ES2929829T3 - Alcoxipiridinil indolsulfonamidas sustituidas - Google Patents
Alcoxipiridinil indolsulfonamidas sustituidas Download PDFInfo
- Publication number
- ES2929829T3 ES2929829T3 ES19731283T ES19731283T ES2929829T3 ES 2929829 T3 ES2929829 T3 ES 2929829T3 ES 19731283 T ES19731283 T ES 19731283T ES 19731283 T ES19731283 T ES 19731283T ES 2929829 T3 ES2929829 T3 ES 2929829T3
- Authority
- ES
- Spain
- Prior art keywords
- fluoro
- chloro
- indole
- compounds
- sulfonamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- RYMYQAMZUWJAEO-UHFFFAOYSA-N 1h-indole-2-sulfonamide Chemical class C1=CC=C2NC(S(=O)(=O)N)=CC2=C1 RYMYQAMZUWJAEO-UHFFFAOYSA-N 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 314
- 238000011282 treatment Methods 0.000 claims abstract description 60
- 125000001153 fluoro group Chemical group F* 0.000 claims description 109
- -1 chloro, fluoro, fluoromethyl Chemical group 0.000 claims description 86
- 229910052739 hydrogen Inorganic materials 0.000 claims description 75
- 239000001257 hydrogen Substances 0.000 claims description 72
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 45
- 201000006417 multiple sclerosis Diseases 0.000 claims description 41
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 32
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 32
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 32
- 208000016192 Demyelinating disease Diseases 0.000 claims description 28
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 claims description 27
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 26
- 239000000203 mixture Substances 0.000 claims description 24
- 150000003839 salts Chemical class 0.000 claims description 20
- 239000012453 solvate Substances 0.000 claims description 17
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 claims description 15
- 239000013078 crystal Substances 0.000 claims description 14
- 125000001424 substituent group Chemical group 0.000 claims description 12
- 238000002560 therapeutic procedure Methods 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 7
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 claims description 5
- 230000001225 therapeutic effect Effects 0.000 claims description 5
- ZCFNZZAPMKVDPL-UHFFFAOYSA-N 6-chloro-7-cyclopropyl-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1C1CC1)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F ZCFNZZAPMKVDPL-UHFFFAOYSA-N 0.000 claims description 4
- ZPDXHGCEVSGAPB-UHFFFAOYSA-N 6-chloro-N-[5-(2,2-difluoroethoxy)-3,6-difluoropyridin-2-yl]-7-fluoro-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1F)S(=O)(=O)NC1=NC(=C(C=C1F)OCC(F)F)F ZPDXHGCEVSGAPB-UHFFFAOYSA-N 0.000 claims description 4
- XKAOGNQFUFTBPW-UHFFFAOYSA-N 6-chloro-N-[5-(2,2-difluoroethoxy)-3-fluoro-6-methoxypyridin-2-yl]-7-fluoro-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1F)S(=O)(=O)NC1=NC(=C(C=C1F)OCC(F)F)OC XKAOGNQFUFTBPW-UHFFFAOYSA-N 0.000 claims description 4
- DEEFSKOIKOVAJK-UHFFFAOYSA-N 6-chloro-N-[5-[2-(difluoromethoxy)ethoxy]-3-fluoro-6-methoxypyridin-2-yl]-7-fluoro-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1F)S(=O)(=O)NC1=NC(=C(C=C1F)OCCOC(F)F)OC DEEFSKOIKOVAJK-UHFFFAOYSA-N 0.000 claims description 4
- NISLMBBOXNSQPA-UHFFFAOYSA-N 6-(difluoromethyl)-7-fluoro-N-[3-fluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound FC(C1=CC=C2C(=CNC2=C1F)S(=O)(=O)NC1=NC=C(C=C1F)OCCF)F NISLMBBOXNSQPA-UHFFFAOYSA-N 0.000 claims description 3
- POIMTPYVVVXCTL-UHFFFAOYSA-N 6-chloro-7-cyclopropyloxy-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1OC1CC1)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F POIMTPYVVVXCTL-UHFFFAOYSA-N 0.000 claims description 3
- KMRUIIHXOAMPII-UHFFFAOYSA-N 6-chloro-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-7-(trifluoromethoxy)-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1OC(F)(F)F)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F KMRUIIHXOAMPII-UHFFFAOYSA-N 0.000 claims description 3
- WGAUJZDIRXCRBO-UHFFFAOYSA-N 6-chloro-N-[5-(2,2-difluoroethoxy)-3-fluoropyridin-2-yl]-7-(difluoromethyl)-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1C(F)F)S(=O)(=O)NC1=NC=C(C=C1F)OCC(F)F WGAUJZDIRXCRBO-UHFFFAOYSA-N 0.000 claims description 3
- JVDCVGYWYJCQLD-UHFFFAOYSA-N 7-chloro-6-(difluoromethyl)-N-[3-fluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC=1C(=CC=C2C(=CNC=12)S(=O)(=O)NC1=NC=C(C=C1F)OCCF)C(F)F JVDCVGYWYJCQLD-UHFFFAOYSA-N 0.000 claims description 3
- GRFBFXQRAXCHKW-UHFFFAOYSA-N 7-chloro-N-[5-(2,2-difluoroethoxy)-3-fluoro-6-methoxypyridin-2-yl]-6-(difluoromethyl)-1H-indole-3-sulfonamide Chemical compound ClC=1C(=CC=C2C(=CNC=12)S(=O)(=O)NC1=NC(=C(C=C1F)OCC(F)F)OC)C(F)F GRFBFXQRAXCHKW-UHFFFAOYSA-N 0.000 claims description 3
- UBYRVMWLCMBENZ-UHFFFAOYSA-N N-[5-(2,2-difluoroethoxy)-3-fluoropyridin-2-yl]-6-(difluoromethyl)-7-fluoro-1H-indole-3-sulfonamide Chemical compound FC(COC=1C=C(C(=NC=1)NS(=O)(=O)C1=CNC2=C(C(=CC=C12)C(F)F)F)F)F UBYRVMWLCMBENZ-UHFFFAOYSA-N 0.000 claims description 3
- JIHQFMCCVHZQFN-UHFFFAOYSA-N 6-chloro-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-7-(difluoromethyl)-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1C(F)F)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F JIHQFMCCVHZQFN-UHFFFAOYSA-N 0.000 claims description 2
- KAGZNZKDTHETGD-UHFFFAOYSA-N 6-chloro-N-[5-(2,2-difluoroethoxy)-3-fluoro-6-methoxypyridin-2-yl]-7-(difluoromethyl)-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1C(F)F)S(=O)(=O)NC1=NC(=C(C=C1F)OCC(F)F)OC KAGZNZKDTHETGD-UHFFFAOYSA-N 0.000 claims description 2
- 150000002431 hydrogen Chemical group 0.000 claims 2
- 101000829770 Homo sapiens Uracil nucleotide/cysteinyl leukotriene receptor Proteins 0.000 abstract description 121
- 102100023407 Uracil nucleotide/cysteinyl leukotriene receptor Human genes 0.000 abstract description 119
- KSAWZZPUQTWENR-UHFFFAOYSA-N N-pyridin-2-yl-1H-indole-2-sulfonamide Chemical class N1C(=CC2=CC=CC=C12)S(=O)(=O)NC1=NC=CC=C1 KSAWZZPUQTWENR-UHFFFAOYSA-N 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 171
- 239000011541 reaction mixture Substances 0.000 description 132
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 116
- 238000000034 method Methods 0.000 description 110
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 106
- 238000006243 chemical reaction Methods 0.000 description 95
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 87
- 230000015572 biosynthetic process Effects 0.000 description 83
- 235000019439 ethyl acetate Nutrition 0.000 description 83
- 238000003786 synthesis reaction Methods 0.000 description 81
- 239000000243 solution Substances 0.000 description 77
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 70
- 201000010099 disease Diseases 0.000 description 69
- 238000005160 1H NMR spectroscopy Methods 0.000 description 55
- 238000004809 thin layer chromatography Methods 0.000 description 55
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 54
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 48
- 239000007787 solid Substances 0.000 description 47
- 239000012043 crude product Substances 0.000 description 44
- 230000023105 myelination Effects 0.000 description 43
- 239000012044 organic layer Substances 0.000 description 43
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 40
- 229910001868 water Inorganic materials 0.000 description 38
- 208000035475 disorder Diseases 0.000 description 37
- 239000000377 silicon dioxide Substances 0.000 description 36
- 230000000694 effects Effects 0.000 description 35
- 238000004440 column chromatography Methods 0.000 description 34
- 239000011734 sodium Substances 0.000 description 31
- 210000004027 cell Anatomy 0.000 description 28
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- 239000003814 drug Substances 0.000 description 26
- 239000002904 solvent Substances 0.000 description 24
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 22
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 22
- 230000002265 prevention Effects 0.000 description 22
- 239000012267 brine Substances 0.000 description 21
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 21
- 238000012360 testing method Methods 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 239000007832 Na2SO4 Substances 0.000 description 20
- 229940079593 drug Drugs 0.000 description 20
- 229910052938 sodium sulfate Inorganic materials 0.000 description 20
- 235000011152 sodium sulphate Nutrition 0.000 description 20
- 238000002600 positron emission tomography Methods 0.000 description 19
- 238000000746 purification Methods 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- 206010012305 Demyelination Diseases 0.000 description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 18
- 230000014509 gene expression Effects 0.000 description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 17
- 239000002609 medium Substances 0.000 description 16
- 210000004248 oligodendroglia Anatomy 0.000 description 16
- 238000003556 assay Methods 0.000 description 15
- 208000024891 symptom Diseases 0.000 description 15
- 239000000556 agonist Substances 0.000 description 14
- 239000000700 radioactive tracer Substances 0.000 description 14
- 238000005481 NMR spectroscopy Methods 0.000 description 13
- 239000005557 antagonist Substances 0.000 description 13
- 239000000706 filtrate Substances 0.000 description 13
- 239000007788 liquid Substances 0.000 description 13
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 11
- 229910052786 argon Inorganic materials 0.000 description 11
- 238000003745 diagnosis Methods 0.000 description 11
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 10
- 229940126585 therapeutic drug Drugs 0.000 description 10
- 208000024827 Alzheimer disease Diseases 0.000 description 9
- 208000008795 neuromyelitis optica Diseases 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 8
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- RMGJCSHZTFKPNO-UHFFFAOYSA-M magnesium;ethene;bromide Chemical compound [Mg+2].[Br-].[CH-]=C RMGJCSHZTFKPNO-UHFFFAOYSA-M 0.000 description 8
- 239000002953 phosphate buffered saline Substances 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 108090000623 proteins and genes Proteins 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 7
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 7
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 210000003169 central nervous system Anatomy 0.000 description 7
- 230000003247 decreasing effect Effects 0.000 description 7
- 238000000132 electrospray ionisation Methods 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 235000018102 proteins Nutrition 0.000 description 7
- OZMLUMPWPFZWTP-UHFFFAOYSA-N 2-(tributyl-$l^{5}-phosphanylidene)acetonitrile Chemical compound CCCCP(CCCC)(CCCC)=CC#N OZMLUMPWPFZWTP-UHFFFAOYSA-N 0.000 description 6
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 6
- HFEREICVZRUIRE-UHFFFAOYSA-N 3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-amine Chemical compound FC=1C(=NC(=C(C=1)OCCF)F)N HFEREICVZRUIRE-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 208000023105 Huntington disease Diseases 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000013262 cAMP assay Methods 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 230000004069 differentiation Effects 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 230000001771 impaired effect Effects 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 230000006698 induction Effects 0.000 description 6
- 230000002757 inflammatory effect Effects 0.000 description 6
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 229940125425 inverse agonist Drugs 0.000 description 6
- 230000000155 isotopic effect Effects 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 238000004949 mass spectrometry Methods 0.000 description 6
- 230000035800 maturation Effects 0.000 description 6
- 238000012544 monitoring process Methods 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 6
- 208000011580 syndromic disease Diseases 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 239000003643 water by type Substances 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- SAYLBLCAXRNUFT-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3,6-difluoropyridin-2-amine Chemical compound FC(COC=1C=C(C(=NC=1F)N)F)F SAYLBLCAXRNUFT-UHFFFAOYSA-N 0.000 description 5
- GDVJSUCUEJJBAG-UHFFFAOYSA-N 6-[bis[(4-methoxyphenyl)methyl]amino]-2,5-difluoropyridin-3-ol Chemical compound COC1=CC=C(C=C1)CN(C1=C(C=C(C(=N1)F)O)F)CC1=CC=C(C=C1)OC GDVJSUCUEJJBAG-UHFFFAOYSA-N 0.000 description 5
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 5
- 108010074922 Cytochrome P-450 CYP1A2 Proteins 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 102000047918 Myelin Basic Human genes 0.000 description 5
- 101710107068 Myelin basic protein Proteins 0.000 description 5
- 208000008589 Obesity Diseases 0.000 description 5
- QSHWIQZFGQKFMA-UHFFFAOYSA-N Porphobilinogen Natural products NCC=1NC=C(CCC(O)=O)C=1CC(O)=O QSHWIQZFGQKFMA-UHFFFAOYSA-N 0.000 description 5
- 208000006011 Stroke Diseases 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 229960005069 calcium Drugs 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- 238000003384 imaging method Methods 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 208000015122 neurodegenerative disease Diseases 0.000 description 5
- 235000020824 obesity Nutrition 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000008194 pharmaceutical composition Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- YPHQRHBJEUDWJW-UHFFFAOYSA-N porphobilinogen Chemical compound NCC1=NC=C(CCC(O)=O)[C]1CC(O)=O YPHQRHBJEUDWJW-UHFFFAOYSA-N 0.000 description 5
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 5
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 4
- BVRCSCXSBQUNGV-UHFFFAOYSA-N 5-[2-(difluoromethoxy)ethoxy]-3-fluoro-6-methoxypyridin-2-amine Chemical compound FC(OCCOC=1C=C(C(=NC=1OC)N)F)F BVRCSCXSBQUNGV-UHFFFAOYSA-N 0.000 description 4
- NAARPSCDUDESEN-UHFFFAOYSA-N 6-[bis[(4-methoxyphenyl)methyl]amino]-5-fluoropyridin-3-ol Chemical compound COC1=CC=C(CN(C2=C(C=C(C=N2)O)F)CC2=CC=C(C=C2)OC)C=C1 NAARPSCDUDESEN-UHFFFAOYSA-N 0.000 description 4
- YKNACVVFQOGNHK-UHFFFAOYSA-N 6-[bis[(4-methoxyphenyl)methyl]amino]-5-methoxypyridin-3-ol Chemical compound COC1=CC=C(CN(C2=C(C=C(C=N2)O)OC)CC2=CC=C(C=C2)OC)C=C1 YKNACVVFQOGNHK-UHFFFAOYSA-N 0.000 description 4
- NYHQPZPFYRHLAV-UHFFFAOYSA-N 6-chloro-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F NYHQPZPFYRHLAV-UHFFFAOYSA-N 0.000 description 4
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- 102000008144 Cytochrome P-450 CYP1A2 Human genes 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 4
- 208000001089 Multiple system atrophy Diseases 0.000 description 4
- 102000006386 Myelin Proteins Human genes 0.000 description 4
- 108010083674 Myelin Proteins Proteins 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- 108010029485 Protein Isoforms Proteins 0.000 description 4
- 102000001708 Protein Isoforms Human genes 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 229960002749 aminolevulinic acid Drugs 0.000 description 4
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 210000005013 brain tissue Anatomy 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 208000026106 cerebrovascular disease Diseases 0.000 description 4
- XTHPWXDJESJLNJ-UHFFFAOYSA-N chlorosulfonic acid Substances OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- KXZHGRCUIXYVFJ-UHFFFAOYSA-N diethylamino thiohypofluorite Chemical compound CCN(CC)SF KXZHGRCUIXYVFJ-UHFFFAOYSA-N 0.000 description 4
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 4
- 239000000975 dye Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 239000008108 microcrystalline cellulose Substances 0.000 description 4
- 229940016286 microcrystalline cellulose Drugs 0.000 description 4
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 4
- 210000005012 myelin Anatomy 0.000 description 4
- 230000004770 neurodegeneration Effects 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical class O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 4
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 4
- 239000002798 polar solvent Substances 0.000 description 4
- 229960004583 pranlukast Drugs 0.000 description 4
- UAJUXJSXCLUTNU-UHFFFAOYSA-N pranlukast Chemical compound C=1C=C(OCCCCC=2C=CC=CC=2)C=CC=1C(=O)NC(C=1)=CC=C(C(C=2)=O)C=1OC=2C=1N=NNN=1 UAJUXJSXCLUTNU-UHFFFAOYSA-N 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 210000000130 stem cell Anatomy 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- RVOIMTSZEHFJRT-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-bromo-7-chloroindole Chemical compound BrC1=CC=C2C=CN(C2=C1Cl)S(=O)(=O)C1=CC=CC=C1 RVOIMTSZEHFJRT-UHFFFAOYSA-N 0.000 description 3
- IEBCORSZRJLOHC-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-chloro-7-(difluoromethyl)indole Chemical compound ClC1=CC=C2C=CN(C2=C1C(F)F)S(=O)(=O)C1=CC=CC=C1 IEBCORSZRJLOHC-UHFFFAOYSA-N 0.000 description 3
- OXFMACOAPXYXBV-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-chloro-7-ethenylindole Chemical compound ClC1=CC=C2C=CN(C2=C1C=C)S(=O)(=O)C1=CC=CC=C1 OXFMACOAPXYXBV-UHFFFAOYSA-N 0.000 description 3
- MGMVWEIATLXAOE-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-chloro-7-fluoroindole Chemical compound C1(=CC=CC=C1)S(=O)(=O)N1C=CC2=CC=C(C(=C12)F)Cl MGMVWEIATLXAOE-UHFFFAOYSA-N 0.000 description 3
- CRNDPRFVKFSNBR-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-chloro-N-[5-(2,2-difluoroethoxy)-3-fluoro-6-methoxypyridin-2-yl]-7-fluoroindole-3-sulfonamide Chemical compound C1(=CC=CC=C1)S(=O)(=O)N1C=C(C2=CC=C(C(=C12)F)Cl)S(=O)(=O)NC1=NC(=C(C=C1F)OCC(F)F)OC CRNDPRFVKFSNBR-UHFFFAOYSA-N 0.000 description 3
- SKETXSLORQFUMK-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-chloroindole-7-carbaldehyde Chemical compound ClC1=CC=C2C=CN(C2=C1C=O)S(=O)(=O)C1=CC=CC=C1 SKETXSLORQFUMK-UHFFFAOYSA-N 0.000 description 3
- DYULLYIODQUXFF-UHFFFAOYSA-N 1-(benzenesulfonyl)-7-bromo-6-chloroindole Chemical compound BrC=1C(=CC=C2C=CN(C=12)S(=O)(=O)C1=CC=CC=C1)Cl DYULLYIODQUXFF-UHFFFAOYSA-N 0.000 description 3
- YKWGDXBSKWAOKE-UHFFFAOYSA-N 1-(benzenesulfonyl)-7-chloro-6-(difluoromethyl)indole Chemical compound C1(=CC=CC=C1)S(=O)(=O)N1C=CC2=CC=C(C(=C12)Cl)C(F)F YKWGDXBSKWAOKE-UHFFFAOYSA-N 0.000 description 3
- MRWVKGDCUMXXAX-UHFFFAOYSA-N 1-(benzenesulfonyl)-7-chloro-6-ethenylindole Chemical compound ClC=1C(=CC=C2C=CN(C=12)S(=O)(=O)C1=CC=CC=C1)C=C MRWVKGDCUMXXAX-UHFFFAOYSA-N 0.000 description 3
- JGNAVUKDIZAWGR-UHFFFAOYSA-N 1-(benzenesulfonyl)-7-chloroindole-6-carbaldehyde Chemical compound C1(=CC=CC=C1)S(=O)(=O)N1C=CC2=CC=C(C(=C12)Cl)C=O JGNAVUKDIZAWGR-UHFFFAOYSA-N 0.000 description 3
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 3
- LVPDSOXLXWWHNG-UHFFFAOYSA-N 1-(difluoromethyl)-2-fluoro-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(C(F)F)=C1F LVPDSOXLXWWHNG-UHFFFAOYSA-N 0.000 description 3
- GXHFOEOQFJGWSJ-UHFFFAOYSA-N 1-chloro-2-cyclopropyloxy-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=C1OC1CC1 GXHFOEOQFJGWSJ-UHFFFAOYSA-N 0.000 description 3
- VOGSDFLJZPNWHY-UHFFFAOYSA-N 2,2-difluoroethanol Chemical compound OCC(F)F VOGSDFLJZPNWHY-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- NNTFJYGYGZAXNQ-UHFFFAOYSA-N 3,6-difluoro-5-(2-fluoroethoxy)-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC=1C(=NC(=C(C=1)OCCF)F)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC NNTFJYGYGZAXNQ-UHFFFAOYSA-N 0.000 description 3
- UUOJLXQZOQEIFZ-UHFFFAOYSA-N 3,6-difluoro-N,N-bis[(4-methoxyphenyl)methyl]-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine Chemical compound FC=1C(=NC(=C(C=1)B1OC(C(O1)(C)C)(C)C)F)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC UUOJLXQZOQEIFZ-UHFFFAOYSA-N 0.000 description 3
- HMRIDZSIODFQSH-UHFFFAOYSA-N 3,6-difluoropyridin-2-amine Chemical compound NC1=NC(F)=CC=C1F HMRIDZSIODFQSH-UHFFFAOYSA-N 0.000 description 3
- LMJAMUQIFVETCK-UHFFFAOYSA-N 3-fluoro-5-(2-fluoroethoxy)pyridin-2-amine Chemical compound FC=1C(=NC=C(C=1)OCCF)N LMJAMUQIFVETCK-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- ADZDOKQAYDNZGM-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3-fluoro-6-methoxypyridin-2-amine Chemical compound FC(COC=1C=C(C(=NC=1OC)N)F)F ADZDOKQAYDNZGM-UHFFFAOYSA-N 0.000 description 3
- ZRKQWKDLSLNNOR-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3-fluoropyridin-2-amine Chemical compound FC=1C(=NC=C(C=1)OCC(F)F)N ZRKQWKDLSLNNOR-UHFFFAOYSA-N 0.000 description 3
- ONNLETGTMVJVBQ-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3-methoxy-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC(COC=1C=C(C(=NC=1)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)OC)F ONNLETGTMVJVBQ-UHFFFAOYSA-N 0.000 description 3
- PIKWQMPZUUMUGZ-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3-methoxypyridin-2-amine Chemical compound FC(COC=1C=C(C(=NC=1)N)OC)F PIKWQMPZUUMUGZ-UHFFFAOYSA-N 0.000 description 3
- FVPOKSLLPNHXNE-UHFFFAOYSA-N 5-(difluoromethoxy)-3-methoxy-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC(OC=1C=C(C(=NC=1)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)OC)F FVPOKSLLPNHXNE-UHFFFAOYSA-N 0.000 description 3
- SBGQSHGPSYRIEJ-UHFFFAOYSA-N 5-(difluoromethoxy)-3-methoxypyridin-2-amine Chemical compound FC(OC=1C=C(C(=NC=1)N)OC)F SBGQSHGPSYRIEJ-UHFFFAOYSA-N 0.000 description 3
- LHDPSCOQRUYGEY-UHFFFAOYSA-N 5-[2-(difluoromethoxy)ethoxy]-3,6-difluoro-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC(OCCOC=1C=C(C(=NC=1F)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)F)F LHDPSCOQRUYGEY-UHFFFAOYSA-N 0.000 description 3
- REHXNANUSIXWCE-UHFFFAOYSA-N 5-[2-(difluoromethoxy)ethoxy]-3,6-difluoropyridin-2-amine Chemical compound FC(OCCOC=1C=C(C(=NC=1F)N)F)F REHXNANUSIXWCE-UHFFFAOYSA-N 0.000 description 3
- NBHJTRKTYZIRKE-UHFFFAOYSA-N 5-bromo-3,6-difluoro-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound BrC=1C=C(C(=NC=1F)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)F NBHJTRKTYZIRKE-UHFFFAOYSA-N 0.000 description 3
- GGAFJTGKYRSMMY-UHFFFAOYSA-N 5-bromo-3,6-difluoropyridin-2-amine Chemical compound NC1=NC(F)=C(Br)C=C1F GGAFJTGKYRSMMY-UHFFFAOYSA-N 0.000 description 3
- HQDYTZDIDXLELH-UHFFFAOYSA-N 5-bromo-3-fluoro-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound BrC=1C=C(C(=NC=1)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)F HQDYTZDIDXLELH-UHFFFAOYSA-N 0.000 description 3
- ZDDBFSWSEKHJHO-UHFFFAOYSA-N 5-bromo-3-methoxy-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound BrC=1C=C(C(=NC=1)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)OC ZDDBFSWSEKHJHO-UHFFFAOYSA-N 0.000 description 3
- ZJBWTBQUJBHTHS-UHFFFAOYSA-N 5-chloro-3,6-difluoropyridin-2-amine Chemical compound NC1=NC(F)=C(Cl)C=C1F ZJBWTBQUJBHTHS-UHFFFAOYSA-N 0.000 description 3
- UWHWAXCLSNQVAG-UHFFFAOYSA-N 6,7-dichloro-1h-indole Chemical compound ClC1=CC=C2C=CNC2=C1Cl UWHWAXCLSNQVAG-UHFFFAOYSA-N 0.000 description 3
- SVENDZZBKXHUQH-UHFFFAOYSA-N 6-(difluoromethyl)-7-fluoro-1H-indole Chemical compound FC(C1=CC=C2C=CNC2=C1F)F SVENDZZBKXHUQH-UHFFFAOYSA-N 0.000 description 3
- YVACBHRHOBDZIU-UHFFFAOYSA-N 6-bromo-7-chloro-1h-indole Chemical compound ClC1=C(Br)C=CC2=C1NC=C2 YVACBHRHOBDZIU-UHFFFAOYSA-N 0.000 description 3
- PWGWZWTVXVEZIH-UHFFFAOYSA-N 6-chloro-1H-indole-3-sulfonic acid Chemical compound ClC1=CC=C2C(=CNC2=C1)S(=O)(=O)O PWGWZWTVXVEZIH-UHFFFAOYSA-N 0.000 description 3
- BJXHJYJLFZABNQ-UHFFFAOYSA-N 6-chloro-7-(trifluoromethoxy)-1H-indole Chemical compound ClC1=CC=C2C=CNC2=C1OC(F)(F)F BJXHJYJLFZABNQ-UHFFFAOYSA-N 0.000 description 3
- GTGLATAOCDNSDI-UHFFFAOYSA-N 6-chloro-7-(trifluoromethoxy)-1H-indole-3-sulfonic acid Chemical compound ClC1=CC=C2C(=CNC2=C1OC(F)(F)F)S(=O)(=O)O GTGLATAOCDNSDI-UHFFFAOYSA-N 0.000 description 3
- KGYOHJPGXOQUOK-UHFFFAOYSA-N 6-chloro-7-cyclopropyloxy-1H-indole Chemical compound ClC1=CC=C2C=CNC2=C1OC1CC1 KGYOHJPGXOQUOK-UHFFFAOYSA-N 0.000 description 3
- OPEAQMMQYFSXKX-UHFFFAOYSA-N 6-chloro-7-fluoro-1h-indole Chemical compound FC1=C(Cl)C=CC2=C1NC=C2 OPEAQMMQYFSXKX-UHFFFAOYSA-N 0.000 description 3
- GTQDVAKKUCXGNX-UHFFFAOYSA-N 6-chloro-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-7-fluoro-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1F)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F GTQDVAKKUCXGNX-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 201000006474 Brain Ischemia Diseases 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 206010008120 Cerebral ischaemia Diseases 0.000 description 3
- PIVOKDPARKTUGU-UHFFFAOYSA-N Clc1ccc2cc[nH]c2c1Br Chemical compound Clc1ccc2cc[nH]c2c1Br PIVOKDPARKTUGU-UHFFFAOYSA-N 0.000 description 3
- 102000010918 Cysteinyl leukotriene receptors Human genes 0.000 description 3
- 108050001116 Cysteinyl leukotriene receptors Proteins 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 208000003164 Diplopia Diseases 0.000 description 3
- 239000012981 Hank's balanced salt solution Substances 0.000 description 3
- 208000004044 Hypesthesia Diseases 0.000 description 3
- 102000004877 Insulin Human genes 0.000 description 3
- 108090001061 Insulin Proteins 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- MHABMANUFPZXEB-UHFFFAOYSA-N O-demethyl-aloesaponarin I Natural products O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=C(O)C(C(O)=O)=C2C MHABMANUFPZXEB-UHFFFAOYSA-N 0.000 description 3
- 208000018737 Parkinson disease Diseases 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical class O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000008186 active pharmaceutical agent Substances 0.000 description 3
- 150000001413 amino acids Chemical group 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 3
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 208000015114 central nervous system disease Diseases 0.000 description 3
- 206010008118 cerebral infarction Diseases 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 208000029444 double vision Diseases 0.000 description 3
- 239000000835 fiber Substances 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 230000037406 food intake Effects 0.000 description 3
- 235000012631 food intake Nutrition 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000003102 growth factor Substances 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 210000002216 heart Anatomy 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 210000003494 hepatocyte Anatomy 0.000 description 3
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 3
- SHFJWMWCIHQNCP-UHFFFAOYSA-M hydron;tetrabutylazanium;sulfate Chemical compound OS([O-])(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC SHFJWMWCIHQNCP-UHFFFAOYSA-M 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 229940125396 insulin Drugs 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 238000004811 liquid chromatography Methods 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 230000000750 progressive effect Effects 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 230000001737 promoting effect Effects 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 238000013268 sustained release Methods 0.000 description 3
- 239000012730 sustained-release form Substances 0.000 description 3
- 230000009747 swallowing Effects 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 3
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 2
- JTLAIKFGRHDNQM-UHFFFAOYSA-N 1-bromo-2-fluoroethane Chemical compound FCCBr JTLAIKFGRHDNQM-UHFFFAOYSA-N 0.000 description 2
- RBAHXNSORRGCQA-UHFFFAOYSA-N 1-chloro-2-fluoro-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=C1F RBAHXNSORRGCQA-UHFFFAOYSA-N 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- SKQZVCFVOPHIOM-UHFFFAOYSA-N 3-fluoro-5-(2-fluoroethoxy)-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC=1C(=NC=C(C=1)OCCF)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC SKQZVCFVOPHIOM-UHFFFAOYSA-N 0.000 description 2
- CBVJVNALUQTENQ-UHFFFAOYSA-N 3-fluoro-N,N-bis[(4-methoxyphenyl)methyl]-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine Chemical compound FC=1C(=NC=C(C=1)B1OC(C(O1)(C)C)(C)C)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC CBVJVNALUQTENQ-UHFFFAOYSA-N 0.000 description 2
- VHTODXQKBUENCV-UHFFFAOYSA-N 3-methoxy-N,N-bis[(4-methoxyphenyl)methyl]-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine Chemical compound COC=1C(=NC=C(C=1)B1OC(C(O1)(C)C)(C)C)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC VHTODXQKBUENCV-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- VOXWXYBDECHROO-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3,6-difluoro-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC(COC=1C=C(C(=NC=1F)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)F)F VOXWXYBDECHROO-UHFFFAOYSA-N 0.000 description 2
- FDTCWWXZGQSQKN-UHFFFAOYSA-N 5-(2,2-difluoroethoxy)-3-fluoro-N,N-bis[(4-methoxyphenyl)methyl]pyridin-2-amine Chemical compound FC(COC=1C=C(C(=NC=1)N(CC1=CC=C(C=C1)OC)CC1=CC=C(C=C1)OC)F)F FDTCWWXZGQSQKN-UHFFFAOYSA-N 0.000 description 2
- QLXASFJHUCKEHU-UHFFFAOYSA-N 5-bromo-3-fluoropyridin-2-amine Chemical compound NC1=NC=C(Br)C=C1F QLXASFJHUCKEHU-UHFFFAOYSA-N 0.000 description 2
- YBNSMFWOIJEMLT-UHFFFAOYSA-N 6,7-dichloro-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1Cl)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F YBNSMFWOIJEMLT-UHFFFAOYSA-N 0.000 description 2
- LSBWUVXQQOOFSB-UHFFFAOYSA-N 6-chloro-1h-indole-3-sulfonyl chloride Chemical compound ClC1=CC=C2C(S(Cl)(=O)=O)=CNC2=C1 LSBWUVXQQOOFSB-UHFFFAOYSA-N 0.000 description 2
- HZXCRKYZBXFTCI-UHFFFAOYSA-N 6-chloro-7-(difluoromethyl)-N-[3-fluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1C(F)F)S(=O)(=O)NC1=NC=C(C=C1F)OCCF HZXCRKYZBXFTCI-UHFFFAOYSA-N 0.000 description 2
- QOZCYTCRRLBJDG-UHFFFAOYSA-N 6-chloro-N-[5-(difluoromethoxy)-3-methoxypyridin-2-yl]-7-(difluoromethyl)-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1C(F)F)S(=O)(=O)NC1=NC=C(C=C1OC)OC(F)F QOZCYTCRRLBJDG-UHFFFAOYSA-N 0.000 description 2
- LUIMSBCTMMTFOG-UHFFFAOYSA-N 7-bromo-6-chloro-N-[3,6-difluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound BrC=1C(=CC=C2C(=CNC=12)S(=O)(=O)NC1=NC(=C(C=C1F)OCCF)F)Cl LUIMSBCTMMTFOG-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- 208000032194 Acute haemorrhagic leukoencephalitis Diseases 0.000 description 2
- 208000019901 Anxiety disease Diseases 0.000 description 2
- 206010003591 Ataxia Diseases 0.000 description 2
- 102000014461 Ataxins Human genes 0.000 description 2
- 108010078286 Ataxins Proteins 0.000 description 2
- 201000004569 Blindness Diseases 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 208000014644 Brain disease Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 206010008025 Cerebellar ataxia Diseases 0.000 description 2
- 206010008190 Cerebrovascular accident Diseases 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108010005939 Ciliary Neurotrophic Factor Proteins 0.000 description 2
- 102100031614 Ciliary neurotrophic factor Human genes 0.000 description 2
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 2
- 208000028698 Cognitive impairment Diseases 0.000 description 2
- 206010010947 Coordination abnormal Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 208000004986 Diffuse Cerebral Sclerosis of Schilder Diseases 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 208000032274 Encephalopathy Diseases 0.000 description 2
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 2
- 206010016059 Facial pain Diseases 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108091006027 G proteins Proteins 0.000 description 2
- 102000030782 GTP binding Human genes 0.000 description 2
- 108091000058 GTP-Binding Proteins 0.000 description 2
- 108010072051 Glatiramer Acetate Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000007818 Grignard reagent Substances 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000019022 Mood disease Diseases 0.000 description 2
- 208000007101 Muscle Cramp Diseases 0.000 description 2
- 208000010428 Muscle Weakness Diseases 0.000 description 2
- 206010028372 Muscular weakness Diseases 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- 101150003085 Pdcl gene Proteins 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 2
- 101100545004 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) YSP2 gene Proteins 0.000 description 2
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 2
- 208000005392 Spasm Diseases 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000012886 Vertigo Diseases 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 230000036506 anxiety Effects 0.000 description 2
- 239000012911 assay medium Substances 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 201000004562 autosomal dominant cerebellar ataxia Diseases 0.000 description 2
- 230000003376 axonal effect Effects 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000011712 cell development Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 208000009885 central pontine myelinolysis Diseases 0.000 description 2
- 230000002490 cerebral effect Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000005957 chlorosulfonylation reaction Methods 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 235000013985 cinnamic acid Nutrition 0.000 description 2
- 229930016911 cinnamic acid Natural products 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 239000003246 corticosteroid Substances 0.000 description 2
- 229960001334 corticosteroids Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000006735 deficit Effects 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000004807 desolvation Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 235000019700 dicalcium phosphate Nutrition 0.000 description 2
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 206010016256 fatigue Diseases 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000002825 functional assay Methods 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 229960003776 glatiramer acetate Drugs 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 102000049028 human GPR17 Human genes 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 208000034783 hypoesthesia Diseases 0.000 description 2
- 206010022437 insomnia Diseases 0.000 description 2
- 229940047124 interferons Drugs 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 230000000302 ischemic effect Effects 0.000 description 2
- 238000011813 knockout mouse model Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 208000036546 leukodystrophy Diseases 0.000 description 2
- GWNVDXQDILPJIG-NXOLIXFESA-N leukotriene C4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@@H](C(=O)NCC(O)=O)NC(=O)CC[C@H](N)C(O)=O GWNVDXQDILPJIG-NXOLIXFESA-N 0.000 description 2
- YEESKJGWJFYOOK-IJHYULJSSA-N leukotriene D4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@H](N)C(=O)NCC(O)=O YEESKJGWJFYOOK-IJHYULJSSA-N 0.000 description 2
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- TXXHDPDFNKHHGW-UHFFFAOYSA-N muconic acid Chemical compound OC(=O)C=CC=CC(O)=O TXXHDPDFNKHHGW-UHFFFAOYSA-N 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 231100000862 numbness Toxicity 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 210000000535 oligodendrocyte precursor cell Anatomy 0.000 description 2
- SBQLYHNEIUGQKH-UHFFFAOYSA-N omeprazole Chemical compound N1=C2[CH]C(OC)=CC=C2N=C1S(=O)CC1=NC=C(C)C(OC)=C1C SBQLYHNEIUGQKH-UHFFFAOYSA-N 0.000 description 2
- 210000001328 optic nerve Anatomy 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 239000001301 oxygen Chemical group 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 230000009984 peri-natal effect Effects 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- CPJSUEIXXCENMM-UHFFFAOYSA-N phenacetin Chemical compound CCOC1=CC=C(NC(C)=O)C=C1 CPJSUEIXXCENMM-UHFFFAOYSA-N 0.000 description 2
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- 230000004800 psychological effect Effects 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 230000000306 recurrent effect Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 2
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 238000007423 screening assay Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 230000001148 spastic effect Effects 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 210000000278 spinal cord Anatomy 0.000 description 2
- 208000020431 spinal cord injury Diseases 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 229960004793 sucrose Drugs 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 2
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 231100000889 vertigo Toxicity 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- 230000004393 visual impairment Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- GNWCZBXSKIIURR-UHFFFAOYSA-N (2-docosanoyloxy-3-hydroxypropyl) docosanoate Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCCCCCCCCCCCCCCCC GNWCZBXSKIIURR-UHFFFAOYSA-N 0.000 description 1
- YLOCGHYTXIINAI-XKUOMLDTSA-N (2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical class C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 YLOCGHYTXIINAI-XKUOMLDTSA-N 0.000 description 1
- ASWBNKHCZGQVJV-UHFFFAOYSA-N (3-hexadecanoyloxy-2-hydroxypropyl) 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(O)COP([O-])(=O)OCC[N+](C)(C)C ASWBNKHCZGQVJV-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- CMVQZRLQEOAYSW-UHFFFAOYSA-N 1,2-dichloro-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=C1Cl CMVQZRLQEOAYSW-UHFFFAOYSA-N 0.000 description 1
- PWDYWSRHRDZFFM-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-chloro-7-fluoroindole-3-sulfonyl chloride Chemical compound C1(=CC=CC=C1)S(=O)(=O)N1C=C(C2=CC=C(C(=C12)F)Cl)S(=O)(=O)Cl PWDYWSRHRDZFFM-UHFFFAOYSA-N 0.000 description 1
- KIHYPELVXPAIDH-HNSNBQBZSA-N 1-[[4-[(e)-n-[[4-cyclohexyl-3-(trifluoromethyl)phenyl]methoxy]-c-methylcarbonimidoyl]-2-ethylphenyl]methyl]azetidine-3-carboxylic acid Chemical compound CCC1=CC(C(\C)=N\OCC=2C=C(C(C3CCCCC3)=CC=2)C(F)(F)F)=CC=C1CN1CC(C(O)=O)C1 KIHYPELVXPAIDH-HNSNBQBZSA-N 0.000 description 1
- QRADKVYIJIAENZ-UHFFFAOYSA-N 1-[[bromo(difluoro)methyl]-ethoxyphosphoryl]oxyethane Chemical compound CCOP(=O)(C(F)(F)Br)OCC QRADKVYIJIAENZ-UHFFFAOYSA-N 0.000 description 1
- OCQUJZKDKBEBIN-UHFFFAOYSA-N 1-bromo-2-(difluoromethoxy)ethane Chemical compound FC(F)OCCBr OCQUJZKDKBEBIN-UHFFFAOYSA-N 0.000 description 1
- JNIDAGAFFKAPRV-UHFFFAOYSA-N 1-bromo-2-chloro-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(Br)=C1Cl JNIDAGAFFKAPRV-UHFFFAOYSA-N 0.000 description 1
- LWLCRRUIYBNYLC-UHFFFAOYSA-N 1-chloro-3-nitro-2-(trifluoromethoxy)benzene Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=C1OC(F)(F)F LWLCRRUIYBNYLC-UHFFFAOYSA-N 0.000 description 1
- QHSMEGADRFZVNE-UHFFFAOYSA-N 1-hydroxymidazolam Chemical compound C12=CC(Cl)=CC=C2N2C(CO)=NC=C2CN=C1C1=CC=CC=C1F QHSMEGADRFZVNE-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- FRPZMMHWLSIFAZ-UHFFFAOYSA-N 10-undecenoic acid Chemical compound OC(=O)CCCCCCCCC=C FRPZMMHWLSIFAZ-UHFFFAOYSA-N 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- KKFDCBRMNNSAAW-UHFFFAOYSA-N 2-(morpholin-4-yl)ethanol Chemical compound OCCN1CCOCC1 KKFDCBRMNNSAAW-UHFFFAOYSA-N 0.000 description 1
- WKAVKKUXZAWHDM-UHFFFAOYSA-N 2-acetamidopentanedioic acid;2-(dimethylamino)ethanol Chemical compound CN(C)CCO.CC(=O)NC(C(O)=O)CCC(O)=O WKAVKKUXZAWHDM-UHFFFAOYSA-N 0.000 description 1
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 1
- UYTRBIUOIKOGQO-UHFFFAOYSA-N 2-bromo-1-chloro-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=C1Br UYTRBIUOIKOGQO-UHFFFAOYSA-N 0.000 description 1
- WLDHPJSICUOHTH-UHFFFAOYSA-N 2-fluoro-3-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC=CC(C=O)=C1F WLDHPJSICUOHTH-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- SBXDENYROQKXBE-UHFFFAOYSA-N 2-phenylbenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1C1=CC=CC=C1 SBXDENYROQKXBE-UHFFFAOYSA-N 0.000 description 1
- MLMQPDHYNJCQAO-UHFFFAOYSA-N 3,3-dimethylbutyric acid Chemical compound CC(C)(C)CC(O)=O MLMQPDHYNJCQAO-UHFFFAOYSA-N 0.000 description 1
- XLZYKTYMLBOINK-UHFFFAOYSA-N 3-(4-hydroxybenzoyl)benzoic acid Chemical compound OC(=O)C1=CC=CC(C(=O)C=2C=CC(O)=CC=2)=C1 XLZYKTYMLBOINK-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OROKHXKJSDPRET-UHFFFAOYSA-N 3-chloro-2,5,6-trifluoropyridine Chemical compound FC1=CC(Cl)=C(F)N=C1F OROKHXKJSDPRET-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- XBUJOVJQQBVKHW-UHFFFAOYSA-N 3-pyridin-2-yl-1H-indole-2-sulfonamide Chemical class N1=C(C=CC=C1)C1=C(NC2=CC=CC=C12)S(=O)(=O)N XBUJOVJQQBVKHW-UHFFFAOYSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-M 4-aminosalicylate(1-) Chemical compound NC1=CC=C(C([O-])=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-M 0.000 description 1
- RJWBTWIBUIGANW-UHFFFAOYSA-N 4-chlorobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Cl)C=C1 RJWBTWIBUIGANW-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- LUEYUHCBBXWTQT-UHFFFAOYSA-N 4-phenyl-2h-triazole Chemical compound C1=NNN=C1C1=CC=CC=C1 LUEYUHCBBXWTQT-UHFFFAOYSA-N 0.000 description 1
- XRVDGNKRPOAQTN-FQEVSTJZSA-N 5-[3-[(1s)-1-(2-hydroxyethylamino)-2,3-dihydro-1h-inden-4-yl]-1,2,4-oxadiazol-5-yl]-2-propan-2-yloxybenzonitrile Chemical compound C1=C(C#N)C(OC(C)C)=CC=C1C1=NC(C=2C=3CC[C@@H](C=3C=CC=2)NCCO)=NO1 XRVDGNKRPOAQTN-FQEVSTJZSA-N 0.000 description 1
- NFBIWMFMHLPVLT-UHFFFAOYSA-N 5-bromo-3-methoxypyridin-2-amine Chemical compound COC1=CC(Br)=CN=C1N NFBIWMFMHLPVLT-UHFFFAOYSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- CCBJHEWGIDERLA-UHFFFAOYSA-N 6,7-dichloro-1h-indole-3-sulfonyl chloride Chemical compound ClC1=CC=C2C(S(Cl)(=O)=O)=CNC2=C1Cl CCBJHEWGIDERLA-UHFFFAOYSA-N 0.000 description 1
- KTSFYFHVHWIGRE-UHFFFAOYSA-N 6,7-dichloro-N-[3-fluoro-5-(2-fluoroethoxy)pyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1Cl)S(=O)(=O)NC1=NC=C(C=C1F)OCCF KTSFYFHVHWIGRE-UHFFFAOYSA-N 0.000 description 1
- BAJKSVYSXPMUFU-UHFFFAOYSA-N 6,7-dichloro-N-[5-(2,2-difluoroethoxy)-3-fluoro-6-methoxypyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1Cl)S(=O)(=O)NC1=NC(=C(C=C1F)OCC(F)F)OC BAJKSVYSXPMUFU-UHFFFAOYSA-N 0.000 description 1
- PLSZARHMGOOQOH-UHFFFAOYSA-N 6,7-dichloro-N-[5-(2,2-difluoroethoxy)-3-fluoropyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1Cl)S(=O)(=O)NC1=NC=C(C=C1F)OCC(F)F PLSZARHMGOOQOH-UHFFFAOYSA-N 0.000 description 1
- OJMXDHOTTDXNLC-UHFFFAOYSA-N 6,7-dichloro-N-[5-(2,2-difluoroethoxy)-3-methoxypyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1Cl)S(=O)(=O)NC1=NC=C(C=C1OC)OCC(F)F OJMXDHOTTDXNLC-UHFFFAOYSA-N 0.000 description 1
- WHLHKOILKLGOFY-UHFFFAOYSA-N 6,7-dichloro-N-[5-(difluoromethoxy)-3-methoxypyridin-2-yl]-1H-indole-3-sulfonamide Chemical compound ClC1=CC=C2C(=CNC2=C1Cl)S(=O)(=O)NC1=NC=C(C=C1OC)OC(F)F WHLHKOILKLGOFY-UHFFFAOYSA-N 0.000 description 1
- RVVAYIVWEMUUBU-UHFFFAOYSA-N 6-(difluoromethyl)-7-fluoro-1H-indole-3-sulfonyl chloride Chemical compound FC(C1=CC=C2C(=CNC2=C1F)S(=O)(=O)Cl)F RVVAYIVWEMUUBU-UHFFFAOYSA-N 0.000 description 1
- YTYIMDRWPTUAHP-UHFFFAOYSA-N 6-Chloroindole Chemical compound ClC1=CC=C2C=CNC2=C1 YTYIMDRWPTUAHP-UHFFFAOYSA-N 0.000 description 1
- BRGYYEKFVTWLIF-UHFFFAOYSA-N 6-chloro-7-(trifluoromethoxy)-1H-indole-3-sulfonyl chloride Chemical compound ClC1=CC=C2C(=CNC2=C1OC(F)(F)F)S(=O)(=O)Cl BRGYYEKFVTWLIF-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 208000031295 Animal disease Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 238000009020 BCA Protein Assay Kit Methods 0.000 description 1
- 238000000035 BCA protein assay Methods 0.000 description 1
- 201000002827 Balo concentric sclerosis Diseases 0.000 description 1
- 238000006159 Bartoli reaction Methods 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 208000020925 Bipolar disease Diseases 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 208000010693 Charcot-Marie-Tooth Disease Diseases 0.000 description 1
- 201000006868 Charcot-Marie-Tooth disease type 3 Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 206010010252 Concentric sclerosis Diseases 0.000 description 1
- 208000014311 Cushing syndrome Diseases 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 102100038496 Cysteinyl leukotriene receptor 1 Human genes 0.000 description 1
- 108010020070 Cytochrome P-450 CYP2B6 Proteins 0.000 description 1
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 1
- 102100026533 Cytochrome P450 1A2 Human genes 0.000 description 1
- 102100038739 Cytochrome P450 2B6 Human genes 0.000 description 1
- 102100039205 Cytochrome P450 3A4 Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- 208000031972 Dejerine-Sottas syndrome Diseases 0.000 description 1
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 108010052167 Dihydroorotate Dehydrogenase Proteins 0.000 description 1
- 102100032823 Dihydroorotate dehydrogenase (quinone), mitochondrial Human genes 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 206010013887 Dysarthria Diseases 0.000 description 1
- 206010013911 Dysgeusia Diseases 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 206010049020 Encephalitis periaxialis diffusa Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 229940124794 GPR17 inhibitor Drugs 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 208000006411 Hereditary Sensory and Motor Neuropathy Diseases 0.000 description 1
- 101000882759 Homo sapiens Cysteinyl leukotriene receptor 1 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- AKOAEVOSDHIVFX-UHFFFAOYSA-N Hydroxybupropion Chemical compound OCC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 AKOAEVOSDHIVFX-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- HCUARRIEZVDMPT-UHFFFAOYSA-N Indole-2-carboxylic acid Chemical class C1=CC=C2NC(C(=O)O)=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108010005716 Interferon beta-1a Proteins 0.000 description 1
- 108010005714 Interferon beta-1b Proteins 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- 239000002211 L-ascorbic acid Substances 0.000 description 1
- 235000000069 L-ascorbic acid Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 239000002139 L01XE22 - Masitinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102000016267 Leptin Human genes 0.000 description 1
- 108010092277 Leptin Proteins 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 208000016285 Movement disease Diseases 0.000 description 1
- TXXHDPDFNKHHGW-CCAGOZQPSA-N Muconic acid Natural products OC(=O)\C=C/C=C\C(O)=O TXXHDPDFNKHHGW-CCAGOZQPSA-N 0.000 description 1
- 241000872931 Myoporum sandwicense Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- HEUHLLWREUXYOX-UHFFFAOYSA-N N-[6-(difluoromethoxy)-5-fluoro-2-methoxypyridin-3-yl]-6-(difluoromethyl)-1H-indole-3-sulfonamide Chemical compound FC(OC1=C(C=C(C(=N1)OC)NS(=O)(=O)C1=CNC2=CC(=CC=C12)C(F)F)F)F HEUHLLWREUXYOX-UHFFFAOYSA-N 0.000 description 1
- QSACCXVHEVWNMX-UHFFFAOYSA-M N-acetylanthranilate Chemical compound CC(=O)NC1=CC=CC=C1C([O-])=O QSACCXVHEVWNMX-UHFFFAOYSA-M 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 208000003435 Optic Neuritis Diseases 0.000 description 1
- 102000016978 Orphan receptors Human genes 0.000 description 1
- 108070000031 Orphan receptors Proteins 0.000 description 1
- 206010069350 Osmotic demyelination syndrome Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 101150053185 P450 gene Proteins 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 208000037273 Pathologic Processes Diseases 0.000 description 1
- 229910002666 PdCl2 Inorganic materials 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 206010067063 Progressive relapsing multiple sclerosis Diseases 0.000 description 1
- 241000238711 Pyroglyphidae Species 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 208000007400 Relapsing-Remitting Multiple Sclerosis Diseases 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 101100221606 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) COS7 gene Proteins 0.000 description 1
- 208000021235 Schilder disease Diseases 0.000 description 1
- 206010040030 Sensory loss Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229940127530 Sphingosine 1-Phosphate Receptor Modulators Drugs 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 208000030886 Traumatic Brain injury Diseases 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 208000002552 acute disseminated encephalomyelitis Diseases 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000002253 anti-ischaemic effect Effects 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WXBLLCUINBKULX-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1.OC(=O)C1=CC=CC=C1 WXBLLCUINBKULX-UHFFFAOYSA-N 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 229930189065 blasticidin Natural products 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 229960001058 bupropion Drugs 0.000 description 1
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 230000001593 cAMP accumulation Effects 0.000 description 1
- 230000001275 ca(2+)-mobilization Effects 0.000 description 1
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 1
- MKJXYGKVIBWPFZ-UHFFFAOYSA-L calcium lactate Chemical compound [Ca+2].CC(O)C([O-])=O.CC(O)C([O-])=O MKJXYGKVIBWPFZ-UHFFFAOYSA-L 0.000 description 1
- 239000001527 calcium lactate Substances 0.000 description 1
- 235000011086 calcium lactate Nutrition 0.000 description 1
- 229960002401 calcium lactate Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920003086 cellulose ether Polymers 0.000 description 1
- 230000031736 central nervous system myelination Effects 0.000 description 1
- 210000001627 cerebral artery Anatomy 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- LPAUOXUZGSBGDU-STDDISTJSA-N chembl1096146 Chemical compound O=C1N(C=2C(=CC=CC=2)C)C(=N/CCC)/S\C1=C/C1=CC=C(OC[C@H](O)CO)C(Cl)=C1 LPAUOXUZGSBGDU-STDDISTJSA-N 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229960002152 chlorhexidine acetate Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000002591 computed tomography Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 210000000877 corpus callosum Anatomy 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- 229940109275 cyclamate Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- YOXHCYXIAVIFCZ-UHFFFAOYSA-N cyclopropanol Chemical compound OC1CC1 YOXHCYXIAVIFCZ-UHFFFAOYSA-N 0.000 description 1
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 206010061811 demyelinating polyneuropathy Diseases 0.000 description 1
- 238000000326 densiometry Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- RBLGLDWTCZMLRW-UHFFFAOYSA-K dicalcium phosphate dihydrate Substances O.O.[Ca+2].[Ca+2].[O-]P([O-])([O-])=O RBLGLDWTCZMLRW-UHFFFAOYSA-K 0.000 description 1
- JXTHNDFMNIQAHM-UHFFFAOYSA-M dichloroacetate Chemical compound [O-]C(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-M 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Substances CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 1
- LDCRTTXIJACKKU-ONEGZZNKSA-N dimethyl fumarate Chemical compound COC(=O)\C=C\C(=O)OC LDCRTTXIJACKKU-ONEGZZNKSA-N 0.000 description 1
- 229960004419 dimethyl fumarate Drugs 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 238000007908 dry granulation Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000001523 electrospinning Methods 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- XBRDBODLCHKXHI-UHFFFAOYSA-N epolamine Chemical compound OCCN1CCCC1 XBRDBODLCHKXHI-UHFFFAOYSA-N 0.000 description 1
- 229950008932 epolamine Drugs 0.000 description 1
- 239000003759 ester based solvent Substances 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 230000001815 facial effect Effects 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960000556 fingolimod Drugs 0.000 description 1
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 1
- 238000010579 first pass effect Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 239000003269 fluorescent indicator Substances 0.000 description 1
- 150000002221 fluorine Chemical class 0.000 description 1
- 125000004428 fluoroalkoxy group Chemical group 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000006005 fluoroethoxy group Chemical group 0.000 description 1
- 125000003784 fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 229960002737 fructose Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 230000005021 gait Effects 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000012268 genome sequencing Methods 0.000 description 1
- 229940114119 gentisate Drugs 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960001031 glucose Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 229940046533 house dust mites Drugs 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 208000013403 hyperactivity Diseases 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 229940124622 immune-modulator drug Drugs 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940124589 immunosuppressive drug Drugs 0.000 description 1
- 238000000126 in silico method Methods 0.000 description 1
- 238000011503 in vivo imaging Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 208000016290 incoordination Diseases 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 229960004461 interferon beta-1a Drugs 0.000 description 1
- 229960003161 interferon beta-1b Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 208000028756 lack of coordination Diseases 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000000832 lactitol Substances 0.000 description 1
- 235000010448 lactitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-JVCRWLNRSA-N lactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-JVCRWLNRSA-N 0.000 description 1
- 229960003451 lactitol Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004577 laquinimod Drugs 0.000 description 1
- GKWPCEFFIHSJOE-UHFFFAOYSA-N laquinimod Chemical compound OC=1C2=C(Cl)C=CC=C2N(C)C(=O)C=1C(=O)N(CC)C1=CC=CC=C1 GKWPCEFFIHSJOE-UHFFFAOYSA-N 0.000 description 1
- 150000002611 lead compounds Chemical class 0.000 description 1
- 229960000681 leflunomide Drugs 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 1
- 229940039781 leptin Drugs 0.000 description 1
- 229940065725 leukotriene receptor antagonists for obstructive airway diseases Drugs 0.000 description 1
- 239000003199 leukotriene receptor blocking agent Substances 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 208000018769 loss of vision Diseases 0.000 description 1
- 231100000864 loss of vision Toxicity 0.000 description 1
- 229940031703 low substituted hydroxypropyl cellulose Drugs 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000845 maltitol Substances 0.000 description 1
- 235000010449 maltitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 1
- 229940035436 maltitol Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 1
- 229960004655 masitinib Drugs 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 229960001810 meprednisone Drugs 0.000 description 1
- PIDANAQULIKBQS-RNUIGHNZSA-N meprednisone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)CC2=O PIDANAQULIKBQS-RNUIGHNZSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 1
- 229960003793 midazolam Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 230000037230 mobility Effects 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 229960005027 natalizumab Drugs 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 208000004296 neuralgia Diseases 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 208000021722 neuropathic pain Diseases 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 229950005751 ocrelizumab Drugs 0.000 description 1
- 229960002450 ofatumumab Drugs 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229960000381 omeprazole Drugs 0.000 description 1
- 229950010704 opicinumab Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical compound OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 description 1
- 108091008880 orphan GPCRs Proteins 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229950008141 ozanimod Drugs 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000009054 pathological process Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229960001291 peginterferon beta-1a Drugs 0.000 description 1
- 108010027737 peginterferon beta-1a Proteins 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 210000001428 peripheral nervous system Anatomy 0.000 description 1
- 201000005936 periventricular leukomalacia Diseases 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000009038 pharmacological inhibition Effects 0.000 description 1
- 229960003893 phenacetin Drugs 0.000 description 1
- 229960002695 phenobarbital Drugs 0.000 description 1
- PDTFCHSETJBPTR-UHFFFAOYSA-N phenylmercuric nitrate Chemical compound [O-][N+](=O)O[Hg]C1=CC=CC=C1 PDTFCHSETJBPTR-UHFFFAOYSA-N 0.000 description 1
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229960005141 piperazine Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 108010017843 platelet-derived growth factor A Proteins 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 108010055896 polyornithine Proteins 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229950009275 ponesimod Drugs 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229960003975 potassium Drugs 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229920003124 powdered cellulose Polymers 0.000 description 1
- 235000019814 powdered cellulose Nutrition 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 201000009395 primary hyperaldosteronism Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 206010036807 progressive multifocal leukoencephalopathy Diseases 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- MCSINKKTEDDPNK-UHFFFAOYSA-N propyl propionate Chemical compound CCCOC(=O)CC MCSINKKTEDDPNK-UHFFFAOYSA-N 0.000 description 1
- 210000004129 prosencephalon Anatomy 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 210000002804 pyramidal tract Anatomy 0.000 description 1
- 229940043131 pyroglutamate Drugs 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 230000027425 release of sequestered calcium ion into cytosol Effects 0.000 description 1
- 230000020874 response to hypoxia Effects 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- 201000008628 secondary progressive multiple sclerosis Diseases 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 235000019615 sensations Nutrition 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229950005693 siponimod Drugs 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229940083542 sodium Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- UDYFLDICVHJSOY-UHFFFAOYSA-N sulfur trioxide-pyridine complex Substances O=S(=O)=O.C1=CC=NC=C1 UDYFLDICVHJSOY-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960000331 teriflunomide Drugs 0.000 description 1
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 229940078499 tricalcium phosphate Drugs 0.000 description 1
- 229910000391 tricalcium phosphate Inorganic materials 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- 229940035722 triiodothyronine Drugs 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 229940075466 undecylenate Drugs 0.000 description 1
- 229940045136 urea Drugs 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000003799 water insoluble solvent Substances 0.000 description 1
- 239000003021 water soluble solvent Substances 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 229950000339 xinafoate Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
La presente solicitud se refiere a piridinil indolsulfonamidas sustituidas con alcoxi. Los compuestos son moduladores negativos de GPR17 y tienen utilidad en el tratamiento de diversos trastornos asociados con GPR17. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Alcoxipiridinil indolsulfonamidas sustituidas
Antecedentes
Los receptores acoplados a proteína G (GPCR) constituyen la familia más grande de receptores de membrana en la célula. Transducen señales extracelulares a sistemas efectores intracelulares y están involucrados en una gran variedad de fenómenos fisiológicos, por lo que representan los objetivos más comunes de los fármacos, aunque solo un pequeño porcentaje de los GPCR son el objetivo de las terapias actuales.
Los GPCR responden a una amplia gama de ligandos. Debido al progreso en la secuenciación del genoma humano, para aproximadamente el 25% de los más de 400 GPCR (sin incluir los GPCR olfativos) que se han identificado, todavía falta un ligando fisiológicamente relevante definido. Estos receptores se conocen como "GPCR huérfanos". Se espera que la "desorfanización" y la identificación de sus funciones in vivo aclaren nuevos mecanismos reguladores y, por lo tanto, revelen nuevos objetivos farmacológicos. Si GPR17 es un receptor huérfano es todavía un tema de debate. Filogenéticamente, GPR17 está estrechamente relacionado con los receptores de nucleótidos P2Y y los receptores de cisteinileucotrieno (CysLT1, CysLT2), con una identidad de secuencia de aminoácidos de entre aproximadamente 30 y aproximadamente 35%, respectivamente.
Los análisis mediante transferencia de Northern y RT-PCR de múltiples tejidos indican una expresión predominante de GPR17 en el sistema nervioso central (SNC) (Ciana et al., 2006, Em Bo J 25(19): 4615; Blasius et al., 1998, J Neurochem 70(4): 1357) y, además, en el corazón y el riñón, es decir, órganos que normalmente sufren daño isquémico. Se han identificado dos isoformas de GPR17 humano que difieren solo en la longitud de su extremo N-terminal. La isoforma corta GPR17 codifica una proteína de 339 residuos de aminoácidos con siete motivos transmembrana típicos de tipo rodopsina. La isoforma larga codifica un receptor con un extremo N-terminal con una longitud de 28 aminoácidos más (Blasius et al., 1998). GPR17 está altamente conservado en las especies de vertebrados (~ 90% de identidad de secuencia de aminoácidos con los ortólogos de ratón y rata), lo que puede constituir una característica ventajosa para el desarrollo de ligandos de moléculas pequeñas y modelos animales en el ámbito del descubrimiento de fármacos.
En el informe original de desorfanización, GPR17 se identificó como un receptor dual para nucleótidos de uracilo y cisteinil-leucotrienos (cysLT) LTC4 y LTD4, respectivamente, en base a los ensayos de unión de 35SGTPyS e inhibición de cAMP, así como la formación de imágenes de calcio de células individuales (Ciana et al., 2006, ibidem). Se proporcionaron pruebas de la funcionalidad de GPR17 en diferentes entornos celulares, como células 1321N1, COS7, CHO y HEK293 (Ciana et al., 2006, ibidem). Posteriormente, un estudio independiente confirmó la activación de GPR17 por nucleótidos de uracilo, pero no pudo recapitular la activación por CysLTs (Benned-Jensen y Rosenkilde, 2010, Br J Pharmacol, 159(5): 1092). Sin embargo, informes independientes recientes (Maekawa et al., 2009, PNAS 106(28), 11685; Qi et al., 2013, J Pharmacol Ther 347,1, 38; Hennen et al., 2013, Sci Signal 6, 298) sugirieron la falta de respuesta de GPR17 tanto a los nucleótidos de uracilo como a CysLT en diferentes entornos celulares que expresan GPR17 de manera estable (células 1321N1, CHO, HEK293). También se ha propuesto una función reguladora novedosa para GPR17: GPR17, al coexpresarse con el receptor CysLT1, hizo que el receptor CysLT 1 no respondiera a sus mediadores lipídicos endógenos LTC4 y LTD4. Se requieren investigaciones adicionales para probar la farmacología y la función de GPR17 con más profundidad.
Los fármacos que modulan la actividad de GPR17 pueden tener efectos neuroprotectores, antiinflamatorios y antiisquémicos y, por lo tanto, pueden ser útiles para el tratamiento de la isquemia cerebral, cardíaca y renal y del accidente cerebrovascular (documento WO 2006/045476), y/o para mejorar la recuperación de estos eventos (Bonfanti et al., Cell Death and Disease, 2017, 8, e2871).
También se cree que los moduladores de GPR17 están involucrados en la ingesta de alimentos, las respuestas de insulina y leptina y, por lo tanto, se afirma que tienen un papel en el tratamiento de la obesidad (documento WO 2011/113032).
Además, existe una fuerte evidencia de que GPR17 está involucrado en los procesos de mielinización y que los moduladores negativos de GPR17 (antagonistas o agonistas inversos) pueden ser fármacos valiosos para el tratamiento o alivio de los trastornos de la mielinización como la esclerosis múltiple o la lesión de la médula espinal (Chen et al., Nature neuroscience 2009, 12(11):1398-1406; Ceruti et al; Brain: a journal of neurology 2009 132 (Pt 8): 2206-18; Hennen et al., Sci Signal, 6, 2013, 298; Simon et al J Biol Chem 291, 2016, 705; Fumagalli et al., Neuropharmacology 104, 2016, 82). Más recientemente, dos grupos demostraron que los ratones adultos con inactivación génica de GPR17-/- tenían una remielinización más rápida que los compañeros de camada de tipo natural después de la desmielinización inducida por LPC en la médula espinal (Lu et al., Scientific Reports, 2018, 8:4502) o en el cuerpo calloso (Ou et al., J. Neurosci., 2016, 36(41):10560). Esto volvió a confirmar un papel potencialmente crucial en GPR17 en el proceso de remielinización. Por el contrario, se ha demostrado que la activación de GPR17 inhibe la maduración de las células precursoras de oligodendrocitos (CPO) evitando así la mielinización eficaz (Simon et al., anteriormente mencionado). Por lo tanto, la identificación de antagonistas o agonistas inversos de GPR17 potentes y selectivos sería de importancia significativa en el tratamiento de los trastornos de la mielinización.
Se sabe que varias enfermedades graves de la mielinización están causadas por alteraciones en la mielinización, ya sea por una pérdida de mielina (generalmente llamada desmielinización) y/o por la incapacidad del cuerpo de formar adecuadamente la mielina (a veces llamada desmielinización). Las enfermedades de la mielinización pueden ser idiopáticas o secundarias a ciertos eventos desencadenantes como p. ej. una lesión cerebral traumática o infección viral. Las enfermedades de la mielinización pueden afectar principalmente al sistema nervioso central (SNC), pero también pueden afectar al sistema nervioso periférico. Las enfermedades de la mielinización incluyen, entre otras, la esclerosis múltiple, neuromielitis óptica (también conocida como enfermedad de Devic), leucodistrofias, síndrome de Guillain-Barré y muchas otras enfermedades que se describen con más detalle más adelante (véase también, p. ej., Love, J Clin Pathol, 59, 2006, 1151, Fumagalli et al., anteriormente mencionado). Las enfermedades neurodegenerativas como la enfermedad de Alzheimer, enfermedad de Huntington, enfermedad de Parkinson, esclerosis lateral amiotrópica (ELA) y atrofia multisistémica (AMS) también se han asociado recientemente a una disminución de la mielinización (véase, p. ej., Ettle et al., Mol Neurobiol 53, 2016, 3046; Jellinger y Welling, Movement Disorders, 31, 2016; 1767; Kang et al., Nature Neurosci 6, 2013, 571; Bartzokis, Neurochem Res (2007) 32:1655).
La esclerosis múltiple (EM) es un trastorno progresivo crónico. Es una enfermedad autoinmune inflamatoria que causa daño a los oligodendrocitos, desmielinización y, en última instancia, pérdida axonal, lo que da lugar a un amplio espectro de signos y síntomas de una enfermedad neurológica grave, como p. ej. fatiga, mareos, problemas de movilidad y marcha, dificultades para hablar y tragar, dolor y otros. La EM adopta varias formas, con nuevos síntomas que ocurren en ataques aislados (formas recurrentes) o que aumentan con el tiempo (formas progresivas). Si bien ciertos síntomas pueden desaparecer por completo entre ataques aislados, a menudo persisten problemas neurológicos graves, especialmente a medida que la enfermedad avanza a una forma más progresiva. Según la Asociación Estadounidense de Esclerosis Múltiple, aproximadamente 400 000 personas han sido diagnosticadas con EM en los Estados Unidos y hasta 2,5 millones en todo el mundo, con una estimación de 10000 casos nuevos diagnosticados anualmente en los Estados Unidos. La esclerosis múltiple es de dos a tres veces más frecuente en mujeres que en hombres.
No existe un tratamiento causal conocido o una cura para la esclerosis múltiple o muchas otras enfermedades de la mielinización. Los tratamientos suelen ser sintomáticos y tratan de mejorar la función después de un ataque y prevenir nuevos ataques, abordando el componente inflamatorio de la enfermedad. Dichos fármacos inmunomoduladores suelen ser solo moderadamente eficaces, en particular si la enfermedad progresa, pero pueden tener efectos secundarios y ser mal tolerados. Además, la mayoría de los fármacos disponibles, como los interferones p, el acetato de glatirámero o los anticuerpos terapéuticos, solo están disponibles en forma inyectable y/o solo abordan el componente inflamatorio de la enfermedad, pero no la desmielinización directamente. Otros fármacos, como los corticosteroides, muestran efectos inflamatorios e inmunosupresores bastante inespecíficos, que conducen potencialmente a efectos secundarios crónicos, como los que se manifiestan en el síndrome de Cushing, por ejemplo.
Por lo tanto, existe una gran necesidad de un fármaco seguro y eficaz para el tratamiento de enfermedades de la mielinización, como la EM, preferiblemente un fármaco que sea adecuado para la administración oral. Idealmente, tal fármaco revertiría el proceso de desmielinización al disminuir la desmielinización y/o al promover la remielinización de las neuronas afectadas. Un compuesto químico que disminuya de manera eficaz la actividad del receptor GPR17 podría cumplir estos requisitos.
Sin embargo, solo se conocen unos pocos compuestos químicos que modulan eficazmente la actividad de GPR17.
El documento WO 2005/103291 propone moléculas endógenas de ácido 5-aminolevulínico (5-ALA) y porfobilinógeno (PBG) como ligandos activadores de GPR17, describe los efectos analgésicos de un agonista de GPR17 y propone el uso de agonistas de GPR17 para tratar el dolor neuropático y como herramientas en los ensayos de detección de GPR17. Sin embargo, la afinidad informada de 5-ALA y PBG es bastante baja y las cantidades necesarias en los ensayos son significativas, es decir, en el rango micromolar de tres dígitos para 5-ALA o incluso en el rango mM para PBG, lo que hace que ambos compuestos no sean adecuados para su uso en ensayos de cribado rutinarios o incluso para la terapia. Además, el PBG es un compuesto reactivo químicamente inestable que se descompone rápidamente después de la exposición al aire y la luz, por lo que no es práctico manipularlo de forma rutinaria. Por lo tanto, estos compuestos no ofrecen un punto de partida prometedor para desarrollar moduladores de GPR17 negativos terapéuticamente eficaces.
Montelukast y pranlukast se desarrollaron originalmente como antagonistas de los receptores de leucotrienos y recientemente se descubrió que también actúan sobre el receptor GPR17 (Ciana et al., EMBO J. 2006, 25, 4615 4627). Sin embargo, los resultados posteriores en un ensayo funcional fueron contradictorios para montekulast (Hennen et al., 2013, ibidem), mientras que la inhibición farmacológica de GPR17 con pranlukast promueve la diferenciación primaria de oligodendrocitos de ratón (Hennen et al., 2013, ibidem) y de rata (Ou et al., J. Neurosci. 36, 2016, 10560-10573). Pranlukast incluso fenocopia el efecto de la depresión de GPR17 en un modelo de lisolecitina de desmielinización focal porque tanto los ratones de tipo natural tratados con pranlukast como los ratones con inactivación génica de GPR17 muestran un inicio más temprano de la remielinización (Ou, ibidem). Estos resultados apoyan firmemente la hipótesis de que los inhibidores de GPR17 ofrecen potencial para el tratamiento de enfermedades desmielinizantes animales/humanas.
Sin embargo, la afinidad de montekulast y prankulast hacia GPR17 se halla solo en el rango micromolar alto (Kose et al., ACS Med. Chem. Lett. 2014, 5, 326-330). Dada la alta unión a proteínas de ambos compuestos y su escasa penetración en el cerebro, es poco probable que puedan alcanzar concentraciones libres suficientemente altas para unirse a los receptores GPR17 en cantidades adecuadas para la terapia humana. Además, los resultados obtenidos in vivo con estos compuestos son difíciles de interpretar debido a su confusa alta afinidad por los receptores CYSLT1. El documento US 8.623.593 describe ciertos ácidos indol-2-carboxílicos como agonistas de GPR17 y su uso en ensayos de cribado. Sin embargo, estos derivados son todos agonistas potentes y no son adecuados para regular a la baja la actividad de GPR17 según sea necesario en el tratamiento de los trastornos de la mielinización como la EM. Además, esta clase de activadores de GPR17 no atraviesa suficientemente la barrera hematoencefálica debido a los grupos carboxilo fácilmente ionizables y, por lo tanto, no fueron compuestos principales adecuados para desarrollar moduladores negativos de GPR17. Véase también Baqi et al., Med. Chem. Commun., 2014, 5, 86 y Kose et al., 2014, ibidem.
El documento WO 2010/054307 describe métodos y moléculas para promover la diferenciación de precursores de oligodendrocitos para su uso, p. ej. en la esclerosis múltiple. Los compuestos que contienen sulfonamida bifenílica y/o piperazina no contienen el resto piridinilo ni indolilo de los compuestos de la presente descripción.
El documento WO 2013/167177 propone ciertos compuestos de feniltriazol y benzodiazepina como antagonistas de GPR17. Sin embargo, los compuestos descritos se seleccionaron únicamente en base a los resultados del cribado in silico y no se proporcionaron datos biológicos en absoluto. Los inventores de la presente solicitud no han podido confirmar hasta el momento la actividad moduladora del antagonista de GPR17 de ninguno de los supuestos ligandos propuestos por los autores de la anterior solicitud de patente. Por lo tanto, existe la necesidad de identificar moduladores potentes, preferentemente moduladores negativos, más preferentemente agonistas inversos de GPR17, que sean capaces de disminuir eficazmente la actividad de GPR17, preferentemente tras la administración oral. Descripción de la invención
La presente invención se refiere a compuestos que actúan como moduladores negativos del receptor GPR17. En una realización preferida, los compuestos actúan como agonistas negativos del receptor GPR17, inhibiendo así un GPR17 constitutivamente activo.
Los inventores de la presente solicitud han descubierto ahora que las piridin-2-ilindolsulfonamidas con ciertos sustituyentes fluoroalcoxi en la posición para del piridilo en combinación con sustituyentes específicos en la posición "R7" y con al menos dos sustituciones adicionales en otras dos posiciones de la molécula central de indolpiridilsulfonamida, como se define específicamente en la presente memoria, proporcionan muy buena actividad inhibidora de GPR17 junto con propiedades mejoradas. Por ejemplo, la adición de sustituyentes específicos en "R7" como se describe en la presente memoria disminuye de manera eficaz la inducción de CYP 450-1A2, que a menudo se asocia con esta subclase específica de compuestos si R7 es hidrógeno, mientras que el sustituyente fluoroalcoxi en la posición para del piridilo proporciona ciertas ventajas farmacocinéticas, como una menor unión a proteínas plasmáticas en comparación con otros sustituyentes que incluyen, p. ej., los halógenos.
Por consiguiente, la presente invención se refiere a compuestos que tienen la fórmula I

donde
R6 se selecciona de fluoro, cloro, fluorometilo y fluorometoxi,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo, fluoroetilo, fluorometoxi y fluoroetoxi,
R8 es fluoro o metoxi,
R11 es hidrógeno, fluoro o metoxi,
L es un enlace, o un enlazador seleccionado de -CH2- y -CH2-CH2-O-,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Una realización se refiere a compuestos de Fórmula I,
donde
R6 se selecciona de cloro, fluorometilo y fluorometoxi,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro o metoxi,
R11 es hidrógeno, fluoro o metoxi,
L es un enlace, o un enlazador seleccionado de -CH2- y -CH2-CH2-O-,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Una realización se refiere a compuestos de Fórmula I,
donde
R6 se selecciona de cloro, monofluorometilo, difluorometilo y trifluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro o metoxi,
R11 es hidrógeno, fluoro o metoxi,
L es un enlace, o un enlazador seleccionado de -CH2- y -CH2-CH2-O-,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Una realización se refiere a compuestos de fórmula I, en la que R6 es cloro.
Una realización se refiere a compuestos de fórmula I, en la que R6 es fluoro.
Una realización se refiere a compuestos de fórmula I, en la que R6 es fluorometilo, preferiblemente difluorometilo. Una realización se refiere a compuestos de fórmula I, en la que R7 se selecciona de fluorometilo, ciclopropiloxi, fluoro y cloro.
Una realización se refiere a compuestos de fórmula I, en la que R7 es fluorometilo, preferiblemente difluorometilo. Una realización se refiere a compuestos de fórmula I, en la que R7 es fluorometoxi, preferiblemente difluorometoxi o trifluorometoxi.
Una realización se refiere a compuestos de fórmula I, en la que R7 es cloro.
Una realización se refiere a compuestos de fórmula I, en la que R7 es fluoro.
Una realización se refiere a compuestos de fórmula I, en la que R7 es ciclopropiloxi. Una realización se refiere a compuestos de fórmula I, en la que R7 es ciclopropilo.
Una realización se refiere a compuestos de fórmula I, en la que R11 es fluoro o hidrógeno. Una realización se refiere a compuestos de fórmula I, en la que R11 es metoxi.
Una realización se refiere a compuestos de fórmula I, en la que X1 es hidrógeno.
Una realización se refiere a compuestos de Fórmula I, en donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene por tanto una estructura según las Fórmulas Ia, Ib o Ic, en las que cualquier sustituyente tiene el significado descrito anteriormente para la fórmula I.

Una realización se refiere a un compuesto que tiene una de las Fórmulas Ia, Ib o Ic, donde R6 se selecciona de cloro y fluorometilo, preferiblemente de cloro y difluorometilo. R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi
R11 se selecciona de hidrógeno, metoxi y fluoro
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro.
Una realización se refiere a los compuestos de Fórmula I como se describieron anteriormente, en donde R8 es fluoro y en donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene por tanto una estructura según las Fórmulas I Ia, IIb o IIc,

donde todos los demás sustituyentes son como se describieron anteriormente.
Una realización se refiere a compuestos de fórmula I, que tienen una de las fórmulas IIa, IIb o IIc, en donde
R6 se selecciona de fluoro, cloro y fluorometilo, preferiblemente de cloro y difluorometilo, R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R11 es hidrógeno, metoxi o fluoro, preferiblemente metoxi o fluoro,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro, donde preferiblemente X1 es hidrógeno y X2 es fluoro.
Una realización se refiere a los compuestos de fórmula I como se describieron anteriormente, donde R8 y R11 son ambos fluoro y donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene por tanto una estructura de acuerdo con la fórmula IIIa, IIIb o IIIc,

donde todos los demás sustituyentes son como se describieron anteriormente.
Una realización se refiere a compuestos de fórmulas IIIa, IIIb o IIIc, en donde
R6 se selecciona de fluoro, cloro y fluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro, y sus sales, solvatos, isótopos y cocristales farmacéuticamente aceptables.
Una realización se refiere a compuestos de fórmula I, donde R8 es fluoro, R11 es hidrógeno y donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene por tanto una estructura de acuerdo con la fórmula IVa, IVb o iVc,

donde todos los demás sustituyentes son como se describieron anteriormente.
Una realización se refiere a compuestos de fórmula IVa, IVb o IVc, en donde
R6 se selecciona de fluoro, cloro y fluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Una realización se refiere a los compuestos de fórmula I como se describieron anteriormente, donde R11 es metoxi y donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene una estructura según la fórmula Va, Vb o Vc:

donde todos los demás sustituyentes son como se describieron anteriormente.
En una realización, en los compuestos de fórmula Va, Vb y Vc,
R6 se selecciona de fluoro, cloro, fluorometilo y fluorometoxi,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro o metoxi, preferiblemente fluoro, y
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro.
En una realización, en los compuestos de fórmula Va, Vb y Vc,
R6 se selecciona de cloro, monofluorometilo, difluorometilo y trifluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro o metoxi, preferiblemente fluoro,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro, donde, en una realización, X2 es preferiblemente hidrógeno,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
En una realización preferida, en los compuestos de fórmula Va, Vb y Vc,
R6 se selecciona de cloro, monofluorometilo, difluorometilo y trifluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi y fluorometilo, R8 es fluoro,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro, donde, en una realización, X2 es preferiblemente hidrógeno,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Una realización preferida se refiere a compuestos de fórmula I, en donde L es -CH2-, y dicho compuesto tiene una estructura de acuerdo con la fórmula Ib

donde
R6 se selecciona de fluoro, cloro, fluorometilo y fluorometoxi,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro o metoxi,
R11 es hidrógeno, fluoro o metoxi,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Una realización preferida se refiere a los compuestos de fórmula Ib, en la que
R6 se selecciona de cloro y fluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi
R8 es fluoro o metoxi,
R11 se selecciona de hidrógeno, metoxi y fluoro,
X1 es hidrógeno y X2 es hidrógeno o fluoro
Una realización preferida se refiere a los compuestos de fórmula Ib, en la que
R6 se selecciona de cloro y fluorometilo, preferiblemente de cloro y difluorometilo,
R7 se selecciona de cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi
R8 es fluoro,
R11 se selecciona de hidrógeno y fluoro,
X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización preferida se refiere a los compuestos de fórmula Ib, en la que
R6 se selecciona de cloro y difluorometilo,
R7 se selecciona de cloro, ciclopropiloxi, fluorometilo y fluorometoxi
R8 es fluoro,
R11 se selecciona de hidrógeno y fluoro,
X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización preferida se refiere a los compuestos de fórmula Ib, en la que
R6 se selecciona de fluoro, cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropiloxi y fluorometilo,
R8 es fluoro,
R11 se selecciona de metoxi y fluoro,
X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización preferida se refiere a los compuestos de fórmula Ib, en la que
R6 se selecciona de cloro y difluorometilo,
R7 se selecciona de cloro, ciclopropiloxi, fluorometilo y fluorometoxi
R8 es fluoro,
R11 es metoxi,
X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización preferida se refiere a los compuestos de fórmula Ib, en la que
R6 se selecciona de cloro y difluorometilo,
R7 se selecciona de cloro, ciclopropiloxi y difluorometilo,
R8 es fluoro,
R11 se selecciona de hidrógeno, metoxi y fluoro, y es preferiblemente metoxi,
X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización se refiere a compuestos de fórmula Ib, en la que
R6 se selecciona de cloro y difluorometilo,
R7 se selecciona de fluoro, cloro, ciclopropiloxi, difluorometilo, difluorometoxi y trifluorometoxi,
R8 es metoxi,
R11 se selecciona de hidrógeno y fluoro,
X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R6 es difluorometilo.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R7 es cloro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R7 es fluoro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R7 es difluorometilo.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 es fluoro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 es fluoro y R11 es metoxi.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 es fluoro y R11 es hidrógeno.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 y R11 son ambos fluoro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 es metoxi.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 es metoxi y R11 es fluoro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R8 es metoxi y R11 es hidrógeno.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R11 es fluoro.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R11 es hidrógeno.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde R11 es metoxi.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde X1 y X2 son ambos hidrógeno.
Una realización preferida se refiere a compuestos de fórmula Ib como se describieron anteriormente donde X1 es hidrógeno y X2 es fluoro.
Una realización preferida se refiere a compuestos de fórmula I, Ia, Ib, Ic, Ila, IIb, IIc, IIIa, IIIb, IIIc, IVa, IVb, IVc, Va, Vb o Vc, en donde
R6 es cloro,
R7 se selecciona de fluoro, cloro, ciclopropiloxi, fluorometilo y fluorometoxi, y se selecciona preferiblemente de cloro, ciclopropiloxi y difluorometilo,
y donde X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización preferida se refiere a compuestos de fórmula I, Ia, Ib, Ic, Ila, IIb, IIc, IIIa, IIIb, IIIc, IVa, IVb, IVc, Va, Vb o Vc, en donde
R6 es difluorometilo
R7 se selecciona de fluoro, cloro, ciclopropiloxi, fluorometilo y fluorometoxi, y se selecciona preferiblemente de cloro, ciclopropiloxi y difluorometilo,
y donde X1 es hidrógeno y X2 es hidrógeno o fluoro.
Una realización se refiere a cualquiera de los compuestos descritos anteriormente, donde X1 es hidrógeno y X2 es fluoro.
Una realización se refiere a cualquiera de los compuestos descritos anteriormente, donde X1 y X2 son ambos hidrógeno.
Una realización preferida se refiere a cualquiera de los compuestos descritos anteriormente donde R6 es cloro. Una realización preferida se refiere a cualquiera de los compuestos descritos anteriormente donde R6 es difluorometilo.
Una realización preferida se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es cloro. Una realización preferida se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es fluoro. Una realización preferida se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es difluorometilo.
Una realización se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es monofluorometilo. Una realización se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es trifluorometilo.
Una realización se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es ciclopropiloxi. Una realización se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es monofluorometoxi. Una realización se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es difluorometoxi. Una realización se refiere a cualquiera de los compuestos descritos anteriormente donde R7 es trifluorometoxi. Una realización preferida se refiere a un compuesto seleccionado de
6.7- dicloro-N-[5-(2,2-difluoroetoxi)-3-metoxipiridin-2-il]-1H-indol-3-sulfonamida
7-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-6-(difluorometil)-1H-indol-3-sulfonamida 6-cloro-N-[5-[2-(difluorometoxi)etoxi]-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-6-(difluorometil)-7-fluoro-1H-indol-3-sulfonamida
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-(trifluorometoxi)-1H-indol-3-sulfonamida
6-cloro-N-[5-(difluorometoxi)-3-metoxipiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida
6.7- dicloro-N-[5-(difluorometoxi)-3-metoxipiridin-2-il]-1H-indol-3-sulfonamida
6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6 -metoxipiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida 6- cloro-N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida
7- cloro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-6-(difluorometil)-1H-indol-3-sulfonamida
6-(difluorometil)-7-fluoro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6-cloro-7-(difluorometil)-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6.7- dicloro-N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-1H-indol-3-sulfonamida
6.7- dicloro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-fluoro-1H-indol -3-sulfonamida
6-cloro-N-[5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
6-cloro-7-ciclopropil-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida
6-cloro-7-ciclopropiloxi-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
Por claridad, cualquier definición de un compuesto como se describió anteriormente en la presente memoria también incluye cualquier sal, solvato, isótopo y cocristal farmacéuticamente aceptable del mismo.
Una realización se refiere a los compuestos de la presente invención para el uso en terapia.
Una realización se refiere a los compuestos de la presente invención para el uso en el tratamiento o alivio de un trastorno desmielinizante.
Una realización se refiere a los compuestos de la presente invención para el uso en el tratamiento o alivio de la esclerosis múltiple.
Una realización se refiere a un método para tratar un trastorno desmielinizante, que incluye, pero sin limitación, la esclerosis múltiple, que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto como se define en la presente memoria.
Una realización se refiere a composiciones terapéuticas que comprenden al menos un compuesto de la presente invención y un vehículo farmacéuticamente aceptable.
Otra realización preferida se refiere a los compuestos de la presente invención que comprenden al menos un isótopo 18F, preferiblemente en la posición de un átomo de flúor como se indica en uno de los compuestos descritos en la
presente memoria. A modo de ejemplo no limitante, en el compuesto 6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-fluoro-1H-indol-3-sulfonamida, descrito en la presente memoria, al menos uno de los cuatro átomos de flúor puede estar representado por un isótopo 18F. Esto es aplicable igualmente a otros compuestos que contienen flúor descritos en la presente memoria. Estos compuestos que contienen 18F se pueden usar preferiblemente como trazadores de PET.
Otra realización preferida se refiere a los compuestos de la presente invención que comprenden al menos un isótopo 11C, preferiblemente en la posición de un átomo de carbono como se indica en la presente memoria. Estos compuestos que contienen 11C se pueden usar preferiblemente como trazadores de PET.
Otra realización preferida se refiere a los compuestos de la presente invención que comprenden al menos un isótopo 123i, 125i o 131I, preferiblemente en la posición de un átomo de halógeno como se indica en la presente memoria. Los compuestos que contienen 123I, 125I o 131I pueden usarse preferiblemente como trazadores de SPECT.
Aplicación terapéutica y diagnóstica
En un aspecto, la invención se refiere a cualquiera de los compuestos descritos en la presente memoria, para el uso en la terapia o el diagnóstico, particularmente en la terapia de animales, en particular seres humanos.
Debido a sus propiedades de modulación de GPR17, los compuestos de la presente invención pueden usarse como medicina y pueden usarse para el tratamiento y/o la prevención de diversas enfermedades del sistema SNC.
Una realización de la presente descripción, por lo tanto, es un compuesto como se describe en la presente memoria para el uso como medicamento, en particular para el uso como medicamento para el tratamiento y/o la prevención de una enfermedad asociada con GPR17.
Una enfermedad o trastorno asociado con GPR17 es una enfermedad que está asociada con una disfunción del sistema de señalización de GPR17 tal como, por ejemplo, una sobreexpresión y/o hiperactividad de los receptores GPR17. Sin pretender limitarse por ninguna teoría, la actividad de GPR17 puede aumentar, extenderse o alterarse de otro modo en ciertos tejidos, por ejemplo, en las células progenitoras de oligodendrocitos (CPO) o durante la maduración de los oligodendrocitos, potencialmente debido a la activación de estímulos endógenos como, por ejemplo, ejemplo, factores de inflamación. La alta actividad de GPR17 puede impedir la diferenciación de los oligodendrocitos y una mielinización eficiente, promoviendo así la aparición o el desarrollo adicional de una enfermedad de la mielinización (véase Chen et al., anteriormente mencionado). Los moduladores negativos de GPR17 pueden promover así la mielinización al disminuir o apagar la actividad de GPR17 y al apoyar la maduración de las c Po hasta oligodendrocitos productores de mielina (véase, por ejemplo, Simon et al., anteriormente mencionado).
En un aspecto preferido, la invención se refiere a cualquiera de los compuestos descritos en la presente memoria, para el uso en la terapia o el diagnóstico para el uso en la prevención o el tratamiento de un trastorno o síndrome seleccionado de y/o asociado con un trastorno de la mielinización, en particular un trastorno desmielinizante, como el del sistema nervioso central. En una realización, los compuestos de la presente invención se usan para promover, estimular y/o acelerar la remielinización en un animal que lo necesite. En una realización, la remielinización asociada con la administración de un compuesto de la presente invención evitará o tratará una enfermedad desmielinizante tal como, pero sin limitación, la esclerosis múltiple.
Los compuestos de la presente invención también pueden ser útiles en el tratamiento o la prevención de un trastorno o síndrome asociado con daño al tejido cerebral, un trastorno cerebrovascular y ciertas enfermedades neurodegenerativas.
Recientemente, se han asociado estrechamente los trastornos neurodegenerativos con una pérdida de mielinización. En consecuencia, se cree que la funcionalidad conservada oligodendroglial y de la mielina es un prerrequisito crucial para la prevención de la degeneración axonal y neuronal (véase, por ejemplo, Ettle et al., anteriormente mencionado). Por lo tanto, los moduladores negativos de GPR17 pueden representar una excelente opción de tratamiento para cualquier enfermedad neurodegenerativa asociada con la desmielinización y/o con una mielinización alterada, como p. ej. ELA, AMS, enfermedad de Alzheimer, enfermedad de Huntington o enfermedad de Parkinson.
En un aspecto particular preferido, los compuestos de la presente invención pueden usarse por tanto en la prevención y/o el tratamiento de un trastorno de la mielinización central o periférico, en particular de un trastorno de la mielinización del sistema nervioso central. En un aspecto, los compuestos de la presente invención se usan en el tratamiento y/o la prevención y/o el diagnóstico de un trastorno de la mielinización mediante administración oral. En una realización preferida, el trastorno de la mielinización a tratar con los compuestos de la presente invención es un trastorno desmielinizante.
Los ejemplos de dichos trastornos de la mielinización a tratar y/o prevenir con los compuestos actualmente descritos son, en particular,
- esclerosis múltiple (EM), incluidas sus diversas subformas,
- neuromielitis óptica (también conocida como enfermedad de Devic),
- neuritis óptica inflamatoria crónica recidivante, encefalomielitis diseminada aguda,
- leucoencefalitis hemorrágica aguda (LHA),
- leucomalacia periventricular
- desmielinización debida a infecciones virales, p. ej. por VIH o leucoencefalopatía multifocal progresiva, - mielinólisis central pontina y extrapontina,
- desmielinización debida a daño traumático del tejido cerebral, incluida la desmielinización inducida por compresión, p. ej. por tumores
- desmielinización en respuesta a hipoxia, accidente cerebrovascular o isquemia u otras enfermedades cardiovasculares,
- desmielinización debida a la exposición a dióxido de carbono, cianuro u otras toxinas del SNC
- enfermedad de Schilder,
- esclerosis concéntrica de Balo,
- Encefalopatía perinatal y
- Enfermedades Neurodegenerativas que incluyen, en particular,
° Esclerosis lateral amiotrófica (ELA).
° Enfermedad de Alzheimer (EA).
° Atrofia multisistémica
° Enfermedad de Parkinson
° Ataxia espinocerebelosa (AEC), también conocida como atrofia espinocerebelosa ° Enfermedad de Huntington
- trastornos psiquiátricos como la esquizofrenia y el trastorno bipolar (véase, p. ej., Fields, Trends Neurosci 31, 2008, 361; Tkachev et al., Lancet 362, 2003, 798).
- enfermedades de la mielinización periférica como leucodistrofias, neuropatías desmielinizantes periféricas, síndrome de Dejerine-Sottas o enfermedad de Charcot-Marie-Tooth
El tratamiento o la prevención de una enfermedad del SNC tal como una enfermedad desmielinizante también incluye el tratamiento de los signos y síntomas asociados con dicha enfermedad.
Por ejemplo, el uso de los compuestos de la presente invención para el tratamiento y/o la prevención de la EM también incluye el tratamiento y/o la prevención de los signos y síntomas asociados con la EM, como los efectos negativos sobre los nervios ópticos (pérdida de visión, visión doble), columnas dorsales (pérdida de sensibilidad), tracto corticoespinal (debilidad espástica), vías cerebelosas (falta de coordinación, disartria, vértigo, deterioro cognitivo), fascículo longitudinal medial (visión doble en la mirada lateral), tracto espinal del trigémino (entumecimiento o dolor facial), debilidad muscular (alteración de la deglución, control de la vejiga o del intestino, espasmos) o efectos psicológicos asociados con la enfermedad subyacente, como depresión, ansiedad u otros trastornos del estado de ánimo, debilidad general o insomnio.
Por lo tanto, los compuestos de la presente invención son para el uso en el tratamiento de los signos y síntomas de una enfermedad de la mielinización, en particular una enfermedad desmielinizante tal como la esclerosis múltiple; dichos signos y síntomas de la EM incluyen, pero sin limitación, el grupo de pérdida de la visión, deterioro de la visión, visión doble, pérdida o deterioro de la sensibilidad, debilidad tal como debilidad espástica, descoordinación motora, vértigo, deterioro cognitivo, entumecimiento facial, dolor facial, alteración de la deglución, alteración del habla, alteración del control de la vejiga y/o el intestino, espasmos, depresión, ansiedad, trastornos del estado de ánimo, insomnio y fatiga.
En una realización preferida, los compuestos de la presente invención son para el uso en el tratamiento de la esclerosis múltiple. La EM es una enfermedad de la mielinización heterogénea y puede manifestarse en una variedad de formas y etapas diferentes, que incluyen, entre otras, EM recurrente-remitente, EM secundaria progresiva, EM primaria
progresiva, EM recurrente progresiva, cada una según la actividad y la progresión de la enfermedad. Por lo tanto, en una realización, los compuestos de la presente invención son para el uso en el tratamiento de la esclerosis múltiple en sus diversas etapas y formas, como se describe en la presente memoria.
En un aspecto, los compuestos de la presente invención son para el uso en el tratamiento y/o la prevención de la neuromielitis óptica (también conocida como enfermedad de Devic o síndrome de Devic). La neuromielitis óptica es un trastorno complejo caracterizado por inflamación y desmielinización del nervio óptico y la médula espinal. Muchos de los síntomas asociados son similares a los de la EM e incluyen debilidad muscular, en particular de las extremidades, sensibilidad reducida y pérdida del control de la vejiga.
En un aspecto, los compuestos de la presente invención son para el uso en la prevención y/o el tratamiento de la ELA. La ELA se ha asociado recientemente con la degeneración de los oligodendrocitos y una mayor desmielinización, lo que sugiere que la ELA es una enfermedad de interés para los moduladores negativos de GPR17 (Kang et al., anteriormente mencionado; Fumagalli et al., Neuropharmacology 104, 2016, 82).
En un aspecto, los compuestos de la presente invención son para el uso en la prevención y/o el tratamiento de la enfermedad de Huntington. Está bien descrito que la enfermedad de Huntington está asociada con una mielinización alterada (Bartzokis et al., anteriormente mencionado; Huang et al., Neuron 85, 2015, 1212).
En un aspecto, los compuestos de la presente invención son para el uso en la prevención y/o el tratamiento de la atrofia multisistémica. Recientemente, la AMS se asoció estrechamente con la desmielinización (Ettle, anteriormente mencionado; Jellinger, anteriormente mencionado), lo que sugiere las estrategias de remielinización para tratar o prevenir la AMS.
En un aspecto, los compuestos de la presente invención son para el uso en la prevención y/o el tratamiento de la enfermedad de Alzheimer. Recientemente se ha observado que la EA está asociada con un aumento de la muerte celular de los oligodendrocitos y la desmielinización focal, y que representa un proceso patológico en la EA (Mitew et al., Acta Neuropathol 119, 2010, 567),
Un aspecto de la presente invención se refiere a un método de tratamiento de cualquiera de las enfermedades o trastornos descritos en la presente memoria, en particular de una enfermedad de la mielinización como la EM, neuromielitis óptica, ELA, corea de Huntington, enfermedad de Alzheimer u otras, mediante la administración a un sujeto que lo necesite, incluido un paciente humano, de una cantidad terapéuticamente eficaz de un compuesto de la presente invención.
En otro aspecto, el compuesto de la presente invención se puede usar en la prevención y el tratamiento de una lesión de la médula espinal, encefalopatía perinatal, accidente cerebrovascular, isquemia o trastorno cerebrovascular.
En un aspecto, la invención se refiere a un método para la prevención y/o el tratamiento de un síndrome o trastorno asociado con un trastorno de la mielinización, o con un trastorno o síndrome asociado con un daño en el tejido cerebral, que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto como se describe en la presente memoria. Un paciente que necesita dicho tratamiento puede ser cualquier paciente que haya sufrido daños en el tejido cerebral, por ejemplo, por un traumatismo mecánico, químico, vírico o de otro tipo.
En un aspecto, la invención se refiere a un método para la prevención y/o el tratamiento de un síndrome o trastorno asociado con un trastorno de la mielinización, o con un trastorno o síndrome asociado con un accidente cerebrovascular u otra isquemia cerebral, que comprende administrar a un paciente que lo necesite una cantidad terapéuticamente eficaz de un compuesto como se describe en la presente memoria. Un paciente que lo necesite puede ser cualquier paciente que haya experimentado recientemente una isquemia cerebral/accidente cerebrovascular que puede haber sido causado, por ejemplo, por la oclusión de una arteria cerebral por un émbolo o por una trombosis local.
Recientemente también se ha asociado a GPR17 con la ingesta de alimentos, el control de la insulina y la obesidad. Según varios informes, los moduladores negativos de GPR17 pueden ser útiles para controlar la ingesta de alimentos y para tratar la obesidad (véase, p. ej., Ren et al., Diabetes 64, 2015; 3670). Por lo tanto, una realización de la presente invención se refiere al uso de los compuestos de la presente memoria para la prevención y/o el tratamiento de la obesidad, y métodos para tratar la obesidad.
Además, los compuestos de la presente invención se pueden usar para el tratamiento o la prevención de tejidos en los que se expresa GPR17, como p. ej. el corazón, pulmón o riñón. En una realización, los compuestos de la presente invención pueden usarse para tratar o prevenir los trastornos isquémicos del riñón y/o del corazón.
GPR17 también se ha asociado con la inflamación pulmonar y el asma como, por ejemplo, la inducida por ácaros del polvo doméstico (Maekawa, J Immunol 2010, 185(3), 1846-1854). Por lo tanto, los compuestos de la presente invención pueden usarse para el tratamiento del asma u otra inflamación pulmonar.
El tratamiento según la presente invención puede comprender la administración de uno de los compuestos actualmente descritos como tratamiento "independiente" de una enfermedad del SNC, en particular de una
enfermedad o trastorno de la mielinización tal como EM o ELA. Alternativamente, un compuesto descrito en la presente memoria puede administrarse junto con otros fármacos útiles en una terapia de combinación.
En un ejemplo no limitante, un compuesto según la presente invención se combina con otro medicamento para tratar una enfermedad de la mielinización, como la EM, que tiene un modo de acción diferente, como p. ej. un fármaco antiinflamatorio o inmunosupresor. Dichos compuestos incluyen, entre otros: (i) corticosteroides como prednisona, metilprednisona o dexametasona, (ii) interferones beta como interferón beta-1a, interferón beta-1b o peginterferón beta-1a, (iii) anticuerpos anti-CD20 como ocrelizumab, rituximab y ofatumumab, (iv) sales de glatiramer como acetato de glatiramer, (v) fumarato de dimetilo, (vi) fingolimod y otros moduladores del receptor de esfingosina-1-fosfato como ponesimod, siponimod, ozanimod o laquinimod, (vii) inhibidores de la dihidro-orotato deshidrogenasa como teriflunomida o leflunomida, (viii) anticuerpos anti-integrina alfa4 como natalizumab, (ix) anticuerpos anti-CD52 como alemtuzumab, (x) mitoxantrona, (xi) anticuerpos anti-Lingo1 como opicinumab, o (xii) otras terapias inmunomoduladoras como masitinib.
Asimismo, se puede combinar un compuesto de la presente invención con un fármaco analgésico si se va a tratar una afección de la mielinización dolorosa. Además, se puede usar un compuesto de la presente descripción en combinación con un antidepresivo para cotratar los efectos psicológicos asociados con la enfermedad de la mielinización subyacente que se va a tratar.
En las terapias combinadas, los dos o más principios activos pueden proporcionarse a través de la misma formulación o como un "kit de partes", es decir, en unidades galénicas separadas. Además, los dos o más principios activos, incluidos los compuestos de la presente invención, pueden administrarse al paciente al mismo tiempo o posteriormente, p. ej. en una terapia a intervalos. El fármaco adicional puede administrarse mediante el mismo modo de administración o mediante uno diferente. Por ejemplo, el modulador de GPR17 de la presente invención puede administrarse por vía oral, mientras que el segundo medicamento puede administrarse mediante inyección subcutánea.
En un aspecto, los compuestos de la presente invención pueden usarse para el diagnóstico y/o la monitorización de una enfermedad relacionada con GPR17, como se describe más adelante en la presente memoria, en particular una enfermedad desmielinizante, como se describe en la presente memoria, preferiblemente en el diagnóstico y la monitorización de la esclerosis múltiple.
En un aspecto, los compuestos de la presente invención pueden usarse para diagnosticar y/o monitorizar la expresión, distribución y/o activación del receptor GPR17 ya sea en vivo, p.ej. directamente en un sujeto, como mediante el uso de técnicas de formación de imágenes moleculares, o in vitro, como p. ej. mediante el examen de muestras tales como fluidos corporales o tejidos tomados de un sujeto. Cualquier determinación de la actividad, expresión y/o distribución de GPR17 puede usarse para predecir, diagnosticar y/o monitorizar (a) el estado y la progresión de una enfermedad asociada a GPR17 como se describe en la presente memoria, en particular una enfermedad de la mielinización que incluye, entre otros, por ejemplo, la esclerosis múltiple, y (b) la eficacia y/o aplicabilidad y/o dosificación adecuada de un tratamiento asociado con cualquier enfermedad asociada con GPR17.
En un aspecto, los compuestos de la presente invención se pueden usar como trazadores de PET o SPECT, como se describe más adelante en la presente memoria, para realizar in vivo el diagnóstico y/o la monitorización de enfermedades. De este modo, puede medirse directamente la expresión, activación y/o distribución de un receptor GPR17 en un sujeto, p. ej. mediante la formación de imágenes de un paciente humano después de la administración de un trazador de PET o SPECT de GPR17 de la presente invención. Esto puede facilitar un diagnóstico adecuado de la enfermedad, puede ayudar a determinar las opciones de tratamiento aplicables y/o puede usarse para monitorizar la progresión de la enfermedad y/o monitorizar o predecir el éxito de una intervención médica, incluida la selección y administración adecuada y/o dosificación de un fármaco terapéutico.
En una realización, los trazadores de PET o SPECT de la presente invención se pueden usar junto con un fármaco terapéutico, es decir, como un diagnóstico complementario, para controlar y/o predecir la eficacia y/o seguridad de dicho fármaco terapéutico en un sujeto determinado, o para estimar la dosis adecuada de un fármaco.
Una realización se refiere a un trazador de PET o SPECT de la presente invención para su uso como un fármaco complementario junto con un fármaco terapéutico. El fármaco terapéutico a utilizar con el trazador de PET o SPECT de la presente invención puede seleccionarse del grupo de (a) un compuesto no marcado de la presente invención, (b) un compuesto modulador de GPR17 que es diferente de los compuestos de la presente invención y (c) un fármaco para el tratamiento de una enfermedad de la mielinización, que incluye, entre otros, un fármaco para el uso en el tratamiento de la esclerosis múltiple que no es un modulador de GPR17, como se describe más adelante en la presente memoria.
Una realización se refiere a un kit que comprende
(a) como primer componente, un trazador de PET o SPECT de la presente invención, en particular un trazador de PET o SPECT basado en los compuestos de la presente invención, pero que ha incorporado al menos un radionúclido que es adecuado para la formación de imágenes de PET o SPECT, preferiblemente un radionúclido seleccionado de 18F, 11C, 123I, 125I y 131I,
(b) como segundo componente, un fármaco terapéutico seleccionado de
i. un compuesto de la presente invención como se define más adelante en la presente memoria, y que no tiene un radionúclido incorporado,
ii. un compuesto modulador de GPR17 que es diferente de los compuestos de la presente invención, y
iii. un fármaco para el tratamiento de una enfermedad de la mielinización, que incluye, entre otros, un fármaco para el uso en el tratamiento de la esclerosis múltiple, pero que no tiene actividad moduladora de GPR17; tales compuestos son conocidos por un experto en la técnica, incluidos los ejemplos descritos anteriormente.
Alternativamente, los compuestos de la presente invención se pueden usar en un ensayo de diagnóstico in vitro, por ejemplo, para el examen de fluidos corporales adecuados de un sujeto, como p. ej. sangre, plasma, orina, saliva o líquido cefalorraquídeo para cualquier nivel de expresión, actividad y/o distribución de GPR17.
Una realización se refiere a un método para tratar una enfermedad asociada a GPR17, en particular una enfermedad de la mielinización que incluye, pero sin limitación, la esclerosis múltiple, donde dicho método incluye las etapas de (a) determinar la expresión, actividad y/o distribución del receptor GPR17 de un sujeto, (b) comparar la expresión, actividad y/o distribución del receptor GPR17 en dicho sujeto con la expresión, actividad y/o distribución del receptor GPR17 en uno o más sujetos sanos o una población, (c) determinar la necesidad de tratamiento médico o profilaxis de dicho sujeto basándose en una desviación de la expresión, actividad y/o distribución de GPR17 en dicho sujeto respecto de los sujetos sanos o una población y (d) tratar al sujeto con la desviación de la expresión, actividad y/o distribución del receptor GPR17 mediante la administración de un fármaco terapéutico a dicho individuo, cuyo fármaco es adecuado para el tratamiento de enfermedades o trastornos asociados a GPR17, en particular mediante la administración de un modulador de GPR17, preferiblemente mediante la administración de uno o más de los compuestos de la presente invención. En una realización, la determinación (a) de la expresión, actividad y/o distribución de GPR17 se realizará utilizando uno de los compuestos de la presente invención, en particular con un trazador de PET o SPECT, o mediante un examen in vitro de fluidos corporales o tejido de dicho sujeto utilizando un trazador de PET o SPECT de la presente invención.
En un aspecto preferido, la invención se refiere a una composición farmacéutica que comprende un compuesto como se describe en la presente memoria y un vehículo farmacéuticamente aceptable.
Para la administración como medicamento, los compuestos pueden usarse en una composición farmacéutica que comprende un compuesto de la presente descripción y un vehículo farmacéuticamente aceptable, como se define más adelante en la presente memoria. Tal composición farmacéutica se puede adaptar, por ejemplo, para la administración oral, intravenosa, intramuscular, subcutánea, nasal, rectal, transcraneal, bucal o transdérmica y puede comprender vehículos, adyuvantes, diluyentes, estabilizantes y similares farmacéuticamente aceptables.
Por ejemplo, los compuestos de la presente invención se pueden disolver en aceites, propilenglicol u otros disolventes que se usan comúnmente para producir una inyección. Los ejemplos adecuados de los vehículos incluyen, entre otros, solución salina fisiológica, polietilenglicol, etanol, aceites vegetales, miristato de isopropilo, etc. Los compuestos de la presente invención pueden formularse en inyecciones disolviéndolos, suspendiéndolos o emulsionándolos en un disolvente soluble en agua como solución salina y dextrosa al 5%, o en disolventes insolubles en agua como aceites vegetales, glicéridos de ácidos grasos sintéticos, ésteres de ácidos grasos superiores y propilenglicol. Las formulaciones de la invención pueden incluir cualquiera de los aditivos convencionales, tales como agentes de disolución, agentes isotónicos, agentes de suspensión, emulsionantes, estabilizantes y conservantes.
En una realización, los compuestos de la presente invención se pueden administrar por vía oral, p. ej. en forma de un comprimido, cápsula, gragea, polvo, granulado o en forma de un líquido o semisólido, que incluyen, p. ej., jarabes, suspensiones, emulsiones o disoluciones, a modo de ejemplo no limitante.
Las formulaciones orales pueden contener, entre otros, agentes de liberación sostenida, desintegrantes, rellenos, lubricantes, estabilizantes, antioxidantes, sabores, agentes de dispersión, electrolitos, tampones, colorantes o agentes de conservación. Los expertos en la técnica conocen los excipientes y formulaciones adecuados y se describen en monografías habituales, como Remington ("The science and practice of pharmacy", Lippincott, Williams & Wilkins, 2000) o se describen en otras fuentes bien conocidas por los expertos en la técnica.
Se puede preparar un comprimido, por ejemplo, mezclando al menos un compuesto de la presente invención con al menos un excipiente atóxico farmacéuticamente aceptable, como p. ej. un aglutinante, relleno/diluyentes, agentes desintegrantes, plastificantes y similares, y un disolvente opcional (acuoso o no acuoso), y mediante el procesamiento posterior de la mezcla hasta un comprimido mediante un proceso que incluye, entre otros, la compresión en seco, granulación en seco, granulación en húmedo, secado por pulverización o extrusión de una masa fundida. Un comprimido puede estar sin recubrir o recubierto mediante técnicas conocidas para enmascarar el mal sabor de un fármaco de sabor desagradable o retrasar la desintegración y absorción del ingrediente activo en el tracto
gastrointestinal. Un comprimido puede proporcionar una liberación inmediata o una liberación sostenida de los compuestos de la presente invención.
Los agentes de liberación sostenida típicos son, por ejemplo, aquellos que se hinchan al contacto con agua tales como polivinilpirrolidona, hidroxietilcelulosa, hidroxipropilcelulosa, otros éteres de celulosa, almidón, almidón pregelatinizado, polimetacrilato, poli(acetato de vinilo), celulosa microcristalina, dextranos y mezclas de estos. Los ejemplos no limitantes de desintegrantes incluyen almidón pregelatinizado, glicolato de almidón sódico, celulosa microcristalina, carboximetilcelulosa sódica (CMC-Na), CMC-Na reticulada e hidroxipropilcelulosa poco sustituida, así como mezclas de los mismos. Los rellenos y aglutinantes adecuados incluyen, entre otros, celulosa microcristalina, celulosa en polvo, lactosa (anhidra o monohidratada), azúcar comprimible, almidón (por ejemplo, almidón de maíz o almidón de patata), almidón pregelatinizado, fructosa, sacarosa, dextrosa, dextranos, otros azúcares como manitol, maltitol, sorbitol, lactitol y sacarosa, celulosa microcristalina siliconada, hidrogenofosfato de calcio, hidrogenofosfato de calcio dihidrato, fosfato dicálcico dihidrato, fosfato tricálcico, lactato de calcio o mezclas de los mismos. Los lubricantes, antiadherentes y/o deslizantes incluyen ácido esteárico, estearato de magnesio, estearato de calcio, lauril sulfato de sodio, aceite vegetal hidrogenado, aceite de ricino hidrogenado, estearil fumarato de sodio, macrogoles, dibehenato de glicerol, talco, almidón de maíz, dióxido de silicio y similares incluyendo las mezclas.
El compuesto de la presente invención también se puede formular para administración parenteral mediante inyección, p. ej. mediante inyección en bolo o infusión. Las composiciones para inyección pueden proporcionarse listas para usar y pueden tomar formas tales como suspensiones, disoluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener excipientes tales como agentes de suspensión, estabilizantes, conservantes y/o dispersantes. Alternativamente, el ingrediente activo puede estar en forma de polvo para la reconstitución con un vehículo adecuado, p. ej. agua estéril apirógena o solución salina, antes de su uso.
Para la administración nasal o la administración mediante inhalación, los compuestos según la presente invención pueden administrarse convenientemente en forma de una presentación de rociador de aerosoles para envases presurizados o un nebulizador, con el uso de un propulsor adecuado, p. ej. diclorodifluorometano, fluorotriclorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado o mezcla de gases.
Para la administración oftálmica, los compuestos de utilidad en la presente invención pueden formularse convenientemente como suspensiones micronizadas en solución salina isotónica estéril con un pH ajustado, con o sin un conservante, como un agente bactericida o fungicida, por ejemplo, nitrato fenilmercúrico, cloruro de bencilalconio o acetato de clorhexidina. Alternativamente, para la administración oftálmica, los compuestos pueden formularse en un ungüento tal como vaselina.
Para la administración rectal, los compuestos de utilidad en la presente invención pueden formularse convenientemente como supositorios. Estos se pueden preparar mezclando el componente activo con un excipiente no irritante adecuado que sea sólido a temperatura ambiente pero líquido a temperatura rectal y, por lo tanto, se derretirá en el recto para liberar el componente activo. Dichos materiales incluyen, por ejemplo, manteca de cacao, cera de abejas y polietilenglicoles.
En una realización, los compuestos se pueden administrar por vía transdérmica. Este modo de administración impide el llamado efecto del primer paso de la administración oral y, además, permite proporcionar niveles plasmáticos más constantes, lo que es una ventaja particular en algunos casos. El diseño de formas transdérmicas como ungüentos o cremas u otros sistemas transdérmicos como p. ej. parches o dispositivos electroforéticos se conoce en general en la técnica, véase, p. ej. Venkatraman y Gale, Biomaterials 1998, vol. 19, pág. 1119; Prausnitz y Langer, Nat Biotechnology 2008, vol. 26.11 pág. 1261; documentos WO 2001/47503; WO2009/000262; WO99/49852; WO 07/094876.
El nivel de dosis preferible de los compuestos según la presente invención depende de una variedad de factores que incluyen el estado y el peso corporal del paciente, la gravedad de la enfermedad en particular, la forma farmacéutica y la vía y período de administración, pero los expertos en la técnica pueden elegirlo de manera adecuada. En diversas realizaciones, los compuestos se administran en una cantidad que oscila de 0,001 a 10 mg/kg de peso corporal por día, o de 0,03 a 1 mg/kg de peso corporal por día. Las dosis individuales pueden oscilar de aproximadamente 0,1 a 1000 mg de ingrediente activo por día, de aproximadamente 0,2 a 750 mg/día, de aproximadamente 0,3 a 500 mg/día, de 0,5 a 300 mg/día o de 1 a 100 mg/día. Las dosis se pueden administrar una vez al día, o varias veces al día con cada porción dividida.
Otro aspecto de la presente invención es un kit que comprende un medicamento o una composición farmacéutica como se describe en la presente memoria, y las instrucciones para su uso.
Definiciones
Los compuestos de la presente invención incluyen los compuestos abarcados por las fórmulas I, Ia, Ib, Ic, Ila, IIb, IIc, IIIa, IIIb, IIIc, IVa, IVb y IVc como se describe con más detalle en la presente memoria, así como los compuestos individuales finales descritos en la descripción y la sección experimental.
Cualquier referencia a un compuesto según la presente invención también incluye las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de dichos compuestos, a menos que se indique expresamente lo contrario.
La expresión "sales farmacéuticamente aceptables" se refiere a cualquier sal que puedan formar los compuestos y que sea adecuada para la administración a sujetos, en particular a seres humanos, según la presente invención. Dichas sales incluyen, entre otras, sales de adición de ácido, formadas con ácidos inorgánicos como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares, o formadas con ácidos orgánicos como ácido acético, ácido propiónico, ácido hexanoico, ácido ciclopentanopropiónico, ácido glicólico, ácido pirúvico, ácido láctico, ácido malónico, ácido succínico, ácido málico, ácido maleico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido 3-(4-hidroxibenzoil)-benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido 1,2-etanodisulfónico, ácido 2-hidroxietanosulfónico, ácido bencenosulfónico, ácido 4-clorobencenosulfónico, ácido 2-naftalenosulfónico, ácido 4-toluenosulfónico, ácido canforsulfónico, ácido 4-metilbiciclo[2.2.2]oct-2-en-1-carboxílico, ácido glucoheptónico, ácido 3-fenilpropiónico, ácido trimetilacético, ácido tercbutilacético, ácido laurilsulfúrico, ácido glucónico, ácido glutámico, ácido hidroxinaftoico, ácido salicílico, ácido esteárico, y ácido mucónico. Otras sales incluyen 2,2-dicloroacetato, adipato, alginato, ascorbato, aspartato, 2-acetamidobenzoato, caproato, caprato, alcanforato, ciclamato, laurilsulfato, edisilato, esilato, isetionato, formiato, galactato, gentisato, gluceptato, glucuronato, oxoglutarato, hipurato, lactobionato, napadisilato, xinafoato, nicotinato, oleato, orotato, oxalato, palmitato, embonato, pidolato, p-aminosalicilato, sebacato, tanato, rodanida, undecilenato y similares; o sales formadas cuando se reemplaza un protón ácido presente en el compuesto original, como por ejemplo con amoníaco, arginina, benetamina, benzatina, calcio, colina, deanol, dietanolamina, dietilamina, etanolamina, etilendiamina, meglumina, glicina, hidrabamina, imidazol, lisina, magnesio, hidroxietilmorfolina, piperazina, potasio, epolamina, sodio, trolamina, trometamina o zinc.
La presente invención incluye en su alcance los solvatos de los compuestos definidos en la presente memoria. Los "solvatos" son cristales formados por un compuesto activo y un segundo componente (disolvente) que, en forma aislada, es líquido a temperatura ambiente. Estos solvatos se pueden formar con disolventes orgánicos comunes, p. ej. disolventes de hidrocarburos tales como benceno o tolueno; disolventes clorados como cloroformo o diclorometano; disolventes alcohólicos como metanol, etanol o isopropanol; disolventes etéreos tales como éter dietílico o tetrahidrofurano; o disolventes de éster tales como acetato de etilo. Alternativamente, los solvatos de los compuestos de la presente memoria pueden formarse con agua, en cuyo caso serán hidratos.
La presente invención también incluye cocristales en su alcance. El término "co-cristal" se utiliza para describir la situación en la que hay presentes componentes moleculares neutros dentro de un compuesto cristalino en una relación estequiométrica definida. La preparación de cocristales farmacéuticos permite realizar modificaciones en la forma cristalina de un ingrediente farmacéutico activo, lo que a su vez puede alterar sus propiedades fisicoquímicas sin comprometer la actividad biológica prevista. Los ejemplos de formadores de cocristales, que pueden estar presentes en el cocristal junto con el ingrediente farmacéutico activo, incluyen ácido L-ascórbico, ácido cítrico, ácido glutárico, ácido cinámico, ácido mandélico, urea y nicotinamida.
La invención también incluye todas las variaciones isotópicas adecuadas de un compuesto de la invención. Un "variación isotópica", o brevemente "isótopo" de un compuesto de la invención se define como uno en el que al menos un átomo se reemplaza por un átomo que tiene el mismo número atómico pero una masa atómica diferente de la masa atómica que normalmente se encuentra en la naturaleza, prefiriéndose los isótopos más abundantes. Los ejemplos de isótopos que se pueden incorporar a los compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, azufre, flúor y cloro tales como 2H, 3H, 11C, 13C, 14C, 15N, 17O, 18O, 35S, 18F, y 36Cl, respectivamente. Ciertas variaciones isotópicas de la invención, por ejemplo aquellas en las que se incorpora un isótopo radiactivo como 3H o 14C, son útiles en los estudios de distribución tisular de fármacos y/o sustratos. Se prefieren especialmente los isótopos de tritio, es decir 3H, y carbono-14, es decir 14C, por su facilidad de preparación y detectabilidad. Además, la sustitución con isótopos como el deuterio, es decir 2H, puede proporcionar ciertas ventajas terapéuticas resultantes de una mayor estabilidad metabólica, por ejemplo, una mayor semivida in vivo, requisitos de dosificación reducidos y, por lo tanto, puede preferirse en algunas circunstancias. Las variaciones isotópicas de los compuestos de la invención generalmente se pueden preparar mediante procedimientos convencionales usando variaciones isotópicas apropiadas de reactivos adecuados.
También forman parte de la invención aquellos compuestos en los que al menos un átomo ha sido reemplazado por un isótopo radiactivo (radioisótopo) del mismo átomo o de uno diferente que se puede utilizar en técnicas de imagen in vivo como la tomografía computarizada por emisión de fotón único (SPECT) o la tomografía por emisión de positrones (PET).
Los ejemplos de tales variaciones isotópicas de moduladores de GPR17 utilizables en los estudios de SPECT (dichos compuestos se denominan en la presente memoria "trazadores de SPECT") son compuestos en los que se ha introducido un 99mTc, 111In, 82Rb, 137Cs, 123I, 125I, 131I, 67Ga, 192Ir o 201Tl, y preferiblemente 123I. Por ejemplo, para que los compuestos de la presente invención se utilicen como trazadores de SPECT, puede introducirse un isótopo 123I en un modulador de GPR17 como se describe en la presente memoria. A modo de ejemplo no limitante, para que un compuesto se utilice como trazador de SPECT, puede introducirse un radionúclido seleccionado de 123I, 125I y 131I en un compuesto de la presente invención. En una realización, un trazador de SPECT de la presente invención puede basarse en la estructura de un modulador de GPR17 que contiene halógenos descrito en la presente memoria, en el
que se ha introducido uno de los radionúclidos 123I, 125I y 1311 en la posición de un halógeno, preferentemente, un átomo de yodo.
En consecuencia, la expresión "trazador de SPECT de la presente invención", se refiere a compuestos como se describen en la presente solicitud de patente y que tienen una estructura de acuerdo con cualquiera de los compuestos de la presente invención como se define adicionalmente en la presente memoria, en los que se ha introducido al menos un radioisótopo que es adecuado para la formación de imágenes de SPECt . Esto incluye, pero sin limitación, 99mTc, 111In, 82Rb, 137Cs, 123I, 125I, 131I, 67Ga, 192Ir o 201Tl.
Los ejemplos de derivados moduladores de GPR17 utilizables en las aplicaciones de PET (en la presente memoria, "trazadores de PET") son compuestos en los que se ha introducido 11C, 13N, 15O, 18F, 76Br o 124I. Por ejemplo, para que un compuesto se utilice como trazador de PET, puede introducirse un isótopo 18F en un compuesto de la presente invención. En una realización, un trazador de PET puede basarse en la estructura de un modulador de GPR17 que contiene flúor descrito en la presente memoria, en el que se ha introducido el radionúclido 18F respectivo en la posición del átomo de flúor. Esto también es aplicable de forma similar a la introducción de al menos un 11C, 13N, 15O, 76Br o 124I en lugar de un átomo de carbono, nitrógeno, oxígeno, bromo o yodo "no marcado", respectivamente (véase, p. ej., Pimlott y Sutherland, Chem Soc Rev 2011, 40, 149; van der Born et al., Chem Soc Rev 2017, 46, 4709).
En consecuencia, la expresión "trazador de PET de la presente invención" se refiere a compuestos de la presente invención como se define adicionalmente en la presente memoria, en los que se ha introducido al menos un radioisótopo que es adecuado para la formación de imágenes de PET. Esto incluye, pero sin limitación, 11C, 13N, 15O, 18F, 76Br o 124I.
La presente invención incluye dentro de su alcance los profármacos de los compuestos de la presente invención. En general, dichos profármacos serán derivados funcionales de los compuestos descritos en la presente memoria que son fácilmente convertibles in vivo, p. ej. por enzimas endógenas en el intestino o la sangre, hasta los compuestos moduladores de GPR17 requeridos descritos en la presente memoria. Los procedimientos convencionales para la selección y preparación de derivados de profármacos adecuados se describen, por ejemplo, en Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985.
Dependiendo de su patrón de sustitución, los compuestos de la presente invención pueden tener o no uno o más estereocentros ópticos, y pueden existir o no como diferentes enantiómeros o diastereómeros. Cualquiera de tales enantiómeros, diastereómeros u otros isómeros ópticos están incluidos en el alcance de la invención.
El compuesto de la presente invención también puede existir en diferentes formas cristalinas, es decir, como polimorfos, todas las cuales están abarcadas por la presente invención.
Los compuestos de la presente invención pueden incluirse en una composición farmacéutica que también puede incluir un vehículo farmacéuticamente aceptable. "Vehículo farmacéuticamente aceptable" se refiere a un diluyente, adyuvante, excipiente o vehículo, u otro ingrediente con el que se administra un compuesto de la invención y que un experto en la técnica entendería como farmacéuticamente aceptable.
Los compuestos de la presente invención son útiles en la prevención y/o el tratamiento de ciertas enfermedades o trastornos en animales, en particular en humanos, como se describe en la presente memoria.
"Prevenir" o "prevención" se refieren a una reducción del riesgo de adquirir una enfermedad o trastorno (es decir, hacer que al menos uno de los síntomas clínicos de la enfermedad no se desarrolle en un sujeto, en particular un sujeto humano, que puede estar expuesto o predispuesto a la enfermedad, pero aún no experimenta ni muestra síntomas de la enfermedad).
"Tratar" o "tratamiento" de cualquier enfermedad o trastorno incluye, en una realización, mejorar la enfermedad o el trastorno (es decir, detener o reducir el desarrollo de la enfermedad o al menos reducir uno de los síntomas clínicos de la enfermedad). En otra realización, "tratar" o "tratamiento" se refiere a mejorar al menos un parámetro físico, que puede o no ser perceptible por el sujeto, en particular un sujeto humano, pero que se basa o está asociado a la enfermedad o trastorno a tratar. En otra realización más, "tratar" o "tratamiento" se refiere a modular o aliviar la enfermedad o el trastorno, ya sea físicamente (por ejemplo, la estabilización de un síntoma discernible o no discernible), fisiológicamente (por ejemplo, la estabilización de un parámetro fisiológico), o ambos. En otra realización más, "tratar" o "tratamiento" se refiere a retrasar el inicio o la progresión de la enfermedad o trastorno. Por lo tanto, "tratar" o "tratamiento" incluye cualquier tratamiento causal de la enfermedad o trastorno subyacente (es decir, la modificación de la enfermedad), así como cualquier tratamiento de los signos y síntomas de la enfermedad o trastorno (ya sea con o sin la modificación de la enfermedad), así como cualquier alivio o mejora de la enfermedad o trastorno, o sus signos y síntomas.
"Diagnóstico", "diagnósticos" o "diagnosticar" una enfermedad o trastorno incluyen, en una realización, la identificación y medición de los signos y síntomas que están asociados a dicha enfermedad. "Diagnóstico", "diagnósticos" o "diagnosticar" incluyen, entre otros, la detección y/o medición de los receptores GPR17 expresados, activados o distribuidos disminuidos, aumentados o de otro modo expresados incorrectamente (por ejemplo, con respecto al tiempo o el lugar) como indicador de una enfermedad o trastorno relacionado con GPR17, en comparación con los
sujetos sanos. En un ejemplo, los ligandos de GPR17 pueden usarse como trazadores de PET o SPECT para dicho diagnóstico, incluido el diagnóstico de una enfermedad de la mielinización.
Los términos "enfermedad(es)"y "trastorno(s)" se utilizan en gran medida de forma intercambiable en la presente memoria.
"Monitorización" se refiere a la observación de una enfermedad, afección o al menos un parámetro médico durante un cierto período de tiempo. "Monitorización" también incluye las observaciones de los efectos de un fármaco terapéutico con la ayuda de un "Fármaco acompañante".
"Diagnóstico complementario", como se usa en la presente memoria, se refiere a un compuesto que se puede usar junto con un fármaco terapéutico con el objetivo de determinar la aplicabilidad (por ejemplo, con respecto a la seguridad y eficacia) de dicho fármaco terapéutico para un paciente específico. El uso de un "diagnóstico complementario" puede incluir etapas de diagnóstico y monitorización.
El término "animal(es)" y "sujeto(s)" incluye los seres humanos. Los términos "humano", "paciente" y "sujeto humano" normalmente se usan indistintamente en la presente memoria, a menos que se indique claramente.
La invención también se refiere a métodos para tratar una enfermedad o trastorno animal, como se describe con más detalle en la presente memoria, en particular una enfermedad o trastorno humano, que incluye la administración de los compuestos de la presente invención en cantidades terapéuticamente eficaces.
"Cantidad terapéuticamente eficaz" significa la cantidad de un compuesto que, cuando se administra a un sujeto, en particular un sujeto humano, para tratar una enfermedad, es suficiente para efectuar dicho tratamiento de la enfermedad. La "cantidad terapéuticamente eficaz" puede variar dependiendo del compuesto, la enfermedad y su gravedad, y el estado, edad, peso, sexo, etc. del sujeto, en particular un sujeto humano, a tratar.
El término "esclerosis múltiple", tal como se usa en la presente memoria, se refiere a la enfermedad clasificada en la Sección G35 del código de diagnóstico ICD-10-CM de la edición estadounidense de 2018.
El término "moduladores de GPR17", como se usa en la presente memoria, pretende describir los compuestos que son capaces de modular la actividad del receptor de GPR17, en particular los compuestos que son capaces de disminuir la actividad de GPR17. Dichos "moduladores negativos de GPR17" incluyen los antagonistas de GPR17 que son capaces de bloquear los efectos de los agonistas de GPR17, así como los agonistas inversos de GPR17 que también son capaces de inhibir los receptores GPR17 o variantes de los receptores constitutivamente activos. Los moduladores de GPR17 preferidos de la presente invención son los agonistas inversos de GPR17.
El término "fluorometilo", tal como se usa, se refiere a un grupo metilo que está sustituido con uno ("monofluorometilo"), dos (difluorometilo) o tres ("trifluorometilo") átomos de flúor. Un grupo fluoroalquilo particularmente preferido es difluorometilo: -CHF2.
El término "fluorometoxi", tal como se usa, se refiere a un grupo metoxi que está sustituido con uno ("monofluorometoxi"), dos ("difluorometoxi") o tres ("trifluorometoxi") átomos de flúor. Un ejemplo de un grupo fluoroalcoxi es trifluorometoxi -OCF3.
El término "ciclopropilo", tal como se usa en la presente memoria, se refiere a un grupo monovalente derivado de un hidrocarburo saturado cíclico de tres átomos de carbono que forman un anillo, que puede estar sin sustituir o sustituido con uno o más sustituyentes, como se indica más adelante en la presente memoria.
Parte Experimental:
A. Química
Los compuestos de la presente invención y sus rutas sintéticas se describen con más detalle a continuación.
A-I Métodos generales para producir los compuestos
Los compuestos de fórmula I según la invención se pueden preparar de forma análoga a los métodos convencionales, como entiende el experto en la técnica de la química orgánica sintética.
A-I Métodos generales para producir los compuestos
Los compuestos de fórmula I según la invención se pueden preparar de forma análoga a los métodos convencionales, como entiende el experto en la técnica de la química orgánica sintética.
Cualquier referencia a la síntesis de compuestos de fórmula general I en la presente memoria también es aplicable de forma similar a los compuestos aplicables de fórmula subgenérica Ia, Ib, Ic, IIa, IIb, IIc, IIIa, IIIb, IIIc, IVa, IVb y IVc, y los compuestos ejemplares específicos descritos en la presente memoria.
Según una realización, pueden prepararse algunos compuestos de fórmula general I mediante la reacción de un cloruro de sulfonilo de Fórmula x Ii con una anilina de Fórmula X según la ecuación:

Esta reacción se puede realizar en presencia de una base tal como piridina utilizada como disolvente a temperatura ambiente o calentando a una temperatura que oscila de 60 a 100 °C, posiblemente en presencia de una cantidad catalítica de dimetilaminopiridina.
Alternativamente, algunos compuestos de fórmula general I pueden prepararse mediante la desprotección in situ de un compuesto de fórmula I-P, donde P es un grupo protector como fenilsulfonilo (PhSO2) según la ecuación:

Los compuestos de Fórmula I-P pueden prepararse mediante la reacción de un cloruro de sulfonilo de fórmula XII-P con una anilina de Fórmula X. Esta reacción se puede realizar en presencia de una base tal como piridina utilizada como disolvente a temperatura ambiente o calentando a una temperatura que oscila de 60 a 100 °C.
Algunos compuestos de Fórmula X pueden prepararse mediante la desprotección de un compuesto de fórmula X-P, donde P es un grupo protector como p-metoxibencilo (PMB) usando condiciones ácidas como ácido trifluoroacético puro o con un disolvente como diclorometano. Otras condiciones de desprotección también pueden ser el uso de un agente reductor como hidrógeno en presencia de un catalizador como paladio o hidróxido de paladio posiblemente activado sobre carbono en un disolvente como metanol.

Algunos compuestos de Fórmula X-P pueden prepararse mediante la reacción de una anilina de fórmula X-OH y una molécula de Fórmula Y-LG donde LG es un grupo saliente como el bromo, en presencia de una base como K2Co 3 o KOH en un disolvente polar como DMF o ACN.

Alternativamente, algunos compuestos de Fórmula X-P pueden prepararse mediante una reacción de tipo Mitsunobu de una anilina de Fórmula X-OH y una molécula de Fórmula Y-OH en presencia de un agente activador o una mezcla como cianometilentributilfosforano (CMBP) o trifenilfosfina y azodicarboxilato de diisopropilo (DIAD) en un disolvente como tolueno o THF con calentamiento a una temperatura que oscila de 60 a 120 °C.

Las anilinas de Formula X-OH pueden prepararse según cualquier método conocido por el experto en la técnica o utilizando procedimientos descritos en la bibliografía.
Los compuestos de Fórmula XII pueden prepararse mediante la clorosulfonilación de un compuesto de Fórmula XI según la ecuación:

Esta reacción se puede realizar en presencia de un agente sulfonilante como ácido clorosulfónico en un disolvente polar como acetonitrilo a temperatura ambiente.
De manera similar, los compuestos de Fórmula XII-P, donde P es un grupo protector tal como fenilsulfonilo, pueden prepararse mediante la clorosulfonilación de un compuesto de Fórmula XI-P según la ecuación:
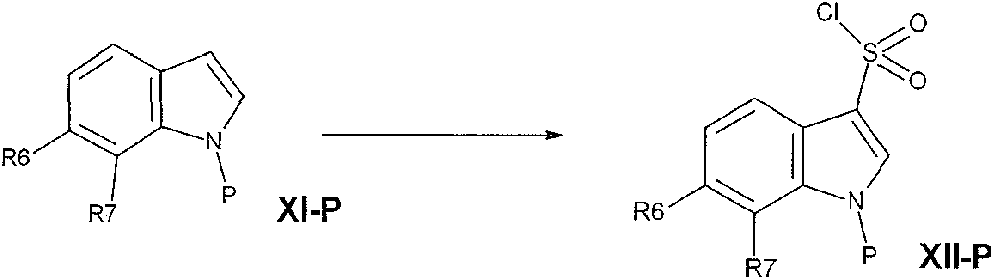
Esta reacción se puede realizar en presencia de ácido clorosulfónico en un disolvente polar tal como acetonitrilo a temperatura ambiente.
Los compuestos de Fórmula XI-P, donde P es un grupo protector tal como fenilsulfonilo, se pueden preparar mediante la protección de un compuesto de Fórmula XI según la ecuación:

Esta reacción se puede realizar según cualquier método conocido por el experto en la técnica.
Los compuestos de Fórmula XI pueden prepararse mediante métodos adecuados bien conocidos por el experto en la técnica.
Alternativamente, algunos compuestos de Fórmula XI pueden prepararse mediante la reacción de un nitroareno ortosustituido XII con un reactivo de Grignard de vinilo XIII (síntesis de indol de Bartoli) según la ecuación:

Esta reacción se puede realizar utilizando un reactivo de Grignard de vinilo, como bromuro de vinil magnesio, en un disolvente polar, como tetrahidrofurano, a una temperatura baja que oscila de -20 a -78 °C.
Alternativamente, algunos compuestos que tienen la fórmula general I pueden prepararse mediante la conversión de grupos funcionales en análogos ya ensamblados de los compuestos que tienen la fórmula general I utilizando procedimientos descritos en la bibliografía o conocidos por el experto en la técnica.
En particular, algunos compuestos de Fórmula I donde R7 es un grupo ciclopropilo pueden prepararse mediante un acoplamiento de tipo Suzuki a partir de un compuesto de Fórmula I donde R7 es un átomo de halógeno, preferentemente bromo, en presencia del correspondiente ácido borónico, una sal de paladio como acetato de paladio, una fosfina como triciclohexilfosfina y una base como fosfato tripotásico en un disolvente como tolueno según métodos conocidos por el experto en la técnica.
A-II. Abreviaturas/reactivos recurrentes
Ac: acetilo
ACN: acetonitrilo
Salmuera: disolución acuosa saturada de cloruro de sodio
NBu: n-butilo
tBu: terc-butilo
CMBP: cianometilentributilfosforano
Ci: ciclohexilo
DAST: fluoruro de dietilaminoazufre
DCM: diclorometano
DIAD: azodicarboxilato de diisopropilo
DMAC: N,N-dimetilacetamida
DMAP: 4-dimetilaminopiridina
DMF: W,W-dimetilformamida
DMSO: sulfóxido de dimetilo
dppf: 1,1 '-bis(difenilfosfino)ferroceno
ES+: ionización positiva por electropulverización
ES-: ionización negativa por electropulverización
ESI: ionización por electropulverización
EtOAc: acetato de etilo
H: hora
LC: cromatografía líquida
LCMS: cromatografía líquida/espectrometría de masas
Me: metilo
MeOH: metanol
min.: minutos
hm: horno de microondas
NBS: N-bromosuccinimida
RMN: resonancia magnética nuclear
Pin: pinacolato
PMB: para-metoxibencilo
t.a.: temperatura ambiente
TBAHSA: hidrogenosulfato de tetrabutilamonio
TFA: ácido trifluoroacético
THF: tetrahidrofurano
TLC: cromatografía en capa fina
A-III. métodos analíticos
Los disolventes y reactivos comerciales se usaron generalmente sin purificación adicional, incluidos los disolventes anhidros cuando fue apropiado (generalmente productos Sure-Seal™ de Aldrich Chemical Company o AcroSeal™ de ACROS Organics). En general, las reacciones se controlaron mediante cromatografía en capa fina o análisis de cromatografía líquida/espectrometría de masas.
Las mediciones de espectrometría de masas en modo LCMS se realizan utilizando diferentes métodos e instrumentos de la siguiente manera:
- Método 1 de LCMS básica:
Se utiliza un espectrómetro de masas de cuadrupolo simple QDA Waters para el análisis de LCMS. Este espectrómetro está equipado con una fuente de ESI y un UPLC Acquity Hclass con detector de matriz de diodos (200 a 400 nm). Los datos se adquieren en un escaneo de MS completo de m/z 70 a 800 en modo positivo/negativo con una elución básica.
La separación de fase inversa se lleva a cabo a 45 °C en una columna Waters Acquity UPLC BEH C18 de 1,7 pm (2,1 x 50 mm) para elución básica.
La elución en gradiente se realiza con agua/ACN/formiato de amonio (95/5/63 mg/L) (disolvente A) y ACN/agua/formiato de amonio (95/5/63 mg/L) (disolvente B) según la tabla 1.
Volumen de inyección: 1 pL. Caudal completo en MS.
Tabla 1:
- Método 2 de LCMS básica:
Los espectros de la espectrometría de masas (MS) se registraron en un espectrómetro de masas LCMS-2010EV (Shimadzu) con ionización por electropulverización (ESI) acoplado a una HPLC Modular Prominence (Shimadzu) usando una columna Xbridge C18-2,1x30 mm, 2,5 pm (Waters). Se inyectó un volumen de 3 pL de disolución de muestra con una concentración de aprox. 1 mg/ml. La fase móvil para las condiciones básicas fue una mezcla de A) formiato de amonio 5 mM amoníaco al 0,1 % en agua, B) fase móvil A al 5 % amoníaco al 0,1 % en acetonitrilo. El gradiente utilizado fue el siguiente: 5:95 (B/A) a 95:5 (B/A) en 4 min y mantener 95:5 (B/A) durante el siguiente 1 min.
- Método 3 de LCMS neutra:
Los espectros de espectrometría de masas (MS) se registraron en un instrumento LCMS (Applied Biosystems API 2000 LC/MS/MS, HPLC Agilent 1100) usando el siguiente procedimiento:
disolución de los compuestos a una concentración de 1,0 m gm l-1 en ACN (Disolvente A) o agua (que contiene acetato de amonio 2 mM): MeOH 90:10 (Disolvente B), y si es necesario sonicación hasta su completa disolución. Luego, se inyectaron 10 pL de la disolución en una columna HPLC Phenomenex Luna C18 (50 x 2,00 mm, tamaño de partícula 3 pm) y se eluyó con un gradiente de agua: ACN (Gradiente A) o agua: MeOH (Gradiente B) de 90 : 10 a 0 : 100 en 10 min, iniciando el gradiente después de 1 min, seguido de elución en disolvente orgánico puro durante 10 min a un caudal de 300 pLm in-1. La absorción UV se detectó de 220 a 400 nm utilizando un detector de matriz de diodos (DAD).
- Método 4 de LCMS ácida:
Se realizó la HPLC-MS en un sistema LC-MS Agilent 1200-6120 acoplado a detección UV (230 a 400 nm y 215 nm) y un espectrómetro de masas Mass Spec Detection Agilent 6120 (ES) con m/z 120 a 800 utilizando una columna X-Bridge C18 Waters, 2,1 x 20 mm, de 2,5 pM. La elución se realizó con un gradiente que se muestra en la Tabla 2 de la Fase Móvil A (formato de amonio 10 mM en agua ácido fórmico al 0,1 %) y la Fase Móvil B (acetonitrilo agua al 5 % ácido fórmico al 0,1 %) con un caudal de 1 ml /min
Tabla 2:

Los materiales brutos se pudieron purificar mediante cromatografía de fase normal, cromatografía de fase inversa (ácida o básica) o recristalización.
La cromatografía de fase normal se realizó utilizando columnas de gel de sílice (gel de sílice de malla 100:200 o cartuchos para sistemas de cromatografía rápida como Isolera™ Four de Biotage® o Teledyne Isco CombiFlash®).
La cromatografía de fase inversa preparativa se realizó con dos instrumentos diferentes y de acuerdo con los métodos siguientes:
- Método 1 de LCMS preparativa básica:
La purificación mediante LCMS utiliza un espectrómetro de masas de cuadrupolo simple SQD o QM Waters para la detección de MS. Este espectrómetro está equipado con una fuente ESI, una bomba binaria Waters 2525 acoplada con un administrador de muestras 2767 y un detector de matriz de diodos (210 a 400 nm).
Parámetros de EM: Tensión capilar ESI 3 kV. Tensión de cono y extractor 10. Temperatura del bloque de la fuente 120 °C. Temperatura de desolvatación 300 °C. Caudal de gas en el cono 30 L/h (nitrógeno), caudal de gas de desolvatación 650 L/h. Los datos se adquieren en un escaneo de MS completo de m/z 100 a 850 en modo positivo/negativo.
Parámetros de LC: La separación de fase inversa se lleva a cabo a temperatura ambiente en una columna XBridge prep OBD C18 (5 pm, 30 x 50 mm). La elución en gradiente se realiza con el disolvente A1 (H2O NH4HCO310 mM 50 pl/L de NH4OH) y disolvente B1 (100 % de ACN) (pH ~8,5). Caudal de HPLC: 35 ml/min a 45 ml/min, volumen de inyección: 990 pl. La relación de división se ajusta a /-1/6000 a MS (tabla 3).
Tabla 3:

- Método 2 de RP-HPLC neutra:
La purificación mediante HPLC de los productos finales se realizó en un sistema HPLC Knauer Smartline 1050 usando una columna RP-HPLC (Knauer 20 mm de d.i., Eurospher-100 C18). El producto se disolvió en metanol (20 mg por 8 ml) y se sometió a HPLC de fase inversa aplicando un gradiente de metanol/agua (70:30 a 100:0 durante 24 min).
Los espectros de RMN se registraron en diferentes instrumentos:
- un espectrómetro de RMN BRUKER AVANCEIII 400 MHz-Ultrashield equipado con una estación de trabajo con Windows 7 Professional que ejecutaba el software Topspin 3.2 y una sonda de banda ancha de resonancia doble de 5 mm (PaBbI 1H/19F-BB Z-GRD Z82021/0075) o una sonda de resonancia triple de 1 mm (PATXI 1H/ D-13C/15N Z-GRD Z868301/004).
- un espectrómetro de RMN Varian de 400 MHz con tiempo de adquisición (at) = 2,0 s, retardo de relajación (d1) = 2,0 s y ensanchamiento de línea (lb) = 0,5 Hz.
- un espectrómetro de RMN Bruker Avance DRX de 500 MHz
- un espectrómetro de RMN Bruker Avance III de 600 MHz
Para los desplazamientos químicos se usan como referencia las señales derivadas de protones residuales de los disolventes deuterados (DMSO-d6, Benceno-06 o CDCh). Los desplazamientos químicos se dan en partes por millón (ppm) y las constantes de acoplamiento (J) en Hercios (Hz). Las multiplicidades de espín se dan como ancho (br), singlete (s), doblete (d), triplete (t), cuarteto (q) y multiplete (m).
Por lo general, los productos se secaron a vacío antes de los análisis finales y de las pruebas biológicas.
A-IV: Compuestos ejemplares y síntesis
Los nombres de los siguientes compuestos son nombres IUPAC generados por el programa informático Biovia Draw Versión 16.1 para los intermedios de Fórmula X, XI, XII y por Pipeline Pilot 2018 usando OpenEye oemetachem versión 1.4.5 para los ejemplos de compuestos de Fórmula I.
Intermedios
Cuando están disponibles comercialmente, los materiales de partida se identifican por sus números de registro CAS. A. Síntesis de intermedios de Fórmula X
A.1. Síntesis de 3,6-difluoro-5-(2-fluoroetoxi)piridin-2-amina X-1:

8
h m ,80 °C
Etapa 7 X-1g X-1
Etapa 1: Síntesis de 5-cloro-3,6-difluoro-piridin-2-amina X-1a
A una disolución de 3-cloro-2,5,6-trifluoro-piridina (0,50 g, 2,98 mmol) en DMSO (10 ml) se le añadió NH3 acuoso al 25 % (4 ml) y la mezcla de reacción se calentó en un horno de acero a 100 °C durante 12 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. T ras la finalización, la mezcla de reacción se diluyó con H2O (200 ml) y se extrajo con EtOAc (400 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir 5-cloro-3,6-difluoro-piridin-2-amina X-1a (0,41 g de producto bruto) como un sólido amarillo.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
1H RMN (400 MHz, DMSO-de) ó 6,85 (s, 2H), 7,80-7,85 (m, 1H).
Etapa 2: Síntesis de 3,6-difluoropiridin-2-amina X-1b
A una disolución de 5-cloro-3,6-difluoro-piridin-2-amina X-1a (0,50 g, 3,03 mmol) en MeOH (100 ml) se le añadió trietilamina (5 ml) y Pd/C (0,40 g) y la mezcla de reacción se agitó a temperatura ambiente bajo presión de hidrógeno en un agitador Parr durante 10 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se filtró a través de Celite y el filtrado se concentró a vacío. El residuo se diluyó con H2O (200 ml) y se extrajo con MeOH al 10 % en DCM (200 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir 3,6-difluoropiridin-2-amina X-1b (0.21 g de producto bruto) como un sólido blanquecino.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LC-MS básica (ES+): 130 (M)+, 91% de pureza.
1H RMN (400 MHz, DMSO-d6) ó 6,09-6,11 (m, 1H) 6,57 (s ancho, 2H) 7,44-7,50 (m, 1H).
Etapa 3: Síntesis de 5-bromo-3,6-difluoro-piridin-2-amina X-1c
A una disolución de 3,6-difluoropiridin-2-amina X-1b (0,60 g, 4,19 mmol) en CH3CN (40 ml) se le añadió NBS (0,52 g, 2,93 mmol) y la mezcla de reacción se agitó en ausencia de luz a temperatura ambiente durante 30 min. Se añadió disolución de NBS (0,52 g, 2,93 mmol) en CH3CN (10 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 30 min. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se diluyó con H2O (100 ml) y se extrajo con EtOAc (160 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, 5 al 10 % de EtOAc en hexanos) para producir 5-bromo-3,6-difluoropiridin-2-amina X-1c (0,70 g) como un sólido blanquecino.
Rendimiento: 79%
Método 2 de LC-MS básica (ES'): 207 (M-H)', 98% de pureza.
1H RMN (400 MHz, DMSO-de) ó 6,86 (s ancho, 2H) 7,82-7,91 (m, 1H).
Etapa 4: Síntesis de 5-bromo-3,6-difluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-1d
A una disolución de 5-bromo-3,6-difluoropiridin-2-amina X-1c (4,00 g, 19,0 mmol) en DMAC (40 ml), se le añadió NaH (2,29 g, 57,1 mmol) en porciones a 0 °C y la reacción se agitó a la misma temperatura durante 30 min. Se añadió gota a gota cloruro de para-metoxibencilo (5,19 ml, 38,1 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se enfrió a 0 °C, se inactivó con NH4Cl saturado (20 ml), se vertió en H2O (60 ml) y se extrajo con EtOAc (3 x 60 ml). La capa orgánica se separó, se lavó con salmuera (2 x 80 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 4 % en hexanos) para producir 5-bromo-3,6-difluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-1d (8,2 g, 96%) como un sólido blanquecino.
Rendimiento: 96%.
1H RMN (400 MHz, DMSO-cfe) ó 3,72 (s, 6H) 4,57 (s, 4H) 6,87-6,89 (m, 4H) 7,16-7,18 (m, 4H) 7,99-8,07 (m, 1H).
Etapa 5: Síntesis de 3,6-difluoro-N,N-bis[(4-metoxifenil)metil]-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-1e: A una disolución de 5-bromo-3,6-difluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-1d (4,00 g, 8,90 mmol) en dioxano (160 ml), se le añadió bis(pinacolato)diboro (4,52 g, 17,8 mmol) y KOAc (3,06 g, 31,2 mmol) a temperatura ambiente y la mezcla de reacción se purgó con argón durante 20 min, seguido de la adición de PdCh(dppf) (0,65 g, 0,89 mmol).
La mezcla de reacción se purgó con argón durante 10 min y se calentó a 100 °C durante 16 h.
El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se enfrió, se filtró a través de una capa de Celite y se lavó con EtOAc (2 x 80 ml). El filtrado se concentró a vacío, el residuo se diluyó con
H2O (100 ml) y se extrajo con EtOAc (2 x 80 ml). La capa orgánica se separó, se lavó con salmuera (2 x 100 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 8 % en hexanos) para producir 3,6-difluoro-N,N-bis[(4-metoxifenil)metil]-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-1e (2,60 g) como un sólido blanquecino.
Rendimiento: 59%.
1H RMN (400 MHz, DMSO-de) ó 1,26 (s, 12H) 3,73 (s, 6H) 4,64 (s, 4H) 6,89 (d, J=8,31 Hz, 4H) 7,17 (d, J=8,80 Hz, 4H)) 7,51-7,56 (m, 1H).
Etapa 6: Síntesis de 6-[bis[(4-metoxifenil)metil]amino]-2,5-difluoro-piridin-3-ol X-1f:
A una disolución de 3,6-difluoro-N,N-bis[(4-metoxifenil)metil]-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-1e (2,50 g, 5,04 mmol) en THF (30 ml) se le añadió una disolución de H2O2 al 30% en H2O (10 ml) a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 15 min. La mezcla de reacción se agitó a temperatura ambiente durante 1,5 horas. El progreso de la reacción se monitorizó mediante TLC y LCMS. La mezcla de reacción se vertió en una disolución de Na2S2O3 al 5% en H2O fría (250 ml) a 0 °C, se diluyó con H2O (100 ml) y se extrajo con EtOAc (2 x 100 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir 6-[bis[(4-metoxifenil)metil]amino]-2,5-difluoro-piridin-3-ol X-1f (1,74 g de producto bruto) como un semisólido amarillo.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LC-MS básica (ES+): 387 (M+H)+, 93% de pureza.
1H RMN (400 MHz, DMSO-cfe) ó 3,71 (s, 6H) 4,32 (s, 4H) 6,86 (d, J=8,80 Hz, 4H) 7,14 (d, J=8,31 Hz, 4H) 7,22-7,27 (m, 1H) 9,84 (s, 1H).
Etapa 7: Síntesis de 3,6-difluoro-5-(2-fluoroetoxi)-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-1g:
A una disolución de 6-[bis[(4-metoxifenil)metil]amino]-2,5-difluoro-piridin-3-ol X-1f (0,70 g, 1,68 mmol) en DMF (13 ml) se le añadió K2CO3 (0,70 g, 5,04 mmol) y 1-bromo-2-fluoroetano (0,43 g, 3,36 mmol) a temperatura ambiente. La mezcla de reacción se calentó en microondas a 80 °C durante 15 min. El progreso de la reacción se monitorizó mediante TLC y LCMS. T ras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en H2O (30 ml) y se extrajo con EtOAc (3 x 25 ml). La capa orgánica se separó, se lavó con salmuera (2 x 30 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 25 % en hexanos) para producir 3,6-difluoro-5-(2-fluoroetoxi)-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-1g (0,52 g) como un líquido marrón.
Rendimiento: 71%.
Método 2 de LC-MS básica (ES+): 433 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-cfe) ó 3,72 (s, 6H) 4,21-4,25 (m, 1H) 4,29-4,32 (m, 1H) 4,42 (s, 4H) 4,63 - 4,66 (m, 1H) 4,75 4,78 (m, 1H) 6,87 (d, J=8,86 Hz, 4H) 7,15 (d, J=8,37 Hz, 4H) 7,69-7,76 (m, 1H).
Etapa 5: Síntesis de 3,6-difluoro-5-(2-fluoroetoxi)piridin-2-amina X-1:
A 3,6-difluoro-5-(2-fluoroetoxi)-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-1g (0,50 g, 1,15 mmol) se le añadió TFA (5 ml) a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 30 min. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O (25 ml), se alcalinizó con una disolución acuosa de NaHCO3 (10 ml) y se extrajo con EtOAc (2 x 25 ml). La capa orgánica se separó, se lavó con salmuera (2 x 40 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante trituración con Et2O (10 ml) y se secó a vacío para producir 3,6-difluoro-5-(2-fluoroetoxi)piridin-2-amina X-1 (0,258 g) como un sólido blanquecino.
Rendimiento: 80%.
Método 2 de LC-MS básica (ES+): 193 (M+H)+, 69% de pureza.
1H RMN (400 MHz, DMSO-d6) ó 4,13-4,17 (m, 1H) 4,20-4,25 (m, 1H) 4,59-4,64 (m, 1H) 4,71-4,76 (m, 1H) 6,10 (s, 2H) 7,57-7,62 (m, 1H).
A.2. Síntesis de 3-fluoro-5-(2-fluoroetoxi)piridin-2-amina X-2:
Br
PdCI 2 (dppf), KOAc, Pin2B2

Etapa 2
CAS: 748812-37-5 X-2a

Etapa 5
X-2d X-2
Etapa 1: Síntesis de 5-bromo-3-fluoro-N,N-bis(4-metoxibencil)piridin-2-amina X-2a:
A una disolución de 5-bromo-3-fluoro-piridin-2-amina (2,00 g, 10,5 mmol) en DMAC (30 ml) se le añadió NaH (1,26 g, 31,4 mmol) por lotes a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 30 min. Se añadió cloruro de p-metoxibencilo (2,85 ml, 20,9 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se enfrió a 0 °C, se inactivó con una disolución saturada de NH4Cl (200 ml), se vertió en H2O enfriada con hielo (400 ml) y se extrajo con EtOAc (2 x 300 ml). La capa orgánica se separó, se lavó con salmuera (2 x 100 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 3 % en hexanos) para producir 5-bromo-3-fluoro-N,N-bis(4-metoxibencil)piridin-2-amina X-2a (4,10 g) como un semisólido de color amarillo pálido.
Rendimiento: 87%
Método 2 de LC-MS básica (ES+): 431 (M+H)+, 95% de pureza.
1H RMN (400 MHz, DMSO-^) 53,72 (s, 6H), 4,57 (s, 4H), 6,87 (d, J=8,31 Hz, 4H), 7,15 (d, J=8,80 Hz, 4H), 7,81 (dd, J=12,96, 1,71 Hz, 1H), 8,06 (s, 1H).
Etapa 2: Síntesis de 3-fluoro-N,N-bis(4-metoxibencil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-2b:
A una disolución de 5-bromo-3-fluoro-N,N-bis(4-metoxibencil)piridin-2-amina X-2a (2,00 g, 4,43 mmol) en dioxano (60 ml), se le añadió bis(pinacolato)diboro (2,25 g, 8,86 mmol) y KOAc (1,52 g, 15,5 mmol) a temperatura ambiente y la mezcla de reacción se purgó con argón durante 20 min, seguido de la adición de PdCl2(dppf) (0,32 g, 0,44 mmol). La mezcla de reacción se purgó con argón durante 10 min y se calentó a 100 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se filtró a través de una capa de Celite, se lavó con EtOAc (2 x 300 ml) y el filtrado se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, 5 al 10 % de EtOAc en hexanos). El producto obtenido se agitó en pentano (10 ml), se decantó y se secó a vacío para proporcionar 3-fluoro-N,N-bis(4-metoxibencil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-2b (1,31 g) como un sólido blanquecino.
Rendimiento: 56%
Método 2 de LC-MS básica (ES+): 479 (M+H)+, 90% de pureza.
1H RMN (400 MHz, DMSO-^) 5 1,27 (s, 12H), 3,72 (s, 6H), 4,65 (s, 4H), 6,87 (d, J=8,80 Hz, 4H), 7,15 (d, J=8,31 Hz, 4H), 7,44 (d, J=15,16 Hz, 1H), 8,15 (s, 1H).
Etapa 3: Síntesis de 6-(bis(4-metoxibencil)amino)-5-fluoropiridin-3-ol X-2c:
A una disolución de X-2b (1,31 g, 2,46 mmol) en THF (20 ml) se le añadió una disolución de H2O2 al 30 % en H2O (7 ml) a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 15 min.
La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. El progreso de la reacción se monitorizó mediante TLC y LCMS. La mezcla de reacción se vertió en una disolución fría de Na2S2O3 al 5% en H2O (350 ml) a 0 °C, se diluyó con H2O (100 ml) y se extrajo con Et2O (2 x 200 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío (baja temperatura) para producir 6-(bis(4-metoxibencil)amino)-5-fluoropiridin-3-ol X-2c (0,90 g de producto bruto) como un semisólido amarillo pálido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LC-MS básica (ES+): 369 (M+H)+, 94% de pureza.
Etapa 4: Síntesis de 3-fluoro-5-(2-fluoroetoxi)-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-2d:
A una disolución de 6-(bis(4-metoxibencil)amino)-5-fluoropiridin-3-ol X-2c (1,70 g, 4,43 mmol) en DMF (10 ml) se le añadió K2CO3 (0,77 g, 5,56 mmol) y 1-bromo-2-fluoroetano (0,67 g, 5,31 mmol) a temperatura ambiente. La mezcla de reacción se calentó en microondas a 70 °C durante la noche. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en H2O (100 ml) y se extrajo con EtOAc (2 x 50 ml). La capa orgánica se separó, se lavó con agua enfriada con hielo (2 x 25 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, 5 al 8 % de EtOAc en hexanos) para proporcionar 3-fluoro-5-(2-fluoroetoxi)-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-2d (0,80 g) como un líquido marrón.
Rendimiento: 43%
Método 2 de LC-MS básica (ES+): 415 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-de) 53,71 (s, 6H), 4,14 - 4,22 (m, 1 H), 4,25 - 4,29 (m, 1 H), 4,40 (s, 4 H), 4,63 - 4,68 (m, 1 H), 4,74 - 4,80 (m, 1 H), 6,85 (d, J=8,80 Hz, 4 H), 7,14 (d, J=8,31 Hz, 4 H), 7,39 (dd, J=14,18, 2,45 Hz, 1 H), 7,78 (d, J=1,96 Hz, 1 H).
Etapa 5: Síntesis de 3-fluoro-5-(2-fluoroetoxi)piridin-2-amina X-2:
A una disolución de X-2d (800 mg, 1,92 mmol) en MeOH (3 ml) se le añadió hidróxido de paladio (160 mg) y la mezcla de reacción se agitó a temperatura ambiente durante 6 h bajo presión de hidrógeno. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se filtró a través de Celite, se lavó con MeOH (2 x 25 ml) y el filtrado se concentró a vacío. El residuo se purificó mediante cromatografía en columna (sílice, malla 100-200, 10 al 15 % de EtOAc en hexanos) para producir 3-fluoro-5-(2-fluoroetoxi)piridin-2-amina X-2 (100 mg) como un sólido marrón.
Rendimiento: 29%
Método 2 de LC-MS básica (ES+): 175 (M+H)+, 97% de pureza.
1H RMN (400 MHz, DMSO-de) 54,11 - 4,16 (m, 1 H), 4,19 - 4,23 (m, 1 H), 4,60 - 4,64 (m, 1 H), 4,72 - 4,77 (m, 1 H), 5,72 (s, 2 H), 7,27 (dd, J=12,23, 2,45 Hz, 1 H), 7,59 (d, J=2,45 Hz, 1 H).
A.3. Síntesis de 3-fluoro-5-(2,2-difluoroetoxi)piridin-2-amina X-3:

Tolueno
X-2c Etapa 2
Etapa 1
Etapa 1: Síntesis de 5-(2,2-difluoroetoxi)-3-fluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-3a:
A una disolución de 6-(bis(4-metoxibencil)amino)-5-fluoropiridin-3-ol X-2c (1,40 g, 3,69 mmol) y 2,2-difluoroetanol (0,66 g, 8,11 mmol) en tolueno (25 ml) se le añadió lentamente (cianometilen)tributilfosforano (1,96 g, 8,11 mmol) a temperatura ambiente. La mezcla de reacción se calentó a 100 °C durante 4 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en H2O (80 ml)
y se extrajo con EtOAc (3 x 100 ml). La capa orgánica se separó, se secó sobre Na2SÜ4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 10 % en hexanos) para producir 5-(2,2-difluoroetoxi)-3-fluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-3a (1,22 g) como un líquido amarillo pálido.
Rendimiento: 59%
Método 2 de LC-MS básica (ES+): 433 (M+H)+, 77% de pureza.
Etapa 2: Síntesis de 3-fluoro-5-(2,2-difluoroetoxi)piridin-2-amina X-3:
A una disolución de X-3a (1,20 g, 2,19 mmol) en MeOH (30 ml) se le añadió hidróxido de paladio sobre carbono (500 mg) y la mezcla de reacción se agitó a temperatura ambiente durante 6 h bajo presión de hidrógeno. El progreso de la reacción se monitorizó mediante TLC y lCm S. Tras la finalización, la mezcla de reacción se filtró a través de Celite, se lavó con MeOH (2 x 50 ml) y el filtrado se concentró a vacío para producir 3-fluoro-5-(2,2-difluoroetoxi)piridin-2-amina X-3 (590 mg) como un semisólido de color amarillo pálido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 77%
Método 2 de LC-MS básica (ES+): 193 (M+H)+, 55% de pureza.
A.4. Síntesis de 5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-amina X-4:

Etapa 1: Síntesis de 5-(2,2-difluoroetoxi)-3,6-difluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-4a
A una disolución de 6-[bis[(4-metoxifenil)metil]amino]-2,5-difluoro-piridin-3-ol X-1f (100 mg, 0,16 mmol), 2,2 difluoroetanol (27 mg, 0,33 mmol) y trifenilfosfina (218 mg, 0,83 mmol) en THF (5 ml) se le añadió lentamente a temperatura ambiente azodicarboxilato de diisopropilo (168 mg, 0,829 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se diluyó con H2O (20 ml) y se extrajo con EtOAc (20 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir 5-(2,2-difluoroetoxi)-3,6-difluoro-N,N-bis[(4-metoxifenil)metil]piridin-2-amina X-4a (20 mg) como un semisólido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 27%
1H RMN (400 MHz, DMSO-cfe) 53,72 (s, 6 H), 4,44 (s, 4H), 4,79-4,85 (m, 2 H), 6,01-6,31 (m, 1 H), 6,87 (d, J=8,31 Hz, 4 H), 7,15 (d, J=8,31 Hz, 4 H), 7,76-7,81 (m, 1 H).
Etapa 2: Síntesis de 5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-amina X-4:
A una disolución de X-4a (540 mg, 1,20 mmol) en MeOH (10 ml), se le añadió hidróxido de paladio sobre carbón (54 mg) y la mezcla de reacción se agitó a temperatura ambiente durante 1 h bajo presión de hidrógeno. El progreso de la reacción se monitorizó mediante TLC y lCm S. Tras la finalización, la mezcla de reacción se filtró a través de Celite, se lavó con MeOH (2 x 50 ml) y el filtrado se concentró a vacío para producir 5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-amina X-4 (300 mg) como un sólido marrón.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 71%
Método 2 de LC-MS básica (ES+): 211 (M+H)+, 60% de pureza.
A.5. Síntesis de 5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-amina X-5:
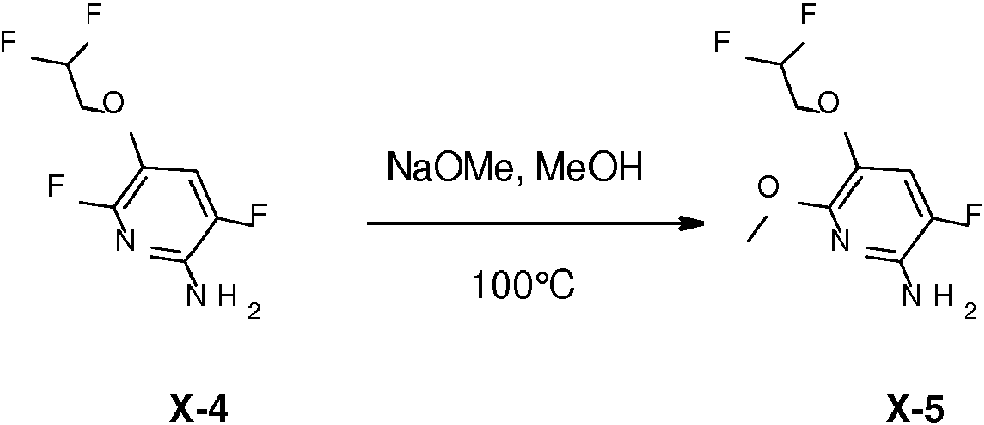
A una disolución de 5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-amina X-4 (0,39 g, 1,74 mmol) en MeOH (14 ml) se le añadió NaOMe (0,48 g, 8,79 mmol) y la mezcla de reacción se calentó a 100 °C durante 30 h. El progreso de la reacción se monitorizó mediante t Lc y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O (500 ml) y se extrajo con EtOAc (600 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir 5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-amina X-5 (0,37 g) como un sólido marrón.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 67%
Método 2 de LC-MS básica (ES-): 221 (M-H)-, 70% de pureza.
1H RMN (400 MHz, DMSO-d6) 5 3,78 (s, 3H) 4,13 (td, J=14,67, 3,42 Hz, 2H) 5,72 (s ancho, 2H) 6,13-6,45 (m, 1H) 7,34 (d, J= 10,76 Hz, 1H).
A.6. Síntesis de 5-(difluorometoxi)-3-metoxipiridin-2-amina X-6:
Br
PdCI 2 (dppf), KOAc, Pin2B2

Etapa 2
C A S : 42409 - 58 - 5 
Etapa 4
X-6c
TFA


X-6d X-6
Etapa 1: Síntesis de 5-bromo-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-6a:
A una disolución de 5-bromo-3-metoxi-piridin-2-amina (5,00 g, 24,6 mmol) en DMF (75 ml) se le añadió NaH (2,95 g, 73,9 mmol) por lotes a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 30 min. Se añadió gota a gota cloruro de p-metoxibencilo (6,71 ml, 49,3 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 2 h.
El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se enfrió a 0 °C, se inactivó con H2O fría (250 ml) y una solución saturada de NH4Cl (250 ml) y se extrajo con EtOAc (2 x 500 ml). La capa orgánica se separó, se lavó con salmuera (2 x 100 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 25 % en
hexanos) para producir 5-bromo-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-6a (9,65 g) como un sólido blanquecino.
Rendimiento: 75%
Método 2 de LC-MS básica (ES+): 443 (M+H)+, 85% de pureza.
1H RMN (400 MHz, DMSO-cfe) ó 3,69 (s, 6H) 3,82 (s, 3H) 4,42 (s, 4H) 6,79-6,85 (m, 4H) 7,11 (d, J=8,31 Hz, 4H) 7,39 (d, J=1,96 Hz, 1H) 7,76 (d, J=1,96 Hz, 1H).
Etapa 2: Síntesis de 3-metoxi-N,N-bis(4-metoxibencil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-6b:
A una disolución de 5-bromo-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-6a (9,60 g, 18,3 mmol) en dioxano (300 ml) se le añadió bis(pinacolato)diboro (9,31 g, 36,7 mmol) y KOAc (6,30 g, 64,1 mmol) a temperatura ambiente y la mezcla de reacción se purgó con argón durante 20 min, seguido de la adición de PdCh(dppf) (1,34 g, 1,83 mmol). La mezcla de reacción se purgó con argón durante 10 min y se calentó a 100 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se filtró a través de una capa de Celite, se lavó con EtOAc (2 x 300 ml) y el filtrado se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, 5 al 10 % de EtOAc en hexanos). El producto obtenido se agitó con pentano (50 ml), se decantó y se secó a vacío para proporcionar 3-metoxi-N,N-bis(4-metoxibencil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-amina X-6b (5,10 g) como un sólido blanquecino.
Rendimiento: 47%
Método 2 de LC-MS básica (ES+): 491 (M+H)+, 83% de pureza.
1H RMN (400 MHz, DMSO-cfe) ó 1,26 (s, 12H) 3,69 (s, 6H) 3,77 (s, 3H) 4,55 (s, 4H) 6,82 (d, J=8,80 Hz, 4H) 7,11 (d, J=8,31 Hz, 4H) 7,21 (s, 1H) 7,94 (s, 1H).
Etapa 3: Síntesis de 6-(bis(4-metoxibencil)amino)-5-metoxipiridin-3-ol X-6c:
A una disolución de 3-metoxi-N,N-bis(4-metoxibencil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)piridin-2-amina X-6b (5,00 g, 8,49 mmol) en THF (100 ml) se le añadió una disolución de H2O2 al 30 % en H2O (40 ml) lentamente a 0 °C. Después de 15 min, la mezcla de reacción se agitó a temperatura ambiente durante 2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se vertió en una disolución fría de Na2S2O3 al 6 % en H2O (600 ml) a 0 °C, se diluyó con H2O (500 ml) y se extrajo con Et2O (2 x 400 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío (a baja temperatura, 20 a 25 °C). El residuo se lavó con pentano (15 ml) para producir 6-(bis(4-metoxibencil)amino)-5-metoxipiridin-3-ol X-6c (3,50 g de producto bruto) como un sólido amarillo pálido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LC-MS básica (ES+): 381 (M+H)+, 84% de pureza.
1H RMN (400 MHz, DMSO-de) ó 3,69 (s, 6H) 3,81 (s, 3H) 4,18 (s, 4H) 6,77 (d, J=2,45 Hz, 1H) 6,80 (d, J=8,31 Hz, 4H) 7,12 (d, J=8,80 Hz, 4H) 7,28 (d, J=2,45 Hz, 1H) 9,21 (s, 1H).
Etapa 4: Síntesis de 5-(difluorometoxi)-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-6d:
A una disolución de 6-(bis(4-metoxibencil)amino)-5-metoxipiridin-3-ol X-6c (3,30 g, 7,31 mmol) en CH3CN (60 ml) se le añadió una disolución de KOH (2,26 g, 40,2 mmol) en H2O (20 ml) gota a gota a 0 °C, seguido de la adición de dietilfosfonato de bromodifluorometilo (10,7 g, 40,2 mmol) a 0 °C. Después de 15 min, la mezcla de reacción se agitó a temperatura ambiente durante 2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con H2O (200 ml) y se extrajo con EtOAc (2 x 250 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 5 % en hexanos) para proporcionar 5-(difluorometoxi)-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-6d (3,21 g) como un semisólido de color amarillo pálido.
Rendimiento: 92%
Método 2 de LC-MS básica (ES+): 431 (M+H)+, 90% de pureza.
1H RMN (400 MHz, DMSO-cfe) ó 3,70 (s, 6H) 3,85 (s, 3H) 4,39 (s, 4H) 6,83 (d, J=8,80 Hz, 4H) 7,11-7,16 (m, 4H) 7,14 (t, J=74 Hz, 1H) 7,19 (d, J=2,45 Hz, 1H) 7,62 (d, J=1,96 Hz, 1H).
Etapa 5: Síntesis de 5-(difluorometoxi)-3-metoxipiridin-2-amina X-6:
A 5-(difluorometoxi)-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-6d (2,50 g, 5,26 mmol) se le añadió TFA (15 ml) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O (400 ml) y una disolución saturada de NaHCO3 (250 ml) y se extrajo con EtOAc (2 x 300 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 40 % en hexanos) para producir 5-(difluorometoxi)-3-metoxipiridin-2-amina X-6 (0,52 g) como un sólido blanquecino.
Rendimiento: 52%
Método 2 de LC-MS básica (ES+): 191 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-de) 5 3,79 (s, 3H) 5,77 (s, 2H) 6,98 (d, J=1,96 Hz, 1H) 7,01 (t, J= 74 Hz, 1H) 7,42 (d, J = 1,96 Hz, 1H).
A.7. Síntesis de 5-(2,2-difluoroetoxi)-3-metoxipiridin-2-amina X-7:

Tolueno Etapa 2
X-6c Etapa 1
Etapa 1: Síntesis de 5-(2,2-difluoroetoxi)-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-7a:
A una disolución de 6-(bis(4-metoxibencil)amino)-5-metoxipiridin-3-ol X-6c (2,80 g, 7,01 mmol) y 2,2-difluoroetanol (0,89 ml, 14,0 mmol) en tolueno (30 ml) se le añadió cianometilentributilfosforano (3,67 ml, 14,0 mmol) lentamente a temperatura ambiente y la mezcla de reacción se calentó a 100 °C durante 2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se diluyó con H2O (30 ml) y se extrajo con EtOAc (3 x 40 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 30 % en hexanos) para producir 5-(2,2-difluoroetoxi)-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-7a (2,10 g) como un aceite de color amarillo pálido.
Rendimiento: 67%
Método 2 de LC-MS básica (ES+): 445 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-CÍ6) 53,68 (s, 6H) 3,85 (s, 3H) 4,24-4,34 (m, 6H) 6,19-6,49 (m, 1H) 6,80 (d, J=8,80 Hz, 4H) 7,03 (d, J=1,96 Hz, 1H) 7,11 (d, J=8,31 Hz, 4H) 7,48 (d, J=2,45 Hz, 1H).
Etapa 2: Síntesis de 5-(2,2-difluoroetoxi)-3-metoxipiridin-2-amina X-7:
A 5-(2,2-difluoroetoxi)-3-metoxi-N,N-bis(4-metoxibencil)piridin-2-amina X-7a (1,70 g, 3,82 mmol) en DCM (15 ml) se le añadió gota a gota t Fa (10 ml) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O (20 ml), NaHCO3 saturado (20 ml) y se extrajo con EtOAc (2 x 30 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 30 % en hexanos) para producir 5-(2,2-difluoroetoxi)-3-metoxipiridin-2-amina X-7 (0,605 g) como un sólido de color amarillo pálido.
Rendimiento: 74%
Método 2 de LC-MS básica (ES+): 205 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-^) 53,77 (s, 3H) 4,23 (td, J=14,80, 3,67 Hz, 2H) 5,33 (s, 2H) 6,18-6,50 (m, 1H) 6,88 (d, J=2,45 Hz, 1H) 7,31 (d, J=2,45 Hz, 1H).
A.8. Síntesis de 5-(2-(difluorometoxi)etoxi)-3-fluoro-6-metoxipiridin-2-amina X-8:


Etapa 1: Síntesis de 5-(2-(difluorometoxi)etoxi)-3,6-difluoro-N,N-bis(4-metoxibencil)piridin-2-amina X-8a:
A una disolución de 6-[bis[(4-metoxifenil)metil]amino]-2,5-difluoro-piridin-3-ol X-1f (0,80 g, 1,52 mmol) en DMF (10 ml) se le añadió K2CO3 (0,63 g, 4,56 mmol) y 1-bromo-2-(difluorometoxi)etano (0,29 g, 1,67 mmol) a temperatura ambiente. La mezcla de reacción se calentó en microondas a 85 °C durante 1,2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en H2O (60 ml) y se extrajo con EtOAc (3 x 40 ml). La capa orgánica se separó, se lavó con salmuera (2 x 50 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. La reacción se repitió con 0,70 g y 2 x 0,80 g y el producto bruto obtenido de las 4 reacciones se agrupó en DCM (200 ml) y se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 8% en hexanos) para producir 5-(2-(difluorometoxi)etoxi)-3,6-difluoro-N,N-bis(4-metoxibencil)piridin-2-amina X-8a (1,31 g) como un líquido marrón.
Rendimiento: 34%
Método 2 de LC-MS básica (ES+): 481 (M+H)+, 99% de pureza.
Etapa 2: Síntesis de 5-(2-(difluorometoxi)etoxi)-3,6-difluoropiridin-2-amina X-8b:
A una disolución de 5-(2-(difluorometoxi)etoxi)-3,6-difluoro-N,N-bis(4-metoxibencil)piridin-2-amina X-8a (1,30 g, 2,36 mmol) en MeOH (25 ml) se le añadió Pd/C al 20 % (0,13 g) a temperatura ambiente y la mezcla de reacción se agitó a temperatura ambiente durante 2 h bajo presión de hidrógeno. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se filtró a través de una capa de Celite y se lavó con MeOH (2 x 30 ml). El filtrado se concentró a vacío. El producto bruto obtenido se purificó lavando con pentano (2 x 20 ml) para producir 5-(2-(difluorometoxi)etoxi)-3,6-difluoropiridin-2-amina X-8b (0,525 g) como un sólido de color marrón pálido.
Rendimiento: 92%
Método 2 de LC-MS básica (ES+): 241 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-cfe) 54,03-4,07 (m, 2H) 4,08-4,10 (m, 2H) 6,07 (s, 2H) 6,68 (t, J=76 Hz 1H) 7,53-7,57 (m, 1H).
Etapa 3: Síntesis de 5-(2-(difluorometoxi)etoxi)-3-fluoro-6-metoxipiridin-2-amina X-8
A una disolución de 5-(2-(difluorometoxi)etoxi)-3,6-difluoropiridin-2-amina X-8b (0,70 g, 2,91 mmol) en MeOH (15 ml) se le añadió NaOMe (25 % en MeOH, 1,57 ml, 7,29 mmol) a 0 °C y la mezcla de reacción se calentó a 70 °C durante 24 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O (20 ml) y se extrajo con EtOAc (3 x 25 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía combi-flash (EtOAc al 10 % en hexanos) para proporcionar 5-(2-(difluorometoxi)etoxi)-3-fluoro-6-metoxipiridin-2-amina X-8 (0,45 g) como un sólido amarillo pálido.
Rendimiento: 59%
Método 2 de LC-MS básica (ES+): 253 (M+H)+, 94% de pureza.
1H RMN (400 MHz, DMSO-de) 53,77 (s, 3H) 4,03-4,06 (m, 4H) 5,61 (s, 2H) 6,72 (t, J=76 Hz, 1H) 7,26 (d, J=11,25 Hz, 1H).
B. Síntesis de intermedios de Fórmula XI
B.1. Síntesis de 6,7-dicloro-1H-indol XI-1:

A una disolución de 1,2-dicloro-3-nitro-benceno (4,00 g, 20,8 mmol) en THF (60 ml) se le añadió bromuro de vinilmagnesio (1 M, 83,3 ml, 83,3 mmol) a -78 °C y el la mezcla de reacción se agitó a la misma temperatura durante 3 horas. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (80 ml) y se extrajo con EtOAc (3 * 100 ml). La capa orgánica se separó, se lavó con salmuera (100 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 2 % en hexanos) para producir 6,7-dicloro-1H-indol XI-1 (1,50 g) como un sólido blanquecino.
Rendimiento: 37%.
Método 2 de LCMS básica (ES+): 186 (M+H)+, 95% de pureza.
1H RMN (400 MHz, DMSO-de) 56,56-6,57 (m, 1H), 7,30 (d, J=8,4 Hz, 1H), 7,43 (t, J=2,4 Hz, 1H), 7,48 (d, J= 8,4 Hz, 1H), 11,62 (s ancho, 1H).
B.2. Síntesis de 6-cloro-7-ciclopropoxi-1H-indol XI-2:

Etapa 1: Síntesis de 1-cloro-2-ciclopropoxi-3-nitrobenceno XI-2a:
A una suspensión agitada de NaH (0,91 g, 22,8 mmol) en THF (30 ml) se le añadió ciclopropanol (1,08 ml, 17,1 mmol) en una atmósfera inerte a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 30 min. Se añadió gota a gota una disolución de 1-cloro-2-fluoro-3-nitrobenceno (2,00 g, 11,4 mmol) en THF (10 ml) a 0 °C y la mezcla de reacción se calentó a 75 °C durante 3 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en hielo picado y se extrajo con EtOAc (2 x 80 ml). La capa orgánica se separó, se lavó con H2O (60 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 10 % en hexanos) para producir 1-cloro-2-ciclopropoxi-3-nitrobenceno XI-2a (1,20 g) como un aceite de color amarillo pálido. Rendimiento: 49%.
1H RMN (400 MHz, DMSO-de) 50,60-0,66 (m, 2H), 0,72-0,77 (m, 2H), 4,28-4,33 (m, 1H), 7,35-7,39 (m, 1H), 7,85-7,92 (m, 2H).
Etapa 2: Síntesis de 6-cloro-7-ciclopropoxi-1H-indol XI-2:
A una disolución de 1-cloro-2-ciclopropoxi-3-nitrobenceno XI-2a (1,15 g, 5,38 mmol) en THF (22 ml) se le añadió gota a gota bromuro de vinilmagnesio (1 M, 21,5 ml, 21,5 mmol) a -78 °C y la mezcla de reacción se agitó a la misma temperatura durante 2 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (70 ml) y se extrajo con EtOAc (3 * 40 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. La reacción se repitió con 0,92 g y el producto bruto obtenido de
las 2 reacciones se agrupó y se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 10 % en hexanos) para producir 6-cloro-7-ciclopropoxi-1H-indol XI-2 (0,69 g) como un líquido amarillo pálido.
Rendimiento: 41%.
Método 2 de LCMS básica (ES+): 208 (M+H)+, 91% de pureza.
1H RMN (400 MHz, DMSO-cfe) 50,52-0,61 (m, 2H), 0,87-0,95 (m, 2H), 4,39-4,43 (m, 1H), 6,46-6,49 (m, 1H), 6,99 (d, J=8,31 Hz, 1H), 7,30 (d, J=8,31 Hz, 1H), 7,34-7,37 (m, 1H), 11,35 (s ancho, 1H).
B.3. Síntesis de 6-cloro-7-(difluorometil)-1-(fenilsulfonil)-1H-indol XI-3:

Etapa 1: Síntesis de 7-bromo-6-cloro-1H-indol XI-3a
A una disolución de 2-bromo-1-cloro-3-nitro-benceno (4,50 g, 19,0 mmol) en THF (90 ml) se le añadió gota a gota bromuro de vinilmagnesio (9,99 g, 76,1 mmol) a -78 °C y se la mezcla de reacción se agitó a la misma temperatura durante 2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. La reacción se repitió a una escala de 4,5 g y la mezcla bruta de las 2 reacciones se agrupó. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (500 ml), se diluyó con H2O (500 ml) y se extrajo con EtOAc (1000 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 2 % en hexanos) para producir 7-bromo-6-cloro-1H-indol XI-3a (3,05 g) como un sólido amarillo.
Rendimiento: 33%
Método 2 de LCMS básica (ES-): 228 (M-H)-, 96% de pureza.
1H RMN (400 MHz, DMSO-CÍ6) 56,59 (d, J=1,96 Hz, 1H) 7,18 (d, J=8,31 Hz, 1H) 7,41-7,45 (m, 1H) 7,57 (d, J=8,31 Hz, 1H) 11,48 (s ancho, 1H)
Etapa 2: Síntesis de 7-bromo-6-cloro-1-(fenilsulfonil)-1H-indol XI-3b:
A una suspensión agitada de NaH (2,42 g, 60,5 mmol) en THF seco (30 ml) se le añadió 7-bromo-6-cloro-1H-indol XI-3a (7,00 g, 30,2 mmol) en porciones a 0 °C y la mezcla de reacción se agitó durante 20 min. Se añadió gota a gota una disolución de cloruro de fenilsulfonilo (5,79 ml, 45,4 mmol) en THF (10 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se diluyó con EtOAc (30 ml) y se lavó con una disolución saturada de NaHCO3 (25 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto orgánico obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para producir 7-bromo-6-cloro-1-(fenilsulfonil)-1H-indol XI-3b (7,50 g) como un sólido blanquecino.
Rendimiento: 54%
Método 2 de LCMS básica (ES+): 370 (M+H)+, 81% de pureza.
1H RMN (400 MHz, DMSO-cfe) 56,88 (d, J=3,42 Hz, 1H), 7,44 (d, J=8,80 Hz, 1H), 7,58-7,64 (m, 3H), 7,70-7,75 (m, 1H), 7,87 (d, J= 3,42 Hz, 1H), 8,01 (d, J=7,82 Hz, 2H).
Etapa 3: Síntesis de 6-cloro-1-(fenilsulfonil)-7-vinil-1H-indol XI-3c:
A una disolución de 7-bromo-6-cloro-1-(fenilsulfonil)-1H-indol XI-3b (4,00 g, 8,72 mmol) en DMF (15 ml) se le añadió tributil(vinil)estannano (3,07 ml, 10,5 mmol), trifenilfosfina (0,11 g, 0,44 mmol) y LiCl (1,11 g, 26,2 mmol) y la mezcla
de reacción se purgó con argón durante 10 min. Se añadió PdCl2(PPh3)2 (0,31 g, 0,44 mmol) y la mezcla de reacción se calentó en un tubo sellado a 100 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOAc (2 x 40 ml). El filtrado se lavó con salmuera (20 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para producir 6-cloro-1-(fenilsulfonil)-7-vinil-1H-indol XI-3c (2,80 g) como un sólido amarillo pálido.
Rendimiento: 98%
1H RMN (400 MHz, DMSO-^) 54,88 (d, J=18,10 Hz, 1H), 5,29 (d, J=12,23 Hz, 1H), 6,81-6,89 (m, 1H), 6,93 (d, J=3,91 Hz, 1H), 7,39 (d, J=8,31 Hz, 1H), 7,54-7,63 (m, 3H), 7,70 (d, J=7,34 Hz, 1H), 7,74 (d, J=7,83 Hz, 2H), 7,93 (d, J=3,42 Hz, 1H).
Etapa 4: Síntesis de 6-cloro-1-(fenilsulfonil)-1H-indol-7-carbaldehído XI-3d:
A una disolución de 6-cloro-1-(fenilsulfonil)-7-vinil-1H-indol XI-3c (2,50 g, 7,63 mmol) en THF (20 ml) y H2O (5 ml) se le añadió OsO4 (0,04 M en agua, 3,77 ml, 0,15 mmol) a 0 °C y la mezcla de reacción se agitó durante 30 min. Se añadió NaIO4 (4,08 g, 19,1 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOAc (2 x 20 ml). El filtrado se lavó con salmuera (20 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para proporcionar 6-cloro-1-(fenilsulfonil)-1H-indol-7-carbaldehído XI-3d (0,60 g) como un sólido amarillo pálido.
Rendimiento: 24%
1H RMN (400 MHz, DMSO-*) 5 7,01 (d, J=3,91 Hz, 1H), 7,48 (d, J=8,31 Hz, 1H), 7,56-7,62 (m, 2H), 7,68-7,75 (m, 3H), 7,78 (d, J= 8,80 Hz, 1H), 7,93 (d, J=3,91 Hz, 1H), 10,43 (s, 1H).
Etapa 5: Síntesis de 6-cloro-7-(difluorometil)-1-(fenilsulfonil)-1H-indol XI-3:
A una disolución de 6-cloro-1-(fenilsulfonil)-1H-indol-7-carbaldehído XI-3d (0,60 g, 1,84 mmol) en DCM (15 ml) se le añadió fluoruro de dietilaminoazufre (1,06 ml, 7,36 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se vertió en hielo H2O (15 ml), se alcalinizó con una disolución saturada de NaHCO3 (15 ml) y se extrajo con EtOAc (2 x 20 ml). La capa orgánica se separó, se lavó con salmuera (15 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para producir 6-cloro-7-(difluorometil)-1-(fenilsulfonil)-1H-indol XI-3 (0,55 g) como un sólido blanquecino.
Rendimiento: 81%
1H RMN (400 MHz, CDCls) 56,67 (d, J=3,91 Hz, 1H), 7,35-7,44 (m, 4H), 7,50-7,55 (m, 1H), 7,58 (d, J=8,31 Hz, 2H), 7,62 (d, J= 3,42 Hz, 1H), 7,86 (t, J=52 Hz, 1H).
B.4. Síntesis de 6-cloro-7-fluoro-1H-indol XI-4:

A una disolución de 1-cloro-2-fluoro-3-nitro-benceno (2,50 g, 14,2 mmol) en THF (50 ml) se le añadió bromuro de vinilmagnesio (5,61 g, 42,7 mmol) a -78 °C y la mezcla de reacción se agitó a la misma temperatura durante 1 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (100 ml), se diluyó con H2O (400 ml) y se extrajo con EtOAc (500 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 5 % en hexanos) para producir 6-cloro-7-fluoro-1H-indol XI-4 (0,60 g) como un líquido rojo.
Rendimiento: 17%
Método 2 de LCMS básica (ES-): 168 (M-H)-, 66% de pureza.
B.5. Síntesis de 6-(difluorometil)-7-fluoro-1H-indol XI-5:

Etapa 1: Síntesis de 1-(difluorometil)-2-fluoro-3-nitro-benceno XI-5a:
A una disolución de 2-fluoro-3-nitro-benzaldehído (2,0 g, 11,8 mmol) en DCM (20 ml) se le añadió fluoruro de dietilaminoazufre (7,63 g, 47,3 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 4 horas El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se vertió en una disolución saturada de NaHCÜ3 enfriada con hielo (50 ml) y se extrajo con DCM (2 * 200 ml). La capa orgánica se separó, se lavó con salmuera (15 ml), se secó sobre Na2SÜ4 anhidro y se concentró a vacío para producir 1-(difluorometil)-2-fluoro-3-nitro-benceno XI-5a (2,23 g) como un líquido de color marrón pálido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 96%
1H RMN (400 MHz, DMSO-^) 57,35 (t, J=52 Hz, 1 H), 7,60 (t, J=8,07 Hz, 1 H), 8,05 (t, J=6,85 Hz, 1 H), 8,35 (t, J=7,82 Hz, 1H).
Etapa 2: Síntesis de 6-(difluorometil)-7-fluoro-1H-indol XI-5:
A una disolución de 1-(difluorometil)-2-fluoro-3-nitro-benceno XI-5a (2,21 g, 11,2 mmol) en THF (20 ml) se le añadió bromuro de vinilmagnesio (disolución 1 M en THF, 45 ml, 45 mmol) a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 3 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (40 ml) a 0 °C y se extrajo con EtüAc (2 * 200 ml).
La capa orgánica se lavó con salmuera (30 ml), se secó sobre Na2SÜ4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 10 % en hexanos) para proporcionar 6-(difluorometil)-7-fluoro-1H-indol XI-5 (0,54 g) como un aceite marrón.
Rendimiento: 25%
Método 2 de LCMS básica (ES): 184 (M-H)-, 95% de pureza.
1H RMN (400 MHz, DMSO-^) 56,57 - 6,62 (m, 1 H), 7,14-7,17 (m, 1 H), 7,29 (t, J=56 Hz 1 H), 7,48 (d, J=8,19 Hz, 1 H), 7,56 (t, J=2,69 Hz, 1 H), 11,94 (s ancho, 1 H).
B.6. Síntesis de 1-(bencenosulfonil)-7-cloro-6-(difluorometil)indol XI-6:

Etapa-1: Síntesis de 6-bromo-7-cloro-1H-indol XI-6a
A una disolución de 1-bromo-2-cloro-3-nitro-benceno (5,0 g, 21,1 mmol) en THF (80 ml) se le añadió bromuro de vinilmagnesio (disolución 1 M en THF, 84,6 ml, 84,6 mmol) gota a gota a -78 °C y la mezcla de reacción se agitó a la misma temperatura durante 3 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (30 ml) y se extrajo con EtOAc (3 * 100 ml). La capa orgánica se separó, se lavó con salmuera (70 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto
obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, 3 al 5 % de EtOAc en hexanos) para producir 6-bromo-7-cloro-1H-indol XI-6a (2,5 g) como un sólido amarillo.
Rendimiento: 49%
Método 2 de LCMS básica (ES'): 228 (M-H)', 96% de pureza.
Etapa 2: Síntesis de 6-bromo-7-cloro-1-(fenilsulfonil)-1H-indol XI-6b:
A una suspensión agitada de NaH (0,87 g, 21,7 mmol) en THF seco (30 ml) se le añadió 6-bromo-7-cloro-1H-indol XI-6a (2,5 g, 10,8 mmol) en porciones a 0 °C y la mezcla de reacción se agitó durante 10 min. Se añadió gota a gota cloruro de fenilsulfonilo (2,3 g, 13 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se vertió en agua enfriada con hielo (100 ml) y se extrajo con EtOAc (3 x 50 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir 6-bromo-7-cloro-1-(fenilsulfonil)-1H-indol XI-6b (3,0 g) como un sólido de color marrón pálido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 71%
Método 2 de LCMS básica (ES-): 368 (M-H)-, 95% de pureza.
Etapa 3: Síntesis de 7-cloro-1-(fenilsulfonil)-6-vinil-1H-indol XI-6c:
A una disolución de 6-bromo-7-cloro-1-(fenilsulfonil)-1H-indol XI-6b (1,5 g, 3,86 mmol) en DMF (10 ml) se le añadió tributil(vinil)estannano (1,47 g, 4,63 mmol), trifenilfosfina (50 mg, 0,19 mmol) y LiCl (0,49 g, 11,6 mmol) y la mezcla de reacción se purgó con argón durante 10 min. Se añadió PdCl2(PPh3)2 (0,13 g, 0,19 mmol) y la mezcla de reacción se calentó en un tubo sellado a 100 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOAc (2 x 100 ml). El filtrado se lavó con salmuera (100 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para producir 7-cloro-1-(fenilsulfonil)-6-vinil-1H-indol XI-6c (0,80 g) como un sólido blanquecino.
Rendimiento: 64%
Método 2 de LCMS básica (ES+): 318 (M+H)+, 98% de pureza.
Etapa 4: Síntesis de 1-(bencenosulfonil)-7-cloro-indol-6-carbaldehído XI-6d:
A una disolución de 7-cloro-1-(fenilsulfonil)-6-vinil-1H-indol XI-6c (1,40 g, 4,19 mmol) en THF (70 ml) y H2O (15 ml) se le añadió OsO4 (0,04 M en agua, 2,07 ml, 0,08 mmol) a 0 °C y la mezcla de reacción se agitó durante 3 horas. Se añadió NaIO4 (2,69 g, 12,6 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 8 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se diluyó con agua (100 ml). El precipitado se filtró y se secó a vacío para producir 1-(bencenosulfonil)-7-cloro-indol-6-carbaldehído XI-6d (1,30 g) como un sólido marrón.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 82%
Método 2 de LCMS básica (ES+): 320 (M+H)+, 82% de pureza.
Etapa 5: Síntesis de 1-(bencenosulfonil)-7-cloro-6-(difluorometil)indol XI-6:
A una disolución de 1-(bencenosulfonil)-7-cloro-indol-6-carbaldehído XI-6d (1,20 g, 3,1 mmol) en DCM (30 ml) se le añadió fluoruro de dietilaminoazufre (1,78 ml, 12,4 mmol) a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se vertió en hielo H2O (50 ml), se alcalinizó con una disolución saturada de NaHCO3 (40 ml) y se extrajo con EtOAc (2 x 50 ml). La capa orgánica se separó, se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para producir 1-(bencenosulfonil)-7-cloro-6-(difluorometil)indol XI-6 (0,65 g) como un sólido blanquecino.
Rendimiento: 60%
Método 2 de LCMS básica (ES): 340 (M-H)-, 97% de pureza.
B.7. Síntesis de 6-cloro-7-(trifluorometoxi)-1H-indol XI-7:

A una disolución de 1-cloro-3-nitro-2-(trifluorometoxi)benceno (2,10 g, 8,69 mmol) en THF (30 ml) se le añadió gota a gota bromuro de vinilmagnesio (1 M en THF, 34,8 ml, 34,8 mmol) a -78 °C y la mezcla de reacción se agitó a la misma temperatura durante 3 h. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 16 horas. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con NH4Cl saturado (100 ml) y se extrajo con EtoAc (3 x 80 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, 2 al 4 % de EtOAc en hexanos) para producir 6-cloro-7-(trifluorometoxi)-1H-indol XI-7 (1,10 g) como un líquido amarillo pálido.
Rendimiento: 49%.
Método 2 de LCMS básica (ES-): 234 (M-H)-, 92% de pureza.
1H RMN (400 MHz, DMSO-d6) ó 6,61 (t, J=2,4 Hz, 1H) 7,15 (d, J=8,0 Hz, 1H) 7,54 (t, J=2,4 Hz, 1H) 7,62 (d, J=8,8 Hz, 1H) 11,8 (s ancho, 1H).
C. Síntesis de intermedios de fórmula XII
C.1. Método A. Síntesis de cloruro de 6,7-dicloro-1H-indol-3-sulfonilo XII-1

XI-1 XII-1
A una disolución de 6,7-dicloro-1H-indol XI-1 (1,35 g, 6,89 mmol) en CH3CN (15 ml) se le añadió gota a gota ácido clorosulfónico (1,60 ml, 24,1 mmol) a 0 °C y la mezcla de reacción se agitó a la misma temperatura durante 1 h. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se vertió en H2O enfriada con hielo (50 ml) y se extrajo con EtOAc (3 x 50 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir cloruro de 6,7-dicloro-1H-indol-3-sulfonilo XII-1 (0,80 g) como un sólido marrón.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Rendimiento: 39%.
Método 2 de LCMS básica (ES-): 282 (M-H)-, 50% de pureza.
Los siguientes intermedios de la Tabla 4 pueden sintetizarse según un método análogo al Método A.
Tabla 4:


Cloruro de 6-cloro-7-ciclopropoxi-1H-indol-3-sulfonilo XII-2

Método 2 de LCMS básica (ES-): 304 (M-H)-, 16% de pureza.
Cloruro de 6-cloro-7-(difluorometil)-1H-indol-3-sulfonilo XII-3

1H RMN (400 MHz, CDCl3) 5 7,33 (t, J=54 Hz, 1H), 7,44-7,50 (m, 1H), 8,08 (d, J=2,93 Hz, 1H), 8,11 (d, J=8,31 Hz, 1H), 9,51 (s ancho, 1H).
Cloruro de 7-bromo-6-cloro-1H-indol-3-sulfonilo XII-4

1H RMN (400 MHz, DMSO-cfe) 57,23 (d, J=8,37 Hz, 1H), 7,41 (d, J=2,46 Hz, 1H), 7,73 (d, J=8,37 Hz, 1H), 11,50 (s ancho, 1H).
Cloruro de 6-(difluorometil)-7-fluoro-1H-indol-3-sulfonilo XII-5

1H RMN (400 MHz, DMSO-^) 5 7,17-7,21 (m, 1 H), 7,29 (t, J=54 Hz 1 H), 7,52 (d, J=2,45 Hz, 1 H), 7,66 (d, J=8,31 Hz, 1 H), 11,92 (s ancho, 1 H).
Cloruro de 7-cloro-6-(difluorometil)-1H-indol-3-sulfonilo XII-6

Producto bruto
C.2. Síntesis de cloruro de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol-3-sulfonilo XII-7

Etapa 1: Síntesis de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol XII-7a
A una suspensión agitada de hidróxido de sodio finamente pulverizado (3,54 g, 0,088 mol) en diclorometano (60 ml) previamente enfriada sobre un baño de hielo se le añadió 6-cloro-7-fluoro-1H-indol XI-4 (5 g, 0,029 mol) como una sola porción, seguido de hidrogenosulfato de tetrabutilamonio (0,501 g, 0,001 mol). Se continuó agitando durante 10 minutos más, luego se añadió gota a gota una disolución de cloruro de bencenosulfonilo (4,2 ml, 0,033 mol) en diclorometano (15 ml) durante 20 minutos y la mezcla de reacción se agitó a 0 °C durante 1 hora. Se retiró el baño de hielo y la mezcla se agitó durante 1 hora más a temperatura ambiente. La mezcla de reacción se filtró sobre una capa de Kieselguhr, enjuagando la torta de filtración con diclorometano (2 x 50 ml). El filtrado se lavó con agua (4 * 50 ml) y salmuera (50 ml), se secó sobre sulfato de sodio anhidro, se filtró y el disolvente se concentró a vacío para producir 1-(bencenosulfonil)-6-cloro-7-fluoro-indol XII-7a (8,57 g) como un sólido beige oscuro.
Rendimiento: 90%.
1H RMN (400 MHz, DMSO-d6) 56,95 (dd, J = 3,7, 2,3 Hz, 1H), 7,37 (dd, J = 8,4, 6,2 Hz, 1H), 7,48 (d, J = 8,6 Hz, 1H), 7,66 (tt, J = 6,9, 1,9 Hz, 2H), 7,80 - 7,71 (m, 1H), 7,98 - 7,91 (m, 2H), 7,99 (d, J = 3,7 Hz, 1H).
Etapa 2: Síntesis de cloruro de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol-3-sulfonilo XII-7
A una disolución agitada de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol XII-7a (8,50 g, 0,027 mol) en acetonitrilo (85 ml) previamente enfriado sobre un baño de hielo se le añadió ácido clorosulfónico (9,12 ml, 0,137 mol) gota a gota durante 20 min y la mezcla de reacción se agitó durante 16 horas a temperatura ambiente. La mezcla de reacción se vertió lentamente con agitación en hielo-agua (340 ml) durante 20 minutos. El sólido precipitado se recogió por filtración, enjuagando la torta de filtración con agua enfriada con hielo (3 x 50 ml) y ciclohexano (50 ml). Luego, la torta de filtración se secó bajo un flujo de nitrógeno durante 2 horas y luego en una estufa de vacío a 40 °C durante 16 horas para producir cloruro de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol-3-sulfonilo XII-7 (7,82 g) como un sólido de color rosa claro.
Rendimiento: 66%.
Método 4 de LCMS ácida (ES+): 388 (M+H)+, 95% de pureza.
1H RMN (400 MHz, DMSO-CÍ6) 57,44 (dd, J = 8,5, 6,3 Hz, 1H), 7,72 - 7,61 (m, 3H), 7,80-7,72 (m, 1H), 7,81 (s, 1H), 8,05 - 7,98 (m, 2H).
C.3. Síntesis de cloruro de 6-cloro-7-(trifluorometoxi)-1H-indol-3-sulfonilo XII-8

Etapa 1: Síntesis de ácido 6-cloro-7-(trifluorometoxi)-1H-indol-3-sulfónico XII-8a:
A una disolución de 6-cloro-7-(trifluorometoxi)-1H-indol XI-7 (0,10 g, 0,39 mmol) en CH3CN (3 ml) se le añadió ClSO3H (0,10 ml) gota a gota a 0 °C y la mezcla de reacción se agitó a temperatura ambiente durante 3 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se vertió en H2O enfriada con hielo (25 ml) y se extrajo con EtOAc (2 x 20 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir ácido 6-cloro-7-(trifluorometoxi)-1H-indol-3-sulfónico XI I-8a (0,11 g de producto bruto) como un líquido de color marrón pálido.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LCMS básica (ES-): 314 (M-H)-, 91% de pureza.
Etapa 2: Síntesis de cloruro de 6-cloro-7-(trifluorometoxi)-1H-indol-3-sulfonilo XII-8:
A una disolución de ácido 6-cloro-7-(trifluorometoxi)-1H-indol-3-sulfónico XII-8a (0,11 g, 0,28 mmol) en CH3CN (3 ml) se le añadió POCh (0,1 ml, 1,12 mmol) a 0 °C y la mezcla de reacción se calentó a 60 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O enfriada con hielo (50 ml) y se extrajo con EtOAc (3 x 40 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido (0,1 g, 0,3 mmol) se disolvió en CH3CN (6 ml) y POCl3 (0,11 ml, 1,22 mmol) gota a gota a 0 °C y la mezcla de reacción se calentó a 60 °C durante 16 h. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se diluyó con H2O enfriada con hielo (60 ml) y se extrajo con EtOAc (3 x 50 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío para producir cloruro de 6-cloro-7-(trifluorometoxi)-1H-indol-3-sulfonilo XII-8 (0,13 g de producto bruto) como un líquido marrón.
Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LCMS básica (ES-): 332 (M-H)-Compuestos de ejemplo
D. Síntesis de compuestos de Fórmula general I
Todos los compuestos de la presente invención descritos específicamente en la presente memoria se denominan "I-x", donde "x" se refiere a un número que identifica los compuestos individuales. En consecuencia, los compuestos de ejemplo se denominan I-1, I-2, I-3, etc. Esto es independiente de si algún compuesto se pudiera describir también mediante cualquier fórmula subgenérica de la presente memoria, p. ej. mediante la Fórmula II, III o IV, y similares. D.1. Método B. Síntesis de 6,7-dicloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-1

A una disolución de 3,6-difluoro-5-(2-fluoroetoxi)piridin-2-amina X-1 (0,10 g, 0,44 mmol) en piridina (5 ml) se le añadió cloruro de 6,7-dicloro-1H-indol-3-sulfonilo XII-1 (0,33 g, 1,10 mmol) en porciones a 0 °C durante un período de 20 min, seguido de la adición de DMAP (0,01 g, 0,09 mmol) a la misma temperatura. La mezcla de reacción se calentó a 100
°C durante 30 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se concentró a vacío. El residuo se trituró con HCl 2 N (10 ml), se diluyó con H2O (20 ml) y se extrajo con EtOAc (3 x 25 ml). La capa orgánica se separó, se lavó con salmuera (2 * 30 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 30 % en hexanos) para producir 6,7-dicloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-1 (0,065 g) como un sólido blanco.
Rendimiento: 34%.
Método 2 de LCMS básica (ES+): 440 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-Ó6) ó 4,22-4,25 (m, 1H), 4,31-4,34 (m, 1H), 4,62-4,65 (m, 1H), 4,75-4,79 (m, 1H), 7,39 (d, J=8,80 Hz, 1H), 7,70-7,76 (m, 2H), 7,97 (s, 1H), 10,64 (s ancho, 1H), 12,52 (s ancho, 1H).
Los siguientes compuestos de la Tabla 5 se pueden sintetizar de acuerdo con un método análogo al Método B.
Tabla 5:

6-cloro-7-ciclopropiloxi-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-2

Método 2 de LCMS básica (ES+): 462 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-d6) 50,54-0,56 (m, 2H), 0,86-0,88 (m, 2H), 4,25-4,27 (m, 1H), 4,32-4,37 (m, 1H), 4,40-4,43 (m, 1H), 4,63-4,65 (m, 1H), 4,75-4,78 (m, 1H), 7,20 (d, J=8,31 Hz, 1H), 7,48 (d, J=8,80 Hz, 1H), 7,79 (s, 1H), 7,92 (s, 1H), 10,56 (s ancho, 1H), 12,31 (s ancho, 1H).
6-doro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-(difluorometil)-1H-mdol-3-sulfonamida I-3

Método 2 de LCMS básica (ES+): 456 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-de) 54,27-4,32 (m, 1H), 4,35-4,41 (m, 1H), 4,64-4,69 (m, 1H), 4,74-4,80 (m, 1H), 7,38 (d, J=8,86 Hz, 1H), 7,53 (t, J=56 Hz, 1H), 7,83 (t, J=9,11 Hz, 1H), 7,90-7,96 (m, 2H), 10,65 (s, 1H), 12,28 (s ancho, 1H).
7-bromo-6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)-2-piridil]-1H-indol-3-sulfonamida I-4

1H RMN (400 MHz, DMSO-de) 54,27-4,30 (m, 1H), 4,34-4,39 (m, 1H), 4,64-4,67 (m, 1H), 4,76-4,80 (m, 1H), 7,41 (d, J=8,31 Hz, 1H), 7,75 (d, J=8,80 Hz, 1H), 7,80-7,86 (m, 1H), 7,97 (d, J=2,93 Hz, 1H), 10,64 (s, 1H), 12,47 (s ancho, 1H).
6,7-dicloro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-5

Método 2 de LCMS básica (ES+): 422 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-^) 54,21 - 4,26 (m, 1H), 4,30 - 4,34 (m, 1H), 4,62 - 4,67 (m, 1H), 4,74 - 4,79 (m, 1H), 7,40 (d, J=8,67 Hz, 1H), 7,52 (dd, J=11,27, 2,60 Hz, 1H), 7,71 (d, J=8,67 Hz, 1H), 7,83 (d, J=2,60 Hz, 1H), 7,97 (s, 1H), 10,43 (s, 1H), 12,60 (s ancho, 1H).
6,7-dicloro-N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-1H-indol-3-sulfonamida I-6

Método 2 de LCMS básica (ES+): 440 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-^) 54,37 (td, J=14,74, 3,47 Hz, 2 H), 6,22 - 6,52 (m, 1 H), 7,40 (d, J=8,24 Hz, 1 H), 7,58 (dd, J=11,27, 2,60 Hz, 1 H), 7,73 (d, J=8,67 Hz, 1 H), 7,86 (d, J=2,17 Hz, 1 H), 7,98 (s, 1 H), 10,52 (s ancho, 1 H), 12,61 (s ancho, 1 H).
6-cloro-7-(difluorometil)-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-7

Método 2 de LCMS básica (ES+): 438 (M+H)+, 98% de pureza.
1H RMN (400 MHz, DMSO-^) 54,20 - 4,26 (m, 1 H), 4,28 - 4,35 (m, 1 H), 4,60 - 4,66 (m, 1 H), 4,71 - 4,79 (m, 1 H), 7,35 (d, J=8,80 Hz, 1 H), 7,48-7,50 (m, 1 H), 7,52 (t, J=52 Hz, 1 H), 7,82 (d, J=1,96 Hz, 1 H), 7,87 - 7,93 (m, 2 H), 10,43 (s, 1 H), 12,21 (s ancho, 1 H).
6-(difluorometil)-7-fluoro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-8

Método 2 de LCMS básica (ES+): 422 (M+H)+, 98% de pureza.
7-cloro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-6-(difluorometil)-1H-indol-3-sulfonamida I-9

Método 2 de LCMS básica (ES+): 456 (M+H)+, 98% de pureza.
6,7-dicloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-1H-indol-3-sulfonamida I-13

Método 2 de LCMS básica (ES+): 470 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-cfe) 53,43 (s, 3H) 4,28 (td, J=14,43, 3,42 Hz, 2H) 6,19-6,50 (m, 1H) 7,39 (d, J=8,80 Hz, 1H) 7,53 (d, J=10,27 Hz, 1H) 7,71 (d, J=8,31 Hz, 1H) 8,00 (d, J=1,96 Hz, 1H) 10,36 (s ancho, 1H) 12,58 (s ancho, 1H).
6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida I-14

Método 2 de LCMS básica (ES+): 456 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-^) 54,37 (td, J=14,67, 3,42 Hz, 2H) 6,22-6,52 (m, 1H) 7,37 (d, J=8,80 Hz, 1H) 7,53 (t, J=54 Hz, 1H) 7,56-7,62 (m, 1H) 7,87 (d, J=1,96 Hz, 1H) 7,89-7,96 (m, 2H) 10,54 (s, 1H) 12,24 (s ancho, 1H).
6-doro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida I-15

Método 2 de LCMS básica (ES+): 486 (M+H)+, 97% de pureza.
1H RMN (400 MHz, DMSO-CÍ6) 53,41 (s, 3H) 4,29 (td, J=14,43, 3,42 Hz, 2H) 6,19-6,50 (m, 1H) 7,37 (d, J=8,31 Hz, 1H) 7,52-7,57 (m, 1H) 7,53 (t, J=54 Hz, 1H) 7,90-7,95 (m, 2H) 10,35 (s, 1H) 12,21 (s ancho, 1H).
6,7-dicloro-N-[5-(difluorometoxi)-3-metoxipiridin-2-il]-1H-indol-3-sulfonamida I-16

Método 2 de LCMS básica (ES+): 438 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-^) 5 3,78 (s, 3H) 7,16 (t, J=74 Hz, 1H) 7,25 (d, J=2,45 Hz, 1H) 7,41 (d, J=8,80 Hz, 1H) 7,62 (d, J=1,96 Hz, 1H) 7,92 (d, J=8,80 Hz, 1H) 8,09 (s, 1H) 10,28 (s ancho, 1H) 12,59 (s ancho, 1H).
6-cloro-N-[5-(difluorometoxi)-3-metoxipiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida I-17

Método 2 de LCMS básica (ES+): 454 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-cfe) 53,78 (s, 3H) 7,16 (t, J=74 Hz, 1H) 7,25 (d, J=1,47 Hz, 1H) 7,39 (s, 1H) 7,52 (t, J=54 Hz, 1H) 7,62 (d, J=1,47 Hz, 1H) 8,02 (s, 1H) 8,13 (d, J=8,31 Hz, 1H) 10,30 (s ancho, 1H) 12,21 (s ancho, 1H).
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-(trifluorometoxi)-1H-indol-3-sulfonamida 1-18

Método 2 de LCMS básica (ES+): 490 (M+H)+, 99% de pureza.
1H RMN (400 MHz, DMSO-cfe) 54,23-4,25 (m, 1H) 4,31-4,33 (m, 1H) 4,61-4,67 (m, 1H) 4,73-4,79 (m, 1H) 7,36 (d, J=8,31 Hz, 1H) 7,71-7,79 (m, 1H) 7,83 (d, J=8,80 Hz, 1H) 8,05 (s ancho, 1H) 10,69 (s, 1H) 12,70 (s ancho, 1H). N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-6-(difluorometil)-7-fluoro-1H-indol-3-sulfonamida 1-19

Método 2 de LCMS básica (ES+): 440 (M+H)+, 82% de pureza.
1H RMN (400 MHz, DMSO-d6) 54,36 (td, J=14,68, 3,47 Hz, 2H) 6,22-6,51 (m, 1H) 7,33 (t, J=54 Hz, 1H) 7,33-7,38 (m, 1H) 7,58 (dd, J=11,10, 2,31 Hz, 1H) 7,69 (d, J=8,32 Hz, 1H) 7,86 (d, J=2,31 Hz, 1H) 8,11 (s, 1H) 10,54 (s ancho, 1H) 12,92 (s ancho, 1H).
7-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-6-(difluorometil)-1H-indol-3-sulfonamida I-20

Método 2 de LCMS básica (ES+): 486 (M+H)+, 98% de pureza.
1H RMN (400 MHz, DMSO-d6) 53,41 (s, 3H) 4,28 (td, J=14,55, 3,18 Hz, 2H) 6,19-6,49 (m, 1H) 7,33 (t, J=56 Hz, 1H) 7,48 (d, J=8,80 Hz, 1H) 7,54 (d, J=10,27 Hz, 1H) 7,86 (d, J=8,31 Hz, 1H) 8,12 (s, 1H) 10,40 (s ancho, 1H) 12,73 (s ancho, 1H).
6,7-dicloro-N-[5-(2,2-difluoroetoxi)-3-metoxipiridin-2-il]-1H-indol-3-sulfonamida I-21

Método 2 de LCMS básica (ES+): 452 (M+H)+, 98% de pureza.
1H RMN (400 MHz, DMSO-da) 53,67 (s, 3 H) 4,29 (td, J=14,67, 2,93 Hz, 2 H) 6,15 - 6,46 (m, 1 H) 7,04 (d, J=1,47 Hz, 1 H) 7,36 (d, J= 8,80 Hz, 1 H) 7,48 (s, 1 H) 7,83 (d, J=8,31 Hz, 1 H) 7,96 (s, 1 H) 9,89 (s ancho, 1 H) 12,48 (s ancho, 1 H)
D.2. Síntesis de 6-doro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-10

A una disolución de 3,6-difluoro-5-(2-fluoroetoxi)piridin-2-amina X-1 (50 mg, 0,26 mmol) en piridina (1 ml) se le añadió cloruro de 1-(bencenosulfonil)-6-doro-7-fluoro-indol-3-sulfonilo XII-7 (106 mg, 0,26 mmol) y luego se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se evaporó a sequedad y luego se vertió en agua y se extrajo con acetato de etilo (dos veces). Las fases orgánicas se secaron sobre MgSO4 y se evaporaron. El residuo se purificó mediante cromatografía en columna (sílice, malla 100-200, 0 al 10 % de MeOH en DCM) seguido de un Método 1 de LCMS preparativa básica para proporcionar 6 mg de 6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-fluoro-1H-indol-3-sulfonamida 1-10 como un sólido amarillo pálido.
Rendimiento: 5%.
Método 1 de LCMS básica (ES-): 422 (M-H)-, 97% de pureza.
D.3. Síntesis de 6-cloro-N-[5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-11

A una disolución de 5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-amina X-4 (154 mg, 0,37 mmol) en piridina (1 ml) se le añadió cloruro de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol-3-sulfonilo XII-7 (150 mg, 0,37 mmol) y luego se agitó a 70 °C durante 2 h. La mezcla de reacción se evaporó a sequedad y luego se vertió en agua y se extrajo con acetato de etilo (dos veces). Las fases orgánicas se secaron sobre MgSO4 y se evaporaron. El residuo se purificó mediante cromatografía en columna (sílice, malla 100-200, 0 al 5 % de MeOH en DCM) seguido de un Método 1 de LCMS preparativa básica para proporcionar 32 mg de 6-cloro-N-[5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-11 como un sólido amarillo pálido.
Rendimiento: 20%.
Método 1 de LCMS básica (ES-): 440 (M-H)-, 97% de pureza.
1H RMN (400 MHz, DMSO-^) 54,41 (td, J = 14,6, 3,5 Hz, 2H), 6,38 (t, J = 3,5 Hz, 1H), 7,31 (dd, J = 8,6, 6,5 Hz, 1H), 7,58 (d, J = 8,6 Hz, 1H), 7,89 (t, J = 9,1 Hz, 1H), 8,06 (d, J = 2,4 Hz, 1H), 10,72 (s, 1H), 12,84 (s, 1H).
D.4. Síntesis de 6-cloro-7-ciclopropil-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-12
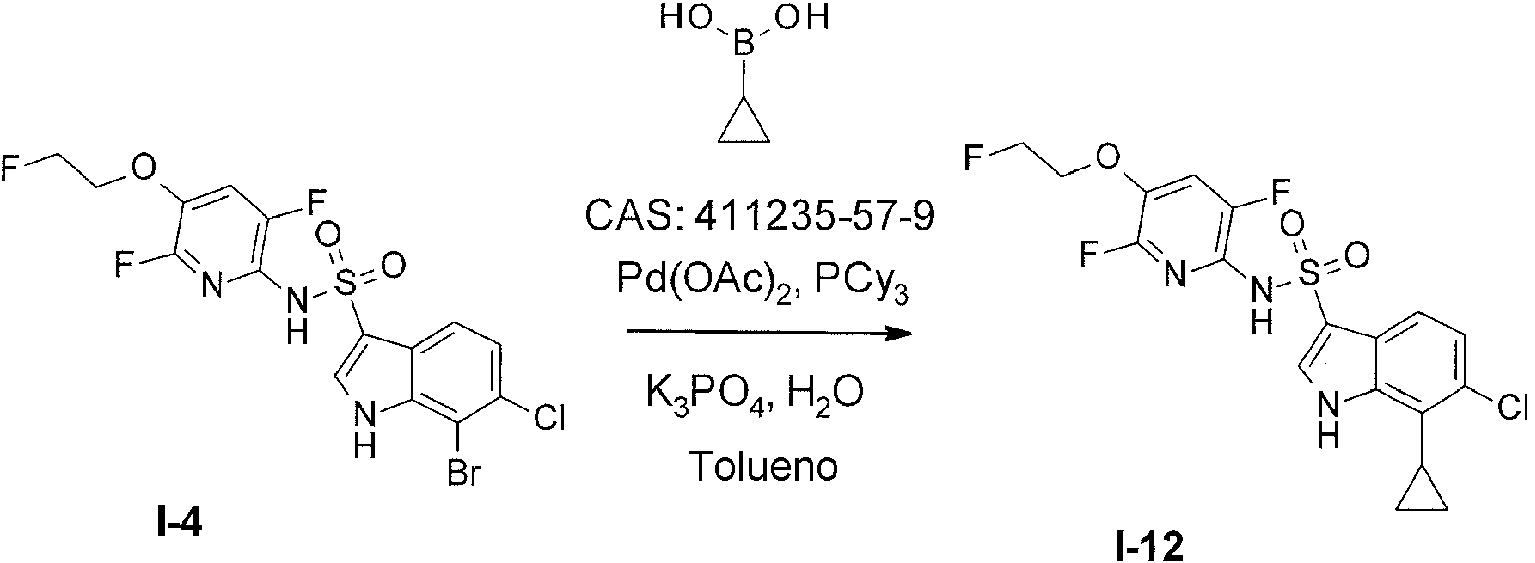
A una disolución de 7-bromo-6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)-2-piridil]-1H-indol-3-sulfonamida I-4 (0,25 g, 0,51 mmol) en tolueno (8 ml) y H2O (0,8 ml) se le añadió Kí Po 4 (0,22 g, 1,01 mmol), ácido ciclopropilborónico (0,07 g, 0,76 mmol) y triciclohexilfosfina (0,03 g, 0,10 mmol). La mezcla de reacción se purgó con argón durante 15 min seguido de la adición de Pd(OAc)2 (0,01 g, 0,05 milimoles). La mezcla de reacción se purgó con argón durante 5 min y se calentó a 100 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOAc (100 ml). El filtrado se lavó con H2O (50 ml). La capa orgánica se separó, se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 20 % en hexanos) para producir 6-cloro-7-ciclopropil-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida I-12 (0,04 g) como un sólido blanquecino.
Rendimiento: 17%.
Método 2 de LCMS básica (ES+): 446 (M+H)+, 95% de pureza.
1H RMN (400 MHz, DMSO-cfe) 50,68-0,70 (m, 2H), 1,09-1,17 (m, 2H), 1,93-2,00 (m, 1H), 4,25-4,29 (m, 1H), 4,32- 4,37 (m, 1H), 4,62-4,67 (m, 1H), 4,74-4,79 (m, 1H), 7,19 (d, J=8,80 Hz, 1H), 7,60 (d, J=8,31 Hz, 1H), 7,77-7,84 (m, 1H), 7,87 (d, J=2,93 Hz, 1H), 10,55 (s, 1H), 11,90 (s ancho, 1H).
D.5. Síntesis de 6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-22
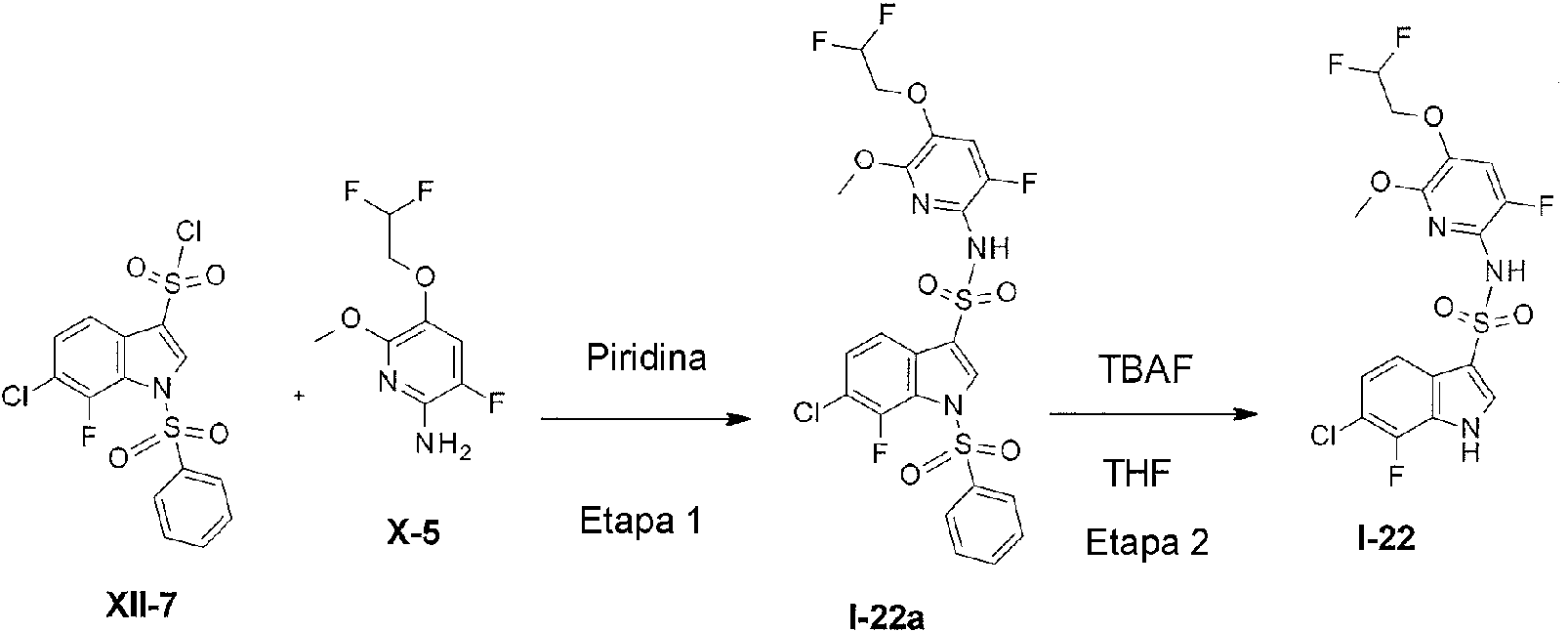
Etapa 1: Síntesis de 1-(bencenosulfonil)-6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxi-2-piridil]-7-fluoro-indol-3-sulfonamida I-22a
En un vial sellado se disolvió 5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-amina X-5 (54 mg, 0,24 mmol) en piridina (2 ml) bajo argón. Se añadió cloruro de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol-3-sulfonilo XII-7 (100 mg, 0,24 mmol) a 0 °C y luego se agitó a 70 °C durante la noche. La mezcla de reacción se evaporó a sequedad. El residuo se purificó mediante cromatografía ultrarrápida sobre sílice (eluyendo con un gradiente de DCM y metanol de 100/0 a 98/2) para proporcionar 96 mg de 1-(bencenosulfonil)-6-cloro-N-[5-(2, 2-difluoroetoxi)-3-fluoro-6-metoxi-2-piridil]-7-fluoro-indol-3-sulfonamida I-22a como un aceite marrón.
Rendimiento: 66%.
Método 1 de LCMS básica (ES-): 592 (M-H)-Etapa 2: Síntesis de N-[6-(difluorometoxi)-5-fluoro-2-metoxipiridin-3-il]-6-(difluorometil)-1H-indol-3-sulfonamida I-16
En un tubo sellado se disolvió 1-(bencenosulfonil)-6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxi-2-piridil]-7-fluoroindol-3-sulfonamida I-22a (96 mg, 0,16 mmol) en THF (1 ml). Se añadió fluoruro de tetrabutilamonio (430 mg, 0,48 mmol) y la mezcla de reacción se agitó a 70 °C durante 3 días. La mezcla de reacción se evaporó a sequedad. El residuo se disolvió en AcOEt y se lavó con agua. La fase orgánica se secó sobre MgSO4 y se evaporó. El residuo se purificó mediante HPLC preparativa básica (Método 1) para proporcionar 27 mg de 6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-22 como un sólido blanco.
Rendimiento: 37%.
Método 1 de LCMS básica (ES-): 452 (M-H)-, 96% de pureza.
1H RMN (400 MHz, DMSO-cfe) 53,45 (s, 3H), 4,30 (t, J = 14,8 Hz, 2H), 6,36 (s, 1H), 7,30 (t, J = 7,6 Hz, 1H), 7,56 (t, J = 11,5 Hz, 2H), 8,06 (s, 1H), 10,36 (s, 1H), 12,78 (s, 1H).
D.6. Síntesis de 6-cloro-N-[5-[2-(difluorometoxi)etoxi]-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-23
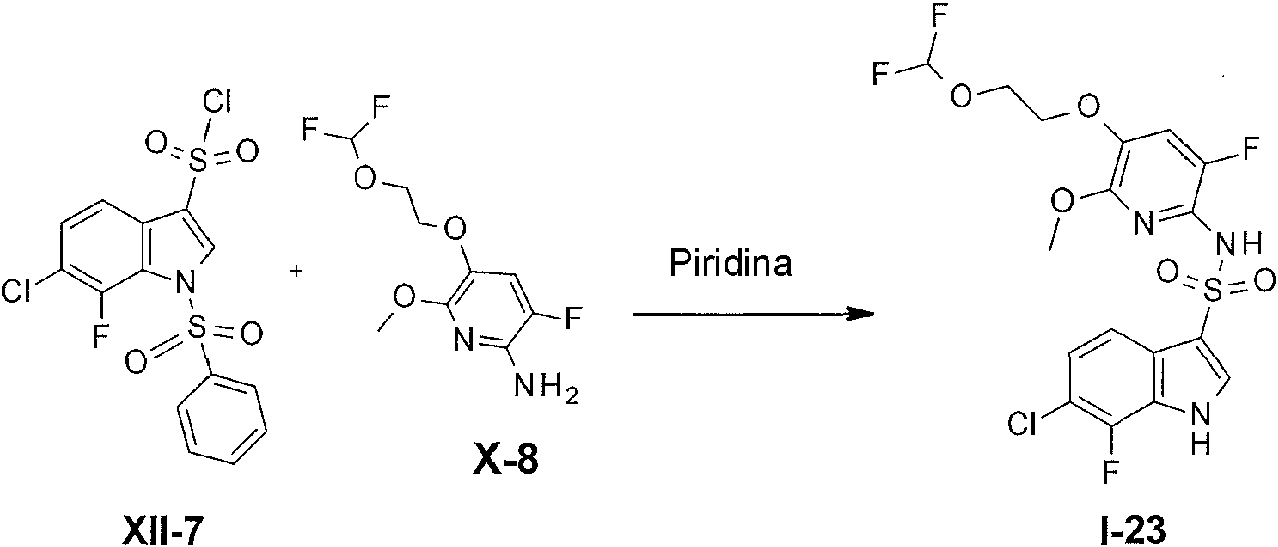
A una disolución de 5-(2-(difluorometoxi)etoxi)-3-fluoro-6-metoxipiridin-2-amina X-8 (74 mg, 0,29 mmol) en piridina (2 ml) se le añadió cloruro de 1-(bencenosulfonil)-6-cloro-7-fluoro-indol-3-sulfonilo XII-7 (100 mg, 0,24 mmol) y luego se agitó a 70 °C durante 2 h. La mezcla de reacción se evaporó a sequedad y luego el residuo se purificó mediante un Método 1 de LCMS preparativa básica para proporcionar 54 mg de 6-cloro-N-[5-[2-(difluorometoxi)etoxi]-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida I-23 como un sólido de color rosa pálido.
Rendimiento: 45%.
Método 1 de LCMS básica (ES-) 482 (M-H)-, 99% de pureza.
1H RMN (400 MHz, DMSO-cfe) 53,40 (s, 3H), 4,21 - 4,03 (m, 4H), 6,71 (s, 1H), 7,29 (dd, J = 8,6, 6,5 Hz, 1H), 7,56 (d, J = 8,6 Hz, 1H), 7,45 (d, J = 10,3 Hz, 1H), 8,03 (s, 1H), protones de NH no visibles.
Síntesis del ejemplo comparativo 1
Ejemplo comparativo 1: 6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida

Etapa 3
Ejemplo comparativo 1
Etapa-1: Síntesis de ácido 6-cloro-1H-indol-3-sulfónico:
A una disolución de 6-cloroindol (1,00 g, 6,62 mmol) en piridina (10 ml) se le añadió complejo de trióxido de azufre y piridina (1,57 g, 9,93 mmol) y la mezcla de reacción se calentó a reflujo durante 16 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se diluyó con H2O (100 ml) y se extrajo con Et2O (250 ml). La capa acuosa se separó y se concentró a vacío. El producto bruto obtenido se co-evaporó con tolueno para proporcionar ácido 6-cloro-1H-indol-3-sulfónico (2,30 g de producto bruto) como un semisólido pardo. Este compuesto se usó tal como estaba para la siguiente reacción sin purificación adicional.
Método 2 de LCMS básica (ES-): 230 (M-H)-, 98% de pureza.
1H RMN (400 MHz, DMSO-^) 5 6,98-7,04 (m, 1H), 7,12 - 7,26 (m, 1H), 7,44 (s, 1H), 7,69 - 7,75 (m, 1H), 11,13 (s ancho, 1 H).
Etapa 2: Síntesis de cloruro de 6-cloro-1H-indol-3-sulfonilo:
A una disolución de ácido 6-cloro-1H-indol-3-sulfónico (2,00 g, 6,45 mmol) en sulfolano (5 ml) y CH3CN (5 ml) se le añadió POCl3 (1,30 ml, 14,2 mmol) gota a gota a 0 °C y la mezcla de reacción se calentó a 70 °C durante 3 h. El progreso de la reacción se monitorizó mediante TLC y LCMS. Tras la finalización, la mezcla de reacción se inactivó con H2O enfriada con hielo (100 ml) y se extrajo con EtOAc (2 * 50 ml). La capa orgánica se separó, se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100-200, EtOAc al 30 % en hexanos) para proporcionar cloruro de 6-cloro-1H-indol-3-sulfonilo (1,00 g) como un sólido rosa claro.
Rendimiento: 62%.
1H RMN (400 MHz, DMSO-^) 57,32 (dd, J=8,56, 1,22 Hz, 1H), 7,71 (s, 1H), 8,03 (d, J=8,80 Hz, 1H), 8,45 (d, J=2,93 Hz, 1H), 12,38 (s ancho, 1H).
Etapa 3: Síntesis de 6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
A una disolución agitada de 3,6-difluoro-5-(2-fluoroetoxi)piridin-2-amina X-1 (250 mg, 0,893 mmol) en piridina (10,00 ml) se le añadió cloruro de 6-cloro-1H-indol-3-sulfonilo (558 mg, 2,23 mmol) en porciones a 0 °C y luego se añadió DMAP (5 mg, 0,04 mmol) a misma temperatura. La mezcla de reacción se calentó a 100 °C durante 16 h. El progreso de la reacción se monitorizó mediante TLC. Tras la finalización, la mezcla de reacción se concentró a vacío, el residuo se trituró con HCl 2 N (10 ml), se diluyó con agua (20 ml) y la capa acuosa se extrajo con EtOAc (3 x 25 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a vacío. El producto bruto obtenido se purificó mediante cromatografía en columna (sílice, malla 100 200, EtOAc al 35 % en hexanos) para proporcionar el Ejemplo comparativo 1, 6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida (57 mg) como un sólido blanquecino.
Rendimiento: 11%.
Método 2 de LC-MS básica (ES+): 406 (M+H)+, 97 % de pureza.
1H RMN (400 MHz, DMSO-da) 54,24-4,29 (m, 1H) 4,31-4,36 (m, 1H) 4,60-4,65 (m, 1H) 4,72-4,77 (m, 1H) 7,18 (dd, J=8,61, 1,72 Hz, 1H) 7,51 (d, J=1,48 Hz, 1H) 7,70 (d, J=8,86 Hz, 1H) 7,78 (dd, J=9,84, 8,37 Hz, 1H) 7,95 (d, J=2,95 Hz, 1H) 10,49 (s, 1H) 12,06 (ss, 1H).
B. Biología/farmacología:
B-I. Cultivos celulares
Línea celular recombinante para GPR17:
Se cultivaron células Flp-In T-REx CHO que expresan de forma estable el receptor GPR17 humano (CHO hGPR17) del laboratorio de Evi Kostenis (Universidad de Bonn, Alemania) a 37 °C en una atmósfera humidificada con un 5 % de CO2. Las células se cultivaron en DMEM con una mezcla de nutrientes F-12 suplementada con higromicina B (500 |jg/ml) y blasticidina (30 jg/ml). La expresión del locus Flp-In se indujo mediante tratamiento con doxiciclina (1 jg/m l) durante 16-20 h antes de los ensayos.
Oligodendrocitos primarios:
Se aislaron células progenitoras de oligodendrocitos primarios (CPO) de los prosencéfalos de crías de rata Wistar en los días postnatales 0 a 2. Los cerebros se disociaron mecánicamente con una jeringa y dos agujas huecas diferentes (primero 1,2 * 40 y luego 0,60 * 30). La suspensión de células sin agrupamientos se filtró a través de un filtro de células de 70 jm y se colocó en matraces de cultivo de 75 cm2 recubiertos con poli-D-lisina con DMEM suplementado con un 10 % (v/v) de suero de ternera fetal inactivado térmicamente, penicilina (100 unidades/ml) y estreptomicina (0,1 mg/ml) con intercambio del medio cada dos días. Después de 8 a 11 días a 37 °C en una atmósfera humidificada con un 5% de CO2, los cultivos mixtos se agitaron a 240 rpm durante 14-24 h para desprender las CPO de los astrocitos y la microglía. Para enriquecer aún más las CPO, la suspensión se sembró en placas de Petri sin recubrir durante 45 min. Luego, las CPO se sembraron en placas recubiertas con poli-L-ornitina y se mantuvieron a 37 °C en una atmósfera humidificada con un 5 % de CO2 en medio Neurobasal de proliferación suplementado con B27 al 2% (v/v), GlutaMAX 2 mM, 100 unidades/ml de penicilina, 0,1 mg/ml de estreptomicina, 10 ng/ml de PDGF-AA y 10 ng/ml de FGF básico cambiando el medio cada dos días.
B-II: Ensayos funcionales de GPR17 in vitro
B-II-A: Ensayo funcional de movilización de calcio
GPR17 es un receptor acoplado a proteína G. La activación de GPR17 desencadena la señalización de la proteína G de tipo Gq que da como resultado la liberación de los almacenes de calcio (Ca2+) del retículo endoplásmico al citosol, que se puede medir usando el colorante Calcium 5, un colorante indicador fluorescente de los niveles de Ca2+ citosólico. Los compuestos de la presente invención se evaluaron en el ensayo de Ca2+ o en el ensayo de cAMP de GPR17, descrito más adelante. Se midieron algunos ejemplos representativos en ambas pruebas de actividad como se indica en la Tabla 5, más adelante.
Descripción del ensayo de Ca2+:
Se descongelaron CHO hGPR17 y se sembraron a una densidad de 20 000 células por pocillo en placas negras de 384 pocillos con fondo transparente. Las células se incubaron durante la noche a 37 °C en una atmósfera humidificada del 5% de CO2. Dieciséis a veinte horas después de la siembra, se cargaron las CHO hGPR17 durante 60 min con colorante Calcium 5, un colorante fluorescente indicador del Ca2+ citosólico, de acuerdo con las instrucciones del fabricante. Se registró la señal fluorescente relativa a la concentración de Ca2+ citosólico a lo largo del tiempo a temperatura ambiente en un lector FLIPR Tetra. Las células se incubaron primero durante 30 minutos a temperatura ambiente en tampón HBSS Hepes de pH 7,4 que contenía concentraciones crecientes de compuestos de prueba (típicamente 10'11 M a 10'6 M). Luego, se añadió a las células MDL29,951 50 nM, un agonista de GPR17. Se midieron los efectos inhibidores de los compuestos de prueba a concentraciones variables y se determinaron las pCI50 resultantes. Todas las incubaciones se realizaron por duplicado y los resultados se compararon con una curva de respuesta a la concentración de compuestos de referencia antagonistas y agonistas de GPR17. El análisis y el ajuste de la curva se realizaron en ActivityBase XE utilizando la ecuación logística de 4 parámetros XLfit y = A ((B-A)/(1+((C/x)AD))), donde A, B, C y D representan la y mínima, la y máxima, la CI50 y la pendiente, respectivamente.
Resultados del ensayo de Ca2+:
Cuando se prueban en un ensayo de movilización de Ca2+, los compuestos de los Ejemplos típicamente exhiben valores de pCl50 mayores o iguales a 6,5; más preferiblemente, mayores o iguales a 7,5, e incluso más preferiblemente mayores o iguales a 8,5. Las actividades de los compuestos de los Ejemplos ensayados se representan en la tabla de la Sección B2B a continuación. Los rangos de actividad A, B y C se refieren a los valores de pCl50 en el ensayo de Ca2+ como sigue: "A": pCl50 <7,5, "B": pCl507,5 < x < 8,5, "C": 8,5 < pCl50
B-IIB. Ensayo funcional de acumulación de cAMP
La activación de GPR17 también puede reclutar la señalización de proteína G de tipo Gi, lo que resulta en una disminución del monofosfato de adenosina cíclico (cAMP) intracelular. Los cambios intracelulares del cAMP se pueden medir utilizando el kit de ensayo dinámico HTRF cAMP de CisBio (Codolet, Francia). Usando la tecnología de fluorescencia homogénea resuelta en el tiempo (HTRF), el ensayo se basa en la competición entre el cAMP nativo producido por las células y el cAMP marcado con el colorante d2. La unión del trazador se determinó mediante un anticuerpo anti-cAMP marcado con criptato.
Descripción del ensayo de cAMP
Se desprendieron células CHO hGPR17 con PBS que contenía EDTA y se distribuyeron en placas negras de 384 pocillos con 5000 células por pocillo. Las células se incubaron primero durante 30 minutos a temperatura ambiente en HBSS Hepes (pH 7,4) que contenía vehículo o concentraciones variables de compuestos antagonistas/agonistas inversos de GPR17 de prueba. Luego, se añadió una curva de respuesta a la dosis del agonista de GPR17 MDL29,951 (típicamente de 10-5 M a 10-10 M) al vehículo y en cada concentración de compuesto de prueba antagonista/agonista inverso de GPR17 en un volumen final de 20 pl de tampón HBSS Hepes (pH 7,4) que contenía DMSO al 1 %, forskolina 5 pM e IBMX 0,1 mM. Después de 60 minutos de incubación a temperatura ambiente, se termina la reacción y se lisan las células añadiendo el reactivo de detección d2 y el reactivo de criptato en 10 pL de tampón de lisis, cada uno según las instrucciones del fabricante. Después de 60 minutos de incubación, se miden los cambios en las concentraciones de cAMP según las instrucciones del fabricante utilizando un lector de placas Envision con excitación láser. Todas las incubaciones se realizaron por duplicado. Los datos se analizaron usando el software GraphPad Prism usando la ecuación logística de 4 parámetros para medir las pCE50 de MDL29,951 en ausencia y presencia de los compuestos de ensayo antagonistas/agonistas inversos de GPR17. La relación de dosis (DR) se representó frente a las concentraciones de antagonistas y el análisis de Schild proporcionó la afinidad estimada pA2 de los compuestos de ensayo antagonistas/agonistas inversos de GPR17.
Resultados del ensayo de cAMP:
Cuando se prueban en un ensayo de cAMP, los compuestos de ejemplo típicamente exhiben valores de pA2 mayores o iguales a 6,5; preferentemente mayores o iguales a 7,5; más preferentemente mayores o iguales a 8,5. Las actividades de los compuestos de los ejemplos ensayados se representan en la siguiente tabla. Los rangos de actividad A, B y C se refieren a los valores de pA2 en el ensayo de cAMP como sigue: "A": pA2 <7,5, "B": pA27,5 < x < 8,5, "C": 8,5 < pA2.
La siguiente Tabla 6 muestra los valores de pCl50 y de pA2 de los compuestos de ejemplo probados en el ensayo de Ca2+ y de cAMP. Los espacios en blanco en la columna de pA2 indican que los compuestos respectivos aún no se probaron, o que el resultado aún no estaba disponible.
Tabla 6:

B-IIC: Ensayos de maduración/mielinización de oligodendrocitos
Se pueden evaluar los efectos de los moduladores negativos de GPR17 en la maduración/mielinización de oligodendrocitos primarios in vitro mediante inmunoensayos utilizando anticuerpos dirigidos contra la proteína básica de mielina (MBP), como marcador de oligodendrocitos maduros.
Descripción del ensayo de transferencia de Western de MBP/oligodendrocitos/mielinización
Después de 3-4 días en medio de proliferación, se sembraron CPO primarias de rata a 25 000 células por cm2 en placas de cultivo tisular de 12 pocillos y se cambió a medio Neurobasal sin factores de crecimiento para inducir la diferenciación y expresión in vitro de la proteína GPR17. Para los análisis de diferenciación terminal y cuantificación de la expresión de proteínas, después de 24-48 h, el medio sin factores de crecimiento se complementó con 0,20 ng/ml de triyodotironina (T3) y 10 ng/ml de factor neurotrófico ciliar junto con los compuestos de prueba antagonistas/agonistas inversos de GPR17 1 pM o vehículo durante 3 días adicionales. Después del tratamiento con los compuestos, las células se lavaron dos veces con PBS helado y se lisaron en tampón de lisis enfriado con hielo (Tris 25 mM, pH 7,4, NaCl 150 mM, EDTA 1 mM, Triton X-100 al 1 %, IGEPAL al 1 %) complementado con una mezcla de inhibidores de proteasas. Los lisados se rotaron 20 min a 4 °C y se centrifugaron a 15000 * g a 4 °C durante 10 min. La concentración de proteína se determinó utilizando el ensayo de proteína de Pierce BCA de acuerdo con las instrucciones del fabricante. Se separaron 7,5-15 pg de proteína mediante electroforesis en gel de poliacrilamida-SDS al 10 % y se transfirieron a una membrana de nitrocelulosa mediante electrotransferencia. Después del lavado, las membranas se bloquearon con Roti-Block durante 1 h a temperatura ambiente y se incubaron durante la noche a 4 °C en Roti-Block con anticuerpo MBP (1:5000, LifeSpan BioSciences). Las membranas se lavaron 3 veces con PBS que contenía Tween al 0,1 % y luego se incubaron durante 1 hora a temperatura ambiente con un anticuerpo IgG anti ratón de cabra conjugado con peroxidasa de rábano en Roti-Block. Las proteínas inmunorreactivas se visualizaron mediante quimioluminiscencia utilizando el reactivo de detección de transferencia de Western ECL Prime de Amersham Biosciences y se cuantificaron mediante densitometría utilizando el programa informático Gelscan. Para normalizar la carga y la transferencia de proteínas por igual, las membranas se volvieron a analizar con un anticuerpo contra p-actina (1:2500, BioLegend; anticuerpo secundario anti-IgG conejo de cabra conjugado a HRP (ABIN)). Los cambios en el nivel de expresión de MBP en presencia de los compuestos de prueba se compararon con la expresión de MBP en las condiciones de control.
Descripción del ensayo de placas de fibras de MBP/maduración de oligodendrocitos/mielinización
Se sembraron CPO a 16000-22 000 células por cm2 en placas de fibra Mimetix Aligned de 96 pocillos (Electrospinning Company). Después de 2 días en medio de proliferación y 2 días en medio Neurobasal sin factores de crecimiento para inducir la diferenciación y expresión de la proteína GPR17 in vitro, se añadió vehículo o los compuestos de ensayo de antagonista/agonista inverso 1 pM en medio de diferenciación terminal suplementado con 0,20 ng/ml de triyodotironina y 10 ng/ml de factor neurotrófico ciliar durante 6 días, cambiando el medio después de 3 días. Luego, las células se fijaron en paraformaldehído al 4 %, seguido de lavados con PBS, permeabilización en Triton X-100 al 0,1 % en PBS y bloqueo con suero de cabra al 10 % y albúmina de suero bovino al 1 % en solución salina tamponada con fosfato. El anticuerpo hacia MBP se diluyó en tampón de bloqueo (1:2000) y se incubó durante 1 hora a 37 °C. Las células se lavaron nuevamente en PBS y se incubaron 1 h con anticuerpos secundarios conjugados con Cy2 contra IgG de ratón (Millipore, 1:500). Después de los lavados con PBS, las células se tiñeron con 0,2 pg/ml de DAPi, se lavaron de nuevo y se montaron con Mowiol. Se tomaron imágenes fluorescentes utilizando un microscopio Zeiss AxioObserver.Z1 con el sistema de formación de imágenes ApoTome y un objetivo Plan-Apochromat 20x/0.8, con un filtro eGFP (excitación 470/40 nm; emisión 525/50 nm) y filtro DAPI (excitación 365 nm; emisión 445/50 nm). Se tomaron imágenes de al menos 15 áreas aleatorias para el control (medio de diferenciación terminal con DMSO al 0,1 %) y para los compuestos de prueba utilizando los mismos ajustes procesados con el programa informático Zeiss ZEN2.3. Se informó de los cambios en el número de fibras mielinizadas por grupo de longitudes de fibra (0 a 40 pm, 41 a 60 pm, 61 a 80, 81 a 100, 101 a 120 y >120 pm) en ausencia o presencia del modulador negativo de GPR17.
Ensayo de inducción de CYP 450-1A2
Se siembran hepatocitos humanos crioconservados de un solo donante en una placa recubierta de colágeno de 96 pocillos de modo que la densidad de siembra final sea de 0,1 * 106 células/pocillo (volumen final por pocillo 0,1 ml). Luego, las células se incuban en medio de siembra a 37 °C, 95 % de humedad, 5 % de CO2 para permitir que las células se adhieran. Después de 4 h, el medio de siembra se reemplaza con 0,1 ml de medio Williams E sin suero precalentado que contiene 100 Ul/ml de penicilina, 100 pg/ml de estreptomicina, 10 pg/ml de insulina, de glutamina 2 mM e hidrocortisona 0,1 pM.
Al día siguiente, se administra a las células el compuesto de prueba en medio de ensayo (concentración final de compuesto de prueba 10 pM; concentración final de DMSO del 0,1%). Los inductores de control positivo, omeprazol (50 pM) para CYP1A2, fenobarbital (500 pM) para CYP2B6 y rifampicina (10 pM) para CYP3A4, se incuban junto con el compuesto de prueba. Se incluyen pocillos de control negativo en los que el compuesto de prueba se sustituye por el disolvente del vehículo (DMSO al 0,1 % en el medio de ensayo). Cada compuesto de ensayo se administra por triplicado. Las células se exponen a las disoluciones durante 72 h y se añade disolución fresca cada 24 h.
Para la determinación de la actividad catalítica, se prepara una disolución de los sustratos de la sonda, fenacetina (concentración final de 25 pM), bupropión (concentración final de 100 pM) y midazolam (concentración final de 2,5
|jM) en medio de ensayo precalentado. Al final del período de exposición de 72 h, el medio se reemplaza con el cóctel de sustratos de la sonda. Los hepatocitos se incuban durante 30 min. Se extrae una alícuota al final del período de incubación y se coloca en un patrón interno que contiene metanol en una proporción de 1:2. Las muestras se centrifugan a 2500 rpm a 4 °C durante 20 min. Se diluye una alícuota del sobrenadante con agua desionizada y se cuantifican los niveles de acetaminofeno, hidroxibupropión y 1-hidroximidazolam utilizando los métodos genéricos de LC-MS/MS de Cyprotex.
Los hepatocitos se solubilizan en hidróxido de sodio 0,1 M a temperatura ambiente y se determina el contenido de proteínas en cada pocillo utilizando el kit de ensayo de proteínas Pierce™ BCA (Thermo Scientific) que utiliza albúmina de suero bovino como patrón.
Para la determinación de la actividad catalítica, la concentración del metabolito formado en cada réplica del compuesto de prueba se convierte en actividad de CYP y se normaliza respecto de la proteína. El cambio proporcional se determina comparándolo con los pocillos de control de vehículo. El cambio proporcional observado a cada concentración de compuesto de prueba se expresa como un porcentaje de los compuestos de control positivo para cada isoforma de P450.
Los compuestos de la presente invención típicamente no muestran una inducción de CYP 1A2, o muestran una inducción de CYP 1A2 al menos significativamente menor que los compuestos comparativos con un hidrógeno en la posición R7.
Como ilustración, los compuestos ejemplares I-1, I-2, I-3 e I-12, donde R7 es cloro, ciclopropiloxi, difluorometilo y ciclopropilo, respectivamente, no mostraron inducción de CYP 1A2 en comparación con el control de vehículo, mientras que el Ejemplo Comparativo 1 correspondiente (6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida), en el que R7 es hidrógeno, mostró una inducción de CYP 1A2 16 veces mayor.
Claims (17)
1. Un compuesto que tiene la fórmula I

donde
R6 se selecciona de cloro, fluoro, fluorometilo y fluorometoxi,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro o metoxi,
R11 es hidrógeno, fluoro o metoxi,
L es un enlace, o un enlazador seleccionado de -CH2- y -CH2-CH2-O-,
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
2. Un compuesto según la reivindicación 1, donde R6 es cloro.
3. Un compuesto según la reivindicación 1, donde R6 es fluorometilo, preferiblemente difluorometilo.
4. Un compuesto según cualquiera de las reivindicaciones anteriores, donde R7 se selecciona de fluorometilo, ciclopropiloxi, fluoro y cloro.
5. Un compuesto según cualquiera de las reivindicaciones anteriores, donde X1 es hidrógeno.
6. Un compuesto según las reivindicaciones 1 a 5, donde R8 es fluoro y donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene una estructura según las Fórmulas IIa, IIb o IIc,

donde todos los demás sustituyentes son como se describieron en las reivindicaciones 1 a 5.
7. Un compuesto según la reivindicación 6,
donde
R6 se selecciona de cloro y fluorometilo, preferiblemente de cloro y difluorometilo
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi
R11 se selecciona de hidrógeno, metoxi y fluoro
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro.
8. Un compuesto según cualquiera de las reivindicaciones 1 a 5, donde R8 y R11 son ambos fluoro y donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene por tanto una estructura según la fórmula IIIa, IIIb o IIIc,

donde todos los demás sustituyentes son como se describieron en las reivindicaciones 1 a 5.
9. Un compuesto según una cualquiera de las reivindicaciones 1 a 5, donde R11 es metoxi y donde L se selecciona de (a) un enlace (b) -CH2- y (c) -CH2-CH2-O-, y dicho compuesto tiene por tanto una estructura según la fórmula Va, Vb o Vc:

donde todos los demás sustituyentes son como se describieron anteriormente.
10. Un compuesto según la reivindicación 9, donde
R6 se selecciona de fluoro, cloro, fluorometilo y fluorometoxi,
R7 se selecciona de fluoro, cloro, ciclopropilo, ciclopropiloxi, fluorometilo y fluorometoxi,
R8 es fluoro, y
X1 y X2 se seleccionan independientemente de hidrógeno y fluoro.
11. Un compuesto según cualquiera de las reivindicaciones 1, 4 y 6 a 10, donde
R6 y R7 se seleccionan independientemente de cloro y difluorometilo
y donde X1 es hidrógeno y X2 es hidrógeno o fluoro.
12. Un compuesto según cualquiera de las reivindicaciones anteriores, donde X1 es hidrógeno y X2 es fluoro.
13. Un compuesto según la reivindicación 1 seleccionado de
6.7- dicloro-N-[5-(2,2-difluoroetoxi)-3-metoxipiridin-2-il]-1H-indol-3-sulfonamida
7-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-6-(difluorometil)-1H-indol-3-sulfonamida 6-cloro-N-[5-[2-(difluorometoxi)etoxi]-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-6-(difluorometil)-7-fluoro-1H-indol-3-sulfonamida
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-(trifluorometoxi)-1H-indol-3-sulfonamida
6-cloro-N-[5-(difluorometoxi)-3-metoxipiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida
6.7- dicloro-N-[5-(difluorometoxi)-3-metoxipiridin-2-il]-1H-indol-3-sulfonamida
6-cloro-N-[5-(2,2-difluoroetoxi)-3-fluoro-6-metoxipiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida 6- cloro-N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida
7- cloro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-6-(difluorometil)-1H-indol-3-sulfonamida
6-(difluorometil)-7-fluoro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6-cloro-7-(difluorometil)-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6.7- dicloro-N-[5-(2,2-difluoroetoxi)-3-fluoropiridin-2-il]-1H-indol-3-sulfonamida
6.7- dicloro-N-[3-fluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
6-cloro-N-[5-(2,2-difluoroetoxi)-3,6-difluoropiridin-2-il]-7-fluoro-1H-indol-3-sulfonamida
6-cloro-7-ciclopropil-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida
6-cloro-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-7-(difluorometil)-1H-indol-3-sulfonamida
6-cloro-7-ciclopropiloxi-N-[3,6-difluoro-5-(2-fluoroetoxi)piridin-2-il]-1H-indol-3-sulfonamida,
y las sales, solvatos, isótopos y cocristales farmacéuticamente aceptables de los mismos.
14. Un compuesto según cualquiera de las reivindicaciones anteriores para el uso en la terapia.
15. Un compuesto según cualquiera de las reivindicaciones anteriores para el uso en el tratamiento o alivio de un trastorno desmielinizante.
16. Un compuesto según cualquiera de las reivindicaciones anteriores para el uso en el tratamiento o alivio de la esclerosis múltiple.
17. Composición terapéutica que comprende un compuesto según cualquiera de las reivindicaciones anteriores y un vehículo farmacéuticamente aceptable.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18178773.0A EP3584244A1 (en) | 2018-06-20 | 2018-06-20 | Substituted alkoxypyridinyl indolsulfonamides |
| PCT/EP2019/066148 WO2019243398A1 (en) | 2018-06-20 | 2019-06-19 | Substituted alkoxypyridinyl indolsulfonamides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2929829T3 true ES2929829T3 (es) | 2022-12-01 |
Family
ID=62715911
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19731283T Active ES2929829T3 (es) | 2018-06-20 | 2019-06-19 | Alcoxipiridinil indolsulfonamidas sustituidas |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US11976056B2 (es) |
| EP (3) | EP3584244A1 (es) |
| JP (1) | JP7371031B2 (es) |
| CN (1) | CN112292375B (es) |
| CA (1) | CA3101086A1 (es) |
| DK (1) | DK3810587T3 (es) |
| ES (1) | ES2929829T3 (es) |
| HR (1) | HRP20221289T1 (es) |
| HU (1) | HUE060181T2 (es) |
| LT (1) | LT3810587T (es) |
| MX (1) | MX2020012823A (es) |
| PL (1) | PL3810587T3 (es) |
| PT (1) | PT3810587T (es) |
| WO (1) | WO2019243398A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020254289A1 (en) * | 2019-06-17 | 2020-12-24 | Ucb Pharma Gmbh | N-(phenyl)-indole-3-sulfonamide derivatives and related compounds as gpr17 modulators for treating cns disorders such as multiple sclerosis |
| US20230167406A1 (en) * | 2020-05-01 | 2023-06-01 | The Children's Medical Center Corporation | Compositions and methods of promoting myelination |
| CN112375027B (zh) * | 2020-12-07 | 2023-03-31 | 中国药科大学 | 吲哚磺酰胺类衍生物及其医药用途 |
| JP2024507176A (ja) * | 2021-02-26 | 2024-02-16 | エフ. ホフマン-ラ ロシュ アーゲー | 新規なピリミジン-2-イルスルホンアミド誘導体 |
| WO2023241960A1 (en) * | 2022-06-15 | 2023-12-21 | Basf Se | A process for the photolytic chlorination of 3-halopyridine with molecular chlorine |
| EP4311828A1 (en) * | 2022-07-26 | 2024-01-31 | Basf Se | A process for the photolytic chlorination of 3-halopyridine with molecular chlorine |
| JP2025523939A (ja) * | 2022-07-20 | 2025-07-25 | エフ. ホフマン-ラ ロシュ アーゲー | 新規のナフチルおよびイソキノリンスルホンアミド誘導体 |
| WO2024017857A1 (en) | 2022-07-20 | 2024-01-25 | F. Hoffmann-La Roche Ag | Novel imidazopyridine and pyrazolopyridine sulfonamide derivatives |
| EP4558496A1 (en) * | 2022-07-20 | 2025-05-28 | F. Hoffmann-La Roche AG | Novel pyrimidinyl and triazinyl sulfonamide derivatives |
| US20260028335A1 (en) * | 2022-07-20 | 2026-01-29 | Hoffmann-La Roche Inc. | Novel isothiazol-3-yl and isoxazol-3-yl sulfonamide compounds |
| CN119546576A (zh) | 2022-07-20 | 2025-02-28 | 豪夫迈·罗氏有限公司 | 新颖的异喹啉酮、吡咯并吡啶酮和噻吩并吡啶酮磺酰胺衍生物 |
| EP4562003A1 (en) * | 2022-07-28 | 2025-06-04 | F. Hoffmann-La Roche AG | Novel 7-substituted indole sulfonamide derivatives |
| MA71601A (fr) * | 2022-07-28 | 2025-05-30 | F. Hoffmann-La Roche Ag | Nouveaux dérivés d'indole sulfonamide substitués en position 7 |
| CR20250058A (es) * | 2022-08-26 | 2025-05-09 | Hoffmann La Roche | Derivados novedosos deuterados de pirimidin-2-il sulfonamida |
| AR131263A1 (es) * | 2022-12-02 | 2025-03-05 | Rewind Therapeutics Nv | Compuestos de pirrolil-sulfonamida fusionados |
| WO2024153233A1 (en) * | 2023-01-19 | 2024-07-25 | Nanjing Immunophage Biotech Co., Ltd | Compounds and their uses as gpr17 antagonists |
| GB2700773A (en) * | 2024-05-23 | 2026-03-11 | Pheno Therapeutics Ltd | Compounds |
| GB2701173A (en) | 2024-05-23 | 2026-04-22 | Pheno Therapeutics Ltd | Compounds |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19814084B4 (de) | 1998-03-30 | 2005-12-22 | Lts Lohmann Therapie-Systeme Ag | D2-Agonist enthaltendes transdermales therapeutisches System zur Behandlung des Parkinson-Syndroms und Verfahren zu seiner Herstellung |
| SE9904750D0 (sv) | 1999-12-23 | 1999-12-23 | Pharmacia & Upjohn Ab | New formulation, use and method |
| DE602005024537D1 (de) | 2004-04-27 | 2010-12-16 | Takeda Pharmaceutical | Neuer ligand eines g-protein-konjugierten rezeptorproteins und verwendung davon |
| ITMI20042007A1 (it) | 2004-10-21 | 2005-01-21 | Consiglio Nazionale Ricerche | "modulatori del ricettore gpr17 e loro impieghi terapeutici" |
| CA2633588A1 (en) | 2005-12-20 | 2007-08-23 | Teikoku Pharma Usa, Inc. | Methods of transdermally administering an indole serotonin receptor agonist and transdermal compositions for use in the same |
| WO2009000262A1 (de) | 2007-06-25 | 2008-12-31 | Acino Ag | Elektrophoretisches transdermales applikationssystem |
| AU2009313315A1 (en) * | 2008-11-07 | 2011-06-30 | The Cleveland Clinic Foundation | Compounds and methods of promoting oligodendrocyte precursor differentiation |
| GB2491520A (en) | 2010-03-11 | 2012-12-05 | Jerry Swinford | Method and apparatus for washing downhole tubulars and equipment |
| EP2567698B1 (en) * | 2011-09-07 | 2014-02-12 | Rheinische Friedrich-Wilhelms-Universität Bonn | GPR 17 agonists and screening assay |
| KR20150008468A (ko) | 2012-05-09 | 2015-01-22 | 폰다지오네 이탈리아나 스클레로시 멀티플라 - 에프아이에스엠 온러스 | Gpr17 수용체 조절제 |
| JP7044789B2 (ja) | 2016-09-24 | 2022-03-30 | ワシントン・ユニバーシティ | Sarm1 nadアーゼ活性の阻害剤およびその使用 |
| TWI754702B (zh) | 2016-12-28 | 2022-02-11 | 德商Ucb製藥有限公司 | (氮雜)吲哚-和苯並呋喃-3-磺醯胺類 |
-
2018
- 2018-06-20 EP EP18178773.0A patent/EP3584244A1/en not_active Ceased
-
2019
- 2019-06-19 EP EP19731283.8A patent/EP3810587B1/en active Active
- 2019-06-19 HU HUE19731283A patent/HUE060181T2/hu unknown
- 2019-06-19 PT PT197312838T patent/PT3810587T/pt unknown
- 2019-06-19 PL PL19731283.8T patent/PL3810587T3/pl unknown
- 2019-06-19 ES ES19731283T patent/ES2929829T3/es active Active
- 2019-06-19 WO PCT/EP2019/066148 patent/WO2019243398A1/en not_active Ceased
- 2019-06-19 HR HRP20221289TT patent/HRP20221289T1/hr unknown
- 2019-06-19 CN CN201980039867.9A patent/CN112292375B/zh active Active
- 2019-06-19 LT LTEPPCT/EP2019/066148T patent/LT3810587T/lt unknown
- 2019-06-19 US US17/058,567 patent/US11976056B2/en active Active
- 2019-06-19 MX MX2020012823A patent/MX2020012823A/es unknown
- 2019-06-19 CA CA3101086A patent/CA3101086A1/en active Pending
- 2019-06-19 EP EP22174561.5A patent/EP4086248A1/en not_active Withdrawn
- 2019-06-19 JP JP2020570978A patent/JP7371031B2/ja active Active
- 2019-06-19 DK DK19731283.8T patent/DK3810587T3/da active
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019243398A1 (en) | 2019-12-26 |
| CA3101086A1 (en) | 2019-12-26 |
| JP2021527696A (ja) | 2021-10-14 |
| HUE060181T2 (hu) | 2023-02-28 |
| JP7371031B2 (ja) | 2023-10-30 |
| DK3810587T3 (da) | 2022-10-31 |
| US11976056B2 (en) | 2024-05-07 |
| CN112292375B (zh) | 2023-12-05 |
| CN112292375A (zh) | 2021-01-29 |
| EP3810587B1 (en) | 2022-08-10 |
| US20210214337A1 (en) | 2021-07-15 |
| MX2020012823A (es) | 2021-02-15 |
| EP4086248A1 (en) | 2022-11-09 |
| EP3810587A1 (en) | 2021-04-28 |
| LT3810587T (lt) | 2022-11-25 |
| HRP20221289T1 (hr) | 2022-12-23 |
| PL3810587T3 (pl) | 2023-02-06 |
| EP3584244A1 (en) | 2019-12-25 |
| PT3810587T (pt) | 2022-11-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2929829T3 (es) | Alcoxipiridinil indolsulfonamidas sustituidas | |
| ES2929992T3 (es) | Piridinil y pirazinil-(aza)indolsulfonamidas | |
| ES2969383T3 (es) | Derivados de N-(fenil)-indol-3-sulfonamida y compuestos relacionados como moduladores de GPR17 para el tratamiento de trastornos del SNC tales como la esclerosis múltiple | |
| RU2799321C2 (ru) | Пиридинил- и пиразинил(аза)индолсульфонамиды | |
| WO2024140754A1 (zh) | 一种萘酰胺类化合物、其制备方法及其应用 | |
| HK40062458A (en) | N-(phenyl)-indole-3-sulfonamide derivatives and related compounds as gpr17 modulators for treating cns disorders such as multiple sclerosis | |
| HK40046165A (en) | Pyridinyl and pyrazinyl-(aza)indolsulfonamides |