WO1999055724A1 - NOUVEAUX DERIVES DE L'ACIDE OCTAHYDRO-6,10-DIOXO-6H-PYRIDAZINO[1,2-a] [1,2] DIAZEPINE-1-CARBOXYLIQUE, LEUR PROCEDE DE PREPARATION ET LEUR APPLICATION A LA PREPARATION DE COMPOSES THERAPEUTIQUEMENT ACTIFS - Google Patents
NOUVEAUX DERIVES DE L'ACIDE OCTAHYDRO-6,10-DIOXO-6H-PYRIDAZINO[1,2-a] [1,2] DIAZEPINE-1-CARBOXYLIQUE, LEUR PROCEDE DE PREPARATION ET LEUR APPLICATION A LA PREPARATION DE COMPOSES THERAPEUTIQUEMENT ACTIFS Download PDFInfo
- Publication number
- WO1999055724A1 WO1999055724A1 PCT/FR1999/000981 FR9900981W WO9955724A1 WO 1999055724 A1 WO1999055724 A1 WO 1999055724A1 FR 9900981 W FR9900981 W FR 9900981W WO 9955724 A1 WO9955724 A1 WO 9955724A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- aryl
- compounds
- dioxo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(CCC1)N(C(*(CC2)N(*)*)=O)N1C2=C Chemical compound *C(CCC1)N(C(*(CC2)N(*)*)=O)N1C2=C 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
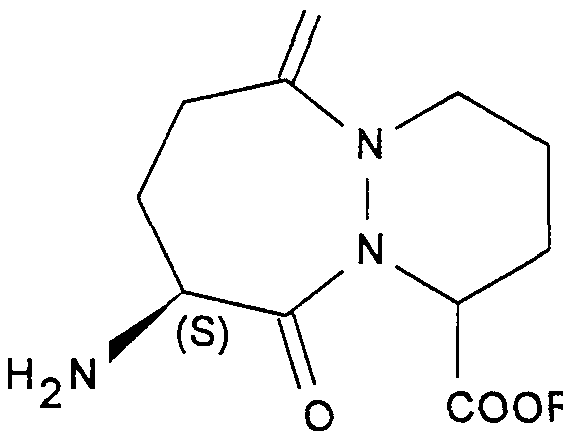
- New derivatives of octahydro-6,10-dioxo-6H-pyridazino [1, 2-a] [1,2] diazepine-1-carboxylic acid their preparation process and their application to the preparation of therapeutically active compounds.
- the present invention relates to new derivatives of octahydro- ⁇ , 10-dioxo-6H-pyridazino [1, 2-a] [1,2] diazepine-1-carboxylic acid, their preparation process and their application to the preparation of therapeutically active compounds.
- R represents a hydrogen atom, an alkyl or aralkyl radical containing up to 18 carbon atoms, the amino function possibly being free or protected.
- R represents for example a radical H, methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl or tertbutyl, or a benzyl or naphthyl radical.
- the protection can be done according to the conventional methods of amino protection.
- a subject of the invention is in particular the compounds corresponding to formula (IA):
- Ra, Rb, Rc and Rd representing an alkyl or aryl radical containing up to 18 carbon atoms, or a mono or polycyclic radical containing one or more heteroatoms
- X representing a hydrogen atom, an alkyl radical containing up to 8 carbon atoms or an aryl radical containing up to 14 carbon atoms
- R 2 represents a hydrogen atom, or else R ⁇ _ and R 2 together form a mono or polycyclic radical containing one or more heteroatoms.
- cyclic compounds for example radicals, can be used to protect the amines:
- a more particular subject of the invention is the compounds of formula (IA) in which R-_ and R 2 together form a polycyclic radical containing one or more heteroatoms and in particular the compounds corresponding to the formula (IA-L):
- a subject of the invention is in particular the compounds of formula (I) in which R represents a methyl radical, of SR configuration or in the form of a SR + SS mixture,.
- the subject of the invention is also a method characterized in that a compound of formula (II) is subjected:
- Hal- ] _ represents a halogen atom and Ar represents an aryl or aralkyl radical containing up to 18 carbon atoms, R- j _ and R retaining the same definition as above, to obtain the compound of formula (VIII ):
- Hal- L represent a chlorine atom
- - aie represents an alkyl radical containing up to 4 carbon atoms
- - Aryl represents a phenyl or naphthyl radical
- - aralkyl represents a benzyl radical
- the basic agent which is reacted on the compound of formula (IV) is sodium or potassium hydroxide
- the alkylating agent which is reacted on the compound of formula (V) is an alcohol, for example methanol, the condensation between the compounds (VI) and (VII) is carried out in the presence of a base such as pyridine, TEA, diisopropylamine,
- the hydrogenation agent is for example hydrogen in the presence of palladium on carbon, palladium dihydroxide in the presence of talc, rhodium in the presence of alumina, ruthenium on carbon, or in the presence of nickel of Raney,
- the cyclization is carried out in the presence of SOCl 2 or PC1 5 or of activated esters or in the presence of dehydrating agents such as APTS, the liberation of the amine can be carried out by means of hydrazine.
- the products (IV), (VII), (VIII) and (IX) used during the process are new products and are in themselves an object of the present invention.
- a more particular subject of the invention is the products whose preparation is given below in the experimental part and in particular the racemic mixture.
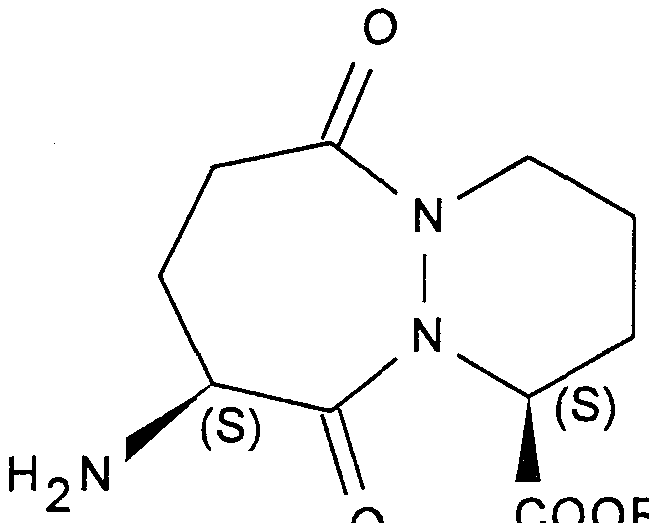
- the invention also relates to the application characterized in that a compound of formula (I) is subjected in the form of a mixture SS, SR, or in SR form, to the action of an asymmetric carbon deracemization agent carried by the 6-ring, to obtain the compound of formula (Iopt):
- a more specific subject of the invention is the application of the compounds of formula (IA) defined above, to the preparation of the compounds of formula (IAopt):
- R represents a methyl radical, and that in which the amino function is protected in the form of phthalimido.
- a more particular subject of the invention is the application characterized in that the deracemization agent is a base, more especially a strong base, for example an alkali or alkaline earth alcoholate such as sodium or potassium methylate, terbutylate sodium or 8 potassium, or a lithiated amine such as LDA.
- a base more especially a strong base, for example an alkali or alkaline earth alcoholate such as sodium or potassium methylate, terbutylate sodium or 8 potassium, or a lithiated amine such as LDA.
- the invention particularly relates to the application described below in the experimental part for preparing the: - (lS-cis) -9- (1,3-dihydro-1,3, dioxo-2H-isoindol-2 -yl) -
- the products of formula (I) can be generally used for the synthesis of medicaments as indicated in the above patent.
- EXAMPLE 1 (IS-cis) -9- (1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl) octahydro-6,10-dioxo-6H-pyridazino [1,2-a] [1, 2] methyl diazepine-1- carboxylate and
- the suspension obtained is heated to 90 ° C. Stirring is continued for 48 hours. Cooled to 20 ° C, poured into a solution containing 50 ml of 2N hydrochloric acid and 150 ml of a mixture of water and ice. Extracted with ethyl acetate, washed with water, dried. It is filtered, rinsed with ethyl acetate and dried. The product obtained is chromatographed on silica (elution heptane 40, AcOEt 20) and 10.71 g of sought product is obtained.
- a solution containing 23.25 g of the product of the preceding stage and 80 ml of ethanol is introduced into 338 ml of a 10 solution of sodium hydroxide in ethanol at 40 g per liter. Stirring is continued for 5 hours 30 minutes and 57 ml of 2N sodium hydroxide are added. The reaction mixture is kept under stirring for 30 hours. 141 ml of a 2N hydrochloric acid solution are added. 260 ml of reaction mixture are distilled under 80-90 millibars. Extracted with dichloromethane, added 20 ml of ethanol, washed with a mixture of water and normal sodium hydroxide solution. The aqueous phases are extracted with dichloromethane.
- 1, 3 (2H) -pyridazinedicarboxylate of 3-methyl 1- (phenylmethyl) 220 ml of methanol and paratoluene sulfonic acid dehydrate (prepared from APTS monohydrate and 12 ml of dichloromethane) are added to 11.05 g of product prepared in the previous stage.
- the suspension obtained is kept under stirring for 15 hours.
- the mixture is heated to 65 ° C. and the stirring is continued for 6 hours 30 minutes.
- the mixture is cooled to 5 ° C., 5.5 ml of a 10 ⁇ sodium bicarbonate solution is added. Concentration is carried out under reduced pressure and the residue is taken up in a mixture of 100 ml of dichloromethane and 100 ml of water.
- a solution containing 11.01 g of the product prepared in the preceding stage and 50 ml of dichloromethane in a solution containing 19.88 g of (S) -gamma- (chlorocarbonyl) -1.3 is introduced over 1 hour at 4 ° C. -dihydro-1,3-dioxo-2H- phenylmethyl isoindole-2-butanoate and 100 ml of dichloromethane.
- the mixture is stirred for half an hour at 4 ° C. and 4.15 ml of pyridine in 25 ml of dichloromethane are introduced over the course of 1.5 hours. Stirring is continued for 15 hours, leaving to return slowly to room temperature.
- the hydrogen is passed through for 3 hours, again adding 3.03 g of catalyst. The hydrogenation is continued for 22 hours. It is filtered, washed with THF and evaporated. 25 ml of isopropanol are added, concentrated, the THF is removed, 15 ml of isopropanol are added. A suspension is obtained to which 100 ml of isopropyl ether are added. The mixture is stirred under nitrogen for 2 hours, drained, washed with isopropyl ether containing 5% isopropanol. It is drained, dried and 9.5 g of sought product is obtained.
- a solution containing 1 ml of thionyl chloride and 40 ml of methylene chloride is added at 5 ° C. in a mixture containing 4.038 g of product from the preceding stage, 40 ml of dichloromethane and 0.4 ml of dimethylformamide. The mixture is stirred for 3.5 hours. The temperature is allowed to rise to 20 ° C, stirred for an hour and a half. We concentrate. A solution containing 0.15 ml of thionyl chloride and 5 ml of methylene chloride is added. The reaction mixture is kept stirring at around 20 ° C for 16 hours. Cooled to 5 ° C and introduced 27 ml of a saturated aqueous solution of sodium hydrogen carbonate.
- a solution containing 0.029 g of potassium terbutylate and 0.3 ml of DMF is introduced at a temperature of -45 ° / -48 ° C in 1 hour 30 minutes, in a mixture containing 0.194 g of the product of Example 1, 1.5 ml of dimethylformamide and 0.75 ml of terbutanol.
- the mixture is kept stirring for 1 hour and, after cooling to -50 ° C., 0.4 g of powdered ammonium chloride is introduced.
- ⁇ D -75.3 ° (1% in methanol).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description












Claims











Priority Applications (25)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69934790T DE69934790T2 (de) | 1998-04-27 | 1999-04-26 | Octahydro-6, 10-dioxo-6h-pyridazino/1,2-a/ /1,2/ diazepin-1-carbonsäure-derivate, verfahren zu deren herstellung und deren verwendung zur herstellung pharmazeutischer wirkstoffe |
| HR20000733A HRP20000733B1 (hr) | 1998-04-27 | 1999-04-26 | NOVI DERIVATI OKTAHIDRO-6,10-DIOKSO-6H-PIRIDAZINO [1,2-a] [1,2]DIAZEPIN-1-KARBOKSILNE KISELINE, POSTUPAK NJIHOVOG DOBIVANJA I UPOTREBA U DOBIVANJU TERAPEUTSKI AKTIVNIH SPOJEVA |
| EEP200000619A EE04682B1 (et) | 1998-04-27 | 1999-04-26 | Oktahdro-6,10-diokso-6H-pridasino[1,2-a][1,2]diasepiin-1-karbokslhappe derivaadid, nende ratseemiline segu, valmistamismeetod ja kasutamine |
| JP2000545882A JP4332298B2 (ja) | 1998-04-27 | 1999-04-26 | オクタヒドロ−6,10−ジオキソ−6H−ピリダジノ[1,2−a][1,2]ジアゼピン−1−カルボン酸の新規な誘導体、それらの製造方法及びそれらの治療学的に活性な化合物の製造への使用 |
| EP99915834A EP1073673B1 (fr) | 1998-04-27 | 1999-04-26 | Derives de l'acide octahydro-6, 10-dioxo-6h-pyridazino/1,2-a/ /1,2/ diazepine-1-carboxylique, leur procede de preparation et leur application a la preparation de composes therapeutiquement actifs |
| CA2330492A CA2330492C (fr) | 1998-04-27 | 1999-04-26 | Nouveaux derives de l'acide octahydro-6,10-dioxo-6h-pyridazino[1,2-a] [1,2] diazepine-1-carboxylique, leur procede de preparation et leur application a la preparation de composes therapeutiquement actifs |
| NZ507618A NZ507618A (en) | 1998-04-27 | 1999-04-26 | New derivatives of octohydro-6,10-dioxo-6H-pyridazino[1,2-a][1,2]diazepine-1-carboxylic acid, preparation process and their use in the preparation of therapeutically active compounds |
| SK1600-2000A SK288050B6 (sk) | 1998-04-27 | 1999-04-26 | Octahydro-6,10-dioxo-6H-pyridazino[1,2-a] [1,2]diazepin-1- carboxylic acid derivatives, preparation method, intermediates and use for preparing therapeutically active compounds |
| SI9930952T SI1073673T1 (sl) | 1998-04-27 | 1999-04-26 | Derivati oktahidro-6,10-diokso-6H-piridazino(1,2-a)(1,2)diazepin-1-karboksilne kisline, postopek za njihovo pripravo in njihova uporaba za pripravo terapevtsko aktivnih spojin |
| DK99915834T DK1073673T3 (da) | 1998-04-27 | 1999-04-26 | Hidtil ukendte octahydro-6, 10-dioxo-6H-pyridazino [1,2-a] [1,2] - diazepin-1-carboxylsyre-derivater, fremgangsmåde til deres fremstilling og deres anvendelse til fremstilling af terapeutisk aktive forbindelser |
| MEP-2008-408A ME00300B (me) | 1998-04-27 | 1999-04-26 | NOVI DERIVATI OKT AHIDR0-6, 10-DIOKS0-6N-PIRIDAZINO/l,2-a/ /l,2/DIAZEPIN-1-KARBOKSILNE KISELINE, NJIHOV POSTUPAK DOBIJANJA I NJIHOVA PRIMENA ZA DOBIJANJE TERAPEUTSKI AKTIVNIH JEDINJENJA |
| US09/674,327 US6548664B1 (en) | 1998-04-27 | 1999-04-26 | Octahydro-6, 10-dioxo-6h-pyridazino/1,2-a/ /1,2/diazepin-1-carboxylic acid, derivatives, preparation method and use for preparing therapeutically active compounds |
| UA2000116680A UA71913C2 (uk) | 1998-04-27 | 1999-04-26 | ПОХІДНІ ОКТАГІДРО-6,10-ДІОКСО-6Н-ПІРИДАЗИНО[1,2-а][1,2]ДІАЗЕПІН-1- КАРБОНОВОЇ КИСЛОТИ, СПОСІБ ЇХ ОДЕРЖАННЯ ТА ОДЕРЖАННЯ ТЕРАПЕВТИЧНО АКТИВНИХ СПОЛУК |
| BR9910020-7A BR9910020A (pt) | 1998-04-27 | 1999-04-26 | Derivados do ácido octa-hidros-6,10-dioxo-6h-piridazino(1,2-a)(1,2)diazepi na-1-carboxìlico, seu processo de preparação e sua aplicação à preparação de compostos terapeuticamente ativos |
| AU34274/99A AU755286B2 (en) | 1998-04-27 | 1999-04-26 | Novel octahydro-6,10-dioxo-6h-pyridazino/1,2-a/ /1,2/diazepin-1-carboxylic acid derivatives, preparation method and use for preparing therapeutically active compounds |
| HU0102447A HU230060B1 (hu) | 1998-04-27 | 1999-04-26 | Új oktahidro-6,10-dioxo-6H-piridazino[1,2-a][1,2]diazepin-1-karbonsav-származékok, eljárás a vegyületek előállítására és alkalmazásuk gyógyászati készítmények előállítására |
| EA200001107A EA003280B1 (ru) | 1998-04-27 | 1999-04-26 | ПРОИЗВОДНЫЕ ОКТАГИДРО-6,10-ДИОКСО-6H-ПИРИДАЗИНО[1,2-a][1,2]ДИАЗЕПИН-1-КАРБОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ТЕРАПЕВТИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ |
| IL13910299A IL139102A0 (en) | 1998-04-27 | 1999-04-26 | Novel octahydro-6, 10-dioxo-6h-pyridazino/1,2-a//diazepin-1-carboxylic acid derivatives, preparation method and use for preparing therapeutically active compounds |
| MEP-408/08A MEP40808A (en) | 1998-04-27 | 1999-04-26 | NOVEL OCTAHYDRO-6,10-DIOXO-6H-PYRIDAZINO/1,2-a/ /1,2/DIAZEPIN-1-CARBOXYLIC ACID DERIVATIVES, PREPARATION METHOD AND USE FOR PREPARING THERAPEUTICALLY ACTIVE COMPOUNDS |
| APAP/P/2000/001973A AP1518A (en) | 1998-04-27 | 1999-04-26 | Novel octahydro-6, 10-dioxo-6h-pyridazino/1,2-a/ /1,2/diazepin-1-carboxylic acid derivatives, preparation method and use for preparing therapeutically active compounds. |
| HK02100645.7A HK1039131B (en) | 1998-04-27 | 1999-04-26 | Derivatives of octahydro-6,10-dioxo-6h-pyridazino(1,2-a)(1,2)diazepine-1-carboxylic acid, the preparation process and the use thereof |
| IL139102A IL139102A (en) | 1998-04-27 | 2000-10-18 | Preparation of Acid-9-Amino Octahydro-6, 10-Dioxo-6H-Pyridazino / 2,1 - // a / 2,1 Diazepine-1-Carboxylic Acid Preparation |
| NO20005391A NO328864B1 (no) | 1998-04-27 | 2000-10-26 | Oktahydro-6,10-diokso-6H-pyridazino[1,2-a][1,2]diazepin-1-karboksylsyrederivater, fremgangsmate for fremstilling derav og anvendelser derav |
| US10/102,591 US6570012B2 (en) | 1998-04-27 | 2002-03-20 | Derivatives of octahydro-6,10-dioxo-6H-pyridazino [1,2-a] [1,2] diazepine-1-carboxylic acid, their preparation process and their use in the preparation of therapeutically active compounds |
| IL190120A IL190120A (en) | 1998-04-27 | 2008-03-12 | Preparation of a history of 9-amino-octahydro-10,6-dioxo-6H-pyridazino / 2a // 2,1 / diazepine-1-carboxylic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9805243A FR2777889B1 (fr) | 1998-04-27 | 1998-04-27 | Nouveaux derives de l'acide octahydro-6,10-dioxo-6h- pyridazino[1,2-a][1,2]diazepine-1-carboxylique, leur procede de preparation et leur application a la preparation de composes therapeutiquement actifs |
| FR98/05243 | 1998-04-27 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/674,327 A-371-Of-International US6548664B1 (en) | 1998-04-27 | 1999-04-26 | Octahydro-6, 10-dioxo-6h-pyridazino/1,2-a/ /1,2/diazepin-1-carboxylic acid, derivatives, preparation method and use for preparing therapeutically active compounds |
| US10/102,591 Division US6570012B2 (en) | 1998-04-27 | 2002-03-20 | Derivatives of octahydro-6,10-dioxo-6H-pyridazino [1,2-a] [1,2] diazepine-1-carboxylic acid, their preparation process and their use in the preparation of therapeutically active compounds |
| US10/313,422 Division US20030130269A1 (en) | 1998-04-27 | 2002-12-06 | Derivatives of octahydro-6, 10-dioxo-6H-pyridazino [1,2-a] [1,2] diazepine-1-carboxylic acid, their preparation process and their use in the preparation of therapeutically active compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999055724A1 true WO1999055724A1 (fr) | 1999-11-04 |
Family
ID=9525705
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR1999/000981 Ceased WO1999055724A1 (fr) | 1998-04-27 | 1999-04-26 | NOUVEAUX DERIVES DE L'ACIDE OCTAHYDRO-6,10-DIOXO-6H-PYRIDAZINO[1,2-a] [1,2] DIAZEPINE-1-CARBOXYLIQUE, LEUR PROCEDE DE PREPARATION ET LEUR APPLICATION A LA PREPARATION DE COMPOSES THERAPEUTIQUEMENT ACTIFS |
Country Status (37)
| Country | Link |
|---|---|
| US (3) | US6548664B1 (fr) |
| EP (1) | EP1073673B1 (fr) |
| JP (2) | JP4332298B2 (fr) |
| KR (1) | KR100561582B1 (fr) |
| CN (1) | CN1167711C (fr) |
| AP (1) | AP1518A (fr) |
| AR (1) | AR016466A1 (fr) |
| AU (1) | AU755286B2 (fr) |
| BG (1) | BG65282B1 (fr) |
| BR (1) | BR9910020A (fr) |
| CA (1) | CA2330492C (fr) |
| CU (1) | CU23155A3 (fr) |
| CY (1) | CY1107599T1 (fr) |
| DE (1) | DE69934790T2 (fr) |
| DK (1) | DK1073673T3 (fr) |
| EA (1) | EA003280B1 (fr) |
| EE (1) | EE04682B1 (fr) |
| ES (1) | ES2279617T3 (fr) |
| FR (1) | FR2777889B1 (fr) |
| GE (1) | GEP20033119B (fr) |
| HR (1) | HRP20000733B1 (fr) |
| HU (1) | HU230060B1 (fr) |
| ID (1) | ID26292A (fr) |
| IL (3) | IL139102A0 (fr) |
| ME (2) | MEP40808A (fr) |
| NO (1) | NO328864B1 (fr) |
| NZ (1) | NZ507618A (fr) |
| PL (1) | PL203762B1 (fr) |
| PT (1) | PT1073673E (fr) |
| RS (1) | RS50423B (fr) |
| SI (1) | SI1073673T1 (fr) |
| SK (1) | SK288050B6 (fr) |
| TR (1) | TR200003142T2 (fr) |
| TW (1) | TWI249537B (fr) |
| UA (1) | UA71913C2 (fr) |
| WO (1) | WO1999055724A1 (fr) |
| ZA (1) | ZA200006081B (fr) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6177565B1 (en) * | 1998-08-19 | 2001-01-23 | Vertex Pharmaceuticals Inc. | Process for synthesizing piperazic acid |
| EP1468993A1 (fr) * | 2003-04-16 | 2004-10-20 | Isochem | Procédé de préparation de dérivés de l'acide hexahydropyridazine-3-carboxylique |
| WO2005028449A1 (fr) * | 2003-06-26 | 2005-03-31 | Honeywell Specialty Chemicals Seelze Gmbh | Procede ameliore de fabrication d'acides hexahydropyridazine-3-carboxyliques 1,2-disubstitues et leurs esters |
| WO2005122682A2 (fr) | 2004-06-18 | 2005-12-29 | Ranbaxy Laboratories Limited | Procede de preparation d'esters d'acide piperazique |
| CN100404516C (zh) * | 2004-02-13 | 2008-07-23 | 大连绿源药业有限责任公司 | 六氢哒嗪三羧酸酯的制备方法 |
| WO2012049646A1 (fr) | 2010-10-12 | 2012-04-19 | Ranbaxy Laboratories Limited | Procédé de préparation d'un intermédiaire de cilazapril |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6201118B1 (en) | 1998-08-19 | 2001-03-13 | Vertex Pharmaceuticals Inc. | Process for forming an N-acylated, N,N-containing bicyclic ring from piperazic acid or an ester thereof especially useful as an intermediate in the manufacture of a caspase inhibitor |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4512924A (en) * | 1982-05-12 | 1985-04-23 | Hoffmann-La Roche Inc. | Pyridazo[1,2-a][1,2]diazepines |
| WO1993023403A1 (fr) * | 1992-05-15 | 1993-11-25 | Merrell Dow Pharmaceuticals Inc. | NOUVEAUX DERIVES DE MERCAPTOACETYLAMIDO PYRIDAZO[1,2]PYRIDAZINE, DE PYRAZOLO[1,2]PYRIDAZINE, DE PYRIDAZO[1,2-a][1,2]DIAZEPINE ET DE PYRAZOLO[1,2-a][1,2]DIAZEPINE UTILISES COMME INHIBITEURS D'ENCEPHALINASE ET D'ACE |
| WO1995033751A1 (fr) * | 1994-06-08 | 1995-12-14 | Sanofi Winthrop, Inc. | INHIBITEURS AU LACTAME BICYCLIQUE DE L'ENZYME DE CONVERSION DE L'INTERLEUKINE-1-$g(b) |
| WO1997022619A2 (fr) * | 1995-12-20 | 1997-06-26 | Vertex Pharmaceuticals Incorporated | INHIBITEURS DE L'ENZYME CONVERTISSANT L'INTERLEUKINE-1$g(b) |
| US5656627A (en) * | 1994-06-17 | 1997-08-12 | Vertex Pharmaceuticals, Inc. | Inhibitors of interleukin-1β converting enzyme |
| US5723602A (en) * | 1992-05-18 | 1998-03-03 | E. R. Squibb & Sons, Inc. | Dual action inhibitors containing a pyridazinodiazepine or pyrazolodiazepine lactam ring |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6177565B1 (en) * | 1998-08-19 | 2001-01-23 | Vertex Pharmaceuticals Inc. | Process for synthesizing piperazic acid |
| US6201118B1 (en) * | 1998-08-19 | 2001-03-13 | Vertex Pharmaceuticals Inc. | Process for forming an N-acylated, N,N-containing bicyclic ring from piperazic acid or an ester thereof especially useful as an intermediate in the manufacture of a caspase inhibitor |
-
1998
- 1998-04-27 FR FR9805243A patent/FR2777889B1/fr not_active Expired - Lifetime
-
1999
- 1999-04-07 TW TW088105540A patent/TWI249537B/zh not_active IP Right Cessation
- 1999-04-22 AR ARP990101859A patent/AR016466A1/es not_active Application Discontinuation
- 1999-04-26 ID IDW20002188A patent/ID26292A/id unknown
- 1999-04-26 AU AU34274/99A patent/AU755286B2/en not_active Ceased
- 1999-04-26 PT PT99915834T patent/PT1073673E/pt unknown
- 1999-04-26 SK SK1600-2000A patent/SK288050B6/sk not_active IP Right Cessation
- 1999-04-26 WO PCT/FR1999/000981 patent/WO1999055724A1/fr not_active Ceased
- 1999-04-26 SI SI9930952T patent/SI1073673T1/sl unknown
- 1999-04-26 IL IL13910299A patent/IL139102A0/xx active IP Right Grant
- 1999-04-26 UA UA2000116680A patent/UA71913C2/uk unknown
- 1999-04-26 JP JP2000545882A patent/JP4332298B2/ja not_active Expired - Fee Related
- 1999-04-26 PL PL343720A patent/PL203762B1/pl unknown
- 1999-04-26 GE GEAP19995634A patent/GEP20033119B/en unknown
- 1999-04-26 KR KR1020007011895A patent/KR100561582B1/ko not_active Expired - Fee Related
- 1999-04-26 EE EEP200000619A patent/EE04682B1/xx unknown
- 1999-04-26 TR TR2000/03142T patent/TR200003142T2/xx unknown
- 1999-04-26 AP APAP/P/2000/001973A patent/AP1518A/en active
- 1999-04-26 ME MEP-408/08A patent/MEP40808A/xx unknown
- 1999-04-26 ES ES99915834T patent/ES2279617T3/es not_active Expired - Lifetime
- 1999-04-26 DK DK99915834T patent/DK1073673T3/da active
- 1999-04-26 CA CA2330492A patent/CA2330492C/fr not_active Expired - Fee Related
- 1999-04-26 HU HU0102447A patent/HU230060B1/hu not_active IP Right Cessation
- 1999-04-26 CN CNB998076139A patent/CN1167711C/zh not_active Expired - Fee Related
- 1999-04-26 EP EP99915834A patent/EP1073673B1/fr not_active Expired - Lifetime
- 1999-04-26 ME MEP-2008-408A patent/ME00300B/me unknown
- 1999-04-26 BR BR9910020-7A patent/BR9910020A/pt not_active IP Right Cessation
- 1999-04-26 RS YUP-658/00A patent/RS50423B/sr unknown
- 1999-04-26 EA EA200001107A patent/EA003280B1/ru not_active IP Right Cessation
- 1999-04-26 NZ NZ507618A patent/NZ507618A/en not_active IP Right Cessation
- 1999-04-26 DE DE69934790T patent/DE69934790T2/de not_active Expired - Lifetime
- 1999-04-26 HR HR20000733A patent/HRP20000733B1/xx not_active IP Right Cessation
- 1999-04-26 US US09/674,327 patent/US6548664B1/en not_active Expired - Lifetime
-
2000
- 2000-10-18 IL IL139102A patent/IL139102A/en not_active IP Right Cessation
- 2000-10-25 CU CU229A patent/CU23155A3/es unknown
- 2000-10-26 NO NO20005391A patent/NO328864B1/no not_active IP Right Cessation
- 2000-10-26 BG BG104891A patent/BG65282B1/bg unknown
- 2000-10-27 ZA ZA200006081A patent/ZA200006081B/xx unknown
-
2002
- 2002-03-20 US US10/102,591 patent/US6570012B2/en not_active Expired - Lifetime
- 2002-12-06 US US10/313,422 patent/US20030130269A1/en not_active Abandoned
-
2007
- 2007-03-22 CY CY20071100404T patent/CY1107599T1/el unknown
-
2008
- 2008-03-12 IL IL190120A patent/IL190120A/en not_active IP Right Cessation
-
2009
- 2009-04-10 JP JP2009096241A patent/JP5101558B2/ja not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4512924A (en) * | 1982-05-12 | 1985-04-23 | Hoffmann-La Roche Inc. | Pyridazo[1,2-a][1,2]diazepines |
| WO1993023403A1 (fr) * | 1992-05-15 | 1993-11-25 | Merrell Dow Pharmaceuticals Inc. | NOUVEAUX DERIVES DE MERCAPTOACETYLAMIDO PYRIDAZO[1,2]PYRIDAZINE, DE PYRAZOLO[1,2]PYRIDAZINE, DE PYRIDAZO[1,2-a][1,2]DIAZEPINE ET DE PYRAZOLO[1,2-a][1,2]DIAZEPINE UTILISES COMME INHIBITEURS D'ENCEPHALINASE ET D'ACE |
| US5723602A (en) * | 1992-05-18 | 1998-03-03 | E. R. Squibb & Sons, Inc. | Dual action inhibitors containing a pyridazinodiazepine or pyrazolodiazepine lactam ring |
| WO1995033751A1 (fr) * | 1994-06-08 | 1995-12-14 | Sanofi Winthrop, Inc. | INHIBITEURS AU LACTAME BICYCLIQUE DE L'ENZYME DE CONVERSION DE L'INTERLEUKINE-1-$g(b) |
| US5656627A (en) * | 1994-06-17 | 1997-08-12 | Vertex Pharmaceuticals, Inc. | Inhibitors of interleukin-1β converting enzyme |
| WO1997022619A2 (fr) * | 1995-12-20 | 1997-06-26 | Vertex Pharmaceuticals Incorporated | INHIBITEURS DE L'ENZYME CONVERTISSANT L'INTERLEUKINE-1$g(b) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6177565B1 (en) * | 1998-08-19 | 2001-01-23 | Vertex Pharmaceuticals Inc. | Process for synthesizing piperazic acid |
| EP1468993A1 (fr) * | 2003-04-16 | 2004-10-20 | Isochem | Procédé de préparation de dérivés de l'acide hexahydropyridazine-3-carboxylique |
| FR2853901A1 (fr) * | 2003-04-16 | 2004-10-22 | Isochem Sa | Procede de preparation de derives de l'acide hexahydropyridazine-3-carboxylique |
| US7132535B2 (en) | 2003-04-16 | 2006-11-07 | Isochem | Process for preparing hexahydropyridazine-3-carboxylic acid derivatives |
| WO2005028449A1 (fr) * | 2003-06-26 | 2005-03-31 | Honeywell Specialty Chemicals Seelze Gmbh | Procede ameliore de fabrication d'acides hexahydropyridazine-3-carboxyliques 1,2-disubstitues et leurs esters |
| CN100404516C (zh) * | 2004-02-13 | 2008-07-23 | 大连绿源药业有限责任公司 | 六氢哒嗪三羧酸酯的制备方法 |
| WO2005122682A2 (fr) | 2004-06-18 | 2005-12-29 | Ranbaxy Laboratories Limited | Procede de preparation d'esters d'acide piperazique |
| WO2012049646A1 (fr) | 2010-10-12 | 2012-04-19 | Ranbaxy Laboratories Limited | Procédé de préparation d'un intermédiaire de cilazapril |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0307303A1 (fr) | [(Pyrimidinyl-2)-aminoalkyl]-1 pipéridines, leur préparation et leur application en thérapeutique | |
| JP5101558B2 (ja) | オクタヒドロ−6,10−ジオキソ−6H−ピリダジノ[1,2−a][1,2]ジアゼピン−1−カルボン酸の新規な誘導体 | |
| EP0309324B1 (fr) | Procédé de synthèse d'alpha amino N alkyles et de leurs esters. Application à la synthèse de carboxyalkyl dipeptides | |
| EP0306375A1 (fr) | Dérivés de[(pipéridinyl-4)méthyl]-2 tétrahydro-1,2,3,4 isoquinoléine, leur préparation et leur application en thérapeutique | |
| EP0383686B1 (fr) | Carboxyalkyl-ethers de la 2-amino-7-hydroxytetraline | |
| FR2584399A1 (fr) | Procede de synthese stereospecifique d'acides amino-4 hydroxy-3 carboxyliques | |
| EP1144436B1 (fr) | Procede de preparation de composes bicycliques et l'application de ce procede pour la preparation d'un compose inhibiteur de ice | |
| EP0103500B1 (fr) | Dérivés de phénéthyl-1alpha-phényl-pipéridine-3-propanenitrile, leur préparation et leur application en thérapeutique | |
| EP0955310B1 (fr) | Nouveaux dérivés de l'acide (3,4,7,8,9,10) -hexahydro-6,10-dioxo-6H-pyridazino /1,2-a/ /1,2/ diazépine-1-carboxylique, leur procédé de préparation et leur application à la préparation de médicaments | |
| AU2003200034B2 (en) | New derivatives of octahydro-6, 10-dioxo-6H-pyridazino [1,2-A][1,2]diazepine-1-carboxylic acid, their preparation process and their use in the preparation of therapeutically active compounds | |
| EP1400531B1 (fr) | Procédé de synthèse de la N-[(S)-1-carbéthoxybutyl]-(S)-alanine et application à la synthèse du perindopril | |
| EP0347313B1 (fr) | Ethers de la 2-amino-7-hydroxytétraline | |
| MXPA00010457A (en) | NOVEL OCTAHYDRO-6,10-DIOXO-6H-PYRIDAZINO/1,2-a/ /1,2/DIAZEPIN-1-CARBOXYLIC ACID DERIVATIVES, PREPARATION METHOD AND USE FOR PREPARING THERAPEUTICALLY ACTIVE COMPOUNDS | |
| BE817382A (fr) | Procede de preparation de derives de l'octahydro-oxazolo(3 | |
| CZ20003956A3 (cs) | Nové deriváty oktahydro-6,10-dioxo-6Hpyridazino[ 1,2-a][1,2]diazepin-l-karboxylové kyseliny, způsob jejich přípravy a jejich použití při přípravě terapeuticky aktivních sloučenin. | |
| CH331208A (fr) | Procédé pour la préparation de N-benzyl- ou N,N-dibenzyl-peptides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: P-658/00 Country of ref document: YU Ref document number: 99807613.9 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AU BA BB BG BR CA CN CU CZ EE GD GE HR HU ID IL IN IS JP KP KR LC LK LR LT LV MG MK MN MX NO NZ PL RO SG SI SK SL TR TT UA US UZ VN YU ZA |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 139102 Country of ref document: IL Ref document number: 507618 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 16002000 Country of ref document: SK Ref document number: 34274/99 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2330492 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2000/010457 Country of ref document: MX Ref document number: 1999915834 Country of ref document: EP Ref document number: PV2000-3956 Country of ref document: CZ Ref document number: IN/PCT/2000/00543/MU Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007011895 Country of ref document: KR Ref document number: 2000/03142 Country of ref document: TR Ref document number: 1200000965 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000/06081 Country of ref document: ZA Ref document number: P20000733A Country of ref document: HR Ref document number: 200006081 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09674327 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200001107 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999915834 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-3956 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007011895 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 34274/99 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020007011895 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1999915834 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 190120 Country of ref document: IL |