BRPI0717367B1 - Di-hidropirazolonas substituídas,seus usos e seu processo de preparação, e medicamento - Google Patents
Di-hidropirazolonas substituídas,seus usos e seu processo de preparação, e medicamento Download PDFInfo
- Publication number
- BRPI0717367B1 BRPI0717367B1 BRPI0717367-9A BRPI0717367A BRPI0717367B1 BR PI0717367 B1 BRPI0717367 B1 BR PI0717367B1 BR PI0717367 A BRPI0717367 A BR PI0717367A BR PI0717367 B1 BRPI0717367 B1 BR PI0717367B1
- Authority
- BR
- Brazil
- Prior art keywords
- alkoxy
- carbonyl
- compound
- mixture
- alkylamino
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 29
- 150000001875 compounds Chemical class 0.000 claims abstract description 284
- 238000011282 treatment Methods 0.000 claims abstract description 37
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 30
- 201000010099 disease Diseases 0.000 claims abstract description 27
- 238000011321 prophylaxis Methods 0.000 claims abstract description 24
- 239000003814 drug Substances 0.000 claims abstract description 16
- 229940079593 drug Drugs 0.000 claims abstract description 10
- 208000024172 Cardiovascular disease Diseases 0.000 claims abstract description 8
- -1 trifluoromethoxy, hydroxy-carbonyl Chemical group 0.000 claims description 134
- 238000006243 chemical reaction Methods 0.000 claims description 53
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 48
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 45
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 44
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 42
- 229910052731 fluorine Inorganic materials 0.000 claims description 42
- 239000011737 fluorine Substances 0.000 claims description 42
- 125000001072 heteroaryl group Chemical group 0.000 claims description 42
- ORTFAQDWJHRMNX-UHFFFAOYSA-N hydroxidooxidocarbon(.) Chemical group O[C]=O ORTFAQDWJHRMNX-UHFFFAOYSA-N 0.000 claims description 41
- 239000001257 hydrogen Substances 0.000 claims description 40
- 229910052739 hydrogen Inorganic materials 0.000 claims description 40
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 40
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 39
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 38
- 150000003839 salts Chemical class 0.000 claims description 37
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 36
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 33
- 239000000460 chlorine Substances 0.000 claims description 33
- 229910052801 chlorine Inorganic materials 0.000 claims description 33
- 239000012453 solvate Substances 0.000 claims description 33
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 32
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 32
- 229910052794 bromium Inorganic materials 0.000 claims description 32
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 26
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 26
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 24
- 239000000126 substance Substances 0.000 claims description 24
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 20
- 125000004043 oxo group Chemical group O=* 0.000 claims description 20
- 125000001424 substituent group Chemical group 0.000 claims description 16
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 14
- 208000007502 anemia Diseases 0.000 claims description 14
- 150000002431 hydrogen Chemical class 0.000 claims description 13
- 239000003112 inhibitor Substances 0.000 claims description 11
- 239000002253 acid Substances 0.000 claims description 9
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 8
- 206010028980 Neoplasm Diseases 0.000 claims description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 7
- 229910052742 iron Inorganic materials 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 206010019280 Heart failures Diseases 0.000 claims description 5
- 239000003242 anti bacterial agent Substances 0.000 claims description 5
- 208000020832 chronic kidney disease Diseases 0.000 claims description 5
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims description 4
- 229940088710 antibiotic agent Drugs 0.000 claims description 4
- 238000002512 chemotherapy Methods 0.000 claims description 4
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims description 4
- 239000013589 supplement Substances 0.000 claims description 4
- 239000005541 ACE inhibitor Substances 0.000 claims description 3
- 229940127291 Calcium channel antagonist Drugs 0.000 claims description 3
- 229940083712 aldosterone antagonist Drugs 0.000 claims description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 claims description 3
- 239000002934 diuretic Substances 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 231100000252 nontoxic Toxicity 0.000 claims description 3
- 230000003000 nontoxic effect Effects 0.000 claims description 3
- 206010002383 Angina Pectoris Diseases 0.000 claims description 2
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 claims description 2
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 claims description 2
- 208000007530 Essential hypertension Diseases 0.000 claims description 2
- 208000006011 Stroke Diseases 0.000 claims description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 claims description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 claims description 2
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 2
- 210000001367 artery Anatomy 0.000 claims description 2
- 208000022831 chronic renal failure syndrome Diseases 0.000 claims description 2
- 238000006482 condensation reaction Methods 0.000 claims description 2
- 208000029078 coronary artery disease Diseases 0.000 claims description 2
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 claims description 2
- 229960005156 digoxin Drugs 0.000 claims description 2
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 claims description 2
- 229940030606 diuretics Drugs 0.000 claims description 2
- 229960003284 iron Drugs 0.000 claims description 2
- 201000005857 malignant hypertension Diseases 0.000 claims description 2
- 239000002394 mineralocorticoid antagonist Substances 0.000 claims description 2
- 230000002093 peripheral effect Effects 0.000 claims description 2
- 230000002685 pulmonary effect Effects 0.000 claims description 2
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 2
- 229940082632 vitamin b12 and folic acid Drugs 0.000 claims description 2
- 241000208011 Digitalis Species 0.000 claims 1
- 206010061216 Infarction Diseases 0.000 claims 1
- 239000013543 active substance Substances 0.000 claims 1
- 102000012740 beta Adrenergic Receptors Human genes 0.000 claims 1
- 108010079452 beta Adrenergic Receptors Proteins 0.000 claims 1
- 230000007574 infarction Effects 0.000 claims 1
- 230000002107 myocardial effect Effects 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 157
- 230000008569 process Effects 0.000 abstract description 8
- 230000002526 effect on cardiovascular system Effects 0.000 abstract description 4
- 208000014951 hematologic disease Diseases 0.000 abstract description 3
- 230000029663 wound healing Effects 0.000 abstract description 3
- 208000017169 kidney disease Diseases 0.000 abstract description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 274
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 240
- 239000000203 mixture Substances 0.000 description 235
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 198
- 239000000243 solution Substances 0.000 description 138
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 125
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 120
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 109
- 238000005160 1H NMR spectroscopy Methods 0.000 description 108
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 92
- 238000001914 filtration Methods 0.000 description 87
- 239000007787 solid Substances 0.000 description 82
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 80
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 80
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 69
- 239000000047 product Substances 0.000 description 64
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 60
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 58
- 239000012071 phase Substances 0.000 description 58
- 238000002953 preparative HPLC Methods 0.000 description 49
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 45
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 45
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 45
- 101000610640 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp3 Proteins 0.000 description 43
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 43
- 101001110823 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-A Proteins 0.000 description 43
- 101000712176 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-B Proteins 0.000 description 43
- 102100040374 U4/U6 small nuclear ribonucleoprotein Prp3 Human genes 0.000 description 43
- 239000011541 reaction mixture Substances 0.000 description 43
- 239000002904 solvent Substances 0.000 description 43
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 41
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 40
- 235000019253 formic acid Nutrition 0.000 description 40
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 39
- 238000010992 reflux Methods 0.000 description 34
- 239000003480 eluent Substances 0.000 description 33
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 30
- 238000004128 high performance liquid chromatography Methods 0.000 description 29
- 238000000746 purification Methods 0.000 description 28
- 238000012360 testing method Methods 0.000 description 27
- 150000003254 radicals Chemical class 0.000 description 22
- 206010021143 Hypoxia Diseases 0.000 description 20
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 20
- 239000002244 precipitate Substances 0.000 description 20
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 19
- 238000001816 cooling Methods 0.000 description 19
- 239000000706 filtrate Substances 0.000 description 19
- 210000004027 cell Anatomy 0.000 description 18
- 229910052736 halogen Inorganic materials 0.000 description 18
- 150000002367 halogens Chemical class 0.000 description 18
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- 102000003951 Erythropoietin Human genes 0.000 description 16
- 108090000394 Erythropoietin Proteins 0.000 description 16
- 239000012482 calibration solution Substances 0.000 description 16
- 229940105423 erythropoietin Drugs 0.000 description 16
- 229910052760 oxygen Inorganic materials 0.000 description 16
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 16
- 238000003756 stirring Methods 0.000 description 16
- 238000000825 ultraviolet detection Methods 0.000 description 16
- 210000004369 blood Anatomy 0.000 description 15
- 239000008280 blood Substances 0.000 description 15
- 108090000623 proteins and genes Proteins 0.000 description 15
- 239000000725 suspension Substances 0.000 description 15
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 14
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 14
- 230000007954 hypoxia Effects 0.000 description 14
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 14
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 13
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 13
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- 239000013078 crystal Substances 0.000 description 12
- 230000014509 gene expression Effects 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 11
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 239000000741 silica gel Substances 0.000 description 11
- 229910002027 silica gel Inorganic materials 0.000 description 11
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 239000001301 oxygen Substances 0.000 description 10
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 9
- 241000282414 Homo sapiens Species 0.000 description 9
- 108010043005 Prolyl Hydroxylases Proteins 0.000 description 9
- 102000004079 Prolyl Hydroxylases Human genes 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 125000001607 1,2,3-triazol-1-yl group Chemical group [*]N1N=NC([H])=C1[H] 0.000 description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 238000007911 parenteral administration Methods 0.000 description 8
- 238000001556 precipitation Methods 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- 108090000223 Hypoxia-inducible factor-proline dioxygenases Proteins 0.000 description 7
- 102000003856 Hypoxia-inducible factor-proline dioxygenases Human genes 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 7
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 7
- 238000010908 decantation Methods 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 239000011877 solvent mixture Substances 0.000 description 7
- 239000011550 stock solution Substances 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 229960000583 acetic acid Drugs 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 238000004587 chromatography analysis Methods 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 6
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- 239000006228 supernatant Substances 0.000 description 6
- 239000011534 wash buffer Substances 0.000 description 6
- 239000003643 water by type Substances 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 206010058116 Nephrogenic anaemia Diseases 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 5
- 238000009835 boiling Methods 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 239000006196 drop Substances 0.000 description 5
- 150000004675 formic acid derivatives Chemical class 0.000 description 5
- 210000002216 heart Anatomy 0.000 description 5
- 238000005534 hematocrit Methods 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 238000005805 hydroxylation reaction Methods 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 230000006698 induction Effects 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 5
- 238000007363 ring formation reaction Methods 0.000 description 5
- 239000012488 sample solution Substances 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 229910052938 sodium sulfate Inorganic materials 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 0 CC1=*N=NC1 Chemical compound CC1=*N=NC1 0.000 description 4
- 108020004635 Complementary DNA Proteins 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 4
- 102100037248 Prolyl hydroxylase EGLN2 Human genes 0.000 description 4
- 102100037247 Prolyl hydroxylase EGLN3 Human genes 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- 238000010804 cDNA synthesis Methods 0.000 description 4
- 239000002299 complementary DNA Substances 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 231100000673 dose–response relationship Toxicity 0.000 description 4
- 210000003743 erythrocyte Anatomy 0.000 description 4
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 4
- 229910052737 gold Inorganic materials 0.000 description 4
- 239000010931 gold Substances 0.000 description 4
- 230000033444 hydroxylation Effects 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000013067 intermediate product Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 239000002480 mineral oil Substances 0.000 description 4
- 235000010446 mineral oil Nutrition 0.000 description 4
- 239000012452 mother liquor Substances 0.000 description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- 239000000902 placebo Substances 0.000 description 4
- 229940068196 placebo Drugs 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- GOXYBEXWMJZLJB-UHFFFAOYSA-N (6-chloropyridin-3-yl)methanol Chemical compound OCC1=CC=C(Cl)N=C1 GOXYBEXWMJZLJB-UHFFFAOYSA-N 0.000 description 3
- 125000006582 (C5-C6) heterocycloalkyl group Chemical group 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 3
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 3
- XJPZKYIHCLDXST-UHFFFAOYSA-N 4,6-dichloropyrimidine Chemical compound ClC1=CC(Cl)=NC=N1 XJPZKYIHCLDXST-UHFFFAOYSA-N 0.000 description 3
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 3
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 3
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 3
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 239000012148 binding buffer Substances 0.000 description 3
- 210000004204 blood vessel Anatomy 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000005018 casein Substances 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 229960000958 deferoxamine Drugs 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 230000010437 erythropoiesis Effects 0.000 description 3
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 3
- SURXNMWQQYCEDX-UHFFFAOYSA-N ethyl 6-hydrazinylpyridine-3-carboxylate Chemical compound CCOC(=O)C1=CC=C(NN)N=C1 SURXNMWQQYCEDX-UHFFFAOYSA-N 0.000 description 3
- 239000010408 film Substances 0.000 description 3
- 238000005755 formation reaction Methods 0.000 description 3
- 230000035876 healing Effects 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 230000000302 ischemic effect Effects 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 210000001995 reticulocyte Anatomy 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 238000000967 suction filtration Methods 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000004885 tandem mass spectrometry Methods 0.000 description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- GNMVDRDYUPNLKB-UHFFFAOYSA-N (6-piperidin-1-ylpyrimidin-4-yl)hydrazine Chemical compound C1=NC(NN)=CC(N2CCCCC2)=N1 GNMVDRDYUPNLKB-UHFFFAOYSA-N 0.000 description 2
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 description 2
- XBYRMPXUBGMOJC-UHFFFAOYSA-N 1,2-dihydropyrazol-3-one Chemical class OC=1C=CNN=1 XBYRMPXUBGMOJC-UHFFFAOYSA-N 0.000 description 2
- IGERFAHWSHDDHX-UHFFFAOYSA-N 1,3-dioxanyl Chemical group [CH]1OCCCO1 IGERFAHWSHDDHX-UHFFFAOYSA-N 0.000 description 2
- BMEMBBFDTYHTLH-UHFFFAOYSA-N 1-(2-methoxyethyl)piperazine Chemical compound COCCN1CCNCC1 BMEMBBFDTYHTLH-UHFFFAOYSA-N 0.000 description 2
- NLTWFUFMHBZOBY-UHFFFAOYSA-N 1-(6-chloropyrimidin-4-yl)azetidin-3-ol Chemical compound C1C(O)CN1C1=CC(Cl)=NC=N1 NLTWFUFMHBZOBY-UHFFFAOYSA-N 0.000 description 2
- YDYBTRNQUBEIER-UHFFFAOYSA-N 1-(6-hydrazinylpyrimidin-4-yl)azetidin-3-ol Chemical compound C1=NC(NN)=CC(N2CC(O)C2)=N1 YDYBTRNQUBEIER-UHFFFAOYSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 2
- GNIKAAGREJCDIK-UHFFFAOYSA-N 2-(6-chloropyrimidin-4-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=NC(Cl)=CC(N2C(C(N3N=NC=C3)=CN2)=O)=N1 GNIKAAGREJCDIK-UHFFFAOYSA-N 0.000 description 2
- OFCRVHFAFKTUNE-UHFFFAOYSA-N 2-(6-cyclopropylpyrimidin-4-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one Chemical compound O=C1C(N2N=NC=C2)=CNN1C(N=CN=1)=CC=1C1CC1 OFCRVHFAFKTUNE-UHFFFAOYSA-N 0.000 description 2
- ZASSNNNRCCOARP-UHFFFAOYSA-N 2-chloro-5-[(2-methylpropan-2-yl)oxymethyl]pyridine Chemical compound CC(C)(C)OCC1=CC=C(Cl)N=C1 ZASSNNNRCCOARP-UHFFFAOYSA-N 0.000 description 2
- NURQLCJSMXZBPC-UHFFFAOYSA-N 3,4-dimethylpyridine Chemical compound CC1=CC=NC=C1C NURQLCJSMXZBPC-UHFFFAOYSA-N 0.000 description 2
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N 4-methylimidazole Chemical compound CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 2
- HBBSDZXXUIHKJE-UHFFFAOYSA-N 6-hydrazinylpyridine-3-carboxylic acid Chemical compound NNC1=CC=C(C(O)=O)C=N1 HBBSDZXXUIHKJE-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 208000032467 Aplastic anaemia Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- YQWPBJQSPKVKJB-UHFFFAOYSA-N CC(C)(C)N1N=C(C)C=C1C1=C(O)N(C=2SC=C(N=2)C=2C=CC(Cl)=CC=2)N=C1C Chemical compound CC(C)(C)N1N=C(C)C=C1C1=C(O)N(C=2SC=C(N=2)C=2C=CC(Cl)=CC=2)N=C1C YQWPBJQSPKVKJB-UHFFFAOYSA-N 0.000 description 2
- 101100011377 Caenorhabditis elegans egl-9 gene Proteins 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 102000016680 Dioxygenases Human genes 0.000 description 2
- 108010028143 Dioxygenases Proteins 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 102100037249 Egl nine homolog 1 Human genes 0.000 description 2
- 108010003751 Elongin Proteins 0.000 description 2
- 102000004662 Elongin Human genes 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 2
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 208000031886 HIV Infections Diseases 0.000 description 2
- 208000037357 HIV infectious disease Diseases 0.000 description 2
- 101000881650 Homo sapiens Prolyl hydroxylase EGLN2 Proteins 0.000 description 2
- 101000881678 Homo sapiens Prolyl hydroxylase EGLN3 Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 108010028501 Hypoxia-Inducible Factor 1 Proteins 0.000 description 2
- 102000016878 Hypoxia-Inducible Factor 1 Human genes 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 108060001084 Luciferase Proteins 0.000 description 2
- 239000005089 Luciferase Substances 0.000 description 2
- MPCRDALPQLDDFX-UHFFFAOYSA-L Magnesium perchlorate Chemical compound [Mg+2].[O-]Cl(=O)(=O)=O.[O-]Cl(=O)(=O)=O MPCRDALPQLDDFX-UHFFFAOYSA-L 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 101000987583 Mus musculus Eosinophil peroxidase Proteins 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- BIMZLRFONYSTPT-UHFFFAOYSA-N N-oxalylglycine Chemical compound OC(=O)CNC(=O)C(O)=O BIMZLRFONYSTPT-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 101710170760 Prolyl hydroxylase EGLN2 Proteins 0.000 description 2
- 101710170720 Prolyl hydroxylase EGLN3 Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 206010052428 Wound Diseases 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- FTCAGZYQHBJGKD-UHFFFAOYSA-N [5-(2,2-dimethylpropoxy)pyridin-2-yl]hydrazine Chemical compound CC(C)(C)COC1=CC=C(NN)N=C1 FTCAGZYQHBJGKD-UHFFFAOYSA-N 0.000 description 2
- GVCNGRWHHFCRLY-UHFFFAOYSA-N [5-[(2-methylpropan-2-yl)oxymethyl]pyridin-2-yl]hydrazine Chemical compound CC(C)(C)OCC1=CC=C(NN)N=C1 GVCNGRWHHFCRLY-UHFFFAOYSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 2
- 125000002393 azetidinyl group Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000002876 beta blocker Substances 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- 210000000748 cardiovascular system Anatomy 0.000 description 2
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 2
- 235000021240 caseins Nutrition 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethanethiol Chemical compound CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 125000004634 hexahydroazepinyl group Chemical group N1(CCCCCC1)* 0.000 description 2
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 230000001146 hypoxic effect Effects 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- GKTNLYAAZKKMTQ-UHFFFAOYSA-N n-[bis(dimethylamino)phosphinimyl]-n-methylmethanamine Chemical compound CN(C)P(=N)(N(C)C)N(C)C GKTNLYAAZKKMTQ-UHFFFAOYSA-N 0.000 description 2
- 125000001715 oxadiazolyl group Chemical group 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 125000003566 oxetanyl group Chemical group 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- 230000009038 pharmacological inhibition Effects 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229950008882 polysorbate Drugs 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229960003975 potassium Drugs 0.000 description 2
- 239000001103 potassium chloride Substances 0.000 description 2
- 235000011164 potassium chloride Nutrition 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- NWELCUKYUCBVKK-UHFFFAOYSA-N pyridin-2-ylhydrazine Chemical compound NNC1=CC=CC=N1 NWELCUKYUCBVKK-UHFFFAOYSA-N 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 238000003753 real-time PCR Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000003319 supportive effect Effects 0.000 description 2
- 238000011477 surgical intervention Methods 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- 229940094937 thioredoxin Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- 210000003556 vascular endothelial cell Anatomy 0.000 description 2
- 208000021331 vascular occlusion disease Diseases 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- CEMAWMOMDPGJMB-UHFFFAOYSA-N (+-)-Oxprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1OCC=C CEMAWMOMDPGJMB-UHFFFAOYSA-N 0.000 description 1
- MHCBYPRBJFPLPP-UHFFFAOYSA-N (2-chloropyrimidin-5-yl)methanamine Chemical compound NCC1=CN=C(Cl)N=C1 MHCBYPRBJFPLPP-UHFFFAOYSA-N 0.000 description 1
- NXWGWUVGUSFQJC-GFCCVEGCSA-N (2r)-1-[(2-methyl-1h-indol-4-yl)oxy]-3-(propan-2-ylamino)propan-2-ol Chemical compound CC(C)NC[C@@H](O)COC1=CC=CC2=C1C=C(C)N2 NXWGWUVGUSFQJC-GFCCVEGCSA-N 0.000 description 1
- BAVDEDVBIHTHJQ-UVJOBNTFSA-N (2s)-1-[(2s)-6-amino-2-[[(1s)-1-carboxy-3-phenylpropyl]amino]hexanoyl]pyrrolidine-2-carboxylic acid;hydrate Chemical compound O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 BAVDEDVBIHTHJQ-UVJOBNTFSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- FNFPJUDYICAQSZ-UHFFFAOYSA-N (4-methylpyridin-2-yl)hydrazine Chemical compound CC1=CC=NC(NN)=C1 FNFPJUDYICAQSZ-UHFFFAOYSA-N 0.000 description 1
- QYQLEYTXFMOLEI-UHFFFAOYSA-N (5-bromopyridin-2-yl)hydrazine Chemical compound NNC1=CC=C(Br)C=N1 QYQLEYTXFMOLEI-UHFFFAOYSA-N 0.000 description 1
- PNVWNMCAGZBUNK-UHFFFAOYSA-N (5-methylpyridin-2-yl)hydrazine Chemical compound CC1=CC=C(NN)N=C1 PNVWNMCAGZBUNK-UHFFFAOYSA-N 0.000 description 1
- BLDRCJXTMYOKCJ-UHFFFAOYSA-N (5-methylsulfonylpyridin-2-yl)hydrazine Chemical compound CS(=O)(=O)C1=CC=C(NN)N=C1 BLDRCJXTMYOKCJ-UHFFFAOYSA-N 0.000 description 1
- RRCPILCYBUGPAL-UHFFFAOYSA-N (6-cyclopropylpyrimidin-4-yl)hydrazine Chemical compound C1=NC(NN)=CC(C2CC2)=N1 RRCPILCYBUGPAL-UHFFFAOYSA-N 0.000 description 1
- CPNKYIWDFIZINI-UHFFFAOYSA-N (6-hydrazinylpyridin-3-yl)methanol Chemical compound NNC1=CC=C(CO)C=N1 CPNKYIWDFIZINI-UHFFFAOYSA-N 0.000 description 1
- XWDNLTCJJYYYCI-UHFFFAOYSA-N (6-morpholin-4-ylpyrimidin-4-yl)hydrazine Chemical compound C1=NC(NN)=CC(N2CCOCC2)=N1 XWDNLTCJJYYYCI-UHFFFAOYSA-N 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- XALCOJXGWJXWBL-UHFFFAOYSA-N 1-(6-chloropyridin-3-yl)-n-methylmethanamine Chemical compound CNCC1=CC=C(Cl)N=C1 XALCOJXGWJXWBL-UHFFFAOYSA-N 0.000 description 1
- KHCKXUAITWGDAT-UHFFFAOYSA-N 1-(6-hydrazinylpyridin-3-yl)-n-methylmethanamine Chemical compound CNCC1=CC=C(NN)N=C1 KHCKXUAITWGDAT-UHFFFAOYSA-N 0.000 description 1
- MWCKLGLEIKIPTE-UHFFFAOYSA-N 1-[2-(6-morpholin-4-ylpyrimidin-4-yl)-3-oxo-1h-pyrazol-4-yl]imidazole-4-carbonitrile;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.O=C1C(N2C=C(N=C2)C#N)=CNN1C(N=CN=1)=CC=1N1CCOCC1 MWCKLGLEIKIPTE-UHFFFAOYSA-N 0.000 description 1
- VHDRGZXIHAWAEA-UHFFFAOYSA-N 1-[2-(6-morpholin-4-ylpyrimidin-4-yl)-3-oxo-1h-pyrazol-4-yl]imidazole-4-carboxamide;hydrochloride Chemical compound Cl.C1=NC(C(=O)N)=CN1C(C1=O)=CNN1C1=CC(N2CCOCC2)=NC=N1 VHDRGZXIHAWAEA-UHFFFAOYSA-N 0.000 description 1
- JPSRCLOEZKUWPF-UHFFFAOYSA-N 1-[3-oxo-2-(6-piperidin-1-ylpyrimidin-4-yl)-1h-pyrazol-4-yl]imidazole-4-carbonitrile;hydrochloride Chemical compound Cl.O=C1C(N2C=C(N=C2)C#N)=CNN1C(N=CN=1)=CC=1N1CCCCC1 JPSRCLOEZKUWPF-UHFFFAOYSA-N 0.000 description 1
- CXBDYQVECUFKRK-UHFFFAOYSA-N 1-methoxybutane Chemical compound CCCCOC CXBDYQVECUFKRK-UHFFFAOYSA-N 0.000 description 1
- NWVGXXPWOYZODV-UHFFFAOYSA-N 1h-imidazole-5-carbonitrile Chemical compound N#CC1=CN=CN1 NWVGXXPWOYZODV-UHFFFAOYSA-N 0.000 description 1
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 1
- XNWYPYFQYFUUTE-UHFFFAOYSA-N 2,4-diphenyl-1h-pyrazol-3-one Chemical class O=C1C(C=2C=CC=CC=2)=CNN1C1=CC=CC=C1 XNWYPYFQYFUUTE-UHFFFAOYSA-N 0.000 description 1
- GQAWEWZCGAMAKC-UHFFFAOYSA-N 2,4-dipyridin-2-yl-1h-pyrazol-3-one Chemical class O=C1C(C=2N=CC=CC=2)=CNN1C1=CC=CC=N1 GQAWEWZCGAMAKC-UHFFFAOYSA-N 0.000 description 1
- IBEOGICJOHYRCR-UHFFFAOYSA-N 2,5-bis(methylsulfonyl)pyridine Chemical compound CS(=O)(=O)C1=CC=C(S(C)(=O)=O)N=C1 IBEOGICJOHYRCR-UHFFFAOYSA-N 0.000 description 1
- TXQDHQBSNAJSHQ-UHFFFAOYSA-N 2,5-dimethyl-2,5-dihydro-1h-pyrrole Chemical compound CC1NC(C)C=C1 TXQDHQBSNAJSHQ-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- RMLJAGXWLFREGR-UHFFFAOYSA-N 2-(4,5-dimethylpyridin-2-yl)-4-[4-(trifluoromethyl)imidazol-1-yl]-1h-pyrazol-3-one;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C1=C(C)C(C)=CN=C1N1C(=O)C(N2C=C(N=C2)C(F)(F)F)=CN1 RMLJAGXWLFREGR-UHFFFAOYSA-N 0.000 description 1
- GBERXKHXWQCKAS-UHFFFAOYSA-N 2-(4,5-dimethylpyridin-2-yl)-4-[4-(trifluoromethyl)imidazol-1-yl]-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=C(C)C(C)=CN=C1N1C(=O)C(N2C=C(N=C2)C(F)(F)F)=CN1 GBERXKHXWQCKAS-UHFFFAOYSA-N 0.000 description 1
- ZHJHZGVKHJEUEW-UHFFFAOYSA-N 2-(4,5-dimethylpyridin-2-yl)-4-imidazol-1-yl-1h-pyrazol-3-one Chemical compound C1=C(C)C(C)=CN=C1N1C(=O)C(N2C=NC=C2)=CN1 ZHJHZGVKHJEUEW-UHFFFAOYSA-N 0.000 description 1
- QSAZAVUPKAUGKF-UHFFFAOYSA-N 2-(4,5-dimethylpyridin-2-yl)-4-imidazol-1-yl-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=C(C)C(C)=CN=C1N1C(=O)C(N2C=NC=C2)=CN1 QSAZAVUPKAUGKF-UHFFFAOYSA-N 0.000 description 1
- OCFRILXPXYIVOK-UHFFFAOYSA-N 2-(4-methylpyridin-2-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one Chemical compound CC1=CC=NC(N2C(C(N3N=NC=C3)=CN2)=O)=C1 OCFRILXPXYIVOK-UHFFFAOYSA-N 0.000 description 1
- BPVRJCXGRHUNKF-UHFFFAOYSA-N 2-(4-methylpyridin-2-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.CC1=CC=NC(N2C(C(N3N=NC=C3)=CN2)=O)=C1 BPVRJCXGRHUNKF-UHFFFAOYSA-N 0.000 description 1
- MCEWYDWEDMPKHF-UHFFFAOYSA-N 2-(4-methylpyridin-2-yl)-4-[4-(trifluoromethyl)imidazol-1-yl]-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.CC1=CC=NC(N2C(C(N3C=C(N=C3)C(F)(F)F)=CN2)=O)=C1 MCEWYDWEDMPKHF-UHFFFAOYSA-N 0.000 description 1
- XTGWZVKYWQHZJU-UHFFFAOYSA-N 2-(4-methyltriazol-1-yl)acetic acid Chemical compound CC1=CN(CC(O)=O)N=N1 XTGWZVKYWQHZJU-UHFFFAOYSA-N 0.000 description 1
- JEILSJPIUOYGJD-UHFFFAOYSA-N 2-(5-bromopyridin-2-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.N1=CC(Br)=CC=C1N1C(=O)C(N2N=NC=C2)=CN1 JEILSJPIUOYGJD-UHFFFAOYSA-N 0.000 description 1
- VHFRECQLDGIYTM-UHFFFAOYSA-N 2-(5-bromopyridin-2-yl)-4-[4-(trifluoromethyl)imidazol-1-yl]-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=NC(C(F)(F)F)=CN1C(C1=O)=CNN1C1=CC=C(Br)C=N1 VHFRECQLDGIYTM-UHFFFAOYSA-N 0.000 description 1
- VXCBQLBVABSTCB-UHFFFAOYSA-N 2-(5-bromopyridin-2-yl)-4-imidazol-1-yl-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.N1=CC(Br)=CC=C1N1C(=O)C(N2C=NC=C2)=CN1 VXCBQLBVABSTCB-UHFFFAOYSA-N 0.000 description 1
- GNCUCOFPNYDIAW-UHFFFAOYSA-N 2-(6-chloropyrimidin-4-yl)-4-imidazol-1-yl-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=NC(Cl)=CC(N2C(C(N3C=NC=C3)=CN2)=O)=N1 GNCUCOFPNYDIAW-UHFFFAOYSA-N 0.000 description 1
- SNFNBCNKNBMJLP-UHFFFAOYSA-N 2-(6-cyclopropylpyrimidin-4-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.O=C1C(N2N=NC=C2)=CNN1C(N=CN=1)=CC=1C1CC1 SNFNBCNKNBMJLP-UHFFFAOYSA-N 0.000 description 1
- ZOPUNWRIJJLXBS-UHFFFAOYSA-N 2-(6-ethylsulfanylpyrimidin-4-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=NC(SCC)=CC(N2C(C(N3N=NC=C3)=CN2)=O)=N1 ZOPUNWRIJJLXBS-UHFFFAOYSA-N 0.000 description 1
- GBBVMHMPOYDIHQ-UHFFFAOYSA-N 2-(6-methoxypyrimidin-4-yl)-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.C1=NC(OC)=CC(N2C(C(N3N=NC=C3)=CN2)=O)=N1 GBBVMHMPOYDIHQ-UHFFFAOYSA-N 0.000 description 1
- FKDQEGVYCGHULR-UHFFFAOYSA-N 2-(6-morpholin-4-ylpyrimidin-4-yl)-1h-pyrazol-3-one Chemical compound O=C1C=CNN1C1=CC(N2CCOCC2)=NC=N1 FKDQEGVYCGHULR-UHFFFAOYSA-N 0.000 description 1
- CKKTYPNSKNHMPI-UHFFFAOYSA-N 2-(6-piperidin-1-ylpyrimidin-4-yl)-4-[5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl]-1h-pyrazol-3-one Chemical compound S1C(C(F)(F)F)=NN=C1C(C1=O)=CNN1C1=CC(N2CCCCC2)=NC=N1 CKKTYPNSKNHMPI-UHFFFAOYSA-N 0.000 description 1
- YBDSNEVSFQMCTL-UHFFFAOYSA-N 2-(diethylamino)ethanethiol Chemical compound CCN(CC)CCS YBDSNEVSFQMCTL-UHFFFAOYSA-N 0.000 description 1
- YIFCWYUSYGFFMH-UHFFFAOYSA-N 2-(ethoxymethylidene)propanedioic acid Chemical compound CCOC=C(C(O)=O)C(O)=O YIFCWYUSYGFFMH-UHFFFAOYSA-N 0.000 description 1
- YUHHBTFHHCASIC-UHFFFAOYSA-N 2-(triazol-1-yl)acetic acid Chemical compound OC(=O)CN1C=CN=N1 YUHHBTFHHCASIC-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- FWHYJTLHEDHRHV-UHFFFAOYSA-N 2-[5-(2,2-dimethylpropoxy)pyridin-2-yl]-4-(triazol-1-yl)-1h-pyrazol-3-one Chemical compound N1=CC(OCC(C)(C)C)=CC=C1N1C(=O)C(N2N=NC=C2)=CN1 FWHYJTLHEDHRHV-UHFFFAOYSA-N 0.000 description 1
- RJKVBHNFPMAEIC-UHFFFAOYSA-N 2-[5-(2,2-dimethylpropoxy)pyridin-2-yl]-4-imidazol-1-yl-1h-pyrazol-3-one Chemical compound N1=CC(OCC(C)(C)C)=CC=C1N1C(=O)C(N2C=NC=C2)=CN1 RJKVBHNFPMAEIC-UHFFFAOYSA-N 0.000 description 1
- TWMGVOPPRSJSCC-UHFFFAOYSA-N 2-[5-(aminomethyl)pyridin-2-yl]-4-(triazol-1-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.N1=CC(CN)=CC=C1N1C(=O)C(N2N=NC=C2)=CN1 TWMGVOPPRSJSCC-UHFFFAOYSA-N 0.000 description 1
- INJRFTMGZSFDGR-UHFFFAOYSA-N 2-[5-(hydroxymethyl)pyridin-2-yl]-4-(triazol-1-yl)-1h-pyrazol-3-one Chemical compound N1=CC(CO)=CC=C1N1C(=O)C(N2N=NC=C2)=CN1 INJRFTMGZSFDGR-UHFFFAOYSA-N 0.000 description 1
- HMZQDVXZMGUJBE-UHFFFAOYSA-N 2-[5-(hydroxymethyl)pyridin-2-yl]-4-[4-(trifluoromethyl)imidazol-1-yl]-1h-pyrazol-3-one Chemical compound N1=CC(CO)=CC=C1N1C(=O)C(N2C=C(N=C2)C(F)(F)F)=CN1 HMZQDVXZMGUJBE-UHFFFAOYSA-N 0.000 description 1
- NHCHSBDWPWYYHX-UHFFFAOYSA-N 2-[5-(hydroxymethyl)pyridin-2-yl]-4-imidazol-1-yl-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.N1=CC(CO)=CC=C1N1C(=O)C(N2C=NC=C2)=CN1 NHCHSBDWPWYYHX-UHFFFAOYSA-N 0.000 description 1
- NERJXXFLRICHNM-UHFFFAOYSA-N 2-[6-(2,5-dimethyl-2,5-dihydropyrrol-1-yl)pyrimidin-4-yl]-4-imidazol-1-yl-1h-pyrazol-3-one Chemical compound CC1C=CC(C)N1C1=CC(N2C(C(N3C=NC=C3)=CN2)=O)=NC=N1 NERJXXFLRICHNM-UHFFFAOYSA-N 0.000 description 1
- TWVLPKUWXIHOTE-UHFFFAOYSA-N 2-[6-(3-hydroxyazetidin-1-yl)pyrimidin-4-yl]-4-(triazol-1-yl)-1h-pyrazol-3-one Chemical compound C1C(O)CN1C1=CC(N2C(C(N3N=NC=C3)=CN2)=O)=NC=N1 TWVLPKUWXIHOTE-UHFFFAOYSA-N 0.000 description 1
- UCNAPMCEWHQTHT-UHFFFAOYSA-N 2-[6-(dimethylamino)pyrimidin-4-yl]-4-[4-(trifluoromethyl)imidazol-1-yl]-1h-pyrazol-3-one Chemical compound C1=NC(N(C)C)=CC(N2C(C(N3C=C(N=C3)C(F)(F)F)=CN2)=O)=N1 UCNAPMCEWHQTHT-UHFFFAOYSA-N 0.000 description 1
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 1
- MLONYBFKXHEPCD-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(N)(CO)CO.OCC(N)(CO)CO MLONYBFKXHEPCD-UHFFFAOYSA-N 0.000 description 1
- PUAQLLVFLMYYJJ-UHFFFAOYSA-N 2-aminopropiophenone Chemical compound CC(N)C(=O)C1=CC=CC=C1 PUAQLLVFLMYYJJ-UHFFFAOYSA-N 0.000 description 1
- PPXUUPXQWDQNGO-UHFFFAOYSA-N 2-azidoacetic acid Chemical compound OC(=O)CN=[N+]=[N-] PPXUUPXQWDQNGO-UHFFFAOYSA-N 0.000 description 1
- MUTMUJVXPZWDPY-UHFFFAOYSA-N 2-bromo-3,4-dimethylpyridine Chemical compound CC1=CC=NC(Br)=C1C MUTMUJVXPZWDPY-UHFFFAOYSA-N 0.000 description 1
- OTVKPSUMDAFRQC-UHFFFAOYSA-N 2-bromo-4,5-dimethylpyridine Chemical compound CC1=CN=C(Br)C=C1C OTVKPSUMDAFRQC-UHFFFAOYSA-N 0.000 description 1
- YZVZMDUURBUGRG-UHFFFAOYSA-N 2-chloro-5-(2,2-dimethylpropoxy)pyridine Chemical compound CC(C)(C)COC1=CC=C(Cl)N=C1 YZVZMDUURBUGRG-UHFFFAOYSA-N 0.000 description 1
- VXLYOURCUVQYLN-UHFFFAOYSA-N 2-chloro-5-methylpyridine Chemical compound CC1=CC=C(Cl)N=C1 VXLYOURCUVQYLN-UHFFFAOYSA-N 0.000 description 1
- OYUNTGBISCIYPW-UHFFFAOYSA-N 2-chloroprop-2-enenitrile Chemical compound ClC(=C)C#N OYUNTGBISCIYPW-UHFFFAOYSA-N 0.000 description 1
- GELVZYOEQVJIRR-UHFFFAOYSA-N 2-chloropyrazine Chemical compound ClC1=CN=CC=N1 GELVZYOEQVJIRR-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- ZBFAXMKJADVOGH-UHFFFAOYSA-N 2-fluoro-4-methylpyridine Chemical compound CC1=CC=NC(F)=C1 ZBFAXMKJADVOGH-UHFFFAOYSA-N 0.000 description 1
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 1
- UAPKLGLVGBGMRA-UHFFFAOYSA-N 2-pyridin-2-yl-1h-pyrazol-5-one Chemical class N1C(=O)C=CN1C1=CC=CC=N1 UAPKLGLVGBGMRA-UHFFFAOYSA-N 0.000 description 1
- FJVASRBZQALVIU-UHFFFAOYSA-N 2-pyridin-2-yl-4-(triazol-1-yl)-1h-pyrazol-3-one Chemical compound O=C1C(N2N=NC=C2)=CNN1C1=CC=CC=N1 FJVASRBZQALVIU-UHFFFAOYSA-N 0.000 description 1
- MOTQBOGFDRADCG-UHFFFAOYSA-N 2H-triazole-4-carbonitrile hydrochloride Chemical compound C1=NNN=C1C#N.Cl MOTQBOGFDRADCG-UHFFFAOYSA-N 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical compound CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- IDWRJRPUIXRFRX-UHFFFAOYSA-N 3,5-dimethylpiperidine Chemical compound CC1CNCC(C)C1 IDWRJRPUIXRFRX-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- GMLBBBCZOOGMMU-UHFFFAOYSA-N 4-(1H-pyrazol-5-yl)-1,2-dihydropyrazol-3-one Chemical class N1N=CC(C2=NNC=C2)=C1O GMLBBBCZOOGMMU-UHFFFAOYSA-N 0.000 description 1
- CFOBQJWOZVMWBR-UHFFFAOYSA-N 4-(6-chloropyrimidin-4-yl)morpholine Chemical compound C1=NC(Cl)=CC(N2CCOCC2)=N1 CFOBQJWOZVMWBR-UHFFFAOYSA-N 0.000 description 1
- QJOJYKKMOIGXJG-UHFFFAOYSA-N 4-(triazol-1-yl)-2-[4-(trifluoromethyl)pyridin-2-yl]-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.FC(F)(F)C1=CC=NC(N2C(C(N3N=NC=C3)=CN2)=O)=C1 QJOJYKKMOIGXJG-UHFFFAOYSA-N 0.000 description 1
- QQWGJFXERXLUHI-UHFFFAOYSA-N 4-chloro-6-cyclopropylpyrimidine Chemical compound C1=NC(Cl)=CC(C2CC2)=N1 QQWGJFXERXLUHI-UHFFFAOYSA-N 0.000 description 1
- MJONHQHUVJXBRU-UHFFFAOYSA-N 4-chloro-6-piperidin-1-ylpyrimidine Chemical compound C1=NC(Cl)=CC(N2CCCCC2)=N1 MJONHQHUVJXBRU-UHFFFAOYSA-N 0.000 description 1
- ZPZXAPRUOLEKKH-UHFFFAOYSA-N 4-imidazol-1-yl-2-(4-methylpyridin-2-yl)-1h-pyrazol-3-one Chemical compound CC1=CC=NC(N2C(C(N3C=NC=C3)=CN2)=O)=C1 ZPZXAPRUOLEKKH-UHFFFAOYSA-N 0.000 description 1
- PGCMDLWXEWELLL-UHFFFAOYSA-N 4-imidazol-1-yl-2-(6-morpholin-4-ylpyrimidin-4-yl)-1h-pyrazol-3-one;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.O=C1C(N2C=NC=C2)=CNN1C(N=CN=1)=CC=1N1CCOCC1 PGCMDLWXEWELLL-UHFFFAOYSA-N 0.000 description 1
- OQZXLBOVVDLUIB-UHFFFAOYSA-N 4-imidazol-1-yl-2-(6-morpholin-4-ylpyrimidin-4-yl)-1h-pyrazol-3-one;hydrochloride Chemical compound Cl.O=C1C(N2C=NC=C2)=CNN1C(N=CN=1)=CC=1N1CCOCC1 OQZXLBOVVDLUIB-UHFFFAOYSA-N 0.000 description 1
- UECBGPSHIWJXPJ-UHFFFAOYSA-N 4-imidazol-1-yl-2-(6-piperidin-1-ylpyrimidin-4-yl)-1h-pyrazol-3-one Chemical compound O=C1C(N2C=NC=C2)=CNN1C(N=CN=1)=CC=1N1CCCCC1 UECBGPSHIWJXPJ-UHFFFAOYSA-N 0.000 description 1
- RGUKYNXWOWSRET-UHFFFAOYSA-N 4-pyrrolidin-1-ylpyridine Chemical compound C1CCCN1C1=CC=NC=C1 RGUKYNXWOWSRET-UHFFFAOYSA-N 0.000 description 1
- DFLGRTIPTPCKPJ-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-imidazole Chemical compound FC(F)(F)C1=CN=CN1 DFLGRTIPTPCKPJ-UHFFFAOYSA-N 0.000 description 1
- PEAOEIWYQVXZMB-UHFFFAOYSA-N 5-bromo-2-chloropyridine Chemical compound ClC1=CC=C(Br)C=N1 PEAOEIWYQVXZMB-UHFFFAOYSA-N 0.000 description 1
- DEKFGDPQJRYLQM-UHFFFAOYSA-N 5-methyl-4-(5-methyl-2-pyridin-2-ylpyrazol-3-yl)-2-pyridin-2-yl-1H-pyrazol-3-one Chemical compound C=1C=CC=NC=1N1N=C(C)C=C1C(C1=O)=C(C)NN1C1=CC=CC=N1 DEKFGDPQJRYLQM-UHFFFAOYSA-N 0.000 description 1
- RLIOOWXWEFSKCM-UHFFFAOYSA-N 6-(4-imidazol-1-yl-3-oxo-1h-pyrazol-2-yl)pyridine-3-carboxylic acid Chemical compound N1=CC(C(=O)O)=CC=C1N1C(=O)C(N2C=NC=C2)=CN1 RLIOOWXWEFSKCM-UHFFFAOYSA-N 0.000 description 1
- UAWMVMPAYRWUFX-UHFFFAOYSA-N 6-Chloronicotinic acid Chemical compound OC(=O)C1=CC=C(Cl)N=C1 UAWMVMPAYRWUFX-UHFFFAOYSA-N 0.000 description 1
- MRJSXXCYZJCFSP-UHFFFAOYSA-N 6-[3-oxo-4-(triazol-1-yl)-1h-pyrazol-2-yl]pyridine-3-carboxylic acid Chemical compound N1=CC(C(=O)O)=CC=C1N1C(=O)C(N2N=NC=C2)=CN1 MRJSXXCYZJCFSP-UHFFFAOYSA-N 0.000 description 1
- MJABNXLMCIHCCW-UHFFFAOYSA-N 6-[3-oxo-4-(triazol-1-yl)-1h-pyrazol-2-yl]pyridine-3-carboxylic acid;hydrochloride Chemical compound Cl.N1=CC(C(=O)O)=CC=C1N1C(=O)C(N2N=NC=C2)=CN1 MJABNXLMCIHCCW-UHFFFAOYSA-N 0.000 description 1
- KVCOOWROABTXDJ-UHFFFAOYSA-N 6-chloropyridin-3-ol Chemical compound OC1=CC=C(Cl)N=C1 KVCOOWROABTXDJ-UHFFFAOYSA-N 0.000 description 1
- KPBJKEHFILEPQO-UHFFFAOYSA-N 6-hydrazinylpyridine-3-carbonitrile Chemical compound NNC1=CC=C(C#N)C=N1 KPBJKEHFILEPQO-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 230000002407 ATP formation Effects 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 102000003984 Aryl Hydrocarbon Receptors Human genes 0.000 description 1
- 108090000448 Aryl Hydrocarbon Receptors Proteins 0.000 description 1
- 206010003497 Asphyxia Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000000412 Avitaminosis Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- LASVXNJMPRBVRH-UHFFFAOYSA-N C=1C=C(Cl)C=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C(SC=1)=NC=1C1=CC=CC=C1 Chemical compound C=1C=C(Cl)C=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C(SC=1)=NC=1C1=CC=CC=C1 LASVXNJMPRBVRH-UHFFFAOYSA-N 0.000 description 1
- OIRLBCUEDFJAEH-UHFFFAOYSA-N C=1C=CC=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C(SC=1)=NC=1C1=CC=CC=C1 Chemical compound C=1C=CC=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C(SC=1)=NC=1C1=CC=CC=C1 OIRLBCUEDFJAEH-UHFFFAOYSA-N 0.000 description 1
- PXNAFOCTBXKETK-UHFFFAOYSA-N C=1C=CC=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C(SC=1)=NC=1C1=CC=CS1 Chemical compound C=1C=CC=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C(SC=1)=NC=1C1=CC=CS1 PXNAFOCTBXKETK-UHFFFAOYSA-N 0.000 description 1
- YLVGUHBQJFHIMV-UHFFFAOYSA-N C=1C=CC=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C1=NC=CS1 Chemical class C=1C=CC=CC=1N1N=C(C)C=C1C(=C1O)C(C)=NN1C1=NC=CS1 YLVGUHBQJFHIMV-UHFFFAOYSA-N 0.000 description 1
- GXZJBYKOOZRZNF-UHFFFAOYSA-N CC1=CSC(N2C(=C(C(C)=N2)C=2N(N=C(C)C=2)C=2C=CC=CC=2)O)=N1 Chemical compound CC1=CSC(N2C(=C(C(C)=N2)C=2N(N=C(C)C=2)C=2C=CC=CC=2)O)=N1 GXZJBYKOOZRZNF-UHFFFAOYSA-N 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- RDNNLYVVOGIJEP-UHFFFAOYSA-N CNCc1ccc(N(C2=O)NC=C2[n]2nncc2)nc1 Chemical compound CNCc1ccc(N(C2=O)NC=C2[n]2nncc2)nc1 RDNNLYVVOGIJEP-UHFFFAOYSA-N 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 206010007558 Cardiac failure chronic Diseases 0.000 description 1
- JOATXPAWOHTVSZ-UHFFFAOYSA-N Celiprolol Chemical compound CCN(CC)C(=O)NC1=CC=C(OCC(O)CNC(C)(C)C)C(C(C)=O)=C1 JOATXPAWOHTVSZ-UHFFFAOYSA-N 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 241000050051 Chelone glabra Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 102100039445 Cortexin-3 Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 230000033616 DNA repair Effects 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 102100030012 Deoxyribonuclease-1 Human genes 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- 206010054044 Diabetic microangiopathy Diseases 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 240000001879 Digitalis lutea Species 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 101710111663 Egl nine homolog 1 Proteins 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 206010016880 Folate deficiency Diseases 0.000 description 1
- 208000010188 Folic Acid Deficiency Diseases 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 102100032742 Histone-lysine N-methyltransferase SETD2 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101100061721 Homo sapiens CTXN3 gene Proteins 0.000 description 1
- 101000881648 Homo sapiens Egl nine homolog 1 Proteins 0.000 description 1
- 101000987586 Homo sapiens Eosinophil peroxidase Proteins 0.000 description 1
- 101000920686 Homo sapiens Erythropoietin Proteins 0.000 description 1
- 101000654725 Homo sapiens Histone-lysine N-methyltransferase SETD2 Proteins 0.000 description 1
- 101001046870 Homo sapiens Hypoxia-inducible factor 1-alpha Proteins 0.000 description 1
- 101000739905 Homo sapiens Sestrin-2 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 206010021074 Hypoplastic anaemia Diseases 0.000 description 1
- 206010021135 Hypovitaminosis Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 208000015710 Iron-Deficiency Anemia Diseases 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- KLDXJTOLSGUMSJ-JGWLITMVSA-N Isosorbide Chemical compound O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 KLDXJTOLSGUMSJ-JGWLITMVSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- 102100034671 L-lactate dehydrogenase A chain Human genes 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 108010088350 Lactate Dehydrogenase 5 Proteins 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102000008109 Mixed Function Oxygenases Human genes 0.000 description 1
- 108010074633 Mixed Function Oxygenases Proteins 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 101000920670 Mus musculus Erythropoietin Proteins 0.000 description 1
- GZDWLMYUMRYDLB-UHFFFAOYSA-N O=C1N(c2ncnc(Cl)c2)NC=C1[n]1cncc1 Chemical compound O=C1N(c2ncnc(Cl)c2)NC=C1[n]1cncc1 GZDWLMYUMRYDLB-UHFFFAOYSA-N 0.000 description 1
- QCAZUHAFTVNPNC-UHFFFAOYSA-N O=C1N(c2ncnc(N3CCOCC3)c2)NC=C1[n]1cnc(C(F)(F)F)c1 Chemical compound O=C1N(c2ncnc(N3CCOCC3)c2)NC=C1[n]1cnc(C(F)(F)F)c1 QCAZUHAFTVNPNC-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- 101000622060 Photinus pyralis Luciferin 4-monooxygenase Proteins 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- CYLWJCABXYDINA-UHFFFAOYSA-N Polythiazide Polymers ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CSCC(F)(F)F)NC2=C1 CYLWJCABXYDINA-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 206010036590 Premature baby Diseases 0.000 description 1
- 229940078467 Prolyl hydroxylase inhibitor Drugs 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 206010039163 Right ventricular failure Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 102100037576 Sestrin-2 Human genes 0.000 description 1
- 201000001880 Sexual dysfunction Diseases 0.000 description 1
- 206010072170 Skin wound Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000010513 Stupor Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102000002933 Thioredoxin Human genes 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 102000006275 Ubiquitin-Protein Ligases Human genes 0.000 description 1
- 108010083111 Ubiquitin-Protein Ligases Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 206010053648 Vascular occlusion Diseases 0.000 description 1
- 229930003779 Vitamin B12 Natural products 0.000 description 1
- DAAZHANFWTZKAM-UHFFFAOYSA-N [6-(4-pyrrolidin-1-ylpiperidin-1-yl)pyrimidin-4-yl]hydrazine Chemical compound C1=NC(NN)=CC(N2CCC(CC2)N2CCCC2)=N1 DAAZHANFWTZKAM-UHFFFAOYSA-N 0.000 description 1
- SNPBAGLHMCDOEY-UHFFFAOYSA-N [6-[4-(2-methoxyethyl)piperazin-1-yl]pyrimidin-4-yl]hydrazine Chemical compound C1CN(CCOC)CCN1C1=CC(NN)=NC=N1 SNPBAGLHMCDOEY-UHFFFAOYSA-N 0.000 description 1
- BZKPWHYZMXOIDC-UHFFFAOYSA-N acetazolamide Chemical compound CC(=O)NC1=NN=C(S(N)(=O)=O)S1 BZKPWHYZMXOIDC-UHFFFAOYSA-N 0.000 description 1
- 229960000571 acetazolamide Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- IPGLIOFIFLXLKR-AXYNENQYSA-N adaprolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CC(=O)OCCC1(C2)C[C@@H](C3)C[C@H]2C[C@@H]3C1 IPGLIOFIFLXLKR-AXYNENQYSA-N 0.000 description 1
- 229950000221 adaprolol Drugs 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 1
- 150000008046 alkali metal hydrides Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960002213 alprenolol Drugs 0.000 description 1
- PAZJSJFMUHDSTF-UHFFFAOYSA-N alprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1CC=C PAZJSJFMUHDSTF-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- RNLQIBCLLYYYFJ-UHFFFAOYSA-N amrinone Chemical compound N1C(=O)C(N)=CC(C=2C=CN=CC=2)=C1 RNLQIBCLLYYYFJ-UHFFFAOYSA-N 0.000 description 1
- 229960002105 amrinone Drugs 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- UQUPQEUNHVVNKW-UHFFFAOYSA-N azetidin-1-ium-3-ol;chloride Chemical compound Cl.OC1CNC1 UQUPQEUNHVVNKW-UHFFFAOYSA-N 0.000 description 1
- GMWFCJXSQQHBPI-UHFFFAOYSA-N azetidin-3-ol Chemical compound OC1CNC1 GMWFCJXSQQHBPI-UHFFFAOYSA-N 0.000 description 1
- 229960003515 bendroflumethiazide Drugs 0.000 description 1
- HDWIHXWEUNVBIY-UHFFFAOYSA-N bendroflumethiazidum Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1=CC=CC=C1 HDWIHXWEUNVBIY-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- NWIUTZDMDHAVTP-UHFFFAOYSA-N betaxolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CCOCC1CC1 NWIUTZDMDHAVTP-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000029918 bioluminescence Effects 0.000 description 1
- 238000005415 bioluminescence Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- 229960000330 bupranolol Drugs 0.000 description 1
- HQIRNZOQPUAHHV-UHFFFAOYSA-N bupranolol Chemical compound CC1=CC=C(Cl)C(OCC(O)CNC(C)(C)C)=C1 HQIRNZOQPUAHHV-UHFFFAOYSA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- UJVLDDZCTMKXJK-WNHSNXHDSA-N canrenone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CCC(=O)C=C3C=C2)C)CC[C@@]11C)C[C@@]11CCC(=O)O1 UJVLDDZCTMKXJK-WNHSNXHDSA-N 0.000 description 1
- 229960005057 canrenone Drugs 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 235000013877 carbamide Nutrition 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 210000001715 carotid artery Anatomy 0.000 description 1
- 229960001222 carteolol Drugs 0.000 description 1
- LWAFSWPYPHEXKX-UHFFFAOYSA-N carteolol Chemical compound N1C(=O)CCC2=C1C=CC=C2OCC(O)CNC(C)(C)C LWAFSWPYPHEXKX-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 229960002320 celiprolol Drugs 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- JIVPVXMEBJLZRO-UHFFFAOYSA-N chlorthalidone Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-UHFFFAOYSA-N 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- GVPFVAHMJGGAJG-UHFFFAOYSA-L cobalt dichloride Chemical compound [Cl-].[Cl-].[Co+2] GVPFVAHMJGGAJG-UHFFFAOYSA-L 0.000 description 1
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 125000004652 decahydroisoquinolinyl group Chemical group C1(NCCC2CCCCC12)* 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000007257 deesterification reaction Methods 0.000 description 1
- 238000005661 deetherification reaction Methods 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 201000009101 diabetic angiopathy Diseases 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- GJQPMPFPNINLKP-UHFFFAOYSA-N diclofenamide Chemical compound NS(=O)(=O)C1=CC(Cl)=C(Cl)C(S(N)(=O)=O)=C1 GJQPMPFPNINLKP-UHFFFAOYSA-N 0.000 description 1
- 229960005081 diclofenamide Drugs 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000005052 dihydropyrazolyl group Chemical group N1(NCC=C1)* 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000007336 electrophilic substitution reaction Methods 0.000 description 1
- 229950006127 embusartan Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 108010018033 endothelial PAS domain-containing protein 1 Proteins 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 229960001208 eplerenone Drugs 0.000 description 1
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 description 1
- 230000000913 erythropoietic effect Effects 0.000 description 1
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 description 1
- 229960003745 esmolol Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- SSVFMICWXDVRQN-UHFFFAOYSA-N ethanol;sodium Chemical compound [Na].CCO SSVFMICWXDVRQN-UHFFFAOYSA-N 0.000 description 1
- 238000006266 etherification reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- KOXLIKDTQXSKIE-UHFFFAOYSA-N ethyl 2-(1,2,4-triazol-1-yl)acetate Chemical compound CCOC(=O)CN1C=NC=N1 KOXLIKDTQXSKIE-UHFFFAOYSA-N 0.000 description 1
- BQVYJKKTPCABLS-UHFFFAOYSA-N ethyl 2-(4-cyanoimidazol-1-yl)-3-(dimethylamino)prop-2-enoate Chemical compound CCOC(=O)C(=CN(C)C)N1C=NC(C#N)=C1 BQVYJKKTPCABLS-UHFFFAOYSA-N 0.000 description 1
- RICHBBLPKXJZLP-UHFFFAOYSA-N ethyl 2-(4-cyanoimidazol-1-yl)acetate Chemical compound CCOC(=O)CN1C=NC(C#N)=C1 RICHBBLPKXJZLP-UHFFFAOYSA-N 0.000 description 1
- PDRSVDAPBJIQNT-UHFFFAOYSA-N ethyl 2-(4-methylimidazol-1-yl)acetate Chemical compound CCOC(=O)CN1C=NC(C)=C1 PDRSVDAPBJIQNT-UHFFFAOYSA-N 0.000 description 1
- DAWFJGMMMBJQBB-UHFFFAOYSA-N ethyl 2-(triazol-1-yl)acetate Chemical compound CCOC(=O)CN1C=CN=N1 DAWFJGMMMBJQBB-UHFFFAOYSA-N 0.000 description 1
- OKTBOSQQRWPUNX-UHFFFAOYSA-N ethyl 2-[4-(trifluoromethyl)imidazol-1-yl]acetate Chemical compound CCOC(=O)CN1C=NC(C(F)(F)F)=C1 OKTBOSQQRWPUNX-UHFFFAOYSA-N 0.000 description 1
- ZSIUZSJEAIUQBM-UHFFFAOYSA-N ethyl 2-[5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl]acetate Chemical compound CCOC(=O)CC1=NN=C(C(F)(F)F)S1 ZSIUZSJEAIUQBM-UHFFFAOYSA-N 0.000 description 1
- OITZNDMCFHYWLX-UHFFFAOYSA-N ethyl 2-imidazol-1-ylacetate Chemical compound CCOC(=O)CN1C=CN=C1 OITZNDMCFHYWLX-UHFFFAOYSA-N 0.000 description 1
- MZYBVBKJQVFPIK-UHFFFAOYSA-N ethyl 3-(dimethylamino)-2-(1,2,4-triazol-1-yl)prop-2-enoate Chemical compound CCOC(=O)C(=CN(C)C)N1C=NC=N1 MZYBVBKJQVFPIK-UHFFFAOYSA-N 0.000 description 1
- BWHAFFALKZHREW-UHFFFAOYSA-N ethyl 3-(dimethylamino)-2-(4-methylimidazol-1-yl)prop-2-enoate Chemical compound CCOC(=O)C(=CN(C)C)N1C=NC(C)=C1 BWHAFFALKZHREW-UHFFFAOYSA-N 0.000 description 1
- XCWWIALUSMNFNR-UHFFFAOYSA-N ethyl 3-(dimethylamino)-2-(triazol-1-yl)prop-2-enoate Chemical compound CCOC(=O)C(=CN(C)C)N1C=CN=N1 XCWWIALUSMNFNR-UHFFFAOYSA-N 0.000 description 1
- SMHMZCONMRESKF-UHFFFAOYSA-N ethyl 3-(dimethylamino)-2-[4-(trifluoromethyl)imidazol-1-yl]prop-2-enoate Chemical compound CCOC(=O)C(=CN(C)C)N1C=NC(C(F)(F)F)=C1 SMHMZCONMRESKF-UHFFFAOYSA-N 0.000 description 1
- YPRNLCTVULJHDR-UHFFFAOYSA-N ethyl 3-(dimethylamino)-2-imidazol-1-ylprop-2-enoate Chemical compound CCOC(=O)C(=CN(C)C)N1C=CN=C1 YPRNLCTVULJHDR-UHFFFAOYSA-N 0.000 description 1
- 125000004705 ethylthio group Chemical group C(C)S* 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000000855 fungicidal effect Effects 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 210000004051 gastric juice Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 229960005150 glycerol Drugs 0.000 description 1
- 230000034659 glycolysis Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 210000003709 heart valve Anatomy 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 230000002949 hemolytic effect Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 239000004009 herbicide Substances 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 102000044890 human EPO Human genes 0.000 description 1
- 102000049131 human HIF1A Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 125000000717 hydrazino group Chemical group [H]N([*])N([H])[H] 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- IBTWUVRCFHJPKN-UHFFFAOYSA-N hydron;pyridine-3-carboxylic acid;chloride Chemical compound Cl.OC(=O)C1=CC=CN=C1 IBTWUVRCFHJPKN-UHFFFAOYSA-N 0.000 description 1
- 230000001096 hypoplastic effect Effects 0.000 description 1
- 230000002989 hypothyroidism Effects 0.000 description 1
- 208000003532 hypothyroidism Diseases 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- NDDAHWYSQHTHNT-UHFFFAOYSA-N indapamide Chemical compound CC1CC2=CC=CC=C2N1NC(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 NDDAHWYSQHTHNT-UHFFFAOYSA-N 0.000 description 1
- 229960004569 indapamide Drugs 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000035987 intoxication Effects 0.000 description 1
- 231100000566 intoxication Toxicity 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N iron oxide Inorganic materials [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- QXEWZXJUGTUHSQ-UHFFFAOYSA-L iron(2+) 2-oxopentanedioate Chemical compound [Fe+2].[O-]C(=O)CCC(=O)C([O-])=O QXEWZXJUGTUHSQ-UHFFFAOYSA-L 0.000 description 1
- BAUYGSIQEAFULO-UHFFFAOYSA-L iron(2+) sulfate (anhydrous) Chemical compound [Fe+2].[O-]S([O-])(=O)=O BAUYGSIQEAFULO-UHFFFAOYSA-L 0.000 description 1
- VBMVTYDPPZVILR-UHFFFAOYSA-N iron(2+);oxygen(2-) Chemical class [O-2].[Fe+2] VBMVTYDPPZVILR-UHFFFAOYSA-N 0.000 description 1
- 229910000359 iron(II) sulfate Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- 229960004592 isopropanol Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229960002479 isosorbide Drugs 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 229960001632 labetalol Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- WMDSZGFJQKSLLH-RBBKRZOGSA-N landiolol Chemical compound O1C(C)(C)OC[C@H]1COC(=O)CCC(C=C1)=CC=C1OC[C@@H](O)CNCCNC(=O)N1CCOCC1 WMDSZGFJQKSLLH-RBBKRZOGSA-N 0.000 description 1
- 229950005241 landiolol Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229960003134 mepindolol Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- FLOSMHQXBMRNHR-DAXSKMNVSA-N methazolamide Chemical compound CC(=O)\N=C1/SC(S(N)(=O)=O)=NN1C FLOSMHQXBMRNHR-DAXSKMNVSA-N 0.000 description 1
- 229960004083 methazolamide Drugs 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- LYVGOAYMIAQLHI-UHFFFAOYSA-N methyl 2-butyl-1-[[2-fluoro-4-[2-(2h-tetrazol-5-yl)phenyl]phenyl]methyl]-6-oxopyridine-4-carboxylate Chemical compound CCCCC1=CC(C(=O)OC)=CC(=O)N1CC1=CC=C(C=2C(=CC=CC=2)C2=NNN=N2)C=C1F LYVGOAYMIAQLHI-UHFFFAOYSA-N 0.000 description 1
- OVXWRTMLZCMTSG-UHFFFAOYSA-N methyl 3-(dimethylamino)-2-[5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl]prop-2-enoate Chemical compound COC(=O)C(=CN(C)C)C1=NN=C(C(F)(F)F)S1 OVXWRTMLZCMTSG-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- BQIPXWYNLPYNHW-UHFFFAOYSA-N metipranolol Chemical compound CC(C)NCC(O)COC1=CC(C)=C(OC(C)=O)C(C)=C1C BQIPXWYNLPYNHW-UHFFFAOYSA-N 0.000 description 1
- 229960002704 metipranolol Drugs 0.000 description 1
- AQCHWTWZEMGIFD-UHFFFAOYSA-N metolazone Chemical compound CC1NC2=CC(Cl)=C(S(N)(=O)=O)C=C2C(=O)N1C1=CC=CC=C1C AQCHWTWZEMGIFD-UHFFFAOYSA-N 0.000 description 1
- 229960002817 metolazone Drugs 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 206010062198 microangiopathy Diseases 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- PZRHRDRVRGEVNW-UHFFFAOYSA-N milrinone Chemical compound N1C(=O)C(C#N)=CC(C=2C=CN=CC=2)=C1C PZRHRDRVRGEVNW-UHFFFAOYSA-N 0.000 description 1
- 229960003574 milrinone Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 description 1
- 229960003702 moxifloxacin Drugs 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- AEXITZJSLGALNH-UHFFFAOYSA-N n'-hydroxyethanimidamide Chemical compound CC(N)=NO AEXITZJSLGALNH-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001298 n-hexoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 108010087904 neutravidin Proteins 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 230000007959 normoxia Effects 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 125000005061 octahydroisoindolyl group Chemical group C1(NCC2CCCCC12)* 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 229940100692 oral suspension Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960004570 oxprenolol Drugs 0.000 description 1
- 150000002926 oxygen Chemical class 0.000 description 1
- 230000019039 oxygen homeostasis Effects 0.000 description 1
- 230000008058 pain sensation Effects 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960002035 penbutolol Drugs 0.000 description 1
- KQXKVJAGOJTNJS-HNNXBMFYSA-N penbutolol Chemical compound CC(C)(C)NC[C@H](O)COC1=CC=CC=C1C1CCCC1 KQXKVJAGOJTNJS-HNNXBMFYSA-N 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 150000002960 penicillins Chemical class 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000000813 peptide hormone Substances 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960002164 pimobendan Drugs 0.000 description 1
- GLBJJMFZWDBELO-UHFFFAOYSA-N pimobendane Chemical compound C1=CC(OC)=CC=C1C1=NC2=CC=C(C=3C(CC(=O)NN=3)C)C=C2N1 GLBJJMFZWDBELO-UHFFFAOYSA-N 0.000 description 1
- 229960002508 pindolol Drugs 0.000 description 1
- PHUTUTUABXHXLW-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=NC=C[C]12 PHUTUTUABXHXLW-UHFFFAOYSA-N 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229960005483 polythiazide Drugs 0.000 description 1
- 229920000046 polythiazide Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 150000003147 proline derivatives Chemical class 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- IVRLZJDPKUSDCF-UHFFFAOYSA-N pyrazin-2-ylhydrazine Chemical compound NNC1=CN=CC=N1 IVRLZJDPKUSDCF-UHFFFAOYSA-N 0.000 description 1
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical compound O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 1
- WDFVICOGUPEJEQ-UHFFFAOYSA-N pyrazol-3-one;hydrochloride Chemical compound Cl.O=C1C=CN=N1 WDFVICOGUPEJEQ-UHFFFAOYSA-N 0.000 description 1
- 150000003217 pyrazoles Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- CLKZWXHKFXZIMA-UHFFFAOYSA-N pyrinuron Chemical group C1=CC([N+](=O)[O-])=CC=C1NC(=O)NCC1=CC=CN=C1 CLKZWXHKFXZIMA-UHFFFAOYSA-N 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000007660 quinolones Chemical class 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 230000000552 rheumatic effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 231100000872 sexual dysfunction Toxicity 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 1
- 229960002256 spironolactone Drugs 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000012192 staining solution Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 1
- 229960000835 tadalafil Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-N tert-butylcarbamic acid Chemical compound CC(C)(C)NC(O)=O XBXCNNQPRYLIDE-UHFFFAOYSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001166 thiolanyl group Chemical group 0.000 description 1
- 108060008226 thioredoxin Proteins 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 239000000225 tumor suppressor protein Substances 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 235000019163 vitamin B12 Nutrition 0.000 description 1
- 239000011715 vitamin B12 Substances 0.000 description 1
- 208000030401 vitamin deficiency disease Diseases 0.000 description 1
- 102100035070 von Hippel-Lindau disease tumor suppressor Human genes 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Virology (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Vascular Medicine (AREA)
- Rheumatology (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Tropical Medicine & Parasitology (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Plant Substances (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description



São preferidos radicais de dialquilamino de cadeia linear ou ramificada, tendo, em cada caso, 1 a 4 átomos de carbono. Podem ser mencionados, por exemplo, e de preferência: N,N-dimetilamino, N,N-dietilamino, N-etil-N-metilamino, N-metil-N-n-propilamino, N-isopropil-N-n-propilamino, N,N-di-isopropilamino, N-n-butil-N-metilamino, N-t-butil-N-metilamino, N-metil-N-n-pentilamino e N-n-hexil-N-metilamino.








e seus sais, solvatos e solvatos dos sais.









A. Exemplos
Abreviações e acrônimos:
aq. solução aquosa
cat. catalítico(a)
d dia(s)
DCI ionização química direta (em MS)
DMF dimetilformamida
DMSO sulfóxido de dimetila
do t. do teórico (rendimento)
EI ionização por impacto de elétrons (em MS)
ESI ionização por spray de elétrons (em MS)
Et etila
GC-MS espectroscopia de massa acoplada à cromatografia em gás
h hora(s)
HPLC cromatografia em líquido de elevado desempenho, em alta pressão
conc. concentrado(a)
LC-MS espectroscopia de massa acoplada à cromatografia em líquido
Met. método
min minuto(s)
MS espectroscopia de massa
RMN espectroscopia de ressonância magnética nuclear
Rt tempo de retenção (em HPLC)
TA temperatura ambiente
TFA ácido trifluroacético
THF tetra-hidrofurano









Rendimento: 6,4 g (47 % do teórico)








Rendimento: 7,0 g (88 % do teórico)


Rendimento: 2,2 g (68 % do teórico)


Rendimento: 42,0 g (99 % do teórico)








Rendimento: 1,5 g (80 % do teórico)


Rendimento: 0,8 g (69% do teórico)



Rendimento: 3,0 g (82% do teórico)

Rendimento: 0,8 g (42% do teórico)























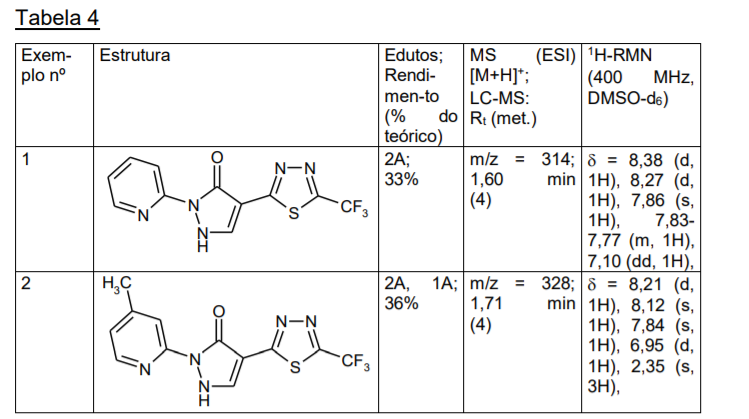






















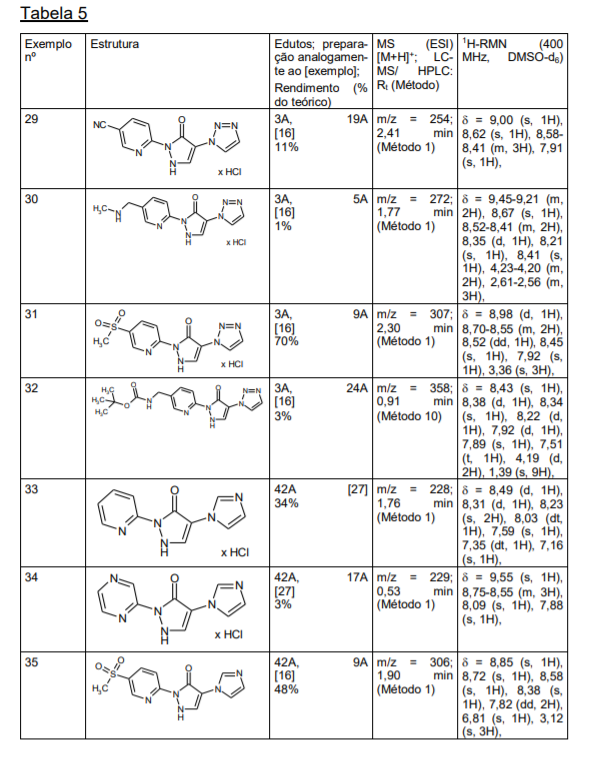






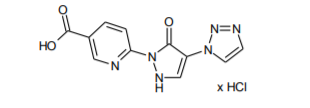






























Estágio b)
Cloridrato de 2-[6-(azetidin-3-ilóxi) pirimidin-4-il1-4-(1H-1,2,3-triazol-1-il)-1,2-di-hidro-3H-pirazol-3-ona






























Abreviações:
DMEM Meio Eagle modificado por Dulbecco
FCS soro de bezerro fetal
TMB 3,3',5,5'-tetrametil-benzidina
Tris tris (hidróxi-metil)-aminometano
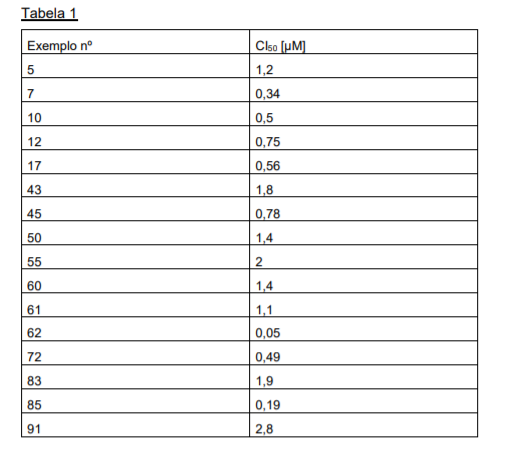

Claims (11)
- Composto, caracterizado pelo fato de que apresenta a Fórmula (I):na qual

R1 representa um grupo heteroarila da Fórmula:nas quais
* significa o ponto de ligação com o anel de di-hidropirazolona, e
R4 significa hidrogênio, flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroximetila, (C1-C4)-alcóxi, trifluorometóxi, hidróxi-carbonila ou (C1-C4)-alcóxi-carbonila,
R2 representa um grupo heteroarila da Fórmula:nas quais
# significa o ponto de ligação com o anel de di-hidropirazolona, e
R6, R6A e R6B são iguais ou diferentes e, independentemente um do outro, significam hidrogênio ou um substituinte escolhido a partir da série consistindo em flúor, cloro, bromo, ciano, (C1-C6)-alquila, trifluorometila, hidroxila, (C1-C6)-alcóxi, trifluorometóxi, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila, (C1-C4)-alcóxi-carbonila, heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros, sendo que
(C1-C6)-alquila, por sua vez, pode estar substituído com hidroxila, (C1-C4)-alcóxi ou amino, e
heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros, por sua vez, podem estas substituídas, em cada caso, uma vez ou duas vezes, de uma maneira igual ou diferente, com flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroxila, (C1-C4)-alcóxi, trifluorometóxi, oxo, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila e/ou (C1-C4)-alcóxi-carbonila, e
R3 representa hidrogênio,
e seus sais, solvatos e solvatos dos sais. - Composto da Fórmula (I), de acordo com a reivindicação 1, caracterizado pelo fato de que:
R1 representa um grupo heteroarila da Fórmula:na qual
* significa o ponto de ligação com o anel de di-hidropirazolona, e
R4 significa hidrogênio, flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroximetila, (C1-C4)-alcóxi, trifluorometóxi, hidróxi-carbonil ou (C1-C4)-alcóxi-carbonila,
R2 representa um grupo heteroarila da Fórmula:nas quais
# significa o ponto de ligação com o anel de di-hidroplrazolona, e
R6, R6A e R6B são iguais ou diferentes e, independentemente um do outro, significam hidrogênio ou um substituinte escolhido a partir da série consistindo em flúor, cloro, bromo, ciano, (C1-C6)-alquila, trifluorometila, hidroxila, (C1-C6)-alcóxi, trifluorometóxi, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila, (C1-C4)-alcóxi-carbonila, heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros, sendo que
(C1-C6)-alquila, por sua vez, pode estar substituída com hidroxila, (C1-C4)-alcóxi ou amino, e
heterocicloalquila de 4 a 6 membros, por sua vez, pode estar substituída, uma vez ou duas vezes, de uma maneira igual ou diferente, com flúor, ciano, (C1-C4)-alquila, trifluorometila, hidroxila, (C1-C4)-alcóxi, oxo, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila e/ou (C1-C4)-alcóxi-carbonila, e
R3 representa hidrogênio,
e seus sais, solvatos e solvatos dos sais. - Composto da Fórmula (I), de acordo com a reivindicação 1, caracterizado pelo fato de que:
R1 representa um grupo heteroarila da Fórmula:nas quais
* significa o ponto de ligação com o anel de di-hidropirazolona, e
R4 significa hidrogênio, flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroximetila, (C1-C4)-alcóxi, trifluorometóxi, hidróxi-carbonil ou (C1-C4)-alcóxi-carbonila,
R2 representa um grupo heteroarila da Fórmula:na qual
# significa o ponto de ligação com o anel de di-hidropirazolona, e
R6 significa hidrogênio ou um substituinte escolhido a partir da série consistindo em flúor, cloro, bromo, ciano, (C1-C6)-alquila, trifluorometila, hidroxila, (C1-C6)-alcóxi, trifluorometóxi, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila, (C1-C4)-alcóxi-carbonila, heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros, sendo que
(C1-C6)-alquila, por sua vez, pode estar substituída com hidroxila, (C1-C4)-alcóxi ou amino, e
heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros por sua vez, pode estar substituída, uma vez ou duas vezes, de uma maneira igual ou diferente, com flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroxila, (C1-C4)-alcóxi, trifluorometóxi, oxo, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila e/ou (C1-C4)-alcóxi-carbonila, e
R3 representa hidrogênio,
e seus sais, solvatos e solvatos dos sais. - Composto da Fórmula (I), de acordo com qualquer uma das reivindicações 1 a 3, caracterizado pelo fato de que:
R1 representa um grupo heteroarila da Fórmula:na qual
* significa o ponto de ligação com o anel de di-hidropirazolona, e
R4 significa hidrogênio, flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroximetila, (C1-C4)-alcóxi, trifluorometóxi, hidróxi-carbonil ou (C1-C4)-alcóxi-carbonila,
R2 representa um grupo heteroarila da Fórmula:na qual
# significa o ponto de ligação com o anel de di-hidropirazolona, e
R6 significa hidrogênio ou um substituinte escolhido a partir da série consistindo em flúor, cloro, bromo, ciano, (C1-C6)-alquila, trifluorometila, hidroxila, (C1-C6)-alcóxi, trifluorometóxi, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila, (C1-C4)-alcóxi-carbonila, heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros, sendo que
(C1-C6)-alquila, por sua vez, pode estar substituída com hidroxila, (C1-C4)-alcóxi ou amino, e
heterocicloalquila de 4 a 6 membros, fenila e heteroarila de 5 ou 6 membros por sua vez, pode estar substituída, uma vez ou duas vezes, de uma maneira igual ou diferente, com flúor, cloro, bromo, ciano, (C1-C4)-alquila, trifluorometila, hidroxila, (C1-C4)-alcóxi, trifluorometóxi, oxo, amino, mono-(C1-C4)-alquilamino, di-(C1-C4)-alquilamino, hidróxi-carbonila e/ou (C1-C4)-alcóxi-carbonila, e
R3 representa hidrogênio,
e seus sais, solvatos e solvatos dos sais. - Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que é 2-(6-Morfolin-4-ilpirimidin-4-il)-4-(1H-1,2,3-triazol-1-il)-1,2-di-hidro-3H-pirazol-3-ona, e que apresenta a seguinte Fórmula:e seus sais, solvatos e solvatos dos sais.

- Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que é 2-(6-Morfolin-4-ilpirimidin-4-il)-4-(1H-1,2,3-triazol-1-il)-1,2-di-hidro-3H-pirazol-3-ona, e que apresenta a seguinte Fórmula:

- Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que é cloridrato de 2-(6-morfolin-4-ilpirimidin-4-il)-4-(1H-1,2,3-triazol-1-il)-1,2-di-hidro-3H-pirazol-3-ona, e que apresenta a seguinte Fórmula:

- Processo para preparação de um composto da Fórmula (I), como definido em qualquer uma das reivindicações 1 a 7, na qual R3 representa hidrogênio, caracterizado pelo fato de que um composto da Fórmula (V):na qual

R1 é como definido em qualquer uma das reivindicações 1 a 7, e
Z1 representa metila ou etila,
é primeiramente submetido a uma reação de condensação com um composto da Fórmula (VI):na qual:
Z2 representa metila ou etila,
para formar compostos da Fórmula (VII):na qual
Z1 e R1 são como definidos em qualquer uma das reivindicações 1 a 7,
os quais são, então, reagidos, na presença de um ácido, com um composto da Fórmula (III),na qual
R2 é como definido em qualquer uma das reivindicações 1 a 7,
para formar compostos da Fórmula (IV-A):na qual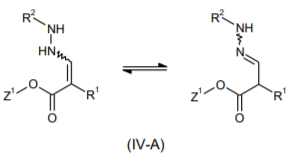
Z1, R1 e R2 são como definidos em qualquer uma das reivindicações 1 a 7,
que se ciclizam já sob estas condições de reação ou em uma etapa de reação subsequente, sob a influência de uma base, para formar os compostos da Fórmula (I), na qual R3 representa hidrogênio. - Uso de um composto, como definido em qualquer uma das reivindicações 1 a 7, caracterizado pelo fato de que é para preparação de um medicamento para tratamento e/ou a profilaxia de doenças cardiovasculares selecionadas dentre insuficiência cardíaca, doença cardíaca coronariana, angina pectoris, infarto do miocárdio, derrame, arterioesclerose, hipertensão essencial, pulmonar e maligna e doença oclusiva de artérias periféricas; anemia, doenças renais crônicas e insuficiência renal.
- Medicamento, caracterizado pelo fato de que compreende um composto, como definido em qualquer uma das reivindicações 1 a 7, em combinação com uma substância auxiliar farmaceuticamente adequada, não tóxica, inerte.
- Medicamento, caracterizado pelo fato de que compreende um composto, como definido em qualquer uma das reivindicações 1 a 7, em combinação com uma ou mais outras substâncias ativas escolhidas a partir do grupo consistindo em inibidores de ACE, antagonistas de receptor de angiotensina II, bloqueadores de receptor beta, antagonistas de cálcio, inibidores de PDE, antagonistas de receptor de mineralocorticoides, diuréticos, aspirina, suplementos de ferro, suplementos de vitamina B12 e de ácido fólico, estatinas, derivados de digitalis (digoxina), quimioterápicos de tumor e antibióticos.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102006050516.6 | 2006-10-26 | ||
| DE102006050516A DE102006050516A1 (de) | 2006-10-26 | 2006-10-26 | Substituierte Dihydropyrazolone und ihre Verwendung |
| PCT/EP2007/008877 WO2008067871A1 (de) | 2006-10-26 | 2007-10-12 | Substituierte dihydropyrazolone zur behandlung kardiovaskulärer und hämatologischer erkrankungen |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0717367A2 BRPI0717367A2 (pt) | 2013-10-22 |
| BRPI0717367B1 true BRPI0717367B1 (pt) | 2020-09-08 |
| BRPI0717367B8 BRPI0717367B8 (pt) | 2021-05-25 |
Family
ID=38734870
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0717367A BRPI0717367B8 (pt) | 2006-10-26 | 2007-10-12 | di-hidropirazolonas substituídas, seus usos e seu processo de preparação, e medicamento |
Country Status (40)
| Country | Link |
|---|---|
| US (5) | US8389520B2 (pt) |
| EP (3) | EP2084151B1 (pt) |
| JP (2) | JP5247712B2 (pt) |
| KR (1) | KR101455879B1 (pt) |
| CN (2) | CN101631785B (pt) |
| AR (1) | AR063392A1 (pt) |
| AU (1) | AU2007328052B2 (pt) |
| BR (1) | BRPI0717367B8 (pt) |
| CA (1) | CA2667382C (pt) |
| CO (1) | CO6170359A2 (pt) |
| CR (2) | CR10750A (pt) |
| CU (2) | CU23743B7 (pt) |
| CY (1) | CY1114272T1 (pt) |
| DE (1) | DE102006050516A1 (pt) |
| DK (2) | DK2546253T3 (pt) |
| ES (3) | ES2421454T3 (pt) |
| GT (1) | GT200900095A (pt) |
| HN (1) | HN2009000784A (pt) |
| HR (2) | HRP20130779T1 (pt) |
| IL (2) | IL198289A (pt) |
| MA (1) | MA30914B1 (pt) |
| MX (1) | MX2009004324A (pt) |
| MY (1) | MY169564A (pt) |
| NO (1) | NO343308B1 (pt) |
| NZ (2) | NZ576458A (pt) |
| PE (3) | PE20080895A1 (pt) |
| PH (1) | PH12013500578B1 (pt) |
| PL (2) | PL2546253T3 (pt) |
| PT (2) | PT2084151E (pt) |
| RS (1) | RS52924B (pt) |
| RU (2) | RU2469031C2 (pt) |
| SG (1) | SG176421A1 (pt) |
| SI (1) | SI2084151T1 (pt) |
| SV (1) | SV2009003236A (pt) |
| TN (1) | TN2009000132A1 (pt) |
| TW (3) | TW201630605A (pt) |
| UA (1) | UA98125C2 (pt) |
| UY (1) | UY30663A1 (pt) |
| WO (1) | WO2008067871A1 (pt) |
| ZA (1) | ZA200902705B (pt) |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR045047A1 (es) * | 2003-07-11 | 2005-10-12 | Arena Pharm Inc | Derivados arilo y heteroarilo trisustituidos como moduladores del metabolismo y de la profilaxis y tratamiento de desordenes relacionados con los mismos |
| DE102008020113A1 (de) | 2008-04-23 | 2009-10-29 | Bayer Schering Pharma Aktiengesellschaft | Substituierte Dihydropyrazolone und ihre Verwendung |
| DE102006050516A1 (de) * | 2006-10-26 | 2008-04-30 | Bayer Healthcare Ag | Substituierte Dihydropyrazolone und ihre Verwendung |
| DE102006050513A1 (de) | 2006-10-26 | 2008-04-30 | Bayer Healthcare Ag | Substitiuierte Dihydropyrazolone und ihre Verwendung |
| DE102006050515A1 (de) * | 2006-10-26 | 2008-04-30 | Bayer Healthcare Ag | Substituierte Dipyridiyl-dihydropyrazolone und ihre Verwendung |
| PT2294066E (pt) | 2008-04-28 | 2014-11-21 | Janssen Pharmaceutica Nv | Benzoimidazoles como inibidores da prolil-hidroxilase |
| FR2940651B1 (fr) * | 2008-12-29 | 2013-05-03 | Sanofi Aventis | Derives de 2-pyridin-2-yl-pyrazol-3(2h)-one,leur preparation et leur application en therapeutique comme activateurs de hif |
| FR2949466A1 (fr) * | 2009-08-28 | 2011-03-04 | Sanofi Aventis | Derives de 2-pyridin-2-yl-pyrazol-3(2h)-one, leur preparation et leur application en therapeutique comme activateurs de hif |
| MA32976B1 (fr) * | 2008-12-29 | 2012-01-02 | Sanofi Sa | Derives de 2-pyridin-2-yl-pyrazol-3(2h)-one, leur preparation et leur application en therapeutique comme activateurs de hif |
| AU2009334570B2 (en) | 2008-12-29 | 2016-06-09 | Sanofi | Derivatives of 2-pyridin-2-yl-pyrazol-3(2h)-one, preparation and therapeutic use thereof |
| IT1393337B1 (it) | 2009-03-06 | 2012-04-20 | Italiana Sint Spa | Sintesi di (4as, 7as)-ottaidro-1h-pirrolo[3,4-b]piridina |
| DE102010044131A1 (de) * | 2010-11-18 | 2012-05-24 | Bayer Schering Pharma Aktiengesellschaft | Substituiertes Natrium-1H-pyrazol-5-olat |
| GB201102659D0 (en) | 2011-02-15 | 2011-03-30 | Isis Innovation | Assay |
| GB201113101D0 (en) | 2011-07-28 | 2011-09-14 | Isis Innovation | Assay |
| WO2013063221A1 (en) | 2011-10-25 | 2013-05-02 | Janssen Pharmaceutica Nv | Meglumine salt formulations of 1-(5,6-dichloro-1h-benzo[d]imidazol-2-yl)-1h-pyrazole-4-carboxylic acid |
| EP2628722A1 (en) * | 2012-02-16 | 2013-08-21 | Bayer CropScience AG | CF3O-containing enaminoketones and their utilization for the preparation of CF3O-containing pyrazoles |
| CN104321318A (zh) * | 2012-03-30 | 2015-01-28 | 第一三共株式会社 | 4-烷酰基氨基-3-吡唑啉酮衍生物 |
| PL2847183T3 (pl) * | 2012-05-08 | 2017-11-30 | Bayer Pharma Aktiengesellschaft | Sposób wytwarzania związków triazolowych |
| CA2873674A1 (en) | 2012-05-29 | 2013-12-05 | Momentive Performance Materials Gmbh | Preparation of isocyanato silanes |
| EA031647B1 (ru) | 2013-03-29 | 2019-02-28 | Такеда Фармасьютикал Компани Лимитед | 6-(5-гидрокси-1h-пиразол-1-ил)никотинамидные ингибиторы phd |
| LT3057580T (lt) * | 2013-10-17 | 2021-04-12 | Bayer Pharma Aktiengesellschaft | Farmacinės dozavimo formos, kurių sudėtyje yra natrio-1-[6-(morfolin-4-il)pirimidin-4-il]-4-(1h-1,2,3-triazol-1-il)-1h-pirazoil-5-olatas |
| WO2016034108A1 (zh) | 2014-09-02 | 2016-03-10 | 广东东阳光药业有限公司 | 喹啉酮类化合物及其应用 |
| CN107778262A (zh) * | 2016-08-24 | 2018-03-09 | 尹玉新 | 1,5‑双取代四氮唑类化合物及其制备方法和应用 |
| CN109694380B (zh) * | 2017-10-23 | 2022-09-27 | 中国医药研究开发中心有限公司 | 二氢吡唑啉酮类化合物及其制备方法和医药用途 |
| CN111825690B (zh) * | 2019-04-17 | 2022-09-27 | 中国医药研究开发中心有限公司 | 一种phd抑制剂的新晶型及其制备方法 |
| US11579118B2 (en) | 2019-06-03 | 2023-02-14 | Tdw Delaware, Inc. | Single point contact triaxial sensor head for an inline inspection tool |
| US10702525B1 (en) | 2019-09-04 | 2020-07-07 | United Arab Emirates University | Pyrimidine derivatives as anti-diabetic agents |
| EP4103281A1 (en) * | 2020-02-11 | 2022-12-21 | Acurastem Inc. | Pikfyve kinase inhibitors |
| PY2112437A (es) | 2020-02-18 | 2022-08-16 | Bayer Ag | Nuevos compuestos de heteroaril-triazol como pesticidas |
| JP2023518543A (ja) * | 2020-03-20 | 2023-05-02 | アケビア セラピューティクス インコーポレイテッド | Phd阻害剤化合物、組成物、及びそれらの使用 |
| EP4121426A1 (en) * | 2020-03-20 | 2023-01-25 | Akebia Therapeutics Inc. | Phd inhibitor compounds, compositions, and use |
| EP3888684A1 (en) | 2020-03-31 | 2021-10-06 | Bayer Animal Health GmbH | Composition having improved voluntary acceptance |
| WO2021216530A1 (en) | 2020-04-20 | 2021-10-28 | Akebia Therapeutics, Inc. | Treatment of viral infections, of organ injury, and of related conditions using a hif prolyl hydroxylase inhibitor or a hif-alpha stabilizer |
| WO2022150623A1 (en) | 2021-01-08 | 2022-07-14 | Akebia Therapeutics, Inc. | Compounds and composition for the treatment of anemia |
| EP4347022A1 (en) | 2021-05-27 | 2024-04-10 | Keryx Biopharmaceuticals, Inc. | Pediatric formulations of ferric citrate |
| DE102021206771B4 (de) | 2021-06-29 | 2025-12-24 | Volkswagen Aktiengesellschaft | Verfahren zum Ausgeben eines Startbereichs für einen Parkvorgang eines Kraftfahrzeugs, elektronische Recheneinrichtung sowie Kraftfahrzeug |
| CN116120340B (zh) * | 2021-11-15 | 2023-10-31 | 艾立康药业股份有限公司 | 一种吡啶并噁嗪类化合物及其制备方法、组合物和用途 |
| CN116063233B (zh) * | 2022-10-31 | 2025-08-15 | 广州市联瑞制药有限公司 | 一种1h-咪唑-1-乙酸烃酯的制备方法 |
| GB202301059D0 (en) | 2023-01-25 | 2023-03-08 | Univ Oxford Innovation Ltd | PHD inhibitors |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE424016C (de) | 1923-05-20 | 1926-01-14 | Allg Vergasungs Ges M B H Fa | Schachtofen mit Schwelaufsatz zur Vergasung und Verschwelung bituminoeser Stoffe |
| FR1519069A (fr) | 1966-04-13 | 1968-03-29 | Geigy Ag J R | Dérivés du sulfanilamide et leur préparation |
| GR63123B (en) | 1975-12-11 | 1979-09-11 | Lilly Co Eli | Preparation process of novel 1,4-diphenyl-3-pyrazolin-5-ones |
| US4075003A (en) | 1975-12-11 | 1978-02-21 | Eli Lilly And Company | Novel herbicidal method utilizing 1,4-diphenyl-3-pyrazolin-5-ones |
| PL103509B1 (pl) * | 1976-09-20 | 1979-06-30 | Lilly Co Eli | Srodek chwastobojczy |
| US4663327A (en) | 1984-05-23 | 1987-05-05 | Bayer Aktiengesellschaft | 1-heteroaryl-4-aryl-pyrazolin-5-ones |
| DE3443308A1 (de) | 1984-11-28 | 1986-05-28 | Bayer Ag, 5090 Leverkusen | 1-heteroaryl-4-aryl-pyrazolin-5-one zur verwendung als arzneimittel |
| DE3527157A1 (de) | 1985-07-30 | 1987-02-12 | Bayer Ag | 1-heteroaryl-4-aryl-pyrazol-derivate |
| DE4204919A1 (de) | 1992-02-19 | 1993-08-26 | Bayer Ag | Verfahren zur herstellung von 2-chlor-5-alkylaminomethyl-pyridinen |
| DE4240168A1 (de) | 1992-11-30 | 1994-06-01 | Bayer Ag | Cumarin-Derivate, Verfahren zu ihrer Herstellung und ihrer Verwendung |
| FR2725988B1 (fr) | 1994-10-24 | 1997-01-24 | Roussel Uclaf | Nouveaux derives de pyrazolones et pyrazoles acides, leur procede de preparation, les nouveaux intermediaires obtenus, leur application a titre de medicaments et les compositions pharmaceutiques les renfermant |
| JPH10306077A (ja) * | 1997-05-07 | 1998-11-17 | Daiichi Rajio Isotope Kenkyusho:Kk | ビピラゾール誘導体並びにこれを有効成分とする医薬および試薬 |
| DE19909237A1 (de) | 1999-03-03 | 2000-09-07 | Merck Patent Gmbh | Pyrazol-3-on-derivate |
| AU783393B2 (en) * | 2000-03-20 | 2005-10-20 | N-Gene Research Laboratories Inc. | Propenecarboxylic acid amidoxime derivatives, a process for the preparation thereof, and pharmaceutical compositions containing the same |
| US6878729B2 (en) * | 2001-05-04 | 2005-04-12 | The Procter & Gamble Company | Medicinal uses of dihydropyrazoles |
| WO2002092573A2 (en) | 2001-05-16 | 2002-11-21 | Vertex Pharmaceuticals Incorporated | Heterocyclic substituted pyrazoles as inhibitors of src and other protein kinases |
| DE60223720T2 (de) | 2001-12-18 | 2008-10-30 | Merck & Co., Inc. | Heteroaryl-substituierte pyrazol-modulatoren des metabotropen glutamatrezeptors-5 |
| WO2003074550A2 (en) | 2002-03-01 | 2003-09-12 | Pintex Pharmaceuticals, Inc. | Pin1-modulating compounds and methods of use thereof |
| US8124582B2 (en) | 2002-12-06 | 2012-02-28 | Fibrogen, Inc. | Treatment of diabetes |
| WO2004087066A2 (en) | 2003-03-27 | 2004-10-14 | Emory University | Hif-1 inhibitors |
| CN100387594C (zh) | 2003-04-03 | 2008-05-14 | 麦克公司 | 二芳基取代的吡唑类代谢型谷氨酸受体-5调节剂 |
| EP1626718A4 (en) * | 2003-05-01 | 2010-05-05 | Panacea Pharmaceuticals Inc | METHOD FOR THE TREATMENT OF ISCHEMIC ILLNESSES |
| JP4499721B2 (ja) | 2003-06-30 | 2010-07-07 | ヒフ バイオ,インク. | 化合物、組成物および方法 |
| AT7634U1 (de) | 2004-06-29 | 2005-06-27 | Binder Co Ag | Detektiervorrichtung und sortiervorrichtung |
| WO2006088491A2 (en) * | 2004-06-29 | 2006-08-24 | Massachusetts Institute Of Technology | Methods and compositions related to the modulation of intercellular junctions |
| DE102008020113A1 (de) * | 2008-04-23 | 2009-10-29 | Bayer Schering Pharma Aktiengesellschaft | Substituierte Dihydropyrazolone und ihre Verwendung |
| CA2601098C (en) | 2005-03-16 | 2011-07-05 | Aventis Pharmaceuticals Inc. | Dipyrazoles as central nervous system agents |
| US8017631B2 (en) * | 2005-04-26 | 2011-09-13 | Neurosearch A/S | Oxadiazole derivatives and their medical use |
| DE102005019712A1 (de) * | 2005-04-28 | 2006-11-09 | Bayer Healthcare Ag | Dipyridyl-dihydropyrazolone und ihre Verwendung |
| WO2007008541A2 (en) | 2005-07-08 | 2007-01-18 | Kalypsys, Inc. | Cellular cholesterol absorption modifiers |
| DE102006050515A1 (de) * | 2006-10-26 | 2008-04-30 | Bayer Healthcare Ag | Substituierte Dipyridiyl-dihydropyrazolone und ihre Verwendung |
| DE102006050513A1 (de) | 2006-10-26 | 2008-04-30 | Bayer Healthcare Ag | Substitiuierte Dihydropyrazolone und ihre Verwendung |
| DE102006050516A1 (de) * | 2006-10-26 | 2008-04-30 | Bayer Healthcare Ag | Substituierte Dihydropyrazolone und ihre Verwendung |
| DE102010044131A1 (de) | 2010-11-18 | 2012-05-24 | Bayer Schering Pharma Aktiengesellschaft | Substituiertes Natrium-1H-pyrazol-5-olat |
-
2006
- 2006-10-26 DE DE102006050516A patent/DE102006050516A1/de not_active Withdrawn
-
2007
- 2007-10-12 EP EP07818948.7A patent/EP2084151B1/de active Active
- 2007-10-12 HR HRP20130779TT patent/HRP20130779T1/hr unknown
- 2007-10-12 ES ES07818948T patent/ES2421454T3/es active Active
- 2007-10-12 EP EP12188156.9A patent/EP2546253B1/de active Active
- 2007-10-12 AU AU2007328052A patent/AU2007328052B2/en active Active
- 2007-10-12 JP JP2009533691A patent/JP5247712B2/ja active Active
- 2007-10-12 UA UAA200905266A patent/UA98125C2/ru unknown
- 2007-10-12 DK DK12188156.9T patent/DK2546253T3/en active
- 2007-10-12 DK DK07818948.7T patent/DK2084151T3/da active
- 2007-10-12 PT PT78189487T patent/PT2084151E/pt unknown
- 2007-10-12 NZ NZ576458A patent/NZ576458A/en unknown
- 2007-10-12 RS RS20130347A patent/RS52924B/sr unknown
- 2007-10-12 CA CA2667382A patent/CA2667382C/en active Active
- 2007-10-12 SI SI200731293T patent/SI2084151T1/sl unknown
- 2007-10-12 EP EP11175197.0A patent/EP2383269B1/de active Active
- 2007-10-12 PT PT121881569T patent/PT2546253E/pt unknown
- 2007-10-12 MX MX2009004324A patent/MX2009004324A/es active IP Right Grant
- 2007-10-12 BR BRPI0717367A patent/BRPI0717367B8/pt active IP Right Grant
- 2007-10-12 PL PL12188156T patent/PL2546253T3/pl unknown
- 2007-10-12 WO PCT/EP2007/008877 patent/WO2008067871A1/de not_active Ceased
- 2007-10-12 ES ES11175197.0T patent/ES2621787T3/es active Active
- 2007-10-12 SG SG2011078359A patent/SG176421A1/en unknown
- 2007-10-12 KR KR1020097010634A patent/KR101455879B1/ko active Active
- 2007-10-12 CN CN2007800482623A patent/CN101631785B/zh active Active
- 2007-10-12 MY MYPI20091696A patent/MY169564A/en unknown
- 2007-10-12 CN CN201310122718.3A patent/CN103342696B/zh not_active Expired - Fee Related
- 2007-10-12 US US12/447,192 patent/US8389520B2/en active Active
- 2007-10-12 PL PL07818948T patent/PL2084151T3/pl unknown
- 2007-10-12 ES ES12188156.9T patent/ES2534872T3/es active Active
- 2007-10-12 NZ NZ596583A patent/NZ596583A/xx unknown
- 2007-10-12 RU RU2009119431/04A patent/RU2469031C2/ru active
- 2007-10-25 TW TW104139543A patent/TW201630605A/zh unknown
- 2007-10-25 PE PE2007001451A patent/PE20080895A1/es not_active Application Discontinuation
- 2007-10-25 TW TW096139977A patent/TWI437991B/zh active
- 2007-10-25 AR ARP070104726A patent/AR063392A1/es active IP Right Grant
- 2007-10-25 PE PE2011001768A patent/PE20120620A1/es active IP Right Grant
- 2007-10-25 TW TW102116012A patent/TWI561237B/zh not_active IP Right Cessation
- 2007-10-25 PE PE2012000031A patent/PE20120635A1/es active IP Right Grant
- 2007-10-29 UY UY30663A patent/UY30663A1/es active IP Right Grant
-
2009
- 2009-04-10 TN TNP2009000132A patent/TN2009000132A1/fr unknown
- 2009-04-20 ZA ZA200902705A patent/ZA200902705B/xx unknown
- 2009-04-22 IL IL198289A patent/IL198289A/en active IP Right Grant
- 2009-04-23 GT GT200900095A patent/GT200900095A/es unknown
- 2009-04-23 CU CU20090066A patent/CU23743B7/es active IP Right Grant
- 2009-04-23 SV SV2009003236A patent/SV2009003236A/es unknown
- 2009-04-23 HN HN2009000784A patent/HN2009000784A/es unknown
- 2009-04-24 CR CR10750A patent/CR10750A/es unknown
- 2009-04-24 CO CO09041439A patent/CO6170359A2/es active IP Right Grant
- 2009-04-27 NO NO20091655A patent/NO343308B1/no unknown
- 2009-05-21 MA MA31909A patent/MA30914B1/fr unknown
-
2010
- 2010-10-12 JP JP2010229627A patent/JP5300813B2/ja active Active
-
2011
- 2011-05-26 CU CU2011000119A patent/CU24056B1/es active IP Right Grant
-
2012
- 2012-01-23 US US13/356,256 patent/US8653111B2/en active Active
- 2012-08-10 RU RU2012134284A patent/RU2611012C2/ru active
- 2012-09-13 IL IL221928A patent/IL221928A/en active IP Right Grant
-
2013
- 2013-02-18 US US13/769,513 patent/US8987261B2/en active Active
- 2013-03-26 PH PH12013500578A patent/PH12013500578B1/en unknown
- 2013-08-21 CY CY20131100718T patent/CY1114272T1/el unknown
-
2014
- 2014-08-21 CR CR20140392A patent/CR20140392A/es unknown
-
2015
- 2015-02-04 US US14/613,367 patent/US9168249B2/en active Active
- 2015-04-10 HR HRP20150396TT patent/HRP20150396T1/hr unknown
- 2015-09-25 US US14/866,323 patent/US20160229858A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DK2546253T3 (en) | SUBSTITUTED DIHYDROPYRAZOLONES FOR TREATMENT OF CARDIOVASCULAR AND HEMATOLOGICAL DISEASES | |
| AU2009240311B2 (en) | Substituted dihydropyrazolones as inhibitors of HIF-prolyl-4-hydroxylases | |
| CA2667392C (en) | Substituted dihydropyrazolones and use thereof as hif-prolyl-4 -hydroxylase inhibitors | |
| ES2595501T3 (es) | Dipiridil-dihidropirazolonas sustituidas y su uso | |
| HK1140478B (en) | Substituted dihydropyrazolones for treating cardiovascular and haematological diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B25D | Requested change of name of applicant approved |
Owner name: BAYER PHARMA AKTIENGESELLSCHAFT (DE) |
|
| B25A | Requested transfer of rights approved |
Owner name: BAYER INTELLECTUAL PROPERTY GMBH. (DE) |
|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001, QUE MODIFICOU A LEI NO 9279/96, A CONCESSAO DA PATENTE ESTA CONDICIONADA A ANUENCIA PREVIA DA ANVISA. CONSIDERANDO A APROVACAO DOS TERMOS DO PARECER NO 337/PGF/EA/2010, BEM COMO A PORTARIA INTERMINISTERIAL NO 1065 DE 24/05/2012, ENCAMINHA-SE O PRESENTE PEDIDO PARA AS PROVIDENCIAS CABIVEIS. |
|
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 08/09/2020, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 12/10/2007 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |
|
| B25G | Requested change of headquarter approved |
Owner name: BAYER INTELLECTUAL PROPERTY GMBH. (DE) |