WO1989010341A1 - Procede de production d'un compose d'organofluor - Google Patents
Procede de production d'un compose d'organofluor Download PDFInfo
- Publication number
- WO1989010341A1 WO1989010341A1 PCT/JP1989/000020 JP8900020W WO8910341A1 WO 1989010341 A1 WO1989010341 A1 WO 1989010341A1 JP 8900020 W JP8900020 W JP 8900020W WO 8910341 A1 WO8910341 A1 WO 8910341A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- reaction
- hydrogen fluoride
- temperature
- alumina
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/63—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B39/00—Halogenation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/07—Preparation of halogenated hydrocarbons by addition of hydrogen halides
- C07C17/087—Preparation of halogenated hydrocarbons by addition of hydrogen halides to unsaturated halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/093—Preparation of halogenated hydrocarbons by replacement by halogens
- C07C17/20—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms
- C07C17/202—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms two or more compounds being involved in the reaction
- C07C17/206—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms two or more compounds being involved in the reaction the other compound being HX
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/093—Preparation of halogenated hydrocarbons by replacement by halogens
- C07C17/20—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms
- C07C17/21—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms with simultaneous increase of the number of halogen atoms
Definitions
- the present invention relates to a method for producing an organic fluorine compound.
- the organic fluorine compound obtained by the method of the present invention can be used for applications such as a refrigerant, an aerosol propellant, a foaming agent, and a detergent. Background art
- chromium (I) compounds such as chromium chloride ( ⁇ ), chromium fluoride (I), and chromium fluoride It is well known to use chromium fluoride obtained by reacting oxygen with chromium or chromium oxide or hydroxide with hydrogen fluoride.
- the latter is the power synthesized by the fluorination reaction of 1-color ⁇ -2,2,2—triflerged roetane '; JP-A-43-10601, JP-A-53-105404 and JP-A-56-24050 disclose the use of a chromium-based catalyst in such cases.
- a catalyst for synthesizing an organic fluorine compound by a gas phase reaction a catalyst other than a chromium catalyst is also known, but as can be seen from the above example, a chromium catalyst is frequently used in practical use.
- an object of the present invention is to provide a method for producing an organic fluorine compound in a gas phase using a fluorination catalyst which does not have the above-mentioned problems and has excellent conversion and productivity. . Disclosure of the invention
- One aspect of the present invention is to use an active alumina having a uniform pore distribution as a catalyst carrier, and to use a metal fluoride selected from nickel, cobalt, iron, manganese, chromium, copper and silver as a catalyst component.
- activated alumina occupying 70 to 90% of pores having an average pore diameter of 40 to 500 persons is used as a catalyst carrier, and nickel, Organochlorine or unsaturated compounds and fluoride water in the presence of a catalyst carrying at least one metal fluoride selected from cobalt, iron, manganese, chromium, copper and silver
- a method for producing an organic fluorine compound characterized by reacting with an element.
- the active alumina useful as a carrier for the catalyst used in the method of the present invention will be described in detail.
- the activated alumina is a hydrated alumina, which has many amorphous (amorphous) parts according to X-ray diffraction, and has little progress in crystallization, such as boehmite or pseudoboehmite or the like.
- Intermediate aluminas such as, 6,,, ⁇ , ⁇ , ⁇ , and ⁇ , which are also fired products, have been identified.
- Activated alumina is usually produced by thermal decomposition of alumina hydrate, ie, controlled heating of the hydrate to remove most of the water.
- hydrate raw materials include aluminum trihydrate, aluminum salt, alkali aluminate, aluminum alcohol, and metal aluminum produced by the Bayer method. If Shimese as a hydrate, Jibusai door, Ba Yi turbocharger La wells, Bruno Le Dos door run-die door (more than A £ (0H) 3), Bemai door, die Asuboa (more than A £ ⁇ 0 3 ⁇ H 20 or A £ 00H).
- alumina hydrates are produced alone or as a mixture depending on conditions such as the ability to form an alumina gel, the temperature, pH, time, and the raw material concentration. Each of these hydrates decomposes into a different arna phase, and accordingly C 'with different pore structure, surface area and physical properties.
- the methods for producing activated alumina include (A) heating and dehydration of alumina hydrates such as dibsite and boehmite, and (B) aqueous sodium aluminate and aqueous aluminum sulfate or carbon dioxide or alumina.
- alumina hydrate obtained by the reaction between sodium acid powder and sulfur dioxide gas, and dehydration of aqueous solution of (C) aluminum salt (for example, aluminum sulfate, aluminum nitrate, aluminum chloride).
- C) aluminum salt for example, aluminum sulfate, aluminum nitrate, aluminum chloride.
- ⁇ Alumina is obtained by subjecting the obtained jibsite to about 400 in air stream. It is activated by heat treatment, and is X7-alumina that contains a small amount of boehmite in X-rays. It has a specific surface area of about 250 mg.g, a pore diameter distribution of 35 A to 10 ⁇ m, and a large number of small pores with a pore diameter of 35 mm or less. You.
- the pores consist of hollow holes such as cylindrical and spherical.
- This pore size distribution is usually measured using an Hg-one porosimeter of 50,000 psi (measuring the pore diameter of 35 A to 177: 1: 1). Also 0 compositionally to ⁇ and S i 0 2 and F e 2 0 3 of about 0.9% of N a z 0 hundreds ppm, and the content of N a 2 0 0. 05% or less Difficult to do.
- Activated alumina can also be obtained by rapidly activating the alumina hydrate of the buyer method at 400 to 800. In this process, the formation of boehmite and decomposition products is significantly reduced.
- T) The pattern of T-lumina is weak, and it is possible to manufacture alumina that is almost amorphous. By agglomerating or rehydrating this alumina, various forms such as spheres can be obtained, and those having crystallites and pore sizes smaller than the former are produced.
- the specific surface area is 300-350 / z.
- the activated alumina thus obtained has a pore volume of 0.3 to 0.8 / g, a specific surface area of 150 to 350 nf / g, and an average pore diameter of about 40 to 150 mm.
- the range is wide, and if the pore size is limited to 40 to 500 A, only about 20% is distributed.
- the activated alumina used in the catalyst of the present invention those having a pore diameter of 40 to 500 A concentrated in a range of 70 to 90% are suitable. Those having a pore size of 40 A or less and having a concentrated distribution are difficult to obtain industrially, and when used as a catalyst carrier, the activity is improved, but the selectivity is poor and there is concern about the catalyst life.
- the pore size is 500 A and the pore distribution is concentrated, when used as a carrier, sufficient catalytic activity cannot be obtained, and the yield is low, which is not preferable.
- the pore volume of activated alumina is determined by the specific surface area and the pore size distribution. If the value is 0.55 ⁇ g or less, an appropriate specific surface area and pore structure cannot be obtained, and the catalytic activity is low. Yield may be poor. Therefore, in the activated alumina used in the present invention, the volume of pores having a pore diameter of 40 A or more is preferably 0.55 fl2 fig or more, particularly preferably 0.55 to: 1.6 ⁇ g.
- An example of an industrial production method of an activated alumina that can be particularly preferably used in the present invention is a Sol-gel Z oil-mouthed topping method (similar to the above-mentioned method C).
- the bulk density, specific surface area, pore volume, pore diameter, and pore size of the active alumina to be generated are adjusted by adjusting the chemical properties of the spherical sol or changing the chemical operation of the sol-gel.
- the pore distribution can be adjusted arbitrarily.
- a preferred activated alumina is one in which trialkylalumina is decomposed into aluminum monohydrate (similar to the above-mentioned method E), and then this alumina hydrate is calcined into r-alumina. Press or extrude this alumina to make pellets. As a result, it is possible to obtain a material with higher purity than alumina produced in nature and uniform pore size and good pore size distribution.
- Active alumina used for such a catalyst carrier is commercially available, and an active alumina having 70 to 90% of pores having an average pore diameter of 40 to 500 A may be selected.
- This active alumina is used as a catalyst in the form of particles, beads or extrudates of less than 20 MI in diameter, preferably a few mm, for convenient handling during filling and unloading of the reactor. It is better to
- the content of sodium should be 100 ppm or less, preferably as small as possible. Further, it is preferable to select an activated alumina in which silicon is less than 300 ppm and iron is less than 100 ppm, and the purity of alumina is preferably 99.9% or more.
- silicon, silicon and iron are present in the carrier as oxides, they have an activity inhibitory effect, a reaction inhibitory effect such as the formation of silicon fluoride by hydrofluoric anhydride, an isomerization or a disproportionation reaction. Therefore, it is preferable not to exist in the catalyst as much as possible.
- Examples of the metal constituting the catalyst component used in the present invention include nickel, cobalt, iron, manganese, chromium, copper and silver.
- these metal sources inorganic or organic salts of each metal can be suitably used.
- nickel chloride, nickel nitrate, nickel sulfate, etc. which are particularly water-soluble, can be suitably used. Similar salts or compounds can be used for other cobalt, iron, manganese, chromium, copper, silver, etc. as long as they are practically available.
- water-soluble ones such as chlorides, nitrates and sulfates can be suitably used for these metals.
- activated alumina is soaked in an aqueous solution of a metal salt, dried and calcined, and then treated with hydrogen fluoride, or the metal salt is reduced to metal with hydrogen and then treated with hydrogen fluoride.
- the method is also applicable. That is, after the above-mentioned metal or its compound is supported on a carrier, this is reacted with hydrogen fluoride in a gas phase to convert at least a part of the metal into fluoride (hydrofluoric acid treatment). Is good.
- the treatment with the hydrogen fluoride may not be necessary in some cases.
- the above-mentioned active alumina is immersed in an aqueous solution of a metal salt or activated alumina is absorbed in the entire amount of a metal salt aqueous solution corresponding to the amount of water absorbed, dried with hot air, and then air-flowed. Baking.
- Drying is preferably carried out at a temperature of 50 to 120 ° C, and calcination is carried out under oxygen or an inert gas containing oxygen, preferably under a stream of air. [SV]. (Space velocity in terms of 0 and 1 atm) 100-1000hr- It is preferable to flow air in the range of 200-500, and it is preferable to carry out in the temperature range of 200-500 hours. After drying the activated alumina supporting the metal salt, When calcination is performed under air flow, rapid heat generation occurs at a temperature of 150 ° C or higher. Therefore, it is preferable to control the temperature and maintain the catalyst temperature at 500 ° C or lower.
- the catalytic activity is low, and when the calcination temperature is 500 ° C or higher, the catalytic effective surface area decreases (so-called sintering occurs), so that the catalytic activity also decreases.
- the amount of metal in the catalyst can be arbitrarily changed, but is preferably in the range of 5 to 30% by weight in consideration of the activity and the limit of the amount of metal supported.
- the metal oxide-alumina catalyst prepared in this manner was used before the treatment for hydrogen fluoride treatment.
- Hydrofluoric anhydride concentration 10 to: 100% (If less than 100%, adjust by diluting hydrofluoric anhydride with inert gas)
- hydrofluoric anhydride hydrogen hydride: HF
- the temperature be sufficiently controlled and the catalyst temperature be 450 or less.
- the reaction between the organochlorine compound and hydrogen fluoride in the present invention involves the following reactions.
- the major atomic group forming an organic chlorine compound CH 2 C £, CHC £, CCi .. CHC ⁇ 2: CC £ ⁇ , and CC £ 3 The basic reaction with hydrogen fluoride (HF)
- Representative organic chlorine compounds include carbon tetrachloride, chlorophorem, and salt.Dragon methylene, hexacrocretan, pentachloretane, tetrachlorene, and ⁇ ⁇ ⁇ ⁇ ⁇ ⁇ ⁇ Hex corp.Corp mouth pan, pen crepe Pronodin, Tetrachloronoethylene, Tricyclonoethylene, Hexacroleno 'mouth pen, Penta pi Zolef "" Mouth pen, Tetrakuronolepropene, Trichloronolepropene, Hexachloronolebutadiene, Hexachloronolebutene, Pentachlorbutene, Tetrachloronobutene, Pentachlorbutane, Tetrachloronolebutane, Benzotricrine Chlorinated benzotrichloride, chlorinated benzotrichloride, trichlormethyl Benzotrichloride, trichlorme Ether
- the organic fluorine compound obtained by the method of the present invention refers to a substance in which some or all of the chlorine atoms of the above-mentioned organic chlorine compounds have been replaced by fluorine atoms, and the fluorine by elemental fluorine gas is used.
- the number of carbon atoms in the basic skeleton is not significantly increased or decreased in the oxidation, and in principle, in the method of the present invention, the basic structure of a given compound is maintained before and after the reaction, and chlorine atoms are fluorinated. Mainly, changes in molecular weight and properties due to substitution by elemental atoms are observed.
