WO1995011902A1 - Quinolinecarboxylic acid derivative and salt thereof - Google Patents
Quinolinecarboxylic acid derivative and salt thereof Download PDFInfo
- Publication number
- WO1995011902A1 WO1995011902A1 PCT/JP1994/001815 JP9401815W WO9511902A1 WO 1995011902 A1 WO1995011902 A1 WO 1995011902A1 JP 9401815 W JP9401815 W JP 9401815W WO 9511902 A1 WO9511902 A1 WO 9511902A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- carbon atoms
- atom
- salt
- acid derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
- C07D207/09—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to a novel quinoline carboxylic acid derivative and a salt thereof having an antibacterial activity useful as a medicament, an animal drug, a marine medicine, an agrochemical, a preservative, an industrial bactericide, and the like, and a synthetic intermediate for a quinoline carboxylic acid derivative. And a salt thereof.
- quinoline antibacterial agents have the disadvantage that they do not have strong antibacterial activity against Gram-positive bacteria such as Streptococcus phenococcus, and their antibacterial activity against resistant bacteria such as MRSA is still satisfactory. Instead, the development of new drugs was desired.
- the present inventors have conducted intensive studies to provide an excellent synthetic antibacterial agent with the above-mentioned requirements improved, and as a result, the following quinoline carboxylic acid derivatives and salts thereof have a wide antibacterial activity, In particular, they have found that they have strong antibacterial activity against Gram-positive bacteria and their drug-resistant bacteria, especially MRSA, and have completed the present invention. Disclosure of the invention
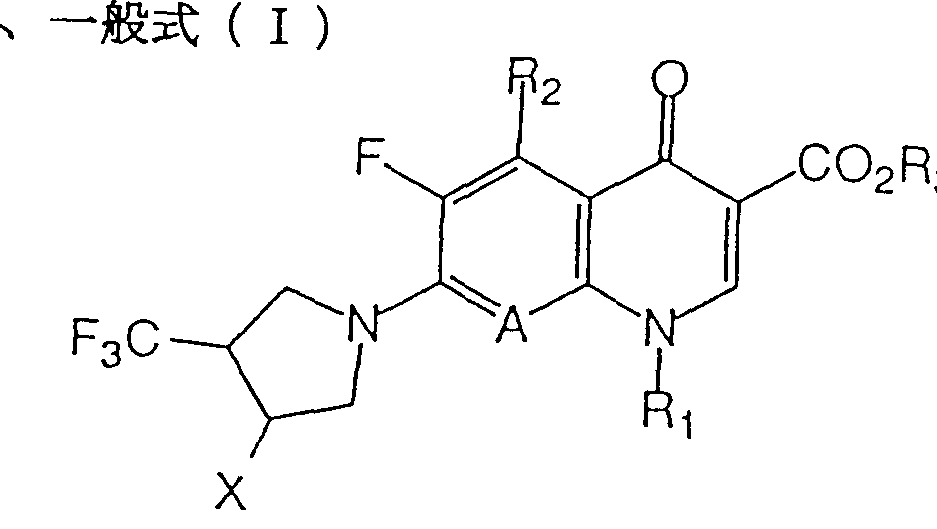
- the present invention is a.
- R 2 is a hydrogen atom; halogen
- hydroxyl group which may be protected, an amino group or Arukiruamino group having 1 to 6 carbon atoms; dialkyl ⁇ amino group or an alkyl group having 1 to 6 carbon atoms having 1 to 6 carbon atoms
- R 3 is hydrogen or C A is a nitrogen atom or
- A is a hydrogen atom, a halogen atom, an alkyl group having 1 to 6 carbon atoms, an alkoxyl group having 1 to 6 carbon atoms which may be substituted, a cyano group or a nitro group, and A ′ is May form a ring together with the ring, and the ring may further contain an oxygen atom, a nitrogen atom or a sulfur atom as a constituent atom, and may be replaced by an alkyl group having 1 to 6 carbon atoms.
- X represents a hydroxymethyl group; an aminomethyl group or an amino group which may be protected.
- X represents a hydroquinmethyl group; an aminomethyl group or an amino group which may be protected.
- a salt thereof. is there.
- the present invention provides an antibacterial agent containing the compound represented by the general formula (I) or a salt thereof as an active ingredient, and a bacterial infection using the compound represented by the general formula (I). It provides a method of treatment.
