TW202000435A - 樹脂膜之製造方法 - Google Patents
樹脂膜之製造方法 Download PDFInfo
- Publication number
- TW202000435A TW202000435A TW108121549A TW108121549A TW202000435A TW 202000435 A TW202000435 A TW 202000435A TW 108121549 A TW108121549 A TW 108121549A TW 108121549 A TW108121549 A TW 108121549A TW 202000435 A TW202000435 A TW 202000435A
- Authority
- TW
- Taiwan
- Prior art keywords
- film
- resin
- resin film
- raw material
- manufacturing
- Prior art date
Links
- 229920005989 resin Polymers 0.000 title claims abstract description 229
- 239000011347 resin Substances 0.000 title claims abstract description 229
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 75
- 229920001721 polyimide Polymers 0.000 title claims abstract description 59
- 239000004642 Polyimide Substances 0.000 title claims abstract description 42
- 229920002647 polyamide Polymers 0.000 title claims abstract description 15
- 239000002994 raw material Substances 0.000 claims abstract description 99
- 238000010438 heat treatment Methods 0.000 claims abstract description 78
- 238000007664 blowing Methods 0.000 claims abstract description 63
- 238000000034 method Methods 0.000 claims description 42
- 230000003749 cleanliness Effects 0.000 claims description 17
- 230000003287 optical effect Effects 0.000 abstract description 19
- 230000000704 physical effect Effects 0.000 abstract description 8
- 230000007547 defect Effects 0.000 abstract description 7
- 239000010408 film Substances 0.000 description 391
- -1 amide imine Chemical class 0.000 description 55
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 49
- 239000010410 layer Substances 0.000 description 33
- 239000002966 varnish Substances 0.000 description 32
- GTDPSWPPOUPBNX-UHFFFAOYSA-N ac1mqpva Chemical compound CC12C(=O)OC(=O)C1(C)C1(C)C2(C)C(=O)OC1=O GTDPSWPPOUPBNX-UHFFFAOYSA-N 0.000 description 30
- 239000002904 solvent Substances 0.000 description 28
- 238000000576 coating method Methods 0.000 description 24
- 239000002245 particle Substances 0.000 description 22
- 238000001035 drying Methods 0.000 description 21
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- 239000011248 coating agent Substances 0.000 description 18
- 239000000463 material Substances 0.000 description 18
- 239000000377 silicon dioxide Substances 0.000 description 17
- 239000013585 weight reducing agent Substances 0.000 description 17
- 238000005259 measurement Methods 0.000 description 15
- 239000009719 polyimide resin Substances 0.000 description 15
- 238000003756 stirring Methods 0.000 description 14
- 150000004985 diamines Chemical class 0.000 description 13
- 150000002430 hydrocarbons Chemical group 0.000 description 13
- 239000012071 phase Substances 0.000 description 13
- 235000012239 silicon dioxide Nutrition 0.000 description 13
- 125000006158 tetracarboxylic acid group Chemical group 0.000 description 13
- 239000012790 adhesive layer Substances 0.000 description 12
- 150000001875 compounds Chemical class 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 10
- 125000000962 organic group Chemical group 0.000 description 10
- 229920000139 polyethylene terephthalate Polymers 0.000 description 10
- 239000005020 polyethylene terephthalate Substances 0.000 description 10
- 229920000642 polymer Polymers 0.000 description 10
- 230000001681 protective effect Effects 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 239000006096 absorbing agent Substances 0.000 description 9
- 239000007921 spray Substances 0.000 description 9
- 125000001931 aliphatic group Chemical group 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 7
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 7
- 239000000654 additive Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000011342 resin composition Substances 0.000 description 7
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 6
- 239000000853 adhesive Substances 0.000 description 6
- 150000004984 aromatic diamines Chemical class 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- CEIPQQODRKXDSB-UHFFFAOYSA-N ethyl 3-(6-hydroxynaphthalen-2-yl)-1H-indazole-5-carboximidate dihydrochloride Chemical compound Cl.Cl.C1=C(O)C=CC2=CC(C3=NNC4=CC=C(C=C43)C(=N)OCC)=CC=C21 CEIPQQODRKXDSB-UHFFFAOYSA-N 0.000 description 6
- 125000001153 fluoro group Chemical group F* 0.000 description 6
- 239000000049 pigment Substances 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 230000001070 adhesive effect Effects 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- 238000005227 gel permeation chromatography Methods 0.000 description 5
- 125000005843 halogen group Chemical group 0.000 description 5
- 239000002346 layers by function Substances 0.000 description 5
- 238000000691 measurement method Methods 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- 229920006122 polyamide resin Polymers 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical group C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 5
- LXEJRKJRKIFVNY-UHFFFAOYSA-N terephthaloyl chloride Chemical compound ClC(=O)C1=CC=C(C(Cl)=O)C=C1 LXEJRKJRKIFVNY-UHFFFAOYSA-N 0.000 description 5
- 229920001187 thermosetting polymer Polymers 0.000 description 5
- 230000002087 whitening effect Effects 0.000 description 5
- NVKGJHAQGWCWDI-UHFFFAOYSA-N 4-[4-amino-2-(trifluoromethyl)phenyl]-3-(trifluoromethyl)aniline Chemical compound FC(F)(F)C1=CC(N)=CC=C1C1=CC=C(N)C=C1C(F)(F)F NVKGJHAQGWCWDI-UHFFFAOYSA-N 0.000 description 4
- FKNQCJSGGFJEIZ-UHFFFAOYSA-N 4-methylpyridine Chemical compound CC1=CC=NC=C1 FKNQCJSGGFJEIZ-UHFFFAOYSA-N 0.000 description 4
- 239000004925 Acrylic resin Substances 0.000 description 4
- 229920000178 Acrylic resin Polymers 0.000 description 4
- 229920002799 BoPET Polymers 0.000 description 4
- PJANXHGTPQOBST-VAWYXSNFSA-N Stilbene Natural products C=1C=CC=CC=1/C=C/C1=CC=CC=C1 PJANXHGTPQOBST-VAWYXSNFSA-N 0.000 description 4
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000011164 primary particle Substances 0.000 description 4
- 235000021286 stilbenes Nutrition 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- GEWWCWZGHNIUBW-UHFFFAOYSA-N 1-(4-nitrophenyl)propan-2-one Chemical compound CC(=O)CC1=CC=C([N+]([O-])=O)C=C1 GEWWCWZGHNIUBW-UHFFFAOYSA-N 0.000 description 3
- MXPYJVUYLVNEBB-UHFFFAOYSA-N 2-[2-(2-carboxybenzoyl)oxycarbonylbenzoyl]oxycarbonylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(=O)OC(=O)C1=CC=CC=C1C(=O)OC(=O)C1=CC=CC=C1C(O)=O MXPYJVUYLVNEBB-UHFFFAOYSA-N 0.000 description 3
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 3
- 229920000089 Cyclic olefin copolymer Polymers 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 239000004820 Pressure-sensitive adhesive Substances 0.000 description 3
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 3
- 239000012346 acetyl chloride Substances 0.000 description 3
- 150000008065 acid anhydrides Chemical class 0.000 description 3
- XECAHXYUAAWDEL-UHFFFAOYSA-N acrylonitrile butadiene styrene Chemical compound C=CC=C.C=CC#N.C=CC1=CC=CC=C1 XECAHXYUAAWDEL-UHFFFAOYSA-N 0.000 description 3
- 229920000122 acrylonitrile butadiene styrene Polymers 0.000 description 3
- 239000004676 acrylonitrile butadiene styrene Substances 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 238000005266 casting Methods 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 239000012788 optical film Substances 0.000 description 3
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 3
- 229920003207 poly(ethylene-2,6-naphthalate) Polymers 0.000 description 3
- 239000011112 polyethylene naphthalate Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 239000002356 single layer Substances 0.000 description 3
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 3
- 238000002834 transmittance Methods 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 238000004804 winding Methods 0.000 description 3
- JYYNAJVZFGKDEQ-UHFFFAOYSA-N 2,4-Dimethylpyridine Chemical compound CC1=CC=NC(C)=C1 JYYNAJVZFGKDEQ-UHFFFAOYSA-N 0.000 description 2
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 2
- MFEIKQPHQINPRI-UHFFFAOYSA-N 3-Ethylpyridine Chemical compound CCC1=CC=CN=C1 MFEIKQPHQINPRI-UHFFFAOYSA-N 0.000 description 2
- ITQTTZVARXURQS-UHFFFAOYSA-N 3-methylpyridine Chemical compound CC1=CC=CN=C1 ITQTTZVARXURQS-UHFFFAOYSA-N 0.000 description 2
- LJMPOXUWPWEILS-UHFFFAOYSA-N 3a,4,4a,7a,8,8a-hexahydrofuro[3,4-f][2]benzofuran-1,3,5,7-tetrone Chemical compound C1C2C(=O)OC(=O)C2CC2C(=O)OC(=O)C21 LJMPOXUWPWEILS-UHFFFAOYSA-N 0.000 description 2
- HYDATEKARGDBKU-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenyl]phenoxy]aniline Chemical group C1=CC(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)C=C1 HYDATEKARGDBKU-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 125000002015 acyclic group Chemical group 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 2
- 229940073608 benzyl chloride Drugs 0.000 description 2
- 239000007844 bleaching agent Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000008199 coating composition Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000012024 dehydrating agents Substances 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 2
- 239000003822 epoxy resin Substances 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- GAEKPEKOJKCEMS-UHFFFAOYSA-N gamma-valerolactone Chemical compound CC1CCC(=O)O1 GAEKPEKOJKCEMS-UHFFFAOYSA-N 0.000 description 2
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 238000007689 inspection Methods 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- BPUBBGLMJRNUCC-UHFFFAOYSA-N oxygen(2-);tantalum(5+) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Ta+5].[Ta+5] BPUBBGLMJRNUCC-UHFFFAOYSA-N 0.000 description 2
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 229920000962 poly(amidoamine) Polymers 0.000 description 2
- 229920000647 polyepoxide Polymers 0.000 description 2
- 229920001225 polyester resin Polymers 0.000 description 2
- 239000004645 polyester resin Substances 0.000 description 2
- 229920005672 polyolefin resin Polymers 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000001226 reprecipitation Methods 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 229910052814 silicon oxide Inorganic materials 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 229910001936 tantalum oxide Inorganic materials 0.000 description 2
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 2
- 229910001887 tin oxide Inorganic materials 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- ARCGXLSVLAOJQL-UHFFFAOYSA-N trimellitic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 ARCGXLSVLAOJQL-UHFFFAOYSA-N 0.000 description 2
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 2
- 239000011787 zinc oxide Substances 0.000 description 2
- 229910001928 zirconium oxide Inorganic materials 0.000 description 2
- OLQWMCSSZKNOLQ-ZXZARUISSA-N (3s)-3-[(3r)-2,5-dioxooxolan-3-yl]oxolane-2,5-dione Chemical compound O=C1OC(=O)C[C@H]1[C@@H]1C(=O)OC(=O)C1 OLQWMCSSZKNOLQ-ZXZARUISSA-N 0.000 description 1
- YJTKZCDBKVTVBY-UHFFFAOYSA-N 1,3-Diphenylbenzene Chemical compound C1=CC=CC=C1C1=CC=CC(C=2C=CC=CC=2)=C1 YJTKZCDBKVTVBY-UHFFFAOYSA-N 0.000 description 1
- GRJWOKACBGZOKT-UHFFFAOYSA-N 1,3-bis(chloromethyl)benzene Chemical compound ClCC1=CC=CC(CCl)=C1 GRJWOKACBGZOKT-UHFFFAOYSA-N 0.000 description 1
- XNFGDDFDPXGEGL-UHFFFAOYSA-N 1,3-dioxobenzo[f][2]benzofuran-6-carboxylic acid Chemical compound C1=C2C(=O)OC(=O)C2=CC2=CC(C(=O)O)=CC=C21 XNFGDDFDPXGEGL-UHFFFAOYSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- JVCBVWTTXCNJBJ-UHFFFAOYSA-N 1-azabicyclo[2.2.1]heptane Chemical compound C1CC2CCN1C2 JVCBVWTTXCNJBJ-UHFFFAOYSA-N 0.000 description 1
- STHHLVCQSLRQNI-UHFFFAOYSA-N 1-azabicyclo[3.2.1]octane Chemical compound C1C2CCN1CCC2 STHHLVCQSLRQNI-UHFFFAOYSA-N 0.000 description 1
- AXWLKJWVMMAXBD-UHFFFAOYSA-N 1-butylpiperidine Chemical compound CCCCN1CCCCC1 AXWLKJWVMMAXBD-UHFFFAOYSA-N 0.000 description 1
- JSHASCFKOSDFHY-UHFFFAOYSA-N 1-butylpyrrolidine Chemical compound CCCCN1CCCC1 JSHASCFKOSDFHY-UHFFFAOYSA-N 0.000 description 1
- QLEIDMAURCRVCX-UHFFFAOYSA-N 1-propylpiperazine Chemical compound CCCN1CCNCC1 QLEIDMAURCRVCX-UHFFFAOYSA-N 0.000 description 1
- VTDIWMPYBAVEDY-UHFFFAOYSA-N 1-propylpiperidine Chemical compound CCCN1CCCCC1 VTDIWMPYBAVEDY-UHFFFAOYSA-N 0.000 description 1
- PGZVFRAEAAXREB-UHFFFAOYSA-N 2,2-dimethylpropanoyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC(=O)C(C)(C)C PGZVFRAEAAXREB-UHFFFAOYSA-N 0.000 description 1
- VOZKAJLKRJDJLL-UHFFFAOYSA-N 2,4-diaminotoluene Chemical compound CC1=CC=C(N)C=C1N VOZKAJLKRJDJLL-UHFFFAOYSA-N 0.000 description 1
- IYAZLDLPUNDVAG-UHFFFAOYSA-N 2-(benzotriazol-2-yl)-4-(2,4,4-trimethylpentan-2-yl)phenol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(O)C(N2N=C3C=CC=CC3=N2)=C1 IYAZLDLPUNDVAG-UHFFFAOYSA-N 0.000 description 1
- NRGGMCIBEHEAIL-UHFFFAOYSA-N 2-ethylpyridine Chemical compound CCC1=CC=CC=N1 NRGGMCIBEHEAIL-UHFFFAOYSA-N 0.000 description 1
- TWBPWBPGNQWFSJ-UHFFFAOYSA-N 2-phenylaniline Chemical group NC1=CC=CC=C1C1=CC=CC=C1 TWBPWBPGNQWFSJ-UHFFFAOYSA-N 0.000 description 1
- HUWXDEQWWKGHRV-UHFFFAOYSA-N 3,3'-Dichlorobenzidine Chemical compound C1=C(Cl)C(N)=CC=C1C1=CC=C(N)C(Cl)=C1 HUWXDEQWWKGHRV-UHFFFAOYSA-N 0.000 description 1
- RGBSGRUHELUMOF-UHFFFAOYSA-N 3,4-cyclopentenopyridine Natural products C1=NC=C2CCCC2=C1 RGBSGRUHELUMOF-UHFFFAOYSA-N 0.000 description 1
- GWHLJVMSZRKEAQ-UHFFFAOYSA-N 3-(2,3-dicarboxyphenyl)phthalic acid Chemical compound OC(=O)C1=CC=CC(C=2C(=C(C(O)=O)C=CC=2)C(O)=O)=C1C(O)=O GWHLJVMSZRKEAQ-UHFFFAOYSA-N 0.000 description 1
- LXJLFVRAWOOQDR-UHFFFAOYSA-N 3-(3-aminophenoxy)aniline Chemical compound NC1=CC=CC(OC=2C=C(N)C=CC=2)=C1 LXJLFVRAWOOQDR-UHFFFAOYSA-N 0.000 description 1
- ZBMISJGHVWNWTE-UHFFFAOYSA-N 3-(4-aminophenoxy)aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(N)=C1 ZBMISJGHVWNWTE-UHFFFAOYSA-N 0.000 description 1
- TYKLCAKICHXQNE-UHFFFAOYSA-N 3-[(2,3-dicarboxyphenyl)methyl]phthalic acid Chemical compound OC(=O)C1=CC=CC(CC=2C(=C(C(O)=O)C=CC=2)C(O)=O)=C1C(O)=O TYKLCAKICHXQNE-UHFFFAOYSA-N 0.000 description 1
- UCFMKTNJZCYBBJ-UHFFFAOYSA-N 3-[1-(2,3-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C=1C=CC(C(O)=O)=C(C(O)=O)C=1C(C)C1=CC=CC(C(O)=O)=C1C(O)=O UCFMKTNJZCYBBJ-UHFFFAOYSA-N 0.000 description 1
- UCPOEBHXOCKECV-UHFFFAOYSA-N 3-[2-(2,3-dicarboxyphenyl)ethyl]phthalic acid Chemical compound OC(=O)C1=CC=CC(CCC=2C(=C(C(O)=O)C=CC=2)C(O)=O)=C1C(O)=O UCPOEBHXOCKECV-UHFFFAOYSA-N 0.000 description 1
- PAHZZOIHRHCHTH-UHFFFAOYSA-N 3-[2-(2,3-dicarboxyphenyl)propan-2-yl]phthalic acid Chemical compound C=1C=CC(C(O)=O)=C(C(O)=O)C=1C(C)(C)C1=CC=CC(C(O)=O)=C1C(O)=O PAHZZOIHRHCHTH-UHFFFAOYSA-N 0.000 description 1
- NYRFBMFAUFUULG-UHFFFAOYSA-N 3-[4-[2-[4-(3-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=C(OC=2C=C(N)C=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=CC(N)=C1 NYRFBMFAUFUULG-UHFFFAOYSA-N 0.000 description 1
- FREZLSIGWNCSOQ-UHFFFAOYSA-N 3-methylbutanoyl 3-methylbutanoate Chemical compound CC(C)CC(=O)OC(=O)CC(C)C FREZLSIGWNCSOQ-UHFFFAOYSA-N 0.000 description 1
- YBRVSVVVWCFQMG-UHFFFAOYSA-N 4,4'-diaminodiphenylmethane Chemical compound C1=CC(N)=CC=C1CC1=CC=C(N)C=C1 YBRVSVVVWCFQMG-UHFFFAOYSA-N 0.000 description 1
- QYIMZXITLDTULQ-UHFFFAOYSA-N 4-(4-amino-2-methylphenyl)-3-methylaniline Chemical compound CC1=CC(N)=CC=C1C1=CC=C(N)C=C1C QYIMZXITLDTULQ-UHFFFAOYSA-N 0.000 description 1
- HLBLWEWZXPIGSM-UHFFFAOYSA-N 4-Aminophenyl ether Chemical compound C1=CC(N)=CC=C1OC1=CC=C(N)C=C1 HLBLWEWZXPIGSM-UHFFFAOYSA-N 0.000 description 1
- IWXCYYWDGDDPAC-UHFFFAOYSA-N 4-[(3,4-dicarboxyphenyl)methyl]phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1CC1=CC=C(C(O)=O)C(C(O)=O)=C1 IWXCYYWDGDDPAC-UHFFFAOYSA-N 0.000 description 1
- DZIHTWJGPDVSGE-UHFFFAOYSA-N 4-[(4-aminocyclohexyl)methyl]cyclohexan-1-amine Chemical compound C1CC(N)CCC1CC1CCC(N)CC1 DZIHTWJGPDVSGE-UHFFFAOYSA-N 0.000 description 1
- IJJNNSUCZDJDLP-UHFFFAOYSA-N 4-[1-(3,4-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C=1C=C(C(O)=O)C(C(O)=O)=CC=1C(C)C1=CC=C(C(O)=O)C(C(O)=O)=C1 IJJNNSUCZDJDLP-UHFFFAOYSA-N 0.000 description 1
- APXJLYIVOFARRM-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(C(O)=O)C(C(O)=O)=C1 APXJLYIVOFARRM-UHFFFAOYSA-N 0.000 description 1
- DDVYLSFEKWFRFS-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1CCC1=CC=C(C(O)=O)C(C(O)=O)=C1 DDVYLSFEKWFRFS-UHFFFAOYSA-N 0.000 description 1
- GEYAGBVEAJGCFB-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)propan-2-yl]phthalic acid Chemical compound C=1C=C(C(O)=O)C(C(O)=O)=CC=1C(C)(C)C1=CC=C(C(O)=O)C(C(O)=O)=C1 GEYAGBVEAJGCFB-UHFFFAOYSA-N 0.000 description 1
- JYZPDAUOQGFBKT-UHFFFAOYSA-N 4-[2-[2-[2-(3,4-dicarboxyphenoxy)phenyl]propan-2-yl]phenoxy]phthalic acid Chemical compound C=1C=CC=C(OC=2C=C(C(C(O)=O)=CC=2)C(O)=O)C=1C(C)(C)C1=CC=CC=C1OC1=CC=C(C(O)=O)C(C(O)=O)=C1 JYZPDAUOQGFBKT-UHFFFAOYSA-N 0.000 description 1
- WUPRYUDHUFLKFL-UHFFFAOYSA-N 4-[3-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(OC=2C=CC(N)=CC=2)=C1 WUPRYUDHUFLKFL-UHFFFAOYSA-N 0.000 description 1
- JCRRFJIVUPSNTA-UHFFFAOYSA-N 4-[4-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC(C=C1)=CC=C1OC1=CC=C(N)C=C1 JCRRFJIVUPSNTA-UHFFFAOYSA-N 0.000 description 1
- KMKWGXGSGPYISJ-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=C(OC=2C=CC(N)=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=C(N)C=C1 KMKWGXGSGPYISJ-UHFFFAOYSA-N 0.000 description 1
- VJXRKZJMGVSXPX-UHFFFAOYSA-N 4-ethylpyridine Chemical compound CCC1=CC=NC=C1 VJXRKZJMGVSXPX-UHFFFAOYSA-N 0.000 description 1
- WXAIEIRYBSKHDP-UHFFFAOYSA-N 4-phenyl-n-(4-phenylphenyl)-n-[4-[4-(4-phenyl-n-(4-phenylphenyl)anilino)phenyl]phenyl]aniline Chemical compound C1=CC=CC=C1C1=CC=C(N(C=2C=CC(=CC=2)C=2C=CC=CC=2)C=2C=CC(=CC=2)C=2C=CC(=CC=2)N(C=2C=CC(=CC=2)C=2C=CC=CC=2)C=2C=CC(=CC=2)C=2C=CC=CC=2)C=C1 WXAIEIRYBSKHDP-UHFFFAOYSA-N 0.000 description 1
- YGYCECQIOXZODZ-UHFFFAOYSA-N 4415-87-6 Chemical compound O=C1OC(=O)C2C1C1C(=O)OC(=O)C12 YGYCECQIOXZODZ-UHFFFAOYSA-N 0.000 description 1
- HTMGQIXFZMZZKD-UHFFFAOYSA-N 5,6,7,8-tetrahydroisoquinoline Chemical compound N1=CC=C2CCCCC2=C1 HTMGQIXFZMZZKD-UHFFFAOYSA-N 0.000 description 1
- VQVIHDPBMFABCQ-UHFFFAOYSA-N 5-(1,3-dioxo-2-benzofuran-5-carbonyl)-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(C(C=2C=C3C(=O)OC(=O)C3=CC=2)=O)=C1 VQVIHDPBMFABCQ-UHFFFAOYSA-N 0.000 description 1
- QQGYZOYWNCKGEK-UHFFFAOYSA-N 5-[(1,3-dioxo-2-benzofuran-5-yl)oxy]-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(OC=2C=C3C(=O)OC(C3=CC=2)=O)=C1 QQGYZOYWNCKGEK-UHFFFAOYSA-N 0.000 description 1
- QHHKLPCQTTWFSS-UHFFFAOYSA-N 5-[2-(1,3-dioxo-2-benzofuran-5-yl)-1,1,1,3,3,3-hexafluoropropan-2-yl]-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(C(C=2C=C3C(=O)OC(=O)C3=CC=2)(C(F)(F)F)C(F)(F)F)=C1 QHHKLPCQTTWFSS-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- UMRZSTCPUPJPOJ-UHFFFAOYSA-N C(C1)C2CC1CC2 Chemical compound C(C1)C2CC1CC2 UMRZSTCPUPJPOJ-UHFFFAOYSA-N 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- 229920002430 Fibre-reinforced plastic Polymers 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 239000004640 Melamine resin Substances 0.000 description 1
- 229920000877 Melamine resin Polymers 0.000 description 1
- MWCLLHOVUTZFKS-UHFFFAOYSA-N Methyl cyanoacrylate Chemical compound COC(=O)C(=C)C#N MWCLLHOVUTZFKS-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- ABTLLKINSDJQHL-UHFFFAOYSA-N NC1=CC=C(OC2=CC=C(C=C2)C2=C(C=3C=CC4=CC=CC=C4C3C=C2)C2=CC=C(C=C2)OC2=CC=C(C=C2)N)C=C1 Chemical compound NC1=CC=C(OC2=CC=C(C=C2)C2=C(C=3C=CC4=CC=CC=C4C3C=C2)C2=CC=C(C=C2)OC2=CC=C(C=C2)N)C=C1 ABTLLKINSDJQHL-UHFFFAOYSA-N 0.000 description 1
- NYZFNIUYLPDHPL-UHFFFAOYSA-N NC=1C=C(OC2=CC=C(C=C2)C2=C(C=3C=CC4=CC=CC=C4C3C=C2)C2=CC=C(C=C2)OC2=CC(=CC=C2)N)C=CC1 Chemical compound NC=1C=C(OC2=CC=C(C=C2)C2=C(C=3C=CC4=CC=CC=C4C3C=C2)C2=CC=C(C=C2)OC2=CC(=CC=C2)N)C=CC1 NYZFNIUYLPDHPL-UHFFFAOYSA-N 0.000 description 1
- SJEYSFABYSGQBG-UHFFFAOYSA-M Patent blue Chemical compound [Na+].C1=CC(N(CC)CC)=CC=C1C(C=1C(=CC(=CC=1)S([O-])(=O)=O)S([O-])(=O)=O)=C1C=CC(=[N+](CC)CC)C=C1 SJEYSFABYSGQBG-UHFFFAOYSA-M 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- NRCMAYZCPIVABH-UHFFFAOYSA-N Quinacridone Chemical compound N1C2=CC=CC=C2C(=O)C2=C1C=C1C(=O)C3=CC=CC=C3NC1=C2 NRCMAYZCPIVABH-UHFFFAOYSA-N 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- NRTOMJZYCJJWKI-UHFFFAOYSA-N Titanium nitride Chemical compound [Ti]#N NRTOMJZYCJJWKI-UHFFFAOYSA-N 0.000 description 1
- QLBRROYTTDFLDX-UHFFFAOYSA-N [3-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCCC(CN)C1 QLBRROYTTDFLDX-UHFFFAOYSA-N 0.000 description 1
- FDLQZKYLHJJBHD-UHFFFAOYSA-N [3-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC(CN)=C1 FDLQZKYLHJJBHD-UHFFFAOYSA-N 0.000 description 1
- OXIKYYJDTWKERT-UHFFFAOYSA-N [4-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCC(CN)CC1 OXIKYYJDTWKERT-UHFFFAOYSA-N 0.000 description 1
- ISKQADXMHQSTHK-UHFFFAOYSA-N [4-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=C(CN)C=C1 ISKQADXMHQSTHK-UHFFFAOYSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000000980 acid dye Substances 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000005577 anthracene group Chemical group 0.000 description 1
- 239000001000 anthraquinone dye Substances 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 239000002519 antifouling agent Substances 0.000 description 1
- 229910000410 antimony oxide Inorganic materials 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- IRERQBUNZFJFGC-UHFFFAOYSA-L azure blue Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Al+3].[Al+3].[Al+3].[Al+3].[Al+3].[Al+3].[S-]S[S-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-] IRERQBUNZFJFGC-UHFFFAOYSA-L 0.000 description 1
- 239000000981 basic dye Substances 0.000 description 1
- 238000005452 bending Methods 0.000 description 1
- FYXKZNLBZKRYSS-UHFFFAOYSA-N benzene-1,2-dicarbonyl chloride Chemical compound ClC(=O)C1=CC=CC=C1C(Cl)=O FYXKZNLBZKRYSS-UHFFFAOYSA-N 0.000 description 1
- 150000001559 benzoic acids Chemical class 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- BKDVBBSUAGJUBA-UHFFFAOYSA-N bicyclo[2.2.2]oct-7-ene-2,3,5,6-tetracarboxylic acid Chemical compound C1=CC2C(C(O)=O)C(C(=O)O)C1C(C(O)=O)C2C(O)=O BKDVBBSUAGJUBA-UHFFFAOYSA-N 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- WKDNYTOXBCRNPV-UHFFFAOYSA-N bpda Chemical compound C1=C2C(=O)OC(=O)C2=CC(C=2C=C3C(=O)OC(C3=CC=2)=O)=C1 WKDNYTOXBCRNPV-UHFFFAOYSA-N 0.000 description 1
- YHASWHZGWUONAO-UHFFFAOYSA-N butanoyl butanoate Chemical compound CCCC(=O)OC(=O)CCC YHASWHZGWUONAO-UHFFFAOYSA-N 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- UFWIBTONFRDIAS-UHFFFAOYSA-N c1cc2ccccc2cc1 Chemical compound c1cc2ccccc2cc1 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 239000003660 carbonate based solvent Substances 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 238000001723 curing Methods 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- WOSVXXBNNCUXMT-UHFFFAOYSA-N cyclopentane-1,2,3,4-tetracarboxylic acid Chemical compound OC(=O)C1CC(C(O)=O)C(C(O)=O)C1C(O)=O WOSVXXBNNCUXMT-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- CRHLEZORXKQUEI-UHFFFAOYSA-N dialuminum;cobalt(2+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Al+3].[Al+3].[Co+2].[Co+2] CRHLEZORXKQUEI-UHFFFAOYSA-N 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- QUFHSZWWLQQDCL-UHFFFAOYSA-N dichloromethane;naphthalene Chemical compound ClCCl.C1=CC=CC2=CC=CC=C21 QUFHSZWWLQQDCL-UHFFFAOYSA-N 0.000 description 1
- 238000007607 die coating method Methods 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000001227 electron beam curing Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003759 ester based solvent Substances 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000011151 fibre-reinforced plastic Substances 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 108010025899 gelatin film Proteins 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 238000007756 gravure coating Methods 0.000 description 1
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 125000005462 imide group Chemical group 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 229910003437 indium oxide Inorganic materials 0.000 description 1
- PJXISJQVUVHSOJ-UHFFFAOYSA-N indium(iii) oxide Chemical compound [O-2].[O-2].[O-2].[In+3].[In+3] PJXISJQVUVHSOJ-UHFFFAOYSA-N 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 239000011256 inorganic filler Substances 0.000 description 1
- 229910003475 inorganic filler Inorganic materials 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- LSACYLWPPQLVSM-UHFFFAOYSA-N isobutyric acid anhydride Chemical compound CC(C)C(=O)OC(=O)C(C)C LSACYLWPPQLVSM-UHFFFAOYSA-N 0.000 description 1
- PXZQEOJJUGGUIB-UHFFFAOYSA-N isoindolin-1-one Chemical compound C1=CC=C2C(=O)NCC2=C1 PXZQEOJJUGGUIB-UHFFFAOYSA-N 0.000 description 1
- YWXYYJSYQOXTPL-SLPGGIOYSA-N isosorbide mononitrate Chemical compound [O-][N+](=O)O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 YWXYYJSYQOXTPL-SLPGGIOYSA-N 0.000 description 1
- 239000005453 ketone based solvent Substances 0.000 description 1
- 239000005001 laminate film Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229940018564 m-phenylenediamine Drugs 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- NYGZLYXAPMMJTE-UHFFFAOYSA-M metanil yellow Chemical group [Na+].[O-]S(=O)(=O)C1=CC=CC(N=NC=2C=CC(NC=3C=CC=CC=3)=CC=2)=C1 NYGZLYXAPMMJTE-UHFFFAOYSA-M 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 239000000983 mordant dye Substances 0.000 description 1
- BBDGYADAMYMJNO-UHFFFAOYSA-N n-butyl-n-ethylbutan-1-amine Chemical compound CCCCN(CC)CCCC BBDGYADAMYMJNO-UHFFFAOYSA-N 0.000 description 1
- VEBPYKMCKZTFPJ-UHFFFAOYSA-N n-butyl-n-propylbutan-1-amine Chemical compound CCCCN(CCC)CCCC VEBPYKMCKZTFPJ-UHFFFAOYSA-N 0.000 description 1
- KQSABULTKYLFEV-UHFFFAOYSA-N naphthalene-1,5-diamine Chemical compound C1=CC=C2C(N)=CC=CC2=C1N KQSABULTKYLFEV-UHFFFAOYSA-N 0.000 description 1
- GOGZBMRXLADNEV-UHFFFAOYSA-N naphthalene-2,6-diamine Chemical compound C1=C(N)C=CC2=CC(N)=CC=C21 GOGZBMRXLADNEV-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 150000004767 nitrides Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000012860 organic pigment Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- VTRUBDSFZJNXHI-UHFFFAOYSA-N oxoantimony Chemical compound [Sb]=O VTRUBDSFZJNXHI-UHFFFAOYSA-N 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- HVNWRBWNOPYOER-UHFFFAOYSA-N pentane-1,2,3,4-tetracarboxylic acid Chemical compound OC(=O)C(C)C(C(O)=O)C(C(O)=O)CC(O)=O HVNWRBWNOPYOER-UHFFFAOYSA-N 0.000 description 1
- 125000002080 perylenyl group Chemical group C1(=CC=C2C=CC=C3C4=CC=CC5=CC=CC(C1=C23)=C45)* 0.000 description 1
- CSHWQDPOILHKBI-UHFFFAOYSA-N peryrene Natural products C1=CC(C2=CC=CC=3C2=C2C=CC=3)=C3C2=CC=CC3=C1 CSHWQDPOILHKBI-UHFFFAOYSA-N 0.000 description 1
- 239000005011 phenolic resin Substances 0.000 description 1
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical compound N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 1
- 239000001007 phthalocyanine dye Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920005575 poly(amic acid) Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001707 polybutylene terephthalate Polymers 0.000 description 1
- 229920005668 polycarbonate resin Polymers 0.000 description 1
- 239000004431 polycarbonate resin Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920006290 polyethylene naphthalate film Polymers 0.000 description 1
- 229920013716 polyethylene resin Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 238000007781 pre-processing Methods 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- IZMJMCDDWKSTTK-UHFFFAOYSA-N quinoline yellow Chemical compound C1=CC=CC2=NC(C3C(C4=CC=CC=C4C3=O)=O)=CC=C21 IZMJMCDDWKSTTK-UHFFFAOYSA-N 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000007665 sagging Methods 0.000 description 1
- 229920002050 silicone resin Polymers 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000004381 surface treatment Methods 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- 150000000000 tetracarboxylic acids Chemical class 0.000 description 1
- 229920002803 thermoplastic polyurethane Polymers 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 239000001003 triarylmethane dye Substances 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 235000013799 ultramarine blue Nutrition 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000037303 wrinkles Effects 0.000 description 1
- 238000004383 yellowing Methods 0.000 description 1
Images





Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C71/00—After-treatment of articles without altering their shape; Apparatus therefor
- B29C71/02—Thermal after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C35/00—Heating, cooling or curing, e.g. crosslinking or vulcanising; Apparatus therefor
- B29C35/02—Heating or curing, e.g. crosslinking or vulcanizing during moulding, e.g. in a mould
- B29C35/04—Heating or curing, e.g. crosslinking or vulcanizing during moulding, e.g. in a mould using liquids, gas or steam
- B29C35/045—Heating or curing, e.g. crosslinking or vulcanizing during moulding, e.g. in a mould using liquids, gas or steam using gas or flames
- B29C2035/046—Heating or curing, e.g. crosslinking or vulcanizing during moulding, e.g. in a mould using liquids, gas or steam using gas or flames dried air
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C55/00—Shaping by stretching, e.g. drawing through a die; Apparatus therefor
- B29C55/02—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2379/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen, or carbon only, not provided for in groups C08J2361/00 - C08J2377/00
- C08J2379/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08J2379/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Physics & Mathematics (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Thermal Sciences (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Mechanical Engineering (AREA)
- Polarising Elements (AREA)
- Processing And Handling Of Plastics And Other Materials For Molding In General (AREA)
- Heating, Cooling, Or Curing Plastics Or The Like In General (AREA)
- Moulding By Coating Moulds (AREA)
Abstract
本發明之目的在於提供一種並無可對視認性產生影響之損傷等缺陷,且具備可撓性顯示裝置之前面板所要求之光學性質及物理性質之樹脂膜之製造方法。
本發明係一種樹脂膜之製造方法,其係至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者之樹脂膜之製造方法,並且具有以熱風加熱原料膜之加熱步驟,上述加熱步驟係藉由來自相互對向之一對噴嘴之吹出口之熱風而進行,關於上述噴嘴,吹出口之吹出風速為2 m/秒以上25 m/秒以下,來自每一個噴嘴之吹出口之吹出風量於沿上述原料膜之寬度方向之噴嘴之每1 m長度中為0.1~3 m3
/秒。
Description
本發明係關於一種至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者之樹脂膜之製造方法。
近年來,要求圖像顯示裝置之薄型化、輕量化及可撓性化,但先前使用之玻璃基材或玻璃前面板不具有可應對上述要求之充分之材質特性。故而,代替玻璃之材料或基材之開發得以進展。其中之一為含有聚醯亞胺樹脂之樹脂膜。就柔軟性、透明性及耐熱性之觀點而言,含有聚醯亞胺樹脂之樹脂膜可用於各種用途。(專利文獻1)。
[先前技術文獻]
[專利文獻]
[專利文獻1]日本專利特開2009-286826號公報
[發明所欲解決之問題]
於將含有聚醯亞胺系樹脂之膜用作圖像顯示裝置之前面板之情形時,出於表面硬度之提高等目的,進行將膜於高溫條件下加熱之步驟。然而,存在因高溫條件下之加熱而導致膜黃變從而損害膜品質之問題。又,前面板係配置於圖像顯示裝置之視認側,故而要求無可對視認性產生影響之損傷等缺陷,又具有前面板所要求之光學性質及物理性質。
進而,就製造之觀點而言,亦需求一種可高效地製造此種物性之樹脂膜之方法。
[解決問題之技術手段]
本發明係鑒於上述課題而完成者,提供一種抑制因製造過程中之膜之晃動而引起之損傷等缺陷,具備前面板所要求之光學性質與物理性質之樹脂膜之製造方法。又,提供一種可高效地製造此種樹脂膜之製造方法。即,本發明提供以下態樣之樹脂膜之製造方法。
[1]一種樹脂膜之製造方法,其係至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者之樹脂膜之製造方法,並且
具有以熱風加熱原料膜之加熱步驟,
上述加熱步驟係藉由來自相互對向之一對噴嘴之吹出口之熱風而進行,
關於上述噴嘴,吹出口之吹出風速為2 m/秒以上25 m/秒以下,來自每一個噴嘴之吹出口之吹出風量於沿上述原料膜之寬度方向之噴嘴之每1 m長度中為0.1~3 m3
/秒。
[2]如[1]之樹脂膜之製造方法,其中上述吹出口之吹出風速為2 m/秒以上且未達25 m/秒。
[3]如[1]或[2]之樹脂膜之製造方法,其中上述噴嘴係具有於上述原料膜之寬度方向上延伸之狹縫狀之吹出口之噴嘴、或具有於上述原料膜之搬送方向及上述原料膜之寬度方向上分別配置有複數個開口之吹出口之噴嘴。
[4]如[1]至[3]中任一項之樹脂膜之製造方法,其中於上述加熱步驟中,對上述原料膜吹送熱風之各個上述噴嘴之吹出口之熱風之於上述膜之寬度方向上之最大吹出風速與最小吹出風速之差為4 m/秒以下。
[5]如[1]至[4]中任一項之樹脂膜之製造方法,其中上述加熱步驟係於無塵室內進行。
[6]如[1]至[5]中任一項之樹脂膜之製造方法,其中上述加熱步驟係於清潔度等級為1,000以下之潔淨度之烘箱中進行。
[發明之效果]
根據本發明,提供一種抑制損傷等缺陷,具備前面板所要求之光學性質(均勻之面內相位差及較低之黃度等)與物理性質(均勻之厚度及較高之撓曲性等)之樹脂膜之製造方法。又,提供一種可高效地製造此種樹脂膜之製造方法。
藉由本發明而獲得之樹脂膜可用作可撓性顯示器之前面板等光學膜。
以下,詳細說明本發明之實施形態。再者,本發明之範圍並不限定於此處說明之實施形態,可於不脫離本發明之主旨之範圍內進行各種變更。
<樹脂膜之製造方法>
本實施形態之樹脂膜之製造方法具有以熱風加熱原料膜之加熱步驟,加熱步驟係利用藉由來自相互對向之一對噴嘴之吹出口之熱風而進行膜之加熱之方法而進行。關於上述噴嘴,吹出口之吹出風速為2 m/秒以上25 m/秒以下,來自每一個噴嘴之吹出口之吹出風量於沿上述原料膜之寬度方向之噴嘴之每1 m長度中為0.1~3 m3
/秒。
上述吹出口之吹出風速較佳為2 m/秒以上且未達25 m/秒。
加熱步驟可於設置有相互對向之一對噴嘴之加熱區域中進行,例如可於烘箱內進行。於本說明書中,所謂烘箱係指可對膜進行加熱之機器,包含加熱爐、乾燥爐及拉幅爐(將膜寬度方向之兩端固定而進行加熱之爐)。加熱爐可為熱風處理或輻射熱線處理之任一種,亦可為併用該等之加熱爐。
關於本實施形態之樹脂膜之製造方法,參照附圖而加以說明。圖1係模式地表示本發明之樹脂膜之製造方法之較佳實施形態之步驟剖視圖。參照圖1,至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者之原料膜20被搬入至烘箱100,於烘箱100內之加熱區域中加熱,其後自烘箱100搬出。於本說明書中,將經過加熱步驟之前與存在溶劑量等之經時變化之加熱步驟中或搬送至烘箱中之膜稱為原料膜,將經過加熱步驟並自烘箱搬出之膜稱為樹脂膜。
原料膜20可自捲取有原料膜之捲筒捲出而搬入至烘箱,亦可自其之前最近之步驟連續地搬入至烘箱。圖2係模式地表示本發明之樹脂膜之製造方法中之加熱步驟之較佳實施形態之步驟剖視圖。參照圖2,原料膜20較佳為與膜之搬送方向(亦稱為MD方向)垂直之方向(亦稱為TD方向、寬度方向)之膜之兩端被固定而於烘箱內搬送。
兩端之固定可使用針板、夾具及膜夾盤等通常於膜之製造裝置中使用之固持裝置而進行。固定之兩端可根據所使用之固持裝置而適宜調整,較佳為於自膜端部起50 cm以內之距離固定。參照圖2,可一面以複數個固持裝置18將膜之兩端固持,一面進行搬送。作為設置於膜之一端之複數個固持裝置18,其鄰接之固持裝置間之距離較佳為可抑制因膜之晃動或加熱所導致之尺寸變化而引起之破裂等缺陷之距離。鄰接之固持裝置間之距離較佳為1~50 mm,更佳為3~25 mm,進而較佳為5~10 mmm。又,固持裝置較佳為以如下方式設置:於與膜搬送軸正交之直線與膜之一端之任意之固持裝置之固持部中央對齊時,該直線和膜之另一端之交點、與離該交點最近之固持裝置之固持部中央之距離較佳為3 mm以下,更佳為2 mm以下,進而較佳為1 mm以下。藉此,可減小對抗之膜兩端部之各自之應力之差,故而所得之樹脂膜可抑制光學性質產生不均。
作為以固持裝置固定膜之兩端之操作例,可列舉如下方法:於搬入至烘箱前或搬入至烘箱後之適當時機,於膜之寬度方向上,藉由以對向之方式設置之複數個膜夾盤將膜之寬度方向之兩端固定。藉由該等操作,可抑制膜之晃動等,獲得厚度不均或損傷等缺陷得到充分抑制之樹脂膜。於進行了加熱步驟之後適時解除膜兩端之固定即可,可於烘箱內進行,亦可自烘箱搬出後進行。
加熱步驟中所使用之烘箱之膜搬送方向之全長可為10~100 m,較佳為15~80 m,更佳為15~60 m。關於烘箱,其內部可為一個空間,亦可分為複數個空間。上述空間可為可控制溫度條件或風速條件等之空間,可不具有間隔板等物理性邊界。於烘箱之內部分為複數個空間之情形時,可與膜之搬送方向垂直或平行地分為複數個空間。空間數可為2~20個,較佳為3~18個,更佳為4~15個,進而較佳為5~10個。不依賴於烘箱之內部構造,可為烘箱整體成為加熱區域,亦可為內部之一部分成為加熱區域。參照圖1,可為區域10、12及14之3個全部成為加熱區域,亦可為該等中之1個,例如區域14成為加熱區域。
烘箱亦可使用複數個。該情形時之烘箱數並無特別限定,例如可為2~12個。各烘箱之內部可為之前所述之構造。複數個烘箱可以膜不與外部氣體接觸而搬送之方式連續設置。於使用複數個烘箱之情形時,可為全部烘箱成為加熱區域,亦可為一部分烘箱成為加熱區域。
關於烘箱內部之空氣循環與排氣,於烘箱之內部分為複數個空間之情形時,較佳為於各空間中進行,於具有複數個烘箱之情形時,較佳為於各烘箱中進行。烘箱內部之溫度(烘箱中之環境溫度)較佳為可對每個烘箱進行調整,於烘箱之內部分為複數個空間之情形時,較佳為可於各空間中獨立調整溫度。各個空間之溫度設定可相同,亦可不同。但各個烘箱或空間之溫度較佳為滿足下述溫度範圍。
參照圖1,作為烘箱100,於其內部之上表面100a設置有複數個上側噴嘴30,於其內部之下表面100b設置有複數個下側噴嘴32。上側噴嘴30與下側噴嘴32係以於上下方向對向之方式設置。噴嘴例如可如圖1之區域14,設置4對噴嘴(共計8個),亦可如圖1之區域12,設置有10對噴嘴(共計20個),可根據烘箱之構造而適宜設置。作為鄰接之噴嘴之間隔,就使烘箱之構造簡單化並且均勻加熱原料膜之觀點而言,較佳為0.1~1 m,更佳為0.1~0.5 m,進而較佳為0.1~0.3 m。
於烘箱之內部劃分為複數個區間之情形時,設置於各空間之熱風吹出用之噴嘴之個數通常可為5~30個。就獲得光學均勻性更優異之樹脂膜之觀點而言,噴嘴之個數較佳為8~20個。若噴嘴個數為上述範圍,則存在浮動之膜之曲率不易變得過大之傾向,又存在膜容易於噴嘴間浮起,即易於浮動之傾向。
設置於烘箱100之上表面100a之上側噴嘴30於下部具有吹出口,可向下方向(箭頭B方向)吹出熱風。另一方面,分別設置於烘箱100之下表面之下側噴嘴32於上部具有吹出口,可向上方向(箭頭C方向)吹出熱風。再者,上側噴嘴30及下側噴嘴32以可於寬度方向均勻加熱原料膜之方式於與圖1之紙面垂直之方向上具有特定尺寸之深度,但未示於圖1。
於本實施形態之樹脂膜之製造方法中,於沿原料膜之寬度方向之噴嘴之每1 m長度中,來自每一個噴嘴30或32之吹出口之吹出風量為0.1~3 m3
/秒。作為吹出風速,就獲得光學均勻性進一步更優異之樹脂膜之觀點而言,較佳為2~23 m/秒,更佳為8~20 m/秒。又,作為吹出風量,就獲得光學均勻性進一步更優異之樹脂膜之觀點而言,於沿膜之寬度方向之噴嘴之每1 m長度中,較佳為0.1~2.5 m3
/秒,更佳為0.2~2 m3
/秒。
若來自噴嘴之吹出風速及風量為上述範圍內,則可均勻地進行原料膜之加熱,故而存在易於獲得膜整個面成為光學及物理上均勻物性之膜之傾向,故而較佳。具體而言,若於上述條件下進行加熱步驟,則膜寬度方向之面內相位差值之不均變小,易於獲得膜整個面具有更均勻之面內相位差值之樹脂膜。因此,於應用於顯示裝置時,對比度之不均得以抑制,成為視認性更優異之前面板。
又,若於上述條件下進行加熱步驟,則得以均勻加熱,故而膜中殘存之溶劑量之不均變小,故而易於獲得膜整個面為更均勻之彈性模數之樹脂膜。因此,膜整個面不易產生撓曲性之不均,可抑制因膜面之撓曲性之不同而引起之破損之產生。
於烘箱內,原料膜20自室溫被加熱至原料膜中所含之溶劑蒸發之溫度,但以原料膜之寬度方向之長度幾乎不變之方式被固持裝置18保持,故而存在因熱膨脹而易於下垂之傾向。若吹出風速及吹出風量為上述範圍,則可充分加熱原料膜20,且可抑制原料膜20之下垂或晃動。
熱風之吹出風速可於噴嘴30、32之熱風吹出口中,使用市售之熱式風速計而測定。又,來自吹出口之吹出風量可藉由吹出風速與吹出口面積之乘積而求得。再者,作為熱風之吹出風速,就測定精度之觀點而言,較佳為於各噴嘴之吹出口進行10點左右之測定,取其平均值。
熱風之吹出風速及吹出風量可根據所製造之樹脂膜之物性(光學特性、機械物性等)而適宜調整,較佳為於任一形態中均為上述範圍內。藉此,可獲得相位差更充分均勻,具有更充分高之軸精度之樹脂膜。於加熱區域中,於所有加熱區域中,吹出風速為2 m/秒以上25 m/秒以下,較佳為2 m/秒以上且未達25 m/秒,又,吹出風量於沿原料膜之寬度方向之噴嘴之每1 m長度中為0.1~3 m3
/秒,較佳為0.1 m3
/秒以上2 m3
/秒以下。
於本實施形態中,於未於烘箱100內導入原料膜20之狀態下,應保持原料膜20之位置之熱風之風速較佳為5 m/秒以下,更佳為至少於加熱區域中為此種風速。藉由使用此種熱風加熱原料膜20,可獲得光學均勻性更充分優異之樹脂膜。
於加熱區域中,各個噴嘴30、32之吹出口之熱風之吹出風速之寬度方向(與圖1之紙面垂直之方向)之最大值與最小值之差較佳為4 m/秒以下。藉由如此使用寬度方向上風速之不均較少之熱風,可獲得寬度方向之光學均勻性更高之樹脂膜。藉由如此使用風速之不均較少之熱風,可獲得光學均勻性進一步更高之樹脂膜。
於烘箱100之加熱區域中,相互對向之上側噴嘴30與下側噴嘴32之間隔L(最短距離)較佳為150 mm以上,更佳為150~600 mm,進而較佳為150~400 mm。藉由以此種間隔L配置上側噴嘴與下側噴嘴,可進一步確實地抑制各步驟中之膜之晃動。
又,加熱區域中設置之各個噴嘴30、32之吹出口之熱風之寬度方向(與圖1之紙面垂直之方向)之最高溫度與最低溫度之差(ΔT)較佳為全部2℃以下,更佳為全部1℃以下。藉由如此使用寬度方向之溫度差充分小之熱風對膜進行加熱,可進一步抑制寬度方向之配向性之不均。再者,熱風之溫度較佳為150~400℃,更佳為150~300℃,進而較佳為150~250℃。
作為樹脂膜之製造方法中可使用之噴嘴之例,可列舉:具有於原料膜之寬度方向上延伸之狹縫狀之吹出口之噴嘴、及具有於原料膜之搬送方向及原料膜之寬度方向分別配置有複數個開口之吹出口之噴嘴。於本說明書中,將具有於原料膜之寬度方向上延伸之狹縫狀之吹出口之噴嘴稱為噴射噴嘴(狹縫噴嘴),將具有於原料膜之搬送方向及原料膜之寬度方向分別配置有複數個開口之吹出口之噴嘴稱為穿孔噴嘴(多孔噴嘴)。
圖3係表示本發明之樹脂膜之製造方法中較佳使用之噴射噴嘴之形狀之一例之模式剖視圖。圖4係表示本發明之樹脂膜之製造方法中較佳使用之穿孔噴嘴之形狀之一例之模式剖視圖。圖5係表示本發明之樹脂膜之製造方法中較佳使用之穿孔噴嘴之形狀之其他例之模式剖視圖。本實施形態中之烘箱100較佳為具備如圖3所示之噴射噴嘴與如圖4及圖5所示之穿孔噴嘴之一者或兩者。
圖3表示噴射噴嘴34,圖4及圖5分別表示穿孔噴嘴36、38。再者,圖3之噴射噴嘴34、圖4之穿孔噴嘴36、圖5之穿孔噴嘴38成為設置於烘箱100內之上表面100a並向下(箭頭B方向)吹出熱風之構造。又,噴射噴嘴34、穿孔噴嘴36、穿孔噴嘴38成為設置於烘箱100內之下表面100b並向上(圖3中箭頭C方向)吹出熱風之構成。噴嘴34、36、38於與圖1之紙面垂直之方向上具有特定尺寸之深度,但未示於圖3~4。深度之長度較佳為長於原料膜20之寬之長度。
噴射噴嘴34具有於膜之寬度方向上延伸之狹縫40作為熱風之吹出口。狹縫40之狹縫寬D較佳為5 mm以上,更佳為5~20 mm。藉由將狹縫寬D設為上述範圍,可更進一步提高所得樹脂膜之光學均勻性。再者,每一個噴射噴嘴34之吹出口之面積可藉由噴射噴嘴34之噴嘴之寬度方向(圖3之深度方向)之長度與狹縫寬D之乘積而求得。該每一個噴嘴之吹出口之面積與吹出風速之乘積成為每一個噴嘴之熱風之吹出風量。藉由將該熱風之吹出風量除以沿膜之寬度方向之狹縫40之長度,可求出沿膜之寬度方向之噴嘴之每1 m長度之熱風之吹出風量。
關於穿孔噴嘴36,如圖4中所示與其長度方向垂直之剖面,為長方形之形狀。穿孔噴嘴36於作為與原料膜20對向之面之下側之面36a具有複數個開口(例如圓形之開口)42。穿孔噴嘴36之熱風之吹出口係包含設置於面36a之複數個開口42。複數個開口42為熱風之吹出口,熱風自開口42以特定風速吹出。開口42於原料膜20之搬送方向上配置有複數個,並且於寬度方向上亦配置有複數個。開口42例如可以鋸齒狀配置。再者,每一個穿孔噴嘴36之吹出口之面積可藉由設置於一個穿孔噴嘴36之全部之開口42之面積之和而求出。該每一個噴嘴之吹出口之面積與吹出風速之乘積成為每一個噴嘴之熱風之吹出風量。藉由將該熱風之吹出風量除以沿膜之寬度方向之狹縫之長度,可求出沿膜之寬度方向之噴嘴之每1 m長度之熱風之吹出風量。
關於穿孔噴嘴38,如圖5中所示與其長度方向垂直之剖面,為作為朝向與原料膜20對向之面38a逐漸擴大狀之梯形形狀。穿孔噴嘴38於作為與膜對向之面之下側之面38a具有複數個開口(例如圓形之開口)44。穿孔噴嘴38之熱風之吹出口係包含設置於面38a之複數個開口44。複數個開口44為熱風之吹出口,熱風自開口44以特定風速吹出。開口44於原料膜20之搬送方向上配置有複數個,並且於寬度方向上亦配置有複數個。開口44例如可以鋸齒狀配置。再者,每一個穿孔噴嘴38之吹出口之面積可藉由設置於一個穿孔噴嘴38之全部之開口44之面積之和而求出。該每一個噴嘴之吹出口之面積與吹出風速之乘積成為每一個噴嘴之熱風之吹出風量。沿膜之寬度方向之噴嘴之每1 m長度之熱風之吹出風量可藉由與穿孔噴嘴36相同之方法而求得。
於使用穿孔噴嘴36或38之情形時,噴嘴之吹出口之熱風之寬度方向之最大吹出風速與最小吹出風速之差可作為自設置於同一噴嘴36或38上之複數個開口42或44吹出之熱風之最大吹出速度與最小吹出速度之差而求得。噴嘴之吹出口之熱風之寬度方向之最高溫度與最低溫度之差亦可以相同之方式求得。
若設置於烘箱100內之噴嘴全部為穿孔噴嘴36或38,則可增大烘箱100整體之熱風吹出口之總面積。故而可減小吹至原料膜20上之熱風之風壓,可進一步減小原料膜20之晃動。藉此,可進一步提高所得樹脂膜之光學均勻性。於烘箱內或加熱區域中,原料膜20自室溫被加熱至原料膜中所含之溶劑蒸發之溫度,但以原料膜之寬度方向之長度幾乎不變之方式被固持裝置18保持,故而存在因熱膨脹而易於下垂之傾向。藉由於加熱區域10中使用穿孔噴嘴36或38,可進一步抑制原料膜20之下垂或晃動。
穿孔噴嘴36、38之面36a、38a上設置之開口42、44之各自之尺寸及數量可於如下範圍內適宜調整:各開口42、44之熱風之吹出風速成為2 m/秒以上且未達25 m/秒,且來自各個噴嘴之吹出風量於沿膜之寬度方向之噴嘴之每1 m長度中成為0.1~3 m3
/秒。
就使來自穿孔噴嘴36、38之各開口之吹出風速變得更均勻之觀點而言,開口42、44之形狀較佳為圓形。於該情形時,開口42、44之直徑較佳為2~10 mm,更佳為3~8 mm。
於使用穿孔噴嘴36、38之情形時,每一個噴嘴之面36a、38a之膜搬送方向之長度較佳為50~300 mm。進而鄰接之穿孔噴嘴之間隔較佳為0.3 m以下。又,穿孔噴嘴36、38之開口42、44之面積之總和(吹出口之面積)相對於穿孔噴嘴36、38之膜寬度方向之長度之比(穿孔噴嘴之開口之面積之總和(m2
)/穿孔噴嘴之膜寬度方向之長度(m))較佳為0.008 m以上。
藉由使用此種穿孔噴嘴36、38,可增大熱風之吹出口之面積。藉此,可充分降低熱風之風速且以充分之風量吹出熱風,可進一步更均勻地加熱膜。因此,可製造相位差進一步更均勻,且具有進一步更高軸精度之膜。
除上述噴射噴嘴及穿孔噴嘴外,亦可併用IR(Infrared,紅外線)加熱器(輻射熱線處理)。IR加熱器可設置於烘箱內或加熱區域,亦可將IR加熱器爐用作一個烘箱。
於本實施形態中,吹送至膜之熱風之風速較佳為剛搬入烘箱後之風速大於烘箱內之其他搬送路徑之風速。所謂剛搬入烘箱後(以下,稱為搬送路徑1),於烘箱之內部未分隔為複數個之情形時,係指自烘箱搬入口未達烘箱長度(自烘箱之搬入口至搬出口之長度)之1/10之距離。於烘箱之內部分為複數個空間之情形時,搬送路徑1係指膜最初通過之空間。於使用複數個烘箱之情形時,可根據最初使用之烘箱之內部構造而與之前之記載相同,亦可設定為最初通過之烘箱內之風速大於第2個以後之烘箱內之風速。
所謂其他搬送路徑,於烘箱之內部未分隔為複數個之情形時,係指自烘箱搬入口至烘箱長度之1/10以後之搬送路徑部。於烘箱之內部分為複數個空間之情形時,係指膜通過之第2個以後之任意空間。於使用複數個烘箱之情形時,可根據最初使用之烘箱之內部構造而與之前之記載相同,亦可設定為第2個以後之烘箱中任意之烘箱內之風速小於最初通過之烘箱內之風速。
搬送路徑1之風速與烘箱內之其他搬送路徑之風速之差較佳為0.1以上,更佳為0.2以上,較佳為15以下,更佳為8以下,進而較佳為5以下,尤佳為3以下。上述上限與下限可任意組合。若以風速之差成為上述範圍之方式使剛搬入烘箱後之風速大於烘箱內之其他搬送路徑之風速,則存在可更高效地去除膜中之溶劑之傾向。若風速之差為上述範圍,則存在不易產生因風速差而引起之膜之晃動,所得樹脂膜之表面形狀之缺陷或相位差等光學特性之不均變小之傾向。
搬送路徑1之風速與烘箱內之其他搬送路徑之風速之差可作為來自設置於搬送路徑1之噴嘴之熱風之吹出風速與來自設置於其他搬送路徑之噴嘴之熱風之吹出風速之差而求得。於吹送至膜之熱風之風速與來自噴嘴之熱風之吹出風速存在2 m/秒以上之差之情形時,亦可作為搬送路徑1及其他搬送路徑之各自之膜附近之熱風之風速之差而求得。
其他搬送路徑較佳為位於搬送路徑1之下一個之搬送路徑(稱為搬送路徑2)。作為搬送路徑2,於烘箱之內部未分隔為複數個之情形時,係指位於自烘箱搬入口至烘箱長度之2/10之搬送路徑部。作為搬送路徑2,於烘箱之內部分為複數個空間之情形時,係指膜通過之第2個空間。於使用複數個烘箱之情形時,可根據最初使用之烘箱之內部構造而與之前之記載相同,亦可設定為第2個烘箱之風速小於最初通過之烘箱之風速。
於如上述般設定搬送路徑1與搬送路徑2之風速之差之情形時,搬送路徑2以後之搬送路徑之風速只要為上述熱風之吹出風速之範圍內即可。搬送路徑2以後之搬送路徑之風速與搬送路徑1或搬送路徑2之各自之風速之差較佳為0.1~12 m/秒,更佳為0.2~8 m/秒。若為此種範圍之風速之差,則存在可抑制因風速差而引起之膜之晃動,又易於將所得樹脂膜之重量減少率調整為所期望之範圍之傾向。
作為上述風速之差,於烘箱之內部未分為複數個空間之情形時,可藉由調整設置噴嘴之位置、噴嘴之熱風之吹出速度及風量、烘箱內之氣流之流動等而調整。於烘箱之內部分為複數個空間之情形時,可於最初之空間與其以後之空間中,藉由調整設置噴嘴之位置、噴嘴之熱風之吹出速度及風量、烘箱內之氣流之流動等而調整。於使用複數個烘箱之情形時,可根據最初之烘箱之構造而以與之前之記載相同之方式進行,亦可以最初之烘箱與第2個以後之烘箱中風速不同之方式設定設置噴嘴之位置、噴嘴之熱風之吹出速度及風量、烘箱內之氣流等。
於本實施形態中,加熱步驟通常於150~350℃之範圍內進行,較佳為170℃以上,進而較佳為180℃以上,又較佳為300℃以下,更佳為250℃以下,進而較佳為230℃以下。於本發明之實施形態中若加熱步驟為該溫度範圍,則存在易於以原料膜成為下述重量減少率M之方式進行調整之傾向,又,存在所得樹脂膜之黃度不易成為3以上之傾向。作為進行加熱步驟之烘箱內之溫度,只要加熱區域為上述範圍即可。於具有複數個烘箱之情形及烘箱內分為複數個空間之情形時,可適宜調整,較佳為全部烘箱或空間為上述範圍內。
烘箱100內之原料膜20之移動速度通常可於0.1~50 m/分鐘之範圍內適宜調整。上述移動速度較佳為20 m/分鐘以下,更佳為15 m/分鐘以下,較佳為0.2 m/分鐘以上,更佳為0.5 m/分鐘以上,進而較佳為0.7 m/分鐘以上,尤佳為0.8 m/分鐘以上。若移動速度為上述範圍,則存在不會為確保所期望之乾燥時間而使烘箱長度變得過長、設備變得過大之傾向,又,存在易於以原料膜成為下述重量減少率M之方式進行調整之傾向,進而存在膜之晃動得以抑制,從而可抑制膜面產生損傷之傾向。
加熱步驟之處理時間通常於60秒~2小時之範圍內調整即可,較佳為於10分鐘~1小時之範圍內調整。處理時間可考慮上述烘箱之溫度、移動速度、熱風之風速及風量等條件而適宜調整。
本實施形態之樹脂膜之製造方法可於加熱步驟中進行改變膜寬之操作或保持膜寬而搬送之操作。作為改變膜寬之操作之例,可列舉:使膜之寬度方向延伸之操作。於使膜之寬度方向延伸之操作中,延伸倍率較佳為0.7~1.3倍,更佳為0.8~1.2倍,進而較佳為0.8~1.1倍。作為保持膜寬而搬送之操作之例,可列舉:以膜之寬度方向之長度幾乎不變之方式保持之操作。進行有該等操作之樹脂膜可具有相對於原料膜之寬度方向之長度為0.7~1.3倍左右之長度,可為自原料膜之寬度方向之長度延伸、等倍或收縮之長度。延伸倍率可作為延伸後之膜寬(除固持部分外)相對於除固持部分外之膜寬之比而求出。
再者,圖2中,將於使膜之寬度方向延伸之操作中延伸倍率超過1倍之情形以實線表示,將延伸倍率為等倍或未達1倍之情形以虛線表示。
樹脂膜可用於可撓性顯示裝置等圖像顯示裝置之前面板等光學用途中,故而較佳為異物等之附著量較少。故而,加熱步驟之潔淨度較佳為10000以下,更佳為1,000以下。具體而言,較佳為進行加熱步驟之烘箱100設置於無塵室內,較佳為烘箱100之內部之潔淨度高於無塵室內之潔淨度(空氣中之異物較少)。無塵室較佳為10,000之潔淨度。又,烘箱100中之潔淨度較佳為清潔度等級1,000以下。本說明書中之所謂「清潔度等級」係指美國聯邦標準(USA FED.STD)209D中規定之清潔度等級,所謂「清潔度等級1000」係指空氣中所含之粒徑0.5 μm以下之微粒子於每1立方英尺(1 ft3
)中不超過1,000個之環境。附帶一提,美國聯邦標準209D中規定之清潔度等級1,000係相當於JIS B9920「無塵室之空氣清浄度之評價方法」中規定之清潔度等級6。
經過加熱步驟之樹脂膜自烘箱搬出後,可連續供給至下一步驟,亦可捲取為卷狀而供給至下一步驟。於將樹脂膜捲取為捲筒之情形時,可積層表面保護膜及其他光學膜等其他膜而捲取。作為積層於樹脂膜之表面保護膜,可使用與下述積層於原料膜之表面保護膜相同者。積層於樹脂膜之表面保護膜之厚度通常為10~100 μm,較佳為10~80 μm。
<原料膜>
供給至加熱步驟之原料膜至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者。原料膜較佳為含有與下述原料膜之形成中所使用之清漆中所含之成分相同之成分,但由於可發生成分之結構變化或溶劑之一部分之蒸發,故而可不相同。原料膜可為自支撐膜,亦可為凝膠膜。
原料膜無論含不含無機材料,藉由熱重量-示差熱測定(以下有時稱為「TG-DTA測定」)而求得之自120℃至250℃之重量減少率M較佳為1~40%左右,更佳為3~20%,進而較佳為5~15%,尤佳為5~12%。原料膜之重量減少率M可使用市售之TG-DTA之測定裝置藉由以下方法而測定。作為TG-DTA之測定裝置,可使用Hitachi High-Tech Science(股)製造之TG/DTA6300。
首先,自原料膜獲取約20 mg之試樣,一面將試樣於以10℃/分鐘之升溫速度自室溫升溫至120℃,於120℃下保持5分鐘後,以10℃/分鐘之升溫速度升溫至400℃之條件下加熱,一面測定試樣之重量變化。其次,自TG-DTA測定之結果,藉由下述式算出自120℃至250℃之重量減少率M(%)即可。於下述式中,W0
表示於120℃下保持5分鐘後之試樣之重量,W1
表示250℃下之試樣之重量。
M(%)=100-(W1
/W0
)×100
若原料膜之重量減少率M大到某程度,則將原料膜作為與基材或表面保護膜之積層體而捲取時,存在積層體之彎曲等變形得以抑制,積層體之捲取性提高之傾向。
若原料膜之重量減少率M小到某程度,則將原料膜作為與基材或表面保護膜之積層體而捲取時,存在原料膜難以貼附於基材或表面保護膜之傾向。故而,可維持原料膜之均勻之透明性,並且可將積層體自捲筒容易地捲出。
(聚醯亞胺系樹脂及聚醯胺系樹脂)
所謂原料膜及樹脂膜中所含之聚醯亞胺系樹脂,係指選自由含有包含醯亞胺基之重複結構單元之聚合物(以下,有時記為聚醯亞胺)、以及含有包含醯亞胺基及醯胺基之兩者之重複結構單元之聚合物(以下,有時記為聚醯胺醯亞胺)所組成之群中之至少一種聚合物。又,所謂聚醯胺系樹脂係指含有包含醯胺基之重複結構單元之聚合物。
聚醯亞胺系樹脂較佳為具有式(10)所表示之重複結構單元。此處,G為4價之有機基,A為2價之有機基。聚醯亞胺系樹脂可含有G及/或A不同之兩種以上之式(10)所表示之重複結構單元。
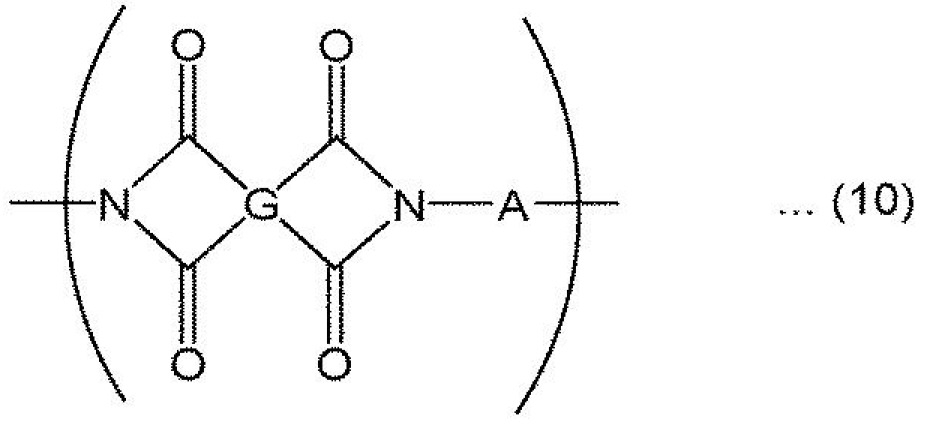
聚醯亞胺系樹脂可於不損害樹脂膜之各種物性之範圍內,含有選自由式(11)、式(12)及式(13)所表示之重複結構單元所組成之群中之一種以上。

式(10)及式(11)中,G及G1
分別獨立為4價之有機基,較佳為可經烴基或氟取代烴基取代之有機基。作為G及G1
,可例示:式(20)、式(21)、式(22)、式(23)、式(24)、式(25)、式(26)、式(27)、式(28)或式(29)所表示之基以及4價之碳數6以下之鏈式烴基。就易於抑制樹脂膜之黃度(YI值)之方面而言,其中,較佳為式(20)、式(21)、式(22)、式(23)、式(24)、式(25)、式(26)或式(27)所表示之基。
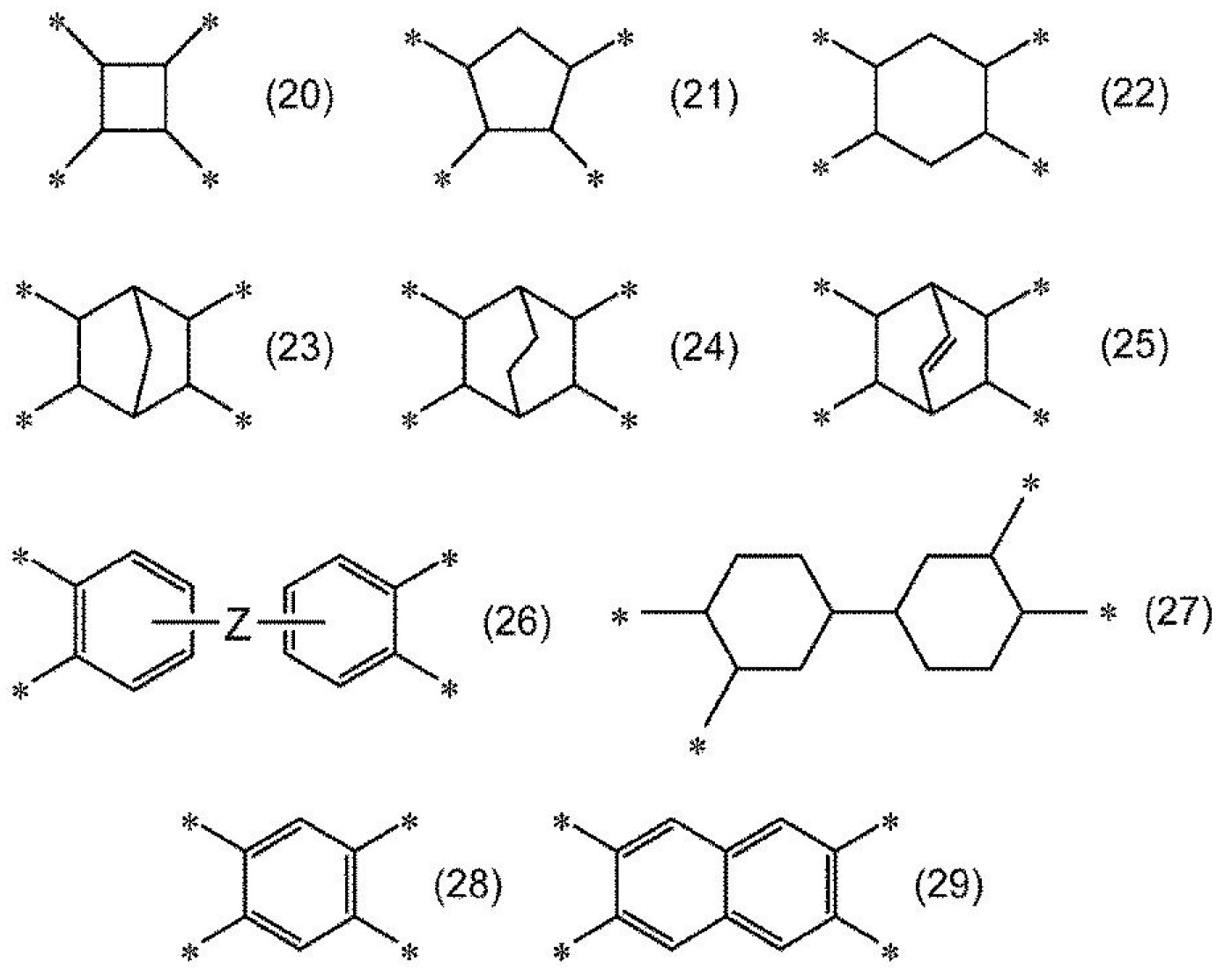
式(20)~式(29)中,
*表示鍵結鍵,
Z表示單鍵、-O-、-CH2
-、-CH2
-CH2
-、-CH(CH3
)-、-C(CH3
)2
-、-C(CF3
)2
-、-Ar-、-SO2
-、-CO-、-O-Ar-O-、-Ar-O-Ar-、-Ar-CH2
-Ar-、-Ar-C(CH3
)2
-Ar-或-Ar-SO2
-Ar-。Ar表示可經氟原子取代之碳數6~20之伸芳基,作為具體例,可列舉伸苯基。
式(12)中,G2
為3價之有機基,較佳為可經烴基或氟取代烴基取代之有機基。作為G2
,可例示:式(20)、式(21)、式(22)、式(23)、式(24)、式(25)、式(26)、式(27)、式(28)或式(29)所表示之基之鍵結鍵之任一個被氫原子取代而成之基以及3價之碳數6以下之鏈式烴基。
式(13)中,G3
為2價之有機基,較佳為可經烴基或氟取代烴基取代之有機基。作為G3
,可例示:式(20)、式(21)、式(22)、式(23)、式(24)、式(25)、式(26)、式(27)、式(28)或式(29)所表示之基之鍵結鍵中不鄰接之兩個被氫原子取代而成之基及碳數6以下之鏈式烴基。
式(10)~式(13)中,A、A1
、A2
及A3
分別獨立為2價之有機基,較佳為可經烴基或氟取代烴基取代之有機基。作為A、A1
、A2
及A3
,可列舉:式(30)、式(31)、式(32)、式(33)、式(34)、式(35)、式(36)、式(37)或式(38)所表示之基;該等經甲基、氟基、氯基或三氟甲基取代而成之基;以及碳數6以下之鏈式烴基。
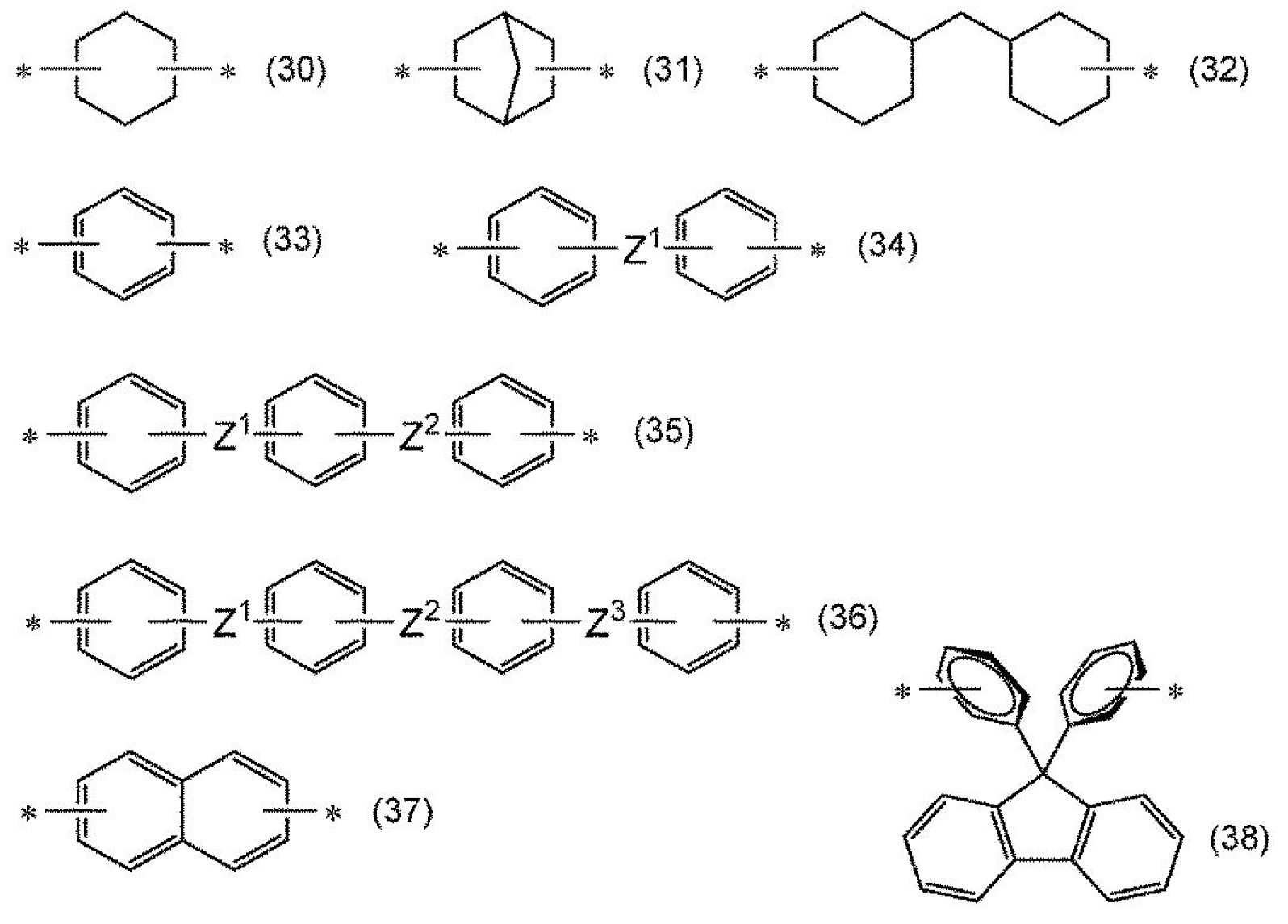
式(30)~式(38)中,
*表示鍵結鍵,
Z1
、Z2
及Z3
分別獨立表示單鍵、-O-、-CH2
-、-CH2
-CH2
-、-CH(CH3
)-、-C(CH3
)2
-、-C(CF3
)2
-、-SO2
-或-CO-。
一個例為:Z1
及Z3
為-O-,且Z2
為-CH2
-、-C(CH3
)2
-、-C(CF3
)2
-或-SO2
-。Z1
與Z2
之對各環之鍵結位置、及Z2
與Z3
之對各環之鍵結位置分別較佳為對各環為間位或對位。
作為聚醯亞胺系樹脂,就易於提高視認性之觀點而言,較佳為至少具有式(10)所表示之重複結構單元與式(13)所表示之重複結構單元之聚醯胺醯亞胺。又,聚醯胺系樹脂較佳為至少具有式(13)所表示之重複結構單元。
於本發明之一實施態樣中,聚醯亞胺系樹脂係使二胺及四羧酸化合物(醯氯化合物、四羧酸二酐等四羧酸化合物相關物)、以及視需要之二羧酸化合物(醯氯化合物等二羧酸化合物相關物)、三羧酸化合物(醯氯化合物、三羧酸酐等三羧酸化合物相關物)等進行反應(縮聚)而獲得之縮合型高分子。式(10)或式(11)所表示之重複結構單元通常係自二胺及四羧酸化合物衍生。式(12)所表示之重複結構單元通常係自二胺及三羧酸化合物衍生。式(13)所表示之重複結構單元通常係自二胺及二羧酸化合物衍生。
於本發明之一實施態樣中,聚醯胺系樹脂係使二胺與二羧酸化合物進行反應(縮聚)而獲得之縮合型高分子。即,式(13)所表示之重複結構單元通常係自二胺及二羧酸化合物衍生。
作為四羧酸化合物,可列舉:芳香族四羧酸二酐等芳香族四羧酸化合物;及脂肪族四羧酸二酐等脂肪族四羧酸化合物。四羧酸化合物可單獨使用,亦可併用兩種以上。四羧酸化合物除二酐外,亦可為醯氯化合物等四羧酸化合物相關物。
作為芳香族四羧酸二酐之具體例,可列舉:4,4'-氧二鄰苯二甲酸二酐、3,3',4,4'-二苯甲酮四羧酸二酐、2,2',3,3'-二苯甲酮四羧酸二酐、3,3',4,4'-聯苯四羧酸二酐、2,2',3,3'-聯苯四羧酸二酐、3,3',4,4'-二苯碸四羧酸二酐、2,2-雙(3,4-二羧基苯基)丙烷二酐、2,2-雙(2,3-二羧基苯基)丙烷二酐、2,2-雙(3,4-二羧基苯氧基苯基)丙烷二酐、4,4'-(六氟亞異丙基)二鄰苯二甲酸二酐(6FDA)、1,2-雙(2,3-二羧基苯基)乙烷二酐、1,1-雙(2,3-二羧基苯基)乙烷二酐、1,2-雙(3,4-二羧基苯基)乙烷二酐、1,1-雙(3,4-二羧基苯基)乙烷二酐、雙(3,4-二羧基苯基)甲烷二酐、雙(2,3-二羧基苯基)甲烷二酐及4,4'-(對伸苯基二氧基)二鄰苯二甲酸二酐及4,4'-(間伸苯基二氧基)二鄰苯二甲酸二酐。該等可單獨使用或組合兩種以上使用。
作為脂肪族四羧酸二酐,可列舉:環式或非環式之脂肪族四羧酸二酐。所謂環式脂肪族四羧酸二酐係指具有脂環式烴結構之四羧酸二酐,作為其具體例,可列舉:1,2,4,5-環己烷四羧酸二酐、1,2,3,4-環丁烷四羧酸二酐、1,2,3,4-環戊烷四羧酸二酐等環烷烴四羧酸二酐、雙環[2.2.2]辛-7-烯-2,3,5,6-四羧酸二酐、二環己基-3,3',4,4'-四羧酸二酐及該等之位置異構體。該等可單獨使用或組合兩種以上使用。作為非環式脂肪族四羧酸二酐之具體例,可列舉:1,2,3,4-丁烷四羧酸二酐、1,2,3,4-戊烷四羧酸二酐等,該等可單獨使用或組合兩種以上使用。又,可組合使用環式脂肪族四羧酸二酐及非環式脂肪族四羧酸二酐。
上述四羧酸二酐中,就高透明性及低著色性之觀點而言,較佳為1,2,4,5-環己烷四羧酸二酐、雙環[2.2.2]辛-7-烯-2,3,5,6-四羧酸二酐及4,4'-(六氟亞異丙基)二鄰苯二甲酸二酐、以及該等之混合物。又,作為四羧酸,可使用上述四羧酸化合物之酸酐之水加成物。
作為三羧酸化合物,可列舉:芳香族三羧酸、脂肪族三羧酸及該等之相關之醯氯化合物、酸酐等,可併用兩種以上。
作為具體例,可列舉:1,2,4-苯三羧酸之酸酐;2,3,6-萘三羧酸-2,3-酐;鄰苯二甲酸酐與苯甲酸以單鍵、-CH2
-、-C(CH3
)2
-、-C(CF3
)2
-、-SO2
-或伸苯基連結而成之化合物。
作為二羧酸化合物,可列舉:芳香族二羧酸、脂肪族二羧酸及該等之相關之醯氯化合物、酸酐等,可將該等併用兩種以上。
作為該等之具體例,可列舉:對苯二甲醯二氯(對苯二甲醯氯(TPC));間苯二甲醯二氯;萘二甲醯二氯;4,4'-聯苯二甲醯二氯;3,3'-聯苯二甲醯二氯;4,4'-氧基雙(苯甲醯氯)(OBBC);碳數8以下之鏈式烴之二羧酸化合物及2個苯甲酸以單鍵、-CH2
-、-C(CH3
)2
-、-C(CF3
)2
-、-SO2
-或伸苯基連結而成之化合物。
作為二胺,例如可列舉:脂肪族二胺、芳香族二胺或該等之混合物。再者,於本實施形態中所謂「芳香族二胺」係指胺基直接鍵結於芳香環之二胺,其結構之一部分中可含有脂肪族基或其他取代基。芳香環可為單環亦可為縮合環,可例示苯環、萘環、蒽環及茀環等,但並不限定於該等。該等之中,較佳為芳香環為苯環。又,所謂「脂肪族二胺」係指胺基直接鍵結於脂肪族基之二胺,其結構之一部分中可含有芳香環或其他取代基。
作為脂肪族二胺,例如可列舉:己二胺等非環式脂肪族二胺及1,3-雙(胺基甲基)環己烷、1,4-雙(胺基甲基)環己烷、降𦯉烷二胺、4,4'-二胺基二環己基甲烷等環式脂肪族二胺等。該等可單獨使用或組合兩種以上使用。
作為芳香族二胺,例如可列舉:對苯二胺、間苯二胺、2,4-甲苯二胺、間苯二甲胺、對苯二甲胺、1,5-二胺基萘、2,6-二胺基萘等具有1個芳香環之芳香族二胺;4,4'-二胺基二苯基甲烷、4,4'-二胺基二苯基丙烷、4,4'-二胺基二苯醚、3,4'-二胺基二苯醚、3,3'-二胺基二苯醚、4,4'-二胺基二苯碸、3,4'-二胺基二苯碸、3,3'-二胺基二苯碸、1,4-雙(4-胺基苯氧基)苯、1,3-雙(4-胺基苯氧基)苯、4,4'-二胺基二苯碸、雙[4-(4-胺基苯氧基)苯基]碸、雙[4-(3-胺基苯氧基)苯基]碸、2,2-雙[4-(4-胺基苯氧基)苯基]丙烷、2,2-雙[4-(3-胺基苯氧基)苯基]丙烷、2,2'-二甲基聯苯胺、2,2'-雙(三氟甲基)聯苯胺(2,2'-雙(三氟甲基)-4,4'-二胺基聯苯(TFMB))、4,4'-雙(4-胺基苯氧基)聯苯、9,9-雙(4-胺基苯基)茀、9,9-雙(4-胺基-3-甲基苯基)茀、9,9-雙(4-胺基-3-氯苯基)茀、9,9-雙(4-胺基-3-氟苯基)茀等具有2個以上芳香環之芳香族二胺。該等可單獨使用或組合兩種以上使用。
上述二胺之中,就高透明性及低著色性之觀點而言,較佳為使用選自由具有聯苯結構之芳香族二胺所組成之群中之一種以上,更佳為使用選自由2,2'-二甲基聯苯胺、2,2'-雙(三氟甲基)聯苯胺、4,4'-雙(4-胺基苯氧基)聯苯及4,4'-二胺基二苯醚所組成之群中之一種以上,進而較佳為使用2,2'-雙(三氟甲基)聯苯胺。
聚醯亞胺系樹脂係藉由如下方式獲得:藉由慣用之方法,例如攪拌等方法將上述二胺、四羧酸化合物、三羧酸化合物、二羧酸化合物等各原料混合後,於醯亞胺化觸媒及視需要之脫水劑之存在下將所得中間物進行醯亞胺化。聚醯胺系樹脂係藉由如下方式獲得:藉由慣用之方法,例如攪拌等方法將上述二胺、二羧酸化合物等各原料混合。
作為醯亞胺化步驟中所使用之醯亞胺化觸媒,並無特別限定,例如可列舉:三丙基胺、二丁基丙基胺、乙基二丁基胺等脂肪族胺;N-乙基哌啶、N-丙基哌啶、N-丁基吡咯啶、N-丁基哌啶及N-丙基六氫氮呯等脂環式胺(單環式);氮雜雙環[2.2.1]庚烷、氮雜雙環[3.2.1]辛烷、氮雜雙環[2.2.2]辛烷及氮雜雙環[3.2.2]壬烷等脂環式胺(多環式);以及2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-乙基吡啶、3-乙基吡啶、4-乙基吡啶、2,4-二甲基吡啶、2,4,6-三甲基吡啶、3,4-環戊烯并吡啶、5,6,7,8-四氫異喹啉及異喹啉等芳香族胺。
作為醯亞胺化步驟中所使用之脫水劑,並無特別限定,例如可列舉:乙酸酐、丙酸酐、異丁酸酐、三甲基乙酸酐、丁酸酐、異戊酸酐等。
於各原料之混合及醯亞胺化步驟中,反應溫度並無特別限定,例如為15~350℃,較佳為20~100℃。反應時間亦無特別限定,例如為10分鐘~10小時左右。視需要,可於惰性環境或減壓之條件下進行反應。又,反應可於溶劑中進行,作為溶劑,例如可列舉作為清漆之製備中所使用之溶劑而例示者。反應後精製聚醯亞胺系樹脂或聚醯胺系樹脂。作為精製方法,例如可列舉如下方法等:於反應液中添加不良溶劑,藉由再沈澱法而使樹脂析出,加以乾燥並取出沈澱物,視需要以甲醇等溶劑清洗沈澱物並使之乾燥。
再者,聚醯亞胺系樹脂之製造例如可參照日本專利特開2006-199945號公報或日本專利特開2008-163107號公報中記載之製造方法。又,聚醯亞胺系樹脂亦可使用市售品,作為其具體例,可列舉:三菱瓦斯化學(股)製造之Neopulim(註冊商標)、河村產業(股)製造之KPI-MX300F等。
聚醯亞胺系樹脂或聚醯胺系樹脂之重量平均分子量較佳為200,000以上,更佳為250,000以上,進而較佳為300,000以上,較佳為600,000以下,更佳為500,000以下。聚醯亞胺系樹脂或聚醯胺系樹脂之重量平均分子量越大,越存在易於表現膜化時之較高之耐撓曲性之傾向。故而,就提高樹脂膜之耐撓曲性之觀點而言,重量平均分子量較佳為上述下限以上。另一方面,聚醯亞胺系樹脂或聚醯胺系樹脂之重量平均分子量越小,越存在易於降低清漆之黏度,易於提高加工性之傾向。又,存在易於提高聚醯亞胺系樹脂或聚醯胺系樹脂之延伸性之傾向。故而,就加工性及延伸性之觀點而言,重量平均分子量較佳為上述上限以下。再者,於本案中,重量平均分子量可進行凝膠滲透層析(GPC)測定,藉由標準聚苯乙烯換算而求得,例如可藉由實施例中記載之方法而算出。
聚醯亞胺系樹脂之醯亞胺化率較佳為95~100%,更佳為97~100%,進而較佳為98~100%,尤佳為100%。就清漆之穩定性、所得樹脂膜之機械物性之觀點而言,醯亞胺化率較佳為上述下限以上。再者,醯亞胺化率可藉由IR法、NMR(nuclear magnetic resonance,核磁共振)法等而求得。就上述觀點而言,清漆中所含之聚醯亞胺系樹脂之醯亞胺化率較佳為上述範圍內。
於本發明之較佳之一實施形態中,本發明之樹脂膜中所含之聚醯亞胺系樹脂或聚醯胺系樹脂可含有例如可藉由上述含氟取代基等而導入之氟原子等鹵素原子。於聚醯亞胺系樹脂或聚醯胺系樹脂含有鹵素原子之情形時,易於提高樹脂膜之彈性模數且減低黃度(YI值)。若樹脂膜之彈性模數較高,則易於抑制該膜之損傷及皺褶等之發生,又,若樹脂膜之黃度較低,則易於提高該膜之透明性。鹵素原子較佳為氟原子。作為用以使氟原子含有於聚醯亞胺系樹脂或聚醯胺系樹脂中之較佳含氟取代基,例如可列舉氟基及三氟甲基。
作為聚醯亞胺系樹脂或聚醯胺系樹脂中之鹵素原子之含量,以聚醯亞胺系樹脂或聚醯胺系樹脂之質量為基準,較佳為1~40質量%,更佳為5~40質量%,進而較佳為5~30質量%。若鹵素原子之含量為上述範圍,則易於進一步提高膜化時之彈性模數,降低吸水率,進一步減低黃度(YI值),進一步提高透明性,又,有時合成變得容易。
於本發明之一實施形態中,作為樹脂膜中之聚醯亞胺系樹脂及/或聚醯胺系樹脂之含量,以樹脂膜之總質量為基準,較佳為40質量%以上,更佳為50質量%以上,進而較佳為70質量%以上。就易於提高耐撓曲性等之觀點而言,較佳為聚醯亞胺系樹脂及/或聚醯胺系樹脂之含量為上述下限以上。再者,作為樹脂膜中之聚醯亞胺系樹脂及/或聚醯胺系樹脂之含量,以樹脂膜之總質量為基準,通常為100質量%以下。
(添加劑)
本發明之樹脂膜可進而含有添加劑。作為此種添加劑,例如可列舉:二氧化矽粒子、紫外線吸收劑、增白劑、二氧化矽分散劑、抗氧化劑、pH值調整劑及調平劑。
(二氧化矽粒子)
本發明之樹脂膜可進而含有二氧化矽粒子作為添加劑。作為二氧化矽粒子之含量,以該樹脂膜之總質量為基準,較佳為1質量%以上,更佳為3質量%以上,進而較佳為5質量%以上,較佳為60質量%以下,更佳為50質量%以下,進而較佳為45質量%以下。又,二氧化矽粒子之含量可於該等上限值及下限值中,選擇組合任意之下限值與上限值。若二氧化矽粒子之含量為上述上限值及/或下限值之數值範圍,則存在於本發明之樹脂膜中,二氧化矽粒子不易凝集,以一次粒子之狀態均勻分散之傾向,故而可抑制本發明之樹脂膜之視認性之下降。
二氧化矽粒子之粒徑較佳為1 nm以上,更佳為3 nm以上,進而較佳為5 nm以上,尤佳為8 nm以上,較佳為30 nm以下,更佳為28 nm以下,進而較佳為25 nm以下,尤佳為20 nm以下。二氧化矽粒子之粒徑可於該等上限值及下限值中,選擇組合任意之下限值與上限值。若二氧化矽粒子之含量為上述上限值及/或下限值之數值範圍,則於本發明之樹脂膜中,不易與白色光中之特定波長之光相互作用,故而可抑制本發明之樹脂膜之視認性之下降。於本說明書中,二氧化矽粒子之粒徑表示平均一次粒徑。樹脂膜內之二氧化矽粒子之粒徑可自使用有穿透式電子顯微鏡(TEM)之攝像而測定。製作樹脂膜之前(例如,添加至清漆之前)之二氧化矽粒子之粒徑可藉由雷射繞射式粒度分佈計而測定。二氧化矽粒子之粒徑之測定方法於實施例中詳細說明。
作為二氧化矽粒子之形態,例如可列舉:二氧化矽粒子分散於有機溶劑等中之矽溶膠、及藉由氣相法而製備之二氧化矽粉末。該等之中,就作業性之觀點而言,較佳為矽溶膠。
二氧化矽粒子可實施表面處理,例如,可為自水溶性醇分散矽溶膠進行溶劑(更具體而言,γ-丁內酯等)置換所得之二氧化矽粒子。水溶性醇係於該水溶性醇分子1個中每1個羥基之碳數為3以下之醇,可列舉:甲醇、乙醇、1-丙醇及2-丙醇等。通常,若二氧化矽粒子經表面處理,則存在與樹脂膜中所含之聚醯亞胺系樹脂之相溶性提高,二氧化矽粒子之分散性提高之傾向,故而可抑制本發明之視認性之下降,但亦依存於二氧化矽粒子與聚醯亞胺系樹脂或聚醯胺系樹脂之種類之親和性。
(紫外線吸收劑)
本發明之樹脂膜可進而含有紫外線吸收劑。例如可列舉;三𠯤系紫外線吸收劑、二苯甲酮系紫外線吸收劑、苯并三唑系紫外線吸收劑、苯甲酸酯系紫外線吸收劑及氰基丙烯酸酯系紫外線吸收劑等。該等可單獨使用,亦可併用兩種以上。作為較佳之市售之紫外線吸收劑,例如可列舉:Sumika Chemtex(股)製造之Sumisorb(註冊商標)340、ADEKA(股)製造之Adekastab(註冊商標)LA-31、及BASF JAPAN(股)製造之TINUVIN(註冊商標)1577等。作為紫外線吸收劑之含量,以本發明之樹脂膜之質量為基準,較佳為1 phr以上10 phr以下,更佳為3 phr以上6 phr以下。
(增白劑)
本發明之樹脂膜可進而含有增白劑。增白劑例如於添加有增白劑以外之添加劑之情形時,可以調整色調為目的而添加。作為增白劑,可列舉:單偶氮系染料、三芳基甲烷系染料、酞菁系染料及蒽醌系染料。該等之中,較佳為蒽醌系染料。作為較佳之市售之增白劑,例如可列舉:Lanxess公司製造之MACROLEX(註冊商標)Violet B、Sumika Chemtex(股)製造之Sumiplast(註冊商標)Violet B、及三菱化學(股)製造之Diaresin(註冊商標)Blue G等。該等可單獨使用,亦可併用兩種以上。於含有增白劑之情形時,其含量以本發明之樹脂膜之質量為基準,較佳為1~50 ppm,更佳為1~45 ppm,進而較佳為3~40 ppm,尤佳為5~35 ppm。
(原料膜之製造方法)
原料膜並無特別限定,例如可藉由包含以下步驟之方法而製造。
(a)製備含有上述樹脂及上述填料之液體(以下,有時記為清漆)之清漆製備步驟;
(b)將清漆塗佈於基材而形成塗膜之塗佈步驟;及
(c)使上述塗膜乾燥,形成原料膜之形成步驟。
於清漆製備步驟中,將上述樹脂溶解於溶劑,添加上述填料及視需要之其他添加劑並加以攪拌混合,藉此製備清漆。再者,於使用二氧化矽作為填料之情形時,可將以上述樹脂可溶解之溶劑,例如下述清漆之製備中所使用之溶劑將含有二氧化矽之矽溶膠之分散液進行置換所得之矽溶膠添加至樹脂。
清漆之製備中所使用之溶劑只要可溶解上述樹脂,則並無特別限定。作為該溶劑,例如可列舉:N,N-二甲基乙醯胺(DMAc)、N,N-二甲基甲醯胺(DMF)等醯胺系溶劑;γ-丁內酯(GBL)、γ-戊內酯等內酯系溶劑;二甲基碸、二甲基亞碸、環丁碸等含硫系溶劑;碳酸乙二酯、碳酸丙二酯等碳酸酯系溶劑;及該等之組合。該等之中,較佳為醯胺系溶劑或內酯系溶劑。該等溶劑可單獨使用或組合兩種以上使用。又,清漆中可含有水、醇系溶劑、酮系溶劑、非環狀酯系溶劑、醚系溶劑等。清漆之固形物成分濃度較佳為1~25質量%,更佳為5~20質量%。
於塗佈步驟中,藉由公知之塗佈方法,將清漆塗佈於基材上而形成塗膜。作為公知之塗佈方法,例如可列舉:線棒塗佈法、反向塗佈、凹版塗佈等輥塗法、模嘴塗佈法、卡馬(comma)塗佈法、模唇塗佈法、網版塗佈法、噴注式塗佈法、噴霧法、流延成形法等。
於形成步驟中,將塗膜乾燥,自基材剝離,藉此可形成原料膜。塗膜之乾燥通常可於50~350℃之溫度下進行。視需要,可於惰性環境或減壓之條件下進行塗膜之乾燥。所得原料膜供給至上述加熱步驟,可連續搬送而供給至加熱步驟,亦可暫時捲取後供給。
作為基材之例,若為金屬系,則可列舉:SUS(Steel Use Stainless,日本不鏽鋼標準)環帶,若為樹脂系,則可列舉:PET(polyethylene terephthalate,聚對苯二甲酸乙二酯)膜、PEN(polyethylene naphthalate,聚萘二甲酸乙二酯)膜、其他聚醯亞胺系樹脂或聚醯胺系樹脂膜、環烯烴系聚合物(COP)膜、丙烯酸系膜等。其中,就平滑性、耐熱性優異之觀點而言,較佳為PET膜、COP膜等,進而就與樹脂膜之密接性及成本之觀點而言,更佳為PET膜。
原料膜可於其表面積層表面保護膜而製為積層體,積層之表面保護膜於進行加熱步驟前剝離即可。表面保護膜積層於原料膜之與基材相反側之面。於積層體捲取為卷狀時,存在黏連等捲取性之問題之情形時,可進而於基材之與原料膜相反側之面追加積層表面保護膜。貼合於原料膜之表面保護膜係用以暫時保護原料膜之表面之膜,只要為可保護原料膜之表面且可剝離之膜,則並無特別限定。例如,可列舉:聚對苯二甲酸乙二酯、聚對苯二甲酸丁二酯、聚萘二甲酸乙二酯等聚酯系樹脂膜;聚乙烯、聚丙烯膜等聚烯烴系樹脂膜、丙烯酸系樹脂膜等,較佳為自由聚烯烴系樹脂膜、聚對苯二甲酸乙二酯系樹脂膜及丙烯酸系樹脂膜所組成之群中選擇。於積層體之兩面貼合表面保護膜之情形時,各面之表面保護膜相互可相同,亦可不同。
表面保護膜之厚度並無特別限定,通常為10~100 μm,較佳為10~80 μm,更佳為10~50 μm。於積層體之兩面貼合表面保護膜之情形時,各面之表面保護膜之厚度可相同,亦可不同。
將上述積層體(基材、原料膜及視需要之表面保護膜)以卷狀捲繞於捲芯者稱為積層體膜卷。積層體膜卷於連續製造中,由於空間之其他制約,暫時以膜卷之形態保管之情形較多,積層體膜卷亦為其之一。
作為構成積層體膜卷之捲芯之材料,例如可列舉:聚乙烯樹脂、聚丙烯樹脂、聚氯乙烯樹脂、聚酯樹脂、環氧樹脂、酚系樹脂、三聚氰胺樹脂、矽樹脂、聚胺基甲酸酯樹脂、聚碳酸酯樹脂、ABS(acrylonitrile-butadiene-styrene resin,丙烯腈-丁二烯-苯乙烯樹脂)樹脂等合成樹脂;鋁等金屬;纖維強化塑膠(FRP(fiber reinforced plastice):使玻璃纖維等纖維含有於塑膠中從而提高強度之複合材料)等。捲芯形成為圓筒狀或圓柱狀等形狀,其直徑例如為80~170 mm。又,膜卷之捲取後之直徑並無特別限定,通常為200~800 mm。
<樹脂膜>
本發明之製造方法中所得之樹脂膜至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者。樹脂膜之厚度較佳為10 μm以上,更佳為20 μm以上,進而較佳為30 μm以上,較佳為120 μm以下,更佳為100 μm以下,進而較佳為80 μm以下,尤佳為60 μm以下。若厚度為上述範圍,則就將樹脂膜組入顯示裝置時之內部之保護之觀點而言有利,又,就耐折性、成本、透明性等觀點而言有利。測定方法於實施例中詳細說明。
經過加熱步驟之樹脂膜之藉由TG-DTA測定而求得之自120℃至250℃之重量減少率M較佳為3%以下,更佳為2%以下,進而較佳為1.5%以下,尤佳為1%以下。又,重量減少率M之下限值並無特別限定,例如可為0.1%。
若樹脂膜之重量減少率M為上述範圍,則可獲得具有可撓性顯示裝置之前面板所要求之充分之硬度及撓曲性之兩者之膜,又,膜表面不會過於柔軟,不易產生損傷,故而存在於後續步驟中膜之操作變得容易之情形。
作為樹脂膜之霧度,就視認性之觀點而言,較佳為1%以下,更佳為0.8%以下,進而較佳為0.5%以下,尤佳為0.3%以下。樹脂膜之霧度可依據JIS K 7136:2000而測定。測定方法於實施例中詳細說明。若樹脂膜之霧度為上述範圍內,則可較佳用於可撓性顯示裝置之前面板。
樹脂膜之全光線透過率較佳為85%以上,更佳為87%以上,進而較佳為89%以上。樹脂膜之全光線透過率可依據JIS K 7361-1:1997而測定。測定方法於實施例中詳細說明。若樹脂膜之全光線透過率為上述數值範圍,則組入於圖像顯示裝置中時,可確保充分之膜外觀。
樹脂膜之黃度較佳為3.0以下,更佳為2.7以下,進而較佳為2.5以下。樹脂膜之黃度可依據JIS K 7373:2006而測定。測定方法於實施例中詳細說明。若為該範圍,則視認性優異,可較佳用作前面板等顯示器構件。
作為樹脂膜之面內相位差,以波長590 nm測定之面內相位差值之比Re(P1/P2)及比Re(P1/P3)分別較佳為0.75以上。Re(P1/P2)及比Re(P1/P3)係藉由如下方法而算出。首先,將樹脂膜於膜寬度方向均分為5份,將膜寬度方向之中心點設為P1,將P1之兩側之點分別設為P2及P3。其次,測定各個點之面內相位差值,可將P1之面內相位差值相對於P2或P3之面內相位差值之比分別作為Re(P1/P2)及Re(P1/P3)而算出。比Re(P1/P2)及比Re(P1/P3)分別較佳為0.80以上。若該等比為上述範圍,則存在樹脂膜之相位差值之不均進一步減低,膜之相位差值變得更均勻之傾向。
樹脂膜之面內相位差可藉由市售之裝置而測定。作為市售之裝置之例,可列舉:大塚電子(股)製造之相位差膜・光學材料檢查裝置(商品名“RETS100”)。
藉由本發明之製造方法而製造之樹脂膜可以樹脂膜之單層使用,亦可以積層有其他層之積層體而使用。該樹脂膜或包含其之積層體具有優異之品質,故而作為圖像顯示裝置等中之光學膜,尤其可撓性顯示器之前面板(視窗膜(window film))而有用。
作為可積層於樹脂膜之其他層,可列舉:硬塗層、紫外線吸收層、黏著層、折射率調整層、底塗層等具有各種功能之層(功能層)。樹脂膜可具備單數或複數之功能層。又,一個功能層可具有複數個功能。
又,作為功能層以外之其他層,可列舉:偏光膜、偏光板、觸控感測器、包圍具有單層或複數層之形態之框而印刷之有色之遮光圖案等顯示裝置所具備之光學構件。
硬塗層較佳為配置於膜之視認側表面。硬塗層可為單層構造,亦可為多層構造。硬塗層可藉由含有照射光或熱能而形成交聯結構之反應性材料之硬塗用組合物之硬化而形成。
作為反應性材料,可列舉光或熱硬化性樹脂。作為其例,可列舉:(甲基)丙烯酸酯單體及低聚物等丙烯酸系樹脂、環氧系樹脂、胺基甲酸酯系樹脂、苄基氯系樹脂、乙烯系樹脂,聚矽氧系樹脂或該等之混合樹脂等紫外線硬化型、電子束硬化型或熱硬化型之樹脂。就表面硬度等機械物性及工業上之觀點而言,硬塗用組合物較佳為含有丙烯酸系樹脂。
硬塗用組合物除上述樹脂外,亦可視需要含有溶劑、光聚合起始劑。又,可於不損害發明之效果之範圍內於硬塗用組合物中含有無機填料、調平劑材料、穩定劑、抗氧化劑、UV(ultraviolet,紫外線)吸收劑、界面活性劑、潤滑劑、防污劑等添加劑。
硬塗層可藉由將硬塗用組合物塗佈於藉由本發明而獲得之樹脂膜之至少一面並使之硬化而形成。硬塗層之厚度並無特別限定,例如可為5~100 μm。若硬塗層之厚度為上述範圍,則可確保充分之表面硬度,又存在耐撓曲性良好,不易發生因硬化收縮而發生捲縮之問題之傾向。
紫外線吸收層係具有紫外線吸收之功能之層,例如包含選自紫外線硬化型之透明樹脂、電子束硬化型之透明樹脂及熱硬化型之透明樹脂中之主材料、以及分散於該主材料之紫外線吸收劑。藉由設置紫外線吸收層作為功能層,可容易地抑制因光照射而導致之黃度之變化。
黏著層係具有黏著性之功能之層,具有使本發明之膜接著於其他構件之功能。作為黏著層之形成材料,可使用通常已知者。例如可使用熱硬化性樹脂組合物或光硬化性樹脂組合物。
黏著層可包含含有具有聚合性官能基之成分之樹脂組合物。於該情形時,可於使膜密接於其他構件後進而使構成黏著層之樹脂組合物聚合,藉此實現牢固之接著。本發明之膜與黏著層之接著強度可為0.1 N/cm以上或0.5 N/cm以上。
黏著層可含有熱硬化性樹脂組合物或光硬化性樹脂組合物作為材料。於該情形時,可藉由於事後供給能量而使樹脂組合物高分子化從而硬化。
黏著層亦可為包含被稱為感壓型接著劑(Pressure Sensitive Adhesive,PSA)之藉由按壓而貼合於對象物上之接著劑之層。感壓型接著劑可為黏著劑,即「於常溫下具有黏著性,以較輕之壓力接著於被接著體上之物質」(JIS K 6800),亦可為膠囊型接著劑,即「特定成分容納於保護覆膜(微膠囊)中,直至藉由適當之方法(壓力、熱等)將覆膜破壞為止均可保持穩定性之接著劑」(JIS K 6800)。
色相調整層係具有色相調整功能之層,其係可將本發明之膜調整為目標色相之層。色相調整層例如為含有樹脂及著色劑之層。作為該著色劑,例如可列舉:氧化鈦、氧化鋅、紅丹、氧鈦系煅燒顏料、群青、鋁酸鈷及碳黑等無機顏料;偶氮系化合物、喹吖啶酮系化合物、蒽醌系化合物、苝系化合物、異吲哚啉酮系化合物、酞菁系化合物、喹酞酮系化合物、士林(threne)系化合物及吡咯并吡咯二酮系化合物等有機顏料;硫酸鋇及碳酸鈣等體質顏料;以及鹼性染料、酸性染料及媒染染料等染料。
折射率調整層係具有折射率調整功能之層,其係具有與本發明之膜中之含有聚醯胺醯亞胺樹脂A之層不同之折射率,可對本發明之膜賦予特定折射率之層。折射率調整層例如可為含有適宜選擇之樹脂、及視情況進而含有顏料之樹脂層,亦可為金屬之薄膜。
作為調整折射率之顏料,例如可列舉:氧化矽、氧化鋁、氧化銻、氧化錫、氧化鈦、氧化鋯及氧化鉭。顏料之平均一次粒徑可為0.1 μm以下。藉由將顏料之平均一次粒徑設為0.1 μm以下,可防止透過折射率調整層之光之漫反射,防止透明度下降。
作為折射率調整層中所使用之金屬,例如可列舉:氧化鈦、氧化鉭、氧化鋯、氧化鋅、氧化錫、氧化矽、氧化銦、氮氧化鈦、氮化鈦、氮氧化矽、氮化矽等金屬氧化物或金屬氮化物。
積層於樹脂膜上之光學構件可經由黏著層或接著層而積層於樹脂膜上,亦可並不經由黏著層或接著層而積層。
[實施例]
以下,藉由實施例進而詳細說明本發明。例中之「%」及「份」若無特別記載,則表示質量%及質量份。於實施例中,各項目之測定方法及評價方法係藉由以下方法進行。
(樹脂膜之厚度)
使用測微計(Mitutoyo(股)製造之「ID-C112XBS」),測定10點以上之樹脂膜之厚度,算出其平均值。
(熱重量-示差熱(TG-DTA)測定)
TG-DTA之測定裝置使用Hitachi High-Tech Science(股)製造之TG/DTA6300。自所製作之透明樹脂膜(聚醯亞胺膜)獲取約20 mg之試樣。一面於如下條件下試樣進行加熱,一面測定試樣之重量變化,即,以10℃/分鐘之升溫速度自室溫升溫至120℃,於120℃下保持5分鐘後,以10℃/分鐘之升溫速度升溫至400℃。圖1係表示下述實施例1中所製作之聚醯亞胺膜之TG-DTA測定結果。
根據TG-DTA測定結果,藉由下述式算出自120℃至250℃之重量減少率M(%)。
M(%)=100-(W1/W0)×100
此處,W0表示於120℃下保持5分鐘後之試樣之重量,W1表示250℃下之試樣之重量。
(樹脂膜之面內相位差值之測定)
樹脂膜之面內相位差係使用大塚電子(股)製造之相位差膜・光學材料檢查裝置(商品名“RETS100”),測定波長590 nm之面內相位差值Re。測定係對以樹脂膜之寬度方向中央為中心取寬700 mm之範圍,以其20 mm間隔進行分割之合計36點進行,作為該等值之平均值而算出。
表2中之面內相位差值之比係藉由如下方式算出之值。首先,將樹脂膜之膜寬之中心點設為P1,將自P1朝向膜兩端離開120 mm之點分別設為P2及P3。其次,測定各個點之面內相位差值,將P1之面內相位差值相對於P2或P3之面內相位差值之比分別作為Re(P1/P2)及Re(P1/P3)而求出。
(清漆之黏度)
清漆之黏度(cps)係依據JIS K 8803:2011,使用E型黏度計,於25℃下測定。又,清漆之樹脂濃度係表示於清漆中含有之樹脂之濃度(質量%),係根據基於清漆之總質量之於清漆中含有之樹脂之質量而算出。
(二氧化矽粒子之粒徑)
二氧化矽粒子之粒徑係依據JIS Z 8830,根據藉由BET(Brunauer-Emmett-Teller,布厄特)吸附法所得之比表面積測定值而算出。使用比表面積測定裝置(Yuasa-ionics(股)製造之「Monosorb(註冊商標)MS-16」),測定使矽溶膠於300℃下乾燥而成之粉末之比表面積。
(重量平均分子量)
凝膠滲透層析(GPC)測定
(1)前處理方法
使試樣溶解於γ-丁內酯(GBL)製為20質量%溶液後,藉由DMF溶離液稀釋至100倍,將以0.45 μm膜濾器進行過濾者作為測定溶液。
(2)測定條件
管柱:TSKgel SuperAWM-H×2+SuperAW2500×1(6.0 mm I.D.×150 mm×3根)
溶離液:DMF(添加10 mmol/L之溴化鋰)
流量:0.6 mL/分鐘
檢測器:RI檢測器
管柱溫度:40℃
注入量:20 μL
分子量標準:標準聚苯乙烯
(樹脂膜之搬送性之評價方法)
自拉幅式乾燥爐入口至乾燥爐出口,以目視確認有無膜之晃動(縱方向之振動)。
(樹脂膜外觀不良)
確認所得膜之夾具部未產生破裂、及對膜照射LED(light-emitting diode,發光二極體)光確認於乾燥爐前後是否有損傷之增加。
<製造例1:聚醯亞胺系樹脂1之製造>
準備於可分離式燒瓶中安裝有矽膠管、攪拌裝置及溫度計之燒瓶、及油浴。於該燒瓶內投入4,4'-(六氟亞異丙基)二鄰苯二甲酸酐(6FDA)75.6 g、及2,2'-雙(三氟甲基)-4,4'-二胺基聯苯(TFMB)54.5 g。一面以400 rpm對其進行攪拌一面添加N,N-二甲基乙醯胺(DMAc)530 g,持續攪拌直至燒瓶之內容物成為均勻之溶液。繼而,一面使用油浴以容器內溫度成為20~30℃之範圍之方式進行調整一面進而持續攪拌20小時,使之反應而生成聚醯胺酸。30分鐘後,將攪拌速度變更為100 rpm。攪拌20小時後,將反應系溫度返回至室溫,添加DMAc 650 g,以聚合物濃度成為10質量%之方式進行調整。進而,添加吡啶32.3 g、乙酸酐41.7 g,於室溫下攪拌10.5小時進行醯亞胺化。自反應容器取出聚醯亞胺清漆。將所得之聚醯亞胺清漆滴加至甲醇中進行再沈澱,將所得之粉體加熱乾燥而去除溶劑,獲得作為固形物成分之聚醯亞胺系樹脂1。對所得之聚醯亞胺系樹脂1進行GPC測定,結果重量平均分子量為365,000。又,聚醯亞胺之醯亞胺化率為99.0%。
<製造例2:聚醯亞胺系樹脂2之製造)
於氮氣環境下,於具備攪拌葉之1 L可分離式燒瓶中添加TFMB 45 g(140.52 mmol)及DMAc 768.55 g,一面於室溫下攪拌一面使TFMB溶解於DMAc。其次,於燒瓶中添加6FDA 18.92 g(42.58 mmol),於室溫下攪拌3小時。其後,於燒瓶中添加4,4'-氧基雙(苯甲醯氯)(OBBC)4.19 g(14.19 mmol),繼而添加對苯二甲醯氯(TPC)17.29 g(85.16 mmol),於室溫下攪拌1小時。繼而,於燒瓶中添加4-甲基吡啶4.63 g(49.68 mmol)與乙酸酐13.04 g(127.75 mmol),於室溫下攪拌30分鐘後,使用油浴升溫至70℃,進而攪拌3.5小時,獲得反應液。
將所得反應液冷卻至室溫,以線狀投入至大量之甲醇中,取出所析出之沈澱物,以甲醇浸漬6小時後,以甲醇清洗。其次,於100℃下進行沈澱物之減壓乾燥,獲得聚醯亞胺系樹脂2。聚醯亞胺系樹脂2之重量平均分子量為455,000。聚醯亞胺之醯亞胺化率為98.9%。
<製造例3:分散液1之製造>
將甲醇分散有機化處理二氧化矽(以BET法測定之粒徑:27 nm)置換為γ-丁內酯(GBL),獲得GBL分散有機化處理二氧化矽(固形物成分30.3質量%)。將該分散液作為分散液1。
<製造例4:清漆(1)之製造>
清漆(1)係以表1所示之組成,將聚合物溶解於溶劑中,獲得根據添加量算出之固形物成分為15.5質量%,25℃下之黏度為36,500 cps之清漆(1)。
<製造例5:清漆(2)之製造>
清漆(2)係於室溫下於GBL溶劑中以聚合物與填料之組成比成為60:40之方式混合,於其中以相對於聚合物與二氧化矽之總質量成為5.7 phr或35 ppm之方式添加Sumisorb 340(UVA)、Sumiplast Violet B(BA),攪拌直至均勻。獲得自添加量算出之固形物成分為10.3質量%,25℃下之黏度為38,500 cps之清漆(2)。
[表1] 
<製造例6:原料膜1之製膜>
將清漆(1)於PET(聚對苯二甲酸乙二酯)膜(東洋紡(股)製造之「COSMOSHINE(註冊商標)A4100」,厚度188 μm,厚度分佈±2 μm)上藉由流延成形而形成塗膜。此時,線速度為0.4 m/分鐘,於如下條件下乾燥塗膜:於70℃下加熱8分鐘後,於100℃下加熱10分鐘,繼而於90℃下加熱8分鐘,最後於80℃下加熱8分鐘。其後,自PET膜剝離塗膜,獲得寬度700 mm、厚度86 μm之原料膜1。原料膜1之重量減少率M為9.6%。
<製造例7:原料膜2之製膜>
將清漆(2)於PET(聚對苯二甲酸乙二酯)膜(東洋紡(股)製造之「COSMOSHINE(註冊商標)A4100」,厚度188 μm,厚度分佈±2 μm)上藉由流延成形而形成塗膜。此時,線速度為0.3 m/分鐘。又,於如下條件下乾燥塗膜:於80℃下加熱10分鐘後,於100℃下加熱10分鐘,繼而於90℃下加熱10分鐘,最後於80℃下加熱10分鐘。其後,自PET膜剝離塗膜,獲得寬度700 mm、厚度58 μm之原料膜2。原料膜2之重量減少率M為9.2%。
<實施例1:樹脂膜1之製作>
對製造例6中獲得之原料膜1利用使用夾具作為固持裝置之圖1所示之拉幅式乾燥爐(內部分為共6室之構成),加熱原料膜,於爐內之共6室中,分別使用噴射噴嘴。拉幅式乾燥爐之開口部面積為0.0055 m2
。去除溶劑獲得厚度79 μm之樹脂膜1。此時,乾燥爐內之條件為:乾燥爐內之溫度為200℃,夾具之固持寬度為25 mm,膜之搬送速度為1.1 m/分鐘,乾燥爐入口之膜寬(夾具間距離)與乾燥爐出口之膜寬之比為1.0,及表2所示之風速。其後,撕去(切斷)夾具部,於該膜貼合PET系表面保護膜,捲取至ABS製造之6英吋之捲芯,獲得卷膜。此時之搬送性、拉幅式乾燥爐後之膜之外觀及所得樹脂膜1之重量減少率M示於表2。
<實施例2:樹脂膜2之製作>
除使用製造例7中獲得之原料膜2,分別將膜搬送速度變更為0.9 m/分鐘,將乾燥爐入口之膜寬(夾具間距離)與乾燥爐出口之膜寬之比變更為0.98倍,及將風速變更為表2所示之條件以外,於與實施例1相同之條件下進行加工,獲得厚度49.5 μm之樹脂膜2。此時之搬送性、拉幅式乾燥爐後之膜之外觀及所得樹脂膜2之重量減少率M示於表2。
<比較例1:樹脂膜3之製作>
除將風速變更為表2所示之條件以外,以與實施例2相同之方法,自原料膜2獲得厚度49 μm之樹脂膜3。此時之搬送性、拉幅式乾燥爐後之膜之外觀及所得樹脂膜3之重量減少率M示於表2。因存在損傷或破裂,故而未進行面內相位差之測定。
[表2] 
10、12、14‧‧‧區域
18‧‧‧固持裝置
20‧‧‧原料膜
30‧‧‧上側噴嘴(噴嘴)
32‧‧‧下側噴嘴(噴嘴)
34‧‧‧噴射噴嘴
36、38‧‧‧穿孔噴嘴
36a、38a‧‧‧面
40‧‧‧狹縫
42、44‧‧‧開口
100‧‧‧烘箱
100a‧‧‧上表面
100b‧‧‧下表面
A‧‧‧膜之搬送方向
圖1係模式地表示本發明之樹脂膜之製造方法之較佳實施形態之步驟剖視圖。
圖2係模式地表示本發明之樹脂膜之製造方法之加熱步驟之較佳實施形態之步驟剖視圖。
圖3係表示本發明之樹脂膜之製造方法中較佳使用之噴射噴嘴之形狀之一例之模式剖視圖。
圖4係表示本發明之樹脂膜之製造方法中較佳使用之穿孔噴嘴之形狀之一例之模式剖視圖。
圖5係表示本發明之樹脂膜之製造方法中較佳使用之穿孔噴嘴之形狀之其他例之模式剖視圖。
10、12、14‧‧‧區域
20‧‧‧原料膜
30‧‧‧上側噴嘴(噴嘴)
32‧‧‧下側噴嘴(噴嘴)
100‧‧‧烘箱
100a‧‧‧上表面
100b‧‧‧下表面
A‧‧‧膜之搬送方向
Claims (11)
- 一種樹脂膜之製造方法,其係至少含有聚醯亞胺系樹脂或聚醯胺系樹脂之任一者之樹脂膜之製造方法,並且 具有以熱風加熱原料膜之加熱步驟, 上述加熱步驟係藉由來自相互對向之一對噴嘴之吹出口之熱風而進行, 關於上述噴嘴,吹出口之吹出風速為2 m/秒以上25 m/秒以下,來自每一個噴嘴之吹出口之吹出風量於沿上述原料膜之寬度方向之噴嘴之每1 m長度中為0.1~3 m3 /秒。
- 如請求項1之樹脂膜之製造方法,其中上述吹出口之吹出風速為2 m/秒以上且未達25 m/秒。
- 如請求項1之樹脂膜之製造方法,其中上述噴嘴係具有於上述原料膜之寬度方向上延伸之狹縫狀之吹出口之噴嘴、或具有於上述原料膜之搬送方向及上述原料膜之寬度方向上分別配置有複數個開口之吹出口之噴嘴。
- 如請求項2之樹脂膜之製造方法,其中上述噴嘴係具有於上述原料膜之寬度方向上延伸之狹縫狀之吹出口之噴嘴、或具有於上述原料膜之搬送方向及上述原料膜之寬度方向上分別配置有複數個開口之吹出口之噴嘴。
- 如請求項1至4中任一項之樹脂膜之製造方法,其中於上述加熱步驟中,對上述原料膜吹送熱風之各個上述噴嘴之吹出口之熱風之於上述膜之寬度方向上之最大吹出風速與最小吹出風速之差為4 m/秒以下。
- 如請求項1至4中任一項之樹脂膜之製造方法,其中上述加熱步驟係於無塵室內進行。
- 如請求項5之樹脂膜之製造方法,其中上述加熱步驟係於無塵室內進行。
- 如請求項1至4中任一項之樹脂膜之製造方法,其中上述加熱步驟係於清潔度等級為1000以下之潔淨度之烘箱中進行。
- 如請求項5之樹脂膜之製造方法,其中上述加熱步驟係於清潔度等級為1000以下之潔淨度之烘箱中進行。
- 如請求項6之樹脂膜之製造方法,其中上述加熱步驟係於清潔度等級為1000以下之潔淨度之烘箱中進行。
- 如請求項7之樹脂膜之製造方法,其中上述加熱步驟係於清潔度等級為1000以下之潔淨度之烘箱中進行。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018118554 | 2018-06-22 | ||
| JP2018-118554 | 2018-06-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| TW202000435A true TW202000435A (zh) | 2020-01-01 |
Family
ID=69098269
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW108121549A TW202000435A (zh) | 2018-06-22 | 2019-06-20 | 樹脂膜之製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP2020001383A (zh) |
| KR (1) | KR20200000360A (zh) |
| TW (1) | TW202000435A (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN217094252U (zh) * | 2022-01-25 | 2022-08-02 | 宁德时代新能源科技股份有限公司 | 烘干装置和极片制造设备 |
| CN119800656B (zh) * | 2025-02-14 | 2025-12-23 | Tcl家用电器(合肥)有限公司 | 洗衣设备控制方法、装置、洗衣机及计算机可读存储介质 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04292934A (ja) * | 1991-03-22 | 1992-10-16 | Toray Ind Inc | 熱可塑性樹脂フイルムの製造方法 |
| JPH0820655A (ja) * | 1994-07-06 | 1996-01-23 | Nippon Paint Co Ltd | 表面溶融処理装置 |
| JP2002240048A (ja) * | 2001-02-16 | 2002-08-28 | Asuriito Fa Kk | 樹脂硬化方法および樹脂硬化装置 |
| US20100276826A1 (en) * | 2007-09-21 | 2010-11-04 | Hiroaki Takahata | Process for producing retardation film |
| JP5228834B2 (ja) * | 2008-03-28 | 2013-07-03 | 東レ株式会社 | エア噴出ノズルおよびそれを用いたテンターオーブン |
| US10829601B2 (en) * | 2015-06-18 | 2020-11-10 | Kaneka Corporation | Process for producing polymer film |
-
2019
- 2019-05-28 JP JP2019099152A patent/JP2020001383A/ja active Pending
- 2019-06-20 KR KR1020190073431A patent/KR20200000360A/ko not_active Withdrawn
- 2019-06-20 TW TW108121549A patent/TW202000435A/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| JP2020001383A (ja) | 2020-01-09 |
| KR20200000360A (ko) | 2020-01-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI813705B (zh) | 樹脂膜及其製造方法 | |
| JP7120819B2 (ja) | ポリイミドフィルムとフレキシブルガラスとの積層体 | |
| CN111267433A (zh) | 层叠体及其制造方法 | |
| JPWO2017010178A1 (ja) | 偏光板、その製造方法、液晶表示装置及び有機エレクトロルミネッセンス表示装置 | |
| TW202010641A (zh) | 積層體及其製造方法 | |
| CN112817069A (zh) | 光学膜及柔性显示装置 | |
| TW202000435A (zh) | 樹脂膜之製造方法 | |
| CN110154474A (zh) | 膜卷 | |
| TW202200672A (zh) | 光學膜及可撓式顯示裝置 | |
| CN110039867B (zh) | 层叠体及其制造方法 | |
| KR102103487B1 (ko) | 투명 수지 필름의 제조 방법 | |
| KR102096323B1 (ko) | 수지 필름 롤의 제조 방법 | |
| TW202146532A (zh) | 光學膜及可撓式顯示裝置 | |
| CN110628065B (zh) | 透明树脂膜的制造方法 | |
| TW202010776A (zh) | 透明樹脂膜之製造方法 | |
| TW202124119A (zh) | 膜之缺陷之檢出系統 | |
| JP2020196249A (ja) | 透明樹脂フィルムの製造方法 | |
| JP2023132813A (ja) | ポリイミド系フィルムの製造方法 | |
| TW202126466A (zh) | 經乾燥之膜捲筒之製造方法 | |
| TW201930400A (zh) | 樹脂膜之製造方法 |