WO2015080177A1 - 抗血栓性材料 - Google Patents
抗血栓性材料 Download PDFInfo
- Publication number
- WO2015080177A1 WO2015080177A1 PCT/JP2014/081307 JP2014081307W WO2015080177A1 WO 2015080177 A1 WO2015080177 A1 WO 2015080177A1 JP 2014081307 W JP2014081307 W JP 2014081307W WO 2015080177 A1 WO2015080177 A1 WO 2015080177A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- polymer
- atoms
- material according
- antithrombotic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L33/00—Antithrombogenic treatment of surgical articles, e.g. sutures, catheters, prostheses, or of articles for the manipulation or conditioning of blood; Materials for such treatment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L33/00—Antithrombogenic treatment of surgical articles, e.g. sutures, catheters, prostheses, or of articles for the manipulation or conditioning of blood; Materials for such treatment
- A61L33/0005—Use of materials characterised by their function or physical properties
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L33/00—Antithrombogenic treatment of surgical articles, e.g. sutures, catheters, prostheses, or of articles for the manipulation or conditioning of blood; Materials for such treatment
- A61L33/0005—Use of materials characterised by their function or physical properties
- A61L33/0011—Anticoagulant, e.g. heparin, platelet aggregation inhibitor, fibrinolytic agent, other than enzymes, attached to the substrate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L33/00—Antithrombogenic treatment of surgical articles, e.g. sutures, catheters, prostheses, or of articles for the manipulation or conditioning of blood; Materials for such treatment
- A61L33/0005—Use of materials characterised by their function or physical properties
- A61L33/0011—Anticoagulant, e.g. heparin, platelet aggregation inhibitor, fibrinolytic agent, other than enzymes, attached to the substrate
- A61L33/0041—Anticoagulant, e.g. heparin, platelet aggregation inhibitor, fibrinolytic agent, other than enzymes, attached to the substrate characterised by the choice of an antithrombatic agent other than heparin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L33/00—Antithrombogenic treatment of surgical articles, e.g. sutures, catheters, prostheses, or of articles for the manipulation or conditioning of blood; Materials for such treatment
- A61L33/0076—Chemical modification of the substrate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L33/00—Antithrombogenic treatment of surgical articles, e.g. sutures, catheters, prostheses, or of articles for the manipulation or conditioning of blood; Materials for such treatment
- A61L33/06—Use of macromolecular materials
Definitions
- the present invention relates to an antithrombotic material.
- Medical devices and instruments that come into contact with blood are required to have high antithrombogenicity in order to prevent functional deterioration due to blood coagulation.
- a method for imparting antithrombogenicity to medical devices and medical devices a method of applying or chemically bonding heparin or a heparin derivative, which is an anticoagulant, to the surface of a substrate is common.
- a technique for binding heparin or heparin derivative to the surface of the substrate mainly, 1) a method of fixing by covalent bonding with a functional group introduced on the surface of the substrate, and 2) on the surface of the substrate.
- a method of immobilizing an introduced positively charged cationic compound with an ionic bond There is known a method of immobilizing an introduced positively charged cationic compound with an ionic bond.
- Patent Document 1 a method of covalently bonding amidated heparin oxidized by nitrous acid treatment to the surface of the aminated substrate (Patent Document 1), polyethyleneimine which is aminated heparin and a cationic compound (hereinafter, “PEI ”) and a covalent bond with the surface of the substrate into which radicals have been introduced (Patent Document 2), and a method in which PEI and heparin introduced on the surface of the substrate are covalently bonded in the presence of a reducing agent (Patent Document) 3) has been reported.
- PEI polyethyleneimine which is aminated heparin and a cationic compound
- PET polyethylene terephthalate
- polyamide which is a base material in aminolysis or amide formation reaction
- polyamine which is a cationic compound
- heparin is ion-bonded there to obtain an antithrombotic material.
- organic cation mixtures such as quaternary ammonium salts or quaternary phosphonium compounds and heparin or heparin derivatives to form ionic complexes, dissolved in an organic solvent
- Patent Documents 6 and 7 for obtaining an antithrombotic material by applying to the surface have been reported.
- an antithrombotic material is obtained by applying a polymer containing a tertiary amino group to the surface of a substrate, converting the amino group to quaternary ammonium and ion-bonding heparin (patent) Document 8) and a method of obtaining an antithrombotic material by ion-bonding heparin after introducing PEI, which is a cationic compound, onto the surface of a substrate by ozone treatment or plasma treatment (Patent Documents 9 and 9). 10).
- Patent Document 11 a method has been reported in which adsorption to a cell surface is inhibited by binding a protein non-adsorbing substance having a negative charge such as heparin to the surface of a substrate.
- Japanese Patent No. 4152075 Japanese Patent No. 3497612 Japanese National Patent Publication No. 10-513074 Japanese Patent Publication No. 60-041947 Japanese Patent Publication No. 60-047287 Japanese Patent No. 4273965 JP-A-10-151192 Japanese Patent No. 3341503 Japanese Patent No. 3497612 Japanese Patent No. 3834602 Japanese Patent No. 4982752
- Patent Documents 2 to 5 are compounds having an anionic anticoagulant activity with respect to the cationic compound by introducing a positively charged cationic compound such as polyamine into the surface of the substrate. Although a method for immobilizing heparin or a heparin derivative by ionic bonding is described, there is no description about an appropriate amount of heparin or heparin derivative to be introduced.
- an ionic complex containing heparin or the like is dissolved in an organic solvent and applied to a substrate.
- the organic solvent to be used is a base that dissolves the ionic complex.
- the material must not dissolve, and even in the drying process after coating, the highly hydrophilic part of the ionic complex agglomerates avoiding the organic solvent and causes phase separation. The present condition is that it cannot be applied.
- organic cation mixtures such as quaternary ammonium salts and low molecular weight compounds such as quaternary phosphonium compounds are not covalently bonded to the substrate only by coating, so when used as an antithrombotic material, Elution is facilitated by contact with body fluids, and the elution rate of heparin or heparin derivatives cannot be controlled.
- Patent Documents 8 to 10 describe a method in which the surface of a substrate is coated with a cationic polymer having an amino group, and then heparin is ionically bonded to the cationic polymer.
- the proper amount of polymer introduced to the surface has not been studied. If the amount of the polymer to be coated is too small, high antithrombogenicity cannot be obtained, and if it is too large, the structure of the surface of the substrate may be buried.
- Patent Document 11 conventionally, it is known that heparin or the like adheres to the base material, and thus cell adhesion to the surface of the base material is reduced.
- the antithrombotic material used is used for an artificial blood vessel, a stent, a stent graft, or the like, the thrombus can be prevented, but there is a case where the biologicalization due to adhesion / proliferation of endothelial cells or the like may be inhibited.
- an object of the present invention is to provide an antithrombotic material that exhibits enhanced safety against low hemolytic toxicity and that exhibits high antithrombogenicity for a long period of time.
- an object of the present invention is to provide an antithrombotic material that has antithrombogenicity and does not reduce cell adhesion to the substrate surface.
- a polymer containing as a constituent monomer a compound selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride, and a compound having an anionic anticoagulant activity containing a sulfur atom.
- a coating material comprising: a substrate having a surface coated with the coating material, wherein the polymer is covalently bonded to the substrate and the presence of all atoms as measured by X-ray electron spectroscopy (XPS) on the surface
- XPS X-ray electron spectroscopy
- An antithrombogenic material wherein the abundance ratio of nitrogen atoms to the amount is 6.0 to 12.0 atomic percent.
- the coating material is an anionic polymer containing a compound selected from the group consisting of acrylic acid, methacrylic acid, ⁇ -glutamic acid, ⁇ -glutamic acid and aspartic acid as a constituent monomer, or oxalic acid, malonic acid, succinic acid
- the abundance ratio of the n2 component which is a split peak of nitrogen atoms with respect to all components of the N1s peak measured by X-ray electron spectroscopy (XPS) on the surface is 20 to 70 atomic%, (1) to ( The antithrombotic material according to any one of 8).
- the abundance ratio of the c3 component which is a split peak of carbon atoms with respect to all components of the C1s peak measured by X-ray electron spectroscopy (XPS) on the surface is 2.0 atomic% or more.
- XPS X-ray electron spectroscopy
- a coating material comprising: a substrate having a surface coated with the coating material, wherein the polymer is covalently bonded to the substrate and the presence of all atoms as measured by X-ray electron spectroscopy (XPS) on the surface An antithrombogenic material, wherein the abundance ratio of sulfur atoms to the amount is 3.0 to 6.0 atomic%.
- the antithrombogenic material of the present invention maintains the structure of the surface of the base material and is free from components other than the compound having an anionic anticoagulant activity containing a sulfur atom through a polymer covalently bonded to the base material. Elution can be suppressed and anticoagulant activity can be exhibited over a long period of time, so that medical devices and instruments that require antithrombogenicity (eg, artificial kidney, artificial lung, artificial blood vessel, artificial valve, stent, stent graft) , Catheters, free thrombus capture devices, vascular endoscopes, sutures, blood circuits, tubes, cannulas, blood bags, syringes, etc.).
- medical devices and instruments that require antithrombogenicity eg, artificial kidney, artificial lung, artificial blood vessel, artificial valve, stent, stent graft
- Catheters, free thrombus capture devices vascular endoscopes, sutures, blood circuits, tubes, cannulas, blood bags, s
- the antithrombogenic material of the present invention includes a polymer containing as a constituent monomer a compound selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride, and an anionic anticoagulant containing a sulfur atom.
- the polymer is covalently bonded to the substrate, and X-ray electron spectroscopy (hereinafter referred to as “XPS”) on the surface. )),
- XPS X-ray electron spectroscopy
- antithrombogenicity is a property that blood does not coagulate on the surface in contact with blood, and inhibits blood coagulation that progresses by, for example, platelet aggregation or activation of blood coagulation factors typified by thrombin.
- the antithrombotic material is a material having antithrombogenicity, and is not particularly limited, but is not limited to medical devices and medical instruments (for example, an artificial kidney, an artificial lung, an artificial blood vessel, an artificial valve, Stents, stent grafts, catheters, free thrombus capture devices, vascular endoscopes, sutures, blood circuits, tubes, cannulas, blood bags, syringes, etc.). Since these medical devices and medical devices often come into contact with blood and blood coagulation tends to proceed on the surfaces of the medical devices and medical devices, it is necessary to use an antithrombotic material as the material.
- medical devices and medical instruments for example, an artificial kidney, an artificial lung, an artificial blood vessel, an artificial valve, Stents, stent grafts, catheters, free thrombus capture devices, vascular endoscopes, sutures, blood circuits, tubes, cannulas, blood bags, syringes, etc.
- “Substrate” refers to a substance that constitutes a surface to be coated with a coating material defined below among the materials constituting the antithrombogenic material.
- the material of the base material in the present invention is not particularly limited.
- polyester, stretched porous polytetrafluoroethylene (hereinafter referred to as “ePTFE”), polyurethane, polyether urethane, polyamide, vinyl chloride, polycarbonate, polystyrene Polyethylene, polypropylene, polymethylpentene, polymethyl methacrylate, and the like are preferable as the base material.
- a highly versatile polyester is preferable as a base material for the antithrombotic material, and a polymer having at least an ester as a constituent monomer is more preferable.
- a polymer having at least an ester as a constituent monomer examples thereof include PET, polytrimethylene terephthalate, polybutylene terephthalate, polyethylene naphthalate, and polybutylene naphthalate.
- PET is more preferred as a base material for an antithrombotic material.
- the coating material is a material that covers at least a part of the surface of the substrate.
- the coating material is selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine, and diallyldimethylammonium chloride.
- a polymer containing a compound as a constituent monomer, and a compound having an anionic anticoagulant activity containing a sulfur atom are included.
- the polymer constituting the coating material is a polymer containing as a constituent monomer a compound selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride. Since these constituent monomers have a cationic nitrogen atom, the polymer becomes cationic.
- the compound having an anticoagulant activity containing a sulfur atom is anionic and can be ionically bonded.
- anionic anticoagulant compound containing a sulfur atom examples include heparin or heparin derivatives, dextran sulfate, polyvinyl sulfonic acid and polystyrene sulfonic acid, and heparin or heparin derivatives are more preferable.
- the heparin or heparin derivative is not particularly limited as long as it can inhibit the blood coagulation reaction, and heparin, unfractionated heparin and low molecular weight heparin which are generally widely used in clinical practice, and antithrombin III. High affinity heparin is also included.
- the polymer constituting the coating material Since the polymer constituting the coating material has a cationic property and may develop cytotoxicity, it is not preferable to elute it in a body fluid such as blood. Therefore, the polymer constituting the coating material is preferably covalently bonded to the surface of the substrate.
- the covalent bond refers to a chemical bond generated by sharing electrons between atoms.
- it is a covalent bond between atoms such as carbon, nitrogen, oxygen, sulfur, etc. on the surface of the polymer constituting the coating material and the substrate, and may be a single bond or a multiple bond.
- the kind of covalent bond is not limited, For example, an amine bond, an azide bond, an amide bond, an imine bond etc. are mentioned. Among these, an amide bond is more preferable from the viewpoint of easy formation of a covalent bond and stability after bonding.
- an amide bond is formed between the polymer constituting the coating material and the surface of the base material, so that the configuration of the polymer base material is an anion containing a sulfur atom. It has been found that the state of ion binding with a compound having sexual anticoagulant activity, such as heparin or a derivative of heparin, is optimized. The confirmation of the covalent bond can be determined from the fact that it does not elute even if it is washed with a solvent that dissolves the polymer.
- the polymer constituting the coating material may be a homopolymer or a copolymer.
- the polymer When the polymer is a copolymer, it may be a random copolymer, a block copolymer, a graft copolymer or an alternating copolymer.
- a block copolymer In the case of a block copolymer, it contains a nitrogen atom.
- it is more preferable to interact with a block portion having repeating units and an anionic anticoagulant-containing compound containing a sulfur atom, so that a block copolymer is more preferable because strong ionic bonding occurs.
- the homopolymer means a polymer compound obtained by polymerizing one kind of constituent monomer
- the copolymer means a polymer compound obtained by copolymerization of two or more kinds of monomers.
- the block copolymer is a copolymer having a molecular structure in which at least two kinds of polymers having different repeating units are connected by covalent bonds to form a long chain.
- a block constitutes a block copolymer.
- the polymer structure may be linear or branched.
- branched compounds are more preferred because they can form more stable ionic bonds at multiple points with an anionic anticoagulant compound containing a sulfur atom.
- the polymer has at least one functional group among primary to tertiary amino groups and quaternary ammonium groups.
- the quaternary ammonium group is a primary group.
- the ionic interaction with an anionic anticoagulant compound containing a sulfur atom rather than a tertiary amino group is stronger, and the elution rate of the anionic anticoagulant compound containing a sulfur atom is controlled. Because it is easy to do, it is preferable.
- the number of carbon atoms of the three alkyl groups constituting the quaternary ammonium group is not particularly limited. However, if the number is too large, the hydrophobicity is high and the steric hindrance increases. Thus, an anionic anticoagulant compound containing a sulfur atom can not be ionically bonded. In addition, since too much cytotoxicity is likely to occur, the number of carbon atoms per alkyl group bonded to the nitrogen atom constituting the quaternary ammonium group is preferably 1 to 12, and more preferably 2 to 6 Is preferred. The three alkyl groups bonded to the nitrogen atom constituting the quaternary ammonium group may all have the same carbon number or may be different.
- polyalkyleneimine is preferably used as the polymer because of the large amount of adsorption based on ionic interaction with an anionic anticoagulant compound containing a sulfur atom.
- examples of the polyalkyleneimine include PEI, polypropyleneimine and polybutyleneimine, and further alkoxylated polyalkyleneimine. Among them, PEI is more preferable.
- PEI examples include “LUPASOL” (registered trademark) (manufactured by BASF) and “EPOMIN” (registered trademark) (manufactured by Nippon Shokubai Co., Ltd.). It may be a copolymer with another monomer or a modified product.
- the modified body as used herein refers to a polymer in which the repeating units of the monomers constituting the polymer are the same, but, for example, a part thereof undergoes radical decomposition, recombination, etc. by irradiation with radiation described later.
- the constituent monomer for forming the copolymer used in addition to alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride is not particularly limited.
- Ions of an anionic anticoagulant compound containing a sulfur atom if too many constituent monomers are used in addition to alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride. Since the bond becomes weak, the content is preferably 10% by weight or less.
- the weight-average molecular weight of the polymer constituting the coating material is too small, the molecular weight is smaller than that of an anionic anticoagulant compound containing a sulfur atom, so that stable ionic bonds are formed on the surface of the substrate. It is not formed, and the desired antithrombogenicity is difficult to obtain.
- the weight average molecular weight of the polymer is too large, an anionic anticoagulant compound containing a sulfur atom is encapsulated by the polymer and is not exposed on the outermost surface of the coating material.
- the weight average molecular weight of the polymer constituting the coating material is preferably 600 to 2,000,000, more preferably 1,000 to 1,000,000, and even more preferably 10,000 to 1,000,000.
- the weight average molecular weight of the polymer can be measured by, for example, a gel permeation chromatography method or a light scattering method.
- the heparin or heparin derivative constituting the coating material may or may not be purified. Any material can be used as long as it can inhibit the blood coagulation reaction, and heparin generally used in clinical practice, unfractionated heparin, low molecular weight heparin, and high affinity heparin for antithrombin III are also included. Specific examples of heparin include “heparin sodium” (Organon API).
- an anionic anticoagulant activity containing a sulfur atom while maintaining the structure of the surface of the substrate and suppressing the elution of components other than the compound having an anionic anticoagulant activity containing a sulfur atom.
- the abundance ratio of sulfur atoms to the abundance of all atoms by XPS on the surface of the antithrombotic material is preferably 3.0 to 6.0 atom%, and 3.2 to 5.5 atom number. % Is more preferable, and 3.5 to 5.0 atomic% is even more preferable.
- the ratio of sulfur atoms to the total amount of atoms is less than 3.0% by number, the amount of the anionic anticoagulant compound containing sulfur atoms decreases, so the desired antithrombogenicity is obtained. I can't.
- the coating amount of an anionic anticoagulant compound containing sulfur atoms becomes an appropriate amount. Cell adhesion is enhanced.
- the abundance ratio of sulfur atoms relative to the abundance of all atoms on the surface of the antithrombotic material can be determined by XPS.
- Apparatus ESCALAB 220iXL (manufactured by VG Scientific)
- the surface of the antithrombotic material here refers to the X electron escape angle under the XPS measurement conditions, that is, when the inclination of the detector with respect to the surface of the antithrombotic material is measured at 90 °, and is detected from the measurement surface.
- the depth is up to 10 nm.
- the substrate may contain a sulfur atom or may not contain a sulfur atom.
- the base material may or may not contain a nitrogen atom.
- the atomic information on the surface of the antithrombotic material can be obtained from the binding energy value of the bound electrons in the substance obtained by irradiating the surface of the antithrombotic material with X-rays and measuring the energy of the photoelectrons generated.
- Information on the valence and bonding state can be obtained from the energy shift of the peak of the bonding energy value. Furthermore, quantitative determination using the area ratio of each peak, that is, the existence ratio of each atom, valence, and bonding state can be calculated.
- the S2p peak indicating the presence of a sulfur atom is observed at a binding energy value near 161 eV to 170 eV, and in the present invention, the area ratio of the S2p peak to the total peak is 3.0 to 6.0 atomic%. It was found that it is preferable.
- the abundance ratio of sulfur atoms to the abundance of all atoms is calculated by rounding off the second decimal place.
- the abundance ratio of nitrogen atoms is preferably 6.0 to 12.0 atomic% in order to increase the antithrombogenicity. 0 to 12.0 atomic% is more preferable, 7.5 to 11.0 atomic% is more preferable, and 8.0 to 10.0 atomic% is even more preferable.
- the abundance ratio of nitrogen atoms to the total abundance of atoms is less than 6.0 atomic%, the amount of polymer covalently bonded to the surface of the substrate is small and the structure of the substrate surface is retained, but the polymer The target antithrombogenicity cannot be obtained because the coating amount of an anionic anticoagulant compound containing a sulfur atom, such as heparin or a derivative of heparin, that is ionically bonded via the nucleoside is reduced.
- the abundance ratio of nitrogen atoms with respect to the abundance of all atoms is 12.0 atomic% or less, the coating amount of an anionic anticoagulant compound containing a sulfur atom becomes an appropriate amount. Cell adhesion is enhanced.
- the abundance ratio of nitrogen atoms to the total abundance of the atoms measured by XPS on the surface of the antithrombogenic material is preferably 6.0 to 12.0 atomic%. 6.0 to 9.5 atomic% is more preferable, and 8.0 to 9.5 atomic% is even more preferable.
- the N1s peak indicating the presence of a nitrogen atom is observed in the vicinity of a binding energy value of 396 eV to 403 eV.
- the area ratio of the N1s peak to the total peak is 6.0 to 12.0 atomic%. It was found that it is preferable.
- the N1s peak is mainly composed of an n1 component (near 399 eV) attributed to a carbon-nitrogen (hereinafter “CN”) bond, an ammonium salt, CN (a structure different from n1), or a nitrogen oxide.
- the peak can be divided into n2 components (near 401 to 402 eV) belonging to (hereinafter “NO”).
- the existence ratio of each divided peak component is calculated by the following formula 1.
- the abundance ratio of nitrogen atoms to the abundance of all atoms and the abundance ratio of each divided peak component are calculated by rounding off the first decimal place.
- Division ratio N1s ratio ⁇ (division percent / 100) Equation 1
- Split ratio Abundance ratio of each split peak component (%)
- N1s ratio abundance ratio of nitrogen atom to abundance of all atoms (%)
- Divided percent Ratio of each divided peak component in N1s peak (%)
- the n2 component attributed to NO obtained by splitting the N1s peak indicates the presence of a quaternary ammonium group, and the ratio of the n2 component to the total components of the N1s peak, that is, split percent (n2) It was found that 20 to 70 atomic% is preferable, 25 to 65 atomic% is more preferable, and 30 to 60 atomic% is more preferable. When the divided percent (n2) is less than 20 atomic%, the amount of quaternary ammonium groups is small, so that the ionic interaction with an anionic anticoagulant compound containing a sulfur atom is weak, and the dissolution rate is low. It becomes faster and it becomes difficult to obtain the desired antithrombogenicity.
- the divided percent (n2) exceeds 70 atomic%, the ionic interaction with the anionic anticoagulant compound containing a sulfur atom is too strong, so the degree of freedom is reduced due to the formation of an ionic complex. Therefore, the anticoagulant activity cannot be expressed for a long time, and the elution rate tends to be slow.
- the abundance ratio of the n2 component, that is, the division ratio (n2) is calculated by the equation 1, it is preferably 1.4 to 8.4 atomic% for the above reasons, and 1.8 to 7.2 atoms. Several percent is more preferred, and 2.4 to 6.0 atomic percent is even more preferred.
- the C1s peak indicating the presence of carbon atoms is seen in the vicinity of a binding energy value of 282 to 292 eV, and the C1s peak is mainly a carbon-hydrogen (hereinafter “CHx”) bond that suggests the presence of saturated hydrocarbons or the like.
- CHx carbon-hydrogen
- the c3 component attributed to the C ⁇ O bond obtained by splitting the C1s peak indicates the presence of an amide group in the present invention.
- the ratio of the c3 component to the total components of the C1s peak ie, the present
- the abundance ratio of the amide group measured by XPS on the surface of the antithrombotic material is preferably 2.0 atomic% or more, more preferably 3.0 atomic% or more.
- the abundance ratio of the amide group is less than 2.0 atomic%, the covalent bond due to the amide bond is small between the polymer constituting the coating material and the surface of the substrate, and the coating amount of the coating material is reduced. Since the ionic bond state with an anionic anticoagulant compound containing a sulfur atom is deteriorated due to the influence of the configuration on the surface of the polymer substrate, it is difficult to obtain the desired antithrombogenicity.
- the antithrombotic material of the present invention is a medical device and a medical device (for example, artificial kidney, artificial lung, artificial blood vessel, artificial valve, stent, stent graft, catheter, free thrombus capturing device, vascular endoscope, suture thread, blood circuit) , Tubes, cannulas, blood bags, syringes, etc.), but it is particularly preferable to use them as materials for free thrombus capture devices and artificial blood vessels.
- a medical device for example, artificial kidney, artificial lung, artificial blood vessel, artificial valve, stent, stent graft, catheter, free thrombus capturing device, vascular endoscope, suture thread, blood circuit
- the antithrombotic material of the present invention is used for a free thrombus capture device, it is preferable to use the antithrombotic material of the present invention for all the components of the free thrombus capture device, but the component for capturing the free thrombus Therefore, it is sufficient that at least the porous material is used as a base material and the porous material is coated with a coating material.
- a porous material which is a base material For example, a porous film, a mesh, etc. are mentioned, A mesh is preferable from the uniformity of a hole and an opening size being higher.
- the material is not particularly limited, but metals such as nickel-titanium alloys, polyurethane and polyester are preferably used, and polyester PET is more preferably used.
- the single yarn diameter of the fibers constituting the mesh is preferably 10 ⁇ m to 50 ⁇ m, and more preferably 20 ⁇ m to 40 ⁇ m.
- the mesh opening is preferably 10 ⁇ m to 200 ⁇ m, and more preferably 50 ⁇ m to 150 ⁇ m.
- the antithrombotic material of the present invention is used for an artificial blood vessel
- the material constituting the inner surface of the artificial blood vessel that is the base material is not particularly limited, but for example, a woven structure made of warp and weft composed of monofilament, multifilament or the like is preferable. Although it does not specifically limit as a material, Nylon, a polyester type, ePTFE, etc. are used suitably, PET which is a polyester type is used more suitably.
- monofilaments and multifilaments having a single yarn diameter of 15 ⁇ m or less are preferable, and monofilaments and multifilaments having a single yarn diameter of 10 ⁇ m or less. Filaments are more preferred, and monofilaments and multifilaments having a single yarn diameter of 5 ⁇ m or less are even more preferred.
- the accuracy of thrombus capture may be lowered by covering the mesh as a base material with a coating material, thereby destroying the mesh opening, which is the fine structure of the mesh.
- the destruction of the woven fabric structure composed of warp and weft which is the fine structure of the inner surface of the artificial blood vessel may affect blood flow and promote the formation of thrombus.
- the polymer is coated so that the abundance ratio of nitrogen atoms to the total abundance of all atoms by XPS on the surface of the antithrombogenic material is 12.0 atomic% or less.
- the coating material is coated with an anionic anticoagulant compound containing sulfur atoms so that the abundance ratio of sulfur atoms to the total amount of atoms by XPS is 6.0 atomic% or less.
- Thickness is 1 to 600 nm, and it exhibits high and long antithrombogenicity without destroying the fine structure of mesh openings used in free thrombus capture devices and the fine structure of fabric structures used on the inner surface of artificial blood vessels. Can do.
- the average thickness of the coating material for coating the base material is preferably 1 to 600 nm, more preferably 1 to 200 nm, because the fine structure on the surface of the base material is destroyed if it is too thick. Even more preferred is ⁇ 100 nm.
- the average thickness here is a thickness at which atoms derived from the coating material are observed with, for example, a scanning transmission electron microscope (hereinafter, “STEM”) described later, and indicates an average value of at least three points.
- the method for producing the antithrombotic material of the present invention is shown below.
- a target substrate is added to a solution containing a polymer selected from the group as a constituent monomer and a solution containing an anionic anticoagulant activity containing a sulfur atom, coating with a coating material is performed.
- the surface of the base material is coated with a coating material after all or any of them is reacted in advance between the polymer and an anionic anticoagulant compound containing a sulfur atom. Also good.
- a compound selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride as the first coating step in order to efficiently develop antithrombogenicity on the surface of the substrate. More preferably, after covalently bonding a polymer containing as a constituent monomer to the surface of the substrate, a compound having an anionic anticoagulant activity containing a sulfur atom is ionically bonded to the polymer as the second coating step.
- the polymer contains a primary to tertiary amino group
- the ionic interaction with the anionic anticoagulant compound containing a sulfur atom is strengthened, and the anionic antistatic agent containing a sulfur atom is strengthened.
- a step of quaternizing the polymer after the first coating step may be added.
- a polymer containing a compound selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride as a constituent monomer is covalently bonded to the surface of the substrate.
- a production method in the case of using a method in which a compound having an anionic anticoagulant activity containing a sulfur atom is ion-bonded to the above polymer as the coating step is shown below.
- the method for covalently bonding the polymer to the surface of the substrate is not particularly limited, but the substrate has functional groups (hydroxyl group, thiol group, amino group, carboxyl group, aldehyde group, isocyanate group, thioisocyanate, etc.).
- the substrate has functional groups (hydroxyl group, thiol group, amino group, carboxyl group, aldehyde group, isocyanate group, thioisocyanate, etc.).
- a polymer having a hydroxyl group, a thiol group, an amino group, or the like may be covalently bonded to the surface of the substrate, or a compound having a hydroxyl group, a thiol group, an amino group, or the like And a method of covalently bonding to the surface of a substrate having a carboxyl group and the like.
- radicals are generated on the surface of the substrate and the polymer by a method of covalently bonding the polymer or by irradiating with radiation.
- radiation ⁇ rays and electron beams are mainly used.
- the amount of ⁇ rays is preferably 2.5 million to 10 million Ci, and more preferably 3 million to 7.5 million Ci.
- the acceleration voltage of the electron beam is preferably 5 MeV or more, and more preferably 10 MeV or more.
- the radiation dose is preferably 1 to 50 kGy, more preferably 5 to 35 kGy.
- the irradiation temperature is preferably 10 to 60 ° C, more preferably 20 to 50 ° C.
- an antioxidant may be used to control the amount of radicals generated.
- the term “antioxidant” refers to a molecule that has the property of easily giving electrons to other molecules. Antioxidants to be used are not particularly limited.
- water-soluble vitamins such as vitamin C, polyphenols, alcohols such as methanol, ethanol, propanol, ethylene glycol, propylene glycol and glycerin, glucose and galactose Sugars such as mannose and trehalose, inorganic salts such as sodium hydrosulfite, sodium pyrosulfite, sodium dithionate, uric acid, cysteine, glutathione, bis (2-hydroxyethyl) iminotris (hydroxymethyl) methane (hereinafter referred to as “ Bis-Tris ”) and the like.
- Bis-Tris bis (2-hydroxyethyl) iminotris (hydroxymethyl) methane
- methanol, ethanol, propylene glycol, and Bis-Tris are particularly preferable from the viewpoint of handleability and persistence, and propylene glycol and Bis-Tris are more preferable.
- These antioxidants may be used alone or in combination of two or more. The antioxidant is preferably added to the aqueous solution.
- an anionic polymer containing a compound selected from the group consisting of acrylic acid, methacrylic acid, ⁇ -glutamic acid, ⁇ -glutamic acid and aspartic acid as a constituent monomer in order to develop higher and longer antithrombogenicity, or At least one anionic selected from the group consisting of oxalic acid, malonic acid, succinic acid, fumaric acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, malic acid, tartaric acid and citric acid
- the first additional step of covalently bonding at least one of the compounds to the surface of the polymer is preferably performed after the first coating step.
- a compound selected from the group consisting of alkyleneimine, vinylamine, allylamine, lysine, protamine and diallyldimethylammonium chloride is used as a constituent monomer.
- a second additional step of covalently bonding the cationic polymer comprising as an anionic polymer or an anionic compound followed by cationizing a compound having an anionic anticoagulant activity containing a sulfur atom, such as heparin or a derivative of heparin More preferably, a second coating step for covalently bonding to the conductive polymer is performed. If necessary, the third and fourth additional steps may be performed using an anionic polymer or an anionic compound and a cationic polymer.
- the anionic polymer is not particularly limited. However, the higher the weight ratio of the anionic functional group, the greater the coating amount due to the covalent bond with the base material or the coating material, and therefore polyacrylic acid (hereinafter referred to as “PAA”). )) Or polymethacrylic acid, poly ⁇ -glutamic acid, poly ⁇ -glutamic acid, or polyaspartic acid is preferred, and PAA is more preferred.
- PAA examples include “polyacrylic acid” (manufactured by Wako Pure Chemical Industries, Ltd.) and the like, but may be a copolymer with other monomers as long as the effects of the present invention are not hindered. It may be a body.
- the anionic polymer is not particularly limited, but may form a copolymer with other constituent monomers, such as ethylene glycol, propylene glycol, vinyl pyrrolidone, vinyl alcohol, vinyl caprolactam, vinyl acetate. , Styrene, methyl methacrylate, hydroxyethyl methacrylate, siloxane and the like. If the amount of the constituent monomer that forms a copolymer with the anionic polymer is too large, the coating amount due to the covalent bond with the base material or the coating material decreases, so that the amount is preferably 10% by weight or less.
- the weight average molecular weight of the anionic polymer is preferably from 600 to 2,000,000, more preferably from 10,000 to 1,000,000.
- the anionic compound is not particularly limited, but the higher the weight ratio of the anionic functional group, the greater the coating amount due to the covalent bond with the base material or the coating material. Therefore, oxalic acid, malonic acid, succinic acid , Fumaric acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, malic acid, tartaric acid and citric acid are preferred, and succinic acid is more preferred.
- the polyester material is used as the material of the base material, it is not particularly limited, but a method of covalently bonding by an aminolysis reaction by contacting a polymer under heating conditions can also be used.
- the ester bond on the surface of the substrate can be hydrolyzed by acid and alkali treatment, and a carboxyl group generated on the surface of the substrate can be subjected to a condensation reaction to form a covalent bond.
- the polymer may be brought into contact with the surface of the substrate to cause the reaction, but it may be brought into contact with the polymer in the state of being dissolved in the solvent.
- the solvent water, alcohol and the like are preferable, but water is particularly preferable from the viewpoints of handleability and persistence.
- the heating means is not particularly limited, and examples thereof include electric heating, microwave heating, and far infrared heating.
- the heating temperature is too low, the aminolysis reaction to the polyester base material by the polymer is difficult to proceed. Preferably there is.
- the temperature is too high, the aminolysis reaction proceeds sufficiently, but the skeletal structure of the polyester base material is broken, and therefore the heating temperature is preferably below the melting point.
- the step of hydrolyzing and oxidizing the ester bond on the surface of the substrate is important before the first coating step.
- a method of treating with an acid or alkali and an oxidizing agent is preferably used.
- a sufficient amount of polymer is not coated on the surface of the substrate by the method of treating with only acid or alkali. The reason for this is that, in the method of treating with only an acid or alkali, a hydroxyl group and a carboxyl group produced by hydrolysis of an ester bond are mixed, and the condensation reaction with the amino group of the polymer does not proceed efficiently.
- a hydroxyl group is not preferable because it is known that complement is easily activated when it comes into contact with blood. That is, a method of treating with an acid or alkali and an oxidizing agent is particularly preferably used in order to enhance the antithrombogenicity by increasing the coating amount of the polymer without activating the complement.
- the present inventors have found that a method of treating with an acid and an oxidizing agent is optimal as a combination of the step of hydrolyzing and oxidizing the ester bond on the surface of the substrate with an acid or alkali and an oxidizing agent in the present invention. Moreover, after processing the surface of a base material with an alkali, you may process with an acid and an oxidizing agent.
- the type of acid used is not particularly limited.
- Inorganic acids such as phosphoric acid, hexafluoroantimonic acid, tetrafluoroboric acid, chromic acid and boric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, trifluoromethanesulfonic acid and polystyrenesulfone Sulfonic acids such as sodium acid, acetic acid, citric acid, formic acid, gluconic acid, carboxylic acids such as lactic acid, oxalic acid and tartaric acid, vinyl carboxylic acids such as ascorbic acid and meldrum acid, nucleic acids such as deoxyribonucleic acid and rib
- the type of base used is not particularly limited.
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide, potassium hydroxide, rubidium hydroxide and cesium hydroxide, tetramethyl hydroxide Tetraalkylammonium hydroxides such as ammonium and tetraethylammonium hydroxide, hydroxides of alkaline earth metals such as calcium hydroxide, strontium hydroxide, barium hydroxide, europium hydroxide and thallium hydroxide, guanidine compounds, diammines Examples thereof include hydroxides of ammine complexes such as silver (I) hydroxide and tetraammine copper (II) hydroxide, trimethylsulfonium hydroxide and diphenyliodonium hydroxide.
- lithium hydroxide, sodium hydroxide, potassium hydroxide, and the like are more preferable from the viewpoint of handleability and the like.
- the kind of the oxidizing agent used is not particularly limited, but for example, potassium nitrate, hypochlorous acid, chlorous acid, perchloric acid, halogen such as fluorine, chlorine, bromine and iodine, potassium permanganate, Permanganate such as sodium permanganate trihydrate, ammonium permanganate, silver permanganate, zinc permanganate hexahydrate, magnesium permanganate, calcium permanganate and barium permanganate, nitric acid Examples include peroxides such as cerium ammonium, chromic acid, dichromic acid, and hydrogen peroxide, torence reagents, and sulfur dioxide. Among them, the strength of oxidizing agents and the deterioration of antithrombotic materials can be appropriately prevented. Permanganate is more preferable from the viewpoint of being able to do so.
- the type of the dehydrating condensing agent to be used is not particularly limited.
- the dehydration condensation agent may be used together with a dehydration condensation accelerator.
- the dehydration condensation accelerator used is not particularly limited.
- pyridine 4-dimethylaminopyridine (hereinafter “DMAP”), triethylamine, isopropylamine, 1-hydroxybenzotriazole, or N-hydroxysuccinic acid.
- DMAP 4-dimethylaminopyridine
- Examples include imide.
- the polymer, the dehydration condensation agent and the dehydration condensation accelerator may be reacted in the form of a mixed aqueous solution, or they may be added in order to carry out the reaction.
- ePTFE When ePTFE is used as the material of the base material, it is not particularly limited, but a method of functionalizing the surface of the base material with plasma or corona can be used.
- fluorine atoms present on the surface of the substrate are extracted using a fluororesin surface treatment agent and react with oxygen, hydrogen, water vapor, etc. in the air to form, for example, hydroxyl groups, carboxyl groups, carbonyl groups, etc.
- a method can also be used.
- a first coating step for covalently bonding the polymer to the surface of the ePTFE substrate can be performed.
- the polymer contains a primary to tertiary amino group
- the ionic interaction with the anionic anticoagulant compound containing a sulfur atom is strengthened, and the anionic antistatic agent containing a sulfur atom is strengthened.
- a step of quaternizing the polymer may be added.
- quaternary ammonium conversion may be performed before the polymer is covalently bonded to the surface of the substrate, or quaternary ammonium conversion after the polymer is covalently bonded to the surface of the substrate.
- the quaternary ammonium group of the polymer is present on the outermost surface of the coating material. Therefore, it is preferable to form a quaternary ammonium after covalent bonding to the surface of the substrate.
- an alkyl halide compound such as ether chloride or ethyl bromide or a glycidyl group-containing quaternary ammonium salt may be directly contacted, or an aqueous solution or organic solvent. You may make it melt
- the second coating step for ionically bonding a compound having an anionic anticoagulant activity containing a sulfur atom to the polymer is not particularly limited, but a method of contacting it in the state of an aqueous solution is preferable.
- the anti-factor Xa activity on the surface of the antithrombotic material was measured.
- the anti-factor Xa activity is an index representing the degree to which the activity of factor Xa that promotes the conversion of prothrombin to thrombin is inhibited.
- an anionic anticoagulant containing a sulfur atom in an antithrombotic material for example, an anionic anticoagulant containing a sulfur atom in an antithrombotic material.
- the compound having activity is heparin or a heparin derivative
- the surface amount of the active unit can be known.
- “Test Team (registered trademark) Heparin S” manufactured by Sekisui Medical Co., Ltd.
- the anti-factor Xa activity is preferably 25 mIU / cm 2, more preferably 30mIU / cm 2, and more preferably 50 mIU / cm 2.
- the surface amount by anti-factor Xa activity here refers to the numerical value measured after being immersed in physiological saline for 30 minutes.
- the antithrombotic material of the present invention has an initial surface amount after being immersed in physiological saline for 30 minutes, although the total coating amount due to the anti-factor Xa activity of heparin or a heparin derivative coated on the surface of the substrate is small. It is characteristic of being high.
- the total coating amount is the total amount of all elution amounts measured by anti-factor Xa activity and the surface amount remaining on the surface of the antithrombotic material. If it is too large, the fine structure on the surface of the substrate is destroyed. On the other hand, if it is too low, it is difficult to obtain the desired antithrombogenicity.
- the total coating amount due to anti-factor Xa activity on the surface of the antithrombotic material is 10,000 mIU / cm 2 or less
- the initial surface amount after being immersed in physiological saline for 30 minutes is 25 mIU / cm 2 or more.
- the initial surface amount after being immersed in physiological saline for 30 minutes is more preferably 50 mIU / cm 2 or more.
- the thrombus weight when human whole blood was brought into contact was quantified as an index indicating antithrombogenicity.
- the test was carried out three times each using an antithrombogenic material coated with the coating material of the present invention and the same type of substrate containing no coating material as a positive control. If the relative value of the thrombus weight is calculated by the following formula 3 and the average value of the three times is 10% or more, the amount of thrombus attached to the antithrombotic material is preferably small in the present invention.
- the antithrombotic material continues to be used, a compound having an anionic anticoagulant activity containing a sulfur atom elutes.
- the hemolysis rate calculated by the following formula 4 was used as an index indicating hemolysis toxicity.
- the hemolytic toxicity is determined according to the hemolytic toxicity test in accordance with the guideline issued by the Ministry of Health, Labor and Welfare, “Basic Concept of Biological Safety Evaluation Necessary for Application for Marketing Approval of Medical Devices” as shown in Table 1. Graded by value.
- the hemolytic toxicity in the present invention is preferably graded into non-hemolytic and mild hemolytic, and more preferably graded into non-hemolytic.
- Hemolysis rate (%) [(At-An) / (Ap-An)] ⁇ 100 Formula 4 At: Absorbance of specimen An: Absorbance of negative control Ap: Absorbance of positive control
- the antithrombogenic material of the present invention is coated with a coating material composed of a polymer and a compound having an anionic anticoagulant activity containing a sulfur atom, based on the interface of the substrate as in the prior art.
- the coating material is also present in the depth direction from the interface of the substrate.
- the STEM includes detectors such as an energy dispersive X-ray spectrometer (hereinafter referred to as “EDX”) and an electron energy loss spectrometer (hereinafter referred to as “EELS”).
- EDX energy dispersive X-ray spectrometer
- EELS electron energy loss spectrometer
- the surface of the antithrombotic material here means up to a depth of 10 nm from the measurement surface measured by XPS, and the interface of the antithrombotic material is embedded at the time of sample preparation before measurement by STEM. It refers to the boundary with acrylic resin.
- Whether the coating material is present in the depth direction from the interface of the base material can be determined from, for example, STEM measurement. Atoms derived from the coating material, which is a compound having an anionic anticoagulant activity containing polymer and sulfur atoms in the depth direction from the interface of the antithrombotic material, were observed, and atoms derived from the substrate were observed It suffices if atoms derived from the coating material are observed at a position deeper than the position.
- the base material is polyester or ePTFE, it has an anionic anticoagulant activity containing a nitrogen atom or a sulfur atom derived from the polymer rather than a position where an oxygen atom or a fluorine atom derived from the base material is observed.
- a sulfur atom derived from a compound may be observed.
- the interface of a base material means the position of the depth direction where the atom derived from a base material was observed.
- the presence of atoms is determined by whether or not the peak intensity derived from each atom is recognized by subtracting the background in the spectrum obtained from the STEM measurement.
- the coating material which is a compound having an anionic anticoagulant activity containing a polymer and a sulfur atom at a position further away from the interface of the antithrombotic material in the depth direction from the position of the interface of the substrate.
- the coating material which is a compound having an anionic anticoagulant activity containing a polymer and a sulfur atom at a position further away from the interface of the antithrombotic material in the depth direction from the position of the interface of the substrate.
- nitrogen atoms and sulfur atoms are preferably present at least in the depth direction from 20 to 100 nm, more preferably from 50 to 90 nm from the interface of the substrate.
- the coating material exists at least in the depth direction of 20 to 100 nm from the interface of the base material, the elution amount and elution rate of the anionic anticoagulant compound containing the eluting sulfur atom are optimal.
- a coating material is bonded to a depth direction of 20 to 100 nm in a base material that is not a porous material having pores.
- PET mesh (diameter: 27 ⁇ m, interfiber distance: 100 ⁇ m) as a base material is 5.0 wt% potassium permanganate (manufactured by Wako Pure Chemical Industries, Ltd.), 0.6 mol / L sulfuric acid (Wako Pure Chemical Industries, Ltd.) And the PET mesh was hydrolyzed and oxidized (hydrolysis and oxidation step). The aqueous solution after the reaction was removed and washed with hydrochloric acid (Wako Pure Chemical Industries, Ltd.) and distilled water.
- the PET mesh was 0.5% by weight DMT-MM (manufactured by Wako Pure Chemical Industries, Ltd.) and an aqueous solution of 5.0% by weight PEI (LUPASOL (registered trademark) P; manufactured by BASF) which is a part of the coating material.
- PEI was covalently bonded to the PET mesh by a condensation reaction (first coating step). The aqueous solution after the reaction was removed and washed with distilled water.
- the PET mesh was immersed in a 1% by weight methanol aqueous solution of ethyl bromide (manufactured by Wako Pure Chemical Industries, Ltd.) or pentyl bromide (manufactured by Wako Pure Chemical Industries, Ltd.) and reacted at 35 ° C. for 1 hour.
- the PEI covalently bonded to the surface of the PET mesh was subjected to quaternary ammonium conversion by heating to 50 ° C. for 4 hours (quaternary ammonium conversion step).
- the aqueous solution after the reaction was removed and washed with methanol or distilled water.
- sample 1 is a PET mesh that has been subjected to the second coating step without performing the quaternary ammonium conversion step
- sample 2 is a PET mesh that has been subjected to the quaternary ammonium conversion step using ethyl bromide
- Sample 3 was a PET mesh subjected to a quaternary ammonium conversion step using pentyl chloride.
- Example 2 After performing the same operation as in Example 1 and performing the first coating step, the PET mesh was dimethylacetamide of 0.5 wt% DMT-MM, 40 wt% succinic anhydride (manufactured by Wako Pure Chemical Industries, Ltd.). And was allowed to react at 50 ° C. for 17 hours (first additional step). The solution after the reaction was removed and washed with methanol or distilled water. Further, the PET mesh was immersed in an aqueous solution of 0.5 wt% DMT-MM and 5.0 wt% PEI and reacted at 30 ° C for 2 hours (second additional step). The aqueous solution after the reaction was removed and washed with distilled water. The same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- the PET mesh which performed the 2nd additional process with PEI (LUPASOL (trademark) P; made by BASF) is the sample 4 and the 2nd additional process with PEI (LUPASOL (trademark) SK; made by BASF).
- the PET mesh subjected to the above was designated as Sample 5.
- Example 3 After performing the same operation as in Example 1 and carrying out the first coating step, the PET mesh was dissolved in an aqueous solution of 0.5 wt% DMT-MM and 0.5 wt% PAA (manufactured by Wako Pure Chemical Industries, Ltd.). It was immersed and reacted at 30 ° C. for 2 hours (first additional step). The aqueous solution after the reaction was removed and washed with an aqueous sodium carbonate solution or distilled water.
- aqueous solution 0.5 wt% DMT-MM and 0.5 wt% PAA (manufactured by Wako Pure Chemical Industries, Ltd.). It was immersed and reacted at 30 ° C. for 2 hours (first additional step). The aqueous solution after the reaction was removed and washed with an aqueous sodium carbonate solution or distilled water.
- the PET mesh was immersed in an aqueous solution of 0.5 wt% DMT-MM and 5.0 wt% PEI and reacted at 30 ° C. for 2 hours (second additional step).
- the aqueous solution after the reaction was removed and washed with distilled water.
- the same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- the PET mesh subjected to the second additional step with PEI (average molecular weight of about 600; manufactured by Wako Pure Chemical Industries, Ltd.) is sample 6, PEI (LUPASOL (registered trademark) P; manufactured by BASF) is used as the second.
- the PET mesh subjected to the additional step was sample 7, and the PET mesh subjected to the second additional step with sample 8 was polyallylamine hydrochloride (hereinafter referred to as “PAH”) (weight average molecular weight 900,000; manufactured by Sigma-Aldrich). did.
- PAH polyallylamine hydrochloride
- Example 4 The same operation as in Example 1 was carried out, and PEI (LUPASOL (registered trademark) P; manufactured by BASF) was polyallylamine hydrochloride (hereinafter referred to as “PAH”) (weight average molecular weight 900,000; manufactured by Sigma-Aldrich), or The first coating step was carried out by changing to poly-L-lysine hydrobromide (hereinafter PLys) (weight average molecular weight 30,000 to 70,000; manufactured by Sigma-Aldrich). The same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- PAH polyallylamine hydrochloride
- PLys poly-L-lysine hydrobromide
- PEI (LUPASOL (registered trademark) P; manufactured by BASF) was changed to PAH and the PET mesh subjected to the first coating step was sample 9, PEI (LUPASOL (registered trademark) P; manufactured by BASF).
- a PET mesh that was changed to PLys and subjected to the first coating step was designated as Sample 10.
- Example 5 The PET mesh was immersed in a 5% PEI aqueous solution and irradiated with 5 kGy of ⁇ -rays (JS-8500 type cobalt 60 ⁇ -ray irradiator; manufactured by Nordion International Co., Ltd.) and covalently bonded (first coating step). After the reaction, the aqueous solution was removed and washed with Triton-X100 (manufactured by Sigma-Aldrich), physiological saline or distilled water. The same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- Triton-X100 manufactured by Sigma-Aldrich
- Example 6 The same operation as in Example 1 was performed, and heparin sodium (manufactured by Organon API) was changed to dextran sodium sulfate (manufactured by Wako Pure Chemical Industries, Ltd.), and a PET mesh subjected to the second coating step was used as sample 12. .
- Sample 12 was evaluated by human whole blood test and hemolytic toxicity. The results are shown in Table 2. As shown in Table 2, there was no thrombus adhesion ( ⁇ ) in the evaluation by the human whole blood test, and the hemolytic toxicity evaluation was also non-hemolytic ( ⁇ ).
- Example 1 The PET mesh was immersed in a 5% PEI aqueous solution and irradiated with 5 kGy of ⁇ -rays (JS-8500 type cobalt 60 ⁇ -ray irradiator; manufactured by Nordion International Co., Ltd.) and covalently bonded (first coating step). After the reaction, the aqueous solution was removed and washed with Triton-X100 (manufactured by Sigma-Aldrich), physiological saline or distilled water. The same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- Triton-X100 manufactured by Sigma-Aldrich
- the PET mesh that was not subjected to the quaternary ammonium conversion step was sample 13, PEI (average molecular weight).
- the sample 15 was subjected to the first coating process with PEI (LUPASOL (registered trademark) P; manufactured by BASF), and then the second coating process was performed with sodium dextran sulfate (manufactured by Wako Pure Chemical Industries, Ltd.).
- the mesh was Sample 16.
- Comparative Example 2 The PET mesh was immersed in an aqueous solution of 5% PEI, heated at 80 ° C. for 2 hours, and PEI was covalently bonded to the PET mesh by an aminolysis reaction (first coating step). The aqueous solution after the reaction was removed and washed with distilled water. The same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- a PET mesh subjected to the first coating step with PEI (average molecular weight of about 600; manufactured by Wako Pure Chemical Industries, Ltd.) was sample 17 and PEI (LUPASOL (registered trademark) P; manufactured by BASF) was used as the first.
- the PET mesh subjected to the coating step was Sample 18, and the PET mesh subjected to the first coating step with PEI (LUPASOL (registered trademark) SK; manufactured by BASF) was Sample 19.
- Example 3 The same operation as in Example 1 was performed, and PEI (LUPASOL (registered trademark) P; manufactured by BASF) was changed to PEI (average molecular weight of about 600; manufactured by Wako Pure Chemical Industries, Ltd.), and the first coating step was performed. did.
- PEI LPASOL (registered trademark) P; manufactured by BASF
- PEI average molecular weight of about 600; manufactured by Wako Pure Chemical Industries, Ltd.
- a PET mesh subjected to the second coating step was used as Sample 20.
- Sample 20 was subjected to measurement of the surface amount by anti-factor Xa activity after immersion in physiological saline for 30 minutes, evaluation by human whole blood test, and hemolysis toxicity evaluation. The results are shown in Table 2. As shown in Table 2, the hemolytic toxicity evaluation of sample 20 was non-hemolytic ( ⁇ ), but thrombus adhesion was found (+) in the human whole blood test. Further, the surface amount due to the anti-factor Xa activity was small.
- Example 4 After performing the same operation as in Example 1 and carrying out the first coating step, the PET mesh was dissolved in an aqueous solution of 0.5 wt% DMT-MM and 0.5 wt% PAA (manufactured by Wako Pure Chemical Industries, Ltd.). It was immersed and reacted at 30 ° C. for 2 hours (first additional step). The aqueous solution after the reaction was removed and washed with an aqueous sodium carbonate solution or distilled water.
- aqueous solution 0.5 wt% DMT-MM and 0.5 wt% PAA (manufactured by Wako Pure Chemical Industries, Ltd.). It was immersed and reacted at 30 ° C. for 2 hours (first additional step). The aqueous solution after the reaction was removed and washed with an aqueous sodium carbonate solution or distilled water.
- the PET mesh was immersed in an aqueous solution of 0.5 wt% DMT-MM and 5.0 wt% PEI and reacted at 30 ° C. for 2 hours (second additional step).
- the aqueous solution after the reaction was removed and washed with distilled water.
- the PET mesh was immersed in an aqueous solution of 0.5 wt% DMT-MM and 0.5 wt% PAA (Wako Pure Chemical Industries, Ltd.) and reacted at 30 ° C. for 2 hours (third additional step). .
- the aqueous solution after the reaction was removed and washed with an aqueous sodium carbonate solution or distilled water.
- the PET mesh was immersed in an aqueous solution of 0.5% by weight DMT-MM and 5.0% by weight PEI and reacted at 30 ° C. for 2 hours (fourth additional step).
- the aqueous solution after the reaction was removed and washed with distilled water.
- the same operation as in Example 1 was performed, and after performing a quaternary ammonium conversion step using ethyl bromide, a second coating step was performed.
- PET mesh which performed the 4th additional process by PEI (LUPASOL (trademark) P; made by BASF Corporation) is the sample 21 and the 4th additional process by PEI (LUPASOL (trademark) SK; made by BASF company).
- a PET mesh subjected to the above was designated as Sample 22.
- Example 7 Here, by changing the base material to a PET film, the same operation as in Example 1 was performed, and the PET film in which the second coating step was performed without performing the quaternary ammonium conversion step as in Sample 1 was performed.
- the film was designated as Sample 25. Samples 23 to 25 were evaluated by a cell adhesion test. The results are shown in Table 3. As shown in Table 3, the cell adhesion evaluation of samples 23 to 25 was (++).
- Example 8 Further, the base material was changed to a PET film, the same operation as in Example 3 was performed, and the second additional step was performed with PEI (LUPASOL (registered trademark) P; manufactured by BASF) similarly to Sample 7. The film was designated as sample 26. Sample 26 was evaluated by a cell adhesion test. The results are shown in Table 3. As shown in Table 3, the cell adhesion evaluation of Sample 26 was (+).
- Example 9 Further, the base material was changed to a PET film, the same operation as in Example 5 was performed, and the first coating step was performed with PEI (LUPASOL (registered trademark) P; manufactured by BASF) similarly to Sample 11. The film was designated as sample 27. Sample 27 was evaluated by a cell adhesion test. The results are shown in Table 3. As shown in Table 3, the cell adhesion evaluation of Sample 27 was (++).
- the surface amount by anti-factor Xa activity the evaluation by human whole blood test, and the evaluation method of hemolysis toxicity are shown below.
- the mesh was taken out, rinsed with PBS (-) (manufactured by Nissui Pharmaceutical Co., Ltd.), and the attached thrombus weight was quantified.
- the thrombus weight was obtained by measuring the dry weights of the mesh before the test and the mesh after the rinsing, respectively. Each sample and positive control were tested three times, and if the average value of three relative values of thrombus weight calculated by Equation 3 was 10% or more, there was a thrombus adhesion (+) and less than 10% If there was no thrombus adhesion, it was determined as ( ⁇ ).
- Cell adhesion is a property indicating the ease of cell adhesion to a material, and is measured by the following evaluation method. Samples 23 to 31 were punched into a disk sample having a diameter of 15 mm with a punching punch. One plate was placed in a well of a 24-well microplate for cell culture (manufactured by Sumitomo Bakelite Co., Ltd.) with the inner wall face up, and a metal pipe weight having a thickness of 1 mm was placed on the well.
- Normal human umbilical vein endothelial cells (Takara Bio Inc.) suspended in 2% FBS endothelial cell culture medium kit-2 (Takara Bio Inc.) were added at 4 ⁇ 10 4 cells per well. The cells were cultured in 1 mL of medium at 37 ° C. and 5% CO 2 for 24 hours. Then, after rinsing with PBS ( ⁇ ) (manufactured by Nissui), 100 ⁇ L of Cell Counting Kit-8 (manufactured by Dojindo) was added, and the cells were cultured for 4 hours in an environment of 37 ° C. and 5% CO 2 .
- a cell adhesion score was determined based on the absorbance As. Specifically, if As is less than 0.5, cell adhesion is weak ( ⁇ ), and if As is 0.5 or more, cell adhesion is strong (+), As is 0.7 or more. If there was, cell adhesion was stronger and determined as (++).
- the antithrombotic material of the present invention can be used in medical fields for medical devices and instruments that require high antithrombogenic properties for a long period of time.
Landscapes
- Health & Medical Sciences (AREA)
- Hematology (AREA)
- Surgery (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Materials For Medical Uses (AREA)
Abstract
Description
(1) アルキレンイミン、ビニルアミン、アリルアミン、リジン、プロタミン及びジアリルジメチルアンモニウムクロライドからなる群から選択される化合物を構成モノマーとして含むポリマー、並びに、硫黄原子を含むアニオン性の抗凝固活性を有する化合物、を含む被覆材料と、上記被覆材料によって表面が被覆された基材と、を備え、上記ポリマーは、上記基材と共有結合され、表面におけるX線電子分光法(XPS)で測定した全原子の存在量に対する窒素原子の存在比率が、6.0~12.0原子数%である、抗血栓性材料。
(2) 表面におけるX線電子分光法(XPS)で測定した全原子の存在量に対する硫黄原子の存在比率が、3.0~6.0原子数%である、(1)記載の抗血栓性材料。
(3)上記ポリマーは、第4級アンモニウム基を有する、(1)又は(2)記載の抗血栓性材料。
(4) 上記第4級アンモニウム基は、窒素原子に結合する炭素鎖がアルキル基で構成され、該アルキル基1つあたりの炭素数が1~12個である、(3)記載の抗血栓性材料。
(5) 上記被覆材料は、アクリル酸、メタクリル酸、α-グルタミン酸、γ-グルタミン酸及びアスパラギン酸からなる群から選択される化合物を構成モノマーとして含むアニオン性ポリマー、又は、シュウ酸、マロン酸、コハク酸、フマル酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、リンゴ酸、酒石酸及びクエン酸からなる群から選択されるアニオン性化合物、を含む、(1)~(4)のいずれか記載の抗血栓性材料。
(6) 上記硫黄原子を含むアニオン性の抗凝固活性を有する化合物は、ヘパリン又はヘパリンの誘導体である、(1)~(4)のいずれか記載の抗血栓性材料。
(7) 上記ポリマーの重量平均分子量は、600~2000000である、(1)~(6)のいずれか記載の抗血栓性材料。
(8) 上記アニオン性ポリマーの重量平均分子量は、600~2000000である、(5)記載の抗血栓性材料。
(9) 表面におけるX線電子分光法(XPS)で測定したN1sピークの全成分に対する窒素原子の分割ピークであるn2成分の存在比率が、20~70原子数%である、(1)~(8)のいずれか記載の抗血栓性材料。
(10) 表面におけるX線電子分光法(XPS)で測定したC1sピークの全成分に対する炭素原子の分割ピークであるc3成分の存在比率が、2.0原子数%以上である、(1)~(9)のいずれか記載の抗血栓性材料。
(11) 上記被覆材料の平均の厚みは、1~600nmである、(1)~(10)のいずれか記載の抗血栓性材料。
(12) 上記被覆材料は、上記基材の界面から深さ方向20~100nmに配置されている、(1)~(11)のいずれか記載の抗血栓性材料。
(13) 細胞接着性を有する、(1)~(12)のいずれか記載の抗血栓性材料。
(14) アルキレンイミン、ビニルアミン、アリルアミン、リジン、プロタミン及びジアリルジメチルアンモニウムクロライドからなる群から選択される化合物を構成モノマーとして含むポリマー並びにヘパリン若しくはヘパリンの誘導体を含む被覆材料と、上記被覆材料によって表面が被覆された基材と、を備え、上記ポリマーは、上記基材と共有結合し、表面におけるX線電子分光法(XPS)で測定した全原子の存在量に対する窒素原子の存在比率が、7.0~12.0原子数%である、抗血栓性材料。
(15) アルキレンイミン、ビニルアミン、アリルアミン、リジン、プロタミン及びジアリルジメチルアンモニウムクロライドからなる群から選択される化合物を構成モノマーとして含むポリマー、並びに、硫黄原子を含むアニオン性の抗凝固活性を有する化合物、を含む被覆材料と、上記被覆材料によって表面が被覆された基材と、を備え、上記ポリマーは、上記基材と共有結合され、表面におけるX線電子分光法(XPS)で測定した全原子の存在量に対する硫黄原子の存在比率が、3.0~6.0原子数%である、抗血栓性材料。
(16) 生理食塩水に30分浸漬した後の抗ファクターXa活性による表面量が、30mIU/cm2以上である、(14)又は(15)記載の抗血栓性材料。
(17) 抗ファクターXa活性による総被覆量が、10000mIU/cm2以下である、(14)~(16)のいずれか記載の抗血栓性材料。
[測定条件]
装置 :ESCALAB220iXL(VG Scientific社製)
励起X線 :monochromaticAlKα1,2線(1486.6eV)
X線径 :1mm
X電子脱出角度 :90°(抗血栓性材料の表面に対する検出器の傾き)
分割ratio : 各分割ピーク成分の存在比率(%)
N1sratio : 全原子の存在量に対する窒素原子の存在比率(%)
分割percent : N1sピークにおける各分割ピーク成分の割合(%)
分割ratio : 各分割ピーク成分の存在比率(%)
C1sratio : 全原子の存在量に対する炭素原子の存在比率(%)
分割percent : C1sピークにおける各分割ピーク成分の割合(%)
Bt : 検体の血栓重量
Bp : 陽性対照の血栓重量
At : 検体の吸光度
An : 陰性対照の吸光度
Ap : 陽性対照の吸光度

[測定条件]
装置 :電界放出型透過電子顕微鏡JEM-2100F(JEOL社製)
EELS検出器 :GIF Tridiem(GATAN社製)
EDX検出器 :JED-2300T(JEOL社製)
画像取得 :Digital Micrograph(GATAN社製)
試料調整 :超薄切片法(銅製マイクログリッドに懸架し、包埋樹脂はアクリル系樹脂を使用。)
加速電圧 :200kV
ビーム径 :直径0.7nm
エネルギー分解能 :約1.0eVFWHM
基材であるPETメッシュ(径:27μm、繊維間距離:100μm)を5.0重量%過マンガン酸カリウム(和光純薬工業株式会社製)、0.6mol/L硫酸(和光純薬工業株式会社製)の水溶液に浸漬し、60℃で3時間反応させてPETメッシュを加水分解及び酸化した(加水分解及び酸化する工程)。反応後の水溶液を除去し、塩酸(和光純薬工業株式会社製)及び蒸留水で洗浄した。
実施例1と同様の操作を行い、第1の被覆工程を実施した後、PETメッシュを0.5重量%DMT-MM、40重量%無水コハク酸(和光純薬工業株式会社製)のジメチルアセトアミドに浸漬し、50℃で17時間反応させた(第1の追加工程)。反応後の溶液を除去し、メタノールや蒸留水で洗浄した。さらにPETメッシュを、0.5重量%DMT-MM、5.0重量%PEIの水溶液に浸漬し、30℃で2時間反応させた(第2の追加工程)。反応後の水溶液を除去し、蒸留水で洗浄した。実施例1と同様の操作を行い、臭化エチルを用いて第4級アンモニウム化工程を実施した後、第2の被覆工程を実施した。
実施例1と同様の操作を行い、第1の被覆工程を実施した後、PETメッシュを0.5重量%DMT-MM、0.5重量%PAA(和光純薬工業株式会社製)の水溶液に浸漬し、30℃で2時間反応させた(第1の追加工程)。反応後の水溶液を除去し、炭酸ナトリウム水溶液や蒸留水で洗浄した。
実施例1と同様の操作を行い、PEI(LUPASOL(登録商標) P;BASF社製)をポリアリルアミン塩酸塩(以下、「PAH」)(重量平均分子量90万;シグマ-アルドリッチ社製)、もしくはポリ-L-リシン臭化水素酸塩(以下、PLys)(重量平均分子量3~7万;シグマ-アルドリッチ社製)に変更して第1の被覆工程を実施した。実施例1と同様の操作を行い、臭化エチルを用いて第4級アンモニウム化工程を実施した後、第2の被覆工程を実施した。
PETメッシュを5%PEIの水溶液に浸漬し、5kGyのγ線を照射(JS-8500型コバルト60γ線照射装置;ノーディオン・インターナショナル社製)し共有結合させた(第1の被覆工程)。反応後の水溶液を除去し、Triton-X100(シグマ-アルドリッチ社製)、生理食塩水や蒸留水で洗浄した。実施例1と同様の操作を行い、臭化エチルを用いて第4級アンモニウム化工程を実施した後、第2の被覆工程を実施した。
実施例1と同様の操作を行い、ヘパリンナトリウム(Organon API社製)をデキストラン硫酸ナトリウム(和光純薬工業株式会社製)に変更して第2の被覆工程を実施したPETメッシュをサンプル12とした。
PETメッシュを5%PEIの水溶液に浸漬し、5kGyのγ線を照射(JS-8500型コバルト60γ線照射装置;ノーディオン・インターナショナル社製)し共有結合させた(第1の被覆工程)。反応後の水溶液を除去し、Triton-X100(シグマ-アルドリッチ社製)、生理食塩水や蒸留水で洗浄した。実施例1と同様の操作を行い、臭化エチルを用いて第4級アンモニウム化工程を実施した後、第2の被覆工程を実施した。
PETメッシュを5%PEIの水溶液に浸漬し、80℃で2時間加熱し、PETメッシュにPEIをアミノリシス反応により共有結合させた(第1の被覆工程)。反応後の水溶液を除去し、蒸留水で洗浄した。実施例1と同様の操作を行い、臭化エチルを用いて第4級アンモニウム化工程を実施した後、第2の被覆工程を実施した。
実施例1と同様の操作を行い、PEI(LUPASOL(登録商標) P;BASF社製)をPEI(平均分子量約600;和光純薬工業株式会社製)に変更して第1の被覆工程を実施した。実施例1と同様の操作を行い、臭化エチルを用いて第4級アンモニウム化工程を実施した後、第2の被覆工程を実施したPETメッシュをサンプル20とした。
実施例1と同様の操作を行い、第1の被覆工程を実施した後、PETメッシュを0.5重量%DMT-MM、0.5重量%PAA(和光純薬工業株式会社製)の水溶液に浸漬し、30℃で2時間反応させた(第1の追加工程)。反応後の水溶液を除去し、炭酸ナトリウム水溶液や蒸留水で洗浄した。
ここで、基材をPETフィルムに変更して、実施例1と同様の操作を行い、サンプル1と同様に第4級アンモニウム化工程を実施せずに第2の被覆工程を実施したPETフィルムをサンプル23、サンプル2と同様に臭化エチルを用いて第4級アンモニウム化工程を実施したPETフィルムをサンプル24、サンプル3と同様に臭化ペンチルを用いて第4級アンモニウム化工程を実施したPETフィルムをサンプル25とした。サンプル23~25について、細胞接着性試験による評価を実施した。結果を表3に示す。表3に示すとおり、サンプル23~25の細胞接着性評価は(++)であった。
また、基材をPETフィルムに変更して、実施例3と同様の操作を行い、サンプル7と同様にPEI(LUPASOL(登録商標) P;BASF社製)で第2の追加工程を実施したPETフィルムをサンプル26とした。サンプル26について、細胞接着性試験による評価を実施した。結果を表3に示す。表3に示すとおり、サンプル26の細胞接着性評価は(+)であった。
また、基材をPETフィルムに変更して、実施例5と同様の操作を行い、サンプル11と同様にPEI(LUPASOL(登録商標) P;BASF社製)で第1の被覆工程を実施したPETフィルムをサンプル27とした。サンプル27について、細胞接着性試験による評価を実施した。結果を表3に示す。表3に示すとおり、サンプル27の細胞接着性評価は(++)であった。
また、基材をPETフィルムに変更して、比較例1と同様の操作を行い、サンプル13と同様にPEI(平均分子量約600;和光純薬工業株式会社製)で第1の被覆工程を実施した後、第4級アンモニウム化工程を実施しなかったPETフィルムをサンプル28、サンプル14と同様にPEI(平均分子量約600;和光純薬工業株式会社製)で第1の被覆工程を実施したPETフィルムをサンプル29とした。サンプル28及び29について、細胞接着性試験による評価を実施した。結果を表3に示す。表3に示すとおり、サンプル28及び29の細胞接着性評価は(++)であった。
また、基材をPETフィルムに変更して、比較例4と同様の操作を行い、サンプル21と同様にPEI(LUPASOL(登録商標) P;BASF社製)で第4の追加工程を実施したPETフィルムをサンプル30、サンプル22と同様にPEI(LUPASOL(登録商標) SK;BASF社製)で第4の追加工程を実施したPETフィルムをサンプル31とした。サンプル30及び31について、細胞接着性試験による評価を実施した。結果を表3に示す。表3に示すとおり、サンプル30及び31の細胞接着性評価は(-)であった。
被覆材料を被覆した抗血栓性材料(例えばPETメッシュ)を0.5×0.5cmのサイズにカットし、生理食塩水を用いて37℃で30分間洗浄した。洗浄後のPETメッシュを“テストチーム(登録商標) ヘパリンS”(積水メディカル株式会社製)の操作手順に従って反応させ、405nmの吸光度をマイクロプレートリーダ(MTP-300;コロナ電気株式会社製)で測定して、テストチーム ヘパリンSの操作手順に従って抗ファクターXa活性による表面量を算出した。表面量は高い程よく、25mIU/cm2以上であることが好ましく、50mIU/cm2以上であることがより好ましい。
被覆材料を被覆した抗血栓性材料(例えばPETメッシュ)、および被覆材料を含まない同種の基材(陽性対照)を有効表面積1.0cm2にカットし、生理食塩水で37℃、30分間洗浄してから2mLのマイクロチューブに入れた。ヒト新鮮血に0.5U/mLとなるようにヘパリンナトリウム注(味の素製薬株式会社製)を添加した後、このヒト血液を2mL添加し、37℃で2時間インキュベートした。インキュベート後にメッシュを取り出し、PBS(-)(日水製薬株式会社製)でリンスした後、付着した血栓重量を定量した。血栓重量は、試験前のメッシュとリンス後のメッシュの乾燥重量をそれぞれ測定し、その差によって求めた。各サンプルおよび陽性対照についてそれぞれ試験を3回実施し、式3により算出した血栓重量の相対値の3回の平均値が10%以上であれば血栓付着は有りとして(+)、10%未満であれば血栓付着は無しとして(-)と判定した。
ヒト新鮮血をガラスビーズ入り三角フラスコの壁面を伝うように入れた。手のひらの上で、水平に円を描くように、およそ1秒間に2回の間隔で約5分間振とうし、脱線維血を調製した。被覆材料を被覆した抗血栓性材料(例えばPETメッシュ)を1.0×2.0cmのサイズにカットし、生理食塩水で37℃、30分間洗浄してから2mLのマイクロチューブに入れた。メッシュが入ったマイクロチューブに生理食塩水で50倍希釈した脱線維血を1mL添加し、37℃で4時間インキュベートした。インキュベート後、750Gで5分間遠心分離した。上清を採取し、576nmでのUV吸収を測定した。式4より算出した値が2より大きい値、すなわち溶血性有りであれば(+)、2以下の値、すなわち非溶血性であれば(-)と判定した。溶血毒性は無い方がよく、非溶血性であることが好ましい。
細胞接着性とは、材料に対する細胞の接着のしやすさを示す性質であり、以下の評価法で測定される。サンプル23~31を打抜ポンチで直径15mmの円板サンプルに打ち抜いた。細胞培養用の24ウェル・マイクロプレート(住友ベークライト社製)のウェルに内壁面を上にして1枚入れ、上から肉厚1mmの金属パイプ状錘を乗せた。2%FBS内皮細胞培地キット-2(タカラバイオ社製)に懸濁した正常ヒト臍帯静脈内皮細胞(タカラバイオ社)を1ウェル当たり4×104個になるように添加した。1mLの培地中で37℃、5%CO2の環境下で24時間培養した。その後、PBS(-)(ニッスイ社製)でリンスした後に、Cell Counting Kit-8(同仁化学社製)を100μL添加し、37℃、5%CO2の環境下で4時間培養した。その後、450nmの吸光度をマイクロプレートリーダ(MTP-300;コロナ電気株式会社製)で測定して、以下の式5に示すように、吸光度を算出した。
As = At-Ab ・・・式5
At : 測定値の吸光度
Ab : ブランク溶液の吸光度(培地及びCell Counting Kit-8の溶液のみで細胞なし。)
As : 算出された吸光度
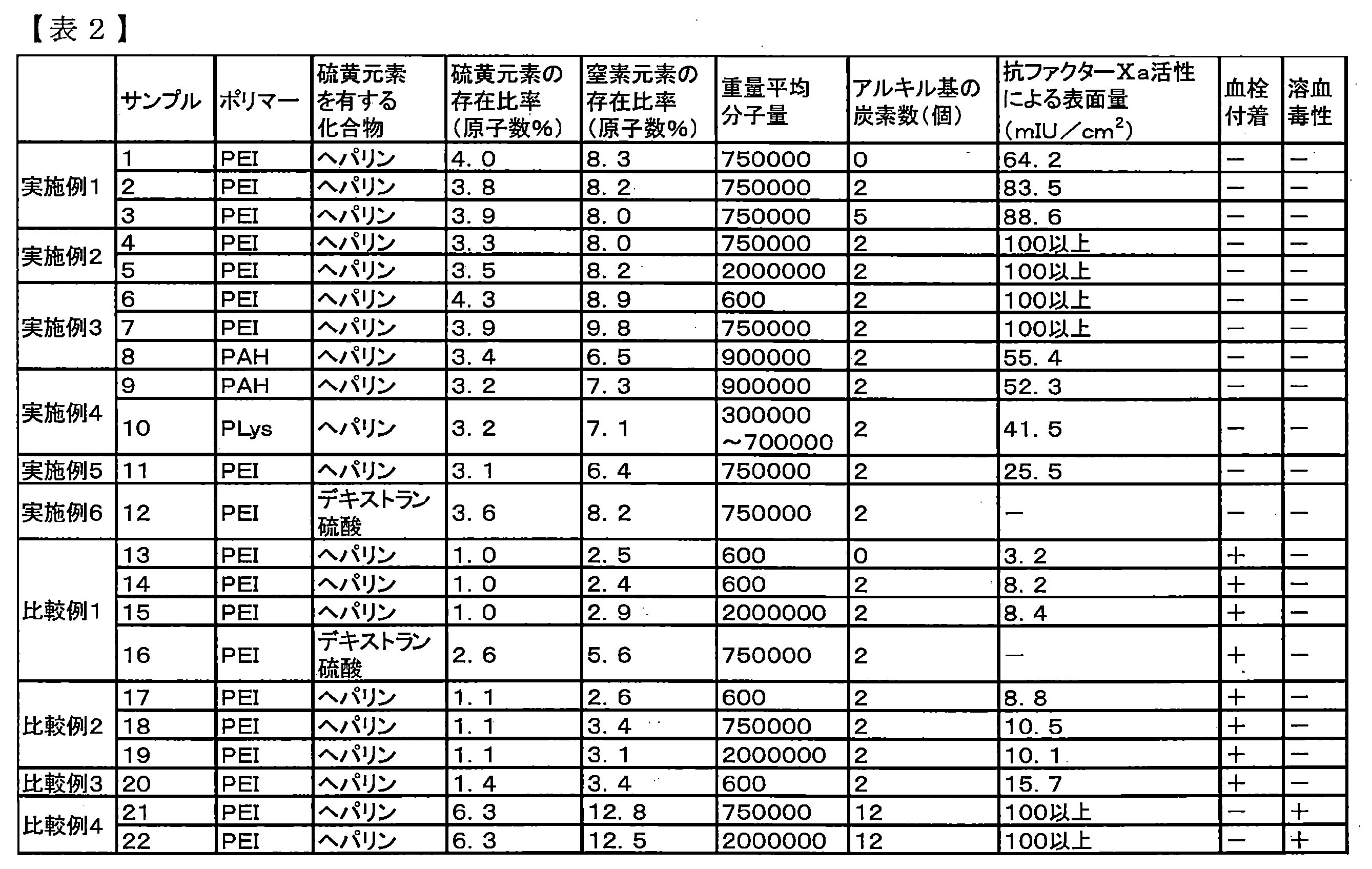
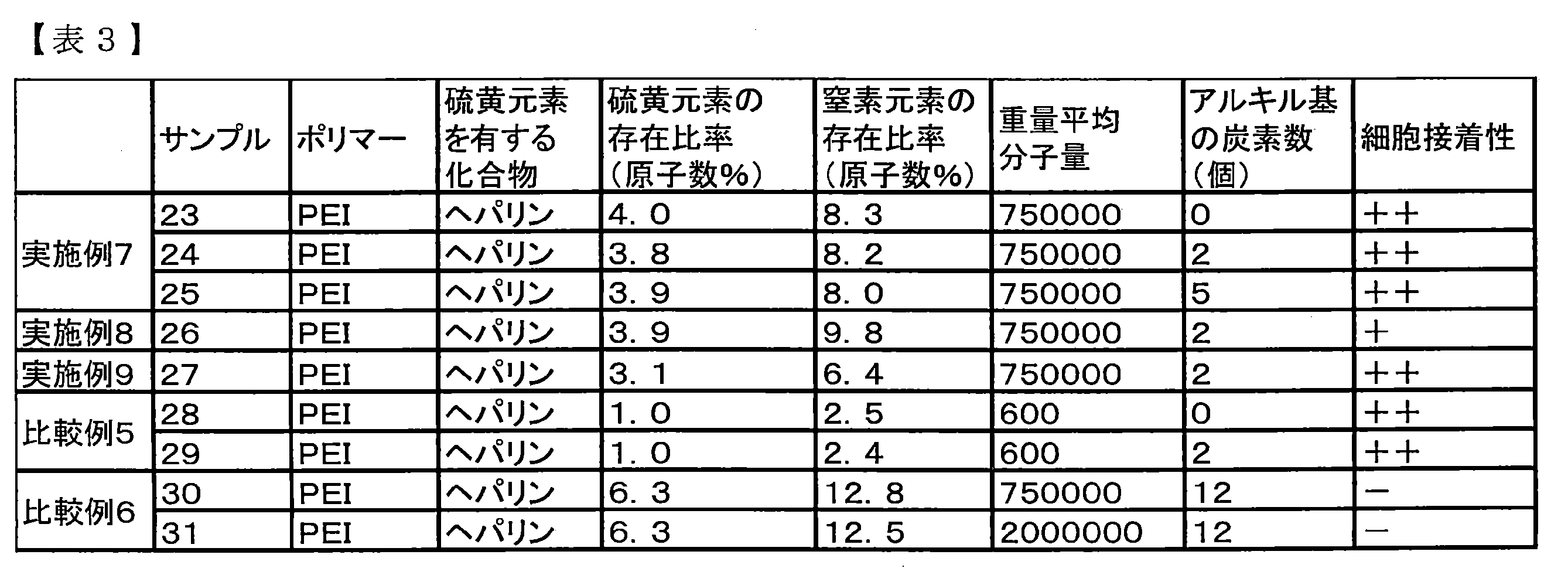
Claims (13)
- アルキレンイミン、ビニルアミン、アリルアミン、リジン、プロタミン及びジアリルジメチルアンモニウムクロライドからなる群から選択される化合物を構成モノマーとして含むポリマー、並びに、硫黄原子を含むアニオン性の抗凝固活性を有する化合物、を含む被覆材料と、該被覆材料によって表面が被覆された基材と、を備え、
前記ポリマーは、前記基材と共有結合され、
表面におけるX線電子分光法(XPS)で測定した全原子の存在量に対する窒素原子の存在比率が、6.0~12.0原子数%である、抗血栓性材料。 - 表面におけるX線電子分光法(XPS)で測定した全原子の存在量に対する硫黄原子の存在比率が、3.0~6.0原子数%である、請求項1記載の抗血栓性材料。
- 前記ポリマーは、第4級アンモニウム基を有する、請求項1又は2記載の抗血栓性材料。
- 前記第4級アンモニウム基は、窒素原子に結合する炭素鎖がアルキル基で構成され、該アルキル基1つあたりの炭素数が1~12個である、請求項3記載の抗血栓性材料。
- 前記被覆材料は、アクリル酸、メタクリル酸、α-グルタミン酸、γ-グルタミン酸及びアスパラギン酸からなる群から選択される化合物を構成モノマーとして含むアニオン性ポリマー、又は、シュウ酸、マロン酸、コハク酸、フマル酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、リンゴ酸、酒石酸及びクエン酸からなる群から選択されるアニオン性化合物、を含む、請求項1~4のいずれか一項記載の抗血栓性材料。
- 前記硫黄原子を含むアニオン性の抗凝固活性を有する化合物は、ヘパリン又はヘパリンの誘導体である、請求項1~4のいずれか一項記載の抗血栓性材料。
- 前記ポリマーの重量平均分子量は、600~2000000である、請求項1~6のいずれか一項記載の抗血栓性材料。
- 前記アニオン性ポリマーの重量平均分子量は、600~2000000である、請求項5記載の抗血栓性材料。
- 表面におけるX線電子分光法(XPS)で測定したN1sピークの全成分に対する窒素原子の分割ピークであるn2成分の存在比率が、20~70原子数%である、請求項1~8のいずれか一項記載の抗血栓性材料。
- 表面におけるX線電子分光法(XPS)で測定したC1sピークの全成分に対する炭素原子の分割ピークであるc3成分の存在比率が、2.0原子数%以上である、請求項1~9のいずれか一項記載の抗血栓性材料。
- 前記被覆材料の平均の厚みは、1~600nmである、請求項1~10のいずれか一項記載の抗血栓性材料。
- 前記被覆材料は、前記基材の界面から深さ方向20~100nmに配置されている、請求項1~11のいずれか一項記載の抗血栓性材料。
- 細胞接着性を有する請求項1~12のいずれか一項記載の抗血栓性材料。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2928629A CA2928629C (en) | 2013-11-28 | 2014-11-27 | Antithrombotic material |
| CN201480064996.0A CN105744967B (zh) | 2013-11-28 | 2014-11-27 | 抗血栓性材料 |
| EP14866413.9A EP3078389B1 (en) | 2013-11-28 | 2014-11-27 | Antithrombotic material |
| RU2016125453A RU2679615C1 (ru) | 2013-11-28 | 2014-11-27 | Антитромботический материал |
| KR1020167010811A KR101851960B1 (ko) | 2013-11-28 | 2014-11-27 | 항혈전성 재료 |
| AU2014355414A AU2014355414B2 (en) | 2013-11-28 | 2014-11-27 | Antithrombotic material |
| US15/038,560 US9795721B2 (en) | 2013-11-28 | 2014-11-27 | Antithrombotic material |
| JP2014557940A JP6435863B2 (ja) | 2013-11-28 | 2014-11-27 | 抗血栓性材料 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-246580 | 2013-11-28 | ||
| JP2013246580 | 2013-11-28 | ||
| JP2013-265834 | 2013-12-24 | ||
| JP2013265834 | 2013-12-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015080177A1 true WO2015080177A1 (ja) | 2015-06-04 |
Family
ID=53199116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/081307 Ceased WO2015080177A1 (ja) | 2013-11-28 | 2014-11-27 | 抗血栓性材料 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9795721B2 (ja) |
| EP (1) | EP3078389B1 (ja) |
| JP (1) | JP6435863B2 (ja) |
| KR (1) | KR101851960B1 (ja) |
| CN (1) | CN105744967B (ja) |
| AU (1) | AU2014355414B2 (ja) |
| CA (1) | CA2928629C (ja) |
| RU (1) | RU2679615C1 (ja) |
| WO (1) | WO2015080177A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015170733A1 (ja) * | 2014-05-09 | 2015-11-12 | 東レ株式会社 | 血管内治療補助具 |
| JP2018053361A (ja) * | 2016-09-16 | 2018-04-05 | 大日本塗料株式会社 | 表面上の第四級アンモニウムカチオン並びに/又は金及び/若しくは銀のハロゲン化物の量が低減された金ナノ粒子の懸濁液 |
| WO2019150937A1 (ja) | 2018-01-30 | 2019-08-08 | 東レ株式会社 | 平織物、その製造方法およびステントグラフト |
| JP2019177061A (ja) * | 2018-03-30 | 2019-10-17 | 東レ株式会社 | 抗血栓性材料 |
| WO2020158847A1 (ja) | 2019-01-30 | 2020-08-06 | 東レ株式会社 | 心血管留置デバイス用の医療基材 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI660722B (zh) * | 2014-05-09 | 2019-06-01 | 日商東麗股份有限公司 | 血管內治療輔助器具 |
| WO2016190407A1 (ja) * | 2015-05-27 | 2016-12-01 | 東レ株式会社 | 抗血栓性材料 |
| BR112020002382A2 (pt) * | 2017-09-29 | 2020-09-01 | Toray Industries, Inc. | material médico antitrombogênico |
| CN108853584B (zh) * | 2018-08-30 | 2021-09-03 | 湖南博隽生物医药有限公司 | 一种抗血栓人造血管及其制备方法 |
| EP3659634B1 (en) | 2018-11-29 | 2023-02-22 | Cook Medical Technologies LLC | Bioactive agent coated medical device and method of coating such a device |
| US11752019B2 (en) | 2018-11-29 | 2023-09-12 | Cook Medical Technologies Llc | Bioactive agent coated medical device and method of coating such a device |
| CN110665071B (zh) * | 2019-10-14 | 2021-04-23 | 中国科学院长春应用化学研究所 | 一种抗菌抗凝血型涂层、具有抗菌抗凝血型涂层的功能材料及其制备方法 |
| CN111701077B (zh) * | 2020-06-24 | 2021-05-11 | 四川大学 | 一种具有抗血栓抗钙化功能的瓣膜及其制备方法和应用 |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5413694A (en) * | 1977-07-01 | 1979-02-01 | Sumitomo Electric Industries | Composite blood vessel prosthesis and method of producing same |
| JPS6041947A (ja) * | 1983-01-04 | 1985-03-05 | マルフア− リミテツド | 視力検査方法及びその装置 |
| JPS6047287A (ja) * | 1983-08-24 | 1985-03-14 | Matsushita Electric Ind Co Ltd | 磁気記録再生装置 |
| JPS6041947B2 (ja) | 1977-12-09 | 1985-09-19 | ユニチカ株式会社 | ポリエステル成形体表面に抗血栓性を付与する方法 |
| JPS6047287B2 (ja) | 1977-12-15 | 1985-10-21 | ユニチカ株式会社 | 抗血栓性ポリアミドの製造方法 |
| JPH01262869A (ja) * | 1988-04-15 | 1989-10-19 | Ube Ind Ltd | 抗血液凝固性医療用具 |
| JPH02144070A (ja) * | 1988-11-25 | 1990-06-01 | Toray Ind Inc | 易滑性医療材料 |
| JPH0686808A (ja) * | 1992-09-07 | 1994-03-29 | Terumo Corp | オリゴ糖または多糖体と基材の結合体 |
| JPH06510783A (ja) * | 1991-09-26 | 1994-12-01 | コルリーネ・システムズ・アクチエボラーグ | 新規接合体、その調製および使用ならびにその接合体を用いて調製された基体 |
| JPH07178161A (ja) * | 1993-12-24 | 1995-07-18 | Terumo Corp | 医療器具 |
| JPH08336587A (ja) * | 1995-06-12 | 1996-12-24 | Terumo Corp | 医療用材料及びその製造方法 |
| JPH09169801A (ja) * | 1995-10-17 | 1997-06-30 | Terumo Corp | ヘパリン複合体およびそれを被覆した医療器具 |
| JPH09276394A (ja) * | 1996-02-15 | 1997-10-28 | Jinkou Ketsukan Gijutsu Kenkyu Center:Kk | 抗血栓性人工血管 |
| JPH10151192A (ja) | 1996-11-26 | 1998-06-09 | Toyobo Co Ltd | 抗血栓性組成物および医用材料 |
| JPH10513074A (ja) * | 1995-02-01 | 1998-12-15 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | 表面改質方法 |
| JPH11235381A (ja) * | 1998-02-23 | 1999-08-31 | Create Medic Co Ltd | 抗血栓性材料およびその製造方法 |
| JPH11511355A (ja) * | 1995-08-22 | 1999-10-05 | メドトロニック・インコーポレーテッド | ヘパリン化生体材料の製造法 |
| JP2002514126A (ja) * | 1997-07-01 | 2002-05-14 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | 水不溶性ポリマーおよびポリアルキレンイミンの反応混合物を使用した表面改質方法 |
| JP3341503B2 (ja) | 1994-11-09 | 2002-11-05 | 東レ株式会社 | 抗血栓性医療材料およびそれを用いたカテーテル |
| JP3834602B2 (ja) | 1999-03-30 | 2006-10-18 | 独立行政法人産業技術総合研究所 | 抗血栓性多糖類をコーティングした高分子材料からなる医療用具 |
| JP4152075B2 (ja) | 1998-09-09 | 2008-09-17 | カルメダ アクチボラゲット | 表面改質物質を調整する新方法 |
| JP4273965B2 (ja) | 2001-06-05 | 2009-06-03 | 東洋紡績株式会社 | 抗血栓性組成物およびそれを有する医療用具 |
| JP4982752B2 (ja) | 2007-02-01 | 2012-07-25 | 国立大学法人東北大学 | タンパク質および細胞が内壁に固定された中空構造体 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6041948B2 (ja) * | 1977-12-15 | 1985-09-19 | ユニチカ株式会社 | 固体表面に抗血栓性を付与する方法 |
| US4521564A (en) * | 1984-02-10 | 1985-06-04 | Warner-Lambert Company | Covalent bonded antithrombogenic polyurethane material |
| JPH04152075A (ja) | 1990-10-17 | 1992-05-26 | Hitachi Ltd | 歩行機械 |
| JPH04273965A (ja) | 1991-02-27 | 1992-09-30 | Sanyo Electric Co Ltd | 氷片供給装置 |
| DE4216798A1 (de) | 1992-05-21 | 1993-11-25 | Metallgesellschaft Ag | Verfahren zur Aufarbeitung von vanadiumhaltigen Rückständen |
| JPH0641947A (ja) | 1992-07-27 | 1994-02-15 | Tokyo Electric Power Co Inc:The | 薬液地盤注入の自動品質管理方法及び薬液地盤注入管理装置 |
| US5672638A (en) * | 1995-08-22 | 1997-09-30 | Medtronic, Inc. | Biocompatability for solid surfaces |
| CA2197375C (en) | 1996-02-15 | 2003-05-06 | Yasuhiro Okuda | Artificial blood vessel |
| JP3014324B2 (ja) * | 1996-02-15 | 2000-02-28 | 株式会社人工血管技術研究センター | 人工血管 |
| US5811151A (en) * | 1996-05-31 | 1998-09-22 | Medtronic, Inc. | Method of modifying the surface of a medical device |
| AU4928399A (en) * | 1998-07-27 | 2000-02-21 | M & M Laboratory Co., Ltd. | Ion complex, coating material, and coating method |
| JP4258703B2 (ja) * | 2001-05-22 | 2009-04-30 | 東洋紡績株式会社 | 血液適合性組成物およびそれをコートした医療用具 |
| CN101011605A (zh) * | 2006-11-28 | 2007-08-08 | 武汉理工大学 | 抗凝血聚氨酯材料及其制备和用途 |
| US8383156B2 (en) * | 2007-04-30 | 2013-02-26 | Cordis Corporation | Coating for a medical device having an anti-thrombotic conjugate |
| GB0816783D0 (en) * | 2008-09-15 | 2008-10-22 | Carmeda Ab | Immobilised biological entities |
| WO2010081884A2 (en) * | 2009-01-16 | 2010-07-22 | Institut Polytechnique De Grenoble | Process for preparing a surface coated by crosslinked polyelectrolyte multilayer films as a biomimetic reservoir for proteins |
| ES2955183T3 (es) * | 2011-03-11 | 2023-11-29 | Gore & Ass | Mejoras para las entidades biológicas inmovilizadas |
| EP2985042B1 (en) * | 2013-04-12 | 2019-03-06 | Toray Industries, Inc. | Antithrombotic artificial blood vessel |
-
2014
- 2014-11-27 EP EP14866413.9A patent/EP3078389B1/en active Active
- 2014-11-27 WO PCT/JP2014/081307 patent/WO2015080177A1/ja not_active Ceased
- 2014-11-27 KR KR1020167010811A patent/KR101851960B1/ko not_active Expired - Fee Related
- 2014-11-27 US US15/038,560 patent/US9795721B2/en active Active
- 2014-11-27 CN CN201480064996.0A patent/CN105744967B/zh active Active
- 2014-11-27 AU AU2014355414A patent/AU2014355414B2/en not_active Ceased
- 2014-11-27 RU RU2016125453A patent/RU2679615C1/ru active
- 2014-11-27 JP JP2014557940A patent/JP6435863B2/ja active Active
- 2014-11-27 CA CA2928629A patent/CA2928629C/en active Active
Patent Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5413694A (en) * | 1977-07-01 | 1979-02-01 | Sumitomo Electric Industries | Composite blood vessel prosthesis and method of producing same |
| JPS6041947B2 (ja) | 1977-12-09 | 1985-09-19 | ユニチカ株式会社 | ポリエステル成形体表面に抗血栓性を付与する方法 |
| JPS6047287B2 (ja) | 1977-12-15 | 1985-10-21 | ユニチカ株式会社 | 抗血栓性ポリアミドの製造方法 |
| JPS6041947A (ja) * | 1983-01-04 | 1985-03-05 | マルフア− リミテツド | 視力検査方法及びその装置 |
| JPS6047287A (ja) * | 1983-08-24 | 1985-03-14 | Matsushita Electric Ind Co Ltd | 磁気記録再生装置 |
| JPH01262869A (ja) * | 1988-04-15 | 1989-10-19 | Ube Ind Ltd | 抗血液凝固性医療用具 |
| JPH02144070A (ja) * | 1988-11-25 | 1990-06-01 | Toray Ind Inc | 易滑性医療材料 |
| JPH06510783A (ja) * | 1991-09-26 | 1994-12-01 | コルリーネ・システムズ・アクチエボラーグ | 新規接合体、その調製および使用ならびにその接合体を用いて調製された基体 |
| JPH0686808A (ja) * | 1992-09-07 | 1994-03-29 | Terumo Corp | オリゴ糖または多糖体と基材の結合体 |
| JPH07178161A (ja) * | 1993-12-24 | 1995-07-18 | Terumo Corp | 医療器具 |
| JP3341503B2 (ja) | 1994-11-09 | 2002-11-05 | 東レ株式会社 | 抗血栓性医療材料およびそれを用いたカテーテル |
| JPH10513074A (ja) * | 1995-02-01 | 1998-12-15 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | 表面改質方法 |
| JPH08336587A (ja) * | 1995-06-12 | 1996-12-24 | Terumo Corp | 医療用材料及びその製造方法 |
| JP3497612B2 (ja) | 1995-06-12 | 2004-02-16 | テルモ株式会社 | 医療用材料及びその製造方法 |
| JPH11511355A (ja) * | 1995-08-22 | 1999-10-05 | メドトロニック・インコーポレーテッド | ヘパリン化生体材料の製造法 |
| JPH09169801A (ja) * | 1995-10-17 | 1997-06-30 | Terumo Corp | ヘパリン複合体およびそれを被覆した医療器具 |
| JPH09276394A (ja) * | 1996-02-15 | 1997-10-28 | Jinkou Ketsukan Gijutsu Kenkyu Center:Kk | 抗血栓性人工血管 |
| JPH10151192A (ja) | 1996-11-26 | 1998-06-09 | Toyobo Co Ltd | 抗血栓性組成物および医用材料 |
| JP2002514126A (ja) * | 1997-07-01 | 2002-05-14 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | 水不溶性ポリマーおよびポリアルキレンイミンの反応混合物を使用した表面改質方法 |
| JPH11235381A (ja) * | 1998-02-23 | 1999-08-31 | Create Medic Co Ltd | 抗血栓性材料およびその製造方法 |
| JP4152075B2 (ja) | 1998-09-09 | 2008-09-17 | カルメダ アクチボラゲット | 表面改質物質を調整する新方法 |
| JP3834602B2 (ja) | 1999-03-30 | 2006-10-18 | 独立行政法人産業技術総合研究所 | 抗血栓性多糖類をコーティングした高分子材料からなる医療用具 |
| JP4273965B2 (ja) | 2001-06-05 | 2009-06-03 | 東洋紡績株式会社 | 抗血栓性組成物およびそれを有する医療用具 |
| JP4982752B2 (ja) | 2007-02-01 | 2012-07-25 | 国立大学法人東北大学 | タンパク質および細胞が内壁に固定された中空構造体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3078389A4 |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015170733A1 (ja) * | 2014-05-09 | 2015-11-12 | 東レ株式会社 | 血管内治療補助具 |
| US10098725B2 (en) | 2014-05-09 | 2018-10-16 | Toray Industries, Inc. | Endovascular treatment aid |
| JP2018053361A (ja) * | 2016-09-16 | 2018-04-05 | 大日本塗料株式会社 | 表面上の第四級アンモニウムカチオン並びに/又は金及び/若しくは銀のハロゲン化物の量が低減された金ナノ粒子の懸濁液 |
| WO2019150937A1 (ja) | 2018-01-30 | 2019-08-08 | 東レ株式会社 | 平織物、その製造方法およびステントグラフト |
| US12053367B2 (en) | 2018-01-30 | 2024-08-06 | Toray Industries, Inc. | Plain-weave fabric, method for manufacturing same, and stent graft |
| JP2019177061A (ja) * | 2018-03-30 | 2019-10-17 | 東レ株式会社 | 抗血栓性材料 |
| JP7043936B2 (ja) | 2018-03-30 | 2022-03-30 | 東レ株式会社 | 抗血栓性材料 |
| WO2020158847A1 (ja) | 2019-01-30 | 2020-08-06 | 東レ株式会社 | 心血管留置デバイス用の医療基材 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3078389A4 (en) | 2017-08-09 |
| CA2928629A1 (en) | 2015-06-04 |
| US20160296679A1 (en) | 2016-10-13 |
| CA2928629C (en) | 2018-04-17 |
| US9795721B2 (en) | 2017-10-24 |
| EP3078389A1 (en) | 2016-10-12 |
| KR20160065136A (ko) | 2016-06-08 |
| JPWO2015080177A1 (ja) | 2017-03-16 |
| KR101851960B1 (ko) | 2018-04-25 |
| RU2016125453A (ru) | 2018-01-10 |
| AU2014355414B2 (en) | 2017-06-01 |
| CN105744967B (zh) | 2018-12-25 |
| CN105744967A (zh) | 2016-07-06 |
| EP3078389B1 (en) | 2020-04-01 |
| RU2679615C1 (ru) | 2019-02-12 |
| JP6435863B2 (ja) | 2018-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6435863B2 (ja) | 抗血栓性材料 | |
| JPWO2015080176A1 (ja) | 抗血栓性材料 | |
| JP6788796B2 (ja) | 抗血栓性金属材料 | |
| CA2987236C (en) | Antithrombotic material | |
| JP2016220717A (ja) | 抗血栓性材料 | |
| JP7043936B2 (ja) | 抗血栓性材料 | |
| CN110944687B (zh) | 使用镍钛合金的抗血栓性医疗材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014557940 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14866413 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2928629 Country of ref document: CA Ref document number: 20167010811 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014866413 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014866413 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014355414 Country of ref document: AU Date of ref document: 20141127 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15038560 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2016125453 Country of ref document: RU Kind code of ref document: A |