- a feature of the compound of the present invention is that a pyrrolidine residue having a trifluoromethyl group binds to quinoline carboxylic acid, whereby the compound can be used for a wide range of antibacterial spectrum, particularly against Gram-positive bacteria containing MRSA. It has a strong antibacterial activity.
- examples of the optionally substituted alkyl group having 1 to 6 carbon atoms represented by include a methyl group, an ethyl group, an isopropyl group, and a tert-butyl group.
- examples include a halogen atom such as a fluorine atom, a chlorine atom and a bromine atom, a hydroxyl group and a lower alkoxyl group.
- Specific examples of the alkyl group having 1 to 6 carbon atoms which may be substituted include, for example, a difluoromethyl group, a 2-fluoroethyl group, a 2-hydroquinethyl group, and a 2-methoxethyl group.
- Examples of the optionally substituted alkenyl group having 2 to 6 carbon atoms include an isopropyl group and a 2-fluoroisopropenyl group.
- Examples of the optionally substituted cycloalkyl group having 3 to 7 carbon atoms include a cyclopropyl group, a cyclobutyl group and a 2-fluorocyclopropyl group.
- the aryl group which may be substituted is selected from the group consisting of, for example, a halogen atom such as a fluorine atom, a chlorine atom and a bromine atom, a lower alkoxyl group, a hydroxyl group, an amino group, a lower alkyl group and a nitro group.
- Examples thereof include a phenyl group which may be substituted by up to three groups, and specific examples thereof include a phenyl group, a 2-fluorophenyl group, a 4-fluorophenyl group, a 2,4-difluorophenyl group, and a 2-phenyl group. Examples thereof include a droxyphenyl group, a 4-methoxyphenyl group, a 4-methylphenyl group, a 412 trophenyl group, and a 2,4-dinitrophenyl group.
- Examples of the halogen atom represented by R 2 include a fluorine atom and a chlorine atom.
- the protecting group for the hydroxyl group which may be protected include, for example, an acetyl group
- Examples of the protecting group for an amino group which may be protected and an alkylamino group having 1 to 6 carbon atoms include a acetyl group and a tert-butoxycarbonyl group.
- Examples of the optionally protected alkylamino group having 1 to 6 carbon atoms and dialkylamino group having 1 to 6 carbon atoms include a methylamino group, an ethylamino group, a dimethylamino group and the like.
- Examples of the alkyl group having 1 to 6 carbon atoms include a methyl group, an ethyl group, an isopropyl group and a tert-butyl group.
- Examples of the alkyl group having 1 to 6 carbon atoms represented by R 3 include a methyl group, an ethyl group, and an isopropyl group.
- Examples of the halogen atom represented by A ′ include a fluorine atom, a chlorine atom, and a bromine atom.
- Examples of the alkyl group having 1 to 6 carbon atoms include a methyl group and an ethyl group.
- Examples of the optionally substituted alkoxy group having 1 to 6 carbon atoms include a methoxy group, a fluoromethoxy group, a difluoromethoxy group, an ethoxy group, a 1-fluoroethoxy group, a 2-fluoroethoxy group and the like.
- Examples of the tricyclic quinoline skeleton containing a ring which A 'can form together with include 2,3-dihydro-7-hydroxyl7H-pyrido [1,2,3-de] [1, 41 Benzoxazines, 2,3-dihydro-7-oxo-17H-pyrido [1,2,3-de] [1,4] benzothiazines and the like.
- Examples of the optionally protected aminomethyl or amino group-protecting group represented by X include, for example, an acetyl group and a tert-butynecarbonyl group.
- the quinolinecarboxylic acid derivative (I) of the present invention can form an acid addition salt or a base addition salt.
- Examples of the acid addition salts include salts with mineral acids such as hydrochloric acid and sulfuric acid, and salts with organic acids such as acetic acid, trifluoroacetic acid, p-toluenesulfonic acid, and methanesulfonic acid.
- base addition salts include salts with alkali metals such as sodium and potassium, salts with alkaline earth metals such as magnesium and calcium, ammonium salts, trimethylamine, pyridine, N-methylbiperidine, and N-methyl.
- Examples thereof include salts with nitrogen-containing organic bases such as rumorpholine, getylamine, benzylamine, and N, N-dimethylethanolamine.
- the quinoline carboxylic acid derivative (I) of the present invention has an asymmetric carbon at the 3- and 4-positions of the pyrrolidine ring, and thus has an optically active form. Frequently, all the optically active substances and mixtures thereof are NH-containing in the compounds of the present invention, and can be separated by applying a known method.
