WO2017142002A1 - Formes cristallines de composé macrolide c-4"-substitué libre et sel de celui-ci, et leurs procédés de production - Google Patents
Formes cristallines de composé macrolide c-4"-substitué libre et sel de celui-ci, et leurs procédés de production Download PDFInfo
- Publication number
- WO2017142002A1 WO2017142002A1 PCT/JP2017/005629 JP2017005629W WO2017142002A1 WO 2017142002 A1 WO2017142002 A1 WO 2017142002A1 JP 2017005629 W JP2017005629 W JP 2017005629W WO 2017142002 A1 WO2017142002 A1 WO 2017142002A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal
- degrees
- compound
- formula
- compound represented
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
Definitions
- the present invention relates to a novel antibiotic having an erythromycin-like skeleton. More specifically, the present invention relates to a crystal form of a macrolide compound having a methyl group substituted with a substituent having a nitrogen atom at the 4 ′′ position of cladinose, a process for producing the same, and a specific form of the macrolide compound. The present invention relates to a crystal form of a salt and a production method thereof.
- Erythromycin A is an antibiotic widely used as a therapeutic agent for infectious diseases caused by Gram-positive bacteria, mycoplasma and the like.
- erythromycin is decomposed by gastric acid, there is a disadvantage that pharmacokinetics is not constant. Accordingly, derivatives having increased stability to acids have been studied, and as a result, macrolide agents with stable pharmacokinetics such as clarithromycin, azithromycin (Patent Documents 1 and 2), and roxithromycin have been developed.
- These macrolide agents for treating external respiratory infections need to have strong antibacterial activity against pneumococci, streptococci and Haemophilus influenzae, which are frequently clinically isolated.
- macrolide-resistant pneumococci are frequently isolated from community-acquired pneumonia, it is important to be effective against resistant pneumococci.
- Patent Document 3 As a result of extensive research in recent years, Agouridas et al. In 1995 as HMR3647 (Terithromycin, Patent Document 3) and Or et al. 1998 as effective macrolides against both erythromycin-resistant pneumococci and erythromycin-resistant streptococci. In the year, ABT-773 (Cesromycin, Patent Document 4) was found one after another. Thereafter, 2-fluoroketolide (Patent Document 5), which is further enhanced in drug efficacy, has been reported.
- the macrolide compound having a methyl group substituted with a substituent having a nitrogen atom at the 4 ′′ position of cladinose is an azalide type having a structural feature of having a nitrogen atom in the lactone ring. Most of the compounds are (Patent Document 6).
- the object of the present invention is effective not only for conventional erythromycin-sensitive bacteria but also for erythromycin-resistant bacteria (for example, resistant pneumococci, resistant streptococci, and mycoplasma), and is reproducible as a single crystal having a certain quality.
- Crystalline forms of novel compounds that are well-obtained and can be stably supplied as crystals of drug substances used in the manufacture of pharmaceuticals and pharmaceutical raw materials, and have physical properties with excellent storage stability and their production It is to provide a method.
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 4.1 degrees, 10.0 degrees, 10.6 degrees and 15.1 degrees; or
- another aspect of the present invention is as follows: (2) According to (1), the compound represented by the formula [1] is added with ethyl acetate, hexane or a mixed solution thereof to form a solution, which is then crystallized and the resulting crystal is dried. It is a manufacturing method of the described crystal.
- A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 4.0 degrees, 7.1 degrees, 8.1 degrees and 12.1 degrees;
- Another aspect of the present invention is as follows: (4) The compound represented by the formula [1] is added with methanol or a water-methanol mixed solution to form a solution, crystallized, and the obtained crystal is dried. It is a manufacturing method of a crystal.
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 3.3 degrees, 4.6 degrees, 11.2 degrees and 15.5 degrees; or
- Another aspect of the present invention is as follows: (6) The production of the crystal according to (5), wherein water is added to the crystal of the compound represented by the formula [1] to form a suspension, and the crystal obtained by stirring is dried. Is the method.
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 5.4 °, 6.6 °, 10.9 ° and 16.6 °; or
- Another aspect of the present invention is as follows: (8) The compound represented by the formula [1] is characterized by adding ethanol or a water-ethanol mixed solution to form a solution, followed by crystallization, and drying the obtained crystal under ice-cooling ( 7) A method for producing a crystal as described in 7).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 5.0 degrees, 5.9 degrees, 10.9 degrees and 16.7 degrees; or
- Another aspect of the present invention is as follows: (10) The method for producing a crystal according to (9), characterized in that acetonitrile is added to the compound represented by the formula [1] to form a solution, followed by crystallization, and drying the obtained crystal. .
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 11.0 degrees, 11.3 degrees, 13.3 degrees and 16.8 degrees; or
- Another aspect of the present invention is as follows: (12) Water is added to the crystal of the compound represented by the formula [1] shown in claim 3, 5, 7 or 9 to form a suspension, and then the crystal obtained by stirring is dried. (11) The crystal production method according to (11).
- Another aspect of the present invention is as follows: (15) After adding malonic acid to an ethyl acetate solution of the compound represented by the formula [1] and allowing it to act, the resulting crystals are collected by filtration and dried. It is a manufacturing method.
- Another aspect of the present invention is as follows: (16) A methanesulfonate salt of the compound represented by the formula [1].
- another aspect of the present invention is as follows: (17) A crystal of the salt according to (16) having at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 9.7 °, 11.1 °, 12.9 ° and 13.4 °; or
- Another aspect of the present invention is as follows: (18) After adding methanesulfonic acid to an acetone solution of the compound represented by the formula [1], the resulting crystals are collected by filtration and dried. It is a manufacturing method.
- another aspect of the present invention is as follows: (20) The salt crystal according to (19), which has at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 8.2 degrees, 10.9 degrees, 12.8 degrees, 14.7 degrees, 16.5 degrees and 19.2 degrees;
- another aspect of the present invention is as follows: (21) After adding benzenesulfonic acid to an acetone solution of the compound represented by the formula [1] and allowing it to act, the resulting crystals are collected by filtration and dried. It is a manufacturing method.
- the present invention it is possible to provide a crystal having excellent physical properties in the production and use environment of a compound represented by the formula [1] (hereinafter referred to as “compound [1]”) as a pharmaceutical product. It was.
- the crystal is in a stable crystal form at a temperature near room temperature and has excellent storage stability.
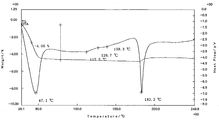
- 2 shows a differential thermal analysis / thermal mass measurement curve of Compound A crystal A.
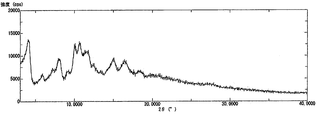
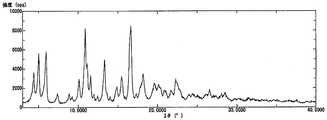
- 1 shows a powder X-ray diffraction pattern of a B-form crystal of Compound [1].
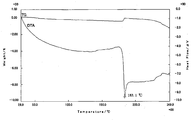
- 2 shows a differential thermal analysis / thermal mass measurement curve of a form B crystal of the compound [1].
- 2 shows an infrared absorption spectrum (ATR method, crystal: diamond) of a B-form crystal of compound [1].
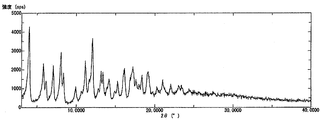
- 1 shows a powder X-ray diffraction pattern of a C-form crystal of Compound [1].
- 2 shows a differential thermal analysis / thermal mass measurement curve of a C-form crystal of compound [1].
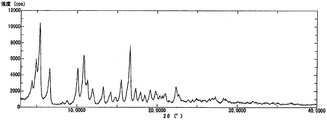
- 1 shows a powder X-ray diffraction pattern of a D-form crystal of Compound [1] hydrate.
- 2 shows a differential thermal analysis / thermal mass measurement curve of Compound [1] D-form crystal.
- 1 shows a powder X-ray diffraction pattern of Form F crystal of Compound [1] hydrate.
- 2 shows a differential thermal analysis / thermal mass measurement curve of Compound [1] hydrate F-form crystal.
- 1 shows a powder X-ray diffraction pattern of Form E crystal of Compound [1] hydrate.
- 2 shows a differential thermal analysis / thermal mass measurement curve of E type crystal of Compound [1] hydrate.
- 1 shows a powder X-ray diffraction pattern of crystals of compound [1] malonate.
- 1 shows a differential thermal analysis / thermal mass measurement curve of a crystal of compound [1] malonate.
- 1 shows a powder X-ray diffraction pattern of crystals of compound [1] methanesulfonate.
- 1 shows a differential thermal analysis / thermal mass measurement curve of a crystal of compound [1] methanesulfonate.
- 1 shows a powder X-ray diffraction pattern of crystals of compound [1] benzenesulfonate.
- 1 shows a differential thermal analysis / thermal mass measurement curve of a crystal of compound [1] benzenesulfonate. It is a figure which shows the result of the therapeutic example test in the test example 3 influenza virus infection animal.
- Test Example 4 shows the results of a therapeutic effect test in erythromycin resistant (erm (B) gene possessed) pneumococcal infected animals.
- Test Example 5 shows the results of a therapeutic effect test in erythromycin-resistant (mef (A) gene possessed) pneumococcal-infected animals.
- the compound [1] according to the present invention has the chemical structural formula shown above.
- the crystal of compound [1] (hereinafter sometimes referred to as “the crystal of the present invention”) is obtained as a single crystal having a certain quality with good reproducibility as described above, and is a drug substance used in the manufacture of pharmaceuticals. It can be stably supplied as crystals and has excellent storage stability.
- the crystal of compound [1] can be produced, for example, by the following method.
- room temperature refers to 20 to 30 ° C.
- the compound [1] can be obtained by dissolving the compound [1] in a predetermined solvent, then precipitating crystals, separating the precipitated crystals from the solvent by filtration, centrifugation, or the like, and drying.
- recrystallization may be repeated not only once but twice or more, it is usually recrystallized only once.
- seed crystals can be used for crystallization.
- the seed crystal can be obtained by a method well known to those skilled in the art, such as rubbing the wall of a container containing a solution for crystallization with a spatula.
- Form A crystal of compound [1] will be described below.
- Form A crystals of compound [1] have at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 4.1 degrees, 10.0 degrees, 10.6 degrees and 15.1 degrees; or
- the powder X-ray diffraction pattern of the form A crystal of the compound [1] is as shown in FIG. 1, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG.
- the characteristic peak by powder X-ray diffraction may change with measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray diffraction of the compound of the present invention.
- the A-form crystal of the compound [1] produced by the production method of the present invention is basically a high-purity crystal. High crystal purity is desirable, and is preferably substantially free of other crystal forms.
- the form A crystal of the compound [1] produced by the production method of the present invention can be obtained as a single crystal having a certain quality with good reproducibility. It can be stably supplied as a drug substance crystal used for the production of raw materials, and has physical properties excellent in storage stability.
- the form A crystal of compound [1] can be produced, for example, by the following method.
- Compound A is obtained by adding ethyl acetate, hexane or a mixture thereof to Compound [1] to form a solution, and then crystallizing and drying the resulting crystal to obtain Form A crystals of Compound [1].
- the starting compound [1] before being dissolved in the solvent is amorphous or crystalline.
- the solvent include ethyl acetate, hexane, or a mixed solution thereof.
- the mixing ratio in the mixed solution of ethyl acetate and hexane can be appropriately changed.
- Crystallization of Form A crystal of Compound [1] is usually performed at 0 ° C. to reflux temperature. Preferably, it is 20 ° C to 30 ° C.
- the precipitated Form A of compound [1] can be separated from the solvent by filtration, centrifugation or the like from the solution. Drying of Form A crystals of Compound [1] is usually performed at 100 ° C. or lower. Preferably, it is 20 ° C to 30 ° C.
- the form B crystal of the compound [1] has at least one of the following physical properties (a) to (c).
- A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 4.0 degrees, 7.1 degrees, 8.1 degrees and 12.1 degrees;
- the powder X-ray diffraction pattern of the B-form crystal of compound [1] is shown in FIG. 3, the differential thermal analysis / thermal mass measurement curve is shown in FIG. 4, and the infrared absorption spectrum (ATR method, crystal: diamond) is shown in FIG. Street.
- the characteristic peak by powder X-ray diffraction may change with measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray diffraction of the compound of the present invention.
- the characteristic peak of the infrared absorption spectrum may vary depending on the measurement conditions. Therefore, an error may occur or the peak of the infrared absorption spectrum of the compound of the present invention may not be clear.
- the form B crystal of the compound [1] produced by the production method of the present invention is basically a high purity crystal. High crystal purity is desirable, and is preferably substantially free of other crystal forms. Further, as shown in the examples described later, the B-form crystal of the compound [1] produced by the production method of the present invention can be obtained as a single crystal having a certain quality with good reproducibility. It can be stably supplied as a drug substance crystal used for the production of raw materials, and has physical properties excellent in storage stability.
- the form B crystal of compound [1] can be produced, for example, by the following method. Methanol or a water-methanol mixture is added to compound [1] to form a solution, which is then crystallized, and the resulting crystals are dried to obtain B-form crystals of compound [1].
- the starting compound [1] before being dissolved in the solvent is amorphous or crystalline.
- the solvent may be methanol or a water-methanol mixed solution as long as the compound [1] is dissolved.
- the mixing ratio in the water-methanol mixture can be changed as appropriate.
- Crystallization of the B-form crystal of compound [1] is usually performed at 0 ° C. to reflux temperature. Preferably, it is 20 ° C to 30 ° C.
- the precipitated B-form crystals of compound [1] can be separated from the solvent by filtration, centrifugation or the like from the solution. Drying of the B-form crystals of compound [1] is usually performed at 100 ° C. or lower. Preferably, it is 20 ° C to 30 ° C.
- the C-type crystal of compound [1] will be described below.
- the form C crystal of the compound [1] has at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 3.3 degrees, 4.6 degrees, 11.2 degrees and 15.5 degrees; or
- the powder X-ray diffraction pattern of the C-form crystal of compound [1] is as shown in FIG. 6, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG.
- the characteristic peak by powder X-ray diffraction may change with measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray diffraction of the compound of the present invention.
- the C-form crystal of the compound [1] produced by the production method of the present invention is basically a high-purity crystal. High crystal purity is desirable, and is preferably substantially free of other crystal forms. Further, as shown in the examples described later, the C-form crystal of the compound [1] produced by the production method of the present invention can be obtained with good reproducibility as a single crystal having a certain quality, and can be used for pharmaceuticals and pharmaceuticals. It can be stably supplied as a drug substance crystal used for the production of raw materials, and has physical properties excellent in storage stability.
- the C-type crystal of compound [1] can be produced, for example, by the following method. Water is added to form A crystals of compound [1] to form a suspension, and the crystals obtained by stirring are collected by filtration, separated from a solvent by centrifugation, etc., and dried to obtain C of compound [1]. Shape crystals can be obtained.
- the starting compound [1] before being dissolved in the solvent is a crystal.
- Specific examples of the predetermined solvent include water.
- Crystallization of the C-form crystal of compound [1] is usually carried out at 0 ° C to 100 ° C. Preferably, it is 20 ° C to 30 ° C.
- the precipitated C-form crystals of compound [1] can be separated from the solvent by filtration, centrifugation or the like from the solution.
- the C-form crystals of compound [1] are usually dried at 100 ° C. or lower. Preferably, it is 20 ° C to 30 ° C.
- the D form crystal of compound [1] hydrate will be described below.
- the D-form crystal of compound [1] hydrate has at least one of the following physical properties (a) to (b).
- (A) It has peaks at 2 ⁇ 5.4 degrees, 6.6 degrees, 10.9 degrees, and 16.6 degrees in powder X-ray diffraction (Cu—K ⁇ );
- the powder X-ray diffraction pattern of the D-form crystal of compound [1] hydrate is as shown in FIG. 8, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG.
- the characteristic peak by powder X-ray diffraction may change with measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray diffraction of the compound of the present invention.
- the D-form crystal of the hydrate of compound [1] produced by the production method of the present invention is basically a high-purity crystal. High crystal purity is desirable, and is preferably substantially free of other crystal forms. Further, as shown in the examples described later, the D-form crystal of the compound [1] hydrate produced by the production method of the present invention can be obtained with good reproducibility as a single crystal having a certain quality, It can be stably supplied as a drug substance crystal used in the manufacture of pharmaceuticals and pharmaceutical raw materials, and has physical properties excellent in storage stability.
- Compound [1] hydrate D-form crystals can be produced, for example, by the following method.
- Compound [1] is dissolved in a predetermined solvent, and then crystals are precipitated.
- the precipitated crystals are collected by filtration, separated from the solvent by centrifugation or the like, and then dried to form D crystals of compound [1] hydrate. Can be obtained.
- the starting compound [1] before being dissolved in the solvent is amorphous or crystalline.
- the solvent may be ethanol or a water-ethanol mixed solution as long as the compound [1] is dissolved.
- the mixing ratio in the water-ethanol mixed solution can be appropriately changed.
- Crystallization of D-form crystals of compound [1] hydrate is usually carried out at 0 ° C. to reflux temperature. Preferably, it is 20 ° C to 30 ° C.
- the precipitated D-form crystals of compound [1] hydrate can be separated from the solvent by filtration, centrifugation, or the like.
- the D-form crystals of compound [1] hydrate are usually dried at 100 ° C. or lower. Preferably, it is 20 ° C to 30 ° C.
- Form F crystals of compound [1] hydrate have at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 5.0 degrees, 5.9 degrees, 10.9 degrees and 16.7 degrees; or
- the powder X-ray diffraction pattern of the F-form crystal of compound [1] hydrate is as shown in FIG. 10, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG.
- the characteristic peak by powder X-ray diffraction may change with measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray diffraction of the compound of the present invention.
- the F-form crystal of the compound [1] hydrate produced by the production method of the present invention is basically a high-purity crystal. High crystal purity is desirable and preferably substantially free of other crystal forms. Further, as shown in the examples described later, the F-form crystal of the compound [1] hydrate produced by the production method of the present invention can be obtained with good reproducibility as a single crystal having a certain quality, It can be stably supplied as a drug substance crystal used in the manufacture of pharmaceuticals and pharmaceutical raw materials, and has physical properties excellent in storage stability.
- the F form crystal of compound [1] hydrate can be produced, for example, by the following method. Acetonitrile is added to compound [1] to form a solution, which is then crystallized. The obtained crystals are collected by filtration, separated from a solvent by centrifugation, etc., and dried to obtain F-form crystals of compound [1]. Can do.
- the starting compound [1] before being dissolved in the solvent is amorphous or crystalline.
- Crystallization of Form F crystals of Compound [1] hydrate is usually carried out at 0 ° C to 100 ° C. Preferably, it is 20 ° C to 30 ° C.
- the precipitated F-form crystals of compound [1] hydrate can be separated from the solvent by filtration, centrifugation, or the like. Drying of the F-form crystals of compound [1] hydrate is usually carried out at 100 ° C. or lower. Preferably, it is 20 ° C to 30 ° C.
- Form E crystals of compound [1] hydrate have at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 11.0 degrees, 11.3 degrees, 13.3 degrees and 16.8 degrees; or
- the powder X-ray diffraction pattern of the E-form crystal of compound [1] hydrate is as shown in FIG. 12, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG.
- the characteristic peak by powder X-ray diffraction may change with measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray diffraction of the compound of the present invention.
- the E-form crystal of the compound [1] hydrate produced by the production method of the present invention is basically a high-purity crystal. High crystal purity is desirable, and is preferably substantially free of other crystal forms.
- the E-form crystal of the compound [1] hydrate produced by the production method of the present invention can be obtained with good reproducibility as a single crystal having a certain quality, It can be stably supplied as a drug substance crystal used in the manufacture of pharmaceuticals and pharmaceutical raw materials, and has physical properties excellent in storage stability.
- the E-type crystal of compound [1] hydrate can be produced, for example, by the following method. After adding water to the A-form crystal, B-form crystal, C-form crystal, D-form crystal or F-form crystal, which will be described later, to form a suspension, the crystal obtained by stirring is filtered and centrifuged. Form E crystals of compound [1] hydrate can be obtained by separating from a solvent by separation or the like and then drying.
- Crystallization of the E-form crystal of compound [1] hydrate is usually carried out at 0 to 100 ° C. Preferably, it is 20 ° C to 30 ° C.
- the precipitated E-form crystals of compound [1] hydrate can be separated from the solvent by filtration, centrifugation, or the like.
- the E-form crystals of compound [1] hydrate are usually dried at 100 ° C. or lower. Preferably, it is 20 ° C to 30 ° C.
- the salt of the compound [1] according to the present invention is obtained as a single salt having a certain quality with good reproducibility, can be stably supplied as a salt used in the production of pharmaceuticals, and has storage stability. It is an excellent one.
- the salt of compound [1] can be produced, for example, by the following method.
- Compound [1] is dissolved in a predetermined solvent.
- a predetermined acid is added thereto, and the mixture is stirred for 1 hour to overnight, and then the precipitated crystals are collected by filtration, separated from a solvent by centrifugation, and then dried to obtain crystals of various salts of the compound [1]. .
- the predetermined solvent include organic solvents such as ethyl acetate, water and the like.
- organic solvent include ethyl acetate, ethanol, acetone, methyl t-butyl ether and the like.
- the salt of compound [1] is usually produced at 0 to 100 ° C. Preferably, it is 20 ° C to 30 ° C.
- the precipitated crystals of various salts of the compound [1] can be separated from the solvent by filtration, centrifugation, or the like from the solution. Drying of crystals of various salts of compound [1] is usually carried out at 100 ° C. or lower. Preferably, it is 20 ° C to 40 ° C.
- the crystals of compound [1] malonate will be described below.
- the crystal of compound [1] malonate has at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 8.5 degrees, 10.0 degrees and 15.6 degrees; or
- the powder X-ray diffraction pattern of the crystal of the compound [1] malonate is as shown in FIG. 14, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG. Note that the characteristic peak due to powder X-ray crystal diffraction may vary depending on the measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray crystal diffraction of the compound of the present invention.
- the crystals of the compound [1] malonate produced by the production method of the present invention are basically high-purity crystals.
- the purity of the crystal is desirably high, and is preferably substantially free from other crystal forms.
- the crystal of the compound [1] malonate produced by the production method of the present invention can be obtained with good reproducibility as a single crystal having a certain quality. It can be stably supplied as a drug substance crystal used in the production of pharmaceutical raw materials, and has physical properties excellent in storage stability.
- the crystal of compound [1] methanesulfonate has at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 9.7 °, 11.1 °, 12.9 ° and 13.4 °; or
- the powder X-ray diffraction pattern of the compound [1] methanesulfonate crystal is as shown in FIG. 16, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG. Note that the characteristic peak due to powder X-ray crystal diffraction may vary depending on the measurement conditions. Therefore, an error may occur or it may not be clear about the peak of the powder X-ray crystal diffraction of the compound of the present invention.
- the crystals of the compound [1] methanesulfonate produced by the production method of the present invention are basically high-purity crystals.
- the purity of the crystal is desirably high, and is preferably substantially free from other crystal forms.
- the crystals of the compound [1] methanesulfonate produced by the production method of the present invention can be obtained as a single crystal having a certain quality with good reproducibility, In addition, it can be stably supplied as a crystal of a drug substance used for manufacturing a pharmaceutical raw material, and has physical characteristics excellent in storage stability.
- the crystal of the compound [1] benzenesulfonate has at least one of the following physical properties (a) to (b).
- (A) In powder X-ray diffraction (Cu-K ⁇ ), it has peaks at 2 ⁇ 8.2 degrees, 10.9 degrees, 12.8 degrees, 14.7 degrees, 16.5 degrees and 19.2 degrees;
- the powder X-ray diffraction pattern of the compound [1] benzenesulfonate crystal is as shown in FIG. 18, and the differential thermal analysis / thermal mass measurement curve is as shown in FIG.
- the crystals of the compound [1] benzenesulfonic acid produced by the production method of the present invention are basically crystals of high purity.
- the purity of the crystal is desirably high, and is preferably substantially free from other crystal forms.
- the crystals of the compound [1] benzenesulfonic acid produced by the production method of the present invention can be obtained with good reproducibility as a single crystal having a certain quality. It can be stably supplied as a drug substance crystal used in the production of pharmaceutical raw materials, and has physical properties excellent in storage stability.
- antibacterial agent means a substance having the ability to act on bacteria such as gram positive bacteria, gram negative bacteria and mycoplasma to suppress or sterilize their growth. It may be something that suppresses the growth of bacteria or kills some bacteria to reduce their number.
- Gram-positive bacteria include, for example, Staphylococcus (S. aureus, Staphylococcus epidermidis, etc.), Streptococcus (S. pyogenes, Group B Streptococcus, Streptococcus pneumoniae, etc.), Enterococcus (Enterococcus faecalis, Enterococcus Fesium etc.).
- Gram-negative bacteria include, for example, Pseudomonas genus (such as Pseudomonas aeruginosa), Escherichia genus (such as Escherichia coli), Klebsiella (such as Klebsiella pneumoniae, Klebsiella oxytoca), Haemophilus (such as Haemophilus influenzae and Parainfluenza), Bordetella genus (Such as Bordetella pertussis and Bacterial sepsis), Serratia (such as Serratia marcescens), Proteus (such as Proteus mirabilis), Enterobacter (such as Enterobacter cloaca), Campylobacter (such as Campylobacter jejuni), Citrobacter Genus, Vibrio (Vibrio parahaemolyticus, Cholera, etc.), Morganella (Morganella, Morgani, etc.), Salmonella (Typhi, Paratyphi, etc.), Shig
- Mycoplasmas include M.Mgallisepticum, M. genitalium, M. hominis, M. hyopneumoniae, M. laboratorium, M. mycoides, M. ovipneumoniae, M.Mpneumonia.
- Compound [1] has excellent antibacterial activity against erythromycin-resistant bacteria (for example, resistant pneumococci, resistant streptococci, and mycoplasma) that have not been able to obtain sufficient antibacterial activity, particularly with conventional macrolide antibiotics. It has the feature of showing.
- erythromycin-resistant bacteria for example, resistant pneumococci, resistant streptococci, and mycoplasma
- Compound [1] may have optical isomers, but compound [1] includes these optical isomers and mixtures of optical isomers.
- the “solvent” of the “solvate” in the present invention includes, for example, water, polar solvents (for example, alcohol solvents such as methanol, ethanol, 1-propanol, 2-propanol, butanol, ethyl acetate, etc. Etc.), inert solvents (for example, halogenated hydrocarbon solvents such as chloroform or methylene chloride, ether solvents such as diethyl ether, tetrahydrofuran or dioxane, amide solvents such as dimethylformamide, dimethylacetamide, dimethyl sulfoxide, acetonitrile, etc.
- polar solvents for example, alcohol solvents such as methanol, ethanol, 1-propanol, 2-propanol, butanol, ethyl acetate, etc. Etc.
- inert solvents for example, halogenated hydrocarbon solvents such as chloroform or methylene chloride, ether solvents such as die
- Aprotic solvents aromatic hydrocarbons such as toluene, or hydrocarbons such as cyclohexane), 2-butanone, hexane, isopropyl ether, acetone, dichloromethane, etc., or a mixed solvent of the solvents exemplified here I mean, And it is not limited to these.
- Compound [1] or a salt thereof, or a hydrate or a solvate thereof exhibits excellent safety.
- the safety is evaluated by various tests, and can be evaluated by, for example, a cytotoxicity test, a hERG test, a cytochrome P450 (CYP) activity inhibition test, and the like.
- Compound [1] or a salt thereof, or a hydrate or a solvate thereof exhibits excellent metabolic stability.
- Metabolic stability is evaluated by various tests, and can be evaluated by, for example, a human liver microsomal metabolic stability test.
- Compound [1] or a salt thereof, or a hydrate or a solvate thereof may be combined with one or more pharmaceutically acceptable carriers, excipients or diluents to form a pharmaceutical preparation. it can.
- Compound [1] or a salt thereof, or a hydrate or a solvate thereof is prepared as a general pharmaceutical preparation.
- a pharmaceutical composition is prepared by mixing, dissolving, and / or dispersing with a pharmaceutically acceptable carrier (excipient, binder, disintegrant, corrigent, emulsifier, diluent, solubilizer, etc.).
- This pharmaceutical composition is suitable for oral or parenteral preparations such as tablets, pills, powders, granules, capsules, solutions, emulsions, suspensions, injections, suppositories, inhalants, and transdermal absorption agents. Administered in the form.
- Oral preparations include solid preparations and liquid preparations.
- the solid preparation in the present invention refers to a preparation having a form in which each element constituting the whole preparation or aggregate has at least a certain shape. Specific examples include tablets, pills, capsules, granules, powders, and powders.
- a capsule whose content is a liquid is included in a solid preparation when one capsule constituting the whole or an aggregate of a plurality of capsules has a certain shape.
- a dry syrup that is dissolved or suspended at the time of use is also included in the solid preparation when the whole preparation or individual particles of powder or granules have a certain shape at the time of storage.
- the liquid preparation in the present invention refers to a preparation that is dissolved or dispersed in a liquid solvent or dispersion medium from the time of storage to the time of administration and is handled as a liquid because it does not have a certain shape.
- excipients, diluents, binders, disintegrants, lubricants, antioxidants, stabilizers, preservatives, solvents, solubilizers, tonicity agents, etc. should be added. Can do.
- Examples of the pharmaceutically acceptable excipient or diluent include lactose, sucrose, glucose, maltose, fructose, mannitol, xylitol, sorbitol, erythritol, starch, starch, sodium carboxymethyl starch, powdered cellulose, crystalline cellulose , Carmellose, crystalline cellulose / carmellose sodium, hydroxypropyl cellulose, hydroxypropyl methylcellulose, calcium hydrogen phosphate, sodium hydrogen phosphate, potassium hydrogen phosphate, potassium dihydrogen phosphate, calcium carbonate, light anhydrous silicic acid, titanium oxide, Examples include magnesium aluminate metasilicate.
- binder examples include hydroxypropylcellulose, hypromellose, starch, starch, pregelatinized starch, partially pregelatinized starch, and polyvinylpyrrolidone.
- disintegrants include powdered cellulose, crystalline cellulose, carmellose, carmellose potassium, carmellose calcium, carmellose sodium, crystalline cellulose / carmellose sodium, croscarmellose sodium, low-substituted hydroxypropylcellulose, starch, and partial alpha. Modified starch, sodium carboxymethyl starch, povidone, crospovidone and the like.
- Examples of the lubricant include stearic acid, magnesium stearate, calcium stearate, polyoxyl stearate, talc, hydrogenated oil, sucrose fatty acid ester, cetanol, beeswax, and white beeswax.
- Examples of the antioxidant include dibutylhydroxytoluene (BHT), propyl gallate, butylhydroxyanisole (BHA), tocopherol, citric acid, edetate and the like.
- Examples of the solvent include water, physiological saline, and ethanol.
- Examples of the solubilizer include isotonic agents such as polyoxyethylene hydrogenated castor oil, polysorbates, sodium lauryl sulfate, macrogol, and sucrose fatty acid ester.
- Examples of the solubilizer include sodium chloride, citric acid, sodium citrate, glycerin, sorbitol, glucose, propylene glycol, macrogols, boric acid, borax, phosphoric acid,
- the dose of compound [1] or a salt thereof, or a hydrate or a solvate thereof is determined based on the results of animal experiments so that it does not exceed a certain amount when administered once and repeatedly. Based on the animal experiment data disclosed in the test examples, 1 to 10000 mg, preferably 5 to 1000 mg as a daily dose is administered to an adult patient once or several times a day orally or parenterally. It is assumed that Furthermore, the appropriate amount and the number of administrations can be determined by a specialist or the like in consideration of various factors such as the administration method, age, weight, sex, sensitivity, and the degree of symptoms of the patient or treated animal. Compound [1] can also be used in combination with other drugs.
- Powder X-ray diffraction was measured with a Rigaku RINT2200 Ultimate.
- Differential thermal analysis / thermal mass measurement was measured by Rigaku Thermo plus EvoTG8120.
- the infrared absorption spectrum was measured with Shimadzu Corporation IRAffinity-1.
- N-chlorosuccinimide 99.7 g was dissolved in chloroform (1 L) and cooled to ⁇ 25 ° C.
- a solution of dimethyl sulfide (210 mL) in chloroform (0.2 L) was added dropwise to the reaction mixture over 20 minutes, and the mixture was stirred for 15 minutes, and then the carbonate form chloroform (1 L) obtained in Reference Example 3- (2) above.
- the solution was added dropwise over 30 minutes and stirred for 15 minutes.
- a solution of triethylamine (136 mL) in chloroform (0.2 L) was added to the reaction solution, and the mixture was stirred for 30 minutes.
- Trimethylsulfoxonium iodide (210 g) was dissolved in a 5: 1 mixed solvent (1.2 L) of dimethylsulfoxide and tetrahydrofuran, 70% sodium hydride (32.6 g) was added little by little, and the mixture was brought to room temperature. And stirred for 1.5 hours. Under ice cooling, a solution of the ketone body (155 g) obtained in Reference Example 3- (3) above in tetrahydrofuran (0.8 L) was added dropwise and stirred at room temperature for 30 minutes. The reaction solution was ice-cooled, distilled water was added, the mixture was extracted with ethyl acetate, and the resulting organic layer was washed with distilled water.
- Ethyl acetate (50 mL) and hexane (600 mL) were added to the resulting crude product, and the mixture was stirred for 30 minutes, and the resulting solid was collected by filtration to obtain a deacetylated product (62.8 g).
- Example 2 Production of Form B Crystal of Compound [1] Methanol (19 mL) was added to and dissolved in the solid (1.0 g) of compound [1] obtained in Example 1- (3), and then water (9 0.5 mL) and stirred at room temperature overnight. The precipitated solid was collected by filtration and dried under reduced pressure to obtain crystals (619 mg) of the compound represented by the formula [1]. The powder X-ray diffraction pattern, differential thermal analysis / thermal mass measurement (TG / DTA), and infrared absorption spectrum of the obtained compound [1] crystal were measured, and it was a B-form crystal.
- Differential thermal analysis / thermal mass measurement (TG / DTA) was increased from room temperature to about 250 ° C. at 10 ° C./min in the atmosphere using a differential thermal balance (Thermo plus EVO TG8120) manufactured by Rigaku and equivalent equipment. Done at speed. As a result, an endothermic peak was observed at 181 to 186 ° C.
- the infrared absorption spectrum was measured using a Fourier transform infrared spectrophotometer (IRAffinity-1) manufactured by Shimadzu Corporation under the total reflection method (ATR method) with 20 integrations and a resolution of 4 cm-1. 1769cm -1, 1685cm -1, 1521cm -1 , 1458cm -1, a peak was observed near 1165 cm -1 and 1111cm -1.
- IRAffinity-1 Fourier transform infrared spectrophotometer manufactured by Shimadzu Corporation under the total reflection method (ATR method) with 20 integrations and a resolution of 4 cm-1. 1769cm -1, 1685cm -1, 1521cm -1 , 1458cm -1, a peak was observed near 1165 cm -1 and 1111cm -1.
- Example 3 Production of Form C Crystal of Compound [1]
- a solid (30 mg) of compound [1] obtained in Example 1- (3) was suspended in water (2 mL) (25 ° C.) for 10 days. Then, the mixture was centrifuged (3000 rpm, 10 minutes) to remove water, and the precipitate was dried at room temperature under reduced pressure.
- TG / DTA Differential thermal analysis / thermal mass measurement
- Example 4 Production of Form D Crystal of Compound [1] Hydrate After dissolving ethanol (6 mL) in the solid (1.0 g) of compound [1] obtained in Example 1- (3), Water (4.5 mL) was added and stirred at room temperature overnight. The precipitated solid was collected by filtration and dried under reduced pressure to obtain Form D crystals (612 mg) of Compound [1].
- TG / DTA Differential thermal analysis / thermal mass measurement
- Example 5 Production of Form E Crystal of Compound [1] Hydrate A solid (105 mg) of compound [1] obtained in Example 4 was suspended in water (10 mL) and stirred at room temperature for 3 days. The solid was collected by filtration and dried under reduced pressure to give Form E crystals (612 mg) of Compound [1].
- TG / DTA Differential thermal analysis / thermal mass measurement
- crystallization of compound [1] hydrate can be obtained also with the following method.
- Example 1 (A-form crystal), Example 2 (B-form crystal) and Example 4 (D-form crystal) (5 mg each) were weighed and mixed, then water (2 mL) was added and 10 days at 25 ° C. Shake and stir. Water was removed by centrifugation (3000 rpm, 10 minutes), and the precipitate was obtained by drying under reduced pressure for 1 day at room temperature.
- the powder X-ray diffraction pattern of the E-form crystal of the obtained compound [1] was measured using a Rigaku powder X-ray diffractometer (Ultima III) and Cu—K ⁇ ray as an X-ray source.
- the E-form crystal of compound [1] hydrate can be obtained by the following method.
- Example 3 (C-form crystals) and Example 4 (D-form crystals) (5 mg each) were weighed and mixed, then water (0.5 mL) was added, and the mixture was shaken and stirred at 25 ° C. for 5 days. Water was removed by centrifugation (3000 rpm, 10 minutes), and the precipitate was obtained by drying under reduced pressure for 1 day at room temperature.
- the powder X-ray diffraction pattern of the E-form crystal of the obtained compound [1] was measured using a Rigaku powder X-ray diffractometer (Ultima III) and Cu—K ⁇ ray as an X-ray source.
- the E-form crystal of compound [1] hydrate can be obtained by the following method.
- the title compounds Example 5 (E-form crystals) and Example 6 (F-form crystals) (5 mg each) were weighed and mixed, then 1 mL of water was added and the mixture was shaken and stirred at 25 ° C. for 3 weeks. Water was removed by centrifugation (3000 rpm, 10 minutes), and the precipitate was obtained by drying under reduced pressure for 1 day at room temperature.
- the powder X-ray diffraction pattern of the E-form crystal of the obtained compound [1] was measured using a Rigaku powder X-ray diffractometer (Ultima III) and Cu—K ⁇ ray as an X-ray source.
- Example 6 Preparation of Form F Crystal of Compound [1] Hydrate 30 ⁇ L of acetonitrile was added to the solid (20 mg) of compound [1] obtained in Example 1- (3) while heating at 80 ° C. to room temperature. And left to stand. The supernatant was removed, and the crystals were dried at room temperature with a vacuum dryer.
- TG / DTA Differential thermal analysis / thermal mass measurement
- Example 7 Production of Compound [1] Malonate Crystals Malonic acid (101 mg) was added to a solution of compound [1] (500 mg) in ethyl acetate (1.5 mL) at room temperature and stirred overnight. The precipitate was collected by filtration and dried at 40 ° C. under reduced pressure to give the title compound as colorless crystals (310 mg).
- the solid powder X-ray diffraction pattern and differential thermal analysis / thermal mass measurement were measured to find malonate crystals.
- Example 8 Production of Crystal of Compound [1] Methanesulfonate Salt methanesulfonic acid (29 ⁇ L) was added to a solution of compound [1] (302 mg) in acetone (4.5 mL) and stirred for 2 hours and 15 minutes. The reaction mixture was filtered, and the resulting residue was washed twice with acetone (2 mL) and dried at 40 ° C. under reduced pressure to give the title compound (213 mg) as colorless crystals.
- the solid powder X-ray diffraction pattern and differential thermal analysis / thermal mass measurement were measured to find crystals of methanesulfonate.
- the solid powder X-ray diffraction pattern and differential thermal analysis / thermal mass measurement were measured to find crystals of benzenesulfonate.
- Differential thermal analysis / thermal mass measurement (TG / DTA) using a differential thermal balance (Thermo plus EVO TG8120) manufactured by Rigaku and an equivalent device, from room temperature to about 250 ° C. in the atmosphere, at a rate of 10 ° C./min. Done at speed. As a result, an endothermic peak was observed at 208 to 214 ° C.
- Test Example 1 In Vitro Antibacterial Activity
- the compound [1] of Example 1 against various test bacteria was measured according to the micro liquid dilution method (CLSI method).
- the compound represented by the formula [3] in Reference Example 4 was also measured in the same manner.
- the test bacteria used are shown in Table 1.
- Table 2 shows the MIC values (microbe growth minimum inhibitory concentration ⁇ g / ml) for the test bacteria having the cell numbers A, B, C, D, E, F, G, H, I, J, K, and L.
- Test Example 2 Haemophilus susceptibility test Using 39 kinds of clinical isolates of Haemophilus influenzae, the drug sensitivity was evaluated using the same method as Test Example 1. Table 3 shows the results.
- Test Example 3 Therapeutic effect test in H. influenzae-infected animals The method shown below was used for evaluation of the pharmacological effect.
- Haemophilus influenzae ATCC43095 strain (cell number A) was used. The cells cultured overnight on a chocolate agar medium were scraped off, suspended in a hemophilus sensitivity test medium or a brain heart infusion medium supplemented with Phils enrichment and cultured overnight. This was diluted with a hemophilus sensitivity test culture medium or a brain heart infusion medium supplemented with Phils enrichment to obtain an inoculum.
- Mice ICR line, male, 4 weeks old) were infected by inoculating 0.05 ml of the inoculum in the respiratory tract.
- the amount of inoculum was 2.25 ⁇ 10 6 CFU / mouse or 9.00 ⁇ 10 5 CFU / mouse.
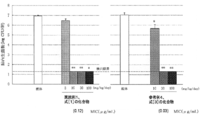
- the number of viable bacteria in the lung 3 days after the inoculation (6 cases per group, mean ⁇ standard error) is shown in FIG.
- the pulmonary viable count as a test result is expressed as a common logarithm of the pulmonary viable count (CFU / lung) (the common logarithm is hereinafter referred to as log).
- the viable bacterial count in the vehicle administration group was 5.88 ⁇ 0.14 [log (CFU / lung)].
- the number of living bacteria in the lungs of the compound [1] 100 and 200 mg / kg administration groups of Example 1 was 3.54 ⁇ 0.49 [log (CFU / lung)] and 2.83 ⁇ 0.53 [log ( CFU / lung)], which was significantly reduced compared to the vehicle administration group.
- the numbers of viable bacteria in the lungs of the compound 100 and 200 mg / kg administration groups of Reference Example 4 were 4.37 ⁇ 0.27 [log (CFU / lung)] and 2.53 ⁇ 0, respectively. .23 [log (CFU / lung)], which was significantly reduced compared to the vehicle administration group. From the above, the compound [1] of Example 1 showed the same therapeutic effect as the compound represented by Formula [3] of Reference Example 4 against the strain.
- Test Example 4 Treatment Effect Test in Erythromycin-Resistant (Erm (B) Gene Carrying) Pneumococcal Infected Animal
- Erm (B) Gene Carrying
- the evaluation of the pharmacological effect was carried out by the following method.
- Streptococcus pneumoniae 1101 strain (clinical isolate) was used.
- the cryopreservation solution of the strain used was added to Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum and cultured until the turbidity (OD600) was about 0.3. This was diluted with Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum to obtain an inoculum.
- mice CBA / JN system, male, 5 weeks old were infected with 0.05 mL of the inoculum by nasal inoculation.
- the amount of inoculum was 7.50 ⁇ 10 4 CFU / mouse or 1.65 ⁇ 10 5 CFU / mouse.
- the number of viable bacteria in the lung 3 days after the inoculation (5 to 6 cases per group, mean ⁇ standard error) is shown in FIG.
- the number of living bacteria in the lungs of the sputum medium administration group was 5.83 ⁇ 0.08 [log (CFU / lung)].
- the numbers of viable bacteria in the lungs of the compound [1] 30 and 100 mg / kg administration groups of Example 1 were 4.14 ⁇ 0.19 [log (CFU / lung)] and 2.28 ⁇ 0.24 [log ( CFU / lung)], which was significantly reduced compared to the vehicle administration group.
- Test Example 5 Therapeutic effect test in erythromycin resistant (mef (A) gene possessed) pneumococcal infected animals
- the evaluation of the pharmacological effect was carried out by the following method.
- Streptococcus pneumoniae 1028 strain (clinical isolate) was used.
- the cryopreservation solution of the strain used was added to Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum and cultured until the turbidity (OD600) was about 0.3. This was diluted with Todd Hewitt liquid medium supplemented with 30 vol% inactivated horse serum to obtain an inoculum.
- mice CBA / JN system, male, 5 weeks old were infected with 0.05 mL of the inoculum by nasal inoculation.
- the inoculum was 3.45 ⁇ 10 4 CFU / mouse or 3.90 ⁇ 10 4 CFU / mouse.
- the compound [1] of Example 1 (3, 10, 30 and 100 mg / kg) or vehicle (0.1 mol / L lactobionic acid solution and 0.5 w / v% sodium bicarbonate) once a day for 2 days from the day after the inoculation An equal volume of solution) was orally administered.
- the number of viable bacteria in the lung 3 days after inoculation (5 to 6 cases per group, mean ⁇ standard error) is shown in FIG.
- the number of viable bacteria in the group administered with sputum medium was 6.94 ⁇ 0.07 [log (CFU / lung)].
- the numbers of viable bacteria in the lungs of the compound [1] 3, 10, 30 and 100 mg / kg administration groups of Example 1 were 6.45 ⁇ 0.18 [log (CFU / lung)] and 1.30 ⁇ 0. 00 [log (CFU / lung)], 1.30 ⁇ 0.00 [log (CFU / lung)] and 1.30 ⁇ 0.00 [log (CFU / lung)], the compound of Example 1 [ 1]
- the number of viable bacteria in the lungs was below the detection limit value in all cases, and was significantly reduced as compared with the vehicle administration group.
- the compound of the present invention or a pharmaceutically acceptable salt thereof has strong antibacterial activity against gram-positive bacteria, gram-negative bacteria and mycoplasma, and in particular, sufficient antibacterial activity was not obtained with conventional macrolide antibiotics. Since it has excellent antibacterial activity against erythromycin-resistant bacteria (for example, resistant pneumococci, streptococci, and mycoplasma), it can be used as a pharmaceutical product.
- the compound [1] free form or salt crystals provided by the present invention can be obtained as a single crystal having a certain quality with good reproducibility and stable as crystals of the drug substance used in the manufacture of pharmaceuticals and pharmaceutical raw materials. It is useful as a drug substance because it has a physical property that is excellent in storage stability.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
La présente invention concerne, par exemple, une forme cristalline A, etc., d'un composé représenté par la formule [1], qui présente une propriété physique (a) et/ou une propriété physique (b), et présente d'excellentes propriétés physiques dans des environnements dans lesquels elle est produite ou utilisée en tant que médicament : (a) ayant des pics à 2θ de 4,1 degrés, 10,0 degrés, 10,6 degrés et 15,1 degrés en diffraction des rayons X sur poudre (Cu-Kα); ou (b) ayant un pic endothermique à 143-148 °C, comme mesuré en analyse thermique différentielle/mesure thermogravimétrique (ATD/TG).
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-027740 | 2016-02-17 | ||
| JP2016027737A JP2019064921A (ja) | 2016-02-17 | 2016-02-17 | C−4”位置換マクロライド化合物の結晶形及びそれらの製造方法 |
| JP2016027740A JP2019064922A (ja) | 2016-02-17 | 2016-02-17 | C−4”位置換マクロライド化合物の塩、それらの結晶形及びそれらの製造方法 |
| JP2016-027737 | 2016-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017142002A1 true WO2017142002A1 (fr) | 2017-08-24 |
Family
ID=59625119
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/005629 Ceased WO2017142002A1 (fr) | 2016-02-17 | 2017-02-16 | Formes cristallines de composé macrolide c-4"-substitué libre et sel de celui-ci, et leurs procédés de production |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017142002A1 (fr) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007019255A2 (fr) * | 2005-08-04 | 2007-02-15 | Novartis Ag | Nouveaux composes |
| WO2007141283A2 (fr) * | 2006-06-08 | 2007-12-13 | Boehringer Ingelheim International Gmbh | Nouveaux sels et formes de sels cristallins d'un dérivé de l'indolinone |
| WO2010072776A1 (fr) * | 2008-12-23 | 2010-07-01 | Boehringer Ingelheim International Gmbh | Formes de sel d'un composé organique |
| WO2010079045A2 (fr) * | 2008-12-15 | 2010-07-15 | Boehringer Ingelheim International Gmbh | Nouveaux sels |
| JP2014058509A (ja) * | 2012-08-20 | 2014-04-03 | Taisho Pharmaceutical Co Ltd | C−4”位置換マクロライド誘導体を含有する医薬 |
| US9139609B2 (en) * | 2011-02-21 | 2015-09-22 | Taisho Pharmaceutical Co., Ltd. | C-4″ position substituted macrolide derivative |
| WO2016027755A1 (fr) * | 2014-08-18 | 2016-02-25 | 大正製薬株式会社 | Composé de macrolide substitué en c-4'' |
-
2017
- 2017-02-16 WO PCT/JP2017/005629 patent/WO2017142002A1/fr not_active Ceased
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007019255A2 (fr) * | 2005-08-04 | 2007-02-15 | Novartis Ag | Nouveaux composes |
| WO2007141283A2 (fr) * | 2006-06-08 | 2007-12-13 | Boehringer Ingelheim International Gmbh | Nouveaux sels et formes de sels cristallins d'un dérivé de l'indolinone |
| WO2010079045A2 (fr) * | 2008-12-15 | 2010-07-15 | Boehringer Ingelheim International Gmbh | Nouveaux sels |
| WO2010072776A1 (fr) * | 2008-12-23 | 2010-07-01 | Boehringer Ingelheim International Gmbh | Formes de sel d'un composé organique |
| US9139609B2 (en) * | 2011-02-21 | 2015-09-22 | Taisho Pharmaceutical Co., Ltd. | C-4″ position substituted macrolide derivative |
| JP2014058509A (ja) * | 2012-08-20 | 2014-04-03 | Taisho Pharmaceutical Co Ltd | C−4”位置換マクロライド誘導体を含有する医薬 |
| WO2016027755A1 (fr) * | 2014-08-18 | 2016-02-25 | 大正製薬株式会社 | Composé de macrolide substitué en c-4'' |
Non-Patent Citations (1)
| Title |
|---|
| KAZUHIDE ASHIZAWA ET AL.: "Polymorphism and crystallization of the pharmaceutical drugs", 20 September 2002 (2002-09-20), pages 272 - 317 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0581552B1 (fr) | Hydrochlorure de triazolylthiométhylthiocéphalosporine, son hydrate cristallin et la préparation de celui-ci | |
| KR20150006082A (ko) | 거대고리 다형체, 이런 다형체를 포함하는 조성물, 및 이의 사용 및 제조방법 | |
| JPH09176182A (ja) | 新規なエリスロマイシン、それらの製造法及び薬剤としての用途 | |
| KR20010099806A (ko) | 아지트로마이신의 에탄올레이트, 이것의 제조 방법 및약학 조성물 | |
| BRPI0702875A2 (pt) | formas cristalinas de tigeciclina e seus processos de preparação | |
| EP3448375B1 (fr) | Dérivés de benzoylglycine et procédés de production et d'utilisation de ces dérivés | |
| EP3037428B1 (fr) | Composé macrolide | |
| US20160031925A1 (en) | Azithromycin antimicrobial derivatives with non-antibiotic pharmaceutical effect | |
| JP5874871B1 (ja) | C−4”位置換マクロライド化合物 | |
| WO2015056799A1 (fr) | Dérivé d'acide hydroxamique | |
| WO2017142002A1 (fr) | Formes cristallines de composé macrolide c-4"-substitué libre et sel de celui-ci, et leurs procédés de production | |
| WO2015056800A1 (fr) | Dérivé d'acide hydroxamique | |
| CN116514775B (zh) | 一种徳拉沙星葡甲胺盐新晶型及其制备方法 | |
| CN101558078B (zh) | 具有抗炎活性的大环内酯化合物 | |
| JP2017145247A (ja) | C−4”位置換マクロライド誘導体を含有する感染症の予防および/又は治療剤 | |
| JP2019064921A (ja) | C−4”位置換マクロライド化合物の結晶形及びそれらの製造方法 | |
| JP2009516704A (ja) | 新規なプリューロムチリン誘導体およびその使用 | |
| CN100427499C (zh) | 一种阿奇霉素的可溶性盐及其制备方法 | |
| JP2019064922A (ja) | C−4”位置換マクロライド化合物の塩、それらの結晶形及びそれらの製造方法 | |
| JP2016199499A (ja) | (2s)−2−メチルアミノ−n−ヒドロキシ−n’,2−ジメチルプロパンジアミドを有する化合物の結晶形及びそれらの製造方法 | |
| CN101233141A (zh) | 子囊霉素晶形及其制备 | |
| WO2008050871A1 (fr) | Composés de carbapénème cristallin | |
| CN117466958A (zh) | 假三糖氨基糖苷及其中间体的大规模制备 | |
| WO2006121151A1 (fr) | Cristal de compose 1-methylcarbapenem | |
| WO2015056798A1 (fr) | Dérivé d'acide hydroxamique |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17753258 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17753258 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |