PARA A PREPARAÇÃO DE (1 R,2R)-3-(3-DIMETILAMINO-1-ETIL-2METIL-RROPILJ-FENOL11.
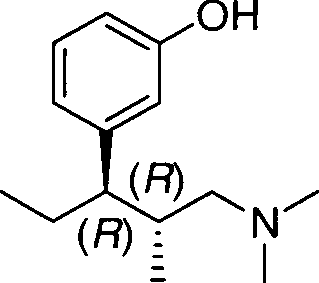
A presente invenção refere-se a um processo para a preparação de (1 R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol.
Uma classe de ingredientes ativos tendo excelente eficácia analgésica e tolerabilidade muito boa são os compostos dimetil-(3-aril-butil)amina substituídos, que são conhecidos, entre outros, a partir de EP 0 693 475. Em particular, (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol tem 10 provado ser um candidato muito promissor no desenvolvimento de um analgésico em exames clínicos.
Um objetivo da presente invenção foi, então, fornecer um processo que permita a preparação de (1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)-fenol através de uma via curta com bom rendimento geral sob condi15 ções ambientalmente aceitáveis.
Em particular, no processo da presente invenção, todos os estereocentros podem ser estabelecidos através de o controle de substrato com formação quase exclusiva de somente um único diastereômero economizando assim etapas de purificação elaboradas para separar os estereoisô20 meros e reagentes, catalisadores ou ligantes quirais dispendiosos. Como não existe nenhum subproduto indesejado formado no processo da presente invenção, cada batelada pode trabalhar em sua capacidade máxima.
O objetivo da presente invenção é alcançado fornecendo um processo para preparar o (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)25 fenol, ou um sal com adição de ácido deste, que compreende as etapas de:
(a) reagir um composto da fórmula geral (I),
O' 1 (I), em que R representa -Ci_6-alquila, -C3-8-cicloalquila, -C1-3 alquileno-fenila, -Ci-3-alquileno-naftila, tetrahidropiranila ou -C(=O)-C-i.6-alquila, com haleto de etilmagnésio em um meio reacional inerte sob condições Grignard.
Preferencialmente, R representa metila, etila, n-propila, isopropila, n-butila, isobutila, tert-butila, n-pentila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, cicloheptila, benzila, fenetila, tetrahidropiranila, -C(=O)-CH3, C(=O)-C2H5, -C(=O)-CH(CH3)2 ou -C(=O)-C(CH3)3 nos compostos da fórmula geral (I). Particularmente preferencialmente, R representa metila, etila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, benzila, fenetila, tetrahidropiranila, -C(=O)-CH3, nos compostos de fórmula geral (I). Mais particularmente, R representa metila, benzila ou tetrahidropiranila nos compostos de fórmula geral (I).
Ainda mais preferencialmente, R na fórmula geral (I) representa metila. Assim, muito preferencialmente, (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona é reagido com o haleto de etilmagnésio em um meio reacional inerte sob condições Grignard.
Preferencialmente, brometo de etilmagnésio ou cloreto de etilmagnésio é usado como haleto de etilmagnésio na etapa (a).
A reação de acordo com a etapa (a) é preferencialmente executada em um meio reacional inerte, preferencialmente em um éter orgânico, por exemplo, selecionado a partir do grupo que consiste em éter dietílico, tetrahidrofurano, 2-metiltetrahidrofurano, tert-butilmetil éter ou qualquer mistura destes. A reação é particularmente preferencialmente executada em tetrahidrofurano com cloreto de etilmagnésio em uma concentração de 0,5 M a 2M do cloreto de etilmagnésio. Particularmente preferencialmente, a reação é executada em uma concentração de 1M ou 2M do cloreto de etilmagnésio.
Outro objetivo da presente invenção é um processo para preparar (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol, ou sal com adição de ácido deste, compreendendo a etapa (a) de reagir um composto de fórmula geral (I),
R
I
em que R representa -Cve-alquila, -C3-8-cicloalquila, -Cra-alquileno-fenila, -Cvs-alquileno-naftila, tetrahidropiranila ou -C(=O)-Ci-e-alquila, com haleto de etilmagnésio em um meio reacional inerte sob condições Grignard, (b) transferir o composto assim obtido de fórmula geral (II),
em que R tem o significado definido acima, para um composto de fórmula geral (III),
onde R tem um significado definido acima, opcionalmente na forma de um sal com adição de ácido, (c) desproteger o composto assim obtido de fórmula geral (III) para obter o (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV),
(d) converter opcionalmente o (1 R,2R)-3-(3-dimetilamino-1-etil-
2-metil-propil)-fenol assim obtido em um sal com adição de ácido.
Preferencialmente R representa metila, etila, n-propila, isopropi la, n-butila, isobutila, tert-butila, n-pentila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, cicloheptila, benzila, fenetila, tetrahidropiranila ou -C(=O)-CH3, C(=O)-C2H5, -C(=O)-CH(CH3)2 ou -C(=O)-C(CH3)3 nos compostos de fórmulas gerais (I), (II) e (III). Particularmente preferencialmente, R representa metila, etila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, benzila, fenetila, tetrahidropiranila, -C(=O)-CH3, nos compostos de fórmulas gerais (I), (II) e (III). Mais particularmente preferencialmente, R representa metila, benzila ou tetrahidropiranila nos compostos de fórmulas gerais (I), (II) e (III).
Ainda mais particularmente preferencialmente R representa metila nos compostos de fórmulas gerais (I), (II) e (III). Assim, (S)-3(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Ia) é transformado em (1 R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol através da seguinte sequência de etapas (esquema 1).
(a)
(IV)
No caso em que R representa metila na fórmula geral (III), o composto (llla) é preferencialmente reagido com ácido bromídrico ou ácido metanossulfônico e metionina ou hidreto de diisobutilalumínio em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, tert-butil-metiléter e misturas destes para produzir (1R,2R)-
3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
No caso em que R representa Ci-6-alquila, exceto metila, na fórmula geral (III), o respectivo composto de fórmula geral (III) é preferencialmente reagido com ácido bromídrico ou hidreto de diisobutilalumínio em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2metiltetrahidrofurano, dioxano, tert-butil-metiléter e misturas destes para produzir (1 R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
No caso em que R representa tetrahidropiranila na fórmula geral (III), o respectivo composto de fórmula geral (III) é preferencialmente reagido com ao menos um ácido inorgânico, preferencialmente com ao menos um ácido inorgânico selecionado a partir do grupo que consiste em ácido clorídrico, ácido bromídrico, ácido sulfúrico e ácido fosfórico, opcionalmente na presença de ao menos um sal, preferencialménte ao menos úm sal selecionado a partir do grupo que consiste em cloreto de amônio e hidrogenossulfato de potássio, em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, tert-butil-metiléter, água e mistura destes para produzir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)fenol de fórmula (IV).
No caso em que R representa C3-8-cicloalquila na fórmula geral (III), o respectivo composto de fórmula geral (III) é preferencialmente reagido com ácido bromídrico ou hidreto de diisobutilalumínio em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, tert-butil-metiléter e misturas destes para produzir (1R,2R)-3-(3dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
No caso em que R representa -Ci-3-alquileno-fenila, OU-C1-3alquileno-naftila, um composto de fórmula geral (III) é reagido com ácido bromídrico ou hidreto de diisobutilalumínio em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, tertbutil-metiléter e mistura destes ou na presença de hidrogênio e ao menos um catalisador, preferencialmente na presença de ao menos um catalisador baseado em paládio ou platina, mais preferencialmente na presença de paládio em carvão, em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, tert-butil-metiléter, e mistura destes para produzir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
No caso em que R representa o -C(=O)-Ci_6-alquila na fórmula geral (III), o respectivo composto de fórmula geral (III) é preferencialmente reagido com ao menos um ácido inorgânico, preferencialmente com ao menos um ácido inorgânico selecionado a partir do grupo que consiste em ácido clorídrico, ácido bromídrico, ácido sulfúrico e ácido fosfórico, ou com ao menos uma base inorgânica, preferencialmente com ao menos uma base inorgânica selecionada a partir do grupo que consiste em hidróxido de sódio, hidróxido de potássio, carbonato de sódio e carbonato de potássio em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2metiltetrahidrofurano, dioxano, tert-butil-metiléter, água e misturas destes para produzir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
Em outra modalidade da presente invenção, o agente para desproteger de acordo com a etapa (c) o processo inventivo é selecionado a partir do grupo que consiste em iodotrimetilsilano, sulfeto de etilsódio, iodeto de lítio e ácido bromídrico, preferencialmente o ácido bromídrico.
O composto (1 R,2R)-3-(3-dimetilamino-1 -etil-2-metil-propil)-fenol pode estar presente na forma de um sal com adição de sal, segundo o qual qualquer ácido adequado capaz de formar tal sal de adição pode ser usado.
A conversão do composto (1 R,2R)-3-(3-dimetilamino-1-etil-2metil-propil)-fenol em um sal de adição correspondente através de a reação com um ácido adequado pode ser efetuada de uma forma bem-conhecida àqueles versados na técnica. Os ácidos adequados incluem, mas não estão limitados a ácido clorídrico, ácido bromídrico, ácido sulfúrico, ácido metanos sulfônico, ácido fórmico, ácido acético, ácido oxálico, ácido succínico, ácido tartárico, ácido mandélico, ácido fumárico, ácido tático, ácido cítrico, ácido glutâmico e ácido aspártico. Em uma modalidade preferencial, o sal com adição de ácido é o sal de cloridrato.
A formação de sal pode preferencialmente ser efetuada em um solvente adequado incluindo éter dietílico, éter diisopropílico, acetatos de alquila, 2-butanona ou qualquer mistura destes. Também preferencialmente, a reação com o trimetilclorossilano em um solvente adequado pode ser usa do para a preparação do sal de adição de cloridrato.
Preferencialmente, um composto de fórmula geral (I) pode ser obtido (a1) reagindo-se um composto de fórmula geral (V),
em que R representa -Ci.6-alquila, -C3.8-cicloalquila, -Ci-3alquileno-fenila, -Ci.3-alquileno-naftila, tetrahidropiranila ou -C(=O)-Ci-6 alquila, com cloridrato de dimetilamina e de paraformaldeído em um meio reacional inerte sob condições Mannich e, (a) resolução subsequente do composto assim obtido de fórmula geral (VI),
R
I
em que R tem o significado definido acima.
Preferencialmente, R representa metila, etila, n-propila, isopropila, n-butila, isobutila, tert-butila, n-pentila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, cicloheptila, benzila, fenetila, tetrahidropiranila, -C(=O)-CH3, C(=O)-C2H5, -C(=O)-CH(CH3)2 ou -C(=O)-C(CH3)3 nos compostos de fórmulas gerais (V) ou (VI). Particularmente preferencialmente, R representa metila, etila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, benzila, fenetila, te trahidropiranila, -C(=O)-CH3, nos compostos de fórmulas gerais (V) ou (VI). Mais particularmente preferencialmente, R representa metila, benzila ou tetrahidropiranila nos compostos de fórmulas gerais (V) ou (VI).
Ainda mais particularmente preferencialmente, R representa metila nas fórmulas gerais (V) e (VI). Assim, 1-(3-metoxifenil)propan-1-ona é convertido em 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Através de) com cloridrato de dimetilamina e paraformaldeído em um meio reacional inerte sob condições Mannich.
Preferencialmente, a resolução na etapa (a) é executada reagindo-se um composto da fórmula geral (VI) com um ácido quiral selecionado a partir do grupo que consiste em ácido L-(-)-dibenzoil tartárico, ácido L(-)-dibenzol tartárico.H2O, e ácido D-(-)-tartárico, a separação subsequente do sal assim obtido e a liberação do composto correspondente de fórmula geral (I) na forma de base livre.
É preferencial que a resolução seja executada em um meio reacional alcoólico selecionado a partir do grupo que consiste em metanol, etanol, 1-propanol, 2-propanol e qualquer mistura destes ou em uma mistura de um meio reacional alcoólico selecionado a partir do grupo que consiste em metanol, etanol, 1-propanol, 2-propanol e acetona.
Preferencialmente, a transferência de acordo coma etapa (b) é executada (b1) submetendo-se o composto de fórmula geral (II) à desidratação e (b) hidrogenação do composto assim obtido de fórmula geral (VII),
R
ã I (VII), em que R representa -Ci-6-alquila, -C3.8-cicloalquila, -Ci.3-alquileno-fenila, -Cvs-alquileno-naftila, tetrahidropiranila ou -C(=O)-Ci-6-alquila, usando um catalisador adequado em um meio reacional inerte na presença de hidrogênio.
Preferencialmente, R representa metila, etila, n-propila, isopropi la, n-butila, isobutila, tert-butila, n-pentila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, cicloheptila, benzila, fenetila, tetrahidropiranila, -C(=O)-CH3, C(=O)-C2H5, -C(=O)-CH(CH3)2 ou -C(=O)-C(CH3)3 nos compostos de fórmula gerais (II). Particularmente preferencialmente, R representa metila, etila, ciclopropila, ciclobutila, ciclopentila, ciclohexila, benzila, fenetila, tetrahidropiranila, -C(=O)-CH3, nos compostos de fórmula geral (II). Mais particularmente preferencialmente, R representa metila, benzila ou tetrahidropiranila nos compostos de fórmula geral (II).
Ainda mais particularmente preferencialmente, R representa metila na fórmula geral (I). Assim, (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2metilpentan-3-ol é transferido para (2S,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina através da desidratação (etapa (b1)) e subsequente hidrogenação (etapa (b)).
Preferencialmente, a hidrogenação da etapa (b) é efetuada através de catálise homogênea na presença de hidrogênio após a etapa de desidratação (b‘). O hidrogênio está preferencialmente na forma gasosa, embora seja possível também que ao menos parte dele possa ser dissolvida em uma fase líquida.
Preferencialmente, o catalisador homogêneo usado para a hidrogenação na etapa (b) de acordo com a presente invenção é um complexo de metais de transição de ródio, irídio ou rutênio, particularmente preferencialmente um complexo de metais de transição de ródio com ligantes de difosfina.
Os ligantes de difosfina que podem preferencialmente ser usados são, por exemplo, conhecidos a partir das seguintes referências de literatura: a) H. Brunner, W. Zettlmeier, Handbook of Enantioselective Catalysis. VHC Weinheim, 1993, vol. 2; b) R. Noyori et a!., Catalytic Asymmetric Synthesis, Segunda edição, (I. Ojima, Ed), Wiley-VCH, Weinheim, 2000; c) Ε. N. Jacobsen, A. Pfaltz, H. Yamamoto (Eds), Comprehensive Asymmetric Catalysis Vol l-lll, Springer Berlin, 1999, e as referências citadas nestes.
Particularmente preferencialmente, o catalisador é escolhido a partir do grupo que consiste em ródio (-)-DIPAMP [(R,R)-(-)-1,2-Bis[(2 metoxi-fenil)(fenil)fosfino]etano], ródio (+)-DIPAMP [(S,S)-(+)-1,2-Bis[(2-metoxifenil) (fenil)fosfino]etano], ródio R-Solphos [R-(+)-N,N'-Dimetil-7,7'bis(difenilfosfino)-3,3',4,4'-tetrahidro-8,8'-bi-2H-1,4-benzoxazina] e ródio SSolphos [S-(-)-N,N'-Dimetil-7,7'-bis difenilfosfino)-3,3',4,4'-tetrahidro-8,8'-bi2H-1,4-benzoxazina]. Os parâmetros de reação para a hidrogenação homogênea na etapa (b), tais como, por exemplo, pressão, temperatura ou tempo de reação, podem variar em uma ampla faixa.
Preferencialmente, a temperatura durante a hidrogenação homogênea na etapa (b) pode ser, em cada caso, de 0 a 250°C, particularmente preferencialmente de 10 a 40°C e muito particularmente preferencialmente de 15 a 25°C.
A hidrogenação homogênea na etapa (b) pode preferencialmente ser executada em pressão reduzida, em pressão normal ou em pressão elevada, preferencialmente na faixa de 1 KPa a 30MPa (0,01 a 300 bar). E particularmente preferencial executar as reações sob pressão em uma faixa de 300KPa a 2,0MPa (3 a 20 bar), em particular de 800KPa a 1,2MPa (8 a 12 bar).
O tempo de reação pode variar na dependência de vários parâmetros, tais como, por exemplo, temperatura, pressão, natureza do composto a ser reagido ou das propriedades do catalisador, e pode ser determinado para o processo em questão pela pessoa versada na técnica usando testes preliminares.
A etapa de desidratação (b1) é preferencialmente catalisada por ácido. Preferencialmente, o ácido é selecionado a partir do grupo que consiste em ácido fórmico, ácido clorídrico, ácido acético, ácido sulfúrico, ácido bromídrico, ácido metanossulfônico, ou qualquer mistura destes. É preferencial se o ácido for empregado em uma alta concentração. Particularmente preferencialmente, a concentração de ácido clorídrico é > 20%, preferencialmente > 30%, particularmente preferencialmente > 35% em peso. Alternativamente, o ácido pode também ser usado na forma gasosa.
Os compostos das fórmulas gerais II e VII usados na etapa (b1) de acordo com a presente invenção estão preferencialmente na fase líquida e para esse fim são preferencialmente misturados ou dissolvidos em um meio reacional que é líquido sob as condições de reação particulares.
Exemplos de meios reacionais adequados são água, ácido acético, ácido fórmico, tolueno, ácido clorídrico, ácido sulfúrico, ácido bromídrico, ácido metanossulfônico, ou qualquer mistura destes. Certamente, é também possível usar misturas ou sistemas de múltiplas fases que compreendem dois ou mais líquidos mencionados acima nos processos de acordo com a presente invenção. Uma reação em CO2 supercrítico como solvente é também possível.
Os parâmetros de reação para a desidratação na etapa (b1), tais como, por exemplo, pressão, temperatura, ou tempo de reação, podem variar em uma ampla faixa.
É preferencial se a temperatura de reação na etapa (b1) estiver entre 35 e 100°C, particularmente preferencialmente entre 45 e 80°C, mais particularmente preferencialmente entre 50 e 60°C.
