DE68929520T2 - Chinolin-Derivate als Leukotrien-D4-Antagonisten, diese enthaltende Zusammensetzungen und Verfahren zu deren Herstellung - Google Patents
Chinolin-Derivate als Leukotrien-D4-Antagonisten, diese enthaltende Zusammensetzungen und Verfahren zu deren Herstellung Download PDFInfo
- Publication number
- DE68929520T2 DE68929520T2 DE68929520T DE68929520T DE68929520T2 DE 68929520 T2 DE68929520 T2 DE 68929520T2 DE 68929520 T DE68929520 T DE 68929520T DE 68929520 T DE68929520 T DE 68929520T DE 68929520 T2 DE68929520 T2 DE 68929520T2
- Authority
- DE
- Germany
- Prior art keywords
- alkyl
- tetrazolyl
- groups
- chemical bond
- hydroxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000000034 method Methods 0.000 title claims description 19
- 238000002360 preparation method Methods 0.000 title claims description 15
- 239000000203 mixture Substances 0.000 title description 28
- 125000002943 quinolinyl group Chemical class N1=C(C=CC2=CC=CC=C12)* 0.000 title description 4
- 239000005557 antagonist Substances 0.000 title description 3
- 229940027991 antiseptic and disinfectant quinoline derivative Drugs 0.000 title 1
- YEESKJGWJFYOOK-IJHYULJSSA-N leukotriene D4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@H](N)C(=O)NCC(O)=O YEESKJGWJFYOOK-IJHYULJSSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 71
- -1 aralkoxy Chemical group 0.000 claims description 40
- 125000000217 alkyl group Chemical group 0.000 claims description 35
- 229910052739 hydrogen Inorganic materials 0.000 claims description 31
- 239000000126 substance Substances 0.000 claims description 31
- 239000001257 hydrogen Substances 0.000 claims description 30
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 28
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 24
- 125000003545 alkoxy group Chemical group 0.000 claims description 23
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 21
- 125000001424 substituent group Chemical group 0.000 claims description 21
- 125000001188 haloalkyl group Chemical group 0.000 claims description 17
- 125000001475 halogen functional group Chemical group 0.000 claims description 17
- 229910052760 oxygen Inorganic materials 0.000 claims description 17
- 150000003839 salts Chemical class 0.000 claims description 17
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 16
- 229910052717 sulfur Inorganic materials 0.000 claims description 15
- 125000002252 acyl group Chemical group 0.000 claims description 12
- 238000006243 chemical reaction Methods 0.000 claims description 11
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 10
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 9
- 125000003342 alkenyl group Chemical group 0.000 claims description 8
- 125000004181 carboxyalkyl group Chemical group 0.000 claims description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 8
- 238000011282 treatment Methods 0.000 claims description 8
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 7
- 125000004442 acylamino group Chemical group 0.000 claims description 6
- 125000001691 aryl alkyl amino group Chemical group 0.000 claims description 6
- 229910052736 halogen Inorganic materials 0.000 claims description 5
- 150000002367 halogens Chemical class 0.000 claims description 5
- 239000003199 leukotriene receptor blocking agent Substances 0.000 claims description 5
- 239000003814 drug Substances 0.000 claims description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 4
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 claims description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims 15
- 206010027654 Allergic conditions Diseases 0.000 claims 1
- 206010061218 Inflammation Diseases 0.000 claims 1
- 229940122142 Lipoxygenase inhibitor Drugs 0.000 claims 1
- 230000001419 dependent effect Effects 0.000 claims 1
- 229940079593 drug Drugs 0.000 claims 1
- 230000004054 inflammatory process Effects 0.000 claims 1
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 230000002265 prevention Effects 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 34
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 31
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 27
- 239000000243 solution Substances 0.000 description 27
- 239000000047 product Substances 0.000 description 23
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000011541 reaction mixture Substances 0.000 description 16
- 150000002431 hydrogen Chemical class 0.000 description 15
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 239000012043 crude product Substances 0.000 description 12
- 238000003818 flash chromatography Methods 0.000 description 12
- LCPDWSOZIOUXRV-UHFFFAOYSA-N phenoxyacetic acid Chemical compound OC(=O)COC1=CC=CC=C1 LCPDWSOZIOUXRV-UHFFFAOYSA-N 0.000 description 11
- 210000001519 tissue Anatomy 0.000 description 11
- 239000002253 acid Substances 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- 230000008018 melting Effects 0.000 description 8
- 238000002844 melting Methods 0.000 description 8
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 239000002585 base Substances 0.000 description 7
- UFINMQZIYXBSNL-UHFFFAOYSA-N 3-(quinolin-2-ylmethoxy)phenol Chemical compound OC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 UFINMQZIYXBSNL-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 150000001299 aldehydes Chemical class 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 150000002617 leukotrienes Chemical class 0.000 description 6
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 6
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 6
- DDEAEWMDOSXKBX-UHFFFAOYSA-N 2-(chloromethyl)quinoline Chemical compound C1=CC=CC2=NC(CCl)=CC=C21 DDEAEWMDOSXKBX-UHFFFAOYSA-N 0.000 description 5
- 125000003118 aryl group Chemical group 0.000 description 5
- 239000012131 assay buffer Substances 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- JJAZWAMCKRENCJ-UHFFFAOYSA-N 2-[2-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]pentanedioic acid Chemical compound OC(=O)CCC(C(O)=O)OC1=CC=CC=C1COC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 JJAZWAMCKRENCJ-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- KEOCSCWHUMKIAJ-UHFFFAOYSA-N 2-[2-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]propanoic acid Chemical compound OC(=O)C(C)OC1=CC=CC=C1COC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 KEOCSCWHUMKIAJ-UHFFFAOYSA-N 0.000 description 3
- JELDFLOBXROBFH-UHFFFAOYSA-N 2-[[4-[[2-(2h-tetrazol-5-ylmethyl)phenyl]methoxy]phenoxy]methyl]quinoline Chemical compound C=1C=C2C=CC=CC2=NC=1COC(C=C1)=CC=C1OCC1=CC=CC=C1CC=1N=NNN=1 JELDFLOBXROBFH-UHFFFAOYSA-N 0.000 description 3
- OVWHFYRFUFIJTB-UHFFFAOYSA-N 2-[[4-[[2-(chloromethyl)phenyl]methoxy]phenoxy]methyl]quinoline Chemical compound ClCC1=CC=CC=C1COC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 OVWHFYRFUFIJTB-UHFFFAOYSA-N 0.000 description 3
- NDLCXMUVSVJHCV-UHFFFAOYSA-N 4-(quinolin-2-ylmethoxy)phenol Chemical compound C1=CC(O)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 NDLCXMUVSVJHCV-UHFFFAOYSA-N 0.000 description 3
- KWSHXUGIGTUDKC-UHFFFAOYSA-N 5-[2-[[2-(carboxymethoxy)phenyl]methoxy]-5-(quinolin-2-ylmethoxy)phenyl]pentanoic acid Chemical compound OC(=O)CCCCC1=CC(OCC=2N=C3C=CC=CC3=CC=2)=CC=C1OCC1=CC=CC=C1OCC(O)=O KWSHXUGIGTUDKC-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzenecarbonitrile Natural products N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- 229940073608 benzyl chloride Drugs 0.000 description 3
- 230000027455 binding Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- RJCLWJRANDFSKD-UHFFFAOYSA-N diethyl 2-(2-methylphenoxy)pentanedioate Chemical compound CCOC(=O)CCC(C(=O)OCC)OC1=CC=CC=C1C RJCLWJRANDFSKD-UHFFFAOYSA-N 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 229960001340 histamine Drugs 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- SUSQOBVLVYHIEX-UHFFFAOYSA-N phenylacetonitrile Chemical compound N#CCC1=CC=CC=C1 SUSQOBVLVYHIEX-UHFFFAOYSA-N 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- PMRFBLQVGJNGLU-UHFFFAOYSA-N -form-1-(4-Hydroxyphenyl)ethanol Natural products CC(O)C1=CC=C(O)C=C1 PMRFBLQVGJNGLU-UHFFFAOYSA-N 0.000 description 2
- LOSLUMRVUKFVDQ-UHFFFAOYSA-N 2-(2-chloroethyl)quinoline Chemical compound C1=CC=CC2=NC(CCCl)=CC=C21 LOSLUMRVUKFVDQ-UHFFFAOYSA-N 0.000 description 2
- UQJTUVOPJMBREK-UHFFFAOYSA-N 3-[2-[3-(carboxymethoxy)phenyl]-1-[3-(quinolin-2-ylmethoxy)phenyl]ethyl]sulfanylpropanoic acid Chemical compound C=1C=CC(OCC=2N=C3C=CC=CC3=CC=2)=CC=1C(SCCC(=O)O)CC1=CC=CC(OCC(O)=O)=C1 UQJTUVOPJMBREK-UHFFFAOYSA-N 0.000 description 2
- KXNUDCTZHFMOHX-UHFFFAOYSA-N 3-[[3-(dimethylamino)-3-oxopropyl]sulfanyl-[3-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenyl]methyl]sulfanylpropanoic acid Chemical compound CN(C)C(=O)CCSC(SCCC(O)=O)C1=CC=CC(COC=2C=C(OCC=3N=C4C=CC=CC4=CC=3)C=CC=2)=C1 KXNUDCTZHFMOHX-UHFFFAOYSA-N 0.000 description 2
- LULAYUGMBFYYEX-UHFFFAOYSA-N 3-chlorobenzoic acid Chemical compound OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 2
- FNYDIAAMUCQQDE-UHFFFAOYSA-N 4-methylbenzene-1,3-diol Chemical compound CC1=CC=C(O)C=C1O FNYDIAAMUCQQDE-UHFFFAOYSA-N 0.000 description 2
- YHBHZZUQHVYBFD-UHFFFAOYSA-N 5-(2,5-dihydroxyphenyl)pentanoic acid Chemical compound OC(=O)CCCCC1=CC(O)=CC=C1O YHBHZZUQHVYBFD-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- 206010002198 Anaphylactic reaction Diseases 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 239000004342 Benzoyl peroxide Substances 0.000 description 2
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- CJQWLNNCQIHKHP-UHFFFAOYSA-N Ethyl 3-mercaptopropanoic acid Chemical compound CCOC(=O)CCS CJQWLNNCQIHKHP-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 229960000583 acetic acid Drugs 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- COJRWHSKVYUZHQ-UHFFFAOYSA-N alpha-methyl-3-hydroxybenzyl alcohol Natural products CC(O)C1=CC=CC(O)=C1 COJRWHSKVYUZHQ-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 230000036783 anaphylactic response Effects 0.000 description 2
- 208000003455 anaphylaxis Diseases 0.000 description 2
- 230000003266 anti-allergic effect Effects 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 235000019400 benzoyl peroxide Nutrition 0.000 description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 2
- 150000005524 benzylchlorides Chemical class 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- MXQXFPSPHGRNMQ-UHFFFAOYSA-N diethyl 2-[2-(bromomethyl)phenoxy]pentanedioate Chemical compound CCOC(=O)CCC(C(=O)OCC)OC1=CC=CC=C1CBr MXQXFPSPHGRNMQ-UHFFFAOYSA-N 0.000 description 2
- QLKZPYYUIWVAQG-UHFFFAOYSA-N diethyl 2-[2-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]pentanedioate Chemical compound CCOC(=O)CCC(C(=O)OCC)OC1=CC=CC=C1COC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 QLKZPYYUIWVAQG-UHFFFAOYSA-N 0.000 description 2
- OXJSWZSWGJACGU-UHFFFAOYSA-N diethyl 2-bromopentanedioate Chemical compound CCOC(=O)CCC(Br)C(=O)OCC OXJSWZSWGJACGU-UHFFFAOYSA-N 0.000 description 2
- 239000002612 dispersion medium Substances 0.000 description 2
- LOZWAPSEEHRYPG-UHFFFAOYSA-N dithiane Natural products C1CSCCS1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 2
- SLUUHAFCXRVNPS-UHFFFAOYSA-N ethyl 2-(2-methylphenoxy)propanoate Chemical compound CCOC(=O)C(C)OC1=CC=CC=C1C SLUUHAFCXRVNPS-UHFFFAOYSA-N 0.000 description 2
- VJOIXAALPHWLCK-UHFFFAOYSA-N ethyl 2-[2-(bromomethyl)phenoxy]propanoate Chemical compound CCOC(=O)C(C)OC1=CC=CC=C1CBr VJOIXAALPHWLCK-UHFFFAOYSA-N 0.000 description 2
- DLSGMPHBLWGXAH-UHFFFAOYSA-N ethyl 2-bromo-5-(2-methylpropanoylamino)pentanoate Chemical compound CCOC(=O)C(Br)CCCNC(=O)C(C)C DLSGMPHBLWGXAH-UHFFFAOYSA-N 0.000 description 2
- ARFLASKVLJTEJD-UHFFFAOYSA-N ethyl 2-bromopropanoate Chemical compound CCOC(=O)C(C)Br ARFLASKVLJTEJD-UHFFFAOYSA-N 0.000 description 2
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- WVDDGKGOMKODPV-UHFFFAOYSA-N hydroxymethyl benzene Natural products OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000004968 inflammatory condition Effects 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- UXBJXJHRVOEYPA-UHFFFAOYSA-N n,n-dimethyl-3-sulfanylpropanamide Chemical compound CN(C)C(=O)CCS UXBJXJHRVOEYPA-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- OIPPWFOQEKKFEE-UHFFFAOYSA-N orcinol Chemical compound CC1=CC(O)=CC(O)=C1 OIPPWFOQEKKFEE-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- IWDCLRJOBJJRNH-UHFFFAOYSA-N p-cresol Chemical compound CC1=CC=C(O)C=C1 IWDCLRJOBJJRNH-UHFFFAOYSA-N 0.000 description 2
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- JPZMNVPVVYVXAD-UHFFFAOYSA-M (4-ethoxy-4-oxobutyl)-triphenylphosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCCC(=O)OCC)C1=CC=CC=C1 JPZMNVPVVYVXAD-UHFFFAOYSA-M 0.000 description 1
- JRNVQLOKVMWBFR-UHFFFAOYSA-N 1,2-benzenedithiol Chemical compound SC1=CC=CC=C1S JRNVQLOKVMWBFR-UHFFFAOYSA-N 0.000 description 1
- MDSFYGBXGRMKQS-UHFFFAOYSA-N 1,2-bis(3-hydroxyphenyl)ethanone Chemical compound OC1=CC=CC(CC(=O)C=2C=C(O)C=CC=2)=C1 MDSFYGBXGRMKQS-UHFFFAOYSA-N 0.000 description 1
- FMGGHNGKHRCJLL-UHFFFAOYSA-N 1,2-bis(chloromethyl)benzene Chemical compound ClCC1=CC=CC=C1CCl FMGGHNGKHRCJLL-UHFFFAOYSA-N 0.000 description 1
- YUIOPHXTILULQC-UHFFFAOYSA-N 1,4-Dithiane-2,5-diol Chemical compound OC1CSC(O)CS1 YUIOPHXTILULQC-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- DMYYTMVFUMDOGC-UHFFFAOYSA-N 2,4-bis(phenylmethoxy)benzaldehyde Chemical compound C1=C(OCC=2C=CC=CC=2)C(C=O)=CC=C1OCC1=CC=CC=C1 DMYYTMVFUMDOGC-UHFFFAOYSA-N 0.000 description 1
- SPBQMQLTSMVQKI-UHFFFAOYSA-N 2-(1-chloroethyl)quinoline Chemical compound C1=CC=CC2=NC(C(Cl)C)=CC=C21 SPBQMQLTSMVQKI-UHFFFAOYSA-N 0.000 description 1
- VWBVIHOSXPNQSB-UHFFFAOYSA-N 2-(1-hydroxybutyl)phenol Chemical compound CCCC(O)C1=CC=CC=C1O VWBVIHOSXPNQSB-UHFFFAOYSA-N 0.000 description 1
- LGRIXGWEPBUFKH-UHFFFAOYSA-N 2-(2-bromoethyl)quinoline Chemical compound C1=CC=CC2=NC(CCBr)=CC=C21 LGRIXGWEPBUFKH-UHFFFAOYSA-N 0.000 description 1
- JMRYQHZADMWCBL-UHFFFAOYSA-N 2-(2-chloro-2-phenylethyl)quinoline Chemical compound C=1C=C2C=CC=CC2=NC=1CC(Cl)C1=CC=CC=C1 JMRYQHZADMWCBL-UHFFFAOYSA-N 0.000 description 1
- BUDKTSZMYKWZEN-UHFFFAOYSA-N 2-(2-chloropropyl)quinoline Chemical compound C1=CC=CC2=NC(CC(Cl)C)=CC=C21 BUDKTSZMYKWZEN-UHFFFAOYSA-N 0.000 description 1
- RQPCKGQTCALFDI-UHFFFAOYSA-N 2-(2-hydroxy-4-methylphenyl)-n,n-dimethylacetamide Chemical compound CN(C)C(=O)CC1=CC=C(C)C=C1O RQPCKGQTCALFDI-UHFFFAOYSA-N 0.000 description 1
- HOQTWYHGNQUMPO-UHFFFAOYSA-N 2-(3-hydroxy-5-methylphenoxy)-n,n-dimethylacetamide Chemical compound CN(C)C(=O)COC1=CC(C)=CC(O)=C1 HOQTWYHGNQUMPO-UHFFFAOYSA-N 0.000 description 1
- YCCILVSKPBXVIP-UHFFFAOYSA-N 2-(4-hydroxyphenyl)ethanol Chemical compound OCCC1=CC=C(O)C=C1 YCCILVSKPBXVIP-UHFFFAOYSA-N 0.000 description 1
- NNAYPIDFVQLEDK-UHFFFAOYSA-N 2-(bromomethyl)quinoline Chemical compound C1=CC=CC2=NC(CBr)=CC=C21 NNAYPIDFVQLEDK-UHFFFAOYSA-N 0.000 description 1
- FHGYGKQYTPEOPT-UHFFFAOYSA-N 2-(chloromethyl)-4-methylquinoline Chemical compound C1=CC=C2C(C)=CC(CCl)=NC2=C1 FHGYGKQYTPEOPT-UHFFFAOYSA-N 0.000 description 1
- HJCDOCDHYGMKFX-UHFFFAOYSA-N 2-(chloromethyl)-6,8-dimethylquinoline Chemical compound N1=C(CCl)C=CC2=CC(C)=CC(C)=C21 HJCDOCDHYGMKFX-UHFFFAOYSA-N 0.000 description 1
- IFOZFMPOHZGLBK-UHFFFAOYSA-N 2-(chloromethyl)-6-methoxyquinoline Chemical compound N1=C(CCl)C=CC2=CC(OC)=CC=C21 IFOZFMPOHZGLBK-UHFFFAOYSA-N 0.000 description 1
- CSLGYHQUXRUXPX-UHFFFAOYSA-N 2-(chloromethyl)-6-methylquinoline Chemical compound N1=C(CCl)C=CC2=CC(C)=CC=C21 CSLGYHQUXRUXPX-UHFFFAOYSA-N 0.000 description 1
- BWRZMNAKJWZHKT-UHFFFAOYSA-N 2-(chloromethyl)-6-nitroquinoline Chemical compound N1=C(CCl)C=CC2=CC([N+](=O)[O-])=CC=C21 BWRZMNAKJWZHKT-UHFFFAOYSA-N 0.000 description 1
- LMYOLCBSTGQMIB-UHFFFAOYSA-N 2-(chloromethyl)-7-methylquinoline Chemical compound C1=CC(CCl)=NC2=CC(C)=CC=C21 LMYOLCBSTGQMIB-UHFFFAOYSA-N 0.000 description 1
- WNPVMZMSLGANAE-UHFFFAOYSA-N 2-(chloromethyl)-7-nitroquinoline Chemical compound C1=CC(CCl)=NC2=CC([N+](=O)[O-])=CC=C21 WNPVMZMSLGANAE-UHFFFAOYSA-N 0.000 description 1
- OVSFGFRGUYAGHH-UHFFFAOYSA-N 2-(chloromethyl)-8-methylquinoline Chemical compound C1=C(CCl)N=C2C(C)=CC=CC2=C1 OVSFGFRGUYAGHH-UHFFFAOYSA-N 0.000 description 1
- LXUNZSDDXMPKLP-UHFFFAOYSA-N 2-Methylbenzenethiol Chemical compound CC1=CC=CC=C1S LXUNZSDDXMPKLP-UHFFFAOYSA-N 0.000 description 1
- KMSAGGAYFSEOFK-UHFFFAOYSA-N 2-[2-[[3-(quinolin-2-ylmethoxy)phenyl]sulfanylmethyl]phenoxy]propanoic acid Chemical compound OC(=O)C(C)OC1=CC=CC=C1CSC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 KMSAGGAYFSEOFK-UHFFFAOYSA-N 0.000 description 1
- PMLSUZLRSAMKRK-UHFFFAOYSA-N 2-[2-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]propanoic acid Chemical compound OC(=O)C(C)OC1=CC=CC=C1COC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 PMLSUZLRSAMKRK-UHFFFAOYSA-N 0.000 description 1
- BRJCDJVZZAFPAM-UHFFFAOYSA-N 2-[2-chloro-6-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]-2-phenylacetic acid Chemical compound ClC=1C=CC=C(COC=2C=C(OCC=3N=C4C=CC=CC4=CC=3)C=CC=2)C=1OC(C(=O)O)C1=CC=CC=C1 BRJCDJVZZAFPAM-UHFFFAOYSA-N 0.000 description 1
- GDJUSMZMWNMQIB-UHFFFAOYSA-N 2-[2-chloro-6-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]propanoic acid Chemical compound OC(=O)C(C)OC1=C(Cl)C=CC=C1COC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 GDJUSMZMWNMQIB-UHFFFAOYSA-N 0.000 description 1
- WCJVBNZBEKMDBT-UHFFFAOYSA-N 2-[2-chloro-6-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]-2-phenylacetic acid Chemical compound ClC=1C=CC=C(COC=2C=CC(OCC=3N=C4C=CC=CC4=CC=3)=CC=2)C=1OC(C(=O)O)C1=CC=CC=C1 WCJVBNZBEKMDBT-UHFFFAOYSA-N 0.000 description 1
- ZBESOMPYNDOMJQ-UHFFFAOYSA-N 2-[2-chloro-6-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]propanoic acid Chemical compound OC(=O)C(C)OC1=C(Cl)C=CC=C1COC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 ZBESOMPYNDOMJQ-UHFFFAOYSA-N 0.000 description 1
- UUWHVMPFAKEIRV-UHFFFAOYSA-N 2-[3-fluoro-4-[[4-(quinolin-2-ylmethoxy)phenyl]methoxy]phenyl]acetic acid Chemical compound FC1=CC(CC(=O)O)=CC=C1OCC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 UUWHVMPFAKEIRV-UHFFFAOYSA-N 0.000 description 1
- BIPRDWHONIIMKU-UHFFFAOYSA-N 2-[4-(carboxymethoxy)-3-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]acetic acid Chemical compound OC(=O)COC1=CC=C(OCC(O)=O)C(COC=2C=C(OCC=3N=C4C=CC=CC4=CC=3)C=CC=2)=C1 BIPRDWHONIIMKU-UHFFFAOYSA-N 0.000 description 1
- KYMAJZQUPQXWGL-UHFFFAOYSA-N 2-[4-chloro-2-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]-2-phenylacetic acid Chemical compound C=1C=C(Cl)C=C(COC=2C=C(OCC=3N=C4C=CC=CC4=CC=3)C=CC=2)C=1OC(C(=O)O)C1=CC=CC=C1 KYMAJZQUPQXWGL-UHFFFAOYSA-N 0.000 description 1
- ZADBLSIKLIGEDN-UHFFFAOYSA-N 2-[4-chloro-2-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]-4-methylpentanoic acid Chemical compound CC(C)CC(C(O)=O)OC1=CC=C(Cl)C=C1COC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 ZADBLSIKLIGEDN-UHFFFAOYSA-N 0.000 description 1
- NLMFNJOZXCIOIU-UHFFFAOYSA-N 2-[[2-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenyl]methyl]pentanoic acid Chemical compound CCCC(C(O)=O)CC1=CC=CC=C1COC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 NLMFNJOZXCIOIU-UHFFFAOYSA-N 0.000 description 1
- NNWWOIPEHSLORU-UHFFFAOYSA-N 2-bromo-n,n-dimethylbutanamide Chemical compound CCC(Br)C(=O)N(C)C NNWWOIPEHSLORU-UHFFFAOYSA-N 0.000 description 1
- RSTGBTUVISMDCM-UHFFFAOYSA-N 2-bromo-n,n-dimethylpentanamide Chemical compound CCCC(Br)C(=O)N(C)C RSTGBTUVISMDCM-UHFFFAOYSA-N 0.000 description 1
- ZEDHSSVDQTYBRV-UHFFFAOYSA-N 2-bromo-n,n-dimethylpropanamide Chemical compound CC(Br)C(=O)N(C)C ZEDHSSVDQTYBRV-UHFFFAOYSA-N 0.000 description 1
- GAHGEBXNGMDRME-UHFFFAOYSA-N 2-hydroxy-n,n,4-trimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=C(C)C=C1O GAHGEBXNGMDRME-UHFFFAOYSA-N 0.000 description 1
- YZJIRUGAHGFNJP-UHFFFAOYSA-N 2-methyl-2-[2-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenyl]pentanoic acid Chemical compound N1=C(C=CC2=CC=CC=C12)COC1=CC=C(OCC2=C(C=CC=C2)C(C(=O)O)(CCC)C)C=C1 YZJIRUGAHGFNJP-UHFFFAOYSA-N 0.000 description 1
- SYGDWQUKURMBGS-UHFFFAOYSA-N 2-phenyl-2-[2-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]acetic acid Chemical compound C=1C=CC=C(COC=2C=CC(OCC=3N=C4C=CC=CC4=CC=3)=CC=2)C=1OC(C(=O)O)C1=CC=CC=C1 SYGDWQUKURMBGS-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- VMKYTRPNOVFCGZ-UHFFFAOYSA-N 2-sulfanylphenol Chemical compound OC1=CC=CC=C1S VMKYTRPNOVFCGZ-UHFFFAOYSA-N 0.000 description 1
- DBQDZRNWPDEEQC-UHFFFAOYSA-N 3-(1-hydroxybutyl)phenol Chemical compound CCCC(O)C1=CC=CC(O)=C1 DBQDZRNWPDEEQC-UHFFFAOYSA-N 0.000 description 1
- RJUXLAONNABKPY-UHFFFAOYSA-N 3-(2-carboxyethylsulfanyl)-2-hydroxy-3-[2-[[4-(quinolin-2-ylmethoxy)phenoxy]methyl]phenyl]propanoic acid Chemical compound OC(=O)CCSC(C(O)C(O)=O)C1=CC=CC=C1COC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 RJUXLAONNABKPY-UHFFFAOYSA-N 0.000 description 1
- AMQIPHZFLIDOCB-UHFFFAOYSA-N 3-(2-hydroxyethyl)phenol Chemical compound OCCC1=CC=CC(O)=C1 AMQIPHZFLIDOCB-UHFFFAOYSA-N 0.000 description 1
- DSOGFOBIDOVCHN-UHFFFAOYSA-N 3-(chloromethyl)quinoline Chemical compound C1=CC=CC2=CC(CCl)=CN=C21 DSOGFOBIDOVCHN-UHFFFAOYSA-N 0.000 description 1
- JZHFMDIXOSLIDO-UHFFFAOYSA-N 3-(quinolin-2-ylmethoxy)benzenethiol Chemical compound SC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 JZHFMDIXOSLIDO-UHFFFAOYSA-N 0.000 description 1
- IQCNXCYYDZQEHB-UHFFFAOYSA-N 3-[1-[3-(dimethylamino)-3-oxopropyl]sulfanyl-2-[3-[3-(quinolin-2-ylmethoxy)phenoxy]phenyl]ethyl]sulfanylpropanoic acid Chemical compound CN(C)C(=O)CCSC(SCCC(O)=O)CC1=CC=CC(OC=2C=C(OCC=3N=C4C=CC=CC4=CC=3)C=CC=2)=C1 IQCNXCYYDZQEHB-UHFFFAOYSA-N 0.000 description 1
- NKUOQDOEWMZTNQ-UHFFFAOYSA-N 3-bromo-n,n-dimethylpentanamide Chemical compound CCC(Br)CC(=O)N(C)C NKUOQDOEWMZTNQ-UHFFFAOYSA-N 0.000 description 1
- 125000006291 3-hydroxybenzyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(=C1[H])C([H])([H])* 0.000 description 1
- WRXOZRLZDJAYDR-UHFFFAOYSA-N 3-methylbenzenethiol Chemical compound CC1=CC=CC(S)=C1 WRXOZRLZDJAYDR-UHFFFAOYSA-N 0.000 description 1
- DOFIAZGYBIBEGI-UHFFFAOYSA-N 3-sulfanylphenol Chemical compound OC1=CC=CC(S)=C1 DOFIAZGYBIBEGI-UHFFFAOYSA-N 0.000 description 1
- GXNGGBMNYUHZOZ-UHFFFAOYSA-N 4-(1-hydroxybutyl)phenol Chemical compound CCCC(O)C1=CC=C(O)C=C1 GXNGGBMNYUHZOZ-UHFFFAOYSA-N 0.000 description 1
- OLSKQWRLRYMCAO-UHFFFAOYSA-N 4-(chloromethyl)quinoline Chemical compound C1=CC=C2C(CCl)=CC=NC2=C1 OLSKQWRLRYMCAO-UHFFFAOYSA-N 0.000 description 1
- 125000003143 4-hydroxybenzyl group Chemical group [H]C([*])([H])C1=C([H])C([H])=C(O[H])C([H])=C1[H] 0.000 description 1
- KJYKFOPHIUCRAW-UHFFFAOYSA-N 4-methyl-2-[2-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenoxy]pentanoic acid Chemical compound CC(C)CC(C(O)=O)OC1=CC=CC=C1COC1=CC=CC(OCC=2N=C3C=CC=CC3=CC=2)=C1 KJYKFOPHIUCRAW-UHFFFAOYSA-N 0.000 description 1
- WLHCBQAPPJAULW-UHFFFAOYSA-N 4-methylbenzenethiol Chemical compound CC1=CC=C(S)C=C1 WLHCBQAPPJAULW-UHFFFAOYSA-N 0.000 description 1
- IDHANRUXEVOWGK-UHFFFAOYSA-N 4-oxo-3-[[3-[[3-(quinolin-2-ylmethoxy)phenoxy]methyl]phenyl]methyl]-4-sulfanylbutanoic acid Chemical compound N1=C(C=CC2=CC=CC=C12)COC=1C=C(OCC=2C=C(C=CC=2)CC(C(=S)O)CC(=O)O)C=CC=1 IDHANRUXEVOWGK-UHFFFAOYSA-N 0.000 description 1
- DTRIDVOOPAQEEL-UHFFFAOYSA-N 4-sulfanylbutanoic acid Chemical compound OC(=O)CCCS DTRIDVOOPAQEEL-UHFFFAOYSA-N 0.000 description 1
- BXAVKNRWVKUTLY-UHFFFAOYSA-N 4-sulfanylphenol Chemical compound OC1=CC=C(S)C=C1 BXAVKNRWVKUTLY-UHFFFAOYSA-N 0.000 description 1
- GUBQPALMKUPKGM-UHFFFAOYSA-N 7-bromo-2-(chloromethyl)quinoline Chemical compound C1=CC(Br)=CC2=NC(CCl)=CC=C21 GUBQPALMKUPKGM-UHFFFAOYSA-N 0.000 description 1
- VDRLGRBSQTUUOX-UHFFFAOYSA-N 7-chloro-2-(chloromethyl)quinoline Chemical compound C1=CC(Cl)=CC2=NC(CCl)=CC=C21 VDRLGRBSQTUUOX-UHFFFAOYSA-N 0.000 description 1
- 235000001674 Agaricus brunnescens Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 102000001381 Arachidonate 5-Lipoxygenase Human genes 0.000 description 1
- 108010093579 Arachidonate 5-lipoxygenase Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- AIYVPUUYGUNNIO-UHFFFAOYSA-N C1=C(P(C2=CC=CC=C2)(CCCC(=O)OCC)C2=CC=CC=C2)C=CC=C1 Chemical compound C1=C(P(C2=CC=CC=C2)(CCCC(=O)OCC)C2=CC=CC=C2)C=CC=C1 AIYVPUUYGUNNIO-UHFFFAOYSA-N 0.000 description 1
- BYHBLUGHLMUDMP-VIFPVBQESA-N CCOC(=O)CC[C@@](C(=O)OCC)(N)Br Chemical compound CCOC(=O)CC[C@@](C(=O)OCC)(N)Br BYHBLUGHLMUDMP-VIFPVBQESA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- BHDKXXOGPCSBQI-UHFFFAOYSA-N Methyl 3-mercaptobutanoate Chemical compound COC(=O)CC(C)S BHDKXXOGPCSBQI-UHFFFAOYSA-N 0.000 description 1
- 241000238367 Mya arenaria Species 0.000 description 1
- CWRVKFFCRWGWCS-UHFFFAOYSA-N Pentrazole Chemical compound C1CCCCC2=NN=NN21 CWRVKFFCRWGWCS-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical class N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 238000007239 Wittig reaction Methods 0.000 description 1
- 238000005644 Wolff-Kishner reduction reaction Methods 0.000 description 1
- NVMBYUCFCPUQAV-UHFFFAOYSA-L [Hg]=O.Cl[Hg]Cl Chemical compound [Hg]=O.Cl[Hg]Cl NVMBYUCFCPUQAV-UHFFFAOYSA-L 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- 235000019568 aromas Nutrition 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- IZALUMVGBVKPJD-UHFFFAOYSA-N benzene-1,3-dicarbaldehyde Chemical compound O=CC1=CC=CC(C=O)=C1 IZALUMVGBVKPJD-UHFFFAOYSA-N 0.000 description 1
- WYLQRHZSKIDFEP-UHFFFAOYSA-N benzene-1,4-dithiol Chemical compound SC1=CC=C(S)C=C1 WYLQRHZSKIDFEP-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 125000000664 diazo group Chemical group [N-]=[N+]=[*] 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- RUIJMAUNJVDAEE-UHFFFAOYSA-N diethyl 2-bromobutanedioate Chemical compound CCOC(=O)CC(Br)C(=O)OCC RUIJMAUNJVDAEE-UHFFFAOYSA-N 0.000 description 1
- BORVABMIWYCBSC-UHFFFAOYSA-N diethyl 2-bromohexanedioate Chemical compound CCOC(=O)CCCC(Br)C(=O)OCC BORVABMIWYCBSC-UHFFFAOYSA-N 0.000 description 1
- FNJVDWXUKLTFFL-UHFFFAOYSA-N diethyl 2-bromopropanedioate Chemical compound CCOC(=O)C(Br)C(=O)OCC FNJVDWXUKLTFFL-UHFFFAOYSA-N 0.000 description 1
- DNDSUVRBUGVSLQ-UHFFFAOYSA-N diethyl 3-bromohexanedioate Chemical compound CCOC(=O)CCC(Br)CC(=O)OCC DNDSUVRBUGVSLQ-UHFFFAOYSA-N 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Natural products CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- XUYQCWAEYPNCOL-UHFFFAOYSA-N ethyl 2-bromo-2-(2-methylpropanoylamino)acetate Chemical compound CCOC(=O)C(Br)NC(=O)C(C)C XUYQCWAEYPNCOL-UHFFFAOYSA-N 0.000 description 1
- WLAAGADGXMKVTO-UHFFFAOYSA-N ethyl 2-bromo-3-(2-methylpropanoylamino)propanoate Chemical compound CCOC(=O)C(Br)CNC(=O)C(C)C WLAAGADGXMKVTO-UHFFFAOYSA-N 0.000 description 1
- GORLTPVIBQVFMB-UHFFFAOYSA-N ethyl 2-bromo-3-methylpentanoate Chemical compound CCOC(=O)C(Br)C(C)CC GORLTPVIBQVFMB-UHFFFAOYSA-N 0.000 description 1
- OCIRBAFSIFFTIO-UHFFFAOYSA-N ethyl 2-bromo-4-(2-methylpropanoylamino)butanoate Chemical compound CCOC(=O)C(Br)CCNC(=O)C(C)C OCIRBAFSIFFTIO-UHFFFAOYSA-N 0.000 description 1
- XIMFCGSNSKXPBO-UHFFFAOYSA-N ethyl 2-bromobutanoate Chemical compound CCOC(=O)C(Br)CC XIMFCGSNSKXPBO-UHFFFAOYSA-N 0.000 description 1
- ORSIRXYHFPHWTN-UHFFFAOYSA-N ethyl 2-bromopentanoate Chemical compound CCCC(Br)C(=O)OCC ORSIRXYHFPHWTN-UHFFFAOYSA-N 0.000 description 1
- QABFSRANYBCWPK-UHFFFAOYSA-N ethyl 2-hydroxy-4-methylbenzoate Chemical compound CCOC(=O)C1=CC=C(C)C=C1O QABFSRANYBCWPK-UHFFFAOYSA-N 0.000 description 1
- JDFYLNFIQHZQQG-UHFFFAOYSA-N ethyl 3-bromo-4-(2-methylpropanoylamino)butanoate Chemical compound CCOC(=O)CC(Br)CNC(=O)C(C)C JDFYLNFIQHZQQG-UHFFFAOYSA-N 0.000 description 1
- DCVMKBCRTYHBTO-UHFFFAOYSA-N ethyl 3-bromo-5-(2-methylpropanoylamino)pentanoate Chemical compound CCOC(=O)CC(Br)CCNC(=O)C(C)C DCVMKBCRTYHBTO-UHFFFAOYSA-N 0.000 description 1
- OPXQLUFLTHEZST-UHFFFAOYSA-N ethyl 3-bromobutanoate Chemical compound CCOC(=O)CC(C)Br OPXQLUFLTHEZST-UHFFFAOYSA-N 0.000 description 1
- VOEAEELHRMARKD-UHFFFAOYSA-N ethyl 3-bromopentanoate Chemical compound CCOC(=O)CC(Br)CC VOEAEELHRMARKD-UHFFFAOYSA-N 0.000 description 1
- NOMZQEBZVIWRST-UHFFFAOYSA-N ethyl 4-bromo-5-(2-methylpropanoylamino)pentanoate Chemical compound CCOC(=O)CCC(Br)CNC(=O)C(C)C NOMZQEBZVIWRST-UHFFFAOYSA-N 0.000 description 1
- YWZOHQWIRAFZCD-UHFFFAOYSA-N ethyl 4-methyl-2-sulfanylbenzoate Chemical compound CCOC(=O)C1=CC=C(C)C=C1S YWZOHQWIRAFZCD-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- HDVRLUFGYQYLFJ-UHFFFAOYSA-N flamenol Chemical compound COC1=CC(O)=CC(O)=C1 HDVRLUFGYQYLFJ-UHFFFAOYSA-N 0.000 description 1
- 230000009969 flowable effect Effects 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000011539 homogenization buffer Substances 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- JUINSXZKUKVTMD-UHFFFAOYSA-N hydrogen azide Chemical compound N=[N+]=[N-] JUINSXZKUKVTMD-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 229960004337 hydroquinone Drugs 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- QLNWXBAGRTUKKI-UHFFFAOYSA-N metacetamol Chemical compound CC(=O)NC1=CC=CC(O)=C1 QLNWXBAGRTUKKI-UHFFFAOYSA-N 0.000 description 1
- SUHLUMKZPUMAFP-UHFFFAOYSA-N methyl 2-hydroxy-3-methylbenzoate Chemical compound COC(=O)C1=CC=CC(C)=C1O SUHLUMKZPUMAFP-UHFFFAOYSA-N 0.000 description 1
- UITFCFWKYAOJEJ-UHFFFAOYSA-N methyl 2-hydroxy-4-methylbenzoate Chemical compound COC(=O)C1=CC=C(C)C=C1O UITFCFWKYAOJEJ-UHFFFAOYSA-N 0.000 description 1
- SNWKNPMDQONHKK-UHFFFAOYSA-N methyl 2-sulfanylpropanoate Chemical compound COC(=O)C(C)S SNWKNPMDQONHKK-UHFFFAOYSA-N 0.000 description 1
- LDTLDBDUBGAEDT-UHFFFAOYSA-N methyl 3-sulfanylpropanoate Chemical compound COC(=O)CCS LDTLDBDUBGAEDT-UHFFFAOYSA-N 0.000 description 1
- YRIAPEVGKLRGHX-UHFFFAOYSA-N methyl 4-sulfanylbutanoate Chemical compound COC(=O)CCCS YRIAPEVGKLRGHX-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 1
- XKBGEWXEAPTVCK-UHFFFAOYSA-M methyltrioctylammonium chloride Chemical compound [Cl-].CCCCCCCC[N+](C)(CCCCCCCC)CCCCCCCC XKBGEWXEAPTVCK-UHFFFAOYSA-M 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- RAZOZUOOHWFOBM-UHFFFAOYSA-N n,n,4-trimethyl-2-sulfanylbenzamide Chemical compound CN(C)C(=O)C1=CC=C(C)C=C1S RAZOZUOOHWFOBM-UHFFFAOYSA-N 0.000 description 1
- WJYAUBVHDUUTGY-UHFFFAOYSA-N n,n-diethyl-3-sulfanylpropanamide Chemical compound CCN(CC)C(=O)CCS WJYAUBVHDUUTGY-UHFFFAOYSA-N 0.000 description 1
- OSGMGPAQURXYQR-UHFFFAOYSA-N n,n-dimethyl-2-(3-methyl-5-sulfanylphenoxy)acetamide Chemical compound CN(C)C(=O)COC1=CC(C)=CC(S)=C1 OSGMGPAQURXYQR-UHFFFAOYSA-N 0.000 description 1
- KHFHDWIWDWWSCY-UHFFFAOYSA-N n,n-dimethyl-2-(4-methyl-2-sulfanylphenyl)acetamide Chemical compound CN(C)C(=O)CC1=CC=C(C)C=C1S KHFHDWIWDWWSCY-UHFFFAOYSA-N 0.000 description 1
- KJIZMZQPWFGTKV-UHFFFAOYSA-N n,n-dimethyl-2-sulfanylacetamide Chemical compound CN(C)C(=O)CS KJIZMZQPWFGTKV-UHFFFAOYSA-N 0.000 description 1
- XWRNVVHLXBWGDT-UHFFFAOYSA-N n,n-dimethyl-2-sulfanylpropanamide Chemical compound CC(S)C(=O)N(C)C XWRNVVHLXBWGDT-UHFFFAOYSA-N 0.000 description 1
- BYQAZMUEZZUDEI-UHFFFAOYSA-N n,n-dimethyl-3-sulfanylbutanamide Chemical compound CC(S)CC(=O)N(C)C BYQAZMUEZZUDEI-UHFFFAOYSA-N 0.000 description 1
- FNXMIAHUQJZWLY-UHFFFAOYSA-N n,n-dimethyl-4-sulfanylbutanamide Chemical compound CN(C)C(=O)CCCS FNXMIAHUQJZWLY-UHFFFAOYSA-N 0.000 description 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000006396 nitration reaction Methods 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000005429 oxyalkyl group Chemical group 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- ZJLMKPKYJBQJNH-UHFFFAOYSA-N propane-1,3-dithiol Chemical compound SCCCS ZJLMKPKYJBQJNH-UHFFFAOYSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- ARDPWQJVWYEJKF-UHFFFAOYSA-N propyl 2-[3-fluoro-4-[[4-(quinolin-2-ylmethoxy)phenyl]methoxy]phenyl]acetate Chemical compound FC1=CC(CC(=O)OCCC)=CC=C1OCC(C=C1)=CC=C1OCC1=CC=C(C=CC=C2)C2=N1 ARDPWQJVWYEJKF-UHFFFAOYSA-N 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- KOUKXHPPRFNWPP-UHFFFAOYSA-N pyrazine-2,5-dicarboxylic acid;hydrate Chemical compound O.OC(=O)C1=CN=C(C(O)=O)C=N1 KOUKXHPPRFNWPP-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000003248 quinolines Chemical class 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- CNHDIAIOKMXOLK-UHFFFAOYSA-N toluquinol Chemical compound CC1=CC(O)=CC=C1O CNHDIAIOKMXOLK-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 239000009637 wintergreen oil Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/12—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/12—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D215/14—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/18—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Quinoline Compounds (AREA)
Description
- Diese Erfindung betrifft bestimmte chemische Verbindungen und ihre Verwendung als wertvolle pharmazeutische Mittel, insbesondere als Lipoxygenase-Inhibitoren und/oder Leukotrien-Anta-gonisten, die antientzündliche und antiallergische Eigenschaften besitzen.
- Erfindungsgemäß werden die Verbindungen, die durch die allgemeine Formel I beschrieben werden, und therapeutische Zusammensetzungen bereitgestellt, die als Wirkbestandteil eine Verbindung mit der allgemeinen Formel Iumfassen, in der

A O oder S ist;
B eine chemische Bindung oderist;
D O, S, NR1,oder eine chemische Bindung ist;
E eine chemische Bindung oderist;
a 0 bis 2 ist;
b 0 bis 1 ist;
c 0 bis 4 ist;
d 0 bis 5 ist;
e 0 bis 4 ist;
f 0 bis 5 ist;
n 0 bis 2 ist;
jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist;
jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist;
jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist;
jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist); -S-(CH2)x-X oder -NR1-(CH2)x-X ist;
wobei x 0 bis 3 ist, und
X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamido ist;
vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird;
geminale R1- und R-Gruppen zusammen einen Spirosubstituenten bilden können, -(CH2)z-, wobei z 2 bis 5 ist;
geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten bilden können, =CHR1;
Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann;
und pharmazeutisch akzeptable Salze derselben. - Die vorliegende Erfindung liefert auch eine Verbindung mit der allgemeinen Formel Iin der

A O oder S ist;
B eine chemische Bindung, O, S, SO, SO2, NR1,ist;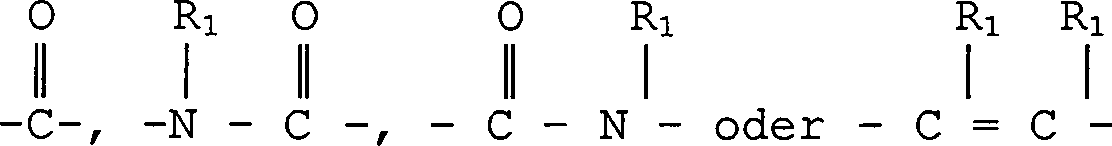
D O, S, NR1,oder eine chemische Bindung ist;
E eine chemische Bindung oderist;
a 0 bis 2 ist;
b 0 bis 1 ist;
c 0 bis 4 ist;
d 0 bis 5 ist;
e 0 bis 4 ist;
f 0 bis 5 ist;
n 0 bis 2 ist;
jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist;
mindestens ein R'' -CH2R oder -CH2-O-(CH2)x-X oder R ist, und das andere unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist;
jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist;
jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist); -S-(CH2)x-X oder -NR1-(CH2)x-X ist;
wobei x 0 bis 3 ist, und
X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Aryl, Aralkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamid ist;
vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird;
geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist;
geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten bilden können, =CHR1;
Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann;
und pharmazeutisch akzeptable Salze derselben. - Die vorliegende Erfindung liefert ferner eine Verbindung mit der allgemeinen Formel I:in der

A O oder S ist;
B eine chemische Bindung, O, S, SO, SO2, NR1,ist;
D O, S, NR1,oder eine chemische Bindung ist;
E eine chemische Bindung oderist;
a 0 bis 2 ist;
b 0 bis 1 ist;
c 0 bis 4 ist;
d 0 bis 5 ist;
e 0 bis 4 ist;
f 0 bis 5 ist;
n 0 bis 2 ist;
jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist;
jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, =CH2R oder -CH2-O-(CH2)x-X oder R ist;
jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist;
jedes R unabhängig -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist); -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und
X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Aryl, Aralkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamido ist;
vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird;
geminale R1- und R-Gruppen zusammen einen Spirosubstituenten bilden können, -(CH2)z-, wobei z 2 bis 5 ist;
geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten bilden können, =CHR1;
Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann;
und pharmazeutisch akzeptable Salze derselben. - Die vorliegende Erfindung liefert ferner eine Verbindung mit der allgemeinen Formel I:in der

A O oder S ist;
B eine chemische Bindung, O, S, SO, SO2, NR1,ist;
D O, S, NR1,oder eine chemische Bindung ist;
E eine chemische Bindung oderist;
a 0 bis 2 ist;
b 0 bis 1 ist;
c 0 bis 4 ist;
d 0 bis 5 ist;
e 0 bis 4 ist;
f 0 bis 5 ist;
n 0 bis 2 ist;
jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist;
jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist;
jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist;
jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist); -S-(CH2)x-X oder -NR1-(CH2)x-X ist;
wobei x 0 bis 3 ist, und
X CN ist;
vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird;
geminale R1- und R-Gruppen zusammen einen Spirosubstituenten bilden können, -(CH2)z-, wobei z 2 bis 5 ist;
geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten bilden können, =CHR1;
Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann;
und pharmazeutisch akzeptable Salze derselben. - Die Verbindungen der Formel I enthalten mindestens drei aromatische Ringe. Für die Zwecke dieser Erfindung können sie wie in Formel II gezeigt bezeichnet werden. Das Substitutionsmuster dieser Ringe entlang der Kette in Bezug zueinander ist wie folgt:
- Formel II

- Das Substitutionsmuster des Chinolinrings, das ist Ring I, ist vorzugsweise an der 2-Position, um die Seitenkette zu verlängern. Wenn diese Seitenkette vom Chinolinring ihren Lauf nimmt, können die beiden Phenylringe, bezeichnet als Ring II und Ring III, entlang der Kette in den ortho-, meta- oder para-Positionen in Bezug zueinander substituiert sein, und Ring II kann in den ortho-, meta- und para-Positionen in Bezug zu dem Chinolinring auch substituiert sein.
- Die bevorzugten Substitutionsmuster für Ring II sind meta oder para, das heißt:
-

- Ring III kann jedoch gleichermaßen in ortho-, meta- oder para-Positionen substituiert sein, das heißt:oder

-

- Weitere bevorzugte Verbindungen dieser Erfindung sind in der folgenden Formel V beschrieben:Formel V in der eines von R -(CH2)x-X; -S-(CH2)x-X; -O-(CH2)x-X (wenn nicht geminal zu B oder D, wenn B oder D O ist); -NR1-(CH2)x-X ist und/oder eines von R'' -CH2R, R oder -CH2-O-(CH2)x-X ist;
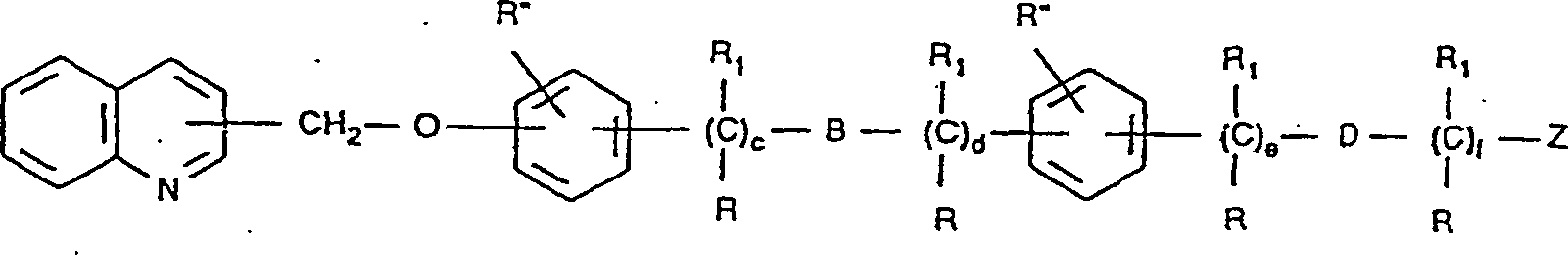
X ausgewählt ist aus -CONR1R1, -COOR1, -CN, Tetrazolyl oder Acylsulfonamido; und
R1, A, B, C, D, Z, c, d, e und f wie für Formel (I) definiert sind. - Die besonders bevorzugten Verbindungen der Formel V sind jene, in denen B O, S,oder eine chemische Bindung ist;

Z COOR1, -CON(R1)2 oder Tetrazolyl ist, und R und R'' wie oben beschrieben sind. - Es ist am meisten bevorzugt, dass das Molekül etwas enthält, was als zwei Seitenketten angesehen werden kann, die aus R- und/oder R''-Anteilen in Kombination mit dem -(C)e-D-(C)f-E-Z Anteil des Moleküls gebildet werden, oder wenn e und f beide 0 sind, und D und E beide chemische Bindungen sind, dann die beiden Seitenketten aus jeder Kombination von R- und R''-Anteilen gebildet werden. Es ist am meisten bevorzugt, dass diese Seitenketten saure und/oder basische Funktionen enthält. Dies wird deutlicher, wenn die Erfindung detaillierter beschrieben wird.
- Die vorliegende Erfindung betrifft zudem die Verwendung dieser Verbindungen als Lipoxygenase-Inhibitoren und/oder Leukotrien-Antagonisten, die antientzündliche und antiallergische Eigenschaften besitzen.
- Wie oben und in der gesamten Offenbarung verwendet, sollen die folgenden Begriffe, solange nicht anders angegeben, die folgenden Bedeutungen haben:
- "Alkyl", entweder allein oder mit verschiedenen hier definierten Substituenten, bedeutet einen gesättigten aliphatischen Kohlenwasserstoff, entweder verzweigt oder geradkettig. "Niederes Alkyl" mit etwa 1 bis etwa 6 Kohlenstoffatomen ist bevorzugt. Beispiele für Alkyl schließen Methyl, Ethyl, n-Propyl, Isopropyl, Butyl, sek.-Butyl, t-Butyl, Amyl, Hexyl, usw. ein.
- "Alkoxy" bezieht sich auf eine niedere Alkyl-O-Gruppe.
- "Alkenyl" bezieht sich auf einen Kohlenwasserstoff mit mindestens einem Punkt von Ungesättigtheit und kann verzweigt oder geradkettig sein. In bevorzugten Alkenylgruppen sind sechs oder weniger Kohlenstoffatome vorhanden, wie Vinyl, Allyl, Ethinyl, Isopropenyl, usw.
- "Aralkyl" bedeutet eine Alkylgruppe, die mit einem Arylrest substituiert ist. Die bevorzugten Aralkylgruppen sind Benzyl oder Phenethyl.
- "Cycloalkyl" bedeutet einen gesättigten monocyclischen Kohlenwasserstoffring mit 3 bis etwa 6 Kohlenstoffatomen, wie Cyclopropyl, Cyclohexyl, usw.
- "Acyl" bedeutet einen organischen Rest, der von einer organischen Säure durch Entfernung ihrer Hydroxylgruppe abgeleitet ist. Bevorzugte Acylgruppen sind Acetyl, Propionyl, Benzoyl, usw.
- "Halo" bedeutet ein Halogen. Bevorzugte Halogene schließen Chlorid, Bromid und Fluorid ein. Die bevorzugte Halogenalkylgruppe ist Trifluormethyl.
- Die erfindungsgemäßen Verbindungen können in Segmenten hergestellt werden, wie es für ein langkettiges Molekül üblich ist. Es ist somit zweckmäßig, diese Moleküle unter Verwendung von Kondensationsreaktionen an den A-, B- und D-Stellen des Moleküls zu synthetisieren. Aus diesem Grund können die vorliegenden Verbindungen durch im Stand der Technik bekannte Verfahren aus bekannten Verbindungen oder leicht herstellbaren Zwischenstufen hergestellt werden. Beispielhafte allgemeine Verfahren sind wie folgt. Zur Herstellung der Verbindungkönnen die folgenden Reaktionen oder Kombinationen von Reaktionen verwendet werden:

 in denen:
in denen:
R, R', R'', R, a, b, c, d, e, f, n, A und D wie oben definiert sind; B O oder S ist; E eine chemische Bindung ist; Z -CN, -C(=O)N(R1), -COOR1 oder Tetrazolyl ist und L eine Abgangsgruppe ist, wie Halogen, Tosylat oder Mesylat,
wobei B O oder S ist, jede Base verwendet werden kann, die normalerweise zum Deprotonieren eines Alkohols oder Thiols verwendet wird, wie Natriumhydrid, Natriumhydroxid, Triethyl-amin, Natriumbicarbonat oder Diisopropyl/Ethylamin. - Die Reaktionstemperaturen liegen im Bereich von Raumtemperatur bis Rückfluss, und die Reaktionszeiten variieren von 2 bis 96 Stunden. Die Reaktion wird üblicherweise in einem Lösungsmittel durchgeführt, das beide Reaktanden löst und auch gegenüber beiden inert ist. Lösungsmittel schließen Diethylether, Tetrahydrofuran, N,N-Dimethylformamid, Dimethylsulfoxid, Dioxan und dergleichen ein, sind jedoch nicht darauf eingeschränkt.
- Wenn B SO oder SO2 ist, dann führt die Behandlung der Thiover-bindung mit m-Chlorbenzoesäure oder Natriumperiodat zu der Sulfinylverbindung. Herstellung der Sulfonylverbindung kann nach bekannten Verfahren bewirkt werden, wie Auflösen der Sulfinylverbindung in Essigsäure und Behandlung mit 30% H2O2.
- Jene Verbindungen, in denen B -C(=O)- ist, können durch die folgende Reaktionssequenz hergestellt werden:
-

- Kondensation des Aldehyds mit 1,3-Propandithiol führt zu der Dithianverbindung. Dies kann in Chloroform bei verminderten Temperaturen (–20°C) durchgeführt werden, während HCl-Gas in die Reaktionsmischung geblasen wird. Die Dithianverbindung wird dann mit N-Butyllithium in unpolarem Lösungsmittel bei –78°C behandelt und dann mit dem substituierten Benzylchlorid umgesetzt. Dies führt zur Addition des Rings III an das Molekül. Der Dithiananteil wird dann mit einer Quecksilber(II)chlorid- Quecksilber(II)oxid-Mischung behandelt, um den Komplex zu bilden, der dann abgespalten wird, wodurch die gewünschte Verbindung zurückbleibt.
- Wittig-Kondensation kann auch an der B-Position des Vorläufers des Moleküls der Formel II stattfinden, die Ringe I und II enthält, wie folgt.
-
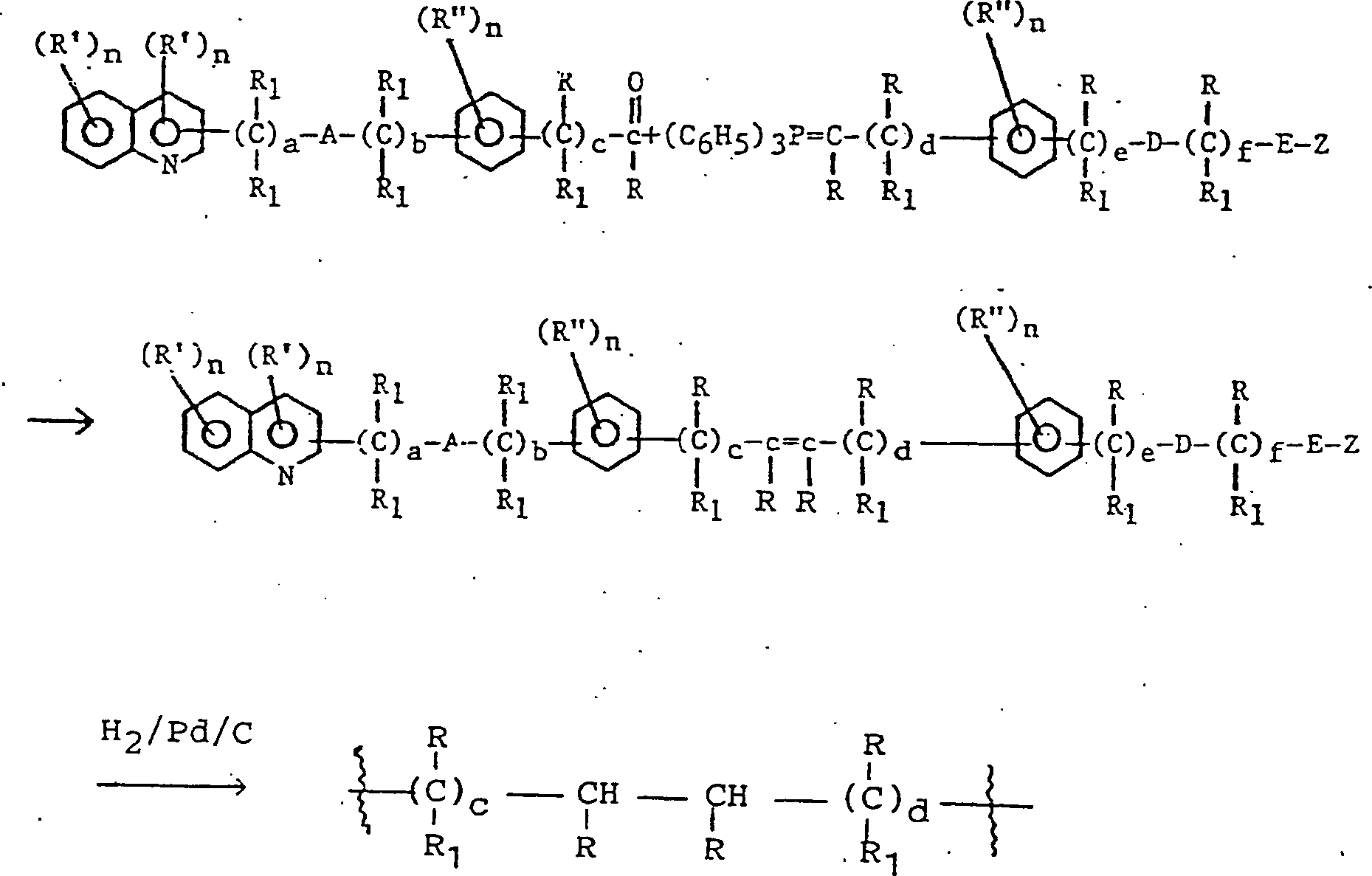
- Dies kann unter Verwendung normaler Wittig-Reaktionsbedingungen durchgeführt werden. Wenn der geeignete Aldehyd oder das geeignete Keton mit einem Wittig-Reagenz umgesetzt wird, führt die Kondensation zur Bildung der Doppelbindung. Das Kondensationsprodukt kann dann katalytisch nach bekannten Verfahren reduziert werden, wie Pd/C oder jede andere geeignete Hydrierbedingung.
- Natürlich kann diese Wittig-Kondensation auch stattfinden, wenn das Wittig-Reagenz am Ring II-Anteil des Moleküls gebildet wird, das dann mit dem Aldehyd von dem Ring I-Anteil kondensiert wird.
- Das Wittig-Reagenz wird nach im Stand der Technik anerkannten Verfahren hergestellt. Beispielsweise führt eine Reaktion von Triphenylphosphin oder Diethylphosphon mit substituiertem Alkylbromid, gefolgt von einer Behandlung mit einer starken organometallischen oder Alkoxidbase, wie n-BuLi oder NaOH, zu dem gewünschten Ylid.
-

- Wenn B -N(R1)-C(=O)- oder -C(=O)-N(R1)- ist, ergibt eine Kondensation des Säurehalogenids mit dem passenden Anilin die gewünschte Verbindung.
-

- Diese Verbindungen, bei denen D und/oder E -C(R1)=C(R1)- sind, werden hergestellt, indem das geeignete Aldehyd oder Keton mit einem substituierten Wittig-Reagenz der Formelreagiert, wobei Z Cyano oder Carbalkoxy ist.

- Das Tetrazol kann in verschiedenen Stufen der Synthese durch Behandlung mit Stickstoffwasserstoffsäure, die in situ aus Natri umazid und einer Säure gebildet wird, aus dem Nitril gebildet werden.
- Die Produkte dieser Erfindung können als racemische Mischungen ihrer d- und l-Isomere erhalten werden, da mindestens ein asymmetrisches Kohlenstoffatom vorhanden sein kann. Wenn zwei asymmetrische Kohlenstoffatome vorhanden sind, kann das Produkt als Mischung zweier Diastereomere basierend auf syn- und anti-Konfigurationen vorliegen. Diese Diastereomere können durch fraktionierte Kristallisation getrennt werden. Jedes Diastereomer kann dann durch konventionelle Verfahren in optische d- und l-Isomere gespalten werden. Es können auch chromatographische Verfahren verwendet werden.
- Eine Trennung kann am Besten in der Zwischenstufe durchgeführt werden, wo es zweckmäßig ist, die racemische Verbindung mit einer optisch aktiven Verbindung durch Salzbildung, Esterbildung oder Amidbildung zu kombinieren, um zwei diastereomere Produkte zu bilden. Falls einer optisch aktiven Base eine Säure zugefügt wird, dann werden zwei diastereomere Salze produziert, die unterschiedliche Eigenschaften und unterschiedliche Löslichkeiten besitzen und durch fraktionierte Kristallisation getrennt werden können. Wenn die Salze durch wiederholte Kristallisation vollständig getrennt worden sind, wird die Base durch Säurehydrolyse abgespalten und es werden die reinen d- und l-Säuren erhalten.
- Die vorliegenden Verbindungen bilden Salze mit Säuren, wenn eine basische Aminofunktion vorhanden ist, und Salze mit Basen, wenn eine Säurefunktion, d. h. Carboxyl, vorhanden ist. Alle solchen Salze sind zur Isolierung und/oder Reinigung der neuen Produkte brauchbar. Von besonderem Wert sind die pharmazeutisch akzeptablen Salze mit sowohl Säuren als auch Basen. Geeignete Säuren schließen beispielsweise Salz-, Schwefel-, Salpeter-, Benzolsulfon-, Toluolsulfon-, Essig-, Malein-, Weinsäure und dergleichen ein, die pharmazeutisch akzeptabel sind. Basische Salze, die zum pharmazeutischen Gebrauch akzeptabel sind, sind die Na-, K-, Ca- und Mg-Salze.
- Verschiedene Substituenten an den vorliegenden neuen Verbindungen, z. B. wie in R, R1 und R2 definiert, können in den Ausgangsverbindungen vorhanden sein, irgendeiner der Zwischenstufen zugefügt oder nach Bildung der Endprodukte durch bekannte Verfahren der Substitutions- oder Umwandlungsbedingungen zugefügt werden. Falls die Substituenten selbst reaktiv sind, dann können die Substituenten selbst gemäß im Stand der Technik bekannten Techniken geschützt werden. Eine Vielfalt von Schutzgruppen kann verwendet werden, die in der Technik bekannt sind. Beispiele für viele dieser möglichen Gruppen finden sich in "Protective Groups in Organic Synthesis" von T. W. Green, John Wiley and Sons, 1981. Beispielsweise können dem aromatischen Ring durch Nitrierung Nitrogruppen hinzugefügt werden, und die Nitrogruppe kann in andere Gruppen umgewandelt werden, wie durch Reduktion in Amino und durch Diazotierung der Aminogruppe und Ersetzen der Diazogruppe durch Halogen. Acylgruppen können durch Friedel-Krafts-Acylierung an die Arylgruppen substituiert werden. Die Acylgruppen können dann nach verschiedenen Methoden in die entsprechenden Alkylgruppen umgewandelt werden, einschließlich der Wolff-Kishner-Reduktion und Clemmenson-Reduktion. Aminogruppen können alkyliert werden, um Mono- und Dialkylaminogruppen zu bilden, und Mercapto- und Hydroxygruppen können unter Bildung entsprechender Ether alkyliert werden. Primäre Alkohole können durch im Stand der Technik bekannte Oxidationsmittel unter Bildung von Carbonsäuren oder Aldehyden oxidiert werden, und sekundäre Alkohole können unter Bildung von Ketonen oxidiert werden. Somit können Substitutions- oder Veränderungsreaktionen verwendet werden, um in dem Molekül des Ausgangsmaterials, der Zwischenstufen oder des Endprodukts eine Vielfalt von Substituenten zu liefern.
- Die erfindungsgemäßen Verbindungen haben potente Aktivität als Leukotrien-Antagonisten und besitzen als solche therapeuti schen Wert zur Behandlung von Entzündungszuständen und allergischen Reaktionen, wie Anaphylaxie und Asthma.
- PROTOKOLL FÜR SRS-A (LANGSAM REAGIERENDE ANAPHYLAXIESUBSTANZ)-ANTAGONISTEN
- Leukotriene, die Produkte des 5-Lipoxygenasewegs des Arachidonsäuremetabolismus, sind bei einer Vielfalt glatter Muskelpräparate potente kontraktile Mittel. Es wurde daher die Hypothese aufgestellt, dass die Leukotriene erheblich zu der Pathophysiologie von Asthma beitragen. Dieses Protokoll beschreibt einen in vitro-Assay, der zum Testen von Verbindungen verwendet wird, die die Wirkungen von Leukotrienen spezifisch antagonisieren.
- Periphere Streifen von Meerschweinchenlungen wurden gemäß dem veröffentlichten Verfahren (Proc. Nat'l. Acad. Sci., USA Band 77, Seiten 4354 bis 4358, 1980) präpariert und in Gewebebäder gehängt (Metro Nr. ME-5505, 10 ml). Die Streifen wurden gründlich in Assay-Puffer gespült und dann mit chirurgischen Seidenfäden mit Haltestangen von den Gewebebädern verbunden. Die Stäbe wurden in den Bädern justiert und die Streifen mit den Druckmesswertwandlern (Grass FT 103 oder Gould US-3) verbunden. Die Gewebebäder wurden mit 95% Sauerstoff – 5% Kohlendioxid belüftet und auf 37°C gehalten. Der Assay-Puffer wurde wie folgt hergestellt: Auf jeden Liter Puffer wurde das folgende zu ungefähr 800 ml Wasser gegeben, das in Glas destilliert wurde – 6,87 g NaCl, 0,4 g MgSO4·7H2O und 2,0 g D-Glukose. Dann wurde eine Lösung von 0,368 g CaCl2·H2O in 100 ml glasdestilliertem Wasser langsam zu dem Puffer gegeben. Es wurde ausreichend Wasser zugegeben, um das Volumen auf 1 Liter einzustellen, und die Lösung wurde mit 95% Sauerstoff – 5% Kohlendioxid belüftet. Üblicherweise wurden für ein Experiment mit 4 Geweben 10 Liter Puffer verwendet. Nachdem die Gewebe wiederholt gewaschen und in dem Gewebebad ins Gleichgewicht kommen gelassen wurden, wurden sie mit 1 M Histamin belastet. Nachdem maximale Kontraktionen erhalten worden waren, wurden die Gewebe gewaschen und zurück zu ihrer Grundlinienspannung relaxieren gelassen. Dieses Histaminbelastungsverfahren wurde mindestens 1 bis 2 weitere Male wiederholt, um eine wiederholbare Kontrollreaktion zu erhalten. Die durchschnittliche Reaktion auf 1 M Histamin für jedes Gewebe wurde verwendet, um alle anderen Belastungen zu normalisieren.
- Reaktionen von jedem Gewebe bis zu einer festgelegten Konzentration von Leukotrien wurden dann erhalten. Üblicherweise wurden Testverbindungen anfangs mit 30 M auf Restspannung der Gewebe ohne irgendwelchen zugesetzten Agonisten oder Antagonisten untersucht, um zu bestimmen, ob die Verbindung irgendwelche mögliche innewohnende Aktivität hat. Die Gewebe wurden gewaschen, und die Testverbindung wurde wieder zugesetzt. Leukotrien wurde nach der gewünschten Vorinkubationszeit zugesetzt. Die innewohnende Aktivität der Verbindungen und ihre Wirkung auf Leukotrieninduzierte Kontraktionen wurden dann aufgezeichnet.
- Die Ergebnisse dieses Tests für die Verbindungen dieser Erfindung zeigen, dass diese Verbindungen als brauchbare Leukotrien-Antagonisten angesehen werden.
- INHIBIERUNGEN VON (3H)-LTD4-BINDUNGSMEMBRANEN AUS MEERSCHWEINCHENLUNGE
- A. Herstellung der rohen Rezeptorfraktion
- Dieses Verfahren wurde von Mong et al. 1984) adaptiert. Männliche Meerschweinchen wurden durch Enthauptung getötet, und ihre Lungen wurden rasch entfernt und in ein Becherglas gegeben, das eiskalten Homogenisierungspuffer enthielt. Die Lungen wurden von Verbindungsgewebe getrennt, mit Scheren zerschnitten, trokkengetupft und gewogen. Das Gewebe wurde dann in 40 Volumina (Gew./Vol.) Homogenisierungspuffer mit einem Polytron 30 Sekunden lang bei einer Einstellung von 6 homogenisiert. Das Homogenisat wurde mit 1000 g 10 Minuten lang zentrifugiert (z. B. 3500 UpM, SS-34 Rotor). Der Überstand wurde durch zwei Lagen Käsetuch fil triert und mit 30 000 g 30 Minuten lang zentrifugiert (z. B. 18 500 UpM, SS-34 Rotor), danach wurde das resultierende Pellet erneut in 20 Volumina Assay-Puffer von Hand suspendiert. Das fertige Pellet wurde in 10 Volumina Assay-Puffer erneut suspendiert und bis zum Gebrauch auf 4°C gehalten.
- B. Bindungsassay
- Jedes Assay-Röhrchen (16 × 100 mm) enthielt das folgende:
490 μl Assay-Puffer
10 μl Testverbindung oder Lösungsmittel
100 μl 3H-LTD4 (etwa 17 500 DMP)
400 μl Proteinzubereitung - Die Inkubationen erfolgten 20 Minuten lang bei 25°C in einem geschüttelten Wasserbad. Die Reaktionen wurden durch Zugabe der Proteinzubereitung gestartet. Am Ende der Inkubationszeit wurden 4,0 ml kalter Waschpuffer zu dem Röhrchen gegeben. Nach dem Vortexieren wurde der Inhalt des Röhrchens sofort über einen Whatman GF/C-Filter (25 mm Durchmesser) gegossen, der sich in einem Vakuumverteiler befand (z. B. Millipore Modell Nr. 3025 Verteiler), an den ein partielles Vakuum angelegt wurde. Die Filter wurden sofort mit weiteren 15 ml kaltem Puffer gewaschen. Die Filter wurden in 7 ml Kunststoff-Szintillations-ampullen überführt, denen 6,0 ml geeignete Szintillationsflüssigkeit (z. B. Scintiverse) zugefügt wurde. Nachdem sie 4 bis 6 Stunden lang ins Gleichgewicht kommen gelassen wurden, wurde die Radioaktivität mit einem Flüssigszintillationszähler gezählt, der geeignet für Tritium eingestellt worden war.
- Die erforderlichen Kontroll-Assay-Röhrchen schlossen das folgende ein:
- Gesamtbindung: es wurde keine Testverbindung zugefügt, Puffer ist substituiert.
- (a) unspezifische Bindung: Nicht markierter Ligand wurde mit einer Konzentration von 1 M zugesetzt.
- (b) Lösungsmittelkontrollen: Falls Testverbindung in einem Lösungsmittel gelöst wird, sind Kontrollen sowohl für Gesamtbindung als auch für unspezifische Bindung erforderlich, welche Lösungsmittel, jedoch keine Verbindungen enthalten.
- Die Ergebnisse dieses Tests zeigen, dass die erfindungsgemäßen Verbindungen wertvolle Eigenschaften zeigen, die zur Behandlung von Entzündungszuständen und allergischen Reaktionen brauchbar sind.
- Die erfindungsgemäßen Verbindungen können einem Säugetierwirt in vielerlei Formen verabreicht werden, die an den gewählten Verabreichungsweg angepasst sind, d. h. oral oder parenteral. Parenterale Verabreichung schließt in dieser Hinsicht Verabreichung durch die folgenden Wege ein: intravenös, intramuskulär, subkutan, intraokular, intrasynovial, transepithelial (einschließlich transdermal), ophthalmisch, sublingual und bukkal; topisch einschließlich ophthalmisch, dermal, Okular, rektal und nasale Inhalation durch Einblasen und Aerosol und rektal-systemisch.
- Die aktive Verbindung kann oral verabreicht werden, beispielsweise mit inertem Verdünnungsmittel oder einem assimilierbaren essbaren Träger, oder kann in Hart- oder Weichschalen-Gelatinekapseln eingeschlossen werden, oder kann zu Tabletten komprimiert werden, oder kann direkt mit dem Nahrungsmittel der Diät eingenommen werden. Für die orale therapeutische Verabreichung kann die aktive Verbindung mit Hilfsmittel eingenommen und in Form von essbaren Tabletten, Bukkal-Tabletten, Pastillen, Kapseln, Elixieren, Suspensionen, Sirups, Oblaten und dergleichen verwendet werden. Solche Zusammensetzungen und Zubereitungen sollten mindestens 0,1% aktive Verbindung enthalten. Der Prozentsatz der Zusammensetzungen und Zubereitungen kann natürlich variieren und kann konventionell zwischen etwa 2 und etwa 6 Gew.-% der Einheit liegen. Die Menge an aktiver Verbindung in diesen therapeutisch brauchbaren Zusammensetzungen ist so, dass eine geeignete Dosis erhalten wird. Bevorzugte erfindungsgemäße Zusammensetzungen oder Zubereitungen werden so hergestellt, dass eine orale Dosiereinheitsform zwischen etwa 50 und 300 mg aktive Verbindung enthält.
- Die Tabletten, Pastillen, Pillen, Kapseln und dergleichen können auch die folgenden enthalten: Bindemittel wie Gum Tragacanth, Akaziengummi, Maisstärke oder Gelatine, Hilfsstoffe wie Dicalciumphosphat, Sprengmittel wie Maisstärke, Kartoffelstärke, Alginsäure und dergleichen; Schmiermittel wie Magnesiumstearat und Süßungsmittel wie Sucrose, Lactose oder Saccharin können zugegeben werden, oder Aromatisierungsmittel wie Pfefferminz, Wintergrünöl oder Kirscharoma. Wenn die Dosiereinheitsform eine Kapsel ist, kann sie zusätzlich zu Materialien des obigen Typs einen flüssigen Träger enthalten. Verschiedene andere Materialien können als Beschichtungen vorhanden sein oder anders die physikalische Form der Dosiereinheit modifizieren. Beispielsweise können Tabletten, Pillen oder Kapseln mit Schellack, Zucker oder beiden beschichtet werden. Ein Sirup oder Elixier kann die aktive Verbindung, Sucrose als Süßungsmittel, Methyl- und Propylparabene als Konservierungsmittel, Farbstoff und Aromatisierungsmittel wie Kirsch- oder Orangenarome enthalten. Natürlich sollte jegliches Material, das zur Herstellung einer beliebigen Dosiereinheitsform verwendet wird, pharmazeutisch rein und im Wesentlichen in den verwendeten Mengen nicht toxisch sein. Zudem kann die aktive Verbindung auch in Zubereitungen und Formulierungen mit verzögerter Freisetzung eingebracht werden.
- Die aktive Verbindung kann auch parenteral oder intraperitoneal verabreicht werden. Lösungen der aktiven Verbindung als freie Base oder pharmazeutisch annehmbares Salz können in Wasser hergestellt werden, das geeignet mit Tensid wie Hydroxypropylcellulose gemischt werden. Dispersion kann auch in Glycerin, flüssigen Polyethylenglykolen und Mischungen davon und in Ölen hergestellt werden. Unter gewöhnlichen Lager- und Gebrauchsbedingungen enthalten diese Zubereitungen Konservierungsmittel, um das Wachstum von Mikroorganismen zu verhindern.
- Die pharmazeutischen Formen, die zur injizierbaren Verwendung geeignet sind, schließen sterile wässrige Lösungen oder Dispersionen und sterile Pulver zur improvisierten Zubereitung steriler injizierbarer Lösungen und Dispersionen ein. In allen Fällen muss die Form steril sein und in dem Maße fließfähig sein, dass sie sich leicht mittels Spritze verabreichen lässt. Sie muss unter den Fertigungs- und Lagerungsbedingungen stabil sein und muss gegen verunreinigende Wirkung von Mikroorganismen wie Bakterien und Pilzen konserviert werden. Der Träger kann Lösungsmittel oder Dispersionsmedium sein, das beispielsweise Wasser, Ethanol, Polyol (beispielsweise Glycerin, Propylenglykol und flüssiges Polyethylenglykol und dergleichen), geeignete Mischungen davon und pflanzliche Öle enthält. Die richtige Fließfähigkeit kann beispielsweise durch die Verwendung einer Beschichtung wie Lecithin, durch Aufrechterhalten der erforderlichen Partikelgröße im Fall einer Dispersion und durch die Verwendung von Tensiden aufrechterhalten werden. Die Konservierung gegen Wirkung von Mikroorganismen kann durch verschiedene antibakterielle und antifungale Mittel bewirkt werden, beispielsweise Parabene, Chlorbutanol, Phenol, Sorbinsäure, Thimeral und dergleichen. Es ist in vielen Fällen bevorzugt, isotone Mittel einzuschließen, beispielsweise Zucker oder Natriumchlorid. Verlängerte Absorption der injizierbaren Zusammensetzungen durch Mittel, die die Absorption verzögern, liefern beispielsweise Aluminiummonostearat und Gelatine.
- Sterile injizierbare Lösungen werden hergestellt, indem die aktive Verbindung nach Bedarf in der erforderlichen Menge in das geeigneten Lösungsmittel mit verschiedenen der anderen oben auf gezählten Bestandteile eingebracht wird, gefolgt von filtrierter Sterilisation. Dispersionen werden im Allgemeinen hergestellt, indem die verschiedenen sterilisierten Wirkbestandteile in ein steriles Vehikel eingebracht werden, welches das Basisdispersionsmedium und die erforderlichen anderen Bestandteile von den oben aufgezählten enthält. Im Falle von sterilen Pulvern zur Herstellung steriler injizierbarer Lösungen sind die bevorzugten Herstellungsverfahren Vakuumtrocknen und die Gefriertrocknungstechnik, die ein Pulver des Wirkbestandteils plus jeglichen zusätzlichen Bestandteils aus der zuvor steril filtrierten Lösung davon ergibt.
- Die erfindungsgemäßen therapeutischen Verbindungen können einem Säugetier allein oder in Kombination mit pharmazeutisch akzeptablen Trägern verabreicht werden, deren Anteil durch die Löslichkeit und chemische Beschaffenheit der Verbindung, den gewählten Verabreichungsweg und pharmazeutische Standardpraxis bestimmt wird.
- Der Arzt wird die Dosis der vorliegenden therapeutischen Mittel bestimmen, die für die Prophylaxe oder Behandlung am geeignetsten sind, und sie variiert mit der Form der Verabreichung und der speziellen gewählte Verbindung und variiert auch gemäß dem speziellen behandelten Patienten. Er wird die Behandlung im Allgemeinen mit geringen Dosen mit kleinen Steigerungen beginnen wollen, bis die Optimalwirkung unter den Umständen erreicht ist. Die therapeutische Dosis ist im Allgemeinen 0,1 bis 100 M/Tag oder etwa 0,1 mg bis etwa 50 mg/kg Körpergewicht pro Tag und darüber, obwohl sie in mehreren verschiedenen Dosiereinheiten verabreicht werden kann. Für die orale Verabreichung sind höhere Dosierungen erforderlich.
- Die erfindungsgemäßen Verbindungen können durch die folgenden repräsentativen Beispiele hergestellt werden.
- BEISPIEL 1
- (3-(Chinolin-2-yl)methyloxy)phenol
- Eine Mischung aus (0,06 Mol) 2-Chinolinylmethylchlorid·HCl, (0,06 Mol) 1,3-Benzoldiol und 18 g Kaliumcarbonat in 50 ml DMF wurde über Nacht auf 70°C erhitzt. Die Reaktionsmischung wurde in Wasser gegossen und das ausgefallene Produkt wurde aufgefangen, filtriert und getrocknet, um (3-(Chinolin-2-yl)methyl-oxy)phenol zu ergeben.
- BEISPIEL 2
- Wenn 2-Chinolinylmethylchlorid von dem obigen Beispiel 1 durch die Chinolinverbindungen der folgenden Tabelle I ersetzt wird, dann wird das entsprechende Produkt erhalten.
- TABELLE I
-
- 2-Chlormethylchinolin
- 2-Brommethylchinolin
- 2-(1-Chlorethyl)chinolin
- 2-(2-Chlorethyl)chinolin
- 2-Bromethylchinolin
- 3-Chlormethylchinolin
- 4-Chlormethylchinolin
- 2-(β-Chlorethyl)chinolin
- 2-(β-Chlorpropyl)chinolin
- 2-(β-Chlor-β-phenethyl)chinolin
- 2-Chlormethyl-4-methylchinolin
- 2-Chlormethyl-6-methylchinolin
- 2-Chlormethyl-8-methylchinolin
- 2-Chlormethyl-6-methoxychinolin
- 2-Chlormethyl-6-nitrochinolin
- 2-Chlormethyl-6,8-dimethylchinolin
- 2-Chlormethyl-7-chlorchinolin
- 2-Chlormethyl-7-bromchinolin
- 2-Chlormethyl-7-nitrochinolin
- 2-Chlormethyl-7-methylchinolin
- BEISPIEL 3
- Wenn 1,3-Benzoldiol-Alkohol von dem obigen Beispiel 1 durch die folgenden Verbindungen der Tabelle II ersetzt wird, dann wird das entsprechende Produkt erhalten.
- TABELLE II
-
- 1,2-Benzoldiol
- 1,4-Benzoldiol
- 2-Mercaptophenol
- 3-Mercaptophenol
- 4-Mercaptophenol
- 1,3-Dimercaptobenzol
- 1,4-Dimercaptobenzol
- 3-Hydroxybenzylalkohol
- 3-Hydroxyethylphenol
- 4-Hydroxybenzylalkohol
- 4-Hydroxyethylphenol
- 2-Methylresorcinol
- 5-Methylresorcinol
- 5-Methoxyresorcinol
- 5-Methyl-1,4-dihydroxybenzol
- 3-(N-Acetylamino)phenol
- 3-(N-Acetylamino)benzylalkohol
- 2-Hydroxy-α-methylbenzylalkohol
- 2-Hydroxy-α-ethylbenzylalkohol
- 2-Hydroxy-α-propylbenzylalkohol
- 3-Hydroxy-α-methylbenzylalkohol
- 3-Hydroxy-α-ethylbenzylalkohol
- 3-Hydroxy-α-propylbenzylalkohol
- 4-Hydroxy-α-methylbenzylalkohol
- 4-Hydroxy-α-ethylbenzylalkohol
- 4-Hydroxy-α-propylbenzylalkohol
- BEISPIEL 4
- Wenn die Verbindungen von Tabelle I, Beispiel 2, mit den Verbindungen von Tabelle II, Beispiel 3, unter den Bedingungen von Beispiel 1 umgesetzt werden, dann werden entsprechende Produkte erhalten.
- BEISPIEL 5
- 2-[2-(3-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]propionsäure
- A. Ethyl-2-(2-methylphenoxy)propionat
- Eine Mischung aus 10,8 g (0,1 Mol) o-Cresol, 18,1 g (0,1 Mol) Ethyl-2-brompropionat und 14,5 g K2CO3 in 50 ml DMF wurde 3 Tage lang auf 70°C erhitzt. Die Reaktionsmischung wurde in H2O gegossen und mit Ethylacetat extrahiert. Die organische Lösung wurde getrocknet und eingedampft, um Ethyl-2-(2-methylphenoxy)propionat zu ergeben, das direkt in der nächsten Stufe verwendet wurde.
- B. Ethyl-2-(2-brommethylphenoxy)propionat
- Eine Mischung aus 6 g (0,029 Mol) Ester, der in Stufe A erhalten wurde, 5,6 g (0,031 Mol) N-Bromsuccinimid und 100 mg Benzoylperoxid in 100 ml CCl4 wurden eine Stunde auf Rückfluss erhitzt, während mit einer Sonnenlampe bestrahlt wurde. Die Reaktionsmischung wurde abgekühlt und zur Trockne filtriert, um Ethyl-2-(2-brommethylphenoxy)propionat zu ergeben, das direkt in der nächsten Stufe verwendet wurde.
- C. Ethyl-2-[2-(3-(2-chinolinylmethoxy)phenoxymethyl)phenoxy]propionat
- Zu einer Lösung von (5,23 mmol) 3-(2-Chinolinylmethyloxy)phenol und (5,23 mmol) des Bromids aus Stufe B in 90 ml 8 : 1 Aceton : DMF wurden 1,52 g (10,97 mmol, 2,1 Äquiv.) K2CO3 gegeben. Die Mischung wurde über Nacht unter Rückfluss gehalten, dann zwischen EtOAc und H2O partitioniert. Die organischen Materialien wurden getrocknet, filtriert und im Vakuum konzentriert. Dies wurde dann durch Flash-Säulenchromatographie gereinigt, um Ethyl-2-[2-(3-(2-Chinolinylmethyloxy)phenoxymethyl)phen-oxy]propionat zu ergeben, das direkt in der nächsten Stufe verwendet wurde.
- D. 2-[2-(3-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]propionsäure
- Zu einer Lösung von (2,65 mmol) des Esters aus Stufe C in 25 ml EtOH wurden 10 ml (10,0 mmol, 3,75 Äquiv.) 1 N NaOH gegeben. Die Mischung wurde eine Stunde lang bei Raumtemperatur gerührt, dann wurde die Mischung auf pH 4 angesäuert, aus heißem MeOH-Et2O umkristallisiert, um 2-[2-(3-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]propionsäure zu ergeben.
- Wenn 3-(2-Chinolinylmethyloxy)phenol der obigen Stufe C durch 3-(2-Chinolinylmethyloxy)mercaptobenzol oder 4-(2-Chinolinylmethyloxy)phenol ersetzt wird, dann sind die hergestellten Produkte 2-[2-(3-(2-Chinolinylmethyloxy)phenylthiomethyl)phenoxy]propionsäure oder 2-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]propionsäure.
- BEISPIEL 6
- Wenn die durch Beispiele 2 bis 4 hergestellten Verbindungen anstelle von 3-(2-Chinolinylmethyloxy)phenol in Beispiel 5 verwendet werden, dann wird das entsprechende Produkt hergestellt.
- BEISPIEL 7
- 5-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzyl]tetrazol
- A. 2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzylchlorid
- Eine Lösung von (10,11 mmol) 4-(2-Chinolinylmethyloxy)phenol, (30,33 mmol, 3 Äquiv.) 2-Chlormethylbenzylchlorid und 4,2 g (30,33 mmol, 3 Äquiv.) K2CO3 in 100 ml 8 : 1 Aceton : DMF wurde 24 Stunden lang unter Rückfluss gehalten. Die Mischung wurde in Eiswasser gegossen und 1 1/2 Stunden lang stehen gelassen. Der sich bildende Niederschlag wurde abfiltriert, in EtOAc wieder aufgelöst, getrocknet und im Vakuum konzentriert. Das Rohprodukt wurde durch Flash-Säulenchromatographie gereinigt, um 2-(4-(2- Chinolinylmethyloxy)phenoxymethyl)benzylchlorid zu ergeben, das direkt in der nächsten Stufe verwendet wurde.
- B. 2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzylnitril
- Eine Mischung aus (4,9 mmol) 2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzylchlorid (22,3 mmol, 2 Äquiv.) KCN und 100 mg Adogen 464 Phasentransferkatalysator in 70 ml Toluol und 70 ml H2O wurde 24 Stunden lang unter Rückfluss gehalten. Die Reaktionsmischung wurde zwischen EtOAc und H2O partitioniert. Die organische Phase wurde abgetrennt, getrocknet und im Vakuum konzentriert, um einen Niederschlag zu ergeben, der aus CH2Cl2-pet umkristallisiert wurde, um 2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzylnitril zu ergeben.
- C. 5-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzyl]tetrazol
- Eine Lösung von (3,19 mmol) 2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzylnitril (63,07 mmol, 20 Äquiv.) NaN3 und 3,4 g (63,07 mmol, 20 Äquiv.) NH4Cl in 20 ml DMF wurde 5 Tage lang unter einer inerten Atmosphäre auf 110°C erhitzt. Die Reaktionsmischung wurde in Eiswasser gegossen und eine Stunde lang stehen gelassen. Der sich bildende Niederschlag wurde abfiltriert und erneut in CH2Cl2 gelöst. Hierzu wurde 1 N NaOH gegeben, und das sich bildende Natriumsalz wurde abfiltriert. Das Salz wurde erneut in heißem H2O gelöst, dann unter Verwendung von 1 N HCl auf pH 4 angesäuert. Der sich bildende Niederschlag wurde abfiltriert, mit heißem MeOH trituriert und filtriert, um 5-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)benzyl]tetrazol zu ergeben.
- REFERENZBEISPIEL 9
- Ethyl-4-[2-(4-(2-chinolinylmethyloxy)phenoxymethyl)phenoxy]-4-carbethoxybutyrat (Referenzverbindung)
- 4-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]-4-carboxybuttersäure (Referenzverbindung)
- A. Ethyl-4-carbethoxy-4-(2-methylphenoxy)butyrat
- Eine Mischung aus o-Cresol (0,1 Mol) und Diethyl-2-bromglutarat (0,1 Mol) und K2CO3 (0,11 Mol) in 50 ml DMF wurde über Nacht auf 70°C erhitzt. Die Reaktion wurde in Wasser gegossen und mit Ethylacetat extrahiert. Die organische Lösung wurde mit Wasser, 0,1 N NaOH-Lösung gewaschen, getrocknet und eingedampft, um Ethyl-4-carbethoxy-4-(2-methylphenoxy)butyrat zu ergeben, das direkt in der nächsten Stufe verwendet wurde.
- B. 2-(1,3-Dicarbethoxypropoxy)benzylbromid
- Eine Mischung aus Ethyl-4-carbethoxy-4-(2-methylphenoxy)butyrat (0,05 Mol) und NBS (0,055 Mol) und 150 mg Benzoylperoxid in 150 ml CCl4 wurde unter Rückfluss gehalten und 2 Stunden lang mit einer Sonnenlampe angestrahlt. Filtration und Verdampfen des Lösungsmittels ergab 2-(1,3-Dicarbethoxypropoxy)-benzylbromid, das ohne Reinigung verwendet wurde.
- C. Ethyl-4-[2-(4-(2-chinolinylmethyloxy)phenoxymethyl)phenoxy]-4-carbethoxybutyrat (Referenzverbindung)
- 4-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]-4-carb-oxybuttersäure (Referenzverbindung)
- Eine Mischung aus 4-(2-Chinolinylmethyloxy)phenol (0,01 Mol) und 0,011 Mol K2CO3 in 50 ml DMF wurde über Nacht auf 70°C erhitzt. Die Reaktionsmischung wurde in Wasser gegossen und anschließend mit Ethylacetat extrahiert. Die organische Lösung wurde mit Wasser gewaschen, getrocknet und zur Trockne eingedampft.
- Das Rohprodukt wurde durch Säulenchromatographie gereinigt, um Ethyl-4-[2-(4-(2-chinolinylmethyloxy)phenoxymethyl)phenoxy]-4- carbethoxybutyrat zu ergeben. Der Ester wurde mit 1 N NaOH-Lösung (30 ml) über Nacht in Dioxan behandelt. Die Reaktionsmischung wurde dann angesäuert und durch Flash-Chromatographie gereinigt, die 4-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)-phenoxy]-4-carboxybuttersäure ergab.
- BEISPIEL 10
- Wenn das Verfahren von Referenzbeispiel 9 nachgearbeitet wurde und Diethyl-2-bromglutamat durch die folgenden Verbindungen der Tabelle I ersetzt werden, dann wird das entsprechende Produkt hergestellt.
- Tabelle I
-
- Diethyl-2-brommalonat
- Diethyl-2-bromsuccinat
- Diethyl-2-bromglutarat
- Diethyl-2-bromadipat
- Diethyl-3-bromadipat
- Ethyl-2-brom-2-dimethylacetamidoacetat
- Ethyl-2-brom-3-dimethylacetamidopropionat
- Ethyl-2-brom-4-dimethylacetamidobutyrat
- Ethyl-2-brom-5-dimethylacetamidovalerat
- Ethyl-3-brom-4-dimethylacetamidobutyrat
- Ethyl-2-brom-5-dimethylacetamidovalerat
- Ethyl-3-brom-5-dimethylacetamidovalerat
- Ethyl-4-brom-5-dimethylacetamidovalerat
- BEISPIEL 11
- Wenn das Verfahren von Beispiel 5 nachgearbeitet wird und Ethyl-2-brompropionat durch die folgenden Verbindungen der Tabelle IV ersetzt wird, dann wird das entsprechende Produkt erhalten.
- Tabelle IV
-
- Ethyl-2-bromacetat
- Ethyl-2-brombutyrat
- Ethyl-3-brombutyrat
- Ethyl-2-bromvalerat
- Ethyl-2-brom-3-methylvalerat
- Ethyl-3-bromvalerat
- N,N-Dimethyl-2-brompropionamid
- N,N-Dimethyl-2-brombutyramid
- N,N-Dimethyl-2-bromvaleramid
- N,N-Dimethyl-3-bromvaleramid
- BEISPIEL 12
- Wenn die Verfahren von Beispiel 5, Referenzbeispiel 9, Beispielen 10 und 11 nachgearbeitet werden und o-Cresol durch die folgenden Verbindungen der Tabelle A ersetzt wird, dann werden die entsprechenden Produkte hergestellt.
- Tabelle A
-
- m-Cresol, p-Cresol
- o-Mercaptotoluol, m-Mercaptotoluol, p-Mercaptotoluol
- 3-Hydroxy-4-carbethoxytoluol
- 3-Hydroxy-4-dimethylcarbamyltoluol
- 3-Hydroxy-4-dimethylcarbamylmethyltoluol
- 3-Hydroxy-5-dimethylcarbamylmethoxytoluol
- 3-Mercapto-4-carbethoxytoluol
- 3-Mercapto-4-dimethylcarbamyltoluol
- 3-Mercapto-4-dimethylcarbamylmethyltoluol
- 3-Mercapto-5-dimethylcarbamylmethoxytoluol
- Methyl-4-methylsalicylat, Methyl-3-methylsalicylat
- Wenn das Verfahren von Beispiel 5 wiederholt wird und o-Cresol durch 5-Methylresorcinol, 4-Methylresorcinol oder Methylhy-drochinon ersetzt wird, dann werden die entsprechenden Produkte hergestellt.
- BEISPIEL 13
- 3-[2-(3-(2-Chinolinylmethyloxy)phenyl)-2-(2-carboxyethylthio)ethyl]phenoxyessigsäure
- A. Ethyl-3-[2-(3-hydroxyphenyl)-2-oxoethyl]phenoxyacetat und
- Ethyl-3-[2-(3-hydroxyphenyl)-1-oxoethyl]phenoxyacetat
- Eine Mischung aus 3,3'-Dihydroxydeoxybenzoin (0,05 Mol), Ethylbromacetat (0,05 Mol) und K2CO3 (0,05 Mol) in 50 ml DMF wurde bei Raumtemperatur über Nacht gerührt. Die Reaktionsmischung wur de in Wasser gegossen und mit EtOAc extrahiert. Die organische Lösung wurde getrocknet und zur Trockne eingedampft. Die Rohprodukte wurden durch Flash-Chromatographie getrennt, um Ethyl-3-[2-(3-hydroxyphenyl)-2-oxoethyl]phenoxyacetat und Ethyl-3-[2-(3-hydroxyphenyl)-1-oxoethyl]phenoxyacetat zu ergeben.
- B. Ethyl-3-[2-(3-(2-chinolinylmethyloxy)phenyl)-2-oxoethyl]phenoxyacetat
- Eine Mischung aus Ethyl-3-[2-(3-hydroxyphenyl)-2-oxoethyl]phenoxyacetat (0,01 Mol), 2-Chlormethylchinolin (0,01 Mol) und K2CO3 (0,01 Mol) in 20 ml DMF wurde über Nacht auf 60°C erwärmt. Nach der Aufarbeitung wurde das Rohprodukt durch Flash-Chromatographie gereinigt, um Ethyl-3-[2-(3-(2-chinolinylmethyloxy)phenyl)-2-oxoethyl]phenoxyacetat zu ergeben.
- C. Ethyl-3-[2-(3-(2-chinolinylmethyloxy)phenyl)-2-hydroxyethyl]phenoxyacetat
- Das in Stufe B (5 mmol) in 50 ml EtOH erhaltene Produkt wurde bei Raumtemperatur mit (2 mmol) NaBH4 behandelt. Nach 30 Minuten wurde die Reaktionsmischung mit Wasser verdünnt und mit EtOAc extrahiert. Die organische Lösung wurde getrocknet und eingedampft und durch Trockensäulenchromatographie gereinigt, um Ethyl-3-[2-(3-(2-chinolinylmethyloxy)phenyl)-2-hydroxyethyl]phenoxyacetat zu ergeben.
- D. Ethyl-3-[2-(3-(2-chinolinylmethyloxy)phenyl)-2-(carbethoxyethylthio)ethyl]phenoxyacetat
- Das aus Stufe C erhaltene Produkt wurde in 50 ml CH2Cl2 mit 2 ml SOCl2 behandelt. Die Reaktionsmischung wurde bei Raumtemperatur zwei Stunden lang gerührt und dann zur Trockne eingedampft. Das so in 2 ml CH2Cl2 erhaltene Rohprodukt wurde mit Ethyl-3-mercaptopropionat und Triethylamin (1 : 1) 16 Stunden lang bei Raumtemperatur behandelt. Nach Verdampfen des Lösungsmittels wurde das Rohprodukt gereinigt, um Ethyl-3-[2-(3-(2-chinolinylmethyloxy)phenyl)-2-(carbethoxyethylthio)ethyl]phenoxyacetat zu erhalten.
- E. 3-[2-(3-(2-Chinolinylmethyloxy)phenyl)-2-(2-carboxyethylthio)ethyl]phenoxyessigsäure
- Das aus Stufe D erhaltene Produkt wurde mit 1 N NaOH-Lösung hydrolysiert. Ansäuern und anschließende Reinigung ergab 3-[2-(3-(2-Chinolinylmethyloxy)phenyl)-2-(2-carboxyethylthio)ethyl]phenoxyessigsäure.
- Wenn Ethyl-3-mercaptopropionat in Stufe D durch die Verbindung der Tabelle V ersetzt wird, dann wird das entsprechende Produkt hergestellt.
- TABELLE V
-
- Methyl-2-mercaptopropionat
- Methyl-3-mercaptopropionat
- Methyl-4-mercaptobutyrat
- Methyl-3-mercaptobutyrat
- 2-Mercapto-N,N-dimethylpropionamid
- 3-Mercapto-N,N-dimethylbutyramid
- 3-Mercapto-N,N-diethylpropionamid
- REFERENZBEISPIEL 14
- 3-[(1-[3-(3-(2-Chinolinylmethyloxy)phenoxy)methylphenyl]-1-(N,N-dimethylcarbamylethylthio)methyl)thio]propionsäure (Referenzverbindung)
- A. 3-[1-Carbomethyloxyethylthio-1-(N,N-diethylcarbamylethylthio)methyl]benzaldehyd
- Eine Lösung von Isophthaldehyd (0,1 Mol), N,N-Dimethyl-3-mercaptopropionamid (0,1 Mol), Thioessigsäure (0,1 Mol) und p-Toluolsulfonsäure (0,1 Mol) in 100 ml CH2Cl2 wurden 24 Stunden lang gerührt. Die Reaktionsmischung wurde mit Wasser gewaschen, getrocknet und zur Trockne eingedampft. Das Rohprodukt wurde durch Flash-Chromatographie gereinigt. Das so erhaltene Produkt (0,05 Mol) wurde mit 0,05 Mol Natriummethoxid in Gegenwart von 0,06 Mol Methylacrylat in 50 ml Methanol behandelt. Nach Verdampfen des Lösungsmittels wurde das Rohprodukt durch Flash-Chromatographie gereinigt, um 3-[1-Carbomethyloxyethylthio-1-(N,N-diethylcarbamylethylthio)methyl]benzaldehyd zu ergeben.
- B. 3-[1-Carbomethyloxyethylthio-1-(N,N-dimethylcarbamylethylthio)methyl]benzylchlorid
- Der aus Stufe A erhaltene Aldehyd (0,05 Mol) wurde in 100 ml EtOH mit 0,02 Mol NaBH4 bei 5°C eine Stunde lang behandelt. Überschüssiges Hydrid wurde mit verdünnter Säure zersetzt, gefolgt von Extraktion mit Ethylacetat. Die organische Lösung wurde getrocknet und zur Trockne eingedampft. Das so erhaltene Rohprodukt in 50 ml CH2Cl2 wurde mit Thionylchlorid behandelt, um 3-[1-Carbomethyloxyethylthio-1-(N,N-dimethylcarbamylethylthio)methyl]benzylchlorid zu ergeben, das direkt in der nächsten Stufe verwendet wurde.
- C. 3-[(1-[3-(3-(2-Chinolinylmethyloxy)phenoxy)methylphenyl]-1-[N,N-dimethylcarbamylethylthio]methyl)thio]propionsäure
- Eine Mischung aus 3-(2-Chinolinylmethyloxy)phenol (0,01 Mol), dem zuvor erhaltenen Benzylchloridderivat von Stufe B (0,01 Mol) und K2CO3 (0,012 Mol) in 50 ml DMF wurde über Nacht auf 50°C erhitzt. Die Reaktionsmischung wurde in Wasser gegossen und mit Ethylacetat extrahiert. Nach Verdampfen des Lösungsmittels wurde das Rohprodukt durch Flash-Chromatographie gereinigt. Die gereinigte Verbindung wurde dann mit 1,1 Äquivalent NaOH-Lösung 2 Stunden lang hydrolysiert. Die Reaktionsmischung wurde dann in Ethylacetat angesäuert. Die organische Lösung wurde getrocknet und zur Trockne eingedampft. Reinigung durch Flash-Chromatographie ergab 3-[(1-[3-(3-(2-Chinolinylmethyloxy)phenoxy)benzyl]-1-[N,N-dimethylcarbamylethylthio]methyl)thio]propionsäure.
- Wenn N,N-Dimethyl-3-mercaptopropionamid in Stufe A durch die Verbindungen der Tabelle VI ersetzt wird, wird das entsprechende Produkt hergestellt.
- TABELLE VI
-
- N,N-Dimethyl-2-mercaptoacetamid
- N,N-Dimethyl-3-mercaptoacetamid
- N,N-Diethyl-3-mercaptoacetamid
- N,N-Dimethyl-4-mercaptobutyramid
- Wenn Thioessigsäure in Stufe A durch die Verbindung von Tabelle VII ersetzt wird, wird das entsprechende Produkt erhalten.
- TABELLE VII
-
- 3-Thiopropionsäure
- 4-Thiobuttersäure
- 3-Thiobuttersäure
- 2-Thiopropionsäure
- BEISPIEL 15
- Methyl-o-[4-(2-chinolinylmethyloxy)-2-(carbomethoxybutyl)phenoxymethyl]phenoxyacetat
- o-[4-(2-Chinolinylmethyloxy)-2-(carboxybutyl)phenoxymethyl]phenoxyessigsäure
- A. Ethyl-5-(2,5-dibenzyloxyphenyl)valerat
- Eine Lösung von 2,4-Dibenzyloxybenzaldehyd (0,1 Mol) in 30 ml DMF wurde tropfenweise zu einer Lösung von (Carbethoxypro pyl)triphenylphosphoran (0,11 Mol, hergestellt aus (Carbethoxypropyl)triphenylphosphoniumbromid und Natriumhydrid) in 100 ml DMF über einen Zeitraum von 20 Minuten gegeben. Die Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt und in Wasser gegossen und mit EtOAc extrahiert. Die organische Lösung wurde gut mit H2O gewaschen, getrocknet und zur Trockne eingedampft. Reinigung durch Flash-Chromatographie ergab Ethyl-2-(2,5-dibenzyloxyphenyl)valerat.
- B. 5-(2,5-Dihydroxyphenyl)valeriansäure
- Ethyl-5-(2,5-dibenzyloxyphenyl)valerat (0,05 Mol) in 200 ml EtOH wurde mit 2 g 5% Pd/C bei 50 psi 5 Stunden lang hydriert. Filtration und Verdampfen von Lösungsmittel ergab 5-(2,5-Dihydroxyphenyl)valeriansäure.
- C. Ethyl-5-[5-(2-chinolinylmethyloxy)-2-hydroxyphenyl]valerat
- Eine Mischung aus 2-Chlormethylchinolin (0,03 Mol), 0,03 Mol des in Stufe B erhaltenen Hydrochinolinderivats und K2CO3 (0,06 Mol) in 50 ml DMF wurden über Nacht auf 60°C erhitzt. Die Reaktionsmischung wurde in Wasser gegossen und mit EtOAc extrahiert. Die kombinierten organischen Extrakte wurden gut mit Wasser gewaschen, getrocknet und zur Trockne eingedampft. Das Rohprodukt wurde durch Flash-Chromatographie gereinigt, um Ethyl-5-[5-(2-chinolinylmethyloxy)-2-hydroxyphenyl]valerat zu ergeben.
- D. Methyl-o-[4-(2-chinolinylmethyloxy)-2-(carbomethoxybutyl)phenoxymethyl]phenoxyacetat
- o-[4-(2-Chinolinylmethyloxy)-2-(carboxybutyl)phenoxymethyl]phenoxyessigsäure
- Eine Mischung der in Stufe C erhaltenen phenolischen Verbindung (0,01 Mol), Methyl-2-(chlormethyl)phenoxyacetat (0,01 Mol) und K2CO3 (0,011 Mol) in 50 ml DMF wurde über Nacht auf 60°C erhitzt. Nach dem Aufarbeiten wurde das Rohprodukt durch Flash-Chromatographie gereinigt, um Methyl-o-[4-(2-chinolinylmethyloxy)-2-(carbomethoxybutyl)phenoxymethyl]phenoxyacetat zu ergeben. Der Diester wurde mit 1 N NaOH-Lösung in Dioxan hydrolysiert, auf pH 5 angesäuert und in EtOAc extrahiert, um o-[4-(2-Chinolinylmethyloxy)-2-(carboxybutyl)phenoxymethyl]phenoxyessigsäure zu ergeben.
- Die folgenden erfindungsgemäßen Verbindungen wurden gemäß den hier beschriebenen Verfahren unter Verwendung analoger Ausgangsmaterialien hergestellt, die kommerziell erhältlich sind oder nach Verfahren hergestellt werden, die literaturbekannt sind.
- 2-Hydroxy-3-[2-carboxyethylthio]-3-[2-{4-(2-chinolinylmethyloxy)phenoxymethyl}phenyl]propionsäure (Schmp. 87 bis 89°C)
- 3,4-Dihydroxyacetat-3'-(2-chinolinylmethyloxy)-(E)-stilbenmononatriumsalz (Schmelzpunkt > 180°C, Zersetzung))
-
- berechnet: C, 66,85; H, 4,45; N, 2,78; Na, 3,66
- gefunden: C, 60,73; H, 5,03; N, 2,37; Na, 3,23
- berechnet: C, 60,37; H, 5,10; N, 2,51; Na, 3,30 (als Trihydrat)
- 2-Phenyl-2-[6-chlor-2-[4-(2-chinolinylmethyloxy)phenoxymethyl]phenoxy]ethansäure (Schmelzpunkt 156 bis 157°C)
-
- berechnet: C, 70,79; H, 4,60; N, 2,66
- gefunden: C, 70,51; H, 4,79; N, 2,63
- 2-Phenyl-2-[4-chlor-2-[3-(2-chinolinylmethyloxy)phenoxymethyl]phenoxy]ethansäure (Schmelzpunkt 128 bis 130°C)
-
- berechnet: C, 70,79; H, 4,60; N, 2,66
- gefunden: C, 70,06; H, 4,70; N, 2,47
- berechnet: C, 70,19; H, 4,66; N, 2,64 (als Viertelhydrat)
- 4-Methyl-2-[4-chlor-2-[3-(2-chinolinylmethyloxy)phenoxymethyl]phenoxy]pentansäure (Schmelzpunkt 123 bis 125°C)
-
- berechnet: C, 68,84; H, 5,58; N, 2,77
- gefunden: C, 68,46; H, 5,55; N, 2,63
- 4-Methyl-2-[4-chlor-2-(4-(2-chinolinylmethyloxy)phenoxymethyl]phenoxy]pentansäure (Schmelzpunkt 143 bis 147°C)
-
- berechnet: C, 68,84; H, 5,58; N, 2,77
- gefunden: C, 68,43; H, 5,64; N, 2,52
- 2-Phenyl-2-[4-chlor-2-(4-(2-chinolinylmethyloxy)phenoxymethyl]phenoxy]ethansäure (Schmelzpunkt 173 bis 175°C)
-
- berechnet: C, 70,79; H, 4,60; N, 2,66
- gefunden: C, 68,72; H, 5,18; N, 2,34
- berechnet: C, 68,45; H, 4,82; N, 2,57 (als Hydrat)
- m-Fluor-p-[4-(2-chinolinylmethyloxy)phenylmethyloxy]phenylessigsäure-n-propylester (Schmelzpunkt 107°C)
-
- berechnet: C, 73,19; H, 5,70; N, 3,05
- gefunden: C, 72,13; H, 5,74; N, 2,90
- berechnet: C, 72,48; H, 5,76; N, 3,02 (als Viertelhydrat)
- p-(4-(2-Chinolinylmethyloxy)phenoxymethyl]phenoxy]acetamid (Schmelzpunkt 182 bis 184°C)
-
- berechnet: C, 72,45; H, 5,35; N, 6,76
- gefunden: C, 71,81; H, 5,41; N, 6,73
- berechnet: C, 71,67; H, 5,41; N, 6,69 (als Viertelhydrat)
- m-Fluor-p-[4-(2-chinolinylmethyloxy)phenylmethyloxy]phenylessigsäure (Schmelzpunkt 173 bis 174°C (Zersetzung))
-
- berechnet: C, 71,93; H, 4,83; N, 3,36
- gefunden: C, 72,15; H, 5,01; N, 3,28
- 4-[3-Fluor-4-(4-(2-chinolinylmethyloxy)benzyloxy]phenyl]-4-hydroxybutansäure-methylester (Schmelzpunkt 122 bis 123°C)
-
- berechnet: C, 70,72; H, 5,51; N, 2,95
- gefunden: C, 69,63; H, 5,51; N, 2,77
- berechnet: C, 69,41; H, 5,62; N, 2,89 (als Hemihydrat)
- α-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]glutarsäuredimethylester
-
- berechnet: C, 69,89; H, 5,67; N, 2,72
- gefunden: C, 68,68; H, 5,71; N, 2,86
- berechnet: C, 68,69; H, 5,76; N, 2,67 (als Hemihydrat)
- 2-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)phenyl]methylpentansäure (Schmelzpunkt 128 bis 132°C)
-
- berechnet: C, 76,46; H, 6,42; N, 3,07
- gefunden: C, 76,70; H, 6,61; N, 3,46
- 2-[6-Propyl-2-[3-(2-chinolinylmethyloxy)phenoxymethyl)phenoxy]propionsäure (Schmelzpunkt 150 bis 157°C)
-
- berechnet: C, 73,87; H, 6,20; N, 2,97
- gefunden: C, 72,10; H, 5,77; N, 2,81
- berechnet: C, 72,48; H, 6,29; N, 2,91 (als Hemihydrat)
- p-Chlor-o-[4-(2-chinolinylmethyloxy)phenoxymethyl)phenoxyessigsäure (Schmelzpunkt 177 bis 180°C)
-
- berechnet: C, 66,74; H, 4,48; N, 3,11
- gefunden: C, 66,67; H, 4,61; N, 2,94
- o-Chlor-o-[3-(2-chinolinylmethyloxy)phenoxymethyl)phenoxyessigsäure (Schmelzpunkt 179 bis 181°C)
-
- berechnet: C, 66,74; H, 4,48; N, 2,88
- gefunden: C, 67,36; H, 4,75; N, 2,78
- o-Chlor-o-[4-(2-chinolinylmethyloxy)phenoxymethyl)phenoxyessigsäure (Schmelzpunkt 188 bis 191°C)
-
- berechnet: C, 66,74; H, 4,48; N, 3,11
- gefunden: C, 66,88; H, 4,62; N, 2,99
- p-Chlor-o-[3-(2-chinolinylmethyloxy)phenoxymethyl)phenoxyessigsäure (Schmelzpunkt 189 bis 191°C)
-
- berechnet: C, 66,74; H, 4,48; N, 3,11
- gefunden: C, 66,51; H, 4,62; N, 2,97
- m-[2-(3-(2-Chinolinylmethyloxy)phenyl)-2-hydroxyethyl]phenoxyessigsäure-t-butylester (Schmelzpunkt 141 bis 142°C)
-
- berechnet: C, 74,21; H, 6,44; N, 2,88
- gefunden: C, 72,97; H, 6,26; N, 2,62
- berechnet: C, 73,12; H, 6,50; N, 2,84 (als Viertelhydrat)
- 3-[3-(2-Chinolinylmethyloxy)phenoxymethyl]phenylmethylthiobernsteinsäure (Schmelzpunkt 198 bis 200°C (Zersetzung))
-
- berechnet: C, 66,78; H, 5,00; N, 2,78; S, 6,37
- gefunden: C, 62,86; H, 4,61; N, 2,50; S, 6,52
- berechnet: C, 62,85; H, 5,37; N, 2,62; S, 5,99 (als 1,75-Hydrat)
- 4-Carboxy-4-[2-(4-(2-chinolinylmethyloxy)phenoxymethyl)phenoxy]buttersäure-dimethylamid (Schmelzpunkt 91 bis 92°C)
-
- berechnet: C, 70,02; H, 5,88; N, 5,44
- gefunden: C, 61,70; H, 6,02; N, 4,72
- berechnet: C, 61,90; H, 6,49; N, 4,81 (als 3,75-Hydrat)
- 1,4-Dihydroxy-1-[3-fluor-4-(3-(2-chinolinylmethyloxy)benzoxy)phenyl]butan (Schmelzpunkt 93 bis 94°C)
-
- berechnet: C, 72,47; H, 5,86; N, 3,13
- gefunden: C, 71,63; H, 5,87; N, 3,18
- berechnet: C, 71,60; H, 5,92; N, 3,09 (als 0,3-Hydrat)
- 4-Methyl-2-[2-(3-(2-chinolinylmethyloxy)phenoxymethyl)phenoxy]pentansäure (Schmelzpunkt 76 bis 87°C)
-
- berechnet: C, 73,87; H, 6,20; N, 2,97
- gefunden: C, 73,67; H, 6,24; N, 2,88
- 2-[2-(4-(2-Chinolinylmethyloxy)phenoxymethyl)phenoxy]-2-phenylethansäure (Schmelzpunkt 206 bis 210°C)
-
- berechnet: C, 75,75; H, 5,13; N, 2,85
- gefunden: C, 74,55; H, 5,74; N, 3,16
- berechnet: C, 74,39; H, 5,24; N, 2,80 (als Hemihydrat)
- 2-[2-(3-(2-Chinolinylmethyloxy)phenoxymethyl)-6-chlorphenoxy]-2-phenylethansäure (Schmelzpunkt 180 bis 181°C)
-
- berechnet: C, 70,79; H, 4,60; N, 2,66
- gefunden: C, 70,72; H, 4,71; N, 2,58
- 2-[2-Chlor-6-(4-(2-chinolinylmethoxy)phenoxymethyl)phenoxy]propionsäure (Schmelzpunkt 149 bis 152°C)
-
- berechnet: C, 67,32; H, 4,78; N, 3,02
- gefunden: C, 66,89; H, 4,94; N, 2,96
- berechnet: C, 66,66; H, 4,84; N, 2,99 (als Viertelhydrat)
- 2-[2-Chlor-6-(3-(2-chinolinylmethoxy)phenoxymethyl)phenoxy]propionsäure (Schmelzpunkt 156 bis 159°C)
-
- berechnet: C, 67,32; H, 4,78; N, 3,02
- gefunden: C, 66,67; H, 4,88; N, 2,89
- berechnet: C, 66,66; H, 4,84; N, 2,99 (als Viertelhydrat)
- p-Carboxymethoxy-o-[3-(2-chinolinylmethyloxy)phenoxymethyl]phenoxyessigsäure (Schmelzpunkt 195 bis 198°C)
-
- berechnet: C, 66,25; H, 4,74; N, 2,86
- gefunden: C, 65,18; H, 5,07; N, 2,67
- berechnet: C, 65,64; H, 4,79; N, 2,83 (als Viertelhydrat)
- 2-[2-Chlor-6-(4-(2-(8-fluorchinolinyl)methyloxy)phenoxymethyl)phenoxy]pentansäure (Schmelzpunkt 173 bis 177°C)
- 5-[2-(4-(2-(8-Fluorchinolinyl)methyloxy)phenoxymethyl)phenylmethyl]tetrazol (Schmelzpunkt 150 bis 153°C)
- 2-[2-(3-(2-Chinolinylmethyloxy)phenoxymethyl)benzyl]pentansäure (Schmelzpunkt 78 bis 81°C)
- 2-[2-(3-(2-(8-Fluorchinolinyl)methyloxy)phenoxymethyl)benzyl]pentansäure
- 2-[3-(3-(2-Chinolinylmethyloxy)phenoxymethyl]benzyl]propansäure
Claims (11)
- Verbindung mit der allgemeinen Formel I:in der A O oder S ist; B eine chemische Bindung oder
 ist; D O, S, NR1,
ist; D O, S, NR1, oder eine chemische Bindung ist; E eine chemische Bindung oder
oder eine chemische Bindung ist; E eine chemische Bindung oder ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamido ist vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamido ist vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
- Verbindung mit der allgemeinen Formel I:in der A O oder S ist; B eine chemische Bindung, O, S, SO, SO2, NR1,
 ist; D O, S, NR1,
ist; D O, S, NR1, oder eine chemische Bindung ist; E eine chemische Bindung oder
oder eine chemische Bindung ist; E eine chemische Bindung oder ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; mindestens ein R'' -CH2R oder -CH2-O-(CH2)x-X oder R ist, und das andere unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamid ist; vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; mindestens ein R'' -CH2R oder -CH2-O-(CH2)x-X oder R ist, und das andere unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamid ist; vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
- Verbindung mit der allgemeinen Formel I:in der A O oder S ist; B eine chemische Bindung, O, S, SO, SO2, NR1,
 ist; D O, S, NR1,
ist; D O, S, NR1, oder eine chemische Bindung ist; E eine chemische Bindung oder
oder eine chemische Bindung ist; E eine chemische Bindung oder ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamido ist; vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X Wasserstoff, Alkyl, Alkenyl, Cycloalkyl, Arylalkyl, Hydroxy, Alkoxy, Aralkoxy, Amino, Mono- und Dialkylamino, Aralkylamino, Acylamino, -CONR1R1, -COOR1, CN, Tetrazolyl oder Acylsulfonamido ist; vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.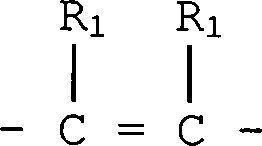
- Verbindung mit der allgemeinen Formel I:in der A O oder S ist; B eine chemische Bindung, O, S, SO, SO2, NR1,
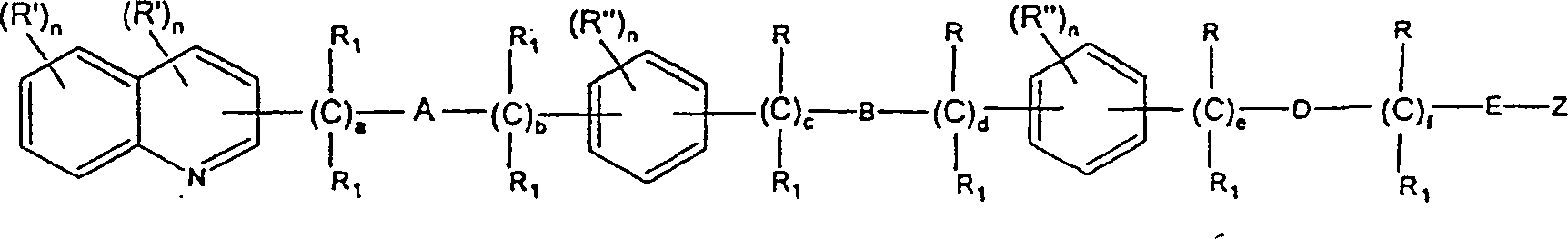 ist; D O, S, NR1,
ist; D O, S, NR1, oder eine chemische Bindung ist; E eine chemische Bindung oder
oder eine chemische Bindung ist; E eine chemische Bindung oder ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X CN ist; vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
ist; a 0 bis 2 ist; b 0 bis 1 ist; c 0 bis 4 ist; d 0 bis 5 ist; e 0 bis 4 ist; f 0 bis 5 ist; n 0 bis 2 ist; jedes R' unabhängig Wasserstoff, Alkyl, Hydroxy, Alkoxy, Carboxy, Carbalkoxy, Halogen, Nitro, Halogenalkyl, Cyano oder Acyl ist; jedes R'' unabhängig Wasserstoff, Hydroxy, Alkoxy, Halogen, Halogenalkyl, -CH2R oder -CH2-O-(CH2)x-X oder R ist; jedes R1 unabhängig Wasserstoff, Alkyl oder Aralkyl ist; jedes R unabhängig -(CH2)x-X; -O-(CH2)x-X; (wenn nicht geminal zu B oder D, wenn B oder D O ist) -S-(CH2)x-X oder -NR1-(CH2)x-X ist; wobei x 0 bis 3 ist, und X CN ist; vicinale R-Gruppen zusammen (CH2)y sein können, wobei y 1 bis 4 ist, wodurch ein 3- bis 6-gliedriger Ring gebildet wird; geminale R1- und R-Gruppen zusammen einen Spirosubstituenten -(CH2)z- bilden können, wobei z 2 bis 5 ist; geminale R1- oder R1- und R-Gruppen zusammen einen Alkylidenylsubstituenten =CHR1 bilden können; Z -CN oder Tetrazolyl oder substituiertes Tetrazolyl ist, wobei der Substituent Alkyl, Carboxyalkyl oder Carbalkoxyalkyl sein kann; und pharmazeutisch akzeptable Salze davon.
- Verbindung nach einem der Ansprüche 1 oder 2, bei der einer von R -(CH2)x-X; -S-(CH2)x-X oder NR1-(CH2)x-X ist und/oder einer von R'' -CH2R; -CH2-O-(CH2)x-X oder R ist und X -CONR1R1, -COOR1, -CN, Tetrazolyl oder Acylsulfonamido ist.
- Verbindung nach einem der Ansprüche 2 bis 4 oder Anspruch 5 in Abhängigkeit von Anspruch 2, bei der B eine chemische Bindung, O, S, oderist; und Z Tetrazolyl ist.

- Verfahren zur Herstellung einer Verbindung gemäß einem der vorhergehenden Ansprüche, das eine oder mehrere der folgenden Reaktionen umfasst, nämlich;
 worin R, R', R'', a, b, c, d, e, f, n, A und D wie in irgendeinem der Ansprüche 1 bis 4 definiert sind, B O oder S ist; E eine chemische Bindung ist; Z -COOR1, -CN,
worin R, R', R'', a, b, c, d, e, f, n, A und D wie in irgendeinem der Ansprüche 1 bis 4 definiert sind, B O oder S ist; E eine chemische Bindung ist; Z -COOR1, -CN, oder Tetrazolyl ist; und L eine Abgangsgruppe ist, wie Halogen, Tosylat oder Mesylat.
oder Tetrazolyl ist; und L eine Abgangsgruppe ist, wie Halogen, Tosylat oder Mesylat.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 6 oder Produkt des Verfahrens nach Anspruch 7 zur Herstellung eines Medikaments.
- Verwendung nach Anspruch 8, bei der das Medikament zur Verwendung als Lipoxygenaseinhibitor und/oder Leukotrienantagonist ist.
- Verwendung nach Anspruch 8, bei der das Medikament zur Verwendung zur Behandlung oder Vorbeugung von Entzündung und/oder allergischen Zuständen ist.
- Pharmazeutische Zusammensetzung, die eine Verbindung nach einem der Ansprüche 1 bis 6 oder das Produkt des Verfahrens nach Anspruch 7 zusammen mit einem pharmazeutisch akzeptablen Träger dafür umfasst.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US209428 | 1988-06-21 | ||
| US07/209,428 US4920131A (en) | 1987-11-03 | 1988-06-21 | Quinoline derivatives and use thereof as antagonists of leukotriene D4 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE68929520D1 DE68929520D1 (de) | 2004-10-07 |
| DE68929520T2 true DE68929520T2 (de) | 2005-09-29 |
Family
ID=22778718
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE68928992T Expired - Lifetime DE68928992T2 (de) | 1988-06-21 | 1989-06-20 | Chinolin-Derivate als Leukotrien-D4-Antagonisten, diese enthaltende Zusammensetzungen und Verfahren zu deren Herstellung |
| DE68929520T Expired - Lifetime DE68929520T2 (de) | 1988-06-21 | 1989-06-20 | Chinolin-Derivate als Leukotrien-D4-Antagonisten, diese enthaltende Zusammensetzungen und Verfahren zu deren Herstellung |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE68928992T Expired - Lifetime DE68928992T2 (de) | 1988-06-21 | 1989-06-20 | Chinolin-Derivate als Leukotrien-D4-Antagonisten, diese enthaltende Zusammensetzungen und Verfahren zu deren Herstellung |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US4920131A (de) |
| EP (2) | EP0784052B1 (de) |
| DE (2) | DE68928992T2 (de) |
| ES (2) | ES2223071T3 (de) |
| WO (1) | WO1989012628A1 (de) |
Families Citing this family (94)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6124343A (en) * | 1919-01-27 | 2000-09-26 | Rhone-Poulenc Rorer Limited | Substituted phenyl compounds with a substituent having a thienyl ring |
| US5017583A (en) * | 1983-04-21 | 1991-05-21 | Merck Frosst Canada, Inc. | Leukotriene antagonists |
| US4920132A (en) * | 1987-11-03 | 1990-04-24 | Rorer Pharmaceutical Corp. | Quinoline derivatives and use thereof as antagonists of leukotriene D4 |
| US4920130A (en) * | 1987-11-02 | 1990-04-24 | Rorer Pharamceutical Corp. | Quinoline derivatives and use thereof as antagonists of leukotriene D4 |
| US5051427A (en) * | 1987-11-03 | 1991-09-24 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Quinoline derivatives as antagonists of leukotriene D4 |
| US5204358A (en) * | 1987-11-25 | 1993-04-20 | Merck Frosst Canada, Inc. | Hetaryl styryl quinolines as leukotriene inhibitors |
| US5104882A (en) * | 1987-11-25 | 1992-04-14 | Merck Frosst Canada, Inc. | Diarylstrylquinoline diacids and pharmaceutical compositions thereof |
| US5232916A (en) * | 1988-06-27 | 1993-08-03 | Merck Frosst Canada, Inc. | Quinoline ether alkanoic acids |
| GB8927287D0 (en) * | 1988-12-23 | 1990-01-31 | Ici Plc | Cyclic ether derivatives |
| NZ231735A (en) * | 1988-12-23 | 1992-04-28 | Ici Plc | Alcohol/ether derivatives, preparation and pharmaceutical compositions thereof |
| IL92620A0 (en) * | 1988-12-23 | 1990-08-31 | Ici Pharma | Cycloalkane derivatives |
| US5214069A (en) * | 1988-12-23 | 1993-05-25 | Imperial Chemical Industries Plc | Alcohol and ether derivatives |
| GB8926981D0 (en) * | 1988-12-23 | 1990-01-17 | Ici Plc | Heterocyclic derivatives |
| US5219881A (en) * | 1988-12-23 | 1993-06-15 | Imperial Chemical Industries Plc | Cyclic ether derivatives |
| US5202326A (en) * | 1989-02-28 | 1993-04-13 | Imperial Chemical Industries Plc | Heterocyclic ethers |
| US5196419A (en) * | 1989-02-28 | 1993-03-23 | Imperial Chemical Industries Plc | Heterocyclic cyclic ethers |
| US5134148A (en) * | 1989-02-28 | 1992-07-28 | Imperial Chemical Industries Plc | Heterocycles for use as inhibitors of leukotrienes |
| US5236919A (en) * | 1989-02-28 | 1993-08-17 | Imperial Chemical Industries Plc | Quinoxalinyl derivatives suitable for use in leukotriene mediated disease |
| US5217977A (en) * | 1989-02-28 | 1993-06-08 | Imperial Chemical Industries Plc | Heterocyclic cycloalkanes |
| US4977162A (en) * | 1989-07-13 | 1990-12-11 | Rorer Pharmaceutical Corporation | Quinolinyl-chromone derivatives and use for treatment of hypersensitive ailments |
| IE902113A1 (en) * | 1989-07-18 | 1991-06-19 | Ici Plc | Diaryl ether cyclic ethers |
| US5214070A (en) * | 1989-07-18 | 1993-05-25 | Imperial Chemical Industries Plc | Diaryl ether cycloalkanes |
| IE64358B1 (en) * | 1989-07-18 | 1995-07-26 | Ici Plc | Diaryl ether heterocycles |
| DE69009887T2 (de) * | 1989-07-26 | 1994-09-22 | Ici Pharma | Bizyklische Derivate. |
| HUT54991A (en) * | 1989-08-11 | 1991-04-29 | Ici Plc | Process for producing quinoline derivatives and pharmaceutical compositions comprising such compounds |
| GB8919504D0 (en) * | 1989-08-29 | 1989-10-11 | Leo Pharm Prod Ltd | Chemical compounds |
| GB9018134D0 (en) * | 1989-09-29 | 1990-10-03 | Ici Plc | Heterocyclic derivatives |
| GB9010394D0 (en) * | 1990-05-09 | 1990-06-27 | Ici Plc | Heterocyclic compounds |
| US5041453A (en) * | 1990-05-30 | 1991-08-20 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Quinolinyl-benzoheterobicyclic derivatives as antagonists of leukotriene D4 |
| ES2083526T3 (es) * | 1990-06-21 | 1996-04-16 | Zeneca Ltd | Derivados piranicos biciclicos y su uso como inhibidores de 5-lipoxigenasa. |
| US5254581A (en) * | 1990-06-21 | 1993-10-19 | Imperial Chemical Industries Plc | Pyran derivatives and their use as inhibitors of 5-lipoxygenase |
| IE911853A1 (en) * | 1990-06-21 | 1992-01-01 | Ici Plc | Heterocyclene derivatives |
| IE911919A1 (en) * | 1990-06-21 | 1992-01-01 | Zeneca Ltd | Bicyclic heterocyclic compounds |
| GB9113137D0 (en) * | 1990-07-13 | 1991-08-07 | Ici Plc | Thioxo heterocycles |
| KR930702304A (ko) * | 1990-09-13 | 1993-09-08 | 스튜어트 알. 슈터 | 피리딜티오 또는 피리딜옥시 알칸산 |
| CA2053216C (en) * | 1990-10-12 | 2003-04-08 | Michel L. Belley | Saturated hydroxyalkylquinoline acids as leukotriene antagonists |
| US5266568A (en) * | 1990-10-12 | 1993-11-30 | Merck Frosst Canada, Inc. | Hydroxyalkylquinoline ether acids as leukotriene antagonists |
| US5179105A (en) * | 1990-10-16 | 1993-01-12 | Terumo Kabushiki Kaisha | Phenoxyacetic acid compounds, method for production thereof, and pharmaceutical preparations containing same |
| GB9025123D0 (en) * | 1990-11-19 | 1991-01-02 | Ici Plc | Nitrogen compounds |
| US5272173A (en) * | 1990-11-28 | 1993-12-21 | Imperial Chemical Industries Plc | 5-lipoxygenase inhibitors |
| IE913866A1 (en) * | 1990-11-28 | 1992-06-03 | Ici Plc | Aryl derivatives |
| IE914005A1 (en) * | 1990-12-14 | 1992-06-17 | Zeneca Ltd | Novel intermediates |
| CA2058254A1 (en) * | 1991-01-15 | 1992-07-16 | John Francis Kingston | Benzodioxole derivatives |
| AU645159B2 (en) * | 1991-01-17 | 1994-01-06 | Ici Pharma | Sulphonamide derivatives |
| US5258399A (en) * | 1991-01-17 | 1993-11-02 | Imperial Chemical Industries Plc | Sulphonamide derivatives |
| US6004979A (en) * | 1991-02-07 | 1999-12-21 | Hoechst Marion Roussel | Nitrogenous bicycles |
| MX9200299A (es) * | 1991-02-07 | 1992-12-01 | Roussel Uclaf | Nuevos derivados biciclicos nitrogenados, su procedimiento de preparacion los nuevos compuestos intermedios obtenidos su aplicacion como medicamentos y las composiciones farmaceuticas que los contienen. |
| IL100555A (en) * | 1991-02-07 | 2000-08-31 | Hoechst Marion Roussel Inc | N-substituted quinoline derivatives their preparation their use for the preparation of medicaments and the pharmaceutical compositions containing them |
| GB9102804D0 (en) * | 1991-02-11 | 1991-03-27 | Ici Plc | Heterocyclic derivatives |
| EP0505122A1 (de) * | 1991-03-21 | 1992-09-23 | Zeneca Limited | Alpha, Alpha-Dialkylbenzylderivate |
| US5157040A (en) * | 1991-04-05 | 1992-10-20 | Merck & Co., Inc. | Substituted quinolines as angiotensin ii antagonists |
| IL101860A0 (en) * | 1991-05-31 | 1992-12-30 | Ici Plc | Heterocyclic derivatives |
| GB9113628D0 (en) * | 1991-06-25 | 1991-08-14 | Ici Plc | Heterocyclic derivatives |
| GB9113626D0 (en) * | 1991-06-25 | 1991-08-14 | Ici Plc | Heterocyclic compounds |
| GB9121727D0 (en) * | 1991-10-14 | 1991-11-27 | Ici Plc | Heterocyclic compounds |
| US6117885A (en) * | 1992-02-14 | 2000-09-12 | E. R. Squibb & Sons, Inc. | Biphenyl-substituted quinoline derivatives |
| US5438141A (en) * | 1993-05-21 | 1995-08-01 | Merck Frosst Canada, Inc. | Heteroaryl and haloaryl quinoline derivatives of cyclopropaneacetic acid as leukotriene antagonists |
| ES2103181B1 (es) * | 1994-08-01 | 1998-04-01 | Menarini Lab | Amidas naftalenicas con accion antagonista de los leucotrienos. |
| ES2103180B1 (es) * | 1994-08-01 | 1998-04-01 | Menarini Lab | Fenilacetamidas con accion antagonista de los leucotrienos. |
| US5542795A (en) * | 1995-01-30 | 1996-08-06 | Kennametal Inc. | Plunge and face milling cutter with universal insert seats |
| EP0725063A1 (de) * | 1995-02-01 | 1996-08-07 | Ciba-Geigy Ag | Chinolin-Derivate |
| US5750539A (en) * | 1995-06-07 | 1998-05-12 | Merck Frosst Canada | Heteroaryl diol acids as leukotriene antagonists |
| US6388076B1 (en) | 1995-06-19 | 2002-05-14 | Ontogen Corporation | Protein tyrosine phosphatase-inhibiting compounds |
| US5770620A (en) * | 1995-06-19 | 1998-06-23 | Ontogen Corporation | Aryl acrylic acid derivatives useful as protein tyrosine phosphatase inhibitors |
| ES2117551B1 (es) * | 1995-12-29 | 1999-04-01 | Menarini Lab | Quinolinas naftalenicas con accion antagonista de los leucotrienos, procedimiento para su preparacion y utilizacion de las mismas. |
| US5783586A (en) * | 1996-10-01 | 1998-07-21 | Abbott Laboratories | Heteroarylmethoxyphenylthioalkyl carboxylates as inhibitors of leukotriene biosynthesis |
| EP1177176B1 (de) * | 1999-04-28 | 2006-04-19 | Aventis Pharma Deutschland GmbH | Tri-aryl-säurederivate als ppar rezeptor liganden |
| KR20080042170A (ko) * | 2005-09-07 | 2008-05-14 | 플렉시콘, 인코퍼레이티드 | Ppar 활성 화합물 |
| WO2012004743A1 (en) | 2010-07-09 | 2012-01-12 | Pfizer Limited | Benzenesulfonamides useful as sodium channel inhibitors |
| EP2593433B1 (de) | 2010-07-12 | 2014-11-26 | Pfizer Limited | N-sulfonylbenzamide als inhibitoren von spannungsabhängigen natriumkanälen |
| JP2013532184A (ja) | 2010-07-12 | 2013-08-15 | ファイザー・リミテッド | 電位開口型ナトリウムチャネル阻害剤として有用なn−スルホニルベンズアミド誘導体 |
| US9102621B2 (en) | 2010-07-12 | 2015-08-11 | Pfizer Limited | Acyl sulfonamide compounds |
| ES2532357T3 (es) | 2010-07-12 | 2015-03-26 | Pfizer Limited | Derivados de sulfonamida como inhibidores de Nav1.7 para el tratamiento del dolor |
| JP2013532185A (ja) | 2010-07-12 | 2013-08-15 | ファイザー・リミテッド | 化合物 |
| ES2643403T3 (es) | 2011-12-28 | 2017-11-22 | Global Blood Therapeutics, Inc. | Compuestos de benzaldehído sustituidos y métodos para su uso en el aumento de la oxigenación tisular |
| HK1203412A1 (en) | 2011-12-28 | 2015-10-30 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| WO2014150258A1 (en) | 2013-03-15 | 2014-09-25 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9422279B2 (en) | 2013-03-15 | 2016-08-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| CN105073728A (zh) | 2013-03-15 | 2015-11-18 | 全球血液疗法股份有限公司 | 化合物及其用于调节血红蛋白的用途 |
| US10266551B2 (en) | 2013-03-15 | 2019-04-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9802900B2 (en) | 2013-03-15 | 2017-10-31 | Global Blood Therapeutics, Inc. | Bicyclic heteroaryl compounds and uses thereof for the modulation of hemoglobin |
| US9458139B2 (en) | 2013-03-15 | 2016-10-04 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| MX379235B (es) | 2013-03-15 | 2025-03-11 | Global Blood Therapeutics Inc | Compuestos y sus usos para modular la hemoglobina. |
| US8952171B2 (en) | 2013-03-15 | 2015-02-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10100043B2 (en) | 2013-03-15 | 2018-10-16 | Global Blood Therapeutics, Inc. | Substituted aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US20140274961A1 (en) | 2013-03-15 | 2014-09-18 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9604999B2 (en) | 2013-03-15 | 2017-03-28 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| EA201992707A1 (ru) | 2013-11-18 | 2020-06-30 | Глобал Блад Терапьютикс, Инк. | Соединения и их применения для модуляции гемоглобина |
| CN114181195A (zh) | 2014-02-07 | 2022-03-15 | 全球血液疗法股份有限公司 | 一种化合物的结晶多晶型物 |
| MA41841A (fr) | 2015-03-30 | 2018-02-06 | Global Blood Therapeutics Inc | Composés aldéhyde pour le traitement de la fibrose pulmonaire, de l'hypoxie, et de maladies auto-immunes et des tissus conjonctifs |
| SG11201804647TA (en) | 2015-12-04 | 2018-06-28 | Global Blood Therapeutics Inc | Dosing regimens for 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| TWI663160B (zh) | 2016-05-12 | 2019-06-21 | 全球血液治療公司 | 用於合成2-羥基-6-((2-(1-異丙基-1h-吡唑-5-基)-吡啶-3-基)甲氧基)苯甲醛之方法 |
| TW202332423A (zh) | 2016-10-12 | 2023-08-16 | 美商全球血液治療公司 | 包含2-羥基-6-((2-(1-異丙基-1h-吡唑-5-基)吡啶-3-基)甲氧基)-苯甲醛之片劑 |
| US11014884B2 (en) | 2018-10-01 | 2021-05-25 | Global Blood Therapeutics, Inc. | Modulators of hemoglobin |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4282230A (en) * | 1979-11-15 | 1981-08-04 | E. R. Squibb & Sons, Inc. | Imidazolylethoxy derivatives of quinoline-2- or 4-methanols, antimicrobial compositions containing them and method for treating bacterial or fungal infections with them |
| US4407803A (en) * | 1981-08-17 | 1983-10-04 | Abbott Laboratories | Antiinflammatory 1-(quinolinyl)-2-pyrazoline derivatives |
| US4839369A (en) * | 1985-04-16 | 1989-06-13 | Rorer Pharmaceutical Corporation | Aryl and heteroaryl ethers as agents for the treatment of hypersensitive ailments |
| IE861607L (en) * | 1985-06-18 | 1986-12-18 | Bunce Roger A | 2-substituted quinolines |
| US4661499A (en) * | 1985-06-18 | 1987-04-28 | Merck Frosst Canada, Inc. | 2-[(substituted)-phenoxymethyl]quinolines |
| US4732978A (en) * | 1985-10-03 | 1988-03-22 | American Home Products Corporation | Novel substituted naphthyloxymethyl quinoline derivatives as anti-inflammatory and antiallergy agents |
| EP0219308B1 (de) * | 1985-10-16 | 1991-10-23 | Merck Frosst Canada Inc. | 2-Substituierte Chinoline |
| IE59889B1 (en) * | 1986-02-14 | 1994-04-20 | Merck Frosst Canada Inc | 2-substituted quinoline dioic acids |
| DE3783105T2 (de) * | 1986-03-13 | 1993-04-22 | Rhone Poulenc Rorer Int | Chinolinylaether tetrazole als mittel zur behandlung von hyperempfindlichen krankheiten. |
| US4769461A (en) * | 1986-09-16 | 1988-09-06 | American Home Products Corporation | Quinolinyl benzene hydroxamic acids as anti-inflammatory/antiallergic agents |
| US4920132A (en) * | 1987-11-03 | 1990-04-24 | Rorer Pharmaceutical Corp. | Quinoline derivatives and use thereof as antagonists of leukotriene D4 |
-
1988
- 1988-06-21 US US07/209,428 patent/US4920131A/en not_active Expired - Lifetime
-
1989
- 1989-06-20 ES ES97200638T patent/ES2223071T3/es not_active Expired - Lifetime
- 1989-06-20 DE DE68928992T patent/DE68928992T2/de not_active Expired - Lifetime
- 1989-06-20 ES ES89306232T patent/ES2134755T3/es not_active Expired - Lifetime
- 1989-06-20 DE DE68929520T patent/DE68929520T2/de not_active Expired - Lifetime
- 1989-06-20 EP EP97200638A patent/EP0784052B1/de not_active Expired - Lifetime
- 1989-06-20 EP EP89306232A patent/EP0348155B1/de not_active Expired - Lifetime
- 1989-06-20 WO PCT/US1989/002691 patent/WO1989012628A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| ES2223071T3 (es) | 2005-02-16 |
| DE68928992D1 (de) | 1999-06-17 |
| EP0784052A1 (de) | 1997-07-16 |
| EP0784052B1 (de) | 2004-09-01 |
| EP0348155A1 (de) | 1989-12-27 |
| ES2134755T3 (es) | 1999-10-16 |
| EP0348155B1 (de) | 1999-05-12 |
| US4920131A (en) | 1990-04-24 |
| DE68929520D1 (de) | 2004-10-07 |
| DE68928992T2 (de) | 2000-01-27 |
| WO1989012628A1 (en) | 1989-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE68929520T2 (de) | Chinolin-Derivate als Leukotrien-D4-Antagonisten, diese enthaltende Zusammensetzungen und Verfahren zu deren Herstellung | |
| DE3855501T2 (de) | Chinolin-derivate als antagonisten von leukotrien d 4? | |
| DE3854890T2 (de) | Chinolinderivate, ihre Verwendung zur Behandlung der überempfindlichen Krankheit und eine pharmazeutische Zusammensetzung davon | |
| DE69118388T2 (de) | Substituierte bizyklische arylderivate mit selektiver leukotrien b4 antagonistischer wirkung | |
| EP0041673B1 (de) | Imidazolderivate, deren Herstellung sowie diese enthaltende Präparate | |
| DE69110254T2 (de) | Hydroxychinolonderivate. | |
| EP0344519A1 (de) | Substituierte 4-(Chinolin-2-yl-methoxy)phenyl-essigsäure-Derivate | |
| EP0499926B1 (de) | 2-Substituierte Chinoline, Verfahren zu ihrer Herstellung sowie ihre Verwendung in Arzneimitteln | |
| DE69112062T2 (de) | Substituierte monocyclische anylderivate mit selektiver leukotrien b4 antagonistischer wirkung. | |
| DE3783105T2 (de) | Chinolinylaether tetrazole als mittel zur behandlung von hyperempfindlichen krankheiten. | |
| DE60008921T2 (de) | Kristalline form von (s)-2-ethoxy-3-4-(2-4-methansulfonyloxyphenyl-ethoxy)phenyl]propansäure | |
| DE3617183A1 (de) | Substituierte benzylether | |
| DE69112074T2 (de) | Disubstituierte arylverbindungen welche eine selektive leukotrien-b4 antagonistische aktivität aufweisen. | |
| DE3927931A1 (de) | Disubstituierte (chinolin-2-yl-methoxy)phenylessigsaeure-derivate | |
| EP0161599A2 (de) | Neue Benzazepinderivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung | |
| EP1268422A1 (de) | Indole zur behandlung von krankheiten die mit schilddrüsenhormonen behandeln werden können | |
| DE3927930A1 (de) | Cyclisch substituierte (chinolin-2-yl-methoxy)phenyl-essig-saeure-derivate | |
| EP0071059B1 (de) | 6-(5-(Omega-(1-Imidazolyl)-alkyl)-thien-2-yl)-3-oxo-2,3,4,5-tetrahydropyridazine und deren Säureadditionssalze, Verfahren zu ihrer Herstellung und diese enthaltende pharmazeutische Präparate | |
| EP0008645A1 (de) | Alkoxyphenylpyrrolidone, Verfahren zu ihrer Herstellung und Arzneimittel auf Basis dieser Verbindungen | |
| EP0549879A2 (de) | Thiazolyl-substituierte Chinolylmethoxyphenylessigsäurederivate | |
| EP0072960B1 (de) | 1,5-Diphenylpyrazolin-3-on-Verbindungen sowie Verfahren und Zwischenprodukte zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| DE60129445T2 (de) | Quinolin-derivate als antientzündungsmittel | |
| EP0582908B1 (de) | 2-Substituierte Chinolylmethoxy-phenylessigsäure- und -benzylacyl Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Leukotriensynthese Hemmer | |
| DE2819873C2 (de) | ||
| DE69007572T2 (de) | Chalkonderivate. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |