WO2020171073A1 - ベンゾアゼピン誘導体の製造方法及びその中間体 - Google Patents
ベンゾアゼピン誘導体の製造方法及びその中間体 Download PDFInfo
- Publication number
- WO2020171073A1 WO2020171073A1 PCT/JP2020/006305 JP2020006305W WO2020171073A1 WO 2020171073 A1 WO2020171073 A1 WO 2020171073A1 JP 2020006305 W JP2020006305 W JP 2020006305W WO 2020171073 A1 WO2020171073 A1 WO 2020171073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- compound
- substituted
- halogen atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 NCC*(CCC1O)c(cccc2)c2C1=O Chemical compound NCC*(CCC1O)c(cccc2)c2C1=O 0.000 description 1
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/16—Benzazepines; Hydrogenated benzazepines
Definitions
- the present invention relates to a novel method for producing an optically active benzazepine derivative and an intermediate thereof.
- Patent Document 1 discloses that 4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide is a compound having a V2 receptor agonizing effect and is effective in preventing or treating nocturia. ing.
- Patent Document 1 N-[(S)-1-hydroxypropan-2-yl]-4-methyl-1-[2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl]-2,3
- the method for producing 4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide is described in Patent Document 1.
- the compound is characterized in that it has a methyl group at the carbon located at the ⁇ -position of the ester group at the 4-position of the benzoazepine ring.
- (R)- An optical resolution method using Phenylglycinol is known. Further, intermediates of the above compounds are reported in Patent Document 2, Patent Document 3 and Non-Patent Document 1.
- Patent Document 4 As a method for asymmetric methylation of the carbon located at the ⁇ -position of the ester group, a method of alkylating the ⁇ -carbon of ⁇ -ketoester stereoselectively is known (Patent Document 4).
- the present inventors have conducted an optical resolution method using (R)-Phenylglycinol described in Patent Document 1 to obtain N-[(S)-1-hydroxypropan-2-yl]-4-methyl-1-[2
- An optically active form of -methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl]-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide was prepared.
- there is a problem of inefficiency because half of the compound having an undesired three-dimensional structure is produced.
- an object of the present invention is to provide an optically active N-[(S)-1-hydroxypropan-2-yl]-4-methyl-1-[2-methyl-4-(3-methyl-1H-pyrazole- It is to provide a novel production method capable of efficiently producing 1-yl)benzoyl]-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide.
- the main configuration of the present invention is as follows.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy optionally substituted with a group
- C 7 Represents a C 12 aralkyl group.
- R 2 is a hydrogen atom, a C 2 -C 6 alkenyl group, a C 1 -C 4 alkyl group or a C 7 -C 12 aralkyl group which may be substituted with a C 1 -C 4 alkoxy group, —COR 3 , — It represents SO 2 R 4 or —CO 2 R 5 .
- R 3 is selected from the group consisting of a halogen atom, an aromatic heterocyclic group optionally substituted with a C 1 -C 4 alkyl group, and a C 1 -C 6 alkyl group optionally substituted with a halogen atom.
- R 4 represents a C 1 -C 4 alkyl group or a phenyl group which may be substituted with a C 1 -C 4 alkoxy group, or a C 1 -C 6 alkyl group which may be substituted with a halogen atom.
- R 5 is substituted with a C 1 -C 6 alkyl group, a C 2 -C 6 alkenyl group, a C 1 -C 4 alkyl group or a C 1 -C 4 alkoxy group which may be substituted with a halogen atom. Also represents a C 7 to C 12 aralkyl group.
- each R 6 independently represents a C 1 -C 4 alkyl group optionally substituted by a halogen atom, or an aryl group optionally substituted by a halogen atom.
- R 7's each independently represent a C 1 -C 4 alkylene group or a naphthalenediyl group.
- R 8 represents a single bond hydrogen atom or together.
- X ⁇ represents a halide anion.
- Formula (V) [In formula, R ⁇ 2 > is synonymous with the above and * represents an asymmetric center. ] Or a method for producing a solvate of the optically active compound, which comprises recrystallizing the optically active compound with a solvent.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents.
- R 2 is a hydrogen atom, an allyl group, a C 1 -C 4 alkyl group or a benzyl group which may be substituted with a C 1 -C 4 alkoxy group, —COR 3 , —SO 2 R 4 , or —CO 2 R Represents 5 .
- R 3 is selected from the group consisting of a halogen atom, an aromatic heterocyclic group optionally substituted with a C 1 -C 4 alkyl group, and a C 1 -C 6 alkyl group optionally substituted with a halogen atom.
- R 4 represents a phenyl group which may be substituted with a C 1 -C 4 alkyl group.
- R 5 is optionally substituted by a halogen atom C 1 ⁇ C 4 alkyl group, an allyl group, or a C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represent ]
- a halogen atom C 1 ⁇ C 4 alkyl group, an allyl group, or a C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represent ]
- an organic solvent the compound represented by
- Formula (II-1) Methylated with a methylating agent in the presence of an asymmetric quaternary ammonium salt represented by, and a base, Formula (III): [In the formula, R 1 and R 2 have the same meanings as described above, and * represents an asymmetric center. ]
- R 1 is a methyl group or an ethyl group.
- R 2 is —COR 3 or —SO 2 R 4 , and R 3 is a halogen atom, an aromatic heterocyclic group which may be substituted with a C 1 -C 4 alkyl group, and a halogen atom.
- R 2 is —COR 3
- R 3 is [In formula, R ⁇ 9 > means the aromatic heterocyclic group which may be substituted by the methyl group. ]
- R 2 is —COR 3
- R 3 is The production method according to [12], which is a substituted phenyl group represented by: and the steric acid at the 4-position of the benzazepine ring of the compound represented by formula (III) is R.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents.
- a pharmaceutically acceptable salt thereof
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents.
- a pharmaceutically acceptable salt thereof [17] The compound or a pharmaceutically acceptable salt thereof according to [15] or [16], wherein R 1 is a methyl group or an ethyl group.
- the optically active N-[(S)-1-hydroxypropan-2-yl]-4-methyl-1-[2-methyl-4-(3-methyl-1H-pyrazole-1- Il)benzoyl]-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide preferably (S)-N-[(S)-1-hydroxypropan-2-yl]- 4-Methyl-1-[2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl]-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide (Hereinafter, referred to as compound A.
- compound (I), compound (II) or the like a compound represented by formula (I), formula (II) or the like is hereinafter referred to as compound (I), compound (II) or the like). It can.
- compound (V) and compound (VI) which are synthetic intermediates of compound A
- the chemical purity and the yield of the optically active substance are improved, and the number of production steps is shortened in the production of compound A. can do.
- the asymmetric methylation reaction can be accelerated.
- compound A can be produced via the steps shown below.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy optionally substituted with a group C 7 Represents a C 12 aralkyl group.
- R 2 is a hydrogen atom, a C 2 -C 6 alkenyl group, a C 1 -C 4 alkyl group or a C 7 -C 12 aralkyl group which may be substituted with a C 1 -C 4 alkoxy group, —COR 3 , — It represents SO 2 R 4 or —CO 2 R 5 .
- R 3 represents a phenyl group which may be substituted with one or more substituents selected from the substituent group A, a C 1 -C 6 alkyl group which may be substituted with a halogen atom, or a hydrogen atom.
- R 4 represents a C 1 -C 4 alkyl group or a phenyl group which may be substituted with a C 1 -C 4 alkoxy group, or a C 1 -C 6 alkyl group which may be substituted with a halogen atom.
- R 5 is substituted with a C 1 -C 6 alkyl group, a C 2 -C 6 alkenyl group, a C 1 -C 4 alkyl group or a C 1 -C 4 alkoxy group which may be substituted with a halogen atom.
- a C 7 to C 12 aralkyl group also represents a C 7 to C 12 aralkyl group. * Represents an asymmetric center.
- the substituent group A is composed of a halogen atom, an aromatic heterocyclic group which may be substituted with a C 1 to C 4 alkyl group, and a C 1 to C 6 alkyl group which may be substituted with a halogen atom. It is a group. ]
- halogen atom means a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom.
- aromatic heterocyclic group means a monocyclic to tricyclic aromatic ring group containing at least one hetero atom such as nitrogen, oxygen or sulfur, and examples thereof include pyridyl group, thienyl group and furyl group.
- the “C 1 -C 6 alkyl group” means a linear or branched saturated hydrocarbon group having 1 to 6 carbon atoms. Examples thereof include methyl group, ethyl group, propyl group, butyl group, pentyl group, hexyl group, and various branched chain isomers thereof.
- the “C 1 -C 4 alkyl group” means a linear or branched saturated hydrocarbon group having 1 to 4 carbon atoms, for example, a methyl group, an ethyl group, a propyl group, a butyl group, And various branched chain isomers thereof.
- the “C 1 -C 4 alkoxy group” means a group in which an oxygen atom is bonded to the above “C 1 -C 4 alkyl group”, and examples thereof include a methoxy group, an ethoxy group, a propoxy group, a butoxy group, and these groups. And various branched chain isomers thereof.
- the “C 7 -C 12 aralkyl group” means a group in which an aryl group is bonded to an alkyl group and has 7 to 12 carbon atoms, and examples thereof include a benzyl group and a phenethyl group.
- the “C 2 -C 6 alkenyl group” means a linear or branched unsaturated hydrocarbon group having 2 to 6 carbon atoms and having at least one double bond, for example, vinyl group. , Allyl group, 1-propenyl group and the like.
- One embodiment of the present invention is a compound represented by formula (VI).
- Compound (VI) is a novel synthetic intermediate of compound A.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents. ]
- Compound (VI) can be obtained by the following first step. That is, R 2 of compound (I) is —COR 3 and R 3 is
- the compound (VI) can be obtained by In the compound (VI), the stereo structure at the 4-position of the benzazepine ring is controlled to the R configuration.
- the absolute configuration at the 4-position of the benzazepine ring of compound (VI) is determined by synthesizing compound A from compound (VI), converting it to an alcohol solvate, and performing single crystal X-ray structural analysis. be able to.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy optionally substituted with a group C 7 Represents a C 12 aralkyl group.
- R 2 is a hydrogen atom, a C 2 -C 6 alkenyl group, a C 1 -C 4 alkyl group or a C 7 -C 12 aralkyl group which may be substituted with a C 1 -C 4 alkoxy group, —COR 3 , — It represents SO 2 R 4 or —CO 2 R 5 .
- R 3 represents a phenyl group which may be substituted with one or more substituents selected from the substituent group A, a C 1 -C 6 alkyl group which may be substituted with a halogen atom, or a hydrogen atom.
- R 4 represents a C 1 -C 4 alkyl group or a phenyl group which may be substituted with a C 1 -C 4 alkoxy group, or a C 1 -C 6 alkyl group which may be substituted with a halogen atom.
- R 5 is substituted with a C 1 -C 6 alkyl group, a C 2 -C 6 alkenyl group, a C 1 -C 4 alkyl group or a C 1 -C 4 alkoxy group which may be substituted with a halogen atom. Also represents a C 7 to C 12 aralkyl group.
- the substituent group A is composed of a halogen atom, an aromatic heterocyclic group which may be substituted with a C 1 to C 4 alkyl group, and a C 1 to C 6 alkyl group which may be substituted with a halogen atom. It is a group. ]
- the first step is a step of asymmetrically methylating the compound (I).
- Compound (I) can be produced according to a known method described in JP-A-2016-40338 or JP-A-2016-185993.
- the R 1 is preferably a methyl group or an ethyl group, more preferably an ethyl group.
- the R 2 is preferably —COR 3 or —SO 2 R 4 , and more preferably —COR 3 .
- R 3 is preferably a phenyl group which may be substituted with one or more substituents selected from the substituent group A.
- the substituent group A is preferably an aromatic heterocyclic group which may be substituted with a C 1 -C 4 alkyl group or a C 1 -C 6 alkyl group which may be substituted with a halogen atom, and C 1 An aromatic heterocyclic group which may be substituted with a to C 4 alkyl group is more preferable.
- R 3 is a phenyl group shown below.
- R 4 is preferably a phenyl group which may be substituted with a C 1 -C 4 alkyl group or a C 1 -C 4 alkoxy group.
- R 5 is preferably a C 1 -C 6 alkyl group optionally substituted with a halogen atom or a C 7 -C 12 aralkyl group optionally substituted with a C 1 -C 4 alkoxy group.
- the first step can be carried out, for example, by methylating the 4-position of the benzazepine ring of compound (I) with an methylating agent in the presence of a catalyst and a base in an organic solvent.
- This methylation reaction is preferably carried out in the presence of an inorganic salt in order to accelerate the reaction.
- the crude product can be isolated and purified by a method well known to those skilled in the art. This method is excellent in stereoselectivity, and the compound (III) thus obtained has excellent optical purity. Furthermore, since the compound (III) can be obtained in good yield by this method, it is suitable for industrial production.
- the catalyst used in the first step is the compound (II) of an asymmetric quaternary ammonium salt.
- R 6's each independently represent a C 1 -C 4 alkyl group optionally substituted with a halogen atom or an aryl group optionally substituted with a halogen atom.
- R 7's each independently represent a C 1 -C 4 alkylene group or a naphthalenediyl group.
- R 8 represents a hydrogen atom or together represents a single bond.
- X ⁇ represents a halide anion.
- R 6 is preferably a 3,5-bistrifluoromethylphenyl group or a 3,4,5-trifluorophenyl group, more preferably a 3,5-bistrifluoromethylphenyl group.
- R 7 is preferably a normal butanediyl group or a naphthalenediyl group, more preferably a naphthalenediyl group. It is preferable that the R 8 s together form a single bond.
- the compound (II) of the asymmetric quaternary ammonium salt include (R,R)-3,5-bistrifluoromethylphenyl-NAS bromide and (R,R)-3,4 as follows. ,5-Trifluorophenyl-NAS bromide, (11bR)-( ⁇ )-4,4-dibutyl-4,5-dihydro-2,6-bis(3,5-ditrifluoromethylphenyl-3H-dinaphtho[2 ,1-c:1′,2′-e]azepinium bromide, and (11bR)-( ⁇ )-4,4-dibutyl-4,5-dihydro-2,6-bis(3,4,5) -Trifluorophenyl-3H-dinaphtho[2,1-c:1',2'-e]azepinium bromide, preferably (R,R)-3,5-bistrifluoromethylphenyl-NAS bromide.
- the compound (II) of the asymmetric quaternary ammonium salt can be used in an amount of 0.0001 to 1 equivalent based on 1 equivalent of the compound (I), but 0.001 to 0.1 equivalent. It is preferably used, and more preferably 0.005 to 0.02 equivalent. Setting the amount of the compound (II) of the asymmetric quaternary ammonium salt to be within this range contributes to completion of asymmetric methylation and suppression of formation of a by-product of a methylated product.
- the organic solvent used in the first step is not particularly limited as long as it does not hinder the progress of the reaction, for example, toluene, xylene, mesitylene, cumene, heptane, ethyl acetate, methyl isobutyl ketone, methyl tertiary butyl ether, Tetrahydrofuran, methylene chloride, chlorobenzene, bromobenzene, nitrobenzene, anisole, 1-butyl-3-methylimidazolium tetrafluoroborate and the like can be mentioned. These solvents may be used alone, or two or more kinds may be mixed or used with water.
- a highly soluble aromatic organic solvent is preferable, and toluene, xylene, chlorobenzene, or bromobenzene is more preferable. By using these solvents, generation of impurities can be suppressed and the asymmetric methylation reaction can be completed.
- the amount of the solvent used is preferably 3 to 20 equivalents, and more preferably 5 to 10 equivalents, relative to 1 equivalent of compound (I).
- the base used in the first step may be an organic base or an inorganic base.
- the organic base include triethylamine, pyridine, N,N'-dimethylaminopyridine and the like.
- the inorganic base include sodium hydroxide, potassium hydroxide, lithium hydroxide, cesium hydroxide, barium hydroxide, rubidium hydroxide, potassium carbonate, cesium carbonate, rubidium carbonate and the like.
- An inorganic base is preferred, and sodium hydroxide, potassium hydroxide, lithium hydroxide, cesium hydroxide, barium hydroxide, rubidium hydroxide, potassium carbonate, cesium carbonate, or rubidium carbonate is preferred. More preferably potassium hydroxide, potassium carbonate, cesium carbonate, or cesium hydroxide.
- the methylating agent used in the first step is not particularly limited as long as it is a commonly used methylating agent, and is preferably trimethyloxonium tetrafluoroborate, methyl trifluoromethanesulfonate, dimethylsulfate, methyl bromide, or iodide. Methyl, more preferably methyl bromide or methyl iodide.
- the amount of the methylating agent used is preferably 1 to 20 equivalents, and more preferably 1 to 10 equivalents, relative to 1 equivalent of compound (I).
- the inorganic salt used in the first step is preferably an alkali metal salt, for example, cesium fluoride, cesium chloride, cesium bromide, cesium iodide, lithium chloride, lithium bromide, potassium chloride, potassium bromide, potassium iodide. , Sodium chloride, sodium bromide, rubidium chloride, and rubidium bromide. More preferably, it is cesium fluoride, cesium chloride, potassium bromide, sodium bromide, rubidium chloride, or rubidium bromide.
- the amount of the inorganic salt used is preferably 0.01 to 1 equivalent, and more preferably 0.05 to 0.5 equivalent, relative to 1 equivalent of compound (I).
- the reaction temperature in the first step is not particularly limited, but is preferably ⁇ 20 to 20° C., more preferably 1 to 10° C. By adjusting the reaction temperature within this range, it contributes to the completion of asymmetric methylation and the suppression of the formation of by-products in the methylated form.
- the compound (III) obtained in the first step can be isolated by a conventional method. For example, it can be purified by recrystallizing the obtained crude product or by column chromatography.
- the solvent used for recrystallization is preferably methanol, ethanol, 2-propanol, ethyl acetate, toluene, or acetonitrile, and more preferably ethyl acetate or acetonitrile.
- Compound (VII) is a novel synthetic intermediate of Compound A.
- R 1 is optionally substituted by a halogen atom C 1 ⁇ C 6 alkyl group, or C 1 ⁇ C 4 alkyl or C 1 ⁇ C 4 alkoxy benzyl group which may be substituted with a group Represents.
- Compound (VII) can be obtained by the following second step.
- R 2 is the same as the compound (VI) described above.
- the stereo structure at the 4-position of the benzazepine ring does not change.
- [Second step] [In the formula, R 1 and R 2 have the same meanings as those in the first step, and * represents an asymmetric center. ]
- the second step is a step of reducing compound (III) to compound (IV).
- it can be obtained by reducing the carbonyl group at the 5-position of the benzazepine ring of compound (III) using an reducing agent in an organic solvent or a mixed solvent with water.
- the reducing agent used in the second step is preferably a borane compound, which allows selective reduction of carbonyl groups.
- the borane compound is preferably sodium borohydride, potassium borohydride, lithium borohydride, or a borane-ammonia complex, more preferably a borane-ammonia complex.
- the borane-ammonia complex those prepared in advance by the method described in the literature (Inorganic Chemistry 2007, 46, 7810) can be used.
- the amount of borane compound used is preferably 0.5 to 2 equivalents, and more preferably 1.0 to 1.5 equivalents, relative to 1 equivalent of compound (III).
- the organic solvent used in the reaction is preferably toluene, tetrahydrofuran, ethyl acetate, or a mixed solution of these organic solvents and water, more preferably a mixed solution of toluene and water.
- the reaction temperature in the second step is not particularly limited, but is preferably 20 to 100°C, more preferably 40 to 60°C. By adjusting the reaction temperature of the second step within this range, the reduction reaction proceeds smoothly and carbonyl group-selective reduction can be performed. Isolation and purification of the resulting compound (IV) can be carried out by a conventional method.
- the compound A can be obtained as the compound (V) from the compound (IV) through the following third step to sixth step.
- the steric structure at the 4-position of the benzazepine ring does not change.
- the third step is a step of chlorinating the hydroxyl group at the 5-position of the benzazepine ring in an organic solvent.
- the chlorination reaction can be carried out, for example, by dissolving the compound (IV) in an organic solvent and adding a chlorinating agent to cause the reaction.
- a chlorinating agent an electrophilic chlorinating agent is preferable. It is also preferable to carry out chlorination in the presence of a base.
- the chlorinating agent is preferably thionyl chloride, oxalyl chloride, phosphorus oxychloride, phosphorus trichloride, phosphorus pentachloride or N-chlorosuccinimide, more preferably thionyl chloride or phosphorus oxychloride.
- the amount of the chlorinating agent used is preferably 0.5 to 3 equivalents, and more preferably 1 to 2 equivalents, relative to 1 equivalent of compound (IV).
- the organic solvent used in the reaction is preferably toluene, methylene chloride, or chloroform, more preferably toluene.
- the reaction temperature is preferably 20 to 120°C, more preferably 60 to 100°C.
- the compound (VIII) thus obtained can be isolated and purified by a conventional method.
- the fourth step is a step of reductively dechlorinating the chloro group at the 5-position of the benzoazepine ring with hydrogen in an organic solvent in the presence of a metal catalyst.
- the dechlorination can be carried out, for example, by dissolving the compound (VIII) in an organic solvent, adding a metal catalyst to the resulting solution, and further carrying out the reaction under a hydrogen gas atmosphere.
- the metal catalyst is preferably a palladium catalyst, a platinum catalyst, or a nickel catalyst.
- the palladium catalyst include palladium carbon and palladium hydroxide.
- platinum catalysts include platinum carbon and platinum oxide hydrate.
- the nickel catalyst include Raney nickel and nickel carbon.
- the metal catalyst is more preferably a palladium catalyst.
- the amount of the metal catalyst used is preferably 0.01 to 1 equivalent, and more preferably 0.05 to 0.2 equivalent, relative to 1 equivalent of compound (VIII).
- the organic solvent used in the reaction is preferably methanol, ethanol, tetrahydrofuran, or ethyl acetate, more preferably methanol or ethanol.
- the hydrogen pressure is preferably 1 to 5 atm, more preferably 1 to 3 atm.
- the reaction temperature is not particularly limited, but is preferably 20 to 100°C, more preferably 20 to 60°C.
- Isolation and purification of the resulting compound (IX) can be carried out by a conventional method.
- the carbonyl group at the 5-position of the benzazepine ring can be regioselectively reduced.
- the reaction can be performed according to a conventional method.
- compound (IX) is treated with a base (eg, sodium hydroxide, potassium hydroxide, etc.) in a suitable solvent (eg, alcohol solvent such as methanol, ethanol, etc., water), usually at room temperature to an organic solvent.
- a base eg, sodium hydroxide, potassium hydroxide, etc.
- a suitable solvent eg, alcohol solvent such as methanol, ethanol, etc., water
- the carboxylic acid compound of the compound (X) can be obtained by reacting at the boiling point of the solvent for 30 minutes to 1 day.
- the obtained carboxylic acid compound is subjected to amidation with L-alaninol to obtain the compound (V).
- a method using a condensing agent for example, the carboxylic acid compound and L-alaninol are condensed in a suitable organic solvent (chloroform, dimethylformamide, etc.) in the presence of a base (eg, diisopropylethylamine, triethylamine, etc.) (for example, 1 , 3-dicyclohexylcarbodiimide (DCC), 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC), etc.) alone or in combination with 1-hydroxybenztriazole (HOBt).
- a base eg, diisopropylethylamine, triethylamine, etc.
- a mixed acid anhydride for example, a carboxylic acid compound in an appropriate organic solvent (eg, dichloromethane, toluene, etc.) in the presence of a base (eg, pyridine, triethylamine, etc.), an acid chloride (eg, pivaloyl chloride, Tosyl chloride, etc.) or an acid derivative (eg, ethyl chloroformate, isobutyl chloroformate, etc.), and the resulting mixed acid anhydride is reacted with L-alaninol usually at 0° C. to room temperature to give compound (V). Can be obtained.
- an appropriate organic solvent eg, dichloromethane, toluene, etc.
- a base eg, pyridine, triethylamine, etc.
- an acid chloride eg, pivaloyl chloride, Tosyl chloride, etc.
- an acid derivative eg, ethyl chloroformat
- an acid chloride is obtained by using a chlorinating agent (eg, thionyl chloride, oxalyl chloride, etc.) in a suitable organic solvent (eg, toluene, xylene, etc.)
- a chlorinating agent eg, thionyl chloride, oxalyl chloride, etc.
- a suitable organic solvent eg, toluene, xylene, etc.
- Acid chloride in the presence of a base eg sodium carbonate, triethylamine etc.
- a suitable organic solvent eg ethyl acetate, toluene etc.
- L-alaninol usually at 0° C. to room temperature
- Compound (V) can also exist as a solvate.
- the solvate of compound (V) can be obtained by a conventional method for producing a solvate. Specifically, it can be obtained by mixing the compound (V) with a solvent while heating, if necessary, and then cooling the mixture while stirring or leaving it to crystallize. It is desirable that the cooling be carried out while adjusting the cooling rate as necessary in consideration of the influence on the crystal quality and grain size. For example, cooling at a cooling rate of 20 to 1° C./hour is preferable, and cooling at a cooling rate of 10 to 3° C./hour is more preferable.
- the organic solvent used in these methods is preferably an alcohol solvent such as methanol, ethanol, propanol, isopropanol, normal propanol, and tertiary butanol.
- the amount of the organic solvent used is preferably 3 to 20 times by weight, more preferably 5 to 10 times by weight, relative to the compound (V).
- Compound A can be prepared as a pharmaceutical preparation by mixing with a pharmaceutically acceptable additive. These can be prepared by a known method (for example, the method described in the general rules for formulation of Japanese Pharmacopoeia 17th edition).
- Compound A acts as a V2 receptor agonist, it is a pharmaceutical composition for the prevention or treatment of central diabetes insipidus, nocturnal enuresis, nocturia, overactive bladder, hemophilia, or von Willebrand disease. It is already known that can be used as.
- Thermogravimetric measurement The TGA thermogram was performed using Perkin Elmer (Pyris1-TGA). The measurement conditions were 20 mL/min of nitrogen gas, isothermal for 1 minute at an initial temperature of 80° C., then increased to 170° C. at 5° C./minute, and isothermal for 1 minute at 170° C.
- Example 12 1420 mL of acetonitrile was added to the crude wet crystals (72% ee), and the mixture was heated and dissolved at 60°C and then stirred at 5°C for 3 hours. The precipitated crystals were collected by filtration, washed with 230 mL of cooled acetonitrile, and then dried at 50° C. overnight to obtain 134 g of a white solid (99% ee).
- the 1 H-NMR and LC-MS measurement results of the product obtained in Example 12 were the same as those shown in Example 1.
- Example 13 4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4,5-tetrahydro-1H- Synthesis of benzo[b]azepine-4-carboxylic acid t-butyl ester (first step) [In the formula, * represents an asymmetric center. ]
- Example 16 The asymmetric yield of Example 16 was found to be 1-t-butyl-4-ethyl-4-methyl-5-oxo-2,3,4,5-tetrahydro-1H-benzo[b]azepinedicarboxylate obtained. Convert ester to 4-methyl-5-oxo-1-(2,2,2-trifluoroacetyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester And measured by HPLC. As a result, the asymmetric yield was 63% ee.
- Examples 17 to 30 are 1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4,5-tetrahydro-1H of Example 1.
- -Benzo[b]azepine-4-carboxylic acid ethyl ester was changed to the substrate shown in Table 3, and the reaction was performed according to the reaction conditions shown in Table 3.
- the isolated yield and the asymmetric yield (% ee) are shown in Tables 3 and 4, and the data of the synthesized compounds are shown in Table 5.
- the organic layer was washed with 0.5 L of 10% saline solution, 0.5 L of 10% sodium hydrogen carbonate solution and 0.5 L of 5% saline solution in this order, and the organic layer was concentrated under reduced pressure to obtain 70 g of a pale yellow amorphous substance.
- Example 36 Isopropanol solvate of compound A To 5.0 g of compound A obtained in Example 35, 65 mL of isopropanol was added, and the mixture was stirred at room temperature for 30 minutes. The precipitated suspension was heated and dissolved, and then allowed to cool to room temperature and stirred overnight at 5°C. The suspension was collected by filtration, washed with cold isopropanol, and dried at 40° C. overnight to obtain 4.9 g of a white solid. When the obtained compound was analyzed by a thermogravimetric apparatus, the content of isopropanol was 8.2% with respect to the compound A, and the molar ratio was 0.7 times that of the compound A.
- Example 37 Ethanol solvate of compound A To 0.15 g of compound A obtained in Example 35, 2 mL of ethanol was added, and the mixture was stirred at room temperature for 30 minutes. The precipitated suspension was heated and dissolved, and then allowed to cool to room temperature and stirred overnight at 5°C. The suspension was collected by filtration, washed with cold ethanol, and dried at 40° C. overnight to obtain 0.07 g of a white solid.
- the obtained compound was analyzed by a thermogravimetric apparatus, the content of ethanol was 5.7% with respect to the compound A, and the molar ratio was 0.6 times the amount with respect to the compound A.
- Example 38 Normal propanol solvate of compound A To 5.0 g of the compound A obtained in Example 35, 31 mL of normal propanol was added, and the mixture was stirred at room temperature for 15 minutes. The precipitated suspension was heated and dissolved, and then allowed to cool to room temperature and stirred overnight at 5°C. The suspension was collected by filtration, washed with cold normal propanol, and dried at 40° C. overnight to obtain 4.1 g of a white solid. When the obtained compound was analyzed by a thermogravimetric apparatus, the content of normal propanol was 8.1% with respect to the compound A, and the molar ratio was 0.7 times the amount of the compound A.
- the method of the present invention is a method for industrially and safely producing compound (VI) and compound (VII) which are useful as pharmaceutical intermediates, and a compound A useful as a pharmaceutical is industrially safe and inexpensive. It is useful as a manufacturing method.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
本出願には、1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルを、有機溶媒中で、特定の不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、(R)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルを製造する方法が開示される。本製造方法によって、光学活性なN-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミド及びその中間体を効率的に製造することができる。
Description
本発明は、光学活性なベンゾアゼピン誘導体の新規製造方法及びその中間体に関する。
N-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミドは、V2受容体作動作用を有する化合物であり、夜間頻尿等の予防又は治療に有効であることが特許文献1に開示されている。
N-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミドの製造方法は、特許文献1に記載がある。前記化合物は、ベンゾアゼピン環の4位でエステル基のα位に位置する炭素にメチル基を有することが特徴であり、その光学活性体を得る方法として、特許文献1に記載の(R)-Phenylglycinolを用いた光学分割法が知られている。また、前記化合物の中間体が特許文献2、特許文献3及び非特許文献1に報告されている。
一方、エステル基のα位に位置する炭素の不斉メチル化法としては、β-ケトエステルのα炭素を立体選択的にアルキル化する方法が知られている(特許文献4)。
一方、エステル基のα位に位置する炭素の不斉メチル化法としては、β-ケトエステルのα炭素を立体選択的にアルキル化する方法が知られている(特許文献4)。
発明協会公開技報 公技番号2014-502240号
本発明者らが、特許文献1に記載の(R)-Phenylglycinolを用いた光学分割法により、N-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミドの光学活性体を製造したところ、望まない立体構造の化合物が半量生成するため、非効率であるという課題があった。また、特許文献1に記載の製造方法においては、官能基の保護反応や脱保護反応を行う必要があり、製造工程数が多く非効率であるという課題があった。したがって、本発明の課題は、光学活性なN-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミドを効率的に製造できる、新規な製造方法を提供することである。
上記課題に鑑み、本発明者らが鋭意検討したところ、ジベンゾアゼピン型4級アンモニウム塩を相間移動触媒として用いることで、非常に効率的に不斉メチル化を達成できることを見出し、本発明を完成するに至った。本発明の主な構成は以下の通りである。
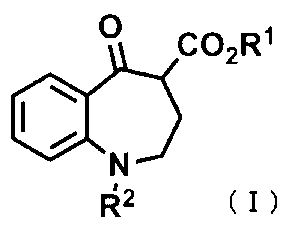
[1] 式(I):

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。〕
で示される化合物を、有機溶媒中で、
R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。〕
で示される化合物を、有機溶媒中で、
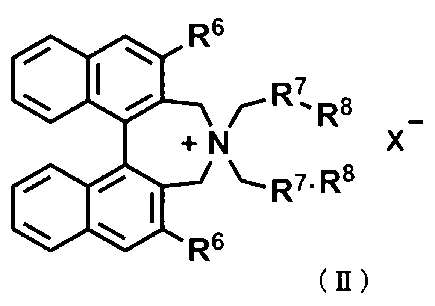
式(II)
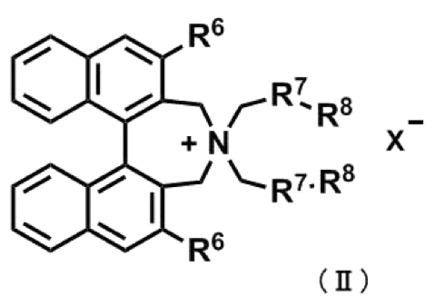
〔式中、R6は、それぞれ独立にハロゲン原子で置換されていてもよいC1~C4アルキル基、又はハロゲン原子で置換されていてもよいアリール基を表す。
R7は、それぞれ独立にC1~C4アルキレン基又はナフタレンジイル基を表す。
R8は、水素原子又は一緒になって単結合を表す。
X-は、ハロゲン化物アニオンを表す。〕
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
R7は、それぞれ独立にC1~C4アルキレン基又はナフタレンジイル基を表す。
R8は、水素原子又は一緒になって単結合を表す。
X-は、ハロゲン化物アニオンを表す。〕
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
式(III):
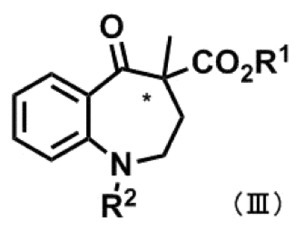
〔式中、R1及びR2は、前記と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物を製造し、次いで当該化合物のカルボニル基を、有機溶媒中で還元剤により還元し、
で示される光学活性化合物を製造し、次いで当該化合物のカルボニル基を、有機溶媒中で還元剤により還元し、
式(IV):

〔式中、R1及びR2は、前記と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物(IV)を製造し、次いで当該化合物のヒドロキシル基をクロル化剤を用いてクロル化し、得られた化合物を金属触媒存在下で水素を用いて還元的脱クロル化し、得られた化合物のベンゾアゼピン環第4位のカルボン酸エステルを塩基性溶液中で加水分解し、得られたカルボン酸をクロル化剤を用いて酸クロリドに変換し、更に塩基の存在下、L-アラニノールと反応させることを特徴とする、
で示される光学活性化合物(IV)を製造し、次いで当該化合物のヒドロキシル基をクロル化剤を用いてクロル化し、得られた化合物を金属触媒存在下で水素を用いて還元的脱クロル化し、得られた化合物のベンゾアゼピン環第4位のカルボン酸エステルを塩基性溶液中で加水分解し、得られたカルボン酸をクロル化剤を用いて酸クロリドに変換し、更に塩基の存在下、L-アラニノールと反応させることを特徴とする、
式(V)

〔式中、R2は、前記と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物を製造する方法、又は前記光学活性化合物を更に溶媒で再結晶することを特徴とする、前記光学活性化合物の溶媒和物を製造する方法。
で示される光学活性化合物を製造する方法、又は前記光学活性化合物を更に溶媒で再結晶することを特徴とする、前記光学活性化合物の溶媒和物を製造する方法。
[2] 式(I):
〔式中、R1及びR2は、[1]における意義と同義である。〕
で示される化合物を、有機溶媒中で、
で示される化合物を、有機溶媒中で、
式(II)

〔式中、R6~R8及びX-は、[1]における意義と同義である。〕
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
式(III):

〔式中、R1及びR2は、[1]における意義と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物を製造する方法。
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
式(III):
で示される光学活性化合物を製造する方法。
[3] [2]に記載の製造方法により得られた前記式(III)で示される化合物のカルボニル基を、有機溶媒中で還元剤により還元し、
式(IV):
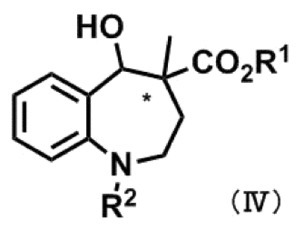
〔式中、R1及びR2は、[1]における意義と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物(IV)を製造する方法。
式(IV):
で示される光学活性化合物(IV)を製造する方法。
[4] [3]に記載の製造方法により得られた前記式(IV)で示される化合物のヒドロキシル基をクロル化剤を用いてクロル化し、得られた化合物を金属触媒存在下で水素を用いて還元的脱クロル化し、得られた化合物のベンゾアゼピン環第4位のカルボン酸エステルを塩基性溶液中で加水分解し、得られたカルボン酸をクロル化剤を用いて酸クロリドに変換し、更に塩基の存在下、L-アラニノールと反応させることを特徴とする、
式(V)

〔式中、R2は、[1]における意義と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物を製造する方法又は前記光学活性化合物を更に溶媒で再結晶することを特徴とする、前記光学活性化合物の溶媒和物を製造する方法。
式(V)
で示される光学活性化合物を製造する方法又は前記光学活性化合物を更に溶媒で再結晶することを特徴とする、前記光学活性化合物の溶媒和物を製造する方法。
[5] 式(I):

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。
R2は、水素原子、アリル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基で置換されていてもよいフェニル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C4アルキル基、アリル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
で示される化合物を、有機溶媒中で、
R2は、水素原子、アリル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基で置換されていてもよいフェニル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C4アルキル基、アリル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
で示される化合物を、有機溶媒中で、
式(II-1)
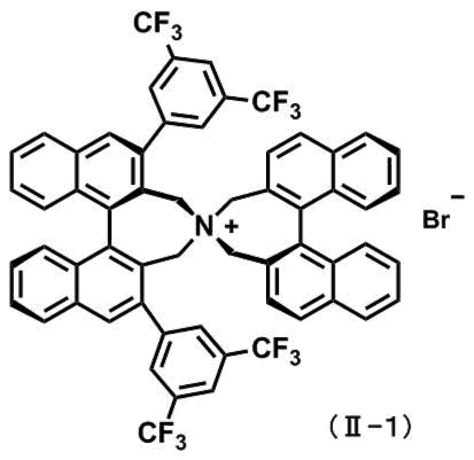
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
式(III):
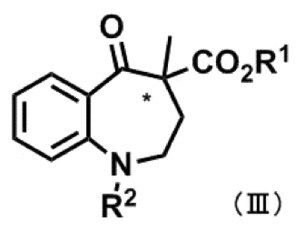
〔式中、R1及びR2は、前記と同義であり、*は不斉中心を表す。〕
で示される光学活性化合物を製造する方法。
式(III):
で示される光学活性化合物を製造する方法。
[6] 前記不斉四級アンモニウム塩及び前記塩基とともに、無機塩の存在下でメチル化反応を行うことを特徴とする、[1]、[2]又は[5]に記載の製造方法。
[7] 前記有機溶媒が、トルエン、キシレン、クロロベンゼン、及びブロモベンゼンからなる群より選択される1種以上である、[1]、[2]又は[5]に記載の製造方法。
[8] 前記塩基が、炭酸カリウム、炭酸セシウム、水酸化カリウム、及び水酸化セシウムからなる群より選択される1種以上である、[1]、[2]又は[5]に記載の製造方法。
[9] 前記無機塩が、フッ化セシウム、塩化セシウム、臭化カリウム、塩化ルビジウム、臭化ルビジウム、及び臭化ナトリウムからなる群より選択される1種以上である、[6]に記載の製造方法。
[10] 前記還元剤がボラン化合物である、[1]又は[3]に記載の製造方法。
[8] 前記塩基が、炭酸カリウム、炭酸セシウム、水酸化カリウム、及び水酸化セシウムからなる群より選択される1種以上である、[1]、[2]又は[5]に記載の製造方法。
[9] 前記無機塩が、フッ化セシウム、塩化セシウム、臭化カリウム、塩化ルビジウム、臭化ルビジウム、及び臭化ナトリウムからなる群より選択される1種以上である、[6]に記載の製造方法。
[10] 前記還元剤がボラン化合物である、[1]又は[3]に記載の製造方法。
[11] 前記R1がメチル基又はエチル基である、[1]~[10]のいずれか一項に記載の製造方法。
[12] 前記R2が-COR3又は-SO2R4であり、前記R3が、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、又は水素原子であり、R4が、C1~C4アルキル基で置換されていてもよいフェニル基である、[1]~[11]のいずれか一項に記載の製造方法。
[12] 前記R2が-COR3又は-SO2R4であり、前記R3が、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、又は水素原子であり、R4が、C1~C4アルキル基で置換されていてもよいフェニル基である、[1]~[11]のいずれか一項に記載の製造方法。
[13] 前記R2が-COR3であり、前記R3が、
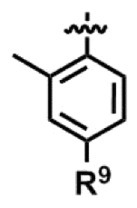
〔式中、R9は、メチル基で置換されていてもよい芳香族ヘテロ環基を意味する。〕
で表される置換フェニル基である、[12]に記載の製造方法。
[14] 前記R2が-COR3であり、前記R3が
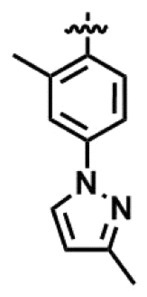
で表される置換フェニル基であり、前記式(III)で示される化合物のベンゾアゼピン環の4位の立体がRである、[12]に記載の製造方法。
で表される置換フェニル基である、[12]に記載の製造方法。
[14] 前記R2が-COR3であり、前記R3が
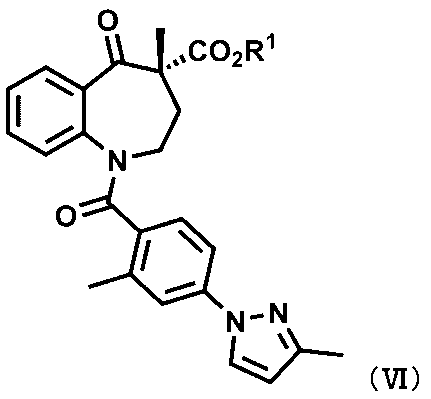
[15] 式(VI):

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
で示される化合物又はその薬学的に許容される塩。
で示される化合物又はその薬学的に許容される塩。
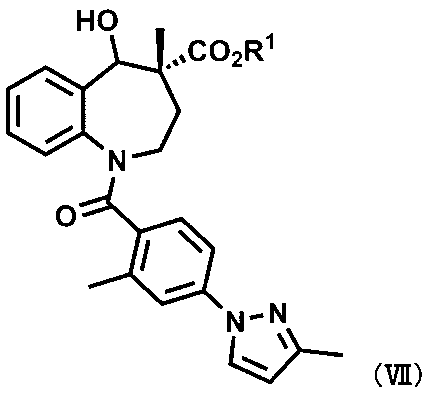
[16] 式(VII):

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
で示される化合物又はその薬学的に許容される塩。
[17] 前記R1が、メチル基又はエチル基である、[15]又は[16]に記載の化合物又はその薬学的に許容される塩。
で示される化合物又はその薬学的に許容される塩。
[17] 前記R1が、メチル基又はエチル基である、[15]又は[16]に記載の化合物又はその薬学的に許容される塩。
本発明によれば、光学活性なN-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミド、好ましくは(S)-N-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミド(以下、化合物Aという。また、式(I)、式(II)等で示される化合物を、以下、化合物(I)、化合物(II)等という。)の新たな製造方法を提供することができる。特に、化合物Aの合成中間体である化合物(V)及び化合物(VI)の製造において、化学純度の向上、光学活性体の収率向上を達成し、化合物Aの製造において、製造工程数を短縮することができる。また、本発明の好ましい態様によれば、不斉メチル化反応を加速することができる。

以下に、本発明の実施形態を更に詳細に説明する。ただし、本発明は以下の実施形態に限定されるものではない。
本発明において化合物Aは、以下に示す工程を介して製造することができる。

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、置換基群Aから選択される1又は複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
*は不斉中心を表す。
尚、置換基群Aは、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群である。〕
R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、置換基群Aから選択される1又は複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
*は不斉中心を表す。
尚、置換基群Aは、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群である。〕
本願明細書において用語の定義は以下の通りである。
「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子、又はヨウ素原子を意味する。
「芳香族ヘテロ環基」とは、窒素、酸素、又は硫黄等のヘテロ原子を少なくとも一つ含んでいる単環~3環の芳香環基を意味し、例えば、ピリジル基、チエニル基、フリル基、ピラジニル基、ピリダジニル基、チアゾリル基、ピリミジニル基、ピラゾリル基、ピロリル基、オキサゾリル基、オキサジアゾリル基、イソチアゾリル基、イソオキサゾリル基、イミダゾリル基、キノリン基、キナゾリン基、プリン基、及びアクリジン基等が挙げられる。
「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子、又はヨウ素原子を意味する。
「芳香族ヘテロ環基」とは、窒素、酸素、又は硫黄等のヘテロ原子を少なくとも一つ含んでいる単環~3環の芳香環基を意味し、例えば、ピリジル基、チエニル基、フリル基、ピラジニル基、ピリダジニル基、チアゾリル基、ピリミジニル基、ピラゾリル基、ピロリル基、オキサゾリル基、オキサジアゾリル基、イソチアゾリル基、イソオキサゾリル基、イミダゾリル基、キノリン基、キナゾリン基、プリン基、及びアクリジン基等が挙げられる。
「C1~C6アルキル基」とは、炭素数1~6個の直鎖状又は分枝鎖状の飽和炭化水素基を意味する。例えば、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、及びこれらの各種分岐鎖異性体等が挙げられる。
「C1~C4アルキル基」とは、炭素数1~4個の直鎖状又は分枝鎖状の飽和炭化水素基を意味し、例えば、メチル基、エチル基、プロピル基、ブチル基、及びこれらの各種分枝鎖異性体等が挙げられる。
「C1~C4アルコキシ基」とは、前記の「C1~C4アルキル基」に酸素原子が結合した基を意味し、例えば、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、及びこれらの各種分枝鎖異性体が挙げられる。
「C1~C4アルキル基」とは、炭素数1~4個の直鎖状又は分枝鎖状の飽和炭化水素基を意味し、例えば、メチル基、エチル基、プロピル基、ブチル基、及びこれらの各種分枝鎖異性体等が挙げられる。
「C1~C4アルコキシ基」とは、前記の「C1~C4アルキル基」に酸素原子が結合した基を意味し、例えば、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、及びこれらの各種分枝鎖異性体が挙げられる。
「C7~C12アラルキル基」とは、アリール基がアルキル基に結合し、炭素数7~12個となった基を意味し、例えば、ベンジル基、及びフェネチル基等が挙げられる。
「C2~C6アルケニル基」とは、少なくとも1個の二重結合を有する炭素数2~6個の直鎖状又は分枝鎖状の不飽和炭化水素基を意味し、例えば、ビニル基、アリル基、及び1-プロペニル基等が挙げられる。
「C2~C6アルケニル基」とは、少なくとも1個の二重結合を有する炭素数2~6個の直鎖状又は分枝鎖状の不飽和炭化水素基を意味し、例えば、ビニル基、アリル基、及び1-プロペニル基等が挙げられる。
本発明の一実施形態は、式(VI)で示される化合物である。化合物(VI)は、化合物Aの新規な合成中間体である。

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
化合物(VI)は以下の第1工程により得ることができる。すなわち、化合物(I)のR2が-COR3であり、R3を
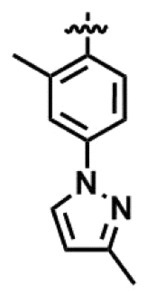
とすることにより、化合物(VI)は得られる。化合物(VI)においては、ベンゾアゼピン環の4位の立体をR体に制御されている。また、化合物(VI)のベンゾアゼピン環の4位の絶対立体配置は、化合物(VI)から化合物Aを合成し、更にアルコール和物に変換し、単結晶X線構造解析を行うことで決定することができる。
[第1工程]

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、置換基群Aから選択される1又は複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
尚、置換基群Aは、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群である。〕
R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、置換基群Aから選択される1又は複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。
尚、置換基群Aは、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群である。〕
第1工程は、化合物(I)を不斉メチル化する工程である。化合物(I)は、特開2016-40338号公報又は特開2016-185993号公報に記載の公知の方法等に従って製造することができる。
前記R1は、メチル基又はエチル基が好ましく、エチル基がより好ましい。
前記R2は、-COR3又は-SO2R4が好ましく、-COR3がより好ましい。
前記R3は、前記置換基群Aから選択される1又は複数の置換基で置換されていてもよいフェニル基が好ましい。
尚、前記置換基群Aは、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基又はハロゲン原子で置換されていてもよいC1~C6アルキル基が好ましく、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基がより好ましい。
前記R2は、-COR3又は-SO2R4が好ましく、-COR3がより好ましい。
前記R3は、前記置換基群Aから選択される1又は複数の置換基で置換されていてもよいフェニル基が好ましい。
尚、前記置換基群Aは、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基又はハロゲン原子で置換されていてもよいC1~C6アルキル基が好ましく、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基がより好ましい。
最も好ましいR3は、以下に示されるフェニル基である。
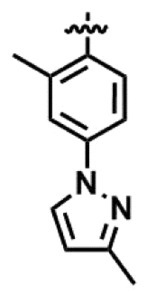
前記R4は、C1~C4アルキル基又はC1~C4アルコキシ基で置換されていてもよいフェニル基が好ましい。
前記R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基又はC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基が好ましい。
前記R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基又はC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基が好ましい。
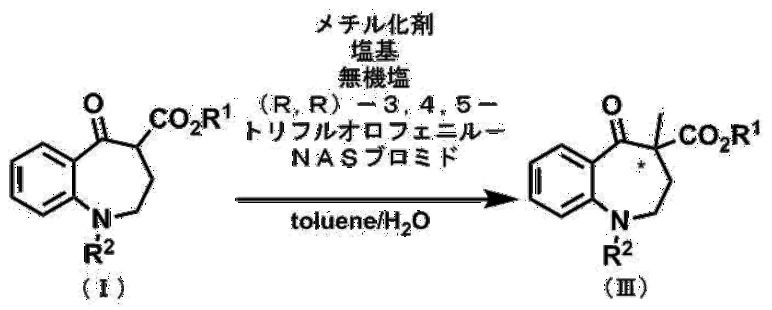
第1工程は、例えば、化合物(I)のベンゾアゼピン環の4位を、有機溶媒中で、触媒及び塩基の存在下、メチル化剤によりメチル化することで実施することができる。このメチル化反応は、反応を加速するために、更に無機塩の存在下で実施するのが好ましい。反応終了後、粗生成物を当業者に周知の方法で単離精製することができる。この方法は立体選択性に優れ、このようにして得られた化合物(III)は優れた光学純度を有する。更に、この方法では化合物(III)が収率よく得られるため、工業的生産に適している。
第1工程で用いられる触媒は、不斉四級アンモニウム塩の化合物(II)である。

〔式中、R6は、それぞれ独立に、ハロゲン原子で置換されていてもよいC1~C4アルキル基、又はハロゲン原子で置換されていてもよいアリール基を表す。
R7は、それぞれ独立にC1~C4アルキレン基又はナフタレンジイル基を表す。
R8は水素原子又は一緒になって単結合を表す。
X-はハロゲン化物アニオンを表す。〕
R7は、それぞれ独立にC1~C4アルキレン基又はナフタレンジイル基を表す。
R8は水素原子又は一緒になって単結合を表す。
X-はハロゲン化物アニオンを表す。〕
前記R6は、3,5-ビストリフルオロメチルフェニル基又は3,4,5-トリフルオロフェニル基が好ましく、3,5-ビストリフルオロメチルフェニル基がより好ましい。
前記R7は、ノルマルブタンジイル基又はナフタレンジイル基が好ましく、ナフタレンジイル基がより好ましい。
前記R8は、一緒になって単結合を形成することが好ましい。
前記R7は、ノルマルブタンジイル基又はナフタレンジイル基が好ましく、ナフタレンジイル基がより好ましい。
前記R8は、一緒になって単結合を形成することが好ましい。
前記不斉四級アンモニウム塩の化合物(II)としては、具体的に以下のように、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド、(R,R)-3,4,5-トリフルオロフェニル-NASブロミド、(11bR)-(-)-4,4-ジブチル-4,5-ジヒドロ-2,6-ビス(3,5-ジトリフルオロメチルフェニル-3H-ジナフト[2,1-c:1’,2’-e]アゼピニウムブロミド、及び(11bR)-(-)-4,4-ジブチル-4,5-ジヒドロ-2,6-ビス(3,4,5-トリフルオロフェニル-3H-ジナフト[2,1-c:1’,2’-e]アゼピニウムブロミドが挙げられ、好ましくは(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドである。これら触媒を相間移動触媒として用いることで、効率的に不斉メチル化することができる。
(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド

(R,R)-3,4,5-トリフルオロフェニル-NASブロミド

(11bR)-(-)-4,4-ジブチル-4,5-ジヒドロ-2,6-ビス(3,5-ジトリフルオロメチルフェニル-3H-ジナフト[2,1-c:1’,2’-e]アゼピニウムブロミド

(11bR)-(-)-4,4-ジブチル-4,5-ジヒドロ-2,6-ビス(3,4,5-トリフルオロフェニル-3H-ジナフト[2,1-c:1’,2’-e]アゼピニウムブロミド

前記不斉四級アンモニウム塩の化合物(II)の使用量は、1当量の化合物(I)に対して、0.0001~1当量を用いることができるが、0.001~0.1当量を用いることが好ましく、0.005~0.02当量を用いることがより好ましい。不斉四級アンモニウム塩の化合物(II)の使用量をこの範囲とすることにより、不斉メチル化の完結及びメチル化体の副生成物の生成抑制に寄与する。
第1工程で用いられる有機溶媒は、反応の進行を阻害しないものであれば特に制限はないが、例えば、トルエン、キシレン、メシチレン、クメン、ヘプタン、酢酸エチル、メチルイソブチルケトン、メチルターシャリーブチルエーテル、テトラヒドロフラン、塩化メチレン、クロロベンゼン、ブロモベンゼン、ニトロベンゼン、アニソール、及び1-ブチル-3-メチルイミダゾリウムテトラフルオロボラート等が挙げられる。これらの溶媒は単独で使用してもよいし、2種以上を混合して又は水と混合して使用してもよい。好ましくは高溶解性の芳香族有機溶媒であり、より好ましくはトルエン、キシレン、クロロベンゼン、又はブロモベンゼンである。これら溶媒を使用することにより、不純物の生成を抑制し、不斉メチル化反応を完結することができる。前記溶媒の使用量は、1当量の化合物(I)に対して、好ましくは3~20当量であり、より好ましくは5~10当量である。
第1工程で用いられる塩基は、有機塩基でもよいし、無機塩基でもよい。有機塩基としては、トリエチルアミン、ピリジン、及びN,N’-ジメチルアミノピリジン等が挙げられる。無機塩基としては、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化セシウム、水酸化バリウム、水酸化ルビジウム、炭酸カリウム、炭酸セシウム、及び炭酸ルビジウム等が挙げられる。無機塩基の方が好ましく、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化セシウム、水酸化バリウム、水酸化ルビジウム、炭酸カリウム、炭酸セシウム、又は炭酸ルビジウムが好ましい。より好ましくは水酸化カリウム、炭酸カリウム、炭酸セシウム、又は水酸化セシウムである。
第1工程で用いられるメチル化剤は、通常用いられるメチル化剤であれば制限はなく、好ましくはテトラフルオロホウ酸トリメチルオキソニウム、トリフルオロメタンスルホン酸メチル、ジメチル硫酸、臭化メチル、又はヨウ化メチルであり、より好ましくは臭化メチル又はヨウ化メチルである。前記メチル化剤の使用量は、1当量の化合物(I)に対して、好ましくは1~20当量であり、より好ましくは1~10当量である。
第1工程で用いられる無機塩は、アルカリ金属塩が好ましく、例えば、フッ化セシウム、塩化セシウム、臭化セシウム、ヨウ化セシウム、塩化リチウム、臭化リチウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、塩化ナトリウム、臭化ナトリウム、塩化ルビジウム、及び臭化ルビジウム等が挙げられる。より好ましくはフッ化セシウム、塩化セシウム、臭化カリウム、臭化ナトリウム、塩化ルビジウム、又は臭化ルビジウムである。前記無機塩の使用量は、1当量の化合物(I)に対して、好ましくは0.01~1当量であり、より好ましくは0.05~0.5当量である。これら無機塩を添加することにより、光学純度を保ったままで不斉メチル化反応を約5倍以上加速することができる。
第1工程における反応温度は、特に制限はないが、好ましくは-20~20℃であり、より好ましくは1~10℃である。反応温度をこの範囲に調整することで、不斉メチル化の完結及びメチル化体の副生成物の生成抑制に寄与する。
第1工程で得られる化合物(III)の単離は、常法により実施することができる。例えば、得られた粗体を再結晶することにより、又はカラムクロマトグラフィーにより精製することができる。再結晶に用いる溶媒は、好ましくはメタノール、エタノール、2-プロパノール、酢酸エチル、トルエン、又はアセトニトリルであり、より好ましくは酢酸エチル又はアセトニトリルである。
本発明の一実施形態は、式(VII)で示される化合物である。化合物(VII)は、化合物Aの新規な合成中間体である。

〔式中、R1は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
化合物(VII)は以下の第2工程により得ることができる。R2については、前述の化合物(VI)と同様である。なお、第2工程では、ベンゾアゼピン環の4位の立体は変化しない。
[第2工程]
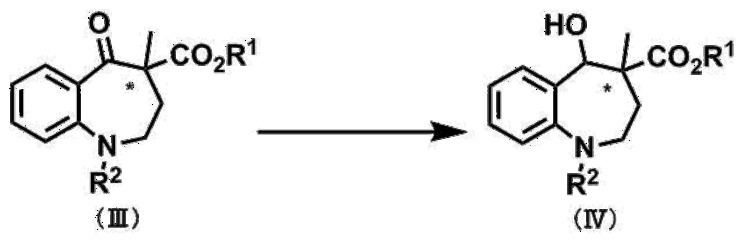
〔式中、R1及びR2は、前記第1工程のそれと同義であり、*は不斉中心を表す。〕
[第2工程]
第2工程は、化合物(III)を化合物(IV)に還元する工程である。例えば、化合物(III)のベンゾアゼピン環5位のカルボニル基を、有機溶媒中あるいは水との混合溶媒中で、還元剤を用いて還元することにより得ることができる。
第2工程で用いられる還元剤としては、ボラン化合物が好ましく、これにより、カルボニル基選択的な還元を行うことができる。
第2工程で用いられる還元剤としては、ボラン化合物が好ましく、これにより、カルボニル基選択的な還元を行うことができる。
前記ボラン化合物としては、好ましくは水素化ホウ素ナトリウム、水素化ホウ素カリウム、水素化ホウ素リチウム、又はボラン・アンモニア錯体であり、より好ましくはボラン・アンモニア錯体である。ボラン・アンモニア錯体は、文献(Inorganic Chemistry 2007,46,7810)に記載の方法で予め調整したものを用いることができる。ボラン化合物の使用量は、1当量の化合物(III)に対して、0.5~2当量を用いることが好ましく、1.0~1.5当量を用いることがより好ましい。反応に使用する有機溶媒は、好ましくはトルエン、テトラヒドロフラン、酢酸エチル、又はこれら有機溶媒と水との混合溶液であり、より好ましくはトルエンと水との混合溶液である。
第2工程の反応温度は特に制限はないが、好ましくは20~100℃であり、より好ましくは40~60℃である。第2工程の反応温度をこの範囲に調整することで、還元反応がスムーズに進行するとともに、カルボニル基選択的な還元を行うことができる。得られる化合物(IV)の単離及び精製は、常法により実施することができる。
化合物Aは、化合物(V)として前記化合物(IV)から以下の第3工程~第6工程を経ることにより、得ることができる。なお、第3工程~第6工程では、ベンゾアゼピン環の4位の立体は変化しない。
[第3工程]

〔式中、R1及びR2は、前記第1工程のそれと同義であり、*は不斉中心を表す。〕
第3工程は、ベンゾアゼピン環5位のヒドロキシル基を、有機溶媒中でクロル化する工程である。当該クロル化反応は、例えば、化合物(IV)を有機溶媒に溶解し、クロル化剤を加えて反応させることで行うことができる。クロル化剤としては、求電子的クロル化剤が好ましい。また、塩基存在下でクロル化を行うことが好ましい。
クロル化剤としては、好ましくは塩化チオニル、塩化オキサリル、オキシ塩化リン、三塩化リン、五塩化リン、又はN-クロロスクシンイミドであり、より好ましくは塩化チオニル又はオキシ塩化リンである。クロル化剤の使用量は、1当量の化合物(IV)に対して、0.5~3当量を用いることが好ましく、1~2当量を用いることがより好ましい。反応に使用する有機溶媒は、好ましくはトルエン、塩化メチレン、又はクロロホルムであり、より好ましくはトルエンである。反応温度は、好ましくは20~120℃であり、より好ましくは60~100℃である。得られる化合物(VIII)の単離及び精製は、常法により実施することができる。
[第4工程]
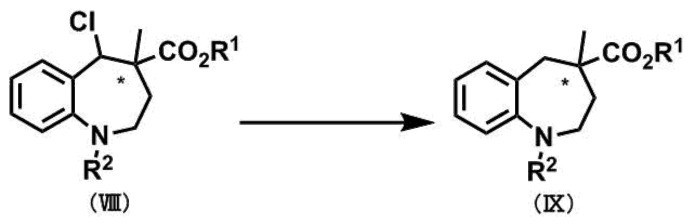
〔式中、R1及びR2は、前記第1工程のそれと同義であり、*は不斉中心を表す。〕
第4工程は、ベンゾアゼピン環5位のクロル基を、金属触媒存在下において、有機溶媒中で水素を用いて還元的脱クロル化する工程である。当該脱クロル化は、例えば、化合物(VIII)を有機溶媒に溶解し、得られた溶液に金属触媒を加え、更に水素ガス雰囲気下で反応を行うことができる。
金属触媒としては、好ましくはパラジウム触媒、白金触媒、又はニッケル触媒である。パラジウム触媒としては、例えば、パラジウム炭素及び水酸化パラジウム等が挙げられる。白金触媒としては、例えば、白金炭素及び酸化白金水和物等が挙げられる。ニッケル触媒としては、例えば、ラネーニッケル及びニッケル炭素等が挙げられる。前記金属触媒として、より好ましくはパラジウム触媒である。金属触媒の使用量は、1当量の化合物(VIII)に対して、0.01~1当量を用いることが好ましく、0.05~0.2当量を用いることがより好ましい。反応に使用する有機溶媒は、好ましくはメタノール、エタノール、テトラヒドロフラン、又は酢酸エチルであり、より好ましくはメタノール又はエタノールである。水素圧は、好ましくは1~5気圧であり、より好ましくは1~3気圧である。反応温度は、特に制限はないが、好ましくは20~100℃であり、より好ましくは20~60℃である。得られる化合物(IX)の単離及び精製は、常法により実施することができる。第2工程~第4工程により、ベンゾアゼピン環5位のカルボニル基の位置選択的な還元を行うことができる。
[第5工程]~[第6工程]

〔式中、R1及びR2は、前記第1工程のそれと同義であり、*は不斉中心を表す。〕
第5工程及び第6工程は、常法に従い反応を行うことができる。
第5工程では、化合物(IX)を適当な溶媒(例えば、メタノール、エタノール等のアルコール溶媒、水等)中、塩基(例えば、水酸化ナトリウム、水酸化カリウム等)を用いて、通常室温から有機溶媒の沸点の温度で30分間から1日間反応させることによって、化合物(X)のカルボン酸体を得ることができる。次に第6工程では、得られたカルボン酸体を、L-アラニノールとアミド化を行うことで化合物(V)を得ることができる。アミド化には縮合剤を用いる方法や、カルボン酸を混合酸無水物や酸クロリドとした後で、L-アラニノールを反応させる方法等を用いることができる。縮合剤を用いる方法では、例えば、カルボン酸体とL-アラニノールを適当な有機溶媒(クロロホルム、ジメチルホルムアミド等)中、塩基(例えば、ジイソプロピルエチルアミン、トリエチルアミン等)の存在下、縮合剤(例えば、1,3-ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-[3-(ジメチルアミノ)プロピル]カルボジイミド(EDC)等)を単独で、又は1-ヒドロキシベンズトリアゾール(HOBt)と組み合わせて用いることで化合物(V)を得ることができる。また、混合酸無水物を用いる方法では、例えば、カルボン酸体を適当な有機溶媒(例えば、ジクロロメタン、トルエン等)中、塩基(例えば、ピリジン、トリエチルアミン等)の存在下、酸クロリド(例えばピバロイルクリド、トシルクリド等)又は酸誘導体(例えば、クロロギ酸エチル、クロロギ酸イソブチル等)と反応させ、得られた混合酸無水物をL-アラニノールと、通常、0℃から室温で反応させることで化合物(V)を得ることができる。また、酸クロリドを経由する方法では、例えば、適当な有機溶媒(例えば、トルエン、キシレン等)中、クロル化剤(例えば、塩化チオニル、塩化オキサリル等)を用いて酸クロリドとした後、得られた酸クロリドを塩基(例えば、炭酸ナトリウム、トリエチルアミン等)存在下で適当な有機溶媒(例えば、酢酸エチル、トルエン等)中、L-アラニノールと、通常、0℃から室温で反応させることで化合物(V)を得ることができる。
第5工程では、化合物(IX)を適当な溶媒(例えば、メタノール、エタノール等のアルコール溶媒、水等)中、塩基(例えば、水酸化ナトリウム、水酸化カリウム等)を用いて、通常室温から有機溶媒の沸点の温度で30分間から1日間反応させることによって、化合物(X)のカルボン酸体を得ることができる。次に第6工程では、得られたカルボン酸体を、L-アラニノールとアミド化を行うことで化合物(V)を得ることができる。アミド化には縮合剤を用いる方法や、カルボン酸を混合酸無水物や酸クロリドとした後で、L-アラニノールを反応させる方法等を用いることができる。縮合剤を用いる方法では、例えば、カルボン酸体とL-アラニノールを適当な有機溶媒(クロロホルム、ジメチルホルムアミド等)中、塩基(例えば、ジイソプロピルエチルアミン、トリエチルアミン等)の存在下、縮合剤(例えば、1,3-ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-[3-(ジメチルアミノ)プロピル]カルボジイミド(EDC)等)を単独で、又は1-ヒドロキシベンズトリアゾール(HOBt)と組み合わせて用いることで化合物(V)を得ることができる。また、混合酸無水物を用いる方法では、例えば、カルボン酸体を適当な有機溶媒(例えば、ジクロロメタン、トルエン等)中、塩基(例えば、ピリジン、トリエチルアミン等)の存在下、酸クロリド(例えばピバロイルクリド、トシルクリド等)又は酸誘導体(例えば、クロロギ酸エチル、クロロギ酸イソブチル等)と反応させ、得られた混合酸無水物をL-アラニノールと、通常、0℃から室温で反応させることで化合物(V)を得ることができる。また、酸クロリドを経由する方法では、例えば、適当な有機溶媒(例えば、トルエン、キシレン等)中、クロル化剤(例えば、塩化チオニル、塩化オキサリル等)を用いて酸クロリドとした後、得られた酸クロリドを塩基(例えば、炭酸ナトリウム、トリエチルアミン等)存在下で適当な有機溶媒(例えば、酢酸エチル、トルエン等)中、L-アラニノールと、通常、0℃から室温で反応させることで化合物(V)を得ることができる。
化合物(V)は、溶媒和物として存在することもできる。化合物(V)の溶媒和物は、溶媒和物を製造するための通常の方法によって得ることができる。詳細には、必要に応じて加熱しながら化合物(V)と溶媒とを混和した後、撹拌又は放置しながらそれを冷却して結晶化させることにより得ることができる。冷却は、結晶の品質、粒度等への影響を考慮し、必要に応じて冷却速度を調節しながら実施することが望ましい。例えば、20~1℃/時間の冷却速度で冷却するのが好ましく、10~3℃/時間の冷却速度で冷却するのがより好ましい。これらの方法で使用される有機溶媒としては、メタノール、エタノール、プロパノール、イソプロパノール、ノルマルプロパノール、及びターシャリーブタノール等のアルコール系溶媒が好ましい。使用する有機溶媒の量は、化合物(V)に対し、3~20倍重量が好ましく、5~10倍重量がより好ましい。
化合物Aは、薬学的に許容される添加剤と混合することによって、医薬製剤として調製することができる。これらは、公知の方法(例えば日本薬局方第17版の製剤総則に記載されている方法)によって調製することができる。
化合物Aは、V2受容体作動薬として作用することから、中枢性尿崩症、夜尿症、夜間頻尿、過活動膀胱、血友病、あるいはフォンウィルブランド病の予防又は治療のための医薬組成物として使用することができることが既に知られている。
以下に、実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
核磁気共鳴スペクトル、LC-MS、HPLC、及び熱重量測定は、以下の測定条件で行った。
[核磁気共鳴スペクトル]
以下の実施例及び参考例における核磁気共鳴(1H-NMR)解析は、Agilent Technologies製(400‐MR)を使用して行った。スペクトルは、テトラメチルシランを標準物質としてケミカルシフト値をδ値(ppm)で記載した。分裂パターンは、一重線を「s」、二重線を「d」、三重線を「t」、四重線を「q」、五重線を「quin」、多重線を「m」、幅広い線を「br」で示した。
[核磁気共鳴スペクトル]
以下の実施例及び参考例における核磁気共鳴(1H-NMR)解析は、Agilent Technologies製(400‐MR)を使用して行った。スペクトルは、テトラメチルシランを標準物質としてケミカルシフト値をδ値(ppm)で記載した。分裂パターンは、一重線を「s」、二重線を「d」、三重線を「t」、四重線を「q」、五重線を「quin」、多重線を「m」、幅広い線を「br」で示した。
[LC-MS]
質量分析は、Waters製(Acquity SQD)を使用し、エレクトロスプレーイオン化法(ESI)で行った。測定条件は、カラムXBridge C18 S3.5 1.0×150mm (Waters製)、カラム温度50℃、移動相0.1%ギ酸水溶液及び0.1%ギ酸アセトニトリル溶液、流速50μL/分、抽出波長210~400nmとした。
質量分析は、Waters製(Acquity SQD)を使用し、エレクトロスプレーイオン化法(ESI)で行った。測定条件は、カラムXBridge C18 S3.5 1.0×150mm (Waters製)、カラム温度50℃、移動相0.1%ギ酸水溶液及び0.1%ギ酸アセトニトリル溶液、流速50μL/分、抽出波長210~400nmとした。
[HPLC]
以下の実施例における不斉収率は、Waters製(Alliance2)を使用した高速液体クロマトグラフィー(HPLC)にて測定した。測定条件は、カラムIC、IE、AS、AD-H、OD-H(DAISEL CHEMICAL INDUSTRIES製)、カラム温度40℃、移動相ヘキサン及びエタノール、流速0.1~2.0mL/分、抽出波長254nmとした。
以下の実施例における不斉収率は、Waters製(Alliance2)を使用した高速液体クロマトグラフィー(HPLC)にて測定した。測定条件は、カラムIC、IE、AS、AD-H、OD-H(DAISEL CHEMICAL INDUSTRIES製)、カラム温度40℃、移動相ヘキサン及びエタノール、流速0.1~2.0mL/分、抽出波長254nmとした。
[熱重量測定]
TGAサーモグラムは、パーキンエルマー製(Pyris1-TGA)を使用して行った。測定条件は、窒素ガス20mL/分、初期温度80℃で1分等温後、5℃/分で170℃まで昇温し、170℃で1分等温とした。
TGAサーモグラムは、パーキンエルマー製(Pyris1-TGA)を使用して行った。測定条件は、窒素ガス20mL/分、初期温度80℃で1分等温後、5℃/分で170℃まで昇温し、170℃で1分等温とした。
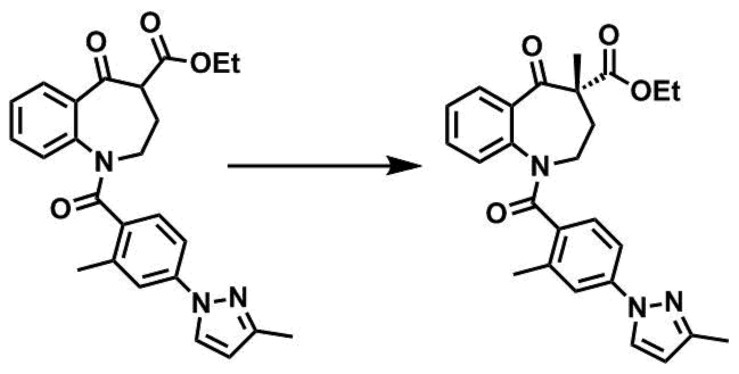
[実施例1](R)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第1工程)

20mLフラスコに1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル108mg、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド5.4mg、炭酸セシウム407mg、水0.02mL及びブロモベンゼン1.6mLを加え、5℃に冷却した。3.5mol/L臭化メチル-ブロモベンゼン溶液0.5mLを5℃で加え、そのまま19時間撹拌した。反応終了後、水2mLを加え、10分間撹拌した後、酢酸エチルを加えて分液した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過した後、ろ液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製し、白色固体56mgを得た(80%ee)。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.70 (s,1H),7.48 (s,2H),7.25-6.98 (m,3H),6.79 (brs,1H),6.66 (brs,1H),6.23-6.17 (m,1H),4.19-4.06 (m,2H),2.46 (s,4H) ,2.33 (s,3H),2.07 (s,1H),1.66-1.55 (m,5H),1.19 (s,3H).
ESI/MS(m/z)446 (M+H) +.
1H-NMR(400Hz,CDCl3) δ7.70 (s,1H),7.48 (s,2H),7.25-6.98 (m,3H),6.79 (brs,1H),6.66 (brs,1H),6.23-6.17 (m,1H),4.19-4.06 (m,2H),2.46 (s,4H) ,2.33 (s,3H),2.07 (s,1H),1.66-1.55 (m,5H),1.19 (s,3H).
ESI/MS(m/z)446 (M+H) +.
[不斉四級アンモニウム塩の検討]
実施例2~4は、実施例1の不斉メチル化反応に用いる触媒を(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドから表1に示す触媒に置き換え、実施例1と同様の条件にて反応を行った。反応後、上澄み溶液20μLをアセトニトリル1mLに希釈しHPLC分析を行った。目的とする生成物が表1に示す不斉収率で得られ、実施例2~4で得られた生成物の1H-NMR、LC-MSの測定結果は実施例1で示したものと同様であった。実施例2~4で用いた触媒を使用した場合は(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドを用いた場合と比べ、不斉収率は低下するものの、不斉メチル化は進行し、40~50%eeで不斉メチル化生成物が得られることが判明した。
実施例2~4は、実施例1の不斉メチル化反応に用いる触媒を(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドから表1に示す触媒に置き換え、実施例1と同様の条件にて反応を行った。反応後、上澄み溶液20μLをアセトニトリル1mLに希釈しHPLC分析を行った。目的とする生成物が表1に示す不斉収率で得られ、実施例2~4で得られた生成物の1H-NMR、LC-MSの測定結果は実施例1で示したものと同様であった。実施例2~4で用いた触媒を使用した場合は(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドを用いた場合と比べ、不斉収率は低下するものの、不斉メチル化は進行し、40~50%eeで不斉メチル化生成物が得られることが判明した。

[無機塩添加による反応の加速]
実施例1から、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドを用いた不斉メチル化では良好な不斉収率でメチル化生成物が得られることが分かった。次に、無機塩の添加による反応の加速への影響について検討を行った。
実施例1から、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミドを用いた不斉メチル化では良好な不斉収率でメチル化生成物が得られることが分かった。次に、無機塩の添加による反応の加速への影響について検討を行った。
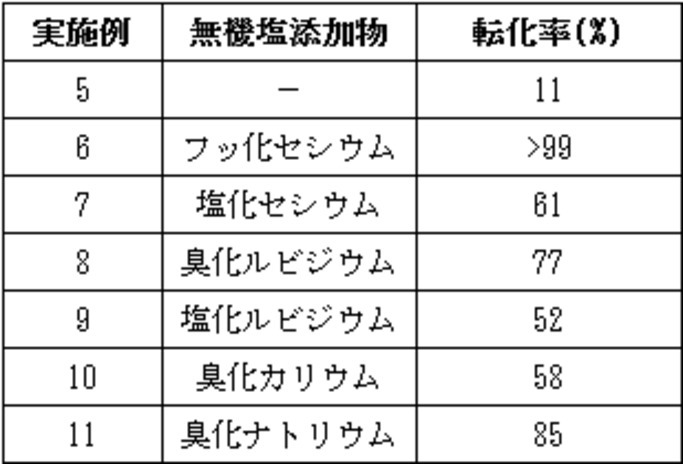
20mLフラスコに1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル108mg、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド3.8mg、炭酸セシウム407mg、表2の無機塩0.025mmolを加えアルゴン置換した。トルエン1.62mL及びヨウ化メチル101μLを加え、5℃で30分撹拌後、水22.5μLを加えた。そのまま24時間撹拌後、撹拌を停止し、上澄み溶液20μLをアセトニトリル1mLに希釈しHPLC分析を行った。結果を表2に示す。
表2における実施例5~11の転化率は以下の式により計算を行った。
転化率=生成物面積値/(原料面積値+生成物面積値)×100(%)
表2に示す無機塩添加物を加えた場合、全ての場合において、無機塩を加えない場合と比べ約5倍以上の反応の加速が見られ、その中でも特にフッ化セシウムを用いた場合にその効果は顕著であった。実施例5~11で得られた生成物の1H-NMR、LC-MSの測定結果は実施例1で示したものと同様であった。
転化率=生成物面積値/(原料面積値+生成物面積値)×100(%)
表2に示す無機塩添加物を加えた場合、全ての場合において、無機塩を加えない場合と比べ約5倍以上の反応の加速が見られ、その中でも特にフッ化セシウムを用いた場合にその効果は顕著であった。実施例5~11で得られた生成物の1H-NMR、LC-MSの測定結果は実施例1で示したものと同様であった。
[実施例12](R)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第1工程)
2Lフラスコに1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル180g、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド0.9g、炭酸セシウム544g、フッ化セシウム6.3g、水68mL、及びブロモベンゼン630mLを加え、5℃に冷却した。臭化メチル198gを0℃で加え、そのまま20時間撹拌した。反応終了後、塩化メチレン271mL及び水540mLを加え、15℃で1時間撹拌した。反応液に酢酸エチル及びアセトンを加えて分液後、有機層を減圧濃縮した。粗液にヘプタン2700mLを加え、室温で2時間撹拌し、析出した粗結晶をろ取し、ヘプタン及びアセトニトリルを用いて洗浄した。粗体の湿結晶(72%ee)にアセトニトリル1420mLを加え、60℃で加熱溶解した後、5℃で3時間撹拌した。析出した結晶をろ取し、冷却したアセトニトリル230mLで洗浄後、50℃で一晩乾燥することで、白色固体134gを得た(99%ee)。実施例12で得られた生成物の1H-NMR、LC-MSの測定結果は実施例1で示したものと同様であった。
[実施例13]4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸t-ブチルエステルの合成(第1工程)

〔式中、*は不斉中心を表す。〕
30mLフラスコに1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸t-ブチルエステル150mg、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド4.9mg、50%水酸化カリウム水溶液123μL、及びトルエン1.3mLを加え、5℃に冷却した。2.5M臭化メチル-トルエン溶液を加え、0℃で20時間撹拌した。反応終了後、水1.3mLを加え、10分撹拌した後、酢酸エチルを加えて分液した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過した後、ろ液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製し、白色固体127mgを得た(50%ee)。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.71 (s,1H),7.50 (s,2H),7.19-7.03 (m,2H),6.93-6.49 (m,2H),6.20 (s,1H),4.12 (q,J = 7.1 Hz,1H),3.90 (brs,1H),2.45 (s,3H),2.34 (s,2H),2.14-1.93 (m,1H),1.60-1.48 (m,3H),1.48-1.29 (m,6H),1.29-1.20 (m,1H).
ESI/MS(m/z)475(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.71 (s,1H),7.50 (s,2H),7.19-7.03 (m,2H),6.93-6.49 (m,2H),6.20 (s,1H),4.12 (q,J = 7.1 Hz,1H),3.90 (brs,1H),2.45 (s,3H),2.34 (s,2H),2.14-1.93 (m,1H),1.60-1.48 (m,3H),1.48-1.29 (m,6H),1.29-1.20 (m,1H).
ESI/MS(m/z)475(M+H) +.
[実施例14]4-メチル-1-(4-ブロモベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第1工程)

〔式中、*は不斉中心を表す。〕
15mL試験管に1-(4-ブロモベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル433mg、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド2.3mg、炭酸セシウム1.36g、5℃に冷却した3.27mol/L臭化メチル-ブロモベンゼン溶液1.59mL、及び水0.171mLを加え、そのまま5℃で24時間撹拌した。反応終了後、アセトン0.69mL及び水1.35mLを加え、10分撹拌した後、酢酸エチルを加えて分液した。有機層を無水硫酸ナトリウムで乾燥し、ろ過した後、ろ液を減圧濃縮した。得られた粗体(78%ee)にアセトニトリル2.24mLを加え、60℃で加熱溶解した後、5℃で3時間撹拌した。析出した結晶をろ取し、冷却したアセトニトリルで洗浄後、50℃で一晩乾燥することで、白色固体279mgを得た(95%ee)。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.85-7.61 (m,1H),7.59-7.40 (m,1H),7.35-7.26 (m,2H),7.25-7.15 (m,1H),7.10 (d,J = 8.3 Hz,2H),6.65 (m,1H),4.40-4.14 (m,1H),4.14-4.00 (m,2H),3.84 (m,1H),2.55 (dd,J = 14.9,5.8 Hz,1H),2.01 (dt,J = 14.4,6.8 Hz,1H),1.55 (d,J = 7.2 Hz,3H),1.26-1.07 (m,3H).
ESI/MS(m/z)430(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.85-7.61 (m,1H),7.59-7.40 (m,1H),7.35-7.26 (m,2H),7.25-7.15 (m,1H),7.10 (d,J = 8.3 Hz,2H),6.65 (m,1H),4.40-4.14 (m,1H),4.14-4.00 (m,2H),3.84 (m,1H),2.55 (dd,J = 14.9,5.8 Hz,1H),2.01 (dt,J = 14.4,6.8 Hz,1H),1.55 (d,J = 7.2 Hz,3H),1.26-1.07 (m,3H).
ESI/MS(m/z)430(M+H) +.
[実施例15]4-メチル-1-ベンゾイル-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第1工程)

〔式中、*は不斉中心を表す。〕
15mL試験管に1-ベンゾイル-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル351mg、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド2.3mg、炭酸セシウム1.36g、フッ化セシウム15.8mg、5℃に冷却した3.27mol/L臭化メチル-ブロモベンゼン溶液1.59mL、及び水0.171mLを加え、そのまま5℃で24時間撹拌した。反応終了後、アセトン0.69mL及び水1.35mLを加え、10分撹拌した後、酢酸エチルを加えて分液した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過した後、ろ液を減圧濃縮した。得られた粗体(75%ee)にアセトニトリル1.82mLを加えて60℃で加熱溶解した後、5℃まで冷却して析出した結晶懸濁液を一晩撹拌した。ろ過した後、ろ液を減圧濃縮して、無色オイル197mgを得た(94%ee)。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.67 (brs,1H),7.34-7.05 (m,6H),6.66 (d,J = 6.7 Hz,1H),4.27 (d,J = 7.1 Hz,1H),4.10 (qq,J = 10.8,7.1 Hz,2H),3.86 (m,1H),2.56 (dt,J = 14.8,6.0 Hz,1H),2.03 (dt,J = 14.3,6.1 Hz,1H),1.57 (s,3H),1.15 (t,J = 7.1 Hz,3H).
ESI/MS(m/z)352(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.67 (brs,1H),7.34-7.05 (m,6H),6.66 (d,J = 6.7 Hz,1H),4.27 (d,J = 7.1 Hz,1H),4.10 (qq,J = 10.8,7.1 Hz,2H),3.86 (m,1H),2.56 (dt,J = 14.8,6.0 Hz,1H),2.03 (dt,J = 14.3,6.1 Hz,1H),1.57 (s,3H),1.15 (t,J = 7.1 Hz,3H).
ESI/MS(m/z)352(M+H) +.
[実施例16]4-メチル-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-1,4-ジカルボン酸-1-t-ブチル-4-エチルエステルの合成(第1工程)

〔式中、*は不斉中心を表す。〕
15mL試験管に5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-1,4-ジカルボン酸-1-t-ブチル-4-エチルエステル346.7mg、(R,R)-3,5-ビストリフルオロメチルフェニル-NASブロミド2.3mg、50%水酸化カリウム水溶液466.8mg、3.27mol/Lヨウ化メチル-ブロモベンゼン溶液1.59mL、及び水0.171mLを加え、そのまま室温で40時間、40℃で8時間撹拌した。反応終了後、アセトン0.69mL及び水1.35mLを加え、10分撹拌した後、酢酸エチルを加えて分液した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過した後、ろ液を減圧濃縮し、黄色固体611mgを得た。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.38 (dd,J = 7.8,1.7 Hz,1H),7.31 (ddd,J = 8.8,7.2,1.7 Hz,1H),6.91-6.89 (m,2H),4.27 (q,J = 7.1 Hz,2H),3.51 (t,J = 6.2 Hz,2H),2.88 (s,3H),2.59 (t,J = 6.2 Hz,2H),1.47 (s,9H),1.34 (t,J = 7.1 Hz,3H).
ESI/MS(m/z)348(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.38 (dd,J = 7.8,1.7 Hz,1H),7.31 (ddd,J = 8.8,7.2,1.7 Hz,1H),6.91-6.89 (m,2H),4.27 (q,J = 7.1 Hz,2H),3.51 (t,J = 6.2 Hz,2H),2.88 (s,3H),2.59 (t,J = 6.2 Hz,2H),1.47 (s,9H),1.34 (t,J = 7.1 Hz,3H).
ESI/MS(m/z)348(M+H) +.
実施例16の不斉収率は、得られた4-メチル-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピンジカルボン酸-1-t-ブチル-4-エチルエステルを4-メチル-5-オキソ-1-(2,2,2-トリフルオロアセチル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルに変換し、HPLCで測定した。その結果、不斉収率は63%eeであった。
実施例17~30は実施例1の1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルを表3に示す基質に変更し、表3に示す反応条件に従い反応を行った。単離収率、及び不斉収率(%ee)を表3及び表4に、合成した化合物のデータを表5に示した。



[実施例31](4R)-5-ヒドロキシ-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第2工程)

3Lフラスコに水素化ホウ素ナトリウム53g、硫酸アンモニウム462g、及びテトラヒドロフラン1575mLを加え、40℃で4時間撹拌した。反応液を室温まで放冷後、水577mLを加え分液した。水294mLを用いて分液洗浄後、有機層を減圧濃縮しテトラヒドロフランを除去した。残渣に(R)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-5-オキソ-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル105g、トルエン1575mLを加え、60℃で9時間撹拌した。反応液を室温まで放冷後、酢酸エチル500mLを加え分液し、水525mL及び10%食塩水525mLを用いて順に分液洗浄した。有機層を減圧濃縮し、無色アモルファス体105gを得た。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.88-7.29 (m,3H),7.21-6.75 (m,5H),6.61-6.52 (m,1H),6.31-6.15 (m,4H),5.55-4.06 (m,2H),3.58 (6H),2.55-2.33 (m,6H),2.04-1.62 (m,2H),1.40-1.20 (m,3H),1.16-1.00 (m,3H).
ESI/MS(m/z)448(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.88-7.29 (m,3H),7.21-6.75 (m,5H),6.61-6.52 (m,1H),6.31-6.15 (m,4H),5.55-4.06 (m,2H),3.58 (6H),2.55-2.33 (m,6H),2.04-1.62 (m,2H),1.40-1.20 (m,3H),1.16-1.00 (m,3H).
ESI/MS(m/z)448(M+H) +.
[実施例32](4S)-5-クロロ-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第3工程)
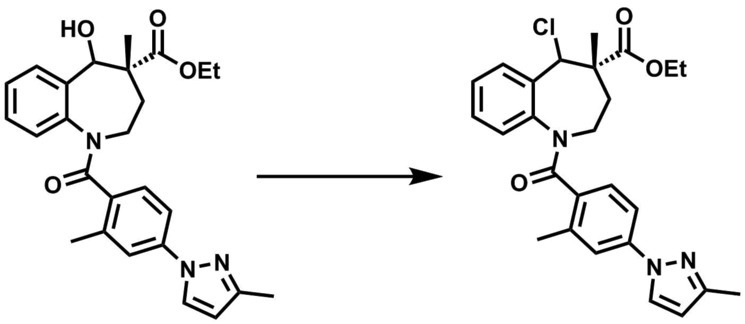
3Lフラスコに(4R)-5-ヒドロキシ-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル105gのトルエン溶液を加え、室温でオキシ塩化リン261g及びピリジン451gを加えた。反応液を60℃で19時間撹拌した後、酢酸エチル1L、1N塩酸水溶液1Lを室温で加えて分液した。有機層を10%食塩水0.5L、10%炭酸水素ナトリウム水溶液0.5L、及び5%食塩水0.5Lで順に洗浄後、有機層を減圧濃縮し、淡黄色アモルファス70gを得た。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.84-7.33 (m,2H),7.19-6.53 (m,6H),6.19 (d,J = 2.5 Hz,1H),5.42-2.72 (m,6H),2.53-1.92 (m,8H),1.43 (m,3H),1.17-0.98 (m,3H).
ESI/MS(m/z)466(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.84-7.33 (m,2H),7.19-6.53 (m,6H),6.19 (d,J = 2.5 Hz,1H),5.42-2.72 (m,6H),2.53-1.92 (m,8H),1.43 (m,3H),1.17-0.98 (m,3H).
ESI/MS(m/z)466(M+H) +.
[実施例33](S)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステルの合成(第4工程)

2Lフラスコに(4S)-5-クロロ-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル70gのメタノール溶液を加え、室温で10%パラジウム-炭素7g及びメタノール770mLを加えた。水素ガスを微加圧の条件下で吹き込み、室温で3時間撹拌した。反応液をろ過し、メタノール70mLで洗浄後、ろ液を減圧濃縮し、無色アモルファス65gを得た。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.95-7.41 (m,2H),7.19-6.53 (m,7H),6.19 (d,J = 2.4 Hz,1H),4.94-4.50 (m,2H),3.56-2.77 (m,2H),2.46 (m,6H),1.09 (m,5H).
ESI/MS(m/z)432(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.95-7.41 (m,2H),7.19-6.53 (m,7H),6.19 (d,J = 2.4 Hz,1H),4.94-4.50 (m,2H),3.56-2.77 (m,2H),2.46 (m,6H),1.09 (m,5H).
ESI/MS(m/z)432(M+H) +.
[実施例34](S)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸の合成
(第5工程)

(第5工程)
2Lフラスコに30%水酸化ナトリウム60mL、水188mL、メタノール130mL、及び(S)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸エチルエステル65gのメタノール溶液を60℃で加え、そのまま2時間撹拌した。反応液を室温まで放冷後、水230mL及びトルエン325mLを加え、分液した。有機層を35%塩酸水溶液60mL及び10%食塩水330mLで順に洗浄後、有機層を減圧濃縮し、白色固体59gを得た。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.98-7.30 (m,4H),7.22-6.84 (m,3H),6.69-6.38 (m,1H),6.33-6.11 (m,1H),5.40-4.92 (m,1H),4.50-4.19 (m,1H),4.00 (qq,J = 7.1,3.7 Hz,1H),3.63-2.66 (m,2H),2.56 (q,J = 6.9,6.0 Hz,3H),2.47-2.28 (m,3H),1.58-1.28 (m,3H),1.20-0.98 (m,3H).
ESI/MS(m/z)404(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.98-7.30 (m,4H),7.22-6.84 (m,3H),6.69-6.38 (m,1H),6.33-6.11 (m,1H),5.40-4.92 (m,1H),4.50-4.19 (m,1H),4.00 (qq,J = 7.1,3.7 Hz,1H),3.63-2.66 (m,2H),2.56 (q,J = 6.9,6.0 Hz,3H),2.47-2.28 (m,3H),1.58-1.28 (m,3H),1.20-0.98 (m,3H).
ESI/MS(m/z)404(M+H) +.
[実施例35](S)-N-[(S)-1-ヒドロキシプロパン-2-イル]-4-メチル-1-[2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル]-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボキサミド(化合物A)の合成(第6工程)
(第6工程)

(第6工程)
0.5Lフラスコに(S)-4-メチル-1-(2-メチル-4-(3-メチル-1H-ピラゾール-1-イル)ベンゾイル)-2,3,4,5-テトラヒドロ-1H-ベンゾ[b]アゼピン-4-カルボン酸59g、トルエン300mLを加え、塩化チオニル12.5mLを60℃で加えた。反応液を45℃で1時間撹拌後、室温まで放冷した。反応液を減圧濃縮後、酢酸エチル48mLを加え、酸クロリド体の酢酸エチル溶液を調整した。1Lフラスコに炭酸ナトリウム30.5g、水196mL、酢酸エチル59mL、L-アラニノール11.9gを加え、5℃以下まで冷却後、別途調整した酸クロリド体の酢酸エチル溶液を滴下した。反応液を5℃以下で30分間撹拌後、10℃で16時間撹拌した。反応液に酢酸エチル196mL、水196mLを加えて分液後、水層を酢酸エチルで分液抽出した。有機層を減圧濃縮し、淡黄色アモルファス状の化合物A70gを得た。
得られた表記化合物の測定結果を以下に示す。
1H-NMR(400Hz,CDCl3) δ7.98 (br,1H),7.50 (br,1H),7.30-6.90 (m,4H),7.21 (d,1H),6.73-6.69 (m,1H),6.25 (br,1H),4.70-2.80 (m,2H),4.07-3.80 (m,1H),3.60-3.20 (m,2H),3.17 (m,2H),2.47 (s,3H),2.28 (s,3H),2.40-1.60 (m,2H),1.41-1.04 (m,6H).
ESI/MS(m/z)461(M+H) +.
1H-NMR(400Hz,CDCl3) δ7.98 (br,1H),7.50 (br,1H),7.30-6.90 (m,4H),7.21 (d,1H),6.73-6.69 (m,1H),6.25 (br,1H),4.70-2.80 (m,2H),4.07-3.80 (m,1H),3.60-3.20 (m,2H),3.17 (m,2H),2.47 (s,3H),2.28 (s,3H),2.40-1.60 (m,2H),1.41-1.04 (m,6H).
ESI/MS(m/z)461(M+H) +.
[実施例36]化合物Aのイソプロパノール和物
実施例35で得られた化合物A5.0gに対し、イソプロパノール65mLを加え、室温で30分間撹拌した。析出した懸濁液を加温溶解させた後、室温まで放冷し、5℃で一晩撹拌した。懸濁液をろ取し、冷却したイソプロパノールで洗浄し、40℃で一晩乾燥することで、白色固体4.9gを得た。得られた化合物を熱重量装置で分析したところ、イソプロパノールの含有量は、化合物Aに対して8.2%であり、モル比で化合物Aに対して0.7倍量であった。
実施例35で得られた化合物A5.0gに対し、イソプロパノール65mLを加え、室温で30分間撹拌した。析出した懸濁液を加温溶解させた後、室温まで放冷し、5℃で一晩撹拌した。懸濁液をろ取し、冷却したイソプロパノールで洗浄し、40℃で一晩乾燥することで、白色固体4.9gを得た。得られた化合物を熱重量装置で分析したところ、イソプロパノールの含有量は、化合物Aに対して8.2%であり、モル比で化合物Aに対して0.7倍量であった。
[実施例37]化合物Aのエタノール和物
実施例35で得られた化合物A0.15gに対し、エタノール2mLを加え、室温で30分間撹拌した。析出した懸濁液を加温溶解させた後、室温まで放冷し、5℃で一晩撹拌した。懸濁液をろ取し、冷却したエタノールで洗浄し、40℃で一晩乾燥することで、白色固体0.07gを得た。得られた化合物を熱重量装置で分析したところ、エタノールの含有量は、化合物Aに対して5.7%であり、モル比で化合物Aに対して0.6倍量であった。
実施例35で得られた化合物A0.15gに対し、エタノール2mLを加え、室温で30分間撹拌した。析出した懸濁液を加温溶解させた後、室温まで放冷し、5℃で一晩撹拌した。懸濁液をろ取し、冷却したエタノールで洗浄し、40℃で一晩乾燥することで、白色固体0.07gを得た。得られた化合物を熱重量装置で分析したところ、エタノールの含有量は、化合物Aに対して5.7%であり、モル比で化合物Aに対して0.6倍量であった。
[実施例38]化合物Aのノルマルプロパノール和物
実施例35で得られた化合物A5.0gに対し、ノルマルプロパノール31mLを加え、室温で15分間撹拌した。析出した懸濁液を加温溶解させた後、室温まで放冷し、5℃で一晩撹拌した。懸濁液をろ取し、冷却したノルマルプロパノールで洗浄し、40℃で一晩乾燥することで、白色固体4.1gを得た。得られた化合物を熱重量装置で分析したところ、ノルマルプロパノールの含有量は、化合物Aに対して8.1%であり、モル比で化合物Aに対して0.7倍量であった。
実施例35で得られた化合物A5.0gに対し、ノルマルプロパノール31mLを加え、室温で15分間撹拌した。析出した懸濁液を加温溶解させた後、室温まで放冷し、5℃で一晩撹拌した。懸濁液をろ取し、冷却したノルマルプロパノールで洗浄し、40℃で一晩乾燥することで、白色固体4.1gを得た。得られた化合物を熱重量装置で分析したところ、ノルマルプロパノールの含有量は、化合物Aに対して8.1%であり、モル比で化合物Aに対して0.7倍量であった。
今回開示された実施の形態はすべての点で例示であって、制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなく特許請求の範囲によって示され、特許請求の範囲と均等の意味、及び範囲内でのすべての変更が含まれることが意図される。
本発明方法は、医薬品合成中間体として有用な化合物(VI)及び化合物(VII)を安全且つ安価に工業的に製造する方法として、また、医薬品として有用な化合物Aを安全且つ安価に工業的に製造する方法として有用である。
Claims (17)
- 式(I):

R2は、水素原子、C2~C6アルケニル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいフェニル基、又はハロゲン原子で置換されていてもよいC1~C6アルキル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C6アルキル基、C2~C6アルケニル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいC7~C12アラルキル基を表す。〕
で示される化合物を、有機溶媒中で、
式(II)

R7は、それぞれ独立にC1~C4アルキレン基又はナフタレンジイル基を表す。
R8は、水素原子又は一緒になって単結合を表す。
X-は、ハロゲン化物アニオンを表す。〕
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
式(III):

で示される光学活性化合物を製造し、次いで当該化合物のカルボニル基を、有機溶媒中で還元剤により還元し、
式(IV):

で示される光学活性化合物(IV)を製造し、次いで当該化合物のヒドロキシル基をクロル化剤を用いてクロル化し、得られた化合物を金属触媒存在下で水素を用いて還元的脱クロル化し、得られた化合物のベンゾアゼピン環第4位のカルボン酸エステルを塩基性溶液中で加水分解し、得られたカルボン酸をクロル化剤を用いて酸クロリドに変換し、更に塩基の存在下、L-アラニノールと反応させることを特徴とする、
式(V)

で示される光学活性化合物を製造する方法、又は前記光学活性化合物を更に溶媒で再結晶することを特徴とする、前記光学活性化合物の溶媒和物を製造する方法。 - 式(I):
で示される化合物を、有機溶媒中で、
式(II)
で示される不斉四級アンモニウム塩、及び塩基の存在下でメチル化剤によりメチル化し、
式(III):

で示される光学活性化合物を製造する方法。 - 請求項2に記載の製造方法により得られた前記式(III)で示される化合物のカルボニル基を、有機溶媒中で還元剤により還元し、
式(IV):

で示される光学活性化合物(IV)を製造する方法。 - 請求項3に記載の製造方法により得られた前記式(IV)で示される化合物のヒドロキシル基をクロル化剤を用いてクロル化し、得られた化合物を金属触媒存在下で水素を用いて還元的脱クロル化し、得られた化合物のベンゾアゼピン環第4位のカルボン酸エステルを塩基性溶液中で加水分解し、得られたカルボン酸をクロル化剤を用いて酸クロリドに変換し、更に塩基の存在下、L-アラニノールと反応させることを特徴とする、
式(V)

で示される光学活性化合物を製造する方法、又は前記光学活性化合物を更に溶媒で再結晶することを特徴とする、前記光学活性化合物の溶媒和物を製造する方法。 - 式(I):

R2は、水素原子、アリル基、C1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基、-COR3、-SO2R4、又は-CO2R5を表す。
R3は、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、ハロゲン原子で置換されていてもよいC1~C6アルキル基、又は水素原子を表す。
R4は、C1~C4アルキル基で置換されていてもよいフェニル基を表す。
R5は、ハロゲン原子で置換されていてもよいC1~C4アルキル基、アリル基、又はC1~C4アルキル基若しくはC1~C4アルコキシ基で置換されていてもよいベンジル基を表す。〕
で示される化合物を、有機溶媒中で、
式(II-1)

式(III):

で示される光学活性化合物を製造する方法。 - 前記不斉四級アンモニウム塩及び前記塩基とともに、無機塩の存在下でメチル化反応を行うことを特徴とする、請求項1、2又は5に記載の製造方法。
- 前記有機溶媒が、トルエン、キシレン、クロロベンゼン、及びブロモベンゼンからなる群より選択される1種以上である、請求項1、2又は5に記載の製造方法。
- 前記塩基が、炭酸カリウム、炭酸セシウム、水酸化カリウム、及び水酸化セシウムからなる群より選択される1種以上である、請求項1、2又は5に記載の製造方法。
- 前記無機塩が、フッ化セシウム、塩化セシウム、臭化カリウム、塩化ルビジウム、臭化ルビジウム、及び臭化ナトリウムからなる群より選択される1種以上である、請求項6に記載の製造方法。
- 前記還元剤がボラン化合物である、請求項1又は3に記載の製造方法。
- 前記R1がメチル基又はエチル基である、請求項1~10のいずれか一項に記載の製造方法。
- 前記R2が-COR3又は-SO2R4であり、前記R3が、ハロゲン原子、C1~C4アルキル基で置換されていてもよい芳香族ヘテロ環基、及びハロゲン原子で置換されていてもよいC1~C6アルキル基からなる群から選択される1若しくは複数の置換基で置換されていてもよいフェニル基、又は水素原子であり、R4が、C1~C4アルキル基で置換されていてもよいフェニル基である、請求項1~11のいずれか一項に記載の製造方法。
- 前記R2が-COR3であり、前記R3が、

で表される置換フェニル基である、請求項12に記載の製造方法。 - 前記R2が-COR3であり、前記R3が

- 式(VI):
で示される化合物又はその薬学的に許容される塩。 - 式(VII):
で示される化合物又はその薬学的に許容される塩。 - 前記R1が、メチル基又はエチル基である、請求項15又は16に記載の化合物又はその薬学的に許容される塩。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019026983A JP2022055368A (ja) | 2019-02-19 | 2019-02-19 | ベンゾアゼピン誘導体の製造方法及びその中間体 |
| JP2019-026983 | 2019-02-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020171073A1 true WO2020171073A1 (ja) | 2020-08-27 |
Family
ID=72144346
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/006305 Ceased WO2020171073A1 (ja) | 2019-02-19 | 2020-02-18 | ベンゾアゼピン誘導体の製造方法及びその中間体 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2022055368A (ja) |
| WO (1) | WO2020171073A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114950492A (zh) * | 2022-05-31 | 2022-08-30 | 安徽大学 | 一种用于光电催化杀灭细菌的1,2-二氨基蒽醌-二硫化钼复合抗菌材料 |
| CN116178271A (zh) * | 2021-11-26 | 2023-05-30 | 四川大学 | 一种氮杂芳环化合物的选择性n-甲基化方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62164668A (ja) * | 1985-12-12 | 1987-07-21 | イ−・ア−ル・スクイブ・アンド・サンズ・インコ−ポレイテツド | ベンズアゼピン誘導体 |
| JPS6440462A (en) * | 1987-07-27 | 1989-02-10 | Squibb & Sons Inc | Benzazepine derivative |
| JPS6471859A (en) * | 1987-08-26 | 1989-03-16 | Squibb & Sons Inc | Benzazepine derivative |
| JP2003514022A (ja) * | 1999-11-18 | 2003-04-15 | アンテックスファーマ・インコーポレーテッド | 置換された1−ベンズアゼピンとその誘導体 |
| JP2003529572A (ja) * | 2000-03-31 | 2003-10-07 | ポリメーリ エウローパ ソシエタ ペル アチオニ | 置換多環式シクロペンタジエン及びその製造方法 |
| JP2004175758A (ja) * | 2002-11-28 | 2004-06-24 | Nagase & Co Ltd | β−ケトエステルを用いてアルキル化物を立体選択的に製造するための方法 |
| JP2004536079A (ja) * | 2001-05-16 | 2004-12-02 | アンテックスファーマ・インコーポレーテッド | 置換1−ベンズアゼピンおよびその誘導体 |
| WO2014104209A1 (ja) * | 2012-12-26 | 2014-07-03 | 株式会社 三和化学研究所 | 新規ベンゾアゼピン誘導体及びその医薬用途 |
| JP2016040338A (ja) * | 2015-12-18 | 2016-03-24 | 株式会社三和化学研究所 | 1−ベンゾイル−5−オキソ−2,3,4,5−テトラヒドロ−1H−ベンゾ[b]アゼピン−4−カルボン酸エステルの製造方法 |
| JP2016185993A (ja) * | 2016-08-02 | 2016-10-27 | 株式会社三和化学研究所 | 1−ベンゾイル−5−オキソ−2,3,4,5−テトラヒドロ−1H−ベンゾ[b]アゼピン−4−カルボン酸エステルの製造方法 |
-
2019
- 2019-02-19 JP JP2019026983A patent/JP2022055368A/ja active Pending
-
2020
- 2020-02-18 WO PCT/JP2020/006305 patent/WO2020171073A1/ja not_active Ceased
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62164668A (ja) * | 1985-12-12 | 1987-07-21 | イ−・ア−ル・スクイブ・アンド・サンズ・インコ−ポレイテツド | ベンズアゼピン誘導体 |
| JPS6440462A (en) * | 1987-07-27 | 1989-02-10 | Squibb & Sons Inc | Benzazepine derivative |
| JPS6471859A (en) * | 1987-08-26 | 1989-03-16 | Squibb & Sons Inc | Benzazepine derivative |
| JP2003514022A (ja) * | 1999-11-18 | 2003-04-15 | アンテックスファーマ・インコーポレーテッド | 置換された1−ベンズアゼピンとその誘導体 |
| JP2003529572A (ja) * | 2000-03-31 | 2003-10-07 | ポリメーリ エウローパ ソシエタ ペル アチオニ | 置換多環式シクロペンタジエン及びその製造方法 |
| JP2004536079A (ja) * | 2001-05-16 | 2004-12-02 | アンテックスファーマ・インコーポレーテッド | 置換1−ベンズアゼピンおよびその誘導体 |
| JP2004175758A (ja) * | 2002-11-28 | 2004-06-24 | Nagase & Co Ltd | β−ケトエステルを用いてアルキル化物を立体選択的に製造するための方法 |
| WO2014104209A1 (ja) * | 2012-12-26 | 2014-07-03 | 株式会社 三和化学研究所 | 新規ベンゾアゼピン誘導体及びその医薬用途 |
| JP2016040338A (ja) * | 2015-12-18 | 2016-03-24 | 株式会社三和化学研究所 | 1−ベンゾイル−5−オキソ−2,3,4,5−テトラヒドロ−1H−ベンゾ[b]アゼピン−4−カルボン酸エステルの製造方法 |
| JP2016185993A (ja) * | 2016-08-02 | 2016-10-27 | 株式会社三和化学研究所 | 1−ベンゾイル−5−オキソ−2,3,4,5−テトラヒドロ−1H−ベンゾ[b]アゼピン−4−カルボン酸エステルの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| VANDORMAEL BART ET AL.: "Asymmetric Synthesis and Conformational Analysis by NMR Spectroscopy and MD of Aba- and alpha-MeAba-Containing Dermorphin Analogues", CHEMMEDCHEM, vol. 6, no. 11, 2011, pages 2035 - 2047, XP008166307, DOI: 10.1002/cmdc.201100314 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116178271A (zh) * | 2021-11-26 | 2023-05-30 | 四川大学 | 一种氮杂芳环化合物的选择性n-甲基化方法 |
| CN114950492A (zh) * | 2022-05-31 | 2022-08-30 | 安徽大学 | 一种用于光电催化杀灭细菌的1,2-二氨基蒽醌-二硫化钼复合抗菌材料 |
| CN114950492B (zh) * | 2022-05-31 | 2023-09-08 | 安徽大学 | 一种用于光电催化杀灭细菌的1,2-二氨基蒽醌-二硫化钼复合抗菌材料 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022055368A (ja) | 2022-04-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TW201210990A (en) | Manufacturing method for compounds having HIV integrase inhibitory activities | |
| PT2471780E (pt) | Sais oxalato de ivabradina cristalinos e seus polimorfos | |
| JP2014514292A (ja) | アミン中間体化合物を使用してドロネダロンを製造するための還元アミノ化方法 | |
| JP2014514291A (ja) | メシル化によるドロネダロンの製造方法 | |
| JP2014509640A (ja) | N−ブチル化によるドロネダロンの調製方法 | |
| CA2773064C (fr) | Nouveau procede de synthese de l'ivabradine et de ses sels d'addition a un acide pharmaceutiquement acceptable | |
| WO2009056993A2 (en) | A process for the synthesis of ramelteon and its intermediates | |
| WO2020171073A1 (ja) | ベンゾアゼピン誘導体の製造方法及びその中間体 | |
| EP3585388A1 (en) | An improved process for preparation and purification of vortioxetine hydrobromide | |
| CN104837817B (zh) | 制备3‑氨基‑哌啶化合物的合成路线 | |
| KR100753353B1 (ko) | 졸미트립탄 화합물의 제조방법 | |
| CN112679411B (zh) | 一种手性5-(甲胺基)六氢环戊并[c]吡咯-2(1H)-Boc甲磺酸盐的制备方法 | |
| CA2503172A1 (en) | Benzoxazocines and their use as monoamine-reuptake inhibitors | |
| US9745264B2 (en) | Method for preparing silodosin and intermediate thereof | |
| JP3831954B2 (ja) | 4−ヒドロキシ−2−ピロリドンの製法 | |
| KR101166280B1 (ko) | 시탈로프램 및 에난티오머의 제조 방법 | |
| JP2009533370A (ja) | 8−アリール−オクタノイル誘導体の製造方法 | |
| JP4011819B2 (ja) | インドール誘導体の製造法およびその中間体 | |
| CN104955803B (zh) | 通过硝基‑四氢吡啶前体制备3‑氨基‑哌啶化合物 | |
| CN115784922B (zh) | 一种(2s)-2-氨基-4-(环丙基/环丁基)丁酸的制备方法 | |
| IE913936A1 (en) | Chemical process | |
| JPS635087A (ja) | (+)−トランス−1a,2,3,4a,5,6−ヘキサヒドロ−9−ヒドロキシ−4−プロピル−4H−ナフト〔1,2−b〕−1,4−オキサジンのキラル合成 | |
| JP2022114189A (ja) | 6-ハロゲノイソインドリノン誘導体の製造方法 | |
| KR20120069710A (ko) | 4?(5?메틸피리딘?2?일아미노)피페리딘?1?카복실산 유도체의 제조방법 | |
| CA3214107A1 (en) | New process for the synthesis of 5-{5-chloro-2-[(3s)-3- [(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1h)- carbonyl]phenyl}-1,2-dimethyl-1h-pyrrole-3-carboxylic acid derivatives and its application for the production of pharmaceutical compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20759466 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20759466 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |