(54) Título: COMPOSTO, COMPOSIÇÃO FARMACÊUTICA, E, USO DA MESMA (73) Titular: H. LUNDBECK A/S, Companhia Dinamarquesa. Endereço: 9, Ottiliavej, DK-2500 Copenhagen-Valby, DINAMARCA(DK) (72) Inventor: CHRISTIAN WENZEL TORNOE; MARIO ROTTLÃNDER; NIKOLAY KHANZHIN; ANDREAS RITZÈN; WILLIAM PATRICK WATSON.
Prazo de Validade: 10 (dez) anos contados a partir de 21/11/2018, observadas as condições legais
Expedida em: 21/11/2018
Assinado digitalmente por:
Alexandre Gomes Ciancio
Diretor Substituto de Patentes, Programas de Computador e Topografias de Circuitos Integrados
»· ·«·« ·
Itf tf · • tf tf tf “COMPOSTO, COMPOSIÇÃO FARMACÊUTICA, E, USO DA MESMA” Campo da Invenção
A presente invenção diz respeito aos novos derivados de morfolina e tiomorfolina substituídos sendo abridores de canais de íon de 5 potássio da família KCNQ. Os compostos são úteis no tratamento de distúrbios e doenças que são responsivos por abrir os canais de íon de potássio da família KCNQ, uma tal doença é a epilepsia.
Fundamentos da Invenção
Os canais de íons são proteínas celulares que regulam o fluxo de íons, incluindo potássio, cálcio, cloreto e sódio dentro e fora das células. Tais canais estão presentes em todas as células animais e humanas e afetam uma variedade processos incluindo transmissão neuronal, contração muscular, e secreção celular.
Os seres humanos têm mais do que 70 genes codificando os subtipos de canais de potássio (Jentsch Nature Reviews Neuroscience 2000, 1, 21-30) com uma enorme diversidade com respeito tanto a estrutura quanto a função. Os canais de potássio neuronais, que são encontrados no cérebro, são primariamente responsáveis por manter um potencial de membrana latente negativo, bem como controlar a repolarização da membrana seguindo uma 20 ação potencial.
Um subconjunto de genes de canal de potássio é a família KCNQ. Mutações em quatro dos cinco genes KCNQ foram mostrados para doenças adjacentes incluindo arritmias cardíacas, surdez e epilepsia (Jentsch Nature Reviews Neuroscience 2000, 1, 21-30).
O gene KCNQ4 é considerado a codificar o correlativo molecular dos canais de potássio encontrados em células pilosas externas da cóclea e nas células pilosas Tipo I do aparelho vestibular, em que as mutações podem levar a uma forma de surdez hereditária.
O KCNQ1 (KvLQTl) é co-montada com o produto do gene
KCNE1 (proteína do canal K(+) mínima) no coração para formar uma corrente K(+) semelhante ao retificador de atraso cardíaco. Mutações neste canal podem causar uma forma de síndrome de QT longa extensa do tipo 1 hereditária (LQT1), bem como sendo associada com uma forma de surdez 5 (Robbins Pharmacol Ther 2001,90, 1-19).
Os genes KCNQ2 e KCNQ3 foram descobertos em 1988 e afiguram-se mutados em uma forma de epilepsia hereditária conhecida como convulsões neonatais familiares benignas (Rogawski Trends in Neurosciences 2000, 23, 393-398). As proteínas codificadas pelos genes KCNQ2 e KCNQ3 10 estão localizadas nos neurônios piramidais do córtex e hipocampo humano, regiões do cérebro associadas com geração e propagação de ataques (Cooper et al. Proceedings National Academy of Science USA 2000, 97, 4914-4919).
Os KCNQ2 e KCNQ3 são duas subunidades de canal de potássio que formam correntes M” quando expressadas in vitro. A corrente 15 M é uma corrente de potássio não inativante encontrada em muitos tipos de células neuronais. Em cada tipo de célula, esta é dominante no controle da excitabilidade da membrana sendo a única corrente sustentada na faixa da iniciação potencial de ação (Marrion Annual Review Physiology 1997, 59, 483-504). A modulação da corrente M tem efeitos dramáticos na 20 excitabilidade neuronal, por exemplo, a ativação da corrente reduzirá excitabilidade neuronal. Os abridores destes canais KCNQ ou ativadores da corrente M, reduzirão a atividade neuronal excessiva e podem deste modo ser de uso no tratamento de ataques e outras doenças e distúrbios caracterizadas pela atividade neuronal excessiva, tal como hiperexcitabilidade neuronal 25 incluindo distúrbios convulsivos, epilepsia e dor neuropática.
A retigabina (D-23129; éster etílico do ácido N-(2-amino-4-(4fluorobenzilamino)-fenil)carbâmico) e análogos destes são divulgados na EP554543. A retigabina é um composto anticonvulsivo com propriedades anticonvulsivas potentes e espectro amplo, tanto in vitro quanto in vivo. Esta é ativa após a administração oral e intraperitoneal em ratos e camundongos em uma faixa de testes anticonvulsivos incluindo: ataques eletricamente induzidos, ataques quimicamente induzidos por pentilenotetrazol, picrotoxina e N-metil-D-aspartato (NMDA) e em um modelo animal genético, o 5 camundongo DBA/2 (Rostock et al. Epilepsy Research 1996, 23, 211-223).
Além disso, a retigabina é ativa no modelo abrasamento de amígdala de ataques parciais complexos, indicando que este composto tem potencial para a terapia anti-convulsiva. Em testes clínicos, a retigabina tem recentemente mostrado eficácia em reduzir a incidência dos ataques em pacientes 10 epilépticos (Bialer et al. Epilepsy Research 2002, 51,31-71).
A retigabina foi mostrada ativar uma corrente K(+) nas células neuronais e a farmacologia desta corrente induzida manifesta concordância com a farmacologia publicada do canal M, que recentemente foi correlacionada ao heteromultímero do canal K(+)KCNQ2/3. Isto sugere que a 15 ativação dos canais KCNQ2/3 podem ser responsáveis por algumas das atividades anticonvulsivas deste agente (Wickenden et al. Molecular Pharmacology 2000, 58, 591-600) - e aqueles outros agentes operando pelo mesmo mecanismo que tem usos similares.
Os canais KCNQ 2 e 3 também foram relatados ser super 20 regulados em modelos de dor neuropática (Wickenden et al. Society for Neuroscience Abstracts 2002, 454.7), e os moduladores de canal de potássio foram supostos ser ativos tanto na dor neuropática quanto na epilepsia (Schroder et al. Neuropharmacology 2001, 40, 888898).
A retigabina também foi mostrada ser benéfica em modelos 25 animais de dor neuropática (Blackbum-Munro e Jensen European Journal of Pharmacology 2003, 460, 109-116), e é deste modo proposto, que os abridores de canais de KCNQ serão de uso no tratamento de distúrbios de dor incluindo dor neuropática.
A localização do mRNA do canal KCNQ é relatada no cérebro e outras áreas do sistema nervoso central associadas com a dor (Goldstein et al. Society for Neuroscience Abstracts 2003, 53.8).
Além de um papel na dor neuropática, a expressão do mRNA para KCNQ de 2 a 5 nos gânglios da raiz trigeminal e dorsal e no núcleo trigeminal caudal implica que os abridores destes canais também podem afetar o processamento sensorial da dor de cabeça de enxaqueca (Goldstein et al. Society for Neuroscience Abstracts 2003, 53.8).
Relatórios recentes revelaram que o mRNA para KCNQ 3 e 5, além daqueles para KCNQ2, são expressados astrócitos e células gliais. Deste 10 modo os canais KCNQ 2, 3 e 5 podem ajudar a modular a atividade sináptica no CNS e contribuir com os efeitos neuroprotetores dos abridores de canal KCNQ (Noda et al., Society for Neuroscience Abstracts 2003, 53.9).
A retigabina e outros moduladores KCNQ podem deste modo exibir proteção contra os aspectos neurodegenerativos da epilepsia, como a 15 retigabina foi mostrada prevenir a neurodegeneração límbica e a expressão de marcadores de apoptose seguindo estados epilépticos induzidos por ácido caínico no rato (Ebert et al. Epilepsy 2002, 43 Suppl 5, 86-95). Isto pode ter relevância para prevenir a progressão da epilepsia em pacientes, isto é, ser anti-epileptogênico. A retigabina também mostrou atrasar a progressão do 20 abrasamento hipocampal no rato, um outro modelo de desenvolvimento da epilepsia (Tober et al. European Joumal Of Pharmacology 1996, 303, 163169).
É deste modo sugerido, que estas propriedades da retigabina e outros moduladores KCNQ podem prevenir o dano neuronal induzido por 25 ativação neuronal excessiva, e tais compostos podem ser de uso no tratamento de doenças neurodegenerativas, e podem ser modificadores da doença (ou anti-epileptogênico) em pacientes com epilepsia.
Dado que os compostos anticonvulsivos tais como benzodiazepinas e clormetiazol são clinicamente usados no tratamento da /' síndrome de retirada do etanol e que outros compostos anticonvulsivos por exemplo, gabapentina, são muito eficazes em modelos de animais desta i síndrome (Watson et al. Neuropharmacology 1997, 36, 1369-1375), supomos que outros compostos anticonvulsivos tais como os abridores de KCNQ sejam deste modo esperados ser eficazes nesta condição.
O mRNA para subunidades KCNQ 2 e 3 é encontrado nas regiões cerebrais associadas com a ansiedade e comportamentos emocionais tais como distúrbio bipolar por exemplo, o hipocampo, a amígdala (Saganich et al. Joumal of Neuroscience 2001, 21, 4609-4624), e a retigabina é segundo notícias ativa em alguns modelos de animais de comportamento semelhante a ansiedade (Hartz et al. Joumal of Psychopharmacology 2003, 17 supl 3, A 28, B 16), e outros compostos anticonvulsivos clinicamente usados são usados no tratamento do distúrbio bipolar. Deste modo, os abridores KCNQ podem ser úteis para o tratamento de distúrbios da ansiedade e distúrbio bipolar.
A WO 200196540 divulga o uso de modulares da corrente M formados pela expressão dos genes KCNQ2 e KCNQ3 para insônia, enquanto a WO 2001092526 divulga que os moduladores de KCNQ5 podem ser utilizados para o tratamento de distúrbios do sono.
A WO 01/022953 descreve o uso da retigabina para a profilaxia e tratamento da dor neuropática tal como alodinia, dor hiperalgésica, dor fantasma, dor neuropática relacionada com neuropatia diabética e dor neuropática relacionada com enxaqueca.
A WO 02/049628 descreve o uso da retigabina para tratamento de distúrbios da ansiedade tal como ansiedade, distúrbio da ansiedade generalizada, ansiedade do pânico, distúrbio obsessivo compulsivo, fobia social, ansiedade de desempenho, distúrbio de tensão pós traumático, reação de tensão aguda, distúrbios de ajuste, distúrbios hipocondríacos, distúrbio de ansiedade separação, agorafobia e fobias específicas.
A WO 97/15300 descreve o uso da retigabina para o íf tratamento de distúrbios neurodegenerativos tais como mal de Alzheimer; coréia de Huntington; esclerose tal como esclerose múltipla e esclerose lateral amiotrófica; doença de Creutzfeld-Jakob; mal de Parkinson; encefalopatia induzida por AIDS e outras encefalopatias relacionadas com infecção sendo 5 causadas por vírus da rubéola, vírus da herpes, borrelia e por patógenos desconhecidos, neurodegeneraçoes induzidas por trauma, estados de hiperexcitação neuronal tais como de retirada de medicamento ou intoxicação, e distúrbios neurodegenerativos do sistema nervoso periférico tal como polineuropatias e polineuritidas.
Consequentemente, há um grande desejo por novos compostos, que são abridores potentes de canais de potássio da família KCNQ.
Também desejados são novos compostos com propriedades melhoradas relativo aos compostos conhecidos, que são abridores de canais de potássio da família KCNQ, tal como retigabina. O melhoramento de um ou mais dos seguintes parâmetros são desejados: meia-vida, liberação, seletividade, interações com outras medicações, biodisponibilidade, potência, formulabilidade, estabilidade química, estabilidade metabólica, permeabilidade de membrana, solubilidade e índice terapêutico. O melhoramento de tais parâmetros pode levar a melhoramentos tais como:
• um regime de dosagem melhorado pela redução do número de doses requeridas em um dia, • facilidade de administração aos pacientes em medicações múltiplas, • efeitos colaterais reduzidos, · índice terapêutico ampliado, • tolerabilidade melhorada ou • complacência melhorada.
Sumário da Invenção
Um objetivo da presente invenção é fornecer novos compostos, que são abridores potentes de canais de potássio da família KCNQ.
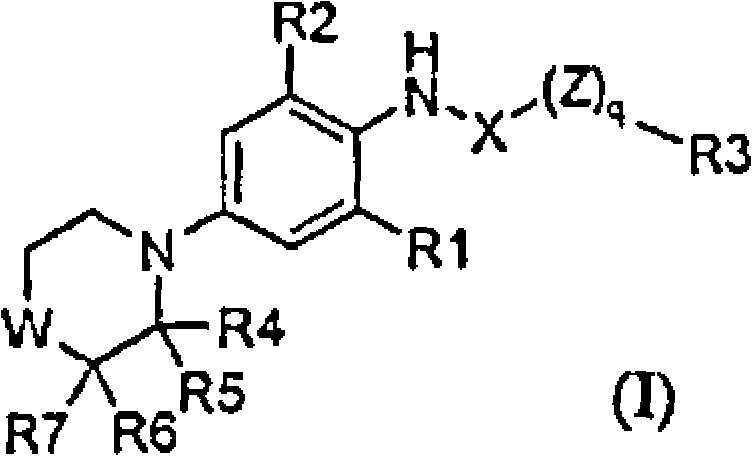
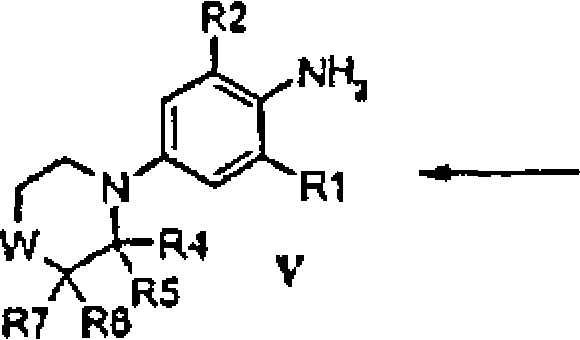
Os compostos da invenção são derivados de morfolina e tiomorfolina substituídos da fórmula geral I ou sais destes
em que q, W, X, Z, R1, R2, R3, R4, R5, R6 e R7 são como definidos abaixo.
A invenção fornece um composto da fórmula I para o uso como um medicamento.
A invenção ainda diz respeito a uma composição farmacêutica 10 que compreende um composto da fórmula I, e o uso deste.
A invenção deste modo fornece um composição farmacêutica que compreende um composto da fórmula I e um carreador ou diluente farmaceuticamente aceitáveis.
A invenção fornece o uso de um composto da fórmula I para a preparação de um medicamento para o tratamento de distúrbios de ataque, distúrbios de ansiedade, dor neuropática e distúrbios de dor de cabeça de enxaqueca ou distúrbios neurodegenerativos.
A invenção, além disso, diz respeito ao uso de um composto da fórmula I em um método de tratamento de distúrbios de ataque, distúrbios 20 de ansiedade, dor neuropática e distúrbios de dor de cabeça de enxaqueca ou distúrbios neurodegenerativos.
Descrição Detalhada da Invenção
A presente invenção diz respeito aos derivados de morfolina e tiomorfolina substituídos que são abridores potentes de canais de potássio de 25 KCNQ.
rí
Consequentemente, a presente invenção diz respeito aos derivados de morfolina e tiomorfolina substituídos da fórmula geral I
em que q é 0 ou 1;
WéOouS;
XéCO;
Zé O;
R1 é selecionado do grupo que consiste de halogênio, ciano, alqu(en/in)ila Ci_6, cicloalqu(en)ila C3.8, cicloalqu(en)ila C3.8 alqu(en/in)ila Cj. 10 6, halo-alqu(en/in)ila Cj.6, halo-cicloalqu(en)ila C3.8, halo-cicloalqu(en)ila C3.
8-alqu(en/in)ila C|«6, alqu(en/in)ilóxi Ci_6, cicloalqu(en)ilóxi C3.8 e cicloalqu(en)ila C3.8-alqu(en/in)ilóxi Ci^;
R2 é selecionado do grupo que consiste de halogênio, ciano, alqu(en/in)ila Ci_6, cicloalqu(en)ila C3.s, cicloalqu(en)ila C3.8-alqu(en/in)ila 15 Ci.6, halo-alqu(en/in)ila C]_6, halo-cicloalqu(en)ila C3.8, cicloalqu(en)ila C3.8 halo-alqu(en/in)ila Ci.6, alqu(en/in)ilóxi Ci.6, cicloalqu(en)ilóxi C3_8, cicloalqu(en)ila C3.8-alqu(en/in)ilóxi Ct.6, fenila opcionalmente substituído e piridila opcionalmente substituído; em que fenila e piridila são opcionalmente substituídos com um ou mais substituintes independentemente sendo 20 halogênio, alqu(en/in)ila C1.6, cicloalqu(en)ila C3.8 ou C3.8-cicloalqu(en)ilaalqu(en/in)ila Cj.6;
R3 é selecionado do grupo que consiste de alqu(en/in)ila Cj.io, cicloalqu(en)ila C3.8, cicloalqu(en)ila C3.s-alqu(en/in)ila Ci-q, Ar-alqu(en/in)ila
C^ô, Ar-cicloalqu(en)ila C3.8, Ar-cicloalqu(en)ila C3.8-alqu(en/in)ila Ct_6 e Ar;
Cada um dos R4, R5, Ró e R7 é independentemente selecionado do grupo que consiste de hidrogênio e Ar;
como a base livre ou sais destes.
Em uma forma de realização do composto da fórmula I, q é 0;
em uma outra forma de realização do composto da fórmula I, q é 1.
Em uma outra forma de realização do composto da fórmula I,
W é um átomo de oxigênio;
em uma outra forma de realização W é um átomo de enxofre.
Em uma outra forma de realização do composto da fórmula I,
R1 é selecionado do grupo que consiste de cicloalqu(en)ila C3.8, cicloalqu(en)ila C3.8-alqu(en/in)ila Ci-6, halo-cicloalqu(en)ila C3.8, halocicloalqu(en)ila C3_8-alqu(en/in)ila C^, cicloalqu(en)ilóxi C3.8 c cicloalqu(en)ila C3_8-alqu(en/in)ilóxi Ci.6í em uma outra forma de realização
R1 é selecionado do grupo que consiste de halogênio, halo-alqu(en/in)ila Cj.6, alqu(en/in)ila Ci_6 e ciano. Tipicamente, R1 é selecionado do grupo que consiste de halogênio, ciano, alqu(en/in)ila Cj.6, halo-alqu(en/in)ila e alqu(en/in)ilóxi Ci_6· Para ilustrar mais sem limitar a invenção uma forma de realização de R1 é halogênio;
uma outra forma de realização de R1 é ciano;
uma outra forma de realização de R1 é alqu(en/in)ila Cj.ô;
uma outra forma de realização de R1 é halo-alqu(en/in)ila Ci-óí uma outra forma de realização de R1 é alqu(en/in)ilóxi Ci_6. Em uma outra forma de realização do composto da fórmula I, 25 R2 é selecionado do grupo que consiste de cicloalqu(en)ila C3-8, cicloalqu(en)ila C3.8-alqu(en/in)ila Cm, halocicloalqu(en)ila C3.8, halocicloalqu(en)ila C3.8-alqu(en/in)ila Ci.g, cicloalqu(en)ilóxi C3.s e cicloalqu(en)ila C3.8-alqu(en/in)ilóxi Cmí em uma outra forma de realização R2 é selecionado do grupo que consiste de halogênio, halo-alqu(en/in)ila C^, alqu(en/in)ila Ci_6 e ciano.
Tipicamente, R é selecionado do grupo que consiste de halogênio, ciano, * alqu(en/in)ila Cp6, halo-alqu(en/in)ila Ci.6> alqu(en/in)ilóxi Cj-β, fenila opcionalmente substituído e piridila opcionalmente substituído. Para ilustrar mais sem limitar a invenção uma forma de realização de R2 é halogênio;
uma outra forma de realização de R2 é ciano;
uma outra forma de realização de R2 é alqu(en/in)ila Ci^J uma outra forma de realização de R2 é halo-alqu(en/in)ila Ci_6; uma outra forma de realização de R2 é alqu(en/in)ilóxi Ci.6Í
A uma outra forma de realização de R é fenila opcionalmente substituído;
uma outra forma de realização de R2 é piridila opcionalmente substituído.
Em uma outra forma de realização do composto de R2, o fenila 15 opcionalmente substituído e o piridila opcionalmente substituído podem ser substituídos com um ou mais substituintes independentemente selecionados do grupo que consiste de halogênio ou alqu(en/in)ila Ci.g;
em uma outra forma de realização de R2, fenila e piridila não são substituídos;
ainda em uma outra forma de realização de R , o fenila opcionalmente substituído e o piridila opcionalmente substituído são substituídos com um substituinte;
ainda em uma outra forma de realização de R, o fenila opcionalmente substituído e o piridila opcionalmente substituído são 25 substituídos com dois substituintes;
ainda em uma outra forma de realização de R , o fenila opcionalmente substituído e o piridila opcionalmente substituído são substituídos com três substituintes.
Em uma outra forma de realização do composto da fórmula I,
Í7
R3 é selecionado do grupo que consiste de cicloalqu(en)ila C3.8, Arcicloalqu(en)ila C3_s e Ar-cicloalqu(en)ila C3.g-alqu(en/in)ila Ci_6.
• Tipicamente, R é selecionado do grupo que consiste de alqu(en/in)ila C|.io, cicloalqu(en)ila C3.8-alqu(en/in)ila Ci.6, Ar-alqu(en/in)ila
Ci_6 e Ar.
Para ilustrar mais sem limitar a invenção uma forma de realização de R3 é alqu(en/in)ila C1.1 oi uma outra forma de realização de R3 é cicloalqu(en)ila C3_8alqu(en/in)ila C1.6;
uma outra forma de realização de R3 é Ar-alqu(en/in)ila C^;
uma outra forma de realização de R3 é Ar.
Em uma outra forma de realização do composto da fórmula I, Ar é selecionado do grupo que consiste de furano opcionalmente substituído, tiazol opcionalmente substituído, quinolina opcionalmente substituído, indol 15 opcionalmente substituído, pirimidina opcionalmente substituído, pirrol opcionalmente substituído e oxazol opcionalmente substituído; em uma outra forma de realização Ar é selecionado do grupo que consiste de fenila opcionalmente substituído, tiofeno opcionalmente substituído e naftila opcionalmente substituído;
em uma outra forma de realização Ar é selecionado do grupo que consiste de fenila opcionalmente substituído, tiofeno opcionalmente substituído, naftila opcionalmente substituído e 2,3-diidro-benzofurano opcionalmente substituído;
em uma outra forma de realização Ar é selecionado do grupo que consiste de fenila opcionalmente substituído e piridina opcionalmente substituído.
Em uma forma de realização da invenção, Ar representa fenila opcionalmente substituído, naftila opcionalmente substituído, tiofeno opcionalmente substituído ou 2,3-diidro-benzofurano opcionalmente
Μ substituído.
Tipicamente, Ar é selecionado do grupo que consiste de fenila opcionalmente substituído, naftila opcionalmente substituído, piridina opcionalmente substituído, 2,3-diidro-benzofurano opcionalmente substituído 5 e tiofeno opcionalmente substituído.
Para ilustrar mais sem limitar a invenção, uma forma de realização de Ar é fenila opcionalmente substituído;
uma outra forma de realização de Ar é naftila opcionalmente substituído;
uma outra forma de realização de Ar é piridina opcionalmente substituído;
uma outra forma de realização de Ar é 2,3-diidro-benzofurano opcionalmente substituído;
uma outra forma de realização de Ar é tiofeno opcionalmente substituído.
Em uma outra forma de realização do composto da fórmula I, Ar se refere a sistemas aromáticos opcionalmente substituídos de 5 a 1 átomos de carbono.
Tipicamente, tais sistemas aromáticos opcionalmente 20 substituídos de 5 a 1 átomos de carbono são selecionados de fenila opcionalmente substituído e naftila opcionalmente substituído.
Em uma outra forma de realização do composto da fórmula I, Ar se refere aos sistemas aromáticos opcionalmente substituídos de 5 a 10 átomos de carbono em que 1, 2, 3 ou 4 átomos de carbono são substituídos por heteroátomos independentemente selecionados de N, S, ou O.
Em uma outra forma de realização do composto da fórmula I, tal sistema aromático opcionalmente substituído de 5 a 1 átomos de carbono em que 1, 2, 3 ou 4 átomos de carbono são substituídos por heteroátomos é selecionado do grupo que consiste de piridina opcionalmente substituído, tiofeno opcionalmente substituído, furano opcionalmente substituído, tiazol opcionalmente substituído, quinolina opcionalmente substituído, indol » opcionalmente substituído, 2,3-diidro-benzofurano opcionalmente substituído, * pirimidina opcionalmente substituído, pirrol opcionalmente substituído e oxazol opcionalmente substituído.
Tipicamente, tal sistema aromático opcionalmente substituído de 5 a 1 átomos de carbono em que 1, 2, 3 ou 4 átomos de carbono sào substituídos por heteroátomos é selecionado do grupo que consiste de piridina opcionalmente substituído, 2,3-diidrobenzofurano opcionalmente substituído 10 e tiofeno opcionalmente substituído.
Em uma outra forma de realização do composto da fórmula I, Ar é opcionalmente substituído com um ou mais substituintes independentemente sendo hidróxi, acila, nitro ou ciano, -CO-NHalqu(en/in)ila Ci^, -CO-N(alqu(en/in)ila Ci_6)2, -NH2, -NH-alqu(en/in)ila Ci.6, 15 -N(alqu(en/in)ila C 1.6)2, -S-alqu(en/in)ila Cj_6, -SO2-alqu(en/in)ila Ci_6, SON(alqu(en/in)ila Ci_6)2 e -SONH-alqu(en/in)ila C].6, ou dois substituintes adjacentes podem juntos com 0 grupo aromático a que estes estão ligados formar um anel de 4 a 8 membros, que opcionalmente contem um, dois ou três heteroátomos;
em uma outra forma de realização Ar é opcionalmente substituído com um ou mais substituintes independentemente sendo halogênio, alqu(en/in)ila Cj.6, cicloalqu(en)ila C3.$, cicloalqu(en)ila C3.8alqu(en/in)ila Ci_6, halo-alqu(en/in)ila Ci_6, alqu(en/in)ilóxi Ci.ó ou alqu(en/in)ilóxi C3-8’, em uma outra forma de realização Ar é opcionalmente substituído com um ou mais substituintes independentemente sendo halogênio ou halo-alqu(en/in)ila Ci.ó.
Em uma outra forma de realização da invenção, Ar é substituído com um ou mais substituintes independentemente sendo halogênio, alqu(en/in)ila Ci.6, halo-alqu(en/in)ila C|_6, alqu(en/in)ilóxi Ci_6.
Para ilustrar mais sem limitar a invenção uma forma de «
f realização de Ar é substituído com halogênio;
uma outra forma de realização de Ar é substituída com 5 alqu(en/in)ila C^; uma outra forma de realização de Ar é substituída com halo-alqu(en/in)ila C^;
uma outra forma de realização de Ar é substituída com alqu(en/in)ilóxi Cj .6; uma outra forma de realização de Ar não é substituída;
uma outra forma de realização de Ar é substituída com 1 10 substituinte;
uma outra forma de realização de Ar é substituída com 2 substituintes;
uma outra forma de realização de Ar é substituída com 3 substituintes.
Uma forma de realização diz respeito a compostos da fórmula geral I em que pelo menos um do R4 e R5 não é Ar.
Uma outra forma de realização diz respeito a compostos da fórmula geral I em que pelo menos um dos Ró e R7 não é Ar.
Ainda uma outra forma de realização diz respeito a compostos 20 da fórmula geral I em que pelo menos três dos R4, R5, R6 e R7 não são Ar.
Ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que no máximo um dos R3, R4, R5, R6 e R7 compreendem Ar;
ainda uma outra forma de realização diz respeito a compostos 25 da fórmula geral I em que no máximo dois do R3, R4, R5, R6 e R7 compreendem Ar;
ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que no máximo três dos R3, R4, R5, Ró e R7 compreendem Ar;
ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que R não compreende Ar;
* • ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que R4 não compreende Ar;
ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que R5 não compreende Ar;
ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que R6 não compreende Ar;
ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que R7 não compreende Ar.
Uma forma de realização diz respeito a compostos da fórmula geral I em que a configuração estéreo no átomo de carbono a que R4 e R5 estão ligados é a configuração S.
Uma outra forma de realização diz respeito a compostos da 15 fórmula geral I em que a configuração estéreo no átomo de carbono a que R4 e R5 estão ligados é a configuração R.
Ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que a configuração estéreo no átomo de carbono a que R6eR7 estão ligados é a configuração S.
Ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que a configuração estéreo no átomo de carbono a que R6eR7 estão ligados é a configuração R.
Ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que a configuração estéreo no átomo de carbono a que 25 R4 e R5 estão ligados é idêntica à configuração estéreo no átomo de carbono a que R6 e R7 estão ligados.
Ainda uma outra forma de realização diz respeito a compostos da fórmula geral I em que a configuração estéreo no átomo de carbono a que R4 e R5 estão ligados é diferente da configuração estéreo no átomo de carbono a que R6 e R7 estão ligados.
Os compostos da lista seguinte e sais destes exemplificam a to • invenção, a lista não é de maneira alguma intencionada ser interpretada como ‘ limitante:
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-2-(4fluoro-fenil)-acetamida;
2-Ciclopentil-N-(2-bromo-6-trifluorometil-4-morfolin-4-ilfenil)-acetamida;
N-(2-Bromo-4-morfolin-4-il-6-triíluorometil-fenil)-310 ciclopentilpropionamida;
N-(2-Cloro-6-ciano-4-morfolin-4-il-fenil)-3-ciclo-hexilpropionamida;
2-Ciclopentil-N-(2,6-dimetil-4-tiomorfolin-4-il-fenil)- acetamida;
2-Ciclopentil-N-[2,6-dimetil-4-(2-fenil-morfolin-4-il)-fenilJacetamida;
2-Ciclopentil-N-[2,6-dimetil-4-(2-fenil-tiomorfolin-4-il)fenil]acetamida;
2-Ciclopentil-N-[2,6-dimetil-4-(3-piridin-3-il-tiomorfolin-420 il)fenil]acetamida;
2-Ciclopentil-N-(2,6-dimetil-4-[2-(4-trifluorometil-fenil)-tiomorfolin-4-il]fenil)-acetamida;
N-{4-[2-(2-Cloro-fenil)-tiomorfolin-4-il]-2,6-dimetil-fenil}-2ciclopentilacetamida;
2-Biciclo[2,2,1 ]hept-2-il-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
2-Ciclo-hexil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida;
-(3,4-Difluoro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil) propionamida;
2- Ciclopentil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida;
Ácido butílico do éster (2,6-Dimetil-4-morfolin-4-il-fenil)* carbâmico;
‘ 2-(4-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)5 acetamida;
(2,6-dimetil-4-morfolin-4-il-fenil)amida do ácido 2,3-diidrobenzofuran-2-carboxílico;
3- Ciclo-hexil-N-(2,6-dimetil-4-morfolin-4-il-fenil) propionamida;
3-Ciclopentil-N-(2,6-dimetil-4-morfolin-4-il-fenil) propionamida;
N-(2,6-Dimetil-4-morfblin-4-il-fenil)-2-(4-fluoro-fenil)acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-tiofen-2-il-acctamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-3,3-dimetil-butiramida;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido hexanóico;
2-Ciclo-heptil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetarnida;
Éster benzílico do ácido (2,6-Dimetil-4-morfolin-4-il-fenil)carbâmico;
Éster 2-cloro-benzílico do ácido (2,6-Dimetil-4-morfolin-4-ilfenil)-carbâmico;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 3,5,5Trimetil-hexanóico;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido octanóico;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido heptanóico;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-fenil-acetamida;
2-(3,4-Dicloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
2-(4-Alilóxi-3-cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il18 fenil)-acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2*(3-trifluorometil fenil)-acetamida;
N-(2,6-Dimetizil-4-morfolin-4-il-fenil)-2-naftalen-2-il5 acetamida;
3-(3-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil) propionamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(3,4-dimetil’fenil)acetamida;
2-(3-Bromo-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
2-(3-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-p-tolil-acetamida;
N-(2J6-Dimetil-4-morfolin-4-il-fenil)-2-m-tolil-acetamida;
2-(3,4-Difluoro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)acetamida;
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-3-ciclohexil propionamida;
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-2-(3fluoro-fenil) acetamida;
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil25 fenil)propionamida;
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)butiramida;
N-(2-Cloro-4-morfolin-4-il-6-trifluorometil-fenil)-2-(3-fluorofenil)acetamida;
N-(2-Cloro-4-morfolin-4-il-6-trifluorometil-fenil)-2ciclopentil-acetamida;
2-Ciclopentil-N-{2,6-dimetil-4-[2-(4-trifIuorometil-fenil)morfolin-4-il]fenil}-acetamida;
N-{4’[2-(2-Cloro-fenil)-morfolin-4-il]-2,6-dimetil-fenil}-2ciclopentilacetamida;
2-Ciclopentil-N-{4-[2-(4-fluoro-fenil)-morfolin-4-il]-2,6dimetil-fenil} acetamida;
2-(2-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)10 acetamida;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido pentanóico; (2,6-dimetil-4-morfolin-4-il-fenil)-amÍda do ácido 4-metilpentanóico;
2-Ciclopent-2-enil-N-(2,6-dimetil-4-morfolin-4-il-fenil)15 acetamida;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 5-metilhexanóico;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 3-metil pentanóico;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido hex-5enóico;
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 3-etil pentanóico;
2-Ciclopentil-N-(4-morfolin-4-il-2-piridin-3-il-625 trifluorometil-fenil)acetamida;
2-Ciclopentil-N-(5-morfolin-4-il-3-trifluorometil-bifenil-2-il)acetamida;
2-Ciclopentil-N-(4’-fluoro-5-morfolin-4-il-3-trifIuorometilbifenil-2-il)acetamida;
2-Ciclopentil-N-(4’-metil-5-morfolin-4-il-3’trifluorometilbifenil-2-il)acetamida;
2-CicIopentil-N-(3’-metil-5-morfolin-4-il-3-trifluorometilbifenil-2-il)acetamida;
2-Ciclopentil-N-(3\4’-difluoro-5-morfolin-4-il-3trifluorometil-bifenil-2-il)acetamida;
2-(4-Fluoro-fenil)-N-(4-morfolin-4-il-2-piridin-3-il-6trifluorometil-fenil)acetamida;
2-Ciclopentil-N-(2,6-dietil-4-morfolin-4-il-fenil)-acetamida;
2-Ciclopentil-N-(2,6-diisopropil-4-morfolin-4-il-fenil)acetamida;
2-Ciclopentil-N-(2,6-difluoro-4-morfolin-4-il-fenil)acetamida;
(2,6-difluoro-4-morfolin-4-il-fenil)-amida do ácido hexanóico;
N-(2í6-Difluoro-4-morfolin-4-il-fenil)-3,3-dimetil-butiramida;
N-(2,6-Difluoro-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)acetamida;
2-Ciclopent-2-enil-N-(2,6-difluoro-4-morfolin-4-il-fenil)acetamida;
2-Biciclo[2,2,1 ]hept-2-il-N-(2,6-difluoro-4-morfolin-4-ilfenil)-acetamida;
2-Biciclo[2,2,l]hept-2-il-N-(2-metil-4-morfolin-4-il-6trifluorometil-fenil)acetamida;
(2-metil“4-morfolin-4-il-6-trifluorometil-feml)amida;
(2-metil-4-morfolin-4-il-6-trifluorometil-feml)-amida do ácido
5-metil hexanóico;
2-Ciclopent-2-enil-N-(2-metil-4-morfolin-4-il-6-trifluorometilfenil)acetamida;
2-Ciclopentil-N-(2-metil“4-morfolin-4-il-6-trifluorometil21
Η fenil)-acetamida;
(2-metil-4-morfolin-4-il-6-trifluorometil-fenil)-amida do ácido hexanóico;
3,3-Dimetil-N-(2-metil-4-morfolin-4-il-6‘trifluorometil-fenil)butiramida;
2-(3,4-Difluoro-fenil)-N-(2-metil-4-morfolin-4-il-6trifluorometil-fenil)acetamida;
(2-metóxi-6-metil-4‘morfolin-4-il-fenil)-amida do ácido hexanóico;
2-Ciclopentil-N-(2-metóxi-6-metil-4-morfolin-4-il-fenil)acetamida;
N-(2-Metóxi-6-metil-4-rnorfolin-4-il-fenil)-3,3-dimetilbutiramida;
2-(3,4-Difluoro-fenil)-N-(2-metóxi-6-metil-4-morfolin-4-ilfenil)-acetamida;
2-Ciclopent-2-enil-N-(2-metóxi-6-metil-4-rnorfolin-4-il-fenil)acetamida;
2-(3-Fluoro-fenil)-N-(2-metóxi-6-metil-4-rnorfolin-4-il-fenil)acetamida;
2-Biciclo[2,2,l]hept-2-il-N-(2-metóxi-6-metil-4-morfolin-4-ilfenil)acetamida;
(2-metóxi-6-metil-4-morfolin-4-il-fenil)-amida do ácido 4metil pentanóico;
(2-metóxi-6-metil-4-morfolin-4-il-fenil)-amida do ácido 5metil-hexanóico;
N-(2-Cloro-6-metíl-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)acetamida;
e
N-(2-ClorO’6-metil-4-morfolin-4-il-fenil)-2-ciclopentil22 β
acetamida;
como a base livre ou um sal deste. Cada destes compostos é considerado uma forma de realização específica e podem ser sujeitados a reivindicações a individuais.
A presente invenção também compreende sais dos compostos da invenção, tipicamente, sais farmaceuticamente aceitáveis. Os sais da invenção incluem sais de adição de ácido, sais metálicos, sais de amônio e amônio alquilado.
Os sais da invenção são preferivelmente sais de adição de ácido. Os sais de adição de ácido da invenção são preferivelmente sais farmaceuticamente aceitáveis dos compostos da invenção formados com ácidos não tóxicos. Os sais de adição de ácido incluem os sais de ácidos inorgânicos bem como ácidos orgânicos. Os exemplos de ácidos inorgânicos adequados incluem os ácidos clorídrico, bromídrico, iodídrico, fosfórico, sulfurico, sulfamico, nítrico e outros. Os exemplos de ácidos orgânicos adequados incluem os ácidos fórmico, acético, tricloroacético, trifluoroacético, propiônico, benzóico, cinâmico, cítrico, fumárico, glicólico, itacônico, lático, metanossulfônico, maleico, málico, malônico, mandélico, oxálico, pícrico, pirúvico, salicíclico, succínico, metanossulfônico, 20 etanossulfônico, tartárico, ascórbico, pamóico, bismetileno salicílico, etanodissulfônico, glicônico, citracônico, aspártico, esteárico, palmítico, EDTA, glicólico, p-aminobenzóico, glutâmico, benzenossul fônico, ptoluenossulfônico, ácidos teofilino acéticos, bem como as 8-haloteofilinas, por exemplo 8-bromoteofilina e outros. Outros exemplos de sais de adição de ácido inorgânicos e orgânicos farmaceuticamente aceitáveis incluem os sais farmaceuticamente aceitáveis listados na J. Pharm. Sei. 1977,66,2, que é aqui incorporada por referência.
Também intencionados como sais de adição de ácido são os hidratos, que os presentes compostos, são capazes de formar.
Os exemplos de sais metálicos incluem sais de lítio, sódio, potássio, magnésio e outros.
Os exemplos de sais de amônio e amônio alquilado incluem sais de amônio, metil-, dimetil-, trimetil-, etil-, hidroxietil-, dietil-, n-butil-, 5 sec-butil-, terc-butil-, tetrametilamônio e outros.
Além disso, os compostos desta invenção podem existir em formas insolúveis bem como em solúveis com solventes farmaceuticamente aceitáveis tais como água, etanol e outros. No geral, as formas solúveis são consideradas equivalentes às formas não solúveis para os propósitos desta 10 invenção.
Os compostos da presente invenção podem ter um ou mais centros assimétricos e isto é intencionado que quaisquer isômeros óticos (isto *
é, enanciômeros ou diastereoisômeros), como isômeros óticos separados, puros ou parcialmente purificados e quaisquer misturas destes incluindo 15 misturas racêmicas, isto é, uma mistura de estereoisómeros, estão incluídos dentro do escopo da invenção.
As formas racêmicas podem ser resolvidas nas formas antípodas por métodos conhecidos, por exemplo, pela separação de sais diastereoisoméricos destes com um ácido oticamente ativo, e que libera o 20 composto amina oticamente ativo tratando-se com uma base. Um outro método para resolver os racematos nas antípodas óticas é fundamentado em cromatografia em uma matriz oticamente ativa. Os compostos racêmicos da presente invenção também podem ser resolvidos nas suas antípodas óticas, por exemplo, por cristalização fracional. Os compostos da presente invenção 25 também podem ser resolvidos pela formação de derivados diastereoisoméricos. Os métodos adicionais para a resolução de isômeros óticos, conhecidos àqueles habilitados na técnica, podem ser usados. Tais métodos incluem aqueles debatidos por J. Jaques, A. Collet e S. Wilen in “Enantiomers, Racemates, and Resolution”, John Wiley and Sons, Nova
Iorque (1981). Os Compostos oticamente ativos também podem ser preparados a partir de materiais de partida oticamente ativos, ou por síntese estereosseletiva.
Além disso, quando uma ligação dupla ou um sistema de anel completa ou parcialmente saturado está presente na molécula, isômeros geométricos podem ser formados. E entendido que quaisquer isômeros geométricos, como isômeros geométricos separados, puros ou parcialmente purificados ou misturas destes são incluídos dentro do escopo da invenção. Do mesmo modo, as moléculas tendo uma ligação com rotação restrita podem 10 formar isômeros geométricos. Estes também são intencionados a estar inclusos dentro do escopo da presente invenção.
Além disso, alguns dos compostos da presente invenção podem existir em diferentes formas tautoméricas e é intencionado que quaisquer formas tautoméricas que os compostos são capazes de formar estão 15 incluídas dentro do escopo da presente invenção.
A invenção também abrange pró-medicamentos dos presentes compostos, que na administração são submetidos à conversão química por processos metabólicos antes de se tomarem substâncias farmacologicamente ativas. No geral, tais pró-medicamentos serão derivados funcionais dos 20 compostos da fórmula geral I, que são prontamente conversíveis in vivo no composto da fórmula I requerido. Os procedimentos convencionais para a seleção e preparação de derivados de pró-medicamentos adequados estão descritos por exemplo, em “Design of Prodrugs”, ed. H. Bundgaard, Elsevier, 1985.
A invenção também abrange metabólitos ativos dos presentes compostos. Um aspecto da invenção fornece um composto da fórmula I ou um sal deste para o uso como um medicamento.
Em uma forma de realização, a invenção diz respeito ao uso de um ou mais compostos de acordo com a invenção em um método de tratamento. O distúrbio ou doença a ser tratado é responsivo a um fluxo de íon melhorado em um canal de potássio tal como os canais de íon de potássio da * família KCNQ. Tal distúrbio ou doença é preferivelmente um distúrbio ou doença do sistema nervoso central.
Ainda em uma outra forma de realização, a invenção fornece uma composição farmacêutica que compreende um ou mais carreadores ou diluentes farmaceuticamente aceitáveis e um ou mais compostos da fórmula I ou um sal deste.
Uma outra forma de realização da invenção diz respeito ao uso de um composto da fórmula I ou um sal deste para a preparação de uma composição farmacêutica para o tratamento de uma doença ou distúrbio em que um abridor de canal de potássio KCNQ tal como um abridor de canal de potássio KCNQ2 é benéfico. Tipicamente, tal distúrbio ou doença é selecionado do grupo que consiste de distúrbios de ataque, distúrbios de ansiedade, dor neuropática e distúrbios de dor de cabeça de enxaqueca ou distúrbios neurodegenerativos.
Em uma forma de realização, os compostos da invenção podem ser administrados como o único composto terapeuticamente eficaz.
Em uma outra de realização os compostos da invenção podem ser administrados como uma parte de uma terapia de combinação, isto é, os compostos da invenção podem ser administrados em combinação com outros compostos terapeuticamente eficazes tendo por exemplo, propriedades anticonvulsivas. Os efeitos de tais outros compostos tendo propriedades anticonvulsivas podem incluir mas não são limitados a atividades:
· nos canais de íons tais como canais de sódio, potássio, ou cálcio.
• nos sistemas de aminoácidos excitatórios por exemplo, bloqueio ou modulação de receptores NMDA.
• nos sistemas de sistemas de neurotransmissores inibitórios por exemplo, aumento da liberação de GABA, ou bloqueio da captação &
de GABA ou • efeitos de estabilização de membrana.
As medicações anti-convulsivas correntes incluem, mas não são limitadas a, tiagabina, carbamazepina, valproato de sódio, lamotrigina, 5 gabapentina, pregabalina, etosuximida, levetiracetam, fenitoína, topiramato, zonisamida bem como membros das classes benzodiazepina e barbiturato.
Os compostos da invenção são considerados úteis para aumentar o fluxo de íon em um canal de potássio dependente de voltagem em um mamífero tal como um ser humano. Os compostos são deste modo 10 considerados úteis no tratamento de um distúrbio ou doença sendo responsivo a um fluxo de íon melhorado em um canal de potássio dependente de voltagem, tal como os canais de íons de potássio da família KCNQ. Tal distúrbio ou doença é preferivelmente um distúrbio ou doença do sistema nervoso central.
Em uma forma de realização, o distúrbio ou doença é selecionado do grupo que consiste de distúrbios de ataque; tal como ataques agudos, convulsões, estados epilépticos e epilepsia tal como síndromes epilépticas e ataques epilépticos; em particular convulsões, epilepsia e estados epilépticos.
Em uma outra de realização o distúrbio ou doença é selecionado do grupo que consiste de dor neuropática e distúrbios de dor de cabeça de enxaqueca; tal como alodinia, dor hiperalgésica, dor fantasma, dor neuropática relacionada com neuropatia diabética, dor neuropática relacionada com neuralgia trigeminal e dor neuropática relacionada com enxaqueca; em particular alodinia, dor hiperalgésica, dor fantasma, dor neuropática relacionada com neuropatia diabética e dor neuropática relacionada com enxaqueca.
Ainda em uma outra forma de realização, o distúrbio ou doença é selecionado do grupo que consiste de distúrbios da ansiedade; tal como ansiedade, distúrbios e doenças relacionados com ataque do pânico, agorafobia, distúrbio do pânico com agorafobia, distúrbio do pânico sem > agorafobia, agorafobia sem histórico de distúrbio do pânico, fobia específica, fobia social e outras fobias específicas, distúrbio obsessivo compulsivo, 5 distúrbio de tensão pós traumático, distúrbios de tensão agudos, distúrbio da ansiedade generalizada, distúrbio de ansiedade devido a condições médicas gerais, distúrbio de ansiedade induzido por substância, distúrbio de ansiedade de separação, distúrbios de ajuste, ansiedade de desempenho, distúrbios hipocondríacos, distúrbio de ansiedade devido a condição médica geral e 10 distúrbio de ansiedade induzido por substância e distúrbio de ansiedade de outro modo não especificado; em particular ansiedade, distúrbio da ansiedade generalizada, ansiedade do pânico, distúrbio compulsivo obsessivo, fobia social, ansiedade de performance, distúrbio de tensão pós-traumática, reação de tensão aguda, distúrbios de ajuste, distúrbios hipocondríacos, distúrbio de 15 ansiedade de separação, agorafobia, fobias específicas, distúrbio de ansiedade devido a condição médica geral e distúrbio de ansiedade induzido por substância.
Ainda em uma outra forma de realização, o distúrbio ou doença é selecionado do grupo que consiste de distúrbios neurodegenerativos;
tal como mal de Alzheimer, coréia de Huntington, esclerose múltipla, esclerose lateral amiotróflca, doença de Creutzfeld-Jakob, mal de Parkinson, encefalopatias induzidas pela AIDS ou infecção pelo vírus da rubéola, vírus da herpes, borrelia e patógenos desconhecidos, neurodegenerações induzidas por trauma, estados de hiperexcitação neuronal tal como na retirada de 25 medicamento ou intoxicação e doenças neurodegenerativas do sistema nervoso periférico tal como polineuropatias e polineuritides; em particular a mal de Alzheimer, coréia de Huntington, esclerose múltipla, esclerose lateral amiotróflca, encefalopatia induzida pela AIDS e outras encefalopatias relacionadas com infecção sendo causadas pelo vírus da rubéola, vírus da

herpes, borrelia e por patógenos desconhecidos, doença de Creutzfeld-Jakob, mal de Parkinson, e neurodegenerações induzidas por trauma. Ainda em uma outra forma de realização, o distúrbio ou doença é selecionado do grupo que consiste de estados de hiperexcitação neuronal tal como na retirada de 5 medicamento ou por intoxicação. Ainda em uma outra forma de realização, o distúrbio ou doença é selecionado do grupo que consiste de distúrbios bipolares.
Ainda em uma outra forma de realização, o distúrbio ou doença é selecionado do grupo que consiste de distúrbios do sono; tal como 10 insônia.
O termo “tratamento” como aqui usado em conexão com uma doença ou distúrbios inclui também a prevenção, inibição e aperfeiçoamento como o caso pode ser.
Em uma forma de realização, os compostos da invenção 15 podem ser encontrados para ter efeito nos canais de potássio da família KCNQ, em particular a subunidade KCNQ2.
A invenção fomece compostos mostrando efeito em um ou mais dos seguintes testes:
• “Efluxo relativo através do canal de KCNQ2 “
Que é uma medida da potência do composto no canal alvo • “Eletrochoque máximo”
Que é uma medida de ataques induzidos por estimulação do CNS não específica por meios elétricos • “Ataques induzidos por pilocarpina “
Os ataques induzidos por pilocarpina são freqüentemente difíceis de tratar com muitas medicações anti-ataque existentes e assim reflete um modelo de “ataques resistentes a medicamentos” • “Testes de limiar de ataque elétrico” e “Teste de limiar de atraque químico”
Estes modelos medem o limiar em que os ataques são iniciados, deste modo sendo modelos que detectam se os compostos podem retardar o início dos ataques.
• “abrasamento de amígdala”
Que é usado como uma medida da progressão da doença, em animais normais os ataques neste modelo são mais severos do que no animal recebe outros estímulos • “registros eletrofisiológicos de clampe de contato em células CHO“ e “registros eletrofisiológicos de canais KCNQ2, KCNQ2/KCNQ3 ou KCNQ5 em oócito”
Nestes testes as correntes KCNQ2, KCNQ2/KCNQ3 ou KCNQ5 de voltagem ativada são registradas.
Em uma forma de realização, os compostos são KCNQ2 ativos com um EC50 de menos do que 15.000 nM tal como menos do que 10.000 nM 15 como medido pelo teste “Efluxo relativo através do canal KCNQ2”. Em uma outra de realização, os compostos são KCNQ2 ativos com um EC50 de menos do que 2.000 nM tal como menos do que 1.500 nM como medido pelo teste “Efluxo relativo através do canal KCNQ2”. Ainda em uma outra forma de realização, os compostos são KCNQ2 ativos com um EC50 de menos do que 20 200 nM tal como menos do que 150 nM como medido pelo teste “Efluxo relativo através do canal KCNQ2”. O teste “Efluxo relativo através do canal KCNQ2” é descrito abaixo.
Em uma forma de realização, os compostos têm um ED50 de menos do que 15 mg/kg no teste “eletrochoque máximo”. Em uma outra de 25 realização, os compostos têm um ED50 de menos do que 5 mg/kg no teste “eletrochoque máximo”. O teste “eletrochoque máximo” é descrito abaixo.
Em uma forma de realização, os compostos têm um ED50 de menos do que 5 mg/kg no “Teste de limiar de ataque elétrico” e “Teste de limiar de ataque químico” que são descritos abaixo.
Alguns compostos têm poucos ou clinicamente insignificantes efeitos colaterais. Alguns dos compostos são deste modo testados em modelos « de calmante indesejado, ações hipotérmicas e atáxicas dos compostos.
Alguns dos compostos têm um grande índice terapêutico entre eficácia anticonvulsivas e efeitos colaterais tais como dano da atividade locomotora ou efeitos atáxicos como medido pela desempenho em uma haste rotativa. Tais compostos serão esperadamente bem tolerados em pacientes permitindo altas doses a ser usadas após os efeitos colaterais serem vistos. Por isso a complacência com a terapia será esperadamente boa e a administração de altas doses podem ser permitidas fazendo um tratamento mais eficaz em pacientes que de outro modo teriam efeitos colaterais com outras medicações.
Um aspecto não limitante da invenção diz respeito a compostos de acordo com as formas de realização abaixo de 1 a 10:
1. Os derivados de morfolina ou tiomorfolina substituídos da fórmula geral I
em que q é 0 ou 1;
W é 0 ou S;
XéCO;
Zé O;
R1 e R2 são independentemente selecionados do grupo que consiste de halogênio, halo-alqu(en/in)ila Ci_6, alqu(en/in)ila e ciano;
R3 é selecionado do grupo que consiste de alqu(en/in)ila Cj.io, cicloalqu(en)ila C3_s-alqu(en/in)ila Ci.6, Ar-alqu(en/in)ila Cj.6 e Ar;
| |
R4, R5, R6 e R7 são independentemente selecionados do grupo que |
| |
consiste de hidrogênio e Ar; |
|
« |
como a base livre ou sais destes. |
| |
2. Um composto de acordo com a forma de realização 1, o dito |
|
5 |
composto sendo selecionado do grupo que consiste de:
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-2-(4-fluorofenil)-acetamida;
2-Ciclopentil-N-(2-bromo-6-trifluorometil-4-morfolin-4-il-fenil)acetamida; |
|
10 |
N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-3-ciclopentilpropionamida;
N-(2-Cloro-6-ciano-4-morfolin-4-il-fenil)-3-ciclo-hexil propionamida;
2-Ciclopentil-N-(2,6-dimetil-4-tiomorfolin-4-il-fenil)-acetamida; |
|
15 |
2-Ciclopentil-N-[2,6-dimetil-4-(2-fenil-morfolin-4-il)fenil]-
acetamida;
2-Ciclopentil-N-[26-dimetil-4-(2-fenil-tiomorfolin-4-il) fenil]-
acetamida;
2-Ciclopentil-N-[2,6-dimetil-4-(3-piridin-3-il-tiomorfolin-4- |
|
20 |
il)fenil]acetamida;
2-Ciclopentil-N-{2,6-dimetil-4-(2-(4-trifluorometil-fenil)tiomorfolin-4-il] fenil} -acetamida;
N-{4-[2-(2-Cloro-fenil)-tiomorfolin-4-il]-2,6-dimetil-fenil}-2ciclopentil-acetamida; |
|
25 |
2-Biciclo[2,2,l]hept-2-il-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
2- CiclO'hexil-N-(2,6-dimetil-4-morfolin-4-il'fenil)-acetamida;
3- (3,4-Difluoro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)propionamida; |
|
5 |
2-Ciclopentil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida; Ácido butílico do éster (2,6-Dimetil-4-morfolin-4-il-fenil)carbâmico;
2-(4-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida;
Ácido 2,3-diidro-benzofuran-2-carboxílico (2,6-dimetil-4-rnorfolin- |
|
10 |
4-il-fenil)-amida;
3-Ciclo-hexil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-propionamida;
3-Ciclopentil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-propionamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(4-fluoro-fenil)-acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-tiofen-2-il-acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-3,3-dimetil-butiramida;
Ácido hexanóico(2,6-dimetil-4-morfolin-4-il-fenil)-airiida;
2-Ciclo-heptil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida;
Ácido benzílico do éster (2,6-Dimetil-4-naorfolin-4-il-fenil)- |
|
15 |
carbâmico;
Éster 2-cloro-benzílico do ácido (2,6-Dimetil-4-morfolin-4-il-fenil)-
carbâmico;
(2,6-dimetil-4-morfolin-4-il-fenil)'amida do ácido 3,5,5-Trimetil- |
|
20 |
hexanóico;
(2,6-dimctil-4-morfolin-4-il'fcnil)-amida do ácido octanóico; |
|
25 |
(2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido heptanóico; N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-fenil-acetamida 2-(3,4-Dicloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
2-(4-Alilóxi-3-cloro-fenil)-N-(2>6-dimetil-4-morfolin-4-il-fenil)acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fcnil)-2-(3-trifluorometil-fenil)acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-naftalen-2-il-acetamida; |
3-(3-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)propionamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(3,4-dimetil-fenil)acetamida;
2-(3-Bromo-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida; 2-(3-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida; N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-p-tolil-acetamida;
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-m-tolil-acetamida; 2-(3,4-Difluoro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida;
e N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)-acetamida.
3. Uma composição farmacêutica que compreende um ou mais carreadores ou diluentes farmaceuticamente aceitáveis e um ou mais compostos de acordo com qualquer uma das formas de realização 1 e 2.
4. O uso de uma composição farmacêutica de acordo com a forma de realização 3 para aumentar o fluxo de íon em um canal de potássio dependente de voltagem de um mamífero tal como um ser humano.
5. O uso de acordo com a forma de realização 4 no tratamento de um distúrbio ou doença sendo responsivo por um fluxo de íon em um canal de potássio dependente de voltagem, tal distúrbio ou doença é preferivelmente um distúrbio ou doença do sistema nervoso central.
6. O uso de acordo com a forma de realização 5 caracterizado em que o distúrbio ou doença é selecionado do grupo que consiste de distúrbios de ataque tais como convulsões, epilepsia e estados epilépticos.
7. O uso de acordo com a forma de realização 5 caracterizado em que o distúrbio ou doença é selecionado do grupo que consiste de distúrbios neuropáticos e de dor de cabeça de enxaqueca tais como alodinia, dor hiperalgésica, dor fantasma, dor neuropática relacionada a neuropatia diabética e dor neuropática relacionada com enxaqueca.
8. O uso de acordo com a forma de realização 5 caracterizado em que o distúrbio ou doença é selecionado do grupo que consiste de distúrbios da ansiedade tal como ansiedade, distúrbio da ansiedade generalizada, ansiedade do pânico, distúrbio compulsivo obsessivo, fobia social, ansiedade de performance, distúrbio de tensão pós traumático, reação de tensão aguda, distúrbios de ajuste, distúrbios hipocondríacos, distúrbio de ansiedade de separação, agorafobia, fobias específicas, distúrbio de ansiedade devido a condição médica geral e distúrbio de ansiedade induzido por substância.
9. O uso de acordo com a forma de realização 5 caracterizado em que o distúrbio ou doença é selecionado do grupo que consiste de distúrbios neurodegenerativos tal como mal de Alzheimer, coréia de Huntington, esclerose múltipla, esclerose lateral amiotrófica, encefalopatia induzida pela AIDS e outras encefalopatias relacionadas com uma infecção sendo causada por vírus da rubéola, vírus da herpes, borrelia e por patógenos desconhecidos, doença de Creutzfeld-Jakob, mal de Parkinson, neurodegenerações induzidas por trauma.
10. O uso de acordo com a forma de realização 5 caracterizado em que o distúrbio ou doença é selecionado do grupo que consiste de estados de hiperexcitação neuronal tal como em retirada de medicamento ou por intoxicação.
Definições
O termo heteroátomo se refere a um átomo de nitrogênio, oxigênio ou de enxofre.
/Π
Halogênio significa flúor, cloro, bromo ou iodo.
A expressão alqu(en/in)ila Ci.6 significa um alquila Ci_6, alquenila C2.6 ou um grupo alquinila C2-6· O termo alquila Ci_6 se refere a um grupo alquila ramificado ou não ramificado tendo de um a seis átomos de 5 carbono inclusive, incluindo mas não limitando a metila, etila, 1-propila, 2propila, 1-butila, 2-butila, 2-metil-2-propila e 2-metil-1-propila. Similarmente, alquenila Ci.6 e alquinila C2_6, respectivamente, designam tais grupos tendo de dois a seis átomos de carbono, incluindo uma ligação dupla e um ligação tripla respectivamente, incluindo mas não limitando a etenila, 10 propenila, butenila, etinila, propinila e butinila.
A expressão alqu(en/in)ila Cmo significa um alquila Cmo, alquenila C2.io ou um grupo alquinila C2-io- O termo alquila Cmo se refere a um grupo alquila ramificado ou não ramificado tendo de um a seis átomos de carbono inclusive, incluindo mas não limitando a metila, etila, 1-propila, 215 propila, 1-butila, 2-butila, 1-pentila, 1-hexila, 1-heptila, 1-octila, 1-nonila, 1decila, 2-metil-2-propila e 2-metil-1-propila. Similarmente, alquenila C2-10 e alquinila C2.i0, respectivamente, designam tais grupos tendo de dois a seis átomos de carbono, incluindo uma ligação dupla e uma ligação tripla respectivamente, incluindo mas não limitando a etenila, propenila, butenila, 20 pentenila, hexenila, heptenila, octenila, nonenila, decenila, etinila, propinila, butinila, pentinila, hexinila, heptinila, octinila, noninila, e decinila.
A expressão cicloalqu(en)ila C3.8 significa um grupo cicloalquila ou cicloalquenila C3_8. O termo cicloalquila C3.8 designa um carbociclo monociclo ou biciclo tendo de três a oito átomos C, incluindo mas 25 não limitando a ciclopropila, ciclopentila, ciclo-hexila, ciclo-heptila, ciclooctila, [ 1,1,1 jbiciclopentila, biciclo[2,2,l]heptila, [2.2.2]bicicloctila e [3,3,0]bicicloctila, etc. O termo cicloalquenila C3.8 designa um carbociclo monociclo ou biciclo tendo de três a oito átomos C, incluindo uma ligação dupla.
O termo halo-alqu(en/in)ila designa alqu(en/in)ila Ci.6 sendo substituído com um ou mais átomos halogênios, incluindo mas não limitando a trifluorometila. Similarmente, halo-cicloalqu(en)ila C3.8 designa cicloalqu(en)ila C3.8 sendo substituído com um ou mais átomos halogênio.
termos alqu(en/in)ila Ci_6 e halo-cicloalqu(en)ila C3_8 são como definidos acima.
Quando dois substituintes adjacentes juntos com o grupo aromático a que estes estão ligados formam um anel de 4 a 8 membros, que opcionalmente contem um, dois ou três heteroátomos, então um sistema de anel é formado por de 4 a 8 átomos selecionados de 4 a 8 átomos de carbono e de 0 a 3 heteroátomos selecionados de N, S, ou O. Tais dois substituintes adjacentes podem juntos formar: -(CH2)a CH2-, -CH=CH-(CH2)b-, -CH2CH=CH-(CH2)c, -CH=CH-CH=CH-, -(CH2)a-O-, -O-(CH2)b-O- -CH2-O15 (CH2)c-O-, -CH2-O-CH2-O-CH2-, -(CH2)a-S-, -S-(CH2)b-S-, -CH2-S-(CH2),-S-,
-CH2-S-CH2-S-CH2-, -(CH2)a NH-, NH-(CH2)b-NH-, -CH2-NH-(CH2)c-NH-, CH=CH-NH-, -O-(CH2)b-NH-, -CH2-O-(CH2)c-NH- ou -O-(CH2)c-NH-CH2-,
-S-(CH2)b-NH-, -N=CH-NH-, -N=CH-O- ou -N=CH-S- ou -N=N-NH-, em que b é 1, 2 ou 3, a é 2, 3 ou 4 e c é 1 ou 2.
O termo Ar se refere a sistemas aromáticos opcionalmente substituídos de 5 a 1 átomos de carbono, em que 0, 1, 2, 3 ou 4 átomos de carbono podem ser substituídos por heteroátomos independentemente selecionados de N, S, ou O. Os exemplos de tais grupos Ar são fenila opcionalmente substituído, naftila opcionalmente substituído, piridina opcionalmente substituído, tiofeno opcionalmente substituído, furano opcionalmente substituído, tiazol opcionalmente substituído, quinolina opcionalmente substituído, indol opcionalmente substituído, 2,3-diidrobenzofurano opcionalmente substituído, pirimidina opcionalmente substituído, pirrol opcionalmente substituído e oxazol opcionalmente üõ substituído. O Ar pode ser substituído com um ou mais substituintes independentemente sendo hidróxi, halogênio, alqu(en/in)ila Cj.6, * cicloalqu(en)ila C3.8, cicloalqu(en)ila C3.8-alqu(en/in)ila Ci-6, haloalqu(en/in)ila Ci.g, alqu(en/in)ilóxi C^, alqu(en/in)ilóxi C3.8, acila, nitro ou ciano, -CO-NH-alqu(en/in)ila Cj.6, -CO-N(alqu(en/in)ila C 1.0)2, -NH2, -NHalqu(en/in)ila C^, -N(alqu(en/in)ila CI.Ó)2, -S-alqu(en/in)ila Ci.6, -SO2alqu(en/in)ila Ci.6, -SO2N(alqu(en/in)ila C1.0)2 e -SO2NH-alqu(en/in)ila Ct.6; ou dois substituintes adjacentes podem juntos com o grupo aromático a que estes estão ligados formar um anel de 4 a 8 membros, que opcionalmente contem um, dois ou três heteroátomos.
Quando Ar é substituído com CO-NH-alqu(en/in)ila Ci-6 ou CO-N(alqu(en/in)ila C 1.6)2, então o átomo de carbono do grupo CO é ligado a Ar.
Quando Ar é substituído com NH2, NH-alqu(en/in)ila C].6 ou 15 N(alqu(en/in)ila C 1.6)2, então 0 átomo de nitrogênio é ligado a Ar.
Quando Ar é substituído com -S-alqu(en/in)ila Cj.6, -SO2alqu(en/in)ila Ci.6, -SO2N(alqu(en/in)ila C 1.0)2 ou -SO2NH-alqu(en/in)ila Cj_6 então 0 átomo de enxofre é ligado a Ar.
O termo acila se refere a formila, alqu(en/in)ilcarbonila Ci_6, 20 cicloalqu(en)ilcarbonila C3.8, Ar-carbonila, Ar-alqu(en/in)ilcarbonila Ci.6 ou um grupo cicloalqu(en)ila C3.8-alqu(en/in)ila-carbonila Cj.6, em que o alqu(en/in)ila Ci.6, cicloalqu(en)ila C3.8 e Ar são como definidos acima.
Os termos cicloalqu(en)ila C3.8-alqu(en/in)ila Cj.6, Aralqu(en/in)ila C1.6, alqu(en/in)ilóxi C1.6 e cicloalqu(en)ilóxi C3.s; designam 25 tais grupos em que o alqu(en/in)ila Ci.6, cicloalqu(en)ila C3.8 e Ar são como definidos acima. Similarmente, o cicloalqu(en)ila C3.8-alqu(en/in)ilóxi Cj.6 designam tais grupos em que 0 cicloalqu(en)ila C3.8 e alqu(en/in)ilóxi Ci^ são como definidos acima.
As expressões Ar-cicloalqu(en)ila C3.8 e Ar-cicloalqu(en)ila
C3,8-alqu(en/in)ila C].6 designam tais grupos em que o alqu(en/in)ila Cm, cicloalqu(en)ila C3.8 e Ar são como definidos acima.
Composições Farmacêuticas
A presente invenção também diz respeito a uma composição farmacêutica. Os compostos da invenção ou sais destes podem ser administrados sozinhos ou em combinação com carreadores ou diluentes farmaceuticamente aceitáveis, em doses única ou múltiplas. As composições farmacêuticas de acordo com a invenção podem ser formuladas com os carreadores ou diluentes farmaceuticamente aceitáveis bem como quaisquer 10 outros adjuvantes e excipientes conhecidos de acordo com as técnicas convencionais tais como aquelas divulgadas em Remington: The Science e Practice of Pharmacy, 19° Edição, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
As composições farmacêuticas podem ser especificamente 15 formuladas para administração por qualquer via adequada tal como a via oral, retal, nasal, pulmonar, tópica (incluindo bucal e sublingual), transdérmica, intracistemal, intraperitoneal, vaginal e parenteral (incluindo subcutânea, intramuscular, intratecal, intravenosa e intradérmica), a via oral sendo preferida. Será apreciado que a via preferida dependerá da condição geral e 20 idade do paciente a ser tratado, a natureza do distúrbio ou doença a ser tratada e o ingrediente ativo escolhido.
As composições farmacêuticas formadas combinando-se o composto da invenção e os carreadores farmacêuticos aceitáveis são então prontamente administradas em uma variedade de formas de dosagem 25 adequadas para as vias de administração divulgadas. As formulações podem convenientemente estar presentes na forma única de dosagem por métodos conhecidos na técnica da farmácia.
Os compostos desta invenção são no geral utilizados como a substância livre ou como um sal farmaceuticamente aceitáveis desta. Um
Llb exemplo é um sal de adição de ácido de um composto tendo a utilidade de 1 uma base livre. Quando um composto da invenção contem uma base livre, tais ♦ sais são preparados de uma maneira convencional tratando-se uma solução ou suspensão de uma base livre da invenção com um equivalente químico de um ácido farmaceuticamente aceitável. Os exemplos representativos estão mencionados acima.
As composições farmacêuticas para a administração oral podem ser sólidas ou líquidas. As formas de dosagem sólidas para a administração oral incluem por exemplo, cápsulas, tabletes, dragas, pílulas, pastilhas, pós, grânulos e comprimidos por exemplo, colocados em uma cápsula de gelatina dura na forma de pó ou pelota ou por exemplo, na forma de um comprimido ou pastilha. Onde apropriado, as composições farmacêuticas para a administração oral podem ser preparadas com revestimentos tais como revestimentos entéricos ou estes podem ser formulados de modo a fornecer liberação controlada do ingrediente ativo tal como liberação sustentada ou prolongada de acordo com os métodos bem conhecidos na técnica. As formas de dosagem líquidas para a administração oral incluem por exemplo, soluções, emulsões, suspensões, xaropes e elixires.
As formulações da presente invenção adequadas para a administração oral podem ser apresentadas como unidades discretas tais como cápsulas ou tabletes, cada um contendo uma quantidade pré-determinada do ingrediente ativo, e que podem incluir um excipiente adequado. Além disso, as formulações oralmente disponíveis podem estar na forma de um pós ou grânulos, um solução ou suspensão em uma emulsão líquida não aquosa, ou líquida óleo em água ou água óleo.
