ES2901953T3 - Procedimiento de preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol - Google Patents
Procedimiento de preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol Download PDFInfo
- Publication number
- ES2901953T3 ES2901953T3 ES17161440T ES17161440T ES2901953T3 ES 2901953 T3 ES2901953 T3 ES 2901953T3 ES 17161440 T ES17161440 T ES 17161440T ES 17161440 T ES17161440 T ES 17161440T ES 2901953 T3 ES2901953 T3 ES 2901953T3
- Authority
- ES
- Spain
- Prior art keywords
- ethyl
- acid
- process according
- general formula
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- KWTWDQCKEHXFFR-SMDDNHRTSA-N tapentadol Chemical compound CN(C)C[C@H](C)[C@@H](CC)C1=CC=CC(O)=C1 KWTWDQCKEHXFFR-SMDDNHRTSA-N 0.000 title claims abstract description 24
- 238000000034 method Methods 0.000 title claims description 43
- 238000002360 preparation method Methods 0.000 title description 18
- 150000001875 compounds Chemical class 0.000 claims abstract description 53
- 239000012429 reaction media Substances 0.000 claims abstract description 37
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 35
- 239000002253 acid Substances 0.000 claims abstract description 24
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims abstract description 22
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims abstract description 21
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 19
- 150000003839 salts Chemical class 0.000 claims abstract description 19
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims abstract description 14
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims abstract description 14
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims abstract description 14
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims abstract description 14
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 14
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 12
- -1 ethyl magnesium halide Chemical class 0.000 claims abstract description 11
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims abstract description 11
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims abstract description 7
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims abstract description 7
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims abstract description 7
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims abstract description 7
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims abstract description 7
- 238000004519 manufacturing process Methods 0.000 claims abstract description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 43
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 28
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 27
- 239000000203 mixture Substances 0.000 claims description 25
- 239000003054 catalyst Substances 0.000 claims description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 21
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 18
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 17
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 16
- 238000005984 hydrogenation reaction Methods 0.000 claims description 16
- 230000018044 dehydration Effects 0.000 claims description 15
- 238000006297 dehydration reaction Methods 0.000 claims description 15
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 13
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 13
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 13
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 13
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 12
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 12
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 11
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 239000001257 hydrogen Substances 0.000 claims description 10
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 10
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 claims description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 6
- 235000019253 formic acid Nutrition 0.000 claims description 6
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 6
- 229910052763 palladium Inorganic materials 0.000 claims description 5
- 229910052697 platinum Inorganic materials 0.000 claims description 5
- 229930040373 Paraformaldehyde Natural products 0.000 claims description 4
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 claims description 4
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 claims description 4
- 238000007210 heterogeneous catalysis Methods 0.000 claims description 4
- 229920002866 paraformaldehyde Polymers 0.000 claims description 4
- YONLFQNRGZXBBF-ZIAGYGMSSA-N (2r,3r)-2,3-dibenzoyloxybutanedioic acid Chemical compound O([C@@H](C(=O)O)[C@@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 YONLFQNRGZXBBF-ZIAGYGMSSA-N 0.000 claims description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 claims description 3
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 claims description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 claims description 3
- 230000001476 alcoholic effect Effects 0.000 claims description 3
- 239000007868 Raney catalyst Substances 0.000 claims description 2
- 229910000564 Raney nickel Inorganic materials 0.000 claims description 2
- 239000012458 free base Substances 0.000 claims description 2
- 238000007172 homogeneous catalysis Methods 0.000 claims description 2
- AUONNNVJUCSETH-UHFFFAOYSA-N icosanoyl icosanoate Chemical compound CCCCCCCCCCCCCCCCCCCC(=O)OC(=O)CCCCCCCCCCCCCCCCCCC AUONNNVJUCSETH-UHFFFAOYSA-N 0.000 claims description 2
- 238000000926 separation method Methods 0.000 claims description 2
- 238000012546 transfer Methods 0.000 claims description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 claims 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 33
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- 238000006243 chemical reaction Methods 0.000 description 25
- 239000011541 reaction mixture Substances 0.000 description 24
- YHCVGGJYRMYIGG-JTQLQIEISA-N (2s)-3-(dimethylamino)-1-(3-methoxyphenyl)-2-methylpropan-1-one Chemical compound COC1=CC=CC(C(=O)[C@@H](C)CN(C)C)=C1 YHCVGGJYRMYIGG-JTQLQIEISA-N 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000001816 cooling Methods 0.000 description 10
- 239000003456 ion exchange resin Substances 0.000 description 10
- 229920003303 ion-exchange polymer Polymers 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- 239000007789 gas Substances 0.000 description 9
- 238000010438 heat treatment Methods 0.000 description 9
- 229910052703 rhodium Inorganic materials 0.000 description 9
- 239000010948 rhodium Substances 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 230000007704 transition Effects 0.000 description 8
- YHCVGGJYRMYIGG-UHFFFAOYSA-N 3-(dimethylamino)-1-(3-methoxyphenyl)-2-methylpropan-1-one Chemical compound COC1=CC=CC(C(=O)C(C)CN(C)C)=C1 YHCVGGJYRMYIGG-UHFFFAOYSA-N 0.000 description 7
- 238000009529 body temperature measurement Methods 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 230000035484 reaction time Effects 0.000 description 7
- 229910052723 transition metal Inorganic materials 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- PZNRRUTVGXCKFC-SWLSCSKDSA-N (2s,3r)-1-(dimethylamino)-3-(3-methoxyphenyl)-2-methylpentan-3-ol Chemical compound CN(C)C[C@H](C)[C@](O)(CC)C1=CC=CC(OC)=C1 PZNRRUTVGXCKFC-SWLSCSKDSA-N 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 150000003624 transition metals Chemical class 0.000 description 6
- 239000007791 liquid phase Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 229940095064 tartrate Drugs 0.000 description 5
- ZELFLGGRLLOERW-YECZQDJWSA-N 3-[(2r,3r)-1-(dimethylamino)-2-methylpentan-3-yl]phenol;hydrochloride Chemical compound Cl.CN(C)C[C@H](C)[C@@H](CC)C1=CC=CC(O)=C1 ZELFLGGRLLOERW-YECZQDJWSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 239000003377 acid catalyst Substances 0.000 description 4
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 4
- 238000006555 catalytic reaction Methods 0.000 description 4
- 229920001429 chelating resin Polymers 0.000 description 4
- VURFVHCLMJOLKN-UHFFFAOYSA-N diphosphane Chemical compound PP VURFVHCLMJOLKN-UHFFFAOYSA-N 0.000 description 4
- 238000009905 homogeneous catalytic hydrogenation reaction Methods 0.000 description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 4
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- YCCXQARVHOPWFJ-UHFFFAOYSA-M magnesium;ethane;chloride Chemical compound [Mg+2].[Cl-].[CH2-]C YCCXQARVHOPWFJ-UHFFFAOYSA-M 0.000 description 4
- 150000007522 mineralic acids Chemical class 0.000 description 4
- 239000011148 porous material Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- JKVBTSJLQLSTHJ-SWLSCSKDSA-N (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentan-1-amine Chemical compound CN(C)C[C@H](C)[C@@H](CC)C1=CC=CC(OC)=C1 JKVBTSJLQLSTHJ-SWLSCSKDSA-N 0.000 description 3
- XKSJVTDZSYZBGF-SBKWZQTDSA-N (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentan-1-amine;hydrochloride Chemical compound Cl.CN(C)C[C@H](C)[C@@H](CC)C1=CC=CC(OC)=C1 XKSJVTDZSYZBGF-SBKWZQTDSA-N 0.000 description 3
- JHKJGWJFBYPMCY-CJTQKWKHSA-N (z,2r)-3-(3-methoxyphenyl)-n,n,2-trimethylpent-3-en-1-amine Chemical compound COC1=CC=CC(C(=C/C)\[C@@H](C)CN(C)C)=C1 JHKJGWJFBYPMCY-CJTQKWKHSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 239000011149 active material Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 238000009904 heterogeneous catalytic hydrogenation reaction Methods 0.000 description 3
- 229910052741 iridium Inorganic materials 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 229910052707 ruthenium Inorganic materials 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- JKVBTSJLQLSTHJ-WFASDCNBSA-N (2r,3s)-3-(3-methoxyphenyl)-n,n,2-trimethylpentan-1-amine Chemical compound CN(C)C[C@H](C)[C@H](CC)C1=CC=CC(OC)=C1 JKVBTSJLQLSTHJ-WFASDCNBSA-N 0.000 description 2
- JWPQTVRDQHIIHD-SBKWZQTDSA-N (2s,3r)-1-(dimethylamino)-3-(3-methoxyphenyl)-2-methylpentan-3-ol;hydrochloride Chemical compound Cl.CN(C)C[C@H](C)[C@](O)(CC)C1=CC=CC(OC)=C1 JWPQTVRDQHIIHD-SBKWZQTDSA-N 0.000 description 2
- QKZWXPLBVCKXNQ-ACHIHNKUSA-N (s)-(2-methoxyphenyl)-[2-[(2-methoxyphenyl)-phenylphosphanyl]ethyl]-phenylphosphane Chemical compound COC1=CC=CC=C1[P@](C=1C=CC=CC=1)CC[P@](C=1C(=CC=CC=1)OC)C1=CC=CC=C1 QKZWXPLBVCKXNQ-ACHIHNKUSA-N 0.000 description 2
- LPDJHUUWTGXTCU-UHFFFAOYSA-N 1-(3-methoxyphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=CC(OC)=C1 LPDJHUUWTGXTCU-UHFFFAOYSA-N 0.000 description 2
- WWILHZQYNPQALT-UHFFFAOYSA-N 2-methyl-2-morpholin-4-ylpropanal Chemical compound O=CC(C)(C)N1CCOCC1 WWILHZQYNPQALT-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 230000000202 analgesic effect Effects 0.000 description 2
- 238000010923 batch production Methods 0.000 description 2
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- QKZWXPLBVCKXNQ-ROJLCIKYSA-N dipamp Chemical compound COC1=CC=CC=C1[P@@](C=1C=CC=CC=1)CC[P@@](C=1C(=CC=CC=1)OC)C1=CC=CC=C1 QKZWXPLBVCKXNQ-ROJLCIKYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Chemical compound [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 2
- FRIJBUGBVQZNTB-UHFFFAOYSA-M magnesium;ethane;bromide Chemical compound [Mg+2].[Br-].[CH2-]C FRIJBUGBVQZNTB-UHFFFAOYSA-M 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- ZKATWMILCYLAPD-UHFFFAOYSA-N niobium pentoxide Chemical compound O=[Nb](=O)O[Nb](=O)=O ZKATWMILCYLAPD-UHFFFAOYSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 1
- ZVXORNFUYLTVBM-UHFFFAOYSA-N 3-(dimethylamino)-1-(3-methoxyphenyl)-2-methylpropan-1-one;hydrochloride Chemical compound Cl.COC1=CC=CC(C(=O)C(C)CN(C)C)=C1 ZVXORNFUYLTVBM-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229910010413 TiO 2 Inorganic materials 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052793 cadmium Inorganic materials 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000012351 deprotecting agent Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- CHRLNRKNUAPHRT-UHFFFAOYSA-N ethylsulfanylethane;sodium Chemical compound [Na].CCSCC CHRLNRKNUAPHRT-UHFFFAOYSA-N 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 238000002309 gasification Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000002638 heterogeneous catalyst Substances 0.000 description 1
- 239000002815 homogeneous catalyst Substances 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229910003455 mixed metal oxide Inorganic materials 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000002114 nanocomposite Substances 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- 239000005051 trimethylchlorosilane Substances 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J25/00—Catalysts of the Raney type
- B01J25/02—Raney nickel
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B57/00—Separation of optically-active compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/26—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing at least one six-membered aromatic ring
- C07C211/27—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing at least one six-membered aromatic ring having amino groups linked to the six-membered aromatic ring by saturated carbon chains
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/26—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing at least one six-membered aromatic ring
- C07C211/28—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing at least one six-membered aromatic ring having amino groups linked to the six-membered aromatic ring by unsaturated carbon chains
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/08—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions not involving the formation of amino groups, hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/46—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C215/48—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by hydroxy groups
- C07C215/54—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by hydroxy groups linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/62—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/64—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms
- C07C217/66—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain
- C07C217/72—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C221/00—Preparation of compounds containing amino groups and doubly-bound oxygen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C225/00—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones
- C07C225/02—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton
- C07C225/04—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated
- C07C225/08—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated and containing rings
- C07C225/10—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated and containing rings with doubly-bound oxygen atoms bound to carbon atoms not being part of rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Un procedimiento para preparar (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol, o una sal de adición de ácido del mismo, que comprende la etapa de (a) hacer reaccionar un compuesto de fórmula general (I), **(Ver fórmula)** donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2H5, - C(=O)-CH(CH3)2 o -C(=O)-C(CH3)3, con haluro de etil magnesio en un medio de reacción inerte en condiciones de Grignard, (b) transferir el compuesto de fórmula general (II) así obtenido, **(Ver fórmula)** donde R tiene el significado definido anteriormente, a un compuesto de fórmula general (III), **(Ver fórmula)** donde R tiene el significado definido anteriormente,v opcionalmente en forma de sal de adición de ácido, (c) desproteger el compuesto así obtenido de fórmula general (III) para obtener (1R,2R)-3-(3-dimetilamino-1-etil-2- metil-propil)-fenol de fórmula (IV), **(Ver fórmula)** (d) convertir opcionalmente el (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol así obtenido en una sal de adición de ácido.
Description
DESCRIPCIÓN
Procedimiento de preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol
La presente invención se refiere a un procedimiento para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)-fenol.
Una clase de ingredientes activos con excelente efectividad analgésica y muy buena tolerabilidad son los compuestos de dimetil-(3-aril-butil)-amina sustituidos, que se conocen entre otros de e P 0693 475. En particular, (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol ha demostrado ser un candidato muy prometedor para el desarrollo de un analgésico en ensayos clínicos.
Un objetivo de la presente invención fue, por lo tanto, proporcionar un procedimiento que permita la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol mediante una ruta corta con buen rendimiento global en condiciones medioambientalmente aceptables.
En particular, en el procedimiento de la presente invención, todos los estereocentros pueden establecerse mediante el control de sustrato con la formación casi exclusiva de un solo diastereoisómero ahorrando así etapas de purificación elaborados para separar estereoisómeros y reactivos, catalizadores o ligandos quirales costosos. Como no se forman productos secundarios no deseados en el procedimiento de la presente invención, cada lote puede funcionar a su capacidad óptima.
El objetivo de la presente invención se cumple proporcionando un procedimiento para preparar (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol, o una sal de adición de ácido del mismo, que comprende la etapa de (a) hacer reaccionar un compuesto de fórmula general (I),

donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2Hs, -C(=O)-CH(CHs)2 o -C(=O)-C(CH3)3,
con haluro de etil magnesio en un medio de reacción inerte en condiciones de Grignard,
(b) transferir el compuesto de fórmula general (II) así obtenido,

donde R tiene el significado definido anteriormente,
a un compuesto de fórmula general (III),

donde R tiene el significado definido anteriormente,
opcionalmente en forma de sal de adición de ácido,
(c) desproteger el compuesto así obtenido de fórmula general (111) para obtener (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV),
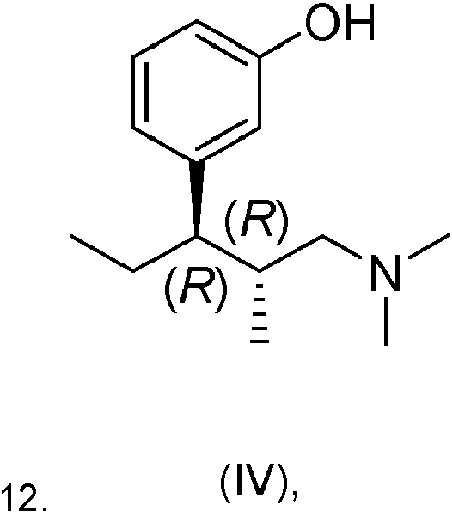
(d) convertir opcionalmente el (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol así obtenido en una sal de adición de ácido.
Particularmente preferiblemente, R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3 en los compuestos de fórmula general (I). Más particularmente preferiblemente, R representa metilo, bencilo o tetrahidropiranilo en los compuestos de fórmula general (I).
Aún más preferiblemente, R en la fórmula general (I) representa metilo. Por tanto, muy preferiblemente se hace reaccionar (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona con haluro de etil magnesio en un medio de reacción inerte en condiciones de Grignard.
Preferiblemente, se usan bromuro de etil magnesio o cloruro de etil magnesio como haluro de etil magnesio en la etapa a).
La reacción según la etapa (a) se lleva a cabo en un medio de reacción inerte, preferiblemente en un éter orgánico, por ejemplo, seleccionado de entre el grupo que consiste en dietil éter, tetrahidrofurano, 2-metiltetrahidrofurano, tercbutilmetil éter o cualquier mezcla. del mismo. La reacción se lleva a cabo particularmente preferiblemente en tetrahidrofurano con cloruro de etil magnesio a una concentración de 0,5 M a 2 M del cloruro de etil magnesio. Particularmente preferiblemente, la reacción se lleva a cabo a una concentración de 1 M o 2 M del cloruro de etil magnesio.
Particularmente preferiblemente, R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3 en los compuestos de fórmulas generales (I), (II) y (III). Más particularmente preferiblemente, R representa metilo, bencilo o tetrahidropiranilo en los compuestos de fórmulas generales (I), (II) y (III).
Aún más particularmente preferiblemente, R representa metilo en las fórmulas generales (I), (II) y (III). Por tanto, (S) -3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Ia) se transforma en (1R,2R)-3-(3-dimetilamino-1-etil- 2-metilpropil)-fenol mediante la siguiente secuencia de etapas (esquema 1).

Esquema 1
En el caso de que R represente metilo en la fórmula general (111), el compuesto (Illa) se hace reaccionar preferiblemente con ácido bromhídrico o ácido metanosulfónico y metionina o hidruro de diisobutilaluminio en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter y mezclas de los mismos para producir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
En el caso de que R represente C1-6-alquilo excepto metilo en la fórmula general (III), el compuesto respectivo de fórmula general (III) se hace reaccionar preferiblemente con ácido bromhídrico o hidruro de diisobutilaluminio en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter y mezclas de los mismos para producir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
En caso de que R represente tetrahidropiranilo en la fórmula general (III), el compuesto respectivo de fórmula general (III) se hace reaccionar preferiblemente con al menos un ácido inorgánico, preferiblemente con al menos un ácido inorgánico seleccionado de entre el grupo que consiste en ácido clorhídrico, ácido bromhídrico , ácido sulfúrico y ácido fosfórico, opcionalmente en presencia de al menos una sal, preferiblemente al menos una sal seleccionada de entre el grupo que consiste en cloruro de amonio e hidrogenosulfato de potasio, en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter, agua y mezclas de los mismos para producir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
En el caso de que R represente -C- 3-8-alquilo excepto metilo en la fórmula general (III), el compuesto respectivo de fórmula general (III) se hace reaccionar preferiblemente con ácido bromhídrico o hidruro de diisobutilaluminio en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter y mezclas de los mismos para producir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
En el caso en que R represente -C1-3-alquileno-fenilo o -C1-3-alquileno-naftilo, se hace reaccionar un compuesto de fórmula general (III) con ácido bromhídrico o hidruro de diisobutilaluminio en un medio de reacción, preferiblemente en un medio de reacción. seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter y mezclas de los mismos o en presencia de hidrógeno y al menos un catalizador, preferiblemente en presencia de al menos un catalizador a base de paladio o platino, más preferiblemente en presencia de paladio sobre carbón, en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter y mezclas de los mismos para producir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
En el caso de que R represente -C(=O)-Ci-6-alquilo en la fórmula general (111), el compuesto respectivo de fórmula general (III) se hace reaccionar preferiblemente con al menos un ácido inorgánico, preferiblemente con al menos un ácido inorgánico seleccionado de entre el grupo que consiste en ácido clorhídrico, ácido bromhídrico, ácido sulfúrico y ácido fosfórico, o con al menos una base inorgánica, preferiblemente con al menos una base inorgánica seleccionada de entre el grupo que consiste en hidróxido de sodio, hidróxido de potasio, carbonato de sodio y carbonato de potasio en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter, agua y mezclas de los mismos para producir (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV).
