-
Die
vorliegende Erfindung bezieht sich auf ein Verfahren zur Herstellung
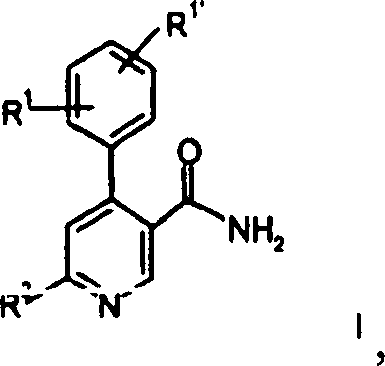
von Verbindungen der allgemeinen Formel
worin
R
1 und
R
1' unabhängig voneinander
Wasserstoff, Niederalkyl, Niederalkoxy, Halogen, Cyano oder Alkylamino sind;
R
2 -N(R
3)
2,
-N(R
3)(CH
2)
nOH, -N(R
3)S(O)
2-Niederalkyl, -N(R
3)S(O)
2-Phenyl, -N=CH-N(R
3)
2, -N(R
3)C(O)R
3 oder ein cyclisches tertiäres Amin
der Gruppe
R
3 unabhängig voneinander
Wasserstoff, C
3-6-Cycloallyl, Benzyl oder
Niederalkyl ist;
R
4 Wasserstoff, Hydroxy,
Niederalkyl, -(CH
2)
nCOO-Niederalkyl,
-N(R
3)CO-Niederalkyl, Hydroxy-niederalkyl, Cyano,
-(CH
2)
nO(CH
2)
nOH, -CHO oder
eine 5- oder 6gliedrige heterocyclische Gruppe, gegebenenfalls über eine
Alkylengruppe gebunden, ist.
-
Die
Verbindungen der Formel I sind wertvolle Zwischenprodukte für die Herstellung
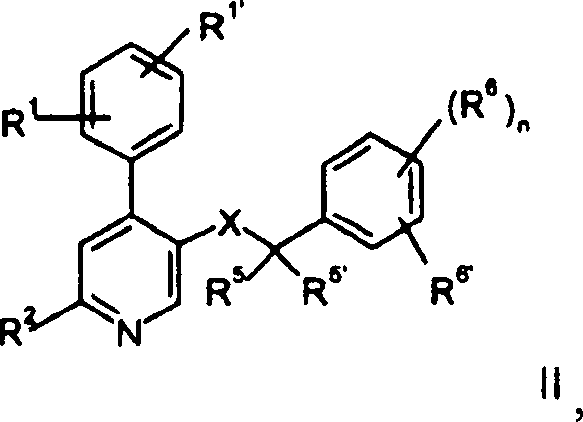
von therapeutisch aktiven Verbindungen der allgemeinen Formel
worin
R
1 und
R
1' unabhängig voneinander
Wasserstoff, Niederalkyl, Alkoxy, Halogen, Cyano oder Alkylamino
sind;
R
2 -N(R
3)
2, -N(R
3)(CH
2)
nOH, -N(R
3)S(O)
2-Niederalkyl,
-N(R
3)S(O)
2-Phenyl,
-N=CH-N(R
3)
2, -N(R
3)C(O)R
3 oder ein
cyclisches tertiäres
Amin der Gruppe
R
3 unabhängig voneinander
Wasserstoff, C
3-6-Cycloalkyl, Benzyl oder
Niederalkyl ist;
R
4 Wasserstoff, Hydroxy,
Niederalkyl, -(CH
2)
nCOO-Niederalkyl,
-N(R
3)CO-Niederalkyl, Hydroxy-niederalkyl, Cyano,
-(CH
2)
nO(CH
2)
nOH, -CHO oder
eine 5- oder 6gliedrige heterocyclische Gruppe, gegebenenfalls über eine
Alkylengruppe gebunden, ist;
R
5 und
R
5' unabhängig voneinander
Wasserstoff, Niederalkyl sind oder zusammen mit dem Kohlenstoffatom eine
Cycloalkylgruppe bilden;
R
6 und R
6' unabhängig voneinander
Wasserstoff, Halogen, Trifluormethyl, Niederalkoxy oder Cyano sind;
oder
R
6 und R
6' zusammen -CH=CH-CH=CH-,
gegebenenfalls substituiert durch einen oder zwei Substituenten, ausgewählt aus
Niederalkyl oder Niederalkoxy, sein können;
X -C(O)N(R
3)-, -(CH
2)
mO-, -(CH
2)
mN(R
3)-, -N(R
3)C(O)- oder -N(R
3)(CH
2)
m- ist;
n
0 bis 4 ist und
m 1 oder 2 ist.
-
Verbindungen
der Formel II sind in
EP-A-1035115 beschrieben,
wie
N-(3,5-Bis-trifluormethyl-benzyl)-N-methyl-6-[methyl-(2-morpholin-4-yl-ethyl)-amino]-4-o-tolyl-nicotinamid,
N-(3,5-Bis-trifluormethyl-benzyl)-N-methyl-6-morpholin-4-yl-4-o-tolyl-nicotinamid,
N-(3,5-Bis-trifluormethyl-benzyl)-N-methyl-6-thiomorpholin-4-yl-4-o-tolyl-nicotinamid,
N-(3,5-Bis-trifluormethyl-benzyl)-N-methyl-6-(1-oxo-1λ
6-4-thiomorpholin-4-yl)-4-o-tolyl-nicotinamid,
N-(3,5-Bis-trifluormethyl-benzyl)-6-(1,1-dioxo-1λ
6-6-thiomorpholin-4-yl)-N-methyl-4-o-tolyl-nicotinamid,
N-(3,5-Bis-trifluormethyl-benzyl)-N-methyl-6-piperazin-1-yl-4-o-tolyl-nicotinamid,
N-(3,5-Bis-trifluormethyl-benzyl)-6-[4-(2-hydroxy-ethyl)-piperazin-1-yl]-N-methyl-4-o-tolyl-nicotinamid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-N-methyl-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-(6-morpholin-4-yl-4-o-tolyl-pyridin-3-yl)-N-methyl-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-[4-(4-fluor-2-methyl-phenyl)-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-N-methyl-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-[4-(2-chlor-phenyl)-pyridin-3-yl]-N-methyl-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-methyl-N-(4-o-tolyl-pyridin-3-yl)-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-methyl-N-[6-(4-pyrimidin-2-yl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-(6-morpholin-4-yl-4-o-tolyl-pyridin-3-yl)-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-[4-(2-chlor-phenyl)-6-dimethylamino-pyridin-3-yl]-isobutyramid,
2-(3,5-Bis-trifluormethyl-phenyl)-N-methyl-N-(6-piperazin-1-yl-4-o-tolyl-pyridin-3-yl)-isobutyramid
und
2-(3,5-Bis-trifluormethyl-phenyl)-N-[6-(1,1-dioxo-1λ
6-thiomorpholin-4-yl)-4-o-tolyl-pyridin-3-yl]-N-methyl-isobutyramid.
