WO2010050628A2 - Composition de caoutchouc expansé pour semelle de chaussure ainsi que semelle - Google Patents
Composition de caoutchouc expansé pour semelle de chaussure ainsi que semelle Download PDFInfo
- Publication number
- WO2010050628A2 WO2010050628A2 PCT/JP2010/051833 JP2010051833W WO2010050628A2 WO 2010050628 A2 WO2010050628 A2 WO 2010050628A2 JP 2010051833 W JP2010051833 W JP 2010051833W WO 2010050628 A2 WO2010050628 A2 WO 2010050628A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- weight
- parts
- rubber
- foaming agent
- shoe sole
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
-
- A—HUMAN NECESSITIES
- A43—FOOTWEAR
- A43B—CHARACTERISTIC FEATURES OF FOOTWEAR; PARTS OF FOOTWEAR
- A43B13/00—Soles; Sole-and-heel integral units
- A43B13/02—Soles; Sole-and-heel integral units characterised by the material
- A43B13/04—Plastics, rubber or vulcanised fibre
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0061—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof characterized by the use of several polymeric components
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/06—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent
- C08J9/10—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent developing nitrogen, the blowing agent being a compound containing a nitrogen-to-nitrogen bond
- C08J9/102—Azo-compounds
- C08J9/103—Azodicarbonamide
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/32—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof from compositions containing microballoons, e.g. syntactic foams
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L21/00—Compositions of unspecified rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/22—Expandable microspheres, e.g. Expancel®
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2321/00—Characterised by the use of unspecified rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2409/00—Characterised by the use of homopolymers or copolymers of conjugated diene hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
Definitions
- the present invention is a rubber foam composition for a shoe sole, which is lightweight and has an appropriate hardness, and excellent in tensile strength, tear strength, impact absorbability, in-mold fluidity, and dimensional stability after foaming, and further its rubber
- the present invention relates to an outsole using the composition.
- Patent Document 1 In order to achieve low hardness and light weight, a chemical foaming agent (Patent Document 1) and a physical foaming agent (Patent Document 2) are used, and low hardness and light weight are achieved by mixing bubbles in the product.
- Patent Document 2 a chemical foaming agent
- Patent Document 2 a physical foaming agent
- problems such as destruction of the foam due to abnormal foaming, deterioration of the surface of the foamed product, and unstable product dimensions.
- thermoplastic resin having a glass transition temperature (Tg) lower than that of conventional 1,2-polybutadiene and vinyl cis polybutadiene (VCR) are used together and crosslinked with an organic oxide to increase the thickness accuracy.
- Tg glass transition temperature
- VCR vinyl cis polybutadiene
- excellent, moderate hardness, excellent smoothness, and further improved workability Patent Document 3
- 1,2-polybutadiene, polyethylene-based polymer, ethylene-vinyl acetate polymer resin is blended with natural rubber (isoprene) as a third component, which is rich in rubber elasticity, light weight and excellent hardness, Improves impact resistance and tear strength.
- the present invention relates to a foam rubber composition for a shoe sole having a tensile strength, a tear strength, and an impact absorption property with a light weight and an appropriate hardness by using a pyrolytic foaming agent and a thermal expansion foaming agent in combination.
- the outsole used is provided.
- the flowability in the mold because the temperature dependency during pressurization is low, the viscosity is low and there is no air bubble inclusion.
- air voids the product is free from chipping and becomes a molded product.
- a foam rubber composition for a shoe sole which is free from defects and excellent in dimensional stability after foaming.
- a vinyl cis-butadiene rubber (VCR) and a thermoplastic resin (C) are combined with a foaming agent comprising (D1) a thermal expansion foaming agent and (D2) a thermal decomposition foaming agent, and (A) + (B) +
- a shoe characterized in that (D1) is 0.5 to 3.0 parts by weight and (D2) is 0.5 to 7.0 parts by weight with respect to 100 parts by weight of the total rubber component comprising (C).
- the present invention relates to a foam rubber composition for a bottom.
- the present invention also relates to an outsole characterized by using the foam rubber composition for a shoe sole.
- the foam rubber composition for shoe soles and the outsole using the same obtained in the present invention are lightweight and have an appropriate hardness, and have tensile strength, tear strength, and impact absorption.
- the fluidity in the mold is improved (because the temperature dependency at the time of pressurization is low, the viscosity decrease is small and the air void is also improved). Therefore, it is possible to provide a foam rubber composition for a shoe sole that is free from defects in the product, has no defects in the molded product, and is excellent in dimensional stability after foaming.
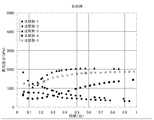
- FIG. 4 shows the relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 1 to 4.
- FIG. FIG. 6 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 5 to 8.
- FIG. FIG. 5 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Comparative Examples 1 to 5.
- FIG. FIG. 4 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Comparative Examples 6 to 10.
- FIG. 4 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 9 to 12.
- FIG. FIG. 6 shows the relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Examples 13 to 15.
- FIG. FIG. 6 is a relationship between the foaming pressure in the reactor and the time at the initial stage of vulcanization (heating and pressurization) in Comparative Examples 11 to 14.
- FIG. It is a relationship between the foaming pressure in a reactor in the initial stage of vulcanization (heating and pressurization) in Comparative Example 15 and time.
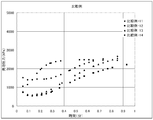
- the in-reactor foaming pressure from the start to the end of vulcanization in Examples 9-12.
- FIG. 6 shows the relationship between the in-reactor foaming pressure and time at the initial stage of vulcanization (heating and pressurization) in Examples 16 to 18 and Comparative Example 16.
- Component (A) As vulcanizable rubber of (A), natural rubber (NR), isoprene rubber (IR), butadiene rubber (BR), styrene-butadiene rubber (SBR), acrylonitrile-butadiene rubber (NBR) ), Butyl rubber (IIR), acrylonitrile-chloroprene rubber, acrylonitrile-isoprene rubber, styrene-chloroprene rubber, styrene-isoprene rubber and other diene rubbers, ethylene-propylene rubber, ethylene-butene rubber, ethylene-vinyl acetate copolymer (EVA) And ethylene- ⁇ -olefin copolymer rubbers such as ethylene-propylene-diene rubber (EPDM). These may be used alone or in combination.
- NR natural rubber
- IR isoprene rubber
- BR butadiene rubber
- SBR styrene-buta
- (A-2) As the styrene-butadiene rubber, it is particularly preferable to use a water-added styrene-butadiene rubber. Further, as the (a-2) styrene-butadiene rubber, it is particularly preferable to use a styrene-butadiene rubber (S-SBR) obtained by solution polymerization. Use of SBR other than S-SBR is not preferable because it causes problems such as deterioration of shrinkage and deterioration of wear resistance (or use of S-SBR exhibits excellent characteristics in terms of shrinkage and wear resistance). Preferred to use).
- S-SBR styrene-butadiene rubber
- the blending amount of (A) is 2 to 30 parts by weight, preferably 5 to 25 parts by weight, particularly preferably 7 to 17 parts by weight of vulcanizable rubber in 100 parts by weight of all rubber components.
- properties necessary for roll processability sheet skin shape appearance
- various physical properties of footwear tensile strength, elongation, tear strength
- the blending amount is less than 2 parts by weight, problems such as roughness of the sheet and workability occur, and conversely if it exceeds 30 parts by weight, problems such as an increase in specific gravity and deterioration of shrinkage occur.
- the blending amount of (a-1) natural rubber (NR) is: It is preferable that 2 to 30 parts by weight, preferably 5 to 20 parts by weight, particularly preferably 7 to 15 parts by weight are blended in 100 parts by weight of the total rubber component.
- properties necessary for roll processability sheet skin shape appearance
- various physical properties of footwear tensile strength, elongation, tear strength
- the amount of the solution-polymerized styrene butadiene rubber (S-SBR) is 2 to 30 parts by weight, preferably 5 to 25 parts by weight, particularly preferably 8 to 20 parts by weight based on 100 parts by weight of the total rubber. What mix
- blends a part is preferable. When blended, properties necessary for roll processability (sheet skin shape appearance, viscosity adjustment) can be imparted.
- the blending ratio (a-1) / (a-2) is preferably 0.07 to 15 More preferably, it is 0.2 to 4, particularly preferably 0.35 to 1.88. Use within the above-mentioned ratio range is preferable because it exhibits excellent properties in terms of shrinkage and wear resistance.
- VCR vinyl cis-butadiene rubber
- SPB 2-polybutadiene resin
- the boiling n-hexane insoluble matter mentioned here means the part recovered as an insoluble matter when the VCR is refluxed in boiling n-hexane, and the boiling n-hexane soluble matter means that the VCR is refluxed in boiling n-hexane. It is the part that dissolves when The boiling n-hexane insoluble matter has a reduced viscosity (135 ° C, concentration 0.20 g / dl orthodichlorobenzene solution) measured with an orthodichlorobenzene solution of 0.5 to 4, preferably 0.8 to 3 is there. When the reduced viscosity of boiling n-hexane insolubles is less than 0.5, the die swell of the formulation is not improved sufficiently.
- the boiling n-hexane insoluble matter is SPB having a melting point of preferably 170 ° C. or higher, particularly preferably 190 to 220 ° C. If the melting point of SPB is less than 170 ° C., problems such as deterioration in workability and mechanical strength (such as tensile stress and tensile strength) occur due to the decrease in SPB reinforcement.
- the weight average molecular weight of the boiling n-hexane soluble part is preferably in the range of 300,000 to 800,000, and if it is less than 300,000, the durability and impact resilience of the vulcanizate are lowered, which is not preferred.
- Exceeding 800,000 is not preferable because the Mooney viscosity of the compound becomes too high and processing becomes difficult.
- VCR production methods are disclosed in, for example, JP-B-49-17666, JP-B-49-17667, JP-B-61-57858, JP-B-62-171, JP-B-63-36324, The methods described in JP-A-2-37927, JP-B-2-38081, JP-B-3-63566 and the like can be used.
- the manufacturing method of the VCR used in the present invention is not limited to these methods.
- the blending amount of the VCR of the component (B) is preferably 40 to 80 parts by weight, and more preferably 50 to 70 parts by weight with respect to 100 parts by weight of all rubber components.
- various physical properties of the footwear tensile strength, elongation, tear strength, abrasion resistance
- the blending amount is less than 40 parts by weight, the problem of deterioration of wear resistance occurs, and conversely if it exceeds 80 parts by weight, problems such as an increase in specific gravity and an increase in hardness occur.
- thermoplastic resin examples include syndiotactic 1,2-polybutadiene resin (SPB), polyethylene and its maleic anhydride graft polymer, polyisobutylene, ethylene vinyl acetate copolymer, Ethylene acrylate, ethylene acrylic acid copolymer, polypropylene and its maleic anhydride graft polymer, chlorinated polypropylene, 4-methylpentene-1 resin, polystyrene, ABS resin, AS resin, acrylic resin, methacrylic resin, vinyl chloride resin, Examples include vinylidene chloride resin, polyamide resin, and polycarbonate. These thermoplastic resins (C) can be used alone or in combination of two or more.
- SPB syndiotactic 1,2-polybutadiene resin
- polyisobutylene polyisobutylene
- ethylene vinyl acetate copolymer Ethylene acrylate
- ethylene acrylic acid copolymer polypropylene and its maleic anhydride graft polymer
- the thermoplastic resin is preferably a polyolefin resin.
