造的化合物(5),與脫離基導入試藥反應,而製造化合物(6)的步驟。
就使用的溶媒而言,可列舉例如,二甲基乙醯胺、二甲基甲醯胺之類的醯胺類;二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙酸甲酯、乙酸乙酯、乙酸丙酯之類的酯類,但適合地為二甲基乙醯胺。
就使用的鹼而言,可列舉例如,三乙基胺、吡啶、二甲基胺基吡啶1-甲基咪唑之類的鹼,但適合地為1-甲基咪唑。
就使用的脫離基導入試藥而言,可列舉例如,對甲苯磺醯氯、甲磺醯氯、三氟甲磺酸酐等,但適合地為對甲苯磺醯氯。
反應溫度雖依使用的脫離基導入試藥而不同,但通常為0℃至50℃,適合地為20至30℃。
反應時間雖依使用的脫離基導入試藥之種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(6)係藉由例如於反應液中添加水,使結晶化而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-3步驟)
本步驟係於溶媒中,使C-2步驟所製造的化合物(6)與氫化物還原劑反應,而製造化合物(7)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;但適合地為四氫呋喃。
就使用的氫化物還原劑而言,可列舉例如,雙(2-甲氧基乙氧基)氫化鋁鈉、氫化鋰鋁、二異丁基氫化鋁,但適合地為雙(2-甲氧基乙氧基)氫化鋁鈉。
反應時間雖依氫化物還原劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(7)係可藉由例如於反應液中添加丙酮、L-酒石酸鈉鉀水溶液後,將含有目的化合物的有機層分離,水洗後,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-4步驟)
本步驟係於溶媒中,鹼存在下,將保護基選擇性地導入C-3步驟所製造的化合物(7)之1級羥基,而製造化合物(8)的步驟。
就使用的溶媒而言,可列舉例如,二甲基乙醯胺、二甲基甲醯胺之類的醯胺類;苯、甲苯等之烴類;二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;四氫呋喃、二甲基醚之類的醚類;乙酸甲酯、乙酸乙酯、乙酸丙酯之類的酯類,但適合地為甲苯。
就使用的鹼而言,可列舉例如,三乙基胺、吡啶、二甲基胺基吡啶1-甲基咪唑、4-甲基啉之類的鹼,但適合地為4-甲基啉。
使用的1級羥基之保護試藥而言,可列舉例如,三苯氯甲烷、4,4’-二甲氧基三苯氯甲烷等,但適合地為三苯氯甲烷。
反應溫度雖依使用的保護試藥而不同,但通常為0℃至50℃,適合地為20至30℃。
反應時間雖依1級羥基之保護試藥的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(8)可藉由例如於反應液中添加水,將含有目的化合物的有機層分離,水洗後,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-5步驟)
本步驟係於溶媒中,鹼、氧化劑、氧化觸媒及輔觸媒存在下,將C-4步驟所製造的化合物(8)之3位的2級羥基氧化,而製造化合物(9)之步驟。
就使用的溶媒而言,可列舉例如,二甲基乙醯胺、二甲基甲醯胺之類的醯胺類;苯、甲苯等之烴類;二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;四氫呋喃、二甲基醚之類的醚類;乙酸甲酯、乙酸乙酯、乙酸丙酯之類的酯類,但適合地為甲苯。
就使用的鹼而言,可列舉例如,氫氧化鈉、氫氧化鉀、碳酸氫鈉、碳酸鈉等之無機鹼;三乙基胺、吡啶、二甲基胺基吡啶1-甲基咪唑、4-甲基啉之類的有機鹼,但適合地為碳酸氫鈉。
就使用的氧化觸媒而言,可列舉例如,2,2,6,6-四甲基哌啶1-氧基、2-氮雜金剛烷-N-氧基、9-氮雜降金剛烷-N-氧基等,但適合地為9-氮雜降金剛烷-N-氧基。
就使用的輔觸媒而言,可列舉溴化鉀、四丁基溴化銨等,但適合地為溴化鉀。
就使用的氧化劑而言,可列舉次氯酸鈉、碘苯二乙酸等,但適合地為次氯酸鈉。
反應溫度雖依使用的氧化觸媒及輔觸媒而不同,但通常為0℃至50℃,適合地為0至10℃。
反應時間雖依氧化觸媒及輔觸媒的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(9)之甲苯溶液係可例如將反應液靜置後,藉由移除水層,將包含目的化合物的有機層分離,經由水洗而獲得,並直接用於下一步驟。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-6步驟)
本步驟係於溶媒中,鹼及烷基化劑存在下,藉由將C-5步驟所製造的化合物(9)之4位的碳,一邊伴隨表異構物化(epimerisation)一邊立體選擇性地羥基甲基化,而製造化合物(10)的步驟。
就使用的溶媒而言,可列舉例如,二甲基乙醯胺、二甲基甲醯胺之類的醯胺類;苯、甲苯等之烴類;二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;四氫呋喃、二甲基醚之類的醚類;乙酸甲酯、乙酸乙酯、乙酸丙酯之類的酯類,但適合地為甲苯。
就使用的鹼而言,可列舉例如,氫氧化鈉、氫氧化鉀、碳酸氫鈉、碳酸鈉等之無機鹼;三乙基胺、吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼,但適合地為1,8-二氮雜雙環[5.4.0]十一-7-烯。
就使用的烷基化劑而言,可列舉例如,聚甲醛、甲醛水溶液等,但適合地為甲醛水溶液。
反應溫度雖依使用的鹼而不同,但通常為0℃至50℃,適合地為20至30℃。
反應時間雖依鹼的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(10)之甲苯溶液可藉由例如將反應液靜置後,藉由去除水層,將含目的化合物的有機層分離,經水洗而獲得,且可直接利用於下一步驟。又,此時一部分生成的環化體(11)係於下一步驟中轉換為化合物(12)。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-7步驟)
本步驟係於溶媒中,使C-6步驟所製造的化合物(10),與氫化物還原劑反應,而製造3位之羥基經立體控制的化合物(12)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;但適合地為甲苯。
就使用的氫化物還原劑而言,可列舉例如,雙(2-甲氧基乙氧基)氫化鋁鈉、氫化鋰鋁、二異丁基氫化鋁、氫化硼鈉,但適合地為氫化硼鈉。
反應溫度雖依使用的氫化物還原劑而不同,但通常為0℃至50℃,適合地為20至30℃。
反應時間雖依氫化物還原劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(12)之甲苯溶液係可例如將反應液靜置後,藉由去除水層,將含目的化合物的有機層分離,經由水洗而獲得,且可直接利用於下一步驟。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(C-8步驟)
本步驟係於溶媒中,鹼及觸媒存在下,使羥基之保護試藥反應,將C-7步驟所製造的化合物(12)之2個羥基加以保護,而製造化合物(13)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;但適合地為甲苯。
就使用的羥基之保護試藥而言,可列舉例如,三苯氯甲烷、三級丁基二苯基矽基氯、三級丁基二甲基矽基氯、乙酸酐、苯甲醯氯等,但適合地為氯甲苯或溴甲苯,更適合地為溴甲苯。
就使用的觸媒而言,可列舉例如四丁基碘化銨、碘化鉀、碘化鈉等,但適合地為四丁基碘化銨。
就使用的鹼而言,可列舉例如,氫氧化鈉、氫氧化鉀、碳酸氫鈉、碳酸鈉等之無機鹼;三乙基胺、吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼,但適合地為氫氧化鉀。
反應溫度雖依使用的羥基之保護試藥而不同,但通常為20℃至100℃,適合地為60至80℃。
反應時間雖依羥基之保護試藥的種類及使用量而不同,但通常為1小時至48小時,適合地為10小時至24小時。
反應結束後,本反應之目的化合物(13)之甲苯溶液,可例如將反應液靜置後,藉由去除水層,將含目的化合物的有機層分離,藉由水洗而獲得,且可將獲得的甲苯層溶媒取代為1-丙醇而使結晶化。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
前述之化合物(13A)(n=2至4之情形),可藉由以下所述的D法而製造。
(D法)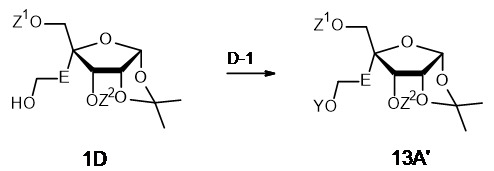
D法中,E為伸乙基、三亞甲基或四亞甲基,Y、Z1
及Z2
表示與前述同意義。
以下,對於D法之各步驟詳細說明。
(D-1步驟)
本步驟係於溶媒中,鹼存在下,使按照WO00/47599之化合物(3b)之製造方法而製造的化合物(1D),與羥基之保護試藥反應,而製造化合物(13A’)的步驟。
就使用的溶媒而言,可列舉例如,二甲基乙醯胺、二甲基甲醯胺之類的醯胺類;苯、甲苯等之烴類;二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;四氫呋喃、二甲基醚之類的醚類;乙酸甲酯、乙酸乙酯、乙酸丙酯之類的酯類;但適合地為甲苯。
就使用的鹼而言,可列舉例如,三乙基胺、吡啶、二甲基胺基吡啶1-甲基咪唑、4-甲基啉之類的鹼,但適合地為4-甲基啉。
就使用的羥基之保護試藥而言,可列舉例如,三苯氯甲烷、4,4’-二甲氧基三苯氯甲烷等,但適合地為三苯氯甲烷。
反應溫度雖依使用的保護試藥而不同,但通常為0℃至50℃,適合地為20至30℃。
反應時間雖依保護試藥的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(13A’)係可例如於反應液中添加水,將含目的化合物的有機層分離,水洗後,藉由餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
本發明之化合物(5E)係可藉由以下所述的E法而製造。
(E法)
E法中,R1
、R2
、X1
、Z1
、Z2
及P1
表示與前述同意義。
以下,對於E法之步驟詳細說明。
(E-1步驟)
本步驟係藉由使用化合物(2A),於溶媒中,鹼存在下,使與活化劑反應,而製造化合物(2E)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;但適合地為甲苯或乙腈。
使用的活化劑可列舉乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑、氯磷酸二苯酯,但適合地為乙酸酐或三氯乙腈。
使用的鹼雖依活化劑而不同,但可列舉三乙基胺、吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼,適合地為吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯。反應結束後,本反應之目的化合物(2E),例如可用於直接醣基化,或將反應液中和,並將反應混合物濃縮,藉由餾除溶媒而獲得。
反應溫度雖依使用的活化劑而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依活化劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(E-2步驟)
本步驟係製造化合物(3E)的步驟,其係藉由將E-1步驟所獲得的化合物(2E),於溶媒中,鹵化劑存在下,與藉由矽化反應而經矽化的嘧啶鹼基(胸腺嘧碇或經醯基保護的胞嘧啶等)之醣基化反應而製造。
就於矽化反應及醣基化反應所使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈,但適合地為乙腈。
使用的矽化劑可列舉N,O-雙三甲基矽基乙醯胺、六甲基二矽氮烷等,但適合地為N,O-雙三甲基矽基乙醯胺。
嘧啶鹼基之矽化反應之溫度雖依矽化劑而不同,但通常為0℃至50℃,適合地為20℃至40℃。
矽化反應之反應時間雖依矽化劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至10小時。
醣基化反應所使用的鹵化劑可列舉氯三甲基矽烷(TMSCl)、溴三甲基矽烷(TMSBr)、碘三甲基矽烷(TMSI),但適合地為TMSI。
醣基化反應之反應溫度雖依化合物(2E)之結構或使用的鹵化劑而不同,但通常為0℃至50℃,適合地為20℃至40℃。
醣基化反應之反應時間雖依鹵化劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為10小時至20小時。
反應結束後,本反應之目的化合物(3E),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(E-3步驟)
本步驟係使化合物(3E),於溶媒中,與羥基之去保護試藥反應,藉由去保護反應而製造化合物(4E)的步驟。
(i)R2
為羥基,Z1
及Z2
為苄基之情形,去保護試藥為承載於碳上的金屬觸媒及還原劑。
就使用的溶媒而言,可列舉例如,苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;甲醇、乙醇、丙醇等之醇類;惟適合地為甲醇。
使用的金屬觸媒可列舉鈀、氫氧化鈀、鉑等,但適合地為鈀。
使用的還原劑可列舉氫、甲酸、甲酸銨等,但適合地為氫。
反應溫度雖依金屬觸媒而不同,但通常為0℃至70℃,適合地為40℃至60℃。
反應時間雖依金屬觸媒、還原劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(4E),例如藉由將反應液過濾而去除金屬觸媒,將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(ii)R2
為經醯基經保護的胺基,Z1
及Z2
為苄基之情形,使用去保護試藥而進行去保護反應。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;惟適合地為二氯甲烷。
使用的去保護試藥,可列舉氯化鐵(III)、三氯化硼等,適合地為三氯化硼。
反應溫度雖依去保護試藥而不同,但通常為-20℃至30℃,適合地為-20℃至20℃。
反應時間雖依去保護試藥而不同,但通常為10分鐘至10小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(4E),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(E-4步驟)
本步驟係藉由使化合物(4E),於溶媒中,鹼存在下,與1級羥基之保護試藥反應,而製造化合物(5E)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙酸乙酯、乙酸丙酯之類的酯類;乙腈;惟適合地為四氫呋喃、或乙酸乙酯。
就使用的1級羥基之保護試藥而言,可列舉例如,三苯氯甲烷、4-甲氧基三苯氯甲烷、4,4’-二甲氧基三苯氯甲烷等,適合地為4,4’-二甲氧基三苯氯甲烷。
使用的鹼可列舉三乙基胺、N-甲基啉等之脂肪族胺、咪唑、吡啶等之芳香族胺類,適合地為吡啶。
反應之溫度雖依保護試藥而不同,但通常為0℃至50℃,適合地為0℃至20℃。
反應時間雖依保護試藥而不同,但通常為10分鐘至10小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(5E),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
本發明之化合物(4F)可藉由以下所述的F法而製造。
(F法)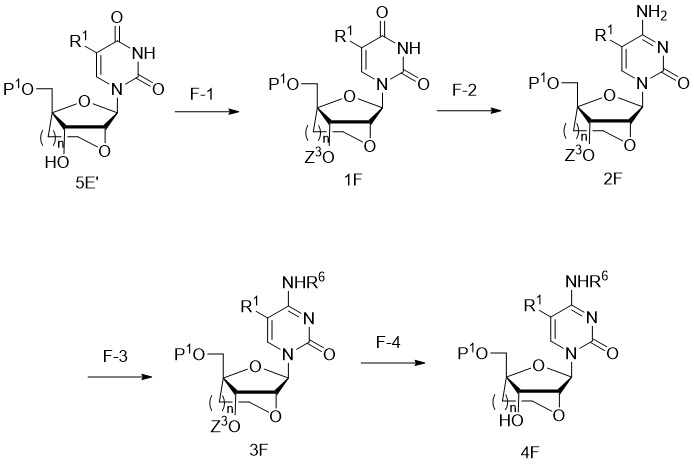 F法中,R1
、R6
、Z3
及P1
表示與前述同意義。
F法中,R1
、R6
、Z3
及P1
表示與前述同意義。
以下,對於F法之各步驟進行詳細說明。
(F-1步驟)
本步驟係藉由使化合物(5E’),於溶媒中,鹼、觸媒存在下,與羥基之保護試藥反應,而製造化合物(1F)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為乙腈。
就使用的羥基之保護試藥而言,可列舉乙酸酐、乙醯氯、苯甲酸酐、苯甲醯氯等,適合地為乙酸酐。
使用的鹼雖依活化劑而不同,但可列舉三乙基胺、吡啶之類的有機鹼,適合地為三乙基胺。
就使用的觸媒而言,可列舉N,N-二甲基胺基吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯等,適合地為N,N-二甲基胺基吡啶。反應溫度雖依使用的鹼而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依保護試藥的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(1F),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(F-2步驟)
本步驟係將化合物(1F),於溶媒中,鹼、觸媒存在下,使用活化劑而將羥基活性化,接著藉由使與胺基化劑反應,而獲得化合物(2F)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為乙腈。
使用的鹼雖依活化劑而不同,但可列舉三乙基胺、吡啶之類的有機鹼,適合地為三乙基胺。
就使用的觸媒而言,可列舉N,N-二甲基胺基吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯等,適合地為N,N-二甲基胺基吡啶。
就使用的活化劑而言,可列舉對甲苯磺醯氯、2,4,6-三異丙基苯磺醯氯等,適合地為2,4,6-三異丙基苯磺醯氯。
反應溫度雖依使用的活化劑而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依活化劑的種類及使用量而不同,但通常為10分鐘至8小時,適合地為1小時至3小時。
就使用的胺基化劑而言,可列舉氨、氨水溶液或碳酸銨或乙酸銨之類的氨鹽類,適合地為氨水溶液。
反應溫度雖依使用的胺基化劑而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依胺基化劑的種類及使用量而不同,但通常為10分鐘至8小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(2F),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(F-3步驟)
本步驟係藉由將化合物(2F),於溶媒中,鹼存在下,使用醯化劑而將胺基進行醯化,而製造化合物(3F)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為乙腈。
就使用的鹼而言,可列舉例如,氫氧化鈉、氫氧化鉀、碳酸氫鈉、碳酸鈉等之無機鹼;三乙基胺、吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼;惟適合地為氫氧化鈉。
就使用的醯化劑而言,可列舉苯甲醯氯、苯甲酸酐等,適合地為苯甲酸酐。
反應溫度雖依使用的醯化劑而不同,但通常為0℃至50℃,適合地為10℃至40℃。
反應時間雖依醯化劑的種類及使用量而不同,但通常為1至48小時,適合地為2小時至20小時。
反應結束後,本反應之目的化合物(3F),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(F-4步驟)
本步驟係將化合物(3F),於溶媒中,藉由鹼將3位之醯基選擇性地水解,而製造化合物(4F)的步驟。
就使用的溶媒而言,可列舉例如,甲醇、乙醇、丙醇等之醇類、水、乙腈;惟適合地為水、乙腈。
就使用的鹼而言,可列舉例如,氫氧化鈉、氫氧化鉀、碳酸氫鈉、碳酸鈉等之無機鹼,適合地為氫氧化鈉。
反應溫度雖依使用的鹼而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依使用的鹼而不同,但通常為10分鐘至24小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(4F),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
本發明之化合物(3G)可藉由以下所述的G法而製造。
(G法)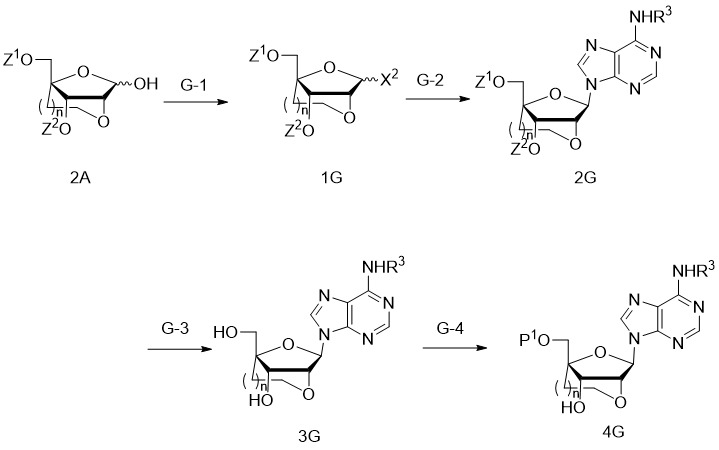 G法中,R3
、X2
、Z1
、Z2
及P1
表示與前述同意義。
G法中,R3
、X2
、Z1
、Z2
及P1
表示與前述同意義。
以下,對於G法之步驟進行詳細說明。
(G-1步驟)
本步驟係藉由使用化合物(2A),於溶媒中,鹼存在下,使活化劑反應,而製造化合物(1G)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為甲苯、乙腈。
反應溫度雖依使用的活化劑而不同,但通常為0℃至50℃,適合地為0℃至30℃。
使用的活化劑可列舉乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑、氯磷酸二苯酯,適合地為乙酸酐、苯甲酸酐。
使用的鹼雖依活化劑而不同,但可列舉三乙基胺、吡啶、N,N-二甲基胺基吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼,適合地為N,N-二甲基胺基吡啶。
反應溫度雖依使用的活化劑而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依活化劑的種類及使用量而不同,但通常為10分鐘至8小時,適合地為1小時至3小時。
反應結束後,本反應之目的化合物(1G),例如可用於直接醣基化,或將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(G-2步驟)
本步驟係對化合物(1G),於溶媒中,酸試藥存在下,進行與胺基經保護的6-胺基嘌呤-9-基的醣基化反應,之後伴隨異構物化之製造化合物(2G)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃之類的醚類;乙腈;惟適合地為乙腈。
使用的酸試藥可列舉二氯二甲基矽烷與三氟甲磺酸、三氟甲磺酸三甲基矽酯與三氟甲磺酸、甲磺酸、三氟乙酸等,適合地為三氟甲磺酸三甲基矽酯與三氟乙酸。
醣基化及異構物化之反應溫度雖依化合物(1G)之結構或使用的酸試藥而不同,但通常為30℃至70℃,適合地為40℃至60℃。
反應時間雖依路易士酸試藥的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(2G),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(G-3步驟)
本步驟係使化合物(2G),於溶媒中,與去保護試藥反應,藉由將Z1
及Z2
去保護而製造化合物(3G)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;惟適合地為二氯甲烷。
Z1
及Z2
為苄基之情形之去保護試藥可列舉氯化鐵(III)、三氯化硼等,適合地為三氯化硼。
反應溫度雖依去保護試藥而不同,但通常為-20℃至30℃,適合地為-20℃至20℃。
反應時間雖依去保護試藥而不同,但通常為10分鐘至10小時,適合地為1小時至5小時。
反應結束後,本反應之目的化合物(3G),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(G-4步驟)
本步驟之目的化合物(4G)可按照E法E-4步驟,藉由保護化合物(3G)之1級羥基而製造。
又,本發明之化合物(5H),亦可藉由以下所述的H法(使用二氯嘌呤的醣基化)而製造。
(H法) H法中,R3
、Z1
、Z2
及P1
表示與前述同意義。
H法中,R3
、Z1
、Z2
及P1
表示與前述同意義。
以下,對於H法之步驟進行詳細說明。
(H-1步驟)
本步驟係藉由使化合物(1G),於溶媒中,鹵化劑存在下,與藉由矽化反應而經矽化的二氯嘌呤與醣基化反應,而製造化合物(1H)的步驟。
就醣基化反應及矽化反應所使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃之類的醚類;乙腈;適合地為乙腈。
矽化反應所使用的矽化劑可列舉N,O-雙三甲基矽基乙醯胺、六甲基二矽氮烷等,適合地為N,O-雙三甲基矽基乙醯胺。
二氯嘌呤之矽化反應之反應溫度雖依矽化劑而不同,但通常為0℃至90℃,適合地為50℃至80℃。
矽化反應之反應時間雖依矽化劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至10小時。
醣基化反應所使用的鹵化劑可列舉氯三甲基矽烷(TMSCl)、溴三甲基矽烷(TMSBr)、碘三甲基矽烷(TMSI),適合地為TMSI。醣基化反應之反應溫度雖依化合物(2B)之結構或使用的鹵化劑而不同,但通常為0℃至90℃,適合地為50℃至80℃。
醣基化反應之反應時間雖依鹵化劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為10小時至5小時。
反應結束後,本反應之目的化合物(1H),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(H-2步驟)
本步驟為藉由使化合物(1H),於溶媒中,與胺基化劑反應,而製造化合物(2H)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為四氫呋喃。
就使用的胺基化劑而言,可列舉氨、氨水溶液或碳酸銨或乙酸銨之類的氨鹽類,適合地為氨水溶液。
反應溫度通常為0℃至90℃,適合地為30℃至60℃。
反應時間雖依溶媒及胺基化劑之使用量而不同,但通常為10分鐘至24小時,適合地為1小時至10小時。
反應結束後,本反應之目的化合物(2H),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(H-3步驟)
本步驟係於溶媒中,藉由化合物(2H)之Z1
及Z2
之去保護反應及嘌呤環之2位之氯原子之氫化反應,而製造化合物(3H)的步驟。
就使用的溶媒而言,可列舉例如,水、甲醇、乙醇、1-丙醇、2-丙醇等之醇類;苯、甲苯等之烴類;四氫呋喃、二乙基醚之類的醚類;乙腈;惟適合地為乙醇。
Z1
及Z2
為苄基之情形,於金屬觸媒存在下,使用還原劑,可同時進行Z1
及Z2
之去保護反應與嘌呤環之2位之氫化反應。
使用的金屬觸媒可列舉鈀、氫氧化鈀、鉑(特別是承載於碳上的鈀、氫氧化鈀、或鉑)等,適合地為鈀(特別是承載於碳上的鈀)。
使用的還原劑可列舉氫、甲酸、甲酸銨等,適合地為氫。
反應溫度可依金屬觸媒而不同,但通常為0℃至70℃,適合地為40℃至60℃。
反應時間可依金屬觸媒、還原劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至10小時。
反應結束後,本反應之目的化合物(3H),例如藉由將反應液過濾而去除金屬觸媒,將反應混合物濃縮,並餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(H-4步驟)
本步驟係化合物(3H),於溶媒中,將羥基以羥基之保護試藥保護後,將嘌呤環上之胺基以醯化劑進行醯化,接著藉由氨使羥基上之保護基脫離,而製造化合物(4H)的步驟。
就使用的溶媒而言,可列舉例如,吡啶、乙腈;惟適合地為吡啶。
就使用的羥基之保護試藥而言,可列舉氯三甲基矽烷、三氟甲磺醯基三甲基矽烷等,適合地為氯三甲基矽烷。
反應之溫度通常為0℃至90℃,適合地為0℃至30℃。
反應時間雖依羥基之保護試藥的種類及使用量而不同,但通常為10分鐘至2小時,適合地為30分鐘至1小時。
就使用的醯化劑而言,可列舉乙酸酐、乙醯氯、苯甲酸酐、苯甲醯氯等,適合地為苯甲醯氯。
醯化反應之溫度通常為0℃至90℃,適合地為0℃至30℃。
反應時間雖依羥基之保護試藥的種類及使用量而不同,但通常為1小時至8小時,適合地為1小時至3小時。
本反應之目的化合物(4H),例如藉由於反應液中添加氨水後,將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(H-5步驟)
本步驟之目的化合物(5H)可按照E法E-4步驟,藉由將化合物(4H)之1級羥基加以保護而製造。
本發明之化合物(4I)可藉由以下所述的I法而製造。
(I法)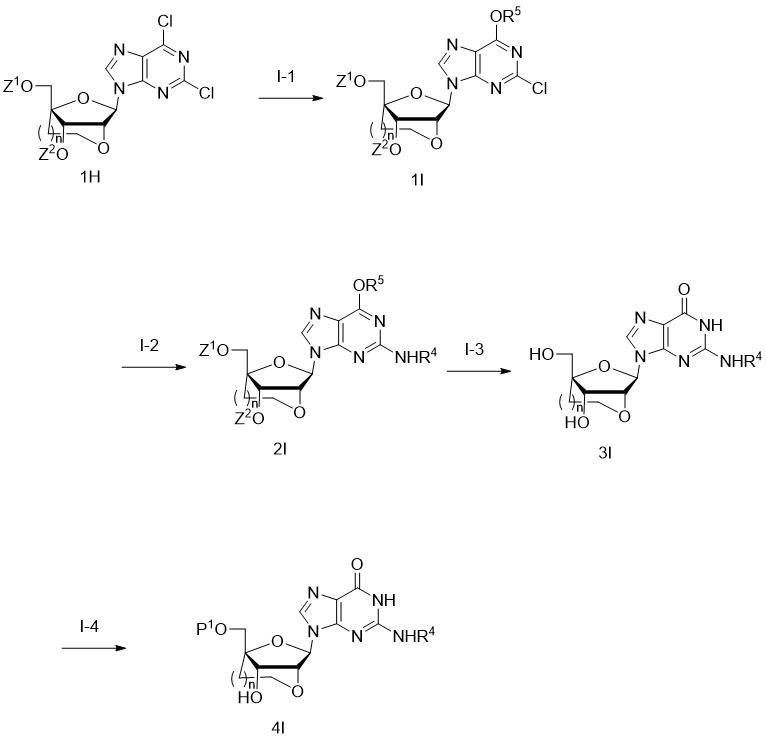 I法中,R4
、R5
、Z1
、Z2
及P1
表示與前述同意義。
I法中,R4
、R5
、Z1
、Z2
及P1
表示與前述同意義。
以下,對於I法之各步驟進行詳細說明。
(I-1步驟)
本步驟係將化合物(1H),於溶媒中,鹼存在下,藉由與可具有取代基的芐醇之取代反應而製造化合物(1I)的步驟。
就可具有取代基的芐醇而言,可列舉例如,芐醇、4-甲基芐醇、2,4,6-三甲基芐醇、3,4,5-三甲基芐醇、4-甲氧基芐醇、4-氯芐醇、4-溴芐醇或4-氰基芐醇,適合地為芐醇。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為四氫呋喃。
就使用的鹼而言,可列舉例如,氫化鈉、碳酸鈉等之無機鹼;三乙基胺、吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼;惟適合地為氫化鈉。
反應溫度通常為0℃至90℃,適合地為0℃至30℃。
反應時間雖依溶媒及鹼之使用量而不同,但通常為10分鐘至24小時,適合地為1小時至10小時。
反應結束後,本反應之目的化合物(1I),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(I-2步驟)
本步驟係將化合物(1I),於溶媒中,鹼、鈀觸媒、膦配位子存在下,藉由與醯胺化劑之交叉偶合反應,而製造化合物(2I)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;苯、甲苯等之烴類;四氫呋喃、二甲基醚之類的醚類;乙腈;惟適合地為甲苯。
就使用的鹼而言,可列舉例如,氫氧化鈉、碳酸鈉、碳酸銫等之無機鹼;三乙基胺、吡啶、1,8-二氮雜雙環[5.4.0]十一-7-烯之類的有機鹼;惟適合地為碳酸銫。
就使用的鈀觸媒而言,可列舉參(二亞苄基丙酮)(氯仿)二鈀、乙酸鈀(II)、參(二亞苄基丙酮)二鈀(0)等,適合地為參(二亞苄基丙酮)(氯仿)二鈀。
就使用的膦配位子而言,可列舉4,5’-雙(二苯基膦基)-9,9’-二甲基𠮿、1,1’-雙(二苯基膦基)二茂鐵、1,2-雙(二苯基膦基)乙烷、2-二環己基膦基-2’-(N,N-二甲基胺基)聯苯等,適合地為4,5’-雙(二苯基膦基)-9,9’-二甲基𠮿。
就使用的醯胺化劑而言,可列舉乙醯基醯胺、苄醯基醯胺、異丁基醯胺等,適合地為異丁基醯胺。
反應溫度通常為20℃至150℃,適合地為90℃至110℃。
反應時間雖依溶媒及鈀觸媒之使用量而不同,但通常為10分鐘至24小時,適合地為5小時至15小時。
反應結束後,本反應之目的化合物(2I),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(I-3步驟)
本步驟係使化合物(2I),於溶媒中,與羥基之去保護試藥反應,藉由Z1
、Z2
及R5
之去保護而製造化合物(3I)的步驟。
Z1
及Z2
為苄基之情形,去保護試藥為承載於碳上的金屬觸媒及還原劑。
使用的金屬觸媒可列舉鈀、氫氧化鈀、鉑等,適合地為氫氧化鈀。
使用的還原劑可列舉氫、甲酸、甲酸銨等,適合地為氫。
反應溫度雖依金屬觸媒而不同,但通常為0℃至70℃,適合地為40℃至60℃。
反應時間雖依金屬觸媒、還原劑的種類及使用量而不同,但通常為10分鐘至24小時,適合地為1小時至10小時。
反應結束後,本反應之目的化合物(3I),例如藉由將反應液過濾而去除金屬觸媒,將反應混合物濃縮,並餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
(I-4步驟)
本步驟之目的化合物(4I)可藉由按照E法E-4步驟將化合物(3I)之1級羥基加以保護製造。
化合物(2J)可藉由以下所述的J法而製造。
(J法) J法中,P1
及B表示與前述同意義。
J法中,P1
及B表示與前述同意義。
以下,對於J法之步驟進行詳細說明。
(J-1步驟)
本步驟係使化合物(1J),於溶媒中,乾燥劑、活化劑存在下,與AMIDITE化試藥反應,並藉由AMIDITE化反應,而製造化合物(2J)的步驟。
就使用的溶媒而言,可列舉例如,二氯甲烷、氯仿、1,2-二氯乙烷之類的鹵化烴類;乙酸乙酯、乙酸丙酯、乙酸丁基等之酯類;苯、甲苯等之烴類;四氫呋喃之類的醚類;乙腈;惟適合地為二氯甲烷。
使用的乾燥劑可列舉分子篩3A、分子篩4A、或分子篩5A,適合地為分子篩4A。
使用的活化劑可列舉5-苄硫基四唑、5-苯基四唑、二溴咪唑、二氰基咪唑、N-烷基咪唑三氟乙酸鹽等,適合地為4,5-二氰基咪唑。
使用的AMIDITE化試藥可列舉2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺、2-氰基乙基二異丙基氯亞磷醯胺,適合地為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺。
反應溫度雖依使用的AMIDITE化試藥而不同,但通常為0℃至50℃,適合地為0℃至30℃。
反應時間雖依使用的AMIDITE化試藥而不同,但通常為10分鐘至24小時,適合地為1小時至16小時。
反應結束後,本反應之目的化合物(2J),例如藉由將反應液中和,並將反應混合物濃縮,餾除溶媒而獲得。
獲得的化合物,若需要,可藉由通常方法,例如再結晶、矽膠層析等而進一步純化。
本發明,作為一個態樣,提供一種製造寡核苷酸之方法,以上述之方法所製造的各ENA單體及市售之核酸或對應修飾核酸的核苷之亞磷醯胺化合物、及將配位體單元之亞磷醯胺化合物藉由磷酸二酯鍵或硫代磷酸酯鍵而連結,使單元鏈伸長,而具有所期望的序列・結構。此種寡核苷酸之製造,可藉由使用市售之合成機(例如,Perkin Elmer公司之利用亞磷醯胺法的模型392)等,依據文獻(Nucleic Acids Research, 12, 4539 (1984))記載之方法合成而進行。此時所使用的各ENA之核苷(即,2’-O,4’-C-伸乙基交聯鳥苷、2’-O,4’-C-伸乙基交聯腺苷、2’-O,4’-C-伸乙基交聯胞苷、2’-O,4’-C-伸乙基交聯胸苷)之亞磷醯胺化合物可藉由上述之方法而被製造的化合物。關於天然型之核苷及2’-O-甲基核苷(即,2’-O-甲基鳥苷、2’-O-甲基腺苷、2’-O-甲基胞苷、2’-O-甲基尿苷)之亞磷醯胺化合物,可使用市售之試藥而製造。
於本發明,核酸鹼基序列係可各自將腺嘌呤記載為(A)或(a),將鳥嘌呤記載為(G)或(g),將胞嘧啶記載為(C)或(c),將胸腺嘧碇記載為(T)或(t),及將尿嘧啶記載為(U)或(u)。可使用5-甲基胞嘧啶替代胞嘧啶。核酸鹼之中,尿嘧啶(U)或(u)與胸腺嘧碇(T)或(t)有互換性。尿嘧啶(U)或(u)與胸腺嘧碇(T)或(t)任一者皆可使用於與互補鏈的腺嘌呤(A)或(a)進行鹼基配對。
將亞磷醯胺化合物偶合後,藉由使硫、二硫化四乙基秋蘭姆(tetraethylthiuram disulfide(TETD),Applied Biosystems公司)、Beaucage試藥(Glen Research公司)、或氫化黃原膠(xanthan hydride)等之試藥反應,可合成具有硫代磷酸酯鍵的寡核苷酸(Tetrahedron Letters, 32, 3005 (1991), J. Am. Chem. Soc. 112, 1253 (1990), PCT/WO98/54198)。
就於合成機所使用的定孔玻璃珠(Controlled pore glass(CPG))而言,2’-O-甲基核苷之鍵結者可利用市售者。又,對於2’-O,4’-C-亞甲基鳥苷、腺苷、5-甲基胞苷及胸苷,按照WO99/14226記載之方法,對於上述之方法所製造的2’-O, 4’-C-伸乙基鳥苷、腺苷、5-甲基胞苷及胸苷,按照文獻(Oligonucleotide Synthesis, Edited by M.J.Gait, Oxford University Press, 1984),可與CPG結合。藉由使用經修飾的CPG(記載於日本特開平7-87982之實施例12b),可合成於3’末端鍵結有2-羥基乙基磷酸基的寡核苷酸。又,若使用3’-amino-Modifier C3 CPG、3’-amino-Modifier C7 CPG, Glyceryl CPG (Glen Research)、3’-specer C3 SynBase CPG 1000、3’-specer C9 SynBase CPG 1000(link technologies),可合成於3’末端鍵結有羥基烷基磷酸基、或胺基烷基磷酸基的寡核苷酸。
藉由本發明所製造的寡核苷酸,亦可含有適合對核酸之組織進行輸送的配位體單元。配位體單元係,配位體部透過連接子(linker)而合成包含磷酸部的AMIDITE化合物,藉此藉由與核苷酸之伸長同樣的方法,可與寡核苷酸之5’末端結合。於對肝臓的輸送,使用GalNAc藉由連接子與磷酸部結合的寡核苷酸,於該寡核苷酸之製造,使用對應以下之X18
或X20
等的亞磷醯胺化合物。可各自使用:
為GalNAc單元的對應X18
的亞磷醯胺化合物,WO2019/172286之參考例39之化合物39D ([(2R,3R,4R,5R,6R)-5-乙醯胺-6-[3-[[2-[[3-[[2-[3-[(2R,3R,4R,5R,6R)-3-乙醯胺-4,5-二乙醯氧基-6-[[雙(4-甲氧基苯基)-苯基-甲氧基]甲基]四氫哌喃-2-基]氧基丙基胺基]-2-側氧基-乙基]胺甲醯氧基]-2-[2-氰基乙氧基-(二異丙基胺基)磷烷基]氧基-丙氧基]羰基胺基]乙醯基]胺基]丙氧基]-4-乙醯氧基-2-[[雙(4-甲氧基苯基)-苯基-甲氧基]甲基]四氫哌喃-3-基] 乙酸酯)、 對應X20
的亞磷醯胺化合物,WO2019/172286之參考例41之化合物41D([(2R,3R,4R,5R,6R)-5-乙醯胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙醯胺-4,5-二乙醯氧基-6-[[雙(4-甲氧基苯基)-苯基-甲氧基]甲基]四氫哌喃-2-基]氧基丙基胺基]-3-側氧基-丙基]胺甲醯氧基]-2-[2-氰基乙氧基-(二異丙基胺基) 磷烷基l]氧基-丙氧基]羰基胺基]丙醯基胺基]丙氧基]-4-乙醯氧基-2-[[雙(4-甲氧基苯基)-苯基-甲氧基]甲基]四氫哌喃-3-基] 乙酸酯)、
對應X20
的亞磷醯胺化合物,WO2019/172286之參考例41之化合物41D([(2R,3R,4R,5R,6R)-5-乙醯胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙醯胺-4,5-二乙醯氧基-6-[[雙(4-甲氧基苯基)-苯基-甲氧基]甲基]四氫哌喃-2-基]氧基丙基胺基]-3-側氧基-丙基]胺甲醯氧基]-2-[2-氰基乙氧基-(二異丙基胺基) 磷烷基l]氧基-丙氧基]羰基胺基]丙醯基胺基]丙氧基]-4-乙醯氧基-2-[[雙(4-甲氧基苯基)-苯基-甲氧基]甲基]四氫哌喃-3-基] 乙酸酯)、
 |
就本發明之方法所製造的寡核苷酸之一個態樣而言,可例示具有以下之式所表示的序列及結構,且有用於杜興氏肌肉失養症(Duchenne muscular dystrophy)之治療的寡核苷酸(參照WO2004/048570)。
(DMD AO01)
HO-Ce2s
-Am1s
-Gm1s
-Te2s
-Te2s
-Um1s
-Gm1s
-Ce2s
-Ce2s
-Gm1s
-Ce2s
-Te2s
-Gm1s
-Ce2s
-Ce2s
-Ce2s
-Am1s
-Am1s
- CH2
CH2
OH(序列識別號1)
(DMD AO02)
HO-Te2s
-Gm1s
-Te2s
-Te2s
-Ce2s
-Te2s
-Gm1s
-Am1s
-Ce2s
-Am1s
-Am1s
-Ce2s
-Am1s
-Gm1s
-Te2s
-Te2s
-Te2s
-Gm1s
- CH2
CH2
OH(序列識別號2)
(DMD AO03)
HO-Ce2s
-Gm1s
-Ce2s
-Te2s
-Gm1s
-Cm1s
-Ce2s
-Ce2s
-Am1s
-Am1s
-Te2s
-Gm1s
-Ce2s
-Ce2s
-Am1s
-Um1s
-Ce2s
-Ce2s
- CH2
CH2
OH(序列識別號3)
(DMD AO04)
HO-Ce2s
-Am1s
-Te2s
-Am1s
-Am1s
-Te2s
-Gm1s
-Am1s
-Ae2s
-Am1s
-Am1s
-Ce2s
-Gm1s
-Cm1s
-Ce2s
-Gm1s
-Ce2s
-Ce2s
- CH2
CH2
OH(序列識別號4)
(DMD AO05)
HO-Te2s
-Um1s
-Ce2s
-Cm1s
-Ce2s
-Am1s
-Am1s
-Te2s
-Um1s
-Cm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Gm1s
-Ae2s
-Am1s
-Te2s
- CH2
CH2
OH(序列識別號5)
(DMD AO06)
HO-Ce2s
-Ce2s
-Am1s
-Um1s
-Te2s
-Um1s
-Gm1s
-Te2s
-Am1s
-Um1s
-Te2s
-Te2s
-Am1s
-Gm1s
-Ce2s
-Am1s
-Te2s
-Gm1s
- CH2
CH2
OH(序列識別號6)
(DMD AO07)
HO-Gm1s
-Gm1s
-Ce2s
-Te2s
-Gm1s
-Cm1s
-Te2s
-Te2s
-Um1s
-Gm1s
-Ce2s
-Cm1s
-Cm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Ce2s
-CH2
CH2
OH(序列識別號7)
(DMD AO08)
HO-Gm1s
-Ce2s
-Te2s
-Am1s
-Gm1s
-Gm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Gm1s
-Te2s
-Gm1s
-Cm1s
-Te2s
-Te2s
-Um1s
-CH2
CH2
OH(序列識別號8)
(DMD AO09)
HO-Am1s
-Ce2s
-Ce2s
-Gm1s
-Cm1s
-Ce2s
-Te2s
-Um1s
-Cm1s
-Ce2s
-Am1s
-Cm1s
-Te2s
-Ce2s
-Am1s
-Gm1s
-Ae2s
-Gm1s
-CH2
CH2
OH; (序列識別號9)
(DMD AO10)
HO-Ge2s
-Ge2s
-Ce2s
-Ae2s
-Te2s
-Um1s
-Um1s
-Cm1s
-Um1s
-Am1s
-Gm1s
-Um1s
-Um1s
-Te2s
-Ge2s
-Ge2s
-Ae2s
-Ge2s
- CH2
CH2
OH(序列識別號10)
(DMD AO11)
HO-Gm1s
-Gm1s
-Ce2s
-Am1s
-Te2s
-Te2s
-Um1s
-Ce2s
-Te2s
-Am1s
-Gm1s
-Um1s
-Te2s
-Te2s
-Gm1s
-Gm1s
-Ae2s
-Gm1s
- CH2
CH2
OH(序列識別號11)
(DMD AO12)
HO-Ae2s
-Gm1s
-Te2s
-Um1s
-Te2s
-Gm1s
-Gm1s
-Ae2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Ae2s
-Gm1s
-Te2s
-Te2s
- CH2
CH2
OH(序列識別號12)
(DMD AO13)
HO-Ce2s
-Te2s
-Cm1s
-Ce2s
-Te2s
-Um1s
-Ce2s
-Ce2s
-Am1s
-Te2s
-Gm1s
-Am1s
-Ce2s
-Te2s
-Ce2s
-Am1s
-Am1s
-Gm1s
- CH2
CH2
OH(序列識別號13)
(DMD AO14)
HO-Ce2s
-Te2s
-Gm1s
-Am1s
-Am1s
-Gm1s
-Gm1s
-Te2s
-Gm1s
-Te2s
-Te2s
-Ce2s
-Te2s
-Te2s
-Gm1s
-Te2s
-Am1s
-Ce2s
- CH2
CH2
OH(序列識別號14)
(DMD AO15)
HO-Te2s
-Te2s
-Cm1s
-Ce2s
-Am1s
-Gm1s
-Ce2s
-Ce2s
-Am1s
-Te2s
-Te2s
-Gm1s
-Te2s
-Gm1s
-Te2s
-Te2s
-Gm1s
-Am1s
- CH2
CH2
OH(序列識別號15)
[於上述式中,左側表示5’末端,右側表示3’末端,A、G、C、U及T各自表示D-核呋喃糖經修飾且5’位之碳原子與左側所表示的結構單元進行硫代磷酸酯鍵結而成的腺苷、鳥苷、胞苷、尿苷及胸苷。附於各核苷酸或核苷之e2s表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯且3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷的5’位碳原子鍵結,e2t表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯且3’位以-O-與3’末端之氫原子鍵結,m1s表示D-核呋喃糖經2’-O-甲基化且3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,m1t表示D-核呋喃糖經2’-O-甲基化且3’位以-O-與3’末端之氫原子鍵結。]
又,就本發明之方法所製造的寡核苷酸之一個態樣而言,可例示具有以下之式所表示的序列及結構,且有用於肝醣貯積病1a型之治療的寡核苷酸(參照WO2019/172286)。
(GSD AO01)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號16)
(GSD AO02)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號17)
(GSD AO03)
X18
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號18)
(GSD AO04)
X18
-Am1s
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號19)
(GSD AO05)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號20)
(GSD AO06)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Am1s
-Te2s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號21)
(GSD AO07)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號22)
(GSD AO08)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號23)
(GSD AO09)
X18
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號24)
(GSD AO10)
X18
-Am1s
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號25)
(GSD AO11)
X18
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H1(序列識別號26)
(GSD AO12)
X18
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2s
-Um1t
-H(序列識別號27)
(GSD AO13)
X20
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號28)
(GSD AO14)
X20
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號29)
(GSD AO15)
X20
-Am1s
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1t
-H(序列識別號30)
(GSD AO16)
X20
-Am1s
-Ae2s
-Um1s
-Cm1s
-Ce2s
-Gm1s
-Ae2s
-Um1s
-Gm1s
-Gm1s
-Ce2s
-Gm1s
-Am1s
-Ae2s
-Gm1s
-Ce2t
-H(序列識別號31)
[於上述式中,左側表示5’末端,右側表示3’末端,A、G、C、U及T各自表示D-核呋喃糖經修飾且5’位之碳原子與左側所表示的結構單元進行硫代磷酸酯鍵結而成的腺苷、鳥苷、胞苷、尿苷及胸苷。附於各核苷酸或核苷之e2s表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯且3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷的5’位碳原子鍵結,e2t表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯且3’位以-O-與3’末端之氫原子鍵結,m1s表示D-核呋喃糖經2’-O-甲基化且3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,m1t表示D-核呋喃糖經2’-O-甲基化且3’位以-O-與3’末端之氫原子鍵結。
於上述式中,X18
及X20
表示下述之式所表示的GalNAc單元。於下述之式中,與磷酸基鍵結的鍵結肢係表示與寡核苷酸之5’末端的碳原子鍵結,而形成磷酸二酯鍵。]
[實施例]
以下參照實施例同時進一步具體地說明本發明,但本發明之範圍並未受彼等實施例限制。
(實施例1)2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖(2,6-anhydro-3-O-benzyl-4-[(benzyloxy)methyl]-5-deoxy-α-L-lyxo-hexofuranose)化合物2之製造
(實施例1-1)(3aR,3bR,6S,6aS,7aR)-6-羥基-2,2-二甲基四氫-2H-呋喃并[2’,3’:4,5]呋喃并[2,3-d][1,3]二氧呃-5(3bH)-酮(化合物5)之二甲基乙醯胺溶液之製造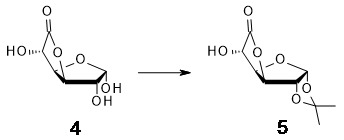
於丙酮(11.9kg)中依序添加市售之(2R)-2-[(2S,3R,4S)-3,4-二羥基-5-側氧基-四氫呋喃-2-基]-2-羥基-乙醛(化合物4)(1.00kg、5.678 mol)及濃硫酸(114g、1.162 mol),於40~50℃攪拌17小時。將反應液冷卻至20~30℃,添加碳酸氫鈉(0.19kg、2.271 mol)而攪拌2小時後,濾除生成的白色固體,將該白色固體以丙酮(2.37kg)洗淨,將洗淨液加至濾液中。重複2次於如此獲得的溶液中添加二甲基乙醯胺(7.50kg)及甲苯(4.34kg)並進行減壓濃縮,進一步添加甲苯(4.34kg)並進行減壓濃縮的操作,獲得化合物5之二甲基乙醯胺溶液。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。1
H NMR (CDCl3
):δ = 1.36 (3H, s), 1.53 (3H, s), 2.72 (1H, d, J = 9.5 Hz), 4.50 (1H, dd, J = 4.5, 9.5 Hz),4.84 (2H, d, J = 2.5 Hz), 4.95 (1H, dd, J = 2.5, 4.5 Hz), 5.99 (1H, d, J = 3.5 Hz).
(實施例1-2)(3aR,3bR,6S,6aR,7aR)-2,2-二甲基-5-側氧基六氫-2H-呋喃并[2’,3’:4,5]呋喃并[2,3-d][1,3]二氧呃-6-基=4-甲基苯-1-磺酸酯(化合物6)之製造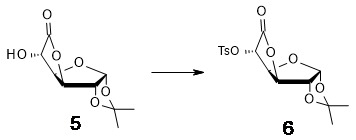
於實施例1-1所獲得的化合物5之二甲基乙醯胺溶液中,依序添加1-甲基咪唑(0.56kg、6.814 mol)及對甲苯磺醯氯(1.24kg、6.530 mol),於20~30℃攪拌3小時。於反應溶液中添加水(5kg)而攪拌3小時後,冷卻至0~5℃而進一步攪拌2小時。濾取於反應溶液中生成的白色固體,依序以水/二甲基乙醯胺(1/1、4kg)及水(8kg)洗淨。將獲得的固體進行減壓乾燥(40℃),而獲得化合物6(1.66kg、產率79.4%)。1
H NMR (CDCl3
):δ = 1.33 (3H, s), 1.49 (3H, s), 2.45 (3H, s), 4.78 (1H, d, J = 3.5 Hz), 4.83 (1H, d, J = 3.0 Hz), 4.97 (1H, dd, J = 3.0, 4.5 Hz), 5.22 (1H, d, J = 4.5 Hz), 5.97 (1H, d, J = 3.5 Hz), 7.37 (2H, d, J = 8.0 Hz), 7.89 (2H, m).
(實施例1-3)5-去氧-1,2-O-(1-甲基亞乙基)-α-D-木-己呋喃糖(化合物7)之甲苯溶液之製造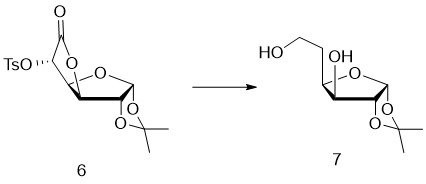
於調節至0~10℃的四氫呋喃(22.1kg、15.0v/w)中,依序添加雙(2-甲氧基乙氧基)氫化鋁鈉(4.53kg、15.69 mol)及實施例1-2所獲得的化合物6(1.66kg、4.48 mol),攪拌30分鐘後,再於20~30℃攪拌1.5小時。反應後,將反應液冷卻至0~10℃,滴加丙酮(1.05kg、0.8v/w)及50%L-酒石酸鈉鉀水溶液(17.0kg、8.0v/w),將反應液升高至40~50℃,攪拌14小時後,於40~50℃進行分液。於獲得的有機層中,於40~50℃,添加20%食鹽水(3.81kg、2.0v/w),藉由分液而獲得四氫呋喃層。藉由重複進行2次相同的分液操作,於獲得的溶液中添加甲苯(7.20kg、5.0v/w),進行減壓濃縮直到容積成為3.3L。於藉由進行3次同樣的減壓濃縮操作所獲得的溶液中添加甲苯(20.0kg、13.6w/w),而獲得化合物7之甲苯溶液。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。1
H NMR (CDCl3
):δ = 1.32 (3H, s), 1.51 (3H, s), 1.94-2.06 (2H, m), 2.46 (1H, br), 3.26 (1H, br), 3.75 (1H, td, J = 3.5, 9.5 Hz), 3.87-3.91 (1H, m), 4.11 (1H, d, J = 7.5 Hz), 4.26 (1H, td, J = 3.0, 6.0 Hz), 4.55 (1H, d, J = 4.0 Hz), 5.91 (1H, d, J = 4.0 Hz).
(實施例1-4)5-去氧-1,2-O-(1-甲基亞乙基)-6-O-(三苯基甲基)-α-D-木-己呋喃糖(化合物8)之甲苯溶液之製造
於實施例1-3所獲得的化合物7之甲苯溶液中,依序添加三苯氯甲烷(0.87 kg、3.14 mol)及4-甲基啉(0.54 kg、5.38 mol),於50~60℃攪拌3.5小時。反應後,將反應液冷卻至20~30℃,添加8%碳酸氫鈉水(3.42 kg、2.0v/w)而進行分液,獲得甲苯層。於獲得的甲苯層中添加5%食鹽水(10.26 kg、6.0v/w),並進行分液。對於獲得的甲苯層,進行3次同樣之分液操作,藉此獲得化合物8之甲苯溶液。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。1
H NMR (CDCl3
):δ = 1.31 (3H, s), 1.48 (3H, s), 1.99-2.02 (2H, m), 2.77 (1H, d, J = 4.0 Hz), 3.15-3.19 (1H, m), 3.40 (1H, ddd, J = 4.5, 5.0, 10.0 Hz), 4.08 (1H, t, J = 2.5 Hz), 4.25 (1H, ddd, J = 2.5, 7.0, 7.5 Hz), 4.53 (1H, d, J = 3.5 Hz), 5.88 (1H, d, J = 3.5 Hz), 7.23-7.26 (3H, m), 7.29-7.32 (6H, m), 7.39-7.41 (6H, m).
(實施例1-5a)5-去氧-1,2-O-(1-甲基亞乙基)-6-O-(三苯基甲基)-α-D-赤蘚-己呋喃-3-酮糖(化合物9)之甲苯溶液之製造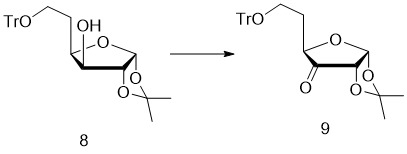
實施例1-4所獲得的化合物8之甲苯溶液中,於室溫添加8%碳酸氫鈉水(5.13kg、3.0v/w)、溴化鉀(53.0 g、0.45 mol)及9-氮雜降金剛烷-N-氧基(6.2 g、44.82 mmol),將此液溫度調節至0~10℃後,添加次氯酸鈉・5水合物(2.21 kg、13.45 mol),進行攪拌1.5小時。反應後,將反應液分液並獲得甲苯層。將獲得的甲苯層,重複2次依序藉由5%亞硫酸鈉水溶液(5.22 kg、3.0v/w)及20%食鹽水(3.80 kg、2.0v/w)進行洗淨的操作,而獲得化合物9之甲苯溶液。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。1
H NMR (CDCl3
):δ = 1.36 (3H, s), 1.44 (3H, s), 1.96-2.01 (1H, m), 2.21-2.28 (1H, m), 3.06 (1H, ddd, J = 3.0, 4.5, 10.0 Hz), 3.30 (1H, ddd, J = 4.0, 4.5, 9.0 Hz), 4.33 (1H, dd, J = 1.0, 4.5 Hz), 4.49 (1H, dd, J = 3.5, 5.5 Hz), 5.60 (1H, d, J = 4.5), 7.21-7.25 (3H, m), 7.29-7.32 (6H, m), 7.35-7.37 (6H, m).
高速液體層析(HPLC)分析條件
管柱:Xbridge C18 3.5 ⌠m, 4.6x150 mm、管柱溫度:40℃
移動相A:10 mM 乙酸銨水溶液、移動相B:乙腈
梯度條件:B(%) 30% (0-3 min), 30→95% (3-20 min), 95% (20-23 min)
流速:1.0 mL/min、檢測波長:210 nm
化合物9之滯留時間:13.9 min及17.5 min
(實施例1-5b)5-去氧-1,2-O-(1-甲基亞乙基)-6-O-(三苯基甲基)-α-D-赤蘚-己呋喃-3-酮糖(化合物9)之甲苯溶液之製造
於實施例1-4所獲得的化合物8之甲苯溶液(30 mL)中,於室溫添加8%碳酸氫鈉水(6.0 mL、3.0v/w)、溴化鉀(60.0 mg、0.54 mmol)及9-氮雜降金剛烷-N-氧基(3.7 mg、0.03 mmol),將此液溫度調節成0~10℃後,於10℃以下添加次氯酸鈉水溶液(6.48 g、10.80 mmol),於0℃進行攪拌2小時。反應結束後,添加15%亞硫酸鈉水溶液(12.0 mL、6.0v/w),於室溫攪拌1小時。將反應液分液並獲得甲苯層。將獲得的甲苯層,以20%食鹽水(4.0 mL、2.0v/w)洗淨,獲得化合物9之甲苯溶液。
獲得的化合物之HPLC分析的滯留時間係與(實施例1-5a)所獲得的化合物之HPLC分析的滯留時間一致。
(實施例1-6)5-去氧-4-(羥基甲基)-1,2-O-(1-甲基亞乙基)-6-O-(三苯基甲基)-β-L-蘇-己呋喃-3-酮糖(化合物10)及(3aS,3bR,7aR,8aR)-2,2-二甲基-7a-[2-(三苯基甲氧基)乙基]四氫-2H,3bH,5H-[1,3]二氧呃并[4,5]呋喃并[3,2-d][1,3]二 英-3b-醇(化合物11)之混合甲苯溶液之製造
英-3b-醇(化合物11)之混合甲苯溶液之製造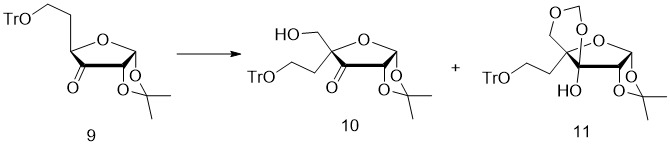
於實施例1-5所獲得的化合物9之甲苯溶液中,添加37%甲醛水溶液(1.81 kg、1.0v/w)及1,8-二氮雜雙環[5.4.0]十一-7-烯(2.05 kg、13.45 mol),於20~30℃進行1.5小時攪拌。反應後,將反應液分液並獲得甲苯層。於獲得的甲苯層中添加20%檸檬酸水溶液(3.59 kg、2.0v/w)並進行分液,獲得甲苯層。於獲得的甲苯層中添加8%碳酸氫鈉水(1.71 kg,1.0v/w)並進行分液,獲得化合物10及11之甲苯溶液。
(實施例1-7)5-去氧-4-(羥基甲基)-1,2-O-(1-甲基亞乙基)-6-O-(三苯基甲基)-β-L-來蘇-己呋喃糖(化合物12)之甲苯溶液之製造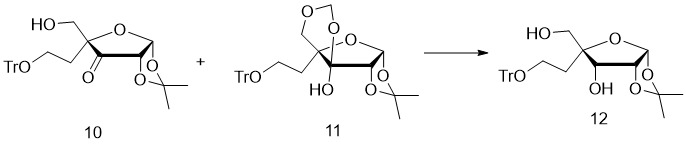
實施例1-6所獲得的化合物10及11之甲苯溶液中,於20~30℃,添加水(4.98 kg、3.0v/w)及氫化硼鈉(0.34 kg、8.96 mol),進行攪拌2小時。反應後,將反應液冷卻至0~10℃,於相同溫度下添加20%檸檬酸水溶液(5.37 kg、3.0v/w)並進行分液,獲得甲苯層。於獲得的甲苯層中添加8%碳酸氫鈉水(3.42 kg、2.0v/w)而進行分液,獲得化合物12之甲苯溶液。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。。1
H NMR (CDCl3
):δ = 1.33 (3H, s), 1.55 (3H, s), 1.99 (1H, ddd, J = 6.5, 8.0, 14.0 Hz), 2.20 (1H, dt, J = 6.0, 14.5 Hz), 2.85 (1H, br), 3.20-3.25 (1H, m), 3.35-3.42 (2H, m), 3.51 (1H, d, J = 12.0 Hz), 4.21 (1H, d, J = 6.0 Hz), 4.63 (1H, dd, J = 4.0, 6.0 Hz), 5.73 (1H, d, J = 4.0), 7.21-7.25 (3H, m), 7.27-7.30 (6H, m), 7.41-7.44 (6H, m).
(實施例1-8)3-O-苄基-4-[(苄氧基)甲基]-5-去氧-1,2-O-(1-甲基亞乙基)-6-O-(三苯基甲基)-β-L-來蘇-己呋喃糖(化合物13)之製造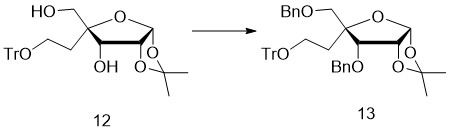
於實施例1-7所獲得的甲苯溶液中,添加48%氫氧化鉀水溶液(7.27 kg、3.0v/w)、四丁基碘化銨(0.20 kg、0.54 mol)及溴甲苯(1.53 kg、8.96 mol),於65~75℃進行攪拌23小時。反應後,於45~55℃,於反應液中添加水(6.64 kg、4.0v/w)及N-乙醯基-L-半胱胺酸(0.50 kg、0.3w/w)並攪拌2小時。將反應液冷卻至20~30℃後,添加水(4.98 kg、3.0v/w)並進行分液而獲得甲苯層。於獲得的甲苯層中添加8%碳酸氫鈉水(3.42 kg、2.0v/w)而進行分液,獲得甲苯層。於獲得的甲苯溶液中添加10%食鹽水(3.55 kg、2.0v/w)並分液,獲得甲苯層。將獲得的甲苯層濃縮直到成為3.3L,添加1-丙醇(5.34 kg、4.0v/w)並過濾不溶物,進一步減壓濃縮至成為3.0L,再添加1-丙醇(0.60 kg)後,於45~55℃攪拌0.5小時,藉此確認結晶的析出。接著於20~30℃攪拌12.5小時及於0~10℃攪拌2小時後,濾取析出的結晶。獲得的結晶使用預先冷卻至0℃的1-丙醇(4.00 kg、3.0v/w)而進行洗淨,藉由進行減壓乾燥(40℃),獲得化合物13(1.25 kg、1.90 mol、產率42.5%)。1
H NMR (CDCl3
):δ = 1.29 (3H, s), 1.51 (3H, s), 1.90-1.96 (1H, m), 2.43-2.49 (1H, m), 3.21-3.32 (3H, m), 3.47 (1H, d, J = 10.5 Hz), 4.10 (1H, d, J = 5.5 Hz), 4.30 (1H, d, J = 12.0 Hz), 4.42 (1H, d, J = 11.5 Hz), 4.47 (1H, d, J = 12.0 Hz), 4.57 (1H, dd, J = 5.5, 4.0 Hz), 4.69 (1H, d, J = 12.0 Hz), 5.69 (1H, d, J = 4.0 Hz), 7.15-7.34 (19H, m), 7.39-7.41 (6H, m).
(實施例1-9)甲基=3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖苷(化合物14)之甲苯溶液之製造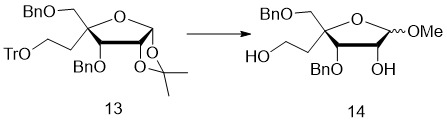
於實施例1-8所獲得的化合物13(250.0 g、380.6 mmol)之甲醇(1250 mL)溶液中添加濃硫酸(3.73 g、0.0380 mmol),將反應液於40~50℃攪拌20小時。將反應液冷卻至20~30℃,添加甲氧基三苯基甲烷(25 mg)及三乙基胺(13.56 g、134.0 mmol)而攪拌2小時後,歷經1.5小時滴加水(250 mL)。將反應液攪拌50分鐘後,濾除生成的白色固體,以甲醇/水(5/1、500 mL)進行洗淨。於獲得的溶液中添加正庚烷(1250 mL)而進行分液後,將正庚烷(500 mL)加至下層(水層)而進行分液。對下層添加20%食鹽水(1250 mL)及甲苯(1250 mL)而分液,獲得甲苯層及水層後,於水層中添加甲苯(1250 mL)而進行分液,合併獲得的甲苯層,減壓濃縮而獲得化合物14之甲苯溶液(625 mL)。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。1
H NMR (β體, CDCl3
):δ = 2.00-2.05 (1H, m), 2.10-2.16 (1H, m), 3.29 (3H, s), 3.35 (1H, d, J = 9.0 Hz), 3.55 (1H, d, J = 9.0 Hz), 3.76-381 (1H, m), 3.85-3.90 (1H, m), 4.08 (2H, dd, J = 5.0, 15.0 Hz), 4.54 (2H, s), 4.63 (2H, s), 4.87 (1H, s), 7.26-7.36 (10H, m).
(實施例1-10)甲基=2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖苷(化合物15)之甲苯溶液之製造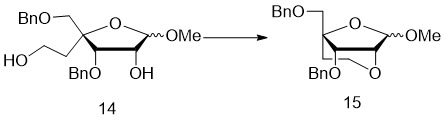
於實施例1-9所獲得的化合物14之甲苯溶液中添加三苯基膦(89.85 g、342.6 mmol),冷卻至0~5℃後,歷經20分鐘滴加偶氮二甲酸二異丙酯(69.27 g、342.6 mmol)之甲苯(180.3 mL)溶液,於20~30℃攪拌反應液3小時。於反應液中添加氯化鎂(90.60 g、951.6 mmol)而攪拌6小時後,添加正庚烷(1500 mL)而攪拌23小時,冷卻至0~5℃而攪拌2小時。濾除生成的白色固體,以甲苯/正庚烷(1/2、750 mL)洗淨後,於獲得的溶液中添加甲醇/水(3/2、750 mL)而進行分液。於獲得的有機層中添加甲醇/水(3/2、750 mL)而進行分液後,將有機層減壓濃縮而調製化合物15之甲苯溶液(625 mL)。
結構的確認係實施部分矽膠管柱純化(己烷/乙酸乙酯),並以NMR確認。1
H NMR (β體, CDCl3
):δ = 1.51 (1H, dd, J = 3.5, 13.0 Hz), 2.19 (1H, ddd, J = 7.5, 11.0, 13.5 Hz), 3.41 (3H, s), 3.53 (2H, dd, J = 10.5, 30.5 Hz), 3.86-3.95 (3H, m), 4.08 (1H, d, J = 3.0 Hz), 4.53 (1H, d, J = 11.5 Hz), 4.61 (2H, dd, J = 12.0, 27.5 Hz), 4.71 (1H, d, J = 6.5 Hz), 5.10 (1H, s), 7.26-7.35 (10H, m).
(實施例1-11)化合物2之製造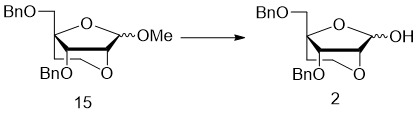
於實施例1-10所獲得的化合物15之甲苯溶液(625 mL)中添加水(625 mL)後,進行減壓濃縮而獲得化合物15之水混合物(625 mL)。於此混合物中依序添加乙酸(750 mL)及濃鹽酸(125 mL),於20~30℃攪拌3小時後,添加化合物2(25 mg),冷卻至0~5℃而攪拌17小時。於此混合物中添加水(750 mL),於0~5℃攪拌17小時後,濾取析出的結晶,依序以預先冷卻至0~5℃的乙酸/水(1/2、500 mL)及水(500 mL)洗淨,獲得粗化合物2之濕產物。使獲得的粗化合物2於35~45℃溶解於甲苯(1250 mL),將生成的水層進行分液而去除。將獲得的有機層減壓濃縮而作成化合物2之甲苯溶液(750 mL)後,添加正庚烷(375 mL)而冷卻至20~30℃。於此溶液中,添加化合物2之晶種(25 mg),於20~30℃攪拌2小時後,添加正庚烷(1125 mL),於0~5℃攪拌19小時。濾取析出的結晶,以甲苯/正庚烷(1/2、750 mL)洗淨後,以正庚烷(750 mL)進行洗淨。將獲得的結晶減壓乾燥(40℃)而獲得化合物2(76.84 g、215.6 mmol、產率56.6%)。
實施例1-11所使用的化合物2之晶種係由以下方法獲得。藉由與實施例1-11之前半步驟同樣的方法而獲得呈結晶之粗化合物2。使用獲得的粗化合物2,藉由與實施例1-11之後半步驟同樣的方法而獲得呈結晶之化合物2。於此,使用先前獲得的粗化合物2之結晶作為化合物2之晶種。將上述之方法所獲得的化合物2之結晶作為實施例1-11中的化合物2之晶種而使用。1
H NMR (CDCl3
):δ = 1.30 (0.39H, d, J = 10.8 Hz), 1.45 (0.59H, dd, J = 10.6, 3.2 Hz), 1.97-2.07 (0.39H, m), 2.13-2.22 (0.64H, m), 2.60 (0.34H, d, J = 1.2 Hz), 2.80 (0.49H, brs), 3.22 (0.36H, d, J = 7.2 Hz), 3.45-3.59 (1.41H, m), 3.71 (0.37H, d, J = 7.2 Hz), 3.78 (0.37H, brs), 3.82 (0.37H, ddd, J = 9.2, 4.0, 1.6 Hz), 3.85-3.95 (1.23H, m), 4.01-4.13 (1.30H, m), 4.16-4.27 (0.83H, m), 4.44-4.75 (4.24H, m), 5.36 (0.07H, dd, J = 7.0, 1.6 Hz), 5.57 (0.57H, s), 7.24-7.39 (10H, m), 9.89 (0.36H, s).
(實施例1-12)化合物2之純化
使實施例1-11所獲得的粗化合物2(50 g)溶解於甲苯(300 mL),添加正庚烷(300 mL)而於20~30℃攪拌3小時後,再添加正庚烷(300 mL)而攪拌16小時。濾取析出的結晶,以甲苯/正庚烷(1/2、250 mL)洗淨後,以正庚烷(250 mL)進行洗淨。將獲得的結晶減壓乾燥(40℃)而獲得化合物2(40.06 g、產率80.1%)。
(實施例2)1-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-5-甲基嘧啶-2,4(1H,3H)-二酮(化合物1t)之製造
(實施例2-1)1-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-5-甲基嘧啶-2,4(1H,3H)-二酮(化合物3t)之甲醇溶液之製造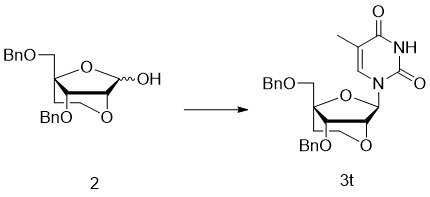
於乙腈(50 mL)中添加胸腺嘧碇(8.85 g、70.18 mmol)及N,O-雙三甲基矽基乙醯胺(28.6 g、140.6 mmol),於20~30℃攪拌1小時,作為溶液A。於另外的反應容器於乙腈(100 mL)中添加實施例1所獲得的化合物2(20.00 g、56.12 mmol)及三氯乙腈(15.20 g、105.27 mmol)後,添加1,8-二氮雜雙環[5.4.0]十一-7-烯(1.07 g、7.02 mmol),於20~30℃攪拌1小時,作為溶液B。於溶液B中添加溶液A後,添加碘三甲基矽烷(17.55 g、87.73 mmol)而於20~30℃攪拌2小時。於反應液中滴加於5%碳酸氫鈉水溶液(160 mL)溶解亞硫酸鈉(17.68 g、175.45 mmol)而成的水溶液後,添加甲苯(200 mL)及甲醇(80 mL),於35~45℃攪拌後,並進行分液。於獲得的有機層中添加40%甲醇水(200 mL),於35~45℃攪拌後,進行分液。再次以40%甲醇水(200 mL)之分液操作後,將獲得的有機層減壓濃縮而作成甲醇溶液(50 mL)後,添加甲醇(200 mL),再次減壓濃縮而作為化合物3t之甲醇溶液(50 mL)。
獲得的化合物之NMR譜與WO00/47599之實施例6記載之化合物的NMR譜一致。
(實施例2-2)1-[2,6-脫水-5-去氧-4-(羥基甲基)-α-L-來蘇-己呋喃糖基]-5-甲基嘧啶-2,4(1H,3H)-二酮(化合物16)之製造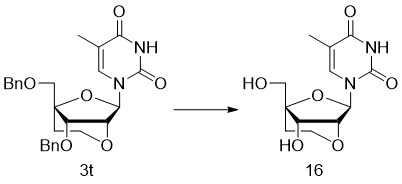
於實施例2-1所獲得的化合物3t之甲醇溶液(50 mL)中,進一步添加甲醇(50 mL)及氫氧化鈀(1.0 g),氫氣環境下於55~65℃攪拌2小時後,於相同溫度下過濾觸媒,將獲得的濾液減壓濃縮而作成甲醇溶液(50 mL)後,添加丙酮(200 mL)而減壓濃縮成50 mL。進一步添加丙酮(100 mL),冷卻至0~5℃而攪拌2小時。濾取生成的結晶,將獲得的結晶以丙酮(50 mL)進行洗淨。將獲得的結晶減壓乾燥(40℃)而獲得化合物16(9.95 g、產率62.4%(來自化合物2))。
獲得的化合物之NMR譜與WO00/47599之實施例7記載的化合物的NMR譜一致。
(實施例2-3)1-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-5-去氧-α-L-來蘇-己呋喃糖基)-5-甲基嘧啶-2,4(1H,3H)-二酮(化合物17)之製造
於四氫呋喃(47.5 mL)中添加實施例2-2所獲得的化合物16(9.50 g、33.42 mmol)及吡啶(10.57 g、133.68 mmol),接著添加4,4’-二甲氧基三苯氯甲烷(13.59 g、40.10 mmol),將反應液於20~30℃攪拌2小時。於反應液中添加甲醇(1.62 mL、40.10 mmol),並依序添加15%碳酸鈉水溶液(28.5 mL)、乙酸乙酯(133 mL)及水(67 mL),進行分液洗淨後,將獲得的有機層以20%檸檬酸水溶液(47.5 mL)進行二次洗淨,進一步依序以15%碳酸鈉水溶液(47.5 mL)及水(47.5 mL)進行洗淨後,進行減壓濃縮至47.5 mL。於獲得的濃縮物中添加乙酸乙酯(100 mL),進行減壓濃縮至100 mL。將獲得的濃縮物於55~65℃攪拌16小時,進一步於20~30℃攪拌5小時,添加正庚烷(50 mL),再攪拌16小時後,濾取析出的固體。將獲得的固體,以乙酸乙酯/正庚烷(2/1、47.5 mL)洗淨後,進行減壓乾燥(40℃),藉此獲得化合物17(17.35 g、88.5%)。
獲得的化合物之NMR譜與WO00/47599之實施例8記載的化合物的NMR譜一致。
(實施例2-4)化合物1t之製造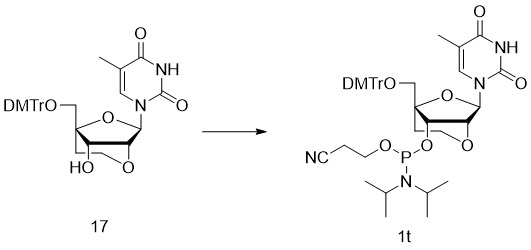
於實施例2-3所獲得的化合物17(5.00 g、8.17 mmol)之乙酸乙酯(40 mL)溶液中,添加2-氰基乙基 N,N,N’,N’-四異丙基亞磷醯二胺(2.96 g、9.82 mmol)及三氟乙酸・吡啶鹽(1.74 g、9.01 mmol),於20~30℃攪拌24小時。於獲得的溶液中,添加20%食鹽水(17.5 mL)而進行分液後,於獲得的有機層中添加5%碳酸氫鈉水溶液(12.5 mL)及20%食鹽水(10 mL)而進行分液。將獲得的有機層冷卻至0~5℃,添加10%磷酸二氫鉀水溶液(20 mL)而進行分液。於獲得的有機層中添加5%碳酸氫鈉水溶液(12.5 mL)及20%食鹽水(10 mL)而進行分液,於獲得的有機層中添加20%食鹽水(17.5 mL)而進行分液。將獲得的有機層減壓濃縮,溶解於乙酸乙酯(15 mL),於此溶液中添加乙酸乙酯(25 mL)及鹼性氧化鋁(10.00 g),於20~30℃攪拌2小時。自反應混合物濾除氧化鋁,氧化鋁以乙酸乙酯(25 mL)進行洗淨後,將濾液與洗淨液合併而獲得的溶液進行減壓濃縮,將濃縮物溶解於乙酸乙酯(15 mL),將獲得的乙酸乙酯溶液於20~30℃,歷經35分鐘滴加至正庚烷(45 mL)及二異丙基醚(80 mL)之混合溶液後,添加正庚烷(90 mL),於相同溫度下攪拌30分鐘。濾取析出的固體,將獲得的固體以正庚烷(25 mL)洗淨,進行減壓乾燥(40℃、12小時之後,50℃、8小時),藉此獲得標題化合物1t(5.18 g、6.58 mmol、80.6%)。
獲得的化合物之NMR譜與WO00/47599之實施例9記載之化合物的NMR譜一致。
(實施例3)1-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-4-苯甲醯胺-5-甲基嘧啶-2(1H)-酮(化合物1c)之製造
(實施例3-1)1-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-4-苯甲醯胺-5-甲基嘧啶-2(1H)-酮(化合物3c)之製造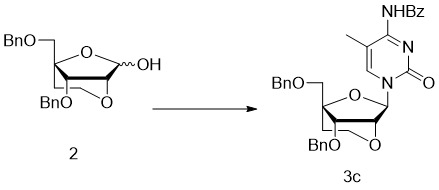
於乙腈(25 mL)中添加N4
-苄醯基-5-甲基胞嘧啶(8.04 g、35.08 mmol)、N,O-雙三甲基矽基乙醯胺(14.27 g、70.16 mmol),於20~30℃攪拌1小時,作為溶液A。於另外的反應容器,於乙腈(50 mL)中添加實施例1所獲得的化合物2(10.00 g、28.06 mmol)、三氯乙腈(6.08 g、42.09 mmol)後,添加1,8-二氮雜雙環[5.4.0]十一-7-烯(0.41 g、2.81 mmol),並於20~30℃攪拌1小時,作為溶液B。於溶液B中添加溶液A後,添加碘三甲基矽烷(7.02 g、35.08 mmol)而於5~15℃攪拌16小時。於反應液中滴加將亞硫酸鈉(8.84 g、87.73 mmol)溶解於5%碳酸氫鈉水溶液(80 mL)而成的水溶液後,添加甲苯(100 mL),分液後,濾除少量的固體。對於獲得的有機層,以50%甲醇水(50 mL)進行二次洗淨。將獲得的有機層減壓濃縮而作成甲醇溶液(50 mL)後,於20~30℃攪拌16小時後,將析出的固體過濾,以甲醇(50 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物3c(9.35 g、58.7%)。
獲得的化合物之NMR譜與WO00/47599之實施例19記載之化合物的NMR譜一致。
(實施例3-2)1-[2,6-脫水-5-去氧-4-(羥基甲基)-α-L-來蘇-己呋喃糖基]-4-苯甲醯胺-5-甲基嘧啶-2(1H)-酮(化合物18)之製造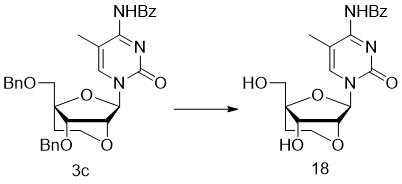
於二氯甲烷(20 mL)中添加實施例3-1所獲得的化合物3c(5.00 g、8.81 mmol),於-20~-10℃滴加1M-三氯化硼/二氯甲烷溶液(40.0 mL、39.65 mmol),並攪拌16小時。於反應液中滴加50%羅謝爾鹽(Rochelle salt)水溶液(20.0 mL),並添加乙酸乙酯(20 mL)、水(20 mL),於20~30℃攪拌16小時後,將析出的固體過濾,以水(20 mL)、乙酸乙酯(20 mL)洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物18(1.59 g、46.6%)。
獲得的化合物之NMR譜與WO00/47599之實施例11記載之化合物的NMR譜一致。
(實施例3-3)1-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-5-去氧-α-L-來蘇-己呋喃糖基)-4-苯甲醯胺-5-甲基嘧啶-2(1H)-酮(化合物19)之製造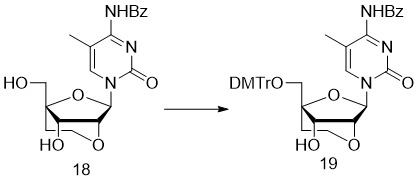
於乙酸乙酯(40 mL)中添加實施例3-2所獲得的化合物18(1.00 g、2.581 mmol),減壓濃縮至10 mL後,添加吡啶(0.815 g、10.324 mmol),添加4,4’-二甲氧基三苯氯甲烷(1.05 g、3.098 mmol),於20~30℃攪拌16小時。於反應液中添加甲醇(1.0 mL)後,添加水(10 mL)、甲醇(10 mL),並於20~30℃攪拌16小時後,將析出的固體過濾,以水(5 mL)、乙酸乙酯(5 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物19之水合物結晶(1.53 g、86.0%)。
獲得的化合物之NMR譜與WO00/47599之實施例21記載之化合物的NMR譜一致。
(實施例3-4)化合物1c之製造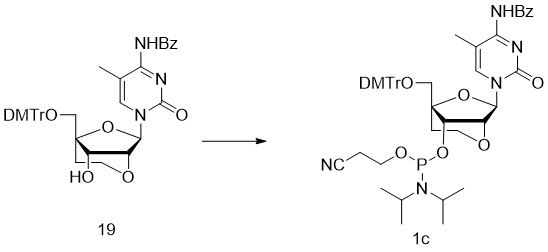
於實施例3-3所獲得的化合物19之水合物(3.00 g、4.24 mmol)中添加二氯甲烷(30 mL),減壓濃縮而作成二氯甲烷溶液(9 mL)。重複二次於獲得的溶液中添加二氯甲烷(30 mL)並進行減壓濃縮而作成二氯甲烷溶液(9 mL)的操作。於獲得的溶液中添加二氯甲烷(15 mL)、N,N,N’,N’-四異丙基亞磷醯二胺(1.54 g、5.11 mmol)、三氟乙酸・吡啶鹽(0.902 g、4.67 mmol),於20~30℃攪拌7小時。將獲得的溶液以5%碳酸氫鈉水溶液(10.5 mL)洗淨二次後,以5%食鹽水(12 mL)洗淨二次,進行減壓濃縮而獲得二氯甲烷溶液(9 mL)。於獲得的溶液中添加二氯甲烷(15 mL)、鹼性氧化鋁(6.00 g),並於20~30℃攪拌30分鐘。濾除氧化鋁,以二氯甲烷(15 mL)進行洗淨後,將獲得的溶液進行減壓濃縮而獲得化合物1c之二氯甲烷溶液(9 mL)。將獲得的溶液於20~30℃歷經35分鐘滴加至正庚烷(66 mL)與二異丙基醚(15.6 mL)之混合溶液中。直接於此溫度下攪拌2小時後,將析出的固體過濾,以正庚烷(15 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物1c(3.33 g、3.74 mmol、88.3%)。
獲得的化合物之NMR譜與WO00/47599之實施例6記載之化合物的NMR譜一致。
(實施例4)9-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-N-苄醯基-9H-嘌呤-6-胺(化合物1a)之製造
(實施例4-1-1)9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-N-苄醯基-9H-嘌呤-6-胺(化合物3a)之製造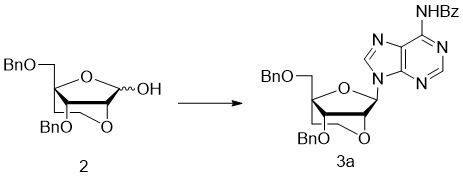
於三氟甲磺酸(7.44 mL、84.18 mmol)中添加二氯二甲基矽烷(5.44 g、42.09 mmol),並於20~30℃攪拌0.5小時後,添加乙腈(80 mL)、N-苄醯基腺嘌呤(10.06 g、42.09 mmol),於20~30℃攪拌3小時,作為溶液A。於另外的反應容器,於乙腈(40 mL)中添加實施例1所獲得的化合物2(10.00 g、28.06 mmol)、三氯乙腈(4.2 mL、42.09 mmol)後,添加1,8-二氮雜雙環[5.4.0]十一-7-烯(0.22 g、1.40 mmol),於20~30℃攪拌1小時,作為溶液B。於溶液A中添加溶液B後,於45~55℃攪拌2小時。將溫度調整至0-10℃後,於反應液中添加甲苯(100 mL)、20%碳酸氫鉀水溶液(70 mL)並進行分液,獲得甲苯層。於獲得的甲苯層中添加20%檸檬酸水溶液(50 mL)並進行分液,獲得甲苯層。藉由重複進行2次相同的分液操作,於獲得的甲苯層中添加水(30 mL)、20%碳酸氫鉀水溶液(25 mL),分液並獲得甲苯層。於獲得的甲苯層中添加50%甲醇水溶液(100 mL),進行分液並獲得甲苯層。將獲得的甲苯層進行減壓濃縮,作成甲苯溶液(80 mL)。於獲得的溶液中添加異丁基醇(100 mL),進行減壓濃縮,藉此獲得甲苯-異丁基醇溶液(100 mL)。於獲得的溶液中添加異丁基醇(50 mL),進行減壓濃縮,藉此獲得甲苯-異丁基醇溶液(50 mL)。於獲得的溶液中,添加異丁基醇(50 mL)後,於20~30℃攪拌2小時,藉此確認結晶之析出。歷經0.5小時將溫度調整至0-10℃後,攪拌1小時,過濾析出的結晶,使用預先冷卻至0℃的異丁基醇(50 mL)而進行洗淨,並進行減壓乾燥(40℃),藉此獲得化合物3a(7.82 g、13.54 mmol、產率48.3%)。
獲得的化合物之NMR譜與WO00/47599之實施例6記載之化合物的NMR譜一致。
(實施例4-1-2)1-O-乙醯基-2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-甘油-己呋喃糖(化合物22)之製造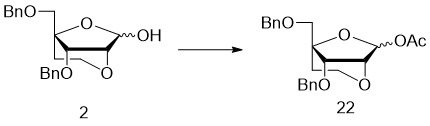
添加甲苯(1497 mL)、實施例1所獲得的化合物2(149.73 g、375.78 mmol)、乙酸酐(64.33 g、563.67 mmol)、吡啶(49.85 g、563.67 mmol)、4-二甲基胺基吡啶(2.57 g、18.79 mmol),於20~30°C攪拌2小時後,於反應液中添加水(749 mL),進行分液並獲得甲苯層。於獲得的甲苯層中添加20%檸檬酸水溶液(749 mL)並進行分液,獲得甲苯層。藉由再次重複同樣的分液操作,於獲得的甲苯層中添加8%碳酸氫鉀水溶液(449 mL),進行分液並獲得甲苯層。於獲得的甲苯層中添加5%食鹽水(449 mL),進行分液並獲得甲苯層。將獲得的甲苯層減壓濃縮,作成甲苯溶液(449 mL)。於獲得的溶液中加入乙腈(1497 mL),並進行減壓濃縮,藉此獲得甲苯-乙腈溶液(449 mL)。於獲得的溶液中添加乙腈(1497 mL),並進行減壓濃縮,藉此獲得化合物22之甲苯-乙腈溶液(449 mL)。
(實施例4-1-3a)9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-N-苄醯基-9H-嘌呤-6-胺(化合物3a)之製造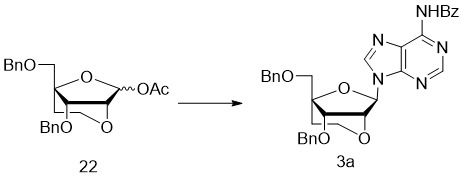
添加乙腈(51 mL)、N-苄醯基腺嘌呤(5.09 g、21.30 mmol)、三氟甲磺酸三甲基矽酯(9.78 g、44.01 mmol),於20~30°C攪拌40分鐘。添加包含實施例4-1-2所獲得的化合物22的甲苯-乙腈溶液(11.65 g、14.20 mmol)後,於20~30°C攪拌16小時。接著,添加三氟乙酸(2.43 g、21.30 mmol),於20~30°C攪拌1小時後,使升溫至45~55°C並攪拌3小時。將溫度調整至20-30℃後,於反應液中添加甲苯(51 mL)、25%氫氧化鈉(10 mL)、8%碳酸氫鈉水溶液(10 mL),並於20~30°C攪拌1小時。接著,添加10%三氟乙酸水溶液(25 mL),進行分液並獲得甲苯層。於藉由重複3次同樣的分液操作所獲得的甲苯層中,添加8%碳酸氫鈉水溶液(51 mL),進行分液並獲得甲苯層。於獲得的甲苯層中添加50%甲醇水溶液(20 mL)、20%食鹽水(5 mL),進行分液並獲得甲苯層。將藉由重複1次同樣的分液操作所獲得的甲苯層進行減壓濃縮,作成甲苯溶液(15 mL)。於獲得的溶液中添加異丁基醇(40 mL),進行減壓濃縮,藉此獲得甲苯-異丁基醇溶液(40 mL)。於獲得的溶液中添加異丁基醇(15 mL),接種化合物3a之結晶(0.1 wt%),於35~45℃攪拌30分鐘,藉此確認結晶的析出。接著,進行減壓濃縮並作成甲苯-異丁基醇溶液(40 mL)後,添加異丁基醇(5 mL),歷經2小時將溫度調整至0-10℃,並攪拌17.5小時。過濾析出的結晶,使用預先冷卻至0℃的異丁基醇(25 mL)加以洗淨,進行減壓乾燥(40℃),藉此獲得化合物3a(6.37 g、11.03 mmol、產率77.7 %)。
(實施例4-1-3b) 9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-N-苄醯基-9H-嘌呤-6-胺(化合物3a)之製造
於乙腈(30 mL)中添加N-苄醯基腺嘌呤(3.02 g、12.63 mmol)、三氟甲磺酸三甲基矽酯(5.61 g、25.25 mmol),將反應混合物於20~30°C攪拌30分鐘。於獲得的反應混合物中,添加含有實施例4-1-2所獲得的化合物22的乙腈溶液(6 mL)及乙腈(3 mL),將反應混合物於20~30℃攪拌2小時。接著,添加三氟乙酸(1.44 g、12.63 mmol),於35~45℃將反應混合物攪拌6小時。將獲得的反應混合物冷卻至0℃後,將反應液使用25%氫氧化鈉(3 mL)調整至pH6.5~7.5。之後,於反應混合物中,於20~30℃,添加水(9 mL),接種化合物3a之結晶(0.1 wt%),於20~30℃將反應混合物攪拌19小時後,進一步添加水(27 mL),並於20~30℃攪拌4小時。過濾析出的結晶,使用50%乙腈水(15 mL)進行洗淨,並進行減壓乾燥(40℃),藉此獲得化合物3a(3.59 g、6.21 mmol、產率73.8 %)。
獲得的化合物之NMR譜與WO00/47599之實施例6記載之化合物的NMR譜一致。
(實施例4-2)9-[2,6-脫水-5-去氧-4-(羥基甲基)-α-L-來蘇-己呋喃糖基]-N-苄醯基-9H-嘌呤-6-胺(化合物20)之製造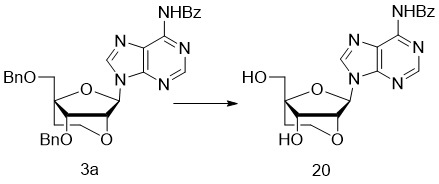
於-20~-10℃,於1M-三氯化硼/二氯甲烷溶液(50.0 mL、51.94 mmol)中添加實施例4-1-3所獲得的化合物3a(2.00 g、3.46 mmol),並攪拌0.5小時。確認反應結束後,藉由減壓濃縮,獲得二氯甲烷溶液(20 mL)。
於獲得的溶液中滴加50%氫氧化鉀水溶液(6.0 mL),添加正庚烷(6 mL),於20~30℃攪拌15分鐘後,將析出的固體過濾,以二氯甲烷/正庚烷(5/1、4 mL)進行洗淨。於獲得的固體中添加乙腈(30 mL)、水(4 mL)。攪拌0.5小時後,將固體過濾,以乙腈(4 mL)進行洗淨。於獲得的固體中添加甲醇(30 mL)後,濾除固體,使用甲醇(10 mL)而進行洗淨,獲得甲醇溶液。將獲得的溶液進行減壓濃縮,調整於甲醇溶液(10 mL)。對於獲得的溶液中,添加乙腈(20 mL),進行減壓濃縮,藉此獲得乙腈-甲醇混合溶液(10 mL)。進行2次同樣之減壓濃縮操作,對獲得的乙腈-甲醇混合溶液(10 mL)添加乙腈(10 mL),攪拌16小時後,過濾固體,並以乙腈(6 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物20(0.61 g、44.3%)。
獲得的化合物之NMR譜與WO00/47599之實施例11記載之化合物的NMR譜一致。
(實施例4-3)9-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-5-去氧-α-L-來蘇-己呋喃糖基)-N-苄醯基-9H-嘌呤-6-胺(化合物21)之製造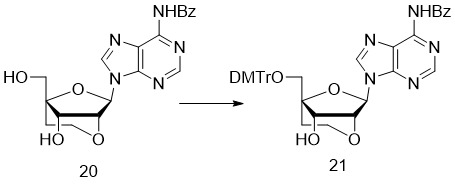
於四氫呋喃(7.5 mL)中添加實施例4-2所獲得的化合物20(0.50 g、1.26 mmol)、4,4’-二甲氧基三苯氯甲烷(0.64 g、1.89 mmol),添加吡啶(0.60 g、7.55 mmol),並於50℃攪拌2小時。於反應液中添加甲苯(5.0 mL)、水(2.5 mL),進行分液並獲得甲苯層。將獲得的甲苯層以20%檸檬酸水溶液(2.5 mL)進行三次分液,添加20%碳酸鉀水溶液(1.0 mL)而進行分液並獲得甲苯層。於獲得的甲苯層中添加水(2.5 mL)而進行分液洗淨後,進行減壓濃縮,藉此獲得甲苯溶液(2.5 mL)。於獲得的溶液中添加甲苯(5.0 mL),並進行減壓濃縮,藉此獲得甲苯溶液(2.5 mL)。藉由重複二次同樣的操作,獲得甲苯溶液(2.5 mL)。於獲得的溶液中添加甲苯(2.5 mL)、水(50μL),並於20~30℃攪拌3小時後,將析出的固體過濾,以甲苯(2.5 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得粗化合物21(0.51 g、58.0%)。
於2-丙醇(1.8 mL)中添加粗化合物21(0.36 g、0.51 mmol)、水(1.8 mL)並於20~30℃攪拌0.5小時。再添加水(1.8 mL),攪拌15分鐘後,過濾固體,以2-丙醇-水(1/3、1.8 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物21之水合物(0.31 g、86.1%)。
獲得的化合物之NMR譜與WO00/47599之實施例13記載之化合物的NMR譜一致。
(實施例4-4)化合物1a之製造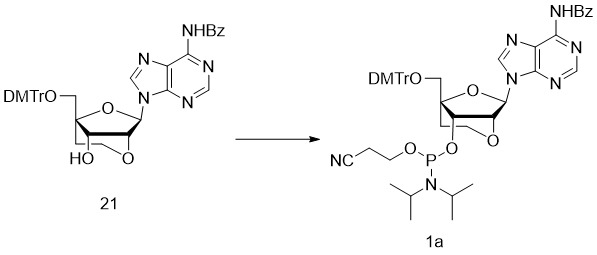
於實施例4-3所獲得的化合物21之水合物(150 mg、0.209 mmol)中添加二氯甲烷(1.5 mL),進行減壓濃縮而乾固。於獲得的泡沫中添加二氯甲烷(1.5 mL),進行減壓濃縮而乾固後,添加二氯甲烷(1.2 mL)、N,N,N’,N’-四異丙基亞磷醯二胺(77.9 mg、0.258 mmol)、三氟乙酸・吡啶鹽(44.4 mg、0.230 mmol),於20~30℃攪拌4小時。獲得的溶液以5%碳酸氫鈉水溶液(0.525 mL)洗淨二次後,以5%食鹽水(0.600 mL)洗淨二次,進行減壓濃縮而乾固。於獲得的泡沫中添加乙酸乙酯(1.2 mL)、中性矽膠(300 mg),於20~30℃攪拌1小時。濾除矽膠,以乙酸乙酯(6 mL)進行洗淨後,將獲得的溶液進行減壓濃縮而乾固。將獲得的泡沫之乙酸乙酯(0.600 mL)溶液於20~30℃歷經20分鐘滴加至正庚烷(1.35 mL)與二異丙基醚(2.4 mL)之混合溶液中。添加正庚烷(3.15 mL)而直接於此溫度下攪拌1小時後,將析出的固體過濾,以正庚烷(6 mL)進行洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物1a(132.5 mg、0.147 mmol、70.4%)。
獲得的化合物之NMR譜與WO00/47599之實施例14記載之化合物的NMR譜一致。
(實施例5)化合物20之製造
(實施例5-1)化合物22之純化
將實施例4-1-2所獲得的溶液濃縮乾固而獲得的化合物22之粗體混合物(6.57 g),以矽膠管柱層析純化(己烷:乙酸乙酯=94:6~50:50),獲得化合物22之β體(4.68 g)。1
H NMR (β體, CDCl3
):δ = 1.43 (1H, dd, J = 3.5, 13.5 Hz), 2.01 (3H, s), 2.17-2.24 (1H, m), 3.57 (2H, dd, J = 11.5, 22.0 Hz), 3.94-3.97 (2H, m), 4.07 (1H, d, J = 3.0 Hz), 4.14 (1H, d, J = 3.0 Hz), 4.54 (1H, d, J = 12.5 Hz), 4.61 (2H, dd, J = 12.5, 18.0 Hz), 4.74 (1H, d, J = 11.5 Hz), 6.33 (1H, s), 7.26-7.37 (10H, m).
(實施例5-2)9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-2,6-二氯-9H-嘌呤(化合物23)之製造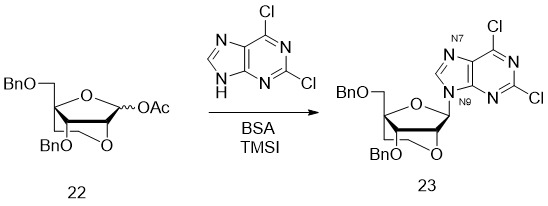
於實施例5-1-2所獲得的化合物22(60.0 mg, 150.58 ⌠mol)之甲苯溶液(0.6 mL)中添加N,O-雙三甲基矽基乙醯胺(40.5 ⌠L, 165.64 ⌠mol)、2,6-二氯嘌呤(31.3 mg, 165.64 ⌠mol),於75℃攪拌1小時後,添加碘三甲基矽烷(22.5 ⌠L, 165.64 ⌠mol)並攪拌3小時。確認反應結束後,冷卻至室溫,添加乙酸乙酯(18.0 mL)5%碳酸氫鈉水(3.0 mL)並進行分液。將獲得的有機層濃縮乾固後,將殘渣以矽膠層析(己烷: 乙酸乙酯= 2:1)純化,進行減壓濃縮,藉此取得呈無色油狀化合物之化合物23(52.0 mg, 65.4%)。1
H-NMR (CDCl3
):δ = 8.79 (1H, s), 7.23-7.39 (10H, m), 6.43 (1H, s), 4.57-4.65 (2H, m), 4.42-4.45 (1H, m), 4.15 (1H, d, J = 3.0 Hz), 4.06-4.09 (2H, m), 3.71 (1H, d, J = 10.5 Hz), 3.56 (1H, d, J = 10.5 Hz), 2.27-2.36 (2H, m), 1.41-1.44 (2H, m)
(實施例5-3) 9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-2-氯-9H-嘌呤-6-胺(化合物24)之製造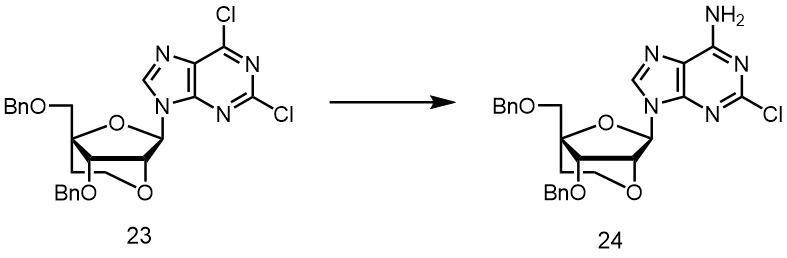
於實施例5-2所獲得的化合物23(52.0 mg, 98.60 ⌠mol)之四氫呋喃溶液(0.75 mL)中添加28%氨水溶液(0.5 mL),並於50℃攪拌7小時。確認反應結束後,冷卻至室溫,添加乙酸乙酯(2.0 mL)並進行分液。獲得的有機層以20%食鹽水(1.0 mL)進行洗淨後,進行濃縮乾固。將殘渣以矽膠層析(己烷: 乙酸乙酯= 4:6)純化,並進行減壓濃縮,藉此取得呈白色結晶之化合物24(44.7 mg, 89.2%)。1
H-NMR (CDCl3
):δ = 8.36 (1H, s), 7.27-7.38 (10H, m), 6.39 (1H, s), 5.69 (2H, brs), 4.56-4.68 (2H, m), 4.44-4.48 (1H, m), 4.20 (1H, d, J = 2.5 Hz), 4.05-4.14 (2H, m), 3.70 (1H, d, J = 10.8 Hz), 3.57 (1H, d, J = 10.8 Hz), 2.28-2.36 (1H, m), 1.53 (1H, m), 1.42-1.44 (1H, m), 1.25-1.27 (1H, m)
(實施例5-4) 9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-9H-嘌呤-6-胺(化合物25)之製造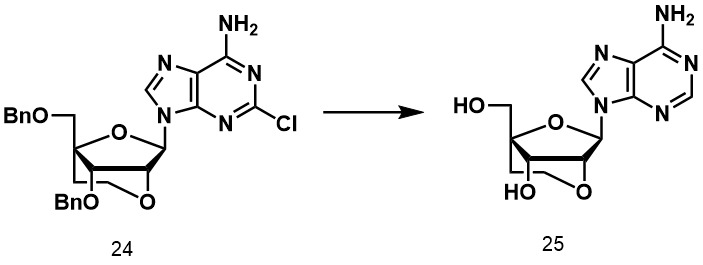
於實施例5-3所獲得的化合物24(26.2 mg, 51.58 ⌠mol)之乙醇溶液(2.0 mL)中添加20%氫氧化鈀碳(20.0 mg)、1N氫氧化鈉水溶液(103.2 ⌠L, 103.16 ⌠mol),3.5 bar之氫壓下,於50℃攪拌5小時。反應後,冷卻至室溫,添加1N鹽酸水溶液(51.6 ⌠L, 51.58 ⌠mol)並進行過濾。於濾液中添加乙醇(10 mL),並進行濃縮乾固,藉此取得呈白色結晶之化合物25(9.7 mg, 64.1%)。1
H-NMR (CD3
OD):δ = 8.72 (1H, s), 8.35 (1H, s), 6.48 (1H, s), 4.37 (1H, d, J = 3.5 Hz), 4.32 (1H, d, J = 3.0 Hz), 4.01-4.13 (2H, m), 3.78 (1H, d, J = 12.5 Hz), 3.72 (1H, d, J = 12.0 Hz), 2.17-2.24 (1H, m), 1.44 (1H, dd, J = 13.0 Hz, 4.0 Hz)
(實施例5-5) 化合物20之製造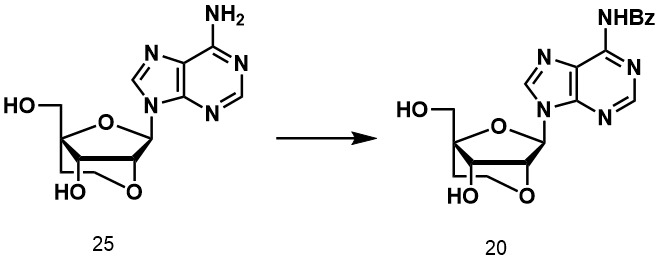
於實施例5-4所獲得的化合物25之吡啶溶液(30v/w)中添加氯三甲基矽烷(5當量),攪拌30分鐘,添加苯甲醯氯(5當量),於室溫使反應2小時。反應後,冷卻至0℃,添加氨水使成為2M-氨溶液。30分鐘後,進行減壓濃縮,添加乙腈(30v/w)並濃縮至(3v/w),並濾取析出的結晶,藉此獲得化合物20。
(實施例6)9-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-2-(2-甲基丙烷醯胺)
-1,9-二氫-6H-嘌呤-6-酮(化合物1 g)之製造
(實施例6-1)9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-6-(苄氧基)-2-氯-9H-嘌呤(化合物26)之製造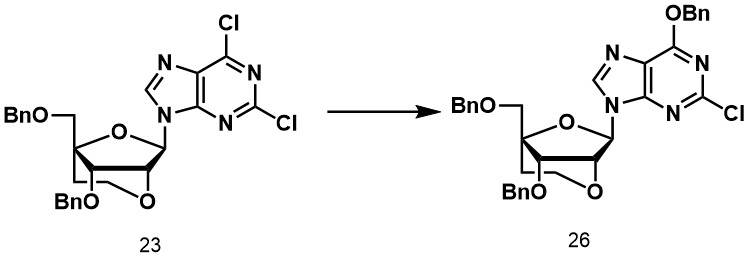
於四氫呋喃(0.5 mL)中添加芐醇(4.63 ⌠L, 66.84 ⌠mol)、氫化鈉(2.5 mg, 57.93 ⌠mol),並於0℃攪拌30分鐘後,添加實施例5-2所獲得的化合物23(23.5 mg, 44.56 ⌠mol)並攪拌3小時。確認反應結束後,添加乙酸(3.6 ⌠L, 62.38 ⌠mol)、20%食鹽水(0.5 mL)、乙酸乙酯(2.0 mL)並進行分液。將獲得的有機層濃縮乾固後,將殘渣以矽膠層析(己烷: 乙酸乙酯= 2:1)純化,並進行減壓濃縮,藉此取得呈無色油狀化合物之化合物26(20.9 mg, 78.3%)。1
H-NMR (CD3
Cl):δ = 8.50 (1H, s), 7.55-7.60 (2H, m), 7.24-7.40 (13H, m), 6.42 (1H, s), 5.68 (1H, d, J = 12.0 Hz), 5.63 (1H, d, J = 11.5 Hz), 4.55-4.64 (3H, m), 4.43-4.44 (2H, m), 4.17 (1H, d, J = 2.5 Hz), 4.06-4.08 (2H, m), 3.69 (1H, d, J = 10.0 Hz), 3.55 (1H, d, J = 10.5 Hz), 2.26-2.32 (1H, m), 1.41-1.44 (1H, m)
(實施例6-2) 9-{2,6-脫水-3-O-苄基-4-[(苄氧基)甲基]-5-去氧-α-L-來蘇-己呋喃糖基}-6-(苄氧基)-N-(2-甲基丙醯基)-9H-嘌呤-2-胺(化合物27)之製造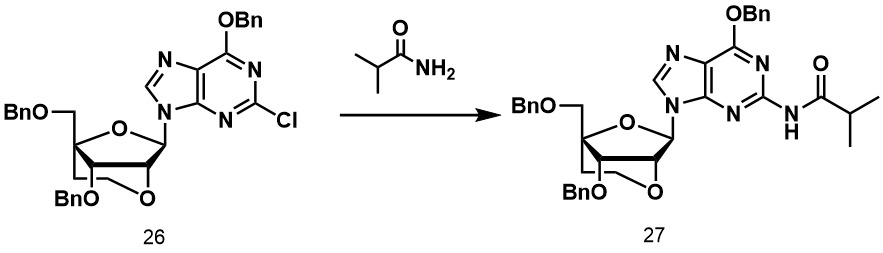
將實施例6-1所獲得的化合物26(34.9 mg, 58.26 ⌠mol)、異丁基醯胺(7.6 mg, 87.39 ⌠mol)、參(二亞苄基丙酮)(氯仿)二鈀(3.0 mg, 2.91 ⌠mol)、4,5’-雙(二苯基膦基)-9,9’-二甲基𠮿(3.4 mg, 5.83 ⌠mol)、碳酸銫(36.1 mg, 110.69 ⌠mol)添加至反應容器後,於氮氣環境下,添加脫氣的甲苯(0.7 mL)。將反應溶液升溫至110℃,攪拌12小時後,添加自來水(1.0 mL)、乙酸乙酯(4.0 mL)並進行分液。將獲得的有機層濃縮乾固後,將殘渣以矽膠層析(己烷: 乙酸乙酯= 1:1)純化,並進行減壓濃縮,藉此取得呈無色油狀化合物之化合物27(18.1 mg, 47.8%)。1
H-NMR (CD3
Cl):δ = 8.37 (1H, s), 7.83 (1H, brs), 7.53-7.55 (2H, m), 7.24-7.39 (13H, m), 6.35 (1H, s), 5.67 (1H, d, J = 12.0 Hz), 5.62 (1H, d, J = 12.0 Hz), 4.56-4.64 (3H, m), 4.43-4.45 (2H, m), 4.18 (1H, d, J = 2.5 Hz), 4.07-4.12 (2H, m), 3.70 (1H, d, J = 11.0 Hz), 3.56 (1H, d, J = 10.5 Hz), 3.22 (1H, brs), 2.27-2.23 (1H, m), 1.43-1.46 (1H, m), 1.28 (6H, dd, J = 6.8 Hz, 2.3 Hz)
(實施例6-3) 9-[2,6-脫水-5-去氧-4-(羥基甲基)-α-L-來蘇-己呋喃糖基]-2-(2-甲基丙烷醯胺)-1,9-二氫-6H-嘌呤-6-酮(化合物28)之製造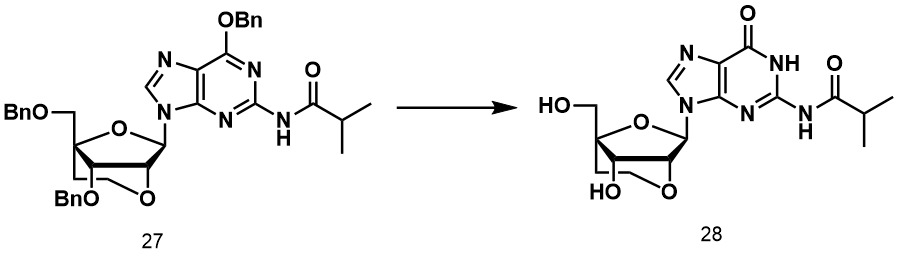
於實施例6-2所獲得的化合物27(17.7 mg, 27.24 ⌠mol)之乙醇溶液(2.0 mL)中,添加20%氫氧化鈀碳(18.0 mg),3.5 bar之氫壓下,於45℃攪拌3小時。確認反應結束後,冷卻至室溫,並進行過濾。藉由將濾液濃縮乾固,取得呈白色結晶之化合物28(11.7 mg, 101.0%)。1
H-NMR (DMSO-d6):δ = 12.10 (1H, s), 11.79 (1H, s), 8.31 (1H, s), 6.21 (1H, s), 5.39 (1H, brs), 5.25 (1H, brs), 4.14 (1H, d, J = 3.5 Hz), 4.10 (1H, d, J = 3.0 Hz), 3.88-3.90 (2H, m), 3.61 (1H, d, J = 12.5 Hz), 2.75-2.81 (1H, m), 2.00-2.07 (1H, m), 1.12 (6H, d, J = 6.5 Hz)
(實施例6-4)9-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-5-去氧-α-L-來蘇-己呋喃糖基)-2-(2-甲基丙烷醯胺)-1,9-二氫-6H-嘌呤-6-酮(化合物29)之製造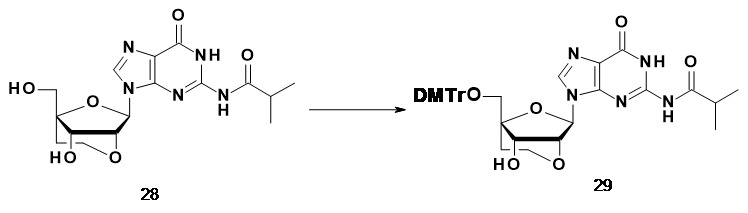
使實施例6-3所獲得的化合物28之四氫呋喃溶液(5v/w)與二甲氧基三苯基氯甲烷(1.5當量)反應。於反應液中添加甲苯(10v/w)、水(5v/w),分液後濃縮,並以矽膠管柱層析進行純化,藉此獲得化合物29。
(實施例6-5)化合物1 g之製造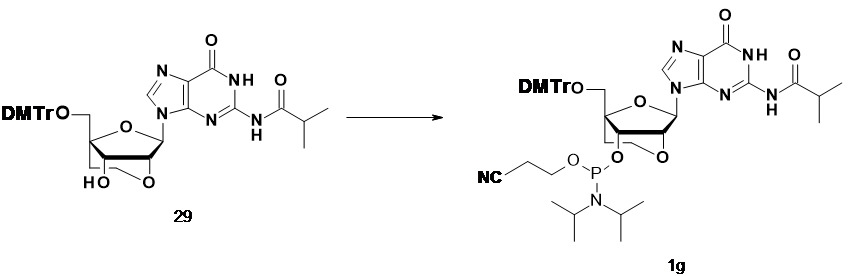
於實施例6-4所獲得的化合物29中添加二氯甲烷(10v/w)、分子篩4A(0.5w/w)、2-氰基乙基N, N, N’, N’-四異丙基磷醯二胺(1.5當量)、4, 5-二氰基咪唑(0.5當量),並於室溫攪拌24小時。於反應液中添加乙酸乙酯(900 mL)水(5v/w),分液後濃縮,並以矽膠管柱層析進行純化,藉此獲得化合物1 g。
(實施例7) 化合物17之製造(經由化合物22的醣基化)
(實施例7-1) 化合物22之乙腈溶液之製造
於化合物2(3.00 g、8.42mmol)之甲苯(30 mL)溶液中,添加N,N-二甲基胺基吡啶(0.05 g、0.42 mmol)、吡啶(1.02 mL、12.63 mmol)及乙酸酐(1.20 mL、12.63 mmol),於20~30℃攪拌1小時後,添加水(15 mL)並進行分液,將獲得的有機層各以20%檸檬酸水溶液(15 mL)2次、以8%碳酸氫鈉水溶液(9 mL)及水(9 mL)1次進行分液洗淨。將獲得的有機層減壓濃縮至6 mL為止後,進行2次以乙腈(30 mL)利用減壓濃縮的溶媒取代,獲得化合物22之乙腈溶液(6 mL)。
(化合物7-2) 化合物3t之1-丙醇溶液之製造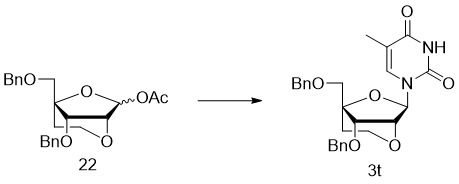
於乙腈(7.5 mL)中添加胸腺嘧碇(1.33 g、10.52 mmol)及N,O-雙三甲基矽基乙醯胺(5.15 mL、21.04 mmol),於20~30℃攪拌1小時的溶液中,添加實施例7-1所獲得的化合物22之乙腈溶液(6 mL)、乙腈(9 mL)及碘三甲基矽烷(1.50 mL、10.52 mmol),而於45~55℃攪拌4小時。於反應液中添加8%碳酸氫鈉水溶液(24 mL)及亞硫酸鈉(1.20 g)後,添加甲苯(30 mL),於20~30℃攪拌30分鐘後,進行分液。將獲得的有機層以20%甲醇水(18 mL)分液洗淨3次後,將獲得的有機層減壓濃縮至成為9 mL為止。於獲得的濃縮液中添加1-丙醇(30 mL),再次減壓濃縮而獲得化合物3t之1-丙醇溶液(9 mL)。
獲得的化合物之NMR譜與WO00/47599之實施例6記載之化合物的NMR譜一致。
(實施例7-3)化合物16之製造
於化合物3t之1-丙醇溶液(9 mL)中,進一步添加1-丙醇(15 mL)、水(6 mL)及5%鈀碳(0.67 g),氫氣環境下於55~65℃攪拌2小時後,於相同溫度下過濾觸媒,以80%1-丙醇水(6 mL)將觸媒洗淨。將獲得的濾液減壓濃縮而作為1-丙醇溶液(9 mL)後,添加乙腈(30 mL)而對9 mL之乙腈溶液(A)進行減壓濃縮。進一步添加乙腈(6 mL),於20~30℃攪拌2小時。濾取生成的結晶,將獲得的結晶以乙腈(9 mL)洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物16(1.57 g、產率65.6%(來自化合物2))。於上述順序,乙腈溶液(A)中之水分含量超過4%的情形,追加乙腈而重複濃縮的操作直到9 mL,藉由將水分含量控制在4%以下,可維持利用晶析的回收的再現性。又,水分含量係利用卡爾-費雪法(Carl Fischer method)而確認。
獲得的化合物之NMR譜與WO00/47599之實施例7記載的化合物的NMR譜一致。
(實施例7-4a) 化合物17之製造
於化合物16(30 g)之四氫呋喃溶液(150 mL)中,添加吡啶(33.39 g)及4,4’-二甲氧基三苯氯甲烷(42.91 g),並於20~30℃攪拌3小時後,添加甲醇(6 mL),進一步於20~30℃攪拌30分鐘。於獲得的溶液中添加15%碳酸鈉水溶液(150 mL)、乙酸乙酯(300 mL)及水(210 mL),並進行分液。將獲得的有機層以20%檸檬酸水溶液(150 mL)2次、各以5%碳酸氫鈉水溶液(150 mL)及水(150 mL)1次進行分液洗淨。減壓濃縮獲得的有機層至成為150 mL為止後,以乙酸乙酯(300 mL)進行利用減壓濃縮的溶媒取代2次,添加乙酸乙酯(90 mL)並獲得化合物17之乙酸乙酯溶液(240 mL)。於獲得的溶液中添加正庚烷(90 mL),並升溫至50℃後,添加晶種(30 m g),於相同溫度攪拌2小時。將獲得的漿液冷卻至20~30℃後,於相同溫度攪拌15小時。於獲得的漿液中添加正庚烷(120 mL),於相同溫度進一步攪拌1小時。濾取析出的結晶,將獲得的結晶以乙酸乙酯與正庚烷之1:1混合液(100 mL)洗淨。將獲得的結晶減壓乾燥(50℃)而獲得化合物17(60.7 g、產率98%)。
獲得的化合物之NMR譜與WO00/47599之實施例8記載的化合物的NMR譜一致。
又,化合物17之晶種係使用將實施例2-3所獲得的化合物17之乙酸乙酯溶液靜置而析出者。
(實施例8a) 化合物1a之製造
於二氯甲烷(675 mL)中添加實施例4-3所獲得的化合物21之水合物(45.00 g、64.31 mmol),藉由減壓濃縮而獲得二氯甲烷溶液(225 mL)。於獲得的二氯甲烷溶液中添加二氯甲烷(450 mL),並進行減壓濃縮,藉此獲得二氯甲烷溶液(225 mL)。再次進行相同的操作,於獲得的二氯甲烷溶液(225 mL)中添加二氯甲烷(225 mL)、分子篩4A(22.50 g)、二丁基羥基甲苯(1.42 g、6.44 mmol)、2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(21.32 g、70.73 mmol)、4, 5-二氰基咪唑(0.91 g、7.71 mmol),於20~30℃攪拌16.5小時。確認反應結束後,於反應液中添加乙酸乙酯(900 mL),於-5~5℃添加中性矽膠(67.50 g),攪拌15分鐘後,進行中性矽膠的濾除,並以二氯甲烷-乙酸乙酯(1/2, 225 mL)進行洗淨,藉此獲得二氯甲烷-乙酸乙酯溶液。將獲得的二氯甲烷-乙酸乙酯溶液減壓濃縮,作成乙酸乙酯溶液(225 mL)。藉由於獲得的溶液中添加甲基三級丁基醚(135 mL),獲得溶液A(360 mL)。於另外的反應容器中添加甲基三級丁基醚(315 mL)、正庚烷(1350 mL),於-5~5°C添加溶液A(360 mL),藉此確認固體的析出。以乙酸乙酯(23 mL)進行洗淨,攪拌0.5小時後,將析出的固體過濾,以預先冷卻至0℃的甲基三級丁基醚-正庚烷(1/3, 225 mL)、正庚烷(225 mL)將固體洗淨。藉由將獲得的固體進行減壓乾燥(40℃)而獲得化合物1a(48.69 g、84.1%)。
獲得的化合物之NMR譜與WO00/47599之實施例14記載之化合物的NMR譜一致。
(實施例8b)化合物1a之製造
於二氯甲烷(45 mL)中添加實施例4-3所獲得的化合物21之水合物(3.57 g),並進行減壓濃縮,藉此獲得二氯甲烷溶液(15 mL)。於獲得的二氯甲烷溶液中添加二氯甲烷(15 mL)、分子篩4A(22.50 g)、2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(1.42 g)、及4,5-二氰基咪唑(100 mg),將反應混合物於20~30℃攪拌24小時。確認反應結束後,於反應液中添加乙酸乙酯(60 mL)。將獲得的溶液冷卻至-5~5℃後,使通過填充中性矽膠(4.5 g)的管柱內,藉由以二氯甲烷-乙酸乙酯(1/2, 30 mL)洗滌,獲得二氯甲烷-乙酸乙酯溶液。將獲得的二氯甲烷-乙酸乙酯溶液減壓濃縮,作成乙酸乙酯溶液(15 mL)後,添加甲苯(60 mL),再次進行濃縮而獲得甲苯溶液(15 mL)。藉由於獲得的溶液中添加甲基三級丁基醚(9 mL)而獲得溶液A(24 mL)。於另外的反應容器中添加甲基三級丁基醚(6 mL)、正庚烷(90 mL),於-5~5°C滴加溶液A(24 mL),藉此確認固體之析出。將於滴加使用的器具以甲苯(1.5 mL)洗滌,攪拌2小時後,將析出的固體過濾,以正庚烷(24 mL)將固體洗淨。藉由將獲得的固體進行減壓乾燥(40℃)而獲得化合物1a(3.97 g、86.5%)。
獲得的化合物之NMR譜與WO00/47599之實施例14記載之化合物的NMR譜一致。
(實施例9) 化合物1t之製造
於乙酸乙酯(40 mL)中添加實施例2-3所獲得的化合物17(5.00 g、8.52 mmol),添加分子篩4A(2.5 g)、2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(2.83 g、9.38 mmol)、4,5-二氰基咪唑(0.20 g、1.70 mmol),並於20~30℃攪拌24小時。確認反應結束後,於反應液中添加中性矽膠(10.00 g),攪拌30分鐘後,進行中性矽膠的濾除,並以乙酸乙酯(100 mL)進行洗淨,藉此獲得乙酸乙酯溶液。將獲得的乙酸乙酯溶液減壓濃縮,作成乙酸乙酯溶液(35 mL)。於獲得的乙酸乙酯溶液中添加三級丁基醚(100 mL),並進行減壓濃縮,藉此獲得甲基三級丁基醚溶液(35 mL)。於獲得的甲基三級丁基醚溶液中添加化合物1t之晶種(2.5 mg),攪拌2小時後,添加正庚烷(100 mL)並冷卻至0~5℃而進一步攪拌2小時。將析出的固體過濾,以預先冷卻至0℃的正庚烷(100 mL)將固體洗淨。將獲得的固體進行減壓乾燥(40℃)而獲得化合物1t(6.12 g、91.3%)。
獲得的化合物之NMR譜與WO00/47599之實施例9記載之化合物的NMR譜一致。
又,化合物1t之晶種係使用將於實施例2-4獲得的化合物1t之乙酸乙酯、二異丙基醚及正庚烷之混合溶液靜置而析出者。
(實施例10) 化合物1c之製造
(實施例10-1) 1-(3-O-乙醯基-2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-5-去氧-α-L-來蘇-己呋喃糖基)-5-甲基嘧啶-2,4(1H,3H)-二酮(化合物30)之製造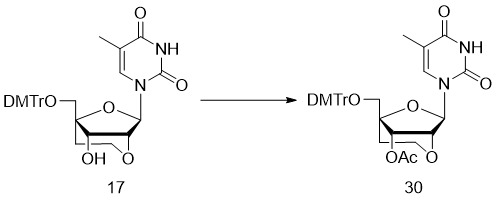
於乙酸乙酯(320 mL)中添加實施例2-3所獲得的化合物17(80.00 g、136.37 mmol)、N,N-二甲基胺基吡啶(1.67 g、13.64 mmol)及三乙基胺(27.60 g、272.74 mmol),冷卻至0~5℃後,於相同溫度下添加乙酸酐(16.71 g、163.64 mmol)。於0~5℃攪拌1小時後,添加水(160 mL)並進行分液,將獲得的有機層以10%檸檬酸水溶液(160 mL)、5%碳酸氫鈉水溶液(160 mL)及10%食鹽水(160 mL)進行分液洗淨。將獲得的有機層減壓濃縮至成為240 mL後,以乙腈(400 mL)進行2次利用減壓濃縮的溶媒取代,獲得化合物30之乙腈溶液(240 mL)。
(實施例10-2)1-(3-O-乙醯基-2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-5-去氧-α-L-來蘇-己呋喃糖基)-4-胺基-5-甲基嘧啶-2(1H)-酮(化合物31)之製造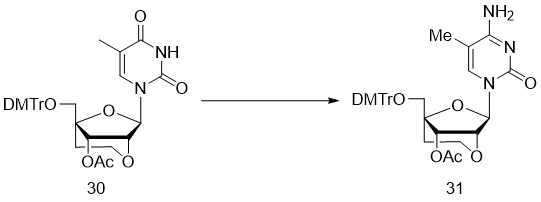
於實施例10-1所獲得的化合物30之溶液中添加乙腈(160 mL)、N,N-二甲基胺基吡啶(3.33 g、27.27 mmol)及三乙基胺(55.20 g、545.51 mmol),冷卻至5~15℃後,於相同溫度添加2,4,6-三異丙基苯磺醯氯(47.50 g、156.83 mmol)。於10℃攪拌2小時後,添加25%氨水(100 kg、156.83 mmol),並於20℃攪拌2小時。將獲得的溶液減壓濃縮至成為約320 mL為止後,添加乙酸乙酯(400 mL)及水(120 mL)進行分液。將獲得的有機層以10%食鹽水(160 mL)進行2次分液洗淨後,添加乙腈(400 mL),並減壓濃縮至成為約320 mL為止。於獲得的濃縮液中添加乙腈(400 mL)並減壓濃縮,獲得化合物31之乙腈溶液(B)(240 mL)。又,於上述步驟,於乙腈溶液(B)中之水分含量超過0.4%的情形,藉由重複實施於乙腈溶液(B)中添加乙腈而濃縮脫水的步驟使乙腈溶液(B)中之水分含量成為0.4%以下,可使下一反應快速地進行。
又,水分含量係利用卡爾-費雪法而確認。
(實施例10-3a) 化合物19之製造
於實施例10-2所獲得的化合物31之乙腈溶液中添加THF(80 mL)及苯甲酸酐(67.87 g、300.01 mmol),並於40℃攪拌5小時後,冷卻至25℃並進一步攪拌15小時。將獲得的漿液,於5~15°C歷經1小時以上滴加至水(80 mL)、25%氫氧化鈉水溶液(176 kg)及THF(80 mL)之混合液。於獲得的混合液中添加THF(160 mL),於20°C攪拌2小時後,使用乙酸而將溶液之pH調整為6.8。於獲得的溶液中添加作為晶種之實施例3-3所獲得的化合物19之水合物結晶(80 mg),並攪拌10小時後,於15~25°C歷經2小時以上滴加水(400 mL)。將獲得的漿液於20℃攪拌1小時後,過濾析出的結晶,以40%THF水(400 mL)將結晶洗淨。將獲得的結晶於50℃減壓乾燥,獲得化合物19(78.35 g、83.3%G/G(來自化合物17))。
獲得的化合物之NMR譜與WO00/47599之實施例21記載之化合物的NMR譜一致。
(實施例10-3b)化合物19之製造
於實施例10-2所獲得的化合物31之乙腈溶液(90 mL)中添加THF(80 mL)及苯甲酸酐(23.14 g、102.3 mmol),並於40℃攪拌5小時後,冷卻至25℃並進一步攪拌20小時。於獲得的漿液中,添加THF(30 mL)及乙酸鉀(23.09 g、235.3 mmol),攪拌1小時後,添加8%食鹽水(135 mL)並進行分液。於獲得的有機層中添加THF(15 mL)、水(15 mL)及25%氫氧化鈉水溶液(33 g),冷卻至0~10℃後,進一步添加25%氫氧化鈉水溶液(15 g)及水(6 mL),並於0~10℃攪拌4小時。確認反應結束後,使用乙酸調整獲得的溶液溶液之pH為6.5。於獲得的溶液中添加食鹽(6.0 g)及THF(60 mL)並進行分液,於獲得的有機層中添加THF(30 mL)、水(45 mL)及作為晶種之實施例3-3所獲得的化合物19之水合物結晶(30 mg),攪拌16小時後,於20~30℃歷經2小時以上滴加水(180 mL)。將獲得的漿液於20~30℃攪拌3小時後,過濾析出的結晶,使用40%THF水(150 mL)而將結晶洗淨。獲得的結晶於50℃減壓乾燥,獲得化合物19(28.48 g、80.7%G/G(來自化合物17))。
於上述步驟,藉由使用乙酸鉀,可將晶析時於pH調整所使用的乙酸之量控制為小於實施例10-3a記載之步驟的情形,因而可更縮短晶析所需要的時間。
獲得的化合物之NMR譜與WO00/47599之實施例21記載之化合物的NMR譜一致。
(實施例11) 化合物1c之製造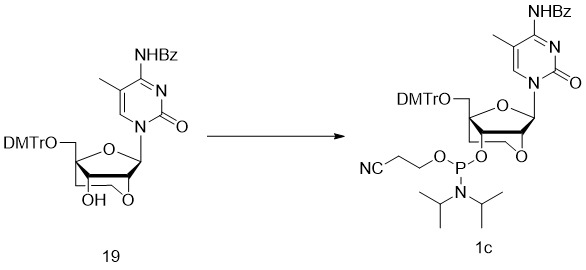
於二氯甲烷(400 mL)中添加實施例10-3所獲得的化合物19(20.00 g、29.00 mmol),並進行減壓濃縮,藉此獲得二氯甲烷溶液(200 mL)。於獲得的二氯甲烷溶液中添加二氯甲烷(200 mL),並進行減壓濃縮,藉此獲得二氯甲烷溶液(200 mL)。再次進行相同的操作,於獲得的二氯甲烷溶液(200 mL)中添加分子篩4A(10.00 g)、二丁基羥基甲苯(0.64 g、2.90 mmol)、2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(9.61 g、31.89 mmol)、4, 5-二氰基咪唑(1.03 g、8.70 mmol),於20~30°C攪拌4小時。確認反應結束後,於反應液中添加乙酸乙酯(100 mL),並於-5~5°C添加中性矽膠(40.00 g),攪拌30分鐘後,進行中性矽膠的濾除,並以二氯甲烷-乙酸乙酯(2/1, 100 mL)進行洗淨,藉此獲得二氯甲烷-乙酸乙酯溶液。將獲得的二氯甲烷-乙酸乙酯溶液減壓濃縮,作成二氯甲烷-乙酸乙酯溶液(100 mL)。於獲得的二氯甲烷-乙酸乙酯溶液中添加乙酸乙酯(100 mL),並進行減壓濃縮,藉此獲得乙酸乙酯溶液(100 mL)。再次進行同樣的操作,於獲得的溶液中添加甲基三級丁基醚(100 mL),並進行減壓濃縮,藉此作成乙酸乙酯-甲基三級丁基醚溶液(100 mL)。於獲得的乙酸乙酯-甲基三級丁基醚溶液中添加甲基三級丁基醚(100 mL),並進行減壓濃縮,藉此獲得甲基三級丁基醚溶液(100 mL)。再次進行同樣的操作,於獲得的溶液中添加正庚烷(200 mL)並攪拌1.5小時後,進一步添加正庚烷(100 mL)並攪拌1小時後,再次添加正庚烷(200 mL)而攪拌1.5小時,並確認固體的析出。將析出的固體過濾,以預先冷卻至0℃的正庚烷(200 mL)將固體洗淨。藉由將獲得的固體進行減壓乾燥(40℃),而獲得化合物1c(22.40 g、86.8%)。
獲得的化合物之NMR譜與WO00/47599之實施例6記載的化合物之NMR譜一致。
(實施例12) 化合物22之立體選擇性的醣基化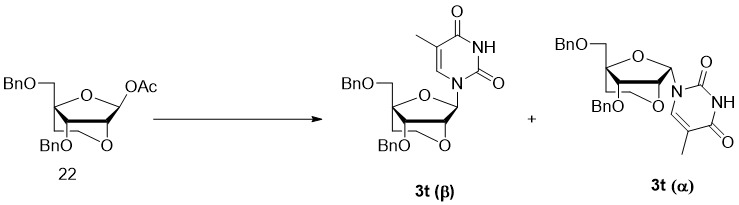
於實施例5-1所獲得的化合物22(30 mg、0.075 mmol)、雙三甲基矽基胸腺嘧碇(40.8 mg、0.151 mmol)中,添加1,2-二氯乙烷(0.3 mL),並於室溫進行攪拌。之後,各自添加相對於原料為2.0當量(0.151 mmol)之作為活化劑之氯三甲基矽烷(TMSCl)、溴三甲基矽烷(TMSBr)、碘三甲基矽烷(TMSI)、三氟甲磺酸三甲基矽酯(TMSOTf),以表中所示溫度進行攪拌。將反應液取樣,以HPLC實施原料、α-加成體、β-加成體之分析。將結果示於表1。
[表1]
| 活化劑 | 反應溫度 (℃) | 反應時間 (h) | HPLC P. A. (%)/210nm | β:α | ||
| 22 | 3t(β) | 3t(α) | ||||
| TMSCl | 70 | 18 | 97.62 | N. D. | N. D. | - |
| TMSBr | 70 | 28 | 7.03 | 67.98 | 21.80 | 75.7:24.3 |
| TMSI | 25 | 4 | 4.30 | 74.38 | 16.62 | 81.7:18.3 |
| TMSOTf | 25 | 45 | 4.14 | 3.49 | 90.19 | 3.7:96.3 |
研討的結果,於使用TMSOTf的條件,立體選擇性地獲得α-加成體,但於使用TMSBr及TMSI的條件下,立體選擇性地獲得β-加成體。其中尤以使用TMSI時,即使於室溫,反應亦良好地進行。
(實施例13) AMIDITE化步驟活化劑篩選
於二氯甲烷(1 mL)中,各自添加相對於原料為1.15當量(0.16 mmol)之實施例4-3所獲得的化合物21水合物(100 mg、0.14 mmol)、2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(51.7 mg、0.17 mmol),作為活化劑之5-苄硫基四唑、5-苯基四唑、4,5-二氰基咪唑、2,4,5-四溴咪唑,於20~30℃進行攪拌。將以HPLC確認反應結束的結果示於表2。
[表2]
| 活化劑 | 反應時間 (h) | HPLC面積比 (%) |
| 1a | ||
| 5-苄硫基四唑 | 3 | 93.57 |
| 5-苯基四唑 | 22 | 88.94 |
| 4,5-二氰基咪唑 | 3 | 95.36 |
| 2,4,5-四溴咪唑 | 48 | 96.45 |
研討的結果,於使用4,5-二氰基咪唑、2,4,5-三溴咪唑的條件獲得良好的結果。
(實施例14) 分子篩效果之驗證
於二氯甲烷(10 v/w)中添加實施例2-3所獲得的化合物17、二丁基羥基甲苯(0.10當量)、2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(1.10當量)、4,5-二氰基咪唑(1.0當量),於分子篩4A(0.5 wt %)存在下或非存在下,於20~30℃進行攪拌。將以HPLC確認反應之進行的結果示於表3。
[表3]
| 分子篩4A (wt %) | HPLC面積比 (%) | |
| 17 | 1t | |
| 無 | 2.8 | 96.5 |
| 0.5 | 0.07 | 98.5 |
藉由使用分子篩,化合物17不殘留地將其轉換成化合物1t。
(實施例15)寡核苷酸之合成
包含期望的序列及結構的寡核苷酸可利用以下之方法而合成。
使用核酸自動合成機(「ABI 394 DNA/RNA Synthesizer」Applied Biosystems製),並使用亞磷醯胺法(Nucleic Acids Research, 12, 4539 (1984))而進行合成。就試藥而言,使用活化劑溶液-3(0.25 mol/L 5-苄硫基-1H-四唑・乙腈溶液、和光純藥工業製、產品編號013-20011)、AKTA用CAP A(1-甲基咪唑・乙腈溶液、Sigma-Aldrich製、產品編號L040050)、AKTA用Cap B1(乙酸酐・乙腈溶液、Sigma-Aldrich製、產品編號L050050)、AKTA用Cap B2(吡啶・乙腈溶液、Sigma-Aldrich製、產品編號L050150)、或DCA Deblock(二氯乙酸・甲苯溶液、Sigma-Aldrich製、產品編號L023050)。作為用以形成硫代磷酸酯鍵的硫化試藥,以成為0.2 M之方式使用乙腈(脫水、關東化學製、產品編號01837-05)、及吡啶(脫水、關東化學製、產品編號11339-05)1:1(v/v)溶液溶解苯基乙醯基二硫醚(CARBOSYNTH製、產品編號FP07495)而使用。就AMIDITE試藥而言,使用2’-O-Me核苷之亞磷醯胺(腺苷體產品編號ANP-5751,胞苷體產品編號ANP-5752;鳥苷體產品編號ANP-5753,尿苷體產品編號ANP-5754)係使用ChemGenes製者。ENA單體 9-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-N-苄醯基-9H-嘌呤-6-胺(化合物1a)、1-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-4-苯甲醯胺-5-甲基嘧啶-2(1H)-酮(化合物1c)、及1-(2,6-脫水-4-{[雙(4-甲氧基苯基)(苯基)甲氧基]甲基}-3-O-{(2-氰基乙氧基)[二(丙烷-2-基)胺基]磷烷基}-5-去氧-α-L-來蘇-己呋喃糖基)-5-甲基嘧啶-2,4(1H,3H)-二酮(化合物1t)係使用於上述之實施例所合成的化合物。作為固相載體,使用Glen Unysupport 0.1μmol(GlenResearch製),可合成具有期望之序列的寡核苷酸。程序為使用附屬於核酸自動合成機的0.2μmol規模用,但AMIDITE體之縮合所需時間設為600秒,硫化所需時間設為150秒。
於5’末端具有配位體單元的寡核苷酸係可按照WO2019/172286記載的方法,接續核苷酸鏈的合成,使配位體單元的亞磷醯胺,同樣地反應而藉此進行合成。對應為GalNAc單元的X18
及X20
的亞磷醯胺化合物係各自使用WO2019/172286之參考例39之化合物39D、及參考例41之化合物41D。
藉由將具有目的序列的經保護的寡核苷酸類似物以300μL之濃氨水進行處理,而將寡聚物自支持體切出的同時,移除磷原子上之保護基氰基乙基與核酸鹼上之保護基。使用Clarity QSP(Phenomenex製),按照所附步驟準則進行純化。
[產業上利用之可能性]
根據本發明,藉由用以製造各種ENA單體之有用的結晶性之2,4-交聯的共通中間體、及該中間體之立體選擇性的製造方法,成為可有效率地製造各種ENA單體。
無。
無。








無。
Claims (111)
- 一種通式(I)所表示的化合物,[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,R表示氫原子或脂肪族醯基,n表示0至4之整數]。
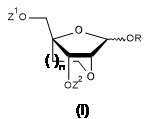
- 如請求項1之化合物,其中R為氫原子或乙醯基。
- 如請求項1或2之化合物,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項1或2之化合物,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項1或2之化合物,其中Z1 及Z2 為苄基。
- 如請求項1至5中任一項之化合物,其中n為1。
- 一種式(I’)所表示的化合物,。
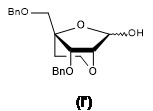
- 一種式(I’’)所表示的化合物,。

- 一種製造通式(II)所表示的化合物之方法,[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數], 其特徵為包含: (i)將通式(III)所表示的化合物之縮醛部位,於酸觸媒存在下,低級烷基醇溶媒中,進行溶媒分解,而將Y去保護的步驟,
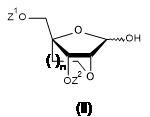 [式中,Z1 、Z2 及n表示與前述同意義,Y表示經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基、低級烷氧基甲基、四氫哌喃基或矽基], (ii)將步驟(i)中所獲得的通式(IV)所表示的化合物之二醇部位進行環化的步驟,
[式中,Z1 、Z2 及n表示與前述同意義,Y表示經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基、低級烷氧基甲基、四氫哌喃基或矽基], (ii)將步驟(i)中所獲得的通式(IV)所表示的化合物之二醇部位進行環化的步驟,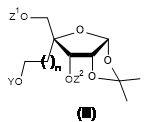 [式中,Z1 、Z2 及n表示與前述同意義,A表示低級烷基], (iii)進行步驟(ii)中所獲得的通式(V)所表示的化合物之變旋異構物(anomer)位之水解的步驟,
[式中,Z1 、Z2 及n表示與前述同意義,A表示低級烷基], (iii)進行步驟(ii)中所獲得的通式(V)所表示的化合物之變旋異構物(anomer)位之水解的步驟, [式中,Z1 、Z2 、A及n表示與前述同意義]。
[式中,Z1 、Z2 、A及n表示與前述同意義]。
- 如請求項9之方法,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項9之方法,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項9之方法,其中Z1 及Z2 為苄基。
- 如請求項9至12中任一項之方法,其中A為甲基、乙基或丙基。
- 如請求項9至12中任一項之方法,其中A為甲基。
- 如請求項9至14中任一項之方法,其中Y為三級丁基二苯基矽基、三級丁基二甲基矽基、四氫哌喃-2-基或三苯甲基。
- 如請求項9至14中任一項之方法,其中Y為三苯甲基。
- 如請求項9至16中任一項之方法,其中n為1。
- 如請求項9至17中任一項之方法,其中酸觸媒為硫酸、對甲苯磺酸或甲磺酸。
- 如請求項9至18中任一項之方法,其中步驟(ii)使用三價磷試藥及偶氮二甲酸酯進行。
- 如請求項19之方法,其中三價磷試藥為三苯基膦或三(正丁基)膦。
- 如請求項19或20之方法,其中偶氮二甲酸酯為偶氮二甲酸二乙酯、偶氮二甲酸二異丙酯或偶氮二甲酸二(三級丁酯)。
- 如請求項9至21中任一項之方法,其中步驟(iii)使用酸進行。
- 如請求項22之方法,其中酸為鹽酸、硫酸、三氟乙酸、甲磺酸或對甲苯磺酸。
- 一種製造通式(VI)所表示的化合物或其鹽之方法,[式中,R1 表示低級烷基或氫原子,R2 表示羥基、胺基或經脂肪族醯基或芳香族醯基保護的胺基,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數], 其特徵為包含: (i)使通式(II)所表示的化合物,於溶媒中,與活化劑反應,將1位之羥基轉化為形成脫離基的基的步驟,
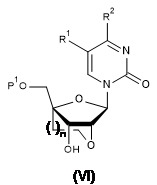 [式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數],及 (ii)使步驟(i)中所獲得的通式(VII)所表示的化合物、與通式(VIII)所表示的化合物或其鹽,於溶媒中,鹵化劑存在下反應,而立體選擇性地獲得通式(IX)所表示的化合物或其鹽的步驟,
[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數],及 (ii)使步驟(i)中所獲得的通式(VII)所表示的化合物、與通式(VIII)所表示的化合物或其鹽,於溶媒中,鹵化劑存在下反應,而立體選擇性地獲得通式(IX)所表示的化合物或其鹽的步驟,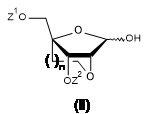 [式中,Z1 、Z2 及n表示與前述同意義,X1 表示形成脫離基的基],
[式中,Z1 、Z2 及n表示與前述同意義,X1 表示形成脫離基的基],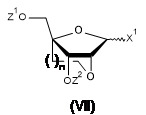 [式中,R1 及R2 表示與前述同意義],
[式中,R1 及R2 表示與前述同意義], [式中,Z1 、Z2 、R1 、R2 及n表示與前述同意義]。
[式中,Z1 、Z2 、R1 、R2 及n表示與前述同意義]。
- 如請求項24之方法,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項24之方法,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項24之方法,其中Z1 及Z2 為苄基。
- 如請求項24至27中任一項之方法,其中P1 為4,4’-二甲氧基三苯甲基。
- 如請求項24至28中任一項之方法,其中X1 為鹵素原子、脂肪族醯氧基、鹵素取代的低級烷基亞胺氧基或鹵素取代的低級烷基磺醯氧基。
- 如請求項24至28中任一項之方法,其中X1 為碘原子、乙醯氧基或三氯乙醯亞胺氧基。
- 如請求項24至30中任一項之方法,其中n為1。
- 如請求項24至31中任一項之方法,其中R1 為甲基或氫原子。
- 如請求項24至32中任一項之方法,其中R2 為羥基或苄醯基胺基。
- 如請求項24至31中任一項之方法,其中R1 為甲基,R2 為羥基。
- 如請求項24至31中任一項之方法,其中R1 為甲基,R2 為苄醯基胺基。
- 如請求項24至35中任一項之方法,其包含: (iii)使步驟(ii)中所獲得的通式(IX)所表示的化合物或其鹽,於溶媒中,與羥基之去保護試藥反應,並將Z1 及Z2 去保護的步驟,[式中,Z1 、Z2 、R1 、R2 及n表示與前述同意義],及 (iv)使步驟(iii)所獲得的二醇化合物或其鹽與1級羥基之保護試藥反應,而獲得通式(VI)所表示的化合物或其鹽的步驟,
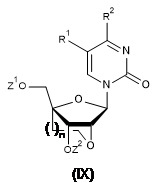 [式中,P1 、R1 、R2 及n表示與前述同意義]。
[式中,P1 、R1 、R2 及n表示與前述同意義]。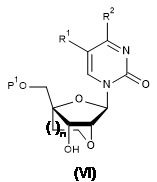
- 如請求項24至36中任一項之方法,其中活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯。
- 如請求項24至37中任一項之方法,其中鹵化劑為氯三甲基矽烷、溴三甲基矽烷或碘三甲基矽烷。
- 一種製造通式(X)所表示的化合物或其鹽之方法,[式中,R3 表示脂肪族醯基或芳香族醯基,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數], 其特徵為包含: (i)使通式(II)所表示的化合物,於溶媒中,與活化劑反應,而將1位之羥基轉化為形成脫離基的基的步驟,
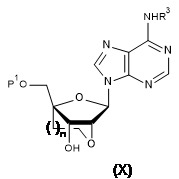 [式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示與前述同意義], (ii)使步驟(i)中所獲得的通式(XI)所表示的化合物與通式(XII)所表示的化合物或其鹽,於溶媒中、酸試藥存在下反應的步驟,
[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示與前述同意義], (ii)使步驟(i)中所獲得的通式(XI)所表示的化合物與通式(XII)所表示的化合物或其鹽,於溶媒中、酸試藥存在下反應的步驟, [式中,Z1 、Z2 及n表示與前述同意義,X2 表示形成脫離基的基],
[式中,Z1 、Z2 及n表示與前述同意義,X2 表示形成脫離基的基], [式中,R3 表示與前述同意義], (iii)接著,使異構物化,立體選擇地獲得通式(XIII)所表示的化合物或其鹽的步驟,
[式中,R3 表示與前述同意義], (iii)接著,使異構物化,立體選擇地獲得通式(XIII)所表示的化合物或其鹽的步驟,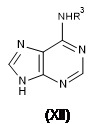 [式中,Z1 、Z2 、R3 及n表示與前述同意義]。
[式中,Z1 、Z2 、R3 及n表示與前述同意義]。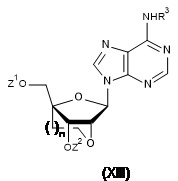
- 如請求項39之方法,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項39之方法,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項39之方法,其中Z1 及Z2 為苄基。
- 如請求項39至42中任一項之方法,其中P1 為4,4’-二甲氧基三苯甲基。
- 如請求項39至43中任一項之方法,其中X2 為鹵素原子、脂肪族醯氧基、鹵素取代的低級烷基亞胺氧基或鹵素取代的低級烷基磺醯氧基。
- 如請求項39至43中任一項之方法,其中X2 為乙醯氧基。
- 如請求項39至45中任一項之方法,其中n為1。
- 如請求項39至46中任一項之方法,其中R3 為乙醯基或苄醯基。
- 如請求項39至46中任一項之方法,其中R3 為苄醯基。
- 如請求項39至48中任一項之方法,其包含: (iv)使步驟(iii)中所獲得的通式(XIII)所表示的化合物或其鹽,於溶媒中,與羥基之去保護試藥反應,並將Z1 及Z2 去保護的步驟,[式中,Z1 、Z2 、R3 及n表示與前述同意義],及 (v)使步驟(iv)中所獲得的二醇化合物或其鹽、與1級羥基之保護試藥反應,選擇性地保護1級羥基,藉此獲得通式(X)所表示的化合物或其鹽的步驟,
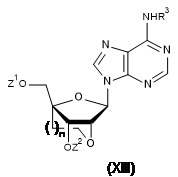 [式中,R3 、n及P1 表示與前述同意義]。
[式中,R3 、n及P1 表示與前述同意義]。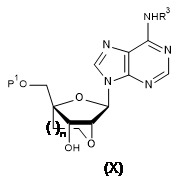
- 如請求項39至49中任一項之方法,其中活化劑為乙酸酐、苯甲酸酐、三氯乙腈、羰基二咪唑或氯磷酸二苯酯。
- 如請求項39至50中任一項之方法,其中酸試藥為三氟甲磺酸三甲基矽酯及三氟乙酸。
- 一種通式(XIV)所表示的化合物或其鹽,[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數]。
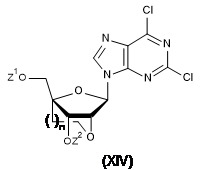
- 如請求項52之化合物或其鹽,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項52之化合物或其鹽,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項52之化合物或其鹽,其中Z1 及Z2 為苄基。
- 如請求項52至55中任一項之化合物或其鹽,其中n為1。
- 一種式(XIV’)所表示的化合物或其鹽,。
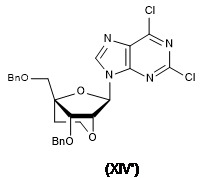
- 一種製造通式(X)所表示的化合物或其鹽之方法,[式中,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,R3 表示脂肪族醯基或芳香族醯基,n表示1至4之整數], 其特徵為包含: (i)使通式(XIV)所表示的化合物或其鹽、與胺基化劑反應,而將嘌呤環之6位之氯原子取代為胺基的步驟,
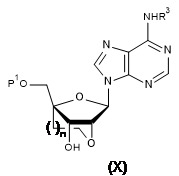 [式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數], (ii)使步驟(i)中所獲得的通式(XV)所表示的化合物或其鹽,於溶媒中、金屬觸媒存在下,與還原劑反應,將嘌呤環之2位之氯原子取代為氫原子,將Z1 及Z2 去保護,藉此獲得通式(XVI)所表示的化合物或其鹽的步驟,
[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數], (ii)使步驟(i)中所獲得的通式(XV)所表示的化合物或其鹽,於溶媒中、金屬觸媒存在下,與還原劑反應,將嘌呤環之2位之氯原子取代為氫原子,將Z1 及Z2 去保護,藉此獲得通式(XVI)所表示的化合物或其鹽的步驟,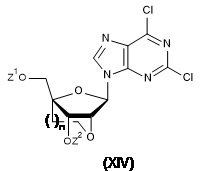 [式中,Z1 、Z2 及n表示與前述同意義],
[式中,Z1 、Z2 及n表示與前述同意義], [式中,n表示與前述同意義]。
[式中,n表示與前述同意義]。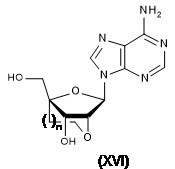
- 如請求項58之方法,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項58之方法,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項58之方法,其中Z1 及Z2 為苄基。
- 如請求項58至61中任一項之方法,其中P1 為4,4’-二甲氧基三苯甲基。
- 如請求項58至62中任一項之方法,其中n為1。
- 如請求項58至63中任一項之方法,其中R3 為乙醯基或苄醯基。
- 如請求項58至63中任一項之方法,其中R3 為苄醯基。
- 如請求項58至65中任一項之方法,其包含: (iii)使步驟(ii)中所獲得的通式(XVI)所表示的化合物或其鹽、與1級羥基之保護試藥反應,而選擇性保護1級羥基的步驟,[式中,n表示與前述同意義], (iv)藉由使步驟(iii)中所獲得的通式(XVII)所表示的化合物或其鹽、與醯化劑反應,而獲得通式(X)所表示的化合物或其鹽的步驟,
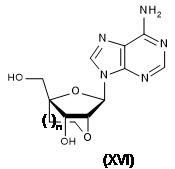 [式中,P1 及n表示與前述同意義],
[式中,P1 及n表示與前述同意義],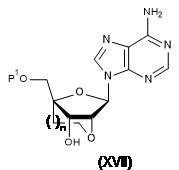 [式中,P1 、R3 及n表示與前述同意義]。
[式中,P1 、R3 及n表示與前述同意義]。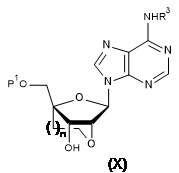
- 如請求項58至66中任一項之方法,其中胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨。
- 如請求項58至67中任一項之方法,其中金屬觸媒為鈀、氫氧化鈀或鉑。
- 如請求項58至68中任一項之方法,其中還原劑為氫、甲酸或甲酸銨。
- 如請求項66至69中任一項之方法,其中醯化劑為苯甲醯氯或苯甲酸酐。
- 一種製造通式(XVIII)所表示的化合物或其鹽之方法,[式中,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,R4 表示脂肪族醯基或芳香族醯基,n表示1至4之整數], 其特徵為包含: (i)使通式(XIV)所表示的化合物或其鹽,於溶媒中,鹼存在下,與可經低級烷基、低級烷氧基、鹵素或氰基取代的芐醇反應,將嘌呤環之6位之氯原子取代為可經低級烷基、低級烷氧基、鹵素或氰基取代的苄氧基的步驟,
 [式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數], (ii)使步驟(i)中所獲得的通式(XIX)所表示的化合物或其鹽,於溶媒中,鈀觸媒、膦配位子存在下,與醯胺化劑交叉偶合,而獲得通式(XX)所表示的化合物或其鹽的步驟,
[式中,Z1 及Z2 為相同或相異,表示羥基之保護基,n表示0至4之整數], (ii)使步驟(i)中所獲得的通式(XIX)所表示的化合物或其鹽,於溶媒中,鈀觸媒、膦配位子存在下,與醯胺化劑交叉偶合,而獲得通式(XX)所表示的化合物或其鹽的步驟,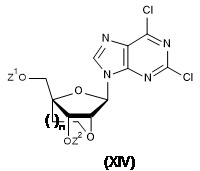 [式中,Z1 、Z2 及n表示與前述同意義,R5 表示可經低級烷基、低級烷氧基、鹵素或氰基取代的苄基],
[式中,Z1 、Z2 及n表示與前述同意義,R5 表示可經低級烷基、低級烷氧基、鹵素或氰基取代的苄基],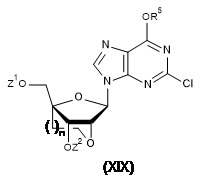 [式中,Z1 、Z2 、R4 、R5 及n表示與前述同意義]。
[式中,Z1 、Z2 、R4 、R5 及n表示與前述同意義]。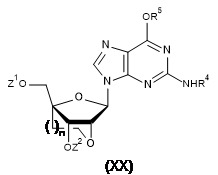
- 如請求項71之化合物或其鹽,其中Z1 及Z2 為相同或相異,為脂肪族醯基、芳香族醯基、經1至3個之芳基取代的甲基、低級烷基、低級烷氧基、芳基環被鹵素或氰基取代之經1至3個之芳基取代的甲基或矽基。
- 如請求項71之方法,其中Z1 及Z2 為相同或相異,為苄基、對甲氧基苄基、三級丁基二苯基矽基或三級丁基二甲基矽基。
- 如請求項71之方法,其中Z1 及Z2 為苄基。
- 如請求項71至74中任一項之方法,其中P1 為4,4’-二甲氧基三苯甲基。
- 如請求項71至75中任一項之方法,其中n為1。
- 如請求項71至76中任一項之方法,其中R4 為異丁醯基。
- 如請求項71至77中任一項之方法,其中R5 為苄基。
- 如請求項71至78中任一項之方法,其包含: (iii)使步驟(ii)中所獲得的通式(XX)所表示的化合物或其鹽,於溶媒中,與羥基之去保護試藥反應,並將Z1 、Z2 及R5 去保護的步驟,[式中,Z1 、Z2 、R4 、R5 及n表示與前述同意義], (iv)使步驟(iii)中所獲得的通式(XXI)所表示的化合物或其鹽、與1級羥基之保護試藥反應,選擇性保護1級羥基,藉此獲得通式(XVIII)所表示的化合物或其鹽的步驟,
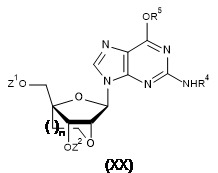 [式中,R4 及n表示與前述同意義],
[式中,R4 及n表示與前述同意義], [式中,P1 、R4 及n表示與前述同意義]。
[式中,P1 、R4 及n表示與前述同意義]。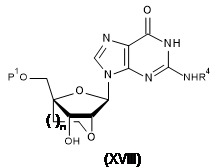
- 如請求項71至79中任一項之方法,其中鹼為氫氧化鈉、碳酸鈉、碳酸銫、三乙基胺、吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯。
- 如請求項71至80中任一項之方法,其中鈀觸媒為參(二亞苄基丙酮)(氯仿)二鈀、乙酸鈀(II)或參(二亞苄基丙酮)二鈀(0)。
- 如請求項71至81中任一項之方法,其中膦配位子為4,5’-雙(二苯基膦基)-9,9’二甲基𠮿、1,1’-雙(二苯基膦基)二茂鐵、1,2-雙(二苯基膦基)乙烷或2-二環己基膦基-2’-(N,N-二甲基胺基)聯苯。
- 如請求項71至82中任一項之方法,其中醯胺化劑為乙醯基醯胺、苄醯基醯胺或異丁基醯胺。
- 如請求項79至83中任一項之方法,其中羥基之去保護試藥為金屬觸媒及還原劑。
- 如請求項84之方法,其中金屬觸媒為鈀、氫氧化鈀或鉑。
- 如請求項84或85之方法,其中還原劑為氫、甲酸或甲酸銨。
- 一種製造通式(XXII)所表示的化合物或其鹽之方法,[式中,R1 表示低級烷基或氫原子,R6 表示脂肪族醯基或芳香族醯基,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,n表示0至4之整數], 其特徵為包含: (i)使通式(XXIII)所表示的化合物或其鹽,於溶媒中,與羥基之保護試藥反應,並保護3’位之羥基的步驟,
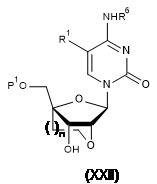 [式中,P1 、R1 及n表示與前述同意義], (ii)使步驟(i)中所獲得的通式(XXIV)所表示的化合物或其鹽,於溶媒中、鹼及觸媒存在下,與活化劑反應的步驟,
[式中,P1 、R1 及n表示與前述同意義], (ii)使步驟(i)中所獲得的通式(XXIV)所表示的化合物或其鹽,於溶媒中、鹼及觸媒存在下,與活化劑反應的步驟,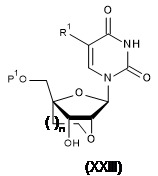 [式中,P1 、R1 及n表示與前述同意義,Z3 表示脂肪族醯基或芳香族醯基], (iii)接著,藉由使與胺基化劑反應,而獲得通式(XXV)所表示的化合物或其鹽的步驟,
[式中,P1 、R1 及n表示與前述同意義,Z3 表示脂肪族醯基或芳香族醯基], (iii)接著,藉由使與胺基化劑反應,而獲得通式(XXV)所表示的化合物或其鹽的步驟,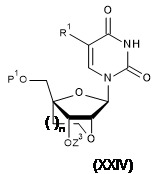 [式中,P1 、R1 、Z3 及n表示與前述同意義]。
[式中,P1 、R1 、Z3 及n表示與前述同意義]。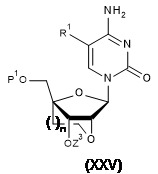
- 如請求項87之方法,其中P1 為三苯甲基。
- 如請求項87或88之方法,其中Z3 為乙醯基。
- 如請求項87至89中任一項之方法,其中n為1。
- 如請求項87至90中任一項之方法,其中R1 為甲基或氫原子。
- 如請求項87至91中任一項之方法,其中R6 為乙醯基或苄醯基。
- 如請求項87至91中任一項之方法,其中R6 為苄醯基。
- 如請求項87至93中任一項之方法,其包含: (iv)使步驟(iii)中所獲得的通式(XXV)所表示的化合物或其鹽,於溶媒中,與醯化劑反應的步驟,[式中,P1 、R1 、Z3 及n表示與前述同意義], (v)使步驟(iv)中所獲得的通式(XXVI)所表示的化合物或其鹽、與羥基之去保護試藥反應,僅將Z3 去保護,藉此獲得通式(XXII)所表示的化合物或其鹽的步驟,
 [式中,P1 、R1 、R6 、Z3 及n表示與前述同意義],
[式中,P1 、R1 、R6 、Z3 及n表示與前述同意義],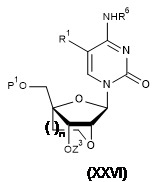 [式中,P1 、R1 、R6 及n表示與前述同意義]。
[式中,P1 、R1 、R6 及n表示與前述同意義]。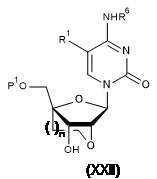
- 如請求項87至94中任一項之方法,其中觸媒為N,N-二甲基胺基吡啶或1,8-二氮雜雙環[5.4.0]十一-7-烯。
- 如請求項87至95中任一項之方法,其中活化劑為對甲苯磺醯氯或2,4,6-三異丙基苯磺醯氯。
- 如請求項87至96中任一項之方法,其中胺基化劑為氨、氨水溶液、碳酸銨或乙酸銨。
- 如請求項94至97中任一項之方法,其中醯化劑為苯甲醯氯或苯甲酸酐。
- 一種製造通式(XXVIII)所表示的化合物或其鹽之方法,其使通式(XXVII)所表示的化合物或其鹽,於溶媒中,活化劑及乾燥劑之存在下,與AMIDITE化試藥反應,[式中,P1 、B及n表示與下述同意義],
 [式中,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,B表示可具有選自下述α群組的1或2個以上之取代基的2-側氧基-嘧啶-1-基或嘌呤-9-基,n表示0至4之整數], (α群組) 羥基、 經保護的羥基、 低級烷氧基、 巰基、 經保護的巰基、 低級烷硫基、 胺基、 經保護的胺基、 低級烷基胺基、 低級烷基、及 鹵素原子。
[式中,P1 表示可經1至3個之低級烷氧基取代的三苯甲基,B表示可具有選自下述α群組的1或2個以上之取代基的2-側氧基-嘧啶-1-基或嘌呤-9-基,n表示0至4之整數], (α群組) 羥基、 經保護的羥基、 低級烷氧基、 巰基、 經保護的巰基、 低級烷硫基、 胺基、 經保護的胺基、 低級烷基胺基、 低級烷基、及 鹵素原子。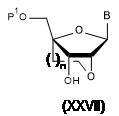
- 如請求項99之方法,其中P1 為4,4’-二甲氧基三苯甲基。
- 如請求項99或100之方法,其中B為2-側氧基-4-羥基-5-甲基嘧啶-1-基、胺基經保護的2-側氧基-4-胺基-嘧啶-1-基、胺基經保護的4-胺基-5-甲基-2-側氧基-嘧啶-1-基、胺基經保護的6-胺基嘌呤-9-基或胺基經保護的2-胺基-6-羥基嘌呤-9-基。
- 如請求項99或100之方法,其中B為2-側氧基-4-羥基-5-甲基嘧啶-1-基、2-側氧基-4-苄醯基胺基-嘧啶-1-基、4-苄醯基胺基-5-甲基-2-側氧基-嘧啶-1-基、6-苄醯基胺基嘌呤-9-基、2-異丁醯基胺基-6-羥基嘌呤-9-基。
- 如請求項99至102中任一項之方法,其中n為1。
- 如請求項99至103中任一項之方法,其中活化劑為吡啶三氟乙酸鹽、N-甲基咪唑三氟乙酸鹽、N-異丙基咪唑三氟乙酸鹽、5-苄硫基四唑、5-苯基四唑、4,5-二氰基咪唑或2,4,5-四溴咪唑。
- 如請求項99至103中任一項之方法,其中活化劑為4,5-二氰基咪唑。
- 如請求項99至105中任一項之方法,其中AMIDITE化試藥為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺(2-cyanoethyl N, N, N’, N’-tetraisopropyl phosphorodiamidite)或2-氰基乙基二異丙基氯亞磷醯胺(2-cyanoethyl diisopropyl chlorophosphoroamidide)。
- 如請求項99至105中任一項之方法,其中AMIDITE化試藥為2-氰基乙基N,N,N’,N’-四異丙基亞磷醯二胺。
- 如請求項99至107中任一項之方法,其中乾燥劑為分子篩3A、分子篩4A、或分子篩5A。
- 一種寡核苷酸之製造方法,其包含以下之步驟: (A)藉由如請求項99至108中任一項之方法,合成ENA單體的步驟,及 (B)使用步驟(A)中所獲得的ENA單體、其他之核酸之亞磷醯胺化合物及/或配位體之亞磷醯胺化合物,依據所期望的序列而使核苷酸鏈延長的步驟。
- 如請求項109之方法,其中寡核苷酸為包含選自以下之DMD AO01~DMD AO15的任一者之式所表示的序列, (DMD AO01) HO-Ce2s -Am1s -Gm1s -Te2s -Te2s -Um1s -Gm1s -Ce2s -Ce2s -Gm1s -Ce2s -Te2s -Gm1s -Ce2s -Ce2s -Ce2s -Am1s -Am1s - CH2 CH2 OH(序列識別號1) (DMD AO02) HO-Te2s -Gm1s -Te2s -Te2s -Ce2s -Te2s -Gm1s -Am1s -Ce2s -Am1s -Am1s -Ce2s -Am1s -Gm1s -Te2s -Te2s -Te2s -Gm1s - CH2 CH2 OH(序列識別號2) (DMD AO03) HO-Ce2s -Gm1s -Ce2s -Te2s -Gm1s -Cm1s -Ce2s -Ce2s -Am1s -Am1s -Te2s -Gm1s -Ce2s -Ce2s -Am1s -Um1s -Ce2s -Ce2s - CH2 CH2 OH(序列識別號3) (DMD AO04) HO-Ce2s -Am1s -Te2s -Am1s -Am1s -Te2s -Gm1s -Am1s -Ae2s -Am1s -Am1s -Ce2s -Gm1s -Cm1s -Ce2s -Gm1s -Ce2s -Ce2s - CH2 CH2 OH(序列識別號4) (DMD AO05) HO-Te2s -Um1s -Ce2s -Cm1s -Ce2s -Am1s -Am1s -Te2s -Um1s -Cm1s -Te2s -Ce2s -Am1s -Gm1s -Gm1s -Ae2s -Am1s -Te2s - CH2 CH2 OH(序列識別號5) (DMD AO06) HO-Ce2s -Ce2s -Am1s -Um1s -Te2s -Um1s -Gm1s -Te2s -Am1s -Um1s -Te2s -Te2s -Am1s -Gm1s -Ce2s -Am1s -Te2s -Gm1s - CH2 CH2 OH(序列識別號6) (DMD AO07) HO-Gm1s -Gm1s -Ce2s -Te2s -Gm1s -Cm1s -Te2s -Te2s -Um1s -Gm1s -Ce2s -Cm1s -Cm1s -Te2s -Ce2s -Am1s -Gm1s -Ce2s -CH2 CH2 OH(序列識別號7) (DMD AO08) HO-Gm1s -Ce2s -Te2s -Am1s -Gm1s -Gm1s -Te2s -Ce2s -Am1s -Gm1s -Gm1s -Te2s -Gm1s -Cm1s -Te2s -Te2s -Um1s -CH2 CH2 OH(序列識別號8) (DMD AO09) HO-Am1s -Ce2s -Ce2s -Gm1s -Cm1s -Ce2s -Te2s -Um1s -Cm1s -Ce2s -Am1s -Cm1s -Te2s -Ce2s -Am1s -Gm1s -Ae2s -Gm1s -CH2 CH2 OH; (序列識別號9) (DMD AO10) HO-Ge2s -Ge2s -Ce2s -Ae2s -Te2s -Um1s -Um1s -Cm1s -Um1s -Am1s -Gm1s -Um1s -Um1s -Te2s -Ge2s -Ge2s -Ae2s -Ge2s - CH2 CH2 OH(序列識別號10) (DMD AO11) HO-Gm1s -Gm1s -Ce2s -Am1s -Te2s -Te2s -Um1s -Ce2s -Te2s -Am1s -Gm1s -Um1s -Te2s -Te2s -Gm1s -Gm1s -Ae2s -Gm1s - CH2 CH2 OH(序列識別號11) (DMD AO12) HO-Ae2s -Gm1s -Te2s -Um1s -Te2s -Gm1s -Gm1s -Ae2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Ae2s -Gm1s -Te2s -Te2s - CH2 CH2 OH(序列識別號12) (DMD AO13) HO-Ce2s -Te2s -Cm1s -Ce2s -Te2s -Um1s -Ce2s -Ce2s -Am1s -Te2s -Gm1s -Am1s -Ce2s -Te2s -Ce2s -Am1s -Am1s -Gm1s - CH2 CH2 OH(序列識別號13) (DMD AO14) HO-Ce2s -Te2s -Gm1s -Am1s -Am1s -Gm1s -Gm1s -Te2s -Gm1s -Te2s -Te2s -Ce2s -Te2s -Te2s -Gm1s -Te2s -Am1s -Ce2s - CH2 CH2 OH(序列識別號14) (DMD AO15) HO-Te2s -Te2s -Cm1s -Ce2s -Am1s -Gm1s -Ce2s -Ce2s -Am1s -Te2s -Te2s -Gm1s -Te2s -Gm1s -Te2s -Te2s -Gm1s -Am1s -CH2 CH2 OH(序列識別號15) [於上述式中,左側表示5’末端,右側表示3’末端,A、G、C、U及T各自表示D-核呋喃糖被修飾且5’位之碳原子與左側所表示的結構單元進行硫代磷酸酯(phosphorothioate)鍵結而成的腺苷、鳥苷、胞苷、尿苷及胸苷;附於各核苷酸或核苷的e2s係表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯且3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,e2t表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯且3’位以-O-與3’末端之氫原子鍵結,m1s表示D-核呋喃糖被2’-O-甲基化且3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,m1t表示D-核呋喃糖被2’-O-甲基化,且3’位以-O-與3’末端之氫原子鍵結]。
- 如請求項109之方法,其中寡核苷酸包含選自以下之GSD AO01~GSD AO16的任一者之式所表示的序列,配位體為以下述式之X18 或X20 所表示者: (GSD AO01) X18 -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1t -H(序列識別號16) (GSD AO02) X18 -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2t -H(序列識別號17) (GSD AO03) X18 -Um1s -Cm1s -Ce2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2s -Um1t -H(序列識別號18) (GSD AO04) X18 -Am1s -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1t -H(序列識別號19) (GSD AO05) X18 -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2t -H(序列識別號20) (GSD AO06) X18 -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Am1s -Te2s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2s -Um1t -H(序列識別號21) (GSD AO07) X18 -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1t -H(序列識別號22) (GSD AO08) X18 -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2t -H(序列識別號23) (GSD AO09) X18 -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2s -Um1t -H(序列識別號24) (GSD AO10) X18 -Am1s -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1t -H(序列識別號25) (GSD AO11) X18 -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2t -H1(序列識別號26) (GSD AO12) X18 -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2s -Um1t -H(序列識別號27) (GSD AO13) X20 -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1t -H(序列識別號28) (GSD AO14) X20 -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2t -H(序列識別號29) (GSD AO15) X20 -Am1s -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1t -H(序列識別號30) (GSD AO16) X20 -Am1s -Ae2s -Um1s -Cm1s -Ce2s -Gm1s -Ae2s -Um1s -Gm1s -Gm1s -Ce2s -Gm1s -Am1s -Ae2s -Gm1s -Ce2t -H(序列識別號31) [於上述式中,左側表示5’末端,右側表示3’末端,A、G、C、U及T各自表示D-核呋喃糖經修飾且5’位之碳原子與左側所表示的結構單元以硫代磷酸酯鍵結而成的腺苷、鳥苷、胞苷、尿苷及胸苷;附於各核苷酸或核苷的e2s表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯,3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷的5’位碳原子鍵結,e2t表示D-核呋喃糖經2’-O,4’-C-伸乙基交聯,3’位以-O-與3’末端之氫原子鍵結,m1s表示D-核呋喃糖經2’-O-甲基化,3’位以-OP(=S)(-OH)-O-與右側鄰接的核苷酸或核苷之5’位碳原子鍵結,m1t表示D-核呋喃糖經2’-O-甲基化,3’位以-O-與3’末端之氫原子鍵結; 於上述式中,X18 及X20 表示下述之式所表示的GalNAc單元;於下述之式中,與磷酸基鍵結的鍵結肢係表示與寡核苷酸之5’末端的碳原子鍵結,而形成磷酸二酯鍵]
 。
。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019191053 | 2019-10-18 | ||
| JP2019-191053 | 2019-10-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| TW202128704A true TW202128704A (zh) | 2021-08-01 |
Family
ID=75538087
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109135908A TW202128704A (zh) | 2019-10-18 | 2020-10-16 | 雙環亞磷醯胺之製造方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20230348522A1 (zh) |
| EP (1) | EP4047005A4 (zh) |
| JP (1) | JP7641905B2 (zh) |
| KR (1) | KR20220083707A (zh) |
| CN (1) | CN114502565A (zh) |
| BR (1) | BR112022007437A2 (zh) |
| CA (1) | CA3155028A1 (zh) |
| IL (1) | IL292286A (zh) |
| TW (1) | TW202128704A (zh) |
| WO (1) | WO2021075538A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL291251B1 (en) * | 2019-09-10 | 2026-02-01 | Daiichi Sankyo Co Ltd | Oligonucleotide-galnac conjugate for targeted delivery to the liver and method for its production |
| CN116178196B (zh) * | 2021-11-26 | 2025-07-25 | 中国科学院大连化学物理研究所 | 一种烯烃和胺类多相催化制酰胺类化合物的方法 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US62519A (en) * | 1867-03-05 | Improved peat machine | ||
| JPH0787982A (ja) | 1993-01-29 | 1995-04-04 | Sankyo Co Ltd | 修飾オリゴデオキシリボヌクレオチド |
| JP3756313B2 (ja) | 1997-03-07 | 2006-03-15 | 武 今西 | 新規ビシクロヌクレオシド及びオリゴヌクレオチド類縁体 |
| US6096881A (en) | 1997-05-30 | 2000-08-01 | Hybridon, Inc. | Sulfur transfer reagents for oligonucleotide synthesis |
| JP4236812B2 (ja) | 1997-09-12 | 2009-03-11 | エクシコン エ/エス | オリゴヌクレオチド類似体 |
| ES2234563T5 (es) * | 1999-02-12 | 2018-01-17 | Daiichi Sankyo Company, Limited | Nuevos análogos de nucleósidos y oligonucleótidos |
| US6525191B1 (en) * | 1999-05-11 | 2003-02-25 | Kanda S. Ramasamy | Conformationally constrained L-nucleosides |
| JP2001247596A (ja) * | 2000-03-07 | 2001-09-11 | Univ Nagoya | オリゴヌクレオチド糖コンジュゲート、オリゴヌクレオチド糖コンジュゲートの製造方法 |
| JP2002322192A (ja) * | 2000-08-10 | 2002-11-08 | Sankyo Co Ltd | 2’−o,4’−c−架橋ヌクレオシドトリリン酸体 |
| ATE287413T1 (de) * | 2001-07-12 | 2005-02-15 | Santaris Pharma As | Verfahren zur herstellung des lna phosphoramidite |
| EP1481983A4 (en) * | 2002-02-13 | 2008-04-02 | Takeshi Imanishi | NUCLEOSIDE ANALOGUE AND OLIGONUCLEOTIDE DERIVATIVES CONTAINING A NUCLEOTIDE ANALOGON THEREOF |
| EP2392660A3 (en) * | 2002-11-25 | 2012-03-28 | Masafumi Matsuo | ENA Nucleic Acid Drugs Modifying Splicing in mRNA Precursor |
| JP4616175B2 (ja) * | 2003-09-02 | 2011-01-19 | 株式会社キラルジェン | 5’−ホスフィチル化モノマーおよびh−ホスホネートオリゴヌクレオチド誘導体の製造方法 |
| DE19216461T1 (de) * | 2011-10-03 | 2021-10-07 | Modernatx, Inc. | Modifizierte nukleoside, nukleotide und nukleinsäuren und verwendungen davon |
| EP2862868B1 (en) | 2012-06-18 | 2020-04-08 | Daiichi Sankyo Company, Limited | Intermediate for production of nucleoside analogue, and method for producing same |
| WO2018049534A1 (en) * | 2016-09-16 | 2018-03-22 | Lcb Pharma Inc. | Nucleoside and nucleotide analogues bearing a quaternary all-carbon stereogenic center at the 2' position and methods of use as a cardioprotective agent |
| KR102398295B1 (ko) * | 2018-03-09 | 2022-05-17 | 다이이찌 산쿄 가부시키가이샤 | 당원병 Ia형 치료약 |
| WO2019224172A1 (en) * | 2018-05-25 | 2019-11-28 | Roche Innovation Center Copenhagen A/S | Novel process for making allofuranose from glucofuranose |
-
2020
- 2020-10-16 IL IL292286A patent/IL292286A/en unknown
- 2020-10-16 CA CA3155028A patent/CA3155028A1/en active Pending
- 2020-10-16 CN CN202080072429.5A patent/CN114502565A/zh active Pending
- 2020-10-16 TW TW109135908A patent/TW202128704A/zh unknown
- 2020-10-16 EP EP20877640.1A patent/EP4047005A4/en active Pending
- 2020-10-16 US US17/769,483 patent/US20230348522A1/en active Pending
- 2020-10-16 WO PCT/JP2020/039050 patent/WO2021075538A1/ja not_active Ceased
- 2020-10-16 JP JP2021552461A patent/JP7641905B2/ja active Active
- 2020-10-16 BR BR112022007437A patent/BR112022007437A2/pt not_active Application Discontinuation
- 2020-10-16 KR KR1020227012762A patent/KR20220083707A/ko not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2021075538A1 (zh) | 2021-04-22 |
| EP4047005A4 (en) | 2024-03-06 |
| BR112022007437A2 (pt) | 2022-10-11 |
| IL292286A (en) | 2022-06-01 |
| CN114502565A (zh) | 2022-05-13 |
| CA3155028A1 (en) | 2021-04-22 |
| EP4047005A1 (en) | 2022-08-24 |
| JP7641905B2 (ja) | 2025-03-07 |
| KR20220083707A (ko) | 2022-06-20 |
| US20230348522A1 (en) | 2023-11-02 |
| WO2021075538A1 (ja) | 2021-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8859755B2 (en) | Method for preparing ribonucleoside phosphorothioate | |
| AU737902B2 (en) | Anti-viral pyrimidine nucleoside analogues | |
| AU780879B2 (en) | Anti-viral pyrimidine nucleoside analogues | |
| CN108137638B (zh) | 桥连型核酸GuNA、其制造方法及中间体化合物 | |
| JP2025138846A (ja) | オリゴヌクレオチドの収束液相合成 | |
| WO2001007455A1 (en) | Novel bicyclonucleoside analogues | |
| EP1745573A2 (en) | METHODS OF MANUFACTURE OF 2 -DEOXY-&bgr;-L-NUCLEOSIDES | |
| WO2009143369A2 (en) | Method of preparing nucleosides and analogs thereof without using chromatography | |
| CN116003496A (zh) | 经修饰的mRNA5`-帽类似物 | |
| MXPA05006865A (es) | Proceso para la produccion de profarmacos 3'-nucleosidos. | |
| US20170073368A1 (en) | Synthesis of bicyclic nucleosides | |
| JP2019508438A (ja) | 核配列に組み込むための5−(n−保護−トリプタミノカルボキシアミド)−2’−デオキシウリジンホスホロアミダイトの化合物及び合成方法 | |
| TW202128704A (zh) | 雙環亞磷醯胺之製造方法 | |
| JP7478660B2 (ja) | 修飾ヌクレオシドホスホロアミダイト | |
| US20040033967A1 (en) | Alkylated hexitol nucleoside analogues and oligomers thereof | |
| CN116178468A (zh) | 5’s-lna核苷酸和寡核苷酸 | |
| CN115151555B (zh) | 桥连型核苷和使用了该桥连型核苷的核苷酸 | |
| Czechtizky et al. | Oligonucleotide Analogues with a Nucleobase‐Including Backbone:, Part 6, 2‐Deoxy‐D‐erythrose‐Derived Phosphoramidites: Synthesis and Incorporation into 14‐Mer DNA Strands | |
| KR20070073958A (ko) | 디플루오로뉴클레오시드 및 이의 제조 방법 | |
| HK40078619A (zh) | 制造二环亚磷酰胺的方法 | |
| JP7818760B2 (ja) | ヌクレオシド誘導体の製造方法 | |
| EP0850248B1 (en) | Compositions and methods for the synthesis of organophosphorus derivatives | |
| TW202434611A (zh) | 合成經靶向配體結合之核苷酸亞磷醯胺之方法 | |
| KR20240131337A (ko) | 폴리뉴클레오티드 제조 방법 | |
| BR112020005067B1 (pt) | Fosforamiditas nucleosídeo modificadas, composição, e métodos para produzir um oligonucleotídeo |