WO1997043343A1 - Composition de resine thermoplastique contenant un composite a base d'argile, et procede de fabrication associe - Google Patents
Composition de resine thermoplastique contenant un composite a base d'argile, et procede de fabrication associe Download PDFInfo
- Publication number
- WO1997043343A1 WO1997043343A1 PCT/JP1997/001605 JP9701605W WO9743343A1 WO 1997043343 A1 WO1997043343 A1 WO 1997043343A1 JP 9701605 W JP9701605 W JP 9701605W WO 9743343 A1 WO9743343 A1 WO 9743343A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- thermoplastic resin
- resin composition
- clay
- group
- swellable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K9/00—Use of pretreated ingredients
- C08K9/04—Ingredients treated with organic substances
- C08K9/06—Ingredients treated with organic substances with silicon-containing compounds
Definitions
- the present invention relates to a thermoplastic resin composition containing a thermoplastic resin and a clay composite, and a method for producing the same.
- layered clay minerals such as talc and mica have been used as fillers in order to improve the mechanical properties and heat resistance of thermoplastic resins (for example, Japanese Patent Publication No. 491-18615, Japanese Patent Application Laid-Open No. 55-16049, Japanese Patent Publication No. Sho 63-3-5322).
- thermoplastic resins for example, Japanese Patent Publication No. 491-18615, Japanese Patent Application Laid-Open No. 55-16049, Japanese Patent Publication No. Sho 63-3-5322.
- the elastic modulus and heat resistance of the obtained resin composition are improved, but the appearance of the molded article obtained from this resin composition is poor, the specific gravity is increased, and the color tone is deteriorated.
- Invite Furthermore, there has been a problem that the strength or toughness of the resin composition is reduced due to poor dispersion of the layered clay mineral or poor adhesion between the layered clay mineral and the resin.
- Surface treatment agents are generally used as a means to improve the adhesion between the filler and the resin and prevent a decrease in strength or toughness.
- surface treatment in which a layered clay mineral is treated with a silane-based surface treatment agent A composite of a filler and a polyester resin is disclosed (for example, JP-A-51-24653, JP-A-51-24654, etc.).
- a layered clay mineral is treated with a silane-based surface treatment agent
- a composite of a filler and a polyester resin is disclosed (for example, JP-A-51-24653, JP-A-51-24654, etc.).
- the effect of improving the strength and the like of the resin composition is still insufficient with the conventional surface treatment method.
- the layered clay mineral itself remains in a laminated structure, so that problems such as poor appearance of the molded product, increase in specific gravity, and deterioration of color tone still occur.
- Layered clay minerals usually have about one to several thousand unit layers with a thickness of about 1 nm. Therefore, in the above-mentioned prior art, the layered clay minerals separated in the thermoplastic resin do not exist independently as unit layers but exist on a coherent structure basis. ing.
- the resin can be strengthened by adding a small amount of the layered clay mineral, and the appearance of the molded product is poor. It is thought that the problems of increase in specific gravity, and deterioration of color shift can be solved.
- the purpose of the present invention is to solve the conventional problems as described above.> Cleaving the cohesive structure of the layered clay mineral, and forming a very fine nm-ordered layer in thermoplastic resin.
- An object of the present invention is to provide a thermoplastic resin composition having excellent mechanical properties (elastic modulus, strength, toughness, etc.), heat resistance, and appearance of a molded product by being independently dispersed.
- the present inventors have conducted intensive studies to achieve the above object, and as a result, completed the present invention.
- thermoplastic resin composition of the present invention contains a thermoplastic resin (A) and a clay complex (B), wherein the clay complex (B) is obtained by adding a swellable gaylate (B 1) to the following general formula: (I)
- Y is each independently a substituted or non- ⁇ substituted hydrocarbon group having 1 to 25 carbon atoms
- X is each independently a hydrolyzable group or It is prepared by introducing a silane compound represented by a hydroxyl group at a) the (B2), and it is [R B 300] value of 20% or more, wherein [R B 300] is equivalent area It is defined as the ratio of the clay complex (B) whose circular diameter [D] is less than 300 nm.
- thermoplastic resin (A) is at least one selected from the group consisting of a thermoplastic polyester resin and a polycarbonate resin.
- swellable gaylate (B1) is at least one selected from the group consisting of smectite-group clays and swellable mica.
- the clay complex (B) is converted by introducing the silane compound (B2) after increasing the base spacing of the expansible gaylate (B1).
- thermoplastic resin composition of the present invention contains 0.1 to 100 parts by weight of the clay composite (B) based on 100 parts by weight of the thermoplastic resin (A).
- the ash fraction from the clay complex (B) is between 0.1 and 50% by weight.
- the [ RB300 ] value is greater than or equal to 50%.
- the [DB] value is less than or equal to 500 nm, where [D B ]
- B is defined as the average of the equivalent area circle diameters of the clay composite (B).
- [D B] / [D B1] value is not more than 0.010, in here [D B] is defined as the average value of the equivalent area circle diameter of the clay compound (B), Then, [D B1 ] is defined as the average value of the equivalent area circle diameter of the swelling gaylate (B 1).
- the number of [N B] / [N B1 ] value is not less than 300, where [N B] is a unit ash content per and clay complex per unit area (B) And [N B1 ] is defined as the number of swellable gay salts (B 1) per unit ash fraction and per unit area.
- a clay composite dispersed in a thermoplastic composition in another embodiment, a clay composite dispersed in a thermoplastic composition
- the average layer thickness of (B) is less than 20 nm.
- a clay composite dispersed in a thermoplastic composition is less than 20 nm.
- More than 20% of (B) has a layer thickness of 5 nm or less.
- [I B] Z [I B1] value is 0.25 or less, where [pi beta] is defined as the diffraction intensity of small angle X-ray diffraction derived from clay complex (B), and [I B1 ] is defined as the small-angle X-ray diffraction intensity derived from the cohesive structure of the swellable silicate (B 1).
- the bottom spacing of the clay composite (B) in the thermoplastic resin composition is at least three times the initial bottom spacing of the swellable silicate (Bl).
- thermoplastic resin composition of the present invention comprises a thermoplastic resin (A) and a clay composite (B), wherein the clay composite (B) is a swellable resin.
- ⁇ is an integer of 0 to 3
- ⁇ is independently a substituted or unsubstituted hydrocarbon group having from 25 to 25 carbon atoms
- X is each independently a hydrolyzable group or a hydroxyl group.
- the thermoplastic resin composition of the present invention contains a thermoplastic resin ( ⁇ ) and a clay complex ( ⁇ ), wherein the clay complex ( ⁇ ) is swelled.
- a thermoplastic resin ( ⁇ ) and a clay complex ( ⁇ ) wherein the clay complex ( ⁇ ) is swelled.
- the silane compound ( ⁇ 2) represented by the above general formula (I) into the neutral gayate (B1), and then dispersed in the thermoplastic resin composition.
- the average layer thickness of ⁇ ) is less than 20 nm.
- the ripened resin composition of the present invention contains a thermoplastic resin (A) and a clay composite (B), wherein the clay composite (B) is a swellable resin.
- the clay compound (B1) is made by introducing the silane compound (B2) represented by the above general formula (I) into the gaylate (Bl), and is dispersed in the thermoplastic resin composition ( More than 20% of B) has an employment thickness of 5 nm or less.
- a thermoplastic resin composition of the present invention comprises a thermoplastic resin (A) and a clay complex (B), wherein the clay complex (B) is silane compounds represented by the swellable Gay salt (B 1) in the general formula (I) and (B 2) is prepared by introducing, and [I B] / C 1 B J value is 0.25 or less , and the origin where [I B] is defined as the diffraction intensity of small angle X-ray diffraction derived from clay complex (B), and the aggregate structure of [I B J swellable Gay salt (B 1) Is defined as the diffraction intensity of small-angle X-ray diffraction.
- a thermoplastic resin composition of the present invention contains a thermoplastic resin (A) and a clay composite (B), wherein the clay composite (B) is a swelling resin.
- a thermoplastic resin composition contains a thermoplastic resin (A) and a clay composite (B), wherein the clay composite (B) is a swelling resin.
- the silane compound (B 2) represented by the above general formula (I) into the neutral gayate (B 1), and the clay complex (B) in the thermoplastic resin composition
- the bottom spacing of is more than three times the initial bottom spacing of the swellable gaylate (B1).
- the method for producing a thermoplastic resin composition containing a thermoplastic resin (A) and a clay complex (B) according to the present invention comprises:
- the swellable silicate (B1) having an increased bottom spacing has the following general formula (I)
- Y is each independently a substituted or unsubstituted hydrocarbon group having 1 to 25 carbon atoms
- X is each independently a hydrolyzable group or a hydroxyl group.
- the method for producing a thermoplastic resin composition of the present invention comprises a step of mixing a clay composite (B) and a polymerizable monomer to obtain a mixture, and the step of mixing the polymerizable monomer in the mixture.
- a thermoplastic resin (A) BRIEF DESCRIPTION OF THE FIGURES
- FIG. 1 shows the thermoplastic polyester resin composition (C) of the present invention obtained in Example 1.
- Fig. 3 is a transmission electron micrograph showing the sensitivity of the clay composite (B) in Fig. 1.
- FIG. 2 is a small-angle X-ray diffraction pattern derived from the clay complex (B) in the thermoplastic polyester resin composition (C) of the present invention obtained in Example 1.
- thermoplastic resin (A) used in the present invention may be any thermoplastic resin.
- thermoplastic resin (A) include thermoplastic polyester resin, polycarbonate resin, polyamide resin, polyolefin resin, polyarylate resin, vinyl polymer compound, polyimide resin, and polyphenylene sulfide.
- any thermoplastic resin such as polyolefin oxide, polyacetal, polysulfone, polyethersulfone, fluororesin, and polyolefin copolymer.
- the thermoplastic resin may be an elastomer or rubber. One or more of these thermoplastic resins can be used.
- Thermoplastic polyester resins, polycarbonate resins, polyamide resins, and polyolefin resins are preferred, with thermoplastic polyester resins and polycarbonate resins being particularly preferred.
- the thermoplastic polyester resin is not particularly limited, and may be prepared from a dicarboxylic acid compound and an ester-forming rust conductor of Z or dicarboxylic acid, and a diol compound and / or an ester-forming derivative of a diol compound. It is a polyester resin. Specific examples include polyethylene terephthalate, polypropylene terephthalate, polybutylene terephthalate, polyhexamethylene terephthalate, polycyclohexane-1,4-dimethyl terephthalate, neopentyl terephthalate, polyethylene isophthalate, polyethylene naphthalate, polyethylene naphthalate, and polybutylene naphthalate.
- polyhexamethylene naphthalate and the like, and copolymerized polyesters thereof may be used alone or in combination of two or more.
- the polycarbonate resin is not particularly limited, and may be a divalent phenol compound and phosgene, or any polycarbonate resin obtained by a reaction of a divalent phenol compound and a carbonic acid diester compound.
- 2,2-bis (4-hydroxyphenyl) propane-type polycarbonate 2,2-bis (3,5-dimethyl-4-hydroxyphenyl) propane-type polycarbonate, 1,1-bis (4- Hydroxyphenyl) cyclohexane-type polycarbonate, 4.4 'dihydroxyphenyl ether-type polycarbonate, 4, 4' dihydroxydiphenyl sulfide-type polycarbonate, 4, 4'-dihydroxydiphenyl sulfone-type polycarbonate, Bis (4-hydroxyphenyl) ketone-type polycarbonate, 1,4-bis (4-hydroxyphenylsulfonyl) benzene and the like can be mentioned. They may be used alone or in combination of two or more.
- the polyamide resin is not particularly limited, and any polyamide resin can be used. Specific examples include polycabroamide (nylon 6), polytetramethylene adipamide (nylon 46), and polyhexamethylene adipamide. (Nylon 66), polyhexamethylene sebacamide (nylon 610), polyhexamethylene dodecamide (nylon 612), polydecamethylene adipamide (nylon 116), polyundecamide (nylon 11), Polydodecamide (nylon 12), polytrimethylhexamethylene terephthalamide (TMHT), polyhexamethylene terephthalamide (nylon 6T), polyhexamethylene isophthalamide (nylon 6I), polybis (4-aminocyclohexane Xyl) methanedodecamide (nylon dimethyl PACM12), polymetaxylylene adipa De (nylon MXD6), Po Li undecamethylene terephthalamide (nylon 1 1 T), the poly ⁇ down decamethylene he
- the polyolefin resin is not particularly limited, and any polyolefin may be used. Wear. Specific examples include homopolymers of ⁇ -olefin olefins containing ethylene, and copolymers of two or more ⁇ -olefins (including any copolymers such as random, block, and graft, and mixtures thereof). Or olefinic elastomers. Examples of ethylene-only polymers include low-density polyethylene (LD ⁇ ), high-density polyethylene (HDPE), and linear low-density polyethylene (L LDPE). be able to. Examples of the polypropylene include not only the homopolymer of polypropylene but also a copolymer of propylene and ethylene.
- the above-mentioned polyethylene or polypropylene may contain an olefin elastomer.
- the olefinic elastomer is a combination of ethylene and one or more ⁇ -olefins other than ethylene (eg, propylene, 1-butene, 11-hexene, 4-methyl-11-pentene, etc.).
- Specific examples include ethylene propylene copolymer (EPR), ethylene butene copolymer (EBR), ethylene-propylene-gen copolymer (EPDM), and the like. They may be used alone or in combination of two or more.
- the molecular weight of the thermoplastic resin (II) used in the present invention is selected in consideration of the molding fluidity in the molding process and various physical properties of the final product, and it is not preferable that the molecular weight is too low or too high. However, since it is mainly determined by the primary structure of the thermoplastic resin ( ⁇ ), it is necessary to set an appropriate molecular weight for each thermoplastic resin ( ⁇ ).
- the molecular weight of the thermoplastic polyester resin that can be suitably used in the present invention is, for example, that the logarithmic viscosity measured at 25 using a mixed solvent of phenol / tetrachloroethane (5/5 weight ratio) is 0.3 to 2; 0 (d 1 Zg).
- thermoplastic resin composition (C) It is preferably 0.35 to 1.9 (dl Zg), and more preferably 0.4 to: 0.8 (d 1 / g).
- the logarithmic viscosity is less than 0.3 (d 1 /)
- the obtained thermoplastic resin composition (C) tends to have low mechanical properties and impact resistance
- the molded article of the thermoplastic resin composition (C) tends to have a low viscosity.
- the ratio is larger than 1 / g), there is a tendency that a problem occurs in workability such as fluidity during molding.
- the molecular weight of the polycarbonate resin that can be suitably used in the present invention is, for example, In gel permeation chromatography (GPC) measurement using a lahydrofuran (THF) solvent, the weight average molecular weight (w) force measured at 40 was converted to a single molecular weight polystyrene dispersion, from 15,000 to 80,000, preferably from 25,000 to 75,000, and Preferably it is 30,000 to 70,000. If the Mw is less than 1500, the molded article of the thermoplastic resin composition (C) obtained tends to have low mechanical properties and impact resistance. Tend to have problems with sex.
- GPC gel permeation chromatography
- the molecular weight of the polyamide resin which can be suitably used in the present invention is, for example, one having a relative viscosity of 1.5 to 5.0 measured at 25 at 98% of sulfuric acid and at 1.0% of oxygen. If the relative viscosity is less than 1.5, the obtained thermoplastic resin composition (C) tends to have low mechanical properties and impact resistance, and if the relative viscosity is more than 5.0, the There is a tendency for problems in processability such as fluidity to occur.
- the molecular weight of polypropylene in the polyolefin resin is, for example, 230
- the melt index measured under a load of 2.16 Kg is preferably 0.3 to 30 gZl 0 minutes, and more preferably 0.5 to 15 gZl 0 minutes. Is preferred. If the melt index is greater than 30 gZl 0 minutes, the mechanical properties and impact resistance of the molded product tend to be low. If the melt index is less than 0.3 gZl 0 minutes, there is a problem with workability such as fluidity during molding. Tends to occur.
- the clay complex (B) used in the present invention refers to a swellable gaylate (Bl) that has the following general formula (I)
- Y is each independently a substituted or unsubstituted hydrocarbon group having 1 to 25 carbon atoms
- X is each independently a hydrolyzable group or a hydroxyl group.
- the silane compound (B2) represented by is introduced.
- Swellable gaylate (B1) As the swellable silicate (B 1), any of the commonly used swelling salts may be used, and it is preferably formed from a tetrahedral sheet mainly composed of gay oxide and an octahedral sheet mainly composed of metal hydroxide. It is a gayate. Examples of swellable gay salts include smectite group clays and swellable mica.
- the smectite clay has the general formula
- X is at least one member selected from the group consisting of K, Na, 1/2 Ca, and 1 2 Mg
- Y is Mg, Fe, Mri. Ni, Zn, L i, a 1.
- Z is at least one selected from the group consisting of 1 or et S i and a.
- H 2 0 is coupled to the interlayer ions Is a natural or synthetic mineral, represented by the following formula: n varies significantly with interlayer ions and relative humidity.
- smectite group clay examples include, for example, montmorillonite, beidellite, nontronite, saponite, iron savonite, hectorite, soconite, stevensite, bentonite, etc. And mixtures thereof.
- the swellable airborne mother is the following general formula
- the swellable mica has the ability to swell in water, a polar solvent compatible with water in an arbitrary ratio, or a mixed solvent of water and the polar solvent.
- the swelling mica include lithium-type teniolite, sodium-type teniolite, lithium-type tetracaine mica, and sodium-type tetracaine mica, or a substitute, a derivative, or a mixture thereof.
- the swellable mica those corresponding to vermiculites may be used. There are three octahedral types and two octahedral types in the bar
- the above-mentioned swellable gay salts (B1) are used alone or in combination of two or more.
- the crystal structure of the swellable gaylate (B1) is preferably stacked in a regular order along the c-axis. Highly pure force is desirable.
- the so-called mixed-layer mineral, in which the crystal cycle is disturbed and multiple types of crystal structures are mixed, is also used. Can be done.
- silane compound (B2) to be introduced into the swellable gaylate (B1) any commonly used silane compound (B2) can be used.
- any commonly used silane compound (B2) can be used.
- n is an integer of 0-3.
- Y is each independently a substituted or unsubstituted hydrocarbon group having 1 to 25 carbon atoms. This substituent may be an ester group, ether group, epoxy group, amino group, carboxyl group, carbonyl group, amide group, mercapto group, sulfonyl group, sulfinyl group, nitro group, nitroso group, nitrile group, halogen atom, and hydroxyl group. At least one selected from the group consisting of: X is each independently a hydrolyzable group or a hydroxyl group.
- the hydrolyzable group is at least one selected from the group consisting of an alkoxy group, an alkenyloxy group, a ketoxime group, an acyloxy group, an amino group, an aminoxy group, an amide group, and a halogen atom.
- n Y and 4—n X may be the same or different, respectively.
- hydrocarbon group refers to a linear or branched (ie, having a side chain) saturated or unsaturated monovalent or polyvalent aliphatic hydrocarbon group, and an aromatic hydrocarbon group. , Means an alicyclic hydrocarbon group.
- an alkyl group an alkenyl group,
- alkyl group J is intended to include a polyvalent hydrocarbon group such as an “alkylene group” unless otherwise specified.
- alkenyl group, alkynyl group, aryl group (phenyl group, naphthyl group, etc.) and cycloalkyl group are alkenylene group, alkynylene group, arylene group (phenylene group, naphthylene group, etc.), and cycloalkylene group, respectively. Etc. are included.
- examples of compounds in which Y is an unsubstituted hydrocarbon group having 1 to 25 carbon atoms include ⁇ having 10 to 25 carbon atoms such as decyltrimethoxysilane.
- lower alkyl groups those in which Y is an unsaturated hydrocarbon group such as 2-hexenyltrimethoxysilane, those in which Y has a side chain such as 2-ethylhexyltrimethoxysilane And phenyltriethoxysilane, in which Y has a phenyl group, 3 in which Y has a naphthyl group, such as 3-3-naphthylpropyltrimethoxysilane, and p-vinylbenzyltrimethoxysilane. And those in which Y has an arylalkyl group.
- examples of the compound in which Y is a vinyl group among the unsaturated hydrocarbon groups include vinyltrimethoxysilane, pinyltrichlorosilane, and vinyltriacetoxysilane.
- examples of the compound when Y is a group having an ester group as a substituent, examples of the compound include methacryloxypropyl trimethoxysilane.
- examples of the compound where Y is a group having an ether group as a substituent include polyoxyethylene propyl trimethoxy silane and 2-ethoxyethyl trimethoxy silane.
- examples of the compound in which Y is a group having an epoxy group as a substituent include glycidoxypropyltrimethoxysilane.
- Y is a group having an amino group as a substituent
- examples include arnaminopropyl trimethoxysilane, arn (2-aminoethyl) aminobutyl virtrimethoxysilane, and arnilinobu mouth pilltrimethoxysilane (NH 2 C 6 H, (C3 ⁇ 4) 3 Si (OCH 3 ) 3 ).
- examples of the compound include ⁇ - (4-carboxyphenyl) propyl trimethoxysilane.
- Examples of compounds in which Y is a group having a carbonyl group as a substituent include ureido propyltriethoxysilane (H 2 NCONH (CH 2 ) 3 Si (OC 2 H s ) 3 )
- Can be Examples of the compound in which Y has an amide group as a substituent include acetylated products of the above compounds having an amino group.
- Examples of the compound in which Y is a group having a mercapto group as a substituent include amercaptoprovir trimethoxysilane.
- Examples of compounds in which Y is a group having a sulfonyl group as a substituent include ⁇ -phenylsulfonylpropyltrimethoxysilane.
- Examples of compounds in which Y is a group having a sulfinyl group as a substituent include phenylsulfinylpropyl propyltrimethoxysilane. Examples of compounds in which Y is a group having a nitro group as a substituent include ⁇ -nitropropyltriethoxysilane. Examples of the compound in which Y is a group having a nitroso group as a substituent include ⁇ -nitrosopropyl triethoxysilane. When Y is a group having a ditril group as a substituent, examples of the compound include ashianoethyltriethoxysilane and ashinovobuchi birutriethoxysilane.
- Examples of the compound in which Y is a group having a halogen as a substituent include ⁇ -chlorobutyryltriethoxysilane.
- Examples of the compound in which Y is a group having a hydroxyl group as an exchange group include N, N-di (2-hydroxyethyl) amino 3-propyltriethoxysilane.
- ⁇ is preferably an integer of 2 to 30 in view of the reactivity with the swellable gaylate (B 1) and the handleability of the silane compound (B 2) itself.
- silane compounds (B2) or a conductor can also be used, and these silane compounds (B2) can be used alone or in combination of two or more.
- the clay complex (B) can be obtained by enlarging the distance between the bottoms of the swellable gaylate (B1) and then introducing the silane compound (B2),
- the bottom surface distance means the distance between the bottom surfaces of the unit layers of the swellable silicate or the obtained clay composite.
- the bottom spacing can be confirmed by small angle X-ray diffraction (SAXS). That is, the X-ray diffraction peak angle value of a dispersion composed of a dispersion medium and a swellable silicate (B1) (or a swellable silicate in an aggregated state before being added to the dispersion medium) was measured by SAXS. The peak angle value is calculated by the equation of B agg
- d is the distance between the bases in the crystal
- 0 is the angle of incidence
- n is a positive integer
- ⁇ is the wavelength of the X-rays
- the step of increasing the distance between the bottom surfaces of the swellable calcium salt (B1) is performed by dispersing the swellable gay salt ( ⁇ 1) in a dispersion medium or physically adding the swellable calcium salt (B1) to the dispersion medium. This can be done by applying force.
- a polar solvent compatible with water at an arbitrary ratio, or a mixed solvent of water and such a polar solvent can be used as the dispersion medium.
- a polar solvent For example, alcohols such as methanol, ethanol, and isopropanol; glycols such as ethylene glycol, propylene glycol, and 1,4-butanediol; ketones such as acetone and methyl ethyl ketone; Ethers such as hydrofuran; amide compounds such as dimethylformamide; and other solvents such as dimethyl sulfoxide and 2-pyrrolidone. These polar solvents may be used alone or in combination of two or more.
- the distance between the bottom surfaces of the swellable gaylate (B 1) in the dispersion medium can be increased by sufficiently stirring and dispersing the swellable silicate (B 1) in the dispersion medium.
- the physical external force is obtained by using a commonly used wet milling method of a filler.
- a commonly used wet milling method of a filler for example, a method using hard particles can be mentioned.
- the hard particles, the swellable gaylate (B 1) and an optional solvent are mixed and mixed, and the swelling is caused by physical collision between the hard particles and the swellable silicate (B 1).
- sex silicate (B 1). Hard particles that are usually used are beads for crushing fillers, for example, glass beads or zirconia beads.
- These grinding beads are selected in consideration of the hardness of the swellable silicate (B 1) or the material of the stirrer, and are not limited to the above-mentioned glass or zirconia.
- the particle size is also not limited to a numerical value because it is determined in consideration of the size of the swellable gaylate (B 1), etc., but the diameter ranges from 0.1 to 6, O mm.
- the solvent used here is not particularly limited, but for example, the same as the above-described dispersion medium is preferable.
- the distance between the bottom surfaces of the layers of the expanded swellable gaylate (B 1) after expansion is preferably at least 3 times, more preferably at least 5 times, as compared with the initial surface of the swellable gay salt (B 1). It is. There is no particular upper limit. If the distance between the bottoms is increased about 10 times or more, Separation becomes difficult, in which case the swelling gaylate (B1) is considered to be present in the subunit layer.
- the initial spacing between the bottom surfaces of the swellable gay salt refers to the spacing between the bottom surfaces of the swellable silicate in which the unit layers are in a mutually cohered state before being added to the dispersion medium.
- the clay complex ( ⁇ ) can be obtained by introducing a silane-based compound ( ⁇ 2) to the surface of the unit layer of the neutral gayate (B 1).
- the silane-based compound is introduced by adding the silane-based compound ( ⁇ 2) to a dispersion containing a swellable gaylate (B1) with an enlarged bottom surface spacing and a dispersed soot. And stirring.
- the stirring speed it is preferable to set the stirring speed to 1000 rpm or more, or to apply a shear rate of 500 (lZs) or more. Stirring at a value greater than 2500 O pm or applying a shear rate greater than 500,000 (1 / s) tends to have no effect, so a higher speed or shear rate must be used. There is no.
- the silane compound (B2) is added to the swellable silicate (B1) while applying a physical external force (for example, wet milling) thereto. ) Can be introduced.
- a swellable gaylate (B1) whose base spacing has been enlarged by physical external force is added to the dispersion medium, and the silane compound (B1) is added to the dispersion medium in the same manner as in the above-described method using the dispersion medium.
- the silane compound (B2) can be introduced.
- the reaction between the swellable gaylate (B 1) and the silane compound (B 2) can proceed sufficiently at room temperature, but is necessary.
- the reaction system may be heated according to the conditions. The maximum temperature during heating depends on the silane compound used.
- the temperature can be arbitrarily set as long as it is lower than the decomposition temperature of (B2) and lower than the boiling point of the dispersion medium.
- the silane-based compound (B2) introduced into the swellable gaylate (B1) further reacts with a reactive group such as a hydroxyl group, a hydroxyl group, an amino group, an epoxy group, or a vinyl group.
- a reactive group such as a hydroxyl group, a hydroxyl group, an amino group, an epoxy group, or a vinyl group.
- a compound capable of reacting with such a reactive group can be further added to react the compound with the reactive group. In this way, the length of the functional group chain of the silane compound (B2) introduced into the swellable silicate (B1) can be increased, and the polarity can be changed.
- the above-mentioned silane-based compound (B2) itself can be used, but the compound is not limited thereto, and any compound can be used according to the purpose. Examples include compounds, amino group-containing compounds, carboxyl group-containing compounds, acid anhydride group-containing compounds, and hydroxyl group-containing compounds.
- the amount of the silane compound (B2) used can be adjusted so that the affinity and dispersibility of the obtained clay composite (B) and the resin (A) used are improved. If necessary, a plurality of types of silane compounds (B2) having different functional groups can be used in combination.
- the amount of the silane compound (B2) to be added is not necessarily limited to a numerical value, but is preferably 0.1 to 200 parts by weight based on 100 parts by weight of the swellable gaylate (B1). Yes, more preferably 0.2 to 160 parts by weight, particularly preferably 0.3 to 120 parts by weight. If the amount of the silane compound (B 2) is less than 0.1 part by weight, the obtained clay composite (B) tends to have insufficient differential sensitizing effect, and is not preferable. It is not necessary to add more than 200 parts by weight as the effect remains the same.
- the base spacing of the clay complex (B) obtained as described above is larger than the initial base spacing of the swellable silicate (B1) due to the presence of the introduced silane compound (B2). I can do it.
- the silicate salt (Bl) returns to a state where the layers are aggregated again when the dispersion medium is removed.
- the distance between the bottom surfaces is increased.
- the bottom spacing of the clay composite (B) is preferably at least 1.5 times, more preferably at least 2 times, the initial spacing of the swellable silicate (B1).
- the introduction of the silane compound (B 2) into the swellable gaylate (B 1) can be confirmed by various methods.
- the following method for example, the following method can be mentioned.
- the clay complex (B) is washed using an organic solvent such as tetrahydrofuran or black form to simply remove the adsorbed silane compound (B2).
- the washed clay complex (B) is made into a powder in a mortar, and then dried sufficiently.
- the clay composite (B) is sufficiently mixed with powdered potassium bromide (KBr) in a predetermined ratio to form a pressurized tablet, and Fourier Transform (FT) —IR is used to obtain a tablet by a transmission method.
- FT Fourier Transform
- the absorption amount derived from the silane compound (B2) is measured.
- a sufficiently dried powdery clay complex (B) can be used as it is by the diffuse reflection method (DR I FT It is desirable to measure with).
- the base spacing of the clay composite (B) is larger than the initial base spacing of the swellable gaylate (B1).
- the following method can be cited. That is, in the same manner as described above, the adsorbed silane compound (B2) is removed from the clay complex (B) by washing with an organic solvent, and after drying, the small-angle X-ray diffraction method (SAXS) is used. You can check with In this method, the X-ray diffraction peak angle value derived from the (0- 1) plane of the powdery clay composite (B) is measured by SAXS, and calculated by applying the Bragg formula to calculate the bottom surface interval. I can ask. Similarly, measure the distance between the bottom surfaces of the initial swellable keic acid 3 ⁇ 4 (B 1) and compare the two. Can confirm the expansion of the bottom gap.
- SAXS small-angle X-ray diffraction method
- the formation of the clay complex (B) can be confirmed.
- the affinity between the clay composite (B) and the resin as the matrix can be increased by introducing the silane compound (B2) and increasing the distance between the bottom surfaces. Can be done.
- thermoplastic resin composition (C) Preparation of thermoplastic resin composition (C)
- the amount of the clay composite (B) is typically 0.1 to 100 parts by weight, preferably 100 to 100 parts by weight of the thermoplastic resin (A). Is prepared to be 0.2 to 85 parts by weight, more preferably 0.5 to 70 parts by weight. If the amount of the clay composite (B) is less than 0.1 part by weight, the effect of improving mechanical properties and heat resistance may be insufficient. If it exceeds 150 parts by weight, the appearance of the molded body and the flow during molding may be insufficient. Properties tend to be impaired. However, this blending amount is not limited to the above range, since it can be appropriately selected according to the final use of the thermoplastic resin composite (C).
- thermoplastic resin composition (C) of the present invention has a ash content of the thermoplastic resin composition (C) derived from the clay composite (B) of typically 0.1 to 50% by weight, preferably 0.2 to 50% by weight. It is adjusted to be about 45% by weight, more preferably 0.5% to 40% by weight. If the ash content is less than 0.1% by weight, the effect of improving the mechanical properties and heat resistance may be insufficient. If the ash content exceeds 50% by weight, the appearance of the molded product and the fluidity during molding tend to be impaired. is there,
- thermoplastic resin composition (C) of the present invention comprises the steps of mixing a previously prepared clay complex (B) and a polymerizable monomer to prepare a mixture (mixing step), and the resulting mixture.
- the polymerization may be carried out by a method including a step of polymerizing this polymerizable monomer to obtain a thermoplastic resin (A) (polymerization step).
- the polymerizable monomer used in the above method is the desired thermoplastic resin (A) It can be any of the monomers commonly used for preparing.
- the dispersion medium used in the preparation of the clay composite '(B) and the polymerizable monomer used here may be the same,
- the polymerizable monomers used include an acid component containing dicarboxylic acid and Z or an ester-forming derivative thereof as a main component, and a diol compound and / or an ester-forming derivative thereof as a main component. And a diol component as follows.
- the above dicarboxylic acids are preferably aromatic dicarboxylic acids.
- oxyacids such as p-oxybenzoic acid and p-hydroxyethoxybenzoic acid, and their ester-forming derivatives may be used. Two or more of these monomers may be used as a mixture. If the amount is small enough not to impair the properties of the resulting polyester resin, one or more aliphatic dicarbonic acids such as adipic acid, azelaic acid, dodecane diacid, sebacic acid, etc. are mixed with these aromatic dicarboxylic acids. Can be used.
- diol component examples include aliphatic diols such as ethylene glycol, propylene glycol, butylene glycol, hexylene glycol, neopentyl glycol, and the like; aliphatics such as 1,4-cyclohexanedimethanol and the like. And aromatic diols such as 2,2-bis (4-hydroxyphenyl) propane and the like, and substituted and derivatives thereof can also be used. Two or more of these may be used in combination. Furthermore, long-chain diols (eg, polyethylene glycol, polytetramethylene glycol) and alkylene glycols of bisphenols may be used in such a small amount that the elastic modulus of the polyester resin is not significantly reduced. At least one kind of oxide addition polymer or the like (for example, ethylene oxide addition polymer of bisphenol A) may be mixed.
- oxide addition polymer or the like for example, ethylene oxide addition polymer of bisphenol A
- a cyclic ester such as ⁇ -cabrolactone can also be used as the polymerizable monomer.
- thermoplastic resin ( ⁇ ) is a polycarbonate resin
- the polymerizable monomer used is a divalent phenol compound and a divalent phenol component mainly composed of a phenolic or ester-forming rust conductor, and a phosgene or carbonate diester compound. It can be
- divalent phenol compound examples include bis (4-hydroxyphenylmethane), 1,1-bis (4-hydroxyphenyl) ethane, 1,1-bis (4-hydroxyphenyl) propane, 2-bis (4-hydroxyphenyl) pu bread (“bisphenol AJ”), 2,2-bis (4-hydroxyphenyl) butane, 2,2-bis (4-hydroxyphenyl) pentane, 2 1,2-bis (4-hydroxyphenyl) -1,3-methylbutane, 2,2-bis (4-hydroxyphenyl) hexane, 2,2-bis (4-hydroxyphenyl) -4-methylpentane, 1,1 —Bis (4-hydroxyphenyl) cyclopentane, 1,1-bis (4-hydroxyphenyl) cyclohexane, bis (4-hydroxy-3-methylphenyl) methane, bis (4-hydro C 3-Methylphenyl) phenylmethane, 1,1-bis (4-hydroxy-13-methylphenyl) ethane, 2,2-bis (4-hydroxy-3
- a polymer obtained by copolymerizing a divalent phenol having a benzotriazole group is used to enhance the flame retardancy.
- Substituents and derivatives of these divalent phenol compounds, alkali metal salts, and alkaline earth metal salts may also be used.A mixture of two or more of these divalent phenol compounds may be used.
- Examples of the carbonic acid diester compound include bisalkyl carbonates such as dimethyl carbonate, methyl carbonate, di-n-propyl carbonate, diisopropyl carbonate, and di-n-butyl carbonate, diphenyl carbonate, and bis (2,4-dichloromethane).
- the step of mixing the clay composite (B) and the polymerizable monomer can be performed, for example, by the following method. That is, the clay composite body (B), which was previously produced and isolated (ie, the dispersion medium was removed) by the above-described method for producing the clay composite (B), was superposed.
- the mixture can be prepared by adding to the miscible monomer and stirring thoroughly.
- the clay composite (B) and the dispersion medium are removed without removing the dispersion medium.
- the mixture can be made from SS directly using the containing dispersion.
- the clay composite (B) is prepared by introducing the silane compound (B2) into the swellable gaylate (B1) in a dispersion medium in accordance with the above-described method for producing the clay composite (B). After obtaining a dispersion composed of the dispersion medium and the clay complex (B) by adding the polymerizable monomer to the dispersion and thoroughly mixing the mixture, a mixture can be obtained.
- the dispersion medium itself may be a polymerizable monomer.
- the polymerizable monomer in the mixture obtained in the mixing step is polymerized by a polymerization method generally used for various thermoplastic resins (A).
- the thermoplastic resin (A) is a thermoplastic polyester resin
- the polymerization step can be carried out using, for example, the following soluble polycondensation method: First, the mixture obtained in the mixing step is used. Introduce into the polymerization reactor If necessary, another monomer composing the thermoplastic polyester resin may be newly added to the system. A catalyst required for the polymerization reaction may be used by adding one or more of gold oxide, carbonated carbonate, acetate, and alcoholate.
- thermoplastic polyester resin having a desired structure is added to the mixture, and the system is heated and mixed to near the melting point of the thermoplastic polyester resin.
- the thermoplastic polyester resin has a logarithmic viscosity of 0.3 to 2.0 (d) Zg measured at 25 in a phenol Z tetrachloroethane (55 weight ratio) mixed solvent. preferable.
- the thermoplastic polyester resin is depolymerized with the diol compound to convert the thermoplastic polyester resin into monomers and oligomers having Z or about 2 to 15 repeating units.
- the clay composite is uniformly dispersed in the system, and then the polycondensation reaction is performed under reduced pressure.
- the catalyst required for the reaction is already contained in the resin as the starting material, but if necessary, one or more of the same catalysts as in the above-mentioned melt polycondensation are newly added. It can also be used.
- a solid phase polymerization method can also be performed.
- the solid phase polymerization method can be performed by the following method. That is, a low polymerization degree of the polyester resin is obtained by a melt polycondensation reaction, and this is cooled and solidified. After the low-polymer is sufficiently dried, the mixture is heated to 150 ° C. or lower of the resin under a stream of inert gas such as nitrogen or under reduced pressure to remove the generated diol compound from the system. Polymerizes in the solid state while removing to obtain a high degree of polymerization,
- the polymerization step can be performed, for example, by utilizing the interfacial polymerization method described below.
- dimethylene salt and phosgene are added to the mixture containing the alkali metal salt of the bisphenol compound obtained in the above mixing step, and while sufficiently stirring, polycondensation is performed at the interface between the alkaline aqueous phase and the methylene chloride phase.
- a reaction can be performed.
- a catalyst required for the interfacial polymerization method one or more kinds of aliphatic tertiary amines, alicyclic tertiary amines, aromatic tertiary amines and the like can be added and used.
- a melt polymerization method can be performed.
- the melt polymerization method include the following methods. First, the bisphenol compound was added to the mixture containing the carbonic acid diester compound obtained in the above mixing step, and the system was heated from about 280 to about 300 "with sufficient stirring to obtain a solution.
- the catalyst required for the solution fiffi polymerization method in which the transesterification reaction is carried out in a state includes alkali metal or alkaline earth metal simple substance, oxide, hydroxide, amide compound, alcohol, phenolate, and Sb 2 ⁇ 3, Z n O, P b O, the organic titanium compound may be used with the addition of one or more of such quaternary Anmoniumu salt.
- thermoplastic resin composition (C) of the present invention is prepared by mixing and polymerizing steps as described above.
- the thermoplastic resin (A) and the clay composite (B) can also be produced by directly melt-kneading them using various general kneaders.
- the kneader include a kneader capable of giving a high shearing force to the system, such as a single-screw extruder, a twin-screw extruder, a Banbury mixer, and a roll.
- a composite twin-screw extruder having a knee disc portion is preferable.
- the dispersion medium used in the production of the clay composite (B) is usually removed in advance.
- the removal of the separating medium can be omitted, and the clay composite (B) containing those can be used.
- the clay composite (B) containing the dispersion medium is preferable because it has good uniformity with respect to the resin.
- thermoplastic resin (A) and the clay composite (B) may be put into the above kneader at once and melt-kneaded, or the thermoplastic resin (A) and the clay composite ( B) may be added for kneading.
- thermoplastic resin composition (C) of the present invention can be produced by polymerization or melt-kneading as described above.
- thermoplastic resin (A) is easily dissolved in a solvent, for example, the thermoplastic resin (A)
- the thermoplastic resin (A) In the case of polycarbonate resin, polyarylate resin, vinyl polymer compound, or polyphenylene oxide resin, after dispersing the clay complex (B) in a solvent, the thermoplastic resin (A) is added to the dispersion. ) Is dissolved and the solvent is removed by drying or the like.
- thermoplastic resin (A) when the above polycarbonate resin is used as the thermoplastic resin (A), methylene chloride or the like can be used as a solvent.
- the clay composite (B) is added to methylene chloride, and the mixture is stirred, mixed and separated, and then the polycarbonate resin is added and dissolved. Then, the methylene chloride is removed by drying to obtain the thermoplastic resin of the present invention.
- a composition (C) is obtained.
- thermoplastic resin composition (C) of the present invention may contain, if necessary, polybutadiene, butadiene-styrene copolymer, acrylic rubber, ionomer, ethylene propylene.
- Hydrogenated butyl rubber, homopolymer of ⁇ -leu-refin, copolymer of two or more ⁇ -leu-refins Hydrogenated butyl rubber, homopolymer of ⁇ -leu-refin, copolymer of two or more ⁇ -leu-refins (Hundam, Zo If it is a u-k, it is possible to add any co-star S-isomer, and these substances may be added.) or an anti-impact property improver such as an olefinic elastomer can be added.
- thermoplastic resin (A) any other resin other than the above thermoplastic resin (A) may be used as long as it does not impair the mechanical properties and moldability of the thermoplastic resin composition ⁇ c>.
- unsaturated polyester resin, polyester resin, liquid crystal polyester resin, polyolefin resin, polyamide resin, rubber polymer reinforced styrenic resin, polyphenylene sulfide resin, polyphenylene ether Fats, polyacetal resins, polysulfone resins, polyarylate resins, and the like alone or in combination of two or more.
- thermoplastic resin composition (C) of the present invention may contain pigments, dyes, heat stabilizers, antioxidants, ultraviolet absorbers, light stabilizers, lubricants, plasticizers, flame retardants, depending on the purpose. And additives such as antistatic agents.
- the structure of the clay complex (B) dispersed in the thermoplastic resin composition (C) of the present invention obtained by the above-described method has a swellable gaylate (B 1) before being mixed. This is completely different from the cohesive structure in which many unit layers are overlapped with each other. That is, by blending the clay composite (B), which has an increased base spacing compared to the cohesive structure of the initial swellable gaylate (B 1), with the thermoplastic resin (A), the layers are further combined. Cleavage increases the base spacing. As a result, the clay composite (B) becomes a number of very fine layers in the thermoplastic resin composition (C). It is dispersed independently of each other.
- the dispersion state of the clay composite (B) in the thermoplastic resin composition (C) can be expressed by various parameters as described below. Ratio of the number of clay composites (B) in which [R B 300 ] is dispersed in the thermoplastic resin composition (C) and the equivalent area circular diameter [D] of the clay composites (B) is 300 nm or less When defined, [R B3 .
- 0 ] value is preferably 20% or more, more preferably 35% or more, even more preferably 50% or more. If the [ RB3 () () ] value is less than 20%, The effect of improving the mechanical properties and heat distortion resistance of the plastic resin composition (C) tends to be insufficient.
- equivalent area circle 3 ⁇ 4 [D] refers to an image obtained with a microscope or the like.
- the area of each dispersed eyebrow dispersed in various shapes and the diameter of a circle having the same area, and the average value means their number average value.
- Equivalent area circle E diameter [D] is measured by selecting an arbitrary area containing 100 or more dispersion layers on an image taken with a microscope or the like, forming an image with an image processing device or the like, and performing computer processing. Can be quantified.
- the average value of the equivalent surface Minoruen diameter of the clay compound (B) [D B] is obtained, for example, by photographing the thermoplastic resin composition (C) of the present invention with a transmission electron microscope (TEM) It can be quantified by using a photograph showing the dispersion state of the clay complex (B).
- TEM transmission electron microscope
- thermoplastic ⁇ composition of the present invention (C) is preferably 500 nm or less, and more preferably not more 450 nm or less, still more preferably below 400 nm. In the range of [R B300] value indicated above, and, when the [D B] value is within this range, the thermoplastic ⁇ composition (C) of mechanical properties and heat distortion resistance improvement effect is further Can be large. Although the lower limit is not particularly [D B] value, the [D B] value effect is not almost less than about 1 0 nm, ⁇ short not be less than 10 nm,
- the size of the cleaved finely divided clay complex (B) can be very small compared to the initial cohesive structure of the swellable gaylate (B1). That is, [DB] is defined as the average value of the equivalent area circle diameter of the clay complex (B) in the thermoplastic resin composition (C) as described above, and [D B1 ] is defined as the swellable gaylate ( Equivalent surface of B 1) When defined as the mean value of Sekien diameter, [D B] / "D B1] value is preferably
- thermoplastic ⁇ fat composition (C) 010 or less, more preferably 0.008 or less, and even more preferably 0.055 or less.
- the equivalent area circle diameter [D B1 ] value can be determined, for example, as follows: the swelling of the thermoplastic resin (A), which has the same ash content as that of the thermoplastic resin composition (C); A resin composite containing an expansive silicate (B1) is separately prepared using a heat press, etc. The swellable silicate (Bl) obtained by photographing the resin composite with an optical microscope is used. can be placed the same way [D B1] value [D B] by using a photograph showing the dispersed state
- the number of clay complexes (B) dispersed in the thermoplastic resin composition (C) is reduced by the initial swellable silicate. It increases compared to (B 1). That is, [N B ] is defined as the number of clay complexes (B) per unit ash fraction and per unit area, and [N Bi ] is defined as the swellable gaylate per unit ash fraction and per unit area.
- thermoplastic resin composition of the present invention when defining the number (C) [N B] in / [N B1] values rather preferably becomes 300 or more, more preferably be 400 or more, more preferably If the [N B ] / [N B1 ] value is within the above range, the number of clay composites (B) that reinforce the physical properties of the thermoplastic resin composition (C) increases. The effect of improving the mechanical properties and thermal deformation resistance of the thermoplastic resin composition (C) is further increased. There is no particular upper limit, but if the [N B ] / [N B1 ] value exceeds about 50,000, the effect will not change, so there is no need to make it larger than 50,000.
- the N ⁇ value which is the number of dispersed layers per unit ash fraction and per unit area, can be obtained, for example, as follows. First, on an image taken with a microscope or the like, An arbitrary region including 100 or more dispersion layers is selected, and the number of dispersion layers existing in the region and the area of the region are determined. Separately, a thermoplastic resin composition derived from the dispersion layer
- [N] value can be obtained by dividing the number of the dispersion layers by the area of the region and the ash content. Therefore, the [N B ] value of the clay complex (B) and the [N B1 ] value of the swellable silicate (B 1) are determined by the equivalent area circle diameter described above.
- thermoplastic resin composition (C) or containing the thermoplastic resin (A) and the swellable gaylate (B1) was used.
- TE transmission electron microscope
- can be quantified by using an optical microscope Mirror photographs free ⁇ composite unit of area is not limited particularly using [N B] values and [N B1] value the same unit together when calculating the, e.g. Any unit can be used, such as, m 2 , nm 2 , or A 2 .
- the clay composite (B) is extremely finely dispersed as compared with the initial aggregate structure of the swellable gaylate (B1). Further, when each of the dispersed clay composites (B1) is in the form of a thin plate, various physical properties such as mechanical properties and heat resistance of the thermoplastic resin composition (C) are more efficiently improved. That is, in the thermoplastic resin composition (C), the average value of the layer thickness of the clay composite (B) is 2 Onm or less, preferably 18 nm or less, and more preferably 15 nm or less. The lower limit is not particularly limited, but is about 1 nm.
- an arbitrary area containing 100 or more clay composites (B) is selected. It can be quantified by measuring the layer thickness of the individual clay composites (B), with the mean being the mean number of them.
- the thermoplastic plastics With respect to the clay composite (B) that is independently dispersed, if the ratio of those with a layer thickness of 5 nm or less is 20% or more, and even 30% or more of the entire clay composite (B), the thermoplastic plastics (4) The mechanical properties and heat deformation resistance of the resin composition (C) can be more efficiently improved.
- the method for measuring the layer thickness is the same as that described above.
- [I When B1] is defined as the diffraction intensity of the initial aggregate structure from which small angle X-ray diffraction of ⁇ of gay salt (B 1)
- [I B ] Z [I B1] value preferably is under 0.25 or less [I B ] [I B1 ]
- the mechanical properties and heat resistance of the thermoplastic resin composition (C) are further improved.
- the lower limit is small angle X-ray diffraction intensity [I B] value is reduced to from not particularly-clay composite (B), when the determining their baseline or noise is difficult [I B ZI B1] value is 0, there clay complex (B) Wahopokan by cleaving all independently.
- [I B1] value as in the case of the [D B1 ⁇ value, A resin composite containing a thermoplastic resin (A) and a swellable gaylate (B1) having the same ash content as the plastic resin composition (C) was separately prepared, and a small-angle X-ray of the resin composite was prepared. It can be determined by performing a diffraction measurement.
- the measurement of the small-angle X-ray diffraction intensity [I] is performed by obtaining the peak intensity or the integrated intensity of the small-angle X-ray diffraction.
- the method of measuring the integrated intensity of the small-angle X-ray diffraction is not particularly limited, and includes a method generally used, for example, a method of obtaining an area from a diffraction pattern of X-ray diffraction measurement, and a method of obtaining an area from a count value.
- Examples of a method for obtaining the area from the above-mentioned diffraction pattern include a method generally used, such as a Blanimeter method, a gravimetric method, and a triangular approximation method (peak height X half width).
- Examples of the method for obtaining from the above count value include a 20-continuous scan method, a 20-step scan method, and a 20-fixed method.
- the bottom spacing of the cleaved clay complex (B) in the thermoplastic resin composition (C) is at least three times the bottom spacing of the initial swellable silicate (B 1), which is the cohesive structure before blending. It is preferable that the ratio be at least 5 times because the mechanical properties and heat resistance of the thermoplastic resin composition (C) can be more efficiently improved.
- the method of measuring the distance between the bottoms is the clay complex (B) May be the same as the method described above with respect to the method of transcribing.
- thermoplastic resin composition (C) of the present invention may be molded by injection molding or hot press molding, and can also be used for blow molding.
- thermoplastic resin composition (C) of the present invention can be used for a biaxially stretched film that maintains transparency and has excellent mechanical properties. Molded articles and films made from the thermoplastic resin composition (C) of the present invention have excellent appearance, mechanical properties, heat-resistant deformation properties, etc., so that, for example, automobile parts, household electric parts, precision machinery parts, etc. Suitable for household daily necessities, packaging and container materials, magnetic recording tape base materials, and other general industrial materials.
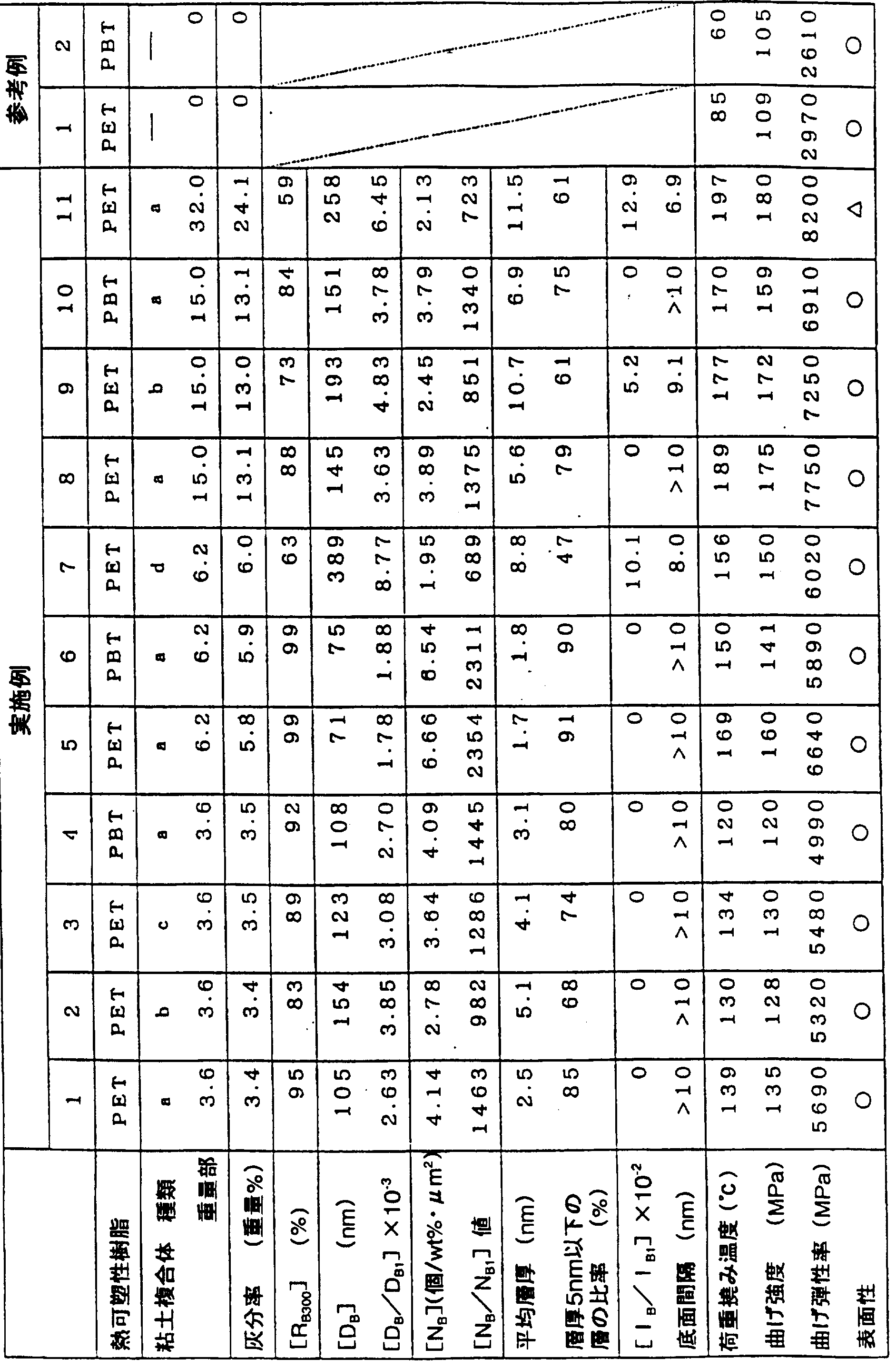
- thermoplastic resin (A) thermoplastic resin
- PET resin Kanebo Co., Ltd., PBK2
- PBT Polybutylene terephthalate
- PC resin Teflon A—2200 manufactured by Idemitsu Petrochemical Co., Ltd.
- BHET NI SSO BHE
- EG Monoethylene glycol manufactured by Nippon Shokubai Co., Ltd.
- 1,4-BD 1,4-butanediol
- the swellable mica used was synthesized as follows.
- the combined clay was added to tetrahydrofuran (THF), and the mixture was stirred for 15 minutes to wash and remove the adsorbed silane-based compound. After centrifugation, the supernatant was separated. This washing operation was repeated three times, and about lmg of the sufficiently dried clay complex and about 20 Omg of KBr powder were thoroughly mixed in a mortar, and then the KBr for measurement was measured from this mixture using a tabletop press. A disc was prepared. Next, measurements were made by the transmission method using an infrared spectrometer. The detector was an MCT detector with a resolution of cn 1 and 100 scans.
- TEM Transmission electron microscope
- the average value of the layer thickness was obtained by measuring the layer thickness of each individual clay aggregate in an arbitrary area where 100 or more clay composites exist in a TEM photograph of the thermoplastic resin composition of the present invention. was determined by number averaging.
- the small-angle X-ray diffraction intensity [I] was obtained from the area of the diffraction pattern. However, when it is difficult to distinguish the diffraction pattern from the baseline, that is, when the diffraction peak is very small, the X-ray diffraction intensity ratio is 0%. And
- the bottom surface interval was calculated by substituting the small-angle X-ray diffraction peak angle value into the equation of Bragg. However, when it is difficult to confirm the small-angle X-ray peak angle value, the layer is sufficiently cleaved and crystallinity substantially disappears as described above, or the peak angle value is approximately 0.8. It was considered difficult to confirm because of the following, and the evaluation result of the bottom surface spacing was> 10 nm.
- thermoplastic resin composition derived from the clay composite conforms to JI SK 7052 was measured.
- the obtained ash content is the ash content derived from the clay composite.
- thermoplastic resin composition After drying the thermoplastic resin composition, use an injection molding machine with a mold clamping pressure of 75 t, and perform injection molding at a resin temperature of about 260 to 280 to obtain a size of about 10 X 100 X 6 mm.
- the test piece was made,
- the deflection temperature at a load of 1.85 MPa of the killed specimen obtained by injection molding was measured according to ASTM D-648.
- the flexural strength and flexural modulus of the test piece obtained by injection molding were measured according to ASTM D-790.
- a part of the slurry was dried and pulverized to obtain a clay composite (clay composite a).
- the distance between the bottom surfaces of the obtained clay composite was 2.6 nm. Further, as a result of FT-IR measurement of the clay complex after washing with THF, absorption bands derived from primary amino groups, secondary amino groups, and ethylene groups were observed.
- the clay composite and slurry obtained in Production Example 1 are referred to as clay composite a and slurry a, respectively.
- a part of the above slurry was dried and pulverized to obtain a clay composite (clay pulp b).
- the distance between the bottom surfaces of the obtained clay composite was 2.0 nm.
- absorption bands derived from epoxy 51 (ethylene oxide group), ether group, and methylene group were observed.
- 150 g of a montmorillon mouth was mixed with 4500 g of pure water by stirring at 5000 rpm for 3 minutes using a high-speed stirrer to disperse. Thereafter, 15 g of ⁇ -polyoxyethylene propyltrimethoxysilane hydrolyzed with water adjusted to pH 4 with hydrochloric acid is added dropwise using a simple pipette, and the mixture is stirred at a shear rate of 20000 (lZs) for 2 hours. As a result, a slurry (slurry c) comprising the clay composite and water was obtained.
- a part of the slurry was dried and pulverized to obtain a clay composite (clay composite c) .
- the distance between the bottom surfaces of the obtained clay composite was 2.4 nm.
- absorption bands derived from an ether group and an ethylene group were observed.
- the clay composite and slurry obtained in Production Example 3 are referred to as clay composite c and slurry c, respectively.
- swellable mica 150 g was dispersed in 3500 g of pure water by stirring for 6 minutes at 6000 rpm using a high-speed It stirrer. Then, 25 g of r- (2-aminoethyl) aminobutyral tritrimethoxysilane was added dropwise using a simple pipette, and the mixture was stirred at a shear rate of 30,000 (1 / s) for 3 hours to obtain a slurry consisting of the clay complex and water. (Slurry d).
- a part of the slurry was dried and pulverized to obtain a clay composite (clay composite d).
- the bottom interval of the clay composite was 1.8 nm.
- the FT-IR measurement of the clay complex after washing with THF showed that absorption bands derived from primary amino groups, secondary amino groups, and methylene groups were observed.
- the clay composite and slurry obtained in Production Example 4 are referred to as clay composite d and slurry d, respectively.
- silane-treated montmorillonite a ′ 15 g of r- (2-aminoethyl) aminobutyral pill with 150 g of montmorillonite Trimethoxysilane was sprayed using a spray and mixed for 1 hour to obtain a silane-treated montmorillonite.
- the bottom spacing of the silane-treated montmorillonite was 1.3 nm, which was the same as the initial bottom spacing of the montmorillonite before the treatment.
- the silane-treated montmorillonite after washing with THF was measured by FT-IR, and as a result, absorption bands derived from primary amino groups, secondary amino groups, and ethylene groups were observed.
- the treated montmorillonite obtained in Production Example 5 is referred to as silane-treated montmorillonite a ′.
- BHET bishydroxyethyl terephthalate
- FIG. 2 shows a small-angle X-ray diffraction pattern of the thermoplastic polyester resin composition obtained in Example 1.
- Fig. 2 shows a small-angle X-ray diffraction pattern of the thermoplastic polyester resin composition obtained in Example 1.
- no crystalline peak derived from the clay complex was observed, indicating that the layers were not aggregated but were dispersed independently.
- the evaluation result of the bottom surface spacing was> 10 nm.
- Optical microscopy and SAXS measurements were performed on the molded product obtained in Production Example 6. After evaluating similarly subjected was calculated [D B] / [D B1 ] value, [N B] / [N B1] ' value, and [I B] Z [I B1 ] value.
- thermoplastic polyester resin composition derived from the clay composite
- deflection temperature under load of the injection-molded test specimen the bending strength
- bending elastic modulus the visual surface property
- Table 2 shows the above evaluation results.
- PET was polymerized in the same manner as in Example 1 except that 2700 g of the slurry b produced in Production Example 2 was used, and a thermoplastic polyester resin composition containing the clay composite b was produced and evaluated. See Table 2.
- the intrinsic viscosity of PET in the thermoplastic polyester resin composition produced in Example 2 was 0.60 (d 1 / g).
- PET was polymerized in the same manner as in Example 1 except that 2700 g of the slurry c produced in Production Example 3 was used, and a thermoplastic polyester resin composition containing the clay composite c was produced and evaluated.
- the logarithmic viscosity of PET in the thermoplastic polyester resin composition produced in Example 3 was 0.60 (dl Zg).
- BHBT bishydroxybutyl terephthalate
- the logarithmic viscosity of PBT in the thermoplastic polyester resin composition produced in Example 4 was 0.82 (dl / g).
- the EG dispersion, 5000 g of PET, and 15 g of a hindered phenol-based stabilizer were charged from the supply port of the horizontal continuous polymerization reactor.
- the mixture was stirred at a temperature of 280 at a rotation speed of 3 Orpm for about 3 hours, and PET was depolymerized while removing ethylene glycol to the outside of the system.
- the PET was polymerized by removal and polycondensation reaction to produce a thermoplastic polyester resin composition containing the clay complex a, which was continuously discharged from the discharge port. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 7 was used instead of the system prepared in Production Example 6. Table 2 shows the results.
- PB was polymerized in the same manner as in Example 5 except that 1,4-BD was used instead of EG, PBT was used instead of PET, and the temperature of the polymerization machine was set to 260.
- the thermoplastic polyester resin composition containing was manufactured. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 13 was used instead of the system prepared in Production Example 6. The results are shown in Table 2.
- the logarithmic viscosity of PBT in the thermoplastic polyester resin composition produced in Example 6 was 0.83 (d 1 / g).
- PET was polymerized in the same manner as in Example 5 except that 7000 g of the slurry d produced in Preparation Example 4 was used instead of the slurry a.
- the thermoplastic polyester resin composition containing the clay-agglomerate d The product was manufactured and evaluated in the same manner as in Example 1 except that the system prepared in Production Example 11 was used instead of the system prepared in Production Example 6. The results are shown in Table 2.
- the logarithmic viscosity of PET in the thermoplastic polyester resin composition produced in the above was 0.65 (dl Zg).
- BHET dispersion (containing a trace amount of water) composed of clay complex a and BHET was produced in the same manner as in Example 1.
- the composition was cooled, cooled and sufficiently dried, and charged into a solid-state polymerization machine.
- thermoplastic polyester resin composition containing the clay complex a was produced by performing solid phase polymerization for 9 hours under the condition of a reduced pressure of about 0.5 torr to increase the molecular weight of the PE.
- the system produced in Production Example 8 was performed in the same manner as in Example 1. Table 2 shows the results.
- the intrinsic viscosity of PET in the thermoplastic polyester resin composition produced in Example 8 was 0.61 (dl / g).
- thermoplastic polyester resin composition containing clay composite b was produced in the same manner as in Example 8, except that 6750 g of slurry b was used instead of slurry a. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 8 was used instead of the system prepared in Production Example 6. Table 2 shows the results.
- the intrinsic viscosity of PET in the thermoplastic polyester resin composition produced in Example 9 was 0.62 (dlZg).
- PBT was polymerized in the same manner as in Example 4 using 10,000 g of slurry a, 2250 g of 1,4-BD, 1330 g of DM, 0.32 of 1 (OBu) 4 and 4.5 g of A060.
- a composition comprising a clay complex a and a low molecular weight g-substance of PBT having an logarithmic viscosity of 0.29 (d 1 / g) was obtained.
- Example 8 the composition was subjected to solid phase polymerization to increase the molecular weight of PBT, thereby producing a thermoplastic polyester resin composition. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 14 was used instead of the system prepared in Production Example 6. Table 2 shows the results.
- the slurry 'a' was dried and pulverized to obtain a powdery clay complex a.
- This clay composite a 4 & 0 g, PET 1500 g, and 4.5 g of AO 60 are dry blended, and the number of rotations is set to 100 rpm by using a unidirectional twin-screw extruder with a kneading disk. Melting and kneading at a temperature of 250 to 27 Ot: Therefore, a thermoplastic polyester resin composition containing the clay composite a was manufactured. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 9 was used instead of the system prepared in Production Example 6. Table 2 shows the results.
- a polyester resin composition was produced in the same manner as in Example 11 except that the clay composite a was 1150 g, the PET was 1000 g, and the AO60 was 3.0 g.
- the evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 10 was used instead of the system prepared in Production Example 6. The results are shown in Table 3.
- PET was polymerized in the same manner as in Example 1 except that 90 g of montmorillonite was used instead of the slurry a, and a composite composed of PET and montmorillonite was produced and evaluated. Table 3 shows the results.
- a composite consisting of PET and montmorillonite was produced in the same manner as in Example 11 except that 640 g of montmorillonite, 2000 g of PET and 6.0 g of AO60 were used. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 9 was used instead of the system prepared in Production Example 6. Table 2 shows the results.
- PET was polymerized in the same manner as in Example 1 except that 155 g of silane-treated montmorillonite a ′ was used instead of slurry a to produce a composite composed of PET and silane-treated montmorillonite a ′. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 7 was used instead of the system prepared in Production Example 6. The results are shown in Table 3. The logarithmic viscosity of PET in the composite produced in Comparative Example 4 was 0.63 (d 1 / g).
- PBT was polymerized in the same manner as in Example 4 except that silane-treated montmorillonite a '124 g was used instead of slurry a.
- a composite consisting of the following kits was produced. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 13 was used instead of the system prepared in Production Example 6. The results are shown in Table 3. In addition, the logarithmic viscosity of PBT in the composite produced in Comparative Example 5 was 0.83 (d 1 / g). Table 3
- the slurry a was dried and pulverized to obtain a powdery clay composite a.
- the clay composite a 12 O g, PC 2000 g and 6.0 g of phosphorus-based stabilizer (Adeka Stab PEP 36 manufactured by Asahi Denka Co., Ltd., hereinafter referred to as PEP 36) were dry-drawn.
- the slurry b and the slurry d were dried and pulverized to obtain a powdery clay composite b and a clay composite d.
- the slurry c was dried and pulverized to obtain a powdery clay composite c.
- Example 1 was repeated except that the system prepared in Production Example 15 was used instead of the system prepared in Production Example 6 to produce a polycarbonate resin composition containing the clay complex c. Was evaluated in the same way as The results are shown in Table 4.
- Test specimens of PC were molded, and the deflection temperature under load, flexural modulus, and surface properties were evaluated. Table 3 shows the results.
- a composite consisting of PC and montmorillonite or silane-treated montmorillonite a ′ was produced in the same manner as in Example 12 except that montmorillonite or silane-treated montmorillonite a ′ was used instead of the clay composite. Evaluation was performed in the same manner as in Example 1 except that the system prepared in Production Example 15 was used instead of the system prepared in Production Example 6. The results are shown in Table 4.
- the clay composite (B) obtained by introducing the silane-based compound (B 2) after enlarging the base spacing of the swellable silicate (B 1) is filled with the thermoplastic resin composition. Used as an agent.
- this clay composite is blended with the thermoplastic resin composition (C), the layers are cleaved and the base spacing is further increased. As a result, the clay composite
- thermoplastic resin composition having excellent properties such as mechanical properties (elastic modulus, strength, toughness, etc.), heat resistance, and surface appearance of a molded article can be obtained by adding a small amount of the clay composite (B).
- Object (C) is provided.