A etapa de desidratação (b1) pode ser preferencialmente executada em pressão reduzida, em pressão normal, ou em pressão elevada, preferencialmente na faixa de 1KPa a 30MPa (0,01 a 300 bar). É particularmente preferencial executar as reações sob pressão em uma faixa de 50 a 500KPa (0,5 a 5 bar), em particular de 50 a 150KPa (0,5 a 1,5 bar).
O tempo de reação pode variar em dependência de vários parâmetros, tais como, por exemplo, temperatura, pressão, natureza do composto a ser reagido ou das propriedades do catalisador, e pode ser determinado para o processo em questão pela pessoa versada na técnica usando testes preliminares. É preferencial se o tempo de reação da etapa (b‘) estiver entre 2 a 10 h, particularmente preferencialmente entre 3 e 8 h, mais particularmente preferencialmente entre 4 e 6 h.
A remoção contínua de amostras de modo a monitorar a reação, por exemplo, por meio de métodos de cromatografia a gás, é também possível, opcionalmente em combinação com a regulação dos parâmetros do processo correspondente.
A concentração de ácido no meio reacional é preferencialmente de 20 a 26 M no caso do ácido fórmico, de 5 a 18 M no caso de ácido acético, de 8 a 14 M no caso de ácido clorídrico e de 4 a 36 M, mais preferencialmente de 4 a 18 M, no caso de ácido sulfúrico.
O composto particular da fórmula geral (VII) obtido pode ser isolado e/ou purificado por métodos convencionais conhecidos pelos versados na técnica.
Alternativamente, a etapa de desidratação (b1) pode também ser executada na presença de ao menos um catalisador ácido, o qual pode preferencialmente ser selecionado a partir do grupo que consiste em resinas de troca de íons, zeólitos, heteropoliácidos, fosfatos, sulfatos, e óxidos de metais opcionalmente misturados.
O termo catalisador, dentro do contexto da presente invenção, inclui ambos os próprios materiais cataliticamente ativos e os materiais inertes que são fornecidos com um material cataliticamente ativo. Consequentemente, o material cataliticamente ativo pode, por exemplo, ser aplicado em um veículo inerte ou pode estar presente em uma mistura com um material inerte. Leva-se em consideração como o veículo inerte ou material inerte, por exemplo, o carbono e outros materiais conhecidos pelos versados na técnica.
Os catalisadores adequados e suas preparações são conhecidos por si mesmos pelos versados na técnica, por exemplo, a partir de Venuto, P.B., Microporous Mater., 1994, 2, 297; Hõlderich, W.F., van Bekkum, H„ Stud. Surf. Sei. Catai., 1991, 58, 631, Hõlderich, W.F., Proceedings of the 10th International Congress on Catalysis, 1992, Budapest, Guczi, L. et al. (editores), New Frontiers in Catalysis, 1993, Elsevier Science Publishers, Kozhenikov, I.V., Catal. Rev. Sci. Eng., 1995, 37, 311, Song, X., Sayari, A., Catal. Rev. Sci. Eng., 1996, 38, 329. As descrições da literatura correspondente são incorporadas aqui por referência e formam parte da descrição.
Eles são adequados para a desidratação, em particular, aquelas resinas de troca de íons que carregam os grupos de ácido sulfônico são usadas.
A preferência é dada às resinas de troca de íons baseadas em copolímeros de tetrafluoroetileno perfluorovinil éter, opcionalmente na forma de suas nanocomposições silicas, como são descritos, por exemplo, nas publicações de literatura de Olah et al., Synthesis, 1996, 513-531 e Harmer et al., Green Chemistry, 2000, 7-14, as descrições correspondentes das quais são incorporadas aqui por referência e formam parte da descrição. Os produtos correspondentes estão disponíveis comercialmente, por exemplo, sob o nome de Nation®, e podem também ser usados nessa forma nos processos de acordo com a presente invenção.
A preferência é adicionalmente dada às resinas trocadoras de íons baseadas nos copolímeros estireno/divinilbenzeno, que podem ser preparadas por processos convencionais conhecidos pelos versados na técnica.
Leva-se em consideração para a desidratação particularmente preferencialmente as resinas trocadoras de íons que carregam o grupo de ácido sulfônico baseadas em copolímeros estireno/divinilbenzeno, que são comercializados, por exemplo, sob o nome de Amberlyst® produzido por Rohm & Haas e que também podem usados como tais nos processos de acordo com a presente invenção. Essas resinas trocadoras de íons são distinguidas em particular por sua estabilidade em direção à água e aos álcoois, mesmo em temperaturas elevadas, por exemplo, de 130 a 160°C.
O grau de reticulação e a estrutura dessas resinas trocadoras de íons podem variar. Por exemplo, menção pode ser feita às resinas trocadoras de íons macroporosas que têm distribuição de diâmetro de poro heterogênea, resinas trocadoras de íons isoporosas que têm distribuição de diâmetro de poro virtualmente uniforme, ou resinas trocadoras de íons como gel que não têm poros ou virtualmente não têm poros. As resinas macroporosas em particular podem ser usadas com particular vantagem para catálise heterogênea na fase líquida.
Particularmente, as resinas macroporosas adequadas que têm um diâmetro de poro médio de 20 a 30 mm e uma concentração mínima de grupos ativos de 4,70 a 5,45 equivalentes por kg de resina estão disponíveis comercialmente sob o nome de Amberlyst® 15, Amberlyst® 35 e Amberlyst® e consequentemente podem também ser usados nos processos de acordo com a presente invenção.
É igualmente preferencial executar a desidratação na presença de um catalisador ácido baseado em óxidos de metais tais como, por exemplo, SiO2) AI2C>3, TiO2, Nb2O5, B2O3 ou baseado em óxidos de metais misturados tais como, por exemplo, AI2O3/SiO2 ou AI2O3/B2O3.
Preferencialmente, a temperatura para a desidratação (b1) quando usando um catalisador ácido como descrito acima é, em cada caso, de 20 a 250°C, particularmente preferencialmente de 50 a 180°C e muito particularmente preferencialmente de 100 a 160°C.
A relação de catalisador ácido e composto da fórmula geral (II) está preferencialmente na faixa de 1:200 a 1:1, em particular de 1:4 a 1:2.
Após a desidratação, o catalisador pode ser separado da mistura reacional de uma forma simples, preferencialmente por filtração. O composto particular de fórmula geral (VII) obtido é isolado e/ou purificado pelos métodos convencionais conhecidos pelos versados na técnica.
Alternativamente, a etapa de desidratação (b1) pode também ser executada submetendo-se um composto da fórmula geral (II) a um excesso de cloreto de tionila, opcionalmente em um meio reacional, preferencialmente em um meio reacional selecionado a partir do grupo que consiste em dietiléter, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, tert-butilmetiléter e misturas destes, e subsequente aquecimento da mistura reacional assim obtida de 40°C a 120°C, preferencialmente de 80°C a 120°C.
A hidrogenação da etapa (b) pode também ser efetuada através de a catálise heterogênea com hidrogênio. O hidrogênio está preferencialmente na forma gasosa, embora seja possível que ao menos em parte dele seja dissolvida em uma fase líquida.
A catálise heterogênea, dentro do contexto da presente invenção, significa que os catalisadores usados na etapa (b) estão em cada caso presentes no estado sólido de agregação.
Preferencialmente, o catalisador heterogêneo usado para a hidrogenação na etapa (b) de acordo com a presente invenção contém um ou mais metais de transição, esses metais podem ser preferencialmente selecionados a partir do grupo que consiste em Cu, Ag, Au, Zn, Cd, Hg, V, Nb, Ta, Cr, Mo, W, Fe, Ru, Os, Co, Rh, Ir, Ni, Pd, Pt, particularmente preferencialmente a partir do grupo que consiste em Ru, Rh, Pd, Pt e Ni.
Os catalisadores correspondentes podem preferencialmente conter um ou mais dos metais de transição mencionados acima nos mesmos estados de oxidação ou em diferentes. Pode também ser preferencial que os catalisadores correspondentes contenham um ou mais dos metais de transição mencionados acima em dois ou mais diferentes estados de oxidação.
A preparação dos catalisadores dopados com metais de transição pode ser executada por processos convencionais conhecidos pelos versados na técnica.
Preferencialmente, o catalisador usado para hidrogenação na etapa (b) é selecionado a partir do grupo que consiste em níquel Raney, paládio, paládio em carbono (1 - 10% em peso, preferencialmente 5% em peso), platina, platina em carbono (1 - 10% em peso, preferencialmente 5% em peso), rutênio em carbono (1 - 10% em peso, preferencialmente 5% em peso) e ródio em carbono (1 - 10% em peso, preferencialmente 5% em peso), mais preferencialmente paládio em carbono (1 -10% do peso, preferencialmente 5% em peso) é usado como o catalisador para a hidrogenação na etapa (b).
Os compostos de fórmula geral VII ou III usados na etapa (b) de acordo com a presente invenção estão preferencialmente na fase líquida e para esse fim são preferencialmente misturados com ou dissolvidos em um meio reacional que é líquido sob as condições de reação particulares.
Exemplos de meios reacionais adequados são o metanol, etanol, isopropanol, n-butanol, n-propanol, tolueno, heptano, hexano, pentano, ácido acético, acetato etílico, ácido fórmico, ácido clorídrico, ácido bromídrico, ácido sulfúrico e misturas destes. Mais preferencialmente, o etanol é usado como o meio reacional na etapa (b). Certamente, é também possível usar as misturas ou sistema de múltiplas fases compreendendo dois ou mais líquidos mencionados acima nos processos de acordo com a presente in venção. A reação em CO2 supercrítico como solvente é também possível.
Os parâmetros da reação para a hidrogenação heterogênea na etapa (b), tais como, por exemplo, pressão, temperatura, ou tempo de reação, podem variar em uma ampla faixa.
Preferencialmente, a temperatura durante a hidrogenação heterogênea na etapa (b) é, em cada caso, de 0 a 250°C, particularmente preferencialmente de 15 a 180°C e muito particularmente preferencialmente de 15 a 30°C.
A hidrogenação heterogênea na etapa (b) pode ser preferencialmente executada em pressão reduzida, em pressão normal ou em pressão elevada, preferencialmente na faixa de 100KPa a 30MPa (1 a 300 bar). É particularmente preferencial executar as reações sob pressão em uma faixa de 200KPa a 1MPa (2 a 10 bar), em particular de 400KPa a 1MPa (4 a 10 bar).
O tempo de reação pode variar em dependência de vários parâmetros, tais como, por exemplo, temperatura, pressão, natureza do composto a ser reagido ou das propriedades do catalisador, e pode ser determinado para o processo em questão pelos versados na técnica que usam testes preliminares.
A remoção contínua de amostras de modo a monitorar a reação, por exemplo, por meio de métodos de cromatografia a gás, é também possível, opcionalmente em combinação com a regulação dos parâmetros do processo correspondente.
A quantidade total de catalisadores usados depende de vários fatores, tal como, por exemplo, a relação do componente cataliticamente ativo para qualquer material inerte presente, ou a natureza da superfície do(s) catalisador(es). A quantidade ótima de catalisador(es) para uma reação particular pode ser determinada pelos versados na técnica usando os testes preliminares.
O composto particular de fórmula geral (III) obtido pode ser isolado e/ou purificado por métodos convencionais conhecidos pelos versados na técnica.
Em outra modalidade da invenção, a etapa (b) (esquema 1) é uma reação de substituição direta do grupo OH pelo H, preferencialmente executada em uma reação em uma única etapa. Mais preferencialmente, um OH’ é substituído por H'.
Cada uma das etapas de acordo com a presente invenção pode ser executada descontinuamente (por bateladas) ou continuamente, preferência sendo dada ao procedimento descontínuo.
Leva-se em consideração como o reator para o procedimento descontínuo, por exemplo, um reator de pasta fluida, e para o procedimento contínuo de um reator de leito fixo ou reator de ciclo.
Na sequência, um processo para a preparação de cloridrato de (1 R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol é descrito.
Exemplo
Preparação de cloridrato de (1R,2R)-3-(3-dimetilamino-1-etil-2-
Etapa (a1): Preparação de 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1ona (Via)
1-(3-Metoxifenil)propan-1-ona (16,42 kg, 100 mols), cloridrato de dimetilamina (8,97 kg, 110 mols), o paraformaldeído (3,30 kg, 110 mols) e ácido clorídrico aquoso (32% em peso, 1,14 kg) foram dissolvidos em etanol sob uma atmosfera de nitrogênio em um recipiente de camisa dupla de 100 L (L = litro) equipado com um agitador elétrico de impulsor, uma linha de transição a gás, equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo. A mistura reacional foi submetida a refluxo por 16 horas, resfriada a 25°C dentro de 3,5 horas e agitada por 1 hora nesta temperatura. A suspensão foi separada através de uma centrífuga e lavada três vezes com 7 L de acetona cada. Cloridrato de 3(Dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona foi dissolvido em água (12,5 L) e em tert-butil-metil-éter (8,5 L) e agitado em temperatura ambiente.
A solução aquosa de hidróxido de sódio (32% em peso) foi adicionada até que o valor de pH entre 10,0 e 10,5 fosse alcançado e até que as fases fossem separadas. A fase orgânica foi destilada sob pressão reduzida até uma temperatura de 40°C até que uma pressão de 500KPa (5 mbar) fosse alcançada. 3-(Dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona foi obtido como um óleo amarelo pálido (20,75 Kg, 94%) que foi usado na próxima etapa sem purificação adicional.
Etapa (a): Preparação de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Ia)
1.a. Preparação de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-me-tilpropan-1ona (2R, 3R)-O,O'-dibenzoiltartarato em acetona
O monohidrato de ácido (2R, 3R)-O,O'-Dibenzoil tartárico (189,1 g, 0,5 mol foi dissolvido em acetona (550 mL) em uma planta de reação equipada com um agitador mecânico, um equipamento que mede a temperatura e um banho de óleo e foi adicionado 3-(dimetilamino)-1-(3-metoxifenil)-
2-metilpropan-1-ona (110,6 g, 0,5 mol). A mistura reacional foi aquecida de 35°C a 40°C por 27 horas e resfriada a 25°C. A suspensão foi sifonada e 3(dimetilamino)-l -(3-metoxifenil)-2-metilpropan-1 -ona (2R,3R)-O,O'-dibenzoiltartarato foi obtido como um sólido incolor (233,2 g, 80,5%, ee 96,9%, ee = excesso enantiomérico).
1.b. Preparação de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-me-tilpropan-1ona (2R, 3R)-O,O'-dibenzoiltartarato em acetona/metanol
O monohidrato de ácido (2R, 3R)-O,O'-Dibenzoil tartárico (2,1 kg, 5,5 rnols) foi dissolvido em uma mistura de metanol (555 mL) e acetona (3340 mL) em um recipiente de camisa dupla de 10 L equipado com um agi tador elétrico de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo e foi adicionado 3-(dimetilamino)-1-(3-metoxifenil)-2metilpropan-1-ona (1,23 kg, 5,56 mols). A mistura reacional foi aquecida a 35°C a 40°C por 24 horas e resfriada a 25°C. A suspensão foi sifonada e (S)-3-(dimetilamino)-1 -(3-metoxifenil)-2-metilpropan-1 -ona (2R, 3R)-O,O'dibenzoiltartarato foi obtido como um sólido incolor (2,38 kg, 74%, ee 98,4%).
2. Preparação de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Ia) (S)-3-(dimetilamino)-1 -(3-metoxifenil)-2-metilpropan-1 -ona (2R, 3R)-O,O'-dibenzoiltartarato (968 g, 1,67 mmol, e 98%) foi suspenso em tertbutilmetil éter (6L) em um recipiente de camisa dupla de 10L equipado com um agitador elétrico de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo e dimetilamina (384 g, 5,25 mols) foi adicionada. A mistura reacional foi agitada de 20°C a 25°C por 90 minutos e um sólido foi sifonado. O filtrado foi concentrado em uma temperatura de 40°C em vácuo até que a pressão de 40KPa (4 mbar) fosse alcançada. (S)-3(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona foi obtido como um óleo incolor (356,7 g, 96,5%, ee 98%).
Etapa (a): Preparação de (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2metilpropan-3-ol (lia)
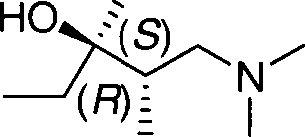
1. Aparas de magnésio (93,57%, 3,85 mols) foram suspensas em éter etílico seco (2L) em um recipiente de camisa dupla de 10 L equipado com um agitador elétrico de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo e brometo etílico (25 g, 0,23 mol) foi adicionado. Depois de a reação ter iniciado, brometo etílico adicional (438,6 g, 4,02 mols) foi adicionado dentro de 90 minutos e abaixo de uma temperatura de 35°C e a mistura reacional foi agitada por mais uma hora. A mistura reacional foi resfriada de 10°C a 15°C, foi adicionado (S)-3-(dimetilamino)-1-(3 metoxifenil)-2-metilpropan-1-ona (774,6 g, 3,5 mols, ee 98%) dissolvido em éter dietilico (0,8 L) e a mistura reacional foi agitada por outras duas horas. A mistura reacional foi resfriada a 5°C e a solução aquosa de hidrogenossulfato de amônio (10% em peso, 2L) foi adicionada. As fases foram separadas e a fase orgânica foi concentrada a vácuo em 40°C até que uma pressão de 500KPa (5 mbar) fosse alcançada. (2S,3R)-1-(Dimetilamino)-3-(3metoxifenil)-2-metilpropan-3-ol (862,3 g, 98%) foi obtido como um óleo incolor (ee 98%).
2. (S)-3-(dimetilamino)-1 -(3-metoxifenil)-2-metilpropan-1 -ona (774,6 g, 3,5 mols, ee 95%) foi dissolvido em tetrahidrofurano seco (800 ml) em um recipiente de camisa dupla de 10 L equipado com um agitador elétrico de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo e brometo de etilmagnésio (2 L, 2 M em THF) foi adicionado a uma temperatura de 15°C dentro de duas horas. A mistura reacional foi agitada por duas horas nesta temperatura, resfriada a 5°C e a solução aquosa de hidrogenossulfato de amônio (10% em peso, 2 L) foi adicionada. As fases foram separadas e a fase orgânica foi concentrada a vácuo em 40°C até que uma pressão de 500KPa (5 mbar) fosse alcançada. (2S,3R)-1(Dimetilamino)-3-(3-metoxifenil)-2-metilpropan-3-ol (871,1 g, 99%) foi obtido como um óleo incolor (ee 95%).
Etapa (b‘): Preparação de (R)-3-(3-metoxifenil)-N,N,2-trimetilpent-3-en-1amina (Vila)
1. (2S,3R)-1 -(Dimetilamino)-3-(3-metoxifenil)-2-metilpropan-3-ol (754,1 g, 3 mols, ee 95%) foi adicionado em acetona (5 L) em um recipiente de camisa dupla de 10 L equipado com um agitador elétrico de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo. O cloreto de hidrogênio (110 g, 3,0 mols) foi transferido dentro de 15 minutos em uma temperatura de 15°C através da mistura reacional. A mistura reacional foi resfriada de 0°C a 5°C e após 24 horas nesta temperatura foi sifonada. O produto foi armazenado a 40°C e 1MPa (10 mbar) por 14 horas em um forno de secagem. Cloridrato de (2S,3R)-1-(Dimetilamino)-3-(3-metoxifenil)-2metilpropan-3-ol foi obtido como um sólido incolor (722,3 g, 83,7%, ee 100%).
2. Cloridrato de (2S,3R)-1-(Dimetilamino)-3-(3-metoxifenil)-2metilpropan-3-ol obtido como descrito acima foi colocado em um frasco de três gargalos de 250 mL equipado com um termômetro, com um agitador de ar comprimido mecânico, um condensador de refluxo e banho de óleo e a solução aquosa de cloreto de hidrogênio (150 mL, 36% em peso) foi adicionada. A mistura reacional foi aquecida a 55°C por 5 horas e resfriada a 20°C. A solução aquosa de hidróxido de sódio (33% em peso) foi adicionada enquanto se resfriava até que um valor de pH de 11 fosse alcançado. O acetato etílico (150 mL) foi adicionado, a mistura reacional foi agitada por 10 minutos, as fases foram separadas e o acetato etílico foi removido a vácuo a 60°C até que a pressão de 1KPa (10 mbar) fosse alcançada. (R)-3-(3metoxifenil)-N,N,2-trimetilpent-3-en-1-amina (21 g, 90%) foi obtido como um resíduo oleoso (relação Z/E 4,5:1).
Etapa (b1): Preparação de Cloridrato de (2R,3R)-3-(3-metoxifenil)-N,N,2trimetilpentan-1-amina (llla)
1. (R)-3-(3-metoxifenil)-N,N,2-trimetilpent-3-en-1-amina (5 kg, 21,43 mmols) foi dissolvido em etanol seco (13 L) em uma temperatura de 25°C e a frequência de agitação rotacional de 850 ±150 por minuto em um aparelho de hidrogenação de camisa dupla equipado com uma tampa estacionária montada que tem uma fonte de hidrogênio e nitrogênio, um agitador de gaseificação elétrico, um equipamento de medição de temperatura Pt100, inspecionando o controlador de gás e vidro Büchi bpc. O aparelho de hidrogenação foi inundado com nitrogênio. O paládio em carvão (375 g, 5% em peso) foi suspenso em cloreto de hidrogênio aquoso (675 g, 32% em peso) e adicionado à mistura reacional. O aparelho de hidrogenação foi inundado novamente com nitrogênio e a reação foi executada em uma pressão primária de hidrogênio de 500KPa (5 bar) e uma pressão interna de hidrogênio de 100KPa (1 bar) até que a reação fosse completa. O aparelho de hidrogenação foi inundado com nitrogênio e o catalisador foi filtrado em um filtro em camadas com terra de filtração. O filtrado foi concentrado a vácuo. O resíduo foi absorvido em acetato etílico e hidróxido de sódio aquoso (10% em peso, 3,7 L) foi adicionado a 20°C até que um pH de 10 a 12 fosse alcançado. A fase orgânica foi concentrada em vácuo de 45°C a 50°C até que a pressão de 500KPa (5 mbar) fosse alcançada. O resíduo oleoso era uma mistura de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina e (2R,3S)-
3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (4,5 kg, 95%, relação de 5,5 (R,R):1 (R,S)).
2. Uma mistura de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetil10 pentan-1-amina e (2R,3S)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (10 kg, 42,56 rnols, relação de 5,5:1) foi dissolvida em acetona (50 L) em um recipiente de camisa dupla de 100 L equipado com um agitador elétrico de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em ó15 leo. Cloreto de hidrogênio (1,55 kg, 42,51 rnols) foi transferido dentro de 15 minutos a uma temperatura de 5°C a 25°C através da mistura reacional. A mistura reacional foi resfriada de 0°C a 5°C e centrifugada após 2 horas de agitação. O sólido úmido foi colocado em um recipiente de agitação, acetona (30 L) foi adicionada e a mistura reacional foi aquecida a refluxo por 15 mi20 nutos. Após o resfriamento de 15°C a 20°C, o produto foi centrifugado e armazenado de 40°C a 50°C em 15 MPa (150 mbar) por 14 horas em um forno de secagem. Cloridrato de (2R,3R)-3-(3-Metoxifenil)-N,N,2-trimetilpentan-
1-amina (7,17 kg, 63%) foi obtido como um sólido incolor com um excesso diastereomérico de 100%.
Etapa (c): Preparação do Cloridrato de (1R,2R)-3-(3-dimetilamino-1-etil-2metil-propil)-fenol (IV)
1. Cloridrato de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1amina (5 kg, 18,4 rnols) foi dissolvido em ácido metanossulfônico (19,5 L) em um recipiente de camisa dupla de 100 L equipado com um agitador elétrico 30 de impulsor, uma linha de transição de gás, um equipamento que mede a temperatura Pt100 e um sistema de resfriamento/aquecimento baseado em óleo e metionina foi adicionada (3,35 kg, 22,5 rnols). A mistura reacional foi agitada em uma temperatura de 75°C a 80°C por 16 horas, resfriada de 15°C a 25°C e água (12,5 L) foi adicionada lentamente nesta temperatura. A solução aquosa de hidróxido de sódio (ca. 28 L, 32% em peso) foi adicionada até que um valor de pH de 10 a 12 fosse alcançado enquanto a temperatura se mantinha abaixo de 50°C. O acetato etílico (15 L) foi adicionado e a mistura reacional foi agitada por 15 minutos em uma frequência de agitação rotacional de 150 por minuto. As fases foram separadas e a fase orgânica foi lavada com água (15 L). Carvão ativado (0,05 kg) foi adicionado à fase orgânica e filtrado após 30 minutos de agitação. O solvente foi removido a vácuo em uma temperatura de 40°C a 50°C até que uma pressão de 5KPa (50 mbar) fosse alcançada. O resíduo foi usado na próxima etapa sem purificação adicional.
2. O resíduo obtido como descrito acima foi dissolvido em acetona (25 L) enquanto agitando e cloreto de hidrogênio (0,78 kg, 21,4 rnols) foi transferido através da mistura reacional em uma temperatura de 20°C a 25°C. A suspensão foi agitada por 3 horas em uma temperatura de 0°C a 5°C e centrifugada. Isopropanol (35 L) foi adicionado ao sólido úmido em um recipiente de reação e a mistura reacional foi aquecida a refluxo por 15 minutos. A mistura reacional foi resfriada de 0°C a 5°C e agitada por 3 horas nesta temperatura. Após a centrifugação, o produto foi armazenado em uma temperatura de 30°C a 40°C a uma pressão de 15KPa (150 mbar) por 16 horas em um forno de secagem. Cloridrato de (1 R,2R)-3-(3-dimetilamino-1etil-2-metil-propil)-fenol (4,18 kg, 88%) foi obtido como um sólido incolor com uma pureza de 100%.


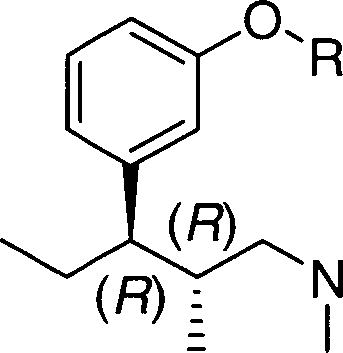







 (IV), ou um sal com adição de ácido do mesmo, o referido processo sendo caracterizado pelo fato de que compreende as etapas de:etapa (a): preparar um composto de fórmula (II):
(IV), ou um sal com adição de ácido do mesmo, o referido processo sendo caracterizado pelo fato de que compreende as etapas de:etapa (a): preparar um composto de fórmula (II): (II), opcionalmente na forma de um sal com adição de ácido, pela reação de um composto da fórmula geral (I),
(II), opcionalmente na forma de um sal com adição de ácido, pela reação de um composto da fórmula geral (I), (I), com brometo de etilmagnésio ou cloreto de etilmagnésio em um éter orgânico selecionado a partir do grupo que consiste em éter dietílico, tetrahidrofurano, 2-metiltetrahidrofurano, tert-butil-metiléter ou qualquer mistura dos mesmos sob condições de Grignard;etapa (b): preparar um composto de fórmula geral (III):
(I), com brometo de etilmagnésio ou cloreto de etilmagnésio em um éter orgânico selecionado a partir do grupo que consiste em éter dietílico, tetrahidrofurano, 2-metiltetrahidrofurano, tert-butil-metiléter ou qualquer mistura dos mesmos sob condições de Grignard;etapa (b): preparar um composto de fórmula geral (III):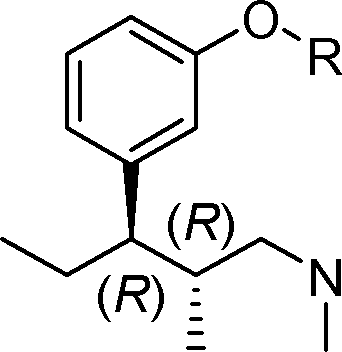 (III), opcionalmente na forma de um sal com adição de ácido, sendoPetição 870180142586, de 19/10/2018, pág. 8/13 que a etapa (b) é executada através de (b') submetendo-se o composto de fórmula geral (II) à desidratação e (b) hidrogenação do composto assim obtido de fórmula geral (VII),
(III), opcionalmente na forma de um sal com adição de ácido, sendoPetição 870180142586, de 19/10/2018, pág. 8/13 que a etapa (b) é executada através de (b') submetendo-se o composto de fórmula geral (II) à desidratação e (b) hidrogenação do composto assim obtido de fórmula geral (VII), ã 1 (vii), usando um catalisador em um meio reacional inerte na presença de hidrogênio;etapa (c): desproteger o composto de fórmula geral (III) para obter o (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV); e opcionalmente uma etapa (d), a saber, converter o composto de fórmula (IV) em um sal com adição de ácido, sendo que nos compostos acima mencionados correspondentes à fórmula geral (I), (II) e (III), R representa metila, benzila ou tetrahidropiranila.
ã 1 (vii), usando um catalisador em um meio reacional inerte na presença de hidrogênio;etapa (c): desproteger o composto de fórmula geral (III) para obter o (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV); e opcionalmente uma etapa (d), a saber, converter o composto de fórmula (IV) em um sal com adição de ácido, sendo que nos compostos acima mencionados correspondentes à fórmula geral (I), (II) e (III), R representa metila, benzila ou tetrahidropiranila. = 1 (VII), etapa (b): submeter o composto de fórmula (VII) à uma reação de hidrogenação via catálise heterogênea na presença de hidrogênio, sendo que o catalisador é selecionado a partir do grupo que consiste em níquel Raney, paládio, paládio em carbono, platina, platina em carbono, rutênio emPetição 870180142586, de 19/10/2018, pág. 9/13 carbono e ródio em carbono.
= 1 (VII), etapa (b): submeter o composto de fórmula (VII) à uma reação de hidrogenação via catálise heterogênea na presença de hidrogênio, sendo que o catalisador é selecionado a partir do grupo que consiste em níquel Raney, paládio, paládio em carbono, platina, platina em carbono, rutênio emPetição 870180142586, de 19/10/2018, pág. 9/13 carbono e ródio em carbono.