ES2821951T3 - Compuestos de benzoxaborol sustituidos en posición 4 y sus usos - Google Patents
Compuestos de benzoxaborol sustituidos en posición 4 y sus usos Download PDFInfo
- Publication number
- ES2821951T3 ES2821951T3 ES16706002T ES16706002T ES2821951T3 ES 2821951 T3 ES2821951 T3 ES 2821951T3 ES 16706002 T ES16706002 T ES 16706002T ES 16706002 T ES16706002 T ES 16706002T ES 2821951 T3 ES2821951 T3 ES 2821951T3
- Authority
- ES
- Spain
- Prior art keywords
- mycobacterium
- compound
- pharmaceutically acceptable
- tuberculosis
- therapeutic agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 4-substituted benzoxaborol compounds Chemical class 0.000 title claims description 93
- 150000001875 compounds Chemical class 0.000 claims abstract description 286
- 150000003839 salts Chemical class 0.000 claims abstract description 157
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 108
- 239000003814 drug Substances 0.000 claims description 132
- 229940124597 therapeutic agent Drugs 0.000 claims description 108
- 241000186359 Mycobacterium Species 0.000 claims description 76
- 201000008827 tuberculosis Diseases 0.000 claims description 76
- 239000008194 pharmaceutical composition Substances 0.000 claims description 59
- 238000011282 treatment Methods 0.000 claims description 42
- 208000027531 mycobacterial infectious disease Diseases 0.000 claims description 41
- 206010062207 Mycobacterial infection Diseases 0.000 claims description 39
- 241000187479 Mycobacterium tuberculosis Species 0.000 claims description 29
- 201000010099 disease Diseases 0.000 claims description 29
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 29
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 18
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 claims description 18
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 claims description 13
- 229960001225 rifampicin Drugs 0.000 claims description 12
- 241001502334 Mycobacterium avium complex bacterium Species 0.000 claims description 11
- 241001302239 Mycobacterium tuberculosis complex Species 0.000 claims description 11
- 239000003085 diluting agent Substances 0.000 claims description 11
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 claims description 11
- 229960003350 isoniazid Drugs 0.000 claims description 10
- 241000186367 Mycobacterium avium Species 0.000 claims description 9
- 229940124522 antiretrovirals Drugs 0.000 claims description 8
- 229960002599 rifapentine Drugs 0.000 claims description 8
- WDZCUPBHRAEYDL-GZAUEHORSA-N rifapentine Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N(CC1)CCN1C1CCCC1 WDZCUPBHRAEYDL-GZAUEHORSA-N 0.000 claims description 8
- 208000019693 Lung disease Diseases 0.000 claims description 7
- 241000186365 Mycobacterium fortuitum Species 0.000 claims description 7
- 239000003903 antiretrovirus agent Substances 0.000 claims description 7
- 229960000285 ethambutol Drugs 0.000 claims description 7
- 208000015181 infectious disease Diseases 0.000 claims description 7
- 229960005206 pyrazinamide Drugs 0.000 claims description 7
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 claims description 7
- 208000023081 Buruli ulcer disease Diseases 0.000 claims description 6
- BXZVVICBKDXVGW-NKWVEPMBSA-N Didanosine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(NC=NC2=O)=C2N=C1 BXZVVICBKDXVGW-NKWVEPMBSA-N 0.000 claims description 6
- 241000187482 Mycobacterium avium subsp. paratuberculosis Species 0.000 claims description 6
- 241000187478 Mycobacterium chelonae Species 0.000 claims description 6
- 241000187484 Mycobacterium gordonae Species 0.000 claims description 6
- 241000186363 Mycobacterium kansasii Species 0.000 claims description 6
- 241000186362 Mycobacterium leprae Species 0.000 claims description 6
- 241001532512 Mycobacterium parafortuitum Species 0.000 claims description 6
- 241000187644 Mycobacterium vaccae Species 0.000 claims description 6
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 claims description 6
- 229960005107 darunavir Drugs 0.000 claims description 6
- CJBJHOAVZSMMDJ-HEXNFIEUSA-N darunavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)C1=CC=CC=C1 CJBJHOAVZSMMDJ-HEXNFIEUSA-N 0.000 claims description 6
- 229960002656 didanosine Drugs 0.000 claims description 6
- 229960002049 etravirine Drugs 0.000 claims description 6
- PYGWGZALEOIKDF-UHFFFAOYSA-N etravirine Chemical compound CC1=CC(C#N)=CC(C)=C1OC1=NC(NC=2C=CC(=CC=2)C#N)=NC(N)=C1Br PYGWGZALEOIKDF-UHFFFAOYSA-N 0.000 claims description 6
- 229960003702 moxifloxacin Drugs 0.000 claims description 6
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 claims description 6
- 241000180044 Mycobacterium avium subsp. avium Species 0.000 claims description 5
- 241000114135 Mycobacterium avium subsp. hominissuis Species 0.000 claims description 5
- 241000178954 Mycobacterium avium subsp. silvaticum Species 0.000 claims description 5
- 241000545499 Mycobacterium avium-intracellulare Species 0.000 claims description 5
- 241000186364 Mycobacterium intracellulare Species 0.000 claims description 5
- 241000187490 Mycobacterium scrofulaceum Species 0.000 claims description 5
- 241000187494 Mycobacterium xenopi Species 0.000 claims description 5
- 239000003443 antiviral agent Substances 0.000 claims description 5
- XDAOLTSRNUSPPH-XMMPIXPASA-N delamanid Chemical compound C([C@]1(C)OC2=NC(=CN2C1)[N+]([O-])=O)OC(C=C1)=CC=C1N(CC1)CCC1OC1=CC=C(OC(F)(F)F)C=C1 XDAOLTSRNUSPPH-XMMPIXPASA-N 0.000 claims description 5
- 238000000371 solid-state nuclear magnetic resonance spectroscopy Methods 0.000 claims description 5
- QUIJNHUBAXPXFS-UHFFFAOYSA-N 1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-2-naphthalen-1-yl-1-phenylbutan-2-ol Chemical compound COC1=NC2=CC=C(Br)C=C2C=C1C(C(O)(CCN(C)C)C=1C2=CC=CC=C2C=CC=1)C1=CC=CC=C1 QUIJNHUBAXPXFS-UHFFFAOYSA-N 0.000 claims description 4
- KIMCGLHTSSZPNS-UHFFFAOYSA-N 2,3-dinitrobenzamide Chemical compound NC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O KIMCGLHTSSZPNS-UHFFFAOYSA-N 0.000 claims description 4
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 claims description 4
- 241001508003 Mycobacterium abscessus Species 0.000 claims description 4
- 241001467553 Mycobacterium africanum Species 0.000 claims description 4
- 241000211133 Mycobacterium caprae Species 0.000 claims description 4
- 241001147828 Mycobacterium haemophilum Species 0.000 claims description 4
- 241000187492 Mycobacterium marinum Species 0.000 claims description 4
- 241000187919 Mycobacterium microti Species 0.000 claims description 4
- 241000187489 Mycobacterium simiae Species 0.000 claims description 4
- 241000187917 Mycobacterium ulcerans Species 0.000 claims description 4
- 239000002671 adjuvant Substances 0.000 claims description 4
- WCWSTNLSLKSJPK-LKFCYVNXSA-N cabotegravir Chemical compound C([C@H]1OC[C@@H](N1C(=O)C1=C(O)C2=O)C)N1C=C2C(=O)NCC1=CC=C(F)C=C1F WCWSTNLSLKSJPK-LKFCYVNXSA-N 0.000 claims description 4
- 229960004287 clofazimine Drugs 0.000 claims description 4
- WDQPAMHFFCXSNU-BGABXYSRSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=N/C(C)C)/C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-BGABXYSRSA-N 0.000 claims description 4
- 229960002542 dolutegravir Drugs 0.000 claims description 4
- RHWKPHLQXYSBKR-BMIGLBTASA-N dolutegravir Chemical compound C([C@@H]1OCC[C@H](N1C(=O)C1=C(O)C2=O)C)N1C=C2C(=O)NCC1=CC=C(F)C=C1F RHWKPHLQXYSBKR-BMIGLBTASA-N 0.000 claims description 4
- 229960003907 linezolid Drugs 0.000 claims description 4
- TYZROVQLWOKYKF-ZDUSSCGKSA-N linezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCOCC1 TYZROVQLWOKYKF-ZDUSSCGKSA-N 0.000 claims description 4
- 210000004072 lung Anatomy 0.000 claims description 4
- 201000003265 lymphadenitis Diseases 0.000 claims description 4
- CNPVJJQCETWNEU-CYFREDJKSA-N (4,6-dimethyl-5-pyrimidinyl)-[4-[(3S)-4-[(1R)-2-methoxy-1-[4-(trifluoromethyl)phenyl]ethyl]-3-methyl-1-piperazinyl]-4-methyl-1-piperidinyl]methanone Chemical compound N([C@@H](COC)C=1C=CC(=CC=1)C(F)(F)F)([C@H](C1)C)CCN1C(CC1)(C)CCN1C(=O)C1=C(C)N=CN=C1C CNPVJJQCETWNEU-CYFREDJKSA-N 0.000 claims description 3
- OKGPFTLYBPQBIX-CQSZACIVSA-N 1-[(2r)-4-benzoyl-2-methylpiperazin-1-yl]-2-(4-methoxy-1h-pyrrolo[2,3-b]pyridin-3-yl)ethane-1,2-dione Chemical compound C1=2C(OC)=CC=NC=2NC=C1C(=O)C(=O)N([C@@H](C1)C)CCN1C(=O)C1=CC=CC=C1 OKGPFTLYBPQBIX-CQSZACIVSA-N 0.000 claims description 3
- CGBYTKOSZYQOPV-ASSBYYIWSA-N 5-chloro-3-[[3-[(e)-2-cyanoethenyl]-5-methylphenyl]-methoxyphosphoryl]-1h-indole-2-carboxamide Chemical compound C1([P@](=O)(C=2C3=CC(Cl)=CC=C3NC=2C(N)=O)OC)=CC(C)=CC(\C=C\C#N)=C1 CGBYTKOSZYQOPV-ASSBYYIWSA-N 0.000 claims description 3
- UXCAQJAQSWSNPQ-XLPZGREQSA-N Alovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](F)C1 UXCAQJAQSWSNPQ-XLPZGREQSA-N 0.000 claims description 3
- AXRYRYVKAWYZBR-UHFFFAOYSA-N Atazanavir Natural products C=1C=C(C=2N=CC=CC=2)C=CC=1CN(NC(=O)C(NC(=O)OC)C(C)(C)C)CC(O)C(NC(=O)C(NC(=O)OC)C(C)(C)C)CC1=CC=CC=C1 AXRYRYVKAWYZBR-UHFFFAOYSA-N 0.000 claims description 3
- 108010019625 Atazanavir Sulfate Proteins 0.000 claims description 3
- 208000006448 Buruli Ulcer Diseases 0.000 claims description 3
- 108010036239 CD4-IgG(2) Proteins 0.000 claims description 3
- QAGYKUNXZHXKMR-UHFFFAOYSA-N CPD000469186 Natural products CC1=C(O)C=CC=C1C(=O)NC(C(O)CN1C(CC2CCCCC2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-UHFFFAOYSA-N 0.000 claims description 3
- 208000011231 Crohn disease Diseases 0.000 claims description 3
- XQSPYNMVSIKCOC-NTSWFWBYSA-N Emtricitabine Chemical compound C1=C(F)C(N)=NC(=O)N1[C@H]1O[C@@H](CO)SC1 XQSPYNMVSIKCOC-NTSWFWBYSA-N 0.000 claims description 3
- 206010018691 Granuloma Diseases 0.000 claims description 3
- KJHKTHWMRKYKJE-SUGCFTRWSA-N Kaletra Chemical compound N1([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=2C=CC=CC=2)NC(=O)COC=2C(=CC=CC=2C)C)CC=2C=CC=CC=2)CCCNC1=O KJHKTHWMRKYKJE-SUGCFTRWSA-N 0.000 claims description 3
- 206010024229 Leprosy Diseases 0.000 claims description 3
- MCPUZZJBAHRIPO-UHFFFAOYSA-N Lersivirine Chemical compound CCC1=NN(CCO)C(CC)=C1OC1=CC(C#N)=CC(C#N)=C1 MCPUZZJBAHRIPO-UHFFFAOYSA-N 0.000 claims description 3
- 208000032376 Lung infection Diseases 0.000 claims description 3
- 241000186366 Mycobacterium bovis Species 0.000 claims description 3
- 241001467552 Mycobacterium bovis BCG Species 0.000 claims description 3
- 241001312372 Mycobacterium canettii Species 0.000 claims description 3
- 241000187496 Mycobacterium szulgai Species 0.000 claims description 3
- 206010066289 Mycobacterium ulcerans infection Diseases 0.000 claims description 3
- 206010035664 Pneumonia Diseases 0.000 claims description 3
- 201000001177 Pyomyositis Diseases 0.000 claims description 3
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 claims description 3
- 206010062255 Soft tissue infection Diseases 0.000 claims description 3
- XNKLLVCARDGLGL-JGVFFNPUSA-N Stavudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1C=C[C@@H](CO)O1 XNKLLVCARDGLGL-JGVFFNPUSA-N 0.000 claims description 3
- SUJUHGSWHZTSEU-UHFFFAOYSA-N Tipranavir Natural products C1C(O)=C(C(CC)C=2C=C(NS(=O)(=O)C=3N=CC(=CC=3)C(F)(F)F)C=CC=2)C(=O)OC1(CCC)CCC1=CC=CC=C1 SUJUHGSWHZTSEU-UHFFFAOYSA-N 0.000 claims description 3
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 claims description 3
- RLAHNGKRJJEIJL-RFZPGFLSSA-N [(2r,4r)-4-(2,6-diaminopurin-9-yl)-1,3-dioxolan-2-yl]methanol Chemical compound C12=NC(N)=NC(N)=C2N=CN1[C@H]1CO[C@@H](CO)O1 RLAHNGKRJJEIJL-RFZPGFLSSA-N 0.000 claims description 3
- 229960004748 abacavir Drugs 0.000 claims description 3
- MCGSCOLBFJQGHM-SCZZXKLOSA-N abacavir Chemical compound C=12N=CN([C@H]3C=C[C@@H](CO)C3)C2=NC(N)=NC=1NC1CC1 MCGSCOLBFJQGHM-SCZZXKLOSA-N 0.000 claims description 3
- 206010000269 abscess Diseases 0.000 claims description 3
- 229960001997 adefovir Drugs 0.000 claims description 3
- 229960003205 adefovir dipivoxil Drugs 0.000 claims description 3
- 229950004424 alovudine Drugs 0.000 claims description 3
- 229950005846 amdoxovir Drugs 0.000 claims description 3
- 229960001830 amprenavir Drugs 0.000 claims description 3
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 claims description 3
- 229960003277 atazanavir Drugs 0.000 claims description 3
- AXRYRYVKAWYZBR-GASGPIRDSA-N atazanavir Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)OC)C(C)(C)C)[C@@H](O)CN(CC=1C=CC(=CC=1)C=1N=CC=CC=1)NC(=O)[C@@H](NC(=O)OC)C(C)(C)C)C1=CC=CC=C1 AXRYRYVKAWYZBR-GASGPIRDSA-N 0.000 claims description 3
- 229950009079 brecanavir Drugs 0.000 claims description 3
- JORVRJNILJXMMG-OLNQLETPSA-N brecanavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=C2OCOC2=CC=1)NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)C(C=C1)=CC=C1OCC1=CSC(C)=N1 JORVRJNILJXMMG-OLNQLETPSA-N 0.000 claims description 3
- YQXCVAGCMNFUMQ-UHFFFAOYSA-N capravirine Chemical compound C=1C(Cl)=CC(Cl)=CC=1SC1=C(C(C)C)N=C(COC(N)=O)N1CC1=CC=NC=C1 YQXCVAGCMNFUMQ-UHFFFAOYSA-N 0.000 claims description 3
- 229950008230 capravirine Drugs 0.000 claims description 3
- XNHZXMPLVSJQFK-UHFFFAOYSA-O dimethyl-[[4-[[3-(4-methylphenyl)-8,9-dihydro-7h-benzo[7]annulene-6-carbonyl]amino]phenyl]methyl]-(oxan-4-yl)azanium Chemical compound C1=CC(C)=CC=C1C1=CC=C(CCCC(=C2)C(=O)NC=3C=CC(C[N+](C)(C)C4CCOCC4)=CC=3)C2=C1 XNHZXMPLVSJQFK-UHFFFAOYSA-O 0.000 claims description 3
- 229960003586 elvitegravir Drugs 0.000 claims description 3
- JUZYLCPPVHEVSV-LJQANCHMSA-N elvitegravir Chemical compound COC1=CC=2N([C@H](CO)C(C)C)C=C(C(O)=O)C(=O)C=2C=C1CC1=CC=CC(Cl)=C1F JUZYLCPPVHEVSV-LJQANCHMSA-N 0.000 claims description 3
- 229960000366 emtricitabine Drugs 0.000 claims description 3
- 229960003142 fosamprenavir Drugs 0.000 claims description 3
- MLBVMOWEQCZNCC-OEMFJLHTSA-N fosamprenavir Chemical compound C([C@@H]([C@H](OP(O)(O)=O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 MLBVMOWEQCZNCC-OEMFJLHTSA-N 0.000 claims description 3
- SWMDAPWAQQTBOG-UHFFFAOYSA-N fostemsavir Chemical compound C1=2N(COP(O)(O)=O)C=C(C(=O)C(=O)N3CCN(CC3)C(=O)C=3C=CC=CC=3)C=2C(OC)=CN=C1N1C=NC(C)=N1 SWMDAPWAQQTBOG-UHFFFAOYSA-N 0.000 claims description 3
- 229950010812 fostemsavir Drugs 0.000 claims description 3
- 229960001936 indinavir Drugs 0.000 claims description 3
- 229960001627 lamivudine Drugs 0.000 claims description 3
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 claims description 3
- 229940121292 leronlimab Drugs 0.000 claims description 3
- 229950004188 lersivirine Drugs 0.000 claims description 3
- 229960004525 lopinavir Drugs 0.000 claims description 3
- CJPLEFFCVDQQFZ-UHFFFAOYSA-N loviride Chemical compound CC(=O)C1=CC=C(C)C=C1NC(C(N)=O)C1=C(Cl)C=CC=C1Cl CJPLEFFCVDQQFZ-UHFFFAOYSA-N 0.000 claims description 3
- 229950006243 loviride Drugs 0.000 claims description 3
- 229960004710 maraviroc Drugs 0.000 claims description 3
- GSNHKUDZZFZSJB-QYOOZWMWSA-N maraviroc Chemical compound CC(C)C1=NN=C(C)N1[C@@H]1C[C@H](N2CC[C@H](NC(=O)C3CCC(F)(F)CC3)C=3C=CC=CC=3)CC[C@H]2C1 GSNHKUDZZFZSJB-QYOOZWMWSA-N 0.000 claims description 3
- 208000004396 mastitis Diseases 0.000 claims description 3
- 229960000884 nelfinavir Drugs 0.000 claims description 3
- QAGYKUNXZHXKMR-HKWSIXNMSA-N nelfinavir Chemical compound CC1=C(O)C=CC=C1C(=O)N[C@H]([C@H](O)CN1[C@@H](C[C@@H]2CCCC[C@@H]2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-HKWSIXNMSA-N 0.000 claims description 3
- CKNAQFVBEHDJQV-UHFFFAOYSA-N oltipraz Chemical compound S1SC(=S)C(C)=C1C1=CN=CC=N1 CKNAQFVBEHDJQV-UHFFFAOYSA-N 0.000 claims description 3
- 229950008687 oltipraz Drugs 0.000 claims description 3
- 229950006460 palinavir Drugs 0.000 claims description 3
- RXBWRFDZXRAEJT-SZNOJMITSA-N palinavir Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)C=1N=C2C=CC=CC2=CC=1)C(C)C)[C@H](O)CN1[C@@H](C[C@@H](CC1)OCC=1C=CN=CC=1)C(=O)NC(C)(C)C)C1=CC=CC=C1 RXBWRFDZXRAEJT-SZNOJMITSA-N 0.000 claims description 3
- 229960004742 raltegravir Drugs 0.000 claims description 3
- CZFFBEXEKNGXKS-UHFFFAOYSA-N raltegravir Chemical compound O1C(C)=NN=C1C(=O)NC(C)(C)C1=NC(C(=O)NCC=2C=CC(F)=CC=2)=C(O)C(=O)N1C CZFFBEXEKNGXKS-UHFFFAOYSA-N 0.000 claims description 3
- YIBOMRUWOWDFLG-ONEGZZNKSA-N rilpivirine Chemical compound CC1=CC(\C=C\C#N)=CC(C)=C1NC1=CC=NC(NC=2C=CC(=CC=2)C#N)=N1 YIBOMRUWOWDFLG-ONEGZZNKSA-N 0.000 claims description 3
- 229960000311 ritonavir Drugs 0.000 claims description 3
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 claims description 3
- 229960001852 saquinavir Drugs 0.000 claims description 3
- QWAXKHKRTORLEM-UGJKXSETSA-N saquinavir Chemical compound C([C@@H]([C@H](O)CN1C[C@H]2CCCC[C@H]2C[C@H]1C(=O)NC(C)(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)C=1N=C2C=CC=CC2=CC=1)C1=CC=CC=C1 QWAXKHKRTORLEM-UGJKXSETSA-N 0.000 claims description 3
- 206010040872 skin infection Diseases 0.000 claims description 3
- 229960001203 stavudine Drugs 0.000 claims description 3
- FNDDDNOJWPQCBZ-ZDUSSCGKSA-N sutezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCSCC1 FNDDDNOJWPQCBZ-ZDUSSCGKSA-N 0.000 claims description 3
- 208000011580 syndromic disease Diseases 0.000 claims description 3
- 210000001258 synovial membrane Anatomy 0.000 claims description 3
- 210000002435 tendon Anatomy 0.000 claims description 3
- 229960004556 tenofovir Drugs 0.000 claims description 3
- VCMJCVGFSROFHV-WZGZYPNHSA-N tenofovir disoproxil fumarate Chemical compound OC(=O)\C=C\C(O)=O.N1=CN=C2N(C[C@@H](C)OCP(=O)(OCOC(=O)OC(C)C)OCOC(=O)OC(C)C)C=NC2=C1N VCMJCVGFSROFHV-WZGZYPNHSA-N 0.000 claims description 3
- 229960000838 tipranavir Drugs 0.000 claims description 3
- SUJUHGSWHZTSEU-FYBSXPHGSA-N tipranavir Chemical compound C([C@@]1(CCC)OC(=O)C([C@H](CC)C=2C=C(NS(=O)(=O)C=3N=CC(=CC=3)C(F)(F)F)C=CC=2)=C(O)C1)CC1=CC=CC=C1 SUJUHGSWHZTSEU-FYBSXPHGSA-N 0.000 claims description 3
- 229950009860 vicriviroc Drugs 0.000 claims description 3
- 229960000523 zalcitabine Drugs 0.000 claims description 3
- QRPZBKAMSFHVRW-UHFFFAOYSA-N 1-(4-benzoylpiperazin-1-yl)-2-[4-methoxy-7-(3-methyl-1,2,4-triazol-1-yl)-1h-pyrrolo[2,3-c]pyridin-3-yl]ethane-1,2-dione Chemical compound C1=2NC=C(C(=O)C(=O)N3CCN(CC3)C(=O)C=3C=CC=CC=3)C=2C(OC)=CN=C1N1C=NC(C)=N1 QRPZBKAMSFHVRW-UHFFFAOYSA-N 0.000 claims description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 2
- 230000000798 anti-retroviral effect Effects 0.000 claims description 2
- 229950010245 ibalizumab Drugs 0.000 claims description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 claims 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims 1
- 229910019142 PO4 Inorganic materials 0.000 claims 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 claims 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 claims 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 claims 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims 1
- CBVCZFGXHXORBI-PXQQMZJSSA-N indinavir Chemical compound C([C@H](N(CC1)C[C@@H](O)C[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H]2C3=CC=CC=C3C[C@H]2O)C(=O)NC(C)(C)C)N1CC1=CC=CN=C1 CBVCZFGXHXORBI-PXQQMZJSSA-N 0.000 claims 1
- 150000004767 nitrides Chemical class 0.000 claims 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims 1
- 239000010452 phosphate Substances 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 142
- GDMJXXLVTYSBBE-UHFFFAOYSA-N 1,2-benzoxaborole Chemical class C1=CC=C2OB=CC2=C1 GDMJXXLVTYSBBE-UHFFFAOYSA-N 0.000 description 89
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 86
- 239000000203 mixture Substances 0.000 description 85
- 239000000243 solution Substances 0.000 description 74
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 55
- 235000019439 ethyl acetate Nutrition 0.000 description 49
- 238000000034 method Methods 0.000 description 49
- 241001465754 Metazoa Species 0.000 description 45
- 230000000052 comparative effect Effects 0.000 description 37
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 33
- 210000004027 cell Anatomy 0.000 description 28
- 239000003208 petroleum Substances 0.000 description 28
- 239000011541 reaction mixture Substances 0.000 description 28
- 238000005160 1H NMR spectroscopy Methods 0.000 description 26
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 26
- 239000007787 solid Substances 0.000 description 26
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 25
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 239000002904 solvent Substances 0.000 description 24
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 23
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 23
- 239000002552 dosage form Substances 0.000 description 23
- 230000002829 reductive effect Effects 0.000 description 22
- 239000000047 product Substances 0.000 description 21
- 238000006243 chemical reaction Methods 0.000 description 20
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 19
- 230000010076 replication Effects 0.000 description 19
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 239000012267 brine Substances 0.000 description 18
- 229940079593 drug Drugs 0.000 description 18
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 18
- 238000010898 silica gel chromatography Methods 0.000 description 18
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 18
- KXCAEQNNTZANTK-UHFFFAOYSA-N stannane Chemical compound [SnH4] KXCAEQNNTZANTK-UHFFFAOYSA-N 0.000 description 18
- 229910000080 stannane Inorganic materials 0.000 description 18
- 239000012044 organic layer Substances 0.000 description 17
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 16
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 16
- 125000003112 azulen-2-yl group Chemical group [H]C1=C2C([H])=C([H])C([H])=C([H])C([H])=C2C([H])=C1* 0.000 description 16
- 239000012453 solvate Substances 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 230000005764 inhibitory process Effects 0.000 description 15
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 15
- 241000124008 Mammalia Species 0.000 description 14
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 14
- 230000000694 effects Effects 0.000 description 13
- 239000000126 substance Substances 0.000 description 13
- 231100000419 toxicity Toxicity 0.000 description 13
- 230000001988 toxicity Effects 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 239000001257 hydrogen Substances 0.000 description 12
- 229910052739 hydrogen Inorganic materials 0.000 description 12
- 230000002401 inhibitory effect Effects 0.000 description 12
- 239000003921 oil Substances 0.000 description 12
- 235000019198 oils Nutrition 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 235000019441 ethanol Nutrition 0.000 description 10
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000003926 antimycobacterial agent Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 241000283690 Bos taurus Species 0.000 description 8
- PLXBWHJQWKZRKG-UHFFFAOYSA-N Resazurin Chemical compound C1=CC(=O)C=C2OC3=CC(O)=CC=C3[N+]([O-])=C21 PLXBWHJQWKZRKG-UHFFFAOYSA-N 0.000 description 8
- 229940034014 antimycobacterial agent Drugs 0.000 description 8
- 210000005260 human cell Anatomy 0.000 description 8
- 150000004677 hydrates Chemical class 0.000 description 8
- 150000002431 hydrogen Chemical class 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 239000000651 prodrug Substances 0.000 description 8
- 229940002612 prodrug Drugs 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 8
- VXKWYPOMXBVZSJ-UHFFFAOYSA-N tetramethyltin Chemical compound C[Sn](C)(C)C VXKWYPOMXBVZSJ-UHFFFAOYSA-N 0.000 description 8
- 239000000814 tuberculostatic agent Substances 0.000 description 8
- 0 *C(*)(COc1c(B(O)OC2CN)c2c(*)cc1)O Chemical compound *C(*)(COc1c(B(O)OC2CN)c2c(*)cc1)O 0.000 description 7
- 241000894006 Bacteria Species 0.000 description 7
- HFLQGDMZFAILFA-SECBINFHSA-N CC1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound CC1=CC=C2C3=C1[C@H](OB3OCCO2)CN HFLQGDMZFAILFA-SECBINFHSA-N 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 7
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 7
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 7
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- 238000001228 spectrum Methods 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 239000003125 aqueous solvent Substances 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 239000003937 drug carrier Substances 0.000 description 6
- 201000009671 multidrug-resistant tuberculosis Diseases 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- 235000015320 potassium carbonate Nutrition 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 6
- 150000003606 tin compounds Chemical class 0.000 description 6
- 241000588724 Escherichia coli Species 0.000 description 5
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 5
- 239000007832 Na2SO4 Substances 0.000 description 5
- 125000000217 alkyl group Chemical group 0.000 description 5
- 239000004599 antimicrobial Substances 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 239000001963 growth medium Substances 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 229910052938 sodium sulfate Inorganic materials 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 241000282472 Canis lupus familiaris Species 0.000 description 4
- 241000283707 Capra Species 0.000 description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 4
- 241000282326 Felis catus Species 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- 241001494479 Pecora Species 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 230000000845 anti-microbial effect Effects 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 231100000517 death Toxicity 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 229910052736 halogen Inorganic materials 0.000 description 4
- 150000002367 halogens Chemical group 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 238000002953 preparative HPLC Methods 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- YMUCKZJGVWMHLN-UHFFFAOYSA-N (5,11,11-trimethyl-2,9,12-trioxa-1-boratricyclo[6.4.1.04,13]trideca-4,6,8(13)-trien-3-yl)methanamine Chemical compound CC1=CC=C2C3=C1C(OB3OC(CO2)(C)C)CN YMUCKZJGVWMHLN-UHFFFAOYSA-N 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 3
- OHXPHMPERMIICA-UHFFFAOYSA-N 2-bromo-3-hydroxybenzaldehyde Chemical compound OC1=CC=CC(C=O)=C1Br OHXPHMPERMIICA-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- GWIRNARPJMQEML-PXCJXSSVSA-N CC1(C)C2CC[C@@]1(C)C(C2)NCc1ccccn1 Chemical compound CC1(C)C2CC[C@@]1(C)C(C2)NCc1ccccn1 GWIRNARPJMQEML-PXCJXSSVSA-N 0.000 description 3
- YMUCKZJGVWMHLN-SNVBAGLBSA-N CC1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN Chemical compound CC1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN YMUCKZJGVWMHLN-SNVBAGLBSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- 241000513886 Mycobacterium avium complex (MAC) Species 0.000 description 3
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 3
- 208000026681 Paratuberculosis Diseases 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 230000000844 anti-bacterial effect Effects 0.000 description 3
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 3
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000000132 electrospray ionisation Methods 0.000 description 3
- 230000001747 exhibiting effect Effects 0.000 description 3
- 230000002147 killing effect Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 238000004808 supercritical fluid chromatography Methods 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- KZBWSRPJMQIDDI-XTEPFMGCSA-N (1S)-2-(dibenzylamino)-1-[5-[[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methoxy]-2-methylphenyl]ethanol Chemical compound C(C1=CC=CC=C1)N(C[C@@H](O)C1=C(C=CC(=C1)OC[C@@H]1OC(OC1)(C)C)C)CC1=CC=CC=C1 KZBWSRPJMQIDDI-XTEPFMGCSA-N 0.000 description 2
- XUEMRISHFIDCSN-GXTWGEPZSA-N (1S)-2-amino-1-[5-[[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methoxy]-2-methylphenyl]ethanol Chemical compound NC[C@@H](O)C1=C(C=CC(=C1)OC[C@@H]1OC(OC1)(C)C)C XUEMRISHFIDCSN-GXTWGEPZSA-N 0.000 description 2
- XTYSXGHMTNTKFH-BDEHJDMKSA-N (2s)-1-[(2s,4r)-4-benzyl-2-hydroxy-5-[[(1s,2r)-2-hydroxy-2,3-dihydro-1h-inden-1-yl]amino]-5-oxopentyl]-n-tert-butyl-4-(pyridin-3-ylmethyl)piperazine-2-carboxamide;hydrate Chemical compound O.C([C@H](N(CC1)C[C@@H](O)C[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H]2C3=CC=CC=C3C[C@H]2O)C(=O)NC(C)(C)C)N1CC1=CC=CN=C1 XTYSXGHMTNTKFH-BDEHJDMKSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- YNGTZRHXRYLWFZ-UHFFFAOYSA-N 11-methyl-3-(nitromethyl)-2,9,12-trioxa-1-boratricyclo[6.4.1.04,13]trideca-4(13),5,7-triene Chemical compound O1C(C)COC2=CC=CC3=C2B1OC3C[N+]([O-])=O YNGTZRHXRYLWFZ-UHFFFAOYSA-N 0.000 description 2
- CYZYBHBEBHVRMJ-UHFFFAOYSA-N 2-methylprop-2-enoxymethylbenzene Chemical compound CC(=C)COCC1=CC=CC=C1 CYZYBHBEBHVRMJ-UHFFFAOYSA-N 0.000 description 2
- DVAATHXFDMCKNM-UHFFFAOYSA-N 3-(2-hydroxy-2-methyl-3-phenylmethoxypropoxy)-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde Chemical compound CC(O)(COCc1ccccc1)COc1cccc(C=O)c1B1OC(C)(C)C(C)(C)O1 DVAATHXFDMCKNM-UHFFFAOYSA-N 0.000 description 2
- LCFCUBURSMCLPP-UHFFFAOYSA-N 3-(2-hydroxy-2-methylpropoxy)-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde Chemical compound CC(C)(O)COc1cccc(C=O)c1B1OC(C)(C)C(C)(C)O1 LCFCUBURSMCLPP-UHFFFAOYSA-N 0.000 description 2
- FPTPNKQUKMCSBK-UHFFFAOYSA-N 3-(2-hydroxypropoxy)-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde Chemical compound CC(O)COC1=CC=CC(C=O)=C1B1OC(C)(C)C(C)(C)O1 FPTPNKQUKMCSBK-UHFFFAOYSA-N 0.000 description 2
- DIDUSPQIIVEBOF-UHFFFAOYSA-N 3-(2-phenylmethoxyethoxy)benzaldehyde Chemical compound O=CC1=CC=CC(OCCOCC=2C=CC=CC=2)=C1 DIDUSPQIIVEBOF-UHFFFAOYSA-N 0.000 description 2
- IAVREABSGIHHMO-UHFFFAOYSA-N 3-hydroxybenzaldehyde Chemical compound OC1=CC=CC(C=O)=C1 IAVREABSGIHHMO-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 241000588626 Acinetobacter baumannii Species 0.000 description 2
- 108700028939 Amino Acyl-tRNA Synthetases Proteins 0.000 description 2
- 102000052866 Amino Acyl-tRNA Synthetases Human genes 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- BDCZTYVHKIXWBF-UHFFFAOYSA-N CC(C)(O)COc1cccc(C=O)c1Br Chemical compound CC(C)(O)COc1cccc(C=O)c1Br BDCZTYVHKIXWBF-UHFFFAOYSA-N 0.000 description 2
- SRRWHQPKQDVXHY-UHFFFAOYSA-N CC(O)(COCc1ccccc1)COc1cccc(C=O)c1Br Chemical compound CC(O)(COCc1ccccc1)COc1cccc(C=O)c1Br SRRWHQPKQDVXHY-UHFFFAOYSA-N 0.000 description 2
- JVBRXNPJABQPCK-UHFFFAOYSA-N CC1(C)COc2cccc3C(C[N+]([O-])=O)OB(O1)c23 Chemical compound CC1(C)COc2cccc3C(C[N+]([O-])=O)OB(O1)c23 JVBRXNPJABQPCK-UHFFFAOYSA-N 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 206010059866 Drug resistance Diseases 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 208000031886 HIV Infections Diseases 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 241000187493 Mycobacterium malmoense Species 0.000 description 2
- GVDXHAREDNSLJZ-SSEXGKCCSA-N N,N-dibenzyl-1-[(3S)-1-hydroxy-7-(2-phenylmethoxyethoxy)-3H-2,1-benzoxaborol-3-yl]methanamine Chemical compound OB1O[C@H](CN(Cc2ccccc2)Cc2ccccc2)c2cccc(OCCOCc3ccccc3)c12 GVDXHAREDNSLJZ-SSEXGKCCSA-N 0.000 description 2
- YIWCQIDDQMFDFM-QGZVFWFLSA-N O[C@H](C[N+]([O-])=O)c1cccc(OCCOCc2ccccc2)c1 Chemical compound O[C@H](C[N+]([O-])=O)c1cccc(OCCOCc2ccccc2)c1 YIWCQIDDQMFDFM-QGZVFWFLSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 229960003496 delamanid Drugs 0.000 description 2
- 239000011903 deuterated solvents Substances 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 229940072185 drug for treatment of tuberculosis Drugs 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 208000036984 extensively drug-resistant tuberculosis Diseases 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 239000002054 inoculum Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000013160 medical therapy Methods 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 238000001243 protein synthesis Methods 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 description 2
- 238000002328 two-dimensional heteronuclear correlation spectroscopy Methods 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- XROSIOTWQBAZRY-UHFFFAOYSA-N (11-methyl-2,9,12-trioxa-1-boratricyclo[6.4.1.04,13]trideca-4(13),5,7-trien-3-yl)methanamine Chemical compound O1C(C)COC2=CC=CC3=C2B1OC3CN XROSIOTWQBAZRY-UHFFFAOYSA-N 0.000 description 1
- KTGRHKOEFSJQNS-BDQAORGHSA-N (1s)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-3h-2-benzofuran-5-carbonitrile;oxalic acid Chemical compound OC(=O)C(O)=O.C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 KTGRHKOEFSJQNS-BDQAORGHSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- DSSYKIVIOFKYAU-XCBNKYQSSA-N (R)-camphor Chemical compound C1C[C@@]2(C)C(=O)C[C@@H]1C2(C)C DSSYKIVIOFKYAU-XCBNKYQSSA-N 0.000 description 1
- 229930007886 (R)-camphor Natural products 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- JNOZGFXJZQXOSU-UHFFFAOYSA-N 1-chloro-2-methylpropan-2-ol Chemical compound CC(C)(O)CCl JNOZGFXJZQXOSU-UHFFFAOYSA-N 0.000 description 1
- YYTSGNJTASLUOY-UHFFFAOYSA-N 1-chloropropan-2-ol Chemical compound CC(O)CCl YYTSGNJTASLUOY-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 1
- WOXFMYVTSLAQMO-UHFFFAOYSA-N 2-Pyridinemethanamine Chemical compound NCC1=CC=CC=N1 WOXFMYVTSLAQMO-UHFFFAOYSA-N 0.000 description 1
- XNCNITOMDXJPNR-UHFFFAOYSA-N 2-bromo-3-(2-hydroxypropoxy)benzaldehyde Chemical compound CC(O)COC1=CC=CC(C=O)=C1Br XNCNITOMDXJPNR-UHFFFAOYSA-N 0.000 description 1
- FWOHDAGPWDEWIB-UHFFFAOYSA-N 2-bromoethoxymethylbenzene Chemical compound BrCCOCC1=CC=CC=C1 FWOHDAGPWDEWIB-UHFFFAOYSA-N 0.000 description 1
- GXKUCQRJGZBZIH-UHFFFAOYSA-N 2-methyl-2-[[2-methyl-1-(2-methylpropylamino)-1-oxopropan-2-yl]diazenyl]-n-(2-methylpropyl)propanamide Chemical compound CC(C)CNC(=O)C(C)(C)N=NC(C)(C)C(=O)NCC(C)C GXKUCQRJGZBZIH-UHFFFAOYSA-N 0.000 description 1
- BYDRTKVGBRTTIT-UHFFFAOYSA-N 2-methylprop-2-en-1-ol Chemical compound CC(=C)CO BYDRTKVGBRTTIT-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- HSBKFSPNDWWPSL-VDTYLAMSSA-N 4-amino-5-fluoro-1-[(2s,5r)-5-(hydroxymethyl)-2,5-dihydrofuran-2-yl]pyrimidin-2-one Chemical compound C1=C(F)C(N)=NC(=O)N1[C@@H]1C=C[C@H](CO)O1 HSBKFSPNDWWPSL-VDTYLAMSSA-N 0.000 description 1
- UHPYRIJDBCLYFO-ZDUSSCGKSA-N 5-[[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methoxy]-2-methylbenzaldehyde Chemical compound CC1(OC[C@@H](O1)COC=1C=CC(=C(C=O)C=1)C)C UHPYRIJDBCLYFO-ZDUSSCGKSA-N 0.000 description 1
- QONBQACIGSEEDX-UHFFFAOYSA-N 5-hydroxy-2-methylbenzaldehyde Chemical compound CC1=CC=C(O)C=C1C=O QONBQACIGSEEDX-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 241000568569 Acinetobacter baumannii ATCC 17978 Species 0.000 description 1
- 241001156739 Actinobacteria <phylum> Species 0.000 description 1
- 229910016455 AlBN Inorganic materials 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-M Aminoacetate Chemical compound NCC([O-])=O DHMQDGOQFOQNFH-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 208000031295 Animal disease Diseases 0.000 description 1
- 101500021166 Aplysia californica Myomodulin-B Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 241001260012 Bursa Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- MPEIKANGCMVBMO-LLVKDONJSA-N C(=C)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN Chemical compound C(=C)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN MPEIKANGCMVBMO-LLVKDONJSA-N 0.000 description 1
- YXPOMCILAKDBQY-SNVBAGLBSA-N C(=C)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound C(=C)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN YXPOMCILAKDBQY-SNVBAGLBSA-N 0.000 description 1
- CSECLKDENBCRHR-GFCCVEGCSA-N C(C)(C)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN Chemical compound C(C)(C)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN CSECLKDENBCRHR-GFCCVEGCSA-N 0.000 description 1
- FITHYAZMQYHLHU-LLVKDONJSA-N C(C)(C)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound C(C)(C)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN FITHYAZMQYHLHU-LLVKDONJSA-N 0.000 description 1
- PRJUXWRSLBUYLS-LLVKDONJSA-N C(C)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN Chemical compound C(C)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN PRJUXWRSLBUYLS-LLVKDONJSA-N 0.000 description 1
- HYMUIOAJICTKDD-SNVBAGLBSA-N C(C)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound C(C)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN HYMUIOAJICTKDD-SNVBAGLBSA-N 0.000 description 1
- WLVIYPGRCAVIPN-GFCCVEGCSA-N C(CC)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN Chemical compound C(CC)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN WLVIYPGRCAVIPN-GFCCVEGCSA-N 0.000 description 1
- OHKHZBQWYGDJQP-LLVKDONJSA-N C(CC)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound C(CC)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN OHKHZBQWYGDJQP-LLVKDONJSA-N 0.000 description 1
- DTOXCHIYVJFIFR-VEGHTAFHSA-N C([C@]\1(C)C2(C)C)CC2CC/1=N/Cc1ccccn1 Chemical compound C([C@]\1(C)C2(C)C)CC2CC/1=N/Cc1ccccn1 DTOXCHIYVJFIFR-VEGHTAFHSA-N 0.000 description 1
- IYCIHHRACTZWBC-GFCCVEGCSA-N C1(CC1)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN Chemical compound C1(CC1)C1=CC=C2C3=C1[C@H](OB3OC(CO2)(C)C)CN IYCIHHRACTZWBC-GFCCVEGCSA-N 0.000 description 1
- SOSJXAPYNRYZBA-LLVKDONJSA-N C1(CC1)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound C1(CC1)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN SOSJXAPYNRYZBA-LLVKDONJSA-N 0.000 description 1
- HSXLBMYUKCLMFL-SNVBAGLBSA-N CC(C)(COc1ccc(C)c2c1B(O)O[C@@H]2CN)O Chemical compound CC(C)(COc1ccc(C)c2c1B(O)O[C@@H]2CN)O HSXLBMYUKCLMFL-SNVBAGLBSA-N 0.000 description 1
- MUEXUKDSSFAYFF-LHIURRSHSA-N CC(COc1ccc(C)c2c1B(O)O[C@@H]2CN)O Chemical compound CC(COc1ccc(C)c2c1B(O)O[C@@H]2CN)O MUEXUKDSSFAYFF-LHIURRSHSA-N 0.000 description 1
- IVFPJYUAFMQZJV-UHFFFAOYSA-N CC(O)=O.CC1(C)COc2cccc3C(CN)OB(O1)c23 Chemical compound CC(O)=O.CC1(C)COc2cccc3C(CN)OB(O1)c23 IVFPJYUAFMQZJV-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- PGZHVOIPMNAJLP-SECBINFHSA-N Cc(c1c2B(O)O[C@@H]1CN)ccc2OCCO Chemical compound Cc(c1c2B(O)O[C@@H]1CN)ccc2OCCO PGZHVOIPMNAJLP-SECBINFHSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- ZDRVQNVFICEHBH-PFEQFJNWSA-N Cl.C1(=CC=CC=C1)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound Cl.C1(=CC=CC=C1)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN ZDRVQNVFICEHBH-PFEQFJNWSA-N 0.000 description 1
- JWOFJDOPVDVRQX-UHFFFAOYSA-N Cl.CC1=CC=C2C3=C1C(OB3OC(CO2)(C)C)CN Chemical compound Cl.CC1=CC=C2C3=C1C(OB3OC(CO2)(C)C)CN JWOFJDOPVDVRQX-UHFFFAOYSA-N 0.000 description 1
- ZODZSWFBXXSVGK-UHFFFAOYSA-N Cl.CC1=CC=C2C3=C1C(OB3OC(CO2)C)CN Chemical compound Cl.CC1=CC=C2C3=C1C(OB3OC(CO2)C)CN ZODZSWFBXXSVGK-UHFFFAOYSA-N 0.000 description 1
- KGJHIQQEYCGBRR-SBSPUUFOSA-N Cl.CC1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound Cl.CC1=CC=C2C3=C1[C@H](OB3OCCO2)CN KGJHIQQEYCGBRR-SBSPUUFOSA-N 0.000 description 1
- CMIJGSMJHSVGGI-SBSPUUFOSA-N Cl.NC[C@H]1OB2OCCOc3cccc1c23 Chemical compound Cl.NC[C@H]1OB2OCCOc3cccc1c23 CMIJGSMJHSVGGI-SBSPUUFOSA-N 0.000 description 1
- MUNOLJCWHNOMKG-JSSVAETHSA-N Cl.O[C@H](CN(Cc1ccccc1)Cc1ccccc1)c1cccc(OCCOCc2ccccc2)c1 Chemical compound Cl.O[C@H](CN(Cc1ccccc1)Cc1ccccc1)c1cccc(OCCOCc2ccccc2)c1 MUNOLJCWHNOMKG-JSSVAETHSA-N 0.000 description 1
- WXZVNQDEPHWMMA-RFVHGSKJSA-N Cl.S1C(=CC=C1)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN Chemical compound Cl.S1C(=CC=C1)C1=CC=C2C3=C1[C@H](OB3OCCO2)CN WXZVNQDEPHWMMA-RFVHGSKJSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- ODKSFYDXXFIFQN-SCSAIBSYSA-N D-arginine Chemical compound OC(=O)[C@H](N)CCCNC(N)=N ODKSFYDXXFIFQN-SCSAIBSYSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 1
- 241001198387 Escherichia coli BL21(DE3) Species 0.000 description 1
- 241001646716 Escherichia coli K-12 Species 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 101000624524 Homo sapiens Leucine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 101000624540 Homo sapiens Leucine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 241000588747 Klebsiella pneumoniae Species 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 239000004395 L-leucine Substances 0.000 description 1
- 235000019454 L-leucine Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 108010071170 Leucine-tRNA ligase Proteins 0.000 description 1
- 102100023339 Leucine-tRNA ligase, cytoplasmic Human genes 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241001332087 Mycobacterium abscessus subsp. bolletii Species 0.000 description 1
- 241001509442 Mycobacterium agri Species 0.000 description 1
- 241000957223 Mycobacterium alvei Species 0.000 description 1
- 241001368179 Mycobacterium arosiense Species 0.000 description 1
- 241000687894 Mycobacterium arupense Species 0.000 description 1
- 241001332085 Mycobacterium aubagnense Species 0.000 description 1
- 241000187473 Mycobacterium aurum Species 0.000 description 1
- 241001532520 Mycobacterium austroafricanum Species 0.000 description 1
- 241001316387 Mycobacterium boenickei Species 0.000 description 1
- 241000567118 Mycobacterium bohemicum Species 0.000 description 1
- 241001662551 Mycobacterium botniense Species 0.000 description 1
- 241001316365 Mycobacterium brisbanense Species 0.000 description 1
- 241001674312 Mycobacterium brumae Species 0.000 description 1
- 241000318680 Mycobacterium canariasense Species 0.000 description 1
- 241001134667 Mycobacterium celatum Species 0.000 description 1
- 241000254210 Mycobacterium chimaera Species 0.000 description 1
- 241000187472 Mycobacterium chitae Species 0.000 description 1
- 241000187913 Mycobacterium chubuense Species 0.000 description 1
- 241000419175 Mycobacterium colombiense Species 0.000 description 1
- 241000761550 Mycobacterium conceptionense Species 0.000 description 1
- 241001134628 Mycobacterium confluentis Species 0.000 description 1
- 241000178318 Mycobacterium conspicuum Species 0.000 description 1
- 241001420379 Mycobacterium cosmeticum Species 0.000 description 1
- 241000187912 Mycobacterium diernhoferi Species 0.000 description 1
- 241000587727 Mycobacterium doricum Species 0.000 description 1
- 241001532524 Mycobacterium duvalii Species 0.000 description 1
- 241001609973 Mycobacterium elephantis Species 0.000 description 1
- 241000187471 Mycobacterium fallax Species 0.000 description 1
- 241001136226 Mycobacterium florentinum Species 0.000 description 1
- 241000235788 Mycobacterium frederiksbergense Species 0.000 description 1
- 241000187485 Mycobacterium gastri Species 0.000 description 1
- 241001509451 Mycobacterium genavense Species 0.000 description 1
- 241000187910 Mycobacterium gilvum Species 0.000 description 1
- 241000202700 Mycobacterium hassiacum Species 0.000 description 1
- 241000142650 Mycobacterium heckeshornense Species 0.000 description 1
- 241000520088 Mycobacterium heidelbergense Species 0.000 description 1
- 241001644172 Mycobacterium holsaticum Species 0.000 description 1
- 241001316369 Mycobacterium houstonense Species 0.000 description 1
- 241001646019 Mycobacterium immunogenum Species 0.000 description 1
- 241001494992 Mycobacterium indicus pranii Species 0.000 description 1
- 241001467535 Mycobacterium interjectum Species 0.000 description 1
- 241001136174 Mycobacterium intermedium Species 0.000 description 1
- 241000516643 Mycobacterium kubicae Species 0.000 description 1
- 241001078572 Mycobacterium kumamotonense Species 0.000 description 1
- 241000439014 Mycobacterium lacus Species 0.000 description 1
- 241001248583 Mycobacterium lentiflavum Species 0.000 description 1
- 241000908167 Mycobacterium lepraemurium Species 0.000 description 1
- 241000178382 Mycobacterium lepromatosis Species 0.000 description 1
- 241001672738 Mycobacterium montefiorense Species 0.000 description 1
- 241001532511 Mycobacterium moriokaense Species 0.000 description 1
- 241000557009 Mycobacterium mucogenicum Species 0.000 description 1
- 241001062465 Mycobacterium murale Species 0.000 description 1
- 241001013798 Mycobacterium nebraskense Species 0.000 description 1
- 241000187469 Mycobacterium neoaurum Species 0.000 description 1
- 241001316374 Mycobacterium neworleansense Species 0.000 description 1
- 241000611872 Mycobacterium novocastrense Species 0.000 description 1
- 241001659709 Mycobacterium palustre Species 0.000 description 1
- 241000961132 Mycobacterium parascrofulaceum Species 0.000 description 1
- 241001101480 Mycobacterium parmense Species 0.000 description 1
- 241000168058 Mycobacterium peregrinum Species 0.000 description 1
- 241000187481 Mycobacterium phlei Species 0.000 description 1
- 241001332086 Mycobacterium phocaicum Species 0.000 description 1
- 241001532509 Mycobacterium porcinum Species 0.000 description 1
- 241001532510 Mycobacterium poriferae Species 0.000 description 1
- 241000089536 Mycobacterium pseudoshottsii Species 0.000 description 1
- 241001509447 Mycobacterium pulveris Species 0.000 description 1
- 241001509458 Mycobacterium rhodesiae Species 0.000 description 1
- 241000409180 Mycobacterium septicum Species 0.000 description 1
- 241001002112 Mycobacterium sherrisii Species 0.000 description 1
- 241000919916 Mycobacterium shottsii Species 0.000 description 1
- 241000187480 Mycobacterium smegmatis Species 0.000 description 1
- 241000187497 Mycobacterium sphagni Species 0.000 description 1
- 241000218972 Mycobacterium triplex Species 0.000 description 1
- 241001646725 Mycobacterium tuberculosis H37Rv Species 0.000 description 1
- 241001293520 Mycobacterium tusciae Species 0.000 description 1
- 241000142559 Mycobacterium vanbaalenii Species 0.000 description 1
- RVQZUUXNBCLRMM-LLVKDONJSA-N NC[C@H]1OB2OCCOC3=C2C1=C(C=C3)C1=CC=CS1 Chemical compound NC[C@H]1OB2OCCOC3=C2C1=C(C=C3)C1=CC=CS1 RVQZUUXNBCLRMM-LLVKDONJSA-N 0.000 description 1
- LHEFBTHSCUKILR-SECBINFHSA-N NC[C@H]1OB2OCCOC3=C2C1=CC=C3 Chemical compound NC[C@H]1OB2OCCOC3=C2C1=CC=C3 LHEFBTHSCUKILR-SECBINFHSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 229940122313 Nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- 229910002666 PdCl2 Inorganic materials 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 241001240958 Pseudomonas aeruginosa PAO1 Species 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 241000282849 Ruminantia Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 101710137500 T7 RNA polymerase Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 208000025087 Yersinia pseudotuberculosis infectious disease Diseases 0.000 description 1
- KGTSLTYUUFWZNW-PPJQWWMSSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,27,29-pentahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-26-[(E)-(4-methylpiperazin-1-yl)iminomethyl]-6,23-dioxo-8,30-dioxa-24-azatetracyclo[23.3.1.14,7.05,28]triaconta-1(29),2,4,9,19,21,25,27-octaen-13-yl] acetate pyridine-4-carbohydrazide Chemical compound NNC(=O)c1ccncc1.CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c(O)c(\C=N\N4CCN(C)CC4)c(NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C KGTSLTYUUFWZNW-PPJQWWMSSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 159000000021 acetate salts Chemical class 0.000 description 1
- HZNGUAOZBLCIIL-UHFFFAOYSA-N acetic acid (11-methyl-2,9,12-trioxa-1-boratricyclo[6.4.1.04,13]trideca-4(13),5,7-trien-3-yl)methanamine Chemical compound CC(O)=O.CC1COc2cccc3C(CN)OB(O1)c23 HZNGUAOZBLCIIL-UHFFFAOYSA-N 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 230000006229 amino acid addition Effects 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 230000001355 anti-mycobacterial effect Effects 0.000 description 1
- 230000002365 anti-tubercular Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229960005475 antiinfective agent Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 208000033354 avian tuberculosis Diseases 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 229960000508 bedaquiline Drugs 0.000 description 1
- QUIJNHUBAXPXFS-XLJNKUFUSA-N bedaquiline Chemical compound C1([C@H](C2=CC3=CC(Br)=CC=C3N=C2OC)[C@@](O)(CCN(C)C)C=2C3=CC=CC=C3C=CC=2)=CC=CC=C1 QUIJNHUBAXPXFS-XLJNKUFUSA-N 0.000 description 1
- ZLVSPMRFRHMMOY-WWCCMVHESA-N bedaquiline fumarate Chemical group OC(=O)\C=C\C(O)=O.C1([C@H](C2=CC3=CC(Br)=CC=C3N=C2OC)[C@@](O)(CCN(C)C)C=2C3=CC=CC=C3C=CC=2)=CC=CC=C1 ZLVSPMRFRHMMOY-WWCCMVHESA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000002306 biochemical method Methods 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 235000014510 cooky Nutrition 0.000 description 1
- 239000013256 coordination polymer Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000002498 deadly effect Effects 0.000 description 1
- 238000001475 decoupling using mind-boggling optimisation pulse sequence Methods 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 208000015355 drug-resistant tuberculosis Diseases 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 229950006528 elvucitabine Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- OLAMWIPURJGSKE-UHFFFAOYSA-N et2o diethylether Chemical compound CCOCC.CCOCC OLAMWIPURJGSKE-UHFFFAOYSA-N 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical class [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- LRDFRRGEGBBSRN-UHFFFAOYSA-N isobutyronitrile Chemical compound CC(C)C#N LRDFRRGEGBBSRN-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229950004697 lasinavir Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 229960003136 leucine Drugs 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 238000005567 liquid scintillation counting Methods 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 1
- 229910000103 lithium hydride Inorganic materials 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-M lysinate Chemical compound NCCCCC(N)C([O-])=O KDXKERNSBIXSRK-UHFFFAOYSA-M 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- 229940055036 mycobacterium phlei Drugs 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 229940042402 non-nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 239000002726 nonnucleoside reverse transcriptase inhibitor Substances 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 244000000019 opportunistic animal pathogen Species 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 230000033116 oxidation-reduction process Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229940124641 pain reliever Drugs 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical class OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- GUUBJKMBDULZTE-UHFFFAOYSA-M potassium;2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid;hydroxide Chemical compound [OH-].[K+].OCCN1CCN(CCS(O)(=O)=O)CC1 GUUBJKMBDULZTE-UHFFFAOYSA-M 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000006514 pyridin-2-ylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 1
- 239000003306 quinoline derived antiinfective agent Substances 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229940048026 sirturo Drugs 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229940056729 sodium sulfate anhydrous Drugs 0.000 description 1
- 239000002689 soil Substances 0.000 description 1
- 238000000279 solid-state nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 238000011272 standard treatment Methods 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- BEUUJDAEPJZWHM-COROXYKFSA-N tert-butyl n-[(2s,3s,5r)-3-hydroxy-6-[[(2s)-1-(2-methoxyethylamino)-3-methyl-1-oxobutan-2-yl]amino]-6-oxo-1-phenyl-5-[(2,3,4-trimethoxyphenyl)methyl]hexan-2-yl]carbamate Chemical compound C([C@@H]([C@@H](O)C[C@H](C(=O)N[C@H](C(=O)NCCOC)C(C)C)CC=1C(=C(OC)C(OC)=CC=1)OC)NC(=O)OC(C)(C)C)C1=CC=CC=C1 BEUUJDAEPJZWHM-COROXYKFSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000004481 total suppression of sideband Methods 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- SYUVAXDZVWPKSI-UHFFFAOYSA-N tributyl(phenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CC=CC=C1 SYUVAXDZVWPKSI-UHFFFAOYSA-N 0.000 description 1
- UKTDFYOZPFNQOQ-UHFFFAOYSA-N tributyl(thiophen-2-yl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CC=CS1 UKTDFYOZPFNQOQ-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- MHNHYTDAOYJUEZ-UHFFFAOYSA-N triphenylphosphane Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 MHNHYTDAOYJUEZ-UHFFFAOYSA-N 0.000 description 1
- VSRBKQFNFZQRBM-UHFFFAOYSA-N tuaminoheptane Chemical compound CCCCCC(C)N VSRBKQFNFZQRBM-UHFFFAOYSA-N 0.000 description 1
- 238000009827 uniform distribution Methods 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 150000003722 vitamin derivatives Chemical class 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/14—Drugs for genital or sexual disorders; Contraceptives for lactation disorders, e.g. galactorrhoea
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/08—Antibacterial agents for leprosy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/04—Esters of boric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Pulmonology (AREA)
- Pregnancy & Childbirth (AREA)
- Rheumatology (AREA)
- Reproductive Health (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Endocrinology (AREA)
- Molecular Biology (AREA)
- Gynecology & Obstetrics (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
Un compuesto que tiene una estructura como la mostrada en la Fórmula III: **(Ver fórmula)** en la que R3 es -CH3; y R1 y R2 son cada uno independientemente H o -CH3, o una de sus sales farmacéuticamente aceptables.
Description
DESCRIPCIÓN
Compuestos de benzoxaborol sustituidos en posición 4 y sus usos
Campo de la invención
Esta invención se refiere a compuestos, composiciones que los contienen, su uso en terapia, incluyendo su uso como antimicobacterianos, por ejemplo, en el tratamiento de la tuberculosis, y métodos para la preparación de estos compuestos.
Antecedentes de la invención
Mycobacterium es un género de la clase de bacterias Actinobacteria con su propia familia distinta conocida como Mycobacteriacae. Mycobacterium contiene diversos patógenos obligados y oportunistas de animales, que también se pueden transmitir a seres humanos y provocar una enfermedad en seres humanos, exhibiendo así un considerable potencial zoonótico. Durante las últimas décadas, miembros del complejo de Mycobacterium aviumintracellulare (MAIC) surgieron como patógenos de enfermedades humanas, incluyendo linfadenitis en niños, pseudotuberculosis pulmonar e infecciones diseminadas (que se presentan predominantemente en personas inmunocomprometidas, particularmente pacientes con sida). De forma similar, importantes enfermedades de animales resultan de infecciones en un animal por miembros de este grupo, p. ej., tuberculosis aviar y paratuberculosis en rumiantes. El MAIC incluye M. intracellulare y 4 subespecies de M. avium, a saber, M. avium subesp. avium, M. avium subesp. hominissuis, M. avium subesp. silvaticum y M. avium subesp. paratuberculosis. Mientras que los miembros del complejo de M. tuberculosis se transmiten mediante contacto directo del hospedador, las especies de MAIC se adquieren predominantemente desde fuentes medioambientales, incluyendo el suelo, el agua, el polvo y el alimento.
Mycobacterium tuberculosis (MTB) es un pequeño bacilo alto en GC inmóvil aeróbico con una "membrana externa" que es inusualmente gruesa, "cerosa", hidrófoba, rica en ácidos micólicos y extremadamente impermeable, haciendo que las infecciones por micobacterias sean difíciles de tratar. Se cree que un tercio de la población mundial está infectado (incluyendo MTB latente), pero este número se incrementa hasta 80% de la población en muchos países de Asia y África. Si no se trata, el índice de mortalidad por infecciones por MTB activas es mayor de 50%. Además, la combinación de VIH y MTB es mortal y números crecientes de cepas de MTB se están volviendo resistentes a fármacos de referencia; se presentan cada año aproximadamente 300.000 nuevos casos de M. tuberculosis resistente a múltiples fármacos (MDR). M. tuberculosis resistentes a múltiples fármacos (MDR) son resistentes a isoniacida y rifampicina, y M. tuberculosis altamente resistentes a fármacos (XDR) también son resistentes a al menos una quinolona y un aminoglicósido. Como se puede observar en la Figura 1, M. tuberculosis XDR se ha presentado en gran parte del mundo.
Añádanse a estos problemas la facilidad de transmisión, según se muestra en la Figura 2, la globalización de los viajes y la reubicación y la emigración en marcha de muchos segmentos de la población mundial y es evidente que la MTB se está convirtiendo en una crisis mundial.
Fármacos sintéticos para tratar la tuberculosis (TB) han estado disponibles durante más de medio siglo, pero las incidencias de la enfermedad continúan aumentando en todo el mundo. Más de 2 mil millones de personas están infectadas actualmente con M. tuberculosis, siendo la mayoría casos latentes, y se estima que cada año se presentan más de 9 millones de nuevos casos, en todo el mundo, dando como resultado de 1,7 a casi 2 millones de muertes al año. Solo en 2004, se registraron aproximadamente 24.500 nuevas infecciones y cerca de 5.500 muertes al día. Véase Zignol, M et al., M. Surveillance of anti-tuberculosis drug resistance in the world: an updated analysis, 2007-2010. Bull. World Health Organ 2012, 90 (2), 111-119D) La coinfección con VIH está conduciendo al incremento en la incidencia (Williams, B. G.; Dye, C. Science, 2003, 301, 1535) y el caso de muerte en 31% de los pacientes con sida en África se puede atribuir a TB. Véase Corbett, E. L et al.,. Arch. Intl. Med., 2003, 163, 1009, Septkowitz, A et al., Clin. Microbiol. Rev. 1995, 8, 180).
Las limitaciones de la terapia y la prevención de la tuberculosis son muy conocidas. La vacuna disponible actual, BCG, se introdujo en 1921 y falla en la protección de la mayoría de las personas después de la niñez. Según un informe de 2006 - "International Standards for Tuberculosis Care", un documento desarrollado por the Tuberculosis Coalition for Technical Assistance (TBCTA) cuyos socios incluyen Centers for Disease Control, American Thoracic Society, Tuberculosis Foundation, KNCV, la Organización Mundial de la Salud y the International Union Against Tuberculosis and Lung Disease - los pacientes que se infectan con enfermedad activa actualmente soportan dos meses de terapia combinada con medicamentos introducidos hace entre 50 y 60 años - isoniacida (1952), rifampina (1963), piracinamida (1954) y etambutol (1961) - seguido por otros 4 meses de isoniacida y rifampina (también conocida como rifampicina). Alternativamente, la fase de continuación podría incluir isoniacida y etambutol durante seis meses cuando no se pueda determinar la observancia terapéutica, pero, según este informe, una fase de continuación más prolongada está asociada con un grado superior de fallo y recaída, especialmente en pacientes con infección por VIH. Por otra parte, según se detalla en este informe, las dosis de fármacos antituberculosos se deben ajustar a la recomendación internacional y se recomiendan intensamente combinaciones de dosis fijas de dos (isoniacida y rifampicina), tres (isoniacida, rifampicina y piracinamida) y cuatro (isoniacida, rifampicina, piracinamida
y etambutol), especialmente cuando no es posible controlar al paciente para asegurar que el tratamiento sea ingerido.
Se requiere una dosificación diaria en estas fases de tratamiento y el escaso cumplimiento terapéutico activa el surgimiento y la extensión de cepas resistentes a múltiples fármacos, que son difíciles de tratar. Se requieren urgentemente tratamientos más cortos de agentes más activos que se puedan tomar menos frecuentemente y que presenten una alta barrera al surgimiento de resistencia, es decir, agentes que sean eficaces frente a cepas resistentes a múltiples fármacos de TB (MDR-TB). Un informe de marzo de 2013 (http://www.aidsmap.com/Onceweekly-continuation-phase-TB-treatment-equals-standard-of-care/page/2589498/) sugiere que una combinación de dos fármacos de rifapentina (un derivado de acción prolongada de rifampicina) con moxifloxacina (un antibiótico de fluoroquinolona que no se ha usado previamente en el tratamiento de la TB) puede permitir que el tratamiento de la tuberculosis (TB) se tome una vez a la semana durante la fase de continuación de cuatro meses y alcance el mismo estándar de atención médica que el tratamiento de continuación tradicional de tratamiento diario con isoniacida y rifampina. Esta fase de tratamiento permitiría que la supervisión del tratamiento se extendiera a través de la fase de continuación, incrementando la observancia terapéutica. Sin embargo, la moxifloxacina todavía no está aprobada para el tratamiento de la TB, y el protocolo de tratamiento de una vez por semana todavía no está avalado o aprobado como un tratamiento de referencia alternativo - se necesitarán grupos de expertos a nivel internacional y nacional para revisar la evidencia publicada para determinar si se debía recomendar la adopción de este protocolo de tratamiento de continuación alternativo. Además, la rifapentina es costosa y las interacciones entre la rifapentina y fármacos antirretrovirales en las clases de inhibidores de transcriptasa inversa no nucleosídicos (NNRTI) e inhibidores de proteasa pueden impedir su uso en pacientes con TB que también son positivos a VIH y que toman medicamentos antirretrovirales. Así, en la actualidad, todavía se tiene que evaluar a fondo el análisis de costes/beneficios de un tratamiento de continuación con rifapentina semanal frente a rifampicina diaria.
El fármaco para la tuberculosis Sirturo™ (bedaquilina) fue aprobado en los Estados Unidos a finales de diciembre de 2012, y otro, la delamanida, está intentando obtener la aprobación reguladora en la UE. Sin embargo, ambos están reservados para tuberculosis resistente a fármacos, que supone solo el 5% de nuevos casos. Un Editorial and News Focus in Nature Medicine de 2007 analiza muchos aspectos de la TB tales como patogénesis, epidemiología, descubrimiento de fármacos y desarrollo de vacunas hasta la fecha (Nature Medicine, 2007, Focus on Tuberculosis, Vol 13(3), páginas 263-312), señalando que, 125 años después del aniversario del descubrimiento de Mycobacterium tuberculosis, más de un tercio de las personas del mundo están infectadas con M. tuberculosis, y, de estas, más de 1 de cada 10 desarrollará la enfermedad conocida como tuberculosis, inicialmente conocida como tisis, a lo largo de su vida.
Cuando está asociado con el surgimiento de cepas resistentes a múltiples fármacos de Mycobacterium tuberculosis (MDR-TB), la escala del problema se amplía. El aumento mundial de bacterias y otros microorganismos resistentes a antibióticos y antimicrobianos en general plantea un reto importante. La extensión de cantidades masivas de agentes antimicrobianos en la ecosfera durante los últimos 60 años ha introducido una potente presión selectiva para el surgimiento y la extensión de patógenos resistentes a antimicrobianos. Por lo tanto, existe una necesidad de descubrir y desarrollar nuevas entidades químicas para tratar la TB (iniciativas recientes se revisan en: Grosset JH, Singer TG, Bishai WR. New Drugs for the Treatment of Tuberculosis: Hope and Reality. Int J Tuberc Lung Dis. agosto de 2012;16(8):1005-14).
La presente invención se refiere a ciertos benzoxaboroles sustituidos que muestran una selectividad inesperada para inhibir la replicación de Mycobacterium tuberculosis (M. tuberculosis) frente a la inhibición (toxicidad) de células humanas en comparación con otros benzoxaboroles sustituidos, y exhiben valores de CIM submicromolares frente a especies de micobacteria, particularmente Mycobacterium tuberculosis y complejo de Mycobacterium tuberculosis (MTC), Mycobacterium avium y complejo de Mycobacterium avium (MAC) y complejo de Mycobacterium avium intracellulare (MAIC). En general, el anillo de benzoxaborol del benzoxaborol sustituido tiene la siguiente estructura que se muestra posteriormente en la Fórmula I, y se puede caracterizar con el siguiente sistema de numeración de sustituyentes:

Fórmula I.
Se entiende que la nomenclatura de la International Union of Pure and Applied Chemistry (IUPAC) puede indicar un sistema de numeración diferente dependiendo de los sustituyentes alrededor del anillo de benzoxaborol. A lo largo de esta solicitud, a menos que se dé el nombre de la IUPAC para un compuesto específico, los benzoxaboroles sustituidos divulgados en la presente memoria se pueden nombrar y numerar usando el esquema de numeración representado en la Fórmula I, mostrada anteriormente.
Moléculas que contienen boro tales como benzoxaboroles que son útiles como antimicrobianos se han descrito previamente, véase, p. ej., "Benzoxaboroles - Old compounds with new applications" Adamczyk-Wozniak, A. et al., Journal of Organometallic Chemistry Volumen 694, Edición 22, 15 de octubre de 2009, páginas 3533-3541, y las Publicaciones de Patente de EE. UU. US20060234981, US20070155699, WO2012033858 y US2013165411. El documento US 2013/0165411 A1 divulga compuestos borados tricíclicos antimicrobianos y sus sales farmacéuticamente aceptables, complejos o tautómeros como agentes antibacterianos. El documento WO 2011/127143 A1 divulga procedimientos para elaborar compuestos de benzoxaborol y sus sales, que se muestran prometedores como agentes antibacterianos.
Ciertos benzoxaboroles sustituidos que están sustituidos en la posición 7 pueden formar un compuesto de benzoxaborol tricíclico (véanse los documentos US20090227541, US2013165411 y WO/KR2015/016558). Los solicitantes han encontrado sorprendentemente que ciertos benzoxaboroles sustituidos, sustituidos en la posición 7 (numerada usando el esquema de numeración representado en la Fórmula I, mostrada anteriormente), también pueden existir como una mezcla en equilibrio de una estructura de benzoxaborol tricíclica y una estructura de benzoxaborol bicíclica en disolventes acuosos. Cuando el benzoxaborol sustituido en 7 resultante está sustituido adicionalmente con un sustituyente alquilo en la posición 4 y un sustituyente aminometilo en la posición 3 (numerada usando el esquema de numeración de la Fórmula I, mostrada anteriormente), estos benzoxaboroles sustituidos son sorprendentemente selectivos hacia y eficaces frente a micobacterias incluyendo M. tuberculosis. La selectividad observada se determina al comparar valores de CIM para estos compuestos con relación a la inhibición (toxicidad) de estos compuestos para células humanas, en comparación con otros benzoxaboroles sustituidos.
El documento US20090227541 divulga una multitud de compuestos, incluyendo dos compuestos de benzoxaborol tricíclicos con diferente actividad antibacteriana frente a un grupo de bacterias gramnegativas (Véanse, p. ej., las Tablas 1 y 2), pero no analiza compuestos de benzoxaborol tricíclicos con sustitución alquílica en la posición 4 en el anillo de benzoxaborol (numerada usando el esquema de numeración representado en la Fórmula I, mostrada anteriormente). El documento WO2012033858 divulga benzoxaboroles sustituidos con actividad frente a Mycobacterium tuberculosis, incluyendo ciertos benzoxaboroles sustituidos (véanse p. ej., los Ejemplos 1.A a 1.V), pero, de nuevo, no se divulga un compuesto de benzoxaborol tricíclico con una sustitución alquílica en la posición 4 en el anillo de benzoxaborol (numerada usando el esquema de numeración representado en la Fórmula I, mostrada anteriormente). El documento US2013165411 divulga compuestos de benzoxaborol tricíclicos que muestran actividad frente a Acinetobacter baumannii, Pseudomonas aeruginosa, Escherichia coli y Klebsiella pneumoniae (véase la Tabla 1), pero apunta específicamente que los compuestos tricíclicos sustituidos con halógeno investigados (Ejemplos 17, 18 y 19) carecen de actividad frente a A. baumannii, con valores de CIM > 16 pg/pl de actividad antibacteriana (véase la Figura 1). Además, nada en el documento US2013165411 sugiere que ninguno de los compuestos de benzoxaborol tricíclicos divulgados sea capaz de existir como una mezcla en equilibrio de una estructura de benzoxaborol tricíclica y una estructura de benzoxaborol bicíclica en condiciones de disolvente acuoso.
Compendio de la invención
Los inventores han encontrado sorprendentemente que los benzoxaboroles sustituidos según se describe en la presente muestran una selectividad inesperada para inhibir la replicación de Mycobacterium tuberculosis (M. tuberculosis) frente a la inhibición (toxicidad) de células humanas en comparación con otros benzoxaboroles sustituidos. Estos benzoxaboroles sustituidos exhiben valores de CIM submicromolares frente a M. tuberculosis, lo que es comparable a o mejor que los valores de CIM para terapias actuales para inhibir M. tuberculosis. Además, en otras realizaciones, los benzoxaboroles sustituidos que se describen en la presente están previstos para el uso en combinación con compuestos antituberculosos actuales y está previsto que alcancen mayor eficacia en el tratamiento de animales, incluyendo seres humanos, infectados con M. tuberculosis.
La resistencia sigue siendo un problema en el tratamiento de la tuberculosis (TB) y una estrategia clínica es enfocarse en la combinación inicial con otros fármacos para la TB y acelerar la evaluación inicial de la eficacia del compuesto en los pacientes. Los compuestos cuya estructura comprende la Fórmula III o la Fórmula IIIa ofrecen una oportunidad única para dirigirse a problemas graves que surgen durante el tratamiento de la TB, tales como resistencia a múltiples fármacos, resistencia extensiva a fármacos, reactividad y/o interacción adversa entre agentes terapéuticos en una combinación de múltiples fármacos y duración del tratamiento, dirigiéndose de ese modo a las necesidades potenciales del paciente.
En ciertas realizaciones de la presente invención, se presentan combinaciones de agentes antituberculosos y ciertos benzoxaboroles sustituidos, para el uso en el tratamiento de infecciones por Mycobacterium tuberculosis en animales, incluyendo seres humanos. En realizaciones particulares, estos benzoxaboroles sustituidos se usan, en combinación con otros agentes antituberculosos conocidos, para tratar a un paciente animal con una infección por Mycobacterium tuberculosis, particularmente en un paciente animal que esté infectado adicionalmente con un retrovirus humano, en particular un virus de inmunodeficiencia humana (VIH).
En una realización ejemplar, la invención es un compuesto como el descrito en la presente memoria, o una de sus sales, incluyendo una de sus sales farmacéuticamente aceptables, o uno de sus hidratos.
Se divulga en la presente memoria un benzoxaborol sustituido como un compuesto o una de sus sales, incluyendo
una de sus sales farmacéuticamente aceptables, cuya estructura comprende la Fórmula III:

Fórmula III
en la que R3 se selecciona de -CH3, -CH2CH3, -CH2=CH2, -CH2CH2CH3, -CH(CH3)2, -CH2CH2=CH2 o ciclopropilo; R1 y R2 se seleccionan cada uno independientemente de H, - CH3, -CH2CH3, -CH2CH2CH3 y -CH(CH3)2.
En realizaciones particulares, el benzoxaborol sustituido puede existir en un equilibrio, según se indica posteriormente, entre una forma cerrada (Fórmula II) y una forma abierta (Fórmula III), en ciertos ambientes y/o disolventes.

Fórmula II Fórmula III Forma Cerrada Forma Abierta
Se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH3 o -CH2CH3; R1 y R2 se seleccionan cada uno independientemente de H, -CH3, -CH2CH3, -CH2CH2CH3 y -CH(CH3)2. También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH3 y R1 y R2 son como se describen en la presente memoria. También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH3 y R1 es H y R2 es como se describe en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH3 y R1 es -CH3 y R2 es como se describe en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH3 y R1 y R2 son como se describen en la presente memoria. También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH3 y R1 y R2 son cada uno independientemente H o -CH3. También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH3 y R1 es H y R2 es -CH3. También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH3 y R1 es -CH3 y R2 es como se describe en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH2CH3 y R1 y R2 son como se describen en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH(CH3)2 y R1 y R2 son como se describen en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH2CH3; R1 y R2 se seleccionan cada uno independientemente de H, -CH3 y -CH2CH3.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH2CH2CH3 y R1 y R2 son cada uno independientemente H o -CH3
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH(CH3)2 y R1 y R2 se seleccionan cada uno independientemente de H, -CH3 y -CH2CH3.
También se divulga un compuesto cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH(CH3)2 y R1 y R2 son cada uno independientemente H o -CH3.
También se divulga un compuesto que comprende una estructura de Fórmula IIIa

en la que R3 es -CH3, -CH2CH3, -CH2=CH2, -CH2CH2CH3, -CH(CH3)2, -CH2CH2=CH2 o ciclopropilo y R1 y R2 se seleccionan cada uno independientemente de H, -CH3, -CH2CH3, - CH2CH2CH3 y -CH(CH3)2, o una de sus sales, incluyendo una de sus sales farmacéuticamente aceptables.
En realizaciones particulares, el compuesto de Fórmula IIIa puede existir en equilibrio, según se indica posteriormente, entre una forma cerrada (Fórmula IIa) y una forma abierta (Fórmula IIIa), en ciertos ambientes y/o disolventes.

Fórmula lia Fórmula Illa
Forma Cerrada Forma Abierta
En realizaciones particulares, el compuesto de Fórmula IIIa puede existir en la forma abierta de Fórmula IIIa en estado sólido. También se divulga un compuesto cuya estructura comprende la Fórmula IIIa en la que R3 es -CH3, -CH2CH3, -CH2X H 2, -CH2CH2CH3, -CH(CH3)2, -CH2CH2X H 2 o ciclopropilo y R1 y R2 se s independientemente de H, -CH3 y -CH2CH3, o una de sus sales farmacéuticamente aceptables o hidratos.
También se divulga un compuesto cuya estructura comprende la Fórmula IIIa en la que R3 es -CH3, -CH2CH3,
-CH2X H 2, -CH2CH2CH3, -Ch (CH3)2, -CH2CH2X H 2 o ciclopropilo y R1 y R2 se seleccionan cada uno independientemente de H y -CH3, o una de sus sales farmacéuticamente aceptables o hidratos.
También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH3 y R1 y R2 son como se describen en la presente memoria. También se divulga un compuesto cuya
estructura comprende la Fórmula Illa o una de sus sales o hidratos, en donde R3 es -CH3 y R1 es H y R2 es como se describe en la presente memoria. También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH3 y R1 es -CH3 y R2 es como se describe en la presente memoria. También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH2CH3 y R1 y R2 son como se describen en la presente memoria. También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH2CH3 y R1 es H y R2 es como se describe en la presente memoria. También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH2CH3 y R1 es -CH3 y R2 es como se describe en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH(CH3)2 y R1 y R2 son como se describen en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula IIIa o una de sus sales o hidratos, en donde R3 es -CH3 y R1 y R2 son como se describen en la presente memoria.
También se divulga un compuesto cuya estructura comprende la Fórmula IIIa en la que R3 es -CH3 y R1 y R2 se seleccionan cada uno independientemente de H, -CH3, - CH2CH3, -CH2CH2CH3 y -CH(CH3)2, o una de sus sales o hidratos, incluyendo una de sus sales farmacéuticamente aceptables.
También se divulga un compuesto cuya estructura comprende la Fórmula IIIa en la que R3 es -CH3 y R1 y R2 se seleccionan cada uno independientemente de H, -CH3 y - CH2CH3, o una de sus sales o hidratos, incluyendo una de sus sales farmacéuticamente aceptables.
En realizaciones particulares, se proporciona un compuesto cuya estructura comprende la Fórmula IIIa en la que R3 es -CH3 y R1 y R2 se seleccionan cada uno independientemente de H y -CH3, o una de sus sales o hidratos, incluyendo una de sus sales farmacéuticamente aceptables.
En realizaciones particulares, los compuestos de Fórmula III y los compuestos de Fórmula IIIa que se describen en la presente memoria existen como un hidrato según se indica por la Fórmula IV o la Fórmula IVa posteriores:

En realizaciones particulares, el benzoxaborol sustituido comprende una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
En realizaciones particulares, el benzoxaborol sustituido es un compuesto que comprende una estructure como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
En otras realizaciones, el benzoxaborol sustituido es un compuesto que comprende una estructura como la indicada posteriormente:

en la que R3 es como se define en la presente memoria, o una de sus sales farmacéuticamente aceptables.
En otras realizaciones, el benzoxaborol sustituido es un compuesto que comprende una estructura como la indicada posteriormente:

en la que R3 es como se define en la presente memoria, o una de sus sales farmacéuticamente aceptables.
En otras realizaciones adicionales, el benzoxaborol sustituido es un compuesto que comprende una estructura como la indicada posteriormente:

y una de sus sales farmacéuticamente aceptables.
En otras realizaciones adicionales, el benzoxaborol sustituido es un compuesto que comprende una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
En otra realización, se proporciona un compuesto, (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, que comprende una estructura como la indicada posteriormente:

En otra realización, se proporciona un compuesto, (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, que comprende una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
Otra realización proporciona una sal farmacéuticamente aceptable de un compuesto, (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, que comprende una estructura como la indicada posteriormente:

Otra realización proporciona una composición farmacéutica que comprende una sal farmacéuticamente aceptable de un compuesto, (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, en donde el compuesto comprende una estructura como la indicada posteriormente:

junto con al menos un excipiente farmacéuticamente aceptable.
En otra realización más, se proporciona un compuesto, (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9aborabenzo[cd]azulen-2-il)metanamina, que comprende una estructura como la indicada posteriormente:

Otra realización adicional proporciona un compuesto, (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9aborabenzo[cd]azulen-2-il)metanamina, que comprende una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
Otra realización proporciona una sal farmacéuticamente aceptable de un compuesto, (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, que comprende una estructura como la indicada posteriormente:

Otra realización proporciona una composición farmacéutica que comprende un compuesto, (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, en donde el compuesto comprende una estructura como la indicada posteriormente:

junto con al menos un excipiente farmacéuticamente aceptable.
Una realización proporciona un benzoxaborol sustituido o una de sus sales farmacéuticamente aceptables, que comprende una estructura cuyo nombre IUPAC es:
3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
(S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
((2S)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
(S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina.
Una realización proporciona un benzoxaborol sustituido o una de sus sales farmacéuticamente aceptables, que comprende una estructura cuyo nombre IUPAC es:
3-(aminometil)-7-(2-hidroxietoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
(S)-3-(aminometil)-7-(2-hidroxietoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
3-(aminometil)-7-(2-hidroxipropoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
(3S)-3-(aminometil)-7-(2-hidroxipropoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
3-(aminometil)-7-(2-hidroxi-2-metilpropoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
(S)-3-(aminometil)-7-(2-hidroxi-2-metilpropoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol.
En una realización relacionada, la sal farmacéuticamente aceptable se selecciona de ácidos clorhídrico, bromhídrico, nítrico, carbónico, monohidrogenocarbónico, fosfórico, monohidrogenofosfórico, dihidrogenofosfórico, sulfúrico, monohidrogenosulfúrico, yodhídrico o fosforoso y similares. En otras realizaciones relacionadas, la sal farmacéuticamente aceptable se deriva de ácidos orgánicos incluyendo ácido acético, ácido propiónico, ácido isobutírico, ácido maleico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido glucarónico, ácido galacturónico, ácido láctico, ácido mandélico, ácido ftálico, ácido bencenosulfónico, ácido ptolilsulfónico, ácido cítrico, ácido tartárico, ácido metanosulfónico y similares. En otras realizaciones relacionadas adicionales, la sal farmacéuticamente aceptable incluye sales de aminoácidos tales como arginato, lisinato, glicinato. En otra realización, la sal farmacéuticamente aceptable incluye sales de ácidos, incluyendo sales de HCI y H2SO4. En aspectos particulares de la invención, el compuesto de Fórmula III o Fórmula IIIa es una mezcla de diastereisómeros. En otros aspectos particulares de la invención, el compuesto de Fórmula III o Fórmula IIIa es un diastereoisómero. En otros aspectos particulares de la invención, el compuesto de Fórmula III es una mezcla racémica de enantiómeros. En otros aspectos particulares adicionales de la invención, el compuesto de Fórmula III es un enantiómero específico. En aspectos particulares de la invención, cuando R1 y R2 son ambos H o CH3, el compuesto de Fórmula III o Fórmula IIIa tiene estereoquímica (S) en el centro quiral en la posición 3 del anillo de benzoxaborol. Una realización proporciona una combinación que comprende: un primer agente terapéutico en donde el primer agente terapéutico es un compuesto como los descritos en la presente memoria, o una de sus sales farmacéuticamente aceptables; opcionalmente un segundo agente terapéutico; opcionalmente un tercer agente terapéutico; opcionalmente un cuarto agente terapéutico; opcionalmente un quinto agente terapéutico; y opcionalmente un sexto agente terapéutico.
Una realización relacionada proporciona una combinación como la descrita en la que el segundo, tercero, cuarto, quinto y sexto agente terapéutico opcional se selecciona independientemente de isoniacida, rifampina, piracinamida, etambutol, moxifloxacina, rifapentina, clofacimina, bedaquilina (TMC207), nitroimidazooxacina PA-824, delamanida (OPC-67683), una oxazolidinona tal como linezolida, tedizolida, radezolida, sutezolida (PNU-100480) o posizolida (AZD-5847), análogo de EMB SQ109, una benzotiacinona, una dinitrobenzamida o un agente antiviral incluyendo un agente antirretroviral.
Una realización relacionada proporciona una combinación como la descrita en la que el agente antirretroviral es cidovudina, didanosina, lamivudina, zalcitabina, abacavir, estavudina, adefovir, adefovir dipivoxilo, focivudina, todoxilo, emtricitabina, alovudina, amdoxovir, elvucitabina, nevirapina, delavirdina, efavirenz, lovirida, immunocal, oltipraz, capravirina, lersivirina, GSK2248761, TMC-278, TMC-125, etravirina, saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, fosamprenavir, brecanavir, darunavir, atazanavir, tipranavir, palinavir, lasinavir, enfuvirtida, T-20, T-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 y BMS-626529, 5-Helix, raltegravir, elvitegravir, GSK1349572, GSK1265744, vicriviroc (Sch-C), Sch-D, TAK779, maraviroc, TAK449, didanosina, tenofovir, lopinavir, o darunavir.
Otra realización de la invención proporciona una combinación como la descrita en la que el segundo, tercero, cuarto, quinto y sexto agente terapéutico se selecciona de un agente terapéutico aprobado o recomendado para el tratamiento de la tuberculosis.
Una realización de la presente invención proporciona una formulación farmacéutica que comprende un primer agente terapéutico, siendo dicho primer agente terapéutico una cantidad terapéuticamente eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa según cualquiera de las realizaciones descritas en la presente memoria o una de sus sales farmacéuticamente aceptables. Una realización relacionada proporciona una combinación como la descrita en la presente memoria y un excipiente adyuvante o diluyente farmacéuticamente aceptable. En otra realización, la formulación farmacéutica puede comprender además un segundo agente terapéutico.
Otra realización proporciona un método para destruir micobacterias y/o inhibir la replicación de micobacterias que
provocan una enfermedad en un animal, que comprende poner en contacto las micobacterias con una cantidad eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables, a fin de destruir las micobacterias y/o evitar la replicación de las micobacterias.
También se divulga un método para tratar una infección por micobacterias en un animal que comprende: administrar al animal una cualquiera de: (i) una cantidad terapéuticamente eficaz de un compuesto cuya estructura comprende la Fórmula III o IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables; (ii) una cantidad terapéuticamente eficaz de una combinación que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables; o (iii) una cantidad terapéuticamente eficaz de una formulación farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables, a fin de tratar la infección por micobacterias en el animal.
También se divulga un método para destruir micobacterias y/o inhibir la replicación de micobacterias o un método para tratar una infección micobacteriana en un animal tal como ganado y animales de compañía, incluyendo vacas, ovejas, cabras, perros y gatos, o un ser humano, incluyendo un ser humano inmunodeprimido, comprendiendo dicho método: poner en contacto las micobacterias con una cantidad eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria, destruyendo de ese modo las micobacterias y/o inhibiendo la replicación de las micobacterias, o comprendiendo dicho método administrar al animal con la infección micobacteriana una cantidad terapéuticamente eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables. En una realización ejemplar, el compuesto de Fórmula III o el compuesto de Fórmula IIIa es parte de una formulación farmacéutica descrita en la presente memoria. En otra realización ejemplar, el contacto se produce bajo condiciones que permitan la entrada de la combinación en la micobacteria.
Otra realización de la invención proporciona un método como el descrito en la presente memoria, en donde las micobacterias se seleccionan de Mycobacterium tuberculosis, Mycobacterium avium incluyendo las subespecies (subesp.) Mycobacterium avium subesp. avium, Mycobacterium avium subesp. hominissuis, Mycobacterium avium subesp. silvaticum y Mycobacterium avium subesp. paratuberculosis; Mycobacterium kansasii, Mycobacterium malmoense, Mycobacterium simiae, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium scrofulaceum, Mycobacterium abscessus, Mycobacterium chelonae, Mycobacterium haemophilum, Mycobacterium leprae, Mycobacterium marinum, Mycobacterium fortuitum, Mycobacterium parafortuitum, Mycobacterium gordonae, Mycobacterium vaccae, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium africanum, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti, Mycobacterium pinnipedi, Mycobacterium leprae, Mycobacterium ulcerans, Mycobacterium intracellulare, complejo de Mycobacterium tuberculosis (MTC), complejo de Mycobacterium avium (MAC), complejo de Mycobacterium avian-intracellulare (MAIC), clado de Mycobacterium gordonae; clado de Mycobacterium kansasii; clado de Mycobacterium chelonae; clado de Mycobacterium fortuitum; clado de Mycobacterium parafortuitum; y clado de Mycobacterium vaccae.
También se divulga un método para tratar una infección micobacteriana en un animal que comprende: administrar al animal una cualquiera de: (i) una cantidad terapéuticamente eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables; (ii) una cantidad terapéuticamente eficaz de una combinación que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables; o (iii) una cantidad terapéuticamente eficaz de una formulación farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables, a fin de tratar la infección micobacteriana en el animal, en donde la infección por micobacterias es una infección por M. tuberculosis.
Otra realización proporciona un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables, para el uso en el tratamiento de una enfermedad resultante de una infección micobacteriana en un animal, incluyendo un ser humano. Otra realización proporciona un compuesto como el descrito en la presente memoria, en donde la enfermedad se selecciona de tuberculosis, lepra, enfermedad de Johne, úlcera de Buruli o Bairnsdale, enfermedad de Crohn, enfermedad pulmonar o infección pulmonar, neumonía, absceso localizado en la bolsa sinovial, la membrana sinovial o las vainas tendinosas, linfadenitis, infecciones de la piel y los tejidos blandos, síndrome de Lady Windermere, enfermedad pulmonar por MAC, complejo de Mycobacterium avium diseminado (DMAC), complejo de Mycobacterium avium intracellulare diseminado (DMAIC), pulmón de jacuzzi, mastitis por MAC, piomiositis por MAC, paratuberculosis por Mycobacterium avum, o granuloma.
También se divulga el uso de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables en la fabricación de un medicamento para el tratamiento de una infección micobacteriana en un animal.
También se divulga un método para tratar una enfermedad resultante de una infección micobacteriana en un animal,
particularmente en un mamífero, más particularmente en un ser humano, método que comprende administrar al animal que necesite este tratamiento una cantidad eficaz de un compuesto de la Fórmula III o la Fórmula IIIa que se describen en la presente memoria o una de sus sales farmacéuticamente aceptables. Otra realización proporciona un método como el descrito, en donde la enfermedad se selecciona de tuberculosis, lepra, enfermedad de Johne, úlcera de Buruli o Bairnsdale, enfermedad de Crohn, enfermedad pulmonar o infección pulmonar, neumonía, absceso localizado en la bolsa sinovial, la membrana sinovial o las vainas tendinosas, linfadenitis, infecciones de la piel y los tejidos blandos, síndrome de Lady Windermere, enfermedad pulmonar por MAC, complejo de Mycobacterium avium diseminado (DMAC), complejo de Mycobacterium avium intracellulare diseminado (DMAIC), pulmón de jacuzzi, mastitis por MAC, piomiositis por MAC, paratuberculosis por Mycobacterium avum, o granuloma.
También se divulga un método para tratar una infección micobacteriana en un animal, particularmente en un mamífero, método que comprende administrar al animal que necesite este tratamiento una cantidad terapéuticamente eficaz de un compuesto descrito en la presente memoria, o una de sus sales farmacéuticamente aceptables. También se divulga un método para tratar una infección micobacteriana en un animal, particularmente un mamífero, en donde la infección micobacteriana es Mycobacterium tuberculosis.
En una realización, se proporciona una formulación farmacéutica que comprende un primer agente terapéutico, siendo dicho primer agente terapéutico una cantidad terapéuticamente eficaz de un compuesto descrito en la presente memoria o una de sus sales farmacéuticamente aceptables y un excipiente, adyuvante o diluyente farmacéuticamente aceptable.
Más particularmente, se proporciona una formulación farmacéutica que comprende un primer agente terapéutico que es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, siendo dicho primer agente terapéutico una cantidad terapéuticamente eficaz de un compuesto como el descrito en la presente memoria o una sal farmacéuticamente aceptable del mismo, en cualquier realización como las descritas en la presente memoria; un excipiente, adyuvante o diluyente farmacéuticamente aceptable; y un segundo agente terapéutico que no es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa. En aspectos relacionados, la formulación farmacéutica comprende un primer agente terapéutico que es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria, o una de sus sales farmacéuticamente aceptables, y opcionalmente comprende un segundo agente terapéutico que no es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa y opcionalmente comprende un tercer agente terapéutico y opcionalmente comprende un cuarto agente terapéutico y opcionalmente comprende un quinto agente terapéutico y opcionalmente comprende un sexto agente terapéutico. En aspectos relacionados, el segundo, tercer, cuarto, quinto y sexto agente terapéutico es un agente antimicobacteriano distinto de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa. En aspectos relacionados, el segundo, tercer, cuarto, quinto y sexto agente terapéutico se selecciona de isoniacida, rifampina, piracinamida, etambutol, moxifloxacina, rifapentina, clofacimina, bedaquilina (TMC207), nitroimidazooxacina PA-824, delamanida (OPC-67683), una oxazolidinona tal como linezolida, tedizolida, radezolida, sutezolida (PNU-100480) y posizolida (AZD-5847), análogo de EMB SQ109, una benzotiacinona, una dinitrobenzamida y un antiviral incluyendo un agente antirretroviral. En aspectos relacionados, el segundo, tercer, cuarto, quinto y sexto agente terapéutico es un agente terapéutico aprobado y/o recomendado para el tratamiento de la tuberculosis.
Una realización relacionada proporciona una formulación farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa o una de sus sales y opcionalmente comprende un segundo, tercer, cuarto, quinto o sexto agente terapéutico, en donde el primer, segundo, tercer, cuarto, quinto o sexto agente terapéutico opcional en un agente antirretroviral seleccionado de cidovudina, didanosina, lamivudina, zalcitabina, abacavir, estavudina, adefovir, adefovir dipivoxilo, focivudina, todoxilo, emtricitabina, alovudina, amdoxovir, elvucitabina, nevirapina, delavirdina, efavirenz, lovirida, immunocal, oltipraz, capravirina, lersivirina, GSK2248761, TMC-278, TMC-125, etravirina, saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, fosamprenavir, brecanavir, darunavir, atazanavir, tipranavir, palinavir, lasinavir, enfuvirtida, T-20, T-1249, PRO-542, PRO-140, TnX-355, BMS-806, BMS-663068 y b Ms -626529, 5-Helix, raltegravir, elvitegravir, GSK1349572, GSK1265744, vicriviroc (Sch-C), Sch-D, TAK779, maraviroc, TAK449, didanosina, tenofovir, lopinavir o darunavir.
Según se describe en la presente memoria, realizaciones de la invención incluyen coadministrar, ya sea simultáneamente, secuencialmente o en combinación, un primer agente terapéutico que es un benzoxaborol sustituido o una de sus sales como los descritos en la presente memoria, preferiblemente un benzoxaborol sustituido de Fórmula III o Fórmula IIIa como los descritos en la presente memoria, o una de sus sales farmacéuticamente aceptables, opcionalmente en combinación con un segundo agente terapéutico, opcionalmente en combinación con un tercer agente terapéutico, opcionalmente en combinación con un cuarto agente terapéutico, opcionalmente en combinación con un quinto y/o un sexto agente terapéutico, a un sujeto expuesto a o infectado con una especie de micobacteria, incluyendo una especie de Mycobacterium tuberculosis. En ciertas realizaciones, el primer agente terapéutico es un benzoxaborol sustituido de Fórmula III o Fórmula IIIa como los descritos en la presente memoria o una de sus sales farmacéuticamente aceptables y el segundo y/o tercer y/o cuarto agente terapéutico es un agente antituberculoso. En ciertas realizaciones, la especie de micobacteria es una variante resistente a fármacos; en ciertas realizaciones la especie de micobacteria es una variante resistente a múltiples fármacos.
En otras realizaciones particulares, se proporciona un método para destruir micobacterias que comprende poner en
contacto las micobacterias o un animal, incluyendo un ser humano, expuesto a o infectado con una micobacteria con un primer agente terapéutico que es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IlIa que se describen en la presente memoria, o una de sus sales farmacéuticamente aceptables, opcionalmente poner en contacto las células o el sujeto con un segundo agente terapéutico, opcionalmente poner en contacto las células o el sujeto con un tercer agente terapéutico, opcionalmente poner en contacto las células o el sujeto con un cuarto agente terapéutico, opcionalmente poner en contacto las células o el sujeto con un quinto y/o un sexto agente terapéutico, de modo que el contacto destruya las células micobacterianas. En realizaciones particulares, el primer agente terapéutico es un benzoxaborol sustituido que es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa que se describen en la presente memoria, o una de sus sales farmacéuticamente aceptables, y el segundo, tercer, cuarto, quinto y/o sexto agente terapéutico opcional en un agente antituberculoso o una de sus sales. En otras realizaciones particulares, el sujeto estuvo expuesto a o está infectado con Mycobacterium tuberculosis.
Otras realizaciones particulares adicionales proporcionan un método para inhibir la replicación de células micobacterianas, comprendiendo el método poner en contacto las células micobacterianas o un an animal, incluyendo un ser humano, expuesto a o infectado con células micobacterianas con un primer agente terapéutico que es un compuesto como los descritos en la presente memoria o una de sus sales, opcionalmente poner en contacto las células micobacterianas o el animal con un segundo agente terapéutico, opcionalmente poner en contacto las células micobacterianas o el animal con un tercer agente terapéutico, opcionalmente poner en contacto las células micobacterianas o el animal con un cuarto agente terapéutico, opcionalmente poner en contacto las células micobacterianas o el animal con un quinto y/o un sexto agente terapéutico, de modo que el contacto inhiba la replicación de las células micobacterianas. En realizaciones particulares, el primer agente terapéutico es un benzoxaborol sustituido que es un compuesto como los descritos en la presente memoria o una de sus sales y el segundo, tercer, cuarto, quinto y/o sexto agente terapéutico opcional es un agente antituberculoso o una de sus sales. En otras realizaciones particulares, el sujeto estuvo expuesto a o está infectado con Mycobacterium tuberculosis.
Breves descripciones de los dibujos
La Figura 1 es un mapa del mundo que indica donde se ha documentado geográficamente XDR-TB.
La Figura 2 muestra la transmisión de la tuberculosis.
La Figura 3A muestra el espectro de RMN en solución de 13C para la estructura de forma cerrada de C2-H.
La Figura 3B muestra el espectro de RMN en solución de 13C para la estructura de forma cerrada de G26-CH3. Las Figuras 3C y 3D muestran el espectro de RMN en solución de 13C para la estructura de forma cerrada de G4-CI. La Figura 4 muestra el espectro de RMNss 13C CP-TOSS para la forma abierta del G26-CH3 en estado sólido.
Las Figuras 5A y 5B son espectros de RMNss que muestran el acoplamiento de 1H-11B HETCOR para C2-H, un compuesto cerrado anular (5A) y G26-CH3, un compuesto abierto anular (5B).
La Tabla 1 proporciona valores de CIM frente a cepas no micobacterianas para benzoxaboroles sustituidos.
La Tabla 2 proporciona valores de CI50 de inhibición de LeuRS, valores de CIM frente a la cepa estándar de M. tuberculosis Mtb H37Rv, valores de toxicidad frente a células HepG2 humanas y valores de selectividad para benzoxaboroles sustituidos de comparación.
La Tabla 3 proporciona valores de CI50 de inhibición de LeuRS, valores de CIM frente a la cepa estándar de M. tuberculosis Mtb H27Rv, valores de toxicidad frente a células HepG2 humanas y valores de selectividad para ciertos compuestos ejemplificados de la invención.
Descripción detallada de realizaciones específicas
"Animal", según se usa en la presente memoria, significa cualquiera de un reino (Animalia) de seres vivos incluyendo organismos multicelulares, incluyendo ganado y animales de compañía, incluyendo vacas, ovejas, cabras, perros y gatos, o un ser humano, incluyendo un ser humano inmunodeprimido.
"Compuesto de la invención", según se usa en la presente memoria, se refiere a un compuesto descrito en la presente memoria para el uso en el tratamiento de una infección micobacteriana y/o que tiene actividad frente a micobacterias y particularmente tiene selectividad para destruir cepas de Mycobacterium tuberculosis, particularmente cuando se compara con la actividad frente a otras cepas no micobacterianas.
"Combinación de la invención," según se usa en la presente memoria, se refiere a las combinaciones de compuestos descritos y/o ejemplificados en la presente y sales (p. ej. sales farmacéuticamente aceptables), profármacos, solvatos e hidratos de estos compuestos.
"Diastereoisómero", según se usa en la presente memoria, se refiere a uno de un par de estereoisómeros que no es imagen especular del otro estereoisómero.
"Enantiómero", según se usa en la presente memoria, se refiere a uno de un par de compuestos racémicos no superponibles (racematos) que es una imagen especular del otro enantiómero. Los enantiómeros tienen la propiedad de hacer girar el plano de luz polarizada en una dirección u otra cuando están en forma pura, pero, como una mezcla racémica, la mezcla no hace girar el plano de luz polarizada.
"Cantidad eficaz" de un compuesto, una de sus combinaciones o una de sus formulaciones significa una cantidad de un compuesto que es el agente activo que inhibe el crecimiento o la proliferación o destruye micobacterias, particularmente Mycobacterium tuberculosis, incluyendo una de sus combinaciones o formulaciones, de modo que la cantidad sea suficiente para proporcionar el efecto local o sistémico deseado. Una cantidad "terapéuticamente eficaz" o "farmacéuticamente eficaz" se refiere a la cantidad de compuesto, incluyendo una de sus combinaciones o formulaciones, suficiente para alcanzar un resultado terapéutico o farmacéutico deseado.
Se entiende que el término "sal farmacéuticamente aceptable" incluye una sal de un compuesto descrito en la presente memoria que se prepara con ácidos o bases relativamente atóxicos, dependiendo de los sustituyentes particulares encontrados en los compuestos descritos en la presente memoria. Cuando los compuestos que se describen en la presente memoria contienen funcionalidades relativamente ácidas, se pueden obtener sales por adición de base al poner en contacto la forma neutra de estos compuestos con una cantidad suficiente de la base deseada, bien puros o bien en un disolvente inerte adecuado. Ejemplos de sales por adición de base farmacéuticamente aceptables incluyen una sal de sodio, potasio, calcio, amonio, amino orgánico (tal como colina o dietilamina o aminoácidos tales como d-arginina, I-arginina, d-lisina o I-lisina) o magnesio, o una sal similar. Cuando los compuestos que se describen en la presente memoria contienen funcionalidades relativamente básicas, se pueden obtener sales por adición de ácido al poner en contacto la forma neutra de estos compuestos con una cantidad suficiente del ácido deseado, bien puros o bien en un disolvente inerte adecuado. Ejemplos de sales por adición de ácido farmacéuticamente aceptables incluyen las derivadas de ácidos inorgánicos como ácidos clorhídrico, bromhídrico, nítrico, carbónico, monohidrogenocarbónico, fosfórico, monohidrogenofosfórico, dihidrogenofosfórico, sulfúrico, monohidrogenosulfúrico, yodhídrico o fosforoso y similares, así como las sales derivadas de ácidos orgánicos relativamente atóxicos como acético, propiónico, isobutírico, maleico, malónico, benzoico, succínico, subérico, fumárico, láctico, mandélico, ftálico, bencenosulfónico, p-tolilsulfónico, cítrico, tartárico, metanosulfónico y similares. También se incluyen sales de aminoácidos tales como arginato y similares y sales de ácidos orgánicos como ácidos glucurónico o galacturónico y similares (véase, por ejemplo, Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Ciertos compuestos específicos como los descritos en la presente memoria contienen funcionalidades tanto básicas como ácidas que permiten que los compuestos se conviertan en sales por adición bien de base o bien de ácido.
Las formas neutras de los compuestos se regeneran preferiblemente al poner en contacto la sal con una base o un ácido y aislar los compuestos originales de modo convencional. La forma original del compuesto difiere de las diversas formas salinas en ciertas propiedades físicas, tales como solubilidad en disolventes polares.
Además de las formas salinas, la divulgación proporciona compuestos que están en forma de profármaco. Los profármacos de los compuestos descritos en la presente memoria sufren fácilmente cambios químicos bajo condiciones fisiológicas para proporcionar los compuestos que se describen en la presente memoria. Adicionalmente, los profármacos se pueden convertir en los compuestos de la invención mediante métodos químicos o bioquímicos en un ambiente ex vivo.
Ciertos de los compuestos de Fórmula III y Fórmula IIIa pueden formar sales por adición de ácido con uno o más equivalentes del ácido. La presente invención incluye dentro de su alcance todas las posibles formas estequiométricas y no estequiométricas.
El término "estructura por RMN 1H" se refiere a una estructura determinada a partir de un espectro de resonancia magnética nuclear de protón (RMN 1H). Un espectro de RMN 1H se puede generar al realizar una espectroscopía de RMN 1H sobre cualquier molécula que tenga átomos de carbono e hidrógenos (protones), tales como los compuestos descritos en los Ejemplos. La espectroscopía de RMN 1H se realiza usando un espectrómetro de RMN de 300 MHz o 400 MHz, en donde el compuesto se disuelve en un disolvente orgánico totalmente deuterado, tal como DMSO-56 o CD3OD. En la espectroscopía de RMN 1H, los protones químicamente equivalentes (los que tienen el ambiente químico y electrónico exacto) dan lugar a señales únicas en un espectro de RMN 1H. La posición de cada señal protónica en un espectro de RMN 1H - su desplazamiento químico - se muestra con relación a un compuesto de referencia - tetrametilsilano (TMS) - y se mide como una delta desde el punto cero - la señal protónica para el TMS. La intensidad y la característica de la señal protónica proporciona información acerca del ambiente de cada protón único y cada uno químicamente equivalente en una molécula, así como información sobre cuántos protones están representados por una señal particular. La señal para protones unidos a C, O y otros átomos de la molécula puede ser asignada por un experto en la técnica usando espectroscopía RMN 1H y a partir de esa señal se puede determinar una estructura para la molécula o el compuesto. En los Ejemplos descritos posteriormente, se registraron espectros de resonancia magnética nuclear de protón (RMN 1H), con desplazamientos químicos presentados en partes por millón (5) por debajo del campo del tetrametilsilano (TMS)
estándar. La señal procedente del pequeño porcentaje de protones no completamente deuterados presentes en el disolvente deuterado usado para la rMn 1H se usa como una referencia. Las abreviaturas para los datos de RMN son como sigue: s = singlete, d = doblete, t = triplete, q = cuadruplete, m = multiplete, dd = doblete de dobletes, dt = doblete de tripletes, ap = aparente, an = ancho.
Así, por ejemplo, la síntesis del Producto intermedio 1, a), posteriormente, indica que un espectro de RMN 1H se registraba a 400 MHz, con el compuesto disuelto en DMSO-afe, de modo que el espectro producía un doblete de picos a 8,47-8,48 asignado como un solo hidrógeno (8,47-8,48 (d, J = 4,4 Hz, 1H)); un triplete de picos a 8,77-8,74 asignado como un solo hidrógeno (8,77-8,74 (t, J = 7,6 Hz, 1H)); un doblete de picos a 7,43-7,41 asignado como un solo hidrógeno (7,43-7,41 (d, J = 8,0 Hz, 1H)); un doblete de dobletes de picos a 7,25-7,22, asignado como un solo hidrógeno (7,25-7,22 (dd, J = 4,8 Hz, 1H)); a doblete de dobletes de picos a 4,49-4,38 asignado a dos hidrógenos (4,49 4,38 (dd, J = 16,4 Hz, 2H)), un multiplete de picos a 2,46-2,42 asignado como un solo hidrógeno (2,46-2,42 (m, 1H)); un multiplete de picos a 1,97-1,93 asignado como dos hidrógenos (1,97-1,93 (m, 2H)); un multiplete de picos como 1,84 1,79 asignado como un solo hidrógeno (1,84-1,79 (m, 1H)); un multiplete de picos a 1,71-1,64 asignado como un solo hidrógeno (1,71-1,64 (m, 1H)); un multiplete de picos a 1,33-1,22 asignado como dos hidrógenos (1,33-1,22 (m, 2H)); un pico de singlete a 0,93 asignado como tres hidrógenos (0,93 (s, 3H)); un pico de singlete a 0,92 asignado como tres hidrógenos (0,92 (s, 3H)); y un pico de singlete a 0,73 asignado como tres hidrógenos (0,73 (s, 3H)).
Ciertos de los benzoxaboroles sustituidos de Fórmula III y Fórmula IIIa descritos en la presente memoria pueden existir en un equilibrio entre una estructura cerrada como la mostrada en la Fórmula II y la Fórmula IIa y una forma abierta, como la mostrada en la Fórmula III y la Fórmula IIIa, en ciertos ambientes de disolvente, tales como en presencia de H2O o cuando están en un disolvente acuoso. Además, los espectros de RMN 1H de ciertos de los benzoxaboroles sustituidos descritos en la presente memoria muestran que estos compuestos, cuando están disueltos en disolventes orgánicos, por ejemplo, DMSO-56 y CD3OD, existen en la forma cerrada, según se indica por los datos de RMN 1H mostrados en los Ejemplos sintéticos posteriormente.
En contraste, los espectros de RMN en estado sólido indican que ciertos de los benzoxaboroles sustituidos descritos en la presente memoria existen en estado sólido en las formas abiertas de Fórmula III y Fórmula IIIa. A lo largo de la solicitud, los benzoxaboroles sustituidos descritos en la presente memoria se pueden mostrar bien en las estructuras en solución para RMN 1H de las formas de anillo cerrado de Fórmula II y Fórmula IIa, o bien en las estructuras en estado sólido de las formas de anillo abierto de Fórmula III y Fórmula IIIa. También se entiende que, en ciertas condiciones, tales como cuando están disueltos en disolventes orgánicos, los benzoxaboroles sustituidos pueden existir en las formas cerradas de Fórmula II y Fórmula IIa; mientras que en otras condiciones, p. ej. tales como cuando está presente agua, los benzoxaboroles sustituidos descritos en la presente memoria pueden existir en un equilibrio entre las formas abiertas de Fórmula III y Fórmula IIIa y las formas cerradas de Formulas II y Fórmula IIa. También se ha mostrado que, en estado sólido, ciertos de los benzoxaboroles sustituidos descritos en la presente memoria pueden existir en las formas abiertas de Fórmula III y Fórmula IIIa.
Los compuestos de Fórmula III y Fórmula IIIa se pueden preparar en forma cristalina o no cristalina y, si son cristalinos, opcionalmente pueden estar solvatados, p. ej. como el hidrato. Esta invención incluye dentro de su alcance solvatos (p. ej. hidratos) estequiométricos, así como compuestos que contienen cantidades variables de disolvente (p. ej. agua). Los compuestos de Fórmula III y los compuestos de Fórmula IIIa que se describen en la presente memoria pueden existir como un hidrato según la estructura de Fórmula IV o de Fórmula IVa posterior:

Fórmula IV Fórmula IVa
en las que R1, R2 y R3 son como se describen en la presente memoria.
La invención en cuestión también incluye compuestos marcados isotópicamente que son idénticos a los citados en la Fórmula III y la Fórmula IIIa excepto por el hecho de que uno o más átomos se reemplazan por un átomo que tiene una masa atómica o un número atómico diferente de la masa atómica o el número atómico encontrado más comúnmente en la naturaleza. Ejemplos de isótopos que se pueden incorporar en compuestos como los descritos en la presente memoria incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, flúor, yodo y cloro tales como 3H, 11C, 14C, 18F, 123I o 125I.
Los compuestos de la presente invención y sales farmacéuticamente aceptables de dichos compuestos que
contienen los susodichos isótopos y/u otros isótopos de otros átomos están dentro del alcance de la presente invención. Compuestos marcados isotópicamente de la presente invención, por ejemplo, aquellos en los que se han incorporado isótopos radiactivos tales como 3H o 14C, son útiles en ensayos de distribución tisular de fármacos y/o sustratos. Los isótopos tritiados, es decir 3H, y de carbono 14, es decir 14C, se prefieren particularmente por su facilidad de preparación y capacidad de detección. Los isótopos de 11C y 18F son particularmente útiles en PET (tomografía de emisión de positrones).
Debido a que los compuestos de Fórmula III y Fórmula IIIb que se describen en la presente memoria están destinados al uso en composiciones farmacéuticas, se entenderá fácilmente que cada uno de ellos se proporciona preferiblemente en forma sustancialmente pura, por ejemplo, al menos 60% pura, más adecuadamente al menos 75% pura y preferiblemente al menos 85%, especialmente al menos 98% pura (los % son en una base de peso en peso). Se pueden usar preparaciones impuras de los compuestos para preparar las formas más puras usadas en las composiciones farmacéuticas.
Se divulga en la presente memoria un benzoxaborol sustituido o una de sus sales que tiene una estructura que comprende la Fórmula III:

en la que R3 se selecciona de -CH3; R1 y R2 son cada uno independientemente H, -CH3, -CH2CH3, -CH2CH2CH3 y -CH(CH3)2;.
Se divulga en la presente memoria un benzoxaborol sustituido cuya estructura comprende la Fórmula III en la que R3 es -CH2CH3 y R1 y R2 se seleccionan cada uno independientemente de H, -CH3, -CH2CH3, -CH2CH2CH3 y -CH(CH3)2.
Se divulga en la presente memoria un benzoxaborol sustituido cuya estructura comprende la Fórmula III o una de sus sales farmacéuticamente aceptables, en donde R3 es -CH3; R1 y R2 se seleccionan cada uno independientemente de H, -CH3 y -CH2CH3;
Se divulga en la presente memoria un benzoxaborol sustituido cuya estructura comprende la Fórmula III o una de sus sales, en donde R3 es -CH3; R1 y R2 se seleccionan cada uno independientemente de H o CH3.
Una realización proporciona un benzoxaborol sustituido cuya estructura comprende la Fórmula III o una de sus sales farmacéuticamente aceptables, en donde R3 es -CH3; R1 y R2 son independientemente H o -CH3.
Una realización proporciona un benzoxaborol sustituido cuya estructura comprende la Fórmula III que se muestra posteriormente:

o una de sus sales farmacéuticamente aceptables, en donde R3 es -CH3; R1 y R2 son cada uno independientemente H o -CH3.
Se divulga en la presente memoria un compuesto cuya estructura comprende la Fórmula III:

en la que R3 se selecciona de -CH3, -CH2CH3, -CH2CH2CH3, -CH(CH3)2, fenilo y tiofenilo; R1 y R2 se seleccionan cada uno independientemente de H, -CH3, -CH2CH3, - CH2CH2CH3 y -CH(CH3)2.
Otra realización proporciona un benzoxaborol sustituido cuya estructura comprende la Fórmula IIIa

en la que R3 es -CH3 y R1 y R2 son cada uno independientemente H o -CH3, o una de sus sales farmacéuticamente aceptables.
En un aspecto, la invención proporciona una composición farmacéutica que comprende un benzoxaborol sustituido cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, y uno o más portadores, excipientes o diluyentes farmacéuticamente aceptables.
También se divulga un método de tratamiento de una infección micobacteriana en un mamífero, particularmente en un ser humano, método que comprende administrar a un mamífero que necesite este tratamiento una cantidad eficaz de un primer agente terapéutico que es un compuesto cuya estructura comprende la Fórmula III o un compuesto cuya estructura comprende la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos. Realizaciones relacionadas comprenden además administrar a un mamífero que necesite este tratamiento una cantidad eficaz de un primer agente terapéutico que es un compuesto cuya estructura comprende la Fórmula III o un compuesto cuya estructura comprende la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables, opcionalmente administrando en combinación con una cantidad eficaz de un segundo agente terapéutico, opcionalmente administrando en combinación con una cantidad eficaz de un tercer agente terapéutico, opcionalmente administrando en combinación con una cantidad eficaz de un cuarto agente terapéutico, opcionalmente administrando en combinación con una cantidad eficaz de un quinto agente terapéutico, opcionalmente administrando en combinación con una cantidad eficaz de un sexto agente terapéutico.
En aspectos relacionados de la realización, el segundo, tercer, cuarto, quinto y sexto agente terapéutico opcional es un agente antimicobacteriano. En aspectos relacionados, la administración del primer agente terapéutico y opcionalmente la administración del segundo, tercer, cuarto, quinto y sexto agente terapéutico se producen al mismo tiempo, o la administración del primer agente terapéutico y opcionalmente la administración del segundo, tercer, cuarto, quinto y sexto agente terapéutico se producen secuencialmente. En otros aspectos relacionados de la invención, uno cualquiera del segundo, tercer, cuarto, quinto o sexto agente terapéutico se selecciona de un agente antimicrobiano, un agente antiviral, un agente antiinfectivo, un analgésico, una vitamina, un complemento nutricional, un agente antiinflamatorio un analgésico y un esteroide.
La invención proporciona por otra parte un compuesto cuya estructura comprende la Fórmula III, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, para el uso en el tratamiento de una infección micobacteriana en un mamífero, particularmente en un ser humano. En aspectos relacionados, el mamífero es un ser humano en el que la infección micobacteriana es una infección por Mycobacterium tuberculosis. En otros aspectos, el ser humano
con una infección por Mycobacterium tuberculosis también está infectado con un retrovirus, incluyendo un virus de inmunodeficiencia humana.
También se divulga el uso de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, en la fabricación de un medicamento para el uso en el tratamiento de una infección micobacteriana en un mamífero, particularmente en un ser humano.
La invención también proporciona una composición farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, y uno o más portadores, excipientes o diluyentes farmacéuticamente aceptables, para el uso en el tratamiento de una infección micobacteriana en un mamífero, particularmente en un ser humano.
La invención también proporciona una composición farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, y uno o más portadores, excipientes o diluyentes farmacéuticamente aceptables, para el uso en el tratamiento de infecciones micobacterianas en un mamífero, particularmente en un ser humano.
En otra realización particular, el benzoxaborol sustituido en la combinación tiene una estructura por RMN en estado sólido que comprende una estructura indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
En una realización particular, el compuesto tiene una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
En una realización particular, el compuesto comprende una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
En una realización particular, el compuesto comprende una estructura como la indicada posteriormente:

o una de sus sales farmacéuticamente aceptables.
Una realización de la invención proporciona un benzoxaborol sustituido cuya forma cerrada es:
3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
(S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
((2S)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
(S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina;
o una de sus sales farmacéuticamente aceptables.
Una realización de la invención proporciona un benzoxaborol sustituido cuya forma abierta es:
3-(aminometil)-7-(2-hidroxietoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
(S)-3-(aminometil)-7-(2-hidroxietoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol;
3-(aminometil)-7-(2-hidroxipropoxi)-4-metilbenzo[c][1,2]oxaborol-1(3H)-ol;
(3S)-3-(aminometil)-7-(2-hidroxipropoxi)-4-metilbenzo[c][1,2]oxaborol-1(3H)-ol;
3-(aminometil)-7-(2-hidroxi-2-metilpropoxi)-4-metilbenzo[c][1,2]oxaborol-1(3H)-ol;
(S)-3-(aminometil)-7-(2-hidroxi-2-metilpropoxi)-4-metilbenzo[c][1,2]oxaborol-1(3H)-ol.
Una realización proporciona un compuesto que tiene un patrón de NMR en estado sólido sustancialmente como el mostrado en la Figura 4, o una de sus sales farmacéuticamente aceptables.
Otra realización proporciona una composición farmacéutica que comprende un compuesto como el descrito en la presente memoria y al menos un excipiente.
Otra realización proporciona un compuesto como el descrito en la presente memoria para el uso en un medicamente para el tratamiento de Mycobacterium tuberculosis.
En otra realización particular, el tratamiento de una infección o afección micobacteriana se produce a través de la inhibición de un dominio de edición de una aminoacil ARNt sintetasa por medio de la unión al sitio activo de edición. En otra realización ejemplar, el tratamiento de una infección o afección micobacteriana se produce a través del bloqueo de un dominio de edición de una aminoacil ARNt sintetasa.
En una realización particular, la infección y/o enfermedad micobacteriana se trata a través de la administración oral
de la combinación de la invención. En una realización ejemplar, la infección y/o enfermedad micobacteriana se trata a través de la administración intravenosa de la combinación de la invención.
Formulaciones farmacéuticas
En otro aspecto, la invención es una formulación farmacéutica que incluye: (a) un compuesto como el divulgado en la presente memoria y b) un excipiente farmacéuticamente aceptable; o (a) una combinación de la invención. En otro aspecto, la formulación farmacéutica incluye: (a) un compuesto como el divulgado en la presente memoria y un excipiente farmacéuticamente aceptable; o (b) una combinación descrita en la presente memoria. En otro aspecto, la formulación farmacéutica incluye: (a) un excipiente farmacéuticamente aceptable; y (b) una combinación descrita en la presente memoria, o uno de sus sales, profármacos, hidratos o solvatos. En otro aspecto, la formulación farmacéutica incluye: (a) un excipiente farmacéuticamente aceptable; y (b) una combinación descrita en la presente memoria, o uno de sus sales, profármacos, hidratos o solvatos. En otro aspecto, la formulación farmacéutica incluye: (a) un excipiente farmacéuticamente aceptable; y (b) una combinación descrita en la presente memoria, o uno de sus sales, profármacos, hidratos o solvatos. En otro aspecto, la formulación farmacéutica incluye: (a) un excipiente farmacéuticamente aceptable; y (b) una sal de una combinación descrita en la presente memoria. En una realización ejemplar, la sal es una sal farmacéuticamente aceptable. En otro aspecto, la formulación farmacéutica incluye: (a) un excipiente farmacéuticamente aceptable; y (b) un profármaco de una combinación descrita en la presente memoria. En otro aspecto, la formulación farmacéutica incluye: (a) un excipiente farmacéuticamente aceptable; y (b) una combinación descrita en la presente memoria. En una realización ejemplar, la formulación farmacéutica es una forma de una sola dosificación unitaria. En una realización ejemplar, la formulación farmacéutica es una forma de una sola dosificación unitaria.
En una realización ejemplar, la formulación farmacéutica es una forma de dosificación unitaria. En una realización ejemplar, la formulación farmacéutica es una forma de una sola dosificación unitaria. En una realización ejemplar, la formulación farmacéutica es una forma de dos dosificaciones unitarias. En una realización ejemplar, la formulación farmacéutica es una forma de tres dosificaciones unitarias. En una realización ejemplar, la formulación farmacéutica es una forma de cuatro dosificaciones unitarias. En una realización ejemplar, la formulación farmacéutica es una forma de cinco dosificaciones unitarias. En una realización ejemplar, la formulación farmacéutica es una forma de seis dosificaciones unitarias. En una realización ejemplar, la formulación farmacéutica es una forma de una, dos, tres, cuatro, cinco, seis o siete dosificaciones unitarias que comprende una primera forma de dosificación unitaria y una segunda, tercera, cuarta, quinta y/o sexta forma de dosificación unitaria, en donde la primera forma de dosificación unitaria incluye a) una cantidad terapéuticamente eficaz de un compuesto como el descrito en la presente memoria y b) un primer excipiente farmacéuticamente aceptable; y la segunda, tercera, cuarta, quinta y/o sexta forma de dosificación unitaria incluye c) una cantidad terapéuticamente aceptable de un agente terapéutico adicional que es un agente antimicobacteriano y d) un segundo excipiente farmacéuticamente aceptable.
Información relativa a excipientes de uso en las formulaciones de la invención se pueden encontrar en Remington: The Science and Practice of Pharmacy, 21a Ed., Pharmaceutical Press (2011).
Combinaciones
En una realización ejemplar, la invención proporciona a) un primer agente terapéutico que es un benzoxaborol sustituido o una de sus sales según se describe en la presente memoria; b) una segunda actividad terapéutica. En ciertas realizaciones, el segundo agente terapéutico es un agente antibacteriano, más específicamente un agente antituberculoso, más específicamente un agente frente a M. tuberculosis.
En una realización ejemplar, la combinación es parte de una formulación farmacéutica descrita en la presente memoria. Estas condiciones son conocidas por los expertos en la técnica y condiciones específicas se indican en los Ejemplos adjuntos a la presente memoria.
Formas de dosificación de la combinación
Los componentes individuales de las combinaciones de la invención, por ejemplo, una combinación descrita en la presente memoria, se pueden administrar bien simultáneamente o bien secuencialmente en una forma de dosificación unitaria. La forma de dosificación unitaria puede ser una forma de dosificación unitaria simple o múltiple. En una realización ejemplar, la invención proporciona una combinación en una forma de una sola dosificación unitaria. Un ejemplo de una forma de una sola dosificación unitaria es una cápsula en la que tanto el benzoxaborol sustituido como el agente terapéutico adicional están contenidos dentro de la misma cápsula. En una realización ejemplar, la invención proporciona una combinación en una forma de dos dosificaciones unitarias. Un ejemplo de una forma de dos dosificaciones unitarias es una primera cápsula que contiene el benzoxaborol sustituido y una segunda cápsula que contiene el agente terapéutico adicional. Así, el término 'una sola unidad' o 'dos unidades' o 'múltiples unidades' se refiere al objeto que ingiere el paciente, no a los componentes interiores del objeto. Las dosis apropiadas de benzoxaborol sustituido serán fácilmente apreciadas por los expertos en la técnica. Las dosis apropiadas de un agente terapéutico adicional que no es un compuesto cuya estructura comprende la Fórmula III o la Fórmula Illa serán fácilmente apreciadas por los expertos en la técnica. En una realización particular, el benzoxaborol sustituido está presente en la combinación en una cantidad terapéuticamente eficaz. En una
realización particular, el agente terapéutico adicional que no es un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa está presente en la combinación en una cantidad suficiente para destruir o reducir la presencia, la cantidad o la velocidad de crecimiento de micobacterias expuestas al benzoxaborol sustituido, incluyendo M. tuberculosis.
Agente o agentes terapéuticos adicionales en la combinación
Las combinaciones de la invención, por ejemplo, una combinación descrita en la presente memoria, también pueden incluir un agente terapéutico o agente terapéuticos adicionales. La invención proporciona así, en un aspecto adicional, una combinación que comprende un benzoxaborol sustituido descrito en la presente memoria o una de sus sales farmacéuticamente aceptables y al menos un agente terapéutico adicional. La invención proporciona así, en un aspecto adicional, una combinación que comprende un benzoxaborol sustituido descrito en la presente memoria o una de sus sales farmacéuticamente aceptables y al menos un agente terapéutico adicional. En una realización ejemplar, el agente terapéutico adicional es un agente antimicobacteriano. En un aspecto, la invención comprende: a) una combinación de la invención; y b) al menos un agente terapéutico adicional. En otra realización ejemplar, la invención comprende: a) una combinación de la invención; b) un primer agente terapéutico adicional; y c) un segundo agente terapéutico adicional. En otra realización ejemplar, la invención comprende: a) una combinación de la invención; b) un primer agente terapéutico adicional; c) un segundo agente terapéutico adicional; y d) un tercer agente terapéutico adicional. El primer agente terapéutico adicional o segundo agente terapéutico adicional o tercer agente terapéutico adicional se puede seleccionar de los agentes terapéuticos adicionales descritos en la presente memoria.
Las combinaciones se pueden presentar convenientemente para el uso en la forma de una formulación farmacéutica. En un aspecto adicional de la presente invención, se proporciona una combinación farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, junto con uno o más agentes terapéuticos adicionales y uno o más portadores, excipientes o diluyentes farmacéuticamente aceptables. Los componentes individuales de estas combinaciones se pueden administrar bien secuencialmente o bien simultáneamente en formulaciones farmacéuticas separadas o combinadas mediante cualquier vía conveniente.
Cuando un agente terapéutico adicional se usa con una combinación como la descrita en la presente memoria frente a el mismo estado patológico, la dosis de cada compuesto puede diferir de aquella cuando el compuesto se usa solo. Las dosis apropiadas serán fácilmente apreciadas por los expertos en la técnica. Se apreciará que la cantidad de un compuesto que se describe en la presente memoria requerida para el uso en el tratamiento variará con la naturaleza de la afección que se trate y la edad y la condición del paciente y estará finalmente a discreción del médico o veterinario responsable.
Preparación de compuestos que contienen boro
Los compuestos de uso en la invención se pueden preparar usando materias primas disponibles comercialmente, productos intermedios conocidos, o al usar los métodos sintéticos descritos en la presente memoria, o publicados en referencias descritas, tales como las Patentes de EE. UU. N° 7.816.344, 8.461.364, US8.703.742, 9.243.003 y sus solicitudes de continuación y divisionarias; los números de publicación de EE. UU. US20100292504, US20140315860 y solicitudes que reivindican la prioridad de las mismas; y los números de solicitud publicados PCT WO2008/157726, WO2010080558, WO2011127143, WO2012/033858 y WO2015/021396 y solicitudes que reivindican la prioridad de las mismas. Los procedimientos generales usados para sintetizar los compuestos de Fórmula III y Fórmula IIIa se describen en los Esquemas de reacción posteriores y se ilustran en los Ejemplos. Ciertos benzoxaboroles sustituidos que se describen en la presente memoria se pueden preparar como se esboza en el Esquema 1.
Esquema 1
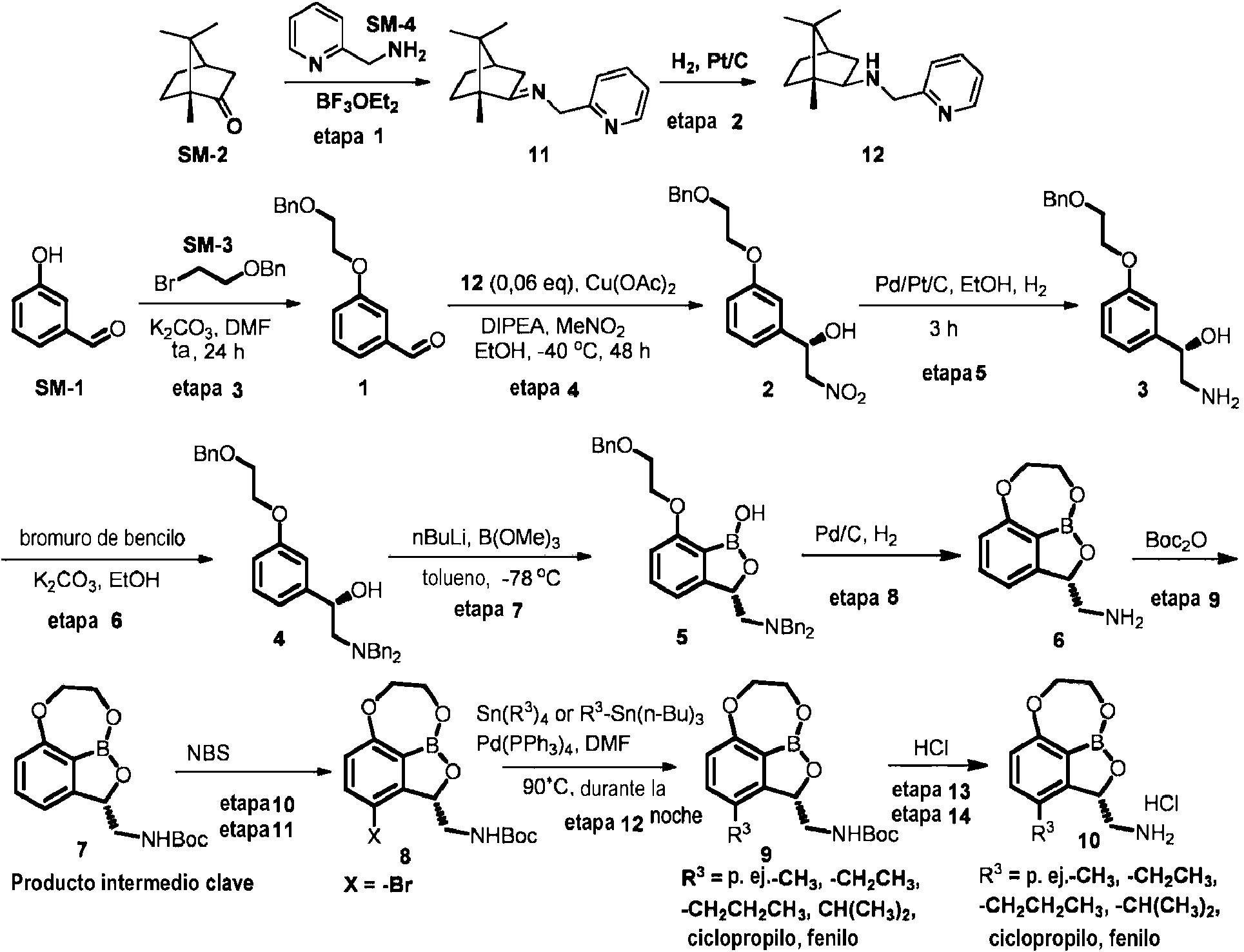
Aunque no se muestran expresamente, los compuestos 6, 7, 8, 9 y 10 del Esquema 1 pueden existir en equilibrio con la estructura abierta correspondiente dependiendo del ambiente. Además, ciertos compuestos de benzoxaborol sustituido divulgados en la presente memoria pueden existir en este equilibrio en ciertos disolventes. Este equilibrio se muestra mediante el ejemplo posterior:
En una realización, se ha encontrado que ciertos benzoxaboroles sustituidos divulgados en la presente memoria existen en la forma abierta en estado sólido. Una combinación de análisis de RMN de 13C simple y en estado sólido confirma que ciertos benzoxaboroles sustituidos divulgados en la presente memoria existen en la forma abierta en estado sólido. Los estudios de RMN en estado de solución también muestran que cuando se disuelven en solución, ciertos compuestos de benzoxaborol sustituido divulgados en la presente memoria existen en equilibrio entre la forma abierta y cerrada y que el balance del equilibrio está afectado por el disolvente usado y la presencia de H2O. Se entiende que los benzoxaboroles sustituidos divulgados en la presente memoria, ya se muestren en la forma cerrada o la forma abierta, pueden existir en la forma cerrada en disolventes orgánicos tales como DMSO y CH3OH, pueden existir en un equilibrio entre la forma cerrada y la forma abierta en un ambiente que comprende H2O y pueden existir en la forma abierta en estado sólido.
Composición y formulaciones
Los compuestos que se describen en la presente memoria se pueden formular para la administración de cualquier modo conveniente para el uso en medicina humana o veterinaria, por analogía con la formulación de agentes antimicobacterianos, o la formulación de otros agentes antituberculosos
Normalmente, pero no necesariamente, los compuestos descritos en la presente memoria se formularán en composiciones farmacéuticas antes de la administración a un paciente. En un aspecto, la invención se dirige a una composición farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una sal farmacéuticamente aceptable. En otro aspecto, la invención se dirige a una composición farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III, un compuesto cuya estructura comprende la Fórmula IIIa, o una sal farmacéuticamente aceptable y uno o más portadores, excipientes o diluyentes farmacéuticamente aceptables. El portador, excipiente o diluyente debe ser "aceptable" en el sentido de ser compatible con los otros ingredientes de la Formulación y no nocivo para su receptor.
Las composiciones farmacéuticas descritas en la presente memoria incluyen aquellas en una forma adaptada para uso oral o parenteral y se pueden usar para el tratamiento de una infección micobacteriana en un mamífero incluyendo un ser humano.
Las composiciones farmacéuticas descritas en la presente memoria incluyen aquellas en una forma adaptada para el uso oral, tópico o parenteral y se pueden usar para el tratamiento de infecciones micobacterianas en un mamífero incluyendo un ser humano.
La composición se puede formular mediante la administración mediante cualquier vía conveniente. Para el tratamiento de la tuberculosis, las composiciones pueden estar en forma de comprimidos, cápsulas, polvos, gránulos, pastillas para chupar, aerosoles o preparaciones líquidas, tales como soluciones o suspensiones orales o parenterales estériles.
Los comprimidos y las cápsulas para administración oral pueden estar en forma de presentación de dosis unitaria y pueden contener excipientes convencionales tales como agentes aglutinantes, por ejemplo jarabe, goma arábiga, gelatina, sorbitol, tragacanto o polivinilpirrolidona; cargas, por ejemplo lactosa, azúcar, almidón de maíz, fosfato cálcico, sorbitol o glicina; lubricantes para la formación de comprimidos, por ejemplo estearato magnésico, talco, polietilenglicol o sílice; desintegrantes, por ejemplo almidón de patata; o agentes humectantes aceptables tales como laurilsulfato sódico. Los comprimidos se pueden revestir según métodos muy conocidos en la práctica farmacéutica normal. Las preparaciones líquidas orales pueden estar en la forma de, por ejemplo, suspensiones, soluciones, emulsiones, jarabes o elixires acuosos u oleosos, o se pueden presentar como un producto seco para reconstitución con agua u otro vehículo adecuado antes de usar. Estas preparaciones líquidas pueden contener aditivos convencionales, tales como agentes de suspensión, por ejemplo sorbitol, metilcelulosa, jarabe de glucosa, gelatina, hidroxietilcelulosa, carboximetilcelulosa, gel de estearato de aluminio o grasas comestibles hidrogenadas, agentes emulsionantes, por ejemplo lecitina, monooleato de sorbitano o goma arábiga; vehículos no acuosos (que pueden incluir aceites comestibles), por ejemplo aceite de almendra, ésteres oleosos tales como glicerina, propilenglicol o alcohol etílico; conservantes, por ejemplo p-hidroxibenzoato de metilo o propilo o ácido sórbico y, si se desea, agentes aromatizantes o colorantes convencionales.
Los supositorios contendrán bases para supositorio convencionales, p. ej. manteca de cacao u otro glicérido.
Para la administración parenteral, se preparan formas de dosificación unitaria utilizando el compuesto y un vehículo estéril, prefiriéndose el agua. El compuesto, dependiendo del vehículo y la concentración usados, puede estar bien suspendido o bien disuelto en el vehículo. Al preparar soluciones, el compuesto se puede disolver en agua para inyección y esterilizarse por filtración antes de cargar en un vial o una ampolla adecuados y sellar.
En un aspecto de la invención, agentes tales como un anestésico local, un conservante y agentes tamponadores se pueden disolver en el vehículo. Para potenciar la estabilidad, la composición se puede congelar después de cargar en el vial y el agua se puede retirar bajo vacío. A continuación, el polvo liofilizado seco se sella en el vial y se puede suministrar un vial adjunto de agua para inyección para reconstituir el líquido antes del uso. Las suspensiones parenterales se preparan sustancialmente del mismo modo, excepto que el compuesto se suspende en el vehículo
en lugar de disolverse y la esterilización no se puede efectuar mediante filtración. El compuesto se puede esterilizar mediante exposición a óxido de etileno antes de suspender en el vehículo estéril. Ventajosamente, se incluye un tensioactivo o agente humectante en la composición para facilitar la distribución uniforme del compuesto.
Las composiciones pueden contener desde 0,1% en peso, preferiblemente de 10-60% en peso, del material activo, dependiendo del método de administración. Cuando las composiciones comprenden unidades de dosificación, cada unidad contendrá preferiblemente de 20-1000 mg del ingrediente activo. La dosificación que se emplea para el tratamiento de seres humanos adultos variará típicamente de 50 a 300 mg al día, a modo de ejemplo de 150 a 200 mg al día dependiendo de la vía y la frecuencia de administración. Esta dosificación corresponde a de 0,5 a 5 mg/kg al día. Preferiblemente, la dosificación es de 0,5 a 2 mg/kg al día y más preferiblemente la dosis es menor de 1 mg/kg al día. El compuesto cuya estructura comprende la Fórmula III, la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, puede ser el único agente terapéutico en las composiciones descritas en la presente memoria, o puede estar presente en la Formulación en combinación con uno o más agentes terapéuticos adicionales. La invención proporciona así, en un aspecto adicional, una combinación que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, junto con uno o más agentes terapéuticos adicionales.
El uno o más agentes terapéuticos adicionales son, por ejemplo, un agente útil para el tratamiento de la tuberculosis en un mamífero. Ejemplos de estos agentes terapéuticos incluyen rifampina, piracinamida, etambutol, moxifloxacina, rifapentina, clofacimina, bedaquilina (TMC207), nitroimidazooxacina PA-824, delamanida (OPC-67683), una oxazolidinona tal como linezolida, tedizolida, radezolida, sutezolida (PNU-100480) y posizolida (AZD-5847), análogo de EMB SQ109, una benzotiacinona, una dinitrobenzamida y un agente antiviral incluyendo un agente antirretroviral, o cualquier agente para la TB que este desarrollado para el tratamiento de la TB con una respuesta positiva en estudios de EBA en Fase IIa, o cualquier agente para la TB bajo desarrollo por the Global Alliance for Tuberculosis. Cuando un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, se usa en combinación con uno o más agentes terapéuticos adicionales, la dosis del compuesto o agente puede diferir de aquella cuando el compuesto o agente se usa solo. Las dosis apropiadas serán fácilmente apreciadas por los expertos en la técnica. Se apreciará que la cantidad de un compuesto descrito en la presente memoria y el uno o más agentes terapéuticos adicionales requeridos para el uso en el tratamiento variará con la naturaleza de la afección que se trate y la edad y la condición del paciente y finalmente estará a la discreción del médico o veterinario responsable.
Las combinaciones se pueden presentar convenientemente para el uso en la forma de una formulación farmacéutica. En un aspecto adicional de la presente invención, se proporciona una combinación farmacéutica que comprende un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables o uno de sus solvatos, junto con uno o más agentes terapéuticos adicionales y uno o más portadores, excipientes o diluyentes farmacéuticamente aceptables. Los componentes individuales de estas combinaciones se pueden administrar bien secuencialmente o bien simultáneamente en formulaciones farmacéuticas separadas o combinadas mediante cualquier vía conveniente.
Cuando la administración es secuencial, bien el compuesto de la presente invención o bien uno o más agentes terapéuticos adicionales se pueden administrar en primer lugar. Cuando la administración es simultánea, la combinación se puede administrar bien en la misma composición farmacéutica o bien en una diferente. Cuando se combinen en la misma formulación, se apreciará que el compuesto y los agentes deben ser estables y compatibles entre sí y con los otros componentes de la formulación. Cuando se formulen separadamente, se pueden proporcionar en cualquier formulación conveniente, convenientemente de un modo tal que sea conocido en la técnica para estos compuestos.
Métodos para inhibir el crecimiento bacteriano o destruir bacterias
Se espera que los compuestos ejemplificados y descritos en la presente memoria y sus combinaciones exhiban potencia frente a micobacterias y por lo tanto tengan el potencial de destruir micobacterias y/o inhibir la replicación de micobacterias. Se espera que las combinaciones que se describen en la presente memoria exhiban potencia frente a micobacterias que poseen resistencia a agentes antimicobacterianos de referencia y así tengan el potencial de destruir micobacterias y/o inhibir la replicación de estas micobacterias "resistentes". En aspectos de la invención, los compuestos que se describen en la presente memoria poseen una actividad notable frente a una selección de aislados micobacterianos sensibles a fármacos, incluyendo aislados clínicos de MDR-TB (TB resistente a múltiples fármacos) y XDR-TB (TB altamente resistente a fármacos), que exhiben valores de CIM de <0,32 pM y la mayoría tienen valores de CIM entre 0,04 - 0,08 pM en 96 aislados investigados.
Un compuesto como el descrito en la presente memoria se puede usar para inhibir o destruir micobacterias. También se divulga un método para destruir micobacterias y/o inhibir la replicación de micobacterias o un método para tratar una infección micobacteriana en un animal tal como ganado y animales de compañía, incluyendo vacas, ovejas, cabras, perros y gatos, o un ser humano, incluyendo un ser humano inmunodeprimido, comprendiendo dicho método: poner en contacto las micobacterias con una cantidad eficaz de un compuesto como los descritos en la
presente memoria, destruyendo de ese modo las micobacterias y/o inhibiendo la replicación de las micobacterias, o comprendiendo dicho método administrar al animal con la infección micobacteriana una cantidad terapéuticamente eficaz de una composición farmacéutica de la invención, en donde la composición farmacéutica comprende un compuesto cuya estructura comprende la Fórmula III o un compuesto cuya estructura comprende la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables. En una realización ejemplar, la combinación es parte de una formulación farmacéutica descrita en la presente memoria. En otra realización ejemplar, el contacto se produce bajo condiciones que permitan la entrada de la combinación en la micobacteria.
También se divulga un método para destruir micobacterias y/o inhibir la replicación de micobacterias o un método para tratar una infección micobacteriana en un animal tal como ganado y animales de compañía, incluyendo vacas, ovejas, cabras, perros y gatos, o un ser humano, incluyendo un ser humano inmunodeprimido, comprendiendo dicho método: poner en contacto las micobacterias con una cantidad eficaz de un compuesto o una combinación como los descritos en la presente memoria, destruyendo de ese modo las micobacterias y/o inhibiendo la replicación de las micobacterias, o comprendiendo dicho método administrar al animal con la infección micobacteriana una cantidad terapéuticamente eficaz de una composición farmacéutica de un compuesto o una combinación como los descritos en la presente memoria, en donde la composición farmacéutica comprende un compuesto cuya estructura comprende la Fórmula III o un compuesto cuya estructura comprende la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables. En una realización ejemplar, la combinación es parte de una formulación farmacéutica descrita en la presente memoria. En otra realización ejemplar, el contacto se produce bajo condiciones que permitan la entrada de la combinación en la micobacteria.
En una realización ejemplar, la micobacteria se destruye o su replicación se inhibe, o la infección micobacteriana se trata, a través de la administración oral de una combinación como la descrita en la presente memoria. En una realización ejemplar, la micobacteria se destruye o su replicación se inhibe, o la infección micobacteriana se trata, a través de la administración intravenosa de una combinación como la descrita en la presente memoria. En una realización ejemplar, la micobacteria se destruye o su replicación se inhibe, o la infección micobacteriana se trata, a través de la administración subcutánea de una combinación como la descrita en la presente memoria, en donde la combinación comprende un compuesto cuya estructura comprende la Fórmula III o un compuesto cuya estructura comprende la Fórmula IIIa, o una de sus sales farmacéuticamente aceptables.
En realizaciones ejemplares, las micobacterias se ponen en contacto o la infección micobacteriana se trata con una combinación como la descrita en la presente memoria que comprende un primer agente terapéutico que es un compuesto cuya estructura comprende la Fórmula III o un compuesto cuya estructura comprende la Fórmula IIIa, o una de sus sales, y opcionalmente que comprende un segundo, tercer, cuarto, quinto y sexto agente terapéutico en una población de micobacterias que comprende una micobacteria resistente con una mutación que confiere resistencia a uno cualquiera o más del segundo, tercer, cuarto, quinto y sexto agente terapéutico opcional. En realizaciones relacionadas, el segundo, tercer, cuarto, quinto y sexto agente terapéutico opcional, o una de sus sales, es un agente antimicobacteriano, particularmente un agente antimicobacteriano conocido, más preferiblemente un agente antimicobacteriano de referencia.
También se divulga un método para destruir y/o inhibir la replicación de micobacterias que provocan o están asociadas con una enfermedad en un animal, o un método para tratar una infección micobacteriana en un animal, comprendiendo el método poner en contacto las micobacterias con una cantidad eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa o una de sus sales, a fin de destruir y/o evitar la replicación de la micobacteria, o administrar al animal una cantidad terapéuticamente eficaz de un compuesto cuya estructura comprende la Fórmula III o la Fórmula IIIa o una de sus sales, en donde las micobacterias se seleccionan de Mycobacterium tuberculosis, Mycobacterium avium incluyendo las subespecies (subesp.) Mycobacterium avium subesp. avium, Mycobacterium avium subesp. hominissuis, Mycobacterium avium subesp. silvaticum y Mycobacterium avium subesp. paratuberculosis; Mycobacterium balnei, Mycobacterium sherrisii, Mycobacterium africanum, Mycobacterium microti, Mycobacterium silvaticum, Mycobacterium colombiense, Mycobacterium indicus pranii, Mycobacterium gastri, Mycobacterium gordonae, Mycobacterium hiberniae, Mycobacterium nonchromagenicum, Mycobacterium terrae, Mycobacterium trivial, Mycobacterium kansasii; Mycobacterium malmoense; Mycobacterium simiae; Mycobacterium triplex, Mycobacterium genavense, Mycobacterium florentinum, Mycobacterium lentiflavum, Mycobacterium palustre, Mycobacterium kubicae, Mycobacterium parascrofulaceum, Mycobacterium heidelbergense, Mycobacterium interjectum, Mycobacterium szulgai; Mycobacterium branden, Mycobacterium cookie, Mycobacterium celatum, Mycobacterium bohemicum, Mycobacterium haemophilum, Mycobacterium lepraemurium, Mycobacterium lepromatosis, Mycobacterium botniense, Mycobacterium chimaera, Mycobacterium conspicuum, Mycobacterium doricum, Mycobacterium forcinogenes, Mycobacterium heckeshornense, Mycobacterium lacus, Mycobacterium monacense, Mycobacterium montefiorense, Mycobacterium murale, Mycobacterium nebraskense, Mycobacterium saskatchewanenese, Mycobacterium scrofulaceum, Mycobacterium shimoidel, Mycobacterium tusciae, Mycobacterium xenopi, Mycobacterium intermedium, Mycobacterium bolletii, Mycobacterium fortuitum, Mycobacterium foruitum subesp. acetamidolyticum, Mycobacterium boenickei, Mycobacterium perigrinum, Mycobacterium porcinum, Mycobacterium senegalense, Mycobacterium septicum, Mycobacterium neworleansense, Mycobacterium houstonense, Mycobacterium mucogenicum, Mycobacterium mageritense, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium parafortuitum, Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium hodieri, Mycobacterium neoaurum, Mycobacterium prederkisbergense, Mycobacterium aurum, Mycobacterium vaccae, Mycobacterium
chitae, Mycobacterium fallax, Mycobacterium confluentis, Mycobacterium flavenscens, Mycobacterium madagascariense, Mycobacterium phlei, Mycobacterium smegmatis, Mycobacterium goodie, Mycobacterium colinskui, Mycobacterium thermoresistbile, Mycobacterium gadium, Mycobacterium kormossense, Mycobacterium obuense, Mycobacterium sphagni, Mycobacterium agri, Mycobacterium aichiense, Mycobacterium alvei, Mycobacterium arupense, Mycobacterium brumae, Mycobacterium canariasense, Mycobacterium chubuense, Mycobacterium conceptionense, Mycobacterium duvalii, Mycobacterium elephantis, Mycobacterium gilvum, Mycobacterium hassiacum, Mycobacterium holsaticum, Mycobacterium immunogenum, Mycobacterium massiliense, Mycobacterium moriokaense, Mycobacterium psychrotoleranse, Mycobacterium pirenivorans, Mycobacterium vanbaalenii, Mycobacterium pulveris, Mycobacterium arosiense, Mycobacterium aubagnense, Mycobacterium caprae, Mycobacterium clorofenolicum, Mycobacterium fluoroanthenivorans, Mycobacterium kumamotonense, Mycobacterium novocastrense, Mycobacterium parmense, Mycobacterium phocaicum, Mycobacterium poriferae, Mycobacterium rhodesiae, Mycobacterium seolense, Mycobacterium tokalense, Mycobacterium xenopi; Mycobacterium scrofulaceum; Mycobacterium abscessus; Mycobacterium chelonae; Mycobacterium haemophilum; Mycobacterium leprae; Mycobacterium marinum; Mycobacterium fortuitum; Mycobacterium bovis; Mycobacterium ulcerans; Mycobacterium pseudoshottsii, Mycobacterium shottsii, Mycobacterium intracellulare; complejo de Mycobacterium tuberculosis (MTC); un miembro del complejo de Mycobacterium avian-intracellulare (MAIC) y un miembro del complejo de Mycobacterium avium (MAC).
En aspectos relacionados, la micobacteria es Mycobacterium tuberculosis. En otros aspectos, la micobacteria es Mycobacterium avium, Mycobacterium kansasii, Mycobacterium malmoense, Mycobacterium simiae, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium scrofulaceum, Mycobacterium abscessus, Mycobacterium chelonae, Mycobacterium haemophilum, Mycobacterium leprae, Mycobacterium marinum, M. fortuitum, Mycobacterium bovis, M. bovis BCG, M. africanum, M. canetti, M. caprae, M. microti, M. pinnipedi, M. leprae o Mycobacterium ulcerans. En realizaciones relacionadas, la micobacteria es una subespecie (subesp.) de Mycobacterium avium, incluyendo Mycobacterium avium subesp. avium, Mycobacterium avium subesp. hominissuis, Mycobacterium avium subesp. silvaticum y Mycobacterium avium subesp. paratuberculosis. En otra realización relacionada, la micobacteria es Mycobacterium intracellulare. En realizaciones relacionadas adicionales, la micobacteria es un miembro del complejo de Mycobacterium tuberculosis (MTC), el complejo de Mycobacterium avium (MAC) o el complejo de Mycobacterium avian-intracellulare (MAIC). En realizaciones relacionadas, la micobacteria es un complejo o clado no tuberculoso, incluyendo: complejo de Mycobacterium avium; clado de Mycobacterium gordonae; clado de Mycobacterium kansasii; clado de Mycobacterium chelonae; clado de Mycobacterium fortuitum; clado de Mycobacterium parafortuitum; y clado de Mycobacterium vaccae. En una realización relacionada, la micobacteria es una Mycobacterium tuberculosis.
En una realización ejemplar, las micobacterias en los métodos descritos en la presente memoria comprenden una micobacteria resistente, particularmente una Mycobacterium tuberculosis resistente o multirresistente. En una realización ejemplar, la micobacteria resistente es una mutación de una micobacteria descrita en la presente memoria.
Métodos para tratar y/o prevenir una enfermedad
Las combinaciones de la presente invención exhiben potencia frente a micobacterias y por lo tanto tienen el potencial de alcanzar eficacia terapéutica en animales, incluyendo seres humanos.
Los compuestos descritos en la presente memoria y/o las formulaciones descritas en la presente memoria exhiben potencia frente a micobacterias y por lo tanto tienen el potencial de alcanzar eficacia terapéutica en animales, incluyendo seres humanos.
También se divulga un método para tratar y/o prevenir una enfermedad. El método incluye administrar al animal una cantidad terapéuticamente eficaz de una combinación de la invención, suficiente para tratar y/o prevenir la enfermedad. En una realización ejemplar, la combinación de la invención se puede usar en terapia médica humana o veterinaria, particularmente en el tratamiento o la profilaxis de una enfermedad asociada a micobacterias. En una realización ejemplar, la combinación se describe en la presente memoria.
También se divulga un método para tratar y/o prevenir una enfermedad. El método incluye administrar al animal una cantidad terapéuticamente eficaz de un compuesto como el descrito en la presente memoria, o una formulación como la descrita en la presente memoria, suficiente para tratar y/o prevenir la enfermedad. En una realización ejemplar, la combinación de la invención se puede usar en terapia médica humana o veterinaria, particularmente en el tratamiento o la profilaxis de una enfermedad asociada a micobacterias. En una realización ejemplar, la combinación se describe en la presente memoria.
En otra realización ejemplar, el animal es como se define en la presente memoria. En otra realización ejemplar, la enfermedad es una enfermedad sistémica o una enfermedad cutánea. En otra realización ejemplar, la enfermedad es una enfermedad respiratoria.
Abreviaturas
Al describir la invención, los elementos químicos se identifican según la Tabla Periódica de los Elementos. Las
abreviaturas y los símbolos utilizados en la presente memoria están de acuerdo con la utilización común de estas abreviaturas y símbolos por los expertos en la técnica química. Las siguientes abreviaturas se usan en la presente memoria:
AcOH ácido acético
Ac2O anhídrido acético
AlBN 2-2'-Azoisobutironitrilo
BOC N-terc-butoxicarbonilo
BOC dicarbonato de di-ferc-butilo anhidro
B2pin2 bis(pinacolato)diborodiboro, también conocido como 4,4,4',4',5,5,5',5'-octametil-2,2'-bi(1,3,2-dioxaborolano) Celite® un adyuvante de filtración compuesto por sílice diatomácea lavada con ácido, (una marca comercial de Manville Corp., Denver, Colorado)
CTAB bromuro de cetiltrimetilamonio
CD3OD metanol deuterado
DCM diclorometano
DIAD azodicarboxilato de diisopropilo
DIBAL-H hidruro de diisobutilaluminio
DME dimetoxietano
DCE dicloroetano
DMF dimetilformamida
DMSO-d6 dimetilsulfóxido deuterado
DMSO dimetilsulfóxido
ESI Ionización por electropulverización
ES MS Espectrometría de masas por electropulverización
Et2O éter dietílico
EtOH etanol
EtOAc, EA acetato de etilo
h horas
HPLC cromatografía de líquidos de alta resolución
KOAc acetato potásico
LCMS Cromatografía de líquidos-espectrometría de masas
mCPBA ácido meta-cloroperbenzoico
MeNO2 nitrometano
MeOH metanol
NBS N-bromosuccinimida
NCS N-clorosuccinimida
NIS N-yodosuccinimida
NXS N-halosuccinimida
NaBH(OAc)3 triacetoxiborohidruro sódico
NMR Espectroscopia de resonancia magnética nuclear
PE éter de petróleo
PPh3 trifenilfosfina
ta o t.a. temperatura ambiente
RT tiempo de retención
SFC cromatografía de fluidos supercríticos
t-BuOMe metil-t-butil-éter
TFA ácido trifluoroacético
THF tetrahidrofurano
uv ultravioleta
Ejemplos
Los siguientes ejemplos ilustran la invención. Estos Ejemplos no pretenden limitar el alcance de la invención, sino que en cambio proporcionan una guía al experto para preparar y usar los compuestos, las composiciones y los métodos de la invención. Aunque se describen realizaciones particulares de la invención, el experto apreciará que se pueden realizar diversos cambios y modificaciones. Las referencias a preparaciones llevadas a cabo de modo similar a, o mediante el método general de, otras preparaciones pueden abarcar variaciones en parámetros habituales tales como el tiempo, la temperatura, las condiciones de trabajo, pequeños cambios en las cantidades de reactivos, etc.
Los espectros de resonancia magnética nuclear de protón (RMN 1H) se registraron y los desplazamientos químicos se presentan en partes por millón (5) por debajo del campo relativo a la señal protónica para el tetrametilsilano (TMS) usando el disolvente no completamente deuterado residual como una referencia Las abreviaturas para los datos de RMN son como sigue: s = singlete, d = doblete, t = triplete, q = cuadruplete, m = multiplete, dd = doblete de dobletes, dt = doblete de tripletes, ap = aparente, an = ancho. Los espectros de masas se obtuvieron usando técnicas de ionización por electropulverización (ES). Todas las temperaturas se presentan en grados centígrados. Determinación de la estructura
Forma abierta
Se realizó RMN en fase de solución sobre G26-CH3 usando RMN 13C y RMN 1H. Espectro de 1H con desacoplamiento homonuclear (DUMBO 100 kHz) Espectro corregido para la adaptación a escala de los desplazamientos químicos.
Las asignaciones de las resonancias de 13C se confirmaron mediante otros datos de RMN del compuesto análogo G4-CI. La resonancia de C8 mostraba acoplamiento J al núcleo 11B cuadripolar próximo, observado como 4 líneas. El desplazamiento químico de C16 a 59,9 ppm es consecuente con una forma abierta anular. Se espera que la forma de anillo cerrado de G26-CH3 dé un desplazamiento químico de aproximadamente 69 ppm para C16. Esto se puede observar al comparar el análogo C4 H en la Figura 3B con el mostrado para C16 para G26-CH3 en la Figura 3A. Este mismo desplazamiento químico (Figuras 3C y 3D) también se observaba experimentalmente en los espectros de RMN en estado de solución del análogo G4-CI que tiene un Cl en la posición 4 del anillo de benzoxaborol en lugar de un CH3 como en G26-CH3 (compárese el pico sombreado en los espectros en las Figuras 3A, 3B, 3C y 3D).
La RMN en estado sólido también se realizó sobre G26-CH3.
Los datos de RMN en estado sólido (véase la Figura 4) apuntan a una estructura de anillo abierto. En el espectro de CP 13C, la resonancia del carbono C16 tiene un valor del desplazamiento químico de alrededor de 60 ppm, que es consecuente con una estructura de anillo abierto. Espectros de 1H-11B HETCOR adquiridos adicionalmente (véase la Figura 5) muestran fuertes correlaciones entre los protones de OH y B1 a valores de ppm altos. Esto también es consecuente con una estructura de anillo abierto con un protón de -OH próximo a B1. El ajuste de la curva cuadripolar de segundo orden del espectro de 11B conducía a valores que son similares a valores de la bibliografía para estructuras de anillo abierto similares.
Las reacciones que implican hidruros metálicos incluyendo hidruro de litio, hidruro de litio y aluminio, hidruro de diisobutilaluminio, hidruro sódico, borohidruro sódico y triacetoxiborohidruro sódico se llevan a cabo bajo argón a menos que se especifique otra cosa.
Síntesis
Producto intermedio 1 (S)-((7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[co(|azulen-2-il)metil)carbamato de tere-butilo
a) (Z)-1-(piridin-2-il)-N-((1 R)-1,7,7-trimetilbiciclo[2,2.1]heptan-2-iliden)metanamina

Una mezcla de (+)-alcanfor (371 g, 2,44 mol), piridin-2-ilmetanamina (277 g, 2,56 mol) y BF3.Et2Ü (17 g, 0,12 mol) en tolueno (3,7 l) se cargó en un matraz de fondo redondo de 5 l equipado con un separador de Dean Stark, un condensador de reflujo, un termómetro y una entrada para nitrógeno. La mezcla se calentó hasta reflujo con retirada azeotrópica de agua durante 20 h. La mezcla se enfrió hasta 15°C y se desactivó con bicarbonato sódico acuoso al 5% (2,5 l), la fase orgánica se separó y se lavó con agua (1,25 l x 2), a continuación la mezcla se concentró hasta 2 l bajo vacío. El residuo se usó en la siguiente etapa sin purificación. RMN 1H (400 MHz, DMSO-d6): 8,47-8,48 (d, J = 4,4 Hz, 1H), 8,77-8,74 (t, J = 7,6 Hz, 1 H), 7,43-7,41 (d, J = 8,0 Hz, 1H), 7,25-7,22 (dd, J = 4,8 Hz, 1 H), 4,49 4,38 (dd, J = 16,4 Hz, 2H), 2,46-2,42 (m, 1H), 1,97-1,93 (m, 2H), 1,84-1,79 (m, 1H), 1,71 -1,64 (m, 1H), 1,33-1,22 (m, 2H), 0,93 (s, 3H), 0,92 (s, 3H), 0,73 (s, 3H). LCMS: [M+H]+ = 243.
b)(1 R)-1,7,7-trimetil-N-(piridin-2-ilmetil)biciclo[2,2.1]heptan-2-amina

Se cargó Pt al 5%/C (40 g) en un recipiente para presión de 5 l, seguido por una solución de (Z)-1-(piridin-2-il)-N-((1R)-1,7,7-trimetilbiciclo[2,2.1]heptan-2-iliden)metanamina (2,44 mol) en tolueno (2 l). El recipiente se presurizó con 689476 Pa (100 psi) de hidrógeno durante un período de 12 h. El sólido se filtró a través de Celite® y la torta se lavó con tolueno (1 l). El filtrado se concentró bajo vacío para obtener el producto deseado (435 g obtenidos, rendimiento total: 73%, a lo largo de dos etapas) como un aceite amarillo claro. RMN 1H (400 MHz, DMSO-d6): 8,49-8,48 (d, J = 4,8 Hz, 1H), 7,75-7,71 (t, J = 7,6 Hz, 1 H), 7,40-7,38 (d, J = 7,6 Hz, 1H), 7,24-7,21 (dd, J = 5,2 Hz, 1 H), 3,79 3,64 (dd, J = 14,4 Hz, 2H), 2,53-2,49 (m, 1 H), 1,99 (s, 1 H), 1,68-1,42 (m, 5H), 1,05 (s, 3H), 0,87 (s, 3H), 0,78 (s, 3H), LCMS: [M+H]+ = 245.
c) 3-(2-(benciloxi)etoxi)benzaldehído

Se añadió K2CO3 (3,83 kg, 27,70 mol) a una solución de 3-hidroxibenzaldehído (2,90 kg, 23,75 mol) y ((2-bromoetoxi)metil)benceno (4,26 kg, 19,79 mol) en DMF (9,3 l). La mezcla de reacción se agitó a t. a. durante 24 h. Se añadieron agua (15 l) y terc-butil-metil-éter (23 l) a la mezcla de reacción. La fase orgánica se separó y se lavó con NaOH 1 N (2X15 l) y agua (15 l) secuencialmente y a continuación se concentró hasta un mínimo. Se añadió etanol (23 l) y la solución se concentró bajo vacío para proporcionar el producto deseado (4,7 kg, 93%) como un aceite incoloro. RMN 1H (400 MHz, DMSO-de): 9,98 (s, 1H), 7,55-7,52 (m, 2H), 7,46 (s, 1H), 7,36-7,34 (m, 4H), 7,32-7,26 (m, 2H), 4,57 (s, 2H), 4,25-4,22 (t, J = 4,4 Hz, 2H), 3,80-3,78 (t, J = 4,4 Hz, 2H). LCMS: [M+Na]+ = 279.
d) (S)-1 -(3-(2-(benciloxi)etoxi)fenil)-2-nitroetanol

Una mezcla de acetato de cobre (II) (167 g, 0,92 mol), (1 R)-1,7,7-trimetil-N-(piridin-2-ilmetil)biciclo[2,2.1 ]heptan-2
amina (269 g, 1,10 mol) en etanol (19 l) se agitó a t. a. durante 1 h, a continuación se añadió una solución de 3-(2-(benciloxi)etoxi)benzaldehído (4,70 kg, 18,34 mol) en etanol (5 l). La mezcla de reacción se enfrió hasta un intervalo de temperatura entre -30°C y -40°C y a continuación se añadió nitrometano (9,9 l, 183,40 mol) gota a gota, manteniendo la temperatura por debajo de -30°C, seguido por la adición de diisopropiletilamina (285 g, 2,20 mol). La reacción se agitó a -30°C durante 24 h y a continuación se desactivó con ácido trifluoroacético (314 g, 2,75 mol). Se añadieron HCl 1 N (24 l) y TBME (47 l) a la solución resultante. La fase orgánica separada se lavó con agua (24 l) y a continuación se concentró bajo vacío. El residuo se añadió a una mezcla de éter de petróleo/acetato de etilo=5:1 (10 l). A continuación, el sólido amarillo se precipitó y se recogió mediante filtración con un embudo de Buchner y se secó bajo vacío a 40°C durante 6 h para proporcionar el producto deseado (5,00 kg, 86%) como un sólido blanco. RMN 1H (400 MHz, DMSO-d6): 7,38-7,25 (m, 6H), 7,03 (s, 1H), 7,01-6,99 (d, J = 7,6 Hz, 1H), 6,90-6,87 (dd, J = 8,0 Hz, 1H), 6,09-6,08 (d, J = 5,2 Hz, 1H), 5,26-5,22 (m, 1H), 4,86-4,82 (dd, J = 12,4 Hz, 1H), 4,57-4,51 (m, 3H), 4,15-4,13 (m, 2H), 3,78-3,76 (t, J = 4,8 Hz, 2H). LC-MS: [M+Na]+ = 340.
e) hidrocloruro de (S)-1-(3-(2-(benciloxi)etoxi)fenil)-2-(dibencilamino)etanol

Se cargaron Pd al 10%/C (800 g) y Pt al 10%/C (200 g) a un recipiente para presión, seguido por una solución de (S)-1-(3-(2-(benciloxi)etoxi)fenil)-2-nitroetanol (5,00 kg, 15,76 mol) en etanol (50 l). El recipiente se presurizó con 689476 Pa (100 psi) de hidrógeno durante 12 h a t. a. El sólido se filtró a través de Celite® y la torta se lavó con etanol (5 l). Se añadieron secuencialmente al filtrado K2CO3 (4,80 kg, 34,67 mol) y bromuro de bencilo (5,93 kg, 34,67 mol). La mezcla de reacción se agitó a t. a. durante 24 h. El sólido se filtró y se lavó con etanol (1 l). El filtrado se diluyó con agua (20 l) y a continuación se calentó hasta 50°C. La solución se agitó a 50°C durante 30 min y a continuación se añadió gota a gota HCl conc. (1,5 l) a lo largo de 1 h. La mezcla se enfrió hasta 0°C y se mantuvo a 0°C durante 30 min adicionales. El producto se filtró y se lavó con etanol acuoso al 20% (1 l) para proporcionar la sal clorhídrica del producto deseado (5,00 kg, 63% a lo largo de dos etapas) como un sólido incoloro. RMN 1H (400 MHz, DMSO-ds): 10,67 (s, 1H), 7,72-7,68 (m, 4H), 7,47-7,45 (m, 6H), 7,38-7,26 (m, 5H), 7,25-7,21 (t, J = 7,6 Hz, 1H), 6,86-6,84 (d, J = 8,0 Hz, 1H), 6,77 (s, 1H), 6,77-6,75 (d, J = 7,2 Hz, 1H), 6,36 (s, 1H), 5,04-5,02 (d, J = 9,2 Hz, 1H), 4,58 (s, 2H), 4,51-4,38 (m, 4H), 4,09-4,07 (t, J = 4,0 Hz, 2H), 3,77-3,75 (t, J = 3,2 Hz, 2H), 3,13-2,96 (m, 2H). LC-MS:
[M+H]+ = 468.
f) (S)-7-(2-(benciloxi)etoxi)-3-((dibencilamino)metil)benzo[c][1,2]oxaborol -1 (3H)-ol

Se añadió n-BuLi (15,3 l, 38,20 mol) gota a gota a lo largo de 6 h a una solución a -30°C de hidrocloruro de (S)-1-(3-(2-(benciloxi)etoxi)fenil)-2-(dibencilamino)etanol (3,85 kg, 7,64 mol) en tolueno seco (39 l) bajo una atmósfera de N2. Después de la adición, la mezcla se agitó a -30°C durante otra 1 h y a continuación se enfrió hasta -70°C; se añadió gota a gota borato de trimetilo (3,97 kg, 38,20 mol) manteniendo la temperatura por debajo de -60°C. Después de la adición, la mezcla de reacción se dejó calentar hasta t. a. y se agitó durante la noche. La reacción se desactivó con NaHCO3 acuoso al 5% (20 l) y se agitó vigorosamente durante 15 min, la suspensión resultante se filtró y el filtrado se separó. La capa orgánica se lavó con agua (20 l x 3) y se concentró bajo vacío y el residuo se purificó mediante cromatografía en gel eluyendo con éter de petróleo/acetato de etilo=5:1 para proporcionar el producto deseado (1,80 kg, 48 %) como un sólido amarillo. RMN 1H (400 MHz, DMSO-d6): 8,81 (s, 1H), 7,39-7,22 (m, 16H), 6,82-6,80 (d, J = 7,6 Hz, 1H), 6,72-6,70 (d, J = 7,6 Hz , 1H), 5,34-5,31 (dd, J = 7,6 Hz, 1H), 4,60 (s, 2H), 4,22-4,19 (t, J = 4,4 Hz, 2H), 3,80-3,72 (m, 6H), 2,88-2,84 (dd, J = 13,6 Hz , 1H), 2,47-2,45 (dd, J = 10 Hz, 1H). LC-MS: [M+H]+ = 494.
g) hidrocloruro de (S)-(7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina
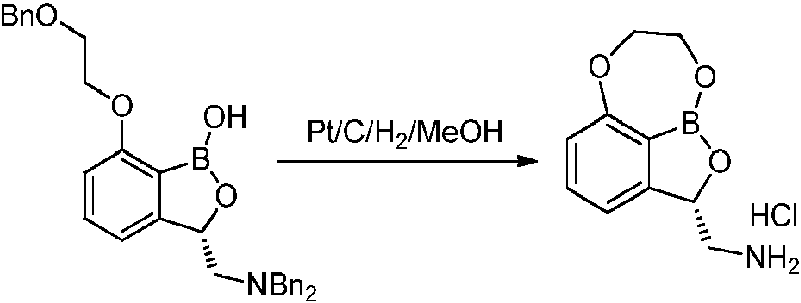
Se cargó Pd al 10%/C (180 g) a un recipiente para presión, seguido por una solución de (S)-7-(2-(benciloxi)etoxi)-3-((dibencilamino)metil)benzo[c][1,2]oxaborol-1(3H)-ol (1,80 kg, 3,65 mol) en metanol (18 l), tolueno (3,6 l) y HCl 1 N (4 l). El recipiente se presurizó con 689476 Pa (100 psi) de hidrógeno durante un período de 12 h a 50°C. El sólido se filtró a través de Celite y la torta se lavó con metanol (1 l). El filtrado se concentró bajo vacío y el residuo se trató con 2-propanol (3,6 l), se agitó a t. a. durante 30 min. El sólido resultante se recogió mediante filtración y se lavó con 2-propanol (500 ml), se secó bajo vacío a 50°C durante 6 h para proporcionar el producto deseado (680 g, 77%) como un polvo amarillo claro. RMN 1H (400 MHz, DMSO-d6): 8,38 (s, 3H), 7,52-7,48 (t, J = 8,0 Hz, 1H), 7,17-7,15 (d, J = 7,6 Hz, 1 H), 6,92-6,90 (d, J = 7,6 Hz, 1H), 5,55 (m, 1H), 4,71 -4,68 (m, 1 H), 4,38-4,22 (m, 3H), 3,53-3,50 (m, 1 H), 2,91-2,86 (m, 1 H). LC-MS: [M+H]+ = 206.
h) (S)-((7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Se añadió (Boc)2O (353,0 g 1,62 mol) gota a gota a lo largo de 2 h a t. a. a una solución de hidrocloruro de (S)-(7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina (390 g, 1,62 mol) y Et3N (163,4 g, 4,85 mol) en DCM (4,6 l). Después de la adición, la mezcla de reacción se agitó a t. a. durante otras 3 h. La reacción se desactivó con HCl 1 N (4 l) y la fase orgánica se separó y se lavó con agua (4 l), se concentró bajo vacío para obtener el producto deseado (460 g, 93%) como un sólido blanco claro. RMN 1H (400 MHz, DMSO-d6): 7,46-7,42 (t, J = 7,6 Hz, 1H), 7,07 (s, 1 H), 7,02-7,00 (d, J = 7,2 Hz, 1H), 6,87-6,85 (d, J = 8,0 Hz, 1 H), 5,27 (m, 1H), 4,68-4,65 (m, 1 H), 4,34-4,18 (m, 3H), 3,41(s, 1H), 3,14-3,08 (m, 1H), 1,38 (s, 9H). LC-MS: [M-55] = 250.
Ejemplo 2 (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina (G26 -CH3)

a) (S)-((3-bromo-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de (S)-((7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (3 g, 9,81 mmol) y NBS (1,92 g, 10,8 mmol) en 50 ml de dicloroetano se agitó a 55°C durante 24 horas. A continuación, la mezcla se vertió en 150 ml de agua, se extrajo con acetato de etilo (150 ml), se lavó con agua (100 ml) y salmuera (100 ml). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró a vacío. El residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:4) para dar el compuesto del epígrafe (3,5 g, 93 %) como un aceite amarillo claro. LC-MS: 384 [M+H]+.
b) (S)-((3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de (S)-((3-bromo-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (500 mg, 1,31 mmol), tetrametilestannano (0,822 g, 4,57 mmol) y Pd(Ph3)4 (151 mg, 0,131 mmol) en 10 ml de DMF se desgasificó con N2 seis veces. A continuación, la mezcla se calentó a 90°C durante 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y a continuación la mezcla se vertió en 25 ml de agua, se extrajo con acetato de etilo (25 ml), se lavó con agua (15 ml) y salmuera (15 ml). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró a vacío. El residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:6) para dar el compuesto del epígrafe (150 mg, 36 %) como un aceite amarillo claro. LC-MS: 320 [M+H]+.
c) hidrocloruro de (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución del compuesto (S)-((3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (150 mg, 0,47 mmol) y TFA (0,6 ml) en 5 ml de DCM se agitó a temperatura ambiente durante 2 horas. El disolvente se evaporó a 40°C a presión reducida y el residuo se trató con HCl 2 M en Et2O, a continuación se lavó con Et2O (10 ml) para dar el compuesto del epígrafe (13,6 mg, 11 %) como un sólido blanco. LC-MS: 220,1 [M+H]+. RMN 1H (400 MHz, CD3OD): 5 7,30 (d, 1H, J=8), 6,87 (d, 1H, J=8), 5,54 (s an, 1H), 4,67 (s an, 1H), 4,46-4,17 (m, 3H), 3,68 (s an, 1 H), 2,99-2,93 (m, 1H), 2,33 (s, 3H).
Ejemplo Comparativo 3 (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina (G27-fenilo)

a) (S)-((3-fenil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamatos de terc-butilo

Una solución del compuesto (S)-((3-bromo-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (400 mg, 1,04 mmol), tributil(fenil)estannano (1,68 g, 4,58 mmol) y Pd(Ph3)4 (151 mg, 0,131 mmol) en 10 ml de DMF se desgasificó con N2 seis veces. A continuación, la mezcla se calentó a 90°C durante 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y a continuación la mezcla se vertió en 20 ml de agua, se extrajo con acetato de etilo (15 ml), se lavó con agua (10 ml) y salmuera (10 ml). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró a vacío. El residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:6) para dar el compuesto del epígrafe (140 mg, 35%) como un aceite amarillo claro. LC-MS: 382 [M+H]+.
b) hidrocloruro de (S)-(3-fenil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución del compuesto (S)-((3-fenil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (140 mg, 0,37 mmol) y TFA (0,5 ml) en 5 ml de DCM se agitó a temperatura ambiente durante 2 horas. El disolvente se evaporó a 40°C a presión reducida y el residuo se trató con HCl 2 M en Et2O, a continuación se lavó con Et2O (10 ml) para dar el compuesto del epígrafe (45 mg, 39 %) como un sólido blanco. LC-MS: 282,1 [M+H]+. RMN 1H (400 MHz, DMSO-d6): 58,17-7,86 (m, 3H), 7,49-7,38 (m, 5H), 7,05 (d, 1H, J=8), 6,01 (s an, 1H), 4,78-4,68 (m, 1 H), 4,46-4,24 (m, 3H), 3,32 (s an, 1 H), 3,01 -2,84 (m, 1H).
Ejemplo Comparativo 4 (S)-(3-(tiofen-2-il)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina (G28-tienilo)

a) (S)-((3-(tiofen-2-il)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución del compuesto (S)-((3-bromo-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (400 mg, 1,04 mmol), tributil(tiofen-2-il)estannano (1,37 g, 3,66 mmol) y Pd(Ph3)4 (120 mg, 0,104 mmol) en 10 ml de DMF se desgasificó con N2 seis veces. A continuación, la mezcla se calentó a 90°C durante 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y a continuación la mezcla se vertió en 20 ml de agua, se extrajo con acetato de etilo (12 ml), se lavó con agua (10 ml) y salmuera (10 ml). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró a vacío. El residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:5) para dar el compuesto del epígrafe (160 mg, 40 %) como un aceite amarillo claro. LC-MS: 388 [M+H]+.
b) hidrocloruro de (S)-(3-(tiofen-2-il)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución del compuesto (S)-((3-(tiofen-2-il)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (160 mg, 0,41 mmol) y TFA (0,6 ml) en 5 ml de DCM se agitó a temperatura ambiente durante 2 horas. El disolvente se evaporó a 40°C a presión reducida y el residuo se trató con HCl 2 M en Et2O, a continuación se lavó con Et2O (10 ml) para dar el compuesto del epígrafe (38,2 mg, 29 %) como un sólido blanco. LC-MS: 288,1 [M+H]+. RMN 1H (400 MHz, DMSO-d6): 57,99 (s an, 3H), 7,67 (d, 1H, J=8), 7,57 (d, 1H, J=8), 7,29 (d, 1 H, J=4), 7,18 (d, 1H, J=4), 7,03 (d, 1H, J=12), 6,02-5,96 (m, 1H), 4,72-4,68 (m, 1H), 4,41 (s an, 2H), 4,29-4,18 (m, 1H), 3,00-2,93 (m, 1H), 2,65-2,52 (m, 1H).
Ejemplo 5 ((2S)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución de 2-bromo-3-hidroxibenzaldehído (6,0 g, 29,85 mmol), 1-cloropropan-2-ol (8,46 g, 89,55 mmol) y K2CO3 (8,24, 59,7 mmol) en DMF (100 ml) se agitó a 100°C durante la noche. A continuación, la mezcla de reacción se desactivó al añadir agua (4 l) y a continuación se extrajo con EtOAc (3x1,5 l). Las capas orgánicas combinadas se lavaron con salmuera (250 ml), se secaron sobre Na2SO4 anhidro y se concentraron hasta sequedad a vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo: acetato de etilo = 5:1 a 2:1) para dar el compuesto buscado en bruto (8,77 g). MS (ESI) m/z =259/261 [M H]+.
b) 3-(2-hidroxipropoxi)-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzaldehído

Una solución de 2-bromo-3-(2-hidroxipropoxi)benzaldehído (8,77 g, 34 mmol), 4,4,4',4',5,5,5',5'-octametil-2,2'-bi(1,3,2-dioxaborolano) (17,27 g, 68 mmol), Pd(dppf)Cl2 (2,49 g, 3,4 mmol) y KOAc (9,99 g, 102 mmol) en dioxano (200 ml) se agitó a 100°C durante la noche. A continuación, la mezcla de reacción se desactivó al añadir agua (200 ml) y a continuación se extrajo con EtOAc (3x200 ml). Las capas orgánicas combinadas se lavaron con salmuera (250 ml), se secaron sobre Na2SO4 anhidro y se concentraron hasta sequedad a vacío. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo: acetato de etilo = 5:1 a 1:1) para dar el compuesto buscado en bruto (6 g). MS (ESI) m/z =307 [M H]+.
c) 8-metil-2-(nitrometil)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azuleno

Se añadió nitrometano (1,2 g, 19,6 mmol) a 5-10°C a una solución de NaOH (261,4 mg, 6,54 mmol) en agua (8 ml). Después de agitar durante 15 min a 5-10°C, se añadió CTAB (0,19 g, 0,52 mmol) a la mezcla de reacción y seguido por la adición de 3-(2-hidroxipropoxi)-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzaldehído (2,0 g, 6,54 mmol) a 5-10°C. La mezcla de reacción se agitó a ta durante 5 h. La mezcla de reacción se acidificó hasta pH=1 usando ácido clorhídrico diluido y se agitó a ta durante la noche. La mezcla de reacción se filtró para dar el compuesto buscado (541 mg, 33%).
d) acetato de (8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución de 8-metil-2-(nitrometil)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azuleno (541 mg, 2,173 mmol) e
hidróxido de paladio (300 mg) en ácido acético (10 ml) se removió bajo 101325 Pa (una atmósfera) de H2 durante la noche a temperatura ambiente. La mezcla se filtró a través de un lecho de Celite y el filtrado se concentró a vacío para dar el compuesto en bruto (350 mg). MS (ESI) m/z = 220 [M H]+.
e) ((8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Se añadió dicarbonato de di-terc-butilo (3,5 g, 16,13 mmol) a la mezcla del compuesto en bruto acetato de (8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina (3,0 g, 10,75 mmol) y trietilamina (6,5 g, 64,5 mmol) en diclorometano (100 ml) a 0°C y la mezcla se agitó durante 2 h a temperatura ambiente. La reacción se desactivó con NaHCO3 sat. (15 ml) y la mezcla resultante se extrajo con EtOAc (3x80 ml), las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a vacío. El residuo se purificó mediante HPLC preparativa usando una columna Daisogel 10p C18 (250 x 50 mm), se eluyó con un gradiente de agua/acetonitrilo (TFA al 0,05%) para dar el producto (700 mg). MS (ESI) m/z = 264 [M-56]+. RMN 1H (300 MHz, DMSO-de): 5 7,44-7,39 (m, 1H), 7,01-6,98 (m, 2H), 6,88-6,85 (m, 1H), 5,24 (m, 1H), 4,52-4,44 (m, 2H), 4,18-4,00 (m, 1H), 3,39 3,36 (m, 1H), 3,15-3,06 (m, 1H), 1,42-1,09 (m, 15H).
f) ((3-bromo-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Se añadió 2,2'-azobis(2-metilpropionitrilo (9,2 mg, 0,056 mmol) a una solución de ((8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (180 mg, 0,564 mmol) y 1-bromopirrolidino-2,5-diona (120 mg, 0,677 mmol) en CH3CN (20 ml) y la mezcla se agitó durante 2 h a 90°C. A continuación, la mezcla de reacción se concentró a alto vacío y el residuo se purificó mediante HPLC preparativa usando una columna Gemini® 5u C18 (150 x 21,2 mm) eluida con un gradiente de agua/acetonitrilo (0,05% TFA) para dar el producto (60 mg). MS (ESI) m/z = 342/344 [M -56]+.
g) ((3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de ((3-bromo-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de tercbutilo (1 eq.), tetrametilestannano (aprox. 3,5-4,5 eq., aprox. 3,9 eq.) y Pd(Ph3)4 (aprox. de 0,01 a 0,5 eq., aprox. 0,1 eq.) en DMF se puede desgasificar con N2. A continuación, la mezcla se puede calentar a entre aproximadamente 50°C y 150°C (aproximadamente 90°C por ejemplo) durante entre aproximadamente 4 horas y 24 horas (aproximadamente 16 horas por ejemplo). La mezcla de reacción se puede enfriar hasta temperatura ambiente y a continuación la mezcla se puede verter en agua, se puede extraer con acetato de etilo y se puede lavar con agua y salmuera. La capa orgánica se puede secar sobre sulfato sódico anhidro, filtrar y concentrar a vacío. El residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo para dar el compuesto del epígrafe.
h) hidrocloruro de (3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución de ((3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamatos de terc-butilo y TFA en 5 ml de DCM se puede agitar a temperatura ambiente durante 2 horas. El disolvente se puede evaporar a presión reducida y el residuo se puede tratar con HCl 2 M en éter, y a continuación se puede lavar con éter para dar el compuesto del epígrafe.
Ejemplo 6 (3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

a) 2-bromo-3-(2-hidroxi-2-metilpropoxi)benzaldehído

Una solución de 2-bromo-3-hidroxibenzaldehído (7,5 g, 37,3 mmol), 1-cloro-2-metilpropan-2-ol (9,4 g, 85,6 mmol) y Na2CO3 (6,7 g, 63,2 mmol) en 70 ml de DMSO se agitó a 140°C durante 3 horas. A continuación, la mezcla se enfrió hasta temperatura ambiente, se vertió en 300 ml de agua, se extrajo con acetato de etilo (600 ml), se lavó con agua (300 ml), salmuera (50 ml), se secó sobre sulfato sódico anhidro. El disolvente se evaporó a 40°C bajo presión reducida y el residuo se purificó mediante cromatografía en gel de sílice, eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:3) para dar el compuesto del epígrafe (9,2 g, 90,3%) como un aceite incoloro. RMN 1H (300 MHz, CDCh): 510,43 (s, 1H), 7,54 (dd, 1H, J1=3,0, J2=7,5), 7,40-7,34 (m, 1H), 7,54 (dd, 1H, J1=3, J2=7,5), 3,90 (s, 2H), 1,2 (s, 6H).
b) 3-(2-hidroxi-2-metilpropoxi)-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzaldehído

Una solución de 2-bromo-3-(2-hidroxi-2-metilpropoxi)benzaldehído (9,2 g, 33,7 mmol), Pin2B2 (17,1 g, 67,4 mmol), KOAc (9,9 g, 101,1 mmol) y Pd(dppf)Cl2 (2,5 g) en 240 ml de 1,4-dioxano se desgasificó con N2 seis veces. A continuación, la reacción se agitó a 99°C bajo nitrógeno durante 16 horas. La reacción se enfrió, se filtró, a continuación se evaporó a 40°C bajo presión reducida y el residuo se purificó mediante cromatografía en gel de sílice, eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:5) para dar el compuesto del epígrafe (10 g, en bruto) incluyendo el subproducto desbromado (usado directamente en la siguiente etapa sin purificación adicional).
c) 8,8-dimetil-2-(nitrometil)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azuleno

Se añadió una solución de NaOH (1,25 g, 31,3 mmol) en 60 ml de agua a temperatura ambiente a una solución
agitada de 3-(2-hidroxi-2-metilpropoxi)-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzaldehído (10 g, 31,3 mmol) y CH3NO2 (5,7 g, 93,8 mmol) en 100 ml de THF. A continuación, la reacción se agitó a temperatura ambiente durante 16 horas. A continuación, la reacción se acidificó mediante HCl conc. hasta pH=1 a 0°C y se agitó a temperatura ambiente durante 1 hora. La mezcla se extrajo con acetato de etilo (100 ml), se lavó con agua (30 ml), a continuación salmuera (30 ml), se secó sobre sulfato sódico anhidro. El disolvente se evaporó a 40°C bajo presión reducida y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:10) para dar el compuesto del epígrafe (3 g, 36,5%) como un aceite incoloro.
d) acetato de (8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución de 8,8-dimetil-2-(nitrometil)-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azuleno (1 g, 3,8 mmol) y Pd(OH)2 (10%, 0,2 g) en 20 ml de ácido acético se hidrogenó a 101325 Pa (1 atm de) H2 a durante 16 horas. A continuación, la mezcla se filtró y el disolvente se evaporó a 40°C bajo presión reducida para dar el compuesto del epígrafe (0,9 g, en bruto) como un aceite (sal de acetato). LC-MS: 234,1 [m H]+.
e) ((8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Se añadió Et3N (0,61 g, 6,0 mmol) a una solución agitada de acetato de (8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9aborabenzo[cd]azulen-2-il)metanamina (0,7 g, 2,39 mmol) en 70 ml de CH2Cl2 enfriado hasta 0°C. A continuación, se añadió Boc2O (0,98 g, 4,5 mmol) en una porción y la reacción se agitó a temperatura ambiente durante 16 horas. La mezcla se lavó con HCl 0,3 N (30 ml), agua (30 ml) y se secó sobre sulfato sódico anhidro. El disolvente se evaporó a 40°C a presión reducida y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:4) para dar el compuesto del epígrafe (0,63 g, 79%) como un aceite. LC-MS: 234,1 [M+H-100]+.
f) ((3-bromo-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de ((8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (232 g, 0,70 mmol), NBS (143 mg, 0,80 mmol) y AIBN (20 mg) en 30 ml de acetonitrilo se agitó a reflujo durante 1 hora. El disolvente se evaporó a 40°C a presión reducida y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:4) para dar el compuesto del epígrafe (260 mg, 88,6%) como un sólido. LC-MS: 312,0/314,0 [M+H-100]+.
g) ((3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de ((3-bromo-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (1 eq.), tetrametilestannano (aprox. 3,5-4,5 eq., aprox. 3,9 eq.) y Pd(Ph3)4 (aprox. de 0,01 a 0,5 eq., aprox.
0,1 eq.) en DMF se puede desgasificar con N2. A continuación, la mezcla se puede calentar a entre aproximadamente 50°C y 150°C (aproximadamente 90°C, por ejemplo) durante entre aproximadamente 4 horas y 24 horas (aproximadamente 16 horas, por ejemplo). La mezcla de reacción se puede enfriar hasta temperatura ambiente y a continuación la mezcla se puede verter en agua, se puede extraer con acetato de etilo y se puede lavar con agua y salmuera. La capa orgánica se puede secar sobre sulfato sódico anhidro, filtrar y concentrar a vacío. El
residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo para dar el compuesto del epígrafe.
h) hidrocloruro de (3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Una solución de ((3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo y TFA en DCM se puede agitar a temperatura ambiente durante 2 horas. El disolvente se puede evaporar a presión reducida y el residuo se puede tratar con HCl 2 M en éter, a continuación se puede lavar con éter para dar el compuesto del epígrafe.
Ejemplo 7 (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

a) (S)-((3-bromo-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de ((8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (5,5 g, 16,5 mmol) y NBS (3,2 g, 18,2 mmol) en 100 ml de dicloroetano se agitó a 50°C durante 18 horas. El disolvente se evaporó a 40°C bajo presión reducida y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo (1:10) para dar el compuesto del epígrafe (5,9 g, 86,5%) como un aceite. El compuesto racémico se separó a través de SFC (columna quiral CHIRALPAK AD-H, eluida con EtOH (20%) y CO2 (80%)) para dar 2,2 g de isómero (S) (isómero que se eluye en primer lugar, RT = 3,0 min) y 2,2 g de isómero (R) (isómero que se eluye en segundo lugar, RT = 4,1 min). LC-MS: 312,0/314,0 [M+H-100]+.
b) (S)-((3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de (S)-((3-bromo-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (1 eq.), tetrametilestannano (aprox. 3,5-4,5 eq., aprox. 3,9 eq.) y Pd(Ph3)4 (aprox. de 0,01 a 0,5 eq., aprox. 0,1 eq.) en DMF se puede desgasificar con N2. A continuación, la mezcla se puede calentar a entre aproximadamente 50°C y 150°C (aproximadamente 90°C, por ejemplo) durante entre aproximadamente 4 horas y 24 horas (aproximadamente 16 horas, por ejemplo). La mezcla de reacción se puede enfriar hasta temperatura ambiente y a continuación la mezcla se puede verter en agua, se puede extraer con acetato de etilo y se puede lavar con agua y salmuera. La capa orgánica se puede secar sobre sulfato sódico anhidro, filtrar y concentrar vacío. El residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo para dar el compuesto del epígrafe.
c) hidrocloruro de (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-y/)metanamina

Una solución de (S)-((3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de tercbutilo y TFA en dCm se puede agitar a temperatura ambiente durante entre aproximadamente 10 minutos y 10 horas, por ejemplo, aproximadamente 2 horas. El disolvente se puede evaporar a una presión reducida y el residuo se puede tratar con HCl 2 M en éter, y a continuación se puede lavar con éter para dar el compuesto del epígrafe.
Ejemplo Comparativo 8 (S)-(3-etil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina
Ejemplo Comparativo 9 (S)-(3-vinil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 10 (S)-(3-propil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 11 (S)-(3-isopropil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 12 (S)-(3-alil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 13 (S)-(3-ciclopropil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Los Ejemplos Comparativos 8-13 se pueden sintetizar utilizando el procedimiento descrito en el Ejemplo 2 -- (S)-(3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, sustituyendo el compuesto de estaño tetrametilestannano en la etapa b) del Ejemplo 2 por los siguientes compuestos de estaño: Ejemplo Comparativo 8-tetraetilestannano o etiltri(n-butil)estannano; Ejemplo Comparativo 9-tetravinilestannano o viniltri(n-butil)estannano; Ejemplo Comparativo 10 -tetrapropilestannano o propiltri(n-butil)estannano; Ejemplo Comparativo 11-tetraisopropilestannano o isopropiltri(n-butil)estannano; Ejemplo Comparativo 12-tetraalilestannano o aliltri(n-butil)estannano; y Ejemplo Comparativo 13-tetraciclopropilestannano o ciclopropiltri(n-butil)estannano.
Ejemplo Comparativo 14 ((2S)-3-etil-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 15 ((2S)-3-vinil-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 16 ((2S)-3-propil-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 17 ((2S)-3-isopropil-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina
Ejemplo Comparativo 18 ((2S)-3-alil-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 19 ((2S)-3-ciclopropil-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Los Ejemplos Comparativos 14-19 se pueden sintetizar utilizando el procedimiento descrito en el Ejemplo 5 -- ((2S)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, sustituyendo el compuesto de estaño tetrametilestannano en la etapa g) del Ejemplo 5 por los siguientes compuestos de estaño: Ejemplo 14-tetraetilestannano o etiltri(n-butil)estannano; Ejemplo 15-tetravinilestannano o viniltri(n-butil)estannano; Ejemplo 16-tetrapropilestannano o propiltri(n-butil)estannano; Ejemplo 17-tetraisopropilestannano o isopropiltri(n-butil)estannano; Ejemplo 18-tetraalilestannano o aliltri(n-butil)estannano; y Ejemplo 19-tetraciclopropilestannano o ciclopropiltri(n-butil)estannano.
Ejemplo Comparativo 20 (S)-(3-etil-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 21 (S)-(3-vinil-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 22 (S)-(3-propil-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina
Ejemplo Comparativo 23 (S)-(3-isopropil-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina
Ejemplo Comparativo 24 (S)-(3-alil-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina Ejemplo Comparativo 25 (S)-(3-ciclopropil-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina

Los Ejemplos Comparativos 20-25 se pueden sintetizar utilizando el procedimiento descrito en el Ejemplo 7 -- (S)-(3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metanamina, sustituyendo el compuesto de estaño tetrametilestannano en la etapa b) del Ejemplo 7 por los siguientes compuestos de estaño: Ejemplo Comparativo 20-tetraetilestannano o etiltri(n-butil)estannano; Ejemplo Comparativo 21-tetravinilestannano o viniltri(n-butil)estannano; Ejemplo Comparativo 22-tetrapropilestannano o propiltri(n-butil)estannano; Ejemplo Comparativo 23-tetraisopropilestannano o isopropiltri(n-butil)estannano; Ejemplo Comparativo 24-tetraalilestannano o aliltri(n-butil)estannano; y Ejemplo Comparativo 25-tetraciclopropilestannano o ciclopropiltri(n-butil)estannano.
Ejemplo 27 ((2S,8R)-2-(aminometil)-3-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-8-il)metanol

a) (S)-5-((2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2- 

Una solución de 5-hidroxi-2-metilbenzaldehído (1 eq., 13,6 mmol), (R)- 4-metilbencenosulfonato de (2,2-dimetil-1,3-dioxolan-4-il)metilo (aprox. 1,1 eq.) y K2CO3 (aprox. 1,25 eq.) en DMSO se puede agitar a entre aproximadamente 50°C y 150°C (aproximadamente 70°C, por ejemplo) durante entre aproximadamente 4 horas y 24 horas (aproximadamente 16 horas, por ejemplo). A continuación, la mezcla se puede verter en agua, se puede extraer con acetato de etilo, se puede lavar con agua y salmuera y se puede secar sobre sulfato sódico anhidro. El disolvente se puede evaporar bajo presión reducida y el residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo para proporcionar el producto.
b) (S)-1 -(5-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilfenil)-2-nitroetan-1 -ol

Una mezcla de acetato de cobre (II) (1 eq.), (1R)-1,7,7-trimetil-N-(piridin-2-ilmetil)biciclo[2,2.1]heptan-2-amina (1,1 eq.) en etanol se puede agitar a t. a. durante aprox. 1 h, a continuación se puede añadir una solución de (S)-5-((2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilbenzaldehído (10 eq.) en etanol. La mezcla de reacción se puede enfriar hasta entre aproximadamente -35°C y aproximadamente -40°C y a continuación se añadió nitrometano (100 eq.) gota a gota, manteniendo la temperatura por debajo de aproximadamente -35°C, seguido por la adición de diisopropiletilamina (2,2 eq.). La reacción se puede agitar a -35°C durante 24 h y a continuación se puede desactivar con ácido trifluoroacético (2,2 eq.). Se puede añadir EtOAc a la solución resultante. La fase orgánica separada se puede lavar con agua y a continuación concentrarse bajo vacío. El residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo para proporcionar el producto. c) (S)-2-amino-1 -(5-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilfenil)etan-1 -ol

Una solución de (S)-1-(5-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilfenil)-2-nitroetan-1-ol y Pd/C en metanol se puede hidrogenar bajo 101325 Pa (1 atm de) H2 a temperatura ambiente durante aproximadamente 48 h. A continuación, se puede filtrar a través de un lecho de Celite y el filtrado se puede concentrar bajo presión reducida para proporcionar el producto en bruto. Se puede usar directamente en la siguiente etapa sin purificación adicional.
d) (S)-2-(dibencilamino)-1 -(5-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilfenil)etan-1 -ol

Se pueden añadir K2CO3 (2 eq.) y BnBr (2 eq.) a una solución agitada de (S)-2-amino-1-(5-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilfenil)etan-1-ol (1 eq.) en EtOH. La mezcla de reacción se puede agitar durante la noche a temperatura ambiente. El disolvente se puede retirar bajo presión reducida y el residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo para proporcionar el producto.
e) (S)-3-((dibencilamino)metil)-7-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-4-metilbenzo[c][1,2]oxaborol-1 (3H)-ol

Se puede añadir n-BuLi (2,5 M en hexano, 7 eq.) gota a gota a lo largo de aproximadamente 30 minutos a una solución de (S)-2-(dibencilamino)-1-(5-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-2-metilfenil)etan-1-ol (1 eq.) en tolueno seco a aproximadamente -30°C bajo atmósfera de N2. Después de la adición, la mezcla se puede agitar a aproximadamente 0°C durante otras aproximadamente 2 h y a continuación enfriar hasta aproximadamente -70°C; se puede añadir gota a gota borato de trimetilo manteniendo la temperatura por debajo de aproximadamente -50°C. Después de la adición, la mezcla de reacción se puede dejar calentar hasta aproximadamente -40°C durante aproximadamente 3 h y a continuación calentar hasta t. a. y agitar durante la noche. La reacción se puede desactivar con NaHCO3 acuoso al 5% y agitar vigorosamente durante aproximadamente 15 min, la suspensión resultante se puede filtrar y el filtrado se puede separar. La capa orgánica se puede lavar con agua y se puede concentrar a vacío para proporcionar el producto en bruto.
f) Compuesto del epígrafe

Una solución de (S)-3-((dibencilamino)metil)-7-(((S)-2,2-dimetil-1,3-dioxolan-4-il)metoxi)-4-metilbenzo[c][1,2]oxaborol-1(3H)-ol (1 eq.) y Pd/C (10 %) en metanol con 2 ml de HCI conc. se puede hidrogenar bajo 101325 Pa (1 atm de) H2 a temperatura ambiente durante aproximadamente 48 h. A continuación, se puede filtrar a través de un lecho de Celite y el filtrado se puede concentrar a presión reducida para dar un aceite. El producto en bruto se puede purificar mediante HPLC preparativa usando una columna Daisogel ^ C18 (250 x 50 mm) y se puede eluir con un gradiente de agua/acetonitrilo (TFA al 0,05%). La fracción recogida se puede concentrar bajo presión reducida. El residuo se puede disolver en éter y HCl sat. (g) en éter y la mezcla se puede agitar a temperatura ambiente durante aproximadamente 1 h. El sólido se puede recoger mediante filtración y lavar con éter para dar el compuesto del epígrafe.
Ejemplo Comparativo 28 ((2S,8R)-2-(aminometil)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-8-il)metanol
Ejemplo Comparativo 29 ((2S,8S)-2-(aminometil)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-8il)metanol

a) ((2 -metilaliloxi)metil)benceno

Una solución de alcohol metalílico (80 g, 1,1 mol) en THF (100 ml) se añadió gota a gota a una suspensión de NaH (66 g, 1,65 mol) en THF (800 ml) a 25°C bajo argón. Después de 1 h, se añadió lentamente una solución de bromuro de bencilo (207 g, 1,2 mol) en THF (100 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 12 h. La mezcla de reacción se desactivó con solución saturada de NH4Cl (200 ml) y se extrajo con acetato de etilo (3 x 200 ml). Las capas orgánicas combinadas se lavaron con agua (100 ml) y salmuera (100 ml), se secaron sobre Na2SO4. El disolvente se retiró bajo presión reducida. El residuo se destiló para proporcionar el producto deseado (134 g, 74%) como un aceite incoloro. RMN 1H (400 MHz, CDCh): ó 7,40 - 7,29 (m, 5H), 5,05 (s, 1H), 4,97 (s, 1H), 4,54 (s, 2H), 3,98 (s, 2H), 1,82 (s, 3H).
b) (2 -(benciloximetil)-2 -metiloxirano

Se disolvió ((2-metilaliloxi)metil)benceno (41,5 g, 256 mmol) en DCM (1200 ml) y se enfrió hasta 0°C. Se añadió m-CPBA (69,7 g, 384 mmol) y la mezcla se agitó durante la noche a temperatura ambiente durante 12 h. Después de que el precipitado blanco se separara por filtración, el filtrado se lavó con solución saturada de Na2CO3 (200 ml), H2O (200 ml) y salmuera. Después de que el disolvente se retirara bajo presión reducida, el residuo en bruto se purificó mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo (1 :20) para proporcionar el producto puro (20 g, 44%) como un aceite incoloro. RMN 1H (400 MHz, CDCh): ó 7,40 - 7,29 (m, 5H), 4,60 (q, J = 12,0 Hz, 2H), 3,61 (d, J = 11,0 Hz, 1H), 3,48 (d, J = 11,0 Hz, 1H), 2,78 (d, J = 4,9 Hz, 1H), 2,66 (d, J = 4,9 Hz, 1 H), 1,43 (s, 3H).
c) 3-(3-(benciloxi)-2-hidroxi-2-metilpropoxi)-2-bromobenzaldehído

Se añadió K2CO3 (42 g, 304,3 mmol), seguido por 2-bromo-3-hidroxibenzaldehído (30 g, 149,3 mmol) a una solución de (2-(benciloximetil)-2-metiloxirano (26 g, 145,9 mmol) en DMF (700 ml). La suspensión se agitó a 90°C durante 6 h. La mezcla se enfrió hasta temperatura ambiente, se diluyó con salmuera y se extrajo con acetato de etilo (200 ml x 3). El disolvente orgánico se retiró bajo vacío y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo (1:20) para proporcionar el producto puro (27 g, 49%) como un aceite amarillo claro. RMN 1H (400 MHz, DMSO-de): ó 10,29 (s, 1H), 7,512 - 7,41 (m, 3H), 7,31 - 7,23 (m, 5H), 4,91 (s, 1H), 4,53 (dd, J1= 12,4 Hz, J2 = 17,2 Hz, 2H), 4,06 (d, J = 9,2 Hz, 1H), 3,91 (d, J = 9,2 Hz, 1H), 3,54 (d, J = 9,3 Hz, 1 H), 3,47 (d, J = 9,3 Hz, 1H), 1,27 (s, 3H).
d) 3-(3-(benciloxi)-2-hidroxi-2-metilpropoxi)-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzaldehído

Una solución de 3-(3-(benciloxi)-2-hidroxi-2-metilpropoxi)-2-bromobenzaldehído (21,3 g, 56,2 mmol), Pin2B2 (28,6 g, 112,4 mmol), KOAc (6,1 g, 61,9 mmol), PdCl2(dppf) - DCM (1,23 g, 1,7 mmol) en DMF (150 ml) se desgasificó 3 veces con nitrógeno. La mezcla se calentó a 90°C durante 16 h. Después de que la reacción se tratara con acetato de etilo y salmuera, el residuo se purificó mediante cromatografía en gel de sílice eluyendo con acetato de etilo y
éter de petróleo (1:20) para proporcionar el producto deseado (15,3 g, 64%) como un aceite amarillo claro. LC-MS: 367,1 [344+Na]+
e) (3-(benciloxi)-2-hidroxi-2-metilpropoxi)-3-(nitrometil)benzo[c][1,2] oxaborol-1 (3H)-ol

Se añadió una solución de NaOH (1,76 g, 44,1 mmol) en agua (100 ml) a una solución enfriada con hielo de 3-(3-(benciloxi)-2-hidroxi-2-metilpropoxi)-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzaldehído (18,8 g, 44,1 mmol) en THF. Después de agitar durante 15 min, se añadió CH3NO2 (3,3 g, 53 mmol) y la mezcla se agitó a temperatura ambiente durante 15 h. La solución de reacción se acidificó con AcOH hasta pH 3-5. La suspensión se extrajo con acetato de etilo (50 ml x 3). La capa orgánica combinada se evaporó bajo vacío y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo (1 :10 ) para proporcionar el producto puro (6,8 g, 40%) como un aceite incoloro. LC-MS: 386,0 [M-1]-f) acetato de (2-(aminometil)-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-8-il)metanol

Se añadió Pd(OH)2/C (200 mg) a una solución de 7-(3-(benciloxi)-2-hidroxi-2-metilpropoxi)-3-(nitrometil)benzo[c][1,2]oxaborol-1(3H)-ol (1 g, en bruto) en AcOH (20 ml). La solución se desgasificó 3 veces con H2 y se agitó a temperatura ambiente durante 12 h. La mezcla de reacción se filtró a través de Celite y el filtrado se concentró bajo vacío para proporcionar el producto en bruto (1 g, en bruto) como un sólido amarillo.
g) ((8-(hidroximetil)-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Se añadió NaHCO3 (437 mg, 5,2 mmol) a una solución de acetato de (2-(aminometil)-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-8-il)metanol (650 mg, 2,1 mmol) en t-BuOH (10 ml) y H2O (10 ml) a temperatura ambiente. Después de agitar durante 15 min, se añadió (Boc)2O (854 mg, 3,9 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. La mezcla se acidificó con AcOH hasta pH 6-7 y se extrajo con DCM (30 ml x 3). Las capas orgánicas combinadas se evaporaron bajo vacío y el residuo se purificó mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo (1:3) para proporcionar el producto deseado (400 mg, 55%) como un aceite incoloro. LC-MS: 294,1 [M-55]+
h) ((3-bromo-8-(hidroximetil)-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Se añadió NBS (102 mg, 0,57 mmol) a una solución de ((8-(hidroximetil)-8-metil-7,8-dihidro-2H-1,6,9-trioxa-9aborabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (200 mg, 0,57 mmol) en ACN (10 ml) y la solución se agitó a 90°C durante 1 h. La reacción se desactivó con solución de NH4Cl, se extrajo con acetato de etilo (20 ml x 3). La capa orgánica se lavó con salmuera, se secó sobre Na2SO4, se concentró a vacío. El residuo en bruto se purificó mediante cromatografía en gel de sílice eluyendo con acetato de etilo y éter de petróleo (1:3) para proporcionar el producto (230 mg, en bruto) como un sólido claro. LC-MS: 328,1 [M-Boc+H]+.
i) ((8-(hidroximetil)-3,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo

Una solución de ((3-bromo-8,8-dimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de terc-butilo (1 eq.), tetrametilestannano (aprox. 3,5-4,5 eq., aprox. 3,9 eq.) y Pd(Ph3)4 (aprox. de 0,01 a 0,5 eq., aprox.
0,1 eq.) en DMF se puede desgasificar con N2. A continuación, la mezcla se puede calentar a entre aproximadamente 50°C y 150°C (aproximadamente 90°C, por ejemplo) durante entre aproximadamente 4 horas y 24 horas (aproximadamente 16 horas, por ejemplo). La mezcla de reacción se puede enfriar hasta temperatura ambiente y a continuación la mezcla se puede verter en agua, se puede extraer con acetato de etilo y se puede lavar con agua y salmuera. La capa orgánica se puede secar sobre sulfato sódico anhidro, filtrar y concentrar a vacío. El residuo se puede purificar mediante cromatografía en gel de sílice eluyendo con una mezcla de acetato de etilo y éter de petróleo para dar el compuesto del epígrafe.
j) Compuestos del epígrafe

Se puede disolver ((3,8,8-trimetil-7,8-dihidro-2H-1,6,9-trioxa-9a-borabenzo[cd]azulen-2-il)metil)carbamato de tercbutilo (en bruto) en una solución de TFA en DCM (de aproximadamente 1:2 a aproximadamente 1:20; 1:10 por ejemplo). La solución se puede agitar a temperatura ambiente durante entre aproximadamente 10 minutos y 10 horas, 1 hora por ejemplo, y a continuación se puede concentrar a vacío. El producto en bruto se puede purificar mediante HPLC preparativa usando una columna Daisogel 10|j C18 y se puede eluir con un gradiente de agua/acetonitrilo (TFA al 0,05%). La fracción recogida se puede concentrar bajo presión reducida para proporcionar los compuestos del epígrafe.
Ensayos in Vitro
Ejemplo 30
Determinación de la CIM frente a micobacterias
La medida de la concentración inhibidora mínima (CIM) frente a cepas de M. tuberculosis para cada compuesto probado se realizó en placas de microvaloración de poliestireno de fondo plano de 96 pocillos en un volumen final de 100 |jl. Se realizaron diez diluciones dobles de fármaco en DMSO puro partiendo de 50 mM. Las soluciones de fármaco se añadieron a medio Middlebrook 7H9 (Difco) y se usó isoniacida (INH) (Sigma Aldrich) como un control positivo con diluciones dobles de INH partiendo de 160 ug/ml. El inóculo se estandarizó hasta aproximadamente 1x107 cfu/ml y se diluyó 1 en 100 en caldo Middlebrook 7H9 (Difco). Este inóculo (100 ^l) se añadió a toda la placa, pero los pocillos G-12 y H-12 se usaron como controles en blanco. Todas las placas se pusieron en una caja cerrada herméticamente para prevenir el secado de los pocillos periféricos y se incubaron a 37°C sin remoción durante seis días. Se preparó una solución de Resazurin al disolver un comprimido de Resazurin (Resazurin Tablets for Milk Testing; Ref 330884Y' VWR International Ltd) en 30 ml de PBS (solución salina tamponada con fosfato) estéril. De esta solución, se añadieron 25 ^l a cada pocillo. La fluorescencia se midió (Spectramax M5 Molecular Devices, Excitación 530 nm, Emisión 590 nm) después de 48 horas para determinar el valor de CIM.
Ejemplo 31
Ensayo general de actividad antimicrobiana
La actividad antimicrobiana en células enteras se determinó mediante microdilución del caldo usando el procedimiento recomendado por the Clinical and Laboratory Standards Institute (CLSI), Documento M7-A7, "Methods for Dilution Susceptibility Tests for Bacteria that Grow Aerobically".
La Tabla 1 proporciona valores de CIM frente a cepas bacterianas K12; E. coli K12 toIC/Tn10; A. baumannii ATCC 17978; y P. aeruginosa PA01 para compuestos de comparación (C1-C15) mostrados en la Tabla 4 y para G26-CH3. Como se puede observar en la Tabla 1, generalmente, los compuestos de comparación no poseen actividad significativa en varias bacterias gramnegativas patógenas, así como una E. coli deficiente en la bomba de descarga.
De forma similar, el compuesto G26-CH3 (Ejemplo 2) tiene escasa actividad frente a estas bacterias gramnegativas. La Tabla 1 proporciona valores de CIM frente a cepas no micobacterianas para compuestos de benzoxaborol sustituido

Ejemplo 32
Expresión y purificación de LeuRS
Para análisis bioquímicos, una LeuRS con seis etiquetas de histidina N-terminales se sobreexpresó en Escherichia coli que era E. coli optimizada codónicamente (GenScript, Piscataway NJ, EE. UU. de A.), a partir de mitocondria y citoplasma humanos y M. tuberculosis. Las proteínas LeuRS con seis etiquetas de histidina N-terminales se sobreexpresaron y purificaron según Novagen (Madison, WI, EE. UU. de A.) usando una cepa de expresión de ARN polimerasa de E. coli BL21 (DE3) T7.
Ejemplo 34
Ensayo de aminoacilación
Los experimentos se realizaron en placas de microvaloración de 96 pocillos, usando 80 pl de mezclas de reacción que contenían HEPES-KOH 50 mM (pH 8,0), MgCl2 30 mM, KCl 3o mM, L-[14C]leucina 13 pM (306 mCi/mmol, Perkin-Elmer), ARNt de E. coli total (Roche, Suiza) 15 pM, bSa al 0,02% (p/v) BSA, DTT 1 mM, LeuRS 0,2 pM y ATP 4 mM a 30°C. Las reacciones se iniciaron mediante la adición de ATP 4 mM. Después de 7 minutos, las reacciones se desactivaron y el ARNt se precipitó mediante la adición de 50 pl de TCA al 10% (p/v) y se transfirieron a placas filtrantes de membrana de nitrocelulosa de 96 pocillos (Millipore Multiscreen HTS, MSHAN4B50). A continuación, cada pocillo se lavó tres veces con 100 pl de TCA al 5%. A continuación, las placas filtrantes se secaron bajo una lámpara térmica y la L-[14C]leucina y Leu de ARNt precipitadas se cuantificaron mediante recuento por centelleo de líquidos usando un contador de centelleo de líquidos Wallac MicroBeta Trilux modelo 1450 (PerkinElmer, Waltham, MA, EE. UU. de A.). La única diferencia era con la LeuRS citoplásmica humana cuando se usaba ARNt aislado de levadura de cerveza (Roche Diagnostics GmbH).
Ejemplo 35
Determinación de CI50
Para determinar la concentración de inhibidor, que reduce la actividad enzimática en 50% (CI50), concentraciones
crecientes de compuesto (Anacor Pharmaceuticals Inc., Palo Alto, CA, EE. UU. de A.) se incubaron con enzima LeuRS, ARNt y L-leucina 20 minutos. Las reacciones se iniciaron mediante la adición de ATP 4 mM. Las reacciones se detuvieron después de 7 minutos y a continuación se precipitaron y se contaron para cuantificar la radiactividad. Los valores de CI50 se determinaron usando el programa informático Graphpad Prism (Graphpad Software Inc. (La Jolla, CA, EE. UU. de A.).
Ejemplo 36
Ensayo de citotoxicidad de HepG2
Se alimentaron células HepG2 (HB-8065) a medio reciente (Essential Minimum Eagle Medium, EMEM, complementado con 5% de suero de ternero fetal y L-glutamina 2 mM) el día antes de subcultivar las placas. El día de la siembra de las placas, se preparó una suspensión celular de 100.000 células/ml en medio de cultivo. La suspensión celular (100 pl) se añadió a cada pocillo de una microplaca negra de 96 pocillos con fondo transparente revestida con colágeno (Becton Dickinson), excepto en la columna 11, en la que se distribuyeron solamente 100 pl de medio de cultivo. Las placas se incubaron durante 24 h. Se formaba una gama de 10 dosis de sustancias de prueba al preparar diluciones en serie 1:2 a partir de la solución madre en DMSO al 100% y se formaba una dilución de 1:200 de cada dosis en medio, para alcanzar una concentración final de 0,5% de DMSO. Después de 24 h, el medio de cultivo se retiró de la placa y 150 ul de diluciones del compuesto de prueba se añadieron en dos réplicas y 150 pl de DMSO al 0,5% en medio de cultivo a las columnas 11 y 12 (control en blando). Las placas se incubaron durante 48 h y a 37°C, CO2 al 5%, 95% de humedad relativa. A continuación, el medio se retiró y se añadieron 200 pl de medio de cultivo reciente y 50 pl de solución de Resazurin a cada pocillo y se incubaron durante 1 h y media. Las placas se retiraron de la incubadora para dejar que la fluorescencia se estabilizara a temperatura ambiente protegidas de la luz durante 15 min. Para cada lectura de la viabilidad de células, se usó Resazurine (BDH). Resazurin se usa como un indicador de oxidación-reducción que da un cambio colorimétrico y una señal fluorescente en respuesta a la actividad metabólica. A medida que la célula crece, la actividad metabólica da como resultado una reducción química de Resazurin indicada por un cambio desde la forma azul no fluorescente hasta la forma rosa fluorescente reducida. Por lo tanto, el grado de fluorescencia de Resazurin es un indicador del número de células viables en el sistema de cultivo. La fluorescencia se midió a una longitud de onda de excitación de 515 nm y una longitud de onda de emisión de 590 nm en un lector de microplacas 1420 Multilabe1HTS counter, Victor 2, (Wallac).
El valor de fluorescencia de cada pocillo se corrige al sustraer el valor de fondo (promedio de la columna 11) del valor absoluto. Los porcentajes de inhibición se calculan con relación a los pocillos de control de DMSO (promedio de la columna 12). Para cada compuesto, el valor promedio de las muestras por duplicado se calcula y la curva se ajusta a un ajuste de curva de regresión no lineal de respuesta a la dosis sigmoidea (pendiente variable) (GraphPad) a fin de calcular la CI50 (Tox50).
Ejemplo 37
El efecto de compuestos descritos en la presente memoria frente a Mycobacterium tuberculosis
El comparador de benzoxaborol sustituido C1-C14 se probó con respecto a la actividad antibacteriana frente a una especie de Mycobacterium tuberculosis y también se probó con respecto a la toxicidad para células hepáticas humanas usando células HepG2. El compuesto ejemplar G26-CH3 de la invención se comparó con los compuestos de comparación C1-H a C14-Br, según se muestra en las Tablas 2 en comparación con la Tabla 3.
La Tabla 2 proporciona valores de CI50 de inhibición de LeuRS, valores de CIM frente a la cepa estándar de M. tuberculosis Mtb H37Rv, valores de toxicidad frente a células HepG2 humanas y valores de selectividad para ciertos benzoxaboroles sustituidos


Denominación Estructura CI50 de CI50 de CI50 de CIM de Tox50 48 h Índice de del del Mtb LeuRS cito. LeuRS mito. Mtb de células Selectividad Compuesto Compuesto LeuRS humana humana H37Rv HepG2 (mM) (A/B)
MM MM MM MM B A

La Tabla 3 proporciona valores de CI50 de inhibición de LeuRS, valores de CIM frente a la cepa estándar de M. tuberculosis Mtb H27Rv, valores de toxicidad frente a células HepG2 humanas y valores de selectividad para ciertos compuestos de la invención ejemplificados.
Tabla 3
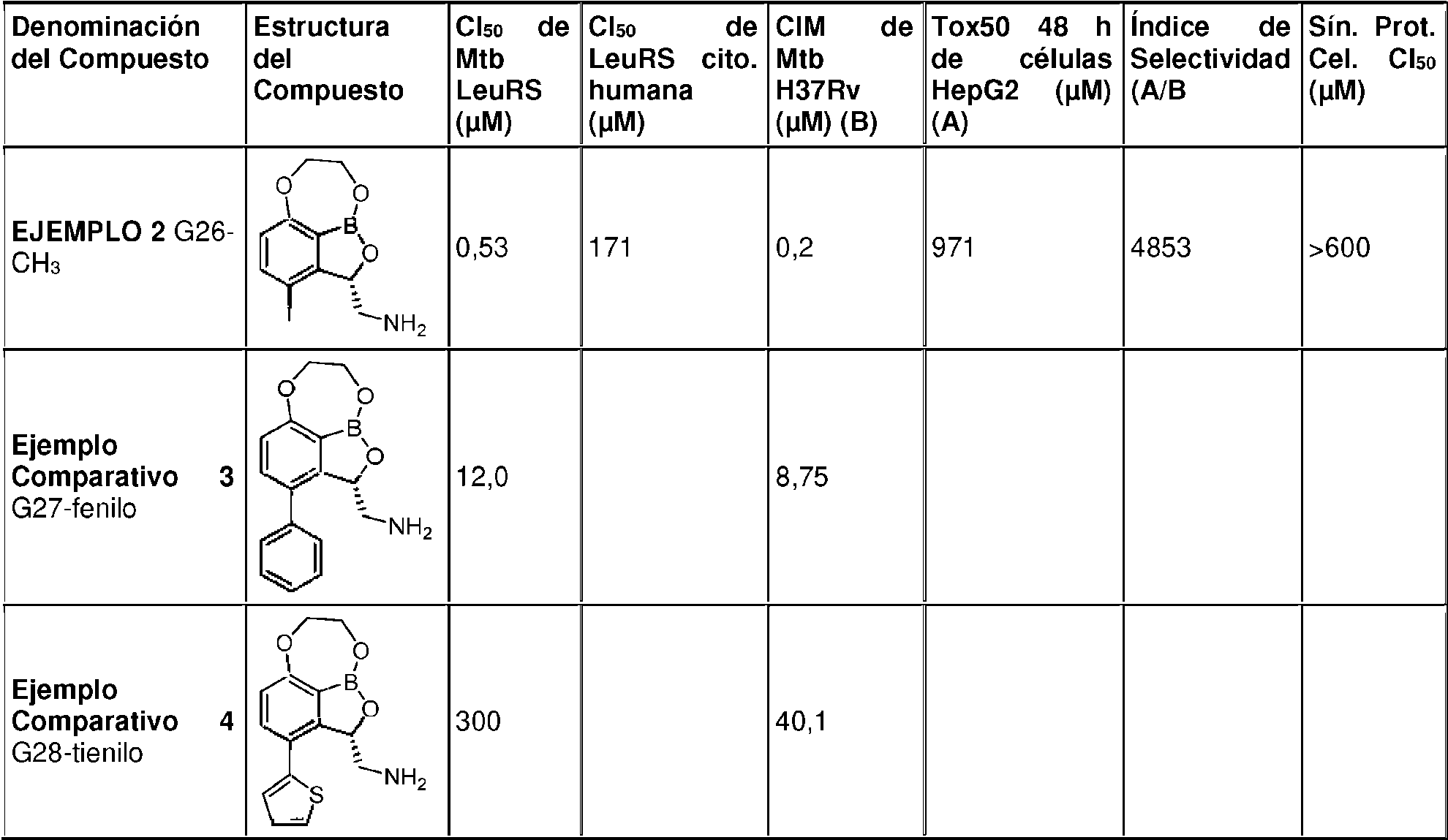
Como se puede observar en la Tabla 3 para los Ejemplos 2 (G26 -CH3), se observaba un enorme incremento de la selectividad para G26-CH3 al inhibir el crecimiento de M. tuberculosis frente a la toxicidad para células HepG2 humanas en comparación con los compuestos de comparación.
Las Tablas 2 y 3 muestran una comparación de ciertos compuestos de comparación de benzoxaborol sustituido (mostrados en forma cerrada) con y sin sustitución con halógeno o alquilo en diversas posiciones en el anillo de benzoxaborol, ciertos benzoxaboroles sustituidos (mostrados en forma cerrada) con y sin sustitución con halógeno o alquilo en la posición 4 de la estructura del anillo de benzoxaborol y ciertos otros compuestos de benzoxaborol sustituidos. A partir de los valores de CIM de Mtb H37Rv (B) y los valores de Tox50 a las 48 h de células HepG2 (A),
es posible determinar la selectividad para la inhibición de M. tuberculosis frente a la inhibición (toxicidad) de células humanas para estos compuestos (véase la segunda columna a la derecha de las Tablas 2 y 3).
Se encontró que el compuesto procedente del Ejemplo 2 G26-CH3 tenía un índice de selectividad (IS) frente a M. tuberculosis de 4853 (véase la Tabla 3) en comparación con los mejores compuestos de comparación C4-Br y C6-CI que exhibían índices de selectividad de 320 y 363, respectivamente (véase la Tabla 2). Además, como se observa en la Tabla 3, se observa que el valor de CI50 para G26-CH3 frente a M. tuberculosis es submicromolar, a 0,53 micromolar. El índice de selectividad (IS) del Ejemplo 2 G26-CH3 frente a M. tuberculosis está inesperadamente mejorado sobre otros benzoxaboroles sustituidos de la Tabla 2. El G26-CH3 del Ejemplo 2, que es un benzoxaborol sustituido que tiene un sustituyente metilo en la posición C-4 del anillo de benzoxaborol y un sustituyente aminometilo en la posición C3 del anillo de benzoxaborol tiene estereoquímica relativa "(S)" en el estereocentro C con el sustituyente aminometilo y sorprendentemente es más selectivo con respecto a la actividad frente a M. tuberculosis que otros benzoxaboroles sustituidos que carecen de algunas de estas características en comparación con la inhibición (toxicidad) de células humanas para estos compuestos. Además, el valor de CIM frente a la cepa de M. tuberculosis H37Rv para el Ejemplo 2 G26-CH3 es 0,2 pM, en contraste con otros benzoxaboroles sustituidos que se comparan.
Así, como se observa en la Tabla 3, se encontró sorprendentemente que los compuestos G26-CH3 del Ejemplo 2 tenían un IS frente a Mycobacterium tuberculosis de 4853. Estos valores del IS son inesperadamente mejores que cualquiera de los compuestos de comparación indicados en la Tabla 2.
La adición de un sustituyente alquilo en el C4 del anillo de benzoxaborol confiere así un incremento en el índice de selectividad en comparación con otros benzoxaboroles sustituidos sin un alquilo en el C4 del anillo de benzoxaborol. La Tabla 3 proporciona valores de CI50 de inhibición de LeuRS, valores de CIM frente a la cepa estándar de M. tuberculosis Mtb H37Rv, valores de toxicidad frente a células HepG2 humanas, valores del índice de selectividad (IS) y valores de la inhibición de la síntesis de proteínas en células de mamífero para un benzoxaborol sustituido de la invención en donde el compuesto (G26-CH3, mostrado en la forma tricíclica cerrada) tiene un metilo en el C4 del anillo de benzoxaborol y un sustituyente aminometilo en la posición C3 del anillo de benzoxaborol que tienen estereoquímica relativa "(S)" en ese estereocentro.
Como se puede observar en la Tabla 3, también se encontró que G26-CH3 tiene buena actividad frente a leucil ARNt sintetasa procedente de M. tuberculosis y exhibía un bajo impacto sobre la síntesis de proteínas celulares de mamífero.
Sorprendentemente, se ha encontrado que ciertos benzoxaboroles sustituidos que son capaces de existir en un equilibrio, en ciertas condiciones de disolvente, entre una forma abierta y una forma cerrada, en donde el compuesto en la forma cerrada tiene un tercer anillo que implica a las posiciones 1 y 7 del anillo de benzoxaborol, exhiben un incremento inesperado en el índice de selectividad. C4-Br, el enantiómero (S) de un compuesto de comparación de benzoxaborol sustituido con un Br en la posición C4 del anillo de benzoxaborol que no es capaz de existir en un equilibrio entre una forma abierta y una forma cerrada en disolventes acuosos, tiene un IS de 320, mientras que C6-CI, el enantiómero (S) de un benzoxaborol sustituido con un Cl en la posición C-4, que tampoco es capaz de existir en un equilibrio entre una forma abierta y una forma tricíclica cerrada en disolventes acuosos, tiene un IS de 363. Esto está en marcado contraste con G26-CH3 del Ejemplo 2, el enantiómero (S) de un benzoxaborol sustituido con un -CH3 en la posición C-4, que es capaz de existir en un equilibrio entre una forma abierta y una forma cerrada en disolventes acuosos, tiene un IS de >4850.
Si se compara el IS de G26-CH3 del Ejemplo 2 con el IS de C5-H, el enantiómero (S) de un compuesto de comparación de benzoxaborol sustituido con un H en la posición C4 del anillo de benzoxaborol, se puede observar que el IS de este compuesto sin un sustituyente metilo en C4 es solamente 3, indicando que este compuesto tiene muy poca selectividad para inhibir M. tuberculosis en comparación con la destrucción de células humanas. En contraste, C26-CH3 del Ejemplo 2, un compuesto con -CH3 en la posición C4 del anillo de benzoxaborol que se ha observado que existe en equilibrio entre una forma abierta y una forma cerrada en disolventes acuosos, tiene un índice de selectividad (IS) de >4850.
Ciertas sustituciones de los benzoxaboroles sustituidos capaces de existir, en ciertas realizaciones, en equilibrio entre una forma abierta y una forma cerrada confieren así un incremento inesperado en el índice de selectividad. En contraste, los compuestos de comparación C9-CI (un compuesto de benzoxaborol tricíclico con un sustituyente cloro en C4 y sustitución de -CH3 en R3 y R4 del anillo de 7 miembros) y C10-H (un compuesto de benzoxaborol tricíclico con un hidrógeno en C4 y sustitución de -CH3 en R3 y R4 del anillo de 7 miembros) tienen índices IS de 10. Posiblemente, esto indica que la sustitución en las posiciones R3 y R4 no está favorecida con respecto a la selectividad para M. tuberculosis frente a la inhibición (toxicidad) de células humanas para estos compuestos particulares. También sugiere que la presencia de un halógeno en la posición C4 del anillo de benzoxaborol (véase C9-CI) no es suficiente para vencer el efecto negativo de la sustitución de metilo tanto en R3 como en R4 del anillo tricíclico de 7 miembros en la posición R3/R4.
Así, los benzoxaboroles sustituidos de la invención, particularmente G26-CH3 del Ejemplo 2, muestran sorprendentemente IS sorprendentemente superiores con relación a los IS de benzoxaboroles sustituidos similares
para M. tuberculosis frente a células humanas.
Se debe entender que la invención cubre todas las combinaciones de aspectos con todos los otros aspectos adecuados y/o realizaciones ejemplares descritos en la presente memoria. Se debe entender que la invención también cubre todas las combinaciones de realizaciones ejemplares con todos los otros aspectos adecuados y/o realizaciones ejemplares descritos en la presente memoria.
Claims (15)
1. Un compuesto que tiene una estructura como la mostrada en la Fórmula III:

Fórmula III
en la que
R3 es -CH3; y
R1 y R2 son cada uno independientemente H o -CH3,
o una de sus sales farmacéuticamente aceptables.
2. Un compuesto según la reivindicación 1 o una de sus sales farmacéuticamente aceptables, en donde R3 es -CH3 y en donde R1 es H y R2 es H.
3. Un compuesto según la reivindicación 1, que tiene una estructura como la mostrada en la Fórmula IIIa:
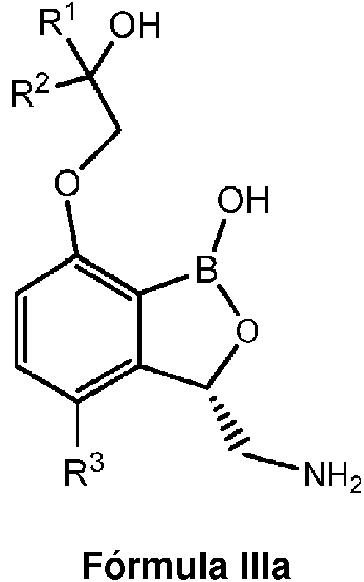
en la que R3 es -CH3; y
R1 es H y R2 es H, o una de sus sales farmacéuticamente aceptables, de modo que el compuesto tiene la siguiente estructura:

4. Un compuesto según la reivindicación 1, o una de sus sales farmacéuticamente aceptables, cuya estructura es:

5. Un compuesto según una cualquiera de las reivindicaciones precedentes, que tiene un patrón de RMN en estado sólido sustancialmente como el mostrado en la Figura 4, en donde el compuesto tiene la siguiente estructura:

6. Una formulación farmacéutica que comprende:
un compuesto según una cualquiera de las reivindicaciones precedentes; y
un excipiente, adyuvante o diluyente farmacéuticamente aceptable.
7. Una combinación que comprende:
un primer agente terapéutico o una de sus sales farmacéuticamente aceptables, en donde el primer agente terapéutico tiene la estructura que se muestra posteriormente:

y un segundo agente terapéutico.
8. La combinación según la reivindicación 7, en la que la sal farmacéuticamente aceptable se selecciona de una sal de hidrocloruro, hidrobromuro, hidroyoduro, nitruro, carbonato, monohidrogenocarbonato, fosfato, monohidrogenofosfato, dihidrogenofosfato, sulfato, monohidrogenosulfato, dihidrogenosulfato, fosfonato, preferiblemente una sal de hidrocloruro o una sal de dihidrogenosulfato.
9. La combinación según la reivindicación 7 o la reivindicación 8, que comprende además un tercer, cuarto, quinto y sexto agente terapéutico opcional, en donde el segundo, tercer, cuarto, quinto y sexto agente terapéutico es un agente terapéutico aprobado o recomendado para el tratamiento de la tuberculosis, preferiblemente en donde el segundo, tercer, cuarto, quinto y sexto agente terapéutico se selecciona independientemente de isoniacida, rifampina, piracinamida, etambutol, moxifloxacina, rifapentina, clofacimina, bedaquilina (TMC207), nitroimidazooxacina PA-824, delamanida (OPC-67683), una oxazolidinona, preferiblemente seleccionada de linezolida, tedizolida, radezolida, sutezolida (PNU-100480) o posizolida (AZD-5847), el análogo de EMB SQ109, una benzotiacinona, una dinitrobenzamida y un agente antiviral incluyendo un agente antirretroviral.
10. Una combinación según la reivindicación 9, en la que el agente antirretroviral es cidovudina, didanosina, lamivudina, zalcitabina, abacavir, estavudina, adefovir, adefovir dipivoxilo, focivudina, todoxilo, emtricitabina, alovudina, amdoxovir, elvucitabina, nevirapina, delavirdina, efavirenz, lovirida, immunocal, oltipraz, capravirina, lersivirina, GSK2248761, TMC-278, TMC-125, etravirina, saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, fosamprenavir, brecanavir, darunavir, atazanavir, tipranavir, palinavir, lasinavir, enfuvirtida, T-20, T-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 y BMS-626529, 5-Helix, raltegravir, elvitegravir, GSK1349572,
GSK1265744, vicriviroc (Sch-C), Sch-D, TAK779, maraviroc, TAK449, didanosina, tenofovir, lopinavir o darunavir, preferiblemente en donde el agente antirretroviral es GSK1349572 o GSK1265744.
11. Un compuesto según una cualquiera de las reivindicaciones 1 a 5 o una de sus sales farmacéuticamente aceptables, o una formulación farmacéutica según la reivindicación 6 , o una combinación según una cualquiera de las reivindicaciones 7 a 10, para el uso en el tratamiento de una enfermedad resultante de una infección micobacteriana,
en donde la infección micobacteriana es una infección de una micobacteria seleccionada de Mycobacterium tuberculosis, Mycobacterium avium incluyendo las subespecies (subesp.) Mycobacterium avium subesp. avium, Mycobacterium avium subesp. hominissuis, Mycobacterium avium subesp. silvaticum y Mycobacterium avium subesp. paratuberculosis; Mycobacterium kansasii, Mycobacterium malmoense, Mycobacterium simiae, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium scrofulaceum, Mycobacterium abscessus, Mycobacterium chelonae, Mycobacterium haemophilum, Mycobacterium leprae, Mycobacterium marinum, Mycobacterium fortuitum, Mycobacterium parafortuitum, Mycobacterium gordonae, Mycobacterium vaccae, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium africanum, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti, Mycobacterium pinnipedi, Mycobacterium leprae, Mycobacterium ulcerans, Mycobacterium intracellulare, complejo de Mycobacterium tuberculosis (MTC), complejo de Mycobacterium avium (MAC), complejo de Mycobacterium avian-intracellulare (MAIC), clado de Mycobacterium gordonae; clado de Mycobacterium kansasii; clado de Mycobacterium chelonae; clado de Mycobacterium fortuitum; clado de Mycobacterium parafortuitum; y clado de Mycobacterium vaccae, preferiblemente en donde la infección micobacteriana se selecciona de Mycobacterium tuberculosis o Mycobacterium avium.
12. Un compuesto o una sal farmacéuticamente aceptable del mismo, una formulación farmacéutica o una combinación para el uso según la reivindicación 11, en donde la infección micobacteriana es una infección por Mycobacterium tuberculosis.
13. Un compuesto o una sal farmacéuticamente aceptable del mismo, una formulación farmacéutica o una combinación para el uso según la reivindicación 11, en donde la enfermedad se selecciona de tuberculosis, lepra, enfermedad de Johne, úlcera de Buruli o Bairnsdale, enfermedad de Crohn, enfermedad pulmonar o infección pulmonar, neumonía, absceso localizado en la bolsa sinovial, la membrana sinovial o las vainas tendinosas, linfadenitis, infecciones de la piel y los tejidos blandos, síndrome de Lady Windermere, enfermedad pulmonar por MAC, complejo de Mycobacterium avium diseminado (DMAC), complejo de Mycobacterium avium intracellulare diseminado (DMAIC), pulmón de jacuzzi, mastitis por MAC, piomiositis por MAC, paratuberculosis por Mycobacterium avium, o granuloma.
14. Un compuesto o una sal farmacéuticamente aceptable del mismo, una formulación farmacéutica o una combinación para el uso según la reivindicación 13, en donde la enfermedad es tuberculosis.
15. Un compuesto o una sal farmacéuticamente aceptable del mismo, una formulación farmacéutica o una combinación para el uso según una cualquiera de las reivindicaciones 11 a 14, en donde el compuesto para el uso tiene la siguiente estructura:
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15382054 | 2015-02-12 | ||
| EP15382055 | 2015-02-12 | ||
| EP15382056 | 2015-02-12 | ||
| PCT/IB2016/050775 WO2016128948A1 (en) | 2015-02-12 | 2016-02-12 | 4 -substituted benzoxaborole compounds and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2821951T3 true ES2821951T3 (es) | 2021-04-28 |
Family
ID=55409879
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16706002T Active ES2821951T3 (es) | 2015-02-12 | 2016-02-12 | Compuestos de benzoxaborol sustituidos en posición 4 y sus usos |
Country Status (36)
| Country | Link |
|---|---|
| US (3) | US20180037595A1 (es) |
| EP (2) | EP3256479B1 (es) |
| JP (2) | JP2018506540A (es) |
| KR (2) | KR20170117158A (es) |
| CN (2) | CN107548398A (es) |
| AU (3) | AU2016217508A1 (es) |
| BR (2) | BR112017017211B1 (es) |
| CA (2) | CA2976030C (es) |
| CL (2) | CL2017002059A1 (es) |
| CO (2) | CO2017008809A2 (es) |
| CR (2) | CR20170371A (es) |
| CY (1) | CY1123687T1 (es) |
| DK (1) | DK3256479T3 (es) |
| DO (2) | DOP2017000188A (es) |
| EA (2) | EA036516B1 (es) |
| ES (1) | ES2821951T3 (es) |
| HR (1) | HRP20201597T1 (es) |
| HU (1) | HUE051113T2 (es) |
| IL (2) | IL253864B (es) |
| LT (1) | LT3256479T (es) |
| MA (2) | MA41494B1 (es) |
| MX (2) | MX379057B (es) |
| MY (1) | MY194004A (es) |
| NZ (1) | NZ734379A (es) |
| PE (2) | PE20171348A1 (es) |
| PH (2) | PH12017501428A1 (es) |
| PL (1) | PL3256479T3 (es) |
| PT (1) | PT3256479T (es) |
| RS (1) | RS60885B1 (es) |
| SG (2) | SG11201706511TA (es) |
| SI (1) | SI3256479T1 (es) |
| SM (1) | SMT202000565T1 (es) |
| TW (2) | TW201702250A (es) |
| UA (1) | UA120527C2 (es) |
| WO (2) | WO2016128948A1 (es) |
| ZA (1) | ZA201705456B (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA032844B1 (ru) * | 2013-08-09 | 2019-07-31 | Глаксосмитклайн Интеллекчуал Проперти (№2) Лимитед | Трициклические соединения бензоксаборола и их применение |
| JP7123074B2 (ja) | 2017-05-08 | 2022-08-22 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッド | マイコバクテリア感染の処置において使用するためのサンフェトリネム、その塩、またはそのエステル |
| US11834466B2 (en) | 2017-11-30 | 2023-12-05 | 5Metis, Inc. | Benzoxaborole compounds and formulations thereof |
| US11236115B2 (en) | 2018-08-18 | 2022-02-01 | 5Metis, Inc. | Solid forms of substituted benzoxaborole and compositions thereof |
| CN110441344B (zh) * | 2019-06-25 | 2020-09-29 | 北京大学 | 一种基于固体核磁共振技术检测rna结构的方法 |
| AU2023287202A1 (en) * | 2022-06-23 | 2024-06-06 | Shanghai Micurx Pharmaceutical Co., Ltd. | Methods and uses of boron compounds in the treatment of nontuberculous mycobacterium infections and pharmaceutical compositions for treatment of same |
| CN119233977B (zh) * | 2022-06-23 | 2026-03-17 | 上海盟科药业股份有限公司 | 硼化合物的前药及其在治疗细菌感染中的用途 |
| KR20240150582A (ko) | 2023-04-08 | 2024-10-15 | 김태서 | 맥세이프 배터리팩을 포함한 아이폰 케이스 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9411587D0 (en) * | 1994-06-09 | 1994-08-03 | Zeneca Ltd | Compound, composition and use |
| AU4967997A (en) | 1996-12-02 | 1998-06-29 | Chisso Corporation | Optically active nitro alcohol derivatives, optically active amino alcohol derivates, and process for preparing the same |
| US7446236B2 (en) | 2002-12-02 | 2008-11-04 | Solvias Ag | Catalytic hydrogenation of carbon-heteroatom double bonds |
| NZ578297A (en) | 2005-02-16 | 2010-11-26 | Anacor Pharmaceuticals Inc | Boron-containing small molecules |
| JP5084717B2 (ja) | 2005-03-08 | 2012-11-28 | エージェンシー フォー サイエンス,テクノロジー アンド リサーチ | キラル・ビスオキサゾリン触媒 |
| EP1976536A4 (en) | 2005-12-30 | 2011-03-02 | Anacor Pharmaceuticals Inc | SMALL MOLECULES CONTAINING BORON |
| TWM325238U (en) * | 2006-12-29 | 2008-01-11 | Universal Scient Ind Co Ltd | Voltage regulator and voltage regulating system |
| KR100848491B1 (ko) | 2007-01-16 | 2008-07-28 | 영진약품공업주식회사 | 베타아미노기를 갖는 2-싸이아졸리딘 유도체, 이의약학적으로 허용 가능한 염 및 이의 제조 방법 |
| JO3396B1 (ar) | 2007-06-20 | 2019-10-20 | Anacor Pharmaceuticals Inc | جزيئات صغيرة تحتوي على البورون |
| MX2011006334A (es) | 2008-12-17 | 2011-09-22 | Anacor Pharmaceuticals Inc | Polimorfos de (s)-3-amino-metil-7-(3-hidroxi-propoxi)-3h-benzo-[c] [1,2]-oxaborol-1-ol. |
| US20110124597A1 (en) * | 2009-09-25 | 2011-05-26 | Anacor Pharmaceuticals, Inc. | Boron containing small molecules |
| MX352607B (es) * | 2010-04-07 | 2017-11-30 | Glaxosmithkline Llc Star | Proceso para la preparacion de benzoxaboroles. |
| WO2012033858A2 (en) | 2010-09-07 | 2012-03-15 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| US8530452B2 (en) | 2011-12-22 | 2013-09-10 | Micurx Pharmaceuticals, Inc. | Tricyclic boron compounds for antimicrobial therapy |
| CN102739587B (zh) | 2012-06-16 | 2014-05-21 | 天地融科技股份有限公司 | 音频数据传输方法 |
| KR101636431B1 (ko) | 2013-07-30 | 2016-07-05 | 동아에스티 주식회사 | 트리사이클릭 벤즈옥사보롤 화합물, 이의 제조방법 및 용도 |
| EA032844B1 (ru) | 2013-08-09 | 2019-07-31 | Глаксосмитклайн Интеллекчуал Проперти (№2) Лимитед | Трициклические соединения бензоксаборола и их применение |
-
2016
- 2016-02-11 MA MA41494A patent/MA41494B1/fr unknown
- 2016-02-11 MA MA041495A patent/MA41495A/fr unknown
- 2016-02-12 LT LTEP16706002.9T patent/LT3256479T/lt unknown
- 2016-02-12 JP JP2017542053A patent/JP2018506540A/ja active Pending
- 2016-02-12 HU HUE16706002A patent/HUE051113T2/hu unknown
- 2016-02-12 NZ NZ734379A patent/NZ734379A/en not_active IP Right Cessation
- 2016-02-12 SM SM20200565T patent/SMT202000565T1/it unknown
- 2016-02-12 MY MYPI2017702943A patent/MY194004A/en unknown
- 2016-02-12 AU AU2016217508A patent/AU2016217508A1/en not_active Abandoned
- 2016-02-12 PL PL16706002T patent/PL3256479T3/pl unknown
- 2016-02-12 CR CR20170371A patent/CR20170371A/es unknown
- 2016-02-12 EA EA201791671A patent/EA036516B1/ru unknown
- 2016-02-12 UA UAA201708405A patent/UA120527C2/uk unknown
- 2016-02-12 BR BR112017017211-9A patent/BR112017017211B1/pt active IP Right Grant
- 2016-02-12 JP JP2017541999A patent/JP6771472B2/ja active Active
- 2016-02-12 PE PE2017001401A patent/PE20171348A1/es not_active Application Discontinuation
- 2016-02-12 WO PCT/IB2016/050775 patent/WO2016128948A1/en not_active Ceased
- 2016-02-12 US US15/550,658 patent/US20180037595A1/en not_active Abandoned
- 2016-02-12 MX MX2017010413A patent/MX379057B/es unknown
- 2016-02-12 US US15/550,693 patent/US10774096B2/en active Active
- 2016-02-12 HR HRP20201597TT patent/HRP20201597T1/hr unknown
- 2016-02-12 WO PCT/IB2016/050776 patent/WO2016128949A1/en not_active Ceased
- 2016-02-12 CA CA2976030A patent/CA2976030C/en active Active
- 2016-02-12 RS RS20201204A patent/RS60885B1/sr unknown
- 2016-02-12 PT PT167060029T patent/PT3256479T/pt unknown
- 2016-02-12 EP EP16706002.9A patent/EP3256479B1/en active Active
- 2016-02-12 MX MX2017010414A patent/MX2017010414A/es unknown
- 2016-02-12 ES ES16706002T patent/ES2821951T3/es active Active
- 2016-02-12 SI SI201630941T patent/SI3256479T1/sl unknown
- 2016-02-12 SG SG11201706511TA patent/SG11201706511TA/en unknown
- 2016-02-12 DK DK16706002.9T patent/DK3256479T3/da active
- 2016-02-12 CN CN201680021488.3A patent/CN107548398A/zh active Pending
- 2016-02-12 CR CR20170372A patent/CR20170372A/es unknown
- 2016-02-12 CN CN201680021538.8A patent/CN107531730B/zh active Active
- 2016-02-12 KR KR1020177025539A patent/KR20170117158A/ko not_active Ceased
- 2016-02-12 CA CA2976308A patent/CA2976308A1/en not_active Abandoned
- 2016-02-12 AU AU2016217507A patent/AU2016217507B2/en not_active Ceased
- 2016-02-12 EP EP16706003.7A patent/EP3256480A1/en not_active Withdrawn
- 2016-02-12 BR BR112017017213-5A patent/BR112017017213A2/pt not_active Application Discontinuation
- 2016-02-12 SG SG11201706510YA patent/SG11201706510YA/en unknown
- 2016-02-12 KR KR1020177025535A patent/KR20170117156A/ko not_active Withdrawn
- 2016-02-12 PE PE2017001400A patent/PE20171432A1/es not_active Application Discontinuation
- 2016-02-12 EA EA201791667A patent/EA201791667A1/ru unknown
- 2016-02-15 TW TW105104422A patent/TW201702250A/zh unknown
- 2016-02-15 TW TW105104376A patent/TWI730953B/zh not_active IP Right Cessation
-
2017
- 2017-08-07 IL IL253864A patent/IL253864B/en unknown
- 2017-08-07 IL IL253865A patent/IL253865A0/en unknown
- 2017-08-09 PH PH12017501428A patent/PH12017501428A1/en unknown
- 2017-08-09 PH PH12017501427A patent/PH12017501427A1/en unknown
- 2017-08-10 DO DO2017000188A patent/DOP2017000188A/es unknown
- 2017-08-10 DO DO2017000189A patent/DOP2017000189A/es unknown
- 2017-08-11 ZA ZA201705456A patent/ZA201705456B/en unknown
- 2017-08-11 CL CL2017002059A patent/CL2017002059A1/es unknown
- 2017-08-11 CL CL2017002060A patent/CL2017002060A1/es unknown
- 2017-08-29 CO CONC2017/0008809A patent/CO2017008809A2/es unknown
- 2017-08-29 CO CONC2017/0008816A patent/CO2017008816A2/es unknown
-
2020
- 2020-09-11 US US17/018,301 patent/US11214582B2/en not_active Expired - Fee Related
- 2020-10-14 AU AU2020256369A patent/AU2020256369A1/en not_active Abandoned
- 2020-10-15 CY CY20201100978T patent/CY1123687T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2821951T3 (es) | Compuestos de benzoxaborol sustituidos en posición 4 y sus usos | |
| US10526352B2 (en) | Tricyclic benzoxaborole compounds and uses thereof | |
| HK1240232B (en) | 4-substituted benzoxaborole compounds and uses thereof | |
| HK1240232A1 (en) | 4-substituted benzoxaborole compounds and uses thereof | |
| HK1219939B (en) | Tricyclic benzoxaborole compounds and uses thereof |