- the compound of the general formula (I) is produced by condensing a quinoline derivative (III) with a pyrrolidine derivative (II).
- examples of the halogen atom represented by Y include a fluorine atom and a chlorine atom
- examples of the sulfonic acid residue include a methanesulfonic acid residue, a p-toluenesulfonic acid residue, and 2,4,6-triisopropyl Examples include benzenesulfonic acid residues.
- the condensation reaction includes aromatic hydrocarbons such as benzene, toluene and xylene, lower alcohols such as methanol, ethanol and isopropanol,
- the reaction is carried out in a non-protonic polar solvent such as ethers such as hydrofuran, dioxane or monoglyme, and acetonitrile, dimethylformamide, dimethylsulfoxide or sulfolane.
- the reaction temperature is usually from 0 ° C to 200 ° C, and the reaction time is usually from 10 minutes to 24 hours.
- the condensation reaction is usually carried out in the presence of a deoxidizing agent using 1 to 5 equivalents of the pyrrolidine derivative (II) per 1 equivalent of the quinoline derivative (III).
- the deoxidizing agent include alkaline metal hydroxides such as sodium hydroxide and hydroxide hydroxide, alkaline earth metal hydroxides such as magnesium hydroxide and calcium hydroxide, sodium carbonate and hydrogen carbonate.
- Alkali metal carbonates such as sodium carbonate carbonate or hydrogen carbonate carbonate, triethylamine, pyridine, N-methylmorpholine or 1,8-diazabicyclo [5,4,0] indene-7-ene (DBU) Organic bases.
- the amount of the deoxidizing agent used varies depending on the amount of the pyrrolidine derivative (II), and is usually 1 to 7 equivalents to 1 equivalent of the quinoline derivative (III).
- [Production Method 2] Production method of compound of general formula (I) wherein R 3 is a hydrogen atom
- R 2 , A and X are the same as described above, and R 3 ′ represents an alkyl group having 1 to 6 carbon atoms.
- the compound of the general formula (Ia) can be produced by hydrolyzing a compound of the general formula ( ⁇ ) in which R 3 ′ is an alkyl group having 1 to 6 carbon atoms, It can be carried out under any of acidic and acidic conditions.
- reaction temperature is usually 0'c to 100 ° C, and the reaction time is usually 10 minutes to 5 hours.
- reaction When the reaction is carried out under acidic conditions, hydrochloric acid, sulfuric acid, formic acid, acetic acid, trifluoroacetic acid, etc. or a mixture thereof is used as the acidic reactant, and usually a lower alcohol such as methanol, ethanol, isopropanol or water is used as a solvent. I do.
- the reaction temperature is generally 0 to 130'c, and the reaction time is usually 10 minutes to 10 hours.
- R 2 , R 3 , A, X and Y are the same as described above, and L represents a fluorine atom or an acetoxyl group.
- the compound of the general formula (Ia) is converted into a boron chelate compound (IV) by adding a boron reagent to a quinoline derivative (III), and then condensed with a pyrrolidine derivative (II) to form a boron chelate compound. After (V), it can be produced by treating with a base.
- boron reagent examples include hydrofluoric acid, boron trifluoride-ethyl ether complex, a mixture of boric acid and acetic anhydride, and the like.
- the boron reagent is used in an amount of 1.2 equivalents to a large excess with respect to the quinoline derivative (III), and is used in water or an ether-based solvent such as getyl ether, tetrahydrofuran, dioxane, etc. at 20 ° C. as needed. Performed with heating up to 'C.
- the reaction time is usually 30 minutes to 24 hours.
- the condensation reaction of the boron chelate compound (IV) with the pyrrolidine derivative (II) is carried out by lower alcohols such as methanol, ethanol and isopropanol, ethers such as tetrahydrofuran and monoglyme, acetonitrile, dimethylformamide, dimethyl
- the reaction is carried out in an aprotic polar solvent such as sulfoxide or sulfolane, and is usually carried out in the presence of a deoxidizing agent using 1 to 5 equivalents of a pyrrolidine derivative (II) to 1 equivalent of a boron chelate compound (IV).
- the deoxidizing agent examples include alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, magnesium hydroxide, alkaline earth metal hydroxides such as a hydroxide hydroxide, sodium carbonate, sodium hydrogen carbonate, and carbonate.
- Metal carbonates such as aluminum and hydrogen carbonate, and organics such as triethylamine, pyridine, N-methylmorpholine, 1,8-diazabicyclo [5,4,0] undeca-7-ene (DBU) Bases.
- the amount of the deoxidizing agent to be used is usually 1 to 7 equivalents to 1 equivalent of the boron chelate compound (IV).
- Bases that can be used to remove the boron reagent from the boron chelate form (V) include alkali metal hydroxides such as sodium hydroxide and hydroxide rim, alkalis such as magnesium hydroxide and calcium hydroxide. Earth metal hydroxides, alkali metal carbonates such as sodium carbonate and potassium carbonate, triethylamine, trimethylamine, And tertiary amines such as N-methylmorpholine.
- the amount of the base used is 2 equivalents to a large excess with respect to 1 equivalent of the boron chelate (V). This reaction is usually carried out using a water-containing lower alcohol as a solvent, usually at 20 '(: ⁇ 100'C, and the reaction time is 30 minutes to 12 hours.
- Compound (I— b) can be produced by removing the protective group of compound (I) wherein X obtained by Production methods 1, 2 and 3 is a protected aminomethyl group or a protected amino group.
- the method for removing the protecting group is a conventional method according to the protecting group to be used.
- the protecting group is a tert-butynecarbonyl group
- the protecting group is treated with an acid such as hydrochloric acid, trifluoroacetic acid, or the like.
- the reaction can be carried out by hydrogenation in the presence of a catalyst such as palladium carbon.
- a catalyst such as palladium carbon.
- the pyrrolidine derivative represented by the formula is a novel compound, and can form an acid addition salt.
- examples thereof include salts with mineral acids such as hydrochloric acid and sulfuric acid, and salts with organic acids such as acetic acid, trifluoroacetic acid, ⁇ -toluenesulfonic acid, and methanesulfonic acid.
- the pyrrolidine derivative (II) of the present invention has an asymmetric carbon at the 3- and 4-positions of the pyrrolidine ring, and thus has an optically active form.However, the configuration of the 3- and 4-positions of the pyrrolidine ring may be cis or trans, All optically active substances and their mixtures are contained in the compound of the present invention, and can be separated by applying a known method.
- R ′ and R ′′ represent an amino protecting group, which may be the same or different.
- Examples of the amino protecting group for R ′ and R ′′ include an acetyl group, a t-tert-butoxycarbonyl group, a benzyloxycarbonyl group and the like.
- the compound (VI) is hydrogenated in the presence of a catalyst such as palladium on carbon to give a compound (XII), and an amino protecting group R 'is introduced by a conventional method to give a compound (XIII), which is then hydrolyzed. Then, the compound (XIV) is obtained, and the carboxyl group of the compound (XIV) is converted to acyl azide with an azide reagent, and then heated to effect a transfer accompanied by denitrification, and the resulting isocyanate is reacted with an appropriate alcohol to obtain R Is a compound (XV) having a carbonyl group, and then the amino protecting group R 'is removed to produce the compound (II_c).
- a catalyst such as palladium on carbon
- an amino protecting group R 'is introduced by a conventional method to give a compound (XIII), which is then hydrolyzed.
- the compound (XIV) is obtained, and the carboxyl group of the compound (XIV) is converted to acyl azi
- the quinolinecarboxylic acid derivative (I) of the present invention has a strong antibacterial activity, it can be used as an antibacterial agent for medicines, animals and fish, or as a preservative for foods and a pesticide.
- the dose of the compound of the present invention for use as a medicament usually varies depending on the administration method and dosage form, but is in the range of 1 Omg to 1 g per day for adults. This daily dose is administered once or several times a day. The daily dose may be exceeded if necessary.
- the dosage for animals differs depending on the type and size of the animal and the type of infectious agent, but is usually in the range of 1 mg to 200 mg / kg body weight per day.
- the antibacterial agent comprising the compound of the present invention is prepared by selecting an appropriate preparation method according to the administration method, and preparing the various dosage forms which are usually performed.
- Antibacterial preparations include tablets, powders, granules, capsules, oral preparations such as solutions and syrups, as well as injections, solid preparations, external preparations, eye drops, nasal drops, etc. it can.
- a tablet containing 100 mg of the compound of Example 12a in one tablet is prepared by mixing the compound of Example 12a, corn starch, Avicel and magnesium stearate, and tableting.
- Example 12a After dissolving the compound of Example 12a and glucose in distilled water for injection, the mixture was injected into an ampoule, purged with nitrogen, and then pasteurized at 121 ° C for 15 minutes to obtain an injection having the above composition.