- polyolefin resins include polyethylene (PE), particularly low density PE (LDPE), medium density PE (MDPE), high density PE (HDPE), and linear low density PE (LLDPE), ethylene-propylene elastomer copolymer.
- EPM ethylene-propylene-diene rubber
- EVA ethylene-vinyl ester copolymers
- EVA ethylene-vinyl acetate copolymer
- EVA ethylene-acrylate copolymers
- ethylene- ⁇ -olefin thermoplastic copolymers as well as propylene, butene -1, and copolymers such as pentene-1.
- These polyolefin resins can be used alone or in combination of two or more.
- thermoplastic resin examples include 1,2-polybutadiene resins having a melting point of 70 ° C. to 110 ° C., and ethylene-vinyl acetate copolymer (EVA).
- EVA ethylene-vinyl acetate copolymer
- the blending amount of the thermoplastic resin (C) is preferably 10 to 35 parts by weight, more preferably 15 to 30 parts by weight, and more preferably 18 to 28 parts by weight based on 100 parts by weight of the total rubber component. It is particularly preferred. When blending within this range, various physical properties (gas barrier properties, dimensional stability) of footwear can be imparted. On the other hand, if the blending amount is less than 10 parts by weight, problems such as an increase in specific gravity and dimensional stability (shrinkage) due to a decrease in gas barrier properties occur, and conversely, if it exceeds 35 parts by weight, a problem of an increase in hardness will occur. Absent.
- the total rubber component means (A) + (B) + (C).
- the thermal expansion type foaming agent is not particularly limited as long as it is a foaming agent that expands by heat, but it is preferable to use thermally expandable microcapsules.
- a heat-expandable microcapsule is an encapsulated liquid or gas that expands when heated in a synthetic resin capsule, and is encapsulated by heat of kneading and melting by a screw or the like during extrusion molding or injection molding.
- the microcapsules that form the outer shell are expanded by the expansion of the liquid or gas. However, depending on the temperature conditions during molding, a microcapsule that melts and completes without bursting is used.
- a copolymer having acrylonitrile as one of monomer components is used, and other monomer components that may be copolymerized with acrylonitrile include, for example, acrylic acid, methacrylic acid, acrylate ester, methacrylic ester, and the like. Examples thereof include, but are not limited to, acid esters, styrene, vinyl acetate, and vinylidene chloride.
- the liquid or gas encapsulated in the microcapsule is one that expands as a gas at a temperature below the softening point of the microcapsule.
- Low boiling point liquids such as methane halides such as heptane, petroleum ether, methyl chloride and methylene chloride, chlorofluorocarbons such as CCl 3 F and CCl 2 F 2 , and tetraalkylsilanes such as tetramethylsilane and trimethylethylsilane
- a compound such as AIBN that is thermally decomposed by heating and becomes gaseous can be mentioned.
- the amount and size of the bubbles are determined by the number (mixing amount) and the particle size of the thermally expandable microcapsules after expansion, and the molding is performed from these. Since the specific gravity of the body is determined, by predicting the number of unexpanded thermally expandable microcapsules (mixed amount) to be mixed with the thermoplastic resin raw material and the particle size of the thermally expandable microcapsules after expansion by the heating amount during molding The foaming state can be controlled, and only the gas does not escape to the outside as compared with the case where the conventional gas is directly expanded, and the target specific gravity can be easily achieved.
- a foam can be formed in a state where the thermally expandable microcapsule is expanded and contained in the thermoplastic resin by extrusion molding or injection molding, and each bubble is stable in a state of being encapsulated in the microcapsule.
- extrusion molding even when the pressure is released by being extruded from a die, the gas does not escape directly to the outside of the molded product, and a foam having a good surface appearance can be obtained.
- thermoly expandable microcapsules for example, EXPANCEL 920DU40, EXPANCEL 920DU80, EXPANCEL009DU80, EXPANCEL920DU40 master batch products 920MB40 and 920MB50, Matsumoto Yushi Pharmaceutical Matsumoto Microsphere F-D, etc.
- the particle size of the thermally expandable microcapsule is preferably 8 to 30 ⁇ m, more preferably 8 to 17 ⁇ m, and even more preferably 10 to 16 ⁇ m. In the said range, the effect of low specific gravity and the smoothness of the surface skin of a molded article can be expected.
- the compounding amount of the thermal expansion foaming agent is 0.5 to 3.0 parts by weight, preferably 0.75 to 3.0 parts by weight, more preferably 0.80 to 2. It is 5 parts by weight, more preferably 0.85 to 2.5 parts by weight, still more preferably 0.90 to 2.0 parts by weight, and particularly preferably 1.2 to 2.0 parts by weight.
- air removal, appearance of molded products (surface state smoothness and dimensional stability) with increasing foaming pressure in the initial stage of vulcanization, and various physical properties (tensile strength, elongation at break and tear strength) associated with lower specific gravity. Can be suppressed.
- the blending amount is less than 0.5 parts by weight, there is a problem that the effect of lowering the specific gravity is reduced.
- the blending amount is more than 3.0 parts by weight, the surface roughness of the molded product (smoothness) is reduced. Problems arise.
- D2 Pyrolytic foaming agent (chemical foaming agent)
- thermally decomposable foaming agent examples include organic foaming agents such as azocicarbonamide complex (ADCA), azobisisobutyronitrile (AIBN), dinitrosopentamethylenetetramine (DPT), N, N′-dimethyl.
- ADCA azocicarbonamide complex
- AIBN azobisisobutyronitrile
- DPT dinitrosopentamethylenetetramine
- N, N′-dimethyl N, N′-dimethyl.
- BSH benzenesulfonyl hydrazide
- TSH p-toluenesulfonyl hydrazide
- OBSH 4,4'-oxybis (benzenesulfonyl hydrazide)
- 3,3'-disulfone hydrazide diphenyl Sulfone toluene disulfonyl hydrazine, p-toluene disulfonyl hydrazide, p-toluene sulfonyl semi strength rubazide, diethyl azodicarboxylate and the like can be used.
- an inorganic foaming agent can also be used as the thermally decomposable foaming agent.
- sodium bicarbonate or ammonium bicarbonate can be used.
- thermal decomposable foaming agents may be used alone or in combination.
- An organic foaming agent having a decomposition temperature of about 195 to 210 ° C. and a generated gas amount of 190 to 240 (ml / g) is preferable for use as a thermally decomposable foaming agent.
- azodicarbonamide complex ADCA
- OBSH 4,4′-oxybis (benzenesulfonylhydrazide)
- the foaming effect with the combined thermal expansion foaming agent (D1) can be maximized, and the air accompanying the increase in foaming pressure at the initial stage of vulcanization can be completely removed.
- the compounding amount of the thermally decomposable foaming agent is 0.5 to 7.0 parts by weight, preferably 0.75 to 7.0 parts by weight, more preferably 0.8 to 7. 0 parts by weight, more preferably 0.85 to 7.0 parts by weight, still more preferably 0.85 to 6.0 parts by weight, and particularly preferably 1.0 to 4.5 parts by weight.
- the blending amount is less than 0.5 parts by weight, there is a problem that the effect of reducing the specific gravity and the hardness is reduced. Conversely, if the blending amount is more than 7.0 parts by weight, the dimensional stability is reduced due to high foaming. Problem arises.
- Examples of the rubber reinforcing agent (E) blended in the rubber composition according to the present invention include various types of carbon black, white carbon, silica, activated calcium carbonate, and ultrafine magnesium silicate. . Of these, silica and / or carbon black are preferred. Above all, silica having an average primary particle size of 5 to 100 nm, such as silicic anhydride by a dry method, hydrous silicic acid by a wet method, and synthetic silicate, and a particle size of 90 nm or less and a dibutyl phthalate (DBP) oil supply amount of 70 ml / 100 g or more.
- Carbon black is preferred. Examples of carbon black include furnace black, channel black, thermal black, and the like. 110, 212, N242, S315, N330, N550, N660, N765 and the like.
- Silica can be either wet silica or dry silica, and can be used in combination. Further, silica can be used alone. Moreover, it is preferable that the silica to be used has a silica purity of 97% or more. Further, pulverized natural quartz having a silica purity of 97% or more can be used. Furthermore, particles obtained by hydrolyzing an organosilicon compound such as silane are also preferably used. Silica having a purity of less than 97% does not have a sufficient rubber reinforcing effect, so that a rubber composition blended with such silica does not have sufficient wear resistance.
- the average particle diameter of the silica particles is preferably in the range of 0.1 to 50 ⁇ m, more preferably 1 to 50 ⁇ m, and particularly preferably 5 to 50 ⁇ m.
- a silica particle size can be obtained, for example, by a method of pulverizing a completely melted quartz glass so as to have an appropriate particle size.
- the average particle size is smaller than 0.1 ⁇ m, it is difficult to mix in the rubber, causing problems such as loss and poor dispersion, and conversely if larger than 50 ⁇ m, the reinforcing effect is low, which is not preferable.
- the amount of the rubber reinforcing agent used is 10 to 30 parts by weight, preferably 12 to 25 parts by weight, more preferably 15 to 20 parts by weight based on 100 parts by weight of the total rubber components. Within the above-mentioned range, a sufficient reinforcing effect can be obtained, and an effect of improving wear resistance can be obtained. On the other hand, if the amount is less than 10 parts by weight, the reinforcing effect is low, causing problems of deterioration in various physical properties (mechanical strength and wear resistance). Conversely, if the amount is more than 30 parts by weight, the workability is lowered due to the increase in viscosity. This is not preferable because it causes problems.
- (F) Vulcanization accelerator examples include aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, xanthates and the like.
- TMTD tetramethylthiuram disulfide
- OBS N-oxydiethylene-2-benzothiazolylsulfenamide
- CBS N-cyclohexyl-2-benzothiazylsulfenamide
- MBTS dibenzo Thiazyl sulfide
- ZnBDC zinc di-n-butyldithiocarbide
- ZnMDC zinc dimethyldithiocarbide
- MBTS dibenzothiazyl sulfide
- DAG diortolyl guanidine
- the amount of the vulcanization accelerator is 0.5 to 1.5 parts by weight, preferably 0.7 to 1.3 parts by weight, more preferably 0.8 to 1.2 parts by weight based on 100 parts by weight of the total rubber component. Part. Within the above range, a sufficient vulcanization effect can be obtained. On the other hand, if the amount is less than 0.5 parts by weight, problems such as a delay in vulcanization time and various physical properties due to a decrease in crosslink density are caused. This is not preferable.
- (G) Vulcanizing agent As the vulcanizing agent used in the present invention, sulfur, a compound that generates sulfur by heating, an organic peroxide, a metal oxide such as magnesium oxide, a polyfunctional monomer, a silanol compound, and the like Is mentioned.
- the compound that generates sulfur by heating include tetramethyl thiuram disulfide and tetraethyl thiuram disulfide. Among these, sulfur is particularly preferable.
- the amount of the vulcanizing agent is 0.3 to 2.2 parts by weight, preferably 0.5 to 1.7 parts by weight, more preferably 1.0 to 1.5 parts by weight, based on 100 parts by weight of all rubber components. It is. Within the above range, a sufficient vulcanization effect can be obtained. On the other hand, if the amount is less than 0.3 parts by weight, problems such as a delay in vulcanization time and a decrease in physical properties due to a decrease in crosslink density are caused. This is not preferable.
- Anti-aging agent examples include amine / ketone-based, imidazole-based, amine-based, phenol-based, sulfur-based and phosphorus-based anti-aging agents.
- H2 Vulcanization aids As vulcanization aids, known vulcanization aids such as aldehydes, ammonia, amines, guanidines, thioureas, thiazoles, thiurams, dithiocarbamates, and xanthates Etc. are used.
- H3 Scorch inhibitor (retarder)
- organic acids nitroso compounds, N-cyclohexylthiophthalimide, sulfonamide derivatives and the like are used.
- H4 Process oil may be any of aromatic, naphthenic, and paraffinic.
- (Processing method) (A) Vulcanizable rubber other than (A) (B) Boiling n-hexane insoluble content: 1 to 25% by weight, boiling n-hexane soluble content: 99 to 75% by weight vinyl cisbutadiene
- the rubber (VCR) and (C) thermoplastic resin are used in combination with a foaming agent comprising (D1) a thermal expansion type foaming agent and (D2) a thermal decomposition type foaming agent.
- the rubber reinforcing agent is an open type such as a roll. It can be obtained by kneading using a kneader such as a closed kneader such as a kneader or Banbury mixer, and can be vulcanized after molding and applied to various rubber products.
- Another main point of the present invention is the adjustment of the pressure in the reactor in the vulcanization process.
- the foaming pressure in the reactor at the initial stage of vulcanization (after heating and pressurization 30 seconds) to 2000 kPa or more, preferably 2200 kPa, air voids are not generated, and there is no chipping in the product. It is possible to provide a method for producing a rubber foam composition for a shoe sole that is excellent in dimensional stability after foaming.
- the foaming pressure in the reactor in the initial stage of vulcanization (after heating and pressurization 30 seconds) is less than 2000 kP, sufficient deaeration cannot be performed in the initial stage of vulcanization, causing air voids. Therefore, the foaming pressure in the reactor in the initial stage of vulcanization (after 30 seconds of heating and pressurization) is a major factor for improving the product.
- the foaming pressure in the reactor is larger than 2000 kPa, and the roles of the two types of foaming agents at the initial stage of vulcanization and the final stage of vulcanization are greatly affected.
- the foaming effect of (D1) thermal expansion type foaming agent affects the rapid increase of the foaming pressure in the reactor.
- the foaming effect of (D2) pyrolytic foaming agent is affected.
- the foaming effect by the pyrolytic foaming agent can suppress the increase in the hardness of the resin and realize weight reduction. Therefore, the combined use of (D1) and (D2) is important.
- FIGS. 1 and 2 show the relationship between the vulcanization time (especially at the initial stage of vulcanization) and the foaming pressure in the reactor of Examples 1 to 8.
- the pressure inside the reactor was higher than 2000 kPa at the time of 0.5 minutes from the start of vulcanization (start of vulcanization: 30 seconds after heating and pressurization).
- start of vulcanization start of vulcanization: 30 seconds after heating and pressurization
- FIGS. 3 to 4 where Comparative Examples 1 to 10 are shown, the pressure inside the reactor is less than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
- Figures 5 to 8 show the relationship between the foaming pressure in the reactor from the start to the end of vulcanization.
- the initial pressure of vulcanization (after 30 seconds of heating and pressurization) was maintained even after the in-reactor foaming pressure exceeded 2000 kPa. It can be seen that, even at the end of vulcanization, the in-reactor foaming pressure still exceeds 1000 kPa and the pressure is maintained. It is desirable to take such a course that there is no sudden pressure decrease, and the foaming effect of the pyrolytic foaming agent (D2) is greatly influenced particularly in the late vulcanization stage.
- D2 pyrolytic foaming agent
- Table 3 shows that no air void occurred in Examples 1 to 8.
- FIGS. 9 to 10 show the relationship between the vulcanization time (particularly at the initial stage of vulcanization) and the foaming pressure in the reactor of Examples 9 to 15 in the present invention.
- the pressure in the reactor was higher than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
- 11 to 12 show the relationship between the foaming pressure in the reactor from the start to the end of vulcanization in Comparative Examples 11 to 15.
- the reaction pressure decreases to 2000 kPa or less from the start to the end of vulcanization, there arises a problem of an increase in specific gravity and hardness accompanying a decrease in the reaction pressure.
- FIG. 17 shows the relationship between the vulcanization time (especially at the initial stage of vulcanization) and the foam pressure in the reactor of Examples 16 to 18 and Comparative Example 16 in the present invention.
- the pressure in the reactor was higher than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: 30 seconds after heating and pressurization).
- the pressure in the reactor was less than 2000 kPa at the time of vulcanization start 0.5 minutes (vulcanization start: after 30 seconds of heating and pressurization).
- the specific gravity value at 2 mm is preferably 0.450 to 0.950 (g / cm 3 ), more preferably 0.500 to 0.800 (g / cm 3 ), and 0.550 to More preferably, it is 0.750 (g / cm 3 ).
- the specific gravity is large, there arises a problem that the hardness cannot be increased and the weight cannot be reduced.
- specific gravity is small, the problem of deterioration of various physical properties (mechanical strength, abrasion resistance, shrinkage
- the foaming pressure in the reactor is a combination of the constituent elements in the composition and the initial stage of vulcanization (after heating and pressurization 30 seconds) is 2000 kPa. It turns out that it is important to adjust so that it may become above, and to adjust so that specific gravity may become small.
- the blending ratio of all rubber components (D1) and (D2) is also important in the present invention.
- the foaming pressure in the reactor at the initial stage of vulcanization (after heating and pressurization 30 seconds) is 2000 kPa or more.
- the amount of the foaming agent is necessary, and an appropriate amount of foaming agent is required to suppress the increase in the hardness of the resin at the end of vulcanization and to realize weight reduction. From such a viewpoint, the blending ratio of (D1) of 0.5 to 3.0 parts by weight and (D2) of 0.5 to 7.0 parts by weight is the optimum condition.
- the physical properties of the obtained rubber composition were measured as follows. (1) Specific gravity (density): Measured by method A according to JIS K6268.
- Foaming pressure Using ALPHA TECHNOLOGIES RPA2000 (rotorless type, rubber processability analyzer), the time to reach 10% and 160% vulcanization degree at 160 ° C. and the foaming pressure were measured according to JIS K6300. The higher the pressure value, the better.
- Hardness Measured at room temperature using a type A durometer according to JIS K6253. Moreover, it measured at room temperature using Asker C type (sponge hardness meter) based on SR1S0101. The smaller the value, the softer and better.
- Tear strength measured in accordance with JIS K6252. The higher the value, the better it will be without tearing when the product is peeled from the mold.
- Rebound resilience measured according to JIS K6255 using a trypso type.
- Shrinkage rate Calculated from the length after 5 minutes at room temperature after molding (length after 30 minutes at room temperature). The smaller the value, the better the dimensional stability.
- Example 1 (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin in Table 1 in a blending ratio of 7/70/23 parts by weight, and rubber reinforcing agents such as silica, zinc white and stearic acid Together with a 1.7 liter closed kneader. Thereafter, the vulcanization accelerator and sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 1.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 2 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 7/65/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
- Example 3 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a ratio of 12/60/28 parts by weight, and a vulcanized test piece was prepared in the same manner as in Example 1 below. The physical properties were evaluated. The results are shown in Table 3.
- Example 4 In Table 1, (a-1) natural rubber / (a-2) water-added styrene-butadiene rubber (manufactured by Asahi Kasei Chemicals Co., Ltd.) / (B) VCR / (C) 1,2 polybutadiene resin is blended in a ratio of 7/10/65. / 18 parts by weight, vulcanized test pieces were prepared in the same manner as in Example 1, and the physical properties were evaluated. The results are shown in Table 3.
- Example 5 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, and 2.5 parts by weight of the thermal expansion foaming agent and 0.7 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 6 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator, sulfur, 1.0 part by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 7 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, the vulcanization accelerator and sulfur, 2.5 parts by weight of the thermal expansion foaming agent, and 6.5 parts by weight of the thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 8 In Table 1, (A) natural rubber / (B) VCR / (C) 1,2 polybutadiene resin was blended in a blending ratio of 7/70/23 parts by weight, and then kneaded in the same manner as in Example 1. Thereafter, a vulcanization accelerator, sulfur, 2.5 parts by weight of a thermal expansion foaming agent, and 1.0 part by weight of a thermal decomposition foaming agent were mixed with an open roll. Next, press vulcanization was performed, and the physical properties were evaluated by the obtained vulcanized test pieces. The results are shown in Table 3.
- Example 10 (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 5 to a blending ratio of 7/18/55/20 parts by weight, rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 11 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 12 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 14 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 15 In Table 5, (a-1) natural rubber / (a-2) styrene-butadiene rubber (SBR) / (B) VCR / (C) olefin resin is blended to 7/18/55/20 parts by weight, and rubber It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 11 The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used. The results are shown in Table 8.
- Example 12 The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 5.50 parts by weight. The results are shown in Table 8.
- Example 13 The same procedure as in Example 9 was conducted except that the thermal expansion foaming agent EXPANCEL DU092-40 was not used and the thermal decomposition foaming agent CAP-500 was used in an amount of 6.50 parts by weight. The results are shown in Table 8.
- Example 14 The same procedure as in Example 9 was conducted, except that 5.00 parts by weight of the thermal expansion foaming agent EXPANCEL DU092-40 was used, and the thermal decomposition foaming agent CAP-500 was not used. The results are shown in Table 8.
- Experimental example 3 (Example 16) (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 17 (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 18 (A-1) Natural rubber / (a-2) Styrene-butadiene rubber (SBR) / (B) VCR / (C) Olefin resin in Table 9 to a blending ratio of 7/10/65/18 parts by weight It knead
- additives such as silica which is a reinforcing agent, zinc white, and stearic acid.
- Example 16 The same procedure as in Example 16 was performed except that the thermal expansion foaming agent was not used. The results are shown in Table 10.
- the foam rubber composition obtained in the present invention it is useful as a sole material in the sole field, in particular, footwear in general such as sports shoes, running shoes, and casual shoes.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Manufacture Of Porous Articles, And Recovery And Treatment Of Waste Products (AREA)
- Footwear And Its Accessory, Manufacturing Method And Apparatuses (AREA)
Abstract
L’invention concerne une composition de caoutchouc expansé pour semelle de chaussure, qui se caractérise par un poids réduit, une rigidité adéquate et qui possède des propriétés de résistance à la traction, de résistance de rupture à la traction et d’absorption de choc. L’invention concerne aussi une semelle utilisant cette composition de caoutchouc expansé pour semelle de chaussure. La composition de caoutchouc expansé pour semelle de chaussure se caractérise par le fait qu’elle associe (A) un caoutchouc vulcanisable, (B) un caoutchouc de vinyl-cis-polybutadiène (VCR) différent de (A) et tel qu’il comprend 1 à 25% en poids de n-hexane bouillant insoluble et 99 à 75% en poids de n-hexane soluble, (C) une résine thermoplastique, (D1) un agent moussant à expansion thermique et (D2) un agent moussant à décomposition thermique; et par le fait que, pour 100 parties en poids de la totalité du composant de caoutchouc consistant en (A) + (B) + (C), (D1) représente de 0,5 à 3,0 parties en poids et (D2) représente de 0,5 à 7,0 parties en poids.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010535862A JP5565313B2 (ja) | 2009-06-22 | 2010-02-08 | 靴底用発泡体ゴム組成物及びアウトソール |
Applications Claiming Priority (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009147947 | 2009-06-22 | ||
| JP2009-147947 | 2009-06-22 | ||
| JP2009176118 | 2009-07-29 | ||
| JP2009-176118 | 2009-07-29 | ||
| JP2009-229768 | 2009-10-01 | ||
| JP2009229768 | 2009-10-01 | ||
| JP2009257678 | 2009-11-11 | ||
| JP2009-257679 | 2009-11-11 | ||
| JP2009-257678 | 2009-11-11 | ||
| JP2009258347 | 2009-11-11 | ||
| JP2009-258347 | 2009-11-11 | ||
| JP2009257679 | 2009-11-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010050628A2 true WO2010050628A2 (fr) | 2010-05-06 |
| WO2010050628A3 WO2010050628A3 (fr) | 2010-06-24 |
Family
ID=42129403
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/051833 Ceased WO2010050628A2 (fr) | 2009-06-22 | 2010-02-08 | Composition de caoutchouc expansé pour semelle de chaussure ainsi que semelle |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP5565313B2 (fr) |
| WO (1) | WO2010050628A2 (fr) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013062685A3 (fr) * | 2011-10-25 | 2013-10-17 | Exxonmobil Chemical Patents Inc. | Composition, mousse et article fabriqué avec ladite composition |
| CN104356437A (zh) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | 解放鞋用压延型叶黄色橡胶围条胶及其制备方法及应用 |
| CN104356438A (zh) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | 耐曲折解放鞋用压延型草绿色橡胶围条胶及制备方法及应用 |
| CN104371151A (zh) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | 耐曲折解放鞋用压延型黑色橡胶围条胶及制备方法及应用 |
| CN104371150A (zh) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | 耐曲折解放鞋用压延型叶黄色橡胶围条胶及制备方法及应用 |
| JP2015193783A (ja) * | 2014-03-20 | 2015-11-05 | 宇部興産株式会社 | 発泡体用ゴム組成物とそれを用いた靴底用ゴム発泡体と靴底 |
| CN105131366A (zh) * | 2015-10-09 | 2015-12-09 | 际华三五三七制鞋有限责任公司 | 无味环保型压延型高伸长率鞋用橡胶本色围条胶及其制备方法及应用 |
| WO2016014230A1 (fr) | 2014-07-25 | 2016-01-28 | Exxonmobil Chemical Patents Inc. | Compositions pour articles chaussants comprenant des élastomères à base de propylène |
| EP3178340A4 (fr) * | 2014-08-07 | 2017-09-20 | ASICS Corporation | Semelle pour chaussures, et chaussures |
| CN107922665A (zh) * | 2015-10-19 | 2018-04-17 | 埃拉斯托米克斯株式会社 | 橡胶组合物及交联橡胶制品及其制造方法 |
| CN112812383A (zh) * | 2021-02-07 | 2021-05-18 | 福建美明达鞋业发展有限公司 | 一种防滑效果好的运动鞋鞋底及其制备方法 |
| US11192992B2 (en) | 2016-12-29 | 2021-12-07 | Exxonmobil Chemical Patents Inc. | Thermoplastic vulcanizates for foaming applications |
| CN114085438A (zh) * | 2021-11-19 | 2022-02-25 | 温州市耀阳鞋业有限公司 | 一种防滑男鞋及其制备方法 |
| JPWO2022137394A1 (fr) * | 2020-12-23 | 2022-06-30 | ||
| CN115926266A (zh) * | 2022-12-06 | 2023-04-07 | 中国皮革制鞋研究院有限公司 | 一种抗收缩橡胶发泡材料及其制备方法、应用 |
| DE102021214842A1 (de) | 2021-12-21 | 2023-06-22 | Continental Reifen Deutschland Gmbh | Schuhsohlenmaterial, Laufsohle und Verfahren zur Herstellung des Schuhsohlenmaterials oder der Laufsohle |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101801848B1 (ko) * | 2016-12-08 | 2017-11-27 | 하관형 | 신발 밑창 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5956432A (ja) * | 1982-09-27 | 1984-03-31 | Toyo Rubber Chem Ind Co Ltd | ゴム発泡体の製造方法 |
| JPS62181342A (ja) * | 1986-02-06 | 1987-08-08 | Japan Synthetic Rubber Co Ltd | 架橋発泡体 |
| JP3476543B2 (ja) * | 1994-06-28 | 2003-12-10 | ジーイー東芝シリコーン株式会社 | シリコーンゴムスポンジ組成物 |
| JP2002088693A (ja) * | 2000-09-08 | 2002-03-27 | Matsui Shikiso Chem Co Ltd | ステキヒトサイズ度が30分以上である壁紙及びその製造方法 |
| JP4003964B2 (ja) * | 2003-11-12 | 2007-11-07 | タイガースポリマー株式会社 | フッ素ゴム発泡体の製造方法 |
| JP2006016518A (ja) * | 2004-07-02 | 2006-01-19 | Jsr Corp | 発泡体用組成物 |
| JP5543686B2 (ja) * | 2007-11-13 | 2014-07-09 | 積水化学工業株式会社 | 熱膨張性マイクロカプセル及び発泡成形体 |
-
2010
- 2010-02-08 JP JP2010535862A patent/JP5565313B2/ja not_active Expired - Fee Related
- 2010-02-08 WO PCT/JP2010/051833 patent/WO2010050628A2/fr not_active Ceased
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013062685A3 (fr) * | 2011-10-25 | 2013-10-17 | Exxonmobil Chemical Patents Inc. | Composition, mousse et article fabriqué avec ladite composition |
| JP2015193783A (ja) * | 2014-03-20 | 2015-11-05 | 宇部興産株式会社 | 発泡体用ゴム組成物とそれを用いた靴底用ゴム発泡体と靴底 |
| WO2016014230A1 (fr) | 2014-07-25 | 2016-01-28 | Exxonmobil Chemical Patents Inc. | Compositions pour articles chaussants comprenant des élastomères à base de propylène |
| EP3178340A4 (fr) * | 2014-08-07 | 2017-09-20 | ASICS Corporation | Semelle pour chaussures, et chaussures |
| CN104371150A (zh) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | 耐曲折解放鞋用压延型叶黄色橡胶围条胶及制备方法及应用 |
| CN104371151A (zh) * | 2014-10-30 | 2015-02-25 | 际华三五三七制鞋有限责任公司 | 耐曲折解放鞋用压延型黑色橡胶围条胶及制备方法及应用 |
| CN104356438A (zh) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | 耐曲折解放鞋用压延型草绿色橡胶围条胶及制备方法及应用 |
| CN104356437A (zh) * | 2014-10-30 | 2015-02-18 | 际华三五三七制鞋有限责任公司 | 解放鞋用压延型叶黄色橡胶围条胶及其制备方法及应用 |
| CN105131366A (zh) * | 2015-10-09 | 2015-12-09 | 际华三五三七制鞋有限责任公司 | 无味环保型压延型高伸长率鞋用橡胶本色围条胶及其制备方法及应用 |
| CN107922665A (zh) * | 2015-10-19 | 2018-04-17 | 埃拉斯托米克斯株式会社 | 橡胶组合物及交联橡胶制品及其制造方法 |
| US11192992B2 (en) | 2016-12-29 | 2021-12-07 | Exxonmobil Chemical Patents Inc. | Thermoplastic vulcanizates for foaming applications |
| JPWO2022137394A1 (fr) * | 2020-12-23 | 2022-06-30 | ||
| WO2022137394A1 (fr) * | 2020-12-23 | 2022-06-30 | 株式会社アシックス | Élément d'amortissement et semelle pour chaussure |
| CN116600981A (zh) * | 2020-12-23 | 2023-08-15 | 株式会社爱世克私 | 鞋用的缓冲构件及鞋底 |
| CN112812383A (zh) * | 2021-02-07 | 2021-05-18 | 福建美明达鞋业发展有限公司 | 一种防滑效果好的运动鞋鞋底及其制备方法 |
| CN114085438A (zh) * | 2021-11-19 | 2022-02-25 | 温州市耀阳鞋业有限公司 | 一种防滑男鞋及其制备方法 |
| DE102021214842A1 (de) | 2021-12-21 | 2023-06-22 | Continental Reifen Deutschland Gmbh | Schuhsohlenmaterial, Laufsohle und Verfahren zur Herstellung des Schuhsohlenmaterials oder der Laufsohle |
| WO2023116989A1 (fr) | 2021-12-21 | 2023-06-29 | Continental Reifen Deutschland Gmbh | Matériau de semelle de chaussure, semelle extérieure et procédé de production du matériau de semelle de chaussure ou de la semelle extérieure |
| CN115926266A (zh) * | 2022-12-06 | 2023-04-07 | 中国皮革制鞋研究院有限公司 | 一种抗收缩橡胶发泡材料及其制备方法、应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010050628A3 (fr) | 2010-06-24 |
| JP5565313B2 (ja) | 2014-08-06 |
| JPWO2010050628A1 (ja) | 2012-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5565313B2 (ja) | 靴底用発泡体ゴム組成物及びアウトソール | |
| JP2012213615A (ja) | 靴底用発泡体ゴム組成物及びアウトソール | |
| CN1800255B (zh) | 三元乙丙橡胶连续气泡发泡体 | |
| CN106459535B (zh) | 包含热塑性硫化橡胶的组合物,发泡材料和由其制成的制品 | |
| EP1872924A1 (fr) | Procédé de fabrication de mousse moulée réticulée | |
| JP2007039499A (ja) | タイヤ用ゴム組成物 | |
| Zhang et al. | Effect of carbon black content on microcellular structure and physical properties of chlorinated polyethylene rubber foams | |
| KR20200034072A (ko) | 수축률이 향상된 혼합발포제 포함하는 수지 조성물 | |
| JP6398522B2 (ja) | 発泡体用ゴム組成物とそれを用いた靴底 | |
| JP5396623B2 (ja) | ゴム製品 | |
| CN107922665A (zh) | 橡胶组合物及交联橡胶制品及其制造方法 | |
| JP2006070260A (ja) | オレフィン系熱可塑性エラストマー組成物及びその発泡体 | |
| JP2004091747A (ja) | タイヤ用ゴム組成物およびその製造方法 | |
| WO2020059917A1 (fr) | Composition de résine à retrait amélioré comprenant un agent gonflant mixte | |
| JP7505000B2 (ja) | 高機能性発泡用樹脂組成物及びその製造方法 | |
| JP2001002866A (ja) | 遮音スポンジ用ゴム組成物及び加硫発泡成形体 | |
| JP2005120335A (ja) | 中空スポンジ及び自動車用シール材 | |
| JP2005171092A (ja) | タイヤ用ゴム組成物 | |
| JP2004091746A (ja) | タイヤ用ゴム組成物の製造方法 | |
| JP2006152058A (ja) | ゴム組成物 | |
| JP3980001B2 (ja) | ゴム組成物 | |
| JP2000234038A (ja) | スポンジ用熱硬化性ゴム組成物及びその加硫ゴム発泡成形体 | |
| JPH11279290A (ja) | リサイクルゴム成形体および製造方法 | |
| JP2004091745A (ja) | タイヤ用ゴム組成物 | |
| JP5929070B2 (ja) | タイヤ用発泡性ゴム組成物および発泡体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10714552 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010535862 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10714552 Country of ref document: EP Kind code of ref document: A2 |