- a swellable gaylate (B1) in an aggregated structure state, which is treated with a silane compound (B2) as it is, is used as a filler for the thermoplastic resin composition. Therefore, adding a small amount cannot improve the thermal conductivity and heat resistance, and adding a large amount decreases the surface properties, strength, toughness, etc. of the molded product, making it difficult to balance various properties. there were.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description



Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP54073197A JP3941841B2 (ja) | 1996-05-13 | 1997-05-13 | 粘土複合体含有熱可塑性樹脂組成物およびその製造方法 |
| EP97920955A EP0899308B1 (en) | 1996-05-13 | 1997-05-13 | Thermoplastic resin composition containing silane-treated foliated phyllosilicate and method for producing the same |
| US09/180,546 US6239195B1 (en) | 1996-05-13 | 1997-05-13 | Thermoplastic resin composition containing silan-treated foliated phyllosilicate and method for producing the same |
| AU27126/97A AU2712697A (en) | 1996-05-13 | 1997-05-13 | Thermoplastic resin composition containing clay composite and process for preparing the same |
| DE69731465T DE69731465D1 (de) | 1996-05-13 | 1997-05-13 | Thermoplastische harz-zusammensetzung enthaltend ein mit einem silan behandelten mehrschichtigen phyllosilicat und verfahren zu deren herstellung |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP11807496 | 1996-05-13 | ||
| JP8/118074 | 1996-05-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997043343A1 true WO1997043343A1 (fr) | 1997-11-20 |
Family
ID=14727364
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1997/001605 Ceased WO1997043343A1 (fr) | 1996-05-13 | 1997-05-13 | Composition de resine thermoplastique contenant un composite a base d'argile, et procede de fabrication associe |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US6239195B1 (ja) |
| EP (1) | EP0899308B1 (ja) |
| JP (1) | JP3941841B2 (ja) |
| AU (1) | AU2712697A (ja) |
| DE (1) | DE69731465D1 (ja) |
| WO (1) | WO1997043343A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002060599A (ja) * | 2000-08-23 | 2002-02-26 | Kanegafuchi Chem Ind Co Ltd | 照明部品用ポリエステル樹脂成形体 |
| JP2002138207A (ja) * | 2000-08-25 | 2002-05-14 | Sekisui Chem Co Ltd | テープ基材用樹脂組成物、それを用いたテープ基材及びそれを用いた粘着テープ |
| US6583208B1 (en) * | 1997-10-30 | 2003-06-24 | Kaneka Corporation | Polyester resin compositions and processes for the preparation thereof |
| JP2004323717A (ja) * | 2003-04-25 | 2004-11-18 | Nippon Valqua Ind Ltd | 充填材入りフッ素樹脂シートおよびその製造方法 |
| JP2020002188A (ja) * | 2018-06-25 | 2020-01-09 | Dic株式会社 | 樹脂組成物、成形体、積層体、コーティング材及び接着剤 |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5916685A (en) * | 1996-07-09 | 1999-06-29 | Tetra Laval Holdings & Finance, Sa | Transparent high barrier multilayer structure |
| TW477743B (en) * | 1999-12-17 | 2002-03-01 | Ind Tech Res Inst | Modified clay materials and polymer composites comprising the same |
| US20040249045A1 (en) | 2001-07-18 | 2004-12-09 | Howard Goodman | Clay mineral products and their use in a rubber compositions |
| EP1428657A4 (en) * | 2001-09-18 | 2005-04-13 | Tokuyama Corp | GASPERRFILM AND GASSPERRÜBERZUGSMITTEL AND METHOD FOR THE PRODUCTION |
| GB0207850D0 (en) | 2002-04-04 | 2002-05-15 | Unilever Plc | Fabric treatment compositions |
| JP2003317226A (ja) * | 2002-04-16 | 2003-11-07 | Fuji Photo Film Co Ltd | 磁気記録媒体 |
| JP2003317231A (ja) * | 2002-04-16 | 2003-11-07 | Fuji Photo Film Co Ltd | 磁気記録媒体 |
| CA2450150C (en) * | 2002-11-22 | 2012-01-24 | Minh-Tan Ton-That | Polymeric nanocomposites |
| FR2851567B1 (fr) * | 2003-02-24 | 2005-04-22 | Solvay | Procede de fabrication de nanocomposites |
| JP4570864B2 (ja) * | 2003-11-25 | 2010-10-27 | 株式会社資生堂 | 樹脂組成物及び樹脂成形体 |
| ES2381052T3 (es) * | 2004-07-14 | 2012-05-22 | Agency For Science, Technology And Research | Nanocompuestos y procedimiento para su producción |
| US20100196611A1 (en) * | 2004-07-14 | 2010-08-05 | Nopphawan Phonthammachai | Nanocomposites and process for their production |
| US20080009568A1 (en) * | 2004-10-18 | 2008-01-10 | Nitin Kumar | Methods to disperse and exfoliate nanoparticles |
| FR2895412B1 (fr) * | 2005-12-23 | 2008-05-23 | Saint Gobain Vetrotex | Procede de preparation de nanoparticules en feuillets et nanoparticules obtenues. |
| US20080081865A1 (en) * | 2006-09-29 | 2008-04-03 | Amit Biswas | Method of making polycarbonate nanocomposites |
| US20080167414A1 (en) * | 2006-09-29 | 2008-07-10 | Amit Biswas | Polycarbonate composition comprising nanomaterials |
| US20100003431A1 (en) * | 2008-07-02 | 2010-01-07 | John Raybuck | Composite materials |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05194851A (ja) * | 1991-10-01 | 1993-08-03 | Tosoh Corp | ポリアリーレンスルフィド複合材料およびその製造方法 |
| JPH06322249A (ja) * | 1993-05-17 | 1994-11-22 | Fukuvi Chem Ind Co Ltd | ポリカーボネート樹脂組成物 |
| JPH08269321A (ja) * | 1995-03-30 | 1996-10-15 | Unitika Ltd | 強化ポリアミド樹脂組成物の製造法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2048285B (en) | 1979-04-26 | 1983-10-19 | Gen Electric | Polyester compositions |
| WO1993004118A1 (en) * | 1991-08-12 | 1993-03-04 | Allied-Signal Inc. | Melt process formation of polymer nanocomposite of exfoliated layered material |
| WO1993011190A1 (en) * | 1991-11-26 | 1993-06-10 | Allied-Signal Inc. | Polymer nanocomposites formed by melt processing of a polymer and an exfoliated layered material derivatized with reactive organo silanes |
| WO1995006090A1 (en) | 1993-08-23 | 1995-03-02 | Alliedsignal Inc. | Polymer nanocomposites comprising a polymer and an exfoliated particulate material derivatized with organo silanes, organo titanates and organo zirconates dispersed therein and process of preparing same |
| US5955535A (en) * | 1993-11-29 | 1999-09-21 | Cornell Research Foundation, Inc. | Method for preparing silicate-polymer composite |
-
1997
- 1997-05-13 DE DE69731465T patent/DE69731465D1/de not_active Expired - Lifetime
- 1997-05-13 US US09/180,546 patent/US6239195B1/en not_active Expired - Lifetime
- 1997-05-13 WO PCT/JP1997/001605 patent/WO1997043343A1/ja not_active Ceased
- 1997-05-13 JP JP54073197A patent/JP3941841B2/ja not_active Expired - Fee Related
- 1997-05-13 EP EP97920955A patent/EP0899308B1/en not_active Expired - Lifetime
- 1997-05-13 AU AU27126/97A patent/AU2712697A/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05194851A (ja) * | 1991-10-01 | 1993-08-03 | Tosoh Corp | ポリアリーレンスルフィド複合材料およびその製造方法 |
| JPH06322249A (ja) * | 1993-05-17 | 1994-11-22 | Fukuvi Chem Ind Co Ltd | ポリカーボネート樹脂組成物 |
| JPH08269321A (ja) * | 1995-03-30 | 1996-10-15 | Unitika Ltd | 強化ポリアミド樹脂組成物の製造法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0899308A4 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6583208B1 (en) * | 1997-10-30 | 2003-06-24 | Kaneka Corporation | Polyester resin compositions and processes for the preparation thereof |
| JP2002060599A (ja) * | 2000-08-23 | 2002-02-26 | Kanegafuchi Chem Ind Co Ltd | 照明部品用ポリエステル樹脂成形体 |
| JP2002138207A (ja) * | 2000-08-25 | 2002-05-14 | Sekisui Chem Co Ltd | テープ基材用樹脂組成物、それを用いたテープ基材及びそれを用いた粘着テープ |
| JP2004323717A (ja) * | 2003-04-25 | 2004-11-18 | Nippon Valqua Ind Ltd | 充填材入りフッ素樹脂シートおよびその製造方法 |
| JP2020002188A (ja) * | 2018-06-25 | 2020-01-09 | Dic株式会社 | 樹脂組成物、成形体、積層体、コーティング材及び接着剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| US6239195B1 (en) | 2001-05-29 |
| EP0899308A1 (en) | 1999-03-03 |
| AU2712697A (en) | 1997-12-05 |
| EP0899308B1 (en) | 2004-11-03 |
| JP3941841B2 (ja) | 2007-07-04 |
| EP0899308A4 (ja) | 1999-03-03 |
| DE69731465D1 (de) | 2004-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1997043343A1 (fr) | Composition de resine thermoplastique contenant un composite a base d'argile, et procede de fabrication associe | |
| EP2873684B1 (en) | Method for producing thermoplastic resin composition | |
| TW593538B (en) | Polycarbonate resin composition, molded article and process for producing it | |
| JPWO1997043343A1 (ja) | 粘土複合体含有熱可塑性樹脂組成物およびその製造方法 | |
| JP4177554B2 (ja) | ポリエステル樹脂組成物およびその製造方法 | |
| JP4708572B2 (ja) | 熱可塑性樹脂組成物およびその製造方法 | |
| CN105392684B (zh) | 具有低烟雾和低热释放的列车内部部件及其制造方法 | |
| CN101514256A (zh) | 树脂成型用材料 | |
| US9796847B2 (en) | Method for producing thermoplastic resin composition | |
| JPWO2000060006A1 (ja) | 熱可塑性樹脂組成物およびその製造方法 | |
| JP3767965B2 (ja) | 粘土複合体層間化合物、粘土複合体層間化合物と熱可塑性樹脂からなる熱可塑性樹脂組成物およびそれらの製法 | |
| JP4526865B2 (ja) | ポリエステル樹脂製光反射体 | |
| JP3686260B2 (ja) | 層状無機物含有樹脂フィルム | |
| WO2005082972A1 (ja) | 芳香族ポリカーボネート樹脂組成物およびその製造方法 | |
| JP2002249609A (ja) | 光学用被覆フィルム | |
| JPH10310420A (ja) | 膨潤性雲母層間化合物およびそれを含む熱可塑性樹脂組成物 | |
| JP2011006530A (ja) | セルロース繊維強化ポリブチレンテレフタレート樹脂組成物 | |
| JP3630921B2 (ja) | ポリエステル樹脂組成物の製造方法 | |
| Labde et al. | Improved method for dispersing clay in a monomer or bis (2‐hydroxyl ethyl terephthalate) before in situ polymerization of poly (ethylene terephthalate)/montmorillonite nanocomposites | |
| WO2000022042A1 (fr) | Compositions de resines et leurs procedes de fabrication | |
| JPH0726123A (ja) | 熱可塑性ポリエステル組成物、その製造方法およびその成形品 | |
| JP7171335B2 (ja) | ポリカーボネート樹脂組成物 | |
| TWI897587B (zh) | 熱塑性樹脂組成物、其製備方法以及包括其之模塑物 | |
| JP3470736B2 (ja) | ポリカーボネート樹脂組成物及びその成形品 | |
| JP2005190933A (ja) | 導電性シート |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AU BA BB BG BR CA CN CU CZ EE GE HU IL IS JP KR LC LK LR LT LV MG MK MN MX NO NZ PL RO SG SI SK TR TT UA US UZ VN YU AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1997920955 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997920955 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09180546 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1997920955 Country of ref document: EP |