ES2927954T3 - Compuestos 2,5-dioxo-azolina N-sustituidos para la utilización en el tratamiento del cáncer - Google Patents
Compuestos 2,5-dioxo-azolina N-sustituidos para la utilización en el tratamiento del cáncer Download PDFInfo
- Publication number
- ES2927954T3 ES2927954T3 ES19167943T ES19167943T ES2927954T3 ES 2927954 T3 ES2927954 T3 ES 2927954T3 ES 19167943 T ES19167943 T ES 19167943T ES 19167943 T ES19167943 T ES 19167943T ES 2927954 T3 ES2927954 T3 ES 2927954T3
- Authority
- ES
- Spain
- Prior art keywords
- cells
- compound
- cell
- compounds
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 114
- 238000011282 treatment Methods 0.000 title claims abstract description 111
- 201000011510 cancer Diseases 0.000 title claims abstract description 91
- -1 N-substituted 2,5-dioxo-azoline compounds Chemical class 0.000 title claims description 39
- 150000001875 compounds Chemical class 0.000 claims abstract description 394
- 238000000034 method Methods 0.000 claims abstract description 110
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 70
- 238000011394 anticancer treatment Methods 0.000 claims abstract description 27
- 239000000203 mixture Substances 0.000 claims description 99
- 230000002062 proliferating effect Effects 0.000 claims description 77
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 29
- 125000000217 alkyl group Chemical group 0.000 claims description 20
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 20
- 150000003839 salts Chemical class 0.000 claims description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims description 19
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 18
- 239000012623 DNA damaging agent Substances 0.000 claims description 17
- 229910052736 halogen Inorganic materials 0.000 claims description 10
- 208000032839 leukemia Diseases 0.000 claims description 10
- 125000001072 heteroaryl group Chemical group 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 9
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 9
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 9
- 206010025323 Lymphomas Diseases 0.000 claims description 8
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 150000002367 halogens Chemical class 0.000 claims description 8
- 125000002252 acyl group Chemical group 0.000 claims description 7
- 125000003545 alkoxy group Chemical group 0.000 claims description 7
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 7
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 6
- 125000004414 alkyl thio group Chemical group 0.000 claims description 6
- 238000002360 preparation method Methods 0.000 claims description 6
- 125000004644 alkyl sulfinyl group Chemical group 0.000 claims description 4
- 125000004104 aryloxy group Chemical group 0.000 claims description 4
- 125000003107 substituted aryl group Chemical group 0.000 claims description 4
- 125000005110 aryl thio group Chemical group 0.000 claims description 3
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 125000005415 substituted alkoxy group Chemical group 0.000 claims description 3
- VBIXDLJJOWRQKF-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 VBIXDLJJOWRQKF-UHFFFAOYSA-N 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims 2
- 230000006978 adaptation Effects 0.000 abstract description 51
- 230000022131 cell cycle Effects 0.000 abstract description 46
- 230000037060 G2 phase arrest Effects 0.000 abstract description 42
- 230000004663 cell proliferation Effects 0.000 abstract description 20
- WIFCKLPZYYALGY-UHFFFAOYSA-N 1h-pyrrole-2,3-dione Chemical class O=C1NC=CC1=O WIFCKLPZYYALGY-UHFFFAOYSA-N 0.000 abstract description 10
- 230000012820 cell cycle checkpoint Effects 0.000 abstract description 8
- 230000010261 cell growth Effects 0.000 abstract description 7
- 230000001093 anti-cancer Effects 0.000 abstract description 3
- 239000003153 chemical reaction reagent Substances 0.000 abstract 2
- 210000004027 cell Anatomy 0.000 description 499
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 117
- 241000699670 Mus sp. Species 0.000 description 62
- 208000035475 disorder Diseases 0.000 description 61
- 230000005778 DNA damage Effects 0.000 description 54
- 231100000277 DNA damage Toxicity 0.000 description 54
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 51
- 230000000694 effects Effects 0.000 description 50
- 239000013067 intermediate product Substances 0.000 description 48
- 241000282414 Homo sapiens Species 0.000 description 43
- 108020004414 DNA Proteins 0.000 description 42
- 230000025084 cell cycle arrest Effects 0.000 description 42
- 235000019439 ethyl acetate Nutrition 0.000 description 42
- 239000000243 solution Substances 0.000 description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 42
- 239000002904 solvent Substances 0.000 description 41
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 40
- 230000004083 survival effect Effects 0.000 description 37
- 239000011541 reaction mixture Substances 0.000 description 36
- 239000000543 intermediate Substances 0.000 description 31
- 239000012044 organic layer Substances 0.000 description 27
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 25
- 230000003013 cytotoxicity Effects 0.000 description 25
- 231100000135 cytotoxicity Toxicity 0.000 description 25
- 239000007787 solid Substances 0.000 description 25
- 208000006112 autosomal recessive hypercholesterolemia Diseases 0.000 description 24
- 208000032655 familial 4 hypercholesterolemia Diseases 0.000 description 24
- 239000000047 product Substances 0.000 description 24
- 239000007858 starting material Substances 0.000 description 24
- 230000005855 radiation Effects 0.000 description 23
- 238000002054 transplantation Methods 0.000 description 23
- 239000007832 Na2SO4 Substances 0.000 description 22
- 230000001472 cytotoxic effect Effects 0.000 description 22
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 22
- 229960003957 dexamethasone Drugs 0.000 description 22
- 229910052938 sodium sulfate Inorganic materials 0.000 description 22
- 235000011152 sodium sulphate Nutrition 0.000 description 22
- 238000003786 synthesis reaction Methods 0.000 description 22
- 208000034578 Multiple myelomas Diseases 0.000 description 20
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 19
- 238000001727 in vivo Methods 0.000 description 19
- 230000010190 G1 phase Effects 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 230000015572 biosynthetic process Effects 0.000 description 18
- 230000030833 cell death Effects 0.000 description 18
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 17
- 239000002246 antineoplastic agent Substances 0.000 description 17
- 238000004809 thin layer chromatography Methods 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- 230000011278 mitosis Effects 0.000 description 16
- 239000003208 petroleum Substances 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 15
- 239000012267 brine Substances 0.000 description 15
- 231100000433 cytotoxic Toxicity 0.000 description 15
- 231100000065 noncytotoxic Toxicity 0.000 description 15
- 239000003921 oil Substances 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- 238000004440 column chromatography Methods 0.000 description 14
- 238000000338 in vitro Methods 0.000 description 14
- 230000001629 suppression Effects 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 12
- 238000011284 combination treatment Methods 0.000 description 12
- 230000007423 decrease Effects 0.000 description 12
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 12
- 229920006395 saturated elastomer Polymers 0.000 description 12
- 230000016596 traversing start control point of mitotic cell cycle Effects 0.000 description 12
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 12
- 201000009030 Carcinoma Diseases 0.000 description 11
- 230000010337 G2 phase Effects 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 230000006907 apoptotic process Effects 0.000 description 11
- 230000003247 decreasing effect Effects 0.000 description 11
- 239000012071 phase Substances 0.000 description 11
- 238000010992 reflux Methods 0.000 description 11
- 125000001424 substituent group Chemical group 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- 239000003981 vehicle Substances 0.000 description 11
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 10
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 10
- 230000006378 damage Effects 0.000 description 10
- 239000012894 fetal calf serum Substances 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 150000008064 anhydrides Chemical class 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 9
- 150000003949 imides Chemical class 0.000 description 9
- 230000002147 killing effect Effects 0.000 description 9
- 230000006618 mitotic catastrophe Effects 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 108010033040 Histones Proteins 0.000 description 8
- 102000006947 Histones Human genes 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 230000014509 gene expression Effects 0.000 description 8
- 230000026731 phosphorylation Effects 0.000 description 8
- 238000006366 phosphorylation reaction Methods 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 235000017557 sodium bicarbonate Nutrition 0.000 description 8
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 8
- AQLJVWUFPCUVLO-UHFFFAOYSA-N urea hydrogen peroxide Chemical compound OO.NC(N)=O AQLJVWUFPCUVLO-UHFFFAOYSA-N 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 206010009944 Colon cancer Diseases 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- 230000001594 aberrant effect Effects 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- 208000029742 colonic neoplasm Diseases 0.000 description 7
- 230000034994 death Effects 0.000 description 7
- 231100000673 dose–response relationship Toxicity 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine Substances NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 238000007912 intraperitoneal administration Methods 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 230000017074 necrotic cell death Effects 0.000 description 7
- 230000035755 proliferation Effects 0.000 description 7
- 229910000104 sodium hydride Inorganic materials 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 210000004881 tumor cell Anatomy 0.000 description 7
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 230000004543 DNA replication Effects 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- 230000018199 S phase Effects 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 230000005757 colony formation Effects 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 230000007547 defect Effects 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000000684 flow cytometry Methods 0.000 description 6
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 6
- 150000002429 hydrazines Chemical class 0.000 description 6
- 239000005457 ice water Substances 0.000 description 6
- 239000007928 intraperitoneal injection Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 230000001575 pathological effect Effects 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 230000008439 repair process Effects 0.000 description 6
- 238000012216 screening Methods 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- ORYWGNFMBHMCEE-UHFFFAOYSA-N 1-[4-bromo-3-(trifluoromethyl)anilino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(Br)C(C(F)(F)F)=C1 ORYWGNFMBHMCEE-UHFFFAOYSA-N 0.000 description 5
- DPIKTOVKAQXACA-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-ethyl-4-methylpyrrole-2,5-dione Chemical compound O=C1C(CC)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 DPIKTOVKAQXACA-UHFFFAOYSA-N 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- FYMKAROPOBEDLZ-UHFFFAOYSA-N 6-[(3,4-dimethyl-2,5-dioxopyrrol-1-yl)amino]-3-(trifluoromethyl)pyridine-2-carbonitrile Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(C#N)=N1 FYMKAROPOBEDLZ-UHFFFAOYSA-N 0.000 description 5
- 239000004342 Benzoyl peroxide Substances 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 229940124650 anti-cancer therapies Drugs 0.000 description 5
- 238000011319 anticancer therapy Methods 0.000 description 5
- 229940041181 antineoplastic drug Drugs 0.000 description 5
- 150000001543 aryl boronic acids Chemical class 0.000 description 5
- 235000019400 benzoyl peroxide Nutrition 0.000 description 5
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 125000000753 cycloalkyl group Chemical group 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000012746 preparative thin layer chromatography Methods 0.000 description 5
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 230000001235 sensitizing effect Effects 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- PADWTOLAUDXONP-UHFFFAOYSA-N 1-[[6-(4-chlorophenyl)-5-(trifluoromethyl)pyridin-2-yl]amino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(C=2C=CC(Cl)=CC=2)=N1 PADWTOLAUDXONP-UHFFFAOYSA-N 0.000 description 4
- MZTRIZBHLSXPLR-UHFFFAOYSA-N 1-[[6-benzyl-5-(trifluoromethyl)pyridin-2-yl]amino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(CC=2C=CC=CC=2)=N1 MZTRIZBHLSXPLR-UHFFFAOYSA-N 0.000 description 4
- BHEPCUXRKMFLBW-UHFFFAOYSA-N 1-[[6-bromo-5-(trifluoromethyl)pyridin-2-yl]amino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Br)=N1 BHEPCUXRKMFLBW-UHFFFAOYSA-N 0.000 description 4
- HXDAXUIQFVRGRG-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-(hydroxymethyl)-4-methylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(CO)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 HXDAXUIQFVRGRG-UHFFFAOYSA-N 0.000 description 4
- UPWAAFFFSGQECJ-UHFFFAOYSA-N 2,6-dichloro-3-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=C(Cl)N=C1Cl UPWAAFFFSGQECJ-UHFFFAOYSA-N 0.000 description 4
- SXLBWNSGCIEART-UHFFFAOYSA-N 2-chloro-6-methyl-4-(trifluoromethyl)pyridine Chemical compound CC1=CC(C(F)(F)F)=CC(Cl)=N1 SXLBWNSGCIEART-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 4
- 102100034533 Histone H2AX Human genes 0.000 description 4
- 108020002230 Pancreatic Ribonuclease Proteins 0.000 description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 4
- 102000005891 Pancreatic ribonuclease Human genes 0.000 description 4
- 239000012980 RPMI-1640 medium Substances 0.000 description 4
- 206010039491 Sarcoma Diseases 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical class NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 4
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 239000010432 diamond Substances 0.000 description 4
- 230000002500 effect on skin Effects 0.000 description 4
- 210000002889 endothelial cell Anatomy 0.000 description 4
- 210000002950 fibroblast Anatomy 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 229960002949 fluorouracil Drugs 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 4
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 4
- 230000010534 mechanism of action Effects 0.000 description 4
- 206010061289 metastatic neoplasm Diseases 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 201000002528 pancreatic cancer Diseases 0.000 description 4
- 208000008443 pancreatic carcinoma Diseases 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000001509 sodium citrate Substances 0.000 description 4
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 238000010189 synthetic method Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- BCGYZNZJBCNQTA-UHFFFAOYSA-N 1-[[3-bromo-6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=NC(Cl)=C(C(F)(F)F)C=C1Br BCGYZNZJBCNQTA-UHFFFAOYSA-N 0.000 description 3
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 3
- HKXMZBUQGOTDNS-UHFFFAOYSA-N 3,4-dimethyl-1-[[6-phenyl-5-(trifluoromethyl)pyridin-2-yl]amino]pyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(C=2C=CC=CC=2)=N1 HKXMZBUQGOTDNS-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 230000006820 DNA synthesis Effects 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 238000011579 SCID mouse model Methods 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- UKLGCLOLNJEQIB-UHFFFAOYSA-N [6-chloro-5-(trifluoromethyl)pyridin-2-yl]hydrazine Chemical compound NNC1=CC=C(C(F)(F)F)C(Cl)=N1 UKLGCLOLNJEQIB-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 229940009456 adriamycin Drugs 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 230000032823 cell division Effects 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 3
- 229940126208 compound 22 Drugs 0.000 description 3
- 229940125851 compound 27 Drugs 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 230000007613 environmental effect Effects 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 230000003394 haemopoietic effect Effects 0.000 description 3
- 238000003306 harvesting Methods 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 206010020718 hyperplasia Diseases 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 230000001394 metastastic effect Effects 0.000 description 3
- 230000001613 neoplastic effect Effects 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- BJWVVGYXYLLFFX-UHFFFAOYSA-N 1-[[6-(3-chlorophenyl)-5-(trifluoromethyl)pyridin-2-yl]amino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(C=2C=C(Cl)C=CC=2)=N1 BJWVVGYXYLLFFX-UHFFFAOYSA-N 0.000 description 2
- JMIDKKZEZVZPIE-UHFFFAOYSA-N 1-[[6-bromo-5-(trifluoromethyl)pyridin-2-yl]methylamino]-3,4-dimethylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NCC1=CC=C(C(F)(F)F)C(Br)=N1 JMIDKKZEZVZPIE-UHFFFAOYSA-N 0.000 description 2
- VHQLAASPWVWVAE-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-(2,2-dimethylpropoxymethyl)-4-methylpyrrole-2,5-dione Chemical compound O=C1C(C)=C(COCC(C)(C)C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 VHQLAASPWVWVAE-UHFFFAOYSA-N 0.000 description 2
- IANWJSWBYCDDTK-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-(2-methoxyethoxymethyl)-4-methylpyrrole-2,5-dione Chemical compound O=C1C(COCCOC)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 IANWJSWBYCDDTK-UHFFFAOYSA-N 0.000 description 2
- WSPHDWRGGCALSF-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-(3-hydroxyhexyl)-4-methylpyrrole-2,5-dione Chemical compound O=C1C(CCC(O)CCC)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 WSPHDWRGGCALSF-UHFFFAOYSA-N 0.000 description 2
- SOBLKGPTGGQHOY-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-(ethoxymethyl)-4-methylpyrrole-2,5-dione Chemical compound O=C1C(COCC)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 SOBLKGPTGGQHOY-UHFFFAOYSA-N 0.000 description 2
- IZIAQHGHXDKHEZ-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-methyl-4-(2-methylpropoxymethyl)pyrrole-2,5-dione Chemical compound O=C1C(COCC(C)C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 IZIAQHGHXDKHEZ-UHFFFAOYSA-N 0.000 description 2
- STQBWQZUNHRYCH-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-methyl-4-(2-propoxyethyl)pyrrole-2,5-dione Chemical compound O=C1C(CCOCCC)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 STQBWQZUNHRYCH-UHFFFAOYSA-N 0.000 description 2
- OKIMZYYMFSCDTO-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-methyl-4-[(2-methylpropan-2-yl)oxymethyl]pyrrole-2,5-dione Chemical compound O=C1C(C)=C(COC(C)(C)C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 OKIMZYYMFSCDTO-UHFFFAOYSA-N 0.000 description 2
- GTMTXJRTVMQDLD-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-methyl-4-[2-(2-methyl-1,3-dioxolan-2-yl)ethyl]pyrrole-2,5-dione Chemical compound O=C1N(NC=2N=C(Cl)C(=CC=2)C(F)(F)F)C(=O)C(C)=C1CCC1(C)OCCO1 GTMTXJRTVMQDLD-UHFFFAOYSA-N 0.000 description 2
- YINAPROGQUSAAA-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-methyl-4-[2-(2-methylpropoxy)ethyl]pyrrole-2,5-dione Chemical compound O=C1C(CCOCC(C)C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 YINAPROGQUSAAA-UHFFFAOYSA-N 0.000 description 2
- XDUKGDCFNGHDKE-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-3-methyl-4-[2-(3-methylbutoxy)ethyl]pyrrole-2,5-dione Chemical compound O=C1C(CCOCCC(C)C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 XDUKGDCFNGHDKE-UHFFFAOYSA-N 0.000 description 2
- ZEVUABRZOIAEMG-UHFFFAOYSA-N 1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]methylamino]-3-(methoxymethyl)-4-methylpyrrole-2,5-dione Chemical compound O=C1C(COC)=C(C)C(=O)N1NCC1=CC=C(C(F)(F)F)C(Cl)=N1 ZEVUABRZOIAEMG-UHFFFAOYSA-N 0.000 description 2
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 2
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 2
- LWUAMROXVQLJKA-UHFFFAOYSA-N 2-amino-3-chlorobenzoic acid Chemical compound NC1=C(Cl)C=CC=C1C(O)=O LWUAMROXVQLJKA-UHFFFAOYSA-N 0.000 description 2
- JFZJMSDDOOAOIV-UHFFFAOYSA-N 2-chloro-5-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=C(Cl)N=C1 JFZJMSDDOOAOIV-UHFFFAOYSA-N 0.000 description 2
- BVYRGNDEWRENFL-UHFFFAOYSA-N 3,4-dimethyl-1-(3,4,5-trichloroanilino)pyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC(Cl)=C(Cl)C(Cl)=C1 BVYRGNDEWRENFL-UHFFFAOYSA-N 0.000 description 2
- ZJOUWIDFMYAYBJ-UHFFFAOYSA-N 3,4-dimethyl-1-[[6-(4-methylphenyl)-5-(trifluoromethyl)pyridin-2-yl]amino]pyrrole-2,5-dione Chemical compound O=C1C(C)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(C=2C=CC(C)=CC=2)=N1 ZJOUWIDFMYAYBJ-UHFFFAOYSA-N 0.000 description 2
- IFXWZAPBANIBTK-UHFFFAOYSA-N 3-butyl-1-[[6-chloro-5-(trifluoromethyl)pyridin-2-yl]amino]-4-methylpyrrole-2,5-dione Chemical compound O=C1C(CCCC)=C(C)C(=O)N1NC1=CC=C(C(F)(F)F)C(Cl)=N1 IFXWZAPBANIBTK-UHFFFAOYSA-N 0.000 description 2
- YGHRJJRRZDOVPD-UHFFFAOYSA-N 3-methylbutanal Chemical compound CC(C)CC=O YGHRJJRRZDOVPD-UHFFFAOYSA-N 0.000 description 2
- 125000001331 3-methylbutoxy group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])O* 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 208000016683 Adult T-cell leukemia/lymphoma Diseases 0.000 description 2
- 201000004384 Alopecia Diseases 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- IFIBIMNKJQVOQZ-UHFFFAOYSA-N CC1=C(CCC(NC)=O)ON(NC2=NC(Cl)=C(C(F)(F)F)C=C2)O1 Chemical compound CC1=C(CCC(NC)=O)ON(NC2=NC(Cl)=C(C(F)(F)F)C=C2)O1 IFIBIMNKJQVOQZ-UHFFFAOYSA-N 0.000 description 2
- OUIGEACSAWPRRO-UHFFFAOYSA-N CC1=C(ON(O1)NC2=NC(=C(C=C2)C(F)(F)F)Cl)CCC(=O)N(C)C3=CC=CC=C3 Chemical compound CC1=C(ON(O1)NC2=NC(=C(C=C2)C(F)(F)F)Cl)CCC(=O)N(C)C3=CC=CC=C3 OUIGEACSAWPRRO-UHFFFAOYSA-N 0.000 description 2
- JMPXOQFDSLUMPZ-UHFFFAOYSA-N CCN(CC)C(=O)CCC1=C(ON(O1)NC2=NC(=C(C=C2)C(F)(F)F)Cl)C Chemical compound CCN(CC)C(=O)CCC1=C(ON(O1)NC2=NC(=C(C=C2)C(F)(F)F)Cl)C JMPXOQFDSLUMPZ-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 2
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 2
- 102100021906 Cyclin-O Human genes 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 230000033616 DNA repair Effects 0.000 description 2
- 206010012735 Diarrhoea Diseases 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 229910004373 HOAc Inorganic materials 0.000 description 2
- 101710195517 Histone H2AX Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 2
- 101001067891 Homo sapiens Histone H2AX Proteins 0.000 description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 208000033766 Prolymphocytic Leukemia Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 2
- 229910006124 SOCl2 Inorganic materials 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 2
- 206010042971 T-cell lymphoma Diseases 0.000 description 2
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 2
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 201000006966 adult T-cell leukemia Diseases 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 229940045799 anthracyclines and related substance Drugs 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- 230000009702 cancer cell proliferation Effects 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000005779 cell damage Effects 0.000 description 2
- 208000037887 cell injury Diseases 0.000 description 2
- 230000019522 cellular metabolic process Effects 0.000 description 2
- 210000003679 cervix uteri Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000010293 colony formation assay Methods 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940125877 compound 31 Drugs 0.000 description 2
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 2
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- 229940020967 gemzar Drugs 0.000 description 2
- 238000001415 gene therapy Methods 0.000 description 2
- 208000024963 hair loss Diseases 0.000 description 2
- 230000003676 hair loss Effects 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000003119 immunoblot Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 229940060367 inert ingredients Drugs 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 201000005962 mycosis fungoides Diseases 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 210000003739 neck Anatomy 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 231100000989 no adverse effect Toxicity 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000003463 organelle Anatomy 0.000 description 2
- 229940127084 other anti-cancer agent Drugs 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 150000004031 phenylhydrazines Chemical class 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- NWELCUKYUCBVKK-UHFFFAOYSA-N pyridin-2-ylhydrazine Chemical class NNC1=CC=CC=N1 NWELCUKYUCBVKK-UHFFFAOYSA-N 0.000 description 2
- 238000002271 resection Methods 0.000 description 2
- 230000035939 shock Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 239000001119 stannous chloride Substances 0.000 description 2
- 235000011150 stannous chloride Nutrition 0.000 description 2
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 210000002229 urogenital system Anatomy 0.000 description 2
- 229940099039 velcade Drugs 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- AAFJXZWCNVJTMK-GUCUJZIJSA-N (1s,2r)-1-[(2s)-oxiran-2-yl]-2-[(2r)-oxiran-2-yl]ethane-1,2-diol Chemical compound C([C@@H]1[C@H](O)[C@H](O)[C@H]2OC2)O1 AAFJXZWCNVJTMK-GUCUJZIJSA-N 0.000 description 1
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- HBUBKKRHXORPQB-FJFJXFQQSA-N (2R,3S,4S,5R)-2-(6-amino-2-fluoro-9-purinyl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O HBUBKKRHXORPQB-FJFJXFQQSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- OWALAMKTKVXDJL-LTHGFNDWSA-N (3R,4R,5R)-2-(6-amino-7H-purin-2-yl)-3-fluorooxane-3,4,5-triol Chemical compound N=1C=2N=CNC=2C(N)=NC=1C1OC[C@@H](O)[C@@H](O)[C@@]1(O)F OWALAMKTKVXDJL-LTHGFNDWSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- LOWMYOWHQMKBTM-UHFFFAOYSA-N 1-butylsulfinylbutane Chemical compound CCCCS(=O)CCCC LOWMYOWHQMKBTM-UHFFFAOYSA-N 0.000 description 1
- KJELNOVEWVMXQP-UHFFFAOYSA-N 1-hydroxy-3,4-dimethylpyrrole-2,5-dione Chemical compound CC1=C(C)C(=O)N(O)C1=O KJELNOVEWVMXQP-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- BQCCJWMQESHLIT-UHFFFAOYSA-N 1-propylsulfinylpropane Chemical compound CCCS(=O)CCC BQCCJWMQESHLIT-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- MFGALGYVFGDXIX-UHFFFAOYSA-N 2,3-Dimethylmaleic anhydride Chemical compound CC1=C(C)C(=O)OC1=O MFGALGYVFGDXIX-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- HTFXWAOSQODIBI-UHFFFAOYSA-N 2-benzyl-1,3-dihydropyrrolo[3,4-c]pyridine Chemical compound C1C2=CC=NC=C2CN1CC1=CC=CC=C1 HTFXWAOSQODIBI-UHFFFAOYSA-N 0.000 description 1
- PARCUCWFQOWGFX-UHFFFAOYSA-N 2-butan-2-ylsulfinylbutane Chemical compound CCC(C)S(=O)C(C)CC PARCUCWFQOWGFX-UHFFFAOYSA-N 0.000 description 1
- BQICMLHZXPNIBU-UHFFFAOYSA-N 2-chloro-1-oxido-5-(trifluoromethyl)pyridin-1-ium Chemical compound [O-][N+]1=CC(C(F)(F)F)=CC=C1Cl BQICMLHZXPNIBU-UHFFFAOYSA-N 0.000 description 1
- OHDYNLHQHOFWGR-UHFFFAOYSA-N 2-chloro-4-methyl-1-(trifluoromethyl)benzene Chemical compound CC1=CC=C(C(F)(F)F)C(Cl)=C1 OHDYNLHQHOFWGR-UHFFFAOYSA-N 0.000 description 1
- WFJXYIUAMJAURQ-UHFFFAOYSA-N 2-propan-2-ylsulfinylpropane Chemical compound CC(C)S(=O)C(C)C WFJXYIUAMJAURQ-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- ZLPORNPZJNRGCO-UHFFFAOYSA-N 3-methylpyrrole-2,5-dione Chemical compound CC1=CC(=O)NC1=O ZLPORNPZJNRGCO-UHFFFAOYSA-N 0.000 description 1
- QLILJMYXMPWURR-UHFFFAOYSA-N 3h-pyrrole-4,5-dione Chemical class O=C1CC=NC1=O QLILJMYXMPWURR-UHFFFAOYSA-N 0.000 description 1
- DRGNXDDQVUFCEF-UHFFFAOYSA-N 4-(bromomethyl)-2-chloro-1-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=C(CBr)C=C1Cl DRGNXDDQVUFCEF-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 4-amino-1-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3,5-triazin-2-one Chemical compound O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- QJPJQTDYNZXKQF-UHFFFAOYSA-N 4-bromoanisole Chemical compound COC1=CC=C(Br)C=C1 QJPJQTDYNZXKQF-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- RYYCJUAHISIHTL-UHFFFAOYSA-N 5-azaorotic acid Chemical compound OC(=O)C1=NC(=O)NC(=O)N1 RYYCJUAHISIHTL-UHFFFAOYSA-N 0.000 description 1
- DEZJGRPRBZSAKI-KMGSDFBDSA-N 565434-85-7 Chemical compound C([C@@H](N)C(=O)N[C@H](CO)C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CO)C(=O)N[C@H](CC=1C(=C(F)C(F)=C(F)C=1F)F)C(=O)N[C@H](CC1CCCCC1)C(=O)N[C@H](CCCNC(N)=N)C(=O)N[C@H](CCCNC(N)=N)C(=O)N[C@H](CCCNC(N)=N)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H](CCCNC(N)=N)C(=O)N[C@H](CCCNC(N)=N)C(O)=O)C(C=C1)=CC=C1C(=O)C1=CC=CC=C1 DEZJGRPRBZSAKI-KMGSDFBDSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- ZGCSNRKSJLVANE-UHFFFAOYSA-N Aglycone-Rebeccamycin Natural products N1C2=C3NC4=C(Cl)C=CC=C4C3=C(C(=O)NC3=O)C3=C2C2=C1C(Cl)=CC=C2 ZGCSNRKSJLVANE-UHFFFAOYSA-N 0.000 description 1
- 208000031873 Animal Disease Models Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 201000000274 Carcinosarcoma Diseases 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 102000011727 Caspases Human genes 0.000 description 1
- 108010076667 Caspases Proteins 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 108010089388 Cdc25C phosphatase (211-221) Proteins 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- 206010010099 Combined immunodeficiency Diseases 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102000003915 DNA Topoisomerases Human genes 0.000 description 1
- 108090000323 DNA Topoisomerases Proteins 0.000 description 1
- 230000005971 DNA damage repair Effects 0.000 description 1
- 230000007064 DNA hydrolysis Effects 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 230000020172 G2/M transition checkpoint Effects 0.000 description 1
- ZPLQIPFOCGIIHV-UHFFFAOYSA-N Gimeracil Chemical compound OC1=CC(=O)C(Cl)=CN1 ZPLQIPFOCGIIHV-UHFFFAOYSA-N 0.000 description 1
- 239000007818 Grignard reagent Substances 0.000 description 1
- 241000463109 Haloprofundus marisrubri Species 0.000 description 1
- 108010068250 Herpes Simplex Virus Protein Vmw65 Proteins 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 241000282596 Hylobatidae Species 0.000 description 1
- 206010020843 Hyperthermia Diseases 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 208000035490 Megakaryoblastic Acute Leukemia Diseases 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- IEUVPWMOYIURDZ-UHFFFAOYSA-N N1=CCCC1.N1C=CC=C1 Chemical class N1=CCCC1.N1C=CC=C1 IEUVPWMOYIURDZ-UHFFFAOYSA-N 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229910019201 POBr3 Inorganic materials 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 244000146510 Pereskia bleo Species 0.000 description 1
- 208000027190 Peripheral T-cell lymphomas Diseases 0.000 description 1
- 241000282405 Pongo abelii Species 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- QEHOIJJIZXRMAN-UHFFFAOYSA-N Rebeccamycin Natural products OC1C(O)C(OC)C(CO)OC1N1C2=C3NC4=C(Cl)C=CC=C4C3=C3C(=O)NC(=O)C3=C2C2=CC=CC(Cl)=C21 QEHOIJJIZXRMAN-UHFFFAOYSA-N 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 208000031672 T-Cell Peripheral Lymphoma Diseases 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- YKXJXHVLEAPHFR-UHFFFAOYSA-N [4-bromo-3-(trifluoromethyl)phenyl]hydrazine Chemical compound NNC1=CC=C(Br)C(C(F)(F)F)=C1 YKXJXHVLEAPHFR-UHFFFAOYSA-N 0.000 description 1
- MBJJCIYNRRPIGV-UHFFFAOYSA-N [6-methyl-4-(trifluoromethyl)pyridin-2-yl]hydrazine Chemical compound CC1=CC(C(F)(F)F)=CC(NN)=N1 MBJJCIYNRRPIGV-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 208000021841 acute erythroid leukemia Diseases 0.000 description 1
- 208000013593 acute megakaryoblastic leukemia Diseases 0.000 description 1
- 208000020700 acute megakaryocytic leukemia Diseases 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 125000005360 alkyl sulfoxide group Chemical group 0.000 description 1
- OFCNXPDARWKPPY-UHFFFAOYSA-N allopurinol Chemical compound OC1=NC=NC2=C1C=NN2 OFCNXPDARWKPPY-UHFFFAOYSA-N 0.000 description 1
- 229960003459 allopurinol Drugs 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical group 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 238000011558 animal model by disease Methods 0.000 description 1
- 230000002022 anti-cellular effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940019748 antifibrinolytic proteinase inhibitors Drugs 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 150000003938 benzyl alcohols Chemical class 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- QKSKPIVNLNLAAV-UHFFFAOYSA-N bis(2-chloroethyl) sulfide Chemical compound ClCCSCCCl QKSKPIVNLNLAAV-UHFFFAOYSA-N 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 230000023359 cell cycle switching, meiotic to mitotic cell cycle Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000036978 cell physiology Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 238000009614 chemical analysis method Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000003759 clinical diagnosis Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 229950000758 dianhydrogalactitol Drugs 0.000 description 1
- WVYXNIXAMZOZFK-UHFFFAOYSA-N diaziquone Chemical compound O=C1C(NC(=O)OCC)=C(N2CC2)C(=O)C(NC(=O)OCC)=C1N1CC1 WVYXNIXAMZOZFK-UHFFFAOYSA-N 0.000 description 1
- RDNWVMUUEDXVNS-HJWRWDBZSA-N diethyl (z)-2-ethyl-3-methylbut-2-enedioate Chemical compound CCOC(=O)C(\C)=C(\CC)C(=O)OCC RDNWVMUUEDXVNS-HJWRWDBZSA-N 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- CCAFPWNGIUBUSD-UHFFFAOYSA-N diethyl sulfoxide Chemical compound CCS(=O)CC CCAFPWNGIUBUSD-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229940087477 ellence Drugs 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- JOZGNYDSEBIJDH-UHFFFAOYSA-N eniluracil Chemical compound O=C1NC=C(C#C)C(=O)N1 JOZGNYDSEBIJDH-UHFFFAOYSA-N 0.000 description 1
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 230000000925 erythroid effect Effects 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 229940117360 ethyl pyruvate Drugs 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 210000005095 gastrointestinal system Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229950009822 gimeracil Drugs 0.000 description 1
- 230000000762 glandular Effects 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 210000000442 hair follicle cell Anatomy 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 125000000268 heptanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- 230000002390 hyperplastic effect Effects 0.000 description 1
- 230000036031 hyperthermia Effects 0.000 description 1
- 229940099279 idamycin Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 229950010897 iproplatin Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 208000019420 lymphoid neoplasm Diseases 0.000 description 1
- 208000025036 lymphosarcoma Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 125000006518 morpholino carbonyl group Chemical group [H]C1([H])OC([H])([H])C([H])([H])N(C(*)=O)C1([H])[H] 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 125000006126 n-butyl sulfonyl group Chemical group 0.000 description 1
- 125000006124 n-propyl sulfonyl group Chemical group 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 230000017095 negative regulation of cell growth Effects 0.000 description 1
- 210000005170 neoplastic cell Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- KPMKNHGAPDCYLP-UHFFFAOYSA-N nimustine hydrochloride Chemical compound Cl.CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 KPMKNHGAPDCYLP-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- QNDVLZJODHBUFM-WFXQOWMNSA-N okadaic acid Chemical compound C([C@H](O1)[C@H](C)/C=C/[C@H]2CC[C@@]3(CC[C@H]4O[C@@H](C([C@@H](O)[C@@H]4O3)=C)[C@@H](O)C[C@H](C)[C@@H]3[C@@H](CC[C@@]4(OCCCC4)O3)C)O2)C(C)=C[C@]21O[C@H](C[C@@](C)(O)C(O)=O)CC[C@H]2O QNDVLZJODHBUFM-WFXQOWMNSA-N 0.000 description 1
- VEFJHAYOIAAXEU-UHFFFAOYSA-N okadaic acid Natural products CC(CC(O)C1OC2CCC3(CCC(O3)C=CC(C)C4CC(=CC5(OC(CC(C)(O)C(=O)O)CCC5O)O4)C)OC2C(O)C1C)C6OC7(CCCCO7)CCC6C VEFJHAYOIAAXEU-UHFFFAOYSA-N 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 229950000193 oteracil Drugs 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 210000003899 penis Anatomy 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000000816 peptidomimetic Substances 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- 238000011458 pharmacological treatment Methods 0.000 description 1
- SUSQOBVLVYHIEX-UHFFFAOYSA-N phenylacetonitrile Chemical compound N#CCC1=CC=CC=C1 SUSQOBVLVYHIEX-UHFFFAOYSA-N 0.000 description 1
- 125000003884 phenylalkyl group Chemical group 0.000 description 1
- UXCDUFKZSUBXGM-UHFFFAOYSA-N phosphoric tribromide Chemical compound BrP(Br)(Br)=O UXCDUFKZSUBXGM-UHFFFAOYSA-N 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 239000000419 plant extract Substances 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- 229960001237 podophyllotoxin Drugs 0.000 description 1
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000009993 protective function Effects 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229960005567 rebeccamycin Drugs 0.000 description 1
- INSACQSBHKIWNS-QZQSLCQPSA-N rebeccamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](OC)[C@@H](CO)O[C@H]1N1C2=C3N=C4[C](Cl)C=CC=C4C3=C3C(=O)NC(=O)C3=C2C2=CC=CC(Cl)=C21 INSACQSBHKIWNS-QZQSLCQPSA-N 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 208000002491 severe combined immunodeficiency Diseases 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical class [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000003375 sulfoxide group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 230000008467 tissue growth Effects 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000003934 vacuole Anatomy 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000009278 visceral effect Effects 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 230000037314 wound repair Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/44—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having three double bonds between ring members or between ring members and non-ring members
- C07D207/444—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having three double bonds between ring members or between ring members and non-ring members having two doubly-bound oxygen atoms directly attached in positions 2 and 5
- C07D207/448—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having three double bonds between ring members or between ring members and non-ring members having two doubly-bound oxygen atoms directly attached in positions 2 and 5 with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms, e.g. maleimide
- C07D207/452—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having three double bonds between ring members or between ring members and non-ring members having two doubly-bound oxygen atoms directly attached in positions 2 and 5 with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms, e.g. maleimide with hydrocarbon radicals, substituted by hetero atoms, directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/50—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyrrole Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Se proporcionan nuevas dionas de azol sustituidas que matan células, suprimen la proliferación celular, suprimen el crecimiento celular, anulan el punto de control G2 del ciclo celular y/o provocan la adaptación a la detención del ciclo celular G2. Se proporcionan métodos para preparar y usar los compuestos de la invención. La invención proporciona azoldionas sustituidas para tratar trastornos de proliferación celular. La invención incluye el uso de azoldionas sustituidas para matar o suprimir selectivamente células cancerosas sin tratamiento anticanceroso adicional. La invención incluye el uso de dionas de azol sustituidas que anulan el punto de control G2 del ciclo celular para sensibilizar selectivamente las células cancerosas a los reactivos, tratamientos y/u otros tipos de reactivos anticancerígenos que dañan el ADN. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos 2,5-dioxo-azolina N-sustituidos para la utilización en el tratamiento del cáncer
Campo de la invención
La invención se refiere a compuestos nuevos de azol dionas sustituidas que presentan una actividad anticancerosa y/o una actividad contra células que proliferan en los que los compuestos de azol dionas sustituidas anulan el punto de control G2 del ciclo celular y/u ocasionan adaptación a la detención del ciclo celular. La invención incluye la utilización de compuestos de azol dionas sustituidas para suprimir o eliminar selectivamente células cancerosas, con o sin tratamiento anticanceroso adicional. La invención incluye compuestos de azol dionas sustituidas que anulan el punto de control G2 del ciclo celular proporcionados en la presente memoria para la utilización en métodos para sensibilizar selectivamente las células cancerosas a agentes que dañan el ADN, a tratamientos que dañan el ADN y/o a otros tipos de agentes anticancerosos.
Antecedentes
El ciclo celular comprende la fase S (replicación de ADN), fase M (mitosis), y dos fases de intervalo (fases G1 y G2) entre las fases S y M. Los puntos de control en el ciclo celular garantizan la progresión precisa a través de las etapas del ciclo celular, e incluyen la monitorización de la integridad del ADN, replicación del ADN, tamaño celular, y el ambiente circundante (Maller (1991) Curr. Opin. Cell Biol., 3:26). Los puntos de control del ciclo celular que monitorizan el estado del genoma incluyen el punto de control G1 antes del comienzo de la replicación del ADN y el punto de control G2 antes del comienzo de la mitosis. El punto de control G1 permite la detección y reparación del daño del ADN antes de entrar en la fase S, proporcionando en consecuencia una función protectora crucial dado que, cuando el ADN dañado se replica, a menudo da lugar a mutaciones (Hartwell (1992) Cell 71: 543). El punto de control G2 permite la detección y reparación del daño del ADN antes de entrar en la mitosis (fase M), proporcionando en consecuencia una función crucial dado que la mitosis sin reparación del ADN puede propagar el daño a través de las células hijas dañadas en su ADN, o la mitosis puede fallar por completo. La progresión a través de los puntos de control G1 y G2, sin reparar el daño extenso del ADN, usualmente da como resultado muerte celular.
La inhibición del punto de control G2 del ciclo celular por péptidos, peptidomiméticos, y “moléculas pequeñas” se ha usado para seleccionar selectivamente células cancerosas dado que la mayoría de las células cancerosas tienen defectos en uno o los dos puntos principales de control del ciclo celular que protegen a las células de los efectos del daño del ADN, de tal modo que la inhibición del punto de control G2 permite que las células dañadas en su ADN reingresen en el ciclo celular sin reparar el daño del ADN. (Kawabe T (2004) “G2 checkpoint abrogators as anti-cancer drugs” Mol Cancer Ther 3: 513-519). Aunque el mecanismo molecular del punto de control G2 del ciclo celular se estudió extensamente, en estudios anteriores no se estableció ninguna diana molecular única para la anulación terapéutica del punto de control G2. Un protocolo de selección basado en fenotipos se ha desarrollado para identificar eficientemente inhibidores potenciales del punto de control G2 (Suganuma M. & Kawabe T., Solicitud EP n° 00964563; Sha et al. (2007) Mol Cancer Ther 6: 147-153), en el que la selección, a base de fenotipos del ciclo celular, de péptidos que anulan el punto de control G2 identificó a CBP501 que tiene un mecanismo único de acción en el punto de control G2 del ciclo celular. (Sha et al. (2007) Mol Cancer Ther 6: 147-153). La patente US n° 6.291.447 divulga compuestos de granulatimida y composiciones farmacéuticas de los mismos.
Sumario de la invención
La presente invención se define en las reivindicaciones. Los compuestos de la invención son útiles para tratar trastornos de proliferación celular. En particular, la invención proporciona compuestos que incluyen: 3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)propanoato de terc-butilo (referido de manera intercambiable como S001860, S01860, or S1860, es decir, S001860 = S01860 = S1860); 1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona (S00109 = S0109 = S109); 3-(butoximetil)-1-1[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03518); 1-[[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-3-[(3-metilbutoxi)metil]azolin-2,5-diona (S03405); 3-[(3,3-dimetilbutoxi)metil]-1-1[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03747); y compuestos relacionados. Estos compuestos tienen efectos que pueden incluir eliminar o suprimir células que proliferan indeseablemente asociadas con un trastorno de proliferación celular.
La presente invención proporciona compuestos que eliminan o suprimen células que proliferan indeseablemente, asociadas con un trastorno de proliferación celular. La presente invención proporciona compuestos que anulan el punto de control G2 del ciclo celular y/u ocasionan adaptación a la detención del ciclo celular en G2. La presente invención proporciona compuestos que anulan el punto de control G2 del ciclo celular y/u ocasionan adaptación a la detención del ciclo celular, conduciendo a muerte o supresión de células que proliferan indeseablemente. La presente invención proporciona compuestos que anulan el punto de control G2 del ciclo celular y/u ocasionan adaptación a la detención del ciclo celular, conduciendo a muerte o supresión de células dañadas en su ADN. La presente invención proporciona compuestos que tienen actividad citotóxica contra células cancerosas. La presente invención proporciona compuestos que tienen actividad citotóxica contra células cancerosas, incluyendo, pero sin limitarse a, células cancerosas dañadas
en su ADN. La presente invención proporciona compuestos que tienen actividad citotóxica contra células cancerosas, incluyendo, pero sin limitarse a, células cancerosas en detención del ciclo celular en G2 debido a daño del ADN. La presente invención proporciona compuestos que tienen actividad citotóxica contra células cancerosas, y poca o ninguna actividad citotóxica contra células normales. La presente invención proporciona compuestos que pueden aumentar el efecto citotóxico de otros agentes y tratamientos anticancerosos, especialmente agentes anticancerosos que dañan el ADN y tratamientos anticancerosos que dañan el ADN. La presente invención proporciona compuestos que pueden sensibilizar a las células frente a otros agentes y tratamientos anticancerosos, especialmente agentes anticancerosos que dañan el ADN y tratamientos anticancerosos que dañan el ADN. La presente divulgación proporciona métodos para obtener y usar los compuestos divulgados en la presente memoria. La presente invención proporciona composiciones farmacéuticas que contienen los compuestos de la invención.
La presente invención proporciona compuestos para la utilización en el tratamiento de un trastorno de proliferación celular. La presente invención proporciona compuestos para la utilización en el tratamiento de cáncer, por ejemplo para tratar la proliferación celular indeseable asociada con células tumorales benignas y malignas, células de leucemia, células de linfoma, o células de mieloma múltiple. La presente invención proporciona compuestos para la utilización para anular el punto de control G2 del ciclo celular en células que proliferan indeseablemente, tales como células cancerosas, por ejemplo células tumorales benignas y malignas, células de leucemia, células de linfoma, o células de mieloma múltiple. La presente invención proporciona compuestos para la utilización para ocasionar adaptación a la detención del ciclo celular en G2 en células que proliferan indeseablemente, tales como células cancerosas, por ejemplo células tumorales benignas y malignas, células de leucemia, células de linfoma, o células de mieloma múltiple.
La presente invención proporciona compuestos para la utilización en métodos para tratar un trastorno de proliferación celular al administrar una cantidad eficaz de un compuesto de la invención in vivo, ex vivo, o in vitro. La presente invención proporciona compuestos para la utilización en métodos para tratar un trastorno de proliferación celular al administrar una cantidad eficaz de un compuesto de la invención a un sujeto. La presente invención proporciona compuestos para la utilización en métodos para tratar un trastorno de proliferación celular en los que el trastorno de proliferación celular es cáncer, incluyendo, pero sin limitarse a, linfoma, mieloma, leucemia. La presente invención proporciona compuestos para la utilización en métodos para tratar cáncer al administrar una cantidad eficaz de un compuesto y administrar por lo menos un tratamiento anticanceroso adicional, por ejemplo un agente que daña el ADN o un tratamiento que daña el ADN.
La invención proporciona compuestos o composiciones farmacéuticas para la utilización en métodos para eliminar o suprimir células al poner en contacto las células con un compuesto de la invención o una composición farmacéutica de la invención, en combinación con un agente o tratamiento que daña el ADN. La invención proporciona métodos para sensibilizar selectivamente las células a un agente y/o tratamiento que daña el ADN, al poner en contacto las células con un compuesto de la invención o una composición farmacéutica de la invención, en combinación con el agente o tratamiento que daña el ADN. La invención proporciona compuestos o composiciones farmacéuticas para la utilización en métodos para inducir apoptosis, necrosis, y/o catástrofe mitótica en células que proliferan indeseablemente, que comprenden administrar un compuesto de la invención o una composición farmacéutica de la invención a las células, en una cantidad suficiente para eliminar o suprimir las células que proliferan indeseablemente, con o sin administrar otros tratamientos. La invención proporciona compuestos o composiciones farmacéuticas para la utilización en métodos para inducir apoptosis, necrosis, y/o catástrofe mitótica en células que proliferan indeseablemente en un sujeto, que comprenden administrar un compuesto de la invención o una composición farmacéutica de la invención al sujeto, en una cantidad suficiente para eliminar o suprimir las células que proliferan indeseablemente, con o sin administrar otros tratamientos.
La selección basada en fenotipos se usó para medir la capacidad de los compuestos de la invención para ocasionar adaptación a la detención del ciclo celular en G2 en células detenidas en G2. La adaptación a la detención del ciclo celular en G2 y reingreso en el ciclo celular puede dar como resultado la muerte de la célula previamente detenida en G2, o supresión (inhibición) de la proliferación posterior de la célula previamente detenida en G2. La capacidad para ocasionar adaptación a la detención del ciclo celular en G2 se midió al poner en contacto las células, en las que la detención en G2 se había inducido por irradiación (por ejemplo, radiación gamma (y) o radiación de rayos X), con los compuestos de la invención en diversas concentraciones y, para cada compuesto en cada concentración, determinando el porcentaje de células que escaparon a la detención en G2 y reingresaron en el ciclo celular, al determinar el porcentaje de células en fase G1. El valor IC50 para cada compuesto se calculó como la dosificación (usualmente en |iM) que ocasionó un incremento medio máximo del porcentaje de células en fase G1 (el incremento G1) medido para ese compuesto. Ciertos compuestos de la invención se identificaron inicialmente por selección basada en fenotipos de bibliotecas de pequeñas moléculas para actividad contra células detenidas en G2 como se describe anteriormente.
Como se define en las reivindicaciones, la invención proporciona compuestos que tienen la fórmula de la estructura (IV) o la estructura (VI):

en la que:
uno de R1 y R2 es metilo, y el otro de R1 y R2 es alquilo o alquilo sustituido con alcoxi, hidroxi, carboxi, alcoxicarbonilo, carbamoílo opcionalmente sustituido, o aminocarbonilo cíclico opcionalmente sustituido;
R3 es H o alquilo;
R4 y R5 se seleccionan independientemente de entre H y alquilo;
A es N o CH;
R6, R7, y R9 se seleccionan independientemente de entre H, alquilo, alquilo sustituido, halógeno, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, alcoxi opcionalmente sustituido, ariloxi opcionalmente sustituido, ciano, nitro, alquiltio opcionalmente sustituido, alquilsulfinilo opcionalmente sustituido, alquilsulfonilo opcionalmente sustituido, ariltio opcionalmente sustituido, o carbamoílo opcionalmente sustituido; y R8 es halógeno o un grupo alquilo sustituido con uno o más halógenos;
o una sal del mismo;
en el que además el compuesto no tiene la siguiente estructura:
La presente invención proporciona compuestos que anulan el punto de control G2 del ciclo celular y/u ocasionan adaptación a la detención del ciclo celular en G2, en el que estos compuestos, cuando se administran a células o a un sujeto, tienen efectos que pueden incluir eliminar o suprimir el crecimiento de células que proliferan indeseablemente, los compuestos incluyen:
3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il) propanoato de terc-butilo (S01860); etil 3 -14-{[6-cloro-5-(trifluorometil)(2-piridil)] amino }-4-2,5-dioxoazolin-3-il)propanoato (S01861);
3-(1 -{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)propanoato de metilo (S01648);
3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)-N-metoxi-N-metilpropanamida (S01796);
1-{[3-bromo-6-cloro-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona (S01981);
1-{[6-doro-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona (S00109);
1-{[6-doro-5-(trifluorometil)(2-piridil)]metilamino}-3,4-dimetilazolin-2,5- diona (S00170);
1-{[6-bromo-5-(trifluorometil)(2-piridil)]metilamino}-3,4-dimetilazolin-2,5-diona (S01007);
1-{[6-doro-5-(trifluorometil)(2-piridil)] amino } -4-metil-3-(3-metilbutil)azolin-2,5-diona (S01554);
1- {[6-doro-5-(trifluorometil)(2-piridil)]amino}-3-(metoximetil)-4-metilazolin-2,5-diona (S01599);
3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)-N,N-dietilpropanamida (S01711);
2- [(1- { [6-cloro-5-(trifluorometil)(2-piridil)] amino } -4-metil-2,5-dioxoazolin-3-il)metil]propano-1,3-dioato de dietilo (S01712);
N-(ferc-butil)-3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)] amino}-4-metil-2,5-dioxoazolin-3-il)propanamida (S01758); 1-{[6-bromo-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona (S00994);
3.4- dimetil-1-{[6-fenil-5-(trifluorometil)(2-piridil)]amino}azolin-2,5-diona (S01266);
1-{[6-cloro-5-(trifluorometil)(2-piridil)] amino }-3-(hidroximetil)-4-metilazolin-2,5-diona (S01470);
3- (1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)-N-metilpropanamida (S01883);
3.4- dimetil-1-[(3,4,5-triclorofenil)amino]azolin-2,5-diona (S00832);
1-{[6-(3-cloro-4-fluorofenil)-5-(trifluorometil)(2-piridil)] amino }-3,4-dimetil (3,4-dimetilazolin-2,5-diona (S01313); 3.4- dimetil-1-[[6-(2-metilpropil)-5-(trifluorometil)(2-piridil)]amino}azolin-2,5-diona (S01457);
1-{[6-cloro-5-(trifluorometil)(2-piridil)] amino }-3-etil-4-metilazolin-2,5-diona (S01553);
1-{[4-cloro-6-fenil-5-(trifluorometil)(2-piridil)]amino }-3,4-dimetilazolin-2,5-diona (S01734);
6-[(3,4-dimetil-2,5-dioxoazolinil)amino]-3-(trifluorometil)piridin-2-carbonitrilo (S01475);
1-{[4-bromo-3-(trifluorometil)fenil]amino}-3,4-dimetilazolin-2,5-diona (S00516);
1-{[6-(4-clorofenil)-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona (S01371);
3.4- dimetil-1-{[6-(4-metilfenil)-5-(trifluorometil)(2-piridil)]amino}azolin-2,5-diona (S01393);
1-{[6-(3-clorofenil)-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona (S01474);
1- {[6-cloro-5-(trifluorometil)(2-piridil)]metilamino}-3-(metoximetil)-4-metilazolin-2,5-diona (S01600);
3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)-N-metil-N-fenilpropanamida (S01759); 3.4- dimetil-1-{[6-bencil-5-(trifluorometil)(2-piridil)]amino}azolin-2,5-diona (S01762);
3-(1-1[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)propanoato de metiletilo (S02022); 3-(1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)propanoato de metilpropilo (S02264); 2- (1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-2,5-dioxoazolin-3-il)acetato de ferc-butilo (S02225);
1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-3-(etoximetil)-4-metilazolin-2,5-diona (S02366);
3- butil-1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03448);
1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-3-[2-(2-metilo(1,3-dioxolan-2-il))etil]azolin-2,5-diona (S03456); 1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-3-[(2-metoxietoxi)metil]-4-metilazolin-2,5-diona (S03742);
1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-(3-hidroxihexil)-3-metilazolin-2,5-diona (S03552);
1-{[6-doro-5-(trifluorometil)(2-piridil)]amino}-4-(3-hidroxipentil)-3-metilazolin-2,5-diona (S03745);
1-{[6-doro-5-(trifluorometil)(2-piridil)] amino }-4-metil-3-[(3-metilbutoxi)metil]azolin-2,5-diona (S03405);
3-(butoximetil)-1-{[6-doro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03518);
3-[(3,3-dimetilbutoxi)metil]-1-{[6-doro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03747);
1-{[6-doro-5-(trifluorometil)(2-piridil)]amino}-3-(2-etoxietil)-4-metilazolin-2,5-diona (S03960);
1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metil-3-[(2-metilpropoxi)metil]azolin-2,5-diona (S03963);
3- [(2,2-dimetilpropoxi)metil]-1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03962); 4- [(1,3-dimetilbutoxi)metil]-1-1[6-cloro-5-(trifluorometil)(2-piridil)]amino}-3-metilazolin-2,5-diona (S03964);
4-[(terc-butoxi)metil]-1-([6-cloro-5-(trifluorometil)(2-piridil)]amino}-3-metilazolin-2,5-diona (S03873);
1-{[6-cloro-5-(trifluorometil)(2-piridil)] amino }-4-metil-3-[2-(2-metilpropoxi)etil]azolin-2,5-diona (S03955);
1-{[6-cloro-5-(trifluorometil)(2-piridil)] amino }-4-metil-3-[2-(3-metilbutoxi)etil]azolin-2,5-diona (S03956);
1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-3-metil-4-(2-propoxietil)azolin-2,5-diona (S04034);
o una sal de cualquiera de estos compuestos.
Se han sintetizado y evaluado formas de realización adicionales de la invención, como se define en las reivindicaciones, y los detalles se exponen en la descripción a continuación y en los ejemplos, tablas, y figuras (dibujos) presentados en la presente memoria. Otros objetos, características y ventajas de la invención resultarán evidentes a partir de la descripción, ejemplos, tablas, dibujos, y a partir de las reivindicaciones.
Breve descripción de los dibujos
La figura 1 muestra el porcentaje de células en fase G1 después de que las células Jurkat predetenidas en la fase G2 por irradiación de rayos X (10 Gy) se tratan con los compuestos S00109 (diamantes vacíos) y S01860 (diamantes llenos), a las dosis indicadas, durante 24 horas.
La figura 2 muestra el nivel de fosforilación de histona H3 (%) en células Jurkat predetenidas en la fase G2 por irradiación de rayos X (10 Gy) y tratadas con el compuesto S00109 a 1 |iM (cuadros vacíos) y 0,3 |iM (círculos llenos) durante tiempos de tratamiento de hasta 24 horas.
La figura 3 es una imagen de una inmunotransferencia que muestra niveles de yH2AX fosforilada después del tratamiento secuencial de células Jurkat con irradiación de rayos X (dosis total 10 Gy) y compuesto S00109 a 1 |iM a y durante los tiempos indicados, en el que yH2AX fosforilada se detectó usando antifosfo-histona H2AX y exposición de 10 minutos de la inmunotransferencia, de izquierda a derecha: M muestra estándares etiquetados de peso molecular; la línea 1 muestra células que no recibieron radiación ni tratamiento con S00109 (células de control); la línea 2 muestra células irradiadas a 24 horas después de la irradiación y sin tratamiento con S00109; la línea 3 muestra células irradiadas a 48 horas después de la irradiación y sin tratamiento con S00109; la fila 4 muestra células irradiadas después de 0 horas de tratamiento con S00109 (a 24 horas después de la irradiación); la línea 5 muestra células irradiadas después de 3 horas de tratamiento con S00109 (a 27 horas después de la irradiación); la línea 6 muestra células irradiadas después de 9 horas de tratamiento con S00109 (a 33 horas después de la irradiación); la línea 7 muestra células irradiadas después de 15 horas de tratamiento con S00109 (a 39 horas después de la irradiación); la línea 8 muestra células irradiadas después de 21 horas de tratamiento con S00109 (a 45 horas después de la irradiación); la línea 9 muestra células irradiadas después de 24 horas de tratamiento con S00109 (a 48 horas después de la irradiación).
La figura 4 muestra recuentos de colonias (eje y) después del tratamiento de células HCT 116 con el compuesto S00109 a concentraciones de 0 a 4 |iM (eje x) para las células tratadas con el compuesto S00109 solo y sin irradiación (0 Gy, representado por círculos vacíos) y células tratadas con el compuesto S00109 en combinación con irradiación de rayos X (dosis total 1 Gy, representado por círculos llenos; dosis total 3 Gy, representado por cuadros vacíos), en el que la disminución en los recuentos de colonias es una medida de la supresión del crecimiento celular y/o de muerte celular.
La figura 5 muestra el porcentaje de células en fase subG1 (eje y) después del tratamiento in vitro de células ARH-77 con el compuesto S00109 a concentraciones de 0 a 10 |ig/ml (eje x) como sigue: células ARH-77 tratadas con
el compuesto S00109 solo a las concentraciones indicadas en el eje x (“S109 solamente” representado por diamantes llenos, línea continua); células ARH-77 tratadas con el compuesto S00109 a las concentraciones indicadas en el eje x, en combinación con dexametasona a 2 ng/ml (“Dex2ng/ml” representado por cuadros vacíos, línea de rayas cortas); células ARH-77 tratadas con el compuesto S00109 a las concentraciones indicadas en el eje x, en combinación con dexametasona a 20 ng/ml (“Dex20ng/ml” representado por triángulos vacíos, línea de punto y raya); y células ARH-77 tratadas con el compuesto S00109 a las concentraciones indicadas en el eje x, en combinación con dexametasona a 200 ng/ml (“Dex200ng/ml” representado por círculos vacíos, línea de rayas largas); en el que la fase subG1 indica muerte celular.
La figura 6 muestra un análisis de supervivencia para ratones SCID con trasplante intraperitoneal de 1,9 x 106 células ARH-77, durante hasta 80 días después del trasplante (eje x, días después del trasplante; eje y, % de ratones que sobreviven) para ratones tratados por inyección intraperitoneal en el Día 1, Día 2, y Día 3 después del trasplante como sigue: ratones de control tratados con vehículo solo (“Control” línea de rayas); ratones tratados con 50 mg/kg del compuesto S00109 (“S109” línea continua); y ratones tratados con 2 mg/kg de dexametasona (“Dexa” línea de punto y raya).
La figura 7 muestra un análisis de supervivencia para ratones SCID con trasplante intraperitoneal de 0,8 x 106 células ARH-77, durante hasta 85 días después del trasplante (eje x, días después del trasplante; eje y, % de ratones que sobreviven), para ratones tratados mediante una sola administración oral de compuestos en el Día 1 después del trasplante como sigue: ratones de control tratados por vía oral con vehículo solo (“control” línea continua); ratones tratados por vía oral con 750 mg/kg del compuesto S00109 (“S109” línea punteada); y ratones tratados por vía oral con 750 mg/kg del compuesto S001860 (“S1860” línea de rayas).
La figura 8 muestra un análisis de supervivencia para ratones SCID con trasplante intraperitoneal de 4,1 x 106 células ARH-77, durante hasta 50 días después del trasplante (eje x, días después del trasplante; eje y, % de ratones que sobreviven), para ratones tratados por administración oral una vez al día de los compuestos en el Día 1 y en el Día 2 después del trasplante como sigue: ratones de control tratados por vía oral una vez al día durante dos días con vehículo solo (“CONT” línea continua); ratones tratados por vía oral una vez al día durante dos días con 250 mg/kg del compuesto S003518 (“S3518” línea punteada); ratones tratados por vía oral una vez al día durante dos días con 250 mg/kg del compuesto S003405 (“S3405” línea de rayas); y ratones tratados por vía oral una vez al día durante dos días con 250 mg/kg del compuesto S003747 (“S3747” línea de punto y raya).
Descripción detallada de la invención
Definiciones
A menos que se defina de otra manera, los términos técnicos y científicos usados en la presente memoria tienen el significado comúnmente comprendido por un experto en la materia a la que pertenece esta invención. Como se usan en la presente memoria, los siguientes términos tienen los significados atribuidos a ellos, a menos que se especifique de otra manera.
Las expresiones “tratamiento que daña el ADN” y “agente que daña el ADN” se refieren a cualquier agente o tratamiento que directa o indirectamente daña el ADN, incluyendo, pero sin limitarse a, fármacos (agentes) que dañan el ADN, irradiación con niveles que dañan el ADN de radiación X, gamma (y), o UV, diversos tipos de choque ambiental, y similares. Es conocido que un tratamiento que daña el ADN o agente que daña el ADN puede actuar directamente sobre el ADN, por ejemplo para afectar la estructura del ADN o interferir con la síntesis de ADN, o puede actuar indirectamente sobre el ADN por sus efectos sobre otros sistemas celulares implicados en síntesis y replicación de ADN, por ejemplo para afectar o inhibir las funciones de los microtúbulos o ADN topoisomerasa. ejemplos específicos de agentes que dañan el ADN incluyen, pero no se limitan a, agentes alquilantes, nitrosoureas, antimetabolitos, alcaloides vegetales, extractos vegetales, radioisótopos, hormonas esteroideas. ejemplos adicionales de agentes que dañan el ADN también incluyen, pero no se limitan a, agentes conocidos como “fármacos que dañan el ADN” o “fármacos anticancerosos” o “agentes anticancerosos” o “agentes anticancerosos que dañan el ADN”, por ejemplo 5-fluorouracilo (5-FU), capecitabina, S-1 (Tegafur, 5-cloro-2,4-dihidroxipiridina y ácido oxónico), 5-etiniluracilo, arabinosilcitosina (ara-C), 5-azacitidina (5-AC), 2',2'-difluoro-2'-desoxicitidina (dFdC), antimetabolitos de purina (mercaptopurina, azatiopurina, tioguanina), gemcitabina (Gemzar® ), bortezomib (Velcade® ) pentostatina, alopurinol, 2-fluoro-arabinosil-adenina (2F-ara-A), hidroxiurea, mostaza de azufre (sulfuro de biscloroetilo), mecloretamina, melfalano, melfarano, vincristina, clorambucilo, ciclofosfamida, ifosfamida, tiotepa, AZQ, mitomicina C, dianhidrogalactitol, dibromoducitol, sulfonato de alquilo (busulfano), nitrosoureas (BCNU, CCNU, 4-metil CCNU o ACNU), procarbazina, decarbazina, rebecamicina, antraciclinas tales como doxorrubicina (adriamicina; ADR), daunorrubicina (Cerubicina), idarrubicina (Idamicina) y epirrubicina (Ellence), análogos de antraciclina tales como mitoxantrona, actinimicina D, inhibidores no intercalantes de topoisomerasas tales como epipodofilotoxinas (etopósido = VP16, tenipósido = VM-26), podofilotoxina, bleomicina (Bleo), pepleomicina, taxanos, compuestos que forman aductos con ácido nucleico incluyendo derivados de platino, por ejemplo cisplatino (CDDP), análogo trans de cisplatino, carboplatino, iproplatino, tetraplatino y oxaliplatino, así como camptotecina, topotecán, irinotecán (CPT-11), y SN-38. ejemplos específicos de tratamientos que dañan el ADN incluyen radiación, por ejemplo radiación ultravioleta (UV), infrarroja (IR), rayos X, a (alfa), p (beta), o y (gamma), así como choque ambiental, por ejemplo hipertermia. Un
experto en la materia puede identificar y usar otros agentes y tratamientos que dañan el ADN.
La expresión “compuesto de la invención” está destinada a referirse a una molécula que tiene la estructura y actividad que se describe en la presente memoria. Un compuesto de la invención puede estar aislado, puro, sustancialmente puro, o puede estar en una composición que contiene una mezcla de otros componentes. La pureza de una composición que contiene un compuesto de la invención puede determinarse, por ejemplo, usando técnicas de química analítica, tales como cromatografía de líquidos de alta resolución (HPLC), o cromatografía de líquidos-espectrometría de masas (LC-MS) o cromatografía de gases-espectrometría de masas (GS-MS), u otras técnicas analíticas conocidas por un experto en la materia. Una composición, como se proporciona en la presente memoria, puede contener uno o más compuestos de la invención, en una mezcla con vehículos, portadores, excipientes, ingredientes inertes, y similares adecuados. Si se desea, una composición, como se proporciona en la presente memoria, puede contener ingredientes activos adicionales, incluyendo agentes que dañan el ADN y similares, así como uno o más compuestos de la invención, en una mezcla con vehículos, portadores, excipientes, ingredientes inertes, y similares adecuados.
Las expresiones “composición farmacéutica” o “medicamento” se refieren a una composición adecuada para su uso farmacéutico en un sujeto, por ejemplo como agente anticáncer. El sujeto puede ser cualquier animal en el que los compuestos de la invención anulan el punto de control G2 del ciclo celular y/u ocasionan adaptación a la detención en G2. En particular, el sujeto puede ser un mamífero, por ejemplo un caballo, vaca, perro, gato, o ser humano. Una composición farmacéutica de la invención es una formulación que puede comprender una cantidad farmacológicamente eficaz de por lo menos un compuesto de la invención y un portador farmacéuticamente aceptable.
Las expresiones “trastorno de proliferación celular” o “trastorno proliferativo” o “trastorno proliferativo celular” o “padecimiento proliferativo” o “padecimiento caracterizado por proliferación celular indeseable” o cualquier equivalente gramatical de las mismas se utilizan para referirse a cualquier padecimiento fisiológico patológico o no patológico caracterizado por proliferación aberrante o indeseable de por lo menos una célula, incluyendo, pero sin limitarse a, padecimientos caracterizados por proliferación celular indeseable o no buscada o aberrante, padecimientos caracterizados por supervivencia celular indeseable o no buscada o aberrante, y padecimientos caracterizados por apoptosis deficiente o aberrante. La expresión “proliferación celular” y equivalentes gramaticales de la misma se utilizan para comprender un incremento en el número de células como resultado de la división celular, así como un incremento en la masa total de células como resultado del crecimiento celular, por ejemplo por el crecimiento de células hijas después de la mitosis. Un ejemplo no limitativo de un “trastorno de proliferación celular” o “trastorno proliferativo” o “padecimiento proliferativo” o “padecimiento caracterizado por proliferación celular indeseable” es cáncer, por ejemplo proliferación y supervivencia indeseable o no buscada o aberrante de células cancerosas, tales como células asociadas con linfoma, mieloma, sarcoma, leucemia, u otros trastornos neoplásicos dados a conocer en otra parte en la presente memoria y conocidos por un experto en la materia.
Las expresiones “eliminan o suprimen células” o “eliminar o suprimir células” o “eliminan o suprimen células que proliferan indeseablemente” o “eliminan o suprimen células diana” o cualquier equivalente gramatical de las mismas se utilizan para referirse a los resultados de estar en contacto con una cantidad eficaz de un compuesto de la invención. Los términos “eliminan” o “eliminar” se utilizan para referirse a muerte celular que resulta de los efectos de un compuesto de la invención sobre una célula, en particular la muerte de una célula que prolifera indeseablemente, tal como una célula cancerosa, en el que la muerte puede deberse a apoptosis, catástrofe mitótica, necrosis, u otra causa, dependiendo de las circunstancias de la célula. Los términos “suprimen” o “suprimir” se utilizan para referirse a supresión de la proliferación celular que resulta de los efectos de un compuesto de la invención sobre una célula, en el que la supresión puede ser parcial o completa. Un compuesto de la invención puede ocasionar supresión parcial de una célula, de tal modo que la célula puede dejar de dividirse pero continúa creciendo, o la célula puede dividirse mucho más lentamente, o la célula puede crecer mucho más lentamente, o una célula cancerosa no puede progresar de un estado premetastásico a un estado metastásico, etc. Un compuesto de la invención puede ocasionar supresión completa, en el que una célula no se divide ni crece, en particular, en el que una célula que prolifera indeseablemente no se divide ni crece.
Las expresiones “cantidad eficaz” o “cantidad suficiente” o cualquier equivalente gramatical de las mismas se utilizan para referirse a una cantidad de un compuesto de la invención suficiente para producir por lo menos un efecto deseado. Las cantidades eficaces se determinan por cualquiera de una diversidad de medidas, incluyendo pero sin limitarse a: muerte celular; proliferación celular disminuida; cantidades disminuidas de células; inhibición de crecimiento celular; tamaño celular disminuido; supervivencia celular disminuida; metabolismo celular disminuido; apoptosis; catástrofe mitótica; adaptación a la detención del ciclo celular (es decir, escape de la detención del ciclo celular, conduciendo usualmente a reingreso al ciclo celular); marcadores de daño celular o citotoxicidad; indicadores indirectos de daño celular o citotoxicidad, tal como contracción tumoral; supervivencia mejorada de un sujeto; o desaparición de marcadores asociados con proliferación celular indeseable, no buscada, o aberrante. Por ejemplo, cuando se desea inhibir la proliferación indeseable de una célula o tipo celular particular, una cantidad eficaz será una cantidad que disminuye de manera detectable la división celular, o disminuye el metabolismo celular, o incrementa la muerte celular, o disminuye la supervivencia celular, de esa célula o tipo celular. Un efecto deseado puede ser un efecto selectivo; por ejemplo, una “cantidad eficaz” es una cantidad que elimina o suprime células diana a la vez que tiene poco o ningún efecto citotóxico sobre células no diana, o una cantidad que produce un beneficio terapéutico deseado en un sujeto que tiene un trastorno de proliferación celular, a la vez que tiene poco o ningún efecto adverso sobre el sujeto. Un
trastorno de proliferación celular se puede tratar in vitro o ex vivo administrando una cantidad eficaz de por lo menos un compuesto de la invención, en el que la cantidad eficaz de por lo menos un compuesto puede administrarse in vitro o ex vivo para tratar las células Alternativamente, el compuesto de la invención es para la utilización en un método para tratar un trastorno de proliferación celular, en el que el método comprende administrar el compuesto in vivo a un sujeto que tiene un trastorno de proliferación celular.
Las expresiones “punto de control G2 del ciclo celular” o “punto de control G2” o cualquier equivalente gramatical de las mismas se refieren al punto de control G2 que se presenta al final de la fase G2 del ciclo celular. Durante la fase de “ intervalo” G2 entre la síntesis de ADN (fase S, replicación del ADN en preparación para la mitosis) y la mitosis (fase M, división celular para producir células hijas), la célula continúa creciendo y produce nuevas proteínas. El punto de control G2, al final de la fase G2, es un punto de comprobación de control en el que se verifica una serie de factores para garantizar que la célula está lista para entrar en la mitosis (fase M). Las funciones del punto de control G2 incluyen detectar daño del ADN. Si el punto de control G2 se pasa, entonces se inicia la entrada en la fase M. Si el punto de control G2 detecta daño del ADN, el punto de control G2 puede generar una señal que conduce a “detención del ciclo celular” o “detención del ciclo celular en G2” o “detención en G2”, que restringe el comienzo de la mitosis hasta que la replicación y reparación del ADN se terminan, evitando en consecuencia la transmisión del daño del ADN a las células hijas. Se sabe que, dado que el daño del ADN puede iniciar o activar el punto de control G2 y ciertas actividades celulares relacionadas con el punto de control G2, en ciertos contextos también pueden usarse la expresión “punto de control G2 inducido por daño del ADN”, y equivalentes gramaticales de la misma. Se sabe además que el punto de control G2 inducido por daño del ADN puede inducirse o iniciarse por agentes o tratamientos que dañan el ADN.
Las expresiones “anulan el punto de control G2” o “anulan el punto de control G2 del ciclo celular” o “anulación del punto de control G2” o “anulación de G2” o “anulación del punto de control G2” o “afectan el punto de control G2” o “inhiben el punto de control G2” o “suprimen el punto de control G2” o cualquier equivalente gramatical de las mismas están destinadas a referirse a la capacidad de los compuestos de la invención para anular, afectar, inhibir, reprimir, o suprimir el punto de control G2. Una célula en la que se anula el punto de control G2 puede tener ausencia completa de la actividad del punto de control G2 (detención en el punto de control G2, o anulación completa del punto de control G2). Una célula en la que se anula el punto de control G2 puede exhibir una disminución en la cantidad de tiempo en el que la célula está en el punto de control G2, por ejemplo un punto de control G2 que tiene una disminución en su duración de minutos, horas, días, semanas o más bajo condiciones apropiadas. Por ejemplo, una disminución en la duración de tiempo del punto de control G2 puede significar que una célula que normalmente está en G2 durante cierto tiempo, por ejemplo 4 horas, cuando se pone en contacto con un compuesto de la invención, está en G2 durante menos de 4 horas, por ejemplo 3,5, 3, 2,5, 2,1 o menos horas. De esta manera, “anulación del punto de control G2” se refiere a cualquier cantidad de anulación del punto de control G2. Se entiende que el resultado de la anulación del punto de control G2 es que la célula entrará en mitosis (fase M) sin reparación del ADN, lo cual debe tener poco o ningún efecto perjudicial sobre células sin daño (normales), y lo cual debe dar como resultado severos efectos adversos sobre células dañadas en su ADN, conduciendo a menudo a muerte celular debido a apoptosis, catástrofe mitótica, o necrosis.
“Detención del ciclo celular” o “detención del ciclo celular en G2” o “detención en G2” o “detención en G2-M del ciclo celular” o cualquier equivalente gramatical de las mismas se refieren a un estado en el que una célula no sale de G2 para entrar en la mitosis (fase M), de tal modo que la célula se considera como “detenida” en la fase G2. La detención del ciclo celular en G2 a menudo se observa en células dañadas en su ADN, tales como muchas células cancerosas. La detención del ciclo celular en G2 puede resultar de cualquiera de una serie de actividades celulares, incluyendo, pero sin limitarse a, ciertas actividades del punto de control G2. El daño del ADN encontrado en muchas células cancerosas puede iniciar la detención del ciclo celular en G2. La detención del ciclo celular en G2 puede inducirse o potenciarse al tratar una célula con agentes que dañan el ADN, tales como adriamicina, doxorrubicina, o bendamustina (un agente alquilante), o tratamientos que dañan el ADN, tal como irradiación con una dosis desencadenante de radiación X, gamma (y), o UV (algunas veces referida como “detención en G2 inducida por radiación”).
“Adaptación” o “adaptación a la detención del ciclo celular” o “adaptación a la detención del ciclo celular en G2” o “adaptación a la detención en G2” se refieren a la cancelación o anulación de la detención del ciclo celular en G2, de tal modo que las células anteriormente detenidas reingresan al ciclo celular. La adaptación a la detención del ciclo celular en G2 se refiere asimismo al escape de la detención del ciclo celular en G2. Los compuestos de la presente invención pueden ocasionar adaptación a la detención del ciclo celular en G2 en células detenidas en G2. Según un aspecto de la invención, “adaptación” o “adaptación a la detención del ciclo celular” o “adaptación a la detención del ciclo celular en G2” o “adaptación a la detención en G2” pueden referirse al escape de una condición de detención del ciclo celular en G2 impuesta por la activación del punto de control G2, en particular, la detención del ciclo celular en G2 impuesta por la activación del punto de control G2 inducido por daño a ADN. Se entiende que la adaptación a la detención del ciclo celular en G2 da como resultado que las células anteriormente detenidas en G2 reingresan al ciclo celular sin reparar el daño del ADN que inició la detención en G2. La adaptación a la detención del ciclo celular en G2, en la que las células dañadas en su ADN reingresan al ciclo, a menudo da como resultado muerte celular debido a apoptosis, catástrofe mitótica, o necrosis. Dado que el mecanismo que promueve la detención del ciclo celular en G2 después del daño del ADN parece estar conservado entre las especies, desde levaduras hasta seres humanos, se entiende que los compuestos de la presente invención pueden ocasionar adaptación a la detención del ciclo celular en G2 en numerosas especies, en particular en todas las especies eucarióticas.
A la vez que la actividad de anulación del punto de control G2 de los compuestos de la invención puede relacionarse con la capacidad para ocasionar adaptación a la detención del ciclo celular en G2, también se entiende que los compuestos de la invención pueden ocasionar adaptación a la detención del ciclo celular en G2 por otros mecanismos no relacionados con la anulación del punto de control G2. De esta manera, dependiendo de las circunstancias particulares y sin pretender limitarse por esta definición, la anulación del punto de control G2 puede referirse, por lo menos en parte, a anular la capacidad de una célula para detener el ciclo celular en el punto de control G2, conduciendo a adaptación a la detención del ciclo celular en G2. En particular, la anulación del punto de control G2 inducido por daño del ADN por los compuestos de la invención, bajo condiciones que normalmente pueden iniciar la detención del ciclo celular en G2, puede incluir la anulación de una señal generada por el punto de control G2, implicada en iniciar la detención del ciclo celular en G2.
El término “apoptosis” se refiere a muerte celular programada, y cambios asociados en la fisiología celular, incluyendo fragmentación de ácidos nucleicos, activación de caspasas, condensación de cromosomas, etc., como se entiende en la técnica.
La expresión “catástrofe mitótica” se refiere a muerte celular que resulta de uno o más errores en el proceso mitótico.
El término “necrosis” se refiere a muerte celular, que a menudo resulta de daño o accidente, caracterizada a menudo por hinchamiento celular, digestión de cromatina, ruptura de la membrana plasmática y membranas de orgánulos, hidrólisis de ADN, formación de vacuolas en el retículo endoplásmico, ruptura de orgánulos, y lisis celular.
Se entiende que el término “sujeto” se refiere a animales, típicamente animales mamíferos, tales como primates (seres humanos, simios, gibones, chimpancés, orangutanes, macacos), animales domésticos (perros y gatos), animales de granja (caballos, vacas, cabras, ovejas, cerdos) y animales para experimentación (ratón, rata, conejo, cobaya). Los sujetos incluyen modelos de enfermedades animales (por ejemplo, ratones propensos a tumores, ratones que llevan tumores, o ratones que reciben tumores xenoinjertados).
Como se usan en la presente memoria, las formas singulares “un”, “una”, “el/la”, y “es” incluyen referentes plurales, a menos que el contexto lo indique claramente de otra manera. De esta forma, por ejemplo, la referencia a “un compuesto” incluye una pluralidad de compuestos, y la referencia a “un resto” o “un aminoácido” incluye la referencia a uno o más restos y aminoácidos.
Terminología química
“Alquilo” se refiere a un grupo de hidrocarburo alifático. Un grupo alquilo puede estar opcionalmente sustituido. “Alquilo sustituido” se refiere a un grupo alquilo que está sustituido con uno o más sustituyentes, tales como grupos halógeno (Cl, Br, F, I), cicloalquilo de C3 a C7, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, alcoxi de C1 a C6, ariloxi opcionalmente sustituido, hidroxi, amino opcionalmente sustituido, amino cíclico opcionalmente sustituido, nitro, tio, ciano, oxo, acilo de C1 a C7, aciloxi de C1 a C7, carboxi, alcoxicarbonilo de C1 a C6, carbamoílo opcionalmente sustituido, aminocarbonilo cíclico opcionalmente sustituido, p-mercapto, alquiltio de C1 a C4, alquilsulfinilo de C1 a C4, o alquilsulfonilo de C1 a C4. Los grupos alquilo sustituidos pueden tener uno, dos, tres, cuatro, cinco, o más sustituyentes, y los grupos alquilo sustituidos de forma múltiple pueden sustituirse con los mismos sustituyentes o diferentes. El resto alquilo, ya esté sustituido o no sustituido, puede ser ramificado, de cadena lineal, o cíclico. Grupos alquilo típicos incluyen, pero de ninguna manera se limitan a, metilo, etilo, propilo, isopropilo, butilo, isobutilo, butilo terciario, pentilo, hexilo, etenilo, propenilo, butenilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, y similares.
“Alcoxi” se refiere a un grupo OR, en el que R es un alquilo o alquilo sustituido. Grupos alcoxi preferidos son “alcoxi de C1 a C6”, tales como metoxi, etoxi, n-propoxi, isopropoxl, n-butoxi, t-butoxi, y grupos similares.
El término “alquiltio” se refiere a grupos sulfuro, tales como metiltio, etiltio, n-propiltio, isopropiltio, n-butiltio, t-butiltio, y grupos similares. El término “alquilsulfóxido” indica grupos sulfóxido, tales como metilsulfóxido, etilsulfóxido, npropilsulfóxido, isopropilsulfóxido, n-butilsulfóxido, sec-butilsulfóxido, y similares. El término “alquilsulfonilo” abarca grupos, tales como metilsulfonilo, etilsulfonilo, n-propilsulfonilo, isopropilsulfonilo, n-butilsulfonilo, t-butilsulfonilo, y similares.
“Acilo” quiere decir que incluye grupos alquilo, heteroalquilo, alquenilo, heteroalquenilo, alquinilo, heteroalquinilo, arilo, o heteroarilo acoplados a un grupo adicional mediante un grupo carbonilo, por ejemplo, -C(O)-alquilo, o -C(O)-arilo. Grupos acilo preferidos son acilo de C1 a C7, tales como formilo, acetilo, propionilo, butirilo, pentanoílo, pivaloílo, hexanoílo, heptanoílo, benzoílo, y similares.
El término “amida” se refiere a un grupo con la fórmula C(O)NHR o NHC(O)R, en la que R está opcionalmente sustituido y se selecciona del grupo que consiste en alquilo, cicloalquilo, arilo, heteroarilo (enlazado a través de un carbono anular) y heteroalicíclico (enlazado a través de un carbono anular). Cualquier cadena lateral de amina, hidroxi, o carboxilo en los compuestos de la presente invención puede estar amidada.
“Arilo” o “aromático” se refiere a un grupo con por lo menos una estructura de anillo que tiene un sistema de electrones pi conjugados, es decir, que tiene las características de aromaticidad en términos de distribución de electrones a través del sistema anular. Un arilo puede estar opcionalmente sustituido. Típicamente, los sistemas anulares contienen 5-12 átomos anulares en cada anillo. Un grupo arilo puede ser monocíclico o un arilo policíclico de anillo condensado. Un grupo arilo es un arilo carbocíclico en el que todos los átomos anulares son carbono, por ejemplo fenilo. Un heteroarilo es un grupo arilo que contiene por lo menos un heteroátomo anular, tal como oxígeno, azufre y/o nitrógeno. Ejemplos de grupos heteroarilo incluyen maleimidilo, imidazolilo, indolilo, pirrolidinilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo, pirrolilo, furanilo, oxazolilo, dioxazolilo, isoxazolilo, ftalimidilo, tiazolilo, y similares. Los grupos arilo pueden fusionarse a otros grupos arilo o a grupos no arilo (no aromáticos).
Como ejemplos de los sustituyentes de dicho “amino opcionalmente sustituido” y “carbamoílo opcionalmente sustituido”, pueden mencionarse fenilo, fenilo sustituido, alquilo de C1 a C6, alquilo sustituido de C1 a C6, alquenilo de C2 a C7, alquenilo sustituido de C2 a C7, alquinilo de C2 a C7, alquinilo sustituido de C2 a C7, fenilalquilo de C7 a C12, fenilalquilo sustituido de C7 a C12, heteroarilo, alquilo de C1 a C6, alquilo sustituido de C1 a C6, acilo de C1 a C7, alcoxicarbonilo de C1 a C7, carbamoílo opcionalmente sustituido, alquilsulfonilo de C1 a C4, y similares. El “amino opcionalmente sustituido” y “carbamoílo opcionalmente sustituido” pueden estar mono-sustituidos o di-sustituidos, con los mismos sustituyentes o diferentes.
“Alcoxicarbonilo” se refiere a un grupo “alcoxi” unido a un grupo carbonilo.
“Cicloalquilo” se refiere a un radical monocíclico o policíclico que contiene solamente carbono e hidrógeno, y puede estar saturado, parcialmente insaturado, o completamente insaturado. Un grupo cicloalquilo puede estar opcionalmente sustituido. Grupos cicloalquilo preferidos incluyen grupos que tienen de tres a doce átomos anulares, más preferiblemente de 5 a 10 átomos anulares.
“Amino cíclico” como en “amino cíclico opcionalmente sustituido”, se refiere a grupos cíclicos que contienen por lo menos un nitrógeno anular, incluyendo piperazino, morfolino, piperidino, pirrolidino, y similares.
Ejemplos de “aminocarbonilo cíclico” como en “aminocarbonilo cíclico opcionalmente sustituido”, incluyen piperazinocarbonilo, morfolinocarbonilo, piperidinocarbonilo, pirrolidinocarbonilo, y similares.
Los sustituyentes de “alcoxi opcionalmente sustituido”, “alquiltio opcionalmente sustituido”, “arilo opcionalmente sustituido”, “ariloxi opcionalmente sustituido”, “ariltio opcionalmente sustituido”, “acilo opcionalmente sustituido”, “heteroarilo opcionalmente sustituido”, “alquiltio opcionalmente sustituido”, “alquilsufinilo opcionalmente sustituido” “alquilsulfonilo opcionalmente sustituido”, “alcoxlcarbonilo opcionalmente sustituido”, “amino cíclico opcionalmente sustituido”, y “aminocarbonilo cíclico opcionalmente sustituido” se definen de la misma manera que los sustituyentes de “alquilo sustituido”.
“Halógeno” se refiere a átomos de flúor, cloro, bromo, o yodo. Uno o más halógenos pueden presentarse en un compuesto, en el que los halógenos pueden ser los mismos o diferentes.
Las formas de realización de compuestos de la presente invención pueden poseer uno o más centros quirales, y cada centro puede existir en la configuración R o S, de tal modo que la presente invención incluye todas las formas diastereoméricas, enantioméricas, y epiméricas, así como las mezclas apropiadas de las mismas. Las formas de realización de la presente invención pueden existir como isómeros geométricos, de tal modo que la presente invención incluye todos los isómeros cis, trans, syn, anti, entgegen (opuestos) (E), y zusammen (juntos) (Z), así como las mezclas apropiadas de los mismos.
Los compuestos de la presente invención pueden existir en formas no solvatadas así como solvatadas con disolventes farmacéuticamente aceptables, tales como agua, etanol, y similares. En general, las formas solvatadas se consideran equivalentes a las formas no solvatadas en el contexto de la presente invención.
Cualquier sal, como se da a conocer en la presente memoria, puede incluir sales con bases inorgánicas, sales con bases orgánicas, sales con ácidos inorgánicos, sales con ácidos orgánicos, y sales con aminoácidos básicos o ácidos.
A menos que se indique de otra manera, cuando un sustituyente se estima como “opcionalmente sustituido”, se quiere decir que el sustituyente es un grupo que puede estar sustituido con un grupo o más grupos como se enumera en la presente memoria o como se conoce por un experto en la materia.
Las descripciones de compuestos de la invención están de acuerdo con los principios de enlace químico conocidos por los expertos en la materia. Por consiguiente, cuando un grupo puede estar sustituido por uno o más de una serie de sustituyentes, tales sustituciones se seleccionan para atenerse a los principios del enlace químico y para proporcionar compuestos que no son inestables inherentemente, y/o pueden conocerse por un experto en la materia como probablemente inestables, bajo condiciones ambientales, tales como condiciones acuosas, neutras, fisiológicas.
Se entiende que los compuestos de la invención pueden describirse por un experto en la materia usando terminología diferente a los términos usados en la presente memoria, sin afectar la precisión de la descripción. Los compuestos, estructuras, sustituyentes, grupos, y similares pueden describirse usando cualquiera de: nomenclatura IUPAC; nombre químico “trivial” o nombre “común”; nombre comercial; número de registro CAS; notación SMILES; u otros descriptores. Por ejemplo, los compuestos de la invención descritos en la presente memoria como “azol dionas sustituidas” o “azolin dionas sustituidas” pueden describirse alternativamente como “maleimidas sustituidas” o “2,5-pirroldionas sustituidas” o “pirroles sustituidos” en combinación con otros descriptores, preferiblemente de acuerdo con terminología química estandarizada, para proporcionar una descripción completa de uno o más compuestos de la invención.
Compuestos de azol dionas sustituidas
La invención proporciona compuestos de azol (azolin) dionas sustituidas que pueden usarse para eliminar o suprimir células dañadas en su ADN, o para tratar trastornos de proliferación celular caracterizados por proliferación celular indeseable o no buscada. Los compuestos de la invención se definen en las reivindicaciones.
En las tablas 1, 2, y 3 en la presente memoria se muestran compuestos representativos. Se entiende en general que los compuestos dados a conocer en las tablas 1,2, y 3 son proporcionados únicamente a título ilustrativo y no limitativo del alcance de la invención.
Esquemas de síntesis representativos
A continuación se presentan esquemas representativos para la síntesis de compuestos de la invención que tienen las Estructuras (IV) o (VI) así como los esquemas representativos para la síntesis de compuestos descritos en la presente memoria a modo de ejemplos de referencia solamente. Los esquemas representativos presentados en la presente memoria no restringen el alcance de la invención de ninguna forma. Se entiende que un experto en la materia puede adaptar los métodos presentados en la presente memoria, y/o diferentes métodos conocidos en la técnica, para sintetizar compuestos adicionales dentro del alcance de la presente invención, incluyendo análogos que tienen diferentes sustituciones o patrones de sustituciones. Se entiende además que, aunque se ha observado que ciertas sustituciones producen estructuras con mayor actividad que otras estructuras, la presente invención proporciona compuestos con todas las sustituciones que tienen todos los niveles de actividad.
En el Método 1 (Esquema 1), un anhídrido (1) se hace reaccionar con una hidrazina sustituida (2) o una bencilamina (3) para formar los compuestos que tienen la estructura general mostrada como (4) a continuación:
Esquema 1
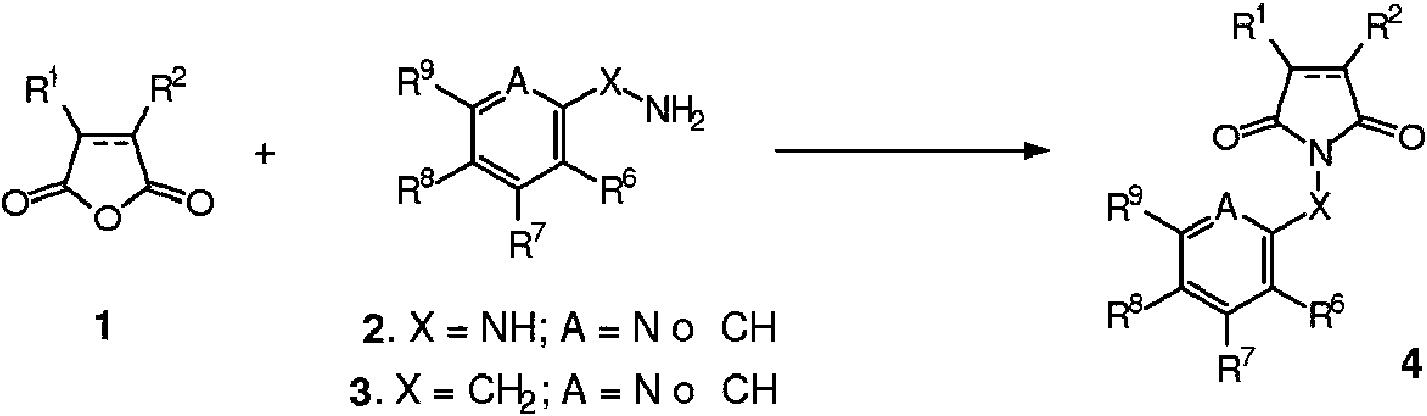
La reacción puede llevarse a cabo en disolventes orgánicos comunes, tales como THF, cloroformo, DMF, ácido acético, etc., a temperaturas que varían de ambientales a elevadas, durante tiempos que varían de varias horas a unos cuantos días. Usualmente, no se necesitan otros aditivos. Los anhídridos e hldrazinas/bencilaminas requeridos se adquieren de fuentes comerciales, o se sintetizan de acuerdo con procedimientos conocidos en la bibliografía. En casos en los que los materiales de partida se desconocen en la bibliografía, se desarrollan métodos sintéticos, como se ilustra por ciertas síntesis descritas en los ejemplos.
En el Método 2 (Esquema 2), una imida (5) se hace reaccionar con un alcohol bencílico (6) bajo condiciones típicas de Mitsunobu para formar compuestos que tienen la estructura general mostrada como (7) a continuación:
Esquema 2

Las condiciones típicas de Mitsunobu incluyen el uso de una fosfina (trifenilfosfina, tributiifosfina etc.), y un compuesto azo (azo-dicarboxilato de dietilo, azo-dicarboxilato de diisopropilo, etc..). La reacción puede llevarse a cabo con una base añadida, usualmente trietilamina, o sin una base añadida, en disolventes tales como THF, a temperatura ambiente o elevada durante varias horas. Las imidas y alcoholes bencílicos requeridos se adquieren de fuentes comerciales, o se sintetizan de acuerdo con procedimientos conocidos en la bibliografía. En casos en los que los materiales de partida se desconocen en la bibliografía, se desarrollan métodos sintéticos, como se ilustra por ciertas síntesis descritas en los ejemplos.
En el Método 3 (Esquema 3), una imida (5) se hace reaccionar con un bromuro de bencilo (8) bajo una condición de reacción típica de sustitución nucleofílica para formar los compuestos que tienen la estructura general mostrada como (9) a continuación:
Esquema 3
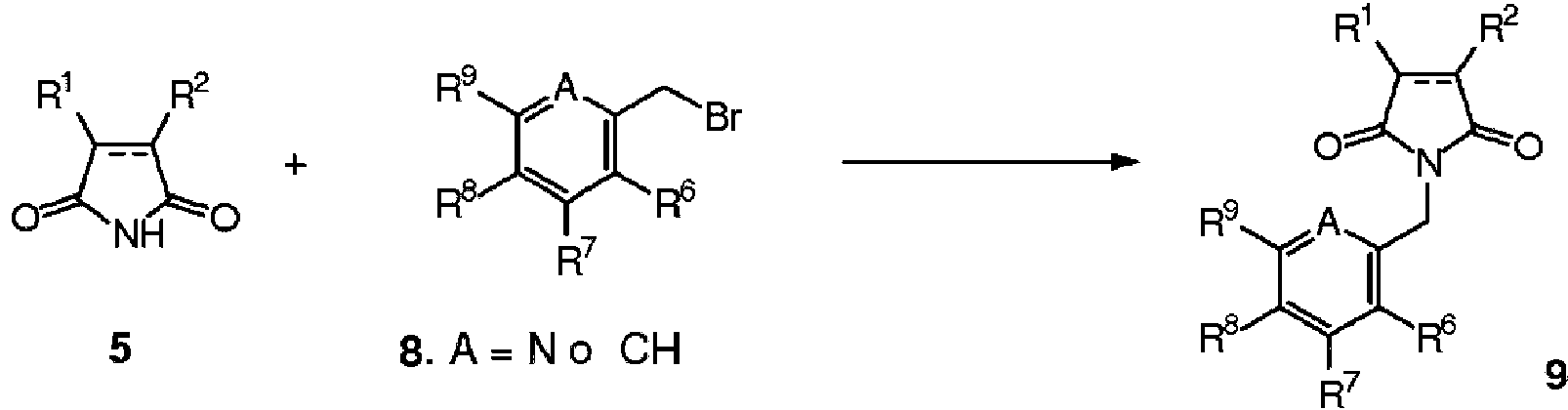
La condición de reacción típica es: someter a reflujo en un disolvente adecuado (acetona, DMF, etc.,) en presencia de una base añadida (carbonato de potasio, carbonato de cesio, etc.) durante varias horas a unos cuantos días. Las imidas y bromuros de bencilo requeridos se adquieren de fuentes comerciales, o se sintetizan de acuerdo con procedimientos conocidos en la bibliografía. En casos en los que los materiales de partida se desconocen en la bibliografía, se desarrollan métodos sintéticos, como se ilustra por ciertas síntesis descritas en los ejemplos.
En el Método 4 (Esquema 4), un ácido arilborónico (10) se hace reaccionar con una N-hidroxiimida (11) bajo una condición de acoplamiento mediado por Cu(I) para formar compuestos que tienen la estructura general mostrada como (12) a continuación:
Esquema 4

La condición de reacción típica es: agitar a temperatura ambiente en disolventes adecuados (DCE, THF, DMF, etc.) en presencia de una base añadida (piridina, trietilamina, etc.), con especies añadidas de Cu(I), tal como CuCl, durante varias horas hasta toda la noche. Los ácidos arilborónicos y N-hidroxiimidas requeridos se adquieren de fuentes comerciales, o se sintetizan de acuerdo con procedimientos conocidos en la bibliografía. En casos en los que los materiales de partida se desconocen en la bibliografía, se desarrollan métodos sintéticos, como se ilustra por ciertas síntesis descritas en los ejemplos.
Las síntesis de compuestos (a título de ejemplo de referencia solamente) o formas de realización particulares se dan a conocer en los ejemplos. Los compuestos sintetizados representativos, presentados como formas de realización en las tablas 1 y 2, se presentan solamente para ilustración, y de ningún modo restringen el alcance de la invención dada a conocer en la presente memoria.
Actividades biológicas de los compuestos de la invención
La presente invención proporciona compuestos para la utilización en el tratamiento de trastornos de proliferación celular. La presente invención proporciona compuestos que eliminan o suprimen células que proliferan indeseablemente. La presente invención proporciona compuestos para la utilización en el tratamiento de trastornos de proliferación celular al eliminar o suprimir selectivamente células que proliferan indeseablemente. La presente
invención proporciona compuestos para la utilización en el tratamiento de trastornos de proliferación celular al eliminar o suprimir selectivamente células que proliferan indeseablemente, que tienen daño acumulado en el ADN (“células dañadas en su ADN”). En poblaciones mixtas de células normales y células que proliferan indeseablemente, los compuestos de la invención pueden usarse para eliminar o suprimir selectivamente células que proliferan indeseablemente a la vez que tienen poco o ningún efecto citotóxico sobre las células normales. En poblaciones mixtas de células normales y células que proliferan indeseablemente dañadas en su ADN, los compuestos de la invención pueden eliminar o suprimir selectivamente células que proliferan indeseablemente dañadas en su ADN a la vez que tienen poco o ningún efecto citotóxico sobre las células normales. En particular, los compuestos de la invención eliminan selectivamente células cancerosas o suprimen la proliferación de células cancerosas, a la vez que tienen poco o ningún efecto citotóxico sobre células “normales” no cancerosas. La presente invención proporciona compuestos para la utilización en métodos para seleccionar selectivamente células dañadas en su ADN. La presente invención proporciona compuestos para la utilización en métodos para seleccionar selectivamente células con un punto de control G1 del ciclo celular alterado. La presente invención proporciona compuestos para la utilización en métodos para seleccionar selectivamente células cancerosas.
Los tratamientos convencionales para trastornos de proliferación celular a menudo incluyen agentes y tratamientos que dañan el ADN, como se describe en otra parte en la presente memoria. Estos tratamientos convencionales, a menudo usados como tratamientos anticancerosos, se han seleccionado para eliminar células que entran rápidamente en el ciclo (que proliferan) esperando eliminar las células que proliferan indeseablemente características del trastorno de proliferación. Ejemplos de tales tratamientos convencionales incluyen, pero no se limitan a, agentes citotóxicos y/o irradiación con radiación a, p, y, X y/o UV. Sin embargo, los tratamientos convencionales a menudo ocasionan que los pacientes sufran efectos adversos sobre las células normales, que también están proliferando, por ejemplo, diarrea debido a daño a células epiteliales intestinales, pérdida de cabello debido a daño a células del folículo piloso, y anemia debido a daño a progenitores de células sanguíneas, todas las cuales están entre las células que proliferan más rápidamente en el cuerpo normal. Estos efectos adversos a menudo obstaculizan los tratamientos. De esta manera, las medicinas que se dirigen específicamente a células que proliferan de forma indeseable, tales como células cancerosas, sin dañar células normales, se han anhelado desde hace mucho en la clínica.
Aunque la invención no se limita por ningún mecanismo de acción particular, y sin pretender limitarse por esta teoría, los compuestos de la invención pueden eliminar o suprimir células que proliferan indeseablemente al anular el punto de control G2 en células que proliferan, y/o al ocasionar adaptación a la detención del ciclo celular en G2 en células detenidas en G2. De esta manera, la invención proporciona compuestos para la utilización en métodos para eliminar o suprimir células que proliferan indeseablemente al poner en contacto las células con por lo menos un compuesto de la invención en una cantidad suficiente para anular el punto de control G2 del ciclo celular y/u ocasionar adaptación a la detención del ciclo celular en G2.
Sin pretender limitarse por esta teoría, la anulación del punto de control G2 en células dañadas en su ADN ocasionaría que las células dañadas en su ADN progresen a través del ciclo celular sin reparar el daño del ADN. Asimismo, la adaptación a la detención del ciclo celular en G2 en células dañadas en su ADN ocasionaría que las células dañadas en su ADN anteriormente detenidas entren en la mitosis sin reparar el daño del ADN. Se entiende en general que las células normales dependen del punto de control G1 como el punto de control principal para detectar daño o defectos en el ADN durante el ciclo celular, y no parecen usar el punto de control G2 en la misma medida para detectar daño o defectos en el ADN, a la vez que las células que tienen un punto de control G1 afectado o defectuoso, por ejemplo la mayoría de las células cancerosas, deben depender del punto de control G2 para detectar daño o defectos en el ADN, y para iniciar la reparación antes de que la célula entre en la mitosis. De esta manera, la anulación del punto de control G2 en células con un punto de control G1 alterado ocasionaría que las células progresen a través del ciclo celular sin reparar ningún daño acumulado en el ADN. Asimismo, la adaptación a la detención del ciclo celular en G2 en células con un punto de control G1 alterado ocasionaría que las células anteriormente detenidas entren en la mitosis sin reparar ningún daño acumulado en el ADN. La expresión “daño del ADN” se utiliza para abarcar daño del ADN no relacionado con el punto de control G1, así como daño del ADN que resulta de un punto de control G1 alterado, de tal modo que las células con un punto de control G1 alterado pueden considerarse “células dañadas en su ADN”. En todas las situaciones descritas anteriormente, se espera que el progreso a través del ciclo celular sin reparar daño del ADN dé como resultado supresión o muerte de las células dañadas en su ADN.
Aunque la invención no se limita a un mecanismo de acción particular, se ha observado que los compuestos de la invención pueden anular el punto de control G2 en células que proliferan, y pueden ocasionar adaptación a la detención del ciclo celular en G2. La anulación del punto de control G2 en células que proliferan dañadas en su ADN permite que la célula dañada en su ADN progrese a través de G2 y entre en la mitosis sin suficiente reparación del daño del ADN. La adaptación a la detención en G2 de células dañadas en su ADN, que da como resultado reingreso de las células anteriormente detenidas dañadas en su ADN en el ciclo celular, permite que la célula dañada en su ADN entre en la mitosis sin suficiente reparación del daño del ADN. Se entiende además que si una célula dañada en su ADN progresa además en el ciclo celular con un punto de control G1 alterado y entra en la fase S, se esperan daños, defectos, y errores adicionales. En todas las situaciones descritas anteriormente, la acumulación de daño podría dar como resultado apoptosis, catástrofe mitótica, necrosis, o supresión celular. Dado que la mayoría de las células cancerosas tienen daño del ADN y/o un punto de control G1 alterado (defectuoso), los compuestos de la invención pueden eliminar o suprimir células cancerosas dado que la anulación del punto de control G2 o adaptación a la
detención en G2 tendrá efectos citotóxicos sobre las células cancerosas, por ejemplo muerte o supresión de las células cancerosas.
Para las células normales sin daño del ADN, se espera que la anulación del punto de control G2 del ciclo celular por los compuestos de la invención tendrá poco o ningún efecto citotóxico. Adicionalmente, puesto que las células normales sin daño del ADN no son susceptibles de estar en detención en G2, se espera que la capacidad de los compuestos de la invención para ocasionar adaptación a la detención en G2 tendrá poco o ningún efecto citotóxico. De esta manera, en una población de células que incluye células que proliferan indeseablemente dañadas en su ADN y células normales, los compuestos de la invención eliminan o suprimen selectivamente células dañadas en su ADN, a la vez que tienen poco o ningún efecto citotóxico sobre las células normales. En una población de células que incluye células cancerosas que proliferan indeseablemente y células normales, los compuestos de la invención eliminan o suprimen selectivamente las células cancerosas, a la vez que tienen poco o ningún efecto citotóxico sobre las células normales.
Compuestos de la invención para la utilización para tratar células
Cualquier célula cuya proliferación no se desea puede tratarse in vitro, ex vivo o in vivo con los compuestos de la invención, aunque los métodos de tratamiento llevados a la práctica en el cuerpo humano o animal no se reivindican. Las células candidatas pueden identificarse al poner en contacto una célula de ensayo con un compuesto de la invención solo, o en combinación con un tratamiento que daña el ADN u otro tratamiento anticanceroso, y determinando si la célula puesta en contacto exhibe proliferación disminuida, muerte celular incrementada, o adaptación a la detención del ciclo celular. Las células candidatas pueden identificarse en base a las características, que incluyen, pero no se limitan a, daño del ADN, crecimiento o proliferación aberrante (in vivo o in vitro), morfología celular, o expresión de marcadores de cáncer. Un diagnóstico clínico de un trastorno proliferativo, tal como cáncer, puede utilizarse como indicio de células que deben tratarse in vitro, ex vivo, o in vivo con los compuestos de la invención.
Las células pueden tratarse in vitro con los compuestos de la invención. A título de ejemplo solamente, las células pueden retirarse de un sujeto, tratarse ex vivo usando los compuestos de la invención, y devolverse al sujeto. Alternativamente, los compuestos de la invención son para la utilización en métodos en los que los trastornos de proliferación celular pueden tratarse in vivo, en los que los compuestos de la invención pueden administrarse de manera sistémica a un sujeto, por ejemplo por vía oral o en forma intravenosa, o por un método de administración dirigida, por ejemplo inyección en un sitio tumoral, inyección intraperitoneal, o al asociar los compuestos de la invención con dispositivos de suministro, tales como ligandos, anticuerpos, jaulas moleculares, o liposomas capaces de seleccionar por lo menos una célula a tratar.
Compuestos de la invención para la utilización en métodos para tratar sujetos
Los sujetos apropiados para el tratamiento usando los compuestos de la invención incluyen sujetos que actualmente se someten a un tratamiento para un trastorno de proliferación celular, o candidatos para el tratamiento para un trastorno de proliferación celular, por ejemplo sujetos que actualmente se someten a, o se designan como candidatos para, tratamiento anticanceroso. Los sujetos incluyen aquellos que tienen un trastorno de proliferación celular, por ejemplo un diagnóstico de cáncer. Los sujetos candidatos incluyen sujetos en riesgo de desarrollar un trastorno de proliferación celular. Por lo tanto, los compuestos de la invención son aplicables para la utilización en métodos para tratar a un sujeto que está en riesgo de desarrollar un trastorno de proliferación celular pero que no ha exhibido todavía síntomas claros del trastorno y/o que no ha recibido todavía un diagnóstico de un trastorno de proliferación celular.
Compuestos de la invención para la utilización en métodos para tratar trastornos de proliferación celular.
Los trastornos de proliferación celular susceptibles al tratamiento usando las composiciones proporcionadas en la presente memoria incluyen condiciones patológicas (enfermedades), tanto benignas como neoplásicas, y condiciones fisiológicas no patológicas, caracterizadas por cantidades de células anormales o indeseables, crecimiento celular o supervivencia celular. Los trastornos o condiciones patológicas pueden constituir un estado de enfermedad, en particular todos los tipos de cáncer, incluyendo crecimientos cancerosos, procesos oncogénicos, metástasis, células y tejidos metastásicos, y células, tejidos, u órganos transformados de manera maligna. Los trastornos de proliferación celular pueden ser no patológicos, incluyendo algunos tipos de crecimiento de tejido fibrótico (por ejemplo, durante la reparación de heridas que da como resultado cicatrización), ciertos trastornos proliferativos de vasos sanguíneos, y ciertas hiperplasias benignas. La presente descripción proporciona suficiente instrucción para permitir que un experto en la materia identifique trastornos de proliferación celular adecuados para el tratamiento usando las composiciones proporcionadas en la presente memoria, y para desarrollar protocolos para tal tratamiento.
Las células que comprenden el trastorno proliferativo pueden agregarse en una masa celular o pueden dispersarse. La expresión “tumor sólido” se refiere a hiperplasias, neoplasias o metástasis que típicamente se agregan en conjunto y forman una masa. Ejemplos particulares incluyen tumores viscerales, tales como cáncer gástrico o de colon, hepatomas, carcinomas venales, tumores/cánceres de pulmón y cerebro. Un “tumor liquido” generalmente se refiere a neoplasias del sistema hematopoyético, tales como linfomas, mielomas y leucemias, o neoplasias que son difusas
en su naturaleza, dado que no forman típicamente una masa sólida. ejemplos particulares de leucemias incluyen linfoblástica aguda y crónica, mieloblástica y mieloma múltiple.
Tales trastornos incluyen neoplasias o cánceres, que pueden afectar virtualmente cualquier tipo de célula o tejido, por ejemplo carcinoma, sarcoma, melanoma, trastornos metastásicos o trastornos neoplásicos hematopoyéticos. Un tumor metastásico puede surgir de una multitud de tipos de tumores primarios, incluyendo pero sin limitarse a de mama, pulmón, tiroides, cabeza y cuello, cerebro, linfoide, gastrointestinal (boca, esófago, estómago, intestino delgado, colon, recto), aparato genitourinario (útero, ovario, cuello uterino, vejiga, testículo, pene, próstata), riñón, páncreas, hígado, hueso, músculo, piel, etc.
Los carcinomas se refieren a neoplasias de tejido epitelial o endocrino, e incluyen carcinomas del sistema respiratorio, carcinomas del sistema gastrointestinal, carcinomas del sistema genitourinario, carcinomas testiculares, carcinomas de mama, carcinomas prostáticos, carcinomas del sistema endocrino, y melanomas. Los carcinomas ejemplificativos incluyen aquellos que se forman en el cuello uterino, pulmón, próstata, mama, cabeza y cuello, colon, hígado y ovario. El término también incluye carcinosarcomas, por ejemplo que incluyen tumores malignos compuestos de tejidos carcinomatosos y sarcomatosos. El adenocarcinoma incluye un carcinoma de un tejido glandular, o en el que el tumor forma una estructura similar a una glándula.
Los sarcomas se refieren a tumores malignos de origen celular mesenquimatoso. Los sarcomas ejemplificativos incluyen, por ejemplo, linfosarcoma, liposarcoma, osteosarcoma, y fibrosarcoma.
Como se usa en la presente memoria, la expresión “trastorno proliferativo hematopoyético” significa una enfermedad que implica células hiperplásicas/neoplásicas de origen hematopoyético, por ejemplo que surgen de los linajes mieloide, linfoide o eritroide, o células precursoras de las mismas. Típicamente, las enfermedades surgen de leucemias agudas pobremente diferenciadas, por ejemplo leucemia eritroblástica y leucemia megacarioblástica aguda. Los trastornos mieloides ejemplificativos adicionales incluyen, pero no se limitan a, leucemia promieloide aguda (APML), leucemia mielógena aguda (AML) y leucemia mielógena crónica (CML); las neoplasias linfoides incluyen, pero no se limitan a, leucemia linfoblástica aguda (ALL), que incluye ALL de linaje p y ALL de linaje T, leucemia linfocítica crónica (CLL), leucemia prolinfocítica (PLL), tricoleucemia (h Ll ) y macroglobulinemia de Waldenstrom (WM). Los linfomas malignos adicionales incluyen, pero no se limitan a, linfoma no de Hodgkin y variantes del mismo, linfomas de células T periféricas, leucemia/linfoma de células T de adultos (ATL), linfoma cutáneo de células T (CTCL), leucemia linfocítica granular grande (LGF), enfermedad de Hodgkin y enfermedad de Reed-Sternberg.
La invención proporciona composiciones para la utilización en métodos para tratar trastornos de proliferación celular usando los compuestos de la invención. La invención proporciona composiciones para la utilización en métodos para tratar cáncer usando los compuestos de la invención. La invención proporciona composiciones para la utilización en métodos para eliminar o suprimir células cancerosas, usando los compuestos de la invención. La invención proporciona composiciones para la utilización en métodos para tratar trastornos de proliferación celular in vivo, in vitro, y ex vivo. La invención proporciona compuestos de la invención para la utilización en métodos para tratar cáncer in vivo, in vitro, y ex vivo. La invención proporciona compuestos farmacéuticos de la invención para la utilización en la eliminación o supresión de células cancerosas in vivo, in vitro, y ex vivo. La invención proporciona composiciones farmacéuticas (medicamentos) que contienen compuestos de la invención, para la utilización como se describe en la presente memoria, incluyendo pero sin limitarse a, tratar trastornos de proliferación celular, eliminar o suprimir células cancerosas, y tratar cáncer.
La invención proporciona composiciones para la utilización en métodos que incluyen por lo menos un compuesto de la invención en combinación con por lo menos un ingrediente activo adicional. La invención proporciona composiciones farmacéuticas (medicamentos) que incluyen por lo menos un compuesto de la invención en combinación con por lo menos un ingrediente activo adicional, para la utilización en métodos para tratar trastornos de proliferación celular. En particular, la invención proporciona composiciones para la utilización en métodos que incluyen compuestos de la invención en combinación con por lo menos un tratamiento anticanceroso. La expresión “tratamiento anticanceroso”, para su uso en combinación con los compuestos de la invención incluye cualquier tratamiento anticanceroso, antiproliferativo, que daña el ADN, o antitumoral como se da a conocer en la presente memoria, incluyendo los “tratamientos que dañan el ADN” y “agentes que dañan el ADN” enumerados anteriormente, o cualquier tratamiento semejante conocido en la técnica. Por ejemplo, un tratamiento anticanceroso (antiproliferativo celular, antitumoral) puede comprender tratamiento con radiación o resección quirúrgica, opcionalmente en combinación con tratamiento farmacológico. El tratamiento puede comprender la administración de una sustancia química, tal como un radioisótopo, un fármaco anticanceroso, tal como un agente quimioterapéutico, o terapia génica, tal como un antioncogén (por ejemplo, Rb, DCC, p53, etc.), un oncogén negativo dominante o un antisentido a un oncogén. Los compuestos de la invención son para la utilización en métodos en los que los compuestos pueden administrarse antes de, al mismo tiempo que o después de otros protocolos de tratamiento. Por ejemplo, los compuestos de la invención son para la utilización en métodos en los que puede administrársele a un sujeto candidato para terapia antiproliferativa celular (por ejemplo, terapia de radiación, quimioterapia, terapia génica, resección quirúrgica, etc.) un compuesto de la invención antes de iniciar la terapia antiproliferativa celular. De esta manera, se proporcionan compuestos de la invención para la utilización en métodos de tratamiento profiláctico.
Como se describe en los ejemplos y se expone a continuación, los compuestos de la invención pueden usarse solos o en combinación, in vivo, ex vivo, e in vitro, en métodos para tratar trastornos de proliferación celular. Como se describe en los ejemplos y se discute a continuación, el compuesto o compuestos de la invención pueden usarse solos o en combinación, in vivo, ex vivo, e in vitro, en métodos para tratar cáncer. Como se describe en los ejemplos y se discute a continuación, los compuestos de la invención pueden usarse solos o en combinación, in vivo e in vitro, en métodos para eliminar o suprimir células cancerosas.
Efectos de los compuestos de la invención
Típicamente se administra una “cantidad eficaz o “cantidad suficiente” de un compuesto de la invención, en el que esa es una cantidad (concentración, dosis, nivel) suficiente para producir un efecto deseado. Las cantidades eficaces se determinan por cualquiera de una diversidad de medidas, incluyendo: muerte celular (por ejemplo, incremento en porcentaje de células en fase subG1), proliferación celular disminuida, cantidades disminuidas de células, masa celular disminuida, apoptosis incrementada, supervivencia disminuida, o adaptación a la detención del ciclo celular (escape de la detención del ciclo celular). Por ejemplo, cuando se desea inhibir la proliferación celular, una cantidad eficaz será una cantidad que disminuye de manera detectable la proliferación celular, o incrementa la muerte celular, o disminuye la supervivencia celular. La cantidad por lo tanto puede ser suficiente para reducir las cantidades de células diana, estabilizar las cantidades de células diana o inhibir los incrementos en cantidades de células diana. Una cantidad eficaz puede ser una cantidad suficiente para incrementar el tiempo de supervivencia de un sujeto que tiene un trastorno de proliferación celular.
Por ejemplo, cuando el trastorno comprende un tumor sólido, una cantidad eficaz de un compuesto de la invención podría reducir el tamaño del tumor, estabilizar el tamaño del tumor, o incrementar el tiempo de supervivencia de un sujeto que tiene el tumor. Como se muestra en el ejemplo 4, cinco (5) compuestos representativos de la invención mostraron citotoxicidad selectiva in vivo contra células cancerosas, y citotoxicidad indetectable in vivo contra células normales, como se ilustra por los drásticos incrementos en la supervivencia del hospedante del tumor.
Cuando el trastorno comprende un “tumor líquido”, una cantidad eficaz de un compuesto de la invención podría reducir las cantidades de células tumorales, estabilizar el número de células tumorales, inhibir incrementos posteriores en el número de células tumorales, u ocasionar que las células cancerosas detenidas en el ciclo celular vuelvan a entrar al ciclo celular (adaptación a la detención del ciclo celular). Además, cantidades eficaces de los compuestos de la invención podrían evitar o inhibir la progresión del trastorno proliferativo, por ejemplo reducir, inhibir, o evitar metástasis.
Una cantidad eficaz de un compuesto de la invención puede ser una cantidad que produce un efecto deseado sin producir un efecto inaceptable o indeseable. Una cantidad eficaz de un compuesto de la invención puede ser una cantidad que elimina o suprime células diana (por ejemplo, células cancerosas) a la vez que tiene poco o ningún efecto citotóxico sobre células no diana (por ejemplo, células normales), o una cantidad que produce un beneficio terapéutico deseado en un sujeto que tiene un trastorno de proliferación celular, a la vez que tiene poco o ningún efecto adverso sobre el sujeto. Además, una cantidad eficaz de un compuesto de la invención puede ser una cantidad que, en combinación con otro tratamiento, produce un efecto deseado sin producir un efecto indeseable inaceptable. Como se muestra en el ejemplo 7 y se ilustra en las tablas 4-11 a continuación, el compuesto S00109 tiene poco o ningún efecto citotóxico sobre las células normales en concentraciones que pueden eliminar o suprimir células cancerosas. Además, como se muestra en el ejemplo 7 y se ilustra en las tablas 4-11 a continuación, el compuesto S00109 puede usarse en combinación con otro tratamiento anticanceroso para eliminar o suprimir células cancerosas, a una concentración de S00109 que tiene poco o ningún efecto citotóxico sobre las células normales.
Las cantidades eficaces de los compuestos de la invención pueden reducir o disminuir objetiva o subjetivamente la gravedad o frecuencia de síntomas asociados con el trastorno o padecimiento. Por ejemplo, una cantidad eficaz de un compuesto de la invención podría reducir el dolor, náusea u otro malestar, o incrementar el apetito o bienestar subjetivo.
Las cantidades eficaces de los compuestos de la invención podrían reducir la cantidad (por ejemplo, dosis) o frecuencia de tratamiento con otro protocolo. Por ejemplo, un paciente con cáncer tratado con un compuesto de la invención puede requerir menos agente o tratamiento anticanceroso que daña el ADN con el fin de alcanzar el nivel deseado de inhibición de la proliferación de células cancerosas, es decir, un nivel deseado de eliminación o supresión de células cancerosas que proliferan.
Los métodos descritos en la presente memoria únicamente a título de ejemplo que conducen a una mejora en la condición del sujeto o un beneficio terapéutico que puede ser permanente, pueden extenderse durante un período de tiempo más prolongado, por ejemplo meses o años, o pueden ser relativamente cortos en duración, por ejemplo la mejoría puede durar varias horas, días o semanas. Una cantidad eficaz no necesita alcanzar una terminación completa de cualquiera o todos los síntomas del padecimiento o trastorno. Una cantidad eficaz para proporcionar uno o más efectos beneficiosos, como se describe en la presente memoria o se conoce en la técnica, se refiere como una “mejora” del estado del sujeto o “beneficio terapéutico” al sujeto.
Una cantidad eficaz de un compuesto de la invención puede determinarse en base a estudios en animales u opcionalmente en pruebas clínicas humanas. El experto apreciará los diversos factores que pueden influenciar la dosificación y duración requerida para tratar un sujeto particular incluyendo, por ejemplo, la salud general, edad, o sexo del sujeto, la gravedad o etapa del trastorno o padecimiento, tratamientos previos, susceptibilidad a efectos secundarios indeseables, desenlace clínico deseado, y la presencia de otros trastornos o padecimientos. Tales factores pueden influir en la dosis y duración requerida para proporcionar una cantidad suficiente para el beneficio terapéutico. El régimen de dosificación también tiene en consideración la farmacocinética, es decir, la velocidad de absorción, biodisponibilidad, metabolismo, y aclaramiento de la composición farmacéutica. Además, las dosis o protocolos de tratamiento pueden ajustarse específicamente al sujeto o modificarse con base en datos farmacogenómicos.
Adaptación a la detención del ciclo celular en G2 inducida por daño del ADN
Aunque la invención no se limita a un mecanismo de acción particular, se ha determinado que los compuestos de la invención pueden ocasionar adaptación a la detención del ciclo celular en G2. De esta manera, la invención proporciona una composición para la utilización en métodos para anular o escapar de la detención del ciclo celular en G2, en particular la detención del ciclo celular en G2 iniciada por daño del ADN. En células dañadas en su ADN, se entiende que las funciones del punto de control G2 inducido por daño a ADN incluyen el reconocimiento del daño del ADN y generación de una señal que produce detención del ciclo celular, de tal modo que las células dañadas en su ADN se detienen en fase G2 hasta que la reparación se termina. Los compuestos de la invención pueden ocasionar que las células en detención del ciclo celular en G2 vuelvan a entrar al ciclo celular, posiblemente al anular el punto de control G2 inducido por daño a ADN en las células detenidas en G2. Como se demuestra en los ejemplos, tablas y figuras de la presente descripción, los compuestos de la invención pueden inducir a las células en detención del ciclo celular en g 2 (es decir, células que tienen daño preexistente en el ADN) a reingresar al ciclo celular, procediendo a través de las fases G2 y M y entrando a las fases G1 (duplicación de ADN) con daño no reparado en el ADN, conduciendo a muerte celular o supresión celular, usualmente por catástrofe mitótica o apoptosis.
Como se describe en el ejemplo 1 y se muestra en la figura 1, células Jurkat (una línea celular derivada de linfoma humano de células T) se “predetuvieron” en la fase G2 por irradiación de rayos X y se trataron con S00109 o S01860 a diversas dosificaciones. La adaptación a la detención del ciclo celular en G2 se determinó al medir el porcentaje de células en fase G1, en el que las células en G1 se identificaron por tener 2N ADN. Las células expuestas a S00109 o S01860 mostraron un incremento dependiente de la dosis en el porcentaje de células en fase G1 (el “incremento G1”). Estos resultados indicaron que la exposición a S00109 o S01860 en concentraciones de 0,019 |iM a 0,625 |iM ocasionó que las células detenidas en G2 volvieran a entrar al ciclo celular y procedieran a través de la fase M y entraran a la fase G1. En contraste, solamente cierto porcentaje de células en la población de células sin tratar detenidas en G2 (es decir, sin exposición a S00109 o S01860) entraron en la fase G1.
La adaptación a la detención del ciclo celular en G2 por los compuestos de la invención se presenta en los ejemplos 5 y 6, y se ilustran en las tablas 1,2 y 3, que exponen las estructuras y valores IC50 (la concentración que ocasiona un incremento G1 medio máximo) para aproximadamente 144 compuestos de la invención. Los abundantes datos proporcionados en las tablas 1, 2 y 3 permitieron determinaciones de estructura-actividad.
Otras medidas de actividad del ciclo celular también pueden utilizarse para demostrar la capacidad de los compuestos de la invención para anular el ciclo celular en G2 y/u ocasionar adaptación a la detención del ciclo celular inducida por daño a ADN. Como se describe en el ejemplo 1 e ilustrada en la figura 2, se midió la fosforilación de histona H3, en la que la fosforilación incrementada de histona H3 indicó adaptación a (escape de) la detención del ciclo celular inducida por daño a ADN. La detención del ciclo celular en G2 se indujo en células Jurkat por daño del ADN, es decir, al suministrar una dosis de 10 Gy de rayos X para producir células “predetenidas”. Las células predetenidas se expusieron al compuesto S00109 (también llamado S109) a 0,3 |iM o 1 |iM, y se observaron incrementos significativos en fosforilación de histona H3 durante un período de tratamiento de 24 horas. Sin pretender limitarse por esta teoría, los compuestos de la invención presumiblemente ocasionaron adaptación a (escape de) la detención del ciclo celular inducida por daño a ADN al anular el punto de control G2 inducido por daño a ADN.
Los compuestos de la invención tienen efectos citotóxicos sobre células cancerosas
Los compuestos de la invención pueden tener un efecto citotóxico sobre células cancerosas, sin ningún tratamiento adicional. De esta manera, la invención proporciona composiciones para la utilización en métodos para eliminar o suprimir células cancerosas sin ningún tratamiento adicional. Como se describe en el ejemplo 2 y se muestra en la figura 4, la exposición de células cancerosas humanas a S00109 solo tiene un efecto citotóxico dependiente de la dosis sobre las células cancerosas cuando se mide por un ensayo de formación de colonias (figura 4, “0 Gy” círculos vacíos, línea continua, que indica sin tratamiento de radiación).
Como se describe en el ejemplo 4 y se muestra en las figuras 6, 7, y 8, se proporcionan compuestos de la invención que tienen un efecto citotóxico sobre células cancerosas, sin ningún tratamiento anticanceroso adicional, y sin efectos citotóxicos sobre las células normales. En la figura 4, los ratones recibieron xenoinjertos de células de mieloma humano por trasplante intraperitoneal, y se midieron los efectos de los compuestos de la invención sobre las tasas de
supervivencia. Como se describe en el ejemplo 4 y se muestra en las figuras 6, 7, y 8, el tratamiento con S00109, S01860, S03518, S03405 o S03747 solo fue suficiente para prolongar la supervivencia de ratones con xenoinjertos de células de mieloma humano, indicando que S00109, S01860, S03518, S03405 o S03747 tuvieron efectos citotóxicos sobre las células cancerosas trasplantadas en el xenoinjerto. Como se muestra en la figura 6, el tratamiento con S00109 (por inyección intraperitoneal) tuvo un efecto terapéutico mucho mayor sobre la supervivencia que el tratamiento “estándar” con dexametasona, o sin tratamiento en absoluto. Como se muestra en las figuras 7 y 8, la administración oral de diversos compuestos representativos de la invención produjo incrementos drásticos en las tasas de supervivencia, en el que algunos tratamientos tuvieron 100% de tasas de supervivencia al final del experimento. Estos resultados demuestran que los compuestos de la invención, como se ilustra por los cinco (5) distintos compuestos representativos ensayados en el ejemplo 4, pueden tener citotoxicidad selectiva contra células cancerosas, y citotoxicidad indetectable contra células normales (es decir, las células normales del hospedante del injerto tumoral de ratón). Estos resultados in vivo demuestran que los compuestos de la invención, como se ilustra por los cinco (5) distintos compuestos representativos ensayados en el ejemplo 4, pueden tener citotoxicidad selectiva in vivo contra células cancerosas, y citotoxicidad indetectable in vivo contra células normales. Estos resultados demuestran que los compuestos de la invención, como se ilustra por los cinco (5) distintos compuestos representativos de la presente invención administrados por diferentes rutas en el ejemplo 4, pueden administrarse a un sujeto en una cantidad eficaz para tratar un trastorno proliferativo en un sujeto.
Los compuestos de la invención pueden sensibilizar células a los tratamientos anticancerosos
Los compuestos de la invención pueden incrementar, o exacerbar, los efectos citotóxicos de otros tratamientos. De esta manera, la invención proporciona composiciones y métodos para sensibilizar células a tratamientos anticancerosos, en particular para sensibilizar células a agentes y tratamientos que dañan el ADN. Como se describe en el ejemplo 2 y se muestra en la figura 4, cuando células cancerosas humanas que se han “predetenido” en G2 por irradiación de rayos X también se exponen a S00109, el tratamiento de combinación tiene un mayor efecto citotóxico sobre las células cuando se mide por un ensayo de formación de colonias. En la figura 4, el efecto de sensibilización de los compuestos de la invención se ilustra mejor para las células que recibieron una dosis de 1 Gy (“1 Gy” círculos llenos, línea de rayas) y tratamiento con diversas dosis de S00109, en el que S00109 mostró un efecto aditivo dependiente de la dosis sobre la citotoxicidad. Como se describe en el ejemplo 3 y se muestra en la figura 5, los tratamientos de combinación de S00109 y dexametasona tienen mucha mayor citotoxicidad que cualquier tratamiento solo, cuando se mide por el porcentaje de células en fase subG1, es decir, el porcentaje de células muertas.
Como se describe en el ejemplo 2 y se muestra en la figura 3, la expresión de y-H2AX fosforilada se midió como un indicador de citotoxicidad. Células tratadas con irradiación de rayos X sola mostraron expresión incrementada de y-H2AX fosforilada, durante un período de 48 horas. El efecto de sensibilización o “aditivo” de los compuestos de la invención puede observarse en células tratadas con irradiación de rayos X seguido de exposición a 1 |iM de S00109 (indicado como “S-109 ” en la leyenda de la figura) dando como resultado niveles significativamente mayores de expresión de y-H2AX fosforilada durante el mismo período de 48 horas, que indica niveles significativamente mayores de citotoxicidad debido a la administración de S00109.
Los compuestos de la invención tienen citotoxicidad selectiva frente a células cancerosas
Según todavía otro aspecto de la invención, los compuestos de la invención pueden eliminar o suprimir selectivamente células diana, en particular células cancerosas, con poco o ningún efecto citotóxico sobre células normales (no diana). La mayoría de los agentes anticancerosos convencionales seleccionan como objetivo células que proliferan independientemente de si son células cancerosas o células normales, con el resultado de que la mayoría de los medicamentos anticancerosos convencionales dan lugar a efectos secundarios tales como náusea, diarrea, o pérdida de cabello. En contraste, los compuestos de la invención seleccionan selectivamente como dianas a células que tienen condiciones tales como un punto de control G1 alterado, detención del ciclo celular en G2, u otros tipos de daño del ADN, eliminando o suprimiendo selectivamente las células diana a la vez que tienen poco o ningún efecto citotóxico sobre las células normales.
La selectividad de los compuestos de la invención se describe en el ejemplo 7 y se muestra en las tablas 4 a 11, en las que los compuestos de la invención no fueron citotóxicos para las células normales en concentraciones en las que los compuestos de la invención tuvieron severos efectos citotóxicos sobre células cancerosas y células dañadas en su ADN (por ejemplo, células irradiadas). De esta manera, la invención proporciona compuestos para la utilización en métodos para seleccionar selectivamente a células dañadas en su ADN tales como células cancerosas, con poco o ningún efecto citotóxico sobre células normales (sin daño), al poner en contacto las células con por lo menos un compuesto de la invención en una cantidad suficiente para anular el punto de control G2. La invención proporciona composiciones farmacéuticas que contienen por lo menos un compuesto de la invención, adecuadas para la utilización en métodos para seleccionar selectivamente a células dañadas en su ADN tales como células cancerosas, con poco o ningún efecto citotóxico sobre células normales (sin daño).
Uso de los compuestos de la invención para cribado
Los compuestos de la invención pueden usarse en protocolos de cribado basados en el fenotipo del ciclo celular, por
ejemplo como se da a conocer por Sha et al. ((2007) Mol Cáncer Ther, 6: 147-153), para identificar compuestos candidatos que pueden interactuar con el punto de control G2 y/o con otros procesos implicados en la adaptación a la detención del ciclo celular en G2. Los compuestos de la invención pueden usarse en protocolos de cribado para identificar compuestos candidatos para la anulación terapéutica del punto de control G2 y/o adaptación terapéutica a la detención en G2. Este protocolo de cribado puede usarse para identificar compuestos que tienen actividad biológica deseada. Los compuestos identificados de esta manera pueden evaluarse además para actividad citotóxica selectiva contra células cancerosas. Los compuestos pueden evaluarse en tratamientos de combinación con agentes anticancerosos convencionales, tal como dexametasona.
Los siguientes ejemplos se ofrecen para ilustrar, pero no para limitar la invención reivindicada.
Ejemplos
Ejemplo 1: Efectos de los compuestos de ensayo S00109 y S01860 sobre células Jurkat detenidas en la fase G2.
Células Jurkat (una línea celular derivada de linfoma humano de células T) se detuvieron en la fase G2 por irradiación de rayos X a l0 Gy, y se cultivaron durante 24 horas en 10% de suero fetal de ternera (FCS)/RPMI164o a 37°C con 5% de CO2/aire (el FCS procedía de Equitech-Bio, Kerrville, TX, y el RPMI1640 procedía de Sigma-Aldrich, St, Louis, MO.). Los compuestos de ensayo se añadieron al medio en las dosis indicadas, y las células se cultivaron bajo las condiciones descritas anteriormente, durante 24 horas adicionales antes de recolectarlas.
Las células recolectadas se tiñeron con amortiguador de Krishan (0,1% de citrato de sodio, 50 pg/ml de yoduro de propidio, 20 pg/ml de RNasa A, 0,5% de Nonidet P-40) y se analizaron por citometría de flujo (BD Biosciences, Franklin Lakes, NJ) para identificar la etapa celular de cada célula en la muestra. Las células en fase G1 se identificaron por tener doble contenido de ADN (2N). La figura 1 muestra el porcentaje de células en fase G1 después del tratamiento con cada compuesto a las dosificaciones indicadas.
El valor de IC50 para cada compuesto se calculó como la dosificación que muestra la actividad media máxima para inducir el incremento del porcentaje de células en fase G1 (el incremento G1). Los valores de IC50 se usaron para medir la actividad de los compuestos y para determinar las interrelaciones estructura-actividad.
Como se muestra en la figura 1, las poblaciones de células Jurkat predetenidas, tratadas con el compuesto S00109 o S01860 durante 24 horas, mostraron un incremento significativo en el número de células en G1, indicando que las células fueron capaces de entrar en el ciclo celular nuevamente.
Fosforilación de histona H3
Las cantidades incrementadas de células en fase G1 (“células en G1 ” detectadas por 2N ADN) después del tratamiento con rayos X indicaron que el punto de control G2 se había anulado y/o que las células se habían adaptado a (escapado de) la detención del ciclo celular en G2 impuesta por la activación del punto de control G2 por el tratamiento con rayos X (es decir, activación del punto de control G2 inducido por daño a ADN por el tratamiento con rayos X). El nivel de fosforilación de histona H3 se midió en células predetenidas tratadas con los compuestos de ensayo, para confirmar que las células predetenidas habían reingresado al ciclo celular y pasado a través de la fase M antes de proceder a la fase G1. La fosforilación incrementada de histona H3 indicó adaptación a (escape de) detención del ciclo celular o anulación del punto de control G2/M inducidas por daño.
Células Jurkat se irradiaron con 10 Gy de rayos X y se cultivaron 24 horas en 10% de FCS-RPMI. El compuesto de ensayo S00109 se añadió al medio de cultivo a 0,3 o 1 pM, y las células se cultivaron con el compuesto de ensayo durante tiempos de tratamiento de 0 a 24 horas. Las células se fijaron con etanol frío, se trataron con 0,1% de saponina/PBS, se tiñeron con antifosfo-histona H3 (Ser10) (Upstate Biotechnology, Uppsala, Suecia) y se analizaron con citometría de flujo (BD Biosciences). En la figura 2, el eje X indica tiempo de tratamiento, es decir, tiempo después de la adición de S00109, y el eje Y indica la relación (%) de células que fueron positivas a fosfo-histona-H3. El nivel de fosforilación de histona H3 (%) después del tratamiento secuencial de células Jurkat con irradiación de rayos X (10 Gy) y compuesto S00109 a 1 pM (cuadros vacíos) y 0,3 pM (círculos llenos) aumentó al aumentar el tiempo de tratamiento hasta 24 horas.
Ejemplo 2: Citotoxicidad de S00109 solo o en combinación con radiación
Expresión de histona H2AX fosforilada
La expresión de histona H2AX fosforilada (yH2AX fosforílada en Ser139) se midió como indicador de citotoxicidad, en particular citotoxicidad relacionada con ADN dañado. Células Jurkat se irradiaron con 10 Gy de rayos X y se cultivaron 24 horas en 10% de FCS-RPMI como se describe anteriormente. Entonces, S00109 se añadió al medio de cultivo a 1 pM durante los tiempos de tratamiento de hasta 24 horas. Las células se lisaron en un amortiguador (100 mM de NaCl,
10 mM de Tris-HCl (pH 8,0), 1 mM de DTT, 0,2% de NP-40,10 mM de NaF, 10 mM de Na 3 VO 4 , 500 nM de ácido okadaico, e inhibidores de proteinasas). Alícuotas del lisado (30 pg de proteína) se sometieron a electroforesis en un gel de SDS PAGE al 15% y se transfirieron a una membrana para análisis de transferencia Western. Un anticuerpo antifosfo-histona H2AX (Ser 139) (Cell Signaling Technology, Beverly, MA) se usó para detectar y-H2AX en la membrana transferida. Como se muestra en la figura 3, los niveles de yH2AX incrementaron el tiempo de tratamiento con S00109, indicando que S00109 ocasionó daño al ADN, además del daño del ADN ocasionado por la radiación.
Análisis de formación de colonias
La actividad citotóxica de S00109 se confirmó además por análisis de formación de colonias usando células HCT-116, una línea celular de cáncer de colon humano, en el que una disminución en los recuentos de colonias es una medida de la supresión del crecimiento celular y/o de muerte celular. Células de cáncer de colon humano HCT-116 se cultivaron en McCoy's 5A (Invitrogen, Carlsbad, Ca ) con 10% de FCS, 5% de CO2/aire a 37°C. Las células se sembraron a 300 células por placa de 6 pocillos por triplicado, se irradiaron con rayos X como se muestra en la leyenda de la figura, y se cultivaron durante 24 horas, entonces se trataron con S00109 a las dosificaciones indicadas y se cultivaron durante 8 días. En el día 8, las colonias se fijaron y tiñeron con violeta cristal (Sigma-Aldrich), y se contó el número de colonias. La figura 4 muestra el efecto de la dosis de S00109 (eje x) sobre las cantidades de colonias (eje y) contadas en el día 8.
Las células que no recibieron radiación (“0 Gy” círculos vacíos, línea continua en la leyenda de la figura 4) se cultivaron bajo las mismas condiciones que las células irradiadas, y se trataron con S00109 para mostrar los efectos de S00109 solo. De esta manera, como se muestra en la figura 4, S00109 solo suprimió la formación de colonias por células HCT-116 de manera dependiente de la dosis, indicando que S00109 solo puede suprimir el crecimiento de células cancerosas y/o eliminar células cancerosas a una dosificación suficientemente alta.
El recuento de colonias “normales” para las células control sin tratar se muestra por el valor para 0 Gy y 0 pM de S00109.
Las células que recibieron una dosis de radiación de 1 Gy (círculos llenos, línea punteada) mostraron una inhibición de la formación de colonias debido a radiación sola. Las células que recibieron una dosis de radiación de 1 y se expusieron a S00109 mostraron además inhibición de la formación de colonias, indicando que S00109 puede aumentar la citotoxicidad del tratamiento con radiación. Bajo estas condiciones de radiación y tratamiento con S00109, se observó un fuerte efecto aditivo dependiente de la dosis de S00109.
Las células que recibieron una dosis de radiación de 3 Gy (“3 Gy” cuadro vacío, línea continua) mostraron una fuerte inhibición de la formación de colonias debido a radiación sola. S00109 parece tener un efecto inhibidor adicional detectable en la concentración más alta (4 pM), indicando nuevamente que S00109 puede aumentar la citotoxicidad del tratamiento con radiación.
Ejemplo 3: Citotoxicidad de S00109 solo y en combinación con dexametasona.
La citotoxicidad de S00109 solo y en combinación con dexametasona se midió al identificar el número de células en fase subG1 después del tratamiento, en el que la fase subG1 indica muerte celular, de tal modo que el número de células en fase subG1 después del tratamiento indica eliminación de células por el tratamiento. Una línea celular derivada de mieloma múltiple humano, ARH-77, se cultivó en presencia o ausencia de S00109, con o sin dexametasona, durante 24 horas en 10% de suero fetal de ternera (FCS)/RPMI1640 a 37°C con 5% de CO2/aire usando materiales y condiciones descritos anteriormente. Las células recolectadas se tiñeron con amortiguador de Krishan (0,1% de citrato de sodio, 50 pg/ml de yoduro de propidio, 20 pg/ml de RNasa A, 0,5% de Nonidet P-40) y se analizaron con citometría de flujo (BD Biosciences, Franklin Lakes, NJ).
La figura 5 muestra el porcentaje de células ARH-77 en fase subG1 (eje y) después del tratamiento con S00109 (eje x) solo o en combinación con dexametasona. El porcentaje “normal” de células en subG1 en una población de células de control sin tratar se muestra por el valor para S00109 solo (diamantes llenos, línea continua, “S109 solamente” en la leyenda de la figura) a 0 pg/ml de S00109. El tratamiento con S00109 solo, a concentraciones de hasta 10 pg/ml, ocasionó muerte celular de manera dependiente de la dosis. El tratamiento con dexametasona solo a 2 ng/ml (cuadros vacíos, línea punteada), 20 ng/ml (triángulos vacíos, línea de puntos/rayas) y 200 ng/ml círculos vacíos, línea de rayas) sin S00109 mostró niveles ligeramente incrementados de células en subG1, es decir, citotoxicidad ligeramente incrementada, en comparación con el control. Sin embargo, el tratamiento con S00109 en combinación con dexametasona dio como resultado un incremento drástico en los niveles de células en subG1. El efecto de la combinación mostró una fuerte dependencia de la concentración de S00109, demostrando un efecto dependiente de la dosis de S00109. La combinación de S00109 y dexametasona dio como resultado un nivel de muerte celular que fue significativamente mayor que el nivel observado con cualquier compuesto solo. De esta manera, S00109 aumentó la citotoxicidad de la dexametasona.
Ejemplo 4: Efectos de los compuestos representativos de la invención, solos y en combinación, sobre la supervivencia de ratones con xenoinjertos de células ARH-77
Ratones con xenoinjertos de ARH-77 (una línea celular derivada de mieloma múltiple humano) se trataron con S00109, S01860, S03518, S03405 o S03747 o con dexametasona, un tratamiento “estándar” reconocido, y su supervivencia se midió y comparó con la supervivencia de ratones con xenoinjertos tratados con vehículo (control). La capacidad de un tratamiento para prolongar la supervivencia se consideró como un indicador de la citotoxicidad del tratamiento frente a las células cancerosas injertadas, sin actividad adversa significativa sobre células normales (ratón) in vivo.
A ratones macho con inmunodeficiencia combinada severa (SCID) de 8 semanas de edad se les trasplantaron, de manera intraperitoneal, 1,9 x 106 (Fig. 6), 0,8 x 106 (Fig. 7), 4,1 x 106 (Fig. 8) células/animal de células ARH-77 (n = 10).
Los animales se alojaron según las directrices de la Association for the Assessment and Accreditation of Laboratory Animal Care International, y los protocolos se aprobaron por el comité del cuidado de animales de institucional de CanBas Co. Ltd.
Para el experimento mostrado en la figura 6, los ratones recibieron 1,9 x 106 células ARH-77 por trasplante intraperitoneal. Los ratones tratados con S00109 (“S109”) recibieron una inyección intraperitoneal de 50 mg/kg de S00109. Los ratones tratados con dexametasona (“Dexa”) recibieron una inyección intraperitoneal de 2 mg/kg de dexametasona. Los animales de control tratados con vehículo recibieron una inyección intraperitoneal de vehículo. Cada inyección se realizó en el día 1, día 2 y día 3 después del trasplante de células ARH-77. La supervivencia (eje y, % de ratones que sobreviven) se midió durante hasta 80 días después del trasplante (eje x) para ratones de control tratados con vehículo solo (“Control” línea de rayas); ratones tratados con 50 mg/kg del compuesto S00109 (“S109” línea continua); y ratones tratados con 2 mg/kg de dexametasona (“Dexa” línea de punto y raya). Los ratones tratados con S00109 tuvieron un plazo significativamente más prolongado de supervivencia que los ratones de control sin tratar. Aunque los ratones tratados con dexametasona tuvieron un plazo más prolongado de supervivencia que los ratones de control sin tratar, el efecto terapéutico de la dexametasona fue mucho menor que el efecto terapéutico de S00109.
Para el experimento mostrado en la figura 7, los ratones recibieron 0,8 x 106 células ARH-77 por trasplante intraperitoneal. Los ratones se trataron por una sola administración oral de compuestos en el Día 1 después del trasplante como sigue: ratones de control tratados por vía oral con vehículo solo (“Control” línea continua); ratones tratados por vía oral con 750 mg/kg del compuesto S00109 (“S109” línea punteada); y ratones tratados por vía oral con 750 mg/kg del compuesto S01860 (“S1860” línea de rayas). Aunque los ratones tratados con S00109 inicialmente mostraron una supervivencia ligeramente inferior a la de los ratones de control, después de alrededor de 64 días los ratones tratados con S00109 mostraron una supervivencia significativamente mayor a la de los ratones de control, con casi 70% de supervivencia a 85 días, en comparación con solamente alrededor de 20% de los ratones de control que sobreviven a 85 días. Los ratones tratados con S01860 mostraron tasas de supervivencia drásticamente mayores que los ratones de control o ratones tratados con S00109, en el que la primera disminución en supervivencia no se observó hasta 70 días después del trasplante, y casi 90% de los ratones tratados con S01860 sobrevivieron a los 85 días.
Para el experimento mostrado en la figura 8, los ratones recibieron 4,1 * 106 células ARH-77 por trasplante intraperitoneal. Los ratones se trataron por administración oral una vez al día de los compuestos en el Día 1 y administración oral una vez al día de los compuestos en el Día 2 después del trasplante como sigue: ratones de control tratados por vía oral una vez al día durante dos días con vehículo solo (“CONT” línea continua); ratones tratados por vía oral una vez al día durante dos días con 250 mg/kg del compuesto S003518 (“S3518” línea punteada); ratones tratados por vía oral una vez al día durante dos días con 250 mg/kg del compuesto S003405 (“S3405” línea de rayas); y ratones tratados por vía oral una vez al día durante dos días con 250 mg/kg del compuesto S003747 (“S3747” línea de punto y raya). Los ratones tratados con S03518, S03405 o S03747 mostraron todos tasas de supervivencia drásticamente mayores que los ratones de control. Los ratones de control mostraron supervivencia disminuida comenzando alrededor de 29 días después del trasplante, y solamente alrededor de 30% de supervivencia a 50 días después del trasplante. En contraste, los ratones tratados con S03518, S03405 o S03747 mostraron muy poca disminución en supervivencia, y aún tuvieron tasas extremadamente altas de supervivencia de entre 80-100% a los 50 días después del trasplante.
Estos resultados demostraron que S00109, S01860, S03518, S03405 o S03747, administrados de manera intraperitoneal o por vía oral, tuvieron citotoxicidad selectiva in vivo contra células cancerosas (células ARH 77 del tumor xenoinjertado) a la vez que tienen citotoxicidad indetectable contra células normales (el hospedante del injerto de ratón). Estos resultados demostraron que cinco compuestos diferentes de la presente invención se administraron a un sujeto en una cantidad eficaz para tratar un trastorno proliferativo en un sujeto.
Ejemplo 5: Capacidad de los compuestos representativos para ocasionar adaptación a la detención del ciclo celular en G2 e inducir células detenidas en G2 para reingresar al ciclo celular
Los compuestos representativos se sintetizaron de acuerdo con los métodos proporcionados en la presente memoria. La estructura y otras propiedades de cada compuesto se determinaron por espectroscopía de RMN 1H para cada compuesto sintetizado.
Células Jurkat predetenidas se prepararon como se describe en el ejemplo 1. Brevemente, las células Jurkat se sometieron a irradiación de rayos X a una dosis de 10 Gy, y se cultivaron durante 24 horas en 10% de suero fetal de ternera (FCS)/RPMI1640 a 37°C con 5% de CO2/aire, después de cuyo tiempo las células se expusieron a diversas concentraciones de compuestos de ensayo, y se cultivaron bajo las condiciones descritas anteriormente durante 24 horas adicionales antes de recolectarlas. Las células recolectadas se tiñeron con amortiguador de Krishan (0,1% de citrato de sodio, 50 p,g/ml de yoduro de propidio, 20 p,g/ml de RNasa A, 0,5% de Nonidet P-40) y se analizaron por citometría de flujo (BD Biosciences, Franklin Lakes, NJ) para identificar la etapa celular de cada célula en la muestra. Las células en fase G1 se identificaron por tener doble contenido de ADN (2N).
El valor de IC50 para cada compuesto se calculó como la dosificación (concentración en p,M) que ocasionó un incremento medio máximo del porcentaje de células en fase G1 (el incremento G1) medido para ese compuesto de ensayo. La tabla 1 a continuación presenta la estructura, masa, valores de RMN 1H, y valores de IC50 para los compuestos representativos.
Tabla 1. Compuestos representativos y valores de IC50


Ejemplo 6: Capacidad de los compuestos representativos para ocasionar adaptación a la detención del ciclo celular en G2 e inducir células detenidas en G2 para que reingresen al ciclo celular
Los compuestos representativos se sintetizaron de acuerdo con los métodos proporcionados en la presente memoria. La estructura y otras propiedades fisicoquímicas de cada compuesto se determinaron por espectroscopia de RMN 1H para cada compuesto sintetizado.
Células Jurkat predetenidas se prepararon como se describe en el ejemplo 1. Brevemente, las células Jurkat se sometieron a irradiación de rayos X a una dosis de 10 Gy, y se cultivaron durante 24 horas en 10% de suero fetal de ternera (FCS)/RPMI1640 a 37°C con 5% de CO2/aire, después de cuyo tiempo las células se expusieron a diversas concentraciones de compuestos de ensayo, y se cultivaron bajo las condiciones descritas anteriormente durante 24 horas adicionales antes de recolectarlas. Las células recolectadas se tiñeron con amortiguador de Krishan (0,1% de citrato de sodio, 50 p,g/ml de yoduro de propidio, 20 p,g/ml de RNasa A, 0,5% de Nonidet P-40) y se analizaron por citometría de flujo (BD Biosciences, Franklin Lakes, NJ) para identificar la etapa celular de cada célula en la muestra. Las células en fase G1 se identificaron por tener doble contenido de ADN (2N).
El valor de IC50 para cada compuesto se calculó como la dosificación (concentración en p,M) que ocasionó un incremento medio máximo del porcentaje de células en fase G1 (el incremento G1) medido para ese compuesto de ensayo. La tabla 2 a continuación presenta estructuras, nombre IUPAC, fórmula molecular, número ID (“SClD”), masa, valores de RMN 1H, y valores de IC50 para los compuestos representativos. La tabla 3 presenta estructuras, nombre IUPAC, fórmula molecular, número ID (“SCID”), masa, valores de RMN 1H, y valores de IC50 para compuestos representativos adicionales.
Tabla 2. Compuestos representativos y valores de IC50





Tabla 3. Compuestos representativos y valores de IC50

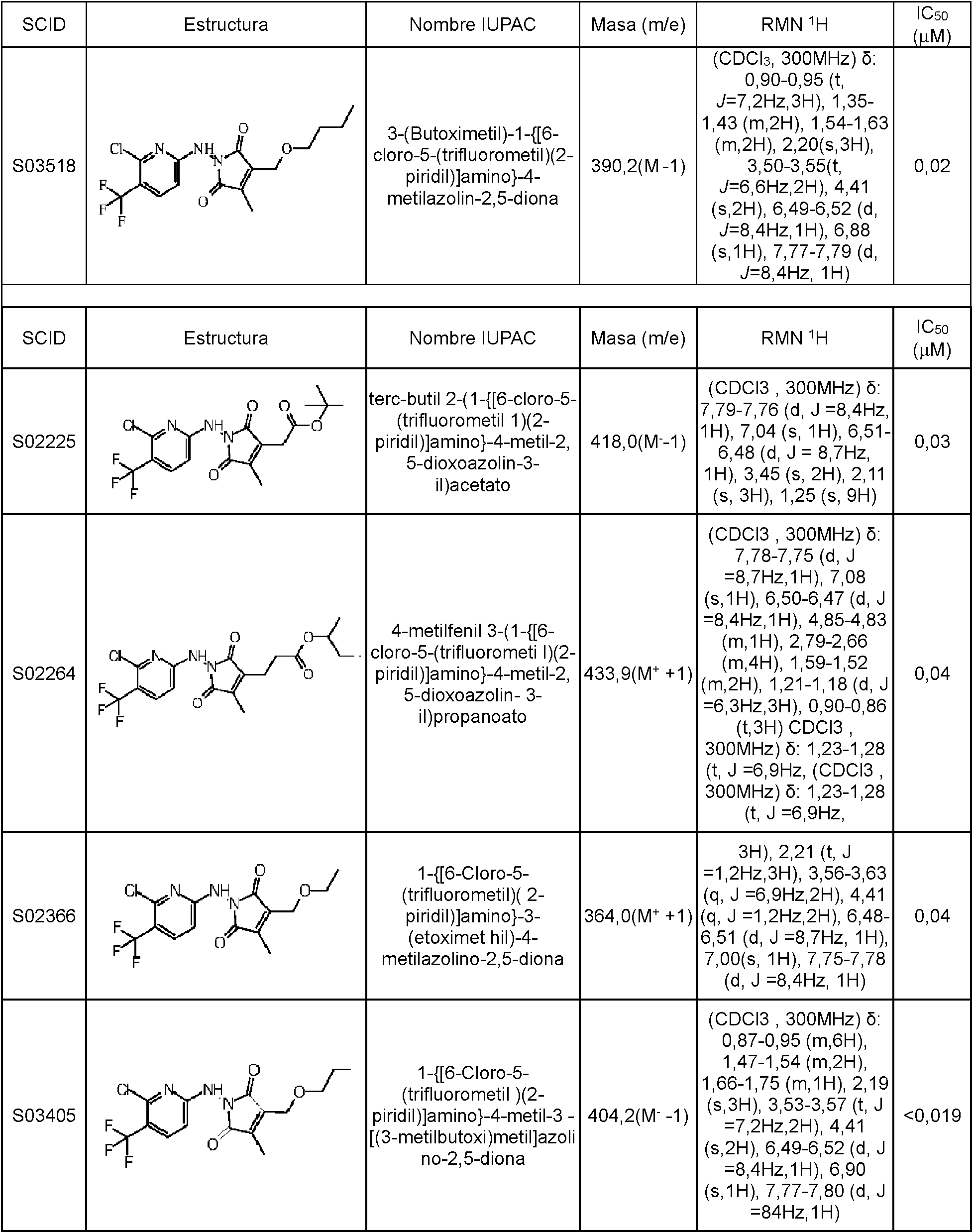





Ejemplo 7: Efectos de S00109 solo, y S00109 en combinación con tratamientos adicionales anticancerosos, sobre células normales y sobre células cancerosas
Los efectos de S00109 solo, y S00109 en combinación con tratamientos anticancerosos bien conocidos, se determinaron para fibroblastos dérmicos humanos normales (NHDF), y células endoteliales umbilicales humanas (HUVEC), y para células MIAPaCa2 (línea celular derivada de cáncer pancreático), células HCT116 (línea celular derivada de cáncer de colon), células IM9 (línea celular derivada de mieloma múltiple), células ARH-77 (línea celular derivada de mieloma múltiple), células RPMI-8226 (línea celular derivada de mieloma múltiple) y células NCI-H929 (línea celular derivada de mieloma múltiple).
Las células se trataron como se describe en las tablas 4-11, luego se recolectaron, se tiñeron con yoduro de propidio para permitir la medida del contenido de ADN, y se analizaron por citometría de flujo para determinar la etapa del ciclo celular de cada célula presente en cada población después del tratamiento. De esta manera, un “fenotipo” o “fenotipo predominante” o “patrón del ciclo celular” se determina en base al porcentaje de células en diversas etapas del ciclo celular (G1, S, G2, M, subG1 (muerta), etc.). Las tablas 4-11 dan a conocer el patrón predominante del ciclo celular, o el cambio más relevante en el patrón del ciclo celular, que corresponde a cada combinación de tratamientos. Por ejemplo, el resultado para una combinación de tratamientos puede darse a conocer como un incremento en el número de células en fase g 2 después de la exposición a cierta concentración de S00109, en comparación con el fenotipo predominante que resulta de la exposición a una concentración inferior de S00109, para el mismo tratamiento anticanceroso. De manera similar, el resultado para un tratamiento anticanceroso, en ausencia de tratamiento con S00109, puede darse a conocer como un incremento en el número de células en fase S, en comparación con el control correspondiente (sin tratamiento anticanceroso, sin S00109, mismas condiciones de cultivo).
Células normales humanas y células cancerosas humanas se expusieron a S00109 (abreviado S109 en las tablas 4 11) a diversas concentraciones desde nada de S00109 hasta 100 |mM de S00109, como se muestra en el encabezado para cada columna.
Células normales humanas y células cancerosas humanas se expusieron a una diversidad de tratamientos anticancerosos incluyendo radiación de rayos X, y los agentes anticancerosos (“fármacos anticancerosos”) metotrexato, CPT-11 (irinotecán, Camptosar® ), 5-FU (5-fluorouracilo), CDDP (cisplatino), adriamicina, Gemzar® (gemcitabina), taxol, Velcade® (bortezomib), vincristina, dexametasona, y melfarano. Los tratamientos incluyeron
tratamiento simultáneo con el agente anticanceroso y la dosis indicada de S00109, así como combinaciones de tratamientos escalonados, en los que las células se trataron primero con un tratamiento anticanceroso y luego con la dosis indicada de S00109. En las tablas 4-11 a continuación se presenta una clave para los tratamientos, como se describe en la leyenda para cada fila.
Los experimentos de control incluyeron experimentos en los que las células se trataron con S109 a la dosis indicada, y sin tratamiento adicional, indicado como “solo” en las tablas 4-11, en las que “S109 a la dosis indicada” incluye nada de S00109 o S00109 a diversas concentraciones de hasta 100 |iM, como se indica en cada encabezado de columna. Los experimentos de control incluyeron experimentos diseñados para evaluar los efectos de tiempos de cultivo de 24 h y 48, así como los efectos de etapas adicionales tales como un cambio del medio de cultivo a 3 horas, o la adición de S00109 en una etapa posterior.

En las tablas 4-11, los resultados se dieron a conocer como sigue


Resultados
Los resultados muestran que S00109 tuvo efectos citotóxicos más graves sobre células cancerosas y poco o ningún efecto citotóxico sobre las células normales. Expresado alternativamente, los resultados muestran que la mayoría de las células cancerosas son más sensibles a S00109 que lo que lo son la mayoría de las células normales. Estos resultados coinciden con los resultados de los experimentos de trasplante de tumor de xenoinjerto ARH-77 del ejemplo 4, en los que el tratamiento de ratones que llevan tumores xenoinjertados con S00109, S01860, S03518, S03405 o S03747 dio como resultado incrementos drásticos en las tasas de supervivencia de los ratones que llevan tumores, indicando que S00109, S01860, S03518, S03405 o S03747 han eliminado o suprimido específicamente las células ARH-77 (mieloma múltiple) en los tumores del xenoinjerto sin que tengan efectos citotóxicos sobre las células u órganos normales del ratón hospedante.
A. Fibroblastos dérmicos humanos normales (NHDF)
Fibroblastos dérmicos humanos normales (NHDF) se trataron como se describe en la primera celda de cada fila de la tabla 4, y se expusieron a nada de S00109 (Columna 2) o a S00109 a las concentraciones listadas en las Columnas 3-9. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 4. Fenotipo de células de fibroblastos dérmicos humanos normales (NHDF) tratadas con S00109 solo, o en combinación con tratamientos anticancerosos


B. Células endoteliales umbilicales humanas normales (HUVEC)
Células normales endoteliales umbilicales humanas (HUVEC) se trataron como se describe en la primera celda de cada fila de la tabla 5, y se expusieron a nada de S00109 (Columna 2) o S00109 a las concentraciones listadas en las Columnas 3-9. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 5. Fenotipo de células normales endoteliales umbilicales humanas (HUVEC) tratadas con S00109 solo o en combinación con tratamientos anticancerosos


C. Línea celular derivada de cáncer pancreático humano MIAPaCa2
Células de la línea celular derivada de cáncer pancreático humano MIAPaCa2 se trataron como se describe en la primera celda de cada fila de la tabla 6, y se expusieron a nada de S00109 (Columna 2) o S00109 a las concentraciones presentadas en las Columnas 3-7. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 6. Fenotipo de la línea celular de cáncer pancreático humano (MIAPaCa2) tratada con S00109 solo o en combinación con tratamientos anticancerosos

D. Línea celular derivada de cáncer de colon humano HCT116
Células de la línea celular derivada de cáncer de colon humano HCT116 se trataron como se describe en la primera celda de cada fila de la tabla 7, y se expusieron a nada de S00109 (Columna 2) o S00109 a las concentraciones listadas en las Columnas 3-8. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 7. Fenotipo de la línea celular de cáncer de colon humano (HCT116) tratada con S00109 solo o en combinación con tratamientos anticancerosos


E. Línea celular derivada de mieloma múltiple humano IM9
Células de la línea celular derivada de mieloma múltiple humano IM9 se trataron como se describe en la primera celda de cada fila de la tabla 8, y se expusieron a nada de S00109 (Columna 2, tabla 8A) o S00109 a las concentraciones listadas en las Columnas 3-7 de la tabla 8A y Columnas 2-7 de la tabla 8B. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 8. Fenotipo de la línea celular de mieloma múltiple humano (IM9) tratada con S00109 solo o en combinación con tratamientos anticancerosos. tabla 8A. Resultados para nada de S00109, y 0,02 a 0,3125 |iM de S00109


Tabla 8B. Resultados para 0,625 a 20 |iM de S00109


F. Línea celular derivada de mieloma múltiple humano ARH-77
Células de la línea celular derivada de mieloma múltiple humano ARH-77 se trataron como se describe en la primera celda de cada fila de la tabla 9, y se expusieron a nada de S00109 (Tabla 9A, Columna 2) o S00109 a las concentraciones listadas en las Columnas 3-7 de la tabla 9A y 2-7 de la tabla 9B. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 9. Fenotipo de la línea celular de mieloma múltiple humano (ARH-77) tratada con S00109 solo o en combinación con tratamientos anticancerosos. tabla 9A. Resultados para nada de S00109, y 0,02 a 0,3125 |iM de S00109

Tabla 9B. Resultados para 0,625 a 20 pM de S00109
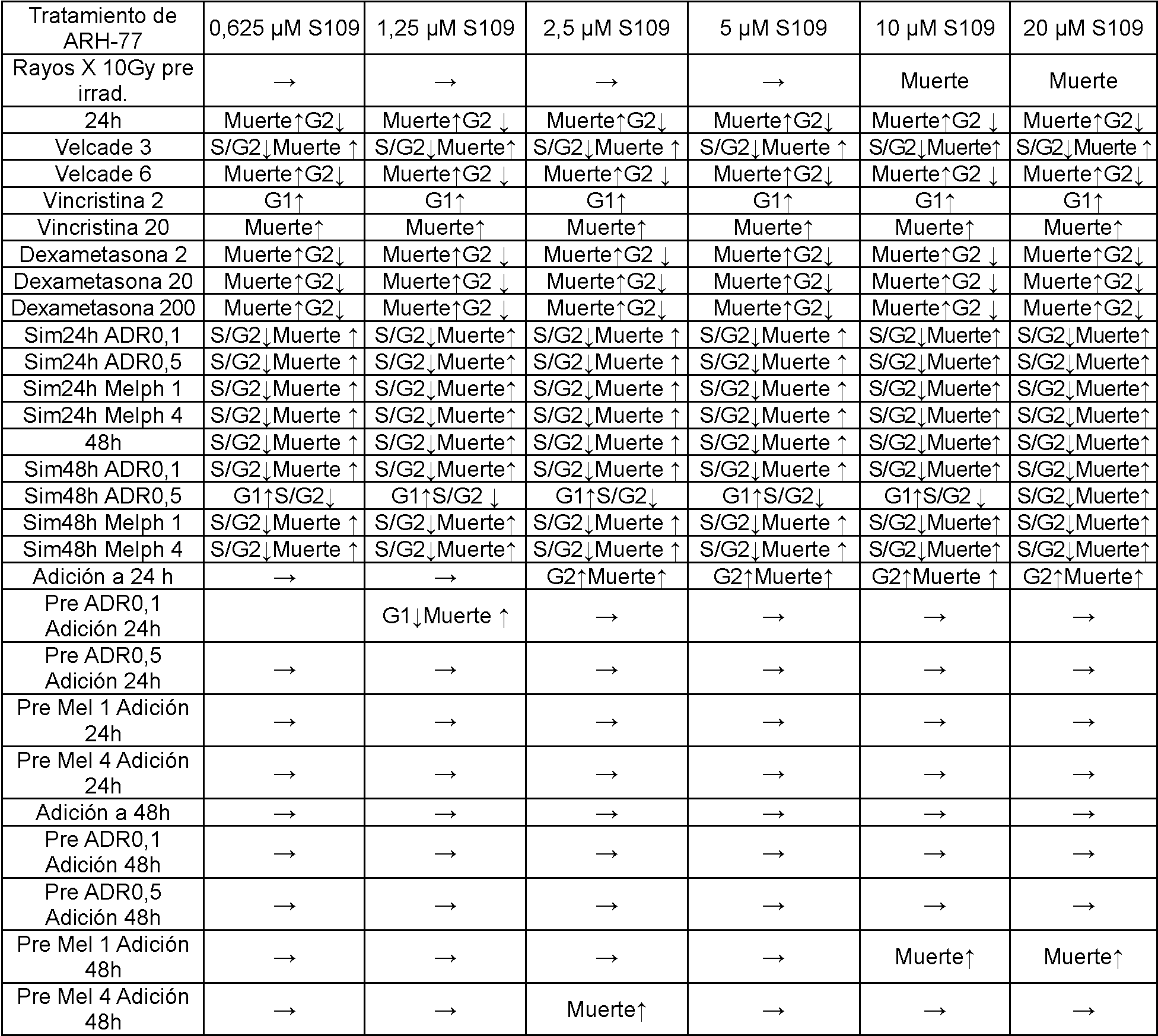
G. Línea celular derivada de mieloma múltiple humano RPMI-8226
Células de la línea celular derivada de mieloma múltiple humano RPMI-8226 se trataron como se describe en la primera celda de cada fila de la tabla 10, y se expusieron a nada de S00109 (Columna 2, tabla 10A) o S00109 a las concentraciones presentadas en las Columnas 3-7 de la Columna 10A y Columnas 2-7 de la Columna 10B. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 10. Fenotipo de la línea celular de mieloma múltiple humano (RPMI-8226) tratada con S00109 solo o en combinación con tratamientos anticancerosos. tabla 10A. Resultados para nada de S00109, y 0,02 a 0,3125 pM de
S00109


Tabla 10B. Resultados para 0,625 a 20 |iM de S00109

H. Línea celular derivada de mieloma humano NCI-H929
Células de la línea celular derivada de mieloma humano NCI-H929 se trataron como se describe en la primera celda de cada fila de la tabla 11, y se expusieron a nada de S00109 (Columna 2, tabla 11A) o S00109 a las concentraciones listadas en las Columnas 3-7 de la tabla 11A y Columnas 2-7 de la tabla 11B. El fenotipo predominante para cada combinación de tratamientos se describe en la celda correspondiente.
Tabla 11. Fenotipo de la línea celular de mieloma múltiple humano (NCI-H929) tratada con S00109 solo o en combinación con tratamientos anticancerosos. tabla 11A. Resultados para nada de S00109, y 0,02 a 0,3125 |iM de
S00109


Tabla 11B. Resultados para 0,625 a 20 pM de S00109

Ejemplo 8: Síntesis de compuestos representativos
Debe apreciarse que los siguientes ejemplos sirven como ilustraciones, y todos los compuestos de esta invención podrían sintetizarse usando métodos similares a los descritos en estos ejemplos.
Procedimiento general para la síntesis de 2-piridilhidrazinas sustituidas

13 14
El procedimiento general para la síntesis de 2-piridilhidrazinas sustituidas se representa en la presente memoria en la síntesis de 4-(trifluorometil)-6-metil-2-piridilhidrazina (14). Un equivalente de 2-cloro-4-(trifluorometil)-6-metilpiridina (13) y 1,5 equivalentes de hidrato de hidrazina se mezclaron en etanol. La disolución se volvió amarilla tras agitarla durante varios minutos. La mezcla de reacción se puso a reflujo hasta que el análisis mediante TLC mostró que no quedaba material de partida. El disolvente se eliminó entonces a vacío, y la suspensión resultante se extrajo con éter tres veces. La disolución etérea resultante se secó sobre MgSO4 anhidro y se evaporó para producir el producto bruto, que se recristalizó después en etanol para proporcionar el compuesto 14.
Síntesis de S00069 (solo para referencia)
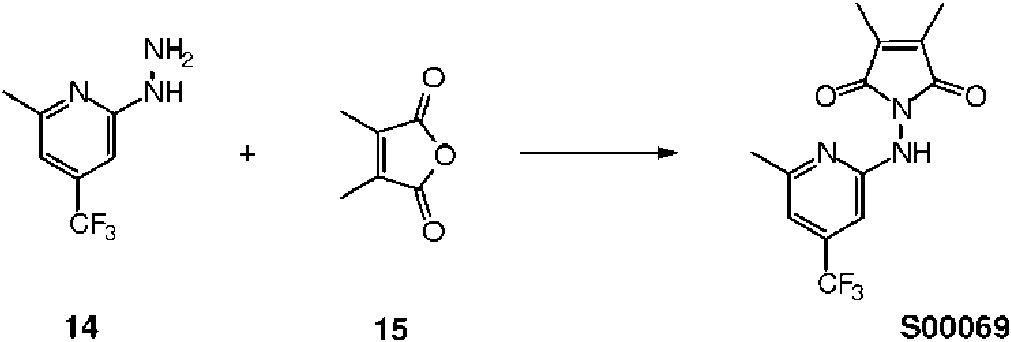
El anhídrido 15 (1 eq.) se añadió a una disolución de la hidrazina 14 (1,0 mmol) en cloroformo y se agitó a reflujo durante 4 h. Se determinó que la reacción estaba terminada mediante TLC (éter de petróleo:acetato de etilo = 3:1). El disolvente se evaporó, y el residuo se purificó mediante cromatografía ultrarrápida (éter de petróleo:acetato de etilo = 2:1) para proporcionar el producto.
Síntesis de S00084 (solo para referencia)

A una disolución de S00069 (35 mg, 0,117 mmoles) en THF (6 ml) a 0°C se le añadió NaH (60% en aceite mineral, 8 mg, 0,12 mmoles). La mezcla se agitó durante 30 min., y después se añadió Mel (20 mg). La mezcla de reacción se agitó durante 2 h a temperatura ambiente, y después se vertió en el NH4C1 acuoso saturado. Éste se extrajo con CHCl 3 . La capa orgánica se secó sobre Na 2 SO 4 anhidro. El disolvente se eliminó, y el residuo se purificó mediante TLC preparativa (éter de petróleo/éter dietílico 5:1) para producir S00084 (3 mg).
S ín te s is de S 00109

Etapa 1: Síntesis de N-óxido de 2-doro-5-trifluorometil-piridina (17):
Se disolvió 2-cloro-5-trifluorometil-piridina (16, 10 mmoles) en CH2Ch (20 ml), y se añadió UHP (compuesto de adición de urea-peróxido de hidrógeno, 21 mmoles). La mezcla se enfrió hasta 0°C, y después se añadió lentamente anhídrido trifluoroacético (20 mmoles) a la mezcla de reacción. Se dejó calentar hasta la temperatura ambiente y se agitó hasta que se juzgó que la reacción se había terminado mediante TLC. La reacción se paralizó con Na2SO3 acuoso, se agitó durante 4 h, se lavó con NaHCO3 acuoso saturado, y se secó sobre MgSO4 anhidro. La cromatografía en columna produjo 1,8 g de compuesto 17 como un aceite.
Etapa 2: Síntesis de 2,6-dicloro-5-trifluorometil-piridina (18):
Se disolvió N-óxido de 2-cloro-5-trifluorometil-piridina (17, 4 mmoles) en POCb recientemente destilado (4,5 ml). La mezcla de reacción se calentó hasta 80°C durante 17 h. Tras enfriar hasta la temperatura ambiente, el disolvente se eliminó a presión reducida. Se añadió hielo, y la mezcla se dejó reposar durante 4 h. La mezcla se repartió entre CH2Cl2 (50 ml) y NaHCO3 acuoso saturado. La cromatografía en columna produjo el compuesto 18 como un aceite amarillo (rendimiento: 50%).
Etapa 3: Síntesis de 6-cloro-5-trifluorometil-2-piridilhidrazina (19):
A la disolución de 2,6-dicloro-5-trifluorometil-piridina (18, 2 g, 9,26 mmoles) en etanol (30 ml) se le añadió hidrato de hidrazina (2,9 g, 46 mmoles). La mezcla de reacción se agitó durante 4 h a temperatura ambiente, después se concentró para eliminar el disolvente, y se añadió acetato de etilo, se lavó con agua. La capa orgánica se secó sobre Na2SO4 anhidro. La cromatografía en columna (sílice, éter de petróleo/acetato de etilo = 4/1 ~3/1) produjo el compuesto 19 como un sólido blanco (rendimiento: 56%) y otro isómero 20 (rendimiento: 18%).
Etapa 4: Síntesis de S00109:
Se añadió anhídrido 2,3-dimetilmaleico (15, 0,126 g, 1,0 mmol) a una disolución de 6-cloro-5-trifluorometil-2-piridilhidrazina (19, 0,211 g, 1,0 mmol) en 5 ml de cloroformo, y la mezcla se puso a reflujo durante 4 horas. El disolvente se eliminó, y el residuo se purificó mediante cromatografía ultrarrápida (éter de petróleo/acetato de etilo 5:1 hasta 2:1) para proporcionar S00109 (0,21 g).
S ín te s is de S 00170

Se suspendieron Compuesto S00109 (40 mg, 0,125 mimóles) y NaH (60% en aceite mineral, 7 mg, 0,188 mimóles) en 2 ml de THF anhidro, y la mezcla se agitó a 0°C durante 30 min. Se añadió lentamente yoduro de metilo (21 mg, 0,150 mimóles) a la disolución a la misma temperatura, y después la mezcla se calentó hasta 25-30°C y se agitó durante toda la noche. El disolvente se evaporó, y se añadió ácido acético para llevar la disolución a pH = 4. Ésta se extrajo con cloroformo tres veces, y la fase orgánica combinada se lavó con HCl 1N, y después con NaHCO3 acuoso saturado. Después se secó sobre Na2SO4 anhidro. El disolvente se eliminó, y el residuo se purificó mediante TLC preparativa (éter de petróleo/éter dietílico 4:1) para proporcionar el compuesto S00170 (4,2 mg).
Síntesis de S00585 (solo para referencia)

El Compuesto 21 se convirtió en el Compuesto 22 usando un procedimiento similar al que se describió en el ejemplo 1. El Compuesto 22 se convirtió en el Compuesto S00585 usando un procedimiento similar al que se describió en el ejemplo 2.
Procedimiento general para la síntesis de fenilhidrazinas sustituidas

El procedimiento general para la síntesis de fenilhidrazinas sustituidas se representa en la presente memoria en la síntesis de 3-(trifluorometil)-4-bromofenilhidrazina (24). Se añadió la bencilamina 23 correspondiente (0,08 moles) a HCl conc. (40 ml). La mezcla se enfrió hasta -5°C mediante hielo y sal con agitación. Después se añadió nitrito de sodio (5,52 g, 0,08 moles) disuelto en agua (20 ml). La agitación se continuó durante 1 h, y se añadió lentamente durante un período de dos horas cloruro estañoso (30g) en HCl conc. (30 ml), mientras se mantiene la temperatura por debajo de 0°C. La mezcla se agitó durante otra hora tras la adición y se filtró. El sólido filtrado se trató con hidróxido de sodio acuoso diluido y después se extrajo con éter. La capa etérea se lavó con agua, se secó sobre Na2SO4 anhidro. El disolvente se eliminó, y el residuo se cristalizó en hexano para proporcionar el Compuesto 24.
Síntesis de S00516

El Compuesto S00516 se sintetizó a partir de la hidrazina 24 correspondiente usando un procedimiento similar al que se describió en el ejemplo 2.

El Compuesto 25 se sintetizó según un procedimiento bibliográfico (Eur. J. Med. Chem. Chim. Ther. 1997, 32(5), 397 408). Se convirtió en el Compuesto S00756 usando un método similar al que se describió en el ejemplo 2.
Síntesis de S00513

Etapa1: bromuro de 3-cloro-4-trifluorometilbencilo 27:
Una mezcla de 2-cloro-4-metil-1-trifluorometilbenceno 26 (0,20 g, 1 mmol), N-bromosuccinimida (0,17 g, 1 mmol) y peróxido de benzoílo (7,4 mg, 0,03 mmoles) en tetracloruro de carbono (2 ml) se calentó a reflujo durante 2 horas. Se añadió otra porción de peróxido de benzoílo (20 mg, 0,08 mmoles). La mezcla se calentó a reflujo durante otras 0,5 horas. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 16 horas. El sólido se eliminó mediante filtración. El disolvente se eliminó a presión reducida. El producto bruto se purificó mediante cromatografía ultrarrápida sobre sílice, usando éter de petróleo como eluyente, para proporcionar 0,22 g (80%) de Compuesto 27.
Etapa 2: Compuesto S00513
A una disolución de 3,4-dimetilmaleimida 28 (43 mg, 0,34 mmoles) en 1,3 ml de acetona se le añadió carbonato de potasio anhidro (50 mg, 0,37 mmoles) y Compuesto 27 (100 mg, 0,37 mmoles). La mezcla de reacción se agitó a temperatura ambiente toda la noche. Se añadió agua, y la mezcla se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó (Na2SO4), y se concentró a presión reducida. El producto bruto se purificó mediante cromatografía sobre sílice, usando éter de petróleo/acetato de etilo (10:1) como eluyente, para proporcionar 70 mg (60%) de Compuesto S00513.
Síntesis de S00628 (solo para referencia)

En una disolución de 1-hidroxi-3,4-dimetilazolin-2,5-diona 30 (56 mg, 0,39 mmoles, 1 equiv.) en 1,2-dicloroetano (2,5 ml), se añadieron CuCl (39 mg, 0,39 mmoles, 1 equiv.), tamices moleculares 4 A recientemente activados (-100 mg), y ácido 4-trifluorometilfenilborónico 29 (150 mg, 0,78 mmoles, 2 equiv.), seguido de piridina (34 mg, 0,43 mmoles, 1,1 equiv.). La suspensión marrón clara resultante se agitó durante 16 h. La mezcla de reacción se filtró. La cromatografía del filtrado (éter de petróleo/acetato de etilo = 7: 1) produjo el compuesto S00628 como un sólido blanco (65 mg, 59%).
Ejemplo 9: Procedimientos sintéticos
Todos los compuestos enumerados en las tablas 1,2, y 3 se sintetizaron usando métodos idénticos a o similares a los descritos en los ejemplos más abajo.
Procedimiento general para la síntesis de análogos de piridina sustituidos con haluro hasta compuestos diana
Esquema 1

Se añadió material de partida disuelto en etanol e hidrato de hidrazina (10,0 eq.) para formar una mezcla, la mezcla se agitó a 50-60°C (temperatura del aceite) durante varias horas (la terminación se comprobó mediante TLC), el disolvente se evaporó, se añadió agua, y la mezcla resultante se extrajo con acetato de etilo, se secó y se concentró para formar una preparación bruta que se usó sin purificación adicional para la siguiente etapa. La preparación bruta se disolvió en cloroformo (o tolueno, ácido acético, u otro disolvente adecuado), se añadió anhídrido (1,0 eq.), la mezcla se calentó a 50-60°C (temperatura del aceite) durante varias horas (la terminación se comprobó mediante TLC), el disolvente se evaporó, y la preparación se purificó mediante TLC prep. para proporcionar el compuesto deseado.

Los materiales de partida estaban comercialmente disponibles, de manera que la ruta sintética de los compuestos S00585, S01098, S01207 fue similar al procedimiento general.
Compuesto S00109
Esquema 2

Producto intermedio 1
Se disolvió 2-cloro-5-trifluorometil-piridina (10 mmoles) en CH2Cl2 (20 ml), y se añadió UHP (compuesto de adición de urea-peróxido de hidrógeno, 21 mmoles). La mezcla se enfrió hasta 0°C, después se añadió lentamente anhídrido trifluoroacético (20 mmoles) a la mezcla de reacción. Se dejó calentar hasta la temperatura ambiente y se agitó hasta que la reacción se terminó mediante monitorización mediante TLC. La reacción se paralizó con Na2S2O3 acuoso, se agitó durante 4 h, se lavó con NaHCO3 acuoso saturado, y se secó sobre MgSO4 anhidro. La cromatografía en columna produjo 1,8 g de compuesto 1 como un aceite.
Producto intermedio 2
Se disolvió 1 (4 mmoles) en POCE recientemente destilado (4,5 ml). La mezcla de reacción se calentó hasta 80°C durante 17 h. Tras enfriar hasta la temperatura ambiente, el disolvente se eliminó a presión reducida. Se añadió hielo, y la mezcla se dejó reposar durante 4 h. La mezcla se repartió entre CH2O 2 (50 ml) y NaHCO3 acuoso saturado. La cromatografía en columna produjo el compuesto 2 como un aceite amarillo (rendimiento: 50%).
Producto intermedio 4
A la disolución de 2 (2 g, 9,26 mimóles) en etanol (30 ml) se le añadió hidrato de hidrazina (2,9 g, 46 mimóles). La mezcla de reacción se agitó durante 4 h a temperatura ambiente, después se concentró para eliminar el disolvente, y se añadió acetato de etilo, se lavó con agua. La capa orgánica se secó sobre Na2SO4 anhidro. La cromatografía en columna (sílice, éter de petróleo/acetato de etilo = 4/1 ~3/1) produjo el compuesto 4 como un sólido blanco (rendimiento: 56%) y otro isómero 3 (rendimiento: 18%).
Compuesto S00109
El procedimiento sintético fue similar al procedimiento general.

Compuesto S00186 (solo para referencia)
El material de partida (anhídrido) estaba comercialmente disponible, de manera que la ruta sintética del compuesto S00186 fue similar al procedimiento general (el anhídrido reacciona con el producto intermedio 4).
Compuesto S00994
Esquema 3

Producto intermedio 5
1 (4 mmoles) se disolvió en POBr3 recientemente destilado (4,5 ml). La mezcla de reacción se calentó hasta 80°C durante 17 h. Tras enfriar hasta la temperatura ambiente, el disolvente se eliminó a presión reducida. Se añadió hielo, y la mezcla se dejó reposar durante 4 h. La mezcla se repartió entre CH2Ch (50 ml) y NaHCO3 acuoso saturado. La cromatografía en columna produjo el compuesto 5 como un aceite amarillo (rendimiento: 50%).
Producto intermedio 6
Se añadió hidrato de hidrazina a la disolución de 5 en etanol. La mezcla de reacción se agitó durante 4 h a temperatura ambiente, después se concentró para eliminar el disolvente, se añadió acetato de etilo, y la mezcla se lavó con agua. La capa orgánica se secó sobre Na2SO4 anhidro. La cromatografía en columna (sílice, éter de petróleo/acetato de etilo = 4/1 ~3/1) produjo el compuesto 6 como un sólido blanco.
Compuesto S00994
El procedimiento sintético fue similar al procedimiento general.
C o m p u e s to S 01860
E squ em a 4

Producto intermedio 7
Una disolución de material de partida (5,0 g, 0,040 moles), NBS (10,6 g, 0,059 moles), BPO (296 mg) en 300 ml de CCI4 se agitó a reflujo durante 5 h. La mezcla de reacción se enfrió después hasta la temperatura ambiente, y se añadió otra porción de BPO (296 mg), y la reacción se agitó a reflujo durante otras 5 h. Después la mezcla de reacción se mantuvo a temperatura ambiente toda la noche. Después se filtró, y el residuo se lavó mediante CCU durante tres (3) veces, y la capa orgánica combinada se lavó con agua y con salmuera, después se secó y se concentró y se purificó mediante cromatografía en columna (PE:EA = 4:1) para proporcionar producto bruto que después se purificó mediante destilación. La segunda fracción obtenida a 128°C~135°C (3 mmHg) fue el producto intermedio 7.
Producto intermedio 8
A la suspensión de hidruro de sodio (60 mg, 1,5 mmoles) en benceno (5 ml), se le añadió gota a gota a temperatura ambiente malonato de dietilo (320 mg, 2,0 mmoles). La mezcla de reacción se agitó durante 5 min., después se añadió una disolución de 7 (210 mg, 1,0 mmol) en benceno (5 ml). La mezcla se agitó a temperatura ambiente durante otras 8 h. Después la mezcla se acidificó con HCl diluido y se extrajo con EtOAc (2*15 ml). Las capas orgánicas combinadas se lavaron con agua, con salmuera y se secaron sobre Na2SO4 anhidro. La concentración de las capas orgánicas a vacío seguido de la purificación cromatográfica en columna en gel de sílice del residuo (éter de petróleo:EtOAc = 4:1) proporcionó el producto como un aceite espeso. El rendimiento fue 200 mg, (74,0%).
Producto intermedio 9
Una disolución de 8 (80 mg, 0,3 mmoles) en hidrocloruro diluido (2 ml, 18%) se puso a reflujo con agitación durante 12 h. La mezcla de reacción se enfrió hasta la temperatura ambiente, y se saturó añadiendo cloruro de sodio sólido. La capa acuosa filtrada se extrajo con EtOAc, se secó sobre Na2SO4 anhidro y se concentró para proporcionar ácido puro. El rendimiento fue 50 mg (90,6%).
Producto intermedio 10
A una disolución agitada de 9 (0,46 g, 2,5 mmoles) y dos gotas de DMF en DCM (10 ml) se le añadió cloruro de oxalilo (0,48 g, 3,75 mmoles) gota a gota. La mezcla se agitó a temperatura ambiente (temperatura del aceite 20-30°C) durante dos horas, después el disolvente se evaporó. Se disolvieron el residuo y ferc-butanol (0,22 g, 3 mmoles) en 10 ml de DCM, se añadió piridina gota a gota a temperatura ambiente (0,3 g, 3,75 mmoles) a esta disolución. La mezcla resultante se agitó a temperatura ambiente durante una hora. Se añadió NH4Cl sat. para detener la reacción, el pH se ajustó a 2 con HCl 1N y se extrajo con acetato de etilo, la capa orgánica combinada se secó sobre Na2SO4, se filtró y se evaporó. El residuo se purificó mediante cromatografía ultrarrápida para proporcionar 10 como un sólido blanco (0,42 g, 70%).
Compuesto S01860
Los Intermedios 10 (119 mg, 0,45 mmoles) y 4 (95 mg, 0,49 mmoles) se añadieron a 5 ml de DCM y se pusieron a reflujo toda la noche, después el disolvente se evaporó, y el residuo se purificó mediante TLC prep. para proporcionar el producto (Rendimiento = 150 mg, 77%).
C o m p u e s to S01861
E squ em a 5

Producto intermedio 11
Se disolvieron 9 (1,0 g, 5,43 mmoles) y 4 (1,15 g, 5,43 mmoles) en 20 ml de cloroformo y se pusieron a reflujo durante 48 h, después se evaporó el disolvente, y el residuo se recristalizó para proporcionar 11 (1,4 g, 68,2%).
Compuesto S01861
El producto intermedio 11 (15 mg, 0,04 mmoles), EDCI (45 mg, 0,24 mmoles), Et3N (1 gota) y etanol (1 ml) se agitaron a temperatura ambiente durante alrededor de 3 h. Después el disolvente se eliminó a vacío. El producto se separó mediante TLC prep. El rendimiento fue 12 mg (76,7%).
Compuestos S01648, S01796, S01711, S01758, S01883, y S01759
La ruta sintética de los compuestos S01648, S01796, S01711, S01758, S01883, y S01759 fue similar a S01861, es decir, el producto intermedio 11 se acopló a diferentes sustancias químicas).
Compuesto S01589 (solo para referencia)
Esquema 6

Producto intermedio 12
La mezcla de material de partida (6,5 g, 28,7 mmoles), ácido malónico (3,3 g, 31,7 mmoles), HOAc (60 ml), NaOAc (2,95 g, 36 mmoles) se agitaron a RT. Tras 6-7 h, se añadió más NaOAc (2,95 g, 36 mmoles), después se puso a reflujo toda la noche. Tras enfriar, la mezcla se filtró, y el filtrado se lavó con agua y con acetato de etilo, después se secó a presión reducida. Se recogieron 5 g de un sólido marrón poco consistente (rendimiento = 65,4%).
Producto intermedio 13
Se añadieron gota a gota cuatro (4) ml de SOCl2 a una suspensión de compuesto 12 y EtOH, en un baño de hielo, y la mezcla se agitó durante 30 min. a temperatura ambiente, después se puso a reflujo durante 6 h. Tras enfriar, la mezcla se filtró y se lavó con EtOH enfriado, y se secó a vacío para obtener 5,25 g un polvo gris pálido (rendimiento = 95%)
Producto intermedio 14
Una mezcla de compuesto 13 y POCI3 (15 ml) se agitó a temperatura ambiente durante 15 min., después se puso a reflujo durante 2 h. La mezcla se concentró a vacío. El residuo se paralizó con agua fría y se extrajo con acetato de etilo, se lavó con NaHCO 3 saturado y con salmuera, se secó sobre MgSO4 , se concentró y se recogieron 4,68 g un sólido marrón poco consistente.
Producto intermedio 15
A una disolución de THF y MeOH, se le añadieron compuesto 14 (4,68 g, 14,9 mmoles) y LiCl con un baño de hielosal, se le añadió NaBH 4 en porciones. Después de la adición, la mezcla de reacción se agitó a temperatura ambiente, se comprobó mediante TLC, se concentró a vacío, y se añadió lentamente HCl diluido al residuo sobre un baño de hielo hasta que la mezcla alcanzó pH 7. Después la mezcla se extrajo con acetato de etilo y se lavó con NaHCO 3 saturado, NH 4 Cl, disoluciones de NaCl (en secuencia), se secó sobre MgSO 4 , se concentró, y se recogieron 4,15 g de un sólido marrón poco consistente.
Producto intermedio 16
El compuesto 15 (4,15 g) se disolvió en SOCl2 y se puso a reflujo toda la noche. El disolvente se evaporó, se añadió agua al residuo, la mezcla se extrajo con acetato de etilo, la capa orgánica combinada se secó sobre Na 2 SO 4 anhidro, el disolvente se evaporó, y se recogieron 4,0 g de compuesto 16.
Producto intermedio 17
El compuesto 16 (200 mg, 0,69 mmoles) se disolvió en dioxano, y se añadió piperazina anhidra (177 mg, 2,05 mmoles), y la mezcla se agitó toda la noche. La mezcla se filtró, el filtrado se concentró a vacío, y se recogieron 250 mg de producto bruto.
Producto intermedio 18
Una disolución de dicarbonato de di-terc -butilo (0,246 g, 1,13 mmoles) en MeOH se añadió gota a gota al compuesto 17 (0,35 g, 1,03 mmoles) en MeOH a temperatura ambiente.
La mezcla de reacción se agitó toda la noche a temperatura ambiente. El disolvente se evaporó, el residuo se extrajo de la manera habitual como se describió anteriormente, y el extracto se purificó mediante cromatografía en columna (EA:PE=1:10). El producto se obtuvo como un sólido blanco.
Compuesto S01589 (solo para referencia)
El procedimiento sintético del producto intermedio 18 al compuesto S01589 fue similar al procedimiento general descrito en la presente memoria.
Compuestos S01037 y S01047 (solo para referencia)

Compuesto S01037 (solo para referencia)
El procedimiento sintético del producto intermedio 15 al compuesto S01037 es similar al procedimiento general. Compuesto S01047 (solo para referencia)
El material de partida (0,145 g, 0,54 mmoles) se disolvió en 5 ml de ácido acético, y la mezcla se calentó con reflujo durante 1 h, después se evaporó y se purificó mediante TLC prep. (éter de petróleo: acetato de etilo = 1:1) para proporcionar el producto.
C o m p u e s to S 01879 (so lo para re fe re nc ia )
E sq u e m a 8

El material de partida (50 mg, 0,09 mmoles) se disolvió en 5 ml de CH2CI2, y se añadió TFA (5 ml) gota a gota a la mezcla agitada sobre un baño de hielo. La mezcla resultante se agitó durante 1 h a temperatura ambiente y se comprobó mediante TLC. El disolvente se evaporó para proporcionar el producto como un sólido amarillo que se usó sin purificación adicional (40 mg).
Compuesto S01879
El compuesto 20 se disolvió en MeCN, y se añadió K2CO3 (3 eq.), después de lo cual, la mezcla se agitó durante alrededor de 30 min., y se añadió ácido benzoico (1 eq.) y EDCI (2 eq.), y la mezcla se agitó toda la noche, después se concentró y se trató de la manera habitual descrita anteriormente. La preparación final se purificó mediante TLC de placa prep., y el producto se obtuvo como un sólido amarillo poco consistente.
Compuestos S01925, S01878, S01877, S01699, S01800, S01801, S01822, S01880, S01683, S01928, S01929 (solo para referencia)
La ruta sintética de los compuestos S01925, S01878, S01877, S01699, S01800, S01801, S01822, S01880, S01683, S01928, S01929 fue similar a la de S01879 (el producto intermedio 20 se acopló con químicas diferentes).
Compuesto S01981
Esquema 9

El material de partida (100 mg, 0,314 mmoles) se disolvió en CCl 4 , se añadieron NBS (112 mg, 0,629 mmoles) y BPO (1,5 mg, 0,0062 mmoles), y la mezcla se puso a reflujo durante alrededor de 4 h. La mezcla de reacción se paralizó con agua, se extrajo con acetato de etilo, la capa orgánica se lavó con salmuera, se secó sobre MgSO 4 y se concentró a vacío, después se purificó mediante placa prep. para obtener el producto.
Compuesto S00170
Esquema 10

Se añadió NaH (8 mg, 0,12 mimóles) gota a gota a una disolución de hidrazina (35 mg, 0,117 mimóles) en THF (6 ml), a 0°C. La mezcla se agitó durante 30 min., después se añadió Mel (20 mg). La mezcla de reacción se agitó durante 2 h a temperatura ambiente, después se vertió en el NH4Cl ac. sat.; se extrajo con CHC13. La capa orgánica se secó sobre Na2SO4, después se cromatografió (PE/AE, 5/1) para obtener el producto (3 mg).
Compuestos S01007, S01473
La ruta sintética de los compuestos S01007, S01473 fue similar a la de S00170.
Compuesto S01470

Producto Intermedio 21
El compuesto 2 (1 g, 4,9 mmoles) se añadió a una disolución enfriada con hielo de KOH ac. 4N (5 ml), y la mezcla se agitó a temperatura ambiente durante 5 h. La mezcla se acidificó lentamente con H2SO46N (5 ml), después se saturó con NaCl sólido y se agitó a temperatura ambiente durante 30 min. La capa acuosa se extrajo con acetato de etilo, y la capa orgánica se lavó con salmuera y se secó. La capa orgánica se concentró a vacío, y el concentrado se aplicó a de gel sílice (PE:EA=1:1) para proporcionar 355 mg de producto.
Compuesto S01470
El procedimiento sintético fue similar al procedimiento general.
Compuestos S01599 y S01600
Esquema 12
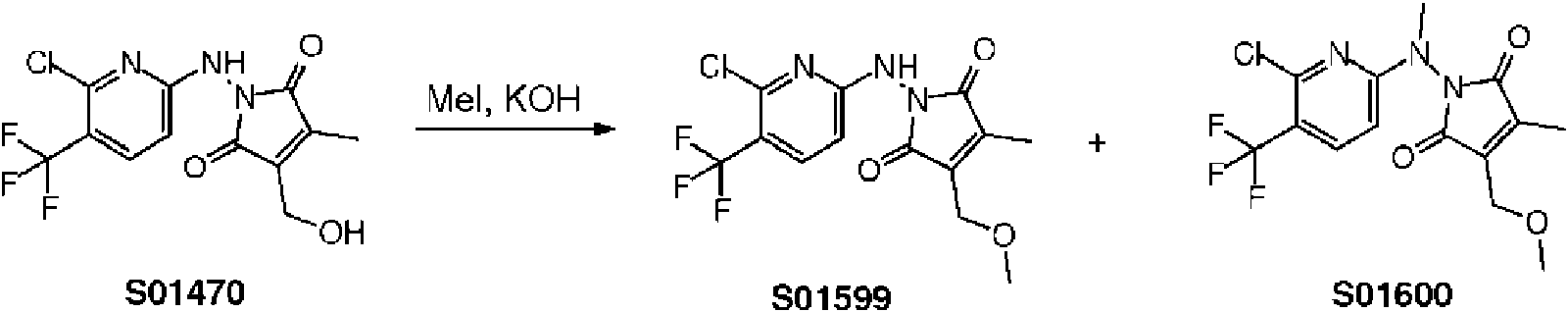
El material de partida (80 mg, 0,24 mmoles), Mel (40 ul, 0,64 mmoles), y KOH (30 mg, 0,54 mmoles) en DMSO (5 ml) se agitaron a temperatura ambiente durante 1 h, después se diluyeron con EtOAc, se lavaron con agua, con salmuera, se secaron sobre Na2SO4 anhidro. El disolvente se eliminó a vacío, y el residuo se purificó mediante TLC prep. para obtener los dos compuestos diana.
Compuesto S01712

El procedimiento sintético fue similar al procedimiento general.
Compuesto S01266
Esquema 14

Se añadió Pd(PPh3)4 (16 mg) a una mezcla de material de partida (50 mg, 0,14 mmoles), ácido bencenoborónico (19 mg, 0,15 mmoles), carbonato de potasio (59 mg, 0,43 mmoles) en 10 ml de tolueno en una atmósfera de nitrógeno. La mezcla resultante se puso a reflujo durante 16 h, después de lo cual el disolvente se evaporó, y el residuo se purificó mediante TLC preparativa para proporcionar 4 mg de producto.
Compuestos S01313, S01457, S01691, S01371, S01393, S01474
La ruta sintética de los compuestos S01313, S01457, S01691, S01371, S01393, S01474 fue similar a la de S01266. Compuesto S01737 (solo para referencia)
Esquema 15

Producto intermedio 22
La piridina (500 mg, 3,4 mmoles) se disolvió en CH2Ch, y se añadió UHP (700 mg, 7,4 mmoles), que se enfrió hasta 0°C, después se añadió lentamente TFAA (1,43g, 6,8 mmoles) a la mezcla de reacción. Después de que la TLC indicó que el material de partida se había consumido, se trató de la manera habitual para producir 420 mg de compuesto diana.
Producto intermedio 23
El compuesto 22 (420 mg, 2,57 mmoles) se disolvió en POCb (3 ml), después se calentó a 90°C toda la noche. La mezcla de reacción se paralizó con agua cuidadosamente, se extrajo mediante CH2Cl2, se lavó con salmuera y se secó sobre MgSO 4 , se concentró a vacío. Se purificó mediante cromatografía en columna (CH 2 Cb:PE=1:3), después se obtuvieron 300 mg de compuesto diana.
Producto intermedio 24
La reacción y el procedimiento de tratamiento fueron los mismos que para el producto intermedio 22, y se obtuvieron 170 mg de compuesto diana.
Producto intermedio 25
La reacción y el procedimiento de tratamiento fueron los mismos que para el producto intermedio 23, y se obtuvieron 120 mg de compuesto diana.
Compuesto S01737 (solo para referencia)
El procedimiento sintético del producto intermedio 25 al compuesto diana fue similar al del procedimiento general. Compuesto S01865 (solo para referencia)
Esquema 16

Producto intermedio 27
Se disolvieron dos materiales de partida en CHCh y se pusieron a reflujo toda la noche, después se concentraron y se purificaron mediante cromatografía en columna (EA:PE = 1:1). El producto se obtuvo como un sólido amarillo claro. Compuesto S01865
El compuesto 27 se disolvió en MeOH anhidro, se añadió EDCI, después se agitó toda la noche. Se concentró a vacío, se trató de la manera habitual y se purificó mediante TLC prep. para obtener el producto final como un sólido amarillo claro.
Compuestos S01734 (compuesto de la invención) y S01688 (solo para referencia)
Esquema 17

Producto intermedio 28
A una disolución de material de partida (9,26 g, 0,05 moles), se le añadió UHP (9,9g, 0,105 moles). Con un baño de hielo, se añadió gota a gota TFAA (21 g, 0,100 moles). Después de la adición, la reacción se mantuvo a temperatura ambiente durante 4 h. La reacción se neutralizó con Na 2 CO 3 (ac.), y la mezcla se extrajo con DCM 3 veces. La capa orgánica se recogió, se secó y se concentró y se purificó mediante cromatografía ultrarrápida (PE:EA = 3:1) para proporcionar el producto puro 8,1 g.
Producto intermedio 29
Una disolución de compuesto 28 (0,8 g, 4,07 mmoles) y en 2 ml de Na 2 CO 3 (ac. 2N) y 3 ml de tolueno se agitó en una atmósfera de N2 y a temperatura ambiente. Después se añadió Pd(PPh 3 ) 4 . La mezcla se agitó a reflujo a una atmósfera de N2 durante 3 h. Después el disolvente se eliminó a vacío. El residuo se trató con agua y se extrajo con EA, y la fase orgánica se recogió, se secó y se concentró para purificarlo mediante recristalización para proporcionar 0,75 g de polvo amarillo pálido.
Producto intermedio 31
Una disolución de compuesto 29 (0,75 g, 3,15 mmoles) en 5 ml de POCb, y la mezcla se agitó a reflujo durante 5 h. Después la mezcla de reacción se vertió en hielo, y la capa acuosa se extrajo con acetato de etilo 3 veces. Después la fase orgánica se recogió, se lavó con disolución acuosa de Na 2 CO 3 y después se secó, se concentró para purificarlo mediante cromatografía en columna para producir 800 mg de producto intermedio 30, que se disolvió en 5 ml de DCM y UHP, seguido de adición de TFAA a la mezcla anterior en un baño de hielo. Después, la mezcla de reacción se agitó a temperatura ambiente toda la noche. Después la mezcla de reacción se neutralizó mediante disolución acuosa de Na 2 CO 3 , y la fase acuosa se extrajo mediante DCM 3 veces. La fase orgánica se recogió, se secó, se concentró y se purificó mediante cromatografía en columna (PE:EA = 5:1) para proporcionar 350 mg de compuesto 31 puro.
Producto intermedio 32
Una disolución de compuesto 31 (350 mg, 1,28 mmoles) en 5 ml POCl 3 se agitó a reflujo durante 4 h. Después la mezcla se vertió en agua con hielo y se extrajo con acetato de etilo. La capa orgánica se lavó con disolución acuosa de Na 2 CO 3 y se secó, se concentró y se purificó para proporcionar 180 mg de compuesto 32 puro.
C o m p u e s to s S 01734 y S 01688
El procedimiento sintético del producto intermedio 32 a los compuestos diana fue similar al del procedimiento general. Compuesto S01864 (solo para referencia)

Producto intermedio 34
El material de partida y el producto intermedio 15 se disolvieron en acetonitrilo y se agitaron a temperatura ambiente toda la noche, después se filtraron, y el disolvente se evaporó. El residuo se purificó mediante TLC preparativa para producir el producto.
Compuesto S01864
El procedimiento sintético del producto intermedio 34 al compuesto diana es similar al procedimiento general.
Compuestos S01268 y S01862 (solo para referencia)
La ruta sintética para los compuestos S01268 y S01862 fue similar a la del compuesto S01864
Compuesto S01475
Esquema 19

Producto intermedio 35
Se añadió cianuro de trimetilsililo (7,44 g, 75 mmoles, 10 ml) a una disolución agitada de producto intermedio 35 (5,92 g, 30 mmoles) y TEA (4,55 g, 45 mmoles, 6,3 ml) en 25 ml de acetonitrilo a temperatura ambiente. Después la mezcla se calentó hasta 110°C (temperatura del baño de aceite) durante 12 h, se enfrió hasta la temperatura ambiente, y el disolvente se evaporó. Se añadieron DCM y NaHCO3 (ac.) saturado, y las capas se separaron. La capa orgánica se secó sobre Na2SO4 anhidro y se evaporó. El residuo se lavó con éter y se filtró, después se evaporó para proporcionar el producto bruto como a aceite negro que después se purificó mediante cromatografía ultrarrápida para proporcionar el producto como un aceite amarillo. El rendimiento fue 4,4 g (71%).
Compuesto S01475
El procedimiento sintético del producto intermedio 35 al Compuesto S01475 fue similar al del procedimiento general.
C o m p u e s to S 01762
Esquema 20

Producto intermedio 36
Se añadió hidruro de sodio (0,264 g, 6,6 mmoles, 60%) a una disolución agitada de cianuro de bencilo (0,645 g, 5,5 mmoles) en 10 ml de DMF a temperatura ambiente. Se añadió material de partida (1,0 g, 5,5 mmoles) a la mezcla tras 30 min., y la mezcla resultante se agitó a temperatura ambiente durante 2 h. Se añadió salmuera para detener la reacción, y la mezcla se extrajo con acetato de etilo. La capa orgánica combinada se secó sobre sulfato de sodio anhidro y se evaporó. El residuo se purificó mediante cromatografía ultrarrápida (se eluyó con éter de petróleo:acetato de etilo = 8:1 hasta 5:1) para proporcionar 0,475 g de producto (rendimiento = 33%).
Producto intermedio 37
El producto intermedio 36 (0,15g, 0,57 mmoles) se mezcló con 5 ml de HCl concentrado y se puso a reflujo toda la noche. Después la mezcla se enfrió hasta la temperatura ambiente, se añadieron 15 ml de agua, el pH se ajustó hasta 8-9 con carbonato de sodio, y la mezcla se extrajo con acetato de etilo (10 ml). La capa orgánica combinada se secó sobre sulfato de sodio anhidro y se evaporó para proporcionar 0,14 g gramos de producto (rendimiento = 100%), que se usó sin purificación adicional para la siguiente etapa.
Producto intermedio 38
El producto intermedio 37 (0,14 g, 0,57 mmoles) se disolvió en 5 ml de DCM, después se añadió UHP (0,17 g, 1,77 mmoles) y después de eso, se añadió gota a gota TFAA (0,36 g, 1,71 mmoles, 0,24 ml) con enfriamiento en un baño de hielo. Después la mezcla se calentó hasta la temperatura ambiente y se agitó toda la noche a la misma temperatura. Se añadieron cinco (5) ml de agua, y la mezcla se neutralizó con carbonato de sodio hasta pH 8-9 y después se extrajo con DCM. La capa orgánica combinada se secó sobre sulfato de sodio anhidro y se evaporó para proporcionar 0,14 g del producto bruto (rendimiento = 95%), que se usó sin purificación adicional para la siguiente etapa.
Producto intermedio 39
El producto intermedio 38 (0,14 g, 0,55 mmoles) se disolvió en 5 ml de POCb, y la mezcla se calentó hasta 80-90°C durante 2 h. Después la mezcla se enfrió hasta la temperatura ambiente, se vertió en agua con hielo, y se extrajo con acetato de etilo. La capa orgánica combinada se lavó con NaHCO3 sat., se secó sobre Na2SO4 y se evaporó. El residuo se purificó mediante TLC preparativa para producir 0,13 g de producto (rendimiento = 87%).
Compuesto S01762
El procedimiento sintético del producto intermedio 39 al compuesto S01762 fue similar al del procedimiento general.
Compuesto S01820 (solo para referencia)
Esquema 21

Una disolución de S01470 en 3 ml de DCM se agitó a temperatura ambiente, y se añadió Et3N. Después se añadió AC2O en un baño de hielo. La mezcla de reacción se calentó hasta la temperatura ambiente y se agitó toda la noche. La reacción se paralizó después y se trató de la manera habitual como se describió anteriormente. El residuo se purificó mediante TLC prep. (PE:EA = 3:1) para proporcionar el compuesto S01820 puro.
Compuesto S00935 (solo para referencia)
Esquema 22

Producto intermedio 40
Se añadió material de partida (9,08 g, 84,4 mmoles) a 19,2 ml de malonato de dietilo, la mezcla se calentó hasta 150°C (temperatura del baño de aceite) durante 6 h, se evaporó, se filtró y se lavó con acetato de etilo para proporcionar 3,7 g de sólido blanco; éste era el producto intermedio 41 (se comprobó mediante LC-MS), el filtrado se evaporó, el residuo se enfrió para producir un segundo lote sólido, se lavó con una disolución de éter de petróleo:acetato de etilo igual a 5:1, se comprobó mediante LC-MS; éste era el producto intermedio 40 (5,42 g).
Producto intermedio 42
A una disolución agitada de producto intermedio 40 (5,42 g, 24,5 mmoles) en THF, se le añadieron 60 ml de LiOH 2N, y la mezcla resultante se agitó a temperatura ambiente durante 3 h. El disolvente se evaporó, el residuo se lavó con acetato de etilo, se filtró, y la torta se añadió a 10 ml de HCl concentrado y se agitó durante 30 min., después la torta se filtró y se secó para proporcionar 3,1 g de producto.
Producto intermedio 43
El producto intermedio 42 (3,1g, 16 mmoles) se añadió a 20 ml de PPA, y la mezcla se calentó hasta 150°C durante 4 h. La mezcla de reacción se vertió en agua con hielo con agitación, después se filtró, y la torta se lavó con agua y se secó para proporcionar 2,92 g de producto.
Producto intermedio 44
El producto intermedio 43 (0,47 g, 2,7 mimóles) se añadió a 10 ml de POCI3, y la mezcla se calentó con reflujo durante 5 h. La mezcla resultante se enfrió hasta la temperatura ambiente y se vertió en agua con hielo, después se extrajo con acetato de etilo. La capa orgánica combinada se secó sobre Na2SO4, y se evaporó para proporcionar el producto bruto (0,45 g) que se usó sin purificación adicional.
Compuesto S00935
A partir del producto intermedio 43 al compuesto S00935, el procedimiento sintético fue similar al del procedimiento general.
Compuestos S00871, S01005, S01078, S01247, y S01311 (solo para referencia)
La ruta sintética de los compuestos S00871, S01005, S01078, S01247, y S01311 es similar a la del compuesto S00935.
Compuesto S00516

Producto intermedio 45
Se añadió material de partida (0,08 moles) a HCl conc. (40 ml). La mezcla se enfrió hasta -5°C mediante hielo y sal con agitación. Después se añadió nitrito de sodio (5,52 g, 0,08 moles) disuelto en agua (20 ml). La agitación se continuó durante 1 h, y se añadió lentamente cloruro estañoso (30 g) en HCl conc. (30 ml) durante un período de dos horas, mientras se mantiene la temperatura por debajo de 0°C. La mezcla se agitó durante otra hora tras la adición y se filtró. El sólido filtrado se trató con hidróxido de sodio acuoso diluido y después se extrajo con éter. La capa etérea se lavó con agua, se secó sobre Na2SO4 anhidro. El disolvente se eliminó, y el residuo se cristalizó en hexano para proporcionar el Compuesto 45.
Compuesto S00516
El procedimiento sintético es similar al procedimiento general.

Compuestos S00738*, S00832, S00942* (* solo para referencia)
Los materiales de partida están comercialmente disponibles, de manera que la ruta sintética de los compuestos S00738, S00832, S00942 fue similar a la de S00516.
C o m p u e s to S01191 (so lo pa ra re fe re nc ia )
Esquema 24

S01191
49
Producto intermedio 46
Una mezcla de ácido 2-amino-3-clorobenzoico (500 mg, 2,91 mmoles) y anhídrido acético (1,2 ml) se calentó con reflujo durante 1 hora, y el anhídrido acético en exceso se eliminó a vacío. El residuo se enfrió y se trató con éter dietílico para proporcionar un precipitado voluminoso, que se separó por filtración, se lavó con éter frío y se secó para proporcionar 550 mg del producto deseado como un sólido amarillo pálido (rendimiento = 97%).
Producto intermedio 47
En un matraz de tres bocas, que se había secado en un horno e inundado con N2, se añadió una pequeña cantidad de I2 a una mezcla de magnesio (59 mg, 2,47 mmoles) en 0,5 ml de THF seco. Cuando la mezcla de reacción se volvió incolora, se añadió a la mezcla una disolución de 4-bromoanisol (440 mg, 2,35 mmoles) en 1,5 ml de THF seco. La mezcla de reacción se agitó a temperatura ambiente hasta que el Mg se eliminó.
El reactivo de Grignard del 4-bromoanisol en 2 ml de THF se trató con compuesto 46 (460 mg, 2,35 mmoles) en 4,5 ml de tolueno seco a 0°C durante 1 hora y a 30°C durante otra 1 hora. La disolución se acidificó cuidadosamente con ácido sulfúrico diluido, y se lavó con NaHCO3 acuoso y agua. La capa orgánica se secó sobre Na2SO4 anhidro y se evaporó para proporcionar un aceite. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 4:1) para proporcionar 450 mg del producto deseado como un sólido marrón pálido (rendimiento = 63%).
Producto intermedio 48
Una mezcla de compuesto 47 (400 mg, 1,32 mmoles), NaH (60% en aceite, 316 mg, 13,20 mmoles) en 1 ml de DMSO se calentó a 60-70°C toda la noche. La mezcla de reacción se vertió en agua con hielo y se extrajo con acetato de etilo, después se lavó con agua y con salmuera. La capa orgánica se secó sobre Na2SO4 anhidro y se evaporó hasta sequedad. El residuo se recristalizó en etanol para proporcionar 80 mg del compuesto deseado 48 como un sólido marrón (rendimiento = 21%).
Compuesto S01191
El procedimiento sintético del producto intermedio 48 al compuesto diana fue similar al del procedimiento general.
Compuesto S01553
Esquema 25

Producto intermedio 50
Una disolución de 2-(dimetoxifosforil)butanonato de etilo (1,0 g, 4,0 mmoles) en 1,2-dimetoxietano (5 ml) se añadió a una suspensión agitada de hidruro de sodio en 1,2-dimetoxietano (10 ml). Cuando cesó la evolución de hidrógeno, se añadió a la disolución piruvato de etilo (480 mg, 4,1 mmoles) en 1,2-dimetoxietano (5 ml). La mezcla se agitó a 50°C toda la noche. Después la disolución se diluyó con EtOAc (100 ml), se lavó con agua y con salmuera, y se secó sobre Na2SO4 anhidro. El disolvente se eliminó a vacío, y el residuo se purificó mediante cromatografía para proporcionar el producto. El rendimiento fue 710 mg (87,2%)
Producto intermedio 51
Una disolución de 2-etil-3-metilmaleato de dietilo (75 mg, 0,35 mmoles) en etanol (0,8 ml) se añadió gota a gota a NaOH acuoso (2M, 0,4 ml) gota a gota. La mezcla se agitó a temperatura ambiente durante 30 min., después se diluyó con agua (10 ml) y se lavó con éter (5 ml). La capa acuosa se acidificó con HCl ac. al 5%, después se extrajo con EtOAc. La capa orgánica se lavó con salmuera y se secó sobre Na2SO4 anhidro. El disolvente se eliminó a vacío, y el residuo se purificó mediante cromatografía sobre una columna de gel sílice. El rendimiento fue 41 mg (83,7%) Compuesto S01553
El procedimiento sintético del producto intermedio 51 al compuesto S01553 fue similar al del procedimiento general. Compuesto S01554
Esquema 26
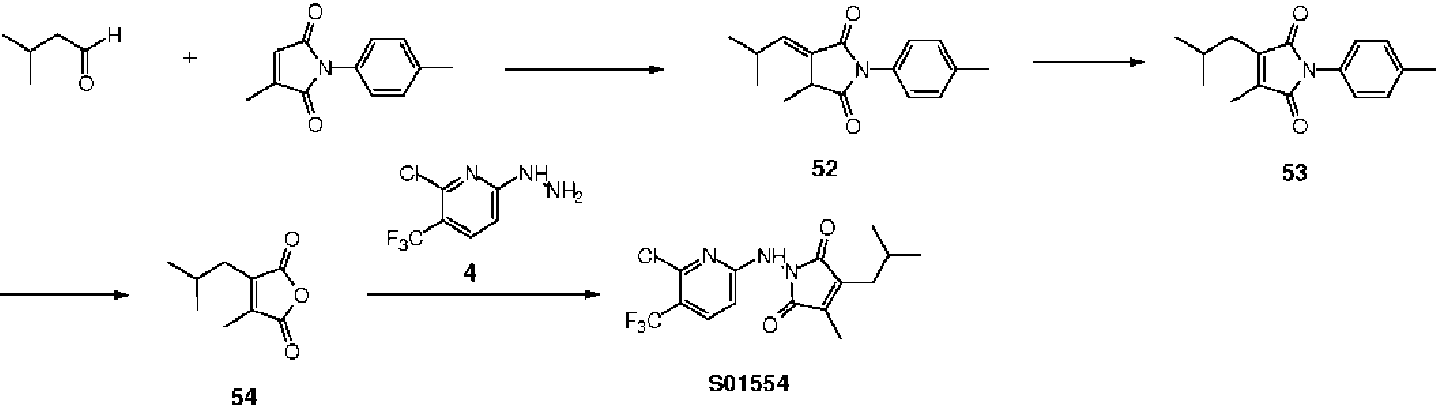
Producto intermedio 52
Una mezcla de citraconimida (200 mg, 1,0 mmol) y PPh3 (320 mg, 1,2 mmoles) en AcOH glacial (7 ml) se agitó a temperatura ambiente durante 1 hora. Se añadió isovaleraldehído (160 |il, 1,5 mmoles), y la mezcla de reacción se puso a reflujo con agitación durante 24 horas. El HOAc se separó mediante destilación a vacío, el residuo se disolvió en EtOAc (30 ml), y la capa orgánica se lavó con H2O, con salmuera, y se secó sobre NaSO4 anhidro. El disolvente se eliminó a vacío, y el residuo se purificó mediante cromatografía sobre una columna de gel sílice. Rendimiento: (90 mg, 35,0%)
Producto intermedio 53
A una disolución agitada de 52 (90 mg) en THF (2 ml) se le añadió Et3N (0,4 ml). La mezcla de reacción se puso a reflujo durante 48 horas, y después se concentró a vacío. El residuo se disolvió en EtOAc, y la capa orgánica se lavó
con agua, con salmuera, y se secó sobre Na2SO4 anhidro. El disolvente se eliminó a vacío, y el residuo se purificó mediante cromatografía sobre una columna de gel sílice. Rendimiento: (85 mg, 94,4%).
Producto intermedio 54
A la disolución de 53 (50 mg, 0,19 mmoles) en THF (0,3 ml) y MeOH (0,6 ml) se le añadió KOH ac. (1 ml, 30%), y la mezcla de reacción se puso a reflujo durante 12 horas con agitación. Después la mezcla de reacción se concentró a vacío, el residuo obtenido se acidificó con HCl ac. diluido y se extrajo con EtOAc (20 ml). La capa orgánica se lavó con agua, con salmuera y se secó sobre Na2SO4 anhidro. El disolvente se eliminó a vacío, y el residuo se purificó mediante cromatografía sobre una columna de gel sílice. Rendimiento: (26 mg, 81,3%).
Compuesto S01554
El procedimiento sintético del producto intermedio 54 al compuesto S01554 fue similar al del procedimiento general. Compuesto S00873 (solo para referencia)
Esquema 27

Producto intermedio 55
A una disolución del éster (5,46 mmoles) y trietilamina (101 g, 10,86 mmoles) en tolueno (5 ml) se le añadió una disolución de anilina (6,52 mmoles) en tolueno (2 ml) a temperatura ambiente. La mezcla de reacción se puso a reflujo hasta que se terminó la reacción. Tras el tratamiento, se obtuvo el compuesto 56, que fue suficientemente puro para usarlo en la siguiente etapa.
Producto intermedio 57
La mezcla de 55 y POCl3 (5 ml) se puso a reflujo durante 5 h, y después se vertió en el agua con hielo. El extracto etéreo se lavó con salmuera y se secó sobre Na2SO4 anhidro, y después se concentró para producir el compuesto 56, que se usó directamente en la siguiente etapa.
La mezcla de 56 e hidrato de hidrazina en 5 ml de etanol se puso a reflujo durante varias horas hasta que el material de partida desapareció. Tras el tratamiento, se obtuvo el compuesto 57.
Compuesto S00873
El procedimiento sintético del producto intermedio 57 al compuesto S00873 fue similar al del procedimiento general. Compuesto S01455 (solo para referencia)
La ruta sintética del compuesto S01455 es similar a la del compuesto S00873.
Claims (12)
1. Compuesto de estructura (IV) o estructura (VI):

en el que:
uno de R1 y R2 es metilo y el otro de R1 y R2 es alquilo, o alquilo sustituido con alcoxi, hidroxi, carboxi, alcoxicarbonilo, carbamoílo opcionalmente sustituido, o aminocarbonilo cíclico opcionalmente sustituido;
R3 es H, o alquilo;
R4 y R5 se seleccionan independientemente de entre H y alquilo;
A es N o CH;
R6, R7, y R9 se seleccionan independientemente de entre H, alquilo, alquilo sustituido, halógeno, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, alcoxi opcionalmente sustituido, ariloxi opcionalmente sustituido, ciano, nitro, alquiltio opcionalmente sustituido, alquilsulfinilo opcionalmente sustituido, alquilsulfonilo opcionalmente sustituido, ariltio opcionalmente sustituido, acilo opcionalmente sustituido, amino opcionalmente sustituido, carboxilo, alcoxicarbonilo opcionalmente sustituido, o carbamoílo opcionalmente sustituido; y
R8 es halógeno o un grupo alquilo sustituido con uno o más halógenos;
o una sal del mismo;
en el que además el compuesto no presenta la estructura siguiente:

2. Compuesto según la reivindicación 1, en el que el compuesto es de estructura (VI):

o una sal del mismo.
3. Compuesto según la reivindicación 1 o 2, en el que el compuesto se selecciona de entre:

, y

, o una sal del mismo.
4. Compuesto según la reivindicación 1, en el que el compuesto es de estructura (IV):

o una sal del mismo.
5. Compuesto según la reivindicación 1 o 4, en el que el compuesto se selecciona de entre:

o una sal del mismo.
6. Compuesto según la reivindicación 1 o 4, en el que el compuesto es:
3-[(3,3-dimetilbutoxi)metil]-1-{[6-doro-5-(trifluorometil)(2-piridil)]amino}-4-metilazolin-2,5-diona (S03747), o una sal del mismo, que presenta la estructura siguiente:

7. Compuesto según la reivindicación 1 o 4, en el que el compuesto es:
1-{[6-cloro-5-(trifluorometil)(2-piridil)]amino}-3,4-dimetilazolin-2,5-diona, o una sal del mismo, que presenta la estructura siguiente:

8. Composición farmacéutica que comprende el compuesto según una cualquiera de las reivindicaciones 1 a 7, o una sal del mismo, en combinación con un excipiente farmacéuticamente aceptable.
9. Compuesto según una cualquiera de las reivindicaciones 1 a 7, o una sal del mismo, o composición farmacéutica según la reivindicación 8 para la utilización en un método para tratar un trastorno de proliferación celular en un sujeto que lo necesita.
10. Utilización del compuesto según una cualquiera de las reivindicaciones 1 a 7, o una sal del mismo, en la preparación de un medicamento para el tratamiento de un trastorno de proliferación celular.
11. Compuesto o composición farmacéutica para la utilización según la reivindicación 9 o la utilización según la reivindicación 10, en el/la que el trastorno de proliferación celular es el cáncer, opcionalmente en el que el cáncer es linfoma, mieloma o leucemia.
12. Compuesto o composición farmacéutica para la utilización según la reivindicación 9 o la utilización según la reivindicación 10, en el/la que el compuesto o la composición es para la administración con por lo menos un tratamiento anticanceroso adicional, en el/la que el tratamiento anticanceroso es opcionalmente un agente que daña el ADN o un tratamiento que daña el ADN.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US91125807P | 2007-04-11 | 2007-04-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2927954T3 true ES2927954T3 (es) | 2022-11-14 |
Family
ID=39939984
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19167943T Active ES2927954T3 (es) | 2007-04-11 | 2008-04-11 | Compuestos 2,5-dioxo-azolina N-sustituidos para la utilización en el tratamiento del cáncer |
| ES16165912T Active ES2732230T3 (es) | 2007-04-11 | 2008-04-11 | Compuestos que anulan el punto de control G2 del ciclo celular para una utilización en el tratamiento del cáncer |
| ES08829410.3T Active ES2594704T3 (es) | 2007-04-11 | 2008-04-11 | Compuestos con actividad anticancerosa |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16165912T Active ES2732230T3 (es) | 2007-04-11 | 2008-04-11 | Compuestos que anulan el punto de control G2 del ciclo celular para una utilización en el tratamiento del cáncer |
| ES08829410.3T Active ES2594704T3 (es) | 2007-04-11 | 2008-04-11 | Compuestos con actividad anticancerosa |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US8084454B2 (es) |
| EP (4) | EP3088397B1 (es) |
| JP (2) | JP5635396B2 (es) |
| KR (1) | KR101475311B1 (es) |
| CN (2) | CN105175394B (es) |
| AU (1) | AU2008294410B2 (es) |
| BR (2) | BRPI0810911B1 (es) |
| CA (4) | CA3188320A1 (es) |
| DK (2) | DK2152692T3 (es) |
| ES (3) | ES2927954T3 (es) |
| HU (2) | HUE029370T2 (es) |
| IL (4) | IL289165B2 (es) |
| MX (3) | MX391154B (es) |
| NZ (1) | NZ580237A (es) |
| RU (1) | RU2482111C2 (es) |
| WO (1) | WO2009031040A2 (es) |
| ZA (1) | ZA200906960B (es) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3088397B1 (en) * | 2007-04-11 | 2019-04-10 | Canbas Co. Ltd. | Compounds that abrogate the cell cycle g2 checkpoint for use in the treatment of cancer |
| EP2304436A1 (en) | 2008-07-10 | 2011-04-06 | Nodality, Inc. | Methods for diagnosis, prognosis and treatment |
| EP2393488B1 (en) * | 2009-02-06 | 2019-06-19 | University Of Southern California | Therapeutic compositions comprising monoterpenes |
| US20100215644A1 (en) * | 2009-02-25 | 2010-08-26 | Nodality, Inc. A Delaware Corporation | Analysis of nodes in cellular pathways |
| EP2542082B8 (en) | 2010-03-03 | 2020-06-17 | Neonc Technologies Inc. | PHARMACEUTICAL COMPOSITIONS COMPRISING (S)-perillyl alcohol |
| US20160038600A1 (en) | 2012-08-03 | 2016-02-11 | Neonc Technologies Inc. | Pharmaceutical compositions comprising poh derivatives |
| DK3685835T3 (da) | 2010-08-27 | 2025-07-21 | Univ Southern California | Farmaceutiske sammensætninger, der omfatter derivater af perillylalkohol |
| ES3056006T3 (en) | 2010-12-17 | 2026-02-17 | Univ Southern California | Methods and devices for using isoperillyl alcohol |
| WO2013020024A2 (en) * | 2011-08-03 | 2013-02-07 | Karyopharm Therapeutics, Inc. | Maleimide compounds and methods of treatment |
| US9814693B2 (en) | 2012-05-09 | 2017-11-14 | Delmar Pharmaceuticals, Inc. | Veterinary use of dianhydrogalactitol, diacetyldianhydrogalactitol, and dibromodulcitol to treat malignancies |
| IN2014MN02512A (es) | 2012-06-14 | 2015-07-17 | Daiichi Sankyo Co Ltd | |
| WO2014118186A1 (en) * | 2013-01-30 | 2014-08-07 | Bayer Pharma Aktiengesellschaft | Amino-substituted isothiazoles |
| US9540462B2 (en) | 2014-12-05 | 2017-01-10 | Massachusetts Institute Of Technology | Catechol-rich polymers from N-substituted maleimides |
| FI3250550T3 (fi) | 2015-01-26 | 2023-07-17 | Ottawa Hospital Res Inst | Koostumuksia ja menetelmiä virusherkistymiseen |
| JP6674957B2 (ja) | 2015-02-12 | 2020-04-01 | ネオンク テクノロジーズ インク. | ペリリルアルコール誘導体を含む医薬組成物 |
| CN109071444A (zh) | 2016-03-16 | 2018-12-21 | 拜耳作物科学股份公司 | 作为农药和植物保护剂的n-(氰苄基)-6-(环丙基-羰基氨基)-4-(苯基)-吡啶-2-羧酰胺衍生物及相关化合物 |
| USD801721S1 (en) | 2016-03-31 | 2017-11-07 | Patricia Grayson Briden | Spooled item holding device |
| USD915100S1 (en) | 2016-03-31 | 2021-04-06 | Ocean 22, Llc | Spooled item holding device |
| TW201811766A (zh) | 2016-08-29 | 2018-04-01 | 瑞士商諾華公司 | N-(吡啶-2-基)吡啶-磺醯胺衍生物及其用於疾病治療之用途 |
| JP2021512165A (ja) | 2018-01-29 | 2021-05-13 | コグノス・セラピューティクス・インコーポレイテッド | ボルテゾミブの腫瘍内送達 |
| MX2020007994A (es) | 2018-02-02 | 2020-09-09 | Boehringer Ingelheim Int | Derivados de triazolopirimidina para usar como inhibidores de ghrelin o-aciltransferasa (goat). |
| WO2019149657A1 (en) | 2018-02-02 | 2019-08-08 | Boehringer Ingelheim International Gmbh | Benzyl-, (pyridin-3-yl)methyl- or (pyridin-4-yl)methyl-substituted oxadiazolopyridine derivatives as ghrelin o-acyl transferase (goat) inhibitors |
| CN111936125B (zh) | 2018-02-08 | 2025-03-04 | 南加州大学 | 穿透血脑屏障的方法 |
| DK4153599T3 (da) | 2020-05-22 | 2024-06-17 | Boehringer Ingelheim Int | Fremgangsmåde til at fremstille alkyl 7-amino-5-methyl-[1,2,5]oxadiazolo[3,4-b]pyridin-carboxylat |
| ES2991340T3 (es) | 2020-05-22 | 2024-12-03 | Boehringer Ingelheim Int | Procedimiento continuo de fabricación de 7-amino-5-metil-[1,2,5]oxadiazolo[3,4-b]piridín-carboxilato de alquilo |
| EP4415700A4 (en) * | 2021-10-15 | 2025-12-24 | Univ Missouri | P1B TYPE ATPASES INHIBITORS |
| CN114425054A (zh) * | 2022-01-20 | 2022-05-03 | 中国人民解放军军事科学院军事医学研究院 | 一种蛋白酶体抑制剂Bortezomib在抗辐射损伤中的用途 |
| CN120247782B (zh) * | 2025-06-04 | 2025-08-19 | 北京颖泰嘉和生物科技股份有限公司 | 氟吡菌酰胺类化合物及其制备方法 |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5564557A (en) * | 1978-11-08 | 1980-05-15 | Mitsui Toatsu Chem Inc | Diphenyl ether compound and herbicide containing the same |
| IL81307A0 (en) * | 1986-01-23 | 1987-08-31 | Union Carbide Agricult | Method for reducing moisture loss from plants and increasing crop yield utilizing nitrogen containing heterocyclic compounds and some novel polysubstituted pyridine derivatives |
| US5635966A (en) * | 1994-01-11 | 1997-06-03 | Hewlett-Packard Company | Edge feed ink delivery thermal inkjet printhead structure and method of fabrication |
| JPH07224032A (ja) | 1994-02-09 | 1995-08-22 | Mitsui Petrochem Ind Ltd | N−アリールオキシフタルイミド誘導体、その製造方 法及びそれを有効成分として含有する除草剤 |
| JPH07244032A (ja) * | 1994-03-01 | 1995-09-19 | Ishikawajima Harima Heavy Ind Co Ltd | 溶接部検査装置 |
| CA2230896A1 (en) * | 1995-09-01 | 1997-03-13 | Signal Pharmaceuticals, Inc. | Pyrimidine carboxylates and related compounds and methods for treating inflammatory conditions |
| US5935966A (en) * | 1995-09-01 | 1999-08-10 | Signal Pharmaceuticals, Inc. | Pyrimidine carboxylates and related compounds and methods for treating inflammatory conditions |
| CA2245029A1 (en) * | 1998-03-13 | 1999-09-13 | University Of British Columbia | Granulatimide compounds as g2 checkpoint inhibitors |
| US6205557B1 (en) | 1998-06-09 | 2001-03-20 | At&T Corp. | Redundant call processing |
| AU2093400A (en) * | 1998-11-24 | 2000-06-13 | Basf Aktiengesellschaft | Fungicides containing pyrrolidones as their active agents |
| JP4610828B2 (ja) | 1999-09-22 | 2011-01-12 | 株式会社 キャンバス | G2期細胞周期停止の阻害、およびdna損傷剤に対する細胞感作のための組成物および方法 |
| EP1257172B1 (de) | 2000-02-26 | 2004-10-13 | Basf Aktiengesellschaft | Fungizide mittel enthaltend als wirkstoffe pyrrolidone und deren verwendung bei der behandlung von pflanzen |
| JP2002318434A (ja) * | 2001-04-23 | 2002-10-31 | Konica Corp | ハロゲン化銀写真感光材料、処理方法、および画像形成方法 |
| FR2826653B1 (fr) * | 2001-06-29 | 2005-10-14 | Servier Lab | Nouveaux derives de pyrido-pyrido-pyrrolo[3,2-g]pyrrolo [3,4-e]-indole et pyrido-pyrrolo[2,3-a]pyrrolo[3,4-c] carbazole, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| WO2003104181A2 (en) * | 2002-06-06 | 2003-12-18 | Canbas Co. Ltd. | Compounds that abrogate dna-damage-induced cell cycle g2 checkpoint and/or augment the anti-cancer activity of dna-damaging treatments |
| WO2005024755A2 (en) * | 2002-12-31 | 2005-03-17 | Deciphera Pharmaceuticals, Llc. | Medicaments for the treatment of neurodegenerative disorders or diabetes |
| US7572816B2 (en) * | 2003-08-01 | 2009-08-11 | Auspex Pharmaceuticals, Inc. | Therapeutic agents for the treatment of cancer, metabolic diseases and skin disorders |
| US7932281B2 (en) * | 2004-03-10 | 2011-04-26 | Kureha Corporation | Amine-based compound and use thereof |
| EP1730128A1 (en) * | 2004-03-31 | 2006-12-13 | Exelixis, Inc. | Anaplastic lymphoma kinase modulators and methods of use |
| AU2006212951A1 (en) * | 2005-02-08 | 2006-08-17 | Merck & Co., Inc. | Inhibitors of checkpoint kinases |
| JP4976394B2 (ja) * | 2005-08-17 | 2012-07-18 | シェーリング コーポレイション | 新規な高親和性のキノリンベースのキナーゼリガンド |
| CA2627910A1 (en) * | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted cycloalkylpyrrolones as allosteric modulators of glucokinase |
| WO2007114338A1 (ja) * | 2006-03-31 | 2007-10-11 | Takeda Pharmaceutical Company Limited | 酸分泌抑制薬 |
| WO2008110891A2 (en) | 2007-03-09 | 2008-09-18 | Orchid Research Laboratories Limited, | New heterocyclic compounds |
| EP3088397B1 (en) | 2007-04-11 | 2019-04-10 | Canbas Co. Ltd. | Compounds that abrogate the cell cycle g2 checkpoint for use in the treatment of cancer |
-
2008
- 2008-04-11 EP EP16165912.3A patent/EP3088397B1/en active Active
- 2008-04-11 CA CA3188320A patent/CA3188320A1/en active Pending
- 2008-04-11 RU RU2009141619/04A patent/RU2482111C2/ru active
- 2008-04-11 CA CA2684037A patent/CA2684037C/en active Active
- 2008-04-11 BR BRPI0810911-7A patent/BRPI0810911B1/pt active IP Right Grant
- 2008-04-11 IL IL289165A patent/IL289165B2/en unknown
- 2008-04-11 JP JP2010502609A patent/JP5635396B2/ja active Active
- 2008-04-11 ES ES19167943T patent/ES2927954T3/es active Active
- 2008-04-11 NZ NZ580237A patent/NZ580237A/en unknown
- 2008-04-11 CA CA3030510A patent/CA3030510C/en active Active
- 2008-04-11 DK DK08829410.3T patent/DK2152692T3/en active
- 2008-04-11 CA CA2913840A patent/CA2913840C/en active Active
- 2008-04-11 MX MX2020009403A patent/MX391154B/es unknown
- 2008-04-11 EP EP22176177.8A patent/EP4074704A1/en not_active Withdrawn
- 2008-04-11 CN CN201510578313.XA patent/CN105175394B/zh active Active
- 2008-04-11 DK DK19167943.0T patent/DK3567035T3/da active
- 2008-04-11 AU AU2008294410A patent/AU2008294410B2/en active Active
- 2008-04-11 ES ES16165912T patent/ES2732230T3/es active Active
- 2008-04-11 HU HUE08829410A patent/HUE029370T2/hu unknown
- 2008-04-11 IL IL309201A patent/IL309201A/en unknown
- 2008-04-11 EP EP19167943.0A patent/EP3567035B1/en active Active
- 2008-04-11 BR BR122022005149-9A patent/BR122022005149B1/pt active IP Right Grant
- 2008-04-11 ES ES08829410.3T patent/ES2594704T3/es active Active
- 2008-04-11 US US12/082,643 patent/US8084454B2/en active Active
- 2008-04-11 WO PCT/IB2008/003036 patent/WO2009031040A2/en not_active Ceased
- 2008-04-11 CN CN200880019414A patent/CN101720323A/zh active Pending
- 2008-04-11 MX MX2015016591A patent/MX374896B/es unknown
- 2008-04-11 EP EP08829410.3A patent/EP2152692B9/en active Active
- 2008-04-11 KR KR1020097023566A patent/KR101475311B1/ko active Active
- 2008-04-11 MX MX2009011025A patent/MX2009011025A/es unknown
- 2008-04-11 HU HUE19167943A patent/HUE059861T2/hu unknown
-
2009
- 2009-10-06 ZA ZA2009/06960A patent/ZA200906960B/en unknown
- 2009-10-11 IL IL201380A patent/IL201380A0/en active IP Right Grant
-
2010
- 2010-11-19 US US12/950,297 patent/US8415357B2/en active Active
-
2013
- 2013-08-28 JP JP2013176324A patent/JP2014012706A/ja not_active Withdrawn
-
2015
- 2015-12-02 IL IL242883A patent/IL242883B/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2927954T3 (es) | Compuestos 2,5-dioxo-azolina N-sustituidos para la utilización en el tratamiento del cáncer | |
| JP6970802B2 (ja) | 二官能性分子によって標的化タンパク質分解を誘導する方法 | |
| ES3038078T3 (en) | Histone acetylation writer inhibitor development and uses thereof | |
| JP7471232B2 (ja) | Polybromo-1(pbrm1)の小分子分解剤 | |
| ES2940263T3 (es) | Compuestos químicos | |
| ES2660914T3 (es) | Derivados de 6-(5-hidroxi-1H-pirazol-1-il)nicotinamida y su uso como inhibidores de PHD | |
| ES2955132T3 (es) | Compuestos heterocíclicos sustituidos con amina como inhibidores de EHMT2 y métodos de uso de los mismos | |
| ES2402395T3 (es) | Compuestos de carbazol y usos terapéuticos de los compuestos | |
| ES2868748T3 (es) | Compuesto de piridina | |
| ES2661733T3 (es) | Compuesto de pirimidina condensado novedoso o sal del mismo | |
| RS57216B1 (sr) | Hemijske vrste | |
| KR20090054984A (ko) | 인돌 화합물 | |
| JP7626721B2 (ja) | 小分子標的ブロモ/アセチルタンパク質およびその使用 | |
| UA121747C2 (uk) | Фармацевтичні сполуки | |
| PT3004112T (pt) | Derivados de pirazolo-pirrolidin-4-ona e sua utilização no tratamento de doença | |
| JP2019038848A (ja) | 白血病を予防および治療するためのマレイミド誘導体の使用 | |
| JP2019529475A (ja) | Dna損傷剤とdna−pk阻害剤との組合せ物を使用する、がんを処置するための方法 | |
| JP2025522816A (ja) | 選択的ヒストンデアセチラーゼ8(hdac8)ディグレーダおよびその使用方法 | |
| WO2017025493A1 (en) | Quinoline ezh2 inhibitors | |
| ES2725881T3 (es) | Nuevos derivados de imidazolidin-2,4-diona | |
| CN121202853A (zh) | 一类mitf转录因子蛋白降解剂及其用途 | |
| HK1139143B (en) | Compounds with anti-cancer activity |