-
Diese
Verbindungen der Formel II sind Antagonisten des Neurokinin-1-Rezeptors
(NK-1, Substanz P). Die zentralen und peripheren Wirkungen der Säuger-Tachykininsubstanz
P stehen mit zahlreichen Entzündungszuständen einschließlich Migräne, Rheumatoidarthritis,
Asthma und entzündlichen
Darmerkrankungen sowie der Vermittlung von Brechreiz und der Modulation
von Erkrankungen des zentralen Nervensystems (ZNS) wie der Parkinson-Krankheit in Verbindung.
Außerdem
sind diese Verbindungen bei der Behandlung von Schmerz, Kopfschmerz,
speziell Migräne,
Alzheimer-Krankheit, multipler Sklerose, Milderung von Morphinentzug,
kardiovaskulären
Veränderungen, Ödemen, wie Ödemen aufgrund
von Verbrennungen, chronischen Entzündungskrankheiten, wie Rheumatoidarthritis,
Asthma/Bronchialhyperreaktivität
und anderen Atemwegserkrankungen, einschließlich allergischer Rhinitis,
Entzündungskrankheiten
des Darms, einschließlich Colitis
ulcerosa und Crohn-Krankheit, Augenverletzungen und Augenentzündungskrankheiten
nützlich.
Außerdem
können
die Verbindungen bei der Behandlung einer Vielzahl von physiologischen
Störungen
nützlich
sein, die Störungen
des zentralen Nervensystems, wie Angst, Depression und Psychose,
umfassen. Die Neurokinin-1-Rezeptorantagonisten sind ferner für die Behandlung
von Bewegungskrankheit, für
die Behandlung von induziertem Erbrechen und für die Reduktion von Cisplatin-induzierter
Emesis nützlich.
-
Die
folgenden Definitionen der in der vorliegenden Beschreibung allgemein
verwendeten Ausdrücke treffen
unabhängig
davon zu, ob die in Frage kommenden Ausdrücke allein oder in Kombination
auftreten.
-
Wie
hierin verwendet, bezeichnet der Ausdruck „Niederalkyl" eine gerad- oder
verzweigtkettige Alkylgruppe, enthaltend 1 bis 7 Kohlenstoffatome,
beispielsweise Methyl, Ethyl, Propyl, Isopropyl, n-Butyl, i-Butyl, t-Butyl
und dergleichen. Bevorzugte Niederalkylgruppen sind Gruppen mit
1 bis 4 Kohlenstoffatomen.
-
Der
Ausdruck „Niederalkoxy" bezeichnet eine
Gruppe, wobei die Alkylreste wie vorstehend definiert sind, und
die über
ein Sauerstoffatom gebunden ist.
-
Der
Ausdruck „Halogen" bezeichnet Chlor,
Iod, Fluor und Brom.
-
Der
Ausdruck „Cycloalkyl" bezeichnet eine
gesättigte
carbocyclische Gruppe, enthaltend 3 bis 6 Kohlenstoffatome.
-
Der
Ausdruck „cyclisches
tertiäres
Amin" bezeichnet
beispielsweise Pyrrol-1-yl, Imidazol-1-yl, Piperidin-1-yl, Piperazin-1-yl,
Morpholin-4-yl, Thiomorpholin-4-yl, 1-Oxo-thiomorpholin-4-yl oder
1,1-Dioxo-thiomorpholin-4-yl.
-
Der
Ausdruck „5-
oder 6gliedrige heterocyclische Gruppe" bezeichnet beispielsweise Pyridinyl,
Pyrimidinyl, Oxadiazolyl, Triazolyl, Tetrazolyl, Thiazolyl, Thienyl,
Furyl, Pyranyl, Pyrrolyl, Imidazolyl, Pyrazolyl, Isothiazolyl, Piperazinyl
oder Piperidyl.
-
Die
Verbindungen der Formel 11 können
z. B. gemäß beispielsweise
EP-A-1035115 hergestellt
werden.
-
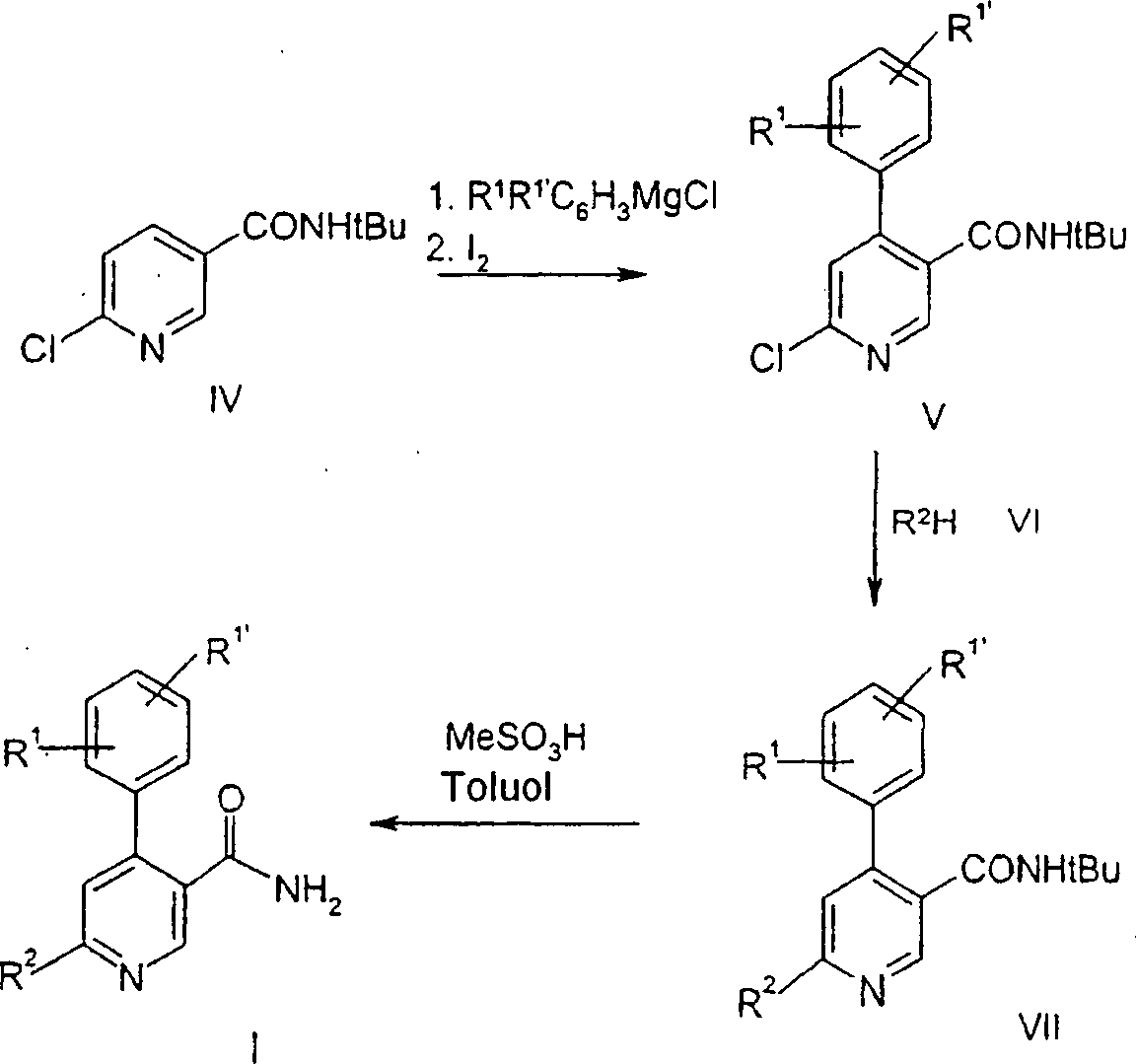
Es
ist bekannt (
EP-A-1035115 ),
daß die
vorliegenden Verbindungen der Formel I beispielsweise durch die
in dem nachstehenden Schema 1 beschriebenen Verfahren hergestellt
werden können: Schema
1
-
Dieses
Verfahren zur Herstellung der Verbindungen der allgemeinen Formel
I ergibt eine hohe Ausbeute, aber erfordert die Verwendung von teuren
Ausgangsmaterialien. Außerdem
ist der Schlüsselschritt
in diesem Verfahren die Substitution des Pyridins mit R1R1'C6H3MgCl durch Grignard-Reaktion.
Der Erfolg dieser Reaktion hängt
von dem Substitutionsmuster an dem aromatischen Ring ab. Falls die
elektronenziehenden Gruppen die Reaktivität des Grignard-Reagens verringern,
muß eine
Suzuki-Reaktion (Suzuki-Kopplung) durchgeführt werden.
-
Das
Problem des Kerns der vorliegenden Erfindung ist daher die Bereitstellung
eines Verfahrens zur Herstellung der Verbindungen der Formel I,
wobei das Verfahren bevorzugt ist, falls die Grignard-Reaktion nicht
funktioniert oder nicht gut funktioniert. Dieses Verfahren ist für die Synthese
von weiteren NK1-Rezeptorantagonisten nützlich.
-
Das
Problem wird gemäß der vorliegenden
Erfindung durch ein Verfahren zur Herstellung der Verbindungen der
Formel I gelöst,
wie in Schema 2 gezeigt: Schema
2
-
In
Schema 2 ist die Definition von Substituenten für R1,
R1' und
R2 oben beschrieben.
-
Die
Verbindung der Formel
wird in guter Ausbeute durch
die Umsetzung von 2-Chloracetamid mit Pyridin erhalten. Die Umsetzung
ist in A. R. Katritzky, N. E. Grzeskowiak und J. Alvarez-Builla,
J. C. S. Perkin I, 1981, 1180–1185
beschrieben.
-
Die
Herstellung der Verbindung der Formel
besteht aus zwei Schritten:
-
Schritt 1)
-
Eine
Lösung
aus 3,3-Dimethoxypropionitril (XII) und einer Verbindung der Formel
XI wird zu Natriummethanolat in einem Alkohol, wie Niederalkylalkohol,
Cycloalkylalkohol, bevorzugt Methanol, durch Halten der Innentemperatur
unter 30°C,
bevorzugt unter 20°C,
zugegeben. Das Reaktionsgemisch wird bei einer Temperatur, die zwischen
20 und 30°C,
bevorzugt zwischen 22 und 25°C,
schwankt, für
5 bis 20 Stunden, bevorzugt 10 bis 12 Stunden, gerührt.
-
Schritt 2)
-
Eine
Säure,
beispielsweise Essigsäure,
H2SO4 oder HCl,
wird dann zu dem Reaktionsgemisch bei einer Temperatur zwischen
10 und 30°C,
bevorzugt zwischen 15 und 25°C,
zugegeben. Nach dem Rühren
des resultierenden Gemisches für
5 bis 180 Minuten, bevorzugt 30 bis 60 Minuten, wird die Verbindung
der Formel XIII in guter Ausbeute erhalten.
-
Das
Verfahren gemäß der vorliegenden
Erfindung zur Herstellung der Verbindungen der Formel I umfaßt die Schritte:
- a) Umsetzen einer Verbindung der Formel mit einer Verbindung der
Formel unter Bildung einer Verbindung
der Formel wobei die Definition der
Substituenten oben beschrieben ist.
-
Die
ausführlichere
Beschreibung von Schritt a) ist folgende:
-
Schritt a1)
-
Die
Reaktion findet in einem organischen Lösungsmittel, wie Ether, Keton
oder Alkohol, bevorzugt Alkohol, am stärksten bevorzugt Methanol,
statt. Das Reaktionsgemisch wird mit einer organischen Base, wie Triethylamin,
bei etwa 10 bis 50°C,
bevorzugt 20 bis 30°C,
behandelt. Das Reaktionsgemisch wird für 0,5 bis 12 Stunden, bevorzugt
2 Stunden, gerührt
und im Vakuum konzentriert.
-
Schritt a2)
-
Der
Rest wird dann in einem organischen Lösungsmittel, wie Dichlormethan,
aufgenommen, mit (Chlormethylen)dimethylammoniumchlorid (Vilsmeier-Reagens)
behandelt und auf 30 bis 60°C,
bevorzugt 45°C,
für 0,5
bis 5 Stunden, bevorzugt 1 Stunde, erhitzt. Um den gesamten flüchtigen
Stoff zu entfernen, wird das Gemisch im Vakuum konzentriert.
-
Schritt a3)
-
Der
resultierende Rest wird, rein oder gelöst in einem Lösungsmittel
mit hoher Siedetemperatur, wie Xylol, Toluol, Diphenylether, auf
150 bis 240°C,
bevorzugt 170 bis 200°C,
am stärksten
bevorzugt 180 bis 190°C,
für 10
bis 60 Minuten, bevorzugt 15 Minuten, erhitzt, auf 10 bis 30°C, bevorzugt
20 bis 25°C,
abgekühlt und
in einem organischen Lösungsmittel
gelöst
und durch Extraktion mit Wasser gereinigt. Die Schichten werden
getrennt. Die Eindampfung der organischen Phase im Vakuum ergibt
das Produkt der Formel
- b) Umwandeln
der OH/=O-Funktion der Verbindungen der Formel XIV/XIVa in eine
Austrittsgruppe P wobei P Halogen oder -O-SO2CF3 ist.
-
Die
Umsetzung der Verbindung der Formel XVI mit Reagenzien, enthaltend
Austrittsgruppen, beispielsweise POCl3,
PBr3, MeI oder (F3CSO2)2O, wird in einem
organischen Lösungsmittel,
beispielsweise Dichlormethan, Trifluormethylbenzol oder Chlorbenzol,
und bei einer Temperatur von etwa 40 bis 80°C, bevorzugt etwa 50°C, für 60 bis
240 Minuten, bevorzugt 80 Minuten, durchgeführt.
- c)
Substituieren der Austrittsgruppe P der Formel XVI durch R2 mit HR2 unter Bildung
einer Verbindung der Formel wobei die Definition der
Substitutionen für
R1, R1' und R2 zuvor
beschrieben ist.
-
Schritt
c) wird bevorzugt in einem organischen Lösungsmittel, beispielsweise
DMF, DMSO, N-Methylpyrrolidon, Chlorbenzol, Toluol oder Gemischen
davon, und bei einer Temperatur von etwa 60 bis 150°C, bevorzugt
90 bis 120°C,
am stärksten
bevorzugt 112°C,
für 10
bis 240 Minuten, bevorzugt 20 bis 120 Minuten, am stärksten bevorzugt
30 bis 60 Minuten, durchgeführt.
Das Gemisch wird abgekühlt
und mit einer Säure,
wie Schwefelsäure,
Essigsäure
oder Hydrochlorid, behandelt.
- d) Hydrolysieren
der Nitrilfunktion der Formel XVI unter Bildung einer Verbindung
der Formel
-
Schritt
d) wird in einem sauren Medium, beispielsweise H2SO4, HCl oder Essigsäure, mit oder ohne ein organisches
Lösungsmittel
bei einer Temperatur von 50 bis 140°C, bevorzugt 60 bis 90°C, am stärksten bevorzugt
70°C, für 1 bis
8 Stunden, bevorzugt 2 Stunden, durchgeführt.
-
Gemäß einer
bevorzugten Ausführungsform
der Erfindung sind R
1 und R
1' unabhängig voneinander Niederalkyl,
Alkoxy, Halogen, Cyano oder Alkylamino; ist P Halogen und ist R
2 ein cyclisches tertiäres Amin der Gruppe
worin R
4 vorstehend
beschrieben ist.
-
Gemäß einer
stärker
bevorzugten Ausführungsform
der Erfindung sind R1 und R1' unabhängig voneinander
Wasserstoff, Niederalkyl, Alkoxy, Halogen, Cyano oder Alkylamino;
ist P Chlor und ist R2 Morpholin-4-yl, 4-Methyl-piperazin-1-yl
oder 1,1-Dioxothiomorpholin-4-yl.
-
Gemäß noch einer
stärker
bevorzugten Ausführungsform
der Erfindung wird das vorliegende Verfahren für die Herstellung von 6-Hydroxy-4-o-tolyl-nicotinonitril,
6-Oxo-4-p-tolyl-1,6-dihydro-pyridin-3-carbonitril, 4-(2-Chlor-phenyl)-6-oxo-1,
6-dihydro-pyridin-3-carbonitril, 4-(4-Chlor-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril,
4-(3-Cyano-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril,
4-(4-Cyano-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril, 4-(4-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril,
4-(3-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril,
4-(2-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril, 4-(4-Dimethylamino-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril,
6-Oxo-4-phenyl-1,6-dihydro-pyridin-3-carbonitril,
N-(6-Oxo-4-phenyl-1,6-dihydro-pyridin-3-yl)-acetamid, 6-Chlor-4-o-tolyl-nicotinonitril,
6-Morpholin-4-yl-4-tolyl-nicotinonitril, 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-nicotinonitril,
6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinonitril, 6-Morpholin-4-yl-4-o-tolyl-nicotinamid,
6-(4-Methylpiperazin-1-yl)-4-o-tolyl-nicotinamid und 6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinamid
verwendet.
-
Bevorzugte
Ausführungsformen
der vorliegenden Erfindung werden durch die folgenden Beispiele
1 bis 14 ausführlicher
beschrieben.
-
Beispiel 1
-
6-Morpholin-4-yl-4-o-tolyl-nicotinamid
-
a) 1-Carbamoylmethyl-pyridiniumchlorid
-
50,00
g (524,01 mmol) 2-Chloracetamid wurden in 100 ml Acetonitril suspendiert.
41,45 g (524,01 mmol) Pyridin wurden zugegeben, und die Suspension
wurde bei 90°C
für 10
Stunden erhitzt. Die Suspension wurde auf 22°C abgekühlt, saugfiltriert und mit
100 ml Hexan gewaschen. Das Produkt 1-Carbamoylmethyl-pyridiniumchlorid
(79,10 g) wurde als farblose Kristalle nach der Umkristallisation
aus Ethanol erhalten, Smp. 205,2°C.
H1 NMR (400 MHz, CDCl3,
ppm). 5,50 (s, 2H), 7,72 (s, 1H), 8,17-8,20 (m, 2H), 8,32 (s, 1H),
8,64-8,68 (m, 1H), 9,04 (d, 2H).
-
b) 2-Formyl-3-o-tolyl-acrylnitril
-
Eine
Lösung
aus 13,19 g (110,00 mmol) 3,3-Dimethoxypropionitril und 12,39 g
(100,00 mmol) o-Tolylaldehyd wurde zu 23,40 g (130,00 mmol) Natriummethanolat
in 22,0 ml Methanol unter Halten der Innentemperatur unter 20°C zugegeben.
Das Reaktionsgemisch wurde bei 22 bis 25°C über Nacht gerührt und
im Vakuum konzentriert (Rotationsverdampfer bei 40°C und 20
mbar). 100,00 ml HCl (25%) wurden bei 15 bis 25°C zugegeben, und das resultierende
Gemisch wurde für
60 Minuten gerührt.
Der Niederschlag wurde saugfiltriert, mit 30 ml Methanol (vorgekühlt auf –20°C) gewaschen
und im Vakuum getrocknet, wodurch 16,14 g 2-Formyl-3-o-tolyl-acrylnitril
als gelbliche Kristalle erhalten wurden, Smp. 81,5°C.
H1 NMR (300 MHz, DMSO, ppm). 2,51 (s, 1H),
7,41-7,58 (m, 3H), 8,06 (d, 1H), 8,76 (s, 1H), 9,74 (s, 1H). MS (EI):
m/e = 171 ([M] 30), 156 (100), 143 (23), 115 (46).
-
c) 6-Hydroxy-4-o-tolyl-nicotinonitril
-
1,726
g (10,0 mmol) 1-Carbamoylmethyl-pyridiniumchlorid und 1,712 g (10,0
mmol) 2-Formyl-3-o-tolyl-acrylnitril in 24,8 ml Methanol wurden
mit 1,05 g (10,4 mmol) Triethylamin bei 20 bis 30°C behandelt.
Das Reaktionsgemisch wurde für
2 Stunden gerührt
und im Vakuum konzentriert (Rotationsverdampfer bei 40°C und 20
mbar). Der Rest wurde in 50 ml Dichlormethan aufgenommen, mit 2,56
g (20,0 mmol) (Chlormethylen)dimethylammoniumchlorid (Vilsmeier-Reagens)
behandelt und bei 45°C
für 1 Stunde
erhitzt. Um den gesamten flüchtigen
Stoff zu entfernen, wurde das Gemisch im Vakuum konzentriert (Rotationsverdampfer
bei 45°C
und 20 mbar). Der Rest wurde auf 180 bis 190°C für 15 Minuten erhitzt, auf 20
bis 25°C
abgekühlt
und in 80,0 ml Dichlormethan und 80,0 ml Wasser vergeteilt. Die
Schichten wurden getrennt. Die Eindampfung der organischen Phase
im Vakuum ergab 1,37 g des amorphen Produktes 6-Hydroxy-4-o-tolyl-nicotinonitril.
H1 NMR (400 MHz, CDCl3,
ppm). 2,30 (s, 3H), 6,54 (s, 1H), 7,18 (d, 1H), 7,28-7,38 (m, 3H),
7,92 (s, 1H). NH ? MS (ISP): 211 ([M+H+]
100).
-
d) 6-Chlor-4-o-tolyl-nicotinonitril
-
Ein
Gemisch aus 2,5 g (11,89 mmol) 6-Hydroxy-4-o-tolyl-nicotinonitril,
3,64 g (23,78 mmol) Phosphor(V)-oxidchlorid in 10,0 ml Dichlormethan
wurde bei 50°C
für 80
Minuten erhitzt. Das Gemisch wurde auf 20 bis 25°C abgekühlt, auf Wasser unter Halten
der Innentemperatur zwischen 20 und 30°C gegossen und durch Zugeben
von weiteren 80,0 ml Dichlormethan extrahiert. Die Eindampfung der
organischen Phase im Vakuum ergab 2,9 g des Rohproduktes 6-Chlor-4-o-tolyl-nicotinonitril,
das durch Chromatographie über
Kieselgel (Ethylacetat:Hexan = 4:1) gereinigt wurde, wodurch 2,4
g 6-Chlor-4-o-tolyl-nicotinonitril erhalten wurden, Smp. 112,4°C.
H1 NMR (400 MHz, CDCl3,
ppm). 2,24 (s, 3H), 7,16 (s, 1H), 7,30-7,41 (m, 4H), 8,74 (s, 1H).
MS (ISP): 229 ([M+H+] 100).
-
e) 6-Morpholin-4-yl-4-tolyl-nicotinonitril
-
500
mg (2,1865 mmol) 6-Chlor-4-o-tolyl-nicotinonitril wurden in 10,0
ml Toluol gelöst
und auf 112°C erhitzt.
Bei dieser Temperatur wurden 762 mg (8,746 mmol) Morpholin zugegeben,
und das Reaktionsgemisch wurde für
weitere 30 Minuten gerührt.
Das Gemisch wurde auf 20 bis 25°C
abgekühlt
und mit 900 mg Schwefelsäure
(95%) behandelt. Die organische Phase wurde mit 5 ml Wasser gewaschen
(pH der Wasserphase 7–7,5).
Die Eindampfung im Vakuum ergab 530 mg 6-Morpholin-4-yl-4-tolyl-nicotinonitril
als weißen
Schaum.
H1 NMR (300 MHz, CDCl3, ppm). 2,24 (s, 3H), 3,65-3,68 (m, 4H),
3,77-3,82 (m, 4H), 6,47 (s, 1H), 7,15-7,35 (m, 4H), 8,48 (s, 1H).
MS (ISP): 280 ([M+H+] 100).
-
f) 6-Morpholin-4-yl-4-o-tolyl-nicotinamid
-
Ein
Gemisch aus 500 mg (1,79 mmol) rohem 6-Morpholin-4-yl-4-tolyl-nicotinonitril,
0,5 ml Toluol und 475 mg Schwefelsäure (95%) wurde bei 70°C für 2 Stunden
erhitzt. Die Suspension wurde auf 20 bis 25°C abgekühlt und mit 5 ml Wasser gequencht.
5 ml Ethylacetat wurden zugegeben, gefolgt von einer Lösung aus 710
mg Natriumhydroxid in 2 ml Wasser. Die Eindampfung der organischen
Phase im Vakuum ergab 700 mg farblosen Feststoff. Nach der Reinigung
durch Chromatographie über
Kieselgel (Ethylacetat/Hexan 1:2) wurden 490 mg 6-Morpholin-4-yl-4-o-tolyl-nicotinamid
als farblose Kristalle erhalten, Smp. 144–145°C.
H1 NMR
(300 MHz, CDCl3, ppm). 2,15 (s, 3H), 3,62-3,64
(m, 4H), 3,80-3,82 (m, 4H), 5,0-5,3 (br, 2H), 6,30 (s, 1H), 7,2-7,37
(m, 4H), 8,94 (s, 1H). MS (EI): m/e = 297 ([M] 64), 266 ([M-CH2OH] 100).
-
Beispiel 2
-
6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-nicotinamid
-
a) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-nicotinonitril:
-
500
mg (2,1865 mmol) 6-Chlor-4-o-tolyl-nicotinonitril wurden in 10,0
ml Toluol gelöst
und auf 112°C erhitzt.
Bei dieser Temperatur wurden 2,19 g (21,865 mmol) 1-Methylpiperazin
zugegeben, und das Reaktionsgemisch wurde für weitere 60 Minuten gerührt. Das
Gemisch wurde auf 50°C
abgekühlt
und unter reduziertem Druck konzentriert. 5 ml Toluol wurden zu
dem erhaltenen Rest bei einer Temperatur von 20 bis 25°C zugegeben,
gefolgt von 900 mg Schwefelsäure
(95%). Die organische Phase wurde mit 5 ml Wasser gewaschen. Die Eindampfung
im Vakuum ergab 520 mg 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-nicotinonitril
als beigefarbenen Schaum.
H1 NMR (300
MHz, CDCl3, ppm). 2,25 (s, 3H), 2,35 (s,
3H), 2,46-2,52 (m, 4H), 3,70-3,73 (m, 4H), 6,48 (s, 1H), 7,15-7,37
(m, 4H), 8,46 (s, 1H). MS (ISP): 293 ([M+H+]
100).
-
b) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-nicotinamid
-
480
mg (1,642 mmol) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-nicotinonitril
wurden mit 3,8 ml Schwefelsäure
(90%) behandelt und bei 80°C
für 1 Stunde
erhitzt. Das Gemisch wurde auf 20 bis 25°C abgekühlt und mit 20 ml Ethylacetat
behandelt. 2,0 g Natriumhydroxidlösung (28%) wurden zugegeben,
und die organische Phase wurde mit 6 ml Wasser gewaschen. Die Eindampfung
im Vakuum ergab 380 mg 6-(4-Methylpiperazin-1-yl)-4-o-tolyl-nicotinamid
als hellgelben kristallinen Schaum.
H1 NMR
(400 MHz, CDCl3, ppm). 2,15 (s, 3H), 2,34
(s, 3H), 2,45-2,52 (m, 4H), 3,67-3,73 (m, 4H), 5,01-5,28 (b, 2H),
6,31 (s, 1H), 7,20-7,36 (m, 4H), 8,93 (s, 1H). MS (ISP): 332 ([M+H+] 100).
-
Beispiel 3
-
6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinamid
-
a) 6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinonitril
-
Ein
Gemisch aus 500 mg (2,1865 mmol) 6-Chlor-4-o-tolyl-nicotinonitril,
1,478 g (10,9325 mmol) Thiomorpholin-1,1-dioxid und 5 ml Ethylacetat
wurde bei 80°C
für 12
Stunden erhitzt. Das Gemisch wurde auf 20 bis 25°C abgekühlt und mit 7,5 ml Ethylacetat
behandelt, gefolgt von 5,0 ml Wasser. Die organische Phase wurde
mit 5,0 ml Wasser gewaschen und unter reduziertem Druck konzentriert.
Die Kristallisation aus Dichlormethan/Hexan 1:2 ergab 450 mg 6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinonitril
als beigefarbene Kristalle, Smp. 182,7°C.
H1 NMR
(300 MHz CDCl3, ppm). 2,24 (s, 3H), 3,07-3,11
(m, 4H), 4,24-4,28 (m, 4H), 6,63 (s, 1H), 7,14-7,41 (m, 4H), 8,52
(s, 1H). MS (ISP): 328 ([M+H+] 100).
-
b) 6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinamid
-
400
mg (1,222 mmol) 6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinonitril
wurden mit 400 mg Schwefelsaure (95%) verdünnt und bei 70°C für 2 Stunden
erhitzt. Das Gemisch wurde auf 20 bis 25°C abgekühlt, mit 5 ml Ethylacetat behandelt,
gefolgt von einer Lösung
aus 600 mg Natriumhydroxid in 2 ml Wasser. Die organische Phase
wurde zweimal mit 2 ml Wasser gewaschen, unter reduziertem Druck
konzentriert, wodurch 360 mg 6-(1,1-Dioxo-1λ6-6-thiomorpholin-4-yl)-tolyl-nicotinamid
als weiße
Kristalle erhalten wurden, Smp. 239,7°C.
H1 NMR
(300 MHz, DMSO, ppm). 2,11 (s, 1H), 3,07-3,18 (m, 4H), 4,06-4,17
(m, 4H), 6,77 (s, 1H), 7,06-7,26 (m, 6H), 8,40 (s, 1H). MS (ISP):
346 ([M+H+] 100).
-
Beispiel 4
-
6-Oxo-4-p-tolyl-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-p-tolyl durchgeführt, wodurch 6-Oxo-4-p-tolyl-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
DMSO, ppm). 2,37 (s, 3H), 6,40 (s, 1H), 7,33 (d, 2H), 7,45 (d, 2H),
8,35 (s, 1H), 12,71 (s, 1H). MS (EI): m/e = 210 ([M] 15), 86 (100),
58 (30).
-
2-Propennitril-2-formyl-3-p-tolyl
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und p-Tolylaldehyd synthetisiert.
-
Beispiel 5
-
4-(2-Chlor-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(2-chlor-phenyl) durchgeführt, wodurch
4-(2-Chlor-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
CDCl3, ppm). 6,59 (s, 1H), 7,26-7,55 (m,
5H), 7,92 (s, 1H). MS (ISP): 231 ([M+H+]
100).
-
2-Propennitril-2-formyl-3-(2-chlor-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und o-Chlor-benzaldehyd synthetisiert.
-
Beispiel 6
-
4-(4-Chlor-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(4-chlor-phenyl) durchgeführt, wodurch
4-(4-Chlor-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril hergestellt
wurde.
H1 NMR (400 MHz, DMSO, ppm).
6,46 (s, 1H), 7,59 (s, 4H), 8,38 (s, 1H), 12,81 (s, 1H). MS (ISP):
231 ([M+H+] 100).
-
2-Propennitril-2-formyl-3-(4-chlor-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und o-Chlor-benzaldehyd synthetisiert.
-
Beispiel 7
-
4-(3-Cyano-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(3-cyano-phenyl) durchgeführt, wodurch
4-(3-Cyano-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
DMSO, ppm). 6,55 (s, 1H), 7,74 (t, 1H), 7,91 (d, 1H), 7,99 (d, 1H),
8,06 (s, 1H), 8,42 (s, 1H), 12,72 (s, 1H). MS (ISN): 220 ([M-H]
100).
-
2-Propennitril-2-formyl-3-(3-cyano-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und m-Cyano-benzaldehyd synthetisiert.
-
Beispiel 8
-
4-(4-Cyano-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(4-cyano-phenyl) durchgeführt, wodurch
4-(4-Cyano-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
DMSO, ppm). 6,52 (s, 1H), 7,76 (d, 2H), 8,01 (d, 2H), 8,42 (s, 1H),
12,87 (s, 1H). MS (ISN): 220 ([M-H] 100).
-
2-Propennitril-2-formyl-3-(4-cyano-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und p-Cyano-benzaldehyd synthetisiert.
-
Beispiel 9
-
4-(4-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(4-methoxy-phenyl) durchgeführt, wodurch
4-(4-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
DMSO, ppm). 3,82 (s, 3H), 6,33 (s, 1H), 7,06 (d, 2H), 7,51 (d, 2H),
8,31 (s, 1H), 12,54 (s, 1H). MS (ISN): m/e = 226 (32), 225 (M-H,
100).
-
2-Propennitril-2-formyl-3-(4-methoxy-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und p-Methoxy-benzaldehyd synthetisiert.
-
Beispiel 10
-
4-(3-Methoxy-phenyl)-o-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(3-methoxy-phenyl) durchgeführt, wodurch
4-(3-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
CDCl3, ppm). 3,87 (s, 3H), 6,66 (s, 1H),
7,04-7,07 (m, 2H), 7,12-7,41 (m, 3H), 7,94 (s, 1H). MS (ISP): 227
([M+H+] 100).
-
2-Propennitril-2-formyl-3-(3-methoxy-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und m-Methoxy-benzaldehyd synthetisiert.
-
Beispiel 11
-
4-(2-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(2-methoxy-phenyl) durchgeführt, wodurch
4-(2-Methoxy-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
CDCl3, ppm). 3,89 (s, 3H), 6,59 (s, 1H),
7,02-7,46 (m, 4H), 7,84 (s, 1H), 12,91 (s, 1H). MS (ISP): 227 ([M+H+] 100).
-
2-Propennitril-2-formyl-3-(2-methoxy-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und o-Methoxy-benzaldehyd synthetisiert.
-
Beispiel 12
-
4-(4-Dimethylamino-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Propennitril-2-formyl-3-(4-dimethylamino-phenyl) durchgeführt, wodurch
4-(4-Dimethylamino-phenyl)-6-oxo-1,6-dihydro-pyridin-3-carbonitril
(amorph) hergestellt wurde.
H1 NMR
(400 MHz, DMSO, ppm). 2,97 (s, 6H), 6,26 (s, 1H), 6,79 (d, 2H),
7,43 (d, 2H), 8,25 (s, 1H), 9,18 (s, 1H). MS (ISP): 240 ([M+H+] 100), 262 ([M+Na+],
10).
-
2-Propennitril-2-formyl-3-(4-dimethylamino-phenyl)
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und p-Dimethylamino-benzaldehyd synthetisiert.
-
Beispiel 13
-
6-Oxo-4-phenyl-1,6-dihydro-pyridin-3-carbonitril
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Cyano-zimtsäurealdehyd
durchgeführt,
wodurch 6-Oxo-4-phenyl-1,6-dihydro-pyridin-3-carbonitril (amorph)
hergestellt wurde.
H1 NMR (300 MHz,
DMSO, ppm). 6,44 (s, 1H), 7,51-7,58 (m, 5H), 8,39 (s, 1H), 12,60
(s, 1H). MS (ISN): 195 ([M-H] 100).
-
2-Cyano-zimtsäurealdehyd
wurde analog zu Beispiel 1b unter Verwendung von 3,3-Dimethoxypropionitril
und Benzaldehyd synthetisiert.
-
Beispiel 14
-
N-(6-Oxo-4-phenyl-1,6-dihydro-pyridin-3-yl)-acetamid
-
Die
Synthese wurde analog zu Beispiel 1c unter Verwendung von 1-Carbamoylmethyl-pyridiniumchlorid
und 2-Acetamido-zimtsäurealdehyd
durchgeführt.
In diesem Fall war die Behandlung mit Vilsmeier-Reagens nicht erforderlich.
Der erhaltene Rest nach der Michael-Additionsreaktion wurde direkt bei 190°C für 30 min
erhitzt, wodurch N-(6-Oxo-4-phenyl-1,6-dihydro-pyridin-3-yl)-acetamid (amorph)
hergestellt wurde.
H1 NMR (400 MHz,
DMSO, ppm). 1,78 (s, 3H), 6,27 (s, 1H), 7,35-7,43 (m, 6H), 9,01
(s, 1H), 11,5 (s, 1H). MS (ISP): 229 ([M+H+]
100), 187 (15).
-
2-Acetamido-zimtsäurealdehyd
wurde analog zu einer beschriebenen Verfahrensweise synthetisiert (K.
Eiter, E. Sackl, Monatshefte für
Chemie 1952, 123–136).



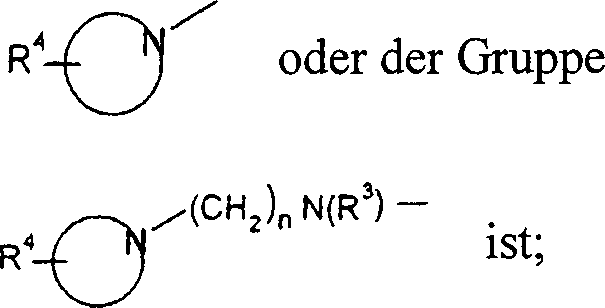



 unter Bildung einer Verbindung der Formel
unter Bildung einer Verbindung der Formel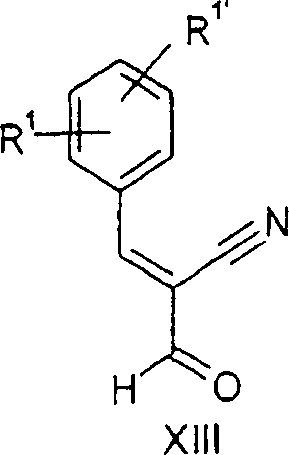 wobei die Definition der Substituenten oben beschrieben ist.
wobei die Definition der Substituenten oben beschrieben ist.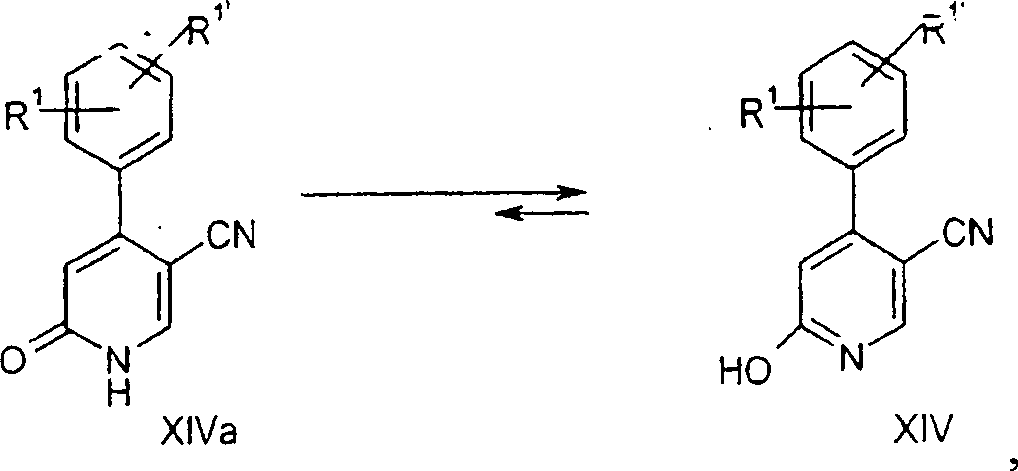





 R3 unabhängig voneinander Wasserstoff, C3-6-Cycloalkyl, Benzyl oder C1-7-Alkyl ist; R4 Wasserstoff, Hydroxy, C1-7-Alkyl, -(CH2)nCOO-C1-7-Alkyl, -N(R3)CO-C1-7-Alkyl, Hydroxy-C1-7-alkyl, Cyano, -(CH2)nO(CH2)nOH, -CHO oder eine 5- oder 6gliedrige heterocyclische Gruppe, gegebenenfalls über eine Alkylengruppe gebunden, ist; dadurch gekennzeichnet, daß es die Schritte a) Umsetzen einer Verbindung der Formel
R3 unabhängig voneinander Wasserstoff, C3-6-Cycloalkyl, Benzyl oder C1-7-Alkyl ist; R4 Wasserstoff, Hydroxy, C1-7-Alkyl, -(CH2)nCOO-C1-7-Alkyl, -N(R3)CO-C1-7-Alkyl, Hydroxy-C1-7-alkyl, Cyano, -(CH2)nO(CH2)nOH, -CHO oder eine 5- oder 6gliedrige heterocyclische Gruppe, gegebenenfalls über eine Alkylengruppe gebunden, ist; dadurch gekennzeichnet, daß es die Schritte a) Umsetzen einer Verbindung der Formel mit einer Verbindung der Formel
mit einer Verbindung der Formel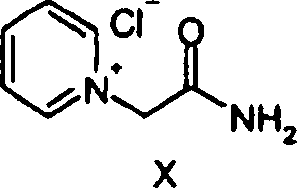 wobei R1 und R1' die oben angegebenen Bedeutungen haben, zu einer Verbindung der Formel
wobei R1 und R1' die oben angegebenen Bedeutungen haben, zu einer Verbindung der Formel wobei die Substituenten wie oben beschrieben sind, b) Umwandeln der OH/=O-Funktion der Verbindungen der Formel XIV/XIVa zu einer Austrittsgruppe P mit einem Reagens, enthaltend eine Austrittsgruppe, ausgewählt aus der Gruppe, umfassend POCl3, PBr3, MeI oder (F3CSO2)2O, unter Bildung einer Verbindung der Formel
wobei die Substituenten wie oben beschrieben sind, b) Umwandeln der OH/=O-Funktion der Verbindungen der Formel XIV/XIVa zu einer Austrittsgruppe P mit einem Reagens, enthaltend eine Austrittsgruppe, ausgewählt aus der Gruppe, umfassend POCl3, PBr3, MeI oder (F3CSO2)2O, unter Bildung einer Verbindung der Formel wobei P Halogen oder Trifluormethansulfonat ist und R1 und R1' die oben angegebenen Bedeutungen haben, c) Substituieren der Austrittsgruppe P durch R2 mit HR2 unter Bildung einer Verbindung der Formel
wobei P Halogen oder Trifluormethansulfonat ist und R1 und R1' die oben angegebenen Bedeutungen haben, c) Substituieren der Austrittsgruppe P durch R2 mit HR2 unter Bildung einer Verbindung der Formel wobei R1 und R1' und R2 die oben angegebenen Bedeutungen haben, d) Hydrolysieren der Nitrilfunktion in einem sauren Medium, umfassend H2SO4, HCl oder Essigsäure, zu einer Verbindung der Formel
wobei R1 und R1' und R2 die oben angegebenen Bedeutungen haben, d) Hydrolysieren der Nitrilfunktion in einem sauren Medium, umfassend H2SO4, HCl oder Essigsäure, zu einer Verbindung der Formel wobei die Definition der Substituenten wie oben angegeben ist, umfaßt.
wobei die Definition der Substituenten wie oben angegeben ist, umfaßt.

 R3 unabhängig voneinander Wasserstoff, C3-6-Cycloalkyl, Benzyl oder C1-7-Alkyl ist; R4 Wasserstoff, Hydroxy, C1-7-Alkyl, -(CH2)nCOO-C1-7-Alkyl, -N(R3)CO-C1-7-Alkyl, Hydroxy-C1-7-alkyl, Cyano, -(CH2)nO(CH2)nOH, -CHO oder eine 5- oder 6gliedrige heterocyclische Gruppe, gegebenenfalls über eine Alkylengruppe gebunden, ist; R5 und R5' unabhängig voneinander Wasserstoff, C1-7-Alkyl sind oder zusammen mit dem Kohlenstoffatom eine Cycloalkylgruppe bilden; R6 und R6' unabhängig voneinander Wasserstoff, Halogen, Trifluormethyl, C1-7-Alkoxy oder Cyano sind; oder R6 und R6' zusammen -CH=CH-CH=CH-, gegebenenfalls substituiert durch einen oder zwei Substituenten, ausgewählt aus C1-7-Alkyl oder C1-7-Alkoxy, sein können; X -C(O)N(R3)-, -(CH2)mO-, -(CH2)mN(R3)-, -N(R3)C(O)- oder -N(R3)(CH2)m- ist; n 0 bis 4 ist und m 1 oder 2 ist, dadurch gekennzeichnet, daß die Zwischenverbindung der Formel I durch die Schritte a) Umsetzen einer Verbindung der Formel
R3 unabhängig voneinander Wasserstoff, C3-6-Cycloalkyl, Benzyl oder C1-7-Alkyl ist; R4 Wasserstoff, Hydroxy, C1-7-Alkyl, -(CH2)nCOO-C1-7-Alkyl, -N(R3)CO-C1-7-Alkyl, Hydroxy-C1-7-alkyl, Cyano, -(CH2)nO(CH2)nOH, -CHO oder eine 5- oder 6gliedrige heterocyclische Gruppe, gegebenenfalls über eine Alkylengruppe gebunden, ist; R5 und R5' unabhängig voneinander Wasserstoff, C1-7-Alkyl sind oder zusammen mit dem Kohlenstoffatom eine Cycloalkylgruppe bilden; R6 und R6' unabhängig voneinander Wasserstoff, Halogen, Trifluormethyl, C1-7-Alkoxy oder Cyano sind; oder R6 und R6' zusammen -CH=CH-CH=CH-, gegebenenfalls substituiert durch einen oder zwei Substituenten, ausgewählt aus C1-7-Alkyl oder C1-7-Alkoxy, sein können; X -C(O)N(R3)-, -(CH2)mO-, -(CH2)mN(R3)-, -N(R3)C(O)- oder -N(R3)(CH2)m- ist; n 0 bis 4 ist und m 1 oder 2 ist, dadurch gekennzeichnet, daß die Zwischenverbindung der Formel I durch die Schritte a) Umsetzen einer Verbindung der Formel mit einer Verbindung der Formel
mit einer Verbindung der Formel wobei R1 und R1' die oben angegebenen Bedeutungen haben, zu einer Verbindung der Formel
wobei R1 und R1' die oben angegebenen Bedeutungen haben, zu einer Verbindung der Formel wobei die Substituenten wie oben beschrieben sind, b) Umwandeln der OH/=O-Funktion der Verbindungen der Formel XIV/XIVa zu einer Austrittsgruppe P mit einem Reagens, enthaltend eine Austrittsgruppe, ausgewählt aus der Gruppe, umfassend POCl3, PBr3, MeI oder (F3CSO2)2O unter Bildung einer Verbindung der Formel
wobei die Substituenten wie oben beschrieben sind, b) Umwandeln der OH/=O-Funktion der Verbindungen der Formel XIV/XIVa zu einer Austrittsgruppe P mit einem Reagens, enthaltend eine Austrittsgruppe, ausgewählt aus der Gruppe, umfassend POCl3, PBr3, MeI oder (F3CSO2)2O unter Bildung einer Verbindung der Formel wobei P Halogen oder Trifluormethansulfonat ist und R1 und R1' die oben angegebenen Bedeutungen haben, c) Substituieren der Austrittsgruppe P durch R2 mit HR2 unter Bildung einer Verbindung der Formel
wobei P Halogen oder Trifluormethansulfonat ist und R1 und R1' die oben angegebenen Bedeutungen haben, c) Substituieren der Austrittsgruppe P durch R2 mit HR2 unter Bildung einer Verbindung der Formel wobei R1 und R1' und R2 die oben angegebenen Bedeutungen haben, d) Hydrolysieren der Nitrilfunktion in einem sauren Medium, umfassend H2SO4, HCl oder Essigsäure, zu einer Verbindung der Formel
wobei R1 und R1' und R2 die oben angegebenen Bedeutungen haben, d) Hydrolysieren der Nitrilfunktion in einem sauren Medium, umfassend H2SO4, HCl oder Essigsäure, zu einer Verbindung der Formel wobei die Definition der Substituenten wie oben angegeben ist, erhalten wird.
wobei die Definition der Substituenten wie oben angegeben ist, erhalten wird.