RU2201932C2 - Модифицированные в боковой цепи эпотилоны - Google Patents
Модифицированные в боковой цепи эпотилоны Download PDFInfo
- Publication number
- RU2201932C2 RU2201932C2 RU99120378/04A RU99120378A RU2201932C2 RU 2201932 C2 RU2201932 C2 RU 2201932C2 RU 99120378/04 A RU99120378/04 A RU 99120378/04A RU 99120378 A RU99120378 A RU 99120378A RU 2201932 C2 RU2201932 C2 RU 2201932C2
- Authority
- RU
- Russia
- Prior art keywords
- epothilone
- modified
- oxide
- epothilones
- protected
- Prior art date
Links
- 238000000034 method Methods 0.000 claims abstract description 32
- 150000001204 N-oxides Chemical class 0.000 claims abstract description 12
- 238000006243 chemical reaction Methods 0.000 claims abstract description 11
- 125000003158 alcohol group Chemical group 0.000 claims abstract description 3
- 238000006735 epoxidation reaction Methods 0.000 claims abstract description 3
- 150000002118 epoxides Chemical class 0.000 claims abstract description 3
- 238000001465 metallisation Methods 0.000 claims abstract description 3
- 229930013356 epothilone Natural products 0.000 claims description 31
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 claims description 30
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 claims description 10
- 150000003883 epothilone derivatives Chemical class 0.000 claims description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 8
- 238000010934 O-alkylation reaction Methods 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 5
- 150000001875 compounds Chemical class 0.000 claims description 5
- 239000003153 chemical reaction reagent Substances 0.000 claims description 4
- ASQQEOXYFGEFKQ-UHFFFAOYSA-N dioxirane Chemical compound C1OO1 ASQQEOXYFGEFKQ-UHFFFAOYSA-N 0.000 claims description 4
- 150000004965 peroxy acids Chemical class 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 3
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 claims description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 2
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 claims description 2
- 150000008064 anhydrides Chemical class 0.000 claims description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 2
- 239000003054 catalyst Substances 0.000 claims description 2
- 238000003776 cleavage reaction Methods 0.000 claims description 2
- 229910000071 diazene Inorganic materials 0.000 claims description 2
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 claims description 2
- 230000002255 enzymatic effect Effects 0.000 claims description 2
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 125000005842 heteroatom Chemical group 0.000 claims description 2
- 230000003301 hydrolyzing effect Effects 0.000 claims description 2
- 229910052751 metal Inorganic materials 0.000 claims description 2
- 239000002184 metal Substances 0.000 claims description 2
- 238000007254 oxidation reaction Methods 0.000 claims description 2
- 230000007017 scission Effects 0.000 claims description 2
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 claims 3
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 claims 2
- 150000002440 hydroxy compounds Chemical class 0.000 claims 1
- 239000000126 substance Substances 0.000 abstract 2
- 230000015572 biosynthetic process Effects 0.000 abstract 1
- 230000000694 effects Effects 0.000 abstract 1
- 238000005984 hydrogenation reaction Methods 0.000 abstract 1
- 238000003786 synthesis reaction Methods 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 96
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- 239000000243 solution Substances 0.000 description 14
- 239000000047 product Substances 0.000 description 13
- 239000002904 solvent Substances 0.000 description 11
- 238000005160 1H NMR spectroscopy Methods 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 238000012746 preparative thin layer chromatography Methods 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- FFHWGQQFANVOHV-UHFFFAOYSA-N dimethyldioxirane Chemical compound CC1(C)OO1 FFHWGQQFANVOHV-UHFFFAOYSA-N 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000010265 sodium sulphite Nutrition 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- -1 trimethyloxonium tetrafluoroborate Chemical compound 0.000 description 2
- FCCNKYGSMOSYPV-DEDISHTHSA-N (-)-Epothilone E Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(CO)sc2)/C)OC(=O)C[C@H](O)C1(C)C FCCNKYGSMOSYPV-DEDISHTHSA-N 0.000 description 1
- UWKQJZCTQGMHKD-UHFFFAOYSA-N 2,6-di-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=CC(C(C)(C)C)=N1 UWKQJZCTQGMHKD-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- YNJSNEKCXVFDKW-UHFFFAOYSA-N 3-(5-amino-1h-indol-3-yl)-2-azaniumylpropanoate Chemical compound C1=C(N)C=C2C(CC(N)C(O)=O)=CNC2=C1 YNJSNEKCXVFDKW-UHFFFAOYSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000002451 electron ionisation mass spectrometry Methods 0.000 description 1
- FCCNKYGSMOSYPV-UHFFFAOYSA-N epothilone E Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC2OC2CC1C(C)=CC1=CSC(CO)=N1 FCCNKYGSMOSYPV-UHFFFAOYSA-N 0.000 description 1
- FCCNKYGSMOSYPV-OKOHHBBGSA-N epothilone e Chemical compound C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(CO)=N1 FCCNKYGSMOSYPV-OKOHHBBGSA-N 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical compound COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 description 1
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 description 1
- 235000012141 vanillin Nutrition 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Plural Heterocyclic Compounds (AREA)
- Epoxy Compounds (AREA)
- Polyesters Or Polycarbonates (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Silicon Polymers (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Other Resins Obtained By Reactions Not Involving Carbon-To-Carbon Unsaturated Bonds (AREA)
Abstract
Изобретение относится к способу получения модифицированных в 16,17-положении эпотилонов, согласно которому защищенные в положении 3,7 или незащищенные эпотилоны А или В а) гидрируют по двойной связи в положении 16,17 либо в) по двойной связи в положении 16,17 проводят эпоксидирование и в случае необходимости полученный эпоксид восстанавливают до спирта в положении 16, к способу получения эпотилон-N-оксидов, при котором защищенные в положении 3,7 или незащищенные эпотилоны А или В переводят в N-оксид, полученный N-оксид при необходимости подвергают реакции Катара; способу получения модифицированных в С-19 положении эпотилонов путем металлизирования в положении С-19 защищенных или незащищенных эпотилонов А или В, а также к модифицированным эпотилонам общей формулы I. 5 с. и 12 з.п.ф-лы, 1 табл.
Description
Известны эпотилоны А и В (см., например, заявки DE 4138042, WO 9310121, WO 9719086).
Известный уровень техники предлагает эпотилоны А и В в качестве терапевтических средств. В журнале PNAS USA, 95 (1998) 1369-1374, эпоталоны упоминаются в качестве ценных терапевтических средств. Благодаря их терапевтическим действиям в журнале Angew. Chem. Int. Ed., 36 (1997) 2097-2103, предусмотрено даже создание обширной библиотеки подобного рода соединений (extensive library of compounds).
Итак, настоящее изобретение относится к способу получения модифицированных в положении 16, 17 эпотилонов, согласно которому защищенные в положении 3, 7 или незащищенные эпотилоны А и В
а) либо гидрируют по двойной связи в положении 16, 17, либо
б) к двойной связи в положении 16, 17 присоединяют атом галогена, либо
в) по двойной связи в положении 16, 17 проводят эпоксидирование и в случае необходимости полученный эпоксид восстанавливают до спирта в положении 16.
а) либо гидрируют по двойной связи в положении 16, 17, либо
б) к двойной связи в положении 16, 17 присоединяют атом галогена, либо
в) по двойной связи в положении 16, 17 проводят эпоксидирование и в случае необходимости полученный эпоксид восстанавливают до спирта в положении 16.
Предлагаемый согласно изобретению способ отличается тем, что в случае
- способа (а) гидрируют диимином или водородом в присутствии гетерогенного или гомогенного металлического катализатора или в случае
- способа (в) эпоксидируют надкислотой или диоксираном.
- способа (а) гидрируют диимином или водородом в присутствии гетерогенного или гомогенного металлического катализатора или в случае
- способа (в) эпоксидируют надкислотой или диоксираном.
Далее, изобретение относится к способу получения 2,3-ненасыщенных эпотилон -N-оксидов, при котором либо
(i) защищенные в положении 3, 7 эпотилоны А или В известным образом переводят в N-оксид и с помощью основания удаляют заместитель в положении 3 с получением двойной связи в положении 2, 3, либо
(ii) защищенные или незащищенные в положении 7 эпотилоны А или В, имеющие в положении 2, 3 двойную связь, известным образом переводят в N-оксид и
в случае необходимости полученный N-оксид подвергают O-алкилированию, в результате чего получают соответствующий продукт О-алкилирования.
(i) защищенные в положении 3, 7 эпотилоны А или В известным образом переводят в N-оксид и с помощью основания удаляют заместитель в положении 3 с получением двойной связи в положении 2, 3, либо
(ii) защищенные или незащищенные в положении 7 эпотилоны А или В, имеющие в положении 2, 3 двойную связь, известным образом переводят в N-оксид и
в случае необходимости полученный N-оксид подвергают O-алкилированию, в результате чего получают соответствующий продукт О-алкилирования.
Далее, настоящее изобретение относится к способу получения эпотилон-N-оксидов, при котором защищенные в положении 3, 7 или незащищенные эпотилоны А или В известным образом переводят в N-оксид, и полученный N-оксид в случае необходимости подвергают O-алкилированию, в результате чего получают соответствующий продукт O-алкилирования.
Предлагаемый согласно изобретению способ может отличаться тем, что N-оксидирование осуществляют с помощью надкислоты или диоксирана, а для необязательного 0-алкилирования используют электрофильные алкильные, арильные или гетероарильные реагенты, в частности, метилйодид или триметилоксоний-тетрафторборат.
Далее, предлагаемый способ может отличаться тем, что полученный N-оксид подвергают реакции Катада, в частности, в соответствии со справочником Houben-Weyl, т. Е7b, стр. 646.
Далее, предлагаемый способ может отличаться тем, что реакцию Катада осуществляют с помощью активированного производного карбоновой кислоты, в частности ее ангидрида или хлорангидрида.
Далее, предлагаемый способ может отличаться тем, что реакцию каталитического гидрирования Катада осуществляют с помощью ацетангидрида и полученные 21-ацетоксиэпотилоны в случае необходимости известным образом расщепляют до 21-оксиэпотилонов А или В (эпотилоны Е и F соответственно).
Далее, предлагаемый способ может отличаться тем, что необязательное расщепление проводят гидролитическим или ферментативным путем.
Далее, изобретение относится к способу получения модифицированных в С-19 положении эпотилонов, согласно которому защищенные в положении 3, 7 или незащищенные эпотилоны А или В металлизируют в положении С-19 и известным образом с помощью электрофильных реагентов улавливают в виде модифицированных в положении С-19 алкил-, арил-, гетероарил-, галоген-, кислород- или серозамещенных эпотилонов.
Предлагаемый способ может отличаться тем, что металлизацию проводят с помощью бутиллития.
Далее, изобретение относится к способу получения модифицированных в С-27 положении эпотилонов, при котором аллильную группу в положениях С-17, С-16 и С-27 известным образом по С-27-метильной группе замещают гетероатомом.
Предлагаемый способ может отличаться тем, что С-27-метильную группу замещают атомом брома, в частности, с помощью N-бромсукцинимида, и полученный бромид в случае необходимости переводят в С-27-окси-соединение.
Наконец, настоящее изобретение относится к соединениям, полученным по предлагаемому способу.
ПРИМЕР 1: ДИЭПОКСИЭПОТИЛОН А. (1а)
К раствору эпотилона А (5 мг, 10 мкмоль) в ацетоне (1 мл) при 0oС добавляют диметилдиоксиран (0,4 мл, 28 мкмоль, 0,07 М в ацетоне). Температуру полученного раствора в течение нескольких часов доводят до комнатной температуры, при которой продолжают перемешивать в течение 20 ч. Поскольку по данным ТСХ устанавливают наличие остатка исходного продукта, добавляют потом еще диметилдиоксиран (0,25 мл, 17 мкмоль) и перемешивают реакционную смесь еще в течение 20 часов при комнатной температуре. Наконец, удаляют растворитель и остаток очищают с помощью метода препаративной тонкослойной хроматографии (ПСХ) (0,25 х 200 х 200 мм, 10% МеОН:CH2Cl2). В результате выделяют следующие продукты:
1. 1,4 мг (27%) диэпоксиэпотилона А (смесь 3:2 эпимеров в положениях 16 и 17). Rf 0,63 (10% МеОН:CH2Cl2); Rt: 6,79 (изомер 1) и 7,39 (изомер 2) мин (RP 18, 250 х 4 мм, МеОН:H2O 65:35, 1 мл/мин); МС: (m/z)=510 (M+); 1H-ЯМР (400 МГц, CDCl3, выбранные сигналы, изомер 1): δ=6,96 (с, 1Н, Н-19), 5,48 (дд, J=12,2 и 2,5 Гц, 1Н, Н-15), 4,37 (дш, J=10,7 Гц, 1Н, Н-3), 4,10 (с, 1Н, Н-17), 3,67 (дд, J=5,6 и 2,5 Гц, 1Н, Н-7), 3,14 (квд, J=6,6 и 2,5 Гц, 1Н, Н-6), 3,00 (ддд, J=9,7, 3,6 и 2,5 Гц, 1Н, Н-13), 2,88 (дт, J=8,6 и 3,6 Гц, 1Н, Н-12), 2,71 (с, 3Н, Н-21), 2,53 (дд, J=13,7 и 11,7 Гц, 1Н, Н-2а), 1,41 (с, 3Н, Н-22), 1,27 (с, 3Н, Н-26), 1,17 (д, J=6,6 Гц, 3Н, Н-24), 1,08 (с, 3Н, Н-23), 0,97 (д, J=7,1 Гц, 3Н, Н-25); (изомер 2) δ=6,98 (с, 1Н, Н-19), 5,11 (дд, J=11,7 и 2,5 Гц, 1Н, Н-15) 4,27 (дш, J=10,7 Гц, 1Н, Н-3), 4,14 (с, 1Н, Н-17), 3,06 (квд, J=6,6 и 2,9 Гц, 1Н, Н-6), 2,96 (ддд, J=9,7, 3,6 и 2,5 Гц, 1H, Н-13), 2,31 (дт, J=14,7 и 2,0 Гц, 1Н, H-14a), 1,36 (с, 3Н, Н-22), 1,15 (д, J=6,6 Гц, 3Н, Н-24), 1,14 (с, 3Н, Н-26), 1,07 (с, 3Н, Н-23).
К раствору эпотилона А (5 мг, 10 мкмоль) в ацетоне (1 мл) при 0oС добавляют диметилдиоксиран (0,4 мл, 28 мкмоль, 0,07 М в ацетоне). Температуру полученного раствора в течение нескольких часов доводят до комнатной температуры, при которой продолжают перемешивать в течение 20 ч. Поскольку по данным ТСХ устанавливают наличие остатка исходного продукта, добавляют потом еще диметилдиоксиран (0,25 мл, 17 мкмоль) и перемешивают реакционную смесь еще в течение 20 часов при комнатной температуре. Наконец, удаляют растворитель и остаток очищают с помощью метода препаративной тонкослойной хроматографии (ПСХ) (0,25 х 200 х 200 мм, 10% МеОН:CH2Cl2). В результате выделяют следующие продукты:
1. 1,4 мг (27%) диэпоксиэпотилона А (смесь 3:2 эпимеров в положениях 16 и 17). Rf 0,63 (10% МеОН:CH2Cl2); Rt: 6,79 (изомер 1) и 7,39 (изомер 2) мин (RP 18, 250 х 4 мм, МеОН:H2O 65:35, 1 мл/мин); МС: (m/z)=510 (M+); 1H-ЯМР (400 МГц, CDCl3, выбранные сигналы, изомер 1): δ=6,96 (с, 1Н, Н-19), 5,48 (дд, J=12,2 и 2,5 Гц, 1Н, Н-15), 4,37 (дш, J=10,7 Гц, 1Н, Н-3), 4,10 (с, 1Н, Н-17), 3,67 (дд, J=5,6 и 2,5 Гц, 1Н, Н-7), 3,14 (квд, J=6,6 и 2,5 Гц, 1Н, Н-6), 3,00 (ддд, J=9,7, 3,6 и 2,5 Гц, 1Н, Н-13), 2,88 (дт, J=8,6 и 3,6 Гц, 1Н, Н-12), 2,71 (с, 3Н, Н-21), 2,53 (дд, J=13,7 и 11,7 Гц, 1Н, Н-2а), 1,41 (с, 3Н, Н-22), 1,27 (с, 3Н, Н-26), 1,17 (д, J=6,6 Гц, 3Н, Н-24), 1,08 (с, 3Н, Н-23), 0,97 (д, J=7,1 Гц, 3Н, Н-25); (изомер 2) δ=6,98 (с, 1Н, Н-19), 5,11 (дд, J=11,7 и 2,5 Гц, 1Н, Н-15) 4,27 (дш, J=10,7 Гц, 1Н, Н-3), 4,14 (с, 1Н, Н-17), 3,06 (квд, J=6,6 и 2,9 Гц, 1Н, Н-6), 2,96 (ддд, J=9,7, 3,6 и 2,5 Гц, 1H, Н-13), 2,31 (дт, J=14,7 и 2,0 Гц, 1Н, H-14a), 1,36 (с, 3Н, Н-22), 1,15 (д, J=6,6 Гц, 3Н, Н-24), 1,14 (с, 3Н, Н-26), 1,07 (с, 3Н, Н-23).
2. 0,8 мг (16%) N-оксида эпотилона A. Rf: 0,44 (10% МеОН:СН2Сl2); Rt: 4,25 мин (RP 18, 250 х 4 мм, МеОН:Н2O 65:35, 1 мл/мин); МС: (m/z)=510 (M+); 1H-ЯМР: см. Способ 1.
ПРИМЕР 2: ДИГИДРОЭПОТИЛОН А. (1в)
К раствору эпотилона А (11 мг, 22 мкмоля) в этаноле (2 мл) добавляют палладий на активированном угле (5 мг, 10%), после чего полученную черную суспензию в течение 24 ч при комнатной температуре подвергают воздействию атмосферы водорода. Поскольку реакция по данным ТСХ проходит неполностью, добавляют еще одну порцию палладия на угле, после чего реакционную смесь в течение еще 20 ч перемешивают в атмосфере водорода. Разделение продуктов осуществляют с помощью ПСХ (1 х 200 х 200 мм, 10% МеОН:CH2Cl2). В результате выделяют следующие продукты:
1. 0,5 мг (5%; дигидроэпотилона A. Rf: 0,60 (10% МеОН:СН2Сl2); Rt: 10,80 мин (RP 18, 250 х 4 мм, МеОН:Н2O 65:35, 1 мл/мин); MC (m/z)=496 (M+), 478, 408, 308; 1H-ЯМР (400 МГц), CDCl3, выбранные сигналы): δ=7,05 (д, J=6,6 Гц, 1H, ОН), 6,77 (с, 1H, H-19), 5,23 (дд, J=12,4 и 2,3 Гц, 1Н, H-15) 4,42 (ддд, J= 11,7, 6,6 и 3,0 Гц, 1H, Н-3), 3,70 (ддд, J=5,3 и 2 Гц, 1H, Н-7), 3,12 (квд, J= 6,6 и 3,0 Гц, 1H, Н-6), 3,07 (д, J=12,7 Гц, 1H, Н-17а), 2,96 (ддд, J= 9,7, 3,6 и 2,0 Гц, 1H, Н-13), 2,91 (ддд, J=9,7, 3,6 и 2,6 Гц, 1H, Н-12), 2,68 (с, 3Н, Н-21), 2,51 (дд, J=13,7 и 11,7 Гц, 1H, Н-2а), 2,24 (д; J=12,7 Гц, 1H, H-17b), 2,19 (м, 1H, Н-16), 2,13 (дд, J=13,7 и 3,0 Гц, 1H, Н-2b), 1,35 (с, 3Н, Н-22), 1,15 (д, J=6,6 Гц, 3Н, Н-24), 1,09 (с, 3Н, Н-23), 0,99 (д, J=7,1 Гц, 3Н, Н-25), 0,93 (д, J=6,6 Гц, 3Н, Н-26).
К раствору эпотилона А (11 мг, 22 мкмоля) в этаноле (2 мл) добавляют палладий на активированном угле (5 мг, 10%), после чего полученную черную суспензию в течение 24 ч при комнатной температуре подвергают воздействию атмосферы водорода. Поскольку реакция по данным ТСХ проходит неполностью, добавляют еще одну порцию палладия на угле, после чего реакционную смесь в течение еще 20 ч перемешивают в атмосфере водорода. Разделение продуктов осуществляют с помощью ПСХ (1 х 200 х 200 мм, 10% МеОН:CH2Cl2). В результате выделяют следующие продукты:
1. 0,5 мг (5%; дигидроэпотилона A. Rf: 0,60 (10% МеОН:СН2Сl2); Rt: 10,80 мин (RP 18, 250 х 4 мм, МеОН:Н2O 65:35, 1 мл/мин); MC (m/z)=496 (M+), 478, 408, 308; 1H-ЯМР (400 МГц), CDCl3, выбранные сигналы): δ=7,05 (д, J=6,6 Гц, 1H, ОН), 6,77 (с, 1H, H-19), 5,23 (дд, J=12,4 и 2,3 Гц, 1Н, H-15) 4,42 (ддд, J= 11,7, 6,6 и 3,0 Гц, 1H, Н-3), 3,70 (ддд, J=5,3 и 2 Гц, 1H, Н-7), 3,12 (квд, J= 6,6 и 3,0 Гц, 1H, Н-6), 3,07 (д, J=12,7 Гц, 1H, Н-17а), 2,96 (ддд, J= 9,7, 3,6 и 2,0 Гц, 1H, Н-13), 2,91 (ддд, J=9,7, 3,6 и 2,6 Гц, 1H, Н-12), 2,68 (с, 3Н, Н-21), 2,51 (дд, J=13,7 и 11,7 Гц, 1H, Н-2а), 2,24 (д; J=12,7 Гц, 1H, H-17b), 2,19 (м, 1H, Н-16), 2,13 (дд, J=13,7 и 3,0 Гц, 1H, Н-2b), 1,35 (с, 3Н, Н-22), 1,15 (д, J=6,6 Гц, 3Н, Н-24), 1,09 (с, 3Н, Н-23), 0,99 (д, J=7,1 Гц, 3Н, Н-25), 0,93 (д, J=6,6 Гц, 3Н, Н-26).
2. 8 мг (72%) 15-деокси-дигидроэпотилоновой кислоты. Rf: 0,10 (10% МеОН: СН2Сl2).
ПРИМЕР 3: 16-ОКСИЭПОТИЛОН А. (1б)
К раствору диэпоксиэпотилона А (7 мг, 14 мкмолей) (смесь (1:1) эпимеров в положении С-16) в этаноле (2 мл) добавляют палладий на активированном угле (10 мг, 10%), после чего полученную черную суспензию в течение 24 ч при комнатной температуре подвергают воздействию атмосферы водорода. Поскольку реакция по данным ТСХ проходит неполностью, добавляют еще одну порцию палладия на угле, после чего реакционную смесь в течение еще 80 ч перемешивают в атмосфере водорода. Разделение продуктов осуществляют с помощью ПСХ (1 х 200 х 200 мм, 10% МеОН:СН2Сl2). В результате выделяют следующие продукты:
1. 3 мг (43%) 16-оксиэпотилона А (изомера 1). Rf: 0,38 (10% МеОН: СН2Сl2); Rt: 6,65 мин (RP 18, 250 х 4 мм, MeOH:H2O 65:35, 1 мл/мин); 1Н-ЯМР (400 МГц, СDСl3, выбранные сигналы): δ=6,85 (с, 1Н, Н-19), 5,02 (дд, J=11,7 и 2,0 Гц, 1Н, Н-15), 4,38 (дш, J=11,2 Гц, 1Н, Н-3), 3,67 (дд, J=4 и 3 Гц, 1Н, Н-7), 3,14 (квд, J=6,8 и 3,0 Гц, 1Н, Н-6), 2,95 (д, J=15,3 Гц, 1Н, Н-17а), 2,89 (д, J=15,3 Гц, 1Н, Н-17b), 2,89 (ддд, J=10,2, 3,6 и 2,0 Гц, 1Н, Н-13), 2,81 (ддд, J=9,7, 3,6 и 2,5 Гц, 1Н, Н-12), 2,70 (с, 3Н, Н-21), 2,53 (дд, J=15,8 и 11,7 Гц, 1Н, Н-2а), 2,14 (дд, J=15,8 и 2,0 Гц, 1Н, Н-2b), 2,08 (дт, J= 14,3 и 2,0 Гц, 1Н, Н-14а), 1,39 (с, 3Н, Н-22), 1,25 (с, 3Н, Н-26), 1,19 (д, J= 6,6 Гц, 3Н, Н-24), 1,05 (с, 3Н, Н-23), 0,99 (д, J=7,1 Гц, 3Н, Н-25).
К раствору диэпоксиэпотилона А (7 мг, 14 мкмолей) (смесь (1:1) эпимеров в положении С-16) в этаноле (2 мл) добавляют палладий на активированном угле (10 мг, 10%), после чего полученную черную суспензию в течение 24 ч при комнатной температуре подвергают воздействию атмосферы водорода. Поскольку реакция по данным ТСХ проходит неполностью, добавляют еще одну порцию палладия на угле, после чего реакционную смесь в течение еще 80 ч перемешивают в атмосфере водорода. Разделение продуктов осуществляют с помощью ПСХ (1 х 200 х 200 мм, 10% МеОН:СН2Сl2). В результате выделяют следующие продукты:
1. 3 мг (43%) 16-оксиэпотилона А (изомера 1). Rf: 0,38 (10% МеОН: СН2Сl2); Rt: 6,65 мин (RP 18, 250 х 4 мм, MeOH:H2O 65:35, 1 мл/мин); 1Н-ЯМР (400 МГц, СDСl3, выбранные сигналы): δ=6,85 (с, 1Н, Н-19), 5,02 (дд, J=11,7 и 2,0 Гц, 1Н, Н-15), 4,38 (дш, J=11,2 Гц, 1Н, Н-3), 3,67 (дд, J=4 и 3 Гц, 1Н, Н-7), 3,14 (квд, J=6,8 и 3,0 Гц, 1Н, Н-6), 2,95 (д, J=15,3 Гц, 1Н, Н-17а), 2,89 (д, J=15,3 Гц, 1Н, Н-17b), 2,89 (ддд, J=10,2, 3,6 и 2,0 Гц, 1Н, Н-13), 2,81 (ддд, J=9,7, 3,6 и 2,5 Гц, 1Н, Н-12), 2,70 (с, 3Н, Н-21), 2,53 (дд, J=15,8 и 11,7 Гц, 1Н, Н-2а), 2,14 (дд, J=15,8 и 2,0 Гц, 1Н, Н-2b), 2,08 (дт, J= 14,3 и 2,0 Гц, 1Н, Н-14а), 1,39 (с, 3Н, Н-22), 1,25 (с, 3Н, Н-26), 1,19 (д, J= 6,6 Гц, 3Н, Н-24), 1,05 (с, 3Н, Н-23), 0,99 (д, J=7,1 Гц, 3Н, Н-25).
2. 3 мг (43%) 16-Оксиэпотилона А (изомера 2). Rf: 0,31 (10% MeOH: CH2Cl2); Rt: 6,10 мин (RP 18, 250 х 4 мм, МеОН:Н2О 65:35, 1 мл/мин); 1Н-ЯМР (300 МГц, СDСl3, выбранные сигналы): δ=6,85 (с, 1Н, Н-19), 5,21 (дд, J=11,3 и 1,9 Гц, 1Н, Н-15), 4,42 (дш, J=10,5 Гц, 1Н, Н-3), 3,71 (сш, 1Н, Н-7), 3,21 (д, J=14,3 Гц, 1Н, Н-17а), 3,13 (квд, J=6,8 и 3,0 Гц, 1Н, Н-6), 3,09 (дт, J= 9,8 и 3,4 Гц, 1Н, Н-13), 2,87 (дт, J=9,4 и 3,0 Гц, 1Н, Н-12), 2,73 (д, J= 14,3 Гц, 1Н, Н-17b), 2,68 (с, 3Н, Н-21), 2,63 (дд, J=16,6 и 11,7 Гц, 1Н, Н-2а), 2,27 (дт, J=14,7 и 2,3 Гц, 1Н, Н-14а), 2,24 (дд, J=16,6 и 2,6 Гц, 1Н, Н-2b), 1,39 (с, 3Н, Н-22), 1,22 (с, 3Н, Н-26), 1,19 (д, J=6,8 Гц, 3Н, Н-24), 1,05 (с, 3Н, Н-23), 0,99 (д, J=7,2 Гц, 3Н, Н-25).
ЭПОТИЛОН A-N-оксид (2а):
К 100 мг эпотилона А в 1 мл дихлорметана добавляют 100 мг 70%-ной м-хлорнадбензойной кислоты в 0,5 мл дихлорметана. После перемешивания в течение 6 ч при комнатной температуре продукт разбавляют дихлорметаном и последовательно экстрагируют путем встряхивания раствором сульфита натрия для разложения избыточной надкислоты и раствором бикарбоната натрия. Потом растворитель упаривают в вакууме, а остаток разделяют с помощью препаративной ВЭЖХ на RP-18-колонке с нуклеосилом (250 х 20 мм, растворитель - смесь метанола и воды 60:40). Выход - 60 мг бесцветного масла.
К 100 мг эпотилона А в 1 мл дихлорметана добавляют 100 мг 70%-ной м-хлорнадбензойной кислоты в 0,5 мл дихлорметана. После перемешивания в течение 6 ч при комнатной температуре продукт разбавляют дихлорметаном и последовательно экстрагируют путем встряхивания раствором сульфита натрия для разложения избыточной надкислоты и раствором бикарбоната натрия. Потом растворитель упаривают в вакууме, а остаток разделяют с помощью препаративной ВЭЖХ на RP-18-колонке с нуклеосилом (250 х 20 мм, растворитель - смесь метанола и воды 60:40). Выход - 60 мг бесцветного масла.
Rf - 0,60 (силикагель, ТХ, алюминиевая фольга, растворитель - смесь дихлорметана и метанола 9:1),
ESI-MC (отрицательные ионы) m/z 510;
УФ (метанол); λмакс 240 нм;
13С-ЯМР (CDCl3): С-1 70,5, С-2 39,9, С-3 70,8, С-4 55,1, С-5 221,4, С-6 40,9, С-7 72,9, С-8 37,6, С-9 31,8, С-10 22,8, С-11, 28,0, С-12 58,0, С-13 55,8, С-14 32,2, С-15 75,5, С-16 144,5, С-17 111,4, С-18 143,4, С-19 110,3, С-20 145,6, С-21 13,5, С-22 15,4, С-23 23,3, С-24 12,0, С-25 16,5, С-27 18,2 ppm;
1Н-ЯМР (СDCl3); 2а-Н 2,12 дд, 2b-Н 2,47 дд, 3-Н 4,55 дд, 3-ОН 6,48 ш, 6-Н 3,25 дкв, 7-Н 3,72 дд, 8-Н 1,81 м, 9а-Н 1,34 м, 9b-Н 1,56 м, 10-Н2 1,48 м, 11а-Н 1,27 м, 11b-H 1,87 м, 12-Н 2,92 ддд, 13-Н 2,98 м, 14а-Н 1,67 ддд, 14b-Н 2,23 д, 15-Н 5,33 д, 17-Н 6,82 с, 19-Н 7,09 с, 21-Н3 2,61с, 22-Н3 1,02 с, 23-Н3 1,42 с, 24-Н3, 1,18 д, 25-Н3, 0,99 д, 27-Н3 2,04 с ppm.
ESI-MC (отрицательные ионы) m/z 510;
УФ (метанол); λмакс 240 нм;
13С-ЯМР (CDCl3): С-1 70,5, С-2 39,9, С-3 70,8, С-4 55,1, С-5 221,4, С-6 40,9, С-7 72,9, С-8 37,6, С-9 31,8, С-10 22,8, С-11, 28,0, С-12 58,0, С-13 55,8, С-14 32,2, С-15 75,5, С-16 144,5, С-17 111,4, С-18 143,4, С-19 110,3, С-20 145,6, С-21 13,5, С-22 15,4, С-23 23,3, С-24 12,0, С-25 16,5, С-27 18,2 ppm;
1Н-ЯМР (СDCl3); 2а-Н 2,12 дд, 2b-Н 2,47 дд, 3-Н 4,55 дд, 3-ОН 6,48 ш, 6-Н 3,25 дкв, 7-Н 3,72 дд, 8-Н 1,81 м, 9а-Н 1,34 м, 9b-Н 1,56 м, 10-Н2 1,48 м, 11а-Н 1,27 м, 11b-H 1,87 м, 12-Н 2,92 ддд, 13-Н 2,98 м, 14а-Н 1,67 ддд, 14b-Н 2,23 д, 15-Н 5,33 д, 17-Н 6,82 с, 19-Н 7,09 с, 21-Н3 2,61с, 22-Н3 1,02 с, 23-Н3 1,42 с, 24-Н3, 1,18 д, 25-Н3, 0,99 д, 27-Н3 2,04 с ppm.
21-АЦЕТОКСИЭПОТИЛОН А (=21-АЦЕТИЛЭПОТИЛОН Е) (3а):
К 50 мг эпотилона А-Н-оксида (2а) в 0,5 мл дихлорметана добавляют 0,05 мл 2,6-ди-трет-бутилпиридина и 0,1 мл ацетангидрида. После нагревания массы в течение 15 мин до 75oС упаривают в вакууме растворитель и реагенты, а остаток разделяют методом препаративной ВЭЖХ на RP-18-колонке с нуклеосилом (250 х 20 мм, растворитель - смесь метанола и воды 60:40). Выход - 30 мг бесцветного масла.
К 50 мг эпотилона А-Н-оксида (2а) в 0,5 мл дихлорметана добавляют 0,05 мл 2,6-ди-трет-бутилпиридина и 0,1 мл ацетангидрида. После нагревания массы в течение 15 мин до 75oС упаривают в вакууме растворитель и реагенты, а остаток разделяют методом препаративной ВЭЖХ на RP-18-колонке с нуклеосилом (250 х 20 мм, растворитель - смесь метанола и воды 60:40). Выход - 30 мг бесцветного масла.
Rf - 0,50 (силикагель, ТХ, алюминиевая фольга, растворитель - смесь дихлорметана и метанола 95:5),
ESI-MC (отрицательные ионы) m/z 552;
УФ (метанол); λмакс 210, 250 нм;
1H-ЯMP (СDСl3, сигналы, отличающиеся от 2а): 15-Н 5,45 дд, 17-Н 6,60 с, 19-Н 7,15 с, 21-Н2 5,25 с, СН3СО 2,15 с ррm.
ESI-MC (отрицательные ионы) m/z 552;
УФ (метанол); λмакс 210, 250 нм;
1H-ЯMP (СDСl3, сигналы, отличающиеся от 2а): 15-Н 5,45 дд, 17-Н 6,60 с, 19-Н 7,15 с, 21-Н2 5,25 с, СН3СО 2,15 с ррm.
ЭПОТИЛОН Е (3б):
К 10 мг 21-ацетоксиэпотилона А (3а) в 0,5 мл метанола добавляют одну каплю концентрированного раствора аммиака, нагревают 1 ч до 40oС и выпаривают в вакууме досуха. Затем полученный остаток разделяют препаративной тонкослойной хроматографией. Выход - 6 мг. Продукт идентичен с оригинальной пробой эпотилона Е.
К 10 мг 21-ацетоксиэпотилона А (3а) в 0,5 мл метанола добавляют одну каплю концентрированного раствора аммиака, нагревают 1 ч до 40oС и выпаривают в вакууме досуха. Затем полученный остаток разделяют препаративной тонкослойной хроматографией. Выход - 6 мг. Продукт идентичен с оригинальной пробой эпотилона Е.
ПРИМЕР 4: 19-МЕТИЛЭПОТИЛОН А (4б)
К раствору эпотилона А (15 мг, 30 мкмолей) в ТГФ (1 мл) при -90oС добавляют н-бутиллития (10 мкл, 160 мкмолей, 1,6 М в гексане). В результате раствор моментально окрашивается в золотисто-оранжевый цвет. После перемешивания в течение 15 мин при -90oС к реакционному раствору добавляют метилйодид (100 мкл, 1,6 ммоля). Полученный в результате зеленовато-желтый раствор нагревают до -30oС и завершают реакцию 2 мл буфера (рН - 7,0). Затем эмульсию с помощью 0,1 н. соляной кислоты доводят до значения рН 6. После насыщения эмульсии твердым хлористым натрием водную фазу экстрагируют CH2Cl2 (2 х 5 мл) и этилацетатом (5 мл), после чего собранные органические фазы высушивают сульфатом магния, фильтруют и, наконец, растворитель удаляют на ротационном испарителе. Очистка осуществляется методом ПСХ (1 х 200 х 200 мм, 10% MeOH:CH2Cl2) и ВЭЖХ (RР 18, 250 х 16 мм МеОН: Н2О 65:35). Выделяют следующие продукты:
1. 2,5 мг (17%) 19-метилэпотилона A. Rf: 0,50 (10% МеОН:СН2Сl2); Rt: 11,70 мин (RP 18, 250 х 4 мм, МеОН:Н2О 65:35, 1 мл/мин); МС: (m/z)=508 (М+), 420, 320; 1H-ЯМР (300 МГц, СDСl3, выбранные сигналы): δ=6,41 (с, 1Н, Н-17), 5,46 (дд, J= 9,0 и 2,3 Гц, 1Н, Н-15), 4,15 (дд, J=10,5 и 3,0 Гц, 1Н, Н-3), 3,77 (дд, J= 8 и 4 Гц, 1Н, Н-7), 3,20 (квд, J=6,8 и 4,5 Гц, 1Н, Н-6), 3,04 (дт, J= 7,5 и 3,8 Гц, 1Н, Н-13), 2,91 (дт, J=7,5 и 3,8 Гц, 1Н, Н-12), 2,61 (с, 3Н, Н-21), 2,51 (дд, J=14,4 и 10,5 Гц, 1Н, Н-2а), 2,38 (дд, J=14,4 и 3,0 Гц, 1Н, Н-2b), 2,32 с, 3Н, Н-27), 2,15 (ддд, J=15,1, 3,8 и 3,0 Гц, 1Н, Н-14а), 2,01 (д, J=1,5 Гц, 3Н, Н-26), 1,91 (дт, J=15,1 и 8,8 Гц, 1Н, Н-14b), 1,34 (с, 3Н, Н-22), 1,16 (д, J=6,8 Гц, 3Н, Н-24), 1,10 (с, 3Н, Н-23), 1,00 (д, J=6,8 Гц, 3Н, Н-25).
К раствору эпотилона А (15 мг, 30 мкмолей) в ТГФ (1 мл) при -90oС добавляют н-бутиллития (10 мкл, 160 мкмолей, 1,6 М в гексане). В результате раствор моментально окрашивается в золотисто-оранжевый цвет. После перемешивания в течение 15 мин при -90oС к реакционному раствору добавляют метилйодид (100 мкл, 1,6 ммоля). Полученный в результате зеленовато-желтый раствор нагревают до -30oС и завершают реакцию 2 мл буфера (рН - 7,0). Затем эмульсию с помощью 0,1 н. соляной кислоты доводят до значения рН 6. После насыщения эмульсии твердым хлористым натрием водную фазу экстрагируют CH2Cl2 (2 х 5 мл) и этилацетатом (5 мл), после чего собранные органические фазы высушивают сульфатом магния, фильтруют и, наконец, растворитель удаляют на ротационном испарителе. Очистка осуществляется методом ПСХ (1 х 200 х 200 мм, 10% MeOH:CH2Cl2) и ВЭЖХ (RР 18, 250 х 16 мм МеОН: Н2О 65:35). Выделяют следующие продукты:
1. 2,5 мг (17%) 19-метилэпотилона A. Rf: 0,50 (10% МеОН:СН2Сl2); Rt: 11,70 мин (RP 18, 250 х 4 мм, МеОН:Н2О 65:35, 1 мл/мин); МС: (m/z)=508 (М+), 420, 320; 1H-ЯМР (300 МГц, СDСl3, выбранные сигналы): δ=6,41 (с, 1Н, Н-17), 5,46 (дд, J= 9,0 и 2,3 Гц, 1Н, Н-15), 4,15 (дд, J=10,5 и 3,0 Гц, 1Н, Н-3), 3,77 (дд, J= 8 и 4 Гц, 1Н, Н-7), 3,20 (квд, J=6,8 и 4,5 Гц, 1Н, Н-6), 3,04 (дт, J= 7,5 и 3,8 Гц, 1Н, Н-13), 2,91 (дт, J=7,5 и 3,8 Гц, 1Н, Н-12), 2,61 (с, 3Н, Н-21), 2,51 (дд, J=14,4 и 10,5 Гц, 1Н, Н-2а), 2,38 (дд, J=14,4 и 3,0 Гц, 1Н, Н-2b), 2,32 с, 3Н, Н-27), 2,15 (ддд, J=15,1, 3,8 и 3,0 Гц, 1Н, Н-14а), 2,01 (д, J=1,5 Гц, 3Н, Н-26), 1,91 (дт, J=15,1 и 8,8 Гц, 1Н, Н-14b), 1,34 (с, 3Н, Н-22), 1,16 (д, J=6,8 Гц, 3Н, Н-24), 1,10 (с, 3Н, Н-23), 1,00 (д, J=6,8 Гц, 3Н, Н-25).
2. Около 50% эпотилона А.
ПРИМЕР 5: 19-БРОМЭПОТИЛОН А (4 а)
К раствору эпотилона А (25 мг, 50 мкмолей) в ТГФ (2,5 мл) при -90oС добавляют н-бутиллития (160 мкл, 225 мкмолей, 1,6 М в гексане). Раствор сразу же окрашивается в золотисто-оранжевый цвет. После перемешивания в течение 15 мин при -90oС добавляют N-бромсукцинимида (27 мг, 150 мкмолей) в ТГФ (0,5 мл). Полученный раствор постепенно обесцвечивается. Полученную в результате коричневатую реакционную смесь нагревают до -30oС и с помощью 0,1 н. соляной кислоты (1 мл) доводят до значения рН 6,5. После насыщения твердым хлоридом натрия водную фазу экстрагируют CH2Cl2 (2 х 5 мл) и этилацетатом (5 мл), собранные органические фазы высушивают сульфатом магния, фильтруют, а растворитель удаляют на ротационном испарителе. Очистка осуществляется методом ПСХ (1 х 200 х 200 мм, 10% MeOH:CH2Cl2) и ВЭЖХ (RP 18, 250 х 16 мм, MeOH:H2O 65:35). Выделяют следующие продукты:
1. 2,6 мг (9%) 19-бромэпотилона A. Rf: 0,53 (10% MeOH:CH2Cl2); Rt: 20,78 мин (RP 18, 250•4 мм, МеОН:Н2О 65:35, 1 мл/мин); MC: (m/z)=574 и 572 (М+), 556, 554, 468, 466, 386, 384, 341; 1H-ЯМР (300 МГц, СDСl3, выбранные сигналы): δ=6,43 (с, 1Н, Н-17), 5,46 (дд, J=8,7 и 2,3 Гц, 1Н, Н-15), 4,13 (ддд, J= 9,4, 6,0 и 3,8 Гц, 1Н, H-3), 3,80 (дд, J=8 и 4 Гц, 1H, H-7), 3,38 (д, J= 6,0 Гц, 1Н, ОН), 3,22 (квд, J=6,8 и 5,3 Гц, 1H, Н-6), 3,05 (дт, J=8,3 и 4,1 Гц, 1H, Н-13), 2,91 (дт, J=7,5 и 3,7 Гц, 1H, Н-12), 2,66 (с, 3Н, Н-21), 2,55 (дд, J=14,7 и 9,4 Гц, 1H, Н-2а), 2,47 (дд, J=14,7 и 3,8 Гц, 1H, Н-2b), 2,16 (д, J=1,1 Гц, 3Н, Н-26), 2,14 (дт, J=14,7 и 3,8 Гц, 1H, H-14a), 1,90 (дт, J= 15 и 8,3 Гц, 1Н, H-14b), 1,34 (с, 3Н, Н-22), 1,17 (д, J=6,8 Гц, 3Н, Н-24), 1,11 (с, 3Н, Н-23), 1,01 (д, J=6,8 Гц, 3Н, Н-25).
К раствору эпотилона А (25 мг, 50 мкмолей) в ТГФ (2,5 мл) при -90oС добавляют н-бутиллития (160 мкл, 225 мкмолей, 1,6 М в гексане). Раствор сразу же окрашивается в золотисто-оранжевый цвет. После перемешивания в течение 15 мин при -90oС добавляют N-бромсукцинимида (27 мг, 150 мкмолей) в ТГФ (0,5 мл). Полученный раствор постепенно обесцвечивается. Полученную в результате коричневатую реакционную смесь нагревают до -30oС и с помощью 0,1 н. соляной кислоты (1 мл) доводят до значения рН 6,5. После насыщения твердым хлоридом натрия водную фазу экстрагируют CH2Cl2 (2 х 5 мл) и этилацетатом (5 мл), собранные органические фазы высушивают сульфатом магния, фильтруют, а растворитель удаляют на ротационном испарителе. Очистка осуществляется методом ПСХ (1 х 200 х 200 мм, 10% MeOH:CH2Cl2) и ВЭЖХ (RP 18, 250 х 16 мм, MeOH:H2O 65:35). Выделяют следующие продукты:
1. 2,6 мг (9%) 19-бромэпотилона A. Rf: 0,53 (10% MeOH:CH2Cl2); Rt: 20,78 мин (RP 18, 250•4 мм, МеОН:Н2О 65:35, 1 мл/мин); MC: (m/z)=574 и 572 (М+), 556, 554, 468, 466, 386, 384, 341; 1H-ЯМР (300 МГц, СDСl3, выбранные сигналы): δ=6,43 (с, 1Н, Н-17), 5,46 (дд, J=8,7 и 2,3 Гц, 1Н, Н-15), 4,13 (ддд, J= 9,4, 6,0 и 3,8 Гц, 1Н, H-3), 3,80 (дд, J=8 и 4 Гц, 1H, H-7), 3,38 (д, J= 6,0 Гц, 1Н, ОН), 3,22 (квд, J=6,8 и 5,3 Гц, 1H, Н-6), 3,05 (дт, J=8,3 и 4,1 Гц, 1H, Н-13), 2,91 (дт, J=7,5 и 3,7 Гц, 1H, Н-12), 2,66 (с, 3Н, Н-21), 2,55 (дд, J=14,7 и 9,4 Гц, 1H, Н-2а), 2,47 (дд, J=14,7 и 3,8 Гц, 1H, Н-2b), 2,16 (д, J=1,1 Гц, 3Н, Н-26), 2,14 (дт, J=14,7 и 3,8 Гц, 1H, H-14a), 1,90 (дт, J= 15 и 8,3 Гц, 1Н, H-14b), 1,34 (с, 3Н, Н-22), 1,17 (д, J=6,8 Гц, 3Н, Н-24), 1,11 (с, 3Н, Н-23), 1,01 (д, J=6,8 Гц, 3Н, Н-25).
2. Около 60% эпотилона А (см. примеры 1а-5а в конце описания).
Пример на получение эпотилон B-N-оксида
К раствору 2 ммоль м-хлорпербензойной кислоты в 11 мл дихлорметана добавили 507 мг (1 ммоль) эпотилона В и перемешивали 3 часа при комнатной температуре. Добавляли этилацетат и избыток пербензойной кислоты восстанавливали раствором сульфита натрия. Органический слой сушили посредством сульфата магния и выпаривали до получения 0,6 г сырого продукта, содержащего 390 мг (75%) желаемого продукта, определенного путем ВЭЖХ-хроматографии. Разделение проводили хроматографически на HD-SIL С-18, 35-70 мкм, колонка (диаметр 9 см, длина 83 см) посредством системы растворителей 45% ацетонитрил / 55% аммонийно-ацетатный буфер 50 мМ при рН 7,5, определение при 254 нм, выход 207 мг. DC: Rf=0,19 (силикагель Si60, дихлорметан/метанол 95:5, определение посредством системы ванилин/серная кислота, серо-голубое окрашивание при нагревании до 120oС);
ВЭЖХ: Rf= 4,1 мин (нуклеосил, С-18, 7 мкм, колонка 250 х 4 мм, система растворителей метанол/вода 70:30, 1 мл/мин, определение при 254 нм);
УФ (МеОН): λмакс(ε) = 283 sh (log ε 3,22), 264 sh (3,63) 236 nm (4,19);
ИК (KBr):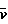 =3439, 2963, 2936, 2877, 1740, 1689 см-1;
=3439, 2963, 2936, 2877, 1740, 1689 см-1;
1Н-ЯМР (CDCl3): δ= 2,13 (dd, J=12,9, 12,0, 2-На); 2,47 (t=12,0, 2-Hb); 4,58 (m, 3-H); 3,29 (dq, J=2,0, 5,8, 6-Н); 3,70 (d, J=5,8, 7-Н); 1,20-1,90 (m, 8-H, 9-H2, 10-H2, 11-H2); 2,75 (m, 13-H); 1,66 (m, 14-Ha); 2,23 (dbr, J= 15,6); 5,32 (d, J=11,6, 15-Н); 6,79 (sbr, 17-H); 7,08 (s, 19-H); 2,60 (s, 21-Н3); 1,02 (s, 22-Н3); 1,27 (s, 23-H3); 1,16 (d, J=6,8, 24-Н3); 0,99 (d, J=7,1, 25-Н3); 1,41 (s, 26-Н3); 2,07 (s, 27-Н3);
13С-ЯМР (150 МГц, CDCl3): δ=170,5 (С-1), 40,0 (С-2), 70,9 (С-3), 55,1 (С-4), 221,4 (С-5), 40,7 (С-6), 72,5 (С-7), 37,3 (С-8), 31,5 (С-9), 22,0 (С-10), 33,3 (С-11), 62,3 (С-12), 63,0 (С-13), 33,4 (С-14), 75,6 (С-15), 144,3 (С-16), 111,1 (С-17), 143,3 (С-18), 110,3 (С-19), 144,7 (С-20), 13,4 (С-21), 15,3 (С-22), 23,2 (С-23), 12,5 (С-24), 16,5 (С-25), 22,2 (С-26), 18,2 (С-27); [a] EI-MS (70 eV): m/z (%) 523,2604 (40 M+), ber. (C27H41NO7S) 523,2629, 424 (38), 336 (42), 194 (18), 182 (75), 154 (100), 126 (42).
К раствору 2 ммоль м-хлорпербензойной кислоты в 11 мл дихлорметана добавили 507 мг (1 ммоль) эпотилона В и перемешивали 3 часа при комнатной температуре. Добавляли этилацетат и избыток пербензойной кислоты восстанавливали раствором сульфита натрия. Органический слой сушили посредством сульфата магния и выпаривали до получения 0,6 г сырого продукта, содержащего 390 мг (75%) желаемого продукта, определенного путем ВЭЖХ-хроматографии. Разделение проводили хроматографически на HD-SIL С-18, 35-70 мкм, колонка (диаметр 9 см, длина 83 см) посредством системы растворителей 45% ацетонитрил / 55% аммонийно-ацетатный буфер 50 мМ при рН 7,5, определение при 254 нм, выход 207 мг. DC: Rf=0,19 (силикагель Si60, дихлорметан/метанол 95:5, определение посредством системы ванилин/серная кислота, серо-голубое окрашивание при нагревании до 120oС);
ВЭЖХ: Rf= 4,1 мин (нуклеосил, С-18, 7 мкм, колонка 250 х 4 мм, система растворителей метанол/вода 70:30, 1 мл/мин, определение при 254 нм);
УФ (МеОН): λмакс(ε) = 283 sh (log ε 3,22), 264 sh (3,63) 236 nm (4,19);
ИК (KBr):
1Н-ЯМР (CDCl3): δ= 2,13 (dd, J=12,9, 12,0, 2-На); 2,47 (t=12,0, 2-Hb); 4,58 (m, 3-H); 3,29 (dq, J=2,0, 5,8, 6-Н); 3,70 (d, J=5,8, 7-Н); 1,20-1,90 (m, 8-H, 9-H2, 10-H2, 11-H2); 2,75 (m, 13-H); 1,66 (m, 14-Ha); 2,23 (dbr, J= 15,6); 5,32 (d, J=11,6, 15-Н); 6,79 (sbr, 17-H); 7,08 (s, 19-H); 2,60 (s, 21-Н3); 1,02 (s, 22-Н3); 1,27 (s, 23-H3); 1,16 (d, J=6,8, 24-Н3); 0,99 (d, J=7,1, 25-Н3); 1,41 (s, 26-Н3); 2,07 (s, 27-Н3);
13С-ЯМР (150 МГц, CDCl3): δ=170,5 (С-1), 40,0 (С-2), 70,9 (С-3), 55,1 (С-4), 221,4 (С-5), 40,7 (С-6), 72,5 (С-7), 37,3 (С-8), 31,5 (С-9), 22,0 (С-10), 33,3 (С-11), 62,3 (С-12), 63,0 (С-13), 33,4 (С-14), 75,6 (С-15), 144,3 (С-16), 111,1 (С-17), 143,3 (С-18), 110,3 (С-19), 144,7 (С-20), 13,4 (С-21), 15,3 (С-22), 23,2 (С-23), 12,5 (С-24), 16,5 (С-25), 22,2 (С-26), 18,2 (С-27); [a] EI-MS (70 eV): m/z (%) 523,2604 (40 M+), ber. (C27H41NO7S) 523,2629, 424 (38), 336 (42), 194 (18), 182 (75), 154 (100), 126 (42).
Определена также цитотоксическая активность эпотилон B-N-оксида, полученные данные приведены в таблице.
Claims (15)
1. Способ получения модифицированных в 16,17-положении эпотилонов, согласно которому защищенные в положении 3,7 или незащищенные эпотилоны А или В а) гидрируют по двойной связи в положении 16,17 либо в) по двойной связи в положении 16,17 проводят эпоксидирование и, в случае необходимости, полученный эпоксид восстанавливают до спирта в положении 16.
2. Способ по п.1, отличающийся тем, что в случае способа (а) гидрируют диимином или водородом в присутствии гетерогенного или гомогенного металлического катализатора или в случае способа (в) эпоксидируют надкислотой или диоксираном.
3. Способ получения эпотилон-N-оксидов, при котором защищенные в положении 3,7 или незащищенные эпотилоны А или В известным образом переводят в N-оксид, и полученный N-оксид при необходимости подвергают реакции Катада.
4. Способ по п.3, отличающийся тем, что N-оксидирование осуществляют надкислотой или диоксираном.
5. Способ по п.3, отличающийся тем, что реакцию Катада осуществляют с помощью активированного производного карбоновой кислоты, в частности, ее ангидрида или хлорангидрида.
6. Способ по п.5, отличающийся тем, что реакцию Катада осуществляют с помощью ацетангидрида и полученные 21-ацетоксиэпотилоны, в случае необходимости, известным образом расщепляют на 21-оксипотилоны А или В (эпотилоны Е и F соответственно).
7. Способ по п.6, отличающийся тем, что необязательное расщепление проводят гидролитическим или ферментативным путем.
8. Способ получения модифицированных в С-19 положении эпотилонов, согласно которому защищенные в положении 3,7 или незащищенные эпотилоны А или В металлизируют в положении С-19 и известным образом с помощью электрофильных реагентов улавливают в виде модифицированных в положении С-19 алкил-, галоген-, серозамещенных эпотилонов.
9. Способ по п.8, отличающийся тем, что металлизацию проводят с помощью бутиллития.
10. Способ получения модифицированных в С-27 положении эпотилонов, при котором алкильную группу в положениях 17,16 и 27 известным образом по С-27-метильной группе замещают гетероатомом.
11. Способ по п.10, отличающийся тем, что С-27-метильную группу замещают атомом брома, в частности, с помощью N-бромсукцинимида, и полученный бромид, в случае необходимости, переводят в С-27-оксисоединение.
12. Модифицированные эпотилоны общей формулы I

где А обозначает R3OCH2 или метил;
V обозначает Н, алкил, атом галогена, S-алкил, S-арил или Si(алкил)3;
W обозначает Н, ОН, атом галогена;
R обозначает Н или метил;
R1 обозначает Н, ацетил, или силил;
R2 обозначает Н, ацетил, или силил;
R3 обозначает Н или ацетил;
Х обозначает ОН или Н;
Y обозначает Н или ОН, или
Х и Y вместе обозначают двойную связь или -О-, в случае необходимости Z обозначает О-.
где А обозначает R3OCH2 или метил;
V обозначает Н, алкил, атом галогена, S-алкил, S-арил или Si(алкил)3;
W обозначает Н, ОН, атом галогена;
R обозначает Н или метил;
R1 обозначает Н, ацетил, или силил;
R2 обозначает Н, ацетил, или силил;
R3 обозначает Н или ацетил;
Х обозначает ОН или Н;
Y обозначает Н или ОН, или
Х и Y вместе обозначают двойную связь или -О-, в случае необходимости Z обозначает О-.
13. Модифицированные эпотилоны общей формулы I по п.12, полученные способом по любому из пп.1-11.
14. Модифицированный эпотилон общей формулы I по п.12, представляющий собой эпотилон-N-оксид (эпотилон А-N-оксид), полученный тем, что незамещенный в положении 3,7 эпотилон А известным образом переводят в N-оксид, полученный N-оксид при необходимости подвергают О-алкилированию с получением продукта О-алкилирования.
15. Модифицированный эпотилон по п.12, представляющей собой соединение формулы
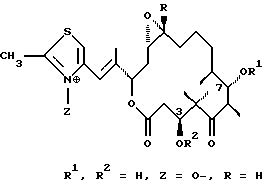
16. Модифицированный эпотилон по п.12, представляющий собой соединение формулы

17. Модифицированный эпотилон общей формулы I по п.12, представляющий собой эпотилон-N-оксид (эпотилон В-N-оксид), полученный тем, что незащищенный в положении 3,7 эпотилон B известным образом переводят в N-оксид, полученный N-оксид при необходимости подвергают О-алкилированию с получением продукта О-алкилирования.
16. Модифицированный эпотилон по п.12, представляющий собой соединение формулы
17. Модифицированный эпотилон общей формулы I по п.12, представляющий собой эпотилон-N-оксид (эпотилон В-N-оксид), полученный тем, что незащищенный в положении 3,7 эпотилон B известным образом переводят в N-оксид, полученный N-оксид при необходимости подвергают О-алкилированию с получением продукта О-алкилирования.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19707505 | 1997-02-25 | ||
| DE19707505.3 | 1997-02-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU99120378A RU99120378A (ru) | 2001-07-20 |
| RU2201932C2 true RU2201932C2 (ru) | 2003-04-10 |
Family
ID=7821415
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU99120378/04A RU2201932C2 (ru) | 1997-02-25 | 1998-02-25 | Модифицированные в боковой цепи эпотилоны |
Country Status (24)
| Country | Link |
|---|---|
| US (1) | US6359140B1 (ru) |
| EP (2) | EP1201666A3 (ru) |
| JP (1) | JP2001513098A (ru) |
| KR (1) | KR100494179B1 (ru) |
| CN (2) | CN1544436A (ru) |
| AR (1) | AR011878A1 (ru) |
| AT (1) | ATE221888T1 (ru) |
| AU (1) | AU736062B2 (ru) |
| BR (1) | BR9807742B1 (ru) |
| CA (1) | CA2281105A1 (ru) |
| CZ (1) | CZ298027B6 (ru) |
| DE (2) | DE59805110D1 (ru) |
| DK (1) | DK0975638T3 (ru) |
| ES (1) | ES2183338T3 (ru) |
| HU (1) | HU228851B1 (ru) |
| IL (1) | IL131343A (ru) |
| NO (1) | NO327211B1 (ru) |
| NZ (1) | NZ337195A (ru) |
| PL (1) | PL190422B1 (ru) |
| PT (1) | PT975638E (ru) |
| RU (1) | RU2201932C2 (ru) |
| TW (1) | TW480263B (ru) |
| WO (1) | WO1998038192A1 (ru) |
| ZA (1) | ZA981575B (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2343155C2 (ru) * | 2002-09-13 | 2009-01-10 | Новартис Аг | Способ получения производных эпотилона |
Families Citing this family (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4183099B2 (ja) | 1995-11-17 | 2008-11-19 | ゲゼルシャフト・フュア・ビオテヒノロジッシェ・フォルシュング・ミット・ベシュレンクテル・ハフツング(ゲー・ベー・エフ) | エポチロンcおよびd、製造法ならびに組成物 |
| ES2312695T3 (es) * | 1996-11-18 | 2009-03-01 | Gesellschaft Fur Biotechnologische Forschung Mbh (Gbf) | Epotilones e y f. |
| US6204388B1 (en) | 1996-12-03 | 2001-03-20 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US20050043376A1 (en) * | 1996-12-03 | 2005-02-24 | Danishefsky Samuel J. | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| CA2273083C (en) | 1996-12-03 | 2012-09-18 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| US6867305B2 (en) | 1996-12-03 | 2005-03-15 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US6660758B1 (en) | 1996-12-13 | 2003-12-09 | The Scripps Research Institute | Epothilone analogs |
| US6441186B1 (en) | 1996-12-13 | 2002-08-27 | The Scripps Research Institute | Epothilone analogs |
| US6380394B1 (en) | 1996-12-13 | 2002-04-30 | The Scripps Research Institute | Epothilone analogs |
| JP2001513098A (ja) * | 1997-02-25 | 2001-08-28 | ゲゼルシャフト フュア バイオテクノロギッシェ フォーシュンク エム ベー ハー(ゲー ベー エフ) | 側鎖を修飾したエポチロン |
| US6605599B1 (en) | 1997-07-08 | 2003-08-12 | Bristol-Myers Squibb Company | Epothilone derivatives |
| US6320045B1 (en) | 1997-12-04 | 2001-11-20 | Bristol-Myers Squibb Company | Process for the reduction of oxiranyl epothilones to olefinic epothilones |
| US6365749B1 (en) | 1997-12-04 | 2002-04-02 | Bristol-Myers Squibb Company | Process for the preparation of ring-opened epothilone intermediates which are useful for the preparation of epothilone analogs |
| US6683100B2 (en) | 1999-01-19 | 2004-01-27 | Novartis Ag | Organic compounds |
| US6399638B1 (en) | 1998-04-21 | 2002-06-04 | Bristol-Myers Squibb Company | 12,13-modified epothilone derivatives |
| US6498257B1 (en) | 1998-04-21 | 2002-12-24 | Bristol-Myers Squibb Company | 2,3-olefinic epothilone derivatives |
| DE19826988A1 (de) * | 1998-06-18 | 1999-12-23 | Biotechnolog Forschung Gmbh | Epothilon-Nebenkomponenten |
| PT987268E (pt) * | 1998-08-05 | 2002-09-30 | Biotechnolog Forschung Mbh Gbf | Agentes farmaceuticos contendo n-oxido de epotilona a e/ou n-oxido de epotilona b |
| US6780620B1 (en) | 1998-12-23 | 2004-08-24 | Bristol-Myers Squibb Company | Microbial transformation method for the preparation of an epothilone |
| US6596875B2 (en) | 2000-02-07 | 2003-07-22 | James David White | Method for synthesizing epothilones and epothilone analogs |
| DE19907588A1 (de) * | 1999-02-22 | 2000-08-24 | Biotechnolog Forschung Gmbh | C-21 Modifizierte Epothilone |
| CZ301498B6 (cs) | 1999-02-22 | 2010-03-24 | Gesellschaft Fuer Biotechnologische Forschung Mbh (Gbf) | C-21 modifikované epothilony |
| US20020058286A1 (en) * | 1999-02-24 | 2002-05-16 | Danishefsky Samuel J. | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US6291684B1 (en) | 1999-03-29 | 2001-09-18 | Bristol-Myers Squibb Company | Process for the preparation of aziridinyl epothilones from oxiranyl epothilones |
| US7125875B2 (en) | 1999-04-15 | 2006-10-24 | Bristol-Myers Squibb Company | Cyclic protein tyrosine kinase inhibitors |
| EP1169038B9 (en) | 1999-04-15 | 2013-07-10 | Bristol-Myers Squibb Company | Cyclic protein tyrosine kinase inhibitors |
| US7125893B1 (en) * | 1999-04-30 | 2006-10-24 | Schering Ag | 6-alkenyl-, 6-alkinyl- and 6-epoxy-epothilone derivatives, process for their production, and their use in pharmaceutical preparations |
| PE20010116A1 (es) * | 1999-04-30 | 2001-02-15 | Schering Ag | Derivados de 6-alquenil-, 6-alquinil- y 6-epoxi-epotilona, procedimientos para su preparacion |
| AU775373B2 (en) | 1999-10-01 | 2004-07-29 | Immunogen, Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| US6518421B1 (en) | 2000-03-20 | 2003-02-11 | Bristol-Myers Squibb Company | Process for the preparation of epothilone analogs |
| US6593115B2 (en) | 2000-03-24 | 2003-07-15 | Bristol-Myers Squibb Co. | Preparation of epothilone intermediates |
| US6589968B2 (en) * | 2001-02-13 | 2003-07-08 | Kosan Biosciences, Inc. | Epothilone compounds and methods for making and using the same |
| UA75365C2 (en) | 2000-08-16 | 2006-04-17 | Bristol Myers Squibb Co | Epothilone analog polymorph modifications, a method for obtaining thereof (variants), a pharmaceutical composition based thereon |
| ES2304240T3 (es) | 2001-01-25 | 2008-10-01 | Bristol-Myers Squibb Company | Procedimientos para la preparacion de preparaciones farmaceuticas que contienen analogos de epotilona para el tratamiento de cancer. |
| WO2002058699A1 (en) * | 2001-01-25 | 2002-08-01 | Bristol-Myers Squibb Company | Pharmaceutical forms of epothilones for oral administration |
| EE200300323A (et) | 2001-01-25 | 2003-10-15 | Bristol-Myers Squibb Company | Epotilooni analoogi sisaldav parenteraalne ravimpreparaat, meetod selle valmistamiseks ning kasutamine |
| US6893859B2 (en) * | 2001-02-13 | 2005-05-17 | Kosan Biosciences, Inc. | Epothilone derivatives and methods for making and using the same |
| KR20040028720A (ko) | 2001-02-20 | 2004-04-03 | 브리스톨-마이어스스퀴브컴파니 | 치료불응성 종양 치료용 에포틸론 유도체 |
| CN1774253A (zh) | 2001-02-20 | 2006-05-17 | 布里斯托尔-迈尔斯斯奎布公司 | 用环氧丙酯酮衍生物治疗顽固性肿瘤 |
| NZ527557A (en) * | 2001-02-27 | 2005-05-27 | Biotechnolog Forschung Gmbh | Degradation of epothilones and ethynyl substituted epothilones |
| IL157443A0 (en) | 2001-03-14 | 2004-03-28 | Bristol Myers Squibb Co | Pharmaceutical compositions for the treatment of cancer including an epothilone analog and a chemotherapeutic agent |
| MXPA03010909A (es) | 2001-06-01 | 2004-02-17 | Bristol Myers Squibb Co | Derivados de epotilona. |
| TW200303202A (en) | 2002-02-15 | 2003-09-01 | Bristol Myers Squibb Co | Method of preparation of 21-amino epothilone derivatives |
| CA2478223C (en) * | 2002-03-08 | 2012-05-15 | Novartis Ag | Combinations comprising epothilone derivatives and alkylating agents |
| SI1483251T1 (sl) | 2002-03-12 | 2010-03-31 | Bristol Myers Squibb Co | C cian epotilonski derivati |
| AU2003218107A1 (en) | 2002-03-12 | 2003-09-29 | Bristol-Myers Squibb Company | C12-cyano epothilone derivatives |
| TW200403994A (en) | 2002-04-04 | 2004-03-16 | Bristol Myers Squibb Co | Oral administration of EPOTHILONES |
| TW200400191A (en) | 2002-05-15 | 2004-01-01 | Bristol Myers Squibb Co | Pharmaceutical compositions and methods of using C-21 modified epothilone derivatives |
| US7405234B2 (en) | 2002-05-17 | 2008-07-29 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| WO2003105828A1 (en) | 2002-06-14 | 2003-12-24 | Bristol-Myers Squibb Company | Combination of epothilone analogs and chemotherapeutic agents for the treatment of proliferative diseases |
| EP2186811A1 (en) | 2002-08-23 | 2010-05-19 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| US6921769B2 (en) | 2002-08-23 | 2005-07-26 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US7649006B2 (en) | 2002-08-23 | 2010-01-19 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| AU2003275068B2 (en) | 2002-09-23 | 2009-09-17 | Bristol-Myers Squibb Company | Methods for the preparation, isolation and purification of epothilone B, and X-Ray crystal structures of epothilone B |
| WO2004045518A2 (en) | 2002-11-15 | 2004-06-03 | Bristol-Myers Squibb Company | Open chain prolyl urea-related modulators of androgen receptor function |
| US7820702B2 (en) | 2004-02-04 | 2010-10-26 | Bristol-Myers Squibb Company | Sulfonylpyrrolidine modulators of androgen receptor function and method |
| US7378426B2 (en) | 2004-03-01 | 2008-05-27 | Bristol-Myers Squibb Company | Fused heterotricyclic compounds as inhibitors of 17β-hydroxysteroid dehydrogenase 3 |
| US7625923B2 (en) | 2004-03-04 | 2009-12-01 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US7696241B2 (en) | 2004-03-04 | 2010-04-13 | Bristol-Myers Squibb Company | Bicyclic compounds as modulators of androgen receptor function and method |
| EP1824458A1 (en) * | 2004-11-18 | 2007-08-29 | Bristol-Myers Squibb Company | Enteric coated bead comprising epothilone or an epothilone analog, and preparation and administration thereof |
| AR052142A1 (es) * | 2004-11-18 | 2007-03-07 | Bristol Myers Squibb Co | Perla recubierta enterica que comprende ixabepilona, y preparacion y administracion de la misma |
| JP4954983B2 (ja) | 2005-05-18 | 2012-06-20 | ファーマサイエンス・インコーポレイテッド | Birドメイン結合化合物 |
| CN101535300B (zh) | 2006-05-16 | 2014-05-28 | 埃格拉医疗公司 | Iap bir域结合化合物 |
| JP2010511408A (ja) | 2006-12-04 | 2010-04-15 | ザ・ボード・オブ・トラスティーズ・オブ・ザ・ユニバーシティ・オブ・イリノイ | 癌をCpGリッチDNAおよびキュプレドキシンで治療するための組成物および方法 |
| US9284350B2 (en) | 2010-02-12 | 2016-03-15 | Pharmascience Inc. | IAP BIR domain binding compounds |
| US20110300150A1 (en) | 2010-05-18 | 2011-12-08 | Scott Eliasof | Compositions and methods for treatment of autoimmune and other disease |
| JP5889337B2 (ja) | 2011-01-20 | 2016-03-22 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | Mriマーカー、送達および抜取りシステムならびにこれらの製造方法および使用方法 |
| WO2012171020A1 (en) | 2011-06-10 | 2012-12-13 | Mersana Therapeutics, Inc. | Protein-polymer-drug conjugates |
| CN102863474A (zh) | 2011-07-09 | 2013-01-09 | 陈小平 | 一类治疗细胞增殖性疾病的铂化合物、其制备方法和应用 |
| CN102993239A (zh) | 2011-09-19 | 2013-03-27 | 陈小平 | 离去基团含氨基或烷胺基的丁二酸衍生物的铂类化合物 |
| WO2014075391A1 (zh) | 2012-11-17 | 2014-05-22 | 北京市丰硕维康技术开发有限责任公司 | 离去基团是含氨基或烷氨基的丙二酸衍生物的铂类化合物 |
| CA2892863C (en) | 2012-12-10 | 2022-03-15 | Mersana Therapeutics, Inc. | Polymeric scaffold based on phf for targeted drug delivery |
| WO2014093640A1 (en) | 2012-12-12 | 2014-06-19 | Mersana Therapeutics,Inc. | Hydroxy-polmer-drug-protein conjugates |
| TN2015000543A1 (en) | 2013-06-11 | 2017-04-06 | Bayer Pharma AG | Combinations for the treatment of cancer comprising a mps-1 kinase inhibitor and a mitotic inhibitor |
| KR102087850B1 (ko) | 2013-10-11 | 2020-03-12 | 메르사나 테라퓨틱스, 인코포레이티드 | 단백질-고분자-약물 접합체 |
| ES2754397T3 (es) | 2013-10-11 | 2020-04-17 | Asana Biosciences Llc | Conjugados de proteína-polímero-fármaco |
| WO2018004338A1 (en) | 2016-06-27 | 2018-01-04 | Tagworks Pharmaceuticals B.V. | Cleavable tetrazine used in bio-orthogonal drug activation |
| US11135307B2 (en) | 2016-11-23 | 2021-10-05 | Mersana Therapeutics, Inc. | Peptide-containing linkers for antibody-drug conjugates |
| AU2018290330A1 (en) | 2017-06-22 | 2020-01-02 | Mersana Therapeutics, Inc. | Methods of producing drug-carrying polymer scaffolds and protein-polymer-drug conjugates |
| CA3099421C (en) | 2018-05-04 | 2025-05-06 | Tagworks Pharmaceuticals B.V. | COMPOUNDS INCLUDING A BINDER TO INCREASE TRANSCYCLOOCTENE STABILITY |
| AU2019262520B2 (en) | 2018-05-04 | 2025-07-10 | Tagworks Pharmaceuticals B.V. | Tetrazines for high click conjugation yield in vivo and high click release yield |
| CN113365664A (zh) | 2018-10-29 | 2021-09-07 | 梅尔莎纳医疗公司 | 具有含肽接头的半胱氨酸工程化的抗体-药物缀合物 |
| IL289094A (en) | 2019-06-17 | 2022-02-01 | Tagworks Pharmaceuticals B V | Tetrazines for increasing the speed and yield of the "click release" reaction |
| DK3983363T3 (da) | 2019-06-17 | 2024-06-24 | Tagworks Pharmaceuticals B V | Forbindelser til hurtig og effektiv klikfrigivelse |
| US20250327057A1 (en) | 2021-09-06 | 2025-10-23 | Veraxa Biotech Gmbh | Novel aminoacyl-trna synthetase variants for genetic code expansion in eukaryotes |
| WO2023094525A1 (en) | 2021-11-25 | 2023-06-01 | Veraxa Biotech Gmbh | Improved antibody-payload conjugates (apcs) prepared by site-specific conjugation utilizing genetic code expansion |
| DK4186529T3 (da) | 2021-11-25 | 2025-08-25 | Veraxa Biotech Gmbh | Forbedrede antistof-payload-konjugater (apc) fremstillet ved stedspecifik konjugering ved hjælp af genetisk kodeudvidelse |
| US20250135011A1 (en) | 2021-12-08 | 2025-05-01 | European Molecular Biology Laboratory | Hydrophilic tetrazine-functionalized payloads for preparation of targeting conjugates |
| US20250114489A1 (en) | 2022-02-15 | 2025-04-10 | Tagworks Pharmaceuticals B.V. | Masked il12 protein |
| CA3261603A1 (en) | 2022-07-15 | 2024-01-18 | Pheon Therapeutics Ltd | ANTIBODY-DRUG CONJUGATES |
| WO2024080872A1 (en) | 2022-10-12 | 2024-04-18 | Tagworks Pharmaceuticals B.V. | Strained bicyclononenes |
| WO2024153789A1 (en) | 2023-01-20 | 2024-07-25 | Basf Se | Stabilized biopolymer composition, their manufacture and use |
| WO2024191293A1 (en) | 2023-03-10 | 2024-09-19 | Tagworks Pharmaceuticals B.V. | Trans-cyclooctene with improved t-linker |
| KR20260046464A (ko) | 2023-07-27 | 2026-04-07 | 베락사 바이오테크 게엠베하 | 친수성 트랜스-시클로옥텐(hyTCO) 화합물, 이를 포함하는 구조체 및 접합체 |
| WO2025056807A1 (en) | 2023-09-15 | 2025-03-20 | Basf Se | Stabilized biopolymer composition, their manufacture and use |
| WO2025149667A1 (en) | 2024-01-12 | 2025-07-17 | Pheon Therapeutics Ltd | Antibody drug conjugates and uses thereof |
| WO2025174248A1 (en) | 2024-02-16 | 2025-08-21 | Tagworks Pharmaceuticals B.V. | Trans-cyclooctenes with "or gate" release |
| WO2026043376A1 (en) | 2024-08-22 | 2026-02-26 | Tagworks Pharmaceuticals B.V. | Trans-cyclooctene formulations |
| WO2026078060A1 (en) | 2024-10-08 | 2026-04-16 | Basf Se | Tocopherol alkoxylates for biopolymer stabilization |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4138042C2 (de) * | 1991-11-19 | 1993-10-14 | Biotechnolog Forschung Gmbh | Epothilone, deren Herstellungsverfahren sowie diese Verbindungen enthaltende Mittel |
| JP4183099B2 (ja) * | 1995-11-17 | 2008-11-19 | ゲゼルシャフト・フュア・ビオテヒノロジッシェ・フォルシュング・ミット・ベシュレンクテル・ハフツング(ゲー・ベー・エフ) | エポチロンcおよびd、製造法ならびに組成物 |
| US6441186B1 (en) * | 1996-12-13 | 2002-08-27 | The Scripps Research Institute | Epothilone analogs |
| JP2001513098A (ja) * | 1997-02-25 | 2001-08-28 | ゲゼルシャフト フュア バイオテクノロギッシェ フォーシュンク エム ベー ハー(ゲー ベー エフ) | 側鎖を修飾したエポチロン |
-
1998
- 1998-02-25 JP JP53730098A patent/JP2001513098A/ja not_active Withdrawn
- 1998-02-25 AT AT98912388T patent/ATE221888T1/de active
- 1998-02-25 AU AU67249/98A patent/AU736062B2/en not_active Ceased
- 1998-02-25 DK DK98912388T patent/DK0975638T3/da active
- 1998-02-25 CN CNA031597866A patent/CN1544436A/zh active Pending
- 1998-02-25 CA CA002281105A patent/CA2281105A1/en not_active Abandoned
- 1998-02-25 RU RU99120378/04A patent/RU2201932C2/ru not_active IP Right Cessation
- 1998-02-25 PL PL98335329A patent/PL190422B1/pl unknown
- 1998-02-25 PT PT98912388T patent/PT975638E/pt unknown
- 1998-02-25 BR BRPI9807742-2A patent/BR9807742B1/pt not_active IP Right Cessation
- 1998-02-25 IL IL13134398A patent/IL131343A/en not_active IP Right Cessation
- 1998-02-25 CN CN98802842A patent/CN1128803C/zh not_active Expired - Fee Related
- 1998-02-25 CZ CZ0286599A patent/CZ298027B6/cs not_active IP Right Cessation
- 1998-02-25 EP EP02001063A patent/EP1201666A3/de not_active Ceased
- 1998-02-25 WO PCT/EP1998/001060 patent/WO1998038192A1/de not_active Ceased
- 1998-02-25 EP EP98912388A patent/EP0975638B1/de not_active Expired - Lifetime
- 1998-02-25 ES ES98912388T patent/ES2183338T3/es not_active Expired - Lifetime
- 1998-02-25 NZ NZ337195A patent/NZ337195A/en unknown
- 1998-02-25 KR KR10-1999-7007773A patent/KR100494179B1/ko not_active Expired - Fee Related
- 1998-02-25 ZA ZA981575A patent/ZA981575B/xx unknown
- 1998-02-25 HU HU0002189A patent/HU228851B1/hu unknown
- 1998-02-25 DE DE59805110T patent/DE59805110D1/de not_active Expired - Lifetime
- 1998-02-25 DE DE19880193T patent/DE19880193D2/de not_active Expired - Lifetime
- 1998-02-26 AR ARP980100851A patent/AR011878A1/es unknown
- 1998-05-28 TW TW087102758A patent/TW480263B/zh not_active IP Right Cessation
-
1999
- 1999-08-17 US US09/376,754 patent/US6359140B1/en not_active Expired - Lifetime
- 1999-08-24 NO NO19994071A patent/NO327211B1/no not_active IP Right Cessation
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2343155C2 (ru) * | 2002-09-13 | 2009-01-10 | Новартис Аг | Способ получения производных эпотилона |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2201932C2 (ru) | Модифицированные в боковой цепи эпотилоны | |
| KR100721488B1 (ko) | 6-알케닐-,6-알키닐- 및 6-에폭시-에포틸론 유도체, 그의제조 방법 및 제약 제제로서 그의 용도 | |
| KR100718616B1 (ko) | 16-할로겐-에포틸론 유도체, 그 제조 방법 및 제약학적 용도 | |
| Baxter et al. | Formal total synthesis of deserpidine demonstrating a versatile amino-Claisen rearrangement/Wenkert cyclization strategy for the preparation of functionalized yohimbane ring systems | |
| Riefert et al. | Approach to the Core Structure of 15-epi-Exiguolide | |
| HUP0303895A2 (hu) | Epotilonok lebontása | |
| MXPA99007546A (en) | Epothilones with a modified side chain | |
| EP0987268B1 (en) | Pharmaceutical agents containing epothilone A-N-oxide and/or epothilone B-N-oxide | |
| DE19930111A1 (de) | C-21 Modifizierte Epothilone | |
| Toth et al. | Synthesis of Vinca Alkaloids and Related Compounds. Part 107. An Efficient Convergent Synthetic Pathway to Build up the Ibophyllidine Skeleton III. Total Synthesis of ((plus/minus))-Ibophyllidine and | |
| HK1023774B (en) | Epothilone b-n-oxide and preparation method thereof | |
| RU2397177C2 (ru) | СПОСОБ ПОЛУЧЕНИЯ 2-ДЕГИДРО-3-ЭПИ-20-ГИДРОКСИЭКДИЗОНА, МИНОРНОГО ЭКДИСТЕРОИДА СЕМЯН РАСТЕНИЙ Froelichia floridana | |
| HK1070897A (en) | 3,7-protected epothilones n-oxides and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20070226 |