Os carreadores farmacêuticos aceitáveis incluem diluentes ou enchedores sólidos inertes, solução aquosa estéril e vários solventes orgânicos. Os exemplos de carreadores sólidos são lactose, terra alba, sacarose, ciclodextrina, talco, gelatina, ágar, pectina, acácia, estearato de uQ magnésio, ácido esteárico, éteres alquílicos inferiores de celulose, amido de ’ milho, amido de batata, gomas e outros. Os exemplos de carreadores líquidos *
* são xarope, óleo de amendoim, óleo de oliveira, fosfolipídeos, ácidos graxos, aminas de ácido graxo, polioxietileno e água.
O carreador ou diluente ou podem incluir qualquer material de liberação sustentada conhecido na técnica, tal como monoestearato de glicerila ou diestearato de glicerila, sozinhos ou misturados com uma cera.
Quaisquer adjuvantes ou aditivos geralmente usados para tais propósitos tal como corantes, agentes de sabor, conservantes etc. podem ser usados contanto que estes são compatíveis com os ingredientes ativos.
A quantidade de carreador sólido pode variar mas usualmente será de cerca de 25 mg a cerca de 1 g.
Se um carreador líquido é usado, a preparação pode estar na forma de um xarope, emulsão, cápsula de gelatina macia ou líquido injetável estéril tal como uma suspensão ou solução aquosa ou não aquosa líquida.
Os tabletes podem se preparados misturando-se o ingrediente ativo com adjuvantes ou diluentes comuns e subsequentemente comprimindo a mistura em uma máquina de tabletagem convencional.
As composições farmacêuticas para a administração parenteral incluem soluções injetáveis, dispersões, suspensões ou emulsões bem como pós estéreis para ser reconstituído em soluções ou dispersões injetáveis estéreis antes do uso. As formulações injetáveis de depósito também são consideradas como estando dentro do escopo da presente invenção.
Para a administração parenteral, as soluções do composto da invenção em solução estéril aquosa, propileno glicol aquoso, vitamina e aquosa ou óleo de gergelim ou amendoim podem ser utilizadas. Tais soluções aquosas devem ser adequadamente tamponadas se necessário e o diluente líquido primeiro rende um isotônico com sal ou glicose suficiente. As soluções aquosas são particularmente adequadas para a administração
Μ intravenosa, intramuscular, subcutânea e intraperitoneal. Os meios estéreis aquosos utilizados são todos prontamente disponíveis por técnicas padrão *
♦ conhecidas àqueles habilitados na técnica.
As soluções para injeções podem ser preparadas dissolvendo5 se o ingrediente ativo e possíveis aditivos em uma parte do solvente para injeção, preferivelmente água estcril, ajustando a solução ao volume desejado, esterilizando a solução e a enchendo em ampolas ou fracos adequados. Qualquer aditivo adequado convencionalmente usado na técnica pode ser adicionado, tal como agentes de tonicidade, conservantes, antioxidantes, etc.
Outras formas de administração adequadas incluem supositórios, pulverizadores, ungüento, cremes, géis, inalantes, emplastos dérmicos, implantes, etc.
Uma dosagem oral típica é na faixa de cerca de 0,001 a cerca de 100 mg/kg por peso corporal por dia, preferivelmente de cerca de 0,01 a 15 cerca de 50 mg/kg por peso corporal por dia, e mais preferido de cerca de
0,05 a cerca de 10 mg/kg por peso corporal por dia administrados em um ou mais dosagens tais como de 1 a 3 dosagens. A dosagem exata dependerá da freqüência e modo de administração, do sexo, idade, peso e condição geral do paciente tratado, da natureza e da severidade do distúrbio ou doença tratados e 20 quaisquer doenças concomitantes a serem tratadas e outros fatores evidentes àqueles habilitados na técnica.
As formulações podem ser convenientemente apresentadas na forma única de dosagem por métodos conhecidos àqueles habilitados na técnica. Uma forma única de dosagem típica para a administração oral, uma 25 ou mais vezes por dia tal como de 1 a 3 vezes por dia pode conter de 0,01 a cerca de 1000 mg, tal como cerca de 0,01 a 100 mg, preferivelmente de cerca de 0,05 a cerca de 500 mg, e mais preferido de cerca de 0,5 mg a cerca de 200 mg.
Para vias parenterais tais como administração intravenosa, intratecal, intramuscular e similar, as doses tipicamente são na faixa de cerca de metade da dose utilizada para a administração oral.
Os exemplos típicos de receitas para a formulação da invenção são como segue:
1) Tabletes contendo 5,0 mg de um composto da invenção calculado como a base livre:
Compostos da fórmula 1 5,0 mg
Lactose 60 mg
Amido de Milho 30 mg
Hidroxipropilcelulose 2,4 mg
Celulose microcristalina 19,2 mg
Croscarmelose de Sódio Tipo A 2,4 mg
Estearato de magnésio 0,84 mg
2) Tabletes contendo 0,5 mg de um composto da invenção calculado como a base livre:
Compostos da fórmula 1 0,5 mg
Lactose 46,9 mg
Amido de Milho 23,5 mg
Povidona 1,8 mg
Celulose microcristalina 14,4 mg
Croscarmelose de Sódio Tipo A 1,8 mg
Estearato de magnésio 0,63 mg
3) Xarope contendo por mililitro:
Composto da fórmula I25 mg
Sorbitol 500 mg
Hidroxipropilcelulose15 mg
Glicerol50 mg metil-parabeno1 mg
Propil-parabeno 0,1 mg
0,005 ml
0,05 mg
0,5 mg ad 1 ml
Etanol
Sabor
Sacarina de sódio
Agua
4) Solução para injeção contendo por mililitro:
Composto da fórmula I 0,5 mg
Sorbitol 5,1 mg
Ácido acético 0,05 mg
Sacarina de sódio 0,5 mg
Água ad 1 ml
Pela expressão, um composto da invenção significa qualquer uma das formas de realização da fórmula I como aqui descrito.
Em um outro aspecto, a presente invenção diz respeito a um método de preparar um composto da invenção como descrito no seguinte.
Preparação dos compostos da invenção
Os compostos da invenção da fórmula geral 1, em que q, W, X, Z, R1, R2, R3, R4, R5, R6 e R7 são como definidos acima podem ser preparados pelos métodos como representados nos esquemas e como descrito abaixo:
Esquema 1
R2 R2 R2
|
nr -
U |
— Χλ
III |
- ώ'-
(V |
v
R7 R6 |
| |
|
|
|
R2 H |
| |
|
|
wVrR4 rtV5 |
Esquema 2
Esquema 3
Esquema 4
Esquema 5
Esquema 6
XIO
XIV
Esquema 7
XI
VI
Nos compostos das fórmulas gerais de I a XV, q, W, X, Z, R1, R2, R3, R4, R5, R6 e R7 são como definidos sob a fórmula I.
Os compostos das fórmulas gerais II, III, VI, VIII e XI são obtidas a partir de fontes comerciais, ou preparadas por métodos padrão conhecidos por químicos habilitados na técnica.
Altemativamente, os compostos da fórmula geral III, onde R2 5 é halogênio tal como Cl, Br ou I (esquema 1), são obtidos por meio de substituição aromática eletrofílica regiosseletiva, bem conhecida aos químicos habilitados na técnica, com eletrófilos apropriados tal como Nclorossuccinimida, N-bromossuccinimida, bromo, iodo ou cloreto de iodo em um solvente adequado tal como ácido acético, como descrito em 10 “Electrophilic halogenations” por P.B.D. de la Mare, Cambridge University Press, Cambridge, 1976.
O grupo nitro em compostos das fórmulas gerais III, IX e XIII (esquemas 1, 4 e 6) pode ser reduzido com agentes de redução adequados tais como pó de zinco ou ferro na presença de ácido tal como ácido acético ou 15 ácido clorídrico aquoso, ou por gás hidrogênio ou formiato de amônio na presença de um catalisador de hidrogenação adequado tal como paládio em carbono ativado em solventes adequados tal como metanol, etanol, ou tetraidrofurano, em temperaturas adequadas ou sob irradiação ultrassônica, para obter anilinas das fórmulas gerais IV, V e XIV, respectivamente.
Altemativamente, cloreto de estanho (II) ou ditioneto de sódio pode ser usado como agentes de redução sob condições bem conhecidas aos químicos habilitados na técnica.
Os compostos das fórmulas gerais I e VII (esquemas 1, 2, 3 e
4) podem ser preparados reagindo-se os compostos das fórmulas gerais V e 25 VI, respectivamente, com reagentes eletrofilicos, tais como, mas não limitando a, cloretos de ácido carboxílico, brometos de ácido carboxílico, iodetos de ácido carboxílico, anidretos de ácido carboxílico, ésteres ativados, formiatos de cloro adequadamente substituídos, e com ou sem a adição de bases, tais como piridina, trialquilaminas, carbonato de potássio, óxido de magnésio ou alcolatos de lítio, sódio, ou potássio, em um solvente adequado, tal como acetato de etila, dioxano, tetraidrofurano, acetonitrila ou éter dietílico, em uma temperatura adequada, tal como a temperatura ambiente ou temperatura de refluxo.
Altemativamente, os compostos das fórmulas gerais I e V (esquemas 2 e 3) podem ser preparados por reação que forma uma ligação ΟΝ catalisada por paládio entre compostos adequadamente substituídos das fórmulas gerais VII e VI e morfolinas ou tiomorfolinas adequadamente substituídas, como descrito por S. L. Buchwald et al. (M. C. Harris, X. Hang e 10 S. L. Buchwald, Organic Letters, 2002, 4, 2885).
Os compostos das fórmulas gerais V e XV (esquemas 1 e 6) podem ser preparados reagindo-se os compostos das fórmulas gerais IV e XIV com éteres bis(2-haloetílico) adequadamente substituídos e com ou sem a adição de bases, tais como trialquil aminas, carbonato de potássio ou 15 alcolatos de lítio, sódio, ou potássio, em um solvente adequado, tal como dimetil sulfóxido ou Ν,Ν-dimetilformamida, em uma temperatura adequada, tal como na temperatura ambiente ou na temperatura de refluxo.
Altemativamente, os compostos da fórmula geral V (esquema
3) podem ser preparados reagindo-se os compostos da fórmula geral VI 20 com derivados de morfolina ou tiomorfolina adequadamente substituídos na presença de um catalisador de paládio, tal como bis(dibenzilidenoacetona)paládio com a adição de um ligando de fosfma adequado, tal como (±)-2,2’-bis(difenilfosfino)-l,r-binaftil na presença de uma base, tal como carbonato de potássio ou alcolatos de lítio, sódio, ou 25 potássio, em um solvente adequado, tal como tolueno ou tetraidrofurano, em uma temperatura adequada, tal como na temperatura ambiente ou na temperatura de refluxo.
Os compostos da fórmula geral IX (esquema 4) podem ser preparados reagindo-se os compostos da fórmula geral VIII com morfolinas ou tiomorfolinas adequadamente substituídas e com ou sem a adição de bases, tais como carbonato de potássio, em um solvente adequado, tal como dimetil sulfóxido ou Ν,Ν-dimetilformamida, em uma temperatura adequada, tal como na temperatura ambiente ou na temperatura de refluxo.
Os compostos da fórmula geral I, em que R2 é Ar ou Het (esquema 5), podem ser preparados a partir dos compostos da fórmula geral X, por meios de reações de ligação cruzada conhecida aos químicos habilitados na técnica, tal como ligação de Suzuki, ligação de Stille, ou outras reações de ligação cruzada catalisadas por metal de transição (D.W. Knight, 10 “Coupling reactions between sp2 carbon centers” em Comprehensive Organic Synthesis, v. 3, pp. 481-520, Pergamon Press, 1991).
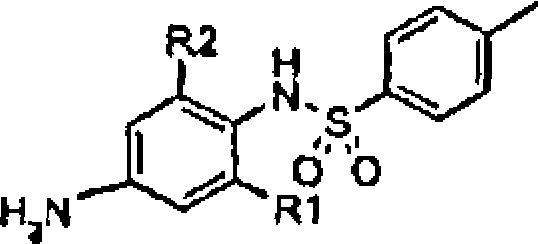
Os compostos da fórmula geral VI (esquema 7), podem ser preparados a partir dos compostos da fórmula geral XI, por meios de substituição aromática eletrofllica bem conhecida aos químicos habilitados na 15 técnica, com eletrófilos apropriados tais como N-bromossuccinimida ou bromo em um solvente adequado tal como ácido acético, como descrito por
P.B.D. de la Mare c J.H. Ridd, “Preparativo methods of aromatic halogenation” em Aromatic substitutions, pp. 105-115, Butterworths Scientific Publications, Londres, 1959.
Os compostos da fórmula geral XII (esquema 6) podem ser preparados reagindo-se os compostos da fórmula geral XI com cloreto ptoluenossulfonila com ou sem a adição de bases, tais como piridina, trialquilaminas, carbonato de potássio, hídrogeno carbonato de sódio, óxido de magnésio ou alcolatos de lítio, sódio, ou potássio, em um solvente 25 adequado, tal como piridina, acetato de etila, dioxano, tetraídrofurano, acetonitrila ou éter dietílico, em temperaturas adequadas, tais como na temperatura ambiente ou na temperatura de refluxo.
Os compostos da fórmula geral XIII (esquema 6) podem ser preparados a partir dos compostos da fórmula geral XII, por reações de nitração conhecidas aos químicos habilitados na técnica, tais como reação com ácido nítrico concentrado, nitrito de sódio ou nitrato de sódio, em um solvente adequado, tal como ácido acético glacial, anidreto acético, ácido trifluoroacético, ácido sulfúrico concentrado ou misturas destes, em 5 temperaturas apropriadas, por exemplo como descrito por P.B.D. de la Mare e
J.H. Ridd, “Preparative methods of nitration” em Aromatic substitutions, pp. 48-56, Butterworths Scientific Publications, Londres, 1959.
Os compostos da fórmula geral V (esquema 6) podem ser preparados tratando-se os compostos da fórmula geral XV sob condições 10 ácidas fortes tais como ácido sulfurico aquoso ou ácido clorídrico aquoso, em temperaturas adequadas, tais como na temperatura ambiente ou na temperatura de refluxo.
Exemplos
Os dados de LC-MS analíticos foram obtidos em um instrumento PE Sciex API 150EX equipado com fotoionização na pressão atmosférica e um sistema Shimadzu LC3_8A/SLC-10A LC. Coluna: Coluna C18 Waters Symmetry de 30 X 4,6 mm com tamanho de partícula de 3,5 pm; Sistema de Solvente: A = água/ácido trifluoroacético (100:0,05) e B = água/acetonitrila/ácido trifluoroacético (5:95:0,03); Método: Eluição de 20 gradiente linear com A a 90% a B a 100% em 4 minutos e com um taxa de fluxo de 2 ml/minuto. A pureza foi determinada pela integração da UV (254 nm) e traço ELSD. O tempo de retenção (tR) são expressados em minutos. A purificação LC-MS preparativa foi realizada no mesmo instrumento com ionização química de pressão atmosférica. Coluna: YMC ODS-A de 50 X 20 25 mm com tamanho de partícula de 5 pm; Método: Eluição gradiente linear com A a 80% a B a 100 em 7 minutos e com um taxa de fluxo de 22,7 ml/minuto. A coleta da fração foi realizada por detecção MS de fluxo dividido.
Os dados LC-MS-TOF analíticos (TOF = tempo de vôo) foram
obtidos em um LCT de micromassa de 4 vias equipado com MUX com um sistema de detecção Waters 2488/Sedex 754. Coluna: coluna Cl8 Waters Symmetry de 30 X 4.6 mm com tamanho de partícula de 3,5 pm; Sistema de solvente: A = água/ácido trifluoroacético (100:0,05) e B = 5 água/acetonitrila/ácido trifluoroacético (5:95:0,03); Método: Eluição gradiente linear com A a 90% a B a 100% em 4 minutos e com uma taxa de fluxo de 2 ml/minuto. A pureza foi determinada pela integração do UV (254 nm) e do traço ELSD. O tempo de retenção (tR) são expressados em minutos.
Os dados GC-MS foram obtidos em um cromatógrafo a gás 10 Varian CP 3800 equipado com uma coluna Phenomenex (Zebron ZB-5, extensão: 15 metros, diâmetro interno: 0,25 mm) ligado a um espectômetro de massa capturador de íons Varian Satum 2000. Método: Duração de 15 minutos, fluxo de coluna 1,4 ml/minuto (o gás carreador foi hélio), gradiente de forno: de 0 a 1 minuto, 60° C; de 1 a 13 minutos, 60 a 300° C; de 13 a 15 15 minutos, 300 °C.
O espectro ’H RMN foram registrados a 500,13 MHz em um Instrumento Bruker Avarice DRX500. Clorofórmio deuterado (a 99,8% D) ou sulfóxido dimetila (a 99,8% D) foram usados como solventes. A TMS foi usada como padrão de referência interna. Os valores de substituição química 20 são expressados em valores ppm. As seguintes abreviações são usadas para a multiplicidade de sinais RMN: s = singleto, d = dupleto, t = tripleto, q = quarteto, qui = quinteto, h = hepteto, dd - dupleto duplo, ddd = dupleto duplo , dt = tripleto duplo, dq = quarteto duplo, tt ~ tripleto de tripletos, m = multipleto e b = singleto amplo.
Os experimentos de microondas foram realizados em frascos ou reatores de processo lacrados usando um Sintetizador Emrys ou um Otimizador EXP Emrys da Personal Chemistry ou um instrumento Milestone Microsynth da Milestone. Quando uma reação foi aquecida em um instrumento microondas, este foi resfriado a 25° C antes da próxima etapa do
processo.
Preparação de intermediários
2,6-Dimetil'4-morfolin-4-ilfenilarnina.
Bis(dibenzilidenoacetona)paládio (2,88 g) e (±)-2,2’5 bis(difenilfosfíno)l,r-binaftil (4069 g) foram adicionados a tolueno seco (175 ml purificado com argônio) e agitado por 15 minutos sob argônio. Tercbutóxido de potássio (7,06 g), morfolina (8,7 ml) e 4-bromo-2,6dimetilanilina (10,03 g) foram adicionados subsequentemente, a mistura de reação foi aquecida a refluxo por 16 horas sob argônio, resfriada e filtrada 10 através de sílica (200 g). Salmoura (250 ml) foi adicionada e a mistura foi extraída com acetato de etila (3 x 200 ml). As fases orgânicas combinadas foram secadas em sulfato de magnésio e concentradas no vácuo. O produto bruto foi dissolvido em éter dietílico (250 ml), filtrado através de sílica (200 g) e concentrado no vácuo para fornecer 8,5 g (rendimento de 41%) do 15 composto do título como um óleo escuro. O produto foi usado sem purificação adicional. GC-MS (m/z) 206 (M+); tR = 6,90. lH RMN (500 MHz, CDC13): 2,18 (s, 6H), 3,02 (m, 4H), 3,85 (m, 4H), 6,62 (b, 2H).
N-(4-Bromo-2,6-dimctil-fenil)-2-ciclopentil-acetamida.
4-Bromo-2,6-dimetilanilina (5,92 g) e cloreto de ciclopentil20 acetila (4,87 ml) foram dissolvidos em acetonitrila (26 ml) e aquecidos a 150° C por 10 minutos em um frasco de processo de microondas lacrado.
A reação foi resfriada a 0° C, 0 produto retirado por filtração e lavado com acetonitrila gelada (50 ml) produzindo 8,43 g (rendimento de 92%) do composto do título como um sólido marrom. O produto bruto foi 25 usado sem purificação adicional. LC-MS (m/z) 312 (MH+); tR = 3,10, (UV, ELSD) 89%, 99%. *H RMN (500 MHz, CDC13): 1,22 (m, 2H), 1,52 (m, 2H), 1,63 (m, 2H), 1,77 (m, 2H), 2,06 (s, 6H), 2,31 (m, 1H), 2,64 (d, 2H), 7,15 (s, 2H), 9,98 (b, 1H).
2-Bromo-4~mtro~6-trifluorometil fenilamina.
Bromo (0,60 ml) dissolvido em ácido acético (11 ml) foi adicionado às gotas a uma solução de 4-nitro-2-trifluorometil-fenilamina (2,4 g) em ácido acético (12 ml). A mistura de reação foi aquecida a 120° C por 2 horas e 30 minutos, vertido em água (400 ml) e filtrada. O sólido coletado foi 5 lavado com água (200 ml) e secado no vácuo para fornecer 3,03 g (rendimento de 91%) do composto do título como um sólido amarelo. RMN (500 MHz, DMSO-d6): 7,08 (s, 2H), 8,23 (d, 1H), 8,51 (d, 1H). 2~Bromo-6-trifluorometil-benzeno~l, 4-dicimiria.
Ácido clorídrico aquoso (2 M, 45 ml) foi adicionado 10 lentamente a uma mistura de pó de zinco (8,6 g) e 2-bromo-4-nitro-6trifluorometil-fenilamina (2,5 g) em tetraidrofurano (50 ml). A mistura de reação foi agitada por 1 hora, filtrada e neutralizada com bicarbonato de sódio aquoso saturado (100 ml). Água (100 ml) foi adicionada e a mistura foi extraída com acetato de etila (2 x 100 ml). As fases orgânicas combinadas 15 foram lavadas com água (2 x 200 ml) e salmoura (200 ml), secadas em sulfato de sódio e concentradas no vácuo para fornecer 2,22 g (rendimento de 98 %) do composto do título como um sólido vermelho. *H RMN (500 MHz, DMSO-dg): 4,55 (s, 2H), 4,91 (s, 2H), 6,76 (d, 1H), 7,01 (d, 1H). 2-Bromo-4-morfolin-4-il-6-trifluorometil fenilamina.
2-Bromo-6-trifluorometil-benzeno-l,4-diamina (2,21 g), éter bis-(2-bromoetil) (1,30 ml) e N,N-diisopropil-etilamina (4,64 ml) foram misturados em N,N-dimetilformamida (19 ml) e aquecidos até 180° C por 30 minutos em um frasco de processo de microondas lacrado. O bicarbonato aquoso saturado (100 ml) foi adicionado e a mistura bruta foi extraída com acetato de etila (100 ml). A fase orgânica foi lavada com água (100 ml) e salmoura (100 ml), secada em sulfato de sódio e concentrada no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 1,78 g (63%) do composto do título como um sólido amarelo. LC-MS (m/z) 326 (MH+); tR = 2,54, (UV, ELSD) 83 %, 75%. 'H RMN (500
MHz, DMSO-de): 2,99 (m, 4H), 3,70 (m, 4H), 5,00 (s, 2H), 7,00 (d, 1 H), 7,38 (d, 1 H).
2,5-Diamino~3-cloro-benzonitrila.
Ácido clorídrico aquoso (12 M, 5,3 ml) foi adicionado lentamente a uma mistura de pó de zinco (2,01 g) e 2-amino-3-cloro-5-nitrobenzonitrila (0,50 g) em tetraidrofurano (40 ml). A mistura de reação foi agitada por 2 horas, neutralizada com carbonato de sódio saturado aquoso (50 ml), e extraída com acetato de etila (3 x 50 ml). As fases orgânicas combinadas foram secadas em sulfato de sódio e concentradas no vácuo para 10 fornecer 0,42 g (rendimento de 99 %) do composto do título como um sólido vermelho. 'H RMN (500 MHz, DMSO-de): 4,89 (s, 2H), 5,13 (s, 2H), 6,64 (d, 1H), 6,89 (d, 1H).
2-Amino-3-cloro-5-morfolin-4~il-benzonitrila.
2,5-Diamino-3-cloro-benzonitrila (387 mg), éter bis-(215 bromoetil) (0,35 ml) e N,N-diisopropil-etilamina (1,25 ml) foram misturados em N,N-dimetilformamida (4 ml) e aquecido a 180° C por 30 minutos em um frasco de processo de microondas lacrado. Bicarbonato saturado aquoso (20 ml) foi adicionado e a mistura bruta foi extraída com acetato de etila (20 ml). A fase orgânica foi lavada com água (20 ml) e salmoura (20 ml), secada em 20 sulfato de sódio e concentrada no vácuo para fornecer 0,50 g (91 %) do composto do título como um sólido marrom. LC-MS (m/z) 238 (MH+); tR 2,31, (UV, ELSD) 85%, 95%. *H RMN (500 MHz, DMSO-d6): 2,97 (m, 4H), 3,69 (m, 4H), 5,59 (s, 2H), 7,04 (d, 1 H), 7,29 (d, 1 H).
Ester terc-butílico do ácido [2~(4-Fluoro-feml)-2~hidróxi-etil]-carbâniico.
A uma solução agitada de éster terc-butílico do ácido carbâmico (0,22 g) em acetonitrila (6 ml) foi adicionado em seqüência a 0o C: Hidróxido de sódio (52 mg) em água (5 ml); após 2 minutos, hipocloreto tercbutílico (139 μΐ); após 10 minutos, diidrato de osmiato de potássio (VI) (9 mg) em água (1 ml); após 1 minuto, hidroquinina (antraquinonal,4-diil)diéter (26 mg) em acetonitrila (4 ml); após 3 minutos, acetonitrila (6,7 ml) e tampão de fosfato (3,3 ml, 0,5 M pH 7,65); após 5 minutos bifosfato de sódio » monoidratado suficiente para fazer pH = 7,65; e finalmente 4-fluoroestireno.
A reação foi extinguida após agitar 3 horas a 25° C com sulfito de sódio (0,20
g) em água (2 ml) a 0o C. as fases foram separadas e a camada aquosa extraída com acetato de etila (3 x 20 ml). As fases orgânicas combinadas foram lavadas com água (1 x 50 ml), secadas em sulfato de sódio, concentradas no vácuo e purificadas por cromatografia por vaporização instantânea para fornecer 90 mg (57 %) do composto do título como um óleo incolor. lH RMN (500 MHz, CDCI3): 1,45 (s, 9H), 3,18 (m, 1H), 3,23 (m, 1H), 3,44 (m, 1H),
4,83 (m, 1H), 4,92 (b, 1H), 7,04 (t, 2H), 7,34 (m, 2H).
Os seguintes compostos foram preparados analogamente: Éster terc-butílico do ácido [2-Hidróxi-2-(4-trifluorometil-fenil)-etil]carbâmico.
Rendimento de: 82%. 'H RMN (500 MHz, CDC13): 1,44 (s,
9H), 3,26 (m, 1H), 3,52 (m, 2H), 4,40 (b, 1 H), 4,92 (b, 1 H), 7,49 (d, 2H), 7,61 (d, 2H).
Éster terc-butílico do ácido [2-(2-Cloro-feml)-2-hidróxi-etil]-carbâmico.
Rendimento de: 61%. lH RMN (500 MHz, CDCbj): 1,44 (s, 20 9H), 3,32 (m, 1H), 3,51 (m, 1H), 4,23 (m, 1 H), 5,0 8 (b, 1 H), 5,19 (b, 1 H),
7,20 (m, 1 H), 7,28 (m, 1 H), 7,5 9 (m, 1 H).
2-(4-Fluoro-fenil)-morfolina.
Uma solução de éster terc-butílico do ácido [2-(4-fluoro-fenil)2-hidróxi-etil]-carbâmico (90 mg) em diclorometano (1 ml) e ácido 25 trifluoroacético (1 ml) foi agitada a 25° C por l hora e depois concentrada no vácuo. O resíduo foi dividido entre acetato de etila (5 ml) e carbonato de potássio saturado aquoso (5 ml). A fase orgânica foi secada em sulfato de sódio, concentrada no vácuo e redissolvido em tetraidrofurano seco (3 ml) e trietilamina (54 pL). Cloreto de cloroacetila (31 pL) em tetraidrofurano seco $
(1 ml) foi adicionado às gotas a 0o C. Após 30 minutos a reação foi diluída com acetato de etila (10 ml) e lavada com água/salmoura (1:1, 3 x 10 ml). A » fase orgânica foi secada em sulfato de sódio, concentrada no vácuo e redissolvida em terc-butanol (5 ml). Terc-butóxido de potássio (79 mg) foi adicionado e a reação agitada a 25° C por 1,5 hora. A reação foi extinguida com cloreto de amônio saturado aquoso (30 ml) e extraída com acetato de etila (2 x 30 ml). As fases orgânicas combinadas foram secadas em sulfato de sódio, concentradas no vácuo e co-evaporadas com tolueno (2x5 ml). O resíduo foi dissolvido em tolueno seco (5 ml) sob argônio e tratado com bis(210 metoxietóxi)alumínio hidreto de sódio (70% em tolueno, 205 pL) às gotas e agitado a 25° C por 5 horas. A reação foi extinguida a 0o C com hidróxido de sódio aquoso a 10% (5 ml), e a mistura foi extraída com éter dietílico (2x15 ml). As fases orgânicas combinadas foram secadas em sulfato de sódio e concentradas no vácuo para fornecer 60 mg (94 %) do composto do título como um óleo incolor. LC-MS (m/z) 182 (MH+); tR = 1,06, (UV, ELSD) 78 %, 98 %.
Os seguintes compostos foram preparados analogamente: 2-(4-Trifliioro-fenil)-morfolina.
Rendimento de: 85%. LC-MS (m/z) 232 (MH+); tR = 1,59, (UV, ELSD) 79 %, 99 %.
2-(2-Cloro-fenil)-morfolina.
Rendimento de: 86%. LC-MS (m/z) 198 (MH+); tR = 1,21, (UV, ELSD) 66 %, 99 %.
4-Bromo-2-metóxi-6-metil-fenilamina.
A 2-metóxi-6-metil-fenilamina (10,0 g) dissolvido em acetonitrila (200 ml) foi adicionado, N-bromossuccinimida (14,3 g) e a mistura de reação foi aquecida a 145° C por 15 minutos em um recipiente de processo dc microondas lacrado. A mistura bruta foi filtrada através de Celite, diluída com éter dietílico (200 ml) e lavada com hidróxido de sódio (2 M, 2 x &
100 ml) e salmoura (1 x 100 ml). A fase orgânica foi secada em sulfato de magnésio, concentrada no vácuo e purificada por cromatografia por • vaporização instantânea para fornecer 3,4 g (26 %) do composto do título como um sólido preto. RMN (500 MHz, DMSO-dó): 2,06 (s, 3H), 3,77 (s,
3H), 4,55 (s, 2H), 6,78 (d, 1H), 6,82 (d, 1H).
Os seguintes compostos foram preparados analogamente:
4-Bromo-2-metil-6-trifliiorometil-fenilamina.
Rendimento de: 80%. GC-MS (m/z) 254 (M+); tR = 3,73. 2-metòxi-6-metil-4-morfolin-4-il-fenilamincL
Bis(dibenzilidenoacetona)paládio (0,63 g) e (2’-diciclo-hexilfosfanilbifenil-2-il)-dimetil-amina (0,68 g) foram adicionados a tolueno seco (100 ml purificado com argônio) e agitados por 15 minutos sob argônio. Tercbutóxido de potássio (3,70 g), morfolina (4,0 ml) e 4-bromo-2-metóxi-6metil-fenilamina (3,40 g) foram adicionados subsequentemente. A mistura de reação foi aquecida a refluxo por 16 horas sob argônio, resfriada e filtrada através de sílica (50 g). Hidróxido de sódio (2 M, 200 ml) foi adicionado e a mistura foi extraída com acetato de etila (3 x 200 ml). As fases orgânicas combinadas foram lavadas com salmoura (1 x 200 ml), secadas em sulfato de magnésio e concentradas no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 1,0 g (rendimento de
%) do composto do título como um óleo preto. lH RMN (500 MHz, DMSO-d6): 2,05 (s, 3H), 2,91 (t, 4H), 3,69 (t, 4H), 3,74 (s, 3H), 3,95 (s, 2H), 6,23 (d, 1 H), 6,3 9 (d, 1 H).
Os seguintes compostos foram preparados analogamente:
2-metil-4-morfolin-4-il-6-trifliiorometil-fenilamina.
Rendimento de: 28%. !H RMN (500 MHz, DMSO-d6): 2,13 (s, 3H), 2,93 (t, 4H), 3,70 (t, 4H), 4,67 (s, 2H), 6,75 (d, 1 H), 6,99 (d, 1 H).
4-(3,5^ifluoro-4-nitro-fenil)-morfolina.
2,4,6-Trifluoronitrobenzeno (4,95 g) e carbonato de potássio (4,63 g) foram misturados em sulfóxido de dimetila seco (40 ml) e resfriados a 10° C sob argônio. Morfolina (2,56 ml) foi adicionado e a mistura de reação foi deixada aquecer até 25° C e agitada sob argônio por 16 horas. A mistura de reação foi concentrada no vácuo. Salmoura (50 ml) foi adicionada e o produto foi extraído com acetato de etila (3 x 50 ml). As fases orgânicas combinadas foram secadas em sulfato de magnésio e concentradas no vácuo. O material bruto foi purificado por cromatografia por vaporização instantânea para fornecer 2,49 g (rendimento de 37 %) do composto do título como um sólido amarelo. 'H RMN (500 MHz, DMSO-d6): 3,43 (t, 4H), 3,69 (t, 4H), 6,87 (d, 2H).
2,6-DifiuorO’4-morfolin-4-il-fenilamina.
Ácido clorídrico concentrado (4,2 ml) foi adicionado lentamente a uma mistura de pó de zinco (3,3 g) e 4-(3,5-difluoro-4-nitrofenil)-morfolina (2,49 g) em tetraidrofurano (40 ml) resfriada a 0o C. A mistura de reação foi depois agitada por 1 hora a 0o C e 2 horas a 25° C. A mistura de reação foi filtrada através de Celite (10 g), concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 1,96 g (rendimento de 90 %) do composto do título como um sólido branco. GC-MS (m/z) 214 (M+); tR = 5,83, 'H RMN (500 MHz, DMSO-d6): 2,94 (t, 4H), 3,69 (t, 4H), 4,53 (s, 2H), 6,57 (m, 2H).
2-Cloro-4-nitro-6-trifluorometil fenilamina.
4-Nitro-2-trifluorometil-fenilamina (5,6 g) e N-clorosuccinimida (4,0 g) foram colocados em suspensão em acetonitrila (15 ml) e aquecidos a 150° C por 10 minutos em um frasco de processo de microondas lacrado. Acetato de etila (80 ml) foi adicionado e a fase orgânica foi lavada com NaOH aquoso a 5% (2 x 50 ml), água (2 x 50 ml) e salmoura (2 x 50 ml). A fase orgânica foi secada em sulfato de magnésio e concentrada no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 4,9 g (rendimento de 75 %) do composto do título como um
6$ sólido amarelo. ’Η RMN (500 MHz, CDC13): 5,35 (b, 2H), 8,35 (b, 1 H), 8,37 (b, 1 H).
Os seguintes compostos foram preparados analogamente: 2-Cloro-6-metil-4-nitro fenilamina.
Rendimento de: 95%. *H RMN (500 MHz, DMSO-dó): 2,22 (s,
3H), 6,56 (b, 2H), 7,89 (d, 1H), 8,00 (d, 1H).
N-(2-Cloro-6-metil-4-nitro-fenil)-2-ciclopentil-acetamida.
2-Cloro-6-metil-4-nitro-fenilamina (6,0 g) e cloreto de ciclopentilacetila (5,1 g) foram dissolvidos em acetonitrila (45 ml) e aquecidos a 10 150° C por 20 minutos em um recipiente de processo de microondas lacrado.
A mistura de reação foi resfriada a 0o C e o produto precipitado coletado por filtração e lavado com acetonitrila gelada para fornecer 5,5 g (58 %) do composto do título como um sólido amarelo. lH RMN (500 MHz, DMSO-dó): 1,24 (m, 2H), 1,58 (m, 4H), 1,78 (m, 2H), 2,27 (m, 1H), 2,30 (s, 3H), 2,38 (d,
2H), 8,15 (d, 1 H), 8,18 (d, 1 H), 9.83 (s, 1 H).
Os seguintes compostos foram preparados analogamente:
N~(2-Cloro-6-metil-4-nitro-fenil)-2-(3-fluoro-fenil)-acetamida.
Rendimento de: 72 %. *H RMN (500 MHz, DMSO-d6): 2,24 (s, 3H), 3,77 (s, 2H), 7,10 (dt, 1H), 7,21 (m, 2H), 7,39 (m, 1 H), 8,14 (d, 1 H), 20 8,19 (d, 1 H), 10,15 (s, 1 H).
2-Cloro-6-trijluorometil-benzeno-l ,4-diamina.
Ácido acético (13 ml) foi adicionado lentamente a uma mistura de pó de zinco (12,4 g) e 2-cloro-4-vitro-6-trifluorometil-fenilamina (4 g) em tetraidrofurano (40 ml). A mistura de reação foi agitada por 1 hora, filtrada 25 através de sílica e concentrada no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 2,9 g (83 %) do composto do título como um sólido preto. *H RMN (500 MHz, CDC13): 3,44 (b, 2H), 4,16 (b, 2H), 6,77 (d, 1 H), 6,86 (d, 1 H).
Os seguintes compostos foram preparados analogamente:
N-(4-Amino-2-cloro-6-metil-feiiil)-2-ciclopeiitil-acetamida.
Rendimento de: 69 %. ]H RMN (500 MHz, DMSO-d6): 1,21 (m, 2H), 1,57 (m, 4H), 1,76 (m, 2H), 2,00 (s, 3H), 2,23 (d, 2H), 2,26 (m, 1H), 5,23 (s, 2H), 6,36 (d, 1H), 6,48 (d, 1H), 8,98 (s, 1H). N-(4-Amino-2-cloro-6-rnctil-fcnil)-2-(3-Jluoro-fenil)-acetamida.
Rendimento de: 88 %. *H RMN (500 MHz, DMSO-d6): 1,90 (s, 3H), 3,62 (s, 2H), 5,26 (s, 2H), 6,3 5 (d, 1 H), 6,49 (d, 1 H), 7,07 (dt, 1 H), 7,20 (m, 2H), 7,37 (m, 1 H), 9.34 (s, 1 H). 2~Cloro-4-morfolm-4dl-6~trifluorometil-fenilamina.
2-Cloro-6-trifluorometil-benzeno-l,4-diamina (2,9 g), éter bis(2-cloroetílico) (1,7 ml) e iodeto de sódio (516 mg) foram misturados em acetonitrila (45 ml) e aquecidos até 165° C por 1 hora em um recipiente de processo de microondas lacrado. A mistura bruta foi concentrada no vácuo, redissolvida em acetato de etila (100 ml) e lavada com água (2 x 100 ml) e salmoura (l x 100 ml). A fase orgânica foi secada em sulfato de magnésio e concentrada no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 940 mg (24 %) do composto do título como um óleo preto. *H RMN (500 MHz, CDC13): 3,02 (m, 4H), 3,84 (m, 4H), 4,30 (b, 2H), 6,97 (d, 1H), 7,06 (d, 1 H). N-(2,6-Diisopropil-4-nitro-fenil)-4-metil-benzenossidfonamida.
2,6-Diisopropil-fenilamina (1,80 ml) e um cloreto de paratoluenossulfonila (2,00 g) foram dissolvidos em piridina (4 ml) e aquecidos a 160° C por 10 minutos em um frasco de processo de microondas lacrado. A pasta fluida resultante foi diluída com acetato de etila (10 ml) e lavada com ácido clorídrico (2 M, 10 ml) e salmoura (10 ml). A fase orgânica foi secada em sulfato de sódio, concentrada no vácuo e colocada em suspensão em ácido nítrico a 65 % (15 ml) e água (60 ml). Ácido acético (60 ml) e nitrito de sódio (0,99 g) foram adicionados sucessivamente e a mistura de reação foi aquecida a 100° C por 12 horas. A mistura foi resfriada a 25° C e vertida em água
gelada (200 ml) e filtrada para fornecer 2,07 g (58 %) do composto do título como um sólido amarelo. RMN (500 MHz, CDC13): 1,06 (d, 12H), 2,43 (s, 3H), 3,19 (m, 2H), 6,29 (s, 1H), 7,28 (d, 2H), 7,59 (d, 2H), 7,97 (s, 2H). N-(4-Amino-2>6-(iiisopropil-fenil)-4-metil-benzenossulfonamida.
A uma suspensão de N-(2,6-diisopropil-4-nitro-fenil)-4-metilbenzenossulfonamida (3,72 g) em etanol (50 mi) foi adicionado diidrato de cloreto estanoso (II) (11,2 g) e a mistura foi aquecida a 80° C por 1,5 hora. Esta foi então vertida em gelo (300 ml), feito fortemente básico com hidróxido de sódio sólido (20 g) e diluída com acetato de etila (100 ml). A suspensão foi filtrada e extraída com acetato de etila (3 x 100 ml). As fases orgânicas combinadas foram secadas em sulfato de sódio e concentradas no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 2,60 g (76 %) do composto do título como um sólido branco amarelado. *H RMN (500 MHz, CDCI3): 0,95 (d, 12H), 2,40 (s, 3H), 3,02 (m, 2H), 3,68 (b, 2H), 5,74 (s, 1H), 6,39 (s, 2H), 7,23 (d, 2H), 7,60 (d, 2H).
2,6-Diisopropil-4-morfolin-4-il-fenilamina.
A mistura de N-(4-amino-2,6-diisopropil-fenil)-4-metilbenzenossulfonamida (346 mg), éter bis-(2-bromoetílico) (151 pL), N,Ndiisopropil-etilamina (0,53 ml) e N-metilpirrolidina (1,0 ml) foi aquecida até 180° C por 20 minutos em um frasco de processo de microondas lacrado. A mistura foi diluída com acetato de etila (20 ml), lavada com salmoura (30 ml) e carbonato de potássio saturado aquoso (30 ml) e secada em sulfato de sódio. A fase orgânica foi concentrada no vácuo e redissolvida em uma mistura de ácido sulfúrico (1,9 ml) e água (0,1 ml) e agitada a 40° C por 3 horas. Gelo (30 ml) e água (30 ml) foram adicionados e a mistura foi basificada com carbonato de potássio sólido. A mistura foi extraída com acetato de etila (3 x 20 ml) e as fases orgânicas combinadas foram secadas em sulfato de sódio e concentradas no vácuo para fornecer 260 mg (99 %) do composto do título como um sólido branco. *H RMN (500 MHz, CDCI3): 1,26 (d, 12H), 2,95 (m, 2H), 3,06 (m, 4H), 3,49 (b, 2H), 3,87 (m, 4H), 6,69 (s, 2H). N-(2,6-Dietil-4~nitro-fenil)-4-metil-benzenossulfonamida.
2,6-Dietil-fenilamina (1,57 ml) e cloreto de para-tolueno- sulfonila (2,00 g) foram dissolvidos em piridina (4 ml) e aquecidos a 160° C por 10 minutos em um frasco de processo de microondas lacrado. A pasta fluida resultante foi diluída com acetato de etila (10 ml) e lavada com ácido clorídrico (2 M, 10 ml) e salmoura (10 ml). A fase orgânica foi secada em sulfato de sódio, concentrada no vácuo e colocada em suspensão em ácido 10 nítrico a 65% (15 ml) e água (60 ml). Ácido acético (60 ml) e nitrito de sódio (0,99 g) foram adicionados sucessivamente e a mistura de reação foi aquecida até 100° C por 12 horas. A mistura foi resfriada a 25° C e vertida em água gelada (200 ml), basificada com hidróxido de sódio sólido e extraída com acetato de etila (3 x 50 ml). As fases orgânicas combinadas foram lavadas 15 com salmoura (l x 100 ml), secadas em sulfato de sódio e concentradas no vácuo para fornecer 0,31 g (9 %) do composto do título como um xarope amarelo. *H RMN (500 MHz, DMSO-d6): 0,98 (t, 6H), 2,34 (s, 3H), 3,30 (m, 4H), 7,25 (d, 2H), 7,52 (d, 2H), 7,80 (s, 2H).
N-(4’Amino-2,6-dietil-feml)-4-metil-benzenossulfonamida.
Uma solução de ditioneto de sódio (772 mg) em água (6 ml) foi adicionada a uma solução de N-(2,6-dietil-4-nitro-fenil)-4-metilbenzenossulfonamida (309 mg) em tetraidrofurano (6 ml) e a mistura resultante foi agitada a 50° C por 20 horas. Após resfriar, a água foi saturada com carbonato de potássio e extraída com acetato de etila (2 x 10 ml). As fases orgânicas combinadas foram secadas em sulfato de sódio, concentradas no vácuo e purificadas por cromatografia por vaporização instantânea para fornecer 70 mg (25%) do composto do título como um sólido branco amarelado. O produto foi usado diretamente na seguinte reação sem caracterização espectral.
&
N-(2,6-Dietil-4-morfolin-4~il-fenil)-4-metil-benzenossulfonamida.
Uma mistura de N-(4-amino-2,6-dietil-fenil)-4-metilbenzenossulfonamida (70 mg), éter bis-(2-bromoetílico) (33 pL), N,Ndiisopropil-etílamina (115 pL) e N-metilpirrolidina (0,3 ml) foi aquecida a
180° C por 20 minutos em um frasco de processo de microondas lacrado. A mistura foi diluída com acetato de etila (20 ml), lavada com salmoura (30 ml) e carbonato de potássio saturado aquoso (30 ml), e secada em sulfato de sódio. A fase orgânica foi concentrada no vácuo para fornecer 78 mg (91 %) do composto do título. LC-MS (m/z) 389 (MH+); tR = 2,77, (UV, ELSD) 57 %, 98 %. O produto bruto foi usado sem purificação adicional.
Cloreto de (3t4-Difliioro~fenil)-acetila.
Ao ácido (3,4-difluoro-fenil)-acético (2,0 g) dissolvido em 1,2dicloroetano (5 ml) foi adicionado cloreto de tionila (5 ml) e a mistura foi submetida a refluxo por 16 horas sob argônio. A mistura bruta foi concentrada 15 no vácuo para fornecer 2,2 g (100 %) do composto do título como um óleo amarelo. 'H RMN (500 MHz, CDClj): 4,10 (s, 2H), 7,00 (m, 1 H), 7,11 (m, 1 H), 7,17 (m, 1 H).
Os seguintes compostos foram preparados analogamente: Cloreto de (3-Fluoro-fenil)-acetila.
Rendimento de: 99 %. 'H RMN (500 MHz, CDC13): 4,13 (s,
2H), 6,92 (m, 1H), 7,01 (m, 2H), 7,34 (m, 1 H).
Compostos da invenção
Os sais de adição de ácido dos compostos da invenção podem facilmente ser formados por métodos conhecidos ao técnico habilitado na 25 técnica.
Exemplo 1 la N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-2-(4-fluoro-fenil)acetamida.
2-Bromo-4-morfolin-4-il-6-trifluorometil-fenilamina (0,236 g) &
e cloreto de 4-fluorofenilacetila (0,105 ml) foram dissolvidos em acetonitrila (5 ml) e aquecidos a 150° C por 10 minutos em um frasco de processo de microondas lacrado. Água (25 ml) foi adicionada e o produto foi extraído com acetato de etila (3 x 25 ml). As fases orgânicas foram lavadas com salmoura (50 ml), secadas em sulfato de magnésio e concentrada no vácuo, O produto bruto foi purificado por cromatografia por vaporizaçao instantânea para fornecer 0,027 g (9 %) do composto do título como um sólido branco amarelado. LC-MS (m/z) 462 (MH+); tR = 2,84, (UV, ELSD) 96%, 100%. 'H RMN (500 MHz, DMSO-dí): 3,23 (m, 4H), 3,62 (s, 2H), 3,72 (m, 4H), 7,14 (dd, 2H), 7,19 (d, 1H), 7,35 (dd, 2H), 7,46 (d, 1H), 9.78 (s, 1H).
Os seguintes compostos foram preparados analogamente:
lb 2-Ciclopentil-N-(2-bromo-6-trifluorometil-4-morfolin-4-il-fenil)acetamida.
Rendimento de: 22%. LC-MS (m/z) 436 (MH+); tR = 2,95, (UV, ELSD) 97 %, 98 %. ’H RMN (500 MHz, DMSO-d6): 1,20 (m, 2H), 1,50 (m, 2H), 1,60 (m, 2H), 1,77 (m, 2H), 2,24 (m, 1 H), 2,26 (d, 2H), 3,23 (m, 4H), 3,72 (m, 4H), 7,19 (d, 1 H), 7,46 (d, 1 H), 9.46 (s, 1 H).
lc N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-3-ciclo-pentilpropionamida.
Rendimento de: 20%. LC-MS (m/z) 450 (MH+); tR = 3,20, (UV, ELSD) 99 %, 98 %. 'H RMN (500 MHz, DMSO-d6): 1,09 (m, 2H), 1,49 (m, 2H), 1,59 (m, 4H), 1,76 (m, 2H), 1,80 (m, 1 H), 2,27 (t, 2H), 3,22 (m, 4H), 3,72 (m, 4H), 7,18 (d, 1 H), 7,46 (d, 1 H), 9.48 (s, 1 H).
Id N-(2-Cloro-6-ciano-4-morfolin-4-il-fenil)-3-ciclo-hexil-propionamida.
Rendimento de: 24 %. LC-MS (m/z) 376 (MH+); tR = 3,10, (UV, ELSD) 98 %, 100%. ’H RMN (500 MHz, DMSO-d6): 0,90 (m, 2H), 1,23 (m, 4H), 1,51 (m, 2H), 1,64 (m, 1H), 1,70 (m, 4H), 2,33 (t, 2H), 3,22 (m, 4H), 3,71 (m, 4H), 7,37 (s, 2H), 9,79 (s, 1H).
le N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-3-ciclo-hexil63 propionamida.
Rendimento de: 19 %. LC-MS (m/z) 464 (MH+); tR - 3,38, (UV, ELSD) 97 %, 99 %. ‘H RMN (500 MHz, DMSO-d6): 0,86 (dq, 2H), 1,16 (m, 3H), 1,27 (m, 1H), 1,48 (q, 2H), 1,61 (m, 1 H), 1,70 (m, 4H), 2,27 (t, 2H), 3 .23 (m, 4H), 3,72 (m, 4H), 7,18 (d, 1 H), 7,46 (d, 1H), 9,47 (s, 1H).
lf N-(2-Bromo-4-morfolin-4-il-6-triffuorometil-fenil)-2-(3-fluoro-fenil)acetamida.
Rendimento de: 44 %. LC-MS (m/z) 462 (MH+); tR = 2,85, (UV, ELSD) 98 %, 99 %.'H RMN (500 MHz, DMSO-d6): 3,23 (m, 4H), 3,67 (s, 2H), 3,72 (m, 4H), 7,10 (m, 2H), 7,18 (m, 2H), 7,36 (m, 1H), 7,46 (m, 1H), 9,82 (s, 1H).
lg N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-propionamida.
Rendimento de: 41 %. LC-MS (m/z) 382 (MH+); tR = 2,16, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-dó): 1,09 (t, 3H), 2,27 (q, 2H), 3,23 (m, 4H), 3,72 (m, 4H), 7,19 (d, 1 H), 7,47 (d, 1 H), 9,46 (s, 1 H). 1 h N-(2-Bromo-4-morfolin-4-il-6-trifluorometil-fenil)-butiramida.
Rendimento de: 76%. LC-MS (m/z) 396 (MH+); tR = 2,43, (UV, ELSD) 99 %, 96%. 'H RMN (500 MHz, DMSO-d6): 0,93 (t, 3H), 1,60 (m, 2H), 2,24 (t, 2H), 3,23 (m, 4H), 3,72 (m, 4H), 7,18 (d, 1 H), 7,46 (d, 1 H), 9,45 (s, 1 H).
li N-(2-Cloro-4-morfolin-4-il-6-trifluorometil-fenil)-2-(3-fluoro-fenil)- acetamida.
Rendimento de: 21 %. LC-MS (m/z) 417 (MH+); tR = 2,84, (UV, ELSD) 97 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 3,23 (m, 4H), 3,67 (s, 2H), 3,72 (m, 4H), 7,07 (dt, 1H), 7,15 (m, 3 H), 7,32 (d, 1 H), 7,36 (m, 1 H), 9,76 (s, 1 H).
lj N-(2-Cloro-4-morfolin-4-il-6-trifluorometil-fenil)-2-ciclo-pentil-acetamida.
Rendimento de: 76%. LC-MS (m/z) 391 (MHj; tR = 2,97, (UV, ELSD) 99 %, 99 %. *H RMN (500 MHz, DMSO-d6): 1,19 (m, 2H), 1,50 (m, 2H), 1,60 (m, 2H), 1,76 (m, 2H), 2,23 (m, IH), 2,26 (d, 2H), 3,23 (m, 4H), 3,72 (m, 4H), 7,15 (d, 1H), 7,32 (d, 1H), 9,40 (s, 1 H).
Exemplo 2
2a 2-Ciclopentil-N-(2,6-dimetil-4-tiomorfolin-4-il-fenil)-acetamida.
Bis(dibenzilidenoacetona)paládio (37 mg) e (2’-diciclo-hexilfosfanilbifenil-2-il)-dimetil-amina (38 mg) foram misturados em tolueno desgaseificado seco (2 ml) sob argônio por 5 minutos. A esta mistura foram adicionados N-(4-bromo-2,6-dimetilfenil)-2-ciclopentil-acetamida (200 mg), terc-butóxido de potássio (90 mg) e tiomorfolina (80 mg) e a mistura de reação foi aquecida a 90° C em um frasco de 4 ml lacrado sob argônio por 16 horas, resfriada e filtrada através de sílica (2 g). Água/salmoura (1:1, 4 ml total) foram adicionadas e a mistura foi extraída com acetato de etila (3x2 ml). As fases orgânicas combinadas foram secadas em sulfato de magnésio e concentradas no vácuo. O produto bruto foi purificado por LC-MS preparativo para fornecer 5,6 mg (rendimento de 3 %) do composto do título. LC-MS-TOF (m/z) 333 (MH+); tR - 2,03, (UV, ELSD) 98 %, 100%.
Os seguintes compostos foram preparados analogamente: 2b 2-Ciclopentil-N-[2,6-dimetil-4-(2-fenil-morfolin-4-il)fenil]-acetamida.
Rendimento de: 3 %. LC-MS-TOF (m/z) 393 (MH+); tR 3,11, (UV, ELSD) 96%, 98 %.
2c 2-Ciclopentil-N-[2,6-dimetil-4-(2-fenil-tiomorfolin-4-il)-fenil]acetamida.
Rendimento de: 4 %. LC-MS-TOF (m/z) 409 (MH+); tR = 3,30, (UV, ELSD) 99 %, 98 %.
2d 2-Ciclopentil-N-(2,6-dimetil-4-(3-piridin-3-il-tiomorfolin-4-il)-fenil]acetamida.
Rendimento de: 12%. LC-MS-TOF (m/z) 410 (MH+); tR 2,00, (UV, ELSD) 99 %, 100%.
2e 2-Ciclopentil-N-{2,6-dimetil-4-(2-(4-trifluorometil-fenil)-tiomorfolin-4il ]-fenil} -acetamida.
Rendimento de: 6%. LC-MS (m/z) 477 (MH); tR = 3,64, (UV, ELSD) 95%, 100%.
2f N-{4-[2-(2-Cloro-feniI)-tiomorfolin-4-il]-2,6-dimetil-fenil}2-ciclopentilacetamida.
Rendimento de: 20%. LC-MS-TOF (m/z) 444 (MH+); tR = 3,59, (UV, ELSD) 89 %, 100%.
2g 2-Ciclopentil-N-{2,6-dimetil-4-[2-(4-trifluorometil-fenil)-morfolin-4-il]fenilj-acetamida.
Rendimento de: 26%. LC-MS (m/z) 461 (MH+); tR = 3,55, (UV, ELSD) 90%, 95%.
2h N- {4-[2-(2-Cloro-fenil)-morfolin-4-il]-2,6-dimetil-fenil} -2-ciclopentilacetamida.
Rendimento de: 35%. LC-MS (m/z) 427 (MH+); tR = 3,44, (UV, ELSD) 77 %, 95%.
2i 2-Ciclopentil-N-{4-[2-(4-fluoro-fenil)-morfolin-4-il]-2,6-dimetil-fenil)acetamida.
Rendimento de: 17 %. LC-MS (m/z) 411 (MH+); tR = 3,17, (UV, ELSD) 98 %, 99 %.
Exemplo 3
3a 2-Biciclo[2,2,1 ]hept-2-il-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Ácido Biciclo[2,2,l]hept-2-il-acético (0,41 g) foi aquecido a 50° C por 2 horas sob argônio em uma mistura de 1:1 de cloreto de tionila e 1,2-dicloroetano (10 ml totais). Os solventes foram removidos no vácuo e o cloreto ácido resultante foi redissolvido em acetonitrila (5 ml) e 2,6-dimetil-4morfolin-4-il-fenilamina (0,50 g) foi adicionado. A mistura de reação foi aquecida a 150° C por 10 minutos em um frasco de processo de microondas lacrado. Água (25 ml) foi adicionada e o produto foi extraído com acetato de etila (3 x 25 ml). As fases orgânicas combinadas foram lavadas com salmoura (50 ml), secadas em sulfato de magnésio e concentrada no vácuo. O produto bruto foi purificado por cromatografia por vaporizaçào instantânea para fornecer 0,074 g (9 %) do composto do título como um sólido branco amarelado. LC-MS-TOF (m/z) 343 (MH+); tR = 2,21, (UV, ELSD) 98 %, 100%. ]H RMN (500 MHz, DMSO-dó): 1,14 (m, 4H), 1,41 (m, 4H), 1,90 (m, 1H), 2,02 (m, 1 H), 2,06 (s, 6H), 2,10 (m, 1 H), 2,20 (m, 2H), 3,05 (m, 4H),
3,71 (m, 4H), 6,62 (s, 2H), 8,92 (s, 1H).
Os seguintes compostos foram preparados analogamente:
3b 2-Ciclo-hexil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Rendimento de: 19 %. LC-MS-TOF (m/z) 331 (MH+); tR = 2,21, (UV, ELSD) 97 %, 100%. RMN (500 MHz, DMSO-d6): 0,98 (m, 2H), 1,22 (m, 4H), 1,68 (in, 6H), 2,07 (s, 6H), 2,15 (d, 2H), 3,05 (m, 4H),
3,71 (m, 4H), 6,63 (s, 2H), 8,93 (s, 1H).
3c 3-(3,4-Difluoro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil) propionamida.
Rendimento de: 40%. LC-MS-TOF (m/z) 375 (MH+); tR = 2,39, (UV, ELSD) 97 %, 100%. lH RMN (500 MHz, DMSO-dõ): 1,95 (s, 6H), 2,60 (t, 2H), 2,91 (t, 2H), 3,04 (m, 4H), 3,71 (m, 4H), 6,60 (s, 2H), 7,10 (m, 1 H), 7,34 (m, 2H), 8,97 (s, 11-3).
Exemplo 4
4a 2-Ciclopentil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
2,6-Dimetil-4-morfolin-4-il-fenilamina (0,50 g) e cloreto de ciclopentilacetila (0,53 ml) foram dissolvidos em acetonitrila (5 ml) e aquecidos a 150° C por 10 minutos em um frasco de processo de microondas lacrado. Água (25 ml) foi adicionada e o produto foi extraído com acetato de etila (3 x 25 ml). As fases orgânicas foram lavadas com salmoura (50 ml), secadas em sulfato de magnésio e concentradas no vácuo. O produto bruto foi purificado por cromatografia por vaporizaçào instantânea para fornecer 0,138 g (20%) do composto do título como um sólido branco amarelado. LC-MSTOF (m/z) 317 (MH+); tR =1,93, (UV, ELSD) 95%, 100%. 111 RMN (500 MHz, DMSO-d6): 1,21 (m, 2H), 1,52 (m, 2H), 1,61 (m, 2H), 1,76 (m, 2H),
2,07 (s, 6H), 2,11 (m, 1 H), 2,25 (d, 2H), 3 .05 (dd, 4H), 3,71 (dd, 4H), 6,63 (s, 2H), 8,94 (s, 1 H).
Os seguintes compostos foram preparados analogamente:
4b Ácido butílico do éster (2,6-Dimetil-4-morfolin-4-il-fenil)-carbâmico.
Rendimento de: 2%. LC-MS-TOF (m/z) 307 (MH+); tR = 2,25, (UV/ELSD) 99 %, 100%.
4c 2-(4-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Rendimento de: 8 %. LC-MS-TOF (m/z) 359 (MH); tR = 2,16, (UV, ELSD) 98 %, 100%?Η RMN (500 MHz, DMSO-de): 2,01 (s, 6H), 3,04 (dd, 4H), 3,60 (s, 2H), 3,71 (dd, 4H), 6,62 (s, 2H), 7,39 (m, 4H), 9,24 (s, 1H). 4d (2,6-dimetil-4-morfolin-4-ilfenil)-amida do ácido 2,3-diidro-benzofuran-2carboxílico.
Rendimento de: 13 %. LC-MS-TOF (m/z) 353 (MH+); tR = 2,11, (UV, ELSD) 97 %, 100%. *H RMN (500 MHz, DMSO-d6): 2,01 (s, 6H), 3,05 (dd, 4H), 3,30 (m, 1H), 3,55 (dd, 1H), 3,71 (dd, 4H), 5,31 (dd, 1H), 6,64 (s, 2H), 6,88 (t, 2H), 7,15 (t, 1H), 7,25 (d, 1H), 9,33 (s, 1 H).
4e 3-Ciclo-hexil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-propionamida.
Rendimento de: 8 %. LC-MS-TOF (m/z) 345 (MIT); tR = 2,64, (UV, ELSD) 97 %, 98 %. 'H RMN (500 MHz, DMSO-d6): 0,88 (m, 2H), 1,16 (m, 2H), 1,25 (m, 2H), 1,49 (q, 2H), 1,63 (m, 1 H), 1,69 (m, 4H), 2,06 (s, 6H), 2,27 (t, 2H), 3,05 (dd, 4H), 3,71 (dd, 4H), 6,63 (s, 2H), 8,95 (s, 1H). 4f 3-Ciclopentil-N-(2,6-dimetil-4-morfolin-4-il-fenil)propionamida.
Rendimento de: 34 %. LC-MS (m/z) 331 (MH+); tR = 2,33, (LTV, ELSD) 97 %, 100%. *H RMN (500 MHz, DMSO-dú): 1,11 (m, 2H), 1,49 (m, 2H), 1,60 (m, 4H), 1,77 (m, 2H), 2,07 (s, 6H), 2,28 (t, 2H), 3,11 (dd, 4H), 3,73 (dd, 4H), 6,74 (s, 2H), 9,01 (s, 1 H).
4g N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(4-íluoro-fenil)-acetamida.
Rendimento de: 13 %. LC-MS-TOF (m/z) 343 (MH+); tR = 2,05, (UV, ELSD) 98 %, 100%. 'H RMN (500 MHz, DMSO-d6): 2,01 (s,
7^
6Η), 3,05 (m, 4H), 3,59 (s, 2H), 3,70 (m, 4H), 6,61 (s, 2H), 7,16 (dd, 2H), 7,38 (dd, 2H), 9,22 (s, 1H).
4h N-(2,6-dimetil-4-morfolin-4-il-fenil)-2-tiofen-2-il-acetamida.
Rendimento de: 3 %. LC-MS-TOF (m/z) 331 (MH); tR = 1,94, (UV, ELSD) 97 %, 100%.
4i N-(2,6-Dimetil-4-morfolín-4-il-fenil)-3,3-dimetilbutiramida.
Rendimento de: 43 %. LC-MS-TOF (m/z) 305 (MW); tR = 2,14, (UV, ELSD) 99 %, 100%. RMN (500 MHz, DMSO-d6): 1,05 (s, 9H), 2,09 (s, 6H), 2,17 (s, 2H), 3,05 (m, 4H), 3,72 (m, 4H), 6,63 (s, 2H), 8,89 (s, 1H).
4j Ácido (2,6-dimetil-4-morfolin-4-il-fenil)-amida hexanóico.
Rendimento de: 14 %. LC-MS (m/z) 305 (MH); tR =1,99, (UV, ELSD) 95%, 97 %. !H RMN (500 MHz, DMSO-d6): 0,88 (t, 3H), 1,31 (m, 4H), 1,60 (m, 2H), 2,06 (s, 6H), 2,26 (t, 2H), 3,05 (m, 4H), 3,71 (m, 4H), 6,63 (s, 2H), 8,94 (s, 1H).
Exemplo 5
5a 2-Ciclo-heptil-N-(2,6-dimetil-4-morfolin-4-il-fenil) acetamida.
Ácido ciclo-heptil acético (0,45 g) e uma gota de N,Ndimetilformamida foram agitados a 25° C por 2 horas sob argônio em uma mistura de 1:1 de cloreto de oxalila (2 M em diclorometano) e 1,2dicloroetano (12 ml totais). Os solventes foram removidos no vácuo e o cloreto ácido resultante foi redissolvido em acetonitrila (8 ml) e 2,6-dimetil-4morfolin-4-il-fenilamina (0,50 g) e óxido de magnésio (0,20 g) foram adicionados. A mistura de reação foi agitada a 25° C por 16 horas sob argônio e depois filtrada através de Celite (10 g). A fase orgânica foi concentrada no vácuo e o produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 0,133 g (16%) do composto do título como um sólido branco amarelado. LC-MS (m/z) 345 (MH+); tR = 2,36, (UV, ELSD) 97 %, 100%. lH RMN (500 MHz, DMSO-dó): 1,23 (m, 2H), 1,44 (m, 4H), 1,60 (m, 4H), 1,73 (m, 2H), 1,99 (m, 1H), 2,07 (s, 61-1), 2,18 (d, 2H), 3,05 (m, 4H), 3,71 (m, 4H), 6,63 (s, 2H), 8,94 (s, 1 H).
Exemplo 6
6a Ácido benzílico do éster (2,6-Dimetil-4-morfolin-4-il-fenil)-carbâmico.
Cloroformiato de benzila (32 mg) foi adicionado a uma solução de 2,6-dimetil4-morfolin-4-il-fenilamina 0,15 M e N,N-diisopropiletilamina 0,30 M em 1,2-dicloroetano (1 ml). O frasco foi agitado por 16 horas e concentrado no vácuo. Hidróxido de sódio aquoso (1 M, 2 ml) foi adicionado e a mistura bruta foi extraída com acetato de isopropila/tetraidrofurano (4:1, 2,5 ml). A fase orgânica foi concentrada no vácuo e redissolvida em sulfóxido de dimetila (0,5 ml) de que 0,2 ml foi sujeito a purificação de LC-MS preparativo para fornecer 9,5 mg (rendimento de 47 %) do composto do título como um óleo. LC-MS (m/z) 341 (MH+); tR = 2,28, (UV, ELSD) 100%, 100%.
Os seguintes compostos foram preparados analogamente: 6b Éster 2-cloro-benzílico do ácido (2,6-Dimetil-4-morfolin-4-il-fenil)carbâmico.
Rendimento de: 33 %. LC-MS (m/z) 375 (MH+); tR = 2,53, (UV, ELSD) 99 %, 100%.
6c (2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 3,5,5-trimetilhexanóico.
Rendimento de: 36%. LC-MS (m/z) 347 (MH+); tR = 2,53, (UV, ELSD) 99 %, 100%.
6d Ácido (2,6-dimetil-4-morfolin-4-il-fenil)-amida octanóico.
Rendimento de: 47 %. LC-MS (m/z) 333 (MH+); tR = 2,47, (UV, ELSD) 99 %, 100%.
6e Ácido (2,6-dimetil-4-morfolin-4-il-fenil)-amida heptanóico.
Rendimento de: 40%. LC-MS (m/z) 319 (MH+); tR - 2,20, (UV, ELSD) 91 %, 99 %.
6f N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-fenil-acetamida.
Rendimento de: 35%. LC-MS (m/z) 325 (MH+); tR = 1,80, (UV, ELSD) 99 %, 100%.
Exemplo 7
7a 2-(3,4-Dicloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Ácido 3,4-Diclorofenil acético (39 mg) foi agitado a 25° C por 2 horas sob argônio em uma mistura de 1:1 de cloreto de oxalila (2 M em diclorometano) e 1,2-dicloroetano (1 ml total). Os solventes foram removidos no vácuo e uma solução de 2,6-dimetil-4-morfolin-4-il-fenilamina 0,15 M e N,N-diisopropil-etilamina 0,30 M em 1,2-dicloroetano (1 ml) foi adicionada ao cloreto ácido resultante. O frasco foi agitado por 16 horas e concentrado no vácuo. Hidróxido de sódio aquoso (1 M, 2 ml) foi adicionado e a mistura bruta foi extraída com acetato de isopropila/tetraidrofurano (4:1, 2,5 ml). A fase orgânica foi concentrada no vácuo e redissolvida em sulfóxido de dimetila (0,5 ml) da qual 0,2 ml, foi submetida a purificação por LC-MS preparativo para fornecer 2,7 mg (rendimento de 11 %) do composto do título como um óleo. LC-MS (m/z) 394 (MH+); tR = 2,40, (UV, ELSD) 80%, 100%.
Os seguintes compostos foram preparados analogamente:
7b 2-(4-Alilóxi-3-cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)acetamida.
Rendimento de: 14 %. LC-MS (m/z) 415 (MHO); tR - 2,40, (UV, ELSD) 91 %, 100%.
7c N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(3-trifluorometil-fenil)acetamida.
Rendimento de: 18 %. LC-MS (m/z) 393 (ME); tR - 2,31, (UV, ELSD) 97 %, 100%.
7d N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-naftaleno-2-il-acetamida.
Rendimento de: 16%. LC-MS (m/z) 375 (MM); tR ~ 2,27, (UV, ELSD) 83 %, 100%.
7e 3-(3-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil) propionamida.
Rendimento de: 10%. LC-MS (m/z) 373 (MH+); tR = 2,23, <77 (UV, ELSD) 71 %, 94 %.
7f N-(2,6-Dimetil-4-rnorfolin-4-il-fenil)-2-(3,4-dimetil-fenil)-acetamida.
Rendimento de: 44 %. LC-MS (m/z) 353 (MH+); tR = 2,21, (UV, ELSD) 80%, 100%.
7g 2-(3-Bromo-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Rendimento de: 22%. LC-MS (m/z) 404 (MH+); tR = 2,19, (UV, ELSD) 95%, 100%.
7h 2-(3-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Rendimento de: 25%. LC-MS (m/z) 359 (MH+); tR = 2,13, (UV, ELSD) 95%, 100%.
7i N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-p-tolil-acetamida.
Rendimento de: 26%. LC-MS (m/z) 339 (MH); tR = 2,04, (UV, ELSD) 99 %, 100%.
7j N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-m-tolil-acetamida.
Rendimento de: 24 %. LC-MS (m/z) 339 (MH+); tR = 2,03, (UV, ELSD) 88 %, 100%.
7k 2-(3,4-Difluoro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Rendimento de: 24 %. LC-MS (m/z) 361 (MH+); tR = 2,03, (UV, ELSD) 99 %, 100%.
N-(2,6-Dimetil-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)-acetamida.
Rendimento de: 12%. LC-MS (m/z) 343 (MH+); tR -1,90, (UV, ELSD) 88 %, 97 %.
7m 2-(2-Cloro-fenil)-N-(2,6-dimetil-4-morfolin-4-il-feniÍ)-acetamida.
Rendimento de: 2%. LC-MS (m/z) 359 (MH); tR - 2,01, (UV, ELSD) 98 %, 99 %.
7n (2,6-dimetil-4-morfolin-4-il-fenii)-amida do ácido pentanóico.
Ácido pentanóico (0,37 g), N,N-diisopropil-etilamina (1,51 ml) e N-[(dimetilamino)-lH-l,2,3-triazolo-[4,5-b]piridin-l-il-metileno]-Nmetil-metanaminio hexafluoro-fosfato N-óxido (1,66 g) foram misturados em
7?
Ν,Ν-dimetilformamida seco (3 ml) e agitados sob argônio por 2 minutos. 2,6Dimetil-4-morfolin-4-il-fenilamina (0,50 g) dissolvido em Ν,Νdimetilformamida seco (2 ml) foi adicionado à mistura de reação, que foi agitada a 25° C sob argônio por 16 horas. Acetato de etila (20 ml) foi adicionado e a fase orgânica foi lavada com cloreto de amônio saturado aquoso/água (1:1, 20 ml), água (20 ml), salmoura/água (1:1, 20 ml), secada em sulfato de magnésio, concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 0,28 g (rendimento de 40%) do composto do título como um sólido branco. LC-MS (m/z) 291 (MH+); tR = 1,61, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 0,91 (t, 3H), 1,34 (m, 2H), 1,58 (qui, 2H), 2,06 (s, 6H), 2,27 (t, 2H), 3,04 (t, 4H), 3,71 (t, 4H), 6,62 (s, 2H), 8,94 (s, 1 H).
Os seguintes compostos foram preparados analogamente:
7o (2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 4-metil pentanóico.
Rendimento de: 28 %. LC-MS (m/z) 305 (MH+); tR =1,88, (UV, ELSD) 98 %, 99 %. ’H RMN (500 MHz, DMSO-d6): 0,91 (d, 6H), 1,49 (m, 2H), 1,57 (m,l 13), 2,06 (s, 6H), 2,27 (t, 2H), 3,05 (t, 4H), 3,71 (t, 4H), 6,63 (s, 2H), 8,95 (s, 1 H).
7p 2-Ciclopent-2-enil-N-(2,6-dimetil-4-morfolin-4-il-fenil)-acetamida.
Rendimento de: 69 %. LC-MS (m/z) 315 (MH+); tR = 1,82, (UV, ELSD) 97 %, 99 %. *H RMN (500 MHz, DMSO-d6): 1,51 (m, 2H), 2,06 (m, 1H), 2,08 (s, 6M), 2,23 (m, 1H), 2,29 (m, 1 H), 2,34 (m, 2H), 3,05 (m, 4H), 3,72 (m, 4H), 5,73 (tn, L H), 5,76 (m, 1 H), 6,63 (s, 2H), 8,98 (s, 1 H). 7q (2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 5-metil-hexanóico.
Rendimento de: 41 %. LC-MS (m/z) 319 (MH+); tR = 2,18, (UV, ELSD) 96%, 99 %. 'H RMN (500 MHz, DMSO-d6): 0,87 (d, 6H), 1,21 (m, 2H), 1,59 (m, 3H), 2,07 (s, 6H), 2,24 (t, 2H), 3,05 (t, 4H), 3,72 (t, 4H), 6,63 (s, 2H), 8,94 (s, 1 H).
7r (2,6-dimetil-4-naorfolin-4-il-fenil)-amida do ácido 3-metil pentanóico.
Rendimento de: 31 %. LC-MS (m/z) 305 (MH+); tR =1,86, (UV, ELSD) 98 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 0,89 (t, 3H), 0,93 (d, 3H), 1,22 (m, 1H), 1,39 (m, 1H), 1,88 (m, 1H), 2,07 (s, 6H), 2,26 (m, 1H), 3,05 (t, 4H), 3,72 (t, 4H), 6,63 (s, 2H), 8,95 (s, 1 H).
7s (2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido hex-5-enóico.
Rendimento de: 68 %. LC-MS (m/z) 303 (MH+); tR = 1,71, (UV, ELSD) 99 %, 99 %. *H RMN (500 MHz, DMSO-dó): 1,69 (qui, 2H), 2,07 (s, 6H), 2,09 (m, 2H), 2,28 (t, 2H), 3,05 (t, 4H), 3,71 (t, 4H), 4,99 (dd, 1H), 5,04 (dd, 1H), 5,84 (m, 1H), 6,63 (s, 2H), 8,95 (s, 1 H).
7t (2,6-dimetil-4-morfolin-4-il-fenil)-amida do ácido 3-etil pentanóico.
Ácido 3-etilpentanóico (0,79 g) e cloreto de tionila(0,44 ml) foram misturados em acetonitrila (10 ml) e aquecidos a 110° C por 5 minutos em um frasco de processo de microondas lacrado. 2,6-dimetil-4-morfolin-4-ilfenilamina (1,25 g) dissolvido em acetonitrila (10 ml) foi adicionado à mistura de reação e aquecido a 150° C por 15 minutos em um frasco de processo de microondas lacrado. Bicarbonato de sódio saturado aquoso/salmoura/água (1:1:1, 50 ml) foram adicionados à mistura bruta e esta foi extraída com acetato de etila (3 x 50 ml). As fases orgânicas combinadas foram secadas em sulfato de magnésio e concentradas no vácuo. O produto bruto foi recristalizado a partir do tolueno quente, e o produto foi coletado por filtração, lavado com tolueno gelado e secado no vácuo para fornecer 0,59 g (rendimento de 30%) do composto do título como um sólido branco amarelado. LC-MS (m/z) 319 (MH+); tR = 2,04, (UV, ELSD) 98 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 0,87 (t, 6H), 1,35 (qui, 4H), 1,76 (m, 1H), 2,07 (s, 6H), 2,20 (d, 2H), 3,05 (t, 4H), 3,71 (t, 4H), 6,62 (s, 2H), 8,94 (s, 1 H). Exemplo 8
8a 2-Ciclopentil-N-(4-morfolin-4-il-2-piridin-3-il-6-triíluoro-metil-fenil)acetamida.
2-Ciclopentil-N-(2-bromo-6-triíluorometil-4-morfolin-4-il74 fenil)-acetamida (lb, 15 mg), ácido 3-piridilborônico (21 mg), carbonato de potássio aquoso (5 M, 90 pL) e acetato de paládio (II) (1 mg) foram misturados em acetona (2 ml) e aquecidos a 130° C por 20 minutos em um frasco de processo de microondas lacrado. A mistura de reação foi filtrada através de sílica (500 mg), concentrada no vácuo, redissolvida em sulfóxido de dimetila (0,5 ml) e submetida a purificação por LC-MS preparativo para fornecer 2,7 mg (rendimento de 18 %) do composto do título como um óleo incolor. LC-MS (m/z) 434 (MH+); tR - 1,89, (UV, ELSD) 99 %, 99 %.
Os seguintes compostos foram preparados analogamente:
8b 2-Ciclopentil-N-(5-morfolin-4-il-3-trifluorometil-bifenil-2-il)-acetamida.
Rendimento de: 46%. LC-MS (m/z) 433 (MH+); tR = 3,16, (UV, ELSD) 96%, 99 %.
8c 2-Ciclopentil-N-(4,-fluoro-5-morfolin-4-il-3-trifluorometil-bifenil-2-il)acetamida.
Rendimento de: 20%. LC-MS (m/z) 451 (MH+); tR = 3,18, (UV, ELSD) 99 %, 99 %.
8d 2-Ciclopentil-N-(4’-metil-5-morfolin-4-il-3-trifluorometil-bifenil-2-il)acetamida.
Rendimento de; 51 %. LC-MS (m/z) 447 (MH+); tR = 3,32, (UV, ELSD) 97 %, 99 %.
8e 2-Ciclopentil-N-(3’-metil-5-morfolin-4-il-3-trifluorometil-bifenil-2-il)acetamida.
Rendimento de: 37 %. LC-MS (m/z) 447 (MH+); tR = 3,33, (UV, ELSD) 99 %, 99 %.
8f 2-Ciclopentil-N-(3’-4’-difluoro-5-morfolin-4-il-3-trifluoro-metil’bifenil-2il)acctamida.
Rendimento de: 51 %. LC-MS (m/z) 469 (MH+); tR = 3,29, (UV, ELSD) 99 %, 99 %.
8g 2-(4-Fluoro-fenil)-N-(4-morfolin-4-il-2-piridin-3-il-6-trifluorometil-fenil)75 acetamida.
Rendimento de: 25%. LC-MS (m/z) 460 (MH+); tR = 1,81, (UV, ELSD) 94 %, 99 %.
Exemplo 9
9a 2-Ciclopentil-N-(2,6-dietil-4-morfolin-4-il-fenil)-acetamida.
N-(2,6-Dietil-4-morfolin-4-il-fenil)-4-metil-benzenossulfonamida (78 mg), ácido sulfürico (0,95 ml) e água (50 pL) foram agitados a 40° C por 3 horas. Gelo (30 ml) e água (30 ml) foram adicionados e a mistura foi basifícada com carbonato de potássio sólido. A mistura foi extraída com acetato de etila (3 x 20 ml), as fases orgânicas combinadas foram secadas em sulfato de sódio e concentradas no vácuo. O resíduo foi redissolvido em tetraidrofurano (1 ml) e misturado com piridina (49 pL) e cloreto de ciclopentilacetila (44 pL. A mistura foi agitada por 1 hora a 25° C, diluída com acetato de etila (20 ml) e lavada com bicarbonato de sódio aquoso a 10% (20 ml) e salmoura (20 ml). A fase orgânica foi secada em sulfato de sódio, concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 35 mg (51 %) do composto do título como um marrom claro sólido. LC-MS (m/z) 345 (MIT); tR = 2,27, (UV, ELSD) 84 %, 98 %.
9b 2-Ciclopentil-N-(2,6-diisopropil-4-morfolin-4-il-fenil)-acetamida.
A uma solução de 2,6-diisopropil-4-morfolin-4-il-fenilamina (279 mg) e piridina (245 pL) em tetraidrofurano (2 ml) foi adicionado cloreto de ciclopentilacetila (210 pL) e a mistura foi agitada por 1 hora a 25° C. A mistura de reação foi diluída com acetato de etila (20 ml) e lavada com bicarbonato de sódio aquoso a 10% (20 ml) e salmoura (20 ml). A fase orgânica foi secada em sulfato de sódio, concentrada no vácuo e purificada por cromatografia por vaporização instantânea. Recristalização do material marrom bruto a partir do acetato de etila quente/heptano forneceu 122 mg (33 %) do composto do título como um sólido branco. LC-MS (m/z) 373 (MH+);
tR = 2,58, (UV, ELSD) 98 %, 99 %. lH RMN (500 MHz, CDC13): 1,19 (d, 12H), 1,27 (m, 2H), 1,60 (m, 2H), 1,68 (m, 2H), 1,93 (m, 2H), 2,40 (m, 1H), 2,41 (m, 2H), 3,05 (m, 2H), 3,17 (m, 4H), 3,87 (m, 4H), 6,49 (s, 1 H), 6,70 (s, 2H).
Exemplo 10
10a 2-Ciclopentil-N-(2,6-difluoro-4-morfolin-4-il-fenil)-acetamida.
2,6-Difluoro-4-morfolin-4-il-fenilamina (0,20 g) e cloreto de ciclopentilacetila (149 pL) foram dissolvidos em acetonitrila (4 ml) e aquecidos a 150° C por 10 minutos em um frasco de processo de microondas lacrado. A mistura de reação foi concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 228 mg (75%) do composto do título como um sólido branco. LC-MS (m/z) 325 (MH+); tR = 2,61, (UV, ELSD) 99 %, 99 %. ‘H RMN (500 MHz, DMSO-d6): 1,19 (m, 2H), 1,51 (m, 2H), 1,60 (m, 2H), 1,74 (m, 2H), 2,19 (m, 1H), 2,26 (d, 2H), 3,14 (m, 4H), 3,70 (m, 4H), 6,6S (d, 2H), 9,20 (s, 1H).
Os seguintes compostos foram preparados analogamente:
10b (2,6-difluoro-4-morfolin-4-il-fenil)-amida do ácido hexanóico.
Rendimento de: 84 %. LC-MS (m/z) 313 (MH+); tR = 2,60, (UV, ELSD) 99 %, 98 %. 'H RMN (500 MHz, DMSO-d6): 0,87 (t, 3H), 1,29 (m, 4H), 1,57 (qui, 2H), 2,27 (t, 2H), 3,14 (t, 4H), 3,70 (t, 4H), 6,68 (d, 2H), 9,22 (s, 1 H).
10c N-(2,6-Difluoro-4-morfolin-4-il-fenil)-3,3 -dimetil-butiramida.
Rendimento de: 58 %. LC-MS (m/z) 313 (MH+); tR = 2,49, (UV, ELSD) 99 %, 99 %. Ή RMN (500 MHz, DMSO-d6): 1,02 (s, 9H), 2,15 (s, 2H), 3,14 (m, 4H), 3,71 (m, 4H), 6,68 (d, 2H), 9,15 (s, 1 H). lOd N-(2,6-Difluoro-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)-acetamida.
Rendimento de: 68 %. LC-MS (m/z) 351 (MH+); tR = 2,52, (UV, ELSD) 96%, 99 %. ‘H RMN (500 MHz, DMSO-dó): 3,14 (t, 4H), 3,67 (s, 2H), 3,70 (t, 4H), 6,69 (d, 2H), 7,09 (m, 1 H), 7,14 (m, 2H), 7,3 7 (m, 1 H),
9,5 8 (s, 1 Η). lOe 2-Ciclopent-2-enil-N-(2,6-difIuoro-4-morfolin-4-il-fenil)-acetamida.
Ácido ciclopent-2-enilacético (0,17 ml), N,N-diisopropiletilamina (0,50 ml) e N-[(dimetilamino)-lH-l,2,3-triazolo-[4,5-b]-piridin-lil-metileno]-N-metilmetanaminio hexafluoro-fosfato N-óxido (0,55 g) foram misturados em Ν,Ν-dimetilformamida seco (3 ml) e agitados sob argônio por 2 minutos. 2,6-Difluoro-4-morfolin-4-il-fenilamina (0,20 g) dissolvido em NN-dimetilformamida seco (2 ml) foi adicionado à mistura de reação, que foi agitada a 25° C sob argônio por 16 horas. Acetato de etila (20 ml) foi adicionado e a fase orgânica foi lavada com cloreto de amônio saturado aquoso/água (1:1, 20 ml), água (20 ml), salmoura/água (1:1, 20 ml), secada em sulfato de magnésio, concentrada no vácuo e purificada por cromatografia por vaporizaçao instantânea para fornecer 0,21 g (rendimento de 71 %) do composto do título como um sólido branco. LC-MS (m/z) 323 (MH+); tR = 2,49, (UV, ELSD) 96%, 99 %. !H RMN (500 MHz, DMSO-d6): 1,48 (m, 1H), 2,02 (m, 1H), 2,29 (m, 4H), 3,03 (m, 1H), 3,14 (t, 4H), 3,71 (t, 4H), 5,71 (m,
I H), 5,76 (m, 1 H), 6,68 (d, 2H), 9,25 (s, 1 H).
Os seguintes compostos foram preparados analogamente: lOf 2-Biciclo[2,2,l]hept-2-il-N-(2,6-difluoro-4-morfolin-4-il-fenil)acetamida.
Rendimento de: 56%. LC-MS (m/z) 351 (MH+); tR = 2,90, (UV, ELSD) 98 %, 99 %. ‘H RMN (500 MHz, DMSO-d6): 1,11 (m, 4H), 1,43 (m, 4H), 1,85 (m, 1H), 2,00 (m, 1H), 2,11 (m, 1 H), 2,21 (m, 2H), 3,14 (t, 4H), 3,71 (t, 4H), 6,67 (d, 2H), 9,19 (s, 1 H).
Exemplo 11
II a 2-Biciclo[2,2,1 ]hept-2-il-N-(2-metil-4-morfolin-4-il-6-trifluorometilfenil)-acetamida.
Ácido biciclo[2,2,l]hept-2-il-acético (160 mg), N,N-diisopropiletilamina (0,44 ml) e N-[(dimetilamino)-lH-l,2,3-triazolo-[4,5-b]78 piridin-l-il-metileno]-N-metilmetanaminio hexafluoro-fosfato N-óxido (0,47 g) foram misturados em Ν,Ν-dimetilformamida seco (3 ml) e agitado sob argônio por 2 minutos. 2-metil-4-morfolin-4-il-6-trifluorometil-fenilamina (0,18 g) dissolvido em Ν,Ν-dimetilformamida seco (2 ml) foi adicionado à mistura de reação, que foi agitada a 25° C sob argônio por 16 horas. Acetato de etila (20 ml) foi adicionado e a fase orgânica foi lavada com cloreto de amônio saturado aquoso/água (1:1, 20 ml), água (20 ml), salmoura/água (1:1, 20 ml), secada em sulfato de magnésio, concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 16 mg (rendimento de 6%) do composto do título como um sólido branco. LC-MS (m/z) 397 (MH+); tR = 3,12, (UV, ELSD) 91 %, 98 %.
Os seguintes compostos foram preparados analogamente:
11b (2-metil-4-morfolin-4-il-6-trifluorometil-fenil)amida do ácido 5-metil pentanóico.
Rendimento de: 6%. LC-MS (m/z) 359 (MH+); tR = 2,76, (UV, ELSD) 94 %, 99 %.
11c (2-metil-4-morfolin-4-il-6-trifluorometil-fenil)amida do ácido 5-metilhexanóico.
Rendimento de: 6%. LC-MS (m/z) 373 (MH+); tR = 3,06, (UV, ELSD) 85%, 99 %.
d 2-Ciclopent-2-enil-N-(2-metil-4-morfolin-4-il-6-trifluorometil-fenil)acetamida.
Rendimento de: 25%. LC-MS (m/z) 369 (MH+); tR = 2,70, (UV, ELSD) 96%, 99 %. !H RMN (500 MHz, DMSO-dó): 1,48 (m, 1H), 2,04 (m, 1H), 2,25 (m, 2H), 2,34 (m, 2H), 3,06 (m, 1 H), 3,16 (t, 4H), 3,74 (t, 4H),
5,71 (m, 1 H), 5,76 (m, 1 H), 7,00 (m, 1 H), 7,13 (m, 1 H), 9,23 (s, 1 H).
le 2-Ciclopentil-N-(2-metil-4-morfolin-4-il-6-trifluorometiLfenil)acetamida.
2-metil-4-morfolin-4-il-6-trifluorometil-fenilamina (0,18 g) e cloreto de ciclopentilacetila (112 mg) foram dissolvidos em acetonitrila (4 ml) e aquecidos a 150° C por 10 minutos em um frasco de processo de microondas lacrado. A mistura de reação foi diluída com acetato de etila (20 ml) e lavada com água (2 x 20 ml) e salmoura (1 x 20 ml). A fase orgânica foi secada em sulfato de magnésio, concentrada no vácuo e purificada por cromatografia por vaporizaçao instantânea para fornecer 132 mg (52%) do composto do título como um sólido branco. LC-MS (m/z) 371 (MH+); tR = 2,87, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-dó): 1,20 (m, 2H), 1,51 (m, 2H), 1,60 (m, 2H), 1,76 (m, 2H), 2,11 (s, 3H), 2,24 (m, 1 H), 2,26 (d, 2H), 3,16 (t, 4H), 3,73 (t, 4H), 7,00 (d, 1 H), 7,12 (d, 1 H), 9,16 (s, 1 H).
Os seguintes compostos foram preparados analogamente:
lf (2-metil-4-morfolin-4-il-6-trifluorometil-fenil)-amida do ácido hexanóico. Rendimento de: 64 %. LC-MS (m/z) 359 (MH); tR = 2,86, (UV, ELSD) 95%, 99 %. 'H RMN (500 MHz, DMSO-d6): 0,88 (t, 3H), 1,30 (m, 4H), 1,58 (qui, 2H), 2,11 (s, 3H), 2,26 (t, 2H), 3,16 (t, 4H), 3,73 (t, 4H), 6,99 (d, 1 H), 7,11 (d, 1 H), 9,17 (s, 1 H).
I lg 3,3-Dimetil-N-(2-metil-4-morfolin-4-il-6-trifluorometil-fenil)butiramida,
Rendimento de: 58 %. LC-MS (m/z) 359 (MH+); tR = 2,76, (UV, ELSD) 97 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 1,03 (s, 9H), 2,13 (s, 3H), 2,18 (s, 2H), 3,16 (t, 4H), 3,73 (t, 4H), 7,00 (d, 1 H), 7,12 (d, 1 H), 9,12 (s,lH).
II h 2-(3,4-Difluoro-fenil)-N-(2-metil-4-morfolin-4-il-6-trifluorometil-fenil)acetamida.
Rendimento de: 23 %. LC-MS (m/z) 415 (MH+); tR = 2,77, (UV, ELSD) 97 %, 99 %. *H RMN (500 MHz, DMSO-d6): 2,03 (s, 3H), 3,16 (t, 4H), 3,63 (d, 2H), 3,72 (t, 4H), 7,00 (d, 1 H), 7,11 (d, 1 H), 7,17 (m, 1 H), 7,3 8 (m, 2H), 9,52 (b, 1 H).
li (2-metóxi-6-metil-4-morfolin-4-il-fenil)-amida do ácido hexanóico.
Rendimento de: 87 %. LC-MS (m/z) 321 (MH+); tR = 2,04, (UV, ELSD) 99 %, 99 %. *H RMN (500 MHz, DMSO-d6); 0,88 (t, 3H), 1,31 (m, 4H), 1,57 (qui, 2H), 2,04 (s, 3H), 2,23 (t, 2H), 3,13 (t, 4H), 3,71 (s, 3H), 3,75 (t, 4H), 6,43 (b, 1H), 6,49 (b, 1H), 8,78 (s, 1H).
lj 2-Ciclopentil-N-(2-metóxi-6-metil-4-rnorfolin-4-il-fenil)-acetamida.
Rendimento de: 81 %. LC-MS (m/z) 333 (MH+); tR = 2,06, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 1,21 (m, 2H), 1,51 (m, ZH), 1,60 (m, 2H), 1,75 (m, 2H), 2,05 (s, 3H), 2,21 (m, 1H), 2,23 (d, 2H), 3,17 (m, 4H), 3,71 (s, 3H), 3,77 (m, 4H), 6,48 (b, 1H), 6,55 (b, 1H), 8,80 (s, 1H).
I lk N-(2-metóxi-6-metil-4-morfolin-4-il-fenil)-3,3-dimetil-butiramida.
Rendimento de: 90%. LC-MS (m/z) 321 (MH4-); tR =1,95, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 1,03 (s, 9H), 2,08 (s, 3H), 2,14 (s, 2H), 3,17 (m, 4H), 3,71 (s, 3H), 3,78 (m, 4H), 6,49 (b, 1H), 6,55 (b, 1H), 8,73 (s, 1H).
111 2-(3,4-Difluoro-fenil)-N-(2-metóxi-6-metil-4-morfolin-4-il-fenil)- acetamida.
Rendimento de: 41 %. LC-MS (m/z) 377 (MH4); tR = 2,12, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 1,96 (s, 3H), 3,09 (t, 4H), 3,59 (s, 2H), 3,71 (s, 3H), 3,72 (t, 4H), 6,34 (d, 1 H), 6,42 (d, 1 H), 7,19 (m, 1 H), 7,40 (m, 2H), 9,13 (s, 1 H).
II m 2-Ciclopent-2-enil-N-(2-metóxi-6-metil-4-morfolin-4-il-fenil)- acetamida.
Rendimento de: 27 %. LC-MS (m/z) 331 (MH); tR = 1,91, (UV, ELSD) 96%, 99 %. 'H RMN (500 MHz, DMSO-d6): 1,51 (m, 1H), 2,04 (m, 1H), 2,05 (s, 3H), 2,22 (m, 2H), 2,29 (m, 2H), 3,04 (m, 1H), 3,13 (t, 4H),
3,71 (s, 3H), 3,75 (t, 4H), 5,74 (m, 2H), 6,43 (b, 1 H), 6,49 (b, 1 H), 8,82 (s, 1 H).
ln 2-(3-fluoro-fenil)-N-(2-metóxi-6-metil-4-morfolin-4-il-fenil)acetamida.
Rendimento de: 14 %. LC-MS (m/z) 359 (MH+); tR = 2,02, (UV, ELSD) 99 %, 99 %. *H RMN (500 MHz, DMSO-dó): 1,96 (s, 3H), 3,09 (t, 4H), 3,61 (s, 2H), 3,71 (s, 3H), 3,72 (t, 4H), 6,34 (d, 1H), 6,42 (d, 1H), 7,07 (dt, 1H), 7,19 (m, 2H), 7,36 (m, 1H), 9,13 (s, 1 H).
o 2-Biciclo[2,2,1 ]hept-2-il-N-(2-metóxi-6-metil-4-morfolin-4-il-fenil)acetamida.
Ácido biciclo[2,2,l]hept-2-il-acético (0,17 g) dissolvido em cloreto de oxalila (2 M em diclorometano, 0,7 ml) foi agitado a 25° C por 2 horas sob argônio. Os solventes foram removidos no vácuo e o cloreto ácido resultante foi redissolvido em acetonitrila (4 ml) e 2-metóxi-6-metil-4morfolin-4-il-fenilamina (50 mg) foi adicionado. A mistura de reação foi aquecida a 150° C por 10 minutos em um frasco de processo de microondas lacrado e depois diluída com acetato de etila (20 ml) e lavada com água (2 x 20 ml) e salmoura (1 x 20 ml). A fase orgânica foi secada em sulfato de magnésio, concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 20 mg (25%) do composto do título como um sólido branco. LC-MS (m/z) 359 (MH+); tR = 2,30, (UV, ELSD) 99 %, 99 %. ‘H RMN (500 MHz, DMSO-dõ): 1,14 (m, 4H), 1,43 (m, 4H), 1,86 (m, 1H), 2,03 (s, 3H), 2,06 (m, 2H), 2,19 (m, 2H), 3,11 (t, 4H), 3,70 (s, 3H), 3,74 (t, 4H), 6,3 8 (b, 1 H), 6,45 (b, 1 H), 8,74 (s, 1 H).
Os seguintes compostos foram preparados analogamente:
lp (2-metóxi-6-metil-4-morfolin-4-il-fenil)-amida do ácido 4-metil pentanóico.
Rendimento de: 66%. LC-MS (m/z) 321 (MH); tR = 1,99, (UV, ELSD) 99 %, 99 %. 'H RMN (500 MHz, DMSO-d6): 0,90 (d, 6H), 1,47 (m, 2H), 1,58 (m, 1H), 2,03 (s, 3H), 2,24 (t, 2H), 3,13 (m, 4H), 3,70 (s, 3H), 3,75 (m, 4H), 6,42 (b, 1H), 6,48 (b, 1H), 8,79 (s, 1H).
lq (2-metóxi-6-metil-4-morfolin-4-il-fcnil)-amida do ácido 5-metilhexanóico.
Rendimento de: 54 %. LC-MS (m/z) 335 (MH+); tR = 2,26, (UV, ELSD) 94 %, 99 %. *H RMN (500 MHz, DMSO-dó): 0,88 (d, 6H), 1,22 (m, 4H), 1,57 (m, 3H), 2,03 (s, 3H), 2,21 (t, 2H), 3,09 (t, 4H), 3,70 (s, 3H), 3,73 (t, 4H), 6,35 (d, 1H), 6,41 (d, 1H), 8,74 (s, 1H).
Exemplo 12
12a N-(2-Cloro-6-metil-4-morfolin-4-il-fenil)-2-(3-fluoro-fenil)-acetamida.
N-(4-Amino-2-clorO’6-metil-fenil)-2-(3-fluoro-fenil)acetamida (616 mg), éter bis-(2-cloroetílico) (260 pL) e iodeto de potássio (400 mg) foram misturados em Ν,Ν-dimetilformamida seco (11 ml) e aquecidos a 180° C por 30 minutos em um frasco de processo de microondas lacrado. Bicarbonato de sódio aquoso a 5% (100 ml) foi adicionado e a mistura foi extraída com acetato de etila (3 x 80 ml). As fases orgânicas combinadas foram lavadas com água (2 x 100 ml), salmoura (1 x 100 ml), secadas em sulfato de sódio e concentradas no vácuo. O produto bruto foi purificado por cromatografia por vaporização instantânea para fornecer 236 mg (31 %) do composto do título como um sólido branco. LC-MS (m/z) 363 (MH+); tR = 2,56, (UV, ELSD) 96%, 99 %.'H RMN (500 MHz, DMSO-d6): 2,04 (s, 3H), 3,10 (m, 4H), 3,65 (s, 2H), 3,70 (m, 4H), 6,79 (d, 1H), 6,85 (d, 1H), 7,07 (dt, 1H), 7,19 (m, 2H), 7,37 (m, 1H), 9,52 (s, 1H).
12b N-(2-Cloro-6-metil-4-morfolin-4-il-fenil)-2-ciclopentil-acetamida.
N-(4-amino-2-cloro-6-metil-fenil)-2-ciclopentil-acetamida (830 mg), éter bis-(2-cloroetílico) (3,35 ml) e iodeto de potássio (470 mg) foram misturados em etanol absoluto (33 ml) e aquecidos a 170° C por 1 hora em um frasco de processo de microondas lacrado. A mistura bruta foi concentrada no vácuo e purificada por cromatografia por vaporização instantânea para fornecer 390 mg (41 %) do composto do título como um sólido branco. LC-MS (m/z) 337 (MH+); tR =2,61, (UV, ELSD) 97 %, 99 %. 'H RMN (500 MHz, DMSO-dó): 1,22 (m, 2H), 1,51 (m, 2H), 1,61 (m, 2H), 1,77 (m, 2H), 2,11 (s, 3H), 2,25 (tn, 1H), 2,26 (m, 2H), 3,10 (m, 4H), 3,71 (m, 4H), 6,80 (d, 1 H), 6,84 (d, 1 H), 9,18 (s, 1 H).
Tabela 1, Reagentes usados para a preparação dos compostos nos Exemplos de 1 a 12,
|
Nome |
Fornecedor |
CAS no. |
CaL no. |
|
(±)-2,2 ’-Bis(difenilfosfino)-1,1*binaftila |
Aldrich |
76189-55-4 |
48,108-4 |
|
(2’-diciclo-hexilfosfanil-bifenil-2-il)dimetil-amina |
STREM |
213697-53-1 |
15-1145 |
|
Hidroquinina(antraquinona-1,4diil)diéter |
Aldrich |
- |
45,670-5 |
|
Cloridreto de 2-(2-
clorofenil)tiomorfolina |
Array |
- |
2TMA-S02-1 |
|
Cloreto de 2,3-diidro-l-benzofuran-2carbonila |
Maybridge |
27347-32-6 |
CC23902 |
|
2,4,6-Trifluoronitrobenzeno |
Aldrich |
315-14-0 |
26,180-7 |
|
2,6-Dietilfenilamina |
Aldrich |
579-66-8 |
14,938-1 |
|
2,6-Diisopropil-fenilamina |
Aldrich |
24544-04-5 |
37,473-3 |
|
Cloridreto de 2-[4-
(trifluorometil)fenil]tiomorfolina |
Array |
- |
2TMA-Q07-1 |
|
2-Amino-3-cloro-5-nitro-benzonitrila |
Acros |
20352-84-5 |
34367-0050 |
|
Cloroformiato de 2-clorobenzila |
Aldrich |
39545-31-8 |
49,379-1 |
|
Ácido 2-clorofcniIacético |
Aldrich |
2444-36-2 |
19,063-2 |
|
2-Cloroestircno |
Aldrich |
2039-87-4 |
16,067-9 |
|
Ácido 2-Ciclopcnteno-l- acético |
Aldrich |
13668-61-6 |
Cl 1,285-2 |
|
2-Metóxi-6-metilfenilamina |
Aldrich |
50868-73-0 |
36,009-0 |
|
2-metil-6-(trifluorometil)fenilamina |
ABCR |
- |
F05171PF |
|
Ácido 2-Naftilacctico |
Aldrich |
581-96-4 |
31,791-8 |
|
Cloridreto de 2-fenilmorfolina |
Array |
- |
2FMA-0-1 |
|
Cloridreto de 2-Feniltiomorfolina |
Array |
- |
2TMA-0-1 |
|
Cloridreto de 2-Piridin-3-
iltiomorfolina |
Array |
- |
2TMA-P03-1 |
|
Ácido 3-(3,4-difluorofenil)-propionila |
Aldrich |
161712-75-0 |
45,702-7 |
|
Ácido 3-(3-Clorofenil)propiônico |
ABCR |
21640-48-2 |
TWC2925 |
|
Ácido 3-(Trifluorometil)fenil-acético |
Aldrich |
351-35-9 |
19,335-6 |
|
Ácido 3,4-diclorofenilacético |
Aldrich |
5807-30-7 |
28,000-3 |
|
Ácido 3,4-difluorofenilacético |
ABCR |
658-93-5 |
F02874E |
|
Ácido 3,4-difluorofenilborônico |
Aldrich |
168267-41-2 |
46,508-9 |
|
Ácido3,4-dimetilfenilacético |
Vitas-M |
17283-16-8 |
TBB000367 |
|
Ácido 3,5,5-trimetilexanóico |
Acros |
3302-10-1 |
26944-0250 |
|
Ácido 3-bromofenilacético |
Aldrich |
1878-67-7 |
28,886-1 |
|
Ácido 3-clorofenilacético |
Aldrich |
1878-65-5 |
C6,335-9 |
|
Cloreto de 3-ciclo-hexilpropionila |
Acros |
39098-75-4 |
35071-0250 |
|
Cloreto de 3-ciclo-hexilpropionila |
Acros |
39098-75-4 |
35071-0250 |
|
Cloreto de 3-ciclopentilpropionila |
Aldrich |
104-97-2 |
26,859-3 |
|
Ácido 3-etilpentanóíco |
Narchem |
58888-87-2 |
58888-87-2 |
|
Ácido 3-fluorofenilacctico |
Aldrich |
331-25-9 |
24,804-5 |
|
Ácido 3-metilpentanóico |
Aldrich |
105-43-1 |
22,245-3 |
|
Ácido 3-metilfenilborônico |
Aldrich |
17933-03-8 |
39,361-4 |
|
4-(trifluorometil)estireno |
Aldrich |
402-50-6 |
36,960-8 |
|
4-bromo-2,6-dimetilanilina |
Aldrich |
24596-19-8 |
19,237-6 |
|
Cloreto de 4-clorofenilacetila |
Lancaster |
25026-34-0 |
6317 |
|
Cloreto de 4-fluorofenilacetila |
Aldrich |
459-04-1 |
46,695-6 |
|
Ácido 4-fluorofenilborônico |
Aldrich |
1765-93-1 |
41,755-6 |
|
4-fluoroestireno |
Aldrich |
405-99-2 |
15,579-9 |
|
Ácido 4-metilpentanóico |
Aldrich |
646-07-1 |
27,782-7 |
|
4-nitro-2-(trifluorometil)-fenilarnina |
Aldrich |
121-01-7 |
19,657-6 |
|
4-nitro-2-(metil)-fenilamina |
Aldrich |
99-52-5 |
14,643-9 |
|
Ácido 4-tolilborônico |
Aldrich |
5720-05-8 |
39,362-2 |
|
Ácido 5-hexanóico |
Lancaster |
1577-22-6 |
12863 |
|
Ácido 5-metilexanóico |
Matrix |
628-46-6 |
3527 |
|
Cloroformiato de benzila |
Aldrich |
501-53-1 |
11,993-8 |
|
Ácido biciclo[2,2,1 ]hept-2-il-acético |
Aldrich |
1007-01-8 |
12,726-4 |
|
Éter bis-(2-bromoetílico) |
Aldrich |
5414-19-7 |
38,220-5 |
|
Éter bis-(2-cloroetílico) |
Aldrich |
111-44-4 |
C4,113-4 |
|
Bis-(dibenzilidenoacetona)-paládio |
Acros |
32005-36-0 |
29197-0050 |
|
Bromo |
Aldrich |
7726-95-6 |
20,788-8 |
|
Cloroformiato de butila |
Aldrich |
592-34-7 |
18,446-2 |
|
Cloreto de butirila |
Aldrich |
141-75-3 |
23,634-9 |
|
Éster terc-butílico do ácido carbâmico |
Aldrich |
4248-19-5 |
16,739-8 |
|
Cloreto de cloroacetila |
Aldrich |
79-04-9 |
10,449-3 |
|
Ácido ciclo-hcptilacético |
Lancaster |
4401-20-1 |
15553 |
|
Ácido clicloexil-acético |
Aldrich |
5992-21-7 |
C10,450-7 |
|
Cloreto de ciclopentilacetila |
Lancaster |
1122-99-2 |
14562 |
|
Ácido heptanóico |
Aldrich |
111-14-8 |
14,687-0 |
|
Cloreto de hexanoíla |
Aldrich |
142-61-0 |
29,465-9 |
|
Morfolina |
Aldrich |
110-91-8 |
25,236-0 |
|
Ácido m-toliacético |
Aldrich |
621-36-3 |
T3,809-1 |
|
Hexafluorofosfato de N-
[(Dimetilamino)-1 Η-1,2,3triazolo[4,5-b]piridin-l-ilmetileno]N-metilmetanamínio N-óxido |
Fluka |
148893-10-1 |
11373 |
|
N-bromossuccinimida |
Aldrich |
128-08-5 |
B8,125-5 |
|
Ácido octanóico |
Aldrich |
124-07-2 |
15375-3 |
|
Cloreto de oxalila |
Aldrich |
79-37-8 |
32,042-0 |
|
Acetato de paládio (II) |
Aldrich |
3375-31-3 |
20,586-9 |
|
Cloreto de pentanoíla |
Aldrich |
638-29-9 |
15,714-7 |
|
Cloreto de fenilacetila |
Aldrich |
103-80-0 |
Pl,675-3 |
|
Ácido fenilborônico |
Aldrich |
98-80-6 |
P2,000-9 |
|
Diidrato de osmiato de potássio (VI) |
Aldrich |
10022-66-9 |
20,910-4 |
|
Cloreto de propionila |
Aldrich |
79-03-8 |
P5,155-9 |
|
Cloreto de p-toluenossulfonila |
Aldrich |
98-59-9 |
T3,595-5 |
|
Ácido p-tolilacctila |
Aldrich |
622-47-9 |
T3,810-5 |
|
Ácido piridino-3-borônico |
Asymchem |
1692-25-7 |
111347 |
|
bis(2-mctoxietóxi)alumínio hidrcto de sódio (70% em tolueno) |
Aldrich |
22722-98-1 |
19,619-3 |
|
Ditioneto de sódio |
Aldrich |
7775-14-6 |
15,795-3 |
|
Diidrato de cloreto estanoso (II) |
Aldrich |
10025-69-1 |
20,803-5 |
Hipocloreto de terc-butila VWR 507-40-4 081328
|
Cloreto de terc-butilacetila |
Aldrich |
7065-46-5 |
B8,880-2 |
|
Tiomorfolina |
Aldrich |
123-90-0 |
19,627-4 |
|
Cloreto de tionila |
Aldrich |
7719-09-7 |
23,046-4 |
|
Cloreto de tiofeno-2-acetila |
Aldrich |
39098-97-0 |
19,599-5 |
Testes in vitro e in vivo
Os compostos da invenção foram testados e mostraram efeito em um ou mais dos modelos abaixo:
Efluxo relativo através do canal KCNQ2,
Este exemplifica um protocolo de triagem de KCNQ2 para avaliar os compostos da presente invenção. O ensaio mede o efluxo relativo através do canal KCNQ2, e foi realizado de acordo com um método descrito por Tang et al. (Tang, W. et al., J. Biomol. Screen. 2001, 6, 325-331) para canais de potássio hERG com as modificações descritas abaixo.
Um número adequado de células CHO expressando estavelmente os canais KCNQ2 acionados por voltagem foi plaqueado a uma densidade suficiente para produzir uma camada mono-confluente no dia da experiência. As células foram cultivadas no dia anterior à experiência e carregadas com 1 pCi/ml [S6Rb] durante a noite. No dia da experiência as células foram lavadas com um tampão contendo HBSS. As células foram préincubadas com o medicamento por 30 minutos e o efluxo 86Rb+ foi estimulado por uma concentração submáxima de KCI 15 mM na presença contínua do medicamento por 30 minutos iniciais. Após um período de incubação adequado, o sobrenadante foi removido e contado em um contador de cintilação líquido (Tricarb). As células foram lisadas com NaOH 2 mM e a quantidade de 86Rb+ foi contada. O efluxo relativo foi calculado ((CPMsuper/(CPMSuper + CPMcéluta))Comp/(CPMsuper/(CPMsupcr + CPMcélula)) I5mM)* 100-100
Os compostos da invenção têm um EC50 de menos do que 20000 nM, na maioria dos casos menos do que 2000 nM e em muitos casos menos do que 200 nM. Consequentemente, os compostos da invenção são considerados como sendo úteis no tratamento de doenças associadas com os canais de potássio da família KCNQ.
Registros de clampe de contato eletroíisiológicos em células CHO
As correntes KCNQ2 de voltagem ativada foram registradas a partir das células CHO mamíferas pelo uso de técnicas registros de clampe de contato convencionais na configuração de clampe de contato da célula inteira (Hamill OP et al., Pflügers Arch 1981; 391: 85-100). As células de CHO com expressão estável de canais KCNQ2 ativados por de voltagem foram cultivadas sob condições de cultura de célula normais em incubadoras de CO2 e usadas para registros eletrofisiológicos de 1 a 7 dias após o plaqueamento. Os canais de potássio KCNQ2 foram ativados por etapas de voltagem de até + 80 mV em diferenciais de 5 a 20 mV (ou com um protocolo de elevação) de um potencial que influência a membrana entre - 100 mV e - 40 mV (Tatulian L et al. J Neuroscience 2001; 21 (15): 5535-5545).
Os efeitos eletrofisiológicos induzidos pelos compostos foram avaliados em vários parâmetros da corrente de KCNQ2 de voltagem ativada. Especialmente os efeitos no limiar de ativação para a corrente e na corrente induzida máxima foram estudados.
Alguns dos compostos da invenção foram testados neste teste. Uma mudança para a esquerda do limiar de ativação ou um aumento na corrente de potássio induzida máxima é esperada diminuir a atividade nas redes neuronais e assim toma os compostos úteis em doenças com atividade neuronal aumentada - como a epilepsia.
Registros eletrofisiológicos de canais KCNQ2, KCNQ2/KCNQ3 ou KCNQ5 em oócitos
Os canais KCNQ2, KCNQ2/KCNQ3 ou KCNQ5 ativados por voltagem correntes foram registrados a partir de oócitos de Xenopus injetados com mRNA que codifica os canais de íon KCNQ2, KCNQ2+KCNQ3 ou KCNQ5 (Wang et al., Science 1998, 282, 1890-1893; Lerche et al., J Biol Chem 2000, 275, 22395-400). Os canais de potássio KCNQ2, KCNQ2/KCNQ3 ou KCNQ5 foram ativados pelas etapas de voltagem do potencial que influencia a membrana (entre - 100 mV e - 40 mV) até + 40 mV em incrementos de 5 a 20 mV (ou por um protocolo de elevação). Os efeitos eletrofisiológicos induzidos pelos compostos foram avaliados em vários parâmetros das correntes de KCNQ2, KCNQ2/KCNQ3 ou KCNQ5 ativados por voltagem. Especialmente efeitos no limiar de ativação para a corrente e na corrente induzida máxima foram estudados. Os efeitos hiperpolarizantes de alguns dos compostos também foram testados diretamente no potencial de membrana durante o clampe de corrente.
Eletrochoque máximo
O teste foi conduzido em grupos de camundongos macho usando elétrodos córneos e administrando uma corrente de onda quadrada de 26 mA por 0,4 segundos de modo a induzir uma convulsão caracterizada por uma extensão do membro traseiro tônico (Wlaz et al. Epilepsy Research 1998,30,219-229). Ataques induzidos por Pilocarpina
Os ataques induzidos por pilocarpina são induzidos pela injeção intraperitoneal de pilocarpina 250 mg/kg aos grupos de camundongos machos e observando quanto a atividade de ataque resultante na perda de postura dentro de um período de 30 minutos (Stan et al. Pharmacology Biochemistry and Behavior 1993, 45, 321-325).
Teste de limiar de ataque elétrico
Uma modificação do método up-and-down (Kimball et al. Radiation Research 1957, 1-12) foi usada para determinar o limiar médio para induzir a extensão de membro traseiro tônico em resposta ao eletrochoque córneo em grupos de camundongos machos. O primeiro camundongo de cada grupo recebeu um eletrochoque a 14 mA, (0,4 s, 50 Hz) e foi observado quanto à atividade de ataque. Se um ataque foi observado a corrente foi reduzida em 1 mA para o camundongo seguinte, entretanto, se nenhum ataque foi observado então a corrente foi aumentada em 1 mA. Este procedimento foi repetido para todos os 15 camundongos no grupo de tratamento.
Teste de limiar de ataque químico
A dose limiar de pentilenotetrazol requerido para induzir uma convulsão clônica foi medida pela infusão controlado no tempo de pentilenotetrazol (5 mg / ml a 0,5 ml / minuto) em uma veia lateral da cauda de grupos de camundongos machos (Nutt et al. J Pharmacy and Phaimacology 1986,38,697-698).
Abrasamento de Amígdala
Os ratos passaram por cirurgia para a implantação de elétrodos tri-polares na amígdala dorsolateral. Após a cirurgia os animais foram deixados recuperar antes que os grupos de ratos recebessem doses variáveis de composto de teste ou o veículo de medicamento. Os animais foram estimulados com seu limiar inicial após descarga + 25 μΑ diariamente por 3 a 5 semanas e em cada ocasião a severidade do ataque, duração do ataque, e duração da eletricidade após a descarga foram anotados. (Racine, Electroencephalography and Clinicai Neurophysiology 1972, 32, 281-294). Efeitos colaterais
Os efeitos colaterais ao sistema nervoso central foram medidos medindo-se o tempo em que os camundongos permaneceríam no aparelho rotarod (Capacio et al. Drug and Chemical Toxicology 1992, 15, 177-201); ou medindo-se a sua atividade locomotora pela contagem do número de feixes de infravermelho cruzados em uma gaiola de teste (Watson et al, Neuropharmacology 1997, 36, 1369-1375). As ações hipotérmicas na temperatura corporal de núcleo dos animais do composto foram medidas pela sonda retal ou transmissores de radiotelemetria implantados capazes de medir a temperatura (Keeney et al. Physiology and Behaviour 2001, 74, 177-184). Farmacocinéticas
As propriedades farmacocinéticas do composto foram determinadas por intermédio da dosagem i.v. e p.o. aos ratos Sprague Dawley, e, depois disso, drenando as amostras de sangue em 20 horas, as concentrações plasmáticas foram determinadas com LC/MS/MS.
1/8