- — N MR spectrum is, d 6 - using dimethyl sulfoxide (DMSO-d 6) or heavy chloroform to (CDC 13) solution in tetramethylsilane (TMS) as an internal standard, JNM- EX270 Katasupeku Torometa ( 270 MHz, manufactured by JEOL Ltd.), and the ⁇ value was shown in ppm.
- Mass spectra were measured with a QP 100 OEX type spectrometer (manufactured by Shimadzu Corporation). Melting points were measured with a micro melting point analyzer (manufactured by Yanagimoto Seisakusho) without correction.
- Trans-1-benzyl-1-hydroxymethyl-4-trifluoromethylpiperidine 2.20 g (8.49 mmo 1) was dissolved in ethanol 2 Om 1, and concentrated hydrochloric acid (lml) was added. Add 440 mg of palladium carbon and hydrogenate at 40 ° C and 1 atmosphere. After 4 hours, the catalyst was removed by filtration and the filtrate was concentrated under reduced pressure to obtain 1.74 g (yield 99.7%) of the desired product as a colorless powder.
- Z represents a benzyloxycarbonyl group.
- the obtained residue was purified by silica gel column chromatography (elution solvent: n-hexane-ethyl acetate (5: 3 ⁇ 5: 4)) to obtain 8.04 g of the desired product as a slightly yellow oil ( Yield 85.9%).
- Trans-1-1-benzyloxycarbonyl-2-3-phthalimid-methyl-1-41-trifluoromethylpyrrolidine 9.33 g (21.6 mmo 1) was dissolved in methanol 15 Oml, and hydrazine monohydrate 2. Add lml (43.2 mmol) and stir at room temperature for 12 hours. The precipitate is removed by filtration, the filtrate is concentrated under reduced pressure, the residue is dissolved in dichloromethane, washed with water, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- trans-1-benzyloxycarbonyl-2- (tert-butoxycarbonylaminomethyl) —4— Trifluoromethylpiperidine 5.59 g (19.5 mm 01) of trans-1-benzyloxycarbonyl-1-aminomethyl-4-aminotrimethylpyrrolidine was dissolved in 6 Om 1 of dichloromethane, and 6.38 g of di-tert-butyl dicarbonate was dissolved. (29.3 mmo 1) and stirred at room temperature for 18 hours.
- reaction solution was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (eluent: chloroform) to give 5.94 g (yield: 75.8%) of the desired product as a pale yellow oil. .
- reaction solution is extracted with 150 ml of Jetil ether, and the organic layer is washed with saturated saline, dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- the residue was purified by silica gel column chromatography (eluent: n ⁇ xane-ethyl acetate (10: 1 ⁇ 5: 1)) to give 15.87 g of the desired product as a colorless oil (yield 92.3). %)Obtained.
- Example 10a 1-1-cyclopropyl-1 6,8-difluoro-1,4-dihydro-4-1 Suspension of 33-mg of oxoquinoline-1-carboxylic acid (0.606 mmol) in 2 ml of dichloromethane, ⁇ ) ⁇ 8 ⁇ ( ⁇ ' ⁇ ) ⁇ ⁇ -or '( ⁇ ' ⁇ ) ⁇ -96 ⁇ ⁇ '( ⁇ ' ⁇ ) ⁇ 8 ⁇ ⁇ '( ⁇ ' ⁇ ) ⁇ ⁇ - ⁇ ' ⁇ ' ⁇ ) m 'oi2'n ( + Yimr- (z / w) sw
- the antibacterial activity of the compound of the present invention was measured as a minimum inhibitory concentration (MIC) by an agar plate dilution method according to the standard method of the Japanese Society of Chemotherapy. Ofloxacin (0 FLX) was used as a control compound. The results are shown in Table 4.
- Example 2 la An acute toxicity test was performed using 6-week-old ICR mice.
- the compound of Example 2 la was used as a test compound, suspended in a 0.5% methylcellulose solution, and orally administered.
- Micronucleus tests were performed using 8-week-old ICR mice to confirm the presence of mutagenicity.
- Example 21 The compound a was suspended in olive oil and administered intraperitoneally, and peripheral blood smears were prepared by acridine orange staining before administration, 24, 48 and 72 hours after administration. When 1000 reticulocytes of each specimen were observed and the frequency of micronuclei was recorded, no change was observed even with 200 mgZ: kg administration.
- the quinoline carboxylic acid derivative of the present invention has a strong antibacterial activity against Gram-positive bacteria such as Streptococcus pentelococcus and also has a strong antibacterial activity against MRSA. It can also be used as a food preservative or pesticide.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Quinoline Compounds (AREA)
Description








































Claims





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69428206T DE69428206T2 (de) | 1993-10-28 | 1994-10-27 | Chinolincarbonsäurederivate und ihre salze |
| AU80035/94A AU8003594A (en) | 1993-10-28 | 1994-10-27 | Quinolinecarboxylic acid derivative and salt thereof |
| EP94931176A EP0726269B1 (en) | 1993-10-28 | 1994-10-27 | Quinolinecarboxylic acid derivatives and salts thereof |
| US08/632,474 US5668147A (en) | 1993-10-28 | 1994-10-27 | Quinolinecarboxylic acid derivatives and salts thereof |
| AT94931176T ATE205209T1 (de) | 1993-10-28 | 1994-10-27 | Chinolincarbonsäurederivate und ihre salze |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP29258793 | 1993-10-28 | ||
| JP5/292587 | 1993-10-28 | ||
| JP5/353780 | 1993-12-28 | ||
| JP35378093 | 1993-12-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995011902A1 true WO1995011902A1 (en) | 1995-05-04 |
Family
ID=26559054
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1994/001815 Ceased WO1995011902A1 (en) | 1993-10-28 | 1994-10-27 | Quinolinecarboxylic acid derivative and salt thereof |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US5668147A (ja) |
| EP (1) | EP0726269B1 (ja) |
| AT (1) | ATE205209T1 (ja) |
| AU (1) | AU8003594A (ja) |
| CA (1) | CA2175351A1 (ja) |
| DE (1) | DE69428206T2 (ja) |
| WO (1) | WO1995011902A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996033992A1 (en) * | 1995-04-27 | 1996-10-31 | Kaken Pharmaceutical Co., Ltd. | Optically active quinolinecarboxylic acid derivatives and salts thereof |
| RU2167867C2 (ru) * | 1998-07-28 | 2001-05-27 | Пфайзер Продактс Инк. | Способ получения тровафлоксацина |
| WO2005073238A1 (ja) * | 2004-02-02 | 2005-08-11 | Daiichi Pharmaceutical Co., Ltd. | ピリドベンズオキサジン誘導体 |
| JPWO2005026147A1 (ja) * | 2003-09-10 | 2007-11-08 | 杏林製薬株式会社 | 7−(4−置換−3−シクロプロピルアミノメチル−1−ピロリジニル)キノロンカルボン酸誘導体 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8367086B1 (en) * | 2004-02-02 | 2013-02-05 | S.S. Steiner, Inc. | Process and product for inhibiting or preventing bacterial infections |
| US20060025468A1 (en) * | 2004-07-29 | 2006-02-02 | Whitten Jeffrey P | Chiral pyrrolidine derivatives, and methods for preparing compounds thereof |
| CA2688008A1 (en) * | 2007-05-24 | 2008-11-27 | Kyorin Pharmaceutical Co., Ltd. | Mutilin derivative having heterocyclic aromatic ring carboxylic acid structure in substituent at 14-position |
| US8501988B2 (en) * | 2008-04-17 | 2013-08-06 | Ecolab Usa Inc. | Synthesis and applications of amino carboxylates |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6219583A (ja) * | 1985-07-17 | 1987-01-28 | Dainippon Pharmaceut Co Ltd | ピリドンカルボン酸誘導体、そのエステルおよびその塩 |
| JPS6345261A (ja) * | 1986-04-25 | 1988-02-26 | Dainippon Pharmaceut Co Ltd | 新規キノロン誘導体およびその塩 |
| JPS63152318A (ja) * | 1986-08-07 | 1988-06-24 | Dainippon Pharmaceut Co Ltd | 抗マイコプラズマ剤 |
| JPH03188074A (ja) * | 1989-03-31 | 1991-08-16 | Wakunaga Pharmaceut Co Ltd | 新規キノロン誘導体及びこれを含有する抗菌剤 |
| JPH04282384A (ja) * | 1990-10-13 | 1992-10-07 | Bayer Ag | 7−(2,7−ジアザビシクロ〔3.3.0〕オクチル)−3−キノロン及びナフチリドンカルボン酸誘導体 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5468861A (en) * | 1984-06-04 | 1995-11-21 | Bayer Aktiengesellschaft | 8-chloro-1-cyclopropyl-6,7-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid and alkyl esters thereof |
| DE3509546A1 (de) * | 1985-03-16 | 1986-09-25 | Bayer Ag, 5090 Leverkusen | 7-amino-1-(subst.cyclopropyl)-1,4-dihydro-4-oxo-3-chinolincarbonsaeuren, verfahren zu ihrer herstellung sowie diese enthaltende antibakterielle mittel |
| DK164287A (da) * | 1986-03-31 | 1987-10-01 | Sankyo Co | Quinolin-3-carboxylsyrederivater og fremgangsmaade til fremstilling deraf og deres anvendelse |
| JPH07108898B2 (ja) * | 1986-08-20 | 1995-11-22 | 大日本製薬株式会社 | 3−アミノピロリジン誘導体およびその塩の製造法 |
| US5563138A (en) * | 1987-04-16 | 1996-10-08 | Otsuka Pharmaceutical Company, Limited | Benzoheterocyclic compounds |
| US5290934A (en) * | 1987-04-16 | 1994-03-01 | Otsuka Pharmaceutical Company, Limited | Benzoheterocyclic compounds |
| ATE117685T1 (de) * | 1988-05-19 | 1995-02-15 | Chugai Pharmaceutical Co Ltd | Chinoloncarbonsäure-derivate. |
| US5252734A (en) * | 1989-04-03 | 1993-10-12 | Bayer Aktiengesellschaft | Antibacterial 5-alkylquinolonecarboxylic acids |
| CN1054980A (zh) * | 1990-02-19 | 1991-10-02 | 杏林制药株式会社 | 具有旋光活性的8-甲氧基喹诺酮羧酸衍生物,它们的制备方法以及它们的中间体 |
| KR960704887A (ko) * | 1993-10-14 | 1996-10-09 | 챨스 엠. 브록 | 퀴놀리지논형 화합물 (Quinolizinone type compounds) |
-
1994
- 1994-10-27 AT AT94931176T patent/ATE205209T1/de not_active IP Right Cessation
- 1994-10-27 AU AU80035/94A patent/AU8003594A/en not_active Abandoned
- 1994-10-27 CA CA002175351A patent/CA2175351A1/en not_active Abandoned
- 1994-10-27 WO PCT/JP1994/001815 patent/WO1995011902A1/ja not_active Ceased
- 1994-10-27 US US08/632,474 patent/US5668147A/en not_active Expired - Fee Related
- 1994-10-27 EP EP94931176A patent/EP0726269B1/en not_active Expired - Lifetime
- 1994-10-27 DE DE69428206T patent/DE69428206T2/de not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6219583A (ja) * | 1985-07-17 | 1987-01-28 | Dainippon Pharmaceut Co Ltd | ピリドンカルボン酸誘導体、そのエステルおよびその塩 |
| JPS6345261A (ja) * | 1986-04-25 | 1988-02-26 | Dainippon Pharmaceut Co Ltd | 新規キノロン誘導体およびその塩 |
| JPS63152318A (ja) * | 1986-08-07 | 1988-06-24 | Dainippon Pharmaceut Co Ltd | 抗マイコプラズマ剤 |
| JPH03188074A (ja) * | 1989-03-31 | 1991-08-16 | Wakunaga Pharmaceut Co Ltd | 新規キノロン誘導体及びこれを含有する抗菌剤 |
| JPH04282384A (ja) * | 1990-10-13 | 1992-10-07 | Bayer Ag | 7−(2,7−ジアザビシクロ〔3.3.0〕オクチル)−3−キノロン及びナフチリドンカルボン酸誘導体 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996033992A1 (en) * | 1995-04-27 | 1996-10-31 | Kaken Pharmaceutical Co., Ltd. | Optically active quinolinecarboxylic acid derivatives and salts thereof |
| RU2167867C2 (ru) * | 1998-07-28 | 2001-05-27 | Пфайзер Продактс Инк. | Способ получения тровафлоксацина |
| JPWO2005026147A1 (ja) * | 2003-09-10 | 2007-11-08 | 杏林製薬株式会社 | 7−(4−置換−3−シクロプロピルアミノメチル−1−ピロリジニル)キノロンカルボン酸誘導体 |
| US7514451B2 (en) | 2003-09-10 | 2009-04-07 | Kyorin Pharmaceutical Co., Ltd. | 7-(4-Substituted-3-cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| JP4639149B2 (ja) * | 2003-09-10 | 2011-02-23 | 杏林製薬株式会社 | 7−(4−置換−3−シクロプロピルアミノメチル−1−ピロリジニル)キノロンカルボン酸誘導体 |
| WO2005073238A1 (ja) * | 2004-02-02 | 2005-08-11 | Daiichi Pharmaceutical Co., Ltd. | ピリドベンズオキサジン誘導体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0726269A4 (en) | 1997-01-22 |
| EP0726269A1 (en) | 1996-08-14 |
| DE69428206T2 (de) | 2002-06-13 |
| AU8003594A (en) | 1995-05-22 |
| US5668147A (en) | 1997-09-16 |
| CA2175351A1 (en) | 1995-05-04 |
| DE69428206D1 (de) | 2001-10-11 |
| ATE205209T1 (de) | 2001-09-15 |
| EP0726269B1 (en) | 2001-09-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20060235037A1 (en) | Heterocyclic inhibitors of protein arginine methyl transferases | |
| US20180362543A1 (en) | Hiv replication inhibitor | |
| NO175939B (no) | Analogifremgangsmåte for fremstilling av terapeutisk aktiv N1-(1,2-cis-2-halogencyklopropyl)-substituerte pyridonkarboksylsyrederivater, eller et farmasöytisk akseptabelt salt derav, eventuelt i en stereoisomerisk form | |
| EP1666477B1 (en) | 7-(4-substituted 3- cyclopropylaminomethyl-1- pyrrolidinyl) q uinolonecarboxylic acid derivative | |
| IL301225A (en) | Heterocyclic compounds as CBP/EP300 bromodomain inhibitors | |
| CN101511833A (zh) | 1,4,5,6,7,8-六氢-1,2,5-三氮杂-甘菊环烃衍生物 | |
| CN101711243A (zh) | 3-氨基-6-(1-氨基-乙基)-四氢吡喃衍生物 | |
| CA3057431A1 (en) | 2(1h)-quinolinone derivative | |
| CZ288493B6 (en) | Derivatives of 7-isoindolinylquinolone and 7-isoindolinylnaphthyridone, process of their preparation, intermediates for their preparation, their use as well as medicaments in which the derivatives are comprised | |
| AU661999B2 (en) | Pyridonecarboxylic acid derivative | |
| JP2673937B2 (ja) | 5−アミノ−8−メチル−7−ピロリジニルキノリン−3−カルボン酸誘導体 | |
| KR101307717B1 (ko) | 트리―, 테트라― 치환―3―아미노피롤리딘 유도체 | |
| WO1995011902A1 (en) | Quinolinecarboxylic acid derivative and salt thereof | |
| Lv et al. | Synthesis and in vitro antibacterial activity of quinolone/naphthyridone derivatives containing 3-alkoxyimino-4-(methyl) aminopiperidine scaffolds | |
| JP7623294B2 (ja) | 化合物、組成物および方法 | |
| TW201132645A (en) | Tricyclic antibiotics | |
| US20240000951A1 (en) | Gpx4 protein degradation-inducing compound | |
| WO2002040478A1 (en) | Dehalogeno compounds | |
| JPH0673056A (ja) | キノリンカルボン酸誘導体およびその塩 | |
| CN107635991B (zh) | 作为tdp2的抑制剂的呋喃并喹啉二酮 | |
| CN101374831A (zh) | 四氢吡喃抗生素 | |
| JPH02292289A (ja) | 置換アゼチジニルイソチアゾロピリドン誘導体、その製造法及びその医薬用途 | |
| WO1995021163A1 (en) | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor | |
| CZ211394A3 (en) | DERIVATIVES OF PYRIDO/1,2,3-d,e//1,3,4/BENZOXADIAZINE, PROCESS OF THEIR PREPARATION, USE AND MEDICAMENTS CONTAINING SUCH COMPOUNDS | |
| US20210323981A1 (en) | Free amino compounds for the treatment and prophylaxis of bacterial infection |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AM AU BB BG BR BY CA CN CZ EE FI GE HU JP KG KR KZ LK LR LT LV MD MG MN NO NZ PL RO RU SI SK TJ TT UA US UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE MW SD SZ AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 08632474 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2175351 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994931176 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994931176 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 1997 858536 Country of ref document: US Date of ref document: 19970519 Kind code of ref document: A |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1994931176 Country of ref document: EP |