ES2782248T3 - Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila - Google Patents
Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila Download PDFInfo
- Publication number
- ES2782248T3 ES2782248T3 ES13847461T ES13847461T ES2782248T3 ES 2782248 T3 ES2782248 T3 ES 2782248T3 ES 13847461 T ES13847461 T ES 13847461T ES 13847461 T ES13847461 T ES 13847461T ES 2782248 T3 ES2782248 T3 ES 2782248T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- compound
- group
- amino acid
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[C@@](C1)Cc2c([C@](CCc3c(C)c(F)c4)NC(CC*)=O)c3c4nc2C1=CC Chemical compound C[C@@](C1)Cc2c([C@](CCc3c(C)c(F)c4)NC(CC*)=O)c3c4nc2C1=CC 0.000 description 2
- ZVYVPGLRVWUPMP-FYSMJZIKSA-N CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)N)c4c5nc13)=C(CO1)C2=O)(C1=O)O Chemical compound CC[C@](C(C=C1N2Cc3c([C@H](CCc4c(C)c(F)c5)N)c4c5nc13)=C(CO1)C2=O)(C1=O)O ZVYVPGLRVWUPMP-FYSMJZIKSA-N 0.000 description 1
- NXUBPJOMIBZQST-UHFFFAOYSA-N CN(C1OC1CC1)C1=O Chemical compound CN(C1OC1CC1)C1=O NXUBPJOMIBZQST-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68037—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a camptothecin [CPT] or derivatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2875—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF/TNF superfamily, e.g. CD70, CD95L, CD153, CD154
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2878—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Cell Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un conjugado de anticuerpo y fármaco en el que un compuesto antitumoral representado por la siguiente fórmula: **(Ver fórmula)** está conjugado a un anticuerpo a través de un engarce, en el que la estructura de fármaco y engarce en el conjugado de anticuerpo y fármaco tiene una estructura seleccionada entre los siguientes grupos de estructura de fármaco y engarce: - (Succinimid-3-il-N) -CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O) - (NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O) - (NH-DX), - (Succinimid-3-il-N)-(CH2)5-C(=O)-N[-(CH2CH2-O)3-CH2CH2-OH]-CH2 - (C=O)-GGFG-NH-(CH2)3-C(=O)-(NH-DX), - (Succinimid-3-il-N)-CH[-(CH2)2-COOH]-C(=O)-NH-(CH2)2-C(=O)-GG FG-NH-(CH2)3-C(=O)-(NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-(NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFGG-(NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFGGG-(NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX), - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-(NH-DX), en los que -(Succinimid-3-il-N)- tiene una estructura representada por la siguiente fórmula: **(Ver fórmula)** que está conectada al anticuerpo en la posición 3 del mismo y está conectada a un grupo metileno en la estructura de engarce que contiene esta estructura en el átomo de nitrógeno en la posición 1, - (NH-DX) es un grupo representado por la siguiente fórmula: **(Ver fórmula)** en la que el átomo de nitrógeno del grupo amino en la posición 1 es la posición de conexión.
Description
DESCRIPCIÓN
Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila
Campo técnico
La presente invención se refiere a un conjugado de anticuerpo y fármaco que tiene un compuesto antitumoral conjugado con un anticuerpo capaz de dirigirse a células tumorales a través de una fracción de estructura enlazadora que tiene una estructura hidrófila, siendo el conjugado útil como fármaco antitumoral.
Antecedentes de la técnica
Un conjugado de anticuerpo y fármaco (ADC, por las siglas del inglés antibody-drug conjúgate) que tiene un fármaco con citotoxicidad conjugado con un anticuerpo, cuyo antígeno se expresa en una superficie de células cancerosas y que también se une a un antígeno capaz de internalización celular y, por lo tanto, puede suministrar el fármaco selectivamente a las células cancerosas y, por tanto, se espera que cause la acumulación del fármaco dentro de las células cancerosas y las destruya (véanse las referencias no de patente 1 a 3). Como un ADC, Mylotarg (Gemtuzumab ozogamicin), en el que la caliqueamicina se conjuga con un anticuerpo anti CD33, está autorizado como agente terapéutico para la leucemia mieloide aguda. Además, Adcetris (Brentuximab vedotin), en el que la auristatina E se conjuga con un anticuerpo anti CD30, ha sido recientemente autorizado como agente terapéutico para el linfoma de Hodgkin y el linfoma anaplásico de células grandes (véase la referencia no de patente 4). Los fármacos contenidos en los ADC que han sido autorizados hasta ahora se dirigen al ADN o a la tubulina.
En lo que respecta a un antitumoral, se conocen compuestos de bajo peso molecular, derivados de la camptotecina, compuestos que inhiben la topoisomerasa I que muestran un efecto antitumoral. Entre ellos, un compuesto antitumoral representado por la siguiente fórmula
[Fórmula 1]

(exatecán, nombre químico: (1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-10,13(9H, 15H)-diona) es un derivado soluble en agua de la camptotecina (referencias de patente 1 y 2). A diferencia del irinotecán, utilizado actualmente en entornos clínicos, no es necesaria una activación por una enzima. Además, la actividad inhibidora sobre la topoisomerasa I es mayor que la de SN-38, que es una sustancia principal farmacéuticamente activa de irinotecán y topotecán también utilizada en entornos clínicos, y que produce mayor actividad citocida in vitro contra diversas células cancerosas. En particular, muestra el efecto contra células cancerosas que tienen resistencia a SN-38 o similar debido a la expresión de la glucoproteína P. Además, en un modelo de ratón de tumor humano trasplantado por vía subcutánea, mostró un fuerte efecto antitumoral, y por tanto se sometió a estudios clínicos, aunque aún no se ha comercializado (véanse las referencias no de patente 5 a 10). Sin embargo, no está claro si el exatecán funciona o no de manera eficaz como un ADC.
DE-310 es un complejo en el que el exatecán se conjuga con un polímero de polialcohol carboximetildextrano biodegradable a través de un separador peptídico GGFG (referencia de patente 3). El exatecán se transforma en una forma de profármaco polimérico, de modo que se puede conservar una propiedad de retención en sangre alta y también aumentar pasivamente una propiedad muy direccionable a una zona tumoral utilizando el aumento de permeabilidad de los vasos sanguíneos recién formados dentro del tumor y la propiedad de retención en los tejidos tumorales. Con DE-310, a través de una escisión con enzima del separador peptídico, tanto exatecán como exatecán con glicina conectada a un grupo amino, se liberan continuamente como sustancia activa principal. Como resultado, los parámetros farmacocinéticos mejoran y se descubrió que DE-310 tenía mayor eficacia que exatecán administrado solo, aunque la dosificación de exatecán era menor que en el caso de la administración de exatecán solo según varios modelos de evaluación tumoral en estudios no clínicos. Se realizó un estudio clínico para DE-310, y se confirmaron casos eficaces en seres humanos, en el que un informe sugería que la sustancia activa principal se acumulaba en un tumor en comparación con la que estaba presente en los tejidos normales, sin embargo, también hay un informe que indica que la acumulación de DE-310 y la sustancia activa principal en un tumor no es muy diferente de la acumulación en tejidos normales en seres humanos y, por lo tanto, no se observa direccionamiento pasivo en seres humanos (véanse las referencias no de patente 11 a 14). Como resultado, DE-310 tampoco se comercializó, y no está claro si
exatecán funciona o no de manera eficaz como un fármaco orientado a dicho direccionamiento.
Como un compuesto relacionado con DE-310, también se conoce un complejo en el que entre el separador -GGFG y el exatecán se inserta -NH(CH2)4C(= O) para formar la estructura separadora -GGFG-NH(CH2)4C(=O) (referencia de patente 4). Sin embargo, el efecto antitumoral del complejo no se conoce en absoluto.
IListado de citas!
Referencias de patente!
[Referencia de patente 1] patente japonesa abierta a inspección pública N° 5-59061
[Referencia de patente 2] patente japonesa abierta a inspección pública N° 8-337584
[Referencia de patente 3] publicación internacional N°WO 1997/46260
[Referencia de patente 4] publicación internacional N° WO 2000/25825
Referencias de no de patente!
[Referencia no de patente 1] Ducry, L., y col. Bioconjugate Chem. (2010) 21, 5-13.; Antibody-Drug Conjugates: Linking cytotoxic payloads to monoclonal antibodies.
[Referencia no de patente 2] Alley, S. C., y col. Current Opinion in Chemical Biology (2010) 14, 529-537.; Antibodydrug conjugates: targeted drug delivery for cancer.
[Referencia no de patente 3] Damle N.K. Expert Opin. Biol. Ther. (2004) 4, 1445-1452.; Tumour-targeted chemotherapy with immunoconjugates of calicheamicin.
[Referencia no de patente 4] Senter P. D., y col. Nature Biotechnology (2012) 30, 631-637.; The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma.
[Referencia no de patente 5] Kumazawa, E., Tohgo, A., Exp. Opin. Invest. Drugs (1998) 7, 625-632.; Antitumour activity of DX-8951f: a new camptothecin derivative.
[Referencia no de patente 6] Mitsui, I., Kumazawa, E., Hirota, Y., y col. Jpn J. Cancer Res. (1995) 86, 776-782.; A new water-soluble camptothecin derivative, DX-8951f, exhibits potent antitumor activity against human tumors in vitro and in vivo.
[Referencia no de patente 7] Takiguchi, S., Tohgo, A., y col. Jpn J. Cancer Res. (1997) 88, 760-769.; Antitumor effect of DX-8951, a novel camptothecin analog, on human pancreatic tumor cells and their CPT-11-resistant variants cultured in vitro and xenografted into nude mice.
[Referencia no de patente 8] Joto, N. y col. Int J Cancer (1997) 72, 680-686.; DX-8951f, a water-soluble camptothecin analog, exhibits potent antitumor activity against a human lung cancer cell line and its SN-38-resistant variant. [Referencia no de patente 9] Kumazawa, E. y col. Cancer Chemother. Pharmacol. (1998) 42, 210-220.; Potent and broad antitumor effects of DX-8951f, a water-soluble camptothecin derivative, against various human tumors xenografted in nude mice.
[Referencia no de patente 10] De Jager, R., y col. Ann NY Acad Sci (2000) 922, 260-273.; DX-8951f: summary of phase I clinical trials.
[Referencia no de patente 11] Inoue, K. y col. Polymer Drugs in the Clinical Stage, Editado por Maeda y col. (2003), 145-153.; CM-dextran-polyalcohol-camptothecin conjugate, DE-310 with a novel carrier system and its preclinical data.
[Referencia no de patente 12] Kumazawa, E. y col. Cancer Sci (2004) 95, 168-175.; DE-310, a novel macromolecular carrier system for the camptothecin analog DX-8951f: Potent antitumor activities in various murine tumor models.
[Referencia no de patente 13] Soepenberg, O. y col. Clinical Cancer Research, (2005) 11, 703-711.; Phase I and pharmacokinetic study of DE-310 in Patients with Advanced Solid Tumors.
[Referencia no de patente 14] Wente M. N. y col. Investigational New Drugs (2005) 23, 339-347.; DE-310, a macromolecular prodrug of the topoisomerase-I-inhibitor exatecan (DX-8951), in patients with operable solid tumors.
ISumario de la invención!
Rroblema técnico!
Con respecto al tratamiento de tumores con un anticuerpo, se puede observar un efecto antitumoral insuficiente incluso cuando el anticuerpo reconoce un antígeno y se une a las células tumorales, y hay un caso en el que se necesita un anticuerpo antitumoral más eficaz. Además, muchos compuestos antitumorales de bajo peso molecular tienen problemas de seguridad, tales como efectos secundarios y toxicidad, aunque los compuestos tengan un excelente efecto antitumoral, sigue siendo un problema conseguir un efecto terapéutico superior potenciando adicionalmente la seguridad. Por tanto, un objeto de la presente invención es proporcionar un fármaco antitumoral que tenga un excelente efecto terapéutico, que sea excelente en cuanto a su efecto antitumoral y que sea inocuo.
rMedios para resolver el problema!
Los inventores pensaron que, cuando un compuesto antitumoral, exatecán, se transforma en un conjugado de
anticuerpo y fármaco, a través de un enlazador, por conjugación con el anticuerpo, que es capaz de dirigirse a las células tumorales, que tiene una propiedad de reconocer células tumorales, una propiedad de unirse a células tumorales, una propiedad de internalizarse en células tumorales, una actividad citocida contra células tumorales, o propiedades similares, el compuesto antitumoral puede suministrase con mayor seguridad a las células tumorales para mostrar específicamente el efecto antitumoral del compuesto en las células tumorales, y así se puede mostrar con seguridad el efecto antitumoral y también se espera un mayor efecto citocida del anticuerpo, y se puede reducir una dosis del compuesto antitumoral en comparación con un caso de administración del compuesto solo, y por tanto se puede aliviar la influencia del compuesto antitumoral en las células normales de manera que se pueda lograr una mayor seguridad.
A este respecto, los inventores crearon un enlazador con una estructura específica.
Los presentes inventores han construido particularmente un enlazador que tiene:
una estructura enlazadora en la que un aminoácido hidrofílico distinto de glicina está conectado en el terminal N de una fracción peptídica del enlazador;
una estructura enlazadora en la que la glicina o la glicilglicina están conectadas en el terminal C de una fracción peptídica del enlazador; o una estructura enlazadora en la que un elemento enlazador que tiene una estructura hidrófila se inserta entre una fracción peptídica en el enlazador y un anticuerpo;
y obtuvieron de manera satisfactoria un conjugado de anticuerpo y fármaco que tenía exatecán conjugado con un anticuerpo a través de dicho enlazador. Los presentes inventores han descubierto además que este conjugado de anticuerpo y fármaco es excelente en cuanto a la liberación del componente farmacológico que tiene un efecto antitumoral, y como resultado, el conjugado de anticuerpo y fármaco de la presente invención, ejerce un excelente efecto antitumoral, conduciendo a la finalización de la presente invención.
Específicamente, la presente invención se refiere a los siguientes.
[1] Un conjugado de anticuerpo y fármaco en el que un compuesto antitumoral representado por la siguiente fórmula:
[Fórmula 2]

está conjugado a un anticuerpo a través de un engarce, en el que la estructura de fármaco y engarce en el conjugado de anticuerpo y fármaco tiene una estructura seleccionada entre los siguientes grupos de estructura de fármaco y engarce:
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
- (Succinimid-3-il-N)-(CH2)5-C(=O)-N[-(CH2CH2-O)3-CH2CH2-OH]-CH2-(C=O)-GGFG-NH-(CH2)a-C(=O)-(NH-DX),
- (Succinimid-3-il-N)-CH[-(CH2)2-COOH]-C(=O)-NH-(CH2)2-C(=O)-GGFG-NH-(CH2)3-C(=O)-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFGG-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFGGG-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX),
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-(NH-DX), en los que -(Succinimid-3-il-N)- tiene una estructura representada por la siguiente fórmula:

que está conectada al anticuerpo en la posición 3 del mismo y está conectada a un grupo metileno en la estructura
de engarce que contiene esta estructura en el átomo de nitrógeno en la posición 1,
- (NH-DX) es un grupo representado por la siguiente fórmula:

en la que el átomo de nitrógeno del grupo amino en la posición 1 es la posición de conexión, en la que F = fenilalanina (Phe), G = glicina (Gly), K = lisina (Lys), D = ácido aspártico (Asp).
La presente invención también se refiere a cada uno de los siguientes.
[2] El conjugado de anticuerpo y fármaco de acuerdo con [1], en el que un número promedio de compuestos antitumorales conjugados por anticuerpo está en el intervalo de 1 a 10.
[3] El conjugado de anticuerpo y fármaco de acuerdo con [1], en el que un número promedio de compuestos antitumorales conjugados por anticuerpo está en el intervalo de 1 a 8.
[4] El conjugado de anticuerpo y fármaco de acuerdo con [1], en el que un número promedio de compuestos antitumorales conjugados por anticuerpo está en el intervalo de 3 a 8.
[5] El conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [4], en el que el anticuerpo es un anticuerpo que tiene una o más de una propiedad de reconocer una célula diana, una propiedad de unirse a una célula diana, una propiedad de internalizarse en una célula diana y una propiedad de dañar una celda diana.
[6] El conjugado de anticuerpo y fármaco según [5], en el que la célula diana es una célula tumoral.
[7] El conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [6], en el que el anticuerpo es un anticuerpo anti-A33, un anticuerpo anti-B7-H3, un anticuerpo anti-CanAg, un anticuerpo anti-CD20, un anticuerpo anti-CD22, un anticuerpo anti-CD30, un anticuerpo anti-CD33, un anticuerpo anti-CD56, un anticuerpo anti-CD70, un anticuerpo anti-CEA, un anticuerpo anti-Cripto, un anticuerpo anti-EphA2, un anticuerpo anti-G250, un anticuerpo anti-MUC1, un anticuerpo anti-GPNMB, un anticuerpo anti-integrina, un anticuerpo anti-PSMA, un anticuerpo antitenascina-C, un anticuerpo anti-SLC44A4, o un anticuerpo anti-mesotelina.
[8] El conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [6], en el que el anticuerpo es un anticuerpo anti-B7-H3, un anticuerpo anti-CD30, un anticuerpo anti-CD33 o un anticuerpo anti-CD70.
[9] El conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [6], en el que el anticuerpo es un anticuerpo anti-B7-H3.
[10] Un fármaco que contiene el conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [9], una sal del mismo o un hidrato del mismo.
[11] Un fármaco antitumoral y/o fármaco contra el cáncer que contiene el conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [9], una sal del mismo o un hidrato del mismo.
[12] El fármaco antitumoral y/o el fármaco contra el cáncer según [11], para su uso en el tratamiento de cáncer de pulmón, cáncer de riñón, cáncer urotelial, cáncer colorrectal, cáncer de próstata, glioblastoma multiforme, cáncer de ovario, cáncer de páncreas, cáncer de mama, melanoma, cáncer de hígado, cáncer de vejiga, cáncer de estómago o cáncer de esófago.
[13] Una composición farmacéutica que contiene el conjugado de anticuerpo y fármaco según uno cualquiera de [1] a [9], una sal del mismo o un hidrato del mismo como componente activo, y un componente de formulación farmacéuticamente aceptable.
[14] La composición farmacéutica según [13], para su uso en el tratamiento de cáncer de pulmón, cáncer de riñón, cáncer urotelial, cáncer colorrectal, cáncer de próstata, glioblastoma multiforme, cáncer de ovario, cáncer de páncreas, cáncer de mama, melanoma, cáncer de hígado, cáncer de vejiga, cáncer de estómago o cáncer de esófago.
[15] Un compuesto intermedio de fármaco y engarce, que se selecciona entre el siguiente grupo:
(maleimid-N-il)-(CH2)5-C(=O)-DGGFG-NH-(CH2)3-C(=O)-(NH-DX),
(maleimid-N-il)-(CH2)5-C(=O)-DGGFG-NH-(CH2)2-C(=O)-(NH-DX),
(maleimid-N-il)-(CH2)5-C(=O)-KGGFG-NH-(CH2)3-C(=O)-(NH-DX),
(maleimid-N-il)-(CH2)5-C(=O)-N[-(CH2CH2-O)3-CH2CH2-OH]-CH2-(C=O)-GGFG-NH-(CH2)3-C(=O)-(NH-DX), (maleimid-N-il)-CH[-(CH2)2-COOH]-C(=O)-NH-(CH2)2-C(=O)-GGFG-NH-(CH2)3-C(=O)-(NH-DX), (maleimid-N-il)-(CH2)5-C(=O)-GGFG-(NH-DX),
(maleimid-N-il)-(CH2)5-C(=O)-GGFGG-(NH-DX),
(maleimid-N-il)-(CH2)5-C(=O)-GGFGGG-(NH-DX),
(maleimid-N-il)-(CH2)5-C(=O)-DGGFG-(NH-DX), y
(maleimid-N-il)-(CH2)5-C(=O)-KGGFG-(NH-DX),
en el que (maleimid-N-il)- es un grupo representado por la siguiente fórmula:

en la que el átomo de nitrógeno está en la posición de conexión y -(NH-DX) es un grupo representado por la siguiente fórmula:

en la que el átomo de nitrógeno del grupo amino en la posición 1 es la posición de conexión,
en la que F = fenilalanina (Phe), G = glicina (Gly), K = lisina (Lys), D = ácido aspártico (Asp).
[16] El compuesto de acuerdo con [15], que es cualquiera de los siguientes:
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFGG-(NH-DX),
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFGGG-(NH-DX).
Efectos ventajosos de la invención
Con un conjugado de anticuerpo y fármaco que tiene un compuesto antitumoral de exatecan conjugado a través de un engarce con una estructura específica, se consigue un excelente efecto antitumoral.
Breve descripción de los dibujos
[Figura 1] La Figura 1 muestra una secuencia de aminoácidos de la B7-H3 variante 1 (SEQ ID NO: 1).
[Figura 2] La Figura 2 muestra una secuencia de aminoácidos de la B7-H3 variante 2 (SEQ ID NO: 2).
[Figura 3] La Figura 3 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H1 (SEQ ID NO: 9) .
[Figura 4] La Figura 4 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H2 (SEQ ID NO: 10) .
[Figura 5] La Figura 5 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H3 (SEQ ID NO: 11).
[Figura 6] La Figura 6 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H4 (SEQ ID NO: 12).
[Figura 7] La Figura 7 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L1 (SEQ ID NO: 13) .
[Figura 8] La Figura 8 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L2 (SEQ ID NO: 14) .
[Figura 9] La Figura 9 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L3 (SEQ ID NO: 15) .
[Figura 10] La Figura 10 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L4 (SEQ ID NO: 16).
[Figura 11] La Figura 11 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L5 (SEQ ID NO: 17).
[Figura 12] La Figura 12 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L6 (SEQ ID NO: 18).
[Figura 13] La Figura 13 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L7 (SEQ ID NO: 19).
[Figura 14] La Figura 14 muestra una secuencia de aminoácidos de una cadena pesada de anticuerpo M30 (SEQ ID NO: 20).
[Figura 15] La Figura 15 muestra una secuencia de aminoácidos de una cadena ligera de anticuerpo M30 (SEQ ID NO: 21).
[Figura 16] La Figura 16 muestra una secuencia de nucleótidos de la B7-H3 variante 1 (SEQ ID NO: 26).
[Figura 17] La Figura 17 muestra los efectos de un anticuerpo M30-H1-L4P y de los conjugados de anticuerpo y fármaco (1), (2), (18) y (19) administrados a una dosis de 10 mg/kg en células A375 de la línea de melanoma humano trasplantadas en ratones. La línea con rombos blancos representa resultados sobre tumor no tratado, la línea con rombos negros representa el efecto del anticuerpo M30-H1-L4P, la línea con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (1), la línea con cuadrados blancos representa el efecto del conjugado de anticuerpo y fármaco (2), la línea con triángulos negros representa el efecto del conjugado de anticuerpo y fármaco (18), y la línea con triángulos blancos representa el efecto del conjugado de anticuerpo y fármaco (19).
[Figura 18] La Figura 18 muestra los efectos de los conjugados de anticuerpo y fármaco (2) y (19) administrados a una dosis de 1 mg/kg y 3 mg/kg en células A375 de la línea de melanoma humano trasplantadas en ratones. La línea con rombos blancos representa resultados sobre tumor no tratado, la línea con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (2) administrado a una dosis de 1 mg/kg, la línea con cuadrados blancos representa el efecto del conjugado de anticuerpo y fármaco (2) administrado a una dosis de 3 mg/kg, la línea con círculos negros representa el efecto del conjugado de anticuerpo y fármaco (19) administrado a una dosis de 1 mg/kg y la línea con círculos blancos representa el efecto del conjugado de anticuerpo y fármaco (19) administrado a una dosis de 3 mg/kg.
[Figura 19] La Figura 19 muestra los efectos de un anticuerpo M30-H1-L4P y de los conjugados de anticuerpo y fármaco (1), (2), (18) y (19) administrados a una dosis de 10 mg/kg, sobre células Calu-6 de la línea de cáncer de pulmón humano no microcítico trasplantadas en ratones. La línea con rombos blancos representa resultados sobre tumor no tratado, la línea con rombos negros representa el efecto del anticuerpo M30-H1-L4P, la línea con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (1), la línea con cuadrados blancos representa el efecto del conjugado de anticuerpo y fármaco (2), la línea con triángulos negros representa el efecto del conjugado de anticuerpo y fármaco (18), y la línea con triángulos blancos representa el efecto del conjugado de anticuerpo y fármaco (19).
[Figura 20] La Figura 20 muestra los efectos de los conjugados de anticuerpo y fármaco (3), (20) y (30) administrados a una dosis de 3 mg/kg y 10 mg/kg en células A375 de la línea de melanoma humano trasplantadas en ratones. La línea con rombos blancos representa resultados sobre tumor no tratado, la línea de puntos con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (3) administrado a una dosis de 3 mg/kg, la línea continua con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (3) administrado a una dosis de 10 mg/kg, La línea de puntos con triángulos negros representa el efecto del conjugado de anticuerpo y fármaco (20) administrado a una dosis de 3 mg/kg, la línea continua con triángulos rellenos representa el efecto del conjugado de anticuerpo y fármaco (20) administrado a una dosis de 10 mg/kg, la línea de puntos con círculos negros representa el efecto del conjugado de anticuerpo y fármaco (30) administrado a una dosis de 3 mg/kg y la línea continua con círculos negros representa el efecto del conjugado de anticuerpo y fármaco (30) administrado a una dosis de 10 mg/kg.
Descripción de las realizaciones
El conjugado de anticuerpo y fármaco de la presente invención es un fármaco antitumoral en el que un anticuerpo antitumoral se conjuga con un compuesto antitumoral a través de una fracción de estructura enlazadora y se explica en detalle a continuación.
[Anticuerpo]
El anticuerpo usado en el conjugado de anticuerpo y fármaco de la presente invención, es una inmunoglobulina, que es una molécula que contiene un sitio de unión a antígeno que se une inmunoespecíficamente a un antígeno. La clase del anticuerpo de la presente invención puede ser cualquiera de IgG, IgE, IgM, IgD, IgA e IgY y es, preferentemente, IgG. La subclase del anticuerpo de la presente invención puede ser cualquiera de IgG1, IgG2, IgG3, IgG4, IgA1 e IgA2 y es, preferentemente, IgG1 o IgG2. El anticuerpo puede proceder de cualquier especie, y como ejemplos preferidos de especie pueden incluirse los seres humanos, ratas, ratones y conejos. En caso de que se proceda de otra especie que no sea la humana, preferentemente, se quimeriza o humaniza usando una técnica muy conocida. El anticuerpo de la presente invención puede ser un anticuerpo policlonal o un anticuerpo monoclonal y es, preferentemente, un anticuerpo monoclonal.
El anticuerpo de la presente invención puede ser uno que sea capaz de dirigirse a células tumorales. Dado que el anticuerpo de la presente invención se conjuga con un fármaco que tiene actividad antitumoral a través de un enlazador, el anticuerpo posee, preferentemente, una o más de una propiedad de reconocer una célula tumoral, una propiedad de unirse a una célula tumoral, una propiedad de internalizarse en una célula tumoral y una propiedad de dañar una célula tumoral.
La actividad de unión del anticuerpo contra células tumorales se puede confirmar mediante citometría de flujo. La internalización del anticuerpo en las células tumorales se puede confirmar usando (1) un ensayo de visualización de un anticuerpo incorporado en las células con un microscopio de fluorescencia usando un anticuerpo secundario (marcado con fluorescencia) que se une al anticuerpo terapéutico (Cell Death and Differentiation (2008) 15, 751-761), (2) un ensayo para medir la cantidad de fluorescencia incorporada en las células usando un anticuerpo secundario (marcado con fluorescencia) que se une al anticuerpo terapéutico (Molecular Biology of the Cell, Vol. 15, 5268-5282, diciembre de 2004), o (3) un ensayo Mab-ZAP utilizando una unión de inmunotoxina al anticuerpo terapéutico en el
que la toxina se libera al incorporarse a las células para inhibir el crecimiento celular (Bio Techniques 28: 162-165, enero de 2000).
La actividad antitumoral del anticuerpo se refiere a una actividad citotóxica o efecto citocida contra las células tumorales y puede confirmarse in vitro determinando la actividad inhibidora contra el crecimiento celular. Por ejemplo, se cultiva una línea celular de cáncer que sobreexpresa una proteína diana para el anticuerpo, y el anticuerpo se añade a diversas concentraciones al sistema de cultivo para determinar una actividad inhibidora contra la formación de focos, formación de colonias y crecimiento de esferoides. La actividad antitumoral se puede confirmar in vivo, por ejemplo, administrando el anticuerpo a un ratón lampiño con una línea celular tumoral trasplantada que exprese en grandes cantidades la proteína diana y determinando el cambio en la célula cancerosa. Dado que el fármaco conjugado en el conjugado de anticuerpo y fármaco ejerce un efecto antitumoral, es más preferido, pero no esencial, que el propio anticuerpo tenga un efecto antitumoral. Para que el fármaco ejerza el efecto antitumoral y también para que produzca daños de manera específica y selectiva a las células tumorales, también es importante y también prefiere que el anticuerpo tenga la propiedad de internalizarse para migrar hacia las células tumorales.
Los ejemplos de dicho anticuerpo pueden incluir, pero sin limitación, un anticuerpo anti-A33, un anticuerpo anti-B7-H3, un anticuerpo anti-CanAg, un anticuerpo anti-CD20, un anticuerpo anti-CD22, un anticuerpo anti-CD30, un anticuerpo anti-CD33, un anticuerpo anti-CD56, un anticuerpo anti-CD70, un anticuerpo anti-CEA, un anticuerpo anti-Cripto, un anticuerpo anti-EphA2, un anticuerpo anti-G250, un anticuerpo anti-MUC1, un anticuerpo anti-GPNMB, un anticuerpo anti-integrina, un anticuerpo anti-PSMA, un anticuerpo anti-tenascina-C, un anticuerpo anti-SLC44A4 y un anticuerpo anti-mesotelina.
El anticuerpo de la presente invención es, preferentemente, un anticuerpo anti-CD30, un anticuerpo anti-CD33, un anticuerpo anti-CD70 o un anticuerpo anti-B7-H3, y más preferentemente, un anticuerpo anti-B7-H3.
El anticuerpo de la presente invención puede producirse usando un procedimiento que habitualmente se lleva a cabo en la técnica, que implica inmunizar a animales con un polipéptido antigénico y recoger y purificar anticuerpos producidos en vivo. El origen del antígeno no se limita al de un ser humano, y los animales pueden inmunizarse con un antígeno procedente de un animal no humano, tal como un ratón, una rata y animales similares. En este caso, la reactividad cruzada de los anticuerpos que se unen al antígeno heterólogo producido, con antígenos humanos, puede analizarse para explorar un anticuerpo aplicable a una enfermedad humana.
Como alternativa, las células productoras de anticuerpos que producen anticuerpos contra el antígeno se fusionan con células de mieloma según un procedimiento conocido en la técnica (por ejemplo, Kohler y Milstein, Nature (1975) 256, págs. 495-497; y Kennet, R. ed., Monoclonal Antibodies, págs. 365-367, Plenum Press, N.Y. (1980)) para establecer hibridomas, a partir de los cuales pueden producirse a su vez anticuerpos monoclonales.
El antígeno puede producirse modificando con ingeniería genética células hospedadoras para producir un gen que codifique la proteína antigénica. Específicamente, lse preparan vectores que permiten la expresión del gen antigénico y transfieren a células hospedadoras para que se exprese el gen. El antígeno así expresado puede purificarse.
El anticuerpo anti-CD30, el anticuerpo anti-CD33 y el anticuerpo anti-CD70, pueden producirse mediante una estrategia conocida en la técnica con referencia al documento WO2002/043661, a la patente de Estados Unidos N.° 5.773.001 y al documento WO2006/113909, respectivamente.
El anticuerpo B7-H3 usado en la presente invención es, preferentemente, uno que tiene las propiedades que se describen a continuación.
(1) Un anticuerpo que tiene las siguientes propiedades:
(a) unirse específicamente con B7-H3,
(b) tener actividad de fagocitosis celular dependiente de anticuerpos (FCDA), y
(c) tener actividad antitumoral in vivo.
(2) El anticuerpo según (1), en el que B7-H3 es una molécula que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 1 o 2.
(3) El anticuerpo según (1) o (2), en el que el anticuerpo tiene, como regiones determinantes de complementariedad de cadena pesada, la CDRH1 que comprende la secuencia de aminoácidos representada por la s Eq ID NO: 3, la CDRH2 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 4 y la CDRH3 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 5, y como regiones determinantes de complementariedad de cadena ligera, la CDRL1 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 6, la CDRL2 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 7 y la CDRL3 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 8.
(4) El anticuerpo según cualquiera de (1) a (3), en el que la región constante del mismo es una región constante procedente de ser humano.
(5) El anticuerpo según cualquiera de (1) a (4), en el que el anticuerpo es un anticuerpo humanizado.
(6) El anticuerpo según (5), en el que el anticuerpo tiene una región variable de cadena pesada que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en (a) una secuencia de aminoácidos descrita
en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9, (b) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 10, (c) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 11, (d) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 12, (e) una secuencia de aminoácidos que tiene una homología de al menos 95 % o mayor con cualquiera de las secuencias (a) a (d), y (f) una secuencia de aminoácidos procedente de cualquiera de las secuencias (a) a (d) por deleciones, reemplazos o adiciones de al menos un aminoácido, y una región variable de cadena ligera que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en (g) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 13, (h) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 14, (i) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 15, (j) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 16, (k) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 17, (l) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 18, (m) una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 19, (n) una secuencia de aminoácidos que tiene una homología de al menos 95 % o mayor con cualquiera de las secuencias (g) a (m), y (o) una secuencia de aminoácidos procedente de cualquiera de las secuencias (g) a (m) por deleciones, reemplazos o adiciones de al menos un aminoácido. (7) El anticuerpo según (6), en el que el anticuerpo tiene una región variable de cadena pesada y una región variable de cadena ligera seleccionada del grupo que consiste en una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de la cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 13, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 14, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 15, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 16, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 17, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 18, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 19, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 13, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 14, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 15, y una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 128 en la SEQ ID NO: 16.
(8) El anticuerpo según (6) o (7), en el que el anticuerpo comprende una cadena pesada y una cadena ligera seleccionada del grupo que consiste en una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 13, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 14, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 15, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 16, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 17, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la
SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 18, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 19, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 13, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 14, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 15, y una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 16.
(9) El anticuerpo según cualquiera de (6) a (8), en el que el anticuerpo comprende una cadena pesada y una cadena ligera seleccionadas del grupo que consiste en una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 13, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 14, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 15, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 16, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 17, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 18, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 19, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 13, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 14, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 15, y una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y un cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 16.
(10) El anticuerpo según (8) o (9), en el que, en la cadena pesada, el anticuerpo carece de un aminoácido en el extremo carboxilo de la secuencia de aminoácidos representada por la SEQ ID NO: 9 o 12.
(11) Un anticuerpo producido por un procedimiento para producir el anticuerpo según cualquiera de (1) a (10), comprendiendo el procedimiento las siguientes etapas: cultivar una célula hospedadora transformada con un vector de expresión que contiene un polinucleótido que codifica el anticuerpo; y recoger el anticuerpo de interés de los cultivos producidos en la etapa anterior.
(12) El anticuerpo según cualquiera de (1) a (11), en el que la modificación de un glucano está regulada para potenciar la actividad citotóxica dependiente de anticuerpos.
En lo sucesivo, se describe el anticuerpo B7-H3 usado en la invención.
Los términos "cáncer" y "tumor" como se usan en el presente documento se usan con el mismo significado.
El término "gen", como se usa en el presente documento, incluye no sólo ADN, sino también ARNm, ADNc y ARNc de los mismos.
El término "polinucleótido" como se usa en el presente documento se usa con el mismo significado que un ácido nucleico y también incluye ADN, ARN, sondas, oligonucleótidos y cebadores.
Los términos "polipéptido" y "proteína" como se usan en el presente documento se usan sin distinción.
El término "célula" como se usa en el presente documento también incluye células de un animal individual y células cultivadas.
La expresión "B7-H3", como se usa en el presente documento, se usa con el mismo significado que proteína B7-H3, y también se refiere a B7-H3 variante 1 y/o B7-H3 variante 2.
El término "CDR", como se usa en el presente documento, se refiere a una región determinante de complementariedad (CDR), y se sabe que cada cadena pesada y ligera de una molécula de anticuerpo tiene tres regiones determinantes de complementariedad (CDR).
La CDR también se denomina región hipervariable, y está presente en una región variable de cada cadena pesada y
ligera de un anticuerpo. Es un sitio que en su estructura primaria tiene una variabilidad inusualmente alta, y en la estructura primaria de cada cadena polipeptídica pesada y ligera, hay tres CDR distintas. En la presente memoria descriptiva, en cuanto a las CDR de un anticuerpo, las CDR de la cadena pesada se representan como CDRH1, CDRH2 y CDRH3 a partir del lado amino terminal de la secuencia de aminoácidos de la cadena pesada, y las CDR de la cadena ligera se representan como CDRL1, CDRL2 y CDRL3 a partir del lado amino terminal de la secuencia de aminoácidos de la cadena ligera. Estos sitios están próximos entre sí en la estructura terciaria y determinan la especificidad por un antígeno al que se une el anticuerpo.
La expresión "la hibridación se realiza en condiciones rigurosas", como se usa en el presente documento, se refiere a un procedimiento en el que la hibridación se realiza en condiciones en las que puede lograrse la identificación realizando la hibridación a 68° C en una solución de hibridación disponible en el comercio, solución de hibridación ExpressHyb (fabricada por Clontech, Inc.) o realizando la hibridación a 68° C en presencia de NaCl de 0,7 a 1,0 M usando un filtro que tiene ADN inmovilizado sobre el mismo, seguido de lavado a 68° C usando solución de SSC de 0. 1.a 2 x (la solución de SSC 1 x está compuesta por NaCl 150 mM y citrato de sodio 15 mM) o en condiciones equivalentes a la misma.
1. B7-H3
B7-H3 es un miembro de la familia B7 que se expresa en células presentadoras de antígeno como una molécula coestimuladora, y se considera que actúa sobre un receptor en linfocitos T para mejorar o suprimir la actividad inmunitaria.
B7-H3 es una proteína que tiene una estructura transmembrana de un solo paso, y el dominio extracelular N-terminal de B7-H3 contiene dos variantes. La proteína B7-H3 variante 1 (4Ig-B7-H3) contiene un dominio de Ig de tipo V o de tipo C en dos sitios, respectivamente, y la B7-H3 variante 2 (2Ig-B7-H3) contiene un dominio de Ig de tipo V o de tipo C en un sitio, respectivamente.
En cuanto a la proteína B7-H3 utilizada en la invención, la proteína B7-H3 puede usarse y purificarse directamente de células de un mamífero humano o no humano (tal como una rata o un ratón) que expresan B7-H3, o puede usarse y prepararse una fracción de membrana celular de las células descritas anteriormente. Además, la proteína B7-H3 puede producirse mediante síntesis de la misma in vitro o producción de la misma en una célula hospedadora mediante modificación con ingeniería genética. Específicamente, en la modificación con ingeniería genética, después de que el ADNc de B7-H3 se integre en un vector capaz de expresar ADNc de B7-H3, la proteína B7-H3 puede producirse sintetizándola en una solución que contiene una enzima, un sustrato y una sustancia energética necesaria para la transcripción y traducción, o expresando la B7-H3 en otra célula hospedadora procariota o eucariota transformada.
La secuencia de aminoácidos de un marco abierto de lectura (ORF, open reading frame) de un gen de la B7-H3 variante 1 humana se representa en la SEQ ID NO: 1 en el Listado de Secuencias. Además, la secuencia de la SEQ ID NO: 1 se muestra en la figura 1.
La secuencia de aminoácidos de un ORF de un gen B7-H3 variante 2 humano se representa en la SEQ ID NO: 2 en el Listado de secuencias. Además, la secuencia de la SEQ ID NO: 2 se muestra en la figura 2.
Además, una proteína que consiste en una secuencia de aminoácidos en la que uno o varios aminoácidos se sustituyen, delecionan y/o añaden en cualquiera de las secuencias de aminoácidos descritas anteriormente de B7-H3 y que también tiene una actividad biológica equivalente a la de la proteína, también se incluye en B7-H3.
La B7-H3 variante 1 humana madura de la que se ha eliminado la secuencia señal, corresponde a una secuencia de aminoácidos que consiste en los restos de aminoácidos 27 a 534 de la secuencia de aminoácidos representada por la SEQ ID NO: 1. Además, La B7-H3 variante 2 humana madura de la que se ha eliminado la secuencia señal, corresponde a una secuencia de aminoácidos que consiste en los restos de aminoácidos 27 a 316 de la secuencia de aminoácidos representada por la SEQ ID NO: 2.
2. Producción de anticuerpo anti-B7-H3
El anticuerpo contra B7-H3 de la invención puede producirse inmunizando a un animal con B7-H3 o con un polipéptido arbitrario seleccionado de la secuencia de aminoácidos de B7-H3, y recogiendo y purificando el anticuerpo producido in vivo según un procedimiento habitual. La especie biológica de B7-H3 que se utilizará como antígeno, no se limita al ser humano, y un animal procedente de uno que no sea un ser humano, tal como un ratón o una rata, puede inmunizarse con B7-H3. En este caso, examinando la reactividad cruzada entre un anticuerpo que se une a la proteína B7-H3 heteróloga producida y a la B7-H3 humana, se puede seleccionar un anticuerpo aplicable a una enfermedad humana.
Además, a partir de un hibridoma establecido, se puede producir un anticuerpo monoclonal fusionando células productoras de anticuerpos que producen un anticuerpo contra B7-H3 con células de mieloma según un procedimiento conocido (por ejemplo, Kohler y Milstein, Nature, (1975) 256, págs. 495-497; Kennet, R. ed., Monoclonal Antibodies, págs. 365-367, Plenum Press, N.Y. (1980)).
Para utilizar la proteína B7-H3 como un antígeno, ésta puede producirse expresando el gen de B7-H3 en una célula hospedadora usando modificación con ingeniería genética.
Específicamente, se produce un vector capaz de expresar el gen de B7-H3, y el vector resultante se transfecta en una célula hospedadora para expresar el gen, y después, el gen de B7-H3 expresado se purifica. En lo sucesivo, se describe específicamente un procedimiento para producir un anticuerpo contra B7-H3.
(1) Preparación de antígeno
Los ejemplos del antígeno que se usará para producir el anticuerpo anti-B7-H3 incluyen B7-H3, un polipéptido que consiste en una secuencia parcial de aminoácidos que comprende al menos 6 aminoácidos consecutivos de B7-H3, y un derivado producido añadiendo a la misma una secuencia de aminoácidos o un vehículo determinado.
B7-H3 puede purificarse directamente de tejidos tumorales humanos o de células tumorales humanas y usarse. Además, B7-H3 puede producirse sintetizándolo in vitro o produciéndolo en una célula hospedadora mediante modificación con ingeniería genética.
Con respecto a la modificación con ingeniería genética, específicamente, después de que el ADNc de B7-H3 se integre en un vector capaz de expresar ADNc de B7-H3, la proteína B7-H3 puede producirse sintetizándola en una solución que contiene una enzima, un sustrato y una sustancia energética necesaria para la transcripción y traducción, o expresando la B7-H3 en otra célula hospedadora procariota o eucariota transformada.
Además, el antígeno también puede producirse como una proteína secretora al expresar una proteína de fusión producida al ligar el dominio extracelular de B7-H3, que es una proteína de membrana, con la región constante de un anticuerpo en un sistema de hospedador y vector apropiado.
El ADNc de B7-H3 puede obtenerse, por ejemplo, mediante el denominado procedimiento de PCR en el que se realiza una reacción en cadena de la polimerasa (en lo sucesivo denominada "PCR") utilizando una biblioteca de ADNc que expresa el ADNc de B7-H3 como molde y cebadores que amplifican específicamente el ADNc de B7-H3 (véase Saiki, R. K., y col., Science, (1988) 239, págs. 487-489).
Como ejemplo de síntesis in vitro del polipéptido, puede ilustrase, aunque sin limitación, por ejemplo, el Sistema de Traducción Rápida (STR) fabricado por Roche Diagnostics, Inc.
Los ejemplos de células hospedadoras procariotas incluyen Escherichia coli y Bacillus subtilis. Para transformar las células hospedadoras con un gen diana, las células hospedadoras se transforman con un vector plasmídico que comprende un replicón, es decir, un origen de replicación procedente de una especie compatible con el hospedador y una secuencia reguladora. Además, el vector tiene, preferentemente, una secuencia capaz de imponer selectividad fenotípica en la célula transformada.
Los ejemplos de células hospedadoras eucariotas incluyen células de vertebrados, células de insectos y células de levadura. Como células de vertebrados, se utilizan con frecuencia, por ejemplo, células COS de simio (Gluzman, Y., Cell, (1981) 23, págs. 175-182, ATCC CRL-1650), fibroblastos murinos NIH3T3 (ATCC N.°CRL-1658) y cepas deficientes en dihidrofolato reductasa (Urlaub, G. y Chasin, L. A., Proc. Natl. Acad. Sci. Estados Unidos (1980) 77, págs. 4126-4220) de células de ovario de hámster chino (células CHO; ATCC CCL-61); y similares, sin embargo, las células no están limitadas a estas.
El transformante así producido puede cultivarse según un procedimiento habitual, y cultivando el transformante, se produce un polipéptido diana intracelular o extracelularmente.
Para el cultivo se puede usar un medio adecuado seleccionado de varios medios de cultivo comúnmente usados dependiendo de las células hospedadoras empleadas. Si se emplea Escherichia coli, por ejemplo, se puede usar un medio LB complementado con un antibiótico, tal como ampicilina o IPMG, según sea necesario.
Una proteína recombinante producida intracelular o extracelularmente por el transformante a través de dicho cultivo puede separarse y purificarse mediante cualquiera de los diversos procedimientos de separación conocidos que utilizan la propiedad física o química de la proteína.
Los ejemplos específicos de los procedimientos incluyen el tratamiento con un precipitante proteico habitual, ultrafiltración, varios tipos de cromatografía líquida, tal como cromatografía de tamiz molecular (filtración en gel), cromatografía de adsorción, cromatografía de intercambio iónico y cromatografía de afinidad, diálisis y una combinación de los mismos.
Además, uniendo una etiqueta de seis restos de histidina a una proteína recombinante que se va a expresar, la proteína se puede purificar eficazmente con una columna de afinidad de níquel. Como alternativa, uniendo la región Fc de IgG a una proteína recombinante que se va a expresar, la proteína se puede purificar eficazmente con una columna de proteína A.
Al combinar los procedimientos descritos anteriormente, se puede producir fácilmente una gran cantidad de un
polipéptido diana con alto rendimiento y alta pureza.
(2) Producción de anticuerpo monoclonal anti-B7-H3
Como ejemplos de unión específica de anticuerpos a B7-H3 se incluyen una unión específica de anticuerpos monoclonales a B7-H3, y a continuación se describe un procedimiento para producir el anticuerpo.
La producción de un anticuerpo monoclonal generalmente requiere las siguientes etapas operativos de:
(a) purificar un biopolímero para su uso como antígeno;
(b) preparar células productoras de anticuerpo inmunizando a un animal mediante inyección del antígeno, recoger la sangre, analizar su título de anticuerpo para determinar cuándo se extirpa el bazo;
(c) preparar células de mieloma (en lo sucesivo "mieloma");
(d) fusionar las células productoras de anticuerpo con el mieloma;
(e) explorar un grupo de hibridomas que produce un anticuerpo deseado;
(f) dividir los hibridomas en clones de células individuales (clonación);
(g) opcionalmente, cultivar el hibridoma o criar un animal al que se la implantado el hibridoma para producir una gran cantidad de un anticuerpo monoclonal;
(h) examinar el anticuerpo monoclonal producido de este modo para determinar la actividad biológica y la especificidad de unión, o analizar las propiedades del mismo como un reactivo marcado; y similares.
En lo sucesivo, el procedimiento para producir un anticuerpo monoclonal se describirá en detalle siguiendo las etapas anteriores, sin embargo, el procedimiento no está limitado a esto, y, por ejemplo, se pueden usar células productoras de anticuerpos distintas de esplenocitos y de mieloma.
(a) Purificación del antígeno
Como antígeno, se puede usar B7-H3 preparado mediante el procedimiento descrito anteriormente o un péptido parcial del mismo.
Además, como antígeno también puede usarse una fracción de membrana preparada a partir de células recombinantes que expresan B7-H3 o de la propias células recombinantes que expresan B7-H3, y también un péptido parcial de la proteína de la invención sintetizado químicamente mediante un procedimiento conocido por los expertos en la técnica.
(b) Preparación de células productoras de anticuerpo
El antígeno producido en la etapa (a) se mezcla con un adyuvante tal como adyuvante completo o incompleto de Freund o sulfato de aluminio y potasio y la mezcla resultante se usa como inmunógeno para inmunizar a un animal de laboratorio. Como animal de laboratorio, puede usarse sin ningún problema cualquier animal que se utilice en un procedimiento de producción de hibridomas conocido. Específicamente, puede usarse, por ejemplo, un ratón, una rata, una cabra, una oveja, una vaca, un caballo o similar. Sin embargo, desde el punto de vista de la facilidad de la disponibilidad de las células de mieloma para fusionarse con las células productoras de anticuerpos extraídas, como animal a inmunizar se usa preferentemente un ratón o una rata.
Además, la cepa de un ratón o de una rata que se vaya a utilizar no está particularmente limitada, y en el caso de un ratón, pueden usarse varias cepas, tales como, por ejemplo, las cepas A, AKR, BALB/c, BDP, BA, CE, C3H, 57BL, C57BL, C57L, DBA, FL, HTH, HT1, LP, NZB, NZW, RF, R III, SJL, SWR, WB, 129 y similares, y en el caso de una rata, puede usarse, por ejemplo, las cepas Wistar, Low, Lewis, Sprague, Dawley, ACI, BN, Fischer y similares.
Estos ratones y ratas están disponibles en el comercio de criadores/distribuidores de animales de laboratorio, por ejemplo, CLEA Japón, Inc. y Charles River Laboratories Japón, Inc.
Entre estos, teniendo en cuenta la compatibilidad de la fusión con las células de mieloma descritas a continuación, como animal a inmunizar en el caso de un ratón, se prefiere la cepa BALB/c, y en el caso de una rata, las cepas Wistar y Low son las cepas particularmente preferidas.
Además, teniendo en cuenta la homología antigénica entre los seres humanos y los ratones, también se prefiere usar un ratón que tenga una función biológica disminuida para eliminar autoanticuerpos, es decir, un ratón con una enfermedad autoinmunitaria.
La edad de dicho ratón o rata en el momento de la inmunización varia, preferentemente, de 5 a 12 semanas de vida, más preferentemente de 6 a 8 semanas de vida.
Para inmunizar a un animal con B7-H3 o con un recombinante del mismo, puede usarse, por ejemplo, un procedimiento conocido descrito con detalle, por ejemplo, por Weir, D. M. en Handbook of Experimental Immunology Vol. I. II. III., Blackwell Scientific Publications, Oxford (1987), Kabat, E. A. y por Mayer, M. M., en Experimental Immunochemistry, Charles C Thomas Publisher Springfield, Illinois (1964) o similares.
Entre estos procedimientos de inmunización, un procedimiento específico preferido en la invención es, por ejemplo, el siguiente.
Es decir, en primer lugar, una fracción de proteína de membrana que sirve como el antígeno o las células causadas para expresar el antígeno, se administra(n) por vía intradérmica o intraperitoneal a un animal.
Sin embargo, se prefiere la combinación de ambas vías de administración para aumentar la eficacia de la inmunización, y cuando la administración intradérmica se realiza en la primera mitad y la administración intraperitoneal se realiza en la segunda mitad o solo en la última dosis, la eficacia de la inmunización puede aumentarse particularmente.
El programa de administración del antígeno varía según el tipo de animal que se vaya a inmunizar, diferencias individuales o similares. Sin embargo, en general, se prefiere un programa de administración en el que la frecuencia de administración del antígeno sea de 3 a 6 veces y el intervalo de dosificación sea de 2 a 6 semanas, y se prefiere más un programa de administración en el que la frecuencia de administración del antígeno sea de 3 a 4 veces y el intervalo de dosificación sea de 2 a 4 semanas.
Además, la dosis del antígeno varía según el tipo de animal, diferencias individuales o similares, sin embargo, generalmente, la dosis se ajusta a de 0,05 a 5 mg, preferentemente, de aproximadamente 0,1 a 0,5 mg.
Se realiza una inmunización de refuerzo de 1 a 6 semanas, preferentemente de 2 a 4 semanas, más preferentemente de 2 a 3 semanas después de la administración del antígeno como se ha descrito anteriormente.
La dosis del antígeno en el momento de realizar la inmunización de refuerzo varía según el tipo o tamaño del animal o similar, sin embargo, en el caso de, por ejemplo, un ratón, generalmente, la dosis se ajusta a de 0,05 a 5 mg, preferentemente, de 0,1 a 0,5 mg, más preferentemente de aproximadamente 0,1 a 0,2 mg.
Los esplenocitos o los linfocitos, incluidas las células productoras de anticuerpos, se eliminan asépticamente del animal inmunizado en un intervalo de 1 a 10 días, preferentemente de 2 a 5 días, más preferentemente de 2 a 3 días después de la inmunización de refuerzo. En este momento, se mide el título de anticuerpos, y si como fuente de suministro de células productoras de anticuerpos se usa un animal que tiene un título de anticuerpos suficientemente aumentado, el procedimiento posterior puede llevarse a cabo de manera más eficaz.
Como ejemplos del procedimiento para medir el título de anticuerpos que se utilizarán en el presente documento se incluyen un procedimiento RIA y un procedimiento ELISA, pero el procedimiento no se limita a los anteriores.
Por ejemplo, si se emplea un procedimiento ELISA, la medición del título de anticuerpos en la invención puede llevarse a cabo según los procedimientos que se describen a continuación.
En primer lugar, un antígeno purificado o parcialmente purificado, se adsorbe a la superficie de una fase sólida, tal como una placa de 96 pocillos de ensayo ELISA, y la superficie de la fase sólida que no tiene antígeno adsorbido en ella, se reviste con una proteína, tal como seroalbúmina bovina (en lo sucesivo "BSA", por las siglas del inglés bovine serum albumin), no relacionada con el antígeno. Después de lavar la superficie, dicha superficie se pone en contacto con una muestra diluida en serie (por ejemplo, suero de ratón) como anticuerpo primario para permitir que el anticuerpo en la muestra se una al antígeno.
Además, como un anticuerpo secundario, se añade un anticuerpo marcado con una enzima contra un anticuerpo de ratón y se deja que se una al anticuerpo de ratón. Después del lavado, se añade un sustrato para la enzima y se mide un cambio en la absorbancia que se produce debido al revelado del color inducido por la degradación del sustrato o similar y el título del anticuerpo se calcula basándose en la medición.
La separación de las células productoras de anticuerpos de los esplenocitos o de los linfocitos del animal inmunizado se puede llevar a cabo según un procedimiento conocido (por ejemplo, Kohler y col., Nature (1975), 256, pág. 495; Kohler y col., Eur. J. Immunol. (1977), 6, pág. 511; Milstein y col., Nature (1977), 266, pág. 550; Walsh, Nature (1977), 266, pág. 495). Por ejemplo, en el caso de los esplenocitos, se puede emplear un procedimiento general en el que las células productoras de anticuerpos se separan homogeneizando el bazo para producir las células mediante filtración con una malla de acero inoxidable y suspendiendo las células en medio esencial mínimo de Eagle (MEM).
(c) Preparación de células de mieloma (en lo sucesivo "mieloma")
Las células de mieloma que se utilizarán para la fusión celular no están particularmente limitadas y pueden seleccionarse células adecuadas de líneas celulares conocidas. Sin embargo, teniendo en cuenta la conveniencia cuando se selecciona un hibridoma de células fusionadas, se prefiere usar una cepa deficiente en HGPRT (hipoxantina-guanina fosforribosil transferasa) cuyo procedimiento de selección se haya establecido.
Más específicamente, como ejemplos de la cepa deficiente en HGPRT se incluyen X63-Ag8 (X63), NS1-ANS/1(NS1), P3X63-Ag8.U1(P3U1), X63-Ag8.653(X63.653), SP2/0-Ag14(SP2/0), MPC11-45.6TG1.7(45.6TG), FO, S149/5XXO y BU.1 procedentes de ratones; 210.RSY3.Ag.1.2.3(Y3) procedente de ratas; y U266AR (SKO-007), GM1500 GTG-A12(GM1500), UC729-6, LICR-LOW-HMy2(HMy2) y 8226AR/NIP4-1(NP41) procedentes de seres humanos. Estas cepas deficientes en HGPRT están disponibles, por ejemplo, en la American Type Culture Collection (ATCC) o similar.
Estas cepas celulares se subcultivan en un medio apropiado, tal como un medio de 8-azaguanina [un medio obtenido añadiendo 8-azaguanina a un medio RPMI 1640 complementado con glutamina, 2-mercaptoetanol, gentamicina y suero de ternera fetal (en lo sucesivo "FCS")], medio de Dulbecco modificado por Iscove (en lo sucesivo "IMDM ") o medio de Eagle modificado por Dulbecco (en lo sucesivo "DMEM"). En este caso, 3 a 4 días antes de realizar la fusión celular, las células se subcultivan en un medio normal [por ejemplo, un medio ASF104 (fabricado por Ajinomoto Co., Ltd.) que contiene FCS al 10 %] para garantizar que el día de la fusión celular haya una cantidad no inferior a 2 x 107 células.
(d) Fusión celular
La fusión entre las células productoras de anticuerpos y las células de mieloma se puede realizar adecuadamente según un procedimiento conocido (Weir, D. M Handbook of Experimental Immunology Vol. I. II. III., Blackwell Scientific Publications, Oxford (1987), Kabat, E. A. y Mayer, M. M., Experimental Immunochemistry, Charles C Thomas Publisher, Springfield, Illinois (1964), etc.), en condiciones tales que la tasa de supervivencia de las células no se reduzca excesivamente.
Como procedimiento de este tipo, puede usarse, por ejemplo, un procedimiento químico en el que las células productoras de anticuerpos y las células de mieloma se mezclan en una solución que contiene un polímero, tal como polietilenglicol, a una alta concentración, un procedimiento físico usando estimulación eléctrica, o similar. Entre estos procedimientos, un ejemplo específico del procedimiento químico es como se describe a continuación.
Es decir, en el caso en el que se use polietilenglicol en la solución que contiene un polímero a una alta concentración, las células productoras de anticuerpos y las células de mieloma se mezclan en una solución de polietilenglicol que tenga un peso molecular de 1500 a 6000, más preferentemente de 2000 a 4000 a una temperatura de 30 a 40° C, preferentemente de 35 a 38° C durante 1 a 10 minutos, preferentemente de 5 a 8 minutos.
(e) Selección de un grupo de hibridomas
El procedimiento de selección de hibridomas producidos por la fusión celular descrita anteriormente no está particularmente limitado. Habitualmente, se usa un procedimiento de selección HAT (hipoxantina, aminopterina, timidina) (Kohler y col., Nature (1975), 256, pág. 495; Milstein y col., Nature (1977), 266, pag. 550).
Este procedimiento es eficaz cuando se producen hibridomas usando las células de mieloma de una cepa deficiente en HGPRT que no puede sobrevivir en presencia de aminopterina.
Es decir, cultivando células e hibridomas no fusionados en un medio HAT, solo los hibridomas que son resistentes a la aminopterina pueden sobrevivir y proliferar selectivamente.
(f) División en clon unicelular (clonación)
Como procedimiento de clonación para hibridomas, puede usarse un procedimiento conocido tal como un procedimiento con metilcelulosa, un procedimiento con agarosa blanda o un procedimiento de dilución limitante (véase, por ejemplo, Barbara, B. M. y Stanley, M. S.: Selected Methods in Cellular Immunology, W. H. Freeman and Company, San Francisco (1980)). Entre estos procedimientos, se prefiere, particularmente, un procedimiento de cultivo tridimensional, tal como un procedimiento con metilcelulosa. Por ejemplo, el grupo de hibridomas que se produce mediante fusión celular se suspende en un medio con metilcelulosa, tal como medio de selección D de ClonaCell-HY (fabricado por StemCell Technologies, Inc., n.° 03804) y se cultiva. Después, las colonias de hibridoma formadas se recogen, por lo que pueden producirse hibridomas monoclonales. Las colonias de hibridoma respectivas recogidas se cultivan, y un hibridoma que se ha confirmado que tiene un título de anticuerpo estable en un sobrenadante de cultivo de hibridoma producido, se selecciona como una cepa de hibridoma productora de anticuerpos monoclonales B7-H3.
Como ejemplos de la cepa de hibridoma establecida de esta manera se incluyen el hibridoma M30 de B7-H3. En la presente memoria descriptiva, un anticuerpo producido por el hibridoma M30 de B7-H3 se denomina "anticuerpo M30" o simplemente "M30".
La cadena pesada del anticuerpo M30 tiene una secuencia de aminoácidos representada por la SEQ ID NO: 20 en el Listado de secuencias. Además, la cadena ligera del anticuerpo M30 tiene una secuencia de aminoácidos representada por la SEQ ID NO: 21 en el Listado de secuencias. En la secuencia de aminoácidos de la cadena pesada representada por la SEQ ID NO: 20 en el Listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácidos 1 a 19 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 es una región variable y una secuencia de aminoácidos que consiste en los restos de aminoácidos 142 a 471 es una región constante. Además, en la secuencia de aminoácidos de la cadena ligera representada por la SEQ ID NO: 21 en el Listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácidos 1 a 22 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácidos 23 a 130 es una región variable, y una secuencia de aminoácidos que consiste en los restos de aminoácidos 131 a 235 es una región constante.
(g) Preparación de anticuerpo monoclonal mediante cultivo de hibridoma
Al cultivar el hibridoma seleccionado de esta manera, se puede producir un anticuerpo monoclonal de manera eficaz. Sin embargo, antes de cultivar, se prefiere realizar la exploración de un hibridoma que produzca un anticuerpo monoclonal diana.
En dicha exploración, puede emplearse un procedimiento conocido.
La medición del título de anticuerpos en la invención puede llevarse a cabo, por ejemplo, mediante un procedimiento ELISA explicado en el apartado (b) descrito anteriormente.
El hibridoma producido por el procedimiento descrito anteriormente puede conservarse en estado congelado en nitrógeno líquido o en un congelador a una temperatura de -80° C o inferior.
Después de finalizar la clonación, el medio se cambia de un medio HT a un medio normal, y se cultiva el hibridoma.
El cultivo a gran escala se realiza mediante cultivo de rotación usando un frasco de cultivo grande o mediante cultivo giratorio. A partir del sobrenadante producido por el cultivo a gran escala, puede producirse un anticuerpo monoclonal que se une específicamente a la proteína de la invención mediante purificación usando un procedimiento conocido por los expertos en la técnica, tal como filtración en gel.
Además, para que el hibridoma prolifere, este se inyecta en la cavidad abdominal de un ratón de la misma cepa que la del hibridoma (por ejemplo, el ratón BALB/c descrito anteriormente) o de un ratón Nu/Nu, por lo que se puede producir la ascitis que contiene una gran cantidad del anticuerpo monoclonal de la invención.
En el caso de que el hibridoma se administre en la cavidad abdominal, si en un periodo de tiempo de 3 a 7 días antes de esto se administra un aceite mineral, tal como 2,6,10,14-tetrametil pentadecano (pristano), se puede producir una mayor cantidad de ascitis.
Por ejemplo, para inactivar los linfocitos T, previamente se inyecta un inmunosupresor en la cavidad abdominal de un ratón de la misma cepa que la del hibridoma. Después de 20 días, en un medio sin suero (0,5 ml), se suspenden de 106 a 107 células clonadas de hibridoma y la suspensión se administra en la cavidad abdominal del ratón. En general, cuando el abdomen se dilata y se llena con la ascitis, la ascitis se extrae del ratón. Mediante este procedimiento, el anticuerpo monoclonal puede producirse a una concentración que es aproximadamente 100 veces o mucho más alta que la de la solución de cultivo.
El anticuerpo monoclonal producido mediante el procedimiento descrito anteriormente se puede purificar mediante un procedimiento descrito, por ejemplo, en Weir, D. M.: Handbook of Experimental Immunology Vol. I, II, III, Blackwell Scientific Publications, Oxford (1978).
El anticuerpo monoclonal producido de esta manera tiene alta especificidad antigénica por B7-H3.
(h) Ensayo de anticuerpo monoclonal
El isotipo y la subclase del anticuerpo monoclonal así producido puede determinarse de la siguiente manera.
En primer lugar, como ejemplos del procedimiento de identificación se incluyen un procedimiento de Ouchterlony, un procedimiento de ELISA y un procedimiento de RIA.
Un procedimiento de Ouchterlony es sencillo, pero cuando la concentración del anticuerpo monoclonal es baja, se requiere una operación de condensación.
Por otra parte, cuando se usa un procedimiento de ELISA o RIA, el isotipo y la subclase del anticuerpo monoclonal pueden identificarse haciendo reaccionar directamente el sobrenadante de cultivo con una fase sólida adsorbida en antígeno y usando, como anticuerpos secundarios, anticuerpos correspondientes contra diversos tipos de isotipos y subclases de inmunoglobulina.
Además, como un procedimiento más sencillo, puede usarse un kit de identificación disponible en el comercio (por ejemplo, Kit Mouse Typer fabricado por Bio-Rad Laboratories, Inc.) o similar.
Además, la determinación cuantitativa de una proteína puede realizarse mediante el procedimiento de Folin Lowry y un procedimiento de cálculo basado en la absorbancia a 280 nm [1,4 (DO 280) = Inmunoglobulina 1 mg/ml].
Además, incluso cuando el anticuerpo monoclonal se produce por separado e independientemente realizando nuevamente las etapas (a) a (h) del apartado (2), es posible producir un anticuerpo que tenga una actividad citotóxica equivalente a la del anticuerpo M30. Como un ejemplo de dicho anticuerpo, puede ilustrarse un anticuerpo que se une al mismo epítopo que el anticuerpo M30. El M30 reconoce un epítopo en el dominio IgC1 o IgC2, que es un dominio en el dominio extracelular de B7-H3, y se une al dominio IgC1 o al dominio IgC2 o a ambos. Por lo tanto, como epítopo para el anticuerpo de la invención, en particular, puede ilustrarse un epítopo presente en el dominio IgC1 o IgC2 de B7-H3. Si un anticuerpo monoclonal recién producido se une a un péptido parcial o a una estructura terciaria parcial a la que se une el anticuerpo M30, se puede determinar que el anticuerpo monoclonal se une al mismo epítopo que el
anticuerpo M30. Además, al confirmar que el anticuerpo monoclonal compite con el anticuerpo M30 por la unión a B7-H3 (es decir, el anticuerpo monoclonal inhibe la unión entre el anticuerpo M30 y B7-H3), se puede determinar que el anticuerpo monoclonal se une al mismo epítopo que el anticuerpo M30 incluso si no se ha determinado la secuencia o estructura específica del epítopo. Cuando se confirma que el anticuerpo monoclonal se une al mismo epítopo que el anticuerpo M30, se espera del todo que el anticuerpo monoclonal tenga una actividad citotóxica equivalente a la del anticuerpo M30.
(3) Otros anticuerpos
El anticuerpo de la invención no solo incluye el anticuerpo monoclonal descrito anteriormente contra B7-H3 sino también un anticuerpo recombinante producido mediante modificación artificial con el fin de disminuir la antigenicidad heteróloga contra humanos, tal como un anticuerpo quimérico, un anticuerpo humanizado y un anticuerpo humano. Estos anticuerpos se pueden producir utilizando un procedimiento conocido.
Como anticuerpo quimérico, puede ilustrarse un anticuerpo en el que las regiones variables y constantes del anticuerpo proceden de diferentes especies, por ejemplo, un anticuerpo quimérico en el que una región variable de anticuerpo procedente de ratón o rata está conectada a una región constante de anticuerpo procedente de ser humano (véase Proc. Natl. Acad. Sci. USA, 81,6851-6855, (1984)).
Como anticuerpo humanizado, puede ilustrarse un anticuerpo producido integrando solo una región determinante de complementariedad (CDR) en un anticuerpo procedente de ser humano (véase Nature (1986) 321, págs. 522-525), y puede ilustrarse un anticuerpo producido injertando una parte de los restos de aminoácidos de la secuencia estructural así como de la secuencia de CDR a un anticuerpo humano mediante un procedimiento de injertación de CDR (documento WO 90/07861).
Sin embargo, el anticuerpo humanizado procedente del anticuerpo M30 no está limitado a un anticuerpo humanizado específico siempre que el anticuerpo humanizado tenga los 6 tipos de secuencias CDR del anticuerpo M30 y tenga una actividad antitumoral. La región variable de la cadena pesada del anticuerpo M30 tiene CDRH1 (NYVMH) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 3 en el Listado de secuencias, CDRH2 (YINPYNDDVKYNEKFKG) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 4 en el Listado de Secuencias, y CDRH3 (WGYYGSPLYYFDY) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 5 en el Listado de Secuencias. Además, la región variable de la cadena ligera del anticuerpo M30 tiene CDRL1 (RASSRLIYMH) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 6 en el Listado de secuencias, CDRL2 (ATSNLAS) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 7 en el Listado de secuencias, y CDRL3 (QQWNSNPPT) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 8 en el Listado de secuencias.
Como un ejemplo del anticuerpo humanizado de un anticuerpo de ratón M30, puede ilustrase una combinación arbitraria de una cadena pesada que comprende una región variable de la cadena pesada que consiste en una cualquiera de (1) una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de las SEQ ID NO: 9, 10, 11 o 12 en el Listado de secuencias, (2) una secuencia de aminoácidos que tiene una homología de al menos el 95% o mayor con la secuencia de aminoácidos (1) descrita anteriormente, y (3) una secuencia de aminoácidos en la que uno o varios aminoácidos en la secuencia de aminoácidos (1) descrita anteriormente, se delecionan, sustituyen o añaden, y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una cualquiera de (4) una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 128 de las SEQ ID NO: 13, 14, 15, 16, 17, 18 o 19 en el Listado de Secuencias, (5) una secuencia de aminoácidos que tiene una homología de al menos el 95 % o mayor con la secuencia de aminoácidos (4) descrita anteriormente, y (6) una secuencia de aminoácidos en la que uno o varios aminoácidos en la secuencia de aminoácidos (4) descrita anteriormente se delecionan, sustituyen o añaden.
El término "varios" como se usa en el presente documento, se refiere a de 1 a 10, de 1 a 9, de 1 a 8, de 1 a 7, de 1 a 6, de 1 a 5, de 1 a 4, de 1 a 3 o 1 o 2.
En esta memoria descriptiva como sustitución de aminoácidos se prefiere una sustitución conservativa de aminoácidos. La sustitución conservativa de aminoácidos se refiere a una sustitución que se produce dentro de un grupo de aminoácidos relacionados con cadenas laterales de aminoácidos. Los grupos de aminoácidos preferidos son los siguientes: un grupo ácido (ácido aspártico y ácido glutámico); un grupo básico (lisina, arginina e histidina); un grupo no polar (alanina, valina, leucina, isoleucina, prolina, fenilalanina, metionina y triptófano); y una familia polar sin carga (glicina, asparagina, glutamina, cisteína, serina, treonina y tirosina). Los grupos de aminoácidos más preferidos son los siguientes: un grupo hidroxilo alifático (serina y treonina); un grupo que contiene amida (asparagina y glutamina); un grupo alifático (alanina, valina, leucina e isoleucina); y un grupo aromático (fenilalanina, triptófano y tirosina). Dicha sustitución de aminoácidos se realiza preferentemente dentro de un intervalo que no perjudica las propiedades de una sustancia que tiene la secuencia de aminoácidos original.
Como un anticuerpo que tiene una combinación preferida de una cadena pesada y una cadena ligera descritas anteriormente, puede ilustrarse, un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a
141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 15; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 16; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 17; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 18; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 19; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste de los restos de aminoácidos 21 a 128 de la SEQ ID NO: 15; y un anticuerpo que consiste en una cadena pesada que comprende una región variable de la cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de la cadena ligera que consiste en una secuencia de aminoácidos que consiste en restos de aminoácidos 21 a 128 de la SEQ ID NO: 16.
Además, como un anticuerpo que tiene una combinación más preferida de una cadena pesada y una cadena ligera descritas anteriormente, puede ilustrarse, un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 15; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 16; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la s Eq ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 17; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 18; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 19; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 14; un anticuerpo que consiste en
una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la s Eq ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEQ ID NO: 15; y un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 471 de la SEC ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 233 de la SEC ID NO: 16.
Adicionalmente, como un anticuerpo que tiene otra combinación más preferida de una cadena pesada y una cadena ligera descritas anteriormente, puede ilustrarse, un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 15; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 16; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 17; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 18; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 19; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de SEQ ID NO: 15; y un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 16.
Combinando una secuencia que tiene una alta homología con la secuencia de aminoácidos de cadena pesada descrita anteriormente, con una secuencia que tiene una alta homología con la secuencia de aminoácidos de cadena ligera descrita anteriormente, es posible seleccionar un anticuerpo que tenga una actividad citotóxica equivalente a la de cada uno de los anticuerpos descritos anteriormente. Dicha homología es, generalmente, una homología del 80 % o mayor, preferentemente una homología del 90 % o mayor, más preferentemente una homología del 95 % o mayor, más preferentemente una homología del 99 % o mayor. Además, combinando una secuencia de aminoácidos en la que en la secuencia de aminoácidos de la cadena pesada o de la cadena ligera se sustituyen, delecionan o añaden de uno a varios restos de aminoácidos, también es posible seleccionar un anticuerpo que tenga una actividad citotóxica equivalente a la de cada uno de los anticuerpos descritos anteriormente.
La homología entre dos secuencias de aminoácidos puede determinarse utilizando los parámetros predeterminados del algoritmo Blast versión 2.2.2 (Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller y David J. Lipman (1997), "Gap BLAST y PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25: 3389-3402). El algoritmo Blast puede usarse también a través de Internet accediendo al sitio www.ncbi.nlm.nih.gov/blast.
En la secuencia de aminoácidos de la cadena pesada representada por las SEQ ID NO: 9, 10, 11 o 12 en el Listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácidos 1 a 19 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácidos 20 a 141 es una región variable y una secuencia de aminoácidos que consiste en los restos de aminoácidos 142 a 471 es una región constante. En las figuras 3, 4, 5 y 6, se muestran las secuencias de las SEQ ID NO: 9, 10, 11 y 12, respectivamente. Además, en la secuencia de aminoácidos de la cadena ligera representada por las SEQ ID NO: 13, 14, 15, 16, 17, 18 o 19 en el Listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácidos 1 a 20 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácidos 21 a 128 es una región variable y una secuencia de aminoácidos que consiste en los restos de aminoácidos 129 a 233 es una región constante. En las figuras 7, 8, 9, 10, 11, 12 y 13 se muestra la secuencia de las SEQ ID NO: 13, 14, 15, 16, 17, 18 y 19, respectivamente.
Además, el anticuerpo de la invención incluye un anticuerpo humano que se une al mismo epítopo que el anticuerpo M30. Un anticuerpo humano anti-B7-H3 se refiere a un anticuerpo humano que solo tiene una secuencia de un anticuerpo procedente de un cromosoma humano. El anticuerpo humano anti-B7-H3 puede producirse mediante un procedimiento que utiliza un ratón productor de anticuerpos humanos que tiene un fragmento de cromosoma humano que comprende genes de cadena pesada y ligera de un anticuerpo humano (véase Tomizuka, K. y col., Nature Genetics (1997) 16, págs. 133-143; Kuroiwa, Y. y col., Nucl. Acids Res. (1998) 26, págs. 3447-3448; Yoshida, H. y col., Animal Cell Technology: Basic and Applied Aspects vol. 10, págs. 69-73 (Kitagawa, Y., Matuda, T. e Iijima, S. eds.), Kluwer Academic Publishers, 1999; Tomizuka, K. y col., Proc. Natl. Acad. Sci. Estados Unidos (2000) 97, págs.
722-727, etc.).
Dicho ratón productor de anticuerpos humanos puede crearse específicamente de la siguiente manera. Un animal modificado genéticamente, en el que se han alterado los locus de los genes de cadenas pesadas y ligeras de inmunoglobulinas endógenas, y en su lugar se han introducido locus de genes de cadenas pesadas y ligeras de inmunoglobulinas humanas a través de un vector de cromosoma artificial de levadura (YAC, por las siglas del inglés yeast artificial chromosome) o similar, se crea produciendo un animal genosuprimido y un animal transgénico y emparejando estos animales.
Además, según una técnica de ADN recombinante, usando los ADNc que codifican cada una de dichas cadenas pesadas y ligeras de un anticuerpo humano, y preferentemente, un vector que comprende dichos ADNc, se transforman células eucariotas y se cultiva una célula transformada que produce un anticuerpo monoclonal humano recombinante, por lo que el anticuerpo también se puede obtener a partir del sobrenadante de cultivo.
En este caso, como hospedador puede usarse, por ejemplo, células eucariotas, preferentemente células de mamífero, tales como células CHO, linfocitos o células de mieloma.
Además, también se conoce un procedimiento para producir un anticuerpo humano procedente de la presentación en fagos, seleccionado de una biblioteca de anticuerpos humanos (véase Wormstone, I.M. y col., Investigative Ophthalmology & Visual Science. (2002) 43 (7), págs. 2301-2308; Carmen S. y col., Briefings in Functional Genomics and Proteomics (2002), 1 (2), págs. 189-203; Siriwardena D. y col., Ophthalmology (2002) 109 (3), págs. 427-431, etc.).
Por ejemplo, puede utilizarse un procedimiento de presentación en fagos en el que una región variable de un anticuerpo humano se expresa en la superficie de un fago como un anticuerpo monocatenario (scFv), y se selecciona un fago que se une a un antígeno (Nature Biotechnology (2005), 23, (9), págs. 1105-1116).
Analizando el gen del fago seleccionado basándose en la unión a un antígeno, se puede determinar una secuencia de ADN que codifica la región variable de un anticuerpo humano que se une a un antígeno.
Si se determina la secuencia de ADN de scFv que se une a un antígeno, se puede obtener un anticuerpo humano preparando un vector de expresión que comprenda la secuencia e introducir el vector en un hospedador apropiado para expresarlo (documentos WO 92/01047, WO 92/20791, WO 93/06213, WO 93/11236, WO 93/19172, WO 95/01438, WO 95/15388, Annu. Rev. Immunol. (1994) 12, págs. 433-455, Nature Biotechnology (2005) 23 (9), págs.
1105-1116).
Si un anticuerpo humano recién producido se une a un péptido parcial o a una estructura terciaria parcial con la que se une el anticuerpo M30, se puede determinar que el anticuerpo humano se une al mismo epítopo que el anticuerpo M30. Además, confirmando que el anticuerpo humano compite con el anticuerpo M30 por la unión a B7-H3 (es decir, el anticuerpo humano inhibe la unión entre el anticuerpo M30 y B7-H3), se puede determinar que el anticuerpo humano se une al mismo epítopo que el anticuerpo M30 incluso si no se ha determinado la secuencia o estructura específica del epítopo. Cuando se confirma que el anticuerpo humano se une al mismo epítopo que el anticuerpo M30, se espera del todo que el anticuerpo humano tenga una actividad citotóxica equivalente a la del anticuerpo m 30.
Los anticuerpos quiméricos, los anticuerpos humanizados o los anticuerpos humanos producidos por el procedimiento descrito anteriormente, se evalúan en cuanto a la propiedad de unión a un antígeno mediante un procedimiento conocido o similar, y se puede seleccionar un anticuerpo preferido.
Como un ejemplo de otro índice para su uso en la comparación de las propiedades de los anticuerpos, puede ilustrarse la estabilidad de los anticuerpos. La calorimetría de barrido diferencial (DSC, por las siglas del inglés differential scanning calorimetry) es un dispositivo capaz de medir de forma rápida y precisa una temperatura de punto medio de desnaturalización térmica (Tm) para su uso como un índice favorable de la estabilidad conformacional relativa de las proteínas. Midiendo los valores de Tm usando DSC y comparando los valores, se puede comparar una diferencia en la estabilidad térmica. Se sabe que la estabilidad de almacenamiento de los anticuerpos muestra cierta correlación con la estabilidad térmica de los anticuerpos (Lori Burton, y col., Pharmaceutical Development and Technology (2007) 12, págs. 265-273), y usando la estabilidad térmica como índice, puede seleccionarse un anticuerpo preferido. Los ejemplos de otros índices para seleccionar anticuerpos incluyen las siguientes características: el rendimiento en una célula hospedadora apropiada es alto; y la capacidad de agregación en una solución acuosa es baja. Por ejemplo, un anticuerpo que muestra el mayor rendimiento no siempre muestra la mayor estabilidad térmica y, por lo tanto, es necesario seleccionar el anticuerpo que sea el más adecuado para la administración a seres humanos haciendo una evaluación exhaustiva basada en los índices descritos anteriormente.
En la invención, también se incluye una variante modificada del anticuerpo. La variante modificada se refiere a una variante producida sometiendo el anticuerpo de la invención a modificación química o biológica. Como ejemplos de variante modificada químicamente se incluyen variantes modificadas químicamente uniendo una fracción química con un esqueleto de aminoácidos, variantes modificadas químicamente con una cadena de hidratos de carbono ligada a N o ligada a O, etc. Como ejemplos de variante modificada biológicamente se incluyen variantes producidas por modificación postraduccional (tal como glucosilación ligada a N o ligada a O, procesamiento de extremo terminal N o C, desamidación, isomerización de ácido aspártico u oxidación de metionina), y variantes en las que se ha añadido un resto de metionina al extremo N al expresarse en una célula hospedadora procariota.
Además, en el significado de variante modificada, también se incluye un anticuerpo marcado para permitir la detección o el aislamiento del anticuerpo o un antígeno de la invención, por ejemplo, un anticuerpo marcado con enzima, un anticuerpo marcado con fluorescencia y un anticuerpo con afinidad marcado. Dicha variante modificada del anticuerpo de la invención es útil para mejorar la estabilidad y retención en sangre del anticuerpo original de la invención, reducir su antigenicidad, detectar o aislar dicho anticuerpo o antígeno, y etcétera.
Además, regulando la modificación de un glucano que está ligado al anticuerpo de la invención (glucosilación, desfucosilación, etc.), Es posible potenciar una actividad citotóxica celular dependiente de anticuerpos. Como técnica para regular la modificación de un glucano de los anticuerpos, se conoce la indicada en los documentos WO 99/54342, WO 00/61739, WO 02/31140, etc. Sin embargo, la técnica no se limita a esta. En el anticuerpo de la invención, también se incluye un anticuerpo en el que se regula la modificación de un glucano.
En el caso en el que se produzca un anticuerpo aislando primero un gen de anticuerpo y después introduciendo el gen en un hospedador apropiado, se puede usar una combinación de un hospedador apropiado y un vector de expresión apropiado. Los ejemplos específicos de gen del anticuerpo incluyen una combinación de un gen que codifica una secuencia de cadena pesada de un anticuerpo descrito en esta memoria descriptiva y un gen que codifica una secuencia de cadena ligera del mismo. Cuando se transforma una célula hospedadora, es posible insertar el gen de secuencia de cadena pesada y el gen de secuencia de cadena ligera en el mismo vector de expresión, y también por separado en diferentes vectores de expresión.
En el caso en el que como hospedador se utilicen células eucariotas, pueden usarse células animales, vegetales y microorganismos eucariotas. Son ejemplos de células animales, células de mamífero, por ejemplo, células COS de simio (Gluzman, Y., Cell, (1981) 23, págs. 175-182, ATCC CRL-1650), fibroblastos murinos NIH3T3 (ATCC N.° CRL-1658) y cepas deficientes en dihidrofolato reductasa (Urlaub, G. y Chasin, L. A., Proc. Natl. Acad. Sci. Estados Unidos (1980) 77, págs. 4126-4220) de células de ovario de hámster chino (células CHO; ATCC CCL-61).
En el caso en el que se utilicen células procariotas, pueden ilustrase, como ejemplos, Escherichia co liy Bacillus subtilis.
El anticuerpo puede obtenerse introduciendo un gen de anticuerpo deseado en estas células a través de la transformación y cultivando las células así transformadas in vitro. En el procedimiento de cultivo descrito anteriormente, algunas veces, el rendimiento puede variar dependiendo de la secuencia del anticuerpo y, por lo tanto, es posible seleccionar un anticuerpo que se produzca fácilmente como un producto farmacéutico utilizando el rendimiento como un índice entre los anticuerpos que tienen una actividad de unión equivalente. Por lo tanto, en el anticuerpo de la invención, también se incluye un anticuerpo producido mediante un procedimiento para producir un anticuerpo, caracterizado porque se incluye una etapa de cultivo de la célula hospedadora transformada y una etapa de recogida de un anticuerpo deseado de un producto cultivado producido en la etapa de cultivo.
Se sabe que en el extremo carboxilo de la cadena pesada de un anticuerpo producido en una célula de mamífero cultivada se deleciona un resto de lisina (Journal of Chromatography A, 705: 129-134 (1995)) y también se sabe que en el extremo carboxilo de la cadena pesada de un anticuerpo producido en una célula de mamífero cultivada dos restos de aminoácidos (glicina y lisina) se delecionan y que se amida un resto de prolina recién ubicado en el extremo carboxilo (Analytical Biochemistry, 360: 75-83 (2007)). Sin embargo, dicha deleción y modificación de la secuencia de la cadena pesada no afecta a la afinidad de unión al antígeno y a la función efectora (la activación de un complemento, la citotoxicidad celular dependiente de anticuerpos, etc.) del anticuerpo. Por lo tanto, en la invención, también se incluye un anticuerpo sometido a dicha modificación, y puede ilustrarse una variante de deleción en la que en el extremo carboxilo de la cadena pesada se han eliminado uno o dos aminoácidos, una variante producida por la amidación de la variante de deleción (por ejemplo, una cadena pesada en la que se ha amidado el resto de prolina del extremo carboxilo), y similares. El tipo de variante de deleción que tiene una deleción en el extremo carboxilo de la cadena pesada del anticuerpo según la invención no se limita a las variantes anteriores siempre que se conserve la afinidad de unión al antígeno y la función efectora. Las dos cadenas pesadas que constituyen el anticuerpo según la invención pueden ser de un tipo seleccionado del grupo que consiste en una cadena pesada de longitud completa y la variante de deleción descrita anteriormente, o pueden ser de dos tipos en combinación seleccionados de las mismas. La proporción de la cantidad de cada variante de deleción puede verse afectada por el tipo de células de mamífero cultivadas que producen el anticuerpo según la invención y las condiciones de cultivo, sin embargo, puede ilustrarse un caso en el que en el extremo carboxilo un resto de aminoácido se ha delecionado en las dos cadenas pesadas contenidas como componentes principales en el anticuerpo según la invención.
Como isotipo del anticuerpo de la invención, puede ilustrarse, por ejemplo, IgG (IgG1, IgG2, IgG3, IgG4) y, preferentemente, IgG1 o IgG2.
Como la función del anticuerpo, puede ilustrarse, generalmente, una actividad de unión a antígeno, una actividad de neutralizar la actividad de un antígeno, una actividad de mejorar la actividad de un antígeno, una actividad de citotoxicidad celular dependiente de anticuerpos (CCDA) y una actividad de citotoxicidad dependiente del complemento (CDC). La función del anticuerpo de la invención es una actividad de unión a B7-H3, preferentemente una actividad de fagocitosis celular dependiente de anticuerpos (FCDA), más preferentemente, una actividad de citotoxicidad (actividad antitumoral) contra células tumorales mediada por una actividad FCDA. Además, además de una actividad FCDA, el anticuerpo de la invención puede tener una actividad CCDA y/o una actividad CDC.
El anticuerpo producido puede purificarse hasta la homogeneidad. La separación y purificación del anticuerpo se puede realizar empleando un procedimiento convencional de separación y purificación de proteínas. Por ejemplo, el anticuerpo se puede separar y purificar seleccionando y combinando apropiadamente las siguientes estrategias, cromatografía en columna, filtración con filtro, ultrafiltración, precipitación salina, diálisis, electroforesis preparativa en gel de poliacrilamida, electroforesis de isoelectroenfoque y similares (Strategies for Protein Purification and Characterization: A Laboratory Course Manual, Daniel R. Marshak y col. eds., Cold Spring Harbor Laboratory Press (1996); Antibodies: A Laboratory Manual. Ed Harlow y David Lane, Laboratorio Cold Spring Harbor (1988)), pero el procedimiento no se limita a los anteriores.
Los ejemplos de dicha cromatografía incluyen cromatografía de afinidad, cromatografía de intercambio de iones, cromatografía hidrófoba, cromatografía de filtración en gel, cromatografía de fase inversa y cromatografía de adsorción.
Dicha cromatografía se puede realizar empleando cromatografía líquida tal como HPLC o FPLC.
Como columna para su uso en cromatografía de afinidad, se puede ilustrar una columna de Proteína A y una columna de Proteína G. Por ejemplo, como una columna que utiliza una columna de proteína A, pueden ilustrarse Hyper D, POROS, Sepharose Ff (Pharmacia) y similares.
Además, utilizando un vehículo que tenga un antígeno inmovilizado sobre el mismo, el anticuerpo también se puede purificar utilizando la propiedad de unión del anticuerpo con el antígeno.
[Compuesto antitumoral]
Se explica el compuesto antitumoral que se conjuga con el conjugado de anticuerpo y fármaco de la presente invención. El compuesto antitumoral es un compuesto que tiene un efecto antitumoral y un grupo sustituyente o una estructura parcial que permite conectarse a una estructura enlazadora. Cuando un conector parcial o completo se escinde en las células tumorales, la fracción del compuesto antitumoral se libera para mostrar el efecto antitumoral del compuesto antitumoral. A medida que el enlazador se escinde en una posición de conexión con el fármaco, el compuesto antitumoral se libera en su estructura intrínseca para mostrar su efecto antitumoral intrínseco.
En el conjugado de anticuerpo y fármaco de la presente invención, se usa exatecán como un derivado de la camptotecina (((1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H, 12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolina-10,13(9H,15H)-diona; mostrado en la siguiente fórmula).
[Fórmula 29]

Aunque tiene un excelente efecto antitumoral, exatecán no se ha comercializado como un fármaco antitumoral. El compuesto se puede obtener fácilmente mediante un procedimiento conocido y el grupo amino en la posición 1 se puede usar, preferentemente, como una posición de conexión con la estructura enlazadora. Además, aunque exatecán también puede liberarse en las células tumorales al mismo tiempo que parte del enlazador sigue unida al mismo, es un compuesto excelente que incluso en este caso muestra un excelente efecto antitumoral.
Con respecto al conjugado de anticuerpo y fármaco, la cantidad de moléculas conjugadas con fármaco por molécula de anticuerpo es un factor clave que influye en la eficacia y la seguridad. La producción del conjugado de anticuerpo y fármaco se realiza definiendo las condiciones de reacción incluyendo las cantidades de uso de materias primas y reactivos para la reacción para tener un número constante de moléculas conjugadas con fármaco, a diferencia de la reacción química de un compuesto de bajo peso molecular, generalmente se obtiene una mezcla que contiene diferentes cantidades de moléculas conjugadas con fármaco. La cantidad de fármacos conjugados en una molécula de anticuerpo se expresa o especifica mediante el valor promedio, es decir, el número promedio de moléculas conjugadas con fármaco. A menos que se describa específicamente lo contrario como un principio, la cantidad de moléculas conjugadas con fármaco significa un valor promedio, excepto en un caso en el que representa un conjugado de anticuerpo y fármaco que tiene una cantidad específica de moléculas conjugadas con fármaco que se incluye en una mezcla de conjugado de anticuerpo y fármaco que tiene una cantidad diferente de moléculas conjugadas con fármaco. La cantidad de moléculas de exatecán conjugadas con una molécula de anticuerpo es controlable y, como un número promedio de moléculas conjugadas con fármaco por anticuerpo, se pueden unir de 1 a 10 exatecanos.
Preferentemente, es de 2 a 8, y más preferentemente de 3 a 8. Por otra parte, basándose en la descripción de los Ejemplos de la presente solicitud, un experto en la técnica puede diseñar una reacción para conjugar un número requerido de moléculas de fármaco con una molécula de anticuerpo y puede obtener un anticuerpo conjugado con un número controlado de moléculas de exatecán.
Dado que el exatecán tiene una estructura de camptotecina, se sabe que en un medio ácido acuoso (por ejemplo, que tenga un pH de 3 o similar), el equilibrio cambia a una estructura con un anillo de lactona cerrado (anillo cerrado) pero cambia a una estructura con un anillo de lactona abierto (anillo abierto) en un medio básico acuoso (por ejemplo, que tenga un pH de 10 o similar). También se espera que al introducir un conjugado de fármaco con un resto de exatecán correspondiente a la estructura de anillo cerrado y a la estructura de anillo abierto, tenga el mismo efecto antitumoral y no hace falta decir que ninguno de ellos está dentro del ámbito de la presente invención.
[Estructura de engarce]
1. Engarce que tiene estructura hidrófila
El conjugado de anticuerpo y fármaco de la presente invención se caracteriza por una estructura de engarce en la que se forma un resto de estructura hidrófila.
El número conectado promedio del resto de estructura de fármaco y engarce por anticuerpo puede ser de 1 a 10. Preferentemente, es de 2 a 8, y más preferentemente de 3 a 8. Los ejemplos preferidos del resto de estructura de fármaco y engarce pueden incluir los siguientes.
Los ejemplos del resto de estructura de fármaco y engarce para conjugar el resto de estructura de fármaco y engarce al anticuerpo a través de un enlace de tioéter pueden incluir las siguientes fórmulas:
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX)
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
o las siguientes fórmulas:
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX)
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-(NH-DX)
Entre ellas, adicionalmente se representa preferentemente mediante las siguientes fórmulas:
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX)
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
o las siguientes fórmulas:
- (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX)
- (Succinimid-3-il-N)-CH(CH2CH2-COOH)-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX) - (Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
[Procedimiento de producción]
A continuación, se dan explicaciones para el procedimiento representativo para la producción del conjugado de anticuerpo y fármaco de la presente invención o un intermedio de producción del mismo. Entre tanto, los compuestos se describen más adelante en el presente documento con el número mostrado en cada fórmula de reacción. Específicamente, se denominan "compuesto de fórmula (1)", un "compuesto (1)", o similares. Los compuestos con números distintos de estos también se describen e un modo similar.
1. Procedimiento de producción 1
El conjugado de anticuerpo y fármaco representado por la fórmula (1) en la que el anticuerpo está conectado a la estructura de engarce a través de un tioéter puede producirse por el siguiente procedimiento, por ejemplo.
[Fórmula 34]

En la fórmula, AB representa un anticuerpo con un grupo sulfhidrilo, y L1’ representa una estructura de engarce L1 en la que el terminal del engarce tiene un grupo maleimidilo (fórmula mostrada más adelante) (en la fórmula, el átomo de nitrógeno es la posición de conexión)
[Fórmula 35]

o el terminal tiene un halógeno, y representa un grupo en el que el resto -(Succinimid-3-il-N)- en -(Succinimid-3-N-N)-(CH2 )n2-C(=O)- entre L1 es un grupo maleimidilo o un grupo halógeno-CH2C(=O)NH-(CH2)n3-C(=O)- en el que el terminal metileno en -CH2C(=O)NH-(CH2 )n4-C(=O)- entre L1 se halogena para formar haloacetamida. Además, el -(NH-DX) representa una estructura representada por la siguiente fórmula:
[Fórmula 36]

y representa un grupo que se produce retirando un átomo de hidrógeno del grupo amino en la posición 1 del fármaco antitumoral. Entre tanto, el compuesto de fórmula (1) en la fórmula de reacción anterior se describe como una estructura en la que un resto de la estructura del fármaco al terminal del engarce está conectado a un anticuerpo. Sin embargo, esto es solo la descripción dada por motivos de conveniencia y, en realidad, hay muchos casos en los que una pluralidad de los restos estructurales está conectada a una molécula de anticuerpo. Lo mismo también se aplica a la explicación del procedimiento de producción descrito más adelante.
Específicamente, el conjugado de anticuerpo y fármaco (1) puede producirse haciendo reaccionar el compuesto (2), que puede obtenerse por el procedimiento descrito más adelante, con el anticuerpo (3a) que tiene un grupo sulfhidrilo.
El anticuerpo (3a) que tiene un grupo sulfhidrilo puede obtenerse por un procedimiento bien conocido en la técnica (Hermanson, G.T, Bioconjugate Techniques, págs. 56-136, págs. 456-493, Academic Press (1996)). Los ejemplos incluyen: un reactivo de Traut se hace reaccionar con el grupo amino del anticuerpo; se hacen reaccionar S-acetiltioalcanoatos de N-succinimidilo se con el grupo amino del anticuerpo, seguido de reacción con hidroxilamina; después de hacer reaccionar con 3-(piridilditio)propionato de N-succinimidilo, se hace reaccionar con un agente reductor; el anticuerpo se hace reaccionar con un agente reductor, tal como ditiotreitol, 2-mercaptoetanol y clorhidrato de tris(2-carboxietil)fosfina (TCEP) para reducir el enlace disulfuro en una parte bisagra en el anticuerpo, pero no se limita al mismo.
Específicamente, el conjugado de anticuerpo y fármaco (1) puede producirse añadiendo una solución de dimetilsulfóxido del compuesto (2) en una solución acuosa de cloruro sódico tamponada con fosfato (pH 7,2) que contiene el anticuerpo (3a) que tiene un grupo sulfhidrilo. Después, como se usa habitualmente en la reacción para la producción de la formación del enlace de fármaco y anticuerpo, el compuesto (2) sin reaccionar se desactiva mediante la adición de N-acetil-L-cisteína (NAC). El conjugado de anticuerpo y fármaco (1) producido puede someterse a los
siguientes procedimientos, tales como, concentración, intercambio de tampón, realizar purificación, medición de la concentración de anticuerpo y número promedio de moléculas de fármaco conjugadas por molécula de anticuerpo, y cálculo del contenido de agregado, identificación del conjugado de anticuerpo y fármaco (1).
Procedimiento común A: Concentración de solución acuosa de anticuerpo o conjugado de anticuerpo y fármaco
En un recipiente Amicon Ultra (50.000 MWCO, Millipore Corporation), se añadió una solución del anticuerpo o conjugado de anticuerpo y fármaco y la solución del anticuerpo o conjugado de anticuerpo y fármaco se concentró mediante centrifugación (centrifugación durante 5 a 20 minutos de 2000 G a 3800 G) usando un centrifugador (Allegra X-15R, Beckman Coulter, Inc.).
Procedimiento común B: Medición de la concentración de anticuerpo
Usando un detector UV (Nanodrop 1000, Thermo Fisher Scientific Inc), se realizó la medición de la concentración de anticuerpo de acuerdo con el procedimiento definido por el fabricante. En ese momento, se usó un coeficiente de absorción de 280 nm diferente para cada anticuerpo (de 1,3 a 1,8 mg/ml).
Procedimiento común C-1: Una columna NAP-25 (Cat. N.° 17-0852-02, GE Healthcare Japan Corporation) usando un portador Sephadex G-25 se equilibró con tampón fosfato (10 mM, pH 6,0) (se le hace referencia como PBS6,0/EDTA en la memoria descriptiva) que contenía cloruro sódico (137 mM) y ácido etilendiaminotetraacético (EDTA, 5 mM) de acuerdo con el procedimiento definido por el manual de instrucciones del fabricante. Una solución acuosa del anticuerpo se aplicó en una cantidad de 2,5 ml a una sola columna NAP-25 y después se recogió la fracción (3,5 ml) eluída con 3,5 ml de PBS6,0/EDTA. La fracción resultante se concentró mediante el Procedimiento común A. Después de medir la concentración del anticuerpo usando el Procedimiento común B, la concentración del anticuerpo se ajustó a 10 mg/ml usando PBS6,0/EDTA.
Procedimiento común C-2: Intercambio de tampón para anticuerpo
Una columna NAP-25 (Cat. N.° 17-0852-02, GE Healthcare Japan Corporation) usando un portador Sephadex G-25 se equilibró con tampón fosfato (50 mM, pH 6,5) (se le hace referencia como PBS6,5/EDTA en la memoria descriptiva) que contenía cloruro sódico (50 mM) y EDTA (2 mM) de acuerdo con el procedimiento definido por el fabricante. Una solución acuosa del anticuerpo se aplicó en una cantidad de 2,5 ml a una sola columna NAP-25 y después se recogió la fracción (3,5 ml) eluída con 3,5 ml de PBS6,5/EDTA. La fracción resultante se concentró mediante el Procedimiento común A. Después de medir la concentración del anticuerpo usando el Procedimiento común B, la concentración del anticuerpo se ajustó a 20 mg/ml usando PBS6,5/EDTA.
Procedimiento común D-1: Purificación de conjugado de anticuerpo y fármaco
Una columna NAP-25 se equilibró con cualquier tampón seleccionado entre tampón fosfato disponible en el mercado (PBS7,4, Cat. N.° 10010-023, Invitrogen), tampón fosfato sódico (10 mM, pH 6,0; se denomina PBS6,0) que contiene cloruro sódico (137 mM), y tampón acetato que contiene sorbitol (5% ) (10 mM, pH 5,5; se denomina ABS en la memoria descriptiva). Una solución acuosa de la reacción de conjugado de anticuerpo y fármaco se aplicó en una cantidad de aproximadamente 1,5 ml a la columna NAP-25 y después se eluyó con el tampón en una cantidad definida por el fabricante para recoger la fracción de anticuerpo. La fracción recogida se aplicó de nuevo a la columna NAP-25 y, repitiendo de 2 a 3 veces en total el procedimiento de purificación por filtración en gel para eluir con tampón, se obtuvo el conjugado de anticuerpo y fármaco excluyendo el engarce de fármaco no conjugado y un compuesto de bajo peso molecular, clorhidrato de (tris(2-carboxietil)fosfina (TCEP), N-acetil-L-cisteína (nAc ) y dimetilsulfóxido).
Procedimiento común D-2: Purificación de anticuerpo derivatizado con 4-(N-maleimidilmetil)-ciclohexano-1-carboxilato succinimidilo (SMCC
Una columna NAP-25 se equilibró con PBS6,5/EDTA. A la columna de NAP-25, se aplicó una solución de reacción (aproximadamente 0,5 ml) que contenía el anticuerpo derivatizado con 4-(N-maleimidilmetil)-ciclohexano-1-carboxilato de succinimidilo (en el presente documento, denominado SMCC), y después se eluyó con el tampón en una cantidad definida por el fabricante para recoger la fracción de anticuerpo para purificación.
Procedimiento común E: Medición de concentración de anticuerpo en conjugado de anticuerpo y fármaco y número promedio de moléculas de fármaco conjugado por molécula de anticuerpo.
La concentración de fármaco conjugado en el conjugado de anticuerpo y fármaco puede calcularse midiendo la absorbancia de UV de una solución acuosa del conjugado de anticuerpo y fármaco a dos longitudes de onda de 280 nm y 370 nm, seguido de realización del cálculo que se muestra más adelante.
Debido a que la absorbancia total a cualquier longitud de onda es igual a la suma de la absorbancia de cada especie química que absorbe luz que están presentes en un sistema [aditividad de absorbancia], cuando los coeficientes de absorción molar del anticuerpo y el fármaco permanecen iguales antes y después de la conjugación entre el anticuerpo y el fármaco, la concentración de anticuerpo y la concentración de fármaco en el conjugado de anticuerpo y fármaco se expresan con las siguientes ecuaciones.
A280 = Ad,280 Aa,280 = £d,28oCd + £a,28oCa Ecuación (1)
A370 = Ad,370 Aa,370 =£d,37oCd + £a,37oCa Ecuación (2)
En lo anterior, A280 representa la absorbancia de una solución acuosa del conjugado de anticuerpo y fármaco a 280 nm; A370 representa la absorbancia de una solución acuosa del conjugado de anticuerpo y fármaco a 370 nm; Aa,280 representa la absorbancia de un anticuerpo a 280 nm; Aa,370 representa la absorbancia de un anticuerpo a 370 nm; Ad,280 representa la absorbancia de un precursor de conjugado a 280 nm; Ad,370 representa la absorbancia de un precursor de conjugado a 370 nm; £a,280 representa el coeficiente de absorción molar de un anticuerpo a 280 nm; £a,370 representa el coeficiente de absorción molar de un anticuerpo a 370 nm; £d,280 representa el coeficiente de absorción molar de un precursor de conjugado a 280 nm; £d,370 representa el coeficiente de absorción molar de un precursor de conjugado a 370 nm; Ca representa la concentración de anticuerpo en un conjugado de anticuerpo y fármaco; y Cd representa la concentración de fármaco en un conjugado de anticuerpo y fármaco.
£a,280, £a,370, £d,280 y £d,370 en lo anterior son conocidos de las mediciones previas. Midiendo A280 y A370 de una solución acuosa del conjugado de anticuerpo y fármaco y resolviendo las ecuaciones simultáneas (1) y (2) usando los valores, pueden obtenerse Ca y Cd. Además, dividiendo Cd entre Ca, puede obtenerse el número de conjugado promedio por anticuerpo.
El compuesto representado por la fórmula (2) en el Procedimiento de producción 1 es cualquier compuesto representado por la siguiente fórmula:
[Fórmula 37]

Halógeno-CH2C(=O)NH-(CH2)n4-C(=O)-L2-Lp-(NH-DX)
Halógeno-CH2C(=O)NH-(CH2)n4-C(=O)-NH-[(CH2CH2-O)n7-CH2CH2-OH]-CH2-C(=O)-Lp-(NH-DX)
En la fórmula, n1, n2 , n3 , n4, n, L2, LP, La, Lb y Lc son como ya se han definido, y LP o Lc es una posición de conexión al fármaco.
Como un intermedio útil en la producción de dicho conjugado de anticuerpo y fármaco de la presente invención, preferentemente, n2 es 5, y se representa mediante las siguientes fórmulas:
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
o las siguientes fórmulas:
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-(NH-DX)
Entre ellos, adicional y preferentemente, n2 es 5 y se representa mediante la siguiente fórmula:
(maleimid-N-il) -CH2CH2CH2CH2CH2-C (=O)-GGFGG-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C (=O)-GGFGGG-(NH-DX)
En la fórmula anterior, n3 es preferentemente 2 y se representa preferentemente mediante la siguiente fórmula: (maleimid-N-il)-CH(CH2CH2-COOH)-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
Adicional y preferentemente, n2 es 5, n7 es 3 o 4, y más preferentemente 3, y se representa mediante la siguiente fórmula; (maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
Para asegurar la cantidad de conjugado, puede prepararse una pluralidad de conjugados obtenidos en condiciones de producción similares que tengan un número equivalente de fármacos (por ejemplo, aproximadamente ± 1) para preparar nuevos lotes. En este caso, el número promedio de fármacos está entre los números promedio de fármacos en los conjugados antes del mezclado.
2. Procedimiento de producción 2
El conjugado de anticuerpo y fármaco representado por la fórmula (1) o una sal farmacológicamente aceptable del mismo, en la que el enlace al anticuerpo es un grupo amida y tiene un enlace de tioéter dentro del engarce, específicamente, una estructura en la que -L1-L2- es -C(=O)-cyc.Hex(1,4)-CH2-(N-ly-3-dimiccuS)-S-(CH2)n8-C(=O)-, también pueden producirse por el siguiente procedimiento.
[Fórmula 38]

[En la fórmula, AB-L1' representa que el anticuerpo está conectado al engarce L1, y el terminal de L1 se convierte en un grupo N-maleimidilo. Específicamente, este grupo tiene una estructura en la que -(N-ly-3-dimiccuS)-in AB-C(=O)-cyc.Hex(1,4)-CH2-(N-ly-3-dimiccuS)- se convierte en un grupo maleimidilo. L2' representa un grupo HS-(CH2)n8-C(=O)-en el que el terminal es un grupo mercapto, y AB representa el anticuerpo].
Específicamente, el conjugado de anticuerpo y fármaco (1) puede producirse haciendo reaccionar el compuesto (2a), que puede obtenerse por el procedimiento descrito más adelante, con el anticuerpo (3b) al que está conectado el engarce que tiene un grupo maleimidilo.
El anticuerpo (3b) que tiene un grupo maleimidilo también puede obtenerse por un procedimiento bien conocido en la técnica (Hermanson, G.T, Bioconjugate Techniques, págs. 56-136, págs. 456-493, Academic Press (1996)). El ejemplo incluye un procedimiento en el que un engarce bifuncional, tal como 4-(N-maleimidometil)ciclohexano-1-carboxilato de succinimidilo (SMCC), que es capaz de conectarse a un grupo amino o un grupo hidroxilo, se permite que actúe en el grupo amino del anticuerpo para introducir un grupo maleimidilo, pero no se limita al mismo.
Por ejemplo, puede usarse preferentemente un compuesto que tiene un resto reactivo de grupo amino y un resto reactivo de grupo tiol se conectan mediante un engarce. En este caso, el resto reactivo de grupo amino puede ser éster activado, éster de imida o similares, y el resto reactivo de tiol puede ser maleimidilo, acetilo halogenado, alquilo halogenado, ditiopiridilo o similares.
Como un procedimiento para construir el engarce a través de un enlace de amida con el grupo amino o el grupo hidroxi, particularmente, el grupo amino, de un aminoácido que constituye un anticuerpo, el compuesto que va a
hacerse reaccionar en primer lugar con el anticuerpo puede ser un compuesto representado por la siguiente fórmula: Q l -L1a-Q2.
En la fórmula, Q1 representa (pirrolidin-2,5-diona-N-il)-O-C(=O)-, (3-sulfo-pirrolidin-2,5-diona-N-il)-O-C(=O)-, RQ-O-C(=N)- u O=C=N-,
L1a- representa -cic.Hex(1,4)-CH2-, un grupo alquileno que tiene de 1 a 10 átomos de carbono, un grupo fenileno, -(CH2)n4-C(=O)-, -(CH2)n4a-NH-C(=O)-(CH2)n4b- o -(CH2)n4a-NH-C(=O)-cic.Hex(1,4)-CH2-,
Q2 representa (maleimid-N-ilo), un átomo de halógeno o -S-S-(2-Piridilo),
RQ representa un grupo alquilo que tiene de 1 a 6 átomos de carbono,
n4 representa un número entero de 1 a 8, n4a representa un número entero de 0 a 6, y n4b representa un número entero de 1 a 6.
En lo anterior, RQ es un grupo alquilo que tiene de 1 a 6 átomos de carbono, y más preferentemente un grupo metilo o un grupo etilo.
El grupo alquileno de L1a puede ser aquellos que tienen de 1 a 10 átomos de carbono. El grupo fenileno puede ser cualquiera de orto, meta y para, es más preferentemente un grupo para- o meta-fenileno.
Los ejemplos preferidos de L1a pueden incluir -cic.Hex(1,4)-CH2-, -(CH2)5-NH-C(=O)-cic.Hex(1,4)-CH2-, -(CH2)2-NH-C(=O)-CH2-, -(CH2)5-NH-C(=O)-(CH)2-, -CH2-, -(CH2)2-, -(CH2)3-, -(C H )5-, -(CH2)10-, -(para-Ph)-, -(meta-Ph)-, -(para-Ph)-CH(-CHa)-, -(CH2)3-(meta-Ph)- y -(meta-Ph)-NH-C(=O)-CH2-.
Q1 es preferentemente (pirrolidin-2,5-diona-N-il)-O-C(=O)- y Q2 es preferentemente (maleimid-N-ilo) o puede usarse -S-S-(2-piridilo) cuando va a formarse un enlace disulfuro.
En lo anterior, (pirrolidin-2,5-diona-N-il)- es un grupo representado por la siguiente fórmula:
[Fórmula 39]

en la que el átomo de nitrógeno está en la posición de conexión, (3-sulfo-pirrolidin-2,5-diona-N-il)- es un grupo representado por la siguiente fórmula:
[Fórmula 40]

en la que el átomo de nitrógeno está en la posición de conexión, y este ácido sulfónico es capaz de formar una sal de litio, sal sódica o sal potásica, y preferentemente sal sódica,
cic.Hex(1,4) representa un grupo 1,4-ciclohexileno, (maleimid-N-il) es un grupo representado por la siguiente fórmula:
[Fórmula 41]

en la que el átomo de nitrógeno está en la posición de conexión, (2-piridil) representa un grupo 2-piridilo, (para-Ph) representa un grupo para-fenileno y (meta-Ph) representa un grupo meta-fenileno.
En cuando a dicho compuesto distinto de los compuestos descritos anteriormente, pueden usarse sulfosuccinimidil-4-(N-maleimidilmetil)ciclohexano-1-carboxilato (sulfo-SMCC), N-succinimidil-4-(N-maleimidilmetil)-ciclohexano-1-carboxi-(6-amidocaproato) (LC-SMCC), N-succinimidil éster del ácido K-maleimidil undecanoico (KMUA), Nsuccinimidil éster del ácido Y-maleimidil butírico (GMBS), éster de N-hidroxisuccinimida del ácido £-maleimidil caproico (EMCS), éster de m-maleimidilbenzoil-N-hidroxisuccinimida (MBS), éster de N-(a-maleimidilacetoxi)-succinimida [AMAS], 6-(p-maleimidilpropionamida)hexanoato de succinimidilo (SMPH), 4-(p-maleimidilfenil)-butirato de N-succinimidilo (SMPB), N-(p-maleimidilfenil)isocianato (PMPI), 4-(yodoacetil)-aminobenzoato de N-succinimidilo (SIAB), yodoacetato de N-succinimidilo (SIA), bromoacetato de N-succinimidilo (SBA), 3-(bromoacetamida)propionato de N-succinimidilo (SBAP), 3-(2-piridoditio)propionato de N-succinimidilo (Sp-DP) y succinimidiloxicarbonil-a-metil-a-(2-piridilditio)tolueno (SMPT).
Específicamente, por ejemplo, haciendo reaccionar de 2 a 6 equivalentes de SMCC con el anticuerpo (3) en un tampón fosfato de pH 6 a 7 a temperatura ambiente de 1 a 6 horas, el éster activo de SMCC puede reaccionar con el anticuerpo para producir el anticuerpo (3b) que tiene un grupo maleimidilo. El anticuerpo obtenido (3b) puede purificarse por el Procedimiento común D-2 descrito más adelante, y usarse para la siguiente reacción con el compuesto (2).
El grupo amino y grupo hidroxilo del anticuerpo se refieren a, por ejemplo, un grupo amino N-terminal portado por el anticuerpo y/o un grupo amino portado por un resto de lisina y un grupo hidroxi portado por un resto de serina, respectivamente, pero no se limitan a los mismos.
Para el conjugado de anticuerpo y fármaco (1) producido, la concentración, intercambio de tampón, purificación e identificación del conjugado de anticuerpo y fármaco (1) mediante la medición de la concentración de anticuerpo y un número promedio de moléculas de fármaco conjugadas por anticuerpo y el cálculo del contenido de agregado pueden realizarse de la misma manera que el Procedimiento de producción 1.
El compuesto representado por la fórmula (3b) en el Procedimiento de producción 2 tiene la siguiente estructura (véase la siguiente fórmula; en la estructura de los mismos, "anticuerpo-NH-" se origina desde un anticuerpo).
[Fórmula 42]

Un compuesto que es un intermedio para producir el conjugado de anticuerpo y fármaco de la presente invención y tiene la estructura anterior es como se describe a continuación (en la fórmula, n es un número entero de 1 a 10, preferentemente de 2 a 8, y más preferentemente de 3 a 7).
[Fórmula 43]

Además, los ejemplos del compuesto de la presente invención en el que el terminal es un grupo mercapto pueden incluir los siguientes. Cuando el engarce de péptido tiene un aminoácido hidrófilo en el terminal N, puede usarse preferentemente un compuesto que tiene una estructura representada por la siguiente fórmula como un intermedio de
producción:
HS-(CH2)n8-C(=O)-DGGF-NH-(CH2)n1-La-Lb-Lc-(NH-DX)
HS-(CH2)n8-C(=O)-KGGF-NH-(CH2)n1-La-Lb-Lc-(NH-DX)
HS-(CH2)n8-C(=O)-DGGFG-NH-(CH2)n1-La-Lb-Lc-(NH-DX)
HS-(CH2)n8-C(=O)-KGGFG-NH-(CH2)n1-La-Lb-Lc-(NH-DX)
En la fórmula anterior, preferentemente, n8 es de 2 a 5, y el resto -NH-(CH2)n1-La-Lb-Lc- es -NH-CH2CH2-C(=O)-, -NH-CH2CH2CH2-C(=O)-, -NH-CH2-O-CH2-C(=O)- o -NH-CH2CH2-O-CH2-C(=O)-. También, se prefieren aquellos en los que LP está conectado directamente al fármaco.
Más específicamente, n8 es preferentemente 2 y se representa mediante la siguiente fórmula:
HS-CH2CH2-C(=O)-DGGF-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGF-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGF-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGF-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGF-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGF-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGF-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGF-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-DGGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGFG-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-KGGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
o las siguientes fórmulas:
HS-CH2CH2-C(=O)-DGGF-(NH-DX)
HS-CH2CH2-C(=O)-KGGF-(NH-DX)
HS-CH2CH2-C(=O)-DGGFG-(NH-DX)
HS-CH2CH2-C(=O)-KGGFG-(NH-DX)
Cuando el resto de péptido del engarce tiene un oligopéptido de glicina en el terminal C, puede usarse preferentemente un compuesto que tiene una estructura representada por la siguiente fórmula como un intermedio de producción: HS-(CH2)n8-C(=O)-GGFGG-(NH-DX)
HS-(CH2)n8-C(=O)-GGFGGG-(NH-DX)
En la fórmula anterior, n8 es preferentemente 2 o 5.
Más preferentemente, n8 es 2 y se representa mediante la siguiente fórmula:
HS-CH2CH2-C(=O)-GGFGG-(NH-DX)
HS-CH2CH2-C(=O)-GGFGGG-(NH-DX)
3. Procedimiento de producción 3
El conjugado de anticuerpo y fármaco representado por la fórmula (1) o una sal farmacológicamente aceptable del mismo en el que el anticuerpo está conjugado al resto de engarce de fármaco a través de un enlace de amida puede producirse por un procedimiento descrito mas adelante. Por ejemplo, puede usarse preferentemente L1' en el que L1 es - C(=O)-(CH2)n5-C(=O)-, y este se convierte en éster activo, por ejemplo, (pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n5-C(=O)-. Además, cuando L2 es un enlace sencillo, el conjugado de anticuerpo y fármaco (1) puede producirse mediante el siguiente procedimiento, por ejemplo.
[Fórmula 44]
Específicamente, el conjugado de anticuerpo y fármaco (1) puede producirse haciendo reaccionar el compuesto (2b), que puede obtenerse por el procedimiento descrito más adelante, con el anticuerpo (3).
El compuesto (2b) tiene una propiedad capaz de conectarse al grupo amino o grupo hidroxilo del anticuerpo. El grupo amino y grupo hidroxilo del anticuerpo se refieren a, como se describe en el Procedimiento de producción 2, por ejemplo, un grupo amino N-terminal portado por el anticuerpo y/o un grupo amino portado por un resto de lisina y un grupo hidroxi portado por un resto de serina, respectivamente, pero no se limitan a los mismos.
El compuesto (2b) es un éster activo compuesto de un grupo de éster de N-hidroxisuccinimidilo y, como alternativa, pueden usarse otros ésteres activos, por ejemplo, un grupo éster de sulfosuccinimidilo, éster de N-hidroxiftalimidilo, éster de N-hidroxisulfoftalimidilo, éster de orto-nitrofenilo, éster de para-nitrofenilo, éster de 2,4-dinitrofenilo, éster de 3-sulfonil-4-nitrofenilo, éster de 3-carboxi-4-nitrofenilo y éster de pentafluorofenilo.
Como la reacción entre el compuesto (2b) y el anticuerpo (3), usando de 2 a 20 equivalentes molares del compuesto (2b) por anticuerpo (3) en la reacción del compuesto (2b) con el anticuerpo (3), puede producirse el conjugado de anticuerpo y fármaco (1) en el que se conjugan de 1 a 10 moléculas de fármaco por anticuerpo. Específicamente, la solución que contiene el compuesto (2b) disuelto en la misma puede añadirse a una solución de tampón que contiene el anticuerpo (3) para la reacción, para producir el conjugado de anticuerpo y fármaco (1). En el presente documento, los ejemplos de la solución de tampón que puede usarse incluyen una solución de acetato sódico, fosfato sódico y borato sódico. El pH para la reacción puede ser de 5 a 9, y más preferentemente la reacción se realiza cercana a pH 7. Los ejemplos del disolvente para disolver el compuesto (2b) incluyen un disolvente orgánico, tal como dimetilsulfóxido (DMSO), dimetilformamida (DMF), dimetilacetamida (DMA) y N-metil-2-piridona (NMP). Es suficiente que la solución de disolvente orgánico que contiene el compuesto (2b) disuelto en la misma se añada a de 1 a 20 % v/v a una solución de tampón que contiene el anticuerpo (3) para la reacción. La temperatura de reacción es de 0 a 37 °C, más preferentemente de 10 a 25 °C, y el tiempo de reacción es de 0,5 a 20 horas.
Para el conjugado de anticuerpo y fármaco (1) producido, la concentración, intercambio de tampón, purificación e identificación del conjugado de anticuerpo y fármaco (1) mediante la medición de la concentración de anticuerpo y un número promedio de moléculas de fármaco conjugadas por molécula de anticuerpo pueden realizarse de la misma manera que el Procedimiento de producción 1.
En el Procedimiento de producción 3, (pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n4-C(=O)- tiene la siguiente estructura.
[Fórmula 45]

Los ejemplos del compuesto que tiene la estructura parcial anterior y que tiene el engarce de péptido que tiene un aminoácido hidrófilo en el terminal N pueden incluir los siguientes.
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-NH-CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-NH-CH2CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-NH-CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-NH-CH2CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX).
Entre ellos, se prefieren más los siguientes.
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-NH-CH2CH2CH2-C(=O)-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2-C(=O)-(NH-DX).
Los ejemplos del compuesto que tiene la estructura parcial anterior y que tiene el engarce de péptido que tiene un aminoácido hidrófilo en el terminal N, que está conectado directamente al fármaco pueden incluir los siguientes.
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGF-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGF-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-KGGFG-(NH-DX).
Los ejemplos del compuesto que tiene la estructura parcial anterior y que tiene el engarce de péptido que tiene un oligopéptido de glicina en el terminal C, que está conectado directamente al fármaco pueden incluir los siguientes.
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFGG-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFGGG-(NH-DX).
Los ejemplos del compuesto que tiene la estructura parcial anterior y que tiene la estructura hidrófila en L1 pueden incluir los siguientes.
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH(CH2CH2-COOH)-CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH(CH2CH2-COOH)-CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH(CH2CH2-COOH)-CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX), (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH(CH2CH2-COOH)-CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX).
Los ejemplos del compuesto que tiene la estructura parcial anterior, que tiene la estructura hidrófila en L1 y que tiene el engarce de péptido conectado directamente al fármaco pueden incluir los siguientes. (Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH(CH2CH2-COOH)-CH2-C(=O)-GGFG-(NH-DX).
Los ejemplos del compuesto que tiene la estructura parcial anterior y que tiene la estructura hidrófila en L2 pueden incluir los siguientes.
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C (=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C (=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX),
(Pirrolidin-2,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX).
Los ejemplos del compuesto que tiene la estructura parcial anterior, que tiene la estructura hidrófila en L2 y el engarce de péptido conectado directamente al fármaco pueden incluir los siguientes.
(Pirrolidin-2, 5-diona-N-il) -O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-N(-CH2CH2-O-CH2CH2-O-CH2CH2-O-CH2CH2-OH)-CH2-C(=O)-GGFG-(NH-DX).
4. Procedimiento de producción 4
Entre el compuesto representado por la fórmula (2) o (2b) usado como un intermedio en el procedimiento de producción previo y una sal farmacológicamente aceptable del mismo, aquellos en los que el engarce tiene una estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc-, y LP tiene un aminoácido hidrófilo distinto de glicina en el terminal N, por ejemplo, pueden producirse por el siguiente procedimiento.
[Fórmula 46-1]

En la fórmula, LC es un grupo -C(=O)-, L1' representa la estructura L1 en la que el terminal es un grupo maleimidilo o un grupo haloacetilo, o L1 se convierte en (Pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n5-C(=O)-, Lp representa una estructura de -Lp1-Lp2-, y P1, P2, P3 , P4 , P5, P6 y P7 representan cada uno un grupo protector.
Puesto que LP se forma conectando Lp1 y Lp2, el aminoácido hidrófilo N-terminal de LP se deriva de Lp1, por lo tanto, puede usarse Lp1 que tiene un aminoácido hidrófilo en el terminal N. En el presente documento, puede haber presente una pluralidad de aminoácidos hidrófilos. Si se usa Lp2 que tiene un aminoácido hidrófilo, de acuerdo con su posición, puede producirse LP de modo que contenga una pluralidad de aminoácidos hidrófilos en el terminal N de LP o el terminal N y otras posiciones.
El compuesto (6) puede producirse derivatizando el compuesto de ácido carboxílico (5) que tiene el grupo amino terminal protegido con P1 en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con NH2-DX [que representa exatecan; nombre químico:
(1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3',4':6,7]indolidino[1,2-b]quinolin-10,13(9H,15H)-diona] (es decir, el compuesto farmacéutico descrito en la reivindicación 2 de la patente japonesa abierta a inspección pública N.° 6-87746) (4) o una sal farmacológicamente aceptable del mismo en presencia de una base.
Para esta reacción, pueden emplearse reactivos y condiciones usadas comúnmente para amidación y síntesis de péptidos. Hay varias clases de éster activo, por ejemplo, puede producirse haciendo reaccionar fenoles, tales como pnitrofenol, N-hidroxi benzotriazol, N-hidroxi succinimida, o similares, con el compuesto de ácido carboxílico (5) usando un agente de condensación, tal como N,N'-diciclohexilcarbodiimida o clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida. Además, el éster activo también puede producirse por una reacción del compuesto de ácido carboxílico (5) con trifluoroacetato de pentafluorofenilo o similares; una reacción del compuesto de ácido carboxílico (5) con hexafluorofosfito de 1-benzotriazolil oxitripirrolidinofosfonio; una reacción del compuesto de ácido carboxílico (5) con cianofosfonato de dietilo (procedimiento de Shioiri); una reacción del compuesto de ácido
carboxílico (5) con trifenilfosfina y disulfuro de 2,2'-dipiridilo (procedimiento de Mukaiyama); una reacción del compuesto de ácido carboxílico (5) con un derivado de triazina, tal como cloruro de 4-(4,6-dimetoxi-1,3,5-triazin-2-il)-4-metilmorfolinio (DMTMM); o similares. Además, la reacción también puede realizarse, por ejemplo, por un procedimiento de haluro de ácido por el cual el compuesto de ácido carboxílico (5) se trata con haluro de ácido, tal como cloruro de tionilo o cloruro de oxalilo en presencia de una base. Haciendo reaccionar el éster activo, anhídrido de ácido mixto o haluro de ácido del compuesto de ácido carboxílico (5) obtenido anteriormente con el compuesto (4) en presencia de una base adecuada, en un disolvente que no inhiba la reacción de -78 °C a 150 °C, puede producirse el compuesto (6).
Los ejemplos específicos de la base usada para cada etapa descrita anteriormente incluyen carbonato de un metal alcalino o metal alcalinotérreo, un alcóxido de metal alcalino, hidróxido o hidruro de un metal alcalino incluyendo carbonato sódico, carbonato potásico, etóxido sódico, butóxido potásico, hidróxido sódico, hidróxido potásico, hidruro sódico e hidruro potásico, una base organometálica representada por un alquil litio que incluye n-butillitio, dialquilamino litio, incluyendo diisopropilamida de litio; una base organometálica de bissililamina, incluyendo bis(trimetilsilil)amida de litio; y una base orgánica, incluyendo piridina, 2,6-lutidina, colidina, 4-dimetilaminopiridina, trietilamina, N-metilmorfolina, diisopropiletilamina y diazabiciclo[5,4,0]undec-7-eno (DBU).
Los ejemplos del disolvente, que se usa para la reacción de la presente invención y no inhibe la reacción, incluyen un disolvente de hidrocarburo halogenado, tal como diclorometano, cloroformo y tetracloruro de carbono; un disolvente de éter, tal como tetrahidrofurano, 1,2-dimetoxietano y dioxano; un disolvente aromático, tal como benceno y tolueno; y un disolvente de amida, tal como N,N-dimetilformamida, N,N-dimetilacetamida y N-metilpirrolidin-2-ona. Además de ellos, un disolvente de sulfóxido, tal como dimetilsulfóxido y sulfolano; un disolvente de alcohol, tal como metanol y etanol; y un disolvente de cetona, tal como acetona y metil etil cetona pueden usarse dependiendo de cada caso.
El grupo hidroxi, grupo carboxi, grupo amino, o similares, de La y Lb en el compuesto (6) pueden protegerse mediante un grupo protector que se usa comúnmente en la síntesis de compuestos orgánicos, como se menciona posteriormente. Específicamente, los ejemplos del grupo protector para un grupo hidroxilo incluyen un grupo alcoximetilo, tal como un grupo metoximetilo; un grupo arilmetilo, tal como un grupo bencilo, grupo 4-metoxibencilo y grupo trifenilmetilo; un grupo alcanoílo, tal como un grupo acetilo; un grupo aroílo, tal como un grupo benzoílo; y un grupo sililo, tal como un grupo ferc-butil difenilsililo. El grupo carboxi puede protegerse, por ejemplo, como un éster con un grupo alquilo, tal como un grupo metilo, grupo etilo y grupo ferc-butilo, un grupo alilo o un grupo arilmetilo, tal como un grupo bencilo. El grupo amino puede protegerse con un grupo protector para un grupo amino que se usa generalmente para la síntesis de péptidos, por ejemplo, un grupo alquiloxi carbonilo, tal como un grupo ferc-butiloxi carbonilo, grupo metoxicarbonilo, grupo etoxicarbonilo y grupo 2-(trimetilsilil)etoxicarbonilo; un grupo arilmetilo, tal como aliloxicarbonilo, grupo 9-fluorenilmetiloxi carbonilo, grupo benciloxi carbonilo, grupo parametoxibenciloxi carbonilo y grupo para (u orto)nitroilbenciloxi carbonilo; un grupo alcanoílo, tal como un grupo acetilo; un grupo arilmetilo, tal como un grupo bencilo y un grupo trifenil metilo; un grupo aroílo, tal como un grupo benzoílo; y un grupo aril sulfonilo, tal como un grupo 2,4-dinitrobenceno sulfonilo o grupo ortonitrobenceno sulfonilo. La protección con, y la desprotección del grupo protector pueden realizarse de acuerdo con un procedimiento realizado comúnmente en la técnica.
Como para el grupo protector P1 para el grupo amino terminal del compuesto (6), puede usarse un grupo protector para un grupo amino que se usa generalmente para la síntesis de péptidos, por ejemplo, un grupo ferc-butiloxi carbonilo, un grupo 9-fluorenilmetiloxi carbonilo y un grupo benciloxi carbonilo. Los ejemplos del otro grupo protector para un grupo amino incluyen un grupo alcanoílo, tal como un grupo acetilo; un grupo alcoxicarbonilo, tal como un grupo metoxicarbonilo y un grupo etoxicarbonilo; un grupo arilmetoxicarbonilo, tal como un grupo parametoxibenciloxi carbonilo y un grupo para (u orto)nitroilbenciloxi carbonilo; un grupo arilmetilo, tal como un grupo bencilo y un grupo trifenil metilo; un grupo aroílo, tal como un grupo benzoílo; y un grupo aril sulfonilo, tal como un grupo 2,4-dinitrobenceno sulfonilo y un grupo ortonitrobenceno sulfonilo. El grupo protector P1 puede seleccionarse dependiendo de, por ejemplo, las propiedades de un compuesto que tiene un grupo amino que va a protegerse.
Desprotegiendo el grupo protector P1 para el grupo amino terminal del compuesto (6) obtenido, puede producirse el compuesto (7). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el péptido o aminoácido (8), que está protegido con P2 en su terminal N, en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (7) obtenido, puede producirse el compuesto (9). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el péptido o aminoácido (8) y el compuesto (7) no están limitados siempre y cuando no inhiban una reacción, y pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P2 para un grupo amino puede seleccionarse adecuadamente a partir de los descritos para el grupo protector del compuesto (6), y la selección puede hacerse basándose en, por ejemplo, las propiedades del compuesto. Como se usa generalmente para la síntesis de péptidos, repitiendo secuencialmente la reacción y la desprotección del aminoácido o péptido que constituye el péptido o aminoácido (8) para alargamiento, también puede producirse el compuesto (9).
Desprotegiendo P2 como el grupo protector para el grupo amino del compuesto (9) obtenido, puede producirse el compuesto (10). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el aminoácido o péptido (11) que tiene el terminal N protegido con P3 y un grupo carboxi, grupo hidroxi o grupo amino de cadena lateral protegido con P4 en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (10) obtenido, puede producirse el compuesto (12). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el aminoácido o péptido (11 ) y el compuesto (10) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). Los grupos protectores P3 y P4 pueden seleccionarse adecuadamente entre aquellos descritos para el grupo protector para el grupo amino, grupo carboxi o grupo hidroxi del compuesto (6). Sin embargo, en tal caso, es necesario que el grupo protector P3 para un grupo amino y el grupo protector P4 para un grupo funcional de cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P3 es un grupo 9-fluorenilmetiloxicarbonilo y P4 es un grupo ferc-butilo o similar para un grupo carboxi, un grupo metoximetilo o similar para un grupo hidroxi, o un grupo ferc-butiloxicarbonilo o similar para un grupo amino. El grupo protector P4 para un grupo funcional de cadena lateral es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. Como se usa generalmente para la síntesis de péptidos, repitiendo secuencialmente la reacción y la desprotección del aminoácido o péptido constituyente para alargamiento, también puede producirse el compuesto (12).
Desprotegiendo P3 como el grupo protector para el grupo amino terminal del compuesto (12) obtenido, puede producirse el compuesto (13). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el derivado de ácido carboxílico (14) o (14b) en un éster activo, anhídrido de ácido mixto, haluro de ácido o similar y haciéndolo reaccionar con el compuesto (13) obtenido, puede producirse el compuesto (15) o (15b). En este caso, el derivado de ácido carboxílico (14) es un compuesto que tiene una estructura de L1' en la que el terminal de engarce es un grupo maleimidilo o un grupo haloacetilo, y el derivado de ácido carboxílico (14b) es un compuesto que tiene una estructura de L1' en la que el terminal de engarce tiene (pirrolidin-2,5-diona-N-il)-O-C(=O)-.
Las condiciones de reacción, reactivos, bases y disolventes utilizados para una formación de enlace de péptidos entre el derivado de ácido carboxílico (14) o (14b) y el compuesto (13) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo P4 como el grupo protector para el grupo carboxi, grupo hidroxi o grupo amino de cadena lateral de aminoácido del resto peptídico del compuesto (15) o (15b) obtenido, puede producirse el compuesto (2) o (2b). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (9) también puede producirse por el siguiente procedimiento, por ejemplo.
Derivatizando el péptido o aminoácido (8) que tiene el terminal N protegido con P2 en éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto de amina (16) que tiene el grupo carboxi terminal protegido con P5 en presencia de una base, puede producirse el compuesto (17). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el péptido o aminoácido (8) y el compuesto (16) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P2 para el grupo amino del compuesto (17) puede seleccionarse adecuadamente entre los descritos para el grupo protector del compuesto (6). Como para el grupo protector P5 para un grupo carboxi, puede usarse un grupo protector usado comúnmente como grupo protector para un grupo carboxi en química sintética orgánica, en particular, síntesis de péptidos. Específicamente, este puede seleccionarse adecuadamente entre los descritos para grupo protector del compuesto (6), por ejemplo, ésteres con un grupo alquilo, tal como un grupo metilo, un grupo etilo o un ferc-butilo, alil ésteres y bencil ésteres. En tal caso, es necesario que el grupo protector P2 para un grupo amino y el grupo protector P5 para un grupo carboxi pueda retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P2 es un grupo ferc-butiloxi carbonilo y P5 es un grupo bencilo. Los grupos protectores pueden seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades de un compuesto que tiene un grupo amino y un grupo carboxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo el grupo protector P5 para el grupo carboxi del compuesto (17) obtenido, puede producirse el compuesto (18). Los reactivos y condiciones se seleccionan dependiendo del grupo protector.
Derivatizando el compuesto (18) obtenido en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciendo reaccionar con el compuesto (4) en presencia de una base, puede producirse el compuesto (9). Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (12) también puede producirse por el siguiente procedimiento, por ejemplo.
Desprotegiendo el grupo protector P2 para el grupo amino del compuesto (17), puede producirse el compuesto (19). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el aminoácido o péptido (11) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (19) obtenido en presencia de una base, puede producirse el compuesto (20). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el aminoácido o péptido (11) y el compuesto (19) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). En este caso, es necesario que los grupos protectores P3 y P4 del aminoácido o péptido (11) y el grupo protector P5 del compuesto (19) puedan retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P3 es un grupo 9-fluorenilmetiloxicarbonilo, P4 es un grupo ferc-butiloxicarbonilo, grupo ferc-butilo o grupo metoximetilo, y P5 es un grupo bencilo. Como se ha mencionado anteriormente, el grupo protector P4 para un grupo funcional de cadena lateral es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo el grupo protector P5 para el grupo carboxi del compuesto (20) obtenido, puede producirse el compuesto (21). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el compuesto (21) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (4) en presencia de una base, puede producirse el compuesto (12). Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (15) también puede producirse por el siguiente procedimiento, por ejemplo.
Desprotegiendo el grupo protector P3 para el grupo amino del compuesto (20), puede producirse el compuesto (22). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el derivado de ácido carboxílico (14) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (22) obtenido en presencia de una base, puede producirse el compuesto (23). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el derivado de ácido carboxílico (14) y el compuesto (22) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P5 para el grupo carboxi del compuesto (23) obtenido, puede producirse el compuesto (24). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (15) puede producirse derivatizando el compuesto (24) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (4) en presencia de una base. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (15) también puede producirse por el siguiente procedimiento, por ejemplo.
Derivatizando el derivado de ácido carboxílico (14) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el aminoácido o péptido (25) que tiene un grupo carboxi protegido con P6 y un grupo carboxi, grupo hidroxi o grupo amino de cadena lateral protegido con P4 en presencia de una base, puede producirse el compuesto (26). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el derivado de ácido carboxílico (14) y el compuesto (25) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). En este caso, los grupos protectores P4 y P6 del compuesto (26) pueden seleccionarse adecuadamente entre los descritos para el grupo protector para el grupo carboxi, grupo hidroxi o grupo amino del compuesto (6). Sin embargo, en tal caso, es necesario que el grupo protector P6 para un grupo carboxi y el grupo protector P4 para un grupo funcional de cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P6 es un grupo bencilo y P4 es un grupo ferc-butilo o similar para un grupo carboxi, un grupo metoximetilo o similar para un grupo hidroxi, o un grupo ferc-butiloxicarbonilo o similar para un grupo amino. El grupo protector P4 para un grupo funcional de cadena lateral es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo el grupo protector P6 para el grupo carboxi del compuesto (26) obtenido, puede producirse el compuesto (27). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el compuesto (27) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (10) en presencia de una base, puede producirse el compuesto (15). Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos,
y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Derivatizando el compuesto (27) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el aminoácido o péptido (28) que tiene un grupo carboxi protegido con P7 en presencia de una base, puede producirse el compuesto (29). Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). En este caso, los grupos protectores P4 y P7 del compuesto (29) pueden seleccionarse adecuadamente entre los descritos para el grupo protector para el grupo carboxi, grupo hidroxi o grupo amino del compuesto (6). Sin embargo, en tal caso, es necesario que el grupo protector P7 para un grupo carboxi y el grupo protector P4 para un grupo funcional de cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P7 es un grupo bencilo y P4 es un grupo ferc-butilo o similar para un grupo carboxi, un grupo metoximetilo o similar para un grupo hidroxi, o un grupo ferc-butiloxicarbonilo o similar para un grupo amino. El grupo protector P4 para un grupo funcional de cadena lateral es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. Repitiendo secuencialmente la reacción y la desprotección del aminoácido o péptido constituyente para alargamiento, también puede producirse el compuesto (29).
Desprotegiendo el grupo protector P7 para el grupo carboxi del compuesto (29) obtenido, puede producirse el compuesto (30). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el compuesto (30) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (7) en presencia de una base, puede producirse el compuesto (15). Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (29) también puede producirse por el siguiente procedimiento, por ejemplo.
Derivatizando el aminoácido o péptido (28) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el aminoácido o péptido (11) que tiene el terminal N protegido con P3 y un grupo carboxi, grupo hidroxi o grupo amino de cadena lateral protegido con P4 en presencia de una base, puede producirse el péptido (31). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el aminoácido o péptido (28) y el aminoácido o péptido (11 ) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). En este caso, como se ha mencionado anteriormente, es necesario que el grupo protector P7 para el grupo carboxi del aminoácido o péptido (28) y los grupos protectores P3 y P4 del aminoácido o péptido (11) puedan retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P3 es un grupo 9-fluorenilmetiloxicarbonilo, P4 es un grupo ferc-butilo o similar para un grupo carboxi, un grupo metoximetilo o similar para un grupo hidroxi, o un grupo ferc-butiloxicarbonilo o similar para un grupo amino, y P7 es un grupo bencilo. El grupo protector P4 para un grupo funcional de cadena lateral es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo P3 como el grupo protector para el terminal N de este péptido (31) obtenido, puede producirse el péptido (32). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el derivado de ácido carboxílico (14) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el péptido (32) obtenido en presencia de una base, puede producirse el compuesto (29). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el derivado de ácido carboxílico (14) y el péptido (32) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (12) también puede producirse por el siguiente procedimiento, por ejemplo.
Desprotegiendo P7 como el grupo protector para el terminal C del péptido (31), puede producirse el péptido (33). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el péptido (33) obtenido en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (7) en presencia de una base, puede producirse el compuesto (12). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el péptido (33) y el compuesto (7) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
5. Procedimiento de producción 5
Entre el intermedio de producción representado por la fórmula (2) o (2b) en el que el engarce tiene una estructura representada por -L1-L2-LP-, y LP es un resto de péptido que tiene un aminoácido hidrófilo distinto de glicina en el terminal N también puede producirse por el siguiente procedimiento.
[Fórmula 46-2]

En la fórmula, L1' representa la estructura L1 en la que el terminal es un grupo maleimidilo o un grupo haloacetilo, o L1 se convierte en (pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n5-C(=O)-, LP representa una estructura de -Lp1-Lp2-, y P2, P3 , P4 y P7 representan cada uno un grupo protector.
Puesto que LP se forma conectando Lp1 y Lp2, el aminoácido hidrófilo N-terminal de LP se deriva de Lp1, por lo tanto, puede emplearse Lp1 que tiene un aminoácido hidrófilo en el terminal N. LP puede tener una pluralidad de aminoácidos hidrófilos. Si se usa Lp2 que tiene un aminoácido hidrófilo, de acuerdo con la posición del mismo, LP puede producirse de modo que contenga aminoácidos hidrófilos en el terminal N de LP o en el terminal N y otras posiciones.
Derivatizando el péptido o aminoácido (8) descrito en el Procedimiento de producción 4 que tiene el terminal N protegido con P2 en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (4) o una sal del mismo, puede producirse el compuesto (34). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el péptido o aminoácido (8) y el compuesto (4) no están limitados
siempre y cuando no inhiban una reacción, y pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P2 puede seleccionarse adecuadamente a partir de los descritos para el grupo protector del compuesto (6), y la selección puede hacerse basándose en, por ejemplo, las propiedades del compuesto que tiene un grupo amino que va a protegerse. Como se usa generalmente para la síntesis de péptidos, repitiendo secuencialmente la reacción y la desprotección del aminoácido o péptido que constituye el péptido o aminoácido (8) para alargamiento, también puede producirse el compuesto (34).
Desprotegiendo P2 como el grupo protector para el grupo amino del compuesto (34) obtenido, puede producirse el compuesto (35). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el aminoácido o péptido (11) descrito en el Procedimiento de producción 4 tiene el terminal N protegido con P3 y un grupo carboxi, grupo hidroxi o grupo amino de cadena lateral protegido con P4 en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (35) obtenido, puede producirse el compuesto (36). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el aminoácido o péptido (11) y el compuesto (35) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). Los grupos protectores P3 y P4 son como se describen en el Procedimiento de producción 4. Como se usa generalmente para la síntesis de péptidos, repitiendo secuencialmente la reacción y la desprotección del aminoácido o péptido constituyente para alargamiento, también puede producirse el compuesto (36).
Desprotegiendo P3 como el grupo protector para el grupo amino del compuesto (36) obtenido, puede producirse el compuesto (37). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el derivado de ácido carboxílico (14) o (14b) en un éster activo, anhídrido de ácido mixto, haluro de ácido o similar y haciéndolo reaccionar con el compuesto (37) obtenido, puede producirse el compuesto (38) o (38b). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el derivado de ácido carboxílico (14) o (14b) y el compuesto (37) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo P4 como el grupo protector para el grupo carboxi o grupo hidroxi o grupo amino del compuesto (38) o (38b) obtenido, puede producirse el compuesto (2) o (2b). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (36) también puede producirse por el siguiente procedimiento, por ejemplo.
Derivatizando el péptido (33) descrito en el Procedimiento de producción 4 en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (4) o una sal del mismo, puede producirse el compuesto (36). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el péptido (33) y el compuesto (4) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (38) también puede producirse por el siguiente procedimiento, por ejemplo.
El compuesto (38) puede producirse derivatizando el compuesto (30) descrito en el Procedimiento de producción 4 en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (4) en presencia de una base, o derivatizando el aminoácido o péptido (27) descrito en el Procedimiento de producción 4 en un éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (35) en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
6. Procedimiento de producción 6
Entre los intermedios de producción representados por la fórmula (2), aquellos que tienen una estructura de -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- o -L1-L2-Lp-, y LP es un resto peptídico que tiene un aminoácido hidrófilo distinto de glicina en terminal N también pueden producirse por el siguiente procedimiento, por ejemplo.
[Fórmula 47]

En la fórmula, L1' representa la estructura L1 en la que el terminal es un grupo maleimidilo o un grupo haloacetilo, o L1 se convierte en (pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n5-C(=O)-, LP representa una estructura de -Lp1-Lp2- y P3 y P8 representan cada uno un grupo protector.
El intermedio de producción representado por la fórmula (2) tiene dos formas del engarce: una estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- y una estructura representada por -L1-L2-LP-.
El compuesto (2) en el que el engarce tiene la estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- puede producirse de la siguiente manera.
El compuesto (40) puede sintetizarse de la misma manera que en el compuesto (12) descrito en el Procedimiento de producción 4. A diferencia del compuesto (12), puede no ser necesario que el grupo protector P3 para un grupo amino y el grupo protector P8 para un grupo funcional de cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes. El grupo funcional de la cadena lateral es un grupo carboxi o un grupo hidroxi, y el grupo protector P3 para un grupo amino y el grupo protector P8 para un grupo carboxi o grupo hidroxi de cadena lateral pueden desprotegerse simultáneamente. Por ejemplo, un ejemplo representativo incluye una combinación en la que P3 es un grupo fere-butiloxicarbonilo, y P8 es un grupo tere-butilo o un grupo tritilo, o P3 es un grupo benciloxicarbonilo, y P8 es un grupo bencilo. Estos grupos protectores pueden seleccionarse adecuadamente entre los descritos para el grupo protector del compuesto (6) dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. El compuesto (40) puede sintetizarse de la misma manera que el Procedimiento de producción 4 usando el aminoácido o péptido protegido que satisfaga las propiedades descritas anteriormente.
Desprotegiendo secuencialmente o simultáneamente los grupos protectores P3 y P8 del compuesto (40), puede producirse el compuesto (41). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Aunque el grupo funcional en la cadena lateral hidrófila LP en el compuesto (41) no está particularmente protegido, el compuesto (2) puede producirse haciéndolo reaccionar con el compuesto (14) o (14b) derivatizado en un éster activo, anhídrido de ácido mixto o similares, en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para la formación de un enlace de péptido pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (2) en el que el engarce tiene la estructura representada por -L1-L2-Lp- puede producirse de la siguiente manera.
El compuesto (42) también puede sintetizarse de la misma manera que el compuesto (36) descrito en el Procedimiento de producción 5. A diferencia del compuesto (36), puede no ser necesario que el grupo protector P3 para un grupo amino y el grupo protector P8 para el grupo funcional de la cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes. El grupo funcional de la cadena lateral es un grupo carboxi o un grupo hidroxi, y el grupo protector P3 para un grupo amino y el grupo protector P8 para un grupo carboxi o grupo hidroxi de cadena lateral también pueden desprotegerse simultáneamente.
Por ejemplo, un ejemplo representativo incluye una combinación en la que P3 es un grupo fere-butiloxicarbonilo, y P8 es un grupo fere-butilo o un grupo tritilo, o P3 es un grupo benciloxicarbonilo, y P8 es un grupo bencilo. Estos grupos protectores pueden seleccionarse adecuadamente entre los descritos para el grupo protector del compuesto (6) dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi
que va a protegerse. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. El compuesto (42) puede sintetizarse de la misma manera que el Procedimiento de producción 5 usando el aminoácido o péptido protegido que satisfaga las propiedades descritas anteriormente. Desprotegiendo secuencialmente o simultáneamente los grupos protectores P3 y P8 del compuesto (42), puede producirse el compuesto (43). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. Aunque el grupo funcional en la cadena lateral hidrófila LP en el compuesto (43) no está particularmente protegido, el compuesto (2) puede producirse haciéndolo reaccionar con el compuesto (14) o (14b) derivatizado en un éster activo, anhídrido de ácido mixto o similares, en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
7. Procedimiento de producción 7
El compuesto (36) mostrado en el Procedimiento de producción 5 en el que el engarce -LP- tiene una estructura de -Lp1-Gly-Gly-Phe-Gly también puede producirse por el siguiente procedimiento.
[Fórmula 48]
H-Gly-Gly-Phe-Gly-(NH-DX)
44

p3_LP1(p4)-Gly-Gly-Phe-Gly-(NH-DX)
45
En la fórmula, L1' representa la estructura L1 en la que el terminal es un grupo maleimidilo o un grupo haloacetilo, o L1 se convierte en (pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n5-C(=O)-, -LP- representa una estructura de -Lp1-Gly-Gly-Phe-Gly-, y cada uno de P3 y P4 representa un grupo protector.
El compuesto (45) puede producirse derivatizando el aminoácido o péptido (11) descrito en el Procedimiento de producción 4 en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con glicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolidino[1,2-b]quinolin-1-il]glicinamida (es decir, una forma libre del compuesto farmacéutico descrito en el documento WO97/46260) (44) o una sal del mismo en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el aminoácido o péptido (11) y el compuesto (44) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P3 para el terminal N y el grupo protector P4 para un grupo funcional de cadena lateral son como se han mencionado anteriormente en el Procedimiento de producción 4. El grupo protector P4 para un grupo funcional de cadena lateral puede estar ausente, y el compuesto (45) puede obtenerse mediante la reacción que usa el aminoácido o péptido (11 ) protegido únicamente en el terminal N.
8. Procedimiento de producción 8
Entre los compuestos representados por la fórmula (2) o (2b), aquellos que tienen la estructura de engarce representada por -L1-L2-LP-, y LP es un resto peptídico en el que el terminal C es un oligopéptido que consiste en 2 o 3 o más glicinas y está conectado al fármaco, e incluso en caso de que un aminoácido hidrófilo esté presente en el terminal N, ningún otro aminoácido hidrófilo distinto de glicina esté presente en el mismo, también pueden producirse por el siguiente procedimiento, por ejemplo.
[Fórmula 49]

En la fórmula, L1' representa la estructura L1 en la que el terminal es un grupo maleimidilo o un grupo haloacetilo, o L1 se convierte en (pirrolidin-2,5-diona-N-il)-O-C(=O)-(CH2)n5-C(=O)-, LP representa una estructura de -Lp1-Lp2- y P7 y P9 representan cada uno un grupo protector.
Puesto que LP se forma conectando Lp1 y Lp2, el número de glicinas contenidas en el mismo para constituir el terminal C de LP puede determinarse considerando el número de glicinas terminales en C de LP y además, el número de usos repetitivos de las mismas para la reacción.
El péptido (46) es un oligopéptido en el que el terminal C consiste en 2 o 3 o más glicinas y, en el caso cuando el terminal N es opcionalmente un aminoácido hidrófilo pero no es un aminoácido hidrófilo distinto de glicina, este terminal N está protegido con P9 El péptido (46) puede sintetizarse repitiendo secuencialmente la reacción de condensación y desprotección del aminoácido o péptido que lo constituye, como se usa generalmente para la síntesis de péptidos. Derivatizando el péptido (46) en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (4) o una sal del mismo, puede producirse el compuesto (47). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el péptido (46) y el compuesto (4) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P9 puede seleccionarse adecuadamente entre aquellos descritos para la síntesis del compuesto (6).
El compuesto (47) también puede producirse derivatizando el aminoácido o péptido (48) que tiene el terminal N protegido con P9 en un éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (35) descrito en el Procedimiento de producción 5. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el aminoácido o péptido (48) y el compuesto (35) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P9 puede seleccionarse adecuadamente entre aquellos descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P9 para el grupo amino del compuesto (47) obtenido, puede producirse el compuesto (49). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Derivatizando el derivado de ácido carboxílico (14) o (14b) en un éster activo, anhídrido de ácido mixto, haluro de ácido o similar y haciéndolo reaccionar con el compuesto (49) obtenido, puede producirse el compuesto (2) o (2b). Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el derivado de ácido carboxílico (14) o (14b) y el compuesto (49) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (2) también puede producirse por el siguiente procedimiento.
El compuesto (50) en el que la glicina de terminal N de Lp1 está conectada a L2 puede sintetizarse de la misma manera que el compuesto (27) descrito en el Procedimiento de producción 4. Derivatizando el aminoácido o péptido (28) descrito en el Procedimiento de producción 4 en éster activo, anhídrido de ácido mixto, haluro de ácido o similar, y haciéndolo reaccionar con el compuesto (50), puede producirse el compuesto (51). En este caso, el aminoácido o péptido (28) es glicina o un oligopéptido que tiene el terminal C que consiste en 2 o 3 o más glicinas, y su terminal C está protegido con P7. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el aminoácido o péptido (28) y el compuesto (50) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (51) también puede producirse derivatizando el compuesto (14) en un éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el péptido (52) que tiene el terminal C protegido con P7. En este caso,
el péptido (52) es un oligopéptido en el que el terminal C que consiste en 2 o 3 o más glicinas, y el terminal N es opcionalmente un aminoácido hidrófilo pero ningún otro aminoácido hidrófilo distinto de glicina está presente en el mismo. El péptido (52) puede sintetizarse repitiendo secuencialmente la reacción de condensación y desprotección del aminoácido o péptido que lo constituye, como se usa generalmente para la síntesis de péptidos. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el péptido (52) y el compuesto (14) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P7 es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P7 para el grupo carboxi del compuesto (51) obtenido, puede producirse el compuesto (53). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (2) puede producirse derivatizando el compuesto (53) en un éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (4) o una sal del mismo. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el compuesto (53) y el compuesto (4) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Como alternativa, el compuesto (2) también puede producirse por el siguiente procedimiento.
El compuesto (2) puede producirse derivatizando el compuesto (35) descrito en el Procedimiento de producción 5 en un éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (50) en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el compuesto (50) y el compuesto (35) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
9. Procedimiento de producción 9
El intermedio de producción representado por la fórmula (2a) descrito en el Procedimiento de producción 2 en el que L2' corresponde a L2 que tiene una estructura en la que el terminal se convierte en un grupo mercaptoalcanoílo puede producirse por el siguiente procedimiento.
[Fórmula 50]

En la fórmula, LP representa una estructura de Lp1-Lp2, y P4, P5, P7y P10 representan cada uno un grupo protector.
El intermedio de producción representado por la fórmula (2a) tiene dos formas del engarce: una estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- y una estructura representada por -L1-L2-LP-.
El compuesto (2a) en el que el engarce tiene la estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- puede producirse de la siguiente manera.
El compuesto (55) puede producirse derivatizando el compuesto de ácido carboxílico (54) que tiene un grupo mercapto terminal protegido con P10 en un éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (13) descrito en el Procedimiento de producción 4. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). Como para el grupo protector P10 para un grupo mercapto, puede usarse un grupo protector usado comúnmente como grupo protector para un grupo mercapto en química sintética orgánica. Específicamente, este puede seleccionarse adecuadamente entre grupos sulfuro, tales como un grupo S-metil sulfuro, un grupo S-etil sulfuro y un grupo S-2-piridil sulfuro, grupos éster, tales como un grupo acetilo, grupos aril metil éter, tales como un grupo bencilo, un grupo 9-fluorenilmetilo y un grupo tritilo, grupos etil éter, tales como un grupo S-2-cianoetilo, y similares. En este caso, el grupo protector P4 para el grupo amino, grupo carboxi o grupo hidroxi de cadena lateral de Lp1 es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo, y puede seleccionarse entre los mencionados anteriormente dependiendo de, por ejemplo, las propiedades del compuesto que tiene un grupo amino, grupo carboxi o grupo hidroxi que va a protegerse. Sin embargo, es necesario que el grupo protector P10 para un grupo mercapto y el grupo protector P4 para el grupo carboxi, grupo hidroxi o grupo amino de cadena lateral de Lp1 puedan retirarse por un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que el grupo protector P4 es un grupo ferc-butilo para un grupo carboxi, y el grupo protector P10 es un grupo S-metil sulfuro. El grupo protector P10 puede estar ausente. En este caso, el grupo mercapto del compuesto (55) está sin proteger.
Desprotegiendo el grupo protector P4 para el grupo carboxi, grupo hidroxi o grupo amino de cadena lateral de Lp1 en el compuesto (55) obtenido, puede producirse el compuesto (56). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo el grupo protector P10 para el grupo mercapto del compuesto (56) obtenido, puede producirse el compuesto (2a). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (55) también puede producirse por el siguiente procedimiento.
El compuesto (57) puede producirse derivatizando el compuesto de ácido carboxílico (54) que tiene un grupo mercapto protegido con P10 en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (22) descrito en el Procedimiento de producción 4. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). Los grupos protectores P4 y P10 son como se han mencionado anteriormente. El grupo protector P5 para un grupo carboxi puede seleccionarse adecuadamente entre aquellos descritos para el grupo protector del compuesto (6). Sin embargo, es necesario que el grupo protector P10 para un grupo mercapto y el grupo protector P4 para un grupo funcional de cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes de aquellas para el grupo protector P5 para un grupo carboxi. Por ejemplo, un ejemplo representativo incluye una combinación en la que P4 es un grupo ferc-butilo para un grupo carboxi, P10 es un grupo S-metil sulfuro, y P5 es un grupo alilo. El grupo protector P10 puede estar ausente. En este caso, el grupo mercapto del compuesto (57) está sin proteger.
El compuesto (58) puede producirse desprotegiendo el grupo protector P5 para el grupo carboxi del compuesto (57) obtenido. Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (55) puede producirse derivatizando el compuesto (58) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (4) en presencia de una base. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (2a) en el que el engarce tiene la estructura representada por -L1-L2-LP-, y LP es un resto peptídico que tiene un aminoácido hidrófilo distinto de glicina en el terminal N terminal puede producirse de la siguiente manera.
El compuesto (59) puede producirse derivatizando el compuesto de ácido carboxílico (54) que tiene un grupo mercapto protegido con P10 en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (37) descrito en el Procedimiento de producción 5. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). Los grupos protectores P4 y P10 son como se han mencionado anteriormente.
El compuesto (59) también puede producirse por el siguiente procedimiento, por ejemplo.
El compuesto (60) puede producirse derivatizando el compuesto de ácido carboxílico (54) que tiene un grupo mercapto protegido con P10 en éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (32) descrito en el Procedimiento de producción 4. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis. Los grupos protectores P4, P7 y P10 son como se han mencionado anteriormente y pueden seleccionarse adecuadamente entre aquellos descritos para el grupo protector del compuesto (6). Sin embargo, es necesario que el grupo protector P10 para un grupo mercapto y el grupo protector P4 para un grupo funcional de cadena lateral puedan retirarse por un procedimiento diferente o condiciones diferentes de aquellas para el grupo protector P7 para un grupo carboxi. Por ejemplo, un ejemplo representativo incluye una combinación en la que P4 es un grupo ferc-butilo para un grupo carboxi, P10 es un grupo S-metil sulfuro, y P7 es un grupo alilo. El grupo protector P10 puede estar ausente. En este caso, el grupo mercapto del compuesto (60) está sin proteger.
Desprotegiendo el grupo protector P7 para el grupo carboxi del péptido en el compuesto (60) obtenido, puede producirse el compuesto (61). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (59) puede producirse derivatizando el compuesto (61) obtenido en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (4) o una sal del mismo en presencia de una base. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P4 para el grupo carboxi de LP1 en el compuesto (59) obtenido, puede producirse el compuesto (62). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo el grupo protector P10 para el grupo mercapto del compuesto (62) obtenido, puede producirse el compuesto (2a). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (2a) en el que el engarce tiene la estructura representada por -L1-L2-LP-, y LP es un resto peptídico en el que el terminal C es un oligopéptido que consiste en 2 o 3 o más glicinas y está conectado al fármaco, e incluso en el caso cuando un aminoácido hidrófilo está presente en el terminal N, ningún otro aminoácido hidrófilo distinto de glicina esté presente en el mismo, puede producirse de la siguiente manera.
El compuesto (2a) puede producirse derivatizando el compuesto de ácido carboxílico (54) en éster activo, anhídrido de ácido mixto o similares, y haciéndolo reaccionar con el compuesto (49) descrito en el Procedimiento de producción 8. En este caso, el grupo mercapto puede no estar protegido con P10. Para la reacción, también pueden utilizarse reactivos de reacción y condiciones que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, bases y disolventes utilizados para la reacción pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
10. Procedimiento de producción 10
Entre el intermedio de producción representado por la fórmula (2), L1' en el que L1 se convierte para que tenga una estructura de (maleimid-N-il)-CH[-(CH2)n3-COOH]-C(=O)- terminal puede producirse por el siguiente procedimiento.
[Fórmula 51]

En la fórmula, P11 y P12 representan cada uno un grupo protector.
El intermedio de producción representado por la fórmula (2) tiene dos formas del engarce: una estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- y una estructura representada por -L1-L2-LP-.
El compuesto (2) en el que el engarce tiene la estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- puede producirse de la siguiente manera.
El compuesto de (maleimid-N-il)-(65) puede producirse haciendo reaccionar el aminoácido (63) que tiene un grupo carboxi de cadena protegida por P11 con la N-metoxicarbonilmaleimida (64) de -40 °C a 100 °C en presencia de una base, tal como bicarbonato sódico en agua. El compuesto de maleimidilo pueden sintetizarse a partir de un compuesto que tiene un grupo amino por un procedimiento conocido en la técnica usando N-metoxicarbonilmaleimida (por ejemplo, Keller, O.; Rudinger, J. Helv. Chem. Acta 1975, 58 (2), 531-541.) o un procedimiento equivalente al mismo. Como para el grupo protector P11 para un grupo carboxi, puede usarse un grupo protector usado comúnmente como grupo protector para un grupo carboxi en química sintética orgánica. Este es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo.
El compuesto (67) puede producirse derivatizando el compuesto (66) que tiene un grupo amino terminal protegido con P12 en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (41) descrito en el Procedimiento de producción 6 en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el compuesto (67) y el compuesto (41) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P12 para el grupo amino del compuesto (66) puede seleccionarse adecuadamente entre los descritos para el grupo protector del compuesto (6).
Desprotegiendo el grupo protector P12 para el grupo amino del compuesto (67) obtenido, puede producirse el compuesto (68). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (69) puede producirse derivatizando el compuesto (65) en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (68) obtenido en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el compuesto (65) y el compuesto (68) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P11 para el grupo carboxi del compuesto (69) obtenido, puede producirse el compuesto (2). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (2) en el que el engarce tiene la estructura representada por -L1-L2-LP- puede producirse de la siguiente manera.
De forma análoga, el compuesto (70) puede producirse derivatizando el compuesto (66) que tiene un grupo amino terminal protegido con P12 en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (43) descrito en el Procedimiento de producción 6 en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el compuesto (66) y el compuesto (43) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6). El grupo protector P12 es como se han mencionado anteriormente.
Desprotegiendo el grupo protector P12 para el grupo amino del compuesto (70) obtenido, puede producirse el compuesto (71). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (72) puede producirse derivatizando el compuesto (65) en un éster activo, anhídrido de ácido mixto o similar, y haciéndolo reaccionar con el compuesto (71) obtenido en presencia de una base. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el compuesto (65) y el compuesto (71) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P11 para el grupo carboxi del compuesto (72) obtenido, puede producirse el compuesto (2). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
11. Procedimiento de producción 11
Entre el intermedio de producción representado por la fórmula (2), aquellos que tienen L1' en el que L1 se convierte para que tenga una estructura de grupo maleimidilo terminal o grupo haloacetilo terminal, y L2 es -N[-(CH2CH2-O)n7-CH2CH2-OH]-CH2-C(=O)- puede producirse por el siguiente procedimiento.
[Fórmula 52]
H2N-CH2-COOP13

Lr-N[-(CH2CH2-0)n7-CH2CH2-0H]-CH2-C00P13
77
1
L1'-N[-(CH2CH2-0)n7-CH2CH2-0H]-CH2-C00H
78
H-Lp-NH-(CH2)n1-La-Lb-Lc-(NH-DX)
, 41

En la fórmula, P13 representa un grupo protector, y X representa un grupo saliente.
El intermedio de producción representado por la fórmula (2) tiene dos formas como el engarce: una estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- y una estructura representada por-L1-L2-LP-.
El compuesto (2) en el que el engarce tiene la estructura representada por -L1-L2-LP-NH-(CH2)n1-La-Lb-Lc- puede producirse de la siguiente manera.
El compuesto (75) puede producirse haciendo reaccionar el derivado de glicina (73) que tiene el terminal C protegido
con P13 con el compuesto (74) en presencia de una base. El grupo protector P13 para un grupo carboxi es preferentemente un grupo protector que puede desprotegerse en condiciones ácidas, pero no se limita al mismo. Los ejemplos del grupo saliente X del compuesto (74) pueden incluir ésteres de ácido sulfónico, tales como p-tolueno sulfonato, metil sulfonato y trifluorometil sulfonato, así como haluros, tales como yoduro, bromuro y cloruro. Para esta reacción, también pueden usarse condiciones de reacción que se usan generalmente para N-alquilación, y la base y disolvente utilizados para la reacción pueden seleccionarse entre aquellos descritos para la síntesis del compuesto (6).
El compuesto (77) puede producirse derivatizando el derivado de ácido carboxílico (76) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (75) obtenido. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de péptido entre el derivado de ácido carboxílico (76) y el compuesto (75) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Desprotegiendo el grupo protector P13 para el grupo carboxi del compuesto (77) obtenido, puede producirse el compuesto (78). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (2) puede producirse derivatizando el compuesto (78) obtenido en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (41) descrito en el Procedimiento de producción 6. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el derivado de ácido carboxílico (78) y el compuesto (41) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
El compuesto (2) en el que el engarce tiene la estructura representada por -L1-L2-LP- puede producirse de la siguiente manera.
De forma análoga, el compuesto (2) puede producirse derivatizando el compuesto (78) en éster activo, anhídrido de ácido mixto, haluro de ácido o similares, y haciéndolo reaccionar con el compuesto (43) descrito en el Procedimiento de producción 6. Las condiciones de reacción, reactivos, bases y disolventes utilizados para formar un enlace de amida entre el derivado de ácido carboxílico (78) y el compuesto (43) pueden seleccionarse adecuadamente entre los descritos para la síntesis del compuesto (6).
Todos los compuestos intermedios de los Procedimientos de producción 1 a 11 pueden formar sales.
Entre tanto, el conjugado de anticuerpo y fármaco de la presente invención, cuando se deja al aire o recristaliza, puede absorber humedad para tener agua de adsorción o convertirse en un hidrato, y dicho un compuesto y una sal que contiene agua también se incluyen en la presente invención.
Un compuesto marcado con diversos isótopos radioactivos o no radioactivos también se incluye en la presente invención. Uno o más átomos que constituyen el conjugado de anticuerpo y fármaco de la presente invención pueden contener un isótopo atómico en una proporción no natural. Los ejemplos del isótopo atómico incluyen deuterio (2H), tritio (3H), yodo-125 (125I) y carbono-13 (13C). Además, el compuesto de la presente invención puede estar marcado radioactivamente con un isótopo radioactivo, tal como tritio (3H), yodo-125 (125I), carbono-14 (14C), cobre-64 (64Cu), circonio-89 (89Zr), yodo-124 (124I), flúor-18 (18F), indio-111 (111I), carbono-11 (11C) y yodo-131 (131I). El compuesto marcado con un isótopo radioactivo es útil como un agente terapéutico o profiláctico, un reactivo para investigar, tal como un reactivo de ensayo y un agente para diagnóstico, tal como un agente de formación de imágenes de diagnóstico in vivo. Sin que esté relacionado con la radioactividad, cualquier tipo de variante isotópica del conjugado de anticuerpo y fármaco de la presente invención está dentro del ámbito de la presente invención.
[Fármacos]
El conjugado de anticuerpo y fármaco de la presente invención muestra una actividad citotóxica contra células cancerosas y, por tanto, se puede usar como fármaco, particularmente como agente terapéutico y/o agente profiláctico para el cáncer.
Los ejemplos del tipo de cáncer al que se aplica el conjugado de anticuerpo y fármaco de la presente invención, incluyen cáncer de pulmón, cáncer de riñón, cáncer urotelial, cáncer colorrectal, cáncer de próstata, glioblastoma multiforme, cáncer de ovario, cáncer de páncreas, cáncer de mama, melanoma, cáncer de hígado, cáncer de vejiga, cáncer de estómago o cáncer de esófago, sin embargo, no se limita a ellos siempre que sea una célula cancerosa que exprese, en una célula cancerosa como sujeto de tratamiento, una proteína que el anticuerpo dentro del conjugado de anticuerpo y fármaco puede reconocer.
El conjugado de anticuerpo y fármaco de la presente invención puede administrarse preferentemente a un mamífero, pero se administra más preferentemente a un ser humano.
Las sustancias usadas en una composición farmacéutica que contiene el conjugado de anticuerpo y fármaco de la presente invención, pueden seleccionarse y aplicarse adecuadamente a partir de aditivos de formulación o similares que generalmente se usan en la técnica, teniendo en cuenta la dosificación o concentración de la administración.
El conjugado de anticuerpo y fármaco de la presente invención puede administrarse como una composición farmacéutica que contiene al menos un principio farmacéuticamente adecuado. Por ejemplo, la composición farmacéutica anterior contiene típicamente al menos un vehículo farmacéutico (por ejemplo, líquido esterilizado). por ejemplo, agua y aceite (aceite de petróleo y aceite de origen animal, origen vegetal u origen sintético (el aceite puede ser, por ejemplo, aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo o similar)). Cuando la composición farmacéutica anterior se administra por vía intravenosa, el agua es el vehículo más típico. También pueden utilizarse como vehículos líquidos, solución salina, una solución acuosa de dextrosa y una solución acuosa de glicerol, en particular, para una solución inyectable. En la técnica se conocen vehículos farmacéuticos adecuados. Si se desea, la composición anterior también puede contener una pequeña cantidad de un agente hidratante, un agente emulsionante o un agente tamponador de pH. En "Remington's Pharmaceutical Sciences" de E. W. Martin, se desvelan ejemplos de vehículos farmacéuticos adecuados. Las formulaciones corresponden a un modo de administración.
Se conocen diversos sistemas de suministro y pueden usarse para administrar el conjugado de anticuerpo y fármaco de la presente invención. Como ejemplos de vías de administración se incluyen la intradérmica, intramuscular, intraperitoneal, intravenosa y subcutánea, pero no se limita a estas. La administración puede hacerse, por ejemplo, mediante inyección o inyección en embolada. Según una realización preferida específica, la administración del conjugado de anticuerpo y fármaco se realiza mediante inyección. La administración parenteral es una vía de administración preferida.
Según una realización representativa, la composición farmacéutica se prescribe como una composición farmacéutica adecuada para administración intravenosa a seres humanos, según los procedimientos convencionales. La composición para administración intravenosa es típicamente una solución en una solución tampón acuosa estéril e isotónica. Si es necesario, para aliviar el dolor en el lugar de la inyección, el fármaco puede contener un agente solubilizante y anestésicos locales (por ejemplo, lignocaína). En general, el principio anterior se proporciona individualmente como uno cualquiera de polvo liofilizado o un concentrado anhidro contenido en un recipiente que se produce mediante sellado en una ampolla o una bolsita que tiene una cantidad del agente activo o como una mezcla en una forma de dosificación unitaria. Cuando el fármaco se administrará por inyección, éste puede administrarse desde un frasco de inyección que contiene agua o solución salina de calidad farmacéutica estéril. Cuando el fármaco se administra mediante inyección, se puede proporcionar una ampolla de agua estéril o solución salina para inyección de modo que los principios mencionados anteriormente se mezclen entre sí antes de la administración.
La composición farmacéutica de la presente invención puede ser una composición farmacéutica que solo contenga el conjugado de anticuerpo y fármaco de la presente invención o una composición farmacéutica que contenga el conjugado de anticuerpo y fármaco y al menos un agente para el tratamiento del cáncer que no sea el conjugado. El conjugado de anticuerpo y fármaco de la presente invención puede administrarse con otro agente para el tratamiento del cáncer. Por consiguiente, el efecto contra el cáncer puede potenciarse. Otro agente contra el cáncer utilizado con ese fin puede administrarse a una persona de manera simultánea, individual o después del conjugado de anticuerpo y fármaco, y puede administrarse modificando el intervalo de administración de cada uno de ellos. Como ejemplos del agente para el tratamiento del cáncer se incluyen abraxano, carboplatino, cisplatino, gemcitabina, irinotecán (CPT-11), paclitaxel, pemetrexed, sorafenib, vinorrelbina, fármacos descritos en la publicación internacional N.°WO 2003/038043, análogos de LH-RH (leuprorelina, goserelina o similares), fosfato de estramustina, antagonista de estrógenos (tamoxifeno, raloxifeno o similares) y un inhibidor de aromatasa (anastrozol, letrozol, exemestano, o similares), pero no está limitado a éstos, siempre que sea un fármaco que tenga actividad antitumoral.
La composición farmacéutica puede formularse en una formulación de liofilización o una formulación líquida como una formulación que tiene la composición deseada y la pureza necesaria. Cuando se formula como una formulación de liofilización, puede ser una formulación que contenga aditivos de formulación adecuados que se usen en la técnica. También para una formulación líquida, ésta se puede formular como una formulación líquida que contenga diversos aditivos de formulación que se usan en la técnica.
La composición y concentración de la composición farmacéutica puede variar dependiendo del procedimiento de administración. Sin embargo, el conjugado de anticuerpo y fármaco contenido en la composición farmacéutica de la presente invención puede mostrar el efecto farmacéutico incluso a una pequeña dosificación cuando el conjugado de anticuerpo y fármaco tiene una mayor afinidad por un antígeno, es decir, mayor afinidad (= menor valor de Kd) en términos de la constante de disociación (es decir, valor de Kd) por el antígeno. Por tanto, para determinar la dosificación del conjugado de anticuerpo y fármaco, la dosificación puede determinarse en vista de una situación relacionada con la afinidad entre el conjugado de anticuerpo y fármaco y el antígeno. Cuando el conjugado de anticuerpo y fármaco de la presente invención se administra, por ejemplo, a un ser humano, se pueden administrar aproximadamente de 0,001 a 100 mg/kg una vez o varias veces con un intervalo de una vez durante 1 a 180 días.
[Ejemplos]
La presente invención se describe específicamente en vista de los ejemplos que se muestran a continuación, sin embargo, la presente invención no se limita a ellos, y además, de ninguna manera se interpretan en un sentido limitado. Además, a menos que se indique específicamente otra cosa, el reactivo, disolvente y material de partida que se describe en la memoria descriptiva puede obtenerse fácilmente de un proveedor comercial.
Ejemplo de referencia 1 Anticuerpo M30-H1-L4
Entre los anticuerpos humanizados de un anticuerpo anti-B7-H3, se produjo un anticuerpo compuesto por una cadena pesada que consiste en una secuencia de aminoácidos descrita en las posiciones de aminoácidos 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos descrita en las posiciones de aminoácidos 21 a 233 en la SEQ ID NO: 16 según un procedimiento conocido en la técnica. El anticuerpo anti-B7-H3 humanizado obtenido se denominó anticuerpo M30-H1-L4.
Ejemplo de referencia 2 Anticuerpo M30-H1-L4P
La modificación de un glucano ligado al anticuerpo M30-H1-L4 obtenido anteriormente se reguló por desfucosilación según un procedimiento conocido en la técnica. El anticuerpo obtenido con la modificación regulada de un glucano se denominó anticuerpo M30-H1-L4P.
Ejemplo de referencia 3 Anticuerpo anti-CD30
Se produjo un anticuerpo anti-CD30 con referencia a la publicación nacional de la solicitud de patente internacional N.° 2005-506035. Su secuencia se muestra en las SEQ ID NO: 27 y 28.
Ejemplo de referencia 4 Anticuerpo anti-CD33
Se produjo un anticuerpo anti-CD33 con referencia a la patente japonesa abierta a inspección pública número N° 8 48637. Su secuencia se muestra en las SEQ ID NO: 29 y 30.
Ejemplo de referencia 5 Anticuerpo anti-CD70
Se produjo un anticuerpo anti-CD70 con referencia a la publicación nacional de la solicitud de patente internacional N.° 2008-538292. Su secuencia se muestra en las SEQ ID NO: 31 y 32.
Ejemplo 1: 4-Amino-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]butanamida
[Fórmula 53]

Procedimiento 1: (4-{[(1S,9S)-9-Etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)carbamato de tere-butilo
Se disolvió ácido 4-(tere-butoxicarbonilamino)butanoico (0,237 g, 1,13 mmol) en diclorometano (10 ml), se cargó con N-hidroxisuccinimida (0,130 g, 1,13 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,216 g, 1,13 mmol) y se agitó durante 1 hora. La solución de reacción se añadió gota a gota a una solución de N,N'-dimetilformamida (10,0 ml) cargada con metanosulfonato del compuesto (4) (0,500 g, 0,941 mmol) y trietilamina (0,157 ml, 1,13 mmol) y se agitó a temperatura ambiente durante 1 día. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo: metanol = 8:2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo intenso (0,595 g, cuantitativo). RMN 1H (400 MHz, DMSO-d6) 6: 0,87 (3H, t, J = 7,2 Hz), 1,31 (9H, s), 1,58 (1H, t, J = 7,2 Hz), 1,66 (2H, t, J = 7,2 Hz), 1,82-1,89 (2H, m), 2,12-2,21 (3H, m), 2,39 (3H, s), 2,92 (2H, t, J = 6,5 Hz), 3,17 (2H, s), 5,16 (1H, d, J = 18,8 Hz), 5,24 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,55-5,59 (1H, m), 6,53 (1H, s), 6,78 (1H, t, J = 6,3 Hz), 7,30 (1H, s), 7,79 (1H, d, J = 11,0 Hz), 8,40 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 621 (M+H)+
Procedimiento 2: 4-Amino-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10, 13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]butanamida
El compuesto (0,388 g, 0,626 mmol) obtenido en el Procedimiento 1 anterior se disolvió en diclorometano (9,00 ml). Se añadió ácido trifluoroacético (9,00 ml) para agitarse durante 4 horas. El disolvente se retiró a presión reducida y el residuo obtenido se purificó por cromatografía en columna sobre gel de sílice [cloroformo - capa orgánica repartida de cloroformo: metanol: agua = 7:3:1 (v/v/v)] para producir trifluoroacetato del compuesto del título en forma de un sólido de color amarillo (0,343 g, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 6: 0,87 (3H, t, J = 7,2 Hz), 1,79-1,92 (4H, m), 2,10-2,17 (2H, m), 2,27 (2H, t, J = 7,0 Hz), 2,40 (3H, s), 2,80-2,86 (2H, m), 3,15-3,20 (2H, m), 5,15 (1H, d, J = 18,8 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,42 (2H, s),
5,54-5,61 (1H, m), 6,55 (1H, s), 7,32 (1H, s), 7,72 (3H, s a), 7,82 (1H, d, J = 11,0 Hz), 8,54 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 521 (M+H)+
Ejemplo 2: Conjugado de anticuerpo y fármaco (1)
[Fórmula 54]

Procedimiento 1: N-(ferc-butoxicarbonil)glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
Se disolvió N-(ferc-butoxicarbonil)glicilglicil-L-fenilalanilglicina (80,9 g, 0,185 mmol) en diclorometano (3,00 ml), se cargó con N-hidroxisuccinimida (21,3 mg, 0,185 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (35,5 mg, 0,185 mmol) y se agitó durante 3,5 horas. La solución de reacción se añadió gota a gota a una solución de N,N'-dimetilformamida (1,50 ml) cargada con el compuesto (80,4 mg, 0,154 mmol) del Ejemplo 1 y se agitó a temperatura ambiente durante 4 horas. El disolvente se retiró a presión reducida y el residuo obtenido se purificó por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo: metanol = 8:2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,106 g, 73 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,36 (9H, s), 1,71 (2H, m), 1,86 (2H, t, J = 7,8 Hz), 2,15-2,19 (4H, m), 2,40 (3H, s), 2,77 (1H, dd, J = 12,7, 8,8 Hz), 3,02 (1H, dd, J = 14,1,4,7 Hz), 3,08-3,11 (2H, m), 3,16-3,19 (2H, m), 3,54 (2H, d, J = 5,9 Hz), 3,57-3,77 (4H, m), 4,46-4,48 (1H, m), 5,16 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 6,53 (1H, s), 7,00 (1H, t, J = 6,3 Hz), 7,17-7,26 (5H, m), 7,31 (1H, s), 7,71 (1H, t, J = 5,7 Hz), 7,80 (1H, d, J = 11,0 Hz), 7,92 (1H, t, J = 5,7 Hz), 8,15 (1H, d, J = 8,2 Hz), 8,27 (1H, t, J = 5,5 Hz), 8,46 (1H, d, J = 8,2 Hz). EM (APCI) m/z: 939 (M+H)+
Procedimiento 2: Trifluoroacetato de glicilglicil-L-fenilalanil-N-(4-{[(1S, 9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (72,6 mg, 77,3 |jmol) obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo (64,8 g, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,71-1,73 (2H, m), 1,82-1,90 (2H, m), 2,12-2,20 (4H, m), 2,40 (3H, s), 2,75 (1H, dd, J = 13,7, 9,4 Hz), 3,03-3,09 (3H, m), 3,18-3,19 (2H, m), 3,58-3,60 (2H, m), 3,64 (1H, d, J = 5,9 Hz), 3,69 (1H, d, J = 5,9 Hz), 3,72 (1H, d, J = 5,5 Hz), 3,87 (1H, dd, J = 16,8, 5,9 Hz), 4,50-4,56 (1H, m), 5,16 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 7,17-7,27 (5H, m), 7,32 (1H, s), 7,78-7,81 (2H, m), 7,95-7,97 (3H, m), 8,33-8,35 (2H, m), 8,48-8,51 (2H, m).
EM (APCI) m/z: 839 (M+H)+
Procedimiento 3: (3S,12S)-12-Bencil-21-{[(1S, 9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-3-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-4,7,10,13,16,21-hexaoxo-5,8,11,14,17-pentaazaheneicosan-1-noato de ferc-butilo
Se disolvió ácido (2S)-4-ferc-butoxi-2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-4-oxobutanoico (0,625 g, 1,52 mmol) en diclorometano (10,0 ml), cargado con N-hidroxisuccinimida (0,175 g, 1,52 mol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,291 g, 1,52 mmol) y se agitó durante 1 hora. La solución de reacción se añadió gota a gota a una solución de N,N'-dimetilformamida (10,0 ml) cargada con el compuesto (1,00 g, 1,01 mmol) obtenido en el Procedimiento 2 anterior y se agitó a temperatura ambiente durante 20 horas. El disolvente se retiró a presión reducida y el residuo obtenido se purificó por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 8:2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,873 g, 70 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,88 (3H, t, J = 7,4 Hz), 1,37 (9H, s), 1,68-1,78 (2H, m), 1,81-1,93 (2H, m), 2,10-2,23 (4H, m), 2,41 (3H, s), 2,68-2,85 (3H, m), 2,99-3,22 (5H, m), 3,58-3,81 (6H, m), 4,19-4,36 (3H, m), 4,38-4,52 (2H, m), 5,17 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 19,2 Hz), 5,43 (2H, s), 5,54-5,62 (1H, m), 6,55 (1H, s), 7,15-7,34 (8H, m), 7,41 (2H, t, J = 7,2 Hz), 7,66-7,75 (4H, m), 7,81 (1H, d, J = 11,0 Hz), 7,88 (2H, d, J = 7,4 Hz), 8,01-8,06 (1H, m), 8,14 (1H, d, J = 8,2 Hz), 8,17-8,22 (1H, m), 8,25-8,30 (1H, m), 8,47 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 1232 (M+H)+
Procedimiento 4: (3S,12S)-12-Bencil-3-{[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]amino}-21-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4,7,10,13,16,21-hexaoxo-5,8,11,14,17-pentaazaheneicosan-1-noato de ferc-butilo
El compuesto (0,800 g, 0,649 mmol) obtenido en el Procedimiento 3 anterior se disolvió en N,N'-dimetilformamida (3,00 ml), se cargó con piperidina (0,643 ml, 6,49 mmol) y se agitó durante 1 hora. El disolvente se retiró secando a presión reducida y el residuo obtenido se disolvió en N,N'-dimetilformamida (10 ml). Se le añadió hexanoato de N-succinimidil 6-maleimida (0,300 g, 0,974 mmol) se y se agitó durante 20 horas. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 8:2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,224 g, 29 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,6 Hz), 1,15-1,22 (2H, m), 1,35 (9H, s), 1,44-1,47 (4H, m), 1,71-1,73 (2H, m), 1,80-1,91 (2H, m), 2,08 (2H, t, J = 7,6 Hz), 2,13-2,20 (4H, m), 2,40 (3H, s), 2,67 (1H, dt, J = 11,1,4,8 Hz), 2,78 (1H, dd, J = 13,6, 9,4 Hz), 2,99-3,17 (6H, m), 3,31-3,36 (2H, m), 3,57-3,76 (6H, m), 4,45-4,47 (1H, m), 4,57-4,60 (1H, m), 5,16 (1H, d, J = 18,7 Hz), 5,25 (1H, d, J = 18,7 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,15 7,27 (5H, m), 7,31 (1H, s), 7,70 (1H, t, J = 5,4 Hz), 7,80 (1H, d, J = 10,9 Hz), 7,99 (1H, t, J = 5,7 Hz), 8,09-8,12 (3H, m), 8,25 (1H, t, J = 6,0 Hz), 8,45 (1H, d, J = 9,1 Hz).
EM (APCI) m/z: 1203 (M+H)+
Procedimiento 5: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-a-aspartilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (0,224 g, 0,186 mmol) obtenido en el Procedimiento 4 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo pálido (21,2 mg, 10 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,2 Hz), 1,13-1,21 (2H, m), 1,42-1,45 (6H, m), 1,70-1,72 (2H, m), 1,85-1,88 (2H, m), 2,06-2,20 (6H, m), 2,39 (3H, s), 2,63-2,67 (1H, m), 2,78-2,81 (1H, m), 3,04-3,12 (6H, m), 3,63-3,70 (6H, m), 4,46-4,52 (2H, m), 5,16 (1H, d, J = 18,8 Hz), 5,25 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,55-5,58 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,18-7,23 (6H, m), 7,30 (1H, s), 7,71 (1H, t, J = 5,5 Hz), 7,79 (1H, d, J = 10,9 Hz), 7,99-8,02 (1H, m), 8,10-8,11 (3H, m), 8,27-8,30 (1H, m), 8,47-8,50 (1H, m).
EM (APCI) m/z: 1147 (M+H)+
Procedimiento 6: Conjugado de anticuerpo y fármaco (1)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que
tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (8,0 ml) se recogió en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,124 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,400 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,249 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 anterior y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,050 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 18,5 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 3,56 mg/ml, rendimiento de anticuerpo: 66 mg (83 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,3.
Ejemplo 3: Conjugado de anticuerpo y fármaco (2)
[Fórmula 55]
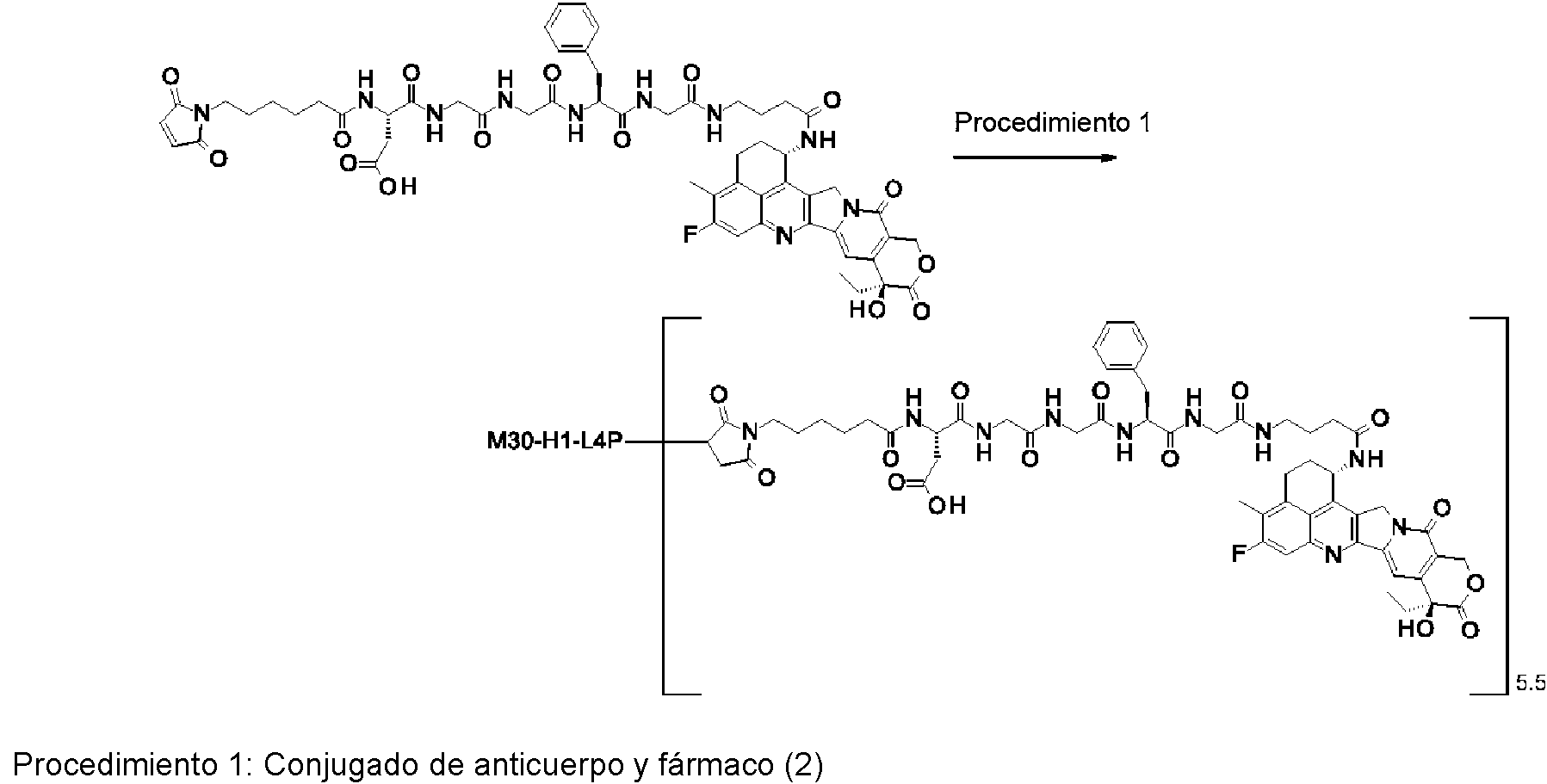
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (8,0 ml) se recogió en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,187 ml; 3,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,400 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,373 ml; 6,9 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,075 ml; 13,8 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20
minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 16 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 3,55 mg/ml, rendimiento de anticuerpo: 57 mg (71 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,5.
Ejemplo 4 : Conjugado de anticuerpo y fármaco (3)
[Fórmula 56]
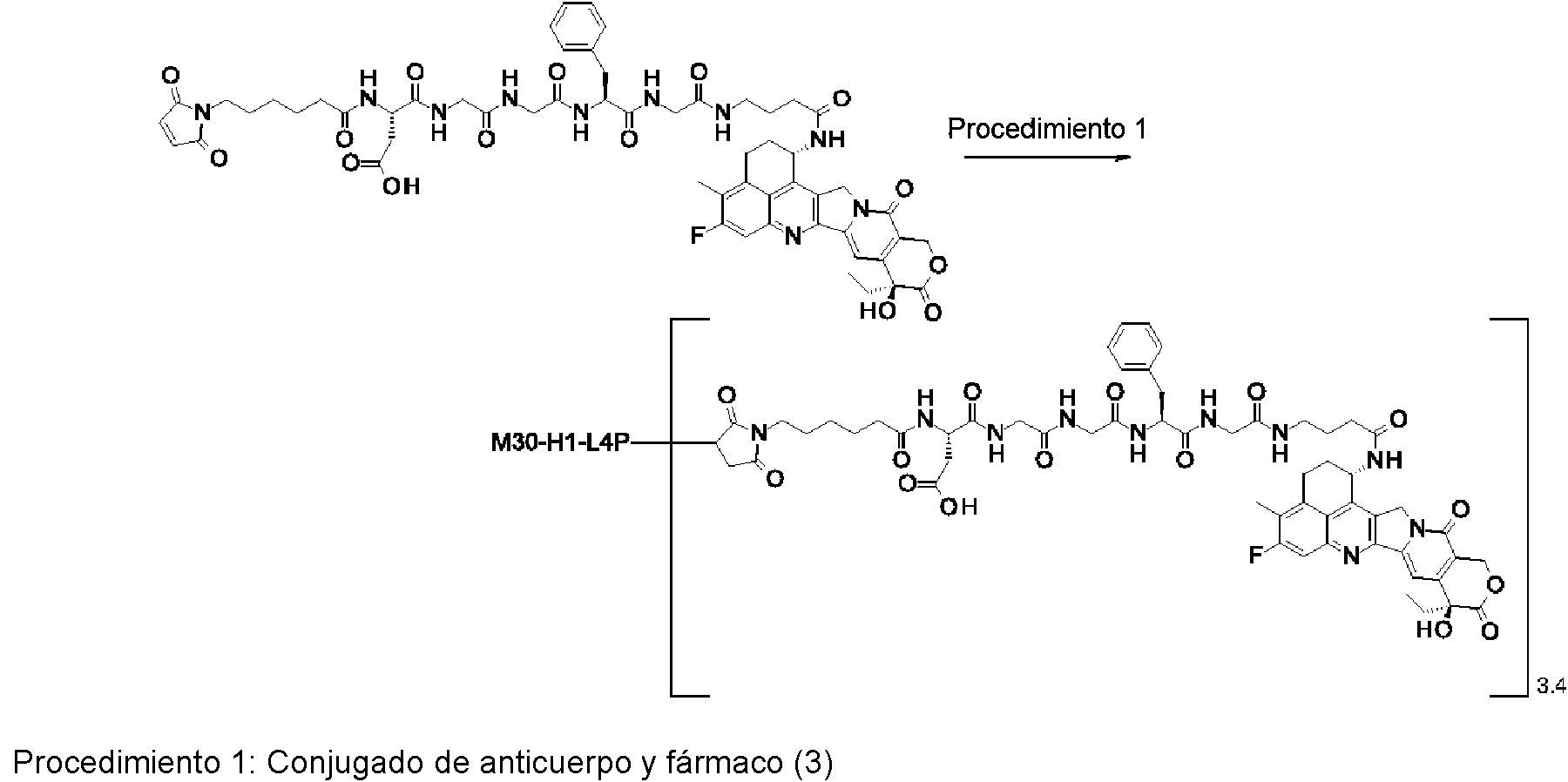
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml reemplazando el medio por PBS6,0/EdTa usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,109ml) y una solución de dimetilsulfóxido que contenía 10 mM (0,039 ml; 4,6 equivalentes por molécula de anticuerpo) del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,008 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título. Después de eso, la solución se concentró por el Procedimiento común A.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 10,63 mg/ml, rendimiento de anticuerpo: 7,4 mg (59%) y un número promedio de
moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 5: Conjugado de anticuerpo y fármaco (4)
[Fórmula 57]

Procedimiento 1: Conjugado de anticuerpo y fármaco (4)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EDTA usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,067 ml) y una solución de dimetilsulfóxido que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 (0,085 ml; 10,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 60 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,013 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,48mg/ml, rendimiento de anticuerpo: 8,88 mg (71 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,8. Ejemplo 6: Conjugado de anticuerpo y fármaco (5)
[F ó rm u la 58 ]

Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EDTA usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,025 ml) y una solución de dimetilsulfóxido que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 (0,127 ml; 15,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 60 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,019 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título. Después de eso, la solución se concentró por el Procedimiento común A.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 0,99 mg/ml, rendimiento de anticuerpo: 5,94 mg (48 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,9.
Ejemplo 7: Conjugado de anticuerpo y fármaco (6)
[F ó rm u la 59]

Casi todas las cantidades de los conjugados de anticuerpo y fármaco de los Ejemplos 5 y 6 se mezclaron y la solución se concentró por el Procedimiento común A para producir el conjugado de anticuerpo y fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 14,36 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,2.
Ejemplo 8: Conjugado de anticuerpo y fármaco (7)
[Fórmula 60]

Procedimiento 1: Conjugado de anticuerpo y fármaco (7)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00593 ml; 9,2 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el
conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 270400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,18mg/ml, rendimiento de anticuerpo: 7,08 mg (71 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,6.
Ejemplo 9: Conjugado de anticuerpo y fármaco (8)
[Fórmula 61]

Procedimiento 1: Conjugado de anticuerpo y fármaco (8)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75mlmg-1cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0119ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 270400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,07 mg/ml, rendimiento de anticuerpo: 6,42 mg (64%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,9.
Ejemplo 10: Conjugado de anticuerpo y fármaco (9)
[Fórmula 62]

Procedimiento 1: Conjugado de anticuerpo y fármaco (9)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00593 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 256400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,09 mg/ml, rendimiento de anticuerpo: 6,54 mg (65%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,4.
Ejemplo 11: Conjugado de anticuerpo y fármaco (10)
[F ó rm u la 63]

Procedimiento 1: Conjugado de anticuerpo y fármaco (10)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 256400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £Dí280 = 5000 (valor promedio medido) y £Dí370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,04 mg/ml, rendimiento de anticuerpo: 6,24 mg (62%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,6.
Ejemplo 12: Conjugado de anticuerpo y fármaco (11)
[Fórmula 64]

Procedimiento 1: Conjugado de anticuerpo y fármaco (11)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 5 del Ejemplo 2 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00593 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,12 mg/ml, rendimiento de anticuerpo: 6,72 mg (67%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,5.
Ejemplo 13: Conjugado de anticuerpo y fármaco (12)
[Fórmula 65]
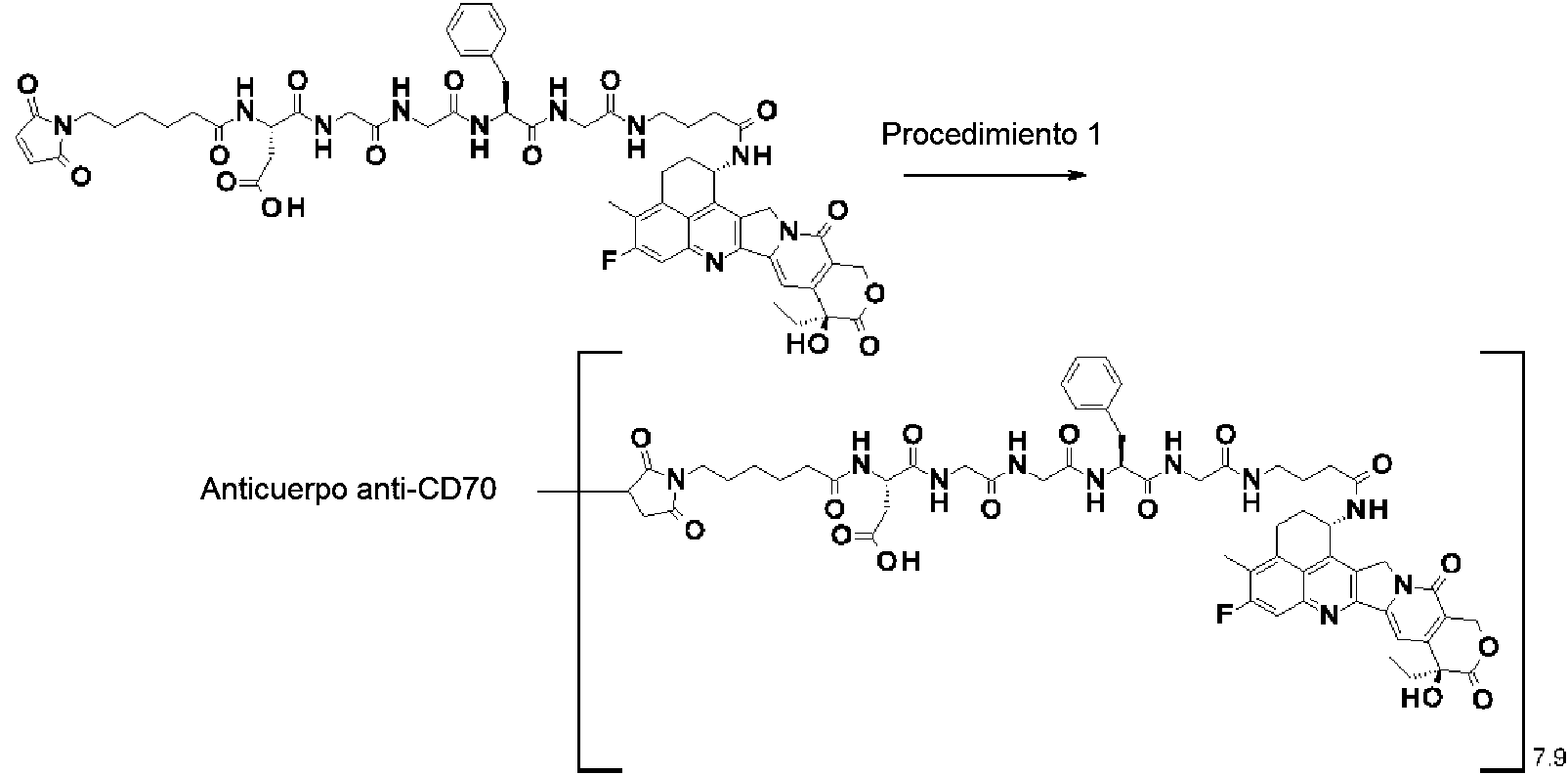
Procedimiento 1: Conjugado de anticuerpo y fármaco (12)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 7 del Ejemplo 1 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,03 mg/ml, rendimiento de anticuerpo: 6,18 mg (62%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,9.
Ejemplo 14: Conjugado de anticuerpo y fármaco (13)
[Fórmula 66]


Procedimiento 1: N-(2-{2-[2-(2-Hidroxietoxi)etoxi]etoxi}etil)glicinato de ferc-butilo
A una solución en N,N-dimetilformamida (50,0 ml) de 4-metilbenceno sulfonato de 2-{2-[2-(2-hidroxietoxi)etoxi]etoxi}etilo (Bioorg. Med. Chem. Lett., 2011, Vol. 21, pág. 550; 1,75 g, 5,00 mmol) y ferc-butil clorhidrato de glicina (1,26 g, 7,52 mmol), se añadió N,N-diisopropiletilamina (1,94 g, 15,0 mmol) y se agitó a 60 °C durante 10 horas. Se añadió cloroformo a la solución de reacción, la capa orgánica se lavó con ácido clorhídrico 1 N y la capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 8:1 (v/v)] para producir el compuesto del título en forma de una sustancia oleosa incolora (426 mg, 28 %).
RMN 1H (400 MHz, CDCh) 8: 1,47 (9H, s), 2,80 (2H, t, J = 5,3 Hz), 3,32 (2H, s), 3,76-3,54 (17H, m).
Procedimiento 2: N-[6-(2,5-Dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-N-(2-{2-[2-(2-hidroxietoxi)etoxi]etoxi}etil)glicinato de ferc-butilo
El compuesto (426 mg, 1,39 mmol) obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 4 del Ejemplo 2 para producir el compuesto del título en forma de una sustancia oleosa incolora (489 mg, 70%).
RMN 1H (400 MHz, CDCla) 8: 1,28-1,36 (2H, m), 1,45 (9H, s), 1,57-1,71 (4H, m), 2,39 (2H, t, J = 7,3 Hz), 3,48-3,76 (3H, m), 4,02 (2H, s), 6,68 (2H, s).
Procedimiento 3: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-N-(2-{2-[2-(2-hidroxietoxi)etoxi]etoxi}etil)glicina El compuesto (489 mg, 0,977 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido incoloro (211 mg, 49 %).
RMN 1H (400 MHz, CDCla) 8: 1,38-1,28 (2H, m), 1,73-1,55 (4H, m), 2,28 (2H, t, J = 7,0 Hz), 3,50-3,79 (18H, m), 4,12 (2H, s), 6,68 (2H, s).
Procedimiento 4: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-N-(2-{2-[2-(2
hidroxietoxi)etoxi]etoxi}etil)glicMglicilglicil-L-fenilalanil-N-(4-{ [(1S,9S)-9-etN-5-fluoro-9-hidroxi-4-metiM0,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indoNzino[1,2-b]quinoNn-1-N]amino}-4-oxobutN)gNcinamida
El compuesto (48,9 mg, 0,110 mmol) obtenido en el Procedimiento 3 anterior se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando el compuesto (84,0 mg, 0,100 mmol) obtenido en el Procedimiento 2 del Ejemplo 2 en lugar de metanosulfonato del compuesto (4) para producir el compuesto del título en forma de un sólido de color amarillo pálido (54,0 mg, 43 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,2 Hz), 1,14-1,26 (2H, m), 1,39-1,51 (4H, m), 1,68-1,76 (2H, m), 1,81-1,91 (2H, m), 2,08-2,23 (4H, m), 2,40 (3H, s), 2,73-2,84 (1H, m), 2,98-3,21 (5H, m), 3,25-3,79 (26H, m), 3,93 (2H, s), 4,43-4,49 (1H, m), 4,54-4,61 (1H, m), 5,21 (2H, c, J = 18,6 Hz), 5,42 (2H, s), 5,54-5,60 (1H, m), 6,53 (1H, s), 7,00 (2H, s), 7,14-7,27 (5H, m), 7,31 (1H, s), 7,68-7,74 (1H, m), 7,80 (1H, d, J = 11,0 Hz), 8,02-8,32 (4H, m), 8,46 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1265 (M+H)+
Procedimiento 5: Conjugado de anticuerpo y fármaco (13)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 4 anterior, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 6 del Ejemplo 2.
Concentración de anticuerpo: 13,13 mg/ml, rendimiento de anticuerpo: 9,2 mg (74%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 15: Conjugado de anticuerpo y fármaco (14)
[Fórmula 67]

Procedimiento 1: N-(ferc-butoxicarbonil)-p-alanilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4oxobutil)glicinamida
El compuesto (0,839 g, 1,00 mmol) obtenido en el Procedimiento 2 del Ejemplo 2 se hizo reaccionar de la misma manera que el Procedimiento 3 del Ejemplo 2 usando N-(terc-butoxicarbonil)-p-alanina en lugar de hexanoato de N-succinimidil 6-maleimida. El producto en bruto obtenido se usó en el siguiente procedimiento sin purificación.
Procedimiento 2: p-Alanilglicilglicil-L-fenilalanil-N-(4-{[(1S, 9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El producto en bruto obtenido en el Procedimiento 1 se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,610 g, 67 %). RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,4 Hz), 1,67-1,77 (2H, m), 1,79-1,92 (2H, m), 2,09-2,22 (4H, m), 2,40 (3H, s), 2,46-2,55 (2H, m), 2,82-2,73 (1H, m), 2,95-3,13 (5H, m), 3,14-3,21 (2H, m), 3,55-3,80 (6H, m), 4,44-4,52 (1H, m), 5,20 (2H, dd, J = 35,0, 19,0 Hz), 5,42 (2H, s), 5,53-5,60 (1H, m), 6,54 (1H, s), 7,14-7,28 (5H, m), 7,31 (1H, s), 7,67 (2H, s a), 7,72-7,78 (1H, m), 7,80 (1H, d, J = 11,0 Hz), 8,10-8,17 (2H, m), 8,29 (1H, t, J = 5,9 Hz), 8,42 (1H, t, J = 5,7 Hz), 8,47 (1H, d, J = 8,6 Hz).
Procedimiento 3: Ácido (2S)-5-terc-butoxi-2-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-5-oxopentanoico
Se disolvió L-glutamato de 5-terc-butilo (1,02 g, 5,00 mmol) en una solución acuosa saturada de hidrogenocarbonato sódico (20,0 ml), cargada con N-metoxicarbonilmaleimida (0,775 g, 5,00 mmol) a 0 °C, y se agitó a 0 °C durante 30 minutos y después se agitó a temperatura ambiente durante 1 hora. La solución de reacción se hizo ácida mediante la adición de ácido clorhídrico 5 N a 0 °C y después se extrajo con acetato de etilo. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida para producir un producto en bruto. El producto en bruto obtenido se usó en el siguiente procedimiento sin purificación.
Procedimiento 4: N-[(2S)-5-terc-butoxi-2-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-5-oxopentanoil]-p-alanilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El producto en bruto (85,0 mg, 0,300 mmol) obtenido en el Procedimiento 3 anterior se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando el compuesto (182 mg, 0,200 mmol) obtenido en el Procedimiento 2 anterior en lugar de metanosulfonato del compuesto (4) para producir el compuesto del título en forma de un sólido de color amarillo pálido (102 mg, 43 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,2 Hz), 1,35 (9H, s), 1,67-1,76 (2H, m), 1,81-1,90 (2H, m), 2,35-2,05 (10H, m), 2,40 (3H, s), 2,75-2,83 (1H, m), 2,99-3,13 (3H, m), 3,14-3,26 (4H, m), 3,55-3,76 (6H, m), 4,36-4,50 (2H, m), 5,21 (2H, c, J = 18,9 Hz), 5,42 (2H, s), 5,54-5,61 (1H, m), 6,53 (1H, s), 7,03 (2H, s), 7,17-7,26 (5H, m), 7,31 (1H, s), 7,68-7,73 (1H, m), 7,80 (1H, d, J = 10,6 Hz), 8,00-8,05 (2H, m), 8,12 (1H, d, J = 7,8 Hz), 8,16-8,20 (1H, m), 8,23-8,28 (1H, m), 8,46 (1H, d, J = 8,6 Hz).
Procedimiento 5: N-[(2S)-4-carboxi-2-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)butanoil]-p-alanilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (102 mg, 86,8 pmol) obtenido en el Procedimiento 4 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo pálido (76,0 mg, 78 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,68-1,75 (2H, m), 1,84-1,91 (2H, m), 2,35-2,05 (10H, m), 2,40 (3H, s), 2,74-2,83 (1H, m), 2,99-3,12 (3H, m), 3,14-3,26 (4H, m), 3,55-3,77 (6H, m), 4,41-4,49 (2H, m), 5,21 (2H, dd, J = 38,7, 18,8 Hz), 5,42 (2H, s), 5,54-5,61 (1H, m), 6,54 (1H, s), 7,03 (2H, s), 7,15-7,27 (5H, m), 7,31 (1H, s), 7,69 7,74 (1H, m), 7,80 (1H, d, J = 10,9 Hz), 8,01-8,07 (2H, m), 8,12 (1H, d, J = 8,2 Hz), 8,19 (1H, t, J = 5,5 Hz), 8,27 (1H, t, J = 6,3 Hz), 8,47 (1H, d, J = 8,6 Hz), 12,12 (1H, s).
EM (IEN) m/z: 1119 (M+H)+
Procedimiento 6: Conjugado de anticuerpo y fármaco (14)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 5 anterior, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 6 del Ejemplo 2.
Concentración de anticuerpo: 12,77 mg/ml, rendimiento de anticuerpo: 8,9 mg (71 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,0.
Ejemplo 16: Conjugado de anticuerpo y fármaco (15)
[Fórmula 68]

Procedimiento 1: N-[(9H-fluoren-9-ilmetoxi)carbonil]gMcMglicil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)-L-fenilalaninamida
El compuesto (300 mg, 0,473 mmol) obtenido en el Procedimiento 2 del Ejemplo 1 se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando N-[(9H-fluoren-9-ilmetoxi)carbonil]glicilglicil-L-fenilalanina (el compuesto descrito en la patente japonesa abierta a inspección pública N.° 2002-60351; 346 mg, 0,691 mmol) en lugar de ácido 4-(fere-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo pálido (230 mg, 40 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,85 (3H, t, J = 7,2 Hz), 1,67-1,68 (2H, m), 1,81-1,84 (2H, m), 2,13 (4H, t, J = 6,8 Hz), 2,39 (3H, s), 2,76 (1H, t, J = 11,4 Hz), 2,96-3,08 (4H, m), 3,16-3,17 (2H, m), 3,59-3,74 (4H, m), 4,22-4,28 (2H, m), 4,39 4,42 (1H, m), 5,16-5,22 (2H, m), 5,36-5,41 (2H, m), 5,56-5,59 (1H, m), 6,52 (1H, s), 7,14-7,20 (5H, m), 7,29-7,31 (3H, m), 7,38-7,41 (2H, m), 7,61 (1H, t, J = 6,0 Hz), 7,69 (2H, d, J = 7,4 Hz), 7,79 (1H, d, J = 11,0 Hz), 7,87 (2H, d, J = 7,8 Hz), 7,95 (1H, s), 8,07 (2H, t, J = 4,3 Hz), 8,42 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 1004 (M+H)+
Procedimiento 2: Glicilglicil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)-L-fenilalaninamida
A una solución en N,N-dimetilformamida (1,00 ml) del compuesto (226 mg, 0,225 mmol) obtenido en el Procedimiento 1, se añadió piperidina (0,223 ml, 2,25 mmol) y se agitó a temperatura ambiente durante 5 horas. El disolvente se retiró a presión reducida para producir una mezcla que contenía el compuesto del título. La mezcla se usó para la siguiente reacción sin purificación adicional.
Procedimiento 3: (3S, 12S)-12-Bencil-18-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-3-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-4,7,10,13,18-pentaoxo-5,8,11,14-tetraazaoctadecan-1-oato de tere-butilo
El compuesto (0,225 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-aspartato de 4-terc-butilo (104 mg, 0,337 mmol) en lugar de ácido 4-(terc-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo pálido (114 mg, 43 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,86 (3H, t, J = 7,2 Hz), 1,35 (9H, s), 1,66-1,69 (2H, m), 1,84-1,85 (2H, m), 2,11-2,13 (4H, m), 2,39 (3H, s), 2,43-2,45 (1H, m), 2,68-2,79 (2H, m), 2,94-3,16 (5H, m), 3,66 (5H, tt, J = 30,5, 10,0 Hz), 4,23 4,30 (3H, m), 4,39-4,41 (1H, m), 5,15 (1H, d, J = 19,2 Hz), 5,21 (1H, d, J = 18,8 Hz), 5,37 (1H, d, J = 17,2 Hz), 5,42 (1H, d, J = 16,0 Hz), 5,53-5,57 (1H, m), 6,54 (1H, s), 7,15-7,22 (5H, m), 7,26-7,34 (3H, m), 7,38-7,40 (2H, m), 7,68 7,70 (2H, m), 7,79 (1H, d, J = 10,9 Hz), 7,86-7,87 (2H, m), 7,88-7,90 (1H, m), 7,96 (1H, t, J = 6,3 Hz), 8,03-8,07 (2H, m), 8,20 (1H, t, J = 5,5 Hz), 8,43 (1H, d, J = 8,6 Hz). EM (APCI) m/z: 1175 (M+H)+
Procedimiento 4: (3S,12S)-3-Amino-12-bencil-18-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4,7,10,13,18-pentaoxo-5,8,11,14-tetraazaoctadecan-1-oato de terc-butilo
El compuesto (110 mg, 0,0936 mmol) obtenido en el Procedimiento 3 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 para producir una mezcla que contenía el compuesto del título. La mezcla se usó para la siguiente reacción sin purificación adicional.
Procedimiento 5: (3S,12S)-12-Bencil-3-{[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]amino}-18-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4,7,10,13,18-pentaoxo-5,8,11,14-tetraazaoctadecan-1-oato de terc-butilo
El compuesto (0,0936 mmol) obtenido en el Procedimiento 4 anterior se hizo reaccionar de la misma manera que el Procedimiento 4 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo pálido (40,2 mg, 38 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,86 (3H, t, J = 7,4 Hz), 1,17-1,19 (2H, m), 1,35 (9H, s), 1,44-1,47 (4H, m), 1,66-1,67 (2H, m), 1,81-1,88 (2H, m), 2,06-2,13 (6H, m), 2,39-2,41 (1H, m), 2,40 (3H, s), 2,67 (1H, dd, J = 16,0, 5,5 Hz), 2,76 (1H, dd, J = 13,3, 9,0 Hz), 2,96 (1H, dd, J = 13,5, 4,9 Hz), 3,04 (2H, td, J = 13,4, 6,6 Hz), 3,18 (2H, s), 3,36 (2H, d, J = 7.0 Hz), 3,58 (1H, dd, J = 16,8, 5,5 Hz), 3,70 (3H, dt, J = 21,5, 7,2 Hz), 4,38-4,41 (1H, m), 4,57-4,59 (1H, m), 5,16 (1H, d, J = 18,8 Hz), 5,24 (1H, d, J = 19,2 Hz), 5,38 (1H, d, J = 16,4 Hz), 5,43 (1H, d, J = 16,0 Hz), 5,57-5,58 (1H, m), 6,54 (1H, s), 6,99 (2H, s), 7,13-7,25 (5H, m), 7,31 (1H, s), 7,80 (1H, d, J = 10,9 Hz), 7,94-8,04 (3H, m), 8,13-8,16 (2H, m), 8,43 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 1146 (M+H)+
Procedimiento 6: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-a-aspartilglicilglicil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)-L-fenilalaninamida
El compuesto (40,0 mg, 0,0349 mmol) obtenido en el Procedimiento 5 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo pálido (33,6 g, 88 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, c, J = 7,2 Hz), 1,14-1,20 (2H, m), 1,46 (4H, td, J = 14,8, 7,3 Hz), 1,67 (2H, td, J = 12,9, 6,3 Hz), 1,84 (2H, cd, J = 25,5, 7,2 Hz), 2,11 (6H, dt, J = 23,4, 7,3 Hz), 2,39 (3H, s), 2,45-2,47 (1H, m), 2,69 (1H, dd, J = 16,5, 5,5 Hz), 2,76 (1H, dd, J = 13,7, 9,3 Hz), 2,94-3,01 (1H, m), 3,05 (2H, cd, J = 25,1,6,4 Hz), 3,17 3,19 (1H, m), 3,34-3,46 (4H, m), 3,59 (1H, dd, J = 16,6, 5,6 Hz), 3,69 (2H, dt, J = 20,1,6,8 Hz), 4,37-4,41 (1H, m), 4,55 (1H, dd, J = 13,5, 7,7 Hz), 5,16 (1H, d, J = 19,0 Hz), 5,22 (1H, d, J = 18,6 Hz), 5,38 (1H, d, J = 16,4 Hz), 5,43 (1H, d, J = 16,4 Hz), 5,55-5,59 (1H, m), 6,54 (1H, s), 6,99 (2H, s), 7,19 (5H, cd, J = 31,6, 7,9 Hz), 7,31 (1H, s), 7,79 (1H, d, J = 11.0 Hz), 7,99 (3H, ddd, J = 25,1, 14,2, 6,2 Hz), 8,11 (1H, t, J = 5,5 Hz), 8,17 (1H, d, J = 7,6 Hz), 8,44 (1H, d, J = 8.5 Hz), 12,32 (1H, s).
EM (APCI) m/z: 1090 (M+H)+
Procedimiento 7: Conjugado de anticuerpo y fármaco (15)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EDTA usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg' 1 cm'1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1.5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,102 ml) y una solución de dimetilsulfóxido que contenía 10 mM del compuesto obtenido en el Procedimiento 6 anterior (0,047 ml; 5,5 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante
40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,009 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título. Después de eso, la solución se concentró por el Procedimiento común A.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 9,16mg/ml, rendimiento de anticuerpo: 6,4 mg (51 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,0.
Ejemplo 17: Conjugado de anticuerpo y fármaco (16)
[Fórmula 69]

Procedimiento 1: N6-(íerc-butoxicarbonil)-N2-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12Hbenzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (167 mg, 0,176 mmol) obtenido en el Procedimiento 2 del Ejemplo 2 se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando NE-(ferc-butoxicarbonil)-Na-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisina (103 mg, 0,22 mmol) en lugar de ácido 4-(ferc-butoxicarbonilamino)butanoico. El producto en bruto obtenido se usó en el siguiente procedimiento sin purificación.
Procedimiento 2: N6-(ferc-butoxicarbonil)-L-lisilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
A una solución en N,N-dimetilformamida (4,00 ml) del producto en bruto obtenido en el Procedimiento 1 anterior, se añadió piperidina (0,400 ml) y se agitó a temperatura ambiente durante 2 horas. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a capa orgánica repartida de cloroformo: metanol: agua = 7:3:1 (v/v/v)], para producir el compuesto del título en forma de un sólido de color amarillo pálido (113 mg, 60 %). RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,4 Hz), 1,18-1,49 (5H, m), 1,36 (9H, s), 1,51-1,60 (1H, m), 1,67-1,76 (2H, m), 1,80-1,91 (2H, m), 2,09-2,20 (4H, m), 2,39 (3H, s), 2,76-2,89
(3H, m), 2,99-3,22 (6H, m), 3,58-3,77 (6H, m), 4,43-4,49 (1H, m), 5,20 (2H, c, J = 18,5 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 6,54 (1H, s), 6,76 (1H, t, J = 5,5 Hz), 7,15-7,26 (5H, m), 7,31 (1H, s), 7,69-7,74 (1H, m), 7,80 (1H, d, J = 10,9 Hz),
8,08 (1H, t, J = 5,7 Hz), 8,14 (1H, d, J = 7,8 Hz), 8,22-8,30 (2H, m), 8,47 (1H, d, J = 8,6 Hz).
Procedimiento 3: N6-(ferc-butoxicarbonil)-N2-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-lisilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (113 mg, 0,106 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera que el Procedimiento 4 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo pálido (102 mg, 61 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,2 Hz), 1,11-1,53 (11H, m), 1,35 (9H, s), 1,56-1,65 (1H, m), 1,68
1,76 (2H, m), 1,81-1,92 (2H, m), 2,06-2,20 (6H, m), 2,40 (3H, s), 2,74-2,90 (3H, m), 2,96-3,39 (7H, m), 3,57-3,74 (6H, m), 4,14-4,21 (1H, m), 4,42-4,49 (1H, m), 5,20 (2H, c, J = 18,9 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 6,54 (1H, s), 6,72 6,78 (1H, m), 7,00 (2H, s), 7,15-7,26 (5H, m), 7,31 (1H, s), 7,69-7,72 (1H, m), 7,80 (1H, d, J = 10,9 Hz), 7,93 (1H, d, J
= 7,4 Hz), 7,99-8,04 (1H, m), 8,10-8,18 (2H, m), 8,26 (1H, t, J = 6,1 Hz), 8,46 (1H, d, J = 8,2 Hz).
Procedimiento 4: N2-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-lisilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
A una solución en diclorometano (4,00 ml) del compuesto (102 mg, 80,9 pmol) obtenido en el Procedimiento 3 anterior, se añadió ácido trifluoroacético (1,00 ml) y se agitó a temperatura ambiente durante 2 horas. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a capa orgánica repartida de cloroformo: metanol: agua = 7:3:1 (v/v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (57,0 mg, 61 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,2 Hz), 1,12-1,35 (4H, m), 1,41-1,55 (7H, m), 1,61-1,77 (3H, m),
1,80-1,91 (2H, m), 2,07-2,22 (6H, m), 2,40 (3H, s), 2,84-2,71 (3H, m), 2,97-3,40 (7H, m), 3,59-3,76 (6H, m), 4,20-4,25
(1H, m), 4,45-4,50 (1H, m), 5,20 (2H, c, J = 18,5 Hz), 5,42 (2H, s), 5,54-5,60 (1H, m), 6,55 (1H, s), 7,01 (2H, s), 7,15 7,26 (5H, m), 7,31 (1H, s), 7,74 (1H, t, J = 5,7 Hz), 7,81 (1H, d, J = 10,9 Hz), 7,97 (1H, d, J = 7,8 Hz), 8,05 (1H, t, J = 6,1 Hz), 8,13-8,18 (2H, m), 8,28 (1H, t, J = 5,7 Hz), 8,47 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1160 (M+H)+
Procedimiento 5: Conjugado de anticuerpo y fármaco (16)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 4 anterior, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 7 del Ejemplo 16.
Concentración de anticuerpo: 19,26 mg/ml, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 18: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
[Fórmula 70]

Procedimiento 1: (3-{[(1S,9S)-9-Etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-3-oxopropil)carbamato de ferc-butilo
El mesilato del compuesto (4) (500 mg, 0,941 mmol) se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando N-(ferc-butoxicarbonil)-p-alanina en lugar de ácido 4-(ferc-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo-pardo (616 mg, cuantitativo).
RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,2 Hz), 1,29 (9H, s), 1,86 (2H, dt, J = 15,1, 7,3 Hz), 2,04-2,22 (2H, m), 2,31 (2H, t, J = 6,8 Hz), 2,40 (3H, s), 3,10-3,26 (4H, m), 5,15 (1H, d, J = 18,8 Hz), 5,26 (1H, d, J = 19,2 Hz), 5,42 (2H, dd, J = 18,8, 16,4 Hz), 5,57 (1H, dt, J = 8,5, 4,2 Hz), 6,53 (1H, s), 6,78 (1H, t, J = 5,5 Hz), 7,30 (1H, s), 7,80 (1H, d, J = 11,0 Hz), 8,46 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 607 (M+H)+
Procedimiento 2: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir trifluoroacetato del compuesto del título en forma de un sólido de color amarillo (499 mg, 86 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,2 Hz), 1,86 (2H, quint. d, J = 14,6, 7,2, 7,2, 7,2, 7,2 Hz), 2,06-2,27 (1H, m), 2,41 (3H, s), 2,46-2,57 (2H, m), 3,08 (2H, t, J = 6,8 Hz), 3,14-3,24 (2H, m), 5,22 (1H, d, J = 18,8 Hz), 5,29 (1H, d, J = 18,8 Hz), 5,43 (2H, s), 5,58 (1H, dt, J = 8,5, 4,5 Hz), 6,55 (1H, s), 7,32 (1H, s), 7,74 (3H, s a), 7,82 (1H, d, J = 11,0 Hz), 8,67 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 507 (M+H)+
Ejemplo 19: Conjugado de anticuerpo y fármaco (17)
[Fórmula 71]

Procedimiento 1: N-(ferc-butoxicarbonil)glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indoNzino[1,2-b]quinoNn-1-N]-p-alaninamida
El compuesto (484 mg, 0,780 mmol) del Ejemplo 18 se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo pálido (626 mg, 87 %). RMN 1H (400 MHz, 400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,4 Hz), 1,27-1,42 (9H, m), 1,77-1,93 (2H, m), 2,06-2,22 (2H, m), 2,36 (2H, t, J = 7,2 Hz), 2,40 (3H, d, J = 1,6 Hz), 2,44-2,54 (2H, m), 2,76 (1H, dd, J = 14,5, 10,2 Hz), 3,02 (1H, dd, J = 13,9, 4,5 Hz), 3,12-3,22 (2H, m), 3,52 (6H, d, J = 6,3 Hz), 4,42-4,54 (1H, m), 5,19 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,4 Hz), 5,42 (1H, dd, J = 18,4, 16,4 Hz), 5,57 (1H, dt, J = 8,7, 4,4 Hz), 6,53 (1H, s), 6,98 (1H, t, J = 5,9 Hz), 7.14- 7,28 (5H, m), 7,31 (1H, s), 7,77-7,84 (1H, m), 7,91 (1H, t, J = 5,5 Hz), 8,16 (1H, d, J = 7,8 Hz), 8,27 (1H, t, J = 5,1 Hz), 8,52 (1H, d, J = 9,0 Hz).
Procedimiento 2: Trifluoroacetato de glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indoNdino[1,2-b]quinoNn-1-N]-p-alaninaamida
El compuesto (624 mg, 0,675 mmol) obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo (626 mg, 92 %).
RMN 1H (400 MHz, 400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,86 (2H, tt, J = 14,5, 7,2 Hz), 2,07-2,22 (2H, m), 2,36 (2H, t, J = 7,2 Hz), 2,40 (3H, s), 2,44-2,54 (2H, m), 2,75 (1H, dd, J = 13,7, 9,8 Hz), 3,04 (1H, dd, J = 13,7, 4,3 Hz), 3,12-3,22 (2H, m), 3,58 (2H, d, J = 4,7 Hz), 3,69 (3H, td, J = 11,2, 5,7 Hz), 3,87 (1H, dd, J = 17,0, 5,7 Hz), 4,54 (1H, m, J = 17,8, 4,5 Hz), 5,19 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,43 (2H, s), 5,51-5,60 (1H, m), 6,55 (1H, s), 7.14- 7,29 (5H, m), 7,32 (1H, s), 7,81 (1H, d, J = 10,9 Hz), 7,88 (1H, t, J = 5,7 Hz), 7,97 (3H, s. a.), 8,29-8,38 (2H, m), 8,50 (1H, t, J = 5,7 Hz), 8,55 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 825 (M+H)+
Procedimiento 3: (9S, 18S)-9-Bencil-1-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-18-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-1,5,8,11,14,17-hexaoxo-4,7,10,13,16-pentaazaicosan-20-noato de terc-butilo
El compuesto (150 mg, 0,182 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 2 usando ácido (2S)-4-ferc-butoxi-2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-4-oxobutanoico (90,0 mg, 0,219 mmol) en lugar de N-(ferc-butoxicarbonil)glicilglicil-L-fenilalanilglicina para producir el compuesto del título en forma de un sólido de color amarillo pálido (84,0 mg, 38 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,82-0,91 (3H, m), 1,35 (9H, s), 1,85 (2H, tt, J = 14,0, 7,3 Hz), 2,06-2,21 (2H, m), 2,39 (3H, s), 2,31-2,53 (5H, m), 2,64-2,73 (1H, m), 2,78 (1H, dd, J = 13,7, 9,8 Hz), 3,02 (1H, dd, J = 13,9, 4,5 Hz), 3,11-3,20 (2H, m), 3,55-3,80 (6H, m), 4,17-4,35 (3H, m), 4,35-4,43 (1H, m), 4,44-4,51 (1H, m), 5,18 (1H, d, J = 19,2 Hz), 5,24 (1H, d, J = 19,2 Hz), 5,41 (2H, dd, J = 18,8, 16,4 Hz), 5,51-5,60 (1H, m), 6,53 (1H, s), 7,13-7,20 (1H, m), 7,20-7,27 (4H, m), 7,27-7,34 (3H, m), 7,39 (2H, t, J = 7,2 Hz), 7,65-7,73 (3H, m), 7,79 (2H, d, J = 10,6 Hz), 7,87 (2H, d, J = 7,4 Hz), 8,00 (1H, t, J = 6,1 Hz), 8,08-8,20 (2H, m), 8,22-8,31 (1H, m), 8,52 (1H, d, J = 8,2 Hz).
EM (IEN) m/z: 1218 (M+H)+
Procedimiento 4: (9S,18S)-9-Bencil-18-{[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]amino}-1-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-1,5,8,11,14,17-hexaoxo-4,7,10,13,16-pentaazaicosan-20-noato de ferc-butilo
El compuesto (81,0 mg, 0,0665 mmol) obtenido en el Procedimiento 3 anterior se hizo reaccionar de la misma manera que el Procedimiento 4 del Ejemplo 2 para producir el compuesto del título (56,0 mg, 71 %).
EM (IEN) m/z: 1189,5 (M+H)+
Procedimiento 5: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-a-aspartilglicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto (52,0 mg, 0,0437 mmol) obtenido en el Procedimiento 4 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo pálido (35,0 mg, 71 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,12-1,22 (2H, m), 1,39-1,51 (4H, m), 1,78-1,92 (2H, m), 2,04-2,19 (2H, m), 2,08 (2H, t, J = 7,2 Hz), 2,40 (3H, s), 2,31-2,46 (6H, m), 2,61-2,72 (1H, m), 2,73-2,85 (1H, m), 3,02 (1H, dd, J = 14,1,4,7 Hz), 3,17 (2H, m, J = 5,5 Hz), 3,26-3,43 (2H, m), 3,55-3,77 (6H, m), 4,42-4,50 (1H, m), 4,51-4,58 (1H, m), 5,19 (1H, d, J = 18,4 Hz), 5,26 (1H, d, J = 18,4 Hz), 5,42 (2H, s a), 5,52-5,60 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,12-7,27 (5H, m), 7,31 (1H, s), 7,80 (2H, d, J = 10,9 Hz), 7,93-8,02 (1H, m), 8,03-8,17 (3H, m), 8,22-8,31 (1H, m), 8,53 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1133 (M+H)+
Procedimiento 6: Conjugado de anticuerpo y fármaco (17)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 5 anterior, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 1 del Ejemplo 4.
Concentración de anticuerpo: 9,56 mg/ml, rendimiento de anticuerpo: 6,7 mg (54 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,5.
Ejemplo 20: Conjugado de anticuerpo y fármaco (18)
[Fórmula 72]



Procedimiento 1: (5S, 14S)-5-Bencil-1-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-14-[[(9H-fluoren-9-ilmetoxi)carbonil]amino}-1,4,7,10,13-pentaoxo-3,6,9,l2-tetraazahexadecan-16-oato de ferc-butilo
En refrigeración con hielo, a una solución en N,N-dimetilformamida (10,0 ml) de glicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-lH,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida (una forma libre del compuesto farmacéutico descrito en el documento WO97/46260; 0,250 g, 0,332 mmol), N-hidroxisuccinimida (57,2 mg, 0,497 mmol) y N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-aspartato de 4-ferc-butilo (0,205 g, 0,497 mmol), se añadió N,N'-diciclohexilcarbodiimida (0,123 g, 0,497 mmol) y se agitó a temperatura ambiente durante 2 días. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 9:1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,278 g, 73 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,86 (3H, t, J = 7,1 Hz), 1,35 (9H, s), 1,79-1,90 (2H, m), 2,03-2,25 (2H, m), 2,40 (3H, s), 2,40-2,51 (2H, m), 2,64-2,82 (2H, m), 2,98 (1H, dd, J = 13,7, 4,6 Hz), 3,16 (2H, s a), 3,55 (1H, dd, J = 16,7, 5,7 Hz), 3,63-3,80 (4H, m), 4,16-4,34 (3H, m), 4,36-4,50 (2H, m), 5,23 (2H, s), 5,37 (1H, d, J = 16,5 Hz), 5,43 (1H, d, J = 16.5 Hz), 5,51-5,62 (1H, m), 6,52 (1H, s), 7,10-7,25 (5H, m), 7,26-7,33 (3H, m), 7,39 (2H, t, J = 7,3 Hz), 7,65-7,72 (3H, m), 7,80 (1H, d, J = 11,0 Hz), 7,86 (2H, d, J = 7,3 Hz), 7,98 (1H, t, J = 5,5 Hz), 8,07 (1H, d, J = 7,8 Hz), 8,15 (1H, t, J = 5.5 Hz), 8,31 (1H, t, J = 5,5 Hz), 8,41 (1H, d, J = 8,7 Hz).
EM (IEN) m/z: 1147 (M+H)+
Procedimiento 2: (5S,14S)-14-Amino-5-bencil-1-{[(1S, 9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-1,4,7,10,13-pentaoxo-3,6,9,12-tetraazahexadecan-16-oato de ferc-butilo
A una solución en N,N-dimetilformamida (2,00 ml) del compuesto (0,279 g, 0,242 mmol) obtenido en el Procedimiento 1 anterior, se añadió piperidina (0,240 ml, 2,42 mmol) y se agitó a temperatura ambiente durante 1 hora. El disolvente
se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 2:1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,265 g, cuantitativo). RMN 1H (400 MHz, DMSO-da) 8: 0,88 (3H, t, J = 7,2 Hz), 1,39 (9H, s), 1,81-1,94 (1H, m), 2,07-2,28 (2H, m), 2,37 (1H, dd, J = 15,8, 8,0 Hz), 2,43 (3H, s), 2,60 (1H, dd, J = 15,8, 4,9 Hz), 2,75-2,82 (1H, m), 3,00 (1H, dd, J = 13,9, 4,5 Hz), 3,16-3,25 (2H, m), 3,50-3,61 (2H, m), 3,65-3,81 (5H, m), 4,40-4,51 (1H, m), 5,27 (2H, dd, J = 24,1, 19,0 Hz), 5,43 (2H, dd, J = 21,3, 16,2 Hz), 5,56-5,65 (1H, m), 6,55 (1H, s), 7,15-7,28 (5H, m), 7,33 (1H, s), 7,83 (1H, d, J = 11,0 Hz), 8,04 (1H, t, J = 5,7 Hz), 8,09 (1H, d, J = 8,2 Hz), 8,26-8,39 (2H, m), 8,44 (1H, d, J = 8,2 Hz).
Procedimiento 3: (5S,14S)-5-Bencil-14-{[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]amino}-1-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-1,4,7,10,13-pentaoxo-3,6,9,12-tetraazahexadecan-16-oato de ferc-butilo
A una solución en N,N-dimetilformamida (2,00 ml) del compuesto (0,100 g, 0,108 mmol) obtenido en el Procedimiento 2 anterior, se añadió hexanoato de N-succinimidil 6-maleimida (40,0 mg, 0,130 mmol) y se agitó a temperatura ambiente durante 2 días. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 9:1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (80,0 mg, 66 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,88 (3H, t, J = 7,2 Hz), 1,13-1,23 (2H, m), 1,37 (9H, s), 1,42-1,54 (4H, m), 1,80-1,96 (2H, m), 2,08-2,25 (4H, m), 2,35-3,76 (15H, m), 2,43 (3H, s), 4,39-4,49 (1H, m), 4,55-4,67 (1H, m), 5,21-5,34 (2H, m), 5,43 (2H, dd, J = 21,1, 16,4 Hz), 5,56-5,64 (1H, m), 6,55 (1H, s), 7,01 (2H, d, J = 0,8 Hz), 7,16-7,26 (5H, m), 7,33 (1H, s), 7,83 (1H, d, J = 11,3 Hz), 8,04-8,18 (3H, m), 8,30-8,37 (1H, m), 8,43 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1118 (M+H)+
Procedimiento 4: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-a-aspartilglicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
En refrigeración con hielo, al compuesto (70,0 mg, 62,6 pmol) obtenido en el Procedimiento 3 anterior, se añadió ácido trifluoroacético (4,00 ml) y se agitó a temperatura ambiente durante 1 hora. El disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido de color amarillo pálido (55,0 mg, 83 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,88 (3H, t, J = 7,4 Hz), 1,14-1,24 (2H, m), 1,41-1,53 (4H, m), 1,79-1,95 (2H, m), 2,08-2,28 (4H, m), 2,37-2,60 (2H, m), 2,42 (3H, s), 2,63-2,82 (2H, m), 2,99 (1H, dd, J = 14,1, 5,1 Hz), 3,12-3,25 (2H, m), 3,29-3,44 (1H, m), 3,52-3,80 (6H, m), 4,38-4,48 (1H, m), 4,56 (1H, dd, J = 13,7, 7,4 Hz), 5,27 (2H, dd, J = 24,3, 18,8 Hz), 5,43 (2H, dd, J = 21,5, 16,4 Hz), 5,57-5,62 (1H, m), 6,55 (1H, s), 7,01 (2H, s), 7,15-7,26 (5H, m), 7,33 (1H, s), 7,82 (1H, d, J = 11,0 Hz), 7,98 (1H, s a), 8,08 (1H, d, J = 6,7 Hz), 8,15 (1H, d, J = 7,8 Hz), 8,34 (1H, s a), 8,44 (1H, d, J = 8,6 Hz), 12,26 (1H, s a).
EM (IEN) m/z: 1062 (M+H)+
Procedimiento 5: Conjugado de anticuerpo y fármaco (18)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1 c irr1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (8,0 ml) se recogió en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,124 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,400 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,249 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 anterior y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,050 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 17,5 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 3,56 mg/ml, rendimiento de anticuerpo: 62 mg (77 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,5.
Ejemplo 21: Conjugado de anticuerpo y fármaco (19)
[Fórmula 73]

Procedimiento 1: Conjugado de anticuerpo y fármaco (19)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (8,0 ml) se recogió en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,187 ml; 3,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,400 ml). Después de confirmar que la solución tenía un pH próximo a 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,373 ml; 6,9 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,075 ml; 13,8 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 16 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 3,66 mg/ml, rendimiento de anticuerpo: 59 mg (74 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,2.
Ejemplo 22: C o n ju g a d o de an ticu e rp o y fá rm a co (20)
[F ó rm u la 74 ]

Procedimiento 1: Conjugado de anticuerpo y fármaco (20)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EDTA usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,109 ml) y una solución de dimetilsulfóxido que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 (0,039 ml; 4,6 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa de NAC 100 mM (Sigma-Aldrich Co. LLC; 0,008 ml) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título. Después de eso, la solución se concentró por el Procedimiento común A descrito en el Procedimiento de producción 1.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 11,14 mg/ml, rendimiento de anticuerpo: 7,8 mg (62%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 23: C o n ju g a d o de an ticu e rp o y fá rm a co (21)
[F ó rm u la 75 ]

Procedimiento 1: Conjugado de anticuerpo y fármaco (21)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EDTA usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,067 ml) y una solución de dimetilsulfóxido que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 (0,085 ml; 10,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 60 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,013 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 0,88 mg/ml, rendimiento de anticuerpo: 5,28 mg (42 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,4.
Ejemplo 24: C o n ju g a d o de a n ticu e rp o y fá rm a co (22 )
[F ó rm u la 76]

Procedimiento 1: Conjugado de anticuerpo y fármaco (22)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EdTa usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,025 ml) y una solución de dimetilsulfóxido (0,127 ml; 15,0 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 60 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,019 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título. Después de eso, la solución se concentró por el Procedimiento común A descrito en el Procedimiento de producción 1.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,19mg/ml, rendimiento de anticuerpo: 7,14 mg (57%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,9.
Ejemplo 25: C o n ju g a d o de a n ticu e rp o y fá rm a co (23 )
[F ó rm u la 77]

Casi todas las cantidades de los conjugados de anticuerpo y fármaco de los Ejemplos 23 y 24 se mezclaron y la solución se concentró mediante el Procedimiento común A descrito en el Procedimiento de producción 1 para producir el conjugado de anticuerpo y fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 9,07 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,6.
Ejemplo 26: Conjugado de anticuerpo y fármaco (24)
[Fórmula 78]
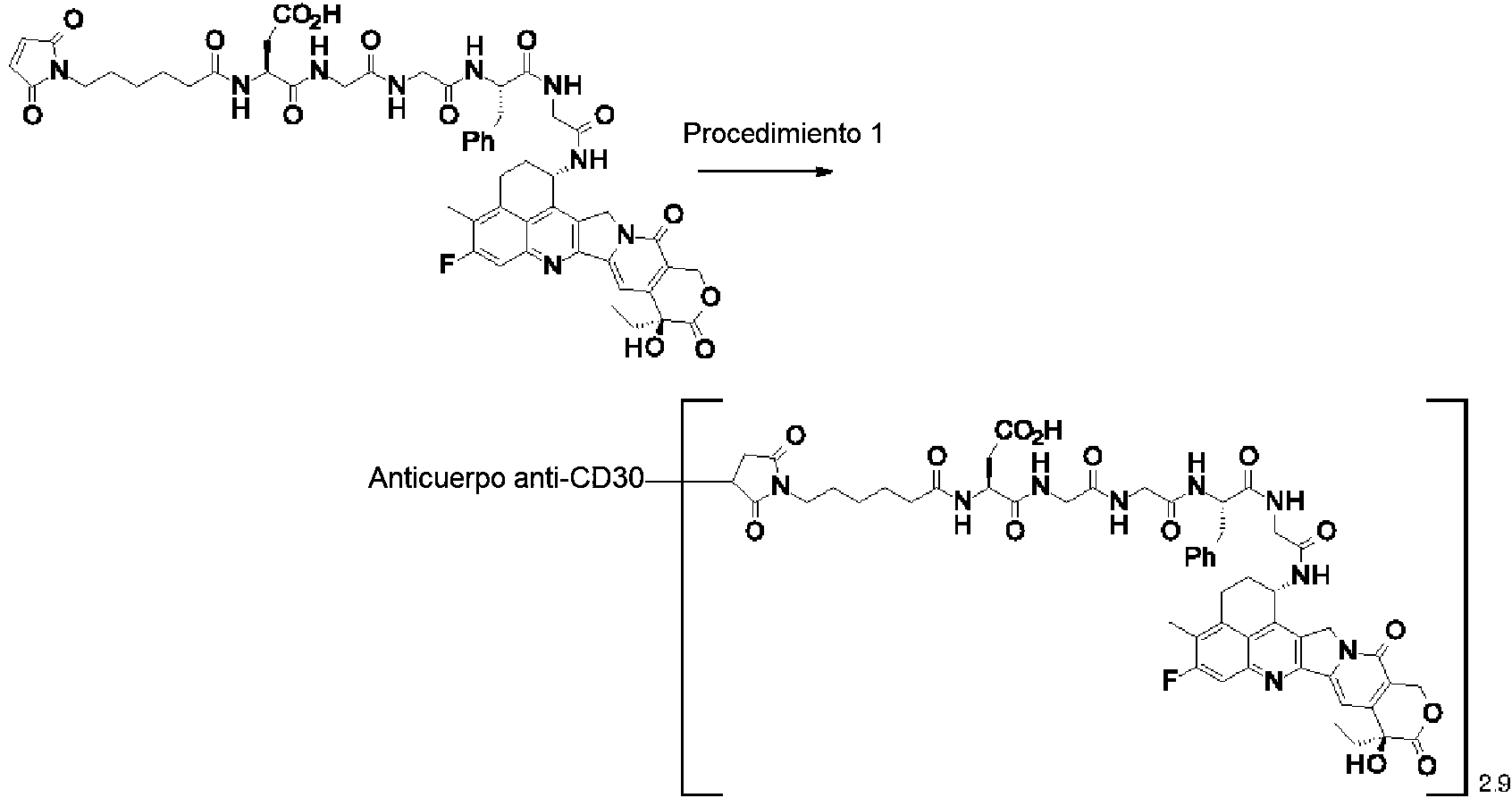
Procedimiento 1: Conjugado de anticuerpo y fármaco (24)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00593 ml; 9,2
equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28o = 270400 (valor de cálculo estimado), £a,37o = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 0,95 mg/ml, rendimiento de anticuerpo: 5,70 mg (57 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,9.
Ejemplo 27: Conjugado de anticuerpo y fármaco (25)
[Fórmula 79]

Procedimiento 1: Conjugado de anticuerpo y fármaco (25)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,o593 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28o = 270400 (valor de cálculo estimado), £a,37o = 0 (valor de cálculo estimado), £d,28o = 5000 (valor promedio medido) y £d,37o = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,04 mg/ml, rendimiento de anticuerpo: 6,24 mg (62%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,8.
Ejemplo 28: Conjugado de anticuerpo y fármaco (26)
[Fórmula 80]

Procedimiento 1: Conjugado de anticuerpo y fármaco (26)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00593 ml; 9,2 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 256400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 0,96 mg/ml, rendimiento de anticuerpo: 5,76 mg (58 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,0. Ejemplo 29: Conjugado de anticuerpo y fármaco (27)
[F ó rm u la 81 ]

Procedimiento 1: Conjugado de anticuerpo y fármaco (27)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 256400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 0,95 mg/ml, rendimiento de anticuerpo: 5,70 mg (57%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,1.
Ejemplo 30: Conjugado de anticuerpo y fármaco (28)
[F ó rm u la 82 ]

Procedimiento 1: Conjugado de anticuerpo y fármaco (28)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00593 ml; 9,2 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,01 mg/ml, rendimiento de anticuerpo: 6,06 mg (61 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,8.
Ejemplo 31: Conjugado de anticuerpo y fármaco (29)
[F ó rm u la 83 ]

Procedimiento 1: Conjugado de anticuerpo y fármaco (29)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 4 del Ejemplo 20 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,18 mg/ml, rendimiento de anticuerpo: 7,08 mg (71 %) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,0.
Ejemplo 32: Conjugado de anticuerpo y fármaco (30)
[F ó rm u la 84]



Procedimiento 1: N6-(ferc-butoxicarbonil)-N2-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilgMcMglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
Se hizo reaccionar glicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida (una forma libre del compuesto farmacéutico descrito en el documento WO97/46260; 0,300 g, 0,397 mmol) de la misma manera que el Procedimiento 1 del Ejemplo 20 usando NE-(ferc-butoxicarbonil)-N°-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisina en lugar de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-aspartato de 4-ferc-butilo para producir el compuesto del título en forma de un sólido de color amarillo (0,471 g, 98 %).
EM (IEN) m/z: 1204 (M+H)+
Procedimiento 2: N6-(íerc-butoxicarbonil)-L-lisilglicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,417 g, 0,391 mmol) obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 20 para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,272 g, 71 %).
EM (IEN) m/z: 1062 (M+H)+
Procedimiento 3: N6-(íerc-butoxicarbonil)-N2-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-lisilglicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,210 g, 0,213 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera
que el Procedimiento 3 del Ejemplo 20 para producir el compuesto del título en forma de un sólido de color amarillo pálido (63,0 mg, 21 %).
EM (IEN) m/z: 1175 (M+H)+
Procedimiento 4: Trifluoroacetato de N2-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-lisilglicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolidino[1,2-b]quinolin-1-il]glicinamida
En refrigeración con hielo, al compuesto (63,0 mg, 53,6 pmol) obtenido en el Procedimiento 3 anterior, se añadió ácido trifluoroacético (2,00 ml) y se agitó a temperatura ambiente durante 1 hora. El disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido de color amarillo (50,0 mg, 78 %).
RMN 1H (400 MHz, DMSO-de) 6: 0,89 (3H, t, J = 7,2 Hz), 1,13-1,39 (4H, m), 1,43-1,58 (7H, m), 1,61-1,73 (1H, m), 1,80-1,94 (2H, m), 2,07-2,28 (4H, m), 2,43 (3H, s), 2,72-2,84 (4H, m), 3,00 (1H, dd, J = 13,7, 3,9 Hz), 3,20 (2H, s a), 3,55-3,80 (6H, m), 4,20-4,30 (1H, m), 4,42-4,52 (1H, m), 5,27 (2H, dd, J = 23,7, 19,8 Hz), 5,43 (2H, dd, J = 21,9, 16,4 Hz), 5,55-5,65 (1H, m), 6,56 (1H, s), 7,02 (2H, s), 7,15-7,27 (5H, m), 7,34 (1H, s), 7,64 (3H, s a), 7,83 (1H, d, J = 10,6 Hz), 7,98-8,04 (2H, m), 8,09-8,20 (2H, m), 8,37 (1H, t, J = 5,5 Hz), 8,47 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1075 (M+H)+
Procedimiento 5: Conjugado de anticuerpo y fármaco (30)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 4, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 1 del Ejemplo 4.
Concentración de anticuerpo: 12,04mg/ml, rendimiento de anticuerpo: 8,4 mg (67%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 33: Conjugado de anticuerpo y fármaco (31)
[Fórmula 85]

Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 4 del Ejemplo 32, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 1 del Ejemplo 5.
Concentración de anticuerpo: 1,79mg/ml, rendimiento de anticuerpo: 10,74mg (86%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,1.
Ejemplo 34: Conjugado de anticuerpo y fármaco (32)
[Fórmula 86]

Procedimiento 1: Conjugado de anticuerpo y fármaco (32)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 4 del Ejemplo 32, el conjugado de anticuerpo y fármaco del título se obtuvo de la misma manera que el Procedimiento 1 del Ejemplo 6.
Concentración de anticuerpo: 1,87 mg/ml, rendimiento de anticuerpo: 11,22 mg (90%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,0.
Ejemplo 35: Conjugado de anticuerpo y fármaco (33)
[Fórmula 87]

Casi todas las cantidades de los conjugados de anticuerpo y fármaco de los Ejemplos 33 y 34 se mezclaron y la solución se concentró mediante el Procedimiento común A descrito en el Procedimiento de producción 1 para producir el conjugado de anticuerpo y fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 22,21 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,9.
Ejemplo 36: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
[Fórmula 88]

Procedimiento 1: (2-{[(1S,9S)-9-Etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetil)carbamato de ferc-butilo
A una solución en diclorometano (3,00 ml) de N-(ferc-butoxicarbonil)-glicina (0,395 g, 2,26 mmol), se añadieron N-hidroxi-succinimida (0,260 g, 2,26 mmol) y clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (0,433 mg, 2,26 mmol) y se agitaron a temperatura ambiente durante 1 hora. Esta solución se añadió a una solución que consistía en metanosulfonato del compuesto (4) (1,00 g, 1,88 mmol), trietilamina (0,315 ml, 2,26 mmol) y N,N-dimetilformamida (3,00 ml) y se agitó a temperatura ambiente durante 16,5 horas. La solución de reacción se diluyó con cloroformo y se lavó con una solución al 10 % de ácido cítrico, y después la capa orgánica se secó sobre sulfato sódico anhidro. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 9:1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo (1,16 g, 99 %). RMN 1H (400 MHz, DIVISOR ) 8: 0,86 (3H, t, J = 7,2 Hz), 1,30 (9H, s), 1,81-1,89 (2H, m), 2,09-2,21 (2H, m), 2,38 (3H, s), 3,15-3,17 (2H, m), 3,55-3,56 (2H, m), 5,15 (1H, d, J = 18,8 Hz), 5,23 (1H, d, J = 19,2 Hz), 5,41 (2H, s), 5,55-5,56 (1H, m), 6,53 (1H, s), 6,95 (1H, t, J = 5,5 Hz), 7,28 (1H, s), 7,77 (1H, d, J = 11,0 Hz), 8,39 (1H, d, J = 8,6 Hz). EM (APCI) m/z: 593 (M+H)+
Procedimiento 2: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,513 g, 1,01 mmol) obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 4 del Ejemplo 17 para producir el compuesto del título en forma de un sólido de color amarillo (0,463 g, 93 %).
RMN 1H (400 MHz, CD3OD) 8: 0,96 (3H, t, J = 7,0 Hz), 1,89-1,91 (2H, m), 2,14-2,16 (1H, m), 2,30 (3H, s), 2,40-2,42 (1H, m), 3,15-3,21 (2H, m), 3,79-3,86 (2H, m), 4,63-4,67 (1H, m), 5,00-5,05 (1H, m), 5,23 (1H, d, J = 16,0 Hz), 5,48 (1H, d, J = 16,0 Hz), 5,62-5,64 (1H, m), 7,40-7,45 (2H, m). EM (APCI) m/z: 493 (M+H)+
Ejemplo 37: Conjugado de anticuerpo y fármaco (34)
[Fórmula 89]


Procedimiento 1: N-(ferc-butoxicarbonil)glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
Se disolvió N-(ferc-butoxicarbonil)-glicilglicil-L-fenilalanilglicina (0,292 mg, 0,669 mmol) en diclorometano (5,00 ml), se cargó con N-hidroxisuccinimida (77,0 mg, 0,0,669 mmol) y N,N'-diciclohexilcarbodiimida (128 mg, 0,669 mmol) y se agitó durante 1 hora y 20 minutos. La solución de reacción se añadió gota a gota a una solución en N,N-dimetilformamida (5,00 ml) del compuesto (0,275 g, 0,558 mmol) del Ejemplo 36 y se agitó a temperatura ambiente durante 1 día. Se le añadió una solución acuosa de ácido cítrico al 10 % (20,0 ml) y se extrajo tres veces con 20 ml de cloroformo. La capa orgánica obtenida se evaporó a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a cloroformo: metanol = 8:2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,430 g, 85 %).
RMN 1H (400 MHz, CD3OD) 8: 0. 94 (3H, t, J = 7,2 Hz), 1,43 (9H, s), 1,83-1,85 (2H, m), 2,20-2,22 (1H, m), 2,29 (3H, s), 2,36-2,39 (2H, m), 2,50-2,53 (1H, m), 2,67 (1H, s), 3,08-3,11 (1H, m), 3,18-3,21 (1H, m), 3,63-3,67 (4H, m), 3,78 3,82 (1H, m), 3,99 (2H, dd, J = 23,5, 16,8 Hz), 4,16 (1H, s), 4,58 (1H, d, J = 18,8 Hz), 5,15 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 16,4 Hz), 5,52 (1H, d, J = 16,4 Hz), 5,59-5,61 (1H, m), 6,89 (2H, d, J = 6,7 Hz), 7,15-7,17 (3H, m), 7,28 (1H, d, J = 10,6 Hz), 7,41 (1H, s).
EM (APCI) m/z: 911 (M+H)+
Procedimiento 2: Glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,227 g, 0,249 mmol) obtenido en el Procedimiento 1 anterior se disolvió en diclorometano (1,00 ml). Se le añadió ácido trifluoroacético (3,00 ml) y se agitó durante 1 hora. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo a capa orgánica repartida de cloroformo: metanol: agua =7:3:1 (v/v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,200 g, 99 %). RMN 1H (400 MHz, CD3OD) 8: 0,93 (3H, t, J = 7,4 Hz), 1,85 (2H, c, J = 7,3 Hz), 2,24-2,45 (5H, m), 2,32 (3H, s), 2,56 (1H, dd, J = 13,7, 5,5 Hz), 3,09-3,25 (2H, m), 3,66-3,76 (6H, m), 4,18-4,24 (1H, m), 4,76 (1H, d, J = 19,2 Hz), 5,18 (1H, d, J = 18,8 Hz), 5,30 (1H, t, J = 18,4 Hz), 5,52 (1H, d, J = 16,0 Hz), 5,63 (1H, t, J = 5,9 Hz), 6,93 (2H, d, J = 6,6 Hz), 7,17 (3H, c, J = 7,3 Hz), 7,30 (1H, d, J = 10,9 Hz), 7,42 (1H, s).
EM (APCI) m/z: 811 (M+H)+
Procedimiento 3: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10, 13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,125 g, 0,154 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera que el Procedimiento 3 del Ejemplo 20 para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,0775 g, 50 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,86 (3H, t, J = 7,2 Hz), 1,18-1,19 (2H, m), 1,45-1,48 (4H, m), 1,83-1,85 (2H, m), 2,12-2,17 (4H, m), 2,39 (3H, s), 2,68 (1H, dd, J = 24,4, 14,7 Hz), 2,83-2,87 (1H, m), 3,17-3,78 (12H, m), 4,42-4,45 (1H, m), 5,23 (2H, s), 5,41 (2H, s), 5,58-5,60 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,15-7,29 (6H, m), 7,76 (1H, d, J = 10,9 Hz), 7,97-8,00 (1H, m), 8,09-8,12 (3H, m), 8,25-8,28 (1H, m), 8,44 (1H, d, J = 8,2 Hz).
EM (APCI) m/z: 1004 (M+H)+
Procedimiento 4: Conjugado de anticuerpo y fármaco (34)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10mg/ml reemplazando el medio por PBS6,0/EDTA usando el Procedimiento común C-1 y el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg' 1 cm'1) descrito en el Procedimiento de producción 1. La solución (1,25 ml) se añadió a un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,102 ml) y una solución de dimetilsulfóxido (0,047 ml; 5,5 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 3 anterior a la solución anterior a temperatura ambiente, esto se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) a temperatura ambiente durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,009 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se agitó a temperatura ambiente durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó ABS como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título. Después de eso, la solución se concentró por el Procedimiento común A descrito en el Procedimiento de producción 1.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo), £a,370 = 0 (valor de cálculo), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron las siguientes características. Concentración de anticuerpo: 12,4 mg/ml, rendimiento de anticuerpo: 8,7 mg (70%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,1.
Ejemplo 38: Conjugado de anticuerpo y fármaco (35)
[Fórmula 90]

Procedimiento 1: N-(terc-butoxicarbonil)glicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10, 13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
Se hizo reaccionar metanosulfonato del compuesto (4) (0,800 g, 1,51 mmol) de la misma manera que el Procedimiento 1 del Ejemplo 1 usando N-(ferc-butoxicarbonil)-glicilglicina (0,419 g, 1,81 mmol) en lugar de ácido 4-(terc-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo (0,965 g, 99 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,4 Hz), 1,23 (9H, s), 1,82-1,89 (2H, m), 2,11-2,19 (2H, m), 2,40 (3H, s), 3,16-3,17 (2H, m), 3,52 (2H, ddd, J = 21,3, 15,5, 4,7 Hz), 3,77 (2H, ddd, J = 24,3, 16,8, 5,9 Hz), 5,23 (2H, s), 5,43
(2H, s), 5,56-5,60 (1H, m), 6,53 (1H, s), 7,04 (1H, t, J = 5,9 Hz), 7,31 (1H, s), 7,80 (1H, d, J = 11,0 Hz), 8,12 (1H, t, J = 5.5 Hz), 8,31 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 650 (M+H)+
Procedimiento 2: Trifluoroacetato de glicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,884 g, 1,36 mmol) obtenido en el Procedimiento 1 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo (0,787 g, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 6: 0,87 (3H, t, J = 7,2 Hz), 1,82-1,89 (2H, m), 2,11-2,18 (2H, m), 2,41 (3H, s), 3,17-3,18 (2H, m), 3,63 (2H, s), 3,88 (2H, d, J = 5,5 Hz), 5,19 (1H, d, J = 18,8 Hz), 5,25 (1H, d, J = 19,2 Hz), 5,42 (2H, s), 5,56 5,61 (1H, m), 6,56 (1H, s), 7,32 (1H, s), 7,81 (1H, d, J = 11,0 Hz), 8,01 (3H, s a), 8,65 (1H, d, J = 8,6 Hz), 8,72 (1H, t, J = 5,5 Hz). EM (APCI) m/z: 550 (M+H)+
Procedimiento 3: N-(terc-butoxicarbonil)glicilglicilfenilalanilglicilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,400 g, 0,728 mmol) obtenido en el Procedimiento 2 anterior se hizo reaccionar de la misma manera que el Procedimiento 1 del Ejemplo 1 usando N-(terc-butoxicarbonil)-glicilglicil-L-fenilalanilglicina (0,381 mg, 0,873 mmol) en lugar de ácido 4-(terc-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,545 g, 77 %).
RMN 1H (400 MHz, DMSO-d6) 6: 0,87 (3H, t, J = 7,2 Hz), 1,37 (9H, s), 1,80-1,90 (2H, m), 2,09-2,11 (1H, m), 2,18-2,21 (1H, m), 2,40 (3H, s), 2,72-2,77 (1H, m), 3,01 (1H, dd, J = 13,7, 4,3 Hz), 3,16-3,17 (2H, m), 3,52-3,83 (10H, m), 4,48 4,51 (1H, m), 5,21 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,43 (2H, s), 5,55-5,59 (1H, m), 6,53 (1H, s), 6,99 (1H, t, J = 5,9 Hz), 7,18-7,24 (5H, m), 7,31 (1H, s), 7,80 (1H, d, J = 11,0 Hz), 7,90 (1H, t, J = 5,3 Hz), 8,02 (1H, t, J = 5.5 Hz), 8,15-8,19 (2H, m), 8,30 (1H, t, J = 5,5 Hz), 8,43 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 968 (M+H)+
Procedimiento 4: Trifluoroacetato de glicilglicilfenilalanilglicilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolidino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,429 g, 0,443 mmol) obtenido en el Procedimiento 3 anterior se hizo reaccionar de la misma manera que el Procedimiento 2 del Ejemplo 1 para producir el compuesto del título en forma de un sólido de color amarillo (0,385 g, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 6: 0,87 (3H, t, J = 7,4 Hz), 1,82-1,89 (2H, m), 2,11-2,19 (2H, m), 2,40 (3H, s), 2,74 (1H, dd, J = 13,7, 9,8 Hz), 3,03 (1H, dd, J = 13,7, 4,3 Hz), 3,16-3,18 (2H, m), 3,57-3,58 (2H, m), 3,67-3,76 (7H, m), 3,82 3,90 (1H, m), 4,53-4,56 (1H, m), 5,23 (2H, s), 5,43 (2H, s), 5,55-5,59 (1H, m), 6,55 (1H, s), 7,17-7,19 (1H, m), 7,22 7,29 (4H, m), 7,31 (1H, s), 7,80 (1H, d, J = 10,9 Hz), 8,00 (3H, s a), 8,07 (1H, t, J = 5,7 Hz), 8,22 (1H, t, J = 5,7 Hz), 8,36 (2H, dd, J = 10,9, 7,0 Hz), 8,47-8,52 (2H, m).
EM (APCI) m/z: 868 (M+H)+
Procedimiento 5: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicilfenilalanilglicilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
El compuesto (0,278 g, 0,320 mmol) obtenido en el Procedimiento 4 anterior se hizo reaccionar de la misma manera que el Procedimiento 3 del Ejemplo 20 para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,166 g, 49%).
RMN 1H (400 MHz, DMSO-d6) 6: 0,87 (3H, t, J = 7,2 Hz), 1,14-1,22 (2H, m), 1,44-1,49 (4H, m), 1,80-1,90 (2H, m), 2,06-2,13 (3H, m), 2,20 (1H, d, J = 14,1 Hz), 2,40 (3H, s), 2,77 (1H, dd, J = 13,3, 8,7 Hz), 3,01 (1H, dd, J = 13,3, 4,3 Hz), 3,17 (2H, t, J = 6,7 Hz), 3,35-3,38 (2H, m), 3,56-3,84 (10H, m), 4,48 (1H, dd, J = 13,1, 9,2 Hz), 5,23 (2H, s), 5,43 (2H, s), 5,55-5,59 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,20-7,24 (5H, m), 7,31 (1H, s), 7,80 (1H, d, J = 11,0 Hz), 8,00 (2H, c, J = 5,5 Hz), 8,06 (1H, t, J = 5,9 Hz), 8,13 (1H, d, J = 8,2 Hz), 8,18 (1H, t, J = 5,7 Hz), 8,28 (1H, t, J = 5,7 Hz), 8,43 (1H, d, J = 8,6 Hz). EM (APCI) m/z: 1061 (M+H)+
Procedimiento 6: Conjugado de anticuerpo y fármaco (35)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el Procedimiento 5 anterior, el conjugado de anticuerpo y fármaco del título se produjo de la misma manera que el Procedimiento 7 del Ejemplo 16.
Concentración de anticuerpo: 11,7mg/ml, rendimiento de anticuerpo: 8,2 mg (66%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,5.
Ejemplo 39: Conjugado de anticuerpo y fármaco (36)
[Fórmula 91]

Procedimiento 1: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -il)hexanoil]glicilgMcil-L-fenilalanMglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-l0, 13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida
Se hizo reaccionar glicilglicil-L-fenilalanil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]glicinamida (una forma libre del compuesto farmacéutico descrito en el documento WO97/46260; 0,150 g, 0,200 mol) de la misma manera que el Procedimiento 3 del Ejemplo 20 para producir el compuesto del título en forma de un sólido de color amarillo pálido (70,0 mg, 37 %). RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,4 Hz), 1,15-1,21 (2H, m), 1,41-1,50 (4H, m), 1,80-1,90 (2H, m), 2,07-2,12 (4H, m), 2,17-2,23 (1H, m), 2,35-2,40 (1H, m), 2,41 (3H, s), 2,73-2,81 (1H, m), 2,98 (1H, dd, J = 13,7, 4,6 Hz), 3,15-3,20 (2H, m), 3,53 (1H, dd, J = 16,6, 5,7 Hz), 3,62-3,77 (5H, m), 4,39-4,45 (1H, m), 5,22 (1H, d, J = 18,9 Hz), 5,27 (1H, d, J = 18,9 Hz), 5,39 (1H, d, J = 16,0 Hz), 5,44 (1H, d, J = 16,0 Hz), 5,55-5,60 (1H, m), 6,53 (1H, s), 6,98 (2H, s), 7,13-7,24 (5H, m), 7,32 (1H, s), 7,81 (1H, d, J = 10,3 Hz), 7,95-8,00 (1H, m), 8,05-8,09 (2H, m), 8,28-8,31 (1H, m), 8,41 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 947 (M+H)+
Procedimiento 2: Conjugado de anticuerpo y fármaco (36)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1 c irr1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0147 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0. 050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 1 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00590 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó PBS7.4 como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo), £a,370 = 0 (valor de cálculo), £d,280 = 50 00 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,23 mg/ml, rendimiento de anticuerpo: 7,38 mg (74%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,0.
Ejemplo 40: Conjugado de anticuerpo y fármaco (37)
[Fórmula 92]

Procedimiento 1: Conjugado de anticuerpo y fármaco (37)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0. 050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0590 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 1 del Ejemplo 39 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0118 ml; 18,4 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó PBS7.4 como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo), £a,370 = 0 (valor de cálculo), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,22 mg/ml, rendimiento de anticuerpo: 7,32 mg (73%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,7.
Ejemplo 41: Conjugado de anticuerpo y fármaco (38)
[Fórmula 93]

Procedimiento 1: Conjugado de anticuerpo y fármaco (38)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/Ed Ta usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0147 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0. 050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 1 del Ejemplo 39 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,00590 ml; 9,2 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó PBS7.4 como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo), £a,370 = 0 (valor de cálculo), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron las siguientes características.
Concentración de anticuerpo: 1,11 mg/ml, rendimiento de anticuerpo: 6,66 mg (67%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 1,8.
Ejemplo 42: Conjugado de anticuerpo y fármaco (39)
[Fórmula 94]

Procedimiento 1: Conjugado de anticuerpo y fármaco (39)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para que tuviera una concentración de anticuerpo de 10 mg/ml con PBS6,0/EdTa usando el Procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1 cm-1) y el Procedimiento común C-1 descrito en el Procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato dipotásico 1 M (Nacalai Tesque, Inc.; 0. 050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y engarce de fármaco: Después de incubar la solución anterior a 22 °C durante 10 minutos, se le añadió una solución de dimetilsulfóxido (0,0590 ml; 9,2 equivalentes por molécula de anticuerpo) que contenía 10 mM del compuesto obtenido en el Procedimiento 1 del Ejemplo 39 y se incubó a 22 °C durante 40 minutos para conjugar el engarce de fármaco al anticuerpo. A continuación, se le añadió una solución acuosa (0,0118 ml; 18,4 equivalentes por molécula de anticuerpo) de nAc 100 mM (Sigma-Aldrich Co. LLC) y se incubó a 22 °C durante 20 minutos más para terminar la reacción de engarce de fármaco.
Purificación: La solución anterior se sometió a purificación usando el Procedimiento común D-1 (se usó PBS7.4 como solución tampón) descrito en el Procedimiento de producción 1 para producir 6 ml de una solución que contenía el conjugado de anticuerpo y fármaco del título.
Caracterización fisicoquímica: Usando el Procedimiento común E descrito en el Procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo), £a,370 = 0 (valor de cálculo), £d,280 = 5000 (valor promedio medido) y £d,370 = 190 00 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,00 mg/ml, rendimiento de anticuerpo: 6,00 mg (60%) y un número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
(Ejemplo de ensayo 1) Producción del vector de expresión de la B7-H3 variante 1 humana de longitud completa El ADNc que codifica la B7-H3 variante 1 humana se amplificó por reacción de PCR usando, como molde, ADNc sintetizado a partir de ARN total de células LNCaP (American Type Culture Collection: ATCC) y el siguiente conjunto de cebadores:
cebador 1:
5'-ctatagggagacccaagctggctagcatgctgcgtcggcggggcag-3' (SEQ ID NO: 22) y
cebador 2:
5' - aacgggccctctagactcgagcggccgctcaggctatttcttgtccatcatcttctt tgctgtcag-3' (SEQ ID NO: 23).
A continuación, el producto obtenido de la PCR se purificó usando el limpiador MagExtractor PCR & Gel (Toyobo Co., Ltd.). El producto purificado se digirió adicionalmente con enzimas de restricción (Nhel/Notl) y después se purificó usando el limpiador MagExtractor PCR & Gel (Toyobo Co., Ltd.). El ADN plasmídico pcDNA3.1 (+) (Life Technologies) se digirió con las mismas enzimas de restricción anteriores (Nhel/Notl) y después se purificó usando el limpiador MagExtractor PCR & Gel (Toyobo Co., Ltd.).
Estas soluciones de ADN purificadas se mezclaron, se cargaron adicionalmente con Ligation high (Toyobo Co., Ltd.) y se incubaron a 16° C durante 8 horas para el ligamiento.
Células competentes DH5a de Escherichia coli (Life Technologies) se transformaron mediante la adición del producto de reacción obtenido.
Las colonias así obtenidas se sometieron a PCR directa de colonias usando cebadores de PCR y el cebador inverso de BGH para seleccionar clones candidatos.
Los clones candidatos obtenidos se cultivaron en un medio líquido (LB/Amp), y el ADN plasmídico se extrajo con MagExtractor-Plasmid-(Toyobo Co., Ltd.).
Cada clon obtenido se comparó con la secuencia CDS proporcionada mediante análisis de secuenciación entre el cebador 3 (cebador promotor de CMV):
5'-cgcaaatgggcggtaggcgtg-3' (SEQ ID NO: 24) y el cebador 4 (cebador inverso de BGH):
5'-tagaaggcacagtcgagg-3'(SEQ ID NO: 25) con el ADN plasmídico obtenido como molde.
Después de confirmar la secuencia, el clon obtenido se cultivó en 200 ml de medio LB/Amp, y usando el kit VioGene Plasmid Midi V-100 se extrajo el ADN plasmídico.
El vector se denominó pcDNA3.1-B7-H3. En las posiciones de nucleótidos 1 a 1602 de la SEQ ID NO: 26 (Figura 16) en el Listado de Secuencias, se muestra la secuencia de un sitio ORF del gen de la B7-H3 variante 1 clonado en el vector. En la SEQ ID NO: 1 en el Listado de secuencias, también se muestra la secuencia de aminoácidos de la B7-H3 variante 1.
(Ejemplo de ensayo 2) Preparación de células CCRF-CEM que expresan de manera estable el gen de la B7-H3 variante 1
El vector pcDNA3.1-B7-H3 producido en el Ejemplo de ensayo 1, se transfectó en células CCRF-CEM (ATCC) mediante electroporación usando Nucleofector II (fabricado por Lonza Group Ltd.). Después, las células se cultivaron adicionalmente durante dos noches en medio RPMI1640 (Life Technologies) que contenía suero bovino fetal (FBS) al 10 % (en lo sucesivo FBS-RPMI1640 al 10 %) a una temperatura de 37° C y con CO2 al 5 %.
Después del día 2 de cultivo, el cultivo se inició en FBS-RPMI1640 al 10% que contenía 750 pg/ml de G418 (Life Technologies) para seleccionar células CCRF-CEM en las que el vector pcDNA3.1-B7-H3 se integró de manera estable.
Después de 1 mes de cultivo, la clonación se realizó mediante el procedimiento de dilución limitante para producir un clon unicelular. Específicamente, las células con resistencia a g 418 se diluyeron en 10 células/ml, se inocularon en una placa de 96 pocillos a una concentración de 100 pl/pocillo, y se cultivaron, y las células se dejaron proliferar y se recuperaron de pocillos individuales.
Para confirmar la expresión de B7-H3 en cada clon recuperado, se usó citometría de flujo. Específicamente, cada clon recuperado se lavó dos veces con PBS que contenía FBS al 5 %, después de lo cual se suspendió añadiendo PBS que contenía FBS al 5 % y 10 pg/ml de m 30, y se dejó en reposo a 4° C durante 30 minutos. El clon se lavó dos veces con PBS que contenía FBS al 5 %, después de lo cual se suspendió añadiendo una fracción de IgG de cabra conjugada con fluoresceína a la IgG de ratón (Molécula entera) (n ° 55493, fabricada por ICN Pharmaceuticals, Inc.) diluida 1000 veces con PBS que contenía FBS al 5 %, y se dejó en reposo a 4° C durante 30 minutos. El clon se lavó dos veces con PBS que contenía FBS al 5 %, después de lo cual se su resuspendió en PBS que contenía FBS al 5 %, y se detectó utilizando un citómetro de flujo (FC500: Beckman Coulter, Inc.).
Las células CCRF-CEM que expresan de manera estable el gen de la B7-H3 variante 1 obtenidas de esta manera mediante estos procedimientos se denominaron células CEM_V1_3.1_2. Las células CCRF-CEM de la línea parental se usaron como una línea celular carente de expresión de B7-H3.
(Ejemplo de ensayo 3) Ensayo de inhibición del crecimiento celular (1) de conjugado de anticuerpo y fármaco
Las células CEM_V1_3.1_2 producidas en el Ejemplo de ensayo 2 o las células CCRF-CEM (ATCC) se cultivaron en RPMI1640 (GIBCO) que contenía suero bovino fetal al 10% (MOREGATE) (en lo sucesivo, medio). Las células CEM_V1_3.1_2 o células CCRF-CEM se prepararon para que tuviesen una concentración de 8 x 104células/ml usando un medio, se añadieron a una concentración de 25 pl/pocillo a una microplaca de 96 pocillos para cultivo celular cargada con 65 pl/pocillo de un medio, y se cultivaron durante la noche. Al día siguiente, cada uno de los anticuerpos M30-H1-L4, M30-H1-L4P y el conjugado de anticuerpo y fármaco diluidos en 1000 nM, 200 nM 40 nM, 8 nM, 1,6 nM, 0,32 nM y 0,064 nM usando un medio, se añadieron a la microplaca una concentración de 10 pl/pocillo. A los pocillos no complementados con la sustancia de ensayo se les añadió un medio a una concentración de 10 pl/pocillo. Las células se cultivaron durante 3 días con CO2 al 5 % a 37° C. Después del cultivo, la microplaca se sacó de la incubadora y se dejó en reposo a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una misma cantidad de CellTiter-Glo Luminescent Cell Viability Assay (Promega) y se agitó. Después de dejar la microplaca en
reposo a temperatura ambiente durante 10 minutos, se midió la cantidad de emisión de luz utilizando un lector de placa (PerkinElmer). El valor de CI50 se calculó según la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) x (LOG-iü b - LO G ^a)/ (d - c) LOG-iüb)
a: Concentración a de la sustancia de ensayo
b: Concentración b de la sustancia de ensayo
c: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración a d: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración b
Las concentraciones a y b establecen la relación a> b cruzando la proporción del 50 % de las células vivas.
La tasa de supervivencia de las células a cada concentración se calculó según la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b x 100
a: Cantidad promedio de emisión de luz de los pocillos complementados con la sustancia de ensayo (n = 2) b: Cantidad promedio de emisión de luz de los pocillos no complementados con la sustancia de ensayo (n = 10)
Los conjugados de anticuerpo y fármaco (6) y (23) mostraron un efecto anticelular de CI50 < 0,1 (nM) contra las células CEM_V1_3.1_2. Los conjugados de anticuerpo y fármaco (3), (33) y (39) mostraron un efecto anticelular de 0,1 <CI50 <1 (nM) contra las células. Los conjugados de anticuerpo y fármaco (13), (14), (15), (16), (17), (20), (30), (35) y (37) mostraron un efecto anticelular de 1 <CI50 <100 (nM) contra las células. Por otra parte, ninguno de los conjugados de anticuerpo y fármaco mostró un efecto anticelular contra las células CCRF-CEM (> 100 (nM)). Ni el anticuerpo M30-H1-L4, ni tampoco el anticuerpo M30-H1-L4P, mostraron actividad citotóxica contra ambas células (> 100 (nM)).
(Ejemplo de ensayo 4) Ensayo de inhibición de crecimiento celular (2) de conjugado de anticuerpo y fármaco
Se cultivaron células positivas a antígeno, células SR (ATCC) o células negativas a antígeno, células Daudi (ATCC) en RPMI1640 (GIBc O) que contenía suero bovino fetal al 10 % (MOREGATE) (en lo sucesivo, medio). Las células SR o células Daudi se prepararon para que tuviesen una concentración de 2,8 x 104 células/ml usando un medio y se añadieron a una concentración de 90 pl/pocillo a una microplaca de 96 pocillos para cultivo celular. Dos horas después, el anticuerpo anti-CD30 y cada uno de los conjugados de anticuerpo y fármaco (7), (8), (24) y (25) se diluyó en 40 nM, 8 nM, 1,6 nM, 320 pM, 64 pM, 12,8 pM y 2,6 pM usando un medio y se añadieron a la microplaca una concentración de 10 pl/pocillo. A los pocillos no complementados con la sustancia de ensayo se les añadió un medio a una concentración de 10 pl/pocillo. Las células se cultivaron durante 3 días con CO2 al 5 % a 37° C. Después del cultivo, la microplaca se sacó de la incubadora y se dejó en reposo a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una misma cantidad de CellTiter-Glo Luminescent Cell Viability Assay (Promega) y se agitó. Después de dejar la microplaca en reposo a temperatura ambiente durante 10 minutos, se midió la cantidad de emisión de luz utilizando un lector de placa (PerkinElmer). El valor de CI50 se calculó según la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) x (LOG-iüb - LOG^ a)/ (d - c) LOG-iüb)
a: Concentración a de la sustancia de ensayo
b: Concentración b de la sustancia de ensayo
c: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración a d: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración b
Las concentraciones a y b establecen la relación a> b cruzando la proporción del 50 % de las células vivas.
La tasa de supervivencia de las células a cada concentración se calculó según la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b x 100
a: Cantidad promedio de emisión de luz de los pocillos complementados con la sustancia de ensayo (n = 2) b: Cantidad promedio de emisión de luz de los pocillos no complementados con la sustancia de ensayo (n = 12)
Los conjugados de anticuerpo y fármaco (7), (8), (24) y (25) mostraron un efecto anticelular de IC50 <0,01 (nM) contra las células SR. Por otra parte, ninguno de los conjugados de anticuerpo y fármaco mostró un efecto anticelular contra las células Daudi (> 4,0 (nM)). El anticuerpo anti-CD30 no mostró ningún efecto anticelular contra ambas células (> 4,0 (nM)).
(Ejemplo de ensayo 5) Ensayo de inhibición de crecimiento celular (3) de conjugado de anticuerpo y fármaco
Células positivas a antígeno, células HL-60 (ATCC) o células negativas a antígeno, células Raji (ATCC), se cultivaron en RPMI1640 (GIBCO) que contenía suero bovino fetal al 10 % (MOREGATE) (en lo sucesivo, medio). Las células HL-60 o las células Raji se prepararon para que tuviesen una concentración de 8 x 104 células/ml usando un medio y se añadieron a una concentración de 25pl/pocillo a una microplaca de 96 pocillos para cultivo celular que contenía 65 pl/pocillo de medio. El anticuerpo anti-CD33 y cada uno de los conjugados de anticuerpo y fármaco (9), (10), (26)
y (27) se diluyó en 1000 nM, 200 nM, 40 nM, 8 nM, 1,6 nM, 0,32 nM y 0,064 nM usando un medio y se añadieron a la microplaca a una concentración de 10 pl/pocillo. A los pocilios no complementados con la sustancia de ensayo se les añadió un medio a una concentración de 10 pl/pocillo. Las células se cultivaron durante 3 días con CO2 al 5 % a 37° C. Después del cultivo, la microplaca se sacó de la incubadora y se dejó en reposo a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una misma cantidad de CellTiter-Glo Luminescent Cell Viability Assay (Promega) y se agitó. Después de dejar la microplaca en reposo a temperatura ambiente durante 10 minutos, se midió la cantidad de emisión de luz utilizando un lector de placa (PerkinElmer). El valor de CI50 se calculó según la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) x (LOG-iü b - LO G ^a)/ (d - c) LOG-iüb)
a: Concentración a de la sustancia de ensayo
b: Concentración b de la sustancia de ensayo
c: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración a d: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración b
Las concentraciones a y b establecen la relación a> b cruzando la proporción del 50 % de las células vivas.
La tasa de supervivencia de las células a cada concentración se calculó según la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b x 100
a: Cantidad promedio de emisión de luz de los pocillos complementados con la sustancia de ensayo (n = 2) b: Cantidad promedio de emisión de luz de los pocillos no complementados con la sustancia de ensayo (n = 5)
El conjugado de anticuerpo y fármaco (10) mostró un efecto citotóxico de CI50 <1 (nM) contra las células HL-60. Los conjugados de anticuerpo y fármaco (9), (26) y (27) mostraron un efecto anticelular de 1 < CI50 <100 (nM). Por otra parte, ninguno de los conjugados de anticuerpo y fármaco mostró efecto anticelular contra las células Raji (> 100 (nM)). El anticuerpo anti-CD33 no mostró ningún efecto anticelular contra ambas células (> 100 (nM)).
(Ejemplo de ensayo 6) Ensayo de citotoxicidad (4) del conjugado de anticuerpo y fármaco
Células positivas a antígeno, células U251 (ATCC) o células negativas a antígeno, células MCF-7 (ATCC), se cultivaron en RPMI1640 (GIBCO) que contenía suero bovino fetal al 10 % (MOREGATE) (en lo sucesivo, medio). Las células U251 y las células MCF-7 se prepararon para que tuviesen una concentración de 2,8 x 104 células/ml usando un medio y se añadieron a una concentración de 90 pl/pocillo a una microplaca de 96 pocillos para cultivo celular, y se cultivaron durante la noche.
Al día siguiente, el anticuerpo anti-CD70 y cada uno de los conjugados de anticuerpo y fármaco (11), (12), (28) y (29) se diluyó en 1000 nM, 200 nM, 40 nM, 8 nM, 1,6 nM, 0,32 nM y 0,064 nM usando un medio y se añadieron a la microplaca a una concentración de 10 pl/pocillo. A los pocillos no complementados con la sustancia de ensayo se les añadió un medio a una concentración de 10 pl/pocillo. Las células se cultivaron durante 6 días con CO2 al 5 % a 37° C. Después del cultivo, la microplaca se sacó de la incubadora y se dejó en reposo a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una misma cantidad de CellTiter-Glo Luminescent Cell Viability Assay (Promega) y se agitó. Después de dejar la microplaca en reposo a temperatura ambiente durante 10 minutos, se midió la cantidad de emisión de luz utilizando un lector de placa (PerkinElmer). El valor de CI50 se calculó según la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) x (LOG-iüb - LOG^ a)/ (d - c) LOG-iüb)
a: Concentración a de la sustancia de ensayo
b: Concentración b de la sustancia de ensayo
c: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración a d: Proporción de células vivas complementadas con la sustancia de ensayo que tiene la concentración b
Las concentraciones a y b establecen la relación a> b cruzando la proporción del 50 % de las células vivas.
La tasa de supervivencia de las células a cada concentración se calculó según la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b x 100
a: Cantidad promedio de emisión de luz de los pocillos complementados con la sustancia de ensayo (n = 2) b: Cantidad promedio de emisión de luz de los pocillos no complementados con la sustancia de ensayo (n = 12)
Los conjugados de anticuerpo y fármaco (12) y (29), mostraron un efecto citotóxico de 1 < CI50 <10 (nM) contra las células U251. Los conjugados de anticuerpo y fármaco (11) y (28), mostraton un efecto citotóxico de 10 < CI50 <100 (nM). Por otra parte, ninguno de los conjugados de anticuerpo y fármaco, mostró efecto citotóxico contra las células MCF-7 (> 90 (nM)). El anticuerpo anti-CD70 no mostró ningún efecto citotóxico contra ambas células (> 100 (nM)).
(Ejemplo de ensayo 7) Ensayo antitumoral (1)
Ratón: Durante 4 a 7 días, antes de su uso en el experimento, se aclimataron ratones desnudos BALB/c hembra de 5 a 6 semanas de vida (Charles River Laboratories Japón, Inc.) en condiciones SPF (siglas del inglés Specific-Pathogen-Free, sin gérmenes patógenos específicos). Los ratones se alimentaron con alimento sólido esterilizado (FR-2, Funabashi Farms Co., Ltd) y recibieron agua corriente esterilizada (preparada añadiendo de 5 a 15 ppm de solución de hipoclorito de sodio).
Ensayo y expresión de cálculo: En todos los estudios, el eje mayor y el eje menor del tumor se midieron dos veces por semana utilizando un calibrador digital electrónico (CD-15C, Mitutoyo Corp.) y se calculó el volumen tumoral (mm3). La expresión de cálculo es como se muestra a continuación.
Volumen tumoral (mm3) = 1/2 x Eje mayor (mm) x [Eje menor (mm)]2
Todos los conjugados de anticuerpo y fármaco se diluyeron con solución salina fisiológica (Otsuka Pharmaceutical Factory, Inc.) y se usaron a un volumen de 10 ml/kg para administración intravenosa en la cola de cada ratón. En la ATCC (American Type Culture Collection) se adquirieron células A375 de la línea de melanoma humano. por vía subcutánea, se trasplantaron 8 x 106 células suspendidas en solución salina fisiológica en el abdomen derecho de cada ratón lampiño hembra (Día 0), y los ratones se agruparon al azar el Día 11. El anticuerpo M30-H1-L4P y cada uno de los conjugados de anticuerpo y fármaco (1), (2), (18) y (19), se administraron por vía intravenosa a una dosis de 10 mg/kg en la cola de cada ratón los Días 11, 18 y 25 siguiendo un programa de administración qw x 3 (una vez por semana x 3).
En la figura 17 se muestran los resultados. En el dibujo, la línea con rombos blancos representa los resultados sobre el tumor no tratado, la línea con rombos negros representa el efecto del anticuerpo M30-H1-L4P, la línea con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (1) administrado, la línea con cuadrados blancos representa el efecto del conjugado de anticuerpo y fármaco (2) administrado, la línea con triángulos negros representa el efecto del conjugado de anticuerpo y fármaco (18) administrado y la línea con triángulos blancos representa el efecto del conjugado de anticuerpo y fármaco (19) administrado.
La administración de los conjugados de anticuerpo y fármaco (1), (2), (18) o (19), disminuyó notablemente el volumen del tumor. En particular, como resultado de la administración de los conjugados de anticuerpo y fármaco (2) o (19), el tumor retrocedió completamente el Día 18 y no se confirmó que reapareciera incluso después del Día 39.
Además, los ratones que recibieron el conjugado de anticuerpo y fármaco (1), (2), (18) o (19) carecían de signos notables tales como pérdida de peso, lo que sugería que estos conjugados de anticuerpo y fármaco eran poco tóxicos y muy inocuos.
(Ejemplo de ensayo 8) Ensayo antitumoral (2)
En la ATCC (American Type Culture Collection) se adquirieron células A375 de la línea de melanoma humano. por vía subcutánea, se trasplantaron 6 x 106 células suspendidas en solución salina fisiológica en el abdomen derecho de cada ratón lampiño hembra (Día 0), y los ratones se agruparon al azar el Día 18. Cada uno de los conjugados de anticuerpo y fármaco (2) y (19), se administraron por vía intravenosa a una dosis de 1,3 mg/kg en la cola de cada ratón los Días 18, 25 y 32 siguiendo un programa de administración qw x 3 (una vez por semana x 3).
En la figura 18 se muestran los resultados. En el dibujo, la línea con rombos blancos representa los resultados sobre el tumor no tratado, la línea con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (2) administrado a una dosis de 1 mg/kg, la línea con cuadrados blancos representa el efecto del conjugado de anticuerpo y fármaco (2) administrado a una dosis de 3 mg/kg, la línea con círculos negros representa el efecto del conjugado de anticuerpo y fármaco (19) administrado a una dosis de 1 mg/kg y la línea con círculos blancos representa el efecto del conjugado de anticuerpo y fármaco (19) administrado a una dosis de 3 mg/kg. Los conjugados de anticuerpo y fármaco (2) y (19) mostraron un efecto inhibidor del crecimiento tumoral de una manera dependiente de la dosis.
(Ejemplo de ensayo 9) Ensayo antitumoral (3)
Las células Calu-6 de la línea de cáncer de pulmón humano no microcítico se adquirieron en la ATCC (American Type Culture Collection). por vía subcutánea, se trasplantaron 5 x 106 células suspendidas en solución salina fisiológica en el abdomen derecho de cada ratón lampiño hembra (Día 0), y los ratones se agruparon al azar el Día 11. El anticuerpo M30-H1-L4P y cada uno de los conjugados de anticuerpo y fármaco (1), (2), (18) o (19), se administraron por vía intravenosa a una dosis de 10 mg/kg en la cola de cada ratón los Días 11, 18 y 25 siguiendo un programa de administración qw x 3.
En la figura 19 se muestran los resultados. En el dibujo, la línea con rombos blancos representa los resultados sobre el tumor no tratado, la línea con rombos negros representa el efecto del anticuerpo M30-H1-L4P, la línea con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (1) administrado, la línea con cuadrados blancos representa el efecto del conjugado de anticuerpo y fármaco (2) administrado, la línea con triángulos negros representa el efecto del conjugado de anticuerpo y fármaco (18) administrado y la línea con triángulos blancos
representa el efecto del conjugado de anticuerpo y fármaco (19) administrado.
La administración de los conjugados de anticuerpo y fármaco (1), (2), (18) o (19) disminuyeron notablemente el volumen del tumor, y no se observó crecimiento tumoral adicional después de la administración final.
Además, los ratones que recibieron el conjugado de anticuerpo y fármaco (1), (2), (18) o (19) carecían de signos notables tales como pérdida de peso, lo que sugería que estos conjugados de anticuerpo y fármaco eran poco tóxicos y muy inocuos.
(Ejemplo de ensayo 10) Ensayo antitumoral (4)
En la ATCC (American Type Culture Collection) se adquirieron células A375 de la línea de melanoma humano. por vía subcutánea, se trasplantaron 8 x 106 células suspendidas en solución salina fisiológica en el abdomen derecho de cada ratón lampiño hembra (Día 0), y los ratones se agruparon al azar el Día 14. Cada uno de los conjugados de anticuerpo y fármaco (3), (20) y (30), se administraron por vía intravenosa a cada una de las dosis (3 y 10 mg/kg) en la cola de cada ratón el Día 14 siguiendo un programa de administración qw x 1 (una vez al día x 1).
En la figura 20 se muestran los resultados. En el dibujo, la línea con rombos blancos representa los resultados sobre el tumor no tratado, la línea de puntos con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (3) administrado a una dosis de 3 mg/kg, la línea continua con cuadrados negros representa el efecto del conjugado de anticuerpo y fármaco (3) administrado a una dosis de 10 mg/kg, La línea de puntos con triángulos negros representa el efecto del conjugado de anticuerpo y fármaco (20) administrado a una dosis de 3 mg/kg, la línea continua con triángulos rellenos representa el efecto del conjugado de anticuerpo y fármaco (20) administrado a una dosis de 10 mg/kg, la línea de puntos con círculos negros representa el efecto del conjugado de anticuerpo y fármaco (30) administrado a una dosis de 3 mg/kg y la línea continua con círculos negros representa el efecto del conjugado de anticuerpo y fármaco (30) administrado a una dosis de 10 mg/kg.
La administración de los conjugados de anticuerpo y fármaco (3), (20) o (30), disminuyó notablemente el volumen del tumor, y todos estos conjugados de anticuerpo y fármaco ejercieron un efecto inhibidor del crecimiento tumoral de una manera dependiente de la dosis.
Además, los ratones que recibieron los conjugados de anticuerpo y fármaco (3), (20) o (30), no presentaron signos notables tales como pérdida de peso, lo que sugería que estos conjugados de anticuerpo y fármaco eran poco tóxicos y muy inocuos.
Texto libre de Listado de Secuencias
SEQ ID NO: 1 - Secuencia de aminoácidos de la B7-H3 variante 1
SEQ ID NO: 2 - Secuencia de aminoácidos de la B7-H3 variante 2
SEQ ID NO: 3 - Secuencia de aminoácidos de CDRH1 del anticuerpo M30
SEQ ID NO: 4 - Secuencia de aminoácidos de CDRH2 del anticuerpo M30
SEQ ID NO: 5 - Secuencia de aminoácidos de CDRH3 del anticuerpo M30
SEQ ID NO: 6 - Secuencia de aminoácidos de CDRL1 del anticuerpo M30
SEQ ID NO: 7 - Secuencia de aminoácidos de CDRL2 del anticuerpo M30
SEQ ID NO: 8 - Secuencia de aminoácidos de CDRL3 del anticuerpo M30
SEQ ID NO: 9 - Secuencia de aminoácidos de la cadena pesada de tipo M30-H1
SEQ ID NO: 10 - Secuencia de aminoácidos de la cadena pesada de tipo M30-H2
SEQ ID NO: 11 - Secuencia de aminoácidos de la cadena pesada de tipo M30-H3
SEQ ID NO: 12 - Secuencia de aminoácidos de la cadena pesada de tipo M30-H4
SEQ ID NO: 13 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L1
SEQ ID NO: 14 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L2
SEQ ID NO: 15 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L3
SEQ ID NO: 16 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L4
SEQ ID NO: 17 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L5
SEQ ID NO: 18 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L6
SEQ ID NO: 19 - Secuencia de aminoácidos de la cadena ligera de tipo M30-L7
SEQ ID NO: 20 - Secuencia de aminoácidos de una cadena pesada del anticuerpo M30
SEQ ID NO: 21 - Secuencia de aminoácidos de una cadena ligera del anticuerpo M30
SEQ ID NO: 22 - cebador 1 de PCR
SEQ ID NO: 23 - cebador 2 de PCR
SEQ ID NO: 24 - cebador del promotor de CMV: cebador 3
SEQ ID NO: 25 - cebador inverso de BGH: cebador 4
SEQ ID NO: 26 - Secuencia de nucleótidos de la B7-H3 variante 1
SEQ ID NO: 27 - Secuencia de aminoácidos de una cadena pesada del anticuerpo anti-CD30
SEQ ID NO: 28 - Secuencia de aminoácidos de una cadena ligera del anticuerpo anti-CD30
SEQ ID NO: 29 - Secuencia de aminoácidos de una cadena pesada del anticuerpo anti-CD33
SEQ ID NO: 30 - Secuencia de aminoácidos de una cadena ligera del anticuerpo anti-CD33
Claims (10)
1. Un conjugado de anticuerpo y fármaco en el que un compuesto antitumoral representado por la siguiente fórmula:

está conjugado a un anticuerpo a través de un engarce, en el que la estructura de fármaco y engarce en el conjugado de anticuerpo y fármaco tiene una estructura seleccionada entre los siguientes grupos de estructura de fármaco y engarce:
-(Succinimid-3-il-N) -CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2-C(=O)-(NH-DX),
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-NH-CH2CH2CH2-C(=O)
-(NH-DX),
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-NH-CH2CH2CH2-C(=O)
-(NH-DX),
-(Succinimid-3-il-N)-(CH2 )5-C(=O)-N[-(CH2CH2-O)3-CH2CH2-OH]-CH2
-(C=O)-GGFG-NH-(CH2)3-C(=O)-(NH-DX),
-(Succinimid-3-il-N)-CH[-(CH2 )2-COOH]-C(=O)-NH-(CH2)2-C(=O)-GG FG-NH-(CH2 )3-C(=O)-(NH-DX), -(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-(NH-DX),
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFGG-(NH-DX),
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFGGG-(NH-DX),
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-DGGFG-(NH-DX),
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-KGGFG-(NH-DX),
en los que -(Succinimid-3-il-N)-tiene una estructura representada por la siguiente fórmula:

que está conectada al anticuerpo en la posición 3 del mismo y está conectada a un grupo metileno en la estructura de engarce que contiene esta estructura en el átomo de nitrógeno en la posición 1,
-(NH-DX) es un grupo representado por la siguiente fórmula:

en la que el átomo de nitrógeno del grupo amino en la posición 1 es la posición de conexión.
2. El conjugado de anticuerpo y fármaco según la reivindicación 1, en el que un número promedio de compuestos antitumorales conjugados por anticuerpo está en el intervalo de 1 a 8.
3. El conjugado de anticuerpo y fármaco según la reivindicación 1, en el que un número promedio de compuestos antitumorales conjugados por anticuerpo está en el intervalo de 3 a 8.
4. El conjugado de anticuerpo y fármaco según la reivindicación 1, en el que el anticuerpo es un anticuerpo anti-B7-H3, un anticuerpo anti-CD30, un anticuerpo anti-CD33 o un anticuerpo anti-CD70.
5. El conjugado de anticuerpo y fármaco según la reivindicación 1, en el que el anticuerpo es un anticuerpo anti-B7-H3.
6. Un fármaco antitumoral y/o fármaco contra el cáncer, que contiene el conjugado de anticuerpo y fármaco según la reivindicación 1 o una sal del mismo.
7. El fármaco antitumoral y/o fármaco contra el cáncer según la reivindicación 6, para su uso en el tratamiento de cáncer de pulmón, cáncer de riñón, cáncer urotelial, cáncer colorrectal, cáncer de próstata, glioblastoma multiforme, cáncer de ovario, cáncer de páncreas, cáncer de mama, melanoma, cáncer de hígado, cáncer de vejiga, cáncer de estómago o cáncer de esófago.
8. Una composición farmacéutica que contiene el conjugado de anticuerpo y fármaco según la reivindicación 1, una sal del mismo o un hidrato del mismo como componente activo, y un componente de formulación farmacéuticamente aceptable.
9. La composición farmacéutica según la reivindicación 8, para su uso en el tratamiento de cáncer de pulmón, cáncer de riñón, cáncer urotelial, cáncer colorrectal, cáncer de próstata, glioblastoma multiforme, cáncer de ovario, cáncer de páncreas, cáncer de mama, melanoma, cáncer de hígado, cáncer de vejiga, cáncer de estómago o cáncer de esófago.
10. Un compuesto intermedio de enlazador y fármaco, que es seleccionado del siguiente grupo:
(maleimid-N-ilo)-(CH2)5-C(=O)-DGGFG-NH-(CH2)3-C(=O)-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-DGGFG-NH-(CH2)2-C(=O)-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-KGGFG-NH-(CH2)3-C(=O)-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-N[-(CH2CH2-O)3-CH2CH2-OH]-CH2-(C=O)-GG FG-NH-(CH2)3-C(=O)-(NH-DX), (maleimid-N-ilo)-CH[-(CH2)2-COOH]-C(=O)-NH-(CH2)2-C(=O)-GGFG-NH-(CH2)3-C(=O)-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-GGFG-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-GGFGG-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-GGFGGG-(NH-DX),
(maleimid-N-ilo)-(CH2)5-C(=O)-DGGFG-(NH-DX) y
(maleimid-N-ilo)-(CH2)5-C(=O)-KGGFG-(NH-DX),
en el que el grupo (maleimid-N-ilo) está representado mediante la siguiente fórmula:

en la que el átomo de nitrógeno es la posición de conexión, y
-(NH-DX) es un grupo representado por la siguiente fórmula:

en la que el átomo de nitrógeno del grupo amino en la posición 1 es la posición de conexión.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012231579 | 2012-10-19 | ||
| PCT/JP2013/006178 WO2014061277A1 (ja) | 2012-10-19 | 2013-10-17 | 親水性構造を含むリンカーで結合させた抗体-薬物コンジュゲート |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2782248T3 true ES2782248T3 (es) | 2020-09-11 |
Family
ID=50487856
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13847461T Active ES2782248T3 (es) | 2012-10-19 | 2013-10-17 | Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US9872924B2 (es) |
| EP (1) | EP2910573B1 (es) |
| JP (1) | JP6272230B2 (es) |
| ES (1) | ES2782248T3 (es) |
| TW (1) | TW201420118A (es) |
| WO (1) | WO2014061277A1 (es) |
Families Citing this family (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2773710T3 (es) | 2012-10-11 | 2020-07-14 | Daiichi Sankyo Co Ltd | Enlazadores para conjugados de anticuerpo - fármaco |
| WO2014061277A1 (ja) | 2012-10-19 | 2014-04-24 | 第一三共株式会社 | 親水性構造を含むリンカーで結合させた抗体-薬物コンジュゲート |
| NZ758049A (en) | 2013-10-15 | 2024-03-22 | Seagen Inc | Pegylated drug-linkers for improved ligand-drug conjugate pharmacokinetics |
| AU2014371934B2 (en) | 2013-12-25 | 2020-01-23 | Daiichi Sankyo Company, Limited | Anti-TROP2 antibody-drug conjugate |
| IL310627B2 (en) | 2014-01-31 | 2026-04-01 | Daiichi Sankyo Co Ltd | Anti-HER2 antibody-drug conjugates, compositions containing them and uses thereof |
| JP2017114763A (ja) * | 2014-03-26 | 2017-06-29 | 第一三共株式会社 | 抗cd98抗体−薬物コンジュゲート |
| KR102445502B1 (ko) * | 2014-04-10 | 2022-09-21 | 다이이찌 산쿄 가부시키가이샤 | 항her3 항체-약물 콘주게이트 |
| EP3130608B1 (en) * | 2014-04-10 | 2019-09-04 | Daiichi Sankyo Co., Ltd. | (anti-her2 antibody)-drug conjugate |
| EP3160513B1 (en) | 2014-06-30 | 2020-02-12 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| JP6971858B2 (ja) | 2015-06-22 | 2021-11-24 | バイエル ファーマ アクチエンゲゼルシャフト | 酵素開裂性基を有する抗体薬物複合体(adc)および抗体プロドラッグ複合体(apdc) |
| CN116059395A (zh) | 2015-06-29 | 2023-05-05 | 第一三共株式会社 | 用于选择性制造抗体-药物缀合物的方法 |
| DK3354729T5 (da) * | 2015-09-24 | 2024-09-30 | Daiichi Sankyo Co Ltd | Anti-garp-antistof |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| KR20180090290A (ko) | 2015-12-04 | 2018-08-10 | 시애틀 지네틱스, 인크. | 사차화 튜불리신 화합물들의 컨쥬게이트들 |
| JP7251981B2 (ja) | 2016-03-24 | 2023-04-04 | バイエル ファーマ アクチエンゲゼルシャフト | 酵素開裂基を有する細胞毒性活性剤のプロドラッグ |
| CN109843919A (zh) | 2016-03-25 | 2019-06-04 | 西雅图基因公司 | 用于制备聚乙二醇化的药物-接头及其中间体的方法 |
| GEP20227398B (en) | 2016-04-15 | 2022-07-25 | Macrogenics Inc | Novel b7-h3 binding molecules, antibody drug conjugates thereof and usage thereof |
| EP3919518A1 (en) | 2016-06-15 | 2021-12-08 | Bayer Pharma Aktiengesellschaft | Specific antibody-drug-conjugates (adcs) with ksp inhibitors and anti-cd123-antibodies |
| US11273155B2 (en) | 2016-12-12 | 2022-03-15 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate and immune checkpoint inhibitor |
| CN110312534B (zh) | 2016-12-21 | 2023-04-04 | 拜耳制药股份公司 | 具有酶促可裂解基团的抗体-药物-缀合物(adc) |
| CN110072556B (zh) | 2016-12-21 | 2023-05-02 | 拜耳制药股份公司 | 具有ksp抑制剂的特异性抗体药物缀合物(adc) |
| US12059472B2 (en) | 2016-12-21 | 2024-08-13 | Bayer Aktiengesellschaft | Prodrugs of cytotoxic active agents having enzymatically cleavable groups |
| JP6679762B2 (ja) | 2017-01-17 | 2020-04-15 | 第一三共株式会社 | 抗gpr20抗体及び抗gpr20抗体−薬物コンジュゲート |
| CN117982673A (zh) | 2017-02-28 | 2024-05-07 | 第一三共株式会社 | 抗her3抗体-药物偶联物的应用 |
| KR20190125310A (ko) | 2017-03-10 | 2019-11-06 | 퀴아펙 파마슈티칼스 에이비 | 방출가능한 접합체 |
| KR102648564B1 (ko) | 2017-03-24 | 2024-03-19 | 씨젠 인크. | 글루쿠로니드 약물-링커의 제조 공정 및 그 중간물 |
| TW202532104A (zh) | 2017-05-15 | 2025-08-16 | 日商第一三共股份有限公司 | 抗體-藥物結合物及其用途 |
| AU2018320470B2 (en) | 2017-08-23 | 2025-04-24 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate preparation and lyophilization for same |
| IL324591A (en) | 2017-08-31 | 2026-01-01 | Daiichi Sankyo Co Ltd | Improved antibody-drug conjugate production methods |
| CA3074208C (en) | 2017-08-31 | 2023-10-03 | Daiichi Sankyo Company, Limited | Novel method for producing antibody-drug conjugate |
| KR20220127380A (ko) | 2018-03-09 | 2022-09-19 | 퀴아펙 파마슈티칼스 에이비 | 방출가능한 항체 접합체 |
| SG11202010496WA (en) | 2018-05-18 | 2020-12-30 | Daiichi Sankyo Co Ltd | Anti-muc1 antibody-drug conjugate |
| KR20210035805A (ko) | 2018-06-15 | 2021-04-01 | 플래그쉽 파이어니어링 이노베이션스 브이, 인크. | 세포후 신호전달 인자의 조절을 통한 면역 활성의 증가 |
| WO2020022475A1 (ja) | 2018-07-27 | 2020-01-30 | 第一三共株式会社 | 抗体-薬物コンジュゲートの薬物部位を認識する蛋白質 |
| CA3108044A1 (en) | 2018-07-31 | 2020-02-06 | Daiichi Sankyo Company, Limited | Treatment of metastatic brain tumor by administration of antibody-drug conjugate |
| EA202190471A1 (ru) | 2018-08-06 | 2021-05-24 | Дайити Санкио Компани, Лимитед | Комбинация конъюгата антитела-лекарственного средства и ингибитора тубулина |
| CN112739826A (zh) | 2018-08-23 | 2021-04-30 | 第一三共株式会社 | 抗体-药物缀合物的敏感性标志物 |
| CA3112473A1 (en) | 2018-09-12 | 2020-03-19 | Quiapeg Pharmaceuticals Ab | Releasable glp-1 conjugates |
| WO2020059772A1 (ja) | 2018-09-20 | 2020-03-26 | 第一三共株式会社 | 抗her3抗体-薬物コンジュゲート投与によるher3変異がんの治療 |
| WO2020063676A1 (zh) * | 2018-09-26 | 2020-04-02 | 江苏恒瑞医药股份有限公司 | 依喜替康类似物的配体-药物偶联物及其制备方法和应用 |
| CA3114474A1 (en) | 2018-09-30 | 2020-04-02 | Jiangsu Hengrui Medicine Co., Ltd. | Anti-b7h3 antibody-exatecan analog conjugate and medicinal use thereof |
| KR102905497B1 (ko) | 2018-12-11 | 2025-12-30 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 컨쥬게이트와 parp 저해제의 조합 |
| US20220040324A1 (en) * | 2018-12-21 | 2022-02-10 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate and kinase inhibitor |
| MX2021011812A (es) | 2019-03-29 | 2021-10-22 | Medimmune Ltd | Compuestos y conjugados de estos. |
| WO2020227159A2 (en) | 2019-05-03 | 2020-11-12 | Flagship Pioneering Innovations V, Inc. | Methods of modulating immune activity |
| US11555019B2 (en) | 2019-07-10 | 2023-01-17 | Cybrexa 3, Inc. | Peptide conjugates of microtubule-targeting agents as therapeutics |
| MX2022000449A (es) | 2019-07-10 | 2022-04-25 | Cybrexa 2 Inc | Conjugados peptídicos de citotoxinas como terapéuticos. |
| CN112125915A (zh) * | 2019-09-18 | 2020-12-25 | 四川百利药业有限责任公司 | 一种喜树碱衍生物及其偶联物 |
| US20230114107A1 (en) | 2019-12-17 | 2023-04-13 | Flagship Pioneering Innovations V, Inc. | Combination anti-cancer therapies with inducers of iron-dependent cellular disassembly |
| JP2023072104A (ja) * | 2020-04-10 | 2023-05-24 | 小野薬品工業株式会社 | 抗体薬物複合体 |
| US20230355804A1 (en) | 2020-06-29 | 2023-11-09 | Flagship Pioneering Innovations V, Inc. | Viruses engineered to promote thanotransmission and their use in treating cancer |
| EP4178559A4 (en) * | 2020-07-10 | 2025-03-05 | Velosbio Inc. | Novel ror1 antibody immunoconjugates |
| JP2023551355A (ja) | 2020-09-30 | 2023-12-08 | デュアリティ バイオロジクス(スーチョウ)カンパニー,リミティド | 抗腫瘍化合物並びにその調製方法及び使用 |
| US20250276078A1 (en) * | 2020-10-12 | 2025-09-04 | Baili-Bio (Chengdu) Pharmaceutical Co., Ltd. | Camptothecin Derivative And Ligand-drug Conjugate Thereof |
| AU2021378152A1 (en) | 2020-11-11 | 2023-06-22 | Daiichi Sankyo Company, Limited | COMBINATION OF AN ANTIBODY-DRUG CONJUGATE WITH ANTI-SIRPα ANTIBODY |
| WO2022099762A1 (zh) * | 2020-11-12 | 2022-05-19 | 博瑞生物医药(苏州)股份有限公司 | 一种抗体偶联物中间体及其制备方法 |
| EP4313109A1 (en) | 2021-03-31 | 2024-02-07 | Flagship Pioneering Innovations V, Inc. | Thanotransmission polypeptides and their use in treating cancer |
| AU2022303363A1 (en) | 2021-06-29 | 2024-01-18 | Flagship Pioneering Innovations V, Inc. | Immune cells engineered to promote thanotransmission and uses thereof |
| TW202320858A (zh) | 2021-07-19 | 2023-06-01 | 美商薩諾管理公司 | 免疫接合物及方法 |
| US11806405B1 (en) | 2021-07-19 | 2023-11-07 | Zeno Management, Inc. | Immunoconjugates and methods |
| WO2023083381A1 (zh) | 2021-11-15 | 2023-05-19 | 成都百利多特生物药业有限责任公司 | 双特异性抗体-喜树碱类药物偶联物及其医药用途 |
| IL312728A (en) | 2021-11-30 | 2024-07-01 | Daiichi Sankyo Co Ltd | Protease-cleavable masked antibodies |
| CN118829450A (zh) * | 2022-01-18 | 2024-10-22 | 甘李药业股份有限公司 | 一种依喜替康衍生物-抗体偶联物及其医药用途 |
| TW202342106A (zh) | 2022-02-09 | 2023-11-01 | 日商第一三共股份有限公司 | 環境應答性遮蔽抗體及其利用 |
| EP4487874A1 (en) * | 2022-02-28 | 2025-01-08 | UBE Corporation | Antibody-drug conjugate precursor and intermediate for synthesis thereof |
| WO2023163227A1 (ja) * | 2022-02-28 | 2023-08-31 | Ube株式会社 | 抗体-薬物コンジュゲート |
| EP4514391A1 (en) | 2022-04-27 | 2025-03-05 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate with ezh1 and/or ezh2 inhibitor |
| CN119403829A (zh) | 2022-05-11 | 2025-02-07 | 第一三共株式会社 | 对于肿瘤抗原特异性的抗体和cd47抑制剂的组合 |
| WO2023234427A1 (ja) * | 2022-06-03 | 2023-12-07 | Ube株式会社 | 抗体・複数薬物コンジュゲート前駆体およびその合成中間体 |
| AU2023279443A1 (en) * | 2022-06-03 | 2025-01-23 | Ube Corporation | Antibody-multidrug conjugate |
| CA3258612A1 (en) * | 2022-06-09 | 2023-12-14 | Beigene, Ltd. | Antibody drug conjugates |
| US20240116945A1 (en) * | 2022-09-02 | 2024-04-11 | Merck Sharp & Dohme Llc | Exatecan-derived topoisomerase-1 inhibitors pharmaceutical compositions, and uses thereof |
| US20240174732A1 (en) | 2022-10-05 | 2024-05-30 | Flagship Pioneering Innovations V, Inc. | Nucleic acid molecules encoding trif and additional polypeptides and their use in treating cancer |
| WO2024078449A1 (en) * | 2022-10-09 | 2024-04-18 | LaNova Medicines Limited | Compounds, compositions and methods |
| CN118307677B (zh) * | 2023-01-09 | 2025-08-12 | 中国医学科学院医药生物技术研究所 | 一种针对间皮素的单克隆抗体及应用 |
| CN116621927B (zh) * | 2023-01-09 | 2024-03-26 | 联宁(苏州)生物制药有限公司 | 带有伊喜替康和C-lock定点偶联基团的抗体偶联中间体、偶联方法及抗体偶联药物 |
| WO2024151687A1 (en) | 2023-01-09 | 2024-07-18 | Flagship Pioneering Innovations V, Inc. | Genetic switches and their use in treating cancer |
| EP4699622A1 (en) | 2023-04-19 | 2026-02-25 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate and other medicine |
| EP4670741A1 (en) * | 2023-04-28 | 2025-12-31 | Pinotbio, Inc. | ANTIBODY-DRUG CONJUGATE IN WHICH ONE OR MORE DRUG-BINDER CONJUGATES ARE LINKED TO AN ANTIBODY AND ITS PREPARATION METHOD |
| EP4721766A1 (en) | 2023-06-02 | 2026-04-08 | Daiichi Sankyo Company, Limited | Combination of (anti-her3 antibody)-drug conjugate and rasg12c inhibitor |
| WO2025002310A1 (zh) * | 2023-06-29 | 2025-01-02 | 石药集团巨石生物制药有限公司 | 抗体-药物偶联物及其用途 |
| US12319747B2 (en) | 2023-07-03 | 2025-06-03 | Medicovestor, Inc. | Methods of using anti-SP17 immunotherapeutics |
| US12364777B2 (en) | 2023-10-20 | 2025-07-22 | Medicovestor, Inc. | Homodimeric antibodies for use in treating cancers and methods of use |
| WO2025106278A1 (en) | 2023-11-17 | 2025-05-22 | Mersana Therapeutics, Inc. | Treatment of cancer using b7-h4-targeted antibody-drug conjugates |
| CN117645608A (zh) * | 2023-11-30 | 2024-03-05 | 昆山新蕴达生物科技有限公司 | 抗体药物偶联物及其应用 |
| WO2025124456A1 (en) * | 2023-12-13 | 2025-06-19 | Beigene, Ltd. | Antibody drug conjugate platforms |
| US12116410B1 (en) | 2023-12-26 | 2024-10-15 | Medicovestor, Inc. | Methods of manufacturing dimeric antibodies |
| US12600795B2 (en) | 2023-12-26 | 2026-04-14 | Medicovestor, Inc. | Oligomeric IgG for immunotherapeutics and diagnostics |
| WO2025209545A1 (zh) * | 2024-04-03 | 2025-10-09 | 上海药明合联生物技术有限公司 | 用于抗体药物偶联物的连接子、其用途和由其制备的抗体药物偶联物 |
| WO2026016639A1 (zh) * | 2024-07-19 | 2026-01-22 | 昆山新蕴达生物科技有限公司 | 一种靶向adam-9的抗体-药物偶联物及其制备方法和用途 |
Family Cites Families (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL162181A (en) | 1988-12-28 | 2006-04-10 | Pdl Biopharma Inc | A method of producing humanized immunoglubulin, and polynucleotides encoding the same |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| DK0585287T3 (da) | 1990-07-10 | 2000-04-17 | Cambridge Antibody Tech | Fremgangsmåde til fremstilling af specifikke bindingsparelementer |
| JP3008226B2 (ja) | 1991-01-16 | 2000-02-14 | 第一製薬株式会社 | 六環性化合物 |
| US5658920A (en) | 1991-01-16 | 1997-08-19 | Daiichi Pharmaceutical Co., Ltd. | Substituted 1H,12H-benz-[DE]pyrano[3',4':6,7] indolizino[1,2-B]quinoline-10,13(9H,15H)-dione compound |
| EP0605522B1 (en) | 1991-09-23 | 1999-06-23 | Medical Research Council | Methods for the production of humanized antibodies |
| PT1024191E (pt) | 1991-12-02 | 2008-12-22 | Medical Res Council | Produção de auto-anticorpos a partir de reportórios de segmentos de anticorpo e exibidos em fagos |
| CA2131151A1 (en) | 1992-03-24 | 1994-09-30 | Kevin S. Johnson | Methods for producing members of specific binding pairs |
| JP3359955B2 (ja) | 1992-07-16 | 2002-12-24 | 第一製薬株式会社 | 抗腫瘍剤 |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| GB9313509D0 (en) | 1993-06-30 | 1993-08-11 | Medical Res Council | Chemisynthetic libraries |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| US5773001A (en) | 1994-06-03 | 1998-06-30 | American Cyanamid Company | Conjugates of methyltrithio antitumor agents and intermediates for their synthesis |
| JPH08337584A (ja) | 1995-04-10 | 1996-12-24 | Dai Ichi Seiyaku Co Ltd | 縮合六環式アミノ化合物、これを含有する医薬及びその製法 |
| US6504029B1 (en) | 1995-04-10 | 2003-01-07 | Daiichi Pharmaceutical Co., Ltd. | Condensed-hexacyclic compounds and a process therefor |
| SG50747A1 (en) | 1995-08-02 | 1998-07-20 | Tanabe Seiyaku Co | Comptothecin derivatives |
| JPH1095802A (ja) * | 1995-12-28 | 1998-04-14 | Tanabe Seiyaku Co Ltd | カンプトテシン誘導体 |
| CA2192725C (en) | 1995-12-28 | 2004-04-20 | Kenji Tsujihara | Camptothecin derivatives |
| TW409058B (en) | 1996-06-06 | 2000-10-21 | Daiichi Seiyaku Co | Method for preparation of a drug complex |
| TW527183B (en) | 1996-06-06 | 2003-04-11 | Daiichi Seiyaku Co | Drug complex |
| JPH1192405A (ja) * | 1997-09-19 | 1999-04-06 | Dai Ichi Seiyaku Co Ltd | 薬物複合体 |
| DK1071700T3 (da) | 1998-04-20 | 2010-06-07 | Glycart Biotechnology Ag | Glykosylerings-modifikation af antistoffer til forbedring af antistofafhængig cellulær cytotoksicitet |
| EA003398B1 (ru) * | 1998-05-22 | 2003-04-24 | Дайити Фармасьютикал Ко., Лтд. | Лекарственный комплекс c полимерным носителем |
| CN1191093C (zh) | 1998-10-30 | 2005-03-02 | 第一制药株式会社 | Dds化合物及其测定方法 |
| CA2704600C (en) | 1999-04-09 | 2016-10-25 | Kyowa Kirin Co., Ltd. | A method for producing antibodies with increased adcc activity |
| JP2002060351A (ja) | 2000-03-22 | 2002-02-26 | Dai Ichi Seiyaku Co Ltd | 水酸基を有する薬物を含むdds化合物 |
| US20030166513A1 (en) | 2000-06-29 | 2003-09-04 | Akihiro Imura | Dds compound and process for the preparation thereof |
| AU2001271037A1 (en) | 2000-07-13 | 2002-01-30 | Daiichi Pharmaceutical Co., Ltd. | Pharmaceutical compositions containing dds compounds |
| CA2953239A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kirin Co., Ltd. | Antibody composition-producing cell |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| TWI313609B (en) | 2001-08-21 | 2009-08-21 | Mitsubishi Tanabe Pharma Corp | Pharmaceutical composition for inhibiting the metastasis or preventing the recurrence of malignant tumor |
| IL161686A0 (en) | 2001-11-01 | 2004-09-27 | Uab Research Foundation | Combinations of antibodies selective for a tumor necrosis factor-related apoptosis-inducing ligand receptor and other therapeutic agents |
| CA2467242A1 (en) | 2001-11-20 | 2003-05-30 | Seattle Genetics, Inc. | Treatment of immunological disorders using anti-cd30 antibodies |
| US8877901B2 (en) | 2002-12-13 | 2014-11-04 | Immunomedics, Inc. | Camptothecin-binding moiety conjugates |
| CA2508831C (en) | 2002-12-13 | 2012-05-01 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| US20040202666A1 (en) | 2003-01-24 | 2004-10-14 | Immunomedics, Inc. | Anti-cancer anthracycline drug-antibody conjugates |
| US20050228007A1 (en) | 2004-02-26 | 2005-10-13 | Prakash Jagtap | Isoquinoline derivatives and methods of use thereof |
| JP4942643B2 (ja) | 2004-03-02 | 2012-05-30 | シアトル ジェネティックス, インコーポレイテッド | 部分的に付加された抗体およびそれらの結合体化方法 |
| JP4806680B2 (ja) * | 2004-05-19 | 2011-11-02 | メダレックス インコーポレイテッド | 自己犠牲リンカー及び薬剤複合体 |
| NZ579482A (en) | 2004-06-01 | 2011-02-25 | Genentech Inc | Antibody drug conjugates and methods |
| JP2008521828A (ja) * | 2004-11-29 | 2008-06-26 | シアトル ジェネティックス, インコーポレイテッド | 操作された抗体およびイムノコンジュゲート |
| DE102005009084A1 (de) | 2005-02-28 | 2006-08-31 | Ktb Tumorforschungsgesellschaft Mbh | Proteinbindende Anthrazyklin-Peptid-Derivate und diese enthaltende Arzneimittel |
| DE102005009099A1 (de) | 2005-02-28 | 2006-08-31 | Ktb Tumorforschungsgesellschaft Mbh | Proteinbindende Camptothecin-Peptid-Derivate und diese enthaltende Arzneimittel |
| JP5122441B2 (ja) | 2005-04-19 | 2013-01-16 | シアトル ジェネティックス, インコーポレイテッド | ヒト化抗cd70結合剤およびその使用 |
| US7833979B2 (en) | 2005-04-22 | 2010-11-16 | Amgen Inc. | Toxin peptide therapeutic agents |
| EP2032606B1 (en) | 2006-05-30 | 2013-11-27 | Genentech, Inc. | Antibodies and immunoconjugates and uses therefor |
| CN101490087B (zh) | 2006-05-30 | 2013-11-06 | 健泰科生物技术公司 | 抗体和免疫偶联物及其用途 |
| ITMI20061473A1 (it) | 2006-07-26 | 2008-01-27 | Indena Spa | Derivati della camptotecina ad attivita antitumorale |
| CA2673410A1 (en) | 2006-12-21 | 2008-07-03 | Schering Corporation | Pyrrolo [3, 2-a] pyridine derivatives for inhibiting ksp kinesin activity |
| US20100143245A1 (en) | 2007-03-22 | 2010-06-10 | Sloan-Kettering Institute For Cancer Research | Uses of monoclonal antibody 8h9 |
| NZ610239A (en) | 2008-04-30 | 2014-11-28 | Immunogen Inc | Cross-linkers and their uses |
| ES3000111T3 (en) | 2009-02-13 | 2025-02-27 | Immunomedics Inc | Intermediates for preparing conjugates with an intracellularly-cleavable linkage |
| JP5829520B2 (ja) | 2009-08-20 | 2015-12-09 | 国立大学法人 千葉大学 | コルヒチン誘導体 |
| JP2013505944A (ja) | 2009-09-24 | 2013-02-21 | シアトル ジェネティックス, インコーポレイテッド | Dr5リガンド薬物結合体 |
| MX2012013001A (es) | 2010-05-13 | 2013-02-26 | Univ Indiana Res & Tech Corp | Peptidos de la superfamilia de glucagon que presentan actividad del receptor nuclear de hormonas. |
| US20110300150A1 (en) | 2010-05-18 | 2011-12-08 | Scott Eliasof | Compositions and methods for treatment of autoimmune and other disease |
| RU2012157167A (ru) | 2010-07-12 | 2014-08-20 | КовЭкс Текнолоджиз Айэлэнд Лимитед | Полифункциональные антительные конъюгаты |
| US20120121615A1 (en) | 2010-11-17 | 2012-05-17 | Flygare John A | Alaninyl maytansinol antibody conjugates |
| SG11201403085PA (en) | 2011-12-14 | 2014-10-30 | Seattle Genetics Inc | Fgfr antibody drug conjugates (adcs) and the use thereof |
| US20130280282A1 (en) | 2012-04-24 | 2013-10-24 | Daiichi Sankyo Co., Ltd. | Dr5 ligand drug conjugates |
| CN104619350A (zh) | 2012-06-14 | 2015-05-13 | Ambrx公司 | 结合到核受体配体多肽的抗psma抗体 |
| ES2773710T3 (es) | 2012-10-11 | 2020-07-14 | Daiichi Sankyo Co Ltd | Enlazadores para conjugados de anticuerpo - fármaco |
| WO2014061277A1 (ja) | 2012-10-19 | 2014-04-24 | 第一三共株式会社 | 親水性構造を含むリンカーで結合させた抗体-薬物コンジュゲート |
| EP2927227A4 (en) | 2013-01-03 | 2015-12-30 | Celltrion Inc | ANTIBODY CONCENTRATOR REMEDY CONJUGATE, METHOD OF MANUFACTURING THEREOF, AND ANTIBODY TOGETHER THEREWITH |
-
2013
- 2013-10-17 WO PCT/JP2013/006178 patent/WO2014061277A1/ja not_active Ceased
- 2013-10-17 EP EP13847461.4A patent/EP2910573B1/en active Active
- 2013-10-17 US US14/436,458 patent/US9872924B2/en active Active
- 2013-10-17 JP JP2014541955A patent/JP6272230B2/ja active Active
- 2013-10-17 ES ES13847461T patent/ES2782248T3/es active Active
- 2013-10-18 TW TW102137701A patent/TW201420118A/zh unknown
-
2017
- 2017-11-22 US US15/821,697 patent/US10729782B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US10729782B2 (en) | 2020-08-04 |
| EP2910573A1 (en) | 2015-08-26 |
| WO2014061277A1 (ja) | 2014-04-24 |
| JP6272230B2 (ja) | 2018-01-31 |
| TW201420118A (zh) | 2014-06-01 |
| US20150352224A1 (en) | 2015-12-10 |
| EP2910573A4 (en) | 2016-06-15 |
| JPWO2014061277A1 (ja) | 2016-09-05 |
| US20180071403A1 (en) | 2018-03-15 |
| US9872924B2 (en) | 2018-01-23 |
| EP2910573B1 (en) | 2020-02-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2782248T3 (es) | Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila | |
| AU2020200548B2 (en) | Antibody-drug conjugate | |
| ES3005311T3 (en) | Anti-her2 antibody-drug conjugate | |
| ES2754348T3 (es) | Conjugado de (anticuerpo anti-HER2)-fármaco | |
| RU2776489C2 (ru) | Конъюгат антитело-лекарственное средство | |
| HK1256631B (en) | Linkers for antibody-drug conjugates | |
| BR122020012304B1 (pt) | Composto ligante para conjugados anticorpo-fármaco, método e intermediários de produção do mesmo |