ES2909399T3 - Proadrenomedulina como marcador de niveles anormales de plaquetas - Google Patents
Proadrenomedulina como marcador de niveles anormales de plaquetas Download PDFInfo
- Publication number
- ES2909399T3 ES2909399T3 ES18765659T ES18765659T ES2909399T3 ES 2909399 T3 ES2909399 T3 ES 2909399T3 ES 18765659 T ES18765659 T ES 18765659T ES 18765659 T ES18765659 T ES 18765659T ES 2909399 T3 ES2909399 T3 ES 2909399T3
- Authority
- ES
- Spain
- Prior art keywords
- proadm
- level
- patient
- sample
- fragment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/74—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving hormones or other non-cytokine intercellular protein regulatory factors such as growth factors, including receptors to hormones and growth factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/86—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving blood coagulating time or factors, or their receptors
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/22—Haematology
- G01N2800/222—Platelet disorders
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/50—Determining the risk of developing a disease
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/52—Predicting or monitoring the response to treatment, e.g. for selection of therapy based on assay results in personalised medicine; Prognosis
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Molecular Biology (AREA)
- Chemical & Material Sciences (AREA)
- Urology & Nephrology (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Food Science & Technology (AREA)
- Microbiology (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Analytical Chemistry (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Physics & Mathematics (AREA)
- Endocrinology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
Método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de los niveles anormales de plaquetas en un paciente, que comprende a. proporcionar una muestra de dicho paciente, b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, c. en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas en dicho paciente.
Description
DESCRIPCIÓN
Proadrenomedulina como marcador de niveles anormales de plaquetas
Descripción
La invención se refiere a un método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de pacientes con niveles anormales de plaquetas, que comprende proporcionar una muestra de dicho paciente, determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas en dicho paciente. En modalidades de la invención, un nivel de gravedad alto de proADM o fragmento(s) de la misma indica niveles bajos de plaquetas en el sujeto, y el posterior inicio o modificación de un tratamiento del paciente para mejorar dicha afección. En algunas modalidades, el método comprende determinar un nivel de uno o más marcadores adicionales en una muestra aislada del paciente, tal como el nivel de plaquetas, el nivel de PCT o fragmento(s) de la misma, uno o más marcadores de trombocitopenia y/o uno o más marcadores de una respuesta inflamatoria.
Antecedentes de la invención
Michels, M. y otros: "High plasma mid-regional pro-adrenomedullin levels in children with severe dengue virus infections", J. OF CLINICAL VIROLOGY, vol. 50, núm. 1, 1 enero de 2011, pages 8-12 describe la proADM como marcador en pacientes con dengue. Se menciona que la mediana del recuento de plaquetas en pacientes con síndrome de choque por Dengue es baja.
A pesar de las mejoras significativas en las medidas de diagnóstico y prevención, la incidencia de sepsis ha seguido aumentando rápidamente en pacientes hospitalizados (1), con tasas de mortalidad que oscilan entre el 10 % y el 54 %, en dependencia del nivel de gravedad de la enfermedad, la definición de disfunción orgánica usada e incidencia específica del país (2, 3). Por lo tanto, una evaluación temprana y precisa tanto de la carga infecciosa como de la gravedad de la enfermedad, en términos de la respuesta fisiopatológica general del huésped, es de crucial importancia en las primeras etapas de la sepsis para poder tomar decisiones rápidas y confiables con respecto a las pruebas diagnósticas y las estrategias de tratamiento, así como también en la fase posterior para orientar de manera confiable el manejo del paciente, seguimiento del tratamiento, decisiones de alta en presencia de recuperación clínica.
Por lo tanto, es sorprendente que actualmente no exista una prueba diagnóstica estándar de oro para la sepsis (4). El uso de procalcitonina (PCT) ha cubierto parcialmente esta necesidad insatisfecha con respecto a la evaluación de la carga infecciosa, con datos de observación e intervención en el campo de la orientación de antibióticos (5-7). Sin embargo, aún no se ha demostrado una medida precisa de la gravedad de la enfermedad.
Como tal, se han propuesto numerosos biomarcadores y puntuaciones clínicas, que incluyen el uso de puntuaciones de gravedad tales como la evaluación secuencial de insuficiencia de órganos (SOFA), la evaluación de salud crónica y fisiológica aguda (APACHE) II y la puntuación fisiológica aguda simplificada (SAPS) II, sin embargo, estos rara vez se calculan diariamente de manera rutinaria debido a la complejidad relativamente alta y los requisitos de recursos de tiempo asociados con cada puntuación. El uso de nuevos biomarcadores puede satisfacer esta necesidad clínica insatisfecha; sin embargo, pocos, si es que alguno, se han convertido con éxito en una rutina clínica generalizada (8).
De estos biomarcadores, la proadrenomedulina de la región media (MR-proADM), un péptido generado por múltiples tejidos para estabilizar la microcirculación y proteger contra la permeabilidad endotelial y la consiguiente insuficiencia de órganos (9-16), se ha mostrado muy prometedor, especialmente en los campos de sepsis (17), infecciones del tracto respiratorio inferior (18-21), trasplante de pulmón (22), cirugía torácica (23) e hipervolemia (WO2017/89474). De hecho, se reconoce ampliamente que el endotelio y la microcirculación desempeñan un papel importante en la respuesta fisiopatológica del huésped a la sepsis (24, 25), siendo de gran importancia la regulación y distribución del flujo sanguíneo dentro de cada órgano (25) y, por lo tanto, pueden proporcionar una indicación alternativa en cuanto a la gravedad de la respuesta general del huésped, en comparación con las puntuaciones de disfunción orgánica individual.
Comprender la respuesta del huésped a la sepsis es crucial para iniciar estrategias de tratamiento adecuadas. Un factor fundamental es comprender los procesos detrás del desarrollo de la disfunción orgánica. Cada sistema de órganos está formado por una red compleja y vasta de capilares, arteriolas y vénulas, que se denomina sistema microvascular. En particular, durante las respuestas inflamatorias, las plaquetas pueden tener efectos nocivos sobre la integridad vascular que, por ejemplo, pueden conducir a un aumento de la permeabilidad de la barrera vascular (59). Sin embargo, los órganos específicos tienen diversos grados de densidad y complejidad microvascular, y la microcirculación desempeña diferentes funciones en dependencia del órgano específico.
La contribución de las plaquetas sanguíneas a la fisiopatología de la sepsis y la insuficiencia de órganos ha sido
objeto de renovada atención. Mediante el uso de umbrales comunes de recuento de plaquetas, la trombocitopenia (un nivel anormalmente bajo de plaquetas en la sangre) representa del 20 al 50 % de los pacientes en la UCI y se asocia con malos resultados (38-47). Las plaquetas son actores bien conocidos en la coagulación y es probable que contribuyan a la coagulación intravascular diseminada (DIC), así como también de ser factores esenciales de la respuesta inmunitaria, reaccionando a la infección y a la alteración de la integridad tisular y contribuyendo a la inflamación, la eliminación de patógenos y la reparación tisular (48-53). Además, el papel de las plaquetas y la trombocitopenia en el contexto de enfermedades críticas existentes y en desarrollo, tal como la sepsis y la insuficiencia de órganos, es un tema de intensa investigación (54-57).
En condiciones vasculares sanas, las plaquetas encuentran señales inhibidoras generadas por las células endoteliales que impiden su activación. Circulan muy cerca de las paredes de los vasos y la interrupción en el revestimiento de las células endoteliales supera estas señales inhibitorias e impulsa la adherencia, activación y agregación de las plaquetas, que obstruyen temporalmente el vaso dañado.
Las plaquetas activadas secretan una profusión de material proinflamatorio y citocinas, que se dirigen a las células endoteliales y los leucocitos. En condiciones normales, el endotelio es una superficie no adhesiva, sin embargo, cuando es activado por las plaquetas sufre cambios profundos que incluyen la expresión de moléculas de adhesión celular y factores tisulares. Las plaquetas se adhieren a las células endoteliales activadas siguiendo un proceso de varias etapas en el que los glicanos juegan un papel fundamental. La activación plaquetaria puede alterar aún más el tono vascular y provocar cambios estructurales, aumentando así la permeabilidad vascular. La formación de agregados plaquetarios en la sangre es un fenómeno temprano en la progresión de la sepsis.
Por lo tanto, comprender la disfunción orgánica en desarrollo, como lo indica la puntuación de evaluación secuencial de insuficiencia de órganos, es crucial para ayudar a desarrollar tratamientos personalizados para la sepsis. Una advertencia temprana de las puntuaciones SOFA crecientes o decrecientes en los primeros momentos después del diagnóstico de sepsis es fundamental. Sin embargo, hasta el momento no hay marcadores diagnósticos y/o pronósticos confiables disponibles que indiquen una puntuación SOFA creciente o decreciente esperada y/o la presencia o desarrollo de niveles anormales de plaquetas, coagulación intravascular diseminada (DIC) y/o insuficiencia de órganos asociada, en particular insuficiencia de órganos específica.
Especialmente para los pacientes gravemente enfermos, el manejo de la trombocitopenia puede ser un desafío, ya que diferentes mecanismos pueden conducir a un número de plaquetas significativamente bajo: hemodilución (infusión de líquidos), aumento del consumo de plaquetas (por ejemplo, debido a la DIC, hemofagocitosis, trombosis), aumento de la destrucción de plaquetas (por ejemplo, debido a autoanticuerpos antiplaquetarios, heparina, fármacos), disminución de la producción de plaquetas (por ejemplo, debido a toxinas bacterianas, fármacos, enfermedad hepática crónica) o aumento del secuestro de plaquetas (por ejemplo, debido a hiperesplenismo, hipotermia). Además, un recuento bajo de plaquetas podría ser insignificante y no patológico debido a las variaciones estacionales e individuales (58). Además, diferentes escenarios provocan un fenotipo trombocitopénico sin ser patológicamente relevante (pseudotrombocitopenia). La coagulación en una muestra de sangre o la acumulación de plaquetas ex vivo inducida por EDTA pueden ser motivos de pseudotrombocitopenia y podrían inducir a error en medidas terapéuticas tales como la transfusión de plaquetas.
Por lo tanto, existe una necesidad urgente de desarrollar herramientas y métodos de diagnóstico y pronóstico que indiquen un desarrollo anormal del nivel de plaquetas y la gravedad asociada de un nivel anormal de plaquetas. Resumen de la invención
A la luz de las dificultades de la técnica anterior, el problema técnico que subyace a la presente invención es la provisión de medios para determinar los niveles anormales de plaquetas en un sujeto. Puede considerarse un objeto de la invención proporcionar los medios para el diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de un evento adverso presente o posterior asociado con niveles anormales de plaquetas, tal como insuficiencia de órganos, insuficiencia de órganos específica y/o muerte en un paciente. Por lo tanto, un objeto de la invención es proporcionar uno o más biomarcadores o combinaciones de biomarcadores para identificar pacientes que tienen un alto riesgo de tal evento adverso.
La solución al problema técnico de la invención se proporciona en las reivindicaciones independientes. Las modalidades preferidas de la invención se proporcionan en las reivindicaciones dependientes.
Por lo tanto, el método se refiere a un método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de niveles anormales de plaquetas en un paciente, que comprende
a. proporcionar una muestra de dicho paciente,
b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra,
c. en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas en dicho paciente.
La invención también se refiere a un método para el diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación de riesgo y/o estratificación de riesgo de trombocitopenia y/o afecciones médicas asociadas, tal como por ejemplo coagulación intravascular diseminada (DIC) o desregulación de órganos o insuficiencia basada en un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra. La invención se refiere por tanto a un método para el diagnóstico, pronóstico, evaluación del riesgo y/o estratificación del riesgo de trombocitopenia y/o coagulación intravascular diseminada (DIC) en base a un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra.
La presente invención se basa en el hallazgo sorprendente de que el nivel de proADM y en particular de MR-proADM en una muestra de un paciente se correlaciona con el recuento de plaquetas de dicho paciente en el momento del aislamiento de la muestra. Aún más sorprendente, se encontró que un nivel creciente de proADM se correlaciona con un número decreciente de trombocitos. Estas correlaciones persisten con el tiempo, de manera que el aumento de los valores de proADM en el día 1 y el día 4 después de la medición al inicio del estudio se correlaciona con la disminución de los niveles de plaquetas y la eventual mortalidad.
En algunas modalidades, la probabilidad de que ocurra un evento adverso posterior, tal como insuficiencia del sistema de coagulación, niveles anormales de plaquetas, DIC y/o trombocitopenia, puede evaluarse comparando el nivel de proADM o fragmento(s) de la misma en la muestra. en comparación con un nivel de referencia (tal como un valor umbral o de corte y/o un promedio poblacional), en donde el nivel de referencia puede corresponder a proADM o fragmentos del mismo en pacientes sanos, o en pacientes que han sido diagnosticados como gravemente enfermos, si es aplicable.
Sin embargo, existe la necesidad en la técnica de desarrollar estrategias de tratamiento personalizadas que se formulen órgano por órgano. En consecuencia, es una gran ventaja del método de la presente invención que es posible predecir con una alta probabilidad una insuficiencia de órganos específica que puede desarrollarse en un futuro próximo, en particular para el riñón, el hígado y el sistema de coagulación sobre la base de un nivel de proADM determinado en el momento 0 (día 0). Los ejemplos proporcionados en la presente descripción demuestran que la MR-proADM puede predecir significativamente mejor las mejoras o deterioros con respecto a un evento adverso, tal como preferentemente las mejoras o deterioros en los sistemas de órganos hepáticos, nefróticos y de coagulación.
En consecuencia, el método de la presente invención puede ayudar a predecir la probabilidad de un evento adverso posterior en la salud del paciente, tal como insuficiencia del sistema de coagulación, niveles anormales de plaquetas, DIC y/o trombocitopenia. Esto significa que el método de la invención puede discriminar pacientes de alto riesgo, que tienen más probabilidades de sufrir complicaciones, o cuyo estado será más crítico en el futuro, de pacientes de bajo riesgo, cuyo estado de salud es estable o incluso mejora, por lo que no se espera que sufran un evento adverso, tal como insuficiencia del sistema de coagulación, niveles anormales de plaquetas, muerte, DIC y/o trombocitopenia, lo que podría requerir determinadas medidas terapéuticas y/o un seguimiento más intenso del paciente.
El método también se refiere a un método para evaluar la gravedad de niveles bajos de plaquetas o afecciones asociadas, tales como trombocitopenia y/o coagulación intravascular diseminada (DIC). La proADM puede usarse de forma cuantitativa o semicuantitativa para evaluar la probabilidad de gravedad o gravedad de una afección existente. Por tanto, la invención se refiere a un método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de un evento adverso presente o posterior en la salud de un paciente, que comprende proporcionar una muestra de dicho paciente, determinar un nivel de adrenomedulina (ADM) o fragmento(s) de la misma en dicha muestra, en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la probabilidad de un evento adverso posterior en la salud de dicho paciente.
En modalidades de la invención, el evento adverso posterior es insuficiencia de órganos, insuficiencia de órganos específica, muerte, muerte dentro de los 28-90 días desde el momento del aislamiento de la muestra, niveles anormales de plaquetas, trombocitopenia, coagulación intravascular diseminada (DIC) y/o insuficiencia hepática o una infección.
En modalidades preferidas, la invención se refiere a un método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de insuficiencia de órganos específica en un paciente, que comprende proporcionar una muestra de dicho paciente, determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la insuficiencia de un órgano específico en dicho paciente.
En modalidades preferidas, la invención se refiere a un método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de insuficiencia hepática en un paciente, que comprende proporcionar una muestra de dicho paciente, determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la insuficiencia del hígado en dicho paciente.
En algunas modalidades, el método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de insuficiencia de un órgano específico en un paciente se lleva a cabo en un paciente con lo que se sospecha que tiene niveles bajos de plaquetas, o una afección relacionada.
El término "insuficiencia de órganos específica" se refiere a la insuficiencia de un órgano específico. Por ejemplo, en el caso de un método de pronóstico, el método de la invención puede usarse para pronosticar insuficiencias no solo de insuficiencias de cualquier órgano, sino de un órgano específico. Por ejemplo, el método puede usarse para pronosticar específicamente la insuficiencia del riñón, hígado y/o el sistema de coagulación de la sangre.
En este caso, la insuficiencia de un órgano o sistema, tal como el hígado o el sistema de coagulación, puede relacionarse tanto con el colapso total del sistema, en el sentido de ausencia o casi ausencia de cualquier función fisiológica del órgano o sistema, y desregulación, que se refiere a un desequilibrio de la homeostasis de un órgano o un sistema. Una desregulación leve puede ocurrir sin síntomas clínicos iniciales, mientras que una desregulación progresiva puede conducir a una pérdida parcial de la función del sistema que conduce a síntomas clínicos y una desregulación fuerte puede ser equivalente a una falla.
Por lo tanto, la invención se refiere a un método para el pronóstico, la evaluación del riesgo y/o la estratificación del riesgo de niveles bajos de plaquetas y/o trombocitopenia y/o coagulación intravascular diseminada (DIC) en un paciente, en donde un nivel de proADM o fragmento(s) de la misma igual o superior a un nivel de gravedad alto (o valor de corte) es indicativo de la probabilidad de desarrollar niveles bajos de plaquetas y/o trombocitopenia y/o DIC dentro de las 12 horas a 120 horas, preferentemente dentro de las 24 horas a las 72 horas, después de obtener una muestra.
Una ventaja particular del método de la presente invención es que un paciente que ha sido identificado como un paciente de bajo riesgo por medio del método de la presente invención podría ser dado de alta más rápidamente, por ejemplo, de una uCi, un departamento de emergencia, una práctica privada o un hospital. Asimismo, para pacientes de bajo riesgo, podría disminuirse la intensidad y/o frecuencia de la observación del estado de salud del paciente. En consecuencia, el hospital u otra institución médica a cargo del paciente podría decidir de manera más eficiente qué pacientes requieren cuidados médicos intensivos y observación. Consecuentemente, el hospital o institución respectiva podría, por ejemplo, ocupar de manera más eficiente las camas de la UCI con pacientes de alto riesgo. Esto conduciría a una mejor atención médica para los pacientes de alto riesgo, ya que el personal médico podría concentrarse en dichos pacientes, mientras que los pacientes de bajo riesgo podrían ser dados de alta. Esto también conduciría a beneficios significativos por costos evitados por medidas innecesarias que de cualquier otra manera se aplicarían a pacientes de bajo riesgo.
Fue completamente sorprendente que el nivel de proADM o fragmentos de la misma en una muestra del paciente pueda proporcionar información crítica sobre la probabilidad de ocurrencia o presencia y gravedad de, por ejemplo, niveles anormales de plaquetas asociados con DIC y/o trombocitopenia o trombocitopenia secundaria a la sepsis. El uso de la proADM o fragmentos de la misma como parámetro único en modalidades de la presente invención es ventajoso sobre el uso de otros parámetros únicos, tal como biomarcadores o puntuaciones clínicas, ya que la proADM es más precisa en la predicción de insuficiencias del sistema de coagulación, anomalías de los niveles de plaquetas, DIC y/o trombocitopenia en comparación con otros marcadores y parámetros clínicos tales como recuento de plaquetas, lactato o puntuaciones clínicas tales como SOFA, qSOFA, SAPS II o APACHE II.
En modalidades de la invención, el paciente que muestra síntomas o no presenta síntomas de cualquier trastorno fisiológico puede examinarse en cualquier entorno médico. De acuerdo con una modalidad preferida, la muestra se aísla de un paciente durante un examen médico.
En modalidades de la invención, el paciente está o ha sido diagnosticado como gravemente enfermo. De acuerdo con otra realización, la muestra se aísla del paciente en el momento del diagnóstico o después. Además, en modalidades del método de la invención, el tratamiento médico se ha iniciado en el momento del diagnóstico o antes. En modalidades, el paciente ha sido diagnosticado como gravemente enfermo y se ha iniciado el tratamiento médico. La muestra puede aislarse del paciente antes, durante o después del diagnóstico y del inicio del tratamiento. En una modalidad, el paciente está, o ha sido diagnosticado como, gravemente enfermo.
En una modalidad, la muestra se aísla del paciente en o después del momento del diagnóstico como gravemente enfermo.
En una modalidad, el paciente se diagnostica con una enfermedad infecciosa.
En una modalidad, el paciente se diagnostica con sepsis, sepsis grave o choque séptico.
En una modalidad, el paciente se diagnostica con una o más insuficiencias orgánicas existentes y/o es un paciente postraumático o postquirúrgico.
En una modalidad, el paciente se diagnostica con una desregulación del sistema de coagulación, tal como coagulación intravascular diseminada (DIC) o trombocitopenia y/o una disfunción de un órgano asociado con el sistema de coagulación, tal como vasos sanguíneos, bazo, médula ósea y/o el sistema inmunológico.
De acuerdo con una modalidad adicional, el paciente está siendo diagnosticado o ha sido diagnosticado de coagulación intravascular diseminada (DIC).
La DIC puede considerarse como un síndrome que puede ocurrir en el contexto de diferentes tipos de enfermedades, tales como por ejemplo tumores sólidos, cáncer de la sangre, linfoma, leucemia, complicaciones obstétricas (tales como (pre)eclampsia, desprendimiento de la placenta, embolia de líquido amniótico, aborto), lesiones masivas (tales como trauma severo, quemaduras, hipertermia, cirugía extensa), sepsis, sepsis severa, choque séptico, infecciones severas (por ejemplo, bacterianas, virales, fúngicas, protozoarias, superinfección/coinfecciones), reacciones transfusionales (tales como reacciones hemolíticas por incompatibilidad a Ob ), reacciones adversas a fármacos (por ejemplo, inducidas por antiinfecciosos, agentes antineoplásicos, agentes antitrombóticos), reacciones alérgicas o tóxicas graves o hemangiomas gigantes.
El experto en la técnica es consciente de otras afecciones y enfermedades que pueden asociarse con la DIC. La DIC puede provocar disfunción/daño de (múltiples) órganos (independientemente de si el problema provino de sangrado o coagulación (en dependencia de la "etapa DIC"). Una combinación de pérdida generalizada del flujo sanguíneo tisular y sangrado simultáneo conduce a un mayor riesgo de muerte. Por lo tanto, la DIC es una emergencia médica que puede estar asociada con complicaciones graves. Cuanto más temprana sea la predicción o el diagnóstico de la DIC, mejor será el pronóstico del paciente.
Se han sugerido varios tratamientos para la DIC, tales como el factor V, el factor XIII-AP, la esfingosina-1-fosfato (S1P), la trombomodulina, los anticuerpos contra el factor tisular o el empalme previo del ARNm del factor tisular, el fragmento del péptido inhibidor de nitrógeno reactivo (RNIP), TAFIa(i), fosfolípido procoagulante, así como también el inhibidor de la trombina.
En las modalidades de la invención, la DIC puede conducir a una disfunción/daño de (múltiples) órganos (independientemente de si el problema provino de sangrado o coagulación (en dependencia de la "etapa DIC"). Una combinación de pérdida generalizada del flujo sanguíneo tisular y sangrado simultáneo conduce a un mayor riesgo de muerte.
En modalidades de la invención, el paciente recibe un tratamiento de DIC, tal como, por ejemplo, el factor V, el factor XIII-AP, la esfingosina-1-fosfato (S1P), la trombomodulina, los anticuerpos contra el factor tisular o el empalme previo del ARNm del factor tisular, el fragmento del péptido inhibidor de nitrógeno reactivo (RNIP), TAFIa(i), fosfolípido procoagulante y/o inhibidor de trombina.
En modalidades adicionales, el tratamiento que recibe el paciente comprende uno o más de tratamiento antibiótico, ventilación mecánica invasiva, ventilación mecánica no invasiva, terapia de reemplazo renal, uso de vasopresores, terapia de fluidos, corticosteroides, transfusión de sangre o plaquetas, esplenectomía, inhibidores directos de trombina (tales como lepirudina o argatroban), anticoagulantes (tales como bivalirudina y fondaparinux), suspensión de heparina en caso de trombocitopenia inducida por heparina, carbonato de litio, folato, purificación extracorpórea de sangre y/o protección de órganos.
En modalidades preferidas de la invención, dicho nivel de proADM se correlaciona inversamente con el nivel de plaquetas.
Preferentemente, la muestra puede ser un fluido corporal. Con mayor preferencia, la muestra se selecciona del grupo que consiste en una muestra de sangre, una muestra de suero, una muestra de plasma y/o una muestra de orina.
Preferentemente, el método se lleva a cabo en algunas modalidades determinando un nivel de proADM o fragmento(s) de la misma, en donde dicha determinación de proADM comprende determinar un nivel de MR-proADM en la muestra. El empleo de determinar MR-proADM se prefiere para cualquier modalidad dada descrita en la presente descripción y puede considerarse en el contexto de cada modalidad, en consecuencia. En modalidades preferidas, el "fragmento de ADM" puede considerarse como MR-proADM. En algunas modalidades, puede emplearse cualquier fragmento o precursor de ADM, tal como pre-pro-ADM, pro-ADM, el péptido conocido como ADM mismo, o fragmentos de la misma, tal como MR-proADM.
De acuerdo con otra modalidad de la invención, determinar un nivel de proADM o fragmento(s) de la misma comprende determinar un nivel de MR-proADM en la muestra.
En una modalidad preferida del método de la invención,
- un nivel de proADM o fragmento(s) de la misma más abajo de un nivel de gravedad alto (valor de corte) es indicativo de niveles de plaquetas normales o altos, o
- un nivel de proADM o fragmento(s) de la misma igual o superior a un nivel de gravedad alto (o valor de corte) es indicativo de la presencia o probabilidad de desarrollar niveles bajos de plaquetas y/o trombocitopenia y/o coagulación intravascular diseminada (DIC),
- en donde un nivel de gravedad alto (o valor de corte) de proADM o fragmento(s) de la misma es un nivel por encima de 6,5 nmol/l, 6,95 nmol/l, o preferentemente 10,9 nmol/l.
De acuerdo con la presente invención, el término "indicar" en el contexto de "indicativo de un evento adverso posterior" e "indicativo de la ausencia de un evento adverso posterior" pretende ser una medida de riesgo y/o probabilidad. Preferentemente, la "indicación" de la presencia o ausencia de un evento adverso pretende ser una evaluación de riesgo, y normalmente no debe interpretarse de manera limitativa como para señalar definitivamente la presencia o ausencia absoluta de dicho evento.
Por lo tanto, el término "indicativo de la ausencia de un evento adverso posterior" o "indicativo de un evento adverso posterior" puede entenderse como indicador de un riesgo bajo o alto de ocurrencia de un evento adverso, respectivamente. En algunas modalidades, un riesgo bajo se relaciona con un riesgo menor en comparación con los niveles de proADM detectados por encima de los valores indicados. En algunas modalidades, un alto riesgo se relaciona con un mayor riesgo en comparación con los niveles de proADM detectados más abajo de los valores indicados.
Teniendo en cuenta lo anterior, la determinación de los niveles de gravedad altos y/o bajos de proADM es, sin embargo, muy confiable con respecto a determinar la presencia o ausencia de un evento adverso posterior cuando se usan los valores de corte descritos en la presente descripción, de manera que la estimación de riesgo permite una actuación adecuada por parte de un profesional médico.
En algunas modalidades, el nivel de gravedad bajo, intermedio o alto de proADM indica la gravedad de una afección física de un paciente con respecto a un evento adverso.
En algunas modalidades, el nivel de gravedad bajo, intermedio o alto de proADM indica la gravedad de una afección física de un paciente con trombocitopenia o síntomas de trombocitopenia.
En algunas modalidades, el nivel de gravedad bajo, intermedio o alto de proADM indica la gravedad de una afección física de un paciente con trombocitopenia o síntomas de trombocitopenia secundarios a sepsis.
Fue completamente sorprendente que un nivel de proADM o fragmentos de la misma pudiera correlacionarse con la probabilidad de la presencia o ausencia de un evento adverso posterior, tal como insuficiencia del sistema de coagulación, niveles anormales de plaquetas, DIC y/o trombocitopenia, también en el contexto de pacientes gravemente enfermos que estaban recibiendo tratamientos en estos momentos.
Los niveles de proADM en las muestras pueden asignarse preferentemente a 3 niveles de gravedad diferentes de proADM. Los niveles altos de proADM indican un nivel de gravedad alto, los niveles intermedios indican un nivel de gravedad intermedio y los niveles bajos indican niveles de gravedad bajos. Las concentraciones respectivas que determinan los valores de corte para los niveles de gravedad respectivos dependen de múltiples parámetros, tal como el momento del aislamiento de la muestra, por ejemplo, después del pronóstico, diagnóstico e inicio del tratamiento del paciente, y el método usado para determinar el nivel de proADM o fragmentos de la misma en dicha muestra.
Los valores de corte descritos en la presente descripción se refieren preferentemente a las mediciones del nivel de proteína de proADM o fragmentos de la misma en una muestra de plasma obtenida de un paciente mediante el ensayo BRAHMS MR proADM KRYPTOR. En consecuencia, los valores descritos en la presente descripción pueden variar hasta cierto punto en dependencia del método de detección/medición empleado, y los valores específicos descritos en la presente descripción también pretenden leer los valores correspondientes determinados por otros métodos.
En una modalidad de la invención, un nivel de gravedad bajo y/o intermedio de proADM o fragmento(s) de la misma es indicativo de la ausencia de un (posterior) evento adverso, tal como insuficiencia del sistema de coagulación, niveles anormales de plaquetas, DIC y /o trombocitopenia, en donde el nivel de gravedad bajo está más abajo de un valor de corte en el intervalo de 1,5 nmol/l y 4 nmol/l. Cualquier valor dentro de estos intervalos puede considerarse
como un valor de corte apropiado para niveles bajos de gravedad de proADM o fragmentos de la misma. Por ejemplo, 1,5, 1,55, 1,6, 1,65, 1,7, 1,75, 1,8, 1,85, 1,9, 1,95, 2,0, 2,05, 2,1, 2,15, 2,2, 2,25, 2,3, 2,35, 2,4, 2,45, 2,5, 2,55, 2,6, 2,65, 2,7, 2,75, 2,8, 2,85, 2,9, 2,95, 3,0, 3,05, 3,1, 3,15, 3,2, 3,25, 3,3, 3,35, 3,4, 3,45, 3,5, 3,55, 3,6, 3,65, 3,7, 3,75, 3,8, 3,85, 3,9, 3,95, 4,0 nmol/l.
En una modalidad de la invención, un nivel de gravedad alto de proADM o fragmento(s) de la misma es indicativo de un evento adverso (posterior), tal como insuficiencia del sistema de coagulación, niveles anormales de plaquetas, DIC y/o trombocitopenia, en donde el nivel de gravedad alto es superior a un valor de corte en el intervalo de 6,5 nmol/l a 12 nmol/l. Cualquier valor dentro de estos intervalos puede considerarse tal como un valor de corte apropiado para niveles de gravedad altos de proADM o fragmentos de la misma. Por ejemplo, 6,5, 6,55, 6,6, 6,65, 6,7, 6,75, 6,8, 6,85, 6,9, 6,95, 7,0, 7,05, 7,1, 7,15, 7,2, 7,25, 7,3, 7,35, 7,4, 7,45, 7,5, 7,55, 7,6, 7,65, 7,7, 7,75, 7,8, 7,85, 7,9, 7,95, 8,0, 8,05, 8,1, 8,15, 8,2, 8,25, 8,3, 8,35, 8,4, 8,45, 8,5, 8,55, 8,6, 8,65, 8,7, 8,75, 8,8, 8,85, 8,9, 8,95, 9.0, 9,05, 9,1, 9,15, 9,2, 9,25, 9,3, 9,35, 9,4, 9,45, 9,5, 9,55, 9,6, 9,65, 9,7, 9,75, 9,8, 9,85, 9,9, 9,95, 10,0, 10,05, 10,1, 10,15, 10,2, 10,25, 10,3, 10,35, 10,4, 10,45, 10,5, 10,55, 10,6, 10,65, 10,7, 10,75, 10,8, 10,85, 10,9, 10,95, 11.0, 11,05, 11,1, 11,15, 11,2, 11,25, 11,3, 11,35, 11,4, 11,45, 11,5, 11,55, 11,6, 11,65, 11,7, 11,75, 11,8, 11,85, 11,9, 11,95, 12,0 nmol/l.
Todos los valores de corte descritos en la presente descripción relacionados con el nivel de un marcador o biomarcador, tal como proADM o PCT, deben entenderse como "igual o superior" a un determinado valor de corte o "igual o inferior" a un determinado valor de corte. Por ejemplo, una modalidad relacionada con un nivel de proADM o fragmento(s) de la misma más abajo de 4 nmol/l, preferentemente más abajo de 3 nmol/l, con mayor preferencia más abajo de 2,7 nmol/l debe entenderse como relacionada con un nivel de proADM o fragmento(s) de la misma igual o inferior a 4 nmol/l, preferentemente igual o inferior a 3 nmol/l, con mayor preferencia igual o inferior a 2,7 nmol/l. Por el contrario, una modalidad relacionada con un nivel de proADM o fragmento(s) de la misma por encima de 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l debe entenderse como relativa a un nivel de proADM o fragmento(s) de la misma igual o superior a 6,5 nmol/l, preferentemente igual o superior a 6,95 nmol/l, con mayor preferencia igual o superior a 10,9 nmol/l.
En otras modalidades descritas en la presente descripción, los niveles de gravedad se definen preferentemente mediante valores de corte, que representan los límites entre los niveles de gravedad bajos, intermedios o altos. Por lo tanto, cualquier modalidad que presente valores de corte puede usar el formato de un único valor de corte como límite entre dos niveles de gravedad, o un único nivel de corte para cada nivel de gravedad.
En algunas modalidades, el valor de corte de la proADM entre los niveles de gravedad bajo e intermedio es:
2,75 nmol/l ± 20 % o 2,75 nmol/l ±15 %, o ±12 %, ±10 %, ±8 % o ±5 %,
y entre los niveles de gravedad intermedios y altos:
10,9 nmol/l ± 20 % o 10,9 nmol/l ±15 %, o ±12 %, ±10 %, ±8 % o ±5 %.
Estos valores de corte son preferentemente relevantes para una evaluación del nivel de gravedad de proADM al inicio del estudio, en otras palabras, al momento del diagnóstico y/o inicio de la terapia y/u hospitalización. Los niveles al inicio del estudio, a través de, por ejemplo, una sola medición o mediciones tomadas en un momento único inicial, pueden indicar la prescripción de terapia de fluidos.
En algunas modalidades, el valor de corte de la proADM entre los niveles de gravedad bajo e intermedio es:
2.80 nmol/l ± 20 %, o 2,80 nmol/l ± 15 %, o ± 12 %, ± 10 %, ± 8 % o ±5 %, y entre niveles de gravedad intermedios y altos:
9,5 nmol/l ± 20 % o 9,5 nmol/l ±15 %, o ±12 %, ±10 %, ±8 % o ±5 %.
Estos valores de corte son preferentemente relevantes para una evaluación del nivel de gravedad de proADM después de 1 día, en otras palabras, aproximadamente 24 horas después del inicio del estudio, es decir, aproximadamente 1 día después del inicio del diagnóstico y/o terapia y/o hospitalización. Por ejemplo, en modalidades en las que la proADM se mide un día después de comenzar la terapia, pueden emplearse los valores de corte para el día 1. Como se desprende de lo anterior, el límite entre intermedio y alto es algo más bajo que al inicio del estudio, es decir, a medida que pasa el tiempo, incluso los niveles algo más bajos (pero todavía relativamente altos) se asocian con un riesgo alto y se clasifican en el nivel de gravedad alto.
En algunas modalidades, el valor de corte de la proADM entre los niveles de gravedad bajo e intermedio es:
2.80 nmol/l ± 20 % o 2,80 nmol/l ±15 %, o ± 12 %, ±10 %, ±8 % o ±5 %,
y entre niveles de gravedad intermedios y altos:
7,7 nmol/l ± 20 % o 7,7 nmol/l ±15 %, o ±12 %, ±10 %, ±8 % o ±5 %.
Estos valores de corte son preferentemente relevantes para una evaluación del nivel de gravedad de la proADM después de 4 días, en otras palabras, aproximadamente 4 días después del inicio del estudio, es decir, aproximadamente 4 días después del inicio del diagnóstico y/o terapia y/o hospitalización. Por ejemplo, en modalidades donde la proADM se mide 4 días después de comenzar la terapia, pueden emplearse los valores de corte para el día 4. Como se desprende de lo anterior, el límite entre intermedio y alto es algo más bajo que al inicio del estudio o en el día 1, es decir, a medida que avanza el tiempo, incluso los niveles algo más bajos (pero todavía relativamente altos) se asocian con alto riesgo y se clasifican en nivel de gravedad alto.
En algunas modalidades, los niveles de corte a emplear en las modalidades descritas anteriormente pueden ajustarse de acuerdo con un nivel apropiado en dependencia del día en que se realice la medición. Cada uno de los valores de corte está sujeto a alguna variación debido a la varianza común, como puede esperar el experto en la técnica. Los valores de corte relevantes se determinan en base a datos extensos, como se presenta más abajo, pero no pretenden ser valores finales o exactos en todas las modalidades posibles. Mediante un valor de corte similar a los citados, es decir, dentro del ± 20 %, ±15 %, ±12 %, ±10 %, ±8 % o ±5 %, como puede determinarlo un experto en la técnica, pueden esperarse resultados similares.
Cualquier modalidad recitando ± 20 % de un valor de corte dado, puede considerarse que también describe ± 15 %, ± 12 %, ± 10 %, ± 8 % o ± 5 %.
Puede considerarse que cualquier modalidad que cite un valor de corte particular para el inicio del estudio, el día 1 o el día 4, también describe los valores de corte correspondientes para los otros días, por ejemplo, puede considerarse que una modalidad que cite un valor de corte de la línea de base también se relaciona con la misma modalidad citando el valor de corte del día 1 o del día 4.
Los valores de corte se aplican en modalidades preferidas a muestras de sangre o muestras derivadas de sangre, pero no se limitan a ellas.
En modalidades de la invención, el nivel de proADM o fragmento(s) de la misma igual o por encima del nivel de alta gravedad (valor de corte) indica el inicio o la modificación de un tratamiento del paciente, tal como el suministro de corticosteroides, transfusión de sangre o plaquetas, transfusión de componentes sanguíneos, tal como suero, plasma o células específicas o combinaciones de los mismos, fármacos que promuevan la formación de trombocitos, tratamiento causal o modalidad de una esplenectomía. Preferentemente, la causa de la desregulación del sistema de coagulación puede identificarse y tratarse (tratamiento causal). De acuerdo con otra modalidad, los pacientes son pacientes de la unidad de cuidados intensivos (UCI), en donde
- el nivel de proADM o fragmento(s) de la misma por debajo del nivel de gravedad bajo (valor de corte) indica el alta de dicho paciente de la UCI, o
- el nivel de proADM o fragmento(s) de la misma igual o superior al nivel de gravedad alto (valor de corte) indica modificar el tratamiento del paciente en la UCI.
Es una ventaja particular de la presente invención que en base a la clasificación de los niveles determinados de proADM o fragmentos de la misma es posible evaluar la probabilidad de ocurrencia de un evento adverso futuro en la salud del paciente. Con base en esta evaluación, es posible ajustar las próximas opciones y decisiones de tratamiento.
Una modificación del tratamiento en el sentido de la presente invención incluiría, sin limitación, un ajuste de la dosis o régimen de administración del medicamento en curso, cambio del tratamiento en curso a un tratamiento diferente, adición de una opción de tratamiento adicional al tratamiento en curso o interrupción de un tratamiento en curso o la identificación y tratamiento de la causa de la disfunción. En la descripción detallada de esta solicitud de patente se han descrito diferentes tratamientos que pueden aplicarse a los pacientes en el contexto de la presente invención. En modalidades preferidas de la invención, el método comprende adicionalmente determinar un nivel de uno o más marcadores adicionales en una muestra aislada del paciente.
El uno o más marcadores adicionales pueden determinarse en la misma muestra o en una diferente de dicho paciente. En caso de una muestra diferente, la muestra puede aislarse al mismo tiempo, antes o después del aislamiento de la muestra para determinar la proADM o fragmento(s) de la misma. Independientemente de si el uno o más marcadores adicionales se determinan en la misma muestra o en una diferente, la medición puede ocurrir en paralelo, simultáneamente, antes y/o después de la medición de la proADM.
En una modalidad preferida, uno o más marcadores adicionales comprenden el nivel de plaquetas en una muestra de sangre.
En una modalidad, uno o más marcadores adicionales comprenden PCT o fragmento(s) de la misma.
Es particularmente ventajoso combinar la determinación de proADM o fragmentos de la misma con la determinación de los niveles de plaquetas en una muestra, en donde la muestra usada para determinar la proADM puede ser la misma o una muestra diferente usada para realizar el recuento de plaquetas.
De acuerdo con otra modalidad preferida de la presente invención, el método descrito en la presente descripción comprende adicionalmente
- determinar un nivel de plaquetas en una muestra aislada del paciente, o
- determinar un nivel de plaquetas en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla antes, en o después del momento del diagnóstico y del inicio del tratamiento,
- determinar un nivel de plaquetas en una segunda muestra aislada de dicho paciente, en donde la segunda muestra ha sido aislada después de la primera muestra, preferentemente dentro de los 30 minutos posteriores al aislamiento de la primera muestra o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después del aislamiento de la primera muestra, y
- determinar una diferencia en el nivel de plaquetas en la segunda muestra en comparación con el nivel de plaquetas en la primera muestra.
En modalidades adicionales, uno o más marcadores adicionales comprenden uno o más marcadores para una desregulación del sistema de coagulación, tal como coagulación intravascular diseminada (DIC) o trombocitopenia, que comprenden micropartículas de membrana, recuento de plaquetas, volumen medio de plaquetas (MPV) sCD14-ST, protrombinasa, antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor von Willebrand (vWF), lipoproteínas en combinación con CRP, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D no desnaturalizado de fibrina, factor plaquetario 4, histonas y un ensayo PT. En modalidades, uno o más marcadores adicionales comprenden una o más histonas.
En modalidades de la invención, la proADM se determina en el contexto de un método para la orientación terapéutica en pacientes con administración de heparina para predecir la trombocitopenia, por ejemplo, adaptación/cambio de la medicación hasta que el paciente muestre recuentos de plaquetas normales y/o los niveles de proADM estén por debajo de un valor de corte descrito en la presente descripción.
En modalidades de la invención, la proADM se determina en el contexto de un método para la orientación terapéutica en pacientes con tratamiento antimicrobiano para predecir la leucocitopenia, por ejemplo, adaptación/cambio de la medicación hasta que el paciente muestre recuentos de plaquetas normales y/o los niveles de proADM estén por debajo de un valor de corte descrito en la presente descripción.
En modalidades adicionales, la PCT se incluye en el control, por ejemplo, con el fin de administración de antibióticos y/o prevención del uso indebido de antibióticos y/o prevención de efectos secundarios, por ejemplo, para reducir el riesgo de contraer trombocitopenia o DIC. En este contexto, los recuentos de plaquetas pueden determinarse como un marcador adicional.
En modalidades de la invención, la proADM se determina en el contexto de la transfusión de plaquetas. En modalidades adicionales, la PCT se incluye en el control, por ejemplo, para la detección de una infección bacteriana, con el propósito de administración de antibióticos y/o prevención del uso indebido de antibióticos y/o prevención de efectos secundarios, por ejemplo, para reducir el riesgo de contraer trombocitopenia o DIC. En este contexto, los recuentos de plaquetas pueden determinarse como un marcador adicional.
En modalidades de la invención, la proADM se determina en el contexto de un método para la predicción y/o diagnóstico de la desregulación del sistema de coagulación, DIC y/o trombocitopenia en un paciente, por ejemplo, un paciente con choque o un paciente con choque séptico, en donde preferentemente la PCT se determina como un marcador adicional.
En modalidades de la invención, el recuento absoluto de plaquetas inmaduras (AIPC) puede determinarse como un marcador adicional.
En modalidades de la invención, la proADM es un marcador mejor que los recuentos de plaquetas para la predicción de la DIC o trombocitopenia. En modalidades, un tratamiento puede ser el manejo de fluidos.
En las modalidades, los pacientes con lesión renal aguda (AKT) /- terapia de reemplazo renal continua (CRRT), neumonía adquirida en la comunidad (CAP), sepsis y pacientes de UCI en general con un proADM más alto tienen un mayor riesgo de mortalidad.
En una modalidad, la invención comprende adicionalmente informar al paciente de los resultados del método descrito en la presente descripción.
En una modalidad, el método permite el pronóstico, la evaluación del riesgo o la estratificación del riesgo de un evento adverso en un paciente con un nivel anormal de plaquetas, que comprende:
a. proporcionar una muestra de dicho paciente,
b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, c. en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas y con la probabilidad de que ocurra un evento adverso en dicho paciente.
En una modalidad, el evento adverso es uno o más de mortalidad, mortalidad relacionada con sepsis, insuficiencia de órganos y/o disfunción orgánica.
En una modalidad, la insuficiencia de órganos o la disfunción orgánica se asocia con trombocitopenia.
En una modalidad, se mide el al menos un marcador o parámetro clínico adicional, preferentemente seleccionado del grupo que consiste en procalcitonina, histona 3, histona 2A, histona 2B, histona 4 o fragmento(s) de la misma, recuento de plaquetas, volumen medio de plaquetas y/o uno o más marcadores de una desregulación del sistema de coagulación.
En una modalidad, el paciente muestra síntomas de una enfermedad infecciosa o sepsis, o se le diagnostica una enfermedad infecciosa y/o una o más insuficiencias orgánicas existentes, o se le ha diagnosticado sepsis, sepsis grave o choque séptico.
En una modalidad adicional, el método permite la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de trombocitopenia séptica en un paciente, que comprende
a. proporcionar una muestra de dicho paciente,
b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, y c. determinar un nivel de procalcitonina (PCT) o fragmento(s) de la misma en dicha muestra,
d. en donde cuando un nivel de procalcitonina (PCT) o fragmento(s) de la misma es >0,5 ng/ml indica la presencia de, o un mayor riesgo de adquirir, sepsis, y en donde cuando un nivel de proADM o fragmento(s) de la misma es > 2,75 nmol/l indica la presencia o un mayor riesgo de adquirir trombocitopenia.
Esta modalidad permite una combinación de funciones beneficiosas que no se derivaban previamente de la técnica que permite a los médicos identificar no solo la presencia de enfermedades sépticas, sino también iniciar un tratamiento dirigido a mejorar los niveles de plaquetas. Los niveles elevados de PCT o fragmentos de la misma indican la presencia de una enfermedad infecciosa, en particular sepsis, mediante el uso de los valores conocidos establecidos en la técnica. La combinación con la medición de ADM, que indica los niveles de plaquetas, seguida preferentemente de las medidas apropiadas para mejorar las plaquetas o los trombocitos, brinda a los profesionales un salto sobre el proceso patológico subyacente en la sepsis, lo que permite un tratamiento mejorado y más rápido. La invención se refiere además a un kit para llevar a cabo el método descrito en la presente descripción, que comprende:
a. reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT o fragmento(s) de la misma y/o uno o más marcadores adicionales como se describe en la presente descripción, en una muestra de un sujeto, y
b. datos de referencia, tales como un nivel de referencia, correspondiente a los niveles de gravedad alto de la proADM, en donde el nivel de gravedad alto es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, y opcionalmente niveles de PCT y/ o niveles de uno o más marcadores adicionales como se describe en la presente descripción, en donde dichos datos de referencia se almacenan en un medio legible por ordenador y/o se emplean en forma de código ejecutable por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT o fragmento(s) de la misma y/o marcadores adicionales como se describe en la presente descripción, a dichos datos de referencia.
En algunas modalidades, el kit comprende agentes terapéuticos adicionales para mejorar los niveles de plaquetas, como se describe con más detalle en la presente descripción.
Los reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente para determinar el nivel de PCT o fragmento(s) de la misma y/o marcadores adicionales de la invención, se seleccionan preferentemente entre los necesarios para realizar el método, por ejemplo, anticuerpos dirigidos a proADM, etiquetas adecuadas, tales como marcadores fluorescentes, preferentemente dos marcadores fluorescentes separadas adecuados para su aplicación en el ensayo BRAHMS KRYPTOR, tubos de recogida de muestras.
En una modalidad del método descrito en la presente descripción, el nivel de proADM o fragmento(s) de la misma y opcionalmente adicionalmente otros biomarcadores tales como por ejemplo PCT o fragmento(s) de la misma se determina mediante el uso de un método seleccionado del grupo que consiste en espectrometría de masas (MS), inmunoensayo de luminiscencia (LIA), radioinmunoensayo (RIA), inmunoensayos de quimioluminiscencia y fluorescencia, inmunoensayo enzimático (EIA), inmunoensayos ligados a enzimas (ELISA), matrices de perlas basadas en luminiscencia, matrices basadas en perlas magnéticas, ensayos de micromatrices de proteínas, prueba rápida de formatos tales como, por ejemplo, pruebas de tiras inmunocromatográficas, análisis de criptatos raros y sistemas/analizadores automatizados.
El método de acuerdo con la presente invención puede realizarse además como un método homogéneo, en donde los complejos tipo sándwich formados por el anticuerpo/anticuerpos y el marcador, por ejemplo, la proADM o un fragmento de la misma, que va a detectarse, permanecen suspendidos en la fase líquida. En este caso, se prefiere que, cuando se usan dos anticuerpos, ambos anticuerpos se marquen con partes de un sistema de detección, lo que conduce a la generación de una señal o a la activación de una señal si ambos anticuerpos se integran en un solo sándwich.
Dichas técnicas deben realizarse en particular como métodos de detección de intensificación de la fluorescencia o inactivación de la fluorescencia. Un aspecto particularmente preferido se refiere al uso de los reactivos de detección que van a usarse por pares, tales como por ejemplo los que se describen en el documento US 4882733 A, EP-B1 0 180492 o EP-B1 0539 477 y el estado de la técnica citado en el mismo. De esta manera, se hacen posibles las mediciones en las que solo se detectan los productos de reacción que comprenden ambos componentes de marcaje en un solo inmunocomplejo directamente en la mezcla de reacción.
Por ejemplo, dichas tecnologías se ofrecen bajo los nombres comerciales TRACE® (Emisión de criptato amplificada resuelta en el tiempo) o KRYPTOR®, implementando las enseñanzas de las aplicaciones citadas anteriormente. Por lo tanto, en aspectos particularmente preferidos, se usa un dispositivo de diagnóstico para llevar a cabo el método proporcionado en la presente descripción. Por ejemplo, se determina el nivel de la proteína proADM o un fragmento de la misma, y/o el nivel de cualquier marcador adicional del método proporcionado en la presente descripción. En aspectos particularmente preferidos, el dispositivo de diagnóstico es k Ry Pt OR®.
En una modalidad del método descrito en la presente descripción, el método es un inmunoensayo y en donde el ensayo se realiza en fase homogénea o en fase heterogénea.
En modalidades adicionales del método descrito en la presente descripción, el método comprende además un análisis molecular de una muestra de dicho paciente para detectar una infección. La muestra usada para el análisis molecular para detectar una infección es preferentemente una muestra de sangre o un fragmento de la misma, tal como suero, plasma o sangre total. En una modalidad preferida, el análisis molecular es un método que tiene como objetivo detectar una o más biomoléculas derivadas de un patógeno. Dicha una o más biomoléculas pueden ser un ácido nucleico, una proteína, un azúcar, carbohidratos, un lípido y/o una combinación de los mismos, tal como una proteína glicosilada, preferentemente un ácido nucleico. Preferentemente, dicha biomolécula es específica para uno o más patógenos. De acuerdo con las modalidades preferidas, dichas biomoléculas se detectan mediante uno o más métodos de análisis de biomoléculas seleccionados del grupo que comprende los métodos de amplificación de ácidos nucleicos tales como PCR, qPCR, RT-PCR, qRT-PCR, secuenciación de alto rendimiento (tal como NGS) o amplificación isotérmica, espectrometría de masas, detección de actividad enzimática y métodos de detección basados en inmunoensayos. Otros métodos de análisis molecular son conocidos por los expertos en la técnica y están comprendidos en el método de la presente invención.
En una modalidad del método descrito en la presente descripción, un primer anticuerpo y un segundo anticuerpo están presentes dispersos en una mezcla de reacción líquida, y en donde un primer componente de marcaje que forma parte de un sistema de marcaje basado en la inactivación o amplificación de la fluorescencia o la quimioluminiscencia se une al primer anticuerpo, y un segundo componente de marcaje de dicho sistema de marcaje se une al segundo anticuerpo de manera que, después de la unión de ambos anticuerpos a dicha proADM o fragmentos de la misma a detectarse, una señal medible que permite la detección de los complejos sándwich resultantes en la solución de medición es generado.
En una modalidad del método descrito en la presente descripción, el sistema de marcaje comprende un criptato o quelato de tierras raras en combinación con un colorante fluorescente o quimioluminiscente, en particular del tipo cianina.
En una modalidad del método descrito en la presente descripción, el método comprende además comparar el nivel
determinado de proADM o fragmento(s) de la misma con un nivel de referencia, valor umbral y/o un promedio de población correspondiente a proADM o fragmentos de la misma en pacientes que han sido diagnosticados como gravemente enfermos y bajo tratamiento médico o que corren el riesgo de contraer o tener un sistema de coagulación desregulado, en donde dicha comparación se lleva a cabo en un procesador informático que usa un código ejecutable por ordenador.
Los métodos de la presente invención pueden implementarse en parte por ordenador. Por ejemplo, la etapa de comparar el nivel detectado de un marcador, por ejemplo, la proADM o fragmentos de la misma, con un nivel de referencia puede realizarse en un sistema informático. En el sistema informático, el nivel determinado de los marcadores puede combinarse con otros niveles de marcadores y/o parámetros del sujeto para calcular una puntuación, que es indicativa para el diagnóstico, pronóstico, evaluación de riesgos y/o estratificación del riesgo, orientación del tratamiento y manejo del paciente. Por ejemplo, los valores determinados pueden introducirse (ya sea manualmente por un profesional de la salud o automáticamente desde el dispositivo o dispositivos en los que se ha determinado el nivel o niveles de marcador respectivos) en el sistema informático. El sistema informático puede estar directamente en el punto de atención (por ejemplo, atención primaria, UCI o ED) o puede estar en una ubicación remota conectada a través de una red informática (por ejemplo, a través de Internet o sistemas médicos especializados en la nube, opcionalmente, combinable con otros sistemas informáticos o plataformas tales como los sistemas de información hospitalaria (HIS)). Normalmente, el sistema informático almacenará los valores (por ejemplo, nivel de marcador o parámetros tales como edad, presión arterial, peso, sexo, etc., o sistemas de puntuación clínica tales como SOFA, qSOFA, IMC, PCT, recuento de plaquetas, etc.) en un medio legible por ordenador y calcular la puntuación en base a niveles de referencia o valores de referencia previamente definidos y/o almacenados previamente. La puntuación resultante se mostrará y/o imprimirá para el usuario (normalmente, un profesional de la salud, tal como un médico). Alternativamente o, además, el pronóstico asociado, el diagnóstico, la evaluación, la orientación del tratamiento, la orientación de manejo del paciente o la estratificación se mostrarán y/o imprimirán para el usuario (típicamente un profesional de la salud tal como un médico o una enfermera).
En una modalidad de la invención, puede emplearse un sistema de software, en donde se evidencia un algoritmo de aprendizaje automático, preferentemente para identificar los pacientes hospitalizados con riesgo de sepsis, sepsis grave y choque séptico mediante el uso de los datos de registros de salud electrónicos (EHR). Puede entrenarse un enfoque de aprendizaje automático en un clasificador de bosque aleatorio mediante los datos de EHR (tales como laboratorios, expresión de biomarcadores, signos vitales y datos demográficos) de los pacientes. El aprendizaje automático es un tipo de inteligencia artificial que proporciona a los ordenadores la capacidad de aprender patrones complejos en los datos sin ser programados explícitamente, a diferencia de los sistemas más simples basados en reglas. Estudios anteriores han usado los datos de registros de salud electrónicos para activar alertas para detectar el deterioro clínico en general. En una modalidad de la invención, el procesamiento de los niveles de proADM puede incorporarse en el software apropiado para compararlos con los conjuntos de datos existentes, por ejemplo, los niveles de proADM también pueden procesarse en el software de aprendizaje automático para ayudar a diagnosticar o pronosticar la aparición de un evento adverso, coagulación desregulada tal como trombocitopenia o DIC.
El empleo combinado de la proADM o fragmentos de la misma en combinación con otro biomarcador tal como PCT o CRP puede realizarse en un solo ensayo multiplex o en dos ensayos separados realizados en una muestra del paciente. La muestra puede estar relacionada con la misma muestra o con muestras diferentes. El ensayo empleado para la detección y determinación de la proADM y, por ejemplo, PCT también puede ser el mismo o diferente, por ejemplo, puede emplearse un inmunoensayo para la determinación de uno de los marcadores anteriores. A continuación, se proporcionan las descripciones más detalladas de ensayos adecuados.
Los valores de corte y otros niveles de referencia de la proADM o fragmentos de la misma en pacientes que han sido diagnosticados como gravemente enfermos y están bajo tratamiento o que están en riesgo de contraer o tener un sistema de coagulación desregulado pueden determinarse mediante los métodos descritos anteriormente. Por ejemplo, los expertos conocen los métodos para usar el coeficiente de variación en la evaluación de la variabilidad de los ensayos cuantitativos para establecer los valores de referencia y/o valores de corte (George F. Reed y otros, Clin Diagn Lab Immunol.2002; 9(6):1235-1239).
Además, la sensibilidad del ensayo funcional puede determinarse para indicar los valores estadísticamente significativos para usar como niveles de referencia o valores de corte de acuerdo con las técnicas establecidas. Los laboratorios son capaces de establecer de forma independiente la sensibilidad funcional de un ensayo mediante un protocolo clínicamente relevante. La "sensibilidad funcional" puede considerarse como la concentración que da como resultado un coeficiente de variación (CV) del 20 % (o algún otro % CV previamente determinado) y, por lo tanto, es una medida de la precisión de un ensayo a niveles bajos de analito. Por lo tanto, el CV es una estandarización de la desviación estándar (SD) que permite la comparación de las estimaciones de variabilidad independientemente de la magnitud de la concentración del analito, al menos en la mayor parte del intervalo de trabajo del ensayo.
Además, los métodos basados en el análisis ROC pueden usarse para determinar las diferencias estadísticamente significativas entre dos grupos clínicos de pacientes. Las curvas de características operativas del receptor (ROC) miden la eficiencia de clasificación de las probabilidades ajustadas del modelo para clasificar los niveles de respuesta. Las curvas ROC también pueden ayudar a establecer los puntos de criterio en las pruebas de
diagnóstico. Cuanto más alta sea la curva desde la diagonal, mejor será el ajuste. Si el ajuste logístico tiene más de dos niveles de respuesta, produce una curva ROC generalizada. En tal gráfico, hay una curva para cada nivel de respuesta, que es la curva ROC de ese nivel frente a todos los demás niveles. Se dispone de software capaz de permitir este tipo de análisis para establecer los niveles de referencia y valores de corte adecuados, por ejemplo, el software estadístico R (versión 3.1.2), JMP 12, JMP 13, Statistical Discovery, de SAS.
Los valores de corte pueden determinarse de manera similar para PCT. La literatura está disponible para una persona capacitada para determinar un valor de corte apropiado, por ejemplo, Philipp Schuetz y otros. (BMC Medicine. 2011; 9:107) describen que a un valor de corte de 0,1 ng/ml, la PCT tenía una sensibilidad muy alta para excluir la infección. Terence Chan y otros. (Expert Rev.Mol.Diagn.2011; 11(5), 487.496) describieron que indicadores como los cocientes de probabilidad positivos y negativos, que se calculan en función de la sensibilidad y la especificidad, también son útiles para evaluar la solidez de una prueba diagnóstica. Los valores se grafican comúnmente para los múltiples valores de corte (CV) como una curva característica operativa del receptor. El valor del área bajo la curva se usa para determinar el mejor CV relevante para el diagnóstico. Esta bibliografía describe la variación de los CV (valores de corte, que dependen del ensayo y del diseño del estudio) y los métodos adecuados para determinar los valores de corte.
Los niveles promedio de la población de proADM o fragmentos de la misma también pueden usarse como valores de referencia, por ejemplo, los valores medios de la población de proADM, por lo que los pacientes diagnosticados como gravemente enfermos, tales como los pacientes con una desregulación del sistema de coagulación, pueden compararse con una población control, en donde el grupo control comprende preferentemente más de 10, 20, 30, 40, 50 o más sujetos.
En una modalidad de la invención, el valor de corte para PCT puede ser un valor en el intervalo de 0,01 a 100,00 ng/ml en una muestra de suero, cuando se usa, por ejemplo, un ensayo Luminex MAC Pix E-Bioscience o el ensayo BRAHMS PCT Kryptor.
En una modalidad preferida, el valor de corte de PCT puede estar en el intervalo de 0,01 a 100, 0,05 a 50, 0,1 a 20 o 0,1 a 2 ng/ml, y con la máxima preferencia > 0,05 a 10 ng/ml. Cualquier valor dentro de estos intervalos puede considerarse como un valor de corte apropiado. Por ejemplo, pueden emplearse 0,01, 0,05, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 or 100 ng/ml. En algunas modalidades, los niveles de PCT para sujetos sanos son de aproximadamente 0,05 ng/ml.
Como se deriva del estudio más abajo, a modo de ejemplo, los datos muestran que los pacientes sépticos con niveles anormalmente bajos de plaquetas tienen un valor de PCT >7 ng/ml.
Las ventajas y modalidades de cada uno de los diversos métodos y kits de la invención descritos en la presente descripción también se aplican y se basan en los respectivos otros métodos y kits.
Modalidades de la invención relativas a la determinación de un método para el seguimiento de una terapia, que comprende el pronóstico, la evaluación del riesgo y/o la estratificación del riesgo de un evento adverso posterior en la salud de un paciente
Como se describió anteriormente, un evento adverso posterior en algunas modalidades es uno o más de trombocitopenia, DIC, infección, insuficiencia de órganos, disfunción orgánica y/o mortalidad.
La invención se refiere a un método para el seguimiento de la terapia, que comprende el pronóstico, la evaluación del riesgo y/o la estratificación del riesgo de un evento adverso posterior en la salud de un paciente, que comprende - proporcionar una muestra de dicho paciente, en donde el paciente ha sido diagnosticado como gravemente enfermo y/o se ha iniciado un tratamiento médico, en donde la muestra se aísla del paciente después del diagnóstico y del inicio del tratamiento,
- determinar un nivel de proADM o fragmento(s) de la misma en dicha muestra,
- en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la probabilidad de un evento adverso posterior en la salud de dicho paciente.
En una modalidad, los pacientes del método de la presente invención ya han sido diagnosticados como gravemente enfermos y ya están recibiendo tratamiento. Por lo tanto, el método de la presente invención puede usarse para controlar el éxito del tratamiento o terapia que se ha iniciado, en base a la determinación de la probabilidad de un evento adverso subsiguiente. El seguimiento de la terapia implica preferentemente el pronóstico de un evento adverso y/o la estratificación del riesgo o evaluación del riesgo del paciente con respecto a un futuro evento adverso, en donde esta evaluación del riesgo y la determinación de dicho riesgo debe considerarse como un medio de seguimiento de la terapia iniciada.
Los médicos o el personal médico que tratan a los pacientes a los que se les ha diagnosticado como gravemente enfermo pueden emplear el método de la presente invención en diferentes entornos clínicos, tal como un entorno de atención primaria o, preferentemente, en un entorno hospitalario, tal como un departamento de emergencias o en una unidad de cuidados intensivos (UCI). El método es muy útil para monitorear el efecto de una terapia que se ha iniciado en un paciente gravemente enfermo y puede usarse para juzgar si un paciente en tratamiento es un paciente de alto riesgo que debe estar bajo observación médica intensa y potencialmente debe recibir terapia adicional o si el paciente es un paciente de bajo riesgo con un estado de salud en mejora que podría no requerir una observación tan intensa y medidas de tratamiento adicionales, posiblemente porque el tratamiento iniciado está mejorando con éxito el estado del paciente. Los tratamientos iniciales de pacientes gravemente enfermos pueden tener un efecto directo sobre la probabilidad de eventos adversos en la salud del paciente. Como tal, la evaluación del riesgo/pronóstico de un evento adverso futuro proporciona retroalimentación o monitoreo de la terapia instigada. La probabilidad de que ocurra un evento adverso posterior puede evaluarse al comparar el nivel de proADM o fragmentos de la misma en la muestra con un nivel de referencia (tal como un umbral o un valor de corte y/o un promedio de la población), en donde el nivel de referencia puede corresponder a la proADM o fragmentos de la misma en pacientes sanos, o en pacientes que han sido diagnosticados como gravemente enfermos.
En consecuencia, el método de la presente invención puede ayudar a predecir la probabilidad de un evento adverso posterior en la salud del paciente. Esto significa que el método de la invención puede discriminar pacientes de alto riesgo, que tienen más probabilidades de sufrir complicaciones, o cuyo estado será más crítico en el futuro, de pacientes de bajo riesgo, cuyo estado de salud es estable o incluso mejora, por lo que no se espera que sufran un evento adverso, tal como la muerte del paciente o un deterioro de los síntomas o signos clínicos del paciente, que puedan requerir determinadas medidas terapéuticas.
Una ventaja particular del método de la presente invención es que un paciente que ha sido identificado como un paciente de bajo riesgo por medio del método de la presente invención podría ser dado de alta más rápidamente de una UCI, del hospital en general o puede requerir menos supervisión frecuente. Asimismo, para pacientes de bajo riesgo, podría disminuirse la intensidad y/o frecuencia de la observación del estado de salud del paciente. En consecuencia, el hospital u otra institución médica a cargo del paciente podría decidir de manera más eficiente qué pacientes requieren cuidados médicos intensivos y observación. En consecuencia, el hospital o institución respectiva podría, por ejemplo, ocupar de manera más eficiente las camas de la UCI con pacientes de alto riesgo. Esto conduciría a una mejor atención médica para los pacientes de alto riesgo, ya que el personal médico podría concentrarse en dichos pacientes, mientras que los pacientes de bajo riesgo podrían ser dados de alta de la UCI. Esto también conduciría a beneficios significativos por costos evitados por medidas innecesarias que de cualquier otra manera se aplicarían a pacientes de bajo riesgo.
El momento en que los pacientes han sido diagnosticados como gravemente enfermos y se inician las primeras medidas de tratamiento se define como "momento 0", que puede ser la referencia para el momento de aislamiento de la muestra usada para la determinación de la proADM o fragmentos de la misma. Si el diagnóstico del paciente y el inicio del tratamiento no ocurren al mismo tiempo, el momento 0 es el momento cuando ocurre el último de los dos eventos de diagnóstico e inicio del tratamiento médico. Por lo general, el diagnóstico de pacientes gravemente enfermos es inmediatamente seguido o concomitante al inicio de la terapia o intervenciones médicas tales como cirugía y/o control de la fuente (por ejemplo, eliminación de tejido necrótico). En el caso de disfunción coagulativa, el punto de partida de las intervenciones puede variar de vez en cuando en dependencia de la gravedad de la afección u otras complicaciones.
Fue completamente sorprendente que el nivel de la proADM o fragmentos de la misma en una muestra del paciente pueda proporcionar información crítica sobre la probabilidad de que ocurra un evento adverso posterior en la salud de dichos pacientes gravemente enfermos. No ha habido indicios de que una sola medición de proADM o fragmentos de la misma después del diagnóstico y el inicio del tratamiento de un paciente gravemente enfermo pueda proporcionar información tan importante con respecto al éxito del tratamiento en curso y el pronóstico del estado de salud del paciente.
El uso de la proADM o fragmentos de la misma como parámetro único en modalidades de la presente invención es ventajoso sobre el uso de otros parámetros únicos, tales como biomarcadores o puntuaciones clínicas, ya que la proADM es más precisa en la predicción de un evento adverso en comparación con otros marcadores tales como el recuento de plaquetas, PCT, PCR, lactato o puntuaciones clínicas tales como SOFA, SAPS II o APACHE II, y marcadores de desregulación del sistema de coagulación, tales como micropartículas de membrana, recuento de plaquetas, volumen plaquetario medio (MPV), sCD14-ST, protrombinasa, antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor de von Willebrand (vWF), lipoproteínas en combinación con PCR, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero de fibrina D no desnaturalizada, factor plaquetario 4, histonas y un ensayo PT.
De acuerdo con una modalidad preferida, la muestra se aísla de un paciente durante un examen médico.
De acuerdo con una modalidad preferida, la muestra se aísla de dicho paciente dentro de los 30 minutos posteriores
a dicho diagnóstico e inicio del tratamiento, o al menos 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 días o 10 días después de dicho diagnóstico e inicio del tratamiento. En otras modalidades, la muestra se aísla de dicho paciente 12-36 horas y/o 3-5 días después del inicio del tratamiento.
El hecho de que el nivel de proADM o fragmentos de la misma en un momento tan breve como unos 30 minutos después del diagnóstico y el inicio del tratamiento pueda proporcionar tal información fue completamente inesperado.
En modalidades preferidas del método de la presente invención dicha muestra se aísla de dicho paciente aproximadamente 30 minutos, 1 hora, 2 horas, 3 horas, 4 horas, 5 horas, 6 horas, 7 horas, 8 horas, 9 horas, 10 horas, 11 horas, 12 horas, 14 horas, 16 horas, 18 horas, 20 horas 22 horas, 24 horas, 30 horas, 36 horas, 42 horas, 48 horas, 60 horas, 72 horas, 84 horas, 4 días, 5 días, 6 días, 7 días, 8 días, 9 días o 10 días después de dicho diagnóstico e inicio del tratamiento.
En otras modalidades, la muestra se aísla en momentos posteriores a dicho diagnóstico e iniciando el tratamiento antibiótico de 30 minutos a 12 horas, 12-36 horas, 3-5 días, 7-14 días, 8-12 días o 9-11 días.
Pueden emplearse intervalos entre cualquiera de los valores anteriores para definir el momento de obtención de la muestra.
En otra modalidad preferida de la presente invención, el paciente ha sido diagnosticado mediante el uso de al menos un biomarcador adicional o una puntuación clínica. Es particularmente ventajoso en el contexto de la presente invención, si el diagnóstico inicial de la enfermedad crítica del paciente en el momento 0 se basó al menos parcialmente en el nivel de al menos un biomarcador o una puntuación clínica determinada.
En determinadas modalidades, la presente invención comprende la determinación de parámetros adicionales, como marcadores, biomarcadores, puntuaciones clínicas o similares.
En otra modalidad preferida de la presente invención, el paciente ha sido diagnosticado mediante el uso de al menos uno de los biomarcadores o puntuaciones clínicas de la procalcitonina (PCT), lactato y proteína C reactiva y/o al menos una de las puntuaciones clínicas de SOFA, APACHE II, SAPS II y marcadores de desregulación del sistema de coagulación, tales como micropartículas de membrana, recuento de plaquetas, volumen plaquetario medio (MPV), sCD14-ST, protrombinasa, antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor von Willebrand (vWF), lipoproteínas en combinación con PCR, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D no desnaturalizado de fibrina, factor plaquetario 4, histonas y un ensayo de PT. En modalidades, uno o más marcadores adicionales comprenden una o más histonas. La determinación de la proADM o fragmentos de la misma en las muestras de pacientes que han sido diagnosticados como gravemente enfermos y están en tratamiento ha demostrado ser especialmente útil para el seguimiento de la terapia si el diagnóstico del paciente se ha basado en estos marcadores, ya que el pronóstico de un evento adverso en tales grupos de pacientes puede ser más preciso en comparación con los pacientes en estado crítico que han sido diagnosticados por otros medios.
En una modalidad de la invención, el paciente gravemente enfermo es un paciente diagnosticado o que corre el riesgo de desarrollar una desregulación del sistema de coagulación, una enfermedad infecciosa, un paciente diagnosticado con una enfermedad infecciosa y una o más insuficiencias inorgánicas existentes, un paciente diagnosticado de sepsis, sepsis grave o choque séptico y/o un paciente postraumático o posquirúrgico. A la luz de los datos presentados en la presente descripción, el valor pronóstico de proADM en muestras de estos grupos de pacientes es particularmente preciso para predecir la probabilidad de un evento adverso en estos pacientes.
En modalidades preferidas de la presente invención, el evento adverso en la salud de dicho paciente es la muerte, preferentemente la muerte dentro de los 28-90 días posteriores al diagnóstico e inicio del tratamiento, una nueva infección, insuficiencia de órganos y/o un deterioro de los síntomas clínicos que requieren un enfoque del procedimiento de limpieza, transfusión de hemoderivados, infusión de coloides, cirugía de emergencia, ventilación mecánica invasiva, reacciones adversas a medicamentos y/o reemplazo renal o hepático.
En modalidades preferidas de la invención, dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la probabilidad de un evento adverso posterior en la salud de dicho paciente dentro de los 28 días posteriores al diagnóstico e inicio del tratamiento. En modalidades preferidas adicionales de la invención, dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la probabilidad de un evento adverso posterior en la salud de dicho paciente dentro de los 90 días posteriores al diagnóstico e inicio del tratamiento.
En determinadas modalidades de la invención, el tratamiento que recibe el paciente comprende uno o más de tratamiento antibiótico o antiinfeccioso, ventilación mecánica invasiva, ventilación mecánica no invasiva, terapia de reemplazo renal, uso de vasopresores, terapia de fluidos, transfusión de plaquetas, transfusión de sangre, purificación de sangre extracorpórea, control de fuentes y/o protección de órganos.
En modalidades preferidas de la invención, la muestra se selecciona del grupo que consiste en una muestra de sangre o una fracción de la misma, una muestra de suero, una muestra de plasma y/o una muestra de orina.
En modalidades adicionales de la invención, el nivel de proADM o fragmento(s) de la misma se correlaciona con la probabilidad de un evento adverso posterior en la salud de dicho paciente. En una modalidad preferida, el nivel de proADM o fragmento(s) de la misma se correlaciona positivamente con la probabilidad de un evento adverso posterior en la salud de dicho paciente. En otras palabras, cuanto mayor sea el nivel de proADM determinado, mayor será la probabilidad de un evento adverso posterior.
De acuerdo con una modalidad preferida de la presente invención,
- un nivel de gravedad bajo de la proADM o fragmento(s) de la misma es indicativo de la ausencia de un evento adverso posterior, o indica un riesgo bajo de un evento adverso posterior, en donde el nivel de gravedad bajo está por debajo de 4 nmol/l, preferentemente por debajo de 3 nmol/l, con mayor preferencia por debajo de 2,7 nmol/l, o
- un nivel de gravedad alto de proADM o fragmento(s) de la misma es indicativo de un evento adverso posterior, o indica un riesgo alto de un evento adverso posterior, en donde el nivel de gravedad alto es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l.
De acuerdo con una modalidad preferida de la presente invención,
- un nivel de proADM o fragmento(s) de la misma por debajo de 4 nmol/l, preferentemente por debajo de 3 nmol/l, con mayor preferencia por debajo de 2,7 nmol/l, es indicativo de la ausencia de un evento adverso posterior, o indica un riesgo bajo de un evento posterior evento adverso, o
- un nivel de proADM o fragmento(s) de la misma por encima de 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, es indicativo de un evento adverso posterior, o indica un alto riesgo de un evento adverso posterior.
De acuerdo con una modalidad preferida de la presente invención,
- un nivel de gravedad bajo de proADM o fragmento(s) de la misma es indicativo de la ausencia de un evento adverso posterior, en donde el nivel de gravedad bajo es inferior a 2,7 nmol/l, o
- un nivel de gravedad alto de proADM o fragmento(s) de la misma es indicativo de un evento adverso posterior, en donde el nivel de gravedad alto es superior a 10,9 nmol/l.
Esta modalidad de la presente invención es particularmente ventajosa cuando los niveles de proADM o fragmentos de la misma se determinan en una muestra que ha sido aislada el día del diagnóstico e inicio del tratamiento del paciente, particularmente alrededor de 30 minutos después del diagnóstico e inicio del tratamiento.
De acuerdo con una modalidad preferida de la presente invención,
- un nivel de gravedad bajo de proADM o fragmento(s) de la misma es indicativo de la ausencia de un evento adverso posterior, en donde el nivel de gravedad bajo es inferior a 2,7 nmol/l, o
- un nivel de gravedad alto de proADM o fragmento(s) de la misma es indicativo de un evento adverso posterior, en donde el nivel de gravedad alto es superior a 10,9 nmol/l,
- en donde el nivel de proADM o fragmentos de la misma se determina en una muestra que ha sido aislada preferentemente el día del diagnóstico e inicio del tratamiento.
De acuerdo con una modalidad preferida de la presente invención,
- un nivel de gravedad bajo de proADM o fragmento(s) de la misma es indicativo de la ausencia de un evento adverso posterior, en donde el nivel de gravedad bajo es inferior a 2,8 nmol/l, o
- un nivel de gravedad alto de proADM o fragmento(s) de la misma es indicativo de un evento adverso posterior, en donde el nivel de gravedad alto es superior a 9,5 nmol/l.
Esta modalidad de la presente invención resulta especialmente ventajosa cuando se determinan los niveles de proADM o fragmentos de la misma en una muestra que ha sido aislada 1 día después de dicho diagnóstico e inicio del tratamiento.
De acuerdo con una modalidad preferida de la presente invención,
- un nivel de gravedad bajo de proADM o fragmento(s) de la misma es indicativo de la ausencia de un evento adverso posterior, en donde el nivel de gravedad bajo es inferior a 2,8 nmol/l, o
- un nivel de gravedad alto de proADM o fragmento(s) de la misma es indicativo de un evento adverso posterior, en donde el nivel de gravedad alto es superior a 9,5 nmol/l,
- en donde el nivel de proADM o fragmentos de la misma se determina en una muestra que ha sido aislada preferentemente 1 día después del diagnóstico e inicio del tratamiento.
Por ejemplo, si el nivel de proADM o fragmentos de la misma cae en la categoría de un nivel de proADM de gravedad baja, el médico tratante puede decidir con más confianza dar de alta a dicho paciente de la UCI, porque es poco probable que un evento adverso en la salud de dicho paciente ocurriría, preferentemente, dentro de los próximos 28 días, con mayor preferencia dentro de los próximos 90 días. En consecuencia, podría no ser necesario mantener a este paciente en la UCI. También podría ser posible concluir que el tratamiento en curso está mejorando con éxito el estado de salud del paciente, según lo evaluado por una medición del riesgo de un evento adverso. Por el contrario, si la determinación del nivel de proADM o fragmentos de la misma de dicho paciente de la UCI indica un nivel de proADM o fragmentos de la misma de alta gravedad, el médico tratante debe mantener al paciente en la UCI. Adicionalmente, debe considerarse ajustar el tratamiento del paciente, ya que es probable que el tratamiento actual no esté mejorando el estado de salud del paciente, por lo que es más probable que el paciente sufra un evento adverso en el futuro.
De acuerdo con una modalidad particularmente preferida de la presente invención, el nivel de gravedad bajo está por debajo de 2,75 nmol/l, dicha muestra se aísla del paciente de la UCI 1 día o más después de dicho diagnóstico e inicio del tratamiento, y el nivel de gravedad bajo de la proADM o fragmento(s) de la misma indica el alta de dicho paciente de la UCI.
La presente invención se refiere además a un método para el seguimiento de la terapia, que comprende el pronóstico, la evaluación del riesgo y/o la estratificación del riesgo de un evento adverso posterior en la salud de un paciente, que comprende
- proporcionar una muestra de dicho paciente, en donde el paciente es un paciente de la unidad de cuidados intensivos (UCI) y se ha iniciado un tratamiento médico, en donde la muestra se aísla del paciente después del ingreso en la UCI y el inicio del tratamiento,
- determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra,
- en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con la probabilidad de un evento adverso posterior en la salud de dicho paciente.
En el contexto del método de la presente invención relacionado con pacientes de la UCI, la referencia para el momento de aislamiento de la muestra usada para determinar la proADM o fragmentos de la misma es el momento cuando los pacientes son admitidos en la UCI y se inician las mediciones para el primer tratamiento (momento 0). Este momento corresponde momento de diagnóstico e inicio del tratamiento en el método de la presente invención en relación con pacientes que han sido diagnosticados como gravemente enfermos.
Todas las modalidades del método de la presente invención relacionadas con pacientes que han sido diagnosticados como gravemente enfermos también se consideran correspondientes a las modalidades del método de la presente invención relacionadas con pacientes de la UCI.
Modalidades de la invención relacionadas con la determinación adicional de un nivel de PCT y/u otros biomarcadores o puntuaciones clínicas en una primera y una segunda muestra (o en el momento del aislamiento de una primera y una segunda muestra)
Una modalidad preferida de la presente invención comprende adicionalmente determinar un nivel de PCT o fragmento(s) de la misma en una muestra aislada del paciente. En una modalidad preferida, la muestra para determinar un nivel de PCT o fragmento(s) de la misma se aísla antes, en o después del momento del diagnóstico y del inicio del tratamiento.
Es particularmente ventajoso combinar la determinación de la proADM o fragmentos de la misma con la determinación de PCT o fragmentos de la misma en una muestra, en donde la muestra usada para determinar la proADM puede ser la misma o una muestra diferente usada para detectar PCT.
La determinación combinada de la proADM o fragmentos de la misma con la determinación de PCT o fragmentos de la misma, ya sea en la misma muestra o en muestras obtenidas en diferentes momentos, proporciona un efecto sinérgico con respecto a la precisión y confiabilidad de determinar el riesgo de un evento adverso posterior. Estos efectos sinérgicos también existen para la evaluación combinada de la proADM o sus fragmentos con otros marcadores o puntuaciones clínicas, tales como recuentos de plaquetas, volumen plaquetario medio (MPV), lactato, PCR, qSOFA, SOFA, SAPS II, APACHE II u otras evaluaciones.
De acuerdo con otra modalidad preferida de la presente invención, el método descrito en la presente descripción comprende adicionalmente
- determinar un nivel de PCT o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla antes, en o después del momento del diagnóstico y del inicio del tratamiento, - determinar un nivel de PCT o fragmento(s) de la misma en una segunda muestra aislada de dicho paciente, en donde la segunda muestra ha sido aislada después de la primera muestra, preferentemente dentro de los 30 minutos posteriores al aislamiento de la primera muestra o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después del aislamiento de la primera muestra, y
- determinar una diferencia en el nivel de PCT o fragmento(s) de la misma en la segunda muestra en comparación con el nivel de PCT o fragmento(s) de la misma en la primera muestra.
Es especialmente ventajoso combinar la determinación de la proADM o fragmentos de la misma en una muestra aislada de un paciente con la determinación de PCT o fragmentos de la misma en una primera muestra y la determinación del nivel de PCT o fragmentos de la misma en una segunda muestra aislada después de la primera muestra, en donde la muestra usada para la determinación de la proADM o fragmentos de la misma puede ser la misma o diferente de la primera muestra o la segunda muestra usada para determinar PCT o fragmentos de la misma.
En una modalidad preferida el método descrito en la presente descripción comprende adicionalmente
- determinar un nivel de PCT o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla en o antes del momento del diagnóstico y el inicio del tratamiento (momento 0), - determinar un nivel de PCT o fragmento(s) de la misma en una segunda muestra (muestra de la reivindicación 1) aislada de dicho paciente después del diagnóstico e inicio del tratamiento, preferentemente dentro de los 30 minutos posteriores a dicho diagnóstico e inicio del tratamiento o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después de dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en el nivel de PCT o fragmento(s) de la misma en la segunda muestra en comparación con el nivel de PCT o fragmento(s) de la misma en la primera muestra.
Es especialmente ventajoso combinar la determinación de la proADM o fragmentos de la misma (en una segunda muestra) con la determinación de PCT o fragmentos de la misma en una muestra anterior (primera muestra) que se aísla de dicho paciente y que puede ser usada para el diagnóstico de dicho paciente como gravemente enfermo en el momento 0 y determinar el nivel de PCT o fragmentos de la misma en una segunda muestra aislada en un determinado momento después del diagnóstico y el inicio del tratamiento, que también es preferentemente el mismo momento cuando se determina la proADM o fragmentos de la misma. Como indican los datos más abajo, determinar una diferencia en el nivel de PCT o fragmentos de la misma en la segunda muestra en comparación con la primera muestra adiciona información adicional a la información obtenida de los niveles de proADM o fragmentos de la misma en la segunda muestra. En base a esta información combinada, podría ser posible predecir con una mayor probabilidad si ocurrirá un evento adverso en la salud de dicho paciente en comparación con predecir la probabilidad de un evento adverso basándose únicamente en la información sobre el nivel de proADM o fragmentos de la misma en la segunda muestra. Esto representa un hallazgo sorprendente, ya que los biomarcadores de sepsis normalmente no son sinérgicos ni complementarios, sino que representan meros marcadores de diagnóstico alternativos.
En una modalidad preferida el método descrito en la presente descripción comprende adicionalmente
- determinar un recuento de plaquetas en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla en o antes del momento del diagnóstico y del inicio del tratamiento (momento 0),
- determinar un recuento de plaquetas en una segunda muestra (muestra de la reivindicación 1) aislada de dicho paciente dentro de los 30 minutos posteriores a dicho diagnóstico e inicio del tratamiento o al menos 30 minutos, preferentemente 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después de dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en el recuento de plaquetas en la segunda muestra en comparación con el recuento de plaquetas en la primera muestra.
Es particularmente ventajoso combinar la determinación de la proADM o fragmentos de la misma con la determinación de los recuentos de plaquetas en una muestra, en donde la muestra usada para determinar la proADM puede ser la misma o una muestra diferente usada para detectar los recuentos de plaquetas.
Una modalidad preferida de la presente invención comprende determinar adicionalmente SOFA o qSOFA. En una modalidad preferida, qSOFA o SOFA se determina antes, en o después del momento del diagnóstico y del inicio del tratamiento.
Es particularmente ventajoso combinar la determinación de proADM o fragmentos de la misma con la determinación de SOFA, en donde el momento de aislamiento de la muestra para determinar la proADM puede ser el mismo o diferente del momento de determinación de SOFA.
De acuerdo con otra modalidad preferida de la presente invención, el método descrito en la presente descripción comprende adicionalmente
- determinar un primer SOFA antes, en o después del momento del diagnóstico y del inicio del tratamiento, - determinar un segundo SOFA dentro de los 30 minutos posteriores a la determinación del primer SOFA o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días posteriores a la determinación del primer SOFA, y
- determinar una diferencia en los dos SOFA determinados.
En una modalidad preferida el método descrito en la presente descripción comprende adicionalmente
- determinar SOFA en o antes del momento de diagnóstico e inicio del tratamiento (momento 0),
- determinar SOFA dentro de los 30 minutos posteriores a dicho diagnóstico e inicio del tratamiento o al menos 30 minutos, preferentemente 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días posteriores a dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en SOFA determinado después de dicho diagnóstico e inicio del tratamiento y SOFA determinado en el momento 0.
Una modalidad preferida de la presente invención comprende la determinación adicional de SAPS II. En una modalidad preferida, SAPS II se determina antes, en o después del momento del diagnóstico y del inicio del tratamiento.
Es particularmente ventajoso combinar la determinación de la proADM o fragmentos de la misma con la determinación de SAPS II, en donde el momento de aislamiento de la muestra para determinar la proADM puede ser el mismo o diferente del momento de determinación de SAPS II.
De acuerdo con otra modalidad preferida de la presente invención, el método descrito en la presente descripción comprende adicionalmente
- determinar un primer SAPS II antes, en o después del momento del diagnóstico y del inicio del tratamiento, - determinar un segundo SAPS II dentro de los 30 minutos posteriores a la determinación del primer SOFA o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días posteriores a la determinación del primer SAPS II, y
- determinar una diferencia en los dos SAPS II determinados.
En una modalidad preferida el método descrito en la presente descripción comprende adicionalmente
- determinar SAPS II en o antes del momento del diagnóstico y el inicio del tratamiento (momento 0),
- determinar SAPS II dentro de los 30 minutos posteriores a dicho diagnóstico e inicio del tratamiento o al menos 30 minutos, preferentemente 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días posteriores a dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en SAPS II determinado después de dicho diagnóstico e inicio del tratamiento y SAPS
II determinado en el momento 0.
Una modalidad preferida de la presente invención comprende además la determinación de APACHE II. En una modalidad preferida, APACHE II se determina antes, durante o después del momento del diagnóstico y del inicio del tratamiento.
Es particularmente ventajoso combinar la determinación de la proADM o fragmentos de la misma con la determinación de APACHE II, en donde el momento de aislamiento de la muestra para determinar la proADM puede ser el mismo o diferente del momento de determinación de APACHE II.
De acuerdo con otra modalidad preferida de la presente invención, el método descrito en la presente descripción comprende adicionalmente
- determinar un primer APACHE II antes, en o después del momento del diagnóstico y del inicio del tratamiento,
- determinar un segundo APACHE II dentro de los 30 minutos posteriores a la determinación del primer APACHE II o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días posteriores a la determinación del primer APACHE II, y
- determinar una diferencia en los dos APACHE II determinados.
En una modalidad preferida el método descrito en la presente descripción comprende adicionalmente
- determinar APACHE II en o antes del momento del diagnóstico y el inicio del tratamiento (momento 0), - determinar APACHE II dentro de los 30 minutos posteriores a dicho diagnóstico e inicio del tratamiento o al menos 30 minutos, preferentemente 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días posteriores a dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en APACHE II determinado después de dicho diagnóstico e inicio del tratamiento y APACHE II determinado en el momento 0.
En una modalidad preferida, es posible identificar pacientes de alto riesgo con niveles crecientes de PCT y un nivel de gravedad alto de proADM o fragmentos de la misma, lo que representa una identificación más precisa de dichos pacientes que probablemente sufrirán un evento adverso en el futuro. En consecuencia, el tratamiento de estos pacientes podría ajustarse al minimizar el riesgo de que este paciente pudiera haber sido un paciente de bajo riesgo. Modalidades de la presente invención relacionadas con la determinación de un nivel de proADM o fragmento(s) de la misma en una primera y una segunda muestra
Una modalidad preferida del método de la presente invención comprende adicionalmente
- determinar un nivel de proADM o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla antes, en o después del momento del diagnóstico y del inicio del tratamiento, y
- determinar un nivel de proADM o fragmento(s) de la misma en una segunda muestra aislada de dicho paciente, en donde dicha segunda muestra ha sido aislada después de la primera muestra y después del momento de diagnóstico e inicio del tratamiento, preferentemente dentro de los 30 minutos posteriores al aislamiento de la primera muestra o 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después del aislamiento de la primera muestra, y
- determinar si es evidente una diferencia en el nivel de proADM o fragmento(s) de la misma en la segunda muestra en comparación con el nivel de proADM o fragmento(s) de la misma en la primera muestra.
La primera y la segunda muestra usadas para determinar el nivel de proADM o fragmento(s) de la misma pueden ser iguales o diferentes de la primera y la segunda muestra usadas para determinar el nivel de PCT o fragmento(s) de la misma.
Una modalidad preferida del método de la presente invención comprende adicionalmente
- determinar un nivel de proADM o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla en o antes del momento del diagnóstico y el inicio del tratamiento (momento 0), y
- determinar un nivel de proADM o fragmento(s) de la misma en una segunda muestra aislada después del diagnóstico e inicio del tratamiento, preferentemente dentro de los 30 minutos, o después de 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después de dicho diagnóstico e inicio del tratamiento, y - determinar si es evidente una diferencia en el nivel de proADM o fragmento(s) de la misma en la segunda
muestra en comparación con el nivel de proADM o fragmento(s) de la misma en la primera muestra.
Una modalidad preferida del método de la presente invención comprende adicionalmente
- determinar un nivel de proADM o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se usa para diagnosticar a dicho paciente como gravemente enfermo (momento 0), y
- determinar un nivel de proADM o fragmento(s) de la misma en una segunda muestra aislada después del diagnóstico e inicio del tratamiento, preferentemente dentro de los 30 minutos, o después de 30 minutos, 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después de dicho diagnóstico e inicio del tratamiento, y - determinar si es evidente una diferencia en el nivel de proADM o fragmento(s) de la misma en la segunda muestra en comparación con el nivel de proADM o fragmento(s) de la misma en la primera muestra.
Otra modalidad preferida del método de la presente invención comprende adicionalmente
- determinar un nivel de proADM o fragmento(s) de la misma y opcionalmente PCT o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se aísla en o antes del momento del diagnóstico y el inicio del tratamiento (momento 0), y
- determinar un nivel de proADM o fragmento(s) de la misma y opcionalmente PCT o fragmento(s) de la misma en una segunda muestra aislada de dicho paciente después de dicho diagnóstico e inicio del tratamiento, preferentemente dentro de los 30 minutos o al menos 30 minutos después del diagnóstico e inicio del tratamiento, preferentemente 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después de dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en el nivel de proADM o fragmento(s) de la misma y/o una diferencia en el nivel de PCT o fragmentos del mismo en la segunda muestra en comparación con el nivel de proADM o fragmento(s) de la misma en la primera muestra.
Otra modalidad preferida del método de la presente invención comprende adicionalmente
- determinar un nivel de proADM o fragmento(s) de la misma y opcionalmente PCT o fragmento(s) de la misma en una primera muestra aislada del paciente, en donde dicha primera muestra se usa para diagnosticar dicho paciente como gravemente enfermo (momento 0), y
- determinar un nivel de proADM o fragmento(s) de la misma y opcionalmente PCT o fragmento(s) de la misma en una segunda muestra aislada de dicho paciente después de dicho diagnóstico e inicio del tratamiento, preferentemente dentro de los 30 minutos o al menos 30 minutos después del diagnóstico e inicio del tratamiento, preferentemente 1 hora, 2 horas, 6 horas, 12 horas, 24 horas, 4 días, 7 o 10 días después de dicho diagnóstico e inicio del tratamiento, y
- determinar una diferencia en el nivel de proADM o fragmento(s) de la misma y/o una diferencia en el nivel de PCT o fragmentos de la misma en la segunda muestra en comparación con el nivel de proADM o fragmento(s) de la misma en la primera muestra.
Resultó sorprendente que la determinación del cambio de los niveles de proADM o fragmentos de la misma desde el momento del diagnóstico e inicio del tratamiento hasta un momento posterior pueda aportar información adicional con respecto a la aparición de un futuro evento adverso en la salud de un paciente que ha sido diagnosticado como gravemente enfermo, por ejemplo, el paciente ha sido diagnosticado con trombocitopenia. Es una gran ventaja de esta modalidad de la presente invención que la misma muestra que se usa para determinar un marcador de diagnóstico en el momento 0 también puede usarse para determinar el nivel al inicio del estudio de proADM o fragmentos del mismo, que puede compararse con el nivel de proADM o fragmentos del mismo en un momento posterior después del diagnóstico y el inicio del tratamiento. Al determinar el cambio del nivel de proADM o fragmentos de la misma durante el curso del tratamiento del paciente, puede aumentarse aún más la precisión de la predicción de la aparición de un evento adverso en la salud del paciente.
En una modalidad del método descrito en la presente descripción, un nivel elevado de proADM o fragmento(s) de la misma en la segunda muestra en comparación con la primera muestra es indicativo de un evento adverso posterior. Fue sorprendente que, basándose en el cambio del nivel de proADM o fragmentos de la misma, es posible predecir con confianza la probabilidad de que ocurra un evento adverso en la salud del paciente sin determinar marcadores adicionales. Un aumento del nivel o nivel de gravedad de proADM o fragmentos del mismo desde el momento del diagnóstico y el inicio del tratamiento indica que es probable que ocurra un evento adverso. Por consiguiente,
basándose en el cambio de proADM o fragmentos de la misma durante el curso del tratamiento, un médico puede decidir si cambia o modifica el tratamiento del paciente o si se mantiene en el tratamiento inicial.
En una modalidad preferida del método de la presente invención
- un nivel elevado de proADM o fragmento(s) de la misma y un nivel elevado de PCT o fragmento(s) de la misma en la segunda muestra en comparación con la primera muestra es indicativo de un evento adverso posterior, y/o En algunas modalidades de la presente invención, un nivel elevado de proADM o fragmentos de la misma en la segunda muestra en comparación con la primera muestra se relaciona con un nivel de gravedad elevado de proADM o fragmentos de la misma. Por el contrario, en algunas modalidades de la presente invención, un nivel más bajo de proADM o fragmentos de la misma en la segunda muestra en comparación con la primera muestra se refiere a un nivel de gravedad más bajo de proADM o fragmentos de la misma en la segunda muestra en comparación con la primera muestra.
Es una gran ventaja que en base al cambio en el nivel de proADM o fragmentos de la misma en combinación con el cambio determinado de PCT o fragmentos de la misma durante el curso del tratamiento de un paciente gravemente enfermo, la probabilidad de un evento adverso en la salud del paciente el paciente puede ser evaluado. En consecuencia, es posible identificar con confianza a pacientes de alto riesgo y pacientes de bajo riesgo en función de los cambios de estos dos marcadores.
Es ventajoso que mediante un análisis combinado del cambio en el nivel de la PCT o fragmentos de la misma y el nivel de gravedad de la proADM o fragmentos de la misma en el momento del aislamiento de la segunda muestra (el último momento) en una UCI el paciente puede decidir si un paciente es un paciente de bajo riesgo que puede ser dado de alta de la UCI mientras se mantiene el tratamiento en curso, o si un paciente es un paciente de alto riesgo que requiere una modificación o ajuste de la terapia actual en la UCI para prevenir la ocurrencia de un evento adverso que está indicado por la combinación respectiva del cambio en los niveles de PCT y el nivel de gravedad actual de proADM.
En otra modalidad, la presente invención se refiere a un kit para llevar a cabo el método de la presente invención, en donde el kit comprende
- reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT, y
- datos de referencia, tales como un nivel de referencia, correspondiente a niveles de gravedad alto y/o bajo de proADM, en donde el nivel de gravedad bajo está por debajo de 4 nmol/l, preferentemente por debajo de 3 nmol/l, con mayor preferencia por debajo de 2,7 nmol/l, y el nivel de gravedad alto es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, y opcionalmente PCT, en donde dichos datos de referencia se almacenan preferentemente en un medio legible por ordenador y/o se emplean en forma de código ejecutable por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT, lactato y/o proteína C reactiva o fragmento(s) de la misma, con dichos datos de referencia.
En otra modalidad, la presente invención se refiere a un kit para llevar a cabo el método de la presente invención, en donde el kit comprende
- reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT y/o uno o más marcadores para una desregulación del sistema de coagulación, tales como micropartículas de membrana, recuento de plaquetas, volumen medio de plaquetas (MPV), sCD14-ST, protrombinasa, actividad antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor de von Willebrand (vWF), lipoproteínas en combinación con CRP, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D de fibrina no desnaturalizado, factor plaquetario 4, histonas y un ensayo PT, en una muestra de un sujeto, y
- datos de referencia, tales como un nivel de referencia, correspondiente a niveles de gravedad alto y/o bajo de proADM, en donde el nivel de gravedad bajo está por debajo de 4 nmol/l, preferentemente por debajo 3 nmol/l, con mayor preferencia por debajo de 2,7 nmol/l, y el nivel de gravedad alto es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, y opcionalmente PCT y/o uno o más marcadores para una desregulación del sistema de coagulación, tales como micropartículas de membrana, recuento de plaquetas, volumen plaquetario medio, sCD14-ST, protrombinasa, antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor von Willebrand (vWF), lipoproteínas en combinación con CRP, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D de fibrina no desnaturalizado, factor plaquetario 4, histonas y un ensayo PT, en donde dichos datos de referencia se almacenan preferentemente en un medio legible por ordenador y/o se emplean en forma de código ejecutable
por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT y/o uno o más marcadores para una desregulación del sistema de coagulación, tales como micropartículas de membrana, recuento de plaquetas, volumen plaquetario medio (MPV), sCD14-ST, protrombinasa, antitrombina y actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor von Willebrand (vWF), lipoproteínas en combinación con PCR, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D de fibrina no desnaturalizado, factor plaquetario 4, histonas y un ensayo PT, a dichos datos de referencia.
En otra modalidad, la presente invención se refiere a un kit para llevar a cabo el método de la presente invención, en donde el kit comprende
- reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT o fragmento(s) de la misma, en una muestra de un sujeto, y - datos de referencia, tales como un nivel de referencia, correspondiente a niveles de gravedad alto y/o bajo de proADM, en donde el nivel de gravedad bajo está por debajo de 4 nmol/l, preferentemente por debajo de 3 nmol/l, con mayor preferencia por debajo de 2,7 nmol/l, y el nivel de alta gravedad es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, y opcionalmente niveles de PCT, en donde dichos datos de referencia se almacenan preferentemente en un medio legible por ordenador y/o se emplean en forma de código ejecutable por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT o fragmento(s) de la misma, con dichos datos de referencia.
En una modalidad, la invención se refiere a un kit para llevar a cabo el método descrito en la presente descripción, que comprende:
- reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT o fragmento(s) de la misma, en una muestra de un sujeto, y - datos de referencia, tales como un nivel de referencia, correspondiente a los niveles de gravedad de proADM de las reivindicaciones 6 y/o 9, y opcionalmente niveles de PCT, en donde dichos datos de referencia se almacenan preferentemente en un medio legible por ordenador y/o se emplean en forma de código ejecutable por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT o fragmento(s) de la misma, con dichos datos de referencia. Descripción detallada de la invención
La presente invención se basa en un hallazgo que identificó una correlación entre los niveles de proADM y los recuentos de plaquetas. Los presentes métodos permiten la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de niveles anormales de plaquetas en un paciente, en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas en dicho paciente.
La presente invención tiene las siguientes ventajas sobre los métodos convencionales: los métodos inventivos y los kits son rápidos, objetivos, fáciles de usar y precisos para el seguimiento de la terapia de pacientes gravemente enfermos. Los métodos y kits de la invención se refieren a marcadores y puntuaciones clínicas que son fácilmente medibles en métodos de rutina en hospitales, debido a que los niveles de proADM, PCT, lactato, proteína c reactiva, SOFA, APACHE II, SAPS II y/o marcadores para una desregulación del sistema de coagulación, tales como micropartículas de membrana, recuento de plaquetas, volumen medio de plaquetas (MPV) sCD14-ST, protrombinasa, antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor de von Willebrand (vWF), lipoproteínas en combinación con CRP, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D de fibrina no desnaturalizado, factor plaquetario 4, histonas y un ensayo PT, pueden determinarse en muestras de sangre obtenidas de forma rutinaria o en otros fluidos o muestras biológicas obtenidos de un sujeto. Como se usa en la presente descripción, el "paciente" o "sujeto" puede ser un vertebrado. En el contexto de la presente invención, el término "sujeto" incluye tanto humanos como animales, particularmente mamíferos y otros organismos.
En el contexto de la presente invención, un "evento adverso en la salud de un paciente" se refiere a eventos que indican complicaciones o empeoramiento del estado de salud del paciente. Dichos eventos adversos incluyen, sin limitación, la muerte del paciente, la muerte de un paciente dentro de los 28-90 días posteriores al diagnóstico y al inicio del tratamiento, la aparición de una infección o una nueva infección, insuficiencia de órganos y deterioro de los signos o síntomas clínicos generales del paciente, tales como hipotensión o hipertensión, taquicardia o bradicardia, desregulación del sistema de coagulación, coagulación intravascular diseminada, niveles anormales de plaquetas, trombocitopenia y funciones orgánicas desreguladas o insuficiencia de órganos asociada con trombocitopenia.
Además, los ejemplos de eventos adversos incluyen situaciones en las que un deterioro de los síntomas clínicos indica la necesidad de medidas terapéuticas, tal como un procedimiento de limpieza del foco, transfusión de hemoderivados, infusión de coloides, ventilación mecánica invasiva, transfusión de plaquetas, ventilación mecánica no invasiva, cirugía de emergencia, terapia de reemplazo de órganos, tal como reemplazo renal o hepático, y terapia vasopresora. Además, los eventos adversos pueden incluir el suministro de corticosteroides, transfusión de sangre o plaquetas, transfusión de componentes sanguíneos, tales como suero, plasma o células específicas o combinaciones de los mismos, medicamentos que promueven la formación de trombocitos, tratamiento causal o modalidad de una esplenectomía.
El paciente descrito en la presente descripción que ha sido diagnosticado como "gravemente enfermo" puede ser diagnosticado como paciente de unidad de cuidados intensivos (UCI), paciente que requiere observación constante y/o intensa de su estado de salud, paciente diagnosticado de sepsis, sepsis grave o choque séptico, un paciente diagnosticado con una enfermedad infecciosa y una o más insuficiencias orgánicas existentes, un paciente pre o posquirúrgico, un paciente intraoperatorio, un paciente postraumático, un paciente traumatizado, tal como un paciente accidentado, un paciente quemado, un paciente con una o más lesiones abiertas. El sujeto descrito en la presente descripción puede estar en el departamento de emergencias o en la unidad de cuidados intensivos, o en otros lugares de atención, tal como en un transportador de emergencia, tal como una ambulancia, o en un médico general, que se enfrenta a un paciente con dichos síntomas. Además, en el contexto de la presente invención, gravemente enfermo puede referirse a un paciente en riesgo de desarrollar o tener un sistema de coagulación desregulado. Por lo tanto, en el contexto de la presente invención, gravemente enfermo se refiere preferentemente a un paciente en riesgo de contraer o tener un bajo número de plaquetas (trombocitopenia). Con mayor preferencia, en el contexto de la presente invención, gravemente enfermo se refiere a un paciente en riesgo de contraer o tener un bajo número de plaquetas (trombocitopenia) como consecuencia de una infección sistémica, sepsis, sepsis grave o choque séptico. Los pacientes de los que se sospecha que padecen SIRS no se consideran necesariamente como gravemente enfermos.
El término "paciente de UCI" se refiere, sin limitación, a un paciente que ha sido ingresado en una unidad de cuidados intensivos. Una unidad de cuidados intensivos también puede denominarse unidad de terapia intensiva o unidad de tratamiento intensivo (ITU) o unidad de cuidados críticos (CCU), es un departamento especial de un hospital o centro de atención médica que brinda medicina de tratamiento intensivo. Los pacientes de la UCI suelen sufrir enfermedades y lesiones severas y potencialmente mortales, que requieren una supervisión estrecha y constante y el apoyo de equipos y medicamentos especializados para garantizar las funciones corporales normales. Las afecciones comunes que se tratan en las UCI incluyen, sin limitación, el síndrome de dificultad respiratoria aguda o del adulto (ARDS), traumatismos, insuficiencia de órganos y sepsis.
Como se usa en la presente descripción, el término "sistema de coagulación" se refiere a los componentes presentes en la sangre que permiten la coagulación. La coagulación (también conocida como coagulación) es el proceso por el cual la sangre cambia de líquido a gel, formando un coágulo de sangre. Potencialmente da como resultado hemostasia, el cese de la pérdida de sangre de un vaso dañado, seguido de reparación. El mecanismo de coagulación implica la activación, adhesión y agregación de plaquetas junto con el depósito y maduración de fibrina. Los trastornos de la coagulación son estados patológicos que pueden provocar sangrado (hemorragia o hematomas) o coagulación obstructiva (trombosis). La coagulación comienza casi instantáneamente después de que una lesión en el vaso sanguíneo haya dañado el endotelio que recubre el vaso. La fuga de sangre a través del endotelio inicia dos procesos: cambios en las plaquetas y la exposición del factor tisular subendotelial al factor VII plasmático, que finalmente conduce a la formación de fibrina. Las plaquetas forman inmediatamente un tapón en el sitio de la lesión; esto se llama hemostasia primaria. La hemostasia secundaria ocurre simultáneamente: factores de coagulación adicionales o factores de coagulación más allá del factor VII responden en una cascada compleja para formar hebras de fibrina, que fortalecen el tapón de plaquetas. Los ejemplos de factores de coagulación comprenden, sin limitación, plaquetas, factor I (fibrinógeno), factor II (protrombina), factor III (factor tisular o tromboplastina tisular), factor IV Calcio, factor V (proacelerina, factor lábil), factor VI, factor VII (factor estable, proconvertina), factor VIII (factor A antihemofílico), factor IX (factor B antihemofílico o factor Christmas), factor X (factor Stuart-Prower), factor XI (antecedente de tromboplastina plasmática), factor XII (factor Hageman), factor XIII (factor estabilizador de fibrina), factor de von Willebrand, precalicreína (factor Fletcher), cininógeno de alto peso molecular (HMWK) (factor de Fitzgerald), fibronectina, antitrombina III, cofactor de heparina II, proteína C, proteína S, proteína Z, inhibidor de la proteasa relacionado con la proteína Z (ZPI), plasminógeno, alfa 2-antiplasmina, activador del plasminógeno tisular (tPA), uroquinasa, inhibidor del activador del plasminógeno-1 (PAI1), inhibidor del activador del plasminógeno-2 (PAI2), procoagulante del cáncer.
Como se usa en la presente descripción, el término "niveles anormales de plaquetas" se refiere a un número o concentración de plaquetas en la sangre de un paciente que es inesperadamente alto o bajo. El valor esperado depende del estado del paciente. En individuos sanos o individuos sin una enfermedad básica conocida, predisposición o diagnóstico, los recuentos de plaquetas normales o esperados están en el intervalo de aproximadamente 150-450 mil millones de plaquetas por L o 150 000 a 450 000 plaquetas por pl. Este intervalo puede variar, por ejemplo, si se sabe que un paciente padece una afección que afecta el número de plaquetas.
La trombocitopenia es una afección caracterizada por niveles anormalmente bajos de trombocitos, también
conocidos como plaquetas, en la sangre. Un recuento normal de plaquetas humanas varía entre 150000 y 450000 plaquetas por microlitro de sangre. Estos límites están determinados por el percentil 2,5 inferior y superior, por lo que los valores fuera de este intervalo no necesariamente indican enfermedad. La trombocitopenia puede requerir tratamiento de emergencia, especialmente si se determina un recuento de plaquetas por debajo de 50 000 por microlitro.
En el contexto de la presente invención, el término trombocitopenia comprende todas las formas y/o causas que conducen a niveles anormalmente bajos de trombocitos, tales como una producción anormalmente baja de plaquetas que puede ser causada por deshidratación, vitamina B12 o deficiencia de ácido fólico, leucemia o síndrome mielodisplásico o anemia aplásica, disminución de la producción de trombopoyetina por el hígado en insuficiencia hepática, sepsis, infección viral o bacteriana sistémica, leptospirosis, síndromes hereditarios, tales como trombocitopenia amegacariocítica congénita, síndrome de radio ausente de trombocitopenia, anemia de Fanconi, síndrome de Bernard-Soulier (asociado con grandes plaquetas), anomalía de May-Hegglin, síndrome de plaquetas de Gray, síndrome de Alport, síndrome de Wiskott-Aldrich; las tasas anormalmente altas de destrucción de plaquetas pueden deberse a afecciones inmunitarias o no inmunitarias, tales como púrpura trombocitopénica inmunitaria, púrpura trombocitopénica trombótica, síndrome urémico hemolítico, coagulación intravascular diseminada, hemoglobinuria paroxística nocturna, síndrome antifosfolípido, lupus eritematoso sistémico, púrpura postransfusional, trombocitopenia aloinmunitaria neonatal, hiperesplenismo, fiebre del dengue, enfermedad de Gaucher, virus zika; trombocitopenia inducida por medicamentos, por ejemplo inducida por ácido valproico, metotrexato, carboplatino, interferón, isotretinoína, panobinostat, heparina, bloqueadores H2 e inhibidores de la bomba de protones; y otras causas tales como mordedura de serpiente, toxicidad por niacina, enfermedad de Lyme y trombocitaféresis (también llamada plaquetaféresis).
El estándar de oro para medir plaquetas/trombocitos es la determinación de recuentos de plaquetas inmaduras (absolutas) ((A)IPC) mediante, por ejemplo, citometría de flujo. Sin embargo, este método está asociado con la desventaja de que la validación técnica de los recuentos de plaquetas a veces es difícil. Los factores de confusión hacen que los resultados no sean confiables, lo que genera el requisito de una validación adicional, lo que cuesta tiempo y personal valiosos. Las interferencias analíticas pueden causar una pseudotrombocitopenia (por ejemplo, por trombocitos gigantes, trombocitos reticulados, agregación de trombocitos o incompatibilidad con EDTA).
El término "trombocitopenia séptica" se relaciona con la presencia asociada de sepsis y niveles bajos de plaquetas. Como se usa en la presente descripción, "diagnóstico" en el contexto de la presente invención se refiere al reconocimiento y detección (temprana) de una afección clínica de un sujeto relacionado con una enfermedad infecciosa. También la valoración de la gravedad de la enfermedad infecciosa puede estar comprendida por el término "diagnóstico".
"Pronóstico" se refiere a la predicción de un resultado o un riesgo específico para un sujeto en base a una enfermedad infecciosa. Esto también puede incluir una estimación de la posibilidad de recuperación o la posibilidad de un resultado adverso para dicho sujeto.
Los métodos de la invención también pueden usarse para el control. "Monitoreo" se refiere al seguimiento de una enfermedad, trastorno, complicación o riesgo infeccioso ya diagnosticado, por ejemplo, para analizar la progresión de la enfermedad o la influencia de un tratamiento o terapia particular en la progresión de la enfermedad de un paciente gravemente enfermo o una enfermedad infecciosa en un paciente.
El término "seguimiento de la terapia" o "control de la terapia" en el contexto de la presente invención se refiere al seguimiento y/o ajuste de un tratamiento terapéutico de dicho sujeto, por ejemplo, mediante la obtención de información sobre la eficacia de la terapia.
En la presente invención, los términos "evaluación del riesgo" y "estratificación del riesgo" se refieren a la agrupación de sujetos en diferentes grupos de riesgo de acuerdo con su pronóstico adicional. La evaluación del riesgo también se relaciona con la estratificación para aplicar medidas preventivas y/o terapéuticas. Los ejemplos de la estratificación del riesgo son los niveles de riesgo bajo, intermedio y alto descritos en la presente descripción.
Como se usa en la presente descripción, el término "orientación de la terapia" se refiere a la aplicación de determinadas terapias o intervenciones médicas basadas en el valor de uno o más biomarcadores y/o parámetros clínicos y/o puntuaciones clínicas.
Se entiende que en el contexto de la presente invención "determinar el nivel de proADM o fragmento(s) de la misma" o similar se refiere a cualquier medio para determinar la proADM o un fragmento de la misma. El fragmento puede tener cualquier longitud, por ejemplo, al menos aproximadamente 5, 10, 20, 30, 40, 50 o 100 aminoácidos, siempre que el fragmento permita la determinación inequívoca del nivel de proADM o fragmento de la misma. En aspectos particularmente preferidos de la invención, "determinar el nivel de proADM" se refiere a determinar el nivel de proadrenomedulina regional media (MR-proADM). La MR-proADM es un fragmento y/o región de la proADM.
El péptido adrenomedulina (ADM) se descubrió como un péptido hipotensor que comprende 52 aminoácidos, que había sido aislado de un fenocromocitoma humano (Kitamura y otros, 1993). La adrenomedulina (ADM) se codifica como un péptido precursor que comprende 185 aminoácidos ("preproadrenomedulina" o "pre proADM"). En la SEQ ID NO: 1 se proporciona una secuencia de aminoácidos ilustrativa de la proADM.
SEQ ID NO: 1: secuencia de aminoácidos de la pre-pro-ADM:
1 MKLVSVALMY LGSLAFLGAD TARLDVASEF RKKWNKWALS RGKRELRMSS
51 SYPTGLADVK AGPAQTLIRP QDMKGASRSP EDSSPDAARI RVKRYRQSMN
101 NFQGLRSFGC RFGTCTVQKL AHQIYQFTDK DKDNVAPRSK ISPQGYGRRR
151 RRSLPEAGPG RTLVSSKPQA HGAPAPPSGS APHFL
La ADM comprende las posiciones 95-146 de la secuencia de aminoácidos de la pre-proADM y es un producto de corte y empalme de la misma. La "proadrenomedulina" ("proADM") se refiere a la pre-proADM sin la secuencia señal (aminoácidos 1 a 21), es decir, a los residuos de aminoácidos 22 a 185 de la pre-proADM. La "proadrenomedulina de la región media" ("MR-proADM") se refiere a los aminoácidos 42 a 95 de la pre-proADM. En la SEQ ID NO: 2 se proporciona una secuencia de aminoácidos ilustrativa de la MR-proADM.
SEQ ID NO: 2: secuencia de aminoácidos de la MR-pro-ADM (AS 45-92 de la pre-pro-ADM):
ELRMSSSYPT GLADVKAGPA QTLIRPQDMK GASRSPEDSS PDAARIRV
También se contempla en la presente descripción que un péptido y un fragmento del mismo de pre-proADM o MR-proADM pueden usarse para los métodos descritos en la presente descripción. Por ejemplo, el péptido o un fragmento del mismo puede comprender los aminoácidos 22-41 de la pre-proADM (péptido PAMP) o los aminoácidos 95-146 de la pre-proADM (adrenomedulina madura, que incluye la forma biológicamente activa, también conocida como bio-ADM). Un fragmento C-terminal de la proADM (aminoácidos 153 a 185 de la pre proADM) se denomina adrenotensina. Los fragmentos de los péptidos de la proADM o los fragmentos de MR-proADM pueden comprender, por ejemplo, al menos aproximadamente 5, 10, 20, 30 o más aminoácidos. En consecuencia, el fragmento de proADM puede, por ejemplo, seleccionarse del grupo que consiste en MR-proADM, PAMP, adrenotensina y adrenomedulina madura, preferentemente en la presente descripción el fragmento es MR-proADM.
La determinación de estas diversas formas de ADM o proADM y fragmentos de las mismas también abarcan medir y/o detectar subregiones específicas de estas moléculas, por ejemplo, empleando anticuerpos u otros reactivos de afinidad dirigidos contra una porción particular de las moléculas, o determinando la presencia y/o cantidad de las moléculas midiendo una porción de la proteína usando espectrometría de masas.
Uno cualquiera o más de los "péptidos o fragmentos de la ADM" descritos en la presente descripción pueden emplearse en la presente invención.
Los métodos y kits de la presente invención también pueden comprender la determinación de al menos otro biomarcador, marcador, puntuación clínica y/o parámetro además de la proADM.
Como se usa en la presente descripción, un parámetro es una característica, característica o factor medible que puede ayudar a definir un sistema en particular. Un parámetro es un elemento importante para las evaluaciones relacionadas con la salud y la fisiología, tal como el riesgo de una enfermedad/trastorno/afección clínica, preferentemente disfunciones de órganos. Además, un parámetro se define como una característica que se mide y evalúa objetivamente como indicador de procesos biológicos normales, procesos patogénicos o respuestas farmacológicas a una intervención terapéutica. Puede seleccionarse un parámetro de ejemplo del grupo que consiste en evaluación de salud crónica y fisiología aguda II (APACHE II), puntuación fisiológica aguda simplificada (puntuación SAPSII), puntuación de evaluación secuencial de insuficiencia de órganos (puntuación SOFA), puntuación de evaluación secuencial rápida de insuficiencia de órganos (qSOFA), índice de masa corporal, peso, edad, sexo, IGS II, ingesta de líquidos, recuento de glóbulos blancos, sodio, recuento de plaquetas, volumen medio de plaquetas (MPV), potasio, temperatura, presión arterial, dopamina, bilirrubina, tasa respiratoria, presión parcial de oxígeno, clasificación de la Federación Mundial de Sociedades de Neurocirugía (WfNs ) y escala de coma de Glasgow (GCS).
Como se usa en la presente descripción, los términos tales como "marcador", "sustituto", "marcador de pronóstico", "factor" o "biomarcador" o "marcador biológico" se usan indistintamente y se relacionan con marcadores biológicos medibles y cuantificables (por ejemplo, concentración específica de proteína o enzima o un fragmento de la misma, concentración de hormona específica o un fragmento de la misma, o presencia de sustancias biológicas o un fragmento de la misma) que sirven como índices para evaluaciones relacionadas con la salud y la fisiología, tales como el riesgo de una enfermedad/trastorno/afección clínica, preferentemente un evento adverso. Un marcador o
biomarcador se define como una característica que puede medirse y evaluarse objetivamente como un indicador de procesos biológicos normales, procesos patogénicos o respuestas farmacológicas a una intervención terapéutica. Los biomarcadores pueden medirse en una muestra (tal como una prueba de sangre, suero, plasma, orina o tejido).
El al menos un marcador y/o parámetro adicional de dicho sujeto puede seleccionarse del grupo que consiste en un nivel de lactato en dicha muestra, un nivel de procalcitonina (PCT) en dicha muestra, la puntuación de evaluación secuencial de insuficiencia de órganos (puntuación SOFA) de dicho sujeto, la puntuación fisiológica aguda simplificada (SAPSII) de dicho sujeto, la puntuación de la evaluación de salud crónica y fisiológica aguda (APACHE II) de dicho sujeto y un nivel de la tirosina quinasa-1 similar a fms soluble (sFlt-1), histona H2A, histona H2B, histona H3, histona H4, calcitonina, endotelina-1 (ET-1), arginina vasopresina (AVP), péptido natriurético auricular (ANP), lipocalina asociada a gelatinasa de neutrófilos (NGAL), troponina, péptido cerebral natriurético (BNP), proteína C reactiva (CRP), proteína de cálculos pancreáticos (PSP), receptor desencadenante expresado en células mieloides 1 (TREM1), interleucina-6 (IL-6), interleucina-1, interleucina-24 (IL-24), interleucina-22 (IL-22), interleucina (IL-20) otras IL, presepsina (sCD14-ST), enlace de lipopolisacárido (LBP), alfa-1-antitripsina, metaloproteinasa de matriz 2 (MMP2), metaloproteinasa 2 (MMP8), metaloproteinasa de matriz 9 (MMP9), metaloproteinasa de matriz 7 (MMP7, factor de crecimiento placentario (PIGF), cromogranina A, proteína S100A, proteína S100B y factor de necrosis tumoral a (TNFa), neopterina, alfa-1-antitripsina, proarginina vasopresina (AVP, proAVP o copeptina), procalcitonina, péptido natriurético auricular (ANP, pro-ANP), endotelina-1, CCL1 /TCA3, CCL11, CCL12/MCP-5, CCL13/MCP-4, CCL14, CCL15, CCL16, CCL17/TARC, CCL18, CCL19, CCL2/MCP-1, CCL20, CCL21, CCL22/MDC, CCL23, CCL24, CCL25, CCL26, CCL27, CCL28, CCL3, CCL3L3, CCL4, CCL4L1/LAG-1, CCL5, CCL6, CCL7, CCL8, CCL9, CX3CL1, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL17, CXCL2 /MIP-2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7/Ppbp, CXCL9, IL8/CXCL8, XCL1, XCL2, FAM19A1, FAM19A2, FAM19A3, FAM19A4, FAM19A5, CLCF1, CNTF, IL11, IL31, IL6, leptina, LIF, OSM, IFNA1, IFNA10, IFNA13, IFNA14, IFNA2, IFNA4, IFNA7, IFNB1, IFNE, IFNG, IFNZ, IFNA8, IFNA5/IFNaG, IFNw/IFNW1, BAFF, 4-1BBL, TNFSF8, CD40LG, CD70, CD95L/CD178, EDA-A1, TNFSF14, LTA/TNFB, LTB, TNFa, TNFSF10, TNFSF11, TNFSF12, TNFSF13, TNFSF15, TNFSF4, IL18, IL18BP, IL1A, IL1B, IL1F10, IL1F3/IL1RA, IL1F5, IL1F6, IL1F7, IL1F8, IL1RL2, IL1F9, IL33 o un fragmento de los mismos. Otros marcadores incluyen micropartículas de membrana, recuento de plaquetas, volumen medio de plaquetas (MPV), sCD14-ST, protrombinasa, antitrombina y/ actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor von Willebrand (vWF), lipoproteínas en combinación con CRP, fibrinógeno, fibrina, B2GP1, GRIib-MIa, dímero D no desnaturalizado de fibrina, factor plaquetario 4, histonas y un ensayo de PT.
Los componentes del sistema de coagulación también pueden considerarse como marcadores de biomarcadores en el sentido de la presente invención y comprenden, sin limitación, plaquetas, factor I (fibrinógeno), factor II (protrombina), factor III (factor tisular o tromboplastina tisular), factor IV Calcio, factor V (proacelerina, factor lábil), factor VI, factor VII (factor estable, proconvertina), factor VIII (factor A antihemofílico), factor IX (factor B antihemofílico o factor Christmas), factor X (factor Stuart-Prower factor), factor XI (antecedente de tromboplastina plasmática), factor XII (factor de Hageman), factor XIII (factor estabilizador de fibrina), factor de von Willebrand, precalicreína (factor Fletcher), cininógeno de alto peso molecular (HMWK) (factor de Fitzgerald), fibronectina, antitrombina III, cofactor de heparina II, proteína C, proteína S, proteína Z, inhibidor de la proteasa relacionado con la proteína Z (ZPI), plasminógeno, alfa 2-antiplasmina, activador del plasminógeno tisular (tPA), uroquinasa, inhibidor del activador del plasminógeno-1 (PAI1), plasma inhibidor del activador de nogen-2 (PAI2), procoagulante del cáncer
Como se usa en la presente descripción, "procalcitonina" o "PCT" se refiere a un péptido que abarca los residuos de aminoácidos 1-116, 2-116, 3-116, o fragmentos de los mismos, del péptido de procalcitonina. La PCT es un péptido precursor de la hormona calcitonina. Por tanto, la longitud de los fragmentos de procalcitonina es de al menos 12 aminoácidos, preferentemente más de 50 aminoácidos, con mayor preferencia más de 110 aminoácidos. La PCT puede comprender las modificaciones postraduccionales tales como glicosilación, liposidación o derivatización. La procalcitonina es un precursor de la calcitonina y la katacalcina. Así, en condiciones normales, los niveles de PCT en la circulación son muy bajos (< de aproximadamente 0,05 ng/ml).
El nivel de PCT en la muestra del sujeto puede determinarse mediante inmunoensayos como se describe en la presente descripción. Como se usa en la presente descripción, también puede determinarse el nivel de ácido ribonucleico o ácidos desoxirribonucleicos que codifican la "procalcitonina" o "PCT". Los métodos para la determinación de PCT son conocidos por un experto en la técnica, por ejemplo, mediante el uso de productos obtenidos de Thermo Fisher Scientific / B r A H M S GmbH.
Se entiende que "determinar el nivel de al menos una histona" o similar se refiere a determinar el nivel de al menos una histona o un fragmento de al menos una histona en la muestra. En particular, se determina en la muestra el nivel de la histona H2B, H3, H2A y/o H4. En consecuencia, la al menos una histona determinada en la muestra puede ser una histona libre o la al menos una histona determinada en la muestra puede presentarse y ensamblarse en un complejo macromolecular, por ejemplo, en el octámero, nucleosoma y/o NET.
El fragmento de al menos una histona puede tener cualquier longitud, por ejemplo, al menos aproximadamente 5, 10, 20, 30, 40, 50 o 100 aminoácidos, siempre que el fragmento permita la determinación inequívoca del nivel de la histona particular. A continuación, se describen diversos fragmentos ilustrativos de las histonas que son adecuados
para determinar el nivel de la histona en la muestra del sujeto. También se entiende en la presente descripción que el nivel de las histonas puede determinarse determinando un fragmento que se extiende por la cola N-terminal o C-terminal de las histonas. Además, la histona o el fragmento de la misma a determinar en el contexto de la presente invención también puede modificarse, por ejemplo, mediante modificación postraduccional. Los ejemplos de modificaciones postraduccionales pueden ser acetilación, citrulinación, desacetilación, metilación, desmetilación, desiminación, isomerización, fosforilación y ubiquitinación. Preferentemente, las histonas o fragmentos de las mismas son circulantes.
En aspectos particulares de la invención, puede determinarse un nivel de una histona o un fragmento de la misma en la muestra que no está ensamblada en un complejo macromolecular, tal como un nucleosoma, un octámero o una trampa extracelular de neutrófilos (NET). Dichas histonas se denominan em la presente descripción como "histonas libres". En consecuencia, el nivel de al menos una histona puede ser particularmente un nivel de al menos una histona libre.
El nivel de tales histonas libres puede determinarse mediante la detección de las secuencias de aminoácidos o epítopos estructurales de histonas que no son accesibles en un complejo macromolecular estequiométrico ensamblado, tal como un mononucleosoma o un octámero. En tales estructuras, las regiones particulares de las histonas están cubiertas y, por lo tanto, son estéricamente inaccesibles como se muestra para las trampas extracelulares de neutrófilos ("NET"). Además, en el octámero o nucleosoma, las regiones de histonas también participan en interacciones intramoleculares, tal como entre las histonas individuales. Por consiguiente, la región/péptido/epítopo de la histona que se determina en el contexto de la invención puede determinar si la histona es una histona libre o una histona ensamblada en un complejo macromolecular. Por ejemplo, en un método basado en inmunoensayos, los anticuerpos utilizados pueden no detectar histonas, por ejemplo, H4, cuando forman parte del núcleo octamérico de los nucleosomas, ya que los epítopos son estructuralmente inaccesibles. Más abajo en la presente descripción, se ejemplifican regiones/péptidos/epítopos de la histona que podrían emplearse para determinar una histona libre. Por ejemplo, pueden emplearse regiones/péptidos/epítopos de la cola N-terminal o C-terminal de las histonas para determinar histonas independientemente de si están ensambladas en el complejo macromolecular o son histonas libres de acuerdo con la presente invención.
"Estequiométrico" en este contexto se refiere a los complejos intactos, por ejemplo, un mononucleosoma o un octámero. Las "proteínas libres de histonas" también pueden comprender histonas no unidas a la cromatina. Por ejemplo, las "proteínas libres de histonas" también pueden comprender las proteínas con histonas individuales o complejos de histonas no octaméricos. Las histonas libres pueden unirse (por ejemplo, transitoriamente) a histonas individuales, por ejemplo, las histonas pueden formar homo o heterodímeros. Las histonas libres también pueden formar homo o heterotetrámeros. El homo o heterotetrámero puede constar de cuatro moléculas de histonas, por ejemplo, H2A, H2B, H3 y/o H4. Un heterotetrámero típico está formado por dos heterodímeros, en donde cada heterodímero consta de H3 y H4. También se entiende de la presente descripción que H2A y H2B pueden formar un heterotetrámero. También se contempla en la presente descripción que un heterotetrámero puede estar formado por un heterodímero que consiste en H3 y H4, y un heterodímero que consiste en H2A y H2B. Por lo tanto, las histonas libres se denominan en la presente descripción y pueden ser histonas monoméricas, heterodiméricas o tetraméricas, que no están ensambladas en un complejo macromolecular ("estequiométrico") que consiste en un octámero de histona unido al ácido nucleico, por ejemplo, un nucleosoma. Además, las histonas libres también pueden estar unidas a ácidos nucleicos, y en donde histonas libres no están ensambladas en un complejo macromolecular ("estequiométrico"), por ejemplo, un nucleosoma intacto. Preferentemente, las histonas libres están esencialmente libres de ácidos nucleicos.
El lactato, o ácido láctico, es un compuesto orgánico con la fórmula CH3CH(OH)COOH, que se encuentra en los fluidos corporales, que incluyen la sangre. Los análisis de sangre para el lactato se realizan para determinar el estado de la homeostasis de la base ácida en el cuerpo. El ácido láctico es un producto del metabolismo celular que puede acumularse cuando las células carecen de suficiente oxígeno (hipoxia) y deben recurrir a un medio menos eficiente de producción de energía, o cuando una afección provoca una producción excesiva o una eliminación deficiente de lactato. La acidosis láctica puede ser causada por una cantidad inadecuada de oxígeno en las células y los tejidos (hipoxia), por ejemplo, si alguien tiene una afección que puede provocar una disminución de la cantidad de oxígeno que llega a las células y los tejidos, como choque, choque séptico o insuficiencia cardíaca, la prueba de lactato puede usarse para ayudar a detectar y evaluar la gravedad de la hipoxia y la acidosis láctica.
La proteína C reactiva (CRP) es una proteína pentamérica, que puede encontrarse en fluidos corporales tal como el plasma sanguíneo. Los niveles de PCR pueden aumentar en respuesta a la inflamación. Medir y graficar los valores de CRP puede resultar útil para determinar el progreso de la enfermedad o la efectividad de los tratamientos.
Tal como se usa en la presente descripción, la "puntuación de evaluación secuencial de insuficiencia de órganos" o "puntuación SOFA" es una puntuación usada para realizar un seguimiento del estado de un paciente durante la estancia en una unidad de cuidados intensivos (UCI). La puntuación SOFA es un sistema de puntuación para determinar el alcance de la función de los órganos de una persona o la tasa de insuficiencia. La puntuación se basa en seis puntuaciones diferentes, una para los sistemas respiratorio, cardiovascular, hepático, de coagulación, renal y neurológico. Tanto la puntuación SOFA media como la más alta son predictores del resultado. Un aumento en la
puntuación SOFA durante las primeras 24 a 48 horas en la UCI predice una tasa de mortalidad de al menos 50 % hasta 95 %. Las puntuaciones inferiores a 9 dan una mortalidad predictiva del 33 %, mientras que las puntuaciones superiores a 14 pueden estar cerca o por encima del 95 %.
Como se usa en la presente descripción, la puntuación SOFA rápida (qSOFA) es un sistema de puntuación que indica la disfunción orgánica o el riesgo de mortalidad de un paciente. La puntuación se basa en tres criterios: 1) una alteración del estado mental, 2) una disminución de la presión arterial sistólica de menos de 100 mm Hg, 3) una frecuencia respiratoria superior a 22 respiraciones por minuto. Los pacientes con dos o más de estas afecciones tienen un mayor riesgo de tener una disfunción orgánica o de morir.
Como se usa en la presente descripción, "APACHE II" o "evaluación de salud crónica y fisiología aguda II" es un sistema de puntuación de clasificación de la gravedad de la enfermedad (Knaus y otros, 1985). Puede aplicarse dentro de las 24 horas posteriores al ingreso de un paciente en una unidad de cuidados intensivos (UCI) y puede determinarse en función de 12 parámetros fisiológicos diferentes: AaDO2 o PaO2 (en dependencia de FiO2), temperatura (rectal), presión arterial media, pH arterial, frecuencia cardíaca, frecuencia respiratoria, sodio (suero), potasio (suero), creatinina, hematocrito, recuento de glóbulos blancos y escala de coma de Glasgow.
Como se usa en la presente descripción, "SAPS II" o "puntuación fisiológica aguda simplificada II" se refiere a un sistema para clasificar la gravedad de una enfermedad o trastorno (véase Le Gall JR y otros, A new Simplified Acute Physiology Score (SAPS II) basada en un estudio multicéntrico europeo/norteamericano. JAMA. 1993;270(24):2957-63.). La puntuación SAPS II se compone de 12 variables fisiológicas y 3 variables relacionadas con la enfermedad. La puntuación se calcula a partir de 12 mediciones fisiológicas de rutina, información sobre el estado de salud previo y alguna información obtenida en el ingreso a la UCI. La puntuación SAPS II puede determinarse en cualquier momento, preferentemente, en el día 2. La "peor" medida se define como la medida que se correlaciona con el mayor número de puntos. La puntuación SAPS II varía entre 0 y 163 puntos. El sistema de clasificación incluye los siguientes parámetros: edad, frecuencia cardíaca, presión arterial sistólica, temperatura, escala de coma de Glasgow, ventilación mecánica o CPAP, PaO2, FiO2, producción de orina, nitrógeno ureico en sangre, sodio, potasio, bicarbonato, bilirrubina, glóbulos blancos, enfermedades crónicas y tipo de ingreso. Existe una relación sigmoidea entre la mortalidad y la puntuación total de SAPS II. La mortalidad de un sujeto es del 10 % con una puntuación SAPSII de 29 puntos, la mortalidad es del 25 % con una puntuación SAPSII de 40 puntos, la mortalidad es del 50 % con una puntuación SAPSII de 52 puntos, la mortalidad es del 75 % con una puntuación SAPSII puntuación de 64 puntos, la mortalidad es del 90 % con una puntuación SAPSII de 77 puntos (Le Gall loc. cit.). Como se usa en la presente descripción, el término "muestra" es una muestra biológica que se obtiene o se aísla del paciente o sujeto. "Muestra" como se usa en la presente descripción puede, por ejemplo, referirse a una muestra de fluido corporal o tejido obtenido con el fin de diagnosticar, pronosticar o evaluar un sujeto de interés, tal como un paciente. Preferentemente en la presente descripción, la muestra es una muestra de un fluido corporal, tal como sangre, suero, plasma, líquido cefalorraquídeo, orina, saliva, esputo, derrames pleurales, células, un extracto celular, una muestra de tejido, una biopsia de tejido, una muestra de heces y similares. Particularmente, la muestra es sangre, plasma sanguíneo, suero sanguíneo u orina.
Las modalidades de la presente invención se refieren al aislamiento de una primera muestra y al aislamiento de una segunda muestra. En el contexto del método de la presente invención, los términos "primera muestra" y "segunda muestra" se refieren a la determinación relativa del orden de aislamiento de las muestras empleadas en el método de la presente invención. Cuando se usan los términos primera muestra y segunda muestra para especificar el presente método, estas muestras no deben considerarse como determinaciones absolutas del número de muestras tomadas. Por lo tanto, pueden aislarse las muestras adicionales del paciente antes, durante o después del aislamiento de la primera y/o la segunda muestra, o entre la primera y la segunda muestras, en donde estas muestras adicionales pueden o no usarse en el método de la presente invención. Por lo tanto, la primera muestra puede considerarse como cualquier muestra obtenida previamente. La segunda muestra puede considerarse como cualquier muestra adicional o posterior.
"Plasma" en el contexto de la presente invención es el sobrenadante prácticamente libre de células de sangre que contiene anticoagulante obtenido después de la centrifugación. Los anticoagulantes ilustrativos incluyen los compuestos que se unen a iones de calcio tales como EDTA o citrato e inhibidores de trombina tales como heparinatos o hirudina. El plasma libre de células puede obtenerse por centrifugación de la sangre anticoagulada (por ejemplo, sangre con citrato, EDTA o heparinizada), por ejemplo, durante al menos 15 minutos a 2000 a 3000 g. "Suero" en el contexto de la presente invención es la fracción líquida de sangre total que se recoge después de que la sangre se deja coagular. Cuando la sangre coagulada (sangre coagulada) se centrifuga, el suero puede obtenerse como sobrenadante.
Como se usa en la presente descripción, "orina" es un producto líquido del cuerpo secretado por los riñones a través de un proceso llamado micción (o micción) y excretado a través de la uretra.
En modalidades preferidas de la presente invención, se ha diagnosticado que el paciente padece sepsis. Más
particularmente, el paciente puede haber sido diagnosticado de sepsis grave y/o choque séptico.
"Sepsis" en el contexto de la invención se refiere a una respuesta sistémica a la infección. Alternativamente, la sepsis puede verse como la combinación de SIRS con un proceso infeccioso confirmado o una infección. La sepsis puede caracterizarse como un síndrome clínico definido por la presencia tanto de infección como de una respuesta inflamatoria sistémica. LevyMM y otros. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003 abril;31(4):1250-6). El término "sepsis" usado en la presente descripción incluye, pero no se limita a, sepsis, sepsis grave, choque séptico.
El término "sepsis" usado en la presente descripción incluye, pero no se limita a, sepsis, sepsis grave, choque séptico. La sepsis grave se refiere a la sepsis asociada con disfunción orgánica, anormalidad de hipoperfusión o hipotensión inducida por sepsis. Las anomalías de hipoperfusión incluyen acidosis láctica, oliguria y alteración aguda del estado mental. La hipotensión inducida por sepsis se define por la presencia de una presión arterial sistólica de menos de aproximadamente 90 mm Hg o su reducción en aproximadamente 40 mm Hg o más desde el inicio del estudio en ausencia de otras causas de hipotensión (por ejemplo, choque cardiogénico). El choque séptico se define como sepsis grave con hipotensión inducida por sepsis que persiste a pesar de la reanimación adecuada con líquidos, junto con la presencia de anomalías de hipoperfusión o disfunción orgánica (Bone y otros, CHEST 101(6): 1644-55, 1992).
El término sepsis puede definirse alternativamente como una disfunción orgánica potencialmente mortal causada por una respuesta desregulada del huésped a la infección. Para la operacionalización clínica, la disfunción orgánica puede representarse preferentemente por un aumento de 2 puntos o más en la puntuación de la evaluación secuencial de insuficiencia de órganos (SOFA), que se asocia con una mortalidad hospitalaria superior al 10 %. El choque séptico puede definirse como un subconjunto de sepsis en el que anomalías metabólicas, celulares y circulatorias particularmente profundas se asocian con un mayor riesgo de mortalidad que con la sepsis sola. Los pacientes con choque séptico pueden identificarse clínicamente por el requerimiento de vasopresores para mantener una presión arterial media de 65 mmHg o más y un nivel de lactato sérico mayor de 2 mmol/l (>18 mg/dl) en ausencia de hipovolemia.
El término "sepsis" usado en la presente descripción se refiere a todas las etapas posibles en el desarrollo de la sepsis.
El término "sepsis" también incluye la sepsis grave o choque séptico según la definición de SEPSIS-2 (Bone y otros, 2009). El término "sepsis" también incluye los sujetos que se encuentran dentro de la definición de SEPSIS-3 (Singer y otros, 2016). El término "sepsis" usado en la presente descripción se refiere a todas las etapas posibles en el desarrollo de la sepsis.
Como se usa en la presente descripción, "infección" dentro del alcance de la invención significa un proceso patológico causado por la invasión de tejido o fluido normalmente estéril por agentes/patógenos, organismos y/o microorganismos patógenos o potencialmente patógenos, y se relaciona preferentemente con infecciones por bacterias, virus, hongos y/o parásitos. Por consiguiente, la infección puede ser una infección bacteriana, una infección viral y/o una infección fúngica. La infección puede ser una infección local o sistémica. A los efectos de la invención, una infección viral puede considerarse como una infección por un microorganismo.
Además, el sujeto que padece una infección puede padecer más de una fuente o fuentes de infección simultáneamente. Por ejemplo, el sujeto que sufre una infección puede sufrir una infección bacteriana y una infección viral; de una infección viral y una infección fúngica; de una infección bacteriana y fúngica, y de una infección bacteriana, una infección fúngica y una infección viral, o padece una infección mixta que comprende una o más de las infecciones enumeradas en la presente descripción, que incluye potencialmente una superinfección, por ejemplo, una o más infecciones bacterianas además de una o más infecciones virales y/o una o más infecciones fúngicas.
Como se usa en la presente descripción, "enfermedad infecciosa" comprende todas las enfermedades o trastornos que están asociados con infecciones bacterianas y/o virales y/o fúngicas.
De acuerdo con la presente invención, los pacientes gravemente enfermos, tal como los pacientes sépticos, pueden necesitar un control muy estricto, con respecto a las funciones vitales y/o la vigilancia de la protección de los órganos, y pueden estar bajo tratamiento médico.
En el contexto de la presente invención, el término "tratamiento médico" o "tratamiento" comprende diversos tratamientos y estrategias terapéuticas, que comprenden, sin limitación, estrategias antiinflamatorias, administración de antagonistas de la proADM tales como anticuerpos terapéuticos, ARNip o ADN, la purificación extracorpórea de la sangre o la eliminación de sustancias nocivas mediante aféresis, diálisis, adsorbedores para evitar la tormenta de citoquinas, eliminación de mediadores inflamatorios, aféresis de plasma, administración de vitaminas tal como la vitamina C, ventilación tal como ventilación mecánica y ventilación no mecánica, para proporcionar al cuerpo suficiente oxígeno, por ejemplo, procedimientos de limpieza de focos, transfusión de productos sanguíneos, infusión
de coloides, reemplazo renal o hepático, tratamiento con antibióticos, ventilación mecánica invasiva, ventilación mecánica no invasiva, terapia de reemplazo renal, uso de vasopresores, terapia con fluido, aféresis y medidas para la protección de órganos, suministro de corticosteroides, sangre o transfusión de plaquetas, transfusión de componentes sanguíneos, tal como suero, plasma o células específicas o combinaciones de los mismos, fármacos que promuevan la formación de trombocitos, control de fuente, cirugías, tratamiento causal o modalidad de una esplenectomía.
Otros tratamientos de la presente invención comprenden la administración de células o productos celulares tales como células madre, sangre o plasma, y la estabilización de la circulación del paciente y la protección del glucocáliz endotelial, por ejemplo, mediante estrategias óptimas de gestión de fluidos, por ejemplo, para alcanzar la normovolemia y prevenir o tratar la hipervolemia o la hipovolemia. Además, los vasopresores o, por ejemplo, las catecolaminas, así como también la inhibición de la albúmina o la heparanasa mediante heparina no fraccionada o heparina re-N-acetilada N-desulfatada son tratamientos útiles para apoyar la circulación y la capa endotelial.
Además, los tratamientos médicos de la presente invención comprenden, sin limitación, la estabilización de la coagulación sanguínea, inhibidores de iNOS, agentes antiinflamatorios tales como hidrocortisona, sedantes y analgésicos, así como también insulina.
"Terapia de reemplazo renal" (TRR) se refiere a una terapia que se emplea para reemplazar la función normal de filtración de la sangre de los riñones. La terapia de reemplazo renal puede referirse a diálisis (por ejemplo, hemodiálisis o diálisis peritoneal), hemofiltración y hemodiafiltración. Dichas técnicas son diversas formas de desviar la sangre a una máquina, limpiarla y luego devolverla al cuerpo. La terapia de reemplazo renal también puede referirse al trasplante de riñón, que es la última forma de reemplazo en la que el riñón viejo se reemplaza por un riñón de donante. La hemodiálisis, la hemofiltración y la hemodiafiltración pueden ser continuas o intermitentes y pueden usar una vía arteriovenosa (en la que la sangre sale de una arteria y regresa por una vena) o una vía venovenosa (en la que la sangre sale de una vena y regresa por una vena). Esto da como resultado diversos tipos de RRT. Por ejemplo, la terapia de reemplazo renal puede seleccionarse del grupo de, pero no se limita a, terapia de reemplazo renal continua (CRRT), hemodiálisis continua (CHD), hemodiálisis arteriovenosa continua (CAVHD), hemodiálisis venovenosa continua (CVVHD), hemofiltración continua (CHF), hemofiltración arteriovenosa continua (CAVH o CAVHF), hemofiltración venovenosa continua (CVVH o CVVHF), hemodiafiltración continua (CHDF), hemodiafiltración arteriovenosa continua (CAVHDF), hemodiafiltración venovenosa continua (CVVHDF), terapia de reemplazo renal intermitente (IRRT), hemodiálisis (IHD), hemodiálisis venovenosa intermitente (IVVHD), hemofiltración intermitente (IHF), hemofiltración venovenosa intermitente (IVVH o IVVHF), hemodiafiltración intermitente (IHDF) y hemodiafiltración venovenosa intermitente (IVVHDF).
La ventilación artificial y mecánica son enfoques efectivos para mejorar el intercambio de gases y la ventilación adecuados y tienen como objetivo salvar vidas durante la hipoxemia grave. La ventilación artificial se relaciona con ayudar o estimular la respiración del sujeto. La ventilación artificial puede seleccionarse del grupo que consiste en ventilación mecánica, ventilación manual, oxigenación por membrana extracorpórea (ECMO) y ventilación no invasiva (NIV). La ventilación mecánica se relaciona con un método para ayudar mecánicamente o reemplazar la respiración espontánea. Esto puede involucrar una máquina llamada ventilador. La ventilación mecánica puede ser ventilación oscilatoria de alta frecuencia o ventilación líquida parcial.
“Manejo de fluidos” se refiere a la monitorización y control del estado de fluidos de un sujeto y la administración de fluidos para estabilizar la circulación o la vitalidad de los órganos, por ejemplo, por administración oral, enteral o intravenosa de fluidos. Comprende la estabilización del equilibrio de líquidos y electrolitos o la prevención o corrección de hiper o hipovolemia, así como también el suministro de hemoderivados.
Las emergencias quirúrgicas/cirugía de emergencia son necesarias si un sujeto tiene una emergencia médica y es posible que se requiera una intervención quirúrgica inmediata para preservar la supervivencia o el estado de salud. El sujeto que necesita cirugía de emergencia puede seleccionarse del grupo que consiste en sujetos que sufren un traumatismo agudo, una infección activa no controlada, trasplante de órganos, cirugía de prevención o estabilización de órganos o cáncer.
Los procedimientos de limpieza son métodos higiénicos para prevenir infecciones, especialmente infecciones nosocomiales, que comprenden la desinfección de todas las superficies orgánicas e inorgánicas que podrían entrar en contacto con un paciente, tales como, por ejemplo, la piel, objetos en la habitación del paciente, dispositivos médicos, dispositivos de diagnóstico o aire de la habitación. Los procedimientos de limpieza incluyen el uso de prendas y unidades de protección, tales como cubrebocas, batas, guantes o candado de higiene, y acciones tales como visitas restringidas de pacientes. Además, los procedimientos de limpieza comprenden la limpieza del propio paciente y la ropa o el paciente.
En el caso de enfermedades críticas, tales como sepsis o infecciones graves, es muy importante tener un diagnóstico temprano, así como también un pronóstico y una evaluación de riesgos para el resultado de un paciente para encontrar la terapia y el manejo óptimos. Los enfoques terapéuticos deben ser muy individuales y variar de un caso a otro. Se necesita un seguimiento terapéutico para una terapia de mejores prácticas y está influenciado por el
momento del tratamiento, el uso de terapias combinadas y la optimización de la dosificación de fármacos. Una terapia o manejo incorrecto u omitido aumentará la tasa de mortalidad cada hora.
Un tratamiento médico de la presente invención puede ser un tratamiento con antibióticos, en donde pueden administrarse uno o más "antibióticos" o "agentes antiinfecciosos" si se ha diagnosticado una infección o se han determinado los síntomas de una enfermedad infecciosa.
Además, los agentes antibióticos comprenden bacteriófagos para el tratamiento de infecciones bacterianas, péptidos antimicrobianos sintéticos o antagonistas del hierro/quelantes del hierro. Además, los anticuerpos terapéuticos o antagonistas contra las estructuras patógenas tales como los anticuerpos anti-VAP, la vacunación con clones antirresistentes, la administración de células inmunitarias, tales como los linfocitos T efectores modulados o estimulados in vitro, son agentes antibióticos que representan opciones de tratamiento para pacientes gravemente enfermos, tales como los pacientes con sepsis. Otros agentes/tratamientos antibióticos o estrategias terapéuticas contra infecciones o para la prevención de nuevas infecciones incluyen el uso de antisépticos, productos de descontaminación, agentes antivirulentos tales como liposomas, saneamiento, cuidado de heridas, cirugía.
También es posible combinar varios de los agentes antibióticos o estrategias de tratamiento mencionados anteriormente con fluidoterapia, transfusión de plaquetas o transfusión de hemoderivados.
De acuerdo con la presente invención, proADM y, opcionalmente, PCT y/u otros marcadores o puntuaciones clínicas se emplean como marcadores para el seguimiento de la terapia, que comprende el pronóstico, la evaluación del riesgo y la estratificación del riesgo de un evento adverso posterior en la salud de un paciente que ha sido diagnosticado como gravemente enfermo.
Un experto en la técnica es capaz de obtener o desarrollar medios para la identificación, medición, determinación y/o cuantificación de cualquiera de las moléculas de proADM anteriores, o fragmentos o variantes de las mismas, así como también los otros marcadores de la presente invención de acuerdo con la práctica biológica molecular estándar.
El nivel de proADM o fragmentos de la misma, así como también los niveles de otros marcadores de la presente invención pueden determinarse mediante cualquier ensayo que determine de forma fiable la concentración del marcador. En particular, pueden emplearse espectrometría de masas (MS) y/o inmunoensayos como se ejemplifica en los ejemplos adjuntos. Como se usa en la presente descripción, un inmunoensayo es una prueba bioquímica que mide la presencia o concentración de una macromolécula/polipéptido en una solución mediante el uso de un anticuerpo o fragmento de unión de anticuerpo o inmunoglobulina.
En la presente invención se pretenden los métodos para determinar la proADM u otros marcadores tales como PCT usados en el contexto de la presente invención. A modo de ejemplo, puede emplearse un método seleccionado del grupo que consiste en espectrometría de masas (MS), inmunoensayo de luminiscencia (LIA), radioinmunoensayo (RIA), inmunoensayos de quimioluminiscencia y fluorescencia, inmunoensayo enzimático (EIA), inmunoensayos ligados a enzimas (ELISA), matrices de perlas basadas en luminiscencia, matrices basadas en perlas magnéticas, ensayos de micromatrices de proteínas, formatos de pruebas rápidas como, por ejemplo, pruebas de tiras inmunocromatográficas, ensayos de criptatos raros y sistemas/analizadores automatizados.
La determinación de la proADM y, opcionalmente, de otros marcadores basados en el reconocimiento de anticuerpos es una modalidad preferida de la invención. Como se usa en la presente descripción, el término "anticuerpo" se refiere a las moléculas de inmunoglobulina y porciones inmunológicamente activas de las moléculas de inmunoglobulina (Ig), es decir, moléculas que contienen un sitio de unión a antígeno que se une específicamente (reacciona inmunológicamente con) un antígeno. De acuerdo con la invención, los anticuerpos pueden ser anticuerpos monoclonales, así como también policlonales. Particularmente, se usan anticuerpos que se unen específicamente a al menos la proADM o fragmentos de la misma.
Se considera que un anticuerpo es específico si su afinidad hacia la molécula de interés, por ejemplo, proADM, o el fragmento de la misma es al menos 50 veces mayor, preferentemente 100 veces mayor, con la máxima preferencia al menos 1000 veces mayor que hacia otras moléculas comprendido en una muestra que contiene la molécula de interés. Es bien conocido en la técnica cómo desarrollar y seleccionar anticuerpos con una especificidad dada. En el contexto de la invención, se prefieren los anticuerpos monoclonales. El anticuerpo o el fragmento de unión del anticuerpo se une específicamente a los marcadores definidos en la presente descripción o fragmentos de los mismos. En particular, el anticuerpo o el fragmento de unión del anticuerpo se une a los péptidos de la proADM definidos en la presente descripción. Por lo tanto, los péptidos definidos en la presente descripción también pueden ser epítopos a los que se unen específicamente los anticuerpos. Además, en los métodos y kits de la invención se usa un anticuerpo o un fragmento de unión de anticuerpo que se une específicamente a la proADM o proADM, particularmente a la MR-proADM.
Además, en los métodos y kits de la invención se usa un anticuerpo o un fragmento de unión de anticuerpo que se une específicamente a la proADM o fragmentos de la misma y, opcionalmente, a otros marcadores de la presente
invención, tal como PCT. Los ejemplos de inmunoensayos pueden ser inmunoensayo de luminiscencia (LIA), radioinmunoensayo (RIA), inmunoensayos de quimioluminiscencia y fluorescencia, inmunoensayo enzimático (EIA), inmunoensayos ligados a enzimas (ELISA), matrices de perlas basadas en luminiscencia, matrices basadas en perlas magnéticas, ensayos de micromatrices de proteínas, formatos de prueba rápida, ensayo de criptato raro. Además, pueden emplearse ensayos adecuados para pruebas en el lugar de atención y formatos de pruebas rápidas tales como, por ejemplo, pruebas en tiras inmunocromatográficas. También se prevén inmunoensayos automatizados, tales como el ensayo KRYPTOR.
Alternativamente, en lugar de anticuerpos, el alcance de la presente invención puede abarcar otras moléculas de captura o estructuras moleculares que reconozcan específica y/o selectivamente la proADM. En la presente descripción, el término "moléculas de captura" o "estructuras moleculares" comprende las moléculas que pueden usarse para unir moléculas diana o moléculas de interés, es decir, analitos (por ejemplo, proADM, proADM, MR-proADM y PCT), de una muestra. Por lo tanto, las moléculas de captura deben moldearse adecuadamente, tanto espacialmente como en términos de características superficiales, tal como carga superficial, hidrofobicidad, hidrofilicidad, presencia o ausencia de donantes y/o aceptores de Lewis, para unirse específicamente a las moléculas diana o moléculas de interés. De este modo, la unión puede estar mediada, por ejemplo, por interacciones iónicas, de van-der-Waals, pi-pi, sigma-pi, hidrofóbicas o de enlace de hidrógeno o una combinación de dos o más de las interacciones antes mencionadas o interacciones covalentes entre la captura de moléculas o armazón molecular y las moléculas diana o moléculas de interés. En el contexto de la presente invención, las moléculas de captura o las estructuras moleculares pueden seleccionarse, por ejemplo, del grupo que consiste en una molécula de ácido nucleico, una molécula de carbohidrato, una molécula de PNA, una proteína, un péptido y una glicoproteína. Las moléculas de captura o estructuras moleculares incluyen, por ejemplo, aptámeros, DARpins (proteínas de repetición de anquirina diseñadas). Se incluyen los afímeros y similares.
En determinados aspectos de la invención, el método es un inmunoensayo que comprende las etapas de:
a) poner en contacto la muestra con
i. un primer anticuerpo o un fragmento de unión a antígeno o derivado del mismo específico para un primer epítopo de dicha proADM, y
ii. un segundo anticuerpo o un fragmento de unión a antígeno o derivado del mismo específico para un segundo epítopo de dicha proADM; y
b) detectar la unión de los dos anticuerpos o fragmentos de unión a antígeno o derivados de los mismos a dicha proADM.
Preferentemente, uno de los anticuerpos puede marcarse y el otro anticuerpo puede unirse a una fase sólida o puede unirse selectivamente a una fase sólida. En un aspecto particularmente preferido del ensayo, uno de los anticuerpos se marca mientras que el otro se une a una fase sólida o puede unirse selectivamente a una fase sólida. El primer anticuerpo y el segundo anticuerpo pueden estar presentes dispersos en una mezcla de reacción líquida, y en donde un primer componente de marcaje que es parte de un sistema de marcaje basado en la extinción o amplificación de fluorescencia o quimioluminiscencia está unido al primer anticuerpo, y un segundo componente de marcaje de dicho sistema de marcaje se une al segundo anticuerpo de forma que, tras la unión de ambos anticuerpos a dicha proADM o fragmentos de la misma a detectar, se genera una señal medible que permite la detección de los complejos sándwich resultantes en la solución de medida. El sistema de marcaje puede comprender un criptato o quelato de tierras raras en combinación con un colorante fluorescente o quimioluminiscente, en particular del tipo cianina.
En una modalidad preferida, el método se ejecuta como inmunoensayo de sándwich heterogéneo, en donde uno de los anticuerpos se inmoviliza en una fase sólida elegida arbitrariamente, por ejemplo, las paredes de tubos de ensayo revestidos (por ejemplo, tubos de ensayo de poliestirol, tubos revestidos, CT) o microtitulación de placas, por ejemplo compuestas de poliestirol, o a partículas, tales como por ejemplo partículas magnéticas, en donde el otro anticuerpo tiene un grupo que se asemeja a un marcador detectable o que permite la unión selectiva a un marcador, y que sirve para la detección de las estructuras sándwich formadas. También es posible una inmovilización retardada temporalmente o posterior mediante el uso de las fases sólidas adecuadas.
El método de acuerdo con la presente invención puede realizarse además como un método homogéneo, en donde los complejos sándwich formados por el anticuerpo/anticuerpos y el marcador, la proADM o un fragmento de la misma, que va a detectarse, permanecen suspendidos en la fase líquida. En este caso, se prefiere que, cuando se usan dos anticuerpos, ambos anticuerpos se marquen con partes de un sistema de detección, lo que conduce a la generación de una señal o a la activación de una señal si ambos anticuerpos se integran en un solo sándwich. Dichas técnicas deben realizarse en particular como métodos de detección de intensificación de la fluorescencia o inactivación de la fluorescencia. Un aspecto particularmente preferido se refiere al uso de los reactivos de detección que van a usarse por parejas, tales como por ejemplo los que se describen en los documentos US4882733, EP0180492 o EP0539477 y el estado de la técnica citado en los mismos. De esta manera, se hacen posibles las
mediciones en las que solo se detectan los productos de reacción que comprenden ambos componentes de mareaje en un solo inmunocomplejo directamente en la mezcla de reacción. Por ejemplo, dichas tecnologías se ofrecen bajo los nombres comerciales TRACE® (Emisión de criptato amplificada resuelta en el tiempo) o KRYPTOR®, implementando las enseñanzas de las aplicaciones citadas anteriormente. Por lo tanto, en aspectos particularmente preferidos, se usa un dispositivo de diagnóstico para llevar a cabo el método proporcionado en la presente descripción. Por ejemplo, se determina el nivel de proADM o fragmentos de la misma y/o el nivel de cualquier marcador adicional del método proporcionado en la presente descripción, tal como PCT. En aspectos particularmente preferidos, el dispositivo de diagnóstico es KRYPTOR®.
El nivel del marcador de la presente invención, por ejemplo, proADM o fragmentos de la misma, PCT o fragmentos de la misma, u otros marcadores, también puede determinarse mediante los métodos basados en espectrometría de masas (MS). Dicho método puede comprender detectar la presencia, cantidad o concentración de uno o más fragmentos de péptidos modificados o no modificados de, por ejemplo, proADM o PCT en dicha muestra biológica o una proteína digerida (por ejemplo, digerido tríptico) de dicha muestra y, opcionalmente, separar la muestra con métodos cromatográficos, y someter la muestra preparada y opcionalmente separada a análisis por MS. Por ejemplo, la espectrometría de masas de monitorización de reacciones seleccionadas (SRM), monitorización de reacciones múltiples (MRM) o monitorización de reacciones paralelas (PRM) puede usarse en el análisis de MS, particularmente para determinar las cantidades de proADM o fragmentos de la misma.
En la presente descripción, el término "espectrometría de masas" o "MS" se refiere a una técnica analítica para identificar los compuestos por su masa. Para mejorar las capacidades de resolución y determinación de masas de la espectrometría de masas, las muestras pueden procesarse antes del análisis de MS. En consecuencia, la invención se refiere a los métodos de detección de MS que pueden combinarse con las tecnologías de inmunoenriquecimiento, métodos relacionados con la preparación de muestras y/o métodos cromatográficos, preferentemente con cromatografía líquida (LC), con mayor preferencia con cromatografía líquida de alta resolución (HPLC) o ultra cromatografía líquida de alta resolución (UHPLC). Los métodos de preparación de muestras comprenden las técnicas de lisis, fraccionamiento, digestión de la muestra en péptidos, empobrecimiento, enriquecimiento, diálisis, desalinización, alquilación y/o reducción de péptidos. Sin embargo, estas etapas son opcionales. La detección selectiva de iones de analitos puede realizarse con espectrometría de masas en tándem (MS/MS). La espectrometría de masas en tándem se caracteriza por una etapa de selección de masas (como se usa en la presente descripción, el término "selección de masas" denota el aislamiento de iones que tienen un m/z específica o un intervalo estrecho de m/z), seguido de la fragmentación de los iones seleccionados y el análisis de masas de los iones del producto resultante (fragmento).
El experto en la técnica sabe cómo cuantificar el nivel de un marcador en la muestra mediante los métodos de espectrometría de masas. Por ejemplo, puede emplearse la cuantificación relativa "rSRM" o la cuantificación absoluta como se ha descrito anteriormente.
Además, los niveles (que incluyen los niveles de referencia) pueden determinarse mediante los métodos basados en espectrometría de masas, tales como los métodos que determinan la cuantificación relativa o determinan la cuantificación absoluta de la proteína o fragmento de la misma de interés.
La cuantificación relativa "rSRM" puede lograrse mediante:
1. Determinar el aumento o la disminución de la presencia de la proteína diana al comparar el área del pico característico de SRM (selección de la reacción) de un péptido de fragmento diana determinado detectado en la muestra con la misma área del pico característico de SRM del péptido del fragmento diana en al menos una segunda, tercera, cuarta o más muestras biológicas.
2. Determinar el aumento o la disminución de la presencia de la proteína diana al comparar el área del pico característico de SRM de un péptido diana dado detectado en la muestra con las áreas del pico característico de SRM desarrolladas a partir de fragmentos de péptidos de otras proteínas, en otras muestras derivadas de fuentes biológicas diferentes y separadas, donde en la comparación del área del pico característico de SRM entre las dos muestras para un fragmento de péptido se normaliza, por ejemplo, para la cantidad de proteína analizada en cada muestra.
3. Determinar el aumento o la disminución de la presencia de la proteína diana al comparar el área del pico característico de SRM para un péptido diana dado con las áreas del pico característico de SRM de otros fragmentos de péptidos derivados de diferentes proteínas dentro de la misma muestra biológica para normalizar los niveles cambiantes de la proteína histona para los niveles de otras proteínas que no cambian sus niveles de expresión bajo diversas afecciones celulares.
4. Estos ensayos pueden aplicarse tanto a fragmentos de péptidos no modificados como a fragmentos de péptidos modificados de las proteínas diana, donde las modificaciones incluyen, pero no se limitan a, fosforilación y/o glicosilación, acetilación, metilación (mono, di, tri), citrulinación, ubiquitinilación y donde los niveles relativos de péptidos modificados se determinan de la misma manera que se determinan las cantidades
relativas de péptidos no modificados.
La cuantificación absoluta de un péptido dado puede lograrse mediante:
1. Comparar del área del pico de la firma SRM/MRM para un péptido de fragmento dado de las proteínas diana en una muestra biológica individual con el área del pico de la firma SRM/MRM de un estándar de péptido de fragmento interno adicionar al lisado de proteínas de la muestra biológica. El estándar interno puede ser una versión sintética marcada del péptido del fragmento de la proteína diana que se interroga o la proteína recombinante marcada. Este estándar se adiciona a una muestra en cantidades conocidas antes (obligatorio para la proteína recombinante) o después de la digestión, y el área del pico característico de SRM/MRM puede determinarse tanto para el estándar de péptido de fragmento interno como para el péptido de fragmento nativo en la muestra biológica por separado, seguido de la comparación de las áreas de ambos picos. Esto puede aplicarse a los péptidos de fragmentos no modificados y péptidos de fragmentos modificados, donde las modificaciones incluyen, pero no se limitan a, fosforilación y/o glicosilación, acetilación, metilación (por ejemplo, mono, di o trimetilación), citrulinación, ubiquitinilación y donde los niveles absolutos de péptidos modificados pueden determinarse de la misma manera que se determinan los niveles absolutos de péptidos no modificados.
2. Los péptidos también pueden cuantificarse mediante el uso de curvas de calibración externas. El enfoque de la curva normal usa una cantidad constante de un péptido pesado como estándar interno y una cantidad variable de péptido sintético ligero añadido a la muestra. Se necesita usar una matriz representativa similar a la de las muestras de prueba para construir las curvas estándar para tener en cuenta un efecto de matriz. Además, el método de la curva inversa evita el problema del analito endógeno en la matriz, donde se agrega una cantidad constante de péptido ligero sobre el analito endógeno para crear un estándar interno y se agregan cantidades variables de péptido pesado para crear un conjunto concentraciones estándares. Las muestras de prueba que van a compararse con las curvas normal o inversa se añaden a la matriz usada para crear la curva de calibración con la misma cantidad de péptido estándar que el estándar interno.
La invención se refiere además a los kits, el uso de los kits y los métodos en donde se usan dichos kits. La invención se refiere a los kits para llevar a cabo los métodos proporcionados anteriormente y más abajo. Las definiciones proporcionadas en la presente descripción, por ejemplo, proporcionadas en relación con los métodos, también se aplican a los kits de la invención. En particular, la invención se refiere a los kits para el seguimiento de terapias, que comprenden el pronóstico, la evaluación del riesgo o la estratificación del riesgo de un evento adverso posterior en la salud de un paciente, en donde dicho kit comprende
- reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT, lactato y/o proteína C reactiva o fragmento(s) de la misma, en una muestra de un sujeto, y - reactivos de detección para determinar dicho nivel de proADM en dicha muestra de dicho sujeto, y
- datos de referencia, tales como un nivel de referencia, correspondiente a niveles de gravedad alto y/o bajo de la proADM, en donde el nivel de gravedad bajo está por debajo de 4 nmol/l, preferentemente por debajo de 3 nmol/l, con mayor preferencia por debajo de 2,7 nmol/l, y el nivel de gravedad alto es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, y opcionalmente niveles de PCT, lactato y/o proteína C reactiva, en donde dichos datos de referencia se almacenan preferentemente en un medio legible por ordenador y/o empleado en forma de código ejecutable por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT, lactato y/o proteína C reactiva o fragmento(s) de la misma, a dichos datos de referencia.
Como se usa en la presente descripción, los "datos de referencia" comprenden los niveles de referencia de la proADM y, opcionalmente, PCT, lactato y/o proteína C reactiva. Los niveles de la proADM y, opcionalmente, PCT, lactato y/o proteína C reactiva en la muestra del sujeto pueden compararse con los niveles de referencia incluidos en los datos de referencia del kit. Los niveles de referencia se describen en la presente descripción anteriormente y también se ejemplifican en los ejemplos adjuntos. Los datos de referencia también pueden incluir una muestra de referencia con la que se compara el nivel de proADM y, opcionalmente, PCT, lactato y/o proteína C reactiva. Los datos de referencia también pueden incluir un manual de instrucciones sobre cómo utilizar los kits de la invención.
El kit puede comprender adicionalmente elementos útiles para la obtención de una muestra, tal como una muestra de sangre, por ejemplo el kit puede comprender un recipiente, en donde dicho recipiente comprende un dispositivo para la fijación de dicho recipiente a una cánula o jeringa, es una jeringa adecuada para aislamiento de sangre, presenta una presión interna inferior a la presión atmosférica, tal que es adecuada para aspirar un volumen previamente determinado de muestra en dicho recipiente, y/o comprende adicionalmente detergentes, sales caotrópicas, inhibidores de ribonucleasas, agentes quelantes, tales como isotiocianato de guanidinio, clorhidrato de guanidinio, dodecilsulfato de sodio, monolaurato de polioxietilensorbitán, proteínas inhibidoras de la ARNasa y mezclas de las mismas, y/o un sistema de filtro que contenga nitrocelulosa, matriz de sílice, esferas ferromagnéticas, un derrame de recuperación de copa, trehalosa, fructosa, lactosa, manosa, polietilenglicol, glicerol, EDTA, TRIS,
limoneno, xileno, benzoilo, fenol, aceite mineral, anilina, pirrol, citrato y mezclas de los mismos.
Como se usa en la presente descripción, el "reactivo de detección" o similar son reactivos que son adecuados para determinar los marcadores descritos en la presente descripción, por ejemplo, de proADM, PCT, lactato y/o proteína C reactiva. Dichos reactivos de detección ilustrativos son, por ejemplo, ligandos, por ejemplo, anticuerpos o fragmentos de los mismos, que se unen específicamente al péptido o epítopos del o los marcadores descritos en la presente descripción. Dichos ligandos podrían usarse en inmunoensayos como se describe anteriormente. Otros reactivos que se emplean en los inmunoensayos para determinar el nivel del marcador o marcadores también pueden estar incluidos en el kit y se consideran en la presente descripción como reactivos de detección. Los reactivos de detección también pueden relacionarse con reactivos que se emplean para detectar los marcadores o fragmentos de los mismos mediante métodos basados en MS. Dicho reactivo de detección también puede ser reactivo, por ejemplo, enzimas, productos químicos, tampones, etc, que se usan para preparar la muestra para el análisis de MS. Un espectrómetro de masas también puede considerarse como un reactivo de detección. Los reactivos de detección de acuerdo con la invención también pueden ser soluciones de calibración, por ejemplo, que pueden emplearse para determinar y comparar el nivel del marcador o marcadores.
La sensibilidad y la especificidad de una prueba diagnóstica y/o pronóstica dependen de algo más que la "calidad" analítica de la prueba, también dependen de la definición de lo que constituye un resultado anormal. En la práctica, las curvas características operativas del receptor (curvas ROC) generalmente se calculan trazando el valor de una variable frente a su frecuencia relativa en poblaciones "normales" (es decir, individuos aparentemente sanos que no tienen una infección) y poblaciones "enfermas", por ejemplo, sujetos que tienen una infección. Para cualquier marcador en particular (como proADM), es probable que se superponga una distribución de niveles de marcador para sujetos con y sin una enfermedad/afección. En tales afecciones, una prueba no distingue absolutamente lo normal de la enfermedad con un 100 % de precisión, y el área de superposición podría indicar dónde la prueba no puede distinguir lo normal de la enfermedad. Se selecciona un umbral, por debajo del cual la prueba se considera anormal y por encima del cual la prueba se considera normal o por debajo o por encima del cual la prueba indica una afección específica, por ejemplo, infección. El área bajo la curva ROC es una medida de la probabilidad de que la medición percibida permita la identificación correcta de una afección. Las curvas ROC pueden usarse incluso cuando los resultados de las pruebas no necesariamente dan un número exacto. Siempre que puedan clasificarse los resultados, puede crearse una curva ROC. Por ejemplo, los resultados de una prueba en muestras de "enfermedad" pueden clasificarse de acuerdo con el grado (por ejemplo, 1=bajo, 2=normal y 3=alto). Esta clasificación puede correlacionarse con los resultados en la población "normal" y crear una curva ROC. Estos métodos son bien conocidos en la técnica; véase, por ejemplo, Hanley y otros. 1982 Radiology 143: 29-36. Preferentemente, se selecciona un umbral para proporcionar un área de la curva ROC superior a 0,5, con mayor preferencia superior a 0,7, aún con mayor preferencia superior a 0,8, incluso con mayor preferencia superior a 0,85 y con la máxima preferencia superior a 0,9. El término "aproximadamente" en este contexto se refiere a /- 5 % de una medida dada.
El eje horizontal de la curva ROC representa (especificidad 1), que aumenta con la tasa de falsos positivos. El eje vertical de la curva representa la sensibilidad, que aumenta con la tasa de verdaderos positivos. Por lo tanto, para un valor de corte particular seleccionado, puede determinarse el valor de (especificidad 1) y puede obtenerse una sensibilidad correspondiente. El área bajo la curva ROC es una medida de la probabilidad de que el nivel del marcador medido permita la identificación correcta de una enfermedad o afección. Por lo tanto, el área bajo la curva ROC puede usarse para determinar la eficacia de la prueba.
En consecuencia, la invención comprende la administración de un antibiótico adecuado para el tratamiento sobre la base de la información obtenida por el método descrito en la presente descripción.
Tal como se usa en la presente descripción, los términos "que comprende" e "que incluye" o variantes gramaticales de los mismos deben interpretarse como una especificación de las características, números enteros, etapas o componentes establecidos, pero no impiden la adición de una o más características adicionales, números enteros, etapas, componentes o grupos de los mismos. Este término abarca los términos "que consiste en" y "que consiste esencialmente en".
Por lo tanto, los términos "que comprende'V'que incluye'V'que tiene" significan que puede/puede estar presente cualquier otro componente (o también características, números enteros, etapas y similares). El término "que consiste en" significa que no está presente ningún otro componente (o características, números enteros, etapas y similares). El término "que consiste esencialmente en" o variantes gramaticales del mismo, cuando se usa en la presente descripción, debe interpretarse como una especificación de las características, números enteros, etapas o componentes establecidos, pero no impide la adición de una o más características, números enteros, etapas, componentes o grupos adicionales de los mismos, pero solo si las características adicionales, números enteros, etapas, componentes o grupos de los mismos no alteran materialmente las características básicas y novedosas de la composición, dispositivo o método reivindicado.
Por lo tanto, el término "que consiste esencialmente en" significa que pueden estar presentes otros componentes
específicos (o características, números enteros, etapas y similares), es decir, aquellos que no afectan materialmente a las características esenciales de la composición, dispositivo o método. En otras palabras, el término "que consiste esencialmente en" (que puede intercambiarse en la presente descripción con el término "que comprende sustancialmente"), permite la presencia de otros componentes en la composición, dispositivo o método además de los componentes obligatorios (o características similares), números enteros, etapas y similares), siempre que las características esenciales del dispositivo o método no se vean afectadas materialmente por la presencia de otros componentes.
El término "método" se refiere a las maneras, los medios, las técnicas y los procedimientos para realizar una tarea determinada, que incluye, pero no se limita a, las maneras, los medios, las técnicas y los procedimientos conocidos o desarrollados fácilmente a partir de las maneras, los medios, las técnicas y los procedimientos conocidos por practicantes de las técnicas químicas, biológicas y biofísicas.
La presente invención se describe adicionalmente con referencia a los siguientes ejemplos no limitantes.
Ejemplos
Métodos de los ejemplos:
Diseño del estudio y pacientes:
Este estudio es un análisis secundario del ensayo controlado con placebo de la terapia antimicrobiana guiada por selenito de sodio y procalcitonina en sepsis grave (SISPCT), que se realizó en 33 unidades de cuidados intensivos (UCI) multidisciplinarias en toda Alemania desde noviembre de 2009 hasta febrero de 2013 (26). Los criterios de elegibilidad incluyeron pacientes adultos >18 años que presentaban sepsis grave o choque séptico de nueva aparición (<24 horas), de acuerdo con la definición de SEPSIS-1 del Comité de la conferencia de consenso ACCP/SCCM, y clasificados adicionalmente de acuerdo con las definiciones de 2016 (sepsis-3 y choque séptico-3) (4). Los detalles del diseño del estudio, la recopilación y el manejo de datos se describieron anteriormente (26). El comité de ética del hospital universitario de Jena y todos los demás centros aprobaron el estudio y se obtuvo el consentimiento informado por escrito cuando fue necesario.
Mediciones de los biomarcadores:
Los pacientes se inscribieron hasta 24 horas después del diagnóstico de sepsis grave o choque séptico y la PCT, PCR y lactato se midieron inmediatamente después. La PCT se midió en los dispositivos con un intervalo de medición de 0,02 - 5000 ng/ml, y una sensibilidad de ensayo funcional y un límite inferior de detección de al menos 0,06 ng/ml y 0,02 ng/ml, respectivamente. Se recopilaron las muestras de sangre adicionales de todos los pacientes y se almacenaron en el laboratorio central del estudio en Jena a -80 °C. Las concentraciones plasmáticas de MR-proADM se midieron retrospectivamente (Kryptor®, Thermo Fisher Scientific, Alemania) con un límite de detección de 0,05 nmol/l. Las puntuaciones de la gravedad clínica, que incluyen la evaluación secuencial de insuficiencia de órganos (SOFA), la evaluación de salud crónica y fisiológica aguda (APACHE) II y la puntuación fisiológica aguda simplificada (SAPS) II, se tomaron al momento de la inscripción en el estudio.
Análisis estadístico:
Las diferencias en las características demográficas y clínicas con respecto a la mortalidad a los 28 días se evaluaron mediante la prueba x2 para variables categóricas y la prueba t de Student o U de Mann-Whitney para variables continuas, de acuerdo con la normalidad de la distribución. Las variables con distribución normal y no normal se expresaron como media (desviación estándar) y mediana [primer cuartil - tercer cuartil], respectivamente. La asociación entre la mortalidad y cada biomarcador y la puntuación clínica en todos los momentos se evaluó mediante el área bajo las curvas características operativas del receptor (AUROC) y el análisis de regresión de Cox, con análisis multivariante corregido por edad y presencia de comorbilidades y choque séptico. Los pacientes se clasificaron además en tres subgrupos de gravedad (baja, intermedia y alta) en función del cálculo de dos valores de corte AUROC en la población total para cada biomarcador y puntuación clínica en cada momento, con una sensibilidad y especificidad previamente definidas de cerca de 90 % Posteriormente se identificó un subgrupo de pacientes clínicamente estables sin ningún procedimiento o complicación asociada a la UCI (que incluyen los procedimientos de limpieza de focos, cirugía de emergencia, aparición de nuevas infecciones, transfusión de hemoderivados, infusión de coloides, ventilación mecánica invasiva, reemplazo renal/hepático) o terapia vasopresora y un deterioro en los signos y síntomas clínicos generales del paciente), y otro grupo identificado con concentraciones bajas de MR-proADM correspondientes que no habían mostrado ningún aumento desde la medición anterior. Se calcularon las tasas de mortalidad y la duración media de la estancia en ambos grupos y se compararon con el grupo de pacientes que fueron dados de alta en cada momento específico.
Por último, se construyeron dos modelos que estratifican a los pacientes con cambios en la PCT del 20 % (desde el inicio del estudio hasta el día 1, según las disminuciones promedio de PCT observadas durante este período) y el 50 % (desde el inicio del estudio hasta el día cuatro, según un modelo construido previamente (26)). Los subgrupos de
pacientes se identificaron posteriormente en función de los niveles de gravedad de MR-proADM y se calcularon las tasas de mortalidad respectivas. El riesgo de mortalidad dentro de cada subgrupo se calculó mediante análisis de regresión de Cox y se ilustró mediante curvas de Kaplan-Meier. El riesgo previsto de desarrollar nuevas infecciones y la necesidad de procedimientos de limpieza de focos y cirugía de emergencia durante los días 4 a 7 se investigaron posteriormente en el modelo del inicio del estudio al día 4. Todos los datos se analizaron mediante el uso del software estadístico R (versión 3.1.2).
Ejemplo 1: características del paciente
Las características de los pacientes al momento de la inscripción en el estudio se resumen en la tabla 1.
Se analizaron un total de 1089 pacientes con sepsis grave (13,0 %) o choque séptico (87,0%), con 445 (41,3 %) y 633 (58,7 %) pacientes que también cumplían los criterios de sepsis-3 y choque séptico-3, respectivamente. Los pacientes inscritos tenían una edad promedio de 65,7 (13,7) años y una puntuación SOFA media de 10,0 (3,3) puntos. La tasa de mortalidad por todas las causas a los 28 días (N = 1076) fue del 26,9 % (sepsis-3: 20,0 %; choque séptico-3: 32,1 %), con una tasa de mortalidad hospitalaria del 33,4 % (sepsis-3: 24,4 %; choque séptico: 40,4 %). Se encontraron infecciones con origen en un solo foco en 836 pacientes (77,7 %), con orígenes más prevalente neumológicos (N = 324; 30,1 %), intraabdominales (N = 252; 23,4 %), urogenitales (N = 57; 5,3%) y óseas/tejido blando (N = 50; 4,6 %). Las tasas de mortalidad correspondientes fueron 26,5 %, 24,6 %, 22,8 % y 28,0 %, respectivamente. Se encontraron múltiples orígenes de infección en 240 (22,3 %) pacientes. Las causas más comunes de mortalidad incluyeron insuficiencia multiorgánica inducida por sepsis (N = 132; 45,7 %), choque séptico refractario (N = 54; 18,7 %), muerte por enfermedad preexistente (N = 35; 12,1 %) e insuficiencia respiratoria aguda (N = 17; 5,9 %). Otras causas, como el choque cardiogénico y hemorrágico, la embolia pulmonar, el edema cerebral, el infarto de miocardio y la arritmia cardíaca, representaron una tasa de mortalidad combinada del 8,6 %. Se aplicó una limitación de la terapia al 3,4 % de los pacientes.
Ejemplo 2: asociación de biomarcadores al inicio del estudio y las puntuaciones clínicas con la mortalidad
El análisis de regresión de Cox univariado y multivariado encontró que la MR-proADM tenía la asociación más fuerte con la mortalidad a los 28 días en la población total de pacientes, así como también dentro de los subgrupos de sepsis-3 y choque séptico-3 (Tabla 2). El análisis AUROc correspondiente encontró diferencias significativas en todas las comparaciones de biomarcadores y puntuaciones clínicas con la MR-proADM, además de APACHE II (subgrupo de pacientes con sepsis-3).
También se encontraron resultados similares para la predicción de mortalidad de 7 días, 90 días, UCI y hospitalaria (Tabla 3), con la adición de MR-proADM a todos los biomarcadores potenciales y combinaciones de la puntuación clínica (N = 63) lo que aumenta significativamente la capacidad pronóstica (Tabla 4).
Ejemplo 3: identificación de pacientes de alto riesgo
La población total de pacientes se estratificó aún más de acuerdo con los niveles de gravedad SOFA existentes y el rendimiento de los biomarcadores y puntuaciones clínicas para predecir la mortalidad a los 28 días evaluada en cada subgrupo. La MR-proADM mostró la precisión más alta de todos los parámetros en los subgrupos de SOFA de gravedad baja (SOFA <7) y moderada (8< SOFA <13) (Tabla 5; tabla 6).
Posteriormente se calcularon dos valores de corte de la MR-proADM correspondientes para identificar los subgrupos de gravedad baja (<2,7 nmol/l) y alta (>10,9 nmol/l) al inicio del estudio. En comparación con SOFA, podría hacerse una reclasificación más precisa en ambos niveles bajos (MR-proADM frente a SOFA: N = 265 frente a 232; 9,8 % frente a 13,8 % de mortalidad) y alta (MR-proADM frente a SOFA: N = 161 frente a 155; 55,9 % frente a 41,3 %) de los valores de corte de la gravedad (Tabla 7).
Un subgrupo de 94 pacientes (9,3 %) con concentraciones altas de MR-proADM y la correspondiente SOFA baja o intermedia tuvo tasas de mortalidad a los 28 y 90 días del 57,4 % y el 68,9 %, respectivamente, en comparación con el 19,8 % y el 30,8 % en la población restante de pacientes con valores SOFA bajos e intermedios. Pueden encontrarse patrones similares para SAPS II, APACHE Il y lactato, respectivamente (Tablas 8-10).
Ejemplo 4: identificación de pacientes de bajo riesgo a lo largo de la estancia en la UCI
La cohorte del estudio comprende un subconjunto de pacientes clínicamente estables que no se enfrentaron a procedimientos o complicaciones relacionados con la UCI, tales como procedimientos de limpieza de focos, cirugía de emergencia, nuevas infecciones, transfusión de hemoderivados, infusión de coloides, ventilación mecánica invasiva, reemplazo renal/hepático, deterioro de los signos y síntomas clínicos generales del paciente. Este grupo de pacientes clínicamente estables se clasificó como pacientes de bajo riesgo.
La MR-proADM mostró la asociación más fuerte con la mortalidad a los 28 días en todos los momentos posteriores (Tabla 11), y podría proporcionar un valor de corte estable de <2,25 nmol/l para identificar una población de
pacientes de bajo riesgo, lo que da como resultado la clasificación de mayor número de pacientes con tasas de mortalidad más bajas en comparación con otros biomarcadores y puntuaciones clínicas (Tabla 12). En consecuencia, pudieron identificarse 290 pacientes de gravedad baja de MR-proADM el día 4, de los cuales 79 (27,2 %) estaban clínicamente estables y no tenían aumento en las concentraciones de MR-proADM desde la última medición (Tabla 13). Pudo encontrarse una concentración de MR-proADM continuamente baja en 51 (64,6 %) pacientes, mientras que se pudo observar una disminución de un nivel de gravedad de nivel intermedio a bajo en 28 (35,4 %) pacientes. La estancia media en la UCI fue de 8 [7 -10] días, con una tasa de mortalidad a los 28 y 90 días del 0,0 % y el 1,4 %, respectivamente. En comparación, solo 43 pacientes fueron dados de alta de la UCI el día 4, con una tasa de mortalidad a los 28 y 90 días del 2,3 % y el 10,0 %. El análisis de las concentraciones de MR-proADM dentro de este grupo de pacientes indicó un intervalo de valores, con 20 (52,6 %), 16 (42,1 %) y 2 (5,3 %) pacientes con concentraciones de gravedad baja, intermedia y alta, respectivamente. Se encontraron resultados similares para los pacientes que permanecieron en la UCI los días 7 y 10.
La MR-proADM con un valor de corte estable de <2,25 nmol/l podría identificar un mayor número de pacientes de bajo riesgo con tasas de mortalidad más bajas en comparación con otros biomarcadores y puntuaciones clínicas. En base a ese hallazgo, más pacientes podrían ser dados de alta de la UCI en comparación con las clasificaciones sin usar ADM. Al dar de alta a más pacientes, el hospital puede ocupar camas de UCI de manera más eficiente y beneficiarse de los costos evitados.
Ejemplo 5: impacto adicional de la MR-proADM en la terapia guiada por procalcitonina
El análisis de regresión de Cox dependiente del tiempo indicó que el aumento adicional significativo más temprano en la información pronóstica a los valores al inicio del estudio de MR-proADM podría observarse el día 1, con mediciones únicas o acumulativas posteriores que dieron como resultado asociaciones significativamente más fuertes con la mortalidad a los 28 días (Tabla 14). Por lo tanto, se construyeron dos modelos de algoritmos guiados por PCT que investigaron los cambios de PCT desde el inicio del estudio hasta el día 1 o el día 4, con el correspondiente análisis de los subgrupos basado en las clasificaciones de gravedad de la MR-proADM.
Se encontró que los pacientes con concentraciones de PCT decrecientes de >20 % desde el inicio del estudio hasta el día 1 (Tabla 15 y tabla 16) o >50 % desde el inicio del estudio hasta el día 4 (Tabla 17 y tabla 18) tenían tasas de mortalidad a los 28 días del 18,3 % (N = 458) y 17,1 % (N = 557), respectivamente. Esto disminuyó al 5,6 % (N = 125) y al 1,8 % (N = 111) cuando los pacientes tenían niveles continuamente bajos de MR-proADM, aunque aumentó al 66,7 % (N = 27) y al 53,8 % (N = 39) en pacientes con valores continuamente altos de MR-proADM (HR [IC de 95 %]: 19,1 [8,0 -45,9] y 43,1 [10,1 -184,0]).
Además, los pacientes con valores de PCT decrecientes de >50 % (inicio del estudio al día 4), pero concentraciones de MR-proADM continuamente altas o intermedias, tenían un riesgo significativamente mayor de desarrollar infecciones nosocomiales posteriores (HR [IC de 95 %]: concentraciones altas: 3,9 [1,5 - 10,5]; concentraciones intermedias: 2,4 [1,1 - 5,1] frente a pacientes con concentraciones continuamente bajas; concentraciones intermedias: 2,9 [1,2 - 6 ,8 ]) frente a concentraciones decrecientes intermedias a bajas), o que requieren cirugía de emergencia (HR [IC de 95 %]: concentraciones intermedias: 2,0 [1,1 - 3,7] frente a concentraciones decrecientes intermedias a bajas). Por el contrario, los pacientes con concentraciones crecientes de intermedias a altas tenían más probabilidades de requerir limpieza del origen infeccioso en comparación con aquellos con niveles de valores continuos intermedios (HR [IC de 95 %]: 3,2 [1,3 - 7,6]) o decrecientes (HR [IC 95 de %]): intermedio a bajo: 8,7 [3,1 - 24,8]); alto a intermedio: 4,6 [1,4 - 14,5]). Cuando los niveles de PCT no lograron disminuir en >50 %, se observó un riesgo significativamente mayor de requerir cirugía de emergencia si las concentraciones de MR-proADM estaban en un nivel continuamente alto (HR [IC de 95 %]: 5,7 [1,5 - 21,9]) o nivel intermedio (HR [IC de 95 %]: 4,2 [1,3 -13,2]), en lugar de ser continuamente bajo.
Ejemplo 6 : asociación de los biomarcadores al inicio del estudio y puntuaciones clínicas con la mortalidad
La MR-proADM mostró la asociación más fuerte en pacientes con infecciones neumológicas e intraabdominales, así como también en pacientes con infecciones Gram positivas, independientemente del origen infeccioso (Tablas 19 20). Cuando los pacientes se agruparon de acuerdo con el historial de cirugía electiva, de emergencia no quirúrgica y de emergencia operatoria que dio lugar al ingreso en la UCI, la MR-proADM proporcionó la asociación más sólida y equilibrada con la mortalidad a los 28 días en todos los grupos (Tabla 21).
Ejemplo 7: correlación de biomarcadores y puntuaciones clínicas con SOFA al inicio del estudio y el día 1
La MR-proADM tuvo la mayor correlación de todos los biomarcadores con la puntuación SOFA al inicio del estudio, que aumentó significativamente cuando los valores iniciales se correlacionaron con las puntuaciones SOFA del día 1. La mayor correlación pudo encontrarse entre MR-proADM y SOFA en el día 10, con diferencias entre las subpuntuaciones SOFA individuales encontradas a lo largo (Tablas 22-24).
Ejemplo 8: identificación de pacientes de alto riesgo
Pudieron encontrarse resultados similares en un subgrupo de 124 pacientes (12,0 %) con concentraciones altas de MR-proADM y valores de SAPS II bajos o intermedios (subgrupo de MR-proADM alto: [54,8 % y 65,6 % de mortalidad]; población restante de SApS II [ 19,7 % y 30,0 % de mortalidad]), así como también en 109 (10,6 %) pacientes con valores de APACHE II bajos o intermedios (subgrupo MR-proADM alto: [56,9 % y 66,7 % de mortalidad]; población restante de APACHE II: [19,5 % y 30,3% de mortalidad]).
Ejemplo 9: terapia guiada por procalcitonina (PCT) mejorada mediante la combinación de PCT y ADM
Se construyeron dos modelos de algoritmos guiados por PCT para investigar los cambios de PCT desde el inicio del estudio hasta el día 1 o el día 4, con el correspondiente análisis de subgrupos basado en las clasificaciones de gravedad de MR-proADM (Tablas 25-30).
Los ejemplos anteriores muestran un valor adicional para ADM en pacientes que tienen una disminución de PCT de <20 % o <50 %, así como también en pacientes en los que la PCT disminuyó en >20 % o >50 %. Sin embargo, un análisis adicional demuestra que ADM puede ser un complemento independientemente del % de disminución o incluso aumento de PCT. Los valores decrecientes de PCT podrían reflejar pacientes en los que el tratamiento con antibióticos parece estar funcionando, por lo tanto, el médico cree que están en una buena forma de supervivencia (es decir, matar la causa raíz de la sepsis, la bacteria, debería hacer que el paciente mejore).
Por ejemplo, algunos pacientes tienen niveles de PCT decrecientes desde el inicio del estudio (día de ingreso) hasta el día 1 con una tasa de mortalidad a los 28 días del 19 %. Al medir adicionalmente la ADM, puede concluir que los pacientes con ADM baja tienen una probabilidad mucho mayor de supervivencia o una probabilidad mucho menor de morir (Tabla 25; compara una tasa de mortalidad del 19 % que PCT decreciente solo frente a 5 % de tasa de mortalidad PCT baja ADM). Al tener un riesgo reducido de morir, los pacientes pueden ser dados de alta de la UCI con más confianza, o se requieren menos pruebas de diagnóstico (es decir, usted sabe que están en un buen camino hacia la recuperación).
Por otro lado, deben considerar nuevas medidas para aquellos con un valor alto de ADM. Tienen un riesgo mucho mayor con respecto a la mortalidad (compara una tasa de mortalidad del 19 % que PCT decreciente solo frente a una tasa de mortalidad del 58,8 % PCT ADM alta). El médico cree que el paciente está mejorando debido a la disminución del valor de PCT, pero en realidad la concentración de ADM sigue siendo la misma. Por lo tanto, puede concluirse que el tratamiento no está funcionando y debe adaptarse lo antes posible).
De manera similar, ADM puede ayudar a estratificar a aquellos pacientes con valores crecientes de PCT (Tabla 25). Desarrollo de nuevas infecciones.
Los cambios de PCT y MR-proADM se analizaron en dos modelos, ya sea desde el inicio del estudio hasta el día 1, o desde el inicio del estudio hasta el día 4. Los pacientes se agruparon de acuerdo con los cambios generales de PCT y los niveles de gravedad de MR-proADM.
Posteriormente se calculó el número de nuevas infecciones durante los días 1, 2, 3 y 4 (Tabla 26) y durante los días 4, 5, 6 y 7 (Tabla 27) en cada paciente que estuvo presente el día 1 o el día 4 respectivamente. En algunos casos, los pacientes fueron dados de alta durante el período de observación. Se supone que no se desarrollaron nuevas infecciones después del lanzamiento. Los pacientes con infecciones múltiples durante los días de observación se contaron como una sola infección nueva.
Como consecuencia clínica, los pacientes con concentraciones elevadas de MR-proADM deberían potencialmente ser tratados con un antibiótico de amplio espectro al ingreso en la UCI, junto con otros, para detener el desarrollo de nuevas infecciones. Debe tenerse especial cuidado con estos pacientes debido a su alta susceptibilidad a contraer nuevas infecciones.
Requisito para la limpieza del foco
Los cambios de PCT y MR-proADM se analizaron en dos modelos, ya sea desde el inicio hasta el día 1, o desde el inicio hasta el día 4. Los pacientes se agruparon de acuerdo con los cambios generales de PCT y los niveles de gravedad de MR-proADM.
Posteriormente se calculó el número de eventos de limpieza de foco durante los días 1, 2, 3 y 4 (Tabla 28) y durante los días 4, 5, 6 y 7 (Tabla 29) en cada paciente que estuvo presente el día 1 o el día 4 respectivamente. En algunos casos, los pacientes fueron dados de alta durante el período de observación.
Requisito de cirugía de emergencia
Los cambios de PCT y MR-proADM se analizaron en dos modelos, ya sea desde el inicio del estudio hasta el día 1, o desde el inicio del estudio hasta el día 4. Los pacientes se agruparon de acuerdo con los cambios generales de
PCT y los niveles de gravedad de MR-proADM.
Posteriormente se calculó el número de necesidades/eventos de cirugía de urgencia durante los días 1, 2, 3 y 4 (Tabla 30) en cada paciente que estuvo presente el día 1. En algunos casos, los pacientes fueron dados de alta durante el período de observación.
Ejemplo 10: requisito de cambio o modificación de antibiótico
Cuando se combina dentro de un algoritmo antibiótico guiado por PCT, MR-proADM puede estratificar a aquellos pacientes que requerirán un cambio o modificación futura en la terapia antibiótica, de aquellos que no lo requerirán. Los cambios de PCT y MR-proADM se analizaron en dos modelos, ya sea desde el inicio del estudio hasta el día 1, 0 desde el inicio del estudio hasta el día 4. Los pacientes se agruparon de acuerdo con los cambios generales de PCT y los niveles de gravedad de MR-proADM.
Posteriormente se calculó el porcentaje de cambios de antibiótico requeridos en el día 4 para cada grupo de pacientes (Tablas 31 y 32).
En pacientes con valores de PCT decrecientes >50 %
Los pacientes con concentraciones crecientes de MR-proADM, de un nivel de gravedad bajo a intermedio, tenían más probabilidades de requerir una modificación en la terapia con antibióticos en el día 4 que aquellos que tenían niveles continuamente bajos (Relación de probabilidades [IC de 95 %]: 1,5 [0,6 -4,1]).
En pacientes con valores de PCT decrecientes <50 %
Los pacientes con concentraciones crecientes de MR-proADM, de un nivel de gravedad intermedio a alto, o concentraciones continuamente altas, también tenían más probabilidades de requerir cambios en su terapia antibiótica en el día 4 que los pacientes con concentraciones continuamente bajas de MR-proADM (Relación de probabilidades [IC de 95 %]: 5,9 [1,9 -18,1] y 2,9 [0,8 -10,4], respectivamente).
Conclusión
A pesar de las concentraciones crecientes de PCT, ya sea desde el inicio del estudio hasta el día 1, o desde el inicio del estudio hasta el día 4, los pacientes con concentraciones continuamente bajas de MR-proADM tuvieron modificaciones significativamente menores en su tratamiento antibiótico prescrito que aquellos con concentraciones continuamente intermedias o altas. Como consecuencia clínica, ante concentraciones crecientes de PCT, el médico debe comprobar los niveles de MR-proADM del paciente antes de decidir cambiar de antibiótico. Aquellos con concentraciones bajas de MR-proADM deben ser considerados para una dosis mayor o una potencia mayor del mismo antibiótico antes de considerar los cambios. Aquellos con concentraciones más altas de MR-proADM deben ser considerados para cambios de antibiótico más tempranos (es decir, en los días 1 a 3, a diferencia del día 4). Ejemplo 11: Identificación de pacientes con niveles anormales de plaquetas e identificación de pacientes de alto riesgo con trombocitopenia (Tablas 33, 34 y 35).
Los niveles de proadrenomedulina y procalcitonina se midieron y analizaron con respecto al recuento de trombocitos, la tasa de mortalidad y la transfusión de plaquetas al inicio del estudio y el día 1. El aumento de las concentraciones de proADM y PCT se correlaciona con la disminución del número de plaquetas (<150 000 por jl) que reflejan trombocitopenia. La disminución más fuerte del recuento de plaquetas se observó en pacientes con los niveles más altos de proADM al inicio del estudio. Además, el aumento de las concentraciones de proADM y PCT estuvo en línea con los pacientes que requirieron una terapia de transfusión de plaquetas. También pudo confirmarse que una mayor tasa de mortalidad está asociada con pacientes que tienen trombocitopenia y niveles elevados de proADM (>6 nmol/l) y PCT (>7 ng/ml).
Los niveles de pro-ADM se investigaron en pacientes que tenían niveles normales de trombocitos al inicio del estudio para ver si el aumento de la proADM podía predecir la trombocitopenia. El 39,4 % de los pacientes con niveles de proADM continuamente elevados al inicio y en el día 1 (proADM >10,9 nmol/l) desarrollaron trombocitopenia. El 25,6 % de los pacientes con niveles elevados de proADM al inicio (proADM >2,75 nmol/l) y el día 1 (proADM >9,5 nmol/l) desarrollaron trombocitopenia. El 14,7 % de los pacientes con un nivel de proADM continuamente bajo al inicio y en el día 1 (proADM <2,75 nmol/l) desarrollaron trombocitopenia. El aumento del nivel de proADM se correlacionó con la gravedad del episodio trombocitopénico y el aumento de la tasa de mortalidad asociada (proADM >10,9 nmol/l tasa de mortalidad del 51 %; proADM <2,75 nmol/l tasa de mortalidad del 9,1 %). El ejemplo 11 se refiere a las tablas 33-35.
Discusión de los ejemplos
Una evaluación precisa y rápida de la gravedad de la enfermedad es crucial para iniciar el tratamiento más adecuado lo antes posible. De hecho, un tratamiento tardío o insuficiente puede conducir a un deterioro general del estado clínico del paciente, lo que hace que el tratamiento adicional se vuelva menos efectivo y una mayor probabilidad de un peor resultado general (8, 27). Como resultado, se han propuesto numerosos biomarcadores y puntuaciones de gravedad clínica para satisfacer esta necesidad clínica insatisfecha, con la puntuación de evaluación secuencial de insuficiencia de órganos (SOFA) actualmente destacado como la herramienta más apropiada, lo que resulta en su papel central en la definición de sepsis-3 de 2016 (4). Este análisis secundario del ensayo SISPCT (26), por primera vez, comparó las medidas secuenciales de los biomarcadores convencionales y puntuaciones clínicas, tales como lactato, procalcitonina (PCT) y SOFA, con las del marcador de disfunción microcirculatoria, MR-proADM, en una gran población de pacientes con sepsis grave y choque séptico.
Nuestros resultados indican que el uso inicial de MR-proADM dentro de las primeras 24 horas después del diagnóstico de sepsis resultó en la asociación más fuerte con la mortalidad a corto, mediano y largo plazo en comparación con todos los demás biomarcadores o puntuaciones. Estudios anteriores confirman en gran medida nuestros hallazgos (17, 28, 29), sin embargo, los resultados contradictorios (30) pueden explicarse en parte por los tamaños de muestra más pequeños analizados, así como también por otros factores destacados en este estudio, tales como las especies microbianas, el origen de la infección y los antecedentes quirúrgicos previos al desarrollo de sepsis, todos los cuales pueden influir en el rendimiento de los biomarcadores, lo que se suma a la posible variabilidad de los resultados en poblaciones de estudio pequeñas. Además, nuestro estudio también confirma estrechamente los resultados de una investigación anterior (17), que destaca el rendimiento superior de MR-proADM en pacientes con disfunción orgánica de gravedad baja e intermedia. De hecho, Andaluz-Ojeda y otros, (17) otorgan una importancia significativa al grupo de pacientes con niveles bajos de disfunción orgánica, ya que "este grupo representa la presentación más temprana en el curso clínico de la sepsis y/o la forma menos grave de la enfermedad". No obstante, pudo mantenerse un rendimiento razonable en todos los grupos de gravedad con respecto a la predicción de mortalidad, lo que también sucedió en ambos grupos de pacientes definidos de acuerdo con los criterios de sepsis-3 y choque séptico-3.
El análisis de las mediciones secuenciales tomadas después del inicio de la sepsis permitió la identificación de grupos de pacientes específicos según la gravedad de la enfermedad. La identificación de pacientes de bajo y alto riesgo fue de gran interés en nuestro análisis. En muchas UCI, la demanda de camas de UCI puede exceder periódicamente la disponibilidad, lo que puede conducir a un triaje inadecuado, un racionamiento de recursos y una disminución posterior en la probabilidad de un ingreso correcto en la UCI (32-35). En consecuencia, una evaluación precisa de los pacientes con bajo riesgo de mortalidad hospitalaria que pueden ser elegibles para un alta temprana de la UCI a una unidad de cuidados intermedios puede ser de gran beneficio. En cada momento medido dentro de nuestro estudio, la MR-proADM podría identificar una mayor cantidad de pacientes de baja gravedad con las tasas más bajas de mortalidad en la UCI, el hospital y los 28 días. Un análisis adicional del grupo de pacientes con una gravedad baja y sin otras terapias específicas de la UCI indicó que se observaron 4 días adicionales de estadía en la UCI en cada momento después de que se tomaron las mediciones de los biomarcadores. En comparación con la población de pacientes que fueron dados de alta en cada momento, un enfoque basado en biomarcadores para identificar con precisión a los pacientes de baja gravedad resultó en una disminución de las tasas de mortalidad a los 28 y 90 días. De hecho, los pacientes que fueron dados de alta tenían una variedad de concentraciones de MR-proADM de gravedad baja, intermedia y alta, lo que posteriormente se reflejó en una tasa de mortalidad más alta. Sin embargo, se desconoce si una cantidad de pacientes dentro de este grupo aún requerían tratamiento adicional en la UCI por problemas no relacionados con la microcirculación y que no ponían en peligro la vida, o si había camas disponibles en una unidad de cuidados intermedios. Sin embargo, tal enfoque basado en biomarcadores para el alta de la UCI, además del juicio médico, puede mejorar la estratificación correcta del paciente, con beneficios clínicos acompañados y posibles ahorros de costos.
Por el contrario, la identificación de pacientes de alto riesgo que pueden requerir un tratamiento temprano y específico para prevenir un deterioro clínico posterior puede tener una relevancia clínica aún mayor. Ya se han observado ahorros sustanciales de costos y reducciones en el uso de antibióticos siguiendo un algoritmo guiado por PCT en el estudio SISPCT y otros ensayos (26, 36, 37), sin embargo, todavía pueden observarse tasas de mortalidad relativamente altas incluso cuando los valores de PCT parecen estar disminuyendo constantemente. Nuestro estudio reveló que la adición de MR-proADM al modelo de disminución de PCT durante los días subsiguientes en la UCI permitió la identificación de grupos de pacientes de riesgo bajo, intermedio y alto, con niveles de gravedad de MR-proADM crecientes y decrecientes desde el inicio del estudio hasta el día 1, lo que proporciona una información sensible y una indicación temprana del éxito del tratamiento. Además, la predicción del requerimiento de limpieza de focos futuros o cirugía de emergencia, así como también la susceptibilidad para el desarrollo de nuevas infecciones, puede ser de gran beneficio para iniciar estrategias terapéuticas e intervencionistas adicionales, intentando de esta manera prevenir cualquier complicación clínica futura en una etapa temprana.
La fortaleza de nuestro estudio incluye el examen exhaustivo de varios subgrupos diferentes con gravedades de enfermedad baja y alta de una base de datos de ensayos aleatorios, al ajustar los posibles factores de confusión e incluir el tamaño de muestra más grande de pacientes con sepsis, caracterizados por las definiciones de SEPSIS 1 y 3, e información sobre la cinética de MR-proADM. En conclusión, la MR-proADM supera a otros biomarcadores y
puntuaciones de gravedad clínica en la capacidad de identificar el riesgo de mortalidad en pacientes con sepsis, tanto en el diagnóstico inicial como en el transcurso del tratamiento en la UCI. En consecuencia, la MR-proADM puede usarse como una herramienta para identificar los pacientes de alta gravedad que pueden requerir intervenciones diagnósticas y terapéuticas alternativas, y pacientes de baja gravedad que potencialmente pueden ser elegibles para un alta temprana de la UCI junto con la ausencia de terapias específicas de la UCI.
Tablas

continuación

Tabla 2. Predicción de la mortalidad a los 28 días tras el diagnóstico de sepsis

N: número; AUROC: área bajo la curva de operación del receptor; LR x2: HR: cociente de riesgos; IQR: intervalo intercuartílico. Todos los análisis multivariados se asociaron por p < 0,0001 con la mortalidad a los 28 días.
Tabla 3. Análisis de supervivencia para 7 días, 90 días, UCI y mortalidad hospitalaria

Todos los valores de p multivariados < 0,0001 excepto PCT y CRP para la mortalidad a los 7 días (0,0015 y 0,0843, respectivamente).
Tabla 4. Análisis de supervivencia para la MR-proADM cuando se adiciona a los biomarcadores individuales o untuaciones clínicas

El HR IQR [IC de 95 %] indica el cociente de riesgos para la MR-proADM en cada modelo bivariado o multivariado. 2 grados de libertad en cada modelo bivariado, comparado con 11 en cada modelo multivariado.
Tabla 5. Análisis AUROC para la predicción de la mortalidad a los 28 días basado en los niveles de gravedad SOFA

N: número; AUROC: área bajo la curva de operación del receptor; LR x2: HR: cociente de riesgos; IQR: intervalo intercuartílico.
Tabla 6. Análisis de supervivencia para la MR-proADM dentro de los diferentes grupos de gravedad de disfunción or ánica cuando se combinan con biomarcadores individuales o untuaciones clínicas

Tabla 7. Gru os de ravedad de la enfermedad SOFA MR- roADM corres ondientes a los 28 días

MR-proADM: proadrenomedulina regional media; SOFA: evaluación secuencial de insuficiencia de órganos
Tabla 8. Gru os de ravedad de la enfermedad SAPS II MR- roADM corres ondientes a los 28 días

MR-proADM: proadrenomedulina regional media; SAPS II: fisiológico agudo simplificado II
Tabla 9. Gru os de ravedad de la enfermedad APACHE II MR- roADM corres ondientes a los 28 días

MR-proADM: proadrenomedulina regional media; APACHE II: evaluación de la salud fisiológica aguda y crónica II
Tabla 10. Gru os de ravedad de la enfermedad lactato MR- roADM corres ondientes a los 28 días

MR-proADM: proadrenomedulina regional media
Tabla 11. Asociación de los biomarcadores SOFA con la mortalidad a los 28 días en los días 1,4, 7 10

Tabla 12. Grupos de gravedad de bajo y alto riesgo y tasas de mortalidad correspondientes a lo largo del tratamiento en la UCI
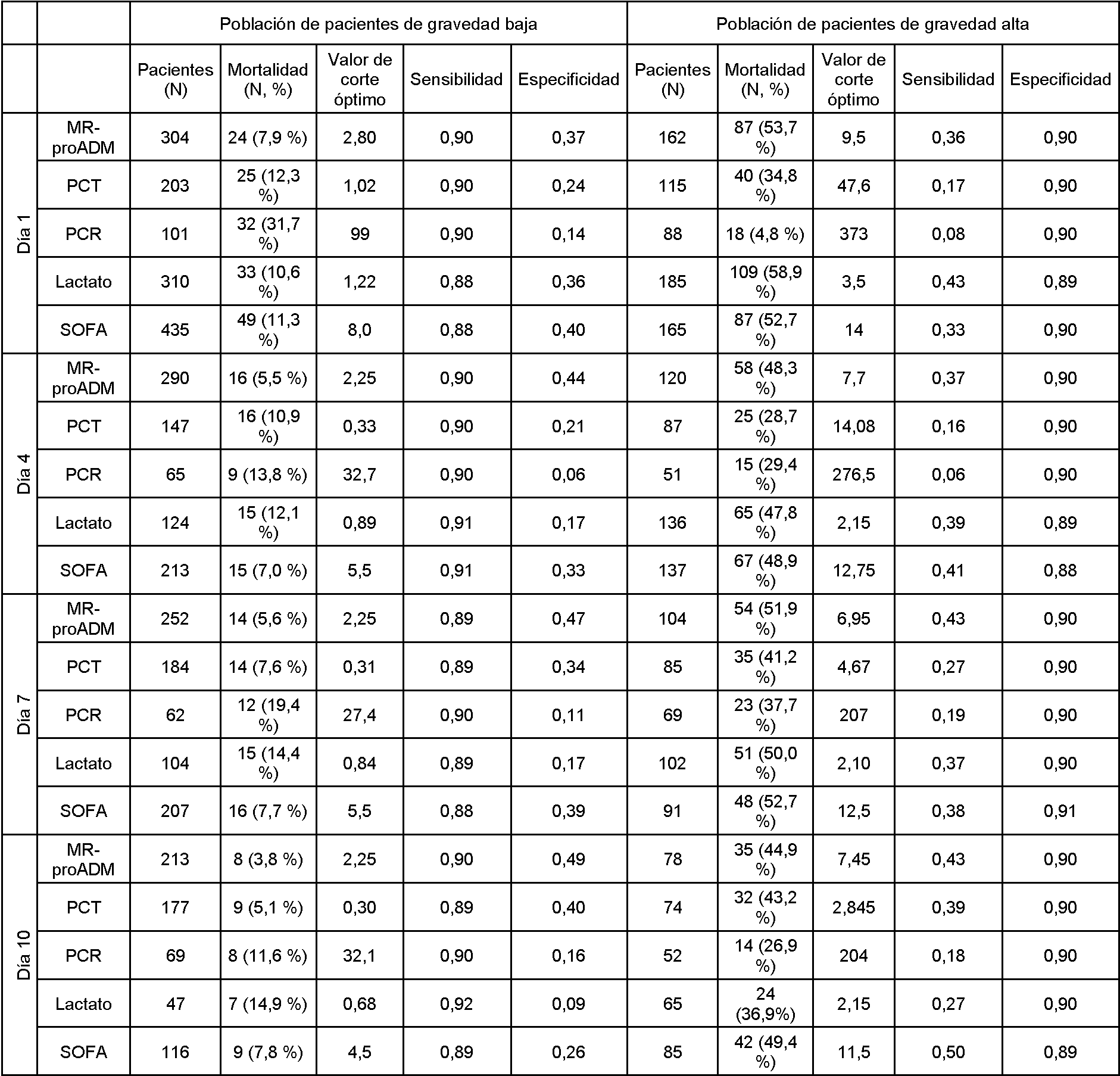
Tabla 13. Mortalidad y duración de la terapia en la UCI según las concentraciones de la MR-proADM y las tera ias es ecíficas de la UCI

* excluye las muertes en el mismo día o al día siguiente
Tabla 14. Regresiones de Cox dependientes del tiempo para adiciones únicas y acumulativas de la MR-roADM
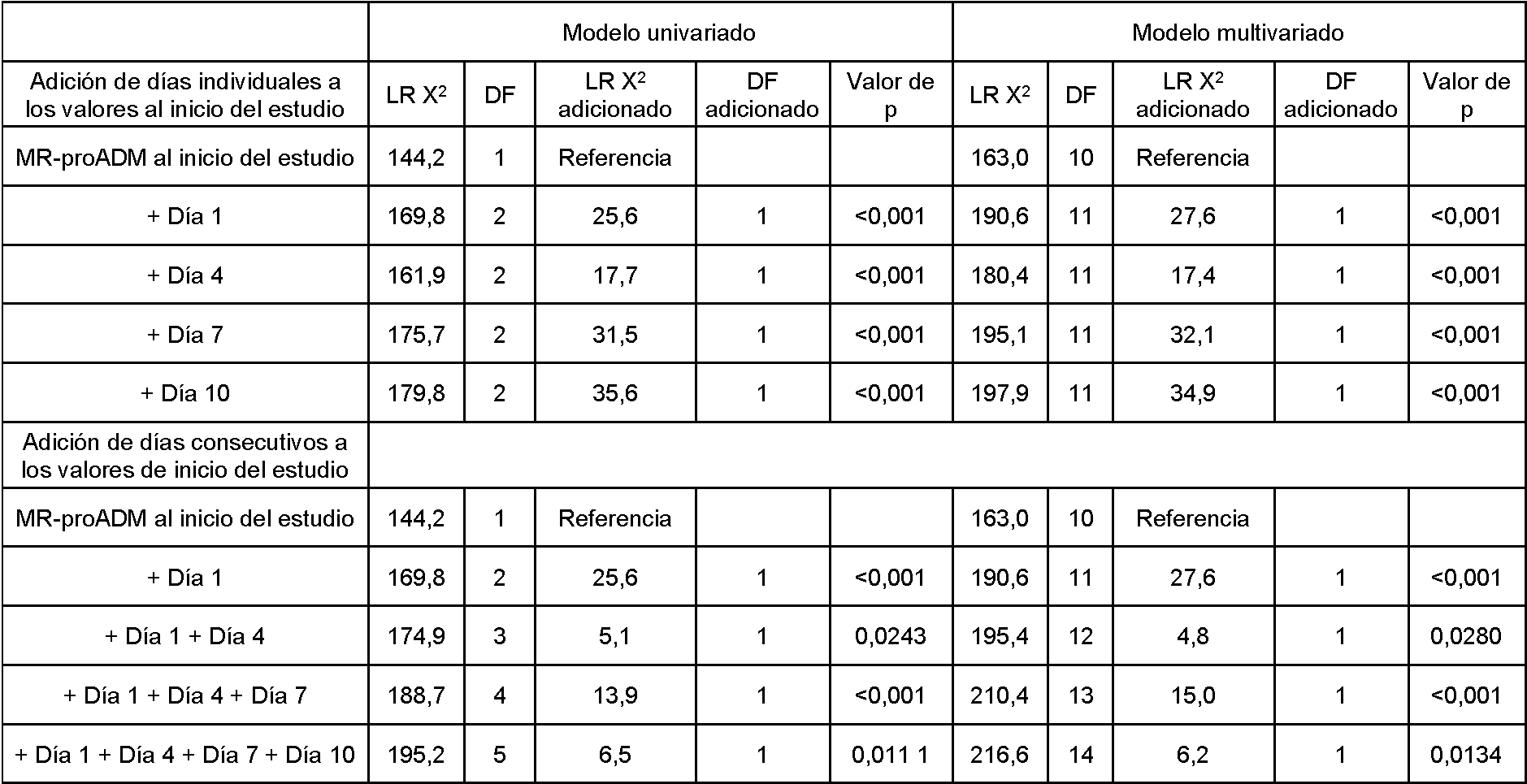
MR-proADM: proadrenomedulina regional media; DF: grados de libertad
Tabla 15. Tasas de mortalidad a los 28 90 días des ués de la cinética de PCT MR- roADM

Cocientes de riesgos para pacientes con: * valores continuamente intermedios frente a bajos; ** valores continuamente altos frente a intermedios *** valores continuamente altos frente a bajos; f valores bajos crecientes a intermedios frente a continuamente bajos; f f valores intermedios crecientes a altos frente a continuamente intermedios; $ valores decrecientes altos a intermedios frente a continuamente altos; $$ valores intermedios decrecientes a bajos frente a intermedios crecientes a altos. Los diagramas de Kaplan Meier ilustran los subgrupos de pacientes individuales o subgrupos crecientes o decrecientes agrupados.
Tabla 16. Tasas de mortalidad luego de los cambios en las concentraciones de PCT y los niveles de gravedad de la MR- roADM

Cocientes de riesgos para pacientes con: * valores continuamente intermedios frente a bajos; ** valores continuamente altos frente a intermedios *** valores continuamente altos frente a bajos; f valores bajos crecientes a intermedios frente a continuamente bajos; f f valores intermedios crecientes a altos frente a continuamente intermedios; $ valores decrecientes altos a intermedios frente a continuamente altos; $$ valores intermedios decrecientes a bajos frente a continuamente intermedios.
Tabla 17. Tasas de mortalidad a 28 y 90 días después de los cambios en las concentraciones de PCT y niveles de ravedad de la MR- roADM

Cocientes de riesgos para pacientes con: * valores continuamente intermedios frente a bajos; ** valores continuamente altos frente a intermedios *** valores continuamente altos frente a bajos; f valores bajos crecientes a intermedios frente a continuamente bajos; f f valores intermedios crecientes a altos frente a continuamente intermedios; $ valores decrecientes altos a intermedios frente a continuamente altos; $$ valores intermedios decrecientes a bajos frente a continuamente intermedios.
Tabla 18. Tasas de mortalidad hospitalaria y en la UCI seguida de los cambios en las concentraciones de PCT niveles de ravedad de la MR- roADM

Cocientes de riesgos para pacientes con: * valores continuamente intermedios frente a bajos; ** valores continuamente altos frente a intermedios *** valores continuamente altos frente a bajos; f valores bajos crecientes a intermedios frente a continuamente bajos; f f valores intermedios crecientes a altos frente a continuamente intermedios; $ valores decrecientes altos a intermedios frente a continuamente altos; $$ valores intermedios decrecientes a bajos frente a continuamente intermedios.
Tabla 19. Influencia del ori en infeccioso en la redicción de mortalidad a los 28 días

Los valores de AUROC de la MR-proADM son significativamente mayores que todos los demás parámetros excepto APACHE II en los orígenes neumológicos de la infección.
Tabla 20. Influencia de las es ecies microbianas en la redicción de mortalidad a los 28 días

Tabla 21. Influencia del modo de in reso a la UCI en la redicción de mortalidad a los 28 días

Tabla 22. Biomarcador al inicio del estudio y correlación de la puntuación clínica con SOFA al inicio del estudio en el día 1

* mediante el uso de los mismos pacientes al inicio del estudio que en el día 1
Tabla 23. Correlaciones de la MR-proADM al inicio del estudio con las subpuntuaciones SOFA al inicio del estudio en el día 1

Tabla 24. Correlaciones de los biomarcadores con las untuaciones SOFA a lo lar o del tratamiento en la UCI

Tabla 25. Mortalidades basadas en la gravedad de la MR-proADM y concentraciones crecientes o decrecientes de PCT - inicio del estudio hasta el día 1

Tabla 26. Cinética de la PCT desde el inicio del estudio hasta el día 1: desarrollo de nuevas infecciones durante los días 1, 2, 3 4.

Tabla 27. Cinética de la PCT desde el inicio del estudio hasta el día 4: desarrollo de nuevas infecciones durante los días 4, 5, 6 7.

Tabla 28. Cinética de la PCT desde el inicio del estudio hasta el día 1: requisito para la limpieza del foco durante los días 1, 2, 3 4.

Tabla 29. Cinética de la PCT desde el inicio del estudio hasta el día 4: requisito para la limpieza del foco durante los días 4, 5, 6 7.

Tabla 30. Cinética de la PCT desde el inicio del estudio hasta el día 1: requisito de la cirugía de emergencia durante los días 1, 2, 3 4.

Tabla 31. PCT creciente desde el inicio del estudio hasta el día 1: cambios de antibiótico en el día 4

Tabla 32. PCT creciente desde el inicio del estudio hasta el día 4: cambios de antibiótico en el día 4




Referencias
1. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 17 de abril de 2003;348(16):1546-1554.
2. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000-2012. JAMA. 02 de abril de 2014;311(13):1308-1316.
3. Vincent JL, Sakr Y, Sprung CL, y otros. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. Febrero de 2006;34(2):344-353.
4. Singer M, Deutschman CS, Seymour CW, y otros. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 23 de febrero de 2016;315(8):801-810.
5. Kopterides P, Siempos, II, Tsangaris I, Tsantes A, Armaganidis A. Procalcitonin-guided algorithms of antibiotic therapy in the intensive care unit: a systematic review and meta-analysis of randomized controlled trials. Crit Care Med. Noviembre de 2010;38(11):2229-2241.
6. de Jong E, van Oers JA, Beishuizen A, y otros. Efficacy and safety of procalcitonin guidance in reducing the duration of antibiotic treatment in critically ill patients: a randomised, controlled, open-label trial. Lancet Infect Dis. Julio de 2016;16(7):819-827.
7. Harbarth S, Holeckova K, Froidevaux C, y otros. Diagnostic value of procalcitonin, interleukin-6 , and interleukin- 8 in critically ill patients admitted with suspected sepsis. Am J Respir Crit Care Med. 1 de agosto de 2001;164(3):396-402.
8. Andriolo BN, Andriolo RB, Salomao R, Atallah AN. Effectiveness and safety of procalcitonin evaluation for reducing mortality in adults with sepsis, severe sepsis or septic shock. Cochrane Database Syst Rev. 10 de enero de 2017;1:CD010959.
9. Temmesfeld-Wollbruck B, Brell B, David I, y otros. Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med. Abril de 2007;33(4):703-710.
10. Muller-Redetzky HC, Will D, Hellwig K, y otros Mechanical ventilation drives pneumococcal pneumonia into lung injury and sepsis in mice: protection by adrenomedullin. Crit Care. 2014;18(2):R73.
11. Vallet B. Endothelial cell dysfunction and abnormal tissue perfusion. Crit Care Med. Mayo de 2002;30(5 Suppl):S229-234.
12. Gonzalez-Rey E, Chorny A, Varela N, Robledo G, Delgado M. Urocortin and adrenomedullin prevent lethal endotoxemia by down-regulating the inflammatory response. Am J Pathol. Junio de 2006;168(6):1921-1930. 13. Carrizo GJ, Wu R, Cui X, Dwivedi AJ, Simms HH, Wang P. Adrenomedullin and adrenomedullin-binding protein- 1 downregulate inflammatory cytokines and attenuate tissue injury after gut ischemia-reperfusion. Surgery. Febrero de 2007;141(2):245-253.
14. Brell B, Hippenstiel S, David I, y otros. Adrenomedullin treatment abolishes ileal mucosal hypoperfusion induced by Staphylococcus aureus alpha-toxin--an intravital microscopic study on an isolated rat ileum. Crit Care Med. Diciembre de 2005;33(12):2810-2016.
15. Brell B, Temmesfeld-Wollbruck B, Altzschner I, y otros. Adrenomedullin reduces Staphylococcus aureus alpha-toxin-induced rat ileum microcirculatory damage. Crit Care Med. April de 2005;33(4):819-826.
16. Vigue B, Leblanc PE, Moati F, y otros. Mid-regional pro-adrenomedullin (MR-proADM), a marker of positive fluid balance in critically ill patients: results of the ENVOL study. Crit Care. 09 de noviembre de 2016;20(1):363.
17. Andaluz-Ojeda D, Cicuendez R, Calvo D, y otros. Sustained value of proadrenomedullin as mortality predictor in severe sepsis. J Infect. Julio de 2015;71(1):136-139.
18. Hartmann O, Schuetz P, Albrich WC, Anker SD, Mueller B, Schmidt T. Time-dependent Cox regression: serial measurement of the cardiovascular biomarker proadrenomedullin improves survival prediction in patients with lower respiratory tract infection. Int J Cardiol. 29 de noviembre de 2012;161(3):166-173.
19. Albrich WC, Dusemund F, Ruegger K, y otros. Enhancement of CURB65 score with proadrenomedullin (CURB65-A) for outcome prediction in lower respiratory tract infections: derivation of a clinical algorithm. BMC
Infect Dis. 2011;11:112.
20. Albrich WC, Ruegger K, Dusemund F, y otros. Optimised patient transfer using an innovative multidisciplinary assessment in Kanton Aargau (OPTIMA I): an observational survey in lower respiratory tract infections. Swiss Med Wkly. 2011;141:w13237.
21. Albrich WC, Ruegger K, Dusemund F, y otros. Biomarker-enhanced triage in respiratory infections: a proof-ofconcept feasibility trial. Eur Respir J. Octubre de 2013;42(4):1064-1075.
22. Riera J, Senna A, Cubero M, Roman A, Rello J, Study Group Investigators TV. Primary Graft Dysfunction and Mortality Following Lung Transplantation: A Role for Proadrenomedullin Plasma Levels. Am J Transplant. 13 de octubre de 2015.
23. Schoe A, Schippers EF, Struck J, y otros. Postoperative pro-adrenomedullin levels predict mortality in thoracic surgery patients: comparison with Acute Physiology and Chronic Health Evaluation IV Score*. Crit Care Med. Febrero de 2015;43(2):373-381.
24. Tyagi A, Sethi AK, Girotra G, Mohta M. The microcirculation in sepsis. Indian J Anaesth. Junio de 2009;53(3):281-293.
25. Hernandez G, Bruhn A, Ince C. Microcirculation in sepsis: new perspectives. Curr Vasc Pharmacol. 01 de marzo de 2013;11(2):161-169.
26. Bloos F, Trips E, Nierhaus A, y otros. Effect of Sodium Selenite Administration and Procalcitonin-Guided Therapy on Mortality in Patients With Severe Sepsis or Septic Shock: A Randomized Clinical Trial. JAMA Intern Med. 01 de septiembre de 2016;176(9):1266-1276.
27. Bloos F, Ruddel H, Thomas-Ruddel D, y otros. Effect of a multifaceted educational intervention for antiinfectious measures on sepsis mortality: a cluster randomized trial. Intensive Care Med. 02 de mayo de 2017. 28. Enguix-Armada A, Escobar-Conesa R, La Torre AG, De La Torre-Prados MV. Usefulness of several biomarkers in the management of septic patients: C-reactive protein, procalcitonin, presepsin and mid-regional pro-adrenomedullin. Clin Chem Lab Med. 17 de junio de 2015.
29. Christ-Crain M, Morgenthaler NG, Struck J, Harbarth S, Bergmann A, Muller B. Mid-regional proadrenomedullin as a prognostic marker in sepsis: an observational study. Crit Care. 2005;9(6):R816-824.
30. Suberviola B, Castellanos-Ortega A, Ruiz Ruiz A, Lopez-Hoyos M, Santibanez M. Hospital mortality prognostication in sepsis using the new biomarkers suPAR and proADM in a single determination on ICU admission. Intensive Care Med. Noviembre de 2013;39(11):1945-1952.
31. Gillmann HJ, Meinders A, Larmann J, y otros. Adrenomedullin Is Associated With Surgical Trauma and Impaired Renal Function in Vascular Surgery Patients. J Intensive Care Med. 01 de enero de 2017:885066616689554.
32. Garrouste-Orgeas M, Montuclard L, Timsit JF, Misset B, Christias M, Carlet J. Triaging patients to the ICU: a pilot study of factors influencing admission decisions and patient outcomes. Intensive Care Med. Mayo de 2003;29(5):774-781.
33. Garrouste-Orgeas M, Montuclard L, Timsit JF, y otros. Predictors of intensive care unit refusal in French intensive care units: a multiple-center study. Crit Care Med. Abril de 2005;33(4):750-755.
34. Mery E, Kahn JM. Does space make waste? The influence of ICU bed capacity on admission decisions. Crit Care. 0 8 de mayo de 2013;17(3):315.
35. Orsini J, Blaak C, Yeh A, y otros. Triage of Patients Consulted for ICU Admission During Times of ICU-Bed Shortage. J Clin Med Res. Diciembre de 2014;6(6):463-468.
36. Kip MM, Kusters R, MJ IJ, Steuten LM. A PCT algorithm for discontinuation of antibiotic therapy is a costeffective way to reduce antibiotic exposure in adult intensive care patients with sepsis. J Med Econ.
2015;18(11):944-953.
37. Wilke MH, Grube RF, Bodmann KF. The use of a standardized PCT-algorithm reduces costs in intensive care in septic patients - a DRG-based simulation model. Eur J Med Res. 02 de diciembre de 2011;16(12):543-548. 38. Baughman RP, Lower EE, Flessa HC, Tollerud DJ. Thrombocytopenia in the intensive care unit. Chest.
1993;104(4):1243-7. 7.
39. Drews RE, Weinberger SE. Thrombocytopenic disorders in critically ill patients. Am J Respir Crit Care Med.
2000;162(2 Pt 1):347-51.
40. Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, y otros. Thrombocytopenia and prognosis in intensive care. Crit Care Med. 2000;28(6):1871-6. 9.
41.Strauss R, Wehler M, Mehler K, Kreutzer D, Koebnick C, Hahn EG. Thrombocytopenia in patients in the medical intensive care unit: bleeding prevalence, transfusion requirements, and outcome. Crit Care Med.
2002;30(8):1765-71. 10.
42. Smith-Erichsen N. Serial determinations of platelets, leucocytes and coagulation parameters in surgical septicemia. Scand J Clin Lab Invest Suppl. 1985;178:7-14. 11.
43. Akca S, Haji-Michael P, de Mendonca A, Suter P, Levi M, Vincent JL. Time course of platelet counts in critically ill patients. Crit Care Med. 2002;30(4):753-6. 12.
44. Crowther MA, Cook DJ, Meade MO, Grifth LE, Guyatt GH, Arnold DM, y otros. Thrombocytopenia in medicalsurgical critically ill patients: prevalence, incidence, and risk factors. J Crit Care. 2005;20(4):348-53. 13.
45. Moreau D, Timsit JF, Vesin A, Garrouste-Orgeas M, de Lassence A, Zahar JR, y otros. Platelet count decline: an early prognostic marker in critically ill patients with prolonged ICU stays. Chest. 2007;131(6):1735-41. 14. 46. Hui P, Cook DJ, Lim W, Fraser GA, Arnold DM. The frequency and clinical signifcance of thrombocytopenia complicating critical illness: a systematic review. Chest. 2011;139(2):271-8. 15.
47. Venkata C, Kashyap R, Farmer JC, Afessa B. Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. J Intensive Care. 2013;1(1):9.
48. Semple JW, Freedman J. Platelets and innate immunity. Cell Mol Life Sci. 2010;67(4):499-511. 17.
49. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol.
2011;11(4):264-74. 18.
50. Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile efector cells in hemostasis, infammation, and the immune continuum. Semin Immunopathol. 2012;34(1):5-30. 19.
51. Herter JM, Rossaint J, Zarbock A. Platelets in infammation and immunity. J Thromb Haemost.
2014;12(11):1764-75. 20.
52. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as imune and infammatory cells. Blood. 2014;123(18):2759-67. 21.
53. Xu XR, Zhang D, Oswald BE, Carrim N, Wang X, Hou Y, y otros. Platelets are versatile cells: new discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci.
2016;53(6):409-30.
54. Yu-Min Shen y otros. Evaluating Thrombocytopenia during heparin therapy. Jama. Febrero de 2018; Egede Johansen y otros. The potential of antimicrobials to induce thrombocytopenia in critically ill patients: data froma arandomized controlled trial. PLOS one. Noviembre de 2013; Vol. 8.
55. Van der Poll y otros. Pathogenisis of DIC in sepsis. Sepsis. 1999; 103-109; Koyama y otros. Time course of immature platelet count and its relation to thrombocytopenia and mortality in patients with sepsis. PLOS one. Enero de 2018.
56. Guru y otros. Association of Thrombocytopenia and Mortality in Critically III Patients on Continuous Renal Replacement Therapy. Nephron. 2016; 133:175-182.
57. Larkin y otros. Sepsis-associated thrombocytopenia. Thrombosis Reserch. 2016;
58. Ali N., Auerbach H. (2017), New-onset acute thrombocytopenia in hospitalized patients: pathophysiology and diagnostic approach, Journal of Community Hospital Internal Medicine Perspectives, 7:3, 157-167.
59. Dewitte y otros. Blood platelets and sepsis pathophysiology: A new therapeutic prospect in critical ill patients?. Ann. Intensive Care. 2017; 7:115.
Claims (17)
1. Método para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de los niveles anormales de plaquetas en un paciente, que comprende
a. proporcionar una muestra de dicho paciente,
b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, c. en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas en dicho paciente.
2. Método de acuerdo con la reivindicación 1, que comprende el diagnóstico, pronóstico, evaluación del riesgo y/o estratificación del riesgo de la desregulación del sistema de coagulación, tal como trombocitopenia y/o coagulación intravascular diseminada (DIC), en base a un nivel de proADM o fragmento(s) de la misma en dicha muestra.
3. Método de acuerdo con la reivindicación 1, en donde el paciente está, o se le ha diagnosticado como, gravemente enfermo, preferentemente en donde la muestra se aísla del paciente en el momento del diagnóstico o después de estar gravemente enfermo.
4. Método de acuerdo con cualquiera de las reivindicaciones anteriores, en donde:
a. el paciente se diagnostica con una enfermedad infecciosa,
b. el paciente se diagnostica con una o más insuficiencias orgánicas existentes, y/o es un paciente postraumático o posquirúrgico, o
c. el paciente se diagnostica con una desregulación del sistema de coagulación, tal como coagulación intravascular diseminada (DIC) o trombocitopenia, y/o con una disfunción de un órgano asociado con el sistema de coagulación, tal como vasos sanguíneos, bazo, médula ósea y/o el sistema inmunológico.
5. Método de acuerdo con cualquiera de las reivindicaciones anteriores, en donde el paciente se diagnostica con sepsis, sepsis grave o choque séptico.
6. Método de acuerdo con cualquiera de las reivindicaciones anteriores, en donde el nivel de proADM se correlaciona inversamente con el nivel de plaquetas.
7. Método de acuerdo con cualquiera de las reivindicaciones anteriores, en donde la muestra se selecciona del grupo que consiste en una muestra de sangre, una muestra de suero, una muestra de plasma y/o una muestra de orina.
8. Método de acuerdo con cualquiera de las reivindicaciones anteriores, en donde determinar un nivel de proADM o fragmento(s) de la misma comprende determinar un nivel de MR-proADM.
9. Método de acuerdo con cualquiera de las reivindicaciones anteriores, en donde
a. un nivel de proADM o fragmento(s) de la misma por debajo de un nivel de gravedad alto (o valor de corte) es indicativo de niveles de plaquetas normales o altos, o
b. un nivel de proADM o fragmento(s) de la misma igual o superior a un nivel de gravedad alto (o valor de corte) es indicativo de la presencia o probabilidad de desarrollar niveles bajos de plaquetas y/o trombocitopenia y/o coagulación intravascular diseminada (DIC),
c. en donde un nivel de gravedad alto (o valor de corte) de la proADM o fragmentos de la misma es un nivel por encima de 6,5 nmol/l, 6,95 nmol/l o preferentemente 10,9 nmol/l.
10. Método de acuerdo con la reivindicación anterior, en donde el nivel de proADM o fragmento(s) de la misma igual o superior al nivel de gravedad alto (valor de corte) indica el inicio o la modificación de un tratamiento del paciente para abordar la presencia o probabilidad de desarrollar, niveles bajos de plaquetas y/o trombocitopenia y/o coagulación intravascular diseminada (DIC), en donde el tratamiento se selecciona preferentemente del grupo que consiste en transfusión de sangre o plaquetas, transfusión de componentes sanguíneos, tales como suero, plasma o células específicas o combinaciones de los mismos, administración de fármacos que promuevan la formación de trombocitos, inicio de un tratamiento causal o realización de una esplenectomía.
11. Método de acuerdo con la reivindicación 9, para el pronóstico, evaluación del riesgo y/o estratificación del riesgo de niveles bajos de plaquetas y/o trombocitopenia y/o coagulación intravascular diseminada (DIC) en un paciente, en donde un nivel de proADM o fragmento(s) de la misma igual o superior a un nivel de gravedad alto (o valor de corte) es indicativo de que se está produciendo un evento adverso, tal como uno o más de trombocitopenia, DIC, infección, insuficiencia de órganos, disfunción orgánica y/o mortalidad
12. Método de acuerdo con la reivindicación 9, en donde los pacientes son pacientes de la unidad de cuidados intensivos (UCI), en donde
a. el nivel de proADM o fragmento(s) de la misma por debajo del nivel de gravedad bajo (valor de corte) indica el alta de dicho paciente de la UCI, y/o
b. el nivel de proADM o fragmento(s) de la misma igual o superior al nivel de gravedad alto (valor de corte) indica modificar el tratamiento del paciente en la UCI.
13. Método de acuerdo con cualquiera de las reivindicaciones anteriores,
a. que comprende determinar un nivel de uno o más marcadores adicionales en una muestra aislada del paciente, tal como el nivel de plaquetas en una muestra de sangre y/o uno o más marcadores para la coagulación intravascular diseminada (DIC), preferentemente en donde el uno o más marcadores para la coagulación intravascular diseminada (DIC) se seleccionan del grupo que consiste en micropartículas de membrana, sCD14-ST, protrombinasa, antitrombina y/o actividad antitrombina, proteína catiónica 18 (CAP18), proteasas que escinden el factor de von Willebrand (vWF), lipoproteínas en combinación con CRP, fibrinógeno, fibrina, B2GP1, GPIIb-IIIa, dímero D no desnaturalizado de fibrina, factor plaquetario 4, histonas y/o un ensayo PT, o
b. en donde se mide al menos un marcador o parámetro clínico adicional, preferentemente seleccionado del grupo que consiste en procalcitonina, histona 3, histona 2A, histona 2B, histona 4 o fragmento(s) de la misma, recuento de plaquetas, volumen plaquetario medio y/o uno o más marcadores de una desregulación del sistema de coagulación.
14. Método de acuerdo con la reivindicación 13, en donde el uno o más marcadores adicionales comprenden PCT o fragmento(s) de la misma.
15. Método de acuerdo con cualquiera de las reivindicaciones anteriores para el pronóstico, evaluación del riesgo o estratificación del riesgo de un evento adverso en un paciente con un nivel anormal de plaquetas, que comprende:
a. proporcionar una muestra de dicho paciente,
b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, c. en donde dicho nivel de proADM o fragmento(s) de la misma se correlaciona con los niveles anormales de plaquetas y con la probabilidad de que ocurra un evento adverso en dicho paciente, en donde preferentemente el evento adverso es uno o más de mortalidad, mortalidad relacionada con sepsis, insuficiencia de órganos y/o disfunción de órganos, en donde preferentemente la insuficiencia de órganos o la disfunción de órganos está asociada con trombocitopenia.
16. Método de acuerdo con cualquiera de las reivindicaciones anteriores para la determinación, diagnóstico, pronóstico, orientación del tratamiento, seguimiento del tratamiento, evaluación del riesgo y/o estratificación del riesgo de trombocitopenia séptica en un paciente, que comprende
a. proporcionar una muestra de dicho paciente,
b. determinar un nivel de proadrenomedulina (proADM) o fragmento(s) de la misma en dicha muestra, y c. determinar un nivel de procalcitonina (PCT) o fragmento(s) de la misma en dicha muestra, d. en donde cuando un nivel de procalcitonina (PCT) o fragmento(s) de la misma es >0,5 ng/ml indica la presencia de, o un mayor riesgo de adquirir, sepsis, y en donde cuando un nivel de proADM o fragmento(s) de la misma es > 2,75 nmol/l indica la presencia o un mayor riesgo de adquirir trombocitopenia.
17. Kit para llevar a cabo el método de cualquiera de las reivindicaciones 1 a 16, que comprende:
a. reactivos de detección para determinar el nivel de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente para determinar el nivel de PCT o fragmento(s) de la misma y/o uno o más marcadores adicionales de acuerdo con la reivindicación 13, en una muestra de un sujeto, y
b. datos de referencia, que comprenden un nivel de referencia, correspondiente a niveles de gravedad alto de la proADM, en donde el nivel de gravedad alto es superior a 6,5 nmol/l, preferentemente superior a 6,95 nmol/l, con mayor preferencia superior a 10,9 nmol/l, y opcionalmente los niveles de PCT y/o niveles de uno o más marcadores adicionales de acuerdo con la reivindicación 13, en donde dichos datos de referencia se almacenan en un medio legible por ordenador y/o se emplean en forma de código ejecutable por ordenador configurado para comparar los niveles determinados de proADM o fragmento(s) de la misma, y opcionalmente adicionalmente los niveles determinados de PCT o fragmento(s) de la misma y/o marcadores adicionales de acuerdo con la reivindicación 13, a dichos datos de referencia.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17190912 | 2017-09-13 | ||
| EP18161091 | 2018-03-09 | ||
| PCT/EP2018/074725 WO2019053118A1 (en) | 2017-09-13 | 2018-09-13 | PROADRÉNOMÉDULLINE AS A MARKER OF ABNORMAL PLATELET RATES |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2909399T3 true ES2909399T3 (es) | 2022-05-06 |
Family
ID=63517904
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18765659T Active ES2909399T3 (es) | 2017-09-13 | 2018-09-13 | Proadrenomedulina como marcador de niveles anormales de plaquetas |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US11327082B2 (es) |
| EP (3) | EP3682247B1 (es) |
| JP (1) | JP7346389B2 (es) |
| CN (1) | CN111094987B (es) |
| DK (1) | DK3682247T3 (es) |
| ES (1) | ES2909399T3 (es) |
| PL (1) | PL3682247T3 (es) |
| WO (1) | WO2019053118A1 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114207436A (zh) * | 2019-05-24 | 2022-03-18 | 机敏医药股份有限公司 | 基于c-c基序趋化因子配体14测量而评估和治疗肾损伤的方法 |
| CN114113580A (zh) * | 2020-08-25 | 2022-03-01 | 张曼 | 尿液血栓调节蛋白及其多肽片段在烧伤中的应用 |
| CN119780441B (zh) * | 2024-12-25 | 2026-02-13 | 首都医科大学附属北京胸科医院 | 一种诊断近期及远期结核感染的生物标志物 |
Family Cites Families (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2570703B1 (fr) | 1984-09-26 | 1988-07-08 | Commissariat Energie Atomique | Complexes macropolycycliques de terres rares et application a titre de marqueurs fluorescents |
| US4882733A (en) | 1987-03-13 | 1989-11-21 | Ford Aerospace Corporation | Method and apparatus for combining encoding and modulation |
| FR2664699B1 (fr) | 1990-07-13 | 1995-08-18 | Cis Bio Int | Procede d'amplification du signal d'emission d'un compose luminescent. |
| DE69602756T2 (de) | 1995-08-18 | 2000-02-10 | The Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services, Rockville | Funktionelle rolle von adrenomedullin(am) und dem gen-verwandten produkt(pamp) in der menschlichen pathologie und physiologie |
| DE19847690A1 (de) * | 1998-10-15 | 2000-04-20 | Brahms Diagnostica Gmbh | Verfahren und Substanzen für die Diagnose und Therapie von Sepsis und sepsisähnlichen systemischen Infektionen |
| KR100425525B1 (ko) | 2000-11-21 | 2004-03-30 | 재단법인서울대학교산학협력재단 | 조산과 태아감염 및 태아손상을 산전 진단하기 위한진단시약 및 진단키트 |
| EP1453529A4 (en) * | 2001-09-19 | 2007-09-26 | Oklahoma Med Res Found | TREATMENT OF SEPSIS WITH TAFI |
| DE10316583A1 (de) * | 2003-04-10 | 2004-10-28 | B.R.A.H.M.S Aktiengesellschaft | Bestimmung eines midregionalen Proadrenomedullin-Teilpeptids in biologischen Flüssigkeiten zu diagnostischen Zwecken, sowie Immunoassays für die Durchführung einer solchen Bestimmung |
| US20060029589A1 (en) * | 2004-06-01 | 2006-02-09 | Hartmut Weiler | Induction of heterozygous FV Leiden carrier status to reduce mortality in sepsis and to prevent organ damage caused by inflammation and/or ischemia-reperfusion injury |
| JP2008505649A (ja) * | 2004-07-09 | 2008-02-28 | アモークス リミテッド | 免疫細胞バイオセンサーおよびその使用方法 |
| ES2300681T3 (es) * | 2004-07-22 | 2008-06-16 | Brahms Aktiengesellschaft | Procedimiento para el diagnostico y tratamiento de pacientes criticos con endotelina, agonistas de endotelina y antagonistas de adrenomedulina. |
| US8906857B2 (en) * | 2005-12-01 | 2014-12-09 | B.R.A.H.M.S. Gmbh | Methods for the diagnosis and treatment of critically ill patients with endothelin, endothelin agonists and adrenomedullin antagonists |
| US20070298034A9 (en) * | 2005-12-09 | 2007-12-27 | Angela Widom | Sulfotyrosine specific antibodies and uses therefor |
| DE102006027818A1 (de) * | 2006-06-16 | 2007-12-20 | B.R.A.H.M.S. Aktiengesellschaft | In vitro Multiparameter-Bestimmungsverfahren zur Diagnose und Frühdiagnose von neurodegenerativen Erkrankungen |
| DE102006034142A1 (de) | 2006-07-24 | 2008-01-31 | B.R.A.H.M.S. Aktiengesellschaft | Verfahren zur Steuerung der Therapie von Patienten mit Herzinsuffizienz anhand der vitro Bestimmung von Schwellenwerten von vasoaktiven Peptiden |
| DE102006052916A1 (de) * | 2006-11-08 | 2008-05-15 | Brahms Aktiengesellschaft | Diagnose und Risikostratifizierung von Diabetes mellitus mittels MR-proADM |
| US9012151B2 (en) * | 2006-11-09 | 2015-04-21 | B.R.A.H.M.S. Gmbh | Methods of diagnosis and risk stratification of adverse events in post myocardial infarction patients using pro-adrenomedullin |
| DE102006060112A1 (de) | 2006-12-20 | 2008-06-26 | Brahms Aktiengesellschaft | Diagnose und Risikostratifizierung mittels dem neuen Marker CT-proADM |
| ATE554395T1 (de) | 2007-03-12 | 2012-05-15 | Biomedica Medizinprodukte Gmbh & Co Kg | Diagnose septischer komplikationen |
| ATE514950T1 (de) | 2007-09-07 | 2011-07-15 | Univ Zuerich | Verfahren zur untersuchung von sepsis bei menschen |
| WO2009055669A2 (en) * | 2007-10-26 | 2009-04-30 | Oklahoma Medical Research Foundation | Monoclonal antibodies against activated and unactivated protein c |
| US20100292131A1 (en) | 2007-11-16 | 2010-11-18 | Pronota N.V. | Biomarkers and methods for diagnosing, predicting and/or prognosing sepsis and uses thereof |
| JP5753159B2 (ja) | 2009-05-05 | 2015-07-22 | ベー.エル.アー.ハー.エム.エス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 内皮性機能/機能不全に関連した疾患に罹患している患者の血管作動性ホルモンに基づいた層化 |
| ITPI20090066A1 (it) * | 2009-05-26 | 2010-11-27 | Consiglio Nazionale Ricerche | Metodo per produrre un dispositivo applicabile a tessuti biologici, in particolare un patch per trattare tessuti danneggiati, e dispositivo ottenuto con tale metodo |
| EP2438447B1 (en) | 2009-06-05 | 2015-02-18 | B.R.A.H.M.S GmbH | Detection of bacterial infections in subjects suffering from dyspnea. |
| WO2011027971A2 (ko) * | 2009-09-01 | 2011-03-10 | 주식회사이언메딕스 | 장내 공생 세균유래 세포밖 소포체, 및 이를 이용한 질병모델, 백신, 후보 약물 탐색 방법, 및 진단 방법 |
| JP5722587B2 (ja) | 2009-10-13 | 2015-05-20 | ベー.エル.アー.ハー.エム.エス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 急性脳卒中または一過性脳虚血発作を有する患者における細菌感染の診断のためのプロカルシトニン及び抗生物質治療のガイダンス |
| JP5798133B2 (ja) | 2010-03-08 | 2015-10-21 | ベー.エル.アー.ハー.エム.エス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 不特定の愁訴を示す患者における細菌感染及び抗生物質治療の指標を診断するために用いられるプロカルシトニン |
| EP2635904A1 (en) | 2010-11-01 | 2013-09-11 | B.R.A.H.M.S GmbH | Prognosis and risk assessment of patients with non-specific complaints |
| DK2781221T3 (da) * | 2011-11-15 | 2020-06-02 | Asahi Kasei Pharma Corp | Medicin til behandling og/eller forbedring af sepsis |
| HRP20170427T1 (hr) * | 2011-11-16 | 2017-06-16 | Sphingotec Gmbh | Testovi na adrenomedulin i postupci određivanja zrelog adrenomedulina |
| ES2734494T3 (es) | 2011-12-08 | 2019-12-10 | Astute Medical Inc | Métodos y usos para el diagnóstico de una lesión renal y de una insuficiencia renal |
| EP2657707A1 (en) * | 2012-04-26 | 2013-10-30 | B.R.A.H.M.S GmbH | Biomakers for the diagnosis, prognosis, assessment and therapy stratification syncope |
| CN104285148B (zh) * | 2012-05-07 | 2016-10-12 | 美迪恩斯生命科技株式会社 | 检测弥散性血管内凝血或感染性弥散性血管内凝血的方法 |
| SG11201507774YA (en) | 2013-03-20 | 2015-10-29 | Sphingotec Gmbh | Adrenomedullin to guide therapy of blood pressure decline |
| US20170128459A1 (en) * | 2014-03-27 | 2017-05-11 | Industrial Cooperation Foundation Chonbuk National University | Pharmaceutical composition containing sirt2 inhibitor |
| JP6944449B2 (ja) | 2015-11-27 | 2021-10-06 | ベー.エル.アー.ハー.エム.エス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 対象の細胞外液量状態のマーカーとしてのMR−proADM |
| RU2019103403A (ru) | 2016-07-08 | 2020-08-10 | Сфинготек Гмбх | Адреномедуллин для оценки застоя у индивидуума с острой сердечной недостаточностью |
| EP3497451B1 (en) | 2016-08-09 | 2025-06-18 | B.R.A.H.M.S GmbH | Histones and/or proadm as markers indicating an adverse event |
-
2018
- 2018-09-13 US US16/646,798 patent/US11327082B2/en active Active
- 2018-09-13 WO PCT/EP2018/074725 patent/WO2019053118A1/en not_active Ceased
- 2018-09-13 EP EP18765659.0A patent/EP3682247B1/en active Active
- 2018-09-13 PL PL18765659T patent/PL3682247T3/pl unknown
- 2018-09-13 CN CN201880059382.1A patent/CN111094987B/zh active Active
- 2018-09-13 DK DK18765659.0T patent/DK3682247T3/da active
- 2018-09-13 ES ES18765659T patent/ES2909399T3/es active Active
- 2018-09-13 JP JP2020514918A patent/JP7346389B2/ja active Active
- 2018-09-13 EP EP21198828.2A patent/EP3971572A1/en active Pending
- 2018-09-13 EP EP21198856.3A patent/EP3971573A1/en active Pending
-
2022
- 2022-03-31 US US17/710,369 patent/US12000845B2/en active Active
- 2022-03-31 US US17/710,329 patent/US12405279B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3682247A1 (en) | 2020-07-22 |
| EP3971573A1 (en) | 2022-03-23 |
| EP3971572A1 (en) | 2022-03-23 |
| EP3682247B1 (en) | 2022-02-09 |
| US20220229076A1 (en) | 2022-07-21 |
| US20220221474A1 (en) | 2022-07-14 |
| US12000845B2 (en) | 2024-06-04 |
| PL3682247T3 (pl) | 2022-04-25 |
| JP7346389B2 (ja) | 2023-09-19 |
| DK3682247T3 (da) | 2022-03-21 |
| CN111094987A (zh) | 2020-05-01 |
| US11327082B2 (en) | 2022-05-10 |
| JP2020533593A (ja) | 2020-11-19 |
| WO2019053118A1 (en) | 2019-03-21 |
| US20200271675A1 (en) | 2020-08-27 |
| US12405279B2 (en) | 2025-09-02 |
| CN111094987B (zh) | 2023-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12405279B2 (en) | Method for preparing a sample | |
| US20250189539A1 (en) | Sample including pro-adm as a therapy monitoring marker for critcally ill patients | |
| JP2023095851A (ja) | 抗生物質治療をモニタリングするためのマーカーとしてのpctおよびpro-adm | |
| BR112021002281A2 (pt) | pro-adm para prognosticar o risco de uma condição médica requerendo hospitalização em pacientes com sintomas de doença infecciosa | |
| HK40069336A (en) | Proadrenomedullin as a marker for abnormal platelet levels | |
| HK40069337A (en) | Proadrenomedullin as a marker for abnormal platelet levels | |
| RU2782305C2 (ru) | Про-адм в качестве маркера мониторинга терапии для критически больных пациентов | |
| HK40032557A (en) | Proadrenomedullin as a marker for abnormal platelet levels | |
| HK40032557B (en) | Proadrenomedullin as a marker for abnormal platelet levels | |
| RU2788885C2 (ru) | Прокальцитонин и про-адм в качестве маркеров для мониторинга лечения антибиотиками |