JP4171071B2 - α4β7インテグリンと反応するヒト化免疫グロブリン - Google Patents
α4β7インテグリンと反応するヒト化免疫グロブリン Download PDFInfo
- Publication number
- JP4171071B2 JP4171071B2 JP50985398A JP50985398A JP4171071B2 JP 4171071 B2 JP4171071 B2 JP 4171071B2 JP 50985398 A JP50985398 A JP 50985398A JP 50985398 A JP50985398 A JP 50985398A JP 4171071 B2 JP4171071 B2 JP 4171071B2
- Authority
- JP
- Japan
- Prior art keywords
- human
- heavy chain
- seq
- humanized immunoglobulin
- variable region
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images




















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2839—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the integrin superfamily
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Cell Biology (AREA)
- Toxicology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
インテグリンレセプターは、リンパ球の再循環とリンパ球の炎症部位への補給の両方の制御に重要である(カルロス(Carlos,T.M.)とハーラン(Harlan,J.M.)、Blood,84:2068-2101(1994))。ヒトα4β7インテグリンは、いくつかのリガンドを有し、そのうちの1つは、腸間膜リンパ節やパイエル斑の高内皮性小静脈で発現している(ストリーター(Streeter,P.R.)ら、Nature 331:41-46(1988))粘膜血管アドレシンMAdCAM−1である(ベルリン(Berlin,C.)ら、Cell 74:185-195(1993);アール(Erle,D.J.)ら、J.Immunol.153:517-528(1994))。このように、α4β7インテグリンは、腸の粘膜リンパ組織へのリンパ球の移動を仲介するホーミングレセプターとして作用する(シュバイクホッファー(Schweighoffer,T.)ら、J.Immunol.151:717-729(1993))。さらに、α4β7インテグリンは、フィブロネクチンおよび血管細胞接着分子−1(VCAM−1)と相互作用する。
例えば、潰瘍性結腸炎やクローン病のような炎症性腸疾患(IBD)は、胃腸管の炎症を伴う衰弱性及び進行性の疾患でありうる。米国だけでも推定200万人の人々を冒している症状には、腹痛、痙攣、下痢および直腸出血などがある。IBD治療には、抗炎症剤(コルチコステロイドおよびスルファサラジンなど)、免疫抑制剤(6−メルカプトプリン、シクロスポリンおよびアザチオプリンなど)および外科手術(結腸切除など)などが用いられている。ポドルスキー(Podolsky)、New Engl.J.Med.,325:928-937(1991)およびポドルスキー(Podolsky)、New Engl.J.Med.,325:1008-1016(1991)。
ネズミモノクローナル抗体(mAb Act−1)のようなヒトα4β7インテグリンに対する抗体は、粘膜リンパ節の高内皮性小静脈に存在する粘膜アドレシン細胞接着分子−1(MAdCAM−1)へのα4β7インテグリン結合を妨害する。Act−1は、ラザロビッツ(Lazarovits,A.I.)ら、J.Immunol.133:1857-1862(1984)により、ヒト破傷風毒素特異的Tリンパ球で免疫したマウスから最初に単離され、マウスIgG1/κ抗体であると報告された。シュバイクホッファー(Schweighoffer,T.)ら、J.Immunol.151:717-729(1993)による該抗体のさらに最近の分析により、該抗体はα4β7インテグリンを選択的に発現するヒトCD4+記憶Tリンパ球のある種のサブセットに結合できることが示唆された。しかしながら、ヒトにおける治療上の適用にネズミ抗体を使用することに関する重大な問題は、それらはヒトにおいて高い免疫原性があり、患者におけるマウス抗体の効力を低下させ、持続投与を妨げうるヒト抗ネズミ抗体応答(HAMA)を迅速に誘導することである。HAMA応答は、マウス抗体の迅速なクリアランスを生じ、いかなる治療の恩恵をも厳密に制限してしまう。
したがって、炎症性腸疾患に対する改良された治療アプローチに関する要求がある。
発明の要約
本発明は、免疫グロブリンが非ヒト起源(例えば、齧歯類)の抗原結合領域およびヒト起源の免疫グロブリンの少なくとも一部(例えば、ヒト枠組み領域、ガンマ型のヒト定常領域)を含有する、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンに関する。1つの態様において、本明細書に記載されるヒト化免疫グロブリンは、α4β7インテグリンへの結合に関して、ネズミAct−1またはLDP−02(実施例4等を参照のこと)と競合することができる。好ましい態様において、ヒト化免疫グロブリンの抗原結合領域は、Act−1モノクローナル抗体に由来する(例えば、LDP−02、図11(配列番号:19)および図12(配列番号:21)それぞれに示された軽鎖および重鎖の可変領域を含有する免疫グロブリン)。
例えば、ヒト化免疫グロブリンは、非ヒト起源の相補性決定領域(CDR)およびヒト枠組み領域に由来する枠組み領域(FR)を含む抗原結合領域を含有することができる。1つの側面において、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンは、α4β7を結合する非ヒト起源の抗体に由来するCDRおよびヒト起源(例えば、GM607’CL)の軽鎖に由来するFRを含む軽鎖、ならびにα4β7を結合する非ヒト起源の抗体に由来するCDRおよびヒト起源(例えば、21/28’CL)の重鎖に由来するFRを含む重鎖を含有する。別の側面において、軽鎖は、Act−1抗体の軽鎖に由来する3つのCDRを含有し、重鎖は、Act−1抗体の重鎖に由来する3つのCDRを含有する。
また、本発明は、ヒト化免疫グロブリン軽鎖(例えば、Act−1抗体の軽鎖のCDR1、CDR2およびCDR3ならびにヒト軽鎖FRを含有する)、ならびにヒト化免疫グロブリン重鎖(例えば、Act−1抗体の重鎖のCDR1、CDR2およびCDR3ならびにヒト重鎖FRを含有する)に関する。好ましい態様において、本発明は、本明細書に記載されたヒト化重鎖および軽鎖(例えば、図7(配列番号:12)に示された軽鎖の可変領域を含有するヒト化軽鎖、図9(配列番号:15)に示された重鎖の可変領域を含有するヒト化重鎖、図12(配列番号:21)に示された軽鎖の可変領域を含有するヒト化軽鎖、図11(配列番号:19)に示された重鎖の可変領域を含有するヒト化重鎖)に関する。また、1以上のヒト化軽鎖および/または重鎖を含有するヒト化免疫グロブリンも含まれる。
さらに、本発明は、本発明のヒト化免疫グロブリン(例えば、単鎖抗体)をコードする配列を含有する単離された核酸、ならびに本発明のヒト化免疫グロブリン軽鎖をコードする配列(例えば、配列番号:20)または重鎖をコードする配列(例えば、配列番号:18)を含有する単離された核酸に関する。例えば、本発明は、ネズミAct−1モノクローナル抗体に由来する抗原結合領域をコードする第1の核酸配列と、ヒト起源の免疫グロブリンの定常領域の少なくとも一部をコードする第2の核酸配列とを含有する、ヒト化免疫グロブリン軽鎖または重鎖をコードする融合遺伝子を提供する。
さらに、本発明は、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンまたはかかる免疫グロブリンの鎖をコードする核酸を含有する構築物に関する。例えば、α4β7インテグリンに対する結合特異性を有する非ヒト抗体の軽鎖に由来するCDRおよびヒト起源の軽鎖に由来する枠組み領域をコードするヌクレオチド配列を含むヒト化免疫グロブリン軽鎖をコードする融合遺伝子を含有する発現ベクターが提供される。かかる構築物の別の例は、α4β7インテグリンに対する結合特異性を有する非ヒト化抗体の重鎖に由来するCDRおよびヒト起源の重鎖に由来する枠組み領域をコードするヌクレオチド配列を含むヒト化免疫グロブリン重鎖をコードする融合遺伝子を含有する発現ベクターである。
また、本発明は、本発明の核酸を含有する構築物の1以上を含む、本発明の核酸を含有する宿主細胞に関する。1つの態様において、本発明は、ヒト化免疫グロブリン軽鎖をコードする第1の組換え核酸およびヒト化免疫グロブリン重鎖をコードする第2の組換え核酸を含有し、前記第1の核酸はネズミAct−1抗体の軽鎖に由来するCDRおよびヒト起源の軽鎖に由来する枠組み領域をコードするヌクレオチド配列を含み;前記第2の核酸はネズミAct−1抗体の重鎖に由来するCDRおよびヒト起源の重鎖に由来する枠組み領域をコードするヌクレオチド配列を含む、宿主細胞に関する。
また、本発明は、本発明の宿主細胞をヒト化免疫グロブリンの発現に適した条件下で維持し、それにより1または複数のヒト化免疫グロブリン鎖を発現させ、ヒト化免疫グロブリンを産生する工程を含むヒト化免疫グロブリンの調製方法を提供する。該方法は、ヒト化免疫グロブリンの単離工程をさらに含むことが可能である。
本発明のヒト化免疫グロブリンは、それらのネズミまたは他の非ヒトカウンターパートよりも免疫原性が弱くなりうる。したがって、本明細書に記載されたヒト化免疫グロブリンは、例えば、粘膜リンパ組織へのリンパ球ホーミングを制御し、それによって腸での炎症応答を低減するために、ヒトにおける治療剤として使用することができる。
さらに、本発明は、診断または(予防を含む)治療における使用のための本発明のヒト化免疫グロブリンに関する。1つの態様において、本発明は、胃腸管(腸に関連する内皮を含む)、他の粘膜組織またはMAdCAM−1分子を発現する組織の白血球浸潤に関連する疾患を含む炎症性疾患の治療等における、組織の白血球浸潤に関連する疾患の治療での使用のための本発明のヒト化免疫グロブリンに関する。特に好ましい態様において、本発明は、潰瘍性結腸炎やクローン病のような炎症性腸疾患(IBD)の治療における使用のための本発明のヒト化免疫グロブリンに関する。
別の側面において、本発明は、胃腸管、他の粘膜組織またはMAdCAM−1分子を発現する組織の白血球浸潤に関連する疾患を含む炎症性疾患の治療等における組織の白血球浸潤に関連する疾患の治療用の医薬の製造のための本発明のヒト化免疫グロブリンの使用に関する。特に好ましい態様において、本発明は、潰瘍性結腸炎やクローン病のような炎症性腸疾患(IBD)の治療用の医薬の製造のための本発明のヒト化免疫グロブリンの使用に関する。
【図面の簡単な説明】
図1は、いくつかの独立したマウス重鎖可変領域クローンから決定した可変領域を含有するコンセンサスDNA配列(配列番号:1)および予想されるアミノ酸配列(配列番号:2)の図である。
図2は、H2B#34と称する独立したマウス重鎖可変領域クローンから決定した可変領域配列の一部を含有するヌクレオチド配列(配列番号:3)および予想されるアミノ酸配列(配列番号:4)の図である。
図3は、いくつかの独立したマウス軽鎖可変領域クローンの可変領域を含有するヌクレオチド配列(配列番号:5)および予想されるアミノ酸配列(配列番号:6)の図である。クローニング用のKasI部位を導入するために作製した2つの変異の位置が示される。
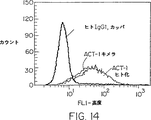
図4Aは、ネズミAct−1 mAbおよびマウスアイソタイプ適合無関係対照抗体(MOPC 21;IgG1、カッパ)の、α4β7インテグリンを発現するHuT78細胞を染色する能力を示す蛍光プロットである。
図4Bは、(i)キメラAct−1抗体、(ii)ヒトアイソタイプ適合無関係対照抗体(IgG1、カッパ)および(iii)COS−7細胞上清の、α4β7インテグリンを発現するHuT78細胞を染色する能力を示す蛍光プロットである。
図5は、マウスAct−1軽鎖可変領域(「Act−1.vl」)のアミノ酸配列(配列番号:7)およびヒトGM 607’CL軽鎖可変領域のアミノ酸配列(配列番号:8)のアラインメントである。同一のアミノ酸を垂直線で示し、類似のアミノ酸を類似性の程度により4個または2個の点で示す。CDRを括弧で標示し、残基に連続番号を付す。
図6は、マウスAct−1重鎖可変領域(「Act−1.vh」)のアミノ酸配列(配列番号:9)およびヒト21/28’CL重鎖可変領域のアミノ酸配列(配列番号:10)のアラインメントである。同一のアミノ酸を垂直線で示し、類似のアミノ酸を類似性の程度により4個または2個の点で示す。CDRを括弧で標示し、残基に連続番号を付す。
図7は、マウスAct−1軽鎖シグナルペプチド配列に結合したマウスAct−1軽鎖可変領域のヌクレオチド配列(配列番号:11)および予想されるアミノ酸配列(配列番号:12)の図である。
図8は、成熟ヒトGM607’CL抗体カッパ軽鎖可変領域のヌクレオチド配列(配列番号:13)およびアミノ酸配列(配列番号:8)の図である。
図9は、マウスAct−1抗体重鎖可変領域のヌクレオチド配列およびアミノ酸配列の図である。可変領域のヌクレオチド配列を、予想されるマウスAct−1重鎖シグナルペプチド配列をコードするヌクレオチド配列に結合させ、混成配列(配列番号:14および15)を得る。(重鎖領域を増幅させるプライマーの同一性は、縮重配列から予想され、シグナルペプチドに対するアミノ酸配列は、プライマー、下流の配列および他のシグナルペプチドの配列に由来した。示されたシグナルペプチドは、Act−1ハイブリドーマのものとは同一ではない。)
図10は、ヒト21/28’CL抗体重鎖可変領域のヌクレオチド配列およびアミノ酸配列の図である。可変領域をコードするヌクレオチド配列は、ヒト抗体HG3’CL(レシャビ(Rechavi,G.)ら、Proc.Natl.Acad.Sci.,USA 80:855-859(1983))のVHに由来するシグナルペプチド配列をコードするヌクレオチド配列に結合させ、混成配列(配列番号:16および17)を得る。
図11は、重鎖シグナルペプチドを有するヒト化Act−1抗体(LDP−02)の重鎖の一部のヌクレオチド配列(配列番号:18)およびアミノ酸配列(配列番号:19)の図である。
図12は、軽鎖シグナルペプチドを有するヒト化Act−1抗体(LDP−02)の軽鎖の一部のヌクレオチド配列(配列番号:20)およびアミノ酸配列(配列番号:21)の図である。
図13は、ヒト化Act−1免疫グロブリン(LDP−02)の軽鎖を作製するために使用したL1〜L6と称する重複相補的オリゴヌクレオチドのヌクレオチド配列(配列番号:22〜27)、およびヒト化Act−1免疫グロブリンの重鎖を作製するために使用したH1〜H10と称する重複相補的オリゴヌクレオチドのヌクレオチド配列(配列番号:28〜37)の図である。
図14は、マウス−ヒトAct−1キメラ免疫グロブリン、ヒト化Act−1免疫グロブリンまたは無関係なヒトアイソタイプ適合対照抗体(IgG1、カッパ)を用いるHuT78細胞の染色を示す蛍光プロットである。
図15は、HuT−78細胞におけるフローサイトメトリーにより行なったビオチニル化ネズミAct−1およびヒト化Act−1(LDP−02/3A9/LOT#1、実施例4)の滴定の結果を示すグラフである。
図16は、対照ネズミIgG1またはヒトIgG1と比較した、ネズミAct−1またはヒト化Act−1免疫グロブリン(LDP−02/3A9/LOT#1、実施例4)によるビオチニル化ネズミAct−1の結合の競合阻害を示すグラフである。
図17は、(a)CAMPATH−1H、(b)CAMPATH−1G、(c)ヒトIgG1、(d)LDP−02/3A9/LOT#1(実施例4)、または(e)LDP−01(ヒト化抗CD18、Fc変異)の、50、25、5、2.5および0.5μg/mlの濃度での存在下で、ヒト末梢血単核細胞の補体介在細胞溶解に関する51クロム放出アッセイの結果を示すグラフである。
図18A〜18Bは、α4β7産生細胞(RPMI8866)のネズミAct−1(図18A)、ネズミIgG1(図18A)、LDP−02/3A9/Lot#1(図18B)またはヒトIgG1(図18B)およびヒトMAdCAM−1−Igキメラ(免疫接着)による接着阻害をモニターする接着アッセイの結果を示すグラフである。
図19は、(a)LDP−02(Fc変異した)、(b)軽鎖に1つの変異(MV4)と重鎖に二重変異(R38K、A40R)を有するLDP−02(Fc変異した)の誘導体、または(c)無関係なヒトアイソタイプ適合対照抗体(IgG1、カッパ)を用いるHuT78細胞の染色を比較したグラフである。
発明の詳細な説明
本発明は、非ヒト起源の抗原結合領域およびヒト起源の免疫グロブリンの少なくとも一部を含有する、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンに関する。ヒト化免疫グロブリンは、少なくとも約107M-1のアフィニティーでα4β7インテグリンを結合しうることが好ましく、好ましくは少なくとも約108M-1、さらに好ましくは少なくとも約109M-1である。1つの態様において、ヒト化免疫グロブリンは、α4β7インテグリンを結合する非ヒト起源の抗原結合領域およびヒト定常領域に由来する定常領域を含む。別の態様において、α4β7インテグリンを結合するヒト化免疫グロブリンは、非ヒト起源の相補性決定領域およびヒト起源の可変枠組み領域、ならびに任意にヒト起源の定常領域を含有する。例えば、ヒト化免疫グロブリンは、軽鎖がα4β7インテグリンを結合する非ヒト起源の抗体に由来する相補性決定領域およびヒト起源の軽鎖に由来する枠組み領域を含み、重鎖がα4β7インテグリンを結合する非ヒト起源の抗体に由来する相補性決定領域およびヒト起源の重鎖に由来する枠組み領域を含む、重鎖ならびに軽鎖を含有しうる。
また、本発明は、ヒト化免疫グロブリン軽鎖またはヒト化免疫グロブリン重鎖に関する。1つの態様において、本発明は、非ヒト起源の軽鎖CDR(即ち、1以上のCDR)およびヒト軽鎖枠組み領域を含むヒト化免疫グロブリン軽鎖に関する。別の態様において、本発明は、非ヒト起源の重鎖CDR(即ち、1以上のCDR)およびヒト重鎖枠組み領域を含むヒト化免疫グロブリン重鎖に関する。CDRは、非ヒト免疫グロブリンに由来することができる。
天然の免疫グロブリンは、2つの同一の軽鎖(約24kD)と2つの同一の重鎖(約55または70kD)が四量体を形成する共通のコア構造を有する。各鎖のアミノ末端部分は、可変(V)領域として知られ、各鎖の残りの部分のより保存された定常(C)領域から区別されうる。J領域として知られるC末端部分は、軽鎖の可変領域内にある。重鎖の可変領域内には、J領域に加え、D領域が存在する。免疫グロブリンにおけるアミノ酸配列の変化の大部分は、抗原結合に直接関与する超可変領域または相補性決定領域(CDR)として知られるV領域中の3つの分離した位置に限られる。アミノ末端から進んで、これらの領域は、それぞれ、CDR1、CDR2およびCDR3と称される。CDRは、より保存された枠組み領域(FR)により適切に保持される。アミノ末端から進んで、これらの領域は、それぞれ、FR1、FR2、FR3およびFR4と称される。CDRおよびFR領域の位置とナンバリングシステムは、カバット(Kabat)らにより定義されている(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991);表3および4も参照のこと)。
ヒト免疫グロブリンは、重鎖のアイソタイプによりクラスおよびサブクラスに分けることができる。クラスは、重鎖がそれぞれ、ガンマ(γ)、ミュー(μ)、アルファ(α)、デルタ(δ)またはイプシロン(ε)型のものであるIgG、IgM、IgA、IgDおよびIgEを含む。サブクラスは、重鎖がそれぞれ、γ1、γ2、γ3、γ4、α1およびα2型のものであるIgG1、IgG2、IgG3、IgG4、IgA1およびIgA2を含む。選択されたクラスまたはサブクラスのヒト免疫グロブリン分子は、カッパ(κ)またはラムダ(λ)軽鎖のいずれかを含んでもよい。例えば、Cellular and Molecular Immunology、ウォンジーヴィクス(Wonsiewicz,M.J.)編、45章、41-50頁、W.B.Saunders Co、フィラデルフィア、PA(1991);ニソノフ(Nisonoff,A.)、Introduction to Molecular Immunology、第2版、4章、45-65頁、Sinauer Associates,Inc.、サンダーランド、MA(1984)を参照のこと。
本明細書で用いる「免疫グロブリン」という用語は、抗体全体およびその生物学的に機能しうる断片を含むものである。かかる生物学的に機能しうる断片は、対応する全長の抗体の少なくとも1つの抗原結合機能(例えば、Act−1抗体のα4β7に対する特異性)を保持し、好ましくはα4β7とその1以上のリガンド(例えば、MAdCAM−1、フィブロネクチン)との相互作用を阻害する能力を保持する。特に好ましい態様において、生物学的に機能しうる断片は、α4β7の粘膜アドレシン(MAdCAM−1)への結合を阻害することができる。使用されうる生物学的に機能しうる抗体断片の例は、単鎖抗体、Fv、Fab、Fab’およびF(ab’)2断片等のα4β7インテグリンに結合可能な断片を含む。かかる断片は、酵素的切断または組換え技術により生成することができる。例えば、パパインまたはペプシン切断を用い、それぞれ、FabまたはF(ab’)2断片を作製することができる。また、1以上の停止コドンを天然の停止部位の上流に導入した抗体遺伝子を用いて、種々の切断型の抗体も生成することができる。例えば、F(ab’)2断片の重鎖をコードするキメラ遺伝子は、重鎖のCH1ドメインおよびヒンジ領域をコードするDNA配列を含むように設計することができる。
本明細書で用いる「ヒト化免疫グロブリン」という用語は、少なくとも一部分がヒト起源のものである、異なる起源の免疫グロブリンの部分を含有する免疫グロブリンをいう。例えば、ヒト化抗体は、必要な特異性を有するマウス等の非ヒト起源の免疫グロブリンおよびヒト起源の免疫グロブリン配列に由来し(例えば、キメラ免疫グロブリン)、通常の技術により化学的に共に連結されるか遺伝子工学技術を用いて連続的なポリペプチドとして調製された(例えば、キメラ抗体のタンパク質部分をコードするDNAを発現させて連続的なポリペプチド鎖を生成することができる)部分を含有することができる。本発明のヒト化免疫グロブリンの別の例は、非ヒト起源の抗体に由来するCDRとヒト起源の軽鎖および/または重鎖に由来する枠組み領域とを含有する免疫グロブリン(例えば、枠組み変化を有するまたは有さないCDRを接ぎ合わせた抗体)である。また、キメラまたはCDRを接ぎ合わせた単鎖抗体は、ヒト化免疫グロブリンという用語に含まれるものである。例えば、キャビリー(Cabilly)ら、米国特許第4,816,567号明細書;キャビリーら、欧州特許第0,125,023 B1号明細書;ボス(Boss)ら、米国特許第4,816,397号明細書;ボスら、欧州特許第0,120,694 B1号明細書;ニューバーガー(Neuberger,M.S.)ら、国際公開第86/01533号パンフレット;ニューバーガーら、欧州特許第0,194,276 B1号明細書;ウインター(Winter)、米国特許第5,225,539号明細書;ウインター、欧州特許第0,239,400 B1号明細書;パドラン(Padlan,E.A.)ら、欧州特許出願第0,519,596 A1号明細書を参照のこと。また、単鎖抗体に関しては、ラドナー(Ladner)ら、米国特許第4,946,778号明細書;ハストン(Huston)ら、米国特許第5,476,786号明細書;およびバード(Bird,R.E.)ら、Science,242:423-426(1988))を参照のこと。
ヒト化免疫グロブリンの抗原結合領域(非ヒト部分)は、α4β7インテグリンに対する結合特異性を有する非ヒト起源の免疫グロブリン(ドナー免疫グロブリンと称する)に由来することができる。例えば、適する抗原結合領域は、ネズミAct−1モノクローナル抗体に由来することができる(ラザロビッツ(Lazarovits,A.I.)ら、J.Immunol.,133(4):1857-1862(1984));例えば、実施例1〜3を参照のこと)。他の供給源は、齧歯類(マウス、ラット等)、ウサギ、ブタ、ヤギまたは非ヒト霊長類(サル等)のような非ヒト供給源から得られたα4β7インテグリン特異的抗体を含む。Act−1抗体と同一または類似のエピトープに結合する抗体のような他のポリクローナルまたはモノクローナル抗体を作製することができる(例えば、コーラーら、Nature,256:495-497(1975);ハーローら、1988、Antibodies:A Laboratory Manual,(Cold Spring Harbor,NY);およびCurrent Protocols in Molecular Biology,第2巻、(補遺27、94年夏)、アウスベルら編、(John Wiley & Sons:New York,NY)、第11章(1991))。
例えば、抗体は、適切な哺乳動物(例えば、マウス、ラット、ウサギまたはヒツジ)において適した免疫原に対して惹起することができる。α4β7を産生する細胞、α4β7を含む膜画分、α4β7の免疫原断片、適当な担体に結合させたβ7ペプチドが適当な免疫原の例である。抗体産生細胞(例えば、リンパ球)は、例えば、免疫した動物のリンパ節または脾臓から単離することができる。次いで、該細胞を適当な不死化細胞(例えば、骨髄腫細胞株)に融合し、それによりハイブリドーマを生成することができる。融合細胞は、選択培養技術を用いて単離することができる。所望の特異性を有する抗体を産生する細胞は、適当なアッセイ(例えば、ELISA)により選択することができる。また、α4β7インテグリンに対する結合特異性を有する非ヒト起源の免疫グロブリンは、抗体ライブラリー(例えば、非ヒトFab分子を含有するファージライブラリー)から得ることもできる。
1つの態様において、ヒト化免疫グロブリンの抗原結合領域は、非ヒト化起源のCDRを含有する。この態様において、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンは、少なくとも1つの非ヒト起源のCDRを含有する。例えば、CDRは、ヒト化免疫グロブリンが非ヒト起源の1以上の免疫グロブリン由来の重鎖CDR1、CDR2および/またはCDR3;および/または軽鎖CDR1、CDR2および/またはCDR3を実質的に含み、生じたヒト化免疫グロブリンがα4β7インテグリンに結合特異性を有するように、非ヒト起源の免疫グロブリンの軽鎖および重鎖可変領域から派生させることができる。好ましくは、選択された鎖の3つのCDRすべてがドナーの対応する鎖のCDRと実質的に同じであり、さらに好ましくは、軽鎖および重鎖の3つのCDRすべてが対応するドナー鎖のCDRと実質的に同じである。
ヒト化免疫グロブリンまたはヒト起源の免疫グロブリン鎖の部分(ヒトの部分)は、いかなる適当なヒト免疫グロブリンまたは免疫グロブリン鎖に由来することができる。もし存在するならば、例えば、ヒト定常領域またはその一部は、アレル変異体を含むヒト抗体のκもしくはλ軽鎖、および/またはγ(例えば、γ1、γ2、γ3、γ4)、μ、α(例えば、α1、α2)、δもしくはε重鎖に由来することができる。特定の定常領域(例えば、IgG1)、変異体またはその一部は、エフェクター機能を調整するために選択することができる。例えば、変異した定常領域(変異体)は、Fcレセプターへの結合および/または補体を固定する能力を最小にするために融合タンパク質に取り込まれうる(例えば、実施例3を参照のこと;ウインター(Winter)ら、英国特許第2,209,757 B号明細書;モリソン(Morrison)ら、国際公開第89/07142号パンフレット;モーガン(Morgan)ら、国際公開第94/29351号パンフレット、1994年12月22日も参照のこと)。
もし存在するならば、ヒト枠組み領域(例えば、軽鎖可変領域のもの)は、抗原結合領域ドナーの類似体または均等の領域(例えば、軽鎖可変領域)に配列類似性を有するヒト抗体可変領域に由来することが好ましい。ヒト化免疫グロブリンのヒト起源の部分に関する枠組み領域の他の供給源は、ヒト可変コンセンサス配列を含む(例えば、実施例2を参照のこと;ケトルボロー(Kettleborough,C.A.)ら、Protein Engineering 4:773-783(1991);カーター(Carter)ら、国際公開第94/04679号パンフレット、1994年3月3日発行も参照のこと))。例えば、非ヒト部分を得るために用いた抗体または可変領域の配列は、カバットら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991)に記載のヒト配列と比較することができる。特に好ましい態様において、ヒト化免疫グロブリン鎖の枠組み領域は、非ヒトドナー(例えば、マウスAct−1抗体)の可変領域と少なくとも全体の約65%の配列同一性を有し、好ましくは少なくとも全体の約70%の配列同一性を有するヒト可変領域に由来する。また、ヒト部分は、非ヒトドナーの均等な部分(例えば、FR)と比較したとき、用いた特定の部分(例えば、FR)内で、少なくとも約65%の配列同一性を有し、好ましくは少なくとも約70%の配列同一性を有するヒト抗体に由来することもできる。例えば、実施例2に記載されるように、マウスAct−1およびヒトGM607’CL軽鎖可変領域間の全体の配列同一性は、71.4%であり、マウスAct−1およびヒト21/28’CL重鎖可変領域間の全体の配列同一性は、68.1%であった。
1つの態様において、ヒト化免疫グロブリンは、ヒト起源の抗体の1以上の鎖に由来する少なくとも1つの枠組み領域(FR)を含有する。したがって、FRは、ヒト起源の1以上の抗体に由来するFR1および/またはFR2および/またはFR3および/またはFR4を含むことができる。好ましくは、選択されたヒト化鎖のヒト部分は、ヒト起源の可変領域に由来(例えば、ヒト免疫グロブリン鎖由来、ヒトコンセンサス配列由来)するFR1、FR2、FR3およびFR4を含む。
本発明に用いられる非ヒト起源およびヒト起源の免疫グロブリン部分は、それらが由来する免疫グロブリンもしくは免疫グロブリン部分またはそれらの変異体と同一の配列を有する。かかる変異体は、1以上の残基の付加、欠失または置換により異なる突然変異体を含む。前に示したように、非ヒト起源のCDRは、非ヒトドナーにおいて実質的に同じであり、好ましくは非ヒトドナーのCDRと同一である。実施例2に記載されるように、ヒト起源の枠組み領域の残基をドナーの対応する部分由来の残基と置換するもの等の枠組み領域における変化をなすことができる。1以上のアミノ酸の欠失、挿入および置換を含む枠組み領域における1以上の突然変異をなすことができる。いくつかのかかる置換は、実施例2においてヒト化Act−1抗体の設計に記載されている。選択されたヒト化抗体または鎖に関して、枠組み突然変異を本明細書に記載のように設計することができる。好ましくは、ヒト化免疫グロブリンは、非ヒトドナーのものと類似かまたはよりよいアフィニティーで、α4β7インテグリンを結合することができる。変異体は、非ヒトドナーまたはアクセプターのヒト鎖の突然変異導入を含む種々の適当な方法で生成することができる。
本発明のヒト化免疫グロブリンは、ヒトα4β7インテグリンに対する結合特異性を有し、ヘテロダイマーのα4および/またはβ7鎖の決定基を結合可能な(断片を含む)ヒト化免疫グロブリンを含む。好ましい態様において、本発明のヒト化免疫グロブリンは、結合機能(例えば、α4β7インテグリンに対する特異性を有する、同一または類似のエピトープ特異性を有する)および/または阻害機能(例えば、イン・ビトロおよび/またはイン・ビボでMAdCAM−1に結合するα4β7インテグリンを阻害する能力、またはα4β7インテグリンを産生する細胞のそのリガンド(例えば、MAdCAM−1を産生する細胞)への結合を阻害する能力のようなイン・ビトロおよび/またはイン・ビボでα4β7依存性接着を阻害する能力)のようなネズミAct−1抗体の少なくとも1つの機能特性を有する。したがって、好ましいヒト化免疫グロブリンは、ネズミAct−1抗体の結合特異性、ネズミAct−1抗体のエピトープ特異性(例えば、α4β7(例えば、α4β7インテグリン産生細胞上)への結合に関して、ネズミAct−1、キメラAct−1抗体(例えば、実施例1を参照のこと)またはヒト化Act−1(例えば、LDP−02)と競合できる)および/または阻害機能を有することができる。
α4β7インテグリンに結合特異性を有するヒト化免疫グロブリンの結合機能は、例えば、ヒト化免疫グロブリンとα4β7インテグリン(例えば、α4β7インテグリンを含有する膜画分、ヒトリンパ球(例えば、CD4+α4hi,β1loサブセットのリンパ球)、ヒトリンパ球細胞株、またはα4β7インテグリンを発現するα4および/またはβ7をコードする核酸を含有する組換え宿主細胞)のようなα4β7インテグリンを産生する細胞上で)との間の複合体の形成をモニターするアッセイを用いる標準的な免疫学的方法により検出することができる。
また、結合および/または接着アッセイまたは他の適当な方法を、所望の特異性で、ヒト化免疫グロブリン(例えば、ライブラリー由来)の同定および/または単離のための手法(例えば、α4β7インテグリンを産生する細胞とそのリガンド(例えば、MAdCAMを発現する第2の細胞、MAdCAM−Igキメラとの接着をモニターするアッセイ(実施例4等を参照のこと)、または他の適当な方法に用いることもできる。
本発明に用いられる非ヒトおよびヒト起源の免疫グロブリン部分は、軽鎖、重鎖ならびに軽鎖および重鎖の一部を含む。これらの免疫グロブリン部分は、免疫グロブリンから(例えば、ある部分のドゥノボの合成により)得ることができるし、免疫グロブリンに由来することができ、または、免疫グロブリンまたは所望の性質(例えば、α4β7インテグリンを結合する、配列類似性)を有するその鎖をコードする核酸を生成して発現することができる。ヒトおよび非ヒト起源の所望の部分(例えば、抗原結合領域、CDR、FR、C領域)を含有するヒト化免疫グロブリンは、所望のヒト化鎖をコードする遺伝子(例えば、cDNA)を調製するための合成および/または組換え核酸を用いて生成することができる。鎖の一部を調製するためには、1以上の停止コドンを所望の位置に導入することができる。例えば、新規に設計したヒト化可変領域をコードする核酸(例えば、DNA)配列は、存在するDNA配列を変化させるためのPCR突然変異導入方法を用いて構築することができる(例えば、カマン(Kamman,M.)ら、Nucl.Acids Res.17:5404(1989)を参照のこと)。新規CDRをコードするPCRプライマーは、同一または非常に類似したヒト可変領域に基づいて前もってヒト化した可変領域のDNA鋳型にハイブリダイズすることができる(サトー(Sato,K.)ら、Cancer Research 53:851-856(1993))。類似のDNA配列が鋳型として利用できないならば、可変領域配列をコードする配列を含有する核酸を合成オリゴヌクレオチドから構築することができる(例えば、コルビンガー(Kolbinger,F.)、Protein Engineering 8:971-980(1993))。また、シグナルペプチドをコードする配列を核酸に組み込む(例えば、合成で、ベクターへの挿入により)こともできる。天然のシグナルペプチド配列を利用できないならば、他の抗体由来のシグナルペプチド配列を用いることができる(例えば、ケトルボロー(Kettleborough,C.A.)ら、Protein Engineering 4:773-783(1991)を参照のこと)。これらの方法、本明細書に記載された方法または他の好適な方法を用いて、変異体を容易に生成することができる(例えば、実施例5を参照のこと)。1つの態様において、クローニングした可変領域(例えば、LDP−02のもの)は、突然変異を導入することができ、所望の特異性を有する変異体をコードする配列を(例えば、ファージライブラリーから;例えば、クレバー(Krobber)ら、米国特許第5,514,548号明細書;フーゲンブーム(Hoogenboom)ら、国際公開第93/06213号パンフレット、1993年4月1日公開)選択することができる。
核酸および該核酸を含有する構築物
また、本発明は、本発明のヒト化免疫グロブリンまたはヒト化免疫グロブリン軽鎖もしくは重鎖をコードする配列を含有する、単離されたおよび/または組換え(例えば、本質的に純粋を含む)核酸に関する。
本明細書における「単離された」核酸とは、その供給源のゲノムDNAまたは細胞RNAの核酸(例えば、細胞中またはライブラリーのような核酸の混合物中に存在するもの)から分離された核酸であり、本質的に純粋な核酸、化学合成により、生物学的方法と化学的方法との組合せにより生成された核酸ならびに単離された組換え核酸を含む、本明細書に記載された方法または他の好適な方法により得られた核酸を含む(例えば、ドウガティ(Daugherty,B.L.)ら、Nucleic Acids Res.,19(9):2471-2476(1991);ルイス(Lewis,A.P.)とクローエ(J.S.Crowe)、Gene,101:297-302(1991))。
本明細書における「組換え」と言及された核酸とは、ポリメラーゼ連鎖反応(PCR)および/または制限酵素を用いるベクターへのクローニングのような人工的な組換え方法に依存する手法により生成する核酸を含む、組換えDNA方法論により生成された核酸である。また、「組換え」核酸は、細胞の天然のメカニズムを通じて発生する組換え事象からも生じるものであるが、所望の組換え事象を起こさせ、おそらく起こすように設計した核酸の細胞への導入後選択されるものでもある。
また、本発明は、より具体的にはヒト化Act−1免疫グロブリン(即ち、非ヒト部分がネズミAct−1モノクローナル抗体に由来する本発明のヒト化免疫グロブリン)またはその鎖をコードするヌクレオチド配列を含有する単離されたおよび/または組換え核酸に関する。1つの態様において、軽鎖は、Act−1抗体の軽鎖に由来する3つの相補性決定領域を含有し、重鎖は、Act−1抗体の重鎖に由来する3つの相補性決定領域を含有する。かかる核酸は、例えば、(a)ヒト化Act−1免疫グロブリンの重鎖可変領域のアミノ酸配列(例えば、図11の重鎖可変領域(配列番号:19)、図9の重鎖可変領域(配列番号:15))を含むポリペプチドをコードする配列を含有する核酸、(b)ヒト化Act−1免疫グロブリンの軽鎖可変領域のアミノ酸配列(例えば、図12の軽鎖可変領域(配列番号:21)、図7の軽鎖可変領域(配列番号:12))を含むポリペプチドをコードする配列を含有する核酸、(c)ヒト化Act−1免疫グロブリンの軽鎖または重鎖可変領域の少なくとも機能性部分(例えば、前記鎖を含有するヒト化免疫グロブリンの抗原結合に充分な部分)をコードする配列を含有する核酸、を含む。遺伝コードの縮重性により、選択されたポリペプチドをコードする種々の核酸を作製することができる。1つの態様において、核酸は、二本鎖または一本鎖のポリヌクレオチドを含む、前記もしくは実質的に前記図11(配列番号:18)、または前記もしくは実質的に前記図12(配列番号:20)の可変領域のヌクレオチド配列を含有する。(種々の図面が可変領域よりも大きいポリペプチド(即ち、シグナルペプチドコーディング配列または定常領域コーディング配列の一部を含む)を図示してもよいが、特定の図面の可変領域への参照は、示された配列の可変領域の部分を含むことを意味する。)これらの基準に適合する単離されたおよび/または組換え核酸は、前記したヒト化Act−1抗体またはその変異体の配列と同一の配列をコードする核酸を含有することができる。
本発明の核酸は、α4β7インテグリンに結合特異性を有するヒト化免疫グロブリンの生成に使用することができる。例えば、本発明のヒト化免疫グロブリンをコードする核酸(例えば、DNA)を、さらなる配列の操作または適当な宿主細胞におけるコード化ポリペプチドの生成のために、適当な構築物(例えば、ベクター)に組み込むことができる。
α4β7インテグリンに対する特異性を有するヒト化免疫グロブリンの製造方法
本発明の別の側面は、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンの調製方法に関する。例えば、ヒト化免疫グロブリンは、例えば、適当な宿主細胞でのα4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンをコードする1以上の組換え核酸の発現により得ることができる。
また、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンの発現に適する構築物または発現ベクターも提供される。構築物は、適当な宿主細胞に導入することができ、本発明のヒト化免疫グロブリンを発現する細胞を製造して培養で維持することができる。好適な宿主細胞は、大腸菌、枯草菌(B.スブチリス)およびまたは他の適当な細菌等の細菌細胞を含む原核細胞、真核細胞、例えばカビ細胞や酵母細胞(例えば、ピチア パストリス、アスペルギルス スピーシーズ、サッカロミセス セレビジエ、シゾサッカロミセス ポンベ、ニューロスポラ クラッサ)または他の下等真核細胞、ならびに昆虫由来の細胞(例えば、Sf9昆虫細胞(国際公開第94/26087号パンフレット、オコンノー(O’Connor)、1994年11月24日公開)または哺乳動物由来の細胞(例えば、COS細胞、NSO細胞、SP2/0、チャイニーズハムスター卵巣細胞(CHO)、HuT78細胞、293細胞)等の高等真核細胞を挙げることができる(例えば、アウスベル(Ausubel,F.M.)ら編、Current Protocols in Molecular Biology,Greene Publishing Associates and John Wiley & Sons Inc.,(1993)を参照のこと)。
α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンを産生する宿主細胞は、以下のように製造できる。例えば、所望のヒト化免疫グロブリンのコーディング配列の全部または一部をコードする核酸を、核酸ベクター、例えばプラスミド、ウイルスまたは他の適当な発現用レプリコン等のDNAベクターに挿入することができる。種々のベクターを利用することができ、それらには単一コピーもしくは多コピーで維持されるベクターまたは宿主細胞染色体に組込まれるベクターが含まれる。
好適な発現ベクターはいくつかの成分を含有でき、限定されないが、下記の1以上:複製起点、選択可能マーカー遺伝子、1以上の発現制御エレメント、例えば、転写制御エレメント(例えば、プロモーター、エンハンサー、ターミネーター)、および/または1以上の翻訳シグナル;膜標的化または分泌のシグナル配列またはリーダー配列が挙げられる。構築物中、シグナル配列を、該ベクターまたは他の供給源により提供することができる。例えば、免疫グロブリンの転写および/または翻訳シグナルを用いて発現を指示することができる。
プロモーターは、適当な宿主細胞での発現用に提供されうる。プロモーターは構成的であってもよいし、誘導性であってもよい。例えば、プロモーターがコードされているポリペプチドの発現を指示するように、ヒト化免疫グロブリンまたは免疫グロブリン鎖をコードする核酸に操作可能に連結することができる。原核生物宿主に適した種々のプロモーター(例えば、大腸菌にはlac、tac、T3、T7プロモーター)および真核生物宿主に適した種々のプロモーター(例えば、酵母アルコールデヒドロゲナーゼ(ADH1)、SV40、CMV)を、利用できる。
さらに、発現ベクターは通例、該ベクターを保有する宿主細胞を選択するための選択可能マーカーおよび複製可能発現ベクターの場合、起点または複製(an origin or replication)を含有する。抗生物質耐性または薬剤耐性を付与する産物をコードする遺伝子は、一般的な選択可能マーカーであり、原核細胞(例えばβ−ラクタマーゼ遺伝子(アンピシリン耐性)、テトラサイクリン耐性用のTet遺伝子)中および真核細胞(例えばネオマイシン(G418またはゼネティシン)、gpt(ミコフェノール酸)、アンピシリン、またはハイグロマイシン耐性遺伝子)中で、使用してもよい。ジヒドロ葉酸レダクターゼマーカー遺伝子は、メトトレキセートによる選択を、種々の宿主で可能にする。宿主の栄養要求性マーカーの遺伝子産物をコードする遺伝子(例えば、LEU2、URA3、HIS3)は、酵母における選択可能マーカーとしてしばしば使用される。ウイルス(例えばバキュロウイルス)またはファージベクターおよび宿主細胞のゲノムに組み込むことができるレトロウイルスベクターなどのベクターの使用も予想される。また本発明は、これらの発現ベクターを保有する細胞にも関する。
例えば、α4β7インテグリンに対する結合特異性を有するヒト化免疫グロブリンの重鎖および軽鎖をコードする核酸(即ち、1以上の核酸)またはかかる1もしくは複数の核酸を含有する構築物(即ち、1以上の構築物)は、1もしくは複数の核酸が1以上の発現制御エレメントに操作可能に(例えば、ベクター中で、細胞中でプロセッシングにより生成された構築物中で、宿主細胞ゲノムに組み込まれて)結合されるように、選択した宿主細胞に適した方法(例えば、形質転換、トランスフェクション、エレクトロポレーション、感染)で適当な宿主細胞に導入することができる。宿主細胞は、発現に適した条件下で(例えば、誘導剤、適切な塩類、成長因子、抗生物質、栄養補給物等を補足した好適な培地の存在下で)維持され、それによりコードされている1もしくは複数のポリペプチドを生成することができる。所望ならば、コードされた蛋白質(例えば、ヒト化Act−1抗体)は、(例えば、宿主細胞、培地、ミルク)から単離できる。この過程は、トランスジェニック動物の宿主細胞での発現を包含する(例えば、国際公開第92/03918号パンフレット、ゲンファーム インターナショナル、1992年3月19日公開を参照のこと)。
ヒト化免疫グロブリンまたは免疫グロブリン鎖が、融合蛋白質のN末端位置、C末端位置または中間で非免疫グロブリン部分(即ち、天然に見出されるような免疫グロブリンには存在しない部分)に結合している融合蛋白質を生成することができる。例えば、いくつかの態様は、免疫グロブリン配列をコードする核酸の適当な発現ベクター、例えば、pETベクター(pET−15b、ノバ−ジェン等)、ファージベクター(pCANTAB 5 E、ファルマシア等)または他のベクター(pRIT2T プロテインA融合ベクター、ファルマシア等)への挿入により生成されうる。得られた構築物を、発現に適した宿主細胞に導入することができる。発現後、適当なアフィニティーマトリックスを利用して、細胞溶解液からいくつかの融合蛋白質を単離または精製できる(例えば、Current Protocols in Molecular Biology(アウスベル(Ausubel,F.M.)ら編、第2巻、補遺26、16.4.1-16.7.8(1991)を参照のこと)。
治療法と組成物
本発明は、(1)α4β7インテグリンをイン・ビトロおよび/またはイン・ビボで結合でき;および/または、(2)α4β7インテグリンの活性または機能、例えば、(a)結合機能(例えば、α4β7インテグリンのMAdCAM−1、フィブロネクチンおよび/またはVCAM−1に結合する能力)および/または(b)組織における白血球の補給および/または蓄積を含む白血球浸潤機能(例えば、腸粘膜組織へのリンパ球移動を阻害する能力)を調節することができるヒト化免疫グロブリンを提供する。好ましくは、ヒト化免疫グロブリンは、イン・ビトロおよび/またはイン・ビボで選択的にα4β7を結合し、α4β7媒介相互作用を阻害することが可能である。1つの態様において、ヒト化免疫グロブリンは、α4β7インテグリンを結合し、α4β7インテグリンの1以上のそのリガンド(例えば、MAdCAM−1、VCAM−1、フィブロネクチン)への結合を阻害することができ、それにより(組織における白血球の補給および/または蓄積を含む)組織の白血球浸潤を好ましくは選択的に阻害する。かかるヒト化免疫グロブリンは、腸関連組織、リンパ節器官または白血球(特にT細胞またはB細胞のようなリンパ球)を含む粘膜組織で、α4β7インテグリンを産生する細胞の血管内皮細胞への細胞接着をイン・ビトロおよび/またはイン・ビボで阻害することができる。特に好ましい態様において、ヒト化免疫グロブリン(例えば、Act−1)は、α4β7のMAdCAM−1および/またはフィブロネクチンとの相互作用を阻害することができる。
本発明のヒト化免疫グロブリンは、研究、診断および治療法における適用を有する様々な過程で有用である。例えば、それらは、α4β7インテグリンまたはその変異体を(例えば、アフィニティー精製または他の好適な方法により)検出、単離および/または精製するため、ならびにα4β7インテグリン構造(例えば、コンフォメーション)および機能を研究するために用いることができる。
また、本発明のヒト化免疫グロブリンは、診断適用(例えば、イン・ビトロ、エクス・ビボ)または治療(予防を含む)適用におけるα4β7インテグリン機能の調節にも使用されうる。
例えば、本発明のヒト化免疫グロブリンは、試料(例えば、炎症滲出物、血液、血清、腸液、α4β7インテグリンを産生する細胞上などの組織または体液)中のα4β7インテグリンのレベルを検出および/または測定するために使用することができる。例えば、試料(例えば、組織および/または体液)は、個体から得ることが可能であり、化学発光アッセイ、ラジオイムノアッセイおよび免疫組織学を含む酵素結合免疫吸着アッセイ(ELISA)等の方法を含む適当な免疫学的方法を用いて、α4β7インテグリン発現を検出および/または測定することができる。1つの態様において、ヒト化免疫グロブリンのα4β7インテグリンへの特異的結合に適した条件下で、試料を本発明のヒト化免疫グロブリンと接触させる工程および形成された抗体−α4β7インテグリン複合体を検出する工程を含む、試料中の選択されたα4β7インテグリンの検出方法が提供される。当該方法の適用において、ヒト化免疫グロブリンは、正常組織と炎症組織(例えば、ヒト由来)とのα4β7インテグリン反応性および/または発現について(例えば、免疫組織学的に)分析し、IBDまたは他の状態とα4β7の発現増加(例えば、冒された組織において)との関連を検出するために用いることができる。本発明のヒト化免疫グロブリンは、正常組織と炎症組織とでα4β7インテグリンの存在の免疫学的評価方法を可能にし、それを通じて、疾患の存在、疾患の進行および/または抗α4β7インテグリン療法の炎症性疾患における効力を評価できる。
また、本発明のヒト化免疫グロブリンは、α4β7インテグリンの結合機能および/または白血球(例えば、リンパ球、単球)浸潤機能を調節するために用いられうる。例えば、α4β7インテグリンのリガンド(即ち、1以上のリガンド)への結合を阻害するヒト化免疫グロブリンは、組織、特にMAdCAM分子を発現する組織の白血球(例えば、リンパ球、単球)浸潤(組織における白血球の補給および/または蓄積を含む)に関連した疾患の治療における方法に従って投与することができる。本発明のヒト化免疫グロブリン(即ち、1以上)の有効量を、かかる疾患を治療するために個体(例えば、ヒトまたは他の霊長類のような哺乳動物)に投与する。例えば、胃腸管(腸関連内皮を含む)、他の粘膜組織、またはMAdCAM分子を発現する組織(例えば、小腸および大腸の固有層の細静脈のような腸関連組織;ならびに乳腺(例えば、泌乳性乳腺))の白血球浸潤に関連する疾患を含む炎症性疾患は、本発明の方法に従って治療することができる。同様に、MAdCAM分子を発現する細胞(例えば、内皮細胞)への白血球の結合の結果としての組織の白血球浸潤に関連した疾患を有する個体を、本発明の方法に従って治療することができる。
特に好ましい態様において、そのように治療できる疾患として、炎症性腸疾患(IBD)、例えば潰瘍性大腸炎、クローン病、回腸炎、腹腔疾患、非熱帯性スプルー、セロネガティブ関節症に関連する腸疾患、微細もしくはコラーゲン性大腸炎、好酸球性胃腸炎または直腸結腸切除術や回腸肛門吻合術後に起こる嚢炎が含まれる。
膵炎とインシュリン依存性真性糖尿病は、本発明の方法を用いて治療できるその他の病気である。MAdCAM−1は、NOD(非肥満糖尿病)マウスならびにBALB/cマウスおよびSJLマウスの外分泌膵臓のいくつかの血管によって発現されると報告されている。MAdCAM−1の発現は、NODマウスの膵臓の炎症を起こした島の内皮上で誘導されると報告されており、MAdCAM−1は、インスリン炎の初期段階にNOD島内皮によって発現される主要アドレシンであった(ハンニネン(Hanninen,A.)ら,J.Clin.Invest.,92:2509-2515(1993))。さらに、島内にはα4β7を発現するリンパ球の蓄積が観察され、MAdCAM−1は、炎症を起こした島由来の血管へのα4β7を介したリンパ腫細胞の結合に関与していた(ハンニネンら,J.Clin.Invest.,92:2509-2515(1993))。
本発明の方法に従って治療できる粘膜組織関連炎症性疾患の例は、乳腺炎(乳腺)、胆嚢炎、胆管炎または胆管周辺炎(胆管と肝臓周辺の組織)、慢性気管支炎、慢性副鼻腔炎、喘息および(例えば、胃腸管における)対宿主性移植片病を含む。クローン病に見られるように、炎症はしばしば粘膜表面を超えて広がるので、間質性繊維症をもたらす肺の慢性炎症性疾患、例えば過敏性肺炎、コラーゲン病、類肉腫症および他の突発性疾患も治療できる。
ヒト化免疫グロブリンは、α4β7インテグリンのそのリガンドへの結合を阻害する有効量で投与される。治療の場合、有効量とは、所望の治療(予防を含む)効果を達成するに足る量(例えば、α4β7インテグリンが介在する結合および/またはシグナル伝達を軽減または妨げ、そうすることによって、白血球の接着および浸潤および/または関連する細胞応答を阻害するに足る量)になるだろう。ヒト化免疫グロブリンは、1回量または多数回量で投与することができる。用量は、当該技術分野で公知の方法によって決定でき、例えば患者の年齢、感受性、耐性および総合的健康状態などに依存することができる。抗体の好適な用量としては、1処置あたり約0.1〜約10.0mg/kg体重であることができる。
本発明の方法に従って、ヒト化免疫グロブリンは、単独でまたは別の薬剤と組み合わせて個体(例えばヒト)に投与できる。ヒト化免疫グロブリンは、その助剤投与の前、助剤投与と同時または助剤投与後に投与できる。1つの態様において、α4β7インテグリンのそのリガンドへの結合を阻害する1を越えるヒト化免疫グロブリンを投与する。別の態様において、内皮リガンドへの白血球の結合を阻害するモノクローナル抗体、例えば、抗MAdCAM−1、抗VCAM−1または抗ICAM−1抗体を、本発明のヒト化免疫グロブリンに追加して投与する。さらに別の態様において、追加の薬学的に活性な成分(例えば、スルファサラジン、他の非ステロイド系抗炎症性化合物またはステロイド系抗炎症性化合物のような抗炎症性化合物)を、本発明のヒト化免疫グロブリンと組み合わせて投与することができる。
様々な投与経路が可能であり、治療対象の疾患または状態に応じて、非経口(静脈内注射、動脈内注射、筋肉内注射、皮下注射など)、経口(食餌法など)、局所、吸入(気管支内吸入、鼻孔内吸入または経口吸入、鼻孔内滴剤など)または直腸投与などが挙げられるが、必ずしもこれらに限られるわけではない。非経口投与が好ましい投与法である。
処方は、選択した投与経路に応じて変化するだろう(例えば液剤、乳剤)。投与対象のヒト化免疫グロブリンを含有する適当な組成物を、生理学的に許容されうる賦形剤または担体中で調製することができる。液剤や乳剤の場合は、適当な担体には、例えば塩水や緩衝媒体を含む水溶液、アルコール/水溶液、乳液または懸濁液が含まれる。非経口用の賦形剤には、塩化ナトリウム溶液、リンゲルデキストロース、デキストロースと塩化ナトリウム、乳酸化リンゲルまたは不揮発性油が含まれうる。静脈内用の賦形剤には、種々の添加物、保存剤、または液体、栄養または電解物質補充剤が含まれうる(一般的には、Remington’s Pharmaceutical Sciences,第17版、Mack Publishing Co.,PA,1985を参照のこと)。吸入法の場合は、化合物を可溶化し、適当な投与用ディスペンサー(例えば、アトマイザー、ネブライザーまたは加圧エアロゾールディスペンサー)に充填することができる。
実施例
以下の実施例によって本発明を説明するが、本発明は、これらの実施例によりなんら限定されるものではない。
実施例1に記載されるように、ネズミAct−1抗体を精製し、該抗体の配列分析を行なった。マウスAct−1抗体の軽鎖および重鎖可変領域をコードするcDNAをPCRクローニングし、シーケンスした。また、Act−1カッパ軽鎖可変領域(VL)のアミノ酸配列は、蛋白質のシーケンスにより決定し、VL遺伝子のDNA配列に由来したアミノ酸配列と正確に適合することがわかった。重鎖可変領域(VH)のアミノ酸配列の大部分は、蛋白質のシーケンスにより決定され、この配列もVH遺伝子のDNA配列から推定されるアミノ酸と適合する。これらの結果により、ハイブリドーマ細胞株から正しいマウスAct−1可変領域がクローニングされたことが示される。正しい配列がクローニングされたことを確認した機能的なキメラAct−1抗体が生成された。特に、マウスAct−1軽鎖および重鎖可変領域をコードするDNAを、ヒトカッパ軽鎖およびヒトガンマ−1もしくはガンマ−4重鎖定常領域それぞれに結合させた。また、キメラ抗体を、ヒト化Act−1 mAb(再形成Act−1 mAb LDP−02)との比較分析に用いた。
α4β7インテグリンとよく結合するヒト化Act−1抗体を作製するために、再形成ヒト可変領域を設計した(実施例2)。設計過程において補助するために、マウスAct−1可変領域の分子モデルを作った。ネズミAct−1抗体の領域は、抗原への結合に直接関与し、相補性決定領域すなわちCDRは、選択された可変領域内に接ぎ合わされた。ヒト可変領域の枠組み領域(FR)内の位置でのいくつかのアミノ酸変化がなされた。再形成ヒトAct−1可変領域は、元のヒト残基が対応するネズミ残基にそれぞれ変化している、選択されたヒト軽鎖可変領域のFR中の1つのアミノ酸変化および選択されたヒト重鎖可変領域のFR中の5つのアミノ酸変化を含んでいた。
実施例3に記載されるように、これらの再形成ヒトAct−1可変領域をコードするDNA配列を構築し、ヒト定常領域をコードするDNA配列に結合させ、得られた核酸を用いてヒト化Act−1免疫グロブリンを生成させた。ヒト化Act−1抗体を、哺乳動物細胞で発現させ(実施例3)、マウスAct−1抗体と比較してヒトα4β7インテグリンへの結合を試験した(実施例4)。表5に示すように、ヒト化Act−1抗体は、ネズミAct−1により認識されるエピトープに対する特異性を保持し、天然のネズミ抗体と比較して予期しない改良された結合アフィニティーを示した。
ヒト化Act−1抗体のいくつかの変異体は、設計過程で同定された(実施例2および5)。例えば、以下の位置の1以上での追加の変化がなされうる:軽鎖変異体M4V(4位でのMet→Val変異)、重鎖変異体R38K(38位でのArg→Lys変異)、重鎖変異体A40R(40位でのAla→Arg変異)。さらに、73位をヒトスレオニン残基に復帰させる重鎖変異体I73T(73位でのIle→Thr復帰突然変異)がヒト枠組み領域中のこの位置で見られた。一本鎖でのこれらの変化の1以上の導入または1を越える鎖でのこれらの変化の種々の組合せがなされうる。
実施例1 Act−1 V H およびV L 領域のクローニングならびにネズミ−ヒトAct−1キメラ免疫グロブリンの構築と発現
Act−1 VHおよびVL領域のクローニング
RNAは、製造業者の示したプロトコールに従ってTRIzol試薬(Gibco/BRL)を用いて、Act−1モノクローナル抗体を産生するハイブリドーマ細胞から得た(ラザロビッツら、J.Immunol.133(4):1857-1862(1984);ラザロビッツとコルビン(R.B.Colvin)により供与された)。
転写された重鎖および軽鎖可変領域は、製造業者の示したプロトコールに従ってIg−Primeキット(Novagen)を用いるポリメラーゼ連鎖反応(PCR)により増幅した。要約すると、1.5μgの全RNAを、2.0μlの5X MMLV緩衝液(5X=250mM Tris−HCl、25℃でpH8.3、375mM KCl、15mM MgCl2)、1.0μlの100mM DTT(ジチオスレイトール)、0.5μlの10mM dNTP混合物(各10mMのdATP、dCTP、dTTP、dGTP)、0.5μlのオリゴdT(1μg/μl)、0.25μlのアセチル化BSA(4mg/ml)、1.0μlの適当なIg−3’プライマー(10pmol/μl)、0.5μlのMMLV逆転写酵素(200ユニット/μl)および10μlの全容量まで添加したRNアーゼ不含有水を含む反応で、cDNAに逆転写した。混合物を37℃で5分間、42℃で30分間および99℃で5分間インキュベートした。各Ig−3’プライマーを別々の反応に用いた。
可変領域は、製造業者のプロトコールに従って、逆転写した材料から増幅した。要約すると、8μlの逆転写した材料を、4μlの2.5mM dNTP、5μlの10X反応緩衝液(10X=100mM Tris−HCl、25℃でpH8.8、500mM KCl、15mM MgCl2、1% Triton X−100)、2.5μlのIg−5’リーダープライマー(10pmol/μl)(各Ig−5’プライマーを別々のPCR反応に用いた)、0.25μl(1.25ユニット)のAmpliTaq▲R▼DNAポリメラーゼ(Perkin-Elmer)および50μlの全容量までの水と混合した。
5’プライマーMuIgVH5’−A、MuIgVH5’−B、MuIgκVL5’−AおよびMuIgκVL5’−Bとの増幅に関して、サイクルパラメーターは、1分、94℃;1分、50℃;2分、72℃の35サイクル後72℃で最後の6分の伸長であった。アニーリング温度を60℃に上げたことを除いて、他のすべての5’プライマーについて同じ反応条件を用いた。
重鎖可変領域は、3’プライマーとしてMuIgGVH3’−2またはMuIgMVH3’−1のいずれか、および5’プライマーとしてMuIgVH5’−BまたはMuIgVH5’−Eのいずれかを用いて増幅が成功した。軽鎖可変領域は、3’プライマーとしてMuIgκVL3’−1および5’プライマーとしてMuIgκVL5’−Gを用いて増幅が成功した。
これらのプライマーの配列は、以下のようであった:

適当なサイズのインサートを含む白色コロニーを、pT7Blueベクターのポリクローニングサイトのすぐ外側のインサートの反対側でアニールするT7プロモータープライマーおよびU−19マープライマーを用いてシーケンスした。シーケンスは、製造業者の推奨したプロトコールに従ってSequenase T7DNAポリメラーゼキット(USB/Amersham Life Science)を用いてミニプレップDNAで行なった。
いくつかの独立した重鎖可変領域クローン由来のコンセンサスDNA配列(配列番号:1)および推定アミノ酸配列(配列番号:2)を図1に示す。縮重プライマーは、配列においてある縮重へ導いた。開始コドンは、ヌクレオチド13〜15によりコードされるMetであり、予想されるリーダーペプチダーゼ切断部位は、ヌクレオチド67〜69によりコードされるSerとヌクレオチド70〜72によりコードされるGlnとの間(ヌクレオチド13〜69はリーダーペプチドをコードする)である。残基433〜435によりコードされるアラニンで始まるネズミ定常領域の部分を示す。
いくつかの独立した軽鎖可変領域クローンのDNA配列(配列番号:5)およびアミノ酸配列(配列番号:6)を図3に示す。重鎖可変領域とは異なり、増幅した配列は縮重していなく、おそらく、使用したプライマーがあまり縮重していなく、可変領域が単一のプライマー対のみから増幅されたためであろう。
キメラ重鎖遺伝子の構築
キメラマウス−ヒト重鎖遺伝子をコードする遺伝子を作製した。ヒト重鎖定常領域の供給源は、野生型ヒトガンマ1(γ1)定常領域を含むクローン(ヘルマン ウォルトマン(Herman Waldman)博士(オックスフォード大学)から得た)であり;構築物は、pEE6発現ベクター(Celltech)中にヒト化抗CD18重鎖遺伝子を含有する3818と称した。定常領域は、その教示がそのまま参照により本明細書に取り込まれる、シムズ(Sims,M.J.)ら、J.Immunol.,151(4):2296-2308(1993)および1993年2月4日に公開された国際公開第93/02191号パンフレットに記載のようにpEE6.hCMVにクローニングされたヒト化CD18重鎖遺伝子の定常領域と一致する。ヒト化抗CD18抗体の重鎖可変領域および定常領域(野生型ガンマ1)をコードする配列は、HindIIIとEcoRIとの消化により発現ベクターから放出された。重鎖遺伝子を含む1.421bp断片を回収し、pCR−ScriptTM(Stratagene)のHindIIIおよびEcoRI部位にサブクローニングし、pCR−CD18Hと称するプラスミドを得た。SpeI制限部位は、抗CD18重鎖遺伝子の可変領域と定常領域との間のジャンクションに位置する。pCR−CD18HをHindIIIとSpeIで消化して、重鎖可変領域を放出させた。この可変領域を、以下のように、縮重したマウスAct−1可変領域と置換した。
新規な制限部位を取り込むために2つのプライマーを合成した。これらのプライマーは:
キメラ軽鎖遺伝子の構築
キメラマウス−ヒト軽鎖遺伝子は、重鎖に関するものと同様な様式で構築した。しかしながら、キメラ軽鎖の場合、新規な制限部位KasIは、KG#87と称するマウスAct−1軽鎖可変領域クローンの1つを鋳型として用いる可変領域断片のPCR増幅、およびヒト化抗CD18カッパ軽鎖遺伝子を含む構築物(ヘルマン ウォルトマン博士(オックスフォード大学)から得た;pEE12発現ベクター中にヒト化抗CD18軽鎖を含む3819と称する構築物)を鋳型として用いるカッパ軽鎖定常領域のPCR増幅により構築物内で工作した。定常領域は、シムズ(Sims,M.J.)ら、J.Immunol.,151(4):2296-2308(1993)および1993年2月4日に公開された国際公開第93/02191号パンフレットに記載のようにpEE12にクローニングされたヒト化CD18軽鎖遺伝子の定常領域と一致する。
可変領域に対するプライマーは:
カッパ定常領域に対するプライマーは:
軽鎖可変領域および定常領域を、それぞれの鋳型とプライマーを用いて別々に増幅し、PCR産物をpCR−ScriptTMに別々にサブクローニングして配列を確認した。次いで、各断片をHindIIIとKasIとの消化により該ベクターから放出させ、ゲルで精製して、HindIII消化によりヒト化抗CD18軽鎖遺伝子をすでに除去しておいた3819 pEE12発現ベクターのHindIII部位にトリプルライゲートした。得られた構築物をPEE12mhACT1Lchiと称する。
キメラ免疫グロブリンの発現
キメラ重鎖および軽鎖遺伝子の両方を含む発現ベクターの構築のために、全重鎖遺伝子とCMVプロモーターを、BglIIとBamHIとの消化によりpEE6発現ベクター(pEE6mhACT1Hchi)から放出させた。次いで、この断片を、pEE12軽鎖遺伝子発現ベクター(pEE12mhACT1Lchi)のBamHI部位にライゲートし、それぞれが別のCMVプロモーターの転写制御下にあるキメラ軽鎖遺伝子およびキメラ重鎖遺伝子を両方とも含むpEE12mhLHchiと称する1つのプラスミドが生成した。
pEE6hCMV−BおよびpEE12発現ベクターならびにCelltech グルタミン シンセターゼ ジーン アンプリフィケーション システムは、以前に記載されている(例えば、その教示がそれぞれ、そのまま参照により本明細書に取り込まれる国際公開第86/05807号パンフレット(Celltech)、国際公開第87/04462号パンフレット(Celltech)、国際公開第89/01036号パンフレット(Celltech)、欧州特許第0 323 997 B1号明細書(Celltech)および国際公開第89/10404号パンフレット(Celltech)を参照のこと)。
キメラ抗体の一過性の発現のために、20μgのpEE12mhLHchiを、以下のようにエレクトロポレーションによりCOS−7細胞(アメリカンタイプカルチャーコレクション、パークローンドライブ12301、ロックビル、MD、20852)にトランスフェクトした。対数増殖期で生育しているCOS−7細胞を、トリプシン−EDTAでの処理により組織培養フラスコから採集した。該細胞をリン酸緩衝化塩水(PBS)で1回、ハンクのバランス塩溶液(HBSS)で1回洗浄し、1mlのHBSS当たり1.5x107個の細胞濃度で再懸濁させた。0.8mlのHBSS中1.2x107個の細胞を20μgのプラスミドDNAと混合し、室温で10分間インキュベートした。次いで、DNA/細胞混合物を0.4cmのエレクトロポレーションキュベットに移し、Bio−Rad GenePulserを用いて250V、960μFで電流を適応した。エレクトロポレーション後室温で10分間のインキュベーション後、該細胞を20mlの培養培地(ダルベッコの改変イーグル培地(DMEM)+10%FCS)に移し、162cm2の組織培養フラスコ(Costar)で培養した。5日後、細胞培養上清を集め、α4β7インテグリンを発現するHuT78細胞を染色する能力を試験した。HuT78細胞(ヒトTリンパ腫細胞株)は、アメリカンタイプカルチャーコレクション、パークローンドライブ12301、ロックビル、MD、20852のアクセッション番号ATCC TIB 161から利用することができる。
100μlの一過性にトランスフェクトしたCOS−7細胞培養上清、モックトランスフェクトしたCOS−7細胞上清、精製したネズミAct−1抗体(10μg/ml)またはそれぞれマウスに対する精製した無関係アイソタイプ適合対照抗体(マウスIgG1、カッパ(MOPC21)、Sigma製の10μg/ml)およびヒトに対する精製した無関係アイソタイプ適合対照抗体(ヒトIgG1、カッパ、Sigma製の10μg/ml)を、1x105個のHuT78細胞と氷上で30分間インキュベートした。該細胞を、2%ウシ胎仔血清(FCS)および0.01%アジ化ナトリウムを含むPBSからなる緩衝液(FACS緩衝液)を氷冷したもので2回洗浄した。次いで、該細胞を適切な蛍光二次抗体(フルオレッセイン(FITC)結合AffiniPure F(ab’)2断片ヤギ抗マウスIgG(H+L)(Jackson ImmunoResearch)またはフルオレッセイン(FITC)結合AffiniPure F(ab’)2断片ヤギ抗ヒトIgG(H+L)(Jackson ImmunoResearch)のいずれか)とともに氷上で30分間インキュベートした。氷上で30分後、該細胞をFACS緩衝液で2回洗浄し、300mlの同じ緩衝液に再懸濁し、Becton Dickinson FACscanでフローサイトメトリーにより分析した。図4Aは、マウスアイソタイプ適合無関係対照抗体、MOPC21(IgG1、カッパ)と比較したネズミAct−1 mAbの染色を示す。図4Bは、ヒトアイソタイプ適合無関係対照抗体(IgG1、カッパ)およびモックトランスフェクトしたCOS−7細胞上清と比較したHuT78細胞のキメラAct−1抗体の染色を示す。したがって、ネズミAct−1抗体により生じた染色と比較して、キメラ抗体は同様にHuT78細胞を染色した。まとめると、これらのデータは、マウスAct−1可変領域に関して適切な配列をうまくクローン化し、かつ、発現したことを示す。
アミノ酸配列分析
アミノ酸配列分析を、精製したネズミAct−1重鎖および軽鎖で行ない、ハイブリドーマから単離された軽鎖および重鎖可変領域のcDNAの同一性を確認した。これは、軽鎖に関しては以下のように達成された:
ネズミAct−1(5mg/ml)を、窒素雰囲気下、0.3M ホウ酸ナトリウム、0.15M 塩化ナトリウム中、37℃で2時間、2mM DTTで還元した。次いで、該溶液をヨードアセトアミドで10mMにし、室温で4時間インキュベートした。非変性条件下でのSDS−PAGE分析により、蛋白質が定量的に還元されていることを確認した。次いで、該蛋白質溶液をPBSで大量に透析し、アリコートをSuperdex 75カラム(16/60、Pharmacia)に適用した(操作1)。重鎖および軽鎖は、排除容量のそれに対応する溶出容量でこのカラムから共溶出し、このことは、2つの鎖がまだ共に保持されていることを示唆する。次いで、新たなアリコートを8M尿素処理し、変性条件下(6M尿素)でsuperdex 75カラムにかけた(操作2)。両鎖は、おそらくフォールディングされないことにより、ボイドボリュームに再び共溶出した。SDS−PAGE分析により、2つのゲル濾過操作から溶出された2つの試料に両鎖が存在することを確認した。これらの試料を、N末端配列分析(Commonwealth Biotechnologies,Inc.)に供し、以下の結果を得た:
内部アミノ酸シーケンスを単純化するために、ペプシンでの切断により抗体からF(ab)’2断片を生成させた。ネズミAct−1を、0.1Mクエン酸ナトリウム、pH3.0中、抗体:ペプシンが1:200の割合でペプシンにより37℃で2時間切断した。SDS−PAGE分析で評価したように、反応は完全であった。次いで、蛋白質をプロテインGおよびプロテインAカラムを通して精製した。次いで、前記したように、試料を還元してアルキル化し、調製用SDS−PAGE(15%)により重鎖断片を軽鎖断片から分離した。重鎖断片を切り出し、ランニング緩衝液を有する1mlの0.1%SDSに2時間電気溶出させた。この試料を、2ngのAsp−Nエンドプロテイナーゼで30分間切断し、SDS−PAGE(17.5%)により断片を分離した。消化産物は、0.1M Hepes pH8.0、0.1%SDSに一晩受動溶出させ、N末端配列分析(Commonwealth Biotechnologies,Inc.)に供した。
17Kda断片から得られた配列は、DYAIDYWG(配列番号:49)であり、これは、重鎖のクローンに存在していた(図1;配列AIDYはJH4領域の開始部位に対応する)。
実施例2 マウスAct−1可変領域の分子モデル化
CDRを接ぎ合わせた可変領域の設計を補助するために、マウスAct−1可変領域の分子モデルを作製した。よくキャラクタライズされた蛋白質ファミリーの構造を免疫グロブリンでモデル化することは、ホモロジーによるモデル化に関して確立された方法を用いて行なった。分子モデル化は、UNIXオペレーティングシステム、分子モデリングパッケージQUANTA(Polygen Corp.,Waltham,MA)および解明された蛋白質構造のBrookhaven結晶学データベース下で稼働するSilicon Graphics IRIS 4Dワークステーションを用いて行なった。第1工程として、新規可変領域の枠組み領域(FR)を、類似の構造的に解明された免疫グロブリン可変領域由来のFRでモデル化した。同一のアミノ酸側鎖は、それらの元の方向で保持されていたが、変異した側鎖は、元のマウスAct−1抗体と同様なカイ(chi)アングルを維持するように最大重複法を用いて置換された。新規可変領域のCDRの大部分は、CDRに関する標準構造に基づいてモデル化された(ショチア(Chothia,C.)とレスク(A.M.Lesk)、J.Mol.Biol.196:901-917(1987);ショチアら、Nature 342:877-883(1989);トラモンタノ(Tramontano,A.)ら、J.Mol.Biol.215:175-182(1990);ショチアら、J.Mol.Biol.227:799-817(1992))。公知の標準構造がない重鎖可変領域のCDR3のような場合、CDRループは、いかなる構造的に解明された蛋白質にも存在する類似のループ構造に基づいてモデル化した。最後に、好ましくない原子の接触を軽減し、ファンデルワールスおよび静電的相互作用を最適化させるために、該モデルをQUANTAで実行したようなCHARMmポテンシャル(ブルックス(Brooks,B.R.)、J.Comp.Chem.4:187-217(1983))を用いるエネルギー極小化に供した。
マウスAct−1可変領域に関して、軽鎖可変領域由来のFRは、マウスモノクローナル抗体4−4−20(ヘロン(Herron,J.N.)ら、Proteins.Structure,Function and Genetics 5:271-280(1989))のFab断片由来のFRでモデル化した。重鎖可変領域由来のFRは、マウスモノクローナル抗体D11.15(キターラ(Chitarra,V.)ら、Proc.Natl.Acad.Sci.,USA 90:7711-7715(1993))のFab断片由来のFRでモデル化した。マウスAct−1抗体とモデルの基礎となった可変領域との間で異なるこれらのアミノ酸側鎖を置換した。次いで、2つの異質可変領域(即ち、4−4−20に基づくカッパ軽鎖可変領域およびD11.15に基づく重鎖可変領域)を互いの観点から正しい方向に置換するために、Fab4−4−20抗体の軽鎖を、(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991)により規定されたように)残基35〜39、43〜47、84〜88および98〜102の空間で整列させることによりD11.15の軽鎖上に重ね合わせた。
mAb Act−1の軽鎖可変領域のCDR1(L1)は、ショチアら、Nature 342:877-883(1989)により提唱されたように、L1標準サブグループ4に適合した。マウスFab4−4−20(前記参照のこと)のL1ループは、アミノ酸の長さが同一であり、アミノ酸配列が類似し、さらに標準サブグループ4にも適合した。その結果として、該L1ループは、Fab4−4−20のL1ループでモデル化された。同様に、mAb Act−1の軽鎖可変領域のCDR2(L2)およびCDR3(L3)は、それぞれの標準サブグループ1ループ構造およびFab4−4−20の対応するCDRの両方に適合した。したがって、Act−1カッパ軽鎖可変領域のL2およびL3ループは、Fab4−4−20のCDR L2およびL3でモデル化された。
mAb Act−1の重鎖可変領域のCDR1(H1)は、マウス mAb D11.15(前記を参照のこと)の対応するH1ループと同様に、ショチアら、Nature 342:877-883(1989)により規定されたように、H1標準サブグループ1に適合した。さらに、mAb D11.15 CDR1ループは、mAb Act−1のH1と長さが同一であり、アミノ酸配列が非常に類似していた。その結果として、軽鎖と同様に、このループは、該モデルの基礎となった重鎖可変領域のCDR1ループでモデル化された。重鎖可変領域のCDR2(H2)を規定するのはより困難であったが、H2標準サブグループ2に対応するように思われた。また、D11.15抗体のH2ループも同一の標準サブグループに適合し、アミノ酸配列が非常に類似していたので、mAb Act−1のH2ループは、D11.15のH2ループでモデル化された。
前記考察したように、重鎖可変領域のCDR3は、高度に変化しており、同定可能な構造グループに分類することができない。H3ループをモデル化するためには、同一の長さと類似のアミノ酸配列のループ−好ましくは別の抗体由来−が同定され、ループのモデル化の基礎として用いられる。ループサイズに関してAct−1のCDR3に適合する、3種の抗体由来のすべてがH3ループである3つのループが存在した。立体的不一致についてすべての3つのループ構造をモデルで試験した後、ヒト抗体Pot(ファン(Fan,Z.C.)ら、J.Mol.Biol.228:188-207(1992))由来のH3ループが選ばれ、mAb Act−1のH3ループをモデル化した。自明な立体的不一致についてモデルの全体を調整した後、該モデルをQUANTAで実行したように、エネルギー極小化に供した。
CDR接合可変領域の設計
CDRを接ぎ合わせた可変領域の設計における第1工程は、ヒト化可変領域の基礎として役立つヒト軽鎖および重鎖可変領域の選択である。ヒト可変領域の選択に関する2つのアプローチを試験して比較した。1つのアプローチでは、ヒト可変領域をヒト可変領域の異なるサブグループに対するコンセンサス配列から選択した(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991))。齧歯類軽鎖および重鎖可変領域を、ヒトコンセンサス配列と比較し、最も類似したヒト軽鎖および重鎖コンセンサス配列を、ヒトラムダ軽鎖可変領域の6つのサブグループ、ヒトカッパ軽鎖可変領域の4つのサブグループおよびヒト重鎖可変領域の3つのサブグループの中から選択した(ケトルボロー、Protein Engineering 4:773-783(1991)を参照のこと)。別のアプローチでは、ヒト可変領域をヒト可変領域に関するすべての公開された配列から選択した(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991))。齧歯類軽鎖および重鎖可変領域のアミノ酸配列を、ヒト配列と比較し、齧歯類可変領域と高い程度の類似性を有するヒト可変領域を選択した。同一のヒト抗体に由来するヒト軽鎖および重鎖可変領域は、2つの可変領域が適切に集合するということを確実にするために用いることができる(クィーン(Queen,C.)ら、Proc.Natl.Acad.Sci.,USA 86:10029-10033(1989))。しかしながら、本明細書に記載されるように、鋳型として選択されたヒト軽鎖および重鎖可変領域は、2つの異なるヒト抗体に由来するものであった。このようにして、齧歯類可変領域に高い程度の類似性を有するヒト可変領域を選択することが可能であった。2つの異なるヒト抗体に由来する可変領域に基づくCDRを接ぎ合わせた抗体の多くの成功例がある。最もよく研究された例の1つは、再形成されたヒトCAMPATH−1抗体である(リーヒマン(Riechmann,L.)ら、Nature 322:323-327(1988))。
再形成ヒトACT−1可変領域を設計するために、マウスACT−1可変領域を、マウスおよびヒト可変領域のすべてのサブグループに関するコンセンサス配列と比較した(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991))。その結果を表1および2に要約する。
マウスAct−1軽鎖可変領域は、全体で83.9%の同一性およびFRのみ内で87.5%の同一性を有し、マウスカッパ軽鎖サブグループIIのコンセンサス配列と最も類似していた(表1)。ヒト抗体配列の観点から、マウスAct−1軽鎖可変領域は、全体で72.3%の同一性およびFRのみ内で78.8%の同一性を有し、ヒトカッパ軽鎖サブグループIIのコンセンサス配列と最も類似していた(表1)。
設計過程の第2の工程は、選択されたヒト軽鎖および重鎖可変領域に齧歯類CDRを挿入することであった。カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、米国保健およびヒトサービス省、米国政府印刷局(1991))により規定されたように、齧歯類CDR全体をヒトFRに結合して単純なCDR接合部を作製した。いくつかの場合において、単純なCDR接合でヒト化される齧歯類抗体は、抗原に対してほとんどまたは全く結合を示さないであろう。これらのアミノ酸残基のいずれが、抗原との相互作用を介して直接的にまたはCDRループの配置を変化させることにより間接的に、抗原への結合に反する影響を及ぼすと考えられるかどうかを決定するために、ヒトFRのアミノ酸配列を研究することが重要である。
第3の工程において、ヒトFRのアミノ酸残基を抗原への良好な結合を達成するために変えるべきかに関する決定がされた。この段階で、齧歯類可変領域のモデルが設計過程において最も有用になる。また、ショチアら、Nature 342:877-883(1989)により規定されたように、CDRの標準構造も有用である。該標準構造の一部である齧歯類アミノ酸残基のいずれかをヒト化可変領域中に保存することが重要である。ある位置のアミノ酸が異常であるかまれであるかを決定するためには、ヒト化対象の齧歯類抗体の配列を他の齧歯類抗体由来の類似の配列と比較することが役に立つ。これは、その位置での齧歯類アミノ酸が抗原結合に重要な役割を有することを示唆するであろう。齧歯類可変領域のモデルを研究することにより、特定の位置のアミノ酸が抗原結合に影響を及ぼしうるか否かを予想することが可能である。個々のヒト抗体由来のヒト可変領域を設計の基礎として用いられつつあるとき、そのときは、個々のヒト配列をヒト可変領域のそのサブグループのコンセンサス配列と比較することが望ましい。特に異常なアミノ酸はいずれも注意すべきである。ほとんどの場合、ヒト可変領域中のその位置に存在するアミノ酸から齧歯類可変領域中のその位置に存在するアミノ酸に変えるべきヒトFRにおけるいくつかのアミノ酸が同定される。
表3および4は、再形成ヒトAct−1可変領域がどのように設計されたかを要約する。表3は、再形成ヒトmAb Act−I VL領域の設計に用いたアミノ酸配列のアラインメントであり、4列にはヒト化対象のマウスAct−1軽鎖可変領域のアミノ酸配列(配列番号:7)、5列(マウスκ−II)にはマウスAct−1可変領域が属するマウス可変領域のサブグループに関するコンセンサス配列(配列番号:50)、6列(ヒトκ−II)にはマウスAct−1可変が最も類似するヒト可変領域のサブグループに対するコンセンサス配列(配列番号:51)、7列には鋳型として供する(即ち、GM607’CL)ヒト可変領域のアミノ酸配列(配列番号:8)および8列(Act−I RHVκ)には設計された再形成ヒトAct−1可変領域のアミノ酸配列(配列番号:52)を掲載する。表4は、再形成ヒトmAb Act−I VH領域の設計に用いたアミノ酸配列のアラインメントであり、4列にはヒト化対象のマウスAct−1重鎖可変領域のアミノ酸配列(配列番号:9)、5列(マウスIIB)にはマウスAct−1可変領域が属するマウス可変領域のサブグループに対するコンセンサス配列(配列番号:53)、6列(ヒトI)にはマウスAct−1が最も類似するヒト可変領域のサブグループに対するコンセンサス配列(配列番号:54)、7列には鋳型として供する(即ち、21/28’CL)ヒト可変領域のアミノ酸配列(配列番号:10)および8列(Act−I RHVH)には設計された再形成Act−1可変領域のアミノ酸配列(配列番号:55)を掲載する。表3および4の終わりから2番目の列は、マウスAct−1と選択されたヒトFRとの間で異なる残基のFRにおける位置(表面または埋没)を示す。表3および4の最後の列は、可変領域中のその位置に関連したコメントを掲載する。
表3において、以下の印を用いる:(*)カバット(Kabat)コンセンサス配列即ち、カバットサブグループ内の95%以上の出現(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、米国保健およびヒトサービス省、米国政府印刷局(1991))(5列および6列の場合)、またはショチアら、Nature 342:877-883(1989)により規定されたようなCDRループ(5列および6列の場合)に対する規定構造の一部としてもしくはCDRループ(8列の場合)に対する規定構造の一部としてのいずれかにより規定された非変異残基;(太字)ヒトアミノ酸残基が対応するマウス残基により置換されたFRおよびCDRでの位置;(下線)ヒト残基が類似のマウス残基番号と異なるFRでの位置;(△)ヒトFRでの変化の番号付け;(マウス Ab Act−1)マウスAct−1抗体由来のVL領域のアミノ酸配列;(マウスκ−II)サブグループII由来のマウスカッパVL領域のコンセンサス配列(カバットら、前述)(ヒトκ−II)サブグループII由来のヒトカッパVL領域のコンセンサス配列(カバットら、前述);(GM607’CL)ヒトGM607’CL抗体由来のアミノ酸配列(クロベックら、Nature 309:73-76(1984));(表面または埋没)抗体可変領域の両鎖における残基の残りに関連したアミノ酸の位置;(Act−I RHVK)再形成ヒトmAb Act−I VL領域のアミノ酸配列。
FR1の4位では、マウス配列においてバリンおよびヒト配列においてメチオニンが存在する。バリンからメチオニンへの変化は、両アミノ酸が非極性で、疎水性残基であるので、それ自体ドラスティックな変化ではなく、したがって、ヒト配列に存在するメチオニンを再形成ヒトAct−1可変領域に使用した。しかしながら、該モデルは、バリンがL1ループとL2ループとの間に埋もれており、蛋白質に埋もれた場合のバリンの平均体積は142Å3であることを示唆するが、一方、メチオニンは約171Å3の空間を占める。より大きなメチオニン残基は、L1ループとL2ループのいずれかまたは両方のコンフォメーション変化を生じさせうるであろう。再形成ヒトAct−1の抗原結合は、再形成ヒトAct−1軽鎖可変領域において4位のメチオニンからバリンへの追加の変化により改良されてもよい。
再形成ヒトAct−1重鎖可変領域の設計(表4)に関して、ヒトFRに存在するアミノ酸から元のマウスFRに存在するアミノ酸に変化したヒトFRの5つの残基が存在した。FR1の24位およびFR3の71位では(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、第5版、米国保健およびヒトサービス省、米国政府印刷局(1991)により規定されたように)、これらの位置はそれぞれH1ループとH2ループの標準構造の一部であるので(ショチアら、Nature 342:877-883(1989))、マウス配列に存在するようなアミノ酸残基が再形成ヒトAct−1重鎖可変領域に保持されていた。これらの位置でのいかなるアミノ酸変化もH1ループとH2ループのパッキングと最終的な構造を破壊しうるので、これらの厳密な位置でのマウス残基は、ヒト化重鎖可変領域で定型的に保存されている。
FR2の48位では、ヒト配列のメチオニンがマウスAct−1配列に存在するようなイソロイシンに変化した。イソロイシンをメチオニンに置換することは異常である。より重要なことには、該モデルは、イソロイシン残基がH2ループの下に隠れて埋もれていることを示す。結論として、この埋もれた位置での変化は、H2ループの構造に影響を及ぼし、故に抗原結合を妨害したかもしれない。
FR3の69位では、ヒト配列のイソロイシンがマウスAct−1配列に存在するようなロイシンに変化した。イソロイシンをロイシンに置換することは異常ではないけれども、該モデルは、ロイシンがH2ループの下に埋もれていることを示す。結論として、48位の残基のように、この位置での変化は、H2ループのコンフォメーションに影響を及ぼし、それにより抗原結合を破壊しうるだろう。
最後に、FR3の73位では、ヒト配列のスレオニンがマウス配列に存在するようなイソロイシンに変化した。FR3のこの位置でのイソロイシンは、マウスサブグループIIBまたはヒトサブグループIでこれまで見られたことがなく(カバット(Kabat,E.A.)ら、Sequences of Proteins of Immunological Interest、米国保健およびヒトサービス省、米国政府印刷局(1991)により規定されたように)、このことは、この位置のイソロイシンが抗原結合に重要な役割を有するかもしれないことを示唆する。
該モデルにおいて、73位のロイシンが結合部位の縁近辺の表面上に存在するようであり、α4β7インテグリンのエピトープの大きさと方向に応じて抗原結合に直接の役割を果たす可能性があるだろう。しかしながら、表面残基の位置として、抗体全体は、もしマウスアミノ酸が再形成ヒト抗体に存在しないならば、より低い免疫原性ポテンシャルを有するだろう。イソロイシンは、再形成抗体誘導体においてヒトスレオニン残基に置換でき、新規な構築物を再試験して、2番目のバージョンが類似のレベルの抗原結合を維持するかどうかを決定した。
再形成ヒトAct−1重鎖可変領域の元の設計でなされたヒトFRでの5つの変化に加え、抗原結合を改良するかもしれない2つの別の変化が存在した。該モデルは、マウスmAb Act−1の重鎖可変領域の38Lysおよび40Arg残基がH2ループの下に隠れて位置し、CDR2の63Pheに近接してパッキングすることを示唆する(表4と同じ番号付け)。しかしながら、これらの残基も重鎖可変領域の核に位置し、それらがそれらの対応するヒトアミノ酸(各38Argおよび40Ala)を置換するために用いられるならば、他の有害な効果を有するかもしれないであろう。したがって、FR2の38位と40位に対する変化は、mAb Act−1の再形成ヒト重鎖可変領域に取り込まれなかった。しかしながら、再形成重鎖の修飾のいずれかまたは両方を誘導体に用いて抗原結合を改良してもよい。
結論
マウスAct−1可変領域のモデルは、主に他の抗体可変領域の溶解した構造に基づいて作られた。該モデルをヒト化Act−1可変領域の設計に用いた。再形成ヒト可変領域の抗原結合部位の構造を保持することに特に重点を置いた。
再形成ヒトAct−1軽鎖可変領域および再形成ヒトAct−1重鎖可変領域を設計した(表3および4)。再形成ヒトAct−1軽鎖可変領域は、マウスAct−1軽鎖可変領域のCDRおよびヒトGM607’CL抗体の軽鎖可変領域由来のFRを基礎とした。1つのアミノ酸変化がヒトFRの2位でなされた。再形成ヒトAct−1重鎖可変領域は、マウスAct−1重鎖可変領域のCDRおよびヒト21/28’CL抗体の重鎖可変領域由来のFRを基礎とした。5つのアミノ酸変化がヒトFRの24、48、69、71および73位でなされた。
さらに、再形成ヒトAct−1可変領域のさらなるバージョンの設計に考慮されるであろうカッパ軽鎖のFR1の4位の1つの部位ならびに重鎖のFR2の38および40位の2つの部位が注目された。また、抗体の表面上の位置という観点から、重鎖のFR3の73位の1つの残基もマウスからヒトアミノ酸への復帰突然変異の候補として同定された。
実施例3 再形成可変領域をコードする核酸の構築
正しい重鎖および軽鎖可変領域が生物化学的に(部分アミノ酸配列)および機能的に(HuT78細胞のキメラ抗体染色)クローニングされたことを確認した後、再形成アミノ酸配列を前記のように設計した。次に、再形成抗体鎖をコードする遺伝子を設計して調製した。
ヒト化ACT−1の設計、構築および発現
実施例2に記載のようにヒト化抗体の一次アミノ酸配列を決定した後、該配列を縮重核酸配列に逆翻訳し、MacVector(Kodak,Scientific Imaging Systems)4.5.3版を用いて可能性のある制限酵素部位を分析した。次いで、制限酵素切断部位を取り込むが一次アミノ酸配列を保存する核酸配列を選択した。サブクローニングに使用される注目の制限酵素部位を有する重鎖核酸配列(配列番号:18)およびアミノ酸配列(配列番号:19)を図11に示し、ならびに軽鎖核酸配列(配列番号:20)およびアミノ酸配列(配列番号:21)を図12に示す。
以下のようにして、ヒト化Act−1重鎖および軽鎖可変領域遺伝子を構築した。軽鎖に対してL1〜L6(それぞれ配列番号:22〜27)、重鎖に対してH1〜H10(それぞれ配列番号:28〜37)と称する重複した相補的なオリゴヌクレオチドを、Applied Biosystems DNA Synthesizer Model 392を用いて合成した(図13)。55℃で一晩脱保護した後、オリゴをSpeed−Vacで乾燥させ、100mlの水に再懸濁させ、Bio−Spin6カラム(Bio-Rad)で脱塩した。該オリゴの濃度は、260nmで吸光度を測定することにより決定し、オリゴを変性ポリアクリルアミドゲル電気泳動により精製した。
各オリゴの100μgを2倍容量の負荷色素(95%ホルムアミド、20mM EDTA、0.05%ブロモフェノールブルー、0.05%キシレンシアノールFF)と混合し、65℃で2分間加熱し、1X TBE中で約3時間、250Vで泳動した。該ゲルを臭化エチジウムで染色し、紫外線下で観察した。次いで、正しい長さのオリゴをゲルから切り出し、水とともに透析チューブに入れ、電気溶出させた。該オリゴを、等容量のフェノール/クロロホルム/イソアミルアルコール(25:24:1 v/v)(Gibco/BRL)で2回抽出し、0.1倍容量の3.0M 酢酸カリウム(pH6)と2倍容量の冷エタノールの添加により沈澱させた。遠心分離後、ペレットを70%エタノールで1回洗浄し、真空乾燥させ、50μlの水に再懸濁させた。
相補性オリゴを、等モル量(50μlの水に約100μg)の精製オリゴを等容量(100μl)の2X アニーリング緩衝液(2X=1M NaCl、pH7.5の40mM Tris−HCl、2mM EDTA)と混合することによりアニーリングした。オリゴは、95℃まで10分間加熱することにより変性させた後、65℃で8時間インキュベーションした。次いで、アニーリングしたオリゴを、以前に記載のようにエタノール沈澱させ、40μlの水に再懸濁した。
アニーリングしたオリゴの伸長は、2μlのラージフラグメントDNAポリメラーゼI(Klenow)、5μlの2.5mM dNTPおよび5μlの10X 緩衝液(10X=10mM Tris−HCl、10mM MgCl2、1mM DTT、25℃でpH7.9)を添加し、最終容量を52μlまで上げることにより達成した。該混合物を室温で1時間インキュベートした。37℃で0.5時間のインキュベーションで1μlのdNTPと1μlのKlenowをさらに添加した。重鎖断片Aを伸長させる必要がなかったことに注目すること。
アニーリングして伸長した断片を、12%非変性ポリアクリルアミドゲルによる電気泳動により一本鎖のアニーリングしていないものから精製した。該ゲルを臭化エチジウムで染色し、紫外線下で観察した。正しい長さの断片を切り出し、前記したように透析チューブ中の電気溶出により回収した。該断片を、等容量のフェノール/クロロホルム/イソアミルアルコールで2回洗浄し、エタノール沈澱し、10μlの水に再懸濁させた。
3つの軽鎖断片(LA、LBおよびLC)ならびに5つの重鎖断片(HA〜HEと称する)を、別々にpCR−ScriptTMにライゲートし、以下に記載したことを除いて、製造業者の推奨したプロトコールに従ってpCR−Scriptキット(Stratagene)を用いてXL−1 Blue Supercompetent Cellsに形質転換した。断片pCR−LAおよびpCR−LBは、制限酵素MscIでの消化を阻止するであろうDcmメチラーゼを避けるために、DM1(Gibco/BRL)コンピテント細胞に形質転換した。白色コロニーを拾い、製造業者の推奨したプロトコールに従ってSequenase T7DNAポリメラーゼキットを用いてミニプレップDNAをシーケンスした。インサートのそれぞれ反対側にアニーリングするT3およびT7プライマーをシーケンスに用いた。
ヒト化重鎖可変領域および軽鎖可変領域断片の複雑サブクローニングは、合成の間に配列に取り込まれた特異的な制限部位を用いて達成された。重鎖断片HA〜HDは、以下に記載するように断片の連続サブクローニングを可能にする、各配列の末端に追加のAgeI制限部位を含む。
pCR−HAおよびpCR−HB由来のミニプレップDNAを制限酵素SpeIとAgeIで消化した。DNAは、1%アガロースゲルで電気泳動した。141bp断片HBを該ゲルから回収し、SpeIおよびAgeI部位でpCR−HAにライゲートし、pCR−HABが生じた。次に、112bp断片HCを、XbaIとAgeIを用いてpCR−HCから放出させ、pCR−HABのXbaIおよびAgeI部位にライゲートし、プラスミドpCR−ACを得た。断片HD(141bp)およびHC(130bp)は、断片HDに対しては制限部位NheIおよびAgeIを、ならびに断片Eに対しては制限部位BstEIIおよびAgeIを用いて、同じ一連の様式でライゲートした。pCR−scriptにすべての5つの重鎖可変領域断片を含む最終プラスミドを、pCR−HAEと称した。すべての消化は、65℃の最適インキュベーション温度を有するBstEIIを用いる場合を除いて、37℃で少なくとも2時間のインキュベーションでミニプレップDNAを用いて行なった。ライゲーションは、1:10のベクター対インサート比でT4DNAリガーゼを用いて16℃で一晩行ない、次の日に、製造業者の推奨したプロトコールに従ってDH5αサブクローニング効率コンピテント細胞(Gibco/BRL)に形質転換した。
pCR−ScriptTM中のAct−1ヒト化重鎖可変領域を、pCR−HAEのHindIIIとAgeIでの消化により放出させた。この411bp断片を用いて、HindIIIとAgeIで消化しておいたpEE6mhACT1Hchi(実施例1)のマウス可変領域配列を置換し、pEE6hCMV−Bにヒト化ACT−1重鎖遺伝子を生成させた。得られたプラスミドをpEE6hACT1Hと称する。正しいDNA配列は、シーケンスにより決定した。
pCR−ScriptTM中の軽鎖断片Aを、BspEIとMscIで消化した。次いで、この153bp断片を用いて、pCR−scriptTM中のマウス可変軽鎖のBspEIからMscIのマウス部分を置換した。このプラスミドをpCR−LhAmBCと称する。MscIとNruIで消化した軽鎖断片BおよびNruIとKasIで消化した軽鎖断片Cを、pCR−LhAmBCのMscIおよびKasI部位にトリプルライゲートし、残りのマウス配列を置換した。消化、ライゲーションおよび形質転換には、DM1コンピテント細胞を最後の形質転換を除くすべてに使用することを除いて、前述したのと同じ手法を用いた。
pCR−ScriptTM中のヒト化軽鎖可変領域およびプラスミドpEE12mhACT1Lchi(実施例1)をHindIIIとKasIで消化した。360bp軽鎖可変領域断片および315bp軽鎖定常領域をゲルで精製し、pEE12のHindIII制限部位にトリプルライゲートし、pEE12hACT1Lを得た。シーケンスを行ない、正しい方向および核酸配列を確認した。
ヒト化重鎖および軽鎖遺伝子の両方を含む発現ベクターは、以下の例外を除いて、キメラ抗体に関して記載されたのと同じ方法(実施例1、キメラ免疫グロブリンの発現を参照のこと)を用いて構築した。ヒト化可変重鎖領域の追加のBglII制限部位により、BglIIでの切断の際には部分消化を用いて正しい断片を得た。ヒト化重鎖および軽鎖遺伝子の両方を含むベクターを、pEE12hACT1LHと称する。
すべてのヒト化抗体構築物の一過性の発現および細胞染色は、キメラ抗体に用いられたものと同じプロトコールを用いて行なった(実施例1、キメラ免疫グロブリンの発現を参照のこと)。図14は、無関係のアイソタイプ適合対照抗体(IgG1、カッパ)と比較して、マウス−ヒトキメラAct−1抗体またはヒト化Act−1抗体を用いるHuT78染色の結果を示す。
骨髄腫細胞株であるNSO細胞(Methods in Enzymol.73(B):3-46(1981);ヨーロピアンコレクション・オブ・アニマルセルカルチャーズ、PHLS CAMR,Porton Down,Salisbury,Wiltshire SP4 OJG,U.K.,ECACC No.85110503)の安定なトランスフェクタントは、NSO細胞のpEE12hACT1LHとのエレクトロポレーションにより得た。
NSO細胞の安定な発現
安定なトランスフェクション用の40μgのpEE12hACT1LHを、構築物の細菌のプラスミド部分内を切断するSalI制限酵素での消化により線状にした。線状DNAを、2倍容量のエタノールと1/10倍容量の酢酸ナトリウムを用いて溶液から沈澱させ、70%エタノールで洗浄し、乾燥させて滅菌水に再懸濁した。
対数増殖中のNSO細胞は、非選択培地(ダルベッコの改変イーグル培地(高グルコース)、2mM L−グルタミン含有、ピルビン酸ナトリウム不含、4500mg/L グルコースおよび25mM HEPES緩衝液(GIBCO/BRL、カタログ番号12430-021)含有、10%ウシ胎仔血清(Gibco/BRL、カタログ番号16000-044)を補足)で維持した。NSO細胞を遠心分離し、洗浄して、DNAの添加後に該細胞が107細胞/mlの濃度で存在するように、冷PBSに再懸濁した。線状プラスミドDNA(40μg)を、氷上のエレクトロポレーションキュベット中の107個の細胞に加えた。該細胞とDNAを泡を生じさせないようにゆっくり混合させ、該混合物を氷上で10分間放置した。該キュベットの外側を拭いて乾燥させ、Gene Pulser(Bio-Rad)を用いて1500V、3μFの2回の連続パルスを加えた。該キュベットを10分間氷上に戻した。
トランスフェクトした細胞を、50μlの非選択培地中3x105、7.5x104および1.5x104細胞/mlの密度で96ウエルプレートに移し、37℃で24時間インキュベートした。その後、150μlの選択培地(グルタミン不含ダルベッコの改変イーグル培地、4500mg/Lのグルコース含有、4mg/LのピリドキシンHCl含有、110mg/Lのピルビン酸ナトリウム含有、硝酸鉄不含、L−グルタミン不含(JRH BioSciences、カタログ番号51435-78P)、1X GSサプルメント(JRH Biosciences、カタログ番号58672-77Pから入手した50X GSサプルメント)と10%透析ウシ胎仔血清(Gibco/BRL、カタログ番号26300-061)を補足)をすべてのウエルに添加した。該プレートを、実質的な細胞死が起こり、不連続な生存コロニーが出現するまでインキュベーターに戻した。一旦グルタミン非依存性トランスフェクタントのコロニーが見られれば、シングルコロニーを有するウエルを選択し、消費した組織培養上清を回収し、以下に記載のようにELISAによりヒトIgG分泌をアッセイした。このようにして、その後の研究に用いた3A9と称する抗体産生クローンを得た。第2のトランスフェクションは、選択をL−メチオニンスルホキシミン(MSX、グルタミンシンテターゼインヒビター)の存在下で行なうことを除いて、前記したように行なった。
陽性のコロニーは、以下のようにしてヒトIgG分泌に対するELISAによりスクリーニングした。ELISAプレート(NUNC Maxisorp)を、炭酸塩緩衝液pH9.5中2.5μg/mlで100μlのAffiniPure F(ab’)2断片ロバ抗ヒトIgG(H+L)(Jackson ImmunoResearch Laboratories)を用いて、4℃で一晩被覆した。プレートをPBS Tween20で4回洗浄し、200μl PBS、1% BSAで、37℃で2時間ブロックした。プレートを洗浄し、100μlの安定なトランスフェクトNSOの上清で37℃で15分間インキュベートした。PBS 1% BSA中1mg/mlのヒトIgG1カッパを標準として用いた。新鮮なNSO培地(DME+GSサプルメント)を陰性対照として用いた。プレートを洗浄し、PBS(Ca2+/Mg2+なし)中0.05μg/mlで100μlのペルオキシダーゼ結合AffiniPure F(ab’)2断片ロバ抗ヒトIgG(H+L)(Jackson ImmunoResearch Laboratories)を用いて、37℃で15分間インキュベートした。5mg O−フェニレンジアミンジヒドロクロリド(OPD)の1錠(Sigma)を12mlのクエン酸緩衝液(0.1M、pH5.0)に溶解し、該錠剤が溶解した後、12μlの30%過酸化水素を加えた。洗浄して二次抗体を除去した後、100μlの溶解したOPD基質を加えた。反応は12.5%の硫酸で停止させ、Dynatech Plate Readerで490nmでプレートを読んだ。陽性のウエルを、ウエル当たり2、1および0.5個の細胞の限界希釈によりクローニングした。シングルクローニング由来のすべてのウエルをELISAによる抗体産生に関して陽性と評価した場合、該株はクローニングされたと見なした。
一過性または安定細胞トランスフェクタント培養物の細胞培養上清由来のヒト化ACT−1抗体の精製は、Bio−Cadワークステーション(Perseptive Biosystems,Inc.)を用いるプロテインAアフィニティークロマトグラフィー(Poros A/M 4.6/100mm,5mL/min)により行なった。該カラムは、PBSで平衡化させた後、前もって0.2ミクロンフィルターで濾過しておいた細胞培養上清を適用した。1回の操作当たり適用した細胞培養上清の体積は、抗体の濃度により変えた。通常、1回の所定の操作で、15mg以下の抗体をカラムに適用した。流速は、精製手法を通して5ml/minであった。結合後、まず、該カラムをOD280nm=0になるまでPBSで大量に洗浄した。次いで、該カラムを最小限50カラム体積でさらに洗浄した。次いで、該カラムを引き続き0.1M 酢酸ナトリウム、pH5.0で洗浄した。溶出は、0.1M クエン酸Na、pH3.5での洗浄により達成された。溶出液は、5mlの画分で回収し、200μlの1.5M Na2CO3 pH12の添加によりpHを中性にした。次いで、抗体含有画分をプールし、限外濾過(セントリコン、30,000KDaカットオフ、Amicon)により所望の濃度に濃縮した。
Fc変異改変体の構築
また、ヒト化Act−1抗体の非Fc結合(Fc変異)版も構築した。この抗体は、ヒト化Act−1抗体と同じ可変領域(図11および図12)ならびに、FcR認識を排除し、Fc結合を除去するように設計されたIgG1重鎖定常領域に2個のアミノ酸の置換(即ち、Leu235→Ala235置換およびGly237→Ala237置換)を除いて同一のヒトIgG1定常領域を有する。Fc変異誘導体の重鎖をコードする核酸を以下のように構築した。pEE12発現ベクター中でヒト化抗CD18抗体(国際公開第93/02191号パンフレット(1993年2月4日公開);シムズら、J.Immunol.151(4):2296-2308(1993))の軽鎖および重鎖をコードするが、部位特異的変異導入(Leu235→Ala235およびGly237→Ala237)により2個のアミノ酸置換をIgG1重鎖定常領域内に導入した3678と称する構築物(ヘルマン・ウォルトマン博士、オックスフォード大学から供与)を、AgeIとEcoRIで消化してガンマ定常領域変異を含む900bpの断片を放出させた。次いで、この断片を用いて、PEE6hACT1HのAgeI/EcoRI部位で重鎖野生型ガンマ1定常領域を置換し、pEE6hACT1H/FCmutを生じさせた。両鎖を含有する他の構築物に対しては、前記したのと同様な方法で、再形成軽鎖遺伝子およびFc変異再形成重鎖遺伝子を含む1つの構築物(pEE12hACT1LH/FCmut)を調製した。
実施例4 ヒト化ACT−1抗体LDP−02のキャラクタライゼーション
最初のキャラクタライゼーション研究は、pEE12hACT1LH/FCmutで一過性にトランスフェクトしたCOS−7細胞から産生された抗体を用いて行なった。この抗体標品は、前記したように産生および精製され、以下、適当なロット番号が後に続く「1°HUM ACT−1」と称する。
前記ネズミ細胞株NSOの安定なトランスフェクタント(線状pEE12hACT1LH/FCmutでトランスフェクト)から産生された抗体を用いて、追加のアッセイを行なった。この抗体標品を以下、「LDP−02/3A9/Lot1」と称する。
「LDP−02/3A9/Lot#1」抗体を以下に記載の次の研究に用いた:SDS−PAGE、ウエスタンブロット分析、等電点電気泳動、アミノ酸組成分析、種交差反応性、滴定、補体介在溶解アッセイ、ADCCアッセイおよび結合阻害アッセイ。「1°HUM ACT−1 Lot#7をアフィニティーアッセイ#1〜2に使用し、1°HUM ACT−1 Lot#8/9をアフィニティーアッセイ#3〜5に使用し、そして1°HUM ACT−1 Lot#8/9をC1q結合アッセイに使用した。
A.物理化学的特性
1.SDS−PAGE
同一性の立証を補助し、最初の標品をキャラクタライズして純度を評価するために、LDP−02/3A9/Lot#1を、非還元および還元条件下でドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS−PAGE)に供し、コロイドクーマシーブルーで染色した。
0.82mg/mlの公称濃度のLDP−02/3A9/Lot#1を80μl、微量濃縮器に加えた。抗体を溶解したクエン酸緩衝液を、160μlのTris緩衝液(0.5mM、pH8.8)で3回交換した。緩衝液交換後の試料の最終容量は、135μlであり、0.486mg/mlの蛋白質濃度を得た。この溶液を非還元および還元緩衝液で2倍に希釈し、0.243mg/mlの濃度を得た。3.16μgの蛋白質を含む0.243mg/ml溶液の13μlアリコートを、SDSゲルの指定された試料のレーンに負荷した。SDS−PAGEを行ない、対照アーティクルは、マーク(Mark)12分子量標準(Novex,#LC5677)を含んでいた。
非還元条件下では、LDP−02/3A9/Lot#1に、200,000ダルトンよりわずかに小さい見かけ上の分子量を有する主要なバンドが存在した。116,300〜200,000ダルトンの間に、いくつかのマイナーな成分が観察された。約97,400ダルトンの分子量、55,400ダルトンよりわずかに大きな分子量および31,000ダルトンより小さい分子量を有する3つの追加のマイナーな成分も観察された。レーザーデンシトメトリーを用いてゲルをスキャンすることにより、染色されたポリペプチドバンドの定量分析、次いで、各可視化バンドに関連したエリアのパーセントの計算を可能にした(表5)。定量分析から得られたデータは、約200,000ダルトンで観察された主要な成分が試験した試料のレーンで全染色バンドの84.4%に相当することを示唆する。この主要なバンドは、インタクトな抗体に相当したが、55,000および、31,000ダルトンの他のバンドはそれぞれ、1本の重鎖および軽鎖に相当した。
還元条件下では、2つの主要な成分が電気泳動ゲル上で観察された。該成分のうちの1つの分子量は、約55,400ダルトンであり、該ゲルレーンで可視化された全染色バンドの68.6%に相当し、一方、31,000ダルトンよりわずかに小さいものに相当する第2の成分は、全染色バンドの30.5%に相当した(表5)。これらの2つの成分の分子量は、免疫グロブリンGの重鎖および軽鎖の予想される分子量とよく一致する。これらのデータは、約99%の標品がインタクトな抗体または1本の重鎖もしくは軽鎖免疫グロブリン鎖のいずれかからなることを示唆する。また、2つの主要な成分以外にも、66,300ダルトンよりわずかに小さい1つのマイナーな成分が観察された。
この分析から、インタクトな免疫グロブリンGに対するものと一致する高分子量種がLDP−02/3A9/Lot#1に主要なバンドとして存在する。また、いくつかのマイナーなバンドもLDP−02/3A9/Lot#1に存在する。還元後、免疫グロブリンG分子の重鎖および軽鎖のものと一致する電気泳動上の移動度を示す2つの主要なバンドが観察された。
前記したように、SDS−PAGEにより試料および標準を分析した。要約すると、還元しないおよび還元した試料を4〜20% Tris−グリシンゲルで分析した。また、ノベックス(Novex)マーク(Mark)12分子量標準もゲルで泳動した。それぞれ0.51および1.09μgの蛋白質を生ずる、0.2143mg/ml溶液の2.1μlおよび4.5μlアリコートの容量をSDSゲルの指定された試料レーンに負荷した。
SDS−PAGE後、ノベックス(Novex)ウエスタン・トランスファー・アパレイタス(Western Transfer Apparatus)の使用説明書に従って、試料蛋白質をゲルからニトロセルロースに移した。用いたトランスファー緩衝液は、20%メタノール中1X Tris−グリシン緩衝液であった。約2時間後、該ニトロセルロースブロットをトランスファー装置から除去し、DDI水でリンスした。次いで、該ニトロセルロースブロットを、3%ゼラチンおよび0.1%Tween20を含むTris緩衝液(20mM)中、37℃で35分間ブロックした。該ブロットをブロッキング溶液から除去し、Tris緩衝液で2回洗浄した。抗マウスIgG抗体ストック溶液を20mM Tris−3% BSA溶液で1000倍に希釈することにより調製したヤギ抗マウスIgG溶液を、該ブロットに添加し、2〜8℃で一晩インキュベートした。インキュベーション後、該ブロットを5分毎にTris緩衝液を4回交換することで洗浄した。抗ヤギIgGアルカリホスファターゼコンジュゲートを20mM Tris−3% BSA溶液で5000倍に希釈することにより調製した抗ヤギIgGアルカリホスファターゼコンジュゲート溶液を、該ブロットに添加し、室温で2時間インキュベートした。インキュベーション後、該ブロットを5分毎にTris緩衝液を4回交換することで洗浄した。BCIP/NBT(5−ブロモ−4−クロロ−3’−インドリルホスフェートp−トルイジン塩/ニトロ−ブルーテトラゾリウムクロリド)基質を、10ml同時に該ブロットに添加した。ブロットを揺動させながら室温で現像した。Tris緩衝液でブロットをリンスすることにより反応を停止させた。次いで、ヤギ抗マウスIgGの代わりにヤギ抗ヒトIgGを用いて前記手法を繰り返した。
抗マウスIgG試薬を用いる非還元および還元の両条件下で、0.51μgおよび1.09μgのIgG試料が、該ニトロセルロースブロット上で明確に検出された。バンドの強度は、濃度が増加するにつれて増加した。非還元条件下で、200,000ダルトンマーカーよりもわずかに速く移動する主要なバンドが検出された。また、いくつかのより弱いバンドも検出された。これらのバンドのうちの2つは、主要バンドよりも遅く移動し、他の3つのバンドは概ねより速く移動した。還元条件下では、免疫グロブリンGの重鎖および軽鎖の特徴を有する2つのバンドが検出された。
非還元および還元の両条件下で抗ヒトIgG試薬を用いて、0.51μgおよび1.09μgのIgG試料を該ニトロセルロースブロット上で明確に検出した。バンドの強度は、濃度が増加するにつれて増加した。非還元条件下で、200,000ダルトンよりもわずかに小さい見かけ上の分子量マーカーを有する種に相当する主要バンドが検出された。また、抗マウスIgGで検出され、該ブロットで観察されたより弱いバンドも検出された。免疫染色の強度は、抗ヒトIgGで検出した際のすべてのバンドに対してより強かった。他のブロットでは観察されないいくつかの追加のバンドが検出された。これらのバンドは、抗マウスIgGで認識されるエピトープを欠くIgG断片に相当すると思われる。還元条件下では、免疫グロブリンGの重鎖の特徴を有するバンドが検出された。該抗体はヒトIgGのFc部分に特異的であったので、軽鎖は検出されなかった。抗ヒトIgGで検出を行なった際に、抗マウスIgGで現像したブロットでは見られないいくつかのマイナーバンドが観察された。2つのブロット間のこの相違は、抗マウスIgG結合に対するエピトープを欠くIgG断片の存在の結果であろう。
3.等電点電気泳動
LDP−02/3A9/Lot#1を等電点電気泳動(IEF)に供し、コロイドクーマシーブルーで染色した。LDP−02/3A9/Lot#1に対して得られた結果を、同一のゲルで泳動したIEF標準と比較した。
0.82mg/mlの公称濃度のLDP−02/3A9/Lot#1を80μl、微量濃縮器に加えた。抗体が入っているクエン酸緩衝液を、160μlのTris緩衝液(0.5mM、pH8.8)で3回交換した。試料の最終容量は、135μlであった。最終濃度は、0.486mg/mlであると計算された。この溶液を、2X IEF試料緩衝液で2倍に希釈して、0.243mg/mlの濃度を得た。3.16μgの蛋白質を生ずる0.243mg/ml溶液に対して13μlのアリコートを、IEFゲルの指定された試料の上に負荷した。対照のアーティクルは、IEF標準pI3.6〜9.3を含んでいた(Sigma、Cat #I-3018)。
標準プロットは、8つのIEF標準の相対的な移動距離の平均をこれらの標準蛋白質のそれぞれに関して公知のpIに対してグラフ化することにより作成した。これらのデータの回帰直線に適合させることにより、0.03459の負の傾斜と8.91857の切片を得た。該適合のR2は、0.99206に等しかった。
表6は、6つのIEF標準およびLDP−02/3A9/Lot#1により移動した平均の距離を含む。また、LDP−02/3A9/Lot#1に関して計算されたpIもこの表に示す。
標準プロット由来の直線回帰パラメーターを用いて、LDP−02/3A9/Lot#1に関する5つのバンドの概略pI値は、8.09のpI値で表される優勢なピークとともに、7.88、7.95、8.09、8.26および8.43であると計算された(表6)。この主要ピークのpI値は、一次アミノ酸配列に基づく予想pI値7.91に都合よく匹敵する。
アミノ酸組成分析を、LDP−02/3A9/Lot#1の蛋白質含有量およびアミノ酸組成を決定して同一性を確認するために行なった。
まず、三連の45μlアリコートを加水分解用に取り除いた。加水分解は、6N HCl蒸気を用いて165℃で60分間行なった。対照として、加水分解容器は、LDP−02/3A9/Lot#1と同時に加水分解される標準蛋白質を含んでいた。また、アミノ酸標準は、LDP−02/3A9/Lot#1分析の前後にクロマトグラフィーに付した。対照のアーティクルは、標準蛋白質としてウシ血清アルブミン(Tektagen Solution Control:310:197A)、およびアミノ酸標準としてアミノ酸加水分解混合物(Tektagen Solution Control:310:199A)を含んでいた。
試験方法には、カラム後ニンヒドリン反応および2波長での吸光度のモニターを伴うイオン交換HPLCによる再懸濁した蛋白質加水分解物または遊離のアミノ酸溶液の分析を用いた。両波長での吸光度は、三連でアミノ酸標準を分析することにより得られた校正表と比較して定量化した。
表7にアミノ酸組成を示す。LDP−02/3A9/Lot#1の蛋白質濃度は、0.709mg/mLであると決定された。WおよびCの定量の欠如に関する補正に基づいて、蛋白質濃度を、0.740mg/mLに修正した。データおよび直接関係のある計算値を表8に要約する。
LDP−02/3A9/Lot#1に関しては、165℃で6N HCl蒸気を用いる単一の加水分解時点(60分)を行なった。標準蛋白質(BSA)に由来する補正係数を、蛋白質含有量の決定に適用した(表8)。
この方法の条件下では、ともに溶出するシステインのピークの存在により、プロリンに関して得られたモルパーセント値(表7)がわずかに高くなるかもしれない。その結果として、プロリンの定量の精度は試料に依存し、試料加水分解物中に存在するシステインの量に基づく。この分析に関して、プロリンの含有量を、BSA由来の補正係数を用いて補正した(表8)。この補正の精度は試料に依存し、BSA(6.0%)および試料中のシステインの相対量に基づく。
重鎖および軽鎖のヌクレオチド配列に基づき、相対パーセント(頻度またはモルパーセント)としてのLDP−02の予想アミノ酸組成(予想%)ならびにアミノ酸分析の実際の結果(実測%)を表9に示す。予想値対実測値の比較は、前記したようにプロリンがともに溶出するシステインのピークにより人為的に高いと思われることを除いて、良好な相関を示す。
LDP−02/3A9/Lot#1をMALDI−TOF MSにより分析して、分子量を決定した。149,808Daを中心とする質量を有する主要ピークが検出された。74,927Daを中心とするピークは、主要ピークに見出される種の+2イオンを表す。+2イオンの質量は、M+Hイオンのちょうど半分ではないことに注目すべきであり;このわずかな格差は実験上の不正確さにより生ずると思われ、これは測定値の+/- 0.2%以内である。
抗体の予測された一次配列に基づくと、予想された分子質量は、147,154Daであるべきである。観測されたおよび予測されたIgG分子質量の間の2,654Daという質量の差は、該分子のグリコシル化によることが最も蓋然性が高いといえる。この観測された差は、約1.8%のグリコシル化レベルを表す。
B.アフィニティー
まず、LDP−02/3A9/Lot#1およびネズミACT−1(Lot#2)の滴定を、ヒト由来のHUT−78細胞でフローサイトメトリーを用いて行なった。要約すると、1.0x106個のHUT−78細胞を、ビオチニル化ネズミACT−1(Lot#2)、ビオチニル化ネズミIgG1(LeukoSite,Inc.で作られたLot#1)、ビオチニル化LDP−02/3A9/Lot#1またはビオチニル化ヒトIgG(Jackson ImmunoResearch,Avondale,PA;Lot 25794)のいずれか100μlの容量に4℃で20分間懸濁した後、抗体を除去した。特に示さない限り、すべての試薬は、0.15M PBS/1.0% FCS/0.1%アジ化ナトリウムで希釈した。両抗体に対する濃度変化には、30μg/ml(ネズミACT−1のみ)、15μg/ml、7.5μg/mlおよびそれぞれの続く1:10希釈が含まれた。一次抗体の除去後、次に、該細胞を、1:200に希釈した100μlのストレプトアビジンフィコエリスリン(Dako Corp.,Carpinteria,CA)に懸濁した。200μlのPBSで洗浄した後、細胞を0.5mlのPBS/1%ホルマリンに再懸濁し、分析するまで冷凍した。試料は、フィコエリスリンを励起させるために488nmのレーザーを用いて、FACScan(Becton Dickinson Corp.,San Jose,CA)で分析した。各試料に対して、最小限10,000個の細胞を分析し、最大平均チャンネルフルオレッセンス(MCF)の半値を計算した。すべての試料は、二連で行なった。
これらの滴定研究により、約1.0μg/mlの濃度で、ネズミACT−1およびLDP−02/3A9/Lot#1の両方を用いて最大の蛍光に近づくことが示唆された(図15)。最大平均チャンネルフルオレッセンスの半値は、ネズミACT−1よりもLDP−02の方が低い濃度で達成された(それぞれ、ビオチニル化ネズミACT−1Lot#2に対して0.1μg/mlおよびLDP−02/3A9/Lot#に対して0.02μg/ml)。
アフィニティー(および特異性)の相対的評価は、フローサイトメトリーならびにLDP−02およびネズミAct−1抗体の交差競合結合を用いて、逆にヒト由来のHuT−78細胞で行なった。要約すると、1.0x106個のHuT−78細胞を、種々の濃度の非コンジュゲート化1°HUM ACT−1または非コンジュゲート化ネズミAct−1のいずれかと0.1μg/mlのビオチニル化ネズミAct−1(Lot#2)の100μlに、4℃で20分間懸濁した後、抗体を除去した。別々の実験において、100μlの0.02μg/mlのビオチニル化LDP−02/3A9/Lot#1を、種々の濃度の非コンジュゲート化ネズミACT−1(Lot#2)および非コンジュゲート化LDP−02/3A9/Lot#1とともに用いた。一定に保たれたビオチニル化抗体の濃度は、前記したように、同一の条件下で染色したHUT−78細胞での最大平均チャンネルフルオレッセンス(MCF)の半値で得られた濃度であった。特に示さない限り、すべての試薬は、0.15M PBS/1.0% FCS/0.1%アジ化ナトリウムで希釈した。両抗体に対する濃度変化は、2.0X10-6M〜5.0X10-11Mの対数増加の半値にわたっていた。一次抗体の除去後、次に、該細胞を、1:200に希釈した100μlのストレプトアビジンフィコエリスリン(Dako Corp.,Carpinteria,CA)に懸濁した。200μlのPBSで洗浄した後、細胞を0.5mlのPBS/1%ホルマリンに再懸濁し、分析するまで冷凍した。試料は、フィコエリスリンを励起させるために488nmのレーザーを用いて、FACScan(Becton Dickinson Corp.,San Jose,CA)で分析した。各試料に対して、最小限10,000個の細胞を分析し、MCFを計算した。すべての試料は、二連で行なった。IC50は、ビオチニル化ホモログ抗体からMCFで50%の低下を生ずる非コンジュゲート化抗体の濃度として決定された。
アフィニティーの見積もりは、LDP−02(1°HUM ACT−1)とネズミACT−1との間の5つの独立した交差競合実験で行なった。ビオチニル化ネズミAct−1を該アッセイで一定に保つ抗体として用いた場合、LDP−02に対する平均IC50値(±1 SEM)(5.43±0.86nM)は、ネズミACT−1に対するもの(7.94±1.17nM;p=.02、両側t−検定:平均に対して対の2個の試料)よりも統計学的に低く、一方、無関係なヒトIgG1またはネズミIgG1は、競合的効果を有していなかった(すべての実験を表10に要約する;1つの実験を図16に示す)。同様に、ビオチニル化LDP−02/3A9/Lot#1が該アッセイで一定に保たれた抗体である場合、HuT−78細胞膜からLDP−02を競合除去するのにLDP−02/3A9/Lot#1よりも高い濃度の非コンジュゲート化ネズミAct−1を要した(それぞれ、IC50=6.3nM対4.3nM)。各実験において、LDP−02は、ネズミAct−1よりも低いIC50を有した。これらの結果は、LDP−02がネズミAct−1により認識されるエピトープこ対して特異的であり、その結合アフィニティーがネズミ抗体のものよりも良好であったことを示唆する。
フローサイトメトリーを用いて、種の交差反応性を評価した。ヒト、イヌ、ネコ、モルモットまたはラットから採取した100μlのEDTA抗凝固血を、FACSチューブに加えた。血漿を除去し、次いで、血液ペレットを、15μg/mlの濃度でビオチニル化LDP−02/3A9/LOT#1、無関係なビオチニル化ヒトIgG(Jackson ImmunoResearch,Avondale,PA)、ビオチニル化ネズミAct−1 Lot#2または無関係なビオチニル化ネズミIgG1(Dako Corp.,Carpinteria,CA)のいずれか100μl中に再懸濁した。特に示さない限り、すべての試薬は、0.15M PBS/1.0% FCS/0.1%アジ化ナトリウムで希釈した。試料を4℃で20分間抗体とともにインキュベートした後、該抗体を洗浄により除去した。次いで、細胞を4℃で20分間、1:200に希釈したストレプトアビジンフィコエリスリン(Southern Biotechnology Associates,Inc.,Birmingham,AL)100μlとともにインキュベートした。次いで、赤血球を、製造業者のプロトコールに従って、市販の溶解試薬(FACS Lysing Solution,Becton Dickinson,San Jose,CA)を用いて溶解した。PBSで洗浄した後、細胞を0.5mlのPBS/1%ホルマリンに再懸濁し、分析するまで冷凍した。試料は、フィコエリスリンを励起させるために488nmのレーザーを用いて、FACScan(Becton Dickinson Corp.,San Jose,CA)で分析した。リンパ球捕捉ゲートを前方に90度の光散乱パラメーターでセットした。各試料に対して、10,000個の細胞を分析した。
ビオチニル化LDP−02/3A9/Lot#1は、ネズミAct−1で生じたものと類似で、ヒトまたはネズミアイソタイプ適合対照での染色により生じたパターンとは異なる異質の染色パターンでヒトリンパ球の亜集団を認識した。さらに、イヌまたはネコ由来のリンパ球で調べたとき、LDP−02/3A9/Lot#1およびネズミAct−1は両方とも、ヒトリンパ球を用いて派生したものと類似の異質な染色パターンを生じた。LDP−02/3A9/Lot#1またはネズミACT−1は、これらの条件下でラットまたはモルモット由来のリンパ球を認識しなかった。
D.C1q結合
以前に記載された技術(シムズら、J.Immunol.151:2296-2308(1993))を用いて、フローサイトメトリーを使用してLPD−02のヒト補体成分C1qを結合する能力を評価した。ヒト末梢血単核細胞(PBMC)を標準フィコール(Ficoll)密度分離により単離した。まず、375,000個の細胞を4℃で10分間、10%正常ウサギ血清/PBSでブロックした。洗浄による除去の後、該細胞を、10μg/mlの(a)CAMPATH−1H(Therapeutic Antibody Center,Cambridge,U.K.)、(b)ヒトIgG1(Sigma Chemial Co.,St.Louis,MO)、(c)LDP−01(2個のアミノ酸置換をIgG1重鎖定常領域に含み(Leu235→Ala235およびGly237→Ala237)、また、「FcRmut CD18」とも称する、国際公開第93/02191号パンフレット(1993年2月4日公開)およびシムズら、J.Immunol.151(4):2296-2308(1993)に記載された抗CD18抗体の誘導体、Therapeutic Antibody Center,Cambridge,U.K.)、または(d)LDP−02(1°C hum ACT−1 Lot#8/9)のいずれか100μlとともに、4℃で20分間インキュベートした。CAMPATH−1Hを陽性対照抗体として供し、一方、LDP−01およびヒトIgG1を陰性対照抗体として用いた。すべての試薬を2%BSA/PBSで希釈した。追加の陰性対照として、2%BSA/PBSも単独で加えた。次いで、抗体を洗浄により除去し、細胞を4℃で30分間、10μg/mlのヒト補体成分C1q(Sigma Chemical Co.,St.Louis,MO)50μlに再懸濁した。次いで、細胞を洗浄して、20μg/mlのFITC結合ウサギ抗ヒトC1q抗体(Dako Corp.,Carpinteria,CA)100μlに4℃で20分間再懸濁した。200μlのPBSで洗浄した後、細胞を0.5mlのPBS/1%ホルマリンに再懸濁し、分析するまで冷凍した。試料は、FITCを励起させるために488nmのレーザーを用いて、FACScan(Becton Dickinson Corp.,San Jose,CA)で分析した。各試料について、最小限10,000個の細胞を分析し、平均チャンネルフルオレッセンス(MCF)を計算した。
ヒトPBMCを、CAMPATH−1H結合ヒトC1qとともにインキュベートし、MCFで有意なシフトが生じたが、PBMCのLDP−01、BSAまたはヒトIgG1とのインキュベーションにより誘発された染色パターンはすべて類似しており、相対的に低いバックグランド染色を特徴とした。LDP−02とのPMBCのプレインキュベーションにより生じた染色のパターンは、これらの陰性対照試料で生じたものと同一であり、LDP−02はこれらの条件下ではC1qを結合しないことを示唆した。
E.補体介在溶解
LDP−02/3A9/Lot#1の補体介在細胞溶解に関与する能力を、ビンドン(Bindon,C.I.)ら(Transplantation,40:538-544(1985))により以前に記載されたプロトコールを用いて調べた。ヘパリン処理したヒト血液を無菌的に吸引し、血漿を回収してすぐに氷上に置いた。末梢血単核細胞(PBMC)は、フィコール−ハイパーク(Ficoll−Hypaque)、密度1.077g/mlの層に重層して15分間遠心分離することにより単離し、RPMI1640/10%FCS/100U/mlペニシリン/100μg/mlストレプトマイシン/2.0mM L−グルタミンからなる完全培地で2回洗浄した。次いで、2500万個の細胞を37℃で1時間、150μCiの51クロム酸ナトリウム(E.I.du Pont de Nemours & Co.Inc.,Wilmington,DE)の滅菌塩水中でインキュベートした。細胞を培地で2回洗浄し、106/mlに再懸濁した。次いで、培地中に50、25、5、2.5および0.5μg/mlの濃度の(a)CAMPATH−1H(Therapeutic Center,Cambridge,U.K.)、(b)CAMPATH−1G(Therapeutic Center,Cambridge,U.K.)、(c)ヒトIgG1(Sigma Chemial Co.,St.Louis,MO)、(d)LDP−02/3A9/Lot#1、または(e)LDP−01(FcRmut CD18、Therapeutic Antibody Center,Cambridge,U.K.(前記参照のこと))のいずれか100μlを含むU底マイクロタイタープレートのウエルに、50μlの懸濁物(5.0x104個の細胞)を添加した。CAMPATH−1抗体を陽性対照抗体としてアッセイに用い、一方、ヒトIgG1およびLDP−01を陰性対照として用いた。追加のウエルは、完全培地中の0.1% Triton−X−100(Fisher Scientific,Fair Lawn,NJ)の100μlに懸濁した細胞を含んでいた。Triton−X−100とインキュベートした細胞を用いて全放出を測定し、一方、抗体を含まない対照ウエルを用いて自発放出を測定した。室温で15分間のインキュベーション後、補体供給源として50μlの自己血漿を各ウエルに添加して20%の最終濃度とした。該細胞を37℃で45分間インキュベートし、次いで、100gで2分間遠心分離し、100ulの上清を回収した。放出された51Crは、CobraIIガンマカウンター(Packard Instruments,Downers Grove,IL)で測定した。すべての試料は、二連で行なった。特異的51Cr放出パーセントは、式:
ビンドン(Bindon)ら(Transplantation,40:538-544(1985))により以前に報告されたように、CAMPATH−1HおよびCAMPATH−1Gは両方とも、用量に依存した様式でヒトPBMCの補体介在溶解を35%まで誘発した。さらに、予想されたように、ヒトIgG1およびLDP−01(Fc−mut CD18)対照は、いかなる検出可能な細胞溶解をも誘発しなかった。LDP−02は、25μg/mlまでを含む調べたいかなる濃度でも細胞溶解を仲介しなかった(図17)。
F.抗体依存性細胞傷害(ADCC)
ヒトCD3+芽球を標的細胞として用い、LDP−02の抗体依存性細胞傷害(ADCC)に関与する能力を評価した。CD3+芽球は、PBSで5μg/mlの濃度に希釈した抗CD3抗体RT66で被覆した24ウエルプレートで生成した。ヒト末梢血単核細胞(PBMCS)は、フィコール−ハイパーク(Ficoll−Hypaque)、密度1.077g/mlの層に重層して15分間遠心分離することにより単離し、前章で記載したように、完全培地で洗浄および再懸濁した。次いで、200万個の細胞を24ウエルプレートの各ウエルに加え、37℃、5%CO2で4日間インキュベートした。次いで、細胞をカルチャーフラスコに移し、10ユニット/mlの濃度のヒト組換えIL−2(Genzyme Corp.,Cambridge,MA)を含む培地で、37℃、5%CO2でインキュベートした。培養3日後、次に、10.0x106個のCD3芽球を37℃で45分間、150μCiの51クロム酸ナトリウム(E.I.du Pont de Nemours & Co.Inc.,Wilmington,DE;Lot#95M682)の滅菌塩水中でインキュベートした。完全培地で2回洗浄した後、細胞を2x105細胞/mlになるように再懸濁し、50μlの懸濁物(10,000個の細胞)をU底96ウエルマイクロタイタープレートのウエルに加えた。該ウエルは、培地中に50、5、2.5、0.5、0.25または0.05μg/mlの最終濃度のCAMPATH−1H(Therapeutic Antibody Center,Cambridge,U.K.)またはLDP−02/3A9/Lot#1のいずれか50μlを含んでいた。細胞を室温で30分間抗体とともにインキュベートした後、異なるドナー由来の0.5x106個の新たに単離したPBMC(ficoll−hypaque勾配、37℃で完全培地で2回洗浄)をエフェクター細胞として各ウエルに加えた(50:1のエフェクター:標的比)。追加のウエルには、培地中5%Triton−X−100(Fisher Scientific,Fair Lawn,NJ)の100μlを加えた。Triton−X−100とインキュベートした細胞を用いて全放出を測定し、一方、抗体およびエフェクター細胞を含まない対照を、自発放射能放出を測定するために算入した。細胞を室温で100g、2分間遠心分離し、37℃、5%CO2で20時間インキュベートした後、細胞をV底96ウエルプレートに移し、室温で沈澱させた。100μlの上清を回収し、放出された放射能をCobraIIガンマカウンター(Packard Instruments,Downers Grove,IL)で測定した。すべての試料は、二連で行なった。特異的51Cr放出パーセントは、式:
シムズら、J,Immunol.,151(4):2296-2308(1993)により以前に示されたように、CAMPATH−1Hは、用量に依存した様式でADCCに関与し、5.0μg/ml以上の濃度で約30%までの特異的51Cr放出を誘発した。調べたいかなる濃度でもLDP−02を含むウエルでは特異的放出を検出しなかった。
G.MAdCAM−1への接着阻害
α4β7のMAdCAM−1への結合を阻害するLDP−02の能力は、蛍光標識したα4β7+RPMI8866細胞(ヒトB細胞リンパ腫)、およびヒトIgG1のFc領域(Fc変異LDP−02の定常領域を作製するために用いた同じ構築物由来の定常領域)に融合したヒトMAdCAM−1の全細胞外ドメインを含有するMAdCAM−1キメラを用いて評価した。
1.MAdCAM−IgGキメラの構築
pcDhuMAd4と称するヒトMAdCAM−1クローン(pCDNA3中のクローン4cDNA;シジャン(Shyjan,A.M.)ら、J.Immunol.,156:2851-2857(1996);その教示は、参照によりそのまま本明細書に取り込まれる)を、1995年2月10日に出願された米国出願第08/386,857号明細書の一部継続出願である、1995年9月1日に出願された米国出願第08/523,004号明細書の一部継続出願である、1996年2月12日に出願された国際出願番号PCT/US96/02153(国際公開第96/24673号パンフレット)に記載されたように、ヒトMAdCAM−1の細胞外領域のPCR増幅用の鋳型として用い、ヒトIgG1の定常領域と融合させた。MAdCAM−IgGキメラを構築するために、ヒトMAdCAM−1コーディング配列の5’末端(ATGコドン、太字)を含むプライマーHUMADIG4/2(配列番号:62)を合成した:
CH1、H(ヒンジ)、CH2およびCH3領域を包含する約1kbの断片を、Fc変異ヒト定常領域を有するヒト免疫グロブリンγ1重鎖をコードする構築物からSpeIとEcoRIとの消化により切り出した。その教示がそれぞれ参照によりそのまま本明細書に取り込まれる、シムズら(J.Immunol.,151:2296-2308(1993))およびウォルトマンら(国際公開第93/02191号パンフレット 1993年2月4日(第23頁))により記載されたように、この構築物中のヒト定常領域は、CAMPATH−1H重鎖(ライヒマンら、Nature,322:323-327(1988))のPCR増幅により得られたものに相当する。この構築物の定常領域中の変異(Leu235→Ala235およびGly237→Ala237)は、ヒトFcγレセプターへの結合を低下させるように設計され、オリゴヌクレオチド特異的変異導入により生成された。したがって、生成したMAdCAM−Ig融合体は、Leu235→Ala235およびGly237→Ala237変異の導入を除いて、シムズら(J.Immunol.,151:2296-2308(1993))およびウォルトマンら(国際公開第93/02191号パンフレット)により記載されたSpeI−EcoRI定常領域断片を含む。
Fc変異IgG1定常領域をコードする1kbのSpeI−EcoRI断片を、Glassmax DNA単離システム(Gibco,Bethesda,MD)を用いるゲル電気泳動により単離した。この定常領域断片およびMAdCAMの全細胞外ドメインを含むHindIII−SpeI断片を、HindIIIとEcoRIで消化しておいたベクターpEE12(ステファンズ(Stephens,P.L.)とコケット(M.L.Cockett)、Nucl.Acids Res.,17:7110(1989)およびベビングトン(Bebbington,C.R.)とヘンチェル(C.C.G.Hentschel)、1987、哺乳動物細胞におけるクローン化遺伝子の発現のための遺伝子増幅に基づくベクターの利用、(Academic Press,N.Y.)に三部ライゲーションで連結した。細菌株DH10Bの形質転換体を得た。コロニーを生育させ、ミニープラスミドプレップを制限マッピングにより分析した。Fc変異IgG1定常領域に融合したMAdCAM−1の全細胞外ドメインを含有する融合蛋白質をコードする構築物(構築物HuMAdIg21)を、全MAdCAM−1部分にわたってシーケンスし、セグメントが適切に融合していることおよびPCRで誘導した変異がないことを確認した。キメラをNSO細胞で生成させ、標準的なプロテインAアフィニティークロマトグラフィーにより精製した。
2.接着アッセイ
高結合型平底96ウエルプレート(Costar)を、炭酸塩緩衝液、pH9.5で2.5μg/mlに希釈した50μlのMAdCAM−1キメラを用いて、37℃で1時間被覆した。次いで、マイクロプレート自動洗浄機(Bio-Tek Instruments,Winooski,VT)を用いて、洗浄緩衝液(50mM Tris HCl、0.14M NaCl、1mM MnCl2、pH7.2)で1回ウエルを洗浄し、PBSに希釈した10%FBS100μlで、37℃で1.5時間ブロックした。
まず、RPMI8866細胞(α4β7を発現する(そしてα4β1を発現しない)ヒトBリンパ腫細胞株(アール(Erle,D.J.)ら、J.Immunol.,153:517(1994);アール博士から供与))を20mlのPBS(4℃)で洗浄し、PBS中4.0x106細胞/mlに再懸濁した。BCECF(2’,7’−ビス−(2−カルボキシエチル)−5−(アンド6)−カルボキシフルオレッセイン,アセトキシメチルエステル;Molecular Probes,Inc.,Eugene,OR)を、DMSO中50μg/mlに再構成し、1:500の最終希釈になるように細胞懸濁物に加えた。37℃で30分間インキュベートした後、次に、細胞をアッセイ緩衝液(2%ウシ胎仔血清を含むHBSS、25mM HEPES、ペニシリン/ストレプトマイシン、pH7.2)で洗浄し、V底96ウエルプレートの各ウエルに50,000個の細胞を加えた。次いで、細胞を、アッセイ緩衝液中に15.0〜0.00075μg/mlの濃度の(a)ネズミAct−1、(b)ネズミIgG1(Sigma Chemical Co.,St.Louis,MO)、(c)LDP−02/3A9/Lot#1、または(d)ヒトIgG1(Sigma Chemical Co.,St.Louis,MO)のいずれか100μlに室温で10分間再懸濁した。MAdCAM−1キメラで被覆したプレートを洗浄してブロッキング緩衝液を除去し、次いで、これらの蛍光標識されたRPMI8866細胞を各ウエルに移した。該プレートをアルミホイルで覆って、室温で30分間、40RPMのプラットホーム振盪機(New Brunswick Scientific Co.,Inc.,Edison,NJ)上に置いた。未結合の細胞を1回の洗浄工程で除去し、続いて洗浄前後に、フルオレッセンス コンセントレーター アナライザー(Fluorescence Concentrator Analyzer)(IDEXX Laboratories,Inc.,Westbrook,ME)で蛍光を測定した(485nmで励起、535nmで読む)。各ウエルについての結合した細胞のパーセントは、式:
LDP−02およびネズミAct−1の両方は、用量に依存した様式で、ヒトMAdCAMへのRPMI8866細胞の接着を阻害した(図18A〜18B)。50%接着を阻害する濃度(IC50)は、ネズミAct−1(0.0018μg/ml)およびLDP−02(0.0014μg/ml)に関して相対的に類似していた。したがって、LDP−02は、少なくともネズミAct−1と同じ程度に有効にMAdCAM−1へのα4β7介在接着を機能的に阻害した。
実施例5 追加のヒト化抗体
前記したように、実施例2で設計された再形成抗体のいくつかの変異を作製してアフィニティーを改良することおよび/または再形成抗体の抗原性を減少させることができる。かかる構築物は、1以上の下記変異:軽鎖のM4V変異、重鎖のR38K変異、重鎖のA40R変異および重鎖のI73T復帰突然変異を有するものを含むがこれに限定されるものではない。変異体は、独立して(例えば、1つの鎖に1つの変異)または種々の組合せで生成させることができる。
例えば、図19は、再形成抗体(実施例2で設計された)または軽鎖に1つの追加の変異(MV4)および重鎖に2つの追加の変異(R38K、A40R)を有する誘導体を用いたHuT78染色の結果を示す。これらの2つの抗体は、HuT78細胞で類似の染色パターンを示す(図19)。該変異は、製造業者の提案するプロトコールに従って、トランスフォーマー サイト−ディレクテッド ミュータゼネシス キット(Clontech)を用いて核酸配列を変化させることにより作製した。重鎖および軽鎖可変領域の両方の変異は、pCR−ScriptTMにクローニングされた可変領域で作製された。トランスオリゴScaI/StuI(Clontech)をトランスオリゴ用に用いた。変異誘発オリゴの配列(配列番号:38〜40)は、下記のようであった:
均等物
当業者であれば、単なる日常的な実験方法によって、本明細書に記載された発明の具体的態様に対する多くの均等物を認識し、あるいは確認することができるであろう。そのような均等物は、以下の請求の範囲の範疇に含まれることを意図されている。
Claims (15)
- 非ヒト起源の軽鎖可変領域の3つの相補性決定領域(CDR1、CDR2およびCDR3)の少なくとも1つおよびヒト起源の軽鎖可変領域に由来する枠組み領域ならびに非ヒト起源の重鎖可変領域の3つの相補性決定領域(CDR1、CDR2およびCDR3)の少なくとも1つおよびヒト起源の重鎖可変領域に由来する枠組み領域を含有する抗原結合領域を含有してなり、該相補性決定領域が以下:
軽鎖:CDR1 配列番号:12のアミノ酸44−59
CDR2 配列番号:12のアミノ酸75−81
CDR3 配列番号:12のアミノ酸114−122
重鎖:CDR1 配列番号:15のアミノ酸50−54
CDR2 配列番号:15のアミノ酸69−85
CDR3 配列番号:15のアミノ酸118−129
に示されるアミノ酸配列を有し、軽鎖が配列番号:21の可変領域を含有し、重鎖が配列番号:19の可変領域を含有してなる、α4β7インテグリンを選択的に結合するヒト化免疫グロブリンまたは抗原結合断片。 - ヒト化免疫グロブリン軽鎖またはその抗原結合断片がネズミAct−1モノクローナル抗体の軽鎖の3つの相補性決定領域(CDR1、CDR2およびCDR3)の少なくとも1つおよびヒト起源の軽鎖の可変領域に由来する枠組み領域を含有し、該相補性決定領域が該ヒト化免疫グロブリン軽鎖またはその抗原結合断片を含む抗体が選択的にα4β7インテグリンを結合するように、以下:
軽鎖:CDR1 配列番号:12のアミノ酸44−59
CDR2 配列番号:12のアミノ酸75−81
CDR3 配列番号:12のアミノ酸114−122
に示されるアミノ酸配列を含有し、ヒト化免疫グロブリン軽鎖またはその抗原結合断片が配列番号:21の可変領域を含有してなる、ヒト化免疫グロブリン軽鎖またはその抗原結合断片。 - 請求項2記載のヒト化免疫グロブリン軽鎖またはその抗原結合断片をコードするヌクレオチド配列を含有してなる単離された核酸であって、前記ヌクレオチド配列が配列番号:20の可変領域コーディング配列を含有してなる単離された核酸。
- 請求項2記載のヒト化免疫グロブリン軽鎖またはその抗原結合断片をコードするヌクレオチド配列を含有してなる発現ベクターであって、前記ヌクレオチド配列が配列番号:20の可変領域コーディング配列を含有してなる発現ベクター。
- ヒト化免疫グロブリン重鎖またはその抗原結合断片がネズミAct−1モノクローナル抗体の重鎖の3つの相補性決定領域(CDR1、CDR2およびCDR3)の少なくとも1つおよびヒト起源の重鎖の可変領域に由来する枠組み領域を含有し、該相補性決定領域が該ヒト化免疫グロブリン重鎖またはその抗原結合断片を含む抗体がα4β7インテグリンを選択的に結合するように、以下:
重鎖:CDR1 配列番号:15のアミノ酸50−54
CDR2 配列番号:15のアミノ酸69−85
CDR3 配列番号:15のアミノ酸118−129
に示されるアミノ酸配列を含有してなり、ヒト化免疫グロブリン重鎖またはその抗原結合断片が配列番号:19の可変領域を含有してなる、ヒト化免疫グロブリン重鎖またはその抗原結合断片。 - 請求項5記載のヒト化免疫グロブリン重鎖またはその抗原結合断片をコードするヌクレオチド配列を含有してなる単離された核酸であって、前記ヌクレオチド配列が配列番号:18の可変領域コーディング配列を含有してなる単離された核酸。
- 請求項5記載のヒト化免疫グロブリン重鎖またはその抗原結合断片をコードするヌクレオチド配列を含有してなる発現ベクターであって、前記ヌクレオチド配列が配列番号:18の可変領域コーディング配列を含有してなる発現ベクター。
- 請求項4又は7記載の発現ベクターを含有してなる宿主細胞。
- 請求項8記載の宿主細胞をヒト化免疫グロブリン軽鎖または重鎖の発現に適した条件下で維持し、それによりヒト化免疫グロブリン軽鎖、ヒト化免疫グロブリン軽鎖抗原結合断片、ヒト化免疫グロブリン重鎖、またはヒト化免疫グロブリン重鎖抗原結合断片を発現させ、産生させ、任意に単離させる工程を含む、ヒト化免疫グロブリン軽鎖、ヒト化免疫グロブリン軽鎖抗原結合断片、ヒト化免疫グロブリン重鎖、またはヒト化免疫グロブリン重鎖抗原結合断片の調製方法。
- ヒト化免疫グロブリン軽鎖またはその抗原結合断片をコードする第1の組換え核酸およびヒト化免疫グロブリン重鎖またはその抗原結合断片をコードする第2の組換え核酸を含有し、該軽鎖またはその抗原結合断片および該重鎖またはその抗原結合断片を含む抗体または抗原結合断片が選択的にα4β7インテグリンを結合する宿主細胞であって、
該第1核酸は以下:
軽鎖:CDR1 配列番号:12のアミノ酸44−59
CDR2 配列番号:12のアミノ酸75−81
CDR3 配列番号:12のアミノ酸114−122
に示されるアミノ酸配列を有する3つの相補性決定領域(CDR1、CDR2およびCDR3)の少なくとも1つおよびヒト起源の軽鎖に由来する枠組み領域をコードするヌクレオチド配列を含み;
該第2核酸は以下:
重鎖:CDR1 配列番号:15のアミノ酸50−54
CDR2 配列番号:15のアミノ酸69−85
CDR3 配列番号:15のアミノ酸118−129
に示されるアミノ酸配列を有する3つの相補性決定領域(CDR1、CDR2およびCDR3)の少なくとも1つおよびヒト起源の重鎖に由来する枠組み領域をコードするヌクレオチド配列を含み、第1核酸が配列番号:21の可変領域をコードし、第2核酸が配列番号:19の可変領域をコードする、宿主細胞。 - 請求項10記載の宿主細胞をヒト化免疫グロブリンの発現に適した条件下で維持し、それによりヒト化免疫グロブリン鎖が発現され、ヒト化免疫グロブリンが産生され、任意に単離される工程を含む、ヒト化免疫グロブリンの調製方法。
- 治療または診断における使用のための請求項1記載のヒト化免疫グロブリンまたは抗原結合断片。
- 請求項1記載のヒト化免疫グロブリンまたは抗原結合断片、および医薬としての使用のための適当な担体を含有してなる、炎症性腸疾患の治療用医薬組成物。
- 炎症性腸疾患の治療用の医薬の製造のための請求項1記載のヒト化免疫グロブリンまたは抗原結合断片の使用。
- 炎症性腸疾患が潰瘍性大腸炎およびクローン病患者における炎症性腸疾患からなる群より選択される、請求項14記載の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/700,737 | 1996-08-15 | ||
| US08/700,737 US7147851B1 (en) | 1996-08-15 | 1996-08-15 | Humanized immunoglobulin reactive with α4β7 integrin |
| PCT/US1997/013884 WO1998006248A2 (en) | 1996-08-15 | 1997-08-06 | HUMANIZED IMMUNOGLOBULIN REACTIVE WITH α4β7 INTEGRIN |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008120608A Division JP4578536B2 (ja) | 1996-08-15 | 2008-05-02 | α4β7インテグリンと反応するヒト化免疫グロブリン |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2001507210A JP2001507210A (ja) | 2001-06-05 |
| JP2001507210A5 JP2001507210A5 (ja) | 2005-04-07 |
| JP4171071B2 true JP4171071B2 (ja) | 2008-10-22 |
Family
ID=24814673
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50985398A Expired - Lifetime JP4171071B2 (ja) | 1996-08-15 | 1997-08-06 | α4β7インテグリンと反応するヒト化免疫グロブリン |
| JP2008120608A Expired - Lifetime JP4578536B2 (ja) | 1996-08-15 | 2008-05-02 | α4β7インテグリンと反応するヒト化免疫グロブリン |
| JP2010118596A Expired - Lifetime JP5166483B2 (ja) | 1996-08-15 | 2010-05-24 | α4β7インテグリンと反応するヒト化免疫グロブリン |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008120608A Expired - Lifetime JP4578536B2 (ja) | 1996-08-15 | 2008-05-02 | α4β7インテグリンと反応するヒト化免疫グロブリン |
| JP2010118596A Expired - Lifetime JP5166483B2 (ja) | 1996-08-15 | 2010-05-24 | α4β7インテグリンと反応するヒト化免疫グロブリン |
Country Status (19)
| Country | Link |
|---|---|
| US (3) | US7147851B1 (ja) |
| EP (2) | EP0918797B2 (ja) |
| JP (3) | JP4171071B2 (ja) |
| CN (1) | CN100406562C (ja) |
| AT (1) | ATE321788T1 (ja) |
| AU (1) | AU730326C (ja) |
| BE (1) | BE2014C068I2 (ja) |
| BR (1) | BRPI9711079B1 (ja) |
| CA (1) | CA2263106C (ja) |
| DE (1) | DE69735596T3 (ja) |
| DK (1) | DK0918797T4 (ja) |
| ES (1) | ES2262186T5 (ja) |
| IL (3) | IL128052A0 (ja) |
| LT (1) | LTC0918797I2 (ja) |
| LU (1) | LU92596I2 (ja) |
| NZ (1) | NZ334226A (ja) |
| PT (1) | PT918797E (ja) |
| SI (1) | SI0918797T2 (ja) |
| WO (1) | WO1998006248A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010246548A (ja) * | 1996-08-15 | 2010-11-04 | Millennium Pharmaceuticals Inc | α4β7インテグリンと反応するヒト化免疫グロブリン |
Families Citing this family (126)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7122636B1 (en) | 1997-02-21 | 2006-10-17 | Genentech, Inc. | Antibody fragment-polymer conjugates and uses of same |
| US6787523B1 (en) * | 1997-12-02 | 2004-09-07 | Neuralab Limited | Prevention and treatment of amyloidogenic disease |
| US7790856B2 (en) | 1998-04-07 | 2010-09-07 | Janssen Alzheimer Immunotherapy | Humanized antibodies that recognize beta amyloid peptide |
| US7964192B1 (en) | 1997-12-02 | 2011-06-21 | Janssen Alzheimer Immunotherapy | Prevention and treatment of amyloidgenic disease |
| US20080050367A1 (en) | 1998-04-07 | 2008-02-28 | Guriq Basi | Humanized antibodies that recognize beta amyloid peptide |
| TWI239847B (en) | 1997-12-02 | 2005-09-21 | Elan Pharm Inc | N-terminal fragment of Abeta peptide and an adjuvant for preventing and treating amyloidogenic disease |
| US7090845B2 (en) * | 1998-05-13 | 2006-08-15 | Genentech, Inc. | Diagnosis and treatment of hepatic disorders |
| AU2011226857B2 (en) * | 1998-05-13 | 2013-09-19 | Genentech, Inc. | Diagnosis and treatment of hepatic disorders |
| WO2000030681A1 (en) * | 1998-11-25 | 2000-06-02 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Antagonists of the alpha e beta 7 integrin as therapeutic agents for inflammatory diseases |
| PT1173201E (pt) * | 1999-04-22 | 2005-08-31 | Biogen Idec Inc | Metodo para o tratamento de fibrose utilizando um antagonista da subunidade alfa 4 de integrina |
| AU2006220426B2 (en) * | 2000-04-14 | 2008-10-30 | Genentech Inc. | Method of administering an antibody |
| US20010046496A1 (en) * | 2000-04-14 | 2001-11-29 | Brettman Lee R. | Method of administering an antibody |
| US7658924B2 (en) * | 2001-10-11 | 2010-02-09 | Amgen Inc. | Angiopoietin-2 specific binding agents |
| US7521053B2 (en) * | 2001-10-11 | 2009-04-21 | Amgen Inc. | Angiopoietin-2 specific binding agents |
| ATE457741T1 (de) * | 2001-12-17 | 2010-03-15 | Univ Cardiff | Enzymatische spaltbare reagenzien zum spezifischen targeting von krankheitsherden |
| CA2473144C (en) | 2002-02-05 | 2013-05-28 | Genentech, Inc. | Protein purification |
| MY139983A (en) | 2002-03-12 | 2009-11-30 | Janssen Alzheimer Immunotherap | Humanized antibodies that recognize beta amyloid peptide |
| WO2003102132A2 (en) | 2002-04-26 | 2003-12-11 | Genetech, Inc. | Non-affinity purification of proteins |
| GB0210121D0 (en) | 2002-05-02 | 2002-06-12 | Celltech R&D Ltd | Biological products |
| SG187991A1 (en) | 2002-05-02 | 2013-03-28 | Wyeth Corp | Calicheamicin derivative-carrier conjugates |
| CA2512729C (en) * | 2003-01-09 | 2014-09-16 | Macrogenics, Inc. | Identification and engineering of antibodies with variant fc regions and methods of using same |
| NZ567324A (en) * | 2003-02-01 | 2009-08-28 | Wyeth Corp | Active immunization to generate antibodies to soluble A-beta |
| DK3095793T3 (da) | 2003-07-28 | 2020-05-25 | Genentech Inc | Reducering af udvaskning af protein A under en protein A-affinitetskromatografi |
| US7850970B2 (en) | 2003-08-26 | 2010-12-14 | The Regents Of The University Of Colorado | Inhibitors of serine protease activity and their use in methods and compositions for treatment of bacterial infections |
| MXPA06000176A (es) | 2003-12-10 | 2006-06-27 | Millennium Pharm Inc | Anticuerpos anti-ccr2 humanizados y sus metodos de uso. |
| MX370489B (es) | 2004-01-09 | 2019-12-16 | Pfizer | Anticuerpos contra madcam. |
| US7662926B2 (en) | 2004-09-02 | 2010-02-16 | Genentech, Inc. | Anti-Fc-gamma receptor antibodies, bispecific variants and uses therefor |
| US7655229B2 (en) | 2004-09-02 | 2010-02-02 | Chan Andrew C | Anti-FC-gamma RIIB receptor antibody and uses therefor |
| WO2006026759A2 (en) * | 2004-09-03 | 2006-03-09 | Genentech, Inc. | Humanized anti-beta7 antagonists and uses therefor |
| JO3000B1 (ar) | 2004-10-20 | 2016-09-05 | Genentech Inc | مركبات أجسام مضادة . |
| EP1838348B1 (en) | 2004-12-15 | 2013-06-26 | Janssen Alzheimer Immunotherapy | Humanized amyloid beta antibodies for use in improving cognition |
| MY146381A (en) | 2004-12-22 | 2012-08-15 | Amgen Inc | Compositions and methods relating relating to anti-igf-1 receptor antibodies |
| WO2007007151A2 (en) * | 2005-07-11 | 2007-01-18 | Pfizer Limited | Use of anti-mad-cam antibody for the treatment of emphysema |
| WO2007007160A2 (en) * | 2005-07-11 | 2007-01-18 | Pfizer Limited | Anti-madcam antibodies to treat fever |
| EP1948691A1 (en) * | 2005-11-17 | 2008-07-30 | Millennium Pharmaceuticals, Inc. | HUMANIZED IMMUNOGLOBULIN REACTIVE WITH a4ß7INTEGRIN |
| US8784810B2 (en) | 2006-04-18 | 2014-07-22 | Janssen Alzheimer Immunotherapy | Treatment of amyloidogenic diseases |
| FR2902799B1 (fr) | 2006-06-27 | 2012-10-26 | Millipore Corp | Procede et unite de preparation d'un echantillon pour l'analyse microbiologique d'un liquide |
| WO2008079302A2 (en) | 2006-12-21 | 2008-07-03 | Millipore Corporation | Purification of proteins |
| US8569464B2 (en) | 2006-12-21 | 2013-10-29 | Emd Millipore Corporation | Purification of proteins |
| US8362217B2 (en) | 2006-12-21 | 2013-01-29 | Emd Millipore Corporation | Purification of proteins |
| RU2474585C2 (ru) | 2007-01-22 | 2013-02-10 | Дженентек, Инк. | Осаждение и очистка белков полиэлектролитами |
| AU2008223541B2 (en) * | 2007-03-02 | 2012-04-05 | Amgen Inc. | Methods and compositions for treating tumor diseases |
| US20100297699A1 (en) * | 2007-03-20 | 2010-11-25 | Millennium Pharmaceuticals, Inc. | Nucleic Acids Encoding Humanized Immunoglobulin That Binds Alpha4Beta7 Integrin |
| US8003097B2 (en) | 2007-04-18 | 2011-08-23 | Janssen Alzheimer Immunotherapy | Treatment of cerebral amyloid angiopathy |
| BRPI0813645A2 (pt) | 2007-06-25 | 2014-12-30 | Esbatech Alcon Biomed Res Unit | Métodos para modificar anticorpos, e anticorpos modificados com propriedades funcionais aperfeiçoadas |
| KR102055873B1 (ko) | 2007-07-09 | 2019-12-13 | 제넨테크, 인크. | 폴리펩티드의 재조합 생산 동안의 디술피드 결합 환원의 방지 |
| DK2182983T3 (da) * | 2007-07-27 | 2014-07-14 | Janssen Alzheimer Immunotherap | Behandling af amyloidogene sygdomme med humaniserede anti-abeta antistoffer |
| JO3076B1 (ar) | 2007-10-17 | 2017-03-15 | Janssen Alzheimer Immunotherap | نظم العلاج المناعي المعتمد على حالة apoe |
| ES2533266T5 (es) | 2007-10-30 | 2018-04-18 | Genentech, Inc. | Purificación de anticuerpos mediante cromatografía de intercambio catiónico |
| RU2503720C2 (ru) * | 2008-01-11 | 2014-01-10 | Астеллас Фарма Инк. | УЛУЧШЕННОЕ ГУМАНИЗИРОВАННОЕ АНТИТЕЛО К ЧЕЛОВЕЧЕСКОМУ α9-ИНТЕГРИНУ |
| JO2913B1 (en) | 2008-02-20 | 2015-09-15 | امجين إنك, | Antibodies directed towards angiopoietin-1 and angiopoietin-2 proteins and their uses |
| US8999702B2 (en) | 2008-06-11 | 2015-04-07 | Emd Millipore Corporation | Stirred tank bioreactor |
| BRPI0823046B1 (pt) | 2008-08-14 | 2021-12-14 | Genentech, Inc | Método para purificar um anticorpo monoclonal de uma composição compreendendo o anticorpo monoclonal e pelo menos uma proteína de ovário de hamster chinês (chop) |
| CN103599541A (zh) | 2008-09-10 | 2014-02-26 | 弗·哈夫曼-拉罗切有限公司 | 用于防止蛋白质氧化降解的组合物和方法 |
| US9067981B1 (en) | 2008-10-30 | 2015-06-30 | Janssen Sciences Ireland Uc | Hybrid amyloid-beta antibodies |
| WO2010074702A1 (en) | 2008-12-16 | 2010-07-01 | Millipore Corporation | Purification of proteins |
| JP2012511929A (ja) | 2008-12-16 | 2012-05-31 | イー・エム・デイー・ミリポア・コーポレイシヨン | 攪拌タンク反応器及び方法 |
| EP2400992B1 (en) | 2009-02-27 | 2015-07-22 | Genentech, Inc. | Methods and compositions for protein labelling |
| AU2016208453B2 (en) * | 2009-03-20 | 2018-02-22 | Amgen Inc. | Alpha-4-beta-7 heterodimer specific antagonist antibody |
| AU2013205341B2 (en) * | 2009-03-20 | 2016-05-05 | Amgen Inc. | Alpha-4-beta-7 heterodimer specific antagonist antibody |
| CN103382222B (zh) * | 2009-03-20 | 2016-12-28 | 安姆根有限公司 | α-4-β-7异二聚体特异性拮抗剂抗体 |
| NZ621170A (en) | 2009-05-13 | 2015-08-28 | Genzyme Corp | Anti-human cd52 immunoglobulins |
| HRP20200768T4 (hr) | 2009-08-11 | 2025-03-28 | F. Hoffmann - La Roche Ag | Proizvodnja proteina u mediju za uzgoj stanica bez glutamina |
| SI2473617T1 (sl) | 2009-09-01 | 2020-07-31 | F. Hoffmann-La Roche Ag | Izboljšano prečiščevanje beljakovin z modificirano elucijo beljakovine A |
| SI2493922T1 (sl) | 2009-10-26 | 2017-06-30 | F. Hoffmann-La Roche Ag | Postopek za proizvodnjo glikoziliranega imunoglobulina |
| EP2550018B1 (en) | 2010-03-22 | 2019-02-27 | F.Hoffmann-La Roche Ag | Compositions and methods useful for stabilizing protein-containing formulations |
| BR112012027828A2 (pt) | 2010-05-03 | 2016-08-09 | Genentech Inc | composição de matéria, artigo de fabricação e método de redução da viscosidade de uma formulação contendo proteína e de preparação de uma formulação aquosa contendo proteína |
| WO2011146394A1 (en) | 2010-05-17 | 2011-11-24 | Millipore Corporation | Stimulus responsive polymers for the purification of biomolecules |
| CA2800728C (en) | 2010-05-28 | 2020-10-27 | Genentech, Inc. | Decreasing lactate level and increasing polypeptide production by downregulating the expression of lactate dehydrogenase and pyruvate dehydrogenase kinase |
| SG186783A1 (en) | 2010-06-24 | 2013-02-28 | Genentech Inc | Compositions and methods containing alkylgycosides for stabilizing protein- containing formulations |
| CA2808185A1 (en) | 2010-08-13 | 2012-02-16 | Genentech, Inc. | Antibodies to il-1.beta. and il-18, for treatment of disease |
| WO2012030512A1 (en) | 2010-09-03 | 2012-03-08 | Percivia Llc. | Flow-through protein purification process |
| MX367097B (es) | 2011-05-02 | 2019-08-05 | Millennium Pharm Inc | FORMULACION PARA ANTICUERPO ANTI-A4ß7. |
| UA116189C2 (uk) | 2011-05-02 | 2018-02-26 | Мілленніум Фармасьютікалз, Інк. | КОМПОЗИЦІЯ АНТИ-α4β7 АНТИТІЛА |
| WO2012178102A2 (en) | 2011-06-24 | 2012-12-27 | The Regents Of The Unversity Of Colorado, A Body Corporate | Compositions, methods and uses for alpha-1 antitrypsin fusion molecules |
| RU2648999C2 (ru) | 2011-12-22 | 2018-03-29 | Дженентек, Инк. | Способы повышения эффективности этапов очистки белка, находящихся ниже по потоку, с использованием мембранной ионообменной хроматографии |
| US10745468B2 (en) | 2011-12-22 | 2020-08-18 | Kota Biotherapeutics, Llc | Compositions and methods for modified B cells expressing reassigned biological agents |
| CA2896951A1 (en) * | 2012-01-10 | 2013-07-18 | The Regents Of The University Of Colorado, A Body Corporate | Compositions, methods and uses for alpha-1 antitrypsin fusion molecules |
| EA034583B1 (ru) * | 2012-01-12 | 2020-02-21 | Милленниум Фармасьютикалз, Инк. | ПРИМЕНЕНИЯ АНТИ-α4β7 АНТИТЕЛА |
| BR112014023176B1 (pt) | 2012-03-27 | 2021-09-28 | Genentech, Inc. | Métodos de produção de uma proteína recombinante |
| CN103374073A (zh) * | 2012-04-26 | 2013-10-30 | 中国科学院上海生命科学研究院 | 识别活化形式整合素α4β7的人源化单克隆抗体 |
| EP2849723B1 (en) | 2012-05-18 | 2018-05-02 | Genentech, Inc. | High-concentration monoclonal antibody formulations |
| CA2898146C (en) | 2012-12-19 | 2020-12-01 | Charles Andrew BOSWELL | Methods and compositions for radiohalogen protein labeling |
| AR095199A1 (es) | 2013-03-15 | 2015-09-30 | Genzyme Corp | Anticuerpos anti-cd52 |
| AU2015287760B2 (en) | 2014-07-09 | 2019-06-20 | Genentech, Inc. | pH adjustment to improve thaw recovery of cell banks |
| WO2016057424A1 (en) | 2014-10-06 | 2016-04-14 | Chemocentryx, Inc. | Combination therapy of inhibitors of c-c chemokine receptor type 9 (ccr9) and anti-alha4beta7 integrin blocking antibodies |
| WO2016086147A1 (en) | 2014-11-26 | 2016-06-02 | Millennium Pharmaceuticals, Inc. | Vedolizumab for the treatment of fistulizing crohn's disease |
| WO2016105572A1 (en) | 2014-12-24 | 2016-06-30 | Millennium Pharmaceuticals, Inc. | PREDICTING OUTCOME OF TREATMENT WITH AN ANTI-α4β7 INTEGRIN ANTIBODY |
| MA41636A (fr) | 2015-03-06 | 2018-01-09 | Millennium Pharm Inc | Méthode de traitement de la cholangite sclérosante primitive |
| SG11201800864XA (en) | 2015-08-11 | 2018-03-28 | Univ Osaka | Antibody |
| JP6951825B2 (ja) | 2015-09-04 | 2021-10-20 | ザ スクリプス リサーチ インスティテュート | インスリン免疫グロブリン融合タンパク質 |
| WO2017117558A1 (en) | 2015-12-31 | 2017-07-06 | The Regents Of The University Of Colorado, A Body Corporate | Methods and uses for alpha-1 antitrypsin or recombinant forms thereof, on steroid-refractory graft versus host disease involving gastrointestinal complications |
| AU2017234010B2 (en) * | 2016-03-14 | 2024-06-13 | Millennium Pharmaceuticals, Inc. | Methods of treating or preventing graft versus host disease |
| CA3017743A1 (en) * | 2016-03-14 | 2017-09-21 | Millennium Pharmaceuticals, Inc. | Method of preventing graft versus host disease |
| WO2017165742A1 (en) | 2016-03-24 | 2017-09-28 | Millennium Pharmaceuticals, Inc. | Methods of treating gastrointestinal immune-related adverse events in anti-ctla4 anti-pd-1 combination treatments |
| JP7069032B2 (ja) | 2016-03-24 | 2022-05-17 | ミレニアム ファーマシューティカルズ, インコーポレイテッド | がん免疫治療における胃腸の免疫関連有害事象の治療方法 |
| EP3436610A1 (en) | 2016-03-29 | 2019-02-06 | Geltor, Inc. | Expression of proteins in gram-negative bacteria wherein the ratio of periplasmic volume to cytoplasmic volume is between 0.5:1 and 10:1 |
| US10918716B2 (en) | 2016-05-04 | 2021-02-16 | Millennium Pharmaceuticals, Inc. | Triple combination therapy for treating Crohn's disease |
| CN109563482A (zh) * | 2016-06-10 | 2019-04-02 | 埃尔瓦有限公司 | 含表达异于分泌免疫球蛋白和细胞溶解功能的膜免疫球蛋白的b淋巴细胞系的组合物和方法 |
| EP3468597A1 (en) | 2016-06-12 | 2019-04-17 | Millennium Pharmaceuticals, Inc. | Method of treating inflammatory bowel disease |
| WO2018104893A1 (en) | 2016-12-06 | 2018-06-14 | Glaxosmithkline Intellectual Property Development Limited | Alpha4-beta7 antibodies with incrased fcrn binding and/or half-life |
| AU2017382281B2 (en) | 2016-12-22 | 2024-07-11 | Genentech, Inc. | Methods and formulations for reducing reconstitution time of lyophilized polypeptides |
| AU2018256840A1 (en) | 2017-04-28 | 2019-11-07 | Millennium Pharmaceuticals, Inc. | Method of treating pediatric disorders with antibodies specific for alpha 4 beta 7 integrin (vedolizumab) |
| EP3652211A1 (en) | 2017-07-14 | 2020-05-20 | Pfizer Inc. | Antibodies to madcam |
| US11180541B2 (en) | 2017-09-28 | 2021-11-23 | Geltor, Inc. | Recombinant collagen and elastin molecules and uses thereof |
| HRP20240584T1 (hr) | 2018-07-03 | 2024-07-19 | Bristol-Myers Squibb Company | Postupak proizvodnje rekombinantnih proteina |
| WO2020076776A1 (en) | 2018-10-10 | 2020-04-16 | Boehringer Ingelheim International Gmbh | Method for membrane gas transfer in high density bioreactor culture |
| JP2022510218A (ja) * | 2018-11-30 | 2022-01-26 | メモリアル スローン ケタリング キャンサー センター | ヘテロ二量体四価特異性抗体およびこれらの使用 |
| TWI856084B (zh) | 2019-04-01 | 2024-09-21 | 美商建南德克公司 | 用於穩定含有蛋白質之配方之組合物及方法 |
| EP3953380A4 (en) | 2019-04-12 | 2023-01-25 | Geltor, Inc. | RECOMBINATION ELASTIN AND ASSOCIATED PRODUCTION |
| JP7778567B2 (ja) * | 2019-04-17 | 2025-12-02 | ミレニアム ファーマシューティカルズ, インコーポレイテッド | α4β7阻害薬及びIL-23阻害薬の併用療法 |
| MX2021015301A (es) * | 2019-06-10 | 2022-02-03 | Takeda Pharmaceuticals Co | Métodos de cultivo celular y composiciones para la producción de anticuerpos. |
| EP3980119A4 (en) * | 2019-06-10 | 2023-06-07 | Takeda Pharmaceutical Company Limited | METHODS FOR PURIFYING ANTIBODIES AND ASSOCIATED COMPOSITIONS |
| EP4007808A1 (en) | 2019-08-01 | 2022-06-08 | Bristol-Myers Squibb Company | Methods of improving protein productivity in fed-batch cell cultures |
| EP4217383A1 (en) | 2020-09-22 | 2023-08-02 | Bristol-Myers Squibb Company | Methods for producing therapeutic proteins |
| US20250115666A1 (en) | 2021-08-05 | 2025-04-10 | Bristol-Myers Squibb Company | Cell culture methods for producing therapeutic proteins |
| US20240390869A1 (en) | 2021-09-21 | 2024-11-28 | Bristol-Myers Squibb Company | Methods of controlling the level of dissolved oxygen (do) in a solution comprising a recombinant protein in a storage container |
| US20240076343A1 (en) | 2022-06-16 | 2024-03-07 | Cephalon Llc | Anti-pd-1 antibody-attenuated il-2 immunoconjugates and uses thereof |
| WO2024249568A1 (en) | 2023-05-30 | 2024-12-05 | Paragon Therapeutics, Inc. | Alpha4beta7 integrin antibody compositions and methods of use |
| WO2024252368A2 (en) | 2023-06-09 | 2024-12-12 | Takeda Pharmaceutical Company Limited | Methods and compositions for treating ulcerative colitis |
| CN121889424A (zh) | 2023-08-14 | 2026-04-17 | 派拉冈医疗公司 | α4β7整联蛋白结合蛋白及使用方法 |
| US20250195677A1 (en) | 2023-12-19 | 2025-06-19 | Cephalon Llc | Uses for attenuated il-2 immunoconjugates |
| CN117777292B (zh) * | 2023-12-28 | 2024-10-18 | 天津大学 | 抗整合素α4β7纳米抗体及其用途 |
| WO2025149974A1 (en) | 2024-01-11 | 2025-07-17 | Takeda Pharmaceutical Company Limited | Combination therapy with an anti-alpha4beta7 antibody |
| WO2025177214A1 (en) | 2024-02-20 | 2025-08-28 | Takeda Pharmaceutical Company Limited | Therapeutic methods |
| WO2026058045A1 (en) | 2024-09-12 | 2026-03-19 | Takeda Pharmaceutical Company Limited | Alpha4beta7 inhibitor and il-23 inhibitor combination therapy |
Family Cites Families (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3220049A1 (de) | 1982-05-27 | 1983-12-01 | Universal Maschinenfabrik Dr. Rudolf Schieber GmbH & Co KG, 7081 Westhausen | Strick- und umhaengeschlosskombination fuer v-bett-flachstrickmaschinen mit schiebernadeln |
| GB8308235D0 (en) | 1983-03-25 | 1983-05-05 | Celltech Ltd | Polypeptides |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| GB8422238D0 (en) | 1984-09-03 | 1984-10-10 | Neuberger M S | Chimeric proteins |
| US4699880A (en) * | 1984-09-25 | 1987-10-13 | Immunomedics, Inc. | Method of producing monoclonal anti-idiotype antibody |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| JP3101690B2 (ja) | 1987-03-18 | 2000-10-23 | エス・ビィ・2・インコーポレイテッド | 変性抗体の、または変性抗体に関する改良 |
| US5258498A (en) * | 1987-05-21 | 1993-11-02 | Creative Biomolecules, Inc. | Polypeptide linkers for production of biosynthetic proteins |
| EP0303463B1 (en) | 1987-08-11 | 1994-11-30 | The Board Of Trustees Of The Leland Stanford Junior University | Method to control leukocyte extravasation |
| US5538724A (en) | 1987-08-11 | 1996-07-23 | The Board Of Trustees For The Leland Stanford Junior Univ. | Method of control leukocyte extravasation |
| US5403919A (en) | 1987-08-11 | 1995-04-04 | Board Of Trustees Of The Leland Stanford Junior University Stanford University | Method to control leukocyte extravasation |
| US5223392A (en) * | 1988-01-25 | 1993-06-29 | Exocell, Inc. | Monoclonal antibodies against glycated albumin, hybrid cell lines producing these antibodies, and use therefore |
| WO1989007142A1 (en) | 1988-02-05 | 1989-08-10 | Morrison Sherie L | Domain-modified constant region antibodies |
| WO1990007321A2 (en) | 1988-12-23 | 1990-07-12 | The Board Of Trustees Of The Leland Stanford Junior University | Lymphocyte receptor homing sequences and their uses |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| IL162181A (en) | 1988-12-28 | 2006-04-10 | Pdl Biopharma Inc | A method of producing humanized immunoglubulin, and polynucleotides encoding the same |
| US5225538A (en) * | 1989-02-23 | 1993-07-06 | Genentech, Inc. | Lymphocyte homing receptor/immunoglobulin fusion proteins |
| IL95501A (en) | 1989-09-01 | 1997-04-15 | Hutchinson Fred Cancer Res | Inhibition by antibodies of lymphocyte adherence to vascular endothelium utilizing an extracellular matrix receptor-ligand interaction, and pharmaceutical compositions containing said antibodies |
| US5859205A (en) | 1989-12-21 | 1999-01-12 | Celltech Limited | Humanised antibodies |
| GB8928874D0 (en) | 1989-12-21 | 1990-02-28 | Celltech Ltd | Humanised antibodies |
| CA2082160C (en) | 1991-03-06 | 2003-05-06 | Mary M. Bendig | Humanised and chimeric monoclonal antibodies |
| US5665595A (en) * | 1991-06-07 | 1997-09-09 | Dowelanco | Immunoglobulins against insect tissue |
| EP1400536A1 (en) | 1991-06-14 | 2004-03-24 | Genentech Inc. | Method for making humanized antibodies |
| GB9115364D0 (en) | 1991-07-16 | 1991-08-28 | Wellcome Found | Antibody |
| US5871734A (en) | 1992-01-13 | 1999-02-16 | Biogen, Inc. | Treatment for asthma with VLA-4 blocking agents |
| JPH07506566A (ja) | 1992-02-12 | 1995-07-20 | バイオゲン インコーポレイテッド | 炎症性胃腸病の処置 |
| US5932214A (en) | 1994-08-11 | 1999-08-03 | Biogen, Inc. | Treatment for inflammatory bowel disease with VLA-4 blockers |
| WO1994012214A1 (en) | 1992-12-01 | 1994-06-09 | Protein Design Labs, Inc. | Humanized antibodies reactive with cd18 |
| AU5675794A (en) | 1992-12-15 | 1994-07-04 | Board Of Trustees Of The Leland Stanford Junior University | Mucosal vascular addressin, dna and expression |
| DE69419721T2 (de) | 1993-01-12 | 2000-04-27 | Biogen, Inc. | Rekombinante anti-vla4 antikörpermoleküle |
| ATE161730T1 (de) | 1993-02-09 | 1998-01-15 | Biogen Inc | Antikörper zur behandlung von insulinabhängigen diabetes |
| JPH06303990A (ja) | 1993-04-24 | 1994-11-01 | Kanebo Ltd | モノクローナル抗体、それを産生するハイブリドーマおよび該抗体の製造方法 |
| AU691811B2 (en) | 1993-06-16 | 1998-05-28 | Celltech Therapeutics Limited | Antibodies |
| US5840299A (en) | 1994-01-25 | 1998-11-24 | Athena Neurosciences, Inc. | Humanized antibodies against leukocyte adhesion molecule VLA-4 |
| HU220799B1 (hu) | 1994-01-25 | 2002-05-28 | Elan Pharmaceuticals, Inc. | Humanizált antitestek VLA-4 leukocita adhéziós molekula ellen |
| US6551593B1 (en) * | 1995-02-10 | 2003-04-22 | Millennium Pharmaceuticals, Inc. | Treatment of Inflammatory bowel disease by inhibiting binding and/or signalling through α 4 β 7 and its ligands and madcam |
| US6037324A (en) * | 1996-01-04 | 2000-03-14 | Leukosite, Inc. | Inhibitors of MAdCAM-1-mediated interactions and methods of use therefor |
| US7147851B1 (en) * | 1996-08-15 | 2006-12-12 | Millennium Pharmaceuticals, Inc. | Humanized immunoglobulin reactive with α4β7 integrin |
-
1996
- 1996-08-15 US US08/700,737 patent/US7147851B1/en not_active Expired - Lifetime
-
1997
- 1997-08-06 CA CA2263106A patent/CA2263106C/en not_active Expired - Lifetime
- 1997-08-06 EP EP97937148.1A patent/EP0918797B2/en not_active Expired - Lifetime
- 1997-08-06 EP EP06006108A patent/EP1710314A1/en not_active Withdrawn
- 1997-08-06 IL IL12805297A patent/IL128052A0/xx unknown
- 1997-08-06 SI SI9730738T patent/SI0918797T2/sl unknown
- 1997-08-06 WO PCT/US1997/013884 patent/WO1998006248A2/en not_active Ceased
- 1997-08-06 NZ NZ334226A patent/NZ334226A/xx not_active IP Right Cessation
- 1997-08-06 AU AU39728/97A patent/AU730326C/en not_active Expired
- 1997-08-06 ES ES97937148.1T patent/ES2262186T5/es not_active Expired - Lifetime
- 1997-08-06 BR BRPI9711079-5A patent/BRPI9711079B1/pt not_active IP Right Cessation
- 1997-08-06 DK DK97937148.1T patent/DK0918797T4/en active
- 1997-08-06 PT PT97937148T patent/PT918797E/pt unknown
- 1997-08-06 DE DE69735596.9T patent/DE69735596T3/de not_active Expired - Lifetime
- 1997-08-06 JP JP50985398A patent/JP4171071B2/ja not_active Expired - Lifetime
- 1997-08-06 CN CNB971972214A patent/CN100406562C/zh not_active Expired - Lifetime
- 1997-08-06 AT AT97937148T patent/ATE321788T1/de active
-
1999
- 1999-01-14 IL IL175570A patent/IL175570A0/en unknown
- 1999-01-14 IL IL128052A patent/IL128052A/en active Protection Beyond IP Right Term
-
2006
- 2006-08-28 US US11/511,164 patent/US7402410B2/en not_active Expired - Fee Related
-
2008
- 2008-05-02 JP JP2008120608A patent/JP4578536B2/ja not_active Expired - Lifetime
- 2008-06-18 US US12/214,304 patent/US20090011464A1/en not_active Abandoned
-
2010
- 2010-05-24 JP JP2010118596A patent/JP5166483B2/ja not_active Expired - Lifetime
-
2014
- 2014-10-13 LT LTPA2014037C patent/LTC0918797I2/lt unknown
- 2014-11-12 BE BE2014C068C patent/BE2014C068I2/fr unknown
- 2014-11-12 LU LU92596C patent/LU92596I2/xx unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010246548A (ja) * | 1996-08-15 | 2010-11-04 | Millennium Pharmaceuticals Inc | α4β7インテグリンと反応するヒト化免疫グロブリン |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4171071B2 (ja) | α4β7インテグリンと反応するヒト化免疫グロブリン | |
| EP0678122B1 (en) | Recombinant anti-vla4 antibody molecules | |
| EP0460171B1 (en) | Cd3 specific recombinant antibody | |
| AU651623B2 (en) | Antibodies directed against CD3 | |
| US5869619A (en) | Modified antibody variable domains | |
| US5770196A (en) | Modified antibody variable domains and therapeutic uses thereof | |
| WO2001064754A1 (en) | Gene recombinant antibody and its fragment | |
| JP2004000249A (ja) | 抗体の調製 | |
| US20070122404A1 (en) | Humanized immunoglobulin reactive with alpha4beta7 integrin | |
| AU2005201265B2 (en) | Humanized immunoglobulin reactive with alpha4beta7 integrin | |
| AU778980B2 (en) | Humanized immunoglobulin reactive with alpha4beta7 integrin | |
| HK1021822B (en) | Humanized immunoglobulin reactive with alpha4beta7 integrin | |
| HK1021822C (en) | Humanized immunoglobulin reactive with alpha4beta7 integrin | |
| HK1097874A (en) | Humanized immunoglobulin reactive with alpha4beta7 integrin | |
| MXPA99001462A (es) | Inmunoglobulina humanizada reactiva con (alfa4beta7) integrina | |
| AU2008203508A1 (en) | Humanized immunoglobulin reactive with a4B7 integrin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040803 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20040803 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070515 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20070810 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20071001 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20070913 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20071029 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20071012 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20071119 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071113 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20080108 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080502 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20080710 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080724 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080808 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110815 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110815 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120815 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130815 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |