WO2002004399A1 - Verfahren zur herstellung von 3,3-diarylpropylaminen - Google Patents
Verfahren zur herstellung von 3,3-diarylpropylaminen Download PDFInfo
- Publication number
- WO2002004399A1 WO2002004399A1 PCT/EP2001/007803 EP0107803W WO0204399A1 WO 2002004399 A1 WO2002004399 A1 WO 2002004399A1 EP 0107803 W EP0107803 W EP 0107803W WO 0204399 A1 WO0204399 A1 WO 0204399A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- mmol
- aroma
- alkyl
- general formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *N1[C@](CO)CCC1 Chemical compound *N1[C@](CO)CCC1 0.000 description 5
- YHXPGNBCQYBCCG-VIFPVBQESA-N CC(O[C@@H](C1CCCCC1)C(O)=O)=O Chemical compound CC(O[C@@H](C1CCCCC1)C(O)=O)=O YHXPGNBCQYBCCG-VIFPVBQESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/04—Formation of amino groups in compounds containing carboxyl groups
- C07C227/10—Formation of amino groups in compounds containing carboxyl groups with simultaneously increasing the number of carbon atoms in the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/70—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form
- C07C45/71—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction with functional groups containing oxygen only in singly bound form being hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/76—Ketones containing a keto group bound to a six-membered aromatic ring
- C07C49/84—Ketones containing a keto group bound to a six-membered aromatic ring containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/08—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms
- C07D295/096—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms with the ring nitrogen atoms and the oxygen or sulfur atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/572—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6571—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and oxygen atoms as the only ring hetero atoms
- C07F9/6574—Esters of oxyacids of phosphorus
- C07F9/65744—Esters of oxyacids of phosphorus condensed with carbocyclic or heterocyclic rings or ring systems
Definitions
- the invention relates to a new process for the preparation of substituted 3, 3-diarylpropylamine derivatives.
- it relates to the preparation of such compounds by hydrocarbonylation / hydrofor ylation with subsequent reductive amination.
- the aforementioned 3, 3-diarylpropylamine derivatives are used for the treatment of urge to urinate incontinence and other spasmogenic conditions (cf. WO 99/58478).
- the processes described there for the preparation of the 3, 3-diphenylpropylamine derivatives are mostly multi-stage and usually require an enantiomer separation for the production of optically active compounds.
- the object of the invention is therefore to provide new processes for the preparation of substituted 3, 3-diarylpropylamine derivatives which are simpler than those described in the prior art, i.e. include fewer steps, and which also allow stereoselective synthesis of the target compounds.
- the invention relates to a process for the preparation of 3, 3-diarylpropylamines of the general formula I.
- Ar represents a substituted or unsubstituted aryl radical
- X represents H, OH or OR 3 ,
- Y represents Cl, Br, I, CN, CHO, CH 2 OR, COOH, COOR, in which R represents Ci-Cio-alkyl or a substituted or unsubstituted aryl radical, or Ci-Cio-alkyl,
- R 1 , R 2 represents Ci-Cio-alkyl or C 3 -C 8 cycloalkyl, where R 1 and R 2 can be linked to form a cyclic structure
- R 3 represents a radical derived from one of the following compounds:
- R 4 for a linear or branched C 1 -C 10 alkyl group or cycloalkyl group or a substituted or unsubstituted aryl radical
- R 5 for C 1 -C 10 alkyl, cycloalkyl, acyl, alkoxycarbonyl, benzoyl, diphenylphosphanyl , Dicyclohexylphosphanyl or diarylphosphanyl,
- R 6 is selected for a substituent from the
- R 7 represents a linear or branched Ci-Cio-alkyl group or a substituted or unsubstituted aryl radical, and stereoisomers thereof
- R 8 represents a substituted or unsubstituted aryl radical and R 9 represents hydrogen or a linear or branched C 1 -C 8 -alkyl radical
- R 3 represents one of the following radicals:
- the substituent X represents hydrogen (H), hydroxy (OH) or the group OR 3 .
