DE60004652T2 - Verfahren zur herstellung von gabapentin - Google Patents
Verfahren zur herstellung von gabapentin Download PDFInfo
- Publication number
- DE60004652T2 DE60004652T2 DE60004652T DE60004652T DE60004652T2 DE 60004652 T2 DE60004652 T2 DE 60004652T2 DE 60004652 T DE60004652 T DE 60004652T DE 60004652 T DE60004652 T DE 60004652T DE 60004652 T2 DE60004652 T2 DE 60004652T2
- Authority
- DE
- Germany
- Prior art keywords
- gabapentin
- retentate
- solution
- membrane
- concentration
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 title claims abstract description 89
- 229960002870 gabapentin Drugs 0.000 title claims abstract description 44
- 238000004519 manufacturing process Methods 0.000 title claims description 4
- 238000000034 method Methods 0.000 claims abstract description 29
- 239000000243 solution Substances 0.000 claims abstract description 24
- 239000012528 membrane Substances 0.000 claims abstract description 21
- 239000012466 permeate Substances 0.000 claims abstract description 18
- 239000012465 retentate Substances 0.000 claims abstract description 17
- 238000011026 diafiltration Methods 0.000 claims abstract description 11
- XBUDZAQEMFGLEU-UHFFFAOYSA-N 2-[1-(aminomethyl)cyclohexyl]acetic acid;hydron;chloride Chemical compound Cl.OC(=O)CC1(CN)CCCCC1 XBUDZAQEMFGLEU-UHFFFAOYSA-N 0.000 claims abstract description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims abstract description 6
- 239000007864 aqueous solution Substances 0.000 claims abstract description 5
- 150000002500 ions Chemical class 0.000 claims abstract description 5
- 150000002894 organic compounds Chemical class 0.000 claims abstract description 5
- 238000002360 preparation method Methods 0.000 claims abstract description 5
- 150000001450 anions Chemical class 0.000 claims abstract description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 3
- 150000007514 bases Chemical class 0.000 claims abstract description 3
- 150000003839 salts Chemical group 0.000 claims description 11
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 5
- 230000014759 maintenance of location Effects 0.000 claims description 5
- 239000002131 composite material Substances 0.000 claims description 4
- 238000002425 crystallisation Methods 0.000 claims description 3
- 230000008025 crystallization Effects 0.000 claims description 3
- 238000001728 nano-filtration Methods 0.000 claims description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical class [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 2
- 239000004677 Nylon Substances 0.000 claims description 2
- 239000004952 Polyamide Substances 0.000 claims description 2
- 229910052783 alkali metal Inorganic materials 0.000 claims description 2
- 150000001340 alkali metals Chemical class 0.000 claims description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 claims description 2
- 229920002301 cellulose acetate Polymers 0.000 claims description 2
- 229920001778 nylon Polymers 0.000 claims description 2
- 229920002647 polyamide Polymers 0.000 claims description 2
- 229920005989 resin Polymers 0.000 claims description 2
- 239000011347 resin Substances 0.000 claims description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims 1
- 238000001704 evaporation Methods 0.000 abstract description 2
- 230000001376 precipitating effect Effects 0.000 abstract description 2
- 210000004379 membrane Anatomy 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- 239000008367 deionised water Substances 0.000 description 6
- 229910021641 deionized water Inorganic materials 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- 239000002033 PVDF binder Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 2
- JAWPQJDOQPSNIQ-UHFFFAOYSA-N 2-Azaspiro[4.5]decan-3-one Chemical compound C1NC(=O)CC21CCCCC2 JAWPQJDOQPSNIQ-UHFFFAOYSA-N 0.000 description 1
- -1 Polypropylene Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- PSLIMVZEAPALCD-UHFFFAOYSA-N ethanol;ethoxyethane Chemical compound CCO.CCOCC PSLIMVZEAPALCD-UHFFFAOYSA-N 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- LELOWRISYMNNSU-UHFFFAOYSA-N hydrogen cyanide Chemical class N#C LELOWRISYMNNSU-UHFFFAOYSA-N 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/38—Separation; Purification; Stabilisation; Use of additives
- C07C227/40—Separation; Purification
- C07C227/42—Crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pyridine Compounds (AREA)
- Separation Using Semi-Permeable Membranes (AREA)
Description
- Die vorliegende Beschreibung betrifft allgemein das Gebiet der pharmazeutischen Chemie.
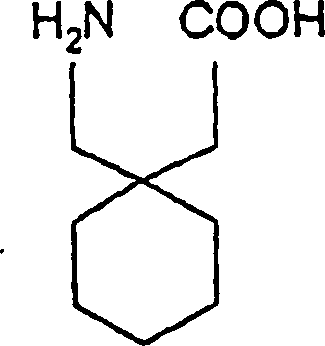
- Insbesondere betrifft die Erfindung ein Verfahren zur Herstellung von 1-(Aminomethyl)-1-cyclohexanessigsäure, ebenso unter dem Namen Gabapentin bekannt, das die folgende chemische Struktur besitzt:
- Gabapentin ist ein aktives Ingredienz, das zur Behandlung verschiedener celebraler Erkrankungen, wie Epilepsie, Hypokinese, kraniale Traumata und dergleichen, verwendet wird und ist Gegenstand der Patentschriften U.S. 4 024 175 und U.S. 4 087 544.
- Eine Anzahl von Verfahren zur Herstellung von Gabapentin sind in der Latur berichtet; beispielsweise beschreibt die U.S.-Patentschrift 4 024 175 einige Herstellungsverfahren, die von Cyclohexyl-1,1-diessigsäure ausgehen.
- Sämtliche Verfahren im Stand der Technik ergeben jedoch GabapentinHydrochlorid, das dann durch Behandlung mit einem basischen Ionenaustauscher und nachfolgender Kristallisation aus einem Ethanol-Äther-Gemisch in die freie Aminosäure überführt werden muss.
- Die U.S.-Patentschrift 4 894 476 beschreibt ein Verfahren zur Überführung von Gabapentin-Hydrochlorid in die freie Aminosäure, das das Durchlaufen einer wässrigen Lösung des Hydrochlorids durch eine Ionenaustauschharzsäule unter Verwendung großer Volumina an Wasser umfasst. Aufgrund der hohen Wasserlöslichkeit von nicht in Salzform vorliegendem Gabapentin müssen große Volumina an Wasser verdampft werden, um diese Verbindung zu gewinnen, wobei bei niedrigen Temperaturen gearbeitet wird, um eine Lactambildung zu verhindern.
- Alternative Verfahren sind zur Überwindung dieser Nachteile vorgeschlagen worden, die die Bildung des Hydrochlorids oder eines anderen Salzes vermeiden. Beispiele dieser Verfahren können in den Patentschriften U.S. 5 132 451, U.S. 5 095 148 und U.S. 5 068 413 gefunden werden und umfassen die Bildung eines Cyanderivats, das daraufhin unter drastischen Bedingungen unter Bildung der freien Aminosäure hydriert wird. Diese Verfahren bringen jedoch die Verwendung von Cyanwasserstoffsäurederivaten mit sich, was sie industriell nicht praktikabel macht.
- In einer neuen Patentanmeldung, WO-A-98/28255, wird ein neuartiges Verfahren zur Herstellung von Gabapentin beschrieben, das vom entsprechenden Hydrochlorid ausgeht und es gestattet, die Probleme der Verfahren aus dem Stand der Technik, insbesondere aus der U.S.-Patentschrift 4 894 476, zu lösen, die mit der Notwendigkeit, große Volumina an Wasser bei niedrigen Temperaturen zu verdampfen, verbunden sind, um freies Gabapentin zu gewinnen. Das Verfahren umfasst die folgenden Schritte:
- – Freisetzung von Gabapentinhydrochlorid aus anorganischen Salzen aus der Synthese durch Lösen von Gabapentinhydrochlorid in organischen Lösungsmitteln, in denen die anorganischen Salze unlöslich sind, und nachfolgende Filtration und Verdampfen des Lösungsmittels,
- – Verdrängung von HCl mit Aminen, wie Tributylamin, Triethylamin und dergleichen, in einem Lösungsmittel, in dem die Hydrochloride dieser Amine löslich sind, freies Gabapentin jedoch nicht löslich ist, das daher in der kristallinen Form III ausfällt, und
- – Umwandlung von Gabapentin der Form III in Gabapentin der Form II.
- Die vorliegende Erfindung zielt auf die Bereitstellung eines Verfahrens zur Herstellung von freiem Gabapentin ausgehend von Gabapentinhydrochlorid, das die oben im Zusammenhang mit dem Verfahren aus der U.S.-Patentschrift 4 894 476 erwähnten Nachteile gemäß alternativer Verfahren zu den in der Patentanmeldung WO-A-98/28255 vorgeschlagenen überwindet.
- Die Aufgabe wird erfindungsgemäß durch ein Verfahren gelöst, das die Schritte:
- – des Herstellens einer wässrigen Lösung von Gabapentinhydrochlorid,
- – des Einstellens des pH-Wertes dieser Lösung auf oder um den isoelektrischen Punkt von Gabapentin durch Zugabe einer basischen Verbindung, die ein einwertiges Anion umfasst,
- – der Diafiltration der Lösung durch eine Membran, die für organische Verbindungen mit einem Molekulargewicht größer als 150 hochselektiv ist und wenig selektiv für anorganische einwertige Ionen ist, um diese Lösung in ein wässriges, Chloridionen-enthaltendes Permeat und ein Retentat aufzutrennen, das nicht in Salzform vorliegendes Gabapentin, im Wesentlichen frei von Chloridionen, enthält,
- – des Konzentrierens des Retentats durch Erhöhung des Drucks auf die Membran, um eine Konzentration von nicht in Salzform vorliegendem Gabapentin in dem Retentat von nicht weniger als 5% zu erhalten,
- – des Einengens des Retentats unter verringertem Druck und bei einer Temperatur T < 35°C und
- – des Ausfällens von Gabapentin durch Zugabe eines Alkohols
- Das erfindungsgemäße Verfahren umfasst geeigneterweise einen weiteren Kristallisationsschritt des ausgefällten, nicht in Salzform vorliegenden Gabapentins mit Methanol.
- Die obige basische Verbindung ist vorzugsweise ausgewählt aus der Gruppe bestehend aus Alkalimetall- und Ammoniumhydroxiden.
- Der pH-Wert der wässrigen Ausgangslösung ist vorzugsweise auf 7,14 eingestellt.
- Während des Diafiltrationsschrittes ist der Druck auf die Membran 10 bis 16 bar, vorzugsweise 14 bis 15 bar.
- Während des Konzentrationsschrittes wird die Konzentration von nicht in Salzform vorliegendem Gabapentin im Retentat vorzugsweise auf einen Wert von etwa 8 bis 10% G/V gebracht. Während dieses Konzentrationsschrittes wird der Druck allmählich erhöht, bis er einen Wert von etwa 18 bis 22 bar erreicht.
- Während der Diafiltration und der Konzentrationsschritte wird die Temperatur vorzugsweise unter 25°C gehalten.
- Der Diafiltrationsschritt wird vorzugsweise mit Hilfe einer Nanofiltration-Mehrschichtverbundmembran durchgeführt, wie die von PERMEARE S.r.l. (Italien) vertriebenen, insbesondere die Membran mit der Bezeichnung ACN2540HS. Derartige Membranen besitzen eine Rückhalterate für anorganische Salze unter oder gleich 50% und eine Rückhalterate für organische Moleküle von über oder gleich 96%. Geeignete Verbundmembranen besitzen eine dünne Schicht aus einem Polyamidmaterial als aktive Schicht. Andere geeignete Membranen für das erfindungsgemäße Verfahren sind diejenigen, welche aus Polysulfonsäureharzen, Nylon, Celluloseacetat, Polypropylen und Polyvinylidenfluorid (PVDF) hergestellt sind.
- Das erfindungsgemäße Verfahren löst die oben unter Bezug auf Verfahren aus dem Stand der Technik erwähnten, auf der Verwendung eines Ionenaustauschharzes beruhenden Probleme durch Verwendung einer einfachen Technik, die keine drastischen Betriebsbedingungen, wie hohe Temperaturen, erfordert, was die unerwünschte Bildung von Gabapentinlactam bewirken würde.
- Weitere Vorteile des erfindungsgemäßen Verfahrens werden aus den folgenden Beispielen klarer.
- BEISPIEL 1
- 5 kg 1-(Aminomethyl)-1-cyclohexanesigsäure.HCl werden in 10 Volumina desionisiertem Wasser bei Raumtemperatur gelöst. Dieser Lösung werden langsam etwa 24 1 1 M NaOH zugegeben, bis der pH-Wert 7,14 (isoelektrischer Punkt von Gabapentin) erreicht, wobei gekühlt wird, um die Temperatur nicht über 20°C ansteigen zu lassen.
- Die erhaltene Lösung wird einer Diafiltration bei einer Temperatur von etwa 22°C in einer etwa 100 1 Pilotanlage unter Verwendung einer Nanofiltration-Mehrschichtverbundmembran PERMEARE ACN2540HS mit einer hohen Selektivität für organische Verbindungen mit einem Molekulargewicht von über 150 (Rückhalterate höher als oder gleich 98%) und niedriger Se lektivität für anorganische, einwertige Ionen (50% Rückhalterate) unterzogen.
- Der Druck auf die Membran ist etwa 14 bar und der Abflussdruck ist etwa 12 bar.
- Dem Permeat wird desionisiertes Wasser zugegeben, wobei das NaCl in dem Permeat dosiert wird.
- Die Werte bleiben in den ersten Permeatanteilen annähernd konstant, erniedrigen sich dann wieder. Die Permeatflussrate wird auf etwa 60 l/h festgesetzt. Nach 4 Stunden beträgt das NaCl im Permeat 98% des berechneten Gehalts. An diesem Punkt wird der Konzentrationsschritt begonnen. Die Leistung bleibt hoch und die in diesem Schritt beobachtete Druckerhöhung (der Einlassdruck ist bis auf etwa 22 bar und der Ausflussdruck auf etwa 20 bar erhöht), um die Permeatflussrate konstant zu halten, beruht auf einer Erhöhung des osmotischen Drucks in Folge der Erhöhung der Konzentration des gelösten Stoffes. Wenn ein Volumen von etwa 50 1 erreicht ist, wird die Retentatlösung, die etwa 10% nicht in Salzform vorliegendes Gabapentin enthält, gewonnen. Letztlich wird die Lösung unter verringertem Druck bei einer Temperatur < 35°C eingedampft, wobei das Volumen von 50 auf 10 1 verringert wird, und Gabapentin wird durch Zugabe von Isopropylalkohol ausgefällt. Der ausgefällte Feststoff wird letztlich durch Filtration gewonnen und aus Methanol unter Erhalt von 3,3 kg reinem Gabapentin kristallisiert.
- BEISPIEL 2
- 5 kg 1-(Aminomethyl)-1-cyclohexanessigsäure.HCl werden in 10 Volumeneinheiten desionisiertem Wasser bei Raumtemperatur gelöst. Dieser Lösung werden langsam etwa 24 1 1 M KOH zugegeben, bis ein pH-Wert von 7,14 (isoelektrischer Punkt von Ga bapentin) erreicht ist, wobei gekühlt wird, um die Temperatur nicht über 20°C ansteigen zu lassen.
- Die resultierende Lösung wird einer Diafiltration bei einer Temperatur von etwa 22°C in derselben Pilotanlage wie oben unter Verwendung der Membran PERMEARE ACN2540HS unterzogen. Der Druck auf die Membran ist etwa 14 bar und der Ausflussdruck ist etwa 12 bar.
- Dem Permeat wird desionisiertes Wasser zugegeben, um das KCl im Permeat zu dosieren.
- Die Werte bleiben in den ersten Permeatanteilen annähernd konstant, erniedrigen sich dann wieder. Die Permeatflussrate wird auf etwa 55 l/h eingestellt. Nach 4 Stunden beträgt das KCl im Permeat 98% des berechneten Gehalts. An diesem Punkt wird der Konzentrationsschritt begonnen, wobei der Einlassdruck auf etwa 22 bar erhöht wird. Wenn ein Volumen von etwa 50 1 erreicht ist, wird die Retentatlösung, die etwa 10% nicht in Salzform vorliegendes Gabapentin enthält, gewonnen und die gleichen Arbeitsgänge wie in Beispiel 1 werden unter Erhalt von 3,25 kg kristallinem, nicht in Salzform vorliegenden Gabapentin durchgeführt.
- BEISPIEL 3
- 5 kg 1-(Aminomethyl)-1-cyclohexanessigsäure.HCl werden in 10 Volumeneinheiten desionisiertem Wasser bei Raumtemperatur gelöst. Dieser Lösung werden langsam etwa 24 l 1 M NH4OH zugegeben, bis ein pH-Wert von 7,14 (isoelektrischer Punkt von Gabapentin) erreicht ist, wobei gekühlt wird, um die Temperatur nicht über 20°C ansteigen zu lassen.
- Die resultierende Lösung wird einer Diafiltration bei einer Temperatur von etwa 20°C in derselben Pilotanlage wie oben unter Verwendung der Membran PERMEARE ACN2540HS unterworfen. Der Druck auf die Membran ist etwa 14 bar und der Ausflussdruck etwa 12 bar.
- Dem Permeat wird desionisiertes Wasser zugegeben, um das NH4Cl im Permeat zu dosieren.
- Die Flussrate des Permeats wird auf etwa 65 l/h eingestellt. Nach 4 Stunden beträgt das NH4Cl im Permeat etwa 98% des berechneten Gehalts. An diesem Punkt wird der Konzentrationsschritt begonnen, wobei der Einlassdruck auf etwa 22 bar erhöht wird. Wenn ein Volumen von etwa 50 1 erreicht ist, wird die Retentatlösung, die 10% nicht in Salzform vorliegendes Gabapentin enthält, gewonnen und dieselben Arbeitsgänge wie in Beispiel 1 werden unter Erhalt von 3,3 kg kristallinem, nicht in Salzform vorliegenden Gabapentin durchgeführt.
umfasst.
Claims (8)
- Verfahren zur Herstellung von Gabapentin ausgehend von Gabapentinhydrochlorid, umfassend die nachfolgenden Schritte: – Herstellung einer wässrigen Lösung von Gabapentinhydrochlorid, – Einstellen des pH-Werts der Lösung auf den isoelektrischen Punkt von Gabapentin durch Zugabe einer basischen Verbindung, die ein einwertiges Anion umfasst, – Diafiltration der Lösung durch eine Membran, die für organische Verbindungen mit einem Molekulargewicht größer als 150 hochselektiv ist und wenig selektiv für anorganische einwertige Ionen ist, um diese Lösung in ein wässriges Chloridionen-enthaltendes Permeat und ein Retentat aufzutrennen, das nicht in Salzform vorliegendes Gabapentin, im Wesentlichen frei von Chloridionen, enthält, – Konzentrieren des Retentats durch Erhöhung des Drucks auf die Membran, um eine Konzentration von nicht in Salzform vorliegendem Gabapentin in dem Retentat von nicht weniger als 5% zu erhalten, – Einengen des Retentats unter vermindertem Druck bei einer Temperatur von T < 35°C, und – Ausfällen von Gabapentin durch Zugabe eines Alkohols.
- Verfahren nach Anspruch 1, umfassend einen weiteren Kristallisationsschritt des ausgefällten Gabapentins mit Methanol.
- Verfahren nach Anspruch 1 oder 2, wobei die basische Verbindung ausgewählt ist aus der Gruppe bestehend aus Alkalimetall- und Ammoniumhydroxiden.
- Verfahren nach Anspruch 3, wobei der pH-Wert der wässrigen Lösung auf 7,14 eingestellt wird.
- Verfahren nach Anspruch 4, wobei die Konzentration des nicht in Salzform vorliegenden Gabapentins in dem Retentat während des Konzentrationsschritts auf einen Wert von 8-10% w/v gebracht wird.
- Verfahren nach einem der vorhergehenden Ansprüche, wobei während der Diafiltrations- und Konzentrationsschritte die Temperatur vorzugsweise unterhalb 25°C gehalten wird.
- Verfahren nach einem der vorhergehenden Ansprüche, wobei die Membran eine Nanofiltration-Mehrschichtverbundmembran mit einer Rückhalterate an organischen Verbindungen mit einem Molekulargewicht oberhalb 150 von 96% oder höher ist.
- Verfahren nach Anspruch 7, wobei die Membran eine Schicht aus Polyamid, Polysulfonsäureharzen, Nylon, Celluloseacetat oder Polyvinylydenfluorid umfasst.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI990625 | 1999-03-26 | ||
| IT1999MI000625A IT1311984B1 (it) | 1999-03-26 | 1999-03-26 | Procedimento per la preparzione di gabapentin. |
| PCT/EP2000/002345 WO2000058268A1 (en) | 1999-03-26 | 2000-03-16 | A process for the preparation of gabapentin |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60004652D1 DE60004652D1 (de) | 2003-09-25 |
| DE60004652T2 true DE60004652T2 (de) | 2004-06-24 |
Family
ID=11382457
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60004652T Expired - Lifetime DE60004652T2 (de) | 1999-03-26 | 2000-03-16 | Verfahren zur herstellung von gabapentin |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US6576790B1 (de) |
| EP (1) | EP1165489B1 (de) |
| AT (1) | ATE247628T1 (de) |
| AU (1) | AU3290400A (de) |
| CA (1) | CA2368476C (de) |
| DE (1) | DE60004652T2 (de) |
| DK (1) | DK1165489T3 (de) |
| ES (1) | ES2204533T3 (de) |
| IT (1) | IT1311984B1 (de) |
| PT (1) | PT1165489E (de) |
| WO (1) | WO2000058268A1 (de) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE271864T1 (de) * | 1998-05-15 | 2004-08-15 | Warner Lambert Co | Aminosäure-stabilisierte gabapentin und pregabalin zubereitungen und verfahren zu ihrer herstellung |
| ITMI20012750A1 (it) | 2001-12-21 | 2003-06-21 | Procos Spa | Processo per la produzione dell'acido 1-(amminometil)-cicloesil-acetico in forma pura |
| CA2478471A1 (en) * | 2002-04-16 | 2003-10-30 | Taro Pharmaceutical Industries Ltd. | Process for preparing gabapentin |
| EP1727784B1 (de) | 2002-11-18 | 2008-12-24 | Nicholas Piramal India Limited | Verbessertes verfahren zur herstellung von gabapentin |
| WO2004046085A1 (en) * | 2002-11-20 | 2004-06-03 | Hikal Ltd. | Process for the preparation of amino methyl cyclo alkane acetic acids |
| US20070043236A1 (en) * | 2003-05-19 | 2007-02-22 | Chandiran Thakashina M | Process for preparation of gabapentin |
| ITMI20031247A1 (it) * | 2003-06-20 | 2004-12-21 | Zambon Spa | Processo per la purificazione di gabapentina |
| US20050187295A1 (en) * | 2004-02-19 | 2005-08-25 | Surendra Kalyan | Processes for the preparation of gabapentin |
| ITMI20040579A1 (it) | 2004-03-25 | 2004-06-25 | Zambon Spa | Processo di preparazione di gabapentina |
| ES2246159B1 (es) * | 2004-07-22 | 2007-03-16 | Menadiona, S.L. | Procedimiento para la obtencion de aminoacidos ciclicos. |
| CN116693410B (zh) * | 2023-06-08 | 2024-06-07 | 浙江竹子制药有限公司 | 一种粒径可控的加巴喷丁制备方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2460891C2 (de) | 1974-12-21 | 1982-09-23 | Gödecke AG, 1000 Berlin | 1-Aminomethyl-1-cycloalkanessigsäuren und deren Ester, Verfahren zu deren Herstellung und diese Verbindungen enthaltende Arzneimittel |
| US4960931A (en) * | 1988-05-02 | 1990-10-02 | Warner-Lambert Company | Gabapentin mohohydrate and a process for producing the same |
| US4894476A (en) * | 1988-05-02 | 1990-01-16 | Warner-Lambert Company | Gabapentin monohydrate and a process for producing the same |
| DE3928184A1 (de) * | 1989-08-25 | 1991-02-28 | Goedecke Ag | Verfahren zur herstellung von cyclischen aminosaeurederivaten sowie zwischenprodukte |
| US5491259A (en) * | 1994-09-13 | 1996-02-13 | The Dow Chemical Company | Process to produce aminocarboxylic acids containing low residual salt |
| IL119890A (en) | 1996-12-24 | 2002-03-10 | Teva Pharma | Gabapentin form iii and preparation of gabapentin form ii |
-
1999
- 1999-03-26 IT IT1999MI000625A patent/IT1311984B1/it active
-
2000
- 2000-03-16 EP EP00910850A patent/EP1165489B1/de not_active Expired - Lifetime
- 2000-03-16 DE DE60004652T patent/DE60004652T2/de not_active Expired - Lifetime
- 2000-03-16 CA CA002368476A patent/CA2368476C/en not_active Expired - Fee Related
- 2000-03-16 US US09/937,378 patent/US6576790B1/en not_active Expired - Lifetime
- 2000-03-16 WO PCT/EP2000/002345 patent/WO2000058268A1/en not_active Ceased
- 2000-03-16 ES ES00910850T patent/ES2204533T3/es not_active Expired - Lifetime
- 2000-03-16 AU AU32904/00A patent/AU3290400A/en not_active Abandoned
- 2000-03-16 DK DK00910850T patent/DK1165489T3/da active
- 2000-03-16 AT AT00910850T patent/ATE247628T1/de active
- 2000-03-16 PT PT00910850T patent/PT1165489E/pt unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ITMI990625A1 (it) | 2000-09-26 |
| EP1165489B1 (de) | 2003-08-20 |
| DE60004652D1 (de) | 2003-09-25 |
| ATE247628T1 (de) | 2003-09-15 |
| IT1311984B1 (it) | 2002-03-22 |
| CA2368476C (en) | 2009-12-08 |
| DK1165489T3 (da) | 2003-12-15 |
| PT1165489E (pt) | 2003-12-31 |
| WO2000058268A1 (en) | 2000-10-05 |
| EP1165489A1 (de) | 2002-01-02 |
| AU3290400A (en) | 2000-10-16 |
| CA2368476A1 (en) | 2000-10-05 |
| ES2204533T3 (es) | 2004-05-01 |
| US6576790B1 (en) | 2003-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60004652T2 (de) | Verfahren zur herstellung von gabapentin | |
| DE69108746T2 (de) | Herstellung und Trennung von Ibuprofen-lysinat. | |
| CH641432A5 (de) | Verfahren zur aufspaltung von racemischer 6-methoxy-alpha-methyl-2-naphthalinessigsaeure in die optischen antipoden. | |
| DE2927672C2 (de) | ||
| DE60115954T2 (de) | Verfahren zur herstellung von gabapentin | |
| DE60007734T2 (de) | Verfahren zur herstellung von wasserfreier gabapentin von pharmazeutischer qualität | |
| DE1493198C3 (de) | Verfahren zur Herstellung eines aliphatischen oder cycloaliphatischen Ketoxims | |
| DE69912773T2 (de) | Nicht hydratisiertes gabapentin polymorph, herstellungsprozess und verwendung zur herstellung von gabapentin pharmazeutischer reinheit. | |
| WO1993016035A1 (de) | Verfahren zur racemattrennung von verapamil | |
| DE60123125T2 (de) | Verfahren zur herstellung von 1-(aminomethyl) cyclohexanessigsäure | |
| DE69103214T2 (de) | Verfahren zur Reinigung von Glycin. | |
| DE69101329T2 (de) | Verfahren zur Herstellung von D-(-)-4-Hydroxyphenylglyzin aus D.L.-4-Hydroxyphenylglyzin. | |
| DE1793779A1 (de) | Verfahren zur herstellung von (+)-2amino-1-butanol-(+)-hydrogentartrat | |
| DE2750376C2 (de) | ||
| DE2814800A1 (de) | Verfahren zur herstellung von guanidin | |
| DE3785886T2 (de) | Methode für Rückgewinnung von Methylester von alpha-L-Aspartyl-L-Phenylalanin. | |
| DE10021790B4 (de) | Verfahren zur Herstellung von ω-Aminoalkansulfonsäuren und deren Verwendung als Biopuffer | |
| EP0492209A1 (de) | Verfahren zur Freisetzung von organischen Sulfonsäuren | |
| DE1493894A1 (de) | Verfahren zur Herstellung von Ornithinasparaginat | |
| EP1187804A1 (de) | Verfahren zur trennung der diastereomeren basen von 2- (dimethylamino)methyl]-1-(3-methoxyphenyl)-cyclohexanol | |
| EP0163222B1 (de) | Verfahren zur Gewinnung wässriger Carnitinamid-Lösungen | |
| DE2710504A1 (de) | Verfahren zur herstellung von optisch aktivem phenylglycin | |
| DE3877641T2 (de) | Isolierung von neutralen aminosaeuren bei hohen ph-werten. | |
| DE10355088A1 (de) | Verfahren zur Herstellung von fremdsalzarmen Sulfinaten | |
| WO1994008950A1 (de) | Verfahren zur racemattrennung von anipamil |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |