ES2749978T3 - Anticuerpos anti-C5 que tienen farmacocinética mejorada - Google Patents
Anticuerpos anti-C5 que tienen farmacocinética mejorada Download PDFInfo
- Publication number
- ES2749978T3 ES2749978T3 ES16177656T ES16177656T ES2749978T3 ES 2749978 T3 ES2749978 T3 ES 2749978T3 ES 16177656 T ES16177656 T ES 16177656T ES 16177656 T ES16177656 T ES 16177656T ES 2749978 T3 ES2749978 T3 ES 2749978T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- seq
- amino acid
- acid sequence
- antigen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/02—Antidotes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/10—Immunoglobulins specific features characterized by their source of isolation or production
- C07K2317/14—Specific host cells or culture conditions, e.g. components, pH or temperature
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/35—Valency
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/526—CH3 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/64—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising a combination of variable region and constant region components
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/72—Increased effector function due to an Fc-modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Cardiology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Urology & Nephrology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pulmonology (AREA)
- Obesity (AREA)
- Dermatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Hospice & Palliative Care (AREA)
- Pain & Pain Management (AREA)
- Reproductive Health (AREA)
Abstract
Un anticuerpo aislado, o fragmento de unión a antígeno del mismo, que: (a) se une al componente C5 del complemento humano; (b) inhibe la escisión de C5 en fragmentos C5a y C5b; y (c) comprende: (i) una CDR1 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 23, (ii) una CDR2 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 19, (iii) una CDR3 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 3, (iv) una CDR1 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 4, (v) una CDR2 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 5, (vi) una CDR3 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 6 y (vii) la región constante de cadena pesada representada en SEQ ID NO: 13.
Description
DESCRIPCIÓN
Anticuerpos anti-C5 que tienen farmacocinética mejorada
Campo técnico
El campo de la invención es la medicina, la inmunología, la biología molecular y la química de proteínas.
Antecedentes
El sistema del complemento actúa junto con otros sistemas inmunológicos del organismo para defenderse contra la intrusión de patógenos celulares y virales. Existen al menos 25 proteínas del complemento, que se encuentran en forma de una colección compleja de proteínas del plasma y cofactores de membrana. Las proteínas del plasma componen aproximadamente 10% de las globulinas en el suero de los vertebrados. Los componentes del complemento logran sus funciones defensivas inmunitarias mediante la interacción en una serie de eventos de escisión enzimática y unión a membrana intrincados pero precisos. La cascada del complemento resultante conduce a la producción de productos con funciones opsónicas, inmunorreguladoras, y líticas. Se proporciona un resumen conciso de las actividades biológicas asociadas con la activación del complemento, por ejemplo, en The Merck Manual, 16a Edición.
La cascada del complemento puede progresar a través de la vía clásica (CP), la vía de la lectina, o la vía alternativa (AP). La vía de las lectinas se inicia típicamente con la unión de la lectina de unión a manosa (MBL) a sustratos de alto contenido de manosa. La AP puede ser un anticuerpo independiente, y puede ser iniciada por ciertas moléculas sobre superficies de patógenos. La CP se inicia típicamente por reconocimiento de anticuerpos de, y la unión a, un sitio antigénico sobre una célula diana. Estas vías convergen en la C3 convertasa - el punto en el que el componente C3 del complemento es escindido por una proteasa activa para producir C3a y C3b.
La AP C3 convertasa es iniciada por la hidrólisis espontánea del componente C3 del complemento, que es abundante en la fracción de plasma de la sangre. Este proceso, también conocido como "activación a ritmo lento" ("tickover"), se produce a través de la escisión espontánea de un enlace tioéster en C3 para formar C3i o C3(H2O). El ritmo lento se ve facilitado por la presencia de superficies que soportan la unión de C3 activado y/o que tienen características de carga neutra o positiva (p. ej., superficies de células bacterianas). Esta formación de C3(H2O) permite la unión de la proteína del plasma Factor B, que a su vez permite que el Factor D escinda el Factor B a Ba y Bb. El fragmento Bb permanece unido a C3 para formar un complejo que contiene C3(H2O)Bb - la C3 convertasa de "fase fluida" o de "iniciación". Aunque solo se produce en pequeñas cantidades, la C3 convertasa de fase fluida puede escindir múltiples proteínas C3 para formar C3a y C3b y da como resultado la generación de C3b y su posterior unión covalente a una superficie (p. ej., una superficie bacteriana). El Factor B unido a C3b unido a la superficie es escindido por el factor D para formar de este modo el complejo de AP C3 convertasa unido a la superficie que contiene C3b,Bb. (Véase, por ejemplo, Müller-Eberhard (1988) Ann Rev Biochem 57: 321-347).
La AP C5 convertasa -(C3b)2,Bb - se forma tras la adición de un segundo monómero C3b a la AP C3 convertasa. (Véanse, por ejemplo, Medicus et al. (1976) J Exp Med 144:1076-1093 y Fearon et al. (1975) J Exp Med 142: 856 863). El papel de la segunda molécula de C3b es unirse a C5 y presentarlo a la escisión por Bb. (Véase, p. ej., Isenman et al. (1980) J Immunol 124:326-331). Las convertasas AP C3 y C5 son estabilizadas mediante la adición de la proteína trimérica properdina como describen, por ejemplo, Medicus et al. (1976), más arriba. Sin embargo, no se requiere la unión de la properdina para formar una vía alternativa a la convertasa C3 o C5 que funcione. (Véanse, p. ej., Schreiber et al. (1978). Proc Natl Acad Sci USA 75: 3948 hasta 3952 y Sissons et al. (1980). Proc Natl Acad Sci USA 77: 559-562).
La CP C3 convertasa se forma tras la interacción del componente C1 del complemento, que es un complejo de C1q, C1r y C1s, con un anticuerpo que se une a un antígeno diana (p. ej., un antígeno microbiano). La unión de la porción de C1q de C1 al complejo anticuerpo-antígeno provoca un cambio conformacional en C1 que activa C1r. C1r activo escinde a continuación el C1s asociado a C1 para generar de este modo una serina proteasa activa. C1s activo escinde el componente C4 del complemento para formar C4b y C4a. Como C3b, el fragmento C4b recién generado contiene un tiol altamente reactivo que forma fácilmente enlaces amida o éster con las moléculas adecuadas sobre una superficie diana (p. ej., una superficie celular microbiana). C1s también escinde el componente C2 del complemento para formar C2b y C2a. El complejo formado por C4b y C2a es la CP C3 convertasa, que es capaz de procesar C3 a C3a y C3b. La CP C5 convertasa -C4b, C2a, C3b - se forma tras la adición de un monómero C3b a la CP C3 convertasa. (Véanse, p. ej., Müller-Eberhard (1988), más arriba y Cooper et al. (1970) J Exp Med 132:775-793).
Además de su papel en las C3 y C5 convertasas, C3b también funciona como una opsonina a través de su interacción con los receptores del complemento presentes en las superficies de células presentadoras de antígeno tales como macrófagos y células dendríticas. Generalmente se considera que la función opsónica de C3b es una de las más importantes funciones anti-infecciosas del sistema del complemento. Los pacientes con lesiones genéticas que bloquean la función de C3b son propensos a infección por una amplia variedad de organismos patógenos,
mientras que se encuentra que los pacientes con lesiones posteriores en la secuencia de la cascada del complemento, es decir, los pacientes con lesiones que bloquean las funciones de C5, son más propensos solo a infección por Neisseria, y después solo algo más propensos.
Las AP y CP C5 convertasas escinden C5 para formar C5a y C5b. La escisión de C5 libera C5a, una potente anafilatoxina y factor quimiotáctico, y C5b, que permite la formación del complejo terminal del complemento lítico, C5b-9. C5b se combina con C6, C7, y C8 para formar el complejo C5b-8 en la superficie de la célula diana. Tras la unión de varias moléculas de C9, se forma el complejo de ataque a la membrana (MAC, C5b-9, complejo del complemento terminal-TCC). Cuando se inserta un número suficiente de MAC en las membranas celulares diana las aberturas que crean (poros de MAC) median la lisis osmótica rápida de las células diana.
Mientras que un sistema del complemento que funcione correctamente proporciona una sólida defensa contra los microbios infectantes, regulación o la activación inapropiadas de las vías del complemento han sido implicadas en la patogénesis de una variedad de trastornos, incluyendo, por ejemplo, artritis reumatoide (AR); nefritis lúpica; asma; lesión por isquemia-reperfusión; síndrome urémico hemolítico atípico (SUHa); enfermedad de depósitos densos (EDD); hemoglobinuria paroxística nocturna (HPN); degeneración macular (p. ej., degeneración macular asociada con la edad (DMAE)); hemólisis, enzimas hepáticas elevadas y bajo recuento de plaquetas (HELLP); púrpura trombocitopénica trombótica (PTT); pérdida fetal espontánea; Vasculitis pauciinmune; epidermolisis bullosa; pérdida fetal recurrente; esclerosis múltiple (EM); lesión cerebral traumática; y daño resultante de infarto de miocardio, derivación cardiopulmonar y hemodiálisis. (Véase, p. ej., Holers et al. (2008) Immunological Reviews 223:300-316). Se ha demostrado que la regulación a la baja de la activación del complemento es eficaz en el tratamiento de varias indicaciones de enfermedades en una variedad de modelos animales. Véase, p. ej., Rother t al. (2007) Nature Biotechnology 25(11):1256-1264; Wang et al. (1996) Proc Natl Acad Sci USA 93:8563-8568; Wang et al. (1995) Proc Natl Acad Sci USA 92:8955-8959; Rinder et al. (1995) J Clin Invest 96:1564-1572; Kroshus et al. (1995) Trasplantation 60:1194-1202; Homeister et al. (1993) J Immunol 150:1055-1064; Weisman et al. (1990) Science 249:146-151; Amsterdam et al. (1995) Am J Physiol 268:H448-H457; y Rabinovici et al. (1992) J Immunol 149:1744 1750.
Igawa et al. (2010) Nat Biotechnol 28: 1203-1207 describen un anticuerpo modificado genéticamente contra el receptor de IL-6 (IL-6R) que se disocia rápidamente de IL-6R en el entorno ácido del endosoma (pH 6,0) mientras que mantiene su afinidad de unión a IL-6R en plasma (pH 7,4).
El documento WO2011/111007 describe anticuerpos con unión a antígeno dependiente del pH que se disocian preferiblemente del antígeno en el endosoma.
Rother et al. (2007) Nat Biotechnol 25: 1256-1264 describen el descubrimiento y desarrollo del inhibidor del complemento eculizumab para el tratamiento de la hemoglobinuria paroxística nocturna.
El documento WO2010/151526 describe anticuerpos biespecíficos que se unen a dos o más proteínas diferentes seleccionadas entre C5a, C5b, un receptor celular para C5a, el complejo C5b-9 o un componente o intermedio del complemento terminal.
Compendio
La descripción se refiere a anticuerpos anti-C5 que tienen una de las características más mejoradas, por ejemplo, con respecto a anticuerpos anti-C5 conocidos utilizados con fines terapéuticos. Por ejemplo, los anticuerpos anti-C5 descritos en la presente memoria muestran aumento de la vida en suero con respecto a la semivida de eliminación en suero del eculizumab. Debido a sus propiedades farmacocinéticas mejoradas, los anticuerpos descritos en la presente memoria incluyen una serie de ventajas, p. ej., ventajas frente a otros anticuerpos anti-C5 que se unen a, e inhiben la escisión de, C5 completa o madura. Al igual que tales anticuerpos anti-C5, los anticuerpos descritos en la presente memoria pueden inhibir la respuesta inflamatoria mediada por C5a y la lisis celular dependiente de C5b (MAC) que resulta de la escisión de C5. Sin embargo, puesto que la concentración de C5 en el plasma humano es de aproximadamente 0,37 pM (Rawal y Pangburn (2001) J Immunol 166(4):2635-2642), el uso de altas concentraciones y/o la administración frecuente de anticuerpos anti-C5, tales como eculizumab, a menudo son necesarios para inhibir eficazmente C5 en un ser humano. La descripción establece en los ejemplos de trabajo datos experimentales que evidencian que mientras que los anticuerpos anti-C5 son muy eficaces en la inhibición del complemento in vitro e in vivo (véase, p. ej., Hillmen et al. (2004). N Engl J Med 350(6):552), los anticuerpos son particularmente susceptibles al aclaramiento mediado por la diana debido a la alta concentración de C5 en la sangre. La descripción también muestra que los nuevos anticuerpos descritos en la presente memoria han reducido la susceptibilidad al aclaramiento mediado por la diana y por lo tanto tienen una semivida de eliminación en suero más larga (semivida), en comparación con los anticuerpos anti-C5 previamente conocidos, en la sangre.
En vista de su vida media más larga, los anticuerpos descritos en la presente memoria se pueden administrar a un ser humano a una dosis mucho más baja y/o con menos frecuencia que los anticuerpos anti-C5 conocidos previamente (tales como, eculizumab) y proporcionar de manera efectiva la misma o mayor inhibición de C5 en un ser humano. La capacidad de administrar una dosis más baja del anticuerpo, en comparación con, por ejemplo, la
dosis de eculizumab, también permite vías de suministro adicionales, tales como, p. ej., administración subcutánea, administración intramuscular, suministro intrapulmonar, y administración a través de la utilización de microesferas biológicamente degradable.
En consecuencia, en un aspecto, la descripción presenta un anticuerpo anti-C5 que tiene una o más propiedades mejoradas (p. ej., propiedades farmacocinéticas mejoradas) con respecto al eculizumab. La invención proporciona un anticuerpo aislado o fragmento de unión al mismo, que: (a) se une al componente C5 del complemento; (b) inhibe la escisión de C5 para formar los fragmentos C5a y C5b; y (c) comprende: (i) una CDR1 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 23, (ii) una CDR2 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 19, (iii) una CDR3 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 3, (iv) una CDR1 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 4, (v) una CDR2 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 5, y (vi) una CDR3 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 6 y (vii) la región constante de cadena pesada representada en la SEQ ID NO: 13.
En algunas realizaciones, el anticuerpo o fragmento de la invención se unen a C5 a pH 7,4 y 25°C con una constante de disociación de afinidad (Kd) que está en el intervalo de 0,1 nM < Kd < 1 nM. El anticuerpo o fragmento se pueden unir a C5 a pH 7,4 y 25°C con una Kd que está en el intervalo de 0,2 nM < Kd < 1 nM, o con una Kd que está en el intervalo de 0,5 nM < Kd < 1 nM.
En algunas realizaciones, el anticuerpo o fragmento de la invención se une a C5 a pH 6,0 y 25°C con una Kd que es > 1 nM (p. ej., > 50 nM, > 100 nm, o > 1 pM).
En algunas realizaciones del anticuerpo o fragmento de la invención, la [(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 6,0 y a 25°C)/(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 7,4 y a 25°C)] es mayor que 25. La Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 6,0 y a 25°C)/(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 7,4 y a 25°C)] puede ser mayor que 100 (p. ej., mayor que 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1200, 1400, 1500, 1600, 1800, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500, 8000 o 8500).
En algunas realizaciones de cualquiera de los anticuerpos o fragmentos descritos en el presente documento, la Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 7,4 y a 25°C es menor que 1 (p. ej., menor que 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, 0,2, o 0,1) nM.
Basándose en la caracterización de diversas moléculas de eculizumab variantes como se describe en los ejemplos de trabajo, los inventores descubrieron un nuevo género de anticuerpos que tienen propiedades farmacocinéticas mejoradas en comparación con el eculizumab. Los anticuerpos dentro de este género tienen una afinidad por C5 que es más débil que la afinidad de eculizumab por C5 a pH 7,4. Sin embargo, los anticuerpos tienen una constante de disociación de afinidad (Kd) para C5 a pH 7,4 que es igual o inferior a 1 nM. Si bien la descripción no está limitada por ninguna teoría o mecanismo de acción particular, los autores de la presente invención creen que reduciendo ligeramente la afinidad de eculizumab por C5 a pH 7,4, y su posterior efecto sobre la afinidad del anticuerpo por C5 a pH 6,0 mientras se mantiene la misma/similar proporción de afinidad a pH 7,4 y pH 6,0, se reducirá sustancialmente el aclaramiento mediado por C5 del anticuerpo sin afectar sustancialmente a la función inhibidora del complemento del anticuerpo resultante en los pacientes. Por lo tanto, los autores de la presente invención han definido un intervalo de afinidad óptima para anticuerpos anti-C5 que dan lugar a propiedades farmacocinéticas mejoradas mientras que preservan las propiedades farmacodinámicas requeridas, cada una relativa al eculizumab. En consecuencia, la descripción presenta un anticuerpo aislado, o fragmento de unión a antígeno del mismo, que: (a) se une al componente del complemento C5 a pH 7,4 y 25°C con una constante de disociación de afinidad (Kd) que es < 1 nM; (b) se une a C5 a pH 6,0 y 25°C con una Kd que es no inferior a 10 nM; (c) inhibe la escisión de C5 para formar los fragmentos C5a y C5b, en donde la [(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 6,0 y 25°C)/(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 a pH 7,4 y 25°C)] es mayor que o igual a 25.
El anticuerpo o fragmento del mismo de la invención comprende una región constante de Fc humana variante (p. ej., una región constante de Fc de IgG humana variante) que se une al receptor de Fc neonatal humano (FcRn) con mayor afinidad que la de la región constante de Fc humana nativa a partir de la cual se obtuvo la región constante de Fc humana variante. La región constante de Fc variante comprende dos sustituciones de aminoácidos con respecto a la región constante de Fc humana nativa a partir de la cual se obtuvo la región constante de Fc humana variante. Las sustituciones están en las posiciones de aminoácidos 428 y 434 (numeración de EU) en relación con la región constante de Fc humana nativa. Las sustituciones son metionina por leucina en la posición 428 y asparagina por serina en la posición 434.
En algunas realizaciones, cualquiera de los anticuerpos o fragmentos de unión a antígeno de los mismos de la invención pueden comprender, o consistir en, un polipéptido de cadena pesada que comprende la secuencia de aminoácidos representada en el SEQ ID NO: 14 y un polipéptido de cadena ligera que comprende la secuencia de
aminoácidos representada en el SEQ ID NO: 11.
La descripción también caracteriza un anticuerpo que comprende la región variable de cadena pesada de eculizumab (SEQ ID NO: 7) o las CDR de la región de cadena pesada de eculizumab (SEQ ID NO: 1-3) y cualquiera de las regiones constantes de Fc humanas variantes descritas en la presente memoria, por ejemplo, la región constante de Fc humana variante que comprende una metionina en la posición 428 y una asparragina en la posición 434, cada una en la numeración e U.
En una realización, el anticuerpo o fragmento de unión a antígeno tiene una mayor semivida en los seres humanos con respecto a la semivida en el suero de eculizumab. La semivida según se utiliza en la presente memoria se define como el tiempo necesario para que la concentración plasmática del fármaco de anticuerpo en el organismo sea reducida a la mitad o 50%. Esta reducción al 50% en la concentración de suero refleja la cantidad de fármaco circulante y no eliminado por los métodos naturales de aclaramiento del anticuerpo. Se ha determinado que la semivida del eculizumab es de 272 82 horas o 11,3 días en los pacientes con HPN y de 12,1 días en pacientes con SUHa (Véase Soliris Prescribing Information). La semivida en seres humanos de los anticuerpos o fragmentos descritos en la presente memoria se puede aumentar con respecto a la semivida en seres humanos de eculizumab. El aumento de la semivida con respecto a eculizumab puede ser al menos 1,5 veces la semivida de eculizumab, al menos 2 veces la semivida de eculizumab, al menos 2,5 veces la semivida de eculizumab o al menos 3 veces la semivida de eculizumab.
Cualquiera de los anticuerpos o fragmentos descritos en la presente memoria, puede tener una semivida en suero en un ser humano que es mayor que, o al menos, 10 (p. ej., mayor que, o al menos, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 o 40) días. Esta semivida (o la extensión de la semivida con respecto a eculizumab) se puede lograr, en algunas realizaciones, mediante un anticuerpo descrito en la presente memoria que contiene una región constante de Fc humana de origen natural. La semivida se puede medir con respecto a un anticuerpo que comprende una región constante de Fc humana variante descrita en la presente memoria. La semivida en seres humanos de los anticuerpos o fragmentos descritos en la presente memoria se puede aumentar con respecto a la semivida en seres humanos de eculizumab. La semivida en seres humanos del anticuerpo descrito en el presente documento es al menos 25 días, al menos 26 días, al menos 27 días, al menos 28 días, al menos 29 días, al menos 30 días, al menos 31 días, al menos 32 días, al menos 33 días, al menos 34 días o al menos 35 días. El anticuerpo de la invención puede tener una semivida en suero en seres humanos de al menos 40 días.
Cualquiera de los anticuerpos o fragmentos descritos aquí pueden ser humanizados, completamente humanos, desinmunizados, o quiméricos. En algunas realizaciones, un anticuerpo o fragmento del mismo descrito en la presente memoria pueden ser, por ejemplo, un anticuerpo recombinante, un anticuerpo de cadena sencilla, un diacuerpo, un intracuerpo, un fragmento Fv, un fragmento Fd, un fragmento Fab, fragmento Fab', y un fragmento F(ab')2.
Cualquiera de los anticuerpos o fragmentos de los mismos descritos en la presente memoria puede comprender un radical heterólogo, por ejemplo, un azúcar. Por ejemplo, el anticuerpo o fragmento del mismo pueden estar glicosilados. El radical heterólogo puede ser también un marcador detectable, por ejemplo, una marca fluorescente, una marca luminiscente, una marca de metales pesados, una marca radiactiva, o una marca enzimática.
En algunas realizaciones, el anticuerpo o fragmento de unión a antígeno del mismo de la invención se pueden elaborar en una célula CHO. En algunas realizaciones, el anticuerpo o fragmento de unión a antígeno del mismo no contiene residuos de ácido siálico detectables.
Cualquiera de los anticuerpos o fragmentos de unión a antígeno de los mismos descritos en la presente memoria pueden ser modificados con un radical que mejora una o ambas de: (a) la estabilización del anticuerpo o fragmento de unión a antígeno del mismo en la circulación y (b) la retención del anticuerpo o fragmento de unión a antígeno del mismo en circulación. Tal radical puede ser PEG (PEGilación).
En otro aspecto, la invención proporciona un ácido nucleico que codifica los polipéptidos de cadena pesada y ligera de cualquiera de los anticuerpos o fragmentos de unión a antígeno de la invención. También se proporciona un vector (p. ej., un vector de clonación o expresión) que comprende el ácido nucleico, preferentemente un vector de expresión, y una célula (p. ej., una célula de insecto, célula bacteriana, célula fúngica, o célula de mamífero) que comprende: (i) el vector de expresión de la invención; (ii) un vector de expresión que codifica el polipéptido de cadena pesada del anticuerpo o fragmento de unión a antígeno del mismo según la invención y un vector de expresión que codifica el polipéptido de cadena ligera del anticuerpo o fragmento de unión a antígeno del mismo según la invención; (iii) un vector de expresión que codifica una región variable de cadena pesada que comprende una CDR1 que comprende la secuencia de aminoácidos representada en la SEQ ID NO: 23, una CDR2 que comprende la secuencia de aminoácidos representada en la SEQ ID NO: 19 y una CDR3 que comprende la secuencia de aminoácidos representada en la SEQ ID NO: 3 y un vector de expresión que codifica una región variable de cadena ligera que comprende una CDR1 que comprende la secuencia de aminoácidos representada en la SEQ ID NO: 4, una CDR2 que comprende la secuencia de aminoácidos representada en la SEQ ID NO: 5 y una
CDR3 que comprende la secuencia de aminoácidos representada en la SEQ ID NO: 6; o (iv) un vector de expresión que codifica una región variable de cadena pesada representada en la SEQ ID NO: 12 y un vector de expresión que codifica una región variable de cadena ligera representada en la SEQ ID NO: 8. El vector puede estar integrado o ser extracromosómico.
La invención proporciona adicionalmente un método para producir el anticuerpo o fragmentos de unión a antígeno de los mismos de la invención, comprendiendo el método cultivar la célula de la invención en condiciones y durante un tiempo suficiente para permitir la expresión por la célula (o cultivo de células) del anticuerpo o fragmento de unión a antígeno del mismo codificados por el ácido nucleico. El método también puede incluir aislar el anticuerpo o fragmento de unión a antígeno del mismo de la célula (o células del cultivo) o de los medios en los que se cultivaron la célula o las células.
En otro aspecto, la invención proporciona una composición farmacéutica que comprende un portador farmacéuticamente aceptable y uno o más de cualquiera de los anticuerpos o fragmentos de unión a antígeno de los mismos de la invención.
En otro aspecto, la invención proporciona un kit terapéutico que comprende: (i) el anticuerpo o fragmento de unión a antígeno del mismo de la invención y (ii) medios para el suministro del anticuerpo o fragmento de unión a antígeno del mismo a un ser humano. Los medios pueden ser, por ejemplo, una jeringa o una bomba.
En otro aspecto más, la invención proporciona un artículo de fabricación que comprende: un recipiente que comprende una etiqueta y una composición que comprende el anticuerpo o fragmento de unión a antígeno del mismo de la invención, en donde la etiqueta indica que la composición se va administrar a un ser humano que tiene, se sospecha que tiene, o en riesgo de desarrollar, una afección asociada al complemento. El artículo de fabricación puede comprender adicionalmente uno o más agentes terapéuticos adicionales activos para uso en el tratamiento de un ser humano que tiene, se sospecha que tiene, o en riesgo de desarrollar, una afección asociada al complemento.
En otro aspecto, la invención proporciona una o más de cualquiera de los anticuerpos o fragmentos de unión a antígeno de los mismos de la invención o la composición farmacéutica de la invención para su uso en un método para el tratamiento de una afección asociada al complemento. La afección asociada al complemento puede ser, p. ej., una seleccionado del grupo que consiste en artritis reumatoide, síndrome de anticuerpos antifosfolípidos, nefritis por lupus, lesión por isquemia-reperfusión, síndrome urémico hemolítico atípico, síndrome urémico hemolítico típico, hemoglobinuria paroxística nocturna, enfermedad por depósitos densos, neuromielitis óptica, neuropatía motora multifocal, esclerosis múltiple, degeneración macular, síndrome de HELLP, pérdida fetal espontánea, púrpura trombocitopénica trombótica, vasculitis pauciinmune, epidermolisis bullosa, pérdida fetal recurrente, lesión cerebral traumática, miocarditis, trastorno cerebrovascular, trastorno vascular periférico, trastorno renovascular, trastorno vascular mesentérico/entérico, vasculitis, púrpura de Henoch-Schonlein, nefritis, vasculitis asociada a lupus eritematoso sistémico, vasculitis asociada a artritis reumatoide, vasculitis por complejos inmunitarios, enfermedad de Takayasu, miocardiopatía dilatada, angiopatía diabética, enfermedad de Kawasaki, embolia de gas venosa, restenosis siguiente a colocación de dispositivo intraluminal, aterectomía rotacional, angioplastia coronaria transluminal percutánea, miastenia gravis, enfermedad por crioaglutininas, dermatomiositis, hemoglobinuria paroxística al frío, síndrome antifosfolípido, enfermedad de Graves, aterosclerosis, enfermedad de Alzheimer, sepsis de respuesta inflamatoria sistémica, choque séptico, lesión de la médula espinal, glomerulonefritis, rechazo de trasplantes (p. ej., trasplante de riñón), tiroiditis de Hashimoto, diabetes tipo I, psoriasis, pénfigo, anemia hemolítica autoinmunitaria, púrpura trombocitopénica idiopática, síndrome de Goodpasture, enfermedad de Degos y síndrome antifosfolípido catastrófico.
Según se utiliza en la presente memoria, el término "anticuerpo" se refiere a un anticuerpo completo que comprende dos polipéptidos de cadena ligera y dos polipéptidos de cadena pesada. Los anticuerpos completos incluyen diferentes isotipos de anticuerpos, incluyendo anticuerpos IgM, IgG, IgA, IgD, e IgE. El término "anticuerpo" incluye un anticuerpo policlonal, un anticuerpo monoclonal, un anticuerpo quimerizado o quimérico, un anticuerpo humanizado, un anticuerpo primatizado, un anticuerpo desinmunizado, y un anticuerpo completamente humano. El anticuerpo se puede preparar en u obtener de cualquiera de una variedad de especies, por ejemplo, mamíferos tales como seres humanos, primates no humanos (p. ej., orangután, babuinos, o chimpancés), caballos, vacas, cerdos, ovejas, cabras, perros, gatos, conejos, cobayas, hámsters, jerbos, ratas y ratones. El anticuerpo puede ser un anticuerpo purificado o recombinante.
Según se utiliza en la presente memoria, el término "fragmento de anticuerpo", "fragmento de unión a antígeno" o términos similares se refieren a un fragmento de un anticuerpo que conserva la capacidad de unirse a un antígeno diana (p. ej., C5 humana) e inhibir la actividad del antígeno diana. Tales fragmentos incluyen, por ejemplo, un anticuerpo de cadena sencilla, un fragmento Fv de cadena única (scFv), un fragmento Fd, un fragmento Fab, un fragmento Fab', o un fragmento F(ab')2. Un fragmento scFv es una cadena sencilla de polipéptido que incluye regiones variables tanto de cadena pesada como ligera del anticuerpo del que deriva el scFv. Además, los intracuerpos, minicuerpos, triacuerpos, y diacuerpos se incluyen también en la definición de anticuerpo y son compatibles para su uso en los métodos descritos en la presente memoria. Véase, por ejemplo, Todorovska et al. (2001) J Immunol Methods 248(1):47-66; Hudson y Kortt (1999) J Immunol Methods 231(1):177-189; Poljak (1994)
Structure 2(12): 1121-23; Rondon y Marasco (1997) Anual Review of Microbiology 51:257-283.
Según se utiliza en la presente memoria, el término "fragmento de anticuerpo" también incluye, p. ej., anticuerpos de dominio único, tales como anticuerpos de dominio único camelizados. Véanse, p. ej., Muyldermans et al. (2001) Trends Biochem Sci 26:230-235; Nuttall et al. (2000) Curr Pharm Biotech 1:253-263; Reichmann et al. (1999) J Immunol Meth 231:25-38; Publicaciones de Solicitud PCT Núm. WO 94/04678 y WO 94/25591; y Patente de Estados Unidos Núm. 6.005.079. En algunas realizaciones, la descripción proporciona anticuerpos de dominio único que comprenden dos dominios VH con modificaciones tales que se forman anticuerpos de dominio único.
Un fragmento de unión a antígeno puede incluir la región variable de un polipéptido de cadena pesada y la región variable de un polipéptido de cadena ligera. Un fragmento de unión al antígeno descrito en la presente memoria comprende las Cd R de la cadena ligera y el polipéptido de cadena pesada de un anticuerpo.
A menos que se defina lo contrario, todos los términos técnicos y científicos utilizados en la presente memoria tienen el mismo significado entendido comúnmente por un experto normal en la técnica a la que pertenece esta descripción. Los métodos y materiales preferidos se describen a continuación, aunque también se pueden utilizar métodos y materiales similares o equivalentes a los descritos en la presente memoria en la práctica o ensayo de los métodos y composiciones descritos actualmente.
Otras características y ventajas de la presente descripción, p. ej., los métodos para tratar o prevenir una afección asociada al complemento, serán evidentes a partir de la siguiente descripción, los ejemplos, y a partir de las reivindicaciones.
Breve descripción de las secuencias
SEQ ID NO: 1 representa la secuencia de aminoácidos de CDR1 de cadena pesada de eculizumab (como se define en las definiciones de Kabat-Chothia combinadas).
SEQ ID NO: 2 representa la secuencia de aminoácidos de CDR2 de cadena pesada de eculizumab (como se define en la definición de Kabat).
SEQ ID NO: 3 representa la secuencia de aminoácidos de CDR3 de cadena pesada de eculizumab (como se define en la definición de Kabat combinada).
SEQ ID NO: 4 representa la secuencia de aminoácidos de CDR1 de cadena ligera de eculizumab (como se define en la definición de Kabat).
SEQ ID NO: 5 representa la secuencia de aminoácidos de CDR2 de cadena ligera de eculizumab (como se define en la definición de Kabat).
SEQ ID NO: 6 representa la secuencia de aminoácidos de CDR3 de cadena ligera de eculizumab (como se define en la definición de Kabat).
SEQ ID NO: 7 representa la secuencia de aminoácidos de la región variable de cadena pesada de eculizumab. SEQ ID NO: 8 representa la secuencia de aminoácidos de la región variable de cadena ligera de eculizumab y el anticuerpo BNJ441.
SEQ ID NO: 9 representa la secuencia de aminoácidos de la región constante de cadena pesada de eculizumab. SEQ ID NO: 10 representa la secuencia de aminoácidos de la cadena pesada de eculizumab.
SEQ ID NO: 11 representa la secuencia de aminoácidos de la cadena ligera completa de eculizumab y el anticuerpo BNJ441.
SEQ ID NO: 12 representa la secuencia de aminoácidos de la región variable de la cadena pesada del anticuerpo BNJ441.
SEQ ID NO: 13 representa la secuencia de aminoácidos de la región constante de la cadena pesada del anticuerpo BNJ441.
SEQ ID NO: 14 representa la secuencia de aminoácidos de la cadena pesada del anticuerpo BNJ441.
SEQ ID NO: 15 representa la secuencia de aminoácidos de una variante de la región constante de cadena pesada de IgG2 que comprende las sustituciones YTE.
SEQ ID NO: 16: representa la secuencia de aminoácidos de la cadena pesada completa de una variante de eculizumab que comprende la región constante de cadena pesada representada en el SEQ ID NO: 15 (anterior). SEQ ID NO: 17 representa la secuencia de aminoácidos de CDR1 de cadena ligera de eculizumab (como se define en la definición de Kabat) con una sustitución de glicina por histidina en la posición 8 con respecto al SEQ ID NO: 4.
SEQ ID NO: 18 representa la secuencia de aminoácidos de la región variable de la cadena ligera del anticuerpo EHG303.
SEQ ID NO: 19 representa la secuencia de aminoácidos de CDR2 de cadena pesada de eculizumab en la que la serina en la posición 8 con respecto al SEQ ID NO: 2 se sustituye por una histidina.
SEQ ID NO: 20 representa la secuencia de aminoácidos de la etiqueta llamada "FLAG".
SEQ ID NO: 21 representa una secuencia de polihistidina utilizada comúnmente como una etiqueta antigénica. SEQ ID NO: 22 representa la secuencia de aminoácidos de la llamada etiqueta de hemaglutinina.
SEQ ID NO: 23 representa la secuencia de aminoácidos de CDR1 de cadena pesada de eculizumab en la que la tirosina en la posición 2 (con respecto al SEQ ID NO: 1) se sustituye por histidina.
SEQ ID NO: 24 representa la secuencia de aminoácidos del polipéptido de cadena pesada del anticuerpo EHG303.
SEQ ID NO: 25 representa la secuencia de aminoácidos del polipéptido de cadena ligera del anticuerpo EHG303. SEQ ID NO: 26 representa la secuencia de aminoácidos del polipéptido de cadena pesada del anticuerpo EHL049.
SEQ ID NO: 27 representa la secuencia de aminoácidos del polipéptido de cadena ligera del anticuerpo EHL049. SEQ ID NO: 28 representa la secuencia de aminoácidos del polipéptido de cadena pesada de EHL000.
SEQ ID NO: 29 representa la secuencia de aminoácidos del polipéptido de cadena ligera del anticuerpo EHL000. SEQ ID NO: 30 representa la secuencia de aminoácidos del polipéptido de cadena ligera de BHL006.
SEQ ID NO: 31 representa la secuencia de aminoácidos del polipéptido de cadena pesada del anticuerpo BHL006.
SEQ ID NO: 32 representa la secuencia de aminoácidos del polipéptido de cadena ligera del anticuerpo BHL009. SEQ ID NO: 33 representa la secuencia de aminoácidos de la cadena pesada del anticuerpo BHL009.
SEQ ID NO: 34 representa la secuencia de aminoácidos de la cadena ligera del anticuerpo BHL0011.
SEQ ID NO: 35 representa la secuencia de aminoácidos de la cadena pesada del anticuerpo BHL011.
Breve descripción de los dibujos
La Fig. 1 es un gráfico de líneas que representa el aclaramiento de eculizumab del suero de ratones transgénicos con FcRn humano en presencia o ausencia de C5 humana exógena. El eje Y representa el porcentaje de anticuerpo restante en el suero y el eje X representa el tiempo en días.
La Fig. 2 es un gráfico de líneas que representa el aclaramiento de una variante de eculizumab que tiene una región constante de IgG2 (Ecu-IgG2) y el anticuerpo Ecu-IgG2 que contiene las sustituciones YTE (Ecu-IgG2 (YTE)) del suero de los ratones. El eje Y representa el porcentaje de anticuerpo restante en el suero y el eje X representa el tiempo en días.
La Fig. 3 es un gráfico de líneas que representa el aclaramiento de una variante de eculizumab que tiene una región constante de IgG2 (Ecu-IgG2) y el anticuerpo Ecu-IgG2 que contiene las sustituciones y Te (Ecu-IgG2 (YTE)) del suero de los ratones. Los experimentos se realizaron en presencia o ausencia de C5 humana exógena. El eje Y representa el porcentaje de anticuerpo que queda en el suero y el eje X representa el tiempo en días.
La Fig. 4 es un diagrama sensograma que representa la cinética de asociación (a pH 7,4) y disociación (a pH 7,4 y pH 6,0) para tres anticuerpos anti-C5: EHL000, EHG303, y EHL049. El eje Y está en unidades arbitrarias, mientras que el eje X representa el tiempo (en segundos).
La Fig. 5A es un diagrama sensograma que representa la cinética de disociación a pH 7,4 y pH 6,0 para el anticuerpo EHG303 (doble sustitución Y27H-S57H), la variante de sustitución única Y27H de eculizumab, y eculizumab (ecu; Ec293F). El eje Y está en nanómetros (nm), mientras que el eje X representa el tiempo (en segundos).
La Fig. 5B es un diagrama sensograma que representa la cinética de disociación a pH 7,4 y pH 6,0 para el anticuerpo EHG304 (doble sustitución I34H-L52H), la variante de sustitución única I34H de eculizumab, y eculizumab (ecu; Ec293F). El eje Y está en nanómetros (nm), mientras que el eje X representa el tiempo (en segundos). El anticuerpo EHG304 no satisfizo el segundo umbral para la selección - a saber, superó la varianza máxima tolerada (de eculizumab) para la disociación a pH 7,4.
La Fig. 5C es un diagrama sensograma que representa la cinética de disociación a pH 7,4 y pH 6,0 para la (doble sustitución Y27H-S57H) EHG303 de anticuerpos y eculizumab (ecu; Ec293F). El eje Y está en nanómetros (nm), mientras que el eje X representa el tiempo (en segundos).
La Fig. 5D es un diagrama sensograma que representa la cinética de disociación a pH 7,4 y pH 6,0 para el anticuerpo EHL049 [G31H (cadena ligera)/doble sustitución Y27H-S57H (cadena pesada)], la variante de doble sustitución Y27H-S57H (EHG303) de eculizumab, y eculizumab (ecu). El eje Y está en nanómetros (nm), mientras que el eje X representa el tiempo (en segundos).
La Fig. 5E es un diagrama sensograma que representa la cinética de disociación a pH 7,4 y pH 6,0 para el anticuerpo EHL058 [G31H (cadena ligera)/doble sustitución L52H-S57H (cadena pesada)], la variante de doble sustitución L52H-S57H (cadena pesada) de eculizumab y eculizumab (ecu). El eje Y está en nanómetros (nm), mientras que el eje X representa el tiempo (en segundos). El anticuerpo EHL058 no satisfizo el segundo umbral para la selección - a saber, superó la varianza máxima tolerada (de eculizumab) para la disociación a pH 7,4. La Fig. 6 es un gráfico de líneas que representa el aclaramiento de EHL000, BNJ421, y BNJ423 del suero de ratones con deficiencia de NOD/scid/C5. El eje Y representa el porcentaje de anticuerpo que queda en el suero y el eje X representa el tiempo en días.
La Fig. 7 es un gráfico de líneas que representa el aclaramiento de EHL000, BNJ421, y BNJ423 del suero de ratones con deficiencia de NOD/scid/C5 en presencia o ausencia de C5 humana. El eje Y representa el porcentaje de anticuerpo que queda en el suero y el eje X representa el tiempo en días.
La Fig. 8 es un gráfico de líneas que representa la actividad de los anticuerpos EHL000, BNJ423, y BNJ421 en un ensayo hemolítico ex vivo. El eje Y representa el porcentaje de hemólisis y el eje X representa el tiempo en días.
La Fig. 9A es un gráfico de líneas que representa la farmacocinética del anticuerpo BHL011 en ratones transgénicos hFcRn. Cada línea representa un animal diferente. El eje Y representa la concentración de anticuerpo en pg/ml. El eje X representa el tiempo en días.
La Fig. 9B es un gráfico de líneas que representa la farmacocinética del anticuerpo BHL011 en ratones hFcRn transgénicos. Cada línea representa un animal diferente. El eje Y representa el % de la concentración de anticuerpo el día 1 que queda en el suero en cada momento específico. El eje X representa el tiempo en días. La Fig. 10 A es un gráfico de líneas que representa la farmacocinética del anticuerpo BHL006 en ratones hFcRn transgénicos. Cada línea representa un animal diferente. El eje Y representa la concentración de anticuerpo en pg/ml. El eje X representa el tiempo en días.
La Fig. 10B es un gráfico de líneas que representa la farmacocinética del anticuerpo BHL006 en ratones hFcRn transgénicos. Cada línea representa un animal diferente. El eje Y representa el % de la concentración de anticuerpo el día 1 que queda en el suero en cada momento específico. El eje X representa el tiempo en días. La Fig. 11A es un gráfico de líneas que representa la farmacocinética del anticuerpo BHL009 en ratones hFcRn transgénicos. Cada línea representa un animal diferente. El eje Y representa la concentración de anticuerpo en pg/ml. El eje X representa el tiempo en días.
La Fig. 11B es un gráfico de líneas que representa la farmacocinética del anticuerpo BHL009 en ratones hFcRn transgénicos. Cada línea representa un animal diferente. El eje Y representa el % de la concentración de anticuerpo el día 1 que queda en el suero en cada momento específico. El eje X representa el tiempo en días.
La Fig. 12 es un gráfico de líneas que representa un diagrama log de la farmacocinética media de los anticuerpos BHL011, BHL006, y BHL009 en ratones transgénicos hFcRn. Cada línea representa un anticuerpo diferente como se indica. El eje Y representa el % de la concentración de anticuerpo el día 1 que queda en el suero en cada momento específico. El eje X representa el tiempo en días.
La Fig. 13 es un gráfico de líneas que representa un diagrama lineal de la farmacocinética media de los anticuerpos BHL011, BHL006, y BHL009 en ratones hFcRn transgénicos. Cada línea representa un anticuerpo diferente como se indica. El eje Y representa el % de la concentración de anticuerpo el día 1 que queda en el suero en cada momento específico. El eje X representa el tiempo en días.
La Fig. 14 es un gráfico de líneas que representa la capacidad de bloqueo del anticuerpo BHL011 en un ensayo hemolítico en suero ex vivo después de una sola dosis. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa el tiempo en días.
La Fig. 15 es un gráfico de líneas que representa la capacidad de bloqueo del anticuerpo BHL006 en un ensayo hemolítico en suero ex vivo después de una sola dosis. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa el tiempo en días.
La Fig. 16 es un gráfico de líneas que representa la capacidad de bloqueo del anticuerpo BHL009 en un ensayo hemolítico en suero ex vivo después de una sola dosis. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa el tiempo en días.
La Fig. 17 es un gráfico que representa la correlación de la concentración en suero de BHL011 y la actividad hemolítica en suero ex vivo después de una sola dosis. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa la concentración de anticuerpos en pg/ml.
La Fig. 18 es un gráfico que representa la correlación de la concentración en suero de BHL006 y la actividad hemolítica en suero ex vivo después de una sola dosis. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa la concentración de anticuerpos en pg/ml.
La Fig. 19 es un gráfico que representa la correlación de la concentración en suero de BHL009 y la actividad hemolítica en suero ex vivo después de una sola dosis. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa la concentración de anticuerpos en pg/ml.
La Fig. 20 es un gráfico lineal que representa la actividad hemolítica media ex vivo después de una dosis única de BHL011, BHL009, o BHL006 en ratones transgénicos hFcRn. Cada línea representa un anticuerpo diferente como se indica. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa el tiempo en días.
Las Figs. 21A y 21B son un par de gráficos de líneas que representan los diagramas semi-log (Fig. 21A) y lineal (Fig. 21B) de la afinidad de BNJ441 y eculizumab como una función del pH. El eje Y representa % de disociación y el eje X es el pH.
La Fig. 22 es un gráfico de líneas que representa la farmacocinética de BNJ441 y eculizumab en los ratones NOD/scid y en ausencia de C5 humana. El eje Y representa la concentración de anticuerpo en pg/ml. El eje X representa el tiempo en días.
La Fig. 23 es un gráfico de líneas que representa la farmacocinética de BNJ441 y eculizumab en los ratones NOD/scid y en presencia de C5 humana. El eje Y representa la concentración de anticuerpo en pg/ml. El eje X representa el tiempo en días.
La Fig. 24 es un gráfico de líneas que representa el porcentaje de BNJ441 y eculizumab que queda en el suero de ratones NOD/scid en presencia de C5 humana como una función del tiempo. El eje Y representa la concentración de anticuerpo en g/ml. El eje X representa el tiempo en días.
La Fig. 25 es un gráfico de líneas que representa la actividad de bloqueo hemolítica suero ex vivo del anticuerpo BNJ441 y eculizumab después de una sola dosis como una función de tiempo. El eje Y representa el porcentaje de hemólisis (niveles con respecto a antes de la dosis) y el eje X representa el tiempo en días.
La Fig. 26 representa los perfiles de concentración-tiempo BNJ441 en suero medios después de la administración intravenosa de una Dosis de 200 mg o 400 mg a voluntarios sanos (panel superior - escala lineal; panel inferior - escala log-lineal).
La Fig. 27 representa los perfiles de hemólisis - tiempo medios en glóbulos rojos de pollo después de la administración intravenosa de placebo, 200 mg de BNJ441, o 400 mg de BNJ441 a voluntarios sanos.
La Fig. 28 describe la relación entre la concentración de BNJ441 y el porcentaje de hemólisis de glóbulos rojos de pollo después de la administración intravenosa de BNJ441 a voluntarios humanos sanos.
La Fig. 29 representa la potencia de BNJ441 comparación con eculizumab en análisis de actividad del complemento terminales.
La Fig. 30 representa la estructura de BNJ441.
La Fig. 31 representa los enlaces disulfuro intercatenarios de BNJ441.
Descripción detallada
La descripción proporciona anticuerpos que son útiles para, entre otras cosas, la inhibición del complemento terminal (p. ej., el ensambaje y/o la actividad del TCC C5b-9) y la inflamación mediada por anafilatoxina C5a y, por lo tanto, el tratamiento de trastornos asociados con el complemento. Los anticuerpos tienen varias propiedades mejoradas con respecto al eculizumab, incluyendo, por ejemplo, el aumento de la semivida en suero en un ser humano. Si bien no se pretende de ninguna manera que sean limitantes, los anticuerpos ilustrativos, los productos conjugados, las composiciones y las formulaciones farmacéuticas, y los métodos para el uso de cualquiera de los anteriores se elaboran a continuación y se ilustran en los Ejemplos de trabajo.
Anticuerpos
Los anticuerpos anti-C5 descritos en la presente memoria se unen al componente C5 del complemento (p. ej., C5 humana) e inhibe la escisión de C5 para formar los fragmentos C5a y C5b. Como se describió anteriormente, tales anticuerpos también tienen, por ejemplo, propiedades farmacocinéticas mejoradas con respecto a otros anticuerpos anti-C5 (p. ej., eculizumab) utilizados para fines terapéuticos.
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende: (i) una CDR1 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 1, (ii) una CDR2 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO : 2, (iii) una CDR3 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 3, (iv) una CDR1 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 4, (v) una CDR2 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 5, y (vi) una CDR3 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 6, en las que al menos uno o varios (p. ej., al menos dos, tres, cuatro, cinco, seis, siete, ocho, nueve, o 10 o más) aminoácidos de (i) - (vi) está sustituido con un aminoácido diferente.
Los límites exactos de las CDR se han definido de manera diferente de acuerdo con diferentes métodos. En algunas realizaciones, las posiciones de las CDR o regiones marco dentro de una cadena ligera o pesada del dominio variable pueden ser las definidas por Kabat et al. [(1991) "Sequences of Proteins of Immunological Interest. "Publicación NIH Núm. 91-3242, Departamento de Salud y Servicios Humanos, Bethesda, MD EE.UU.]. En tales casos, las CDR pueden ser referidas como "CDR de Kabat" (p. ej., "LCDR2 de Kabat" o "HCDR1 de Kabat"). En algunas realizaciones, las posiciones de las CDR de una región variable de cadena pesada o ligera pueden ser las definidas por Chothia et al. (1989) Nature 342: 877-883. Por consiguiente, estas regiones pueden ser referidas como "CDR de Chothia" (p. ej., "LCDR2 de Chothia" o "HCDR3 de Chothia"). En algunas realizaciones, las posiciones de los CDR de las regiones variables de cadena ligera y pesada pueden ser definidas por una definición combinada de Kabat-Chothia. En tales realizaciones, estas regiones pueden ser referidas como "CDR de Kabat-Chothia combinadas". Thomas et al. [(1996) Mol Immunol 33(17/18):1489-01] ilustran la identificación de los límites de CDR de acuerdo con las definiciones de Kabat y Chothia.
Cualquier aminoácido puede ser sustituido por cualquier otro aminoácido. En algunas realizaciones, la sustitución es una sustitución conservativa. Las sustituciones conservativas incluyen típicamente sustituciones dentro de los siguientes grupos: glicina y alanina; valina, isoleucina, y leucina; ácido aspártico y ácido glutámico; asparragina, glutamina, serina y treonina; lisina, histidina y arginina; y fenilalanina y tirosina. En algunas realizaciones, uno o más aminoácidos son sustituidos por histidina.
En algunas realizaciones, al menos un aminoácido (p. ej., al menos dos, tres, cuatro, o cinco) de la CDR1 de cadena pesada es sustituido por un aminoácido diferente. En algunas realizaciones, al menos un aminoácido (p. ej., al menos dos, tres, cuatro, o cinco) de la CDR2 de cadena pesada es sustituido por un aminoácido diferente. En algunas realizaciones, al menos un aminoácido (p. ej., al menos dos, tres, cuatro, o cinco) de la CDR3 de cadena pesada es sustituido por un aminoácido diferente.
En algunas realizaciones, al menos un aminoácido (p. ej., al menos dos, tres, cuatro, o cinco) de la CDR1 de cadena ligera es sustituido por un aminoácido diferente. En algunas realizaciones, al menos un aminoácido (p. ej., al menos dos, tres, cuatro, o cinco) de la CDR2 de cadena ligera es sustituido por un aminoácido diferente. En algunas realizaciones, al menos un aminoácido (p. ej., al menos dos, tres, cuatro, o cinco) de la CDR3 de cadena ligera es
sustituido por un aminoácido diferente.
En algunas realizaciones, se realiza una sustitución en una posición de aminoácido seleccionada del grupo que consiste en: glicina en la posición 1 con respecto al SEQ ID NO: 1, tirosina en la posición 2 con respecto al SEQ ID NO: 1, isoleucina en la posición 3 con respecto al SEQ ID NO: 1, fenilalanina en la posición 4 con respecto al SEQ ID NO: 1, serina en la posición 5 con respecto al SEQ ID NO: 1, asparragina en la posición 6 con respecto al SEQ ID NO: 1, tirosina en la posición 7 con respecto al SEQ ID NO: 1, triptófano en la posición 8 con respecto al SEQ ID NO: 1, isoleucina en la posición 9 con respecto al SEQ ID NO: 1, la glutamina en la posición 10 con respecto al SEQ ID NO: 1, el ácido glutámico en la posición 1 con respecto a SEQ ID NO : 2, isoleucina en la posición 2 con respecto al SEQ ID NO: 2, leucina en la posición 3 con respecto al SEQ ID NO: 2, prolina en la posición 4 con respecto al SEQ ID NO: 2, la glicina en la posición 5 con respecto al SEQ ID NO: 2, serina en la posición 6 con respecto al SEQ ID NO: 2, la glicina en la posición 7 con respecto al SEQ ID NO: 2, serina en la posición 8 con respecto al SEQ ID NO: 2, treonina en la posición 9 con respecto al SEQ ID NO: 2, glutámico ácido en la posición 10 con respecto al SEQ ID NO: 2, tirosina en la posición 11 con respecto al SEQ ID NO: 2, treonina en la posición 12 con respecto al SEQ ID NO: 2, ácido glutámico en la posición 13 con respecto al SEQ ID NO: 2, asparragina en la posición 14 con respecto al SEQ ID NO: 2, fenilalanina en la posición 15 con respecto al SEQ ID NO: 2, la lisina en la posición 16 con respecto al SEQ ID NO: 2, ácido aspártico en la posición 17 con respecto al SEQ ID NO: 2, tirosina en la posición 1 con respecto al SEQ ID NO: 3, fenilalanina en la posición 2 con respecto al SEQ ID NO: 3, fenilalanina en la posición 3 con respecto al SEQ ID NO: 3, la glicina en la posición 4 con respecto al SEQ ID NO: 3, serina en la posición 5 con respecto al SEQ ID NO: 3, serina en la posición 6 con respecto al SEQ ID NO: 3, prolina en la posición 7 con respecto al SEQ ID NO: 3, asparragina en la posición 8 con respecto al SEQ ID NO: 3, triptófano en la posición 9 con respecto al SEQ ID NO: 3, tirosina en la posición 10 con respecto al SEQ ID NO: 3, fenilalanina en la posición 11 con respecto al SEQ ID NO: 3, ácido aspártico en la posición 12 con respecto al SEQ ID NO: 3, y valina en la posición 13 con respecto al SEQ ID NO: 3.
En algunas realizaciones, la glicina en la posición 31 con respecto al SEQ ID NO: 8 es sustituida por un aminoácido diferente. Por ejemplo, la glicina subrayada de la CDR1 de la cadena ligera del eculizumab puede ser sustituida por un aminoácido diferente: GASENIYGALN (SEQ ID NO: 4). La sustitución puede ser una histidina por glicina, es decir, GASENIYHALN (SEQ ID NO: 17).
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende una sustitución de aminoácido en una posición de aminoácido seleccionada del grupo que consiste en: glicina en la posición 26 con respecto al SEQ ID NO: 7, tirosina en la posición 27 con respecto al SEQ ID NO: 7, isoleucina en la posición 28 con respecto al SEQ ID NO: 7, fenilalanina en la posición 29 con respecto al SEQ ID NO: 7, serina en la posición 30 con respecto al SEQ ID NO: 7, asparragina en la posición 31 con respecto al SEQ ID NO: 7, tirosina en la posición 32 con respecto al SEQ ID NO: 7, triptófano en la posición 33 con respecto al SEQ ID NO: 7, isoleucina en la posición 34 con respecto al SEQ ID NO: 7, glutamina en la posición 35 con respecto al SEQ ID NO: 7, el ácido glutámico en la posición 50 con respecto al SEQ ID NO: 7, isoleucina en la posición 51 con respecto al SEQ ID NO: 7, leucina en la posición 52 con respecto al SEQ ID NO: 7, la prolina en la posición 53 con respecto al SEQ ID NO: 7, la glicina en la posición 54 con respecto al SEQ ID NO: 7, serina en la posición 55 con respecto al SEQ ID NO: 7, la glicina en la posición 56 con respecto al SEQ ID NO: 7, serina en la posición 57 con respecto al SEQ ID NO: 7, treonina en la posición 58 relativa al SEQ ID NO: 7, el ácido glutámico en la posición 59 con respecto al SEQ ID NO: 7, tirosina en la posición 60 con respecto al SEQ ID NO: 7, treonina en la posición 61 con respecto al SEQ ID NO: 7, el ácido glutámico en la posición 62 relativa al SEQ ID NO: 7, asparragina en la posición 63 con respecto al SEQ ID NO: 7, fenilalanina en la posición 64 con respecto al SEQ ID NO: 7, lisina en la posición 65 con respecto al SEQ ID NO: 7, ácido aspártico en la posición 66 con respecto al SEQ ID NO: 7, tirosina en la posición 99 con respecto al SEQ ID NO: 7, fenilalanina en la posición 100 con respecto al SEQ ID NO: 7, fenilalanina en la posición 101 con respecto al SEQ ID NO: 7, la glicina en la posición 102 con respecto al SEQ ID NO: 7, serina en la posición 103 con respecto al SEQ ID NO: 7, serina en la posición 104 con respecto al SEQ ID NO: 7, la prolina en la posición 105 con respecto al SEQ ID NO: 7, asparragina en la posición 106 con respecto al SEQ ID NO: 7, triptófano en la posición 107 con respecto al SEQ ID NO: 7, tirosina en la posición 108 con respecto al SEQ ID NO: 7, fenilalanina en la posición 109 con respecto al SEQ ID NO: 7, ácido aspártico en la posición 110 con respecto al SEQ ID NO: 7 y valina en la posición 111 con respecto al SEQ ID NO: 7. En algunas realizaciones, el anticuerpo anti-C5 comprende dos o más (p. ej., al menos dos, tres, cuatro, cinco, seis, siete, ocho, nueve, o 10 o más) de cualquiera de las sustituciones anteriores y en cualquier combinación.
En algunas realizaciones, el anticuerpo anti-C5 comprende al menos una sustitución que cumpla con los siguientes criterios con respecto a eculizumab:
(1) una variación máxima de la cinética de asociación a pH 7,4 de un desplazamiento de fase máximo menor de 33% a 800 segundos en comparación con el desplazamiento de fase máximo promedio a 800 segundos observado para eculizumab;
(2) una variación máxima de la cinética de disociación a pH 7,4 de una reducción de no más de 3 veces en el desplazamiento de fase máximo por encima de 800 segundos en comparación con el desplazamiento de la fase maxíma promedio a 800 segundos observado para eculizumab; y
(3) una variación mínima de la cinética de disociación a pH 6,0 de una reducción de al menos 3 veces en el
desplazamiento de fase máximo por encima de 800 segundos en comparación con el desplazamiento de fase máximo promedio a 800 segundos observado para eculizumab.
Por ejemplo, con respecto al criterio (1) anterior, si el desplazamiento de fase máximo promedio después de 800 segundos de asociación con eculizumab es de aproximadamente 0,75 nm, un anticuerpo de ensayo que tiene un desplazamiento de fase de menos de 0,5 nm (p. ej., reproducido dos o más veces) podría no cumplir los criterios anteriores. Por el contrario, un anticuerpo anti-C5 con un desplazamiento de fase máximo de más de 0,5 nm a 800 segundos cumple el primer criterio. Tales sustituciones dan lugar a anticuerpos anti-C5 que solo se desvían de la ka y kd de eculizumab a pH 7,4 en un grado menor, pero se desvían de la kd de eculizumab a pH 6,0 más significativamente.
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende al menos una (p. ej., al menos dos, tres, o cuatro) sustitución de aminoácido en una posición de aminoácido seleccionada del grupo que consiste en: glicina en la posición 31 con respecto al SEQ ID NO: 8, leucina en la posición 33 con respecto al SEQ ID NO: 8, valina en la posición 91 con respecto al SEQ ID NO: 8, y treonina en la posición 94 con respecto al SEQ ID NO: 8. En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende al menos una (p. ej., dos, tres, cuatro o cinco) sustitución de aminoácido en una posición de aminoácido seleccionada del grupo que consiste en: tirosina en la posición 27 con respecto al SEQ ID NO: 7, isoleucina en la posición 34 con respecto al SEQ ID NO: 7, leucina en la posición 52 con respecto al SEQ ID NO: 7, y serina en la posición 57 con respecto al SEQ ID NO: 7.
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria contiene en su región variable de cadena ligera al menos una sustitución seleccionada entre las siguientes: glicina en la posición 31 con respecto al SEQ ID NO: 8, leucina en la posición 33 con respecto al SEQ ID NO: 8, valina en la posición 91 con respecto al SEQ ID NO: 8, y treonina en la posición 94 con respecto al SEQ ID NO: 8. Véase la Tabla 1 a continuación. En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria contiene en su región variable de cadena pesada al menos una sustitución seleccionada entre las siguientes: tirosina en la posición 27 con respecto al SEQ ID NO: 7, isoleucina en la posición 34 con respecto al SEQ ID NO: 7, leucina en la posición 52 con respecto al SEQ ID NO: 7, y serina en la posición 57 con respecto al SEQ ID NO: 7. Véase la Tabla 1 a continuación.
En algunas realizaciones, un anticuerpo comprende al menos dos (p. ej., al menos tres, cuatro, cinco, seis, siete, ocho, nueve, o 10) sustituciones de aminoácidos con respecto al conjunto de CDR definido por los SEQ ID NO: 1-6. Por lo tanto, en algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende dos o más sustituciones en las combinaciones y en las posiciones de aminoácidos expuestas en la Tabla 1.
Tabla 1. Combinaciones de Sustituciones de Aminoácidos

continuación

continuación

Para ser claros, los anticuerpos anti-C5 variantes mencionados en la Tabla 1 solo tienen que tener las secuencias de aminoácidos de las seis (6) CDR de eculizumab en las que se realizan las sustituciones de aminoácidos proporcionadas, indicadas. Los anticuerpos variantes pueden incluir, opcionalmente, las regiones marco del SEQ ID NO: 7 o del SEQ ID NO: 8.
Las sustituciones descritas en la Tabla 1 pueden ser por cualquier aminoácido que es diferente del residuo de aminoácido indicado. En algunas realizaciones, el aminoácido diferente es una histidina.
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende una sustitución realizada en una posición de aminoácido seleccionada del grupo que consiste en: tirosina en la posición 27 con respecto al SEQ ID NO: 7, isoleucina en la posición 34 con respecto al SEQ ID NO: 7, leucina en la posición 52 con respecto al SEQ ID NO: 7, y serina en la posición 57 con respecto al SEQ ID NO: 7. En algunas realizaciones, tanto la tirosina en la posición 27 con respecto al SEQ ID NO: 7 como la leucina en la posición 52 con respecto al SEQ ID NO: 7 son cada una sustituidas por un aminoácido diferente. En algunas realizaciones, tanto la isoleucina en la posición 34 con respecto al SEQ ID NO: 7 como la serina en la posición 57 con respecto al SEQ ID NO: 7 son cada una sustituidas por un aminoácido diferente. En algunas realizaciones, tanto la isoleucina en la posición 34 con respecto al SEQ ID NO: 7 y la leucina en la posición 52 con respecto al SEQ ID NO: 7 son cada una sustituidas por un aminoácido diferente. En algunas realizaciones, tanto la tirosina en la posición 27 con respecto al SEQ ID NO: 7 como la serina en la posición 57 con respecto al SEQ ID NO: 7 son cada una sustituidas por un aminoácido diferente. En algunas realizaciones de cualquiera de los anticuerpos anti-C5 descritos en la presente memoria, el aminoácido diferente es una histidina. Por ejemplo, la tirosina en la posición 27 y la serina en la posición 57 pueden ser cada una sustituidas por histidina.
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende una CDR1 de cadena pesada que comprende, o está constituida por, la siguiente secuencia de aminoácidos: GHIFSNYWIQ (SEQ ID NO: 23). En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende una cadena pesada de CDR2 que comprende, o está constituida por, la siguiente secuencia de aminoácidos: EILPGSGHTEYTENFKD (SEQ ID NO: 19). En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende una región variable de cadena pesada que comprende la siguiente secuencia de aminoácidos:
QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEWMGEILPGSGH
TEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCARYFFGSSPNWYFDVWGQG
TLVTVSS (SEQ ID NO: 12).
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria comprende una región variable de cadena ligera que comprende la siguiente secuencia de aminoácidos:
DIQMTQSPSSLSASVGDRVTITCGASENIYGALNWYQQKPGKAPKLLIYGATNLADGVP
SRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLTFGQGTKVEIK (SEQ ID NO:8).
Un anticuerpo anti-C5 descrito en la presente memoria puede unirse a C5 a pH 7,4 y 25°C (y, por otra parte, en condiciones fisiológicas) con una constante de disociación de afinidad (Kd) que es al menos 0,1 (p. ej., al menos 0,15, 0,175, 0,2, 0,25, 0,275, 0,3, 0,325, 0,35, 0,375, 0,4, 0,425, 0,45, 0,475, 0,5, 0,525, 0,55, 0,575, 0,6, 0,625, 0,65, 0,675, 0,7, 0,725, 0,75, 0,775, 0,8, 0,825, 0,85, 0,875, 0,9, 0,925, 0,95, o 0,975) nM. En algunas realizaciones, la Kd del anticuerpo anti-C5 no es mayor que 1 (p. ej., no mayor que 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, o 0,2) nM.
En algunas realizaciones de cualquier anticuerpos anti-C5 descrito en la presente memoria, la [(Kd del anticuerpo para C5 a pH 6,0 a C)/(Kd del anticuerpo para C5 a pH 7,4 a 25°C)] es mayor que 21 (p. ej., mayor que 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 350, 400, 450, 500, 600, 700, 800, 900, 1000, 1500,
2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500, o 8000).
Los métodos para determinar si un anticuerpo se une a un antígeno proteico y/o la afinidad de un anticuerpo por un antígeno proteico son conocidos en la técnica. Por ejemplo, la unión de un anticuerpo a un antígeno proteico se puede detectar y/o cuantificar utilizando una variedad de técnicas tales como, pero no limitadas a, la transferencia Western, la transferencia puntual, el método de resonancia de plasmón superficial (RPS) (p. ej., sistema BIAcore; Pharmacia Biosensor AB, Uppsala, Suecia y Piscataway, NJ), o el análisis de inmunoabsorción ligado a enzimas (ELISA). Véase, p. ej., Harlow y Lane (1988) "Antibodies: A Laboratory Manual" Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; Benny K. C. Lo (2004) "Antibody Engineering: Methods and Protocols," Humana Press (ISBN: 1588290921); Borrebaek (1992) "Antibody Engineering, A Practical Guide," W.H. Freeman and Co., NY; Borrebaek (1995) "Antibody Engineering," 2a Edición, Oxford University Press, NY, Oxford; Johne et al. (1993) J Immunol Meth 160:191-198; Jonsson et al. (1993) Ann Biol Clin 51:19-26; y Jonsson et al. (1991) Biotechniques 11:620-627. Además, los métodos para la medición de la afinidad (p. ej., constantes de disociación y asociación) se exponen en los ejemplos de trabajo.
Según se utiliza en la presente memoria, el término "ka" se refiere a la constante de velocidad para la asociación de un anticuerpo a un antígeno. El término "kd" se refiere a la constante de velocidad para la disociación de un anticuerpo del complejo anticuerpo/antígeno. Y el término "Kd" se refiere a la constante de disociación en el punto de equilibrio de una interacción anticuerpo-antígeno. La constante de disociación en el punto de equilibrio se deduce de la razón de las constantes de velocidad cinética, Kd = ka/kd. Tales determinaciones se miden preferiblemente a 25°C o 37°C (véase los ejemplos de trabajo). Por ejemplo, la cinética de la unión del anticuerpo a C5 humana se puede determinar a pH 8,0, 7,4, 7,0, 6,5 y 6,0 por medio de resonancia de plasmón superficial (RPS) en un aparato BIAcore 3000 utilizando un método de captura anti-Fc para inmovilizar el anticuerpo.
El anticuerpo anti-C5 descrito en la presente memoria puede tener actividad en el bloqueo de la generación o actividad de los fragmentos activos C5a y/o C5b de una proteína C5 (p. ej., una proteína C5 humana). A través de este efecto de bloqueo, los anticuerpos inhiben, por ejemplo, los efectos proinflamatorios de C5a y la generación del complejo de ataque a membrana (MAC) C5b-9 en la superficie de una célula.
Los métodos para determinar si un anticuerpo particular descrito en la presente memoria inhibe la escisión de C5 son conocidos en la técnica. La inhibición del componente C5 del complemento humano puede reducir la capacidad de lisis celular del complemento en los fluidos corporales de un sujeto. Tales reducciones de la capacidad de lisis celular del complemento presente en los fluidos corporales se puede medir por métodos bien conocidos en la técnica tales como, por ejemplo, mediante un análisis hemolítico convencional, tal como el análisis de hemólisis descrito por Kabat y Mayer (eds.), "Experimental Immunochemistry, 2a Edición," 135-240, Springfield, IL, CC Thomas (1961), páginas 135-139, o una variación convencional de dicho análisis tal como el método de hemólisis de eritrocitos de pollo descrito, por ejemplo, por Hillmen et al. (2004) en N Engl J Med 350 (6):552. Los métodos para determinar si un compuesto candidato inhibe la escisión de C5 humana para proporcionar las formas C5a y C5b son conocidos en la técnica y son descritos, por ejemplo, por Moongkarndi et al. (1982) en Immunobiol 162: 397; Moongkarndi et al. (1983) Immunobiol 165:323; Isenman et al. (1980) J Immunol 124(1):326-31; Thomas et al. (1996) Mol Immunol 33(17-18):1389-401; y Evans et al. (1995) Mol Immunol 32 (16):1183-95. Por ejemplo, la concentración y/o actividad fisiológica de C5a y C5b en un líquido corporal pueden medirse mediante métodos bien conocidos en la técnica. Los métodos para medir la concentración o actividad de C5a incluyen, p. ej., análisis de quimiotaxis, RIA o ELISA (véanse, p. ej., Ward y Zvaifler (1971) J Clin Invest 50 (3): 606-16 y Wurzner et al. (1991) Complement Inflamm 8:328-340). Para C5b, se pueden utilizar análisis hemolíticos o análisis para C5b-9 soluble como los comentados en la presente memoria. También se pueden utilizar otros análisis conocidos en la técnica. Utilizando análisis de estos u otros tipos adecuados, se pueden escrutar agentes candidatos capaces de inhibir el componente C5 del complemento humano.
Se pueden utilizar técnicas inmunológicas tales como, pero no limitadas a, ELISA para medir la concentración de proteína de C5 y/o sus productos de degradación para determinar la capacidad de un anticuerpo anti-C5 para inhibir la conversión de C5 en productos biológicamente activos. En algunas realizaciones, se mide la generación de C5a. En algunas realizaciones, se utilizan anticuerpos específicos del neoepítopo C5b-9 para detectar la formación del complemento terminal.
Se pueden utilizar análisis hemolíticos para determinar la actividad inhibidora de un anticuerpo anti-C5 sobre la activación del complemento. Con el fin de determinar el efecto de un anticuerpo anti-C5 sobre la hemólisis mediada por la vía clásica del complemento en una solución de ensayo de suero in vitro, por ejemplo, se utilizan eritrocitos de oveja recubiertos con hemolisina o eritrocitos pollo sensibilizados con anticuerpo anti-eritrocitos pollo como células diana. El porcentaje de lisis se normaliza considerando 100% de lisis igual a la lisis que ocurre en ausencia del inhibidor. En algunas realizaciones, la vía clásica del complemento es activada por un anticuerpo IgM humano, por ejemplo, como se utiliza en el Wieslab® Classical Pathway Complement Kit (Wieslab® COMPL CP310, Euro-Diagnostica, Suecia). En pocas palabras, el suero de ensayo se incuba con un anticuerpo anti-C5 en presencia de un anticuerpo IgM humano. La cantidad de C5b-9 que se genera se mide poniendo en contacto la mezcla con un anticuerpo anti-C5b-9 conjugado con enzima y un sustrato fluorogénico y midiendo la absorbancia a la longitud de onda apropiada. Como control, se incuba el suero de ensayo en ausencia del anticuerpo anti-C5. En algunas
realizaciones, el suero de ensayo es un suero con deficiencia de C5 reconstituido con un polipéptido C5.
Para determinar el efecto del anticuerpo anti-C5 sobre la hemólisis mediada por la vía alternativa, se utilizan eritrocitos de conejo o de cobaya no sensibilizados como células diana. En algunas realizaciones, la solución de ensayo de suero es un suero con deficiencia de C5 reconstituido con un polipéptido C5. El porcentaje de lisis se normaliza considerando 100% de lisis igual a la lisis que ocurre en ausencia del inhibidor. En algunas realizaciones, la vía alternativa del complemento es activada por moléculas de lipopolisacáridos, por ejemplo, tal como se utiliza en el Wieslab® Alternative Pathway Complement Kit (Wieslab® COm Pl AP330, Euro-Diagnostica, Suecia). En pocas palabras, el suero de ensayo se incuba con un anticuerpo anti-C5 en presencia de lipopolisacárido. La cantidad de C5b-9 que se genera se mide poniendo en contacto la mezcla con un anticuerpo anti-C5b-9 conjugado con enzima y un sustrato fluorogénico y midiendo la fluorescencia a la longitud de onda apropiada. Como control, el suero de ensayo se incuba en ausencia del anticuerpo anti-C5.
En algunas realizaciones, la actividad de C5, o la inhibición de la misma, se cuantifica utilizando un análisis CH50eq. El análisis CH50eq es un método para medir la actividad clásica del complemento total en el suero. Este ensayo un análisis lítico, que utiliza eritrocitos de anticuerpos sensibilizados como activador de la vía clásica del complemento y diversas diluciones del suero de ensayo para determinar la cantidad requerida para proporcionar 50% de lisis (CH50). El porcentaje de hemólisis se puede determinar, por ejemplo, utilizando un espectrofotómetro. El ensayo CH50eq proporciona una medida indirecta de la formación de complejo de complemento terminal (TCC), ya que los propios TCC son directamente responsables de la hemólisis que se mide.
El análisis es bien conocido y comúnmente puesto en práctica por los expertos en la técnica. En pocas palabras, para activar la vía clásica del complemento, se añaden muestras de suero sin diluir (p. ej., muestras de suero humano reconstituido) a pocillos de microanálisis que contienen los eritrocitos sensibilizados con anticuerpo para generar de ese modo TCC. A continuación, los sueros activados se diluyen en pocillos de microanálisis, que están recubiertos con un reactivo de captura (p. ej., un anticuerpo que se une a uno o más componentes del TCC). El TCC presente en las muestras activadas se une a los anticuerpos monoclonales que recubren la superficie de los pocillos de microanálisis. Se lavan los pocillos y a cada pocillo se le añade un reactivo de detección que está marcado de manera detectable y reconoce el TCC unido. La marca detectable puede ser, por ejemplo, una marca fluorescente o una marca enzimático. Los resultados del análisis se expresan en unidades de CH50 equivalentes por mililitro (U CH50 Eq/ml).
La inhibición, por ejemplo, en lo que respecta a la actividad del complemento terminal, incluye al menos una disminución de 5% (p. ej., al menos 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, o 60%) de la actividad del complemento terminal en, p. ej., un análisis hemolítico o un análisis CH50eq en comparación con el efecto de un anticuerpo de control (o fragmento de unión a antígeno del mismo) en condiciones similares y a una concentración equimolar. Inhibición sustancial, según se utiliza en la presente memoria, se refiere a la inhibición de una actividad dada (p. ej., la actividad del complemento terminal) de al menos 40% (p. ej., al menos 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, o 95% o mayor). En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria contiene una o más sustituciones de aminoácidos con respecto a las CDR de eculizumab (es decir, SEQ ID NO: 1-6), sin embargo, conserva al menos 30% (p. ej., al menos 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90, o 95%) de la actividad inhibidora del complemento de eculizumab en un análisis hemolítico o análisis CH50eq.
Un anticuerpo anti-C5 descrito en la presente memoria tiene una semivida en suero en seres humanos que es de al menos 20 (p. ej., al menos 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, o 36) días. Los métodos para la medición de la semivida en suero de un anticuerpo son conocidos en la técnica y se ilustran en los ejemplos de trabajo. Véanse, p. ej., Dall'Acqua et al. (2006) J Biol Chem 281: 23514-23524; Hinton et al. (2004) J Biol Chem 279:6213-6216; Hinton et al. (2006) J Immunol 176:346-356; y Petkova et al. (2006) Int Immunol 18 (12):1759-69. En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria tiene una semivida en suero que es al menos de 20% (p. ej., al menos 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 125, 150, 175, 200, 250, 300, 400, 500%) mayor que la semivida en suero de eculizumab, por ejemplo, como se mide en uno de los sistemas modelo de ratón descritos en los ejemplos de trabajo (p. ej., el sistema modelo de ratón con deficiencia de C5/NOD/scid o de ratón transgénico hFcRn).
Modificaciones de la región Fc
Un anticuerpo anti-C5 descrito en la presente memoria puede, en algunas realizaciones, comprender una región constante de Fc humana variante que se une al receptor de Fc neonatal humano (FcRn) con mayor afinidad que la de la región constante de Fc humana nativa de la que se obtuvo la región constante de Fc humana variante. Por ejemplo, la región constante de Fc puede comprender una o más (p. ej., dos, tres, cuatro, cinco, seis, siete, u ocho o más) sustituciones de aminoácidos con respecto a la región constante de Fc humana nativa de la que se obtuvo la región constante de Fc humana variante. Las sustituciones pueden aumentar la afinidad de unión de un anticuerpo IgG que contiene la región constante de Fc variante a FcRn a pH 6,0, mientras se mantiene la dependencia del pH de la interacción. Véase, p. ej., Hinton et al. (2004) J Biol Chem 279:6213-6216 y Datta-Mannan et al. (2007) Drug Metab Dispos 35:1-9. Los métodos para someter a ensayo si una o más sustituciones en la región constante de Fc
de un anticuerpo aumentan la afinidad de la región constante de Fc por FcRn a pH 6,0 (mientras se mantiene la dependencia de pH de la interacción) son conocidos en la técnica y se ilustran en los ejemplos de trabajo. Véase, p. ej., Datta-Mannan et al. (2007) J Biol Chem 282(3):1709-17; Publicación Internacional Núm. WO 98/23289; Publicación Internacional Núm. Wo 97/34631; y Patente de Estados Unidos Núm. 6.277.375.
Las sustituciones que mejoran la afinidad de unión de una región constante Fc de anticuerpo contra FcRn se conocen en la técnica e incluyen, por ejemplo, (1) la triple sustitución M252Y/S254T/T256E descrita por Dall'Acqua et al. (2006) J Biol Chem 281:23514-23524; (2) las sustituciones M428L o T250Q/M428L descritas por Hinton et al. (2004) J Biol Chem 279:6213-6216 y Hinton et al. (2006) J Immunol 176:346-356; y (3) las sustituciones N434A o T307/E380A/N434A descritas por Petkova et al. (2006) Int Immunol 18(12):1759-69. Los emparejamientos de sustitución adicionales: P257I/Q311I, P257I/N434H, y D376V/N434H son descritas, por ejemplo, por Datta-Mannan et al. (2007) J Biol Chem 282(3):1709-17.
En algunas realizaciones, la región constante variante tiene una sustitución en el residuo de aminoácido 255 de EU para la valina. En algunas realizaciones, la región constante variante tiene una sustitución en el residuo de aminoácido 309 de EU para la asparragina. En algunas realizaciones, la región constante variante tiene una sustitución en el residuo de aminoácido 312 de EU para la isoleucina. En algunas realizaciones, la región constante variante tiene una sustitución en el residuo de aminoácido 386 de EU.
En algunas realizaciones, la región constante de Fc variante comprende no más de 30 (p. ej., no más de 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, nueve, ocho, siete, seis, cinco, cuatro, tres, o dos) sustituciones, inserciones, o deleciones de aminoácidos con respecto a la región constante nativa de la que se obtuvo. En algunas realizaciones, la región constante de Fc variante comprende una o más sustituciones de aminoácidos seleccionadas del grupo que consiste en: M252Y, S254T, T256e , N434S, M428L, V259I, T250a, y V308F. En algunas realizaciones, la región constante de Fc humana variante comprende una metionina en la posición 428 y una asparragina en la posición 434, cada una en la numeración de EU. En algunas realizaciones, la región constante de Fc variante comprende una doble sustitución 428L/434S como se describe, p. ej., la patente de Estados Unidos Num. 8.088.376.
En algunas realizaciones, la región constante variante comprende una sustitución en la posición del aminoácido 237, 238, 239, 248, 250, 252, 254, 255, 256, 257, 258, 265, 270, 286, 289, 297, 298, 303 ácido, 305, 307, 308, 309, 311, 312, 314, 315, 317, 325, 332, 334, 360, 376, 380, 382, 384, 385, 386, 387, 389, 424, 428, 433, 434 o 436 (numeración EU) con respecto a la región constante de Fc humana nativa. En algunas realizaciones, la sustitución se selecciona del grupo que consiste en: metionina por glicina en la posición 237; alanina por prolina en la posición 238; lisina por serina en la posición 239; isoleucina por lisina en la posición 248; alanina, fenilalanina, isoleucina, metionina, glutamina, serina, valina, triptófano, o tirosina por treonina en la posición 250; fenilalanina, triptófano o tirosina por metionina en la posición 252; treonina por serina en la posición 254; ácido glutámico por arginina en la posición 255; ácido aspártico, ácido glutámico, o glutamina por treonina en la posición 256; alanina, glicina, isoleucina, leucina, metionina, asparragina, serina, treonina, o valina por prolina en la posición 257; histidina por ácido glutámico en la posición 258; alanina por ácido aspártico en la posición 265; fenilalanina por ácido aspártico en la posición 270; alanina, o ácido glutámico por asparragina en la posición 286; histidina por treonina en la posición 289; alanina por asparragina en la posición 297; glicina por serina en la posición 298; alanina por valina en la posición 303; alanina por valina en la posición 305; alanina, ácido aspártico, fenilalanina, glicina, histidina, isoleucina, lisina, leucina, metionina, asparragina, prolina, glutamina, arginina, serina, valina, triptófano, o tirosina por treonina en la posición 307; alanina, fenilalanina, isoleucina, leucina, metionina, prolina, glutamina, o treonina por valina en la posición 308; alanina, ácido aspártico, ácido glutámico, prolina, o arginina por leucina o valina en la posición 309; alanina, histidina, o isoleucina o glutamina en la posición 311; alanina o histidina por ácido aspártico en la posición 312; lisina o arginina por leucina en la posición 314; alanina o histidina por asparragina en la posición 315; alanina por lisina en la posición 317; glicina por asparragina en la posición 325; valina por isoleucina en la posición 332; leucina por lisina en la posición 334; histidina por lisina en la posición 360; alanina por ácido aspártico en la posición 376; alanina por ácido glutámico en la posición 380; alanina por ácido glutámico en la posición 382; alanina por asparragina o serina en la posición 384; ácido aspártico o histidina por glicina en la posición 385; prolina por glutamina en la posición 386; ácido glutámico por prolina en la posición 387; alanina o serina por asparragina en la posición 389; alanina por serina en la posición 424; alanina, ácido aspártico, fenilalanina, glicina, histidina, isoleucina, lisina, leucina, asparragina, prolina, glutamina, serina, treonina, valina, triptófano, o tirosina por metionina en la posición 428; lisina por histidina en la posición 433; alanina, fenilalanina, histidina, serina, triptófano, o tirosina por asparragina en la posición 434; e histidina por tirosina o fenilalanina en la posición 436, todas en la numeración EU.
Un anticuerpo anti-C5 descrito en la presente memoria puede, en algunas realizaciones, comprender un polipéptido de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 12 o 14 y/o un polipéptido de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 8 o 11. Métodos para producir los anticuerpos anti-C5 y fragmentos de unión a antígeno de los mismos
La descripción también incluye métodos para producir cualquiera de los anticuerpos anti-C5 o fragmentos de unión a
antígeno de los mismos descritos en la presente memoria. En algunas realizaciones, los métodos para la preparación de un anticuerpo descrito en la presente memoria pueden incluir la inmunización de un sujeto (p. ej., un mamífero no humano) con un inmunógeno apropiado. Los inmunógenos adecuados para la generación de cualquiera de los anticuerpos descritos en la presente memoria se exponen en la presente memoria. Por ejemplo, para generar un anticuerpo que se une a C5, un experto en la técnica puede inmunizar un sujeto adecuado (p. ej., un mamífero no humano tal como una rata, un ratón, un hámster, un hámster, un perro, un gato, un cerdo, una cabra, un caballo, o un primate no humano) con un polipéptido C5 completo tal como un polipéptido C5 humano completo. En algunas realizaciones, el mamífero no humano tiene deficiencia de C5, por ejemplo, un ratón con deficiencia de C5 es descrito, por ejemplo, por Levy y Ladda (1971) Nat New Biol 229(2):51-52; Crocker et al. (1974) J Clin Pathol 27(2):122-124; Wetsel et al. (1990) J Biol Chem 265:2435-2440; y Jungi y Pepys (1981) Immunology 43(2):271-279.
Un sujeto adecuado (p. ej., un mamífero no humano) puede ser inmunizado con el antígeno apropiado junto con inmunizaciones de refuerzo posteriores un número de veces suficiente para provocar la producción de un anticuerpo por el mamífero. El inmunógeno puede administrarse a un sujeto (p. ej., un mamífero no humano) con un coadyuvante. Los coadyuvantes útiles en la producción de un anticuerpo en un sujeto incluyen, pero no se limitan a, coadyuvantes de proteínas; coadyuvantes bacterianos, por ejemplo, bacterias completas (BCG, Corynebacterium parvum o Salmonella minnesota) y componentes bacterianos que incluyen esqueleto de la pared celular, dimicolato de trehalosa, monofosforil lípido A, residuo extraíble con metanol (MER) de bacilo de la tuberculosis, coadyuvante completo o incompleto de Freund; coadyuvantes virales; coadyuvantes químicos, por ejemplo, hidróxido de aluminio, y yodoacetato y hemisuccinato de colesterilo. Otros coadyuvantes que se pueden utilizar en los métodos para inducir una respuesta inmunitaria incluyen, por ejemplo, la toxina del cólera y las proteínas de parapoxvirus. Véase también Bieg et al. (1999) Autoimmunity 31(1):15-24. Véanse también, p. ej., Lodmell et al. (2000) Vaccine 18:1059-66; Johnson et al. (1999) J Med Chem 42:4640-4649; Baldridge et al. (1999) Methods 19:103-107; y Gupta et al. (1995) Vaccine 13(14):1263-76.
En algunas realizaciones, los métodos incluyen la preparación de una línea celular de hibridoma que secreta un anticuerpo monoclonal que se une al inmunógeno. Por ejemplo, un mamífero adecuado, tal como un ratón de laboratorio se inmuniza con un polipéptido C5 como se ha descrito anteriormente. Las células productoras de anticuerpos (p. ej., las células B del bazo) del mamífero inmunizado se pueden aislar de dos a cuatro días después de al menos una inmunización de refuerzo del inmunógeno y desarrollar a continuación brevemente en cultivo antes de la fusión con células de una línea celular de mieloma adecuada. Las células se pueden fusionar en presencia de un promotor de fusión tal como, p. ej., virus vaccinia o polietilenglicol. Las células híbridas obtenidas en la fusión se clonan, y se seleccionan clones de células que secretan los anticuerpos deseados. Por ejemplo, las células de bazo de ratones Balb/c inmunizados con un inmunógeno adecuado se pueden fusionar con células de la línea celular de mieloma PAI o la línea celular de mieloma Sp2/0-Ag 14. Después de la fusión, las células se expanden en un medio de cultivo adecuado, que se complementa con un medio de selección, por ejemplo medio HAT, a intervalos regulares con el fin de evitar que las células de mieloma normales desborden las células de hibridoma deseadas. Las células híbridas obtenidas se escrutan a continuación para determinar la secreción de los anticuerpos deseados, por ejemplo, un anticuerpo que se une a C5 e inhibe la escisión de C5 en fragmentos C5a y C5b.
En algunas realizaciones, cualquiera de los anticuerpos o fragmentos de unión a antígeno de los mismos descritos en la presente memoria se puede fabricar en una célula CHO. En algunas realizaciones, los anticuerpos o fragmentos de unión a antígeno de los mismos no contienen residuos de ácido siálico detectables.
En algunas realizaciones, un experto en la técnica puede identificar un anticuerpo anti-C5 de una biblioteca sesgada no inmunitaria como se describe, por ejemplo, en la patente de Estados Unidos Núm. 6.300.064 (de Knappik et al.; Morphosys AG) y Schoonbroodt et al. (2005) Nucleic Acids Res 33(9):e81.
Una subpoblación de anticuerpos seleccionados que utilizan los métodos anteriores puede ser caracterizada para determinar su especificidad y afinidad de unión para un inmunógeno concreto (p. ej., C5) utilizando cualquier método basado en inmunología o bioquímica conocido en la técnica. Por ejemplo, la unión específica de un anticuerpo a C5 completo, nativo, en comparación con C5a, se puede determinar, por ejemplo, utilizando métodos basados en inmunología o bioquímica tales como, pero no limitados a, un análisis ELISA, análisis de RPS, análisis de inmunoprecipitación, cromatografía de afinidad, y diálisis en equilibrio como se describe anteriormente. Los inmunoanálisis que se pueden utilizar para analizar la unión inmunoespecífica y la reactividad cruzada de los anticuerpos incluyen, pero no se limitan a, sistemas de análisis competitivos y no competitivos que utilizan técnicas tales como transferencias Western, RIA, ELISA (análisis de inmunoabsorción ligado a enzimas), "inmunoanálisis sándwich", análisis de inmunoprecipitación, análisis de inmunodifusión, análisis de aglutinación, análisis de fijación del complemento, análisis inmunorradiométricos, inmunoanálisis fluorescentes, e inmunoanálisis de proteína A. Tales análisis son rutinarios y bien conocidos en la técnica.
Los anticuerpos también pueden analizarse utilizando cualquier análisis basado en RPS conocido en la técnica para la caracterización de los parámetros cinéticos de la interacción del anticuerpo con C5. Cualquier aparato RPS disponible comercialmente, incluyendo, pero no limitado a, aparatos BIAcore (Biacore AB; Uppsala, Suecia); aparatos LASYS (Affinity Sensors; Franklin, Massachusetts); sistema IBIS (Windsor Scientific Limited; Berks, UK),
sistemas de RPS-CELLIA (Nippon Laser and Electrónics Lab; Hokkaido, Japón), y SPR Detector Spreeta (Texas Instruments; Dallas, Texas) se puede utilizar en los métodos descritos en la presente memoria. Véase, p. ej., Mullett et al. (2000) Methods 22:77-91; Dong et al. (2002) Curr Opin Biotechnol Reviews in Mol Biotech 82:303-323; Fivash et al. (1998) Curr Opin Biotechnol 9:97-101; y Rich et al. (2000) Curr Opin Biotechnol 11:54-61.
Se entiende que los métodos anteriores también se pueden utilizar para determinar si, por ejemplo, un anticuerpo anti-C5 no se une a, proteínas C3 y/o C4 completas nativas.
Como se describe en las referencias anteriores, después de la selección de los fagos, las regiones codificantes de anticuerpo del fago pueden aislarse y utilizarse para generar anticuerpos completos, incluyendo anticuerpos humanos, o fragmentos deseados cualesquiera, y expresarse en cualquier anfitrión deseado, incluyendo células de mamífero, células de insecto, células vegetales, levaduras y bacterias, por ejemplo, como se describe en detalle a continuación. Por ejemplo, las técnicas para producir recombinantemente fragmentos Fab, Fab' y F(ab')2 también pueden emplearse utilizando métodos conocidos en la técnica tales como los descritos en las publicaciones PCT Núm. WO 92/22324; Mullinax et al. (1992) BioTechniques 12(6):864-869; y Sawai et al. (1995) Am J Immunol Repr 34:26-34; y Better et al. (1988) Science 240:1041-1043. Los ejemplos de las técnicas que se pueden utilizar para producir Fv y anticuerpos de cadena sencilla incluyen los descritos en las Patentes de estados Unidos Núm.
4.946.778 y 5.258.498; Huston et al. (1991) Methods in Enzymology 203:46-88; Shu et al. (1993) Proc Nat Acad Sci USA 90:7995-7999; y Skerra et al. (1988) Science 240:1038-1040.
En algunas realizaciones, se puede utilizar el mapeo de epítopos para identificar, por ejemplo, la región de C5 que interactúa con un anticuerpo. Los métodos para identificar el epítopo al que se une un anticuerpo concreto también son conocidos en la técnica y se han descrito anteriormente.
Los anticuerpos y fragmentos de los mismos identificados en la presente memoria pueden ser o se pueden hacer "quiméricos". Los anticuerpos quiméricos y fragmentos de unión a antígeno de los mismos comprenden porciones de dos o más especies diferentes (p. ej., ratón y ser humano). Los anticuerpos quiméricos se pueden producir con las regiones variables de ratón de especificidad deseada fusionadas a los dominios constantes humanos (p. ej., Patente de Estados Unidos Num. 4,816,567). De esta manera, los anticuerpos no humanos pueden ser modificados para que sean más adecuados para la aplicación clínica humana (p. ej., métodos para tratar o prevenir un trastorno mediado por el complemento en un sujeto).
Los anticuerpos monoclonales de la presente divulgación incluyen formas "humanizadas" de los anticuerpos no humanos (p. ej., ratón). Los mAbs humanizados o injertados con CDR son particularmente útiles como agentes terapéuticos para seres humanos debido a que no se eliminan de la circulación tan rápidamente como los anticuerpos de ratón y no suele provocar una reacción inmunitaria adversa. Generalmente, un anticuerpo humanizado tiene uno o más residuos de aminoácidos introducidos en él de una fuente no humana. Estos residuos de aminoácidos no humanos se denominan frecuentemente como residuos "importados", que típicamente se obtienen a partir de un dominio variable "de importación". Los métodos para preparar anticuerpos humanizados son generalmente bien conocidos en la técnica. Por ejemplo, la humanización puede realizarse esencialmente siguiendo el método de Winter y colaboradores (véanse, p. ej., Jones et al. (1986) Nature 321:522-525; Riechmann et al. (1988) Nature 332:323-327; y Verhoeyen et al. (1988) Science 239:1534-1536), mediante la sustitución de los marcos de roedores o secuencias de CDR por las secuencias correspondientes de un anticuerpo humano. Véase también, por ejemplo, Staelens et al. (2006) Mol Immunol 43:1243-57. En algunas realizaciones, las formas humanizadas de anticuerpos no humanos (p. ej., de ratón) son anticuerpos humanos (anticuerpo receptor) en las que los residuos de aminoácido de la región CDR del anticuerpo no humano (p. ej., anticuerpo de ratón, rata, conejo, o primate no humano) que tiene la especificidad, afinidad y capacidad de unión deseadas se injertan en el armazón marco de un anticuerpo humano.
En algunos casos, uno o más residuos de aminoácido de la región marco de la inmunoglobulina humana se remplazan también por los residuos de aminoácidos correspondientes del anticuerpo no humano (denominadas "retromutaciones"). Además, se pueden utilizar bibliotecas de presentación en fagos para variar los aminoácidos en las posiciones elegidas dentro de la secuencia del anticuerpo. Las propiedades de un anticuerpo humanizado también se ven afectadas por la elección del marco humano. Además, los anticuerpos humanizados y quimerizados pueden ser modificados para que comprendan residuos que no se encuentran en el anticuerpo receptor o en el anticuerpo donador con el fin de mejorar adicionalmente las propiedades de los anticuerpos, tales como, por ejemplo, la afinidad o la función efectora.
También se proporcionan en la descripción anticuerpos completamente humanos. El término "anticuerpo humano" incluye anticuerpos que tienen regiones variables y constantes (si estuvieran presentes) derivadas de secuencias de inmunoglobulina humana, preferiblemente secuencias de la línea germinal humana. Los anticuerpos humanos pueden incluir residuos de aminoácidos no codificados por secuencias de inmunoglobulina de la línea germinal humana (p. ej., mutaciones introducidas por mutagénesis aleatoria o específica de sitio in vitro o por mutación somática in vivo). Sin embargo, el término "anticuerpo humano" no incluye anticuerpos en los que las secuencias de CDR derivadas de otras especies de mamíferos, tales como un ratón, han sido injertadas en secuencias marco humanas (es decir, anticuerpos humanizados). Los anticuerpos completamente humanos o humanos pueden
obtenerse de ratones transgénicos que llevan genes de anticuerpos humanos (que llevan los exones variable (V), de diversidad (D), de unión (J) y constante) C) o de células humanas.
Las secuencias humanas pueden codificar las cadenas tanto pesadas como ligeras de los anticuerpos humanos y funcionarían correctamente en los ratones, experimentando reordenación para proporcionar un amplio repertorio de anticuerpos similar al de los seres humanos. Los ratones transgénicos pueden inmunizarse con el inmunógeno proteína diana para crear una amplia gama de anticuerpos específicos y su ARN codificante. Los ácidos nucleicos que codifican los componentes de la cadena de anticuerpo de dichos anticuerpos pueden ser clonados a continuación a partir del animal en un vector de presentación. Típicamente, se clonan poblaciones separadas de ácidos nucleicos que codifican secuencias de cadena pesada y ligera, y las poblaciones separadas son recombinadas a continuación en la inserción en el vector, de tal manera que cualquier copia dada del vector recibe una combinación aleatoria de una cadena pesada y una ligera. El vector está diseñado para expresar cadenas de anticuerpo de modo que puedan ser ensambladas y presentadas en la superficie exterior de un paquete de presentación que contiene el vector. Por ejemplo, las cadenas de anticuerpo pueden expresarse como proteínas de fusión con una proteína de la envoltura del fago de la superficie exterior del fago. Después de eso, los paquetes de visualización se pueden seleccionar y se escrutar para la visualización de anticuerpos que se unen a una diana.
En algunas realizaciones, los anticuerpos anti-C5 descritos en la presente memoria comprenden una región alterada de la cadena pesada constante que tiene la función efectora reducida (o nula) con respecto a su correspondiente región constante inalterada. Las funciones efectoras que implican la región constante del anticuerpo anti-C5 se pueden modular mediante la alteración de propiedades de la región constante o Fc. Las funciones efectoras alteradas incluyen, por ejemplo, una modulación de uno o más de las siguientes actividades: la citotoxicidad celular dependiente de anticuerpos (CCDA), la citotoxicidad dependiente del complemento (CDC), la apoptosis, la unión a uno o más receptores de Fc, y las respuestas pro-inflamatorias. Modulación se refiere a un aumento, disminución o eliminación de una actividad de función efectora exhibida por un anticuerpo sujeto que contiene una región constante alterada en comparación con la actividad de la forma inalterada de la región constante. En realizaciones concretas, la modulación incluye situaciones en las que una actividad se suprime o está completamente ausente.
Una región constante alterada con afinidad de unión a FcR y/o actividad CCDA alteradas y/o actividad CDC alterada es un polipéptido que tiene mayor o menor actividad unión a FcR y/o actividad CCDa y/o actividad CDC en comparación con la forma inalterada de la región constante. Una región constante alterada que presenta aumento de la unión a un FcR se une al menos a un FcR con mayor afinidad que el polipéptido sin alterar. Una región constante alterada que muestra disminución de la unión a un FcR se une al menos a un FcR con menor afinidad que la forma inalterada de la región constante. Tales variantes que exhiben disminución de la unión a un FcR pueden tener poca o ninguna unión apreciable unirse a un FcR, por ejemplo, de 0 a 50% (p. ej., menos de 50, 49, 48, 47, 46, 45, 44, 43, 42, 41, 40, 39, 38, 37, 36, 35, 34, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, o 1%) de la unión al FcR en comparación con el nivel de unión de una secuencia nativa constante de inmunoglobulina o una región Fc al FcR. Del mismo modo, una región constante alterada que presenta actividad CCDA y/o CDC modulada puede mostrar aumento o reducción de la actividad CCDA y/o CDC en comparación con la región constante inalterada. Por ejemplo, en algunas realizaciones, el anticuerpo anti-C5 que comprende una región constante alterada puede exhibir aproximadamente de 0 a 50% (p. ej., menos de 50, 49, 48, 47, 46, 45, 44, 43, 42, 41, 40, 39, 38, 37, 36, 35, 34, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, o 1%) de la actividad CCDA y/o CDC de la forma inalterada de la región constante. Un anticuerpo anti-C5 descrito en la presente memoria que comprende una región constante alterada que presenta CCDA y/o CDC reducidas puede exhibir actividad CCDA y/o CDC reducida o no.
En ciertas realizaciones, la región constante alterada tiene al menos una sustitución, inserción y/o deleción de aminoácido, en comparación con una región constante de secuencia nativa o con la región constante sin alteraciones, por ejemplo, de aproximadamente una a aproximadamente un centenar de sustituciones, inserciones y/o deleciones de aminoácidos en una región constante de secuencia nativa o en la región constante del polipéptido parental. En algunas realizaciones, la región constante alterada en la presente memoria poseerá al menos alrededor de 70% de homología (similitud) o identidad con la región constante inalterada y en algunos casos al menos aproximadamente 75% y en otros casos al menos aproximadamente 80% de homología o identidad con la misma, y en otras realizaciones al menos aproximadamente 85%, 90% o 95% de homología o identidad con la misma. La región constante alterada también puede contener una o más deleciones o inserciones de aminoácidos. Además, la región constante alterada puede contener una o más sustituciones, deleciones, o inserciones de aminoácidos que dan como resultado modificaciones post-traduccionales alteradas, incluyendo, por ejemplo, un patrón de glicosilación alterado (p. ej., la adición de uno o más componentes de azúcar, la pérdida de uno o más componentes de azúcar, o un cambio en la composición de uno o más componentes de azúcar con respecto a la región constante inalterada).
Los anticuerpos con funciones efectoras alteradas o sin las mismas pueden ser generados modificando genéticamente o produciendo anticuerpos con regiones constantes variantes, Fc, o de cadena pesada; se pueden utilizar tecnología de ADN recombinante y/o condiciones de cultivo y expresión celular pueden ser utilizados para producir anticuerpos con función y/o actividad alteradas. Por ejemplo, la tecnología de a Dn recombinante se puede utilizar para diseñar una o más sustituciones, deleciones, o inserciones de aminoácidos en regiones (tales como, por
ejemplo, regiones Fc o constantes) que afectan a la función de los anticuerpos incluyendo las funciones efectoras. Alternativamente, se pueden lograr cambios en modificaciones post-traduccionales, tales como, p. ej., los patrones de glicosilación, mediante la manipulación del cultivo celular y las condiciones de expresión por medio de las cuales se produce el anticuerpo. Los métodos adecuados para la introducción de una o más sustituciones, adiciones o deleciones en una región Fc de un anticuerpo son bien conocidos en la técnica e incluyen, por ejemplo, técnicas de mutagénesis de ADN convencionales como las descritas, por ejemplo, por Sambrook et al. (1989) en "Molecular Cloning: A Laboratory Manual, 2a Edición," Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; Harlow y Lane (1988), más arriba; Borrebaek (1992), más arriba; Johne et al. (1993), más arriba; Publicación PCT Núm. WO 06/53301; y Patente de Estados Unidos Núm. 7.704.497.
En algunas realizaciones, un anticuerpo anti-C5 descrito en la presente memoria muestra función efectora reducida o nula. En algunas realizaciones, un anticuerpo anti-C5 comprende una región constante híbrida, o una porción de la misma, tal como una región constante G2/G4 híbrida (véanse, por ejemplo, Burton et al. (1992) Adv Immun 51:1-18; Canfield et al. (1991) J Exp Med 173:1483-1491; y Mueller et al. (1997) Mol Immunol 34(6):441-452). Véase más arriba.
Además de utilizar un constructo G2/G4 como se describió anteriormente, se puede producir un anticuerpo anti-C5 descrito en la presente memoria que tiene la disminución de la función efectora mediante la introducción de otros tipos de cambios en la secuencia de aminoácidos de ciertas regiones del anticuerpo. Tales cambios en la secuencia de aminoácidos incluyen pero no se limitan a la mutación Ala-Ala descrita, por ejemplo, en las Publicaciones PCT Núm. WO 94/28027 y WO 98/47531; y Xu et al. (2000) Cell Immunol 200:16-26. Por lo tanto, en algunas realizaciones, un anticuerpo anti-C5 con una o más mutaciones dentro de la región constante que incluye la mutación Ala-Ala tiene función efectora reducida o nula. De acuerdo con estas realizaciones, la región constante del anticuerpo puede comprender una sustitución a una alanina en la posición 234 o una mutación a una alanina en la posición 235. Además, la región constante alterada puede contener una mutación doble: una mutación a una alanina en la posición 234 y una segunda mutación a una alanina en la posición 235. En una realización, un anticuerpo anti-C5 comprende un marco de IgG4, en donde la mutación Ala-Ala describiría una o varias mutaciones de fenilalanina a alanina en la posición 234 y/o una mutación de leucina a alanina en la posición 235. En otra realización, el anticuerpo anti-C5 comprende un marco de IgG1, en donde la mutación Ala-Ala describiría una o varia mutaciones de leucina a alanina en la posición 234 y/o una mutación de leucina a alanina en la posición 235. Un anticuerpo anti-C5 puede llevar alternativa o adicionalmente otras mutaciones, incluyendo la mutación puntual K322A en el dominio CH2 (Hezareh et al. (2001) J Virol 75:12161-12168). Un anticuerpo con dicha mutación o dichas mutaciones en la región constante puede ser, además, un anticuerpo bloqueador o no bloqueador.
Las sustituciones adicionales que, cuando se introduce en una región constante de cadena pesada, dan como resultado una disminución de la función efectora son expuestos, por ejemplo, por Shields et al. (2001) en J Biol Chem 276(9):6591-6604. Véase concretamente la Tabla 1 "Binding of human IgG1 variants to human FcRn and FcyR) de Shields et al. Mediante el escrutinio de una biblioteca de anticuerpos anti-IgE, cada anticuerpo de la biblioteca que difiere en una o más sustituciones en la región constante de cadena pesada, para su unión a un panel de receptores de Fc (incluyendo FcRn, FcyRI, FcyRIIA, FcyRIIB, and FcyRIIIA), los autores identificaron una serie de sustituciones que modulan las interacciones específicas de los receptores de Fc-Fc. Por ejemplo, una región constante de cadena pesada de IgG2a variante en la que el dominio CH2 contiene una sustitución D265A (numeración de aminoácidos de cadena pesada de acuerdo con Kabat et al. (más arriba)) da como resultado una pérdida completa de la interacción entre la región constante de la variante y los receptores de Fc de IgG FcyRIIB, FcyRIII, FcyRi, and FcyRIV. Shields et al. (2001) en la página 6595, Tabla 1. Véase también Baudino et al. (2008) J Immunol 181:6664-6669 (más arriba).
Los cambios dentro de la región bisagra también afectan a las funciones efectoras. Por ejemplo, la supresión de la región bisagra puede reducir afinidad por los receptores de Fc y puede reducir la activación del complemento (Klein et al. (1981) Proc Natl Acad Sci USA 78: 524-528). La presente descripción por lo tanto también se refiere a anticuerpos con alteraciones en la región bisagra.
En algunas realizaciones, un anticuerpo anti-C5 puede contener una región constante alterada que exhibe mayor o menor citotoxicidad dependiente del complemento (CDC). La actividad de CDC modulada puede lograrse introduciendo una o más sustituciones, inserciones, o deleciones de aminoácidos en una región Fc del anticuerpo. Véase, p. ej., la patente de Estados Unidos Núm. 6.194.551. Alternativamente o adicionalmente, se pueden introducir uno o varios residuos de cisteína en la región Fc, permitiendo de ese modo la formación de enlaces disulfuro intercatenarios en esta región. El anticuerpo homodimérico así generado puede haber mejorado o reducido la capacidad de internalización y/o aumentado o disminuido la muerte celular mediada por el complemento. Véase, p. ej., Caron et al. (1992) J Exp Med 176:1191-1195 y Shopes (1992) Immunol 148:2918-2922; Publicaciones PCT Núm. WO 99/51642 y WO 94/29351; Duncan y Winter (1988) Nature 322:738-40; y Patentes de Estados Unidos Núm. 5.648.260 y 5.624.821.
Otro medio potencial de modulación de la función efectora de los anticuerpos incluye cambios en la glicosilación, resumidos, p. ej., por Raju (2003) BioProcess International 1(4):44-53. De acuerdo con Wright y Morrison, la microheterogeneidad de oligosacáridos de IgG humana puede afectar a funciones biológicas tales como la CDC y la
CCDA, la unión a varios receptores Fc, y la unión a la proteína Clq. (1997) TIBTECH 15:26-32. Los patrones de glicosilación de anticuerpos pueden variar dependiendo de la célula productora y de las condiciones de cultivo celular (Raju, más arriba). Tales diferencias pueden conducir a cambios tanto en la función efectora como en la farmacocinética. Véase, p. ej., Israel et al. (1996) Immunology 89(4):573-578; y Newkirk et al. (1996) Clin Exp Immunol 106(2):259-264. Las diferencias en la función efectora pueden estar relacionadas con la capacidad de la IgG para unirse a los receptores de Fc gamma (FcyR) en las células efectoras. Shields et al. han demostrado que IgG, con alteraciones en la secuencia de aminoácidos que han mejorado de unión a FcyR, puede exhibir CCDA mejorada hasta 100% utilizando células efectoras humanas. (2001) J Biol Chem 276(9):6591-6604. Aunque estas alteraciones incluyen cambios en los aminoácidos que no se encuentran en la interfase de unión, tanto la naturaleza del componente azúcar, como su patrón estructural también pueden contribuir a las diferencias observadas. Además, la presencia o ausencia de fucosa en el componente oligosacárido de una IgG pueden mejorar la unión y la CCDA. Véase, p. ej., Shields et al. (2002) J Biol Chem 277(30):26733-26740. Una IgG que carecía de un hidrato de carbono fucosilado conectado a Asn297 exhibió una unión normal al receptor, al receptor FcyRI. Por el contrario, la unión al receptor FcRIIIa mejoró 50 veces y estuvo acompañada de CCDA mejorada, especialmente a concentraciones de anticuerpo inferiores.
Aún existen otros enfoques para alterar la función efectora de los anticuerpos. Por ejemplo, las células productoras de anticuerpos pueden ser hipermutagénicas, generando de ese modo anticuerpos con residuos de polipéptidos alterados aleatoriamente a través de una molécula de anticuerpo entera. Véase, p. ej., la publicación PCT Núm. WO 05/011735. Las células anfitrionas hipermutagénicas incluyen células con deficiencia en la reparación de ADN con emparejamientos erróneos. Los anticuerpos producidos de esta manera pueden ser menos antigénicos y/o tener propiedades farmacocinéticas beneficiosas. Además, dichos anticuerpos se pueden seleccionar para determinar propiedades tales como el aumento o la disminución de la función o las funciones efectoras. Los detalles adicionales de las técnicas de biología molecular útiles para preparar un anticuerpo o fragmento de unión a antígeno del mismo descritos en la presente memoria se exponen a continuación.
Expresión y purificación de anticuerpos recombinantes
Los anticuerpos o fragmentos de unión a antígeno de los mismos descritos en la presente memoria se pueden producir utilizando una variedad de mecanismos conocidos en la técnica de la biología molecular y la química de proteínas. Por ejemplo, un ácido nucleico que codifica uno o ambos polipéptidos de cadena pesada y ligera de un anticuerpo se pueden insertar en un vector de expresión que contiene secuencias reguladoras de la transcripción y la traducción, que incluyen, por ejemplo, secuencias promotoras, sitios de unión al ribosoma, secuencias de inicio y parada de la transcripción, secuencias de inicio y parada de la traducción, señales terminadoras de la transcripción, señales de poliadenilación, y secuencias potenciadoras o activadoras. Las secuencias reguladoras incluyen un promotor y secuencias de inicio y parada de la transcripción. Además, el vector de expresión puede incluir más de un sistema de replicación de tal manera que puede mantenerse en dos organismos diferentes, por ejemplo en células de mamífero o insecto para su expresión y en un anfitrión procariótico para su clonación y amplificación.
Se pueden introducir diversas modificaciones, p. ej., sustituciones, en las secuencias de ADN que codifican los polipéptidos de cadena pesada y/o de ligera descritos en la presente memoria utilizando métodos convencionales conocidos por los expertos en la técnica. Por ejemplo, la introducción de una sustitución de histidina en una o más posiciones de la CDR de un anticuerpo puede llevarse a cabo utilizando métodos convencionales, tales como mutagénesis mediada por PCR, en la que los nucleótidos mutados se incorporan a los cebadores de PCR de tal manera que el producto de PCR contiene las mutaciones deseadas o la mutagénesis dirigida al sitio. Se puede introducir una sustitución en una o más de las regiones CDR para aumentar o disminuir la Kd del anticuerpo para el antígeno, por ejemplo, a pH 7,4 o pH 6,0. Los mecanismos en la mutagénesis dirigida al sitio son bien conocidos en la técnica. Véase, p. ej., Sambrook et al., más arriba.
Varios sistemas de vectores posibles están disponibles para la expresión de polipéptidos de cadena pesada y de cadena ligera clonados a partir de ácidos nucleicos en células de mamífero. Una clase de vectores se basa en la integración de las secuencias génicas deseadas en el genoma de la célula anfitriona. Las células que han integrado de forma estable el ADN se pueden seleccionar introduciendo simultáneamente genes de resistencia a fármacos como gtp de E. coli (Mulligan y Berg (1981) Proc Natl Acad Sci USA 78:2072) o Tn5 neo (Southern y Berg (1982) Mol Appl Genet 1:327). El gen marcador seleccionable se puede conectar a las secuencias génicas de ADN que se van a expresar, o introducir en la misma célula mediante co-transfección (Wigler et al. (1979) Cell 16:77). Una segunda clase de vectores utiliza elementos de ADN que confieren capacidades de replicación autónoma a un plásmido extracromosómico. Estos vectores se pueden obtener a partir de virus animales, tales como el virus del papiloma bovino (Sarver et al. (1982) Proc Natl Acad Sci USA, 79:7147), citomegalovirus, virus del polioma (Decanos y col. (1984) Proc Natl Acad Sci USA 81:1292), o virus SV40 (Lusky y Botchan (1981) Nature 293:79).
Los vectores de expresión pueden ser introducidos en las células de una manera adecuada para la posterior expresión del ácido nucleico. El método de introducción es dictado en gran medida por el tipo de célula diana, comentado a continuación. Los métodos ilustrativos incluyen precipitación con CaPO4, fusión de liposomas, liposomas catiónicos, electroporación, infección viral, transfección mediada por dextrano, transfección mediada por polibreno, fusión de protoplastos, y microinyección directa.
Las células anfitrionas adecuadas para la expresión de anticuerpos o fragmentos de unión a antígeno de los mismos incluyen células de levadura, bacterias, insectos, plantas, y mamífero. Tienen un particular interés bacterias tales como E. coli, hongos tales como Saccharomyces cerevisiae y Pichia pastoris, células de insectos tales como SF9, líneas celulares de mamífero (p. ej., líneas de células humanas), así como líneas de células primarias.
En algunas realizaciones, un anticuerpo o fragmento del mismo pueden ser expresados en, y purificados a partir de, animales transgénicos (p. ej., mamíferos transgénicos). Por ejemplo, un anticuerpo puede ser producido en mamíferos transgénicos no humanos (p. ej., roedores) y aislado de la leche como describe, por ejemplo, Houdebine (2002) en Curr Opin Biotechnol 13 (6): 625-629; van Kuik-Romeijn et al. (2000) Transgenic Res 9(2):155-159; y Pollock et al. (1999) J Immunol Methods 231(1-2):147-157.
Los anticuerpos y fragmentos de los mismos pueden ser producidos a partir de las células mediante el cultivo de una célula anfitriona transformada con el vector de expresión que contiene el ácido nucleico que codifica los anticuerpos o fragmentos, en condiciones, y para una cantidad de tiempo, suficiente para permitir la expresión de las proteínas. Tales condiciones para la expresión de la proteína variarán con la elección del vector de expresión y la célula anfitriona, y serán reconocidos fácilmente por un experto en la técnica a través de experimentación de rutina. Por ejemplo, los anticuerpos expresados en E. coli pueden ser replegados a partir de cuerpos de inclusión (véase, por ejemplo, Hou et al. (1998) Cytokine 10:319-30). Los sistemas de expresión bacterianos y métodos para su uso son bien conocidos en la técnica (véase Current Protocols in Molecular Biology, Wiley & Sons y Molecular Cloning - A Laboratory Manual -3a Ed, Cold Spring Harbor Laboratory Press, Nueva York (2001)). La elección de los codones, los vectores de expresión adecuados y las células anfitrionas adecuadas variarán dependiendo de diversos factores, y puede ser optimizada fácilmente según sea necesario. Un anticuerpo (o fragmento del mismo) descrito en la presente memoria puede expresarse en células de mamífero o en otros sistemas de expresión incluyendo pero no limitados a levadura, baculovirus, y sistemas de expresión in vitro (véase, p. ej., Kaszubska et al. (2000) Protein Expression and Purification 18:213-220).
Después de la expresión, se pueden aislar los anticuerpos y fragmentos de los mismos. Los términos "purificado" o "aislado" según se aplican a cualquiera de las proteínas (anticuerpos o fragmentos) descritas en la presente memoria se refieren a un polipéptido que se ha separado o purificado de componentes (p. ej., proteínas u otras moléculas biológicas u orgánicas de origen natural) que naturalmente lo acompañan, p. ej., otras proteínas, lípidos y ácidos nucleicos en un procariota que expresa las proteínas. Típicamente, un polipéptido está purificado cuando constituye al menos 60% (p. ej., al menos 65, 70, 75, 80, 85, 90, 92, 95, 97, o 99%), en peso, de la proteína total en una muestra.
Un anticuerpo o fragmento del mismo se puede aislar o purificar en una variedad de formas conocidas por los expertos en la técnica dependiendo de qué otros componentes estén presentes en la muestra. Los métodos de purificación convencionales incluyen técnicas electroforéticas, moleculares, inmunológicas, y cromatográficas, incluyendo cromatografía de intercambio iónico, hidrófoba, de afinidad, y HPLC de fase inversa. Por ejemplo, un anticuerpo puede ser purificado utilizando una columna de anti-anticuerpo convencional (p. ej., una columna de proteína A o proteína G). También son útiles las técnicas de ultrafiltración y diafiltración, junto con concentración de proteínas, Véase, p. ej., Scopes (1994) "Protein Purification, 3a edición," Springer-Verlag, Nueva York, Nueva York. El grado de purificación necesario variará dependiendo del uso deseado. En algunos casos, no será necesaria la purificación del anticuerpo expresado o fragmentos del mismo.
Los métodos para determinar el rendimiento o la pureza de un anticuerpo purificado o fragmento del mismo son conocidos en la técnica e incluyen, por ejemplo, el análisis de Bradford, espectroscopia UV, análisis de proteínas de Biuret, análisis de proteínas de Lowry, análisis de proteínas con negro-amido, cromatografía líquida de alta presión (HPLC), espectrometría de masas (MS), y métodos electroforéticos en gel (p. ej., utilizando una tinción de proteínas tal como azul de Coomassie o tinción de plata coloidal).
En algunas realizaciones, las endotoxinas se pueden eliminar de los anticuerpos o fragmentos. Los métodos para eliminar las endotoxinas de una muestra de proteína son conocidos en la técnica. Por ejemplo, las endotoxinas se pueden eliminar de una muestra de proteína utilizando una variedad de reactivos disponibles comercialmente, incluyendo, sin limitación, los Kits de Eliminación de Endotoxinas ProteoSpin™ (Norgen Biotek Corporation), el Gel de eliminación de endotoxinas Detoxi-Gel (Thermo Scientific; Protein Pierce Research Products), el Kit de Eliminación de Endotoxinas MiraCLEAN® (Mirus), o la membrana Acrodisc™ - Mustang® E (Pall Corporation). Los métodos para detectar y/o medir la cantidad de endotoxinas presentes en una muestra (tanto antes como después de la purificación) son conocidos en la técnica y están disponibles kits comerciales. Por ejemplo, la concentración de endotoxina en una muestra de proteína se puede determinar utilizando el kit QCL-1000 cromogénico (BioWhittaker) o los kits a base de producto lisado de amebocitos de Limulus (LAL) tales como los kits Pyrotell®, Pyrotell®-T, Pyrochrome®, Chromo-LAL, y el CSE disponibles de Associates of Cape Cod Incorporated. Modificación de los anticuerpos o fragmentos de unión a antígeno de los mismos
Los anticuerpos o fragmentos de unión a antígeno de los mismos pueden ser modificados después de su expresión y purificación. Las modificaciones pueden ser modificaciones covalentes o no covalentes. Tales modificaciones pueden introducirse en los anticuerpos o fragmentos, por ejemplo, haciendo reaccionar residuos de aminoácido diana del polipéptido con un agente derivatizante orgánico que es capaz de reaccionar con cadenas laterales o residuos terminales seleccionados. Los sitios adecuados para la modificación pueden ser elegidos utilizando cualquiera de una variedad de criterios, entre ellos, por ejemplo, el análisis estructural o el análisis de la secuencia de aminoácidos de los anticuerpos o fragmentos.
En algunas realizaciones, los anticuerpos o fragmentos de unión a antígeno de los mismos pueden conjugarse con un radical heterólogo. El radical heterólogo puede ser, por ejemplo, un polipéptido heterólogo, un agente terapéutico (p. ej., una toxina o un fármaco), o una marca detectable tal como, pero no limitada a, una marca radiactiva, una marca enzimática, una marca fluorescente, una marca de metal pesado, una marca luminiscente, o una etiqueta de afinidad tal como biotina o estreptavidina. Los polipéptidos heterólogos adecuados incluyen, por ejemplo, una etiqueta antigénica (p. ej., FLAG (DYKDDDDK (SEQ ID NO: 20)), polihistidina (6-His; HHHHHH (SEQ ID NO: 21)), hemaglutinina (HA; YpYDVPDYA (SEQ ID NO: 22)), glutatión-S-transferasa (GST), o proteína de unión a maltosa (MBP)) para su uso en la purificación de los anticuerpos o fragmentos. Los polipéptidos heterólogos también incluyen polipéptidos (p. ej., enzimas) que son útiles como marcadores de diagnóstico o detectables, por ejemplo, luciferasa, una proteína fluorescente (p. ej., proteína fluorescente verde (GFP)), o cloranfenicol acetil transferasa (CAT). Los marcadores radiactivos adecuados incluyen, p. ej., 32P, 33P, 14C, 125I, 131I, 35S, y 3H. Los marcadores fluorescentes adecuados incluyen, sin limitación, fluoresceína, isotiocianato de fluoresceína (FITC), proteína fluorescente verde (GFP), DyLight 488, ficoeritrina (PE), yoduro de propidio (PI), PerCP, PE-Alexa Fluor® 700, Cy5, aloficocianina, y Cy7. Las etiquetas luminiscentes incluyen, por ejemplo, cualquiera de una variedad de quelatos de lantánidos luminiscentes (p. ej., europio o terbio). Por ejemplo, los quelatos de europio adecuados incluyen el quelato de europio de ácido dietilentriaminopentaacético (DTPA) o ácido tetraazaciclododecano-1,4,7,10-tetraacético (DOTA). Las marcas enzimáticas incluyen, por ejemplo, fosfatasa alcalina, CAT, luciferasa, y peroxidasa de rábano picante.
Dos proteínas (p. ej., un anticuerpo y un radical heterólogo) puede ser entrecruzados utilizando cualquiera de una serie de agentes de entrecruzamiento químico conocidos. Los ejemplos de tales agentes de entrecruzamiento son los que conectan dos residuos de aminoácido mediante una conexión que incluye un enlace disulfuro con "impedimento estérico". En estas conexiones, un enlace disulfuro dentro de la unidad de entrecruzamiento está protegido (por grupos de impedimento estérico a cada lado del enlace disulfuro) de la reducción mediante la acción, por ejemplo, de glutatión reducido o la enzima disulfuro reductasa. Un reactivo adecuado, 4-succinimidiloxicarbonila-metil-a(2-piridilditio)tolueno (SMPT), forma tal conexión entre dos proteínas que utilizan una lisina de terminal en una de las proteínas y una cisteína terminal en la otra. También se pueden utilizar reactivos heterobifuncionales que entrecruzan por medio de un radical de acoplamiento diferente en cada proteína. Otros entrecruzadores útiles incluyen, sin limitación, reactivos que conectan dos grupos amino (p. ej., N-5-azido-2-nitrobenzoiloxisucinimida), dos grupos sulfhidrilo (p. ej., 1,4-bis-maleimidobutano), un grupo amino y un grupo sulfhidrilo (p. ej., éster de mmaleimidobenzoil-N-hidroxisuccinimida), un grupo amino y un grupo carboxilo (p. ej., 4-[pazidosalicilamido]butilamina), y un grupo amino y un grupo guanidinio que está presente en la cadena lateral de la arginina (p. ej., monohidrato de p-azidofenilglioxal).
En algunas realizaciones, se puede conjugar directamente un marcador radioactivo con la cadena principal de aminoácidos del anticuerpo. Alternativamente, el marcador radiactivo puede ser incluido como parte de una molécula más grande, por ejemplo, 125I en meta-[125I]yodofenil-N-hidroxisuccinimida ([125I]mIPNHS) que se une a los grupos amino libres para formar derivados meta-yodofenilo (MIP) de proteínas relevantes (véase, p. ej., Rogers et al. (1997) J Nucl Med 38:1221-1229) o quelato (p. ej., de DOTA o DtpA), que a su vez se une al esqueleto de la proteína. Los métodos de conjugación de las marcas radiactivas o moléculas/quelatos más grandes que las contienen a los anticuerpos o fragmentos de unión a antígeno descritos en la presente memoria son conocidos en la técnica. Tales métodos implican la incubación de las proteínas con la marca radiactiva en condiciones (p. ej., pH, concentración de sal, y/o temperatura) que facilitan la unión de la marca o quelato radiactivos a la proteína (véase, por ejemplo, la patente de Estados Unidos Num. 6.001.329).
Los métodos para conjugar un marcador fluorescente (a veces referido como un "fluoróforo") a una proteína (p. ej., un anticuerpo) son conocidos en la técnica de la química de proteínas. Por ejemplo, los fluoróforos se pueden conjugar con los grupos amino libres (p. ej., de lisinas) o grupos sulfhidrilo (p. ej., cisteína) de proteínas utilizando radicales éster de succinimidilo (NHS) o éster de tetrafluorofenilo (TFP) unidos a los fluoróforos. En algunas realizaciones, los fluoróforos se pueden conjugar con un radical de entrecruzamiento heterobifuncional tal como sulfo-SMCC. Los métodos de conjugación adecuados implican la incubación de una proteína de anticuerpo, o fragmento de la misma, con el fluoróforo en condiciones que facilitan la unión del fluoróforo a la proteína. Véase, p. ej., Welch y Redvanly (2003) "Handbook of Radiopharmaceuticals: Radiochemistry and Applications", John Wiley and Sons (ISBN 0471495603).
En algunas realizaciones, los anticuerpos o fragmentos pueden ser modificados, por ejemplo, con un radical que mejora la estabilización y/o retención de los anticuerpos en la circulación, p. ej., en sangre, suero, u otros tejidos. Por ejemplo, el anticuerpo o fragmento puede ser PEGilado como describen, por ejemplo, Lee et al. (1999) en Bioconjug
Chem 10(6): 973-8; Kinstler et al. (2002) Advanced Drug Deliveries Reviews 54: 477-485; y Roberts et al. (2002) Advanced Drug Delivery Reviews 54:459-476 o HESilados (Fresenius Kabi, Alemania; véase, p. ej., Pavisic et al. (2010) Int J Pharm 387(1-2):110-119). El radical de estabilización puede mejorar la estabilidad o la retención del anticuerpo (o fragmento) en al menos 1,5 (p. ej., al menos 2, 5, 10, 15, 20, 25, 30, 40, o 50 o más) veces.
En algunas realizaciones, los anticuerpos o fragmentos de unión a antígeno de los mismos descritos en la presente memoria pueden ser glicosilados. En algunas realizaciones, un anticuerpo o fragmento de unión a antígeno del mismo descritos en la presente memoria pueden ser sometidos a tratamiento enzimático o químico, o producidos a partir de una célula, de manera que el anticuerpo o fragmento tienen glicosilación reducida o ausente. Los métodos para producir anticuerpos con glicosilación reducida son conocidos en la técnica y se describen, por ejemplo, en la patente de Estados Unidos Núm. 6.933.368; Wright et al. (1991) EMBO J 10(10):2717-2723; y Co et al. (1993) Mol Immunol 30:1361.
Composiciones farmacéuticas y formulaciones
Las composiciones descritas en la presente memoria pueden formularse en forma de una solución farmacéutica, por ejemplo, para la administración a un sujeto para el tratamiento o prevención de un trastorno asociado al complemento. Las composiciones farmacéuticas generalmente incluirán un vehículo farmacéuticamente aceptable. Según se utiliza en la presente memoria, un "vehículo farmacéuticamente aceptable" se refiere a, e incluye, cualquiera y todos los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardantes de absorción, y similares que son fisiológicamente compatibles. Las composiciones pueden incluir una sal farmacéuticamente aceptable, por ejemplo, una sal de adición de ácido o una sal de adición de base (véase, por ejemplo, Berge et al. (1977) J Pharm Sci 66:1-19).
Las composiciones se pueden formular de acuerdo con métodos convencionales. La formulación farmacéutica es una técnica bien establecida, y la describen adicionalmente, por ejemplo, Gennaro (2000) "Remington: The Science and Practice of Pharmacy," 20a Edición, Lippincott, Williams & Wilkins (ISBN: 0683306472); Ansel et al. (1999) "Pharmaceutical Dosage Forms and Drug Delivery Systems," 7a Edicion, Lippincott Williams & Wilkins Publishers (ISBN: 0683305727); y Kibbe (2000) "Handbook of Pharmaceutical Excipients American Pharmaceutical Association," 3a Edición (ISBN: 091733096X). En algunas realizaciones, una composición se puede formular, por ejemplo, en forma de una solución tamponada a una concentración adecuada y conveniente para el almacenamiento a 2-8°C (p. ej., 4°C). En algunas realizaciones, se puede formular una composición para su almacenamiento a una temperatura por debajo de 0°C (p. ej., -20°C o -80°C). En algunas realizaciones, la composición se puede formular para su almacenamiento durante hasta 2 años (p. ej., un mes, dos meses, tres meses, cuatro meses, cinco meses, seis meses, siete meses, ocho meses, nueve meses, 10 meses, 11 meses, 1 año, 1 año y medio o 2 años) a 2-8°C (p. ej., 4°C). Por lo tanto, en algunas realizaciones, las composiciones descritas en la presente memoria son estables durante almacenamiento durante al menos 1 año a 2-8°C (p. ej., 4°C).
Las composiciones farmacéuticas pueden estar en una variedad de formas. Estas formas incluyen, por ejemplo, formas de dosificación líquidas, semisólidas y sólidas, tales como soluciones líquidas (p. ej., soluciones inyectables e infusibles), dispersiones o suspensiones, comprimidos, píldoras, polvos, liposomas y supositorios. La forma preferida depende, en parte, del modo pretendido de administración y aplicación terapéutica. Por ejemplo, las composiciones que contienen una composición destinada a la administración sistémica o local pueden estar en forma de soluciones inyectables o infusibles. De acuerdo con ello, las composiciones se pueden formular para su administración mediante un modo parenteral (p. ej., intravenoso, subcutáneo, intraperitoneal o intramuscular). "Administración parenteral", "administrado parenteralmente", y otras frases gramaticalmente equivalentes, según se utiliza en la presente memoria, se refiere a modos de administración distintos de la administración enteral y tópica, normalmente mediante inyección, e incluyen, sin limitación, intravenosa, intranasal, intraocular, pulmonar, intramuscular, intraarterial, intratecal, intracapsular, intraorbital, intracardíaca, intradérmica, intrapulmonar, intraperitoneal, transtraqueal, subcutánea, subcuticular, intraarticular, subcapsular, subaracnoidea, intraespinal, epidural, intracerebral, intracraneal, intracarotídea e inyección e infusión intraesternal (véase más adelante).
Las composiciones se pueden formular en forma de una solución, microemulsión, dispersión, liposoma, u otra estructura ordenada adecuada para el almacenamiento estable a alta concentración. Las soluciones inyectables estériles se pueden preparar mediante la incorporación de una composición descrita en la presente memoria en la cantidad requerida en un disolvente apropiado con uno o una combinación de ingredientes enumerados anteriormente, según se requiera, seguido de esterilización por filtración. Generalmente, las dispersiones se preparan mediante la incorporación de una composición descrita en la presente memoria a un vehículo estéril que contiene un medio de dispersión alcalino y los otros ingredientes requeridos de los enumerados anteriormente. En el caso de los polvos estériles para la preparación de soluciones inyectables estériles, los métodos para la preparación incluyen secado a vacío y secado mediante congelación que producen un polvo de una composición descrita en la presente memoria más cualquier ingrediente deseado adicional (véase más adelante) a partir de una solución previamente esterilizada mediante filtración de la misma. La fluidez apropiada de una solución se puede mantener, por ejemplo, mediante el uso de un recubrimiento tal como lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersión y mediante el uso de tensioactivos. La absorción prolongada de las composiciones inyectables puede provocarse incluyendo en la composición un reactivo que retrase la absorción, por
ejemplo, sales monoestearato y gelatina.
Las composiciones descritas en la presente memoria también se pueden formular en composiciones de inmunoliposomas. Tales formulaciones se pueden preparar mediante métodos conocidos en la técnica tales como, por ejemplo, los métodos descritos en Epstein et al. (1985) Proc Natl Acad Sci USA 82: 3688; Hwang et al. (1980) Proc Natl Acad Sci USA 77: 4030; y Patentes de Estados Unidos Núm. 4.485.045 y 4.544.545. Los liposomas con aumento del tiempo de circulación se describen, por ejemplo, en la patente de Estados Unidos Num. 5.013.556. En ciertas realizaciones, las composiciones se pueden formular con un vehículo que protegerá el compuesto contra la liberación rápida, tal como una formulación de liberación controlada, que incluye implantes y sistemas de suministro microencapsulados. Se pueden utilizar polímeros biodegradables, biocompatibles, tales como etilenoacetato de vinilo, polianhídridos, ácido poliglicólico, colágeno, poliortoésteres, y poli(ácido láctico). Muchos métodos para la preparación de tales formulaciones son conocidos en la técnica. Véase, p. ej., J. R. Robinson (1978) "Sustained and Controlled Release Drug Delivery Systems," Marcel Dekker, Inc., Nueva York.
En algunas realizaciones, las composiciones se pueden formular en una composición adecuada para la administración intrapulmonar (p. ej., para la administración mediante un inhalador o nebulizador) a un mamífero tal como un ser humano. Los métodos para la formulación de tales composiciones son bien conocidos en la técnica y se describen, por ejemplo, en la publicación de la Solicitud de Patente de Estados Unidos Núm. 20080202513; las Patentes de Estados Unidos Núm. 7.112.341 y 6.019.968; y las Publicaciones PCT Núm. WO 00/061178 y WO 06/122257. Las formulaciones para inhaladores de polvo seco y sistemas adecuados para la administración de las formulaciones se describen, por ejemplo, en la publicación de la Solicitud de Patente de Estados Unidos Núm.
20070235029, la publicación PCT Núm. WO 00/69887; y la patente de Estados Unidos Num. 5.997.848. Las formulaciones adicionales adecuadas para la administración intrapulmonar (así como métodos para la formulación de polipéptidos) se exponen en, por ejemplo, las Publicaciones de las Solicitudes de Patente de Estados Unidos Núm. 20050271660 y 20090110679.
En algunas realizaciones, las composiciones se pueden formular para su suministro al ojo. Según se utiliza en la presente memoria, el término "ojo" se refiere a todos y cada uno de los tejidos y estructuras anatómicas asociados con un ojo. El ojo tiene una pared compuesta de tres capas distintas: la esclerótica exterior, la capa coroides media, y la retina interna. La cámara detrás del cristalino se rellena con un fluido gelatinoso conocido como humor vítreo. En la parte posterior del ojo se encuentra la retina, que detecta la luz. La córnea es un tejido ópticamente transparente, que transmite imágenes a la parte posterior del ojo. La córnea incluye una vía para la penetración de fármacos al ojo. Otras estructuras tisulares anatómicas asociadas con el ojo incluyen el sistema lagrimal de drenaje, que incluye un sistema de secreción, un sistema de distribución y un sistema excretor. El sistema de secreción comprende secretores que son estimulados por medio del parpadeo y el cambio de temperatura debido a la evaporación de las lágrimas y reflejos secretores que tienen un suministro nervioso parasimpático eferente y secretan lágrimas en respuesta a la estimulación física o emocional. El sistema de distribución incluye los párpados y el menisco de lágrimas alrededor de los bordes del párpado de un ojo abierto, que extiende las lágrimas sobre la superficie ocular mediante el parpadeo, lo que reduce la aparición de zonas secas.
En algunas realizaciones, las composiciones se pueden administrar localmente, por ejemplo, a modo de aplicación tópica o inyección intravítrea. Por ejemplo, en algunas realizaciones, las composiciones se pueden formular para su administración por medio de una gota ocular.
La preparación terapéutica para el tratamiento del ojo puede contener uno o más agentes activos a una concentración de aproximadamente 0,01 a aproximadamente 1% en peso, preferiblemente de aproximadamente 0,05 a aproximadamente 0,5% en una solución, suspensión o pomada farmacéuticamente aceptables. La preparación estará preferiblemente en forma de una solución acuosa estéril que contiene, por ejemplo, ingredientes adicionales tales como, pero no limitados a, conservantes, tampones, agentes de tonicidad, antioxidantes y estabilizadores, humectante no iónicos o agentes clarificantes, y agentes que aumentan la viscosidad.
Los conservantes adecuados para su uso en una solución de este tipo incluyen cloruro de benzalconio, cloruro de bencetonio, clorobutanol, timerosal y similares. Los tampones adecuados incluyen, por ejemplo, ácido bórico, bicarbonato de sodio y de potasio, boratos de sodio y de potasio, carbonato de sodio y de potasio, acetato de sodio, y bifosfato de sodio, en cantidades suficientes para mantener el pH entre aproximadamente pH 6 y pH 8, y preferiblemente, entre aproximadamente pH 7 y pH 7,5. Los agentes de tonicidad adecuados son dextrano 40, dextrano 70, dextrosa, glicerina, cloruro de potasio, propilenglicol, y cloruro de sodio.
Los antioxidantes y estabilizadores adecuados incluyen bisulfito de sodio, metabisulfito de sodio, tiosulfito de sodio, y tiourea. Los humectantes y agentes clarificantes adecuados incluyen polisorbato 80, polisorbato 20, poloxámero 282 y tiloxapol. Los agentes que aumentan la viscosidad adecuados incluyen dextrano 40, dextrano 70, gelatina, glicerina, hidroxietilcelulosa, hidroximetilpropilcelulosa, lanolina, metilcelulosa, vaselina, polietilenglicol, poli(alcohol vinílico), polivinilpirrolidona y carboximetilcelulosa. La preparación puede ser administrada por vía tópica al ojo del sujeto que necesite tratamiento (p. ej., un sujeto que padece AMD) por medio de métodos convencionales, por ejemplo, en forma de gotas, o bañando el ojo en una solución terapéutica, que contiene una o más composiciones.
Además, se han desarrollado una variedad de dispositivos para introducir fármacos en la cavidad vitrea del ojo. Por ejemplo, la publicación de la Solicitud de Patente de Estados Unidos Núm. 20020026176 describe un tapón que contiene medicamento que puede ser insertado a través de la esclerótica de tal manera que se proyecta a la cavidad vítrea para suministrar el agente farmacéutico a la cavidad vítrea. En otro ejemplo, la patente de Estados Unidos Núm. 5.443.505 describe un dispositivo implantable para la introducción en un espacio más arribacoroidal o una región avascular para la liberación sostenida del fármaco al interior del ojo. Las Patentes de Estados Unidos Núm.
5.773.019 y 6.001.386 describen cada una un dispositivo implantable de suministro de fármaco que se puede fijar a la superficie escleral de un ojo. El dispositivo comprende un núcleo interno que contiene una cantidad eficaz de un agente de baja solubilidad cubierto por un polímero no bioerosionable que es permeable al agente de baja solubilidad. Durante el funcionamiento, el agente de baja solubilidad impregna la cubierta de polímero bioerosionable para la liberación sostenida fuera del dispositivo. Los métodos y dispositivos adicionales (p. ej., un parche transescleral y el suministro a través de lentes de contacto) para el suministro de un agente terapéutico para el ojo son descritos, por ejemplo, por Ambati y Adamis (2002) en Prog Retin Eye Res 21(2):145-151; Ranta y Urtti (2006) Adv Drug Delivery Rev 58(11):1164-81; Barocas y Balachandran (2008) Expert Opin Drug Delivery 5(1):1-10(10); Gulsen y Chauhan (2004) Invest Ophthalmol Vis Sci 45:2342-2347; Kim et al. (2007) Oftálmica Res 39:244-254; y la publicación PCT Núm. WO 04/073551.
Como se ha descrito anteriormente, se pueden preparar composiciones de concentración relativamente alta. Por ejemplo, las composiciones se pueden formular a una concentración de entre aproximadamente 10 mg/mL y 100 mg/mL (p. ej., entre aproximadamente 9 mg/mL y 90 mg/mL; entre aproximadamente 9 mg/mL y 50 mg/mL; entre aproximadamente 10 mg/mL y 50 mg/mL; entre aproximadamente 15 mg/mL y 50 mg/mL; entre aproximadamente 15 mg/mL y 110 mg/mL; entre aproximadamente 15 mg/mL y 100 mg/mL; entre aproximadamente 20 mg/mL y 100 mg/mL; entre aproximadamente 20 mg/mL y 80 mg/mL; entre aproximadamente 25 mg/mL y 100 mg/mL; entre aproximadamente 25 mg/mL y 85 mg/mL; entre aproximadamente 20 mg/mL y 50 mg/mL; entre aproximadamente 25 mg/mL y 50 mg/mL; entre aproximadamente 30 mg/mL y 100 mg/mL; entre aproximadamente 30 mg/mL y 50 mg/mL; entre aproximadamente 40 mg/mL y 100 mg/ml, o entre aproximadamente 50 mg/mL y 100 mg/ml). En algunas realizaciones, las composiciones se pueden formular a una concentración mayor de 5 mg/mL y menor de 50 mg/mL. Los métodos para la formulación de una proteína en una solución acuosa son conocidos en la técnica y se describen, por ejemplo, en la patente de Estados Unidos Num. 7.390.786; McNally y Hastedt (2007), "Protein Formulation and Delivery," Segunda Edición, Drugs and the Pharmaceutical Sciences, Volume 175, CRC Press; y Banga (2005), "Therapeutic peptides and proteins: formulation, processing, and delivery systems, Segunda Edición" CRC Press. En algunas realizaciones, la solución acuosa tiene un pH neutro, por ejemplo, un pH entre, por ejemplo, 6,5 y 8 (p. ej., entre e incluso 7 y 8). En algunas realizaciones, la solución acuosa tiene un pH de aproximadamente 6,6, 6,7, 6,8, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9, o 8,0. En algunas realizaciones, la solución acuosa tiene un pH mayor que (o igual a) 6 (p. ej., mayor que o igual a 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, o 7,9), pero menor que pH 8.
Los ácidos nucleicos que codifican un polipéptido terapéutico puede ser incorporado a un constructo génico para su uso como parte de un protocolo de terapia génica para suministrar ácidos nucleicos que se pueden utilizar para expresar y producir los agentes dentro de las células. Los constructos de expresión de tales componentes se pueden administrar en cualquier portador terapéuticamente eficaz, por ejemplo, cualquier formulación o composición capaz de suministrar eficazmente el gen componente a las células in vivo. Los enfoques incluyen la inserción del gen sujeto en vectores virales incluyendo retrovirus recombinantes, adenovirus, virus adenoasociados, lentivirus, y virus del herpes simplex tipo 1 (HSV-1), o plásmidos bacterianos o eucarióticos recombinantes. Los vectores virales pueden transfectar células directamente; se puede suministrar ADN plasmídico con la ayuda, por ejemplo, de liposomas catiónicos (lipofectina) o derivatizados, productos conjugados de polilisina, gramicidina S, envolturas virales artificiales u otros de tales portadores intracelulares, así como inyección directa del constructo génico o precipitación con CaPO4 (véase, por ejemplo, WO04/060407) llevada a cabo en vivo. (Véase también, "Enfoques ex vivo", a continuación). Los ejemplos de los retrovirus adecuados incluyen pLJ, pZIP, pWE y pEM que son conocidos por los expertos en la técnica (véase, por ejemplo, Eglitis et al. (1985) Science 230:1395-1398; Danos y Mulligan (1988) Proc Natl Acad Sci USA 85:6460-6464; Wilson et al. (1988) Proc Natl Acad Sci USA 85:3014-3018; Armentano et al. (1990) Proc Natl Acad Sci USA 87:6141-6145; Huber et al. (1991) Proc Natl Acad Sci USA 88:8039-8043; Ferry et al. (1991) Proc Natl Acad Sci USA 88:8377-8381; Chowdhury et al. (1991) Science 254:1802-1805; van Beusechem et al. (1992) Proc Natl Acad Sci USA 89:7640-7644; Kay et al. (1992) Human Gene Terapia 3:641-647; Dai et al. (1992) Proc Natl Acad Sci USA 89:10892-10895; Hwu et al. (1993) J Immunol 150:4104-4115; las Patentes de Estados Unidos Núm. 4.868.116 y 4.980.286; y las Publicaciones PCT Núm. WO89/07136, WO89/02468, WO89/05345 y WO92/07573). Otro sistema viral de suministro de genes utiliza vectores derivados de adenovirus (véase, por ejemplo, Berkner et al. (1988) BioTechniques 6:616; Rosenfeld et al. (1991) Science 252:431-434; y Rosenfeld et al. (1992) Cell 68:143-155). Los vectores adenovirales adecuados derivados de la cepa de adenovirus Ad tipo 5 dl324 u otras cepas de adenovirus (p. ej., Ad2, Ad3, Ad7, etc.) son conocidos por los expertos en la técnica. Sin embargo, otro sistema de vector viral útil para la administración del gen sujeto es el virus adeno-asociado (AAV). Véanse, p. ej., Flotte et al. (1992) Am J Respir Cell Mol Biol 7:349-356; Samulski et al. (1989) J Virol 63:3822-3828; y McLaughlin et al. (1989) J Virol 62:1963-1973.
En algunas realizaciones, las composiciones se pueden formular con uno o más agentes terapéuticos adicionales,
por ejemplo, terapias adicionales para el tratamiento o la prevención de un trastorno asociado al complemento (p. ej., un trastorno asociado con AP o un trastorno asociado con CP) en un sujeto. Los agentes adicionales para el tratamiento de un trastorno asociado al complemento en un sujeto variarán dependiendo del trastorno concreto que se vaya a tratar, pero pueden incluir, sin limitación, un antihipertensivo (p. ej., un inhibidor de la enzima conversora de angiotensina) [para su uso en el tratamiento de, por ejemplo, el síndrome HELLP], un anticoagulante, un corticosteroide (p. ej., prednisona), o un agente inmunosupresor (p. ej., vincristina o la ciclosporina A). Los ejemplos de los anticoagulantes incluyen, por ejemplo, warfarina (Coumadin), aspirina, heparina, fenindiona, fondaparinux, idraparinux, e inhibidores de trombina (p. ej., argatroban, lepirudina, bivalirudina, o dabigatrán). Una composición descrita en la presente memoria también se puede formular con un agente fibrinolítico (p. ej., Ancrod, ácido £-aminocaproico, antiplasmina-al, prostaciclina, y defibrotida) para el tratamiento de un trastorno asociado al complemento. En algunas realizaciones, se puede formular una composición con un agente reductor de lípidos tal como un inhibidor de la hidroximetilglutaril reductasa CoA. En algunas realizaciones, una composición se puede formular con, o para su uso con, un agente anti-CD20, tal como rituximab (Rituxan™; Biogen Idec, Cambridge, MA). En algunas realizaciones, p. ej., para el tratamiento de la AR, la composición se puede formular con uno o ambos de infliximab (Remicade®; Centocor, Inc.) y metotrexato (Rheumatrex®, Trexall®). En algunas realizaciones, una composición descrita en la presente memoria se puede formular con un fármaco anti-inflamatorio no esteroideo (AINE). Están disponibles muchos AINE diferentes, algunos en venta incluyendo ibuprofeno (Advil, Motrin, Nuprin®) y naproxeno (Aleve) y muchos otros están disponibles con receta médica incluyendo meloxicam (Mobic®), etodolaco (Lodine®), nabumetona (Relafen®), sulindaco (Clinoril®), tolementina (Tolectin®), salicilato de magnesio de colina (Trilisate®), diclofenaco (Cataflam®, Voltaren®, Arthrotec®), diflunisal (Dolobid®), indometacina (Indocin®), ketoprofeno (Orudis®, Oruvail®), oxaprozina (Daypro®), y piroxicam (Feldene®). En algunas realizaciones, una composición se puede formular para su uso con un anti-hipertensivo, un agente anti-convulsivo (p. ej., sulfato de magnesio), o un agente anti-trombótico. Los antihipertensivos incluyen, por ejemplo, labetalol, hidralazina, nifedipina, antagonistas de los canales de calcio, nitroglicerina o nitroprusiato de sodio. (Véase, p. ej., Mihu et al. (2007) J Gastrointestin Liver Dis 16(4):419-424). Los agentes anti-trombóticos incluyen, por ejemplo, heparina, antitrombina, prostaciclina, o aspirina a dosis bajas.
En algunas realizaciones, las composiciones formuladas para su administración intrapulmonar pueden incluir al menos un agente activo adicional para el tratamiento de un trastorno pulmonar. El al menos un agente activo puede ser, por ejemplo, un anticuerpo anti-IgE (p. ej., omalizumab), un anticuerpo anti-IL-4 o un anticuerpo anti-IL-5, un inhibidor anti-IgE (p. ej., montelukast sódico), un simpaticomimético (p. ej., albuterol), un antibiótico (p. ej., tobramicina), una desoxirribonucleasa (p. ej., Pulmozyme®), un fármaco anticolinérgico (p. ej., bromuro de ipratropio), un corticosteroide (p. ej., dexametasona), un agonista p-adrenérgico, un inhibidor de leucotrieno (p. ej., zileuton), un inhibidor de 5-lipoxigenasa, un inhibidor de PDE, un antagonista de CD23, un antagonista de IL-13, un inhibidor de la liberación de citoquinas, un antagonista del receptor de histamina H1, un anti-histamínico, un agente anti-inflamatorio (por ejemplo, cromoglicato sódico), o un inhibidor de la liberación de histamina.
En algunas realizaciones, las composiciones se pueden formular para su administración con uno o más agentes terapéuticos adicionales para su uso en el tratamiento de un trastorno asociado del ojo al complemento. Tales agentes terapéuticos adicionales pueden ser, por ejemplo, bevacizumab o el fragmento Fab de bevacizumab o ranibizumab, ambos comercializados por Roche Pharmaceuticals, Inc., y pegaptanib sódico (Mucogen®; Pfizer, Inc.). Dicho kit también puede incluir, opcionalmente, instrucciones para la administración de la composición a un sujeto.
En algunas realizaciones, las composiciones se pueden formular para su administración a un sujeto junto con terapia intravenosa de gammaglobulina (IVIG), plasmaféresis, sustitución de plasma, o intercambio de plasma. En algunas realizaciones, las composiciones se pueden formular para su uso antes, durante, o después de, un trasplante de riñón.
Cuando las composiciones se van a utilizar combinadas con un segundo agente activo, las composiciones se pueden co-formular con el segundo agente o las composiciones se pueden formular por separado de la segunda formulación del agente. Por ejemplo, las respectivas composiciones farmacéuticas se pueden mezclar, por ejemplo, inmediatamente antes de su administración, y se administran juntas o se pueden administrar por separado, por ejemplo, en el mismo o diferentes momentos (véase más adelante).
Aplicaciones
Las composiciones descritas en la presente memoria se pueden utilizar en una serie de aplicaciones de diagnóstico y terapéuticas. Por ejemplo, las moléculas de unión al antígeno marcadas de forma detectable se pueden utilizar en los análisis para detectar la presencia o la cantidad de los antígenos diana en una muestra (p. ej., una muestra biológica). Las composiciones se pueden utilizar en análisis in vitro para estudiar la inhibición de la función del antígeno diana. En algunas realizaciones, p. ej., en las que las composiciones se unen a, e inhiben, una proteína de complemento, las composiciones se pueden utilizar como controles positivos en análisis diseñados para identificar nuevos compuestos adicionales que inhiben la actividad del complemento o de otro modo son útiles para tratar un trastorno asociado al complemento. Por ejemplo, se puede utilizar una composición para la inhibición de C5 como un control positivo en un análisis para identificar compuestos adicionales (p. ej., moléculas pequeñas, aptámeros, o
anticuerpos) que reducen o anulan la producción de C5 o la formación de MAC. Las composiciones también se pueden usar en procedimientos terapéuticos como se detalla a continuación.
Métodos de Tratamiento
Las composiciones descritas en la presente memoria se pueden administrar a un sujeto, por ejemplo, un sujeto humano, utilizando una variedad de métodos que dependen, en parte, de la vía de administración. La vía puede ser, p. ej., inyección o infusión intravenosa (IV), inyección subcutánea (SC), inyección intraperitoneal (IP), o inyección intramuscular (IM).
La administración subcutánea se puede completar por medio de un dispositivo. Los medios de dispositivo puede ser una jeringa, una jeringa precargada, un auto-inyector, ya sea desechable o reutilizable, el inyector tipo pluma, un inyector de parche, un inyector de uso diario, una bomba de infusión de jeringa ambulatoria con equipos de infusión subcutánea u otro dispositivo para la combinación con el fármaco anticuerpo para inyección subcutánea.
La administración se puede lograr, p. ej., infusión local, inyección, o por medio de un implante. El implante puede ser de un material poroso, no poroso, o gelatinoso, incluyendo membranas, tales como membranas de silásticas o fibras. El implante puede ser configurado para la liberación sostenida o periódica de la composición al sujeto. Véase, p. ej., la publicación de la Solicitud de Patente de Estados Unidos Núm. 20080241223; las Patentes de Estados Unidos Núm. 5.501.856; 4.863.457; y 3.710.795; EP488401; y EP 430539. Una composición descrita en la presente memoria puede ser suministrada al sujeto por medio de un dispositivo implantable basado, p. ej., en o sistemas convectivos erosionables difusivos, p. ej., bombas osmóticas, implantes biodegradables, sistemas de electrodifusión, sistemas de electroósmosis, bombas de presión de vapor, bombas electrolíticas, bombas efervescentes, bombas piezoeléctricas, sistemas basados en erosión, o sistemas electromecánicos.
En algunas realizaciones, una composición descrita en la presente memoria se entrega terapéuticamente a un sujeto por medio de administración local. En la presente memoria, "administración local" o "suministro local", se refiere al suministro que no se basa en el transporte de la composición o agente a su tejido o sitio diana deseado a través del sistema vascular. Por ejemplo, la composición se puede administrar por inyección o implantación de la composición o el agente o por inyección o implantación de un dispositivo que contiene la composición o el agente. Después de la administración local en la proximidad de un tejido o sitio diana, la composición o el agente, o uno o más componentes de los mismos, pueden difundirse hacia el tejido o sitio diana previstos.
En algunas realizaciones, una composición descrita en la presente memoria se puede administrar localmente a una articulación (p. ej., una articulación articulada). Por ejemplo, en realizaciones en las que el trastorno es artritis, se puede administrar una composición terapéuticamente apropiada directamente a una articulación (p. ej., a un espacio de la articulación) o en las proximidades de una articulación. Los ejemplos de las articulaciones intraarticulares a la que se puede administrar una composición descrita en la presente memoria localmente incluyen, por ejemplo, la cadera, la rodilla, el codo, la muñeca, la articulación esternoclavicular, la articulación temporomandibular, el carpo, el tarso, el tobillo, y cualquier otra articulación sujeta a afecciones artríticas. Una composición descrita en la presente memoria también se puede administrar a la bursa tal como, p. ej., acromial, bicipitorradial, cubitoradial, deltoides, infrarrotuliana, isquiática, y cualquier otra bursa conocida en la técnica de la medicina.
En algunas realizaciones, una composición descrita en la presente memoria se puede administrar localmente al ojo. Según se utiliza en la presente memoria, el término "ojo" se refiere a todos y cada uno de los tejidos y estructuras anatómicas asociada con un ojo. El ojo tiene una pared compuesta de tres capas distintas: la esclerótica exterior, la capa coroides media, y la retina interna. La cámara detrás del cristalino se rellena con un fluido gelatinoso conocido como humor vítreo. En la parte posterior del ojo se encuentra la retina, que detecta la luz. La córnea es un tejido ópticamente transparente, que transmite imágenes a la parte posterior del ojo. La córnea incluye una vía para la penetración de fármacos en el ojo. Otras estructuras de tejido anatómicas asociadas con el ojo incluyen el sistema lagrimal de drenaje, que incluye un sistema de secreción, un sistema de distribución y un sistema excretor. El sistema de secreción comprende secretores que son estimulados por medio del parpadeo y el cambio de temperatura debido a la evaporación de las lágrimas y reflejos secretores que tienen un suministro nervioso parasimpático eferente y secretan lágrimas en respuesta a la estimulación física o emocional. El sistema de distribución incluye los párpados y el menisco de lágrimas alrededor de los bordes del párpado de un ojo abierto, que extiende las lágrimas sobre la superficie ocular mediante el parpadeo, lo que reduce la aparición de zonas secas.
En algunas realizaciones, una composición descrita en la presente memoria se administra a la cámara posterior del ojo. En algunas realizaciones, una composición descrita en la presente memoria se administra por vía intravítrea. En algunas realizaciones, una composición descrita en la presente memoria se administra por vía trans-escleral.
En algunas realizaciones, por ejemplo, en realizaciones para el tratamiento o la prevención de un trastorno tal como EPOC o asma, se puede administrar a un sujeto una composición descrita en la presente memoria a través del pulmón. La administración pulmonar de fármacos se puede conseguir mediante inhalación, y la administración mediante inhalación en la presente memoria puede ser oral y/o nasal. Los ejemplos de dispositivos farmacéuticos
para la administración pulmonar incluyen inhaladores de dosis, inhaladores de polvo seco (IPS), y nebulizadores. Por ejemplo, una composición descrita en la presente memoria se puede administrar a los pulmones de un sujeto por medio de un inhalador de polvo seco. Estos inhaladores son dispositivos exentos de gas propulsor que proporcionan formulaciones de polvo seco dispersables y estables a los pulmones. Los inhaladores de polvo seco son bien conocidos en la técnica de la medicina, e incluyen, sin limitación: Turbohaler® (AstraZeneca; Londres, Inglaterra), el inhalador AIR® (Alkermes®; Cambridge, Massachusetts); Rotahaler® (GlaxoSmithKline; Londres, Inglaterra); y Eclipse™ (Sanofi-Aventis, París, Francia). Véanse también, p. ej., las Publicaciones PCT Núm. WO 04/026380, WO 04/024156 y WO 01/78693. Los dispositivos IPS se han utilizado para la administración pulmonar de polipéptidos tales como la insulina y la hormona del crecimiento. En algunas realizaciones, se puede administrar una composición descrita en la presente memoria intrapulmonarmente por medio de un inhalador de dosis medida. Estos inhaladores se basan en un propelente para suministrar una dosis discreta de un compuesto a los pulmones. Los ejemplos de los compuestos administrados mediante inhaladores de dosis medidas incluyen, por ejemplo, Astovent® (Boehringer-Ingelheim; Ridgefield, Connecticut) y Flovent® (GlaxoSmithKline). Véanse también, p. ej., las Patentes de Estados Unidos Núm. 6.170.717; 5.447.150; y 6.095.141.
En algunas realizaciones, una composición descrita en la presente memoria se puede administrar a los pulmones de un sujeto por medio de un nebulizador. Los nebulizadores utilizan aire comprimido para suministrar un compuesto en forma de aerosol licuado o neblina. Un nebulizador puede ser, por ejemplo, un nebulizador de chorro (p. ej., nebulizadores de aire o de líquido a chorro) o un nebulizador ultrasónico. Los dispositivos adicionales y los métodos de administración intrapulmonar se exponen en, por ejemplo, las Publicaciones de las Solicitudes de Patente de Estados Unidos Núm. 20050271660 y 20090110679.
En algunas realizaciones, las composiciones proporcionadas en la presente memoria están presentes en forma de dosificación unitaria, que puede ser especialmente adecuada para su auto-administración. Un producto formulado de la presente descripción se puede incluir dentro de un recipiente, típicamente, por ejemplo, un vial, cartucho, jeringa precargada o pluma desechable. También se puede utilizar un dosificador tal como el dispositivo dosificador descrito en la patente de Estados Unidos Num. 6.302.855, por ejemplo, con un sistema de inyección de la presente descripción.
Un sistema de inyección de la presente descripción puede emplear una pluma de suministro como se describe en la patente de Estados Unidos Num. 5.308.341. Los dispositivos de tipo pluma, más comúnmente utilizados para la auto-administración de insulina a los pacientes con diabetes, son bien conocidos en la técnica. Tales dispositivos pueden comprender al menos una aguja de inyección (p. ej., una aguja de calibre 31 de aproximadamente 5 a 8 mm de longitud), típicamente se pre-cargan con una o más dosis unitarias terapéuticas de una solución terapéutica, y son útiles para suministrar rápidamente la solución a un sujeto con el menor dolor posible.
Una pluma de suministro de medicación incluye un soporte de vial dentro del cual se puede recibir un vial de insulina u otro medicamento. El soporte del vial es una estructura alargada generalmente tubular con extremos proximal y distal. El extremo distal del soporte del vial incluye medios de fijación para acoplar una cánula de aguja de doble extremo. El extremo proximal también incluye medios de fijación para acoplar un cuerpo de la pluma que incluye un aparato de ajuste del controlador y de la dosis. Un medicamento desechable (p. ej., una solución de alta concentración de una composición descrita en la presente memoria) que contiene el vial para su uso con el soporte de vial de la técnica anterior incluye un extremo distal que tiene un septo elastomérico perforable que puede ser perforado por un extremo de una cánula de aguja de doble extremo. El extremo proximal de este vial incluye un tapón dispuesto de forma deslizable en acoplamiento hermético de fluido con la pared cilíndrica del vial. Esta pluma de suministro de medicación se utiliza insertando el vial de medicación en el soporte de vial. Un cuerpo de la pluma se conecta a continuación al extremo proximal del soporte de vial. El cuerpo de la pluma incluye un aparato de ajuste de la dosis para designar una dosis de medicación que va a ser suministrado por la pluma y un aparato de accionamiento para empujar el tapón del vial de manera distal para una distancia correspondiente a la dosis seleccionada. El usuario de la pluma monta una cánula de aguja de doble extremo al extremo distal del soporte de vial de tal manera que la punta de la cánula de la aguja perfora el septo proximal en el vial. El paciente selecciona a continuación una dosis y acciona la pluma para empujar el tapón distal para suministrar la dosis seleccionada. El aparato selector de dosis vuelve a cero después de la inyección de la dosis seleccionada. Después, el paciente quita y desecha la cánula de la aguja, y mantiene la pluma de administración de medicación en un lugar conveniente para la siguiente administración de la medicación requerida. El medicamento en el vial se agotará después de varias administraciones de dichos medicamentos. El paciente separa a continuación el soporte de vial del cuerpo de la pluma. El vial vacío puede ser retirado y desechado a continuación. Un nuevo vial se puede insertar en el soporte de vial, y el soporte de vial y cuerpo de la pluma se pueden volver a reensamblar y utilizar como se explicó anteriormente. En consecuencia, una pluma de suministro de medicación en general, tiene un mecanismo de accionamiento para una dosificación precisa y facilidad de uso.
Un mecanismo de dosificación tal como un botón giratorio permite que el usuario ajuste con precisión la cantidad de medicamento que será inyectado por la pluma desde un vial de medicación preenvasado. Para inyectar la dosis de medicación, el usuario inserta la aguja debajo de la piel y presiona el botón de una vez en la medida que se pueda presionar. La pluma puede ser un dispositivo completamente mecánico o puede ser combinado con un circuito electrónico para ajustar y/o indicar con precisión la dosis de la medicación que se inyecta al usuario. Véase, p. ej., la
Patente de Estados Unidos Num. 6.192.891.
En algunas realizaciones, la aguja del dispositivo tipo pluma es desechable y los kits incluyen una o más agujas de recambio desechables. Los dispositivos de tipo pluma adecuados para la administración de una cualquiera de las composiciones actualmente destacadas también se describe, p. ej., en las Patentes de Estados Unidos Núm.
6.277.099; 6.200.296; y 6.146.361. Un dispositivo de tipo pluma basado en microagujas se describe, por ejemplo, en la Patente de Estados Unidos Núm. 7.556.615. Véase también el dispositivo Precision Pen Injector (PPI), Molly™, fabricado por Scandinavian Health Ltd.
La presente descripción también presenta las formulaciones de liberación controlada o de liberación prolongada adecuadas para administración crónica y/o auto-administración de un medicamento como una composición descrita en la presente memoria. Las diferentes formulaciones se pueden administrar a un paciente que necesite tratamiento con la medicación en forma de bolo o mediante infusión continua a lo largo de un período de tiempo.
En algunas realizaciones, se formula una composición de alta concentración descrita en la presente memoria para liberación sostenida, liberación prolongada, de liberación gradual, de liberación controlada, o administración de liberación continua. En algunas realizaciones, se utilizan formulaciones de depósito para administrar la composición al sujeto que lo necesite. En este método, la composición se formula con uno o más portadores que proporciona una liberación gradual de agente activo durante un período de varias horas o días. Tales formulaciones se basan a menudo en una matriz degradante que se dispersa gradualmente en el organismo para liberar el agente activo. En algunas realizaciones, una composición descrita en la presente memoria se administra por medio de administración intrapulmonar a un sujeto que lo necesite. Por ejemplo, una composición descrita en la presente memoria se puede suministrar por medio de un nebulizador o un inhalador para un sujeto (p. ej., un ser humano) que sufre un trastorno tal como asma o EPOC.
Una dosis adecuada de una composición descrita en la presente memoria, cuya dosis es capaz de tratar o prevenir un trastorno en un sujeto, puede depender de una variedad de factores que incluyen, por ejemplo, la edad, el sexo, y el peso de un sujeto que se vaya a tratar y el compuesto inhibidor concreto utilizado. Por ejemplo, puede ser necesaria una dosis diferente de una composición (p. ej., una composición anti-C5) para tratar a un sujeto con AR en comparación con la dosis de una composición diferente (p. ej., una composición anti-TNFa) requerida para tratar al mismo sujeto. Otros factores que afectan a la dosis administrada al sujeto incluyen, p. ej., el tipo o gravedad de la enfermedad. Por ejemplo, un sujeto que tiene AR puede requerir la administración de una dosis diferente de una composición anti-C5 descrita en la presente memoria que un sujeto con PNH. Otros factores pueden incluir, por ejemplo, otros trastornos médicos que afectan concurrentemente o previamente al sujeto, la salud general del sujeto, la predisposición genética del sujeto, la dieta, el tiempo de administración, la velocidad de excreción, la combinación de fármacos, y otros agentes terapéuticos adicionales cualesquiera que se administran al sujeto. También se debe entender que una dosificación específica y régimen de tratamiento para cualquier sujeto concreto también dependerá del criterio del médico que atienda (p. ej., médico o enfermero).
Una composición descrita en la presente memoria se puede administrar como una dosis fija, o en una dosis de miligramo por kilogramo (mg/kg). En algunas realizaciones, la dosis también se puede elegir para reducir o evitar la producción de anticuerpos u otras respuestas inmunitarias del anfitrión contra una o más de las moléculas de unión a antígeno en la composición. Si bien no se pretende de ninguna manera que sean limitantes, las dosis ilustrativas de un anticuerpo, tal como una composición descrita en la presente memoria incluyen, por ejemplo, 1-1000 mg/kg, 1-100 mg/kg, 0,5-50 mg/kg, 0,1-100 mg/kg, 0,5 a 25 mg/kg, 1 a 20 mg/kg, y de 1-10 mg/kg. Las dosis de ejemplo de una composición descrita en la presente memoria incluyen, sin limitación, 0,1 mg/kg, 0,5 mg/kg, 1,0 mg/kg, 2,0 mg/kg, 4 mg/kg, 8 mg/kg, o 20 mg/kg.
Una solución farmacéutica puede incluir una cantidad terapéuticamente eficaz de una composición descrita en la presente memoria. Tales cantidades eficaces pueden ser determinadas fácilmente por un experto normal en la técnica basándose, en parte, en el efecto de la composición administrada, o el efecto combinatorio de la composición y uno o más agentes activos adicionales, si se utiliza más de un agente. Una cantidad terapéuticamente eficaz de una composición descrita en la presente memoria también puede variar de acuerdo con factores tales como el estado de enfermedad, la edad, el sexo y el peso del individuo, y la capacidad de la composición (y uno o más agentes activos adicionales) para obtener una deseada respuesta en el individuo, p. ej., la mejora de al menos un parámetro de estado, por ejemplo, la mejora de al menos un síntoma de la enfermedad mediada por el complemento. Por ejemplo, una cantidad terapéuticamente eficaz de una composición descrita en la presente memoria puede inhibir (reducir la gravedad de, o eliminar la ocurrencia de) y/o prevenir un trastorno concreto, y/o cualquiera de los síntomas del trastorno concreto conocido en la técnica o descrito en la presente memoria. Una cantidad terapéuticamente eficaz también es una en la que cualquier efecto tóxico o perjudicial de la composición son superados por los efectos terapéuticamente beneficiosos.
Las dosis humanas adecuadas de cualquiera de las composiciones descritas en la presente memoria se pueden evaluar adicionalmente, por ejemplo, en estudios de escalamiento de dosis en Fase I. Véase, p. ej., van Gurp et al. (2008) Am J Trasplantation 8(8):1711-18; Hanouska et al. (2007) Clin Cancer Res 13(2, parte 1):523-531; y
Hetherington et al. (2006) Antimicrobial Agents and Chemotherapy 50(10):3499-3500.
Se pretende que los términos "cantidad terapéuticamente eficaz" o "dosis terapéuticamente eficaz" o términos similares utilizados en la presente memoria signifiquen una cantidad de un agente (p. ej., una composición descrita en la presente memoria) que provocará la respuesta biológica o médica deseada (p. ej., una mejora en uno o más síntomas de un trastorno asociado al complemento). En algunas realizaciones, una solución farmacéutica descrita en la presente memoria contiene una cantidad terapéuticamente eficaz de al menos una de dichas composiciones. En algunas realizaciones, las soluciones contienen una o más composiciones y uno o más (p. ej., dos, tres, cuatro, cinco, seis, siete, ocho, nueve, 10, o 11 o más) agentes terapéuticos adicionales de tal manera que la composición como un todo es terapéuticamente eficaz. Por ejemplo, una solución puede contener una composición anti-C5 descrita en la presente memoria y un agente inmunosupresor, en donde la composición y el agente están cada uno a una concentración que cuando se combinan son terapéuticamente eficaces para tratar o prevenir un trastorno asociado al complemento (p. ej., un trastorno inflamatorio asociado al complemento, tal como EPOC, asma, sepsis, o AR) en un sujeto.
La toxicidad y la eficacia terapéutica de tales composiciones se pueden determinar por medio de procedimientos farmacéuticos conocidos en cultivos celulares o animales experimentales (p. ej., modelos animales de cualquiera de los trastornos mediados por el complemento descritos en la presente memoria). Estos procedimientos se pueden utilizar, por ejemplo, para determinar la DL50 (la dosis letal para el 50% de la población) y la DE50 (la dosis terapéuticamente eficaz en el 50% de la población). La razón de dosis entre los efectos tóxicos y terapéuticos es el índice terapéutico y puede expresarse como la razón DL50/DE50. Se prefiere una composición descrita en la presente memoria que presenta un alto índice terapéutico. Aunque se pueden utilizar composiciones que muestran efectos secundarios tóxicos, se debe tener cuidado de diseñar un sistema de administración que dirija dichos compuestos al sitio del tejido afectado y de minimizar el daño potencial a las células normales y, por lo tanto, reducir los efectos secundarios.
Los datos obtenidos a partir de análisis de cultivo celular y estudios animales se pueden utilizar para formular un intervalo de dosificación para su uso en seres humanos. La dosificación de la composición descrita en la presente memoria se encuentra generalmente dentro de un intervalo de concentraciones circulantes de las composiciones que incluye la DE50 con poca o ninguna toxicidad. La dosificación puede variar dentro de este intervalo dependiendo de la forma de dosificación empleada y la vía de administración utilizada. Para una composición descrita en la presente memoria, la dosis terapéuticamente eficaz puede estimarse inicialmente a partir de análisis de cultivo celular. Una dosis puede formularse en modelos animales para conseguir un intervalo de concentración de plasma circulante que incluye la CI50 (es decir, la concentración de anticuerpo que logra una inhibición semimáxima de los síntomas) según se determina en cultivo celular. Dicha información se puede utilizar para determinar con mayor precisión las dosis útiles en seres humanos. Los niveles en plasma se pueden medir, por ejemplo, mediante cromatografía líquida de alto rendimiento. En algunas realizaciones, por ejemplo, cuando se desea la administración local (p. ej., en el ojo o una articulación), se pueden utilizar cultivo celular o modelado de animales para determinar una dosis requerida para alcanzar una concentración terapéuticamente eficaz en el sitio local.
En algunas realizaciones, los métodos pueden llevarse a cabo junto con otras terapias para los trastornos asociado al complemento. Por ejemplo, la composición se puede administrar a un sujeto al mismo tiempo, antes de, o después de, la plasmaféresis, la terapia IVIG, o el intercambio de plasma. Véase, p. ej., Appel et al. (2005) J Am Soc Nephrol 16:1392-1404. En algunas realizaciones, la composición se puede administrar a un sujeto al mismo tiempo, antes de, o después de, un trasplante de riñón.
Un "sujeto", Según se utiliza en la presente memoria, puede ser cualquier mamífero. Por ejemplo, un sujeto puede ser un ser humano, un primate no humano (p. ej., orangután, gorila, macaco, babuino, o chimpancé), un caballo, una vaca, un cerdo, una oveja, una cabra, un perro, un gato, un conejo, una cobaya, un gerbo, un hámster, una rata, o un ratón. En algunas realizaciones, el sujeto es un niño (p. ej., un niño humano).
Según se utiliza en la presente memoria, un sujeto "que necesite prevención", "que necesite tratamiento" o "que lo necesite", se refiere a uno, a juicio de un profesional médico apropiado (p. ej., un médico, una enfermera, o un profesional de enfermería en el caso de los seres humanos; un veterinario en el caso de los mamíferos no humanos), se beneficiaría razonablemente de un tratamiento dado.
El término "prevenir" está reconocido en la técnica, y cuando se utiliza con respecto a una afección, es bien entendido en la técnica, e incluye la administración de una composición que reduce la frecuencia de, o retrasa la aparición de, los síntomas de una afección médica en un sujeto respecto a un sujeto que no recibe una composición descrita en la presente memoria. Por lo tanto, la prevención de un trastorno asociado al complemento tal como el asma incluye, por ejemplo, la reducción de la magnitud o la frecuencia de la tos, las sibilancias, o el dolor de pecho en una población de pacientes que reciben un tratamiento profiláctico con respecto a una población de control sin tratar, y/o el retraso de la aparición de la tos o las sibilancias en una población tratada frente a una población de control sin tratar, por ejemplo, en una cantidad estadísticamente y/o clínicamente significativa.
Como se describió anteriormente, las composiciones descritas en la presente memoria (p. ej., las composiciones
anti-C5) se pueden utilizar para tratar una variedad de trastornos asociados al complemento tales como, pero no limitados a: artritis reumatoide (AR); nefritis lúpica; lesión por isquemia-reperfusión; síndrome urémico hemolítico atípico (SUHa); síndrome urémico hemolítico típico o infeccioso (SUHt); enfermedad de depósitos densos (EDD); hemoglobinuria paroxística nocturna (HPN); esclerosis múltiple (EM); degeneración macular (p. ej., degeneración macular asociada con la edad (DMAE)); hemólisis, enzimas hepáticas elevadas y bajo recuento de plaquetas (HELLP); septicemia; dermatomiositis; retinopatía diabética; púrpura trombocitopénica trombótica (PTT); pérdida fetal espontánea; vasculitis pauciinmune; epidermolisis bullosa; pérdida fetal recurrente; esclerosis múltiple (MS); y lesión cerebral traumática. Véase, p. ej., Holers (2008) Immunological Reviews 223:300-316 y Holers y Thurman (2004) Molecular Immunology 41:147-152. En algunas realizaciones, el trastorno mediado por el complemento es un trastorno vascular mediado por el complemento tal como, pero no limitado a, un trastorno cardiovascular, miocarditis, un trastorno cerebrovascular, un trastorno vascular periférico (p. ej., músculo esquelético), un trastorno renovascular, un trastorno vascular mesentérico/entérico, revascularización de trasplantes y/o replantes, vasculitis, púrpura de Henoch-Schonlein, nefritis, vasculitis asociada a lupus eritematoso sistémico, vasculitis asociada a artritis reumatoide, vasculitis por complejos inmunitarios, transplante de órganos o tejidos, enfermedad de Takayasu, síndrome de extravasación capilar, miocardiopatía dilatada, angiopatía diabética, aneurisma de aorta torácicaabdominal, enfermedad de Kawasaki (arteritis), embolia gaseosa venosa (EGV), y reestenosis después de la colocación de dispositivo intraluminal, aterectomía rotacional, y angioplastia coronaria transluminal percutánea (ACTP). (Véase, p. ej., la Publicación de la Solicitud de Patente de Estados Unidos Núm. 20070172483.) En algunas realizaciones, el trastorno asociado al complemento es la miastenia gravis, la enfermedad por crioaglutininas (CAD), hemoglobinuria paroxística fría (PCH), dermatomiositis, esclerodermia, cálido anemia hemolítica autoinmunitaria, la enfermedad de Graves, la tiroiditis de Hashimoto, la diabetes tipo I, la psoriasis, el pénfigo, la anemia hemolítica autoinmunitaria (AHAI), la púrpura trombocitopénica idiopática (PTI), el síndrome de Goodpasture, el síndrome antifosfolipídico (SAF), enfermedad de Degos, y el SAF catastrófico (CAPS).
En algunas realizaciones, una composición descrita en la presente memoria, sola o combinada con un segundo agente antiinflamatorio, se puede utilizar para tratar un trastorno inflamatorio tal como, pero no limitado a, AR (más arriba), enfermedad inflamatoria intestinal, sepsis (más arriba), choque séptico, lesión pulmonar aguda, coagulación intravascular diseminada (CID), o enfermedad de Crohn. En algunas realizaciones, el segundo agente anti inflamatorio puede ser uno seleccionado del grupo que consiste de AINE, corticosteroides, metotrexato, hidroxicloroquina, agentes anti-TNF, tales como etanercept e infliximab, un agente que agota las células B tal como rituximab, un antagonista interleucina-1, o un agente de bloqueo co-estimulador de células T tal como abatacept.
En algunas realizaciones, el trastorno asociado al complemento es un trastorno neurológico asociado complemento tal como, pero no limitado a, esclerosis lateral amiotrófica (ELA), lesión cerebral, enfermedad de Alzheimer, y neuropatía desmielinizante inflamatoria crónica.
Los trastornos asociados al complemento también incluyen trastornos pulmonares asociados al complemento tales como, pero no limitados a, asma, bronquitis, enfermedad pulmonar obstructiva crónica (EPOC), enfermedad pulmonar intersticial, deficiencia de anti-tripsina a-1, enfisema, bronquiectasia, bronquiolitis obliterante, alveolitis, sarcoidosis, fibrosis pulmonar y trastornos vasculares del colágeno.
En algunas realizaciones, una composición descrita en la presente memoria se administra a un sujeto para tratar, prevenir, o mejorar al menos un síntoma de una respuesta inflamatoria asociada al complemento (p. ej., el aspecto de la respuesta inflamatoria asociada al complemento de un trastorno asociado al complemento) en un sujeto. Por ejemplo, una composición se puede usar para tratar, prevenir y/o mejorar uno o más síntomas asociados con una respuesta inflamatoria asociada al complemento tales como el rechazo de injerto/enfermedad de injerto contra anfitrión (EICA), lesiones de reperfusión (p. ej., siguiente a derivación cardiopulmonar o trasplante de tejido), y daño tisular después de otras formas de lesión traumática, tales como una quemadura (p. ej., una quemadura grave), traumatismo cerrado, lesión de la médula, o congelación. Véase, p. ej., Park et al. (1999) Anesth Analg 99(1):42-48; Tofukuji et al. (1998) J Thorac Cardiovasc Surg 116(6):1060-68; Schmid et al. (1997) Shock 8(2):119-124; y Bendice et al. (1999) Am J Physiol 276(1):L57-L63.
En algunas realizaciones, una composición descrita en la presente memoria se puede administrar a un sujeto en forma de monoterapia. Alternativamente, como se ha descrito anteriormente, la composición se puede administrar a un sujeto como terapia combinada con otro tratamiento, p. ej., otro tratamiento para un trastorno asociado al complemento o una respuesta inflamatoria asociada al complemento. Por ejemplo, la terapia combinada puede incluir la administración al sujeto (p. ej., un paciente humano) uno o más agentes adicionales (p. ej., fármacos anti coagulantes, anti-hipertensivos, o anti-inflamatorios (p. ej., esteroides)) que proporcionan un beneficio terapéutico a un sujeto que tiene o está en riesgo de desarrollar, sepsis. En otro ejemplo, la terapia combinada puede incluir administrar al sujeto uno o más agentes adicionales (p. ej., un anticuerpo anti-IgE, un anticuerpo anti-IL-4, un anticuerpo anti-IL-5, o una anti-histamina) que proporcionan un beneficio terapéutico a un sujeto que tiene, está en riesgo de desarrollar, o se sospecha que tiene un trastorno pulmonar asociado al complemento tal como EPOC o asma. En algunas realizaciones, una composición y los uno o más agentes activos adicionales se administran al mismo tiempo. En otras realizaciones, la composición se administra en primer lugar primero en el tiempo y los uno o más agentes activos adicionales se administran en segundo lugar en el tiempo. En algunas realizaciones, los uno o más agentes activos adicionales se administran en primer lugar en el tiempo y la composición se administra en
segundo lugar en el tiempo.
Una composición descrita en la presente memoria puede reemplazar o aumentar una terapia administrada antes o en este momento. Por ejemplo, después del tratamiento con una composición descrita en la presente memoria, la administración de uno o más agentes activos adicionales puede cesar o disminuir, por ejemplo, administrarse a niveles más bajos, por ejemplo, niveles más bajos de eculizumab tras la administración de una composición anti-C5 descrita en la presente memoria. En algunas realizaciones, la administración de la terapia anterior se puede mantener. En algunas realizaciones, una terapia anterior se mantendrá hasta que el nivel de la composición alcance un nivel suficiente para proporcionar un efecto terapéutico. Las dos terapias pueden administrarse combinadas. La supervisión de un sujeto (p. ej., un paciente humano) para la determinación de una mejora de un trastorno (p. ej., sepsis, quemaduras graves, AR, nefritis por lupus, síndrome de Goodpasture, o asma), como se define en la presente memoria, significa la evaluación del sujeto para determinar un cambio en un parámetro de la enfermedad, p. ej., una mejora en uno o más síntomas de un trastorno dado. Los síntomas de muchos de los trastornos anteriores (trastornos por ejemplo, asociados al complemento) son bien conocidos en la técnica de la medicina. En algunas realizaciones, la evaluación se lleva a cabo al menos una (1) hora, p. ej., al menos 2, 4, 6, 8, 12, 24, o 48 horas, o al menos 1 día, 2 días, 4 días, 10 días, 13 días, 20 días o más, o al menos 1 semana, 2 semanas, 4 semanas, 10 semanas, 13 semanas, 20 semanas o más, después de una administración de una composición descrita en la presente memoria. El sujeto puede evaluarse en uno o más de los siguientes períodos: antes del inicio del tratamiento; durante el tratamiento; o después de que se han administrado uno o más elementos del tratamiento. La evaluación puede incluir la evaluación de la necesidad de tratamiento adicional, por ejemplo, la evaluación de si una dosis, frecuencia de administración, o duración del tratamiento deben ser modificados. También puede incluir la evaluación de la necesidad de añadir o eliminar una modalidad terapéutica seleccionada, p. ej., añadir o eliminar cualquiera de los tratamientos para un trastorno asociado al complemento descrito en la presente memoria.
Los siguientes ejemplos son meramente ilustrativos y no deben interpretarse como limitantes del alcance de esta descripción en modo alguno, puesto que serán evidentes muchas variaciones y equivalentes para los expertos en la técnica al leer la presente descripción.
EJEMPLOS
Ejemplo 1. La semisemivida de Eculizumab es una combinación de varias tasas de aclaramiento
La semivida promedio de eculizumab en pacientes con HPN y SUHa que reciben el régimen de dosificación prescrito es de aproximadamente 11 a 12 días, mientras que se prevé que la semivida esperada para un anticuerpo monoclonal humanizado que tiene un Fc de IgG2/4 Fc sea similar a la de una anticuerpo que contiene un Fc de IgG2 o Fc de IgG4, aproximadamente 21 a 28 días. Morell et al. (1970) J Clin Invest 49(4): 673-680. Para comprender el impacto potencial del aclaramiento mediado por antígenos por sobre la tasa de aclaramiento global de eculizumab, se realizaron los siguientes experimentos utilizando el modelo de ratón de receptor Fc neonatal humano (hFcRn) (los ratones carecen de FcRn endógeno pero son transgénicos para hFcRn (B6.Cg- fc tm1Dcr Tg (FCGRT)32Dcr/DcrJ; Número de inventario 014565, Jackson Laboratories, Bar Harbor, Maine)). El modelo FcRn transgénico ha sido descrito, por ejemplo, por Petkova et al. (2006) Int Inmunology 18(12): 1759-1769; Oiao et al. (2008) Proc Natl Acad Sci USA 105(27): 9337-9342; y Roopenian et al. (2010) Methods Mol Biol 602: 93-104.
Se administró una dosis única de 100 |jg de eculizumab en 200 jL de solución salina tamponada con fosfato (PBS) por inyección intravenosa (iv) para cada uno de cinco ratones transgénicos hFcRn. Se recogieron muestras de sangre de aproximadamente 100 jL de cada uno de los ratones en los días uno, tres, siete, 14, 21, 28 y 35 después de la administración. La concentración de eculizumab en el suero se midió por medio de ELISA. Brevemente, las placas de análisis se recubrieron con un anticuerpo de captura anti-cadena ligera IgK humana de oveja y se bloquearon. Los pocillos de la placa se pusieron en contacto a continuación con las muestras de suero en condiciones que permitían que el eculizumab, si estaba presente en el suero, se uniera al anticuerpo de captura. Se detectó la cantidad relativa de eculizumab ligado a cada pocillo utilizando un anticuerpo anti-IgG4 humano marcado de forma detectable y se cuantificó con relación a una curva patrón generada a partir de suero de ratón no sometido a tratamiento previo que contiene cantidades conocidas de eculizumab.
La semivida en suero del anticuerpo se calculó utilizando la siguiente fórmula:

Donde: T = Tiempo transcurrido, Ao = concentración en suero original del anticuerpo (concentración en el día 1 en el presente estudio) y At = Cantidad del anticuerpo que queda después del tiempo transcurrido T (concentración mínima detectable o último punto temporal de toma de muestra de sangre (día 35) en el presente estudio).
Los resultados del experimento se representan en la Fig. 1. La semivida de eculizumab en el modelo de ratón hFcRn
fue de 13,49 ± 0,93 días.
Para determinar el efecto de C5 humana en la semivida de eculizumab utilizando el modelo de hFcRn, se mezcló previamente el anticuerpo con una razón molar 4:1 de C5 humana (Complement Technology Inc., Número de catálogo: A120) antes de la dosis, una dosis de 100 g de eculizumab fue administrada por vía intravenosa (i.v.) el día 0. Se recogieron aproximadamente 100 pL de sangre en tubos de 1,5 mL Eppendorf para suero mediante sangrado retro-orbital a 1, 3, 7, 14, 21, 28 y 35 días.
Como se muestra en la Fig. 1, la semivida de eculizumab en el modelo de ratón hFcRn en presencia de C5 era de 4,55 ± 1,02 días. Estos resultados indican que, además de los mecanismos de aclaramiento del anticuerpo mediado por endocitosis en el que una larga semivida se rige en gran medida por el reciclaje mediado por FcRn, la semivida de eculizumab puede verse afectada de manera significativa por el aclaramiento mediado por antígeno a través de C5 humana.
Ejemplo 2. Las sustituciones de aminoácidos en el dominio Fc de eculizumab aumentan la semivida de eculizumab, pero no son suficientes para superar el efecto de C5 sobre el aclaramiento de eculizumab Se ha demostrado que ciertas sustituciones de aminoácidos en la región Fc de un anticuerpo IgG disminuyen la tasa de eliminación del anticuerpo de la circulación. Las sustituciones que incrementan la afinidad de unión de un anticuerpo IgG para FcRn a pH 6,0 son ejemplos de semejante efecto biológico. Véase, p. ej., Dall'Acqua et al. (2006) J Immunol 117: 1129-1138 y Ghetie et al. (1997) Nat Biotech 15: 637-640. Zalevsky et al. [(2010) Nat Biotech 28: 157-159] describen una serie de sustituciones de aminoácidos, p. ej., M428L/N434S, capaces de aumentar la semivida de un anticuerpo IgG en el suero. Otras sustituciones de aminoácidos que prolongan la semivida incluyen, p. ej., T250Q/M428L y M252Y/S254T/T256E. Véase, p. ej., la publicación de solicitud de patente internacional núm. WO 2008/048545 y Dall'Acqua et al. (2006) J Biol Chem 281: 23514-23524. Para determinar si una o más sustituciones de aminoácidos en la región constante Fc de eculizumab son capaces de prolongar la semivida de eculizumab en el suero, se introdujeron las siguientes sustituciones en eculizumab: 252Y/S254T/T256E, basada en el índice de numeración de EU (en el presente documento después de esta variante de eculizumab se refiere como la variante YTE). La región constante de cadena pesada consistió en la siguiente secuencia de aminoácidos:
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSS GLYSLSSVVTVTSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSV FLFPPKPKDTLYITREPEVTCWVDVSHEDPEVQFNWYVDGMEVHNAKTKPREEQFNS TFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREE MTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSR WQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO:15). La secuencia de aminoácidos para el polipéptido de la cadena pesada completa de la variante YTE de eculizumab se representa en el SEQ ID NO: 16.
La variante YTE se evaluó junto eculizumab en el modelo de ratón hFcRn descrito en el Ejemplo 1. Esto es, se administraron 100 pg de eculizumab (región Fc de IgG2/4), una variante de eculizumab que contenía un Fc o la variante YTE de eculizumab en 200 pl de solución salina tamponada con fosfato (PBS) por vía inyección intravenosa (i.v.) para cada uno de ocho ratones transgénicos hFcRn. Se recogió el suero de cada uno de los ratones en los días uno, tres, siete, 14, 21, 28 y 35 después de la administración. La concentración de cada anticuerpo en el suero se midió por medio de ELISA y se calculó la semivida como se describe en el Ejemplo 1. Los resultados se representan en la Fig. 2 y en la Tabla 2.
Tabla 2

Como se muestra en la Fig. 2 y en la Tabla 2, la sustitución YTE aumentó la semivida promedio de eculizumab más de 2 veces desde los días 14,28 ± 1 a 29,07 ± 4,7.
Para determinar el efecto de C5 humana en la semivida de la variante YTE de eculizumab, se les administró a los ratones C5 humana como se ha descrito anteriormente en el Ejemplo 1. Una dosis de 100 pg de eculizumab, la variante de eculizumab-IgG2, y la variante YTE de eculizumab-IgG2 se administraron por vía intravenosa en el día 0. Tal como se muestra en la Fig. 3 y en la Tabla 3, la semivida de eculizumab, la variante eculizumab-IgG2, y la variante YTE de eculizumab-IgG2 disminuyó significativamente en presencia de un exceso molar de C5 humana. Por lo tanto, las sustituciones de aminoácidos en el dominio de unión a FcRn de eculizumab fueron insuficientes para superar la contribución al aclaramiento mediado por C5 en la semivida de eculizumab.
Tabla 3.

Ejemplo 3. Efecto de las sustituciones de aminoácidos en las CDR de eculizumab en la semivida
Como se describió anteriormente, la semivida de eculizumab en ratones es significativamente más corta en presencia de su antígeno, C5 humana (hC5). Sin estar ligado a ninguna teoría o mecanismo de acción concretos, se plantea la hipótesis de que el aclaramiento acelerado en presencia del antígeno es, en parte, el resultado de la muy alta afinidad de eculizumab por C5 (Kd ~ 30 pM a pH 7,4 y ~600 pM a pH 6,0) que no permite la disociación eficaz del complejo anticuerpo:C5 en los primeros compartimientos endosomales después de pinocitosis. Sin disociación, el complejo anticuerpo:antígeno es reciclado al compartimiento extracelular a través del receptor Fc neonatal (FcRn) o dirigido a la degradación lisosomal. En cualquier caso, el anticuerpo es incapaz de unirse a más de dos moléculas de C5 en su semivida.
La fuerte afinidad de eculizumab por C5 (Kd ~ 30 pM) permite la unión casi completa de toda la C5 en sangre, asegurando que muy poca C5 se active para formar C5a y TCC. Por tanto, la afinidad de eculizumab por C5 está conectada directamente con la eficacia in vivo del anticuerpo en los pacientes tratados con el anticuerpo. Los autores de la presente invención establecen un debilitamiento de la afinidad de eculizumab para C5, sin comprometer la eficacia de eculizumab in vivo. Si bien la descripción no se limita a un enfoque de este tipo, esto se logró mediante la introducción de histidina en una o más posiciones de las CDR de eculizumab. La histidina tiene un pKa de 6,04. Esto significa que a medida que los valores de pH caen desde 7,4 (sangre) a menos de 6,0 (endosomas tempranos), las histidinas ganan un protón. Por lo tanto, en el endosoma, las histidinas se cargan más positivamente. Los autores de la presente invención plantearon la hipótesis de al introducir histidinas en o cerca del sitio de unión para C5 en eculizumab, el cambio de carga en el endosoma puede interrumpir la unión en el endosoma, a la vez que se preserva la alta afinidad por C5 a pH neutro en la sangre. Se plantea la hipótesis de que tales sustituciones aumentan la semivida al facilitar la disociación de anticuerpo del complejo anticuerpo:C5 en el ambiente ácido del endosoma, permitiendo que el anticuerpo libre sea reciclado, mientras que la C5 se degrada en el lisosoma.
Al utilizar eculizumab como anticuerpo parental, se generó una serie de anticuerpos variantes en la que cada posición de CDR que fue sustituida por una histidina. La región variable de cadena pesada de eculizumab tiene la siguiente secuencia de aminoácidos:
QVQLVQSGAEVKKPGASVKVSCKASGYIFSNYWIQWVRQAPGQGLEWMGEILPGSGST EYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYYCARYFFGSSPNWYFDVWGOGT LVTVSS (SEQ ID NO:7). (Las regiones CDR de la región variable de la cadena pesada están subrayadas).
La región variable de cadena ligera de eculizumab tiene la siguiente secuencia de aminoácidos:
DIOMTOSPSSLSASVGDRVTITCGASENIYGALNWYOOKPGKAPKLLIYGATNLADGVP SRFSGSGSGTDFTLT1SSLOPEDFATYYCONVLNTPLTFGOGTKVE1K (SEQ ID NO:8).
El resultado de este intento de barrido de histidina proporcionó 66 variantes de sustitución de histidina individuales de eculizumab. Las secuencias codficantes de la cadena ligera y la cadena pesada para estas variantes de anticuerpo se clonaron en plásmidos separados de "constructos de un solo gen" adecuados para la expresión en células de mamífero y se confirmó su secuencia. Los anticuerpos que contenían una sola sustitución de aminoácido se expresaron transitoriamente en células HEK293F por transfección simultánea de constructos de un solo gen que codifican una cadena ligera o cadena pesada. También se llevó a cabo una transfección simultánea de cadenas pesadas y ligeras "de tipo salvaje" que representan secuencias de CDR de eculizumab no modificadas (EHL000). Los sobrenadantes de cultivo de tejidos se normalizaron para el nivel de expresión del anticuerpo y se utilizaron para evaluar la unión del anticuerpo a C5 humana, con respecto a EHL000, utilizando interferometría biocapa en un aparato Octet Red (ForteBio Inc.). Brevemente, los anticuerpos fueron capturados sobre un biosensor de anti-Fc de IgG humana (ForteBio, Núm. Cat. 18 a 5001). Las puntas cargadas se expusieron a continuación a una solución tamponada a pH 7,4 que contenía 12,5 nM de C5 humana purificada nativa durante 800 segundos para evaluar la cinética de asociación con respecto al anticuerpo parental. Las cinéticas de disociación se evaluaron mediante transferencia de la punta a una solución tamponada de pH 7,4 o solución tamponada de pH 6,0 durante 800 segundos. Todas las mediciones se repitieron para garantizar la coherencia de las lecturas.
Se seleccionaron variantes de sustitución de histidina individuales de eculizumab basándose en una serie de tres propiedades en relación con eculizumab. Las variantes de histidina preferidas solamente se desviaron de la ka y la
kd de eculizumab a pH 7,4 en un grado menor, pero se desviaron de la kd eculizumab a pH 6,0 más significativamente. Los criterios de selección relativos de umbral fueron los siguientes:
(1) una variación máxima de la cinética de asociación a pH 7,4 de un desplazamiento de fase máximo menor de 33% a 800 segundos en comparación con el desplazamiento de fase máximo promedio a 800 segundos observado para eculizumab;
(2) una variación máxima de la cinética de disociación a pH 7,4 de una reducción de no más de 3 veces en el desplazamiento de fase máximo por encima de 800 segundos en comparación con el desplazamiento de fase máximo promedio a 800 segundos observado para eculizumab; y
(3) una variación mínima de la cinética de disociación a pH 6,0 de una reducción de al menos 3 veces en el desplazamiento de fase máximo por encima de 800 segundos en comparación con el desplazamiento de la máxima promedio a 800 segundos observado para eculizumab.
Por ejemplo, con respecto al apartado (1) anterior, si el desplazamiento de fase máximo promedio después de 800 segundos de asociación con eculizumab es de aproximadamente 0,75 nm, un anticuerpo de ensayo que tenga un desplazamiento de fase máximo de menos de 0,5 nm (p. ej., reproducido dos o más veces) no satisfaría los criterios anteriores. Por el contrario, un anticuerpo de ensayo con un desplazamiento de fase máximo de más de 0,5 nm a 800 segundos satisface el primer criterio.
Las sustituciones individuales en la región variable de cadena ligera que satisfacían estos umbrales fueron las siguientes: G31H, L33H, V91H, y T94H, todas relativas al SEQ ID NO: 8. Las sustituciones individuales en la región variable de cadena pesada que satisfacían estos umbrales fueron las siguientes: Y27H, I34H, L52H, y S57H, todas con relación al SEQ ID NO: 7. Véanse las Figs. 5A, 5B, 5C y 5D.
Se generó una segunda serie de anticuerpos que contenían todas las combinaciones posibles de dos sustituciones de histidina en las posiciones en las que las sustituciones individuales satisfacían los criterios umbral. Véase la Tabla 1. Estas cinéticas de asociación y disociación fueron analizadas a través de los mismos métodos y comparadas tanto con el anticuerpo parental original como con las sustituciones de histidina individuales. Del mismo modo, se generaron una tercera y una cuarta series de anticuerpos que contenían tres o cuatro sustituciones de histidina, respectivamente, y se analizaron las cinéticas de asociación y disociación en comparación con los dos o tres predecesores de sustitución de histidina pertinentes. Véase la Tabla 1. En cada fase se utilizaron los mismos criterios de umbrales mínimos para la cinética de asociación a un pH de 7,4, umbrales máximo s para la cinética de disociación a pH 7,4 y umbrales mínimos para la cinética de disociación a pH 6. Ocho combinaciones de sustituciones satisficieron los criterios anteriores y se seleccionaron para la determinación de la afinidad a pH 7,4 y pH 6,0 a través de RPS. Las afinidades se exponen en la Tabla 4.
Tabla 4

Para estas combinaciones de sustituciones, la afinidad de eculizumab por C5 se redujo en más de 1000 veces a pH 6,0, mientras que la afinidad sufrió una reducción no mayor de 20 veces en la afinidad a pH 7,0. De estas, se seleccionó EHG303 (Tabla 4) para su posterior análisis debido a su alta afinidad a pH 7,4 (0,146 nM) y la razón de (Kd a pH 6,0)/(Kd a pH 7,4) de más de 8.000.
El polipéptido de cadena pesada del anticuerpo EHG303 comprende la siguiente secuencia de aminoácidos:
MGWSCIILFLVATATGVHSLEQVQL VQSGAEVKKPGASVKVSCKASGHIFSNYWIQW RQAPGQGLEWMGEILPGSGHTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYY CARYFFGSSPNWYFDVWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDY FPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNT KVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEV QFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENN YKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK (SEQ ID NO:24). El polipéptido de cadena ligera del anticuerpo EHG303 comprende la siguiente secuencia de aminoácidos:
MGWSCIILFLVATATGVHSRDIQMTQSPSSLSASVGDRVTITCGASENIYGALNWYQQKP GKAPKLLIYGATNLADGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLTFGQ GTKVEIKRTRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSG NSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:25). En las secuencias anteriores, las partes subrayadas corresponden a la secuencia líder de cada polipéptido y las porciones en cursiva son aminoácidos heterólogos introducidos en virtud de la clonación.
También fue seleccionado el anticuerpo EHL049. Su polipéptido de cadena pesada comprende la siguiente secuencia de aminoácidos:
MGWSCIILFLVATATGVHSLEQVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWV RQAPGQGLEWMGEILPGSGHTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYY CARYFFGSSPNWYFDVWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDY FPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNT KVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEV QFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENN YKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK (SEQ ID NO:26). El polipéptido de cadena ligera del anticuerpo EHL049 comprende la siguiente secuencia de aminoácidos:
MGWSCIILFLVATATGVHSRDIQMTQSPSSLSASVGDRVTITCGASENIYHALNWYQQKP GKAPKLLIYGATNLADGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLTFGQ GTKVEIKRTRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSG NSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:27). En las secuencias anteriores, las partes subrayadas corresponden a la secuencia líder de cada polipéptido y las porciones en cursiva son aminoácidos heterólogos introducidos en virtud de la clonación.
Por último, el polipéptido de cadena pesada EHL000 comprende la siguiente secuencia de aminoácidos:
MGWSCIILFLVATATGVHSLEQVQL VQSGAEVKKPGASVKVSCKASGYIFSNYWIQWV RQAPGQGLEWMGEILPGSGSTEYTENFKDRVTMTRDTSTSTVYMELSSLRSEDTAVYY CARYFFGSSPNWYFDVWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDY FPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNT KVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEV QFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENN YKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK (SEQ ID NO:28). El polipéptido de cadena ligera del anticuerpo EHL000 comprende la siguiente secuencia de aminoácidos:
MGWSCIILFLVATATGVHSRDIQMTQSPSSLSASVGDRVTITCGASENIYGALNWYQQKP GKAPKLLIYGATNLADGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLTFGQ GTKVEIKRTRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSG NSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:29). En las secuencias anteriores, las partes subrayadas corresponden a la secuencia líder de cada polipéptido y las porciones en cursiva son aminoácidos heterólogos introducidos en virtud de la clonación.
Ejemplo 4. Las sustituciones de histidina prolongan la semivida de eculizumab en suero
El polipéptido de cadena ligera y el polipéptido de cadena pesada de cada uno de los anticuerpos EHL y EHG anteriores, se expresaron a partir de constructos de genes individuales. Las secuencias codificantes de la cadena pesada y ligera de EHG303 se combinaron en un vector de doble expresión génica, así como las secuencias de cadena ligera y pesada para el anticuerpo EHL049. El clon EHG303 resultante se denominó BNJ421 y el clon EHL049 resultante se denominó BNJ423. La secuencia de aminoácidos de la región variable de cadena pesada de BNJ421 es la siguiente:
QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEWMGEILPGSGHTEYTENFKDRVTMTRDTS TSTVYMELSSLRSEDTAVYYCARYFFGSSPNWYFDVWGQG TLVTVSS (SEQ ID NO: 12). La secuencia de aminoácidos de la región variable de la cadena ligera para BNJ421 es la siguiente:
DIQMTQSPSSLSASVGDRVTTTCGASENIYGALNWYQQKPGKAPKLLIYGATNLADGVP
SRFSGSGSGTDFTLT1SSLQPEDFATYYCQNVLNTPLTFGQGTKVEIK (SEQ ID N0:8).
La región variable de la cadena pesada del anticuerpo BNJ423 comprende la siguiente secuencia de aminoácidos: QVQLVQSGAEVKKPGASVKVSCKASGHIFSNYWIQWVRQAPGQGLEWMGEILPGSGHTEYTENFKDRVTMTRDTS TSTVYMELSSLRSEDTAVYYCARYFFGSSPNWYFDVWGQG TLVTVSS (SEQ ID NO:12). La secuencia de aminoácidos de la cadena ligera para BNJ423 es la siguiente:
DIQMTQSPSSLSASVGDRVTITCGASENIYHALNWYQQKPGKAPKLLIYGATNLADGVP
SRFSGSGSGTDFTLTISSLQPEDFATYYCQNVLNTPLXFGQGTKVEIK (SEQ ID NO: 18).
Estas dos moléculas se evaluaron junto a EHL000 en ratones que eran inmunodeficientes (NOD/scid) y con deficiencia de C5. Se administró una única dosis de 100 pg de EHL000, BNJ421, o BNJ423 en 200 pL de solución salina tamponada con fosfato (PBS) por medio de inyección intravenosa (i.v.) para cada uno de ocho ratones. Se recogió el suero de cada uno de los ratones en los días uno, tres, siete, 14, 21, 28 y 35 después de la administración. La concentración de cada anticuerpo en el suero se midió por medio de ELISA. La semivida en suero de los anticuerpos se calculó utilizando el soporte lógico Pharsight Phoenix® WinNonlin® versión 6.3 utilizando el análisis no compartimental (ANC) y la respuesta directa Emax. El porcentaje del anticuerpo que permanece en el suero se calculó como sigue:
Ct
% de anticuerpo que permanece = ------ X 100
Ci
en donde, Ct = concentración de anticuerpo en un día determinado; y Ci = concentración de anticuerpo en el día 1. Los resultados se representan en la Fig. 6 y en la Tabla 5.
Tabla 5

Para determinar el efecto de C5 humana sobre la semivida de estos anticuerpos utilizando el mismo modelo de ratón, se administró C5 humana a los ratones por vía subcutánea a una dosis de carga de 250 pg en el día -1 (el día antes de que se administraran los anticuerpos a los ratones), seguido de dosis dos veces al día de 50 pg de C5 para mantener la concentración en suero de C5 a aproximadamente 20 pg/mL (como se describe en el Ejemplo 1).
Como se muestra en la Fig. 7 (y en la Tabla 6, a continuación), la semivida de EHL000 (eculizumab-IgG1) en el modelo de ratón en presencia de C5 humana (hC5) (a una concentración que era mayor que una razón molar 1:1 de C5 con respecto a eculizumab) fue de 2,49 ± 0,34 días, mientras que la semivida de los anticuerpos BNJ421 y BNJ423 (que contienen las sustituciones de histidina) fue sustancialmente mayor en 15,25 ± 0,90 días y 22,71 ± 0,71 días, respectivamente. Estos resultados indican que las sustituciones de histidina en las CDR de eculizumab, y la afinidad resultante dependiente del pH para C5, disminuyen significativamente la tasa de aclaramiento de las variantes de eculizumab del suero con respecto al eculizumab.
Tabla 6

Ejemplo 5. Las variantes de eculizumab sustituidas con histidina no pierden la actividad inhibidora del complemento
Además, también se evaluó la actividad hemolítica en suero en cada una de las muestras que contenía C5 humana a partir de los experimentos descritos en el Ejemplo 4. La actividad del complemento terminal en sueros de ratón se determinó mediante la evaluación de su capacidad para lisar los eritrocitos de pollo. Dado que los ratones utilizados tenían deficiencia de C5, la actividad hemolítica refleja directamente la actividad de C5 humana en la muestra. Brevemente, los anticuerpos a 50, 3, y 0 pg/mL en Gelatina Veronal-Solución salina tamponada (GVBS)
(Comptech Núm. Catálogo B100) que contenía gelatina al 0,1%, NaCI 141 mM, MgCb 0,5 mM, CaCb 0,15 mM, y barbital sódico 1,8 mM se utilizaron como control de lisis bajo, medio y del 100%, respectivamente. Las muestras experimentales se prepararon diluyendo el suero de ensayo murino 1:10 en GVBS. Se dispensaron alícuotas de la muestra (50 j L) en los correspondientes pocillos por triplicado de una placa de 96 pocillos (Corning;. Tewksbury, MA Núm. Catálogo 3799) que contenía un volumen igual de suero con deficiencia de C5 de ratón al 20% y suero humano al 20% (Bioreclamation, Núm. Catálogo HMSRM-COMP ) en GVBS en pocillos de control y un volumen igual de suero con deficiencia de C5 de ratón al 20% y suero humano con C5 agotada al 20% (Complement Technologies, Número de catálogo A320) en GVBS en los pocillos con muestra de ensayo. Se añadió EDTA (2 j L a 500 mM, Sigma, Número de catálogo 9884-E) en el tercer pocillo de los pocillos triplicados de control y de la muestra para generar un control de color de suero "sin hemólisis". Los eritrocitos de pollo se lavaron en GVBS, se sensibilizaron para activar la vía clásica del complemento por incubación con un anticuerpo policlonal anti-RBC de pollo (Intercell Technologies; 0,1% v/v) a 4°C durante 15 minutos, se lavaron de nuevo, y se resuspendieron en GVBS a una concentración final de ~ 7,5 * 107 células/mL. Los eritrocitos de pollo sensibilizados (~ 2,5 x 106 células) se añadieron a la placa que contenía los controles y las muestras, se mezclaron brevemente en un aparato de sacudimiento de placas, y se incubaron a 37°C durante 30 min. Los reactivos se mezclaron de nuevo, se centrifugaron a 845 xg durante 3 min, y se transfirieron 85 j L del sobrenadante a los pocillos de una placa de microtitulación de 96 pocillos de fondo plano (Nunc, Penfield, NY, Núm. de catálogo 439454). Se midió la absorbancia a 415 nm utilizando un lector de microplacas y se determinó el porcentaje de hemólisis utilizando la siguiente fórmula:
DO muestra - DO control color muestra
% de hemólisis = ------------------------------------------------------------------------------------- X 100 DO control lisis 100% - DO control color lisis 100%
en donde, DO = densidad óptica
Como se muestra en la Fig. 8, a pesar de la ligera reducción en la afinidad a pH 7,4 con respecto al eculizumab, tanto BNJ421 y BNJ423 todavía eran capaces de unirse casi a toda la C5 humana presente en la circulación y de inhibir la hemólisis. Estos resultados indican la afinidad de eculizumab para C5 puede ser debilitada sin comprometer la eficacia del anticuerpo in vivo, y conferir al anticuerpo un aumento de la semivida en suero.
Ejemplo 6. La unión dependiente del pH a C5 y la mejora del reciclaje mediado por FcRn son aditivas para la semivida en suero de las variantes de eculizumab
Como se ha mostrado anteriormente, en presencia de C5 humana, la semivida de una variante de eculizumab sustituida con histidina se amplió significativamente en los ratones transgénicos. Para evaluar los posibles efectos aditivos de la unión a C5 dependiente del pH y a FcRn sobre la farmacocinética (PK) y la farmacodinámica (PD) de los anticuerpos anti-C5 en presencia de la síntesis de C5 constitutiva y FcRn humano, se realizó una serie de experimentos de PK/PD utilizando anticuerpos anti-C5 de ratón con regiones constantes humanas en ratones transgénicos que expresaban FcRn humano. Estos anticuerpos anti-C5 murinos fueron diseñados a partir de la región variable de b B5.1, un anticuerpo murino, que sirve como sustituto farmacológico para eculizumab ya que se une a C5 de ratón y evita su escisión en los fragmentos metabólicos activos C5a y C5b [De Vries et al. (2003) Trasplantation 3:375-382]. Se diseñó un anticuerpo anti-C5 de ratón de alta afinidad (denominado: BHL011) con una variante de afinidad optimizada de las regiones variables murinas de BB5.1 y regiones constantes de IgK humana e IgG2/Ig4 humana. Se diseñó una variante de BHL011 dependiente del pH que incorporaba tres sustituciones de histidina en las regiones variables murinas (esta variante se denominó: BHL006). Se diseñó un tercer anticuerpo mediante la incorporación de dos sustituciones de aminoácidos en la región constante de la cadena pesada humana (M428L, N434S) para aumentar la afinidad por hFcRn (esta variante fue denominada: BHL009).
La secuencia de aminoácidos del polipéptido de cadena ligera de BHL006 es la siguiente:
NIMMTQSPSSLAVSAGEKVTMSCKSSQSVLYSSNQKNYLAWYQQKPGQSPKLLIYWA TRESGVPDRFTGSGSGTDFTLTISSVQAEDLAVYYCAQHLSHRTFGGGTKLEIKRTVAAP SVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDST YSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:30). La secuencia de aminoácidos del polipéptido de la cadena pesada del anticuerpo BHL006 es la siguiente:
QVQLQQPGAELVRPGTSVKLSCKASGYTFTSSWMHWVKQRPGQGLEWIGVIDPHDSYT NYNQKFKGKATLTVDTSSSTAYMQLSSLTSEDSAVYYCARGGGSSYNRYFDVWGTGTT VTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPA VLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPV AGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREE QFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPP SQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV DKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK (SEQ IDN0:31).
La secuencia de aminoácidos del polipéptido de cadena ligera de BHL009 es la siguiente:
NIMMTQSPSSLAVSAGEKVTMSCKSSQSVLYSSNQKNYLAWYQQKPGQSPKLLIYWAS TRESGVPDRFTGSGSGTDFTLTISSVQAEDLAVYYCAQHLSHRTFGGGTKLEIKRTVAAP SVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDST YSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:32).
La secuencia de aminoácidos del polipéptido de cadena pesada de BHL009 es la siguiente:
QVQLQQPGAELVRPGTSVKLSCKASGYTFTSSWMHWVKQRPGQGLEWIGVIDPHDSYT NYNQKFKGKATLTVDTSSSTAYMQLSSLTSEDSAVYYCARGGGSSYNRYFDVWGTGTT VTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPA VLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPV AGP S VFLFPPKPKD TLMISRTPE VT C V V VD V S QEDPEV QFN W Y VD G VEVHN AKTKPREE QFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPP SQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV DKSRWQEGNVFSCSVLHEALHSHYTQKSLSLSLGK (SEQ ID NO:33).
La secuencia de aminoácidos del polipéptido de cadena ligera de BHL011 es la siguiente:
NIMMTQSPSSLAVSAGEKVTMSCKSSQSVLYSSNQKNYLAWYQQKPGQSPKLLIYWAS TRESGVPDRFTGSGSGTDFTLTISSVQAEDLAVYYCAQYLSSRTFGGGTKLEIKRTVAAP SVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDST YSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO:34).
QYQLQQPGAELYRPGTSYKLSCKASGYTFTSSWMHWVKQRPGQGLEWIGYIDPSDSYT NYNQKFKGKATLTVDTSSSTAYMQLSSLTSEDSAVYYCARGGGSSYNRYFDVWGTGTT VTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPA VLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPV AGP S VFLFPPKPKD TLM I SRTPE VT C V V VD V S QEDPEV QFN W Y VD G VEVHN AKTKPREE QFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPP SQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV DKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK (SEQ ID NO:35).
La cinética de la unión de BHL011, BHL006 y BHL009 a ratón purificado se determinó mediante RPS en un aparato BIACore 3000 utilizando un método de captura anti-Fc humano. Brevemente, se inmovilizó anti-Fc humano (KPL,
Número de catálogo: 01-10-20) diluido a 0,1 mg/mL en acetato de sodio 10 mM pH 5,0, sobre dos células de flujo de un chip CM5 durante 8 minutos por acoplamiento de amina. Los anticuerpos se diluyeron a 0,25 pg/mL en tampón de migración (HBS-EP; HEPES 0,01 M pH 7,4, NaCl 0,15 M, EDTA 3 mM, Tensioactivo P20 al 0,005% v/v; GE Life Sciences, Número de catálogo: BR1001-88). A continuación, se inyectó el anticuerpo diluido en una célula de flujo seguido por una inyección de C5 de ratón 6 nM en ambas células. La segunda célula de flujo se utilizó como una superficie de referencia. La unión se evaluó a pH 7,4 y pH 6,0. La superficie se regeneró cada vez con HCl 20 mM, P20 al 0,01%. Los datos se procesaron con un modelo de Langmuir 1:1 utilizando el soporte lógico BIAevaluation 4.1 con "doble referencia". La disociación de BHL011, BHL006 y BHL009 complejados con C5 de ratón a pH 6,0 se evaluó de manera similar, con una inyección de C5 de ratón 6 nM (pH 7,4) seguido de una inyección de tampón HBS-EP (pH 6,0). Los resultados de estos experimentos se muestran en la Tabla 7.
Tabla 7.

Con el fin de determinar los efectos de la unión a C5 dependiente del pH sobre la farmacocinética (PK) de un anticuerpo anti-C5 en presencia de la síntesis de C5 constitutiva y el potencial para un mayor reciclaje de FcRn para conferir aumentos aditivos en la semivida, se analizaron la concentración total en suero de BHL011, BHL006 y BHL009 utilizando el modelo de ratón transgénico FcRn descrito en el Ejemplo 1. La concentración total en suero de anticuerpo y la concentración en suero como un porcentaje de la concentración el día 1 se muestran en las Fig. 9 11. Los ratones macho se representan como líneas continuas y las hembras como líneas discontinuas. Las concentraciones totales de anticuerpos en suero el día 1 fueron mayores para las hembras que para los machos, proporcionales a las diferencias en la masa corporal y el volumen de distribución. Esta diferencia por género contribuyó a la variabilidad entre animales para la farmacocinética de BHL011, posiblemente debido a la no linealidad dependiente de la dosis que resulta del aclaramiento mediado por C5 (Figs. 9A y 9B). En general, la variabilidad entre animales fue baja para BHL006 (Figs. 10A y 10B) y BHL009 (Figs. 11A y 11B) con la excepción de una hembra en la cohorte con dosis de BHL006 (2939), que mostró un aclaramiento acelerado. Las razones del aclaramiento acelerado en el animal 2939 son desconocidas.
En presencia de la síntesis constitutiva de C5 y hFcRn, el anticuerpo anti-C5 IgG2/4 de alta afinidad (BHL011) tenía una semivida terminal media de 6 días y se aclaró de la circulación ~98% a los 21 días (Figs. 12 y 13, Tabla 8). La tasa de aclaramiento media de un anticuerpo anti-C5 dependiente del pH con una región Fc de IgG2/4 (BHL006) fue atenuada con una semivida media en fase beta de 16-19 días. Se observó un aumento adicional de ~ 2 veces en la semivida de un anticuerpo anti-C5 dependiente del pH con una región Fc de IgG2/4 con una afinidad mejorada para hFcRn (semivida BHL009 ~36 días). Estos parámetros son coherentes con los observados para los anticuerpos IgG2/4 con y sin sustituciones M428L, N434S en ausencia de antígeno en ratones hFcRn. Estos resultados demuestran que la unión a C5 dependiente del pH y el incremento de afinidad por FcRn confieren efectos aditivos para ampliar la exposición PK de los anticuerpos anti-C5.
Tabla 8
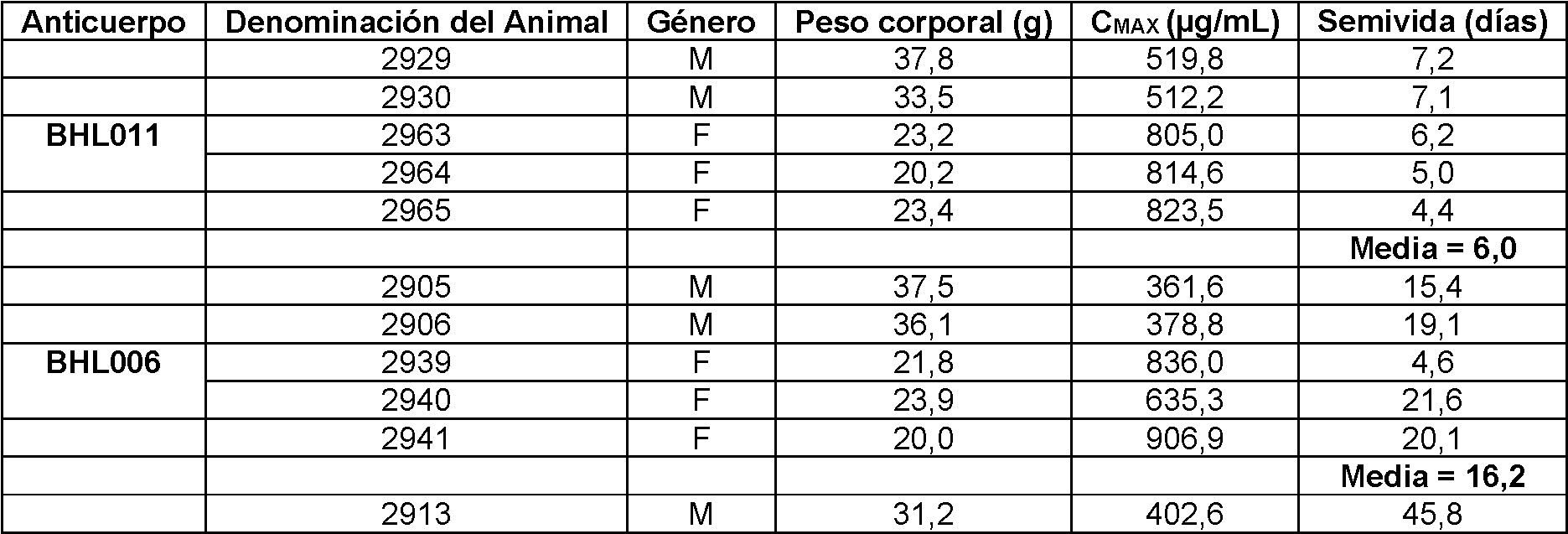
(continuación)

Farmacodinámica de anticuerpos anti-C5 de ratón en ratones transgénicos con FcRn humano
La actividad farmacológica de los anticuerpos anti-C5 de ratón en las muestras de suero se evaluó ex vivo en un análisis de hemólisis con eritrocitos de pollo mediado por la vía clásica del complemento (glóbulos rojos de pollo; cRBC). La actividad hemolítica se calculó como un porcentaje de la actividad en las muestras antes de la dosis y se muestra en las Fig. 14-16. Los machos se representan como líneas continuas y las hembras como líneas discontinuas. El antagonismo de la actividad hemolítica ex vivo es proporcional a la concentración de anticuerpo total en la muestra. La diferencia de género en la duración del antagonismo de la actividad hemolítica fue pronunciada para BHL011 (Fig. 14) que correspondía a la variabilidad entre animales dependiente de la masa corporal para la PK de BHL011 (Fig. 9). En general, la variabilidad entre animales fue baja para BHL006 (Fig. 15) y b Hl 009 (Fig. 16) con la excepción de la hembra en la cohorte con dosis de BHL006 (2939), que mostró un aclaramiento acelerado del anticuerpo (Fig. 10).
Las diferencias en la correlación entre la concentración total en suero de anticuerpos y el antagonismo de la actividad hemolítica ex vivo son proporcionales a la afinidad del anticuerpo para C5. El anticuerpo de alta afinidad (BHL011) casi suprimió completamente la actividad hemolítica a ~ 200 pg/mL (Fig. 17), mientras que los anticuerpos anti-C5 dependientes del pH, de afinidad más débil, requieren concentraciones de 2 a 3 veces más altas para lograr un antagonismo completo ex vivo (Figs. 18 y 19).
A pesar de esta pérdida de potencia en los anticuerpos anti-C5 dependientes del pH; los niveles medios de actividad para la hemólisis de cRBC en los animales de cada cohorte sugieren que podrían apoyar un intervalo de dosificación ampliado. El día 14, los animales tratados con anti-C5 de alta afinidad (BHL011) tenían niveles de actividad hemolítica medios de > 40%, mientras que los animales tratados con anti-C5 dependiente del pH (BHL006 y BHL009) mantenían niveles de actividad hemolíticas media <40% hasta el día 21 y 28, respectivamente (Fig. 20).
La ampliación significativa de la semivida y la correspondiente duración del antagonismo de los anticuerpos con unión dependiente del pH a C5 de ratón (BHL006 y BHL009) con respecto al anticuerpo anti-C5 de ratón de alta afinidad (BHL011) fue compatible con los estudios descritos en los Ejemplos 4 y 7 en los que un anticuerpo anti-C5 humana dependiente del pH (BNJ421, BNJ423 o BNJ441) mostró un aumento de la semivida similar con respecto a su homólogo de alta afinidad (EHL000 o eculizumab) en ratones a los que se había administrado simultáneamente C5 humana. Estos resultados corroboran adicionalmente más la noción de que el diseño de una unión al antígeno dependiente del pH a través de determinadas sustituciones de histidina en las CDR puede atenuar significativamente el aclaramiento mediado por el antígeno aunque C5 permita que el anticuerpo libre sea reciclado de nuevo a la circulación. Además, la combinación de la unión al antígeno dependiente del pH y una mayor afinidad para FcRn en BHL009 era aditiva en los efectos sobre las propiedades de la PK, duplicando la semivida por encima de la unión dependiente del pH solo (BHL006). Estas observaciones son compatibles con la hipótesis de que la unión a C5 dependiente del pH combinada con una mejor afinidad para FcRn puede proporcionar una ampliación significativa en los parámetros de la PK y la duración de la PD terapéutica observado para eculizumab para permitir una dosificación > mensual.
Ejemplo 7. Generación de una variante de eculizumab con unión a C5 dependiente del pH y mejor reciclaje mediado por FcRn
Se generó un anticuerpo utilizando eculizumab como molécula de partida. Con respecto a eculizumab, el anticuerpo variante (denominado BNJ441) contenía cuatro sustituciones de aminoácidos en la cadena pesada, Tyr-27-His, Ser-57-His, Met-429-Leu y Asn-435-Ser (obsérvese que las posiciones 429 y 435 de BNJ441 corresponden a las posiciones 428 y 434 bajo el sistema de numeración de EU). La secuencia de aminoácidos para el polipéptido de cadena pesada se muestra en el SEQ ID NO: 14. La secuencia de aminoácidos para el polipéptido de cadena ligera se representa en el SEQ ID NO: 11. Estas mutaciones fueron diseñados para permitir un intervalo de dosificación ampliado con BNJ441 (véase eculizumab) mediante el aumento de la semivida en circulación a través de dos mecanismos distintos: (1) reducción del aclaramiento del anticuerpo a través del aclaramiento del anticuerpo mediado por la diana y (2) aumento de la eficacia del reciclaje del anticuerpo mediado por FcRn.
Las dos sustituciones de aminoácidos en la primera y segunda regiones determinantes de la complementariedad (CDR) de la región variable de cadena pesada, Tyr-27-His y Ser-57-His, debilitar la constante de disociación de afinidad (Kd ) de BNJ441 para C5 ~ 17 veces a pH 7,4 y ~36 veces a pH 6,0 en comparación con eculizumab. Las dos mutaciones en el tercer dominio de la región constante de la cadena pesada (CH3), Met-429-Leu y Asn-435-Ser,
aumentan la afinidad de BNJ441 para FcRn ~ 10 veces a pH 6,0 en comparación con eculizumab.
Cinética de unión (Anticuerpos para C5)
La cinética de la unión de BNJ441 o eculizumab a C5 se determinó por medio de resonancia de plasmón superficial (RPS) en un aparato BIAcore 3000 utilizando un método de captura anti-Fc a pH 8,0, 7,4, 7,0, 6,5 y 6,0. Se diluyó anticuerpo policlonal anti-IgG (Fc) humana de cabra (KPL Núm. 10-1-20) a 0,1 mg/mL en acetato de sodio 10 mM pH 5,0 y se inmovilizó sobre dos células de flujo de un chip CM5 durante 8 min mediante acoplamiento de amina. El anticuerpo de ensayo (BNJ441 o eculizumab) se diluyó hasta 0,20 pg/mL en tampón de migración (HBS-EP; HEPES 0,01 M pH 7,4, NaCl 0,15 M, EDTA 3 mM, tensioactivo P20 al 0,005% v/v; GE Life Sciences, Número de catálogo: BR1001-88). A continuación, se inyectó el anticuerpo diluido en una célula de flujo (20 pL para el experimento a pH 7,4 experimento y 40 pL para el experimento a pH 6,0) seguido de inyecciones de concentraciones variables de C5 en ambas células. El tampón de migración se tituló con HCl 3M para la cinética a pH 7,0, 6.5 y 6,0 y con NaOH 0,5 M para la cinética a pH 8,0. La superficie se regeneró cada ciclo con HCl 20 mM, P20 al 0,01%. Los datos fueron procesados con un Modelo 1:1 de Langmuir utilizando el soporte lógico BIAevaluation 4.1 (Biacore AB, Uppsala, Suecia) con "doble referencia".
Las velocidades de disociación de C5 de BNJ441 o eculizumab a pH 8,0, 7,4, 7,0, 6,5 y 6,0 se determinaron a través de RPS en un aparato BIAcore 3000 utilizando el método de captura anti-Fc descrito anteriormente con las siguientes modificaciones. Los anticuerpos de ensayo diluidos se inyectaron en una célula de flujo seguido por una inyección de C56 nM en ambas células. Inmediatamente después de la inyección de C5, se inyectaron 250 pL de tampón de migración a diversos pH. Los tampones d migración se prepararon como se ha descrito anteriormente. Los datos fueron procesados utilizando el soporte lógico BIAevaluation 4.1 (Biacore AB, Uppsala, Suecia) con "doble referencia". El % de disociación de C5 de BNJ441 y eculizumab se calculó tomando la diferencia de disociación en t = 0 y t = 300 segundos.
Cinética de unión (Anticuerpos para FcRn)
La cinética de la unión de BNJ441 o eculizumab a FcRn humano se determinó a través de RPS en un aparato BIAcore 3000 utilizando un método de captura F(ab')2 a pH 7,4, y 6,0. Se inmovilizó anti-F(ab')2 de IgG humana de cabra (Rockland Immunochemicals, Número de catálogo 709-1118) diluido a 0,04 mg/mL en acetato de sodio 10 mM pH 5,0, sobre dos células de flujo de un chip CM5 durante 7 minutos por acoplamiento de amina. El anticuerpo de ensayo (BNJ441 o eculizumab) se diluyó a 2 pg/mL en tampón de migración ((HBS-EP; HEPES 0,01 M, pH 7,4, NaCl 0,15 M, EDTA 3 mM, tensioactivo P20 al 0,005% v/v; g E Life Sciences, Núm. Cat. BR1001-88). A continuación se inyectó el anticuerpo diluido en una célula de flujo seguido de inyecciones de FcRn en ambas células. El tampón de migración se tituló con HCl 3M para la cinética a pH 6,0. La superficie se regeneró cada ciclo con glicina HCl 10 mM, pH 1,5). Los datos fueron procesados con un Modelo 1:1 de Langmuir utilizando el soporte lógico BIAevaluation 4.1 (Biacore AB, Uppsala, Suecia) con "doble referencia".
Resultados de los estudios de unión
La cinética de la unión anticuerpo:C5 que era dependiente del pH con efectos sobre las velocidades de asociación y disociación se muestra en la Tabla 9.
Tabla 9

En un intento de modelar las velocidades relativas de disociación de los complejos de anticuerpo:C5 tras la pinocitosis y la acidulación del endosoma temprano, se permitió que se formaran complejos anticuerpo: C5 en un tampón de pH 7,4, a continuación, se cambiaron las condiciones de pH del tampón durante la disociación. Se calculó
el porcentaje de disociación del complejo de anticuerpo (estimado por la disminución en unidades de resonancia [UR]) después de 300 segundos para cada condición de pH (Tabla 10). Solo BNJ441 a pH 6,0 dio como resultado una disociación de anticuerpo:C5 mayor del 50% después de 5 minutos.
Tabla 10

Las Figs. 21A y 21B representan gráficos semi-log y lineales del porcentaje de disociación de los complejos de BNJ441:C5 o de los complejos eculizumab: C5 como una función del pH.
Las dos sustituciones de aminoácidos en la primera y segunda regiones determinantes de complementariedad (CDR) de la región variable de cadena pesada, Tyr-27-His y Ser-57-His, debilitan la constante de disociación de afinidad (Kd ) de BNJ441 para C5 ~17 veces a pH 7,4 y ~36 veces a pH 6,0 en comparación con eculizumab. No está claro si la dependencia del pH en la afinidad de BNJ441 para C5 es el resultado de los cambios en el estado de protonación de las histidinas introducidas en las posiciones 27 y/o 57, o simplemente un debilitamiento general de la afinidad para C5. Se ha observado en otros anticuerpos anti-C5, sin embargo, que estas mutaciones combinadas con sustituciones de histidina adicionales, dan como resultado pérdidas de afinidad mucho más pronunciadas a niveles de pH por debajo de 6,5. Las dos mutaciones en el tercer dominio de la región constante de cadena pesada (CH3), Met-429-Leu y Asn-435-Ser, fortalecen la afinidad de BNJ441 para FcRn ~10 veces a pH 6,0 en comparación con eculizumab.
Propiedades de la PK del anticuerpo BNJ441
El anticuerpo BNJ441 y eculizumab se evaluaron en ratones que eran inmunodeficientes (NOD/SCID) y tenían deficiencia de C5. Se administró una única dosis de 100 |jg de BNJ441 o eculizumab en 200 jL de solución salina tamponada con fosfato (PBS) por inyección intravenosa (i.v.) a cada uno de ocho ratones. Se recogió el suero de cada uno de los ratones en los días uno, tres, siete, 14, 21, 28 y 35 después de la administración. La concentración de cada anticuerpo en suero se midió por medio de ELISA.
Como se muestra en la Fig. 22, en ausencia de C5 humana, las concentraciones de anticuerpo en suero disminuyeron de manera similar en ratones a los que se habían administrado dosis de BNJ441 y eculizumab durante un período de 35 días. Sin embargo, en presencia de C5 humana, las concentraciones en suero de eculizumab disminuyeron rápidamente a niveles no detectables después de 14 días, mientras que la concentración en suero de BNJ441 disminuyó más lentamente y a una velocidad constante a lo largo de la duración del estudio (Fig. 23).
Comparando los perfiles de PK de los dos anticuerpos en la presencia y ausencia de C5 humana, el aclaramiento de eculizumab se aceleró en presencia de C5 humana en comparación con el que se observaba en ausencia de C5 humana, mientras que el perfil de PK de BNJ441 en presencia de C5 humana fue similar al de BNJ441 en ausencia de C5 humana hasta el día 28, y el aclaramiento solamente se aceleró entre los días 28 y 35 (Fig. 24). La semivida de BNJ441 y la semivida de eculizumab fueron comparables en ausencia de C5 humana (25,37 ± 1,02 días para BNJ441 y 27,65 ± 2,28 días para eculizumab). Sin embargo, en la presencia de C5 humana, BNJ441 demostró un aumento de más de tres veces en la semivida en comparación con eculizumab (13,40 ± 2,18 días para BNJ441 vs.
3,93 ± 0,54 días para eculizumab). Cabe señalar que la tasa de aclaramiento de BNJ441 no difirió significativamente en presencia o ausencia de C5 humana hasta el día 28. Véase la Tabla 11.
Tabla 11

(continuación)

Actividad hemolítica en suero
Para determinar el efecto de las sustituciones de histidina sobre la actividad hemolítica del anticuerpo, se realizó un análisis hemolítico ex vivo como se describe en el Ejemplo 6. En presencia de BNJ441, o eculizumab, la actividad del complemento terminal era compatible con los respectivos perfiles de PK de cada anticuerpo (Fig. 25) - es decir, el nivel de inhibición de la actividad hemolítica en suero fue proporcional a la concentración de cada anticuerpo restante en el suero. Ambos anticuerpos confirieron una inhibición casi total de la hemólisis hasta el día 3. Sin embargo, eculizumab no mostró antagonismo el día 14, mientras que el BNJ441 conservó una inhibición de aproximadamente 83% el día 14 y una inhibición parcial del complemento hasta el día 28 días.
Conclusión
Los descubrimientos de este estudio sugieren que, en presencia de C5 humana, BNJ441 mostró una ampliación de más de tres veces en la semivida en comparación con eculizumab. Además, la semivida en suero de BNJ441 con respecto a eculizumab se tradujo en un perfil farmacodinámico prolongado, como se evidencia por la inhibición hemolítica prolongada.
Ejemplo 8. Seguridad, tolerabilidad, PK y PD de BNJ1441 en sujetos sanos humanos
La seguridad, tolerabilidad, PK y PD de BNJ441 se evaluó en un estudio clínico en seres humanos de Fase 1, de dosis única ascendente (SAD), controlado por placebo, ciego, aleatorizado, en donde BNJ441 se administró por vía intravenosa a sujetos sanos.
BNJ441 se formuló en una solución acuosa estéril, sin conservantes, con excipientes de formulación. La formulación de BNJ441 no contenía ningún excipiente inusual, o excipiente de origen animal o humano. La formulación se tamponó con fosfato a un pH de 7,0. Los componentes incluyeron BNJ441 a 10 mg/ml, fosfato de sodio monobásico 3,34 mM, fosfato de sodio dibásico 6,63 mM, cloruro de sodio 150 mM, polisorbato 80 al 0,02% y agua Q.S.
La formulación de BNJ441 se suministró en forma de una solución de anticuerpo a 10 mg/mL en un vial de un solo uso de 20 mL, y fue diseñada para la infusión mediante su dilución en solución salina comercialmente disponible (inyección de cloruro de sodio al 0,9%, Ph Eur) para la administración IV.
Tabla 12: Prueba clínica en Fase 1 en voluntarios sanos

Diez sujetos sanos recibieron una dosis única de BNJ441. Cuatro sujetos recibieron una dosis de 200 mg y seis sujetos recibieron una dosis de 400 mg. Los datos de PK y de seguridad para este estudio se determinaron y se comentan a continuación.
Farmacocinética
Los perfiles de concentración-tiempo de BNJ441 en suero después de la administración IV de dosis de 200 mg y 400 mg se representan en la Figura 26. Se disponía de los datos de concentración-tiempo hasta el Día 90 (2136 horas) y el día 57 (1344 horas), después de las dosis de 200 mg y 400 mg, respectivamente. Las concentraciones medias en suero se mantuvieron por encima de 50 pg/mL durante 2 a 4 días (de 48 a 96 horas) después de la dosis de 200 mg, y 14 a 21 días (336 a 504 horas) después de la dosis de 400 mg.
En la Tabla 13 de más abajo se hace referencia a un resumen de los parámetros PK de BNJ441. La media geométrica (CV) Cmax de BNJ441 fue 78,5 (10,2%) pg/mL después de la dosis de 200 mg, y 139 (16,2%) pg/mL después de la dosis de 400 mg. La mediana observada (rango) tmax fue 2,4 (0,79 a 8,0) horas para la dosis de 200 mg, y 0,58 (0,58 a 1,1) horas para la dosis de 400 mg después del inicio de la infusión. La media geométrica (CV) AUC(0-56 días) es de 32.800 (8,6%) p-hr/mL para la dosis de 200 mg y 58.100 (18,9%) g-hr/mL para la dosis de 400 mg. La media geométrica Cmáx y AuC(0-56 días) indican que la exposición aumenta de manera proporcional a la dosis aparente. La media geométrica tv2 (CV) es de 38,5 (18,4%) días y 32,9 (13,3%) días para las dosis de 200 mg y 400 mg, respectivamente.
En resumen, los datos farmacocinéticos sugieren la Cmáx media y el AUC(0-56 días) de BNJ441 aumentaron de manera proporcional a la dosis, y apoyan un t1/2 medio (desviación típica [DT]) de 35,5 ± 6,1 días después de la administración IV. El análisis de los datos de hemólisis de glóbulos rojos de pollo (cRBC) indican que complemento terminal fue inhibido completamente hasta 2 días después de una sola dosis de 400 mg por vía IV, cuando las concentraciones de BNJ441 fueron superiores a 100 pg/mL.
Tabla 13: Resumen de los parámetros farmacocinéticos para BNJ441 después de la administración IV of 200 ________ ____________ ____________mg o 400 mg a voluntarios sanos_______ _________________________ D i Estadística Cm a x (pg/m L ) Cm a x ! Do a x (h) A U C i° A UC r/D i (día) escriptiv fo /m L/ (hug/m /m L) N 4 4 4 4 4 4 Media 78,5 ,39 2,40b 2,80 164 38,5 eométric Media 10,2 10,2 ,79 - 8,6 8,6 18,4 eométric 8,0c CV% N 6 6 6  6 6 6 Media 139 ,34 0,58b 58,10 145 32,9
6 6 6 Media 139 ,34 0,58b 58,10 145 32,9 
eométric




(continuación)

a AUCt = AUC(0 -56 días)
b mediana
c rango____________
Farmacodinámica
Se evaluó también la capacidad de BNJ441 para inhibir la hemólisis de cRBC con el tiempo, como se ilustra en la Figura 27. La actividad de hemólisis de cRBC media fue relativamente estable en los sujetos que recibieron placebo. El inicio de la inhibición de la hemólisis de cRBC fue rápido, observándose una inhibición completa del complemento terminal al final de la infusión (0,29 horas para la dosis de 200 mg, y 0,58 horas para la dosis de 400 mg). BNJ441 tuvo una duración de la acción dependiente de la dosis, que duró de 4 a 14 días.
La relación entre la concentración de BNJ441 y la hemólisis cRBC se traza y se representa en la Figura. 28. Como se muestra en la Figura 28, la inhibición completa del complemento terminal se produjo a concentraciones de BNJ441 superiores a 50 pg/mL, no se observó inhibición a concentraciones de BNJ441 por debajo de 25 pg/mL. Ejemplo 9. Estudio de dosis única en monos cinomolgos
Se administró una sola dosis IV de BNJ441 a monos cinomolgos a dosis de 60 o 150 mg/kg (n = 4 para cada grupo de dosificación, 2 machos y 2 hembras por grupo de dosificación) en forma de una infusión de 2 horas. Las muestras de sangre para el análisis de BNJ441 se recogieron del Día 1 al Día 112.
Todos los monos tratados con BNJ441 fueron escrutados para determinar la presencia de anticuerpos anti-humanos de Cynomologus (CAHA) antes de la dosificación (0 horas), y en los Días 8, 14, 28, 56, 84, y 112.
Todos los monos del grupo de dosificación de 60 y 150 mg/kg fueron confirmados como positivos en al menos una sola ocasión, excepto el Animal 2002 del grupo de dosificación de 150 mg/kg. La presencia de CAHA en el Animal 2002, o en momentos específicos no positivos para los otros animales, no se puede excluir, debido a la posible interferencia del BNJ441 administrado con el análisis de vinculación de BNJ441 biotinilado y BNJ441 rutenilado. Se observaron resultados de CAHA positivos en el grupo de dosificación de 60 mg/kg desde el día 56 al 112 después de la dosificación, y en el grupo de dosificación de 150 mg/kg desde el día 28 al 112 después de la dosificación. La primera muestra CAHA-positiva confirmada en el grupo de 60 mg/kg fue el Día 56 (animales 1002 y 1503), 2 en el Día 84 (animales 1002 y 1503), y 3 en el Día 112 (animales 1001, 1002, y 1502). El animal 1503, que fue CAHA positivo los Días 56 y 84, ya no era CAHA-positivo el Día 112. La primera muestra CAHA positiva confirmada en el grupo de dosificación de 150 mg/kg fue el animal 2502 el Día 28, seguido de 2 monos el Día 56 (animales 2001 y 2502), 3 monos el Día 84 (animales 2001, 2501 y 2502), y 3 monos el Día 112 (animales 2001, 2501 y 2502).
Se calcularon los perfiles de concentración-tiempo de BNJ441 individuales. En el grupo de dosificación de 60 mg/kg, todos los monos tuvieron concentraciones en plasma cuantificables de BNJ441 hasta la muestra de PK del Día 112, mientras que en el grupo de dosificación de 150 mg/kg, solo el 1 mono (animal 2002) tenía concentraciones de BJN441 cuantificables en plasma hasta el Día 112. Los datos de concentración-tiempo indicaron una residencia prolongado de BNJ441 en la circulación sistémica de los monos.
Los parámetros PK no compartimentales y las estadísticas resumidas para BNJ441 se calcularon para todos los monos por nivel de dosificación, y se muestran en las Tablas 14 y 15 para los niveles de dosificación de 60 mg/kg y 150 mg/kg, respectivamente. En consonancia con la duración de la infusión, la tmax mediana fue de 2 horas para los niveles de dosificación de 60 mg/kg y 150 mg/kg. Un mono en el grupo de dosificación de 150 mg/kg, animal 2501, tuvo tmáx de 12 horas después de la dosificación, y tuvo un perfil relativamente plano de 2 a 12 horas después de la dosificación, con una concentración en la muestra 12 horas después de la dosificación aproximadamente 5% mayor que la observada a las 2 horas después de la dosificación. Todas las medias geométricas de Cmax , AU C , y AUCfinal aumentaron al aumentar la dosis. Las medias geométricas de los valores de Cmax normalizados para la dosis fueron similares en las 2 dosis, lo que indica un aumento proporcional a la dosis de la concentración máxima de BNJ441 con un aumento de la dosis, pero las medias geométricas de los valores de AUC~ normalizados para la dosis fueron diferentes entre los grupos de dosificación. Esta diferencia se debe probablemente al incremento mediado por CAHA en BNJ441 CL en el grupo de dosificación de 150 mg/kg; el aclaramiento de BNJ441 fue aproximadamente 37% mayor en los monos que recibieron dosis de 150 mg/kg en comparación con los monos que recibieron dosis de 60 mg/kg. La media geométrica Vss fue similar (dentro de 12%) entre los 2 grupos de dosificación.


Ejemplo 10. Evaluación Comparativa de BNJ441, eculizumab y h5G1.1 que se unen a los receptores Fc gamma C1q in vitro
Se examinó la unión de tres anticuerpos humanizados, BNJ441, eculizumab y h5G1.1-IgG1 a moléculas conocidas por ser mediadoras de la función efectora de anticuerpos. BNJ441, eculizumab, y h5G1.1-IgG1 tienen cada uno perfiles funcionales y terapéuticos únicos. Sin embargo, los tres son antagonistas de anticuerpos humanizados del complemento terminal, que se unen un epítopo muy similar en el componente C5 del complemento humano y evitan su escisión durante la activación del complemento a sus metabolitos activos, C5a y C5b.
BNJ441, eculizumab, y h5G1.1-IgG1 tienen secuencias de cadena ligera idénticas, teniendo cada uno una región variable humanizada y una región constante IgKappa humana. BNJ441 y eculizumab contienen ambos un Fc IgG2-G4 híbrido humano, que incluye la región CH1, la bisagra y los primeros 29 aminoácidos de la región CH2 de IgG2 humana fusionada con el resto de las regiones CH2 y CH3 de IgG4 humana. Este Fc quimérico combina el emparejamiento de enlaces disulfuro estable de una IgG2 con las propiedades menos efectoras de una IgG4. Puesto que BNJ441 y eculizumab se dirigen contra un antígeno soluble, no fue posible evaluar directamente su capacidad para iniciar la citotoxicidad mediada por células dependiente de anticuerpos (ADCC) o la citotoxicidad dependiente del complemento (CDC). En su lugar, se realizaron mediciones directas de la unión de BNJ441 o eculizumab a receptores Fc gamma (FcyR) y el componente del complemento C1q y se infirió que, en ausencia de unión no pueden mediar en la ADCC o CDC, respectivamente. h5G1.1-IgG1 (un anticuerpo de isotipo IgG1 con la misma región variable humanizada que eculizumab) se incluyó como control. Se espera que la región Fc del isotipo IgG1 se una a moléculas con función efectora totalmente, aunque h5G1.1-IgG1 no provocaría ADCC o CDC en ausencia de un antígeno asociado a la célula.
Como se comentó anteriormente en el Ejemplo 7, BNJ441 fue diseñado a partir de eculizumab para aumentar su semivida in vivo mediante la introducción de 4 sustituciones de aminoácidos en la cadena pesada. Se introdujeron dos cambios de aminoácidos en la región variable de la cadena pesada humanizada, Tyr-27-His y Ser-57-His, respectivamente (numeración de aminoácidos de la cadena pesada de acuerdo con Kabat et al.), para desestabilizar la unión a C5 a pH 6,0 con un mínimo impacto en la unión a C5 a pH 7,4. Las mutaciones en el tercer dominio de la región constante de la cadena pesada (CH3), Met-428-Leu y Asn-434-Ser, se introdujeron para mejorar la unión al receptor Fc neonatal humano (FcRn). Tomadas en conjunto estas mutaciones fueron diseñadas para atenuar significativamente el aclaramiento del fármaco mediado por el antígeno por el aumento de la disociación de los complejos de anticuerpo:C5 para liberar el anticuerpo en el entorno acidulado del endosoma temprano después de pinocitosis, y para aumentar la fracción de anticuerpo reciclada a partir del endosoma temprano de nuevo al compartimento vascular por FcRn.
En estos estudios, se evaluaron las interacciones multiméricas de las subclases de FcyR (FcyRI, FcYRIIa, FcYRIIb, FcYRIIb/c, FcYRIIIa y FcYRIIIb) con los tres anticuerpos en un análisis inmunoenzimático (ELISA) y se evaluaron las interacciones monoméricas con los FcyR utilizando la resonancia de plasmón superficial (RPS). La interferometría de biocapa interferometría y la RPS se utilizaron para examinar la unión de C1q a los tres anticuerpos. Los reactivos utilizados para llevar a cabo estos análisis se muestran en la Tabla 16.
T l 1 . An i r r iv r i

Unión de complejos de anticuerpos multivalentes a FcyR
Se prepararon complejos de anticuerpo mediante incubación de BNJ441, Eculizumab o h5G1.1-HG1 durante la noche con anticuerpo anti-F(ab')2 humano-biotina de cabra (Jackson Immunolabs), a una razón molar de anticuerpo:F (ab')2 de 2.1 en solución salina tamponada con fosfato (PBS) en un tubo de microcentrífuga de 1,5 mL. Las placas de microtitulación pre-recubiertas con Ni-NTA (Qiagen) se incubaron con 50 pL/pocillo de 6X FcyR humanos etiquetados con histidina (FcyRI, FcYRIIa, FcYRIIb, FcYRIIb/c, FcYRIIIa y FcYRIIIb), a una concentración de receptor de 5 pg/mL en PBS, durante la noche a 4°C. La placa se lavó a continuación 3 veces con PBS/Tween-20
al 0,05%. Después del lavado, se incubaron 50 pL de complejos de anticuerpo en PBS/Tween-20 al 0,05% en la placa durante 60 minutos a la temperatura ambiente. Después de lavar la placa con PBS/Tween-20 al 0,05%, se añadieron 50 pL de estreptavidina-HRP (Invitrogen) en PBS/Teween-20 al 0,05% a la placa y se incubaron durante 60 min a RT. Después de esta incubación y lavados, se añadieron 75 pL de sustrato TMB-ELISA (3,3',5,5'-tetrametilbencidina, Thermo Scientific). La reacción se detuvo con 75 pL de H2SO42 M, y la absorbancia se leyó a 450 nm.
Las muestras se ejecutaron por duplicado y los datos se presentan como valores medios. Los resultados se introdujeron en un programa de hoja de cálculo. La absorbancia a 450 nm de cada concentración de anticuerpo del complejo inmunitario de anticuerpo o ausencia de complejos inmunitarios de anticuerpos se representa en forma de representación gráfica. Las constantes de disociación clave se calcularon y se resumen en la Tabla 17 y se comentan a continuación.
Unión de anticuerpos monovalentes a FcyR
Se determinaron las cinéticas de unión de BNJ441, eculizumab, y h5G1.1-IgG a los FcyR mediante RPS en un aparato BIAcore 3000 mediante inmovilización directa. BNJ441, eculizumab, y h5G1.1 se diluyeron en acetato de sodio 10 mM pH 5,0, se inmovilizaron sobre una célula de flujo de un chip CM5 mediante acoplamiento de amina. Una segunda célula de flujo se utilizó como superficie de referencia. Las concentraciones de FcyR diluidos en tampón de migración (HBS-EP, pH 7,4) se inyectaron en ambas células. La superficie se regeneró cada ciclo con HCl 20 mM, P20 al 0,01%. Los datos se analizaron mediante un modelo de afinidad en estado estacionario en el soporte lógico BIAevaluation 4.1 (Biacore AB, Uppsala, Suecia) con "doble referencia".
La cinética de la unión de h5G1.1-IgG1 a FcyRI se evaluó a través de la cinética del ciclo individual debido a su afinidad más fuerte. El anticuerpo se diluyó en acetato de sodio 10 mM a pH 5,0 y directamente se inmovilizó sobre una célula de flujo de un chip CM5 mediante acoplamiento de amina. Se utilizó una segunda célula de flujo como superficie de referencia. Las concentraciones de FcyR1 diluido en tampón de migración (HBS-EP, pH 7,4) se inyectaron en ambas células. Este análisis no requiere regeneración. Los datos fueron analizados utilizando un modelo de cinética de titulación 1:1 en BIAevaluation 4.1 (Biacore AB, Uppsala, Suecia) con el soporte lógico de "doble referencia"'.
Tabla 17: Constantes de disociación para la unión de BNJ441, eculizumab y h5G1.1-IgG1 a FcyR
monoméricos

Se realizaron análisis ELISA para detectar interacciones multiméricas dirigidas por avidez de complejos inmunitarios de anticuerpos y FcyR. Los resultados se resumen en la Tabla 17. BNJ441 y eculizumab no mostraron ninguna unión detectable a FcyRI, FcYRIb/c, FcYRIIIa o FcYRIIIb y una asociación de 4 veces a 8 veces más débil con FcYRIIa, respectivamente. Las constantes de disociación (Kd ) para la unión de FcyR monomérico a BNJ441 y eculizumab obtenidas mediante RPS confirmaron que las interacciones con FcyR son muy débiles y casi indistinguibles entre los dos anticuerpos: FcyRI (~ 4 pM), FcYRIIa (~ 2 pM), FcYRIIb (~ 9 pM), FcYRIIIa (~ 7 pM) y FcYRIIIb (~ 3 pM). Las constantes de disociación para el control de isotipo IgG1 (h5G1.1-IgG1) fueron compatibles con las interacciones de alta afinidad con FcyR1 (123 pM) y modestos aumentos en la unión a los FcyR de baja afinidad con respecto a los anticuerpos de isotipo IgG2-G4: FcYRIIa (~ 1 pM ), FcYRIIb (~ 3 pM), FcYRIIIa (~ 1 pM) y FcYRIIIb (~ 2 pM). Véase la Tabla 17. No se detectaron interacciones entre C1q y BNJ441 o eculizumab mediante interferometría de biocapa. Estos resultados son compatibles con la idea de que Fc de IgG2-G4 quimérico humano de eculizumab tiene poca o ninguna capacidad para lograr la función efectora a través de los FcyR o C1q para mediar en la ADCC o CDC, respectivamente. Además, estos resultados muestran que las sustituciones de aminoácidos en la cadena pesada incorporadas en BNJ441 no alteran significativamente la unión a estos, con respecto a eculizumab.
Ejemplo 11. Estudios de reacción cruzada con tejidos
1. Estudios de reactividad cruzada con GLP humano
Se determinó la reactividad cruzada potencial con los tejidos humanos utilizando BNJ441 con fluoresceína (denominado BNJ441-FITC) y un anticuerpo de control (OX-90G2G4-FITC) con una especificidad antigénica diferente.
BNJ441-FITC produjo una tinción de moderada a intensa del material de control positivo (portas con aplicación de
ultravioleta [UV]-resina con proteína C5 del complemento humano purificada, denominada hC5), pero no reaccionó específicamente con el material de control negativo (portas con aplicación de UV-resina con péptido tumoral humano relacionado con la hipercalcemia humana, residuos de aminoácido 1-34, denominado PTHrP 1-34). El artículo de control, OX-90G2G4-FITC, no reaccionó específicamente con ninguno de los materiales de control positivo o negativo. Las excelentes reacciones específicas de BNJ441-FITC con el material de control positivo y la falta de reactividad específica con el material de control negativo, así como la falta de reactividad del artículo de control, indicaron que el análisis era sensible, específico y reproducible.
La tinción con BNJ441-FITC se observó en el panel de tejidos humanos, como se resume a continuación:
• Material proteico en la mayoría de los tejidos humanos
• Citoplasma y gránulos citoplásmicos en los siguientes elementos tisulares:
-células mononucleares en colon, esófago, ganglio linfático, paratiroides, bazo, y tonsila
-plaquetas en frotis de sangre y médula ósea
-megacariocitos en médula ósea
-epitelio en trompa de Falopio, hígado (hepatocitos), conductos pancreáticos, y cérvix
-mesotelio en el pulmón
Debido a que C5 es una proteína sérica circulante, se esperaba la tinción de material proteico. Se ha informado de que las células mononucleares, tales como monocitos, macrófagos y células dendríticas, así como las plaquetas, secretan C5; por lo tanto, también se espera la tinción de estos tipos de células con BNJ441-FITC. Además, se ha demostrado que las líneas de células mesoteliales producen C5. Sin embargo, no estaba disponible la bibliografía que describe la expresión de C5 por los tipos de células epiteliales teñidas con BNJ441-FITC en el estudio actual, o por los megacariocitos, aunque las plaquetas, que se ha demostrado que producen C5, derivan de megacariocitos. Por lo tanto, la tinción de tipos de células epiteliales podría representar cualquiera de los sitios de expresión de C5 no reconocidos previamente, o reactividad cruzada tisular con una secuencia de proteína o estructura de una proteína similar pero no relacionada u otro constituyente o sustituyentes de las secciones de tejido. Sin embargo, con la excepción de la tinción del material proteico, toda la tinción observada en este estudio fue de naturaleza citoplasmática, y es poco probable que el citoplasma y las estructuras citoplasmáticas sean accesibles para el artículo de ensayo in vivo. En resumen, no se observó reactividad cruzada específica de la tinción BNJ441-FITC que diera lugar a la expectativa de la toxicidad relacionada con el tratamiento.
1. Estudios de reactividad cruzada tisular en mono Cynomolgus
También se realizó un estudio de reactividad cruzada tisular con GLP convencional utilizando un panel de tejidos de mono cinomolgo para examinar la unión tanto fuera de la diana como en la diana, con los mismos reactivos utilizados en los estudios de unión de tejido humano.
Se observó cierta tinción con BNJ441-FITC en el panel de tejidos de mono cinomolgo, como se resume a continuación:
• Material proteico en la mayoría de tejidos de mono cinomolgo
• Citoplasma y/o gránulos citoplasmáticos de los siguientes elementos del tejido:
- células mononucleares en el ganglio linfático, bazo y tonsila
- epitelio en la trompa de Falopio.
El patrón de tinción de BNJ441-FITC observado en el panel de tejidos de mono cinomolgo fue en general menos intenso y menos frecuentes que el observado en el panel de tejidos humanos en el estudio de reactividad cruzada tisular en seres humanos complementario. Además, en el panel de tejidos humanos, se observó tinción de las plaquetas, megacariocitos, epitelio ductal pancreático, epitelio cervical, hepatocitos, y mesotelio, aunque estos elementos de tejido no se tiñeron en el panel de tejidos de mono cinomolgo. Por otra parte, con la excepción de la tinción de material proteico, la tinción observada en este estudio fue de naturaleza citoplasmática, y es poco probable que el citoplasma y las estructuras citoplasmáticas sean accesibles para el artículo de ensayo in vivo. Debido a que BNJ441 ha demostrado ser exquisitamente específico para C5 humana (y no presenta reacción cruzada con C5 de primates no humanos), es probable que la unión limitada observada en este estudio sea debida a la unión no específica con un material de reacción cruzada no identificado
Ejemplo 12. Potencia de BNJ441 en comparación con eculizumab en análisis de actividad del complemento terminal
Las mutaciones diseñadas en BNJ441 para proporcionar la unión dependiente del pH a C5 debilitan su afinidad a pH de 7,4 (aproximadamente 491 pM) aproximadamente 17 veces con respecto a eculizumab (aproximadamente 29,3 pM) y se podría esperar que redujeran la potencia de inhibición de BNJ441 de la actividad del complemento terminal
mediada por C5 en comparación con eculizumab. Para estimar las potencias de BNJ441 y eculizumab en condiciones fisiológicamente relevantes, se evaluó el antagonismo de la hemólisis mediada por el complemento de los glóbulos rojos (RBC) a partir de 3 modelos animales utilizados comúnmente (pollo, ovejas, y conejos) en suero humano normal al 90%.
Los RBC o los RBC de oveja fueron sensibilizados previamente con anticuerpos para iniciar la activación de la vía clásica del complemento (CCP). Los glóbulos rojos de conejo (rRBC) no fueron sensibilizados previamente y se utilizaron como modelo de activación de activación de la vía alternativa del complemento (CAP). Los anticuerpos se pre-incubaron en suero a 100, 200, y 400 nM para proporcionar razones molares de los sitios de unión del antígeno a C5 de aproximadamente 0,5:1, 1:1, y 2:1, respectivamente. El anticuerpo BNJ430 contiene la misma región Fc que BNJ441, pero no se une C5 humana, y se incluyó como control negativo. Se midió el porcentaje de hemólisis a 0, 1, 2, 3, 4, 5, 6, y 8 minutos para asegurarse de que no se observaban reacciones en las condiciones de velocidad iniciales.
Como se muestra en la Figura. 29, ni BNJ441 ni eculizumab mostraron antagonismo a 100 nM en la hemólisis de cRBC. Ambos anticuerpos mostraron un antagonismo parcial a 200 nM (razón molar de aproximadamente 1:1 de los sitios de unión de antígeno a C5), teniendo BNJ441 una potencia reducida con respecto a eculizumab. La inhibición de la hemólisis fue casi completa, para cualquier anticuerpo cuando se incubó a una razón molar 2:1 de los sitios de unión de antígeno a C5 (400 nM). Los resultados de los análisis de hemólisis de sRBC fueron similares, mostrando menos de 20% de hemólisis en presencia de BNJ441 a 200 nM, y una inhibición casi completa con cada anticuerpo a 400 nM (datos no mostrados). Los análisis de hemólisis de rRBC mediada por CAP exhiben mayores niveles de hemólisis en presencia de anticuerpos anti-C5, sin inhibición detectable a 200 nM, y solo una inhibición parcial a 400 nM (datos no mostrados).
En conclusión, la modesta pérdida de potencia de BNJ441 con respecto a eculizumab en estos análisis in vitro de actividad del complemento es compatible con su afinidad más débil para C5. La afinidad de BNJ441 para C5 es todavía aproximadamente 1000 veces menor que las concentraciones de C5 in vivo y es poco probable por lo tanto que los niveles terapéuticos selectivos de BNJ441 comprometan su eficacia terapéutica.
Ejemplo 13. Selectividad de BNJ441 en comparación con eculizumab en análisis de actividad del complemento terminal
Para evaluar la actividad farmacológica de BNJ441 en modelos animales no humanos se midió la capacidad de BNJ441 para ejercer un efecto antagónico sobre la hemólisis mediada por el complemento de cRBC sensibilizados con anticuerpo en suero de chimpancé, babuino, macaco rhesus, macacos cinomolgo, perro beagle, conejo, cobaya, rata y ratón. Se utilizaron eculizumab y un anticuerpo anti-C5 de ratón con un Fc de IgG2/G4 humano (BNJ430) como controles de isotipo.
Los cRBC sensibilizados se prepararon para cada análisis a partir de 400 pL de sangre completa de pollo en solución de Alsever (Lampire Biologicals) y se lavaron 4 veces con 1 mL de GVBS a 4°C y se resuspendieron en GVBS a 5 x 107 células/mL. Para sensibilizar los eritrocitos de pollo, se añadió un anticuerpo policlonal anti-RBC de pollo (Rockland) a las células a 150 pg/mL y se incubó durante 15 min en hielo. Después de lavar con GVBS una vez, las células se resuspendieron en GVBS a un volumen final de 3,6 mL.
Se obtuvieron sueros conservados con complemento de Bioreclamation incluyendo el suero de los siguientes mamíferos: ser humano; chimpancé; babuino; macaco rhesus; macaco cinomolgo; perro beagle; conejo; cobaya; y rata. Los anticuerpos BNJ441 a 10 mg/ml; eculizumab (10 mg/ml); BNJ430 a 0,873 mg/ml se diluyeron a una concentración final de 0, 60, 300 y 600 nM en suero al 30% en GVBS y se incubaron a la temperatura ambiente durante 30 min. Los cRBCs sensibilizados se añadieron a la mezcla de anticuerpos/suero a 30 pL por pocillo (2,5 x 106 células), se incubaron a 37°C durante 30 minutos y las reacciones se detuvieron mediante la adición de 30 pL de EDTA 0,5 M a cada pocillo. Las placas se centrifugaron a 1800 Xg durante 3 min y se transfirieron 80 pL del sobrenadante a una nueva placa de 96 pocillos de fondo plano. La absorbancia se midió a 415 nm.
Puesto que el suero de ratón es una fuente pobre de actividad del complemento de la vía clásica, se mezcló suero de ratón con suero humano con C5 agotada 1:1 para evaluar la actividad farmacológica de BNJ441 potencial en ratones. Los anticuerpos se diluyeron a una concentración final de 0, 60, 300 y 600 nM en el suero total al 50% (suero de ratón al 25%, suero humano con C5 agotada al 25%) en GVBS y se incubaron a la temperatura ambiente durante 30 min. Los cRBCs sensibilizados se añadieron a la mezcla de anticuerpos/suero a 30 pL por pocillo (2,5 x 106 células), se incubaron a 37°C durante 30 minutos y las reacciones se detuvieron mediante la adición de 30 pL de EDTA 0,5 M a cada pocillo. Las placas se centrifugaron a 1800 x g durante 3 min y se transfirieron 80 pL del sobrenadante a una nueva placa de 96 pocillos de fondo plano. La absorbancia se midió a 415 nm.
Las muestras que contenían suero sin anticuerpos anti-C5 con o sin EDTA 10 mM se utilizaron como controles sin lisis o con lisis completa, respectivamente. Las condiciones de la muestra se ejecutaron por triplicado o duplicado. Los resultados se introdujeron en una hoja de cálculo para permitir la sustracción del fondo de los controles sin lisis y
la normalización de porcentaje de hemólisis con respecto a los controles con lisis completa, el cálculo de los valores medios (± d.t.) y la representación gráfica de los datos. Los valores de absorbancia para el fondo medio desde los controles sin lisis se restaron de cada réplica y la absorbancia de la muestra se expresó como el porcentaje de lisis en los controles con lisis completa de acuerdo con la siguiente ecuación: % de hemólisis de cRBC es igual a (valor de A415 en cada réplica de muestra - valor de A415 medio en el control sin lisis) / (valor de A415 medio en el control con lisis completa - valor de A415 medio en el control sin lisis) x 100.
La media y la desviación típica del % de hemólisis de cRBC para las réplicas de la muestra se trazan como una representación gráfica (datos no mostrados).
Se demostró que BNJ441 no tenía ninguna unión detectable a C5 nativa de macaco cinomolgo y ninguna actividad farmacológica in vitro en ningún suero no humano sometido a ensayo a un exceso molar de 8 veces de sitios de unión a antígeno para C5. En conjunto, estos datos son compatibles con la conclusión de que BNJ441 no tiene ninguna actividad farmacológica relevante en ninguna especie no humana de fácil acceso adecuada para el modelado de la farmacocinética o la farmacodinámica en los seres humanos.
Ejemplo 14. Caracterización fisicoquímica de BNJ441
El anticuerpo BNJ441 es un anticuerpo recombinante, humanizado, y consiste en dos cadenas pesadas de 448 aminoácidos idénticas y dos cadenas ligeras de 214 aminoácidos idénticas. Véase la Figura. 30. Las regiones constantes de BNJ441 incluyen la región constante de la cadena ligera kappa humana y la región constante de de la cadena pesada de IgG2-IgG4 humana híbrida (también referida como "G2/G4"). La región constante de IgG2/G4 fue diseñada racionalmente para reducir la activación de la función efectora, la activación del complemento, y la inmunogenicidad del anticuerpo. El dominio CH1 de la cadena pesada, la región bisagra y los primeros 5 aminoácidos del dominio CH2 coinciden con la secuencia de aminoácidos de IgG2, residuos 6 a 36 en la región CH2 y común a la secuencia de aminoácidos tanto de IgG2 como de IgG4 humanas, mientras que el resto de la dominio CH2 y el dominio CH3 coinciden con la secuencia de aminoácidos de IgG4 humana. Las regiones variables de la cadena ligera y pesada que forman el sitio de unión a C5 humana que consisten en las regiones marco humanas se injertaron en las regiones determinantes de complementariedad murinas. Los enlaces disulfuro entre cadenas en el anticuerpo BNJ441 se representan en la Figura. 31. Los números de los residuos se muestran en la Figura 31 para todo el emparejamiento de enlaces disulfuro y sitios de glicanos ligados a N.
La Tabla 18 enumera las propiedades generales del anticuerpo BNJ441. La fórmula química teórica y el peso molecular medio teórico para el componente principal presentados a continuación suponen que el anticuerpo contiene dieciocho enlaces disulfuro, dos piroglutamaciones N-terminales en las cadenas pesadas, el recorte de dos lisinas C terminales de la cadena pesada, y la adición de dos residuos glicano GOF. El número de residuos de aminoácido en BNJ441 ha sido pronosticado por medio de análisis de aminoácidos.
Tabla 18: Propiedades generales del anticuerpo BNJ441

Se desarrolló una línea celular estable de ovario de hámster chino (CHO) que expresaba BNJ441 para la elaboración de BNJ441. Las células CHOK1SV de origen utilizados para generar esta línea celular se obtuvieron del banco de células madre CHOK1SV 269-M Lonza Biologics. Se verificó que esta fuente de células estaba libre de contaminantes bacterianos y fúngicos y todos los virus detectables distintos de las partículas retrovirales endógenas de las células que no son infecciosas. Las células CHOK1SV anfitrionas se transfectaron con plásmido el pBNJ441.1 y los clones estables se seleccionaron con MSX. El clon primario 3A5 fue seleccionado como la línea celular de producción para la elaboración de BNJ441.
Los lotes de diseño y GMP de los lotes de sustancia farmacológica a granel BNJ441 se prepararon y se caracterizaron fisicoquímicamente por medio de los ensayos enumerados en la Tabla 19. El lote de diseño se produjo en una planta piloto utilizando células CHO desarrolladas en un biorreactor de 200 L y el material purificado se utilizó en el estudio de la PK. El lote GMP fue producido utilizando células CHO desarrolladas en la planta piloto utilizando un biorreactor de 200 L. El diseño de BNJ441 y los lotes de sustancia farmacológica a granel GMP se formularon y se sometieron a ensayo a aproximadamente 10 mg/mL. Las propiedades físico-químicas para los lotes se resumen en la Tabla 20.
Tabla 19: Caracterización fisico uímica de BNJ441

(continuación)

Tabla 20: Resumen fisico uímico de BNJ441

continuación

continuación

La Tabla 20 muestra que el peso molecular intacto determinado para el lote de diseño era 147.830,80 Da y para el lote GMP 147.830,72 Da. Los valores fueron compatibles con el valor del peso molecular del componente principal calculado para BNJ441 de 147.827,62 Da en la Tabla 18, y dentro de la exactitud de masa 100 ppm de ESI-ToF-Ms calibrado externamente. No se observaron picos importantes más allá del intervalo de 147.000-149.500 Da. Este método identificó la molécula basándose en el peso molecular intacto. Se inyectaron muestras de ensayo en una columna de RP-HPLC C4 y se eluyeron con un gradiente de disolvente orgánico:acuoso. A continuación, el eluato se sometió a electropulverización en un espectrómetro de masas ToF y se desconvolucionó un espectro de la mitad superior del pico cromatográfico para proporcionar el peso molecular intacto.
La Tabla 20 muestra la secuencia N-terminal determinada para los lotes de BNJ441. Las secuencias N-terminales determinadas de la cadena pesada y de la cadena ligera fueron compatibles con la secuencia de aminoácidos para los lotes de BNJ441. Se encontró que la cadena pesada era bloqueada con PyroQ, como se esperaba, y se desbloqueaba con piroglutamato aminopeptidasa (PGAP). Se determinó la secuencia primaria de la proteína en el extremo N-terminal de la cadena polipeptídica por degradación de Edman secuencial y análisis de HPLC.
La Tabla 20 muestra el análisis de los residuos de aminoácido por molécula determinado para los lotes de BNJ441. Estos valores fueron compatibles con el número calculado de residuos por molécula de BNJ441 basado en la secuencia primaria, mostrada en la primera columna de la Tabla 20. El Análisis de Aminoácidos se adquirió por triplicado. Este método evalúa la estructura primaria de la molécula por hidrólisis ácida de la proteína en sus constituyentes aminoacídicos individuales. Este método no detecta la cisteína no el triptófano. La asparragina y el aspartato se detectaron en un solo pico y se marcaron como Asx. La glutamina y el glutamato también se detectaron en un único pico y se marcaron como Glx. De los 20 aminoácidos convencionales, catorce se detectan únicamente por este método más los grupos Asx y Glx para un total de dieciséis aminoácidos. De los representados, BNJ441 tiene un total de 1262 residuos que pueden ser detectados por estos métodos.
La Tabla 20 muestra los resultados del dicroísmo circular (DC), las características locales de UV Cercano, Las Características de UV Lejano y la desconvolución para los lotes de BNJ441. La desconvolución describe las cantidades de Hélice a, Hélice 3/10, Lámina p, Giros, Poli (Pro) II y estructuras desordenadas determinadas por medio del soporte lógico CDPro frente a un conjunto de referencia dado. Se determinaron los espectros de DC para UV Cercano (estructura terciaria) y para UV Lejano (estructura secundaria) por cada lote. Este método analizó la
estructura molecular de orden superior (2° y 3°) en la molécula por la absorción diferencial de la luz polarizada circularmente a izquierda y derecha mostrada en las bandas de absorción de moléculas ópticamente activas (quirales), tales como las proteínas. e realizó la desconvolución del espectro DC y los resultados se muestran en la Tabla 20.
La Tabla 20 muestra el peso molecular medio para cada glicano determinado. Los pesos moleculares del Oligosacárido unido a N o glicano para los lotes BNJ441 fueron compatibles con los pesos moleculares de glicano teóricos mostrados en la Tabla 21. Los espectros de peso molecular de glicano libre se determinaron por espectrometría de masas MALDI-TOF. Este método identifica los glicanos asociados con la molécula de fármaco por el peso molecular. Los glicanos fueron previamente escindidos enzimáticamente del anticuerpo con PNGasa F. Los glicanos se extrajeron a continuación en fase sólida y se mezclaron con la solución matriz de ácido 3,4-dihidroxibenzoico y se co-precipitaron sobre la diana de MALDI. Esta muestra seca se ionizó con un láser de nitrógeno en un espectrómetro de masas TOF. Se recogió un espectro m/z (M Na)+.
Tabla 21: Peso Molecular de Glicano Teórico

Los porcentajes de oligosacáridos determinados para los lotes de BNJ441 se muestran en la Tabla 20. Se calcularon los totales para diversos tipos de oligosacáridos ligados a N: (G0F Total, GIF), Ácidos, de alto contenido de Manosa, Neutro, Monosialilado y Disialilado. Los oligosacáridos unidos a N solamente contenían oligosacáridos neutros. El nivel de oligosacáridos neutros fue de 99,99 y 100,0% para los lotes de diseño y BPF, respectivamente. Los oligosacáridos se detectaron utilizando HPLC y los cromatogramas se evaluaron cuantitativamente. Este método evalúa el patrón de glicosilación mediante la identificación de los oligosacáridos unidos a N asociados con la molécula de fármaco basándose en el tiempo de retención de los oligosacáridos liberados enzimáticamente y etiquetados fluorescentemente. Este método proporciona la abundancia relativa de cada especie de oligosacárido. Brevemente, los oligosacáridos se escindieron enzimáticamente a partir del anticuerpo con PNGasa F y se etiquetaron con ácido antranílico. El ácido antranílico en exceso se eliminó utilizando una etapa de filtración HILIC. Las muestras se inyectaron en un sistema sAX- HPLC con una Columna Showa Denko Asahipak Amino y los oligosacáridos etiquetados se detectaron con un detector de fluorescencia; 360 nm de excitación y 420 nm de emisión.
Los porcentajes de monosacáridos se determinaron para los lotes de BNJ441 y se muestran en la Tabla 20. Los porcentajes de monosacáridos se determinaron para los cinco monosacáridos (GlcNAc, GalAc, Galactosa, Manosa, Fucosa) utilizando el marcaje con fluorescencia seguido de cromatografía de alta presión de fase inversa (RP-HPLC). Este análisis caracteriza el patrón de glicosilación mediante la determinación de los monosacáridos asociados con la molécula de fármaco basándose en el tiempo de retención de los monosacáridos marcados con fluorescencia. En resumen, la hidrólisis ácida eliminó los oligosacáridos de la proteína y en sus monosacáridos constituyentes. Los monosacáridos libres se marcaron a continuación con ácido antranílico (AA) por aminación reductora. Las muestras fueron inyectadas en un sistema de RP-HPLC con una columna Waters Symmetry® C-18 y los monosacáridos etiquetados con AA se detectaron con un detector de fluorescencia; 360 nm de excitación 420 nm de emisión. Las muestras se sometieron a ensayo por duplicado y el valor referido fue la media de los dos resultados.
A continuación los autores de la presente invención determinaron los ácidos siálicos ácido N-acetilneuramínico (NANA), y ácido N-glicolilneuramínico (NGNA). En cada caso, el contenido de los ácidos siálicos NANA y NGNA determinado de los lotes de BNJ441 estaba por debajo del límite de cuantificación (<6 mmoles/mol) como se muestra en la Tabla 20. No se observó NGNA, para ninguno de los lotes. Los ácidos siálicos se midieron por separado sobre RP-HPLC después del marcaje con fluorescencia y utilizando la calibración de múltiples puntos. Este método evalúa el patrón de glicosilación mediante la determinación del tipo y la cantidad relativa de los ácidos siálicos asociados con la molécula de fármaco. Los ácidos siálicos se escindieron químicamente a partir del anticuerpo por incubación con bisulfato de sodio, luego se etiquetaron con O-fenilendiamina. Las muestras se inyectaron en un sistema de RP-HPLC con una columna Beckman Ultrasphere C18 y los ácidos siálicos etiquetados se detectaron con un detector de fluorescencia (excitación 230 nm; emisión 425 nm). Las muestras se sometieron a ensayo por duplicado y se informó de la media de los dos resultados.
El valor Tm determinado de cada lote de BNJ441 fue de 67,0°C, como se muestra en la Tabla 20. Se realizaron calorimetrías diferenciales de barrido (CDB) y los datos de las calorimetría se adquirieron utilizando Micro-Cal VP-CDB mediante barrido superior a una velocidad de 75°C/hr desde 20°C a 95°C. El eje de la Y y las calibraciones de la temperatura se realizaron antes de someter a ensayo las muestras. El % de error de deflexión del eje de la Y fue
Claims (15)
1. Un anticuerpo aislado, o fragmento de unión a antígeno del mismo, que:
(a) se une al componente C5 del complemento humano;
(b) inhibe la escisión de C5 en fragmentos C5a y C5b; y
(c) comprende: (i) una CDR1 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 23, (ii) una CDR2 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 19, (iii) una CDR3 de cadena pesada que comprende la secuencia de aminoácidos representada en SEQ ID NO: 3, (iv) una CDR1 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 4, (v) una CDR2 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 5, (vi) una CDR3 de cadena ligera que comprende la secuencia de aminoácidos representada en SEQ ID NO: 6 y (vii) la región constante de cadena pesada representada en SEQ ID NO: 13.
2. El anticuerpo aislado, o fragmento de unión a antígeno del mismo, de acuerdo con la reivindicación 1, que comprende un polipéptido de cadena pesada que comprende la secuencia de aminoácidos representada en el SEQ ID NO: 14 y un polipéptido de cadena ligera que comprende la secuencia de aminoácidos representada en el SEQ ID NO: 11.
3. El anticuerpo aislado, o fragmento de unión a antígeno del mismo, de acuerdo con la reivindicación 1 o 2, en donde:
(i) el anticuerpo o fragmento de unión a antígeno del mismo se une a C5 humano a pH 7,4 y 25°C con una constante de disociación de afinidad (Kd) que está en el intervalo de 0,1 nM < Kd < 1 nM;
(ii) el anticuerpo o fragmento de unión a antígeno del mismo se une con C5 humano a pH 6,0 y 25°C con una Kd > 10 nm; y/o
(iii) la [(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 humano a pH 6,0 y a 25°C)/(Kd del anticuerpo o fragmento de unión a antígeno del mismo para C5 humano a pH 7,4 y a 25°C)] es mayor que 25; y/o (iv) el anticuerpo tiene una semivida en suero en seres humanos de al menos 40 días.
4. El anticuerpo aislado de una cualquiera de las reivindicaciones anteriores, en donde el anticuerpo se fabrica en una célula CHO.
5. El anticuerpo aislado de la reivindicación 4, en donde el anticuerpo no contiene residuos de ácido siálico detectables.
6. Un ácido nucleico que codifica:
(i) el polipéptido de cadena pesada del anticuerpo o fragmento de unión a antígeno del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 3 y el polipéptido de cadena ligera del anticuerpo o fragmento de unión a antígeno del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 5;
(ii) una región variable de cadena pesada que comprende una CDR1 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 23, una CDR2 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 19 y una CDR3 que comprende la secuencia de aminoácidos representada en s Eq ID NO: 3, y una región variable de cadena ligera que comprende una CDR1 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 4, una CDR2 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 5 y una CDR3 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 6; o
(iii) una región variable representada en SEQ ID NO: 12 y una región variable de cadena ligera representada en SEQ ID NO: 8.
7. Un vector que comprende el ácido nucleico de acuerdo con la reivindicación 6.
8. El vector de la reivindicación 7, que es un vector de expresión.
9. Una célula que comprende:
(i) el vector de acuerdo con la reivindicación 8; o
(ii) un vector de expresión que codifica el polipéptido de cadena pesada del anticuerpo o fragmento de unión a antígeno del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 3 y un vector de expresión que codifica el polipéptido de cadena ligera del anticuerpo o fragmento de unión a antígeno del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 3;
(iii) un vector de expresión que codifica una región variable de cadena pesada que comprende una CDR1 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 23, una CDR2 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 19 y una CDR3 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 3 y un vector de expresión que codifica una región variable de cadena
ligera que comprende una CDR1 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 4, una CDR2 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 5 y una CDR3 que comprende la secuencia de aminoácidos representada en SEQ ID NO: 6; o
(iv) un vector de expresión que codifica una región variable de cadena pesada representada en SEQ ID NO: 12 y un vector de expresión que codifica una región variable de cadena ligera representada en SEQ ID NO: 8.
10. Un método para producir un anticuerpo o fragmento de unión a antígeno del mismo, comprendiendo el método cultivar la célula de la reivindicación 9 en condiciones y durante un tiempo suficientes para permitir la expresión por la célula del anticuerpo o fragmento de unión a antígeno del mismo codificado por el ácido nucleico y comprendiendo además opcionalmente aislar el anticuerpo o fragmento de unión a antígeno del mismo.
11. Una composición farmacéutica que comprende un portador farmacéuticamente aceptable y el anticuerpo o fragmento de unión a antígeno del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 5.
12. Un kit terapéutico que comprende: (i) el anticuerpo o fragmento de unión a antígeno aislado de acuerdo con una cualquiera de las reivindicaciones 1 a 5 y (ii) medios para el suministro del anticuerpo o fragmento de unión a antígeno del mismo a un ser humano, en donde el medio es opcionalmente una jeringa.
13. Un artículo de fabricación que comprende:
un recipiente que comprende una etiqueta; y
una composición que comprende: (i) el anticuerpo o fragmento de unión a antígeno aislado de acuerdo con una cualquiera de las reivindicaciones 1 a 5, en donde la etiqueta indica que la composición se va a administrar a un ser humano que tiene, se sospecha que tiene, o se encuentra en riesgo de desarrollar, una afección asociada al complemento.
14. Un anticuerpo o fragmento de unión a antígeno del mismo, de acuerdo con una cualquiera de las reivindicaciones 1 a 5 o una composición farmacéutica de acuerdo con la reivindicación 11 para uso en un método de tratamiento de una afección asociada al complemento.
15. El anticuerpo, o fragmento de unión a antígeno del mismo, o composición farmacéutica para su uso de acuerdo con la reivindicación 14, en donde la afección asociada al complemento se selecciona del grupo que consiste en artritis reumatoide, síndrome de anticuerpos antifosfolípidos, nefritis por lupus, lesión por isquemia-reperfusión, síndrome urémico hemolítico atípico, síndrome urémico hemolítico típico, hemoglobinuria paroxística nocturna, enfermedad por depósitos densos, neuromielitis óptica, neuropatía motora multifocal, esclerosis múltiple, degeneración macular, síndrome de HELLP, pérdida fetal espontánea, púrpura trombocitopénica trombótica, vasculitis pauciinmune, epidermolisis bullosa, pérdida fetal recurrente, lesión cerebral traumática, miocarditis, trastorno cerebrovascular, trastorno vascular periférico, trastorno renovascular, trastorno vascular mesentérico/entérico, vasculitis, púrpura de Henoch-Schonlein, nefritis, vasculitis asociada a lupus eritematoso sistémico, vasculitis asociada a artritis reumatoide, vasculitis por complejos inmunitarios, enfermedad de Takayasu, miocardiopatía dilatada, angiopatía diabética, enfermedad de Kawasaki, embolia de gas venosa, restenosis siguiente a colocación de dispositivo intraluminal, aterectomía rotacional, angioplastia coronaria transluminal percutánea, miastenia grave, enfermedad por crioaglutininas, dermatomiositis, hemoglobinuria paroxística al frío, síndrome antifosfolípido, enfermedad de Graves, aterosclerosis, enfermedad de Alzheimer, sepsis de respuesta inflamatoria sistémica, choque séptico, lesión de la médula espinal, glomerulonefritis, rechazo de trasplantes, tiroiditis de Hashimoto, diabetes tipo I, psoriasis, pénfigo, anemia hemolítica autoinmunitaria, púrpura trombocitopénica idiopática, síndrome de Goodpasture, enfermedad de Degos y síndrome antifosfolípido catastrófico.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461949932P | 2014-03-07 | 2014-03-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2749978T3 true ES2749978T3 (es) | 2020-03-24 |
Family
ID=52684742
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16177656T Active ES2749978T3 (es) | 2014-03-07 | 2015-03-06 | Anticuerpos anti-C5 que tienen farmacocinética mejorada |
| ES15710396.1T Active ES2589230T3 (es) | 2014-03-07 | 2015-03-06 | Anticuerpos anti-C5 que tienen farmacocinética mejorada |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15710396.1T Active ES2589230T3 (es) | 2014-03-07 | 2015-03-06 | Anticuerpos anti-C5 que tienen farmacocinética mejorada |
Country Status (28)
| Country | Link |
|---|---|
| US (11) | US9079949B1 (es) |
| EP (3) | EP3594235A1 (es) |
| JP (5) | JP6112469B1 (es) |
| KR (4) | KR20230082690A (es) |
| CN (2) | CN113416254B (es) |
| AR (1) | AR099698A1 (es) |
| AU (4) | AU2015226977B2 (es) |
| BR (1) | BR112016020454B1 (es) |
| CA (2) | CA2942165C (es) |
| CY (1) | CY1118018T1 (es) |
| DK (2) | DK2970455T3 (es) |
| EA (2) | EA202090275A3 (es) |
| ES (2) | ES2749978T3 (es) |
| HR (1) | HRP20161146T1 (es) |
| HU (2) | HUE030944T2 (es) |
| IL (4) | IL296502B1 (es) |
| LT (1) | LT2970455T (es) |
| MA (2) | MA38859B1 (es) |
| ME (1) | ME02629B (es) |
| MX (2) | MX368909B (es) |
| NZ (2) | NZ631007A (es) |
| PL (2) | PL2970455T3 (es) |
| PT (2) | PT2970455T (es) |
| RS (1) | RS55386B1 (es) |
| SI (2) | SI2970455T1 (es) |
| SM (1) | SMT201600366B (es) |
| TW (4) | TW202446794A (es) |
| WO (1) | WO2015134894A1 (es) |
Families Citing this family (133)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2892925T3 (es) | 2006-03-31 | 2022-02-07 | Chugai Pharmaceutical Co Ltd | Métodos para controlar la farmacocinética en sangre de anticuerpos |
| CN101874042B9 (zh) | 2007-09-26 | 2019-01-01 | 中外制药株式会社 | 利用cdr的氨基酸取代来改变抗体等电点的方法 |
| KR102057826B1 (ko) | 2008-04-11 | 2019-12-20 | 추가이 세이야쿠 가부시키가이샤 | 복수 분자의 항원에 반복 결합하는 항원 결합 분자 |
| TWI440469B (zh) | 2008-09-26 | 2014-06-11 | Chugai Pharmaceutical Co Ltd | Improved antibody molecules |
| TWI812066B (zh) | 2010-11-30 | 2023-08-11 | 日商中外製藥股份有限公司 | 具有鈣依存性的抗原結合能力之抗體 |
| CN112812184A (zh) | 2011-02-25 | 2021-05-18 | 中外制药株式会社 | FcγRIIb特异性Fc抗体 |
| CN109517059B (zh) | 2011-06-30 | 2023-03-28 | 中外制药株式会社 | 异源二聚化多肽 |
| JP6322411B2 (ja) | 2011-09-30 | 2018-05-09 | 中外製薬株式会社 | 複数の生理活性を有する抗原の消失を促進する抗原結合分子 |
| TW201817744A (zh) | 2011-09-30 | 2018-05-16 | 日商中外製藥股份有限公司 | 具有促進抗原清除之FcRn結合域的治療性抗原結合分子 |
| JP6124800B2 (ja) | 2011-11-30 | 2017-05-10 | 中外製薬株式会社 | 免疫複合体を形成する細胞内への運搬体(キャリア)を含む医薬 |
| JP6226752B2 (ja) | 2012-02-09 | 2017-11-08 | 中外製薬株式会社 | 抗体のFc領域改変体 |
| WO2013126006A1 (en) | 2012-02-20 | 2013-08-29 | Swedish Orphan Biovitrum Ab (Publ) | Polypeptides binding to human complement c5 |
| CN103382223B (zh) * | 2012-04-01 | 2015-06-10 | 上海益杰生物技术有限公司 | 针对表皮生长因子受体隐蔽表位和t细胞抗原的多功能抗体多肽 |
| TW202237660A (zh) | 2012-08-24 | 2022-10-01 | 日商中外製藥股份有限公司 | FcγRIIb特異性Fc區域變異體 |
| US10766960B2 (en) | 2012-12-27 | 2020-09-08 | Chugai Seiyaku Kabushiki Kaisha | Heterodimerized polypeptide |
| AU2014250434B2 (en) | 2013-04-02 | 2019-08-08 | Chugai Seiyaku Kabushiki Kaisha | Fc region variant |
| NZ631007A (en) | 2014-03-07 | 2015-10-30 | Alexion Pharma Inc | Anti-c5 antibodies having improved pharmacokinetics |
| MX2017004742A (es) * | 2014-10-20 | 2017-07-20 | Nestec Sa | Métodos para la predicción de niveles de farmacos anti-tnf alfa y formación de autoanticuerpos. |
| US20160168237A1 (en) | 2014-12-12 | 2016-06-16 | Alexion Pharmaceuticals, Inc. | Method for treating a complement mediated disorder caused by an infectious agent in a patient |
| CN107207607B (zh) * | 2014-12-19 | 2021-05-04 | 中外制药株式会社 | 抗-c5抗体及使用方法 |
| KR101860280B1 (ko) | 2014-12-19 | 2018-05-21 | 추가이 세이야쿠 가부시키가이샤 | 항-마이오스타틴 항체, 변이체 Fc 영역을 함유하는 폴리펩타이드, 및 사용 방법 |
| CN107428823B (zh) * | 2015-01-22 | 2021-10-26 | 中外制药株式会社 | 两种以上抗-c5抗体的组合与使用方法 |
| CN114773470A (zh) | 2015-02-05 | 2022-07-22 | 中外制药株式会社 | 包含离子浓度依赖性的抗原结合结构域的抗体,fc区变体,il-8-结合抗体及其应用 |
| JP6130983B2 (ja) | 2015-02-27 | 2017-05-17 | 中外製薬株式会社 | Il−6関連疾患治療用組成物 |
| US20180072769A1 (en) * | 2015-03-23 | 2018-03-15 | Alexion Pharmaceuticals, Inc. | Virus filtration |
| US20190135903A1 (en) | 2015-03-31 | 2019-05-09 | Alexion Pharmaceuticals, Inc. | Identifying and treating subpopulations of paroxysmal nocturnal hemoglobinuria (pnh) patients |
| US20180311299A1 (en) | 2015-05-01 | 2018-11-01 | Alexion Pharmaceuticals, Inc. | Efficacy of an anti-c5 antibody in the prevention of antibody mediated rejection in sensitized recipients of a kidney transplant |
| US11207465B2 (en) | 2015-06-04 | 2021-12-28 | West Pharma. Services Il. Ltd. | Cartridge insertion for drug delivery device |
| WO2017044811A1 (en) | 2015-09-11 | 2017-03-16 | Bruce Andrien | Recombinant glycosylated eculizumab and eculizumab variants |
| CA2997745A1 (en) | 2015-09-14 | 2017-03-23 | Children's Hospital Medical Center | Methods and compositions for treatment of gaucher disease via modulation of c5a receptor |
| JP2018530574A (ja) | 2015-10-07 | 2018-10-18 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 患者の加齢黄斑変性を治療する方法 |
| EP3359555B1 (en) | 2015-10-07 | 2023-12-20 | Apellis Pharmaceuticals, Inc. | Dosing regimens |
| US20180311345A1 (en) * | 2015-10-30 | 2018-11-01 | Alexion Pharmaceuticals, Inc. | A method of inhibiting exacerbations of t cell-mediated allograft vasculopathy |
| IL259256B2 (en) * | 2015-12-18 | 2023-02-01 | Chugai Pharmaceutical Co Ltd | Anti-c5 antibodies and methods of use |
| US20200270363A1 (en) | 2015-12-25 | 2020-08-27 | Chugai Seiyaku Kabushiki Kaisha | Antibody having enhanced activity, and method for modifying same |
| US11359009B2 (en) | 2015-12-25 | 2022-06-14 | Chugai Seiyaku Kabushiki Kaisha | Anti-myostatin antibodies and methods of use |
| EP3402816A1 (en) * | 2016-01-11 | 2018-11-21 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treatment |
| EP3408291A1 (en) * | 2016-01-25 | 2018-12-05 | Shire Human Genetic Therapies, Inc. | Anti-c5 antibodies with enhanced ph switch |
| PL234632B1 (pl) * | 2016-03-02 | 2020-03-31 | Inst Immunologii I Terapii Doswiadczalnej Polskiej Akademii Nauk | Przeciwciała oddziałujące z komórkami psich chłoniaków typu B i ich zastosowania |
| MA44780A (fr) | 2016-04-28 | 2019-03-06 | Chugai Pharmaceutical Co Ltd | Préparation contenant un anticorps |
| JP2019517473A (ja) * | 2016-05-27 | 2019-06-24 | アレクシオン ファーマシューティカルズ インコーポレイテッドAlexion Pharmaceuticals, Inc. | 難治性全身型重症筋無力症の処置のための方法 |
| CN109310760A (zh) | 2016-06-07 | 2019-02-05 | 诺华股份有限公司 | 抗c5抗体给药方案 |
| ES2984352T3 (es) | 2016-06-14 | 2024-10-29 | Regeneron Pharma | Anticuerpos anti-C5 y usos de los mismos |
| NZ789212A (en) * | 2016-06-17 | 2026-02-27 | Chugai Pharmaceutical Co Ltd | Anti-c5 antibodies and methods of use |
| KR102591955B1 (ko) | 2016-07-29 | 2023-10-19 | 추가이 세이야쿠 가부시키가이샤 | 증강된 fviii 보인자 기능 대체 활성을 갖는 이중특이성 항체 |
| TWI693940B (zh) | 2016-08-05 | 2020-05-21 | 日商中外製藥股份有限公司 | Il-8相關疾病之治療用或預防用組成物 |
| EP3526250A1 (en) | 2016-10-12 | 2019-08-21 | Alexion Pharmaceuticals, Inc. | Efficacy of an anti-c5 antibody in the prevention of antibody mediated rejection in sensitized recipients of a kidney transplant |
| JP7096240B2 (ja) | 2016-10-19 | 2022-07-05 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 試料中の非結合c5の定量化方法 |
| US20200057046A1 (en) | 2016-10-27 | 2020-02-20 | Alexion Pharmaceuticals, Inc. | Assay for c5b-9 deposition in complement-associated disorders |
| EP3554543A4 (en) * | 2016-12-16 | 2020-09-02 | Samsung Bioepis Co., Ltd. | STABLE Aqueous ANTI-C5 ANTIBODY COMPOSITION |
| JP7191833B2 (ja) | 2017-01-30 | 2022-12-19 | 中外製薬株式会社 | 抗スクレロスチン抗体およびその使用 |
| IL307593A (en) * | 2017-01-31 | 2023-12-01 | Chugai Pharmaceutical Co Ltd | A pharmaceutical composition for use in the treatment or prevention of a c5-related disease and a method for treating or preventing a c5-related disease |
| US11578137B2 (en) * | 2017-03-06 | 2023-02-14 | The Trustees Of The University Of Pennsylvania | Anti-C5 antibodies and uses thereof |
| CN110461358A (zh) | 2017-03-31 | 2019-11-15 | 公立大学法人奈良县立医科大学 | 可用于预防和/或治疗凝血因子ⅸ异常、包含代替凝血因子ⅷ的功能的多特异性抗原结合分子的药物组合物 |
| ES2999550T3 (en) * | 2017-03-31 | 2025-02-26 | Alexion Pharma Inc | Method for simultaneous quantification of alxn1210 and eculizumab in human serum or urine |
| JP2020516607A (ja) | 2017-04-07 | 2020-06-11 | アペリス・ファーマシューティカルズ・インコーポレイテッドApellis Pharmaceuticals,Inc. | 投与レジメンならびに関連組成物および方法 |
| US20200123238A1 (en) | 2017-04-19 | 2020-04-23 | Alexion Pharmaceuticals, Inc. | Efficacy of an anti-c5 antibody in the prevention of antibody mediated rejection in sensitized recipients of a kidney transplant |
| US11643473B2 (en) | 2017-04-24 | 2023-05-09 | Alexion Pharmaceuticals, Inc. | Antibody immune cell inhibitor fusion proteins |
| EP3620531A4 (en) | 2017-05-02 | 2021-03-17 | National Center of Neurology and Psychiatry | METHOD OF PREDICTION AND EVALUATION OF THERAPEUTIC EFFECT IN DISEASES RELATING TO IL-6 AND NEUTROPHILS |
| NZ758605A (en) * | 2017-05-30 | 2022-07-29 | Chong Kun Dang Pharmaceutical Corp | A novel anti-c-met antibody and use thereof |
| BR112019028269A2 (pt) * | 2017-07-11 | 2020-07-14 | Alexion Pharmaceuticals, Inc. | polipeptídeos que se ligam ao componente c5 do complemento ou à albumina sérica e suas proteínas de fusão |
| SI3658184T1 (sl) * | 2017-07-27 | 2024-01-31 | Alexion Pharmaceuticals, Inc., | Formulacije z visoko koncentracijo protiteles proti-C5 |
| AR113142A1 (es) | 2017-09-29 | 2020-01-29 | Chugai Pharmaceutical Co Ltd | Moléculas de unión al antígeno multiespecíficas que tienen actividad de sustitución de la función de cofactor del factor viii de coagulación de sangre (fviii), y formulaciones farmacéuticas que contienen dicha molécula como ingrediente activo |
| WO2019070714A1 (en) * | 2017-10-04 | 2019-04-11 | Alexion Pharmaceuticals, Inc. | ASSAY AND ADMINISTRATION OF ANTI-C5 ANTIBODIES FOR THE TREATMENT OF PATIENTS WITH MEMBRANOPROLIFERATIVE GLOMERULONEPHRIPT |
| CA3080187A1 (en) | 2017-10-26 | 2019-05-02 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treatment of paroxysmal nocturnal hemoglobinuria (pnh) and atypical hemolytic uremic syndrome (ahus) |
| AU2018361430B2 (en) | 2017-11-01 | 2025-08-14 | Chugai Seiyaku Kabushiki Kaisha | Antibody variant and isoform with lowered biological activity |
| EP3710589A4 (en) | 2017-11-14 | 2021-11-10 | Chugai Seiyaku Kabushiki Kaisha | ANTI-C1S ANTIBODIES AND METHOD OF USING |
| BR112020010916A2 (pt) * | 2017-12-04 | 2020-11-17 | Ra Pharmaceuticals, Inc | moduladores da atividade do complemento |
| MA51147A (fr) | 2017-12-13 | 2021-03-24 | Regeneron Pharma | Associations d'anticorps anti-c5 et utilisations associées |
| AU2018386304B2 (en) | 2017-12-15 | 2023-12-07 | Apellis Pharmaceuticals, Inc. | Dosing regimens and related compositions and methods |
| JP7402799B2 (ja) | 2017-12-22 | 2023-12-21 | ウェスト ファーマ サービシーズ イスラエル リミテッド | サイズの異なるカートリッジを利用可能な注射器 |
| WO2019143761A1 (en) * | 2018-01-17 | 2019-07-25 | Children's Hospital Medical Center | Compositions and methods for treatment of hunter's syndrome |
| US20190247560A1 (en) | 2018-02-13 | 2019-08-15 | Gambro Lundia Ab | Extracorporeal devices and methods of treating complement factor related diseases |
| US12404320B2 (en) * | 2018-05-31 | 2025-09-02 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-C5 antibodies for treatment of paroxysmal nocturnal hemoglobinuria (PNH) in pediatric patients |
| JP7577542B2 (ja) | 2018-06-04 | 2024-11-05 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 小児患者における非典型溶血性尿毒症症候群(aHUS)の治療のための抗C5抗体の用量および投与 |
| CN112654394B (zh) * | 2018-06-19 | 2025-07-11 | 阿塔盖有限责任公司 | 针对补体成分5的抗体分子和其用途 |
| WO2020006266A1 (en) | 2018-06-28 | 2020-01-02 | Alexion Pharmaceuticals, Inc. | Methods of producing anti-c5 antibodies |
| EP3835321A4 (en) | 2018-08-10 | 2022-11-02 | Chugai Seiyaku Kabushiki Kaisha | ANTI-CD137 ANTIGEN-BINDING MOLECULE AND USE THEREOF |
| WO2020041301A1 (en) | 2018-08-20 | 2020-02-27 | Achillion Pharmaceuticals, Inc. | Pharmaceutical compounds for the treatment of complement factor d medical disorders |
| CA3111396A1 (en) * | 2018-09-06 | 2020-03-12 | The Trustees Of The University Of Pennsylvania | Humanized anti-c5 antibodies and uses thereof |
| WO2020051532A2 (en) | 2018-09-06 | 2020-03-12 | Achillion Pharmaceuticals, Inc. | Macrocyclic compounds for the treatment of medical disorders |
| EP3852874A1 (en) | 2018-09-17 | 2021-07-28 | Kyoto University | Administration of an anti-c5 agent for treatment of hepatic injury or failure |
| US20200095307A1 (en) | 2018-09-21 | 2020-03-26 | Alexion Pharmaceuticals, Inc. | Compositions and methods for the treatment of neuromyelitis optica |
| US20200115443A1 (en) * | 2018-10-12 | 2020-04-16 | Trican Biotechnology Co., Ltd | Bi-functional fusion proteins and uses thereof |
| US20210388070A1 (en) * | 2018-10-30 | 2021-12-16 | Alexion Pharmaceuticals, Inc. | Co-administration of a hyaluronidase and anti-c5 antibody for treatment of complement-associated conditions |
| CN112996561A (zh) * | 2018-10-30 | 2021-06-18 | 亚力兄制药公司 | 用于治疗阵发性睡眠性血红蛋白尿症(pnh)的抗c5抗体的皮下剂量和施用 |
| US20220002393A1 (en) | 2018-11-20 | 2022-01-06 | Alexion Pharmaceuticals, Inc. | Methods for treatment of refractory generalized myasthenia gravis in pediatric patients |
| CA3127797A1 (en) | 2019-01-25 | 2020-07-30 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treatment of atypical hemolytic uremic syndrome (ahus) |
| US11642470B2 (en) * | 2019-02-11 | 2023-05-09 | West Pharma. Services IL, Ltd. | Anti-C5 antibody dispensing injector and method of injection |
| MX2021009789A (es) * | 2019-02-14 | 2021-11-17 | Alexion Pharma Inc | Dosificacion y administracion de anticuerpos anti-c5 para el tratamiento de miastenia gravis generalizada. |
| JP7654564B2 (ja) * | 2019-04-24 | 2025-04-01 | ザ トラスティーズ オブ ザ ユニバーシティ オブ ペンシルバニア | 二機能性ヒト化抗c5抗体ならびにそのh因子融合タンパク質およびその使用 |
| MX2021013441A (es) | 2019-05-15 | 2021-12-10 | Chugai Pharmaceutical Co Ltd | Molecula de union a antigenos, composicion farmaceutica y metodo. |
| WO2020242849A1 (en) | 2019-05-24 | 2020-12-03 | Alexion Pharmaceuticals, Inc. | Methods of treating vitiligo using an anti-c5 antibody |
| CN113966224A (zh) * | 2019-06-04 | 2022-01-21 | Ra制药公司 | 采用补体抑制剂的炎性疾病治疗 |
| US20220259305A1 (en) | 2019-08-05 | 2022-08-18 | Alexion Pharmaceuticals, Inc. | Methods for treatment of refractory generalized myasthenia gravis with eculizumab |
| WO2021067526A1 (en) | 2019-10-02 | 2021-04-08 | Alexion Pharmaceuticals, Inc. | Complement inhibitors for treating drug-induced complement-mediated response |
| EP4054634A1 (en) * | 2019-11-08 | 2022-09-14 | Alexion Pharmaceuticals, Inc. | High concentration anti-c5 antibody formulations |
| KR20220110827A (ko) * | 2019-12-09 | 2022-08-09 | 알렉시온 파마슈티칼스, 인코포레이티드 | 시신경척수염 스펙트럼 장애의 치료를 위한 항-c5 항체 |
| JP7734673B2 (ja) | 2019-12-23 | 2025-09-05 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 抗c5抗体を使用して妊娠関連非典型溶血性尿毒症症候群を治療する方法 |
| WO2021168320A1 (en) | 2020-02-20 | 2021-08-26 | Achillion Pharmaceuticals, Inc. | Heteroaryl compounds for treatment of complement factor d mediated disorders |
| JP2023522208A (ja) | 2020-04-16 | 2023-05-29 | アシスタンス ピュブリック-オピト ド パリ | ウイルスによって引き起こされる補体媒介性障害を処置する方法 |
| MX2022013606A (es) | 2020-04-30 | 2023-01-16 | Alnylam Pharmaceuticals Inc | Composiciones de ácido ribonucleico interferente (arni) del factor b de complemento (cfb) y métodos de uso de las mismas. |
| BR112022022980A2 (pt) | 2020-05-12 | 2022-12-20 | Alexion Pharma Inc | Uso de inibidores de fator d de complemento sozinhos ou em combinação com anticorpos anti-c5 para tratamento de hemoglobinúria paroxística noturna |
| WO2021231720A1 (en) | 2020-05-15 | 2021-11-18 | Alexion Pharmaceuticals, Inc. | Method of using extracellular vesicles to detect complement activation, and uses thereof to assess and/or monitor treatment of a complement-mediated disease |
| CN113754763B (zh) * | 2020-06-05 | 2024-03-08 | 天辰生物医药(苏州)有限公司 | 分离的抗原结合蛋白及其用途 |
| US20230303670A1 (en) * | 2020-06-24 | 2023-09-28 | Alexion Pharmaceuticals, Inc. | Subcutaneous (sc) administration of anti-c5 antibodies for treatment of complement-associated conditions |
| WO2021263056A1 (en) * | 2020-06-25 | 2021-12-30 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treatment of amyotrophic lateral sclerosis |
| JP2023533030A (ja) | 2020-07-09 | 2023-08-01 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 小児患者において発作性夜間血色素尿症(pnh)を治療するための抗c5抗体の投薬量及び投与 |
| IL300115A (en) * | 2020-08-13 | 2023-03-01 | Alexion Pharma Inc | Dosage and Administration of ANTI-C5 Antibodies for Hematopoietic Stem Cell Transplantation Treatment (HSCT-TMA) |
| CN116261569A (zh) | 2020-09-21 | 2023-06-13 | 阿雷克森制药公司 | 用于治疗包括狼疮性肾炎(LN)和/或IgA肾病(IgAN)的C5介导的肾小球肾炎(GN)的抗C5抗体的剂量和施用 |
| WO2022066774A1 (en) | 2020-09-23 | 2022-03-31 | Achillion Pharmaceuticals, Inc. | Pharmaceutical compounds for the treatment of complement mediated disorders |
| CA3196434A1 (en) | 2020-10-23 | 2022-04-28 | Krista K. JOHNSON | Methods of treating patients having complement disorders using anti-c5 antibodies |
| JP2023552456A (ja) * | 2020-12-07 | 2023-12-15 | インベテックス インコーポレイテッド | 治療薬の家畜動物における半減期を増加させるための組成物及びその使用方法 |
| CN114149501B (zh) * | 2020-12-11 | 2023-05-26 | 天士力生物医药股份有限公司 | 抗c5抗体及其应用 |
| CN117042798A (zh) * | 2020-12-25 | 2023-11-10 | 宾夕法尼亚大学理事会 | 人源化抗-c5抗体和因子h融合蛋白及其用途 |
| KR20230134533A (ko) | 2021-01-22 | 2023-09-21 | 알렉시온 파마슈티칼스, 인코포레이티드 | 항-c5 항체를 이용한 보체 매개 혈전성 미세혈관병증의치료 방법 |
| CA3220629A1 (en) | 2021-05-28 | 2022-12-01 | Tobin J. CAMMETT | Methods for detecting cm-tma biomarkers |
| WO2022265915A1 (en) | 2021-06-14 | 2022-12-22 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treating dermatomyositis (dm) |
| JP2024526742A (ja) | 2021-07-14 | 2024-07-19 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 重症筋無力症の治療のための抗c5抗体の投与量及び投与 |
| WO2023023220A1 (en) | 2021-08-20 | 2023-02-23 | Alexion Pharmaceuticals, Inc. | Methods for treating sickle cell disease or beta thalassemia using a complement alternative pathway inhibitor |
| EP4402174A1 (en) | 2021-09-17 | 2024-07-24 | Novartis AG | Methods for prevention of graft rejection in xenotransplantation |
| TW202333749A (zh) | 2021-10-29 | 2023-09-01 | 美商艾拉倫製藥股份有限公司 | 補體因子b(cfb)irna組成物及其使用方法 |
| CA3237246A1 (en) * | 2021-11-01 | 2023-05-04 | Ipc Research, Llc | Administration of c5-binding proteins |
| CN120035450A (zh) | 2022-09-06 | 2025-05-23 | 阿雷克森制药公司 | 造血干细胞移植物相关血栓性微血管病(hsct-tma)患者的诊断和预后生物标志物谱 |
| TW202426048A (zh) | 2022-09-06 | 2024-07-01 | 美商阿雷希昂製藥公司 | 用於治療造血幹細胞移植相關血栓性(hsct-tma)的抗c5抗體的補充劑量和投與 |
| TW202423479A (zh) | 2022-09-28 | 2024-06-16 | 美商阿雷希昂製藥公司 | 用於預防或最小化患有慢性腎病的患者中的心臟手術相關急性腎損傷(csa-aki)和/或隨後的主要不良腎事件(make)的抗c5抗體之劑量及投與 |
| WO2024238421A1 (en) | 2023-05-12 | 2024-11-21 | Alexion Pharmaceuticals, Inc. | Use of biomarkers for the identification and treatment of complement-mediated disorders |
| WO2024238422A1 (en) | 2023-05-12 | 2024-11-21 | Alexion Pharmaceuticals, Inc. | Use of a panel of lectins for detection of complement biomarkers in urine extracellular vesicles |
| CN116712390B (zh) * | 2023-08-04 | 2023-11-14 | 上海览屹医药科技有限公司 | 一种高浓度高稳定性的抗体制剂及其制备方法 |
| WO2025045250A1 (en) | 2023-09-03 | 2025-03-06 | Kira Pharmaceuticals (Us) Llc | Anti-human factor d antibody constructs and uses thereof |
| AU2025214388A1 (en) | 2024-02-02 | 2026-03-19 | Alexion Pharmaceuticals, Inc. | Dosage and administration of anti-c5 antibodies for treating iga nephropathy (igan) |
| WO2025199107A1 (en) | 2024-03-19 | 2025-09-25 | Alexion Pharmaceuticals, Inc. | Risk evaluation and management strategy involving patient follow-ups relating to the use or discontinuation of a complement inhibitor |
| WO2025221479A1 (en) | 2024-04-16 | 2025-10-23 | Alexion Pharmaceuticals, Inc. | Biomarkers for monitoring effective treatment of neuromyelitis optica spectrum disorder (nmosd) with complement component c5 inhibitors |
Family Cites Families (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3710795A (en) | 1970-09-29 | 1973-01-16 | Alza Corp | Drug-delivery device with stretched, rate-controlling membrane |
| US4485045A (en) | 1981-07-06 | 1984-11-27 | Research Corporation | Synthetic phosphatidyl cholines useful in forming liposomes |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4544545A (en) | 1983-06-20 | 1985-10-01 | Trustees University Of Massachusetts | Liposomes containing modified cholesterol for organ targeting |
| WO1987000201A1 (en) | 1985-07-05 | 1987-01-15 | Whitehead Institute For Biomedical Research | Epithelial cells expressing foreign genetic material |
| US4980286A (en) | 1985-07-05 | 1990-12-25 | Whitehead Institute For Biomedical Research | In vivo introduction and expression of foreign genetic material in epithelial cells |
| EP0245993B1 (en) * | 1986-04-28 | 1993-05-26 | Cetus Oncology Corporation | Monoclonal antibodies against c5a and des-arg74-c5a, their production and use |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US4863457A (en) | 1986-11-24 | 1989-09-05 | Lee David A | Drug delivery device |
| WO1988007089A1 (en) | 1987-03-18 | 1988-09-22 | Medical Research Council | Altered antibodies |
| US5258498A (en) | 1987-05-21 | 1993-11-02 | Creative Biomolecules, Inc. | Polypeptide linkers for production of biosynthetic proteins |
| EP0378576B1 (en) | 1987-09-11 | 1995-01-18 | Whitehead Institute For Biomedical Research | Transduced fibroblasts and uses therefor |
| WO1989005345A1 (en) | 1987-12-11 | 1989-06-15 | Whitehead Institute For Biomedical Research | Genetic modification of endothelial cells |
| JP2917998B2 (ja) | 1988-02-05 | 1999-07-12 | ホワイトヘッド・インスティチュート・フォー・バイオメディカル・リサーチ | 修飾された肝細胞およびその用途 |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5164188A (en) | 1989-11-22 | 1992-11-17 | Visionex, Inc. | Biodegradable ocular implants |
| JP3249516B2 (ja) | 1990-10-31 | 2002-01-21 | ソマティクス セラピー コーポレイション | 遺伝子治療のためのレトロウイルスのベクター |
| KR0185215B1 (ko) | 1990-11-30 | 1999-05-01 | 요시다 쇼오지 | 서방성 안구삽입용 약제 |
| GB9026191D0 (en) | 1990-12-01 | 1991-01-16 | Harris Pharma Ltd | Breath actuated dispensing device |
| CA2110799A1 (en) | 1991-06-14 | 1992-12-23 | Arnold H. Horwitz | Microbially-produced antibody fragments and their conjugates |
| US5714350A (en) | 1992-03-09 | 1998-02-03 | Protein Design Labs, Inc. | Increasing antibody affinity by altering glycosylation in the immunoglobulin variable region |
| BR9306765A (pt) | 1992-07-23 | 1998-12-08 | Minnesota Mining & Mfg | Processo para a preparação de uma partícula abrasiva e artigo abrasivo |
| AU701578B2 (en) | 1992-08-21 | 1999-02-04 | Vrije Universiteit Brussel | Immunoglobulins devoid of light chains |
| US6005079A (en) | 1992-08-21 | 1999-12-21 | Vrije Universiteit Brussels | Immunoglobulins devoid of light chains |
| WO1994025591A1 (en) | 1993-04-29 | 1994-11-10 | Unilever N.V. | PRODUCTION OF ANTIBODIES OR (FUNCTIONALIZED) FRAGMENTS THEREOF DERIVED FROM HEAVY CHAIN IMMUNOGLOBULINS OF $i(CAMELIDAE) |
| US5885573A (en) | 1993-06-01 | 1999-03-23 | Arch Development Corporation | Methods and materials for modulation of the immunosuppressive activity and toxicity of monoclonal antibodies |
| AU691811B2 (en) | 1993-06-16 | 1998-05-28 | Celltech Therapeutics Limited | Antibodies |
| IT1265573B1 (it) | 1993-09-17 | 1996-11-22 | Zanussi Grandi Impianti Spa | Forno di cottura di alimenti con porta perfezionata |
| US5308341A (en) | 1993-09-28 | 1994-05-03 | Becton, Dickinson And Company | Method of testing the dose accuracy of a medication delivery device |
| US5443505A (en) | 1993-11-15 | 1995-08-22 | Oculex Pharmaceuticals, Inc. | Biocompatible ocular implants |
| NZ281112A (en) | 1994-03-07 | 1998-04-27 | Inhale Therapeutic Syst | Powdered insulin delivered as an aerosol |
| US6074642A (en) | 1994-05-02 | 2000-06-13 | Alexion Pharmaceuticals, Inc. | Use of antibodies specific to human complement component C5 for the treatment of glomerulonephritis |
| US6019968A (en) | 1995-04-14 | 2000-02-01 | Inhale Therapeutic Systems, Inc. | Dispersible antibody compositions and methods for their preparation and use |
| AU725609C (en) | 1995-08-18 | 2002-01-03 | Morphosys Ag | Protein/(poly)peptide libraries |
| US5773019A (en) | 1995-09-27 | 1998-06-30 | The University Of Kentucky Research Foundation | Implantable controlled release device to deliver drugs directly to an internal portion of the body |
| JP4046354B2 (ja) | 1996-03-18 | 2008-02-13 | ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム | 増大した半減期を有する免疫グロブリン様ドメイン |
| US6001329A (en) | 1996-05-06 | 1999-12-14 | Uab Research Foundation | Radiolabeled fusion toxins for cancer therapy |
| US6146361A (en) | 1996-09-26 | 2000-11-14 | Becton Dickinson And Company | Medication delivery pen having a 31 gauge needle |
| WO1998023289A1 (en) | 1996-11-27 | 1998-06-04 | The General Hospital Corporation | MODULATION OF IgG BINDING TO FcRn |
| GB9626960D0 (en) | 1996-12-27 | 1997-02-12 | Glaxo Group Ltd | Valve for aerosol container |
| US6277375B1 (en) | 1997-03-03 | 2001-08-21 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| WO1998047531A2 (en) | 1997-04-21 | 1998-10-29 | Arch Development Corporation | Fc receptor non-binding anti-cd3 monoclonal antibodies deliver a partial tcr signal and induce clonal anergy |
| US5954047A (en) | 1997-10-17 | 1999-09-21 | Systemic Pulmonary Development, Ltd. | Methods and apparatus for delivering aerosolized medication |
| ES2292236T3 (es) | 1998-04-02 | 2008-03-01 | Genentech, Inc. | Variantes de anticuerpos y sus fragmentos. |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| US6302855B1 (en) | 1998-05-20 | 2001-10-16 | Novo Nordisk A/S | Medical apparatus for use by a patient for medical self treatment of diabetes |
| HU230769B1 (hu) | 1999-01-15 | 2018-03-28 | Genentech Inc. | Módosított effektor-funkciójú polipeptid-változatok |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| EP1169053A1 (en) | 1999-04-13 | 2002-01-09 | Inhale Therapeutic Systems, Inc. | Pulmonary administration of dry powder formulations for treating infertility |
| WO2000061178A1 (en) | 1999-04-13 | 2000-10-19 | Inhale Therapeutics Systems, Inc. | Pulmonary administration of dry powder formulations for treating infertility |
| US6192891B1 (en) | 1999-04-26 | 2001-02-27 | Becton Dickinson And Company | Integrated system including medication delivery pen, blood monitoring device, and lancer |
| GB9910975D0 (en) | 1999-05-13 | 1999-07-14 | Univ Strathclyde | Rapid dehydration of proteins |
| US6277099B1 (en) | 1999-08-06 | 2001-08-21 | Becton, Dickinson And Company | Medication delivery pen |
| PE20011227A1 (es) | 2000-04-17 | 2002-01-07 | Chiesi Farma Spa | Formulaciones farmaceuticas para inhaladores de polvo seco en la forma de aglomerados duros |
| ATE547080T1 (de) | 2000-08-30 | 2012-03-15 | Univ Johns Hopkins | Vorrichtungen zur intraokularen arzneimittelabgabe |
| PT1355919E (pt) | 2000-12-12 | 2011-03-02 | Medimmune Llc | Moléculas com semivida longa, composições que as contêm e suas utilizações |
| BRPI0211953B8 (pt) * | 2001-08-17 | 2021-05-25 | Genentech Inc | anticorpo monoclonal ou fragmento de ligação a antígeno que se liga a c5 e c5a sem a prevenção da formação de c5b, linhagem celular, composição farmacêutica, bem como método in vitro para inibir a atividade de c5a e método de diagnóstico in vitro |
| AU2002333554C1 (en) | 2001-09-12 | 2008-12-11 | Becton, Dickinson And Company | Microneedle-based pen device for drug delivery and method for using same |
| AU2003217912A1 (en) | 2002-03-01 | 2003-09-16 | Xencor | Antibody optimization |
| AU2003256266A1 (en) | 2002-06-12 | 2003-12-31 | Genencor International, Inc. | Methods and compositions for milieu-dependent binding of a targeted agent to a target |
| US20050271660A1 (en) | 2002-09-06 | 2005-12-08 | Alexion Pharmaceuticals, Inc. | Nebulization of monoclonal antibodies for treating pulmonary diseases |
| US9415102B2 (en) | 2002-09-06 | 2016-08-16 | Alexion Pharmaceuticals, Inc. | High concentration formulations of anti-C5 antibodies |
| US20040102469A1 (en) | 2002-09-13 | 2004-05-27 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Method for reducing the mortality rate |
| GB0222023D0 (en) | 2002-09-21 | 2002-10-30 | Aventis Pharma Ltd | Inhaler |
| WO2004029207A2 (en) | 2002-09-27 | 2004-04-08 | Xencor Inc. | Optimized fc variants and methods for their generation |
| FR2849436B1 (fr) | 2002-12-27 | 2007-01-05 | Patrick Frayssinet | Particules et ceramiques de phosphates de calcium pour transfection in vivo et in vitro |
| WO2004073551A2 (en) | 2003-02-18 | 2004-09-02 | Massachusetts Eye And Ear Infirmary | Transscleral drug delivery device and related methods |
| AU2004261229A1 (en) | 2003-07-29 | 2005-02-10 | Eisai, Inc. | Antibodies and methods for generating genetically altered antibodies with enhanced effector function |
| GB0324368D0 (en) | 2003-10-17 | 2003-11-19 | Univ Cambridge Tech | Polypeptides including modified constant regions |
| US20050249723A1 (en) | 2003-12-22 | 2005-11-10 | Xencor, Inc. | Fc polypeptides with novel Fc ligand binding sites |
| EP1737890A2 (en) | 2004-03-24 | 2007-01-03 | Xencor, Inc. | Immunoglobulin variants outside the fc region |
| US8685435B2 (en) | 2004-04-30 | 2014-04-01 | Allergan, Inc. | Extended release biodegradable ocular implants |
| US7919094B2 (en) | 2004-06-10 | 2011-04-05 | Omeros Corporation | Methods for treating conditions associated with MASP-2 dependent complement activation |
| GB0416328D0 (en) | 2004-07-21 | 2004-08-25 | Univ Cardiff | Use of dry powder compositions for pulmonary delivery |
| US20060074225A1 (en) | 2004-09-14 | 2006-04-06 | Xencor, Inc. | Monomeric immunoglobulin Fc domains |
| US8802820B2 (en) | 2004-11-12 | 2014-08-12 | Xencor, Inc. | Fc variants with altered binding to FcRn |
| US8367805B2 (en) | 2004-11-12 | 2013-02-05 | Xencor, Inc. | Fc variants with altered binding to FcRn |
| DK1817340T3 (da) | 2004-11-12 | 2012-08-13 | Xencor Inc | Fc-varianter med ændret binding til fcrn |
| WO2006094234A1 (en) | 2005-03-03 | 2006-09-08 | Xencor, Inc. | Methods for the design of libraries of protein variants |
| CA2602663A1 (en) | 2005-03-31 | 2006-10-05 | Xencor, Inc. | Fc variants with optimized properties |
| WO2007041635A2 (en) | 2005-10-03 | 2007-04-12 | Xencor, Inc. | Fc variants with optimized fc receptor binding properties |
| BRPI0620316A2 (pt) | 2005-12-21 | 2011-11-08 | Wyeth Corp | formulações de proteìnas com viscosidades reduzida e seus usos |
| RS53864B1 (sr) | 2006-03-02 | 2015-08-31 | Alexion Pharmaceuticals, Inc. | Produžavanje preživljavanja alografta inhibiranjem aktivnosti komplementa |
| PL3167888T3 (pl) * | 2006-03-15 | 2024-08-26 | Alexion Pharmaceuticals, Inc. | Leczenie pacjentów z napadową nocną hemoglobinurią za pomocą inhibitora dopełniacza |
| ES2892925T3 (es) | 2006-03-31 | 2022-02-07 | Chugai Pharmaceutical Co Ltd | Métodos para controlar la farmacocinética en sangre de anticuerpos |
| US8037880B2 (en) | 2006-04-07 | 2011-10-18 | The University Of Western Ontario | Dry powder inhaler |
| US20100034194A1 (en) | 2006-10-11 | 2010-02-11 | Siemens Communications Inc. | Eliminating unreachable subscribers in voice-over-ip networks |
| AU2007313300A1 (en) | 2006-10-16 | 2008-04-24 | Medimmune, Llc. | Molecules with reduced half-lives, compositions and uses thereof |
| WO2008092117A2 (en) | 2007-01-25 | 2008-07-31 | Xencor, Inc. | Immunoglobulins with modifications in the fcr binding region |
| US20090110679A1 (en) | 2007-07-13 | 2009-04-30 | Luk-Chiu Li | Methods and compositions for pulmonary administration of a TNFa inhibitor |
| CN101874042B9 (zh) | 2007-09-26 | 2019-01-01 | 中外制药株式会社 | 利用cdr的氨基酸取代来改变抗体等电点的方法 |
| RU2529951C2 (ru) | 2007-12-26 | 2014-10-10 | Ксенкор, Инк. | ВАРИАНТЫ Fc С ИЗМЕНЕННЫМ СВЯЗЫВАНИЕМ C FcRn |
| KR102057826B1 (ko) | 2008-04-11 | 2019-12-20 | 추가이 세이야쿠 가부시키가이샤 | 복수 분자의 항원에 반복 결합하는 항원 결합 분자 |
| EP3524620A1 (en) | 2008-10-14 | 2019-08-14 | Genentech, Inc. | Immunoglobulin variants and uses thereof |
| LT2894165T (lt) * | 2008-11-10 | 2023-03-10 | Alexion Pharmaceuticals, Inc. | Būdai ir kompozicijos, skirti su komplementu susijusių sutrikimų gydymui |
| EP2373689A1 (en) * | 2008-12-12 | 2011-10-12 | MedImmune, LLC | Crystals and structure of a human igg fc variant with enhanced fcrn binding |
| NZ597259A (en) * | 2009-06-23 | 2014-04-30 | Alexion Pharma Inc | Bispecific antibodies that bind to complement proteins |
| MX2012005151A (es) * | 2009-11-05 | 2012-08-23 | Federico Ii University Of Naples | Tratamiento de hemoglobinuria nocturna paroxismica, anemias hemoliticas y estados de enfermedad que involucran hemolisis intravascular y extravascular. |
| SG183867A1 (en) * | 2010-03-11 | 2012-10-30 | Rinat Neuroscience Corp | ANTIBODIES WITH pH DEPENDENT ANTIGEN BINDING |
| TWI667346B (zh) | 2010-03-30 | 2019-08-01 | 中外製藥股份有限公司 | 促進抗原消失之具有經修飾的FcRn親和力之抗體 |
| SG185107A1 (en) | 2010-04-30 | 2012-12-28 | Alexion Pharma Inc | Antibodies having reduced immunogenicity in a human |
| EP2824111B1 (en) * | 2010-04-30 | 2017-03-22 | Alexion Pharmaceuticals, Inc. | Anti-C5A Antibodies and Methods for Using the Antibodies |
| TWI812066B (zh) | 2010-11-30 | 2023-08-11 | 日商中外製藥股份有限公司 | 具有鈣依存性的抗原結合能力之抗體 |
| WO2012088247A2 (en) * | 2010-12-22 | 2012-06-28 | Medimmune, Llc | Anti-c5/c5a/c5adesr antibodies and fragments |
| EP2698431B1 (en) | 2011-03-30 | 2020-09-09 | Chugai Seiyaku Kabushiki Kaisha | Retention of antigen-binding molecules in blood plasma and method for modifying immunogenicity |
| JP6322411B2 (ja) | 2011-09-30 | 2018-05-09 | 中外製薬株式会社 | 複数の生理活性を有する抗原の消失を促進する抗原結合分子 |
| TW201817744A (zh) | 2011-09-30 | 2018-05-16 | 日商中外製藥股份有限公司 | 具有促進抗原清除之FcRn結合域的治療性抗原結合分子 |
| WO2013165690A1 (en) * | 2012-04-30 | 2013-11-07 | Medimmune, Llc | Molecules with reduced effector function and extended half-lives, compositions, and uses thereof |
| NZ631007A (en) | 2014-03-07 | 2015-10-30 | Alexion Pharma Inc | Anti-c5 antibodies having improved pharmacokinetics |
| US20190135903A1 (en) | 2015-03-31 | 2019-05-09 | Alexion Pharmaceuticals, Inc. | Identifying and treating subpopulations of paroxysmal nocturnal hemoglobinuria (pnh) patients |
| US20180311299A1 (en) | 2015-05-01 | 2018-11-01 | Alexion Pharmaceuticals, Inc. | Efficacy of an anti-c5 antibody in the prevention of antibody mediated rejection in sensitized recipients of a kidney transplant |
| US20180311345A1 (en) | 2015-10-30 | 2018-11-01 | Alexion Pharmaceuticals, Inc. | A method of inhibiting exacerbations of t cell-mediated allograft vasculopathy |
| ES2984352T3 (es) * | 2016-06-14 | 2024-10-29 | Regeneron Pharma | Anticuerpos anti-C5 y usos de los mismos |
| EP3526250A1 (en) | 2016-10-12 | 2019-08-21 | Alexion Pharmaceuticals, Inc. | Efficacy of an anti-c5 antibody in the prevention of antibody mediated rejection in sensitized recipients of a kidney transplant |
-
2014
- 2014-09-11 NZ NZ631007A patent/NZ631007A/en unknown
- 2014-09-11 NZ NZ711451A patent/NZ711451A/en unknown
-
2015
- 2015-03-06 ES ES16177656T patent/ES2749978T3/es active Active
- 2015-03-06 LT LTEP15710396.1T patent/LT2970455T/lt unknown
- 2015-03-06 KR KR1020237018254A patent/KR20230082690A/ko active Pending
- 2015-03-06 TW TW113129415A patent/TW202446794A/zh unknown
- 2015-03-06 EP EP19176561.9A patent/EP3594235A1/en active Pending
- 2015-03-06 EA EA202090275A patent/EA202090275A3/ru unknown
- 2015-03-06 TW TW109133882A patent/TWI845769B/zh active
- 2015-03-06 BR BR112016020454-9A patent/BR112016020454B1/pt active IP Right Grant
- 2015-03-06 SI SI201530009A patent/SI2970455T1/sl unknown
- 2015-03-06 SI SI201530940T patent/SI3095795T1/sl unknown
- 2015-03-06 HR HRP20161146TT patent/HRP20161146T1/hr unknown
- 2015-03-06 PT PT157103961T patent/PT2970455T/pt unknown
- 2015-03-06 JP JP2016555463A patent/JP6112469B1/ja active Active
- 2015-03-06 EA EA201691764A patent/EA035025B1/ru unknown
- 2015-03-06 MA MA38859A patent/MA38859B1/fr unknown
- 2015-03-06 CN CN202110517494.0A patent/CN113416254B/zh active Active
- 2015-03-06 DK DK15710396.1T patent/DK2970455T3/en active
- 2015-03-06 WO PCT/US2015/019225 patent/WO2015134894A1/en not_active Ceased
- 2015-03-06 PL PL15710396T patent/PL2970455T3/pl unknown
- 2015-03-06 CA CA2942165A patent/CA2942165C/en active Active
- 2015-03-06 ES ES15710396.1T patent/ES2589230T3/es active Active
- 2015-03-06 IL IL296502A patent/IL296502B1/en unknown
- 2015-03-06 ME MEP-2016-264A patent/ME02629B/me unknown
- 2015-03-06 EP EP16177656.2A patent/EP3095795B1/en active Active
- 2015-03-06 TW TW104107187A patent/TWI667256B/zh active
- 2015-03-06 EP EP15710396.1A patent/EP2970455B1/en active Active
- 2015-03-06 KR KR1020177032026A patent/KR102304136B1/ko active Active
- 2015-03-06 PT PT161776562T patent/PT3095795T/pt unknown
- 2015-03-06 RS RS20160996A patent/RS55386B1/sr unknown
- 2015-03-06 TW TW108122663A patent/TWI708785B/zh active
- 2015-03-06 MX MX2016011628A patent/MX368909B/es active IP Right Grant
- 2015-03-06 MA MA050373A patent/MA50373A/fr unknown
- 2015-03-06 DK DK16177656.2T patent/DK3095795T3/da active
- 2015-03-06 CN CN201580012081.XA patent/CN106459189B/zh active Active
- 2015-03-06 KR KR1020217011897A patent/KR102539636B1/ko active Active
- 2015-03-06 HU HUE15710396A patent/HUE030944T2/en unknown
- 2015-03-06 AR ARP150100697A patent/AR099698A1/es active IP Right Grant
- 2015-03-06 CA CA3166772A patent/CA3166772A1/en active Pending
- 2015-03-06 KR KR1020167027411A patent/KR101796886B1/ko active Active
- 2015-03-06 AU AU2015226977A patent/AU2015226977B2/en active Active
- 2015-03-06 US US14/641,026 patent/US9079949B1/en active Active
- 2015-03-06 HU HUE16177656A patent/HUE046091T2/hu unknown
- 2015-03-06 PL PL16177656T patent/PL3095795T3/pl unknown
- 2015-06-01 US US14/727,313 patent/US9107861B1/en active Active
- 2015-07-01 US US14/789,329 patent/US9206251B2/en active Active
- 2015-10-27 US US14/923,879 patent/US9371377B2/en active Active
-
2016
- 2016-05-20 US US15/160,364 patent/US9663574B2/en active Active
- 2016-08-17 IL IL247331A patent/IL247331B/en active IP Right Grant
- 2016-09-07 MX MX2019012455A patent/MX2019012455A/es unknown
- 2016-09-16 CY CY20161100922T patent/CY1118018T1/el unknown
- 2016-10-12 SM SM201600366T patent/SMT201600366B/it unknown
-
2017
- 2017-03-03 JP JP2017040585A patent/JP6894265B2/ja active Active
- 2017-04-20 US US15/492,622 patent/US9803007B1/en active Active
- 2017-09-19 US US15/708,658 patent/US10227400B2/en active Active
-
2019
- 2019-01-14 US US16/246,842 patent/US10584164B2/en active Active
- 2019-05-27 JP JP2019098449A patent/JP7450344B2/ja active Active
- 2019-09-24 AU AU2019236617A patent/AU2019236617B2/en active Active
-
2020
- 2020-01-23 US US16/750,173 patent/US11434280B2/en active Active
- 2020-03-17 IL IL273337A patent/IL273337B/en unknown
- 2020-10-14 JP JP2020173023A patent/JP2021036870A/ja active Pending
- 2020-11-16 AU AU2020267324A patent/AU2020267324B2/en active Active
-
2021
- 2021-11-15 IL IL288139A patent/IL288139A/en unknown
-
2023
- 2023-07-07 US US18/219,138 patent/US20240141024A1/en active Pending
-
2024
- 2024-01-29 JP JP2024011008A patent/JP2024038502A/ja active Pending
- 2024-07-16 AU AU2024204884A patent/AU2024204884A1/en active Pending
-
2025
- 2025-01-09 US US19/014,615 patent/US20250145698A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20250145698A1 (en) | Anti-c5 antibodies having improved pharmacokinetics | |
| HK40021039A (en) | Anti-c5 antibodies having improved pharmacokinetics | |
| HK1228405B (en) | Anti-c5 antibodies having improved pharmacokinetics | |
| HK1228405A1 (en) | Anti-c5 antibodies having improved pharmacokinetics | |
| HK1217207B (en) | Anti-c5 antibodies having improved pharmacokinetics | |
| EA046852B1 (ru) | Антитела против с5 с улучшенными фармакокинетическими характеристиками |