- the substituent R 3 can have the meaning given for the substituents R and R 'in claim 1 of WO 99/58478.
- R 3 preferably represents a radical which is derived from the following compounds and alcohols:
- substituent R 3 can be derived from the following amino acid derivatives:
- L-phenylalanine and the alcohols that result from these amino acids by reducing the carboxylic acid function to the hydroxymethylene unit.
- R 3 can be derived from the following ⁇ -hydroxycarboxylic acids:
- R 4 is a linear or branched C 1 -C 8 -alkyl group or cycloalkyl group or a substituted or unsubstituted aryl radical and R 5 is Ci-Cio-alkyl, cycloalkyl, acyl, alkoxycarbonyl, benzoyl, diphenylphosphanyl, dicyclohexylphosphanyl or diarylphosphanyl.
- R 3 can also be derived from the carboxylic acids or alcohols shown below:
- R ⁇ is a substituent selected from the group consisting of PPh 2 , alkyl, acyl, alkoxycarbonyl, benzoyl, arylcarbonyl, diphenylphosphanyl, diarylphosphanyl, dicyclohexylphosphanyl, and stereoisomers thereof.
- R 3 can be derived from the carboxylic acids or alcohols shown below:
- R 7 represents a linear or branched Ci-Cio-alkyl group or a substituted or unsubstituted aryl radical, and stereoisomers thereof.
- substituent R 3 can be derived from one of the carboxylic acids shown below:
- All of the aforementioned acids have one or more asymmetry centers and are used optically actively.
- radical R 3 can also differ from compounds of the general formula
- R 8 represents a substituted or unsubstituted aryl radical and R 9 represents hydrogen or a linear or branched C ⁇ -C ⁇ o-alkyl radical.
- the substituent R 3 can also be derived from ⁇ -naphthol, ⁇ -naphthol or ' (R) - or, - (S) -1- (9-anthryl) -2,2, 2-trifluoroethanol.
- R 3 can be derived from ephedrine, ie 2-methylamino-l-phenylpropan-l-ol. This compound is chiral and all stereoisomeric forms are intended to be included in the context of the present invention.
- R 3 is in turn bound via the oxygen atom, resulting in an ether structure.
- R 3 can also represent a phosphite radical of the general formula -P (OR 10 ) (OR 11 ), in which R 10 and R 11 can be the same or different and represent an optionally polycyclic or bridged aryl radical.
- Preferred examples are phosphite radicals of the formulas
- R can also be used for:
- the substituent Y according to the general formula I represents Cl, Br, I, CN, COOH, COOR, CHO, CH 2 OR or Ci-Cio-alkyl.
- the substituent R represents a linear or branched C 1 -C 8 -alkyl group or a substituted or unsubstituted aryl radical.
- the substituent Y is chosen so that it can be converted into a hydroxymethyl group by simple, generally known methods. For example, an ester or carboxylic acid group is directly reduced, a halide can be converted into the corresponding carboxylic acid via a Grignard intermediate and then reduced, and a nitrile can first be hydrolyzed, for example, to the carboxylic acid and then reduced to a hydroxymethyl group.
- This conversion of the substituent Y into a hydroxymethyl group enables the 3,3-diphenylpropylamine derivatives described in WO 99/58478 to be prepared in a simple manner.
- Corresponding methods for converting the substituent Y into a hydroxymethyl group are also described there.
- Y represents an ester group which is in position to the substituent X.
- substituents R 1 and R 2 which may be the same or different, represent a Ci-Cio-alkyl group or a C 3 -C 8 cycloalkyl group, where R 1 and R 2 can be linked together to form a cyclic structure.
- This cyclic structure can contain heteroatoms, such as nitrogen, oxygen, etc., so that R 1 and R 2 together with the nitrogen atom to which they are attached can form, for example, a morpholine residue.
- R 1 and R 2 are isopropyl and Ar is phenyl.
- alkyl denotes a linear or branched hydrocarbon chain with preferably 1 to 10 carbon atoms.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and corresponding isomers of pentyl and hexyl, with the ' isopropyl group being preferred.
- cycloalkyl or “cycloalkyl group” describes cyclic hydrocarbon radicals with preferably 3 to 10 carbon atoms, which can optionally be substituted.
- substituents are understood to mean groups known per se to those skilled in the art, such as alkyl, alkoxy, halogen (fluorine, chlorine, bromine, iodine), nitro and the like.
- substituted or unsubstituted benzyl describes a benzyl group, the phenyl ring of which is optionally mono- or polysubstituted.
- Substituted benzyl groups are preferably 4-methylbenzyl, 4-methoxybenzyl, 4-nitrobenzyl, 2-nitrobenzyl, 4-chlorobenzyl and 2- Chlorobenzyl.
- R represents an alkyl group.
- Preferred alkylcarbonyl groups are acetyl, propionyl, isobutyryl, valeroyl and pivaloyl.
- aryl denotes an aromatic hydrocarbon radical, such as phenyl (CgHs-), naphthyl (C ⁇ 0 H 7 -) and anthryl (C 14 H 9 -).
- phenyl and naphthyl groups especially phenyl groups, these groups being simple or can be substituted several times, optionally two or more aryl radicals can be bridged or condensed to form polycyclic structures.
- substituted acyl groups include 4-methoxybenzoyl, 2-methoxybenzoyl, 4-chlorobenzoyl, 2 -Chlorbenzoyl, 4-nitrobenzoyl and 2-nitrobenzoyl.
- Preferred alkoxycarbonyl groups are methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, t-butyloxycarbonyl and benzyloxycarbonyl and alicyclic alkoxycarbonyl groups.
- amino acid or “amino acid residue” describes residues which are derived from naturally occurring or synthetic amino acids (all optical antipodes being included). Preferred amino acid residues are valyl, leucyl, isoleucyl, phenylalanyl, prolyl, seryl, threonyl, methionyl and hydroxyprolyl.
- the amino acid residue can be substituted by a suitable group.
- substituted amino acid residues are, for example, N-benzoylprolyl, N-tert. -Butoxycarbonylprolyl, N-alkyl, N-acyl or N-diphenylphosphanylprolyl.
- the process according to the invention is based on the hydrocarbonylation / hydroformylation of compounds of the general formula
- X, Y and Ar are as defined above, and a subsequent reductive amination of the corresponding oxo compounds.
- the oxo compounds can be isolated or can also be converted directly to the corresponding diarylamines in a one-pot reaction.
- the method according to the invention summarizes the hydrocarbonylation / hydroformylation by synthesis gas, ie a mixture of carbon monoxide (CO) and hydrogen (H 2 ) in the presence of suitable catalysts and the reductive amination with amines in the presence of suitable catalysts of the general formula HNR 1 R 2 , wherein R 1 and R 2 are as defined above.
- the hydroaminomethylation leads to secondary amines in the presence of primary amines, while the reaction with secondary amine components leads to tertiary amines.
- Diisopropylamine is preferably used as the amine component in the process according to the invention.
- the substituent X stands for hydroxy (OH). This allows, starting from 1,1-diarylethene compounds, the hydroformylation to chromene / lactol systems according to the general formula shown below
- 1, 1-diarylethene compounds according to general formula II can be prepared by methods known per se, such as those described in Yamaguchi M; Arisawa M; Omata K; Kabuto K; Hirama M; Uchimaru T; Journal of Organic Che istry 1998, 63 (21), 7298-7305 and Yamaguchi M; Hayas-hi A; Hirama M; Journal of the American Chemical Society 1995, 117 (3), 1151-2.
- the process according to the invention is generally carried out at a temperature in the range from 50 to 200 ° C. and preferably 100 to 140 ° C.
- the reaction pressure is 40 to 200 bar and preferably 80 to 120 bar.
- a mixture of carbon monoxide and hydrogen (synthesis gas) is used in the hydroformylation, the ratio of carbon monoxide (CO) to hydrogen (H 2 ) generally being 10/90 to 90/10 and preferably 70/30 to 90/10.
- the breadth of the pressure ratios results from the necessity that the ratios of ligand and catalyst are optimized separately for each substrate and are therefore dependent on the substrate, catalyst precursor and ligand etc.
- the reaction time is generally in the range from 2 hours to 4 days and preferably in the range from 1 to 3 days.
- reaction times are influenced by the respective equipment. With optimal gas introduction, shorter reaction times can also be achieved.
- the catalyst used in the process according to the invention comprises one or more transition metals selected from the group consisting of ruthenium, rhodium, platinum, cobalt, iridium, palladium, nickel, with rhodium being preferred.
- the catalyst is formed in situ from a catalyst precursor and a ligand.
- Suitable catalyst precursors are preferably [Rh (cod) Cl] 2 and / or Rh (acac) (CO) 2 or comparable rhodium complexes.
- Suitable ligands are listed below together with the relevant abbreviation:
- BINAPHOS R-2- (diphenylphosphino) -1,1 '-binaphthalene-2' -yl- (S) -1, 1 '-binaphthalene-2,2'-diylphosphite,
- DIOP (2,2-dimethyl-4,5-diphenylphosphinomethyl) -1,3-dioxolane
- DIOP-DBP (2,2-dimethyl-4,5-bis (5H-dibenzophosphol-5-ylmethyl) -1, 3-dioxolane
- DPPB 1,4-bis (diphenylphosphino) butane
- CHIRAPHOS 2,3-bis (diphenylphosphino) butane
- CBDPP 1,2-bis (diphenylphosphinomethyl) cyclobutane
- CBDBP 1,2-bis (5H-dibenzophosphol-5-ylmethyl) cyclobutane
- CHDPP 1,2-bis (diphenylphosphinomethyl) cyclohexane
- CHDBP 1,2-bis ( 5H-dibenzophosphol-5-ylmethyl) cyclohexane
- CHDPPO 1, 2-bis (diphenylphosphinoxy) cyclohexane
- BzMePhP * benzyl-methyl-phenylphosphine
- CAMP cyclohexyl-o-anisyl-methylphosphine
- NMDPP neomenthyldiphenylphosphine
- PAMP phenyl-o-anisyl-methylphosphine
- BPPM (2S, 4S) -N-tert-butoxycarbonyl-4-diphenylphosphino-2-diphenylphosphinomethylpyrrolidine
- o-DPPB ortho-diphenylphosphanylbenzoyl
- PBu3 tributylphosphine
- BINAP 2, 2'-bis (diphenylphosphino) -1, 1 '-binaphthyl.
- Phosphites and binaphthyl compounds can also be used as ligands.
- Preferred ligands are tributylphosphine, (+) - or (-) - (2, 2-dimethyl-4, 5-diphenylphosphinomethyl) -1, 3-dioxolane, (R) - or (S) -BINAP and / or (R, S) -BINAPHOS.
- the ratio of ligand to rhodium is generally 1: 1 to 25: 1 and preferably 4: 1 to 10: 1.
- the process according to the invention is based on the hydroaminomethylation of 1,1-diarylethenes using a suitable catalyst system. It has the advantage that it can be carried out as a one-pot reaction, which enables the desired diarylpropylamine derivatives to be isolated directly.
- the chirality center in position 3 of the 3, 3-diarylpropylamine derivatives can be generated in a stereoselective manner by appropriate selection of chiral ligands for the metallic catalyst center (ligand control). If one of the aryl groups in the 1,1-diarylethene used as the starting material is substituted in the ortho position by a heteroatom which is modified with chiral groups, the chiral synthesis is controlled by the substrate (substrate control). This can be compared to homoallylic alcohols, the conformation of which is pre-fixed by the planar aromatics. Finally, the combination of these methods (ligand and substrate control) allows double stereo side differentiation.
- the NMR spectra were recorded with a DRX 400 from Bruker. TMS was used as an internal standard.
- Mass spectra were measured on a Finnigan CA 5.
- the elemental composition was determined using a Leco CHNS-932.
- the lactol obtained can be converted by reductive amination according to ⁇ known processes for the desired diaryl prophylaxis.
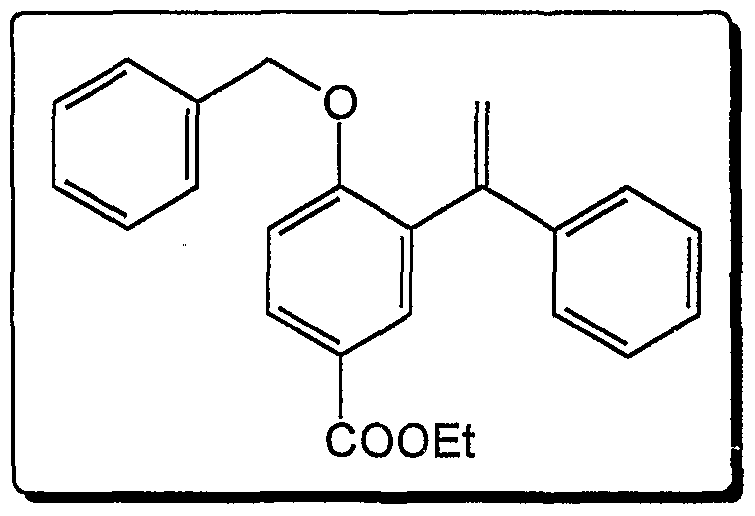
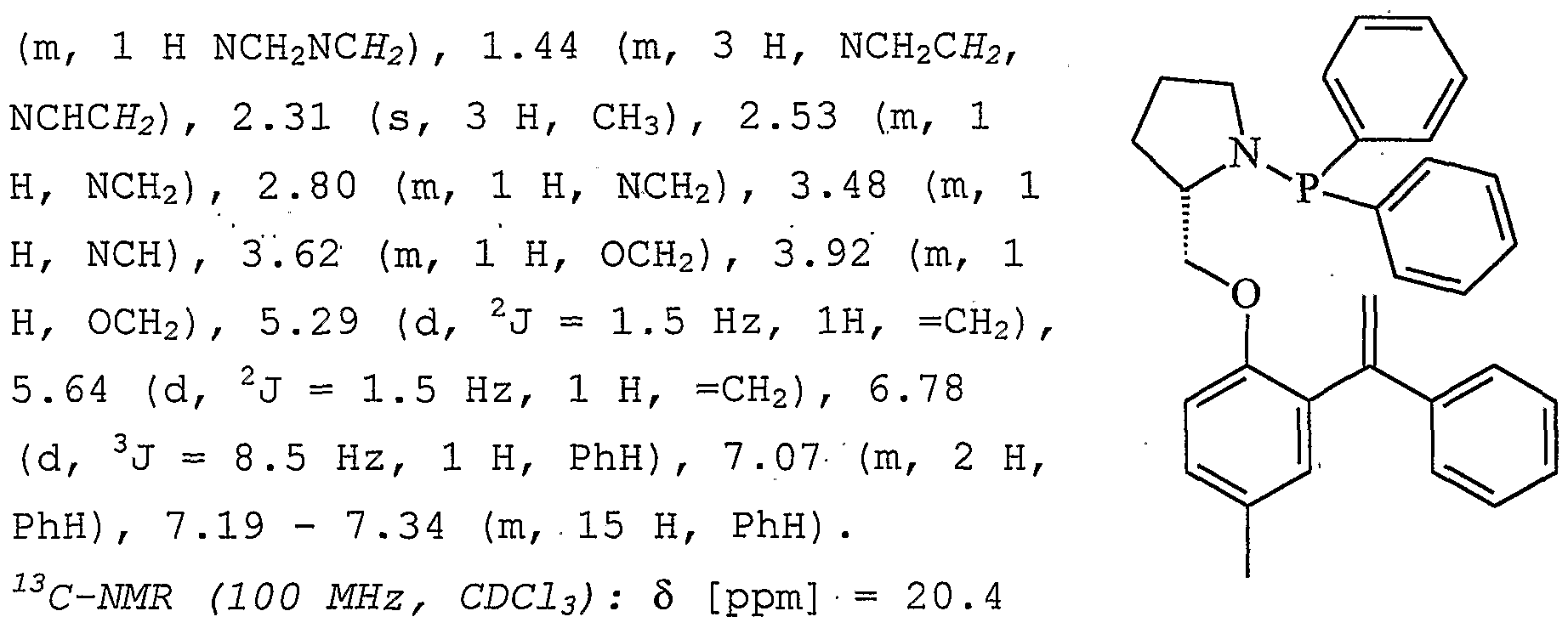
- Example 14 Hydroaminomethylation of l-benzyloxy-4-methyl-2- (1-phenylvinyl) benzene with chiral ligands
- NCH 2 55.7 and 55.9
- NCH 68.0 and 68.3
- OH 2 68.0 and 68.3
- 79.0 and 79.4 CMe 3
- 111.9 CH aroma
- 114.9 and 115.2 CH 2
- 125.7 and 126.0 CH aroma
- 126.3 (2 x CH aEO m
- 128.0 " (2 x CH aEO m)
- the aqueous phase is extracted twice with 10 ml dichloromethane and the combined organic Phases are dried over MgS0 4 .
- 1.31 g (2.7 mmol, 61%) of diphenylphosphanyl- [4-methyl-2- (1-phenyl-vinyl) - phenoxymethyl] pyrrolidine are obtained. Yield: 1.31 g (2.7 mmol, 61%) diphenylphosphanyl- [4-methyl-2- (1-phenyl-vinyl) -phenoxymethyl] pyrrolidine.
- the spectroscopic data correspond to that of the P.G. Andresson, H.E. Schenk, K. ⁇ sterlund J. Org. Chem. 1998, 63, 8067.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pyrane Compounds (AREA)
- Pyrrole Compounds (AREA)
Abstract
Description










































Claims








Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE50112449T DE50112449D1 (de) | 2000-07-07 | 2001-07-06 | Verfahren zur herstellung von 3,3-diarylpropylaminen |
| US10/332,290 US6809225B2 (en) | 2000-07-07 | 2001-07-06 | Method for producing 3,3-diarylpropylamines |
| HK03103416.7A HK1051178B (en) | 2000-07-07 | 2001-07-06 | Method for producing 3,3-diarylpropylamines |
| JP2002509068A JP2004502748A (ja) | 2000-07-07 | 2001-07-06 | 3,3−ジアリールプロピルアミンの製法 |
| AU2001283932A AU2001283932A1 (en) | 2000-07-07 | 2001-07-06 | Method for producing 3,3-diarylpropylamines |
| EP01962840A EP1299342B1 (de) | 2000-07-07 | 2001-07-06 | Verfahren zur herstellung von 3,3-diarylpropylaminen |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10033016.9 | 2000-07-07 | ||
| DE10033016A DE10033016A1 (de) | 2000-07-07 | 2000-07-07 | Verfahren zur Herstellung von 3,3-Diarylpropylaminen |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002004399A1 true WO2002004399A1 (de) | 2002-01-17 |
Family
ID=7648103
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/007803 Ceased WO2002004399A1 (de) | 2000-07-07 | 2001-07-06 | Verfahren zur herstellung von 3,3-diarylpropylaminen |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US6809225B2 (de) |
| EP (1) | EP1299342B1 (de) |
| JP (1) | JP2004502748A (de) |
| CN (1) | CN1269793C (de) |
| AT (1) | ATE361272T1 (de) |
| AU (1) | AU2001283932A1 (de) |
| DE (2) | DE10033016A1 (de) |
| ES (1) | ES2283427T3 (de) |
| WO (1) | WO2002004399A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2364966A1 (de) | 2010-03-09 | 2011-09-14 | LEK Pharmaceuticals d.d. | Verfahren zur Herstellung von 3-(2-hydroxy-5-substituierten phenyl)-3-phenylpropylaminen, Zwischenprodukte zur Herstellung von Hydroxytolterodin |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0957073A1 (de) * | 1998-05-12 | 1999-11-17 | Schwarz Pharma Ag | 3,3-Diphenylpropylaminderivate |
| DE10315917A1 (de) * | 2003-04-08 | 2004-11-18 | Schwarz Pharma Ag | Hochreine Basen von 3,3-Diphenylpropylaminmonoestern |
| JP4513535B2 (ja) * | 2004-12-03 | 2010-07-28 | 住友化学株式会社 | トルテロジンの製造方法 |
| ATE480531T1 (de) * | 2006-05-24 | 2010-09-15 | Pfizer Ltd | Verfahren zur herstellung von benzopyran-2- olderivaten |
| US7767862B2 (en) * | 2008-08-11 | 2010-08-03 | Shell Oil Company | Ligand, catalyst and process for hydroformylation |
| CN104744514B (zh) * | 2013-12-27 | 2018-03-23 | 中国科学院上海药物研究所 | 一种手性磷烯配体、合成方法及其在不对称反应中的应用 |
| CN118496302B (zh) * | 2024-05-17 | 2025-01-24 | 广东医科大学 | 一种多肽色氨酸残基吲哚c2位膦酰化修饰的化合物及其制备方法与应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994011337A1 (en) * | 1992-11-06 | 1994-05-26 | Pharmacia Ab | Novel 3,3-diphenylpropylamines, their use and preparation |
| WO1997044329A1 (en) * | 1996-05-20 | 1997-11-27 | Teijin Limited | Diarylalkyl cyclic diamine derivatives as chemokine receptor antagonists |
| WO1998029402A1 (en) * | 1996-12-31 | 1998-07-09 | Pharmacia & Upjohn Company | Process to prepare tolterodine |
| WO1999058478A1 (en) * | 1998-05-12 | 1999-11-18 | Schwarz Pharma Ag | Novel derivatives of 3,3-diphenylpropylamines |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE8800207D0 (sv) * | 1988-01-22 | 1988-01-22 | Kabivitrum Ab | Nya aminer, deras anvendning och framstellning |
-
2000
- 2000-07-07 DE DE10033016A patent/DE10033016A1/de not_active Ceased
-
2001
- 2001-07-06 ES ES01962840T patent/ES2283427T3/es not_active Expired - Lifetime
- 2001-07-06 CN CNB018125425A patent/CN1269793C/zh not_active Expired - Fee Related
- 2001-07-06 AT AT01962840T patent/ATE361272T1/de not_active IP Right Cessation
- 2001-07-06 EP EP01962840A patent/EP1299342B1/de not_active Expired - Lifetime
- 2001-07-06 US US10/332,290 patent/US6809225B2/en not_active Expired - Fee Related
- 2001-07-06 WO PCT/EP2001/007803 patent/WO2002004399A1/de not_active Ceased
- 2001-07-06 AU AU2001283932A patent/AU2001283932A1/en not_active Abandoned
- 2001-07-06 JP JP2002509068A patent/JP2004502748A/ja active Pending
- 2001-07-06 DE DE50112449T patent/DE50112449D1/de not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994011337A1 (en) * | 1992-11-06 | 1994-05-26 | Pharmacia Ab | Novel 3,3-diphenylpropylamines, their use and preparation |
| WO1997044329A1 (en) * | 1996-05-20 | 1997-11-27 | Teijin Limited | Diarylalkyl cyclic diamine derivatives as chemokine receptor antagonists |
| WO1998029402A1 (en) * | 1996-12-31 | 1998-07-09 | Pharmacia & Upjohn Company | Process to prepare tolterodine |
| WO1999058478A1 (en) * | 1998-05-12 | 1999-11-18 | Schwarz Pharma Ag | Novel derivatives of 3,3-diphenylpropylamines |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2364966A1 (de) | 2010-03-09 | 2011-09-14 | LEK Pharmaceuticals d.d. | Verfahren zur Herstellung von 3-(2-hydroxy-5-substituierten phenyl)-3-phenylpropylaminen, Zwischenprodukte zur Herstellung von Hydroxytolterodin |
| WO2011110556A1 (en) | 2010-03-09 | 2011-09-15 | Lek Pharmaceuticals D.D. | Short synthesis of tolterodine, intermediates and metabolites |
Also Published As
| Publication number | Publication date |
|---|---|
| DE50112449D1 (de) | 2007-06-14 |
| AU2001283932A1 (en) | 2002-01-21 |
| US20040034080A1 (en) | 2004-02-19 |
| ATE361272T1 (de) | 2007-05-15 |
| CN1269793C (zh) | 2006-08-16 |
| EP1299342A1 (de) | 2003-04-09 |
| HK1051178A1 (en) | 2003-07-25 |
| EP1299342B1 (de) | 2007-05-02 |
| CN1441771A (zh) | 2003-09-10 |
| ES2283427T3 (es) | 2007-11-01 |
| JP2004502748A (ja) | 2004-01-29 |
| DE10033016A1 (de) | 2002-01-24 |
| US6809225B2 (en) | 2004-10-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60102356T2 (de) | Asymmetrische synthese von pregabalin | |
| EP0794957B1 (de) | Verfahren zur herstellung optisch aktiver metallocenylphosphine | |
| DE69512415T2 (de) | Heteroaromatische diphospine als chirale liganden | |
| EP0643065A1 (de) | Neue Bisphosphine für asymmetrische Hydrierkatalysatoren | |
| WO2008132057A1 (de) | Verfahren zur herstellung optisch aktiver carbonylverbindungen | |
| DE69114986T2 (de) | Eine Phosphino-Binapthyl-Verbindung und Übergangsmetall-Komplexe davon. | |
| DE69014724T2 (de) | Verbindung des 2,2-bis(Diphenylphosphino)-1,1-binaphtyl und katalytische Komplexe davon. | |
| EP0579797B1 (de) | Diphosphinliganden | |
| DE602004008478T2 (de) | Biphosphinrutheniumkomplexe mit chiralen diaminliganden als katalysatoren | |
| WO2021001240A1 (de) | Hydrierung von estern zu alkoholen in gegenwart eines ru-pnn-komplexes | |
| EP1299342A1 (de) | Verfahren zur herstellung von 3,3-diarylpropylaminen | |
| DE69307706T2 (de) | Verfahren zur enantioselektiven hydrierung der carbonylgruppe unter verwendung von ruthenium komplexen mit biphosphineliganden | |
| DE602005003572T2 (de) | Ferrocenylliganden für homogene, enantioselektive hydrierungskatalysatoren | |
| DE60109484T2 (de) | P-chirale bisphospholane ligande, deren übergangsmetall-komplexe | |
| DE3148098C2 (de) | ||
| DE69300826T2 (de) | Hydrierung. | |
| WO2020094528A1 (en) | Enantioselective process | |
| EP1692149B1 (de) | Verfahren zur herstellung von orthometallierten und orthosubstituierten aromatischen verbindungen | |
| EP1692151A1 (de) | Ferrocenyl-1, 2-diphosphine, deren herstellung und deren verwendung | |
| EP0512416B1 (de) | Neue homochirale Diphosphine | |
| EP1636243B1 (de) | Chirale liganden zur anwendung in asymmetrischen synthesen | |
| KR20190134637A (ko) | 키랄 금속 착화합물 | |
| DE102004012438A1 (de) | Verfahren zur Herstellung von enantiomerenangereicherten Ferrocenylliganden | |
| WO2006082054A1 (de) | 1,4-bis-diphosphines. 1.4-bis-diphosphites and 1,4-bis- diphosphonites from optically active (z)-olefines as chiral ligands | |
| EP1256570A1 (de) | Mehrzähnige unsymmetrische Liganden auf Binaphthylbasis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001962840 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018125425 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001962840 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10332290 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001962840 Country of ref document: EP |