- the method of the present invention can also be advantageously used for a fluorination reaction of an organic unsaturated compound. Tetrachlorethylene and trichloroethylene as exemplified above are also examples of organic unsaturated compounds.For example, in the reaction of trichloroethylene with hydrogen fluoride,
- the organic fluorine compound encompasses a compound generated by the addition of hydrogen fluoride to an organic unsaturated compound.
- reaction types that can be used in the production of an organic fluorine compound by gas phase fluorination of an organic chlorine compound by the method of the present invention include a fixed catalyst bed type and a moving bed type. it can.
- the reaction temperature is between 100 and 600, preferably between 150 and 550 * C.
- the reaction rate is slower, and at higher temperatures, side reactions such as dehydrofluoric acid, disproportionation, isomerization, and coke formation reaction occur at the same time, and the selectivity is remarkably deteriorated, making it impractical.
- the reaction pressure may be any pressure as long as the gaseous phase can be maintained. In general, the pressure is 1 to 10 atm. However, considering operability and productivity, the pressure is particularly at or around atmospheric pressure. I like it. Space velocity [SV; 50 ⁇ 2000H r is - ', that are proportion from 0.5 to 2 0 moles of full Tsu hydrogen to organic chlorine compounds 1 molar No.
- the ratio of hydrogen fluoride is important in determining the selectivity of the reaction, and it is preferable to select the ratio appropriately in consideration of the ratio of substitution of chlorine or fluorine in the organic chlorine compound.
- HF molar ratio the molar ratio of hydrogen fluoride to the organic chlorine compound and Z or unsaturated compound as the raw material.
- Trichloroethylene is used as an organic unsaturated compound, and is reacted at a temperature of 200 to 450 at a molar ratio of hydrogen fluoride to trichloroethylene of 3 to 10 to obtain 11-chloro-2, Produce 2,2—trifluoroethane (F-133a).
- the reaction is carried out at 10 to produce 1> 2, 2, 2 — tetrafluorobenzene (F-134a).
- the productivity of the catalyst deteriorates, which is not preferable.
- the HF molar ratio should be higher than the stoichiometric ratio, but if it is 6 or higher, 1,1,1,1,2—tetrafluorochloro-loroethane (F-124), 1,1,1,2,2-penta Since the generation of Fluorotan (F-125) takes precedence, the F-123 generation selectivity is poor, which is not desirable.
- 1,1,1-tricyclomethane as an organochlorine compound at a temperature of 150 to 400'C
- the reaction is carried out at 1 to 4 to produce 1,1,1 -dichlorofluorene (F-141) and 1, or 1,1,1-difluorochloroethane (F-142b).
- F-141 1,1,1 -dichlorofluorene
- F-142b 1,1,1-difluorochloroethane
- the HF molar ratio is higher than the stoichiometric ratio. However, when the HF molar ratio is 4 or higher, the formation of 1, 1, 1-1. rifluorethane (F-143a) takes precedence. The generation selectivity is poor and unfavorable No.
- Difluoromethane (F—22) is prepared by reacting with chloroform at a temperature of 150 to 350 ° C and a molar ratio of hydrogen fluoride to chloroform of 0.5 to 3, using chloroform as the organochlorine compound. To manufacture. At a reaction temperature of 150 ° C or lower, the conversion is remarkably low, and at a reaction temperature of 350 ° C or higher, the selectivity is extremely deteriorated, and neither is preferred. When the HF molar ratio is less than 0.5, the productivity of the catalyst is deteriorated, which is not preferable. On the other hand, when the HF molar ratio is 3 or more, the formation of trifluormethane (F-23) takes precedence, and the F—22 formation selectivity becomes poor, which is not preferable.
- the temperature is 200 to 460 (: difluorochloromethane).
- the reaction is carried out at a molar ratio of hydrogen fluoride to tan and trifluoro or trifluoro methane at 0.5 to 3 to produce tetrafluorometan (F-1).
- the conversion is extremely low, and at a reaction temperature of 460 ° C. or higher, side reactions such as decomposition occur, and the selectivity is extremely deteriorated.
- the HF molar ratio is less than 0.5, the productivity of the catalyst becomes poor, which is not preferable.
- the HF molar ratio is 3 or more, it is not preferable because the recovery and separation of HF from F-14 are complicated and the productivity per catalyst is deteriorated.
- Trifluorochloroethylene (F-113) is used as the organic unsaturated compound, and the temperature is 200-450.
- C 1,1,1,2,2-reacted with a molar ratio of hydrogen fluoride to trifluorochloroethylene of 1 to 5, 1-tetrafluorobenzene (F-124) And or 1, 1, 1, 2, 2 —Pentafluoroethane (F-125) is manufactured.
- F-124 1-tetrafluorobenzene
- F-125 1, 1, 1, 2, 2 —Pentafluoroethane
- a catalyst was prepared using 1.6 high-purity spherical activated alumina (JGC Universal Co., Ltd., NST-7) having the following physical properties.
- This activated alumina was produced by a sol-gel noil dropping method and shows a distribution of about 80% pore volume around a pore diameter of 80.
- the dried catalyst is filled in a SUS container, and air is flowed at [SV] ⁇ hr- 1 and calcined under circulation. After baking until the heat generation of the catalyst layer stops at 200 days, the temperature is further raised to 400'C, and baking is performed for 3 hours.
- This calcined catalyst was filled into a reaction tube (made by Nickel) having an inner diameter of 25 thighs and a height of 1 m.
- Fluoro-2,2,2 Trifluoroethan (CH 2 C-CF 3 , F-133a) hydrofluoric anhydride and 100% hydrofluoric anhydride diluted with nitrogen before the hydrofluoric treatment Fluorination was performed on the catalyst using an acid to activate the catalyst.
- the conditions for treating the catalyst with hydrofluoric anhydride are shown below.
- a gas-phase fluorination reaction of F-133a was performed.
- the product gas exiting the reactor is passed through an alkaline aqueous solution to absorb unreacted hydrofluoric anhydride and the generated HC, then dehydrated in a molecular sieve 3A tower, weighed with a gas meter, and then generated.
- the gas was analyzed by gas chromatography. The analysis was performed after the reaction reached a steady state after passing hydrofluoric anhydride and F-133a through the catalyst.
- a catalyst was prepared using the following high-purity activated alumina. This activated alumina has a pore volume distribution of about 80% around a pore diameter of 270A.
- the preparation method was the same as in Example 1, and the composition was Cr 2 0 3 24% by weight.
- Example 6 shows the results.
- Example 6 shows that the nitric acid chromium Cr (N0 3) 3 ⁇ 9H 2 0 was used as the chromium raw material in the same manner as in Example 1, Cr 2 0 3 12% - A i 2 0 3 88% of A catalyst was prepared.
- the reaction tube was filled with the catalyst 100, and the fluorination reaction of F-133a was carried out under the same conditions as in Example 2 for the hydrogen fluoride treatment and the fluorination reaction. Table 1 shows the results.
- Example 6 shows that the nitric acid chromium Cr (N0 3) 3 ⁇ 9H 2 0 was used as the chromium raw material in the same manner as in Example 1, Cr 2 0 3 12% - A i 2 0 3 88% of A catalyst was prepared.
- the reaction tube was filled with the catalyst 100, and the fluorination reaction of F-133a was carried out under the same conditions as in Example 2 for the hydrogen fluoride treatment and the fluorination
- a continuous production test of F-134a was performed by fluorination of F-133a under the following reaction conditions, using the catalyst of Example 2 and changing the reaction conditions. .
- activated alumina having the following physical properties (manufactured by Sumitomo Chemical Co., Ltd., KHA-24), a catalyst having a composition of Cr 2 0 3 12%-A £ 2 0 3 88% was prepared in the same manner as in Example 1. did.
- the activated alumina shows only a broad pore distribution in the pore diameter range of 0.0 to 500 A, and has about 20% of pores of 40 or less.
- Particle size 2-4 ram A £ z 0 3 houses owned 98.4% by weight or more
- the reaction tube was charged with the catalyst lOOffifi, and the fluorination reaction of F-133a was performed under the same conditions as in Example 1 for the hydrogen fluoride treatment and the fluorination reaction. Table 1 shows the results.
- a catalyst was prepared using 1.6 fraction spherical high-purity active alumina (JST Universal Co., Ltd., NST-7) having the following physical properties.
- This active alumina has a pore volume distribution of about 80% around a pore diameter of 80A.
- reaction temperature 350'C
- reaction pressure 1 atm
- 123hr- ' molar ratio of HF to trichloroethylene (HF molar ratio) 3.4
- flow rates of HF and trichloroethylene were set to 0.425 and 0.125 mol Zhr, respectively.
- sampling was performed to separate the reaction products.
- the exhaust gas is passed through an absorption tower consisting of two layers of aluminum and toluene: the unreacted HF is absorbed and the product is collected and absorbed, and then the ethanol is passed through an ethanol dry ice drip. The product of the absorption was collected.
- the toluene recovery solution containing the product was analyzed by gas chromatography.
- a catalyst was prepared using the following high-purity active materials.
- This activated alumina has a pore volume distribution of about 80% around a pore diameter of 270 people.
- F-133a was synthesized under the same conditions as in Example 8 for the catalytic hydrogen fluoride treatment and the fluorination reaction of trichloroethylene.
- Example 11 Same as Example 11, except that the reaction temperature was 400, and the ethylene molar ratio in the HFZ tri-mouth was 6.5, [SV].
- the fluorination reaction of ethylene was performed at 208 hr- 1 . Table 2 shows the results.
- Activated alumina (Sumitomo Chemical Co. Ltd., KHA-24) having the following physical using Example 8 in the same manner as 12% Cr 2 0 3 - A i z 0 3 catalyst was prepared, full Tsu hydrogen Processed.
- This active alumina shows only a broad pore distribution in the pore diameter range of 40 to 500 A, and has about 20% of pores of 40 A or less.
- Grain 1 ⁇ 2 2-4 meetings A £ 20 3 Household: 98.4% by weight or more
- a catalyst was prepared using a 3.2-row spherical high-purity active alumina (JST Universal Co., Ltd., NST-3) having the following physical properties.
- This active alumina is manufactured by the sol-gel / oil-dropping method and shows a distribution of pore volume of about 80% around a pore diameter of 270A.
- the catalyst Prior to the reaction, the catalyst was partially hydrofluorinated using hydrofluoric anhydride and 100% hydrofluoric acid diluted with nitrogen to activate the catalyst.
- the conditions for treating the catalyst with hydrofluoric anhydride are shown below.
- a gas-phase fluorination reaction of F-133a was carried out.
- the product gas exiting the reactor is passed through an aqueous solution of sodium hydroxide to absorb the unreacted hydrofluoric anhydride and the product, then dehydrated in a molecular sieve 3A tower, weighed with a gas meter, and the product gas The separation was carried out by chromatography. The analysis was performed after hydrofluoric anhydride and F-133a were passed through the catalyst layer and the reaction was in a steady state.
- a catalyst was prepared using the following high-purity activated alumina.
- This active alumina has a pore volume distribution of about 80% around a pore diameter of about 80 people.
- a i 2 0 3 house owned 99.93% by weight or more
- Ni ( ⁇ 0 3) 2 ⁇ 6H 2 0 As nickel raw salt, 446 g of Ni ( ⁇ 0 3) 2 ⁇ 6H 2 0 , except that used was Example 1 5 and, prepare 23.7% NiO-A 1 2 0 3 catalyst in the same manner. Fill the reaction tube with catalyst 100 and treat the catalyst with hydrofluoric acid. The fluorination reaction of F-133a was carried out under the same conditions as in Example 15 except for the working conditions and the reaction conditions. Table 3 shows the results.
- Activated alumina (Sumitomo Chemical Co. Ltd., KHA-24) having the following physical properties was used to adjust the 23.7% NiO- A £ z 0 3 catalysts in the same manner as in Example 1 5.
- This activated alumina shows only a broad pore distribution in the pore diameter range of 40 to 500 A, and has about 20% of blue pores of 40 A or less.
- the reaction tube was charged with the catalyst 100, and a fluorination reaction of F-133a was carried out under the same conditions as in Example 15 for treating the catalyst with hydrogen fluoride and the reaction. Table 3 shows the results.
- Example 15 Using the same alumina carrier as in Example 15, an Ag, Fe, Co, Mn or Cu catalyst was prepared. Composition same as alumina 1 g per Rino metal loading of Ni, i.e. 4. 2 [mg- atom Metal / g- A £ 2 0 3) and the. Ag and Fe used nitrate, and Co. Mn and Cu used chloride as raw material salts.
- the catalyst preparation method and the treatment of the catalyst with hydrogen fluoride were performed in the same manner as in Example 15.
- the fluorination reaction of F-133a was performed using the same reactor, catalyst charge (150), hydrofluoric treatment conditions for the catalyst, and product analysis method as in Example I5. Was done.
- the reaction conditions were a reaction temperature of 390 °, a molar ratio of HF to F—133a of 8 or 8, 4 and [SV; Ohr- I set it to 1 .
- the results are shown in Table 4. Metal Ag Fe Co Mn Cu
- Example 15 Using the catalyst of Example 15 and the Ag catalyst of Example 22, a fluorination reaction of 1,1,1 trichloroethane (CC £ 3- CH 3 ) was performed.
- the method of treating the catalyst with hydrogen fluoride and the fluorination reaction were carried out according to Example 15.
- the reaction results are shown in Table & together with the reaction conditions.
- F-14 was synthesized according to Example 15 using the Fe catalyst in the catalyst of Example 22.
- Table 7 shows the reaction results together with the reaction conditions.
- Example 22 Using the Co catalyst among the catalysts used in Example 2,
- F-124 CHC i F-CF 3 , 1, 1,! L 2-tetrafluoro ⁇ tan F-125: CHF 2 -CF 3 .1,1, ⁇ -. ⁇ -Ventough ⁇ tan
- F-125 CHF 2 -CF 3 .1,1, ⁇ -. ⁇ -Ventough ⁇ tan
- the present invention can be advantageously used for the production of an organic fluorine compound useful as a refrigerant, an aerosol propellant, a foaming agent, a detergent, and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Description
明 細 書 有機フ ッ素化合物の製造方法 技術分野
本発明は、 有機フ ッ素化合物の製造方法に関する。 本発明 の方法により得られる有機フ ッ素化合物は、 冷媒、 エアゾル 噴射剤、 発泡剤、 洗剤等の用途に用いる こ とができる。 背景技術
有機塩素化合物とフ ッ化水素との反応によって有機フ ッ素 化合物を製造する方法に関しては、 これまで多数の提案がさ れている。 従来の方法は液相法と気相法に大別される力、'、 過 去においては前者が、 また近年においては後者が研究の対象 と して採り上げられてきている。 特に気相法による有機フ ッ 素化合物の製造においては触媒の選択が極めて重要である。
例えば、 1 —ク ロ 口一 2 , 2 , 2 — ト リ フルォ ロェタ ン.又 は 1 , 2 , 2 , 2 —テ ト ラ フルォロェタ ンの合成触媒の例を みる と、 前者を ト リ ク ロ 口ヱチ レンの気相フ ッ素化反応で合 成する場合、 ク ロム ( I ) 化合物、 例えば塩化ク ロム ( ΠΙ )、 フ ッ化ク ロム ( I ) 、 さ らにフ ッ化ク ロムに酸素を作用させ、 あるいは酸化ク ロムまたは水酸化ク α ムにフ ッ化水素を作用 させて得られる ク ロム才キ シフルオ リ ドを用いる こ とがよ く 知られている。 後者は 1 一 ク ロ α— 2 , 2 , 2 — ト リ フ ル才 ロェタ ンのフ ッ素化反応によ り 合成される力';、 例えば、 特公
昭 43 - 10601、 特開昭 53— 105404、 特開昭 56 - 24050には、 かか る場合にクロム系触媒を用いることが開示されている。 気相 反応による有機フ ッ素化合物の合成触媒として、 ク ロム系触 媒以外のものも公知であるが、 上記の例からもわかるように、 実用上はク ロム系触媒が多用されている。
しかしながら、 これら公知のクロム系触媒には、 工業的な 製遣が難しく、 触媒安定性が悪いという問題があり、 また転 化率や生産性の点で改良の余地がある。 従って、 本発明は、 上記の如き問題が無く、 転化率および生産性の優れたフ ッ素 化触媒を用いて、 気相で有機フ ッ素化合物を製造する方法を 提供することを目的とする。 発明の開示
本発明の百的は、 触媒担体として均一な細孔分布を有する 活性アルミ ナを用い、 これに触媒成分としてニッケル、 コバ ルト、 鉄、 マンガン、 ク ロム、 銅および銀から選ばれる金属 のフ ッ化物が担持された触媒を用いることにより達成される, 本発明によれば、 40〜500 人の平均細孔径を有する孔が 70 〜90 %を占める活性アルミナを触媒担体とし、 これにニッケ ル、 コバル ト、 鉄、 マンガン、 ク ロム、 銅および銀から選ば れる少な く とも 1種の金属のフ ッ化物が担持された触媒の存 在下に、 有機塩素化合物または有機不飽和化合物とフ ッ化水 素とを反応させる ことを特徴とする、 有機フ ッ素化合物の製 造方法が提供される。
発明を実施するための最良の形態
本発明の方法で用いられる触媒の担体として有用な活性ァ ルミナについて詳述する。
活性アルミ ナと しては、 水和アルミ ナであって、 X線回折 によると非晶質 (無定形) 部分が多 く 、 結晶化のあまり進ん でいないべ一マイ トまたは擬ベーマイ トやその焼成品でもあ る、 , 6 , , Γ , ^ , χ , ρ等の中間アルミナが同定さ れている。 通常、 活性アルミナと して賞用されるのは r ある いは?? 一アルミナであり、 アルミナ中に r , の両相が存在 する場合には X線的には区別できず、 r Z?? —アルミナと呼 ばれる。
活性アルミ ナは、 通常、 アルミナ水和物の熱分解即ち水和 物を制御加熱して大部分の水分を除去するこ とにより製造さ れる。 水和物の原料としては、 バイ ヤー法で製造されたアル ミナ 3水 物、 アルミ ニウム塩、 アルミ ン酸アルカ リ 、 アル ミ ニゥムアルコ キ シ ド、 金属アルミ ニウム等がある。 水和物 と して示せば、 ジブサイ ト、 バ イ ャ ラ イ ト、 ノ ル ドス ト ラ ン ダイ ト (以上 A £ (0H) 3)、 ベーマイ ト、 ダイ ァスボア (以上 A £ ζ03 · H20 または A £ 00H)等である。
バイ ヤー法によるアルミナ水和物を用いる場合以外はアル ミナゲルを作る力 、 その際の温度、 pH、 時間、 原料濃度など の条件によって各種のアルミナ水和物が単独あるいは混合物 として生成する。 これらの水和物はそれぞれ異なったアル ナ相に分解し、 それに従って綰孔構造、 表面積および物性が 異なって く るも C'である。
活性アルミナの製造方法としては、 (A ) ジブサイ ト、 ベ 一マイ ト等のアルミナ水和物の加熱脱水、 ( B ) アルミ ン酸 ソーダ水溶液と硫酸ァルミユウム水瑢液または炭酸ガスもし く はアルミ ン酸ソーダ粉末と亜硫酸ガスとの反応で得られる アルミ ナ水和物 (ゲル) の脱水、 ( C ) アルミ ニウム塩 (例 えば、 硫酸アルミニウム、 硝酸アルミ ニウム、 塩化アルミ二 ゥム) の水溶液にア ンモニアもし く はアル力 リ金属の水酸化 物もし く は炭酸塩の水溶液を添加して得られるアルミナ水和 物の脱水、 アルミニウム塩に尿素を添加し、 加熱する均一沈 澱法により得られる水和アルミ ナの脱水、 もし く は硫酸アル ミニゥムに炭酸塩を加えて塩基性硫酸アルミニゥム (一種の アルミナヒ ドロゾル) を生成させ、 それから得られるアルミ ナヒ ドロゲルの脱水、 ( D ) アルミニウム塩の加熱分解、 ( E ) アルミ ニウムィ ソプロポキシ ドの加水分解などが工業 的に実施されている。
これらの活性アルミナのう ち、 最も入手が容易なのは、 バ ィャ一法によるアルミナ 3水和物から合成されるものである < このアルミナは、 得られたジブサイ トを空気気流中約 400て で熱処理して活性化したものであり、 X線的には少量のベー マイ トを舍む Z 7?—アルミナである。 比表面積は約 250 m . gであり、 細孔直径 35 A〜: 10 μのブロー ドな細孔径分布を有 し、 さ らに細孔直径 3 5 Α以下の小細孔もかなり多く存在す る。 細孔は円筒状、 球状等の空孔からなる。 この細孔径分布 は、 通常、 50000ps i ( 35 A〜177 の細孔直: 1:を測定) の H g 一ポロシメーターを用いて測定されるものである。 また
0 組成的には約 0. 9 %の N a z 0と数百 ppm の S i 02および F e 20 3 と を舍有し、 N a 20の含有量を 0. 05 %以下とすることは難しい。 活性アルミナは、 バイ ヤー法アルミ ナ水和物を 400〜 800 てで急速に活性化するこ とでも得られ、 この過程でベーマイ ト生成および分解物生成が著し く滅少するため、 Ί / T)一 T ルミナのパターンが弱く 、 非晶質に近いアルミ ナが製造でき る。 このアルミ ナを凝集または再水和するこ とにより、 球状 など各種形態のものが得られ、 結晶子および細孔径とも前者 より小さいものが製造される。 比表面積は 300〜 350 / z て'ある。
アルミナゲルを出発原料と した場合 (前記 Bおよび C法) 、 沈澱を水洗し、 完全に水切り した後ではアルミ ナの X線は擬 ベ一マイ トである。 工業的にはこのケーキを乾燥破砕して押 出しシリ ンダー状とするか、 噴霧乾燥して 5 0 人位の球状微 粒子として球またはペレ ツ トに成型する。 これを熱処理して 活性化し、 活性アルミ ナとする。 X線的には非晶質に近い r ノ?? —アルミ ナであり、 組成的には少量の S i 0 2を舍むかまた は 2 〜 3 %の50 3 を舍有している。 この方法では種々 の構造 を持つゲルを合成できる力^ 比表面積は 300〜 600 ni Z gで あり、 極めて小さな細孔を多 く 有するアルミナである。
このよう に して得られる活性アルミ ナは細孔容積が 0. 3 〜 0. 8 / g . 比表面積が 1 50〜350 nf / g , 平均細孔径カ; 40 〜150 Α程度の細孔を有する。 細孔径分布についてみる と、 その範囲は広く 、 細孔径 40〜500 Aに限定すれば約 2 0 %程 度が分布しているに過ぎない。
本発明の触媒で用いる活性アルミナにおいては、 細孔径 40 〜500 Aの属する割合が 70〜90 %の範囲に集中するものが適 当である。 細孔径が 4 0 A以下で分布が集中するものは、 ェ 業的にも得難く、 触媒担体として用いたときには活性は向上 するが、 選択性が悪く 、 触媒寿命の点で不安がある。 細孔柽 が 500 Aで細孔分布が集中するものは、 担体として用いた時 に十分な触媒活性が得られず、 収率が低く なるという問題が あり、 好ましく ない。
活性アルミナの細孔容積は、 比表面積および細孔径分布に よって决まり、 その値が 0. 55 ^ノ g以下では、 適切な比表面 積および細孔構造が得られず、 触媒活性が低く 、 収率が悪く なることがある。 従って、 本発明に用いる活性アルミナにお いては、 4 0 A以上の細孔直径の孔の容積は 0. 55 fl2fi g以上、 特に 0. 55〜: I. 6 ^ gであるのが好ましい。
本発明で特に好適に用いるこ とのできる活性アルミ ナのェ 業的製造方法の例としては、 ゾルーゲル Zオイ ルド口 ッピン グ法 (前記 C法に類似) がある。 この方法によれば、 球形ゾ ルの化学的性状を調整したり、 ゾルーゲルの化学的操作を変 化させるこ とによって、 生成する活性アルミ ナの嵩密度、 比 表面積、 細孔容積、 細孔径および細孔分布を任意に調整する ことができる。
また、 好ま しい活性アルミ ナとしては、 ト リ アルキルアル ミナをアルミ ニウム一水和物に分解し (前記 E法に類似) 、 次いでこのアルミナ水和物を r 一アルミナに焼成したものが ある。 このアルミ ナをプレスまたは押し出ししてペレ ., トに
すると天然に産出するアルミナより も純度が高く、 細孔径が 揃った細孔径分布のよいものが得られる。
このような触媒担体に用いられる活性アル ミ ナは商業的に も入手可能なものであり、 平均細孔直径 40〜500 Aの孔を 70 〜90 %有する活性アルミ ナを選択すればよい。 こ の活性アル ミナは、 触媒として反応器への充塡ー抜出し時の取り扱いに 便利なように直径が 2 0 MI以下、 好ま し く は数 mmの粒子、 ビ ーズまたは押出し成型品の形にするのがよい。
活性アルミナ中の不純物については、 ナ ト リ ゥムの含有量 が l OOppm以下、 好まし く はできるだけ少ないこ とが必要であ る。 さ らに珪素が 300p pm未満でかつ鉄が l OO p pm未満である活 性アルミナを選択するのがよ く、 アルミナ純度と して 99 . 9 % 以上であるのが好ま しい。 ナ ト リ ウム、 珪素および鉄は、 酸 化物として担体中に存在する場合、 活性阻害作用、 無水フ ッ 酸によるフ ッ化珪素の生成、 異性化、 不均化反応の促進など の反応阻害作用を呈するため、 極力触媒中に存在させないこ とが好ま しい。
本発明に用いる触媒成分を構成する金属と しては、 ニッケ ル、 コ バル ト、 鉄、 マ ンガン、 ク ロム、 銅および銀を挙げる ことができる。 これらの金属源としては各金属の無機または 有機塩類等を好適に使用することができる。 例えば、 ニ ッケ ルの場合を例にとると、 塩化ニ ッ ケル、 硝酸ニ ッ ケル、 水酸 化ニ ッ ケル、 硫酸ニ ッ ケル、 炭酸ニ ッ ケル、 塩基性炭酸ニ ッ ケル、 蟻酸ニ ッ ケ ル'、 修酸ニッケル .、 酢酸ニ ッ ケル、 酸化二 ッゲル、 三二酸化ニ ッ ケル、 二酸化ニ ッ ケル、 水加三二酸化
ニ ッ ケル、 フ ッ化第一ニ ッケル、 フ ッ化第二ニ ッケル、 水加 フ ッ化第一二ッケル、 酸化フ ッ化ニッケル、 硫化ニ ッケル、 等が挙げられ、 所望ならば金属二ッゲルを使用することもで きる。 なかでも、 特に水溶性である塩化ニ ッケル、 硝酸ニッ ケル、 硫酸ニ ッケル等を好適に用いることができる。 その他 のコ バル ト、 鉄、 マ ンガ ン、 ク ロム、 銅、 銀等についても、 実用上入手可能なものである限り、 類似の塩類または化合物 を使用することができる。 また、 ニ ッ ケルの場合と同様に、 これらの金属についても、 塩化物、 硝酸塩、 硫酸塩等の水溶 性であるものを好適に使用することができる。
触媒の製造方法に関しては周知の各種の方法を適用するこ とができる。 例えば、 活性アルミ ナを金属塩水溶液に舍浸後、 乾燥、 焼成した.後フ ツ化水素処理する方法、 またはいつたん 水素で金属塩を金属に還元し、 次いでフ ッ化水素処理するな どの方法も適用できる。 即ち、 上記金属またはそ 0化合物を 担体に担持させた後、 これを気相でフ ッ化水素と反応させて、 該金属の少く とも 1部をフ ッ化物とする (フ ッ化水素処理) のがよい。 担体に上記金属のフ ッ化物が担持される場合には、 前記フ ッ化水素による処理は不要である場合もある。
触媒の製造に際しては、 好ま しく は、 上記活性アル ミ ナを 金属塩の水溶液に舍浸後、 あるいは活性アルミナに吸水量相 当の金属塩水溶液を全量吸収させ-、 熱風乾燥後、 空気流通下 で焼成する。
乾燥は 50〜: 120 'Cの温度で行うのがよ く、 焼成は酸素また は酸素を舍有する不活性ガス、 好ま し く は空気の流 i甬下に
[SV]。( 0 て、 1 気圧換算での空間速度) 100〜 1000hr— 'の範囲 で空気を流し、 200〜500 て の温度範囲で行うのが好ま しい, 金属塩を担持した活性アルミ ナを乾燥後、 空気流通下で焼成 する場合、 150'C以上の温度で急激な発熱を伴う ため、 温度 制御を行って、 触媒温度を 500 'C以下に保持するのがよい。 焼成温度が 200'C以下の場合には触媒活性が低く 、 また 500 て以上とする場合には触媒有効表面積が減少する (いわゆる シ ンタ リ ングを起こす) ため、 やはり触媒活性が低く なる。 触媒中の金属舍有量は、 任意に変化させることができるが、 活性および担持量の限界を考慮すると 5 〜 3 0重量%の範囲 が好適である。
このよう に調製した金属酸化物一アルミ ナ触媒はフ ッ化水 素処理に使用する前段で、
無水フ ッ酸濃度 : 10〜: 100 % (100%以下の場合、 不活性ガ スで無水フ ッ酸を稀釈して調整する)
[SV] 0 : 50~600 hr' 1
温度 : 200 〜450 て
の範囲で無水フ ッ酸 (フ 'ン化水素 : H F ) 処理を行って、 金 属酸化物およびアルミナを部分フ ッ素化して活性化する必要 がある。 この触媒の部分フ ッ化水素処理は発熱を伴うので、 温度制御を十分に行って、 触媒温度を 450て以下とするのが 好ま しい。
本発明における有機塩素化合物とフ ッ化水素との反応は次 の諸反応を舎むものである。 有機塩素化合物を構成する主要 原子団を CH2C£ , CHC£ , CCi .. CHC ί 2 : CC £ ζ , CC £ 3 と
するフ ッ化水素 ( H F ) との基本反応は
CH2C£ +HF CH2F + HC£
CHC£ +HF CHF +HC£
CC& +HF CF +HC£
CHC i z 十 HF " CHF2 + CHC £ F + HC£
CC £ z +HF CFZ +CC£ F + HC£
CC Jl 3 十 HF CF3 +CC£ FZ + CC £ zF-rHC^ で表される。 もっとも中間的なフッ素化物である、 CHC £ F, CC £ F, CC £ F2 l CC £ ZFなる原子団を有する有機塩素化合物 とフ ッ化水素との反応は
CC £ F + HF -- CFZ + HC£
CC £ F2 + HF CF3 +WC&
CC £ 2F + HF CF3 +CC £ FZ + HC£
であり、 もちろんこれらの反応も本発明に包舍される。
本発明にいう有機'塩素化合物とは上記のような CHZC£ , CHC ί , CC ί , CHC £ ζ , CC £ 2 , CC £ 3 , CHC £ F ", CC £ F , CC £ F2および' CC £ 2Fのごとき原子団を舍有するボリハロゲン 化有機化合物であり、 これらは部分的に CH2F CHF , CF , CHFZ , CF2 CF3等の原子団を舍んでいてもよい。
代表的な有機塩素化合物を示すと、 四塩化炭素、 ク ロロホ レム、 塩 ·{匕メ チ レ ン、 へキサク ロクレエタ ン、 ペ ンタ ク ロルェ タ ン、 テ ト ラ ク ロル'ェタ ン、 ト リ ク ロルェタ ン、 ォク タ ク ロ ルプ口 ' ン、 ヘプタク 口 ルプロ \。 ン、 へキサク コルプ口 パ ン 、 ペ ンタ ク レプ π ' ン テ ト ラ ク ロ ルフ' π ン - ト リ ク ロル
プロノヽ ·ン、 テ ト ラ ク ロノレエチ レ ン、 ト リ ク ロ ノレエチ レ ン、 へ キサク ロ ノレフ' 口 ペ ン、 ペ ンタ ク π ゾレフ "" 口 ペ ン、 テ ト ラ ク ロ ノレ プロペ ン、 ト リ ク ロノレプロ ペ ン、 へキ サク ロノレブタ ジエ ン、 へキサク ロノレブテ ン、 ペンタ ク ロルブテ ン、 テ ト ラ ク ロノレブ テ ン、 ペ ンタ ク ロ ルブタ ン、 テ ト ラ ク ロノレブタ ン、 ベンゾ ト リ ク ロ リ ド、 塩素化べンゾ ト リ ク ロ リ ド、 ニ ト ロ化ベ ンゾ ト リ ク ロ リ ド、 ト リ ク ロルメ チル化べンゾ ト リ ク ロ リ ド、 ト リ ク ロルメ チル基を舍むエーテル、 ト リ ク ロ ルメ チル基を舍む ケ ト ン、 ト リ ク ロ ルメ チル基を舍むエステル、 ト リ ク ロノレメ チル基を含む酸のハラ ィ ド、 ト リ ク ロルメ チル基を含むピリ ジ ン、 ト リ ク ロルメ チル基を舍むニ ト リ ノレ、 ある いはこれら の化合物中の ト リ ク ロルメ チル基、 ジク ロルメ チル基の塩素 原子の一部がフ ッ素原子に置換されたポ リ ハ ロゲン化有機化 合物等を挙げる こ とができる。
これらの中で工業的に重要なものは、 低級炭化水素のボリ 塩素化物、 へキサ ク ロルァセ にン、 ベ ンゾ ト リ ク ロ リ ドおよ びそれらの誘導体である。
本発明の方法によって得られる有機フ ッ素化合物は上記し た有機塩素化合物の塩素原子の一部あるいは全部がフ ッ素原 子により置換された物質を示し、 元素状フ ッ素ガスによるフ ッ素化でみられる基本骨格の炭素原子数の増減はあま り行わ れず、 原則と して本発明の方法では反応の前後を通 じて所定 の化合物の基本構造が維持され、 塩素原子がフ ッ素原子によ り置換されたこ とによる分子量、 性質の変化が観察されるの が主である。
本発明の方法は、 また、 有機不飽和化合物のフ ッ素化反応 にも有利に用いることができる。 上記に例示した如きテ トラ ク 口ルェチレンや ト リ ク ロルェチレンは、 有機不飽和化合物 の一例でもあるが、 例えば、 この ト リ ク ロルエチレンとフ ッ 化水素との反応においては、

2HF
CH z C £ - CF 3.
のように段階的に付加反応と置換反応とが進行する。
従って、 本発明において、 有機フ ッ素化合物とは、 有機不 飽和化合物に対するフッ化水素の付加により生じる化合物を も包舍するものである。
本発明の方法により、 有機塩素化合物の気相フ ッ素化によ り有機フ ッ素化合物を製造するに際して用いることのできる 反応型式としては、 触媒固定床式および移動床式を挙げるこ とができる。
通常、 反応温度—は 100〜600 て、 好ま し く は 150〜550 *C の範囲である。 これより低温では反応速度が遅く、 高温では 脱フ ッ酸、 不均化、 異性化、 コーク生成反応等の副反応が併 発し、 著しく選択性が悪化するため、 実用的でな く なる。
反応圧力は気相を維持できる限り任意の圧力でよ く、 一般 には 1 〜 1 0気圧であるけれども、 操作性および生産性を考 盧すると、 特に大気圧かその前後の圧力であるのが好ま しい。 空間速度 [S V;。 は 50〜2000h r - 'であり、 有機塩素化合物 1 モ ルに対するフ ッ化水素の割合 0. 5 〜 2 0 モルてあるのがよ
い。 このフ ッ化水素の割合は反応の選択性を決める上で重要 であり、 有機塩素化合物の塩素またはフ ッ素の置換の割合を 考察して適宜選択するのがよい。
以下においては、 原料の有機塩素化合物および Zまたは不 飽和化合物に対するフ ッ化水素のモル比を 「 H Fモル比」 と いう。
以下に、 本発明の方法における代表的な原料および製品の 例と、 それぞれの好ま しい反応条件を列挙する。
有機不飽和化合物として ト リ ク ロ ロエチレンを用い、 温度 200〜 450て、 ト リ ク ロ ロエチ レ ンに対するフ ッ化水素のモ ル比 3 〜 1 0で反応させて 1 一ク ロロ ー 2 , 2 , 2— ト リ フ ルォロェタ ン ( F — 133 a ) を製造する。
反応温度が 200'C以下では著し く転化率が低く 、 450 'C以 上では極端に選択率が悪化していずれも好ま し く ない。 H F モル比は化学量論比以上とし、 反応速度を落さず且つ F — 133 a の脱 H Fによる CHC £ =CF2 の生成を抑制するため 1 0以下が好ま しい。 また、 H Fモ ル比が 1 0以上では、 触 媒の生産性が悪く なり、 好ま し く ない。
有機塩素化合物として 1 — ク ロ ロ ー 2 , 2 , 2 — ト リ フル ォロェタ ンを用い、 温度 300〜 420て、 1—ク ロ σ — 2 , 2 , 2 — ト リ フルォロエタ ンに対するフ ッ化水素のモル比 1 〜
1 0 で反応させて 1 > 2 , 2 , 2 —テ ト ラ フルォ ロェタ ン ( F - 134 a ) を製造する。
反応温度が 300て以下ては著し く 転化率が低く 、 420て以上 では極端に選択率が悪化していずれも好ま し く ない。 H Fモ
ル比は化学量論比以上とし、 F— 133 a の反応速度を落さず 且つ F— 133 a の脱 H Fによる CHC£ =CF2 の生成を抑制す るため 1 0以下が好ましい。 CHC£ =CF2 は F— 134 a との 分離が難しく生成を極力抑制する必要がある。 また、 H Fモ ル比が 1 0以上では触媒の生産性も悪く なり好ま しく ない。 有機不飽和化合物としてバーク ロロエチレンを用い、 温度 200〜 400て、 パークロロェチレンに対するフ ッ化水素のモ ル比 3〜 6で反応させて、 1 , 1 ージク ロロー 2 , 2 , 2 — ト リ フルォ口ェタ ン ( F— 123)を製造する。
反応温度が 200'C以下では著し く転化率が低く、 400'C以上 では極端に選択率が悪化していずれも好ましく ない。 H Fモ ル比は化学量論比以上とするが、 6以上では、 1 , 1 , 1 , 2 —テ ト ラフルォロク.ロ ロェタ ン ( F— 124)、 1 , 1 , 1 , 2 , 2—ペンタフルォロェタ ン ( F—125)の生成が優先する ため、 F— 123生成選択率が悪くなり、 好まし く ない。
有機塩素化合物として 1 , 1 , 1 一 ト リ クロ口エタ ンを用 い、 温度 150〜 400'C、 1 , 1 , 1 一 ト リ ク ロ 口エタ ンに対 するフ ッ化水素のモル比 1〜 4で反応させて 1 , 1 , 1 —ジ ク ロロフルォロェタ ン ( F— 141 ) およびノまたは 1 , 1 , 1 ージフルォロク ロ ロェタ ン ( F— 142 b ) を製造する。 反 応温度が 150'C以下では著し く転化率が低く、 400'C以上で は極端に選択率が悪化していずれも好ま しく ない。 H Fモル 比は化学量論比以上であるが、 4以上では、 1 , 1 , 1 - 1. リ フルォロェタ ン ( F— 143 a ) の生成が優先するため、 F - 141 bまたは F - 142 b生成選択率が悪く なり好まし く な
い。
有機塩素化合物と してク ロ 口ホルムを用い、 温度 150〜 350 'C、 ク ロ ロ ホルムに対するフ ッ化水素のモル比 0. 5 〜 3 で反応させてジフルォロメ タ ン ( F — 2 2 ) を製造する。 反応温度が 150'C以下では著し く 転化率が低く 、 350 'C以上 では極端に選択率が悪化していずれも好ま し く ない。 H Fモ ル比が 0. 5以下の場合、 触媒の生産性が悪 く なり好ま し く な い。 また H Fモル比が 3以上では、 ト リ フルォロメ タ ン ( F - 2 3 ) の生成が優先するため、 F — 2 2生成選択率が悪く なり好ま し く ない。
有機塩素化合物と してジフルォ ロ ジク ロ ロ メ タ ン ( F — 1 2 ) およびノまたは ト リ フルォロク ロ ロメ タ ン ( F — 1 3 ) を用い、 温度 200〜 460 · (:、 ジフルォロジク ロ ロメ タ ンおよ びノまたは ト リ フルォロク 口ロメ タ ンに対するフ ッ化水素の モル比 0. 5〜 3 で反応させてテ ト ラ フルォ ロメ タ ン ( F — 1 ) を製造する。
反応温度が 200 'C以下では著し く 転化率が低 く 、 460 'C以上 ては分解等の副反応が起るため極端に選択率が悪化して好ま し く ない。 H Fモル比が 0. 5以下の場合、 触媒の生産性が悪 く なり好ま し く ない。 H Fモル比が 3以上の場合、 F — 1 4 からの H Fの回収分離が複雑となる こ と、 触媒当り の生産性 が悪 く なる こ とよ り好ま し く ない。
有機不飽和化合物と して ト リ フルォロ ク ロ ロエチ レ ン ( F — 113)を用い、 温度 200〜 450。C:、 ト リ フルォ ロ ク ロ ロ ェチ レ ンに対するフ ッ化水素のモル比 1 〜 5 で反応させて 1 , 1 , 1 , 2 —テ ト ラ フルォ ロ ェタ ン ( F - 124)および または
1 , 1 , 1 , 2 , 2 —ペンタフルォロェタ ン ( F — 125)を製 造する。 反応温度が 200'C以下では著し く転化率が低く、
450 *C以上では分解等の副反応が起るため極端に選択率が悪 化して好ましく ない。 H Fモル比は化学量論比以上とする力 <、 5以上では F— 124又は F— 125と H Fとの分離が複雑とな り、 且つ触媒当りの生産性が悪く なることより好ましく ない。 次に、 実施例および比較例を示し、 本発明をさらに具体的 に説明する。
実施例 1
次の物性を有する、 1. 6賺の球状の高純度活性アルミナ (日揮ユニバーサル㈱製品、 NST-7)を使用して触媒を調製し た。 この活性アルミナはゾルーゲルノオイ ル ドロ ッ ビング法 で製造されたもので、 細孔直径 8 0 付近に約 8 0 %の細孔 容積の分布を示すものである。
細孔容積 0.6 mi/ g
平均細孔径 80 A
全 B E T比表面積 250 Ίή/ s
充塡密度 0。70 g
权{圣 1.6 mm φ
A 2203 含有量 93.93重量%以上
Na含有量 10 ppm 以下
Si舍有量 150 ppm 以下
Fe含有量 100 pptn 以下
塩化ク ロム、 CrC £ • 6H20、 191.5 gを純水 132ffl£に投 入し、 湯浴上で 70〜80'Cに加熱して溶解する。 溶液を室温ま
で冷却後、 上記活性アルミ ナ 400 gを浸漬して、 アルミナに 触媒液を全量吸収させる。 次いで、 触媒液で濡れた状態のァ ル ミナを 9 0 ての湯浴上で乾燥し、 乾固する。 乾固した触媒 を空気循環型の熱風乾燥器内で 110'Cで 3時間乾燥する。
乾燥触媒を S U S製容器に充填し、 空気を [SV] ^^hr- 1で 流し、 流通下で焼成する。 200てで触媒層の発熱がな く なる まで焼成した後、 さ らに 400 'Cまで昇温し、 3時間焼成する。 触媒組成は計算値で Cr303 12重量%、 A £ 203 88重量%であ る。 こ の焼成触媒 を内径 2 5腿、 高さ 1 mの反応管 (二ッケル製) に充塡した。 1 一ク ロロー 2 , 2 , 2 — ト リ フルォロェタ ン(CH2C -CF3 , F-133a)のフ ッ化水素処理を 行う前段で窒素で稀釈した無水フ ッ酸および 100%無水フ ッ 酸を用いて触媒の部分でフ ッ素化を行い、 触媒を活性化した。 無水フ ッ酸による触媒の処理条件を次に示す。
無水フ ッ酸濃度 25〜100 モル%
.',日
i /又 250〜420 て
[sv] 0 400 hr一 1
処理時間 約 15時間
このようにして得られた触媒を用いて F — 133 a の気相フ ッ素化反応を行った。 反応器を出た生成ガスをアルカ リ 水溶 液に通して未反応無水フ ッ酸および生成 HC の吸収を行い、 次いでモ レキユラ一シーブ 3 A塔で脱水後、 ガスメ 一ターで 計量し、 また生成ガスをガスク ロマ トグラフ ィ ーで分折した, 分析は無水フ ッ酸および F - 133 a を触媒に通じて後、 反応 が定常状態となってから行つた。
反応温度 330'C、 反応圧力 1気圧、 F — 133 aに対する無 水フ ッ酸のモル比 ( H Fモル比) 4. 6、 [SV] olOlhr-1で F— 133 a のフ ッ素化反応を行ったところ、 F — 133 a の転化率 は 20.3%であり、 1 , 2 , 2 , 2—テ トラフルォロェタ ン
(CH2F-CF3 , F- 134a)の収率および選択率はそれぞれ 19.1お よび 94.3%であった。 このとき、 F— 134 a の空時得率 S T Y 〔 5ノ 触媒 ' 111~) は 15.6であった。
実施例 2
高純度活性アルミナとして次のものを用いて触媒を調製し た。 この活性ァルミナは細孔直径 270A付近に約 8 0 %の細 孔容積の分布を示すものである。
細孔容積 1,5 id/ g
平均細孔径 270 A
全 B E T比表面積 190 πί/ g
充填密度 0.37 g /id
粒径 3。 2 mm φ
£ 203 含有量 99.93重量%以上
含有量 10 ppm 以下
Si含有量 150 ppm 以下
Fe含有量 100 ppm 以下
調製方法は実施例 1 と同じに行い、 組成を Cr203 24重量%.
203 76重量%と した β 触媒充塡量、 触媒のフ ッ化水素処 理および F - 133 a のフ ッ素化反応条件を実施例 1 と同じに して F — 134 a の合成を行った。 結果を表 1 に示す。
実施例 3および 4
実施例 1 と同一の触媒を用いて、 反応条件を変更する以外、 実施例 1 と同じにして F — 133 a のフ ッ素化反応を行つた。 結果を反応条件とともに表 1 に示す。
実施例 5
287.5 g の硝酸ク ロム Cr(N03) 3 · 9H20をク ロム原料と して 用いた以外は実施例 1 と同様にして、 Cr 203 12% - A i 203 88%の触媒を調製した。 触媒 100 を反応管に充塡し、 フ ッ 化水素処理およびフ ッ素化反応の条件を実施例 2 と同じにし て、 F — 133 a のフ ッ素化反応を行った。 結果を表 1 に示す。 実施例 6
39.9 g の CrC £ 3 · 6H 20および 31.9 g の(N'H 4 ) 2 CrO 4をク ロ ム原料として用いた以外は実施例 1 と同様にして、 触媒を調 製した。 但し、 ク ロムの含有率を 9. 5重量%と した。 触媒 100 を反応管に充塡し、 フ ッ素化処理および反応の条件を実施 例 1 と同じにして、 F — 133 a のフ ッ素化反応を行った。 結 果を表 1 に示す。
実施例 7
実施例 2 の触媒を用い、 反応条件を変更する以外実施例 2 と同じに して、 次の反応条件で F — 133 a のフ ッ素化による F — 134 a の連続製造テス トを行った。
i ) 反応条件
反応温度 330 "C
反応圧力 1気圧
H Fモル比 4. 2
[SV] 0 250 hr— 1
η ) テス ト結果

次の物性をもつ活性アルミナ (住友化学工業㈱製、 KHA-24 ) を用いて、 実施例 1 と同様にして Cr203 12% - A £ 203 88% の組成をもつ触媒を調製した。 この活性アルミナは細孔直径 . 0〜 500 Aの範囲ではブロー ドな細孔分布しか示さず、 かつ 4 0 人以下の細孔を約 2 0 %もつものである。
細孔容積 0.53 ノ g
全 B E T比表面積 150 in / g
充壚密度 0.72 g / mi
粒径 2 〜 4 ram
A £ z03 舍有量 98.4重量%以上
Na舍有量 1900ppm 以下
Si含有量 140 ppm 以下
Fe含有量 210 ppm 以下
触媒 lOOffifiを反応管に充塡し、 フ ッ化水素処理およびフ ッ 素化反応の条件を実施例 1 と同じにして、 F — 133 a のフ ッ 素化反応を行った。 結果を表 1 に示す。
比較例 2
市販の酸化ク ロム ーアル ミ ナ触媒 (粒径 5咖 X 5卿ペ レ ッ ト、 1 2 %Cr 203 舍有) を用いて、 フ ッ化水素処理および フ ッ素化反応の条件を実施例 1 と同様にして、 F — 133 a の フ ッ素化反応を行った。 結果を表 1 に示す。

次の物性を有する、 1. 6 画の球状の高純度活性アルミ ナ (日揮ユニバーサル㈱製品、 NST-7)を使用して触媒を調製し た。 この活性アルミ ナは細孔直径 8 0 A付近に約 8 0 %の細 孔容積の分布を示すものである。
細孔容積 0.6 id / g
平均細孔径 80 A
全 B E T比表面積 250 πί/ g
充塡密度 0.70 g
粒径 1.6 mm φ
A a 2o3 含有量 99, 93重量%以上
Na含有量 10 ppm 以下
Si含有量 · 150 ppm 以下
Fe含有量 100 ppm 以下
硝酸ク ロム、 Cr (N03) 3 · 9H20、 287.5 gを純水 に投 入し、 湯浴上で 70〜80てに加熱して溶解する。 溶液を室温ま で冷却後、 上記活性アルミ ナ 400 gを浸漬して、 アルミ ナに 触媒液を全量吸収させる。 次いで、 触媒液て '濡れた状態のァ ルミナを 9 0 ての湯浴上で乾燥し、 乾固する。 乾固した触媒 を空気循環型の熱風乾燥器内で 110'Cで 2時間乾燥する。
乾燥触媒を S U S製容器に充塡し、 空気を [SV] 。540hr- 'で 流し、 流通下で焼成する。 200てで触媒の発熱がな く なる迄 焼成した後、 さ らに 400てまで异温し、 3時間焼成する。 触 媒組成は計算値で Cr 303 12重量%、 A £ 203 88重量%である。 こ の触媒 100 を内径 2 5 m 、 高さ i mの反応管 (ニ ッ ケ ル
製) に充填した。 ト リ クロロエチレン(CHC =CC£ 2)のフ ッ 素化反応を行う前段で、 窒素で稀釈したフ ッ化水素および 100%フ ッ化水素を用い、 触媒の部分でフ ッ化水素処理を行 つて、 触媒をフ ッ化物とし、 活性化した。 フ ッ化水素による 触媒の処理条件を次に示す。
H F濃度 : 25〜100 容量%
温度 : 250〜420 'C
[SV] 0 : 400 hr- 1
処理時間 : 約 15時間
このようにして得られた触媒を用いて ト リ ク ロ ロエチレン のフ ッ素化反応を行った。 反応温度 350'C、 反応圧力 1気圧、 [SV]。123hr— '、 H F と ト リ ク ロ ロエチレンのモル比 ( H Fモ ル比) 3. 4、 H Fおよび ト リ ク ロ ロエチレンの流量をそれぞ れ 0.425、 0.125モル Zhrとした。 反応が定常状態となって から、 サンプリ ングし、 反応生成物の分圻を行った。 排出ガ スをアル力 リおよび トルエ ンの二層よりなる吸収塔に通して : 未反応 H Fの吸収および生成物の捕集吸収を行い、 次いでェ タノール ドライ アイ ス ドラ ップに通して、 未吸収の生成物を 捕集した。 生成物を舍有する トルエ ン回収液をガスク ロマ ト グラフ ィ一で分折した。
この結果、 ト リ ク ロロエチレンの反応率 88.4%、 F - 133 a の収率 81.4%、 選択率 92.1%の値が得られた。 また、 F— 133 a の空時得率 S T Y (^ £触媒 . 11:〕 ば 121 であった。 実施例 S
高純度活性ァ几 ミナとして次のものを用いて触媒を調製し
た。 この活性アルミナは細孔直径 270人付近に約 8 0 %の細 孔容積の分布を示すものである。
細孔容積 1.5 id / g
平均細孔径 270 A
全 B E T比表面積 190 ηί/ g
充塡密度 0.37 g /m£
粒径 3.2 讓 ø
A £ z03 含有量 99, 93重量%以上
含有量 10 ppm 以下
Si含有量 150 ppm 以下
Fe舍有量 100 ppm 以下
調製方法は実施例 8 に準じた。 触媒フ ツ化水素処理および ト リ ク ロ ロエチ レ ンのフ ッ素化反応条件を実施例 8 と同じに して F— 133 a の合成を行った。
結果を表 2に示す。
実施例 1 0
実施例 8 と同一触媒を用い、 反応温度を 31G'C と した以外 は実施例' 8 と同一条件で ト リ ク ロ ロエチ レ ンのフ ッ素化反応 を行った。 結果を表 2 に示す。
実施例 1 1
ク ロム原料として 191.5 g の CrC£ 3 · 6Η20を用いた以外 は実施例 8 と同様にして 12% Cr 203 - A £ 203 触媒を調製し た。 触媒 を反応管に充塡し、 触媒のフ ッ化水素処理お よび反応の条件を実施例 8 と同じにして、 ト リ ク ロ コェチレ ンのフ ッ素化反応を行った。 結果を衷 2 に示す。
実施例 1 2
実施例 1 1 と同様であるが、 反応温度 400て、 H F Z ト リ ク ロ口エチレンモル比 6. 5、 [SV]。 208hr— 1として ト リ ク ロ 口エチレンのフ ッ素化反応を行った。 結果を表 2 に示す。
実施例 1 3
39.9 gの CrC 3 · 6H20および 31.9 g の(NH4) 2Cr04をクロ ム原料として用いた以外は実施例 8 と同様にして、 触媒を調 製した。 但し、 ク ロム舍有率を 9. 5重量%とした。 触媒のフ ッ化水素処理および反応の条件を実施例 8 と同じにして、 ト リ クロロエチレンのフ ッ素化反応を行つた。 結果を表 2 に示 す。
実施例 1 4
実施例 1 3で、 [SV] o Ohr-1 H F Zト リ ク ロ ロエチレン モル比 5 とした以外は同一条件で ト リ ク ロ ロエチレンのフ ッ 素化反応を行った。 結果を表 2に示す。
比較例 3
次の物性をもつ活性アルミナ (住友化学工業㈱製、 KHA-24) を用いて、 実施例 8 と同様にして 12%Cr203 — A i z03 触媒 を調製し、 フ ッ化水素処理した。 この活性アルミ ナは細孔直 径 40〜500 Aの範囲ではブロードな細孔分布しか示さず、 か つ 4 0 A以下の細孔を約 2 0 %もつものである。
細孔容積 : 0.53/nje/ g
全 B E T比表面積 : 150 n / g
充¾密度 : 0.72 g Zffl£
粒 ½: ·· 2 〜 4議
A £ 203 舍有量 : 98.4重量%以上
N'a含有量 : 1900ppm
Si含有量 : 140 ppm
Fe含有量 : 210 ppm
実施例 8 と同様にして ト リ ク ロエチ レ ンのフ ッ素化反応を 行った。 結果を表 2 に示す。
比較例 4
市販の酸化ク ロ ム -アル ミ ナ触媒 (12%Cr 203 舍有、 3 nun X 3 讓ペ レ ツ ト ) を用いて、 実施例 8 と同様にしてフ ッ化 水素処理を行い ト リ ク ロ口ヱチ レ ンのフ ッ素化反応を行った, 結果を表 2 に示す。

次の物性を有する、 3. 2讓の球状の高純度活性アル ミ ナ (日揮ユニバーサル㈱製品、 NST-3)を使用して触媒を調製し た。 こ の活性アル ミ ナは、 ゾルーゲル/オ イ ル ドロ ッ ビ ング 法で製造されたもので、 細孔直径 270 A付近に約 8 0 %の細 孔容積の分布を示すものである。
細孔容積 1.5 mi / z
平均細孔径 270 Λ
全 B E T比表面積 190 iri/" g
充塡密度 0.37 g
粒径 3。 2 mm φ
A 203 含有量 99。93重量%以上
Na含有量 10 ppm 以下
Si含有量 150 ppm 以下
Fe含有量 100 ppm 以下
塩化ニ ッ ケル、 NiC £ 2 · 6HZ0 365 gをイ オ ン交換水 370 に投入し、 湯浴上で 70〜80てに加熱して溶解する。 溶液を 室温まで冷却後、 上記活性アル ミ ナ 369 gを浸漬して、 アル ミ ナ に触媒液を全量吸収させる。 次いで、 触媒液で濡れた状 態のアル ミ ナを 9 0 ての湯浴上で乾燥し、 乾固する。 乾固し た触媒を空気循環型の熱風乾燥器内で 120てで 3時間乾燥す る。
乾燥触媒を S U S製容¾に充塡し、 乾燥空気を [SV] O540hr- ' で流し、 流通下で焼成する。 200'Cで触媒層の発熱がな く な る迄焼成した後.、 さ らに 400てまで异温し . 3時間焼成する„
触媒 成は NiO 23.7重量%、 S. z03 76.3重量%である。 こ の触媒 150^2を内径 2 5腿、 高さ 1 mの反応 媒固定床反応 管 (二ッケル製) に充塡した。 F— 133 a のフ ッ素化
反応を行う前段で窒素で稀釈した無水フ ッ酸および 100%無水フ ッ酸を用いて触媒の部分フ ッ化物化を行い、 触 媒を活性化した。 無水フ ッ酸による触媒の処理条件を次に示 す。
無水フ ソ 25~100 モノレ%
250〜420 て
[SV]。 400 hr"1
処理時間 約 15時間
このようにして得られた触媒を用いて F— 133 a の気相フ ッ素化反応を行つた。 反応器を出た生成ガスをアル力 リ水溶 液に通して未反応無水フ ッ酸および生成 の吸収を行い、 次いでモレキユラ一シーブ 3 A塔で脱水後、 ガスメーターで 計量し、 また生成ガスをガスクロマ トグラフ ィ 一で分圻した, 分折は無水フ ッ酸および F— 133 aを触媒層に通じて後、 反 応が定常扰態となってから行った。
反応温度 330'C、 反応圧力 1気圧、 F— 133 aに対する無 水フ ッ酸のモル比 ( H Fモル比) 4. 0、 [SV]。 90hr-'で F— 133 a のフ ッ素化反応を行ったところ、 F— 133 a の転化率 は 13.1 %であり、 1 , 2 , 2 , 2 —テ ト ラ フルォ ロェタ ン (CH2P-CF3 , F-134a) の収率および選択率はそれぞれ 12.0お よび 91.6%であった。 こ。のとき、 F— 13^a の空時得率 S T Y i z / ί触媒 · hr〕 は 9. 8 あった„
実施例 1 6
高純度活性アルミナとして次のものを用いて触媒を調製し た。 この活性アルミ ナは細孔直径 8 0 人付近に約 8 0 %の細 孔容積の分布を示すものである。
細孔容積 0.6 mi/ g
平均細孔径 80 A
全 B E T比表面積 250 irf / g
充塡密度 0.70 g / mi
粒径 1.6 mm φ
A i 203 舍有量 99.93重量%以上
Na舍有量 10 ppm 以下
Si含有量 150 ppm 以下
Fe含有量 100 ppm 以下
調製方法は実施例 1 5 と同じに行い、 組成を NiO 23.7重量 %、 i 203 76.3重量%とした。 触媒充塡量、 触媒フ ッ化水 素処理および F — 133 a のフ ッ素化反応条件を実施例 1 5 と 同じにして F — 13 a の合成を行った。 結果を表 3に示す。 実施例 1 7〜 : I 9
実施例 1 5 と同一の触媒を用いて、 反応条件を変更する以 外、 実施例 1 5 と同じにして、 F — 133 a のフ ッ素化反応を 行った。 結果を反応条件とともに表 3 に示す。
実施例 2 0
ニッケル原料塩として、 446 gの Ni (Ν03) 2 · 6H20を用いた 以外は実施例 1 5 と同様にして 23.7% NiO-A 1 203 触媒を調 製した。 触媒 100 を反応管に充塡し、 触媒のフ ッ化水素処
理および反応の条件を実施例 1 5 と同じにして、 F— 133 a のフ ッ素化反応を行った。 結果を表 3 に示す。
比較例 5
次の物性をもつ活性アルミナ (住友化学工業㈱製, KHA-24) を用いて、 実施例 1 5 と同様にして 23.7% NiO— A£ z03 触 媒を調整した。 この活性アルミナは細孔直径 40〜500 Aの範 囲ではブロードな細孔分布しか示さず、 かつ 4 0 A以下の紺 孔を約 2 0 %もつものである。
細孔容積 0. g
全 B E T比表面積 150 iri/ g
充塡密度 0.72 g /mi
粒径 2〜 4讓 ø
A £ 203 含有量 98.4重量%以上
Na含有量 1900ppm 以下
Si含有量 140 ppm 以下
Pe含有量 210 ppm 以下
触媒 100 を反応管に充塡し、 触媒のフ ツ化水素処理およ び反応の条件を実施例 1 5 と同じにして、 F — 133 a のフ ッ 素化反応を行った。 結果を表 3 に示す。
比較例 6
市販の酸化ニ ッケル- アルミナ触媒 (粒柽 5腿 《5 X 5 ranぺ レツ 卜、 NiO 20%舍有) を用いて、 実施例 1 5 と同様にフ ッ 化水素処理を行った後、 F -- 133 a のフ ッ素化反応を行った 結果を衷 3 に示す。
3 反 応 条 件 反 応 成 績
反温 ί HF/ 〔SV〕 o F- 133 a F— 134 a F— 134 a F-- 134 a 応度 } F- 133 a (hr"1) w (%) 選択率 (%) 収率 (%) STY(g/£
( ±.) モル比 匪 · hr)
1 4.0 90 15.2 92.8 14.1 11.6
1 8.4 90 23.2 95.3 22.1 10.1
1 4.0 235 13.2 92.0 12.1 25.9
1 8.4 430 26.5 91.0 24.1 50.2 1 4.0 90 15.7 90.0 14.1 11.6
1 4.0 90 5.2 52.0 2.7 2.2
1 4.0 90 2.4 56.0 1.35 1.1
実施例 2 1
1 一ク ロ ロー 2 , 2 —ジフルォ口エチレン(CHC£ =CF2)を 2 %舍有する F — 133 a のブッ素化反応を 1 —ク ロ口— 2 , 2 —ジフルォ口エチレンに対する H Fモル比 2、 [ S V] 0
00hr " 温度 200 'C とする以外は実施例 1 5 に準じて行つ た。
生成物 Φに 1 一 ク ロ 口一 2 , 2 —ジフルォロエチレンは検 出できなかったが、 F— 133 a は未反応のまま残留すること が認められた。
かかる低温においてはフ ッ化水素の不飽和二重結合への付 加が行われるのみで、 塩素に対するフ 'ン素の置換反応は起こ らないことがわかる。
実施例 2 2
実施例 1 5 と同一のアルミナ担体を用いて、 Ag、 Fe、 Co、 Mnまたは Cu舍有の触媒を調製した。 組成はアルミナ 1 g 当た りの金属担持量を Niの場合と同じ、 即ち 4. 2 [mg- atom Metal/ g- A£ 203)とした。 原料塩として、 Agおよび Feは硝酸塩を、 Co. Mnおよび Cuは塩化物をそれぞれ用いた。 触媒調製方法お よび触媒のフ ッ化水素処理も実施例 1 5 と同様に行った。 各触媒について、 反応器、 触媒充塡量(150 ) 、 触媒のフ ッ化水素処理条件および生成物分折法を実施例 I 5 と同じに して、 F — 133 a のフ ッ素化反応を行った。 反応条件は反応 温度 390° 、 F — 133 a に対する H Fモル比 8 または 8, 4 お よび [SV;。 Ohr—1とした。 洁果を衷 4 に示す。
金 属 Ag Fe Co Mn Cu
HFZ F- / ¾■ 8.4 8.4 8.0 8.0 8.0
(原料モル比)
F— 134a収率 〔%〕 15.1 11.2 8.1 4。5 3.6
F- 134a選択率 〔%〕 95.0 75.1 64.0 60.1 55.0 実施例 2 3
実施例 1 5 と同一の触媒を用いて、 テ ト ラ フルォ口メ タ ン (CF, , F- 14)、 ク ロ ロ ジフルォロメ タ ン(CHC FZ , F-22)、 および F — 133 a の合成を行った。 触媒のフ ッ化水素処理に ついても実施例 1 5 に準じて行った。 反応結果を反応条件と ともに表 5 に示す。
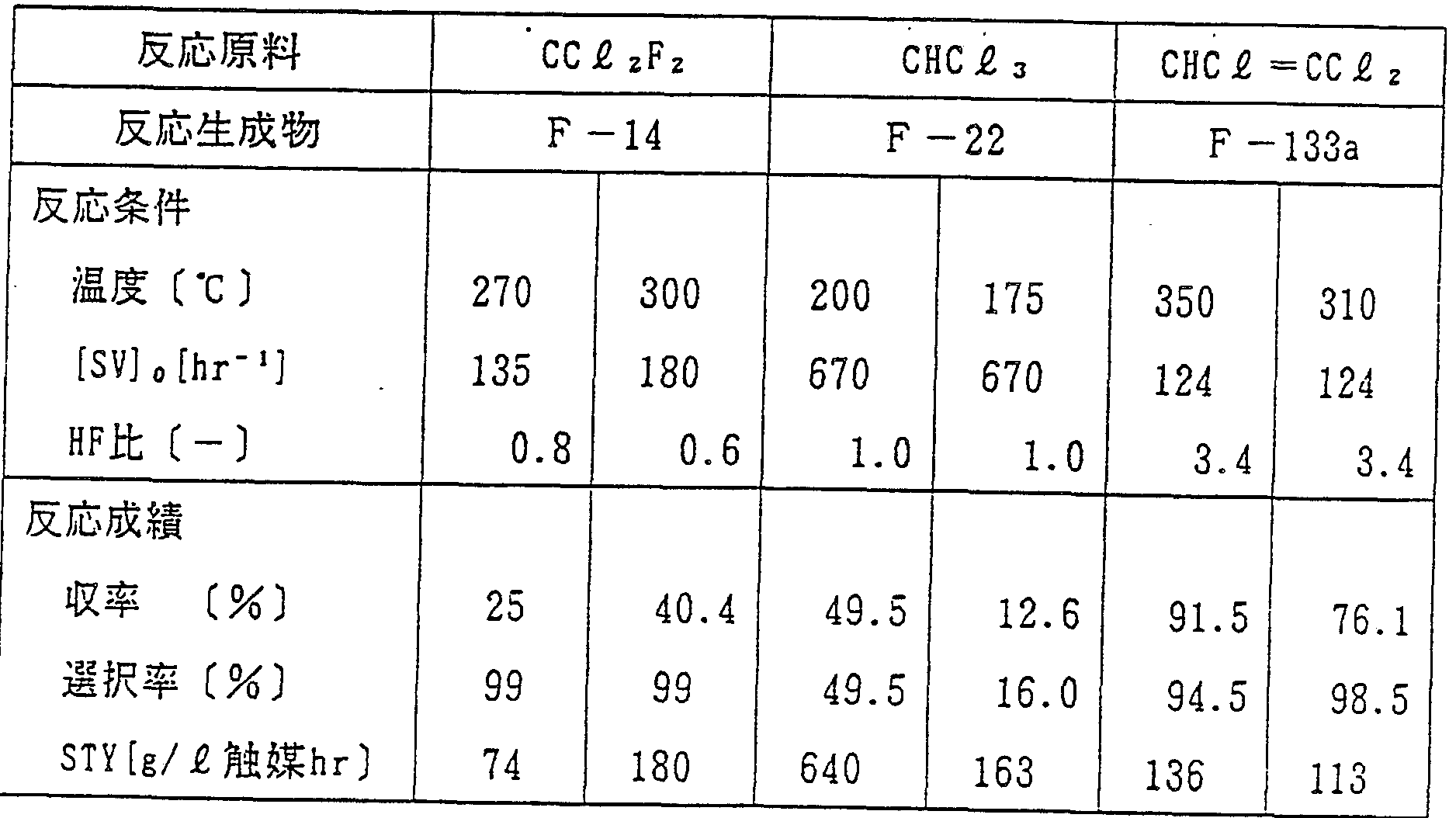
表 5
実施例 1 5の触媒および実施例 2 2中の Ag触媒を用いて、 1 , 1 , 1 一ト リ ク ロロェタ ン(CC£ 3-CH3)のフ ッ素化反応 を行った。 触媒のフ ッ化水素処理およびフッ素化反応の方法 は実施例 1 5に準じて行った。 反応結果を反応条件とともに 表 &に示す。
表 6

*1 ; 1 ーフルォロー 1 , 1 ジク ロ ロェタ ン (CC£ 2F-CH3) *2; 1 ーク α ロー 1 , 1 ージフルォロェタ ン (CC £ F2-CH3) *3 ; 1 , 1 , 1一 ト リ フルォロェタ ン(CF 3- CH 3) 実施例 2 5
ク ロム原料として塩化ク ロム、 CrC SL z . 6H20を用い、 ま た担体として実施例 1 5 と同じアルミナを用いて、 実施例 1 5 と同様の製法により、 Cr203 24重量%、 A SL 202 76重量 %のク ロ ミア一アルミナ触媒を調製し、 この触媒を用いて、 F-14 , F-22. へキサフルォ πエタ ン(CF3-CF3 , F-116) の合
成を行った。 触媒のフ ッ化水素処理、 反応方法は実施例 1 5 に準じて行った。 反応結果を反応条件とともに表 7 に示す。 実施例 2 6
実施例 2 2 の触媒中の F e触媒を用い、 実施例 1 5 に準じて F - 14の合成を行った。 反応結果を反応条件とと もに表 7 に示 す。
実施例番 · 2 5 2 6
反応原料 CC ϋ zVz CHC t 3 CF 3 - CF ZC J? CC £ 2Fz 反応生成物 F ~ 14 F - 22 F ― 116 F - 14 反応条件
温 L¾i C ΐ 〕 274 301 200 175 455 395 274
[SV] o ihr" ' ] 135 181 670 670 100 100 135
'比 [ - 〕 0.79 0, 58 1.0 1.0 1.0 3.9 0.79
CO
反応成績 CO 収率 〔% 3 30.8 45.4 56 16 32.8 5.8 10.1 選択率 ( % ) 99 99 56 18 95.0 80.6 98
sSTV fg/ i触媒 hrj 91 205 720 210 101 7 30
実施例 2 7
実施例 2 5 で使用したク ロ ミア一アルミ ナ触媒を用いて、 パーク ロ ロエチ レ ン (CC£ Z =CC £ 2)のフ ッ素化反応を行つ た。 触媒のフ ッ化水素処理および反応は実施例 2 5 に準じて 行った。 反応条件と得られた結果を次に示す。
反応条件 温度 : 250'C
[SV]。 : 240hr-'
H Fモル比 : 3.7
反応成績
収率 〔%〕 1 , 1 —ジク ロ σ — 2 , 2 , 2 —
ト リ フノレオ ロ ェタ ン
(CHC £ Z— CF3 , F- 123) 20.0 1 一 ク ロ ロ ー 1 , 2 , 2 , 2 - テ ト ラ フノレォ ロ ェタ ン
(CHC £ F-CF3 , F-124) 6.4 CC £ z = CC £ F 十 CHC £ 2 — CC £ F2 4.1
STY123;70 〔 g Z 触媒 · hr〕 実施例 2 8
実施例 2 5 で使用したク ロ ミ ア一アルミ ナ触媒を用いて、 1 , 1 , 1 — ト リ ク ロ 口エタ ン (CC £ 3 — CH3)のフ ッ素化反 応を行った。 触媒のフ ッ化水素処理および反応方法は実施例 2 5 に同じである。 反応条件と得られた結果を次に示す。 反応条 ί牛 温度 :て: : 200
[SV:。 r- ': : 108
H F比 :—: : 1.3
反応成績
収率 〔%〕 141b 80
142 b 10
143a 5
STY141b 196 〔 §ノ £触媒111"〕
実施例 2 9
実施例 2 2で用いた触媒の内の Co触媒を用いて、 実施例
2 5 に準じて温度 200*C、 [SV】。 108hr-'、 ^1 ?モル比 1。 3 で 1 , 1 , 1 一 ト リ グロロェタ ンのフ ッ素反応を行った。
Ulbの収率は 1 6 %であった。
実施例 3 0
実施例 2 5で使用したク 口 ミア一アルミナ触媒を用いて、 ト リ フルォロク ロ πエチレン(CC £ F =CF2 , F-1113) のフ ッ化反応を扦つた。 触媒のフッ化水素処理および反応は実施 例 2 5 に準じて行った。 反応条件と得られた結果を次に示す。
反応条件 温度 〖'C] : 300又は 350
[SV] 0 〖hr_リ : 210
H Fモル比 [一〗 : 1.65
反応成績

F-124 : CHC i F-CF3, 1 , 1 , !L 2-テトラフルォ nク πϋΐタン F-125 : CHF2-CF3.1,1, Ι-.Ι -ベンタフ!け πιタン
実施例 3 1
実施例 1 5 の触媒を用いて、 ベンゾ ト リ ク ロ リ ドのフ ッ素 化反応を行った。 反応生成物を冷却捕集後、 分留して生成物 を得た。 反応温度 300て、 [SV]。 90hr-'、 ベンゾ ト リ ク ロ リ ドに対するフ ッ酸のモル比 9. 5 の反応条件下で合成を行った。 ベンゾ ト リ フルオ リ ド収率は 7 4 %であった。
実施例 3 2
実施例 1 5 の触媒を用いて、 へキサク ロ πアセ ト ンのフ ッ 素化反応を行った。 反応によって生成したガスをフ ッ化ナ ト リ ウム層を通して冷却し、 液化した後、 分留して生成物を得 た。 反応温度 350'C、 [SV]。600hr—'、 へキサク ロ 口ア セ ト ン に対するフ ッ化水素のモル比 1 8 の反応条件下で合成を行つ た。 へキサフルォロアセ ト ン、 ペンタフノレォロモノ ク ロルァ セ ト ンおよびテ ト ラ フルォ ロ ジク πルァセ ト ンがそれぞれ 14 , 22および 26 %の収率で得られた。 産業上の利用可能性
本発明は、 冷媒、 エアゾル噴射剤、 発泡剤、 洗剤等と して 有用な有機フ ッ素化合物の製造に有利に利用する こ とができ る。
Claims
1. 40〜 500人の平均細孔径を有する孔が 70〜90 %を占め る活性アル ミ ナを触媒担体とし、 これにニ ッ ケル、 コ バル ト、 鉄、 マ ンガン、 ク ロム、 銅および銀から選ばれる少な く とも
1 種の金属のフ ッ化物が a持された触媒の存在下に、 有機塩 素化合物または有機不飽和化合物とフ ッ化水素とを反応させ ることを特徴とする、 有機フ ッ素化合物の製造方法。
2. 前記金属のフ ッ化物が、 該金属の化合物を担持後、 気 相でフ ッ化水素処理して得られた、 該金属の部分フ ッ化物で ある、 請求の範囲第 1 項記載の方法。
3. 前記金属が無機または有機塩の溶液の形て'前記担体に 担持し、 次いで酸素または酸素含有ガス下 200〜 500 'Cで加 熱した後、 200〜 450てでフ ッ化水素処理する、 請求の範囲 第 1項、 または第 2項記載の方法。
4. 前記活性アル ミ ナが、 純度 99 . 9重量%以上で、 かつナ ト リ ゥム舍有率が l O Oppm以下である、 請求の範囲第 1 項記載 の方法。 '
5. 前記活性アルミナにおいて、 4 0 A以上の細孔直径の 孔の容積が 0 . 55〜: L 6 rag / £である、 請求の範囲第 1項また は第 4項記載の方法。
G. 前記有機不飽和化会物として ト リ ク ロ口ェチレンを用 I-:、 温度 200〜 450て、 ト リ ク ェチレ ンに対するフ ノ化 水素のモ几-比 3 〜 1 0 で反応させて ί - ク 口 c! - - 2 , 2 , 2 - ト リ フルォコエタ ンを製造する ¾求 Ο範囲第 1 項記載の方
法。
Ί, 前記有機塩素化合物と して 1 —ク ロ 口 — 2 , 2 , 2 , 一 ト リ フルォロェタ ンを用い、 温度 300 420 'C 1 一ク ロ Ό - 2 , 2 , 2 — ト リ フルォロェタ ンに対するフ ッ化水素の モル比 1 1 0 で反応させて 1 , 2 , 2 , 2 —テ ト ラ フルォ πエタ ンを製造する、 請求の範囲第 1 項記載の方法。
8. 前記有機不飽和化合物と してパーク ロ ロエチ レ ンを用 い、 温度 200 400て、 パーク ロ ロ エチ レ ンに対するフ ッ化 水素のモル比 3 6 で反応させて 1 , 1 —ジク ロ ロ — 2 , 2 , 2 — ト リ フルォ ロエタ ンを製造する、 請求の範囲第 1 項記載 の方法。
9. 前記有機塩素化合物と して 1 , 1 , 1 一 ト リ ク ロ ロェ タ ンを用い、 温度 150 400て、 1 , 1 , 1 — ト リ ク ロ ロェ タ ンに対するフ ッ化水素のモル比 1 4 で反応させて 1 , 1 1 — ジク ロ ロフルォロェタ ンおよび Ζまたは 1 , 1 , 1 ー ジ フルォロ ク ロ ェタ ンを製造する、 請求の範囲第 1 項記載の 方法。
10. 前記有機塩素化合物と して ク ロ 口 ホルムを用い、 温度 150 350て、 ク ロ 口 ホルムに対する フ ッ化水素のモ ル比
0. 5 3 で反応させてジフルォ ロ メ タ ンを製造する、 ¾求の 範囲第 1 項記載 C'方法。
11. 前記有機塩素化合物上- し て:'' フ ルォ ロ ジ ク ロ ロ メ タ ン および /または ト リ フルォ ロ ク ロ 口 メ ク ンを用い、 温度 200 460 t . ジフルォ ジ .ク ロ メ ク ンおよび, または ト :! フ ル'ォ 口 ク u メ ク ンに対する フ -:. 化水素 οモル上ヒ 0. π〜: 3 て
反応させてテ トラフルォロメ タ ンを製造する、 請求の範囲第 I 項記載の方法。
12. 前記有機不飽和化合物として ト リ フルォロク ロロェチ レンを用い、 温度 200〜 450 ΐ、 ト リ フルォロク ロロェチレ ンに対するフ フ化水素のモル比 1 〜 5 で反応させて 1 , 1 , 1 , 2 —テ トラフルォロクロロェタ ンおよびノまたは 1 , 1 : 1 , 2 , 2 —ペンタフルォロェタ ンを製造する、 請求の範囲 第 1項記載の方法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP63/104305 | 1988-04-28 | ||
| JP10430588 | 1988-04-28 | ||
| JP63/104306 | 1988-04-28 | ||
| JP10430688 | 1988-04-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1989010341A1 true WO1989010341A1 (fr) | 1989-11-02 |
Family
ID=26444809
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1989/000020 Ceased WO1989010341A1 (fr) | 1988-04-28 | 1989-01-10 | Procede de production d'un compose d'organofluor |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP0366797A4 (ja) |
| WO (1) | WO1989010341A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1050862C (zh) * | 1993-03-04 | 2000-03-29 | 北京大学 | 利用昆虫细胞高效表达人尿激酶原及其它有用蛋白质的方法 |
| US6080900A (en) * | 1991-05-23 | 2000-06-27 | Daikin Industries Limited | Process for fluorinating halogenated hydrocarbon |
| WO2005026090A1 (ja) * | 2003-09-10 | 2005-03-24 | Showa Denko K.K. | ハイドロフルオロカーボンの製造方法、その製品およびその用途 |
| JP2006111611A (ja) * | 2004-09-16 | 2006-04-27 | Showa Denko Kk | フルオロメタンの製造方法およびその製品 |
| CN111635291A (zh) * | 2020-06-09 | 2020-09-08 | 浙江省化工研究院有限公司 | 一种二氟一氯甲烷的制备工艺 |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9007029D0 (en) * | 1990-03-29 | 1990-05-30 | Ici Plc | Chemical process |
| TR25587A (tr) * | 1991-05-16 | 1993-07-01 | Ici Plc | 1,1,1,2-TETRAFLüORETANIN IMALINE MAHSUS ISLEM |
| WO1993019845A1 (en) * | 1992-03-28 | 1993-10-14 | British Technology Group Ltd | Catalyst for promoting halogen exchange |
| US5208395A (en) * | 1992-04-06 | 1993-05-04 | Elf Atochem North America, Inc. | Manufacture of hydrofluorocarbons |
| JP3792051B2 (ja) * | 1997-09-17 | 2006-06-28 | 日本ゼオン株式会社 | パーハロゲン化シクロペンテンの製造方法 |
| RU2152254C1 (ru) * | 1997-09-29 | 2000-07-10 | Институт нефтехимии и катализа АН РБ и УНЦ РАН | Катализатор для гидрохлорирования ненасыщенных соединений |
| RU2151640C1 (ru) * | 1997-09-29 | 2000-06-27 | Институт нефтехимии и катализа АН РБ и УНЦ РАН | Катализатор для гидрохлорирования ненасыщенных соединений |
| CA2403117C (en) * | 2000-03-31 | 2009-01-27 | Council Of Scientific And Industrial Research | A process for the preparation of 1,1,1,2-tetrafluoroethane |
| US7060165B2 (en) * | 2002-02-14 | 2006-06-13 | Pcbu Services, Inc. | Processes for purification and production of fluorocarbons |
| KR101397113B1 (ko) | 2006-10-03 | 2014-05-19 | 멕시켐 아만코 홀딩 에스.에이. 데 씨.브이. | 탄소수 3-6의 (하이드로)플루오로알켄의 생성을 위한 탈수소할로겐화 방법 |
| GB0706978D0 (en) | 2007-04-11 | 2007-05-16 | Ineos Fluor Holdings Ltd | Process |
| GB201207666D0 (en) | 2012-05-02 | 2012-06-13 | Mexichem Amanco Holding Sa | Process |
| CN102964231B (zh) * | 2012-12-17 | 2015-01-14 | 南京信息工程大学 | 一种气相催化制备1,1,1,3,3,3-六氟丙酮的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59108726A (ja) * | 1982-12-15 | 1984-06-23 | Central Glass Co Ltd | 3.3.3.−トリフルオロプロペン−1の製造方法 |
| JPH05318503A (ja) * | 1992-05-15 | 1993-12-03 | Toyota Motor Corp | ウレタンフォーム成形品の製造方法および製造装置 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2885427A (en) * | 1956-11-15 | 1959-05-05 | Dow Chemical Co | Fluorination of trichloroethylene |
| GB881003A (en) * | 1958-05-07 | 1961-11-01 | Ici Ltd | Improvements in the manufacture of organic halogen compounds |
| GB901297A (en) * | 1960-04-12 | 1962-07-18 | Union Carbide Corp | Process for the production of pentafluoroethane |
| JPS53135909A (en) * | 1977-04-28 | 1978-11-28 | Bayer Ag | Process for preparing 1*1*11trifluoroo22chloroethane |
-
1989
- 1989-01-10 EP EP19890901299 patent/EP0366797A4/en not_active Withdrawn
- 1989-01-10 WO PCT/JP1989/000020 patent/WO1989010341A1/ja not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59108726A (ja) * | 1982-12-15 | 1984-06-23 | Central Glass Co Ltd | 3.3.3.−トリフルオロプロペン−1の製造方法 |
| JPH05318503A (ja) * | 1992-05-15 | 1993-12-03 | Toyota Motor Corp | ウレタンフォーム成形品の製造方法および製造装置 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0366797A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6080900A (en) * | 1991-05-23 | 2000-06-27 | Daikin Industries Limited | Process for fluorinating halogenated hydrocarbon |
| CN1050862C (zh) * | 1993-03-04 | 2000-03-29 | 北京大学 | 利用昆虫细胞高效表达人尿激酶原及其它有用蛋白质的方法 |
| WO2005026090A1 (ja) * | 2003-09-10 | 2005-03-24 | Showa Denko K.K. | ハイドロフルオロカーボンの製造方法、その製品およびその用途 |
| JP2006111611A (ja) * | 2004-09-16 | 2006-04-27 | Showa Denko Kk | フルオロメタンの製造方法およびその製品 |
| CN111635291A (zh) * | 2020-06-09 | 2020-09-08 | 浙江省化工研究院有限公司 | 一种二氟一氯甲烷的制备工艺 |
| CN111635291B (zh) * | 2020-06-09 | 2022-09-30 | 浙江省化工研究院有限公司 | 一种二氟一氯甲烷的制备工艺 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0366797A1 (en) | 1990-05-09 |
| EP0366797A4 (en) | 1990-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5731481A (en) | Process for the manufacture of 1,1,1,2-Tetrafluoroethane | |
| WO1989010341A1 (fr) | Procede de production d'un compose d'organofluor | |
| JP2557936B2 (ja) | 触媒の存在下でのフッ化水素化による1,1,1−トリフルオロ−2,2−ジクロロエタンの製法 | |
| US5932776A (en) | Process for fluorination of perchloroethylene or of pentachloroethane | |
| KR100351211B1 (ko) | 크롬계플루오르화촉매,그촉매의제조방법및그촉매를사용한플루오르화방법 | |
| US5494876A (en) | Fluorination catalyst and fluorination process | |
| US2744148A (en) | Process for fluorination of haloalkanes using a hydrogen fluoride activated catalyst containing alumina, a metal fluoride and basic aluminum fluoride | |
| US3087976A (en) | Process for making halogenated organic compounds | |
| JPH11171806A (ja) | ペンタフルオロエタンの製造方法、並びにフッ素化用触媒及びその製造方法 | |
| CN104428273B (zh) | 用于制造含氟烯烃的方法 | |
| JPH09220469A (ja) | クロム酸化物をベースにした触媒とその生成方法及びハロゲン化炭化水素のフッ素化への適用 | |
| JPH0348632A (ja) | 1,1,1,2―テトラフルオロエタンの製造法 | |
| JP2015509096A (ja) | 含フッ素オレフィンの製造方法 | |
| US5616820A (en) | Process for the manufacture of 1,1,1,2-tetrafluoro-2-chloroethane and of pentafluoroethane | |
| JP3558385B2 (ja) | クロム系フッ素化触媒、及びフッ素化方法 | |
| JPH06506221A (ja) | 2−クロロ−1,1,1,2−テトラフルオロエタン及びペンタフルオロエタンの製造法 | |
| CN106748626B (zh) | 一种合成反式1-氯-3,3,3-三氟丙烯的方法及其催化剂的制备 | |
| JP2996598B2 (ja) | クロム系フッ素化触媒、その製法及びフッ素化方法 | |
| US3138559A (en) | Method for the preparation of a catalyst composition by the reaction of a fluorocarbo with activated alumina | |
| JPH04346943A (ja) | ハロゲン化炭化水素のフッ素化方法 | |
| CN102151576B (zh) | 一种用于氟化氯代烯烃的催化剂及其制备方法 | |
| JPH06506232A (ja) | 2−クロロ−1,1,1−トリフルオロエタンの製造法 | |
| JP3300120B2 (ja) | 1,1,1−トリフルオロ−2−クロロエタンの製造方法 | |
| JP3158720B2 (ja) | 1,1,1,2−テトラフルオロエタンの精製方法 | |
| US4273678A (en) | Method of preparing an oxychlorination catalyst |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): DE FR GB IT NL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1989901299 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1989901299 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1989901299 Country of ref document: EP |