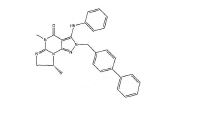
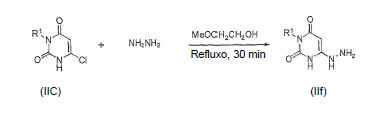Este pedido reivindica prioridade do Pedido de Patente Provisório US N° 60/687,715, depositado em 6 de junho de 2005, cujo conteúdo está incorporado neste por referência.
Campo Técnico
[001] A presente invenção refere-se a novos compostos 7,8-diidro-imidazo [1,2-α] pirazolo[4,3-e]pirimidin-4-ona e compostos 7,8,9-triidro-1H ou 2H]-pirimido[1,2- a]pirazolo[4,3-e]pirimidin-4(5H)-ona, processos para sua produção, sua uso como produtos farmacêuticos e composições farmacêuticas compreendendo os mesmos. São de particular interesse os novos compostos úteis como inibidores de fosfodiesterase 1 (PDE1), por exemplo, no tratamento de doenças envolvendo distúrbios da via intracelular do receptor de dopamina D1, tal como Doença de Parkinson, depressão e dano à função cognitiva, por exemplo, na esquizofrenia.
Antecedentes da Invenção
[002] Onze famílias de fosfodiesterases (PDEs) foram identificadas, mas demonstrou-se que apenas as PDEs na Família I, a fosfodiesterases dependentes de calmodulina-Ca2+ (CaM-PDEs), mediam as vias sinalizadoras de cálcio e nucleotídeo cíclico (por exemplo AMPc e GMPc). Os três genes conhecidos CaM-PDE, PDE1A, PDE1B, e PDE1C, são todos expressos no tecido do sistema nervoso central. PDE1A é expresso através do cérebro com maiores níveis de expressão nas camadas CA1 a CA3 do hipocampo e cerebelo e a um baixo nível no estriato. A PDE1A é também expressa no pulmão e coração. A PDE1B é predominantemente expressa no estriato, giro denteado, trato olfativo e cerebelo, e sua expressão está correlacionada com regiões do cérebro tendo altos níveis de inervação dopaminérgica. Apesar da PDE1B ser primariamente expressa no sistema nervoso central, ela pode ser detectada no coração. A PDE1C é primariamente expressa em epitélio olfativo, células granulares cerebelares, e estriato. A PDE1C é também expressa no coração e músculo liso vascular.
[003] As fosfodiesterases de nucleotídeos cíclicos diminuem AMPc e GMPc sinalizadores intracelulares hidrolisando estes nucleotídeos cíclicos a seus respectivos 5'-monofosfatos (5'AMP e 5'GMP) inativos. As CaM-PDEs desempenham um papel crítico em mediação da transdução do sinal em células cerebrais, particularmente em uma área do cérebro conhecida como o gânglio basal ou estriato. Por exemplo, a ativação do receptor de glutamato do tipo NMDA e/ou a ativação do receptor de dopamina D2 resulta em maiores concentrações intracelulares de cálcio, levando à ativação de efetores tais como quinase II dependente de calmodulina (CaMKII) e calcineurina e à ativação de CaM-PDEs, resultante em AMPc e GMPc reduzidos. a ativação do receptor de dopamina D1, por outro lado, leva à ativação de nucleotídeo ciclases dependentes de cálcio, resultante em maiores AMPc e GMPc. Estes nucleotídeos cíclicos por sua vez ativam a proteína quinase A (PKA; proteína quinase dependente de AMPc) e/ou a proteína quinase G (PKG; proteína quinase dependente de GMPc) que fosforila a transdução de sinais no sentido abaixo via elementos tais como DARPP-32 (dopamina e fosfoproteína regulada por AMPc) e proteína de ligação de elemento responsivo a AMPc (CREB).
[004] CaM-PDEs pode desse modo afetar a via sinalizadora intracelular regulada por dopamina e outras no gânglio basal (estriato), incluindo, mas não limitado a, as vias sinalizadoras intracelulares de óxido nítrico, noradrenérgico, neurotensina, CCK, VIP, serotonina, glutamato (por exemplo, receptor NMDA, receptor AMPA), GABA, acetilcolina, adenosina (por exemplo, receptor A2A), canabinóide receptor, peptídeo natriurético (por exemplo, ANP, BNP, CNP) e endorfina.
[005] A atividade da fosfodiesterase (PDE), em particular, a atividade da fosfodiesterase 1 (PDE1), atua no tecido cerebral como um regulador da atividade locomotora e cognição e memória. A PDE1 é um alvo terapêutico para a regulação de vias sinalizadoras intracelulares, preferen-cialmente no sistema nervoso, incluindo, mas não limitado a, as vias sinalizadoras intracelulares de receptor de dopamina D1, receptor de dopamina D2, óxido nítrico, noradrenérgico, neurotensina, CCK, VIP, serotonina, glutamato (por exemplo, NMDA receptor, AMPA receptor), GABA, acetilcolina, adenosina (por exemplo, Receptor A2A), receptor canabinóide, peptídeo natriurético (por exemplo, ANP, BNP, CNP) ou endorfina. Por exemplo, a inibição de PDE1B deveria atuar para poten-cializar o efeito do agonista de dopamina D1 por proteger GMPc e AMPc de degradação, e deveria similarmente inibir as vias sinalizadoras do receptor de dopamina D2, por inibir a atividade PDE1. a elevação crônica no cálcio intracelular é associada a morte celular em diversos distúrbios, particu-larmente em doenças neurodegenerativas tal como doença de Alzheimer, Parkinson e Huntington, e em distúrbios do sistema circulatório que levam ao AVC e enfarto do miocárdio. Os inibidores PDE1 são desse modo potencialmente úteis em doenças caracterizadas por reduzida atividade sinalizadora de receptor de dopamina D1, tal como Doença de Parkinson, síndrome da perna cansada, depressão, e deficiência cognitiva.
[006] Há assim uma necessidade para compostos que seletivamente inibam a atividade de PDE1, especialmente a atividade de PDE1B.
[007] Resumo da Invenção
[008] A invenção fornece novas, opcionalmente substituídas, 7,8-diidro-[1H ou 2H]-imidazo [1,2-α] pirazol [4,3-e] pirimidin-4 (5H)-ona ou 7,8,9-triidro-[1H ou 2H]- pirimido [1,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona, substituídas na posição 1 ou 2 com C2-9 alquila ou Cicloalquila C3-9, ou opcionalmente substituídos heteroarilalquila ou substituídos arilalquila, na forma livre, de sal ou pró-fármaco (daqui por diante "Compostos da Invenção"). A posição substituinte 1- ou 2- é preferencialmente benzila ou piridilmetila substituída, por exemplo, para-substituídos em relação ao ponto de ligação, por exemplo, com arila, por exemplo, fenila, ou heteroarila, por exemplo, piridila ou tiadiazolila. Descobriu-se surpreendentemente que estes compostos inibem seletivamente a atividade de fosfodiesterase 1 (PDE1), especialmente atividade de PDE1B.
[009] Preferencialmente, os compostos da invenção são 7,8-diidro-[1H ou 2H]- imidazo[1,2-α]pirazol[4,3-e]pirimidin-4(5H)-onas ou 7,8,9-triidro-[1H ou 2H]-pirimido [1,2-α]pirazol[4,3-e]pirimidin-4(5H)-onas, de fórmula I
onde (i) R1 é H ou alquila C1-4 [por exemplo, metila]; (ii) R4 é H ou alquila C1-4 e R2 e R3 são, independentemente, H ou alquila C1-4, arila, heteroarila, heteroarilalcóxi, arilalcóxi, heteroarilalquila, ou arilalquila; ou R2 é H e R3 e R4 juntos formam uma ponte di-, tri-, ou tetra-metileno; (iii) R5 é uma heteroarilalquila substituída, por exemplo, substituída com haloalquila ou R5 é ligado a um dos átomos de nitrogênio na porção pirazol da fórmula A e é uma fração da fórmula A

onde X, I e Z são, independentemente, N ou C; R8, R9, R11 e R12 são independentemente H ou halogênio; e R10 é halogênio, alquila, cicloalquila haloalquila, arila, heteroarila, ou tiadiazolila, diazolila, triazolila, tetra-zolila, arilcarbonila, alquilsulfonila, heteroarilcarbonila, ou alcoxicarbonila; desde que, quando X, I ou Z é nitrogênio, R8, R9, ou R10, respectivamente, é ausente; (iv) R6 é H, alquila, arila, heteroarila, arilalquila, arilamina, heterarilamina, N,N- dialquilamina, N,N-diarilamina, ou N-aril-N-(arilalquila)amina; e (v) n = 0 ou 1; (vi) quando n = 1, A é -C(R13R14)- onde R13 e R14, são, independentemente, H ou alquila C1-4, arila, heteroarila, heteroarilalcóxi, arilalcóxi, hetero-arilalquila ou arilalquila; na forma livre, de sal ou pró- fármaco, incluindo seus enantiômeros, diasteroisômeros e racematos.
[0010] A invenção ainda fornece os compostos de Fórmula I como segue:
[0011] 1.1 Fórmula I onde R1 é metila e n = 0;
[0012] 1.2 Fórmula I ou 1.1 onde R4 é H ou alquila C1-4 e pelo menos um de R2 e R3 é alquila menor, tal que quando o carbono carregando R3 for quiral, ele terá uma configuração R, por exemplo, onde ambos R2 e R3 são metila, ou onde um é hidrogênio e o outro é isopropila;
[0013] 1.3 Fórmula I ou 1.1 onde R4 é H e pelo menos um de R2 e R3 é arilalcóxi;
[0014] 1.4 Fórmula I onde R1 é metila, R2, R3, e R4 são H, n = l, e R13 e R14 são, independentemente, H ou alquila C1-4 (por exemplo, metila ou isopropila);
[0015] 1.5 Fórmula I ou 1.1 onde R2 é H e R3 e R4 juntos formam uma ponte tri- ou tetrametileno, tendo a configuração cis, preferencialmente onde os carbonos carregando R3 e R4 têm a configurações R e S respectivamente;
[0016] 1.6 Fórmula I, 1.1 ou 1.5 onde R5 é uma heteroarilmetila substituída, por exemplo, para-substituídos com haloalquila;
[0017] 1.7 Fórmula I, 1.1. 1.2, 1.3, 1.4 ou 1.5 onde R5 é uma fração de Fórmula A onde R8, R9, R11, e R10 são H e R10 é fenila;
[0018] 1.8 Fórmula I, 1.1. 1.2, 1.3, 1.4 ou 1.5 onde R5 é uma fração de Fórmula A onde R8, R9, R11, e R12 são H e R10 é piridila ou tiadiazolila;
[0019] 1.9 Fórmula I, 1.1. 1.2, 1.3, 1.4 ou 1.5 onde R5 é uma fração de Fórmula A onde R8, R9, R11, e R12 são, independentemente, H ou halogênio, e R10 é haloalquila;
[0020] 1.10 Fórmula 1. 1.1. 1.2, 1.3, 1.4 ou 1.5 onde R5 é uma fração de Fórmula A onde R8, R9, R11, e R12 são, independentemente, H, e R10 é alquil sulfonila;
[0021] 1.11 qualquer uma das fórmulas precedentes onde R5 é ligado ao 2- posição nitrogênio em um anel pirazol;
[0022] 1.12 qualquer uma das fórmulas precedentes onde R6 é benzila;
[0023] 1.13 qualquer uma das fórmulas precedentes onde R6 é fenilamina ou fenilalquilamina (por exemplo, benzilamina);
[0024] 1.14 qualquer uma das fórmulas precedentes onde R6 é fenilamina;
[0025] 1.15 qualquer uma das fórmulas precedentes onde X, I, e Z são todos C;
[0026] 1.16 qualquer uma das fórmulas precedentes onde X, I, e Z são todos C e R10 é fenila ou 2-piridila; e/ou
[0027] 1.17 qualquer uma das fórmulas precedentes onde os compostos inibem hidrólise de GMPc mediada por fosfodiesterase (por exemplo, mediada por PDE1, especialmente mediada por PDE1B), por exemplo, com um IC50 de menos de 1 μM, preferencialmente menos de 25 nM em um ensaio de PDE reagente de uma partícula de afinidade a metal imobilizado, por exemplo, como descrito no Exemplo 24; em forma livre ou de sal.
[0028] Por exemplo, os compostos da invenção incluem 7,8-diidro-[1H ou 2H]- imidazo [1,2-α] pirazol[4,3-e]pirimidin-4-(5H)-onas de Fórmula Ia
onde (i) R1 é H ou alquila C1-4 [por exemplo, metila]; (ii) R4 é H e R2 e R3 são, independentemente, H ou alquila C1-4 [por exemplo, R2 e R3 são ambos metila, ou R2 é H e R3 é isopropila], arila, ou arilalquila; ou R2 é H e R3 e R4 juntos formam uma ponte di-, tri- ou tetrametileno [preferencialmente onde R3 e R4 têm a configuração cis, por exemplo, onde os carbonos carregando R3 e R4 têm as configurações R e S respectivamente]; (iii) R5 é ligado a um dos nitrogênios na porção pirazol de fórmula I e é uma benzila substituída de fórmula B
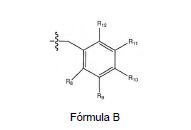
onde R8, R9, R11 e R12 são independentemente H ou halogênio (por exemplo, C1 ou F); e R10 é halogênio, alquila, cicloalquila, haloalquila (por exemplo, trifluormetila) arila (por exemplo, fenila), heteroarila (por exemplo, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1,2,3-tiadiazo1-4-ila), arilcarbonila (por exemplo, benzoíla), alquil sulfonila ou heteroarilcarbonila; e (iv) R6 é H, alquila, arila, heteroarila, arilalquila [por exemplo, benzila], arilamina [por exemplo, fenilamina], heteroarilamina, arilalquilamina, N,N-dialquilamina, N,N- diarilamina, ou N-arila-N-(arilalquil)amina [por exemplo N-fenil-N-(1,1’-bifen-4- ilmetil)amina]; na forma livre, de sal ou pró-fármaco.
[0029] A invenção ainda fornece compostos de Fórmula Ia como segue:
[0030] 1.1: Fórmula Ia onde R1 é metila;
[0031] 1.2: Fórmula Ia ou 1.1 onde R4 é H e pelo menos um de R2 e R3 é alquila menor, tal que quando o carbono carregando R3 seja quiral, tenha uma configuração R, por exemplo, onde ambos R2 e R3 são metila, ou onde um é hidrogênio e o outro isopropila;
[0032] 1.3: Fórmula Ia ou 1.1 onde R2 é H e R3 e R4 juntos formam uma ponte tri- ou tetrametileno, tendo a configuração cis, preferencialmente onde os carbonos carregando R3 e R4 têm as configurações R e S respectivamente;
[0033] 1.4: Fórmula Ia, 1.1. 1.2 ou 1.3 onde R5 é uma fração de fórmula A onde R8, R9, R11, e R12 são H e R10 é fenila;
[0034] 1.5: Fórmula Ia, 1.1. 1.2, ou 1.3 onde R5 é uma fração de fórmula A onde R8, R9, R11 e R12 são H e R10 é piridila ou tiadiazolila;
[0035] 1.6: Fórmula Ia, 1.1. 1.2, 1.3, 1.4, ou 1.5 onde R5 é ligado ao nitrogênio de posição 2 em um anel pirazol;
[0036] 1.7: Fórmula Ia, 1.1. 1.2, 1.3, 1.4, 1.5 ou 1.6 onde R6 é benzila;
[0037] 1.8: Fórmula Ia, 1.1. 1.2, 1.3, 1.4, 1.5 ou 1.6 onde R6 é fenilamina ou fenilalquilamina (por exemplo, benzilamina); e/ou
[0038] 1.9: Fórmula Ia, 1.1. 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, ou 1.8 onde um compostos inibem a hidrólise de GMPc mediada por fosfodiesterase (por exemplo, mediada por PDE1. especialmente mediada por PDE1B), por exemplo, com um IC50 de menos de 1 μM, preferencialmente menos de 25 nM em um ensaio de PDE reagente de uma partícula de afinidade a metal imobilizado, por exemplo, como descrito no Exemplo 15; em forma livre ou de sal.
[0039] Em uma outra modalidade, os compostos da invenção são compostos de Fórmula I onde (i) R1 é metila; (ii) R2, R3 e R4 são H; (iii) n = 1 e Ra e Rb são, independentemente, H ou metila; (iv) R5 é uma fração de Fórmula A onde R8, R9, R11 e R12 são H e R10 é fenila, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1,2,3-tiadiazo1-4-ila); (v) R6 é benzila, fenilamina ou benzilamina; em forma livre ou de sal.
[0040] Em outra modalidade, os compostos da invenção são compostos de Fórmula I onde (i) R1 é metila; (ii) n = 0; (iii) R2 é H e R3 e R4 juntos formam uma ponte tri- ou tetrametileno [preferencialmente com os carbonos carregando R3 e R4 tendo a configuração R e S respectivamente]; ou pelo menos um de R2 e R3 é metila, isopropila ou arilalcóxi e R4 é H; ou R2 e R3 são H e R4 é a alquila C1-4; (iv) R5 é uma heteroarilmetila substituída, por exemplo, para-substituída com haloalquila; ou R5 é uma fração de Fórmula A onde R8, R9, R11 e R12 são H ou halogênio e R10 é haloalquila, fenila, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1,2,3-tiadiazo1-4-ila); e (v) R6 é benzila, fenilamina ou benzilamina; em forma livre ou de sal.
[0041] Em outra modalidade, os compostos da invenção são compostos de Fórmula Ia onde (i) R1 é metila; (ii) R2 é H e R3 e R4 juntos formam uma ponte tri- ou tetrametileno [preferencialmente com os carbonos carregando R3 e R4 tendo a configuração R e S respectivamente]; ou R2 e R3 são cada um metila e R4 é H; ou R2 e R4 são H e R3 é isopropila [preferencialmente o carbono carregando R3 tendo uma configuração R]; (iii) R5 é uma fração de Fórmula A onde R8, R9, R11, e R12 são H e R10 é haloalquila, fenila, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1,2,3- tiadiazo1-4-ila); e (iv) R6 é benzila, fenilamina ou benzilamina; em forma livre ou de sal.
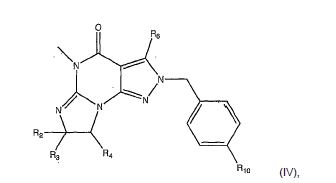
[0042] Por exemplo, os compostos da invenção incluem compostos de acordo com as fórmulas II, III e IV.
onde Ra e Rb são, independentemente, H ou alquila C1-4; R6 é fenilamina ou benzilamina; R10 é fenila, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1.2.3- tiadiazo1-4-ila); em forma livre ou de sal.
onde R2 é H e R3 e R4 juntos formam uma ponte tri- ou tetrametileno [preferencialmente com os carbonos carregando R3 e R4 tendo a configuração R e S respectivamente]; ou pelo menos um de R2 e R3 é metila, isopropila ou arilalcóxi e R4 é H; ou R2 e R3 são H e R4 é a alquila C1-4; R6 é fenilamina ou benzilamina; R10 é haloalquila, fenila, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1,2,3-tiadiazo1-4-ila); em forma livre ou de sal.
onde R2 é H e R3 e R4 juntos formam uma ponte tri- ou tetrametileno [preferencialmente com os carbonos carregando R3 e R4 tendo a configuração R e S respectivamente]; ou pelo menos um de R2 e R3 é metila, isopropila ou arilalcóxi e R4 é H; ou R2 e R3 são H e R4 é a alquila C1-4; R6 é fenilamina ou benzilamina; R10 é fenila, piridila (por exemplo pirid-2-ila), ou tiadiazolila (por exemplo, 1,2,3-tiadiazo1-4-ila); em forma livre ou de sal.
[0043] Os compostos da invenção incluem, por exemplo, os compostos título dos Exemplos 1-23 abaixo.
[0044] Se não for especificado de outro modo ou claro no contexto, os seguintes termos como usado neste têm os seguintes sentidos: a. "Alquila" como usado neste é uma fração de hidrocarboneto saturado ou insaturado, preferencialmente saturado, preferencialmente com um a quatro átomos de carbono de comprimento, que pode ser linear ou ramificado, e pode ser opcionalmente substituído, por exemplo, mono-, di-, ou tri-substituído, por exemplo, com halogênio (por exemplo, cloro ou flúor), hidroxila, ou carboxila. b. "Cicloalquila" como usado neste é uma fração de hidrocarboneto não- aromático saturado ou insaturado, prefe-rencialmente saturado, preferencialmente compreendendo de três a nove átomos de carbono, pelo menos alguns dos quais formam uma estrutura não-aromática mono- ou bicíclica, ou cíclica em ponte, e que pode ser opcionalmente substituída, por exemplo, com halogênio (por exemplo, cloro ou flúor), hidroxila, ou carboxila. c. "Arila" como usado neste é hidrocarboneto aromático mono ou biciclico, preferencialmente fenila, opcionalmente substituído, por exemplo, com alquila (por exemplo, metila), halogênio (por exemplo, cloro ou flúor), haloalquila (por exemplo, trifluormetila), hidroxila, carbo-xila, ou uma arila ou heteroarila adicional (por exemplo, bifenila ou piridilfenila). d. "Heteroarila" como usado neste é uma fração aromática onde um ou mais de um átomo que formam o anel aromático é enxofre ou nitrogênio em detrimento ao carbono, por exemplo, piridila ou tiadiazolila, que pode ser opcio-nalmente substituída, por exemplo, com alquila, halogênio, haloalquila, hidroxila ou carboxila.
[0045] Os compostos da invenção pode existir em forma livre ou de sal, por exemplo, como sais de adição ácida. Nesta especificação, a menos que indicado de outro modo, expressões tais como Os Compostos da Invenção devem ser entendidas como englobando os compostos em qualquer forma, por exemplo, forma livre ou de sal de adição ácida, ou onde os compostos contêm substituintes ácidos, na forma de sais de adição básica. Os compostos da invenção são pretendidos para uso como produtos farmacêuticos, desse modo sais farmaceuticamente aceitáveis são preferidos. Os sais que são impróprios para usos farmacêuticos podem ser úteis, por exemplo, para o isolamento ou purificação dos compostos livres da invenção ou seus sais farmaceuticamente aceitáveis, e são desse modo também incluídos.
[0046] Os compostos da invenção pode em alguns casos também existir na forma de pró-fármaco. Por exemplo quando os compostos contêm substituintes hidroxila ou carboxila, estes substituintes podem formar ésteres fisiologicamente hidrolisáveis e aceitáveis. Como usado neste, "éster fisiologicamente hidrolisáveis e aceitáveis " significa ésteres dos compostos da invenção que são hidrolisáveis sob condições fisiológicas para render ácidos (no caso dos compostos da invenção que têm substituintes hidroxila) ou álcoois (no caso dos compostos da invenção que têm substituintes carboxila) que são eles próprios fisiologi-camente toleráveis nas doses a serem administradas. Como será apreciado o termo assim engloba formas farmacêuticas convencionais de pró-fármacos.
[0047] A invenção também fornece métodos de produzir os Compostos da Invenção, novos intermediários úteis para produzir os compostos da invenção, e métodos de usar os compostos da invenção para o tratamento de doenças e distúrbios como determinados abaixo (especialmente tratamento de doenças caracterizadas por atividade sinalizadora reduzida do receptor de dopamina D1, tal como Doença de Parkinson, síndrome da perna cansada, depressão, e a deficiência cognitiva da esquizofrenia).
Descrição Detalhada da Invenção
[0048] Métodos de Produção dos Compostos da Invenção
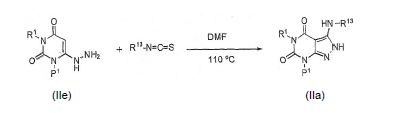
[0049] Os compostos da fórmula I e seus sais farmaceuticamente aceitáveis podem ser produzidos usando os métodos como descrito e exemplificados neste e por métodos similares aos mesmos e por métodos conhecidos na química técnica. Tais métodos incluem, mas não são limitados a, aqueles descrito abaixo. Se não comercialmente disponíveis, os materiais de partida para estes processos podem ser produzidos por procedimentos, que são selecionadas da técnica química usando técnicas que são similares ou análogas à síntese de compostos conhecidos. Todas as referências citadas neste são aqui incorporados em sua totalidade por referência.
[0050] Alguns compostos individuais no âmbito desta invenção podem conter ligações duplas. As representações de ligações duplas nesta invenção devem incluir tanto os isômeros E como Z da ligação dupla. Além disso, alguns compostos no âmbito desta invenção podem conter um ou mais centros assimétricos. Esta invenção inclui o uso de qualquer um dos estereoisômeros oticamente puros assim como qualquer uma combinação de estereoisômeros.
[0051] Os pontos de fusão não são corrigidos e (dec) indica decomposição. As temperaturas são dadas em graus Celsius (°C); a menos que especificado de outro modo, as operações são conduzidas à temperatura ambiente, ou seja, a temperatura em uma faixa de 18-25 °C. Cromatografia significa cromatografia flash em sílica gel; cromatografia de camada fina (TLC) é conduzida em placas de sílica gel. Os dados de RMN estão em valores delta de prótons de diagnóstico principal, dados em partes por milhão (ppm) em relação a tetrametilsilano (TMS) como um padrão interno. As abreviações convencionais para a forma de sinal são usadas. As constantes de acoplamento (J) são dadas em Hz. Para o espectro de massa (EM), a íon principal de menor massa é relatado para moléculas onde a divisão dos isótopos resulta em picos múltiplos de espectro de massa Composições de mistura de solventes são dados como percentuais de volume ou razões de volume. Nos casos onde os espectros de RMN são complexos, apenas os sinais diagnósticos são relatados.
[0052] Termos e abreviações: ButOH = álcool terc-butílico, CAN = nitrato de amônio cério (IV), DIPEA = diisopropiletilamina, DMF = N,N-dimetilforamida, DMSO = dimetil sulfóxido, Et2O = dietil éter, EtOAc = acetato de etila, equiv. = equivalente(s), h = hora(s), HPLC = cromatografia líquida de alto desempenho, K2CO3 = carbonato de potássio, MeOH = metanol, NaHCO3 = bicarbonato de sódio, NH4OH = amônio hidróxido, PMB = p-metoxibenzila, POCl3 = oxicloreto de fósforo, SOCl2 = cloreto de tionila, TFA = trifluoracético ácido, TF = tetraidrofurano.
[0053] Os métodos sintéticos nesta invenção são ilustrados abaixo. Os significados para os grupos R são como determinados acima para a fórmula I a menos que indicado de outro modo.
[0054] Em um aspecto da invenção, os compostos interme-diários de fórmula IIb podem ser sintetizados reagindo um composto de fórmula IIa com um ácido dicarboxílico, anidrido acético e ácido acético misturando com aquecimento por aproximadamente 3 horas e então resfriado:
onde R1 é H ou alquila C1-4 [por exemplo, metila].
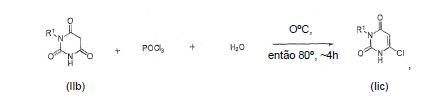
[0055] O intermediário IIc pode ser preparado, por exemplo, reagindo um composto de IIb com, por exemplo, um composto clorado tal como POCl3, algumas vezes com pequenas quantidades de água e aquecido por aproximadamente 4 horas e então resfriado:
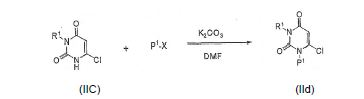
[0056] O intermediário IId pode ser formado reagindo um composto de IIc com, por exemplo, um P1-X em um solvente tal como DMF e a base tal como K2CO3 à temperatura ambiente ou com aquecimento:
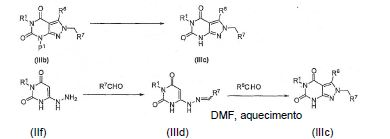
onde P1é um grupo protetor [por exemplo, grupo p-metoxibenzila (PMB)]; X é um grupo de saída tal como o halogênio, mesilato, ou tosilato.
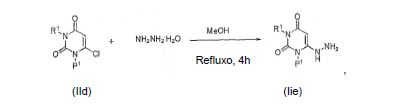
[0057] O intermediário IIe pode ser preparado reagindo um composto IId com hidrazina ou hidrato de hidrazina em um solvente tal como metanol e submetido a refluxo por aproximadamente 4 horas e então resfriado:
[0058] O intermediário IIf pode ser sintetizado reagindo um composto IIc com hidrazina ou hidrato de hidrazina em um solvente tal como metoximetanol e submetido a refluxo por aproximadamente 30 min e então resfriado:
[0059] O intermediário IIg (onde R13 é alquila, arila [por exemplo, fenila], heteroarila, arilalquila, ou heteroari-lalquila), pode ser sintetizado reagindo um composto IIe com, por exemplo, um isotiocianato ou isocianato de arila em um solvente tal como DMF e aquecido a 110 °C por aproximadamente 2 dias e então resfriado:
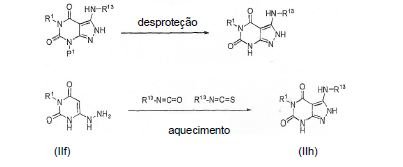
[0060] O intermediário IIh pode ser sintetizado de um composto IIg removendo um grupo protetor P1 com um método apropriado. Por exemplo, se P1 é um grupo p- metoxibenzila, então ele pode ser removido com AlCl3 à temperatura ambiente ou com TFA sob condições de aquecimento. O intermediário IIh pode também ser preparado diretamente de um composto IIf usando métodos similares, mas os rendimentos são relati vamente baixos.
[0061] O intermediário II-I pode ser preparado, por exemplo, reagindo um composto IIh com, por exemplo, um composto clorado tal como POCl3. A reação pode ser conduzida a pressão atmosférica e submetida a refluxo por aproxi-madamente 2 dias, ou aquecida a 150-200 °C por aproxima-damente 10 min em um tubo lacrado com um instrumento de micro-ondas.
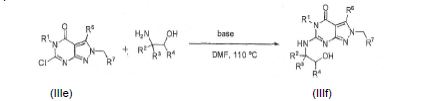
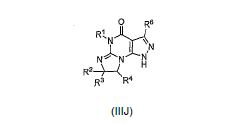
[0062] O intermediário IIJ pode ser preparado reagindo um composto II-I comum amino álcool sob condições básicas em um solvente tal como DMF. A reação podeser aquecida durante a noite e então resfriada:
[0063] A menos que especificado ou definido, R2, R3 e R4 são os mesmos que aqueles definidos anteriormente, por exemplo, em relação à Fórmula 1.
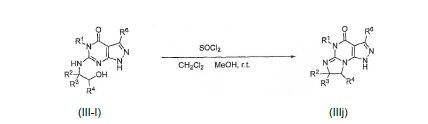
[0064] O intermediário HK pode ser formado reagindo um composto IIJ com, por exemplo, um agente de desidratação tal como SOCl2 em um solvente tal como CH2CI2 à temperatura ambiente durante a noite ou aquecido a 35 °C por aproximadamente 4 horas, e então resfriado:
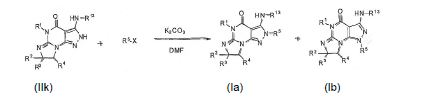
[0065] Composto Ia e Ib pode ser formado reagindo um composto IIk com, por exemplo, a R5-X em um solvente tal como DMF e uma base tal como K2CO3 à temperatura ambiente ou com aquecimento:
onde R5 é como definido anteriormente [por exemplo um grupo benzila opcionalmente substituído]; X é um grupo de saída tal como um halogênio, mesilato, ou tosilato.
[0066] R5 pode também ser introduzido antes, por exemplo, reagindo IIg com R5X e então executando procedimento similar como descrito acima para formar os compostos Ia e Ib, desde que R5 não será clivado em uma etapa de desproteção de P1.
[0067] A segunda rota sintética é designada para a preparação de compostosIa e Ib onde R6 é um grupo alquila, arila ou heteroarila.
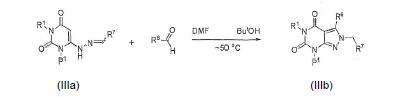
[0068] O intermediário IIIa (onde R7 é arila, preferen-cialmente fenila substituída com R8-12 correspondente à benzila substituída da Fórmula A supra) pode ser formado reagindo um composto de IIe com um aldeído R7CHO em um solvente tal como EtOAc a 0°C ou temperatura ambiente:
[0069] O intermediário IIIb pode ser preparado, por exemplo, reagindo um composto de IIIa com, por exemplo, um aldeído em um solvente tal como DMF e aquecido durante a noite e então resfriado:
[0070] O intermediário IIIc pode ser sintetizado de um composto de IIIb removendo um grupo protetor P1 com um método apropriado. Por exemplo, se P1 é um grupo p-metoxibenzila, então ele pode ser removido com CAN à temperatura ambiente. O intermediário IIIc pode também ser preparado diretamente de um composto IIf usando métodos similares, mas os rendimentos são relativamente baixos.
[0071] O intermediário IIIe pode ser preparado reagindo um composto de IIIc com, por exemplo, um composto clorado tal como POCl3. A reação pode ser conduzida a pressão atmosférica e submetida a refluxo por aproximadamente 2 dias, ou aquecida a 150-200 °C por aproximadamente 10 min em um tubo lacrado com uminstrumento de micro-ondas.
[0072] O intermediário IIIf pode ser formado reagindo um composto de IIIe com um amino álcool sob condições básicas em um solvente tal como DMF eaquecido durante a noite e então resfriado:
[0073] O composto Ia pode ser formado reagindo um composto de IIIf com, por exemplo, um agente de desidratação tal como SOCl2 em um solvente tal como CH2Cl2 à temperatura ambiente durante a noite ou aquecido a 35 °C por aproximadamente 4 horas, e então resfriado:
[0074] Há uma abordagem alternativa para a síntese de composto Ia e Ib onde R6 é um grupo alquila ou arila. Se uma condição adversa é empregada para a desproteção de IIIb, então um grupo R7CH2 pode ser também clivado. Por exemplo, se P1é um grupo p-metoxibenzila e R7 é um grupo fenila substituído, então ambos P1 e R7CH2 pode ser clivado com AlCl3 à temperatura ambiente. Assim, o intermediário Illg pode ser formado com esta abordagem:
[0075] O intermediário lllh pode ser preparado reagindo um composto de lllg com, por exemplo, um composto clorado tal como POCl3. A reação pode ser conduzida a pressão atmosférica e submetida a refluxo por aproximadamente 2 dias, ou aquecida a 150-200 °C por aproximadamente 10 min em um tubo lacrado com um instrumento de micro-ondas.
[0076] O intermediário lll-l pode ser formado reagindo um composto de lllhcom um amino álcool sob condições básicas em um solvente tal como DMF eser aquecida durante a noite e então resfriada:
[0077] O intermediário lllJ pode ser formado reagindo um composto de lll-l com, por exemplo, um agente de desidratação tal como SOCl2 em um solvente tal como CH2Cl2 ou metanol à temperatura ambiente durante a noite ou aquecido a 35 °C por aproximadamente 4 horas, e então resfriado:
[0078] O intermediário IIIJ pode também ser formado reagindo um compostode Ia com, por exemplo, um ácido forte ou ácido de Lewis tal como AICI3:
[0079] O composto Ia e Ib pode ser formado reagindo um composto de IIIJ com, por exemplo, um R5-X em um solvente tal como DMF e uma base tal como K2CO3 à temperatura ambiente ou com aquecimento:
[0080] Uma terceira rota sintética é designada para a preparação do compostoIa e Ib onde R6 é hidrogênio.
[0081] O intermediário IVa pode ser formado, por exemplo, reagindo umcomposto de IIe com POCl3 e DMF:
[0082] O intermediário IVb pode ser formado reagindo um composto de IVacom, por exemplo, um R5-X em um solvente tal como DMF e uma base tal como K2CO3 à temperatura ambiente ou com aquecimento:
[0083] O intermediário IVc pode ser sintetizado de um composto de IVb removendo um grupo protetor P1 com um método apropriado. Por exemplo, se P1 é um grupo PMB, então ele pode ser removido com CAN à temperatura ambiente:
[0084] O intermediário IVd pode ser preparado reagindo um composto de IVc com, por exemplo, um composto clorado tal como POCl3 e submetendo a refluxo por aproximadamente 2 dias, ou aquecendo a 150-200 °C por aproximadamente 10 min em um tubo lacrado com um instrumento de micro-ondas e então resfriado:
[0085] O intermediário IVe pode ser formado reagindo um composto de IVd com um amino álcool sob condições básicas em um solvente tal como DMF e aquecido durante a noite então resfriado:
[0086] O composto Ia pode ser formado reagindo um composto de IVe com, por exemplo, um agente de desidratação tal como SOCl2 em um solvente tal como CH2CI2 à temperatura ambiente durante a noite ou aquecido a 35 °C por aproxi-madamente 4 horas, e então resfriado. Similar aos métodos descrito acima, o grupo R5 em um composto de Ia pode ser clivado usando um método apropriado, e então o intermediário obtido pode reagir com outro R5X para dar composto Ia e Ib.
[0087] A invenção fornece assim métodos de produzir os compostos da invenção como descrito acima, por exemplo, compreendendo (i) reagindo a 7,8-diidro-[1H ou 2H]-imidazo [1,2-α]pirazol[4,3-e]pirimidin- 4(5H)-ona ou 7,8,9-triidro-[1H ou 2H]-pirimido [1,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona com um composto de fórmula X-R5 onde X é um grupo de saída, por exemplo, halogênio, mesilato, ou tosilato, e R5 é alquila C2-9, cicloalquila C3-9, heteroarilalquila, ou arilalquila substituídas, por exemplo, onde R5 é a benzila substituída de fórmula A como definido acima, por exemplo, sob condições básicas, por exemplo, onde a 7,8- diidro-[1H ou 2H]-imidazo [1,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona é um composto de Fórmula IIIJ:
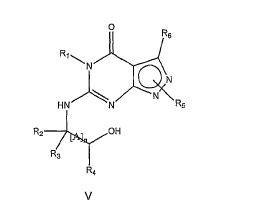
(HU) onde R1-6 são como definido acima, por exemplo, com referência a Fórmula I; e /ou (ii) desidratar um composto de Fórmula V
onde R1-6 e [A]n são como definido acima, por exemplo, com referência a Fórmula I, por exemplo, usando um agente de desidratação, por exemplo, cloreto de tionila; e isolando um composto da invenção assim obtido.
[0088] A os compostos da invenção são úteis em a tratamento de doenças caracterizadas por rompimento de ou dano a vias mediadas por AMPc e GMPc, por exemplo, como resultado de expressão aumentada de PDE1 ou expressão diminuída de AMPc e GMPc devido a inibição ou níveis reduzidos de indutores de síntese de nucleotídeo cíclico, tal como dopamina e óxido nítrico (NO). Evitando a degradação de AMPc e GMPc por PDE1B, aumentando assim os níveis intracelulares de AMPc e GMPc, os compostos da invenção potencializam a atividade de indutores de síntese de nucleotídeo cíclico.
[0089] A invenção fornece métodos de tratamento de qualquer uma ou mais da seguintes condições: (i) Doenças neurodegenerativas, incluindo doença de Parkinson, perna cansada, tremores, discinesias, doença de Huntington, doença de Alzheimer, e distúrbios do movimento induzidos por fármacos; (ii) Distúrbios mentais, incluindo depressão, transtorno do déficit de atenção, transtorno do déficit de atenção e hiperatividade, transtorno bipolar, ansiedade, distúrbios do sono, deficiência cognitiva, demência, síndrome de abstinência de psicoestimulantes, e dependência química; (iii) Distúrbios circulatórios e cardiovasculares, incluindo doença cerebrovascular, AVC, doença cardíaca congestiva, hipertensão, hipertensão pulmonar, e disfunção sexual; (iv) Distúrbios respiratórios e inflamatórios, incluindo asma, doença pulmonar obstrutiva crônica, e rinite alérgica, assim como doenças autoimunes e inflamatórias; e/ou (v) Qualquer uma doença ou condição caracterizadas por baixos níveis de AMPc e/ou GMPc (ou inibição das vias sinalizadoras de AMPc e/ou GMPc) em células expressando PDE1, compreendendo administrar uma quantidade eficaz de um composto da invenção a um paciente humano ou animal em necessidade da mesma.
[0090] A invenção também fornece a método para melhorar ou potencializar a atividade sinalizadora intracelular de dopamina D1 em célula ou tecido compreendendo contatar a referida célula ou tecido com uma quantidade de um composto da invenção suficiente para inibir a atividade de PDE1B.
[0091] A invenção também fornece a método para tratar um distúrbio relacionado a PDE1, especialmente relacionado a PDE1B, ou a via sinalizadora intracelular de receptor de dopamina D1, em um paciente em necessidade do mesmo compreendendo administrar ao paciente uma quantidade eficaz de um composto da invenção que iniba PDE1B, onde a atividade de PDE1B module a fosforilação de DARPP-32 e/ou o receptor AMPA de GluR1.
[0092] A presente invenção também fornece (i) um composto da invenção para uso como um produto farmacêutico, por exemplo, para uso em qualquer método ou no tratamento de qualquer doença ou condição como determinado daqui por diante, (ii) o uso de um composto da invenção na produção de medicamento para tratar qualquer doença ou condição como determinado daqui por diante, e (iii) uma composição farmacêutica compreendendo um composto da invenção em combinação ou associação com um diluente ou veículo farmaceuticamente aceitáveis.
[0093] As palavras "tratamento" e "tratar" devem ser entendidas do mesmo modo como englobando profilaxia e tratamento ou melhora de sintomas de doença assim como o tratamento da causa da doença
[0094] Os compostos da invenção são particularmente úteis para a tratamento de doença de Parkinson.
[0095] Os compostos da invenção podem ser usados como um único agente terapêutico, mas podem também ser usados em combinação ou para coadministração com outros agentes ativos. Por exemplo, como os compostos da invenção potencializam a atividade de agonistas D1, tal como dopamina, eles podem ser administrados simultaneamente, sequencialmente, ou contemporaneamente com medicações dopaminérgicas convencionais, tal como levodopa e adjuntos de levodopa (carbidopa, inibidores COMT, inibidores MAO-B), agonistas dopamina, e anticolinérgicos, por exemplo, no tratamento de paciente tendo doença de Parkinson.
[0096] As dosagens empregadas na prática da presente invenção variarão certamente dependendo, por exemplo, da doença ou condição particular a ser tratada, o composto da invenção particular usado, o modo de administração, e a terapia desejada. os compostos da invenção podem ser administrados por qualquer rota adequada, incluindo oralmente, parenteralmente, transdermicamente, ou por inalação, mas são preferencialmente administradas oralmente. Em geral, indica-se que resultados satisfatórios, por exemplo, para o tratamento de doenças como daqui por diante determinadas são obtidos na administração oral para dosagens na ordem de aproximadamente 0,01 a 2,0 mg/kg. Em mamíferos superiores, por exemplo, seres humanos, uma dosagem indicada diariamente para administração oral estará de acordo em uma faixa de aproximadamente 0,75 a 150 mg, convenientemente administrada uma vez, ou em doses divididas em 2 a 4 vezes, diariamente ou em forma de liberação controlada. Formas de dosagem unitárias para administração oral assim, por exemplo, podem compreender de aproximadamente 0,2 a 75 ou 150 mg, por exemplo, de aproximadamente 0,2 ou 2,0 a 50, 75 ou 100 mg de um composto da invenção, junto com um diluente ou veículo farmaceuticamente aceitável do mesmo.
[0097] As composições farmacêuticas compreendendo os compostos da invenção podem ser preparadas usando diluentes ou veículos convencionais e técnicas conhecidas na técnica galênica. Assim, as formas de dosagem oral podem incluir comprimidos, cápsulas, soluções, suspensões e similares.
Exemplos
[0098] Exemplo 1:
[0099] 2-(Bifeni1-4-ilmetil)-7,8-diidro-5,7,7-trimetil-[2H]-imidazo-[1,2-α]pirazol[4,3-β]pirimidin-4(5H)-ona
[00100] (a) 1-Metilpirimidina-2,4,6(1H,3H,5H)-triona
[00101] A uma solução de ácido malônico (80 g, 0,79 mol) e metiluréia (50 g, 0,68 mol) em 180 mL de ácido acético a 70 °C, anidrido acético (130 mL, 1,37 mol) é adicionada lentamente. Após completar a adição da adição, a mistura reacional é agitada a 90 °C por 3 horas, e então resfriada à temperatura ambiente. O solvente é removido sob pressão reduzida, e o resíduo é tratado com 350 mL de etanol para precipitar um sólido amarelado. Um sólido é recristalizado de etanol para dar 63,1 g de produto como sólidos cristalinos (Rendimento: 65,8%). p.f. = 131,2-133,1 °C [Lit.1: p.f. = 130-131,5 °C].
[00102] (b) 6-Cloro-3-metilpirimidina-2,4(1H,3H)-diona
[00103] Água (2,7 mL) é adicionada por gotejamento à suspensão de 1- metilpirimidina-2,4,6(1H,3H,5H)-triona (14,2 g, 100 mol) em POCl3 (95 mL) a 0 °C. A mistura reacional é então aquecida a 80 °C por 5 horas. A solução marrom resultante é resfriada, e POCl3 é evaporado sob pressão reduzida. O resíduo é tratado com MeOH, e o sólido obtido é recristalizado de etanol para dar 11,5 g de produto (Rendimento: 71,6%). p.f. = 279-282 °C (dec) [Lit.2: 280-282 °C]. 1H RMN (400 MHz, DMSO-d6) (53,10 (S, 3H), 5,90 (S, 1H), 12,4 (br, 1H).
[00104] (c) 6-Cloro-1-(4-metoxibenzil) -3-metilpirimidina-2,4(1H,3H)-diona
[00105] Uma mistura de 6-cloro-3-metilpirimidina-2,4(1H,3H)-diona (16,2 g, 101 mmol), cloreto de p-metoxibenzila (16,5 mL, 122 mmol) e carbonato de potássio (7,0 g, 50,7 mmol) em DMF anidro (200 mL) é aquecida a 60 °C por 3 horas. Mais carbonato de potássio (3,0 g, 21,7 mmol) é adicionado, e a mistura reacional é aquecida a 60 °C por mais 3 horas. Após a filtragem a quente, o filtrado é evaporado à secura sob pressão reduzida. O óleo obtido é diretamente usado para a síntese em uma etapa seguinte. A pequena quantidade de produto é ainda purificada por cromatografia flash em sílica-gel para dar o produto puro como cristais. 1H RMN (400 MHz, MeOH-d4) δ 3,37 (s, 3H), 3,83 (s, 3H), 5,24 (s, 2H), 5,96 (s, 1H), 6,91 e 7,32 (AB, 4H, J = 6,8 Hz). EM (FAB) m/z 281,23 [M+H]+.
[00106] (d) 6-Hidrazinil-1-(4-metoxibenzil)-3-metilpirimidina-2,4(1 H,3H)-diona
[00107] A uma solução de 6-cloro-1- (4-metoxibenzil)-3-metilpirimidina- 2,4(1H,3H)-diona (2,4 g, 8,6 mmol) em EtOH (25 mL) e MeOH (50 mL), hidrazina anidra (1,2 mL) é adicionada lentamente. A mistura reacional é submetida a refluxo por três horas, e então resfriada. Uma grande quantidade de éter é adicionada na mistura reacional, e então filtrada para dar 2,0 g de produto como sólidos cristalinos (Rendimento: 84%). 1H RMN (DMSO-d6) (53,13 (s, 3H), 3,73 (s, 3H), 4,42 (br, 1H), 5,03 (s, 2H), 5,15 (s, 1H), 6,88 e 7,15 (AB, 4H, J = 6,4 Hz), 8,08 (br, 1H). EM (FAB) m/z 277,28 [M+H]+.
[00108] (e) 7-(4-Metoxibenzil)-5-metila-1H-pirazol [3,4-d] pirimidina- 4,6(5H,7H)-diona
[00109] A uma solução de 6-hidrazinil-1-(4-metoxibenzil)-3-metilpirimidina- 2,4(1H,3H)-diona (0,45 g, 1,6 mmol) em DMF (2 mL), POCl3 (0,3 mL, 3,3 mmol) é adicionada por goteja-mento a 0 °C. Após a mistura reacional ser agitada a 0 °C por 1 hora, a mistura é tratada com metanol cuidadosamente para dar um sólido branco. O sólido é ainda purificado por cromatografia para dar 0,4 g do produto (Rendimento: 85%). 1H RMN (DMSO-d6) δ 3,23 (s, 3H), 3,71 (s, 3H), 5,05 (s, 2H), 6,85 e 7,31 (AB, 4H? J = 11,6 Hz), 8,47 (s, 1H), 13,5 (br, 1H). EM (FAB) m/z 287,21 [M+H]+.
[00110] (f) 2-(Bifeni1-4-ilmetil)-7-(4-metoxibenzil)-5-metila-2H-pirazol[3,4- d]pirimidina-4,6(5H,7H)-diona
[00111] Uma mistura de 7- (4- metoxibenzil) -5-metila-1H-pirazol[3,4- d]pirimidina-4,6(5H,7H)-diona (0,312 g, 1,09 mmol), brometo de p-bifenilmetila (0,296 g, 1,20 mmol) e carbonato de potássio (0,151 g, 1,09 mmol) em acetona (20 mL) é agitada à temperatura ambiente durante a noite. O solvente é evaporado sob pressão reduzida. O resíduo é diretamente purificado por cromatografia para dar 0,382 g do produto como um sólido branco (Rendimento: 77,5). 1HRMN (400 MHz, CDCl3) δ 3,37 (s, 3H), 3,75 (s, 3H), 5,15 (s, 2H), 5,34 (s, 2H), 6,81 (m, 2H), 7,27 (m, 3H), 7,47 (m, 4H), 7,60 (m, 4H), 7,87 (s, 1H). EM (FAB) m/z 453,3 [M+H]+.
[00112] (g) 2-(Bifeni1-4-ilmetil)-5-metila-2H-pirazol[3,4-d]pirimidina-4,6(5H,7H)-diona A uma solução de 2-(bifeni1-4-ilmetil)-7-(4-metoxibenzil)-5-metila- 2H-pirazol[3,4-d] piri-midina-4,6(5H,7H)-diona (300 mg, 0,663 mmol) em TF (9 mL), uma solução de nitrato de amônio cério (IV) (1,82 g, 3,32 mmol) em água (3 mL) é adicionado. Uma solução laranja resultante é agitada à temperatura ambiente durante a noite. Outro lote de CAN (1,82 g, 3,32 mmol) é adicionado e uma mistura é agitada por 6 horas, e então um terceiro lote de CAN (1,82 g) é adicionado, e uma mistura é agitada à t.a. durante a noite. A mistura reacional é evaporada à secura. O resíduo é tratado com salmoura, e extraído com cloreto de metileno cinco vezes. Uma fase orgânica é combinada e concentrada. O resíduo é purificado por cromatografia para dar o produto como um sólido branco com alto rendimento. 1H RMN (400 MHz, DMDO- d6) 03,16 (s, 3H), 5,37 (s, 2H), 7,38 (m, 3H), 7,46 (m, 2H), 7,65 (m, 4H), 8,59 (s, 1H), 11,6 (s, 1H). EM (FAB) m/z 333,3 [M+H]+.
[00113] (h) 2-(Bifeni1-4-ilmetil)-6-cloro-5-metila-2H-pirazol [3,4-d]pirimidin- 4(5H)-ona
[00114] 2-(bifeni1-4-ilmetil) -5-metila-2H-pirazol [3,4-d] pirimidina-4,6(5H,7H)- diona (25 mg, 0,075 mmol) é submetida a refluxo em POCl3 (10 mL) por 60 horas, e uma mistura é evaporada à secura. O resíduo é purificado por cromatografia flash em sílica gel para dar 26 mg do produto como um sólido branco (Rendimento: 98,5%). 1H RMN (400 MHz, CDCl3) <53,68 (S5 3H), 5,45 (s, 2H), 7,39 (m, 3H), 7,43 (m, 2H), 7,59 (m, 4H), 8,01 (s, 1H). EM (FAB) m/z 351,2 [M+H]+.
[00115] (i) 2-(Bifeni1-4-ilmetil)-6- (1-hidroxila-2-metil-propan-2-ilamino)-5- metila-2H-pirazol[3,4-d]pirimidin-4(5H)-ona
[00116] Uma solução de 2-(bifeni1-4-ilmetil)-6-cloro-5-metila-2H-pirazol[3,4- d]pirimidin-4(5H)-ona (26 mg, 0,074 mmol) e 2-amino-2-metila-1-propanol (71 μL, 0,74 mmol) em DMF (1 mL) é aquecido a 110 °C durante a noite. A mistura reacional é então purificada por cromatografia para dar 21,1 mg do produto (Rendimento: 71%). EM (FAB) m/z 404,2 [M+H]+.
[00117] (j) 2-(Bifeni1-4-ilmetil)-7,8-diidro-5,7,7-trimetil-[2H]-imidazo-[1,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona
[00118] A uma solução de 2-(bifeni1-4-ilmetil)-6-(1-hidroxila-2-metilpropan-2- ilamino)-5-metila-2H-pirazol[3,4-d]pirimidin-4(5H)-ona (17 mg, 0,042 mmol) em cloreto de metileno (1 mL), é adicionada solução de cloreto de tionila (63 μL, 0,126 mmol) em CH2Cl2 2,0 M sob argônio. A mistura reacional é agitada à t.a. durante a noite. A reação é extinta com 5% NaHCO3, e a mistura resultante é purificada por cromatografia para dar 11 mg do produto final como um sólido branco (Rendimento: 68%).1H RMN (400 MHz, DMSO-d6 + CDCl3) δ 1,36 (s, 6H), 3,30 (s, 3H), 3,69 (s, 2H), 5,30 (s, 2H), 7,36 (m, 3H), 7,43 (m, 2H), 7,58 (m, 4H), 8,10 (s, 1H). EM (FAB) m/z 386,1 [M+H]+.
[00119] Exemplo 2
[00120] Cis-(6aR*,10aS*) -1-(4-Benzoilbenzil)-5,6a,7,8,9,10, 10a-heptaidro-5- metil-3- (fenilamina)ciclo-hex [4,5] imidazo [1,2-α]pirazol[4,3-e]pirimidin-4(1H)-ona
[00121] (a) 7-(4-Metoxibenzil)-5-metila-3-(fenilamina)-1H-pirazol[3,4-d]pirimidina-4,6(5H,7H)-diona
[00122] Isotiocianato de fenila (3,9 mL, 32,7 mmol) é adicionada à suspensão de 6-hidrazini1-1-(4-metoxibenzil)-3-metilpirimidina-2,4(1H,3H)-diona (0,45 g, 1,6 mmol) em DMF (12 mL). A mistura reacional é aquecida a 120 °C por 40 horas, e então evaporada parpara remover o solvente sob pressão reduzida. O resíduo é lavado com hexanos, e então tratado com MeOH (125 mL), e armazenado a -15 °C por 2 dias para dar um sólido cristalino. Um sólido é recristalizado de CH3OH-EtOAc para fornecer 2,5 g do produto (Rendimento: 61%). 1H RMN (400 MHz, DMSO-d6) δ 3,21 (s, 3H), 3,73 (s, 3H), 5,01 (s, 2H), 6,88-7,36 (m, 9H). EM (FAB) m/z 378,3 [M+H]+.
[00123] (b) 5-Metila-3-(fenilamina)-1H-pirazol[3,4-d] piri-midina-4,6(5H,7H)- diona
[00124] AlCl3 (0,733 g, 5,50 mmol) é adicionada a uma solução de 7-(4- metoxibenzil) -5-metila-3- (fenilamina) -1H-pirazol[3,4-d]pirimidina-4,6(5H,7H)-diona (0,692g, 1,83 mmol) e anisol (40 μL, 0,367 mmol) em 1,2-dicloroetano (10 mL) sob argônio. A mistura reacional é agitada à temperatura ambiente por 30 min, e então extinta com água com resfri-amento. A suspensão resultante é filtrada através de uma camada de celite e a celite é lavada com MeOH (20 mL). A produto é eluído da celite com uma grande quantidade de TF. O TF eluente é evaporado para fornecer 0,47 g de produto (Rendimento: 99%). EM (FAB) m/z 258,2 [M+H]+.
[00125] (c) 6-Cloro-5-metila-3-(fenilamina)-1H-pirazol[3,4-d] pirimidin-4(5H)- ona
[00126] 5-metila-3-(fenilamino)-1H-pirazol[3,4-d]pirimidina-4,6(5H,7H)-diona (450 mg, 1,75 mmol) é submetida a refluxo em POCl3 (20 mL) por 60 horas, e a mistura é evaporada à secura. O resíduo é purificado por cromatografia flash em sílica gel para dar 122 mg do produto como um sólido branco e 207 mg material de partida é recuperado (Rendimento: 47%). EM (FAB) m/z 276,1 [M+H]+.
[00127] (d) 6- ((6aR*,10aS*) -2-Hidroxicicloexilamino) -5-metila-3-(fenilamina)-1H-pirazol [3,4-d]pirimidin-4(5H)-ona
[00128] Uma solução de 6-cloro-5-metila-3-(fenilamina)-1H-pirazol[3,4- d]pirimidin-4(5H)-ona (75,8 mg, 0,275 mmol), cloridrato de 6-cloro-2-amino- cicloexanol (83,4 mg, 0,55 mmol) e DIPEA (144 μL, 0,825 mmol) em DMF (3 mL) é aquecido a 110 °C durante a noite. A mistura reacional é evaporada para remover DMF sob pressão reduzida. O resíduo é então purificado por cromatografia para dar 63,1 mg do produto (Rendimento: 64,7%). EM (ESI) m/z 355,0 [M+H]+.
[00129] (e) Cis-(6aR*,10aS*) -5,6a,7,8,9,10,10a-heptaidro-5-metila-3-(fenilamina)cicloex[4,5]imidazo[1,2-α]pirazol[4,3-e] pirimidin-4(1H)-ona
[00130] Solução 2,0 M de cloreto de tionila em CH2Cl2 (267 μL, 0,534 mmol) é adicionada a uma solução de 6-((6aR*,10aS*)-2-hidroxicicloexilamino)-5-metila-3- (fenilamina)-1H-pirazol [3,4-d]pirimidin-4(5H)-ona (63,1 mg, 0,178 mmol) em CH2Cl2 (6 mL) e TF (4 mL). A mistura reacional é agitada à t.a. durante a noite, e então extinta com 100 μL de 28% NH4OH. A mistura resultante é concentrada e purificada por cromato-grafia para dar 25 mg do produto como um sólido branco (Rendimento: 42%). EM (ESI) m/z 337,1 [M+H]+.
[00131] (f) Cis-(6aR*,10aS*) -1-(4-Benzoilbenzila)-5,6a,7, 8,9,10,10a-heptahidro-5-metila-3- (fenilamina) cicloex [4,5] imidazo[1,2-α]pirazol[4,3-e]pirimidin- 4(1H)-ona
[00132] Uma mistura de cis-(6aR*,10aS*)-5,6a,7,8,9,10,10a-heptahidro-5- metila-3-(fenilamina)cicloex[4,5]imidazo[1,2-α] pirazol[4,3-e]pirimidin-4(li<:>f)-ona (7,1 mg, 0,021 mmol), brometo de 4-benzoilbenzila (5,8 mg, 0,021 mmol), e K2CO3 (2,92 mg, 0,021 mmol) em DMF (1 mL) é agitada à temperatura ambiente durante a noite sob argônio. A mistura reacional é purificada por uma HPLC semi-preparativa para dar 3,5 mg do final produto (Rendimento: 31%). EM (ESI) m/z 531,1 [M+H]+.
[00133] Exemplo 3
[00134] 3-Benzila-2-(bifeni1-4-ilmetil)-7,8-diidro-5,7,7-trimetil-[2H]-imidazo- [1,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona
[00135] (a) 6- (2-(Bifeni1-4-ilmetileno) hidrazinil)-1-(4-metoxibenzil)-3-metilpirimidina-2,4(17f,3H)-diona
[00136] Uma solução de 4-fenilbenzaldeído (395 mg, 2,17 mmol) em EtOAc é lentamente adicionada em uma pasta seca resfriada em gelo de 6-hidrazini1-1-(4- metoxibenzil)-3-metilpirimidina-2,4(1 H,3H)-diona (200 mg, 0,724 mmol) em EtOAc. Após a adição, a mistura reacional é agitada à temperatura ambiente por 2 horas. O solvente é evaporado sob pressão reduzida, e o resíduo é triturado com MeOH, seguido por filtragem para dar 256 mg do produto como sólidos amarelo pálido (Rendimento: 80,3%). 1H RMN (400 MHz, DMSO-d6) (53,17 (s, 3H), 3,71 (s, 3H), 5,22 (s, 2H), 5,59 (s, 1H), 6,91 e 7,21 (AB, J = 7,2 Hz, 4H), 7,37-7,81 (m, 9H), 8,36 (s, 1H), 10,67 (s, 1H). EM (FAB) m/z 441,4 [M+H]+.
[00137] (b) 3-Benzila-2- (bifeni1-4-ilmetil) -7-(4-metoxi-benzil) -5-metila-2H- pirazol [3,4-d] pirimidina-4,6 (5H,7H)-diona
[00138] Ácido acético (4,4 mL) é adicionado em a uma solução de 6-(2- (Bifeni1-4-ilmetileno)hidrazinil)-1-(4-meto-xibenzil)-3-metilpirimidina-2,4(1H,3H)-diona (3,2 g, 7,26 mmol) em DMF (50 mL) e Bu1OH (25 mL) a 50 °C. Piperidina (8,7 mL) é misturada com uma solução de 2-fenilacetaldeído (8,5 mL, 72,6 mmol) em DMF (20 mL), e a solução esverdeada resultante é adicionada na solução acima. A mistura reacional é agitada a 40-45 °C por 36 horas sob argônio, e o solvente é evaporado sob alto vácuo. O resíduo é tratado com MeOH (200 mL) para precipitar 1,23 g do produto como sólidos cor de areia (Rendimento: 31,4%). EM (FAB) m/z 543,4 [M+H]+.
[00139] (c) 3-Benzila-2- (bifenil-4-ilmetil) -5-metila-2H-pirazol[3,4-d]pirimidina- 4,6(5H,7H)-diona
[00140] Uma solução de nitrato de amônio cério (IV) (204 mg, 0,371 mmol) em água (0,6 mL) é adicionada a uma solução de 3-benzila-2-(bifeni1-4-ilmetil)-7-(4- metoxibenzil)-5-metila-2H-pirazol[3,4-d]pirimidina-4,6(5H,7H)-diona (40,3 mg, 0,0743 mmol) em TF (2 mL). A solução laranja resultante é agitada à temperatura ambiente durante a noite. Outro lote de CAN (204 mg, 0,371 mmol) é adicionado e a mistura é agitada por 3 horas, e então um terceiro lote de CAN (204 mg) é adicionado, e a mistura é agitada à t.a. durante a noite. A mistura reacional é evaporada à secura. O resíduo é tratado com salmoura, e extraído com cloreto de metileno cinco vezes. A fase orgânica é combinada e concentrada. O resíduo é purificado por cromatografia para dar 11,6 mg do produto como um sólido branco (Rendimento: 36,9%). 1H RMN (400 MHz, acetona-d6) δ 3,27 (s, 3H), 4,51 (s, 2H), 5,33 (s, 2H), 7,13-7,62 (m, 14H), 10,26 (s, 1H). EM (FAB) m/z 423,2 [M+H]+.
[00141] (d) 3-Benzila-2-(bifeni1-4-ilmetil)-6-cloro-5-metila-2H-pirazol[3,4- d]pirimidin-4(5H)-ona
[00142] 3-benzila-2-(bifeni1-4-ilmetil)-5-metila-2H-pirazol [3,4-d]pirimidina- 4,6(5H,7F)-diona (10 mg, 0,024 mmol) é submetida a refluxo em POCl3 (10 mL) por 4 dias, e então a mistura é evaporada à secura. O resíduo é purificado por cromatografia flash em sílica gel para dar 10,4 mg do produto como um sólido branco (Rendimento: 100%). EM (FAB) m/z 441,2 [M+H]+.
[00143] (e) 3-Benzila-2-(bifeni1-4-ilmetil)-6-(1-hidroxila-2-metilpropan-2-ilimino)- 5-metila-6,7-diidro-2H-pirazol[3,4-d]pirimidin-4(5H)-ona
[00144] Uma solução de 3-Benzila-2-(bifeni1-4-ilmetil)-6-cloro-5-metila-2H- pirazol [3,4-d] pirimidin-4(57i)-ona (9,5 mg, 0,022 mmol) e 2-amino-2-metila-1- propanol (21 μL, 0,22 mmol) em DMF (2 mL) é aquecida a 110 °C durante a noite. A mistura reacional é então purificada por cromatografia para dar 5,5 mg do produto (Rendimento: 52%). EM (FAB) m/z 494,4 [M+H]+.
[00145] (f) 3-Benzila-2-(bifenil-4-ilmetil)-7,8-diidro-5,7,7-trimetil-[2H]-imidazo- [1,,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona
[00146] Uma solução de cloreto de tionila 2,0 M (25 μL, 0,050 mmol) em CH2Cl2 é adicionada em uma solução de 3-benzila-2-(bifeni1-4-ilmetil)-6-(1-hidroxila- 2-metilpropan-2-ilimino)-5-metila-6,7-diidro-2H-pirazol[3,4-d]pirimidin-4(5H)-ona (5,0 mg, 0,010 mmol) em cloreto de metileno (1 mL). A mistura reacional é agitada à t.a. durante a noite, e então extinta com 5% NaHCO3, A mistura resultante é purificada por cromatografia para dar 3,2 mg do produto final (Rendimento: 67%).EM (FAB) m/z 476,4 [MH-H]+.
[00147] Exemplo 4
[00148] 1-(Bifenil-4-ilmetil)-7,8-diidro-5,7,7-trimetil-3-(fenilamina)-[1H]- imidazo-[1,2-α] pirazol [4,3-e]pirimidin-4 (5H)-ona
[00149] O método sintético é análogo ao exemplo 2 onde 2-amino-2-metila-1- propanol é adicionada na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de p-bifenilmetila é adicionado na etapa (f) ao invés de brometo de benzoilbenzila.
[00150] Exemplo 5
[00151] 1-(4-(1,2,3-tiadiazo1-4-il)benzila)-7,8-diidro-5,7,7-trimetil-3-(fenilamina)- [1H]-imidazo-[1,2-α] pirazol [4,3-e] pirimidin-4(5H)-ona
[00152] O método sintético é análogo ao exemplo 2 onde 2-amino-2-metila-1- propanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de 4-(1,2,3-tiadiazo1-4il)benzila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00153] Exemplo 6
[00154] 1-(Bifeni1-4-ilmetil)-3-((bifeni1-4-ilmetil)(fenila) amino)-7,8-diidro-5,7,7- trimetil-[1H]-imidazo-[1,2-α] pirazol [4,3-e]pirimidin-4(5H)-ona
[00155] O método sintético é análogo ao exemplo 2 onde 2-amino-2-metila-1- propanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de p-bifenilmetila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00156] Exemplo 7
[00157] Cis-(6aR*,10aS*) -5,6a,7,8,9,10,10a- heptahidro-5-metil-3-(fenilamina)-1-(4-(piridin-2il)benzila)-cicloex[4,5] imidazo[1,2-α]pirazol[4,3-e]pirimidin- 4-(1H)-ona
[00158] O método sintético é análogo ao exemplo 2 onde 2-(4- (bromometil)fenila)piridina é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00159] Exemplo 8
[00160] Cis-(6aR*,10aS*)-2-(4-(Piridin-2il)benzil)-5,6a,7, 8,9,10,10a-heptahidro -5-metil-3- (fenilamina) cicloex [4,5] imidazo[1,2-fl]pirazol[4,3-e]pirimidin- 4(2H)-ona
[00161] O método sintético é análogo ao exemplo 2 onde brometo de 4-pirid- 2-ilbenzila é adicionado na etapa (f) ao invés de brometo de benzoilbenzila.
[00162] Exemplo 9
[00163] Cis- (6aR*,10aS*) -3- (Benzil) -5,6a,7,8,9,10,10a-heptahidro-5-metila-2-(4-(1,2,3-tiadiazol-4-il)benzil)-cicloex [4,5]imidazo[1,2-α]pirazol[4,3-e]pirimidin-4(2H)- ona
[00164] O método sintético é análogo ao exemplo 3 onde 4-(1,2,3-tiadiazo1- 4il)benzaldeído e DMF são adicionados na etapa (a) ao invés de 4-fenilbenzaldeído e aquecidos durante a noite; e cloridrato de trans-2-amino-cicloexanol é adicionado na etapa (e) ao invés de 2-amino-2-metil-1-propanol.
[00165] Exemplo 10
[00166] Cis-(6aR*,10aS*)-3-(Benzila)-2-(4-Bifeni1-4-ilmetil)-5,6a,7,8,9,10,10a- heptahidro-5-metil-cicloex [4,5] imidazo [1,2-α]pirazol[4,3-e]pirimidin-4(2H)-ona
[00167] O método sintético é análogo ao exemplo 3 onde cloridrato de trans- 2-amino-cicloexanol é adicionado na etapa (e) ao invés de 2-amino-2-metil-1- propanol.
[00168] Exemplo 11
[00169] (R)-3-(benzil)-2-(bifeni1-4-ilmetil)-7,8-diidro-7-isopropila-5-metil-[2H]-imidazo-[1,2-α]pirazol[4,3-e]pirimi-din-4(5H)-ona
[00170] O método sintético é análogo ao exemplo 3 onde (R)-2-amino-3- metilbutan-1-ol é adicionado na etapa (e) ao invés de 2-amino-2-metila-1-propanol.
[00171] Exemplo 12
[00172] (6aR,9aS)-5,6a,7,8,9,9a-hexaidro-5-metila-3-(feni-lamina)-2-(4-Piridin-2il)-benzila) -ciclopent [4,5] imidazo [1,2-fl]pirazol[4,3-e]pirimidin-4(2H)-ona
[00173] O método sintético é análogo ao exemplo 2 onde (1R,2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e 2-(4-(bromometil)fenila)piridina é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00174] Exemplo 13
[00175] (6aR,9aS)-5,6a,7,8,9,9a-hexaidro-5-metila-3-(feni-lamina)-1- (4-Piridin-2il)-benzila) -ciclopent [4,5]imidazo-[1,2-α]pirazol[4,3-e]pirimidin-4(1 H)-ona
[00176] O método sintético é análogo ao exemplo 2 onde (1R,2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-amino- cicloexanol; e brometo de 4-pirid-2-ilbenzil é adicionado na etapa (f) ao invés de brometo de benzoilbenzila.
[00177] Exemplo 14
[00178] (6aR,9aS)-3-(benzilamina) -5,6a,7,8,9,9a-hexaidro-5-metila-2-(4-Piridin-2il)-benzila)-ciclopent [4,5] imidazo [1,2-α]pirazol[4,3-e]pirimidin-4(2H)-ona
[00179] O método sintético é análogo ao exemplo 2 onde isotiocianato de benzila é adicionado na etapa (a) ao invés de isotiocianato de fenila; (1R,2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de 4-pirid-2-ilbenzila é adici-onado na etapa (f) ao invés de brometo de benzoilbenzila.
[00180] Exemplo 15
[00181] (6aR,9aS)-3-(fenilamina)-5,6a,7,8,9,9a-hexaidro-5-metil-2-(bifeni1-4- ilmetil) -ciclopent [4,5] imidazo [1,2-α] pirazol[4,3-e]pirimidin-4(2H)-ona
[00182] O método sintético é análogo ao exemplo 2 onde (1R,2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e 4-(bromometil)bifenila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00183] Exemplo 16
[00184] 2-(Bifeni1-4-ilmetil)-7,8,9-triidro-5,8,8-trimetil-3-(fenilamina)-[2H]-pirimido-[1,2-α] pirazol [4,3-e]pirimidin-4(5H)-ona
[00185] O método sintético é análogo ao exemplo 2 onde 3-amino-2,2-dimetil- 1-propanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e 4-(bromometil)bifenila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00186] Exemplo 17
[00187] (7R)-2-(Bifeni1-4-ilmetil)-7,8-diidro-5,7-dimetil-3-(fenilamino)-[2H]-imidazo-[1,2-α] pirazol[4,3-e]pirimidin-4(5H)-ona
[00188] O método sintético é análogo ao exemplo 2 onde (R)-2-aminoprop-1- ol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino-cicloexanol; e 4- (bromometil) bifenila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00189] Exemplo 18
[00190] (8R)-2-(Bifeni1-4-ilmetil)-7,8-diidro-5,8-dimetil-3-(fenilamina)-[2H]- imidazo-[1,2-α]pirazol[4,3-e]pirimidin-4(5H)-ona
[00191] O método sintético é análogo ao exemplo 2 onde (R)-1-aminopropan- 2-ol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino-cicloexanol; e 4-(bromometil) bifenila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00192] Exemplo 19
[00193] (7R)-2-(Bifenil-4-ilmetil)-7,8-diidro-3-(fenilamina)-5-metila-7-(1-metiletil)- [2H]-imidazo-[1,2-α]pirazol[4,3-e] pirimidin-4(5H)-ona
[00194] O método sintético é análogo ao exemplo 2 onde (R)-2-amino-3- metilbutan-1-ol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e 4-(bromometil)bifenila é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00195] Exemplo 20
[00196] (6aR,9aS)-3-(fenilamina)-5,6a,7,8,9,9a-hexaidro-5-metila-2-(4- (trifluormetil)-benzila)-ciclopent [4,5] imidazo [1,2-α]pirazol[4,3-e]pirimidin-4(2H)-ona
[00197] O método sintético é análogo ao exemplo 2 onde (1R,2R)-2-amino-ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de p-trifluormetilbenzila é adicionado na etapa (f) ao invés de brometo de benzoilbenzila.
[00198] Exemplo 21
[00199] (6aR,9aS)-3-(fenilamina)-5,6a,7,8,9,9a-hexaidro-5-metil-2-((6-trifluormetil)-piridin-3-il) metila) -ciclopent [4,5]imidazo[1,2-α]pirazol[4,3-e]pirimidin- 4(2H)-ona
[00200] O método sintético é análogo ao exemplo 2 onde (1R,2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e 5-(bromometil)-2-(trifluormetil)piridina é adicionada na etapa (f) ao invés de brometo de benzoilbenzila.
[00201] Exemplo 22
[00202] (6aR,9aS)-3-(fenilamina)-5,6a,7,8,9,9a-hexaidro-5-metila-2-(3-flúor- 4-(trifluormetil) -benzil)-ciclopent[4,5] imidazo[1,2-α]pirazol[4,3-e]pirimidin-4(2H)-ona
[00203] O método sintético é análogo ao exemplo 2 onde (1R,2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de 3-flúor-4-trifluormetil-benzila é adicionado na etapa (f) ao invés de brometo de benzoilbenzila.
[00204] Exemplo 23
[00205] (6aR,9aS)-3-(fenilamina)-5,6a,7,8,9,9a-hexaidro-5-metil-2-(4-metilsulfonil-benzil)-ciclopent[4,5]imidazo[1,2-α]pirazol[4,3-e]pirimidin-4(2H)-ona
[00206] O método sintético é análogo ao exemplo 2 onde (1R, 2R)-2-amino- ciclopentanol é adicionado na etapa (d) ao invés de cloridrato de trans-2-amino- cicloexanol; e brometo de p-metilsulfonil-benzila é adicionado na etapa (f) ao invés de brometo de benzoilbenzila.
[00207] Exemplo 24
[00208] Medida de inibição de PDE1B in vitro usando Kit de Ensaio de Fosfodiesterase IMAP
[00209] A fosfodiesterase 1B (PDE1B) é uma enzima fosfo-diesterase dependente de cálcio/calmodulina que converte monofosfato de guanosina cíclico (GMPc) a monofosfato de 5'-guanosina (5'-GMP). PDE1B pode também converter um substrato GMPc modificado, tal como uma molécula fluorescente GMPc-fluoresceína, a correspondente GMP-fluoresceína. A geração de GMP-fluoresceína de GMPc- fluoresceína pode ser quanti-ficada, usando, por exemplo, um reagente de uma partícula de afinidade a metal imobilizado IMAP (Molecular Devices, Sunnyvale, CA).
[00210] Em resumo, o reagente IMAP se liga com altos afinidade a 5'-fosfato livre que é encontrado em GMP-fluoresceína e não em GMPc-fluoresceína. O complexo resul-tante GMP-fluoresceína-IMAP é grande em relação a GMPc- fluoresceína. Pequenos fluoróforos que são ligados em um grande e lentamente caído complexo podem ser distintos de fluoróforos não ligados, porque os fótons emitidos a medida que fluorescem retêm a mesma polaridade como os fótons usados para excitar a fluorescência.
[00211] Em um ensaio de fosfodiesterase, GMPc-fluores-ceína, que não pode ser ligada a IMAP, e desse modo retém pouca polarização de fluorescência, é convertido a GMP-fluoresceína, que, quando ligado a IMAP, rende um grande aumento na polarização de fluorescência (Δmp). A inibição de fosfodiesterase, desse modo, é detectada como uma diminuição em Δmp.
[00212] Ensaio Enzimático
[00213] Materiais: Todas as susbstâncias químicas estão disponíveis de Sigma-Aldrich (St. Louis, MO) exceto os reagentes IMAPs (tampão de reação, tampão de ligação, FL-GMP e IMAP grânulos), que estão disponíveis de Molecular Devices (Sunnivale, CA).
[00214] Ensaio: Fosfodiesterase de cérebro bovino 3',5'-cíclico-nucleotídeo- específica (Sigma, St. Louis, MO) é reconstituída com glicerol 50% a 2,5 U/mL. Uma unidade de enzima irá hidrolisar 1,0 μmol de 3',5'-AMPc a 5'-AMP por min a pH 7,5 a 30°C. Uma parte da enzima é adicionada a 1999 partes de tampão de reação (CaCl2 30 μM, 10 U/mL de calmodulina (Sigma P2277), Tris-HCl 10 mM pH 7,2, MgCl2 10 mM, BSA 0,1%, NaN3 0,05%) para render uma concentração final de 1,25 mU/mL. 99 μl de solução de enzima diluída é adicionada em cada poço em uma placa de poliestireno de 96 poços de fundo redondo a qual 1 μl de composto teste dissolvido em DMSO 100% é adicionado. Os compostos são misturados e pré-incubados com a enzima por 10 min à temperatura ambiente.
[00215] A reação de conversão FL-GMP é iniciada pela combinação de 4 partes de enzima e mistura de inibidor com 1 parte de solução de substrato (0,225 μM) em uma placa de microtítulo de 384 poços. A reação é incubada no escuro à temperatura ambiente por 15 min. A reação é interrompida por adição de 60 μl de reagente de ligação (1:400 diluição de grânulos IMAP em tampão de ligação suplementado com diluição 1:1800 de antiespumante) a cada poço de uma placa de 384 poços. A placa é incubada à temperatura ambiente por 1 hora para deixar que a ligação de IMAP continue até completar-se, e então é colocada em um leitor de microplaca multimodo Envision (PerldnElmer, Shelton, CT) para medir a polarização de fluorescência (Δmp).
[00216] A diminuição na concentração de GMP, medida como Δmp diminuído, é indicativa de inibição de atividade PDE. Valores IC50 são determinados pela medida da atividade enzimática na presença de 8 a 16 concentrações de composto variando de 0,0037 nM a 80.000 nM e então traçando a concentração do fármaco versus Δmp, o que permite que valores IC50 sejam estimados usando software de regressão não linear (XLFit; IDBS, Camponte, MA). Os compostos dos Exemplos 1-14 têm valores IC50 de menos de 1 μM nesse ensaio, geralmente menos de 10 nM.