ES2905164T3 - Compuesto de amida peptídico y método de preparación y uso médico del mismo - Google Patents
Compuesto de amida peptídico y método de preparación y uso médico del mismo Download PDFInfo
- Publication number
- ES2905164T3 ES2905164T3 ES18835454T ES18835454T ES2905164T3 ES 2905164 T3 ES2905164 T3 ES 2905164T3 ES 18835454 T ES18835454 T ES 18835454T ES 18835454 T ES18835454 T ES 18835454T ES 2905164 T3 ES2905164 T3 ES 2905164T3
- Authority
- ES
- Spain
- Prior art keywords
- amino
- alkyl
- group
- tert
- heterocyclic group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1024—Tetrapeptides with the first amino acid being heterocyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1016—Tetrapeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Pain & Pain Management (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Ophthalmology & Optometry (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Compuesto de fórmula general (I) o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo: **(Ver fórmula)** en la que **(Ver fórmula)** R1 se selecciona de cada uno de m1, m2 se selecciona independientemente de 1, 2, 3 ó 4; cada uno de m3, m4 se selecciona independientemente de 0, 1, 2, 3 ó 4; con la condición de que m3 y m4 no pueden ser 0 al mismo tiempo; cada uno de n1, n2 se selecciona independientemente de 0, 1, 2, 3 ó 4; Z se selecciona de CRz1Rz2 o NRz3; cada uno de Rz1, Rz2 se selecciona independientemente de H, F, Cl, Br, I, OH, CF3, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, -C(=O)-alquilo C1-6, -(CH2)q-C(=O)O-alquilo C1-6, -(CH2)q-NR1eR1f, -(CH2)q- COOH, -(CH2)q-CONH2, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, alcoxilo, alquenilo, alquinilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de S, S=O o S(=O)2; cada uno de R1e, R1f se selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)O- (CH2)q-grupo carbocíclico C3-8 o -C(=O)O-(CH2)q-grupo heterocíclico de 3 a 8 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; alternativamente, Rz1 y Rz2 forman un anillo heterocíclico que contiene nitrógeno de 3 a 10 miembros con el átomo de carbono al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, =O, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros; cada uno de R1a, R1b se selecciona independientemente de F, CF3, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, alquenilo, alquinilo o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S; Rz3 se selecciona independientemente de H, -C(=O)-alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)-grupo carbocíclico C3-8, -C(=O)O-grupo carbocíclico C3-8, -C(=O)O-(grupo heterocíclico de 3 a 8 miembros), -S(=O)p- alquilo C1-6, -S(=O)p-grupo carbocíclico C3-8, -S(=O)p-(grupo heterocíclico de 3 a 8 miembros), -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S; cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-6; alternativamente, R1g, R1h forman un anillo heterocíclico de 3 a 10 miembros con el átomo de nitrógeno al que están unidos, el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2- 6 o -S(=O)p-alquilo C1-6, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; q se selecciona de 0, 1, 2, 3 ó 4; p se selecciona de 0, 1 ó 2; a se selecciona de 0, 1, 2 ó 3; R4 se selecciona independientemente de H, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o -(CH2)q-grupo carbocíclico C3-8, el grupo alquilo, alquenilo, alquinilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CN, CF3, NO2, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1- 4, -C(=O)O-(CH2)q-grupo carbocíclico C3-8, -C(=O)O-(CH2)q-grupo heterocíclico de 3 a 8 miembros o y el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S; b se selecciona de 0, 1, 2, 3, 4 ó 5; c se selecciona de 0, 1, 2, 3, 4 ó 5; cada uno de R5, R6 se selecciona independientemente de F, Cl, Br, I, CF3, ciano, nitro, alquilo C1-4, - OR5a, -C(O)OR5b, -SR5c, -S(O)R5d, -S(O)2R5e o -NR5fR5g; cada uno de R5a, R5b, R5c, R5d, R5e, R5f y R5g se selecciona independientemente de H o alquilo C1-4; alternativamente, R5f, R5g forman un anillo heterocíclico de 5 a 6 miembros con el átomo de nitrógeno al que están unidos, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S.
Description
DESCRIPCIÓN
Compuesto de amida peptídico y método de preparación y uso médico del mismo
Campo técnico
La presente invención se refiere a un compuesto de amida peptídico que tiene un efecto analgésico, a un método de preparación del mismo y a su uso en medicina.
Antecedentes
Los fármacos opioides se han usado para el tratamiento del dolor durante miles de años y desempeñan un papel fisiológico principalmente al unirse a los tres receptores de opioides clásicos conocidos |i, 8 y k . Estos tres receptores son miembros de la familia de receptores acoplados a proteínas g, distribuidos principalmente en el sistema nervioso central y también en muchos tejidos periféricos. Uno de los fármacos más clásicos es la morfina, que ejerce un efecto analgésico principalmente a través de la acción de los receptores de opioides |i.
Además, los analgésicos clínicos usados habitualmente incluyen otros fármacos receptores de opioides |i, tales como los opioides tradicionales representados por dihidromorfinona y fentanilo.
Sin embargo, los fármacos receptores de opioides |i producen una variedad de efectos secundarios después de un uso prolongado, tales como tolerancia, dependencia y depresión respiratoria, y efectos sobre la motilidad gastrointestinal, lo que no sólo aumenta el coste del tratamiento, sino que también afecta al ciclo del paciente para recuperarse. Algunas inyecciones de fármacos distintos de opioides, tales como paracetamol y los AINE (fármacos antiinflamatorios no esteroideos), tienen un uso y dosificación limitados debido a su escaso efecto analgésico. Además, tienen ciertos efectos secundarios, tales como que el paracetamol aumenta la toxicidad hepática, y los AINE (antiinflamatorios no esteroideos) provocan diversas enfermedades gastrointestinales.
Con la creciente presión de la vida y el trabajo en la sociedad moderna y la llegada de una sociedad envejecida, y ante el papel crítico de los receptores de opioides para el tratamiento de diferentes tipos de dolor, la búsqueda de nuevos opioides con alta actividad analgésica y bajos efectos secundarios tóxicos tienen una significación científica y social importante.
Algunos estudios han hallado que usando agonistas de los receptores de opioides k , los receptores de opioides k pueden usarse como dianas de intervención para tratar el dolor y prevenir una amplia variedad de enfermedades y afecciones. Por ejemplo, en 1993, Woold et al. describieron el uso de agonistas de receptores de opioides k para el tratamiento de la sensibilización al dolor (Anasthesia and Analgesia, 1993, 77, 362-379); en 1999, Wu et al. propusieron agonistas de receptores de opioides k como dianas para la prevención y el tratamiento de enfermedades cardiovasculares (Circulation Res 1999, 84, 1388-1395); en 2003, Kaushik et al. describieron los efectos neuroprotectores de los agonistas de receptores de opioides k (J. Postgraduate Medicine 2003, 49 (1), 90-95); en 2004, Potter et al. describieron el uso de agonistas de receptores de opioides k en trastornos oculares y dolor ocular (Pharmacol. Exp. Ther 2004, 209, 548-553); en 2005, Wikstrom et al. describieron el uso de agonistas k en el tratamiento de la uremia y el prurito inducido por opio (J. Am. Soc. Nephrol 2005, 16, 3742-3747.); en 2006, Bileviciute-Ljungar et al. evaluaron las propiedades de los agonistas de receptores de opioides k para enfermedades inflamatorias tales como osteoartritis y artritis reumatoide (Rheumatology 2006,45, 295-302); en 2006, Lembo evaluó el uso de agonistas de receptores de opioides k en enfermedades gastrointestinales (Diges. Dis. 2006, 24, 91-98); en 2006, Jolivalt et al. describieron el papel del agonista de receptores de opioides k acimadrina en la neuropatía diabética de roedores (Diabetologia 2006, 49 (11), 2775-2785); en 2008, Schteingart, Claudio, D et al. de Cara Therapeutics Co., Ltd. evaluaron los efectos de los agonistas de receptores de opioides k sobre el dolor visceral, el dolor relacionado con la activación de nociceptores sensibles al pH y el dolor ocular inducido por capsaicina en el documento WO2008057608A2.
Sumario
El objeto de la presente invención es proporcionar un agonista de receptores de opioides k que tenga una estructura nueva, mejor actividad biológica y mejor efecto analgésico, y un método de preparación del mismo y su uso en medicina.
La presente invención proporciona un compuesto de fórmula general (I) o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo:

en la que
„ 
R1 se selecciona
cada uno de mi y m2 se selecciona independientemente de 1, 2, 3 ó 4;
cada uno de m3 y m4 se selecciona independientemente de 0, 1,2, 3 ó 4; con la condición de que m3 y m4 no pueden ser 0 al mismo tiempo;
cada uno de n1 y n2 se selecciona independientemente de 0, 1,2, 3 ó 4;
Z se selecciona de CRz1Rz2 o NRz3;
cada uno de Rz1 y Rz2 se selecciona independientemente de H, F, Cl, Br, I, OH, CF3, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, -C(=O)-alquilo C1-6, -(CH2)q-C(=O)O-alquilo C1-6, -(CH2)q-NR1eR1f, -(CH2)q-COOH, -(CH2)q-CONH2, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros. El grupo alquilo, alcoxilo, alquenilo, alquinilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de S, S=O o S(=O)2;
cada uno de R1e, R1f se selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)O-(CH2)q-grupo carbocíclico C3-8 o -C(=O)O-(CH2)q-grupo heterocíclico de 3 a 8 miembros. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
alternativamente, Rz1 y Rz2 forman un anillo heterocíclico que contiene nitrógeno de 3 a 10 miembros con el átomo de carbono al que están unidos. El anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, =O, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros;
cada uno de R1a, R1b se selecciona independientemente de F, CF3, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o grupo heterocíclico de 3 a 8 miembros. El grupo alquilo, alquenilo, alquinilo o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-s o grupo heterocíclico de 3 a 8 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
Rz3 se selecciona independientemente de H, -C(=O)-alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)-grupo carbocíclico C3-8, -C(=O)O-grupo carbocíclico C3-8, -C(=O)O-(grupo heterocíclico de 3 a 8 miembros), -S(=O)p-alquilo C1-6, -S(=O)p-grupo carbocíclico C3-8, -S(=O)p-(grupo heterocíclico de 3 a 8 miembros), -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o grupo heterocíclico de 3 a 8 miembros. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
cada uno de R1g, R1h, R1¡, R1j se selecciona independientemente de H o alquilo Ci-a;
alternativamente, R1g, R1h forman un anillo heterocíclico de 3 a 10 miembros con el átomo de nitrógeno al que están unidos. El anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C ^ , alcoxilo C ^ , alquenilo C2-6, alquinilo C2-6 o -S(=O)p-alquilo C1-6. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
q se selecciona de 0, 1, 2, 3 ó 4;
p se selecciona de 0, 1 ó 2;
a se selecciona de 0, 1, 2 ó 3;
R4 se selecciona independientemente de H, alquilo C ^ , alquenilo C2-6, alquinilo C2-6 o -(CH2)q-grupo carbocíclico C3-8. El grupo alquilo, alquenilo, alquinilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CN, CF3, NO2, alquilo C ^ , alcoxilo C ^ , alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C-i-e, -C(=O)0-alquilo C1-4, -C(=O)0-(CH2)q-NH
> ¡ A Nh
grupo carbocíclico C3-8, -C(=O)0-(CH2)q-grupo heterocíclico de 3 a 8 miembros o 2. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C ^ , alcoxilo C ^ , alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
b se selecciona de 0, 1,2, 3, 4 ó 5;
c se selecciona de 0, 1, 2, 3, 4 ó 5;
cada uno de R5, R6 se selecciona cada uno independientemente de F, Cl, Br, I, CF3, ciano, nitro, alquilo C1-4, -OR5a, -C(O)OR5b, -SR5c, -S(O)R5d, -S(O)2R5e o -NR5fR5g;
cada uno de R5a, R5b, R5c, R5d, R5e, R5f y R5g se selecciona independientemente de H o alquilo C1-4; alternativamente, R5f, R5g forman un anillo heterocíclico de 5 a 6 miembros con el átomo de nitrógeno al que están unidos. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S.
En una realización preferida de la invención, un compuesto de fórmula general (I) o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, en el que:

R1 se selecciona de
cada uno de ith, m2, m3, mu se selecciona independientemente de 1 ó 2;
cada uno de m, n2 se selecciona independientemente de 0, 1 ó 2;
Z se selecciona de CRz1Rz2 o NRz3;
cada uno de Rz1, Rz2 se selecciona independientemente de H, F, Cl, Br, I, OH, CF3, nitro, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, -C(=O)-alquilo C1.4, -(CH2)q-C(=O)O-alquilo C1.4, -(CH2)q-NR1eR1f, -(CH2)q-COOH, -(CH2)q-CONH2, grupo carbocíclico C3-a o un grupo heterocíclico de 3 a 6 miembros, preferiblemente H, alquilo C1-4, -(CH2)q-C(=O)O-alquilo C1.4, -(CH2)q-NR1eR1f, -(CH2)q-COOH, -(CH2)q-CONH2, grupo carbocíclico C3-a o un grupo heterocíclico de 3 a 6 miembros. El grupo alquilo, alcoxilo, alquenilo, alquinilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, =O, carboxilo, ciano, amino, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s)
de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de S, S=O o S(=O)2;
cada uno de R1e, R1f se selecciona independientemente de H, alquilo C1-4, -C(=O)O-alquilo C1-4, -C(=O)O-(CH2)q-grupo carbocíclico C3.6 o -C(=O)O-(CH2)q-grupo heterocíclico de 3 a 6 miembros, preferiblemente H, alquilo C1-4, -C(=O)O-alquilo C1.4 o -C(=O)O-(CH2)q-grupo carbocíclico C3-6. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
alternativamente, Rz1 y Rz2 forman un anillo heterocíclico que contiene nitrógeno de 3 a 10 miembros, preferiblemente forman un anillo heterocíclico que contiene nitrógeno de 4 a 6 miembros, con el átomo de carbono al que están unidos. El anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, =O, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros;
cada uno de R1a, R1b se selecciona independientemente de F, CF3, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o un grupo heterocíclico de 3 a 6 miembros, preferiblemente F, CF3, alquenilo C2-4 o alquinilo C2-4. El grupo alquilo, alquenilo, alquinilo o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
Rz3 se selecciona independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -C(=O)-grupo carbocíclico C3-4, -C(=O)O-grupo carbocíclico C3-6 o -C(=O)O-(grupo heterocíclico de 3 a 6 miembros), -S(=O)p-alquilo C1.4, -S(=O)p-grupo carbocíclico C3-6, -S(=O)p-(grupo heterocíclico de 3 a 6 miembros), -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros, preferiblemente H, -C(=O)-alquilo C1-4, -C(=O)O-alquilo C1.4, -S(=O)p-alquilo C1.4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h o un grupo heterocíclico de 3 a 6 miembros. El grupo alquilo, alquenilo, alquinilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-6, preferiblemente H o alquilo C1-4;
alternativamente, R1g, R1h forman un anillo heterocíclico de 3 a 10 miembros, preferiblemente forman un anillo heterocíclico de 4 a 6 miembros, con el átomo de nitrógeno al que están unidos. El anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4 o -S(=O)p-alquilo C1.4. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
q se selecciona de 0, 1, 2, 3 ó 4; preferiblemente 0 ó 1;
p se selecciona de 0, 1 ó 2; preferiblemente 2;
a se selecciona de 0, 1, 2 ó 3; preferiblemente 3;
R4 se selecciona independientemente de H, alquilo C1-4, alquenilo C2-4, alquinilo C2-4 o -(CH2)q-grupo carbocíclico C3-6, preferiblemente alquilo C1-4. El grupo alquilo, alquenilo, alquinilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CN, CF3, NO2, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-4, -C(=O)0-alquilo C1-4, C(=O)O-(CH2)q-NH
K ’^ N H 2
grupo carbocíclico C3-6, -C(=O)0-(CH2)q-grupo heterocíclico de 3 a 6 miembros o > preferiblemente H, alquilo C1-4, -C(=O)O-alquilo C1-4, -C(=O)O-(CH2)q-grupo carbocíclico C3-6. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
b se selecciona de 0, 1, 2, 3, 4 ó 5, preferiblemente 0 ó 1;
c se selecciona de 0, 1, 2, 3, 4 ó 5, preferiblemente 0 ó 1;
cada uno de R5, R6 se selecciona cada uno independientemente de F, Cl, Br, I, CF3, ciano, nitro, alquilo C1-4 o -NR5fR5g, preferiblemente F, CF3 o alquilo C1-4;
cada uno de R5f y R5g se selecciona independientemente de H o alquilo C1-4.
En una realización preferida de la invención, un compuesto de fórmula general (I) o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, en el que:
cada uno de ith, m2, m3, itu se selecciona independientemente de 1 ó 2;
cada uno de n1, n2 se selecciona independientemente de 0, 1 ó 2;
Z se selecciona de CRz1Rz2 o NRz3;
cada uno de Rz1, Rz2 se selecciona independientemente de H, alquilo C1-4, -(CH2)q-C(=O)O-alquilo C1-4, -(CH2)q-NR1eR1f, -(CH2)q-COOH, -(CH2)q-CONH2, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de S, S=O o S(=O)2;
cada uno de R1e, R1fse selecciona independientemente de H, alquilo C1-4, -C(=O)O-alquilo C1-4 o -C(=O)O-(CH2)qgrupo carbocíclico C3-6. El grupo alquilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, metilo, etilo, metoxilo, etoxilo, fenilo;
alternativamente, Rz1 y Rz2 son capaces de formar un anillo heterocíclico que contiene nitrógeno de 4 a 6 miembros con un átomo de carbono al que están unidos. El anillo está opcionalmente sustituido adicionalmente con un sustituyente de =O;
R1a, R1b se seleccionan independientemente de F, CF3, metilo, etilo, propanoílo o isopropilo;
Rz3 se selecciona cada uno independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -S(=O)p-alquilo C1-4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, metilo, etilo, metoxilo, etoxilo, ciclopropilo o fenilo. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-4;
alternativamente, R1g, R1h forman un anillo heterocíclico de 4 a 6 miembros con el átomo de nitrógeno al que están unidos. El anillo está opcionalmente sustituido adicionalmente con uno o más sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, metilo, etilo, metoxilo, etoxilo o -S(=O)p-alquilo C1-4 (preferiblemente -S(=O)p-metilo, preferiblemente -S(=O)p-etilo). El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
p se selecciona de 2;
q se selecciona de 0 ó 1;
a se selecciona de 3;
R4 se selecciona de propanoílo o isopropilo;
cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-4, -C(=O)O-alquilo C1-4 o -C(=O)O-bencilo;
b se selecciona de 0;
c se selecciona de 0.
En una realización preferida de la invención, la invención proporciona un compuesto de fórmula general (I), en el que el compuesto se selecciona del compuesto de fórmula general (II) o un estereoisómero, hidrato, solvato, sal
farmacéuticamente aceptable o cocristal del mismo, en el que:

R1 se selecciona 
cada uno de m1, m2, m3, m4 se selecciona independientemente de 1 ó 2;
cada uno de n1, n2 se selecciona independientemente de 0 ó 2;
R1a, R1b se seleccionan independientemente de F;
Z se selecciona de CRz1Rz2 o NRz3;

alternativamente, Rz1 y Rz2 son capaces de formar una lactama con el átomo de carbono al que están unidos;
con el átomo de carbono al que están unidos;
Rz3 se selecciona cada uno independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -S(=O)p-alquilo C1-4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros. El grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, metilo, etilo, metoxilo, etoxilo, ciclopropilo o fenilo. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C i-4;
alternativamente, R1g, R1h forman un anillo heterocíclico de 4 a 6 miembros con el átomo de nitrógeno al que están unidos. El anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, CF3, metilo, metoxilo o -S(=O)p-alquilo C1-4. El grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;
p se selecciona de 2;
cada uno de R2, R3, R7, R8 se selecciona independientemente de H, metilo o -C(=O)O-terc-butilo.
En una realización preferida de la invención, la invención proporciona un compuesto de fórmula general (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, en el que
R1 se selecciona 
cada uno de m1, m2, m3, m4 se selecciona independientemente de 1 ó 2;
cada uno de n1, n2 se selecciona independientemente de 0 ó 2;
R1a, R1b se seleccionan de F;
Z se selecciona de CRz1Rz2 o NRz3;

alternativamente, Rz1 y Rz2 son capaces de formar una lactama con el átomo de carbono al que están unidos;
con el átomo de carbono al que están unidos;

cada uno de R2, R3, R7, R8 se selecciona independientemente de H, metilo o -C(=O)O-terc-butilo.
En una realización preferida de la invención, la invención proporciona un compuesto de fórmula general (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, en el que:
R1 se selecciona de 

Z se selecciona de CRz1Rz2 o NRz3;

alternativamente, Rz1 y Rz2 son capaces de formar una lactama con el átomo de carbono al que están unidos;
con el átomo de carbono al que están unidos;
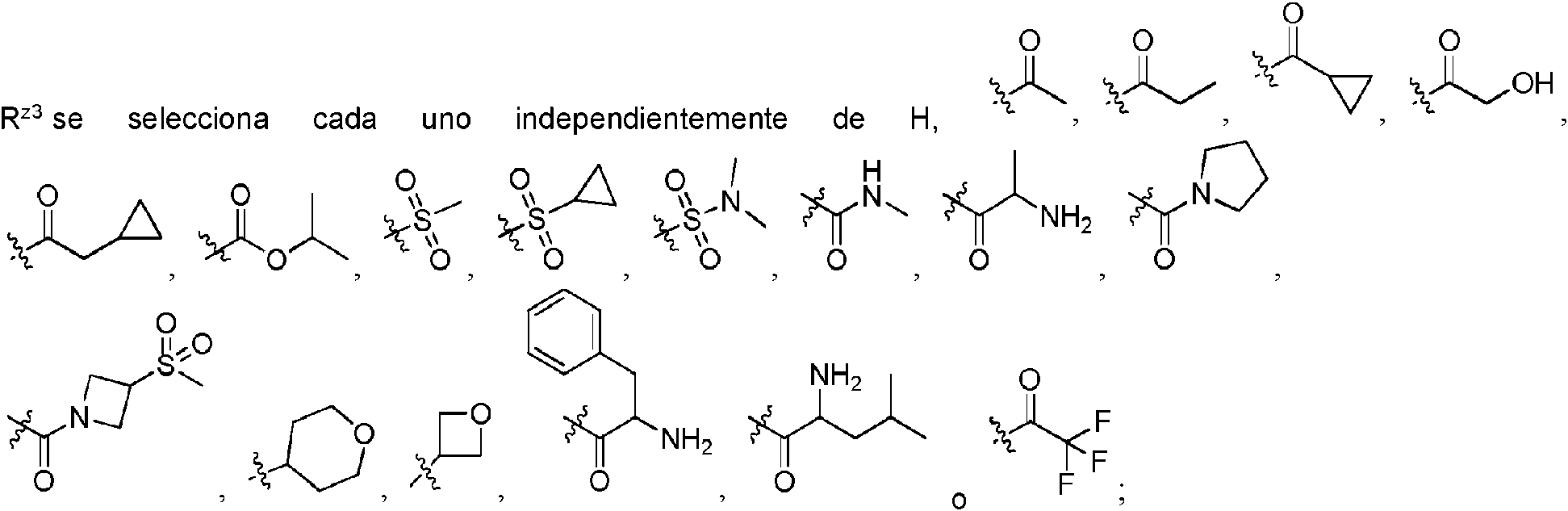
cada uno de R2, R3, R7, R8 se selecciona independientemente de H, metilo o -C(=O)O-terc-butilo.
En una realización preferida de la invención, la invención proporciona un compuesto de fórmula general (I) o (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, en el que el compuesto incluye, pero no se limita a, uno de los compuestos representados por la siguiente fórmula estructural:

Comuesto 11 ompuesto

Compuesto 16

puestos ompuesto

Compuesto 23 Compuesto 24

Com uesto 25

Com uesto 27

Compuesto 29 Compuesto 30

Compuesto 31

Compuesto 36 Compuesto: 35

En una realización preferida de la invención, la invención proporciona un compuesto de fórmula general (I) o (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo. La sal farmacéuticamente aceptable se selecciona de un trifluoroacetato.
La invención proporciona una composición farmacéutica que comprende un compuesto de fórmula general (I) o (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, y uno o más portadores y/o excipientes farmacéuticamente aceptables.
El uso de un compuesto de fórmula general (I) o (II) de la presente invención, o de un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, o de una composición farmacéutica que comprende el compuesto de fórmula general (I) o (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, en la fabricación de un medicamento para el tratamiento o la prevención de una enfermedad o afección asociada con un receptor de opioides k en un mamífero.
En una realización preferida de la invención, en la que la enfermedad o afección asociada al receptor de opioides k se selecciona del grupo que consiste en dolor, inflamación, prurito, edema, hiponatremia, hipopotasemia, íleo, tos y glaucoma.
En una realización preferida de la invención, en la que el dolor se selecciona del grupo que consiste en dolor neuropático, dolor físico, dolor visceral y dermatalgia.
En una realización preferida de la invención, en la que el dolor se selecciona del grupo que consiste en dolor por artritis, dolor por cálculos renales, histeroespasmo, dismenorrea, endometriosis, dispepsia, dolor posquirúrgico, dolor tras tratamiento médico, dolor ocular, dolor por otitis, dolor oncológico fulminante y dolor asociado con trastornos GI.
La invención proporciona un método para tratar o prevenir una enfermedad o afección asociada con un receptor de opioides k en un mamífero, comprendiendo el método administrar el compuesto de fórmula general (I) o (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo, o la composición farmacéutica que comprende el compuesto de fórmula general (I) o (II), o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo. La enfermedad o afección asociada al receptor de opioides k se selecciona preferiblemente del grupo que consiste en dolor, inflamación, prurito, edema, hiponatremia, hipopotasemia, íleo, tos y glaucoma. El dolor se selecciona preferiblemente del grupo que consiste en dolor neuropático, dolor somático, dolor visceral y dermatalgia; o el dolor se selecciona preferiblemente del grupo que consiste en dolor por artritis, dolor por cálculos renales, histeroespasmo, dismenorrea, endometriosis, dispepsia, dolor posquirúrgico, dolor tras tratamiento médico, dolor ocular, dolor por otitis, dolor oncológico fulminante y dolor asociado con trastornos GI (trastornos gastrointestinales).
A menos que se establezcan de otro modo, los términos usados en la memoria descriptiva y las reivindicaciones tienen los siguientes significados.
El carbono, hidrógeno, oxígeno, azufre, nitrógeno o halógeno implicados en los grupos y compuestos de la presente invención incluyen sus isótopos, y el carbono, hidrógeno, oxígeno, azufre, nitrógeno o halógeno implicados en los grupos y compuestos de la presente invención opcionalmente se reemplazan adicionalmente por uno o más de sus isótopos correspondientes, en los que los isótopos de carbono incluyen 12C, 13C y 14C, los isótopos de hidrógeno incluyen protio (H), deuterio (D, también conocido como hidrógeno pesado), tritio (T, también conocido como hidrógeno superpesado), los isótopos de oxígeno incluyen 16O, 17O y 18O, los isótopos del azufre incluyen 32S, 33S, 34S y 36S, los isótopos de nitrógeno incluyen 14N y 15N, los isótopos del flúor incluyen 19F, los isótopos del cloro incluyen 35Cl y 37Cl, los isótopos del bromo incluyen 79Br y 81Br.
Un “alquilo” significa un grupo hidrocarbonado saturado monovalente de cadena lineal y ramificada, y el grupo de cadena lineal y ramificada tiene una cadena principal que comprende de 1 a 10 átomos de carbono, preferiblemente de 1 a 8 átomos de carbono, más preferiblemente de 1 a 6 átomos de carbono, más preferiblemente de 1 a 4 átomos de carbono, lo más preferiblemente de 1 a 2 átomos de carbono. Los ejemplos de alquilo incluyen, pero no se limitan a metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, 2-pentilo, 3-pentilo, 2-metil-2-butilo, 3-metil-2-butilo, n-hexilo, n-heptilo, n-octilo, n-nonilo y n-decilo, etc. El alquilo también puede estar opcionalmente sustituido con 0, 1, 2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, =O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a, en los que cada uno de R19y R19a se selecciona independientemente de H, hidroxilo, amino, carboxilo, alquilo C1-8, alcoxilo C1-8, alquenilo C2-8, alquinilo C2-8, grupo carbocíclico de 3 a 10 miembros, grupo heterocíclico de 4 a 10 miembros, grupo carbocicliloxilo de 3 a 10 miembros o grupo oxilo heterocíclico de 4 a 10 miembros, k se selecciona de 0, 1, 2, 3, 4 ó 5, j se selecciona de 0, 1 ó 2. El alquilo, k, j, R19 y R19a, en el presente documento son tal como se definieron anteriormente.
Un “alquileno” significa un grupo hidrocarbonado saturado divalente de cadena lineal o ramificada, incluyendo -(CH2)v-(v es un número entero de 1 a 10), y los ejemplos de alquileno incluyen, pero no se limitan a, metileno, etileno, propileno, butileno o similar. El alquileno puede estar opcionalmente sustituido adicionalmente con 0, 1,2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, =O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a. Cuando el número de sustituyente(s) en el grupo alquileno es de 2 o más, el/los sustituyente(s) puede(n) condensarse entre sí para formar una estructura cíclica. El alquileno de la presente invención es tal como se definió anteriormente.
Un “alcoxilo” significa un grupo monovalente de un grupo O-alquilo, en el que alquilo es tal como se define en el presente documento, y los ejemplos de alcoxilo incluyen, pero no se limitan a, metoxilo, etoxilo, 1-propoxilo, 2-propoxilo, 1-butoxilo, 2-metil-1-propoxilo, 2-butoxilo, 2-metil-2-propoxilo, 1 -pentiloxilo, 2-pentiloxilo, 3-pentiloxilo, 2-metil-2-butoxilo, 3-metil-2-butoxilo, 3-metil-1-butoxilo y 2-metil-1-butoxilo y similares.
Un “alquenilo” significa un grupo hidrocarbonado insaturado monovalente de cadena lineal o ramificada que tiene al menos 1, generalmente 1,2 ó 3 dobles enlaces carbono-carbono, con una cadena principal del mismo que comprende de 2 a 10 átomos de carbono, más preferiblemente de 2 a 6 átomos de carbono, más preferiblemente de 2 a 4 átomos de carbono en la cadena principal. Los ejemplos de alquenilo incluyen, pero no se limitan a, vinilo, alilo, 1-propenilo, 2-propenilo, 1 -butenilo, 2-butenilo, 3-butenilo, 1-pentenilo, 2-pentenilo, 3-pentenilo, 4-pentenilo, 1 -metil-1-butenilo, 2-metil-1-butenilo, 2-metil-3-butenilo, 1-hexenilo, 2-hexenilo, 3-hexenilo, 4-hexenilo, 5-hexenilo, 1-metil-1-pentenilo, 2-metil-1-pentenilo, 1-heptenilo, 2-heptenilo, 3-heptenilo, 4-heptenilo, 1-octenilo, 3-octenilo, 1-nonenilo, 3-nonenilo, 1-decenilo, 4-decenilo, 1,3-butadieno, 1,3-pentadieno, 1,4-pentadieno y 1,4-hexadieno y similares; el alquenilo puede
estar opcionalmente sustituido adicionalmente con 0, 1, 2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, =O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1.6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a. El alquenilo de la presente invención es tal como se definió anteriormente.
Un “alquinilo” significa un grupo hidrocarbonado insaturado monovalente de cadena lineal o ramificada que tiene al menos 1, generalmente 1, 2 ó 3 triples enlaces carbono-carbono, con una cadena principal que comprende de 2 a 10 átomos de carbono, más preferiblemente de 2 a 6 átomos de carbono, más preferiblemente de 2 a 4 átomos de carbono en la cadena principal. Los ejemplos de alquinilo incluyen, pero no se limitan a etinilo, 1 -propinilo, 2-propinilo, butinilo, 2-butinilo, 3-butinilo, 1 -metil-2-propinilo, 4-pentinilo, 3-pentinilo, 1 -metil-2-butinilo, 2-hexinilo, 3-hexinilo, 2-heptinilo, 3-heptinilo, 4-heptinilo, 3-octinilo, 3-noninilo y 4-decinilo y similares; el alquinilo puede estar opcionalmente sustituido adicionalmente con 0, 1, 2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I,
=O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a. El alquinilo de la presente invención es tal como se definió anteriormente.
Un “cicloalquilo” significa un grupo hidrocarbonado carbocíclico saturado monovalente, que tiene habitualmente de 3 a 10 átomos de carbono, y los ejemplos no limitativos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo o cicloheptilo y similares. El cicloalquilo puede estar opcionalmente sustituido adicionalmente con 0, 1, 2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, =O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a. El cicloalquilo de la presente invención es tal como se definió anteriormente.
Un “carbociclilo” significa un anillo aromático o no aromático, saturado o insaturado. El anillo aromático o no aromático puede ser un anillo monocíclico de 3 a 10 miembros, un anillo bicíclico de 4 a 12 miembros o un sistema de anillos tricíclico de 10 a 15 miembros. El grupo carbocíclico puede estar unido a un anillo con puente o un espiroanillo. Los ejemplos no limitativos incluyen ciclopropilo, ciclobutilo, ciclopentilo, 1-ciclopentil-1-alquenilo, 1 -ciclopentil-2-alquenilo,
1 -ciclopentil-3-alquenilo, ciclohexilo, 1 -ciclohexil-2-alquenilo, 1 -ciclohexil-3-alquenilo, ciclohexenilo, ciclohexadienilo, cicloheptilo, ciclooctilo, ciclononilo, ciclodecilo, cicloundecilo, ciclododecilo, fenilo o naftilo. El grupo carbocíclico puede estar opcionalmente sustituido adicionalmente con 0, 1, 2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, =O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a. El carbociclo de la presente es tal como se definió anteriormente.
Un “heterociclo” significa un anillo aromático o no aromático saturado o insaturado, y el anillo aromático o no aromático puede ser un sistema monocíclico de 3 a 10 miembros, bicíclico de 4 a 12 miembros o tricíclico de 10 a 15 miembros, e incluye de 1 a 4 heteroátomos seleccionados de N, O o S, preferiblemente un grupo heterocíclico de 3 a 8 miembros, y N, S opcionalmente sustituidos en el anillo del grupo heterocíclico pueden oxidarse a diversos estados de oxidación.
El grupo heterocíclico puede estar unido a un heteroátomo o un átomo de carbono, y el grupo heterocíclico puede estar unido a un anillo con puente o espiroanillo. Los ejemplos no limitativos incluyen epoxietilo, epoxipropilo, azaciclopropilo, oxeciclobutilo, azaciclobutilo, tioheterobutilo, 1,3-dioxolanilo, 1,4-dioxolanilo, 1,3-dioxohexilo, azacicloheptilo, oxepanilo, tiocicloheptilo, oxazepinilo, diazepinilo, tiazepinilo, piridilo, piperidinilo, homopiperidinilo, furilo, tienilo, piranilo, N-alquilpirrolilo, pirimidinilo, pirazinilo, piridazinilo, piperazinilo, homopiperazinilo, imidazolilo, piperidinilo, morfolinilo, tiomorfolinilo, oxatianilo, dihidrofuranilo, dihidropiranilo, ditiapentanilo, tetrahidrofuranilo, tetrahidrotienilo, tetrahidropiranilo, tetrahidrotiranilo, tetrahidropirrolilo, tetrahidroimidazolilo, tetrahidrotiazolilo, tetrahidropiranilo, bencimidazolilo, benzopiridilo, pirrolopiridilo, benzodihidrofurilo, 2-pirrolinilo, 3-pirrolinilo, dihidroindolilo, 2H-piranilo, 4H-piranilo, dioxano, 1,3-dioxolilo, pirazolinilo, ditiaalquilo, ditiacenilo, dihidrotienilo, pirazolidinilo, imidazolinilo, imidazolidinilo, 1,2,3,4-tetrahidroisoquinolinilo, 3-azabiciclo[3.1.0]hexilo, 3-azabiciclo[4.1.0]heptilo, azabiciclo[2.2.2]hexilo, 3H-indolilquinazinilo, N-piridilurea, 1,1-dioxotiomorfolinilo, azabiciclo[3.2.1]octilo, azabiciclo[5.2.0]nonanilo, oxatriciclo[5.3.1.1]dodecilo, azaadamantilo y oxaespiro[3.3]heptilo. El grupo heterocíclico puede estar opcionalmente sustituido adicionalmente con 0, 1, 2, 3, 4 ó 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, =O, hidroxilo, -SR19, nitro, ciano, alquilo C1-6, hidroxialquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8, grupo heterocíclico de 3 a 8 miembros, -(CH2)a-C(=O)-R19, -(CH2)k-C(=O)-O-R19, -(CH2)k-C(=O)-NR19R19a, -(CH2)k-S(=O)j-R19, -O-C(=O)-O-R19 o -NR19R19a. Los heterociclos del presente documento se definen tal como se describió anteriormente.
El “opcional” u “opcionalmente” significa que el evento o entorno descrito posteriormente puede producirse, pero no es necesario que se produzca, lo que indica un caso en el que el evento o entorno se produce o no se produce. Por ejemplo, “grupo alquilo opcionalmente sustituido con F” significa que el grupo alquilo puede, pero no es necesario que esté sustituido con F, lo que indica un caso en el que el grupo alquilo está sustituido con F y un caso en el que el grupo alquilo no está sustituido con F.
“Composición farmacéutica” significa una mezcla de uno o más de los compuestos descritos en el presente documento o una sal fisiológica/farmacéuticamente aceptable de los mismos, o un estereoisómero, solvato, sal farmacéuticamente aceptable o cocristal de los mismos y otros constituyentes. Donde otros componentes comprenden portadores y excipientes fisiológica/farmacéuticamente aceptables.
“Estereoisómero” significa isómeros que resultan de la disposición espacial de los átomos en una molécula, incluyendo isómeros cis y trans, enantiómeros e isómeros conformacionales.
“Dosis eficaz” significa la cantidad de un compuesto que provoca una respuesta fisiológica o médica a un tejido, sistema o sujeto, cuya cantidad se busca. Cuando se administra a un sujeto, es suficiente para prevenir la aparición o reducción de uno o más síntomas de las enfermedades o afecciones que están tratándose en cierta medida.
“Solvato” significa un compuesto de la invención o una sal del mismo, que también incluye una cantidad estequiométrica o no estequiométrica de disolvente unido por fuerzas intermoleculares no covalentes. Cuando el disolvente es agua, es un hidrato.
Descripción detallada
Las soluciones técnicas de la presente invención se describen con detalle a continuación con referencia a los dibujos y ejemplos adjuntos, pero el alcance de la presente invención los incluye, pero no los limita. El alcance de la presente invención está definido por las reivindicaciones adjuntas.
La estructura del compuesto se determina mediante resonancia magnética nuclear (RMN) o (y) espectrometría de masas (EM). El desplazamiento de RMN (8) se facilita en unidades de 10-6 (ppm). La RMN se midió usando un aparato magnético nuclear (Avance III 400 de Bruker y Avance 300 de Bruker), y el disolvente para la medición fue dimetilsulfóxido deuterado (DMSO-d6), cloroformo deuterado (CDCh), metanol deuterado (CD3OD) y el patrón interno es tetrametilsilano (TMS).
La EM se mide usando (6120B (ESI) de Agilent y 6120B (APCI) de Agilent).
La HPLC se mide usando un cromatógrafo de líquidos de alta presión 1260DAD de Agilent (Zorbax SB-C18 100 x 4,6 mm).
La placa de gel de sílice para cromatografía en capa fina es una placa de gel de sílice HSGF254 de Yantai Yellow Sea 0 GF254 de Qingdao. La placa de gel de sílice usada para la cromatografía en capa fina (CCF) tiene una especificación de 0,15 mm 0,20 mm, y el producto de purificación y separación mediante cromatografía en capa fina tiene una especificación de 0,4 mm 0,5 mm.
La cromatografía en columna usa generalmente gel de sílice de malla 200-300 de gel de sílice Yantai Huanghai como portador.
Los materiales de partida conocidos de la presente invención pueden sintetizarse mediante o según métodos conocidos en la técnica, o pueden adquirirse de Titan Technology, Energy Chemical, Shanghai DEMO, Chengdu Kelong Chemical, Accela ChemBio Co.Ltd y J&K Scientific Ltd, y similares.
La atmósfera de nitrógeno se refiere al frasco de reacción conectado a un globo de nitrógeno de aproximadamente 1 l de volumen.
La atmósfera de hidrógeno se refiere al frasco de reacción conectado a un globo de hidrógeno de aproximadamente 1 l de volumen.
La reacción de hidrogenación generalmente se evacua y se carga con hidrógeno, y la operación se repite 3 veces. A menos que se indique especialmente en los ejemplos, se permitió que avanzase toda la reacción en una atmósfera de nitrógeno.
A menos que se indique especialmente en los ejemplos, toda las disoluciones significan una disolución acuosa. A menos que se indique especialmente en los ejemplos, todas las temperaturas de reacción son la temperatura ambiente.
La temperatura ambiente es la temperatura de reacción óptima, que oscila entre 20°C y 30°C.
A menos que se indique especialmente en los ejemplos, todo M es mol/l.
Boc se refiere al grupo terc-butiloxicarbonilo.
Cbz se refiere al grupo benciloxicarbonilo.
THP se refiere al grupo tetrahidropiranilo.
Producto intermedio 1:
Ácido (2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]-amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoico (producto intermedio 1)

Etapa 1:
(2R)-2-[[(2R)-2-(Benciloxicarbonilamino)-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1b)

Se disolvió (2R)-2-amino-6-(terc-butoxicarbonilamino)hexanoato de metilo (1a) (2,6 g, 10mmol) en acetato de etilo (50 ml) a temperatura ambiente y se enfrió la temperatura hasta 0°C. Se añadieron secuencialmente a la disolución de reacción ácido (2R)-2-(benciloxicarbonilamino)-4-metil-pentanoico (2,8 g, 11 mmol), 1-hidroxibenzotriazol (1,62 g, 12 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (2,3 g, 12 mmol), y se elevó la temperatura hasta 25°C, y se permitió que avanzase la reacción a esta temperatura durante 15 h. Se añadió una disolución acuosa de ácido clorhídrico 1 M (25 ml) a la disolución de reacción para lavar la reacción y se sometió la mezcla a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de bicarbonato de sodio (25 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y se sometió la mezcla a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa de ácido clorhídrico 1 M (25 ml), una disolución acuosa saturada de bicarbonato de sodio (25 ml), una disolución acuosa saturada de cloruro de sodio (25 ml) en este orden y se secó sobre sulfato de sodio anhidro (2 g). Se filtró y se concentró el filtrado a presión reducida para obtener (2R)-2-[[(2R)-2-(benciloxicarbonilamino)-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1b) como un sólido espumoso de color blanco (5,0 g, rendimiento del 99%).
Etapa 2:
(2R)-2-[[(2R)-2-Amino-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1c)

Se disolvió (2R)-2-[[(2R)-2-(benciloxicarbonilamino)-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1b) (5,0 g, 10 mmol) en acetato de etilo (50 ml) a temperatura ambiente y se añadió paladio sobre carbono al 10% (1 g, al 20% p/p) a la disolución de reacción, y se reemplazó la atmósfera por hidrógeno 3 veces. Se permitió que avanzase la reacción en una atmósfera de hidrógeno a temperatura ambiente durante 5 h. Se filtró la disolución de reacción a través de tierra de diatomeas (3 g) y se concentró el filtrado a presión reducida para obtener (2R)-2-[[(2R)-2-amino-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1c) en bruto como un sólido espumoso de color blanco (3,7 g, rendimiento del 99%) y se usó directamente en la siguiente reacción.
Etapa 3:
(2R)-2-[[(2R)-2-[[(2R)-2-(Benciloxicarbonilamino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]-6-(tercbutoxicarbonilamino)hexanoato de metilo (1d)

Se disolvió (2R)-2-[[(2R)-2-amino-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1c) en bruto (3,7 g, 9,9 mmol) en acetato de etilo (50 ml) a temperatura ambiente y se enfrió la temperatura hasta 0°C. Se añadieron secuencialmente a la disolución de reacción ácido (2R)-2-(benciloxicarbonilamino)-3-fenilpropanoico (3,3 g, 11 mmol), 1-hidroxibenzotriazol (1,62 g, 12 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (2,3 g,
12 mmol), y se elevó la temperatura hasta 25°C, y se permitió que avanzase la reacción a esta temperatura durante 5 h. Se añadió ácido clorhídrico acuoso 1 M (25 ml) para lavar la reacción y se sometió la mezcla a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de bicarbonato de sodio (25 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y se sometió la mezcla a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa de ácido clorhídrico 1 M (25 ml), una disolución acuosa saturada de bicarbonato de sodio (25 ml), una disolución acuosa saturada de cloruro de sodio (25 ml) en este orden y se secó sobre sulfato de sodio anhidro (2 g). Se filtró y se concentró el filtrado a presión reducida para obtener (2R)-2-[[(2R)-2-[[(2R)-2-(benciloxicarbonilamino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1d) como un sólido espumoso de color blanco (3,0 g, rendimiento del 46%), y se usó directamente en la siguiente reacción.
Etapa 4:
(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]-6-(tercbutoxicarbonilamino)hexanoato de metilo (1e)

Se disolvió (2R)-2-[[(2R)-2-[[(2R)-2-(benciloxicarbonilamino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]-6-(tercbutoxicarbonilamino)hexanoato de metilo (1d) en bruto (3,0 g, 4,58 mmol) en acetato de etilo (50 ml) a temperatura ambiente, se añadió paladio sobre carbono al 10% (1 g, al 33% p/p) a la disolución de reacción y se reemplazó la atmósfera por hidrógeno 3 veces. Se permitió que avanzase la reacción a temperatura ambiente durante 5 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas (3 g) y se concentró el filtrado hasta sequedad a presión reducida. Se le añadió acetato de etilo (6 ml) y se calentó la mezcla hasta que se disolvió. Después de añadir éter de petróleo (6 ml), se redujo lentamente la temperatura hasta temperatura ambiente para precipitar un sólido y se filtró. Se secó la torta de filtración a 50°C a presión reducida para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de metilo (1e) como un sólido espumoso de color blanco (2,1 g, rendimiento del 88%).
Etapa 5:
(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoato de metilo (1f)

Se disolvió (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]-6-(tercbutoxicarbonilamino)hexanoato de metilo (1e) (2,1 g, 4,0 mmol) en acetato de etilo (30 ml) a temperatura ambiente y se redujo la temperatura hasta 0°C. Se añadieron secuencialmente a la disolución de reacción ácido (2R)-2-(tercbutoxigencarbonil)-3-fenilpropanoico (1,3 g, 4,9 mmol), 1-hidroxibenzotriazol (0,65 g, 4,8 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (1,1 g, 5,7 mmol), y se elevó la temperatura hasta 25°C y se permitió que avanzase la reacción a esta temperatura durante 5 h. Se añadió ácido clorhídrico acuoso 1 M (15 ml) para lavar la reacción y se sometió la mezcla a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de bicarbonato de sodio (15 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y se sometió la mezcla a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa de ácido clorhídrico 1 M (15 ml), una disolución acuosa saturada de bicarbonato de sodio (15 ml), una disolución acuosa saturada de cloruro de sodio (15 ml) en este orden y se secó sobre sulfato de sodio anhidro. Se filtró y se concentró el filtrado a presión reducida, se separó y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo/acetato de etilo (v/v)
=100:1 ~5:1) para obtener (2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoato de metilo (1f) como un sólido espumoso de color blanco (2,3 g, rendimiento del 74%).
Etapa 6:
Ácido (2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoico (producto intermedio 1)

Se disolvió (2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoato de metilo (1f) (2,3 g, 3,0 mmol) en metanol (20 ml) a temperatura ambiente. Se añadió una disolución acuosa de hidróxido de sodio (200 mg, 5,0 mmol) (20 ml) a la disolución de reacción y se permitió que avanzase la reacción a esta temperatura durante 5 h. Se ajustó la disolución de reacción a pH <4 con una disolución acuosa de ácido clorhídrico 1 M, luego se extrajo con acetato de etilo (40 ml) y se sometió la mezcla a un proceso de separación de líquido. Se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró, y se concentró el filtrado a presión reducida para obtener ácido (2R)-6-(tercbutoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanoil]amino]hexanoico (producto intermedio 1)como un sólido espumoso de color blanco (2,1 g, rendimiento del 93%).
EM m/z (ESI): 752,5 [M-H]-;
1H-RMN (400 MHz, CDCls) 87,38-7,27 (m, 3H), 7,25-7,07 (m, 7H), 4,82-4,62 (m, 1H), 4,61-4,41 (m, 2H), 4,37-4,18 (m, 1H), 3,37-2,67 (m, 6H), 2,00-1,65 (m, 3H), 1,59-1,37 (m, 15H), 1,35-1,26 (m, 9H), 0,90-0,80 (m, 6H).
Ejemplo 1:
Trifluoroacetato del ácido 2-amino-7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxílico

Etapa 1: 2-[(1S)-1-feniletil]imino-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (2B)

Se disolvieron 2-oxo-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (2A) (7,2 g, 30 mmol), (1S)-1-fenetilamina (3,7 g, 31 mmol) en tolueno (100 ml) y se añadió ácido p-toluenosulfónico (300 mg, 1,74 mmol). Se usó un aparato Dean-Stark para calentar el sistema a reflujo para separar el agua. Después de 6 h, se concentró la disolución de reacción hasta sequedad a presión reducida para obtener 2-[(1S)-1-feniletil]imino-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (2B) en bruto como un sólido espumoso de color amarillo (10 g, rendimiento del 97%).
Etapa 2:
2-Ciano-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (2C)

Se disolvió 2-[(1S)-1-feniletil]imino-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (2B) en bruto (10 g, 29,2 mmol) en metanol (90 ml) a temperatura ambiente y se enfrió hasta 0°C en un baño de hielo. Se añadió cloruro de zinc (210 mg, 1,54 mmol) con agitación y se añadió lentamente gota a gota cianuro de trimetilsililo (3 g, 30,2 mmol). Se mantuvo la reacción a 0°C durante 3 h. Se filtró la disolución de reacción y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v/v) = 4:1) para obtener 2-ciano-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-7-carboxilato de terc
butilo (2C) como un sólido espumoso de color amarillo (4,7 g, rendimiento del 44%).
Etapa 3: ácido 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxílico (2D)

Se disolvió 2-ciano-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (2C) (2 g, 5,4 mmol) en ácido clorhídrico concentrado (20 ml) a temperatura ambiente y luego se calentó la mezcla a reflujo durante 40 h. Se redujo la temperatura hasta temperatura ambiente y se concentró la disolución de reacción a presión reducida para obtener ácido 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxílico (2D) en bruto como un líquido oleoso de color amarillo (1,5 g, rendimiento del 96%) y se usó directamente en la siguiente reacción.
Etapa 4:
Ácido 7-terc-butoxicarbonil-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxílico (2E)

Se disolvió ácido 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxílico (2D) en bruto (1 g, 3,47 mmol) en tetrahidrofurano (10 ml) a temperatura ambiente. Se añadió una disolución acuosa de hidróxido de sodio (0,5 g, 12,5 mmol) (10 ml), luego se añadió dicarbonato de di-terc-butilo (908 mg, 4,16 mmol) y se permitió que avanzase la reacción a temperatura ambiente durante 6 h. Se filtró la disolución de reacción para obtener ácido 7-tercbutoxicarbonil-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxílico (2E) en bruto como un sólido de color blanco (0,8 g, rendimiento del 60%).
Etapa 5: 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2,7-dicarboxilato de O7-terc-butilo y O2-metilo (2F)

Se disolvió ácido 7-terc-butoxicarbonil-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxílico (2E) (775 mg, 2,0 mmol) en diclorometano (10 ml) a temperatura ambiente y se añadió metanol (10 ml). Se añadieron secuencialmente a la disolución 1-hidroxibenzotriazol (270 mg, 2,0 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,6 g, 3,13 mmol) con agitación a temperatura ambiente y se permitió que reaccionase el sistema durante 15 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:1) para obtener 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2,7-dicarboxilato de O7-terc-butilo y O2-metilo (2F) como un sólido espumoso de color amarillo claro (560 mg, rendimiento del 70%).
Etapa 6: 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2G)

Se disolvió 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2,7-dicarboxilato de O7-terc-butilo y O2-metilo (2F) (500 mg, 1,24 mmol) en diclorometano (4,5 ml) a temperatura ambiente y se redujo la temperatura hasta 0°C. Se añadió ácido trifluoroacético (1,5 ml) y se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se concentró la disolución de reacción a presión reducida para obtener 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2G) como un líquido oleoso de color amarillo (320 mg, rendimiento 85%).
Etapa 7:
metilo
7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxilato de metilo

Se disolvió 2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2G) (300 mg, 1,0mmol) en diclorometano (10 ml) a temperatura ambiente, y se añadió el producto intermedio 1 (753 mg, 1,0 mmol). Se añadieron secuencialmente 1-hidroxibenzotriazol (135 mg, 1,0 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,23 g, 1,2 mmol) con agitación a temperatura ambiente y se permitió que reaccionase el sistema durante 15 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1). Se recogió el eluyentey se concentró a presión reducida para obtener 7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R))-2-[[(2R)fenil-propanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2H) como un sólido espumoso de color blanco (710 mg, rendimiento del 69%).
Etapa 8:
2-Amino-7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2I)

Se disolvió 7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R))-2-[[(2R)fenilpropanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2-[[(1S)-1-feniletil]amino]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2H) (700 mg, 0,7 mmol) en acetato de etilo (10 ml) a temperatura ambiente y se añadió paladio sobre carbono (0,1 g, al 20% en peso) a la disolución de reacción. Se reemplazó la atmósfera por hidrógeno 3 veces y se permitió que avanzase la reacción a temperatura ambiente durante 5 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado hasta sequedad. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1), para obtener 2-amino-7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2I) como un sólido espumoso de color blanco (370 mg, rendimiento del 60%).
Etapa 9: metilo
2-Amino-7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-p-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxilato de metilo; ácido 2,2,2-trifluoroacético (2j )
Se disolvió 2-amino-7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxilato de metilo (2I) (37o mg, 0,4 mmol) en diclorometano (3 ml) a temperatura ambiente y se redujo la temperatura hasta 0°C. Se añadió ácido trifluoroacético (1 ml) y se elevó la temperatura hasta temperatura ambiente y se permitió que reaccionase el sistema durante 3 h. Se concentró la disolución de reacción hasta sequedad a presión reducida para obtener 2-amino-7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R))-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nona-2-carboxilato; tri-ácido trifluoroacético (2J) en bruto como un líquido oleoso de color amarillo (305 mg, rendimiento del 72%).
Etapa 10:
Ácido 2-amino-7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxílico; tri-ácido trifluoroacético (compuesto 2)

Se disolvió hidróxido de sodio (50 mg, 1,25 mmol) en agua (2 ml) a temperatura ambiente y se añadió 2-amino-7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxilato; tri-ácido trifluoroacético (2J) (305 mg, 0,288 mmol). Se permitió que reaccionase el sistema durante 5 horas a temperatura ambiente. Se concentró la disolución de reacción hasta sequedad a presión reducida, y se separó y purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 mmol) [[(2R)-2-met; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la disolución de preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener ácido 2-amino-7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-carboxílico; tri-ácido trifluoroacético (compuesto 2) (92 mg, rendimiento del 31%).
EM m/z (ESI): 360,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,44-7,18 (m, 10H), 4,65 (t, 1H), 4,33-4,18 (m, 2H), 3,58 (a, 2H), 3,52-3,41 (m, 1H), 3,41 3,29 (m, 1H), 3,17 (d, 2H), 3,10-2,90 (m, 4H), 2,70-2,46 (m, 2H), 2,32-2,18 (m, 2H), 2,05-1,28 (m, 14H), 0,98-0,84 (m, 6H).
Compuesto 2-1 (compuesto 2 en forma libre):
Se hizo pasar el compuesto 2 (7,3 g, 6,88 mmol) a través de una resina de intercambio iónico (300 ml) (se eluyó con amoniaco - al 3,3% en agua) y se concentró la disolución de elución recibida a presión reducida (se concentró a presión reducida hasta 100 ml a 60°C) y se liofilizó adicionalmente para obtener el compuesto 2-1 como un sólido de color blanco (4,5 g, rendimiento del 90,8%).
EM m/z = 720,5 [M+2H]+;
1H-RMN (400 MHz, D2O) 87,34-7,22 (m, 6H), 7,18-7,06 (m, 4H), 4,78-4,72 (m, 1H), 4,55 (t, 1H), 4,25 (t, 1H), 3,65 3,46 (m, 3H), 3,45-3,25 (m, 2H), 3,09-2,97 (m, 1H), 2,95-2,84 (m, 3H), 2,85-2,73 (m, 2H), 2,51-2,33 (m, 2H), 2,00-1,83 (m, 2H), 1,82-1,25 (m, 13H), 0,96-0,78 (m, 6H).
Ejemplo 2:
(2R)-N-[(1R)-5-amino-1-(2-oxo-3,10-diazadiespiro[4,1.5A{7}.1A{5}]tridecano-10-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 3)
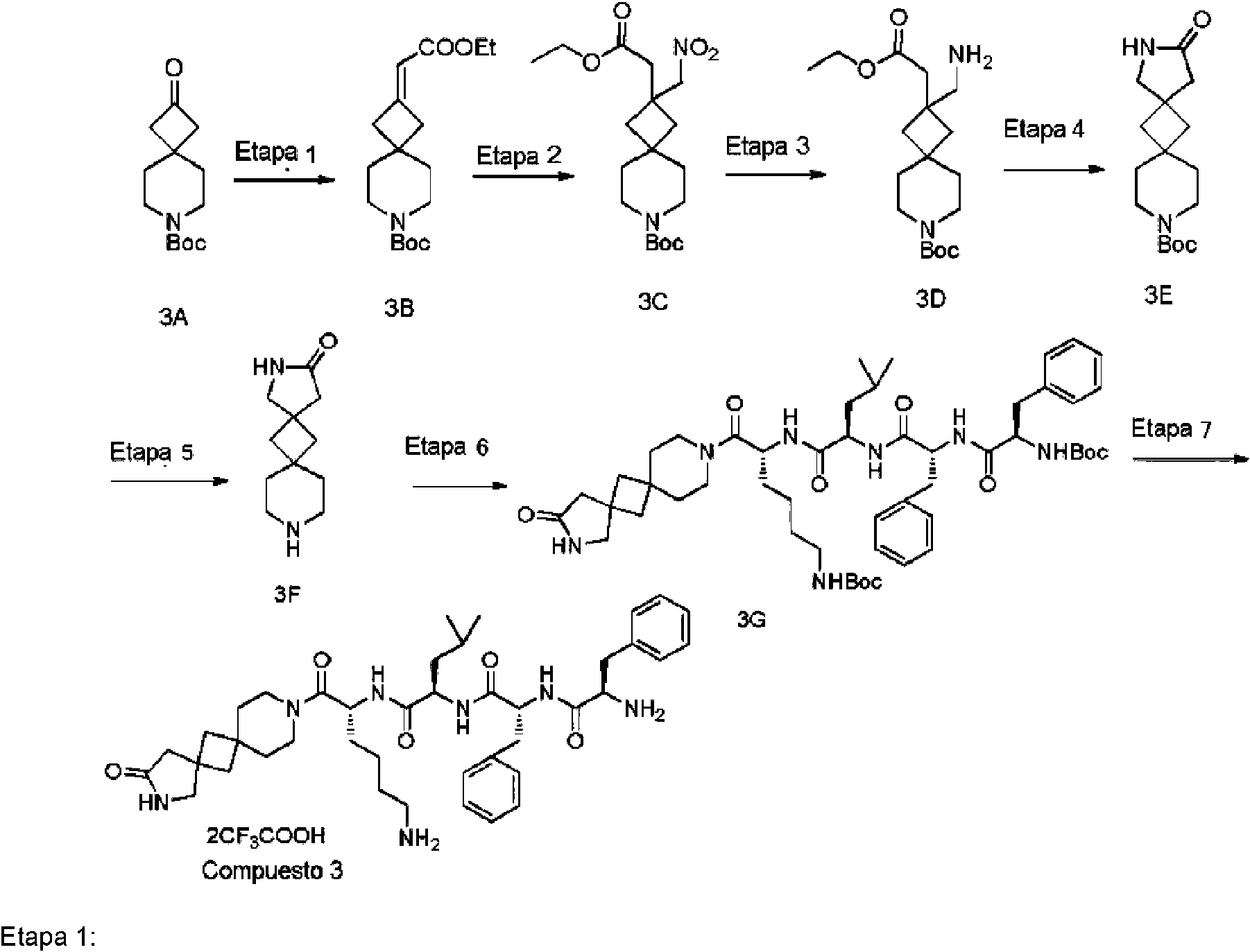
2-(2-Etoxi-2-oxo-etiliden)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3B)

Boc
Se añadió tetrahidrofurano (50 ml) a un matraz de reacción y se añadió hidruro de sodio (1,3 g, 54,2 mmol) bajo protección de nitrógeno. Se enfrió hasta 0°C en un baño de agua con hielo y se añadió lentamente gota a gota fosfonoacetato de trietilo (6,9 g, 31 mmol). Después de la adición, se llevó a cabo la reacción a 0°C durante 20 minutos. Se enfrió hasta de -5 a 0°C y se añadió lentamente gota a gota 2-oxo-7-azaespiro[3.5]nonano-7-carboxilato de tercbutilo (3A) (5 g, 20,9 mmol) en tetrahidrofurano (20 ml). Después de la adición, se elevó la temperatura hasta temperatura ambiente y se hizo reaccionar durante 1 h. Se añadió una disolución acuosa saturada de cloruro de sodio (50 ml) y se extrajo la mezcla con acetato de etilo (50 ml x 2). Se combinaron las fases orgánicas y se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 2-(2-etoxi-2-oxo-etiliden)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3B) en bruto como un líquido oleoso de color amarillo claro (6,0 g, rendimiento del 92,8%), y se usó directamente en la siguiente etapa.
Etapa 2:
2-(2-Etoxi-2-oxo-etil)-2-(nitrometil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3C)

Se añadió 2-(2-etoxi-2-oxo-etiliden)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3B) a un matraz de reacción y se añadió tetrahidrofurano (60 ml). Se disolvió completamente a temperatura ambiente con agitación, luego se añadieron nitrometano (6,0 g, 98,3 mmol) y fluoruro de tetrabutilamonio (7,85 g, 30 mmol). Después de la adición, se calentó la reacción a reflujo durante 5 h. Se enfrió la disolución de reacción hasta temperatura ambiente, se añadió acetato de etilo (150 ml) y se usó una disolución acuosa saturada de cloruro de sodio (100 ml x 1) para el lavado y la separación. Se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró y se concentró hasta sequedad a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 10:1) para obtener 2-(2-etoxi-2-oxo-etil)-2-(nitrometil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3C) como un líquido oleoso transparente incoloro (6,5 g, rendimiento del 90%).
Etapa 3:
2-(Aminometil)-2-(2-etoxi-2-oxo-etil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3D)

Se añadió 2-(2-etoxi-2-oxo-etil)-2-(nitrometil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3C) (6,5 g, 18 mmol) a un matraz de reacción y se añadieron etanol (75 ml) y agua (25 ml), polvo de hierro (4,9 g, 88 mmol) y cloruro de amonio (4,7 g, 88,00 mmol) y se calentó la reacción a reflujo durante 5 h. Se enfrió la temperatura hasta temperatura ambiente y se concentró el sistema de reacción hasta 20 ml. Se añadió agua (30 ml), se ajustó el pH a más de 10 con agua amoniacal y se extrajo con diclorometano (50 ml x 2). Se combinaron las fases orgánicas y se concentraron las fases orgánicas hasta sequedad a presión reducida para obtener 2 -(aminometil)-2 -(2 -etoxi-2 -oxoetil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3D) como un líquido oleoso de color amarillo claro (5,1 g, rendimiento del 85%).
Etapa 4:
2-Oxo-3,10-diazadiespiro[4.1.5A{7}.1A{5}]tridecano-10-carboxilato de terc-butilo (3E)
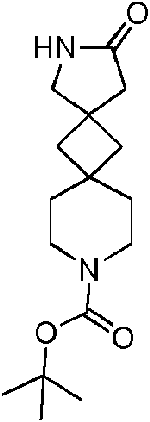
Se añadió 2-(aminometil)-2-(2-etoxi-2-oxo-etil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3D) (3 g, 8,8 mmol) a un matraz de reacción, se añadió una disolución acuosa de hidróxido de sodio (400 mg, 10 mmol) (30 ml) y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se filtró la disolución de reacción para obtener 2-oxo-3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecano-10-carboxilato de terc-butilo (3E) como un sólido de color blanco (2 ,10 g, rendimiento del 81 %).
Etapa 5:
3,10-diazadiespiro[4.1.5A{7}.1 A{5}]tridecan-2-ona (3F)

Se disolvió 2-oxo-3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecano-10-carboxilato (3E) (1 g, 3,4 mmol) en diclorometano (9 ml) y se añadió ácido trifluoroacético (3 ml). Se permitió que avanzase la reacción a temperatura ambiente durante 5 h para que reaccionase por completo. Se concentró la disolución de reacción hasta sequedad a presión reducida, se añadió agua (20 ml) y el sistema se ajustó a pH> 10 con una disolución acuosa de hidróxido de sodio 2 M. Se extrajo con diclorometano (20 ml x 2) y se sometió la mezcla a un proceso de separación de líquido. Se combinaron las fases orgánicas y concentraron a presión reducida para obtener 3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecano-2-ona (3F) en bruto como un líquido oleoso de color amarillo (0,6 g, rendimiento del 85%).
Etapa 6:
N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-oxo-3,10-diazaespiro[A{7}.1A{5}]tridecano-10-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (3G)

Se añadió diclorometano (30 ml) a un matraz de reacción bajo protección de nitrógeno y se añadieron el producto intermedio 1 (2,25g, 2,98 mmol), 3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecan-2-ona (3F) en bruto (0,6 g, 2,88 mmol), 1-hidroxibenzotriazol (0,39 g, 2,89 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,83 g, 4,33 mmol). Se permitió que reaccionase el sistema a temperatura ambiente durante 15 h. Luego se añadió agua (20 ml) y se extrajo con acetato de etilo (30 ml x 3), se separaron las fases y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 20:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-oxo-3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecano-10-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (3G) como un sólido espumoso de color blanco (1,77 g, rendimiento del 73,2%).
Etapa 7:
(2R)-N-[(1R)-5-amino-1-(2-oxo-3,10-diazadiespiro[4.1.5A{7}.1A{5}]tridecano-10-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 3)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)1]-(2-oxo-3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecano-10-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (3G) (0,90g, 1 mmol) y diclorometano (21 ml) a un matraz de reacción bajo protección con nitrógeno, y se añadió ácido trifluoroacético (7 ml) con agitación. Se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se concentró la disolución de reacción hasta sequedad a presión reducida y se separó el residuo y se purificó (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la
muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(2-oxo-3,10-diazaespiro[4.1.5A{7}.1A{5}]tridecano-10-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3 -fenilpropanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 3) como un sólido de color blanco (0,98 g, rendimiento del 54%).
EM m/z (ESI): 365,9 [M+2H]+/2;
1H-RMN (400 MHz, DMSO-da) 88,86-8,67 (m, 1H), 8,40-8,23 (m, 1H), 8,14-8,01 (m, 3H), 7,84-7,72 (m, 2H), 7,52-7,42 (m, 1H), 7,37-7,15 (m, 10H), 4,76-3,94 (m, 4H), 3,59-2,65 (m, 12H), 2,29-2,15 (m, 2H), 1,93-1,19 (m, 17H), 0,88 (dd, 6H).
Ejemplo 3:
(2R)-N-[(1R)-5-amino-1-(9-oxo-2,8-diazadiespiro[3.1.4A{6}.1A{4}]undecano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 4)

Etapa 1: 6-(2-etoxi-2-oxo-etiliden)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4B)

Boc
Se añadió tetrahidrofurano (50 ml) a un matraz de reacción y se añadió hidruro de sodio (2,5 g, 59,2 mmol). Se enfrió hasta 0°C en un baño de agua con hielo y se añadió lentamente gota a gota fosfonoacetato de trietilo (7,96 g, 35,5 mmol). Se permitió que avanzase la reacción a 0°C durante 20 min después de la adición. Luego se enfrió hasta de -5 a 0°C, y se añadió gota a gota lentamente 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4A) (5 g, 23,7 mmol) en tetrahidrofurano (20 ml). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 1 h. Luego, se añadió una disolución acuosa saturada de cloruro de sodio (50 ml) a la disolución de reacción, y se extrajo la mezcla con acetato de etilo (50 ml x 2) y se sometió la mezcla a un proceso de separación de
líquido. Se combinaron las fases orgánicas. Se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida para obtener 6-(2-etoxi-2-oxo-etiliden)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4B) en bruto como un líquido oleoso de color amarillo (5,5 g, rendimiento del 83%), y se usó directamente en la siguiente etapa.
Etapa 2:
6-(2-Etoxi-2-oxo-etil)-6-(nitrometil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4C)

Se añadió 6-(2-etoxi-2-oxo-etiliden)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4B) en bruto a un matraz de reacción y se añadió tetrahidrofurano (60 ml). Se agitó el sistema para que se disolviera por completo a temperatura ambiente, luego se añadieron nitrometano (6,0 g, 98,3 mmol) y fluoruro de tetrabutilamonio (7,85 g, 30 mmol). Una vez completada la adición, se calentó la reacción a reflujo durante 5 h. Se enfrió la disolución de reacción hasta temperatura ambiente, se le añadió acetato de etilo (150 ml) y se lavó la mezcla con una disolución acuosa saturada de cloruro de sodio (100 ml) y se sometió la mezcla a un proceso de separación de líquido. Se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1), para obtener 6-(2-etoxi-2-oxo-etil)-6-(nitrometil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4C) como un líquido oleoso transparente incoloro (5,1 g, rendimiento del 76%).
Etapa 3:
6-(Aminometil)-6-(2-etoxi-2-oxo-etil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4D)

Se añadió 6-(2-etoxi-2-oxo-etil)-6-(nitrometil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4C) (5,1 g, 15 mmol) a un matraz de reacción, se añadieron etanol (75 ml), agua (25 ml), polvo de hierro (4,2 g, 74 mmol) y cloruro de amonio (4,0 g, 74 mmol) y se calentó la reacción a reflujo durante 5 h. Se redujo la temperatura hasta temperatura ambiente y se concentró el sistema de reacción hasta aproximadamente 20 ml. Se añadió agua (30 ml), luego se ajustó el pH a más de 10 con amoniaco, se extrajo con diclorometano (50 ml x 2) y se sometió la mezcla a un proceso de separación de líquido. Se combinaron las fases orgánicas y se concentraron las fases orgánicas a presión reducida para obtener 6-(aminometil)-6-(2-etoxi-2-oxo-etil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4D) en bruto (4,6 g, rendimiento del 99%) y se usó directamente en la siguiente etapa.
Etapa 4:
9-Oxo-2,8-diazadiespiro[3.1.4A{6}.1A{4}]undecano-2-carboxilato de terc-butilo (4E)

N
Boc
Se añadió 6-(aminometil)-6-(2-etoxi-2-oxo-etil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (4D) en bruto (4,6 g, 15 mmol) a un matraz de reacción. Se añadió una disolución acuosa de hidróxido de sodio (600 mg, 15 mmol)
(45 ml) y se hizo reaccionar la mezcla a temperatura ambiente durante 5 h. Se filtró la disolución de reacción para obtener 9-oxo-2,8-diazaespiro[3.1.4A{6}.1A{4}]undecano-2-carboxilato de terc-butilo (4E) como un sólido de color blanco (2,90 g, rendimiento del 74%).
Etapa 5:
Clorhidrato de 2,8-diazadiespiro[3.1.4A{6}.1A{4}]undecan-9-ona (4F)

Se disolvió 9-oxo-2,8-diazaespiro[3.1.4A{6}.1A{4}]undecano-2-carboxilato de terc-butilo (4E) (2 g, 7,5 mmol) en una disolución de HCl 4 N-isopropanol (9 ml) y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción hasta sequedad a presión reducida, para obtener clorhidrato de 2,8-diazaespiro[3.1.4A{6}.1A{4}]undecan-9-ona (4F) en bruto como un sólido de color blanco (0,95 g, rendimiento del 62%) y se usó directamente en la siguiente etapa.
Etapa 6:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(9-oxo-2,8-diazadiespiro[3.1.4A{6}.1A{4}]undecano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (4G);

Se añadió diclorometano (30 ml) a un matraz de reacción bajo protección de nitrógeno y se añadieron el producto intermedio 1 (2,25g, 2,98 mmol), clorhidrato de 2,8-diazaespiro[3.1.4A{6}.1A{4}]undecan-9-ona (4F) (0,6g, 3,0 mmol), 1-hidroxibenzotriazol (0,39 g, 2,89 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,83 g, 4,33 mmol). Se permitió que avanzase la reacción a temperatura ambiente durante 15 h. Luego se añadió agua (20 ml) y se extrajo la mezcla con acetato de etilo (30 ml x 3) y se sometió la mezcla a un proceso de separación de líquido. Se combinaron las fases orgánicas y se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 10:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-(9-oxo-2,8-diazaespiro[3.1.4A{6}.1A{4}]undecano-2-carbonil)pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (4G) como un sólido espumoso de color blanco (1,9 g, rendimiento del 70%).
Etapa 7:
(2R)-N-[(1R)-5-amino-1-(9-oxo-2,8-diazadiespiro[3.1.4A{6}.1A{4}]undecano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 4)

N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)1]-(9-oxo-2,8-diazaespiro[3.1.4A{6}.1 A{4}]undecano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (4G) (1,7g, 1,9 mmol) y diclorometano (21 ml) a un matraz de reacción bajo protección con nitrógeno. Se añadió ácido trifluoroacético (7 ml) con agitación y se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se concentró la disolución de reacción hasta sequedad a presión reducida y se separó el residuo y se purificó mediante cromatografía preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(9-oxo-2,8-diazaespiro[3.1.4A{6}.1A{4}]undecano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 4) como un sólido de color blanco (0,7 g, rendimiento del 40%).
EM m/z (ESI): 351,8 [M+2H]+/2;
1H-RMN (400 MHz, DMSO-De) 88,76 (d, 1H), 8,32 (d, 1H), 8,11-7,97 (m, 4H), 7,85-7,69 (m, 3H), 7,50 (s, 1H), 7,33 7,18 (m, 10H), 4,74-3,74 (m, 8H), 3,24-2,63 (m, 8H), 2,27-2,05 (m, 6H), 1,71-1,17 (m, 9H), 0,89 (dd, 6H).
Ejemplo 4:
(2R)-N-[(1R)-5-Amino-1-(2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida (compuesto 6)

2,7-Diazaespiro[3.5]nonano-2,7-dicarboxilato de 7-bencilo y 2-terc-butilo (6B)

Se añadió gota a gota trietilamina (0,15 ml, 1,1 mmol) a un 2,7-diazaespiro[3.5]nonano-2-carboxilato de tercbutilo (6A) (0,23g, 1 mmol) en una disolución en tetrahidrofurano (5 ml) en un frasco de una sola boca de 50 ml a 0°C. Después de la adición gota a gota, se añadió cloroformiato de bencilo (340 mg, 2 mmol) y se agitó durante 10 min. Se elevó la temperatura hasta temperatura ambiente y se continuó agitando durante 1 h. Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de 7-bencilo y 2-terc-butilo (6B) como un producto oleoso incoloro (362 mg, rendimiento del 99%).
EM m/z = 383,2 [M+Na]+;
1H-RMN (400 MHz, CDCla) 87,34-7,32 (m, 5H), 5,12 (s, 2H), 3,64 (s, 4H), 3,43 (dd, 4H), 1,77-1,61 (m, 4H), 1,44 (s, 9H).
Etapa 2:
2,7-Diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C)

Se añadieron 2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de 7-bencilo y 2-terc-butilo (6B) (0,36 g, 1 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (1 ml) a temperatura ambiente. Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) en bruto como un líquido oleoso de color amarillo (260 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 261,2 [M+H]+;
Etapa 3:
2-Ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6D)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) en bruto (260mg, 1 mmol), trietilamina (200 mg, 2 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml. Se enfrió la disolución hasta 0°C en un baño de hielo y se añadió gota a gota cloruro de ciclopropilsulfonilo (170 mg, 1,2 mmol). Después de la adición, se elevó la temperatura hasta temperatura ambiente durante 4 h. Luego, se extinguió el sistema de reacción con bicarbonato de sodio saturado (10 ml), se extrajo con acetato de etilo (5 ml x 3) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1) para obtener el compuesto 2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6D) como un sólido de color blanco (360 mg, rendimiento del 99%).
EM m/z = 365,2 [M+H]+;
1H-RMN (400 MHz, CDCla) 87,43-7,28 (m, 5H), 5,12 (s, 2H), 3,70 (s, 4H), 3,53-3,36 (m, 4H), 2,41-2,22 (m, 1H), 1,85 1,69 (m, 4H)), 1,19-1,09 (m, 2H), 1,04-0,93 (m, 2H).
Etapa 4:
2-Ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano (6E)

Se añadieron 2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6D) (360 mg, 0,99 mmol), paladio sobre carbono (72 mg, al 20% en peso) y acetato de etilo (10 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se permitió que avanzase la reacción a temperatura ambiente durante 2 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano (6E) como un sólido de color amarillo claro (210 mg, rendimiento del 92%) y se usó directamente en la siguiente reacción.
Etapa 5:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (6f)
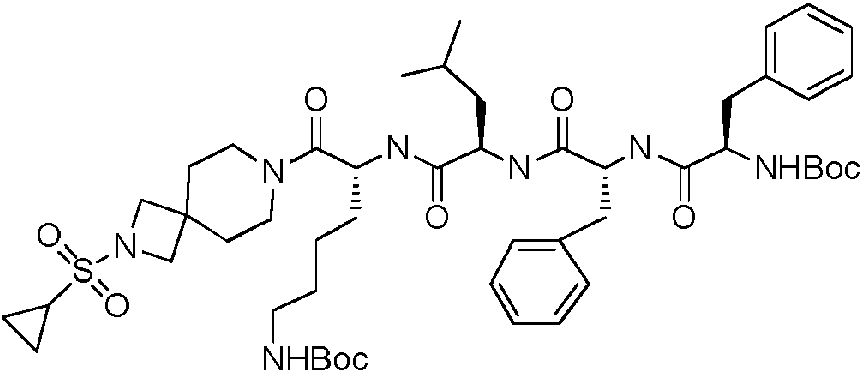
Se añadieron 2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano (6E) (161mg, 0,7 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (124 mg, 0,8 mmol), 1-hidroxibenzotriazol (108 mg, 0,8 mmol), producto intermedio 1 (500 mg, 0,7 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (6F) como un sólido de color blanco (610 mg, rendimiento del 95%).
Etapa 6:
(2R)-N-[(1R)-5-Amino-1-(2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida (compuesto 6)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)1]-(2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (6f) (300 mg, 0,31 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y
se liofilizó para obtener un compuesto pulverulento de color blanco. Luego, se usó la resina de intercambio iónico (se eluyó con agua hasta el 3,3% de amoniaco) para concentrar la disolución de elución recibida a presión reducida (se concentró a presión reducida a 25 ml a 60°C) y se liofilizó adicionalmente para obtener (2R)-N-[(1R)-5-amino-1-(2-ciclopropilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida (compuesto 6) como un sólido de color blanco (153 mg, rendimiento del 63%).
EM m/z = 383,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,40-7,24 (m, 6H), 7,16 (dd, 4H), 4,72-4,73 (m, 1H), 4,58-4,52 (m, 1H), 4,25 (t, 1H), 3,84 (dd'' 4H), 3,67-3,55 (m, 3H), 3,46-3,45 (m, 1H), 3,35-3,34 (m, 1H), 3,03 (dd, 1H), 2,91 (dd, 3H), 2,82 (d, 2H), 2,69 (ddd, 1H), 1,85-1,86 (m, 3H), 1,670-1,66 (m, 5H), 1,51-1,49 (m, 3H), 1,38-1,36 (m, 2H), 1,23-1,06 (m, 4H)), 0,88 (dd, 6H).
Ejemplo 5:
Di-trifluoroacetato de (2R)-N-[(1R)-5-amino-1-(2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida (compuesto 7)

Etapa 1:
2-Metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (7A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (330mg, 1,3mmol), trietilamina (263 mg, 2,6 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml y se agitó para disolverlo. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de metanosulfonilo (164 mg, 1,43 mmol) y se permitió que avanzase la reacción durante 4 h. Luego se elevó la temperatura hasta temperatura ambiente. Se lavó la disolución de reacción con una disolución acuosa saturada de bicarbonato de sodio (60 ml), una disolución acuosa de ácido clorhídrico 3 N (60 ml) y se sometió la mezcla a un proceso de separación de líquido. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:1) para obtener 2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (7A) como una sustancia oleosa de color amarillo claro (236 mg, rendimiento del 54,6%).
EM m/z = 339,0 [M+H]+;
1H-RMN (400 MHz, CDCls) 87,41-7,28 (m, 5H), 5,12 (s, 2H), 3,68 (s, 4H), 3,50-3,38 (m, 4H), 2,86 (s, 3H), 1,83-1,70 (m, 4H).
Etapa 2:
2-Metilsulfonil-2,7-diazaespiro[3.5]nonano (7B)

Se añadieron 2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (7A) (236 mg, 0,7 mmol), paladio sobre carbono (40 mg, al 20% en peso) y metanol (20 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se permitió que avanzase la reacción a temperatura ambiente durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeasy se concentró el filtrado a presión reducida para obtener 2-metilsulfonil-2,7-diazaespiro[3.5]nonano (7B) en bruto como un sólido de color amarillo claro (133 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 205,1 [M+1]+;
Etapa 3:
N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (7c )

Se añadieron 2-metilsulfonil-2,7-diazaespiro[3.5]nonano (7B) en bruto (133mg, 0,65 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (374 mg, 1,95 mmol), 1-hidroxibenzotriazol (96,6 mg, 0,72 mmol), producto intermedio 1 (490 mg, 0,65 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-(2-metilsulfonil-2,7)-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (7C) como un sólido de color amarillo claro (317 mg, rendimiento del 52%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-(2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida (compuesto 7)
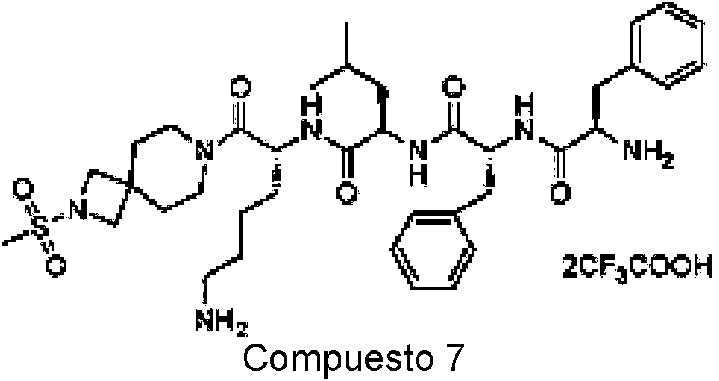
Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)1-(2)-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (7C) (317 mg, 0,34 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó por cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se recogió la preparación y se concentró a presión reducida para eliminar la mayor parte del disolvente orgánico y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]
4-metil-pentanamida (compuesto 7) como un polvo de color blanco (217 mg, rendimiento del 66,5%).
EM m/z = 370,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,43-7,27 (m, 6H), 7,25-7,20 (m, 4H), 4,67-4,62 (m, 2H), 4,33-4,20 (m, 2H), 3,87-3,71 (m, 4H), 3,69-3,56 (m, 2H), 3,53-3,41 (m, 1H), 3,40-3,29 (m, 1H), 3,23-3,12 (m, 2H), 3,11-3,07 (m, 3H), 3,07-2,91 (m, 4H)), 1,94-1,78 (m, 3H), 1,78-1,60 (m, 5H), 1,58-1,48 (m, 3H), 1,46-1,28 (m, 2H), 0,98-0,82 (m, 6H).
Ejemplo 6:
(2R)-N-[(1R)-1-(2-Acetil-2,7-diazaespiro[3.5]nonano-7-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 8)

2-Acetil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (8A)

Se disolvió 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (520 mg, 2,0 mmol) en diclorometano (5 ml) bajo protección de nitrógeno en un matraz de reacción de 50 ml y se añadió trietilamina (607 mg, 6,0 mmol) con agitación. Luego, se redujo la temperatura hasta -20°C y se añadió gota a gota cloruro de acetilo (314 mg, 4,0 mmol). Después de la adición, se elevó de manera natural la temperatura hasta temperatura ambiente y se agitó durante 2 h. Luego, se añadió a la reacción una disolución acuosa diluida de ácido clorhídrico 0,5 M (20 ml), se separaron las fases con agitación y se sometió la mezcla a un proceso de separación de líquido. Se extrajo la fase acuosa con diclorometano (20 ml x 2) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (acetato de etilo puro) para obtener el compuesto de 2-acetil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (8A) como un líquido oleoso de color amarillo claro (440 mg, rendimiento del 72,8%).
Etapa 2:
1-(2,7-Diazaespiro[3.5]nonan-2-il)etanona (8B)

Se añadió 2-acetil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (8A) (440 mg, 1,46 mmol) a una disolución mixta de acetato de etilo (5 ml) y metanol (2 ml) en un matraz de reacción de 50 ml. Luego se añadió paladio sobre carbono (80 mg, al 20% en peso) y se agitó el sistema bajo una atmósfera de hidrógeno (globo) a temperatura ambiente durante 2 h. Luego, se filtró la disolución de reacción y se concentró el filtrado a presión reducida para obtener 1-(2,7-diazaespiro[3.5]nonan-2-il)etanona (8B) en bruto como un líquido oleoso de color amarillo claro (250 mg, rendimiento del 99%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(2-acetil-2,7-diazaespiro[3.5]nonano-7-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (8C)

Se añadió 1-(2,7-diazaespiro[3.5]nonan-2-il)etanona (8B) en bruto (200 mg, 1,19 mmol) en acetato de etilo (10 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno. Se enfrió hasta 0°C en un baño de hielo, luego se añadieron el producto intermedio 1 (867 mg, 1,15 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (331 mg, 1,73 mmol), 1-hidroxibenzotriazol (186 mg, 1,38 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 1,5 h. Posteriormente, se añadió a la disolución de reacción una disolución acuosa de ácido clorhídrico 1 N (15 ml), y se agitó la mezcla y luego se sometió a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de carbonato de sodio (15 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y luego se sometió a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa saturada de cloruro de sodio (15 ml), se secó sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida para obtener N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(2-acetil-2,7-diazaespiro[3.5]nonano-7-carbonil)-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato de terc-butilo (8C) como un sólido espumoso de color amarillo claro (1,04 g, rendimiento del 99%) y se usó directamente en la siguiente reacción.
Etapa 4:
(2R)-N-[(1R)-1-(2-Acetil-2,7-diazaespiro[3.5]nonano-7-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 8)

Se disolvió N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(2-acetil-2,7-diazaespiro[3.5]nonano-7-carbonil)-5-(tercbutox¡carbon¡lam¡no)pent¡l]carbamo¡l]-3-metil-but¡ljam¡no]-í-benc¡l-2-oxo-et¡l]am¡no]-1-benc¡l-2-oxo-et¡l]carbamato de terc-butilo (8C) en bruto (1,04 g, 1,15 mmol) en diclorometano (7,5 ml) y se añadió ácido trifluoroacético (3,5 ml). Se agitó el sistema a temperatura ambiente durante 1 h. Posteriormente, se concentró la disolución de reacción a presión reducida. Después de que se separó el residuo y se purificó mediante cromatografía de líquidos preparativa
(condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja), se recogió la preparación y se concentró a presión reducida para eliminar la mayor parte del disolvente orgánico y se liofilizó para obtener (2R)-N-[(1R)-1-(2-acetil-2,7-diazaespiro[3.5]nonano-7-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 8) como un sólido de color blanco (460 mg, rendimiento en dos etapas del 42,9%).
EM m/z (ESI): 352,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,49-7,00 (m, 10H), 4,66-4,49 (m, 2H), 4,30-3,93 (m, 4H), 3,79-3,54 (m, 4H), 3,52-3,25 (m, 2H), 3,22-2,90 (m, 6H), 1,92-1,27 (m, 16H), 1,02-0,75 (m, 6H).
Compuesto 8-1:
Se hizo pasar el compuesto 8 (1,0 g, 1,07 mmol) a través de una resina de intercambio iónico (60 ml) (se eluyó con agua 3,3% de amoniaco) y se concentró la disolución de elución recibida a presión reducida (se concentró a presión reducida hasta 100 ml a 60°C) y se liofilizó adicionalmente para obtener el compuesto 8-1 como la forma libre del compuesto 8 como un sólido de color blanco (451 mg, rendimiento del 60,0%).
EM m/z (ESI): 352,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,45-7,32 (m, 6H), 7,23 (dd, 4H), 4,85-4,75 (m, 1H), 4,64 (t, 1H), 4,35 (t, 1H), 4,07 (d, 2H), 3,83 (d, 2H), 3,74-3,63 (m, 3H), 3,62-3,50 (m, 1H), 3,50-3,39 (m, 1H), 3,17-2,65 (m, 6H), 2,01-1,66 (m, 9H), 1,65-1,30 (m, 7H), 0,96 (dd, 6H).
Ejemplo 7:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo; di-ácido trifluoroacético (compuesto 9)

Etapa 1:
2,7-Diazaespiro[3.5]nonano-2,7-dicarboxilato de O7-bencilo y O2-isopropilo (9A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (310 mg, 1,2 mmol), trietilamina (364 mg, 3,6 mmol) y diclorometano (20 ml) en un matraz de una sola boca de 50 ml y se disolvió con agitación a temperatura ambiente. Luego se enfrió hasta -10°C y se añadió gota a gota cloroformiato de isopropilo (146 mg, 1,2 mmol). Después de la adición, se elevó la temperatura hasta temperatura ambiente y se permitió que avanzase la reacción durante 4 h. Se lavó el sistema de reacción secuencialmente con una disolución acuosa saturada de bicarbonato de sodio (60 ml), una disolución acuosa de ácido clorhídrico 3 M (60 ml) y se sometió la mezcla a un proceso de
separación de líquido. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:1) para obtener el compuesto 2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de O7-bencilo y O-isopropilo (9A) como un líquido oleoso de color amarillo claro (279 mg, rendimiento del 68%).
EM m/z = 347,2 [M+H]+;
1H-RMN (400 MHz, CDCls) 87,38-7,30 (m, 5H), 5,12 (s, 2H), 4,95-4,80 (m, 1H), 3,68 (s, 4H), 3,47-3,39 (m, 4H), 1,75 1,68 (m, 4H)), 1,23 (d, 6H).
Etapa 2:
2,7-Diazaespiro[3.5]nonano-2-carboxilato de isopropilo (9B)

Se añadieron 2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de O7-bencilo y O-isopropilo (9A) (260 mg, 0,75 mmol), paladio sobre carbono (52 mg, al 20% en peso de l) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo (9B) en bruto como un sólido de color amarillo claro (159 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 213,2 [M+1].
Etapa 3:
7-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-]fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo (9C)

Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo (9B) en bruto (159 mg, 0,75 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (374 mg, 1,95 mmol), 1-hidroxibenzotriazol (110 mg, 0,81 mmol), producto intermedio 1 (565 mg, 0,75 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener 7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo (9c ) como un sólido de color amarillo claro (556 mg, rendimiento del 78%).
Etapa 4:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo; di-ácido trifluoroacético (compuesto 9)

Se añadieron 7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo (9C) (317 mg, 0,334 mmol) y ácido trifluoroacético (2 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se purificó el residuo mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener 7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de isopropilo; di-ácido trifluoroacético (compuesto 9) como un producto pulverulento de color blanco (326 mg, rendimiento del 98%).
EM m/z = 374,9 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,50-7,14 (m, 10H), 4,89-4,78 (m, 1H), 4,66 (t, 1H), 4,30 (t, 1H), 4,22 (t, 1H), 3,89-3,74 (m, 4H), 3,69-3,54 (m, 2H), 3,54-3,41 (m, 1H), 3,41-3,28 (m, 1H), 3,21-3,11 (m, 2H), 3,11-2,90 (m, 4H), 1,93-1,30 (m, 14H), 1,27 (d, 6H), 0,93 (q, 6H).
Ejemplo 8:
(2R)-N-[(1R)-5-Amino-1-(2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 10)
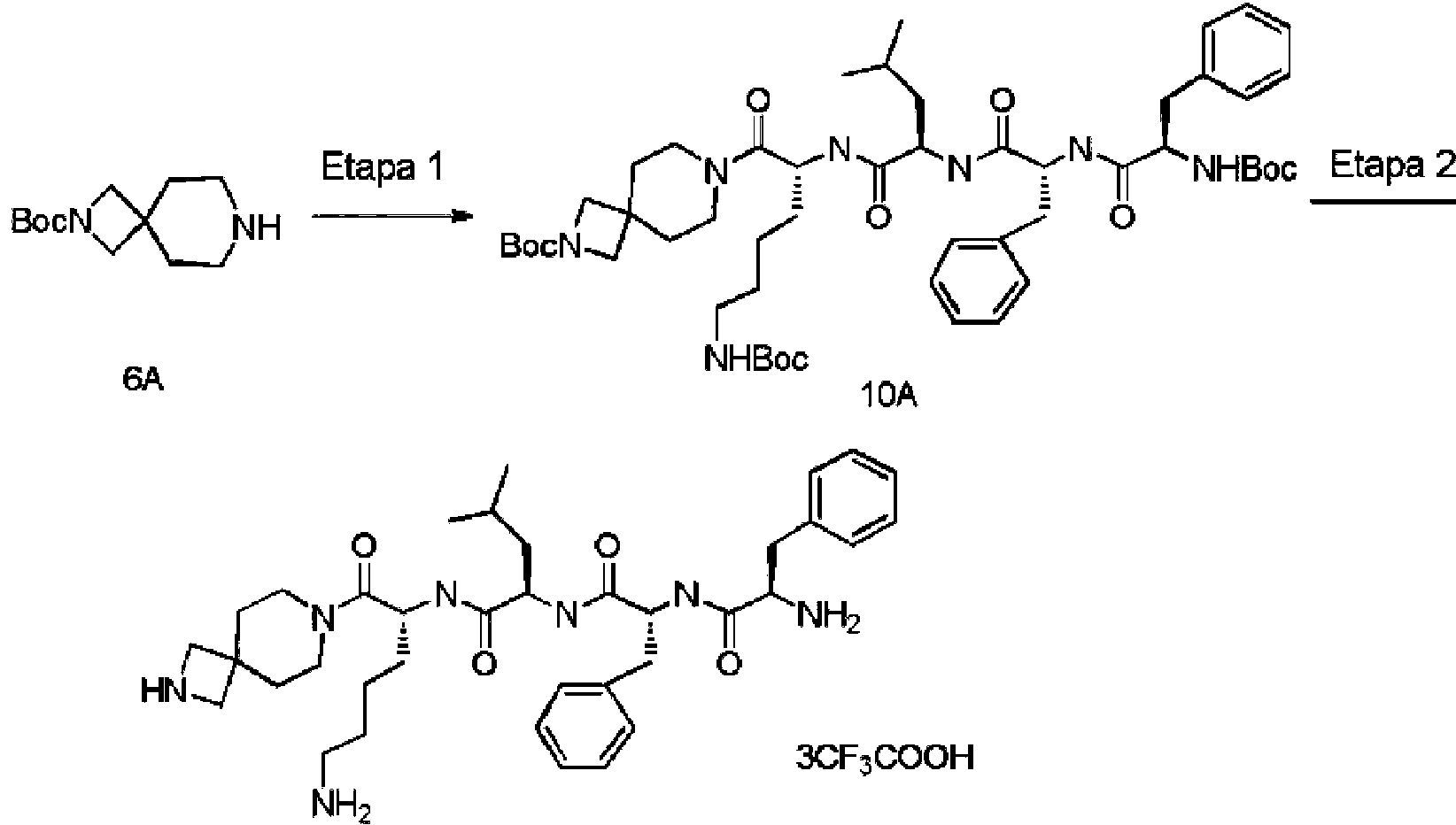
Com puesto 10
Etapa 1:
7-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (10a )

Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (0,11 g, 0,5 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (115 mg, 0,6 mmol), 1-hidroxibenzotriazol (81 mg, 0,6 mmol), producto intermedio 1 (378 mg, 0,5 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener 7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (10A)como un sólido de color blanco (450 mg, rendimiento del 93%).
Etapa 2:
(2R)-N-[(1R)-5-Amino-1-(2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 10)

Se añadieron 7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (10a ) (450 mg, 0,468 mmol) y ácido trifluoroacético (2 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida, se purificó el residuo por cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B Para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 10) como un producto pulverulento de color blanco (358 mg, rendimiento del 76%).
EM m/z = 331,8 [M-2CFsCOOH+2H]+/2;
1H-RMN (400 MHz, D2O) 87,48-7,10 (m, 10H), 4,65-4,61 (m, 2H), 4,28-4,20 (m, 2H), 3,93 (d, 4H), 3,70-3,57 (m, 2H), 3,52-3,39 (m, 1H), 3,39-3,27 (m, 1H), 3,15 (d, 2H), 3,02-2,94 (m, 4H), 1,98-1,87 (m, 3H), 1,82-1,60 (m, 5H), 1,51-1,50 (m, 3H), 1,44-1,36 (m, 2H), 0,89 (dd, 6H).
Ejemplo 9:
(2R)-N-[(1R)-5-Amino-1-(2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 11)

Etapa 1:
2- [(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-]fenil-propanoil]amino]-3- fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (11A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (0,26 g, 1 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (230 mg, 1,2 mmol), 1-hidroxibenzotriazol (162 mg, 1,2 mmol), producto intermedio 1 (0,75g, 1 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener 2-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R))-2-[[(2R)-2-(terc-butoxicarbonilamino]-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (11A) como un sólido de color blanco (850 mg, rendimiento del 85%). Etapa 2:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (11 B)

Se añadieron 2-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R))-2-[[(2R)-2-(terc-butoxicarbonilamino]-3-) fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (11A) (850 mg, 0,85 mmol), paladio sobre carbono (170 mg, 20% en peso) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se permitió que avanzase la reacción a temperatura ambiente durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2,7-diazaespiro[3.5]nonano-2
carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (11B) en bruto como un sólido de color blanco (580 mg, rendimiento del 79%) y se usó directamente en la siguiente reacción. Etapa 3:
(2R)-N-[(1R)-5-Amino-1-(2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 11)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2,7-diazaespiro[3.5]nonano-2-carbonií)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (11B) (0,5 g, 0,58 mmol) y ácido trifluoroacético (2 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se purificó el residuo mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 11) como un producto pulverulento de color blanco (380 mg, rendimiento del 66%).
EM m/z = 331,8 [M-2CFsCOOH+2H]+/2;
1H-RMN (400 MHz, D2O) 87,43-7,13 (m, 10H), 4,61 (t, 1H), 4,28-4,06 (m, 5H), 3,84-3,74 (m, 2H), 3,19-3,14 (m, 6H), 2,99-2,95 (m, 4H), 2,09-1,94 (m, 4H), 1,79-1,61 (m, 4H), 1,55-1,31 (m, 5H), 0,89 (dd, 6H).
Ejemplo 10:
2R)-N-[(1R)-5-Amino-1-(7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 12)
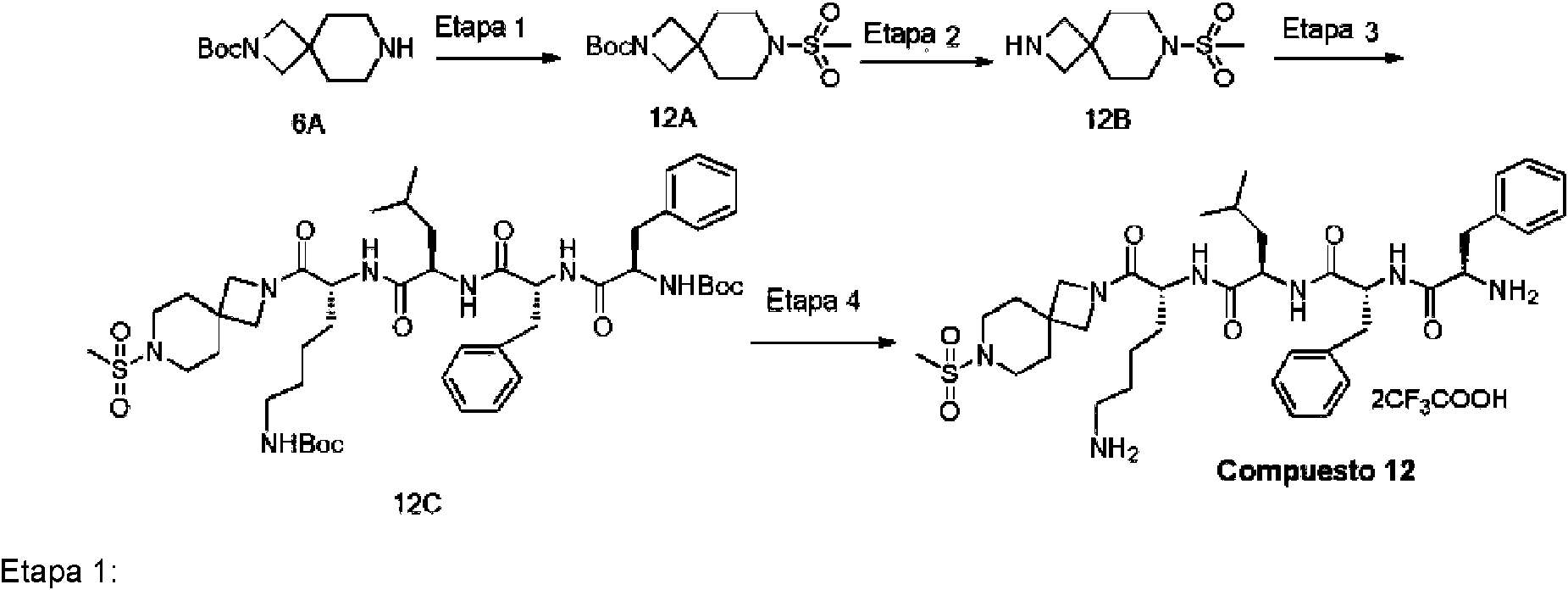
7-Metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (12A)

Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (0,23 g, 1 mmol), trietilamina (210 mg,
2,0 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se disolvieron con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de metanosulfonilo (140 mg, 1,2 mmol) y se permitió que reaccionase el sistema durante 4 h. Luego se elevó la temperatura hasta temperatura ambiente y se extinguió el sistema de reacción con una disolución acuosa saturada de bicarbonato de sodio (10 ml), se extrajo con acetato de etilo (5 ml x 3) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1) para obtener 7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (12A) como una sustancia oleosa de color amarillo claro (250 mg, rendimiento del 81%). EM m/z = 327,2 [M+Na]+.
Etapa 2:
7-Metilsulfonil-2,7-diazaespiro[3.5]nonano (12B)

Se añadieron 7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (12A) (0,25 g, 0,81 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (1 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 7-metilsulfonil-2,7-diazaespiro[3.5]nonano (12B) en bruto como un líquido oleoso de color amarillo claro (165 mg, rendimiento del 100%), y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (12C)

Se añadieron 7-metilsulfonil-2,7-diazaespiro[3.5]nonano (12B) en bruto (165 mg, 0,75 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (192 mg, 1 mmol), 1-hidroxibenzotriazol (135 mg, 1 mmol), producto intermedio 1 (610mg, 0,81 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (12C) como un sólido de color blanco (630 mg, rendimiento del 82,7%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-(7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 12)
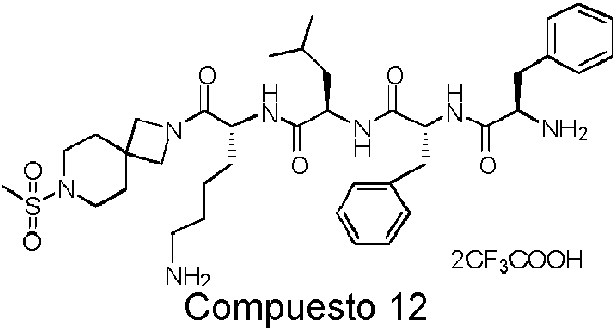
Se añadieron N-[(1R)-1-bencil-2-[[(1 R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de
terc-butilo (12C) (630 mg, 0,34 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(7-metilsulfonil-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di ácido trifluoroacético (compuesto 12) como un polvo de color blanco (410 mg, rendimiento del 63,2%).
EM m/z = 370,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,43-7,15 (m, 10H), 4,61 (t, 1H), 4,33-3,96 (m, 5H), 3,79-3,69 (m, 2H), 3,25-3,13 (m, 7H), 3,02-2,93 (m, 6H), 1,91-1,86 (m, 3H), 1,70-1,64 (m, 3H), 1,56-1,31 (m, 5H), 1,25 (t, 2H), 0,89 (dd, 6H).
Ejemplo 11:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 13)

Cloruro de pirrolidin-1-carbonilo (13B)

Se añadieron NaHCO3 (5,04g, 60 mmol), trifosgeno (5,94 g, 20 mmol) y diclorometano (10 ml) en un matraz de una sola boca de 50 ml. Se enfrió la disolución de reacción hasta 10°Cy luego se añadió lentamente gota a gota pirrolidina (2,16 g, 30,4 mmol). Después de la adición, se devolvió la temperatura a la temperatura ambiente y se hizo reaccionar durante la noche. Se filtró la disolución de reacción y se concentró el filtrado a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 3:1), para obtener cloruro de pirrolidin-1-carbonilo (13B) como una sustancia oleosa incolora (2,07 g, rendimiento del 51,65%).
1H-RMN (400 MHz, CDCls) 83,62-3,56 (m, 2H), 3,54-3,44 (m, 2H), 2,02-1,90 (m, 4H).
Etapa 2:
2-(Pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (13C)
Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (350 mg, 1,3mmol), trietilamina (408 mg, 4,03 mmol) y diclorometano (20 ml) en un matraz de una sola boca de 50 ml y se disolvió con agitación a temperatura ambiente. Luego, se enfrió la disolución de reacción hasta 0°C y se añadió gota a gota cloruro de pirrolidin-1-carbonilo (13B) (123 mg, 0,92 mmol). Después de la adición, se elevó la temperatura hasta temperatura ambiente y se permitió que avanzase la reacción durante 4 h. Se lavó la disolución de reacción secuencialmente con una disolución acuosa saturada de bicarbonato de sodio (60 ml), una disolución acuosa de ácido clorhídrico 3 mol/l (60 ml) y se sometió la mezcla a un proceso de separación de líquido. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (13C) en bruto como un líquido oleoso de color amarillo claro (460 mg, rendimiento del 100%).
EM m/z = 358,2 [M+H]+.
Etapa 3:
2,7-Diazaespiro[3.5]nonan-2-il(pirrolidin-1-il)metanona (13D)

Se añadieron 2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (13C) en bruto (460 mg, 1,3 mmol), hidróxido de paladio/carbono (100 mg, añ 20% en peso) e isopropanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se calentó a 100°C en el baño de aceite durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 2,7-diazaespiro[3.5]nonan-2-il(pirrolidin-1-il)metanona (13D) en bruto como un sólido de color amarillo claro (290 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 224,3 [M+H]+.
Etapa 4:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (13e)

Se añadieron secuencialmente 2,7-diazaespiro[3.5]nonan-2-il(pirrolidin-1-il)metanona (13D) en bruto (167 mg, 0,75 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (374 mg, 1,95 mmol), 1-hidroxibenzotriazol (108 mg, 0,81 mmol), producto intermedio 1 (556 mg, 0,75 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (13E) como un sólido de color blanco (360 mg, rendimiento del 29%).
Etapa 5:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 13)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (13E) (360 mg, 0,38 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (3 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(pirrolidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 13) como un sólido de color blanco (169 mg, rendimiento del 45,6%).
EM m/z = 380,4 [M+2H]+/2.
1H-RMN (400 MHz, DMSO-D) 88,75 (d, 1H), 8,35 (d, 1H), 8,09 (d, 1H), 8,01 (a, 3H), 7,73 (a, 3H), 7,34-7,17 (m, 10H), 4,71-4,60 (m, 2H), 4,40-4,32 (m, 1H), 4,08-3,96 (m, 2H), 3,73-3,23 (m, 8H), 3,16-3,01 (m, 3H), 2,98-2,86 (m, 1H), 2,85 2,69 (m, 3H), 1,82-1,69 (m, 4H), 1,69-1,42 (m, 11H), 1,36-1,22 (m, 2H), 0,89 (dd, 6H).
Ejemplo 12:
(2R)-N-[(1R)-5-amino-1-[2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 14)

3-Cloruro de metilsulfonilazetidin-1-carbonilo (14B)

Se añadieron NaHCO3 (756 mg, 9,0 mmol), trifosgeno (0,89 g, 4,5 mmol) y diclorometano (8 ml) en un matraz de una sola boca de 50 ml. Se enfrió la disolución de reacción hasta-10°Cy luego se añadió lentamente gota a gota clorhidrato de 3-metilsulfonilazetidina (772 mg, 4,5 mmol). Después de la adición, se devolvió la temperatura a la temperatura, se volvió a la temperatura ambiente y se hizo reaccionar durante la noche. Se filtró la disolución de reacción, se concentró
a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:1) para obtener como una sustancia oleosa incolora, cloruro de 3-metilsulfonilazetidin-1-carbonilo (14B) (494 mg, rendimiento del 50%).
1H-RMN (400 MHz, CDCls) 84,62-4,32 (m, 4H), 4,07-3,92 (m, 1H), 2,93 (s, 3H).
Etapa 2:
2-(3-Metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (14C)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (350 mg, 1,3mmol), trietilamina (406 mg, 4,01 mmol) y diclorometano (20 ml) en un matraz de una sola boca de 50 ml y se disolvió con agitación a temperatura ambiente. Luego, se enfrió la disolución de reacción hasta 0°C y se añadió gota a gota cloruro de 3-metilsulfonilazetidin-1-carbonilo (14B) (293 mg, 1,48 mmol). Después de la adición, se elevó la temperatura hasta temperatura ambiente y se permitió que reaccionase el sistema durante 4 h. Se lavó la disolución de reacción secuencialmente con una disolución acuosa saturada de bicarbonato de sodio (60 ml), una disolución acuosa de ácido clorhídrico 3 mol/l (60 ml) y se separó. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja) para obtener 2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (14C) como un sólido de color blanco (300 mg, rendimiento del 55%). EM m/z = 422,2 [M+H]+;
1H-RMN (400 MHz, CDCls) 87,41-7,28 (m, 5H), 5,12 (s, 2H), 4,30-4,19 (m, 4H), 4,00-3,89 (m, 1H), 3,69 (s, 4H), 3,49 3,36 (m, 4H), 2,90 (s, 3H), 1,79-1,67 (m, 4H).
Etapa 3:
2,7-Diazaespiro[3.5]nonan-2-il-(3-metilsulfonilazetidin-1-il)metanona (14D)

Se añadieron secuencialmente 2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (14C) (300 mg, 0,7 mmol), hidróxido de paladio/carbono (60 mg, al 20% en peso) e isopropanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces, y se calentó hasta 100°C en el baño de aceite y se hizo reaccionar durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 2,7-diazaespiro[3.5]nonan-2-il-(3-metilsulfonilazetidin-1-il)metanona (14D) en bruto como un sólido de color amarillo claro (202 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 288,1 [M+H]+.
Etapa 4:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (14E)

Se añadieron secuencialmente 2,7-diazaespiro[3.5]nonan-2-il-(3-metilsulfonilazetidin-1-il)metanona (14D) en bruto (202 mg, 0,7 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (402 mg, 2,10 mmol), 1-hidroxibenzotriazol (115 mg, 0,85 mmol), producto intermedio 1 (536 mg, 0,71 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 40:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (14E) como un sólido de color amarillo claro (686 mg, rendimiento del 96%).
Etapa 5:
(2R)-N-[(1R)-5-Amino-1-[2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; ácido 2,2,2-trifluoroacético (compuesto 14)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etilcarbamato de terc-butilo (14E)(680 mg, 0,66 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (3 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[2-(3-metilsulfonilazetidin-1-carbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 14) como un sólido de color blanco (330 mg, rendimiento del 41,3%).
EM m/z = 412,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,44-7,18 (m, 10H), 4,66 (t, 1H), 4,47-4,16 (m, 7H), 3,90-3,72 (m, 4H), 3,69-3,55 (m, 2H), 3,53-3,41 (m, 1H), 3,39-3,27 (m, 1H), 3,24-3,14 (m, 2H), 3,10 (s, 3H), 3,09-2,90 (m, 4H), 1,94-1,26 (m, 14H), 0,92 (d, 6H).
Ejemplo 13:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[3.5]nona-2-carboxamida; ácido 2,2,2-trifluoroacético (compuesto 15)

Etapa 1:
2-(Metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (15B)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (310 mg, 1,2mmol), trietilamina (364 mg, 3,6 mmol) y diclorometano (20 ml) en un matraz de una sola boca de 50 ml y se disolvió con agitación a temperatura ambiente. Luego, se enfrió la disolución de reacción hasta 0°C y se añadió gota a gota cloruro de metilaminoformilo (15A) (123 mg, 1,32 mmol). Después de la adición, se elevó la temperatura hasta temperatura ambiente y se permitió que avanzase la reacción durante 4 h. Se lavó la disolución de reacción secuencialmente con una disolución acuosa saturada de bicarbonato de sodio (60 ml), una disolución acuosa de ácido clorhídrico 3 mol/l (60 ml) y se separó. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (15B) en bruto como una sustancia oleosa de color amarillo (440 mg, rendimiento del 100%).
EM m/z = 318,2 [M+H]+;
1H-RMN (400 MHz, CDCls) 87,39-7,28 (m, 5H), 5,12 (s, 2H), 3,66 (s, 4H), 3,50-3,36 (m, 4H), 2,79 (s, 3H), 1,82-1,63 (m, 4H).
Etapa 2:
N-Metil-2,7-diazaespiro[3.5]nonano-2-carboxamida (15C)

Se añadieron secuencialmente 2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (15B) en bruto (260 mg, 0,82 mmol), hidróxido de paladio/carbono (50 mg, 20% en peso) e isopropanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se calentó la reacción hasta 100°C en el baño de aceite y se hizo reaccionar durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida (15C) en bruto como un sólido de color amarillo claro (150 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 184,3 [M+H]+.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (15D)

Se añadieron secuencialmente N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida (15C) en bruto (150 mg, 0,82 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (374 mg, 1,95 mmol), 1-hidroxibenzotriazol (110 mg, 0,81 mmol), producto intermedio 1 (565 mg, 0,75 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 40:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (15D)como un sólido de color amarillo claro (480 mg, rendimiento del 63%).
Etapa 4:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida; di-ácido trifluoroacético (compuesto 15)
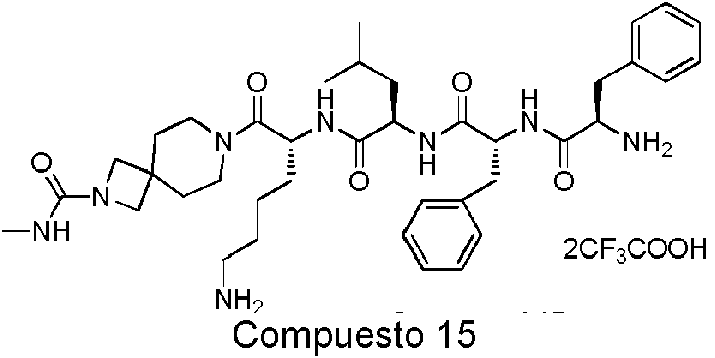
Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (15D) (480 mg, 0,52 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (3 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener 7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida; di-ácido trifluoroacético (compuesto 15) como un sólido de color blanco (260 mg, rendimiento del 53%).
EM m/z = 360,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,43-7,28 (m, 6H), 7,27-7,21 (m, 4H), 4,66 (t, 1H), 4,30 (t, 1H), 4,21 (t, 1H), 3,74 (s, 2H), 3,70 (s, 2H), 3,69-3,59 (m, 2H), 3,55-3,43 (m, 1H), 3,41-3,31 (m, 1H), 3,24-3,12 (m, 2H), 3,11-2,92 (m, 4H), 2,69 (s, 3H), 1,94-1,27 (m, 14H), 0,93 (dd, 6H).
Ejemplo 14:
(2R)-N-[(1R)-5-Amino-1-(5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; ácido 2,2,2-trifluoroacético (compuesto 16)

Etapa 1:
5,5-Difluoro-2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de O7-bencilo y O2-terc-butilo (16B)

Se añadieron secuencialmente trietilamina (0,85 ml, 8,4 mmol), carboxilato de 5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-terc-butilo (16A) (2,1 g, 8,0 mmol) y tetrahidrofurano (15 ml) en un matraz de una sola boca de 50 ml. Se enfrió la disolución de reacción hasta 0°C y luego se añadió lentamente gota a gota cloroformiato de bencilo (1,5 g, 8,8 mmol). Después de la adición, se mantuvo la reacción a 0°C durante 10 minutos y luego se elevó la temperatura de nuevo hasta temperatura ambiente y se continuó agitando durante 5 h. Se filtró la disolución de reacción y se concentró el filtrado a presión reducida para obtener 5,5-difluoro-2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de O7-bencilo y O2-terc-butilo (16B) como un producto oleoso de color amarillo claro (3,5 g, rendimiento del 100%).
EM m/z = 419,3 [M+Na]+;
Etapa 2:
5,5-Difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16C)
Se añadieron 5,5-difluoro-2,7-diazaespiro[3.5]nonano-2,7-dicarboxilato de O7-bencilo y O2-terc-butilo (16B) (3,17 g, 8,0 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (6,0 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se ajustó la disolución de reacción a un pH de aproximadamente 13 con agua amoniacal y luego se sometió la mezcla a un proceso de separación de líquido. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16C) como un líquido oleoso de color amarillo (2,43 g, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 297,1 [M+H]+;
Etapa 3:
5,5-Difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16D)

Se añadieron 5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16C) (310 mg, 1,05 mmol), trietilamina (318 mg, 3,14 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml y se disolvieron con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de metanosulfonilo (156 mg, 1,36 mmol) y se permitió que reaccionase el sistema durante 4 h. Luego, se elevó la temperatura hasta temperatura ambiente y se lavó la disolución de reacción secuencialmente con una disolución acuosa saturada de bicarbonato de sodio (60 ml), una disolución acuosa de ácido clorhídrico 3 M (60 ml) y se separó. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 60:1) para obtener 5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16D) como un sólido de color blanco (233 mg, rendimiento del 59%).
EM m/z = 397,2 [M+Na]+;
1H-RMN (400 MHz, CDCls) 87,42-7,29 (m, 5H), 5,15 (s, 2H), 4,09 (d, 2H), 3,78-3,59 (m, 4H), 3,51 (t, 2H), 2,89 (s, 3H), 2,08 (s, 2H).
Etapa 4:
5,5-Difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano (16E)
Se añadieron 5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16D) (230 mg, 0,5 mmol), paladio/carbono (40 mg, al 20% en peso) e isopropanol (20 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se calentó la reacción hasta 100°C en el baño de aceite y se hizo reaccionar durante 8 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano (16E)como un sólido de color amarillo claro (155 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 241,1 [M+H]+;
Etapa 5:
N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-? 1-(5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (16F)

Se añadieron 5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano (16E) (155 mg, 0,5 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (300 mg, 2,0 mmol), 1-hidroxibenzotriazol (115 mg, 0,85 mmol), producto intermedio 1 (377 mg, 0,5 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión
reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 40:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-(5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (16F)como un sólido de color blanco (380 mg, rendimiento del 78%).
Etapa 6:
(2R)-N-[(1R)-5-Amino-1-(5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; ácido 2,2,2-trifluoroacético (compuesto 16)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (16F) (380 mg, 0,34 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (3 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 p,m; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(5,5-difluoro-2-metilsulfonil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanamida; di-ácido trifluoroacético (compuesto 16) como un polvo de color blanco (270 mg, rendimiento del 71%).
EM m/z = 388,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,44-7,17 (m, 10H), 4,66 (t, 1H), 4,42-4,13 (m, 4H), 4,13-3,49 (m, 7H), 3,25-2,92 (m, 9H), 2,30-2,00 (m, 2H), 1,91-1,26 (m, 9H), 0,93 (q, 6H).
Ejemplo 15:
(2R)-N-[(1R)-5-Amino-1-[2-[(2R)-2-aminopropanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 17)
H2N - Y OH E,aPa 1 B o c H N '^ ^ °ht ,aPa 2  3
3 
NHBoc

Etapa 1:
Ácido (2R-2-(terc-butoxicarbonilamino)propanoico (17B)

Se añadieron D-alanina (17A) (10 g, 112,24 mmol) y agua (56 ml) en un matraz de reacción de 250 ml y se enfrió la disolución de reacción hasta 0°Cy luego se añadió hidróxido de sodio (6,73 g, 168,36 mmol). Después de la adición, se mantuvo la reacción a 0°C durante 10 minutos y luego se añadió gota a gota una disolución de dicarbonato de diterc-butilo (31,85 g, 145,91 mmol) en tetrahidrofurano (50 ml). Después de la adición, se elevó la temperatura hasta temperatura ambiente y se agitó durante la noche. Se extrajo la disolución de reacción con éter de petróleo (100 ml x 2) y se desechó la fase orgánica. Se acidificó la fase acuosa con una disolución de ácido clorhídrico 4 M hasta un pH de aproximadamente 1 y luego se extrajo con acetato de etilo (100 ml x 4). Se combinaron las fases orgánicas y se secaron sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener ácido (2R)-2-(terc-butoxicarbonilamino)propanoico (17B) como una sustancia oleosa incolora (21,2 g, rendimiento:100%).
EM m/z = 212,1 [M+Na]+;
1H-RMN (400 MHz, DMSO-D) 812,34 (a, 1H), 7,05 (d, 1H), 3,98-3,87 (m, 1H), 1,38 (s, 9H), 1,22 (d, 3H).
Etapa 2:
2-[(2R)-2-(terc-Butoxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (17C)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (335 mg, 1,3 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (748 mg, 3,9 mmol), 1-hidroxibenzotriazol (211 mg, 1,56 mmol), ácido (2R)-2-(terc-butoxicarbonilamino)propanoico (17B) (246 mg, 1,3 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 3:1) para obtener 2-[(2R)-2-(terc-butoxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (17C) como un sólido de color blanco (510 mg, rendimiento del 91%).
EM m/Z = 454,3 [M+Na]+;
Etapa 3:
N-[(1R)-2-(2,7-Diazaespiro[3.5]nonan-2-il)-1-metil-2-oxo-etil]carbamato de terc-butilo (17D)
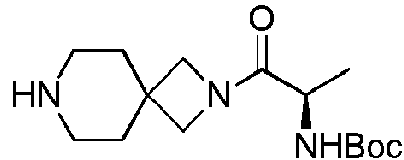
Se añadieron 2-[(2R)-2-(terc-butoxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (17C) (460 mg, 1,1 mmol), hidróxido de paladio/carbono (92 mg, 20% en peso) e isopropanol (20 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se calentó la reacción a 100°C en el baño de aceite durante 8 h bajo una atmósfera de hidrógeno (globo). Luego se enfrió hasta temperatura ambiente, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener N-[(1R)-2-(2,7-diazaespiro[3.5]nonan-2-il)-1-metil-2-oxo-etil]carbamato de terc-butilo (17D)como un sólido de color amarillo claro (252 mg, rendimiento del 77%) y se usó directamente en la siguiente reacción.
EM m/z = 298,3 [M+H]+;
Etapa 4:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-[(2R)-2-(tercbutoxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (17E)

Se añadieron secuencialmente N-[(1R)-2-(2,7-diazaespiro[3.5]nonan-2-il)-1-metil-2-oxo-etil]carbamato de terc-butilo (17D) en bruto (223 mg, 0,75 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (374 mg, 1,95 mmol), 1-hidroxibenzotriazol (110 mg, 0,81 mmol), producto intermedio 1 (565 mg, 0,75 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 40:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-[(2R)-2]-(terc-butoxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (17E)como un sólido de color amarillo claro (560 mg, rendimiento del 69%).
Etapa 5:
(2R)-N-[(1R)-5-Amino-1-[2-[(2R)-2-aminopropanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 17)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-[(2R)-2-(tercbutoxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (17E) (560 mg, 0,52 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (3 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[2-[(2R)-2-aminopropanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 17) como un polvo de color blanco (310 mg, rendimiento del 45%).
EM m/z = 367,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,44-7,28 (m, 6H), 7,26-7,20 (m, 4H), 4,65 (t, 2H), 4,33-4,21 (m, 2H), 4,19-4,01 (m, 3H), 3,96-3,78 (m, 2H), 3,75-3,59 (m, 2H), 3,56-3,42 (m, 1H), 3,42-3,30 (m, 1H), 3,24-3,12 (m, 2H), 3,10-2,92 (m, 4H), 1,99 1,79 (m, 3H), 1,80-1,63 (m, 5H), 1,53 (d, 3H), 1,50-1,30 (m, 5H), 0,92 (dd, 6H).
Ejemplo 16:
(2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-aminopropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 18)

Etapa 1:
N-[(1R)-2-(2,7-Diazaespiro[3.5]nonan-7-il)-1-metil-2-oxo-etil]carbamato de bencilo (18B)

Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (0,45g, 2 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (780 mg, 4,1 mmol), 1-hidroxibenzotriazol (340 mg, 2,5 mmol), ácido (2R)-2-(benciloxicarbonilamino)propanoico (446 mg, 2,0 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-2-(2,7-diazaespiro[3.5]nonan-7-il)-1-metil-2-oxoetil]carbamato de bencilo (18B) como un sólido de color blanco (860 mg, rendimiento del 99%).
EM m/Z = 454,2 [M+Na]+.
Etapa 2:
N-[(1R)-2-(2,7-Diazaespiro[3.5]nonan-7-il)-1-metil-2-oxo-etil]carbamato de bencilo (18C)

Se añadieron N-[(1R)-2-(2,7-diazaespiro[3.5]nonan-7-il)-1-metil-2-oxo-etil]carbamato de bencilo (18B) (0,86 g, 2 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener N-[(1R)-2-(2,7-diazaespiro[3.5]nonan-7-il)-1-metil-2-oxo-etil]carbamato de bencilo (18C) en bruto como un líquido oleoso de color amarillo (660 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
EM m/z = 332,2 [M+H]+;
Etapa 3:
N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2Hbenciloxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (18D)

Se añadieron N-[(1R)-2-(2,7-diazaespiro[3.5]nonan-7-il)-1-metil-2-oxo-etil]carbamato de bencilo (18C) (243 mg, 0,7 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (300 mg, 2,0 mmol), 1-hidroxibenzotriazol (200 mg, 1,0 mmol), producto intermedio 1 (500 mg, 0,7 mmol) y diclorometano (50 ml) en un matraz de reacción de 100 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 30:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-(benciloxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (18D) como un sólido de color amarillo claro (740 mg, rendimiento del 99%).
Etapa 4:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-aminopropanoil]-2,7-]diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (18E)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2]-(benciloxicarbonilamino)propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (18D) (700 mg, 0,7 mmol), paladio sobre carbono (140 mg, al 20% en peso) y metanol (15 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 8 h bajo una atmósfera de hidrógeno. Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener (1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)]-1-[7-[(2R)-2-aminopropanoil]-1,7-diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonil)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de tercbutilo (18E) en bruto como una sustancia oleosa de color amarillo claro (650 mg, rendimiento del 99%) y se usó directamente en la siguiente reacción.
Etapa 5:
(2R)-N-[(1R)-5-Amino-1-[7-[(2R)-2-aminopropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 18)

Se añadieron (1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-aminopropanoil]-1,7]-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonil)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (18E) en bruto (650 mg, 0,7 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 p,m; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-aminopropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 18) como un polvo de color blanco (170 mg, rendimiento del 19%).
EM m/z = 733,5 [M+H]+;
1H-RMN (400 MHz, D2O) 87,44-7,28 (m, 6H), 7,27-7,20 (m, 4H), 4,64 (t, 1H), 4,55-4,46 (m, 1H), 4,32-4,23 (m, 2H), 4,21-4,05 (m, 3H), 3,86-3,71 (m, 2H), 3,63-3,40 (m, 4H), 3,25-3,10 (m, 2H), 3,10-2,90 (m, 4H), 2,00-1,60 (m, 8H), 1,60 1,29 (m, 8H), 0,92 (dd, 6H).
Ejemplo 17:
(2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-aminopropanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 19)

19C
Etapa 1:
2-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (19A)

Se añadieron5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16C) (797 mg, 2,69 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (1,04 g, 5,4 mmol), 1-hidroxibenzotriazol (400 mg, 3 mmol), producto intermedio 1 (2,03 g, 2,69 mmol) y diclorometano (50 ml) en un matraz de reacción de 100 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 30:1) para obtener 2-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (19A) como un sólido de color amarillo claro (2,49 g, rendimiento del 89,6%).
Etapa 2: N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (19b)
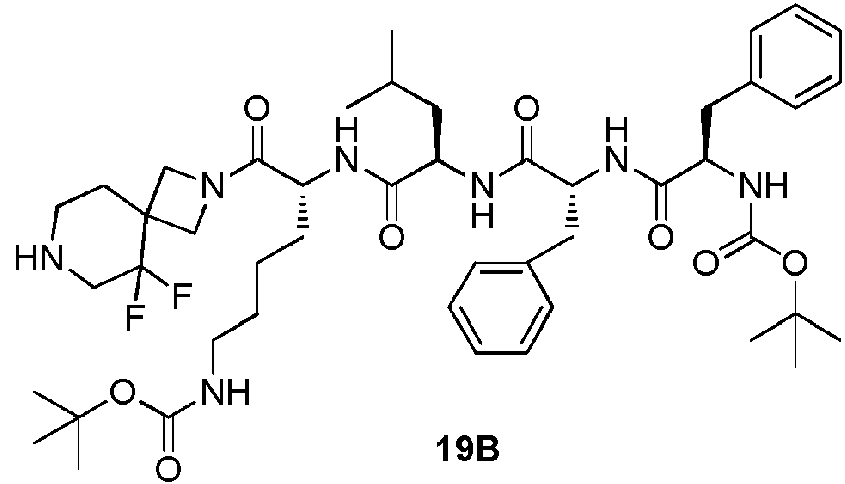
Se añadieron 2-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (19A) (2,49 g, 2,41 mmol), paladio sobre carbono (500 mg, al 20% en peso) y metanol (25 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 4 h bajo una atmósfera de hidrógeno (globo). Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (19B) como un sólido oleoso de color amarillo claro (2,16 g, rendimiento del 99%), y se usó directamente en la siguiente reacción.
EM m/z = 899,5 [M+H]+;
Etapa 3: N-(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-[(2R)-2-(tercbutoxicarbonilamino)propanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (19C)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (19B)en bruto (400 mg, 0,4 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,23 g, 1,2 mmol), 1-hidroxibenzotriazol (70 mg, 0,5 mmol), Boc-D-alanina (78 mg, 0,41 mmol) y diclorometano (50 ml) en un
matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 30:1) para obtener N-(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-[(2R)-2-(terc-butoxicarbonilamino)propanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (19C) como un sólido de color amarillo claro (340 mg, rendimiento del 79%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[7-[(2R)-2-aminopropanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 19)

Se añadieron N-(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-[(2R)-2-(tercbutoxicarbonilamino)propanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (19C) (340 mg, 0,32 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-aminopropanoil]-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 19) como un polvo de color blanco (170 mg, rendimiento del 48%).
EM m/z (ESI): 385,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,43-7,29 (m, 6H), 7,27-7,21 (m, 4H), 4,67-4,47 (m, 3H), 4,31-4,09 (m, 5H), 4,09-3,76 (m, 3H), 3,74-3,46 (m, 2H), 3,18 (d, 2H), 3,10-2,91 (m, 4H), 2,33-1,97 (m, 2H), 1,84-1,61 (m, 4H), 1,61-1,26 (m, 8H), 0,92 (dd, 6H).
Ejemplo 18:
(2R)-N-[(1R)-1-(7-Acetil-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 20)

7-Acetil-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (20B)
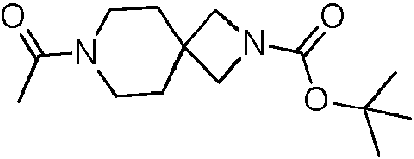
Se disolvió 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (450 mg, 2,0 mmol) en diclorometano (5 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno y se añadió trietilamina (606 mg, 6,0 mmol) con agitación. Luego, se redujo la temperatura hasta -20°C y se añadió gota a gota cloruro de acetilo (310 mg, 4,0 mmol). Después de la adición, se elevó de manera natural la temperatura hasta temperatura ambiente y se agitó el sistema durante 2 h. Posteriormente, se añadió a la reacción una disolución acuosa de ácido clorhídrico diluido 0,5 M (20 ml) y se separaron las fases mediante agitación y se sometió la mezcla a un proceso de separación de líquido. Se extrajo la fase acuosa con diclorometano (20 ml x 2) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (acetato de etilo puro), para obtener 7-acetil-2,7.-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (20B) como un líquido oleoso de color amarillo claro (520 mg, rendimiento del 97,0%).
Etapa 2:1-(2,7-Diazaespiro[3.5]nonan-7-il)etanona (20C)
1-(2,7-Diazaespiro[3.5]nonan-7-il)etanona

Se añadieron 7-acetil-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (20B) (520 mg, 1,9 mmol) y diclorometano (6 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (3 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida y al residuo se le añadieron 4 ml de agua amoniacal concentrada, seguido de secado con sulfato de sodio anhidro, lavado con metanol (20 ml) y concentración de la disolución de lavado para obtener 1-(2,7-diazaespiro[3.5]nonan-7-il)etanona (20C) como un líquido oleoso de color amarillo (250 mg, rendimiento del 77%), y se usó directamente en la siguiente reacción.
Etapa 3:
N-(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato (20D)

Se añadió 1-(2,7-diazaespiro[3.5]nonan-7-il)etanona (20C) (250 mg, 1,49 mmol) en el acetato de etilo (10 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno. Se enfrió hasta 0°C en un baño de hielo y se añadieron el producto intermedio 1 (800 mg, 1,06 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (305 mg, 1,6 mmol), 1-hidroxibenzotriazol (172 mg, 1,27 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 1,5 h. Posteriormente, se añadió una disolución acuosa de ácido clorhídrico 1 M (15 ml) a la disolución de reacción y se agitó la mezcla y luego se sometió a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de carbonato de sodio (15 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y luego se sometió a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa saturada de cloruro de sodio (15 ml), se secó sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida para obtener N-(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-(tercbutox¡carbon¡lam¡no)pent¡l]carbamoil]-3-met¡l-but¡ljam¡no]-í-benc¡l-2-oxo-et¡l]am¡no]-1-benc¡l-2-oxoetil]carbamato (20D) como un sólido espumoso de color amarillo claro (0,85 g, rendimiento del 88%) y se usó directamente en la siguiente reacción.
Etapa 4:
(2R)-N-[(1R)-1-(7-Acetil-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 20)
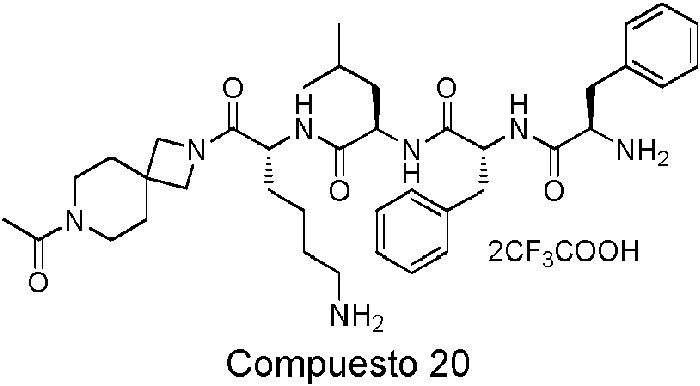
Se disolvió N-(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato (20D) en bruto (0,85 g, 0,94 mmol) en diclorometano (7,5 ml) y se añadió ácido trifluoroacético (3,5 ml). Se agitó el sistema a temperatura ambiente durante 1 h. Posteriormente, se concentró la disolución de reacción a presión reducida. Después de que se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 * 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; ¡socrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja), se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener ((2R)-N-[(1R)-1-(7-acetil-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanamida; di-ácido trifluoroacético (compuesto 20) como un sólido de color blanco (310 mg, rendimiento del 40,0%).
EM m/z (ESI): 352,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,46-7,17 (m, 10H), 4,64 (t, 1H), 4,29-4,05 (m, 5H), 3,83-3,74 (m, 2H), 3,57-3,35 (m, 4H), 3,24-3,11 (m, 2H), 3,09-2,93 (m, 4H), 2,17-1,29 (m, 16H), 0,92 (dd, 6H).
Ejemplo 19:
(2R)-N-[(1R)-5-Amino-1-[2-(1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; ácido trifluoroacético (compuesto 21)

Etapa 1:
2-(1,1-Dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (21A)

Se añadieron secuencialmente 2-oxo-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3A) (129 mg, 0,54 mmol), ácido acético (65 mg, 1,08 mmol), 1,1-dióxido de 1,4-tiazinano (72 mg, 0,54 mmol), triacetoxiborohidruro de sodio (229 m g, 1,08 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 16 h. Se filtró la disolución de reacción con succión y se lavó el filtrado con una disolución saturada de bicarbonato de sodio (50 ml). Después de la separación, se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró con succión y se concentró el filtrado a presión reducida, para obtener de terc-butilo (1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carboxilato (21A) como un polvo de color blanco (147 mg, rendimiento del 76%).
Etapa 2:
1,1 -Dióxido de 4-(7-azaespiro[3.5]nonan-2-il)-1,4-tiazinano (21B)

Se añadieron (1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (21A) (147 mg, 0,41 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 1,1-dióxido de 4-(7-azaespiro[3.5]nonan-2-il)-1,4-tiazinano (21B) en bruto como un líquido oleoso de color amarillo (106 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (21C)

Se añadieron clorhidrato de 1,1-dióxido de 4-(7-azaespiro[3.5]nonan-2-il)-1,4-tiazinano (21B) en bruto (106 mg, 0,41 mmol), 1-(3-dimetilaminopropil)-3-etilcarbodiimida (94 mg, 0,49 mmol), 1-hidroxibenzotriazol (66 mg, 0,49 mmol), producto intermedio 1 (309 mg, 0,41 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml, se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1), para obtener de (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-[2-(1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (21C) como un sólido de color blanco (367 mg, rendimiento del 90%).
Etapa 4:
(2R)-N-[(1R)-5-amino-1-[2-(1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 21)

Se añadieron (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(1,1-dioxo-1,4-tiazinan-4-il)-7-azaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (21C) (367 mg, 0,37 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml, y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 ^m; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[2-(1,1-dioxo-1,4-tiazinan)-4-il)-7-azaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 21) como un polvo de color blanco (295 mg, rendimiento del 70%).
EM m/z = 397,9 [M+2H]+ /2;
1H-RMN (400 MHz, D2O) 57,36-7,19 (m, 10H), 4,63-4,60 (m, 1H), 4,32-4,16 (m, 3H), 3,92-3,78 (m, 1H), 3,63 (d, 9H), 3,53-3,22 (m, 3H), 3,15 (d, 2H), 3,01-2,91 (m, 4H), 2,48-2,32 (m, 2H), 1,98-2,06 (m, 2H), 1,75-1,23 (m, 13H), 0,89 (dd, 6H).
Ejemplo 20:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[4.4]nona-2-carboxamida; di-ácido trifluoroacético (compuesto 22)

Etapa 1:
7-(Metilcarbamoil)-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (22B)

22B
2-Boc-2,7-diazaespiro[4.4]nonano (22A) (452 mg, 2,0 mmol), trietilamina (400 mg, 4,0 mmol) y diclorometano (15 ml) se añadieron en un matraz de reacción de 50 ml y se disolvieron con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de metil amino formilo (188 mg, 2,01 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se añadió ácido clorhídrico diluido 3 M (50 ml) a la disolución de reacción y luego se extrajo con diclorometano (60 ml x 2). Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 7-(metilcarbamoil)-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (22B) como un sólido de color blanco (570 mg, rendimiento del 65,5%).
1H-RMN (400 MHz, CDCls) 83,51-3,16 (m, 8H), 2,82 (s, 3H), 1,98-1,74 (m, 4H), 1,46 (s, 9H).
Etapa 2:
N-Metil-2,7-diazaespiro[4.4]nonano-2-carboxamida (22C)

Se añadieron 7-(metilcarbamoil)-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (22B) (161 mg, 0,568 mmol), diclorometano (10 ml) y ácido trifluoroacético (1,2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida para obtener N-metil-2,7-diazaespiro[4.4]nonano-2-carboxamida (22C) en bruto como una sustancia oleosa de color amarillo claro, y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(metilcarbamoil)-2,7-diazaespiro[4.4]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (22D)

Se añadieron secuencialmente N-metil-2,7-diazaespiro[4.4]nonano-2-carboxamida (22C) en bruto (81 mg, 0,44 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,288 g, 1,5mmol), 1-hidroxibenzotriazol (81 mg, 0,6 mmol), producto intermedio 1 (330 mg, 0,44 mmol) y diclorometano (50 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 30:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-[2-(metilcarbamoil)-2,7]-diazaespiro[4.4]nonano-7-carbonil]pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (22D) como un sólido de color amarillo claro (420 mg, rendimiento del 98%).
Etapa 4:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[4.4]nonano-2-carboxamida; di-ácido trifluoroacético (compuesto 22)

Se añadieron N-[(1R)-1-bencil-2-[[(1 R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(metilcarbamoil)-2,7-diazaespiro[4.4]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (22D) (400 mg, 0,44 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener 7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R))-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[4.4]nonano-2-carboxamida; di-ácido trifluoroacético (compuesto 22) como un polvo de color blanco (130 mg, rendimiento del 31,5%). EM m/z (ESI): 360,3 [M+2H]+72;
1H-RMN (400 MHz, D2O) 87,43-7,19 (m, 10H), 4,68-4,60 (m, 1H), 4,50-4,19 (m, 3H), 3,93-3,62 (m, 2H), 3,55-3,27 (m, 5H), 3,24 (s, 1H), 3,18 (d, 2H), 3,09-2,94 (m, 4H), 2,74-2,66 (m, 3H), 2,10-1,85 (m, 4H), 1,84-1,62 (m, 4H), 1,59-1,43 (m, 4H), 1,43-1,29 (m, 1H), 0,92 (dd, 6H).
Ejemplo 21:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(1-piperidinil)-7-azaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 23)

2-(1-Piperidinil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (23A)

Se añadieron carboxilato de 2-oxo-7-azaespiro[3.5]nonano-7-de terc-butilo (0,48 g, 2 mmol), piperidina (0,25 g, 3 mmol) y diclorometano (10 ml) a un matraz de reacción y se agitó durante 30 minutos, se enfrió hasta 0-5°C y se añadió cianoborohidruro de sodio (0,13 g, 4 mmol). Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 4 h, y se usó CCF para monitorizar si se completaba la reacción hasta que se completó. Se añadieron diclorometano (10 ml) y agua (10 ml) y se agitó durante 5 min. Se dejaron en reposo las fases y se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida para obtener 2-(1-piperidinil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (23A) en bruto como un sólido de color amarillo claro (0,53 g, rendimiento del 86%) y se usó directamente en la siguiente etapa.
Etapa 2:
2-(1-Piperidinil)-7-azaespiro[3.5]nonano; ácido 2,2,2-trifluoroacético (23B)

Se añadió 2-(1 -piperidinil)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (23A, 0,53 g, 1,7 mmol) a diclorometano (5 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno, y se añadió ácido trifluoroacético (2 ml) con agitación. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se usó CCF para monitorizar si se completaba la reacción y se concentró la reacción hasta sequedad a presión reducida para obtener 2-(1-piperidinil)-7-azaespiro[3.5]nonano; ácido trifluoroacético (23B) como una sustancia oleosa de color amarillo claro (0,50 g, rendimiento del 90%) y se usó directamente en la siguiente etapa.
Etapa 3:
N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(1-piperidinil)-7-azaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (23C)

Se añadió ácido 2-(1-piperidinil)-7-azaespiro[3.5]nonanotrifluoroacético (23B, 0,50 g, 1,60 mmol) a diclorometano (10 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno. Se enfrió hasta 0°C en un baño de hielo y se añadieron el producto intermedio 1 (0,50 g, 0,66 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (190 mg, 0,99 mmol), 1-hidroxibenzotriazol (110 mg, 0,81 mmol). Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Posteriormente, se añadió una disolución acuosa de ácido clorhídrico 1 M (15 ml) a la disolución de reacción y se agitó la mezcla y luego se sometió a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de carbonato de sodio (15 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y luego se sometió a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa saturada de cloruro de sodio (15 ml), se secó sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-[2-(1-piperidinil)-7-]azaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (23C) en bruto como un sólido espumoso de color amarillo claro (0,45. G, rendimiento del 72%) y se usó directamente en la siguiente reacción.
Etapa 4:
(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(1-piperidinil)-7-azaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 23)

Se disolvió N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(1-piperidinil)-7-azaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (23C) en bruto (0,45 g, 0,48 mmol) en diclorometano (7,5 ml), se añadió ácido trifluoroacético (3,5 ml) y se agitó el sistema a temperatura ambiente durante 1 h. Posteriormente, se concentró la disolución de reacción a presión reducida. Después de que se separó el residuo y se purificó por cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5^m; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja), se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-N-[(1R)-5-amino-1-[2-(1-piperidinil)-7-azaespiro[3.5]nonano-7-carbonil]pentil]-4-metilpentanamida; tri-ácido trifluoroacético (compuesto 23) como un sólido de color blanco (260 mg, rendimiento del 56%).
EM m/z (ESI): 372,9 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,48-7,09 (m, 10H), 4,68-4,60 (m, 1H), 4,34-4,17 (m, 2H), 3,73-3,52 (m, 3H), 3,52-2,92 (m, 10H), 2,80-2,64 (m, 2H), 2,47-2,24 (m, 2H), 2,05-1,88 (m, 4H), 1,88-1,20 (m, 18H), 1,00-0,81 (m, 6 H).
Ejemplo 22:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]-4-metil-pentanamida; ácido tri-trifluoroacético (compuesto 24)

7-Tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (24A)

Se añadieron 2,7-diazaespiro[3.4]nonano-2-carboxilato de terc-butilo (6A) (0,452 g, 2 mmol), tetrahidropirona (200 mg, 2 mmol), ácido acético (120 mg, 2,0 mmol) y dicloroetano (7 ml) en un matraz de reacción de 50 ml y se agitó durante 0,5 h. Se añadió triacetoxiborohidruro de sodio (0,636 g, 3 mmol) y se permitió que reaccionase el sistema durante 5 h. Luego, se extinguió la disolución de reacción con agua (10 ml), se extrajo con acetato de etilo (5 ml x 3) y se combinaron las fases orgánicas, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1) para obtener 7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (24A) como una sustancia oleosa de color amarillo claro (500 mg, rendimiento del 80,64%).
EM m/z = 311,2 [M+H]+.
Etapa 2:
7-Tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano (24B)

Se añadieron 7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (24A) (0,5 g, 1,61 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (1 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano (24B) en bruto como un líquido oleoso de color amarillo claro (338 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (24C)

Se añadieron 7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano (24B) en bruto (338mg, 1,61 mmol), clorhidrato de 1(3-dimetilaminopropil)-3-etilcarbodiimida (384 mg, 2 mmol), 1-hidroxibenzotriazol (270 mg, 2 mmol), producto intermedio 1 (40omg, 0,53 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-(7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metilbutil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (24C) como un sólido de color blanco (200 mg, rendimiento del 39,93%)
Etapa 4:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 24)
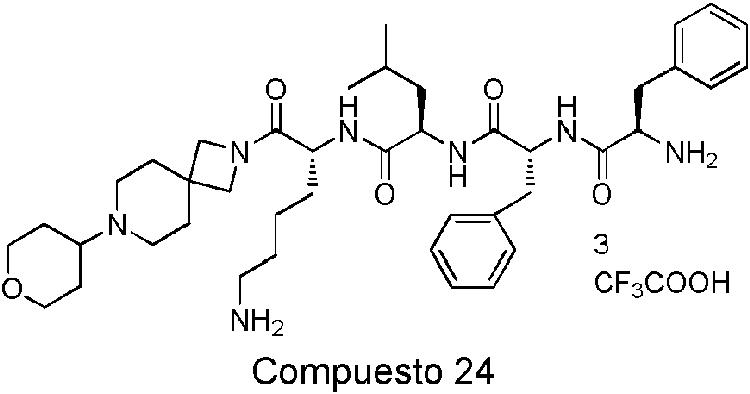
Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (24C) (200 mg, 0,21 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-N-[(1R)-5-amino-1-(7-tetrahidropiran-4-il-2,7-diazaespiro[3.5]nonano2-carbonil)pentil]-4-metilpentanamida; tri-ácido trifluoroacético (compuesto 24) como un polvo de color blanco (135 mg, rendimiento del 86,1%).
EM m/z = 373,9 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,46-7,29 (m, 10H), 4,71 (t, 1H), 4,27-4,16 (m, 7H), 3,93-3,86 (m, 2H), 3,66-3,53 (m, 5H), 3,25-3,05 (m, 8H), 2,34-2,13 (m, 6H), 1,79-1,43 (m, 11H), 1,02-0,95 (m, 6H).
Ejemplo 23:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 25)

Etapa 1:
2-Pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (25A)

Se añadieron secuencialmente 2-oxo-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3A) (131 mg, 0,55 mmol), ácido acético (66 mg, 1,1 mmol), pirrolidina (39 mg, 0,55 mmol), triacetoxiborohidruro de sodio (233 mg, 1,1 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 6 h. Se filtró la disolución de reacción con succión y se lavó el filtrado con una disolución saturada de bicarbonato de sodio (30 ml). Después de la separación del líquido, se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró con succión y se concentró el filtrado a presión reducida para obtener 2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (25A)) como un polvo de color blanco (120 mg, rendimiento del 75%).
Etapa 2:
2-Pirrolidin-1-il-7-azaespiro[3.5]nonano (25B)

Se añadieron (2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (25A) (120 mg, 0,41 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 2-pirrolidin-1-il-7-azaespiro[3.5]nonano (25B) en bruto como un líquido oleoso de color amarillo (80 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (25C)

Se añadieron 2-pirrolidin-1-il-7-azaespiro[3.5]nonano (25B) en bruto (80 mg, 0,41 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (96 mg, 0,5 mmol), 1-hidroxibenzotriazol (67,5 mg, 0,5 mmol), producto intermedio 1 (309 mg, 0,41 mmol) y diclorometano (20 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener de (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (25C) como un sólido de color amarillo claro (344 mg, rendimiento del 90%).
Etapa 4:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 25)

Se añadieron N(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-pirrolidin)-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (25C) (344 mg, 0,37 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener ((2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil)amino)-3-fenilpropanoil]amino]-N-[(1R)-5-amino-1-(2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 25) como un polvo de color blanco (249 mg, rendimiento del 70%).
EM m/z = 365,8 [M+2H]+ /2;
1H-RMN (400 MHz, D2O) 87,43-7,13 (m, 10H), 4,71-4,70 (m, 1H), 4,62 (t, 1H), 4,30-4,17 (m, 2H), 3,82-3,74 (m, 1H), 3,68-3,39 (m, 5H), 3,38-3,21 (m, 1H), 3,15 (d, 2H), 3,06-2,90 (m, 6H), 2,43-2,25 (m, 2H), 2,16-1,89 (m, 6H), 1,78-1,27 (m, 13H), 0,89 (dd, 6H).
Ejemplo 24:
(2R)-1-[7-[(2R)-6-Amino-2-[[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxílico; tri-ácido trifluoroacético (compuesto 26)
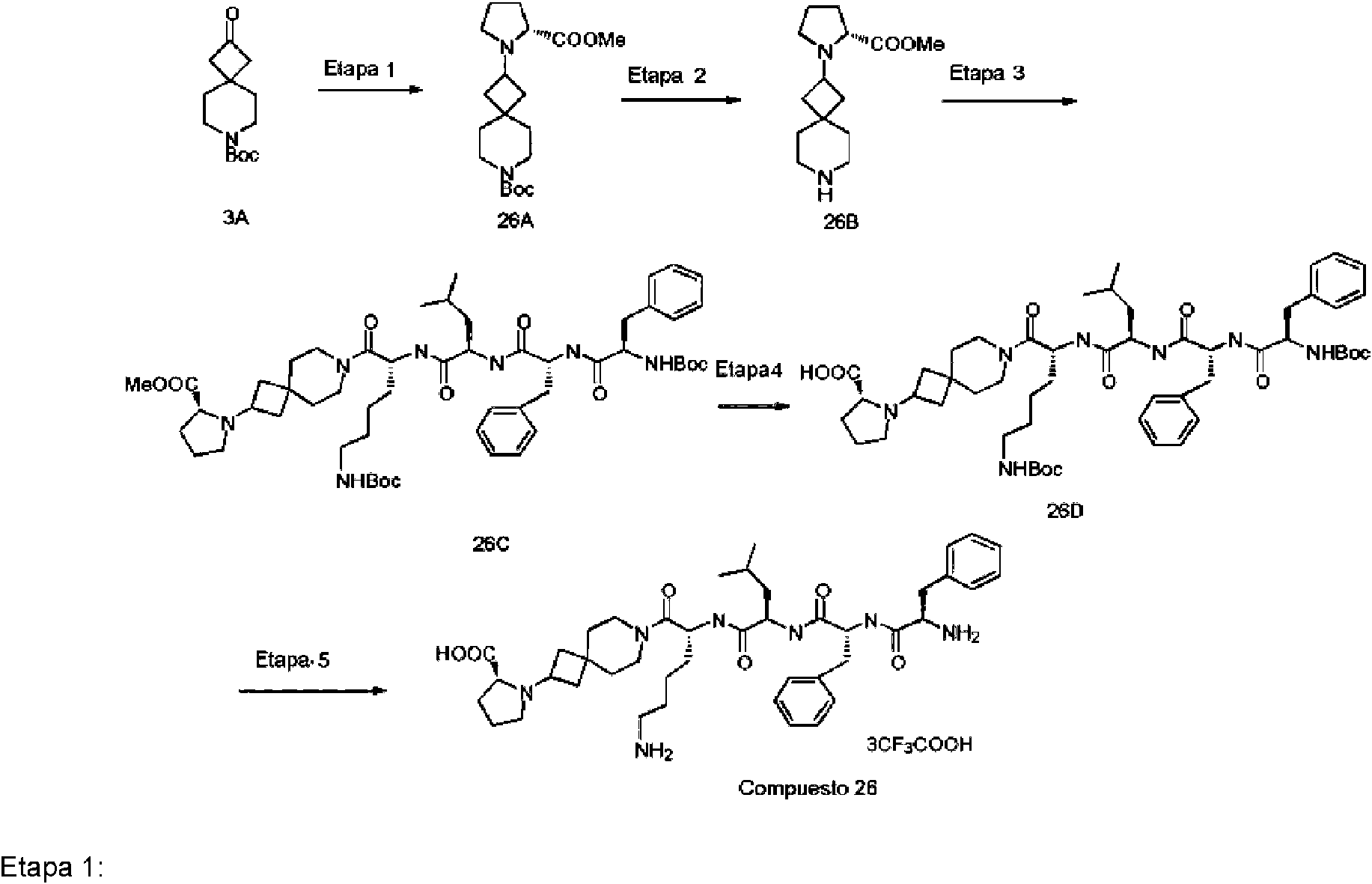
2-[(2R)-2-Metoxicarbonilpirrolidin-1-il]-7-azaespiro[3.5]nonano-7-carboxilato (26A) de terc-butilo
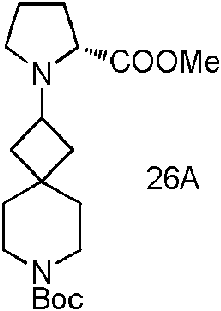
Se añadieron secuencialmente 2-oxo-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (3A) (287 mg, 1,2mmol), ácido acético (144 mg, 2,4 mmol), éster metílico de D-prolina (154 mg, 1,2mmol)), triacetoxiborohidruro de sodio (233 mg, 2,4 mmol) y diclorometano (30 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 6 h. Se filtró la disolución de reacción con succión y se lavó el filtrado con una disolución saturada de bicarbonato de sodio (50 ml). Después de la separación del líquido, se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró con succión y se concentró el filtrado a presión reducida para obtener 2-pirrolidin-1-il-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (26A)) como un polvo de color blanco (275 mg, rendimiento del 65%).
Etapa 2:
(2R)-1-(7-azaespiro[3.5]nonan-2-il)pirrolidin-2-carboxilato de metilo (26B)

Se añadieron 2-[(2R)-2-Metoxicarbonilpirrolidin-1-il]-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (26A) (275 mg, 0,78 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener (2R)-1-(7-azaespiro[3.5]nonan-2-il)pirrolidin-2-carboxilato de metilo (26B) como un líquido oleoso de color
amarillo (196 mg, rendimiento del 100%).) y se usó directamente en la siguiente reacción.
Etapa 3:
(2R)-1-[7-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarboxilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxilato de metilo (26C)

Se añadieron clorhidrato de (2R)-1-(7-azaespiro[3.5]nonan-2-il)pirrolidin-2-carboxilato de metilo (26B) (196 mg, 0,78 mmol), 1-(3-dimetilaminopropil)-3-etilcarbodiimida (180 mg, 0,94 mmol), 1-hidroxibenzotriazol (127 mg, 0,94 mmol), producto intermedio 1 (587 mg, 0,78 mmol) y diclorometano (20 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener (2R)-1-[7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[terc-butoxicarbonilamino)-3-fenil-propanoil)amino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxilato de metilo (26C) como un sólido de color blanco (385 mg, rendimiento del 50%).
Etapa 4:
Ácido (2R)-1-[7-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxílico (26D)

Se disolvió (2R)-1-[7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[terc-butoxicarbonilamino)-3-fenilpropanoil)amino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxilato de metilo (26C) (385 mg, 0,39 mmol) en metanol (5 ml) a temperatura ambiente y se añadió una disolución acuosa de hidróxido de sodio (16 mg, 0,4 mmol) (10 ml) a la disolución de reacción. Se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se ajustó la disolución de reacción a pH <4 con una disolución acuosa de ácido clorhídrico 1 M, se extrajo con acetato de etilo (20 ml) y se sometió la mezcla a un proceso de separación de líquido. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener (2R)-1-[7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(tercbutoxicarbonilamino)-3-fenil-propanoil)amino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxilato de metilo (26D) como un sólido de color blanco (345 mg, rendimiento del 93%).
Etapa 5:
Ácido (2R)-1-[7-[(2R)-6-Amino-2-[[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxílico; ácido tri-ácido trifluoroacético (compuesto 26)
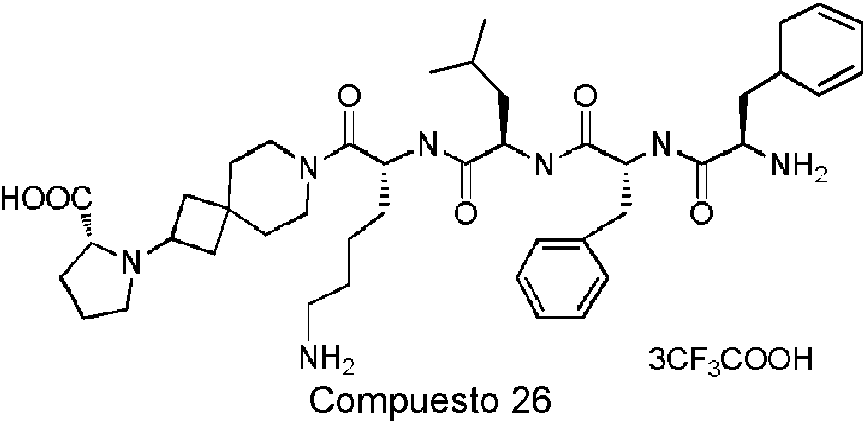
Se añadieron ácido (2R)-1-[7-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(tercbutoxicarbonilamino)-3-fenil-propanoil)amino)-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxílico (26D) (345 mg, 0,36 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja), se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener ácido (2R)-1-[7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]pirrolidin-2-carboxílico; tri-ácido trifluoroacético (compuesto 26) como un polvo de color blanco (240 mg, rendimiento del 60%).
EM m/z = 387,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,44-7,09 (m, 10H), 4,27-4,19 (m, 3H), 3,96-3,84 (m, 2H), 3,67-3,50 (m, 3H), 3,48-3,21 (m, 3H), 3,19-2,89 (m, 7H), 2,50-2,22 (m, 3H), 2,18-1,91 (m, 5H), 1,78-1,24 (m, 13H), 0,88 (dd, 6H).
Ejemplo 25:
2-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[3.5]nona-7-carboxamida; di-ácido trifluoroacético (compuesto 27)

Etapa 1:
7-(Metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (27A)

Se añadieron 2-terc-butoxigencarbonil-2,7-diazaespiro[3.5]nonano (0,45 g, 2 mmol), diclorometano (10 ml) y trietilamina (0,30 g, 3 mmol) a un matraz de reacción bajo protección de nitrógeno; se enfrió hasta 0-5°C y se añadió
cloruro de metilaminoformilo (0,20 g, 2,2 mmol). Después de la adición, se retiró el enfriamiento y se elevó la temperatura hasta temperatura ambiente y se hizo reaccionar durante 1 h. Se usó CCF para monitorizar si se completaba la reacción. Se añadieron diclorometano (10 ml) y agua (10 ml) y, después de agitar durante 5 minutos, se dejó que se separasen las fases; se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano/metanol=(v/v) 20/1) para obtener 7-(metilcarbamoil)-2,7-diazaespiro-[3,5]nonano-2-carboxilato de tercbutilo (27A) como una sustancia aceitosa de color amarillo claro (0,48 g, rendimiento del 85%).
Etapa 2:
N-Metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; ácido 2,2,2-trifluoroacético (27B)

Se añadió 7-(metilcarbamoil)-2,7-diazaespiro-[3,5]nonano-2-carboxilato de terc-butilo (27A, 0,48 g, 1,7 mmol) a diclorometano (5 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno, y se añadió ácido trifluoroacético (2 ml) con agitación. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se usó CCF para monitorizar que se completaba la reacción, y se concentró la reacción hasta sequedad a presión reducida para obtener N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; ácido trifluoroacético (27B) como una sustancia oleosa de color amarillo claro (0,49 g, rendimiento del 97%) y se usó directamente en la siguiente etapa.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (27C)
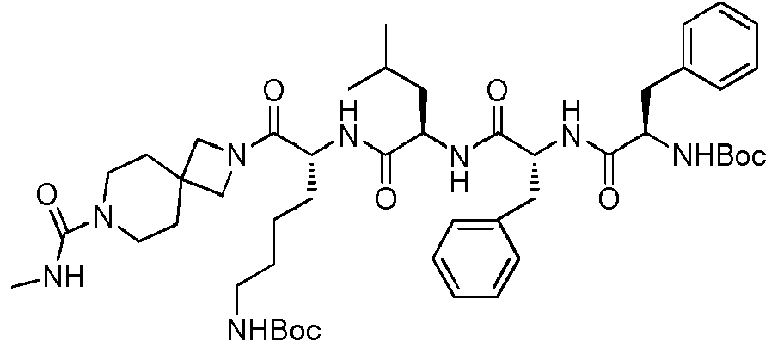
Se añadió N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; ácido trifluoroacético (27B) en bruto (0,48 g, 2,60 mmol) en diclorometano (10 ml) en un matraz de reacción de 50 ml bajo protección de nitrógeno. Se enfrió hasta 0°C en un baño de hielo y se añadieron el producto intermedio 1 (0,50 g, 0,66 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (200 mg, 1,73 mmol), 1-hidroxibenzotriazol (125 mg, 0,93 mmol). Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Posteriormente, se añadió una disolución acuosa de ácido clorhídrico 1 M (15 ml) a la disolución de reacción y se agitó la mezcla y luego se sometió a un proceso de separación de líquido. Se añadió una disolución acuosa saturada de carbonato de sodio (15 ml) a la fase orgánica y se agitó la mezcla durante 30 minutos y luego se sometió a un proceso de separación de líquido. Se lavó la fase orgánica con una disolución acuosa saturada de cloruro de sodio (15 ml), se secó sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1- [[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(metilcarbamoil)-2,7-]diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (27 C) en bruto como un sólido espumoso de color amarillo claro (0,5 g, rendimiento del 80%) y se usó directamente en la siguiente reacción.
Etapa 4:
2- [(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[3.5]nona-7-carboxamida; di-ácido trifluoroacético (compuesto 27)

Se disolvió N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (27C) en bruto (0,5 g, 0,5 mmol) en diclorometano (7,5 ml) y se añadió ácido trifluoroacético (3,5 ml). Se agitó el sistema a temperatura ambiente durante 1 h. Posteriormente, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 pm; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener 2-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; di-ácido trifluoroacético (compuesto 27) como un sólido de color blanco (220 mg, rendimiento del 40%).
EM m/z (ESI): 360,3 [M+1H]+/2;
1H-RMN (400 MHz, D2O) 87,45-7,16 (m, 10H), 4,66-4,57 (m, 1H), 4,32-4,19 (m, 2H), 4,19-3,99 (m, 3H), 3,83-3,66 (m, 2H), 3,40-3,22 (m, 4H), 3,22-3,12 (m, 2H), 3,06-2,99 (m, 4H), 2,69 (s, 3H), 1,80-1,62 (m, 8H), 1,58-1,27 (m, 5H), 1,03 0,79 (m, 6H).
Ejemplo 26:
7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida; di-ácido trifluoroacético (compuesto 28)

Etapa 1:
5,5-Difluoro-2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (28A)

Se añadieron 5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16C) (330 mg, 1,11 mmol), trietilamina (364 mg, 3,6 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml y se disolvió con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de metilaminoformilo (104 mg, 1,11 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se añadió ácido clorhídrico diluido 3 M (50 ml) a la disolución de reacción y se extrajo la mezcla con diclorometano (60 ml x 2). Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 5,5-difluoro-2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (28A) como un sólido de color blanco (268 mg, rendimiento del 68,6%).
EM m/z (ESI): 354,1 [M+H]+;
1H-RMN (400 MHz, CDCls) 87,41-7,28 (m, 5H), 5,14 (s, 2H), 4,09 (d, 2H), 3,65 (dd, 4H), 3,51-3,44 (m, 2H), 2,79 (s, 3H), 2,01 (s, 2H).
Etapa 2:
5,5-Difluoro-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida (28B)

Se añadieron 5,5-difluoro-2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (28A) (269 mg, 0,76 mmol), paladio sobre carbono (54 mg, al 20% en peso) y metanol (5 ml) en un matraz de reacción de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla bajo una atmósfera de hidrógeno (globo) a temperatura ambiente durante 3 h. Luego, se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida (28B) en bruto como una sustancia oleosa de color amarillo claro (164 mg, rendimiento del 98,55%), y se usó directamente en la siguiente reacción.
EM m/z (ESI): 220,2 [M+H]+;
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[5,5-difluoro-2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (28C)

Se añadieron secuencialmente 5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida (28B) en bruto (164 mg, 0,75 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,374 g, 1,95 mmol), 1-hidroxibenzotriazol (110 mg, 0,81 mmol), producto intermedio 1 (565 mg, 0,75 mmol) y diclorometano (50 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener de N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2
[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[5,5-difluoro-2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-1-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (28C) como un sólido de color amarillo claro (600 mg, rendimiento del 84%).
Etapa 4:
7-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida; diácido trifluoroacético (compuesto 28)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[5,5-difluoro-2-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-1-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (28C) (600 mg, 0,6 mmol) y ácido trifluoroacético (3 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener 7-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-2-carboxamida; di-ácido trifluoroacético (compuesto 28) como un polvo de color blanco (100 mg, rendimiento del 13%).
EM m/z (ESI): 755,5 [M+H]+;
1H-RMN (400 MHz, D2O) 87,43-7,18 (m, 10H), 4,86-4,75 (m, 1H), 4,65 (t, 1H), 4,39-4,17 (m, 2H), 4,15-4,05 (m, 2H), 4,04-3,45 (m, 6H), 3,24-3,10 (m, 2H), 3,10-2,90 (m, 4H), 2,77-2,60 (m, 3H), 2,06 (d, 2H), 1,85-1,61 (m, 4H), 1,60-1,46 (m, 3H), 1,45-1,27 (m, 2H), 0,92 (dt, 6H).
Ejemplo 27:
2-[(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; diácido trifluoroacético (compuesto 29)
Etapa 1:
5,5-Difluoro-7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (29B)

Se añadieron 5,5-difluoro-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (16C) (113 mg, 0,43 mmol), trietilamina (43 mg, 0,43 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se disolvió con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de metilaminoformilo (42 mg, 0,45 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se añadió ácido clorhídrico diluido 0,3 M (10 ml) a la disolución de reacción y se extrajo la mezcla con diclorometano (5 ml x 2). Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 5,5-difluoro-7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (29B) como un sólido de color blanco (96 mg, rendimiento del 70%).
Etapa 2:
5,5-Difluoro-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida (29C)

Se añadieron 5,5-difluoro-7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (29B) (96 mg, 0,3 mmol) y diclorometano (8 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida (29C), como un líquido oleoso de color amarillo (66 mg, rendimiento del 100%), y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[5,5-difluoro-7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (29D)

Se añadieron 5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida (29C) (66 mg, 0,3 mmol), clorhidrato de 1- (3-dimetilaminopropil)-3-etilcarbodiimida (239 mg, 0,36 mmol), 1-hidroxibenzotriazol (49 mg, 0,36 mmol), producto intermedio 1 (226 mg, 0,3 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener de (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[5,5-difluoro-7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (29D) como un sólido blanco (286 mg, rendimiento del 90%).
Etapa 4:
2- [(2R)-6-Amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; diácido trifluoroacético (compuesto 29)

Se añadieron (1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[5,5-difluoro-7-(metilcarbamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (29D) (286 mg, 0,3 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener 2-[(2R)-6-amino-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-5,5-difluoro-N-metil-2,7-diazaespiro[3.5]nonano-7-carboxamida; di-ácido trifluoroacético (compuesto 29) como un polvo de color blanco (192 mg, rendimiento del 65%).
EM m/z = 378,3 [M+2H]+ /2;
1H-RMN (400 MHz, D2O) 87,46-7,12 (m, 10H), 4,64-4,58 (m, 1H), 4,53-4,43 (m, 1H), 4,26-4,19 (m, 2H), 4,17-4,02 (m, 3H), 3,80-3,76 (m, 1H), 3,72-3,57 (m, 2H), 3,36 (s, 2H), 3,19-3,11 (m, 2H), 3,03-2,95 (m, 4H), 2,68 (s, 3H), 2,03-1,99 (m 2H), 1,75-1,63 (m, 4H), 1,54-1,26 (m, 5H), 0,89 (dd, 6H).
Ejemplo 28:
(2R)-N-[(1R)-1-(7-Acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 30)

7-Acetil-2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (30A)

Se añadieron 2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (A) (0,414 g, 2 mmol), trietilamina (420 mg, 4,0 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se disolvió con agitación. Después de enfriar hasta -10°C, se añadió cloruro de acetilo (188 mg, 2,4 mmol) y se hizo reaccionar el material resultante durante 10 min.
Luego se elevó la temperatura hasta temperatura ambiente y se agitó el sistema durante 3 h. Luego, se extinguió la reacción con una disolución acuosa saturada de bicarbonato de sodio (10 ml) y se extrajo con acetato de etilo (5 ml x 3) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1) para obtener 7-acetil-2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (30A) como una sustancia oleosa de color amarillo claro (411 mg, rendimiento del 81%).
EM m/z = 255,2 [M+H]+.
Etapa 2:
7-Acetil-2,7-diazaespiro[3.4]octano (30B)

Se añadieron 7-acetil-2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (30A) (0,41 g, 1,62 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (1 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 7-acetil-2,7-diazaespiro[3.4]octano (30B) en bruto como un líquido oleoso de color amarillo claro (249 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-2-[[(1R)-2-[[(1 R)-1-[[(1R)-1-(7-Acetil-2,7-diazaespiro[3.4]octano]-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (30C)

Se añadieron 7-acetil-2,7-diazaespiro[3.4]octano (30B) en bruto (249mg, 1,62 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (384 mg, 2 mmol), 1-hidroxibenzotriazol (270 mg, 2 mmol), producto intermedio 1 (400 mg, 0,53 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (30C) como un sólido de color blanco (200 mg, rendimiento del 42,3%).
Etapa 4:
(2R)-N-[(1R)-1-(7-Acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 30)

Se añadieron N-[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (30C) (200 mg, 0,22 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-1-(7-acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5-aminopentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 30) como un polvo de color blanco (120 mg, rendimiento del 79,1%).
EM m/z = 345,9 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,45-7,29 (m, 10H), 4,71 (t, 1H), 4,37-4,2 (m, 5H), 4,05-4,00 (m, 2H), 3,82-3,52 (m, 4H), 3,26-3,05 (m, 6H), 2,32-2,11 (m, 5H), 1,8-1,76 (m, 4H), 1,61-1,45 (m, 5H), 1,02-0,96 (dd, 6H).
Ejemplo 29:
(2R)-N-[(1R)-1-(7-Acetil-2,7-diazaespiro[4.4]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 31)

Etapa 1:
7-Acetil-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (31A)

Se añadieron 2-Boc-2,7-diazaespiro[4.4]nonano (22A) (452 mg, 2,0 mmol), trietilamina (400 mg, 4,0 mmol) y diclorometano (15 ml) en un matraz de reacción de 50 ml y se disolvió con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de acetilo (160 mg, 2,0 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 4 h. Luego se añadió ácido clorhídrico diluido 1 M (50 ml) a la disolución de reacción y se extrajo la mezcla con diclorometano (60 ml x 2). Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 7-acetil-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (31A) como una sustancia oleosa de color amarillo claro (392 mg, rendimiento del 73%). EM m/z = 291,2 [M+Na]+.
Etapa 2:
1-(2,7-Diazaespiro[4.4]nonan-2-il)etanona (31B)

Se añadieron 7-acetil-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (31A) (161 mg, 0,6 mmol), diclorometano (10 ml) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida para obtener 1-(2,7-diazaespiro[4.4]nonano-2-il)etanona (31B) en bruto como una sustancia oleosa de color amarillo claro (100 mg, rendimiento del 99%).
Etapa 3:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-Acetil-2,7-diazaespiro[4.4]nonano]-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (31C)

Se añadieron secuencialmente 1-(2,7-diazaespiro[4.4]nonano-2-il)etanona (31B) en bruto (100 mg, 0,6 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,288 g, 1,5 mmol), 1-hidroxibenzotriazol (81 mg, 0,6 mmol), producto intermedio 1 (400 mg, 0,5 mmol) y diclorometano (50 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 30:1) para obtener N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[4.4]nonano-2-carbonil)-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxoetil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo(31C) como un sólido de color amarillo claro (120 mg, rendimiento del 22%).
Etapa 4:
(2R)-N-[(1R)-1-(7-Acetil-2,7-diazaespiro[4.4]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 31)

Se añadieron N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[4.4]nonano-2-carbonil)-5-(tercbutox¡carbon¡lam¡no)pent¡l]carbamo¡l]-3-metil-but¡ljam¡no]-í-benc¡l-2-oxo-et¡l]am¡no]-1-benc¡l-2-oxo-et¡l]carbamato de terc-butilo (31C) (120 mg, 0,22 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; ¡socrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-1-(7-acetil-2,7-diazaespiro[4.4]nonano-2-carbonil)-5-aminopentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido
trifluoroacético (compuesto 31) como un polvo de color blanco (77 mg, rendimiento del 85%).
EM m/z (ESI): 352,7 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,43-7,18 (m, 10H), 4,68-4,60 (m, 1H), 4,47-4,19 (m, 3H), 3,91-3,27 (m, 8H), 3,18 (d, 2H), 3,08-2,92 (m, 4H), 2,13-1,86 (m, 7H), 1,85-1,63 (m, 4H), 1,62-1,27 (m, 5H), 0,92 (dd, 6H).
Ejemplo 30:
(2R)-N-[(1R)-1-(7-Acetil-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 32)

Etapa 1:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil-carbamato de terc-butilo (32A)

Se añadieron secuencialmente N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (19B)(430 mg, 0,48 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,18 g, 0,94 mmol), 1-hidroxibenzotriazol (71 mg, 0,53 mmol), ácido acético (28,8 mg, 0,48 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml, y se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (32A) como un sólido de color blanco (430 mg, rendimiento del 95%).
Etapa 2:
(2R)-N-[(1R)-1-(7-Acetil-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 32)

Se añadieron N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxoetil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (32A) (430 mg, 0,46 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-1-(7-acetil-5,5-difluoro-2,7-diazaespiro[3.5]nonano-2-carbonil)-5-amino-pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanamida; di-ácido trifluoroacético (compuesto 32) como un polvo de color blanco (273 mg, rendimiento del 61%).
EM m/z (ESI): 370,8 [M+2H]+72;
1H-RMN (400 MHz, D2O) 87,43-7,19 (m, 10H), 4,67-4,60 (m, 1H), 4,58-4,47 (m, 1H), 4,31-4,08 (m, 5H), 3,98-3,70 (m, 3H), 3,70-3,43 (m, 2H), 3,18 (d, 2H), 3,06-2,94 (m, 4H), 2,23-1,94 (m, 5H), 1,80-1,63 (m, 4H), 1,56-1,29 (m, 5H), 0,91 (dd, 6H).
Ejemplo 31:
(2R)-N-[(1R)-5-Amino-1-[7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 33)

7-(Oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (33A)

Se añadieron secuencialmente 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (0,452 g, 2,0 mmol), ácido acético (0,24 g, 4,0 mmol), 3-oxetanona (0,288 g, 4,0 mmol), triacetoxiborohidruro de sodio (1,48 g, 6,98 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml. Después de la adición, se permitió que avanzase la reacción
a temperatura ambiente durante 16 h. Se filtró la disolución de reacción y se lavó el filtrado con una disolución saturada de bicarbonato de sodio (50 ml). Después de la separación del líquido, se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró y se concentró el filtrado a presión reducida para obtener 7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (33A) como un polvo de color blanco (432 mg, rendimiento del 76%).
1H-RMN (400 MHz, CDCls) 84,67-4,55 (m, 4H), 3,61 (s, 4H), 3,41 (p, 1H), 2,19 (s, 4H), 1,78 (t, 4H), 1,44 (s, 9H). Etapa 2:
7-(Oxetan-3-il)-2,7-diazaespiro[3.5]nonano (33B)

Se añadieron 7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (33A) (0,14 g, 0,5 mmol) y diclorometano (5 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (57 mg, 0,5 mmol) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano (33B) en bruto como un líquido oleoso de color amarillo (70,5 mg, rendimiento del 88%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (33C)

Se añadieron 7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano (33B) en bruto (70,5 mg, 0,44 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (288 mg, 1,5 mmol), 1-hidroxibenzotriazol (81 mg, 0,60 mmol), producto intermedio 1 (330 mg, 0,44 mmol) y diclorometano (50 ml) en un matraz de reacción de 100 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (33C) como un sólido de color amarillo claro (400 mg, rendimiento del 99%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 33)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(oxetan-3-il)-2,7diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-il]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (33C) (400 mg, 0,40 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Luego, se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-(oxetan-3-il)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; ácido tri-trifluoroacético (compuesto 33) como un polvo de color blanco (160 mg, rendimiento del 56%).
EM m/z (ESI): 359,8 [M+2H]+72;
1H-RMN (400 MHz, D2O) 87,42-7,20 (m, 10H), 4,96 (t, 2H), 4,85 (dd, 2H), 4,66-4,56 (m, 2H), 4,48-4,35 (m, 2H), 4,30 4,10 (m, 5H), 3,84 (s, 2H), 3,43 (a, 1H), 3,17 (d, 2H), 3,14 (s, 1H), 3,07-2,93 (m, 4H), 2,12 (a, 4H), 1,80-1,60 (m, 4H), 1,58-1,29 (m, 5H), 0,91 (dd, 6H).
Ejemplo 32:
(2R)-N-[(1R)-5-amino-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 34)

7-(Dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (34A)
Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (0,452 g, 2 mmol), trietilamina (400 mg, 4,0 mmol) y diclorometano (15 ml) en un matraz de reacción de 50 ml y se disolvió con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de dimetilaminosulfonilo (287 mg, 2,0 mmol). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante 3 h. Se añadió ácido clorhídrico diluido 3 M (50 ml) a la disolución de reacción, seguido de extracción con diclorometano (60 ml x 2). Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida para obtener 7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (34A) en bruto como un sólido de color amarillo claro (440 mg, rendimiento del 66%).
Etapa 2:
N,N-Dimetil-2,7-diazaespiro[3.5]nonano-7-sulfonamida (34B)

Se añadieron 7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (34A) en bruto (0,22 g, 0,66 mmol) y diclorometano (5 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener N,N-dimetil-2,7-diazaespiro[3.5]nonano-7-sulfonamida (34B) en bruto como un líquido oleoso de color amarillo (103 mg, rendimiento del 76%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (34C)

Se añadieron N,N-dimetil-2,7-diazaespiro[3.5]nonano-7-sulfonamida (34B) en bruto (103 mg, 0,44 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (288 mg, 1,5 mmol), 1-hidroxibenzotriazol (81 mg, 0,60 mmol), producto intermedio 1 (330 mg, 0,44 mmol) y diclorometano (50 ml) en un matraz de reacción de 100 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(dimetilsulfamoil)-2,7]-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (34C) como un sólido de color amarillo claro (240 mg, rendimiento del 56%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 34)

N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (34C) (400 mg, 0,4 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil- pentanamida; di ácido trifluoroacético (compuesto 34) como un polvo de color blanco (130 mg, rendimiento del 29%).
EM m/z (ESI): 385,3 [M+2H]+72;
1H-RMN (400 MHz, D2O) 87,46-7,29 (m, 6H), 7,29-7,18 (m, 4H), 4,65 (t, 1H), 4,32-3,99 (m, 5H), 3,85-3,68 (m, 2H), 3,35-3,11 (m, 6H), 3,11-2,92 (m, 4H), 2,81 (d, 6H), 1,96-1,79 (m, 4H), 1,71 (dd, 4H), 1,60-1,32 (m, 5H), 0,93 (dd, 6H).
Ejemplo 33:
(2R)-N-[(1R)-5-Amino-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 35)

7-(Dimetilsulfamoil)-2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (35A)

Se añadieron 2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (A) (0,414 g, 2 mmol), trietilamina (420 mg, 4,0 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se disolvió con agitación. Después de enfriar hasta -10°C, se añadió gota a gota cloruro de dimetilsulfamoílo (343 mg, 2,4 mmol) y se permitió que avanzase la reacción durante 10 minutos. Luego se elevó la temperatura hasta temperatura ambiente y se agitó durante 3 h. Se extinguió la disolución de reacción con una disolución acuosa saturada de bicarbonato de sodio (10 ml), se extrajo con acetato de etilo (5 ml x 3) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1) para obtener 7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (35A) como una sustancia oleosa de color amarillo claro (414 mg, rendimiento del 0,65%).
EM m/z = 320,2 [M+H]+;
Etapa 2:
7-(Dimetilsulfamoil)-2,7-diazaespiro[3.4]octano (35B)

Se añadieron 7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano-2-carboxilato de terc-butilo (35A) (0,41 g, 1,3 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (1 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano (35B) en bruto como un líquido oleoso de color amarillo claro (284 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-(7-Dimetilsulfamoil-2,7-diazaespiro[3.4]octano-2-carbonil)-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (35C)

Se añadieron 7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano (35B) en bruto (284mg, 1,3 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (384 mg, 2 mmol), 1-hidroxibenzotriazol (270 mg, 2 mmol), producto intermedio 1 (400mg, 0,53 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (35C) como un sólido de color blanco (201 mg, rendimiento del 39,7%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 35)

Se añadieron N-[(1R)-2-[[(1R)-1-[[(1R)-1-(7-acetil-2,7-diazaespiro[3.4]octano-2-carbonil)-5(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (35C) (201 mg, 0,17 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-(7-(dimetilsulfamoil)-2,7-diazaespiro[3.4]octano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanamida; di ácido trifluoroacético (compuesto 35) como un polvo de color blanco (115 mg, rendimiento del 89,7%).
EM m/z = 378,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,46-7,29 (m, 10H), 4,71 (t, 1H), 4,38-4,21 (m, 5H), 4,07-4,05 (m, 2H), 3,60-3,46 (m, 4H), 3,23-3,05 (m, 6H), 2,9-2,89 (d, 6H), 2,32-2,26 (m, 2H) 1,8-1,59 (m, 9H), 1,02-0,96 (dd, 6H).
Ejemplo 34:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tetra-ácido trifluoroacético (compuesto 36)

2-(4-Benciloxicarbonilpiperazin-1-il)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (36A)
Se añadieron 2-oxo-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (C) (0,478 g, 2 mmol), carbonato de bencil-1-piperazina (440 mg, 2 mmol), ácido acético (120 mg, 2,0 mmol) y dicloroetano (7 ml) en un matraz de reacción de 50 ml y se agitó el sistema durante media hora. Se añadió triacetoxiborohidruro de sodio (0,636 g, 3 mmol) y se hizo reaccionar el material resultante durante 5 h. Se extinguió el sistema de reacción con agua (10 ml), se extrajo con acetato de etilo (5 ml x 3) y se combinaron las fases orgánicas. Se secaron las fases orgánicas sobre sulfato de sodio anhidro, se filtraron y se concentró el filtrado a presión reducida. Se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 4:1) para obtener 2-(4-benciloxicarbonilpiperazin-1-il)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (36A) como una sustancia oleosa de color amarillo claro (450 mg, rendimiento del 50,79%).
EM m/z = 444,2 [M+H]+;
Etapa 2:
4-(7-Azaespiro[3.5]nonan-2-il)piperazin-1-carboxilato de bencilo (36B)

Se añadieron 2-(4-benciloxicarbonilpiperazin-1-il)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (36A) (0,45 g, 1,20 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (1 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener 4-(7-azaespiro[3.5]nonano-2-il)piperazin-1-carboxilato de bencilo (36B) en bruto como un líquido oleoso de color amarillo claro (411 mg, rendimiento del 100%), y se usó directamente en la siguiente reacción.
Etapa 3:
4-[7-[(2R)-6-(terc-Butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-]3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanoil]amino]hexanoil]-7-azaespiro[3.5]nonan-2-il]piperazin-1-carboxilato de bencilo (36C)

Se añadieron 4-(7-azaespiro[3.5]nonano-2-il)piperazin-1-carboxilato de bencilo (36B) en bruto (411 mg, 1,2mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (384 mg, 2 mmol), 1-hidroxibenzotriazol (270 mg, 2 mmol), producto intermedio 1 (400 mg, 0,53 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener 4-[7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-il]piperazin-1-carboxilato de bencilo (36C) como un sólido de color blanco (300 mg, rendimiento del 52,4%).
Etapa 4:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (36D)

Se añadieron 4-[7-[(2R)-6-(terc-butoxicarbonilamino)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-(terc-butoxicarbonilamino)-]3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metilpentanoil]amino]hexanoil]-7-azaespiro[3.5]nonano-2-il]piperazin-1-carboxilato de bencilo (36C) (300 mg, 0,27 mmol) y metanol (5 ml), y Pd/C (30 mg) en un matraz de reacción de 50 ml, y se permitió que reaccionase el sistema a temperatura ambiente durante 4 h bajo una atmósfera de hidrógeno. Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró a presión reducida para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (36D) (255 mg, 100%) y se usó directamente en la siguiente etapa.
Etapa 5:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tetra-ácido trifluoroacético (compuesto 36)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de tercbutilo (36D) en bruto (255 mg, 0,27 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de
columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-N-[(1R)-5-amino-1-(2-piperazin-1-il-7-azaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; tetra-ácido trifluoroacético (compuesto 36) como un polvo de color blanco (160 mg, rendimiento del 77,6%).
EM m/z = 373,4 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,47-7,30 (m, 10H), 4,71 (t, 1H), 4,36-4,29 (m, 2H), 3,90-3,86 (m, 1H), 3,85-3,24 (m, 12H), 3,23-3,04 (m, 6H), 2,52-2,44 (m, 2H), 2,14-2,12 (m, 2H), 1,77-1,47 (m, 14H), 1,03-0,96 (dd, 6H).
Ejemplo 35:
(2R)-N-[(1R)-5-Amino-1-[7-[(2R)-2-amino-3-fenilpropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 37)

7-[(2R)-2-(Benciloxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (37A) Boc

Ph
37A
Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (83 mg, 0,35 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (80 mg, 0,42 mmol), 1-hidroxibenzotriazol (57 mg, 0,42 mmol), ácido (2R)-2-(benciloxicarbonilamino)-3-fenil-propanoico (105 mg, 0,35 mmol) y diclorometano (30 ml) en un matraz de reacción de
50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 7-[(2R)-2-(benciloxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (37A) como un sólido de color blanco (177 mg, rendimiento del 80%).
Etapa 2:
N-[(1R)-1-Bencil-2-(2,7-diazaespiro[3.5]nonan-7-il)-2-oxo-etil]carbamato de bencilo (37B)

37B
Se añadieron 7-[(2R)-2-(benciloxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de tercbutilo (37A) (177 mg, 0,35 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener N-[(1R)-1-bencil-2-(2,7-diazaespiro[3.5]nonan-7-il)-2-oxo-etil]carbamato de bencilo (37B) como un líquido oleoso de color amarillo (143 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2Hbenciloxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (37C)

Se añadieron N-[(1R)-1-bencil-2-(2,7-diazaespiro[3.5]nonan-7-il)-2-oxo-etil]carbamato de bencilo (37B) (143 mg, 0,35 mmol), 1-clorhidrato de (3-dimetilaminopropil)-3-etilcarbodiimida (80 mg, 0,42 mmol), 1-hidroxibenzotriazol (57 mg, 0,42 mmol), producto intermedio 1 (264 mg, 0,35 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-(benciloxicarbonilamino)-3-fenilpropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (37C) como un sólido de color blanco (400 mg, rendimiento del 99%).
Etapa 4:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-Amino-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de terc-butilo (37D)

Se añadieron (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2]-(benciloxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (37C) (400 mg, 0,35 mmol), paladio sobre carbono (80 mg, al 20% en peso de l) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener (1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-amino-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato de terc-butilo (37D) como un sólido de color amarillo claro (353 mg, rendimiento del 100%), y se utilizó directamente en la siguiente reacción.
Etapa 5:
(2R)-N-[(1R)-5-Amino-1-[7-[(2R)-2-amino-3-fenilpropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 37)

Se añadieron (1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-amino-3-fenilpropanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonil)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato de terc-butilo (37d) (353 mg, 0,35 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-amino-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 37) como un polvo de color blanco (282 mg, rendimiento del 70%).
EM m/z = 405,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,52-7,09 (m, 15H), 4,78-4,74 (m, 1H), 4,64-4,58 (m, 1H), 4,26-4,19 (m, 2H), 4,15-3,96 (m, 3H), 3,83 (d, 1H), 3,71-3,63 (m, 1H), 3,59-3,50 (m, 1H) 3,43-2,90 (m, 11H), 1,90-1,18 (m, 13H), 0,98-0,80 (m, 6H).
Ejemplo 36:
(2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-amino-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 38)

7-[(2R)-2-(Benciloxicarbonilamino)-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (38A)

Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (93mg, 0,41 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (94 mg, 0,49 mmol), 1-hidroxibenzotriazol (66 mg, 0,49 mmol), ácido (2R)-2-(benciloxicarbonilamino)propanoico (446 mg, 2,0 mmol) y diclorometano (10 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 7-[(2R)-2-(benciloxicarbonilamino)-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (38A) como un sólido de color blanco (175 mg, rendimiento del 90%).
Etapa 2
N-[(1R)-1-(2,7-Diazaespiro[3.5]nonano-7-carbonil)-3-metil-butil]carbamato de bencilo (38B)

Se añadieron 7-[(2R)-2-(benciloxicarbonilamino)-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carboxilato de tercbutilo (38A) (175 mg, 0,37 mmol) y diclorometano (20 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético (2 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener N-[(1R)-1-(2,7-diazaespiro[3.5]nonano-7-carbonil)-3-metil-butil]carbamato de bencilo (38B) como un líquido oleoso de color amarillo (138 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-H[(1R)-1-[7-[(2R)-2Hbenciloxicarbonilamino)-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxoetil]amino]-2-oxo-etil]carbamato de terc-butilo (38C)

Se añadieron N-[(1R)-1-(2,7-diazaespiro[3.5]nonano-7-carbonil)-3-metil-butil]carbamato de bencilo (38B) (138 mg, 0,37 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (84 mg, 0,44 mmol), 1-hidroxibenzotriazol (59 mg, 0,44 mmol), producto intermedio 1 (279 mg, 0,37 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-(benciloxicarbonilamino)-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (38C) como un sólido de color blanco (366 mg, rendimiento del 90%).
Etapa 4:
N-[(1R)-2-[[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-Amino-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato de terc-butilo (38D)

Se añadieron (1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2]-(benciloxicarbonilamino)-4-metilpentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonilamino)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (38C) (366 mg, 0,33 mmol), paladio sobre carbono (73 mg, al 20% en peso de l) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno. Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener (1R)-2-[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-amino-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(tercbutoxicarbonil)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxo-etil]carbamato de tercbutilo (38D) como un sólido de color amarillo claro (322 mg, rendimiento del 100%), y se usó directamente en la siguiente reacción.
Etapa 5:
(2R)-N-[(1R)-5-Amino-1-[7-[(2R)-2-amino-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 38)
Compuesto 38
Se añadieron (1R)-2-[(1R)-2-[[(1R)-1-[[(1R)-1-[7-[(2R)-2-amino-4-metil-pentanoil]]-2,7-diazaespiro[3.5]nonano-2-carbonil]-5-(terc-butoxicarbonil)pentil]carbamoil]-3-metil-butil]amino]-1-bencil-2-oxo-etil]amino]-1-bencil-2-oxoetil]carbamato de terc-butilo (38d) (322 mg, 0,33 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml, y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-[(2R)-2-amino-4-metil-pentanoil]-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 38) como un polvo de color blanco (240 mg, rendimiento del 65%).
EM m/z = 388,3 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,44-7,14 (m, 10H), 4,61 (t, 1H), 4,50-4,42 (m, 1H), 4,28-4,02 (m, 5H), 3,83-3,70 (m, 2H), 3,66-3,36 (m, 4H), 3,14 (t, 2H), 2,97 (t, 4H), 1,94-1,64 (m, 10H), 1,54-1,30 (m, 6 H), 1,01-0,81 (m, 12H).
Ejemplo 37:
(2R)-N-[(1R)-5-Amino-1-[2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 39)

Etapa 1:
2-(Ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (39A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (390 mg, 1,5 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (760 mg, 4,9 mmol), 1-hidroxibenzotriazol (300 mg, 2,0 mmol), ácido ciclopropilcarboxílico (172 mg, 2,0 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (39A) como una sustancia aceitosa de color amarillo claro (320 mg, rendimiento del 65%).
Etapa 2:
Cidopropil(2,7-diazaespiro[3.5]nonan-2-N)metanona (39B)

Se añadieron 2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (39A) (320 mg, 0,97 mmol), paladio sobre carbono (64 mg, 20% en peso de l) y metanol (10 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener ciclopropil(2,7-diazaespiro[3.5]nonan-2-il)metanona (39B) en bruto como un sólido de color amarillo claro (189 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (39C)

Se añadieron ciclopropil(2,7-diazaespiro[3.5]nonan-2-il)metanona (39B) en bruto (189 mg, 0,75 mmol), clorhidrato de 1- (3-dimetilaminopropil)-3-etilcarbodiimida (216 mg, 1,13 mmol), 1-hidroxibenzotriazol (122 mg, 0,9 mmol), producto intermedio 1 (565 mg, 0,75 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2- oxo-etil]carbamato de terc-butilo (39C) como un sólido de color amarillo claro (410 mg, rendimiento del 45%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 39)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (39C) (410 mg, 0,44 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 p,m; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[2-(ciclopropanocarbonil)-2,7
diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 39) como un polvo de color blanco (330 mg, rendimiento del 78%).
EM m/z = 365,8 [M+2H]+ /2;
1H-RMN (400 MHz, D2O) 87,45-7,18 (m, 10H), 4,66 (t, 1H), 4,26 (dt, 2H), 4,13 (d, 2H), 3,78 (d, 2H), 3,71-3,61 (m, 2H), 3,56-3,45 (m, 1H), 3,42-3,31 (m, 1H), 3,18 (d, 2H), 3,10-2,93 (m, 4H), 1,99-1,30 (m, 15H), 1,01-0,77 (m, 10H).
Ejemplo 38:
(2R)-N-[(1R)-5-Amino-1-[7-(cidopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 40)

7-(Ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (40A)

Se añadieron 2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (6A) (0,453 g, 2,0 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (767 mg, 4,0 mmol), 1-hidroxibenzotriazol (324 mg, 2,4 mmol), ácido ciclopropilcarboxílico (176 mg, 2,2 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 7-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (40A) como una sustancia oleosa de color amarillo claro (590 mg, rendimiento del 96%).
1H-RMN (400 MHz, CDCls) 83,68 (s, 4H), 3,62-3,52 (m, 4H), 1,81-1,69 (m, 5H), 1,45 (s, 9H), 1,00-0,93 (m, 2H), 0,78 0,71 (m, 2H)).
Etapa 2:
Ciclopropil(2,7-diazaespiro[3.5]nonan-7-il)metanona (40B)

Se añadieron 7-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carboxilato de terc-butilo (40A)(0,25 g, 0,83 mmol) y diclorometano (7 ml) en un matraz de reacción de 50 ml y se añadió gota a gota ácido trifluoroacético
(1 ml) a temperatura ambiente. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 3 h. Se concentró la disolución de reacción directamente a presión reducida para obtener ciclopropil(2,7-diazaespiro[3.5]nonan-7-il)metanona (40B) en bruto como un líquido oleoso de color amarillo (161 mg, rendimiento del 100%), y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (40C)

Se añadieron (2,7-diazaespiro[3.5]nonan-7-il)metanona (40B) en bruto (0,16 g, 0,83 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (239 mg, 1,25 mmol), 1-hidroxibenzotriazol (135 mg, 1,0 mmol), producto intermedio 1 (625 mg, 0,83 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(ciclopropanocarbonil)-2,7]-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (40C) como un sólido de color amarillo claro (700 mg, rendimiento del 90%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[7-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 40)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[7-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (40C) (700 mg, 0,75 mmol) y ácido trifluoroacético (3 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[7-(ciclopropanocarbonil)-2,7-diazaespiro[3.5]nonano-2-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 40) como un polvo de color blanco (200 mg, rendimiento del 28%).
EM m/z = 365,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,40-7,17 (m, 10H), 4,62 (t, 1H), 4,32-4,00 (m, 5H), 3,82-3,60 (m, 4H), 3,48 (a, 2H), 3,23 3,08 (m, 2H), 3,07-2,91 (m, 4H), 2,01-1,61 (m, 9H), 1,58-1,27 (m, 5H), 0,97-0,67 (m, 10H).
Ejemplo 39:
(2R)-N-[(1R)-5-Amino-1-[2-[(2R)-2-amino-3-fenilpropanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 41)

2-[(2R)-2-(terc-Butoxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (41A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (390 mg, 1,5mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (760 mg, 4,9 mmol), 1-hidroxibenzotriazol (300 mg, 2,0 mmol), Boc-D-fenilalanina (530 mg, 2,0 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 2-[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (41A), como un sólido de color blanco (514 mg, rendimiento del 67%).
Etapa 2:
N-[(1R)-1-Bencil-2-(2,7-diazaespiro[3.5]nonan-2-il)-2-oxo-etil]carbamato de terc-butilo (41B)

Se añadieron 2-[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (41A) (514 mg, 1,01 mmol), paladio sobre carbono (100 mg, 20% en peso) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener N-[(1R)-1-bencil-2-(2,7-diazaespiro[3.5]nonan-2-il)-2-oxo-etil]carbamato de terc-butilo (41B) en bruto como un sólido de color amarillo claro (377 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-[(2R)-2-(tercbutoxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (41C)

Se añadieron N-[(1R)-1-bencil-2-(2,7-diazaespiro[3.5]nonan-2-il)-2-oxo-etil]carbamato de terc-butilo (41B) en bruto (377 mg, 1,01 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (290 mg, 1,52 mmol), 1-hidroxibenzotriazol (164 mg, 1,21 mmol), producto intermedio 1 (761 mg, 1,01 mmol) y diclorometano (30 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (éter de petróleo:acetato de etilo (v:v) = 1:2) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-[(2R)-2-(terc-butoxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (41C), como un sólido de color amarillo claro (410 mg, rendimiento del 36,6%).
Etapa 4:
(2R)-N-[(1R)-5-Amino-1-[2-[(2R)-2-amino-3-fenilpropanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; tri-ácido trifluoroacético (compuesto 41)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-[(2R)-2-(tercbutoxicarbonilamino)-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (41C) (410 mg, 0,37 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[2-[(2R)-2-amino-3-fenil-propanoil]-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenilpropanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida: ácido tri-trifluoroacético (compuesto 41) como un polvo de color blanco (298 mg, rendimiento del 70%).
EM m/z = 405,4 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,52-7,28 (m, 11H), 7,28-7,20 (m, 4H), 4,67-4,62 (m, 1H), 4,32-4,15 (m, 3H), 3,85-3,38 (m, 6H), 3,37-3,22 (m, 2H), 3,22-3,13 (m, 2H), 3,12-2,90 (m, 6H), 2,65 (dd, 1H), 1,83-1,48 (m, 9H), 1,47-1,28 (m, 3H), 1,25 1,11 (m, 1H), 0,92 (dd, 6H).
Ejemplo 40:
(2R)-N-[(1R)-5-Amino-1-[2-(2-hidroxiacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 42)

Ácido 2-tetrahidropiran-2-iloxiacético (42B)
O
hcA ^ othp
42B
Se añadieron secuencialmente 3,4-dihidro-2H-pirano (840 mg, 10 mmol), glicolato de metilo (42A) (900 mg, 10 mmol), ácido p-toluenosulfónico monohidratado (100 mg, 0,5 mmol) y diclorometano (100 ml) en un matraz de una sola boca de 250 ml. Después de la adición, se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se añadió al residuo una disolución de hidróxido de litio (una disolución mixta de 75 ml de metanol 25 ml de agua). Después de la adición, se permitió que avanzase la reacción a temperatura ambiente durante la noche. Se añadió gota a gota ácido clorhídrico diluido 0,1 M (12 ml) a la disolución de reacción y se comprobó que el pH de la disolución de reacción era de 2-3. Se extrajo la disolución de reacción con diclorometano (100 m lx 2), se secó la fase orgánica sobre sulfato de sodio anhidro, se filtró con succión y se concentró el filtrado a presión reducida para obtener ácido 2-tetrahidropiran-2-iloxiacético (42B) como una sustancia aceitosa de color amarillo claro (510 mg, rendimiento del 30%).
EM m/z = 183,1 [M+Na]+;
Etapa 2: 2-(2-tetrahidropiran-2-iloxiacetil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (42C)
o
ctefr]x> ^ oTHp
42C
Se añadieron ácido 2-tetrahidropiran-2-iloxiacético (42B) (510 mg, 3,2 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (1,2 g, 6,3 mmol), 1-hidroxibenzotriazol (516 mg, 3,82 mmol), 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (830 mg, 3,2 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 2-(2-tetrahidropiran-2-iloxiacetil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (42C) como un sólido de color amarillo claro (580 mg, rendimiento del 45%).
Etapa 3:
1-(2,7-Diazaespiro[3.5]nonan-2-il)-2-tetrahidropiran-2-iloxi-etanona (42D)

Se añadieron 2-(2-tetrahidropiran-2-iloxiacetil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (42C) (580 mg, 1,4 mmol), paladio sobre carbono (120 mg, 20% en peso de l) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas
y se concentró el filtrado a presión reducida para obtener 1-(2,7-diazaespiro[3.5]nonan-2-il)-2-tetrahidropiran-2-iloxietanona (42D) en bruto como un sólido de color amarillo claro (387 mg, rendimiento del 100%) y se usó directamente en la siguiente reacción.
Etapa 4:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(2-tetrahidropiran-2-iloxiacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (42E)

Se añadieron 1-(2,7-diazaespiro[3.5]nonan-2-il)-2-tetrahidropiran-2-iloxi-etanona (42D) en bruto (387 mg, 1,4 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (553 mg, 2,88 mmol), 1-hidroxibenzotriazol (233 mg, 1,72 mmol), producto intermedio 1 (1,09 g, 1,45 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(tercbutoxicarbonilamino)-1-[2-(2-tetrahidropiran-2-iloxiacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (42E) como un sólido de color amarillo claro (590 mg, rendimiento del 41%).
Etapa 5:
(2R)-N-[(1R)-5-Amino-1-[2-(2-hidroxietil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 42)

Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(2-tetrahidropiran-2-iloxiacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (42E) (590 mg, 0,59 mmol) y ácido trifluoroacético (4 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-N-[(1R)-5-amino-1-[2-(2-hidroxietil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 42) como un polvo de color blanco (286 mg, rendimiento del 67%).
EM m/z = 720,3 [M+H]+
1H-RMN (400 MHz, D2O) 87,44-7,29 (m, 6H), 7,24 (d, 4H), 4,66 (t, 1H), 4,34-4,20 (m, 2H), 4,14 (d, 2H), 4,02 (d, 2H), 3,83 (d, 2H), 3,72-3,60 (m, 2H), 3,56-3,42 (m, 1H), 3,42-3,30 (m, 1H), 3,18 (d, 2H), 3,10-2,94 (m, 4H), 1,96-1,29 (m, 14H), 0,92 (dd, 6H).
Ejemplo 41:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-propanoil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 43)

2-Propanoil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (43A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (390 mg, 1,5mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (575 mg, 3,0mmol), 1-hidroxibenzotriazol (243 mg, 1,80 mmol), ácido propanoico (122 mg, 1,65 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 2-propanoil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (43a ) como un sólido de color amarillo claro (245 mg, rendimiento del 52%).
Etapa 2:
1-(2,7-Diazaespiro[3.5]nonan-2-il)propan-1-ona (43B)

Se añadieron 2-propanoil-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (43A) (245 mg, 0,775 mmol), paladio sobre carbono (49 mg, al 20% en peso de l) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno. Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 1-(2,7-diazaespiro[3.5]nonan-2-il)propan-1-ona (43B) en bruto como un sólido de color amarillo claro (141 mg, rendimiento del 99,8%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-propanoil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (43C)

Se añadieron 1-(2,7-diazaespiro[3.5]nonan-2-il)propan-1-ona (43B) en bruto (141 mg, 0,774 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (383 mg, 2,0 mmol), 1-hidroxibenzotriazol (135 mg, 1,0 mmol), producto intermedio 1 (0,565 g, 0,75 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-propanoil-2,7-diazaespiro)[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (43C) como un sólido de color amarillo claro (500 mg, rendimiento del 72,7%).
Etapa 4:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-(2-propanoil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 43)
Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-(2-propanoil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (43C) (260 mg, 0,28 mmol) y ácido trifluoroacético (1,3 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-N-[(1R)-5-amino-1-(2-propanoil-2,7-diazaespiro[3.5]nonano-7-carbonil)pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 43) como un polvo de color blanco (125 mg, rendimiento del 51%).
EM m/z = 359,8 [M+2H]+/2;
1H-RMN (400 MHz, D2O) 87,45-7,28 (m, 6H), 7,24 (d, 4H), 4,80-4,75 (m, 1H) 4,66 (t, 1H), 4,29 (t, 1H), 4,27 (t, 1H), 4,01 (d, 2H), 3,77 (d, 2H), 3,71-3,60 (m, 2H), 3,55-3,42 (m, 1H), 3,41-3,27 (m, 1H), 3,18 (dd, 2H), 3,11-2,93 (m, 4H), 2,19 (qd, 2H), 1,95-1,63 (m, 8H), 1,61-1,49 (m, 3H), 1,48-1,30 (m, 2H), 1,07 (td, 3H), 0,92 (dd, 6H).
Ejemplo 42:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 44)

Etapa 1:
2-(2,2,2-Trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (44A)

Se añadieron 2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (6C) (390 mg, 1,5mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (575 mg, 3,0 mmol), 1-hidroxibenzotriazol (243 mg, 1,80 mmol), ácido trifluoroacético (171 mg, 1,5 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener 2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (44A) como un sólido de color amarillo claro (224 mg, rendimiento del 63%).
Etapa 2:
1-(2,7-Diazaespiro[3.5]nonan-2-il)-2,2,2-trifluoroetanona (44B)

Se añadieron 2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de bencilo (44A) (223 mg, 0,63 mmol), paladio sobre carbono (46 mg, al 20% en peso de l) y metanol (20 ml) en un matraz de una sola boca de 50 ml. Se reemplazó la atmósfera por hidrógeno 3 veces y se hizo reaccionar la mezcla a temperatura ambiente durante 3 h bajo una atmósfera de hidrógeno (globo). Se filtró la disolución de reacción a través de tierra de diatomeas y se concentró el filtrado a presión reducida para obtener 1-(2,7-diazaespiro[3.5]nonan-2-il)-2,2,2-trifluoroetanona (44B) en bruto como un sólido de color amarillo claro (116 mg, rendimiento del 83%) y se usó directamente en la siguiente reacción.
Etapa 3:
N-[(1R)-1-Bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxo-etil]carbamato de terc-butilo (44C)

Se añadieron 1-(2,7-diazaespiro[3.5]nonan-2-il)-2,2,2-trifluoroetanona (44B) en bruto (116 mg, 0,52 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (575 mg, 3,7 mmol), 1-hidroxibenzotriazol (240 mg, 1,3 mmol), producto intermedio 1 (0,47 g, 0,626 mmol) y diclorometano (50 ml) en un matraz de una sola boca de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 5 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol (v:v) = 50:1) para obtener N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(2,2,2-trifluoroacetil))-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (44c ) como un sólido de color amarillo claro (216 mg, rendimiento del 42%).
Etapa 4:
(2R)-2-[[(2R)-2-[[(2R)-2-Amino-3-fenil-propanoil]amino]-3-fenil-propanoil]amino]-N-[(1R)-5-amino-1-[2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-4-metil-pentanamida; di-ácido trifluoroacético (compuesto 44)
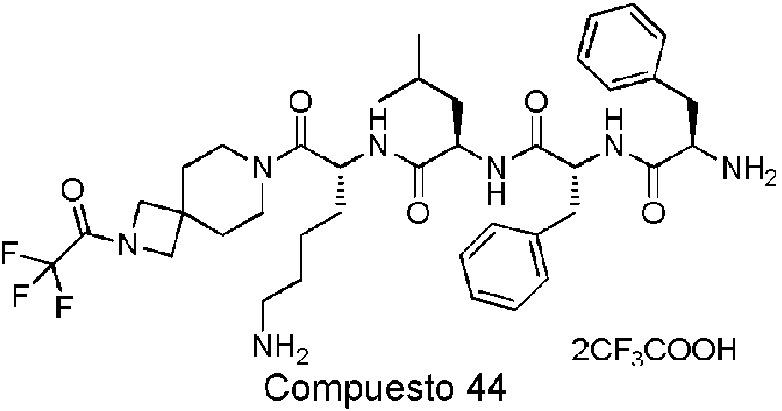
Se añadieron N-[(1R)-1-bencil-2-[[(1R)-1-bencil-2-[[(1R)-1-[[(1R)-5-(terc-butoxicarbonilamino)-1-[2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]carbamoil]-3-metil-butil]amino]-2-oxo-etil]amino]-2-oxoetil]carbamato de terc-butilo (44C) (210 mg, 0,22 mmol) y ácido trifluoroacético (2 ml) en un matraz de reacción de 50 ml y se permitió que reaccionase el sistema a temperatura ambiente durante 2 h. Se concentró la disolución de reacción a presión reducida y se separó el residuo y se purificó mediante cromatografía de líquidos preparativa (condiciones de preparación: instrumento: GX-281 de Gilson; columna: Xbridge C18, 150 x 30 mm de D.I., 5 |am; fase móvil: A para ACN y B para H2O; isocrático: 65% de A; velocidad de flujo: 30 ml/min; contrapresión:1000 psi; temperatura de columna: 30°C; longitud de onda: 210 nm; periodo: 18 min; preparación de la muestra: se disolvió el compuesto en 12 ml de metanol; inyección: 0,9 ml/aguja). Se concentró la preparación a presión reducida para eliminar la mayor parte del disolvente y se liofilizó para obtener (2R)-2-[[(2R)-2-[[(2R)-2-amino-3-fenil-propanoil]amino]-3-fenilpropanoil]amino]-N-[(1R)-5-amino-1-[2-(2,2,2-trifluoroacetil)-2,7-diazaespiro[3.5]nonano-7-carbonil]pentil]-4-metilpentanamida; ácido di-trifluoroacético (compuesto 44) como un polvo de color blanco (111 mg, rendimiento del 51%).
EM m/z = 758,3 [M+H]+;
1H-RMN (400 MHz, D2O) 87,44-7,28 (m, 6 H), 7,24 (d, 4H), 4,64 (t, 1H), 4,38-4,21 (m, 4H), 3,97 (d, 2H), 3,73-3,58 (m, 2H), 3,56-3,43 (m, 1H), 3,40-3,32 (m, 1H), 3,25-3,11 (m, 2H), 3,11-2,93 (m, 4H), 1,99-1,63 (m, 9H), 1,53 (d, 3H), 1,49 1,33 (m, 2H), 0,93 (dd, 6 H).
Ejemplos de pruebas biológicas
Prueba 1: Actividad agonista sobre los receptores de opioides k humanos
La forskolina puede estimular la liberación de AMPc desde una línea celular que sobreexpresa el receptor de opioides k humano, las células OPRK1 (DiscoveRx) y los agonistas de receptores de opioides k pueden inhibir la liberación de AMPc estimulada por la forskolina. Detectando el efecto inhibidor del compuesto de prueba sobre la liberación de AMPc estimulada por forskolina, puede determinarse la actividad agonista del compuesto sobre el receptor de opioides k humano. En primer lugar, se incubaron una determinada concentración de forskolina y diferentes concentraciones del compuesto de prueba con líneas celulares que sobreexpresan el receptor de opioides k humano. Se usó un inmunoensayo de AMPc (LANCE®, PerkinElmer) basado en la transferencia de energía de resonancia de
fluorescencia con resolución temporal (TR-FRET) para determinar los niveles de AMPc en las células OPRK1 estimuladas. El método específico es el siguiente:
Se cultivaron células OPRK1 (DiscoveRx) que expresan altamente receptores de opioides k humanos en medio 5A de McCoy (16600-082 de Gibco) que contenía FBS al 10% (10099-141 de Gibco). El día del experimento, se lavaron las células en la fase de crecimiento exponencial y se separaron con PBS/EDTA 5 mM, se recogieron mediante centrifugación, se resuspendieron con tampón de estimulación y se contaron. Se ajustó la concentración de las células a 3*105 células/ml. Se usó DMSO para disolver la forskolina y el compuesto de prueba respectivamente, de modo que la concentración de las aguas madres fue de 10 mM, y luego se diluyó la forskolina a 4 |iM con tampón de estimulación y diferentes concentraciones del compuesto de prueba (las concentraciones fueron de 80, 16, 3,2, 0,64, 0,128, 0,0256, 0,00512, 0,001024, 0 |iM), se añadieron 5 |il por pocillo a una placa de 384 pocillos. Se añadieron 5 |il de suspensión celular a cada pocillo y se incubaron a temperatura ambiente durante 30 min. Posteriormente, se añadieron a cada pocillo 5 |il de disolución de trabajo de trazador de 4 x Eu-AMPc (dilución de 50 veces de la disolución madre de Eu-AMPc con tampón de detección de AMPc) y 5 |il de disolución de trabajo 4 x Ulight-anti-AMPc (dilución de 150 veces de disolución madre de ULight-anti-AMPc con tampón de detección de AMPc) y se incubaron a temperatura ambiente durante 1 hora. Se sometieron a ensayo placas de 384 pocillos para determinar los niveles de AMPc usando un método de TR-FRET con lector de microplacas (Perkin Elmer, Envision). Se procesaron los datos obtenidos y se ajustaron a CE50 usando el software Origin 7.5. Se midió la actividad agonista de receptores de opioides k humanos del compuesto de la presente invención mediante los experimentos anteriores, y los valores de CE50 medidos se muestran en la tabla 1.
Método de preparación del tampón de estimulación: se disolvieron 14 ml de 1*HBSS(invitrogen, n.° de cat. 14025 092), 75 |il de HEPES 1 M (Invitrogen, n.° de cat. 15630-080), 30 |il de IBMX 250 mM en DMSO (Sigma, n.° de cat.
17018) y se mezcló con 200 |il de estabilizador de BSA al 7,5%. Se ajustó el pH de la disolución a 7,4 con NaOH 0,1 N y se enrasó hasta 15 ml con 1 * HBSS.
Tabla 1 Actividad agonista de los compuestos de prueba sobre los receptores de opioides k humanos

Conclusión: Los compuestos de la invención tienen efectos agonistas significativos sobre los receptores de opioides k humanos.
Prueba 2: Experimento de placa calefactora con ratón
Se adquirieron ratones hembra C57 de 18-22 g de Chengdu Dashuo Experimental Animal Co., Ltd. Se fijó la temperatura del instrumento de placa calefactora a 56°C y, después de alcanzar los 56°C, se estabilizó la temperatura durante 30 minutos antes del experimento. Se sometieron los animales a una prueba de placa calefactora para observar la reacción de lamer las patas traseras, que se usó como indicador de la respuesta al dolor. Se registró el tiempo desde la entrada del animal a la placa calefactora hasta el lamido inducido por calor de las patas traseras. Los animales que cumplen con los criterios de inclusión (el tiempo de respuesta para lamerse la pata trasera es menor de 25 segundos) se incluyen en el número de grupo. Se agruparon los animales por umbral de nivel inicial, con 10 en cada grupo. Se administraron compuestos de diferentes concentraciones por vía subcutánea a 10 ml/kg, y se llevó a cabo una detección después de 15 minutos desde la administración, con 30 segundos como tiempo de corte, y se registró el tiempo de reacción. Se analizaron los resultados estadísticamente y se calculó el valor de % de MPE según la fórmula: % de MPE=(Tn-T0)/(30-T0), (Tn es el tiempo para que el animal se lama la pata trasera después de la administración, Tües el tiempo para que el animal se lama la pata antes de la administración). Se muestran los resultados experimentales en la tabla 2.
T a b la 2

C o n c lu s ión : El e fec to a n a lg é s ico de los c o m p u e s to s de la p re se n te inve n c ió n se logra a tra v é s de los re ce p to re s de o p io id e s k pe rifé ricos.
P ru e b a 3: E xp e rim e n to de co n to rs io n e s del ra tón
La in ye cc ión in tra p e rito n e a l de ác ido a cé tico en ra to n e s p uede p ro v o c a r co n to rs io n e s en ra tones. La re sp u e s ta de c o n to rs io n e s se re fie re a ra to n e s que p re se n ta n re s p u e s ta s de c o m p o rta m ie n to t íp ic a s que son c a ra c te r ís tic a s de la co n tra c c ió n o e x te n s ió n de los m ú scu lo s a b d o m in a le s . La a c tiv ida d a n a lg é s ica de l c o m p u e s to p uede re fle ja rse d e te c ta n d o el e fe c to in h ib id o r de l c o m p u e s to so b re el c o m p o rta m ie n to de co n to rs io n e s de los ra to n e s p ro vo ca d o po r el ác ido a cé tico . El m é to d o e s p e c ífic o es el s igu ien te :
R a to n e s IC R de 8 s e m a n a s de edad (a d q u ir id o s de C h e n g du D a sh u o B io te c h n o lo g y C o m pa n y, n úm ero de licencia : S C X K (S ich u a n ) 2008 -24 (N O : 51203500002150 )). Se d iv id ie ron los ra to n e s a le a to ria m e n te en g rupos de 10 an im a les, m itad m ach o s y m itad hem bras; en e s ta d o de a yu n o pero con libre a cce so a a g u a d u ra n te 12 h a n tes del exp e rim e nto . El d ía del e xp e rim e n to , se a d m in is tró p o r v ía in tra ve n o sa 1,0 m g/kg de l c o m p u e s to de e n sa yo y se a d m in is tró un rea c tivo de b la n co al g ru p o de con tro l. 15 m inu to s d e sp u é s de la a d m in is tra c ió n , se inyectó p o r v ía in tra p e rito n e a l una d iso luc ió n de á c id o a cé tico al 0 ,6 % (v /v) a una d o s is de 0 ,4 m l/ra tón . S e reg is tró el n ú m e ro de c o n to rs io n e s de l ra tón a los 15 m in y 6 h d e s p u é s de la inye cc ión de á c id o a cé tico , re s p e c tiva m e n te , y se c a lcu ló el p o rce n ta je de inh ib ic ión de las co n to rs io n e s p ro v o c a d a s p o r el á c id o acé tico en ra to n e s p o r el c o m p u e s to , re s p e c tiv a m e n te . S e m ue stra n los re s u lta d o s del aná lis is en la ta b la 3.
% de p o rce n ta je de inh ib ic ión = (n ú m e ro de co n to rs io n e s en el g ru p o de co n tro l - n ú m e ro de co n to rs io n e s en el g rupo de a d m in is tra c ió n )/n ú m e ro de co n to rs io n e s en el g ru p o de contro l.
T a b la 3 P o rce n ta je de inh ib ic ión de los co m p u e s to s de p ru e b a con resp e c to al c o m p o rta m ie n to de c o n to rs io n e s in d u c id a s p o r ác ido a cé tico de ra tones


NQ significa no sometido a prueba.

su base libre.
Conclusión: Los compuestos de la presente invención tienen efectos analgésicos significativos. Se sometieron a prueba adicionalmente algunos compuestos de prueba para una prueba de acción prolongada sobre el comportamiento de contorsiones inducidas por ácido acético de ratones. Según el mismo método de prueba tal como se describió anteriormente, el método de administración fue la inyección intravenosa y la dosis fue de 3 mg/kg o 10 mg/kg. Se registró el número de veces que el ratón presentaba contorsiones en el plazo de las 18 h posteriores a la inyección de ácido acético, y se calculó el porcentaje de inhibición del comportamiento de contorsiones inducidas por ácido acético de los ratones por los compuestos de prueba, respectivamente. Se muestran los resultados en la tabla 4.
Tabla 4 Porcentaje de inhibición tras 18 horas de los compuestos de prueba con respecto al comportamiento de contorsiones inducidas por ácido acético de ratones

La estructura 
Conclusión: algunos compuestos de la presente invención tienen efectos analgésicos significativos y tienen la ventaja de efectos duraderos.
4. Prueba PK en ratas


Tabla 5 Resultados PK de rata (1 mg/kg)

5. Prueba PK de ratón

Tabla 6 Resultados PK de ratón (1 mg/kg)

Claims (1)
- REIVINDICACIONESCompuesto de fórmula general (I) o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo:
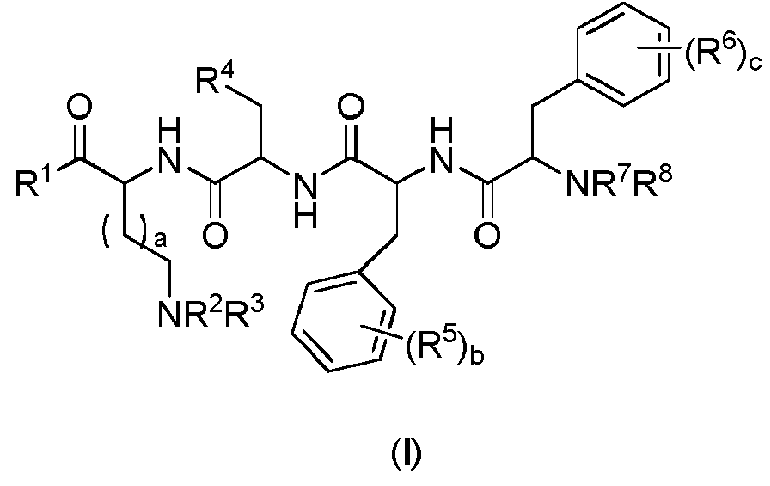 en la queR1 se selecciona
en la queR1 se selecciona cada uno de mi, m2 se selecciona independientemente de 1,2, 3 ó 4;cada uno de m3, m4 se selecciona independientemente de 0, 1,2, 3 ó 4; con la condición de que m3 y m4 no pueden ser 0 al mismo tiempo;cada uno de n1, n2 se selecciona independientemente de 0, 1,2, 3 ó 4;Z se selecciona de CRz1Rz2 o NRz3;cada uno de Rz1, Rz2 se selecciona independientemente de H, F, Cl, Br, I, OH, CF3, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, -C(=O)-alquilo C1-6, -(CH2)q-C(=O)O-alquilo C1-6, -(CH2)q-NR1eR1f, -(CH2V COOH, -(CH2)q-CONH2, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, alcoxilo, alquenilo, alquinilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de S, S=O o S(=O)2;cada uno de R1e, R1fse selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)O-(CH2)q-grupo carbocíclico C3-8 o -C(=O)O-(CH2)q-grupo heterocíclico de 3 a 8 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;alternativamente, Rz1 y Rz2 forman un anillo heterocíclico que contiene nitrógeno de 3 a 10 miembros con el átomo de carbono al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, =O, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros; cada uno de R1a, R1b se selecciona independientemente de F, CF3, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, alquenilo, alquinilo o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-s o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;Rz3 se selecciona independientemente de H, -C(=O)-alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)-grupo carbocíclico C3-8, -C(=O)O-grupo carbocíclico C3-8, -C(=O)O-(grupo heterocíclico de 3 a 8 miembros), -S(=O)palquilo C1.6, -S(=O)p-grupo carbocíclico C3-8, -S(=O)p-(grupo heterocíclico de 3 a 8 miembros), -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-6; alternativamente, R1g, R1h forman un anillo heterocíclico de 3 a 10 miembros con el átomo de nitrógeno al que están unidos, el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6 o -S(=O)p-alquilo C1-6, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; q se selecciona de 0, 1, 2, 3 ó 4;p se selecciona de 0, 1 ó 2;a se selecciona de 0, 1, 2 ó 3;R4 se selecciona independientemente de H, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o -(CH2)q-grupo carbocíclico C3-8, el grupo alquilo, alquenilo, alquinilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CN, CF3, NO2, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3.8 o grupo heterocíclico de 3 a 8 miembros, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1.4, -C =0 0-(CH2)q-grupo carbocíclico C3-8, -C(=O)0-(CH2)q-grupo heterocíclico de 3 a 8 miembros oy el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;
cada uno de mi, m2 se selecciona independientemente de 1,2, 3 ó 4;cada uno de m3, m4 se selecciona independientemente de 0, 1,2, 3 ó 4; con la condición de que m3 y m4 no pueden ser 0 al mismo tiempo;cada uno de n1, n2 se selecciona independientemente de 0, 1,2, 3 ó 4;Z se selecciona de CRz1Rz2 o NRz3;cada uno de Rz1, Rz2 se selecciona independientemente de H, F, Cl, Br, I, OH, CF3, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, -C(=O)-alquilo C1-6, -(CH2)q-C(=O)O-alquilo C1-6, -(CH2)q-NR1eR1f, -(CH2V COOH, -(CH2)q-CONH2, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, alcoxilo, alquenilo, alquinilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de S, S=O o S(=O)2;cada uno de R1e, R1fse selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)O-(CH2)q-grupo carbocíclico C3-8 o -C(=O)O-(CH2)q-grupo heterocíclico de 3 a 8 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;alternativamente, Rz1 y Rz2 forman un anillo heterocíclico que contiene nitrógeno de 3 a 10 miembros con el átomo de carbono al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, =O, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros; cada uno de R1a, R1b se selecciona independientemente de F, CF3, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, alquenilo, alquinilo o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-s o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;Rz3 se selecciona independientemente de H, -C(=O)-alquilo C1-6, -C(=O)O-alquilo C1-6, -C(=O)-grupo carbocíclico C3-8, -C(=O)O-grupo carbocíclico C3-8, -C(=O)O-(grupo heterocíclico de 3 a 8 miembros), -S(=O)palquilo C1.6, -S(=O)p-grupo carbocíclico C3-8, -S(=O)p-(grupo heterocíclico de 3 a 8 miembros), -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 8 miembros, y el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S;cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-6; alternativamente, R1g, R1h forman un anillo heterocíclico de 3 a 10 miembros con el átomo de nitrógeno al que están unidos, el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6 o -S(=O)p-alquilo C1-6, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; q se selecciona de 0, 1, 2, 3 ó 4;p se selecciona de 0, 1 ó 2;a se selecciona de 0, 1, 2 ó 3;R4 se selecciona independientemente de H, alquilo C1-6, alquenilo C2-6, alquinilo C2-6 o -(CH2)q-grupo carbocíclico C3-8, el grupo alquilo, alquenilo, alquinilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CN, CF3, NO2, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3.8 o grupo heterocíclico de 3 a 8 miembros, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S; cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-6, -C(=O)O-alquilo C1.4, -C =0 0-(CH2)q-grupo carbocíclico C3-8, -C(=O)0-(CH2)q-grupo heterocíclico de 3 a 8 miembros oy el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, alquilo C1-6, alcoxilo C1-6, alquenilo C2-6, alquinilo C2-6, grupo carbocíclico C3-8 o grupo heterocíclico de 3 a 8 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S; b se selecciona de 0, 1, 2, 3, 4 ó 5;c se selecciona de 0, 1, 2, 3, 4 ó 5;cada uno de R5, R6 se selecciona independientemente de F, Cl, Br, I, CF3, ciano, nitro, alquilo C1-4, -OR5a, -C(O)OR5b, -SR5c, -S(O)R5d, -S(O)2R5e o -NR5fR5g;cada uno de R5a, R5b, R5c, R5d, R5e, R5f y R5g se selecciona independientemente de H o alquilo C1-4; alternativamente, R5f, R5g forman un anillo heterocíclico de 5 a 6 miembros con el átomo de nitrógeno al que están unidos, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S.Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 1, en el quecada uno de it h , m2, m3, mu se selecciona independientemente de 1 ó 2;cada uno de m, n2 se selecciona independientemente de 0, 1 ó 2;Z se selecciona de CRz1Rz2 o NRz3,cada uno de Rz1, Rz2 se selecciona independientemente de H, alquilo C1-4, -(CH2)q-C(=O)O-alquilo C1.4, -(CH2)q-NR1eR1f, -(CH2)q-COOH, -(CH2)q-CONH2, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros, y el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de 5, S=O o S(=O)2;cada uno de R1e, R1f se selecciona independientemente de H, alquilo C i-4, -C(=O)O-alquilo C i-4 o -C(=O)O-(CH2)q-grupo carbocíclico C3-6, el grupo alquilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, metilo, etilo, metoxilo, etoxilo, fenilo;alternativamente, Rz1 y Rz2 son capaces de formar un anillo heterocíclico que contiene nitrógeno de 4 a 6 miembros con un átomo de carbono al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) de=O;R1a, R1b se seleccionan independientemente de F, CF3, metilo, etilo, propanoílo o isopropilo;Rz3 se selecciona cada uno independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -S(=O)p-alquilo C1-4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, metilo, etilo, metoxilo, etoxilo, ciclopropilo o fenilo, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-4;alternativamente, R1g, R1h forman un anillo heterocíclico de 4 a 6 miembros con el átomo de nitrógeno al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, metilo, etilo, metoxilo, etoxilo o -S(=O)p-alquilo C1-4, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;p se selecciona de 2 ;q se selecciona de 0 ó 1 ;a se selecciona de 3;R4 se selecciona de propanoílo o isopropilo;cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-4, -C(=O)O-alquilo C1-4 o -C(=O)O-bencilo;b se selecciona de 0 ;c se selecciona de 0.Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 2, en el que el compuesto se selecciona del compuesto de fórmula general (II):
b se selecciona de 0, 1, 2, 3, 4 ó 5;c se selecciona de 0, 1, 2, 3, 4 ó 5;cada uno de R5, R6 se selecciona independientemente de F, Cl, Br, I, CF3, ciano, nitro, alquilo C1-4, -OR5a, -C(O)OR5b, -SR5c, -S(O)R5d, -S(O)2R5e o -NR5fR5g;cada uno de R5a, R5b, R5c, R5d, R5e, R5f y R5g se selecciona independientemente de H o alquilo C1-4; alternativamente, R5f, R5g forman un anillo heterocíclico de 5 a 6 miembros con el átomo de nitrógeno al que están unidos, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S.Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 1, en el quecada uno de it h , m2, m3, mu se selecciona independientemente de 1 ó 2;cada uno de m, n2 se selecciona independientemente de 0, 1 ó 2;Z se selecciona de CRz1Rz2 o NRz3,cada uno de Rz1, Rz2 se selecciona independientemente de H, alquilo C1-4, -(CH2)q-C(=O)O-alquilo C1.4, -(CH2)q-NR1eR1f, -(CH2)q-COOH, -(CH2)q-CONH2, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros, y el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 5 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, =O, carboxilo, nitro, ciano, amino, alquilo C1-4, alcoxilo C1-4, alquenilo C2-4, alquinilo C2-4, grupo carbocíclico C3-6 o un grupo heterocíclico de 3 a 6 miembros, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) opcionalmente seleccionado(s) de N, O o S, y cuando el heteroátomo se selecciona de S, está opcionalmente en forma de 5, S=O o S(=O)2;cada uno de R1e, R1f se selecciona independientemente de H, alquilo C i-4, -C(=O)O-alquilo C i-4 o -C(=O)O-(CH2)q-grupo carbocíclico C3-6, el grupo alquilo o carbocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, metilo, etilo, metoxilo, etoxilo, fenilo;alternativamente, Rz1 y Rz2 son capaces de formar un anillo heterocíclico que contiene nitrógeno de 4 a 6 miembros con un átomo de carbono al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) de=O;R1a, R1b se seleccionan independientemente de F, CF3, metilo, etilo, propanoílo o isopropilo;Rz3 se selecciona cada uno independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -S(=O)p-alquilo C1-4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, metilo, etilo, metoxilo, etoxilo, ciclopropilo o fenilo, y el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-4;alternativamente, R1g, R1h forman un anillo heterocíclico de 4 a 6 miembros con el átomo de nitrógeno al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, ciano, nitro, metilo, etilo, metoxilo, etoxilo o -S(=O)p-alquilo C1-4, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;p se selecciona de 2 ;q se selecciona de 0 ó 1 ;a se selecciona de 3;R4 se selecciona de propanoílo o isopropilo;cada uno de R2, R3, R7, R8 se selecciona independientemente de H, alquilo C1-4, -C(=O)O-alquilo C1-4 o -C(=O)O-bencilo;b se selecciona de 0 ;c se selecciona de 0.Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 2, en el que el compuesto se selecciona del compuesto de fórmula general (II): R1 se selecciona
R1 se selecciona cada uno de mi, m2, m3, m4 se selecciona independientemente de 1 ó 2;cada uno de n1, n2 se selecciona independientemente de 0 ó 2;R1a, R1b se seleccionan independientemente de F;Z se selecciona de CRz1Rz2 o NRz3;
cada uno de mi, m2, m3, m4 se selecciona independientemente de 1 ó 2;cada uno de n1, n2 se selecciona independientemente de 0 ó 2;R1a, R1b se seleccionan independientemente de F;Z se selecciona de CRz1Rz2 o NRz3; cada uno de Rz1, Rz2 se selecciona inde endientemente de H, carboxilo,
cada uno de Rz1, Rz2 se selecciona inde endientemente de H, carboxilo, alternativamente, Rz1 y Rz2 son capaces de formar una lactama con el átomo de carbono al que están unidos
alternativamente, Rz1 y Rz2 son capaces de formar una lactama con el átomo de carbono al que están unidos Rz3 se selecciona cada uno independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -S(=O)p-alquilo C1-4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, metilo, etilo, metoxilo, etoxilo, ciclopropilo o fenilo, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-4;alternativamente, R1g, R1h forman un anillo heterocíclico de 4 a 6 miembros con el átomo de nitrógeno al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, CF3, metilo, metoxilo o -S(=O)p-alquilo C1-4, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;p se selecciona de 2;cada uno de R2, R3, R7, R8 se selecciona independientemente de H, metilo o -C(=O)O-terc-butilo.Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 3, en el que el compuesto se selecciona del compuesto de fórmula general (II):
Rz3 se selecciona cada uno independientemente de H, -C(=O)-alquilo C1-4, -C(=O)-grupo carbocíclico C3-6, -C(=O)O-alquilo C1-4, -S(=O)p-alquilo C1-4, -S(=O)p-grupo carbocíclico C3-6, -C(=O)NR1gR1h, -S(=O)p-NR1iR1j o un grupo heterocíclico de 3 a 6 miembros, el grupo alquilo, carbocíclico o heterocíclico está opcionalmente sustituido adicionalmente con de 0 a 3 sustituyente(s) seleccionado(s) del grupo que consiste en F, Cl, Br, I, OH, CF3, nitro, ciano, amino, metilo, etilo, metoxilo, etoxilo, ciclopropilo o fenilo, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;cada uno de R1g, R1h, R1i, R1j se selecciona independientemente de H o alquilo C1-4;alternativamente, R1g, R1h forman un anillo heterocíclico de 4 a 6 miembros con el átomo de nitrógeno al que están unidos, y el anillo está opcionalmente sustituido adicionalmente con sustituyente(s) seleccionado(s) del grupo que consiste en F, CF3, metilo, metoxilo o -S(=O)p-alquilo C1-4, el grupo heterocíclico contiene de 1 a 3 heteroátomo(s) seleccionado(s) de N, O o S;p se selecciona de 2;cada uno de R2, R3, R7, R8 se selecciona independientemente de H, metilo o -C(=O)O-terc-butilo.Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 3, en el que el compuesto se selecciona del compuesto de fórmula general (II): Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 4,
Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 4, 6. Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 5, en el que el compuesto se selecciona de las siguientes estructuras:
6. Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según la reivindicación 5, en el que el compuesto se selecciona de las siguientes estructuras: Compuesto 13 C om puestos
Compuesto 13 C om puestos Compuesto 18 Com uesto 17
Compuesto 18 Com uesto 17 C om puestos Compuesto 20
C om puestos Compuesto 20 Compuesto 23 Compuesto 24
Compuesto 23 Compuesto 24 Com uesto 258
Com uesto 258 Com uesto 27
Com uesto 27 Compuesto 29 Compuesto 30
Compuesto 29 Compuesto 30 Compuesto 31
Compuesto 31 Compuesto 36 Compuesto: 35
Compuesto 36 Compuesto: 35 Compuesto 39Com uesto 40
Compuesto 39Com uesto 40 Compuesto 42
Compuesto 42 7. Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según una cualquiera de las reivindicaciones 1-6, en el que las sales farmacéuticamente aceptables se seleccionan de trifluoroacetatos.8. Composición farmacéutica que comprende el compuesto o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según una cualquiera de las reivindicaciones 1 a 7, y uno o más portadores y/o excipientes farmacéuticamente aceptables.9. Uso del compuesto o de un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según una cualquiera de las reivindicaciones 1 a 7, o de la composición farmacéutica según la reivindicación 8, en la fabricación de un medicamento para tratar o prevenir una enfermedad o afección asociada al receptor de opioides k en un mamífero.10. Uso según la reivindicación 9, en el que la enfermedad o afección asociada al receptor de opioides k se selecciona del grupo que consiste en dolor, inflamación, prurito, edema, hiponatremia, hipopotasemia, íleo, tos y glaucoma.11. Uso según la reivindicación 9, en el que el dolor se selecciona del grupo que consiste en dolor neuropático, dolor somático, dolor visceral y dermatalgia.12. Uso según la reivindicación 9, en el que el dolor se selecciona del grupo que consiste en dolor por artritis, dolor por cálculos renales, histeroespasmo, dismenorrea, endometriosis, dispepsia, dolor posquirúrgico, dolor tras tratamiento médico, dolor ocular, dolor por otitis, dolor oncológico fulminante y dolor asociado a trastornos GI.
7. Compuesto o estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según una cualquiera de las reivindicaciones 1-6, en el que las sales farmacéuticamente aceptables se seleccionan de trifluoroacetatos.8. Composición farmacéutica que comprende el compuesto o un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según una cualquiera de las reivindicaciones 1 a 7, y uno o más portadores y/o excipientes farmacéuticamente aceptables.9. Uso del compuesto o de un estereoisómero, hidrato, solvato, sal farmacéuticamente aceptable o cocristal del mismo según una cualquiera de las reivindicaciones 1 a 7, o de la composición farmacéutica según la reivindicación 8, en la fabricación de un medicamento para tratar o prevenir una enfermedad o afección asociada al receptor de opioides k en un mamífero.10. Uso según la reivindicación 9, en el que la enfermedad o afección asociada al receptor de opioides k se selecciona del grupo que consiste en dolor, inflamación, prurito, edema, hiponatremia, hipopotasemia, íleo, tos y glaucoma.11. Uso según la reivindicación 9, en el que el dolor se selecciona del grupo que consiste en dolor neuropático, dolor somático, dolor visceral y dermatalgia.12. Uso según la reivindicación 9, en el que el dolor se selecciona del grupo que consiste en dolor por artritis, dolor por cálculos renales, histeroespasmo, dismenorrea, endometriosis, dispepsia, dolor posquirúrgico, dolor tras tratamiento médico, dolor ocular, dolor por otitis, dolor oncológico fulminante y dolor asociado a trastornos GI.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710598408 | 2017-07-21 | ||
| CN201810014939 | 2018-01-11 | ||
| PCT/CN2018/096271 WO2019015644A1 (zh) | 2017-07-21 | 2018-07-19 | 肽酰胺类化合物及其制备方法和在医药上的用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2905164T3 true ES2905164T3 (es) | 2022-04-07 |
Family
ID=65015483
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18835454T Active ES2905164T3 (es) | 2017-07-21 | 2018-07-19 | Compuesto de amida peptídico y método de preparación y uso médico del mismo |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US11014964B2 (es) |
| EP (1) | EP3656782B1 (es) |
| JP (1) | JP7008787B2 (es) |
| KR (1) | KR102375543B1 (es) |
| CN (1) | CN110637025B (es) |
| AU (1) | AU2018303080B2 (es) |
| BR (1) | BR112020001157A2 (es) |
| CA (1) | CA3069820C (es) |
| ES (1) | ES2905164T3 (es) |
| MY (1) | MY200925A (es) |
| PT (1) | PT3656782T (es) |
| TW (1) | TWI712615B (es) |
| WO (1) | WO2019015644A1 (es) |
| ZA (1) | ZA202000491B (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114127086B (zh) * | 2019-07-25 | 2023-10-20 | 四川海思科制药有限公司 | 氘代肽酰胺类化合物及其制备方法和在医药上的用途 |
| CN114127085B (zh) * | 2019-07-25 | 2024-08-13 | 西藏海思科制药有限公司 | 一种肽酰胺盐及其制备方法和在医药上的用途 |
| TWI764248B (zh) * | 2019-08-23 | 2022-05-11 | 大陸商四川海思科製藥有限公司 | 肽醯胺類組合物及其製備 |
| CN112442106A (zh) * | 2019-09-03 | 2021-03-05 | 四川海思科制药有限公司 | 一种多肽化合物的固体形式和在医药上的应用 |
| CN112552374B (zh) * | 2019-09-10 | 2024-06-04 | 西藏海思科制药有限公司 | 一种肽酰胺类化合物及其中间体的制备方法 |
| CN114341155B (zh) * | 2019-09-10 | 2025-03-11 | 海思科医药集团股份有限公司 | 一种肽酰胺类化合物及其中间体的制备方法 |
| EP4122483B1 (en) * | 2020-03-18 | 2025-04-30 | Xizang Haisco Pharmaceutical Co., Ltd. | Oral pharmaceutical composition |
| JP7549439B2 (ja) * | 2020-04-30 | 2024-09-11 | メッドシャイン ディスカバリー インコーポレイテッド | フェニルプロピオンアミド系化合物及びその使用 |
| CN117177744A (zh) | 2021-01-29 | 2023-12-05 | 塞迪拉治疗股份有限公司 | Cdk2抑制剂及其使用方法 |
| AU2022259250A1 (en) * | 2021-04-12 | 2023-11-16 | Xizang Haisco Pharmaceutical Co., Ltd. | Use of peptide amide compound in preparation of drug for treating pruritus |
| MX2023015436A (es) | 2021-06-26 | 2024-02-21 | Cedilla Therapeutics Inc | Inhibidores de cdk2 y metodos de uso de los mismos. |
| CN115707711A (zh) * | 2021-08-18 | 2023-02-21 | 天地恒一制药股份有限公司 | 肽酰胺类化合物、其药学上可接受的盐、立体异构体及其应用 |
| CN115873069A (zh) * | 2021-08-18 | 2023-03-31 | 天地恒一制药股份有限公司 | 肽酰胺类化合物、其药学上可接受的盐、立体异构体及其应用 |
| CN119836295A (zh) * | 2022-07-11 | 2025-04-15 | 人福医药美国公司 | 用于治疗医学病症的肽 |
| WO2025077727A1 (zh) * | 2023-10-10 | 2025-04-17 | 海思科医药集团股份有限公司 | 肽酰胺类化合物在制备预防恶心呕吐的药物中的用途 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU614716B2 (en) * | 1985-08-14 | 1991-09-12 | G.D. Searle & Co. | Phenyl substituted dipeptide amides |
| US4822775A (en) * | 1986-02-14 | 1989-04-18 | G. D. Searle & Co. | Phenyl substituted dipeptide amides |
| US4760180A (en) * | 1986-02-14 | 1988-07-26 | G. D. Searle & Co. | N-terminally substituted dipeptide amides |
| NZ520724A (en) * | 1995-02-27 | 2004-03-26 | Gilead Sciences Inc | Tetrahydro-pyridine compounds for inhibiting neuraminidase and treating influenza neuraminidase |
| US5965701A (en) * | 1997-12-23 | 1999-10-12 | Ferring Bv | Kappa receptor opioid peptides |
| FR2814367B1 (fr) * | 2000-09-25 | 2008-12-26 | Inst Nat Sante Rech Med | Ligands du recepteur npff pour le traitement de la douleur et des hyperalgies |
| CN101454338A (zh) * | 2006-05-26 | 2009-06-10 | 卡拉治疗学股份有限公司 | κ阿片样受体肽的N-氧化物 |
| US7842662B2 (en) | 2006-11-10 | 2010-11-30 | Cara Therapeutics, Inc. | Synthetic peptide amide dimers |
| US8236766B2 (en) * | 2006-11-10 | 2012-08-07 | Cara Therapeutics, Inc. | Uses of synthetic peptide amides |
| US8906859B2 (en) | 2006-11-10 | 2014-12-09 | Cera Therapeutics, Inc. | Uses of kappa opioid synthetic peptide amides |
| MX2009005000A (es) * | 2006-11-10 | 2009-10-12 | Cara Therapeutics Inc | Amidas de peptidos sinteticos. |
| US10550150B2 (en) | 2015-05-11 | 2020-02-04 | Cadila Healthcare Limited | Short-chain peptides as Kappa (κ) opioid receptors (KOR) agonist |
| CN106928127B (zh) * | 2015-12-31 | 2021-07-16 | 四川海思科制药有限公司 | 取代哌啶类酰胺衍生物及其制备方法和在药学上的应用 |
-
2018
- 2018-07-19 EP EP18835454.2A patent/EP3656782B1/en active Active
- 2018-07-19 WO PCT/CN2018/096271 patent/WO2019015644A1/zh not_active Ceased
- 2018-07-19 CN CN201880031309.3A patent/CN110637025B/zh active Active
- 2018-07-19 AU AU2018303080A patent/AU2018303080B2/en active Active
- 2018-07-19 ES ES18835454T patent/ES2905164T3/es active Active
- 2018-07-19 KR KR1020207002720A patent/KR102375543B1/ko active Active
- 2018-07-19 TW TW107124926A patent/TWI712615B/zh active
- 2018-07-19 US US16/632,228 patent/US11014964B2/en active Active
- 2018-07-19 BR BR112020001157-6A patent/BR112020001157A2/pt active Search and Examination
- 2018-07-19 PT PT188354542T patent/PT3656782T/pt unknown
- 2018-07-19 CA CA3069820A patent/CA3069820C/en active Active
- 2018-07-19 MY MYPI2020000323A patent/MY200925A/en unknown
- 2018-07-19 JP JP2020501793A patent/JP7008787B2/ja active Active
-
2020
- 2020-01-24 ZA ZA2020/00491A patent/ZA202000491B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA3069820C (en) | 2022-12-06 |
| BR112020001157A2 (pt) | 2020-07-21 |
| TW201920231A (zh) | 2019-06-01 |
| KR20200031114A (ko) | 2020-03-23 |
| CN110637025A (zh) | 2019-12-31 |
| KR102375543B1 (ko) | 2022-03-16 |
| EP3656782B1 (en) | 2021-12-22 |
| AU2018303080B2 (en) | 2020-12-24 |
| AU2018303080A1 (en) | 2020-01-30 |
| EP3656782A4 (en) | 2021-04-21 |
| ZA202000491B (en) | 2021-08-25 |
| PT3656782T (pt) | 2022-02-08 |
| EP3656782A1 (en) | 2020-05-27 |
| MY200925A (en) | 2024-01-24 |
| TWI712615B (zh) | 2020-12-11 |
| CN110637025B (zh) | 2023-07-18 |
| US20200172573A1 (en) | 2020-06-04 |
| CA3069820A1 (en) | 2019-01-24 |
| WO2019015644A1 (zh) | 2019-01-24 |
| US11014964B2 (en) | 2021-05-25 |
| JP7008787B2 (ja) | 2022-01-25 |
| JP2020526571A (ja) | 2020-08-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2905164T3 (es) | Compuesto de amida peptídico y método de preparación y uso médico del mismo | |
| US11052076B2 (en) | Spirocyclic indolines as IL-17 modulators | |
| AU2016215432B2 (en) | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors | |
| WO2023275301A1 (en) | Imidazotriazine derivatives as il-17 modulators | |
| US11643436B2 (en) | Polypeptide compound, pharmaceutical composition, preparation method and application thereof | |
| AU2015299149B2 (en) | Optionally fused heterocyclyl-substituted derivatives of pyrimidine useful for the treatment of inflammatory, metabolic, oncologic and autoimmune diseases | |
| ES2249237T3 (es) | Compuestos de bencimidazol como antagonistas del receptor orl1. | |
| AU2016348847A1 (en) | Oxime compounds as agonists of the muscarinic M1 and/or M4 receptor | |
| BR112020003946A2 (pt) | compostos de espirociclo e métodos para produzir e usar os mesmos | |
| ES2441221T3 (es) | Piperidina espiro pirrolidinona y piperidinonas sustituidas usadas como moduladores de H3 | |
| CA3065904A1 (en) | Pharmaceutical compounds | |
| WO2021170631A1 (en) | Difluorocyclohexyl derivatives as il-17 modulators | |
| CA3179686A1 (en) | Difluorocyclohexyl derivatives as il-17 modulators | |
| RS64836B1 (sr) | Terapijska jedinjenja | |
| TWI838736B (zh) | 做為腺苷A2a受體拮抗劑之化合物及包含該化合物之醫藥組合物 | |
| EP3807296A1 (en) | Peptidomimetic inhibitors of the peptidyl-prolyl cis/trans isomerase (pin1) | |
| US20250230129A1 (en) | Polyamide compounds, method for preparing same, and medical use thereof | |
| EA040570B1 (ru) | Пептидные амидные соединения, способ их получения и медицинского применения | |
| HK40011806B (en) | Peptide amide compounds, preparation method thereof and use in medicine | |
| HK40011806A (en) | Peptide amide compounds, preparation method thereof and use in medicine | |
| AU2013382944A1 (en) | 5-Amino-quinoline-8-carboxamide derivatives as 5-HT4 receptor agonists | |
| JP2025537627A (ja) | セレン含有複素環式化合物およびその使用 | |
| NZ709872B2 (en) | Spiro-lactam nmda receptor modulators and uses thereof | |
| HK1254323A1 (zh) | 作为irak4调节剂的双环稠合杂芳基或芳基化合物 |