JP4411708B2 - ビス(アミノスチリル)ベンゼン化合物 - Google Patents
ビス(アミノスチリル)ベンゼン化合物 Download PDFInfo
- Publication number
- JP4411708B2 JP4411708B2 JP31206999A JP31206999A JP4411708B2 JP 4411708 B2 JP4411708 B2 JP 4411708B2 JP 31206999 A JP31206999 A JP 31206999A JP 31206999 A JP31206999 A JP 31206999A JP 4411708 B2 JP4411708 B2 JP 4411708B2
- Authority
- JP
- Japan
- Prior art keywords
- general formula
- embedded image
- mmol
- compound
- organic electroluminescent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 C*#CCCC1*=NC(C*)CC1 Chemical compound C*#CCCC1*=NC(C*)CC1 0.000 description 14
- XZFBOOCTMFZQMT-LRVMPXQBSA-N COc(cc1)ccc1N(c1ccc(/C=C/c(cc(c(/C=C\c(cc2)ccc2N(c(cc2)ccc2OC)c(c2c3cccc2)ccc3OC)c2)C#N)c2C#N)cc1)c(c1c2cccc1)ccc2OC Chemical compound COc(cc1)ccc1N(c1ccc(/C=C/c(cc(c(/C=C\c(cc2)ccc2N(c(cc2)ccc2OC)c(c2c3cccc2)ccc3OC)c2)C#N)c2C#N)cc1)c(c1c2cccc1)ccc2OC XZFBOOCTMFZQMT-LRVMPXQBSA-N 0.000 description 1
- INSWYKCFIHUDOV-VRONRDDVSA-N COc(cc1)ccc1N(c1ccc(/C=C\c(c(C#N)c(c(/C=C\c(cc2)ccc2N(c(cc2)ccc2[O](C)C)c(cc2)c(cccc3)c3c2OC)c2C#N)C#N)c2C#N)cc1)c(c1c2cccc1)ccc2OC Chemical compound COc(cc1)ccc1N(c1ccc(/C=C\c(c(C#N)c(c(/C=C\c(cc2)ccc2N(c(cc2)ccc2[O](C)C)c(cc2)c(cccc3)c3c2OC)c2C#N)C#N)c2C#N)cc1)c(c1c2cccc1)ccc2OC INSWYKCFIHUDOV-VRONRDDVSA-N 0.000 description 1
- JUXKSOLGLQEGJA-UHFFFAOYSA-N COc(cc1)ccc1N(c1ccc(C=O)cc1)c1c(cccc2)c2ccc1 Chemical compound COc(cc1)ccc1N(c1ccc(C=O)cc1)c1c(cccc2)c2ccc1 JUXKSOLGLQEGJA-UHFFFAOYSA-N 0.000 description 1
- OFUNCHHYACWSBL-UHFFFAOYSA-N Cc(cc1)ccc1N(c1ccccc1)c1ccc(C2OC=C2)cc1 Chemical compound Cc(cc1)ccc1N(c1ccccc1)c1ccc(C2OC=C2)cc1 OFUNCHHYACWSBL-UHFFFAOYSA-N 0.000 description 1
- IULUNTXBHHKFFR-UHFFFAOYSA-N Cc(cc1)ccc1N(c1ccccc1)c1ccccc1 Chemical compound Cc(cc1)ccc1N(c1ccccc1)c1ccccc1 IULUNTXBHHKFFR-UHFFFAOYSA-N 0.000 description 1
- URLKBWYHVLBVBO-UHFFFAOYSA-N Cc1ccc(C)cc1 Chemical compound Cc1ccc(C)cc1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 1
- SENGJJPSALDGMH-UHFFFAOYSA-N Oc(cc1)ccc1N(c1ccc(C=O)cc1)c1c(cccc2)c2ccc1 Chemical compound Oc(cc1)ccc1N(c1ccc(C=O)cc1)c1c(cccc2)c2ccc1 SENGJJPSALDGMH-UHFFFAOYSA-N 0.000 description 1
- DMBHHRLKUKUOEG-UHFFFAOYSA-N c(cc1)ccc1Nc1ccccc1 Chemical compound c(cc1)ccc1Nc1ccccc1 DMBHHRLKUKUOEG-UHFFFAOYSA-N 0.000 description 1
Images













































Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent materials, e.g. electroluminescent or chemiluminescent
- C09K11/06—Luminescent materials, e.g. electroluminescent or chemiluminescent containing organic luminescent materials
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/58—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/631—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/631—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine
- H10K85/633—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine comprising polycyclic condensed aromatic hydrocarbons as substituents on the nitrogen atom
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K2102/00—Constructional details relating to the organic devices covered by this subclass
- H10K2102/10—Transparent electrodes, e.g. using graphene
- H10K2102/101—Transparent electrodes, e.g. using graphene comprising transparent conductive oxides [TCO]
- H10K2102/103—Transparent electrodes, e.g. using graphene comprising transparent conductive oxides [TCO] comprising indium oxides, e.g. ITO
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/321—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3]
- H10K85/324—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3] comprising aluminium, e.g. Alq3
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Electroluminescent Light Sources (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
【発明の属する技術分野】
本発明は、所望の発光色を呈する有機発光材料として好適なビス(アミノスチリル)ベンゼン化合物に関するものである。
【0002】
【従来の技術】
自発光であって、応答速度が高速であり、視野角依存性の無いフラットパネルディスプレイの1候補として、有機電界発光素子(EL素子)等が近時注目されており、その構成材料として、有機発光材料への関心が高まっている。有機発光材料の第一の利点は、分子設計によって材料の光学的な性質をある程度コントロールできるところにあり、これによって赤、青、緑色の3原色発光をすべて各々の有機発光材料で作成したフルカラー有機発光素子の実現が可能である。
【0003】
下記一般式〔A〕で示されるビス(アミノスチリル)ベンゼン化合物は、導入される置換基に依存して、可視部領域に青〜赤の強い発光を呈することから、有機電界発光素子材料に限らず、さまざまな用途に利用可能である。さらに、これら材料は昇華性であり、真空蒸着等のプロセスによって、均一なアモルファス膜を形成しうる利点がある。今日では、分子軌道計算等により、材料の光学的な性質がある程度までは予測可能であるが、実際には要求される材料を高効率に製造する技術が産業上もっとも重要であることは、いうまでもない。
【0004】
【化167】
【0005】
【発明が解決しようとする課題】
これまで、有機発光材料として前記一般式〔A〕に属する多くの化合物が製造されてきたが、これらの材料の蛍光(発光)は多くが青〜緑色であり、黄色〜赤色の発光を呈するものはわずかに報告されているのみであり〔電気情報通信学会、技術研究報告、有機エレクトロニクス,17,7(1992)、Inorganic and Organic Electroluminescence 96 Berlin, 101(1996)等〕、またその高効率な製造法も確立されていなかった。
【0006】
本発明の目的は、上記のような現状に鑑み、強い発光を呈する特に黄色〜赤色の有機発光材料として好適な化合物を提供することにある。
【0007】
【課題を解決するための手段】
本発明者は、上記課題を解決するために鋭意検討した結果、一般式〔I〕、〔II〕、〔III〕又は〔IV〕で表されるビス(アミノスチリル)ベンゼン化合物に含まれる特定の化合物が強い発光を呈し、黄色〜赤色の発光材料となりうることを見い出し、本発明に到達したものである。
【0008】
即ち、本発明はまず、下記一般式〔I〕、〔II〕、〔III〕又は〔IV〕で表されるビス(アミノスチリル)ベンゼン化合物(以下、第1の化合物と称する。)に含まれる特定の化合物に係るものである。
【化168】
【化169】
【化170】
【化171】
【化172】
【化173】
【化174】
【化175】
【化176】
【0009】
上記の第1の化合物及び本発明の化合物は、黄色〜赤色の発光を示す有機発光材料として有効に利用することができ、また、高いガラス転移点及び融点を有する物質であり、電気的、熱的或いは化学的な安定性に優れている上、非晶質でガラス状態を容易に形成し得るので、蒸着等を行うこともできる。
【0010】
本発明の化合物は、下記一般式で表されるものである。
【化177】
【化178】
【0011】
本発明の第1の化合物は、より具体的には、下記一般式(9)、(10)、(11)、(12)、(13)、(14)、(15)又は(15’)で表されるものがよい。
【化179】
【化180】
【化181】
【化182】
【化183】
【化184】
【化185】
【0012】
本発明の第1の化合物は、下記構造式(16)−1、(16)−2、(16)−3、(16)−4、(16)−5、(16)−6、(16)−7、(16)−8、(16)−9、(16)−10、(16)−11、(16)−12又は(16)−13で表されるものが具体的に例示される。
【化186】
これら以外にも、次の化合物も例示することができる。
【化187】
【化188】
【化189】
【化190】
【化191】
【化192】
【化193】
【化194】
なお、上記した構造式(16)−8や(16)−9などの如く、t−ブチルなどといった嵩高い置換基の導入によって、次のような特性改善が図れる可能性がある。
▲1▼強固な分子間相互作用を弱めることにより、安定なアモルファス膜を実現する。
▲2▼ホールのホッピングサイト距離を離し、ホール輸送性をコントロールする。
【0022】
また、下記一般式〔XIX〕で示されるビス(アミノスチリル)ベンゼン化合物(以下、第2の化合物と称する。)が考えられる。
【化195】
【化196】
【0023】
この第2の化合物は、緑色〜赤色の発光を示す有機発光材料として有効に利用することができ、また、高いガラス転移点及び融点を有する物質であり、電気的、熱的或いは化学的な安定性に優れている上、非晶質でガラス状態を容易に形成し得るので、蒸着等を行なうこともできる。
【0024】
この第2の化合物は、下記一般式〔XX〕で示されるものが好ましい。
【化197】
【0025】
そして、前記R90、R91、R92及びR93は少なくとも一つが下記一般式(41)で表されるアリール基であり、残りが無置換のアリール基であるのがよい。
【化198】
【0026】
特に、下記一般式(42)で表されるものがよい。
【化199】
【0027】
この第2の化合物は、下記構造式(40)−1、(40)−2、(40)−3、(40)−4、(40)−5、(40)−6又は(40)−7で示されるものが具体的には例示される。
【化200】
本発明の第1の化合物を高効率に製造する方法として、下記一般式〔V〕又は〔VI〕で表される4−(N,N−ジアリールアミノ)ベンズアルデヒドの少なくとも1種と;下記一般式〔VII〕で表されるジホスホン酸エステルまたは下記一般式〔VIII〕で表されるジホスホニウム塩と;を縮合させることによって、前記一般式〔I〕、〔II〕、〔III〕又は〔IV〕で表されるビス(アミノスチリル)ベンゼン化合物を得る製造方法(以下、第1の製造方法と称する。)を採用するのがよい。
【0029】
【化201】
【化202】
【0030】
この第1の製造方法は、具体的には、前記縮合をウィッティヒ−ホーナー(Wittig-Horner)反応又はウィッティヒ(Wittig)反応によって行い、前記ジホスホン酸エステル及び/又は前記ジホスホニウム塩を溶媒中で塩基で処理することによってカルボアニオンを生成させ、このカルボアニオンと前記4−(N,N−ジアリールアミノ)ベンズアルデヒドと縮合させるものである。
【0031】
例えば下記一般式〔I’〕で表されるビス(アミノスチリル)ベンゼン化合物を得るに際し
【化203】
【化204】
【0032】
この縮合を反応スキームで示すと、例えば次の反応スキーム1のようになる。
【化205】
この反応はまず、一般式(19)又は(20)の化合物を適当な溶媒中で塩基と処理することにより、カルボアニオンを発生させることから始まり、次にこのカルボアニオンが一般式(17)のアルデヒドと縮合することにより完結する。塩基と溶媒の組み合わせとしては、以下のものが考えられる。
【0034】
水酸化ナトリウム/水、炭酸ナトリウム/水、炭酸カリウム/水、ナトリウムエトキシド/エタノールまたはジメチルホルムアミド、ナトリウムメトキシド/メタノール−ジエチルエーテル混合溶媒またはジメチルホルムアミド、トリエチルアミン/エタノールまたはジグライムまたはクロロホルムまたはニトロメタン、ピリジン/塩化メチレンまたはニトロメタン、1,5−ジアザビシクロ[4.3.0] ノン−5−エン/ジメチルスルホキシド、カリウムt−ブトキシド/ジメチルスルホキシドまたはテトラヒドロフラン、ブチルリチウム/ジエチルエーテルまたはテトラヒドロフランまたはベンゼンまたはジメチルホルムアミド、フェニルリチウム/ジエチルエーテルまたはテトラヒドロフラン、ナトリウムアミド/アンモニア、水素化ナトリウム/ジメチルホルムアミドまたはテトラヒドロフラン、トリチルナトリウム/ジエチルエーテルまたはテトラヒドロフラン等。
【0035】
この反応は比較的低温(−30℃〜30℃)で進行し、選択的であるため、クロマトグラフィーによる目的物の精製が容易であることに加え、一般式〔I’〕の本発明の第1の化合物は結晶性が高いため、再結晶により純度を向上させることができる。再結晶の方法については、特に問わないが、アセトンに溶解し、ヘキサンを添加する方法が簡便であり、その後の溶媒留去も容易である。この反応は常温〜30℃、常圧で3〜24時間で行ってよい。
【0036】
この第1の製造方法によって、前記一般式(10)、(11)、(12)、(13)、(14)又は(15)で表されるビス(アミノスチリル)ベンゼン化合物を得ることができ、具体的には、前記構造式(16)−1、(16)−2、(16)−3、(16)−4、(16)−5、(16)−6、(16)−7、(16)−8又は(16)−9で表されるビス(アミノスチリル)ベンゼン化合物を得ることができる。
【0037】
上記した第2の化合物を高効率に製造する方法として、下記一般式〔V’〕又は〔VI’〕で表される4−(N,N−ジアリールアミノ)ベンズアルデヒドの少なくとも1種と;下記一般式〔VII'〕で表されるジホスホン酸エステル又は下記一般式〔VIII’〕で表されるジホスホニウム塩と;を縮合させる、ビス(アミノスチリル)ベンゼン化合物の製造方法(以下、第2の製造方法と称する。)を採用するのがよい。
【化206】
【化207】
【0038】
この第2の製造方法は、具体的には、前記縮合をウィッティヒ−ホーナー(Wittig-Horner)反応又はウィッティヒ(Wittig)反応によって行い、前記ジホスホン酸エステル及び/又は前記ジホスホニウム塩を溶媒中で塩基で処理することによってカルボアニオンを生成させ、このカルボアニオンと前記4−(N,N−ジアリールアミノ)ベンズアルデヒドと縮合させるものである。
【0039】
この縮合を反応スキームで示すと、例えば次の反応スキーム1’のようになる。
【0040】
【化208】
この反応はまず、一般式(19’)又は(20’)の化合物を適当な溶媒中で塩基と処理することにより、カルボアニオンを発生させることから始まり、次にこのカルボアニオンが一般式〔V’〕のアルデヒドと縮合することにより完結する。塩基と溶媒の組み合わせとしては、上記と同様のものが考えられる。
【0042】
この反応は比較的低温(−30℃〜30℃)で進行し、選択的であるため、クロマトグラフィーによる目的物の精製が容易であることに加え、本発明の第2の化合物は結晶性が高いため、再結晶により純度を向上させることができる。再結晶の方法については、特に問わないが、アセトンに溶解し、ヘキサンを添加する方法が簡便であり、その後の溶媒留去も容易である。この反応は常温〜30℃、常圧で3〜24時間で行なってよい。
【0043】
この第2の製造方法によって、具体的には、前記構造式(40)−1、(40)−2、(40)−3、(40)−4、(40)−5、(40)−6又は(40)−7で表されるビス(アミノスチリル)ベンゼン化合物を得ることができる。
【0044】
本発明はまた、本発明の第1の化合物の合成中間体として好適な種々の化合物も提供するものである。
【0045】
即ち、まず、前記一般式〔V〕又は〔VI〕で表され、前記一般式〔I〕、〔II〕、〔III 〕又は〔IV〕で表されるビス(アミノスチリル)ベンゼン化合物の合成中間体として用いられる4−(N,N−ジアリールアミノ)ベンズアルデヒドである。
【0046】
この合成中間体(以下、合成中間体1と称する。)は、前記一般式(17)又は(18)で表され、具体的には下記一般式(21)、(22)、(23)、(24)、(25)、(26)又は(26’)で表され、下記構造式(27)−1、(27)−2、(27)−3、(27)−4、(27)−5、(27)−6、(27)−7、(27)−8、(27)−9、(27)−10、(27)−11、(27)−12、(27)−13、(27)−14、(27)−15として例示される。
【0047】
【化209】
【化210】
【化211】
【化212】
【0048】
【化213】
【0049】
【化214】
【化215】
【0050】
【化216】
上記した第2の化合物の合成中間体として好適な種々の化合物がある。
【0052】
即ち、前記一般式〔V’〕又は〔VI' 〕で表され、前記一般式〔XIX 〕で表されるビス(アミノスチリル)ベンゼン化合物の合成中間体として用いられる4−(N,N−ジアリールアミノ)ベンズアルデヒドである。
【0053】
この合成中間体(以下、合成中間体1’と称する。)は、具体的には下記一般式(43)で表され、下記構造式(41)−1、(41)−2、(41)−3、(41)−4、(41)−5、(41)−6又は(41)−7として例示される。
【0054】
【化217】
【0055】
【化218】
この合成中間体1又は1’は、その前駆体としての合成中間体から次のようにして導くことができる。
【0057】
下記一般式〔IX〕又は〔X〕、或いは下記一般式〔IX’〕又は〔X’〕で表され、前記一般式〔I〕、〔II〕、〔III〕又は〔IV〕で表されるビス(アミノスチリル)ベンゼン化合物の合成中間体として用いられるトリアリールアミン(以下、合成中間体2と称する。)、或いは前記一般式〔XIX〕で表されるビス(アミノスチリル)ベンゼン化合物の合成中間体として用いられるトリアリールアミン(以下、合成中間体2’と称する。)をジメチルホルムアミドとオキシ塩化リンとの付加体によってホルミル化し、前記ビス(アミノスチリル)ベンゼン化合物の合成中間体1又は1’としての前記一般式〔V〕又は〔VI〕、或いは〔V’〕又は〔VI’〕で表される4−(N,N−ジアリールアミノ)ベンズアルデヒドを得る。このホルミル化反応は、ジメチルホルムアミド中で室温(20℃)〜80℃の温度、常圧で3〜24時間で行うことができる。
【0058】
【化219】
【0059】
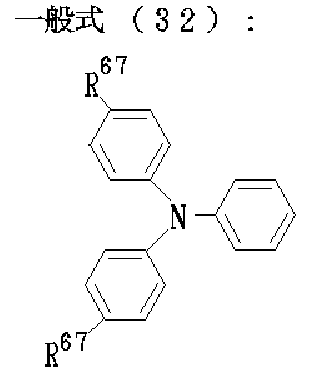
そして、上記の合成中間体2又は2’は、前記一般式〔IX〕又は〔X〕、或いは〔IX’〕又は〔X’〕で表され、具体的には、下記一般式(28)又は(29)で表され、更に具体的には下記一般式(30)、(31)、(32)、(33)、(34)又は(35)で表され、下記構造式(36)−1、(36)−2、(36)−3、(36)−4、(36)−5、(36)−6、(36)−7、(36)−8、(36)−9、(36)−10又は(36)−11として例示される。
【化220】
【0060】
【化221】
【化222】
【化223】
【0061】
【化224】
【0062】
【化225】
【0063】
【化226】
【0064】
【化227】
下記一般式〔IX〕又は〔X〕、〔IX’〕又は〔X’〕で表される合成中間体2又は2’は、次のようにして合成することができる。
【0066】
下記一般式〔XI〕又は〔XI’〕で表されるジアリールアミンと下記一般式〔XII 〕又は〔XII'〕で表されるハロゲン化ベンゼンとを触媒及び塩基の存在下でカップリングさせるか、或いは、下記一般式〔XIII〕又は〔XIII' 〕で表されるジアリールアミンと下記一般式〔XIV 〕又は〔XIV'〕で表されるハロゲン化アリール化合物とを触媒及び塩基の存在下でカップリングさせ、合成中間体2又は2’としてのトリアリールアミンを得る。
【0067】
【化228】
【0068】
この合成中間体2又は2’を合成する反応で用いられる前記触媒とは、Cu、CuX、CuX2、CuO、Pd(CH3COO)2、Pd(PR3)4(Rはフェニル基またはアルキル基)等であり、また前記塩基とは、K2CO3、Ca2CO3、NaOH、BuONa、PrONa、C2H5ONa、CH3ONa等である。また、この反応は、ジメチルホルムアミド、ジメチルスルホキシド、ニトロベンゼン、ジクロロベンゼン、キシレン等の溶媒中で、反応温度は100〜200℃、常圧で反応時間は2〜48時間としてよい。
【0069】
本発明では、上記した第1及び第2の化合物の合成中間体として、前記一般式〔VII〕又は〔VII’〕で表されるジホスホン酸エステルまたは前記一般式〔VIII〕又は〔VIII’〕で表されるジホスホニウム塩(以下、合成中間体3と称する。)を用いるのがよい。
【0070】
この合成中間体3は、具体的には下記一般式(19)又は(20)、或いは下記一般式(19’)又は(20’)で表される。
【0071】
【化229】
【0072】
この合成中間体3は、その前駆体としての合成中間体から次のようにして導くことができる。
【0073】
下記一般式〔XV〕又は〔XV’〕で表されるハロゲン化アリール化合物と、下記一般式〔XVI 〕で表される亜リン酸トリアルキル又はトリフェニルホスフィン(PPh3 )とを反応させることによって、前記一般式〔VII 〕又は〔VIII〕、〔VII'〕又は〔VIII’〕で表されるジホスホン酸エステル又はジホスホニウム塩を合成中間体3として得る。この反応は、無溶媒または過剰の亜リン酸トリアルキルまたはキシレン等の溶媒中で反応温度120〜160℃、常圧で反応時間30分〜12時間としてよい。
【0074】
【化230】
【0075】
一般式〔XVI 〕:
P(OR81)3 又は P(OR82)3
(但し、前記一般式〔XVI 〕において、R81及びR82はそれぞれ、同一の又は異なる炭化水素基、特に炭素数1〜4の飽和又は不飽和の炭化水素基であって、前記R75又はR76、R75’又はR76’に相当する基である。)
【0076】
合成中間体3を得るための合成中間体として、前記一般式〔XV〕又は〔XV’〕で表されるハロゲン化アリール化合物(以下、合成中間体4と称する。)を用いるのがよい。
【0077】
この合成中間体4は、下記一般式〔XVII〕又は〔XVII’〕で表されるキシレン化合物と、下記一般式〔XVIII〕で表されるN−ハロゲン化スクシンイミドとを光照射下に反応させることによって得ることができる。例えば、四塩化炭素、クロロホルム、ベンゼン等の溶媒中、高圧水銀灯、低圧水銀灯、キセノン灯、ハロゲン灯等の光源を用いて100〜500Wの光の照射下に、20〜60℃の温度、常圧で30分〜48時間の反応時間で反応させる。
【0078】
【化231】
【0079】
【化232】
【0080】
以上に述べた各合成中間体1〜4をそれぞれ得る反応は、次の反応スキーム2及び反応スキーム3又は3’で示すことができる。
【0081】
【化233】
本発明の化合物、及び第2の化合物の合成中間体として更に好適な化合物がある。
【0083】
即ち、この合成中間体は、下記一般式(44)、(45)又は(46)で表されるアセタール化合物(以下、合成中間体5と称する。)である。
【化234】
【化235】
【0084】
この合成中間体5を得るに際し、
前記一般式(44)で表されるアセタール化合物を得る場合には、触媒及び塩基の存在下で、下記一般式(48)で表されるアミン化合物と下記一般式(49)で表されるアセタール化合物とをカップリングさせ
【化236】
前記一般式(45)で表されるアセタール化合物を得る場合には、触媒及び塩基の存在下で、下記一般式(49’)で表されるアセタール化合物と下記一般式(50)で表されるアリール化合物とをカップリングさせ
【化237】
前記一般式(46)で表されるアセタール化合物を得る場合には、触媒及び塩基の存在下で、下記一般式(51)で表されるアミン化合物と下記一般式(52)で表されるアセタール化合物とをカップリングさせるのがよい。
【化238】
【0085】
前記カップリング時に用いる前記触媒として、下記のPd(O)−ホスフィン錯体が活性種として働くのがよい。
Pd(O)−ホスフィン錯体
{但し、Pd(O)はPd(O)、Pd(I)、Pd(II) の試薬として添加されてもよく、ホスフィンは下記一般式(53)又は(54)で表される3級ホスフィンである。
【化239】
一般式(55):
−(CH2)n −G −(CH2)n −
一般式(56):
−Ar15−G −Ar16−
(但し、前記一般式(55)及び(56)において、Gは酸素原子、硫黄原子、アミノ基、炭化水素基又は金属原子であり、Ar15及びAr16は置換基を有してもよいアリール基である。)〕}
【0086】
そして、この合成中間体5、即ち、下記一般式(44)、(45)又は(46)で表されるアセタール化合物を、ケトン溶媒中、酸触媒又は塩基触媒の存在下でアセタール交換させることにより、下記一般式(57)、(58)又は(59)で表される4−(N,N−ジアリールアミノ)ベンズアルデヒド化合物を得ることが有利である。
【化240】
【化241】
【化242】
【0087】
ここで、例えば上記の一般式(44)の化合物としては、具体的には以下のものが挙げられる。
【化243】
また、上記の酸触媒と溶媒の組み合わせとして、硫酸/メタノール、塩酸/メタノール、DCC−SnC14 /しゅう酸/アルコール、トリフルオロ酢酸/ジメトキシメタン/ニトロメタン、p−トルエンスルホン酸/ジメトキシメタン/メタノール、塩酸/テトラメトキシシラン/メタノール、p−トルエンスルホン酸/アセトン、トリフルオロ酢酸/クロロフォルム/水、テトラクロロチタン/沃化リチウム/ジエチルエーテル、酢酸/水、蟻酸/ペンタン、酢酸/亜鉛−銀/テトラヒドロフラン、p−トルエンスルホン酸ピリジニウム/アセトン−水、シリカゲル/水−塩化メチレン等が挙げられる。
【0089】
また、上記のカップリング用の触媒については、パラジウム触媒〔Pd(CH3 COO)2〕を使用することにより、カップリングの収率を向上させることができるが、更に反応性の低い系について下記の触媒系を適用したところ、収率が改善された(但し、下記の触媒系は、後述の実施例では用いていない)。
【化244】
パラジウムは反応系中でPd(O)になっていればよく、Pd(CH3 COO)2 −PPh4 の系では、PPh4 によりPd(II)が還元されて、Pd(O)が発生していると思われる(但し、ホスフィンは、リンの周りが嵩高く、C−P−Cの2面角が、大きいほうが良いとされている。上記のようなPd(O)−ホスフィンの反応系中の活性種の具体的な構造については良く分かっていない)。
【0091】
パラジウム触媒系の典型的な組み合わせは、Pd錯体、3級ホスフィン、塩基、キシレン溶媒、還流2〜10時間である。
【0092】
前記一般式(57)又は(58)のベンズアルデヒドは、上述した合成中間体1又は1’と同様、具体的には、前記した構造式(27)−1、(27)−2、(27)−3、(27)−4、(27)−5、(27)−6、(27)−7、(27)−8、(27)−9、(27)−10又は(27)−11で表されるものであってよい。
【0093】
上述した一般式〔V〕又は〔VI〕、或いは〔V’〕又は〔VI’〕で表される本発明の合成中間体1又は1’を得るには、合成中間体2又は2’である3級アミンをアルデヒドに変換するために、オキシ塩化リン(POCl3 )−ジメチルホルムアミド(DMF)付加体を用いたが、この方法では例えば下記の如き問題点が生じる場合がある。
【0094】
3級アミンがシアノ基などの置換基を有していて電子吸引性を帯びているとき、オキシ塩化リン−ジメチルホルムアミド付加体が反応しにくい。
【化245】
オキシ塩化リン−ジメチルホルムアミド付加体による方法では、活性部位が2点以上あるとき、反応の位置選択性が低い。
【化246】
そこで、前記一般式(49)、(49’)又は(52)のアセタール化合物を用い、これを一般式(48)、(50)又は(51)の化合物と反応させることによって、位置選択性良く容易に目的とするアルデヒドを合成することができる。この反応を含む本発明の化合物の合成方法を反応スキームで示すと、例えば次の反応スキーム4、5、6の如くとなる。
【0097】
(1)ビス(アミノスチリル)ベンゼン化合物(構造式(16)−9)の合成
【化247】
(2)ビス(アミノスチリル)ベンゼン化合物(構造式(16)−8)の合成
【化248】
(3)ビス(アミノスチリル)ベンゼン化合物(構造式(16)−3)の合成
【化249】
図45〜図48は、本発明の化合物を有機発光材料として用いる有機電界発光素子(EL素子)の例をそれぞれ示すものである。
【0101】
図45は陰極3を発光光20が透過する透過型有機電界発光素子Aであって、発光20は保護層4の側からも観測できる。図46は陰極3での反射光も発光光20として得る反射型有機電界発光素子Bを示す。
【0102】
図中、1は有機電界発光素子を形成するための基板であり、ガラス、プラスチック及び他の適宜の材料を用いることができる。また、有機電界発光素子を他の表示素子と組み合わせて用いる場合には、基板を共用することもできる。2は透明電極(陽極)であり、ITO(Indium tin oxide)、SnO2 等を使用できる。
【0103】
また、5は有機発光層であり、本発明の化合物を発光材料として含有している。この発光層について、有機電界発光20を得る層構成としては、従来公知の種々の構成を用いることができる。後述するように、例えば、正孔輸送層と電子輸送層のいずれかを構成する材料が発光性を有する場合、これらの薄膜を積層した構造を使用できる。更に本発明の目的を満たす範囲で電荷輸送性能を上げるために、正孔輸送層と電子輸送層のいずれか若しくは両方が、複数種の材料の薄膜を積層した構造、または、複数種の材料を混合した組成からなる薄膜を使用するのを妨げない。また、発光性能を上げるために、少なくとも1種以上の蛍光性の材料を用いて、この薄膜を正孔輸送層と電子輸送層の間に挟持した構造、更に少なくとも1種以上の蛍光性の材料を正孔輸送層若しくは電子輸送層、またはこれらの両方に含ませた構造を使用しても良い。これらの場合には、発光効率を改善するために、正孔または電子の輸送を制御するための薄膜をその層構成に含ませることも可能である。
【0104】
本発明の化合物が、電子輸送性能と正孔輸送性能の両方を持つ場合、素子構成中、電子輸送層を兼ねた発光層としても、或いは正孔輸送層を兼ねた発光層としても用いることが可能である。また、本発明の化合物を発光層として、電子輸送層と正孔輸送層とで挟み込んだ構成とすることも可能である。
【0105】
なお、図45及び図46中、3は陰極であり、電極材料としては、Li、Mg、Ca等の活性な金属とAg、Al、In等の金属との合金、或いはこれらを積層した構造を使用できる。透過型の有機電界発光素子においては、陰極の厚さを調節することにより、用途に合った光透過率を得ることができる。また、図中、4は封止・保護層であり、有機電界発光素子全体を覆う構造とすることにより、その効果が上がる。気密性が保たれれば、適宜の材料を使用することができる。また、8は電流注入用の駆動電源である。
【0106】
この有機電界発光素子において、有機層が、正孔輸送層と電子輸送層とが積層された有機積層構造(シングルヘテロ構造)を有しており、正孔輸送層又は電子輸送層の形成材料として本発明の化合物が用いられてよい。或いは、有機層が、正孔輸送層と発光層と電子輸送層とが順次積層された有機積層構造(ダブルヘテロ構造)を有しており、発光層の形成材料として本発明の化合物が用いられてよい。
【0107】
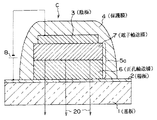
このような有機積層構造を有する有機電界発光素子の例を示すと、図47は、透光性の基板1上に、透光性の陽極2と、正孔輸送層6と電子輸送層7とからなる有機層5aと、陰極3とが順次積層された積層構造を有し、この積層構造が保護膜4によって封止されてなる、シングルヘテロ構造の有機電界発光素子Cである。
【0108】
図47に示すように発光層を省略した層構成の場合には、正孔輸送層6と電子輸送層7の界面から所定波長の発光20を発生する。これらの発光は基板1側から観測される。
【0109】
また、図48は、透光性の基板1上に、透光性の陽極2と、正孔輸送層10と発光層11と電子輸送層12とからなる有機層5bと、陰極3とが順次積層された積層構造を有し、この積層構造が保護膜4によって封止されてなる、ダブルヘテロ構造の有機電界発光素子Dである。
【0110】
図48に示した有機電界発光素子においては、陽極2と陰極3の間に直流電圧を印加することにより、陽極2から注入された正孔が正孔輸送層10を経て、また陰極3から注入された電子が電子輸送層12を経て、それぞれ発光層11に到達する。この結果、発光層11においては電子/正孔の再結合が生じて一重項励起子が生成し、この一重項励起子から所定波長の発光を発生する。
【0111】
上述した各有機電界発光素子C、Dにおいて、基板1は、例えば、ガラス、プラスチック等の光透過性の材料を適宜用いることができる。また、他の表示素子と組み合わせて用いる場合や、図47及び図48に示した積層構造をマトリックス状に配置する場合等は、この基板を共用としてよい。、また、素子C、Dはいずれも、透過型、反射型のいずれの構造も採りうる。
【0112】
また、陽極2は、透明電極であり、ITO(indium tin oxide)やSnO2 等が使用できる。この陽極2と正孔輸送層6(又は正孔輸送層10)との間には、電荷の注入効率を改善する目的で、有機物若しくは有機金属化合物からなる薄膜を設けてもよい。なお、保護膜4が金属等の導電性材料で形成されている場合は、陽極2の側面に絶縁膜が設けられていてもよい。
【0113】
また、有機電界発光素子Cにおける有機層5aは、正孔輸送層6と電子輸送層7とが積層された有機層であり、これらのいずれか又は双方に上記した本発明の化合物が含有され、発光性の正孔輸送層6又は電子輸送層7としてよい。有機電界発光素子Dにおける有機層5bは、正孔輸送層10と上記した本発明の化合物を含有する発光層11と電子輸送層12とが積層された有機層であるが、その他、種々の積層構造を取ることができる。例えば、正孔輸送層と電子輸送層のいずれか若しくは両方が発光性を有していてもよい。
【0114】
また、特に、正孔輸送層6又は電子輸送層7や発光層11が本発明の化合物からなる層であることが望ましいが、これらの層を本発明の化合物のみで形成してもよく、或いは、本発明の化合物と他の正孔又は電子輸送材料(例えば、芳香族アミン類やピラゾリン類等)との共蒸着によって形成してもよい。さらに、正孔輸送層において、正孔輸送性能を向上させるために、複数種の正孔輸送材料を積層した正孔輸送層を形成してもよい。
【0115】
また、有機電界発光素子Cにおいて、発光層は電子輸送性発光層7であってよいが、電源8から印加される電圧によっては、正孔輸送層6やその界面で発光される場合がある。同様に、有機電界発光素子Dにおいて、発光層は層11以外に、電子輸送層12であってもよく、正孔輸送層10であってもよい。発光性能を向上させるために、少なくとも1種の蛍光性材料を用いた発光層11を正孔輸送層と電子輸送層との間に挟持させた構造であるのがよい。または、この蛍光性材料を正孔輸送層又は電子輸送層、或いはこれら両層に含有させた構造を構成してよい。このような場合、発光効率を改善するために、正孔又は電子の輸送を制御するための薄膜(ホールブロッキング層やエキシトン生成層など)をその層構成に含ませることも可能である。
【0116】
また、陰極3に用いる材料としては、Li、Mg、Ca等の活性な金属とAg、Al、In等の金属との合金を使用でき、これらの金属層が積層した構造であってもよい。なお、陰極の厚みや材質を適宜選択することによって、用途に見合った有機電界発光素子を作製できる。
【0117】
また、保護膜4は、封止膜として作用するものであり、有機電界発光素子全体を覆う構造とすることで、電荷注入効率や発光効率を向上できる。なお、その気密性が保たれれば、アルミニウム、金、クロム等の単金属又は合金など、適宜その材料を選択できる。
【0118】
上記した各有機電界発光素子に印加する電流は通常、直流であるが、パルス電流や交流を用いてもよい。電流値、電圧値は、素子破壊しない範囲内であれば特に制限はないが、有機電界発光素子の消費電力や寿命を考慮すると、なるべく小さい電気エネルギーで効率良く発光させることが望ましい。
【0119】
次に、図49は、有機電界発光素子を用いた平面ディスプレイの構成例である。図示の如く、例えばフルカラーディスプレイの場合は、赤(R)、緑(G)及び青(B)の3原色を発光可能な有機層5(5a、5b)が、陰極3と陽極2との間に配されている。陰極3及び陽極2は、互いに交差するストライプ状に設けることができ、輝度信号回路14及びシフトレジスタ内蔵の制御回路15により選択されて、それぞれに信号電圧が印加され、これによって、選択された陰極3及び陽極2が交差する位置(画素)の有機層が発光するように構成される。
【0120】
即ち、図49は例えば8×3RGB単純マトリックスであって、正孔輸送層と、発光層および電子輸送層のいずれか少なくとも一方とからなる積層体5を陰極3と陽極2の間に配置したものである(図47又は図48参照)。陰極と陽極は、ともにストライプ状にパターニングするとともに、互いにマトリクス状に直交させ、シフトレジスタ内蔵の制御回路15および14により時系列的に信号電圧を印加し、その交叉位置で発光するように構成されたものである。かかる構成のEL素子は、文字・記号等のディスプレイとしては勿論、画像再生装置としても使用できる。また陰極3と陽極2のストライプ状パターンを赤(R)、緑(G)、青(B)の各色毎に配し、マルチカラーあるいはフルカラーの全固体型フラットパネルディスプレイを構成することが可能となる。
【0121】
【実施例】
以下、本発明を実施例について具体的に説明するが、本発明は以下の実施例に限定されるものではない。
【0122】
実施例1
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−1)の合成例>
【0123】
【化250】
2,5−ジ(ブロモメチル)−テレフタロニトリル750mg(2.39mmol)に亜リン酸トリエチル794mg(4.78mmol)を滴下後、125℃で30分攪拌し、ジホスホン酸エステル(19a)を得た。反応によって生じた臭化エチルを留去し、無水テトラヒドロフラン(THF)25mlに溶解して保存した。
【0125】
水素化ナトリウム18.5mmolを無水テトラヒドロフラン70mlに懸濁させ、窒素雰囲気下で上記で得られたジホスホン酸エステル(19a)の無水テトラヒドロフラン溶液(2.39mmol相当)を15分かけて滴下し、その後に室温で20分攪拌した。
【0126】
次に、4−[ N−フェニル−N−(4−エトキシフェニル)アミノ] ベンズアルデヒド((27)−1)1.78g(5.60mmol)の無水テトラヒドロフラン溶液(40ml)を15分かけて滴下した後、室温で2時間30分攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウムで乾燥した。
【0127】
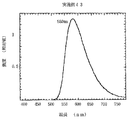
この反応混合物から目的物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:8)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−1)900mgを得た。この収率は51%、ガラス転移点は140℃、融点は180℃であった。テトラヒドロフラン溶液の可視吸収極大は475nm、蛍光極大波長は590nmであった。また、その1 HNMRスペクトルは図1と下記に示す通りであった(なお、図中のTMSは1 HNMRスペクトル測定時に添加する基準物質であるトリメチルシランのピーク;以下、同様)。
NMR(CDC13) δ(ppm):1.32(6H,t),4.03(4H,q),6.83(4H,d),6.98〜7.22(22H,m),7.40(4H,d),7.98(2H,s)
【0128】
実施例2
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−1)の合成例>
【化251】
2,5−ジ(ブロモメチル)−テレフタロニトリル750mg(2.39mmol)とトリフェニルホスフィン1.38g(5.26mmol)をキシレンに溶解し、20時間還流した。反応溶液を室温まで冷却し、生じた沈澱物をろ別し、キシレン5mlで洗った後、減圧乾燥し、無水テトラヒドロフラン25mlに溶解して保存した。
【0130】
水素化ナトリウム18.5mmolを無水テトラヒドロフラン70mlに懸濁させ、窒素雰囲気下で上記で得られたジホスホニウム(20a)の無水テトラヒドロフラン溶液(2.39mmol相当)を15分かけて滴下し、その後室温で48時間攪拌した。
【0131】
次に、4−[ N−フェニル−N−(4−エトキシフェニル)アミノ] ベンズアルデヒド((27)−1)1.78g(5.60mmol)の無水テトラヒドロフラン溶液(40ml)を15分かけて滴下した後、室温で2時間30分攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウムで乾燥した。
【0132】
シリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:8)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−1)558mgを得た。この収率は31%であり、諸物性値は、実施例1で得られたビス(アミノスチリル)ベンゼン化合物((16)−1)と一致した。
【0133】
実施例3
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−2)の合成例>
【0134】
【化252】
水素化ナトリウム11.3mmolを無水テトラヒドロフラン20mlに懸濁させ、窒素雰囲気下で実施例1で得られたジホスホン酸エステル(19a)の無水テトラヒドロフラン溶液(1.13mmol相当)を15分かけて滴下し、その後に室温で20分攪拌した。
【0136】
次に、4−[ N,N−ジ(4−メトキシフェニル)アミノ] ベンズアルデヒド((27)−2)750mg(2.25mmol)の無水テトラヒドロフラン溶液(40ml)を15分かけて滴下した後、室温で1時間攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウムで乾燥した。
【0137】
この反応混合物から目的物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:8)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−2)488mgを得た。この収率は31%、ガラス転移点は130℃、融点は170℃であった。テトラヒドロフラン溶液の可視吸収極大は486nm、蛍光極大波長は620nmであった。また、その1 HNMRスペクトルは図2及び下記に示す通りであった。
NMR(CDC13) δ(ppm):3.81(12H,s),6.84(12H,m),7.05(8H,d),7.19(2H,d),7.39(4H,d),7.98(2H,s)
【0138】
実施例4
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−4)の合成例>
【0139】
【化253】
水素化ナトリウム11.3mmolを無水テトラヒドロフラン20mlに懸濁させ、窒素雰囲気下で実施例1で得られたジホスホン酸エステル(19a)の無水テトラヒドロフラン溶液(1.13mmol相当)を15分かけて滴下し、その後に室温で20分攪拌した。
【0141】
次に、4−[ N−(1−ナフチル)−N−フェニルアミノ] ベンズアルデヒド((27)−4)728mg(2.25mmol)の無水テトラヒドロフラン溶液(12ml)を15分かけて滴下した後、室温で2時間攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウムで乾燥した。
【0142】
この反応混合物から目的物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:8)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−4)546mgを得た。この収率は63%、ガラス転移点は150℃、融点は210℃であった。テトラヒドロフラン溶液の可視吸収極大は461nm、蛍光極大波長は550nmであった。また、その1 HNMRスペクトルは図3及び下記に示す通りであった。
NMR(CDC13) δ(ppm):6.97(4H,d),7.02(2H,s),7.25〜7.49(26H,m),7.81(2H,d),7.92(4H,d),7.97(2H,s)
【0143】
実施例5
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−5)の合成例>
【0144】
【化254】
水素化ナトリウム11.3mmolを無水テトラヒドロフラン20mlに懸濁させ、窒素雰囲気下で実施例1で得られたジホスホン酸エステル(19a)の無水テトラヒドロフラン溶液(1.13mmol相当)を15分かけて滴下し、その後に室温で20分攪拌した。
【0146】
次に、4−[ N−(1−ナフチル)−N−(4−メトキシフェニル)アミノ] ベンズアルデヒド((27)−5)761mg(2.25mmol)の無水テトラヒドロフラン溶液(12ml)を15分かけて滴下した後、室温で2時間攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウムで乾燥した。
【0147】
この反応混合物から目的物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:8)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−5)386mgを得た。この収率は43%、ガラス転移点は130℃、融点は190℃であった。テトラヒドロフラン溶液の可視吸収極大は465nm、蛍光極大波長は555nmであった。
【0148】
実施例6
<4−[ N,N−ジ(4−メトキシフェニル)アミノ] ベンズアルデヒド(構造式(27)−2)の合成例>
【0149】
【化255】
室温にて無水ジメチルホルムアミド中での攪拌下、オキシ塩化リン1.76g(11.5mmol)を滴下し、続いてN,N−ジ(4−メトキシフェニル)アニリン((36)−2)1.75gの無水ジメチルホルムアミド溶液25mlを滴下した後、反応温度を上げ、70℃で90分攪拌した。
【0151】
室温まで冷却し、氷でクエンチした後、反応溶液をトルエンにて抽出し、飽和食塩水で洗い、無水硫酸ナトリウム上で乾燥した。
【0152】
この反応生成物から目的物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4)により精製し、化合物((27)−2)0.750gを得た。この収率は39%であり、また、その1 HNMRスペクトルは図4及び下記に示す通りであった。
NMR(CDCl3) δ(ppm):3.81(6H,s),6.82(2H,d),6.90(4H,d),7.13(4H,d),7.62(2H,d),9.78(1H,s)
【0153】
実施例7
<N,N−ジ(4−メトキシフェニル)アニリン(構造式(36)−2)の合成例>
【0154】
【化256】
N,N−ジ(4−メトキシフェニル)アミン(31a)1.00g(4.46mmol)、ヨードベンゼン(32a)1.00g(4.90mmol)、t−BuONa0.502g(5.23mmol)およびPd(CH3 COO)2 0.010g(0.044mmol)を無水キシレンに溶解し、窒素雰囲気下で還流しながら、さらに0.237MのP(But )3 1.0mlを滴下した後、4時間還流した。
【0156】
この反応生成物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4)により精製し、溶出液をアセトン−ヘキサンから再結晶して化合物((36)−2)を得た。この収量は1.17g(収率88%)であった。また、その1 HNMRスペクトルは図5及び下記に示す通りであった。
NMR(CDCl3) δ(ppm):3.80(6H,s),6.80(4H,d),6.82(1H,t),6.92(2H,d),7.02(4H,d),7.17(2H,t)
【0157】
実施例8
<N−(1−フェニル)−N−(4−エトキシフェニル)アニリン(構造式(36)−1)の合成例>
【化257】
N,N−ジフェニルアミン(31a)8.20g(50mmol)、ヨードアニソール(32a)12.40g(50mmol)、t−BuONa5.76g(60mmol)およびPd(CH3 COO)2 0.224g(1.00mmol)をジクロロベンゼンに溶解し、窒素雰囲気下で還流しながら、さらに0.237MのP(But )3 17mlを滴下した後、4時間還流した。
【0159】
カラムクロマトグラフィー(アルミナ、ヘキサン:トルエン=4:1)により精製し、溶出液をアセトン−ヘキサンから再結晶して目的物を得た。この収量は10.9g(収率79%)であり、1 HNMRスペクトルを図6と下記に示す。
NMR(CDCl3) δ(ppm):2.28(3H,t),4.02(2H,q),6.84(2H,d),6.94(2H,t),7.03(4H,d),7.06(2H,d),7.20(4H,t)
【0160】
実施例9
<2,5−ジ(ブロモトリフェニルホスホメチル)テレフタロニトリル(構造式(20a))の合成例>
【化258】
2,5−ジ(ブロモメチル)テレフタロニトリル(35a)750mg(2.39mmol)とトリフェニルホスフィン1.38g(5.26mmol)をキシレンに溶解し、20時間還流した。反応溶液を室温まで冷却し、生じた沈澱物をろ別し、キシレン5mlで洗った後、減圧乾燥し、無水テトラヒドロフラン25mlに溶解して保存した。これによって、実施例2で述べたジホスホニウム(20a)が得られた。
【0162】
実施例10
<2,5−ジ(ブロモメチル)テレフタロニトリル(構造式(35a))の合成例>
【0163】
【化259】
2,5−ジメチルテレフタロニトリル(36a)1.00g(6.4mmol)とN−ブロモスクシンイミド(NBS)(37a)8.10g(90mmol)を500mlのクロロホルムに溶かし、高圧水銀灯(400W)を照射しながら48時間還流した。
【0165】
溶媒を留去し、反応生成物をシリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4)により精製し、得られた溶出液をアセトン−ヘキサンから2度再結晶することにより、化合物(35a)が白色結晶として選択的に得られた。この収量は698mg(収率34%)であり、また、その1 HNMRスペクトルは図7及び下記に示す通りであった。
NMR(CDCl3)δ(ppm):4.60(4H,s),7.83(2H,s)
【0166】
実施例11
<N−(p−トルイル)−N,N−ジフェニルアミン(構造式(36)−6)の合成例>
【0167】
【化260】
N,N−ジフェニルアミン(31a)9.70g(57.3mmol)、4−ヨードトルエン(32b)12.5g(57.3mmol)、t−BuONa6.61g(68.8mmol)、Pd(CH3 COO)2 260mg(1.15mmol)、およびトリフェニルホスフィン1.20g(4.58mmol)をキシレンに溶解し、窒素雰囲気下で4時間還流した。
【0169】
不溶物をろ別し、アルミナクロマトグラフィー(中性アルミナ300メッシュ、テトラヒドロフラン:ヘキサン=1:4)により精製し、溶出液をアセトン−ヘキサンから再結晶して目的物((36)−6)を定量的に得た。この生成物((36)−6)の1 HNMRスペクトルを図8及び下記に示す。
NMR(CDCl3)δ(ppm):2.31(3H,s),6.94〜7.27(14H,m)
【0170】
実施例12
<4−[ N−(p−トルイル)−N−フェニルアミノ] ベンズアルデヒド(構造式(27)−6)の合成例>
【0171】
【化261】
室温にて攪拌下、無水ジメチルホルムアミド(DMF)50ml中にオキシ塩化リン5.96g(38.9mmol)を滴下し、続いてN−(p−トルイル)−N,N−ジフェニルアミン((36)−6)5.04g(19.4mmol)の無水ジメチルホルムアミド(DMF)溶液50mlを滴下した後、反応温度を上げ、70℃で90分攪拌した。
【0173】
室温まで冷却し、反応混合液を少量の氷でクエンチし、シリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4)により精製し、オイル状物質((27)−6)をほぼ定量的に得た。この生成物の1 HNMRスペクトルを図9と下記に示す。
NMR(CDCl3)δ(ppm):2.35(3H,s),6.96〜7.64(11H,m),7.66(2H,d),9.80(1H,s)
【0174】
実施例13
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−6)の合成例>
【0175】
【化262】
水素化ナトリウム14.5mmolを無水テトラヒドロフラン(THF)20mlに懸濁させ、窒素雰囲気下でジホスホン酸エステル(19a)の無水テトラヒドロフラン溶液(2.33mmol相当)を滴下し、60分攪拌した。次に、4−[ N−(p−トルイル)−N−フェニルアミノ] ベンズアルデヒド((27)−6))1.34g(4.66mmol)の無水テトラヒドロフラン溶液(40ml)を滴下した後、室温で12時間攪拌した。
【0177】
反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウム上で乾燥した。シリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4→1:1)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−6)0.787gを得た。この収率は49%であり、1 HNMRスペクトルを図10と下記に示す。
NMR(CDCl3)δ(ppm):2.34(6H,s),7.01〜7.30(26H,m),7.42(4H,d),7.99(2H,s)
【0178】
この物質((16)−6)のテトラヒドロフラン溶液の可視吸収極大は469nm、蛍光極大波長は568nmであった。
【0179】
実施例14
<N,N−ジ(p−トルイル−N−フェニルアミン(構造式(36)−7)の合成例>
【0180】
【化263】
N,N−ジ(p−トルイル)アミン(31b)10.0g(50.7mmol)、4−ヨードベンゼン10.3g(50.7mmol)、t−BuONa5.85g(60.8mmol)、Pd(CH3 COO)2 300mg(1.34mmol)およびトリフェニルホスフィン1.50g(5.71mmol)をキシレン500mlに溶解し、窒素雰囲気下で4時間還流した。
【0182】
不溶物をろ別し、アルミナクロマトグラフィー(中性アルミナ300メッシュ、テトラヒドロフラン:ヘキサン=1:4)により精製し、溶出液をアセトン−ヘキサンから再結晶して目的物((36)−7)を定量的に得た。この生成物の1 HNMRスペクトルを図11と下記に示す。
NMR(CDCl3)δ(ppm):2.30(6H,s),6.90〜7.07(11H,m),7.16〜7.22(2H,m)
【0183】
実施例15
<4−[ N,N−ジ(p−トルイル)アミノ] ベンズアルデヒド(構造式(27)−7)の合成例>
【0184】
【化264】
室温にて攪拌下、無水ジメチルホルムアミド(DMF)20ml中にオキシ塩化リン5.90g(38.4mmol)を滴下し、続いてN,N−ジ(p−トルイル)−N−フェニルアミン((36)−7)7.00g(25.6mmol)の無水ジメチルホルムアミド溶液50mlを滴下した後、室温で24時間攪拌した。
【0186】
反応混合液を少量の氷でクエンチした後、トルエンで抽出し、飽和食塩水で洗い、Na2 SO4 上で乾燥した。シリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4)により精製し、オイル状物質((27)−7)をほぼ定量的に得た。この生成物の1 HNMRスペクトルを図12と下記に示す。
NMR(CDCl3)δ(ppm):2.35(6H,s),6.93(2H,d),7.06(4H,d),7.15(4H,d),7.64(4H,d),9.78(1H,s)
【0187】
実施例16
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−7)の合成例>
【0188】
【化265】
水素化ナトリウム14.3mmolを無水テトラヒドロフラン(THF)20mlに懸濁させ、窒素雰囲気下でジホスホン酸エステル(19a)750mg(2.39mmol)の無水テトラヒドロフラン溶液20mlを滴下し、次に4−[ N,N−ジ(p−トルイル)アミノ] ベンズアルデヒド((27)−7)(2.39mmol相当)25mlの無水テトラヒドロフラン溶液を滴下した後、室温で48時間攪拌した。
【0190】
反応混合液を少量の氷でクエンチし、飽和食塩水で洗い、無水硫酸ナトリウム上で乾燥した。シリカゲルクロマトグラフィー(WAKO−gel C−300,テトラヒドロフラン:ヘキサン=1:4→1:1)により精製し、アセトン−ヘキサンから再結晶することにより、ビス(アミノスチリル)ベンゼン化合物((16)−7)431mgを得た。この収率は25%であり、1 HNMRスペクトルを図13と下記に示す。
NMR(CDCl3)δ(ppm):2.33(6H,s),6.97〜7.21(24H,m),7.39(4H,d),7.97(2H,s)
【0191】
この物質のテトラヒドロフラン溶液の可視吸収極大は476nm、蛍光極大波長は590nmであった。
【0192】
実施例17
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−9)の合成例>
【化266】
(1)中間体(53)の合成
アセタール化合物(51)2.75g(10.2mmol)、アニリン(52)20ml(215mmol)、t−BuONa1.00g(10.4mmol)及びPd(OAc)2 0.022g(0.047mmol)をキシレン150mlに溶解し、窒素雰囲気下で還流しながら、さらに0.1MのP(t−Bu)3 2.0ml(0.20mmol)を滴下した後、6時間還流した。アルミナカラムクロマトグラフィー(200メッシュ、トルエン:THF=1:1)により原料物質を除き、減圧蒸留にて過剰のアニリンを留去して、中間体(53)を定量的に得た。
【0194】
この生成物は、 1HNMR及びFAB−MS測定により目的物(53)と同定した。
1HNMR(CDCl3)δ(ppm):0.80(3H,s),1.30(3H,s),3.63(2H,d),3.76(2H,d),5.34(1H,s),5.74(1H,brs),6.92(1H,t),7.06(4H,d),7.26(2H,t),7.39(2H,d)
中間体(53)の 1HNMRスペクトルを図14に示す。
【0195】
(2)中間体(55)の合成
化合物(54)5.00g(28.9mmol)を50mlのCHCl3 に溶解し、0℃に冷却して窒素雰囲気下で攪拌しつつ0.2mlのCF3 SO3 Hを添加した後、イソブチレンガスを3時間かけてゆっくりとバブリングさせた。6mlのNEt3 を添加して反応溶液を中和し、ドライアルミナ(300メッシュ、トルエン)を通して原料物質を除き、溶媒を留去して化合物(55)を定量的に得た。
【0196】
この生成物は、 1HNMR及びFAB−MS測定により、目的物(55)と同定した。
1HNMR(CDCl3)δ(ppm):1.33(9H,s),6.86(2H,d),7.37(2H,d)
中間体(55)の 1HNMRスペクトルを図15に示す。
【0197】
(3)中間体(56)の合成
化合物(55)0.809g(3.53mmol)、化合物(53)1.00g(3.53mmol)、t−BuONa0.407g(4.24mmol)、及びPd(OAc)2 7.9mg(0.035mmol)を100mlのキシレンに懸濁させ、窒素雰囲気下、120℃で還流攪拌しつつ、0.1MのP(t−Bu)3 1.4mlを添加してさらに4時間還流した。反応溶液を放冷し、不溶物を除いてろ液を濃縮し、シリカゲルクロマトグラフィー(WAKO−gel
C−300、ヘキサン:THF=20:1)により精製し、アセトン−ヘキサンから再結晶して白色結晶1.44gを得た。
【0198】
この生成物は、 1HNMR及びFAB−MS測定により目的物(56)と同定した(収率95%)。
1HNMR(CDCl3)δ(ppm):0.79(3H,s),1.30(3H,s),1.34(9H,s),3.64(2H,d),3.76(2H,d),5.34(1H,s),6.86(2H,d),6.96-7.06(7H,m),7.22(2H,d),7.36(2H,d)
化合物(56)の 1HNMRスペクトルを図16に示す。
【0199】
(4)中間体(57)の合成
化合物(56)1.44g(3.34mmol)とTPPS(p−トルエンスルホン酸ピリジニウム)0.084g(0.334mmol)をアセトン60ml−水10mlの混合溶媒の溶解し、3時間還流した。溶媒を留去し、トルエンで抽出して飽和食塩水で洗った後、Na2 SO4 上で乾燥した。シリカゲルクロマトグラフィー(WAKO−gel C−300、ヘキサン→ヘキサン:THF=8:1)により精製し、生成物(57)0.940gを得た。
【0200】
この生成物は、 1HNMR及びFAB−MS測定により目的物(56)と同定した(収率81%)。
1HNMR(CDCl3)δ(ppm):1.37(9H,s),6.96(4H,d),7.07 (2H,d),7.16(2H,m),7.33(2H,m),7.66(2H,d),9.79(1H,s),
化合物(57)の 1HNMRスペクトルを図17に示す。
【0201】
(5)ビス(アミノスチリル)ベンゼン化合物((16)−9)の合成
NaH(60%鉱油分散)9.54mmolをヘキサンで2度洗い、無水THF(テトラヒドロフラン)10mlに懸濁させ、氷浴上、窒素雰囲気下でホスホン酸エステル(58)(1.59mmol)と化合物(57)0.940g(2.72mmol)の無水THF溶液50mlを1時間かけて滴下し、その後、氷浴上で3時間攪拌し、室温にて更に12時間攪拌した。反応混合液を少量の氷でクエンチし、トルエンで抽出して飽和食塩水で洗った後、Na2 SO4 上で乾燥した。得られた固体をシリカゲルクロマトグラフィー(WAKO−gel C−300、トルエン)により精製し、トルエンから再結晶して生成物((16)−9)0.856gを得た。
【0202】
この生成物は、 1HNMR及びFAB−MS測定により目的物((16)−9)と同定した(収率66%)。この生成物の 1HNMRスペクトルを図18と下記に示す。
1HNMR(CDCl3)δ(ppm):1.37(18H,s),6.92(4H,d),7.00-7.32(22H,m),7.42(4H,d),7.98(2H,s)
【0203】
この生成物のトルエン溶液の可視吸収極大波長は481nm、蛍光極大波長は540nmであった。
【0204】
実施例18
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−8)の合成例>
【化267】
(1)中間体(61)の合成
1−ブロモ−4−t−ブチルベンゼン(60)12.5g(58.7mmol)、ジフェニルアミン(59)9.93g(58.7mmol)、Pd(OAc)2 263mg(1.17mmol)、トリス(2−メチルフェニル)ホスフィン1.43g(4.69mmol)及びt−BuONa8.45g(88.0mmol)のキシレン懸濁液200mlを3時間、120℃で還流した。反応混合液を冷却した後、これに水を加え、トルエンで抽出(3回)し、有機層を無水硫酸ナトリウムで乾燥、濃縮した。得られた残渣をカラムクロマトグラフィーで精製して、トリアリールアミン(61)18.4gを無色結晶として定量的に得た。
【0206】
この生成物は、 1HNMR及びFAB−MS測定により目的物(61)と同定した。
1HNMR(CDCl3)δ(ppm):1.32(9H,s),6.98-7.09(8H,m),7.19-7.27(6H,m)
この生成物の 1HNMRスペクトルを図19に示す。
【0207】
(2)中間体(62)の合成
オキシ塩化リン27.5g(179mmol)をDMF100mlに滴下し、120℃で5分間攪拌した。得られた赤色溶液を室温まで冷却し、トリアリールアミン(61)18.0g(59.7mmol)を加えた。得られた混合物を80℃で攪拌した。この反応混合物を減圧下で濃縮したのち、NaHCO3 /氷に注意深く注いだ。得られた混合物を酢酸エチルで抽出(3回)し、有機層をNa2 SO4 で乾燥、濃縮した。そして、残渣をカラムクロマトグラフィーで精製し、ジアリールアミノベンズアルデヒド(62)6.69gを淡黄色結晶として得た。
【0208】
この生成物は、 1HNMR及びFAB−MS測定により、目的物(62)と同定した(収率34%)。
1HNMR(CDCl3)δ(ppm):1.33(9H,s),6.99(2H,d),7.08(2H,d),7.17(3H,m),7.33(4H,m),7.66(2H,d)
この生成物の 1HNMRスペクトルを図20に示す。
【0209】
(3)ビス(アミノスチリル)ベンゼン化合物(構造式(16)−8)の合成NaH(60%鉱油分散)9.54mmolをヘキサンで2度洗い、無水THF10mlに懸濁させ、氷浴上、窒素雰囲気下で攪拌した。化合物(58)1.59mmolと(62)1.26g(3.82mmol)の無水THF溶液40mlを15分かけて滴下し、その後氷浴上で6時間攪拌し、室温にて更に6時間攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗った後、Na2 SO4 上で乾燥した。シリカゲルクロマトグラフィー(WAKO−gel C−300、トルエン)により精製し、トルエンから再結晶して生成物((16)−8)1.11gを得た。
【0210】
この生成物は、 1HNMRおよびFAB−MS測定により、目的物((16)−8)と同定した(収率90%)。また、この 1HNMRスペクトルを図21と下記に示す。
1HNMR(CDCl3)δ(ppm):1.33(18H,s),7.02-7.07(10H,m),7.14(4H,d),7.22-7.32(6H,m),7.33(4H,d),7.99(2H,s)
【0211】
この生成物のトルエン溶液の可視吸収極大波長は479nm、蛍光極大波長は535nmであった。
【0212】
実施例19
<ビス(アミノスチリル)ベンゼン化合物(構造式(16)−3)の合成例>
【化268】
(1)中間体(64)の合成
目的物((16)−3)を合成したが、まず、実施例17と同様に合成した化合物(53)2.85g(10.1mmol)、4−ブロモ−N,N−ジメチルアニリン(63)2.00g(10.0mmol)、t−BuONa1.20g(12.0mmol)及びPd(OAc)2 0.066g(0.29mmol)をキシレン150mlに溶解し、窒素雰囲気下で還流しながら、更に0.1MのP(t−Bu)3 12.0ml(0.40mmol)を滴下した後、9時間還流した。シリカゲルクロマトグラフィー(Wakogel C−300、THF:ヘキサン=1:10)により精製し、目的物(64)の黄色結晶2.28gを得た。
【0214】
この生成物は、 1HNMR及びFAB−MS測定により、目的物(64)と同定した(収率57%)。
1HNMR(CDCl3)δ(ppm):0.74(3H,s),1.17(3H,s),2.89(6H,s),3.62(4H,q),5.32(1H,s),6.72(2H,d)6.86(7H,m),7.20-7.27(4H,m)
この生成物の 1HNMRスペクトルを図22に示す。
【0215】
(2)中間体(65)の合成
化合物(64)2.28g(5.71mmol)、p−トルエンスルホン酸一水和物0.133g(0.700mmol)をアセトン300ml、水25mlに溶解し、2時間還流した。アセトンを留去し、Na2 SO4 上で乾燥した後、シリカゲルクロマトグラフィー(Wakogel C−300、トルエン)により精製し、目的物(65)の黄色結晶1.67gを得た。
【0216】
この生成物は、 1HNMR及びFAB−MS測定により目的物(65)と同定した(収率92%)。
1HNMR(CDCl3)δ(ppm):2.97(6H,s),6.71(2H,d),6.93(2H,s),7.07-7.34(7H,m),7.63(2H,d),9.76(1H,s)
この生成物の 1HNMRスペクトルを図23に示す。
【0217】
(3)ビス(アミノスチリル)ベンゼン化合物(構造式(16)−3)の合成NaH(60%鉱油分散)9.54mmolをヘキサンで2度洗い、無水THF10mlに懸濁させ、氷浴上、窒素雰囲気下で攪拌した。化合物(58)1.59mmolと化合物(65)1.14g(3.60mmol)の無水THF溶液70mlを15分かけて滴下し、その後室温にて12時間攪拌した。反応混合液を少量の氷でクエンチし、飽和食塩水で洗った後、Na2 SO4 上で乾燥した。アルミナカラムクロマトグラフィー(300mesh,トルエン:THF=5:1)により原料物質を除き、トルエン−ヘキサンから再結晶して目的物((16)−3)1.02gを得た。
【0218】
この生成物は、 1HNMR及びFAB−MS測定により目的物((16)−3)と同定した(収率88%)。また、その 1HNMRスペクトルを図24と下記に示す。
1HNMR(DMSO−d6)δ(ppm):2.91(12H,s),6.75(4H,d),6.88(4H,s),6.98-7.13(7H,m),7.30(4H,d),7.47(4H,d),7.59(2H,d),8.42(2H,s)
【0219】
この生成物のトルエン溶液の可視吸収極大波長は499nm、蛍光極大波長は620nmであった。
【0220】
実施例20
<1,4−ビス〔2−〔4−(N−4−メトキシフェニル−N−フェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−1)の合成例>
【0221】
【化269】
水素化ナトリウム(60%ミネラルオイル懸濁状、104mg、2.59mmol)のテトラヒドロフラン(THF)懸濁液(3ml)に(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(47.6mg、0.105mmol)を加え、室温下、10分攪拌した。この混合物に、(4−N−メトキシフェニル−N−フェニルアミノ)ベンズアルデヒド(108mg、0.356mmol)を加えて室温下、5時間攪拌した。得られた混合物に、メタノール(0.5ml)を加えた後、飽和塩化アンモニウム水溶液を加え、酢酸エチルで3回抽出した。
【0223】
生じた有機層を水、飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥、ろ過した。ろ液を減圧下、濃縮し、得られた残渣をカラムクロマトグラフィー(シリカゲル、展開溶液:ヘキサン/酢酸エチル=5/1)で精製し、生成物((40)−1)(66.9mg、収率85%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0224】
この生成物のTHF溶液の可視吸収極大波長は438nm、蛍光発光波長は542nmであった。
【0225】
実施例21
<1,4−ビス〔2−〔4−(N−ジフェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−2)の合成例>
【0226】
【化270】
水素化ナトリウム(60%ミネラルオイル懸濁状、153mg、3.84mmol)、(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(115mg、0.256mmol)、N,N−ジフェニルアミノベンズアルデヒド(245mg、0.897mmol)を用い、実施例20 と同様の方法で合成した。
【0228】
その結果、生成物(構造式(40)−2)(150mg、収率85%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0229】
この生成物のTHF溶液の可視吸収極大波長は428nm、蛍光発光波長は522nmであった。
【0230】
実施例22
<1,4−ビス〔2−〔4−(N−4−メチルフェニル−N−フェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−3)の合成例>
【0231】
【化271】
水素化ナトリウム(60%ミネラルオイル懸濁状、160mg、4.01mmol)、(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(90.3mg、0.201mmol)、〔4−(N−4−メチルフェニル)−N−フェニル〕アミノベンズアルデヒド(202mg、0.702mmol)を用い、実施例20と同様の方法で合成した。
【0233】
その結果、生成物(構造式(40)−3)(115mg、収率80%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0234】
この生成物のTHF溶液の可視吸収極大波長は433nm、蛍光発光波長は532nmであった。
【0235】
実施例23
<1,4−ビス〔2−〔4−(N−4−t−ブチルフェニル−N−フェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−4)の合成例>
【0236】
【化272】
水素化ナトリウム(60%ミネラルオイル懸濁状、185mg、4.63mmol)、(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(69.7mg、0.155mmol)、〔4−(N−t−ブチルフェニル)−N−フェニル〕アミノベンズアルデヒド(203mg、0.619mmol)を用い、実施例20と同様の方法で合成した。
【0238】
その結果、生成物(構造式(40)−4)(74.3mg、収率60%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0239】
この生成物のTHF溶液の可視吸収極大波長は433nm、蛍光発光波長は532nmであった。
【0240】
実施例24
<1,4−ビス〔2−〔4−(N−4−t−ブトキシフェニル−N−フェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−5)の合成例>
【0241】
【化273】
水素化ナトリウム(60%ミネラルオイル懸濁状、113mg、2.83mmol)、(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(45.5mg、0.101mmol)、〔N−4−t−ブトキシフェニル〕−N−フェニル〕アミノベンズアルデヒド(132mg、0.385mmol)を用い、実施例20と同様の方法で合成した。
【0243】
その結果、生成物(構造式(40)−5)(54.8mg、収率65%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0244】
この生成物のTHF溶液の可視吸収極大波長は435nm、蛍光発光波長は537nmであった。
【0245】
実施例25
<1,4−ビス〔2−〔4−N,N−ビス(4−メチルフェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−6)の合成例>
【0246】
【化274】
水素化ナトリウム(60%ミネラルオイル懸濁状、237mg、5.78mmol)、(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(260mg、0.578mmol)、〔ビス−N−(4−メチルフェニル)〕アミノベンズアルデヒド(522mg、1.73mmol)を用い、実施例20と同様の方法で合成した。
【0248】
その結果、生成物(構造式(40)−6)(241mg、収率56%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0249】
この生成物のTHF溶液の可視吸収極大波長は440nm、蛍光発光波長は537nmであった。
【0250】
実施例26
<1,4−ビス〔2−〔4−N,N−ビス(4−メトキシフェニル)アミノフェニル〕エテニル〕−2,3,5,6−テトラフルオロベンゼン(構造式(40)−7)の合成例>
【0251】
【化275】
水素化ナトリウム(60%ミネラルオイル懸濁状、186mg、4.65mmol)、(2,3,5,6−テトラフルオロベンゼン)−1,4−ジイル−ビス(メタンホスホン酸ジエチルエステル)(110mg、0.245mmol)、〔ビス−N−(4−メトキシフェニル)〕アミノベンズアルデヒド(253mg、0.759mmol)を用い、実施例20と同様の方法で合成した。
【0253】
その結果、生成物(構造式(40)−7)(118mg、収率62%)を黄橙色結晶として得た。なお、この生成物の同定は 1HNMR及びFAB−MS測定により行った。
【0254】
この生成物のTHF溶液の可視吸収極大波長は446nm、蛍光発光波長は560nmであった。
【0255】
実施例27
本実施例は、一般式〔I〕の上記化合物のうち、R1 、R4 に3−エトキシフェニル基を、R6 、R8 にシアノ基を持った下記構造式(16)−1の化合物を正孔輸送性発光材料として用い、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0256】
【化276】
まず、真空蒸着装置中に、100nmの厚さのITOからなる陽極が一表面に形成された30mm×30mmのガラス基板をセッティングした。蒸着マスクとして、複数の2.0mm×2.0mmの単位開口を有する金属マスクを基板に近接して配置し、真空蒸着法により10-4Pa以下の真空下で上記構造式(16)−1の化合物を例えば50nmの厚さに正孔輸送層(兼発光層)として成膜した。蒸着レートは0.1nm/秒とした。
【0258】
さらに、電子輸送材料として下記構造式のAlq3 (トリス(8−キノリノール)アルミニウム)を正孔輸送層に接して蒸着した。Alq3 からなるこの電子輸送層の膜厚も例えば50nmとし、蒸着レートは0.2nm/秒とした。
【0259】
【化277】
陰極材料としてはMgとAgの積層膜を採用し、これも蒸着により、蒸着レート1nm/秒として例えば50nm(Mg膜)および150nm(Ag膜)の厚さに形成し、実施例27による図47に示した如き有機電界発光素子を作製した。
【0261】
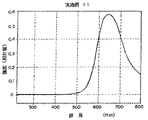
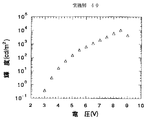
このように作製した実施例27の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、分光測定を行った結果、図25に示すように、620nmに発光ピークを有するスペクトルを得た。分光測定は、大塚電子社製のフォトダイオードアレイを検出器とした分光器を用いた。また、電圧−輝度測定を行ったところ、図27に示すように、8Vで10000cd/m2 の輝度が得られた。
【0262】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで4000時間であった。
【0263】
実施例28
本実施例は、一般式〔I〕の上記化合物のうち、R1 、R4 に3−エトキシフェニル基を、R6 、R8 にシアノ基を持った上記構造式(16)−1の化合物を電子輸送性発光材料として用い、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0264】
まず、真空蒸着装置中に、100nmの厚さのITOからなる陽極が一表面に形成された30mm×30mmのガラス基板をセッティングした。蒸着マスクとして、複数の2.0mm×2.0mmの単位開口を有する金属マスクを基板に近接して配置し、真空蒸着法により10-4Pa以下の真空下で、下記構造式のα−NPD(α−ナフチルフェニルジアミン)を例えば50nmの厚さに正孔輸送層として成膜した。蒸着レートは0.1nm/秒とした。
【0265】
【化278】
さらに、電子輸送材料として上記構造式(16)−1の化合物を正孔輸送層に接して蒸着した。上記構造式(16)−1の化合物からなる電子輸送層(兼発光層)の膜厚も例えば50nmとし、蒸着レートは0.2nm/秒とした。
【0267】
陰極材料としてはMgとAgの積層膜を採用し、これも蒸着により、蒸着レート1nm/秒として例えば50nm(Mg膜)および150nm(Ag膜)の厚さに形成し、実施例28による図47に示した如き有機電界発光素子を作製した。
【0268】
このように作製した実施例28の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、実施例28と同様に分光測定を行った結果、図26に示すように、620nmに発光ピークを有するスペクトルを得た。また、電圧−輝度測定を行ったところ、図28に示すように、8Vで8000cd/m2 の輝度が得られた。
【0269】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで3500時間であった。
【0270】
実施例29
本実施例は、一般式〔I〕の上記化合物のうち、R1 、R4 に3−エトキシフェニル基を、R6 、R8 にシアノ基を持った上記構造式(16)−1の化合物を発光材料として用い、ダブルヘテロ層構造の有機電界発光素子を作製した例である。
【0271】
まず、真空蒸着装置中に、100nmの厚さのITOからなる陽極が一表面に形成された30mm×30mmのガラス基板をセッティングした。蒸着マスクとして、複数の2.0mm×2.0mmの単位開口を有する金属マスクを基板に近接して配置し、真空蒸着法により10-4Pa以下の真空下で、上記構造式のα−NPDを例えば30nmの厚さに正孔輸送層として成膜した。蒸着レートは0.2nm/秒とした。
【0272】
さらに、発光材料として上記構造式(16)−1の化合物を正孔輸送層に接して蒸着した。上記構造式(16)−1の化合物からなる発光層の膜厚も例えば30nmとし、蒸着レートは0.2nm/秒とした。
【0273】
さらに、電子輸送材料として上記構造式のAlq3 を発光層に接して蒸着した。Alq3 の膜厚を例えば30nmとし、蒸着レートは、0.2nm/秒とした。
【0274】
陰極材料としてはMgとAgの積層膜を採用し、これも蒸着により、蒸着レート1nm/秒として例えば50nm(Mg膜)および150nm(Ag膜)の厚さに形成し、実施例30による図48に示した如き有機電界発光素子を作製した。
【0275】
このように作製した実施例29の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、分光測定を行った結果、620nmに発光ピークを有するスペクトルを得た。電圧−輝度測定を行ったところ、8Vで11000cd/m2 の輝度が得られた。
【0276】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで5000時間であった。
【0277】
実施例30
正孔輸送性材料としてα−NPDに替えて下記構造式のTPD(トリフェニルジアミン誘導体)を用いた他は層構成、成膜法とも実施例28に準拠して、有機電界発光素子を作製した。
【0278】
【化279】
本実施例の有機電界発光素子も実施例29と同様の赤色の発光を呈した。分光測定の結果、スペクトルは実施例29の有機電界発光素子のスペクトルと一致した。
【0280】
実施例31
本実施例は、一般式〔II〕の上記化合物のうち、R14、R15、R16、R17に3−メトキシフェニル基を、R19、R21にシアノ基を持った下記構造式(16)−2の化合物を正孔輸送性発光材料として用い、これ以外は実施例27と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0281】
【化280】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、分光測定を行った結果、図29に示すように、650nmに発光ピークを有するスペクトルを得た。分光測定は、大塚電子社製のフォトダイオードアレイを検出器とした分光器を用いた。また、電圧−輝度測定を行ったところ、図31に示すように、9.5Vで1200cd/m2 の輝度が得られた。
【0283】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度200cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで1000時間であった。
【0284】
実施例32
本実施例は、一般式〔II〕の上記化合物のうち、R14、R15、R16、R17に3−メトキシフェニル基を、R19、R21にシアノ基を持った上記構造式(16)−2の化合物を電子輸送性発光材料として用い、これ以外は実施例28と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0285】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、実施例28と同様に分光測定を行った結果、図30に示すように、650nmに発光ピークを有するスペクトルを得た。また、電圧−輝度測定を行ったところ、図32に示すように、10.5Vで600cd/m2 の輝度が得られた。
【0286】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度200cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで700時間であった。
【0287】
実施例33
本実施例は、一般式〔II〕の上記化合物のうち、R14、R15、R16、R17に3−メトキシフェニル基を、R19、R21にシアノ基を持った上記構造式(16)−2の化合物を発光材料として用い、これ以外は実施例29と同様にして、ダブルヘテロ層構造の有機電界発光素子を作製した例である。
【0288】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、分光測定を行った結果、650nmに発光ピークを有するスペクトルを得た。電圧−輝度測定を行ったところ、8.5Vで1800cd/m2 の輝度が得られた。
【0289】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度200cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで1500時間であった。
【0290】
実施例34
正孔輸送性材料としてα−NPDに替えて上記構造式のTPD(トリフェニルジアミン誘導体)を用いた他は層構成、成膜法とも実施例32に準拠して、有機電界発光素子を作製した。
【0291】
本実施例の有機電界発光素子も実施例32と同様の赤色の発光を呈した。分光測定の結果、スペクトルは実施例33の有機電界発光素子のスペクトルと一致した。
【0292】
実施例35
本実施例は、一般式〔III 〕の上記化合物のうち、R27、R30に3−ジメチルアミノフェニル基を、R32、R34にシアノ基を持った下記構造式(16)−3の化合物を正孔輸送性発光材料として用い、これ以外は実施例27と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0293】
【化281】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、分光測定を行った結果、図33に示すように、640nmに発光ピークを有するスペクトルを得た。分光測定は、大塚電子社製のフォトダイオードアレイを検出器とした分光器を用いた。また、電圧−輝度測定を行ったところ、図35に示すように、8Vで6000cd/m2 の輝度が得られた。
【0295】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで3800時間であった。
【0296】
実施例36
本実施例は、一般式〔III 〕の上記化合物のうち、R27、R30に3−ジメチルアミノフェニル基を、R32、R34にシアノ基を持った上記構造式(16)−3の化合物を電子輸送性発光材料として用い、これ以外は実施例28と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0297】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、実施例28と同様に分光測定を行った結果、図34に示すように、640nmに発光ピークを有するスペクトルを得た。また、電圧−輝度測定を行ったところ、図36に示すように、8Vで5300cd/m2 の輝度が得られた。
【0298】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで3200時間であった。
【0299】
実施例37
本実施例は、一般式〔III 〕の上記化合物のうち、R27、R30に3−ジメチルアミノフェニル基を、R32、R34にシアノ基を持った上記構造式(16)−3の化合物を発光材料として用い、これ以外は実施例29と同様にして、ダブルヘテロ層構造の有機電界発光素子を作製した例である。
【0300】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、分光測定を行った結果、640nmに発光ピークを有するスペクトルを得た。電圧−輝度測定を行ったところ、8Vで6800cd/m2 の輝度が得られた。
【0301】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで4500時間であった。
【0302】
実施例38
正孔輸送性材料としてα−NPDに替えて上記構造式のTPD(トリフェニルジアミン誘導体)を用いた他は層構成、成膜法とも実施例36に準拠して、有機電界発光素子を作製した。
【0303】
本実施例の有機電界発光素子も実施例36と同様の赤色の発光を呈した。分光測定の結果、スペクトルは実施例36の有機電界発光素子のスペクトルと一致した。
【0304】
実施例39
本実施例は、一般式〔IV〕の上記化合物のうち、R41、R42に無置換フェニル基を、R40、R43に無置換ナフチル基を、R45、R47にシアノ基を持った下記構造式(16)−4の化合物を正孔輸送性発光材料として用い、これ以外は実施例27と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0305】
【化282】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は黄色であり、分光測定を行った結果、図37に示すように、578nmに発光ピークを有するスペクトルを得た。分光測定は、大塚電子社製のフォトダイオードアレイを検出器とした分光器を用いた。また、電圧−輝度測定を行ったところ、図40に示すように、8Vで6500cd/m2 の輝度が得られた。
【0307】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで4000時間であった。
【0308】
実施例40
本実施例は、一般式〔IV〕の上記化合物のうち、R41、R42に無置換フェニル基を、R40、R43に無置換ナフチル基を、R45、R47にシアノ基を持った上記構造式(16)−4の化合物を電子輸送性発光材料として用い、これ以外は実施例28と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0309】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は黄色であり、実施例39と同様に分光測定を行った結果、図38に示すように、578nmに発光ピークを有するスペクトルを得た。また、電圧−輝度測定を行ったところ、図41に示すように、8Vで5900cd/m2 の輝度が得られた。
【0310】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで3500時間であった。
【0311】
実施例41
本実施例は、一般式〔IV〕の上記化合物のうち、R41、R42に無置換フェニル基を、R40、R43に無置換ナフチル基を、R45、R47にシアノ基を持った上記構造式(16)−4の化合物を発光材料として用い、これ以外は実施例29と同様にして、ダブルヘテロ層構造の有機電界発光素子を作製した例である。
【0312】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は黄色であり、分光測定を行った結果、図39に示すように、578nmに発光ピークを有するスペクトルを得た。電圧−輝度測定を行ったところ、図42に示すように、8Vで7500cd/m2 の輝度が得られた。
【0313】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。また、初期輝度300cd/m2 で電流値を一定に通電して連続発光し、強制劣化させた際、輝度が半減するまで5000時間であった。
【0314】
実施例42
正孔輸送性材料としてα−NPDに替えて上記構造式のTPD(トリフェニルジアミン誘導体)を用いた他は層構成、成膜法とも実施例40に準拠して、有機電界発光素子を作製した。
【0315】
本実施例の有機電界発光素子も実施例40と同様の黄色の発光を呈した。分光測定の結果、スペクトルは実施例40の有機電界発光素子のスペクトルと一致した。
【0316】
実施例43
本実施例は、一般式〔I〕の上記化合物のうち、R1 、R4 に無置換フェニル基を、 R2 、R3 にt−ブチル基を持った下記構造式(16)−8の化合物を電子輸送性発光材料として用い、これ以外は実施例28と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0317】
【化283】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は橙色であり、実施例39と同様に分光測定を行った結果、図43に示すように、580nmに発光ピークを有するスペクトルを得た。また、電圧−輝度測定を行ったところ、8Vで300cd/m2 の輝度が得られた。
【0319】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。
【0320】
実施例44
本実施例は、前記一般式〔I〕の上記化合物のうち、R1 、R4 に無置換フェニル基を、R2 、R3 にターシャリブトキシ基を持った下記構造式(16)−9の化合物を電子輸送性発光材料として用い、これ以外は実施例43と同様にして、シングルヘテロ構造の有機電界発光素子を作製した例である。
【0321】
【化284】
本実施例の有機電界発光素子に、窒素雰囲気下で順バイアス直流電圧を加えて発光特性を評価した。発光色は赤色であり、実施例39と同様に分光測定を行った結果、図44に示すように、628nmに発光ピークを有するスペクトルを得た。また、電圧−輝度測定を行ったところ、7.5Vで15cd/m2 の輝度が得られた。
【0323】
この有機電界発光素子を作製後、窒素雰囲気下に1カ月間放置したが、素子劣化は観察されなかった。
【0324】
【発明の作用効果】
本発明の化合物は、導入される置換基に依存して、黄色〜赤色又は緑色〜赤色の強い発光を示す有機発光材料として有効に利用することができ、高いガラス転移点及び融点を有する物質であり、耐熱性に優れると共に、電気的、熱的或いは化学的な安定性に優れ、また非晶質でガラス状態を容易に形成し得、昇華性もあって真空蒸着等によって均一なアモルファス膜を形成することもできる。また、本発明の化合物は、合成中間体を経て一般的かつ高効率な方法で製造することができる。
【図面の簡単な説明】
【図1】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−1)の1 HNMRスペクトル図である。
【図2】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−2)の1 HNMRスペクトル図である。
【図3】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−4)の1 HNMRスペクトル図である。
【図4】 合成中間体としての4−[N,N−ジ(4−メトキシフェニル)アミノ]ベンズアルデヒド(構造式(27)−2)の1HNMRスペクトル図である。
【図5】 合成中間体としてのN,N−ジ(4−メトキシフェニル)アニリン(構造式(36)−2)の1HNMRスペクトル図である。
【図6】 合成中間体としてのN−(1−フェニル)−N−(4−エトキシフェニル)アニリン((構造式(36)−1)の1HNMRスペクトル図である。
【図7】 合成中間体としての2,5−ジ(ブロモメチル)テレフタロニトリル(構造式(35a))の1HNMRスペクトル図である。
【図8】 合成中間体としてのN−(p−トルイル)−N,N−ジフェニルアミン(構造式(36)−6)の1HNMRスペクトル図である。
【図9】 合成中間体としての4−[N−(p−トルイル)−N−フェニルアミノ]ベンズアルデヒド(構造式(27)−6)の1HNMRスペクトル図である。
【図10】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−6)の1 HNMRスペクトル図である。
【図11】 合成中間体としてのN,N−(p−トルイル−N−フェニルアミン(構造式(36)−7)の1HNMRスペクトル図である。
【図12】 合成中間体としての4−[N,N−ジ(p−トルイル)アミノ]ベンズアルデヒド(構造式(27)−7)の1HNMRスペクトル図である。
【図13】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−7)の1 HNMRスペクトル図である。
【図14】 合成中間体としてのアセタール化合物(構造式(53))の1HNMRスペクトル図である。
【図15】 合成中間体としての化合物(構造式(55))の1HNMRスペクトル図である。
【図16】 合成中間体としてのアセタール化合物(構造式(56))の1HNMRスペクトル図である。
【図17】 合成中間体としてのアルデヒド化合物(構造式(57))の1HNMRスペクトル図である。
【図18】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−9)の 1HNMRスペクトル図である。
【図19】 合成中間体としてのアミン化合物(構造式(61))の1HNMRスペクトル図である。
【図20】 合成中間体としてのアルデヒド化合物(構造式(62))の1HNMRスペクトル図である。
【図21】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−8)の 1HNMRスペクトル図である。
【図22】 合成中間体としてのアセタール化合物(構造式(64))の1HNMRスペクトル図である。
【図23】 合成中間体としてのアルデヒド化合物(構造式(65))の1HNMRスペクトル図である。
【図24】本発明のビス(アミノスチリル)ベンゼン化合物(構造式(16)−3)の 1HNMRスペクトル図である。
【図25】本発明の実施例27による有機電界発光素子の発光スペクトル図である。
【図26】同、実施例28による有機電界発光素子の発光スペクトル図である。
【図27】同、実施例27による有機電界発光素子の電圧−輝度特性図である。
【図28】同、実施例28による有機電界発光素子の電圧−輝度特性図である。
【図29】同、実施例31による有機電界発光素子の発光スペクトル図である。
【図30】同、実施例32による有機電界発光素子の発光スペクトル図である。
【図31】同、実施例31による有機電界発光素子の電圧−輝度特性図である。
【図32】同、実施例32による有機電界発光素子の電圧−輝度特性図である。
【図33】同、実施例35による有機電界発光素子の発光スペクトル図である。
【図34】同、実施例36による有機電界発光素子の発光スペクトル図である。
【図35】同、実施例35による有機電界発光素子の電圧−輝度特性図である。
【図36】同、実施例36による有機電界発光素子の電圧−輝度特性図である。
【図37】同、実施例39による有機電界発光素子の発光スペクトル図である。
【図38】同、実施例40による有機電界発光素子の発光スペクトル図である。
【図39】同、実施例41による有機電界発光素子の発光スペクトル図である。
【図40】同、実施例39による有機電界発光素子の電圧−輝度特性図である。
【図41】同、実施例40による有機電界発光素子の電圧−輝度特性図である。
【図42】同、実施例41による有機電界発光素子の電圧−輝度特性図である。
【図43】同、実施例43による有機電界発光素子の発光スペクトル図である。
【図44】同、実施例44による有機電界発光素子の発光スペクトル図である。
【図45】本発明に基づく有機電界発光素子の要部概略断面図である。
【図46】同、他の有機電界発光素子の要部概略断面図である。
【図47】同、他の有機電界発光素子の要部概略断面図である。
【図48】同、他の有機電界発光素子の要部概略断面図である。
【図49】同、有機電界発光素子を用いたマルチ又はフルカラーの平面ディスプレイの構成図である。
【符号の説明】
1…基板、2…透明電極(陽極)、3…陰極、4…保護膜、
5、5a、5b…有機層、6…正孔輸送層、7…電子輸送層、8…電源、
10…正孔輸送層、11…発光層、12…電子輸送層、14…輝度信号回路、
15…制御回路、20…発光光、A、B、C、D…有機電界発光素子
Claims (4)
- 下記一般式で表されるビス(アミノスチリル)ベンゼン化合物。
- 前記R60の炭素数が2〜6であり、前記R61、R62、R63及びR64の炭素数が1〜6である、請求項1に記載したビス(アミノスチリル)ベンゼン化合物。
- 下記一般式(10)、(11)、(12)、(13)、(14)、(15)又は(15’)で表される、請求項1に記載したビス(アミノスチリル)ベンゼン化合物。
- 下記構造式(16)−1、(16)−2、(16)−3、(16)−4、(16)−5、(16)−6、(16)−7、(16)−8、(16)−9、(16)−10、(16)−11、(16)−12又は(16)−13で表される、請求項1に記載したビス(アミノスチリル)ベンゼン化合物。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP31206999A JP4411708B2 (ja) | 1998-12-07 | 1999-11-02 | ビス(アミノスチリル)ベンゼン化合物 |
| US09/455,724 US6337167B1 (en) | 1998-12-07 | 1999-12-06 | Bis(aminostyryl)benzene compounds and synthetic intermediates thereof, and process for preparing the compounds and intermediates |
| US09/704,960 US6525212B1 (en) | 1998-12-07 | 2000-11-02 | Bis(aminostyryl)benzene compounds and synthetic intermediates thereof, and process for preparing the compounds and intermediates |
| US10/228,019 US20030060652A1 (en) | 1998-12-07 | 2002-08-26 | Bis (aminostyryl) benzene compounds and synthetic intemediates thereof, and process for preparing the compounds and intermediates |
| US10/227,671 US6979746B2 (en) | 1998-12-07 | 2002-08-26 | (1,4-phenylene)bis(methylene) phosphonic acid esters and (1,4-phenylene)bis(methylene) triphenyl phosphonium salt compounds |
| US10/227,711 US20030073863A1 (en) | 1998-12-07 | 2002-08-26 | Bis (aminostyryl) benzene compounds and synthetic intermediates thereof, and process for preparing the compounds and intermediates |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP10-347561 | 1998-12-07 | ||
| JP34756198 | 1998-12-07 | ||
| JP31206999A JP4411708B2 (ja) | 1998-12-07 | 1999-11-02 | ビス(アミノスチリル)ベンゼン化合物 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008232374A Division JP4386140B2 (ja) | 1998-12-07 | 2008-09-10 | ビス(アミノスチリル)ベンゼン化合物 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2000230132A JP2000230132A (ja) | 2000-08-22 |
| JP2000230132A5 JP2000230132A5 (ja) | 2006-05-18 |
| JP4411708B2 true JP4411708B2 (ja) | 2010-02-10 |
Family
ID=26567017
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP31206999A Expired - Fee Related JP4411708B2 (ja) | 1998-12-07 | 1999-11-02 | ビス(アミノスチリル)ベンゼン化合物 |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US6337167B1 (ja) |
| JP (1) | JP4411708B2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11469378B2 (en) | 2018-06-11 | 2022-10-11 | Lg Display Co., Ltd. | Electroluminescent compound and electroluminescent device including the same |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100495038B1 (ko) * | 2002-12-30 | 2005-06-14 | 엘지전자 주식회사 | 유기 전계 발광 소자용 적색 발광 화합물 및 이를 사용한유기 전계 발광 소자 |
| US7402343B2 (en) * | 2003-01-29 | 2008-07-22 | Samsung Sdi Co., Ltd. | Molecular chemical compounds with structures allowing electron displacement and capable of emitting photoluminescent radiation, and photoluminescence quenching device employing the same |
| JP4001118B2 (ja) | 2003-03-24 | 2007-10-31 | ソニー株式会社 | 有機電界発光素子及びアミノモノスチリルナフタレン化合物 |
| KR101112574B1 (ko) | 2004-12-22 | 2012-02-15 | 재단법인서울대학교산학협력재단 | 프탈로니트릴 유도체 |
| JP4623166B2 (ja) | 2008-08-25 | 2011-02-02 | ソニー株式会社 | 標識化合物及びこれを用いた検出方法 |
| CN107601860A (zh) * | 2017-10-13 | 2018-01-19 | 华南农业大学 | 一种红色发光玻璃陶瓷、其制备方法及植物灯 |
| CN113429317A (zh) * | 2021-06-22 | 2021-09-24 | 四川金象赛瑞化工股份有限公司 | 一种苯二甲腈的非液相生产方法 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4111693A (en) * | 1976-12-22 | 1978-09-05 | Eastman Kodak Company | Multilayer aggregate photoconductive elements |
| US4175961A (en) * | 1976-12-22 | 1979-11-27 | Eastman Kodak Company | Multi-active photoconductive elements |
| JP2552695B2 (ja) * | 1987-04-21 | 1996-11-13 | 株式会社リコー | ジオレフィン芳香族化合物 |
| US5100985A (en) * | 1991-03-25 | 1992-03-31 | Hoechst Celanese Corp. | 1,4-bis(4-arylbutadienyl) benzenes exhibiting nonlinear optical response |
| JP3540061B2 (ja) * | 1995-07-21 | 2004-07-07 | 株式会社リコー | ジヒドロキシル基含有ジアミン化合物 |
| WO1998021521A1 (en) * | 1996-11-12 | 1998-05-22 | California Institute Of Technology | Two-photon or higher-order absorbing optical materials and methods of use |
| JP3694591B2 (ja) * | 1997-07-24 | 2005-09-14 | 京セラミタ株式会社 | スチルベン誘導体、その製造方法および電子写真感光体 |
| US6022998A (en) * | 1997-07-24 | 2000-02-08 | Mita Industrial Co., Ltd. | Stilbene derivative and method for producing the same |
| JPH11273859A (ja) * | 1998-03-24 | 1999-10-08 | Sony Corp | 有機電界発光素子及びその製造方法 |
| JP2000017057A (ja) * | 1998-04-30 | 2000-01-18 | Fuji Photo Film Co Ltd | スチリル系化合物及びその製造方法並びにそれを用いた有機発光素子 |
| JP4164174B2 (ja) * | 1998-11-13 | 2008-10-08 | キヤノン株式会社 | 電子写真感光体の製造方法 |
| JP4386140B2 (ja) * | 1998-12-07 | 2009-12-16 | ソニー株式会社 | ビス(アミノスチリル)ベンゼン化合物 |
-
1999
- 1999-11-02 JP JP31206999A patent/JP4411708B2/ja not_active Expired - Fee Related
- 1999-12-06 US US09/455,724 patent/US6337167B1/en not_active Expired - Lifetime
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11469378B2 (en) | 2018-06-11 | 2022-10-11 | Lg Display Co., Ltd. | Electroluminescent compound and electroluminescent device including the same |
Also Published As
| Publication number | Publication date |
|---|---|
| US6337167B1 (en) | 2002-01-08 |
| JP2000230132A (ja) | 2000-08-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100843819B1 (ko) | 안트라센 유도체 및 이를 사용한 유기 전기발광 소자 | |
| KR100846221B1 (ko) | 유기 발광 화합물 및 이를 이용한 유기 전계 발광 소자 | |
| WO2002043449A1 (en) | Luminescent element material and luminescent element comprising the same | |
| JP4164718B2 (ja) | ビス(アミノスチリル)ナフタレン化合物及びその合成中間体、これらの製造方法、並びに有機電界発光素子 | |
| KR100696528B1 (ko) | 트리아릴아민계 화합물, 그 제조방법 및 이를 이용한 유기발광 표시 소자 | |
| JP4411708B2 (ja) | ビス(アミノスチリル)ベンゼン化合物 | |
| US7196225B2 (en) | Bis(aminostyryl) anthracene compound, synthesis intermediate thereof, and process for production thereof | |
| JP4158078B2 (ja) | アミノスチリルフェナントレン化合物及びその合成中間体 | |
| JP2004307472A (ja) | 有機電界発光素子、アミノスチリルナフタレン化合物及びその合成中間体、並びにこれらの製造方法 | |
| KR100717696B1 (ko) | 유기 전계발광 소자 | |
| JP2001288377A (ja) | アミノスチリルアントラセン化合物及びその合成中間体、並びにこれらの製造方法 | |
| US6525212B1 (en) | Bis(aminostyryl)benzene compounds and synthetic intermediates thereof, and process for preparing the compounds and intermediates | |
| JP4386140B2 (ja) | ビス(アミノスチリル)ベンゼン化合物 | |
| US6492557B1 (en) | Bis (aminostyryl) naphthalene compound, synthesis intermediate thereof, and process for production thereof | |
| JP2004091342A (ja) | 芳香族メチリデン化合物、それを製造するための化合物、それらの製造方法、及び芳香族メチリデン化合物を用いた有機電界発光素子 | |
| JP4460704B2 (ja) | ジスチリルアリーレン誘導体および有機電界発光素子 | |
| JP2001131128A (ja) | ビス(アミノスチリル)スチルベン系化合物及びその合成中間体、これらの製造方法、並びに有機電界発光素子 | |
| KR20040026542A (ko) | 신규한 아릴포스핀계 화합물 및 이를 채용한 유기 el 소자 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060328 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060328 |
|
| RD13 | Notification of appointment of power of sub attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7433 Effective date: 20070125 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080731 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080910 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20090522 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A132 Effective date: 20090616 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090810 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090908 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090930 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20091027 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20091109 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121127 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |