明 細 書
新規セ フ ァ ロ スポ リ ン誘導体 技術分野
本発明は有用な抗菌活性を有する新規なセフ ユム化 合物、 詳し く はセフ ァ ロ スポ リ ン誘導体及びその製薬 学的に許容される塩とエス テルに関する。
背景技術
セフ ァ ロ スポ リ ン系抗生物質は細菌に優れた抗菌活 性を有しかつ哺乳類に対して低い毒性を有する こ とか ら哺乳類の細菌感染症の治療に極めて有効な薬剤であ る。
グラ ム陽性菌およびグラ ム陰性菌に対して広い抗菌 活性を示す種々の半合成セフ ァ ロ スポ リ ン誘導体がす でに合成され市販されて、 各種の細菌の感染性疾病の 治療剤 と して臨床に用いられている。 しかしながら、 既知のセ フ ァ α スポ リ ン誘導体のなかで、 緑膿菌ゃ変 形菌に対してすぐれた抗菌活性を示す誘導体は数少な い。 また既知のセフ ァ ロ スポ リ ン誘導体の多 く は耐性 菌によ り産生される ーラ ク タマーゼに対して不安定 であ り 、 現在臨床上問題と されている各種の耐性菌に 対する抗菌活性が低い欠点及びその他の欠点がある。 また、 既存のセフ エム系の抗菌性化合物の多 く は、 注 射剤と して開発されたものが主であるから、 経口投与 した場合、 体内への吸収率が悪く 、 充分な治療有効性 を示さ ない とい う 欠点も有していた。 近年、 セフ エム
環の 7 位にァ ミ ノチアゾリ ルァセチル基を有するセ フ ァ ロ スポ リ ン誘導体の数多 く が強い抗菌力 と ^ ー ラ ク タマーゼに対する安定性を持つこ とから、 研究され開 発されてい.る。
例えば、 セフ エム環の 7位にア ミ ノ チアゾ リ ルァセ チル基を有し且つ 3位に ) 3 —(置換又は非置換) ビニル 基を有する既知のセフ エム化合物には、 次式 (A)
(A)
CHzCOOH で示されるセフ ィ キシム(Cef ixime) と、 次式 (B〉
(B)
で示されるセフジニール(Cef dinir)がある
〔式中、 R1はァ ミ ノ基又は保護されたァ ミ ノ基であ り R2は低級アルキル基、 カルボキ メ チル基又は保護さ れたカルボキシメ チル基であ り ; R3は水素原子、 塩生
成カチオン又はカルボキシル保護基であ り 、 : そ して Aはフ ヱニル基、 低級アルキルー フヱニル基、 低級ァ ルコキシーフエニル基又はハロ ー フ エニル基、 あるい はフ リ ノレ基、' ニ ト ロ ーフ リ ノレ基、 又はハロ ーフ リ ル基、 あるいはチアゾ リ ル基、 低級アルキル一チアゾ リ ル基 又はハロ ーチアゾリ ル基、 若し く はチア ゾ リ ル環上の 3位以外の位置に低級アルキル基の 1 個を置換基と し て有する こ とができ且つチア ゾ リ オ基中の 3位の第 4 級窒素原子に配位する陰イオン性のカ ウ ンターイオン を有する 3—低級アルキル一チア ゾ リ オ基を表わす〕 で示されるセフ エム化合物 ( シ ン異性体) 又はその塩 又はそのエステルが知られている (特公平 3-64503号, 欧州特許第 Q175610号及び米国特許第 4,839, 350号参 照)。 一般式 (C) の化合物の一例には、 7 — 〔 2 —メ ト キシィ ミ ノ ー 2 — ( 2—ァ ミ ノ チアゾールー 4 ーィ ル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 ー メ チルチア ゾ 一ノレ一 5 — ィ ノレ〉 ビニノレ〕 一 3 — セ フ エ ム ー 4 — 力 ノレ ボン酸(シン異性体、 シス異性体) (以下、 ME 1206化合 物 と略記される) がある。 この ME1206化合物の ピパロ ィルォキシメ チルエステル (以下、 ME 1207化合物 と略 記される) は一般名セフジ ト レン ピボキシル
(Cef ditoren pivoxil) と して知られる。
〔式中、 R1は低級(C 〜C6)アルキル基であ り 、 R2は生 体内で加水分解によ り 脱離できるエステル形成基であ り 、 しかも 4-メ チルチアゾ リ ル基とセフエム環と は、 セフ ヱ ム環の 3位にある置換ビニル基の炭素一炭素 2 重結合に関してシス位にある〕 で示されるセフ エム化 合物のシン異性体は経口投与時の吸収性が髙いこ とが 知られる (米国特許第 4, 918, 068号及び欧州特許第 023 6231号参照)。
本発明者らは、 セ フ ィ キシム、 セ フ ジニール及び ME 1206化合物よ り も向上された抗菌活性をも ち且つその 他の優れた性資をも つ新規なセ フ ァ ロ スポ リ ン誘導体 を提供する こ と を 目 的と して鋭意研究を統けた。 その 結果、 セフ エム環の 3位に — ( 4一置換又は非置換 チアゾールー 5 — ィル) ビュル基を有し且つセ フ エ ム 環の 7位に 2 — ( 2 —ァ ミ ノチア ゾールー 4一ィル) ァセ ト ア ミ ド基又は 2 — ( 5—ア ミ ノ ー 1 , 2 , 4 — チアジア ゾールー 3 —ィル) ァセ ト ア ミ ド基を有し、 しかも 7 位の側鎖中のァシル基のひ位に ヒ ドロ キシィ ミ ノ基 (保護されていても よい) を置換基と して導入 してあるセフ ァ ロ スポ リ ン誘導体であって、 後記の一 般式(I ) で総括的に表し う る新規なセフ ァ ロ スポ リ ン
誘導体を合成す.るこ と に成功した。
そして本発明者らは後記の一般式(I) で示される新 規セファ ロスポリ ン系化合物は極めて広範囲の抗菌ス ベク トルを有するこ と、 そしてグラム陽性菌及びグラ ム陰性菌並びに各種の耐性菌に対しても強い抗菌活性 を示し、 またセフィ キシム, セフジニール及び ME1206 化合物よ り も優れた抗菌活性を有する こ と、 特に黄色 ブ ド ウ球菌のみな らず、 前記の 3 種のセフ エム化合 物が抗菌活性を実質上示さ ない腸球菌フ エ 力 リ ス (Enterococcus faecal is ) に対 して も極めて強レヽ抗菌 活性を示すこ とを見出した。 これらの知見に基いて本 発明を完成した。
発明の開示
すなわち、 第 1 の本発明による と、 次の一般式(I)
〔式中、 Xは CHも し く は Nであ り、 R1はァ ミ ノ基又は保 護されたァ ミ ノ基であ り、 R2は水素原子であるか又は ヒ ドロキシィ ミ ノ基の保護基であ り 、. R3は水素原子、 塩形成カ チオ ン又はカ ルボキ シル保護基であ り 、 R4は 水素原子であ り 、 R5は水素原子、 低級アルキル基、 ハ ロ ー (低級) アルキル基又はハロ.ゲン原子を表わす〕 で示されるセフ ヱ ム化合物のシン異性体、 又はその製
薬学的に許容できる塩又はエステルが提供される。
式(I ) の本発明化合物において、 セフ エム環の 3位 に置換したビ ル基の 位に結合しているチア ゾ リ ル 基は、 そのチアゾール環上に全く置換基を持たないチ ァゾールー 5 —ィル基であるか、 若し く は 4一低級ァ ルキルチアゾーノレ一 5 — ィル基、 4一ノヽロ チアゾール 一 5一ィル基あるいは 4一ノヽ ロ (低級)アルキルチアゾ 一ルー 5 —ィル基である こ とができ る。 式(I ) の本発 明化合物には、 3位の置換ビニル基の置換基の位置に 依存する (E)異性体(すなわち ト ラ ンス異性体) と (Z) 異性体(すなわちシス異性体) があ り 、 本発明は(E)異 性体、 又は(Z )異性体又はそれらの混合物を包含する。
上記及び下記の説明において示される種々の用語の 定義と、 それら定義された用語に含まれる適当な例を 詳細に説明する と次の とお り である。
用語 「低級アルキル基」 又は 「低級アルコ キ シ基」 における 「低級 j なる語は特にこ と わ り のないかぎ り 炭素原子 1〜 6個のも のである こ と を意味する も のと する。 式(I ) の化合物において R 1が保護されたァ ミ ノ 基である場合、 ァ ミ ノ基の保護基と は、 第 3級ブ ト キ シカルポニル基のごと き低級アルコ キシカルボニル基、 又はホル ミル基、 ク ロ ロ アセチル基のごと きハロ ー低 級アルカ ノ ィル基、 も し く は ト リ チル基な どの、 酸加 水分解又は加水素分解によ り 容易に脱離でき る通常の ァ ミ ノ保護基である。 R2が ヒ ドロ キシィ ミ ノ基の保護
基である場合、 これは ト リ チル基、 メ ト キシメ チル基 のごと き低級アルコキシ一低級アルキル基、 又はメ ト キシェ ト キシメ チル基のごと き低級アルコキシ一低級 アルコキシ 低級アルキル基のよ う な酸加水分解によ り容易に脱離できる ヒ ドロ キシィ ミ ノ保護基である。
R3が塩形成カチオンである場合、 アルカ リ 金属塩、 アルカ リ 土類金属塩、 アンモニゥム塩等のカチオンが 挙げられる。 また R3がカルボキシル保護基である場合、 かかる保護基と して、 ァ リ ル (al ly l )基、 低級アルコ キシメ チル基、 低級アルキルチオメ チル基、 低級アル 力 ノ ィルォキシメチル基又は低級アルコキシ置換ベン ジル基 (例えば p—メ ト キシベンジル基) である、 セ フ ァ ロ スポ リ ンの 4位カルボキシル基の保護に通常用 いられる カルボキシル保護基が挙げられる。
式(I ) の本発明化合物がエステルの形である場合に は、 R 3は生体内で加水分解できて脱離でき る代謝的に 不安定なエステル形成基と して、 低級アルコキシカル ボニルォキシアルキル基、 低級アルキルカルボニルォ キシアルキル基、 及び置換基 (例えば 5 —低級アルキ ル基) を有しても よい ( 2—ォキ ソ一 1, 3—ジォキ ソ レン一 4一ィル) メ チル基その他が举げられる。 R 5 がハロ ー (低級) アルキル基又はハロ ゲン原子である 場合、 かかるハロ置換基又はハロ ゲン原子と しては、 フ ッ素原子、 塩素原子、 臭素原子、 ヨ ウ素原子が挙げ られるが、 フ ッ素原子または塩素原子が好ま しい。
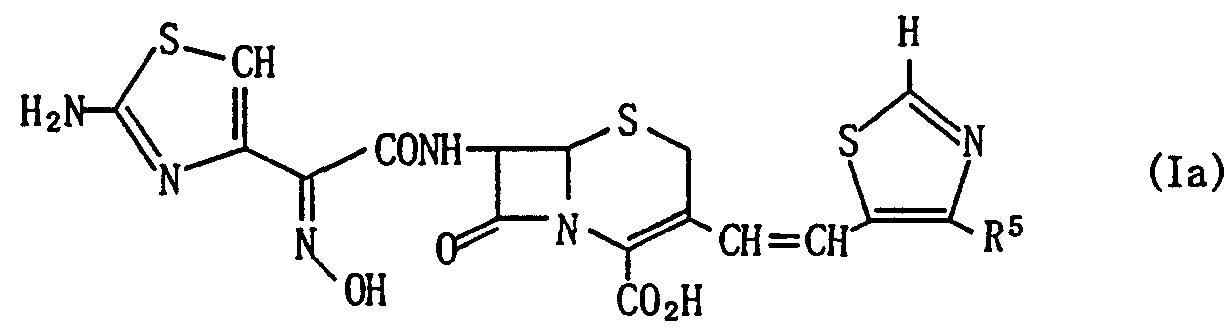
第 1 の本発明によ る式 ( I ) のセフヱム化合物又は その塩又はエステルは、 下記に示す式 (la) のセフエ ム化合物、 式 (lb) のセフ ム化合物及び式 (Ic) の セフエム化合物、 あるいはこれらの塩又はエステルを 包含する。
(1) 次式 (la)
〔式中、 R5は水素原子、 低級アルキル基、 ハロ ー (低 級) アルキル基又はハロ ゲン原子を表わす〕 で示され るセフヱム化合物のシン異性体、 又はその 4位カルボ キシル基における製薬学的に許容でき る塩又はエステ ル。
(2) 次式 (lb)
〔式中、 R
5は水素原子、 低級アルキル基、 ハロ ー (低 級) アルキル基又はハロゲン原子を表わす〕 で示され るセフヱム化合物のシン異性体、 又はその 4位カルボ キシル基における製薬学的に許容でき る塩又はエステ ル
〔式中、 Xは CHも し く は Nであ り 、 Aはアルコキシ力 ルポニル基、 ホルミ ル基、 ノヽロ ー (低級) アルカ ノ ィ ル基及び ト リ チル基から選ばれる酸加水分解又は加水 素分解によ り 容易に脱離でき るァ ミ ノ保護基であ り 、 R2a は ト リ チル基、 低級アルコ キ シ一低級アルキル基 及び低級アルコ キシ一低級アルコ キ シ一低級アルキル 基よ り選ばれる酸加水分解によ り容易に脱離でき る ヒ ド ロ キシィ ミ ノ 保護基であ り 、 R3a はァ リ ル基、 低級 アルキル基、 低級アルコ キ シメ チル基、 低級アルキル チオメ チル基、 低級アルカ ノ ィルォキシメ チル基及び 低級アルコキシ置換べンジル基から選ばれるカルポキ シル保護基であ り 、 R5は水素原子、 低級アルキル基、 ハロ ー (低級) アルキル基又はハロ ゲン原子である〕 で示される官能基が保護されたセフ ヱ ム化合物。
式 (la) のセ フ エ ム化合物又はその塩又はそのエス テルの具体的な例には、 7 — [ 2—ヒ ド ロ キシィ ミ ノ 一 2 — ( 2 — ァ ミ ノ チア ゾールー 4一ィ ル) ァセ ト ァ ミ ド〕 一 3 — 〔 2 — ( 4一ク ロ 口 チア ゾーノレ一 5—ィ ル) ビニノレ 〕 一 3 —セ フ エ ム ー 4 - カルボン酸 (シン 異性体) (シス異性体) ; 7 — 〔 2—ヒ ド π キ シイ ミ
ノ ー 2 — ( 2—ア ミ ノ チアゾール ー 4一ィ ル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4一 ト リ フルォロ メ チルチア ゾールー 5 —ィル) ビニル〕 一 3 —セフ エムー 4 一力 ルボン酸 (シン異性体) (シス異性体) ; 7 — 〔 2 — ヒ ドロ キシイ ミ ノ ー 2 — ( 2 —ァ ミ ノ チア ゾールー 4 一ィル) ァセ ト ア ミ ド〕 ー 3 — 〔 2 — ( 4 ーメ チルチ ァゾールー 5 —ィル) ビニル〕 一 3 —セフエムー 4一 カルボン酸 (シン異性体) (シス異性体) ; 7 — 〔 2 · - ヒ ド ロ キシイ ミ ノ ー 2 — ( 2 —ァ ミ ノ チアゾールー 4一ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2— (チア ゾール 一 5 —ィル) ビニル〕 一 3 —セフ エムー 4 一力ルボン 酸 (シン異性体)(シス異性体) ; 又は 7 — 〔 2—ヒ ド ロ キシイ ミ ノ ー 2 — ( 2 —ァ ミ ノ チアゾールー 4 ーィ ル) ァセ ト ア ミ ド〕 一 3 — 〔 2— (チア ゾールー 5 — ィル) ビニル〕 一 3—セフエムー 4 一力ルボン酸 (シ ン異性体)( ト ラ ンス異性体又はシス異性体と ト ラ ンス 異性体と の混合物) ; あるいはこれらセフ エム化合物 のナ ト リ ゥム塩、 あるいはピパコ イルォキシメ チルェ ステル、 ァセ ト キシメ チルエステル、 1 ーァセ ト キシ ェチルエステル、 1 一 (エ ト キシカルボニルォキシ) ェチノレエステノレ、 ( 2—ォキ ソ一 1 , 3 —ジォキ ソ レ ンー 4一ィル) メ チルエステル又は( 5—メ チルー 2 一ォキ ソ一 1 , 3—ジォキ ソ レン一 4 ー ィノレ) メ チル エステルがある。
式 (lb) のセフエム化合物又はその塩又はそのエス
テルの具体的な例には、 7 — 〔 2 — ヒ ド ロ キシィ ミ ノ 一 2 — ( 5 —ア ミ ノ ー 1 , 2 , 4 ーチアジアゾールー 3 —ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 一 ク ロ 口 チアゾール 5 —ィル) ビエル〕 一 3 —セ フ エ ム ー 4 一力ルボン酸 (シン異性体)(シス異性体) ; 7 — 〔 2 ー ヒ ド ロ キシイ ミ ノ ー 2 — ( 5 —ア ミ ノ ー 1 , 2 , 4 ーチアジアゾールー 3 — ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 一 ト リ フルォ ロ メ チルチアゾールー 5 — ィ ル) ビニル〕 一 3 —セ フ エ ム ー 4 一 力ルボン酸(シン 異性体) (シス異性体) ; 7 — 〔 2 — ヒ ドロ キ シイ ミ ノ ー 2 — ( 5 —ア ミ ノ ー 1 , 2 , 4 ーチア ジア ゾール — 3 —ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 ー メ チ ルチア ゾールー 5 — ィル) ビエル〕 一 3 —セ フ エ ムー 4 一 力ルボン酸 (シン異性体)(シス異性体) ; 又は 7 一 〔 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 5 —ア ミ ノ ー 1 ,
2 , 4 —チア ジア ゾールー 3 —ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — (チア ゾールー 5 —ィル) ビニル〕 一 3 ー セ フ エ ムー 4 一 力 ルボン酸 (シン異性体)(シス異性 体) ; あるいはこれらセフ エム化合物のナ ト リ ゥ ム塩、 あ る いは ピ ノ ロ イルォキ シ メ チルエステル、 ァセ ト キ シ メ チノレエス テノレ 、 1 ー ァセ ト キ シェチノレエス テノレ 、 1 — (エ ト キ シカノレポニノレオキ シ) ェチルエス テノレ、 ( 2 —ォキ ソ 一 1 , 3 —ジォキ ソ レン一 4 一ィル) メ チルエス テル又は ( 5 — メ チルー 2 —ォキ ソ 一 1 , 3 一ジォキ ソ レン一 4 ー ィノレ ) メ チルエス テルがあ る 。
式(Ic) のセ フ エ ム化合物の具体的な例には、 7 — 〔 2 — ト リ チルォキ シイ ミ ノ ー 2 — ( 2 — ト リ チルァ ミ ノ チ ア ゾールー 4 一ィ ル) ァ セ ト ア ミ ド〕 一 3 — 〔 2 — ( 4一ク ロ 口 チアゾールー 5—ィル) ビュル〕 一 3 —セ フ エ ム ー 4 一 力ルボン酸 p — メ ト キ シベンジ ルエス テル (シン異性体)(シス異性体) ; 7 — 〔 2 — ト リ チルォキシイ ミ ノ ー 2 — ( 2 — ト リ チルア ミ ノ チ ァ ゾ一ルー 4 一ィ ル) ァ セ ト ア ミ ド〕 一 3 — 〔 2 — ( 4一 ト リ フ ノレオ ロ メ チルチアゾーノレ一 5 — ィル) ビ -ル〕 一 3—セ フ エ ム ー 4 一 力ルボン酸 p—メ ト キシ ベンジルエステル (シ ン異性体)(シス異性体) ; 7 — 〔 2 — ト リ チルォキ シイ ミ ノ ー 2 — ( 2 — ト リ チルァ ミ ノ チ ア ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — ( 4 ー メ チルチア ゾールー 5—ィル) ビニル〕 一 3—セ フ エ ム ー 4 一 力ルボン酸 p—メ ト キ シベンジ ルエス テル ( シン異性体)(シス異性体) ; 7 — 〔 2 — ト リ チルォキ シィ ミ ノ 一 2 — ( 2 — ト リ チルア ミ ノ チ ァ ゾール ー 4 一ィ ル) ァ セ ト ア ミ ド〕 一 3 — 〔 2 — (チアゾールー 5 — ィル) ビニル〕 一 3 —セ フ エ ム ー 4 一 力ルボン酸 p—メ ト キ シベンジルエス テル (シン 異性体)(シス異性体又は ト ラ ンス異性体又はシス体と ト ラ ンス体 と の混合物) ; 7 — 〔 2— ト リ チルォキ シ ィ ミ ノ 一 2 — ( 5— ト リ チルア ミ ノ ー 1 , 2 , 4 ー チ ア ジア ゾールー 3—ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 一 ( 4 ー メ チルチア ゾールー 5—ィル) ビニル〕 一 3
ーセフエムー 4 一力ルボン酸 p — メ ト キシベンジルェ ス テル (シン異性体)(シス異性体) ; 又は 7 — 〔 2 — ト リ チルォキシイ ミ ノ ー 2 — ( 5— ト リ チルア ミ ノ ー 1, 2, 4 ·^チアジア ゾールー 3—ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4一 ト リ フルォロ メ チルチアゾー ルー 5 — ィル) ビニル〕 一 3 —セ フ エ ム ー 4 一 力ルポ ン酸 P —メ ト キシベンジルエステル(シン異性体) (シ ス異性体)等がある。
第 1 の本発明による一般式(I ) のセフエム化合物は、 式 (l a ) の化合物及び式 (lb ) の化合物を含めて、 そ れらの製薬学的に許容でき る塩の形である こ と ができ る。 そのよ う な製薬学的に許容される塩の適当な例は、 通常の製薬学的に許容でき る非毒性の塩であ り 、 その よ う な塩にはアルカ リ 金属、 た と えばナ ト リ ウ ム、 力 リ ウ ム との塩、 及びアルカ リ 土類金属、 た と えばカル シゥム、 マグネシウ ム との塩のよ う な金属塩、 アンモ ニゥ ム塩、 製薬学的に許容でき る有機塩基、 た と えば ト リ ェチルァ ミ ン、 ト リ メ チルァ ミ ン、 ピ リ ジン、 ピ コ リ ン、 ジシ ク ロへキシルァ ミ ン、 N, N ' -ジベンジル エチ レ ンジァ ミ ン と の塩、 な らびに製薬学的に許容で き る有機酸、 た と えば酢酸、 ト リ フルォロ酢酸、 マ レ イ ン酸、 酒石酸、 メ タ ンスルホ ン酸、 ベンゼンスルホ ン酸、 ギ酸、 トルエンスルホン酸と の塩、 またはア ミ ノ酸、 た と えばアルギニン、 ァスパラギン酸、 グルタ ミ ン酸と の塩が含まれる。
本発明の一般式 (I) のセフ ァ ロ スポ リ ン誘導体は、 各種の病原性細菌類に対する強い抗菌活性を有する も のである。 また経口投与時にかな り 良い吸収性を示し、 さ らに投与後の高い血中濃度を与えて保持する性質を 持っている若干の化合物例をも包含する。 以下に本発 明化合物の有利な薬理学的性質を若干の代表的化合物 について試験例で説明する。
試験例 1
本例では、 本発明の一般式(I) の化合物の う ちの下 記の代表例の試験化合物の抗菌活性を、 常法の倍数希 釈法で測定された各種の細菌に対する最小発育阻止濃 度 (MIC, g/ml)を示すこ と によ り 例証する。 この際 の測定は、 感性ディ ス ク用培地 N ( 日水製薬社製) に 供試菌を 106CFU/ml で接種し、 35 °Cで 18〜 20時間培養 後に評価して行われた。 試験化合物は下記の化合物 A 〜化合物 Fのナ ト リ ゥム塩である。
(1) 化合物 A :
7 - 〔 2 — ヒ ドロ キシィ ミ ノ 一 2 — ( 2 —ア ミ ノ チ ァ ゾールー 4 一ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 一 ク ロ 口チアゾールー 5 — ィ ル) ビ -ノレ〕 一 3 — セフ エムー 4 一力ルボン酸(シン異性体, シス異性体) (後記の実施例 2参照)
(2) 化合物 B :
7 - 〔 2 — ヒ ドロ キシイ ミ ノ ー 2 — ( 2 —ア ミ ノ チ ァ ゾール ー 4 ー ィ ノレ) ァセ ト ア ミ ド 〕 一 3 — 〔 2 —
( 4 一 ト リ フルォロ メ チルチア ゾールー 5 — ィル) ビ ニル〕 一 3 —セ フ エ ム ー 4 一 力 ルボン酸 (シン異性体, シス異性体)(後記の実施例 5参照)
(3) 化合物 C :
7 — 〔 2 — ヒ ド ロ キ シィ ミ ノ 一 2 — ( 2 —ア ミ ノ チ ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — ( 4 ー メ チルチアゾールー 5 — ィル) ビュル〕 一 3 — セ フ エ ム ー 4 一 力ルボン酸 (シ ン異性体, シス異性体) (後記の実施例 8参照)
(4) 化合物 D :
7 — 〔 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2 —ア ミ ノ チ ァ ゾ一ルー 4 一ィ ル〉 ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — (チア ゾールー 5 — ィル) ビニル〕 一 3 —セ フ エ ム ー 4 一力ルボン酸(シン異性体, シス異性体) (後記の実 施例 11参照)
(5) 化合物 E :
7 — 〔 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2 —ア ミ ノ チ ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 ー 3 — 〔 2 — (チアゾールー 5 — ィル) ビニル〕 一 3 —セ フ エ ム ー 4 —カルボン酸(シ ン異性体, ト ラ ンス異性体) (後記 の実施例 13参照)
(6) 化合物 F :
7 - 〔 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 5 —ア ミ ノ ー 1 , 2, 4 ーチア ジア ゾールー 3 —ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 ー メ チルチア ゾールー 5 —ィル)
ビニル〕 一 3 —セ フ エ ムー 4 一 力ルボン酸 (シ ン異性 体, シス異性体)(後記の実施例 16参照)
上記の化合物 A〜 Fの最小発育阻止浪度 (MIC, μ g /ml ) の測定値を表 1 に示す。 また、 比較のため、 同 様に測定さ れた ME 1206化合物(Na塩), セ フ ジニール
(Na塩) (CFDNと略記) 及びセ フ ィ キシム (Na塩)(CFIX と略記) の MIC値 ( iig/ml) も表 1 に示す。
最小癸育阻止濃度 ( g/ ml)
商 Ά ME1206 CFDN CFIX 化合物 A 化合物 B 化合物 C 化合物 D 化合物 E 化合物 F
(比 ¥乂) (比較) (比較) スタ フィ π π ッ カス '了フ レフ 0.10 0.39 0.20 0.10 0.20 0.39 0.39 0.20 12.5
(Sta aureus) 209P JC- i
スタ フイロコッカス.ァゥ レウス 6.25 6.25 6.25 3.13 6.25 3.13 25 6.25 100
(Sta. aureus) M133*
スタフイロコッカス.ァゥレウス 25 25 25 25 25 50 100 > 100 > 100
(Sta. aureus) Ml 26*
ェンテロコッカス.ヒラエ 12.5 6.25 6.25 12.5 25 25 50 50 > 100
(Ent. hirae) ATCC 8043
ェンテロコッカス.フエカ リス 6.25 12.5 12.5 1.56 3.13 25 100 25 > 100
(Ent. faecalis) W-73
ェシヱリキア .コリ(E. coli) 255 12.5 6.25 12.5 6.25 6.25 25 12.5 50 > 100 ェシエリキア .コリ(E. coli) GN206 1.56 1.56 3.13 1.56 0.78 6.25 0.78 25 50 モルガネラ 'モルガ二 12.5 1.56 1.56 12.5 6.25 25 25 25 50
(M. morganii) 1510
ェンテロノ ίクタ一.ク ロァカェ 1.56 1.56 0.78 0.78 0.78 1.56 1.56 6.25 0.78
(E. cloacae) G- 0008
セラチア .マルセセンス 1.56 1.56 1.56 0.78 0.78 3.13 0.39 25 0.78
(oer. marcescens No.l
(注) *印はメチシリ ン耐性黄色ブドゥ球菌 (MRSA)であることを示す <
試験例 2
化合物 Cの ピパロ ィルォキシメ チルエステル (後記 の実施例 9 の化合物) と化合物 Dの ピパロ ィルォキシ メ チルエステル (後記の実施例 12の化合物) を、 供試 化合物 と してマ ウス(ICR系、 雄性、 4週令, 1 群 3 匹) に 1 匹当 り 0.5mgの投与量で経口投与した。 投与方法 と して供試化合物を 0.2 %CMC水溶液に懸濁した液を経 口投与した。
なお、 供試化合物 (エステル体) はいずれも投与後 に消化器から吸収されて生体内で 4位カルボキシル基 上のエステル形成基が容易に脱離して、 対応する遊離 のカルボン酸の形と なる。 投与後 0 〜 4 時間の間に尿 中に排泄された遊離のカルボン酸の形の本発明化合物 を定量し、 尿中回収率を算定した。
定量方法 : 高速液体ク ロ マ ト グラ フ ィ 一法(カ ラ ム: Licrosorb RP18 4 φ X 150ram ; UV - detector 270nm で 検出) で行った。
得られた結果 ( 3例平均) を表 2 に示す。
2
試 3
本例では、 本発明化合物の代表例 と して前記の化合 物 A, B , C , D , E及び Fが皮下注射時に高い血中 濃度を維持する こ とを下記の試験法で例証する。
試験方法は次の通 り である 。 すなわち、 4 週令の Jcl ; ICR (ォス) マウスに、 注射用蒸留水にとかした
2.5mg/ral の供試化合物の水溶液を、 皮下投与で 0.2ml ずつ (0.5mg/マウス) 投与した。 投与後 5, 15, 30, 60, 120 分間の時点で腋下から採血した。 採取した血液は 室温で約 2時間放置し、 遠心分離機で 10分間遠心し、 血清を採取した。 得られた血清にメ タノールを等量加 え混和し、 1200 Orpra 3分間遠心し、 上清をフ ィ ルタ ー (サンプレップ LCR13-LH)に通し、 HPLC用血清サンプ ルと した。 HPLCを行って供試化合物の血清中濃度を測 定した。
供試化合物について、 薬動力学パラメ ーターである 血中半減期と血中濃度曲線下面積 (AUC) (Area under the plasma Concentration - 1 irae curve ) を Gauss - New ton法で算出した。
比較のために、 ME 1206化合物も同様に試験した。 得 られた結果を表 3 に示す。
表 3
第 1 の本発明による一般式(I ) のセフエム化合物の う ち、 官能基の保護基を脱離された化合物、 すなわち 前記の式(l a)と式(l b)のセフエム化合物又はその製薬 学的に許容される塩またはエステルは、 これを細菌感 染症の治療の目的で投与するにあたっては、 上記化合 物を有効成分と して含有し、 経口投与、 非経口投与又 は外用に適した有機または無機質の固体又は液体の担 体と混合されて抗菌剤組成物の形に製剤化されう る。
この組成物は慣用の製剤の形であるこ とができる。 この様な製剤と しては、 カプセル、 錠剤、 軟膏、 座薬 溶液、 懸濁液、 乳剤などが挙げられる。 さ らに必要に よ り前記製剤に捕助剤、 安定剤、 湿潤剤又は乳化剤、 緩衝剤その他の通常使用される添加剤を含有させる こ とができる。
従って、 第 2 の本発明による と、 次式 (la )
(式中、 R
5は水素原子、 低級アルキル基、 ハ口 - (低級) アルキル基又はハロ ゲン原子を表す) で示されるセフ ェム化合物のシン異性体、 又はその 4位カルボキシル 基における製薬学的に許容でき る塩又はエステル、 あ るいは次式 (lb )
(式中、 R5は水素原子、 低級アルキル基、 ハ π - (低級) アルキル基又はハロ ゲン原子を表わす) で示されるセ フヱム化合物のシン異性体、 又はその 4位カルボキシ ル基における製薬学的に許容でき る塩又はエステルの 少な く と も 1 つを、 有効成分と して含み、 しかも有効 成分が製薬学的に許容し得る固体状又は液体状の担体 と混和されている こ と を特徵とする、 抗菌性組成物が 提供される。
第 2 の本発明によ る抗菌性組成物は注射剤、 経口剤 または坐剤な どの製剤の形で投与される。 賦形剤およ び担体と しては製薬学上許容される も のが選ばれ、 そ の種類は投与経路や投与方法によ って決まる。 例えば、 液状担体と して水、 アルコールも し く は大豆油、 ゴマ 油、 ミ ネラル油な どの動植物油、 または合成油な どが 用いられる。 固体担体と してマル ト ース、 シュ ク ロ ー スな どの糖類、 リ ジンな どのア ミ ノ酸類、 ヒ ド ロ キシ
プロ ピルセルロ ース な どのセルロ ース誘導体、 シ ク ロ デキス ト リ ンな どの多糖類、 ステア リ ン酸マグネシゥ シムな どの有機酸塩な どが使用される。
注射剤と して製剤する場合には、 一般に生理食塩水、 各種緩衝液、 グルコ ース、 イ ノ シ ト ール、 マ ンニ ト ー ルな どの糖類溶液、 エチ レ ングリ コール、 ポ リ エチ レ ングリ コールな どのグ リ コール類が望ま しい。 また、 イ ノ シ ト ール、 マ ンニ ト ール、 グルコ ース、 マ ン ノ ー ス、 マル ト ース、 シュ ク ロ ースな どの糖類、 フ エ ニル ァラニンなどのア ミ ノ酸類な どの賦形剤 と共に、 凍結 乾燥製剤 と し、 それを投与時に注射用の適当な溶剤、 例えば滅菌水、 生理食塩水、 ブ ド ウ糖液、 電解質溶液、 ア ミ ノ酸溶液な どの静脈投与用液体に溶解して用いる こ と も でき る 。
製剤された組成物中における式(l a ) 又は式 (l b)の セフ ヱ ム化合物の含量は製剤型によ り 種々異なるが、 通常は 0 . 1〜 9 9重量%、 好ま し く は 1 〜 9 0重量%であ る。 例えば注射液の場合には、 通常 0 . 1〜 1 0重量%の 有効成分化合物を含むよ う にする こ とがよい。 経口投 与の場合には、 前記固体担体も し く は液状担体と共に 錠剤、 カ プセル剤、 粉剤、 顆粒剤、 ドラ イシ ロ ッ プ剤、 液剤、 シロ ッ プ剤な どの形態で用い られる。 カプセル、 錠剤、 顆粒、 粉剤の場合、 一般に有効成分化合物の含 量は 3 〜 9 9重量%、 好ま し く は 5 〜 9 0重量%であ り 、 残部は担体である。
本発明によ る式(I ) の有効成分セフ エ ム化合物また はその塩またはエス テルの投与量は、 患者の年令、 体 重、 症状、 治療目的な どによ り 決定されるが、 投与量 は感染した細菌を殺滅するのに有効な量である。 しか し、 その投与量は動物試験の結果な ど種々の状況を勘 案して総投与量が一定量を越えない範囲で、 連続的ま たは間けつ的に投与でき る。
非経口投与の場合における その総投与量は投与方法、 患者の状況、 例えば年令、 体重、 性別、 食事、 併用薬 剤な どに応じて適宜変更して投与する こ と はも ちろん である。 一定の条件下における適量と投与回数は、 上 記の指針を基に して専門医の決定によ らなければなら ない。 これらの投与条件は経口投与においても 同様で ある。
本発明の別の要旨においては、 前記の式 (l a ) のセ フ エム化合物又は式 (lb ) のセフ エム化合物あるいは これの製薬学的に許容でき る塩又はエス テルを抗菌剤 の製造に用いる使用が提供される。
発明を実施するための最良の形態
次に、 式(I ) の本発明の化合物の製造方法を説明す る。 これは種々の方法によ って製造でき るが、 下記の 反応式で図式的に示した工程 1 、 工程 2 および工程 3 からなる製造法 A、 も し く は後記に示す製造法 Bで製 造するのが便利である。
製造法 A
化合物 ( I )
上記の反応式で X , R1, R2, R3, R 及び R5 は前記 と同じであ り 、 R6はァ リ ール基、 例えばフ エニル基で あ り 、 Wはハロ ゲン原子を意味する。
製造法 Aの各工程について詳し く 説明する と、 次の
とお り である。 '
工程 1
—般式 (I I ) で示される化合物またはその塩に一般 式(I I I ) で示される ト リ 置換ホス フ ィ ンを作用させる。 化合物 (I I ) の適当な塩と しては、 化合物(I ) につい て例示したも の と同じ塩基と の塩類が使用でき る。
この反応は、 ヨ ウ化ナ ト リ ウム、 ヨ ウ化カ リ ウ ム、 臭化ナ ト リ ゥ ム等のアル力 リ 金属ハラ ィ ドの様な金属 ハライ ド共存下に実施する こ と が望ま しい。 反応は、 アセ ト ン、 N, N — ジ メ チルホルムア ミ ド、 ジ メ チル スルホキシ ド、 塩化メ チ レ ン、 テ ト ラ ヒ ド ロ フ ラ ン、 酢酸ェチルまたはこれらの混合溶媒中で行われる。 反 応温度は特に限定されないが、 室温が望ま しい。 反応 生成物 と して一般式 (IV ) で示される化合物が生成し、 これは必要であれば単離しても よい。
工程 2
一般式 (IV ) で示される化合物またはその塩に塩基 を作用させる。 化合物 (IV ) の適当な塩類と しては、 化合物 ( I ) で例示したも の と同じ塩基との塩類が使 用でき る。 こ の工程で使用する塩基と しては炭酸水素 アルカ リ 金属 (た と えば炭酸水素ナ ト リ ウ ム、 炭酸水 素カ リ ウ ムな ど) 、 炭酸アルカル金属 (た と えば炭酸 ナ ト リ ウ ム、 炭酸カ リ ウムな ど) 、 炭酸アルカ リ 土類 金属 (た と えば炭酸カルシウ ムな ど) な どの無機塩基 類、 または ト リ (低級)アルキルア ミ ン (た と えば ト リ
メ チルァ ミ ン、 ト リ ェチルァ ミ ン) 、 ピ リ ジン、 N— (低級〉 アルキルモルホ リ ン、 N, N — ジ (低級) 了 ルキルベンジルァ ミ ン等が挙げられる。 この反応は、 通常、 アセ ト ン、 テ ト ラ ヒ ドロ フ ラ ン、 塩化メ チ レン、 水またはこれらの混合溶媒中で行われる。 反応温度は 特に限定されないが、 室温が望ま しい。 反応生成物と して一般式(V ) で示される化合物が生成し、 これは必 要であれば単離しても よい。
工程 3
一般式(V ) で示される化合物またはその塩に一般式
(V I ) で示される アルデヒ ドを作用 して縮合させる。 化合物 (V ) の適当な塩と しては、 化合物 (I ) につい て例示したも の と同じ塩基との塩類が使用でき る。 こ の反応は、 塩化メチ レン、 テ ト ラ ヒ ドロ フ ラ ン、 ジォ キサンまたはこれらの混合溶媒中で行われる。 反応温 度は特に限定されないが、 反応は通常、 冷却下ない し 室温付近で行われる。 これによつて、 目的とする式(I ) の化合物が生成される。 また工程 2 と工程 3 は順次行 な う こ と な く 、 化合物 (I V ) に塩基とアルデヒ ド (V I ) を同時に作用させても よい。
製造法 B
化合物 ( I )
製造法 Bにおいては、 上記の反応式で示すよ う に式 (VII) (式中、 R3, R4及び R5は前記と 同じ意味である) で示される化合物、 も し く はそのア ミ ノ基における反 応性誘導体またはそれらの塩に、 式(VIII) (式中、 X R1および R2は前記と同じ意味である) で示される化合 物、 も し く はその力ルポキ シル基における反応性誘導 体またはそれらの塩を反応せしめる。
製造法 Bの方法で用いられる化合物 (VII)のァ ミ ノ 基における反応性誘導体の適当な例 と しては、 化合物 (VII ) と 、 アルデ ヒ ド、 ケ ト ンな どのよ う なカルボ二 ル化合物 との反応によって生成したシ ッ フ塩基型のィ ミ ノ誘導体またはその互変異性体であるェナ ミ ン型異 性体 ; あるいは化合物(VII) と ビス ( ト リ メ チルシ リ
ル) ァセ ト ア ミ ドな どのよ う なシ リ ル誘導体 ; あるい は化合物(V I I ) と三塩化リ ンまたはホス ゲン と の反応 に よ って生成した誘導体な どが挙げ られる 。 化合物 (V I I ) および化合物 (VI I I ) の適当な塩と しては、 有 機酸との塩 (た と えば酢酸塩、 マ レイ ン酸塩、 酒石酸 塩、 ベンゼンスルホ ン酸塩、 ト ルエンスルホ ン酸塩な ど) 又は無機酸との塩 (た と えば塩酸塩、 臭化水素酸 塩、 硫酸塩、 リ ン酸塩な ど) のよ う な酸付加塩、 アル カ リ 金属塩 (た と えばナ ト リ ウ ム塩、 カ リ ウ ム塩な ど) 、 アルカ リ 土類金属塩 (た と えばカルシウ ム塩、 マグネシウム塩な ど) のよ う な金属塩 ; アンモニゥム 塩 ; 有機ア ミ ン塩 (た と えば ト リ ェチルァ ミ ン塩、 ジ シク ロへキシルァ ミ ン塩な ど) な どが举げられる。
化合物 (VI I I ) のカルボキシル基における反応性誘 導体の適当な例 と しては、 酸ハロ ゲン化物、 酸アジ ド、 酸無水物、 活性ア ミ ド、 活性エス テルな どが挙げられ る。 さ らに詳細には、 酸塩化物、 酸臭化物、 置換リ ン 酸 (た と えばジアルキル リ ン酸、 ジベンジルリ ン酸、 ノヽロ ゲン化リ ン酸な ど) 、 ジアルキル亜リ ン酸、 亜硫 酸、 チォ硫酸、 硫酸、 炭酸アルキル (たと えば炭酸メ チル、 炭酸ェチルな ど) 、 脂肪族カルボン酸 (た と え ばピパ リ ン酸、 吉草酸、 ィ ゾ吉草酸、 ト リ ク ロ 口酢酸 な ど) 又は芳香族カ ルボン酸 (た と えば安息香酸な ど) のよ う な酸との混合酸無水物 ; ィ ミ ダゾール、 ジメ チ ル ビラ ゾール、 ト リ ァ ゾールま たはテ ト ラ ゾール と の
活性ア ミ ド ; 又は活性エス テル (た と えばシ ァ ノ メ チ ルエステル、 メ ト キシメ チルエステル、 ビエルエステ ノレ、 プロ ノ ノレギノレエステノレ、 p — 二 ト ロ フ エ ニノレエス テル、 2 , 4 —ジニ ト ロ フ エ ニルエス テノレ、 ト リ ク ロ 口 フエニノレエス テノレ、 ペン タ ク ロ ロ フエニノレエス テノレ、 メ シノレフ エ ニノレエス テノレ、 フ エ ニノレア ゾフ エ 二ノレエス テノレ、 フ エ ニノレチォ エス テノレ、 p — 二 ト ロ フ エ ニノレチ ォエステル、 p — ク レジルチオエステル、 カルボキシ メ チルチオエステル、 ビラニルエステル、 ピ リ ジルェ ステルな ど〉 、 も し く は N— ヒ ドロ キシ化合物 (た と えば N , N —ジメ チルヒ ドロ キシルァ ミ ン、 1 — ヒ ド 口 キシー 2 — ( 1 H ) — ピ リ ドン、 N — ヒ ド ロ キシス クシンイ ミ ド、 N— ヒ ドロ キシフ タルイ ミ ド、 1 ー ヒ ドロ キシー 6 — ク ロ ロ ー 1 H —ベンゾ ト リ アゾールな ど) とのエステルな どが挙げられる。 これらの反応誘 導体は使用すべき反応剤 (V I I I ) の種類によって適宜 選択される。
製造法 Bの方法においては、 化合物(V I I ) と化合物 (V I I I ) の反応は、 通常、 水、 アセ ト ン、 ジォキサン、 ァ セ ト ニ ト リ ノレ、 ク ロ 口 ホルム、 塩化メ チ レ ン、 テ ト ラ ヒ ドロ フ ラ ン、 酢酸ェチル、 N, N —ジメ チルホル ムア ミ ド、 ピ リ ジンのよ う な慣用溶媒又はこの反応に 影饗を与えない他の有機溶媒中で行われる。 こ れ ら の 溶媒は水と混合して使用 しても よい。
こ の反応において、 化合物 (V I I I ) を遊離の形また
は塩の形で使用する場合、 縮合剤 と しては、 た と えば N , N ' — ジシ ク ロ へキ シルカノレボジイ ミ ド ; N—シ ク ロ へキ シルー N ' —モルホ リ ノ ェチルカルボジイ ミ ド ; N—シ ク ロ へキシルー N ' — ( 4 ー ジェチルア ミ ノ シク ロへキシル) カルポジイ ミ ド ; N , N ' ージェ チルカルボジイ ミ ド ; N , N ' — ジイ ソ プロ ピル力ノレ ポジイ ミ ド ; N—ェチルー N ' — ( 3 — ジ メ チルア ミ ノ プロ ピル) カルポジイ ミ ド ; N , N ' —カルボニル ビス ( 2 — メ チルイ ミ ダゾール) ; ペン タ メ チ レンケ テ ン一 N—シ ク ロ へキ シノレイ ミ ン ; ジフ エ ニノレケテン 一 N—シ ク ロ へキシノレイ ミ ン ; エ ト キ シアセチ レ ン ; 1 ー ァノレコ キ シ一 1 一 ク ロ 口 エチ レ ン ; 亜 リ ン酸 ト リ アルキル ; ポ リ リ ン酸ェチル ; ポ リ リ ン酸イ ソ プロ ピ ル ; ォキシ塩化 リ ン ; 三塩化リ ン ; 塩化チォニル ; ト リ フ エ二ノレホス フ ィ ン ; N , N—ジメ チルホルムア ミ ド と塩化チォニル、 ホスゲン、 ォキシ塩化 リ ン と の反 応によ って得られるいわゆる ビルスマイヤー試薬な ど が举げられる。
こ の反応は、 また無機塩基又は有機塩基の存在下に 行なっていても よい。 このよ う な塩基の例 と しては、 炭酸水素アルカ リ 金属 (た と えば炭酸水素ナ ト リ ウム、 炭酸水素カ リ ウムな ど) 、 炭酸アルカ リ 土類金属 (た と えば炭酸カルシウムなど) な どの無機塩基類、 また は ト リ (低級) アルキルァ ミ ン (た と えば ト リ メ チル ァ ミ ン、 ト リ ェチルァ ミ ン) 、 ピ リ ジン、 N—(低級)
アルキルモルホ 'リ ン、 N, N —ジ (低級) ァノレキルべ ンジルァ ミ ン等が挙げられる。 反応温度は特に限定さ れず、 反応は通常、 冷却下ないし加温下に行なわれる。 こ の反応によって、 目的とする式(I ) の化合物が生成 される。
なお、 以上の方法で得られた本発明によ る一般式(I ) で表され且つ保護基をもつセフ エ ム化合物において、 必要であれば常法に従い、 保護基の除去を行なっても よい。 カルボキシル保護基又はァ ミ ノ保護基又はヒ ド 口 キシィ ミ ノ保護基の除去の方法は、 脱離される保護 基の種類によ り 適宜選択される。 ァ ミ ノ保護基の脱離 反応には加水分解又は加水素分解を行う こ とができ、 またア ミ ノ保護基がァ シル基である化合物に対しては ィ ミ ノハロ ゲン化剤、 次いでィ ミ ノ エーテル化剤を作 用 させた後、 必要に応じて加水分解する方法等の慣用 される任意の方法を適用でき る。 酸を用いた加水分解 の方法はア ミ ノ保護基を脱離させる一般的な方法の一 つであ り 、 例えばアルコキシ力ノレボニル基、 ホル ミル 基、 ト リ チル基等の基の脱離に適用される。 また使用 される酸と しては、 ギ酸、 ト リ フルォロ酢酸、 塩酸等 がァ ミ ノ 保護基の種類に応じて適宜選択される。 反応 は無溶媒下でも または水、 親水性有機溶媒も し く はそ れらの混合溶媒の存在下でも行う こ と ができ る。 また ト リ フルォロ酢酸を用いる場合はァニソールの存在下 に反応を行っても よい。 カルボキシル保護基の脱離反
応には加水分解、 還元等の慣用される任意の方法を適 用でき る。 酸を用いた加水分解の方法はカルボキシル 保護基を脱離させる一般的な方法の一つであ り 、 例え ばシ リ ル基、' ジフエニルメ チル基及び p —メ ト キシべ ンジル基等のの如き カルボキシル保護基の脱離に適用 される。 ヒ ドロ キシィ ミ ノ 基の保護に用いる メ ト キシ ェ ト キ シメ チル基の よ う な低級アルコ キ シ一低級アル コキシ一低級アルキル基は塩化メ チ レン溶液中で四塩 化チタ ンで処理する こ と によ り脱離でき る。
上記の製造法で得られた式(I) のセ フ エ ム化合物が 遊離カルボン酸の形である場合には、 また必要に応じ てカルボキシル基の代謝上不安定な無毒性エステル基 への変換をおこなっても よい。 代謝上不安定なエステ ルへの変換の方法はそれ自体公知の慣用されるエス テ ル化方法、 た と えばカルボン酸の形のセフ エム化合物 に導入すべきエステル形成基の反応性誘導体 (例えば ョ ーダイ ドな どのハラ イ ド) を溶媒中で反応させる方 法である。
以下、 本発明を参考例 と実施例によ り 説明する。
参考例 1
4 - ト リ フルォ ロ メ チル一 5 —チア ゾールアルデ ヒ ドの製造
4一 ト リ フルォロ メ チルー 5 —チア ゾールカ ルボン 酸ェチノレエス テノレ 〔L. F. Lee et al . , J. Heterocycl . Chem. , 22. 1621 ( 1985 ) 〕 2000mg の ト ルエン溶液 25ml
を一 75 °Cに冷却し、 ジイ ソブチルアルミ ニウ ム ヒ ド リ ド 1.58mlを滴下した。 同温度で 30分間撹拌した後、 水 3 mlを加えて室温まで昇温した。 1N塩酸を加えて撹拌 後酢酸ェチ'ルで 3 回抽出した。 有機層を無水硫酸マグ ネシゥムで乾燥した。 滅圧下濃縮しシ リ カゲルカ ラム ク ロマ ト グラ フ ィ ー (溶出溶媒 : へキサン : 酢酸ェチ ル = 3 : 1 )で精製して、 標題化合物 0.30mg (収率 19%) を得た。
NMR (CDC13) : δ 9.14(1H, s), 10.29(lH,s)
実施例 1
p ー メ ト キ シベンジノレ 7 — [ 2— ト リ チルォキシ イ ミ ノ ー 2 — ( 2— ト リ チルァ ミ ノ チア ゾ一ルー 4一 ィノレ ) ァセ ト ア ミ ド〕 一 3 — [ 2 — ( 4一ク ロ 口 チア ゾールー 5 — ィル) ビニル〕 一 3 — セ フ エ ムー 4 — 力 ルポキシ レー ト ( シ ン異性体)(シ ス異性体) の製造
(製造法 Aによ る)
P — メ ト キ シベンジル 7 — 〔 2— ト リ チルォキシ ィ ミ ノ 一 2 — ( 2— ト リ チルァ ミ ノ チア ゾールー 4一 ィル) ァセ ト ア ミ ド〕 一 3 — ク ロ ロ メ チル一 3 — セ フ ェ ムー 4一カ ルボキ シ レー ト 1532mgと 卜 リ フ エ ニルホ ス フ イ ン 413mg のァセ ト ン溶液 12ralに室温にて ヨ ウ化 ナ ト リ ゥ ム 263mgを加え 2 時間撹拌した。
反応液を滅圧下に濃縮乾固し次に塩化メ チ レ ン 15ml を加える。 得られた溶液に 4一ク ロ ロ ー 5 —チア ゾー ルアルデ ヒ ド〔米国特許第 4, 839, 350号明細書に示され
る〕 243 mgを加え、 次いで 5 %炭酸水素ナ ト リ ウ ム水 溶液 12.6mlを加えた。 室温で 2時間撹拌後に分液し、 水層は塩化メ チ レンにて抽出 した。 抽出液を有機層 と 混合して飽'和食塩水で洗浄し、 無水硫酸マグネシウム で乾燥後に滅圧下濃縮した。 残渣をシ リ カ ゲルカ ラム ク ロマ ト グラ フ ィ ^" (溶出溶媒 : へキサン : 酢酸ェチ ル = 3 : 2 ) で精製して、 標題化合物 1266mg (収率 76 % ) を得た。
NMR (CDC13) : δ 3.04(lH, d, J=18.9Hz) ,
3.43 (1H, d, J=18.9Hz), 5.15(3H,m), 6.11(lH,m),
6.36 (1H, d, J=12.8Hz), 6.44(1H, m),
6.64 (1H, d, J=12.8Hz), 6.84(2H,m), 7.26 ( 30H, in) , 8.43 (1H, s)
実施例 2
7 — 〔 2—ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2—ア ミ ノ チ ァ ゾール ー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — [ 2 — ( 4一ク ロ 口 チア ゾールー 5 — ィル) ビニル〕 一 3 — セ フ エ ム ー 4 一 力ルボン酸ナ ト リ ゥ ム塩(シ ン異性体) (シス異性体) の製造
実施例 1 で得た p — メ ト キ シベンジル 7 — 〔 2 — ト リ チルォキ シイ ミ ノ ー 2 — ( 2 — ト リ チルア ミ ノ チ ァ ゾール— 4 一ィ ル) ァ セ ト ア ミ ド〕 一 3 — 〔 2 — ( 4一ク ロ 口 チア ゾーノレ一 5 — ィノレ ) ビエル〕 一 3 — セ フ ヱ ム ー 4一カルボキシ レー ト (シ ン異性体) (シス 異性体) 920mgと 4 ー メ ト キシフ エ ノ ール 205 Orag のァ
二ソ ール溶液 1.5mlに氷冷下 ト リ フルォロ酢酸 7 ralを 滴下した。 氷冷下にて 1 時間撹拌後に、 冷却したイ ソ プロ ピルエーテルを加えた。
生じた沈'澱を濾取しイ ソプロ ピルエーテルで洗浄し、 乾燥させた後、 5 %炭酸水素ナ ト リ ウム水溶液で中和 した。 これを非イオン性吸着樹脂、 ダイヤイオン H P 一 2 0 のカ ラムで精製し、 目 的物を含む画分を集めて 滅圧下濃縮後に凍結乾燥して、 標題化合物 240mg (収率 57% ) を得た。
NMR (D20) : δ 3.41 (1H, d, J=18.5Hz) ,
3.68 (1H, d, J=18.5Hz), 3.81(3H,s),
5.43 (1H, d, J = 4.8Hz) , 5.92 (1H, d, J = 4.8Hz) ,
6.43(1H, d, J=ll.5Hz), 6.70 ( 1H, d, J = 11.5Hz),
7.03 (1H, s) , 8.87 (1H, s )
実施例 3
7 — [ 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2 —ア ミ ノ チ ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — ( 4 一 ク ロ 口 チア ゾールー 5 — ィル) ビエル ] 一 3 — セ フ エ ム ー 4 一 力ルボン酸 ピパロ ィルォキ シ メ チルェ ステル (シン異性体)(シス異性体) の製造
実施例 2 で得た 7 — 〔 2 — ヒ ド ロ キシイ ミ ノ ー 2 — ( 2 — ァ ミ ノ チア ゾールー 4 一ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 一 ク ロ 口 チア ゾールー 5 —ィル) ピ ニル〕 一 3 —セフ エ ム 一 4 一力ノレボン酸ナ ト リ ウム塩 (シ ン異性体)(シス異性体) 40mgを N, N—ジメ チルホ
ルムア ミ ド 1 ralに溶解した。 得られた溶液に氷冷下ョ 一ドメ チル ピパ レー ト 18.8 mgを加え米冷下の温度で 1 時間撹拌した。
反応液を'酢酸ェチルで希釈し少量の氷水で洗浄し、 無水硫酸マ グネシ ウ ムで乾燥後に 0.5ml まで滅圧下浪 縮した。 濃縮液に冷却したイ ソプロ ピルエーテルを加 え、 生じた沈澱を濂取しイ ソプロ ピルエーテルで洗浄 し、 乾燥させて、 標題化合物 15nig (収率 31 % ) を得た。 醒 (CDC13) : δ 1.14 (9Η, s) , 3.35 (1Η, d, J=18.0Hz) , 3.58 (1H, d, J=18.0Hz), 5.18 (1H, d, J=5. Hz) ,
5.77 (1H, d, J=5.1Hz) , 5.85 (1H, d, J=5.1Hz),
5.98 (1H, dd, J = 5.4, 9.0Hz), 6 · 45 (1H, d, J= 11.7Hz ), 6.72 (1H, d, J=ll.7Hz) , 7.10(lH,br), 8.01 (1H, s) 8.65 (1H, s) , 10.78 (1H, br)
実施例 4
p—メ ト キ シベンジル 7 — 〔 2— ト リ チルォキ シ イ ミ ノ ー 2 — ( 2 — ト リ チルァ ミ ノ チア ゾールー 4一 ィ ル) ァセ ト ア ミ ド: I — 3 — [ 2 — ( 4一 ト リ フルォ α メ チルチア ゾ一ノレ一 5 — ィル) ビニノレ 〕 一 3 —セ フ ェ ム ー 4一カ ルボキ シ レー ト (シン異性体)(シス異性 体) の製造
ρ — メ ト キ シベンジル 7 — 〔 2— ト リ チルォキシ イ ミ ノ ー 2 — ( 2— ト リ チルァ ミ ノ チア ゾールー 4一 ィノレ) ァセ ト ア ミ ド〕 一 3 — ク ロ ロ メ チルー 3 — セ フ ェ ムー 4一カルボキ シ レ一 卜 919mg , ト リ フ エ ニルホ
スフイ ン 259mg; ヨ ウ化ナ ト リ ウム 148rag, 及び参考例 1 によ り製造した 4 一 ト リ フルォロ メ チルー 5—チア ゾールアルデヒ ド 179mg を用いて、 実施例 1 と同様に 反応と精製'を行い、 標題化合物 700mg (収率 64% ) を得 た。
顏 (CDC13) : δ 2.98 (1H, d, J=18.6Hz) ,
3.14(1H, d, J=18.6Hz), 3.90(3H, s),
5.10(1H, d, J = 4.9Hz) , 5.17 (2H, s) ,
6.10(1H, dd, J = 9.2, 4.9Hz) , 6.43 (1H, s) ,
6.53 (1H, d, J=ll.9Hz), 6.88(3H,ra),
7.06 (1H, d, J=9.2Hz) , 7.29 (33H,ra), 8.48(1H, s) 実施例 5
7 — 〔 2—ヒ ドロ キシイ ミ ノ ー 2 — ( 2—ア ミ ノ チ ァ ゾールー 4 一ィル) ァセ ト ア ミ ド ] 一 3 — [ 2 — ( 4 - 卜 リ フ ノレオロ メ チルチアゾールー 5 —ィノレ ) ピ 二ル:! 一 3 —セフ エムー 4 一力ルポン酸ナ ト リ ゥ ム塩 (シン異性体)(シス異性体) の製造
実施例 4 で得た p — メ ト キシベンジル 7 — [ 2 — ト リ チルォキシイ ミ ノ ー 2 — ( 2 — ト リ チルア ミ ノ チ ァ ゾールー 4一ィル) ァセ ト ア ミ ド〕 一 3 — [ 2 —
( 4一 ト リ フルォロ メ チルチアゾールー 5 —ィノレ ) ビ 二ノレ 〕 一 3 —セ フエムー 4 一力ルポキシ レー ト (シン 異性体)(シス異性体) 700mgを用いて、 実施例 2 と 同様 に反応と精製を行い、 標題化合物 166rag (53% ) を得 た。
NMR (D20) : δ 3.36 (1Η, d, J=17.9Hz) ,
3.63 (1H, d, J=17.9Hz), 5.38 (1H, d, J = 4.9Hz) ,
5.90 (1H, d, 4.9Hz) , 6.59 (1H, d, J = ll.9Hz) ,
6.86 (ΙΗ,-d, J=ll.9Hz), 6.99(1H, s), 8.94(lH,s) 実施例 6
7 - 〔 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2 —ア ミ ノ チ ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — ( 4 一 ト リ フルォ ロ メ チルチア ゾーノレ一 5 — ィル) ビ ニル] 一 3 —セ フ エ ム ー 4 一 力ルボン酸 ピ ノ 口 イ ノレオ キ _シ_メ チルエステル (シン異性体 )丄シス ¾ 生体) の! L 実施例 5 で得た 7 — 〔 2 — ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2 —ァ ミ ノ チア ゾールー 4 ー ィノレ ) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 一 ト リ フルォ ロ メ チルチア ゾールー 5 — ィル) ビニル〕 一 3 —セ フ エ ム ー 4 一 力ノレボン酸 ナ ト リ ウ ム塩 (シン異性体)(シス異性体) 40mgを用い て、 実施例 3 と同様に ョー ドメ チル ピ ノ レ ー ト との 反応と精製を行い、 標題化合物 13mg(27% ) を得た。 NMR (DMS0 - d6) : δ 1.13 (9Η, s),
3.68 (1H, d, J=18.0Hz), 3.84(1H, d, J=18.0Hz) , 5.53 (1H, d, J=5.1Hz) , 5.91 (1H, d, J=5.1Hz) ,
5.98 (1H, d, J=5.3Hz) , 6.17 (1H, dd, J=5.3, 9.0Hz) , 6.88 (1H, d, J=ll.7Hz) , 7.07(1H, d, J=ll.7Hz), 7.40(1H, br), 9.44(1H, s) 9.80(1H, d, J=9.0Hz), 11.6 (1H, s)
実施例 7
ΐ) ー メ ト キシベンジル 7 — 〔 2— ト リ チルォキシ イ ミ ノ ー 2 — ( 2— ト リ チルァ ミ ノ チア ゾールー 4一 ィル) ァセ.ト ア ミ ド〕 一 3 — [ 2 — ( 4 ー メ チルチア ゾールー 5 — ィ ル) ビニル ] 一 3 —セ フ エ ム ー 4 一 力 ルポキシ レー ト ( シ ン異性体)(シス異性体) の製造 (製造法 Bのよ る)
2 — ( 2— ト リ チルァ ミ ノ チアゾールー 4一ィル) 一 2 — ト リ チルォキシィ ミ ノ酢酸 (シン異性体) 1000 mgの塩化メ チ レン溶液 15mlに、 氷冷下 N, N—ジシク 口へキシルカルポジイ ミ ド 322mg 及びヒ ドロ キシペン ズ ト リ ァゾール 21 lragを加え、 室温で 3 時間撹拌した。 次に p —メ ト キシベンジル 7—ア ミ ノ ー 3 — 〔 2 — ( 4 ー メ チルチアゾールー 5 — ィル) ビニル〕 一 3 — セ フ エ ム ー 4一カルボキ シ レー ト (シス異性体) 660mg を加え 5 °Cにて 12.5時間放置した。 不溶物を濾去し、 滅圧下に濃縮して得た残渣をシ リ 力 ゲルカ ラム ク ロマ ト グラ フ ィ ー (溶出溶媒 : へキサン : 酢酸ェチル = 2 : 3 ) で精製して、 標題化合物 1128rag (69% ) を得た。
N R (CDC13) : δ 2.45(3H,s), 3.10(1H, d, J=18. Hz) ,
3.44(lH,d, J=18.4Hz), 3.83(3H,s), 5.15(3H,ra), 6.11 (1H, m) , 6.32 (1H, d, J=ll.8Hz), 6.44(1H, s), 6.56 (1H, d, J=ll.8Hz), 6.83(3H,m), 7.30 (33H, m) , 8.49 (1H, s)
実施例 8
7 - 〔 2—ヒ ド π キシイ ミ ノ ー 2 — ( 2—ア ミ ノ チ ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — ( 4 ー メ チルチア ゾールー 5 — ィル) ピエル〕 一 3 — セ フ エ ム ー 4 一 力ルボン酸ナ ト リ ゥ ム塩(シン異性体) (シス異性体) の製造
実施例 7 で得た p —メ ト キシベンジル 7 — 〔 2 — ト リ チルォキシイ ミ ノ ー 2 — ( 2 — ト リ チルア ミ ノ チ ァ ゾールー 4 一ィル) ァ セ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 ー メ チルチア ゾールー 5 — ィル) ビニル〕 一 3 — セ フ エ ム ー 4一カルボキ シ レー ト (シン異性体)(シス 異性体) 1128mgを用いて、 実施例 2 と同様に反応と精 製を行い、 標題化合物 234rog (46 % ) を得た。
NMR (D20) : δ 2.39(3H, s), 3.36 (1H, d, J = 17.9Hz) , 3.62 (1H, d, J=17.9Hz), 5.40 (1H, d, J = 4.9Hz),
5.89 ( 1H, d, J = 4.9Hz) , 6.32 (1H, d, J= 11.6Hz ),
6.68 (1H, d, J=ll.6Hz), 7.00(lH, s), 8.79 (1H, s ) 実施例 9
7 - 〔 2—ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2—ア ミ ノ チ ァ ゾールー 4 一ィル) ァ セ ト ア ミ ド :! 一 3 — 〔 2 — ( 4—メ チルチア ゾールー 5—ィル) ビニル ] 一 3 — セ フ エ ム ー 4 一 力ルボン酸 ピノ ロ イルォキ シメ チルェ ス テル (シン異性体)(シス異性体) の製造
実施例 8 で得た 7 — 〔 2—ヒ ドロ キシイ ミ ノ ー 2 — ( 2—ァ ミ ノ チア ゾールー 4一ィル) ァセ ト ア ミ ド〕 — 3 — 〔 2 — ( 4 ー メ チルチア ゾールー 5—ィル) ビ
ニル〕 一 3 —セフエムー 4 一力ルボン酸ナ ト リ ゥ ム塩 (シン異性体) (シス異性体) 40mgを用いて、 実施例 3 と 同様に ョー ドメ チル ピパレー ト との反応と精製を 行い、 標題化合物 13mg(27% ) を得た。
NMR (DMSO-de) : δ 1.15 (9Η, s) ,
3.68 (1H, d, J=18.0Hz), 3.84(1H, d, J=18.0Hz) , 4.15(1H, d, J=5.3Hz) , 5.72 (1H, d, J = 5.1Hz) ,
5.79 (1H, d, J=5.1Hz) , 5.92 (1H, dd, J=5.3, 9.0Hz) , 6.38 (1H, d, J=ll.5Hz), 6.75 ( 1H, d, J = 11.5Hz), 8.10(lH,br), 8.98(1H, s) 9.60 (1H, d, J=9.0Hz) ,
11.50(1H, s)
実施例 10
p—メ ト キシベンジル 7 — 〔 2— ト リ チルォキシ イ ミ ノ ー 2 — ( 2— ト リ チルァ ミ ノ チア ゾールー 4一 ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2— (チア ゾールー 5 一ィル) ビニル〕 一 3 —セフ エムー 4一カノレポキシ レ 一 ト (シ ン異性体)(シス異性体及び ト ラ ンス異性体) の製造 (製造法 Aによ る)
p —メ トキシベンジル 7 — 〔 2— ト リ チルォキシ イ ミ ノ ー 2 — ( 2— ト リ チルァ ミ ノ チアゾールー 4一 ィル) ァセ ト ア ミ ド〕 一 3 — ク ロ ロ メ チルー 3 —セフ ェムー 4一カルボキシ レー ト 1156mg、 ト リ フ エニルホ ス フ イ ン 256mg、 ヨ ウ化ナ ト リ ウ ム 185mg及び 5—チア ゾールアルデヒ ド 640mg を用いて、 実施例 1 と 同様に 反応を行い、 シ リ カ ゲルカ ラ ム ク ロ マ ト グラ フ ィ ー
(溶出溶媒 : トルエン : 酢酸ェチル = 5 : 1 ) で精製 して、 標題化合物のシス異性体 400mg と、 ト ラ ンス異 性体 200rag と、 シス異性体及び ト ラ ンス異性体の混合 物 300ragと を'得た (総収率 93% ) 。
シス異性体
NMR (CDC13) : S 3.09 (1H, d, J=18.6Hz) ,
3.34(1H, d, J=18.6Hz), 3.80(3H,s), 5.15(3H,ra), 6.10 (1H, dd, J = 5.2, 9.0Hz) , 6.29 (1H, d, J=ll.8Hz) , 6.45(lH, s), 6.65(lH,d, J=ll.8Hz), 6.79(lH,br), 6.85 (2H, d, J=8.8Hz) , 7.25 (33H,m), 7.72(lH,s),
8.58 (1H, s)
ト ラ ンス異性体
NMR (CDCls) : δ 3.49 (2Η, s) , 3.82 (3H, s) ,
5.08 (1H, d, J = 4.9Hz) , 5.28 (2H, s) ,
6.03 (1H, dd, J = 4.9, 8.8Hz), 6.44 (1H, s) ,
6.82 (1H, d, J=8.8Hz) , 6.91 (3H, m) , 7.30 (34H,m), 7.80 (1H, s) , 8.68 (1H, s)
実施例 11
7 — 〔 2 — ヒ ドロ キシイ ミ ノ ー 2 — ( 2 —ア ミ ノチ ァ ゾールー 4 一ィル) ァセ ト ア ミ ド:! 一 3 — 〔 2 —
(チア ゾールー 5 —ィル) ビエル〕 一 3 —セフ エムー 4 一力ルポン酸ナ 卜 リ ゥム塩 (シン異性体)(シス異性 体) の製造
実施例 10で得た P —メ ト キシベンジル 7 — 〔 2 — ト リ チルォキシイ ミ ノ ー 2 — ( 2 — ト リ チルア ミ ノ チ
ァ ゾールー 4 —ィ ル) ァ セ ト ア ミ ド〕 一 3 — 〔 2 — (チアゾールー 5 — ィル) ビニノレ〕 一 3 —セ フ エ ム ー 4 一 力 ルポキ シ レ一 ト (シン異性体)(シス異性体) 400 mgを用いて、' 実施例 2 と同様に反応と精製を行い、 標 題化合物 120rag (68% ) を得た。
NMR (D20) : δ 3.41 (1H, d, J=18.3Hz) ,
3.69 (1H, d, J=18.3Hz), 5.45 (1H, d, J=4.8Hz) ,
5.93 (1H, d, J = 4.8Hz) , 6.33 (1H, d, J=ll.5Hz),
6.77(lH,d, J=11.5Hz), 7.03(lH,s), 7.81(lH,s), 8.89 (1H, s)
実施例 12
7 — 〔 2—ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2—ア ミ ノ チ ァ ゾ一ル ー 4 一ィル) ァ セ ト ア ミ ド ] 一 3 — 〔 2 — (チア ゾールー 5 — ィル) ビエル ] 一 3 —セ フ エ ム ー 4 一 力 ノレボン酸 ピ ノ ロ イノレォキシ メ チルエステル (シ ン異性体)(シス異性体) の製造
実施例 11で得た 7 — 〔 2—ヒ ドロ キ シイ ミ ノ ー 2 — ( 2—ァ ミ ノ チア ゾ一ルー 4 ー ィノレ ) ァセ ト ア ミ ド〕 一 3 — 〔 2— (チア ゾールー 5—ィル) ピニル〕 一 3 ーセフエムー 4 一力ルボン酸ナ ト リ ウ ム塩 (シン異性 体)(シス異性体) 22ragを用いて、 実施例 3 と同様に ョ 一ドメ チル ピパ レー ト と の反応 と精製を行い、 標題 化合物 8 mg(30% ) を得た。
NMR (DMS0-d6 ) : δ 1.14(9H, s),
3.66 ( 1 H, d, J=18.0Hz) , 3.85 ( 1H , d , J = 18.0Hz),
5.38 (1H, d, J=5.3Hz) , 5.68 (1H, d, J=5.1Hz) ,
5.74(1H, d, J=5.1Hz) , 5.90 (1H, dd, J = 5.3, 8.7Hz) , 6.32 (1H, d, J=ll.2Hz), 6.71 (1H, s),
6.82 (1H, d, J=ll.2Hz) , 7.95(1H, br) , 9.05 (1H, s) 9.65 (1 H, d, J=8.7Hz)
実施例 13
7 — [ 2—ヒ ド ロ キ シイ ミ ノ ー 2 — ( 2—ア ミ ノ チ ァ ゾール ー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — (チア ゾールー 5 — ィル) ビニル〕 一 3 —セ フ ヱ ム ー 4 一 力ルボン酸ナ ト リ ウ ム塩 (シン異性体)( 卜 ラ ンス 羼性体体) の製造
実施例 10で得た P — メ ト キ シベンジル 7 — 〔 2 — ト リ チルォキ シイ ミ ノ ー 2 — ( 2 — ト リ チルア ミ ノ チ ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — [ 2 — (チア ゾールー 5—ィル) ビエル〕 一 3 —セ フ エ ム ー
4 一カルボキ シ レー ト (シン異性体)( ト ラ ンス異性体) 200mgを用いて、 実施例 2 と同様に反応と精製を行い、 標題化合物 80mg (93% ) を得た。
NMR (D20) : 5 3.70(1H, d, J=18. OHz),
3.75 ( 1 H, d, J=18. OHz) , 5.29 (1H, d, J = 4.9Hz),
5.87 (1H, d, J = 4.9Hz) , 6.91 (1H, d, J=16.2Hz) ,
6.98 (1H, s) , 7.12 (1H, d, J=16.2Hz), 7.80(1H, s), 8.81 (1H, s )
実施例 14
7— ェ 2—ヒ ド ロ キ シイ ミ ノ ー 2 — _{ 2—ア ミ ノ チ
ァ ゾールー 4 一ィ ル) ァ セ ト ア ミ ド 〕 一 3 — 〔 2 — (チア ゾールー 5 — ィル) ビニル〕 一 3 — セ フ エ ム 一 4 一 力ルボン酸 ピパロ ィルォキ シ メ チルエス テル (シ ン異性体)(ト ラ ンス異性体) の製造
実施例 13で得た 7 — 〔 2 — ヒ ド ロ キシイ ミ ノ ー 2 —
( 2 —ァ ミ ノ チア ゾールー 4 一ィル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — (チア ゾールー 5 —ィル) ビニル〕 一 3 ーセ フ ヱ ムー 4 一 力 ルボン酸ナ ト リ ゥ ム塩 (シン異性 体)( ト ラ ンス異性体) 30mgを用いて、 実施例 3 と同様 に ョー ドメ チル ピパ レー ト との反応と精製を行い、 標題化合物 12mg (34% ) を得た。
醒 (DMS0-d6 ) : δ 1.16(9H,s),
3.74(1H, d, J=18.5Hz), 4.17 (1H, d, J=l 8.5Hz),
5.28 (1H, d, J=5.2Hz) , 5.84 (1H, dd, J = 5.0, 8.7Hz) , 5.86 (1H, d, J=5.4Hz) , 5.9 (1H, d, J = 5.4Hz) ,
6.71 (1H, s) , 7.16 (1H, d, J=16.4Hz) , 7.95(lH,br) , 9.08 (1H, s) , 9.54(1H, d, J=8.7Hz)
実施例 15
p — メ ト キ シベンジル 7 — 〔 2 — メ ト キ シェ ト キ シ メ チルォキ シイ ミ ノ ー 2 — ( 5 — ト リ チルア ミ ノ ー 1 , 2 , 4 —チアジア ゾールー 3 —ィル) ァセ ト ア ミ ド:! 一 3 — [ 2 — ( 4 —メ チルチア ゾールー 5 —ィル) ビニル〕 一 3 —セ フ エ ム ー 4 一 力ノレボキ シ レー ト (シ ン羼性体)(シス異性体) の製造 (製造法 B によ る)
2 — ( 5 — ト リ チルア ミ ノ ー 1 , 2 , 4 —チア ジア
ゾール一 3—ィル) 一 2—メ ト キシエ ト キシメ チルォ キシィ ミ ノ酢酸(シン異性体) 900ragと 7 —ア ミ ノ ー 3 - 〔 2 — ( 4 ー メ チルチアゾールー 5—ィル) ビニル〕 一 3—セフ ムー 4 一力ルボン酸(シス異性体) 827rag の塩化メ チレン溶液 20mlに、 一 20°Cにて ピ リ ジン 0.63 ralおよびォキ シ塩化 リ ン 0.19mlを順次加えた。 同 じ 一 20 °Cの温度にて 1 時間撹拌後、 氷水及び塩化メ チレ ンを加え撹拌後に有機層を分離した。 有機層は飽和食 塩水で洗浄し、 無水硫酸マグネシウ ムで乾燥後に滅圧 下溏縮した。 残渣をシ リ カゲルカ ラ ム ク ロ マ ト グラ フ ィ ー (溶出溶媒 : へキサン : 酢酸ヱチル = 1 : 4 ) で 精製して、 標題化合物 1435rag (88% ) を得た。
NMR (CDC13) : δ 2.41 (3Η, s) , 3 · 23 ( 1H, d, J = 18.2Hz ), 3.30 (3H, s) , 3.47(1H, d, J=18.2Hz), 3.54(2H,m), 3.80 (3H, s) , 3.91 (2H, m) , 5.12 (2H, s) ,
5.13 (1H, d, J = 4.8Hz) , 5.30 (1H, d, J = 7.5Hz) ,
5.41 (1H, d, J = 7.5Hz), 6.01 (1H, dd, J = 5.1, 8. Hz) ,
6.31 (1H, d, J=ll.8Hz) , 6.58 (1H, d, J =l 1.8Hz),
6.82 (2H, d, J=8.7Hz) , 7.28 (17H,ra), 7.48(lH,s), 8.05 (1H, d, J=8.5Hz) , 8.56 (1H, s)
実施例 16
7 — [ 2—ヒ ドロ キシイ ミ ノ ー 2 — ( 5—ア ミ ノ ー 1 , 2 , 4 —チアジア ゾールー 3—ィル) ァセ ト ア ミ ド ] 一 3 — [ 2 — ( 4—メ チルチアゾール 5—ィル) ビニル] 一 3 —セフエム一 4 —力ノレボン酸ナ ト リ ゥ ム
塩 (シ ン異性体)(シス異性体) の製造
実施例 15で得た p —メ ト キシベンジル 7 — 〔 2 — メ ト キシエ ト キシメ チルォキシイ ミ ノ ー 2 — ( 5 — ト リ チルア ミ ノ ー 1 , 2, 4 ーチアジアゾール一 3 —ィ ル) ァセ ト ア ミ ド〕 一 3 — 〔 2 — ( 4 ー メ チルチア ゾ 一ルー 5 — ィル) ビニル〕 一 3 — セ フ エ ム ー 4一カル ボキシ レー ト (シン異性体)(シス異性体) 700mg の塩 化メ チ レ ン溶液 10mlに 1.0M四塩化チタ ンの塩化メ チレ ン溶液 2. Omlを氷冷下で滴下し、 1.5時間撹拌した。 メ ト キシェ ト キシメ チル基の脱離の反応が行われた。 反 応液 5 mlを と り 、 実施例 2 と同様な反応剤を加えて同 様に反応と精製を行い、 標題化合物 35mgを得た。
NMR (D20) : 6 2.41(3H,s), 3.37(1H, d, J=18.2Hz) , 3.65 (1H, d, J=18.2Hz) , 5.41 ( 1H, d , J = 4.8Hz) , 5.94(1H, d, J=4.8Hz) , 6.34(1H, d, J=ll.8Hz),
6.69 (1H, d, J=ll.8Hz), 8.83 (1H, s )
産業上の利用可能性
以上のよ う に、 本発明においては、 各種の細菌に対 して高い抗菌活性を有する新規なセファロ スポ リ ン誘 導体が得られた。 これらセファロ スポ リ ン誘導体は細 菌感染症の治療に抗菌剤と して有用である。