明 細 書
PLK1阻害剤としての新規ァミノピリミジン誘導体 技術分野
本発明は医薬の分野で有用であり、 PLK1阻害作用に基づいて、 腫瘍細胞の増殖 を阻害し、 抗腫瘍効果を発揮する、 新規置換アミノピリミジン誘導体、 及びそれを含 む P L K 1阻害剤並びに杭がん剤に関する。 背景技術
正常細胞と比較してがん細胞は一般的に増殖が活発であるとされており、多くの場 合、細胞周期制御機構の異常による無秩序な増殖ががんの原因であると考えられてい る。 細胞周期において有糸分裂期 (M期) は、 染色体を娘細胞に均等に分配するステ ップであり、 この過程の厳密な制御は細胞の増殖、 生存に不可欠である。 従って、 M 期の進行を阻害することは細胞増殖の抑制に有効な手段であると考えられ、実際に夕 キソ一ルゃビンクリスチン等の M期を標的をした杭がん剤が臨床で有効な成績を収 めている。
M期の進行の多くのステップがタンパク質のリン酸化を行うプロティン ·キナーゼ によって制御されていることが知られている。 PLK (ポロ ライク キナーゼ (p o 1 o l i ke k i n a s e s )) ファミリ一は M期を含む細胞周期の制御に重 要な役割を果たしているセリンスレオニンキナーゼであり、 PLK1、 PLK2、 P LK3、 SAKの 4つの類似したタンパク質から構成される(ネイチヤー ·レビュー · モレキュラー'セル'バイオロジー(Na t. Re v. Mo l . Ce l l B i o l .), 第 5巻、 429 頁、 (2004年))。 その中でも PLK 1は哺乳動物細胞の M期の複 数の重要な段階に関与していることが知られている。即ち、 P L K 1は M期への進入、 中心体の制御、 染色体の分離、 細胞質分裂等の各ステップに、 様々な基質をリン酸化 することによって関与していることが報告されている (ネイチヤー ·レビュー ·モレ キユラ一 ·セル ·バイオロジー (Na t. Rev. Mo l . C e 1 1 B i o l .)、 第 5巻、 429 頁、 (2004年))。
さらに、 PLK 1はヒ卜の様々ながん組織において過剰発現しているという報告が 多数ある。 例えば、 非小細胞肺がん(オンコジーン(On c og en e)、 第 14巻、 543頁、 (1997年))、 頭頸部がん(キャンサ一 · リサ一チ(C a n c e r Re s e a r c h), 第 15巻、 2794頁、 (1999年)) において PLK 1の過剰発 現が認められ、 また PLK1の過剰発現はこれら患者の予後と相関するといぅデ一夕 が得られている。 他に、 大腸がん、 食道がん、 卵巣がん、 メラノ一マといったがん種 においても PLK1の発現が上昇しているという報告がある。 以上の報告は、 PLK 1の過剰発現が細胞の癌化になんらかの形で関与していること、 また PLK1の機能 が特にがん細胞における M期の進行に重要である、 ということを示唆している。
JP2007/075240 これらの事実より、 PLK1を標的とした杭がんアプローチとしての可能性が考え られる。実際に、 様々な実験手法を用いてがん細胞に対する PL K1の機能阻害効果 を調べる実験が複数報告されている。例えば PLK1の機能阻害変異体をウィルスべ クタ一を用いて細胞内に発現させる実験では、 PLK1阻害によるがん細胞選択的な 細胞死の誘導が報告されている (セル'グロウス 'アンド 'ディファレンシエーショ ン(Ce l 1 g r owt h&D i f f.)、 第 11巻、 615頁、 (2000年))。 ま た PLK1に対する s i RNA (ジャーナル ·ォブ ·ナショナル ·キャンサー 'イン スティチュート(J. Na t l . Canc e r I n s t.)、第 94巻、 1863頁、 (2002年)) が、 がん細胞に増殖阻害やアポトーシスを誘導するという報告もあ. る。 さらに、 PLK 1に対する s hRNA (ジャーナル ·ォブ ·ナショナル ·キャン サ一 ·インスティチュート (J. Na t l . Can c e r I n s t.)、 第 96巻、 862頁、 (2004年)) やアンチセンスオリゴヌクレオチド (オンコジーン(On c o g e ne)、 第 21卷、 3162頁、 (2002年)) がマウス Xe n o g r a f tモデルで抗腫瘍効果を示すことが報告されている。 これらの実験結果は、 PLK1 活性の阻害ががん細胞に増殖抑制や細胞死の誘導をもたらすことを示しており、 P L K 1阻害剤が杭がん剤とし有効であることを強く示唆している。 '
本出願の発明者は、先に PL K阻害作用を有する置換イミダゾ一ル誘導体について 出願した (国際公開第 2006/025567号パンフレツト)。 発明の開示
PLK1阻害作用に基づく抗腫瘍剤を開発すべく、 PLK1阻害作用を示し、 それ に基づく細胞増殖抑制作用が優れた新規なアミノピリミジン誘導体を創製すること が本発明の解決課題である。
本発明者等は、 上記課題を解決すべく、 ァミノピリミジン誘導体を広く合成し、 一 般式 [I] で表される化合物が、 優れた PLK1阻害作用、 及びそれに基づく細胞増 殖抑制作用を示すことを見いだして本発明を完成した。
即ち、 本発明は、
一般式 [I] :
[式中、 及び R
2 は、 同一若しくは異なって、 水素原子; <置換基群 Q!>から 選択される置換基; <置換基群 >から選択される置換基で 1個若しくは 2個以上 置換されてもよい低級アルキル基; 又は、 置換されてもよい炭素数 3個ないし 6個 のシクロアルキル基であり ;
R3及び R4 は、 同一若しくは異なって、
a) 水素原子;
b) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 置換 されていてもよい低級アルキル基、 置換されていてもよいべンジル基、 又は置換され ていてもよいシクロアルキル基である。) で置換される低級アルキル基;
c) <置換基群 〉から選択される置換基;
d )ぐ置換基群 /3 >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルキル基;
e )ぐ置換基群 ;6 >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルケニル基;
f ) フエニル基;
g) フエニル基で置換される低級アルキル基; · h) 4員ないし 7員の脂肪族複素環基;
i ) 4員ないし 7員の脂肪族複素環基で置換される低級アルキル基;
j ) 5員ないし 6員の芳香族複素環基;又は、
k) 5員ないし 6員の芳香族複素環基で置換される低級アルキル基 (ここで、 前記 フエニル基、 脂肪族複素環基、 及び芳香族複素環基は、 それぞれ独立して、 下記 1) ないし 4) :
1 ) 低級アルキル基;
2) ぐ置換基群 3>から選択される置換基;
3)ぐ置換基群 j8>から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β>から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若し.くは 2個以上置換されてもよ く、また、前記脂肪族複素環基は、不飽和結合を含んでいてもよく、さらに、前記 b)、 g)、 i)、 及び k) の低級アルキル基は、 適宜置換されてもよい。) であるか、 或い は、
R3及び R4 は、 一緒になつて、 4員ないし 7員の脂肪族複素環基 (ここで、 前記 脂肪族複素環基は、 下記 1) ないし 4) :
1) 低級アルキル基;
2) <置換基群 >から選択される置換基;
3 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 iS >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) を形成し; R 5 は、 水素原子、 シァノ基、 ハロゲン原子、 又は低級アルキル基である。
ぐ置換基群ひ〉及びぐ置換基群 S >は、 下記のように定義される。
ぐ置換基群 α > :
ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル基、 Ύ ミノスルホニル基、 イミノ基、 低級アルキルアミノ基、 ジ低級アルキルアミノ基、 低. 級アルキルスルホニル基、 低級アルキルスルホニルァミノ基、 低級アルコキシ基、 低 級アルコキシカルボ二ル基、 低級アルコキシカルボニルァミノ基、 低級アルカノィル 基、 低級アルカノィルォキシ基、 低級アルキルチオ基、 及び力ルポキシル基
<置換基群 j6 >:
ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル基、 ァ ミノスルホニル基、 イミノ基、 低級アルキルスルホニル基、 低級アルキルスルホニル アミノ基、 低級アルコキシ基、 低級アルコキシカルボ二ル基、 低級アルコキシ力ルポ ニルァミノ基、 低級アルカノィル基、 低級アルカノィルォキシ基、 低級アルキルチオ 基、 カルボキシル基、 及びべンジル基]で示される化合物又はその薬学的に許容され る塩若しくはエステル、 に関する。 なお、 上記式 (I ) で表される化合物には、 当該 化合物のラセミ体のみならず、存在可能なすべてのェナンチォマ一及びジァステレオ マ一を含む。
また、 本発明は、 がん治療において同時に、 別々に、 又は順次に投与するための組 み合わせ製剤であって、 2つの別個の製剤:
* 薬学的に許容し得る担体又は希釈剤と一緒に、上記一般式 [ I ]で示される化合 物又はその薬学的に許容し得る塩若しくはエステルを含む製剤、 並びに
* 薬学的に許容し得る担体又は希釈剤と一緒に、 杭がん性アルキル化剤、 抗がん 性代謝拮抗剤、 抗がん性抗生物質、 植物由来杭がん剤、 抗がん性白金配位化合物、 抗 がん性カンプ卜テシン誘導体、 杭がん性チロシンキナーゼ阻害剤、 モノクローナル抗 体、 インターフェロン、 生物学的応答調節剤、 及びそ 他杭がん剤からなる群から選 択される杭がん剤又はその薬学的に許容し得る塩若しくはエステルを含む製剤(ここ で、
杭がん性アルキル化剤は、 ナイトロジェン マスタ一ド N—才キシド、 シクロ ホスフアミド、ィホスフアサミド、メルファラン、ブスルファン、ミトブロニトール、 カルボコン、 チォテパ、 ラニムスチン、 二ムスチン、 テモゾロミド又はカルムスチン であり、
抗がん性代謝拮抗剤は、 メトトレキサート、 6—メルカプトプリンリポシド、 メ ルカプトプリン、 5—フルォロウラシル、 テガフール、 ドキシフルリジン、 カルモフ ール、 シ夕ラビン、 シ夕ラビンォクホスフアート、 エノシ夕ビン、 S— 1、 ゲムシ夕
ビン、 フルダラビン又はぺメトレクスド ジソディゥムであり、
杭がん性抗生物質は、 ァクチノマイシン D、 ドキソルビシン、 ダウノルビシン、 ネオカルチノスタチン、 ブレオマイシン、 ぺプロマイシン、 マイ卜マイシン C、 ァク ラルビシン、. ピラルビシン、 ェピルビシン、 ジノス夕チンスチマラマー、 イダルビシ ン、 シロリムス、 又はバルルビシンであり、
植物由来杭がん剤は、 ビンクリスチン、 ビンブラスチン、 ビンデシン、 エトポシ ド、 ソブゾキサン、 ドセタキセル、 パクリタキセル、 又はビノレルビンであり、 抗がん性白金配位化合物は、 シスブラチン、 カルポプラチン、 ネダプラチン、 又 はォキザリブラチンであり、
抗がん性カンプトテシン誘導体は、 イリノテカン、 トポテカン、 又はカンプトテ シンンであり、
抗がん性チロシンキナーゼ阻害剤は、 ゲフイチニブ、 イマチニブ、 又はエルロチ ニブであり、
モノクローナル抗体は、 セツキシマブ、 ベバシズマブ、 リツキシマ;/、 ベバシズ マブ、 ァレムッズマブ又はトラスッズマブであり、
インターフェロンは、 インタ一フエロンひ、 インターフェロン α— 2 a、 インタ —フエロン oi—2 b、 インターフェロン jS、 インターフェロンァー 1 a、 又はイン夕 一フエロンァー n 1であり、
生物学的応答調節剤は、クレスチン、レンチナン、シゾフィラン、ピシバニール、 又はウベ二メクスであり、 そして、
その他抗がん剤は、ミトキサントロン、 Lーァスパラギナーゼ、プロカルバジン、 ダカルバジン、 ヒドロキシカルバミド、 ペントス夕チン、 トレチノイン、 ァレファセ プト、 ダルべポェチン アルファ、 ァナス卜ロゾール、 ェキセムスタン、 ビカルタミ ド、 リュープロレリン、 フルタミド、 フルべストラント、 ぺガプ夕ニブ ォクタソデ ィゥム、 デニリューキン ジフティトクス、 アルデスリューキン、 チロトロピン ァ ルファ、 ァルセニック トリオキシド、 ボルテゾミブ、 力ぺシ夕ビン、 又はゴセレリ ンである。) :
からなる組み合わせ製剤、 に関する。
さらに、 本発明は、 薬学的に許容し得る担体又は希釈剤と一緒に、 上記一般式 [ I ] で示される化合物又はその薬学的に許容し得る塩若しくはエステル、並びに抗がん性 アルキル化剤、 抗がん性代謝拮抗剤、 抗がん性抗生物質、 植物由来杭がん剤、 杭がん 性白金配位化合物、 杭がん性カンプトテシン誘導体、 抗がん性チロシンキナーゼ阻害 剤、 モノクローナル抗体、 生物学的応答調節剤、 及びその他杭がん剤 (ここで、 各抗 がん剤の定義は、 上記と同義である。) からなる群から選択される抗がん剤又はその 薬学的に許容し得る塩若しくはエステルを含むことを特徴とする医薬組成物、 に関す る。
また、 本発明は、 治療上有効量の上記一般式 [ I ]で示される化合物又はその薬学的 に許容し得る塩若しくはエステルを、 杭がん性アルキル化剤、 杭がん性代謝拮抗剤、
抗がん性抗生物質、 植物由来杭がん剤、 抗がん性白金配位化合物、 杭がん性カンプト テシン誘導体、 抗がん性チロシンキナーゼ阻害剤、 モノクローナル抗体、 インタ一フ ェロン、 生物学的応答調節剤、 及びその他抗がん剤 (ここで、 各抗がん剤の定義は、 前記と同じである。) からなる群から選択される治療上有効量の杭がん剤又はその薬 学的に許容し得る塩若しくはエステルと組み合わせて、 同時に、 別々に、 又は順次に 投与することを特徴とするがん治療方法、 に関する。
さらに、 本発明は、 がん治療のための医薬を製造するための、 P L K 1阻害剤の使 用、 及び、 がん治療のための医薬を製造するための、 杭がん剤と組み合わせた、 P L K 1阻害剤の使用、 に関する。 また、 本発明は、 哺乳類 (特にヒト) におけるがんを. 治療する方法であって、 当該哺乳類に治療上の有効量の P L K 1阻害剤を投与するこ とを特徴とする方法、 及び、 哺乳類 (特にヒト) におけるがんを治療する方法であつ て、 治療上の有効量の杭がん剤と組み合わせて、 当該哺乳類に治療上の有効量の P L K 1阻害剤を投与することを特徴とする方法、 に関する。
. また、 本発明は、 P L K 1阻害剤を有効成分として含有するがん治療剤、 及び、 抗 がん剤と一緒に、 P L K 1阻害剤を有効成分として含有するがん治療剤、 に関する。 次に、 本明細書に記載された記号及び用語について説明する。
上記式 (I ) 中の 「低級アルキル基」 とは、 炭素数 1ないし 6個の直鎖状又は分岐 状のアルキル基をいい、 例えばメチル基、 ェチル基、 プロピル基、 イソプロピル基、 ブチル基、 イソブチル基、 s e c一ブチル基、 t e r t—ブチル基、 ペンチル基、 へ キシル基等が挙げられる。
上記式 (I ) 中の 「低級アルケニル基」 とは、 炭素数 2ないし 6個の直鎖状又は分 岐状のアルケニル基をいい、 例えばビニル基、 1一プロぺニル基、 ァリル基、 イソプ 口ぺニル基、 1—ブテニル基、 3—ブテニル基、 1, 3—ブタンジェニル基、 2—ぺ ンテニル基、 4一ペンテニル基、 1一へキセニル基、 3—へキセニル基、 5—へキセ ニル基等が挙げられる。
上記式 (I ) 中の 「シクロアルキル基」 とは、 3員ないし 8員の脂肪族環状基であ り、 例えば、 シクロプロピル基、 シクロブチル基、 シクロペンチル基、 シクロへキシ ル基、 シクロへプチル基、 シクロォクチル基等が挙げられ、 好ましくは、 3員ないし 6員の脂肪族環状基である。 例えば、 シクロプロピル基、 シクロブチル基、 シクロべ ンチル基、 シクロへキシル基が好ましい。
上記式 (I ) 中の 「脂肪族複素環基」 とは、 一般には、 炭素原子以外に、 窒素原子 、 酸素原子、 及び硫黄原子から選ばれる少なくも 1個の原子を含み、 単環又は 2環な いし 3環からなる縮合環である、 飽和若しくは不飽和脂肪族複素環基をいい、 例えば 、 ァゼチジル基、 ピロリジニル基、 ピペリジニル基、 ピペラジニル基、 モルホリノ基
、 テトラヒドロフラニル基、 イミダゾリジニル基、 チオモルホリノ基、 テトラヒドロ キノリル基、 テトラヒドロイソキノリル基等が挙げられる。 上記式 (I ) 中の 「4員 ないし 7員の脂肪族複素環基」 とは、 4員ないし 7員の単環である、 前記飽和若しく
は不飽和脂肪族複素環基であり、 例えば、 ァゼチジル基、 ピロリジニル基、 ピベリジ ニル基、 ピペラジニル基、 モルホリノ基、 テトラヒドロフラニル基、 イミダゾリジニ ル基、 チオモルホリノ基等が挙げられる。
上記式 (I ) 中の 「芳香族複素環基」 とは、 一般には、 窒素原子や酸素原子などの 少なくとも 1個のへテロ原子を含む、 芳香族性の複素環基を示し、 例えば、 5員ない し 7員の単環式複素環基、 及び、 これに 3員ないし 8員の環が縮合した縮環式複素環 基などであり、 具体的には、 チェニル基、 ピロリル基、 フリル基、 チアゾリル基、 ィ ミダゾリル基、 ピラゾリル基、 ォキサゾリル基、 ピリジル基、 ビラジニル基、 ピリミ ジニル基、 ピリダジニル基、 イソォキサゾリル基、 イソキノリル基、 イソインドリル. 基、 インダゾリル基、 インドリル基、 キノキサリニル基、 キノリル基、 ベンゾイミダ ゾリル基、 ベンゾフラニル基などが挙げられる。 上記式 (I ) 中の 「5員ないし 6員 の芳香族複素環基」 とは、 5員ないし 6員の前記芳香族性単環式複素環基であり、 例 えば、 チェニル基、 ピロリル基、 フリル基、 チアゾリル基、 イミダゾリル基、 ピラゾ リル基、 ォキサゾリル基、 ピリジル基、 'ビラジニル基、 ピリミジニル基、 ピリダジニ ル基、 イソォキサゾリル基などが挙げられる。
上記式 (I ) 中の 「ハロゲン原子」 としては、 例えばフッ素原子、 塩素原子、 臭素 原子、 ヨウ素原子等が挙げられ、 中でも例えばフッ素原子、 塩素原子、 又は臭素原子 が好ましい。
上記式 (I ) 中の 「低級アルキルアミノ基」 とは、 ァミノ基に上記 「低級アルキル 基」 'が N—置換した置換基をいい、 例えば N—メチルァミノ基、 N—ェチルァミノ 基、 N—プロピルアミノ基、 N— ^ Γソプロピルアミノ基、 N—プチルァミノ基、 N 一イソブチルァミノ基、 N - t e r t 一プチルァミノ基、 N—ペンチルァミノ基、 N—へキシルァミノ基等が挙げられる。
上記式 (I ) 中の 「ジ低級アルキルアミノ基」 とは、 ァミノ基に上記 「低級アルキ ル基」が N, N—ジ置換した置換基をいい、例えば N, N—ジメチルァミノ基、 N, N—ジェチルァミノ基、 N, N—ジプロピルアミノ基、 N, N—ジイソプロピルァ ミノ基、 N, N—ジブチルァミノ基、 N, N—ジイソプチルァミノ基、 N, N— ジ t e r t —プチルァミノ基、 N, N—ジペンチルァミノ基、 N, N—ジへキシル アミノ基、 N— ェチルー N—メチルァミノ基、 N —メ.チル一 N—プロピルアミノ基 等が挙げられる。
上記式 (I ) 中の 「ァミノ低級アルキル基」 とは、 ァミノ基で置換された炭素数 1 ないし 6個の直鎖状又は分岐状のアルキル基をいい、 例えばアミノメチル基、 1—ァ ミノェチル基、 2—アミノエチル基、 1ーァミノプロピル基、 2—ァミノプロピル基、 ァミノイソプロピル基、 1 一アミノブチル基、 2—アミノブチル基、 ァミノイソプチ ル基、ァミノ s e c—プチル基、ァミノ t e r t —ブチル基、 1ーァミノペンチル基、 . 2—ァミノペンチル基、 3—ァミノペンチル基、 1ーァミノへキシル基、 2—ァミノ へキシル基、 3—ァミノへキシル基等が挙げられる。
上記式 (I ) 中の 「低級アルキルスルホニル基」 とは、 スルホニル基の硫黄原子に
上記 「低級アルキル」 が結合した置換基をいい、 例えばメチルスルホニル基、 ェチ ルスホニル基、 プチルスルホニル基等が挙げられる。
上記式 (I ) 中の 「低級アルキルスルホニルァミノ基」 とは、 ァミノ基に上記 「低 級アルキルスルホニル基」 が N—置換した置換基をいい、 例えばメチルスルホニルァ ミノ基、 ェチルスルホニルァミノ基、 プチルスルホニルァミノ基等が挙げられる。 上記式 (I ) 中の 「低級アルコキシ基」 とは、 酸素原子に 「低級アルキル基」 が結 合した基をいい、例えばメトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、 ブトキシ基、 イソブトキシ基、 s e c一ブトキシ基、 t e r t一ブトキシ基、 ペンチ ルォキシ基、 ネオペンチルォキシ基、 へキシルォキシ基、 イソへキシルォキシ基等が. 挙げられる。
上記式 (I ) 中の 「低級アルコキシカルボニル基」 とは、 カルボニル基に上記 「低 級アルコキシ基」 が結合した基をいい、 具体的には例えばメトキシカルボニル基、 ェ トキシカルボニル基、 プロポキシカルポニル基、 ィソプロボキシカルボニル基、 ブト キシカルボ二ル基、 イソブトキシカルポニル基、 s e c一ブトキシカルボニル基、 t e r t 一ブトキシカルボニル基、 ぺンチルォキシカルポニル基、 ネオペンチルォキシ カルボニル基、 へキシルォキシカルポニル基、 ィソへキシルォキシカルポニル基等が 挙げられる。
上記式(I ) 中の「低級アルコキシカルボニルァミノ基」 とは、 ァミノ基に上記「低 級アルコキシカルポニル基」 が N—置換した基をいい、 具体的には例えばメトキシカ ルポニルァミノ基、 エトキシカルボニルァミノ基、 プロポキシ力ルポニルァミノ基、 イソプロポキシ力ルポニルァミノ基、 ブトキシカルボニルァミノ基、 イソブトキシカ ルポニルァミノ基、 s e c—ブトキシカルポニル Tミノ基、 t e r t—ブトキシカル ボニルァミノ基、 ペンチルォキシカルボニルァミノ基、 ネオペンチルォキシカルポ二 ルァミノ基、 へキシルォキシカルボニルァミノ基、 ィソへキシルォキシカルボニルァ ミノ基等が挙げられる。
上記式 (I ) 中の 「低級アルカノィル基」 とは、 カルポニル基に上記 「低級アルキ ル基」 が結合した基をいい、 カルボニル基に炭素数 1ないし 5個のアルキル基が結合 した基が好ましく、 例えばァセチル基、 プロピオニル基、 プチリル基、 イソプチリル 基、 バレリル基、 イソバレリル基、 ピバロイル基、 ペン夕ノィル基等が挙げられる。 上記式 (I ) ·中の 「低級アルカノィルォキシ基」 とは、 酸素原子に上記 「低級アル カノィル基」が結合した基をいい、例えばァセチルォキシ基、プロピオ二ルォキシ基、 プチリルォキシ基、 イソプチリルォキシ基、 バレリルォキシ基、 イソバレリルォキシ 基、 ビバロイルォキシ基、 ペンタノィルォキシ基等が挙げられる。
上記式 (I ) 中の 「低級アルキルチオ基」 とは、 硫黄原子に上記 「低級アルキル」 が結合した置換基をいい、 例えばメチルチオ基、 ェチルチオ基、 プチルチオ基等が 挙げられる。
「P L K」 とは、 ポロ ライク キナーゼ (p o l o l i k e k i n a s e ) を表す。
「P L K 1」 とは、 P L K (ポロ ライク キナーゼ (p o l o l i k e k i n a s e ) ) ファミリ一は P L K 1、 P L K 2、 P L K 3、 S AKから構成されるが、 その中の一つである。
「P L K 阻害剤」 とは、 ポロ ライク キナーゼ 1を阻害する薬剤である。
「その薬学的に許容される塩若しくはエステル」、 及び 「薬学的に許容できる担体 又は希釈剤」 の説明は後述する。
本明細書で用いる 「がん治療」 という用語は、 がん患者に対して、 杭がん剤を投与 することにより、 がん細胞の増殖を阻害することを意味する。 好ましくは、 かかる治 療は、 がん増殖を後退、 即ち、 測定可能ながんの大きさを減縮させることができる。. さらに好ましくは、 かかる治療は、 がんを完全に消失させる。
本明細書で用いる「がん」という用語は、固形がん及び造血器がんである。ここで、 固形がんは、 例えば、 脳腫瘍、 頭頸部がん、 食道がん、 甲状腺がん、 小細胞肺がん、 非小細胞肺がん、 乳がん、 胃がん、 胆のう *胆管がん、 肝がん、 膝がん、 結腸がん、 直腸がん、 卵巣がん、 絨毛上皮がん、 子宮体がん、 子宮頸がん、 腎盂 ·尿管がん、 膀 胱がん、 前立腺がん、 陰茎がん、 睾丸がん、 胎児性がん、 ウィルス腫瘍、 皮膚がん、 悪性黒色腫、 神経芽細胞腫、 骨肉腫、 ユーイング腫、 軟部肉腫などである; ^ 一方、 造 血器がんとしては、 例えば、 急性白血病、 慢性リンパ性白血病、 慢性骨髄性白血病、 真性多血病、 悪性リンパ腫、 多発性骨髄腫、 非ホジキンリンパ腫などである。
本明細書で用いる 「製剤」 という用語は、 経口製剤及び非経口製剤を含む。 経口製 剤としては、 例えば、 錠剤、 カプセル剤、 散剤、 顆粒剤などであり、 好ましくは、 錠 剤、 カプセル剤などである。 一方、 非経口製剤としては、 例えば、 溶液若しくは懸濁 液等の殺菌した液状の製剤、 具体的には、 注射剤、 点滴剤などであり、 好ましくは、 静脈内注射剤又は静脈内点滴剤である。
本明細書で用いる「組み合わせ製剤」という用語は、治療において同時に、別々に、 又は順次に投与するための 2個以上の製剤からなるものをいい、 それらが、 いわゆる キット型の製剤又は医薬組成物になっていてもよい。 上述したような、 がん治療にお いて用いる 2つの別個の製剤からなる組み合わせ製剤に対して、 さらに 1個以上の製 剤を組み合わせたものも、 上記 「組み合わせ製剤」 に含まれる。
上述した 2個の別個の製剤に対して、薬学的に許容し得る担体又は希釈剤と一緒に、 杭がん性アルキル化剤、杭がん性代謝拮抗剤、抗がん性抗生物質、植物由来抗がん剤、 杭がん性白金配位化合物、 杭がん性カンプトテシン誘導体、 杭がん性チロシンキナー ゼ阻害剤、 モノクローナル抗体、 インターフェロン、 生物学的応答調節剤、 及びその 他杭がん剤 (ここで、 各杭がん剤の定義は、 前記と同じである。) からなる群から選 択される杭がん剤少なくとも 1種以上又はその薬学的に許容し得る塩若しくはエス テルを含む製剤 1個以上を、 さらに組み合わせることもできる。 この場合、 さらに加 えられた 1個以上の製剤は、 上記 2つの別個の製剤と、 同時に、 別個に、 又は順次に 投与されてもよい。 例えば、 3つの製剤からなる組み合わせ製剤としては、 上記一般 式 (I ) で表される化合物を含む製剤、 5—フルォロウラシルを含む製剤、 及びロイ
コポリンを含む製剤が挙げられる。
ここで、 上記の組み合わせ製剤において、 2個の別個の製剤の両方が、 経口製剤又 は非経口製剤であってもよく、 また、 一方が経口製剤であり、 もう一方が非経口製剤 (注射剤又は点滴剤) であってもよい。
本発明に係る 「製剤」 においては、 通常、 本発明に係る化合物の治療上の有効量を 薬学的に許容し得る担体又は希釈剤と共に含んでいてもよい。 この製剤化技術は、 当 該技術分野の当業者にとって技術常識であると考えられ、 よく知られている。好まし くは、 薬学的に許容し得る担体又は希釈剤と一緒に、 当業者によく知られている多く の方法で経口製剤用、 静脈内点滴用又は注射用に製剤化することができる。
本明細書で用いる 「投与」 という用語は、 本発明に係る組み合わせ製剤を用いる場 合、 非経口投与及び Z又は経口投与を意味する。 即ち、 組み合わせ製剤を投与する場 合、両方とも非経口投与でもよく、一方が非経口投与でもう一方が経口投与でもよく、 また、 両方とも経口投与でもよい。 ここで、 「非経口投与」 は、 例えば、 静脈内投与、 皮下投与、 筋肉内投与などであり、 好ましくは、 静脈内投与である。 また、 3個以上 の製剤が組み合わされて投与される場合でも、 少なくとも 1個の製剤は、非経口投与 されてもよく、 好ましくは、 静脈内投与されてもよく、 さらに好ましくは静脈内点滴 又は静脈内注射されてもよい。 また、 3個以上の製剤が組み合わされて投与される場 合において、 いずれも経口製剤又は非経口製剤であってもよい。
なお、 本発明の実施において、 上記一般式 (I ) で示される化合物は、 他の杭がん 剤ど同時に投与してもよい。 また、 上記一般式 (I ) で示される化合物を投与してか ら連続して他の杭がん剤を投与してもよいし、他の抗がん剤を投与してから上記一般 式 (I ) で示される化合物を連続して投与してもよい。 さらに、 上記一般式 (I ) で 示される化合物を投与し、 時間をおいて別々に他の杭がん剤を投与してもよいし、 他 の抗がん剤を投与し、 時間を置いて別々に上記一般式 (I ) で示される化合物を投与 してもよい。 かかる投与順序及び投与間隔は、 用いられる上記一般式 (I ) で示され る化合物を含む製剤、 及びそれと併用される杭がん剤を含む製剤、 治療すべきがん細 胞の種類、 患者の状態などに応じて、 当業者が適宜選択することができる。
また、 本明細書で用いる 「同時に」 とは、 ほぼ同じ時間に治療に使用することをい レ 「別々に」 とは、 異なった時間に別々に治療に使用することをいい、 例えば、 1 日目に 1つの薬剤、 2日目にもう 1つの薬剤を治療に使用するような場合をいう。「順 次に」とは、順番に従って使用することをいい、例えば、最初に 1つの薬剤を使用し、 次いで、 決められた時間後に、 他の薬剤を治療に使用するような場合をいう。
本明細書で用いる 「杭がん性アルキル化剤」 は、 杭がん活性を有するアルキル化剤 を意味し、 ここで、 「アルキル化剤」 とは、 一般に、 有機化合物の水素原子をアルキ ル基で置換するアルキル化反応において、 アルキル基を与えるものをいう。 「抗がん 性アルキル化剤」 は、 例えば、 ナイトロジェン マスタード N—ォキシド、 シクロ ホスフアミド、ィホスフアサミド、メルファラン、ブスルファン、ミトブロニトール、 力ルポコン、 チォテパ、 ラニムスチン、 二ムスチン、 テモゾロミド又はカルムスチン
などである。
本明細書で用いる 「杭がん性代謝拮抗物質」 は、 杭がん活性を有する代謝拮抗物質 をいい、 ここで、 「代謝拮抗物質」 とは、 広義には、 生体にとって重要な代謝物 (ビ タミン、 補酵素、 アミノ酸、 糖類など) と構造上又は機能上類似しているために、 正 常な物質代謝を行わなくさせる物質や、電子伝達系を阻害することによって高工ネル ギ一中間体をつくれなくさせる物質を包含する。 「抗がん性代謝拮抗物質」 は、 例え ば、 メトトレキサ一ト、 6—メルカプトプリンリポシド、 メルカプトプリン、 5—フ ルォロウラシル、 テガフール、 ドキシフルリジン、 カルモフール、 シ夕ラビン、 シ夕 ラビンォクホスファー卜、 エノシ夕ビン、 S— 1、 ゲムシ夕ビン、 フルダラビン又は. ぺメトレクスド ジソディウムなどである。
本明細書で用いる 「抗がん性抗生物質」 は、 杭がん活性を有する抗生物質をいい、 ここで、 「抗生物質」 とは、 微生物によってつくられ、 微生物その他の生物細胞の発 育その他の機能を阻害する物質を包含する。 「杭がん性抗生物質」 は、 例えば、 ァク チノマイシン D、 ドキソルビシン、 ダウソルビシン、 ネオカルチノス夕チン、 ブレオ マイシン、 ぺプロマイシン、 マイトマイシン C、 アクラルビシン、 ピラルビシン、 X ピルビシン、 ジノス夕チンスチマラマ一、 イダルビシン、 シロリムス又はバルルビシ ンなどである。
本明細書で用いる 「植物由来杭がん剤」 は、 植物を起源として見いだされた抗がん 活性を有する化合物であるか、 或いは、 その化合物を化学修飾を加えた化合物を包含 する。 「植物由来杭がん剤」 は、 例えば、 ビンクリスチン、 ビンプラスチン、 ビンデ シン、 エトポシド、 ソブゾキサン、 ドセタキセル、 パクリタキセル、 ビノレルビンな どである。
本明細書で用いる 「杭がん性カンプトテシン誘導体」 は、 カンプトテシン自身を含 み、 構造的にカンプトテシンに関連するがん細胞増殖阻害性化合物を意味する。 「抗 がん性カンプトテシン誘導体」 としては、 特に限定されないが、 カンプトテシン、 1 0—ヒドロキシカンプトテシン、 卜ポテカン、 イリノテカン、 9—ァミノカンプ卜テ シンなどが挙げられる。 なお、 イリノテカンは、 生体内で代謝されて S N— 3 8とし て杭がん作用を示す。 カンプトテシン誘導体は、 作用機構および活性はほぼカンプト テシンと同様と考えられる (新田 他、 癌と化学療法、. 1 4, 8 5 0 - 8 5 7 ( 1 9 8 7 ) など)。
本明細書で用いる 「抗がん性白金配位化合物」 は、 杭がん活性を有する白金配位化 合物をいい、 ここで、 「白金配位化合物」 は、 イオンの形態で白金を提供する白金配 位化合物を意味する。 好ましい白金化合物としては、 シスブラチン;シスージアンミ ンジアコ白金 (I I) —イオン;クロ口 (ジエチレントリアミン) 一白金 (I I) クロリ ド;ジクロロ (エチレンジァミン) 一白金 (I I) ;ジアンミン (1 , 1—シクロブ夕 ンジカルポキシラト) 白金 (I I) (カルポプラチン);スピロブラチン;ィプロプラチ ン;ジアンミン (2—ェチルマロナト) —白金 (I I ) ;エチレンジァミンマロナ卜白 金 (I I);アクア (1, 2—ジアミノジシクロへキサン) スルファト白金 (I I);ァク
ァ (1, 2—ジアミノジシクロへキサン) マロナト白金 (II); (1, 2—ジアミノシ クロへキサン) マロナ卜白金 (II) ; (4一力ルポキシフタラト) (1, 2—ジァミノ シクロへキサン) 白金 (II); (1, 2—ジアミノシクロへキサン) 一 (イソシ卜ラト) 白金 (II) ;.(1, 2—ジアミノシクロへキサン) ォキサラト白金 (II) ;オルマブラ チン;テトラプラチン;カルポプラチン;ネダプラチン及びォキザリブラチンである。 また、 本明細書で挙げた他の杭がん性白金配位化合物は、 公知であり、 商業的に入手 可能であり、 及び Z又は、 慣用技術によって当業者が製造することができる。
本明細書で用いる 「抗がん性チロシンキナーゼ阻害剤」 とは、 抗がん活性を有する チロシンキナーゼ阻害剤をいい、 ここで、 「チロシンキナーゼ阻害剤」 とは、 ATP. のアーリン酸基をタンパク質の特定のチロシンのヒドロキシル基に転移する 「チロシ ンキナーゼ」 を阻害する化学物質をいう。 「抗がん性チロシンキナーゼ阻害剤」 とし ては、 ゲフイチニブ、 イマチニブ、 エル口チニブなどが挙げられる。
本明細書で用いる 「モノクローナル抗体」 は、 単クローン性抗体ともいわれ、 単一 クローンの抗体産生細胞が産生する抗体をいい、 例えば、 セツキシマブ、 ベバシズマ ブ、 リツキシマブ、 ァレムッズマブ、 トラスッズマブなどが挙げられる。
本明細書で用いる 「インターフェロン」 とは、 抗がん活性を有するインタ一フエ口 ンをいい、 一般に、 ウィルス感染に際して、 ほとんどすべての動物細胞が生産 ·分泌 する分子量約 2万の糖タンパク質であり、 ウィルス増殖抑制のみならず、 細胞 (特に 腫瘍細胞) の増殖抑制や、 ナチュラルキラ一活性の増強をはじめ多様な免疫エフェク ター作用があり、 サイト力インの 1種と位置づけられる。 「インターフェロン」 とし ては、 例えば、 インターフェロン α、 インタ一フエロン α— 2 a、 イン夕一フエロン α— 2b、 インターフェロン j6、 インターフェロンァ一 1 a、 イン夕一フエロン τ一 η 1などが挙げられる。
本明細書で甩いる 「生物学的応答調節剤」 とは、 いわゆるバイオロジカル ·レスポ ンス ·モディフアイャ一 (b i o l o g i c a l r e s pon s e mod i f i e r ; BRM) であり、 一般に、 生体のもつ防御機構や組織細胞の生存、 増殖、 ま たは分化など生物学的反応を調節することによって、腫瘍や感染あるいはその他の疾 病に対して、個体に利する方向にもっていくことを目的とする物質や薬剤の総称をい う。 「生物学的応答調節剤」 としては、 例えば、 クレス.チン、 レンチナン、 シゾフィ ラン、 ピシバニ一ル、 ウベ二メクスなどが挙げられる。
本明細書で用いる 「その他杭がん剤」 とは、 杭がん活性を有する上記のいずれにも 属しない抗がん剤をいう。 「その他抗がん剤」 としては、 ミトキサントロン、 Lーァ スパラギナーゼ、 プロカルバジン、 ダカルバジン、 ヒドロキシカルバミド、 ペントス 夕チン、 トレチノイン、 ァレファセブト、 ダルべポェチン アルファ、 アナストロゾ ール、 ェキセムスタン、 ビカルタミド、 リュープロレリン、 フルタミド、 フルべス卜 ラント、 ぺガプタニブ ォクタソディウム、 デニリューキン ジフティトクス、 アル デスリューキン、 チロトロピン アルファ、 ァルセニック トリオキシド、 ポルテゾ ミブ、 力ぺシタビン、 ゴセレリン、 などが挙げられる。
上記「杭がん性アルキル化剤」、「杭がん性代謝拮抗物質」、「抗がん性抗生物質」、「植 物由来杭がん剤」、「抗がん性白金配位化合物」、「抗がん性カンプトテシン誘導体」、「抗 がん性チロシンキナーゼ阻害剤」、 「モノクローナル抗体」、 「インタ一フエロン」、 「生 物学的応答調節剤」、 及び 「その他杭がん剤」 は、 いずれも公知であり、 商業的に入 手可能であり、 或いは、 それ自体公知の方法ないし周知 ·慣用的な方法によって当業 者が製造することができる。 また、 ゲフイチニブの製造方法は、 例えば、 米国特許第 5, 7 7 0 , 5 9 9号明細書に;セツキシマブの製造方法は、 例えば、 国際公開 WO 9 6 / 4 0 2 1 0号パンフレツ卜に; ベパシズマブの製造方法は、 例えば、 国際公 開 W0 9 4 Z 1 0 2 0 2号パンフレットに; ォキザリブラチンの製造方法は、 例え. ば、 米国特許第 5, 4 2 0 , 3 1 9号明細書、 同第 5 , 9 5 9 , 1 3 3号明細書に; ゲムシ夕ビンの製造方法は、 例えば、 米国特許第 5 , 4 3 4 , 2 5 4号明細書、 同第 5 , 2 2 3 , 6 0 8号明細書に; カンプトテシンの製造方法は、 米国特許第 5 , 1 6 2 , 5 3 2号明細書、 同第 5, 2 4 7 , 0 8 9号明細書、 同第 5 , 1 9 1 , 0 8 2 号明細書、 同第 5 , 2 0 0 , 5 2 4号明細書、 同第 5, 2 4 3 , 0 5 0号明細書、 同 第 5, 3 2 1 , 1 4 0号明細書に; ィリノテカンの製造方法は、 例えば、 米国特許 第 4, 6 0 4 , 4 6 3号明細書に; トポテカンの製造方法は、 例えば、 米国特許第 5 , 7 3 4 , 0 5 6号明細書に; テモゾ口ミドの製造方法は、 例えば、 日本特許公 報平 4一 5 0 2 9号明細書に; リツキシマブの製造方法は、 日本公表特許公報平 2 - 5 0 3 1 4 3号明細書に、 それぞれ記載されている。
上記の抗がん性アルキル化剤については、 例えば、 ナイトロジェンマスタード N 一才キシドは、 ナイトロミン (商品名) として三菱ゥエルファーマから; シクロホ スフアミドは、 エンドキサン (商品名) として塩野義製薬から; ィホスフアサミド は、ィフォミド (商品名) として塩野義製薬から; メルファランは、 ァルケラン(商 品名) としてダラクソスミスクラインから; ブス レファンは、 マブリン (商品名) として武田薬品から; ミトブロニトールは、 ミエブロール (商品名) として杏林製 薬から; カルボコンは、 エスキノン (商品名) として三共から; チォテパは、 テ スパミン (商品名) として住友製薬から; ラニムスチンは、 シメリン (商品名) と して三菱ゥエルファーマから; 及び二ムスチンは、 ニダラン (商品名) として三共 から; テモゾロミドは、 テモダール (商品名) としてシエリングから; 及びカル ムスチンは、 グリアデル ゥオファー (商品名) としてグリフオードから、 それぞれ 市販で入手することができる。
上記の抗がん性代謝拮抗剤については、 例えば、 メトトレキサートは、 メトトレキ セート (商品名) として武田薬品から; 6—メルカプトプリンリポシドは、 チオイ ノシ (商品名) としてアベンテイスから; メルカプトプリンは、 ロイケリン (商品 名) として武田薬品から; 5—フルォロウラシルは、 5—F U (商品名) として協 和発酵から; テガフールは、 フトラフール (商品名) として大鵬薬品から; ドキ シフルリジンは、 フルツロン (商品名) として日本ロシュから; カルモフールは、 ャマフール(商品名) として山之内製薬から; シ夕ラビンは、 シロサイド (商品名)
として日本新薬から; シ夕ラビンォクホスフアートは、 ストラシド (商品名) とし て日本化薬から; エノシ夕ビンは、 サンラビン (商品名) として旭化成から; S — 1は、 T S— 1 (商品名) として大鵬薬品から; ゲムシ夕ビンは、 ゲザール (商 品名) としてリリーから; フルダラビンは、 フルダラ (商品名) として日本シエー リングから; 及びぺメトレクスド ジソディウムは、 ァリム夕 (商品名) としてィ
—ライリリーから、 それぞれ市販で入手することができる。
上記の抗がん性抗生物質としては、例えば、ァクチノマイシン Dは、コスメゲン(商 品名) として万有製薬から; ドキソルビシンは、 アドリアシン (商品名) として協 和発酵から; ダウノルビシンは、 ダウノマイシン (商品名) として明治製菓から;. ネオカルチノスタチンは、 ネオカルチノス夕チン (商品名) として山之内製薬から; ブレオマイシンは、 プレオ (商品名) として日本化薬から; ペブロマイシンは、 ぺ プロ (商品名) として日本化薬から; マイトマイシン Cは、 マイトマイシン (商品 名) として協和発酵から; アクラルビシンは、 ァクラシノン (商品名) として山之 内製薬から; ピラルビシンは、 ピノルビン (商品名) として日本化薬から; ェピ ルビシンは、 フアルモルビシン (商品名) としてフアルマシアから; ジノス夕チン スチマラマ一は、 スマンクス (商品名) として山之内製薬から; イダルビシンは、 イダマイシン (商品名) としてフアルマシアから; シロリムスは、 ラパムン (商品 名) としてワイスから; 及びバルルビシンは、 バルスター (商品名) としてアンス ラ ファーマシューティカルからそれぞれ市販で入手することができる。
上記の植物由来抗がん剤としては、 例えば、 ビンクリスチンは、 オンコビン (商品 名) として塩野義製薬から; ビンブラスチンは、 ビンブラスチン (商品名) として 杏林製薬から; ビンデシンは、 フィルデシン(商品名) として塩野義製薬から; ェ トポシドは、 ラステツト (商品名) として日本化薬から; ソブゾキサンは、 ペラ ゾリン (商品名) として全薬工業から; ドセタキセルは、 夕キソテール (商品名) としてアベンテイスから; パクリタキセルは、 タキソール (商品名) としてブリス トルから; 及びビノレルビンは、 ナベルビン (商品名) として協和発酵から、 それ ぞれ市販で入手することができる。
上記の抗がん性白金配位化合物としては、 例えば、 シスブラチンは、 ランダ (商品 名) として日本化薬から; カルポプラチンはパラプラチン (商品名) としてブリス トルから; ネダプラチンは、 ァクブラ (商品名) として塩野義製薬から;及びォキ ザリブラチンは、 エロキサチン (商品名) としてサノフィから、 それぞれ市販で入手 することができる。
上記の抗がん性カンプ卜テシン誘導体としては、 例えば、 イリノテカンは、 カンプ ト (商品名) としてヤクルトから; トポテカンは、 ハイカムチン (商品名) として ダラクソスミスクラインから; 及び力ンプトテシンは、 米国アルドリツチケミカル などから、 それぞれ市販で入手することができる。 .
上記の杭がん性チロシンキナーゼ阻害剤としては、 例えば、 ゲフイチニブは、 ィレ ッサ (商品名) としてァストラゼネ力から; イマチニブは、 ダリベック (商品名)
としてノバルテイスから; 及びエル口チニブは、 夕ルセバ (商品名) としてォーェ スアイ ファーマシューティカルから、 それぞれ市販で入手することができる。
上記のモノクローナル抗体としては、例えば、セツキシマプは、ェルビタックス(商 品名)としてブリストルマイヤーズスクイブから; べバシズマブは、ァバスチン(商 品名) としてジエネンテックから; リツキシマブは、 リツキサン (商品名) として バイオジェンから; ァレムッズマブは、 カンパス (商品名) としてベルレックスか ら; 及びトラスッズマブは、 ハーセプチン (商品名) として中外製薬から、 それぞ れ市販で入手することができる。
上記のインターフェロンとしては、 例えば、 インタ一フエロン aは、 スミフエロン (商品名) として住友製薬から; インターフェロン a— 2 aは、 カンフエロン一A (商品名) として武田薬品から; インターフェロン α— 2 bは、 イントロン A (商 品名) としてシエリングプラウから; インターフェロン; 6は、 I F N /3 (商品名) として持田製薬から; インターフェロンァー 1 aは、ィムノマックスーァ (商品名) として塩野義製薬から; 及びインターフェロンァー n 1は、 ォガンマ (商品名) と して大塚製薬から、 それぞれ市販で入手することができる。
上記の生物学的応答調節剤としては、例えば、 クレスチンは、 クレスチン(商品名) として三共から; レンチナンは、 レンチナン (商品名) としてアベンテイスから; シゾフィランは、 ソニフイラン (商品名) として科研製薬から; ピシバニ一ルは、 ピシバニ一ル(商品名) として中外製薬から; 及びウベ二メクスは、べスタチン(商 品名) として日本化薬から、 それぞれ市販で入手することができる。
上記のその他抗がん剤としては、 例えば、 ミトキサントロンは、 ノバントロン (商 品名) として日本ワイスレダリーから; Lーァスパラギナ一ゼは、 ロイナーゼ (商 品名) として協和発酵から; プロカルバジンは、 ナツラン (商品名) として日本口 シュから; ダカルバジンは、 ダカルバジン (商品名) として協和発酵から ; ヒ ドロキシカルバミドは、 ハイドレア (商品名) としてブリストルから; ペントス夕 チンは、 コフォリン (商品名) として化学及び血清療法研究所から; トレチノイン は、べサノイド(商品名) として日本ロシュから; ァレファセブトは、ァメビブ(商 品名) としてバイオジェンから; ダルべポェチン アルファは、 ァラネスプ (商品 名) としてアムジェンから; アナストロゾールは、 アリミデックス (商品名) とし てァストラゼネ力から; ェキセメスタンは、 ァロマシン (商品名) としてフアイザ 一から; ビカル夕ミドは、カソデックス (商品名) としてァストラゼネ力から ; リ ユープロレリンは、 リュープリン (商品名) として武田薬品から ; フル夕ミドは、 ユーレキシン (商品名) としてシエリングプラウから; フルべストラントは、 ファ スロデックス (商品名) としてァストラゼネ力から; ぺガプ夕ニブ ォク夕ソディ ゥムは、 マクゲン (商品名) としてギリードサイエンスから; デニリューキン ジ フティトクスは、 オンタック (商品名) としてリガンドから; アルデスリューキン は、 プロリューキン (商品名) としてキロンから; チロトロピン アルファは、 チ ロゲン (商品名) としてゲンザィムから; ァルセニック 卜リオキシドは、 卜リセ
ノックス (商品名) としてセル セラピューテイクスから; ポルテゾミブは、 ベル ケィド (商品名) としてミレニウムから; 力ぺシタビンは、 ゼロダ (商品名) とし てロシュから; 及びゴセレリンは、 ゾラデックス (商品名) としてァストラゼネカ から、 それぞれ市販で入手することができる。
本明細書で用いる 「杭がん剤」 とは、 上記 「杭がん性アルキル化剤」、 「抗がん性代 謝拮抗物質」、 「杭がん性抗生物質」、 「植物由来杭がん剤」、 「抗がん性白金配位化合物 」、 「抗がん性カンプトテシン誘導体」、 「抗がん性チロシンキナ一ゼ阻害剤」、 「モノク ローナル抗体」、 「インタ一フエロン」、 「生物学的応答調節剤」、 及び 「その他杭がん 剤」 から選ばれる杭がん剤をいう。 上記式 ( I ) で示される化合物の実施の形態についてさらに詳しく説明 する。
R ,及び R 2 は、 同一若しくは異なって、 水素原子; ハロゲン原子、 ヒドロキシ 基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル基、 アミノスルホニル基、 ィミノ 基、 低級アルキルアミノ基、 ジ低級アルキルアミノ基、 低級アルキルスルホニル基、 低級アルキルスルホニルァミノ基、低級アルコキシ基、低級アルコキシカルポニル基、 低級アルコキシ力ルポニルァミノ基、 及び低級アルカノィル基、 低級アルカノィルォ キシ基、 低級アルキルチオ基、 及びカルボキシル基 (以下、 これをぐ置換基群ひ >と いう。) から選択される置換基; <置換基群 α >から選択される置換基で 1個若し くは 2個以上置換されてもよい低級アルキル基; 又は、 置換されてもよい炭素数 3 個ないし 6個のシクロアルキル基である。
R x は、 好ましくは、 <置換基群 >から選択される置換基、 ぐ置換基群 α >から 選択される置換基で 1個若しくは 2個以上置換されてもよい低級アルキル基、 又は、 シクロプロピル基であり、 <置換基群ひ >が、 ハロゲン原子である。
R a は、 さらに好ましくは、 フッ素原子で 1個ないし 3個置換されていてもよい炭 素数 1個ないし 2個の低級アルキル基、 シクロプロピル基又は塩素原子である。
R , は、 とりわけ好ましくは、 メチル基、 ェチル基、 二フッ化メチル基、 Sフッ化 メチル基、 シクロプロピル基、 又は塩素原子である。
R , は、 とりわけ一層好ましくは、 ェチル基、 ニフツ化メチル基、 三フッ化メチル 基、 シクロプロピル基、 又は塩素原子である。
R 2 は、 好ましくは、 水素原子である。
R 3及び R 4は、 同一若しくは異なって、
a ) 水素原子;
b ) N R a R b (ここで、 R a及び R bは、 同一若しくは異なって、 水素原子、 置換 されていてもよい低級アルキル基、 置換されていてもよいべンジル基、 又は置換され ていてもよいシクロアルキル基である。) で置換される低級アルキル基;
c ) ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル 基、 アミノスルホニル基、 イミノ基、 低級アルキルスルホニル基、 低級アルキルスル
ホニルァミノ基、 低級アルコキシ基、 低級アルコキシカルボニル基、 低級アルコキシ カルボニルァミノ基、 低級アルカノィル基、 低級アルカノィルォキシ基、 低級アルキ ルチオ基、 力ルポキシル基、 及びベンジル基 (以下、 これをぐ置換基群 j6 >という。) から選択される置換基;
d )ぐ置換基群^ >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルキル基;
e ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルケニル基;
f ) フエニル基;
g ) フエニル基で置換される低級アルキル基;
h ) 4員ないし 7員の脂肪族複素環基;
i ) 4員ないし 7員の脂肪族複素環基で置換される低級アルキル基;
j ) 5員ないし 6員の芳香族複素環基;又は、
k ) 5員ないし 6員の芳香族複素環基で置換される低級アルキル基 (ここで、 前記 フエニル基、 脂肪族複素環基、 及び芳香族複素環基は、 それぞれ独立して、 下記 1 ) ないし 4 ) : '
1 ) 低級アルキル基;
2 ) <置換基群) 6 >から選択される置換基;
3 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β〉から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、また、前記脂肪族複素環基は、不飽和結合を含んでいてもよく、さらに、前記 b )、 g )、 i )、 及び k ) の低級アルキル基は、 適宜置換されてもよい。) であるか、 或い は、
R 3及び R 4は、 一緒になつて、 4員ないし 7員の脂肪族複素環基 (ここで、 前記 脂肪族複素環基は、 下記 1 ) ないし 4 ) :
1 ) 低級アルキル基; ,
2 ) ぐ置換基群 jS >から選択される置換基;
3 )ぐ置換基群 >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) を形成する。
R 3及び R 4 は、 好ましくは、 同一若しくは異なって、
a ) 水素原子;
b ) N R a R b (ここで、 R a及び R bは、 同一若しくは異なって、 水素原子、 低級 アルキル基、 ベンジル基であり、 又はシクロアルキル基であり、 ここで、 前記べンジ ル基及びシクロアルキル基は、 それぞれ独立して、 下記 1ないし 3 ) :
1 ) 低敏アルキル基;
2 ) <置換基群 ^ >から選択される置換基;及び
3 )ぐ置換基群 β〉から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記シクロアルキル基は、 不飽和結合を含んでいてもよい。) で置換され. る低級アルキル基;
c ) ぐ置換基群 jS >から選択される置換基;
d )ぐ置換基群 ;6〉から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルキル基;
e ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルケニル基;
f ) フエニル基; '
g ) フエニル基で置換される低級アルキル基;
h ) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基;
i ) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基で置換される低級アルキル基; .
j ) ピロリル基、 イミダゾリル基、 ピラゾリル基、 ピリジル基、 ピラジニル基、 及 びピリミジニル基から選択される 5員ないし 6員の芳香族複素環基;又は、
k ) ピロリル基、 イミダゾリル基、 ピラゾリル基、 ピリジル基、 ピラジニル基、 及 びピリミジニル基から選択される 5員ないし 6員の芳香族複素環基で置換される低 級アルキル基 (ここで、 前記フエニル基、 脂肪族複素環基、 及び芳香族複素環基は、 それぞれ独立して、 下記 1 ) ないし 4 ):
1 ) 低級アルキル基;
2 ) ぐ置換基群 i8〉から選択される置換基;
3 ) <置換基群 /3〉から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) であるか; 或いは、 R 3及び R 4が、 一緒になつて、 ァゼチジニル基、 ピロリジニル基、 ピぺ リジニル基、 及びピペラジニル基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、 前記脂肪族複素環基は、 下記 1 ) ないし 4 ) :
1) 低級アルキル基;
2) <置換基群 3>から選択される置換基;
3 )ぐ置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル碁; 及び
4) <置換基群 i8>から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) を形成する。
R3及び R4 に関し、さらに好ましくは、 R3及び R4 の一方が、水素原子であり、 . 尺2'及び1^3 のもう一方が、
a) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 低級 アルキル基、 ベンジル基、 又は炭素数 3個ないし 6個のシクロアルキル基であり、 こ こで、 前記シクロアルキル基は、 下記 1ないし 3 ) :
1) 低級アルキル基;
2) ぐ置換基群 /3>から選択される置換基;及び
3 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記シクロアルキル基は、 不飽和結合を含んでいてもよい。) で置換され る低級アルキル基;
b) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基; 又は、
c) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基で置換される低級アルキル基 (ここで、 前記脂肪族複素環基は、 下記 1) ないし 4) :
1 ) 低級アルキル基;
2) ぐ置換基群 i6>から選択される置換基;
3 )ぐ置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4) <置換基群 |6>から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ レ^) である。
R3及び R4 に関し、 とりわけ好ましくは、 R3及び R4の一方が、 水素原子であ り、 R3及び R4 のもう一方が、
a) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 低級 アルキル基、 又は炭素数 5個ないし 6個のシクロアルキル基であり、 前記シクロアル キル基は、 下記 1ないし 3) :
1 ) 低級アルキル基;
2 ) ぐ置換基群 ]8 >から選択される置換基;及び
3 ) <置換基群 β〉から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル棊:
から選択される同一若しくは異なる置換塞で 1個若しくは 2個以上置換されてもよ レ^ ) で置換される低級アルキル基;又は
b ) ァゼチジニル基、 ピロリジニル基及びピペリジニル基から選択される 4員ない し 6員の脂肪族複素環基 (ここで、 前記脂肪族複素環基は、 下記 1 ) ないし 3 ) : 1 ) 低級アルキル基;
2 ) <置換基群; S >から選択される置換基;及び
3 )く置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ レ^ ) である。
R 3及び R 4 に関する <置換基群 ;6〉は、 好ましくは、 ハロゲン原子、 ヒドロキシ 基、 アミノ基、 低級アルキルスルホニル基、 及び低級アルコキシ基、 であ'る。
R 3 及ぴ R 4 に関し、 特に好ましくは、 R 3及び R 4 の一方が、 水素原子であり、 R 3及び R 4のもう一方が、 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換若しくは N , N—ジ置換されるァミノ低級アルキル基 (ここで、 前記低級ァ ルキル基は、 炭素数 1ないし 3個の直鎖状又は分岐状のアルキル基である。)、 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピペリジニル基、炭 素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピロリジニル 基、炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるァゼチジ ニル基、 又は炭素数 5個ないし 6個のシクロアルキル基である (ここで、 前記ピペリ ジニル基、 ピロリジニル基、 及びァゼチジニル基は、 それぞれ独立して、 炭素数 1な いし 3個の直鎖状又は分岐状のアルキル基で、 さらに置換されていてもよく、 また、 前記シクロアルキル基は、 ヒドロキシ基で置換されてもよい炭素数 1個ないし 3個の アルキル基で置換されていてもよい。)。 ここで、 前記ピペリジニル基は、 N—置換ピ ペリジン一 3—ィル、 N—置換ピぺリジン一 4ーィル どが好ましく ; 前記ピロリ ジニル基は、 N—置換ピロリジン一 2—ィル、 N—置換ピロリジン一 3—ィルなどが 好ましく、 N—置換ピロリジン一 2—ィルがより好ましく; 前記ァゼチジニル基は、 N—置換ァゼチジニルー 3—ィルなどが好ましく ; 前記シクロアルキル基は、 シク 口ペンチル基、 シクロへキシル基が好ましく、 シクロペンチル基がより好ましい。
R 3及び R 4 に関し、 特に一層好ましくは、 R 3及び R 4の一方が、 水素原子であ り、 R 3及び R 4のもう一方が、 ジメチルァミノ基、 イソプロピルアミノ基、 1, 1 ージメチルプロピルアミノ基、 又は t一プチルァミノ基で置換される、 炭素数 1ない し 3個の直鎖状又は分岐状のアルキル基; 炭素数 1ないし 5個の直鎖状又は分岐状 のアルキル基で N—置換されるピペリジニル基; 炭素数 1ないし 5個の直鎖状又は
分岐状のアルキル基で N—置換されるピロリジニル基; 炭素数 1ないし 5個の直鎖 状又は分岐状のアルキル基で N—置換されるァゼチジニル基; 或いは、 メチル基若 しくはヒドロキシメチル基で置換されてもよいシクロペンチル基であり (ここで、 前 記ピペリジ^ル基、 ピロリジニル基、 及びァゼチジニル基は、 炭素数 1ないし 3個の 直鎖状又は分岐状のアルキル基で、 さらに置換されていてもよい。) である。
R 5 は、 水素原子、 シァノ基、 ハロゲン原子、 又は低級アルキル基である。
R 5 は、 好ましくは、 水素原子、 シァノ基、 ハロゲン原子、 又はメチル基であり ; より好ましくは、シァノ基、ハロゲン原子、又はメチル基であり; 特に好ましくは、 シァノ基、 フッ素原子、 又はメチル基であり
Rェないし R 5に関して、 P L K 1阻害作用に基づく細胞増殖抑制作用の観点から、 下記 (A) の場合が好ましく、 下記 (B) の場合がより好ましく、 下記 (C ) がより 一層好ましい。
(A) 力 フッ素原子で 1個ないし 3個置換されていてもよい炭素数 1個な いし 2個の低級アルキル基、 シクロプロピル基又はハロゲン原子であり ;
R 2が、 水素原子であり ;
R 3 及び R 4 の一方が、 水素原子であり、 R 3 及ぴ R 4 のもう一方が、 炭素数 1な いし 5個の直鎖状又は分岐状のアルキル基で N—置換若しくは N, N—ジ置換される ァミノ低級アルキル基 (ここで、 前記低級アルキル基は、 炭素数 1ないし 3個の直鎖 状又は分岐状のアルキル基である。)、炭素数 1ないし 5個の直鎖状又は分岐状のアル キル基で N—置換されるピペリジニル基、炭素数 1ないし 5個の直鎖状又は分岐状の アルキル基で N—置換されるピロリジニル基、炭素数 1ないし 5個の直鎖状又は分岐 状のアルキル基で N—置換されるァゼチジニル基、又は炭素数 5個ないし 6個のシク 口アルキル基であり (ここで、 前記ピペリジニル基、 ピロリジニル基、 及びァゼチジ ニル基は、 それぞれ独立して、 炭素数 1ないし 3個の直鎖状又は分岐状のアルキル基 で、 さらに置換されていてもよく、 また、 前記シクロアルキル基は、 ヒドロキシ基で 置換されてもよい炭素数 1個ないし 3個のアルキル基で置換されていてもよい。) ; R 5が、 シァノ基、 ハロゲン原子、 又はメチル基である場合。
( B ) 1^ 力 メチル基、 ェチル基、 二フッ化メチル基、 三フッ化メチル基、 シ クロプロピル基、 又は塩素原子であり ;
R 2が、 水素原子であり ;
R 3及ぴ1¾ 4の一方が、 水素原子であり、 R 3及び R 4 のもう一方が、 ジメチルァ ミノ基、 イソプロピルアミノ基、 1, 1ージメチルプロピルアミノ基、 又は t 一プチ ルァミノ基で置換される、 炭素数 1ないし 3個の直鎖状又は分岐状のアルキル基; 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピベリジ二 ル基; 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピロ リジニル基; 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換され るァゼチジニル基; 或いは、 メチル基若しくはヒドロキシメチル基で置換されても よいシクロペンチル基であり (ここで、 前記ピペリジニル基、 ピロリジニル基、 及び
ァゼチジニル基は、 それぞれ独立して、 炭素数 1ないし 3個の直鎖状又は分岐状のァ ルキル基で、 さらに置換されていてもよい。) ;
R 5力 シァノ基、 フッ素原子、 又はメチル基である場合。
( C) R x が、 ェチル基、 二フッ化メチル基、 Ξフッ化メチル基、 シクロプロピ ル基、 又は塩素原子であり ;
R 2力 水素原子であり ;
R 3及び R 4 の一方が、 水素原子であり、 R 3及び R 4 のもう一方が、 t一ブチル アミノメチル基、 1ーメチルー 1ージメチルアミノエチル基、 炭素数 1ないし 5個の 直鎖状又は分岐状のアルキル基で N—置換されるピペリジン— 3—ィル基、炭素数 1 . ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピペリジン一 4ーィ ル基、炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピロリ ジン一 2—ィル基、炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換 されるァゼチジニルー 3—ィル基、又はメチル基若しくはヒドロキシメチル基で置換 されてもよいシクロペンチル基であり (ここで、 前記ピぺリジン一 3—ィル基、 ピぺ リジン一 4ーィル基、 ピロリジン一 2一^ Γル基、 及びァゼチジニルー 3—ィル基は、 メチル基で、 さらに置換されていてもよい。); '
R 5力 シァノ基又はフッ素原子である場合。
<置換基群ひ >は、
ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル基、 ァ ミノスルホニル基、 イミノ基、 低級アルキルアミノ基、 ジ低級アルキルアミノ基、 低 級アルキルスルホニル基、 低級アルキルスルホニルァミノ基、 低級アルコキシ基、 低 級アルコキシカルポニル基、 低級アルコキシ力ルポニルァミノ基、 低級アルカノィル 基、低級アルカノィルォキシ基、低級アルキルチオ基、及びカルボキシル基、である。 ぐ置換基群 α >は、 好ましくは、 ハロゲン原子であり、 とりわけ好ましくは、 塩素 原子である。
ぐ置換基群 iS >は、
ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル基、 ァ ミノスルホニル基、 イミノ基、 低級アルキルスルホニル基、 低級アルキルスルホニル アミノ基、 低級アルコキシ基、 低級アルコキシカルボ二ル基、 低級アルコキシカルボ ニルァミノ基、 低級アルカノィル基、 低級アルカノィルォキシ基、 低級アルキルチオ 基、 力ルポキシル基、 及びべンジル基、 である。
<置換基群 ι6 >は、 好ましくは、 ハロゲン原子、 ヒドロキシ基、 アミノ基、 低級ァ ルキルスルホニル基、 及び低級アルコキシ基、 である。 下記部分構造中、矢印の不斉炭素原子に関して、上記式( I )で示される化合物は、 好ましくは S体である。
上記式 (I ) の化合物は、 好ましくは、
(a) 2 - [((1 S) 一 1一 {4一 [2— (t e r t—プチルァミノ) 一 1—ヒド 口キシェチル] フエ二ル} ェチル) ァミノ] ー4一 (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二卜リル (実施例 5および 6 )、
(b) (1 R) — 1— [4一 ((I S) - 1 - {[5—ブロモー 4一 (8—ェチルイ ミダゾ [1, 2 - a] ピリジン一 3—ィル) ピリミジン一 2—ィル] アミノ}ェチル) フエニル] 一 2— ( t e r t一プチルァミノ) エタノール (実施例 1 1)、
(c) 2— [((1 S) 一 1— {4- [ヒドロキシ (ピリジン一 2—ィル) メチル] フエ二ル} ェチル) ァミノ] -4- (8—メチルイミダゾ [1, 2 - a] ピリジン一
3 -ィル) ピリミジン— 5—カルボ二トリル (実施例 1 2)、 ·
(d) 4— (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— ({(1 S) ― 1一 [4一 (4ーヒドロキシー 1ーメチルビペリジン一 4一ィル) フエニル] ェチル } ァミノ) ピリミジン一 5—力ルポ二トリル (実施例 20)、
(e) 4— (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 S) — 1— {4- [ヒドロキシ (1—イソプロピルピぺリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル (実施例 26および 2 7)、
( f ) 4- (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2 - [((1 S) - 1 - {4- [ヒドロキシ (1ーメチルピペリジン一 4一ィル) メチ ル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル (実施例 36)、
(g) 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 s) 一 1一 {4一 [(1ーシクロプロピルピぺリジン一 4一^ rル) (ヒドロキシ) メチ ル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル (実施例 37)、 (h) 4一 ( 8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 S) - 1 - {4— [ヒドロキシ (1ーメチルビペリジン一 3—ィル) メチル] フエ二 ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル (実施例 41、 42、 43お よび 44)、
( i ) 2- {[(I S) - 1 - (4一 {ヒドロキシ [1一メチルピロリジン一 2—ィ ル] メチル } フエニル) ェチル] アミノ} —4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリル (実施例 50, 5 1, 52お よび 53)、
( j ) 2— [((I S) — 1— {4- [2- (t e r t一プチルァミノ) 一 1ーヒド
口キシェチル] フエ二ル} ェチル) ァミノ] 一 4ー (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル(実施例 9および 61)、
(k) 2 - [((1 S) 一 1一 {4一 [2 - (t e r t—ブチルァミノ) 一 1ーヒド 口キシェチル] フエ二ル} ェチル) ァミノ] 一 4一 [8- (ジフルォロメチル) イミ ダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—力ルポ二トリル (実施例 10および 60)、
(1) 4一 (8—シクロプロピルイミダゾ [1, 2 - a] ピリジン一 3—ィル) ― 2— [((1 S) - 1 - {4一 [(1, 2—ジメチルピロリジン一 2—ィル) (ヒドロキ シ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン— 5—力ルポ二トリル (実施例. 63、 64、 65および 66)、
(m) (1 S) - 2 - ( t e r t一プチルァミノ) 一 1一 [4— ((I S) — 1一 {[4 一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—フルォロピリミ ジン一 2—ィル] アミノ} ェチル) フエニル] エタノール (実施例 67)、
(n) (I S) — 1— [4- ((I S) — 1— {[4- (8—クロロイミダゾ [1, 2 - a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチ ル) フエニル] 一 2— [(1, 1ージメチルプロピル) ァミノ] エタノール (実施例 68)、
(o) (1 S) — 1一 [4一 ((1 S) 一 1一 {[4一 (8—クロロイミダゾ [1, 2 - a] ピリジン一 3—ィル) —5—フルォロピリミジン一 2—ィル] アミノ} ェチ ル) フエニル] 一 2— [(1ーメチルシクロペンチル) ァミノ] エタノール (実施例 71)、
(p) (1 S) - 2 - ( t e r t一プチルァミノ) 一 1一 [4一 ((1 S) 一 1一 {[4 一 (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—メチル ピリミジン一 2—ィル] アミノ} ェチル) フエニル] エタノール (実施例 73)、 (q) (1 S) - 2 - ( t e r tーブチルァミノ) 一 1一 [4一 (( 1 S ) — 1一 { [4 一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—メチルピリミジ ンー 2 -ィル] アミノ} ェチル) フエニル] エタノール (実施例 74)、
(r) (l S) — 2— (t e r t一プチルァミノ) 一 1一 [4一 (( 1 S) — 1— { [4 一 ( 8—シクロプロピルイミダゾ [1, 2 - a] ピリジン一 3—^ rル) 一 5—フルォ 口ピリミジン一 2—ィル] アミノ} ェチル) フエニル] エタノール (実施例 79)、
(s) (1 S) -2- (t e r t一プチルァミノ) ー1一 {4一 [(I S) — 1一 ({5 一フルオロー 4一 [8 - (トリフルォロメチル) イミダゾ [1, 2 - a] ピリジン一 3一ィル] ピリミジン— 2—ィル } ァミノ) ェチル] フエ二ル} エタノール (実施例 81)、
(t) [4- (U S) — 1— {[4一 (8—クロロイミダゾ [1, 2— a] ピリジ ン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエ二.ル] (1, 2—ジメチルピロリジン— 2—ィル) メタノール (実施例 82および 83)、 (u) 1一 [4- ((1 S) — 1一 {[4- (8—クロロイミダゾ [1, 2— a] ピ
リジン一 3—ィル) 一 5—フルォロピリミジン— 2—ィル] アミノ} ェチル) フエ二 ル] 一 2— (ジメチルァミノ) 一 2—メチルプロパン一 1一オール (実施例 84およ び 85 )、
(V) [4.— ((1 S) 一 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピリジ ン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1一イソプロピルァゼチジン一 3—ィル) メタノール (実施例 88および 89)、
(w) [4一 ((1 S) — 1一 {[4- (8—クロロイミダゾ [1, 2— a] ピリジ ンー 3一ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1一イソプロピルピぺリジン一4—ィル) メタノール (実施例 90および 91)、 . 又は
(x) (1 S) -2- (t e r t一プチルァミノ) 一 1一 {4一 [(I S) — 1一 ({4 一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 5— フルォロピリミジン— 2—ィル } ァミノ) ェチル] フエ二ル} エタノール (実施例 9 3)、
或いは、 その薬学的に許容される塩若しくはエステルである。 また、 本願発明の好ましい形態は、 次のようにも表現することができる。 即ち、
(1) R, が、 <置換基群 α>から選択される置換基、 ぐ置換基群 α>から選択さ れる置換基で 1個若しくは 2個以上置換されてもよい低級アルキル基、 又は、 シクロ プロピル基であり、 かつ、 <置換基群 〉が、 ハロゲン原子であり ; 及び
R2力 水素原子である、 上記式 (I) の化合物又はその薬学的に許容される塩若 しくはエステル; 又は
(2) R3及び; R4が、 同一若しくは異なって、
a) 水素原子;
b) NRaRb (ここで、 1 3及び1^は、 同一若しくは異なって、 水素原子、 低級ァ ルキル基、 ベンジル基であり、 又はシクロアルキル基であり、 ここで、 前記べンジル 基及びシクロアルキル基は、 それぞれ独立して、 下記 1ないし 3) :
1 ) 低級アルキル基;
2) <置換基群 |6>から選択される置換基;及び .
3)ぐ置換基群 j6>から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記シクロアルキル基は、 不飽和結合を含んでいてもよい。) で置換され る低級アルキル基; '
C) ぐ置換基群 /3>から選択される置換基;
d ) <置換基群 j8 >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルキル基;
e ) ぐ置換基群 /3 >から選択される置換基で 1個若しくは 2個以上置換されてもよ
い低級アルケニル基;
f ) フエニル基;
g) フエニル基で置換される低級アルキル基;
h) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基;
i ) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基で置換される低級アルキル基;
j ) ピロリル基、 イミダゾリル基、 ピラゾリル基、 ピリジル基、 ピラジニル基、 及 びピリミジニル基から選択される 5員ないし 6員の芳香族複素環基;又は、
k) ピロリル基、 イミダゾリル基、 ピラゾリル基、 ピリジル基、 ビラジニル基、 及 びピリミジニル基から選択される 5員ないし 6員の芳香族複素環基で置換される低 級アルキル基 (ここで、 前記フエニル基、 脂肪族複素環基、 及び芳香族複素環基は、 それぞれ独立して、 下記 1) ないし 4) :
1 ) 低級アルキル基;
2) ぐ置換基群 i8>から選択される置換基;
3 )ぐ置換基群 β>から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 )ぐ置換基群 /3 >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) であるか; 或いは、 R3及び R4が、 一緒になつて、 ァゼチジニル基、 ピロリジニル基、 ピぺ リジニル基、 及びピペラジニル基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、 前記脂肪族複素環基は、 下記 1) ないし 4) :
1 ) 低級アルキル基;
2) ぐ置換基群 >から選択される置換基;
3 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 )ぐ置換基群 /3〉から選択される置換基で 1個 しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) を形成する、 上記 (1) 記載の化合物又はその薬学的に許容される塩若しくはエステル; 又は
(3) R5が、水素原子、 シァノ基、ハロゲン原子、又はメチル基である、上記(1) 又は (2) に記載の化合物又はその薬学的に許容される塩若しくはエステル; 又は
(4) R3及び R4の一方が、 水素原子であり、 R3及び R4のもう一方が、 a) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 低級ァ ルキル基、 ベンジル基、 又は炭素数 3個ないし 6個のシクロアルキル基であり、 ここ
で、 前記シクロアルキル基は、 下記 1ないし 3) :
1 ) 低級アルキル基;
2) ぐ置換基群 >から選択される置換基;及び
3 ) <¾換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記シクロアルキル基は、 不飽和結合を含んでいてもよい。) で置換され る低級アルキル基;
b) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から. 選択される 4員ないし 6員の脂肪族複素環基; 又は、
c) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基で置換される低級アルキル基 (ここで、 前記脂肪族複素環基は、 下記 1) ないし 4) :
1 ) 低級アルキル基;
2) <置換基群 〉から選択される置換基;
3 )ぐ置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 )ぐ置換基群 /3 >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ レ^) である、 上記 (1) ないし (3) のいずれか 1項に記載の化合物又はその薬学 的に許容される塩若しくはエステル; 又は
(5) が、 フッ素原子で 1個ないし 3個置換されていてもよい炭素数 1個ない し 2個の低級アルキル基、 シクロプロピル基又は塩素原子、 である、 上記 (1) ない し (4) のいずれか 1項に記載の化合物又はその薬学的に許容される塩若しくはエス テル; 又は
(6) ぐ置換基群 j6>が、 ハロゲン原子、 ヒドロキシ基、 アミノ基、 低級アルキル スルホニル基、 及び低級アルコキシ基、 である、 上記 (1) ないし (5) のいずれか 1項に記載の化合物又はその薬学的に許容される塩若しくはエステル; 又は
(7) R 3及び R4の一方が、 水素原子であり、 R3及び R 4のもう一方が、 a) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 挺級ァ ルキル基、 又は炭素数 5個ないし 6個のシクロアルキル基であり、 前記シクロアルキ ル基は、 下記 1ないし 3) :
1 ) 低級アルキル基;
2) ぐ置換基群 ι8>から選択される置換基;及び
3 )ぐ置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ
レ^) で置換される低級アルキル基;又は
b ) ァゼチジニル基、 ピロリジニル基及びピペリジニル基から選択される 4員ない し 6員の脂肪族複素環基 (ここで、 前記脂肪族複素環基は、 下記 1 ) ないし 3 ) :
1 ) 低敏アルキル基;
2 ) ぐ置換基群 iS >から選択される置換基;及び
3 ) <置換基群 13〉から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ レ^ ) である、 上記 (1 ) ないし (6 ) のいずれか 1項に記載の化合物又はその薬学. 的に許容される塩若しくはエステル; 又は
( 8 ) 1^ が、 フッ素原子で 1個ないし 3個置換されていてもよい炭素数 1個ない し 2個の低級アルキル基、 シクロプロピル基、 又はハロゲン原子であり ;
R 2力 水素原子であり ;
R 3 及び R 4の一方が、 水素原子であり、 R 3及び R 4のもう一方が、 炭素数 1な いし 5個の直鎖状又は分岐状のアルキル基で N—置換若しくは N, N—ジ置換される ァミノ低級アルキル基 (ここで、 前記低級アルキル基は、 炭素数 1ないし' 3個の直鎖 状又は分岐状のアルキル基である。)、炭素数 1ないし 5個の直鎖状又は分岐状のアル キル基で N—置換されるピペリジニル基、炭素数 1ないし 5個の直鎖状又は分岐状の アルキル基で N—置換されるピロリジニル基、炭素数 1ないし 5個の直鎖状又は分岐 状のアルキル基で N—置換されるァゼチジニル基、又は炭素数 5個ないし 6個のシク 口アルキル基であり (ここで、 前記ピペリジニル基、 ピロリジニル基、 及びァゼチジ ニル基は、 それぞれ独立して、 炭素数 1ないし 3個の直鎖状又は分岐状のアルキル基 で、 さらに置換されていてもよく、 また、 前記シクロアルキル基は、 ヒドロキシ基で 置換されてもよい炭素数 1個ないし 3個のアルキル基で置換されていてもよい。) ; R 5力 シァノ基、 ハロゲン原子、 又はメチル基である、 上記 (1 ) ないし (7 ) のいずれか 1項に記載の化合物又はその薬学的に許容される塩若しくはエステル; 又は
( 9 ) 1^ が、 メチル基、 ェチル基、 二フッ化メチル基、 三フッ化メチル基、 シク 口プロピル基、 又は塩素原子であり ;
R 2力 水素原子であり ;
R 3及び R 4の一方が、 水素原子であり、 R 3及び R 4のもう一方が、 ジメチルァ ミノ基、 イソプロピルアミノ基、 1 , 1ージメチルプロピルアミノ基、 又は t 一プチ ルァミノ基で置換される、 炭素数 1ないし 3個の直鎖状又は分岐状のアルキル基; 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピぺリジニ ル基; 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換されるピロ リジニル基; 炭素数 1ないし 5個の直鎖状又は分岐状のアルキル基で N—置換され るァゼチジニル基; 或いは、 メチル基若しくはヒドロキシメチル基で置換されても よいシクロペンチル基であり (ここで、 前記ピペリジニル基、 ピロリジニル基、 及び
ァゼチジニル基は、 それぞれ独立して、 炭素数 1ないし 3個の直鎖状又は分岐状のァ ルキル基で、 さらに置換されていてもよい。);
R5が、 シァノ基、 フッ素原子、 又はメチル基である、 上記 (1) ないし (8) の いずれか 1項に記載の化合物又はその薬学的に許容される塩若しくはエステル、であ また、 本願発明の別の実施形態は、 下記 (l x) ないし (3 x) に示されるように も表現することができる。
1 X) —般式 [I] : '
[式中、 及び R2 は、 同一若しくは異なって、 水素原子; <置換基群 >から 選択される置換基; <置換基群 α >から選択される置換基で 1個若しくは 2個以上 置換されてもよい低級アルキル基; 又は、 置換されてもよい炭素数 3個ないし 6個 のシクロアルキル基であり ;
R3及び R4は、 同一若しくは異なって、
a ) 水素原子;
b) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 置換 されていてもよい低級アルキル基、 又は置換されていてもよいべンジル基である。) で置換される低級アルキル基;
c) <置換基群^〉から選択される置換基;
d) <置換基群 j8>から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルキル基;
e )ぐ置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されてもよ い低級アルケニル基;
f ) フヱニル基;
g) フエニル基で置換される低級アルキル基;
h) 4員ないし 7員の脂肪族複素環基;
i ) 4員ないし 7員の脂肪族複素環基で置換される低級アルキル基;
j ) 5員ないし 6員の芳香族複素環基;又は、
k) 5員ないし 6員の芳香族複素環基で置換される低級アルキル基 (ここで、 前記
フエニル基、 脂肪族複素環基、 及び芳香族複素環基は、 下記 1 ) ないし 4 ) :
1 ) 低級アルキル基;
2 ) <置換基群^ >から選択される置換基;
3 ) <*換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、また、前記脂肪族複素環基は、不飽和結合を含んでいてもよく、さらに、前記 b )、 . g )、 i )、 及び k ) の低級アルキル基は、 適宜置換されてもよい。) であるか、 或い は、
R 3及び R 4は、 一緒になつて、 4員ないし 7員の脂肪族複素環基 (ここで、 前記 脂肪族複素環基は、 下記 1 ) ないし 4 ) :
1 ) 低級アルキル基;
2 ) ぐ置換基群 ι6 >から選択される置換基;
3 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β >から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ く、 また、 前記脂肪族複素環基は、 不飽和結合を含んでいてもよい。) を形成し;
R 5 は、 シァノ基、 ハロゲン原子、 又は低級アルキル基である。
<置換基群ひ >及びぐ置換基群 β >は、 下記のように定義される。
ぐ置換基群ひ > :
ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル基、 ァ ミノスルホニル基、 イミノ基、 低級アルキルアミノ基、 ジ低級アルキルアミノ基、 低 級アルキルスルホニル基、 低級アルキルスルホニルァミノ基、 低級アルコキシ基、 低 級アルコキシカルポニル基、 低級アルコキシカルボニルァミノ基、 低級アルカノィル 基、 低級アルカノィルォキシ基、 低級アルキルチオ基、 .及び力ルポキシル基
<置換基群 i6 > :
ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 力ルバモイル¾、 ァ ミノスルホニル基、 イミノ基、 低級アルキルスルホニル基、 低級アルキルスルホニル アミノ基、 低級アルコキシ基、 低級アルコキシカルボ二ル基、 低級アルコキシ力ルポ ニルァミノ基、 低級アルカノィル基、 低級アルカノィルォキシ基、 低級アルキルチオ 基、 カルボキシル基、 及びべンジル基]で示される化合物又はその薬学的に許容され る塩若しくはエステル; 又は
( 2 x ) R が、 フッ素原子で 1個ないし 3個置換されていてもよい炭素数 1個な いし 2個の低級アルキル基、 シク口プロピル基又は塩素原子であり ;
R2が、 水素原子であり ;
R3及び R4 の一方が、 水素原子であり、 R3及び R4 のもう一方が、
a) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子、 低級 アルキル基、.又はべンジル基である。) で置換される低級アルキル基; ' b) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基; 又は、
c) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及びピペラジニル基から 選択される 4員ないし 6員の脂肪族複素環基で置換される低級アルキル基 (ここで、 前記脂肪族複素環基は、 下記 1) ないし 4) :
1) 低級アルキル基;
2) ぐ置換基群 ;8〉から選択される置換基;
3 )ぐ置換基群 β〉から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基; 及び
4 ) <置換基群 β〉から選択される置換基で 1個若しくは 2個以上置換されても よい炭素数 3個ないし 6個のシクロアルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ レ^) であり ;
R5が、 シァノ基である、
上記 (l x) に記載の化合物又はその薬学的に許容される塩若しくはエステル; 又 は、 '
(3 x) <置換基群 jS〉が、 ハロゲン原子、 ヒドロキシ基、 アミノ基、 低級アルキ ルスルホニル基、 及び低級アルコキシ基、 であり ;
R3及び R4 の一方が、 水素原子であり、 : 3及び R4 のもう一方が、
a) NRaRb (ここで、 Ra及び Rbは、 同一若しくは異なって、 水素原子又は低 級アルキル基である。) で置換される低級アルキル基;又は
b) ピロリジニル基及びピぺリジニル基から選択される 5員ないし 6員の脂肪族複 素環基 (ここで、 前記脂肪族複素環基は、 下記 1) ないし 3) :
1 ) 低級アルキル基;
2) <置換基群 ι6>から選択される置換基;及び ,
3)ぐ置換基群 ι6>から選択される置換基で 1個若しくは 2個以上置換される低 級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されてもよ い。) である、 上記 (l x) 又は (2x) に記載の化合物又はその薬学的に許容され る塩若しくはエステル。 また、 本発明に係る 2つの別個の製剤からなる組み合わせ製剤において、 好ましく は、 2つの別個の製剤のいずれか一方又は両方が、 経口製剤又は非経口製剤である。 本発明に係る 2つの別個の製剤からなる組み合わせ製剤は、 好ましくは、 一方の製
剤が、 薬学的に許容し得る担体又は希釈剤と一緒に、
(a) 2— [((I S) — 1— {4— [2 - (t e r t—プチルァミノ) 一 1ーヒド 口キシェチル] フエ二ル} ェチル) ァミノ] 一 4— (8—ェチルイミダゾ [1, 2― a] ピリジ; >一 3—ィル) ピリミジン一 5—力ルポ二トリル、
(b) (1 R) 一 1一 [4一 ((1 S) - 1 - {[5—ブロモ _4一 (8—ェチルイ ミダゾ [1, 2-a] ピリジン— 3—ィル) ピリミジン一 2 _ィル] アミノ}ェチル) フエニル] 一 2— ( 1: e r t—ブチルァミノ) エタノール、
(c) 2- [((1 S) 一 1— {4- [ヒドロキシ (ピリジン一 2—ィル) メチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—メチルイミダゾ [1 , 2-a] ピリジン一. 3一ィル) ピリミジン一 5—力ルポ二トリル、
(d) 4— (8—ェチルイミダゾ [1 , 2-a] ピリジン— 3—ィル) 一 2— ({(1 S) 一 1一 [4- (4ーヒドロキシー 1ーメチルビペリジン一 4一ィル) フエニル] ェチル } ァミノ) ピリミジン一 5—力ルポ二トリル、
(e) 4— (8—ェチルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 2— [((1 S) -1 - {4- [ヒドロキシ (1一イソプロピルピぺリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、 -
( f ) 4一 ( 8—シクロプロピルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 2— [((1 S) 一 1一 {4- [ヒドロキシ (1ーメチルビペリジン一 4一ィル) メチ ル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、
(g) 4— (8—クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル) —2— [((1 S) — 1一 {4- [(1ーシクロプロピルピぺリジン一 4一ィル) (ヒドロキシ) メチ ル] フエ二ル} ェチル) ァミノ〕 ピリミジン一 5—力ルポ二トリル、
(h) 4- (8—ェチルイミダゾ [1, 2— a] ピリジンー3—ィル) 一 2— [((1 S) 一 1一 {4- [ヒドロキシ (1ーメチルビペリジン一 3—^ Γル) メチル] フエ二 ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル、
( 1) 2— {[(1 S) — 1一 (4一 {ヒドロキシ [1一メチルピロリジン一2—ィ ル] メチル } フエニル) ェチル] アミノ} 一 4一 (8—メチルイミダゾ [1, 2-a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル、
( j ) 2- [((I S) — 1— {4- [2- (t e r t一プチルァミノ) 一 1ーヒド 口キシェチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル、
(k) 2— [((1 S) — 1一 {4- [2- (t e r t—プチルァミノ) 一 1—ヒド 口キシェチル] フエ二ル} ェチル) ァミノ] —4一 [8— (ジフルォロメチル) イミ ダゾ [1, 2— a] ピリジンー 3—ィル] ピリミジン— 5—力ルポ二トリル、
(1) 4— (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2- [((1 S) 一 1一 {4一 [(1, 2—ジメチルピロリジン一 2—ィル) (ヒドロキ シ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、
(m) (1 S) — 2— ( t e r t—プチルァミノ) 一 1一 [4一 ((I S) — 1一 {[4
一 (8—クロロイミダゾ [1, 2- a] ピリジン一 3—ィル) 一 5—フルォロピリミ ジン一 2—ィル] アミノ} ェチル) フエニル] エタノール、
(n) (I S) — 1— [4一 ((1 S) 一 1一 {[4- (8—クロロイミダゾ [1, 2 - a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2一ィル] アミノ} エヂ ル) フエニル] 一 2— [(1, 1ージメチルプロピル) ァミノ] エタノール、
(o) (1 S) 一 1一 [4一 ((1 S) 一 1一 {[4- (8—クロロイミダゾ [1, 2-a] ピリジン— 3—ィル) 一 5—フルォロピリミジン一 2一ィル] アミノ} ェチ ル) フエニル] - 2 - [(1ーメチルシクロペンチル) ァミノ] エタノール、
(P) (1 S) — 2— (t e r t一プチルァミノ) 一 1一 [4一 (( 1 S ) - 1一 {[4. 一 ( 8—シクロプロピルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 5—メチル ピリミジン一 2一^ rル] アミノ} ェチル) フエニル] エタノール、
(q) (1 S) -2- (t e r t一プチルァミノ) 一1— [4— ((I S) — 1— {[4 一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—メチルピリミジ ンー 2一ィル] アミノ} ェチル) フエニル] エタノール、
(r) (l S) — 2— (t e r t—プチルァミノ) 一 1一 [4一 ((1 S) — 1一 {[4 一 (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一' 5—フルォ 口ピリミジン一 2—ィル] ァミノ) ェチル) フエニル] エタノール、
(s) (1 S) — 2— (t e r t一プチルァミノ) 一 1ー {4一 [(I S) — 1— ({5 一フルオロー 4一 [8— (トリフルォロメチル) イミダゾ [1, 2— a] ピリジン— 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] フエ二ル} エタノール、
(t) [4一 ((1 S) —1— {[4一 (8—クロロイミダゾ [1, 2 a] ピリジ ンー 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1 , 2—ジメチルピロリジン— 2—ィル) メタノール、
(u) 1一 [4- ((1 S) — 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピ リジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエ二 ル] 一 2— (ジメチルァミノ) 一 2—メチルプロパン一 1一オール、
(V) [4— ((1 S) - 1 - {[4- (8—クロロイミダゾ [1, 2— a] ピリジ ンー 3一ィル) 一 5—フルォロピリミジン一 2一^ Γル] アミノ} ェチル) フエニル] ( 1一イソプロピルァゼチジン— 3—ィル) メタノール、
(w) [4— . ((1 S) — 1一. {[4一 (8—クロロイミダゾ [1, 2— a] ピリジ ンー 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1一イソプロピルピぺリジン一 4 _ィル) メタノール、
又は
(x) (1 S) -2- (t e r t—プチルァミノ) 一 1一 {4— [(I S) — 1— ({4 一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 5— フルォロピリミジン— 2—ィル } ァミノ) ェチル] フエ二ル} エタノール、 或いは、 その薬学的に許容し得る塩若しくはエステルを含む製剤である。
さらに、 本発明に係る 2つの別個の製剤からなる組み合わせ製剤に対して、 薬学的
に許容し得る担体又は希釈剤と一緒に、杭がん性アルキル化剤、杭がん性代謝拮抗剤、 抗がん性抗生物質、 植物由来抗がん剤、 抗がん性白金配位化合物、 杭がん性カンプ卜 テシン誘導体、 杭がん性チ口シンキナ一ゼ阻害剤、 モノクローナル抗体、 インターフ ェロン、 生物学的応答調節剤、 及びその他杭がん剤 (ここで、 各杭がん剤の定義は、 上記の記載と同じである。) からなる群から選択される杭がん剤又はその薬学的に許 容し得る塩若しくはエステルを含む製剤 1個以上を、 さらに組み合わせてもよい。 また、 本発明に係る医薬組成物は、 好ましくは、 薬学的に許容し得る担体又は希釈 剤と一緒に、
(a) 2— [((1 S) - 1 - {4- [2- (t e r t一プチルァミノ) — 1一ヒド. 口キシェチル] フエ二ル} ェチル) ァミノ] -4- (8—ェチルイミダゾ [1, 2一 a] ピリジン一 3—ィル) ピリミジン— 5—力ルポ二トリル、
(b) (1 R) 一 1一 [4一 ((1 S) 一 1一 {[5—ブロモー 4一 (8—ェチルイ ミダゾ [ 1, 2 - a] ピリジン一 3—^ rル) ピリミジン一 2—ィル] アミノ} ェチル) フエニル] 一 2— ( t e r t—ブチルァミノ) エタノール、
(c) 2— [((1 S) — 1— {4一 [ヒドロキシ (ピリジン一 2—ィル) メチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—メチルイミダゾ [1, 2 -a] ピリジン— 3—ィル) ピリミジン一 5—カルボ二トリル、
(d) 4— (8—ェチルイミダゾ [1, 2- a] ピリジン一 3—ィル) 一 2— ({(1 S) 一 1一 [4- (4ーヒドロキシー 1ーメチルピペリジン一 4一ィル) フエニル] ェチル } ァミノ) ピリミジン一 5—カルボ二トリル、
(e) 4— (8—ェチルイミダゾ [1 , 2 - a] ピリジン一 3—ィル) - 2 - [((1 S) — 1— {4- [ヒドロキシ (1一イソプロピルピぺリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、
(f ) 4一 (8—シクロプロピルイミダゾ [1, 2— a] ピリジン— 3—ィル) 一 2 - [((1 S) 一 1一 {4- [ヒドロキシ (1ーメチルピペリジン一 4—ィル) メチ ル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、
(g) 4- (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 S) 一 1一 {4- [(1ーシクロプロピルピぺリジン一 4一ィル) (ヒドロキシ) メチ ル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、
(h) 4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— [((1 S) - 1 - {4一 [ヒドロキシ (1ーメチルピペリジン一 3—ィル) メチル] フエ二 ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル、
(i) 2— {[(1 S) - 1- (4- {ヒドロキシ [1一メチルピロリジン一 2—ィ ル] メチル } フエニル) ェチル] アミノ} - 4- (8—メチルイミダゾ [1, 2-a] ピリジン一 3—^ rル) ピリミジン— 5—力ルポ二トリル、
( j ) 2- [((I S) 一 1― {4- [2— (t e r. t—プチルァミノ) 一 1ーヒド 口キシェチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン— 5—力ルポ二卜リル、
(k) 2— [((I S) - 1 - {4- [2- (t e r t—プチルァミノ) 一 1ーヒド 口キシェチル] フエ二ル} ェチル) ァミノ] -4- [8 - (ジフルォロメチル) イミ ダゾ [1, 2-a] ピリジン一 3—ィル] ピリミジン— 5—力ルポ二トリル、
(1) 4- ( 8—シクロプロピルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 2- [((1 S) - 1 - {4- [(1, 2—ジメチルピロリジン一 2—ィル) (ヒドロキ シ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル、
(m) (1 S) - 2 - (t e r t—プチルァミノ) 一 1一 [4一 (( 1 S ) — 1一 { [4 一 (8—クロロイミダゾ [1 , 2-a] ピリジン一 3—^ Γル) 一 5—フルォロピリミ ジン一 2一ィル] アミノ} ェチル) フエニル] エタノール、
(n) (I S) — 1一 [4一 ((1 S) 一 1一 {[4一 (8—クロロイミダゾ [1, 2-a] ピリジン一 3—ィル) 一5—フルォロピリミジン一 2—ィル] アミノ} ェチ ル) フエニル] -2- [(1, 1ージメチルプロピル) ァミノ] エタノール、
(o) (1 S) 一 1一 [4- ((1 S) —1— {[4- (8—クロロイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチ ル) フエニル] 一 2— [(1ーメチルシクロペンチル) ァミノ] エタノール、
(P) (1 S) — 2— (t e r t一プチルァミノ) - 1 - [4 - (( 1 S) - 1 - { [4 一 (8—シクロプロピルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 5—メチル ピリミジン一 2—ィル] アミノ} ェチル) フエニル] エタノール、
(q) (I S) — 2— (t e r t一プチルァミノ) 一 1一 [4— (( 1 S ) — 1一 { [4 一 (8—クロロイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 5—メチルピリミジ ン— 2一^ rル] アミノ} ェチル) フエニル] エタノール、
(r) (l S) — 2— (t e r t—プチルァミノ) 一 1一 [4一 (( 1 S) — 1— { [4 一 (8—シクロプロピルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 5—フルォ 口ピリミジン一 2—ィル] アミノ} ェチル) フエニル] エタノール、
(s) (1 S) — 2— (t e r t一プチルァミノ) 一 1一 { 4一 [( 1 S ) — 1一 ({ 5 —フルオロー 4一 [8 - (トリフルォロメチル) イミダゾ [1, 2-a] ピリジン一 3一ィル] ピリミジン一 2—ィル } ァミノ) ェチル] フエ二ル} エタノール、
(t) [4- ((I S) — 1一 {[4一 (8—クロロイミダゾ [1, 2-a] ピリジ ンー 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1, 2—ジメチルピロリジン一 2—ィル) メタノール、
(u) 1一 [4- ((1 S) 一 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピ リジン一 3—ィル) 一5—フルォロピリミジン一 2—ィル] ァミノ) ェチル) フエ二 ル] - 2 - (ジメチルァミノ) 一 2—メチルプロパン一 1一オール、
(V) [4— ((1 S) 一 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピリジ ン一 3—^ fル) 一 5—フルォロピリミジン一 2—^ Γル] アミノ} エヂル) フエ二ル] . (1一^ fソプロピルァゼチジン一 3一ィル) メタノール、
• (w) [4— ((I S) —1— {[4- (8—クロロイミダゾ [1, 2-a] ピリジ ンー 3—ィル) 一 5—フルォロピリミジン一 2—ィル] ァミノ) ェチル) フエニル]
(1一イソプロピルピぺリジン一 4一ィル) メタノール、 又は
(X) (1 S) 一 2— (t e r t一プチルァミノ) 一 1一 { 4一 [( 1 S) - 1 一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン— 3—ィル]
フルォロピリミジン一 2—ィル } ァミノ) ェチル] フエ二ル} エタノール、 或いはその薬学的に許容し得る塩若しくはエステルを含む。 以下に本発明化合物の代表的な製造方法について説明する。 スキーム 1 a :式 (I I a) ) の化合物の製造法
(lla) ("la)
スキーム 1 a 本発明に係る上記式 (I) の化合物 (ここで、 R2 R3 R4、 及び R5は前 記と同義である) は、 まず、 上記式 (I I a) の化合物 (ここで、 R2、 及び R 5は前記と同義である) を酸化反応に付して、上記式(I l i a) の化合物(ここで、 R2、 及び R5は前記と同義であり、 nは 1または 2である) を得て、 次に、 式
(I l i a) の化合物と、 上記式 (I V) で示されるフエネチルァミン (ここで、 R 3 R4は前記と同義である) との置換反応をすることにより合成することができる。 上記式 (I l i a) の化合物は、 上記式 (I I a) の化合物を、 ジクロロメタン、 クロ口ホルム、 N N—ジメチルホルムアミド等の溶媒中、 m—クロ口過安息香酸(m 一 CPBA)、過酸化べンゾィル、過酸化水素水、過ヨウ素酸ナトリウム等の酸化剤、 好ましくは、 m—クロ口過安息香酸を用いて酸化することにより合成することができ る。 当該反応においては、 上記式 (I I a) の化合物 1モルに対して、 m—クロ口過 安息香酸 (m— CPBA) を 2 5モル、 好ましくは 2モル用いる。 反応温度は、 使
用される原料化合物あるいは反応溶媒に応じて当業者が適宜選択することができる が、 通常、 0°Cから室温である。 また、 反応は、 通常 1時間から 24時間で完結する 力 反応時間は適宜増減することができる。 また、 得られた上記式 (I I I a) の化 合物は単離精製することなく、 次の反応に付すことができる。
上記式(I l i a)の化合物と上記式( I V)のフエネチルアミンとの置換反応は、 塩基 (例えば、 炭酸カリウム、 炭酸水素ナトリウム等の無機塩基、 もしくはトリェチ ルァミン、 ジイソプロピルェチルァミン等の有機塩基) の存在下で行うことが好まし レ^反応溶媒はクロ口ホルム、テトラヒドロフラン、 N, N—ジメチルホルムアミド、 ジメチルスルホキシド等を用いることができ、 好ましくはクロ口ホルム、 テトラヒド. 口フランである。当該反応においては、上記式(I I I a)の化合物 1モルに対して、 上記式 (I V) のフエネチルァミンを 0. 5〜3モル用いる。 反応温度は使用される 原料化合物あるいは反応溶媒に応じて、 当業者が適宜選択できるが、 通常、 室温から 溶媒の沸点であり、 好ましくは室温である。 また、 反応は、 通常 1時間から 24時間 で完結するが、 反応時間は適宜増減することができる。 ーム l b :式 (I l b) の化合物から式 (I) の化合物の製造法 ·
スキーム 1 b また本発明に係る上記式 (I) の化合物 (ここで、 Rい R2、 R3、 R4、 及び R5 は前記と同義である) は、 上記式 (I l b) の化合物 (ここで、 R2、 及び R5 は前記と同義である) と、 上記式 (I V) で示されるフエネチルァミン (ここで、 R 3、 R4は前記と同義である) との置換反応をすることにより合成することができる。 上記式 (I l b) の化合物と上記式 (I V) のフエネチルァミンとの置換反応は、 塩基 (例えば、 炭酸カリウム、 炭酸ナトリウム、 炭酸セシウム、 炭酸水素ナトリウム 等の無機塩基、 もしくはトリエヂルァミン、 ジイソプロピルェチルァミン等の有機塩 基)の存在下で行うことが好ましい。反応溶媒はクロロホルム、テトラヒドロフラン、
N, N—ジメチルホルムアミド、 N, N—ジメチルァセトアミド、 N—メチルー 2— ピロリジノン、 ジメチルスルホキシド等を用いることができ、 好ましくは N, N—ジ メチルホルムアミド、 N, N—ジメチルァセトアミド、 N—メチルー 2—ピロリジノ ンである。 当該反応においては、 上記式 (l i b) の化合物 1モルに対して、 上記式
(I V) のフエネチルァミンを 0. 5〜 3モル用いる。 反応温度は使用される原料化 合物あるいは反応溶媒に応じて、 当業者が適宜選択できるが、 通常、 室温から溶媒の 沸点であり、 好ましくは 80〜200 である。 また、 反応は、 通常 1時間から 24 時間で完結するが、 反応時間は適宜増減することができる。 スキーム 2 a :式 (I I a) の化合物の代表的な製造法
(Va) (Via) (Vila)
スキーム 2a
上記式 (Vi a) の化合物 (ここで、 R
5は前記と同義である) は、 ジクロロビス (トリフエニルホスフィン) パラジウム (I I) を触媒とする、 上記式 (Va) の化 合物 (ここで、 R
5は前記と同義である) と、 シス一 1一エトキシー 2—トリー n— ブチルすず (J. Am. Chem. S o c., 1977, 99, 7365に記載され ている方法により合成できる) とのスティル (s t i l l) カップリング反応により 合成することができる。 反応溶媒はァセトニ卜リルが好ましく、 反応温度は、 通常、 室温から溶媒の沸点であり、 好ましくは溶媒の沸点で ¾る。 また、 反応は、 通常 1時 間から 24時間で完結するが、 反応時間は適宜増減することができる。
上記式 (Vi l a) の化合物 (ここで、 R5は前記と同義である) は、 上記式 (V l a) の化合物を、 1, 4一ジォキサン中、 N—ブロモこはく酸イミドと反応させて 調製することができる。 当該反応においては、 上記式 (VI a) の化合物 1モルに対 して、 N—プロモこはく酸イミドを 1〜3モル、 好ましくは 1モル用いる。 反応温度 は使用される原料化合物に応じて当業者が適宜選択することができるが、好ましくは 0°Cから室温である。 また、 反応は、 通常 1時間から 12時間で完結するが、 反応時 間は適宜増減することができる。 また、 得られた上記式 (Vi l a) の化合物は単離 精製することなく、 次の反応に付すことができる。
上記式(I I a) の化合物(ここで、 R2、 及び R5は前記と同義である) は、 1, 4一ジォキサン中、 上記式 (V i l a) の化合物と上記式 (VI I I) の化合物
(ここで、 1^、及び R2は前記と同義である)から合成される。当該反応においては、 上記式 (Vi l a) の化合物 1モルに対して、 上記式 (V I I I) の化合物を 1〜3 モル、 好ましくは 1モル用いる。 反応温度は使用される原料化合物に応じて当業者が 適宜選択することができるが、 通常、 室温から溶媒の沸点であり、 好ましくは室温か ら 50°Cである。 また、 反応は、 通常 1時間から 24時間で完結するが、 反応時間は 適宜増減することができる。
なお、 上記式 (Va) の化合物は、 例えば、 4一クロロー 2— (メチルスルファ二 ル) 一 5—ピリミジン力ルポ二トリル等であり、 また、 上記式 (VI I I) の化合物 は、 例えば、 2—アミノー 3—ピコリン等であり、 市販で入手できるか、 或いは、 巿 販で入手できる化合物から当業者に周知の方法、 または、 それに準じた方法を用いて 合成することができる (文献:国際公開第 2004/043936号パンフレツ卜、 32〜 33ページなど)。 スキーム 2 b :式 ( I I b) の化合物の代表的な製 '
(Vb) (Vlb) (Vllb)
スキーム 2 b
上記式 (I l b) (ここで、 R
2、 及び R
5は前記と同義である) の化合物は、 スキーム 2 aにおける上記式 (Va) の化合物に代えて、 上記式 (Vb) (ここで、 R
5は前記と同義である) の化合物を出発物質として、 スキーム 2 aと同様な方法を 用いて合成することができる (スキーム 2 b参照)。 .
なお、 上記式 (Vb) の化合物は、 例えば、 2, 4—ジクロロー 5—メチルピリミ ジン等であり、 また、 上記式 (V I I I) の化合物は、 例えば、 2—アミノー 3—ピ
コリン等であり、 市販で入手できるか、 或いは、 市販で入手できる化合物から当業者 に周知の方法、 または、 それに準じた方法を用いて合成することができる (文献:国 際公開第 2004/043936号パンフレツ卜、 32〜33ページなど)。 的な製造法
(VIII) (IX)
o
1. n-BuLi 2.R3 R4(XA)
3. NaBH4 スキーム 3 上記式 (I X) の化合物 (ここで、 Bo cは、 t e r t—ブトキシカルポニル基で ある) は、 市販で入手できる (S) 一 (一) 一 1― (4一ブロモフエニル) ェチルァ ミン (上記化合物 (VI I D) を用いて、 当業者に周知慣用な方法 (文献:プロテ クティブ'グループス 'イン 'オーガニック ·シンセシス 第三版 (P r o t e c t i ve Gr oup s i n Or an i c Syn t he s i s, t he t h i r d e d i t i on), T. W. グリーン (T. W. G r e e n e) 著、 J o h n Wi l ey & S o n s社、 518〜 524ページなど) に従って合成するこ とができる。
次に、 上記式 (XI) の化合物 (ここで、 Bo c、 R3、. 及び R4は前記と同義であ る) は、 テトラヒドロフラン中、 上記式 (I X) の化合物に n—ブチルリチウムを付 すことにより、 反応性中間体であるァリ一ルリチウムとした後に、 求電子剤である上 記式(Xa)のケトン(ここで、 R3、及び R4は前記と同義である) または上記式(X b) のアルデヒド (ここで、 R4は前記と同義である) と反応させることにより合成 することができる。 当該反応においては、 上記式 (I X) の化合物 1モルに対して、
n—ブチルリチウムを 2〜 5モル、 好ましくは 2モル用いる。 また、 上記式 (I X) の化合物 1モルに対して、 上記式 (Xa) のケトンまたは上記式 (Xb) のアルデヒ ドを 1〜3モル用いる。 反応温度は、 通常、 一 78T:から 0°Cであり、 好ましくは一 78°Cであ!)。 また、 反応は、 通常 1時間から 24時間で完結するが、 反応時間は適 宜増減することができる。
また、 上記式 (X I) の化合物は、 以下の方法によっても合成することができる。 即ち、 上記式 (I X) の化合物に n—ブチルリチウムを付すことにより、 反応性中間 体であるァリールリチウムとした後に、 求電子剤であるワインレブアミド (We i n r eb ami de) (Xc) (ここで、 R3は前記と同義である) と反応させる。 得. られた化合物を、 さらに水素化ホウ素ナトリウムを用いて還元反応に付して、 上記式 (XI) の化合物を合成することができる。 当該反応においては、 上記式 (I X) の 化合物 1モルに対して、 n—ブチルリチウムを 2〜 5モル、好ましくは 2モル用いる。 また、 上記式 (I X) の化合物 1モルに対して、 上記式 (Xc) のアミドを 1〜3モ ル用いる。反応温度は、通常、一 78°Cから 0°Cであり、好ましくは— 78°Cである。 また、 反応は、 通常 1時間から 24時間で完結するが、 反応時間は適宜増減すること ができる。 また、 還元反応においては、 上記式 (I X) の化合物 1モルに対して、 水 素化ホウ素ナトリウムを 1〜 3モル、 好ましくは 1モルを用いる。
上記式 (I V) の化合物 (ここで、 R3、 及び R4は前記と同義である) は、 上記式 (XI) の化合物を用いて、 当業者の周知慣用な方法 (文献:プロテクティブ ·グル ープス ·イン'オーガニック ·シンセシス 第三版 (P r o t e c t i ve G r o up s i n Or an i c Syn t he s i s, t he t h i r d e d i t i on), T. W. グリーン (T. W. G r e e n e) 著、 J o h n Wi l ey & S on s社、 518〜 524ページなど)に従って容易に合成することができる。 なお、上記式(X a)の化合物は、例えば、 1—メチル—4—ピペリドン等であり、 上記式 (Xb) の化合物は、 例えば、 ベンジル 2一ホルミルピロリジン— 1—カル ポキシレート等である。 また上記式(Xc)の化合物は、例えば、ベンジル 4一 (N ーメトキシー N—メチルカルバモイル) ピぺリジン一 1一力ルポキシレート等であり、 これらの化合物は市販で入手できるか、 或いは、 市販で入手できる化合物から当業者 に周知の方法、 または、 それに準じた方法を用いて合成することができる (文献: B i oo r g. Me d. C h em., 2003, 11, 3153、 国際公開第 03/0 11285号パンフレット、 60〜61ページなど)。 スキーム 4 :式 (XV I) の代表的な製造法
スキーム 4 まず、 上記式 (X I I) の化合物 (ここで、 Bo cは、 t e r t—ブトキシカルポ ニル基である) は、 N, N—ジメチルホルムアミド、 1, 4一ジォキサン、 トルエン、 テトラヒドロフラン、 メタノール、 ジメトキシェタン等の溶媒中、 テトラキス (トリ フエニルホスフィン) パラジウム (0) ゃジクロ口ビス (トリフエニルホスフィン) パラジウム (I I) 等のパラジウム触媒存在下、 上記式 (I X) の化合物 (ここで、 Bo cは、 前記と同義である) とビニルトリフルォロホウ酸カリウム、 もしくはビニ ルトリブチルすずとの力ップリング反応に付すことにより、 合成することができる。 ここで、 ビニルトリフルォロほう酸カリウムを用いる場合は、 炭酸ナトリウム、 炭酸 水素ナトリウム、 炭酸カリウム等の無機塩基、 もしくはトリェチルァミン、 ジイソプ 口ピルェチルァミン等の有機塩基存在下で反応を行うことが好ましい。当該反応にお いては、 反応温度は使用される反応試剤あるいは反応溶媒に応じて、 当業者が適宜選 択することができるが、 通常、 室温から溶媒の沸点である。 また、 反応は通常、 1時 間から 24時間で完結するが、 反応時間は適宜増減することができる。
上記式 (X I I I) の化合物 (ここで、 Bo cは、 前記と同義である) は、 上記式 (X I I) の化合物を、 アセトンと水の混合溶媒中、 四酸化オスミウム及び N—メチ ルモルホリン N—才キシドを用いたジヒドロキシル化反応に付すことにより、合成 することができる。 当該反応は、 通常、 0°Cから室温で行われることが好ましい。 ま た、 反応は通常、 1時間から 48時間で完結するが、 反応時間は適宜増減することが できる。
なお、 四酸化オスミウムに代えて、 AD— mi X—《もしくは (アルドリッチネ土 (A 1 d r i c h) より入手可能である) を酸化剤として用い、 上記式 (X I I ) の 化合物をシャープレス (S h a r p 1 e s s ) の不斉ジヒドロキシル化反応 (文献: C h e m. Re v., 1994, 94, 2483など) に付すことにより、 上記式 (X
I I I) の化合物の光学活性体を合成することもできる。
上記式 (X I V) の化合物 (ここで、 Bo cは、 前記と同義である) は、 ジクロロ メタン、 クロ口ホルム、 トルエン、 テトラヒドロフラン等の溶媒中、 水酸化ナトリウ
ム、 水酸化カリウム、 炭酸カリウム等の無機塩基、 もしくはトリェチルァミン、 ジィ ソプロピルアミン、 ピリジン等の有機塩基の存在下、 上記式 (XI I I) の化合物の 1級水酸基に選択的に脱離基を導入し、その後に加熱することにより環化反応に付し て合成する とができる。 この場合、 脱離基としてはメタンスルホ二ル基ゃ P—トル エンスルホニル基などを用いることができる。 脱離基を導入する反応においては、 上 記式 (XI I I) の化合物 1モルに対して、 塩化メタンスルホニルもしくは塩化 P— トルエンスルホニルを 1〜3モル、好ましくは 1. 1モル用いる。また、反応温度は、 0°Cから室温、 好ましくは 0°Cである。 さらに、 当該反応は、 通常、 1時間から 24 時間で完結するが、 適宜増減することができる。
上記式 (XV) の化合物 (ここで、 Nuは、 求核剤由来の置換基、 例えば t e r t 一プチルァミノ基などであり、 Bo cは、 前記と同義である) は、 メタノール、 エタ ノール、 水等の溶媒中、 上記式 (XI V) で示されるエポキシドを求核剤と反応させ ることにより合成することができる。 当該反応においては、 上記式 (XV) の化合物 1モルに対して、 求核剤を 1モルから大過剰、 好ましくは 10モル程度用いる。 反応 温度は、使用される原料化合物あるいは反応溶媒に応じて当業者が適宜選択すること ができるが、 通常、 室温から溶媒の沸点であり、 好ましくは 40°Cから溶媒の沸点で ある。 また、 反応は、 通常 1時間から 24時間で完結するが、 反応時間は適宜増減す ることができる。 なお、 当該反応における求核試薬とは、 例えば t e r t一プチルァ ミン、 ピぺラジン等であり、 市販で入手できるか、 或いは、 市販で入手できる化合物 から'当業者に周知の方法、または、それに準じた方法を用いて合成することができる。 上記式(XV I) の化合物 (ここで、 Nuは前記と同義である) は、 上記式 (XV) の化合物から、 当業者の周知慣用な方法(文献:プロテクティブ 'グループス 'イン ' オーガニック ·シンセシス 第三版 (P r o t e c t i ve Gr oup s i n Or an i c Syn t he s i s, t he t h i r d e d i t i on , T. W. グリーン(T. W. G r e e n e)著、 J o h n Wi l ey & S o n s社、 518〜 524ページなど) を用いて合成することができる。 なお、 スキーム 1から 4に記載の製造方法において、 必要に応じて、 有機合成化学 で常用とされる方法、 例えば官能基の保護、 脱保護等 〔例えばプロテクティブ ·ダル ープス 'イン'.オーガニック 'シンセシス 第三版 (P r o t e c t i V e Gr o u p s i n Or an i c Syn t he s i s, t he t h i r d e d i t i o n), T. W. グリーン (T. W. G r e e n e) 著、 J ohn Wi l e y & S o n s社参照] を用いることで、 所望の化合物を得ることもできる。 次に、 一般式 (I) の化合物の PLK1阻害作用及び細胞増殖抑制作用について以 下説明する。
1. PLK1活性阻害作用の測定 (方法 A)
(1) PLK1— T210Dの調製
ヒト PLK1の 210番目のコドンは本来スレオニンをコードするがこの部位を ァスパラギン酸に改変することによって活性化型になることが知られている [モレキ ユラ一 'ア ド 'セルラ一バイオロジー (Mo 1. Ce l l . B i o l .), 17巻、 3408項 (1997年)]。 ヒト活性化型 P L K 1タンパク質を得るために、 ヒト P LK1 cDNAの 210番目のコドンで塩基置換を行うことによって、 210番目 のコドンがァスパラギン酸をコードするようになった変異型 PLK1 (PLK1 -T 210D) の cDNAを作成した。 この PLK1— T210D cDNAの N末端に GST (ダルタチオン S—トランスフェラ一ゼ) を融合したバキュロウィルスを作製. した後に、 Spodop t e ra frug ipe rda (スポドプテラ フルギぺ ルダ) (S O 9昆虫細胞に感染させた PLK1— T210Dを GST—融合タンパ クとして高発現させた。 その細胞を回収してリシスバッファ一 (50mMトリス一塩 酸バッファー (pH7. 4) Z15 OmM塩化ナトリウム ZlmM EDTA (ェチ レンジアミンー 4酢酸) /ImMジチオトレイ ] ル /0. 1%ポリオキシエチレン ソルビタンモノラウレート) に懸濁しソニケ一夕一で細胞破碎を行い、 遠心後上清を 回収した。 上清をダルタチオンセファロ一スビーズと反応させ、 ビーズをりシスバッ ファーで洗浄した。 その後ビーズをプレシジョンプロテア一ゼ (GEヘルスケア バ ィォサイエンス社より購入) 入りリシスバッファーと反応させ上清を回収した。
(2) PLK1— T210Dの活性測定
P LK1 -T210Dの活性測定において、基質は PLK1の基質部位として報告 されている CDC 25 Cのアミノ酸配列 198番目のセリン周辺配列 [ェンポ ·レポ —ト (EMB〇 Re p o r t.), 3巻、 341項 (2002年)] を改変した合成 ペプチド (アルギニン—アルギニン一アルギニンーァスパラギン酸一グルタミン酸一 ロイシン一メチォニン一グルタミン酸ーァラニンーセリンーフエ二ルァラニンーァ ラニンーァスパラギン酸一グルタミン一グルタミン酸ーァラニン一リジンーバリン) (SEQ. I D. NO. : 1) を用いた。
反応は豊島一森本 (T o y o s h i ma -Mo r i mo t o) らの方法 [ネーチヤ — (Na t u r e、第 410巻、 215— 220頁、 (2001年)]に準じて行った。 反応液量は 21. 1 1で、 反応バッファーの組成は 20 mMトリス一塩酸バッファ 一 (pH7. 4) /1 OmM塩化マグネシウム 0. 5mMジチオトレイ! ル / 1 mM EGT A (エチレングリコ一ルービス(ベ一夕一アミノエチルェ一テル)— N, N, Ν', N' — 4酢酸) で、 そこに精製した PLK1と 50 Mの基質ペプチドと 5 0 zMの非標識アデノシン三リン酸 (ATP) および 1 C iの [ァ一33 P] 標識 ATP (2000-4000 C i /mmo 1 e) を添加して、 反応温度 25°Cで 20 分間反応させた。 その後、 10// 1の 350mMリン酸バッファーを反応系に添加し て反応を停止させ、その液を 96 we 1 1マルチスクリーンフォスフォセルロースフ ィルターにスポッ卜した。 75mMリン酸バッファーでそのフォスフォセル口一スフ ィルターを洗浄した後、乾燥させて放射活性を液体シンチレーシヨンカウン夕一で測
定した。 非標識 ATP、 [T— 33P] 標識 ATPはアマシャム 'バイオサイエンスか ら、マルチスクリーンフォスフォセルロースフィル夕一はミリポア社からそれぞれ購 入した。
本発明化合物の反応系への添加は、予め終濃度の 20倍濃度でジメチルスルホキシ ドに溶解させた溶液を 1. 1 1加えることによって行った。 反応系へジメチルスル ホキシドを 1. 1 1加えたものを対照とした。
本発明化合物の P LK 1 -T 210 D活性に対する I C50値をもとめたのでその 結果を下記の表 1に示す。 表 1
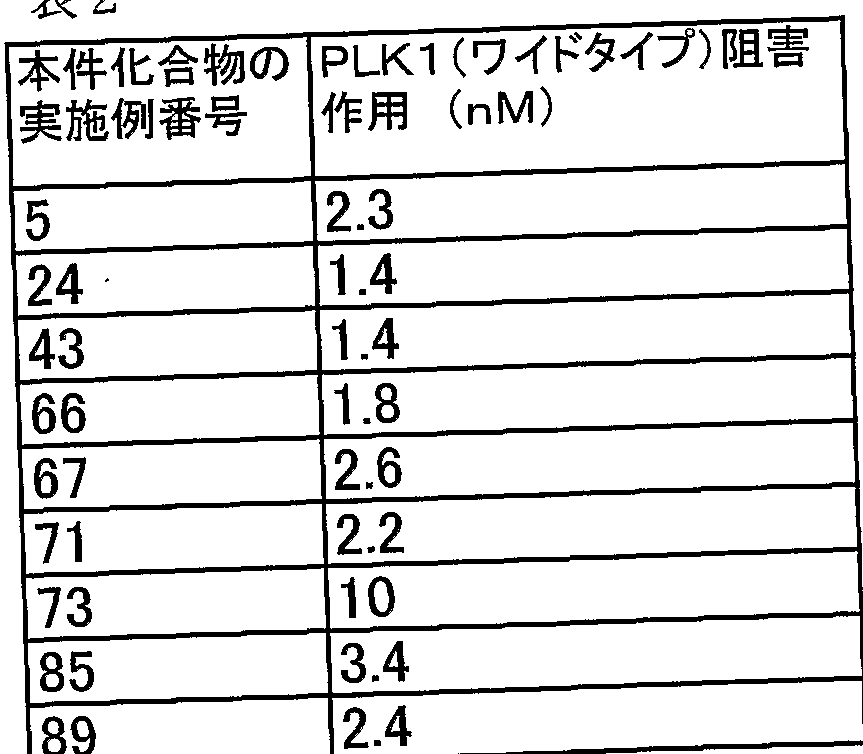
2. PLKl活性阻害作用の測定 (方法
(1) PLK1 (ワイルドタイプ) の調製
ヒト PLK1は、 カルナバイオサイエンス社から購入した。 カルナバイオサイェン ス社のプロダクトインフオメ一シヨン (P r oduc t I n f o rma t i on) によれば、 本酵素は野生型 PLKlの全長 cDNAの N末端に GST (ダル夕チオン S—トランスフェラーゼ) を融合したバキュロウィルスを作製し、 昆虫細胞に感染さ せて PLK1を GST—融合夕ンパクとして高発現させた後に、 グル夕チオンセファ ロースクロマトグラフィーにかけることにより精製したものである。
(2) PLK1 (ワイルドタイプ) の活性測定
PLK1の活性測^において、基質は P LK 1の基質部位として報告されている C DC 25 Cのアミノ酸配列 198番目のセリン周辺配列 [ェンポ*レポート (EMB 〇 Re p o r t.), 3巻、 341項 ( 2002年)] を改変した合成ペプチド (ァ ルギニン—アルギニン一アルギニンーァスパラギン酸—ダル夕ミン酸一口イシン一 メチォニン一グルタミン酸ーァラニン一セリンーフエ二ルァラニンーァラニン一ァ スパラギン酸一グルタミン一グルタミン酸ーァラニン一リジン一バリン) (SEQ. I D. NO. : 1) を用いた。
反応は豊島—森本 (Toyo s h ima— Mo r imo t o) らの方法 [ネーチヤ — (Na t u r e、第 410巻、 215— 220頁、 (2001年)]に準じて行った。 反応液量は 10. 5 i 1で、 反応バッファーの組成は 20 mMトリス一塩酸バッファ 一 (pH7. 4) /1 OmM塩化マグネシウム/ 0. 5mMジチオトレイトール Zl mM EGTA (エチレングリコール一ビス(ベ一夕ーァミノェチルエーテル)一 N, N, Ν', N' 一 4酢酸) で、 そこに精製した PLK1と 20 Μの基質ペプチドと
10 Mの非標識アデノシン三リン酸 (ATP) および 0. 3 xC iの [ァ一33 P] 標識 ATP (> 2500 C i /mmo 1 e) を添加して、 反応温度 25°Cで 120分 間反応させた。 その後、 20 1の 35 OmMリン酸バッファーを反応系に添加して 反応を停止させ、その液を 384we 1 1マルチスクリーンフォスフォセルロースフ ィル夕一にスポットした。 75mMリン酸バッファーでそのフォスフォセルロースフ ィルタ一を洗浄した後、乾燥させて放射活性を液体シンチレーションカウンターで測 定した。 非標識 ATP、 [ァ一33 P] 標識 ATPは GEヘルスケア バイオサイェン ス社から、マルチスクリーンフォスフォセルロースフィル夕一はミリポア社からそれ ぞれ購入した。
本発明化合物の反応系への添加は、予め終濃度の 20倍濃度でジメチルスルホキシ ドに溶解させた溶液を 0. 5 1加えることによって行った。 反応系へジメチルスル ホキシドを 0. 5 1加えたものを対照とした。
本発明化合物の PLK 1活性に対する I C 5。値をもとめたのでその結果を下記の 表 2に示す。 表 2
以上より、 本発明化合物の PLK1阻害活性は、 方法 A及び方法 Bのいずれを用 いても同様の結果が得られ、 かつ、 その阻害活性は著しく高いことが明らかである。
3. 細胞増殖抑制作用の測定: 細胞レベルでの P L K 1阻害活性の測定
(1) 細胞培養の方法
細胞レベルでの化合物の P L K 1阻害活性の測定にはヒト子宮頸がん細胞株 H e L a S 3細胞を用いた。 He L a S 3細胞はアメリカン タイプ カルチヤ コレク シヨン (ATTC : Ame r i c an Typ e Cu l t u r e Co l l e c t i on) より入手し、 1 0%牛胎児血清添加ダルベッコ変法イーグル培地を用いて 37°Cで 5%C〇2存在下、 飽和水蒸気の C02インキュベータ一内にて培養した。 (2) 本発明に係る化合物の阻害活性測定
PLK1は哺乳動物細胞の有糸分裂期 (M期) の様々,な段階に重要な役割を果たし ていることが報告されている (ネィチヤ一 レビュー モレキュラー セル バイオ ロジー (Na t. Rev. Mo 1. Ce l l . B i o l .)、 第 5巻、 429頁、 (2 004年))。.実際、 哺乳動物細胞を PLK1に対する s i RN Aで処理してその発現 レベルを抑制すると、 細胞周期の進行が阻害されて細胞は M期に停止する。 またこの とき、 M期における染色体凝縮に必要と考えられているヒストン H3の 10番目のセ リン残基のリン酸化レベルを調べると、そのレベルが高度に亢進されていることが観 察される。 そこで、 細胞を本発明に係る化合物で処理した後にヒストン H3のリン酸 化レベルを間接蛍光抗体法により調べ、そのレベルを指標に M期細胞を同定して M期. 停止細胞の割合を解析し、 さらには各化合物の E C 50値を算出して細胞レベルでの PLK1阻害活性を評価した。
まず、 HeLaS 3細胞を、 リジン処理した 96ゥエルプレート (ファルコン社、 Fa l c on) に 1ゥエルあたり 3, 000個の割合で播種し、 前述の C〇2インキ ュベータ—内に静置した。 播種 24時間後、 段階希釈した本発明に係る化合物をプレ 一卜の各ゥエルに添加し、 さらに C〇2インキュベータ一中に静置した。 本発明に係 る化合物を添加して 18時間後、 プレートの各ゥエル内の本発明に係る化合物を含む 培地を除去した後、 氷冷した 100%メタノール (和光純薬) を 10 添加して 10分間細胞の固定ならびに膜透過性の亢進処理を行った。 次いで、 メタノールを除 去したのちのゥエルに、 50 の1%83八 ?83を添加して30分間ブロッキ ングを行った後、 一次抗体反応として 50 ALの 2. 5mg/mL 抗 pho s ph o-H i s t on eH3 (S e r l O) 抗体 (アップステート社、 Up s t a t e) を含む 1 %B SA/PB Sをゥエルに添加し、 室温で 90分間プレートを放置した。 反応終了後、 PBSで各ゥエルを 1回洗浄し、 次いで二次抗体反応として 50 の 1. 5mg/mL C y 5標識抗ゥサギ I g G (H+ L) 抗体 (ケミコン社、 C h e m i c o n) ならびに核染色試薬である 10 u g/mL DAP I (シグマ社、 S I GMA) を含む 1 %B S AZPB Sを添加し、 室温でさらに 90分間放置した。 反応 終了後、 ゥエル内の反応液を除去して 100 xLの PBSで置換した後、 イン セル アナライザー 1000 ( I N Ce l l An a l yz e r 1000 ; GE ァ マシャム社製) を用いて蛍光画像を取り込み、 各視野における M期細胞の割合 (M i t o t i c i nd e ) を解析した。 E C 50は、 各薬剤が誘導することのできる M期停止細胞の割合の最高値を 100%とした時に、その 50%を誘導することので きる薬剤濃度として定義した。
上記の方法で求められた E C 50値を以下の表 3に示した。
表 3
本発明の化合物は、表 3に示されるとおり、強い細胞増殖抑制作用を示すことから、 抗腫瘍剤として極めて有用であると考えられる。国際公開第 2 0 0 6 / 0 2 5 5 6 7 号パンフレットに開示された化合物に比べて、 本発明の化合物は、 下記の部分構造を 有することを構造的特徴とすることにより、際立って優れた細胞増殖抑制作用を示す ことは明らかである。
以上より、 本発明に係る化合物は、 優れた P L K 1阻害活性を有するとともに、 さ らに、 強い細胞増殖抑制作用を示しているので、 がん細胞の増殖 ¾強く阻害する杭が ん剤として有用であると考えられる。 即ち、 本発明に係る新規置換アミノビリミジン 誘導体又はその医薬上許容される塩若しくはエステルを含む医薬組成物、 或いは、 本 発明に係る新規置換アミノビリミジン誘導体又はその医薬上許容される塩若しくは エステルを含む抗がん剤は、 がん患者の治療において有効と考えられる。 また、 該医 薬組成物及び該抗がん剤は、薬学的に許容できる担体又は希釈剤を含んでいてもよい。 ここで、 「薬学的に許容できる担体又は希釈剤」 は、 賦形剤 〔例えば、 脂肪、 蜜蝌、 半固体及び液体のポリオール、 天然若しくは硬化オイルなど〕 ; 水 (例えば、 蒸留 水、特に、注射用蒸留水など)、生理学的食塩水、 アルコール(例えば、エタノール)、
グリセロール、 ポリオール、 ブドウ糖水溶液、 マンニトール、 植物オイルなど; 添 加剤〔例えば、 増量剤、崩壊剤、 結合剤、潤滑剤、 湿潤剤、安定剤、 乳化剤、分散剤、 保存剤、 甘味料、 着色剤、 調味料若しくは芳香剤、 濃化剤、 希釈剤、 緩衝物質、 溶媒 若しくは可溶化剤、 貯蔵効果を達成するための薬剤、 浸透圧を変えるための塩、 コー ティング剤、 又は抗酸化剤〕 などを意味する。
さらに、 本発明に係る化合物は、 エステルを含むプロドラッグとして使用すること もできる。 ここで、 「プロドラッグ」 とは、 一般に、 ある薬物分子を化学的に修飾し た誘導体で、 それ自体は生理活性を示さず、 投与後体内で、 元の薬物分子に復元し薬 効を示すものをいう。 本発明に係る化合物のプロドラッグの例として、 例えば、 その. ヒドロキシル基がリン酸基などでァシル化された上記式(I )の化合物が挙げられる。 プロドラッグ ·エステルの製造は、 当業者に周知ないし慣用の方法に従ってすること ができる。
また、 本発明に係る化合物の治療効果が期待される好適な腫瘍としては、 例えばヒ 卜の固形がん等が挙げられる。 ヒトの固形がんとしては、 例えば、 脳がん、 頭頸部が ん、 食道がん、 甲状腺がん、 小細胞がん、 非小細胞がん、 乳がん、 胃がん、 胆のう · 胆管がん、 肝がん、 膝がん、 結腸がん、 直腸がん、 卵巣がん、 絨毛上皮がん、 子宮体 がん、 子宮頸がん、 腎盂 ·尿管がん、 膀胱がん、 前立腺がん、 陰茎がん、 睾丸がん、 胎児性がん、 ウィルムスがん、 皮膚がん、 悪性黒色腫、 神経芽細胞腫、 骨肉腫、 ユー ィング腫、 軟部肉腫などが挙げられる。
次に、 上述した 「その医薬上許容される塩もしくはエステル」 について説明する。 本発明に係る化合物は、 抗がん剤などとして使用される場合には、 その薬学的に許 容しうる塩としても使用することができる。薬学的に許容しうる塩の典型例としては、 例えばナトリウム、 カリウム等のアルカリ金属との塩、 塩酸塩、 硫酸塩、 硝酸塩、 リ ン酸塩、 炭酸塩、 炭酸水素塩、 過塩素酸塩等の無機酸塩;例えば酢酸塩、 プロピオン 酸塩、 乳酸塩、 マレイン酸塩、 フマール酸塩、 酒石酸塩、 リンゴ酸塩、 クェン酸塩、 ァスコルビン酸塩等の有機酸塩;例えばメタンスルホン酸塩、 イセチオン酸塩、 ベン ゼンスルホン酸塩、 トルエンスルホン酸塩等のスルホン酸塩;例えばァスパラギン酸 塩、 グルタミン酸塩等の酸性アミノ酸塩等を挙げることができる。 本発明に係る化合 物の好ましい塩は、 塩酸塩などである。
本発明に係る化合物の薬学的に許容しうる塩の製造法は、有機合成化学分野で通常 用いられる方法を適宜組み合わせて行うことができる。 具体的には、 本発明に係る化 合物の遊離型の溶液をアル力リ溶液あるいは酸性溶液で中和滴定すること等が挙げ られる。
本発明に係る化合物のエステルとしては、 例えば、 メチルエステル、 ェチルエステ ルなどを挙げることができる。 これらのエステルは遊離力ルポキシ基を常法に従って エステル化して製造することができる。
本発明に係る化合物を杭がん剤などとして使用する際の投与形態としては各種の 形態を選択でき、 例えば錠剤、 カプセル剤、 散剤、 顆粒剤、 液剤等の経口剤、 例えば
溶液、 懸濁液等の殺菌した液状の非経口剤等が挙げられる。
ここで、 固体の製剤は、 常法に従い、 そのまま錠剤、 カプセル剤、 顆粒剤又は粉末 の形態として製造することもできるが、適当な添加物を使用して製造することもでき る。 該添加物としては、 例えば乳糖、 ブドウ糖等の糖類; 例えばトウモロコシ、 小 麦、 米等の澱粉類; 例えばステアリン酸等の脂肪酸; 例えばメタケイ酸ナトリウ ム、 アルミン酸マグネシウム、 無水リン酸カルシウム等の無機塩; 例えばポリビニ ルピロリドン、 ポリアルキレングリコール等の合成高分子; 例えばステアリン酸力 ルシゥム、ステアリン酸マグネシウム等の脂肪酸塩; 例えばステアリルアルコール、 ベンジルアルコール等のアルコール類; 例えばメチルセルロース、 カルボキシメチ. ルセルロース、 ェチルセルロース、 ヒドロキシプロピルメチルセルロース等の合成セ ルロース誘導体; その他、 水、 ゼラチン、 タルク、 植物油、 アラビアゴム等通常用 いられる添加物等が挙げられる。
これらの錠剤、 カプセル剤、 顆粒剤、 粉末等の固形製剤は、 一般的には 0 . 1〜1 0 0重量%、 好ましくは 5〜1 0 0重量%の有効成分を含むことができる。
また、 液状製剤は、 水、 アルコール類又は例えば大豆油、 ピーナツ油、 ゴマ油等の 植物由来の油等液状製剤において通常用いられる適当な添加物を使用し、懸濁液、 シ 口ップ剤、 注射剤等の形態として製造することができる。
特に、 非経口的に筋肉内注射、 静脈内注射、 皮下注射で投与する場合の適当な溶剤 又は希釈剤としては、例えば注射用蒸留水、塩酸リドカイン水溶液(筋肉内注射用)、 生理食塩水、 ブドウ糖水溶液、 エタノール、 静脈内注射用液体 (例えばクェン酸、 ク ェン酸ナトリウム等の水溶液)、 電解質溶液 (例えば点滴静注、 静脈内注射用) 等又 はこれらの混合溶液が挙げられる。
また、 これらの注射剤は予め溶解したものの他、 粉末のまま又は適当な添加物を加 えたものを要時溶解する形態もとることができる。 これらの注射液は、 通常 0. 1〜 1 0重量%の有効成分を含むことができる。
また、 経口投与の懸濁剤又はシ口ップ剤等の液剤は、 0 . 5〜 1 0重量%の有効成 分を含むことができる。
本発明に係る治療方法において、 好ましい治療単位は、 上記一般式 (I ) で示され る化合物の投与形態、 使用される上記一般式 (I ) で示される化合物の種類、 使用さ れる上記一般式 (I ) で示される化合物の剤型; 併用される他の抗がん剤の種類、 投与形態、 剤型など;及び治療されるがん細胞、 患者の状態などによって変化しても よい。 所定の条件において最適な治療は、 慣用の治療決定単位を基にして、 及び/又 は、 本明細書を考慮して、 当業者が決定することができる。
本発明に係る化合物の実際に好ましい投与量は、 使用される化合物の種類、 配合さ れた組成物の種類、適用頻度及び治療すべき特定部位及び患者の病状によって適宜増 減することができる。例えば、一日当りの成人一人当りの投与量は、経口投与の場合、 1 0なぃし 5 0 0 m g、 非経口投与、 好ましくは静脈内注射の場合、 1日当り 1 0な いし 1 0 O m gである。なお、投与回数は、投与方法及び症状により異なるが、単回、
又は 2ないし 5回に分けて投与することができる。
また、上記一般式( I )で示される化合物と併用される他の杭がん剤の治療単位は、 特に限定されないが、公知文献などにより当業者が必要に応じて決定することができ る。 例えば、.以下に示す通りである。
5—フルォロウラシル (5— FU) の治療単位は、 経口投与の場合、 例えば、 1日 200〜30 Omgを 1〜3回に連日投与し、 注射剤の場合は、 例えば、 1日 5〜1 5mgZk gを最初の 5日間連日 1日 1回静注又は点滴静注し、 以後、 5〜7. 5m gXk g を隔日に 1日 1回静注又は点滴静注する(投与量は、適宜増減してもよい)。
S— 1 (テガフール ·ギメスタット ·ォスタツトカリウム)の治療単位は、例えば、 . 初回投与量 (1回量) を体表面積に合わせて次の基準量とし、 朝食後及び夕食後の 1 日 2回、 28日間連日経口投与し、 その後 14日間休薬する。 これを 1コースとして 投与を繰り返す。 体表面積当たりの初回基準量 (テガフール相当量) は、 1. 25m 2未満: 4 Omg/回、 1. 25m 2以上〜 1. 5m2未満: 5 OmgZ回、 1. 5 m2以上: 6 Omg/回であり、 患者の状態により適宜増減する。
ゲムシ夕ビンの治療単位は、 例えば、 ゲムシタビンとして 1回 1 gZm2を 30分 かけて点滴静注し、 週 1回投与を 3週連続し、 4週目は休薬する。 これを 1コースと して投与を繰り返す。 年齢、 症状又は副作用の発現に応じて適宜減量する。
ドキソルビシン(例えば、塩酸ドキソルビシン)の治療単位は、静脈内注射の場合、 例えば、 1日 1回 10mg (0. 2mg/k g) (力価) で 4〜6日間連日静脈内ヮ ンショット投与後、 7〜 10日間休薬し、 これを 1コースとし、 2〜 3コース繰り返 す。 ここで、 総投与量は、 50 Omg (力価) /m2 (体表面積) 以下が好ましく、 その範囲内で適宜増減してもよい。
エトポシドの治療単位は、 静脈内注射の場合、 例えば、 1日 60〜10 OmgZm 2 (体表面積) で 5日間連続投与し、 3週間休薬する (投与量は、 適宜増減してもよ レ 。 これを 1コースとして繰り返す。 一方、 経口投与の場合は、 例えば、 1日 17 5〜20 Omgを 5日間連続投与し、 3週間休薬する (投与量は、 適宜増減してもよ い)。 これを 1コースとして繰り返す。
ドセタキセル (ドセタキセル水和物) の治療単位は、 例えば、 1日 1回、 ドセタキ セルとして 60mg/m2 (体表面積) を 1時間以上かけて 3〜 4週間間隔で点滴静 注する (投与量は、 適宜増減することができる)。
パクリタキセルの治療単位は、 例えば、 1日 1回 210mgZm2 (体表面積) を 3時間かけて点滴静注し、 少なくとも 3週間休薬する。 これを 1コースとして、 繰り 返す。 投与量は適宜増減することができる。
シスブラチンの治療単位は、 静脈内注射の場合、 例えば、 1日 1回 50〜70mg Zm2 (体表面積)で投与し、 3週間以上休薬する(投与量は、適宜増減してもよい)。 これを 1コースとして繰り返す。
カルポプラチンの治療単位は、 例えば、 1日 1回 300〜400mgZm2を 30 分以上かけて点滴静注し、少なくとも 4週間休薬する(投与量は適宜増減してもよい)。
これを 1コースとして繰り返す。
ォキザリブラチンの治療単位は、 1日 1回 85mgZm2を静注し、 2週間休薬し、 これを 1コースとして繰り返す。
イリノテカン (例えば、 塩酸イリノテカン) の治療単位は、 例えば、 1日 1回 10 0mg/m2、 1週間間隔で 3〜4回点滴静注し、 少なくとも 2週間休薬する。
トポテカンの治療単位は、例えば、 1日 1回 1. 5mgZm2を 5日間点滴静注し、 少なくとも 3週間休薬する。
シクロホスフアミドの治療単位は、 静脈内注射の場合、 例えば、 1日 1回 100m g連日静注し、 患者が耐えられる場合は 1日 20 Omgに増量してもよく、 総量で 3_ 000〜800 Omg投与するが、 適宜増減してもよい。 また必要に応じて筋肉内、 胸腔内、 又は腫瘍内に注射又は注入してもよい。 一方、 経口投与の場合は、 例えば、 1日 100〜 20 Omgで投与する。
ゲフイチニブの治療単位は、 例えば、 1日 1回 25 Omgを経口投与する。
セツキシマブの治療単位は、 例えば、 第 1日目に 400 m g /m 2を点滴静注し、 その後 25 OmgZmSを毎週点滴静注する。
ベバシズマブの治療単位は、 例えば、 3mgZk gを毎週点滴静注する。 '
トラスッズマブの治療単位は、 例えば、 通常、 成人に対して 1日 1回、 トラスッズ マブとして初回投与時には 4mgZk g (体重) を、 2回目以降は 2mgZk gを 9 0分以上かけて 1週間間隔で点滴静注する。
ェキセメスタンの治療単位は、 例えば、 通常、 成人には 1日 1回 25 mgを食後 に経口投与する。
リュープロレリン(例えば、酢酸リュープロレリン)の治療単位は、例えば、通常、 成人には 12週に 1回として 11. 25 mgを皮下に投与する。
イマチニブの治療単位は、 例えば、 通常、 慢性骨髄性白血病の慢性期の成人には 1 日 1回 40 Omgを食後に経口投与する。
5— FUとロイコポリンを組み合わせた場合の治療単位は、 例えば、 第 1日目から 第 5日目に 5— FU 425mgZm2、 ロイコポリン 200mgZm2を点滴静 注し、 これを 4週間間隔で繰り返す。 実施例
以下に実施例を挙げて本発明を更に具体的に説明する力 もとより本発明はこれら の実施例のみに限定されるものではない。 実施例において、 薄層クロマトグラフィー は、 プレー卜として S i l i c a g e l 6。F254 (Me r c k)、 又は NH (FU J I S I LYS IA CHEMI CAL) を、 検出法として UV検出器を用いた。 カラム用シリカゲルとしては、 Wa k o g e 1 TMC— 300又は C— 200 (和光純 薬)、 又は NH (FUJ I S I LYS IA CHEM I C AL)、 又は B i o t a g e S i、 又は B i o t ag e NH (B i o t a g e )、 又は P u r i f — P a c k (MOR I TEX) を用いた。 逆相分取液体クロマトグラフィーは、 Comb i P
r e p P r o C 18 (YMC) をカラムに用い、 0. 1%トリフルォロ酢酸水溶 液、 0. 1%トリフルォロ酢酸ァセトニトリル溶液を移動相に用いた。 MSスぺクト ルは、 JMS— SX102A (日本電子 ( J EOL))、 QUATTRO I I (マイク 口マス)、 は LC一 MSは ZMD (マイクロマス) を用いて測定した。 NMRスぺ クトルは、重ジメチルスルホキシド溶液で測定する場合には内部基準としてジメチル スルホキシドを、重クロ口ホルム溶液で測定する場合には内部基準としてテトラメチ ルシランを、重メタノール溶液で測定する場合には内部基準としてメタノールを用い、 Me r c u r y400 (400 MH z ; V a r i a n)、 I n o v a 400 (400 MHz ; Va r i a n)、 または J NM— AL 400 (400MHz ; J EOL) 型. スぺクトロメータを用いて測定し、 全 <5値を p pmで示した。 実施例で用いた略号の意味を以下に示す。
s :シンクレツ卜
d :ダブレツ卜
d d :ダブル ダブレツト
t : トリプレツト '
d t :ダブル トリプレツト
Q :クァ レテツ卜
d q :ダブル クアルテツト
qu i n :クインテッ卜
m:マルチプレツ卜
b r :ブロード
b r s :ブロード シングレツ卜
J :カツプリング定数
Hz :ヘルツ
DMSO-d6:重ジメチルスルホキシド
CDC 13:重クロ口ホルム
CD3OD:重メタノール
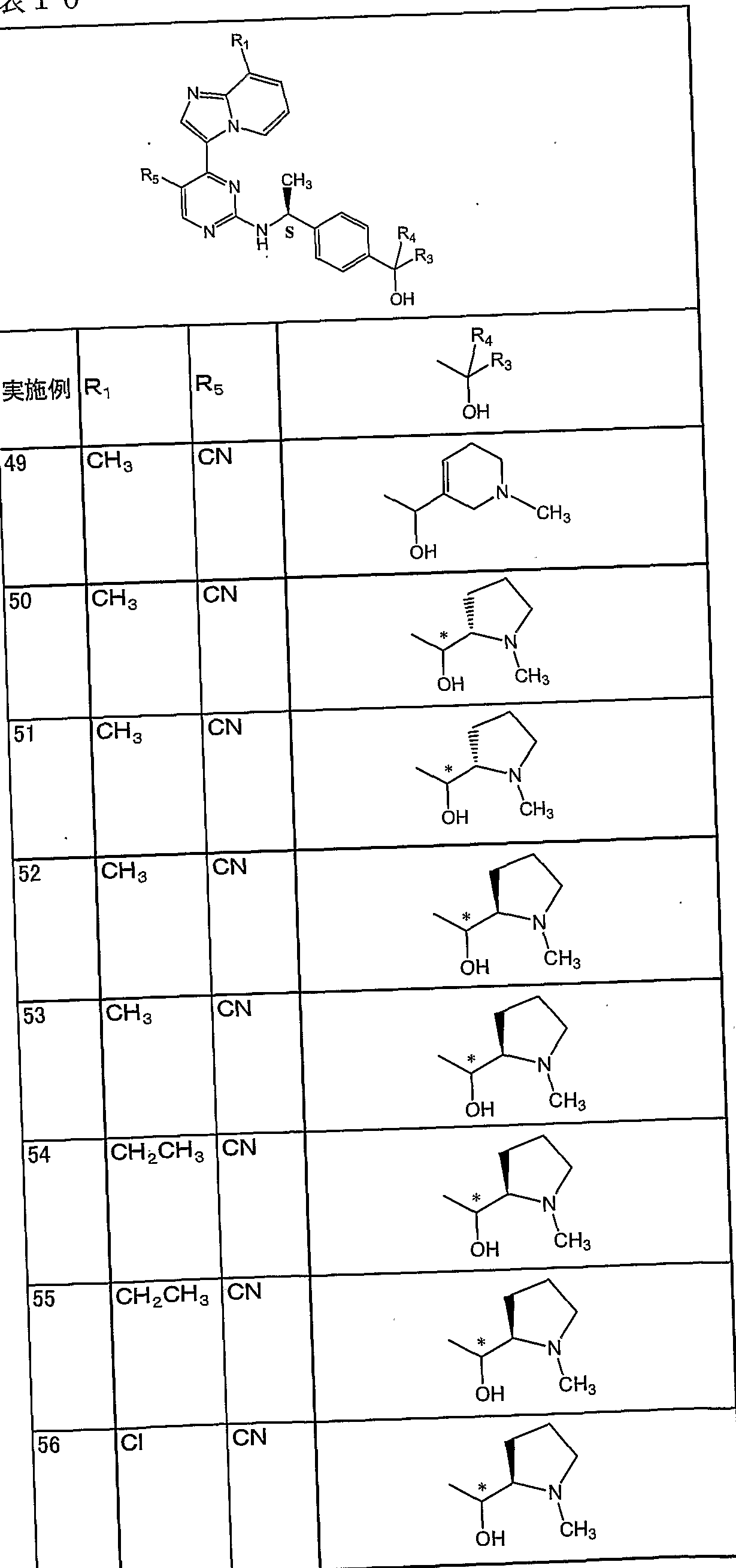
RT: リテンションタイム (保持時間) 以下、 実施例 1から 93の化合物の構造式を表 4から 15に示す。 なお、 表中、 R 3、 R4、 及び OHが結合している不斉炭素原子付近に、記号 *が記載されている化合 物は、 当該不斉炭素原子に関して、 一方が R体であり、 もう一方が S体であることを 示している。
但し、実施例 41から 44については、次の通りである。表 9において、 R3、 R4、 及び OHが結合している不斉炭素原子付近に、 記号 * *が記載されている化合物は、 当該不斉炭素原子に関して、 R体又は S体のいずれか一方の同じ異性体あることを示 す。 一方、 記号 * **が記載されている化合物は、 当該不斉炭素原子に関して、 R体
JP2007/075240 又は S体のいずれか一方の同じ異性体あるが、記号 * *が記載されている化合物と異 なる異性体であることを示す。
6
8拏
O Z^O/LOOZdT/X^d 6g «6I80/800^O
表 9
2— [ ( (I S) —1一 {4- [2— (ジメチルァミノ) 一 1ーヒドロキシェチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) リミジン一 5—力ルポ二トリル [1] (以下、 化合物 [1] という) の 合成。
(1) S— (一) - 1 - (4—プロモフエ二ル) ェチルァミン 2 gをクロ口ホルム 20mLに溶解し、 トリエヂルァミン 4. 18mLを加えた。 氷冷下、 二炭酸ジー t e r t一ブチル 2. 62 gを加え、 室温にて 30分間撹拌した。 反応液を酢酸エヂル. 300mLにて希釈し、 水、 飽和食塩水で順に洗浄し、 得られた有機層を無水硫酸マ グネシゥムにて乾燥した。 不溶物をろ過し、 ろ液を減庄濃縮し、 得られた残渣を少量 のクロ口ホルムに溶解し、 へキサンにて固化し、 t e r t—ブチル [(I S) - 1 - (4一ブロモフエニル) ェチル] カーバメート [1一 1] (以下、 化合物 [1— 1] という) 2. 6 gを白色固体として得た。
(2) 化合物 [1— 1] 600mg、 ジクロロビス (トリフエニルホスフィン) パ ラジウム ( I I ) 75 m g、 トリブチル (1一エトキシビニル) すず 743 jLi L、 お よび 1, 4一ジォキサン 3mLの混合物を 100°Cにて終夜撹拌した。 放冷後、 不溶 物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシリカゲルカラムクロマトグラフ ィ一(溶出液:へキサン/酢酸ェチル =8 Z2) にて精製し、 t e r t一ブチル [(1 S) 一 1一 (4ーァセチルフエニル) ェチル] カーバメート [1 -2] (以下、 化合 物 [1— 2] という) 367mgを無色固体として得た。
(3) 臭化べンジルトリプチルアンモニゥム 984mgをクロ口ホルム 3mLとメ 夕ノール 2mLの混合溶媒に溶解した後、 臭素 141 を加え、 さらに化合物 [1
-2] 363mgをクロ口ホルム 4mLとメタノール 0. 8mLに溶解した溶液を加 えた。 反応混合物を 40°Cにて 30分間撹拌し、 放冷後、 水を加え、 クロ口ホルムに て抽出した。有機層を飽和食塩水にて洗浄した後、無水硫酸ナトリゥムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を、:リカゲルカラムクロマトダラ フィ一(溶出液:へキサン/酢酸ェチル =7 Z3)にて精製し、 t e r t—ブチル {(1 S) 一 1一 [4— (プロモアセチル) フエニル] ェチル } カーバメート [1— 3] (以 下、 化合物 [1— 3] という) 17 Omgを無色油状物として得た。
(4) 化合物 [1一 3] 5 Omgを N, N—ジメチルホルムアミド 0. 5mLに溶 解し、 ジメチルァミン (2. 0Mテトラヒドロフラン溶液) 365 /_iLを加えた。 室 温にて終夜撹拌した後、 飽和炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出 した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物を ろ過し、ろ液を減圧濃縮し、得られた残渣を分取薄層クロマトグラフィ一にて精製し、
t e r t—ブチル {(1 S) — 1一 [4- (N, N—ジメチルダリシル) フエニル] Xチル } カーバメート [1一 4] (以下、 化合物 [1—4] という) 26. 3mgを 無色油状物として得た。 (5) 化合物 [1—4] 26. 3mgをメタノール 1. 5mLに溶解し、 水素化ほ う素ナトリウム 16. 2mgを加え、 室温にて 30分間撹拌した。 反応^?夜に飽和炭酸 水素ナトリウム水溶液を加え、 クロ口ホルムとメタノールの混合溶媒 (混合比: 9Z 1) にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥し た。 不溶物をろ過し、 ろ液を減圧濃縮し、 t e r t—ブチル ((I S) — 1一 {4一
[2 - (ジメチルァミノ) 一 1—ヒドロキシェチル] フエ二ル} ェチル) カーバメ一 ト [1— 5] (以下、 化合物 [1— 5] という) を得た。 化合物 [1一 5〕 はさらに 精製することなく次の反応に付した。
(6) 4一 [(Z) 一 2—エトキシビニル] 一 2— (メチルチオ) 一 5—ピリミジ ンカルポ二トリル(国際公開第 2006/025567号パンフレツト、 90-91 ページに記載されている方法により合成した) 10 gを 1, 4一ジォキサン 150m Lと水 3 OmLの混合溶媒に溶解した後、 氷冷下、 N—ブロモこはく酸イミド 8. 0 4 gを加え、室温にて 1. 5時間撹拌した。反応溶液に 2—アミノー 3—ピコリン 4. 89 gを加え、 さらに 2. 5時間撹拌した。 水 20 OmLを加え、 クロ口ホルムとメ タノールの混合溶媒 (混合比: 9Z1) 2Lで抽出し、 有機層を水にて洗浄した。 無 水硫酸マグネシウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られ た残渣を少量のクロ口ホルムに溶解し、 へキサンを加えて固化し、 4— (8—メチル イミダゾ [1, 2 - a] ピリジン一 3—ィル) 一 2— (メチルチオ) ピリミジン一 5 一力ルポ二トリル [1一 6] (以下、 化合物 [1— 6] という) 4. 02 gを淡褐色 固体として得た。
(7) 化合物 [1— 6] 20. lmgをクロ口ホルム 1. 5 mLに溶解し、 氷冷下、 m—クロ口過安息香酸 24. 7mgを加え、 0 °Cにて 30分間撹拌した。 反応液に飽 和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムに T抽出した。 得られた有機層を 飽和食塩水にて洗浄した後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ 液を減圧濃縮し、化合物 [1— 6] の酸化体を得た。別のフラスコに化合物 [1— 5] をクロ口ホルム 1. 5mLに溶解し、 トリフルォロ酢酸 1. 5mLを加え室温にて 1 時間撹拌した。 反応液を減圧濃縮し、 得られた残渣をメタノールに溶解し、 弱陰ィォ ン交換樹脂 (ポンドェルート ·レギュラータイプ PSA、 ジーエルサイエンス株式 会) に通しトリフルォロ酢酸を除き、 溶媒を減圧留去した。 得られた残渣をテトラヒ ドロフラン 0. 5mLに溶解し、 トリェチルァミン 9. 9 Lと、 先の酸化体をクロ 口ホルム lmLに溶解した溶液を加え、 室温にて 3時間撹拌した。 反応溶液に飽和炭 酸水素ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を飽和
食塩水にて洗浄した後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を 減圧濃縮した後、 分取薄層クロマトグラフィー (展開液:クロ口ホルムノメタノール
= 20/1) にて精製し、 目的化合物 [1] 14. 5mgを黄色固体として得た。 化合物 [1] のスぺクトルデ一夕を以下に示す。
^-NMR (CDC 13) δ 9. 64- 9. 62 (m, 1HX 1/5), 8. 95 一 8. 74 (m, 1 H+ 1HX4/5), 8. 56 (s, 1HX 1/5), 8. 53 (s , 1 HX 4/5), 7. 43-7. 15 (m, 5H), 6. 64-6. 63 (m, 1 H), 6. 02-6. 01 (m, 1HX 4/5), 5. 85 - 5. 76 (m, 1 HX 1/5), . 5. 42 - 5. 33 (m, 1 H+ 1 HX 1/5), 5. 1 6-5. 1 2 (m, 1 HX 4/5), 4. 74-4. 69 (m, 1H), 2. 69 (s, 1 HX 3/5), 2. 6 4 (s , 2H+ 1 HX 2/5), 2. 36 (s , 3H), 2. 03— 1. 80 (m, 2 H), 1. 63 (d, J = 6. 8Hz, 3 H)
ma s s : 442 (M+ 1) +. 実施例 2 ·
2— [((I S) — 1一 {4一 [1ーヒドロキシー 2— (メチルァミノ) ェチル] フエ 二ル} ェチル) ァミノ] 一 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3— ィル) ピリミジン一 5—力ルポ二トリル [2] (以下、 化合物 [2] という) の合成。
(1) Z-S AR-OH (国産化学株式会社より入手可能である) 1. O l g、 ジ イソプロピルェチルァミン 4. 72mL、 1ーヒドロキシベンゾ卜リァゾールー水和 物 1. 83 g、 N, O—ジメチルヒドロキシルァミン塩酸塩 1. 32 を1^, N—ジ メチルホルムアミド 1 5mLに溶解し、 氷冷下、 1ーェチルー 3 - (3—ジメチルァ ミノプロピル)カルポジイミド塩酸塩 2. 6 gを加え、室温にて 2. 5時間撹拌した。 反応溶液に飽和炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 得られ た有機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウムにて乾燥した。 不溶物を ろ過し、ろ液を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(溶 出液:へキサン Z酢酸ェチル =1Z1) にて精製し、 ベンジル {2— [メトキシ (メ チル) ァミノ] 一 2—ォキソェチル } メチルカ一パメート [2- 1] (以下、 化合物
[2- 1] という) 1. 02 gを淡黄色油状物として得た。
(2) 化合物 [1一 1] 20 Omgをテトラヒドロフラン 4mLに溶解し、一 78で にした後、 n_ブチルリチウム (2. 66 Mへキサン溶液) 626 を加えた。 同 温にて 1時間撹拌した後、化合物 [2— 1] 177mgのテトラヒドロフラン溶液 0. 6mLを加え、 さらに 2時間撹拌した。 反応混合物に飽和炭酸水素ナトリウム水溶液 を加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫 酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシ
リカゲルカラムクロマトグラフィー (溶出液:へキサン Z酢酸ェチル =6/4) にて 精製し、 ベンジル [2— (4一 {(1 S) ― 1― [(t e r t一ブトキシカルボニル) ァミノ] ェチル } フエニル) —2—才キソェチル] メチルカーバメート [2— 2] (以 下、 化合物 . [2— 2] という) 36. 2 mgを無色油状物として得た。
(3) 化合物 [2— 2] 159mgをテトラヒドロフラン 4mLとメタノ一ル 2 m Lの混合溶媒に溶解し、 10%パラジウム炭素触媒 48mgを加え、 水素雰囲気下 2 時間撹拌した。 10%パラジウム炭素触媒 95mgを追加し、水素雰囲気下さらに 3. 5時間撹拌した。不溶物をセライトろ過し、ろ液を減圧濃縮し、 t e r t—ブチル {(1. S) — 1— [4- (N—メチルダリシル) フエニル] ェチル }カーバメート [2-3] (以下、 化合物 [2— 3] という) の粗精製物を得た。 化合物 [2— 3] はさらに精 製することなく次の反応に付した。
(4) 化合物 [2— 3] をクロ口ホルム 3mLに溶解し、 ベンズアルデヒド 76 Lとトリァセトキシ水素化ほう素ナトリウム 158mgを加えた。室温にて終夜撹拌 した後、 飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムとメタノールの混合溶 媒 (混合比: 9/1) にて抽出した。 得られた有機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られ た残渣を分取薄層クロマトグラフィーにて精製し、 t e r t—ブチル {(1 S) — 1 一 [4一 (N—ベンジル— N—メチルダリシル) フエニル]ェチル }カーバメート [2 -4] (以下、 化合物 [2— 4] という) 64m gを無色油状物として得た。
(5) 匕合物 [1-6] 39mgとィ匕合物 [2-4] 64mgから、実施例 1— (7) の方法に準じて、 2— {[(1 S) 一 1― (4- {2- [ベンジル (メチル) ァミノ] 一 1ーヒドロキシェチル } フエニル) ェチル] アミノ} 一 4一 (8—メチルイミダゾ [1, 2 -a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [2— 5] (以 下、 化合物 [2— 5] という) 36mgを黄色油状物として得た。
(6) 化合物 [2— 5] 18mgをテトラヒドロフラン lmLとメタノール 0. 5 mLに溶解し、 20%水酸化パラジウム炭素触媒 18mgを加えた。 水素雰囲気下、
4時間撹拌した後、 不溶物をセライトろ過し、 ろ液を減圧濃縮した。 得られた残渣を 分取薄層クロマトグラフィーにて精製し、 目的化合物 [2] 6mgを淡黄色固体とし て得た。 化合物 [2] のスペクトルデ一夕を以下に示す。
1H— NMR (CDC 13) (5 : 9. 74 - 9. 73 (m, 1 HX 1/5), 8. 88 — 8. 80 (m, 1 H+ 1 HX 4/5), 8. 54 (s , 1 HX 1/5), 8. 52 (s , 1HX 4/5), 7. 43- 6. 80 (m, 5H), 6. 70— 6. 65 (m, 1H),
5. 32 - 5. 30 (m, 1H+1HX 1/5), 5. 15— 5. 10 (m, 1 HX 4/5), 4. 82 -4. 79 (m, 1 H), 2. 80-2. 76 (m, 2H), 2. 66 (s , 1 HX 3/5), 2. 62 (s, 2 H+ 1 HX 2X5), 2. 49 (s , 1 HX 3/5), 2. 46 (s, 2H+ 1HX2/5), 1. 62 (d, J = 6. 8Hz, 3H), 1 · 27 (b r s, 1 H)
ma s s : 428 (M+ 1 ) +. 実施例 3
2— ( { (I S) —1一 [4- (1ーヒドロキシー 2—ピロリジン— 1—ィルェチル). フエニル] ェチル } ァミノ) 一 4一 (8—メチルイミダゾ [1, 2 - a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [3] (以下、 化合物 [3] という) の 合成。
(1) 化合物 [1— 6] 30 Omgをクロ口ホルム 6mLに溶解し、 氷冷下、 m— クロ口過安息香酸 368mgを加えた。 同温にて 30分間撹拌した後、 飽和炭酸水素 ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を無水硫酸ナ トリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をク ロロホルム 3mLに溶解し、 S— (一) - 1 - (4一ブロモフエニル) ェチルァミン 257mgとトリェチルァミン 224 ^ Lのテトラヒドロフラン溶液 3 mLに加え、 室温にて終夜撹拌した。 反応混合物に飽和炭酸水素ナトリウム水溶液を加え、 クロ口 ホルムにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウム にて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られた残渣をシリカゲル カラムクロマトグラフィー (溶出液:クロ口ホルム Zメタノール =20Z1) にて精 製し、 2— {[(1 S) 一 1一 (4一ブロモフエニル) ェチル] ァミノ) —4一 (8— メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリ ル [3— 1] (以下、 化合物 [3— 1] という) 252mgを淡黄色油状物として得 た。
(2) 化合物 [3- 1] 252mg、 ジクロロビス (トリフエニルホスフィン) パ ラジウム ( I I ) 20 m g、 トリブチル (1一エトキシビニル) すず 216 xL、 お よび 1, 4一ジォキサン 3. 5mLの混合物を 100でにて終夜撹拌した。 反応混合 物を減圧濃縮した後、 残渣をテトラヒドロフラン 3mLと水 lmLに溶解した。 N— ブロモこはく酸イミド 135mgを加え、 室温にて 30分間撹拌した。 反応混合物に 飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層 を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液 を減圧濃縮した後、 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液: へキサン/クロ口ホルムノ酢酸ェチル =3/ 2/ 5) にて精製し、 2— ({(1 S) 一 1一 [4- (プロモアセチル) フエニル] ェチル } ァミノ) 一 4一 (8—メチルイミ
ダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [3-2] (以下、 化合物 [3— 2] という) 16 Omgを黄色固体として得た。
(3) 化合物 [3— 2] 20mgを N, N—ジメチルホルムアミド 0. 4mLに溶 解し、 ピロリジン 7 / Lを加えた。 反応混合物を室温にて 30分間撹拌した後、 飽和 炭酸水素ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。得られた有機層を飽 和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減 圧濃縮した、 得られた残渣を分取薄層クロマトグラフィーにて精製し、 4一 (8—メ チルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— ({(1 S) ー1一 [4一 (ピ. 口リジン一 1ーィルァセチル) フエニル] ェチル } ァミノ) ピリミジン一 5—力ルポ 二トリル [3— 3] (以下、 化合物 [3— 3] という) 3mgを無色油状物として得 た。
(4) 化合物 [3— 3] 3mgをテ卜ラヒドロフラン 0. 4mLとメタノール 0. 4mLの混合溶媒に溶解し、 20 %水酸化パラジウム炭素触媒 9mgを加えた。 混合 物を水素雰囲気下、 室温にて 2時間撹拌した後、 触媒をセライトろ過した'。 ろ液を減 圧濃縮し、得られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [3] 1. 5 mgを無色固体として得た。 化合物 [3] のスペクトルデ一夕を以下に示す。
— NMR (CDC 13) δ : 9. 66 - 9. 56 (m, 1 HX 1/5), 8. 95 一 8. 74 (m, 1H+ 1HX4ノ 5), 8. 56 (s , 1 HX 1/5), 8. 52 (s , 1 HX 4/5), 7. 45-7. 16 (m, 5 H), 6. 65 -6. 64 (m, 1 H), 6. 08 - 6. 00 (m, 1 HX 4/5), 5. 88— 5. 80 (m, 1 HX 1/5), 5. 36 - 5. 28 (m, 1H+1HX 1Z5), 5. 13-5. 12 (m, 1 HX 4/5), 4. 88 -4. 86 (m, 1H), 2. 92 - 2. 62 (m, 4H), 2. 69 (s , 1 H X 3 / 5 ) , 2. 64 (s , 2H+ 1 HX 2/5), 2. 15— 1. 7 0 (m, 6H), 1. 62 (d, J = 6. 8Hz, 3 H)
ma s s : 468 (M+ 1 ) +. 実施例 4
2- [ ( (I S) - 1- {4- [2- (t e r t—ブチルァミノ) 一 1ーヒドロキシ ェチル] フエ二ル} ェチル) ァミノ] —4一 (8—メチルイミダゾ [1, 2— a] ピ リジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [4] (以下、 化合物 [4] と いう) の合成。 化合物 [3— 2] 4 Omgと N— t e r t一ブチルベンジルアミン 15. 3 Lから、 実施例 3— (3)、 (4) の方法に準じて 目的化合物 [4] 2. 8mgを無色固体と
して得た。 化合物 [4] のスペクトルデータを以下に示す。
iH— NMR (CDC 13) (3 : 9. 6 5 - 9. 6 3 (m, 1HX 1/5), 8. 94 一 8. 7 2 (m, 1 H+ 1 HX 4/5), 8. 56 - 8. 5 1 (m, 1H), 7. 45 一 6. 94 (m, 5H), 6. 6 7 - 6. 64 (m, 1 H), 6. 3 1 - 6. 2 7 (m, 1 HX4/5), 6. 00 - 5. 9 3 (m, 1 HX 1/5), 5. 34- 5. 2 5 (m, 1 HX 1/5), 5. 1 4- 5. 1 0 (m, 1 HX 4/5), 5. 04- 5. 0 1 (m, 1 H), 3. 2 0 - 2. 7 6 (m, 2 H), 2. 6 8 (s, 1 HX 3/5), 2. 6 2 (s , 2H+ 1 HX 2/5), 1. 64- 1. 5 9 (m, 3H), 1. 2 7 (s , 4 H+ 1 HX 1/2), 1. 2 5 (s , 4H+ 1 HX 1/2)
ma s s : 47 0 (M+ 1 ) +. 実施例 5
2— [ ( (I S) 一 1一 {4- [ (I S) - 2 - ( t e r t—プチルァミノ) 一 1— ヒドロキシェチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [5] (以下、 化合 物 [5] という) の合成。 (1) 化合物 [1一 1] 1. 0 g、 ビニルトリフルォロほう酸カリウム 8 9 2mg、 ジクロロビス (卜リフエニルホスフィン) パラジウム (I I ) 468mg、 2. 0 炭酸ナトリウム水溶液 1 0mL、 および N, N—ジメチルホルムアミド 2 OmLの混 合物を、 9 0°Cにて終夜撹拌した。 放冷後、 水を加え、 酢酸ェチルにて抽出した。 得 られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。不溶物を ろ過し、ろ液を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(溶 出液:へキサン/酢酸ェチル = 1 0 0/0 - 7 5/2 5) にて精製し、 t e r t—ブ チル [(I S) — 1— (4一ビニルフエニル)ェチル]カーバメ一卜 [5— 1] (以下、 化合物 [5— 1] という) 48 5mgを白色固体として得た。 (2) AD—. mi X— (アルドリッチ社(A 1 d r i c h) より入手可能である) 2 5. 3 gを t e r t—ブ夕ノール 1 1 OmLと水 1 1 OmLの混合溶媒に加え、 懸 濁液とした。 氷冷下、 化合物 [5— 1] 4. 34 gを加え、 同温にて 24時間撹拌し た。 反応溶液に亜硫酸ナトリウム 2 5 gを加え、 0°Cにて 3 0分間撹拌した後、 クロ 口ホルムにて抽出し、 得られた有機層を無水硫酸ナトリウムにて乾燥した。 不溶物を ろ過し、ろ液を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(溶 出液:クロ口ホルム Zメタノール = 1 0 0/0— 9 0ノ 1 0) にて精製し、 t e r t 一ブチル ((1 S) — 1一 {4- [(1 S) 一 1, 2—ジヒドロキシェチル] フエ二ル} ェチル) カーバメート [5- 2] (以下、 化合物 [5 - 2] という) 4. 3 2 gを白
色固体として得た。
(3) 化合物 [5— 2] 4. 32 gをピリジン 5 OmLに溶解し、 氷冷下、 塩化 p 一トルエンスルホニル 3. 66 gを加えた。 反応混合物を 0 Cにて終夜撹拌した後、 水を加え、 酢酸ェチルにて抽出し、 得られた有機層を無水硫酸ナトリウムにて洗浄し た。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られた残渣をシリカゲルカラムクロ マトグラフィ一 (溶出液:へキサン/酢酸ェチル =50/50 - 0/100) にて精 製し、 (2 S) — 2— (4一 {(1 S) - 1 - [(t e r t—ブトキシカルボニル) ァ ミノ] ェチル } フエニル) —2—ヒドロキシェチル 4一メチルベンゼンスルホネー. ト [5— 3] (以下、 化合物 [5— 3] という) 6. 52 gを無色アモルファスとし て得た。
(4) 化合物 [5— 3] 6. 52 gをクロ口ホルム 44. 4mLに溶解し、 水酸化 ナトリウム 2. 4 gを水 5. 54mLに溶解した溶液を加えた後、 硫酸水素テトラブ チルアンモニゥム 203mgを加えた。 反応混合物を室温にて終夜撹拌した後、 水を 加え、 酢酸ェチルにて抽出し、 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナ トリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られた残渣をシ リカゲルカラムクロマトグラフィー (溶出液: 50%クロ口ホルム一へキサン Z酢酸 ェチル = 100/0 - 7 5/25) にて精製し、 t e r t一ブチル ((1 S) 一 1一 {4.一 [(2 S) 一ォキシラン一 2一ィル] フエ二ル} ェチル) カーバメート [5 - 4] (以下、 化合物 [5— 4] という) 4. 4 gを白色固体として得た。
(5) 化合物 [5— 4] 4. 4 gをエタノール 4 OmLに溶解し、 t e r t—プチ ルァミン 2 OmLを加え、 50°Cにて 3日間撹拌した。 反応液を濃縮し、 得られた残 渣をシリカゲルカラムクロマトグラフィー (溶出液:へキサン/酢酸ェチル = 100 /0-0/100) にて精製し、 t e r t一ブチル ((I S) ― 1― {4- [(I S) 一 2— (t e r t一プチルァミノ) 一 1ーヒドロキシェチル] フエ二ル} ェチル) 力 一バメ一卜 [5— 5] (以下、 化合物 [5— 5] という) 3. l gを白色固体として 得た。 .
(6) 4- [(Z) — 2—エトキシビニル] 一 2— (メチルチオ) —5—ピリミジ ンカルポ二トリル (国際公開第 2006/025567号パンフレツト、 90— 91 ページに記載されている方法により合成した) 4. 89 ^を1, 4一ジォキサン 50 mLと水 5 mLの混合溶媒に溶解した後、 氷冷下、 N—ブロモこはく酸イミド 3. 9 3 gを加え、 室温にて 1時間撹拌した。 反応溶液に 3—ェチルピリジン— 2—ァミン (国際公開第 2006/025567号パンフレツ卜、 142ページに記載されてい る方法により合成した) 2. 7 gを加え、 室温にて終夜撹拌した。 飽和炭酸水素ナト リウム水溶液を加え、 クロ口ホルムにて抽出し、 得られた有機層を無水硫酸ナトリウ
ムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣にジェチルエー テルを加え、 3時間撹拌した。 生じた固体をろ取し、 減圧乾燥し、 4一 (8—ェチル イミダゾ [1, 2— a] ピリジン一 3—ィル) - 2 - (メチルチオ) ピリミジン一 5 一力ルポ二トリル [5— 6] (以下、 化合物 [5— 6] という) 4. 46 gを淡黄色 固体として得た。
(7) 化合物 [5 6] 2 gをクロ口ホルム 1 5 OmLに溶解し、 氷冷下、 m—ク ロロ過安息香酸 1. 9 5 gを加え、 0°Cにて 3 0分間撹拌した。 反応液に飽和炭酸水 素ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を飽和食塩. 水にて洗浄した後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧 濃縮し、 化合物 [5— 6] の酸化体を得た。 別のフラスコに化合物 [5— 5] をクロ 口ホルム 2 OmLに溶解し、 氷冷下、 トリフルォロ酢酸 1 OmLを加え室温にて 1時 間撹拌した。 反応液を減圧濃縮し、 得られた残渣をテ卜ラヒドロフラン 2 OmLに溶 解し、 トリェチルァミン 2 5mLと、 先の酸化体をクロ口ホルム 2 OmLに溶解した 溶液を加え、 室温にて 1時間撹拌した。 反応溶液に飽和炭酸水素ナトリゥム水溶液を 加え、 クロ口ホルムとメタノールの混合溶媒 (混合比: 9/1) にて抽出し、 得られ た有機層を水で洗浄した後、 無水硫酸ナトリウムにて洗浄した。 不溶物をろ過し、 ろ 液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液:ク ロロホルム/メタノール = 1 0 0Z0— 9 0 1 0) にて精製し、 目的化合物 [5] 1. '2 gを淡黄色固体として得た。 化合物 [5] 8 03mgをエタノール 2 OmLに 懸濁させ、 1 N塩酸水溶液 1. 6 6mLを加え、 均一な溶液とした。 混合物を減圧濃 縮し、 得られた残渣にエタノール 2 OmLを加え、 再度減圧濃縮した。 生じた固体を 40°Cにて減圧乾燥し、 化合物 [5] の塩酸塩 8 3 5mgを淡黄色固体として得た。 化合物 [5] のスペクトルデータを以下に示す。
XH-NMR (DMSO— d6) δ 9. 9 9 (d, J = 6. 8Hz, 1 HX 1/2), 9. 1 0 (d, J = 6. 3 Hz , 1 HX 1/2), 9. 0 4 (d, J = 7. 3Hz, 1 HX 1/2), 8. 9 9 (d, J =7. 8Hz, 1 HX 1X2), 8. 8 9 (b r s , 1H), 8. 7 6 (s , 1H), 8. 7 1 (s , 1 HX 1/2), 8. 64 (s , 1 H X 1/2), 8; 42 - 8. 40 (m, 1 H), 7. 46 - 7. 3 8. (m, 4H), 7. 1 9 ( t , J =7. 1Hz, 1 HX 1/2), 7. 0 7 ( t, J = 7. 1 H z,' 1 H X 1/2), 6. 1 0 (m, 1 H), 5. 24 ( t , J = 7. 3Hz , 1 HX 1/2), 5. 1 5 (t , J = 7. 1 Hz , 1 HX 1/2), 4. 8 7 (m, 1H), 3. 0 7— 2. 9 7 (m, 3H), 2. 90 - 2. 8.5 (m, 1 H), 1. 5 3 (d, J = 7. 3 Hz, 3HX 1/2), 1 · 5 0 (d, J = 6. 8Hz, 3 HX 1/2), 1. 3 3 ( t , J = 7. 6Hz, 3 HX 1/2), 1. 3 0 ( t , J = 7. 3Hz , 3HX 1/2), 1. 2 9 (s, 9HX 1/2), 1. 26 (s , 9HX 1/2)
ma s s : 484 (M+ 1) +.
実施例 6
2— [ ( (I S) — 1一 {4一 [ (1 R) 一 2— ( t e r t—プチルァミノ) 一 1一 ヒドロキシ Xチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—ェチルイミダゾ [1, 2 - a] ピリジン— 3—ィル) ピリミジン一 5—力ルポ二トリル [6] (以下、 化合 物 [6] という) の合成。
(1) 化合物 [5— 1] 4. 14 gと AD—m i X— /3 (アルドリッチ社 (A 1 d r i c h) より入手可能である) 24 gから、 実施例 5— (2) 〜 (5) の方法に準. じて、 t e r t—ブチル ((I S) - 1 - {4一 [(1 R) — 2— ( t e r t一ブチル ァミノ) ー1ーヒドロキシェチル] フエ二ル} ェチル) カーバメート [6— 1] (以 下、 化合物 [6— 1] という) 2. 5 gを白色固体として得た。
(2) 化合物 [5— 6] 1. 5 gと化合物 [6— 1] 1. 5 6 gから、 実施例 5— (7) の方法に準じて、 化合物 [6] の塩酸塩 1. 1 7 gを淡黄色泡状物質として得 た。 ' 化合物 [6] のスペクトルデータを以下に示す。
一 NMR (DMSO-d6) δ : 9. 9 8 (d, J = 6. 8Hz, 1HX 1/2), 9. 04 (d, J = 8. 3Hz, 1 H), 9. 0 1 (d, J = 8. 3 Hz, 1 HX 1
Z2), 8. 9 7 - 8. 9 2 (m, 1 H), 8. 7 6 (s, 1 HX 1/2), 8. 7 6 (s, 1 HX 1/2), 8. 7 1 (s , 1 HX 1/2), 7. 49 (d, J =6. 3H z , 1 HX 1/2), 7. 44- 7. 3 7 (m, 5H), 7. 2 1 ( t , J = 7. 3H z , 1 HX 1/2), 7. 0 8 ( t , J = 7. 3Hz, 1HX 1/2), 6. 1 1 (b r s , 1 HX 1/2), 5. 2 7 - 5. 2 0 (m, 1 HX 1/2), 5. 1 7 - 5. 1
0 (m, 1 HX 1/2), 4. 8 9 -4. 8 5 (m, 1 H), 3. 0 5 -2. 9 5 (m,
3H+ 1 HX 1/2), 2. 9 1 - 2. 8 0 (m, 1 HX 1/2), 1. 5 3— 1. 4
9 (m, 3H), 1. 3 4- 1. 3 0 (m, 3H), 1. 2 8 (s , 9HX 1/2),
1. 2 5 (s , 9HX 1/2).
ma s s : 484 (M+ 1 ) +. 実施例 7
2— { [ (I S) — 1— (4— { 2 - [ (1, 1ージメチルプロピル) ァミノ] 一 1 ーヒドロキシェチル }フエニル)ェチル]アミノ}一 4一 (8—ェチルイミダゾ [1, 2 -a] ピリジン— 3—ィル) ピリミジン一 5—力ルポ二トリル [7] (以下、 化合 物 [7] という) の合成。 化合物 [5— 1] 48 5mgをアセトン 1 8mLと水 3mLの混合溶媒に溶
解し、 4一メチルモルホリン 4—ォキシド一水和物 46 Omg、四酸化オスミウム(0. 1 M水溶液) 5 9 0 Lを順に加え、 室温にて終夜撹拌した。 1 0 %亜硫酸ナ卜リウ ム水溶液 1 0mLを加え、 3 0分間撹拌した後、 酢酸ェチルにて抽出した。 得られた 有機層を飽和食塩水にて洗浄し、無水硫酸ナトリゥムにて乾燥した。不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液: へキサン/酢酸ェチル = 1 0 0/0— 0/1 0 0) にて精製し、 t e r t—ブチル {(1 S) 一 1— [4一 (1, 2—ジヒドロキシェチル) フエニル] ェチル } カーバ メート [7— 1] (以下、 化合物 [7— 1] という) 3 5 lmgを無色油状物として 得た。
(2) 化合物 [7— 1] 3 5 lmg力、ら、実施例 5— (3)、 (4) の方法に準じて、 t e r t—ブチル [(1 S) — 1一 (4一ォキシラン一 2—^ fルフエ二ル) ェチル] カーバメート [7— 2] (以下、 化合物 [7— 2] という) 2 5 5mgを白色固体と して得た。
(3) 化合物 [7— 2] 3 Omgをエタノール 2mLに溶解し、 t e r 't—ァミル ァミン 2mLを加え、 50°Cにて終夜撹拌した。 反応液を濃縮した後、 得られた残渣 をシリカゲルカラムクロマトグラフィー (溶出液:へキサン Z酢酸ェチル = 1 0 0/ 0 - 2 0/8 0) にて精製し、 t e r t一ブチル [(I S) — 1一 (4一 { 2— [(1, 1ージメチルプロピル) ァミノ] 一 1ーヒドロキシェチル } フエニル) ェチル] カー バメート [7— 3] (以下、 化合物 [7— 3] という) 3 lmgを無色油状物として 得た。
(4) 化合物 [7— 3] 3 lmgと化合物 [5— 6] 3 lmg力、ら、実施例 5 の方法に準じて、 目的化合物 [7] の塩酸塩 2 Omgを黄色固体として得た。 化合物 [7] のスペクトルデータを以下に示す。
XH-NMR (DMSO— d6) (5 : 9. 9 9 (d, J = 7. 3Hz, 1 HX 1/2), 9. 0 8 - 9. 0 2 (m, 1 HX 1/2), 9. 0 0— 8.. 9 0 (b r s , 1 H), 8. 7 9 - 8. 6 7 (m, 2H), 8. 40 - 8. 3 0 (b r s , 1 H), 7. 54- 7. 3 8 (m, 4H), 7. 2 5 ( t , J = 6. 8Hz , 1HX 1/2), 7. 1 1 (d t , J = 3. 2Hz , 7. 1 Hz , 1 HX 1/2), 5. 24 (t , J = 7. 3Hz , 1 HX 1/2), 5. 1 3 (t , J = 6. 1Hz, 1 HX 1/2), 4. 9 1—4. 8 8 (m, 1H), 3. 0 7— 2. 9 7 (m, 3H), 2. 9 1 - 2. 8 8 (m, 1H), 1. 6 5 (q, J = 7. 6Hz , 2 HX 1/2), 1. 6 2 (q, J = 7. 6Hz, 2HX 1/2), 1. 54 (d, J = 6. 8Hz, 3HX 1/2), 1. 5 0 (d, J =7. 3Hz, 3HX 1/2), 1. 3 3 ( t , J = 7. 6Hz, 3HX 1/2), 1. 3 0 ( t , J = 6. 8Hz, 3HX 1/2), 1. 24 (s , 6HX 1/2), 1. 2
2 (s, 6HX 1/2), 0. 8 5 (q, J = 7. 6 Hz , 3 H)
ma s s : 49 8 (M+ 1) +. 実施例 8 .
4- (8—ェチルイミダゾ [1, 2— a] ピリジン— 3—ィル) 一 2— [ ( (1 S) ― 1一 {4- [1—ヒドロキシー 2 - (4ーメチルピペラジン一 1—ィル) ェチル] フエ二ル} ェチル) ァミノ] ピリミジン— 5—力ルポ二トリル [8] (以下、 化合物 [8] という) の合成。 . (1) 化合物 [7— 2] 3 Omgと N—メチルピペリジン 2mLから、 実施例 7— (3) の方法に準じて t e r t—プチル ((I S) — 1一 {4- [1ーヒドロキシー 2— (4ーメチルビペラジン一 1一ィル) ェチル] フエ二ル} ェチル) カーバメート [8- 1] (以下、 化合物 [8— 1] という) 3 5mgを無色油状物として得た。 (2) 化合物 [8— 1] 3 5mgと化合物 [5— 6] 34mgから、実施例 5—(7) の方法に準じて、 目的化合物 [8] の塩酸塩 1 9mgを黄色固体として得た。 化合物 [8] のスペクトルデータを以下に示す。
^-NMR (DMS〇一d6) (3 : 1 0. 0 0 (d, J = 6. 8Hz , 1 HX 1/2), 9. 0 2 ( t , J = 7. 3Hz , 1 H), 8. 9 8 (d, J = 7. 8Hz, 1 HX 1 /2), 8. 7 6 (s, 1 H), 8. 7 1 (s , 1 HX 1/2), 8. 64 (d, J = 1. 5Hz, 1 HX 1/2), 7. 48 - 7. 3 7 (m, 4H), 7. 1 9 ( t, J = 6. 8Hz , 1 HX 1 /2), 7. 04 (m, 1 HX 1/2), 5. 2 3 ( t, J = 7. 3Hz , 1 HX 1X2), 5. 1 3 ( t , J = 7. 1 Hz , 1 HX 1/2), 5. 1 1 - 5. 0 0 (m, 1 H), 3. 46 - 3. 4 1 (m, 8H), 3. 0 6 - 2. 9 6 (m, 3H), 2. 7 7 (m, 4H), 1. 5 3 (d, J = 7. 3Hz 3 HX 1/2) 5 0 (d, J = 7. 3Hz, 3HX 1/2)., 1. 3 3 ( t , J = 7. 3Hz 3H X 1/2), 1. 3 0 ( t, J = 7. 3Hz , 3HX 1/2)
m a s s : 548 (M+ 1) +. 実施例 9
2— [ ( (I S) - 1 - {4- [ (1 R) — 2— (t e r t—プチルァミノ) 一 1一 ヒドロキシェチル] フエ二ル} ェチル) ァミノ] 一 4— (8—クロロイミダゾ [1, 2— a] ピリジン— 3—ィル) ピリミジンー5—力ルポ二トリル [9] (以下、 化合 物 [9Ί という) の合成。
(1) 2 _アミノー 3—クロ口ピリジン 4 gと 4一 [(Z) — 2—エトキシビニル] — 2— (メチルチオ)— 5—ピリミジン力ルポ二トリル 6. 9 gから、実施例 1一(6)
の方法に準じて 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2 一 (メチルチオ) ピリミジン一 5—力ルポ二トリル [9— 1] (以下、 化合物 [9一 1] という) 6. 3 gを淡褐色固体として得た。 (2) 化合物 [9一 1] 2 Omgと化合物 [6— 1] 2 2mg力、ら、実施例 1一(7) の方法に準じて、 目的化合物 [9] 1 5mgを白色泡状物質として得た。 化合物 [9] のスペクトルデータを以下に示す。
XH-NMR (DMSO-d6) (5 : 1 0. 0 6 (d, J = 7. 3 Hz , 1 HX 1Z. 2), 9. 0 6 (d, J = 7. 3Hz, 1 HX 1/2), 8. 9 6 (d, J = 7. 3H z, 1 H), 8. 7 8 ( s , 1 HX 1/2), 8. 7 5 (s , 1HX 1/2), 8. 7 4 (s , 1HX 1/2), 8. 6 1 (s , 1 HX 1/2), 7. 7 9 (d, J = 7. 3 Hz, 1HX 1/2), 7. 7 1 (d, J = 7. 3Hz, 1 HX 1/2), 7. 3 6 - 7. 2 9 (m, 4H), 7. 1 9 ( t, J = 7. 3Hz , 1 HX 1/2) , 6. 9 9 ( t , J = 7. 3Hz, 1 HX 1/2), 5. 2 8 - 5. 2 5 (m, 2H), 4. 49 -4. 44 (m, 1H), 2. 5 6 - 2. 5 1 (m, 2H), 1. 5 3— 1. 48 (m, 3H), 1. 0 0 (s, 9HX 1/2), 0. 9 3 (s , 9HX 1/2).
m s s : 490 (M+ 1 ) +. 実施例 1 0
2- [ ( (I S) — 1— {4— [ (1 R) 一 2— (t e r t—プチルァミノ) 一 1一 ヒドロキシェチル] フエ二ル} ェチル) ァミノ] 一 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン— 5—力ルポ二トリル [1 0] (以下、 化合物 [1 0] という) の合成。
(1) 2—アミノー 3—ホルミルピリジン 5. 6 9 gと 4一 [(Z) 一 2—ェトキ シビニル] - 2 - (メチルチオ) 一 5—ピリミジン力ルポ二トリル 1 0 gから、 実施 例 1一 (6) の方法に準じて 4— (8—ホルミルイミダゾ [1, 2— a] ピリジン—
3—ィル) — 2— (メチルチオ) ピリミジン— 5—力ルポ二トリル [1 0— 1] (以 下、 化合物 [1 0— 1] という) 7. 1 5 gを淡褐色固体として得た。
(2) 化合物 [1 0— 1] 5 gをクロ口ホルム 20 OmLに懸濁させ、 氷冷下、 ビ ス(2—メトキシェチル)アミノサルファートリフルオリド 7. 8 mLを加え同温にて 撹拌した。 反応混合物は約 3 0分後に透明な褐色溶液となり、 さらに 3 0分間撹拌し た後に、飽和炭酸水素ナトリウム水溶液 3 0 OmLを加えた。有機層と水層を分離し、 水層をクロ口ホルムとメタノールの混合溶媒 (混合比: 9/1) で抽出した。 先の有 機層と合わせて水で洗浄し、 無水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液:
クロ口ホルム Z酢酸ェチル = 1 0 0Z0— 3 0Z7 0) にて精製した。 溶媒を留去し た後、 少量のクロ口ホルムに溶解し、 へキサンを加えて固化させ、 4一 [8— (ジフ ルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] — 2— (メチルチオ) ピリミジン一 5—力ルポ二卜リル [1 0— 2] (以下、 化合物 [1 0— 2] という) 2. 0 3 gを淡褐色固体として得た。
(3) 化合物 [10— 2] 2 Omgと化合物 [6— 1] 2 1mgから、 実施例 1一 (7) の方法に準じて、 目的化合物 [1 0] 1 2mgを白色泡状物質として得た。 化合物 [1 0] のスペクトルデータを以下に示す。
^-NMR (DMSO— d6) 5 : 1 0. 20 (d, J = 7. 3Hz , 1 HX 1/2), 9. 0 9 (d, J = 7. 3Hz , 1 HX 1 /2), 9. 0 5 (d, J = 7. 3Hz, 1 HX 1/2), 8. 9 6 (d, J = 7. 3Hz, 1 HX 1 /2), 8. 7 8 (s , 1 HX 1/2), 8. 7 7 (s , 1 HX 1/2), 8. 74 (s , 1 HX 1/2), 8. 6 3 (s, 1 HX 1/2), 7 · 8 6 (d, J = 7. 3Hz, 1 HX 1/2), 7. 7
8 (d, J = 7. 3 Hz , 1 HX 1/2), 7. 5 1 ( t , J = 54. 6Hz , 1 H X 1/2), 7. 45 ( t , J = 54. 1 Hz, 1 HX 1/2), 7. 3 7 - 7. 2 9
(m, 5H+ 1 HX 1/2), 7. 1 2 ( t , J = 7. 3Hz, 1 HX 1/2), 5. 30 - 5. 04 (m, 2H), 4. 5 1 -4. 45 (m, 1 H), 2. 6 0 - 2. 5 1 (iri, 2H), 1. 54- 1. 49 (m, 3H), 1. 0 0 (s, 9HX 1/2), 0.
94 (s, 9HX 1/2).
ma s s : 506 (M+ 1 ) +. 実施例 1 1
(1 R) — 1ー [4一 ( (I S) — 1一 { [5—プロモ—4 _ (8—ェチルイミダゾ [1, 2 - a] ピリジン— 3—ィル) ピリミジン一 2—ィル] ァミノ) ェチル) フエ ニル] —2— ( t e r t一プチルァミノ) エタノール [1 1] (以下、 化合物 [1 1] という) の合成。 (1) 5—ブロモ一 2, 4ージクロ口ピリミジン 3. 1 1 g、 シス一 1一エトキシ _ 2—トリー n—ブチルス夕ニルエチレン ( J . Am. Ch em. S o c. , 1 9 7 7, 9 9, 7 3 6 5に記載されている方法により合成した) 5. 1 8 g、 ジクロロピ ス (トリフエニルホスフィン) パラジウム (I I ) 0. 47 9 g、 および N, N—ジ メチルホルムアミド 6 OmLの混合物を 7 0°Cにて 6時間撹拌した。 放冷後、 水 6 0 mL、 酢酸ェチル 6 0mL、 およびフッ化カリウム 20 gを加え、 室温にて終夜撹拌 した。 不溶物をセライトろ過し、 ろ液をクロ口ホルムにて抽出した。 得られた有機層 を水、 飽和食塩水にて順に洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過 し、ろ液を減圧濃縮した後、得られた残渣をシリカゲルカラムクロマトグラフィー(溶
出液: 10%クロ口ホルム一へキサン/酢酸ェチル = 100/0 - 0X100) にて 精製し、 5—ブロモ一2—クロロー 4— [(E) —2—エトキシビエル] ピリミジン
[11 - 1] (以下、 化合物 [11— 1] という) 1. 15 gを淡黄色固体として得 た。
(2) 化合物 [11— 1] 347mgと 3—ェチルピリジン一 2—ァミン (国際公 開第 2006/025567号パンフレツト、 142ページに記載されている方法に より合成した) 118mgから、 実施例 1一 (6) の方法に準じて 3— (5—ブロモ 一 2_クロ口ピリミジン一 4—ィル) 一 8—ェチルイミダゾ [1, 2— a] ピリジン.
[11 -2] (以下、 化合物 [11一 2] という) 137mgを淡褐色固体として得 た。
(3) 化合物 [6— 1] 78mgをクロ口ホルム 2mLに溶解し、 トリフル,ォロ酢 酸 2mLを加え、 室温にて 1時間撹拌した。 反応混合物を減圧濃縮した後、 メタノー ルに溶解し、 弱イオン交換樹脂に通しトリフルォロ酢酸を除去した。 溶媒を留去し、 (1 R) 一 1— {4- [(1 S) 一 1一アミノエチル] フエ二ル} 一 2—'(t e r t 一プチルァミノ)エタノール [11一 3] 55mgを白色固体として得た。化合物 [1 1-3] (以下、 化合物 [1 1一 3] という) はさらに精製することなく、 次の反応 に付した。
(4) 化合物 [1 1一 3] 55mg、 化合物 [11一 2] 78mg、 およびジイソ プロピルェチルァミン 81 Lをジメチルスルホキシド 4mLに溶解し、 120°Cに て 12時間撹拌した。 放冷後、 反応混合物を逆相分取液体クロマトグラフィーにて精 製した。 得られた水溶液に飽和炭酸水素ナトリウム水溶液を加え塩基性とした後、 ク ロロホルムとメタノールの混合溶媒 (混合比: 9/1) で抽出した。 得られた有機層 を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液 を減圧濃縮して得られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [11] 78 mgを灰色固体として得た。 化合物 [11] のスペクトルデータを以下に示す。
一 NMR (CDC 13) δ 8. 71 (s, 1 H), 8. 44 (s, 1H), 7. 4 1 -7. 31 (m, 5H), 7. 10 (d, J = 7. 2Hz, 1 H), 6. 65-6. 55 (m, 1H), 5. 58-5. 50 (m, 1H), 5. 10-5. 02 (m, 1H), 4. 59 (dd, J =8. 4Hz, 3. 6Hz, 1 H), 3. 07 (q, J = 7. 2 Hz, 2H), 2. 91 (dd, J =l 1. 6Hz, 3. 6Hz, 1 H), 2. 59 (d d, J = 11. 6Hz, 8. 4Hz, 1 H), 1. 57. (d, J = 6. 8Hz, 3H), 1. 37 ( t, J = 7. 2Hz, 3H), 1. 09 (s , 9 H)
ma s s : 537, 539 (M+ 1) +,
実施例 1 2
2— [ ( (I S) — 1ー {4— [ヒドロキシ (ピリジン一 2—ィル) メチル] フエ二 ル} ェチル) ァミノ] —4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィ ル) ピリミジン一 5—力ルポ二トリル [1 2] (以下、 化合物 [1 2] という) の合 成。
(1) 化合物 [1一 1] 5 0 Omgをテトラヒドロフラン 1 OmLに溶解し一 7 8°C とした後、 n—ブチルリチウム (1. 57Mへキサ.ン溶液) 2. 3 3mLを加えた。. 同温にて 1時間撹拌した後、生じた白色懸濁液にピコリンアルデヒド 193 Lを加 え、 室温まで昇温しながら終夜撹拌した。 反応混合物に水 5 OmLを加え、 酢酸ェチ ル 20 OmLにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸マグ ネシゥムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシ リカゲルカラムクロマトグラフィー (溶出液:へキサンノ酢酸ェチル = 1 0 0/0— 30/7 0) にて精製し、 t e r t一ブチル (( 1 S ) — 1一 { 4一 [ヒドロキシ (ピ リジン一 2—ィル) メチル] フエ二ル} ェチル) カーバメート [1 2 - 1·] (以下、 化合物 [1 2— 1] という) 2 1 2mgを淡褐色油状物として得た。
(2) 化合物 [1 2— 1] 5 8mgと化合物 [1一 6] 4 lmgから、 実施例 (7 ) の方法に準じて目的化合物 [1 2] 24mgを灰色固体として得た。 化合物 [1 2] のスペクトルデータを以下に示す。
XH-NMR (DMSO— d6) (3 : 9. 9 5 (d, J = 6. 8Hz, 1 HX 1/2), 8. 94 (d, J = 7. 1 Hz , 1 HX 1/2), 8. 8 8 (d, J =7. 8Hz, 1 HX 1/2), 8. 7 5 - 8. 7 0 (m, 1 H+ 1 HX 1/2), 8 6 7 (s , 1 HX 1/2), 8. 5 6— 8. 5 5 (m, 1 HX 1/2), 8. 42 - 8. 3 5 (m, 1H), 7. 7 7 - 7. 6 7 (m, 1 H), 7. 56 - 7. 5 1 (m, 1H), 7. 4 2- 7. 2 5 (m, 5H), 7. 2 1 - 7. 1 5 (m, 1 H), 7. 1 1 ( t , J = 6. 8Hz , 1 HX 1/2), 6. 6 8 - 6. 6 1 (m, 1 HX 1/2), 6. 0 3 - 6. 0000 ((mm,, 11 HH)),, 55.. 66 66 -- 55.. 66 55 ((mm,, 11 HH)),, 55. 1 9 (qu i n t , J = 7. 1Hz , 1 HX 1/2), 5. 0 2 (q u i n t , J = 6. SHz, 1 HX 1 /2), 2. 5 8 (s , 3HX 1/2), 2. 5 2 (s , 3HX 1/2), 1. 49 (d, J = 7. 1Ηζ, 3HX 1/2), 1. 45 (d, J = 6. 8Hz , 3HX 1/2) ma s s : 46 2 (M+ 1) +. 実施例 1 3
2- [ ( (1 S) - 1 - {4- [ヒドロキシ (フエニル) メチル] フエ二ル} ェチル) ァミノ] —4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジ ン— 5—力ルポ二トリル [13] (以下、 化合物 [13] という) の合成。 化合物 [1— 1] 100mg、 ベンズアルデヒド 51 L、 および化合物 [1— 6] 38mgから、 実施例 12の方法に準じて目的化合物 [13] l lmgを白色固体と して得た。 化合物 [13] のスペクトルデータを以下に示す。
一 NMR (DMSO - d 6) (5 : 9. 98 - 9. 94 (m, 1 HX 1/2), 8. 96- 8. 94 (m, 1 HX 1/2), 8. 89 - 8. 87 (m, 1 HX 1/2), 8. 80— 8. 78 (m, 1 HX 1/2), 8. 73-8. 71 (m, 1H), 8. 68— 8. 67 (m, 1 HX 1/2), 8. 58 - 8. 56 (m, 1 HX 1/2), 7. 42 -7. 09 (m, 10H+ 1HX 1/2), 6. 69 - 6. 63 (m, 1HX 1/2), 5. 82 (b r s, 1 H), 5. 65 (b r s, 1H), 5. 23-5. 13 (m, 1 HX 1/2), 5. 06 -4. 99 (m, 1 HX 1/2), 2. 57 (s, 3 H X 1 / 2), 2. 52 (s , 3HX 1/2), 1. 49 (d, J = 6. 8 Hz, 3HX 1/2), 1. 46 (d, J = 6. 8 Hz, 3HX 1/2)
ma s s : 46 1 (M+ 1) +. 実施例 14
2 - [ ( (I S) 一 1一 {4- [ヒドロキシ (1ーメチルー 1 H—イミダゾ一ルー 2 —ィル) メチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—メチルイミダゾ [1, 2 -a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリル [14] (以下、 化合 物 [14] という) の合成。 化合物 [1— 1] 1 00mg、 1ーメチルー 2—イミダゾールカルポキシアルデヒド 75mg、 および化合物 [1— 6] 74mgから、 実施例 12の方法に準じて目的化 合物 [14] 23mgを白色固体として得た。 化合物 [14] のスペクトルデータを以下に示す。
^-NMR (DMSO-d6) δ : 9. 97 (d, J = 7. 1Hz, 1HX 1/2), 8. 97 (d, J = 7. lHz, 1 HX 1 /2), 8. 92— 8. 87 (m, 1 HX 1/2), 8. 78 - 8. 69 (m, 1H+ 1HX 1/2), 8. 59 (s , 1 HX 1 /A), 8. 57 (s, 1H+ 1HX 1/4), 7. 43 -7. 24 (m, 5H+ 1 H X 1/2), 712 ( t , J = 7. 2Hz. 1HX 1/2), 7. 0 1 -6. 96 (m, 1H), 6. 87 ( t , J = 7. 0Hz, 1 HX 1/4), 6. 74— 6. 72 (m, 1H), 6. 63 (t, J = 7. 0Hz, 1HX 1/4), 6. 1 1 -6. 08 (m,
1 H), 5. 8 3 - 5. 8 1 (m, 1 H), 5. 2 5 - 5. 2 1 (m, 1HX 1/2), 5. 0 9 - 5. 0 3 (m, 1 HX 1/2), 3. 50 (s, 3HX 1 /2), 3. 3 7 (s , 3HX 1 /2), 2. 5 8 (s, 3HX 1/2), 2. 5 3 (s, 3HX 1/2), 1. 5 2 (d, J = 6. 8Hz , 3 HX 1/2), 1. 49 (d, J = 7. 1Hz, 3HX 1/2),
ma s s : 46 5 (M+ 1 ) +. 実施例 1 5
2- [ ( (I S) — 1— {4一 [ヒドロキシ (ピリジン一 4一ィル) メチル] フエ二. ル} ェチル) ァミノ] -4- (8—メチルイミダゾ [1 , 2— a] ピリジン— 3—ィ ル) ピリミジン一 5—カルボ二トリル [1 5] (以下、 化合物 [1 5] という) の合 成。
(1) 化合物 [1— 1] 5 gをテトラ七ドロフラン 1 0 OmLに溶解し一 7 8°Cと した後、 n—ブチルリチウム (1. 6 Mへキサン溶液) 3 1. 2mLを加え、 同温に て 1時間撹拌した。 生じた白色懸濁液に N, N—ジメチルホルムアミド 774 Lを 加え、 徐々に室温まで昇温しながら 5時間撹拌した。 飽和塩化アンモニゥム水溶液を 加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸 マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシ リガゲルカラムクロマトグラフィー (溶出液:へキサン Z酢酸ェチル = 1 0 0/0 - 5 0/5 0) にて精製し、 t e r t一ブチル [(I S) — 1— (4一ホルミルフエ二 ル) ェチル] カーバメート [1 5— 1] (以下、 化合物 [1 5— 1] という) 5 14 mgを淡黄色アモルファスとして得た。 (2) 4一ブロモピリジン 1 6 3mgをテトラヒドロフラン 3mLに溶解した溶液 に、 塩化イソプロピルマグネシウム (2. 0Mテトラヒドロフラン溶液) 5 を加え、 室温にて 1. 5時間撹拌した後に、 化合物 [1 5— 1] 1 7 1mgを加え、 室温にて終夜撹拌した。 反応液に水 1 0 OmLを加え、 クロ口ホルムにて抽出した。 得られた有機層を無水硫酸マグネシウムにて乾燥し、 不溶物をろ過し、 ろ液を減圧濃 縮した。 得られた残渣を分取薄層クロマトグラフィーにて精製し、 t e r t—ブチル ((I S) - 1 - {4— [(R) —ヒドロキシ (ピリジン _ 4一ィル) メチル] フエ二 ル} ェチル) カーバメート [1 5— 2] (以下、 化合物 [1 5— 2] という) 4 1m gを淡黄色ァモルファスとして得た。 (3) 化合物 [1 5— 2] 4 lmgと化合物 [1一 6] 3 2mgから、 実施例 1一 (7) の方法に準じて、 目的化合物 [1 5] 8 mgを灰色固体として得た。 化合物 [1 5] のスペクトルデータを以下に示す
P T/JP2007/075240
^-NMR (DMSO-d6) δ : 9. 9 5 (d, J = 7. 1 Hz, 1 HX 1/2), 8. 9 5 (d, J = 7. 1 Hz , 1 HX 1/2), 8. 8 9 (d, J = 8. 0Hz , 1 HX 1 /2), 8. 8 0 (d, J = 7. 1Hz , 1 HX 1/4), 8. 7 7 (d, J =7. 1 Hz, 1 HX 1/4), 8. 7 3 (s , 1 HX 1/2), 8. 7 2 (s, 1 H
X 1 /2), 8. 6 8 ( s 1 HX 1/2), 8. 5 7 (s , 1 HX 1/2), 8. 4 6— 8. 44 (m, 1H), 8. 41 - 8. 3 9 (m, 1 H), 8. 42 - 8. 24 (m, 7H), 7. 1 1 (t , J =7. 1 Hz, 1 HX 2/4), 6. 7 1 ( t , J = 7. 1 Hz , 1 HX 1/4), 6 6 0 ( t , J = 7. 1 Hz, 1 HX 1/4), 6. 0 8 - 6. 0 5 (m, 1H), 5 6 7 (b r s , 1 H), 5. 2 0 (q u i n t , J = 7.. 6 Hz , 1 HX 1/2), 5. 0 8— 5. 0 0 (m, 1HX 1/2), 2. 5 8 ( s , 3 HX 1/2), 2. 5 3 (s , 3 HX 1/2), 1. 49 (d, J = 7. 1 Hz , 3 HX 1/2), 1. 46 (d, J 7. 1 Hz , 3HX 1 /2)
ma s s : 46 2 (M+ l) +. 実施例 1 6
2 - ( { (I S) — 1一 [4一 (1ーヒドロキシ— 2—フエニルェチル) 'フエニル] ェチル } ァミノ) 一 4— (8—メチルイミダゾ [1, 2 - a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [1 6] (以下、 化合物 [1 6] という) の合成。 (1) 化合物 [1 5— 1] 7 Omgをテトラヒドロフラン 5mLに溶解し 0°Cとし た後、 塩化べンジルマグネシウム ( 2. 0 Mテトラヒドロフラン溶液) 3 0 0 Lを 加え、 徐々に室温まで昇温しながら終夜撹拌した。 反応液に飽和塩化アンモニゥム水 溶液を加え、 酢酸ェチルにて抽出した。 得られた有機層を水、 飽和食塩水にて順に洗 浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られた残渣を分取薄層クロマトグラフィーにて精製し、 t e r t—ブチル {(1 S) 一 1一 [4一 (1—ヒドロキシー 2—フエニルェチル) フエニル] ェチル } 力一バメ ート [1 6— 1〕 (以下、 化合物 [1 6— 1] という) 1 lmgを無色油状物として 得た。
(2) 化合物 [1 6— 1] 1 lmgと化合物 [1— 6] 1 0 m g力、ら、 実施例 1一 (7) の方法に準じて、 目的化合物 [1 6] 4mgを灰色固体として得た。 化合物 [1 6] のスペクトルデ一夕を以下に示す。
^-NMR (CDC 13) δ : 9. 8 0 (d, J = 7. 2Hz , 1 HX 1/5), 9. 1 2 - 8. 7 5 (m, 1 H+ 1 HX 4/5), 8. 5 5 (s , 1 HX 1/5), 8. 5 0 (s , 1 HX 4/5), 8. 1 0 - 7. 00 (m, 1 1 H), 6. 6 8 - 6. 6 0 (m, 1 H), 5. 8 0 (b r s , 1 H), 5. 1 5 - 5. 1 0 (m, 1 HX 1/5), 4. 94— 4. 8 9 (m, 1 HX 4/5), 2. 64 (s , 3H), 1. 64 (d, J = 6.
8Hz, 3H)
ma s s : 475 (M+ 1) +. 実施例 17 .
2— [ ( (1 S) —1— {4ー [1ーヒドロキシー 2— (1—メチルビペリジン一 4 一ィル) ェチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—メチルイミダゾ [1, 2 -a] ピリジン一 3·—ィル) ピリミジン一 5—力ルポ二トリル [17] (以下、 化合 物 [17] という) の合成。 (1) 化合物 [1— 1] 300mg、 1一ベンジルォキシカルポ二ルー 4一 (ホル ミルメチル) ピペリジン (欧州特許出願公開第 0367110号明細書、 108— 1 09ページに記載されている方法に準じて合成した) 516mg、 および化合物 [1 一 6] 4 lmgから実施例 12の方法に準じて、 ベンジル 4一 {2 - [4一 ((1 S) - 1 - {[5—シァノー 4一 (8—メチルイミダゾ [1, 2— a] ピリジンー3—ィ ル) ピリミジン一 2一ィル] アミノ} ェチル) フエニル] 一 2—ヒドロキシェチル } ピぺリジン一 1—カルポキシレート [17— 1] (以下、 化合物 [17— 1] という) 43 mgを淡黄色固体として得た。
(2) 化合物 [17— 1 ] 2 lmgをテトラヒドロフラン 3mLとメタノール 3m Lの混合溶媒に溶解し、 20%水酸化パラジウム炭素触媒 2 Omgを加え、 水素雰囲 気下、 室温にて 2時間撹拌した。 触媒をセライトろ過し、 ろ液を減圧濃縮した。 得ら れた残渣にジェチルエーテルを加え、生じた固体をろ取し、 2—({(1 S)— 1— [4 ― (1ーヒドロキシー 2—ピペリジン一 4一ィルェチル) フエニル]ェチル }ァミノ) 一 4一 (8—メチルイミダゾ [1, 2 - a] ピリジン一 3—ィル) ピリミジン一 5— カルボ二トリル [17-2] (以下、 化合物 [17-2] という) 6mgを得た。
( 3 ) 化合物 [17-2] 2mgと 37 %ホルムアルデヒド液 100 / Lの混合物 に、塩化亜鉛 4mgとシァノトリヒドロほう酸ナトリウム 4mgのメタノール溶液 2 00 xLを加え、 室温にて終夜撹拌した。 反応混合物に水とクロ口ホルムを加え、 有 機層と水層を分離した。得られた有機層を飽和炭酸水素ナトリゥム水溶液と飽和食塩 水にて順に洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧 濃縮した後、 得られた残渣にジェチルエーテルを加え、 生じた固体をろ取し、 目的ィ匕 合物 [17] 2. 2mgを灰色固体として得た。 化合物 [17] のスペクトルデータを以下に示す。
JH-N R (CDC 13) 0 : 9. 62 (d, J = 7. 2Hz , 1 ΗΧ 1/5), 8. 92- 8. 64 (m, 1H+ 1HX4/5), 8. 55 (s, 1 ΗΧ 1/5), 8. 4 1 (s , 1HX4/5), 7. 45 -6. 55 (m, 6Η), 6. 23 - 6. 20 (m,
TJP2007/075240 1HX4Z5), 5. 95 - 5. 90 (m, 1 HX 1/5), 5. 40 -4. 70 (m, 2H), 3. 00 - 2. 70 (m, 4H), 2. 58 (s, 3H), 2. 30 (s, 3 H), 2. 10-1. 40 (m, 7H), 1. 62 (d, J = 6. 8Hz, 3 H) ma s s : 496 (M+ 1) +. 実施例 18
2一 ( { (1 S) — 1一 [4— (4ーヒドロキシピペリジン一 4—ィル) フエニル] ェチル } ァミノ) —4— (8—メチルイミダゾ [1, 2-a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [18] (以下、 化合物 [18] という) の合成。
(1) 化合物 [1— 1] 20 Omgと N—ベンジルォキシカルポ二ルー 4ーピペリ ドン 186mgから、 実施例 12— (1) の方法に準じてベンジル 4一 (4一 {(1 S) - 1— [( t e r t一ブトキシカルポニル) ァミノ] ェチル } フエニル) —4一 ヒドロキシピペリジン一 1一力ルポキシレ一卜 [18-1] (以下、 化合物 [18- 1] という) 17 lmgを得た。
(2) 化合物 [18— 1] 82mgと化合物 [1一 6] 5 Omg力、ら、 実施例 1 _ (7) の方法に準じて、ベンジル 4一 [4一 ((1 S) — 1一 {[5—シァノー 4一 (8 ーメチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 2—ィル] アミ ノ エチル) フエニル] —4ーヒドロキシピペリジン— 1一力ルポキシレート [18 一 2] (以下、 化合物 [18— 2] という) 18mgを得た。
(3) 化合物 [18— 2] 14mgをテトラヒドロフラン 2mLとメタノール 0. 2 mLの混合溶媒に溶解した溶液に、 10%パラジウム炭素触媒 15mgを加え、 水 素雰囲気下、 室温にて終夜撹拌した。 触媒をセライトろ過し、 ろ液を減圧濃縮し、 得 られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [18] 2mgを 淡黄色ァモルファスとして得た。 化合物 [18] のスペクトルデータを以下に示す。
— NMR (CDC 13) 6 9. 65 - 9. 62 (m, 1 HX.1/5), 8. 95 (s , 1 HX 1/5), 8. 89 (s, 1HX4/5), 8. 79 (d, J = 7. 3 Hz, 1 HX 4/5), 8. 56- 8. 56 (m, 1 HX 1/5), 8. 52 (s , 1 HX 4/5), 7. 57— 7. 52 (m, 2H), 7. 40— 7. 38 (m, 2H), 7. 27 - 7. 25 (m, 1HX 1/5), 7. 15 (d, J = 6. 8Hz , 1 HX 4 / 5), 6. 96 - 6. 92 (m, 1 HX 1X5), 6. 59 ( t , J =6. 8Hz, · 1 HX 4/5), 6. 05 - 6. 03 (m, 1 HX 4/5), 5. 84- 5. 82 (m, 1 HX 1/5), 6. 36 - 6. 32 (m, 1 HX 1/5), 5. 17-5. 10 (m, 1 HX 4/5), 4. 43-4. 42 (m, 1 HX 1/5), 4. 29-4. 27 (m,
1 HX 4/5), 3. 14-3. 08 (m, 2 H), 2. 98 - 2. 95 (m, 2H), 2. 83 (s , 3 HX 1/5), 2. 63 (s , 3 HX 4/5), 2. 06- 1. 99 (m, 2H), 1. 73 - 1. 68 (m, 2H), 1. 28 - 1. 25 (m, 3 H) ma s s : 454 (M+ 1) +. 実施例 19
2- ( { (1 S) - 1 - [4- (3—ヒドロキシー 1—メチルピロリジン一 3—ィル) フエニル] ェチル } ァミノ) 一 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一
3—ィル) ピリミジン一 5—力ルポ二トリル [19] (以下、 化合物 [19] という). の合成。
(1) 1一 N—ベンジルォキシカルポ二ルー 3—ピロリジノン 164m gと化合物 [1-1] 15 Omg、 および化合物 [1一 6] 33mg力、ら、 実施例 12の方法に 準じて、 ベンジル 3— [4— ((1 S) - 1 - {[5—シァノー 4一 (8—メチルイ ミダゾ [1, 2— a] ピリジン— 3—ィル) ピリミジン一 2—ィル] アミノ}ェチル) フエニル]― 3—ヒドロキシピロリジン一 1一力ルポキシレート [19一 1] (以下、 化合物 [19ー 1] という) 23 mgを黄色油状物どして得た。
(2) 化合物 [19— 1] 15mg力、ら、 実施例 17— (2)、 (3) の方法に準じ て、 '目的化合物 [19] 6mgを白色固体として得た。 化合物 [19] のスペクトルデータを以下に示す。
^-NMR (CDC 13) δ : 9. 64 (d, J =6. 8Hz, 1 HX 1/5), 8. 94 (s, 1 HX 1/5), 8. 87 (s, 1HX 4/5), 8. 78 (d, J = 6. 8 Hz, 1 HX 4/5), 8. 56 (s, 1 HX 1/5), 8. 49 (s, 1 HX 4/ 5), 7. 56 - 6. 93 (m, 5H), 6. 64—6. 60 (m, 1 H), 6. 22 -6. 12 (m, 1 HX 4/5), 5. 92 - 5. 85 (m, 1 HX 1/5), 5. 3 8-5. 28 (m, 1 HX 1/5), 5. 1 5— 5. 11 (m, 1 HX 4/5), 3. 20-3. 14 (m, 1H), 2. 98 - 2. 93 (m, 1 H), 2. 68-2. 61 (m, 1 H), 2. 65 (s , 1 HX 3/5), 2. 63 (s , 2 H.+ 1 HX 2 5 ) , 2. 54-2. 17 (m, 3H), 2. 45 (s , 2H+ 1 HX 2/5), 2. 44 (s , 1HX 3/5), 2. 10 (b r s , 1 H), 1. 63 (d, J = 6. 8Hz , 3 H) ma s s : 454 (M+ 1) +. 実施例 20
4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— ( { (1 S) - 1 - [4- (4ーヒドロキシ一 1ーメチルビペリジン一 4一^ Γル) フエニル] ェチ
ル} ァミノ) ピリミジン一 5—力ルポ二トリル [2 0] (以下、 化合物 [2 0] とい う) の合成。
(1) 化合物 [1 8— 1] 1 1 9mgと化合物 [5— 6] 1 1 5mgから、 実施例 1一 (7) の方法に準じて、 ベンジル 4一 [4一 ((1 S) 一 1一 {[5—シァノ一4 一 (8—ェチルイミダゾ [1, 2 - a] ピリジン一 3—ィル) ピリミジン一 2—ィル] アミノ}ェチル)フエニル]一 4ーヒドロキシピペリジン一 1一カルボキシレート [2
0 - 1] (以下、 化合物 [20— 1] という) 1 0 2mgを得た。 (2) 化合物 [2 0— 1] 1 0 2mgから、 実施例 1 7— (2)、 (3) の方法に準 じて、 目的化合物 8mgを白色固体として得た。 化合物 [20] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) δ : 9. 6 1 (d, J = 7. 2Hz , 1 HX 1/4), 8. 9 2 (s , 1 HX 1/4), 8. 8 5 (s, 1 HX 3/4), 8. 7 6 (d, J = 7.
2 Hz , 1 HX 3/4), 8. 5 5 (s , 1 HX 1/4), 8. 43 (s , 1HX 3Z
4), 7. 6 0 - 6. 9 0 (m, 5H+ 1 HX 1/4), 6. 6 1 ( t, J =6. 4 PI z , 1 HX 3/4), 6. 29 - 6. 2 8 (m, 1 HX 3/4), 5. 9 6 - 5. 9 2 (m, 1 HX1/4), 5. 40 - 5. 3 0 (m, 1 HXl/4), 5. 1 8 - 5. 0 8 (m, 1 HX 3/4), 3. 48 (s , 3 H) 3. 1 0— 3. 0 0 (m, 2H), 2.
8 0 - 2. 7 0 (m, 2H), 2. 50 - 2. 40 (m, 2H), 2. 20 - 2. 1 0 (m, 2H), 1. 8 5 - 1. 7 0 (m, 2H), 1. 6 2 (d, J =7. 2Hz , 3
H), 1. 5 6— 1. 5 1 (m, 3H), 1. 43— 1. 3 3 (m, 3 H)
ma s s : 48 2 (M+ 1) +. 実施例 2 1
4- (8—ェチルイミダゾ [1, 2 - a] ピリジン一 3—ィル) — 2 - ( { (1 S) 一 1一 [4- (3—ヒドロキシー 1一メチルァゼチジン— 3—ィル) フエニル] ェチ ル} ァミノ) ピリミジン一 5—力ルポ二トリル [2 1]. (以下、 化合物 [2 1] とい う) の合成。 .
(1) 1一べンズヒドリルァゼチジン— 3—オン 1 3 7mgと化合物 [1— 1] 3 0 Omgから、 実施例 1 2— (1) の方法に準じて、 t e r t—ブチル ((I S) —
1— {4一 [1— (ジフエニルメチル) 一 3—ヒドロキシァゼチジン一 3—ィル] フ ェニル } ェチル) カーバメート [2 1 - 1] (以下、 化合物 [2 1 - 1] という) 1
27 mgを淡黄色固体として得た。
(2) ί匕合物 [2 1 - 1] 9 6mgとィ匕合物 [5— 6」 6 2mgから、 実施例 1一
2007/075240 (7) の方法に準じて、 2— [((1 S) 一 1一 {4- [1— (ジフエニルメチル) 一
3—ヒドロキシァゼチジン— 3—ィル] フエ二ル} ェチル) ァミノ] 一 4— (8—ェ チルイミダゾ [1, 2-a] ピリジン一 3—ィル) ピリミジン— 5—力ルポ二トリル
[21一 2]. (以下、化合物 [21— 2] という) 64mgを淡黄色固体として得た。
(3) 化合物 [21 -2] 64mgから、 実施例 1 7— (2)、 (3) の方法に準じ て、 目的化合物 [21] 6mgを灰色固体として得た。 化合物 [21] のスペクトルデ一夕を以下に示す。
一 NMR (CDC 13) δ : 9. 63 (d, J = 7. 2Hz, 1 HX 1/5), 9. 00 - 8. 70 (m, 1 H+ 1 HX 4/5), 8. 56 (s, 1 HX 1/5), 8. 5 2 (s , 1 HX 4/5), 7. 68 -6. 60 (m, 6 H), 6. 18-6. 10 (m, 1 H), 5. 20-5. 1 5 (m, 1 H), 3. 70— 3. 65 (m, 2H), 3. 4
4- 3. 40 (m, 2H), 3. 10— 3. 00 (m, 2H), 2. 44 (s , 3H), 1. 62 (d, J = 7. 0Hz, 3H), 1. 50— 1. 30 (m, 3 H)
ma s s : 454 (M+ 1 ) +. ' 実施例 22
4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— ( { (1 S) - 1 - [4一 (1ーヒドロキシー 1ーピペリジン一 4一^ fルェチル) フエニル] ェチ ル} ァミノ) ピリミジン一 5—力ルポ二卜リル [22] (以下、 化合物 [22] とい う) の合成。
(1) ベンジル 4一 (N—メトキシー N—メチルカルバモイル) ピぺリジン一 1一 カルボキシレー卜 2. 39 gと化合物 [1一 1] 2. 13 gから、 実施例 12— (1) の方法に準じて、 ベンジル 4一 (4- {(1 S) 一 1— [(t e r t—ブトキシカル ポニル) ァミノ] ェチル } ベンゾィル) ピぺリジン一 1一力ルポキシレート [22— 1] (以下、 化合物 [22— 1] という) 1. 26 gを得た。 (2) 化合物 [22— 1] 1 12mgをテトラヒドロフラン 5mLに溶解し 0°Cと した後、 塩化メチルマグネシウム (3. 0Mテトラヒドロフラン溶液) 320 / Lを 加え、 室温まで昇温しながら 1. 5時間撹拌した。 反応液に飽和塩化アンモニゥム水 溶液を加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無 水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して、 得られた 残渣をシリカゲルカラムクロマトグラフィー (溶出液:へキサン/酢酸ェチル =10 0/0 - 30/70) にて精製し、 ベンジル 4一 [1一 (4- {(1 S) - 1一 [( t e r t一ブトキシカルボニル) ァミノ]ェチル }フエニル) 一 1ーヒドロキシェチル] ピぺリジン— 1—力ルポキシレ一卜 [22-2] (以下、 化合物 [22— 2] という)
53mgを得た。
(3) 化合物 [2 2— 2] 5 3mgと化合物 [5— 6] 3 Omgから、 実施例 1一 (7) の方法に準じてベンジル 4— { 1 - [4- ((1 S) ー 1一 {[5—シァノ— 4一 (8—ェチルイミダゾ [1 , 2— a] ピリジン一 3 Γル) ピリミジン一 2 Γ ル] アミノ} ェチル) フエニル] 一 1ーヒドロキシェチル } ピぺリジン一 1—力ルポ キシレート [2 2 - 3] (以下、 化合物 [2 2 - 3] という) 1 7mgを得た。
(4) 化合物 [2 2— 3] 1 7mgから、 実施例 1 7— (2) の方法に準じて目的. 化合物 [2 2] 5mgを灰色固体として得た。 化合物 [2 2] のスペクトルデ一夕を以下に示す。
^-NM (DMSO - d6) δ : 9. 9 8 (d, J = 7. 1 Hz, 1 HX 1/2), 8. 9 6 (d, J = 6. 8Hz , 1 HX 1/2), 8. 8 8 (d, J = 8. 3Hz, 1 HX 1/2), 8, 8 5 (d, J = 7. 1Hz, 1 HX 1/2), 8. 74- 8. 7 1 (m, 1 H+ 1 HX 1/2), 8. 6 0 (s , 1 HX 1/2), 8. 5 9· (s, 1 H X 1/2), 7. 5 1 - 7. 3 1 (m, 5H), 7. 1 4 ( t , J = 7. 1 Hz , 1 H X 1/2), 6. 9 1 - 6. 8 6 (m, 1 HX 1/2), 3. 46 - 3. 40 (m, 1 H), 3. 0 1 (q, 1 = 7. 6Hz , 1 HX 1/2), 2. 9 6 (q, J = 7. 6 H z, 1 HX 1/2), 2. 9 3 - 2. 1 7 (m, 2H), 1. 5 3— 1. 49 (m, 4 H), 1. 3 6 - 1. 2 2 (m, 7H), 1. 1 3 - 0. 9 6 (m, 4H)
ma s s : 49 6 (M+ 1) +. 実施例 2 3、 24
4- (8—ェチルイミダゾ [1, 2— a] ピリジンー3—ィル) 一 2— [ ( (1 S) 一 1一 {4— [ヒドロキシ (1ーメチルピペリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力.ルポ二トリル [2 3] (以下、 化合物 [2 3] という)、 および [24] (以下、 化合物 [24] という) の合成 (ここで、 化合物 [2 3] と化合物 [24] は、 ジァステレオマ一の関係にある。 表 6参照)。
(1) マグネシウム 9 7 5mg、 ブロモェ夕ン 1 0 0 L、 およびテトラヒドロフ ラン 2 OmLの混合物に触媒量のヨウ素を加え、 ヨウ素の褐色が消失するまで約 1 0 分間撹拌した。 4—クロロー 1—メチルビペリジン 5. 9 gのテトラヒドロフラン溶 液 2 OmLを加え、 2. 5時間加熱還流した。 生じた白色懸濁液を 0 °Cとし、 化合物
[1 5— 1] 2 gのテトラヒドロフラン溶液 1 0 mLを加えた。 徐々に室温まで昇温 しながら終夜撹拌した後、 飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出 した。 得られた有機層を水、 飽和食塩水にて順に洗浄し、 無水硫酸マグネシウムにて 乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムク
P T/JP2007/075240 口マトグラフィー (溶出液:クロ口ホルム Zメタノール = 100/0 - 90/10) にて精製し、 t e r t—ブチル ((I S) — 1— {4- [ヒドロキシ (1—メチルビ ペリジン一 4一ィル) メチル] フエ二ル}ェチル) カーバメート [23— 1] (以下、 化合物 [23— 1] という) 2. 27 gを得た。
(2) 化合物 [23— 1] 1. 42 gと化合物 [5— 6] 1 gから、実施例 1一 (7) の方法に準じて目的化合物 [23] と [24] の混合物 1. 2 gを得た。
(3) 化合物 [23] と [24] の混合物 70 Omgをキラルセル OD— Hにて分 割した。
光学分割条件は以下のとおりである。
カラム:キラルセル OD— H (C h i r a 1 c e 1 OD— H、ダイセル化学工業)、 直径 2 Omm、 長さ 25 Omm
溶出液:へキサン/エタノール Zジェチルァミン =85/15/0. 1
流速: 15m /m i n
得られた溶液を減圧濃縮し、残渣を少量のクロ口ホルムに溶解しジェチルエーテルに て固化させ、 目的化合物 [23] 217mg (RT= 18. 3分) を白色固体、 目的 化合物 [24] 23 Omg (RT=24. 3分) を淡黄色固体として得た。 化合物 [23] および化合物 [24] のスペクトルデ一夕を以下に示す。
化合物 [23]
^-NMR (DMSO— d6) δ : 9. 98 (d, J = 6. 3Hz, 1 HX 2/5), 8. 97 (d, J = 7. 1 Hz , 1 HX 3/5), 8. 90 (d, J = 7. 3Hz, 1H), 8. 73 (s , 1 H), 8. 70 (s , 1 HX 2/5), 8. 60 (s, 1 H X 3/5), 7. 42 (d, J = 6. 1Hz, 1 HX 2/5), 7. 35 - 7. 33 (m, 2H+ 1 HX 3/5), 7. 26-7. 21 (m, 2H), 7. 15 ( t , J = 7. 1 Hz , 1HX 2/5), 6. 89 ( t, J = 7. 1 Hz, 1 HX 3/5), 5. 24 (q u i n t , J = 7. 1 Hz, 1 HX 2/5), 5. 17-5. 05 (m, 1H+ 1H X 3/5), 4. 20-4. 13 (m, 1H), 3. 02 (q, J = 7. 6Hz, 2H X 2/5), 2. 96 (q, J = 7. 6Hz, 2HX 3/5), 2. 7.4-2. 63 (m, 1H+ 1 HX 2/5), 2. 50 - 2. 48 (m, 1 HX 3/5), 2. 06 (s , 3 HX 2/5), 2. 02 (s , 3HX 3/5), 1. 73 - 1. 62 (m, 2H+ 1H X 2/5), 1. 52 (d, J =7. 1 Hz , 3HX 2/5), 1. 49 (d, J = 7. 1 Hz , 3HX 3/5), 1. 32 ( t, J = 7. 6Hz, 3HX 2/5), 1. 28 ( t , J =7. 6Hz , 3HX 3/5), 1. 22— 1. 04 (m, 4H), 0. 97 一 0. 94 (m, 1 HX 3/5)
ma s s : 496 (M+ 1) +.
化合物 [24]
丄11一 NMR (DMSO-d6) δ : 9. 9 8 (d, J = 6. 1Ηζ, 1 HX 1/2),
8. 9 7 (d, J = 7. 1Hz , 1HX 1/2), 8. 9 0— 8. 8 9 (m, 1H),
8. 7 3 (s , 1 HX 1/2), 8. 7 3 (s , 1HX 1/2), 8. 7 0 (s , 1 H X 1/2), 8. 6 0 (s , 1 HX 1/2), 7. 43 (d, J = 6. 1 Hz, 1 HX
1/2), 7. 3 5 - 7. 3 3 (m, 2H+ 1HX 1/2), 7. 2 5 (d, J = 8.
3Hz, 1 HX 1/2), 7. 2 2 (d, J = 8. 0Hz, 1 HX 1/2), 7. 1 5 ( t, J = 7. 1 Hz, 1 HX 1/2), 6. 9 2 ( t, J = 7. 1 Hz ' 1 HX 1
/2), 5. 24 (q u i n t , J =7. 1 Hz , 1 HX 1/2), 5. 0 8 - 5. 0. 5 (m, 1 H+ 1 HX 1/2), 4. 20 -4. 1 6 (m, 1 H), 3. 0 1 (q, J
=7. 6Hz, 2HX 1/2), 2. 9 6 (q, J = 7. 6Hz, 2HX 1/2), 2.
74- 2. 64 (m, 1 H+ 1 HX 1/2), 2. 5 5 - 2. 5 1 (m, 1 HX 1Z
2), 2. 0 6 (s , 3HX 1/2), 2. 0 2 (s , 3HX 1/2), 1. 7 2 - 1.
5 5 (m, 2H+ 1 HX 1/2), 1. 5 2 (d, J =7. 1 Hz, 3HX 1/2), 1. 49 (d, J = 7. 1 Hz , 3 HX 1/2), 1. 3 2 ( t , J = 7. 6Hz,
3HX 1/2), 1. 2 8 ( t , J = 7. 6Hz, 3HX 1/2), 1. 2 2 - 1. 1
1 (m, 4H), 1. 0 3 - 1. 0 0 (m, 1 HX 1/2)
ma s s : 49 6 (M+ 1) +. 実施例 2 5
4一 (8—ェチルイミダゾ [1, 2— a] ピリジン— 3—ィル) — 2— { [ (1 S) - 1 - (4一 {ヒドロキシ [1一 (2—メトキシェチル) ピぺリジン一 4—ィル] メ チル } フエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [2 5] (以下、 化合物 [2 5] という) の合成。
(1) 化合物 [2 2— 1] 2 6 7mgをテトラヒドロフラン 1 OmLとメタノール lmLの混合溶媒に溶解し、 水素化ほう素ナトリウム 2 2mgを加え、 室温にて終夜 撹拌した。 反応液に水を加え、 酢酸ェチルにて抽出した。 得られた有機層を水、 飽和 食塩水にて順に洗浄し、 無水硫酸マグネシウムにて乾罈した。 不溶物をろ過し、 ろ液 を減圧濃縮し、 得られた残渣をシリカゲル力ラムクロマトグラフィー (溶出液:へキ サン Z酢酸ェチル = 1 00Z0— 3 0/7 0)にて精製し、ベンジル 4— [(4— {(1 S) — 1— [( t e r t—ブトキシカルポニル) ァミノ] ェチル } フエニル) (ヒドロ キシ) メチル] ピぺリジン一 1一力ルポキシレート [2 5— 1] (以下、 化合物 [2 5 - 1] という) 2 56mgを得た。
(2) 化合物 [2 5— 1] 2 5 6mgと化合物 [5.— 6] 1 46mgから、 実施例 1一 (7) の方法に準じてベンジル 4— [[4- ((1 S) - 1 - {[5—シァノ一4 一 (8—ェチルイミダゾ [1, 2 - a] ピリジン— 3—ィル) ピリミジン一 2—ィル]
アミノ} ェチル) フエニル] (ヒドロキシ) メチル] ピぺリジン一 1一力ルポキシレ —ト [25— 2] (以下、 化合物 [25— 2] という) 1 18mgを得た。
(3) 化合物 [25— 2] 1 18mgをエタノール 4mLに溶解し、 20 %水酸化 パラジウム炭素触媒 5 Omgを加え、 水素雰囲気下、 室温にて終夜撹拌した。 触媒を セライトろ過し、 ろ液を減圧濃縮した後、 得られた残渣を分取薄層クロマ卜グラフィ 一にて精製し、 4一 (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2 一 [(( 1 S) 一 1一 {4- [ヒドロキシ (ピペリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン— 5—力ルポ二トリル [25-3] (以下、 化合物 [2, 5-3] という) 66mgを得た。
(4) 化合物 [25— 3] 33mgを N, N—ジメチルホルムアミド lmLに溶解 し、 炭酸カリウム 29mgと 2—ブロモェチルメチルエーテル 1 0 を加え、 5 0°Cにて 2. 5時間撹拌した。 放冷後、 反応液をクロ口ホルムにて希釈し、 水で洗浄 した。 有機層を無水硫酸マグネシウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧 濃縮した。得られた残渣を分取薄層クロマトグラフィーにて精製し、 得られた粗精製 物を少量のクロ口ホルムに溶解し、 へキサンにて固化させ、 目的化合物 [25] 21 mgを白色固体として得た。 化合物 [25] のスペクトルデータを以下に示す。
^-NMR (CDC 13) δ : 9. 65 - 9. 63 (m, ΙΗΧ Ι/5·), 8. 94 (s , 1 ΗΧ 1/5), 8. 89 (s , 1HX 4/5), 8. 83 (d, J = 6. 8Η ζ, 1 ΗΧ 4/5), 8. 57 (s , 1HX 1/5), 8. 51 (s, 1HX4/5), 7. 38 - 7. 30 (m, 4Η), 7. 16 (d, J = 6. 8Hz, 1 H), 7. 00 - 6. 96 (m, 1 HX 1/5), 6. 68 - 6. 62 (m, 1 HX 4/5), 6. 0 3 (d, J = 5. 8Hz, 1 HX 4/5), 5. 83-5. 81 (m, 1 HX 1/5), 5. 34 (b r s, 1 HX 1/5), 5. 1 7- 5. 09 (m, 1 HX 4/5), 4. 39 (d, J =6. 3Hz , 1 H), 3. 48— 3. 44 (m, 2H), 3. 32 (s , 3HX 4/5), 3. 3 1 (s , 3HX 1/5), 3. 1 2-2. 79 (m, 4H), 2. 52 - 2. 48 (m, 2H), 1. 93— 1. 88 (m, 2H), 1. 63 (d, J = 7. 1Hz, 3H), 1. 46- 1. 17 (m, 7H)
ma s s : 540 (M+ 1) +. 実施例 26、 27
4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— [ ( (1 S) 一 1一 {4- [ヒドロキシ (1—イソプロピルピぺリジン一 4一ィル) メチル] フエ 二ル}ェチル) ァミノ] ピリミジン— 5—力ルポ二トリル [26] (以下、 化合物 [2
T/JP2007/075240 6] という)、 および [27] (以下、 化合物 [2 7] という) の合成 (ここで、 化合 物 [2 6] と化合物 [2 7] は、 ジァステレオマ一の関係にある。 表 7参照)。
(1) 化合物 [2 5— 3] 7 8 6mgをクロ口ホルム 1 5mLに溶解し、 アセトン 2. 4mLと卜リアセトキシ水素化ほう素ナトリウム 7 2 8mgを加え、 5 0°Cにて 終夜撹拌した。 放冷後、 反応液に飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホル ムにて抽出した。得られた有機層を水で洗浄し、無水硫酸マグネシゥムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトダラ フィー (溶出液:クロ口ホルム Zメタノール = 1 0 0/0 - 9 3/7) にて精製し、. 目的化合物 [26] および [2 7] の混合物 7 3 8mgを得た。 混合物をキラルセル 〇D— Hにて分割した。
光学分割条件は以下のとおりである。
カラム:キラルセル〇D— H (Ch i r a l c e l OD— H、ダイセル化学工業)、 直径 20 mm、 長さ 2 50 mm
溶出液:へキサン Zエタノール/ジェチルァミン =8 5/1 5/0. 1
流速: 1 5mL/m i n '
得られた溶液を減圧濃縮し、残渣を少量のクロ口ホルムに溶解しジェチルエーテルに て固化させ、 目的化合物 [2 6] 1 7 5mg (RT= 1 7分)を淡黄色アモルファス、 目的化合物 [2 7] 3 9 Omg (RT=2 1分) を灰色固体として得た。 化合物 [2 6] および化合物 [2 7] のスペクトルデータを以下に示す。
化合物 [2 6]
XH-NMR (DMSO-d6) δ : 9. 9 8 (d, J = 6. 6Hz, 1 HX 1/2), 8. 9 5 (d, 1 = 7. 1Hz , 1 HX 1/2), 8. 9 0 - 8. 8 8 (m, 1H), 8. 7 3 (s , 1 HX 1/2), 8. 7 2 (s , 1 HX 1/2), 8. 7 0 (s , 1 H X 1/2), 8. 6 1 (s , 1 H X 1 / 2 ) , 7. 42 (d, J = 6. 8Hz , 1 HX 1/2), 7. 3 5 - 7. 3 2 (m, 2H+ 1 HX 1/2), 7. 2 6 - 7. 2 0 (m, 2H), 7. 1 4 ( t, J = 7. 1 Hz , 1 HX 1/2), 6. 8 9 ( t , J =7. 1 Hz , 1 HX 1/2), 5. 24 (q u i n t , J = 7. 3Hz , 1 HX 1/2), 5. 06 (q u i n t, J = 7. 1 Hz , 1 HX 1X2), 5. 0 3 - 5. 01 (m, 1 H), 4. 2 0 -4. 1 8 (m, 1 HX 1/2), 4. 1 5 -4. 1 2 (m, 1HX 1 /2), 3. 0 2 (q, J = 7. 6Hz, 2HX 1/2), 2. 9 6 (q, J = 7. 6 Hz, 2HX 1/2), 2. 7 5 - 2. 6 0 (m, 1 H+ 1 HX 1/2), 2. 5 8 - 2. 48 (m, 1 HX 1/2), 1. 9 5 - 1. 8 8 (m, 1 H+ 1HX 1/2), 1. 7 6 - 1. 7 3 (m, 1 H+ 1 HX 1/2), 1. 5 2 (d, J = 7. 3Hz , 3H X 1/2), 1. 5 0 (d, J = 7. 1 Hz, 3 HX 1/2), 1. 3 8 - 1. 2 5 (m, 1H), 1. 3 2 ( t , J =7. 6Hz, 3HX 1/2), 1. 2 7 (t , J = 7. 6 Hz, 3HX 1/2), 1. 1 8 - 1. 0 9 (m, 2 H), 1. 0 0— 0. 9 6 (m,
1H), 0. 89 - 0. 83 (m, 6 H)
ma s s : 524 (M+ 1) +. 化合物 [27]
XH-NMR (DMSO— d6) 6 : 9. 98 (d, J = 6. 8Hz 1 HX 1/2), 8. 96 (d, J = 7. 1Hz, : 1 HX 1/2), 8. 9 1 - 8. 88 (m, 1H), 8. 73 (s, 1H), 8. 7 1 (s , 1 HX 1/2), 8. 60 (s 1 HX 1/2), 7. 43 (d, J = 7. 8Hz, 1 HX 1/2), 7. 35- 7. 33 (m, 2H + 1HX 1/2), 7. 25- 7. 2 0 (m, 2H), 7. 1 5 ( t J =6. 8Hz,. 1HX 1/2), 6. 92 ( t , J =6. 8Hz, 1 HX 1/2), 5. 26-5. 2 2 (m, 1 HX 1/2), 5. 09 - 5. 06 (m, 1 HX 1/2), 5. 04- 5. 00 (m, 1 H), 4. 21 -4. : L 6 (m, 1H), 3. 02 (q, J = 7. 3Hz, 2HX 1/2), 2. 96 (q, J = 7. 3Hz, 2HX 1/2), 2. 74-2. 7 1 (m, 1 HX 1/2), 2. 67 - -2. 63 (m, 1 H), 2. 55- 2. 48 (m, 1 HX 1/2), 1. 98- 1. 6 5 (m, 3H), 1. 52 (d, J = 7. 1Hz, 3HX 1/2), 1. 50 (d, J =7. 3 Hz , 3HX 1/2), 1. 3 '5— 1. 2 6 (m, 1 H), 1. 32 ( t , J =7. 6Hz, 3HX 1X2), 1. 28 (t, J =7. 6Hz, 3 HX 1/2), 1. 14- 1. 03 (m, 3H), 0. 88 (d, J =6. 3Hz , 6 HX 1/2), 0. 85 (d, J = 6. 6Hz, 6HX 1/2) ma s s : 524 (M+ 1) +. 実施例 28
2- [ ( ( S) - 1 - {4- [ (1 一 4—ィル) ドロキシ) メチル] フエ二ル} ェチル) ァミノ] —4— (8—ェチルイミダゾ 2- a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル
[28] (以下、 化合物 [28] という) の合成。 化合物 [25-3] 5mgとシクロペン夕ノン 5 xLから、 実施例 17— (3) の方 法に準じて目的化合物 [28] 5. 5mgを黄色固体^して得た。 化合物 [28] のスペクトルデ一夕を以下に示す。
^-NMR (CDC 13) δ : 9. 63 (d, J = 8. 0Hz, 1 HX 1/6), 8.. 93 (s, 1 HX 1/6), 8. 85 (d, J = 8. 0Hz , 1HX 5/6), 8. 8 2 - 8. 74 (m, 1 HX 5/6), 8. 56 (s , 1 H X 1 / 6 ) , 8. 45 (s , 1 HX 5/6), 7. 38-7. 12 (m, 5H), 6. 96 ( t , J = 8. 0Hz, 1HX 1/6), 6. 67— 6. 58 (m, 1HX 5/6), 6. 17 (d, J = 8. 0Hz , 1 HX 5/6), 5. 92— 5. 82 (m, 1 HX 1/6), 5. 33 (b r s , 1 HX 1/6), 5. 12 (q, J = 7. 5Hz, 1 HX 5/6), 4. 40— 4.
3 7 (m, 1H), 3. 0 7 - 2. 84 (m, 4H), 2. 40 - 2. 3 6 (m, 1H), 1. 9 7 - 1. 9 5 (m, 1 H), 1. 8 3— 1. 20 (m, 2 1 H)
ma s s : 5 5 0 (M+ 1 ) +. 実施例 2 9
2 - [ ( (1 S) - 1 - {4- [ [1 - (2, 2—ジメチルプロピル) ピぺリジン一 4—ィル] (ヒドロキシ) メチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—ェチル イミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [2 9] (以下、 化合物 [2 9] という) の合成。 化合物 [2 5 - 3] 5mgとトリメチルァセトアルデヒド 5 から、 実施例 1 7 - (3) の方法に準じて目的化合物 [2 9]- 14mgを黄色固体として得た。 化合物 [2 9] のスペクトルデータを以下に示す。
^-NMR (CDC 13) δ : 9. 6 3 (d, J = 8. OHz , 1 HX 1/6), 8. 9 3 (s, 1 HX 1/6), 8. 8 6 (d, J = 8. OHz, 1HX 5/6), 8. 8 1— 8. 7 2 (m, 1 HX 5/6), 8. 5 6 (s , 1 HX 1/6), 8. 45 (s , 1HX 5/6), 7. 40 - 7. 1 3 (m, 5H), 6. 9 6 ( t , J =8. OHz , 1 HX 1/6), 6. 6 7 - 6. 5 8 (m, 1 HX 5/6), 6. 1 5 (d, J = 8. OHz, 1 HX 5/6), 5. 9 2— 5. 8 2 (m, 1 HX 1/6), 5. 34 (b r s , 1 HX 1/6), 5. 1 2 (q, J =7. 5Hz , 1 HX 5/6), 4. 4 1 -4.
37 (m, 1H), 3. 1 2 - 3. 0 0 (m, 3H), 2. 8 2- 1. 8 5 (m, 6H), 1. 7 5- 0. 8 0 (m, 2 0 H)
ma s s : 5 5 2 (M+ 1) +. 実施例 3 0、 3 1
4— (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ー2— [ ( (1 S) - 1 - {4- [ (1ーェチルピペリジン一 4一ィル) (ヒドロキシ) メチル] フエ二 ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [3 0] (以下、 化合物 [3 0] という)、 および [3 1] (以下、 化合物 [3 1] という) の合成 (ここで、 化合 物 [3 0] と化合物 [3 1] は、 ジァステレオマ一の関係にある。 表 7参照)。' 化合物 [2 5— 3] 5 Omgとァセ卜アルデヒド 30 Lから、 実施例 1 7— (3) の方法に準じて、 目的化合物 [3 0] と [3 1] の混合物 48mg得た。 混合物はキ ラルセル〇 D— Hにて分割した。
光学分割条件は以下のとおりである。
カラム:キラルセル OD— H (Ch i r a l c e l 〇D— H、ダイセル化学工業)、 直径 2 Omm、 長さ 2 5 Omm
溶出液:へキサン Zエタノール/ジェチルァミン =85Z15/0. 1 流速: 15 mL/m i n
得られた溶液を減圧濃縮し、残渣を少量のクロ口ホルムに溶解しジェチルエーテルに て固化させ、. 目的化合物 [30〕 6mg (RT= 17. 8分) を黄色固体、 目的化合 物 [31] 14mg (RT=23. 1分) を黄色固体として得た。 化合物 [30] および化合物 [3 1] のスペクトルデータを以下に示す。
化合物 [30]
:H-NMR (CDC 13) δ : 9. 63 (d, J = 8. 0Hz, 1HX 1/6), 8. 93 (s , 1HX 1/6), 8. 82 (s, 1 HX 5/6), 8. 70 (d, J = 8. 0Hz, 1 HX 5/6), 8. 55 (s , 1 HX 5/6), 8. 45 (s, 1 HX 5/ 6), 7. 37-7. 24 (m, 4H), 7. 1 1 (d, J = 8. 0Hz , 1H), 6. 98 ( t , J = 8. 0Hz, 1 HX 1/6), 6. 57 (t, J = 8. 0Hz, 1 H X 5/6), 6. 21 (d, J = 8. 0Hz, 1 HX 5/6), 5. 91 (d, J = 8. 0Hz, 1 HX 1/6), 5. 33 (b r s , 1 HX 1/6), 5. 10 (qu i n t, J = 7. 5Hz, 1HX 5/6), 4. 42 (d, J = 7. 5Hz, 1 H); 3. 13 -2. 82 (m, 5H), 2. 38 (q, J = 7. 5Hz , 2H), 2. 03-1. 7 0 (m, 4H), 1. 62 (d, J = 7. 5Hz, 3H), 1. 49- 1. 24 (m, 3H), 1. 35 (t, J = 7. 5Hz, 3H), 1. 05 ( t , J = 7. 5Hz, 3 H)
ma s s : 51 0 (M+ 1) +. 化合物 [3 1]
XH-NMR (CDC ") 6 : 9. 62 (d, J = 8. 0Hz, 1 HX 1/6), 8. 87 (s , 1 HX 1/6), 8. 8 1 (d, J = 8. 0Hz, 1 HX 5/6), 8. 8 0 (s, 1 HX 5/6), 8. 54 (s, 1 HX 1/6), 8. 36 (s , 1 HX 5/ 6), 7. 39 - 7. 22 (m, 4H), 7. 12 (d, J = 8. 0Hz, 1H), 6. 93 (t, J = 8. 0Hz, 1 HX 1/6), 6. 66 ( t , J = 8. 0Hz, 1H X 5 / 6 ) , 6. 47 (d, J = 8. 0Hz , 1 HX 5/6), 6. 04 (d, J = 8. 0 Hz, 1 HX 1/6), 5. 34 (b r s, 1 HX 1/6), 5. 1 2 (q u i n t, J = 7. 5Hz, 1 HX 5/6), 4. 36
—2. 78 (m, 5H), 2. 34 (q, J
0 (m, 4H), 1. 63 (d, J = 7. 5
3H), 1. 36 (t, J = 7. 5Hz, 3H), 1. 04 ( t , J = 7. 5Hz, 3 H)
ma s s : 510 (M+ 1) +. 実施例 32
7075240
- 4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [ ( (1 S) — 1一 {4- [ヒドロキシ(1一プロピルピぺリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン— 5—カルボ二トリル [3 2] (以下、 化合物 [3 2] という) の合成。 化合物 [2 5— 3] 1 0mgを N, N—ジメチルホルムアミド lmLに溶解し、 炭酸 カリウム 5mgと 1 ョ一ドプロパン 3 Lを加え、 50 °Cにて 2時間撹拌した。 放 冷後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を分取薄層クロマトダラ フィ一にて精製し、 目的化合物 [3 2] 8mgを無色固体として得た。 化合物 [3 2] のスペクトルデータを以下に示す。
XH-NMR (CDC 1 3) δ : 9. 64 (d, J = 8. OHz , 1 HX 1/6), 8. 9 2 (s , 1 HX 1/6), 8. 8 3 (d, J == 8. OHz , 1 HX 5/6), 8. 8 0— 8. 7 2 (m, 1 HX 5/6), 8. 5 5 (s , 1 HX 1/6), 8. 43 (s, J = 8. OHz, 1 HX 5/6), 7. 3 9— 7. 24 (m, 4H), 7. 1 2 (d, J = 8. OHz , 1 H), 6. 9 6 ( t , J = 8. OHz , 1 HX 1/6); 6. 6 7 - 6. 5 7 (m, 1 HX 5/6), 6. 24 (d, J = 8. OHz, 1 HX 5/6), 5. 9 5 - 5. 8 5 (m, 1 HX 1/6), 5. 3 3 (b r s, 1 HX 1/6), 5. 1 1 (q, J = 7. 5Hz , 1 HX 5/6), 4. 44-4. 3 7 (m, 1 H), 3. 1 2 - 2. 74 (m, 5H), 2. 22 - 2. 1 8 (m, 2H), 1. 9 8 - 1. 6 2 (m, 7 H), 1. 46— 1. 2 0 (m, 8H), 0. 8 5 ( t , J = 7.' 5Hz , 3 H)
ma s s : 5 24 (M+ 1) +. 実施例 3 3
4一 (6—ブロモー 8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [ ( (1 S) 一 1一 {4- [ヒドロキシ (1一イソプロピルピぺリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [3 3] (以 , 下、 化合物 [3 3] という) の合成。
(1) 3一ェチルピリジン一 2—ァミン (国際公開第 2 0 0 6/02 5 5 6 7号パ ンフレット、 1 42ページに記載されている方法により合成した) 3 6 0111 を1, 4一ジォキサン 1 2mLと水 4mLの混合溶媒に溶解した溶液に、 0 にて N—ブロ モこはく酸イミド 5 5 3mgを加え、 同温にて 1. 5時間撹拌した。 反応液に水を加 え、 酢酸ェチルにて抽出し、 得られた有機層を水、 飽和食塩水にて順に洗浄した。 無 水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、.ろ液を減圧濃縮した。 得られた 残渣をシリカゲルカラムクロマトグラフィー (溶出液:へキサンノ酢酸ェチル =7 5 /2 5) にて精製し、 5一プロモー 3—ェチルピリジン一 2—ァミン [3 3 - 1] (以
0 下、 化合物 [33— 1] という) 394mgを得た。
(2) 化合物 [33— 1] 388mgから実施例 1一 (6) の方法に準じて、 4一 (6—プロモー 8ーェチルイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— (メ チルチオ) ピリミジン— 5—力ルポ二トリル [33-2] (以下、 化合物 [33-2] という) 53 Omgを得た。
(3) 化合物 [25— 1] 2. 98 gをテトラヒドロフラン 3 OmLと 2—プロパ ノール 1 OmLの混合溶媒に溶解した溶液に 20 %水酸化パラジウム炭素触媒 75. Omgを加え、 水素雰囲気下、 室温にて終夜撹拌した。 触媒をセライトろ過し、 ろ液 を減圧濃縮し、 t e r t—ブチル ((1 S) 一 1一 {4- [ヒドロキシ (ピペリジン 一 4一ィル) メチル] フエ二ル} ェチル) カーバメート [33- 3] (以下、 化合物 [33— 3] という) 2. 3 gを得た。 化合物 [33— 3] はさらに精製することな く次の反応に付した。
(4) 化合物 [33— 3] 2. 3 gとアセトン 15mLから、 実施例 1'7— (3) の方法に準じて t e r t一ブチル ((I S) — 1一 {4一 [ヒドロキシ (1一イソプ 口ピルピぺリジン一 4一ィル) メチル] フエ二ル}ェチル) カーバメート [33-4]
(以下、 化合物 [33— 4] という) 2. 14 gを得た。
(5) 化合物 [33— 4] 7 Omgと化合物 [33— 2] 63mgから、 実施例 1 一 (7) の方法に準じて、 目的化合物 [33] 4 Imgを黄色アモルファスとして得 た。 化合物 [33] のスペクトルデ一夕を以下に示す。
XH-N R (CDC 13) δ : 9. 89 (s, 1HX 5/6), 9. 74 (s , 1 H X 1/6), 8. 93 (s , 1 HX 5/6), 8. 86 (s , 1 HX 1/6), 8. 5 5 (s , 1 HX 1/6), 8. 16 ( s, 1 HX 5/6), 7. 41 -7. 27 (m, 5H), 6. 76 (d, J = 8. OHz, 1 HX 5/6), 6. 29 (d, J = 8. 0 Hz , 1 HX 1X6), 5. 35 (b r s , 1 HX 1/6), 5. .27 (qu i n t, J = 7. 5Hz , 1 HX 5/6), 4. 38 (d, J = 7. 5Hz, 1 H), 3. 10 (q, J = 7. 5Hz, 2H), 2. 98— 2. 72 (m, 5H), 2. 14— 1. 9 2 (m, 3H), 1. 69 (d, J = 7. 5Hz, 3H), 1. 59— 1. 50 (m, 1H), 1. 43 - 1. 25 (m, 2H), 1. 41 (t, J = 7. 5Hz, 3H), 1. 02 (d, J =7. 5Hz , 6H)
ma s s : 602, 604 (M+ 1) +. 実施例 34
7075240 2— [ ( (1 S) - 1一 {4- [ヒドロキシ (1ーメチルー 1, 2, 3, 6一テ卜ラ ヒドロピリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] 一 4— (8—メチ ルイミダゾ [1, 2— a] ピリジン— 3—ィル) ピリミジン一 5—力ルポ二トリル [3 4] (以下、 化合物 [34] という) の合成。
(1) ェチル 1一メチル— 1, 2, 3, 6ーテトラヒドロ 4一ピリジンカルボキシ レート 1. 7 gをテトラヒドロフラン 2 OmLに溶解した溶液に、 水素化リチウムァ ルミニゥム 7 7 3mgを加え、 室温にて 1時間撹拌した。 反応液を 0°Cとした後、 硫 酸ナトリウム · 1 0水和物を気泡が出なくなるまでゆつくり加え、 室温にて終夜撹拌. した。 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をクロ口ホルム 4 OmL に溶解し、 二酸化マンガン 2 gを加え室温にて終夜撹拌した。 不溶物をセライ卜ろ過 し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出 液:クロ口ホルムノメタノール = 1 0 0/0- 9 0/1 0) にて精製し、 1一メチル - 1, 2, 3, 6—テトラヒドロピリジン一 4—カルボアルデヒド [34— 1] (以 下、 化合物 [34— 1] という) 2 1 2mgを得た。
(2) 化合物 [34— 1] 2 1 2mgと化合物 [1一 1] 3 0 Omgから、 実施例 1 2— (1) の方法に準じて t e r t一ブチル ((I S) — 1— {4一 [ヒドロキシ
(1ーメチルー 1, 2, 3, 6—テトラヒドロピリジン一 4一ィル) メチル] フエ二 ル} 'ェチル) カーバメート [34— 2] (以下、 化合物 [34— 2] という) 1 7 7 mgを得た。
(3) 化合物 [34— 2] 1 7 7mgと化合物 [1一 6] 143mgから、 実施例 1一 (7) の方法に準じて目的化合物 [34] 8 8mgを淡黄色固体として得た。 化合物 [34] のスペクトルデータを以下に示す。
一 NMR (CDC 13) 5 : 9. 6 2 (d, J = 7. 2Hz , 1 ΗΧ 1/5), 8. 9 2 - 8. 6 9 (m, 1 Η+ 1 ΗΧ 4/5), 8. 5 5 (s, 1 ΗΧ 1/5), 8. 4 7 (s , 1HX4/5), 7. 74- 6. 5 8 (m, 6Η), 6. 3 0 - 6. 2 0 (m, 1HX 4/5), 5. 9 8 - 5. 8 0 (m, 1H+ 1 HX 1X5),, 5. 3 8 - 5. 0 0 (m, 2H), 3. 48 - 2. 9 9 (m, 2H), 2. 7 0 - 1. 9 0 (m, 4H), 2. 6 0 (s, 3H), 1. 6 0 (d, J = 6. 8Hz , 3 H)
ma s s : 48 0 (M+ 1) +. 実施例 3 5
4- (8—クロロイミダゾ [1, 2— a] ピリジンー3—ィル) 一 2— [ ( (1 S) 一 1一 {4 - [ヒドロキシ (1ーメチルビペリジン一 4一ィル) メチル] フエ二ル}
2007/075240 ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [3 5] (以下、 化合物 [3 5] という) の合成。 化合物 [9— 1] 7 9mgと化合物 [2 3— 1] l O Omgから、 実施例 1一 (7) の方法に準じて目的化合物 [3 5] 44mgを白色固体として得た。 化合物 [3 5] のスペクトルデ一夕を以下に示す。
^-NMR (DMSO— d6) δ: 1 0. 0 6 (d, J = 7. 1Hz, 1 HX 1/2), 9. 04 (d, J = 7. 1 Hz , 1 HX 1/2), 8. 9 6 - 8. 94 (m, 1 H), . 8. 7 8 - 8. 74 (m, 1H+ 1 HX 1/2), 8. 6 2 (s , 1 HX 1/4), 8. 6 1 (s , 1 HX 1/4), 7. 7 9 (d, 1 = 7. 1 Hz , 1 HX 1/2), 7. 7 4- 7. 7 1 (m, 1 HX 1/2), 7. 34— 7. 3 2 (m, 2H), 7. 2 6 - 7. 1 7 (m, 2H+ 1 HX 1/2), 6. 9 5 (q, J = 7. 1 Hz, 1 HX 1/2), 5. 24 (q u i n t , J = 7. 6Hz, 1 HX 1/2), 5. 1 0 - 5. 0 5 (m, 1 H+ 1 HX 1/2), 4. 2 0 -4. 1 3 (m, 1 H), 2. 74- 2. 6 6 (m, 1 H+ 1 HX 1/2), 2. 5 5 - 2. 49 (m, 1 HX 1/2), 2. 0 '6 (s , 3 HX 1/2), 2. 0 3 (s, 3HX 1/2), 1. 7 2 - 1. 5 9 (m, 3H), 1. 5 2 (d, J =7. 1Hz , 3 HX 1/2), 1. 49 (d, J =7. 1 Hz , 3H X 1/2), 1. 3 2 - 0. 94 (m, 4H)
m s s : 50 2, 504 (M+ 1) +. 実施例 3 6
4一 ( 8—シクロプロピルイミダゾ [ 1, 2 - a]ピリジン一 3—ィル)一 2— [ ( (1 S) - 1 - {4- [ヒドロキシ (1ーメチルピペリジン一 4一ィル) メチル] フエ二 ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [3 6] (以下、 化合物 [3 6] という) の合成。
(1) 3—シクロプロピルピリジン一 2—ァミン (国際公開第 2 0 0 6/0 2 5 5 6 7号パンフレット、 142ページに記載されている方法により合成した) 3 9 0m gから、 実施例 1一 (6) の方法に準じて 4一 (8—シクロプロ.ピルイミダゾ [1, 2— a] ピリジン一 3—ィル) - 2 - (メチルチオ) ピリミジン一 5—力ルポ二トリ ル [3 6 - 1] (以下、 化合物 [3 6— 1] という) 7 5 Omgを橙色固体として得 た。 (2) 化合物 [3 6— 1] 1 0 Omgと化合物 [2 3— 1] 1 1 Omgから、 実施 例 1一 (7) の方法に準じて目的化合物 [3 6] 6 6mgを黄色固体として得た。 化合物 [3 6] のスペクトルデータを以下に示す。
7075240 XH-NMR (CD3OD) δ : 9. 85 (d, J = 8. OHz, lHX l/3)' 8. 78 - 8. 73 (m, 1 HX 2/3), 8. 68 (s, 1HX 1/3), 8. 61 (s , 1 HX 2/3), 8. 43 (s , 1 HX 2/3), 8. 36 (s, 1HX 1/3), 7. 40- 7. 25 (m, 4H), 6. 93 ( t , J = 8. OHz, 1HX 2/3), 6. 87 (d, J = 8. OHz, 1 HX 2/3), 6. 71 (d, J = 8. OHz , 1 H X 1/3), 6. 67 (t, J = 8. OHz, 1 HX 1/3), 5. 23 (q, J = 7. 5Hz, 1 HX 1/3), 5. 01 (qu i n t, J = 7. 5Hz, 1 HX 2/3), 4. 28-4. 22 (m, 1 H), 2. 87-2. 71 (m, 2HX 2/3), 2. 5 9-2. 51 (m, 2HX 1/3), 2. 39- 2. 36 (m, 1H), 2. 16 (s , . 3HX 1/3), 2. 12 (s, 3 HX 2/3), 1. 99— 1. 79 (m, 2H), 1. 71 - 1. 04 (m, 7 H), 1. 56 (d, J = 7. 5Hz, 3 HX 2/3), 1. 06 (d, J = 7. 5Hz, 3 HX 1/3), 0. 82— 0. 73 (m, 2 H) ma s s : 508 (M+ 1) +. 実施例 37
4— (8—クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— [· ( (1 S) 一 1― {4— [ (1ーシクロプロピルピぺリジン一 4一ィル) (ヒドロキシ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [37] (以下、 化合 物 [37] という) の合成。
'
(1) 化合物 [25— 1] 1 gをメタノール 1 OmLに溶解し、 20%水酸化パラ ジゥム炭素触媒 20 Omgを加え、 水素雰囲気下、 室温にて 1時間撹拌した。 触媒を セライ卜ろ過し、 ろ液を減圧濃縮して t e r t一ブチル ((I S) — 1一 {4— [ヒ ドロキシ (ピペリジン一 4一ィル) メチル] フエ二ル} ェチル) カーバメート [37 -1] 71 Omgを得た。 化合物 [37- 1] (以下、 化合物 [37 - 1] という) はさらに精製することなく次の反応に付した。
(2) 化合物 [37— 1] 71 Omgをクロ口ホルム 1 OmLに溶解し、 トリェチ ルァミン 1. 2 mLとトリフルォロ酢酸無水物 744 xLを加え、 室温にて終夜撹拌 した。 反応液に水を加え、 クロ口ホルムにて抽出し、 得られた有機層を飽和食塩水に て洗浄した。 無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮 した。 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液:クロ口ホルム Zメタノール- 100/0 - 97X3) にて精製し、 t e r t—プチル [(I S) ― 1一 (4一 {ヒドロキシ [1— (トリフルォロアセチル) ピぺリジン一 4一ィル] メ チル } フエニル) ェチル] カーバメート [37— 2] (以下、 化合物 [37— 2] と いう) 328mgを得た。
(3) 化合物 [37— 2] 328mgと化合物 [9一 1] 21 Omg力、ら、 実施例
1一 (7) の方法に準じて 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3— ィル) —2— {[(1 S) 一 1― (4— {ヒドロキシ [1 - (トリフルォロアセチル) ピぺリジン一 4一ィル] メチル } フエニル) ェチル] アミノ} ピリミジン一 5—カル ポニトリル . [37— 3] (以下、 化合物 [37— 3] という) 237mgを得た。
(4) 化合物 [37— 3] 237mgをテトラヒドロフラン 3mLとメタノール 1 mLの混合溶媒に溶解し、 5 N水酸化ナトリウム水溶液 163 /xLを加え、 50°Cに て 5時間撹拌した。 放冷後、 反応液を減圧濃縮し、 得られた残渣をシリカゲルカラム クロマトグラフィー (溶出液:クロ口ホルム Zメタノール =95Z5— 93Z7) に. て精製し、 4一(8—クロロイミダゾ [1, 2— a]ピリジン一 3—ィル)一 2— [((1 S) 一 1— {4- [ヒドロキシ (ピペリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [37— 4] (以下、 化合物 [37— 4] という) 156mgを得た。 (5) 化合物 [37— 4] 28mgをエタノール 2mLに溶解した溶液に、 (1一 エトキシシクロプロポキシ) トリメチルシラン 58mg、 酢酸 33 L、 およびモレ キュラーシ一ブ 4A1 Omgを加え、 室温にて 10分間撹拌した後、 シァノ水素化ほ う素ナトリウム 1 lmgを加え、 6時間加熱還流した。 放冷後、 不溶物をろ過し、 ろ 液に水酸化ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残 渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [37] 20mgを淡黄色 固体として得た。 化合物 [37] のスペクトルデ一夕を以下に示す。
^-NMR (CDC ") (3 : 9. 68 (d, J = 8. OHz , 1 HX 1/6), 8. 90 (s , 1 HX 1/6), 8. 63 (d, J = 8. OHz, 1 HX 5/6), 8. 5 7 (s , 1 HX 1/6), 8. 44- 8. 34 (m, 1 HX 5/6), 8. 42 ( s , 1 HX 5/6), 7. 47 - 7. 26 (m, 5H), 6. 92 ( t , J = 8. OHz, 1HX 1/6), 6. 57 -6. 43 (m, 2HX 5/6), 6. 05 (d, J = 8. OHz, 1 HX 1/6), 5. 34 (b r s , 1 HX 1/6), 5. 0.3— 4. 98 (m, 1HX 5/6), 4. 47 -4. 39 (m, 1H), 3. 91-3. 65 (b r s , 1 H), 3. 09-2. 91 (m, 2H), 2. 16- 1. 48 (m, 8 H), 1. 37 一 1. 12 (m, 3H), 0. 49— 0. 36 (m, 4H)
ma s s : 528、 530 (M+ 1) +. 実施例 38
4- (8—クロロイミダゾ [1 , 2 - a] ピリジン一 3—ィル) - 2 - ( { (1 S) 一 1一 [4一 (ヒドロキシ {1— [2- (メチルスルホニル) ェチル] ピぺリジン一
4—ィル }メチル) フエニル]ェチル }ァミノ) ピリミジン— 5—力ルポ二トリル [3 8] (以下、 化合物 [38] という) の合成。
(1) 2— (メチルスルホニル) エタノール 20 Omgとトリエチルァミン 450 zLをクロ口ホルム 5mLに溶解し 0°Cとした後、塩化メタンスルホニル 190 を加え、 同温にて 3時間撹拌した。 反応液に飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮し、 2一 (メチルスルホニル) ェチル メタンスル ホネート [38— 1] (以下、 化合物 [38— 1] という) を得た。 化合物 [38—. 1] はさらに精製することなく次の反応に付した。
(2) 化合物 [37— 4] 5mgをクロ口ホルム 0. 5mLとメタノール 0. 5m Lの混合溶媒に溶解し、 トリェチルァミン 4. 5 /iLと化合物 [38— 1] 6mgを 加えた。 室温にて 30分間撹拌した後、 分取薄層クロマトグラフィーにて精製し、 目 的化合物 [38] 6mgを無色固体として得た。 化合物 [38] のスぺクトルデ一夕を以下に示す。
^-NMR (CDC 13) δ 9. 70 (d, J = 8. OHz , 1 HX 1/6), 8. 95 (s, 1HX 1/6), 8. 69 (d, J = 8. OHz, ΓΗΧ 5/6), 8. 5 9 (s , 1 HX 1/6), 8. 50 (s, 1 HX 5/6), 8. 50 - 8. 42 (m, 1 HX 5/6), 7. 53 - 7. 26 (m, 5H), 6. 96 (t, J =8. OHz, 1 HX 1/6), 6. 58 - 6. 46 (m, 1 HX 5/6), 6. 25 - 6. 23 (m, 1 HX 5/6), 5. 95 (d, J = 8. OHz, 1HX 1/6), 5. 34 (b r s , 1 HX 1/6), 5. 05 - 5. 00 (m, 1 HX 5/6), 4. 47— 4. 39 (m, 1 H), 3. 55 - 3. 26 (b r s, 1 H), 3. 12— 3. 09 (m, 2H), 3. 01 (s, 3H), 2. 97-2. 94 (m, 1H), 2. 83 - 2. 79 (m, 3H), 2. 09 - 1. 84 (m, 4H), 1. 64-1. 61 (m, 1 H), 1. 46- 1. 21 (m, 5 H)
ma s s : 594、 596 (M+ 1) +. 実施例 39
4- [8- (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 2 — [ ( (l S) —l— {4— [tドロキシ (1—イソプロピルピぺリジン一 4一ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン— 5—力ルポ二トリル [39] (以 下、 化合物 [39] という) の合成。 化合物 [10— 2] 40mgと化合物 [33-4] 47mgから実施例 1一 (7) の 方法に準じて、 目的化合物 [39] 22mgを黄色固体として得た。
2007/075240
化合物 [3 9] のスペクトルデ一夕を以下に示す。
XH-NMR (CDC 13) δ : 9. 85 (d, J = 8. OHz, 1 HX 1/6), 8. 93 (s, 1.HX 1/6), 8. 89 - 8. 82 (m, 1 HX 5/6), 8. 77-8. 73 (m, 1 HX 5/6), 8. 58 (s, 1 HX 1/6), 8. 44 (d, J = 8. 0Hz, 1HX 5/6), 7. 71 (d, J =8. OHz, 1 HX 1/6), 7. 59 -7. 54 (m, 1 HX 5/6), 7. 41 -7. 21 (m, 4H), 7. 12-7. 09 (m, 1 HX 1/6), 6. 79 - 6. 64 (m, 1 HX 5/6), 6. 45 (d, J = 8. OHz, 1 HX 5/6), 6. 12-6. 02 (m, 1 HX 1/6), 5. 3. 4 (b r s , 1 HX 1/6), 5. 10-5. 03 (m, 1 HX 5/6), 4. 44— 4. 39 (m, 1 H), 2. 93 - 1. 88 (m, 8H), 1. 64- 1. 58 (m, 4H), 1. 40- 1. 13 (m, 3H), 1. 00 (d, J = 7. 5Hz, 6 H) m a s s : 546 (M+ 1) +. 実施例 40
4一 (8—クロ□イミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {' [ (1 S) 一 1一 (4- {ヒドロキシ [1― (2—ヒドロキシェチル) ピぺリジン一 4一ィル] メチル } フエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [40] (以 下、 化合物 [40] という) の合成。 化合物 [3 7 -4] 2 Omgと 2—ブロモエタノール 5 Lから、実施例 2 5— (4) の方法に準じて目的化合物 [40] 16 mgを黄色固体として得た。 化合物 [40] のスペクトルデ一夕を以下に示す。
一 NMR (CD3〇D) <5 : 10. 08 (d, J = 8. OHz , 1 HX 1/3), 8. 90 - 8. 87 (m, 1 HX 2X3), 8. 76 (s , 1 HX 1/3), 8. 65 (s , 1 HX 2/3), 8. 55 (s , 1 HX 2/3), 8. 49 (s , 1 HX 1/3), 7. 63 (d, J = 8. OHz, 1 HX 1/3), 7. 55 (t, J =8. OHz, 1 HX 2/3), 7. 40 - 7. 26 (m, 4H), 7.. 08 ( t, J = 8. OHz, 1 H X 1 / 3 ) , 6. 85 - 6. 80 (m, 1 HX 2/3), 5. .27 (q, J = 7. 5Hz, 1 HX 1/3), 5. 07 (q, J =7. 5Hz, 1 HX 2/3), 4. 30 -4. 25 (m, 1 H), 3. 64- 3. 58 (m, 2H), 2. 97 - 2. 85 (m, 2HX 2/3), 2. 75 - 2. 67 (m, 2 HX 1/3), 2. 47— 2. 40 (m, 2H), 1. 99- 1. 74 (m, 3H), 1. 58 (d, J = 7. 5Hz, 3H), 1. 58 - 1. 03 (m, 4H)
ma s s : 532、 534 (M+ 1) +. 実施例 41、 42
4一 (8—ェチルイミダゾ [1, 2— a] ピリジン— 3—ィル) 一 2— {[(I S) — 1 - (4- {ヒドロキシ [(3 R) — 1—メチルピペリジン一 3—ィル] メチル } フ ェニル) ェチル] アミノ} ピリミジン一 5—力ルポ二トリル [41] (以下、 化合物 [41] という)、 および 4一 (8—ェチルイミダゾ [1, 2— a] ピリジン一 3— ィル) 一2— {[(1 S) — 1— (4一 {ヒドロキシ [(3 S) — 1—メチルピベリジ ンー 3—ィル] メチル } フエニル) ェチル] アミノ} ピリミジン一 5—力ルポ二トリ ル [42] (以下、 化合物 [42] という) の合成 (ここで、 化合物 [41] と化合 物 [42] は、 ジァステレオマーの関係にある。 表 9参照)。 (1) ベンジル (3R) —3—ホルミルピぺリジン一 1一力ルポキシレート (国際 ■ 公開第 02Z461 57号パンフレットに記載されている方法により合成した) 15 gと化合物 [1 - 1] 16. 6 gから、 実施例 12— (1) の方法に準じてベンジル 3— [(4一 {(1 S) — 1一 [( t e r t一ブトキシカルボニル) ァミノ] ェチル } フエニル) (ヒドロキシ)メチル] ピぺリジン一 1一カルボキシレート [41一 1] (以 下、 化合物 [41— 1] という) 1 1. 8 gを 4種の異性体の混合物として得た。
(2) 化合物 [5— 6] 7. 56 gと化合物 [41一 1] 1 1. 8 gから、 実施例 1 - (7) の方法に準じて、 ベンジル 3— [[4一 ((1 S) 一 1— {[5—シァノー 4一 (8—ェチルイミダゾ [1, 2— a] ピリジン— 3—ィル) ピリミジンー2—ィ ル] 'アミノ} ェチル) フエニル] (ヒドロキシ) メチル] ピぺリジン一 1—カルポキ シレート [41一 2] (以下、 化合物 [41一 2] という) 6. 81 gを得た。
(3) 化合物 [41— 2] 6. 8 1 gを、 実施例 17— (2) の方法に準じて 4一 (8—ェチルイミダゾ [1, 2— a] ピリジン— 3—ィル) —2— [((1 S) - 1 - {4- [ヒドロキシ (ピペリジン一 3—ィル) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [41 -3] 5 gを得た。 得られた化合物 [41一 3]は 4種の異性体の混合物であり、シリカゲルカラムクロマトグラフィー(溶出液: クロ口ホルム/メタノール =100/0 - 90/10 )にて精製し、低極性化合物(当 該条件で先に溶出される化合物である) [41— 3 a] . (以下、 化合物 [41一 3 a] という) 2. 2 l gと、 高極性化合物(当該条件で後に溶出される化合物である) [4 1一 3 b] (以下、 化合物 [41一 3 b] という) 1. 88 gを、 それぞれ 2種の異 性体の混合物として得た。 ここで、 化合物 [41一 3 a] は化合物 [41] と [42〕 のそれぞれの前躯体の混合物であり、 化合物 [41一 3 b] は後述の化合物 [43] と [44] のそれぞれの前躯体の混合物である。
(4) 化合物 [41—3 a] 2. 21 gから実施例 1 7— ( 3 ) の方法に準じて、 目的化合物 [41] と [42] の混合物 1. 89 gを得た。 混合物はキラルパック A Dにより分割した。
光学分割条件は以下のとおりである。
カラム:キラルパック AD (Ch i r a 1 p ak AD、 ダイセル化学工業)、 直 径 50 mm、 長さ 5000 mm
溶出液:へキサン Z 2—プロパノール Zジェチルァミン =65/35/0. 1 流速: 10 OmL/m i n
得られた溶液を減圧濃縮し、残渣を少量のクロ口ホルムに溶解しジェチルエーテルに て固化させ、 目的化合物 [41] 892mg (RT=15. 5分) を淡黄色固体、 目 的化合物 [42] 298mg (RT=35分) を淡黄色固体として得た。 化合物 [41] および [42] のスペクトルデータを以下に示す。
化合物 [41]
XH-NMR (DMSO-d6) δ : 9. 98 (d, J = 6. 8Hz, 1HX 1/3), 8. 96 (d, J = 6. 8Hz , 1 HX 2/3), 8. 89 - 8. 86 (m, 1 H), 8. 73 (s , 1 H), 8. 70 (s, 1 HX 1/3), 8. 61 (s, 1 HX 2/3), 7. 41 -7. 16 (m, 5H+ 1HX 1/3), 6. 89 ( t , J = 7. 1Hz, 1 HX 2/3), 5. 30 - 5. 00 (m, 2 H), 4. 29-4. 19 (m, 1 H), 3. 32 (s , 3H), 3. 02 - 2. 93 (m, 2H), 2. 65 -2. 50 (m, 1 H), 2. 32- 2. 28 (m, 1 HX 1/3), 2. 12-2. 06 (m, 1 HX 2/3), 1. 80- 1. 40 (m, 9H), 1. 32 - 1. 20 (m, 3 H) ma s s : 496 (M+ 1 ) +. 化合物 [42]
^-NM (DMS〇—d6) (5 : 9. 90 (m, 1 HX 2/5), 8. 98-8. 60 (m, 3H+ 1HX 3/5), 7. 60 - 6. 85 (m, 6 H), 5. 18- 5. 05 (m, 2H), 4. 30 -4. 20 (m, 1H), 3. 34 (s, 3H), 3. 2 0-2. 80 (m, 2H), 2. 70 - 2. 50 (m, 1 H), 2. 18— 1. 45 (m, 1 OH), 1. 40- 1. 20 (m, 3 H)
ma s s : 496 (M+ 1) +. 実施例 43、 44
4一 (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) 一 1— (4一 {ヒドロキシ [(3R) — 1ーメチルビペリジン一 3—ィル] メチル } フ ェニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [43] (以下、 化合物 [43] という)、 および 4一 (8—ェチルイミダゾ [1, 2— a] ピリジンー3— ィル) 一2— {[(1 S) 一 1一 (4一 {ヒドロキシ [(3 S) —1—メチルピベリジ ン一 3—ィル] メチル } フエニル) ェチル] アミノ} ピリミジン一 5—力ルポ二トリ ル [44] (以下、 化合物 [44] という) の合成 (ここで、 化合物 [43] と化合 物 [44] は、 ジァステレオマ一の関係にある。 表 9参照)。
化合物 [41一 3 b] 917mgから、 実施例 17— (3) の方法に準じて、 目的化 合物 [43] と [44] の混合物 92 lmgを得た。 混合物はキラルセル OD— Hに て分割した。.
光学分割条件は以下のとおりである。
カラム:キラルセル OD— H (Ch i r a l c e l OD— H、ダイセル化学工業)、 直径 20mm、 長さ 250 mm
溶出液:へキサン/エタノール/ジェチルァミン =85/15/0. 1
流速: 20 mL/m i n
得られた溶液を減圧濃縮し、残渣を少量のクロ口ホルムに溶解しジェチルェ一テルに て固化させ、 目的化合物 [43] 51 Omg (RT= 16. 8分) を淡黄色固体、 目 的化合物 [44] 253mg (RT=23. 1分) を黄色固体として得た。 化合物 [43] および [44] のスペクトルデータを以下に示す。
化合物 [43]
XH-NMR (DMSO-d6) δ: 10. 00 (d, J =6. 8 Hz, 1HX2/5), 8. 98 (d, J = 6. 8Hz , 1 HX 3/5), 8. 92— 8. 87 (m, 1 H), 8. 76 (s , 1 H), 8. 73 (s , 1 HX 2/5), 8. 63 (s , 1 HX 3/5), 7. 46-7. 15 (m, 5H+ 1 HX 2/5), 6. 90 ( t , J = 7. 1Hz, 1 HX 3/5), 5. 25 - 5. 05 (m, 2H), 4. 22-4. 20 (m, 1 H), 3. 34 (s, 3H), 3. 10-2. 80 (m, 2H), 2. 70-2: 50 (m, 1 H), 2. 15— 1. 60 (m, 1 OH), 1. 39— 1. 22 (m, 3 H) ma s s : 496 (M+ 1) +. 化合物 [44]
XH-NMR (DMSO - d6) δ : 9. 90 (m, 1 HX 2/5), 9. 00-8. 60 (m, 3H+ 1 HX 3/5), 7. 40 -6. 80 (m, 6 H), 5. 20- 5. 00 (m, 2H), 4. 20-4. 00 (m, 1H), 3. 30 (s, 3H), 3. 4 0-3. 20 (m, 2H), 2. 70一 1. 40 (m, 8H), 1. 40- 1. 20 (m, 5H)
ma s s : 496 (M+ 1) +. 実施例 45
4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一2— [((1 S) - 1- {4- [(1ーェチルピペリジン— 3—ィル) (ヒドロキシ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [45] (以下、 化合物 [45] という) の合成。
JP2007/075240 化合物 [41 - 3 a] 25 mgとァセトアルデヒドから実施例 17— (3) の方法に 準じて、 目的化合物 [45] 23mgを白色固体として得た。 化合物 [45] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) «3 : 9. 62 - 9. 60 (m, 1 HX 1/5), 8. 95 一 8. 75 (m, 1 H+ 1 HX 4/5), 8. 56 (s , 1 HX 1/5), 8. 42 (s , 1 HX 4/5), 7. 40- 6. 60 (m, 6H), 6. 30— 6. 25 (m, 1 HX 4/5), 5. 95 - 5. 90 (m, 1 HX 1/5), 5. 35 -4. 85 (m, 2H), 3. 1 5-3. 00 (m, 2H), 2. 70 - 2. 30 (m, 6 H), 2. 00 - 1.. 80 (m, 2H), 1. 63 (d, J =7. 0Hz, 3H), 1. 55 - 1. 20 (m, 6H), 1. 10-1. 00 (m, 3 H)
m a s s : 510 (M+ 1) +. 実施例 46
4一 (8—ェチルイミダゾ [1, 2— a] ピリジン— 3—ィル) 一2— [((1 S) 一 1一 {4- [(1ーェチルピペリジン一 3—ィル) (ヒドロキシ) メチル] 'フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [46] (以下、 化合物 [46] という) の合成。 化合物 [41一 3 b] 36 mgとァセトアルデヒドから実施例 17— (3) の方法に 準じて、 目的化合物 [46] 28mgを白色固体として得た。 化合物 [46] のスペクトルデータを以下に示す。
^-NMR (CDC 13) 6 : 9. 61— 9: 59 (m, 1 HX 1/5), 8. 90 一 8. 75 (m, 1 H+ 1 HX 4/5), 8. 52 (s, 1 HX 1/5), 8. 43 (s , 1 HX 4/5), 7. 40-6. 60 (m, 6H), 6. 38 - 6. 32 (m, 1 HX 4/5), 5. 95 - 5. 90 (m, 1 HX 1/5), 5. 35-5. 10 (m, 1 H), 4. 68-4. 52 (m, 1 H), 3. 12— 3. 00 (m, 2H), 2. 80— 1. 70 (m, 8H), 1. 62 (d, J = 7. 0Hz, 3H), 1. 60— 1. 20 (m, 6 H), 1. 10- 1. 00 (m, 3H)
ma s s : 510 (M+ 1) +. 実施例 47
4- (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— [ ( (1 S) 一 1一 {4一 [ (1—イソプロピルピぺリジン一 3—^ Γル) (ヒドロキシ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン— 5—力ルポ二トリル [47] (以下、 化合 物 [47] という) の合成。
化合物 [41— 3 a] 25mgとアセトンから実施例 17— (3) の方法に準じて、 目的化合物 [47] 23mgを白色固体として得た。 化合物 [47] のスペクトルデ一夕を以下に示す。
^-NMR (CDC 13) δ : 9. 61 (m, 1HX 1/5), 8. 92-8. 70 (m, 1H+1HX4/5), 8. 56 (s , 1 HX 1/5), 8. 48 (s, 1 HX 4/5), 7. 40— 6. 60 (m, 6 H), 6. 22 -6. 20 (m, 1HX4/5), 5. 95 - 5. 85 (m, 1 HX 1/5), 5. 38— 5. 00 (m, 2H), 3. 1 5-3. 00 (m, 2H), 2. 95 -2. 30 (m, 5H), 2. 00 - 1. 80 (m, . 2H), 1. 63 (d, J = 7. 0Hz, 3H), 1. 55— 1. 10 (m, 6 H) , 1. 03 - 1. 00 (m, 6H)
ma s s : 524 (M+ 1 ) +. 実施例 48
4- (8—ェチルイミダゾ [1, 2 - a] ピリジン— 3—ィル) - 2 - [ ( (1 S) 一 1一 {4- [ (1一イソプロピルピぺリジン一 3—ィル) (ヒドロキシ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [48] (以下、 化合 物 [48] という) の合成。 化合物 [41一 3 b] 36mgとアセトンから実施例 17— (3) の方法に準じて、 目的化合物 [48] 2 lmgを白色固体として得た。 化合物 [48] のスペクトルデータを以下に示す。
— NMR (CDC 13) δ : 9. 62— 9. 60 (m, 1 HX 1/5), 8. 95 一 8. 78 (m, 1 H+ 1 HX 4/5), 8. 56 (s , 1 HX 1/5), 8. 45 (s, 1 HX 4/5), 7. 40-6. 60 (m, 6H), 6. 22 - 6. 20 (m, 1 HX 4/5), 5. 95 - 5. 85 (m, 1 HX 1/5), 5. 35— 5. 10 (m, 1 H), 5. 05-4. 68 (m, 1 H), 3. 18-3. 00 (m, 2H), 2. 80-2. 38 (m, 5H), 2. 05— 1. 80 (m, 2H), 1.. 63 (d, J = 7. 0Hz, 3 H), 1. 60 - 1. 20 (m, 6 H) , 1. 05— 0. 98. (m, 6 H) ma s s : 524 (M+ 1) +. 実施例 49
2 - [ ( (I S) — 1一 {4- [ヒドロキシ (1ーメチルー 1, 2, 5, 6—テトラ ヒドロピリジン一 3—ィル) メチル] フエ二ル} ェチル) ァミノ] 一 4一 (8—メチ. ルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリル [4 9] (以下、 化合物 [49] という) の合成。
T/JP2007/075240 (1) ァレカイジン一塩酸塩 780mg、 トリェチルァミン 3. 07mL、 N, 〇 ージメチルヒドロキシルァミン塩酸塩 644mg、 1—ェチルー 3— (3—ジメチル ァミノプロピル) カルポジイミド塩酸塩 1. 26 g、 および 1ーヒドロキシベンゾト リアゾールー水和物 1. 01 gをクロ口ホルム 1 OmL中に溶解し、 室温にて終夜撹 拌した。 反応液に水を加え、 クロ口ホルムにて抽出し、 得られた有機層を水、 飽和食 塩水にて順に洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減 圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液:クロロホ ルム Zメ夕ノ一ル= 100/0 - 90/10) にて精製し、 N—メトキシー N, 1 - ジメチルー 1, 2, 5, 6—テトラヒドロピリジン一 3—力ルポキシアミド [49一. 1] (以下、 化合物 [49ー 1] という) 857mgを得た。
(2) 化合物 [49一 1] 299mgと化合物 [1一 1] 300mgから、 実施例 2- (2) の方法に準じて t e r t一ブチル ((I S) — 1一 {4- [(1一メチル — 1, 4, 5, 6—テトラヒドロピリジン一 3—ィル) カルボニル] フエ二ル} ェチ ル) カーバメート [49一 2] (以下、 化合物 [49一 2] という) 162mgを得 た。 '
(3) 化合物 [49— 2] 162mgと化合物 [1一 6] l O Omgから、 実施例 1一 (7) の方法に準じて、 4一 (8—メチルイミダゾ [1, 2-a] ピリジン一 3 —ィル) 一 2— [((1 S) 一 1— {4一 [(1ーメチルー 1, 2, 5, 6—テトラヒ ドロピリジン一 3—ィル) カルボニル] フエ二ル} ェチル) ァミノ] ピりミジン一 5 —力ルポ二トリル [49一 3] (以下、 化合物 [49— 3] という) 4 Omgを得た。
(4) 化合物 [49一 3] 4 Omgをテトラヒドロフラン 5mLとメタノール lm Lの混合溶媒に溶解し 0°Cとした後、 水素化ほう素ナトリウム 1 Omgを加え、 室温 まで昇温しながら 1時間撹拌した。反応溶液に飽和炭酸水素ナ卜リゥム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナト リウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を分取薄層 クロマトグラフィーにて精製した。得られた粗精製物を少量のクロ口ホルムに溶解し、 へキサンにて固化させ、 目的化合物 [49] 7mgを白色固体として得た。 化合物 [49] のスペクトルデータを以下に示す。
:H-NMR (CDC ") δ : 9. 62 (d, J = 7. 2Hz, 1 HX 1/5), 8. 95 - 8. 72 (m, 1 H+ 1 HX 4/5), 8. 55 (s , 1 HX 1/5), 8. 5 0 (s , 1 HX 4/5), 7. 45-6. 55 (m, 6H), 6. 10-6. 05 (m, 1 HX 4/5), 5. 90- 5. 80 (m, 1 H+ 1 HX 1/5), 5. 35-5. 0 8 (m, 2H), 3. 05-2. 90 (m, 2H), 2. 80 - 2. 60 (m, 2H), 2. 62 (s, 3H), 2. 40-2. 20 (m, 2H), 1. 62 (d, J = 6. 8
Hz, 3H)
m a s s : 480 (M+ 1) +. 実施例 50、. 51
2— { [ (I S) — 1— (4一 {ヒドロキシ [ (2 S) — 1一メチルピロリジン一 2 一ィル] メチル } フエニル) ェチル] アミノ} 一 4一 (8—メチルイミダゾ [1, 2 -a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [50] (以下、 化合 物 [50] という)、 および [51] (以下、 化合物 [51] という) の合成(ここで、 化合物 [50] と化合物 [51] は、 ジァステレオマーの関係にある。表 10参照)。 .
(1) ベンジル (2 S) — 2—ホルミルピロリジン一 1一力ルポキシレ一ト (B i oo r g. Me d. Ch em. , 2003. 11. 3153— 3164に記載されて いる方法により合成した) 467mgと化合物 [1一 1] 50 Omgから、 実施例 1
2- (1) の方法に準じて、 ベンジル (2 S) — 2— [(4- {(1 S) 一 1一 [(t e r t—ブトキシカルボニル) ァミノ] ェチル } フエニル) (ヒドロキシ) メチル] ピロリジン一 1一力ルポキシレート [50— 1] (以下、 化合物 [50— 1] という) 95mgを得た。
(2) 化合物 [50— 1] 93mgと化合物 [1— 6] 48mgから、 実施例 1一 (7) の方法に準じてベンジル (2 S) —2— [[4一 ((1 S) — 1一 {[5—シァ ノー 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 2 一ィル] アミノ} ェチル) フエニル] (ヒドロキシ) メチル] ピロリジン一 1一カル ポキシレート [50— 2] (以下、 化合物 [50— 2] という) 68mgを得た。 (3) 化合物 [50— 2] 3 Omgから、 実施例 17— (2) の方法に準じて 2— {[(1 S) 一 1一 (4一 {ヒドロキシ [(2 S) —ピロリジン一 2—ィル] メチル } フエニル) ェチル] アミノ} 一 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一
3—ィル) ピリミジン一 5—力ルポ二トリル [50— 3] (以下、 化合物 [50— 3] という) 1 Omgを得た。 .
(4) 化合物 [50— 3] 7mgをクロ口ホルム lmLに溶解し、 37 %ホルムァ ルデヒド液 4mgとトリァセトキシ水素化ほう素ナトリウム 1 Omgをカロえ、室温に て 2時間 30分撹拌した。 反応混合物に飽和炭酸水素ナトリウム水溶液を加え、 クロ 口ホルムにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウ ムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を分取薄層クロ マトグラフィー (展開液:クロ口ホルム/メタノール =20/1) にて精製し、 ジァ ステレオマーである化合物 [50] と化合物 [51] を分離した。 上記の精製条件に おいて、 低極性化合物である目的化合物 [50] 3mg、 高極性化合物である目的化
合物 [51] 2mgをともに白色固体として得た 化合物 [50]、 および [51] のスペクトルデータを以下に示す。
化合物 [50]
XH-NMR (CDC 13) δ : 9. 63 (d, J = 6. 3Hz, 1HX 1/5), 8. 94 (s, 1 HX 1/5), 8. 89 (s, 1 HX 4/5), 8. 76 (d, J = 6. 3Hz , 1 HX 4/5), 8. 57 (s, 1/5), 8. 53 ( s , 1 HX 4/5), 7. 42-7. 15 (m, 5H), 6. 64-6. 60 (m, 1H), 6. 07-6. 05 (m, 1HX 4/5), 5. 88 - 5. 80 (m, 1 HX 1/5), 5. 36-5. - 25 (m, 1H+ 1HX 1/5), 5. 15-5. 11 (m, 1 HX 4/5), 4. 9 4 (s, 1 H), 2. 65 -2. 40 (m, 3 H), 2. 69 (s, 1 HX 3/5), 2. 64 (s, 2H+ 1 HX 2/5), 2. 15— 1. 70 (m, 6H), 2. 53 (s, 3H), 1. 82- 1. 64 (m, 4H), 1. 63 (d, J = 7. 2Hz, 3 H) ma s s : 468 (M+ 1) +. 化合物 [51] '
iH— NMR (CDC ") 5 : 9. 64- 9. 63 (m, 1 HX 1/5), 8. 94 一 8. 88 (m, 1 H+ 1 HX 4/5), 8. 53 (s , 1 HX 1/5), 8. 52 (s, 1 HX 4/5), 7. 43-7. 16 (m, 5 H), 6. 66 - 6. 62 (m, 1 H), 6. 04- 6. 02 (m, 1 HX 4/5), 5. 83 - 5. 82 (m, 1 HX 1/5), 5. 18-5. 13 (m, 1 HX 4/5), 4. 55 -4. 54 (m, 1 H), 3. 4 0— 3. 20 (m, 1 H), 2. 69 (s , 1HX3/5), 2. 65 (s, 2H+ 1 HX 2/5), 2. 57 - 2. 45 (m, 6 H), 2. 38 (s, 3H), 1. 96— 1. 55 (m, 4H), 1. 63 (d, J = 7. 2Hz, 3 H)
ma s s : 468 (M+ 1) +. 実施例 52、 53
2— { [ (1 S) — 1— (4— {ヒドロキシ [ (2R) — 1一メチルピロリジン一 2 —ィル] メチル } フエニル) ェチル] アミノ} 一 4一 (8—メチルイミダゾ [1, 2 -a] ピリジン— 3—ィル) ピリミジン一 5—カルボ二トリル [52] (以下、 化合 物 [52] という)、 および [53] (以下、 化合物 [53] という) の合成 (ここで、 化合物 [52] と化合物 [53] は、 ジァステレオマーの関係にある。表 10参照)。.
(1) ベンジル (2 R) 一 2—ホルミルピロリジン一 1一力ルポキシレート (B i oo r g. Me d. Ch em. , 2003. 11. 3153— 3164に記載されて いる方法により合成した) 1. 06 gと化合物 [1— 1] 1. 14 g力 ^ら、 実施例 1 2— (1) の方法に準じて、 ベンジル (2R) -2- [(4- {(1 S) 一 1一 [(t e r t—ブトキシカルポニル) ァミノ] ェチル } フエニル) (ヒドロキシ) メチル]
ピロリジン— 1一力ルポキシレ一卜 [52- 1] (以下、 化合物 [ 52— 1 ] という) 337mgを得た。
(2) 化合物 [52— 1] 335mgと化合物 [1一 6] 173mgから、 実施例 50、 51— (2) 〜 (4) の方法に準じて、 目的化合物 [52] 18mgと目的化 合物 [53] 19mgを、 ともに白色固体として得た。なお、実施例 50, 51 - (4) に記載の精製条件において、 低極性化合物が化合物 [52] であり、 高極性化合物が 化合物 [53] である。 化合物 [52]、 および [53] のスペクトルデータを以下に示す。
化合物 [52]
XH-NMR (CDC 13) δ : 9. 65 - 9. 50 (m, 1 HX 1/5), 8, 96 -8. 87 (m, 1 H+ 1 HX 4/5), 8. 58 (s , 1 HX 1/5), 8. 5 1 (s , 1 HX 4/5), 7. 42-6. 93 (m, 5H), 6. 67 - 6. 52 (m, 1H), 6. 1 1 -6. 10 (m, 1 HX 4/5), 5. 88 - 5. 80 (m, 1 HX 1/5), 5. 36 - 5. 29 (m, 1 HX 1/5), 5. 1 6-5. 13 (m, 1 HX 4/5), 4. 9 1-4. 90 (m, 1 H), 3. 25— 3. 15 (m, 1 H) , 2. 68 (s, 1 HX 3/5), 2. 64 (s , 2H+ 1 HX 2/5), 2. 50 (s, 1 HX 3/5), 2. 48 (s, 2H+ 1 HX 2/5), 1. 79— 1. 52 (m, 4H), 1. 6 1 (d, J = 7. 2Hz , 3H)
ma s s : 468 (M+ 1) +. . 化合物 [53]
XH-NMR (CDC 13) δ : 9. 65 (d, J =6. 8Hz, 1 HX 1/5), 8. 95 (s, 1 HX 1/5), 8 · 9 1 (s , 1 HX 4/5), 8. 79 (d, J = 6. 8Hz , 1 HX 4/5), 8. 56 (s, 1 HX 1X5), 8. 53 (s , 1 HX 4/ 5), 7. 43-7. 1 5 (m, 5H), 6. 66- 6. 64 (m, 1 H), 6. 04 一 6. 03 (m, 1 HX 4/5), 5. 85 - 5. 82 (m, 1 HX 1/5), 5. 3
3- 5. 30 (m, 1 HX 1/5), 5. 1 8— 5. 1.0 (m, 1 HX 4/5), 4. 45-4. 44 (m, 1H), 3. 22-3. 16 (m, 1H), 2. 89-2. 77
(m, 1 H), 2. 69 (s , 1 HX 3/5), 2. 64 (s , 2H+ 1 HX 2/5), 2. 58 - 2. 34 (m, 2H), 2. 29 (s , 3H), 1. 92 - 1. 52 (m, H), 1. 62 (d, J = 7. 2Hz, 3 H)
m s s : 468 (M+ 1) +. 実施例 54、 55
4— (8—ェチルイミダゾ [1, 2-a] ピリジン一 3—ィル) 一 2— { [ (I S) ― 1一 (4- {ヒドロキシ [ (2 R) 一 1一メチルピロリジン一 2—ィル] メチル }
7075240 フエニル) ェチル] アミノ} ピリミジン—5·—力ルポ二トリル [54] (以下、 化合 物 [54] という)、 および [55] (以下、 化合物 [55] という) の合成 (ここで、 化合物 [54] と化合物 [55] は、 ジァステレオマーの関係にある。表 10参照)。 (1) 化合物 [52— 1] 30 Omgをテトラヒドロフラン 6mLとメタノール 6 mLの混合溶媒に溶解し、 20 %水酸化パラジウム炭素触媒 10 Omgを加え、 水素 雰囲気下、 室温にて 2. 5時間撹拌した。 触媒をセライトろ過し、 ろ液を減圧濃縮し た。 得られた残渣をテトラヒドロフラン 6mLに溶解し、 37%ホルムアルデヒド液 147 とトリァセトキシ水素化ほう素ナトリウム 42 Omgを加え、室温にて 1. 5時間撹拌した。 反応液に飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムとメ 夕ノールの混合溶媒 (混合比: 9/1) にて抽出し、 得られた有機層を飽和食塩水に て洗浄した。 無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮 し、 t e r t一ブチル [(I S) — 1一 (4一 {ヒドロキシ [(2 R) ― 1ーメチルビ 口リジン一 2—ィル] メチル } フエニル) ェチル] カーバメート [54— 1] (以下、 化合物 [54—1] という) 22 Omgを得た。
(2) 化合物 [54— 1] 73mgと化合物 [5— 6] 65mgから、 実施例 1一 (7)の方法に準じて目的化合物 [54] 15mg、 目的化合物 [55] 13mgを、 ともに無色固体として得た。 なお、 実施例 1一 (7) に記載の精製条件において、 低 極性化合物が化合物 [54] であり、 高極性化合物が化合物 [55] である。 化合物 [54]、 および [55] のスペクトルデ一夕を以下に示す。
化合物 [54]
^-NMR (CDC 13) (5 : 9. 67 - 9. 60 (m, 1 HX 1/5), 8. 95 - 8. 89 (m, 1 H+ 1 HX 4/5), 8. 57 (s , 1 HX 1/5), 8. 52 (s , 1 HX 4/5), 7. 45-7. 17 (m, 5 H), 6. 70 - 6. 67 (m, 1 H), 6. 03 - 6. 02 (m, 1 HX 4/5), 5. 85 - 5. 76 (m, 1 HX 1/5), 5. 35 - 5. 34 (m, 1 HX 1/5), 5. 16-5. 13 (m, 1 HX 4/5), 4. 88-4. 87 (m, 1H), 3. 14-3. 05. (m, 3 H), 2. 46 (s , 3 H), 2. 53 - 2. 24 (m, 3H), 1. 75-1. 51 (m, 4H), 1. 6 3 (d, J = 6. 8Hz, 3H), 1. 37 (t, J = 7. 8Hz, 3 H)
m a s s : 482 (M+ 1) +. 化合物 [55]
XH-NMR (CDC 13) δ : 9. 64 (d, J = 6. 8Hz, 1 HX 1/5), 8. 94 (s, 1 HX 1/5), 8. 89 (s, 1 HX 4/5), 8. 80 (d, J = 6. 8Hz, 1 HX 4/5), 8. 56 (s , 1 HX 1/5), 8. 52 (s , 1 HX 4, 5), 7. 43-7. 16 (m, 5H), 6. 70— 6. 66 (m, 1H), 6. 04
一 6, 0 3 (m, 1 HX 4/5), 5. 8 5— 5. 7 8 (m, 1 HX 1/5), 5. 3
6 - 5. 2 6 (m, 1 HX 1/5), 5. 1 6 - 5. 1 1 (m, 1 HX 4/5), 4.
34-4. 3 3 (m, 1 H), 3. 1 0— 3. 04 (m, 3H), 2. 74- 2. 7 1 (m, 1 H)., 2. 44- 2. 1 7 (m, 2H), 2. 2 6 (s , 1 HX 3/5), 2. 1 7 ( s , 2H+ 1 HX 2/5), 1. 9 0— 1. 68 (m, 4H), 1. 64 (d,
J = 6. 8 Hz , 3H), 1. 3 7 ( t , J = 7. 8Hz, 3 H)
m s s : 48 2 (M+ 1) +. 実施例 5 6、 5 7
4- (8—クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— { [ (1 S) — 1— (4一 {ヒドロキシ [ (2 R) — 1一メチルピロリジン一 2—ィル] メチル } フエニル) ェチル〕 アミノ} ピリミジン一 5—力ルポ二トリル [56] (以下、 化合 物 [56] という)、 および [5 7] (以下、 化合物 [5 7] という) の合成(ここで、 化合物 [56] と化合物 [5 7] は、 ジァステレオマーの関係にある。 表 1 0、 表 1 1参照)。 化合物 [54— 1] 7 3mgと化合物 [9— 1] 6 6 m gから、 実施例 1一 ( 7 ) の 方法に準じて、 目的化合物 [5 6] 1 2mg、 目的化合物 [5 7] 1 2mgを、 とも に無色固体として得た。 なお、 実施例 1一 (7) に記載の精製条件において、 低極性 化合物が化合物 [56] であり、 高極性化合物が化合物 [57] である。 化合物 [56]、 および [5 7] のスペクトルデータを以下に示す。
化合物 [56]
— NMR (CDC 13) (5 : 9. 7 1 (d, J = 6. 8Hz , 1 HX 1/5), 8. 9 7 (s , 1 HX 1/5), 8. 9 2 (s, 1 HX 4/5), 8. 8 7 (d, J = 6. 8Hz , 1 HX 4/5), 8. 6 1 (s , 1 HX 1/5), 8. 5 6 (s , 1 HX 4/ 5), 7. 5 6 - 6. 9 6 (m, 5H), 6. 6 6 - 6. 6 2 (m, 1 H), 6. 1 3 一 6. 1 2 (m, 1 HX 4/5), 5. 9 1 - 5. 8 2 (m, 1 HX 1/5), 5. 3 4- 5. 3 2 (m, 1 HX 1/5), 5. 1 5 - 5. 0.8 (m, 1 HX 4/5), 4. 8 6— 4. 8 5 (m, 1H), 3. 6 8 - 3. 5 0 (m, 1 H), 3. 1 8 - 3. 0 5 (m, 1 H), 2. 45 (s , 3H), 2. 5 3 - 2. 2 7 (m, 2H), 1. 64- 1. 5 3 (m, 4H), 1. 64 (d, J = 7. 2 Hz , 3 H)
ma s s : 48 8、 490 (M+ 1) +. 化合物 [5 7]
^-NMR (CDC 13) δ : 9. 7 1 (d, J = 6. 8Hz, 1 HX 1/5), 8. 9 7 (s , 1 HX 1/5), 8. 8 9 (s , 1 HX 4/5), 8. 7 1 (d, J = 6. 8Hz , 1 HX 4/5), 8. 6 0 (s , 1 HX 1/5), 8. 5 6 (s, 1 HX 4/
5), 7. 52 - 6. 9 7 (m, 5H), 6. 66 - 6. 63 (m, 1 H), 6. 1 1 一 6. 09 (m, 1HX4/5), 5. 92- 5. 80 (m, 1 HX 1/5), 5. 3 8-5. 25 (m, 1 HX 1/5), 5. 12-5. 06 (m, 1 HX 4/5), 4.
32-4. 3 1 (m, 1 H), 4. 22 (b r s , 1 H), 3. 10 - 3. 07 (m, 1H), 2. 73 - 2. 69 (m, 1 H), 2. 41 -2. 35 (m, 1H), 2. 2 5 (s, 1 HX 3/5), 2. 18 (s , 2H+ 1 HX 2/5), 1. 93- 1. 60
(m, 4H), 1. 61 (d, J = 7. 2 Hz, 3 H)
ma s s : 488、 490 (M+ 1) +. 実施例 58、 59
4一 [8 - (ジフルォロメチル) イミダゾ [1, 2 - a] ピリジン一 3—ィル] ― 2 ― { [ (1 S) 一 1一 (4一 {ヒドロキシ [ (2R) 一 1—メチルピロリジン一 2— ィル] メチル } フエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [58]
(以下、 化合物 [58] という)、 および [59] (以下、 化合物 [59] という) の 合成(ここで、化合物 [58] と化合物 [59] は、 ジァステレオマ一の関係にある。 表 1 1参照) 化合物 [54— 1] 73mgと化合物 [10— 2] 66mgから、 実施例 1一 (7) の方法に準じて、 目的化合物 [58] l lmg、 目的化合物 [59] 12mgを、 と もに無色固体として得た。 なお、 実施例 1一 (7) に記載の精製条件において、 低極 性化合物が化合物 [58] であり、 高極性化合物が化合物 [59] である。 化合物 [58]、 および [59] のスペクトルデータを以下に示す。
化合物 [58]
λΗ - NMR (CDC 13) 5 : 9. 86 (d, J = 6. 8Hz, 1 HX 1/5), 9. 01 (d, J = 6. 8Hz, 1HX4/5), 9. 00 (s, 1 HX 1/5), 8. 9 3 (s , 1 HX 4/5), 8. 61 (s, 1 HX 1/5), 8. 56 (s , 1 HX 4/ 5), 7. 7 5-7. 1 2 (m, 6H), 6. 80- 6. 77 (m, 1 H), 6. 1 1 -6. 10 (m, 1 HX 4/5), 5. 90 - 5. 82. (m, 1 HX 1/5), 5. 3 4- 5. 33 (m, 1 HX 1/5), 5. 1 5-5. 08 (m, 1 HX 4/5), 4. 87 -4. 86 (m, 1H), 3. 14-3. 10 (m, 1 H), 2. 45 (s , 3H), 2. 48 - 2. 27 (m, 3H), 1. 65 - 1. 53 (m, 4H), 1. 64 (d, J = 7. 2Hz, 3H)
ma s s : 504 (M+ 1) +. 化合物 [59]
一 NMR (CDC 13) (5 : 9. 87 (d, J = 6. 8Hz, 1HX 1/5), 8. 99 (s, 1 HX 1/5), 8. 90 (s , 1HX 4/5), 8. 85 (d, J = 6.
2007/075240 8Hz' 1HX 4/5), 8. 6 0 (s, 1HX 1/5), 8. 5 7 (s, 1 HX 4/ 5), 7. 74- 7. 1 0 (m, 6H), 6. 8 2— 6. 7 8 (m, 1 H), 6. 1 3 - 6. 0 5 (m, 1 HX 4/5), 5. 9 0 - 5. 8 2 (m, 1 HX 1/5), 5. 3 8 - 5. 3 0 (m, 1 HX 1/5), 5. 1 1— 5. 0 6 (m, 1 HX 4/5), 4. 3 3— 4. 3 1 (m, 1H), 3. 1 1 - 3. 0 7 (m, 1H), 2. 74- 2. 6 9 (m, 1H), 2. 45 - 2. 1 7 (m, 2H), 2. 2 5 (s, 1 HX 3/5), 2. 1 9 (s , 2H+ 1 HX 2/5), 1. 9 0— 1. 6 0 (m, 4H), 1. 6 2 (d, J = 7. 2Hz , 3H)
ma s s : 5 04 (M+ 1 ) +· 実施例 60
2— [ ( (I S) — 1— {4- [ (I S) - 2 - ( t e r t一プチルァミノ) — 1一 ヒドロキシェチル] フエ二ル} ェチル) ァミノ] 一 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジンー3—ィル] ピリミジン一 5—力ルポ二トリル [6 0] (以下、 化合物 [6 0] という) の合成。 化合物 [5— 5] 1 0 lmgと化合物 [1 0— 2] 9 5mgから実施例 5— (7) の 方法に準じて、 目的化合物 [6 0] の塩酸塩 2 9mgを白色固体として得た。 化合物 [60] のスペクトルデータを以下に示す。
XH-NMR (DMSO— d6) δ: 1 0. 20 (d, J = 6. 8 H z , 1 H X 1 / 2 ) , 9. 1 1 (d, J = 6. 4Hz , 1 HX 1/2), 9. 0 6 (d, J = 7. 2Hz , 1HX 1/2), 8. 96 (d, J =8. 0Hz, 1 HX 1/2), 8. 7 8 (s , 1 HX 1/2), 8. 7 7 ( s , 1 HX 1/2), 8. 74 ( s , 1 HX 1/2), 8. 64 (s , 1 HX 1/2), 7. 8 6 (d, J = 8. 0Hz, 1 HX 1/2), 7. 7 9 (d, J = 7. 2Hz , 1 HX 1/2), 7. 5 1 (t , J = 54. 4Hz, 1 H X 1/2), 7. 45 (t , J = 54. 4Hz, 1 HX 1/2), 7. 3 7 - 7. 2 9 (m, 5H+ 1 HX 1/2), 7. 1 3 ( t , J = 7. 2Hz , 1 HX 1/2), 5. 3 0 - 5. 04 (m, 2H), 4. 5 1 -4. 45 (m,. 1 H), 2. 6 0 - 2. 5 0 (m, 2H), 1. 54- 1. 48 (m, 3H), 1. 0 0 (s , 9.HX 1/2), 0. 9 3 (s , 9HX 1/2).
ma s s : 5 0 6 (M+ 1) +. 実施例 6 1
2— [ ( (I S) — 1— {4- [ (I S) 一 2— ( t e r t—プチルァミノ) 一 1一 ヒドロキシェチル] フエ二ル} ェチル) ァミノ] —4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [6 1] (以下、 化 合物 [61] という) の合成。
化合物 [5— 5] 2. 2 3 gと化合物 [9— 1] 2 g力、ら、 実施例 5— (7) の方法 に準じて、 目的化合物 [6 1] の塩酸塩 1. 2 gを白色固体として得た。 化合物 [6 1] のスペクトルデータを以下に示す。
一 NMR (DMSO-d6) (5 : 1 0. 06 (d d, J = 6. 8Hz, 0. 8 Hz , 1 HX 1/2), 9. 0 6 (d, J = 6. 8Hz, 1 HX 1/2), 8. 9 9 (d d, J = 7. 2Hz, 1. 2Hz , 1 HX 1/2), 8. 9 7 (d, J = 8 4Hz, 1 HX 1/2), 8. 7 8 (s , 1 HX 1 /2), 8. 7 5 (d, J = 5. 2Hz, 1 H. X 1/2), 8. 6 2 (s , 1 HX 1/2), 7. 7 9 (d d, J =7. 6Hz, 1. 2 Hz , 1 HX 1/2), 7. 7 2 (d d, J = 7. 6Hz, 0. 8H z , 1 HX 1 /2), 7. 3 7 - 7. 2 8 (m, 5H+ 1 HX 1 /2), 7. 1 9 (t , J = 7. 2 Hz, 1 HX 1 /2) , 7. 0 0 ( t, J = 7. 2Hz, HX 1/2), 5. 2 8 -
5. 0 5 (m, 2H), 4. 4 9 -4. 44 (m, 1 H), 2. 5 7 - 2. 5 1 (m, 2H), 1. 54- 1. 48 (m, 3 H) , ] • 0 0 (s , 9HX 1/2), 0. 9 3 (s , 9HX 1/2).
ma s s : 49 0、 49 2 (M+ 1) 実施例 6 2
2— [((I S) — 1— {4- [(I S) — 2— (t e r t—ブチルァミノ) 一 1ーヒ ドロキシェチル] フエ二ル} ェチル) ァミノ] —4一 (8—シクロプロピルイミダソ [1, 2- a] ピリジン一 3—ィル) ピリミジン一 5—力ルポ二トリル [6 2] (以 下、 化合物 [6 2] という) の合成。 化合物 [5— 5] 1 0 2mgと化合物 [3 6— 1] 9 3mgから、 実施例 5— (7) の方法に準じて、 目的化合物 [6 2] の塩酸塩 48mgを黄色固体として得た。 化合物 [6 2] のスペクトルデータを以下に示す。
ー應 R (DMS〇_d6) δ 9. 9 3 (d d, J = 6. 8 Hz, 1. 2Hz, 1 HX 1/2), 9. 0 6 (d d, J = 7. 2Hz, 1. 2Hz, 1 HX 1/2), 9. 0 3 (d, J = 7. 2Hz, 1 HX 1/2), 8. 9 7 (d, J =8. 0Hz, 1 H X 1/2), 8. 9 7 - 8. 8 8 (m, 1 H), 8. 7 5 (s, 1 HX 1/2), 8. 74 (s , 1 HX 1/2), 8. 6 9 (s , 1 HX 1/2), 8 . 64 (s , 1HX 1
/2), 8. 5 0— 8. 3 5 (m, 1 H), 7. 45— 7. 3 6 (m, 4H), 7. 1 7 - 7. 08 (m, 3HX 1 /2) , 7. 0 1 (t, J = 7. 2Hz, 1HX 1/2), 6. 2 0 - 6. 0 0 (m, 1 H), 5. 2 6 - 5. 1 0 (m, 1 H), 4. 9 0— 4. 8 0 (m, 1 H), 3. 0 5 - 2. 7 9 (m, 2H), 2. 6 2 - 2. 5 1 (m, 1 H), 1. 5 2 (d, J = 7. 2Hz , 3HX 1/2), 1. 5 0 (d, J = 7. 2Hz,
0
3HX 1/2), 1. 2 8 (s , 9HX 1/2), 1. 2 5 (s , 9HX 1/2), 1. 1 1 - 0. 9 5 (m, 4H).
ma s s : 49 6 (M+ 1) +. 実施例 63、 64
4一 (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 S) 一 1一 {4一 [[(2 S) — 1, 2—ジメチルピロリジン一 2—ィル] (ヒドロキ シ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [63] (以下、 化合物 [63] という)、 および [64] (以下、 化合物 [64] という) の. 合成(ここで、化合物 [63] と化合物 [64] は、 ジァステレオマーの関係にある。 表 1 2参照)。
(1) H- α-Me -P r o-OH (ケム一インペックス社 (Ch em— Imp e x) より入手可能である) 5 0 Omgと塩化ギ酸ベンジル 6 9 3mgを 1, 4ージォ キサン 1 OmLと水 5mLに溶解した溶液に、室温にて pHが 1 0を保つように 1M 水酸化ナトリウム水溶液を加え、 室温にて 3 0分間攪拌した。 反応液に pHが 2にな るよう 5M塩酸を加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水にて 洗浄し、 無水硫酸マグネシウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 1 一 [(ベンジルォキシ)カルボニル]一 2—メチルー L一プロリン [6 3 - 1] (以下、 化合物 [63— 1] という) 842mgを得た。
(2) 化合物 [6 3— 1] 84 Omgをテトラヒドロフラン 1 6mLに溶解した溶 液に、室温にてポラン一硫化メチル錯体 4. 3 lmL (2 Mテトラヒドロフラン溶液) を加え、 4時間加熱還流した。 反応液に飽和炭酸水素ナトリウム水溶液を加え、 酢酸 ェチルにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシゥ ムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシリカゲル力 ラムクロマ卜グラフィー (溶出液:へキサン/酢酸ェチル 1 0 0/0 - 6 0/40) にて精製し、 ベンジル (2 S) — 2— (ヒドロキシメチル) 一 2—メチルピロリジ ンー 1一力ルポキシラート [6 3- 2] (以下、 化合物. [6 3— 2] という) 49 8 mgを得た。
(3) 化合物 [6 3— 2] 3 7 7mgとピリジン 147mgをクロ口ホルム 7. 5 mLに溶解した溶液に、 室温にてデス一マーチン過よう素酸 6 7 3mgを加え、 1時 間加熱還流した。 反応液に飽和チォ硫酸ナトリウム水溶液を加え、 クロ口ホルムにて 抽出した。 得られた有機層を飽和炭酸水素ナトリウム水溶液、 飽和食塩水にて順に洗 浄し、 無水硫酸ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した。 得ら れた残渣をシリカゲルカラムクロマトグラフィー (溶出液:へキサン /酢酸ェチル 1 0 0/0 - 6 0/40) にて精製し、 ベンジル (2 S) 一 2—ホルミル一 2—メチ
P T/JP2007/075240 ルピロリジン一 1一カルボキシラート [63— 3] (以下、 化合物 [63— 3] とい う) 346mgを得た。
(4) 化合物 [1 - 1] 423mgと N, N, Ν', Ν, ーテトラメチルエチレン ジァミン 508mgをテトラヒドロフラン 22 mLに溶解し一 78°Cとした後、 n— ブチルリチウム (2. 66 Mへキサン溶液) 1. 09mLを加えた。 同温にて 1時間 撹拌した後、 生じた白色懸濁液に化合物 [63— 3] 384mgを溶解したテトラヒ ドロフラン溶液 2. 2mLを加え、 同温にて 1時間半撹拌した。 反応混合物に飽和塩 化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩. 水にて洗浄し、 無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃 縮し、 ベンジル (2 S) — 2— [(4一 {(1 S) 一 1一 [( t e r t一ブトキシカル ポニル) ァミノ] ェチル } フエニル) (ヒドロキシ) メチル] 一 2—メチルピロリジ ンー 1一力ルポキシラート [63— 4] が得られた。 得られた化合物 [63— 4] は 2種の異性体の混合物であり、 シリカゲル力ラムクロマトグラフィー (溶出液:へキ サン Z酢酸ェチル = 100Z0— 40/60) にて精製し、 低極性化合物 (当該条件 で先に溶出される化合物である) [63— 4 a] (以下、 化合物 [63— 4 a] という) 199mgと、 高極性化合物 (当該条件で後に溶出される化合物である) [63— 4 b] (以下、 化合物 [63— 4 b] という) 117mgを得た。 ここで、 化合物 [6 3-4 a] は化合物 [63] の前躯体であり、 化合物 [63— 4b] は後述の化合物 [64] の前躯体である。
(5) 化合物 [36— 1] 38mgと化合物 [63— 4 a] 57. 5mg力 ら、 実 施例 50、 51— (2) 〜 (4) の方法に準じて、 化合物 [63] 23. 3mgを白 色固体として得た。 化合物 [63] のスペクトルデ一夕を以下に示す。
XH-NMR (CDC 13) δ : 9. 70— 9. 62 (m, 1 HX 1/5), 8. 98 (d, J =6. 8Hz, 1 HX 4/5), 8. 92 (s , 1 HX 1/5), 8. 88 (s , 1 HX 4/5), 8. 57 (s , 1 HX 1/5), 8. 52 (s , 1 HX 4/5), 7. 36 - 7. 28 (m, 4H), 6. 96 - 6. 88 (m, 1H+ 1HX 1/5), 6. 71-6. 68 (m, 1HX4/5), 5. 38 - 5. 27 (m, 1 HX 1/5), 5. 15-5. 13 (m, 1 HX 4/5), 4. 39 (s, 1 H), 3. 10-3. 0 7 (m, 1H), 2. 59-2. 45 (m, 2H), 2. 19-2. 03 (m, 2H), 2. 10 (s , 3H), 1. 73-1. 51 (m, 2H), 1. 62 (d, J = 6. 8 Hz , 3H), 1. 31— 1. 26 (m, 1 H), 1. 17— 1. 15 (m, 2 H), 0. 92 - 0. 86 (m, 5 H)
ma s s : 508 (M+ 1 ) +.
2007/075240 (6) 化合物 [36— 1] 38mgと化合物 [63— 4b] 57. 5mg力、ら、 実 施例 50、 51— (2) 〜 (4) の方法に準じて、 目的化合物 [64] 10. 6mg を白色固体として得た。 化合物 [64] のスペクトルデータを以下に示す。
一 NMR (CDC 13) δ : 9. 63— 9. 56 (m, 1HX 1/5), 8. 97 (s , 1 HX 1/5), 8. 90 (s , 1HX4/5), 8. 74 (d, J =6. 8 Hz , 1 HX 4/5), 8. 58 (s, 1 HX 1/5), 8. 53 (s , 1 HX 4/5), 7. 44- 7. 28 (m, 4H), 6. 93— 6. 85 (m, 1H+ 1HX 1/5),. 6. 68 - 6. 64 (m, 1HX4/5), 6. 04-6. 02 (m, 1HX4/5), 5. 85 - 5. 80 (m, 1 HX 1/5), 5. 40— 5. 28 (m, 1 HX 1/5), 5. 14-5. 11 (m, 1 HX 4/5), 4. 43 (s , 1 H), 3. 15-3. 1 0 (m, 1 H), 2 · 62- 2. 49 (m, 2 H) , 2. 28 ( s, 3H), 2. 13 -2. 05 (m, 1 H), 1. 78- 1; 50 (m, 2H), 1. 62 (d, J = 6. 8 Hz, 3H), 1. 38— 1. 25 (m, 1 H), 1. 15-1. 12 (m, 2H), 0 , 94— 0. 85 (m, 5 H) '
ma s s : 508 (M+ 1) +. 実施例 65、 66
4一 (8—シクロプロピルイミダゾ [1, 2 - a] ピリジン一 3—ィル) 一 2— [((1 S) ― 1― {4- [(R) 一 [(2R) — 1, 2—ジメチルピロリジン一 2—ィル] (ヒ ドロキシ) メチル] フエ二ル}ェチル)ァミノ] ピリミジン— 5—力ルポ二トリル [6 5] (以下、化合物 [65] という)、および 4一 (8—シクロプロピルイミダゾ [1, 2 -a] ピリジン一 3—ィル) 一 2— [((1 S) 一 1一 {4- [(S) 一 [(2R) 一 1, 2—ジメチルピロリジン一 2—ィル] (ヒドロキシ) メチル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二卜リル [66] (以下、 化合物 [66] という) の合成 (ここで、 化合物 [65] と化合物 [66] は、 ジァステレオマーの関係にあ る。 表 12参照)。 (1) H—《— Me— D— P r o— OH (ケム一インペックス社 (Chem— Im p e x) より入手可能である) 1 gから、 実施例 63、 64— (1) 〜 (4) の方法 に準じて、 ベンジル (2R) 一 2— [(4一 {(1 S) — 1一 [(t e r t一ブトキシ カルボニル) ァミノ] ェチル } フエニル) (ヒドロキシ) メチル] 一 2—メチルピロ リジン一 1一力ルポキシラート [65— 1]が得られた。得られた化合物 [65— 1] は 2種の異性体の混合物であり、 実施例 63、 64- (4) に記載の精製条件におい て、 [65— l a] (以下、 化合物 [65—1 a] という) 195mgと、 [65— 1 b] (以下、 化合物 [65— l b] という) 134mgを得た。 なお、 実施例低極性 化合物 (当該条件で先に溶出される化合物である) が化合物 [65- 1 a] であり、
高極性化合物 (当該条件で後に溶出される化合物である) が化合物 [65— 1 b] で ある。
(2) 化合物670 21 [365— 1 a] 193mgから、 実施例 54、 55— (1) の方法に 準じて、 t e r t 5— 9 91 -ブチル ((I S) — 1一 {4一 [(R) 一 [(2 R) — 1, 2—ジ
- メチルピロリジン一 21 π Πu ' ( ( ( n n m m m— ¾ 1'' J>ィル] (ヒドロキシ) メチル] フエ二ル} ェチル) 力一バメ —ト [65— 2] (以下、 化合物 [65— 2] という) 14 Omgを得た。
(3) 化合物 [36- 1] 45. 6mg化合物と [65- 2] 49mgから、 実施. 例 1一 (7) の方法に準じて、 目的化合物 [65] (以下、 化合物 [65] という) 21. 4m gを淡黄色固体として得た。 化合物 [65] のスペクトルデータを以下に示す。
'H-NMR (CDC 13) δ : 9. 63 - 9. 55 (m, 1 HX 1/5), 8. 97 (s, 1 HX 1/5), 8. 89 (s, 1 HX 4/5), 8. 7 3 (d, J = 6. 8 Hz, 1 HX 4/5), 8. 58 (s, 1 HX 1/5), 8 3 (s , 1 HX 4/5),
7. 37-7. 4H), 6. 93-6. 84 , 1 H+ 1 HX 1/5), 6. 68-6. L HX 4/5), 6 · 02-6 0 (m, 1 HX 4/5), 5. 84- 5. L HX 1/5), 5. 38- 5 6 (m, 1 HX 1/5), 5. 13- 5. 1 HX 4/5), 4. 67-4. 50 (m, 1H), 4. 3 4 (s , 1 H) -3. 07 (m, 1 H), 2. 62-2 45 Cm, 2H), 2. 1 7-2. 2H), 2. 04 (s , 3H), 1. 74— .. 61 (m 2H), 1 =6. 8Hz, 3H), L . 32- 1. 20 (m, 2H), 1. 15-
1H), 0. 90-0. 85 (m, 5.H)
ma s s : 508 (M + ) +.
(4) 目的化合物 [65]の絶対立体配置は以下の方法により決定した。化合物 [6 5— l a] 9. 2mgに、 室温にて 2 M水酸化カリウム水溶液 1 mLを加え、 同温に て 2時間攪拌した。 反応液を減圧濃縮し、 得られた残渣に水を加え、 クロ口ホルムに て抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムで乾燥し た。 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を分取薄層クロマトグラフ ィ一にて精製し、 t e r t—ブチル ((I S) — 1一 {4— [(1 R, 7aR) -7a ーメチルー 3—才キソテトラヒドロー 1 H—ピロ口 [1, 2— c] [1, 3] ォキサ ゾ一ル— 1一ィル] フエ二ル}ェチル) カーバメート [65— 4] (以下、 化合物 [6 5 -4] という) 6. lmgを得た。 化合物 [65— 4] の NOE (核オーバーハウ ザ一効果) 測定により、 化合物 [65-4] および目的化合物 [65] の絶対立体配 置を決定した。
(5) 化合物 [3 6— 1] 45. 6mg化合物と [6 5— 1 b] 1 3 2mgから、 実施例 6 5、 6 6 - (2)、 (3) の方法に準じて、 目的化合物 [6 6] (以下、 化合 物 [6 6] という) 1 5. 7mgを淡黄色固体として得た。 化合物 [6 6] のスペクトルデータを以下に示す。
XH-NMR (CDC 1 3) «3 : 9. 6 3 - 9. 5 5 (m, 1HX 1/5), 8. 9 7 一 8. 9 0 (m, 1H+ 1 HX 4/5), 8. 5 8 (s , 1 HX 1/5), 8. 5 3 (s ,
1 HX 4/5), 7. 43 - 7. . 3 2 (m, 4H), 6. 9 3 -6. 8 5 (m, 1H + 1 HX 1/5), 6. 6 6— 6. 6 2 (m, 1 HX 4/5), 6. 04- 6. 0 3 (m, 1 HX 4/5), 5. 8 5 - 5. 7 8 (m, 1 HX 1/5), 5. 3 8 - 5. 2 8 (m, 1 HX 1/5), 5. 1 6 - 5. 1 3 (m, 1 HX 4/5), 4. 8 0 -4. 6 7 (m, 1 H), 4. 43 (s , 1 H), 3. 1 5 - 3. 0 7 (m, 1 H), 2. 6 3 - 2. 4 9 2H), 2. 2 7 (s , 3H), 2. 0 9 - 2. 0 5 (m, 1 H), 1. 8 5 5 0 (m, 2H), 1. 6 2 (d, J = 6. 8Hz, 3H), 1. 3 6 - 1. 2
2H), 1. 1 6 - 1. 14 (m, 1 H) , 0. 9 0 - 0. 8 0 (m, 5H) s : 5 0 8 (M+ 1) +
(6) 目的化合物 [6 6]の絶対立体配置は以下の方法により決定した。化合物 [6 5— 1 b] 3 Omg力 ら、 実施例 6 5、 6 6 - (4) の方法に準じて、 t e r t—ブ チル ((I S) — 1一 {4一 [(1 S, 7aR) 一 7 a—メチルー 3—ォキソテトラヒ ドロー 1 H—ピロ口 [1, 2— c] [1, 3] ォキサゾールー 1一ィル] フエ二ル} ェチル) カーバメート [6 5- 6] (以下、 化合物 [6 5 - 6] という) 1 1. 6m gを得た。 化合物 [6 5— 6] の N〇E (核オーバ一ハウザー効果) 測定により、 化 合物 [6 5— 6] および目的化合物 [6 6] の絶対立体配置を決定した。 実施例 6 7
(1 S) — 2 - (t e r t—プチルァミノ) — 1— [4一 ((1 S) — 1一 {[4一 (8 一クロロイミダゾ [1, 2 - a] ピリジン一 3—ィル) - 5—フルォロピリミジン一 2 _ィル] アミノ} ェチル) フエニル] エタノール [6 7] (以下、 化合物 [6 7] という) の合成。
(1) 2, 4ージクロ口一 5—フルォロピリミジン (フルォロケム社 (F l u o r o c h em) より入手可能である) 1 8. 9 gとシス一 1一エトキシー 2—トリ— n —ブチルス夕ニルエチレン (J . Am. C h em. S o c . , 1 9 7 7, 9 9, 7 3 6 5に記載されている方法により合成した) 42. 9 g、 ジクロロビス (トリフエ二 ルホスフィン) パラジウム (I I ) 3. 9 7 g、 およびァセトニトリル 3 8 OmLの 混合物を 8 0度にて 3時間攪拌した。 放冷後、 水 1 1 3mL、 およびフッ化カリウム 5 3 gを加え、 室温にて終夜撹拌した。 不溶物をセライ卜ろ過し、 ろ液を酢酸ェチル
にて抽出した。 得られた有機層を水、 飽和食塩水にて順に洗浄し、 無水硫酸ナトリウ ムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られた残渣をシリカゲ ルカラムクロマトグラフィー (溶出液:へキサン Z酢酸ェチル = 100/0 -40/ 60) にて精製し、 2—クロロー 4一 [(Z) —2—エトキシビニル] —5—フルォ 口ピリミジン [67— 1] (以下、 化合物 [67— 1] という) 14. 0 gを黄色固 体として得た。
(2) 化合物 [67— 1] 25. 2 gを 1, 4一ジォキサン 32 OmLと水 32m Lの混合溶媒に溶解した後、 氷冷下、 N—ブロモこはく酸イミド 22. 2 gを加え、. 室温にて 2時間撹拌した。 反応溶液に 2—ァミノ _3—クロ口ピリジン 16. O gを 加え、 室温にて終夜撹拌した。 飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルム とメタノールの混合溶媒 (混合比: 9/1) にて抽出し、 得られた有機層を無水硫酸 ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣にへキ サンを加え、 30分間撹拌した。 生じた固体をろ取し、 減圧乾燥し、 8—クロロー 3 一 (2—クロロー 5—フルォロピリミジン一 4一ィル) イミダゾ [1, 2 - a] ピリ ジン [67— 2] (以下、 化合物 [67— 2] という) 20. O gを黄色固体として 得た。
(3) 化合物 [5— 5] 1 5. 8 gをクロ口ホルム 5 OmLに溶解し、 氷冷下、 ト リブルォロ酢酸 45mLを加え、 室温にて 1時間撹拌した。 反応液を減圧濃縮し、 得 られた残渣をシリカゲルカラムクロマトグラフィー (溶出液:クロ口ホルムノメタノ ール =100/0 - 90/10) にて精製し、 (1 S) - 1一 {4- [(1 S) 一 1― アミノエチル] フエ二ル} - 2 - ( t e r tーブチルァミノ) エタノール [67-3] (以下、 化合物 [67— 3] という) 10. 7 gを白色固体として得た。
(4) 化合物 [67— 2] 3. 00 g、 化合物 [67— 3] 3. 01 g、 炭酸ナト リウム 1. 12 g、 および N—メチル一2—ピロリジノン 1 OmLの混合物を、 1 7 0°Cにて 2時間撹拌した。反応液に水を加え、 クロ口ホルムと酢酸ェチルの混合溶媒
(混合比: 1/10) にて抽出した。 得られた有機層を水にて洗浄した後、 無水硫酸 ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリ 力ゲルカラムクロマトグラフィー (溶出液:クロ口ホルム Zメタノール = 100/0 -98/2)にて精製した。得られた溶液を減圧濃縮し、残渣を酢酸ェチルに溶解し、. 固化させ、 目的化合物 [67] 3. 79 gを淡黄色固体として得た。 化合物 [67] のスペクトルデ一夕を以下に示す,
^-NMR (CDC 13) 6 9. 22 (b r s 1H), 8. 41 (d, J = 3. 9Hz , 1H), 8. 24 (d, J = 3. 9Hz, ■ H), 7. 42 -7. 37 (m, 5H), 6. 68 (b r s, 1 H), 5. 50 (d J = 5. 9Hz, 1H), 5. 0
3 (qu i n t, J =6. 8Hz, 1 H), 4. 57 (dd, J = 9. 0Hz, 3. 7 Hz , 1 H), 2. 87 (dd, 12. 0Hz 3. 7 Hz , 1H), 2. 53 (d d, J = 12. 0Hz, 9. 0Hz, 1 H), 1. 60 (d, J = 6. 8Hz, 3H), 1. 07 (s , 9H)
ma s s : 483 (M+ 1) +. 実施例 68
(I S) - 1 - [4— ((I S) - 1 - {[4— (8—クロロイミダゾ [1, 2— a] ピリジン— 3—ィル) 一 5—フルォロピリミジン一 2一ィル] アミノ} ェチル) フエ- ニル] 一 2— [(1, 1ージメチルプロピル) ァミノ] エタノール [68] (以下、 化 合物 [68] という) の合成。
(1) 化合物 [5— 4] 500mgと t e r t—アミルァミン 1. 66 gから、 実 施例 7— (3) の方法に準じて、 t e r t—ブチル [( 1 S ) — 1一 (4— { ( 1 S ) 一 2— [(1, 1ージメチルプロピル) ァミノ] — 1—ヒドロキシェチル } フエニル) ェチル] カーバメート [68— 1] (以下、 化合物 [68— 1] という) 509mg を得た。
(2) 化合物 [67— 2] 43. 5mgと化合物 [68— 1] 70mg力、ら、 実施 例 6'7— (3)、 (4) の方法に準じて、 目的化合物 [68] (以下、 化合物 [68] という) 34mgを淡褐色固体として得た。 化合物 [68] のスペクトルデータを以下に示す。
一 NMR (CDC 13) δ : 9. 23 (b r, 1 H), 8. 42 (d, J = 3. 9 Hz, 1H), 8. 25 (d, J = 3. 7Hz, 1 H), 7. 42— 7. 40 (m, 5 H), 7. 26 (s , 1 H), 6. 69 (b r , 1 H), 5. 49 (d, J = 6. 3H z, 1 H), 5. 03 (q u i n t, J = 6. 3Hz, 1 H), 4. 57(d d, J = 9. 0 Hz , 3. 7 Hz, 1 H), 2. 83 (dd, J = 1 1. 7Hz, 3. 7Hz, 1 H), 2. 49 (dd, J =l 1. 7Hz, 9. 0Hz, 1 H), 1. 60 (d, J =6. 8Hz, 3H), 1. 36 (q, J = 7. 6Hz, 2H), 1. 05— 1. 00 (m, 1 H), 1. 00 (s, 6H), 0. 81 (t, J = 7. 6Hz, 3 H) ' ma s s : 497, 499 (M+ 1) +. 実施例 69
(I S) - 1 - [4- ((I S) — 1— {[4- (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエ ニル] —2— (シクロペンチルァミノ) エタノール [69] (以下、 化合物 [69] という) の合成。
(1) 化合物 [5— 4] 44 Omgとシクロペンチルァミン 3. O gから、 実施例 7- (3) の方法に準じて、 t e r t—ブチル ((I S) — 1一 {4一 [(1 S) 一 2— (シクロペンチルァミノ) 一 1ーヒドロキシェチル] フエ二ル} ェチル) カーバ メート [69— 1] (以下、 化合物 [69— 1] という) 45 lmgを得た。
(2) 化合物 [67— 2] 30mgと化合物 [69— 1] 35. 6mg力、ら、 実施 例 67— (3)、 (4) の方法に準じて、 目的化合物 [69] (以下、 化合物 [69] という) 4. 6 mgを淡黄色固体として得た。 化合物 [69] のスぺクトルデ一夕を以下に示す。
^-NMR (CDC 13) δ : 9. 20 (b r s, 1 H), 8. 41 (d, J =3. 9Hz, 1H), 8. 24 (d, J = 3. 4Hz, 1H), 7. 40 (m, 5H), 6. 67 (m, 1 H), 5. 51 (m, 1 H), 5. 03 (m, 1 H), 4. 69 (dd,
J = 9. 0Hz, 3. 2Hz, 1 H), 3. 09 (qu i n t, 6. 6Hz , 1 H); 2. 89 (d d, J = 12. 0 Hz , 3. 7 Hz , 1 H), 2. 63 (dd, J = 1 2. 0 Hz , 9. 0Hz , 1 H), 1. 80 - 1. 65 (m, 4H), 1. 59 (d: J = 6. 8Hz, 3H), 1 · 55- 1. 20 (m, 4 H)
m a s s : 495 (M+ 1) +. 実施例 70
(I S) — 1一 [4- ((I S) — 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピリジン— 3—ィル) —5—フルォロピリミジン— 2—ィル] ァミノ) ェチル) フエ ニル] 一 2— (イソプロピルァミノ) エタノール [70] (以下、 化合物 [70] と いう) の合成。
(1) 化合物 [5— 4] 2. 4 gとイソプロピルアミン 12. 4 gから、 実施例 7 一 (3) の方法に準じて、 t e r t一ブチル ((I S) — 1— {4— [(I S) 一 1 ーヒドロキシー 2— (イソプロピルァミノ) ェチル] フエ二ル} ェチル) カーバメー ト [70— 1] (以下、 化合物 [70— 1] という) 2. 94gを得た。
(2) 化合物 [67— 2] 20mgと化合物 [70— 1] 29. 7mg力、ら、 実施 例 67— (3)、 (4) の方法に準じて、 目的化合物 [70] (以下、 化合物 [70] という) 34mgを黄色固体として得た。 化合物 [70] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) (5 : 9. 20 (b r s, lH), 8. 40 (d, J =3. 9Hz, 1H), 8. 23 (d, J = 3. 4Hz, 1H), 7. 40 (m, 5H), 6.
67 (m, 1 H), 5. 52 (m, 1H), 5. 03 (m, 1H), 4. 67 (d d, J = 9. 3Hz, 3. 4Hz , 1 H), 2. 91 (d d, 12. 2Hz, 3. 4Hz, 1 H), 2. 81 (qu i n t, J = 6. 1 Hz , 1H), 2. 61 (dd, J = 12. 0Hz, 9.. 0Hz, 1H), 1. 59 (d, J = 6. 8Hz, 3H), 1. 05 (d d, J = 6. 3Hz , 3. 4Hz , 6 H)
ma s s : 469 (M+ 1) +. 実施例 71
(I S) —1— [4— ((I S) — 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエ ニル] 一 2— [(1ーメチルシクロペンチル) ァミノ] エタノール [71] (以下、 化 合物 [71] という) の合成。
(1) 化合物 [5— 4] 50 Omg、 1—メチルシクロペンチルァミン一塩酸塩( J . Me d. Ch em. , 2006, 49, 3068に記載されている方法により合成し た) 639mg、 N, N—ジイソプロピルェチルァミン 613mg、 エタノール 5m L、 および水 5mLの混合物を、 終夜加熱還流した。 反応液を濃縮し、 得られた残渣 に水を加え、 酢酸ェチルにて抽出した。 得られた有機層を無水硫酸マグネシウムにて 乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムク 口マトグラフィー (溶出液:クロ口ホルム Zメタノール = 100/0— 95/5) に て精製し、 t e r t—ブチル [(1 S) — 1— (4一 Kl S) — 1ーヒドロキシ— 2一 [(1ーメチルシクロペンチル) ァミノ] ェチル } フエニル) ェチル] カーバメ ート [71— 1] (以下、 化合物 [71— 1] という) 365mgを白色固体として 得た。
(2) 化合物 [67— 2] 45mgと化合物 [71— 1] 150mgから、 実施例 67— (3)、 (4) の方法に準じて、 目的化合物 [71] (以下、 化合物 [71] と いう) 25. 9 mgを淡黄色固体として得た。 化合物 [7 1] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) δ : 9. 21 (b r, 1H), 8. 41 (d, J = 3. 9 Hz, 1 H), 8. 23 (d, J = 3. 4Hz , 1 H), 7. 42- 7. 37 (m, 5 H), 7. 26 (s, 1H), 6. 68 (b r s, 1H), 5. 54 (b r s , 1 H), 5. 07-4. 99 (m, 1H), 4. 58 (dd, J = 3. 9 Hz , 9. 3Hz, 1H), 2. 88 (dd, J = 3. 9Hz , 12. 2Hz , 1H), 2. 53 (dd, J = 9. 3Hz , 12. 2Hz, lH), 1. 65— 1. 52 (m, 6H), 1. 60 (d, J = 6. 8Hz, 3H), 1. 47- 1. 39 (m, 2H), 1. 30-1. 2 7 (m, 1H), 1. 12 (s , 3 H)
ma s s : 509, 51 1 (M+ 1) +. 実施例 72
(I S) — 1一 [4一 ((I S) — 1— {[4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエ ニル] —2— {[1 - (ヒドロキシメチル) シクロペンチル] アミノ}エタノール [7 2] (以下、 化合物 [72] という) の合成。
(1) 化合物 [5— 4] 500mgと 1ーァミノ一 1ーシクロペンタンメタノール. 1. 13 gから、 実施例 7— (3) の方法に準じて、 t e r t—ブチル {(1 S) — 1一 [4- ((1 S) 一 1ーヒドロキシー 2— {[1一 (ヒドロキシメチル) シクロべ ンチル] アミノ} エヂル) フエニル] ェチル } カーバメート [72— 1] (以下、 化 合物 [72— 1] という) 483mgを得た。 (2) 化合物 [67— 2] 55mgと化合物 [72— 1] 1 5 Omgから、 実施例
67- (3)、 (4) の方法に準じて、 目的化合物 [72] (以下、 化合物' [72] と いう) 7 mgを白色固体として得た。 化合物 [72] のスペクトルデ一夕を以下に示す。
^-NMR (CDC 13) (5 : 9. 23 (b r , 1 Η), 8. 39- 8. 37 (m, 1 Η), 8. 23 - 8. 22 (m, 1 Η), 7. 42 - 7. 37 (m, 5Η), 7. 3 0 (s, 1 Η), 6. 74-6. 68 (m, 1 Η), 5. 03-4. 99 (m, 1Η), 4. 72 -4. 70 (m, 1 Η), 3. 43 -3. 36 (m, 2 Η), 2. 80— 2.
70 (m, 1 Η), 2. 64-2. 59 (m, 2Η), 1. 66 - 1. 52 (m, 13 Η)
ma s s : 525, 527 (Μ+ 1) +. 実施例 73
(I S) — 2— (t e r t—プチルァミノ) 一 1一 [4一 ((1 S) 一 1一 {[4一 (8 ーシクロプロピルイミダゾ [1, 2 - a] ピリジン一 3—ィル) 一 5_メチルピリミ ジン一 2—ィル] アミノ} ェチル) フエニル] エタノール [73] (以下、 化合物 [7 3] という) の合成。
(1) 2, 4ージクロロー 5—メチルピリミジン (アルドリツチ社 (A 1 d r i c h)より入手可能である) 1 39 mgと 3—シクロプロピルピリジン一 2—ァミン(国 際公開第 2006/025567号パンフレツト、 142ページに記載されている方 法により合成した) 50. 2mg力、ら、 実施例 67— (1)、 (2) の方法に準じて、 3― (2—クロ口— 5—メチルピリミジン一 4一ィル) 一 8—シクロプロピルイミダ
ゾ [1, 2— a] ピリジン [73— 1] (以下、 化合物 [73— 1] という) 23. lmgを得た。
(2) 化合物 [67— 3] 53mgと化合物 [73— 1] 21. 3mg力ら、 実施 例 67— (4) の方法に準じて、 目的化合物 [73] (以下、 化合物 [73] という) 10. 9mgを白色固体として得た。 化合物 [73] のスペクトルデータを以下に示す。
一 NMR (CDC 13) 5 : 8. 77 (b r s, 1 H), 8. 21 (s , 1 H), 8. 06 (s, 1H), 7. 38 (s, 4H), 6. 77 (d, J = 6. 8Hz, 1H), 6. 54 (b r s, 1 H), 5. 38 (d, J = 5. 9Hz, 1H), 5. 11 (q u i n t, J = 7. 0Hz, 1 H), 4. 60 (dd, J = 9. 0 Hz , 3. 7Hz, 1H), 2. 89 (dd, J = 11. 7Hz, 3. 4Hz, 1H), 2. 64— 2. 5 6 (m, 2H), 2. 35 (s , 3H), 1. 56 (d, J = 6. 8Hz, 3H), 1. 15-1. 10 (m, 2H), 1. 08 (s , 9H), 0. 92— 0. 82 (m, 2 H) ma s s : 485 (M+ 1) +. ' 実施例 74
(1 S) -2- (t e r t一プチルァミノ) 一 1一 [4— ((1 S) 一 1一 {[4一 (8 —グロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—メチルピリミジン一 2 —ィル] アミノ} ェチル) フエニル] エタノール [74] (以下、 化合物 [74] と いう) の合成。
(1) 2, 4—ジクロロー 5—メチルピリミジン 139mgと 3—クロ口ピリジン 一 2—ァミン 1 1 Omgから、 実施例 67— (1)、 (2) の方法に準じて、 8—クロ ロー 3— (2—クロ口一 5—メチルピリミジン一 4一ィル) イミダゾ [1, 2— a] ピリジン [74— 1] (以下、 化合物 [74—1] という) 65. lmgを得た。
(2) 化合物 [67— 3] 55. 4mgと化合物 [74— 1] 21. 8mgから、 実施例 67— (4) の方法に準じて、 目的化合物 [74] (以下、 化合物 [74] と いう) 11. lmgを白色固体として得た。 化合物 [74] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) <5 : 8. 71 (b r s, 1H), 8. 23 (s, 1H), 8. 06 (s , 1 H), 7. 39 (m, 4H), 7. 31 (d, J = 7. OHz, 1 H), 6. 52 (b r s, 1 H), 5. 47 (d, J = 7. OHz, 1H), 5. 07 (q u i n t, J = 6. 7 Hz , 1H), 4. 62 (dd, J = 8. 6Hz, 3. 5Hz, 1H), 2. 90 (d d, J = 11. 7Hz , 3. 5Hz , 1 H), 2. 60 (dd,
J = 1 1. 2Hz , J = 8. 4Hz, 1 H), 2. 33 (s, 3 H), 1. 56 (d, J = 7. 0Hz, 3H), 1. 10 (s, 9H)
m a s s : 479 (M+ 1) +. 実施例 75
(1 S) - 2 - ( t e r t—プチルァミノ) — 1— [4— ((I S) — 1一 {[4一 (8 一クロロイミダゾ [1 , 2 - a] ピリジン一 3—ィル) ピリミジン一 2一ィル] アミ ノ} ェチル) フエニル] エタノール [75] (以下、 化合物 [75] という) の合成。 (1) 2, 4—ジクロロピリミジン 565mgと 3—クロ口ピリジン一 2—ァミン 488mgから、 実施例 67— (1)、 (2) の方法に準じて、 8—クロロー 3— (2 一クロ口ピリミジン一 4一ィル) イミダゾ [1, 2 - a] ピリジン [75- 1] (以 下、 化合物 [75— 1] という) 721mgを得た。 (2) 化合物 [67— 3] 33mgと化合物 [75— 1] 48mgから、 実施例 6 7- (4) の方法に準じて、 目的化合物 [75] (以下、 化合物 [75] という) 6. 6 mgを白色固体として得た。 化合物 [75] のスぺクトルデ一夕を以下に示す。
iH— NMR (CDC 13) δ : 8. 29 (d, J = 5. 4Hz, 1 H), 8. 20 (s, 1 H), 7. 43 - 7. 33 (m, 5H), 7. 26 (s, 1 H), 6. 94 (d, J =5. 4Hz, 1H), 6. 68 (b r, 1 H), 5. 57 (b r s, 1H), 5. 1 1 (b r s, 1 H), 4. 59 (d d, J = 9. 3Hz, 3. 4Hz, 1 H), 2. 8 7(d d, J = 3. 4Hz, 11. 7Hz, 1 H), 2. 55(dd、 J = 11. 7Hz, 9. 3Hz, 1 H), 1. 82 - 1. 72 (m, 2H), 1. 61 (d, J = 6. 8 H z, 3H), 1. 08 (s, 9H)
ma s s : 465, 467 (M+ 1) +. 実施例 76 .
(I S) — 2— (t e r t一プチルァミノ) —1— [4- ((1 S).— 1一 {[4一 (8 —シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 2—ィ ル] アミノ} ェチル) フエニル] エタノール [76] (以下、 化合物 [76] という) の合成。 (1) 2, 4—ジクロ口ピリミジン 3 gと 3—シクロプロピルピリジン一 2—アミ ン (国際公開第 2006/025567号パンフレツト、 142ページに記載されて いる方法により合成した) 1. 19 gから、 実施例 67— (1)、 (2) の方法に準じ て、 3— (2—クロ口ピリミジン一 4一ィル) 一 8—シクロプロピルイミダゾ [1,
2— a] ピリジン [76- 1] (以下、 化合物 [76-1] という) 775mgを得 た。
(2) 化合物 [67— 3] 66, 8mgと化合物 [76— 1] 50mgから、 実施 例 67— (4) の方法に準じて、 目的化合物 [76] (以下、 化合物 [76] という) 62. 9 mgを淡黄色固体として得た。 化合物 [76] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) δ : 9. 12 (b r , 1Η), 8. 25 (d, J = 5. 4. Hz, 1 H), 8. 18 (s , 1 H), 7. 43— 7. 36 (m, 4H), 7. 26 (s, 1H), 6. 93 (d, J = 5. 4Hz, 1 H), 6. 79— 6. 78 (m, 1H), 6. 66 (b r s , 1 H), 5. 49 (b r s, 1 H), 5. 19-5. 10 (m, 1 H), 4. 58 (d d, J = 3. 9Hz, 8. 8Hz , 1 H), 2. 86 (d d, J :; . 9Hz, 11. 7 Hz , 1H), 2. 61 -2. 57 (m, 1H), 2. 55 (d d, J = 8. 8Hz, 11. 7Hz, 1 H), 1. 60 (d, J = 6. 8Hz, 3H), 1. 13-1. 11 (m, 2H), 1. 07 (s, 9H), 0. 88 - 0. 85 (m, 2H)
ma s s : 471 (M+ 1) +. 実施例 77
(I S) — 2— (t e r t—プチルァミノ) 一 1— [4一 ((1 S) - 1 - {[5—ク ロロ一 4— (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリ ミジン一 2—ィル] アミノ} ェチル) フエニル] エタノール [77] (以下、 化合物
[77] という) の合成。
(1) 2, 4, 5—トリクロ口ピリミジン 1. 59 gと 3—シクロプロピルピリジ ン— 2—ァミン (国際公開第 2006/025567号パンフレツト、 142ページ に記載されている方法により合成した) 1. 16 gから、 実施例 67— (1)、 (2) の方法に準じて、 8—シクロプロピル一 3— (2, 5—ジクロロピリミジン一 4—ィ ル) イミダゾ [1, 2— a] ピリジン [77— 1] (以下、 化合物 [77— 1] とい う) 83 lmgを得た。
(2) 化合物 [67— 3] 52. 5mgと化合物 [77— 1] 67. 8mgから、 実施例 67— (4) の方法に準じて、 目的化合物 [77] . (以下、 化合物 [77] と いう) 49. 6mgを白色固体として得た。 化合物 [77] のスペクトルデータを以下に示す。
1H— NMR (CDC 13) δ : 8. 69 (s, 1H), 8. 32 (s , 1H), 7. 3
8 (s , 4H), 7. 2 6 (s , 1 H), 6. 8 0 (d, J = 7. 3Hz, 1H), 6. 5 8 (b r, 1 H), 5. 40 - 5. 3 0 (m, 1 H), 5. 1 3 - 5. 0 3 (m, 1 H), 4. 5 9 (d d, J = 3. 9Hz, 8. 8Hz, 1H), 2. 8 9 (d d, J = 3. 9Hz,. 1 2. 2Hz , 1 H), 2. 6 5 - 2. 6 0 (m, 1 H), 2. 5 7 (d d, J = 8. 8Hz, 1 2. 2Hz , 1 H), 1. 6 1 (b r , 2H), 1. 58 (d, J = 6. 8Hz, 3H), 1. 1 3 (d d, 1 = 2. 0Hz, 8. 3Hz, 2H), 1. 0 8 (s , 9H), 0. 8 9— 0. 8 6 (m, 2 H)
ma s s : 5 0 5, 5 0 7 (M+ 1) +. 実施例 78
(I S) - 2 - ( t e r t—プチルァミノ) 一 1一 [4— ((1 S) — 1一 {[5—ク ロロ一 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 2—ィル] アミノ} ェチル) フエニル] エタノール [7 8] (以下、 化合物 [7 8] という) の合成。
(1) 2, 4, 5—トリクロ口ピリミジン 1. 5 8 gと 3—クロ口ピリジン一 2 _ ァミン 1. 1 1 gから、 実施例 6 7 - (1)、 (2) の方法に準じて、 8—クロ口— 3 一 (2, 5—ジクロ口ピリミジン一 4一ィル) イミダゾ [1, 2 - a] ピリジン [7 8— 1] (以下、 化合物 [7 8— 1] という) 1. 24 gを得た。
(2) 化合物 [6 7 - 3] 5 2. 5mgと化合物 [7 8 - 1] 6 6. '5mgから、 実施例 6 7— (4) の方法に準じて、 目的化合物 [7 8] (以下、 化合物 [7 8] と いう) 6 2. 8mgを白色固体として得た。 化合物 [7 8] のスペクトルデータを以下に示す。
XH-NMR (CDC 1 3) (5 : 8. 6 8 (s , 1 H), 8. 3 5 (s , 1 H), 7. 4 2 - 7. 34 (m, 5H), 7. 2 6 (s , 1H), 6. 56 (b r , 1 H), 5. 5 6 (b r s , 1 H), 5. 0 5 (b r s , 1 H), 4. 6 0 (d d, J = 3. 4Hz , 8. 8Hz, 1 H), 2. 9 0 (d d, J = 3. 4 Hz, 1 1. 7 H z , 1 H), 2. 5 7 (d d, J = 1 1. 7Hz , 9. 3Hz, 1H), 1. 6 3 (b r , 2H), 1. 58 (d, J = 7. 3Hz, 3H), 1. 0 9 (s , 9 H)
ma s s : 49 9, 50 1 (M+ 1) +. 実施例 7 9
(I S) — 2— (t e r t—プチルァミノ) 一 1一 [4— ((I S) — 1— {[4— (8 ーシクロプロピルイミダゾ [1, 2 - a] ピリジン一 3—ィル) 一 5—フルォロピリ ミジン— 2—ィル] アミノ} ェチル) フエニル] エタノール [7 9] (以下、 化合物
[7 9] という) の合成。
(l) 2, 4ージクロロー 5—フルォロピリミジン (フルォロケム社 (F l uo r o c h em) より入手可能である) I I. 5 gと 3—シクロプロピルピリジン一 2— ァミン (国際公開第 2006/025567号パンフレツト、 I 42ページに記載さ れている方法により合成した) 9. 2 l gから、 実施例 67— (1)、 (2) の方法に 準じて、 3— (2—クロ口一 5—フルォロピリミジン一 4一ィル) 一 8—シクロプロ ピルイミダゾ [1, ·2— a] ピリジン [79— 1] (以下、 化合物 [79— 1] とい う) 13. 4 gを得た。 (2) 化合物 [67— 3] 1. 17 gと化合物 [79— 1] 1. l gから、 実施例 67- (4) の方法に準じて、 目的化合物 [79] (以下、 化合物 [79] という)
i
673 mgを橙色固体として得た。 化合物 [79] のスペクトルデータを以下に示す。
XH-NMR (CDC 13) δ : 9. 20 (b r s, 1 H), 8. 41 (d, J =3.
9Hz, 1 H), 8. 19 (d, J = 3. 9Hz, 1H), 7. 39 (q, 4H), 6.
83 (d, J = 6. 8Hz, 1H), 6. 69 (m, 1 H), 5. 44 (d, J = 5.
9Hz, 1 H), 5. 06 (qu i n t, J = 7. 3Hz, 1 H), 4. 57 (d d,
J =8. 8Hz, 3. 4Hz, 1 H), 2. 85 (dd, 12. 0Hz, 3. 7 Hz , 1H), 2. 62 (dd, J = 11. 7Hz, 8. 8Hz, 1H), 1. 59 (d, J
=6. 8Hz, 3H), 1. 13 (d, J = 8. 8Hz, 2H), 1. 06 (s , 9 H),
0. 87 (m, 2H)
ma s s : 489 (M+ 1) +. 実施例 80
(I S) — 2— (t e r t一プチルァミノ) 一 1一 (4— {(1 S) — 1— [(5—フ ルオロー 4—イミダゾ [1, 2 - a] ピリジン一 3—ィルピリミジン— 2—ィル) ァ ミノ] ェチル } フエニル) エタノール [80] (以下、 化合物 [80] という) の合 成。 .
(1) 2, 4ージクロ口一 5—フルォロピリミジン (フルォロケム社 (F l uo r o c hem)より入手可能である) 412mgと 2—アミノピリジン 232mgから、 実施例 67— (1)、 (2) の方法に準じて、 3— (2—クロロー 5—フルォロピリミ ジン一 4一ィル) イミダゾ [1, 2— a] ピリジン [80— 1] (以下、 化合物 [8 0 - 1 ] という) 309mgを得た。
(2) 化合物 [67— 3] 102mgと化合物 [80— 1] 70mgから、 実施例 67- (4) の方法に準じて、 目的化合物 [80] (以下、 化合物 [80] という)
5 lmgを白色固体として得た。 化合物 [80] のスペクトルデ一夕を以下に示す。
^-NMR (CDC 13) <5 : 9. 36 (b r, 1 H), 8. 42 (d, J = 3. 9 Hz, 1H), 8. 21 (d, J = 3. 9Hz, 1 H), 7. 70 (d, J = 9. 3H z, 1H), 7. 44- 7. 38 (m, 4H), 7. 34— 7. 31 (m, 1 H), 7. 26 (s, 1 H), 6. 79 - 6. 75 (m, 1 H), 5. 45 (d, J = 6. 8Hz, 1 H), 5. 07 (qu i n t, J = 73 Hz , 1H), 4. 58(d d, J = 3. 9 Hz, 8. 8Hz, 1 H), 2. 87 (dd, J = 3. 9 Hz , 12. 2Hz , 1 H), 2. 55 (dd, J = 12. 2Hz, 8. 8Hz, 1 H), 1. 60 (d, J = 6. 8Hz, 3H), 1. 07 (s, 9 H)
m s s : 449 (M+ 1) +. 実施例 81
(I S) — 2— (t e r t—プチルァミノ) 一 1_ {4一 [(I S) — 1— ({5—フ ルオロー 4— [8— (トリフルォロメチル) イミダゾ [1, 2 - a] ピリジン一 3— ィル] ピリミジン一 2—ィル } ァミノ) ェチル] フエ二ル} エタノール [81] (以 下、 化合物 [81] という) の合成。 (1) 2, 4ージクロロー 5—フルォロピリミジン (フルォロケム社 (F 1 uo r o c hem) より入手可能である) 113mgと 3—トリフルォロメチルピリジン一 2—ァミン (国際公開第 2006/025567号パンフレツト、 81ページに記載 されている方法により合成した) 104mgから、 実施例 67— (1)、 (2) の方法 に準じて、 3— (2—クロ口一 5—フルォロピリミジン一 4—ィル) 一 8— (トリフ ルォロメチル) イミダゾ [1, 2— a] ピリジン [81 - 1] (以下、 化合物 [81 - 1] という) 143mgを得た。
( 2 ) 化合物 [67— 3] 45. 6mgと化合物 [81— 1] 47 mg力、ら、 実施 例 67— (4) の方法に準じて、 目的化合物 [81] (以下、 化合物 [81] という) 30. 6 mgを淡黄色固体として得た。 化合物 [81] のスペクトルデータを以下に示す。
一 NMR (CDC 13) δ : 9. 55- 9. 25 (m, 1H), 8. 47 (d, J =3. 9Hz, 1 H), 8. 26 (d, J = 3. 9Hz , 1 H), 7. 64 (d, J = 7. 3Hz , 1 H), 7. 43 - 7. 37 (m, H), 6. 85— 6. 73 (m, 1 H), 5. 55 - 5. 53 (m, 1H), 5. 04-5. 01 (m, 1 H), 4. 59 -4. 56 (m, 1 H), 2. 87 (d d, 12. 0Hz, 3. 7Hz, 1H), 2. 53 (dd, J = 12. 0Hz , 8. 8Hz , 1H), 1. 61 (d, J = 6. 8H
z, 3H), 1. 07 (s, 9H)
ma s s : 517 (M+ 1) +. 実施例 82 .
(R) ― [4— ((I S) —1— {[4— (8—クロロイミダゾ [1, 2— a] ピリジ ン一 3—ィル) 一 5—フルォロピリミジン一 2—^ fル] アミノ} ェチル) フエニル] [(2R) — 1, 2—ジメチルピロリジン一 2—ィル] メタノール [82] (以下、 化 合物 [82] という) の合成。 化合物 [65— 4] 34. 2mgと化合物 [67— 2] 30 mgから、 実施例 67— (3)、 (4) の方法に準じて、 目的化合物 [82] (以下、 化合物 [82] という) 15. 6mgを黄色固体として得た。 化合物 [82] のスペクトルデータを以下に示す。
^-NMR (CDC 13) δ : 9. 27 - 9. 05 (m, 1H), 8. 39 (d, J
=3. 9Hz, 1 H), 8. 24 (d, J = 3. 9Hz, 1 H), 7. 42-7. 30 (m, 5H), 6. 78 - 6. 70 (m, 1 H), 5. 01—4. 99 (m, 1H),
4. 36 (s , 1 H), 3. 1 1-3. 07 (m, 1 H), 2. 52— 2. 46 (m,
1 H), 2. 26 ( s , 3H), 2. 09 (s, 3H), 1. 75 - 1. 53 (m, 2 H),. 1. 60 (d, J = 6. 8Hz, 3H), 1. 25— 1. 21 (m, 1 H), 0.
86 (s , 3H)
ma s s : 495, 497 (M+ 1 ) +. 実施例 83
[4— ((1 S) — 1— {[4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3_ ィル) - 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] [(2 S) 一 1, 2—ジメチルピロリジン一 2—ィル] メタノール [83] (以下、 化合物 [8 3] という) の合成。 (1) 化合物 [63— 4 a] 2. 55 gから、 実施例 54、 55- (1) の方法に 準じて、 t e r t—ブチル ((1 S) 一 1一 {4- [[(2 S) - 1, 2—ジメチルピ 口リジン一 2—ィル] (ヒドロキシ) メチル] フエ二ル} ェチル) カーバメート [8 3- 1] (以下、 化合物 [83— 1] という) 1. 69 gを得た。 (2) 化合物 [83— 1] 3 Omgと化合物 [67— 2] 18. 4mgから、 実施 例 67— (3)、 (4) の方法に準じて、 目的化合物 [83] (以下、 化合物 [83] という) 10. 5mgを得た。
化合物 [83] のスペクトル —夕を以下に示す。
XH-NMR (CDC 13) 6 : 9. 48— 9. 35 (m, 1 H), 8. 42 (d, J =3. 4Hz, 1 H), 8. 24 (d, J = 3. 9Hz, 1H), 7. 42-7. 28 (m, 5H)., 6. 78 - 6. 70 (m, 1H), 5. 05 - 5. 03 (m, 1H), 4. 37 (s , 1H), 3. 12-3. 07 (m, 1 H), 2. 51— 2. 47 (m, 1 H), 2. 08 (s, 3H), 1. 85 (s, 3H), 1. 85-1. 71 (m, 3 H), 1. 59 (d, J = 6. 8Hz , 3H), 0. 85 (s, 3 H)
ma s s : 495, 497 (M+ 1) +. 実施例 84、 85
1— [4一 ((I S) —1— {[4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3一^ Γル) —5—フルォロピリミジン— 2—ィル] アミノ} ェチル) フエニル] 一 2 一 (ジメチルァミノ) 一 2—メチルプロパン一 1一オール [84] (以下、 化合物 [8 4] という)、 および [85] (以下、 化合物 [85] という) の合成 (ここで、 化合 物 [84] と化合物 [85] は、 ジァステレオマーの関係にある。 表 14参照)。
(1) ジメチルスルホキシド 51 gをジクロロメタン 7 OmLに溶解し一 78 と した後、 二塩化ォキサリル 59. 1 gを溶解したジクロロメタン溶液 16 OmLを加 えた。 同温にて 30分間撹拌した後、 2— (ジメチルァミノ) —2—メチルプロパン 一 1'一オール 18. 2 gを溶解したジクロロメタン溶液 12 OmLを加えた。 同温に て 20分間撹拌した後、 トリェチルァミン 107 gを加え、 同温にて 40分間撹拌し た。 反応混合物を室温まで昇温した後、 ジクロロメタンで希釈した。 得られた有機層 を飽和炭酸水素ナトリウム水溶液、 飽和食塩水にて順に洗浄し、 無水硫酸マグネシゥ ムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を減圧蒸留 にて精製し、 2— (ジメチルァミノ) 一 2—メチルプロパナール [84— 1] (以下、 化合物 [84—1] という) 11. l gを得た。
(2) 化合物 [1一 1] 24. 1 gと化合物 [84— 1] 11. l gから、 実施例 63、 64- (4) の方法に準じて、 t e r t—ブチル ((I S) — 1一 {4一 [2 - (ジメチルァミノ) 一 1—ヒドロキシー 2—メチルプロピル] フエ二ル} ェチル) カーバメート [84— 2] (以下、 化合物 [84— 2] という) 12. 8 gを得た。
(3) 化合物 [84— 2] 6. 24gと化合物 [67— 2] 2. 34gから、 実施 例 67— (3)、 (4) の方法に準じて、 目的化合物 [84] と [85] の混合物 2. 31 gを得た。
(4) 化合物 [84] と [85] の混合物 70. 2mgをキラルセル AD— Hにて 分割した。
光学分割条件は以下のとおりである。
カラム:キラルセル AD— H (Ch i r a l c e l AD— H、ダイセル化学工業)、 直径 2 0 mm、 長さ 2 50 mm
溶出液:へキサンノ 2—プロパノール Zジェチルァミン = 7 5/2 5/0. 1 流速: 1 2mL/m i n
得られた溶液を減圧濃縮し、 目的化合物 [84] 2 2. 5mg (RT= 1 1. 5分)、 目的化合物 [8 5] 2 lmg (RT=22. 3分) を得た。 化合物 [84] のスペクトルデータを以下に示す。
^-NMR (CD3OD) <5 : 8. 3 7 ( s , 1 H) , 8 9 (s , 1 H) , 7. 7 5 (d, J = 7. 0Hz , 1 H) , 7. 5 1 - 7. 42 5H) , 7. 2 1 - 7. 0 5 (m, 1 H) , 5. 1 2 - 5. 0 1 (m, 1 H) , 9 5 (s , 1 H) , 2. 94 (s , 3H) , 2. 8 0 (s , 3 H) , 1. 5 8 J = 7. 0Hz , 3 H) , 1. 1 6 (s , 3H) , 1. 1 3 ( s , 3 H)
ma s s : 48 3, 48 5 (M+l) + 化合物 [8 5] のスペクトルデ一夕を以下に示す。
— NMR (CD3OD) δ : 8. 3 8 (s , 1 H) , 8 9 (s , 1 H) , 7. 7 5 (d, J = 7. 0Hz, 1H) , 7. 5 1— 7. 43 5H) , 7. 1 9 - 7. 04 (m, 1 H) , 5. 1 0 - 5. 0 1 (m, 1 H) , 9 6 (s, 1 H) , 2. 94 (s , 3H) , 2. 8 0 (s , 3 H) , 1. 5 8 J = 7. 0Hz, 3 H) , 1. 1 7 (s , 3H) , 1. 1 2 (s, 3 H)
ma s s : 48 3, 48 5 (M+ 1) + 実施例 8 6
1 - [4- ((I S) - 1 - {[4- (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] — 2 ーメチルー 2— (メチルァミノ) プロパン一 1一オール [8 6] (以下、 化合物 [8 6] という) の合成。
(1) ベンズアルデヒド 5 9 5mgをエタノール 10mLに溶解した溶液に、 2— アミノー 2—メチルプロパン一 1一オール 5 0 Omgを室温にて加え、 4時間加熱還 流した。 反応混合物に、 室温にて水素化ホウ素ナトリウム 1. 0 6 gを加え、 同温に て 1時間半撹拌した。 反応混合物に 1M水酸化ナトリウム水溶液を加え、 酢酸ェチル にて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウムにて 乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。得られた残渣とパラホルムアル デヒド 6 74mgをメタノール 7mLに溶解した溶液に、室温にてシァノ水素化ホウ
0 5mgを加え、 同温にて 4時間半撹拌した。 反応混合物に 1M水酸
化ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水 にて洗浄し、 無水硫酸マグネシウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃 縮した。 得られた残渣をシリカゲル力ラムクロマトグラフィー (溶出液:へキサン Z 酢酸ェチル = 100/0— 70/30) にて精製し、 2—アミノー 2—メチルプロパ ンー 1一オール [86— 1] (以下、 化合物 [86— 1] という) 632mgを得た。
(2) 化合物 [86— 1] 417 mgとトリエチルァミン 1. 09 gをジメチルス ルホキシド 5 mLに溶解した溶液に、三酸化いおうピリジン錯体 859mgを室温に て加え、 1時間半撹拌した。 反応混合物に飽和炭酸水素ナトリウム水溶液を加え、 酢. 酸ェチルにて抽出した。 得られた有機層を水および飽和食塩水にて順に洗浄し、 無水 硫酸マグネシウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた 残渣をシリカゲルカラムクロマトグラフィー (溶出液:へキサン/酢酸ェチル =10 0/0 - 80/20) にて精製し、 2― [ベンジル (メチル) ァミノ] 一 2—メチル プロパナール [86— 2] (以下、 化合物 [86— 2] という) 305mgを得た。
(3) 化合物 [1— 1] 398mgと化合物 [86— 2] 305mgかち、 実施例 63、 64- (4) の方法に準じて、 t e r t—ブチル [(I S) — 1一 (4— {2 一 [ベンジル (メチル) ァミノ] 一 1ーヒドロキシ一 2—メチルプロピル } フエニル) ェチル] カーバメート [86— 3] (以下、 化合物 [86— 3] という) 12. 8 g を得た。
(4) 化合物 [86— 3] 227mgをメタノール 7mLに溶解し、 20%パラジ ゥム炭素触媒 114mgを加え、 水素雰囲気下、 室温にて 1時間撹拌した。 触媒をセ ライトろ過し、 ろ液を減圧濃縮して、 t e r t—ブチル ((1 S) — 1一 {4— [1 ーヒドロキシー 2—メチルー 2— (メチルァミノ) プロピル] フエ二ル} ェチル) 力 ーバメート [86— 4] (以下、 化合物 [86— 4] という) 188mgを得た。
(5) 化合物 [86— 4] 206mgと化合物 [67— 2] 15 lmgから、 実施 例 67— (3)、 (4) の方法に準じて、 目的化合物 [36] (以下、 化合物 [86] という) 14 Omgを得た。 化合物 [86] のスぺクトルデータを以下に示す。
:H-NMR (CDC ") δ : 9. 43- 9. 26 (m, 1 HX 1/2) , 9. 25
—9. 07 (m, 1 HX 1/2) , 8. 42-8. 39 (m, 1 H) , 8. 25-8. 23 (m, 1 H) , 7. 41-7. 36 (m, 5 H) , 6. 76 - 6. 67 (m, 1
H) , 5. 52 (s , 1 HX 1/2) , 5. 51 (s , 1 HX 1/2) , 5. 09- 4. 98 (m, 1 H) , 4. 44 (s, 1 HX 1/2) , 4. 42 (s , 1HX 1/ 2) , 2. 36 (s , 3H) , 1. 60 (d, J = 7. 0Hz, 3H) , 1. 04 (s,
3HX 1/2) , 1. 0 2 (s , 3 ΗΧ 1 /2) , 0. 8 3 (s , 3HX 1/2) , 0. 8 2 (s , 3ΗΧ 1 /2)
ma s s : 46 9, 47 1 (Μ+ 1) + 実施例 8 7
(1 - t e r t _プチルピペリジン一 4一ィル) [4一 ((1 S) 一 1一 {[4_ (8 一クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—フルォロピリミジン一 2 -ィル] アミノ} ェチル) フエニル] メタノール [8 7] (以下、 化合物 [8 7] という) の合成。
(1) ジイソプロピルアミン 6. 5 2 gをテトラヒドロフラン 24mLに溶解し氷 冷した後、 n—ブチルリチウム (2. 6 6Mへキサン溶液) 2. 9 lmLを加えた。 同温にて 30分間撹拌した後、 一 7 8°Cとした。 反応混合液に同温にてトリメチルシ リルジァゾメタン ( 2 Mへキサン溶液) 3. 8 7mLを加え、 同温にて 1時間撹拌し た。反応混合液に同温にて 1— t e r t—ブチルピリミジン— 4一オン(J . O r g. Ch em. , 2 0 0 5, 7 0, 1 9 3 0に記載されている方法により合成した) l g をテトラヒドロフラン 6 mLに溶解した溶液を加え、 同温にて 1時間半撹拌した後、 終夜加熱還流した。 反応混合物に水を加え、 酢酸ェチル 2 0 OmLにて抽出した。 得 られた有機層を、 無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧 濃縮した。 得られた残渣を酢酸ェチル 1 2 OmLに溶解し、 室温にてシリカゲル 24 gを加えて、 同温にて 1時間半撹拌した。 不溶物をろ過し、 ろ液を減圧濃縮し、 1— t e r t一プチルピペリジン一 4一力ルポアルデヒド [8 7 - 1] (以下、化合物 [8 7 - 1] という) 8 3 0mgを得た。 (2) 化合物 [8 7— 1] 2 7 6mgと化合物 [6 7— 2] 3 Omgから、 実施例 84、 8 5 - (2)、 (3) の方法に準じて、 目的化合物 [8 7] (以下、 化合物 [8 7] という) 5. 5mgを得た。 化合物 [8 7] のスペクトルデータを以下に示す。
一 NMR (CDC 1 3) 5 : 9. 1 1 (b r s , 1 H) , 8. .3 5 (d, J = 3. 9Hz , 1HX 1/2) , 8. 3 0 (d, J = 3. 9Hz, 1HX 1/2) , 8. 2 1 (d, J = 3. 5Hz, 1HX 1/2) , 8. 2 0 (d, J = 3. 5Hz, 1 HX 1/2) , 7. 42 - 7. 2 9 (m, 5 H) , 6. 7 0- 6. 60 (m, 1 H) , 5. 5 7 - 5. 5 1 (m, 1 H) , 5. 0 3 -4. 9 5 (m, 1 H) , 4. 40 (s, 1 HX 1/2) , 4. 3 8 (s , 1 HX 1/2) , 3. 2 2 - 3. 1 3 (m, 1 H) , 3. 0 2 - 2. 9 0 (m, 1 H) , 2. 1 9— 1. 7 8 (m, 6H) , 1. 5 9 (d, J = 7. 0Hz, 3H) , 1. 2 7 - 1. 24 (m, 1 H) , 1. 1 1 (s , 9HX 1/2) , 1. 1 0 (s, 9HX 1/2)
ma s s : 537, 539 (M+ 1) + 実施例 88、 89
[4- ((l.S) - 1 - {[4- (8—クロロイミダゾ [1, 2— a] ピリジン一 3— ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1—ィ ソプロピルァゼチジン一 3—ィル) メタノール [88] (以下、 化合物 [88] とい う)、 および [89] (以下、 化合物 [89] という) の合成 (ここで、 化合物 [88] と化合物 [89] は、 ジァステレオマーの関係にある。 表 1 5参照)。 (1) 1一べンズヒドリルァゼチジン一 3—力ルボン酸から、実施例 1— (1)、(2) の方法に準じて、 t e r t—ブチル [(I S) - 1 - (4- {[1 - (ジフエニルメ チル) ァゼチジン一 3—ィル] 力ルポ二ル} フエニル) ェチル] カーバメート [88 一 1] (以下、 化合物 [88— 1] という) 9. 94 gを得た。 (2) 化合物 [88— 1] 9. 94 gをテトラヒドロフラン 1 00 mLとメタノー ル 2 OmLに溶解し、 室温にて水素化ホウ素ナトリウム 799mgを加え'、 同温にて 2時間半撹拌した。 反応混合物に水を加え、 クロ口ホルムにて抽出した。 得られた有 機層を、 無水硫酸マグネシウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮し た。 得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液:へキサン Z酢酸 ェチル = 100Z0 _ 30Z70) にて精製し、 t e r t—ブチル ((1 S) 一 1一 {4- [[1一 (ジフエニルメチル) ァゼチジン— 3—ィル] (ヒドロキシ) メチル] フエ二ル} ェチル) カーバメート [88-2] (以下、 化合物 [88-2] という) 632 m gを得た。 (3) 化合物 [88— 2] 9. 79 gから、 実施例 33— (3)、 (4) の方法に準 じて、 t e r t—ブチル ((1 S) — 1一 {4一 [ヒドロキシ (1一イソプロピルァ ゼチジン一 3—ィル) メチル] フエ二ル}ェチル) カーバメート [88-3] (以下、 化合物 [88— 3] という) 6. 26 gを得た。 (4) 化合物 [88— 3] 72 Omgと化合物 [67— 2] 20 Omgから、 実施 例 67— (3)、 (4) の方法に準じて、 目的化合物 [88] と [89] の混合物 1 3 5mgを得た。
(5) 化合物 [88] と [89] の混合物 13 Omgをキラルセル AD—Hにて分 割した。
光学分割条件は以下のとおりである。
カラム:キラルセル AD— H (Ch i r a 1 c e 1 AD— H、ダイセル化学工業)、 直径 20 mm、 長さ 250 mm
溶出液:へキサン/エタノール Zジェチルァミン =7 0/3 0/1
流速: 1 2mL/m i n
得られた溶液を減圧濃縮し、 目的化合物 [8 8] 3 6mg (RT = 1. 4分)、 目 的化合物 [8 9] 6 3mg (RT=2 1. 8分) を得た。 化合物 [8 8] のスペクトルデータを以下に示す。
XH-NMR (CDC 1 3) δ : 8. 9 0 (b r, 1 H), 8. 2 2— 8. 1 6 (m, 2H), 7. 43 - 7. 3 8 (m, 4H), 7. 3 2 (d, J = 7. 3Hz, 1 H), 7. 2 7 (s, 1 H), 6. 6 0 (b r s , 1 H), 5. 58 (b r s , 1 H), 4. 9 5 (b r s , 1 H), 4. 8 9 (d, J =6. 8Hz 1 H), 3. 2 2 (d, 5. 9Hz, 2H), 3. 1 5- 3. 1 2 (m, 1 H), 3. 0 5 - 3. 04 (m H), 2. 6 8 - 2. 6 5 (m, 1 H), 2. 30— 2. 2 5 (m, 1 H)
ma s s : 49 5, 49 7 (M+ 1) +. 化合物 [8 9] のスペクトルデータを以下に示す。
XH-NMR (CDC 1 3) δ : 8. 9 9 (b r, 1H), 8. 2 9 (b r 's, 1 H), 8. 1 8— 8. 1 7 (m, 1 H), 7. 4 1 - 7. 3 7 (m, 4H), 7. 3 2 (d, J = 6. 8Hz, 1 H), 7. 2 7 (s , 1 H), 6. 5 7 (b r, 1 H), 5. 6 1 (b r , 1 H), 4. 9 9 -4. 9 5 (m, 1 H), 4. 8 7 (d, J = 6. 3Hz, 1 H), 3. 2 1 (d, J = 6. 3Hz, 2H), 3. 1 4- 3. 1 0 (m, 1H), 3. 0 5 - 3. 0 2 (m, 1 H), 2. 6 6 - 2. 6 2 (m, 1H), 2. 2 7 (q u i n t, J = 6. 3 Hz , 1 H), 1. 5 8 (d, J = 6. 8Hz, 3H), 0. 8 9 (d, J = 5. 9Hz, 6H)
ma s s : 49 5, 49 7 (M+ 1) +. 実施例 9 0、 9 1
[4- ((I S) - 1 - {[4- (8—クロロイミダゾ [1, 2— a] ピリジン一 3— ィル) 一 5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] (1ーィ ソプロピルピぺリジン— 4一ィル) メタノール [90]. (以下、 化合物 [90] とい う)、 および [9 1] (以下、 化合物 [9 1] という) の合成 (ここで、 化合物 [9 0] と化合物 [9 1] は、 ジァステレオマーの関係にある。 表 1 5参照)。
(1) 化合物 [3 3 -4] 4 1 5mgと化合物 [6 7— 2] 24 Omg力、ら、 実施 例 6 7— (3)、 (4) の方法に準じて、 目的化合物 [9 0] と [9 1] の混合物 1 5 6mgを得た。
(2) 化合物 [9 0] と [9 1] の混合物 1 56mgをキラルセル AD— Hにて分 割した。
光学分割条件は以下のとおりである。
カラム:キラルセル AD— H (Ch i r a 1 c e 1 AD— H、ダイセル化学工業)、 直径 20 mm、 長さ 2 5 0 mm
溶出液:へキサン Z 2—プロパノールノジェチルァミン =7 0/3 0/0. 1 流速: 1 2 mL/m i n
得られた溶液を減圧濃縮し、 目的化合物 [9 0] 40. 5mg (RT= 1 3. 5分)、 目的化合物 [9 1] 3 9. 8mg (RT=24. 2分) を得た。 化合物 [9 0] のスペクトルデータを以下に示す。
XH-NMR (DMSO— d6) δ : 8. 44 (b r s, 1 H), 8. 3 2 (s, 1H),
7. 9 9 (d, J = 7. 3Hz, 1 H), 7. 7 1 (s , 1 H), 7. 3 6 (d, J =
8. 0Hz, 2H), 7. 2 1 (d, 8. 0Hz , 2H), 7. 1— 7. 0 (b r s , 1 H), 5. 0 (m, 2H), 4. 2 (m, 1 H), 2. 8 - 2. 5 (m, 2H), 2. 0— 1. 6 (m, 4H), 1. 49 (d, 6. 8Hz, 3H), 1. 4- 0. 9 (m, 4H), 0. 8 7 (d, 5. 9Hz , 6 H)
ma s s : 5 2 3 (M+ 1) +. ' 化合物 [9 1] のスペクトルデータを以下に示す。
一 NMR (DMSO - d6) δ : 8. 45 (b r s , 1 H), 8. 3 2 (s , 1 H), 7. 9 9 (d, J = 7. 3Hz, 1 H), 7. 7 1 (s , 1 H), 7. 3 5 (d, J =
8. 3Hz, 2 H), 7. 2 1 (d, 7. 8Hz , 2H), 7. 1 - 6. 9 (b r s ,
1H), 5. 0 1 (m, 2H), 4. 1 (m, 1H), 2. 7 (m, 1H), 2. 6 - 2.
5 (m, 1H), 2. 0 - 1. 7 (m, 4H), 1. 49 (d, 6. 8Hz, 3H),
1. 4— 0. 9 (m, 4H), 0. 8 6 (m, 6 H)
ma s s : 5 2 3 (M+ 1) +. 実施例 9 2
(1 - t e r t一ブチルァゼチジン一 3—ィル) [4— ((1 S) 一 1一 {[4一 (8 一クロロイミダゾ [1, 2— a] ピリジン一 3—ィル).ー5—フルォロピリミジン一 2—ィル] アミノ} ェチル) フエニル] メタノール [9 2] (以下、 化合物 [9 2] という) の合成。
1 - t e r t一ブチルァゼチジン一 3—力ルポン酸 (C h em. P h a rm. Bu i 1., 1 9 74, 2 2, 149 0に記載されている方法により合成した) 9 8 0mg と化合物 [6 7— 2] 3 5. 5mg力、ら、 実施例 8 8、 8 9— (1) 〜 (4) の方法 に準じて、 目的化合物 [9 2] (以下、 化合物 [9 2] という) 34mgを淡黄色固 体として得た。
化合物 [92] のスペクトルデ一夕を以下に示す。
:H-NMR (CDC 13) δ : 9. 21 (b r, 1H), 8. 40 (d d, J = 3.
9Hz , 7. 3Hz, 1 H), 8. 24 (d, J = 2. 9Hz, 1 H), 7. 40-7.
37 (m, 5 H), 7. 26 (s , 1 H), 6. 68 -6. 64 (m, 1 H), 5. 4 8 (d, J = 5. 9Hz, 1 H), 5. 04— 5. 01 (m, 1H), 4. 92 (dd,
J = 5. 4Hz, 2. 4Hz, 1 H), 3. 28-3. 2 1 (m, 1 H), 3. 1 9一
3. 1 5 (m, 1 H), 3. 1 2-3. 06 (m, 2 H) , 2. 56 - 2. 49 (m,
1H), 1.59 (d, J = 6. 8Hz, 3H), 0. 93 (s, 9 H)
ma s s : 509, 51 1 (M+ 1) +. 実施例 93
(I S) —2— (t e r t一プチルァミノ) 一 1一 {4— [(I S) — 1一 ({4 - [8 一 (ジフルォロメチル) イミダゾ [1, 2 - a] ピリジン一 3—ィル] - 5—フルォ 口ピリミジン一 2—ィル } ァミノ) ェチル] フエ二ル} エタノール [93] (以下、 化合物 [93] という) の合成。
(1) 2, 4—ジクロロー 5—フルォロピリミジン (フルォロケム社 (F 1 u o r o c hem) より入手可能である) 568mgと 2—ァミノニコチンアルデヒド (ァ ルドリツチ社 (A 1 d r i c h) より入手可能である) 415mg力、ら、 実施例 67 一 (1)、 (2) の方法に準じて、 3— (2—クロロー 5—フルォロピリミジン一 4一 ィル) イミダゾ [1, 2-a] ピリジン一 8—カルボアルデヒド [93- 1] (以下、 化合物 [93— 1] という) 180mgを得た。
(2) 化合物 [93— 1] 1 8 Omgから、 実施例 10— (2) の方法に準じて、 3—(2—クロ口一 5—フルォロピリミジン一 4一ィル)一 8— (ジフルォロメチル) イミダゾ [1, 2-a] ピリジン [93-2] (以下、 化合物 [93-2] という) 97mgを得た。
(3) 化合物 [67-3] 33. 5mgと化合物 [9.3— 2] 3 Omgから、 実施 例 67— (4)·の方法に準じて、 目的化合物 [93] (以下、 化合物 [93] という)
12. 9mgを得た。 化合物 [93] のスペクトルデータを以下に示す。
— NMR (CDC 13) 6 : 9. 36 (b r s, 1 H) , 8. 43 - 8. 40 (m, 1 HX 4/5), 8. 25 - 8. 22 (m, 1 HX 4/5) , 7. 97 - 7. 93 (m, 1HX 1/5 + 1HX 1/5) , 7. 63 - 7. 58 (m, 1HX4/5) , 7. 5 0-7. 37 (m, 4H+ 1HX 1/5 + 1HX 1/5) , 7. 32 ( t , J = 55. IHz, 1 H) , 6. 87 - 6. 79 (m, 1HX4/5) , 5. 57— 5. 51 (m,
1 H X 4 / 5 ) , 5. 06 - 5. 00 (m, 1 HX 4/5) , 4. 67 -4. 57 (m, 1HX4/5 + 1HX 1/5 + 1HX 1/5) , 4. 32-4. 26 (m, 1 HX 1 /5) , 2. 98-2. 91 (m, 1 HX 1/5) , 2. 90 -2. 84 (m, 1 H X 4 / 5 ) ,. 2. 59-2. 51 (m, 1 H) , 1. 62 - 1. 58 (m, 3 H) , 1. 12- 1. 10 (m, 9 HX 1/5) , 1. 09 - 1. 06 (m, 9HX4/5) ma s s : 499 (M+ 1 ) + 産業上の利用の可能性
本発明の化合物は、優れた P L K 1阻害作用及び細胞増殖阻害作用を有すること力' ら医薬の分野において有用な抗腫瘍剤として期待される。