DE60006703T2 - 4-hydroxybiphenylhydrazid-derivate - Google Patents
4-hydroxybiphenylhydrazid-derivate Download PDFInfo
- Publication number
- DE60006703T2 DE60006703T2 DE60006703T DE60006703T DE60006703T2 DE 60006703 T2 DE60006703 T2 DE 60006703T2 DE 60006703 T DE60006703 T DE 60006703T DE 60006703 T DE60006703 T DE 60006703T DE 60006703 T2 DE60006703 T2 DE 60006703T2
- Authority
- DE
- Germany
- Prior art keywords
- compound
- mol
- following formula
- base
- concentration
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- PXTZQUZELQEHDH-UHFFFAOYSA-N 5-hydroxy-2-phenylbenzohydrazide Chemical class OC=1C=C(C(=CC1)C1=CC=CC=C1)C(=O)NN PXTZQUZELQEHDH-UHFFFAOYSA-N 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims description 30
- 238000000034 method Methods 0.000 claims description 29
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 24
- 239000002585 base Substances 0.000 claims description 19
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 18
- 239000003960 organic solvent Substances 0.000 claims description 17
- YXVFYQXJAXKLAK-UHFFFAOYSA-N biphenyl-4-ol Chemical group C1=CC(O)=CC=C1C1=CC=CC=C1 YXVFYQXJAXKLAK-UHFFFAOYSA-N 0.000 claims description 14
- 229940125890 compound Ia Drugs 0.000 claims description 11
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 9
- 239000012022 methylating agents Substances 0.000 claims description 9
- 239000011541 reaction mixture Substances 0.000 claims description 9
- 239000002841 Lewis acid Substances 0.000 claims description 8
- 150000007517 lewis acids Chemical class 0.000 claims description 8
- 238000002360 preparation method Methods 0.000 claims description 8
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 6
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 claims description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 claims description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 claims description 4
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 claims description 4
- 239000002904 solvent Substances 0.000 claims description 4
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 230000003301 hydrolyzing effect Effects 0.000 claims description 3
- 230000001035 methylating effect Effects 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- 239000001257 hydrogen Substances 0.000 claims description 2
- 229910052739 hydrogen Inorganic materials 0.000 claims description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 claims description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims 3
- VHLKTXFWDRXILV-UHFFFAOYSA-N bifenazate Chemical compound C1=C(NNC(=O)OC(C)C)C(OC)=CC=C1C1=CC=CC=C1 VHLKTXFWDRXILV-UHFFFAOYSA-N 0.000 description 11
- 239000005653 Bifenazate Substances 0.000 description 8
- 239000000203 mixture Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 238000005576 amination reaction Methods 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 238000007069 methylation reaction Methods 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- 241001120493 Arene Species 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- -1 phenylhydrazine derivative isopropyl-2- (4-methoxy- [1,1'-biphenyl] -3-yl) hydrazine carboxylate Chemical class 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- OTDXAMKMHDGFSP-UHFFFAOYSA-N (2-hydroxy-5-phenylanilino)carbamic acid Chemical compound C1=C(O)C(NNC(=O)O)=CC(C=2C=CC=CC=2)=C1 OTDXAMKMHDGFSP-UHFFFAOYSA-N 0.000 description 1
- LIEOEYTUTSDYKB-BUHFOSPRSA-N 2,2,2-trichloroethyl (ne)-n-(2,2,2-trichloroethoxycarbonylimino)carbamate Chemical compound ClC(Cl)(Cl)COC(=O)\N=N\C(=O)OCC(Cl)(Cl)Cl LIEOEYTUTSDYKB-BUHFOSPRSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- 150000002148 esters Chemical group 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000003129 miticidal effect Effects 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 150000004031 phenylhydrazines Chemical class 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- UANLFUYGOMALOJ-UHFFFAOYSA-N propan-2-yl n-(2-hydroxy-5-phenylphenyl)-n-(propan-2-yloxycarbonylamino)carbamate Chemical compound C1=C(O)C(N(C(=O)OC(C)C)NC(=O)OC(C)C)=CC(C=2C=CC=CC=2)=C1 UANLFUYGOMALOJ-UHFFFAOYSA-N 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/02—Compounds containing any of the groups, e.g. carbazates
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Description
- Die vorliegende Erfindung betrifft bestimmte 4-Hydroxybiphenylhydrazidderivate, die sich als Zwischenprodukte bei der Herstellung von Isopropyl-2-(4-methoxy-[1,1'-biphenyl]-3-yl)hydrazincarboxylat (Bifenazat) eignen.
- Hintergrund der Erfindung
- Die
US 5 367 093 A beschreibt ein Verfahren zur Herstellung des mitiziden Phenylhydrazinderivats Isopropyl-2-(4-methoxy-[1,1'-biphenyl]-3-yl)hydrazincarboxylat (Bifenazat) unter Verwendung eines sechsstufigen Verfahrens, das die unerwünschten Stufen der Herstellung und Reduktion eines Diazoniumsalzes umfasst. - Bestimmte Phenylhydrazinderivate können unter Verwendung der in der
US 4 864 032 A (Aminierung einer Grignard-Verbindung), bei Mitchell, J. Org. Chem. 59: 682 (1994) (Aminierung elektronenreicher Arene), und bei Lenarsic, J. Org. Chem. 64: 2558 (1999) (mittels elektrophiler Azodicarboxylate), beschriebenen Verfahren hergestellt werden. - J. Org. Chem. 60, 4268–4271 (1995), offenbart die para-gerichtete Aminierung von elektronenreichen Arenen mit Bis(2,2,2-trichlorethyl)azodicarboxylat.
- Zweck der vorliegenden Erfindung ist es, neue Zwischenprodukte mit Eignung bei der Herstellung von Bifenazat bereitzustellen. Weiterer Zweck der vorliegenden Erfindung ist es, ein neues Verfahren zur Herstellung von Bifenazat anzugeben.
- Zusammenfassung der Erfindung
- Die vorliegende Erfindung betrifft Verbindungen der folgenden Formel:worin R für Wasserstoff (IA) oder Isopropylester (CO2CH(CH3)2)(IB) steht:


- Die Verbindungen der Formeln IA und 1B eignen sich als Zwischenprodukte bei der Herstellung von Bifenazat.
- Gegenstand der vorliegenden Erfindung ist ferner ein Verfahren zur Herstellung der Verbindung der Formel IA durch Hydrolisieren der Verbindung der Formel 1B in Gegenwart einer wirksamen Menge einer Base in einem geeigneten organischen Lösemittel.
- Gegenstand der vorliegenden Erfindung ist ferner ein Verfahren zur Herstellung von Bifenazat durch Methylieren der Verbindung IA in Gegenwart einer wirksamen Menge eines Methylierungsmittels und einer Base in einem geeigneten Lösemittel.
- Detaillierte Beschreibung der Erfindung
- Die erfindungsgemäßen Verbindungen können gemäß Beschreibung im nachfolgenden Schema 1 hergestellt werden: 1. Aminierung

- 4-Hydroxybiphenyl wird mit Diisopropylazodicarboxylat in Gegenwart einer wirksamen Menge einer Lewis-Säure in einem geeigneten Lösemittel unter Herstellung der Verbindung IB umgesetzt. Geeignete Lewis-Säuren umfassen beispielsweise Bortrifluoridetherat und Aluminiumchlorid. Die Konzentration der Lewis-Säure zu dem 4-Hydroxybiphenyl in dem Reaktionsgemisch kann zwischen 1:0,2 und 1:1,1 (mol/mol), vorzugsweise bei etwa 1:1,1 (mol/mol), liegen. Geeignete organische Lösemittel sind diejenigen organischen Lösemittel, die für die Aminierungsreaktion nicht schädlich sind, und umfassen beispielsweise Ethylacetat, Dichlormethan, Toluol und Glyme. Die Temperatur des Reaktionsgemisches sollte zwischen 0 und 60°C, vorzugsweise bei Raumtemperatur, liegen.
- 2. Selektive Hydrolyse
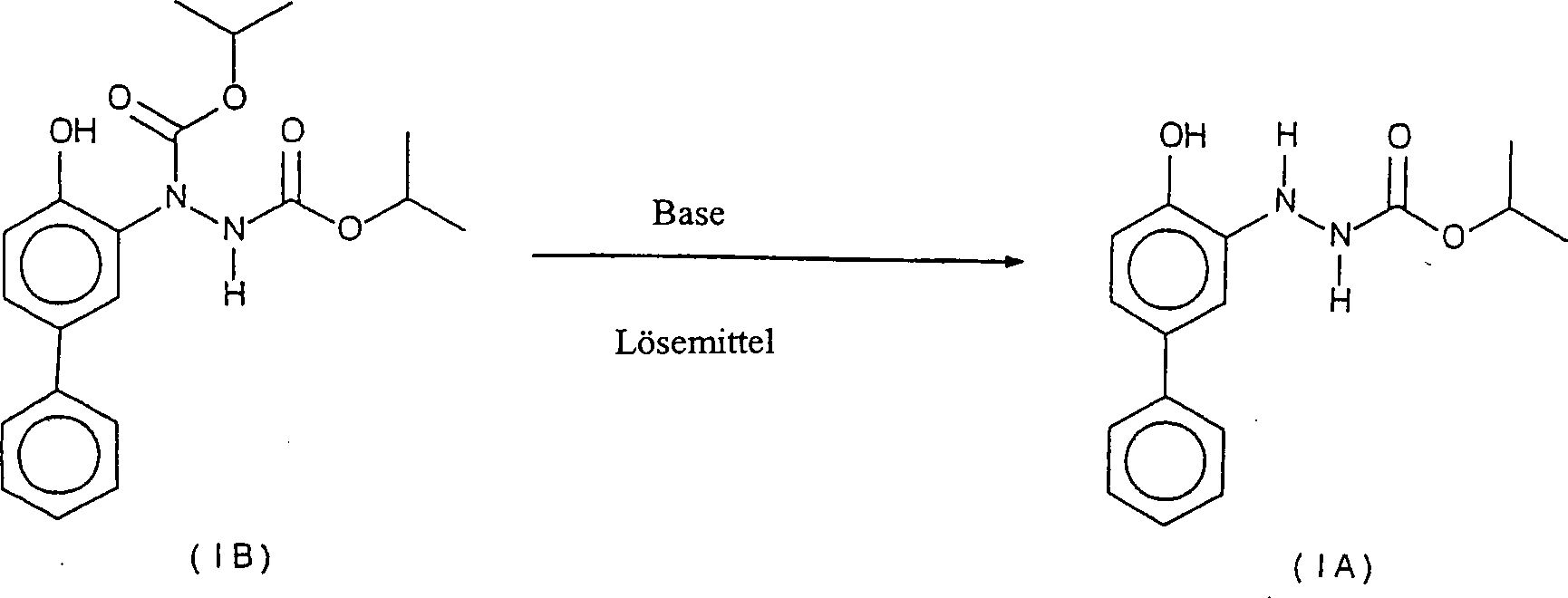
- Die Verbindung IB wird mit einer wirksamen Menge einer Base in einem geeigneten organischen Lösemittel unter Bildung der Verbindung IA behandelt. Geeignete Basenverbindungen sind solche Basenverbindungen, die die Esterfunktionalität hydrolisieren können, und umfassen beispielsweise Natriumhydroxid und Kaliumhydroxid. Die Konzentration der Base zur Verbindung IB kann zwischen 7:1 und 10:1 (mol/mol), vorzugsweise bei 9:1 (mol/mol), liegen. Geeignete organische Lösemittel sind diejenigen organischen Lösemittel, die für die Hydrolysereaktion nicht schädlich sind, und umfassen beispielsweise Toluol, Dimethylsulfoxid und Glyme. Vorzugsweise sollte die Temperatur des organischen Lösemittels oberhalb von Raumtemperatur und unterhalb von 110°C liegen.
- Die Verbindung IA wird anschließend in Gegenwart einer wirksamen Menge eines Methylierungsmittels und einer Base in einem geeigneten organischen Lösemittel methyliert. Für die Zwecke der vorliegenden Erfindung ist ein „Methylierungsmittel" eine beliebige Verbindung, die ein Wasserstoffatom in der 4-Hydroxygruppe der Verbindung IA durch eine Methylgruppe zu substituieren vermag. Geeignete Methylierungsmittel umfassen beispielsweise Dimethylsulfat und Methyliodid. Die Konzentration des Methylierungsmittels zu der Verbindung IA kann zwischen 1:1 und 1:1,2 (mol/mol), vorzugsweise bei 1:1 (mol/mol), liegen. Geeignete Basenverbindungen sind diejenigen Basenverbindungen, die Phenole zu deprotonieren vermögen, und umfassen z.B. Natriumcarbonat und Kaliumcarbonat. Die Konzentration der Base zu der Verbindung IA kann zwischen 1:1 und 3:1 (mol/mol), vorzugsweise bei 2:1 (mol/mol), liegen. Geeignete organische Lösemittel sind diejenigen organischen Lösemittel, die für die Methylierungsreaktion nicht schädlich sind, und umfassen z.B. Toluol und Aceton. Die Methylierungsreaktion kann bei Raumtemperatur durchgeführt werden. Das Methylierungsverfahren ist im nachfolgenden Schema 2 veranschaulicht.
- SCHEMA 2

- Die folgenden Beispiele sollen die vorliegende Erfindung veranschaulichen.
- Beispiel 1
- Herstellung eines 1-(4-Hydroxy-[1,1'-biphenyl]-3-yl)-1,2-hydrazindicarbonsäure-bis(1-methylethyl)esters
- (Verbindung IB)
- Eine Lösung von 4-Hydroxybiphenyl (5,50 g) in Ethylacetat (60 ml) bei Raumtemperatur wurde mit Bortrifluoridetherat (4,1 ml) versetzt. Das erhaltene Gemisch wurde auf –5°C abgekühlt, tropfenweise mit Diisopropylazodicarboxylat (6,3 ml) versetzt und das Ganze 30 min bei dieser Temperatur und anschließend 2 h bei Raumtemperatur verrührt. Das Gemisch wurde anschließend mit Wasser (100 ml) gequenscht und mit Ethylacetat (50 ml) extrahiert. Die organische Phase wurde abgetrennt, über Magnesiumsulfat getrocknet und im Vakuum aufkonzentriert. Man erhielt ein Öl, das auf Silicagel unter Verwendung von 20 bis 30% Ethylacetat/Hexan chromatographiert wurde. Man erhielt die Verbindung IB als beigefarbenen Feststoff (10,65 g, 88% Ausbeute). 1H-NMR (ppm, in CDCl3): m(12) 1,30; m(2) 5,04, m(2) 7,10, m(1) 7,32; dd(2) 7,43, m(3) 7,51–7,55, br s(1) 8,53.
- Beispiel 2
- Herstellung eines 2-(4-Hydroxy-[1,1'-biphenyl]-3-yl)-hydrazincarbonsäure-1-methyleth lesters
- (Verbindung IA)
- Kaliumhydroxid (5,0 g) wurde zu einer gerührten Suspension der oben in Beispiel 1 hergestellten Verbindung IB (5,15 g) in Toluol (50 ml) zugegeben. Durch das erhaltene purpurfarbene Gemisch wurde 20 min Stickstoff perlen gelassen, worauf das Ganze 4 Tage auf 45°C erwärmt wurde. Das Gemisch wurde anschließend auf 0°C abgekühlt, worauf 6M HCl zugegeben wurde, bis der pH-Wert des Gemisches etwa 1 betrug. Das Gemisch wurde anschließend mit Ethylacetat extrahiert. Die organische Phase wurde abgetrennt, mit Kochsalzlösung gewaschen, über Magnesiumsulfat getrocknet und zu einem braunen Feststoff eingeengt. Ein Umkristallisieren aus Toluol lieferte die Verbindung IA in Form eines beigefarbenen Pulvers (3,35 g, 85% Ausbeute). 1H-NMR (ppm, in CDCl3): d(6) 1,81, Septett(1) 5,04, br d(1) 5,91, br s(1) 6,61, d(1) 6,74, dd(1) 7,01, d(1) 7,14, dd(1) 7,32, dd(2) 7,42, dd(2) 7,61.
- Beispiel 3
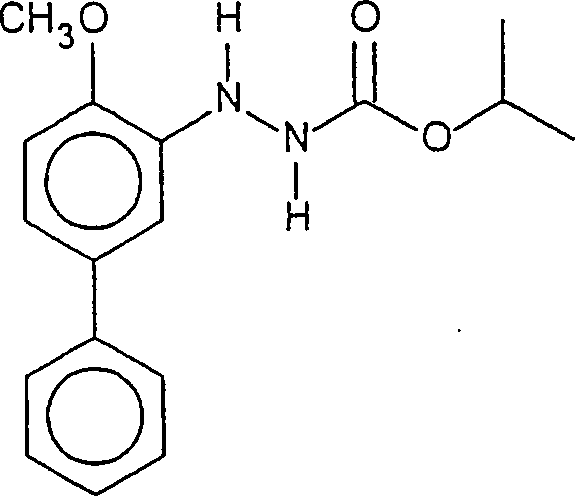
- Herstellung eines 2-(4-Methoxy-[1,1'-biphenyl]-3-yl)-hydrazincarbonsäure-1-methylethylesters
- (Bifenazat)
- Durch eine Suspension der oben in Beispiel 2 hergestellten Verbindung IA (2,63 g) und von Kaliumcarbonat (2,50 g) in Aceton (40 ml) wurde 20 min Stickstoff perlen gelassen. Anschließend wurde die Suspension mit Dimethylsulfat (0,96 ml) bei Raumtemperatur versetzt. Nach 2 h wurde das erhaltene Reaktionsgemisch in einem Eisbad abgekühlt. 2M HCl wurde anschließend vorsichtig zu dem Reaktionsgemisch zugegeben (ca. 30 ml), bis der pH-Wert des Reaktionsgemisches etwa 1 betrug. Das Reaktionsgemisch wurde anschließend eingeengt, um den größten Teil des Acetons zu entfernen. Der aus dem eingeengten Reaktionsgemisch erhaltene Feststoff wurde abfiltriert, mit Wasser und Hexan gewaschen und an Luft unter Saugen getrocknet, um das Bifenazat herzustellen (2,60 g). Die 1H-NMR-Spektrumsdaten stehen im Einklang mit dem für Bifenazat in der
US 5 367 093 A angegebenen 1H-NMR.
Claims (22)
- Verbindung der folgenden Formel:worin R für Wasserstoff oder CO2CH(CH3)2) steht.

- Verbindung nach Anspruch 1 mit der folgenden Formel:

- Verbindung nach Anspruch 1 mit der folgenden Formel:

- Verfahren zur Herstellung einer Verbindung der folgenden Formel:durch Umsetzen von 4-Hydroxybiphenyl mit Diisopropylazodicarboxylat in Gegenwart einer wirksamen Menge einer Lewis-Säure in einem geeigneten Lösemittel.

- Verfahren nach Anspruch 4, wobei die Konzentration der Lewis-Säure zu dem 4-Hydroxybiphenyl in dem Reaktionsgemisch zwischen 1:0,2 und 1:1.1 (mol/mol) liegt.
- Verfahren nach Anspruch 5, wobei die Konzentration der Lewis-Säure zu dem 4-Hydroxybiphenyl in dem Reaktionsgemisch bei 1:1,1 (mol/mol) liegt.
- Verfahren nach Anspruch 5, wobei die Lewis-Säure aus der Gruppe ausgewählt ist, die aus Bortrifluoridetherat und Aluminiumchlorid besteht.
- Verfahren nach Anspruch 4, wobei das organische Lösemittel aus der Gruppe ausgewählt ist, die aus Ethylacetat, Dichlormethan, Toluol, Glyme und Diethylether besteht.
- Verfahren zur Herstellung einer Verbindung der folgenden Formel:durch Hydrolisieren einer Verbindung der folgenden Formel:
 in Gegenwart einer wirksamen Menge einer Base und eines geeigneten organischen Lösemittels.
in Gegenwart einer wirksamen Menge einer Base und eines geeigneten organischen Lösemittels.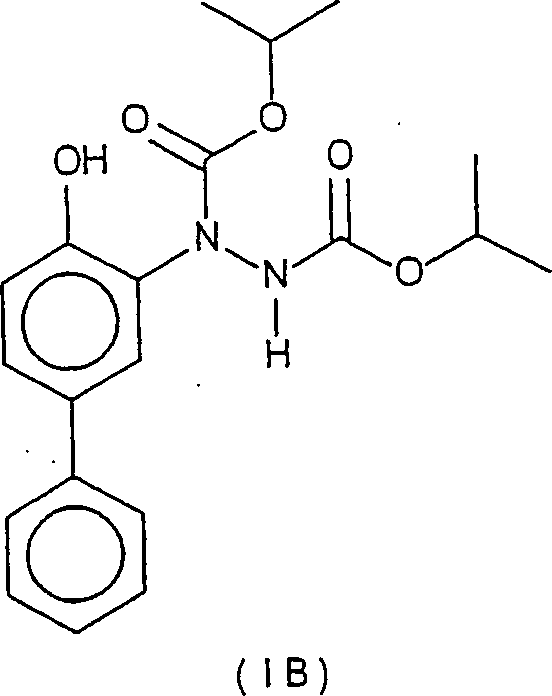
- Verfahren nach Anspruch 9, wobei die Base aus der Gruppe ausgewählt ist, die aus Natriumhydroxid und Kaliumhydroxid ausgewählt ist.
- Verfahren nach Anspruch 9, wobei das organische Lösemittel aus der Gruppe ausgewählt ist, die aus Toluol, Dimethylsulfoxid und Glyme besteht.
- Verfahren nach Anspruch 9, das bei einer Temperatur zwischen Raumtemperatur und 110°C durchgeführt wird. 13. Verfahren nach Anspruch 9, wobei die Konzentration der Base zu der Verbindung IB zwischen 7:1 und 10:1 (mol/mol) liegt.
- Verfahren nach Anspruch 13, wobei die Konzentration der Base zu der Verbindung IB bei 7:1 (mol/mol) liegt. 15. Verfahren zur Herstellung einer Verbindung der folgenden Formeldurch Methylieren einer Verbindung der folgenden Formel:
 in Gegenwart einer wirksamen Menge eines Methylierungsmittels und einer Base und eines geeigneten organischen Lösemittels.
in Gegenwart einer wirksamen Menge eines Methylierungsmittels und einer Base und eines geeigneten organischen Lösemittels.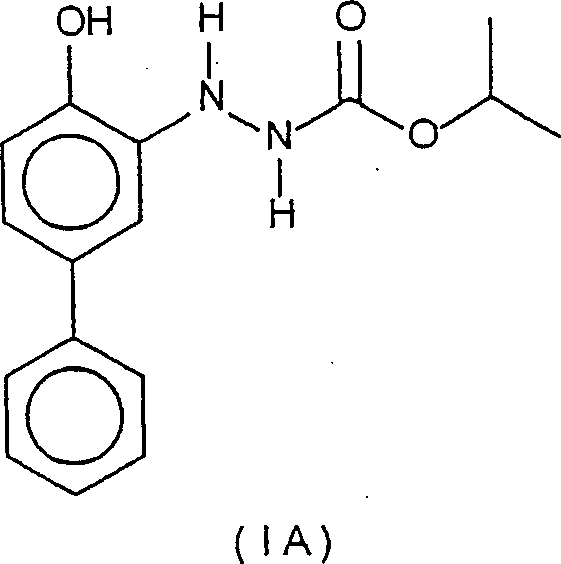
- Verfahren nach Anspruch 15, wobei das Methylierungsmittel aus der Gruppe ausgewählt ist, die aus Dimethylsulfat und Methyliodid besteht.
- Verfahren nach Anspruch 15, wobei die Base aus der Gruppe ausgewählt ist, die aus Natriumcarbonat und Kaliumcarbonat besteht.
- Verfahren nach Anspruch 15, wobei das organische Lösemittel aus der Gruppe ausgewählt ist, die aus Toluol und Aceton besteht.
- Verfahren nach Anspruch 15, das bei Raumtemperatur durchgeführt wird.
- Verfahren nach Anspruch 15, wobei die Konzentration des Methylierungsmittels zur Verbindung IA zwischen 1:1 und 1,2:1 (mol/mol) liegt.
- Verfahren nach Anspruch 20, wobei die Konzentration des Methylierungsmittels zur Verbindung IA bei 1:1 (mol/mol) liegt.
- Verfahren nach Anspruch 15, wobei die Konzentration der Base zur Verbindung IA zwischen etwa 1:1 und 3:1 (mol/mol) liegt.
- Verfahren nach Anspruch 22, wobei die Konzentration der Base zur Verbindung IA bei 2:1 (g/g) liegt.
- Verfahren zur Herstellung einer Verbindung der folgenden Formelwobei das Verfahren die folgenden Stufen umfasst: (a) Umsetzen von 4-Hydroxybiphenyl mit Diisopropylazodicarboxylat in Gegenwart einer wirksamen Menge einer Lewis-Säure in einem geeigneten Lösemittel unter Bildung einer Verbindung der folgenden Formel
 (b) Hydrolisieren der Verbindung der Formel IB in Gegenwart einer wirksamen Menge einer Base und eines geeigneten organischen Lösemittels unter Bildung einer Verbindung der folgenden Formel:
(b) Hydrolisieren der Verbindung der Formel IB in Gegenwart einer wirksamen Menge einer Base und eines geeigneten organischen Lösemittels unter Bildung einer Verbindung der folgenden Formel: und (c) Methylieren der Verbindung der Formel IA in Gegenwart einer wirksamen Menge eines Methylierungsmittels und einer Base in einem geeigneten organischen Lösemittel unter Bildung der Verbindung der folgenden Formel:
und (c) Methylieren der Verbindung der Formel IA in Gegenwart einer wirksamen Menge eines Methylierungsmittels und einer Base in einem geeigneten organischen Lösemittel unter Bildung der Verbindung der folgenden Formel: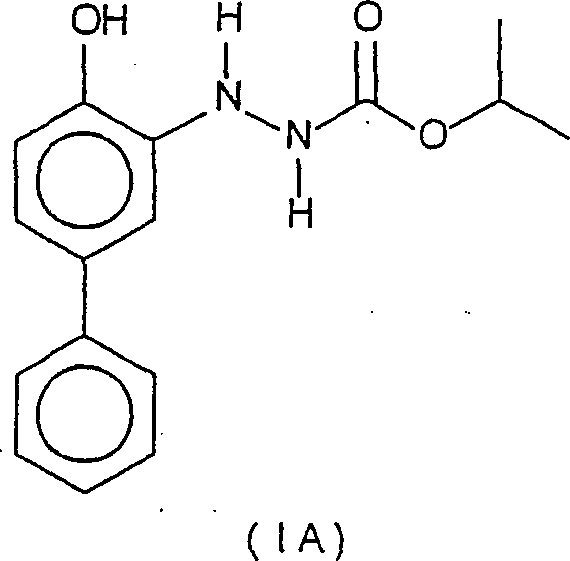

Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US413072 | 1999-10-06 | ||
| US09/413,072 US6093843A (en) | 1999-10-06 | 1999-10-06 | 4-hydroxybiphenyl hydrazide derivatives |
| PCT/US2000/026465 WO2001025191A1 (en) | 1999-10-06 | 2000-09-26 | 4-hydroxybiphenyl hydrazide derivatives |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60006703D1 DE60006703D1 (de) | 2003-12-24 |
| DE60006703T2 true DE60006703T2 (de) | 2004-09-30 |
Family
ID=23635704
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60006703T Expired - Lifetime DE60006703T2 (de) | 1999-10-06 | 2000-09-26 | 4-hydroxybiphenylhydrazid-derivate |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US6093843A (de) |
| EP (1) | EP1218340B1 (de) |
| JP (1) | JP3779616B2 (de) |
| KR (1) | KR100669572B1 (de) |
| CN (1) | CN1168708C (de) |
| AR (2) | AR025951A1 (de) |
| AT (1) | ATE254598T1 (de) |
| AU (1) | AU783729B2 (de) |
| BR (1) | BR0014461B1 (de) |
| CA (1) | CA2385200C (de) |
| CZ (1) | CZ295841B6 (de) |
| DE (1) | DE60006703T2 (de) |
| DK (1) | DK1218340T3 (de) |
| ES (1) | ES2211613T3 (de) |
| HU (1) | HU228227B1 (de) |
| IL (2) | IL148629A0 (de) |
| PL (1) | PL200493B1 (de) |
| PT (1) | PT1218340E (de) |
| RU (1) | RU2242461C2 (de) |
| WO (1) | WO2001025191A1 (de) |
| ZA (1) | ZA200202185B (de) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6706895B1 (en) | 2002-11-14 | 2004-03-16 | Uniroyal Chemical Company, Inc. | 4-methoxybiphenyl hydrazone derivatives |
| US7343196B2 (en) * | 2003-05-09 | 2008-03-11 | Ge Medical Systems Global Technology Company Llc | Cardiac CT system and method for planning and treatment of biventricular pacing using epicardial lead |
| US7592374B2 (en) * | 2005-03-03 | 2009-09-22 | Crompton Corporation | Insecticidal nitromethylene compounds |
| US7399757B1 (en) * | 2007-05-16 | 2008-07-15 | Chemtura Corporation | Pesticidal diazene oxide carboxylates |
| US7511029B2 (en) * | 2007-05-16 | 2009-03-31 | Chemtura Corporation | Pesticidal diazene oxide carboxylates |
| CN102344395A (zh) * | 2011-07-28 | 2012-02-08 | 同济大学 | 一种联苯肼酯的合成方法 |
| CN102885074B (zh) * | 2012-11-06 | 2013-12-11 | 江苏辉丰农化股份有限公司 | 具有增效作用的杀螨剂组合物 |
| CN103155939A (zh) * | 2013-03-22 | 2013-06-19 | 青岛瀚生生物科技股份有限公司 | 噻虫胺与联苯肼酯复配杀虫组合物 |
| WO2015198343A1 (en) * | 2014-06-25 | 2015-12-30 | Council Of Scientific & Industrial Research | Ortho-alkynyl anilines and process for preparation thereof |
| CN105707118B (zh) * | 2016-03-25 | 2018-02-06 | 江西正邦生物化工有限责任公司 | 一种含有亚胺硫磷和联苯肼酯的农药组合物 |
| CN106831387B (zh) * | 2017-01-19 | 2020-12-29 | 上海科技大学 | 一种可见光催化的饱和碳氢键直接氧化方法 |
| CN109988084B (zh) * | 2019-04-23 | 2021-09-10 | 绍兴上虞新银邦生化有限公司 | 一种联苯肼酯合成方法 |
| CN110818596A (zh) * | 2019-11-18 | 2020-02-21 | 合肥锦绣田园化工科技有限公司 | 一种氨基脲类化合物及其制备与应用 |
| CN115974727B (zh) * | 2023-01-17 | 2024-03-12 | 青岛前线生物工程有限公司 | 一种联苯肼酯的合成方法 |
| CN118344267A (zh) * | 2024-04-17 | 2024-07-16 | 广东广康生化科技股份有限公司 | 一种联苯肼酯高效合成新方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4864032A (en) * | 1987-07-02 | 1989-09-05 | Ortho Pharmaceutical Corporation | Process for the preparation of indazoles |
| SK282306B6 (sk) * | 1991-11-22 | 2002-01-07 | Uniroyal Chemical Company, Inc. | Fenylhydrazínové deriváty, pesticídny prostriedok obsahujúci tieto zlúčeniny a spôsob kontrolovania nežiaduceho hmyzu |
| UA41298C2 (uk) * | 1991-11-22 | 2001-09-17 | Юніроял Кемікал Компані Інк. | Фенілгідразинове похідне, спосіб боротьби зі шкідниками сільськогосподарських культур та інсектоакарициднонематоцидна композиція |
-
1999
- 1999-10-06 US US09/413,072 patent/US6093843A/en not_active Expired - Lifetime
-
2000
- 2000-09-26 DE DE60006703T patent/DE60006703T2/de not_active Expired - Lifetime
- 2000-09-26 RU RU2002111656/04A patent/RU2242461C2/ru not_active IP Right Cessation
- 2000-09-26 CN CNB008139210A patent/CN1168708C/zh not_active Expired - Fee Related
- 2000-09-26 DK DK00965464T patent/DK1218340T3/da active
- 2000-09-26 BR BRPI0014461-4A patent/BR0014461B1/pt not_active IP Right Cessation
- 2000-09-26 IL IL14862900A patent/IL148629A0/xx active IP Right Grant
- 2000-09-26 PT PT00965464T patent/PT1218340E/pt unknown
- 2000-09-26 JP JP2001528139A patent/JP3779616B2/ja not_active Expired - Lifetime
- 2000-09-26 AU AU76180/00A patent/AU783729B2/en not_active Ceased
- 2000-09-26 EP EP00965464A patent/EP1218340B1/de not_active Expired - Lifetime
- 2000-09-26 KR KR1020027003783A patent/KR100669572B1/ko not_active Expired - Lifetime
- 2000-09-26 CZ CZ20021185A patent/CZ295841B6/cs not_active IP Right Cessation
- 2000-09-26 CA CA002385200A patent/CA2385200C/en not_active Expired - Lifetime
- 2000-09-26 WO PCT/US2000/026465 patent/WO2001025191A1/en not_active Ceased
- 2000-09-26 PL PL354483A patent/PL200493B1/pl not_active IP Right Cessation
- 2000-09-26 HU HU0203538A patent/HU228227B1/hu not_active IP Right Cessation
- 2000-09-26 ES ES00965464T patent/ES2211613T3/es not_active Expired - Lifetime
- 2000-09-26 AT AT00965464T patent/ATE254598T1/de active
- 2000-10-04 AR ARP000105233A patent/AR025951A1/es active IP Right Grant
-
2002
- 2002-03-12 IL IL148629A patent/IL148629A/en unknown
- 2002-03-18 ZA ZA200202185A patent/ZA200202185B/xx unknown
-
2010
- 2010-07-05 AR ARP100102395A patent/AR079399A2/es not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| CA2385200A1 (en) | 2001-04-12 |
| KR20020033822A (ko) | 2002-05-07 |
| HUP0203538A3 (en) | 2005-10-28 |
| AR025951A1 (es) | 2002-12-26 |
| EP1218340A1 (de) | 2002-07-03 |
| ES2211613T3 (es) | 2004-07-16 |
| BR0014461A (pt) | 2002-08-20 |
| CA2385200C (en) | 2008-11-18 |
| EP1218340B1 (de) | 2003-11-19 |
| BR0014461B1 (pt) | 2011-02-22 |
| WO2001025191A1 (en) | 2001-04-12 |
| AR079399A2 (es) | 2012-01-25 |
| US6093843A (en) | 2000-07-25 |
| ATE254598T1 (de) | 2003-12-15 |
| HU228227B1 (en) | 2013-02-28 |
| PL354483A1 (en) | 2004-01-26 |
| CZ20021185A3 (cs) | 2003-11-12 |
| CZ295841B6 (cs) | 2005-11-16 |
| JP2003511364A (ja) | 2003-03-25 |
| AU7618000A (en) | 2001-05-10 |
| HUP0203538A2 (hu) | 2003-02-28 |
| IL148629A (en) | 2006-10-31 |
| CN1168708C (zh) | 2004-09-29 |
| DE60006703D1 (de) | 2003-12-24 |
| IL148629A0 (en) | 2002-09-12 |
| JP3779616B2 (ja) | 2006-05-31 |
| PL200493B1 (pl) | 2009-01-30 |
| AU783729B2 (en) | 2005-12-01 |
| RU2242461C2 (ru) | 2004-12-20 |
| DK1218340T3 (da) | 2004-03-29 |
| CN1378529A (zh) | 2002-11-06 |
| PT1218340E (pt) | 2004-02-27 |
| ZA200202185B (en) | 2003-08-27 |
| KR100669572B1 (ko) | 2007-01-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60006703T2 (de) | 4-hydroxybiphenylhydrazid-derivate | |
| CH446289A (de) | Verfahren zur Herstellung von Alkenderivaten | |
| EP0454624B1 (de) | Verfahren zur Herstellung von 1,3-Diketonen | |
| DE69912264T2 (de) | Verfahren zur herstellung chiraler (s)-2,3-disubstituierter 1-propylamin-derivate | |
| DE69624796T2 (de) | Aminotetralon-derivate und verfahren zu deren herstellung | |
| DE10160721A1 (de) | Verfahren zur Herstellung von Deoxybenzoinen | |
| DE2535855A1 (de) | Verfahren zur herstellung von cephalosporinen | |
| DE3223877A1 (de) | Isocarbostyrilderivate, verfahren zu deren herstellung und diese enthaltende arzneimittel | |
| DE2409675A1 (de) | Alpha-alkyl(oder -aryl)-thio-5-hydroxytryptophan-derivat und verfahren zu seiner herstellung | |
| DE3233395C1 (de) | Verfahren zur Herstellung von sterisch gehinderten 2-Benzothiazolsulfenamiden | |
| EP0002721B1 (de) | Verfahren zur Herstellung von Hydrazinen | |
| DE1468624B2 (de) | Verfahren zur herstellung von beta-cyanketonen | |
| EP0018540B1 (de) | Ester des Isocamphyl-guajakols, Verfahren zu ihrer Herstellung und ihre Verwendung zur Herstellung von 3-(Iso-camphyl-(5))-cyclohexanol | |
| EP0110116B1 (de) | Verfahren zur Herstellung von Keten-O,N-acetalen | |
| CH635829A5 (de) | Verfahren zur herstellung von indazolderivaten. | |
| EP0110202A2 (de) | Verfahren zur Herstellung von Phenylethanolaminen | |
| DE1518652A1 (de) | Verfahren zur Herstellung von pharmazeutisch wirksamen Derivaten des 2-Aminoindans | |
| AT235269B (de) | Verfahren zur Herstellung von 2-Phenyläthylaminderivaten sowie von deren nicht toxischen Salzen | |
| CH516513A (de) | Verfahren zur Herstellung von Aminderivaten | |
| AT265275B (de) | Verfahren zur Herstellung von 10-Aminoalkyl-5,5-dialkylacridanen und von deren pharmazeutisch verträglichen Salzen | |
| AT351528B (de) | Verfahren zur herstellung von neuen indazol- derivaten | |
| DE19832146B4 (de) | Verfahren zur Herstellung von 3-Methylpyrazol bzw. dessen Salze | |
| CH521345A (de) | Verfahren zur Herstellung von Tetrahydroisochinolinverbindungen | |
| EP0101934A1 (de) | Verfahren zur Herstellung von 1-(Halogenalkyl)-1,2,3,4-tetrahydro-beta-carbolinen | |
| DE2126865A1 (en) | Nitrogen substituents introduction - in methyl and methylene gps using phosgene immonium chloride |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8328 | Change in the person/name/address of the agent |
Representative=s name: KROHER, STROBEL RECHTS- UND PATENTANWAELTE, 80336 |
|
| 8328 | Change in the person/name/address of the agent |
Representative=s name: DR. SCHOEN & PARTNER, 80336 MUENCHEN |