En otra realización de la presente invención, los agentes de desprotección según la etapa c) del procedimiento de la invención se seleccionan de entre el grupo que consiste en yodotrimetilsilano, etilsulfuro de sodio, yoduro de litio y ácido bromhídrico, preferiblemente ácido bromhídrico.
El compuesto (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol puede estar presente en forma de una sal de adición de ácido, por lo que puede ser usado cualquier ácido adecuado capaz de formar tal sal de adición.
La conversión del compuesto (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol en una sal de adición correspondiente, por ejemplo, a través de la reacción con un ácido adecuado, puede efectuarse de una manera bien conocida por los expertos en la técnica. Los ácidos adecuados incluyen, entre otros, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido metanosulfónico, ácido fórmico, ácido acético, ácido oxálico, ácido succínico, ácido tartárico, ácido mandélico, ácido fumárico, ácido láctico, ácido cítrico, ácido glutámico y/o ácido aspártico. En una realización preferida de la invención, la sal de adición de ácido es la sal clorhidrato.
La formación de la sal se puede efectuar preferiblemente en un disolvente adecuado que incluye éter dietílico, éter diisopropílico, acetatos de alquilo, acetona, 2-butanona o cualquier mezcla de los mismos. También preferiblemente, se puede usar la reacción con trimetilclorosilano en un disolvente adecuado para la preparación de la sal de adición de clorhidrato.
Preferiblemente, se puede obtener un compuesto de fórmula general (I) mediante (a') haciendo reaccionar un compuesto de fórmula general (V),

donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2H5, -C(=O)-CH(CH3)2 o -C(=O)-C(CH3)3 , con clorhidrato de dimetilamina y paraformaldehído en un medio de reacción inerte en condiciones de Mannich y
(a") resolución posterior del compuesto así obtenido de fórmula general (VI),

(VI),
donde R tiene el significado definido anteriormente.
Particularmente preferiblemente, R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3 en los compuestos de fórmulas generales (V) o (VI). Más particularmente
preferiblemente, R representa metilo, bencilo o tetrahidropiranilo en los compuestos de fórmulas generales (V) o (VI). Aún más particularmente preferiblemente, R representa metilo en las fórmulas generales (V) y (VI). Por lo tanto, la 1-(3-metoxifenil)propan-1-ona se convierte en 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Via) con clorhidrato de dimetilamina y paraformaldehído en un medio de reacción inerte en condiciones de Mannich.
Preferiblemente, la resolución en la etapa (a") se realiza haciendo reaccionar un compuesto de fórmula general (VI) con un ácido quiral seleccionado de entre el grupo que consiste en ácido L-(-)-dibenzoil tartárico, ácido L-(-)-dibenzoil tartárico ■ H2O y ácido D-(-)-tartárico, la posterior separación de la sal obtenida de este modo y la liberación del compuesto correspondiente de fórmula general (I) en forma de la base libre.
Se prefiere que la resolución se realice en un medio de reacción alcohólico seleccionado de entre el grupo que consiste en metanol, etanol, 1-propanol, 2-propanol y cualquier mezcla de los mismos o en una mezcla de un medio de reacción alcohólico seleccionado de entre el grupo que consiste en metanol, etanol, 1-propanol, 2-propanol y acetona.
Preferiblemente, la transferencia según la etapa (b) se realiza (b') sometiendo el compuesto de fórmula general (II) a deshidratación y (b") hidrogenación del compuesto de fórmula general (VII) así obtenido,

donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2H5, -C(=O)-CH(CH3)2 o -C(=O)-C(CH3)3 , usando un catalizador adecuado en un medio de reacción inerte en presencia de hidrógeno.
Particularmente preferiblemente, R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3 en los compuestos de fórmula general (II).
Más particularmente preferiblemente, R representa metilo, bencilo o tetrahidropiranilo en los compuestos de fórmula general (II).
Incluso más particularmente preferiblemente, R representa metilo en el compuesto de fórmula general (II). Por tanto, (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol se transfiere a (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina por deshidratación (etapa (b')) y posterior hidrogenación (etapa (b")).
Preferiblemente, la hidrogenación en la etapa (b") se efectúa mediante catálisis homogénea en presencia de hidrógeno después de la etapa de deshidratación (b'). El hidrógeno se encuentra preferiblemente en forma gaseosa, aunque también es posible que al menos una parte del mismo se disuelva en una fase líquida.
Preferiblemente, el catalizador homogéneo usado para la hidrogenación en la etapa (b") según la presente invención es un complejo de metal de transición de rodio, iridio o rutenio, particularmente preferiblemente un complejo de metal de transición de rodio o iridio, más particularmente un complejo de metal de transición de rodio con ligandos de difosfina.
Los ligandos de difosfina que se pueden utilizar preferiblemente son, por ejemplo, conocidos de las siguientes referencias bibliográficas: a) H. Brunner, W. Zettlmeier, Handbook of Enantioselective Catalysis. VCH Weinheim, 1993, vol. 2; b) R. Noyori y col. in Catalytic Asymmetric Synthesis Segunda Edición (I. Ojima, Ed.), Wiley-VCH, Weinheim, 2000; c) E. N. Jacobsen, A. Pfaltz, H. Yamamoto (Eds.), Comprehensive Asymmetric Catalysis Vol I-III, Springer Berlin , 1999, y las referencias allí citadas.
Particularmente preferiblemente, el catalizador se elige de entre el grupo que consiste en rodio (-)-DIPAMP [(R,R)-(-)-1,2-Bis[(2-metoxifenil)(fenil)fosfino]etano], rodio (+)-DIPAMP [(S,S)-(+)-1,2-Bis[(2-metoxifenil)(fenil)fosfino]etano], rodio R-Solphos [R-(+)-N,N'-Dimetil-7,7'-bis(difenilfosfino)-3,3',4,4'-tetrahidro-8,8'-bi-2H-1,4-benzoxazina] y rodio S-Solphos [S-(-)-N,N'-Dimetil-7,7'-bis(difenilfosfino)-3,3',4,4'-tetrahidro-8,8'-bi-2H-1,4-benzoxazina].
Los parámetros de reacción para la hidrogenación homogénea en la etapa (b"), tales como, por ejemplo, presión, temperatura o tiempo de reacción, pueden variar en un amplio intervalo.
Preferiblemente, la temperatura durante la hidrogenación homogénea en la etapa (b") puede ser en cada caso de 0 a 250 °C, particularmente preferiblemente de 10 a 40 °C y muy particularmente preferiblemente de 15 a 25 °C.
La hidrogenación homogénea en la etapa (b") se puede llevar a cabo preferiblemente a presión reducida, a presión normal o a presión elevada, preferiblemente en el intervalo de 0,01 a 300 bar. Se prefiere especialmente llevar a cabo las reacciones bajo presión en un intervalo de 3 a 20 bar, en particular de 8 a 12 bar.
El tiempo de reacción puede variar en función de varios parámetros, tales como, por ejemplo, la temperatura, la presión, la naturaleza del compuesto a reaccionar o las propiedades del catalizador, y puede ser determinado para el procedimiento en cuestión por el experto en la materia utilizando pruebas preliminares.
La etapa de deshidratación (b') se cataliza preferiblemente con ácido. Preferiblemente, el ácido se selecciona de entre el grupo que consiste en ácido fórmico, ácido clorhídrico, ácido acético, ácido sulfúrico, ácido bromhídrico, ácido metanosulfónico o cualquier mezcla de los mismos. Es preferible que el ácido se emplee en una concentración elevada. Particularmente preferiblemente, la concentración del ácido clorhídrico es > 20%, preferiblemente > 30%, particularmente preferiblemente > 35% en peso. Alternativamente, el ácido también se puede utilizar en forma gaseosa.
Los compuestos de fórmula general II y VII usados en la etapa (b') según la presente invención están preferiblemente en fase líquida y para ese fin se mezclan o disuelven preferiblemente en un medio de reacción que es líquido en las condiciones de reacción particulares.
Ejemplos de medios de reacción adecuados son agua, ácido acético, ácido fórmico, tolueno, ácido clorhídrico, ácido sulfúrico, ácido bromhídrico, ácido metanosulfónico o cualquier mezcla de los mismos. Por supuesto, también es posible usar mezclas o sistemas multifásicos que comprenden dos o más de los líquidos mencionados anteriormente en los procedimientos según la presente invención. También es posible una reacción en CO2 supercrítico como disolvente.
Los parámetros de reacción para la hidrogenación homogénea en la etapa (b'), tales como, por ejemplo, presión, temperatura o tiempo de reacción, pueden variar en un amplio intervalo.
Es preferible que la temperatura de reacción en la etapa (b') esté entre 35 y 100 °C, particularmente preferiblemente entre 45 y 80 °C, más particularmente preferiblemente entre 50 y 60 °C.
La etapa de deshidratación (b') se puede llevar a cabo preferiblemente a presión reducida, a presión normal o a presión elevada, preferiblemente en el intervalo de 0,01 a 300 bar. Se prefiere particularmente llevar a cabo las reacciones bajo presión en un intervalo de 0,5 a 5 bar, en particular de 0,5 a 1,5 bar.
El tiempo de reacción puede variar en función de varios parámetros, tales como, por ejemplo, la temperatura, la presión, la naturaleza del compuesto a reaccionar o las propiedades del catalizador, y puede ser determinado para el procedimiento en cuestión por el experto en la materia utilizando pruebas preliminares. Es preferible que el tiempo de reacción de la etapa (b') se encuentre entre 2 y 10 h, con particularmente preferiblemente entre 3 y 8 h, más particularmente preferible entre 4 y 6 h.
También es posible la extracción continua de muestras para controlar la reacción, por ejemplo mediante procedimientos de cromatografía de gases, opcionalmente en combinación con la regulación de los correspondientes parámetros del procedimiento.
La concentración del ácido en el medio de reacción es preferiblemente de 20 a 26 M en el caso de ácido fórmico, de 5 a 18 M en el caso de ácido acético, de 8 a 14 M en el caso de ácido clorhídrico y de 4 a 36 M, más preferiblemente de 4 a 18 M, en caso de ácido sulfúrico.
El compuesto particular de fórmula general (VII) obtenido se puede aislar y/o purificar por procedimientos convencionales conocidos por el experto en la materia.
Alternativamente, la etapa de deshidratación (b') también se puede llevar a cabo en presencia de al menos un catalizador ácido, que preferiblemente se puede seleccionar de entre el grupo que consiste en resinas de intercambio iónico, zeolitas, heteropoliácidos, fosfatos, sulfatos y óxidos metálicos opcionalmente mezclados.
El término catalizador dentro del contexto de la presente invención incluye tanto los propios materiales catalíticamente activos como los materiales inertes que están provistos de un material catalíticamente activo. Por consiguiente, el material catalíticamente activo puede, por ejemplo, aplicarse a un vehículo inerte o puede estar presente en una mezcla con un material inerte. Entran en consideración como portador inerte o material inerte, por ejemplo, carbono y otros materiales conocidos por el experto en la materia.
Catalizadores adecuados y su preparación son conocidos per se por el experto en la materia, por ejemplo de Venuto,
P.B., Microporous Mater., 1994, 2, 297; Holderich, W.F., van Bekkum, H., Stud. Surf. Sci. Catal., 1991, 58, 631, Holderich, W.F., Proceedings of the 10th International Congress on Catalysis, 1992, Budapest, Guczi, L. y col. (editores), "New Frontiers in Catalysis", 1993, Elsevier Science Publishers, Kozhenikov, I.V., Catal. Rev. Sci. Eng., 1995, 37, 311, Song, X., Sayari, A., Catal. Rev. Sci. Eng., 1996, 38, 329. Las descripciones de la bibliografía correspondiente se incorporan en esta invención como referencia y forman parte de la divulgación.
Son adecuadas para la deshidratación, en particular se utilizan resinas de intercambio iónico que portan grupos de ácido sulfónico.
Se da preferencia a las resinas de intercambio iónico basadas en copolímeros de tetrafluoroetileno/perfluoroviniléter, opcionalmente en forma de sus nanocompuestos de sílice, como se describen, por ejemplo, en las publicaciones bibliográficas de Olah y col. Synthesis, 1996, 513-531 y Harmer y col. Green Chemistry, 2000, 7-14, cuyas descripciones correspondientes se incorporan en esta invención como referencia y forman parte de la divulgación. Los productos correspondientes están disponibles comercialmente, por ejemplo bajo el nombre Nafion®, y también se pueden utilizar en esa forma en los procedimientos según la presente invención.
Además, se da preferencia a las resinas de intercambio iónico basadas en copolímeros de estireno/divinilbenceno, que pueden prepararse mediante procedimientos convencionales conocidos por el experto en la materia.
Para la deshidratación entran en consideración de forma especialmente preferente resinas de intercambio iónico que portan grupos sulfonicacidos a base de copolímeros de estireno/divinilbenceno, como se comercializan, por ejemplo, bajo el nombre de Amberlyst® por Rohm & Haas y que también se puede utilizar como tales en los procedimientos según la presente invención. Estas resinas de intercambio iónico se distinguen en particular por su estabilidad frente al agua y los alcoholes, incluso a temperaturas elevadas, por ejemplo de 130 a 160 °C.
El grado de reticulación y la estructura de estas resinas de intercambio iónico pueden variar. Por ejemplo, se pueden citar las resinas de intercambio iónico macroporosas que tienen una distribución heterogénea del diámetro de los poros, las resinas de intercambio iónico isoporosas que tienen una distribución del diámetro de los poros prácticamente uniforme o las resinas de intercambio iónico en forma de gel que no tienen o prácticamente no tienen poros. Las resinas macroporosas en particular se pueden utilizar con especial ventaja para catálisis heterogénea en la fase líquida.
Resinas macroporosas particularmente adecuadas que tienen un diámetro de poro medio de 20 a 30 nm y una concentración mínima de grupos activos de desde 4,70 a 5,45 equivalentes por kg de resina están disponibles comercialmente bajo los nombres Amberlyst® 15, Amberlyst® 35 y Amberlyst® 36 y por consiguiente, también se puede utilizar en procedimientos según la presente invención.
Asimismo, se prefiere realizar la deshidratación en presencia de un catalizador ácido a base de óxidos metálicos como, por ejemplo, SiO2 , AbO3, TiO2 , Nb2O5, B2O3 o en base a mezclas óxidos metálicos como, por ejemplo, AbO3/SiO2 o AbO3/B2O3.
Preferiblemente, la temperatura de deshidratación (b') cuando se usa un catalizador ácido como se describió anteriormente es en cada caso de 20 a 250 °C, particularmente preferiblemente de 50 a 180 °C y muy particularmente preferiblemente de 100 a 160 °C.
La relación de catalizador ácido y compuesto de fórmula general (II) está preferiblemente en el intervalo de 1:200 para 1:1, en particular de 1:4 para 1:2.
Después de la deshidratación, el catalizador se puede separar de la mezcla de reacción de forma sencilla, preferiblemente mediante filtración. El compuesto particular de fórmula general (VII) obtenido se puede aislar y/o purificar por procedimientos convencionales conocidos por el experto en la materia.
Alternativamente, la etapa de deshidratación (b') también se puede llevar a cabo sometiendo un compuesto de fórmula general (II) a un exceso de cloruro de tionilo, opcionalmente en un medio de reacción, preferiblemente en un medio de reacción seleccionado de entre el grupo que consiste en éter dietílico, tetrahidrofurano, tolueno, 2-metiltetrahidrofurano, dioxano, terc-butil-metiléter y mezclas de los mismos, y calentamiento posterior de la mezcla de reacción así obtenida de 40 °C a 120 °C, preferiblemente de 80°C a 120°C.
La hidrogenación de la etapa (b") también se puede realizar mediante catálisis heterogénea con hidrógeno. El hidrógeno se encuentra preferiblemente en forma gaseosa, aunque también es posible que al menos una parte del mismo se disuelva en una fase líquida.
La catálisis heterogénea en el contexto de la presente invención significa que los catalizadores utilizados en la etapa (b") están presentes en cada caso en el estado sólido de agregación.
Preferiblemente, el catalizador heterogéneo usado para la hidrogenación en la etapa (b") según la presente invención contiene uno o más metales de transición, estos metales pueden seleccionarse preferiblemente de entre el grupo que
consiste en Cu, Ag, Au, Zn, Cd, Hg, V, Nb, Ta, Cr, Mo, W, Fe, Ru, Os, Co, Rh, Ir, Ni, Pd, Pt, de forma especialmente preferente de entre el grupo formado por Ru, Rh, Pd, Pt y Ni.
Los catalizadores correspondientes pueden contener preferiblemente uno o más de los metales de transición mencionados anteriormente en el mismo o en diferentes estados de oxidación. También puede ser preferible que los catalizadores correspondientes contengan uno o más de los metales de transición mencionados anteriormente en dos o más estados de oxidación diferentes.
La preparación de catalizadores dopados con metales de transición se puede llevar a cabo mediante procedimientos convencionales conocidos por el experto en la materia.
Preferiblemente, el catalizador usado para la hidrogenación en la etapa (b") se selecciona de entre el grupo que consiste en níquel Raney, paladio, paladio sobre carbono (1 - 10 % en peso, preferiblemente 5 % en peso), platino, platino sobre carbono (1-10 % en peso, preferiblemente 5 % en peso), rutenio sobre carbono (1-10 % en peso, preferiblemente 5 % en peso) y rodio sobre carbono (1 - 10 % en peso, preferiblemente 5 % en peso), más preferiblemente paladio sobre carbono (1-10 % en peso, preferiblemente 5 % en peso) se utiliza como catalizador para la hidrogenación en el etapa (b").
Los compuestos de fórmula general VII y III usados en la etapa (b") según la presente invención están preferiblemente en fase líquida y para ese fin se mezclan o disuelven preferiblemente en un medio de reacción que es líquido en las condiciones de reacción particulares.
Ejemplos de medios de reacción adecuados son metanol, etanol, isopropanol, n-butanol, n-propanol, tolueno, heptano, hexano, pentano, ácido acético, acetato de etilo, ácido fórmico, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico y mezclas de los mismos. Más preferiblemente, se usa etanol como medio de reacción en la etapa (b"). Por supuesto, también es posible usar mezclas o sistemas multifásicos que comprenden dos o más de los líquidos mencionados anteriormente en los procedimientos según la presente invención. También es posible una reacción en CO2 supercrítico como disolvente.
Los parámetros de reacción para la hidrogenación heterogénea en la etapa (b"), tales como, por ejemplo, presión, temperatura o tiempo de reacción, pueden variar en un amplio intervalo ambos.
Preferiblemente, la temperatura durante la hidrogenación heterogénea en la etapa (b") puede ser en cada caso de 0 a 250 °C, particularmente preferiblemente de 15 a 180 °C y muy particularmente preferiblemente de 15 a 30 °C.
La hidrogenación heterogénea en la etapa (b") se puede llevar a cabo preferiblemente a presión reducida, a presión normal o a presión elevada, preferiblemente en el intervalo de 1 a 300 bar. Se prefiere particularmente llevar a cabo las reacciones bajo presión en un intervalo de 2 a 10 bar, en particular de 4 a 10 bar.
El tiempo de reacción puede variar en función de varios parámetros, tales como, por ejemplo, la temperatura, la presión, la naturaleza del compuesto a reaccionar o las propiedades del catalizador, y puede ser determinado para el procedimiento en cuestión por el experto en la materia utilizando pruebas preliminares.
También es posible la extracción continua de muestras para controlar la reacción, por ejemplo mediante procedimientos de cromatografía de gases, opcionalmente en combinación con la regulación de los correspondientes parámetros de procedimiento.
La cantidad total del catalizador o catalizadores usados depende de varios factores, tales como, por ejemplo, la relación del componente catalíticamente activo a cualquier material inerte presente, o la naturaleza de la superficie del catalizador o catalizadores. La cantidad óptima de catalizador i catalizadores para una reacción particular puede ser determinada por el experto en la materia usando pruebas preliminares.
El compuesto particular de fórmula general (III) obtenido se puede aislar y/o purificar por procedimientos convencionales conocidos por el experto en la materia.
En otra realización de la invención, la etapa b) (esquema 1) es una reacción de sustitución directa del grupo OH por H, preferiblemente llevada a cabo en una reacción en un solo recipiente. Más preferiblemente, un OH' se reemplaza por H-.
Las etapas según la presente invención pueden realizarse cada una de forma discontinua (por lotes) o de forma continua, dando preferencia al procedimiento discontinuo.
Entran en consideración como reactor para el procedimiento discontinuo, por ejemplo, un reactor de suspensión, y para el procedimiento continuo un reactor de lecho fijo o reactor de bucle.
A continuación se describe un procedimiento para la preparación de clorhidrato de (1R,2R)-3-(3-dimetilamino-1-etil-2metil-propil)-fenol.
Ejemplo
Preparación de clorhidrato de (1R,2R)-3-(3-Dimetilamino-1-etil-2-metil-propil)-fenol

Etapa (a'): Preparación de 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Via)
1-(3-metoxifenil)propan-1-ona (16,42 kg, 100 mol), clorhidrato de dimetilamina (8,97 kg, 110 mol), paraformaldehído (3,30 kg, 110 mol) y ácido clorhídrico acuoso (32 % en peso, 1,14 kg) se disolvieron en etanol bajo atmósfera de nitrógeno en un recipiente de 100 L (L = litro) de doble camisa equipado con un agitador impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento. La mezcla de reacción se calentó a reflujo durante 16 horas, se enfrió a 25 °C en 3,5 horas y se agitó durante 1 hora a esa temperatura. La suspensión se separó mediante una centrífuga y se lavó tres veces con 7 L de acetona cada una. Se disolvió clorhidrato de 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona en agua (12,5 L) y terc-butil-metil-éter (8,5 L) y se agitó a temperatura ambiente.
Se añadió solución acuosa de hidróxido de sodio (32 % en peso) hasta que se alcanzó un valor de pH entre 10,0 y 10,5 y se dejó que las fases se separaran. La fase orgánica se eliminó por destilación a presión reducida hasta que, a una temperatura de 40 °C, se alcanzó una presión de 5 mbar. Se obtuvo 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona como un aceite amarillo pálido (20,75 kg, 94%) que se utilizó en la siguiente etapa sin más purificación.
Etapa (a"): Preparación de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (Ia)
1. a. Preparación de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (2R,3R)-O,O'-dibenzoiltartrato en acetona
Se disolvió monohidrato de ácido (2R,3R)-O,O’-dibenzoil tartárico (189,1 g, 0,5 mol) en acetona (550 mL) en una planta de reacción de 2 L equipada con un agitador mecánico, equipo de medición de temperatura y un baño de aceite y se añadió 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (110,6 g, 0,5 mol). La mezcla de reacción se calentó de 35 °C a 40 °C durante 27 horas y se dejó enfriar a 25 °C. La suspensión se separó por sifón y se obtuvo (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (2R,3R)-O,O'-dibenzoiltartrato como un sólido incoloro ( 233,2 g, 80,5 %, ee 96,9 %, ee = exceso enantiomérico).
1. b. Preparación de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (2R,3R)-O,O'-dibenzoiltartrato en acetona/metanol
Se disolvió monohidrato de ácido (2R,3R)-O,O'-dibenzoil tartárico (2,1 kg, 5,5 mol) en una mezcla de metanol (555 mL) y acetona (3340 ml) en un recipiente de doble camisa de 10 L equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento basado en aceite y se añadió 3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (1,23 kg, 5,56 mol). La mezcla de reacción se calentó de 35 °C a 40 °C durante 24 horas y se dejó enfriar a 25 °C. La suspensión se separó por sifón y se obtuvo (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (2R,3R)-O,O'-dibenzoiltartrato como un sólido incoloro ( 2,38 kg, 74%, ee 98,4%).
2. Preparación de (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (la)
(S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (2R,3R) -O,O'-dibenzoiltartrato (968 g, 1,67 mmol, ee 98 %) se suspendió en terc-butilmetiléter (6 L) en un recipiente de doble camisa de 10 L equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento basado en aceite y de añadió dietilamina (384 g, 5,25 mol). La mezcla de reacción se agitó de 20 °C a 25 °C durante 90 minutos y se separó por sifón un sólido. El filtrado se concentró a una temperatura de 40 °C al vacío hasta que se alcanzó una presión de 4 mbar. Se obtuvo (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona como un aceite incoloro (356,7 g, 96,5 %, ee 98 %).
Etapa (a): Preparación de (2S,3R)-1-(dimeilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol (Ila)
1. Se suspendieron virutas de magnesio (93,57 g, 3,85 mol) en éter etílico seco (2 L) en un recipiente de doble camisa de 10 L equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de refrigeración/calentamiento basado en aceite. y se añadió bromuro de etilo (25 g, 0,23 mol). Una vez iniciada la reacción, se añadió más bromuro de etilo (438,6 g, 4,02 mol) en un plazo de 90 minutos por debajo de una temperatura de 35 °C y la mezcla de reacción se agitó durante otra hora. La mezcla de reacción se enfrió de 10 °C a 15 °C, (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (774,6 g, 3,5 mol, ee 98 %) se disolvió en éter dietílico (0,8 L) y la mezcla de reacción se agitó durante otras dos horas. La mezcla de reacción se enfrió a 5 °C y se añadió una solución acuosa de hidrogenosulfato de amonio (10 % en peso, 2 L). Las fases se separaron y la fase orgánica se concentró al vacío a 40 °C hasta que se alcanzó una presión de 5 mbar. Se obtuvo (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol (862,3 g, 98 %) como un aceite incoloro (ee 98 %).
2. (S)-3-(dimetilamino)-1-(3-metoxifenil)-2-metilpropan-1-ona (774,6 g, 3,5 mol, ee 95 %) se disolvió en tetrahidrofurano seco (800 mL) en un recipiente de doble camisa de 10 L equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento basado en aceite y se añadió bromuro de etil magnesio (2 l, 2 M en THF) a una temperatura de 15 °C en 2 horas. La mezcla de reacción se agitó durante dos horas a esa temperatura, se enfrió a 5 °C y se añadió una solución acuosa de hidrogenosulfato de amonio (10 % en peso, 2 L). Las fases se separaron y la fase orgánica se concentró al vacío a 40 °C hasta que se alcanzó una presión de 5 mbar. Se obtuvo (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol (871,1 g, 99 %) como un aceite incoloro (ee 95 %).
Etapa (b'): Preparación de (R)-3-(3-metoxifenil)-N,N,2-trimetilpent-3-en-1-amina (Vila)
1. Se disolvieron (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol (754,1 g, 3 mol, ee 95 %) en acetona (5 L) en un recipiente de doble camisa de 10 L equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento basado en aceite. Se transfirió cloruro de hidrógeno (110 g, 3,0 mol) en 15 minutos a una temperatura de 15 °C a través de la mezcla de reacción. La mezcla de reacción se enfrió de 0 °C a 5 °C y después de 24 horas a esa temperatura se extrajo por sifón. El producto se almacenó a 40 °C y 10 mbar durante 14 horas en un horno de secado. Se obtuvo clorhidrato de (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol como un sólido incoloro (722,3 g, 83,7 %, ee 100 %).
2. El clorhidrato de (2S,3R)-1-(dimetilamino)-3-(3-metoxifenil)-2-metilpentan-3-ol obtenido como se describió anteriormente se puso en un matraz de tres bocas de 250 mL equipado con un termómetro, un agitador mecánico de aire comprimido, condensador de reflujo y baño de aceite y solución acuosa de cloruro de hidrógeno (150 mL, 36 % en peso). La mezcla de reacción se calentó a 55 °C durante 5 h y se dejó enfriar a 20 °C. Se añadió solución acuosa de hidróxido de sodio (33 % en peso) mientras se enfriaba hasta que se alcanzó un valor de pH de 11. Se añadió acetato de etilo (150 mL), la mezcla de reacción se agitó durante 10 minutos, las fases se separaron y el acetato de etilo se eliminó al vacío a 60 °C hasta que se alcanzó una presión de 10 mbar. Se obtuvo (R)-3-(3-metoxifenil)-N,N,2-trimetilpent-3-en-1-amina (21 g, 90 %) como un residuo aceitoso (Proporción Z/E 4,5:1).
Etapa (b"): Preparación de clorhidrato de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (Mía)
1. Se disolvió (R)-3-(3-metoxifenil)-N, N,2-trimetilpent-3-en-1-amina (5 kg, 21,43 mmol) en etanol seco (13 L) a una temperatura de 25 °C y frecuencia de agitación rotacional de 850 ± 150 por minuto en un aparato de hidrogenación de doble camisa equipado con una tapa montada estacionaria que tiene un suministro de hidrógeno y nitrógeno, agitador de gasificación eléctrico, equipo de medición de temperatura Pt100, vidrio de inspección y controlador de gas "Büchi bpc". El aparato de hidrogenación se llenó con nitrógeno. Se suspendió paladio sobre carbón vegetal (375 g, 5 % en peso) en cloruro de hidrógeno acuoso (675 g, 32 % en peso) y se añadió a la mezcla de reacción. El aparato de hidrogenación se llenó de nuevo con nitrógeno y la reacción se llevó a cabo a una presión primaria de hidrógeno de 5 bar y una presión interna de hidrógeno de 1 bar hasta que se completó la reacción. El aparato de hidrogenación se llenó con nitrógeno y el catalizador se filtró en un filtro de una capa con tierra filtrante. El filtrado se concentró al vacío. El residuo se recogió en acetato de etilo y se añadió hidróxido de sodio acuoso (10 %
en peso, 3,7 L) a 20 °C hasta que se alcanzó un valor de pH de 10 a 12. La fase orgánica se concentró al vacío de 45 °C a 50 °C hasta que se alcanzó una presión de 5 mbar. El residuo aceitoso era una mezcla de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina y (2R,3S)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (4,5 kg, 95 %, relación 5,5 (R,R):1 (R,S)).
2. Una mezcla de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina y (2R,3S)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (10 kg, 42,56 mol, relación 5,5:1) se disolvió en acetona (50 L) en un recipiente de doble camisa de 100 L equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento basado en aceite. Se transfirió cloruro de hidrógeno (1,55 kg, 42,51 mol) en 15 minutos a una temperatura de 5 °C a 25 °C a través de la mezcla de reacción. La mezcla de reacción se enfrió de 0 °C a 5 °C y se centrifugó después de 2 horas de agitación. El sólido húmedo se puso en un recipiente con agitación, se añadió acetona (30 L) y la mezcla de reacción se calentó a reflujo durante 15 minutos. Después de enfriar a 15 °C a 20 °C, el producto se centrifugó y se almacenó a 40 °C a 50 °C y 150 mbar durante 14 horas en un horno de secado. Se obtuvo clorhidrato de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (7,17 kg, 63 %) como un sólido incoloro con un exceso diastereomérico de 100 %.
Etapa (c): Preparación de clorhidrato de (1R,2R)-3-(3-Dimetilamino-1-etil-2-metil-propil)-fenol (IV)
1. Se disolvió clorhidrato de (2R,3R)-3-(3-metoxifenil)-N,N,2-trimetilpentan-1-amina (5 kg, 18,4 mol) en ácido metanosulfónico (19,5 L) en un recipiente de doble de 100 l. recipiente de doble camisa equipado con un agitador de impulsor eléctrico, una línea de transición de gas, equipo de medición de temperatura Pt100 y un sistema de enfriamiento/calentamiento basado en aceite y se añadió metionina (3,35 kg, 22,5 mol). La mezcla de reacción se agitó a una temperatura de 75 °C a 80 °C durante 16 horas, se enfrió de 15 °C a 25 °C y se añadió lentamente agua (12,5 L) a esa temperatura. Se añadió solución acuosa de hidróxido de sodio (ca.28 L, 32 % en peso) hasta que se alcanzó un valor de pH de 10 a 12 mientras que la temperatura se mantuvo por debajo de 50 °C. Se añadió acetato de etilo (15 L) y la mezcla de reacción se agitó durante 15 minutos a una frecuencia de agitación rotacional de 150 por minuto. Se separaron las fases y se lavó la fase orgánica con agua (15 L). Se añadió carbón activado (0,05 kg) a la fase orgánica y se filtró después de 30 minutos de agitación. El disolvente se eliminó al vacío a una temperatura de 40 °C a 50 °C hasta que se alcanzó una presión de 50 mbar. El residuo se usó en la siguiente etapa sin purificación adicional.
2. El residuo obtenido como se describió anteriormente se disolvió en acetona (25 L) mientras se agitaba y se transfirió cloruro de hidrógeno (0,78 kg, 21,4 mol) a través de la mezcla de reacción a una temperatura de 20 °C a 25 °C. La suspensión se agitó durante 3 horas a una temperatura de 0 °C a 5 °C y se centrifugó. Se añadió isopropanol (35 L) al sólido húmedo en un recipiente de reacción y la mezcla de reacción se calentó a reflujo durante 15 minutos. La mezcla de reacción se enfrió de 0 °C a 5 °C y se agitó durante 3 horas a esa temperatura. Después de la centrifugación, el producto se almacenó entre 30 °C y 40 °C y 150 mbar durante 16 horas en un horno de secado. Se obtuvo clorhidrato de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol (4,18 kg, 88 %) como un sólido incoloro con una pureza del 100 %.
Claims (19)
1. Un procedimiento para preparar (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol, o una sal de adición de ácido del mismo, que comprende la etapa de
(a) hacer reaccionar un compuesto de fórmula general (I),

donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2Hs, -C(=O)-CH(CHs)2 o -C(=O)-C(CH3)3,
con haluro de etil magnesio en un medio de reacción inerte en condiciones de Grignard,
(b) transferir el compuesto de fórmula general (II) así obtenido,

donde R tiene el significado definido anteriormente,
a un compuesto de fórmula general (III),

donde R tiene el significado definido anteriormente,
opcionalmente en forma de sal de adición de ácido,
(c) desproteger el compuesto así obtenido de fórmula general (III) para obtener (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol de fórmula (IV),

(d) convertir opcionalmente el (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol así obtenido en una sal de adición de ácido.
2. Un procedimiento según la reivindicación 1, caracterizado porque R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3.
3. Un procedimiento según cualquiera de las reivindicaciones 1 ó 2, caracterizado porque R representa metilo, bencilo o tetrahidropiranilo.
4. Un procedimiento según cualquiera de las reivindicaciones 1 a 3, caracterizado porque el haluro de etil magnesio usado en la etapa (a) es el cloruro o bromuro.
5. Un procedimiento según una cualquiera de las reivindicaciones 1 a 4, caracterizado porque el medio de reacción inerte se selecciona de entre el grupo que consiste en éter dietílico, tetrahidrofurano, 2-metiltetrahidrofurano, terc-butilmetiléter, diisopropiléter o cualquier mezcla de los mismos.
6. Un procedimiento según una cualquiera de las reivindicaciones 1 a 5, caracterizado porque se obtuvo un compuesto de fórmula general (I) mediante (a') haciendo reaccionar un compuesto de fórmula general (V),
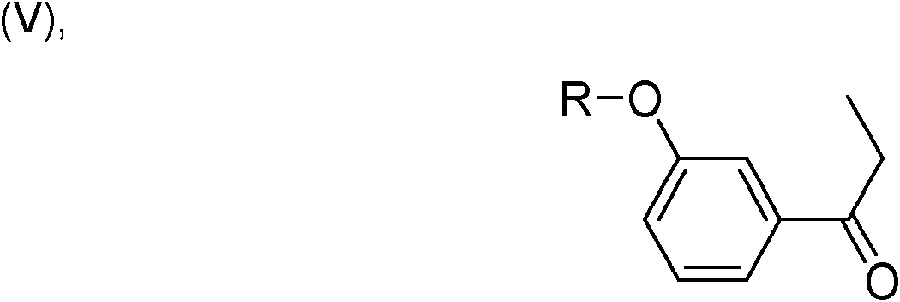
donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2H5, -C(=O)-CH(CH3)2 o -C(=O)-C(CH3)3,
con clorhidrato de dimetilamina y paraformaldehído en un medio de reacción inerte en condiciones de Mannich y (a") resolución posterior del compuesto así obtenido de fórmula general (Vi),

(VI),
donde R tiene el significado definido anteriormente.
7. Un procedimiento según la reivindicación 6, caracterizado porque R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3.
8. Un procedimiento según cualquiera de las reivindicaciones 6 ó 7, caracterizado porque R representa metilo, bencilo o tetrahidropiranilo.
9. Un procedimiento según cualquiera de las reivindicaciones 6 a 8, caracterizado porque la resolución en la etapa (a") se realiza haciendo reaccionar un compuesto de fórmula general (VI) con un ácido quiral seleccionado de entre el grupo que consiste en ácido L-(-)-dibenzoil tartárico, ácido L-(-)-dibenzoil tartárico ■ H2O y ácido D-(-)-tartárico, la posterior separación de la sal obtenida de este modo y la liberación del compuesto correspondiente de fórmula general (I) en forma de la base libre.
10. Un procedimiento según la reivindicación 9, caracterizado porque la resolución se realiza en un medio de reacción alcohólico seleccionado de entre el grupo que consiste en metanol, etanol, 1-propanol, 2-propanol y cualquier mezcla de los mismos.
11. Un procedimiento según cualquiera de las reivindicaciones 1 a 10, caracterizado porque la transferencia según la etapa (b) se realiza (b') sometiendo el compuesto de fórmula general (II) a deshidratación y (b") hidrogenación del así obtenido compuesto de fórmula general (VII),

donde R representa metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, terc-butilo, n-pentilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, bencilo, fenetilo, tetrahidropiranilo, -C(=O)-CH3, -C(=O)-C2H5, -C(=O)-CH(CH3)2 o -C(=O)-C(CH3)3,
usando un catalizador adecuado en un medio de reacción inerte en presencia de hidrógeno.
12. Un procedimiento según la reivindicación 11, caracterizado porque R representa metilo, etilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, bencilo, fenetilo, tetrahidropiranilo o -C(=O)-CH3.
13. Un procedimiento según cualquiera de las reivindicaciones 11 ó 12, caracterizado porque R representa metilo, bencilo o tetrahidropiranilo.
14. Un procedimiento según cualquiera de las reivindicaciones 11 a 13, caracterizado porque después de la etapa de deshidratación (b') se efectúa la hidrogenación en la etapa (b") mediante catálisis homogénea.
15. Un procedimiento según cualquiera de las reivindicaciones 11 a 13, caracterizado porque la etapa de deshidratación (b') es catalizada por ácido.
16. Un procedimiento según la reivindicación 15, caracterizado porque el ácido se selecciona de entre el grupo que consiste en ácido fórmico, ácido clorhídrico, ácido sulfúrico, ácido metanosulfónico, ácido bromhídrico o cualquier mezcla de los mismos.
17. Un procedimiento según cualquiera de las reivindicaciones 11 a 13, caracterizado porque la hidrogenación de la etapa (b") se efectúa mediante catálisis heterogénea.
18. Procedimiento según la reivindicación 17, caracterizado porque el catalizador utilizado para la hidrogenación se selecciona de entre el grupo que consiste en níquel Raney, paladio, paladio sobre carbono, platino, platino sobre carbono, rutenio sobre carbono o rodio sobre carbono.
19. Un procedimiento según una cualquiera de las reivindicaciones 11 a 18, caracterizado porque el medio de reacción se selecciona de entre el grupo que consiste en éter dietílico, tetrahidrofurano, 2-metiltetrahidrofurano, tercbutil-metiléter, diisopropiléter o cualquier mezcla de los mismos.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06015338 | 2006-07-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2901953T3 true ES2901953T3 (es) | 2022-03-24 |
Family
ID=37198995
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES21163457T Active ES2961956T3 (es) | 2006-07-24 | 2007-07-23 | Procedimiento para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol |
| ES17161440T Active ES2901953T3 (es) | 2006-07-24 | 2007-07-23 | Procedimiento de preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol |
| ES07786260.5T Active ES2524252T3 (es) | 2006-07-24 | 2007-07-23 | Proceso para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)fenol |
| ES11004087.0T Active ES2629342T3 (es) | 2006-07-24 | 2007-07-23 | Proceso para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)fenol |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES21163457T Active ES2961956T3 (es) | 2006-07-24 | 2007-07-23 | Procedimiento para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES07786260.5T Active ES2524252T3 (es) | 2006-07-24 | 2007-07-23 | Proceso para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)fenol |
| ES11004087.0T Active ES2629342T3 (es) | 2006-07-24 | 2007-07-23 | Proceso para la preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metilpropil)fenol |
Country Status (26)
| Country | Link |
|---|---|
| US (7) | US8877974B2 (es) |
| EP (4) | EP3868739B1 (es) |
| JP (1) | JP5612308B2 (es) |
| KR (1) | KR101423996B1 (es) |
| CN (1) | CN101495445B (es) |
| AU (1) | AU2007278464C1 (es) |
| BR (1) | BRPI0714558B8 (es) |
| CA (1) | CA2657104C (es) |
| CO (1) | CO6160221A2 (es) |
| CY (3) | CY1115651T1 (es) |
| DK (3) | DK2368871T3 (es) |
| ES (4) | ES2961956T3 (es) |
| HR (3) | HRP20211902T1 (es) |
| HU (2) | HUE034253T2 (es) |
| IL (3) | IL196049A (es) |
| LT (2) | LT3210965T (es) |
| MX (1) | MX2009000667A (es) |
| NO (2) | NO341939B1 (es) |
| NZ (1) | NZ573911A (es) |
| PL (3) | PL2046724T3 (es) |
| PT (3) | PT2046724E (es) |
| RU (1) | RU2466124C2 (es) |
| SI (3) | SI2046724T1 (es) |
| TW (2) | TWI496762B (es) |
| WO (1) | WO2008012047A1 (es) |
| ZA (1) | ZA200900136B (es) |
Families Citing this family (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI496762B (zh) | 2006-07-24 | 2015-08-21 | 製備(1r,2r)-3-(3-二甲胺基-1-乙基-2-甲基-丙基)-酚之方法 | |
| US20100272815A1 (en) * | 2009-04-28 | 2010-10-28 | Actavis Group Ptc Ehf | Amorphous form of tapentadol hydrochloride |
| CN102002065B (zh) * | 2009-09-02 | 2014-09-10 | 上海特化医药科技有限公司 | 他喷他多的制备方法及其中间体 |
| US8288592B2 (en) | 2009-09-22 | 2012-10-16 | Actavis Group Ptc Ehf | Solid state forms of tapentadol salts |
| IT1397189B1 (it) | 2009-12-01 | 2013-01-04 | Archimica Srl | Nuovo processo per la preparazione di tapentadol e suoi intermedi. |
| WO2011080756A1 (en) * | 2009-12-29 | 2011-07-07 | Ind-Swift Laboratories Limited | Process for the preparation of 1-phenyl-3-dimethylaminopropane derivatives |
| PL2519100T3 (pl) * | 2009-12-29 | 2017-09-29 | Mapi Pharma Limited | Związki pośrednie i sposoby wytwarzania tapentadolu i pokrewnych związków |
| EP2542521A2 (en) | 2010-03-05 | 2013-01-09 | Actavis Group Ptc Ehf | Improved resolution methods for isolating desired enantiomers of tapentadol intermediates and use thereof for the preparation of tapentadol |
| CA2793948A1 (en) | 2010-04-05 | 2011-10-20 | Actavis Group Ptc Ehf | Novel process for preparing highly pure tapentadol or a pharmaceutically acceptable salt thereof |
| EP2383255A1 (en) | 2010-04-28 | 2011-11-02 | Lacer, S.A. | New compounds, synthesis and use thereof in the treatment of pain |
| ITMI20100924A1 (it) * | 2010-05-21 | 2011-11-22 | Fidia Farmaceutici | Nuovo metodo di sintesi delle due forme enantiomeriche del tapentadol |
| CN103168025B (zh) | 2010-06-15 | 2015-02-25 | 格吕伦塔尔有限公司 | 用于制备取代的3-(1-氨基-2-甲基戊烷-3-基)苯基化合物的方法 |
| IT1401109B1 (it) * | 2010-07-02 | 2013-07-12 | Archimica Srl | Nuovo processo per la preparazione di tapentadol e suoi intermedi. |
| CN110015955A (zh) | 2010-07-23 | 2019-07-16 | 格吕伦塔尔有限公司 | 3-(3-二甲基氨基-1-乙基-2-甲基-丙基)-苯酚的盐或共晶体 |
| US8853456B2 (en) | 2010-08-16 | 2014-10-07 | Indoco Remedies Limited | Process for the preparation of tapentadol |
| CN101948397A (zh) * | 2010-09-07 | 2011-01-19 | 天津泰普药品科技发展有限公司 | 镇痛药他喷他多重要中间体的制备方法 |
| CZ2010995A3 (cs) * | 2010-12-30 | 2012-04-11 | Zentiva, K.S. | Zpusob výroby (2R,3R)-N,N-dimethyl-3-(3-hydroxyfenyl)-2-methylpentylaminu |
| CZ302992B6 (cs) * | 2010-12-30 | 2012-02-08 | Zentiva, K.S. | Zpusob výroby (2R,3R)-N,N-dimethyl-3-(3-hydroxyfenyl)-2-methylpentylaminu (tapentadolu) |
| EP2545028A4 (en) | 2011-01-27 | 2013-07-03 | Symed Labs Ltd | STEREOSPECIFIC SYNTHESIS OF (-) (2S, 3S) -1-DIMETHYLAMINO-3- (3-METHOXYPHENYL) -2-METHYLPENTAN-3-OLE |
| CN102617501A (zh) * | 2011-01-31 | 2012-08-01 | 中国科学院上海药物研究所 | 取代正戊酰胺类化合物、其制备方法及用途 |
| CN102206164A (zh) * | 2011-04-11 | 2011-10-05 | 中国药科大学 | 一种他喷他多中间体的制备方法 |
| WO2012146978A2 (en) | 2011-04-28 | 2012-11-01 | Actavis Group Ptc Ehf | A novel process for the preparation of tapentadol or a pharmaceutically acceptable salt thereof |
| EP2530072A1 (en) | 2011-06-03 | 2012-12-05 | Lacer, S.A. | New compounds, synthesis and use thereof in the treatment of pain |
| WO2013105109A1 (en) | 2011-11-09 | 2013-07-18 | Indoco Remedies Limited | Process for the preparation of tapentadol |
| WO2013090161A1 (en) | 2011-12-12 | 2013-06-20 | Boehringer Ingelheim International Gmbh | Stereoselective synthesis of tapentadol and its salts |
| CN102557851B (zh) * | 2011-12-13 | 2014-03-26 | 安徽省新星药物开发有限责任公司 | 一种盐酸他喷他多及其类似物的合成方法 |
| WO2013120466A1 (en) | 2012-02-17 | 2013-08-22 | Zentiva, K.S. | A new solid form of tapentadol and a method of its preparation |
| EP2674414A1 (en) | 2012-06-15 | 2013-12-18 | Siegfried AG | Method for the preparation of 1-aryl-1-alkyl-3-dialkylaminopropane compounds |
| CN103159633B (zh) * | 2012-07-06 | 2015-08-12 | 江苏恩华药业股份有限公司 | 他喷他多的制备方法及用于制备他喷他多的化合物 |
| CN102924303B (zh) * | 2012-10-31 | 2013-11-20 | 合肥市新星医药化工有限公司 | 盐酸他喷他多晶型c及其制备方法和应用 |
| CN103553940A (zh) * | 2013-11-07 | 2014-02-05 | 南京艾德凯腾生物医药有限责任公司 | 一种新晶型盐酸他喷他多的制备方法 |
| IN2013MU03670A (es) | 2013-11-21 | 2015-07-31 | Unimark Remedies Ltd | |
| EP3083551B1 (en) | 2013-12-16 | 2018-02-14 | Farma GRS, d.o.o. | Crystalline form of tapentadol intermediate |
| CN104803861B (zh) * | 2014-01-27 | 2017-05-24 | 上海博邦医药科技有限公司 | 一种合成盐酸他喷他多的方法 |
| CZ307492B6 (cs) | 2014-02-04 | 2018-10-17 | Zentiva, K.S. | Pevná forma maleátu tapentadolu a způsob její přípravy |
| CN106278915A (zh) * | 2015-05-19 | 2017-01-04 | 重庆圣华曦药业股份有限公司 | 一种制备他喷他多中间体的方法 |
| EP3377049B1 (en) | 2015-11-17 | 2021-01-06 | MSN Laboratories Private Limited | Crystalline forms of tapentadol salts and process for preparation thereof |
| CN106674029B (zh) * | 2016-12-23 | 2018-08-21 | 四川久凌制药科技有限公司 | 一种他喷他多中间体的制备方法 |
| SK812017A3 (sk) | 2017-08-18 | 2019-03-01 | Saneca Pharmaceuticals A. S. | Spôsob prípravy tapentadolu a jeho medziproduktov |
| CN112262121A (zh) * | 2018-06-15 | 2021-01-22 | 法尔玛赞公司 | 用于制备他喷他多的新方法 |
| WO2020194326A1 (en) | 2019-03-28 | 2020-10-01 | Council Of Scientific And Industrial Research | Process for the preparation of tapentadol and analogs thereof |
| EP4116288A1 (en) | 2021-07-08 | 2023-01-11 | KRKA, d.d., Novo mesto | Racemization of (s) and/or (r)-3-(dimethylamino)-1-(3-methoxyphenyl)-2-methylpropan-1- one and its mixtures |
| EP4269384A1 (en) | 2022-04-26 | 2023-11-01 | KRKA, d.d., Novo mesto | Method for crystallizing tapentadol intermediate, tapentadol intermediate of high purity, method of making tapentadol and tapentadol of high purity |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE124521C (es) | ||||
| DE107260C (es) | ||||
| AT322534B (de) | 1972-03-16 | 1975-05-26 | Thomae Gmbh Dr K | Verfahren zur herstellung von neuen optisch aktiven 1-(4'-amino-3'-halogenphenyl)-2-aminoäthanolen und deren säureadditionssalzen |
| US4012456A (en) * | 1974-11-19 | 1977-03-15 | Chaplits Donat N | Method for separation of isobutylene from C4 hydrocarbon fractions |
| DD124521A1 (es) | 1976-03-16 | 1977-03-02 | ||
| US4276195A (en) * | 1979-12-20 | 1981-06-30 | Iowa State University Research Foundation, Inc. | Converting homogeneous to heterogeneous catalysts |
| US6022637A (en) * | 1984-10-23 | 2000-02-08 | Wilson; John T. R. | High temperature battery |
| JPH0690934B2 (ja) * | 1987-08-07 | 1994-11-14 | 日本電信電話株式会社 | 二次電池およびその製造方法 |
| US5568594A (en) | 1991-04-08 | 1996-10-22 | Kabushiki Kaisha Toshiba | Image forming apparatus with improved ability to emulate other image forming apparatuses |
| US6313240B1 (en) | 1993-02-22 | 2001-11-06 | Tosoh Corporation | Process for producing ethylene/α-olefin copolymer |
| DE59410267D1 (de) * | 1993-02-26 | 2003-05-15 | Syngenta Participations Ag | Ferrocenyldiphosphine als Liganden für homogene Katalysatoren |
| EP0646590B1 (de) * | 1993-10-01 | 1999-08-25 | Novartis AG | Mit Fluoralkyl substituierte Ferrocenyldiphosphine als Liganden für homogene Katalysatoren |
| JPH07326372A (ja) | 1994-05-30 | 1995-12-12 | Yuasa Corp | 固体電解質電池 |
| DE4426245A1 (de) * | 1994-07-23 | 1996-02-22 | Gruenenthal Gmbh | 1-Phenyl-3-dimethylamino-propanverbindungen mit pharmakologischer Wirkung |
| IL117114A (en) | 1995-02-21 | 2000-02-17 | Montell North America Inc | Components and catalysts for the polymerization ofolefins |
| EP0729969B1 (de) * | 1995-02-24 | 2000-08-16 | Novartis AG | Silylierte Ferrocenyldiphosphine, an anorganische oder polymere organische Träger gebundene silylierte Ferrocenyldiphosphine sowie Metallkomplexe davon, ihre Herstellung und Verwendung |
| EP0728768B1 (de) | 1995-02-24 | 2002-08-28 | Syngenta Participations AG | An Seitenketten von Polymeren gebundene Diphosphine und deren Metall-Komplexe |
| DE19609847A1 (de) | 1996-03-13 | 1997-09-18 | Gruenenthal Gmbh | Dimethyl-(3-aryl-but-3-enyl)-aminverbindungen als pharmazeutische Wirkstoffe |
| US6337156B1 (en) * | 1997-12-23 | 2002-01-08 | Sri International | Ion battery using high aspect ratio electrodes |
| JPH11345629A (ja) * | 1998-03-31 | 1999-12-14 | Canon Inc | 二次電池及びその製造方法 |
| JPH11345623A (ja) | 1998-06-01 | 1999-12-14 | Aisin Seiki Co Ltd | 燃料電池装置 |
| DE10000311A1 (de) * | 2000-01-05 | 2001-07-12 | Gruenenthal Gmbh | Aminomethyl-Phonyl-Cyclohexanonderivate |
| GB0010437D0 (en) | 2000-04-28 | 2000-06-14 | Darwin Discovery Ltd | Process |
| DE10049481A1 (de) | 2000-09-29 | 2002-05-02 | Gruenenthal Gmbh | Substituierte C-Cyclohexylmethylamin-Derivate |
| JP4951809B2 (ja) | 2000-11-21 | 2012-06-13 | 日油株式会社 | 二次電池用電解質および二次電池 |
| JP4777593B2 (ja) | 2002-11-29 | 2011-09-21 | 株式会社オハラ | リチウムイオン二次電池の製造方法 |
| JP4366101B2 (ja) * | 2003-03-31 | 2009-11-18 | キヤノン株式会社 | リチウム二次電池 |
| WO2004089920A2 (de) | 2003-04-07 | 2004-10-21 | Solvias Ag | Amin-substituierte diphenyldiphosphine und deren verwendung in metallkomplexen für asymmetrische synthesen |
| DE10326097A1 (de) | 2003-06-06 | 2005-01-05 | Grünenthal GmbH | Verfahren zur Herstellung von Dimethyl-(3-aryl-butyl)-aminverbindungen |
| DE10328316A1 (de) * | 2003-06-23 | 2005-01-20 | Grünenthal GmbH | Verfahren zur Herstellung von Dimethyl-(3-aryl-buthyl)-aminverbindungen als pharmazeutische Wirkstoffe |
| US7258757B2 (en) | 2004-10-28 | 2007-08-21 | Film Technologies International, Inc. | Method of manufacturing an impact resistant and insulated glass unit composite with solar control and low-E coatings |
| EP1854410A4 (en) | 2005-03-02 | 2009-12-02 | Nat Inst Of Advanced Ind Scien | BIOSENSOR WITH COUPLED NEEDLE |
| CN101296708B (zh) * | 2005-10-26 | 2011-12-07 | 伊莱利利公司 | 选择性vpac2受体肽激动剂 |
| TWI496762B (zh) | 2006-07-24 | 2015-08-21 | 製備(1r,2r)-3-(3-二甲胺基-1-乙基-2-甲基-丙基)-酚之方法 |
-
2007
- 2007-07-20 TW TW103106389A patent/TWI496762B/zh not_active IP Right Cessation
- 2007-07-20 TW TW096126478A patent/TWI448447B/zh not_active IP Right Cessation
- 2007-07-23 ES ES21163457T patent/ES2961956T3/es active Active
- 2007-07-23 DK DK11004087.0T patent/DK2368871T3/en active
- 2007-07-23 EP EP21163457.1A patent/EP3868739B1/en active Active
- 2007-07-23 LT LTEP17161440.7T patent/LT3210965T/lt unknown
- 2007-07-23 LT LTEP11004087.0T patent/LT2368871T/lt unknown
- 2007-07-23 ES ES17161440T patent/ES2901953T3/es active Active
- 2007-07-23 PL PL07786260T patent/PL2046724T3/pl unknown
- 2007-07-23 PL PL17161440T patent/PL3210965T3/pl unknown
- 2007-07-23 BR BRPI0714558A patent/BRPI0714558B8/pt not_active IP Right Cessation
- 2007-07-23 CN CN2007800278848A patent/CN101495445B/zh not_active Expired - Fee Related
- 2007-07-23 HU HUE11004087A patent/HUE034253T2/en unknown
- 2007-07-23 NZ NZ573911A patent/NZ573911A/en not_active IP Right Cessation
- 2007-07-23 CA CA2657104A patent/CA2657104C/en active Active
- 2007-07-23 AU AU2007278464A patent/AU2007278464C1/en not_active Ceased
- 2007-07-23 EP EP07786260.5A patent/EP2046724B1/en active Active
- 2007-07-23 EP EP11004087.0A patent/EP2368871B8/en active Active
- 2007-07-23 SI SI200731545T patent/SI2046724T1/sl unknown
- 2007-07-23 SI SI200732181T patent/SI3210965T1/sl unknown
- 2007-07-23 PT PT77862605T patent/PT2046724E/pt unknown
- 2007-07-23 PT PT110040870T patent/PT2368871T/pt unknown
- 2007-07-23 ES ES07786260.5T patent/ES2524252T3/es active Active
- 2007-07-23 US US12/374,874 patent/US8877974B2/en active Active
- 2007-07-23 SI SI200731921A patent/SI2368871T1/sl unknown
- 2007-07-23 EP EP17161440.7A patent/EP3210965B1/en active Active
- 2007-07-23 ES ES11004087.0T patent/ES2629342T3/es active Active
- 2007-07-23 WO PCT/EP2007/006515 patent/WO2008012047A1/en not_active Ceased
- 2007-07-23 HR HRP20211902TT patent/HRP20211902T1/hr unknown
- 2007-07-23 JP JP2009521156A patent/JP5612308B2/ja not_active Expired - Fee Related
- 2007-07-23 PL PL11004087T patent/PL2368871T3/pl unknown
- 2007-07-23 PT PT171614407T patent/PT3210965T/pt unknown
- 2007-07-23 HR HRP20140891TT patent/HRP20140891T1/hr unknown
- 2007-07-23 DK DK17161440.7T patent/DK3210965T3/da active
- 2007-07-23 MX MX2009000667A patent/MX2009000667A/es active IP Right Grant
- 2007-07-23 RU RU2009105817/04A patent/RU2466124C2/ru active
- 2007-07-23 DK DK07786260.5T patent/DK2046724T3/da active
- 2007-07-23 HU HUE17161440A patent/HUE056642T2/hu unknown
- 2007-07-23 KR KR1020097003831A patent/KR101423996B1/ko not_active Expired - Fee Related
-
2008
- 2008-12-18 IL IL196049A patent/IL196049A/en active IP Right Grant
-
2009
- 2009-01-07 ZA ZA2009/00136A patent/ZA200900136B/en unknown
- 2009-01-26 NO NO20090376A patent/NO341939B1/no unknown
- 2009-01-27 CO CO09007191A patent/CO6160221A2/es unknown
-
2014
- 2014-09-05 US US14/478,797 patent/US20140378706A1/en not_active Abandoned
- 2014-10-16 CY CY20141100851T patent/CY1115651T1/el unknown
-
2016
- 2016-04-12 US US15/096,986 patent/US20160221928A1/en not_active Abandoned
- 2016-12-25 IL IL249748A patent/IL249748B/en active IP Right Grant
-
2017
- 2017-04-17 US US15/488,888 patent/US20170217875A1/en not_active Abandoned
- 2017-04-18 HR HRP20170614TT patent/HRP20170614T1/hr unknown
- 2017-05-22 CY CY20171100533T patent/CY1119126T1/el unknown
- 2017-10-22 IL IL255201A patent/IL255201B/en active IP Right Grant
- 2017-11-15 NO NO20171811A patent/NO343951B1/no unknown
-
2019
- 2019-05-13 US US16/409,910 patent/US20190263747A1/en not_active Abandoned
-
2020
- 2020-06-09 US US16/896,754 patent/US20200299225A1/en not_active Abandoned
-
2021
- 2021-01-06 US US17/142,325 patent/US11739049B2/en active Active
- 2021-12-22 CY CY20211101129T patent/CY1124873T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2901953T3 (es) | Procedimiento de preparación de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol | |
| HK1130246B (en) | Process for the preparation of (1r,2r)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol | |
| HK1243056B (en) | Process for the preparation of (1r,2r)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol | |
| HK1162463B (en) | Process for the preparation of (1r,2r)-3-(3-dimethylamino-1-ethyl-2-methylpropyl)-phenol | |
| HK1162463A (en) | Process for the preparation of (1r,2r)-3-(3-dimethylamino-1-ethyl-2-methylpropyl)-phenol |