Antecedentes da Invenção
[001] O presente pedido de patente reivindica prioridade do pedido de patente provisório U.S. número de série 60/647.271 depositado em 26 de janeiro de 2005.
Campo da Invenção
[002] A presente invenção refere-se a derivados de 1-aril-1-hidróxi-2,3-diamino-propil aminas, 1- heteroaril-1-hidróxi-2,3-diamino-propil aminas e a compostos relacionados tendo atividade analgésica e em alguns casos imunoestimulante.
[003] A presente invenção refere-se também a composições farmacêuticas contendo esses compostos como ingrediente ativo para alívio ou eliminação de dor em mamíferos e/ou estimulação do sistema imune em mamíferos e a métodos de uso das ditas composições farmacêuticas como analgésicos ou imunoestimulantes.
Antecedente da Técnica
[004] 1-Fenil-2-decanoilamino-3-morfolino-1- propanol (PDMP) foi encontrado por Vunam, R.R. e Radin, N., Chem. Phys. Lipids, 26, 265-278, 1980. Preparação de PDMP é descrita em Inokuchi, J. e outros, J. Lipids Res. 28, 565-571; Radin, A. e outros, NeuroProtocols, 3(2), 145-55, 1993; Radin, A. e outros, J. Lipid Res., 36, 611621, 1995 e U.S. 5916911.
[005] Esses derivados inibem a formação de glicosilceramida (GlcCer) através da inibição da enzima GlcCer sintase, deste modo diminuindo o nível de glicoesfingolipídeos. Os isômeros mais ativos têm a configuração R,R-(D-treo)-. Quatro enantiômeros são produzidos durante a síntese. Devido ao fato de apenas os enantiômeros D-treo serem ativos na inibição da glicosilceramida sintase, separação dos inibidores de D- treo foi realizada através de cromatografia quiral.
[006] Além disso, D-treo-PDMP tem atividade antitumor através da inibição da biossíntese de glicosfingolipídeo conforme descrito por Inokuchi, J., Cancer Letters, 38 (1-2), 23,30, 1987.
[007] Ainda, foi também descrito que D-treo-PDMP suprime a função sináptica por Mizutani e outros, Biochem. Biphys. Res. Commum., 222, 494-498, 1996.
[008] Preparação de D-treo-PDMP enantiomericamente puro foi descrita por Mitchell, Scott, A. [J. Org. Chem., 63 (24), 8837-8842, 1998]; Miura e outros [Bioorg. Med. Chem., 6, 1481-1489, 1998]; Shin, S. e outros, [Tetrahedron asymmetry, 11, 3293-3301, 2000]; WO 2002012185.
[009] L-treo-PDMP é um agente para tratamento de doenças neuronais WO 95/05177. Este composto é também descrito ser um agente para proteção do cérebro na US 6407064. Além disso, tratamento com L-treo-PDMP após isquemia transitória cerebral anterior em ratos melhorou o déficit de uma memória espacial bem-aprendida de uma tarefa de labirinto de 8 braços, sugerindo um potencial para distúrbios neurodegenerativos conforme descrito por Inokuchi e outros, Ann. N.Y. Acad. Sci., 845(1), 219-224, 1998 e JP 10324671 (Seikagaku Kogyo Co.).
[0010] Uma síntese estereosseletiva de L-treo- PDMP enantiomericamente puro foi também descrita por Shin, S. e outros, Tetrahedron asymmetry, 11, 3293-3301, 2000 e WO 2002012185 a etapa chave é a clivagem regiosseletiva por nucleófilos de nitrogênio, como morfolina, da ligação C(3)-N- de aziridina-2-metanóis enantiomericamente puros não-ativados.
i) TMS-I, CH3CN ii) a) morfolina b) HCl iii) Pd(OH)2, H2, AcOH, MeOH, 40o C iv) NaOH a 10%, cloreto de decanoíla 81%.
[0011] Por outro lado, a síntese de (1S,2S)-1- fenil-2-decanoilamino-3-morfolino-1-propanol enantiomericamente puro (L-treo-PDMP) a partir de L- serina foi também descrita por Mitchell, Scott, A., J. Org. Chem., 63 (24), 8837-8842, 1998.
[0012] Outros métodos conhecidos para se obter L- treo-PDMP são descritos por Miura, T. e outros, Bioorg. Med. Chem., 6, 1481-1498, 1998 e na JP-A-9-216858. L- treo-PDMP é um agente para tratamento de doenças neuronais WO 95/05177. Este composto é também descrito ser um agente para proteção do cérebro na US 6407064. Além disso, tratamento com L-treo-PDMP após isquemia transitório cerebral anterior em ratos melhorou o déficit de uma memória espacial bem aprendida de uma tarefa de labirinto de 8 braços, sugerindo um potencial para distúrbios neurodegenerativos conforme descrito por Inokuchi e outros, Ann. N.Y. Acad. Sci., 845(1), 219-224, 1998 e JP 10324671 (Seikagaku Kogyo Co.).
[0013] Sínteses de (1S,2S)-treo e (1R,2S)-eritro- 1-fenil-2-palmitoilamino-3-N-morfolino-1-propanol (PPMP) de aldeído Garner de L-serina, por Nishida, A., Synlett, 389-390, 1998.
[0014] Compostos com grupos acila graxos de cadeia mais longa (do que a decanoíla) foram verificados ser substancialmente mais eficazes como inibidores de GCS. Análogos de D-treo-1-fenil-2-palmitoilamino-3- pirrolidino-1-propanol (P4 ou PPPP) foram primeiro obtidos através de uma reação Mannich conforme descrito por Abe, A. e outros, J. Biochem., 111, 191-196, 1992 ou US 5916911 e WO 2001004108.
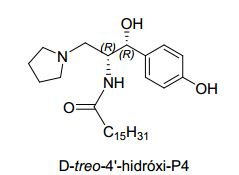
[0015] Preparação de D-treo-4'-hidróxi-P4, um dos inibidores mais potentes de GCS, foi descrita por Lee, L. e outros, J. Biol. Chem., 274, 21, 14662-14669, 1999. Ainda, uma série de substituições de dioxana foi planejada e testada. Essas incluíam 3',4'- metilenodioxifen il-3',4'-etilenodioxifenila e homólogos substituídos com 3',4'-trimetilenodioxifenila.
[0016] Síntese de D-treo-1-fenil-2- benziloxicarbonilamino-3-pirrolidino-1-propanol enantiomericamente puro (PBPP) e D-treo-P4 e seus análogos de N-benziloxicarbonil-D-serina foi descrita por Jimbo, M. e outros, J. Biochem., 127(3), 485-91, 2000 e EP 782992 (Seikagaku Kogyo Co.). PBPPP é descrito como inibidor de GCS potente.
[0017] Pró-fármacos novos de derivados de P4 foram descritos na US 20020198240 e no WO 2002062777.
[0018] Sínteses de D-treo-etilenodióxi-P4 enantiomericamente puro e D-treo-p-metóxi-P4 foram descritas por Husain, A. e Ganem, B., Tetrahedron Lett., 43, 8621-8623, 2002. A etapa-chave é uma adição altamente syn-seletiva de reagentes Grignard ao aldeído Garner.
i) brometo de 3,4-etilenodioxifenilmagnésio, Cul, THF; Me2S, 64%, ii) HCl a 0,1N, THF a 82%, MsCl, Et3N, DCM, 0°C, 85%, iii) pirrolidina, DMF, 45°C, 58% iv) HCl a 3N, 0°C, para temperatura ambiente quando C15H31 COCl, Et3N, DMAP, DCM, -20°C, 87%
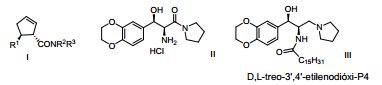
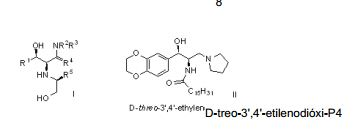
[0019] Sínteses diastereosseletivas de análogos de P4 foram descritas na US 03/0153768 e no WO 2003045928 (Genzyme Corp.); Oxazolinas I [R1 = arila (não)substituídas; R2, R3 = H, alifáticas (não)substituído; NR2R3 = heterocíclicas] são preparadas como intermediários para inibidores de P4 glicosiltranferase de R1CHO e R2R3NCOCH2CN. Deste modo, isocianoacetato de metila CNCH2CO2Me foi tratado com pirrolidina e a amida foi tratada com 1,4-benzodioxano-6- carboxaldeído, seguido por hidrólise da oxazolina usando HCl em metanol, redução do grupo ceto de amida II usando LiAlH4 e acilação com cloreto de palmitoíla para dar D,L- treo-etilenodióxi-P4 III.
[0020] Sínteses de análogos de P4 enantiopuros foram descritas no WO 2003008399 (Genzyme Corp.). Derivados de P4, tal como I [R1, R5 = aromáticos (não)substituídos; R2, R3 = H, alifáticos (não)substituídos; NR2R3 = anel heterocíclico não- aromático (não)substituído; R4 = O, H2], foram preparados para seu uso terapêutico como inibidores de GCS. Deste modo, D-treo-etilenodióxi-P4 foi preparado através de uma seqüência sintética multietapa partindo de S-(+)-Ph glicinol, fenil-α-bromoacetato, 1,4-benzodioxan-6- carboxaldeído, pirrolidina e cloreto de palmitoíla.
[0021] Novos análogos de D-treo-P4 que carregam substituintes éter no anel aromático foram recentemente sintetizados de D-serina e verificados suprimir extensão de neurito em uma linhagem de célula de inseto embriônica conforme descrito por Slavish, J.P. e outros, Bioorg. Med. Chem. Lett., 14, 1487-1490, 2004.
[0022] Referências adicionais que servem como base para a presente invenção são as Patentes U.S. N°s 5.945.442; 5.952.370; 6.030.995 e 6.051.598; Journal of Labelled Compounds & Radiopharmaceuticals (1996), 38(3), 285-97; Pedido de Patente PCT publicado WO 01/38228; e Kastron e outros, Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija (1965) (4), 474-7.
[0023] Significantemente, de acordo com tanto quanto é do conhecimento dos presentes inventores nenhum dos compostos da técnica anterior que são estruturalmente similares aos novos compostos da presente invenção é conhecido na técnica anterior como analgésico ou imunoestimulante.
Sumário da Invenção
[0024] A presente invenção refere-se a compostos da Fórmula I
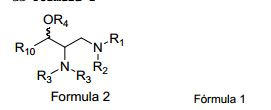
onde R1 é H ou alquila de 1 a 6 carbonos, R2 é H, alquila de 1 a 6 carbonos ou os grupos R1 e R2 junto com o nitrogênio formam um anel de 4, 5, 6 ou 7 membros saturado ou insaturado que inclui opcionalmente um ou dois heteroátomos independentemente selecionados de N, O e S, o dito anel de 4, 5, 6, ou 7 membros sendo opcionalmente substituído com um halogênio, COOH, CH2OH, OH, B(OH)2, ciano ou com um grupo alquila tendo 1 a 6 grupos alquila; R3 é independentemente selecionado de H, alquila de 1 a 20 carbonos, arila ou heteroarila, aril-alquila ou heteroaril-alquila onde a porção alquila é de 1 a 4 carbonos, cicloalquila de 3 a 6 carbonos, os ditos grupos arila ou heteroarila sendo opcionalmente substituídos com 1 a 3 grupos independentemente selecionados do grupo consistindo em halogênio, alquila de 1 a 6 carbono, alcóxi de 1 a 6 carbonos e tióxi de 1 a 6 carbonos, ou R3 é CO-R7 ou CO-O-R7, onde R7 é H, alquila de 1 a 20 carbonos, benzila, alquila de 1 a 20 carbonos substituída com um grupo NH2, com uma NHCOOalquila ou com um grupo NH-COalquila onde o grupo alquila tem um a 6 carbonos, ou R7 é arila, heteroarila, aril-alquila ou heteroaril-alquila onde a porção alquila é ramificada ou não-ramificada e tem 1 a 4 carbonos, os ditos grupos arila ou heteroarila sendo opcionalmente substituídos com 1 a 3 grupos independentemente selecionados do grupo consistindo em halogênio, alquila de 1 6 carbonos, alcóxi de 1 a 6 carbonos e tióxi de 1 a 6 carbonos; R4 é H, alquila de 1 a 6 carbonos ou CO-R8, onde R8 é alquila de 1 a 6 carbonos; as linhas onduladas representam ligações conectadas a carbonos tendo configuração R ou S, e R10 é selecionado dos grupos das fórmulas (i) e (ii)
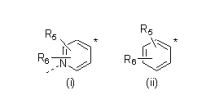
onde o * indica o átomo de carbono ao qual a porção restante da molécula é ligada; R5 e R6 são independentemente H, alquila de 1 a 6 carbonos, halogênio, alcóxi de 1 a 6 carbonos ou os grupos R5 e R6 junto com os átomos aos quais eles estão ligados formam juntamente um anel carbocíclico ou um heterocíclico, o anel carbocíclico tendo 5 ou 6 átomos no anel, o anel heterocíclico tendo 5 ou 6 átomos no anel e 1 a 3 heteroátomos independentemente selecionados de N, O e S, e o dito anel carbocíclico ou heterocíclico juntamente formado por R5 e R6 sendo opcionalmente substituído com 1 a 6 grupos R9 onde R9 é independentemente selecionado de halogênio, alquila de 1 a 6 carbonos, alcóxi de 1 a 6 carbonos, contanto que: quando R10 tiver a fórmula (ii) então a Fórmula 1 não inclua compostos onde R4 é hidrogênio e R1 e R2 juntamente com o nitrogênio formem um anel morfolino ou pirrolidino e onde R5 e R6 forem ambos H ou um de R5 e R6 for OCH3 e o outro seja H, e a presente invenção refere-se também a todos os sais farmaceuticamente aceitáveis dos ditos compostos.
[0025] A presente invenção refere-se também a composições farmacêuticas contendo o composto novo acima mencionado a ser usado como analgésicos e/ou imunoestimulantes em mamíferos e a métodos de uso das ditas composições farmacêuticas como analgésicos e/ou imunoestimulantes.
Descrição Detalhada da Invenção
[0026] Uma descrição geral dos compostos da invenção é provida na Seção Sumário do presente pedido de patente. A maioria dos compostos da invenção contém um ou mais centros assimétricos, de modo que os compostos podem existir em formas enantioméricas bem como diastereoméricas. Na realidade, a maioria dos compostos da presente invenção tem dois carbonos adjacentes assimétricos um ao outro e então pode existir em forma eritro ou treo, com cada uma dessas duas formas tendo enantioméricos dextrogiratórios (D) ou levogiratórios (L). Embora a forma treo seja geralmente preferida de acordo com a presente invenção para atividade analgésica, a menos que seja especificamente mencionado ao contrário, o escopo da presente invenção inclui todos os enantiômeros, diastereômeros e misturas diastereoméricas e racêmicas. À luz do acima, deve ser claramente compreendido que a designação "DL" ou "(+/-)" ou "(±)" neste pedido de patente inclui o enantiômero dextrogiratório puro, o enantiômero levogiratório puro e todas as misturas racêmicas, incluindo misturas onde os dois enantiômeros estão presentes em proporções iguais ou desiguais. Além disso, por questão de simplicidade em muitas das fórmulas estruturais, tal como no exemplo abaixo, apenas um dos enantiômeros é realmente mostrado mas quando a designação "DL" ou "(+/-)" ou "(±)" aparece ela também inclui a forma enantiomérica (imagem de espelho) da estrutura realmente mostrada na fórmula. Por exemplo:

[0027] Deste modo, no exemplo acima, apenas um enantiômero é mostrado, mas devido ao fato da designação "DL" ou "(+/-)" ou "(±)" aparecer abaixo na fórmula, seu isômero óptico
e todas as misturas racêmicas dos dois isômeros ópticos são também incluídos.
[0028] No caso de alguns compostos da presente invenção um enantiômero do treo, e em alguns casos do eritro, é significantemente mais ativo como um analgésico ou imunoestimulante do que o outro enantiômero do mesmo par. Por essa razão, o enantiômero isolado que é significantemente mais ativo do que o outro é considerado uma composição nova e inventiva mesmo se a mistura racêmica ou um enantiômero simples dos mesmos compostos tiver sido descrito na técnica anterior.
[0029] Alguns dos novos compostos da presente invenção podem conter três ou mais centros assimétricos.
[0030] Tendo os exemplos acima em mente uma pessoa versada na técnica deve compreender prontamente o escopo de cada exemplo descrito, embora em um sentido amplo todos os isômeros, enantiômeros e misturas racêmicas estejam dentro do escopo da invenção.
[0031] O termo "alquila" na descrição geral e definição dos compostos inclui grupos alquila de cadeia reta bem como cadeia ramificada.
[0032] Falando de um modo geral os compostos da invenção podem formar sais com ácidos ou bases farmaceuticamente aceitáveis, e tais sais farmaceuticamente aceitáveis dos compostos da Fórmula I estão também dentro do escopo da invenção.
[0033] Com referência agora aos novos compostos da Fórmula 1, os grupos R5 e R6 são de preferência ambos independentemente selecionados de H, alquila, alcóxi e com mais preferência ainda eles são H. Nos compostos preferidos os grupos R3 são de preferência ambos H, ou um dos grupos R3 é H e o outro é um grupo acila ou um grupo arilalquilcarbamoíla. O grupo R4 é de preferência H (mas vide a "condição" na seção Sumário) ou alcanoíla, e os grupos R1 e R2 são de preferência pirrolidino ou morfolino.
[0034] Os compostos mais preferidos da invenção são descritos com suas fórmulas estruturais na Tabela e/ou descrição a seguir, mostrando atividade de compostos exemplares relevante para sua habilidade em agir como analgésicos. Atividade Biológica, Modos de Administração
[0035] Os novos compostos da invenção têm atividade analgésica e/ou imunoestimulante. Alguns dos compostos descritos na introdução que per se são conhecidos na técnica foram verificados pelos presentes inventores ter também efeito analgésico em mamíferos. Tanto quanto é do conhecimento dos presentes inventores a atividade biológica analgésica ou imunoestimulante dos compostos conhecidos não era conhecida antes da presente constatação.
[0036] Um modelo ou ensaio aceito na técnica para medição de um efeito analgésico de um composto em dor crônica (em particular neuropatia periférica) é o modelo conhecido como Kim e Chung, 1992, Pain 150, pp. 355-363 (modelo Chung). Este modelo envolve a ligação cirúrgica dos nervos espinhais L5 (e opcionalmente o L6) em um lado em animais experimentais. Ratos se recuperando da cirurgia ganham peso e mostram um nível de atividade geral similar àquele de ratos normais. No entanto, esses ratos desenvolvem anormalidades da pata, onde a pata traseira é moderadamente evertida e os dedos são mantidos juntos. Mais importante, a pata traseira no lado afetado pela cirurgia parece ficar sensível a estímulos mecânicos de limiar baixo e vão sentir dor ao invés da leve sensação de toque. Esta sensibilidade a toque normalmente não-doloroso, chamada "alodinia tátil", se desenvolve dentro da primeira semana após cirurgia e dura pelo menos dois meses. A resposta de alodinia inclui levantamento da pata traseira afetada para escapar do estímulo, lambida da pata e manutenção dela no ar por muitos segundos. Nenhuma dessas respostas é normalmente vista no grupo de controle.
[0037] Para produzir a alodinia tátil, os ratos são anestesiados antes da cirurgia. O local cirúrgico é depilado e preparado ou com betadina ou Novacaína. Incisão é feita a partir da XIII vértebra toráxica para baixo em direção ao sacro. O tecido muscular é separado da vértebra espinhal (lado esquerdo) nos níveis de L4-S2. A vértebra L6 é localizada e o processo transverso é cuidadosamente removido com uma pinça saca-bocado pequena para expor os nervos espinhais L4-L6. Os nervos espinais L5 e L6 são isolados e firmemente ligados com fio de seda 6-0. O mesmo procedimento é feito no lado direito como um controle, exceto que nenhuma ligação dos nervos espinhais é realizada.
[0038] Após uma hemostase completa ser confirmada, os ferimentos são suturados. Uma pequena quantidade de ungüento antibiótico é aplicada à área que sofreu incisão, e o rato é transferido para a gaiola plástica de recuperação sob uma lâmpada de temperatura de calor regulada.
[0039] No dia do experimento, pelo menos sete dias após a cirurgia, tipicamente seis ratos por grupo de teste são administrados com os fármacos de teste através de injeção intraperitoneal (i.p.) ou gavagem oral (p.o.). Para administração i.p., os compostos são formulados em H2O e dados em um volume de 1 ml/kg de peso do corpo através de injeção na cavidade intraperitoneal. Para administração p.o., os compostos são formulados em H2O e dados em um volume de 1 mg/kg de peso do corpo usando uma agulha de gavagem de 7,62 cm (3 polegadas), 18 gauge, que é lentamente inserida através do esôfago no estômago.
[0040] Alodinia tátil é avaliada através de pêlos von Frey, que são uma série de pêlos finos com diferenças grandes na rigidez. Os ratos são postos em uma gaiola plástica com um fundo de malha de arame e deixados aclimatar por aproximadamente 30 minutos. Para estabelecer a linha de base de pré-fármaco, os pêlos von Frey são aplicados perpendicularmente através da malha à região médio-plantar da pata traseira do rato com força suficiente para causar leve encurvamento e mantidos por 6-8 segundos. A força aplicada foi calculada para faixa de a partir de 0,41 a 15,1 gramas. Se a pata for retirada claramente, ela é considerada uma reposta positiva. Um animal normal não vai responder a estímulos nesta faixa, mas uma pata cirurgicamente ligada será retirada em resposta a um pêlo de 1-2 gramas. O limiar de 50% de retirada de pata é determinado usando o método de Dixon, W.J., Ann. Rev. Pharmacol. Toxicol., 20:441-462 (1980) aqui incorporado a título de referência. Alodinia tátil é medida antes e 15, 30 e 60 minutos após administração do fármaco. O limiar pós-fármaco é comparado com o limitar pré-fármaco e a reversão percentual de sensibilidade tátil é calculada com base em um limiar normal de 15,1 gramas.
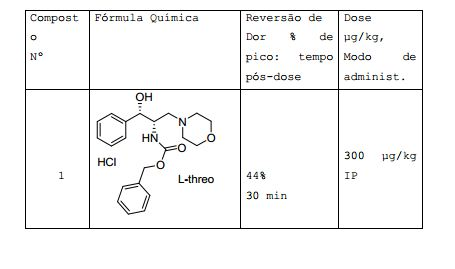
[0041] A Tabela 1 abaixo indica o grau de reversão de dor obtido no modelo Chung com compostos exemplares da invenção. A administração intraperitoneal (i.p.) e/ou intravenosa (i.v.) dos compostos era em doses variando de 1 μg/kg a 300 μg/kg ou 3 mg/kg P.O. e a porcentagem de pico da reversão de alodinia foi medida a 15, 30 ou 60 minutos após administração, como é indicado na tabela. Os dados são expressos como a % mais alta de reversão de alodinia (de 3 momentos: 15 min., 30 min. ou 60 min. pós-fármaco) com um mínimo de reversão de alodinia de 20% no modelo Chung de rato. Comparações entre os grupos (tratado com fármaco vs. solução salina) foram feitas usando um teste t não-emparelhado, de 2 amostras, bilateral. Os compostos que não são mostrados são os que não eram estatisticamente analgésicos seguindo uma dose I.P. de 300 ug/kg, mas podem ainda ser analgésicos. Os compostos que não exibem analgesia significante a 100 mg/kg não são considerados ser analgésicos. Tabela 1
[0042] Um método aceito na técnica para medição de imunoestimulação compreende administração sistêmica de compostos para ensaiar a habilidade em estimular o sistema imune, possivelmente devido à supra-regulagem não-específica do sistema hemolinforreticular. Esta supra-regulagem poderia resultar em números altos de linfócitos de ambas linhagens de célula T e B. Embora a requerente não deseje ser limitada à teoria biológica da estimulação imune, a eficácia imunoestimuladora real dos compostos pode ser demonstrada in vivo através da avaliação do tamanho do baço em resposta à administração do composto de teste a ratos de espécie de teste de laboratório. Falando de um modo geral qualquer composto que exiba aumento do baço seguindo dosagem de 200 mg/kg ou menos pode ser considerado um imunoestimulante.
Modos de Administração
[0043] Os compostos da presente invenção podem ser administrados em dosagens farmaceuticamente eficazes. Tais dosagens são normalmente a dose mínima necessária para atingir o efeito terapêutico desejado; no tratamento de dor crônica, esta quantidade seria mais ou menos aquela necessária para reduzir o desconforto causado pela dor para níveis toleráveis. Para adultos humanos tais doses geralmente estarão na faixa de 0,1-5000 mg/dia; com mais preferência na faixa de 1 a 3000 mg/kg, com mais preferência ainda na faixa de 10 mg a 1000 mg/dia. No entanto, a quantidade real do composto a ser administrada em qualquer caso dado será determinada por um médico levando em consideração as circunstâncias relevantes, tal como a severidade da dor, a idade e o peso do paciente, a condição física geral do paciente, a causa da dor e a através de de administração.
[0044] Os compostos são úteis no tratamento de dor em um mamífero; particularmente um ser humano. De preferência, o paciente receberá o composto oralmente em qualquer forma aceitável, tal como um comprimido, líquido, cápsula, pó e similar. No entanto, outras vias podem ser desejáveis ou necessárias, particularmente se o paciente sofrer de náusea. Tais outras vias podem incluir, sem exceção, modos de aplicação transdermal, intraperitoneal, parenteral, subcutânea, intranasal, intratecal, intramuscular, intravenosa e intratecal. Um outro aspecto da invenção refere-se a composições terapêuticas compreendendo os novos compostos da presente invenção e os sais farmaceuticamente aceitáveis desses compostos e um excipiente farmaceuticamente aceitável. Tal excipiente pode ser um veículo ou um diluente; este é geralmente misturado com o composto ativo ou deixado diluir ou confinar o composto ativo. Se um diluente, o veículo pode ser material sólido, semi-sólido ou líquido que age como um excipiente ou veículo para o composto ativo. As formulações podem também incluir agentes umectantes, agentes emulsificantes, agentes de preservação, agentes adoçantes e/ou agentes aromatizantes. Se usada como em um formato oftálmico ou de infusão, a formulação conterá geralmente um ou mais sais para influenciar a pressão osmótica da formulação.
[0045] Em um outro aspecto, a invenção refere-se a métodos para o tratamento de dor, particularmente dor crônica, através da administração de um ou mais dos compostos novos ou de outro modo conhecidos da invenção ou de seus sais farmaceuticamente aceitáveis a um mamífero com necessidade. Conforme acima indicado, o composto será geralmente formulado em uma forma de acordo com o modo de aplicação desejado.
[0046] Os compostos da invenção que são imunoestimulantes são administrados ao indivíduo para os mesmos princípios básicos que os compostos tendo atividade analgésica, em doses que são melhor determinadas em um caso-para-caso e/ou espécie-para- espécie e, no caso de seres humanos, às vezes em uma base de paciente-para-paciente. Falando de um modo geral a dose eficaz estará na faixa de 10 μg/kg a 200 mg/kg. Métodos Sintéticos para Obtenção dos Compostos da Invenção, Experimentais
[0047] O composto da invenção pode ser sintetizado utilizando os métodos sintéticos descritos no experimento abaixo ou tais modificações dos métodos experimentais descritos abaixo que serão imediatamente aparentes àqueles versados na técnica à luz da presente descrição. Geral
[0048] Espectros de 1H RMN foram registrados em temperatura ambiente com um espectrômetro Avance 300 (Bruker). Os compostos foram analisados através de cromatografia líquida de alto desempenho de fase reversa (HPLC) usando um Waters Autopurification System equipado com uma Bomba Waters 2525, um detector de disposição de diodo Waters 2696 e uma coluna Xterra (Parte N° 186000482, 5 μm, C18, 4,5 x 50 mm).
[0049] O método de HPLC usado era um gradiente de 5% de solvente B a 100% em, 7 minutos. Solvente A era H2O com 0,05% de TFA e solvente B era CH3CN com 0,05% de TFA (Método A).
[0050] Os pontos de fusão foram medidos com um aparelho de ponto de fusão Büchi B-545 e eram não- corrigidos. Para isolar produtos de reação o solvente foi removido através de evaporação usando um evaporador giratório a vácuo, a temperatura do banho de água não excedendo 40°C. Vias Sintéticas Gerais
[0051] O composto da invenção pode ser sintetizado utilizando os métodos sintéticos descritos em um sentido geral imediatamente abaixo e em mais detalhes na seção experimental do presente pedido de patente ou através de tais modificações dos métodos geral e experimental descritos abaixo que serão prontamente aparentes àqueles versados na técnica à luz da presente descrição.
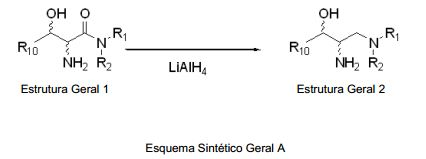
[0052] Uma através de sintética geral para o composto da presente invenção que são "1-hidroxil-propil- aminas" substituídas pode levar à síntese dos compostos "3-hidroxil-propil amida" substituídos correspondentes, seguido pela redução do grupo carbonila da porção "amida do ácido carboxílico" com um agente de redução tal como hidreto de alumínio lítio ou agente de redução similar.
[0053] Esta reação é ilustrada no Esquema Sintético Geral A, onde, falando de um modo geral, as variáveis têm o significado descrito no Seção Sumário do presente pedido de patente. Uma pessoa versada na técnica de síntese orgânica vai não obstante compreender imediatamente que dependendo da natureza dos substituintes designados R1, R2 e R10, certos grupos podem necessitar de proteção para o desempenho da etapa de redução.
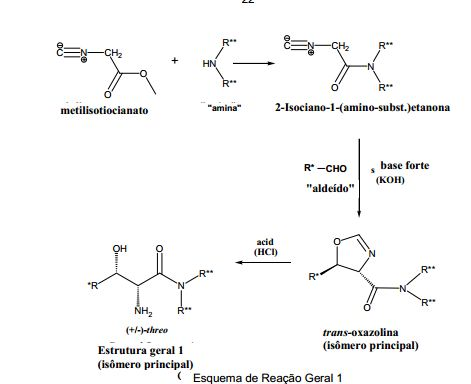
[0054] Os compostos "3-hidroxil-propil amida" substituídos podem, falando de um modo geral, ser sintetizados conforme descrito abaixo no Esquema de Reação Geral 1 e no Esquema de Reação Geral 2.
[0055] Deste modo, de acordo com o Esquema Geral isocianoacetato de metila (ou etil isocianoacetato comercialmente disponível) é reagido com uma "amina" que inclui os grupos R1 e R2 para prover o derivado amida do ácido 2-isocianoacético mostrado no Esquema de Reação Geral 1. Exemplos típicos para as aminas usadas na reação são pirrolidina, piperidina, azetidina, morfolina, 2,5- diidro-1H-pirrol, dialquilaminas tal como dietilamina, pirrolidinas 3-flúor-, 3,3-diflúor- ou 3-hidróxi- substituídas. O derivado amida do ácido 2-isocianoacético é então reagido em metanol na presença de base (tal como KOH) com um "aldeído" que inclui o grupo R10 para prover uma trans "oxazolina" com alta diastereosseletividade (razões trans:cis geralmente >97:3) conforme mostrado no Esquema de Reação Geral 1. A trans oxazolina é então tratada em metanol com um ácido forte, tal como HCl, para abrir o anel e prover os intermediários amida do ácido 3- hidróxi-2-amina-propiônico treo-3-substituído (com razões treo:eritro geralmente >97:3) conforme mostrado no Esquema de Reação Geral 1.
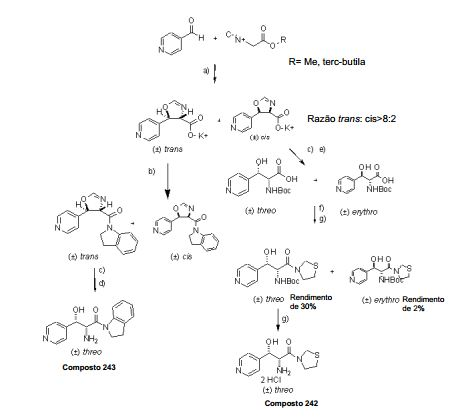
[0056] Os compostos da Fórmula 1 e/ou da Estrutura Geral 1, onde o grupo amina da fórmula NHR1R2 é um nucleófilo mais fraco, tal como indolina, tiomorfolina e similar, podem ser feitos conforme ilustrado no Esquema de Reação 2 para a síntese de compostos intermediários bicloridrato de (±)-treo-2-amina-3-hidróxi-1-(indolin-1- il)-3-(piridin-4-il)propan-1-ona Composto 243 e bicloridrato de (±)-treo-2-amina-3-hidróxi-1-(tiazolidin- 3-il)-3-(piridin-4-il)propan-1-ona Composto 242.
[0057] No Esquema de Reação 2, EDCl significa cloridrato de 1-(3-dimetilaminopropil)-etilcarbodiimida; HOBT significa 1-hidroxibenzotriazol; BOC2O significa di- t-butil-dicarbonato e TEA significa trietilamina. Os compostos 242 e 243 podem ser reduzidos, conforme ilustrado no Esquema Sintético Geral A para prover compostos da invenção.
[0058] Uma outra através de sintética geral pode seguir em termos gerais a síntese do Composto 1, Composto 2 e Composto 3, especificamente descrita em detalhes na seção experimental abaixo, modificada com tais modificações que à luz da presente invenção serão prontamente aparentes a uma pessoa versada na técnica.
[0059] Compostos isomericamente puros e/ou enantiomericamente puros e derivados adicionais dos intermediários amida do ácido 3-hidróxi-2-amina-propiônco 3-substituído ou das 1-hidróxi propilaminas substituídas da invenção são obtidos através de técnicas de separação e reações que, per se, são bem-conhecidas da química sintética. Algumas das técnicas de separação e reações típicas são geralmente descritas abaixo.
[0060] Separação de isômeros treo e eritro, quando ambos são formados nas reações levando aos compostos da invenção, pode ser tipicamente realizada através de métodos cromatográficos. A separação cromatográfica pode acontecer no nível dos compostos intermediários da amida do ácido 3-hidroxil-propiônico ou no nível dos compostos 1-hidroxil propil amina substituídos da invenção.
[0061] Os isômeros treo mais abundantemente formados podem ser também convertidos nos isômeros eritro através da oxidação para o nível cetona do grupo hidroxila na posição 3 da porção de ácido propanóico e subseqüentemente redução da cetona resultante para o nível hidroxila nos compostos amida do ácido 3-hidróxi-2- amina-propiônico 3-substituídos ou nos compostos da invenção.
[0062] Separação de misturas enantioméricas pode ser realizada em colunas Chiralpack que são bem- conhecidas na técnica.
[0063] A função amina na posição 2 da porção propil amina é, falando de um modo geral, mais reativa com relação à acilação e carbamoilação do que o grupo hidroxila na posição 1. Deste modo, derivados acilados da função 2-amina podem ser preparados usando cloreto de acila tal como cloreto de acetila e cloreto de hexanoíla. Ou os grupos 1-hidróxi e 2-amina dos compostos da invenção podem ser acilados na mesma reação. Derivados carbamato da função 2-amina podem ser obtidos usando cloroformiatos, tal como benzilcloroformiato. Uma função butil carbamoíla terciária ou função benzil carbamoíla pode também servir como um grupo de proteção removível da função 2-amina.
[0064] Alquilação da função 2-amina pode ser realizada através da condensação do composto carregando o grupo 2-NH2 com um aldeído para se obter um intermediário de base Schiff que pode ser reduzido, sem isolamento, para prover os compostos N-alquil, arilalquil ou heteroaril-alquila da invenção. Descrição Detalhada da Síntese de Compostos Preferidos (Experimental) Preparação de bicloridrato de D-treo-2-amina-3-morfolino- 1-fenilpropan-1-ol Composto 4 1-((S)-1-feniletil)aziridino-2-carboxilato de (R)-metila EBE 06044B.
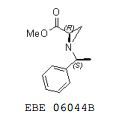
[0065] À solução de 2,3-dibromopropionato de metila (25 mL, 198 mmols) em tolueno a 5°C foi adicionada trietilamina (55 mL, 0,39 mmol) em tolueno (100 mL). Após agitar por 5 minutos (S)-(1)-feniletilamina (25 mL, 198 mmols) em tolueno (100 mL) foi adicionada em gotas. A suspensão foi refluxada por 3 horas e deixada esfriar, filtrada e os voláteis foram evaporados sob pressão reduzida para dar um resíduo que foi purificado através de cromatografia de coluna (950 g de sílica-gel) com um gradiente de 0-20% de EtOAc em cicloexano para dar 1- ((S)-1-feniletil)aziridino-2-carboxilato de (S)-metila EBE 06044A como um óleo amarelo (17,31 g, 43% de rendimento) e 1-((S)-1-feniletil)aziridino-2-carboxilato de (R)-metila EBE 06044B como um óleo amarelo (15,14 g, 37% de rendimento).
[0066] PM: 205,3; Rendimento de EBE 06044B: 37%; Óleo Amarelo: EBE 06044A: 43%, Óleo Amarelo.
[0067] Rf: EBE 06044A = 0,5; Rf: EBE 06044B = 0,35 (EtOAc:cicloexano = 25:75).
[0068] 1H-RMN (CDC13, δ) EBE 06044A: 1,47 (d, 3H, J = 6,6 Hz, CH3), 1,60 (d, 1H, J = 6,4 Hz, CH), 2,13 (d, 1H, J = 2,6 Hz), 2,21 (dd, 1H, J = 3,2 Hz, J = 6,4Hz), 2,54 (q, 1H, J = 6,6 Hz), 3,75 (s, 3H, OCH3) 7,23-7,40 (m, 5H, ArH).
[0069] 1H-RMN (CDC13, δ) EBE 06044B: 1,46 (d, 3H, J = 6,6 Hz, CH3), 1,79 (d, 1H, J = 6,6 Hz, CH), 2,08 (d, 1H, J = 3,1 1Hz, 6,6 Hz)5 2,34 (dd, 1H, J = 3,1 Hz, J = 1,0 Hz), 2,56 (q, 1H, J = 6,6 Hz), 3,67 (s, 3H, OCH3), 7,24- 7,36 (m, 5H, ArH).
[0070] 13C-RMN (CDC13, δ) EBE 06044B: 23,5, 35,0, 36,9, 52,2, 69,8, 126,5, 127,2, 128,5, 143,6, 171,1.
[0071] HPLC: Método A, detecção a 254 nm, EBE 06044B TA = 6,11 min, área de pico 92,9%.
[0072] ((R)-1-((S)-1-Feni1eti1)aziridin-2- i1)metano1 EBE 06046.
[0073] Um frasco de fundo redondo de 250 mL foi carregado com THF anidro (100 mL) e LiAlH4 (2,77 g, 73,1 mmols). Enquanto a suspensão é agitada a 0°C, uma solução de 1-((S)-1-feniletil)aziridino-2-carboxilato de (S)- metila EBE 06044B (10,0 g, 48,7 mmols) em THF (50 mL) foi adicionada em gotas durante 20 minutos. O funil de gotejamento foi lavado com THF (2 x 3 mL) e deixado reagir 20 minutos a 0°C. Mantendo a mistura de reação a 0°C, uma solução de KOH (10%, 20 mL) foi adicionada em gotas por 20 minutos (cuidado a reação é exotérmica). A mistura foi agitada por 0,5 hora a 25°C e o precipitado branco removido através de filtragem em uma almofada de celite que foi lavada com éter de dietila (30 mL). Os filtrados orgânicos combinados foram lavados com NaH2PO4 e a camada aquosa foi extraída com Et2O (3 x 30 mL). A fase orgânica combinada foi seca com Na2SO4 e concentrada para dar ((R)-1-((S)-1-feniletil)aziridin-2-il)metanol EBE 06046 como um sólido branco (10,4 g, 90% de rendimento).
[0074] PM: 177,2; Rendimento: 90%; Sólido branco; Pf (oC): 37,7.
[0075] 1H-RMN (CDC13, δ): 1,43 (d, 3H, J = 6,6 Hz, CH3), 1,49 (d, 1H, J = 6,5 Hz, CH), 1,65-1,71 (m, 1H, CH), 1,92 (d, 1H, J = 3,5 Hz, NCH), 2,26 (s, 1H, OH), 2,53 (q, 1H, J = 6,6 Hz, NCH), 3,32-3,37 (m, 1H, OCH2), 3,56 (m, 1H, OCH2), 7,23-7,35 (m, 5H, ArH).
[0076] 13C-RMN (CDC13, δ): 22,9, 31,4, 39,3, 62,5, 69,4, 126,6, 127,3, 128,6, 144,5.
[0077] (R)-1-((S)-1-Feniletil)aziridino-2- carbaldeído EBE 06048
[0078] Um frasco de fundo redondo de 250 mL, três gargalos, foi equipado com um termômetro de baixa temperatura e dois (2) funis de gotejamento de equalização. Um desses foi conectado a uma linha de nitrogênio e carregado com uma solução de ((R)-1-((S)-1- feniletil)aziridin-2-il)metanol EBE 06046 (7,0 g, 39,5 mmols) em CH2Cl2 (75 mL), o outro foi carregado com uma solução de DMSO (9,25 g, 118,5 mmols) em CH2Cl2 (11 mL). A uma solução de cloreto de oxalila (7,5 g, 59,3 mmols) em CH2Cl2 (90 mL) sob N2 a -78°C, a solução de DMSO foi adicionada em gotas durante 20 minutos e agitada por 20 minutos. EB 06046 (7,0 g, 39,5 mmols) em CH2Cl2 (75 mL) foi adicionado em gotas durante 50 minutos, então o funil de gotejamento foi carregado com DIEA (42,6 mL, 237 mmols) em CH2Cl2 (10 mL) e a mistura de reação foi agitada por 30 minutos a -45°C. A solução de DIEA foi adicionada durante 5 minutos com a mistura de reação a - 78°C e a reação foi deixada aquecer para temperatura ambiente. A mistura de reação foi lavada com H2O (3 x 50 mL), seca em MgSO4, filtrada e evaporada. O produto bruto obtido foi purificado através de cromatografia de coluna em sílica com um gradiente de 0-20% [v/v] de EtOAc em cicloexano para dar (R)-1-((S)-1-feniletil)aziridino-2- carbaldeído EBE 06048 como um óleo amarelo (5,59 g, 81% de rendimento).

[0079] PM: 175,2; Rendimento: 81%; Óleo Amarelo.
[0080] R7: EBE 06048: 0,3 (EtOAc:cicloexano = 20:80),
[0081] 1H-RMN (CDC13, δ): 1,47 (d, 3H, J = 6,6 Hz, CH3), 1,94 (d, 1H, J = 6,7 Hz, NCH2), 2,08 (dt, J = 2,9 Hz, J = 6,4 Hz, NCH), 2,37 (d, 1H, J = 2,6 Hz, NCH2), 2,61 (q, 1H, J = 6,6 Hz, NCH), 7,20-7,38 (m, 5H, ArH), 8,92 (d, 1H, J = 6,2 Hz).
[0082] 13C-RMN (CDC13, δ): 22,7, 32,1, 43,2, 68,1, 125,5, 126,5, 127,6, 142,4, 198,7.
[0083] (R)-Feni1((R)-1-((S)-1-feni1eti1)aziridin- 2-i1)metano1 EBE06066.
[0084] A uma so1ução de bromobenzeno (4,93 g, 31,4 mmo1s) em THF 125 mL sob nitrogênio a -78°C foi adicionado t-BuLi (1,7M em pentano, 50 mL). A mistura foi agitada por 0,5 hora em temperatura ambiente. A mistura foi resfriada para -78o e uma so1ução de (R)-1-((S)-1- feni1eti1)aziridino-2-carba1deído EBE 06048 (2,5 g, 14,3 mmo1s) em THF (16,7 mmo1s) a -78°C foi adicionada em gotas. A mistura de reação foi tratada com H2O (20 mL), a camada orgânica foi separada e a fase aquosa foi extraída com EtOAc. As camada orgânicas combinadas foram secas em MgSO4, fi1tradas e concentradas in vacuo para dar um resíduo que foi purificado através de cromatografia de co1una usando um gradiente de 0-20% [v/v] de EtOAc em cic1oexano para dar (R)-feni1-((R)-1-((S)-1- feni1eti1)aziridin-2-i1)-metano1 EBE 06066 (3,12 g, 86% de rendimento).
[0085] PM: 253,3; Rendimento: 86%.
[0086] Rf = 0,3 (EtOA:cicloexano = 20:80).
[0087] 1H-RMN (CDC13, δ): 1,47 (d, 3H, J = 6,6 Hz, CH3), 1,57 (d, 1H, J = 6,5 Hz, CH), 1,79 (dt, 1H, J = 3,5 Hz, J = 8,7 Hz, CH), 2,04 (d, 1H, J = 3,5 Hz, OCH), 2,35 (s amp1o, 1H, OH), 2,53 (q, 1H, J = 6,5 Hz, CH), 4,23 (d, 1H, J = 5,7 Hz, OCH), 7,07-7,13 (m, 2H, ArH), 7,16-7,20 (m, 3H, ArH), 7,24- 7,34 (m, 5H, ArH).
[0088] 13C-RMN (CDC13, δ): 22,4, 32,0, 44,6, 69,4, 74,1, 125,8(2xC), 126,9 (2xC), 127,3, 127,6, 128,2 (2xC), 128,7 (2xC), 142,0, 144,2.
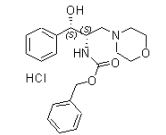
[0089] [α]22D = -71,53 (c = 0,59, CHC13). Bicloridrato de D-treo-2- ( (S) -1-feniletilamino) -3- morfolino-1-fenilpropan-1-ol Composto 5
[0090] A uma so1ução de (R)-feni1((R)-1-((S)-1- feniletil)aziridin-2-il)metanol EBE 06066 (1,5 g, 5,92 mmols) em CH2CN (19 mL) em temperatura ambiente foi adicionado iodotrimetilsilano (3,55 g, 17,8 mmols). A solução foi agitada por 2 horas e morfolina (1,032 g, 11,84 mmols) foi adicionada. Após 2 horas em refluxo, a mistura de reação foi tratada com HCl (1M) para atingir pH = 1 e agitada por 10 minutos. Após uma adição lenta de NaHCO3 para atingir pH = 9, o produto foi extraído com EtOAc, seco em Na2SO4, filtrado para dar após evaporação um óleo marrom bruto que foi purificado através de cromatografia de coluna usando um gradiente de 0-20% [v/v] de MeOH em EtOAc para dar D-treo-2-((S)-1- feniletilamino)-3-morfolino-1-fenilpropan-1-ol EBE 06068A (0,831 g, 42%) como um sólido marrom pálido. A uma solução de D-treo-2-((S)-1-feniletilamino)-3-morfolino-1- fenilpropan-1-ol EBE06068A (0,100 g, 0,294 mmol) em etanol (1 mL) foi adicionada uma solução de HCl (0,8 M, 0,816 mL) em EtOH. Evaporação dos voláteis deu bicloridrato de D-treo-2-((S)-1-feniletilamino)-3- morfolino-1-fenilpropan-1-ol Composto 5 como um sólido branco (0,125 g, 100%).
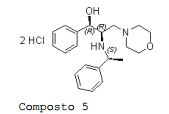
[0091] PM: 412,37; Rendimento: 42%; Sólido branco; Pf (oC): 157,2 (dec).
[0092] Rf: 0,3 (MeOH:EtOAc = 20:80) EBE 06068 A,
[0093] 1H-RMN (CD3OD, δ): 1,19 (t, 2H, J = 7,0 Hz, NCH2), 1,71 (d, 3H, J = 6,8 Hz, CH3), 3,45 (m, 2H, J = 7,1 Hz, NCH2), 3,62 (q, 2H, J = 7,1 Hz, N-CH2), 3,97 (t, 4H, J = 4,5 Hz, OCH2), 4,06 (m, 1H, CH-N), 4,75 (q, 1H, J = 6,8 Hz, CH-N), 5,21 (d, 1H, J = 5,1 Hz, CH-O), 7,44-7,56 (m, 10H, ArH).
[0094] EM-ESI m/z (% Int. rel.): 341,1 ([MH]+, 20).
[0095] 13C-RMN (CD3OD, δ): 24,4, 54,5 (2xC), 55,5, 55,9, 60,0, 67,0 (2xC), 75,6, 126,3 (2xC), 126,5 (2xC), 127,0, 127,1, 128,1 (2xC), 128,5 (2xC), 142,2, 145,3.
[0096] HPLC: Método A, detecção a 254 nm, Composto 5 TA = 4,41 min, área de pico 99%. Bicloridrato de treo-2-amina-3-:morfolino-1-fenilpropan-1- ol Composto 4.
[0097] A uma solução de D-treo-2-((S)-1- feniletilamino)-3-morfolino-1-fenilpropan-1-ol EBE 06068A (0,400 g, 1,17 mmol) em MeOH (6 mL) em temperatura ambiente foi adicionado ácido acético (0,133 mL, 2,35 mmols). O recipiente de reação foi refluxado com nitrogênio e Pd(OH)2 (25% em peso, 0,150 g) foi adicionado. A atmosfera de nitrogênio foi trocada com hidrogênio usando três ciclos de adição de vácuo e hidrogênio usando um balão de hidrogênio. Após agitar por 16 horas sob hidrogênio a mistura de reação foi filtrada em celite para dar EBE 06070A o sal de acetato de (2R)- amina-3-morfolin-4-il-(1R)-fenil-propan-1-ol (0,279 g, 98% de rendimento). A uma solução de EBE 06070A o sal de acetato de (2R)-amina-3-morfolin-4-il-(1R)-fenil-propan- 1-ol (0,100 g, 0,338 mmol) em etanol (1 mL) foi adicionada uma solução de HCl (0,8 M, 0,930 mL) em EtOH. Evaporação dos voláteis deu o bicloridrato de D-treo-2- amina-3-morfolino-1-fenilpropan-1-ol Composto 4 (0,104 g, 100% de rendimento) como um sólido esbranquiçado. (Adaptado de Shin, S-H.; Han, E.Y.; Park, C.S.; Lee, W.K.; Ha, H.J., Tetrahedron Asymmetry, 2000, 11, 32933301).

[0098] PM: 309,23; Rendimento: 99%; Sólido Branco Amarelado; Pf (oC): 183,4.
[0099] 1H-RMN (CD3OD, δ): 3,30-3,77 (m, 6H, CH2N), 3,92-4,05 (m, 4H, CH2O), 4,05-4,16 (m, 1H, CH), 4,85-4,98 (m, 1H, CH), 7,35-7,60 (m, 5H, ArH).
[00100] 13C-RMN (CD3OD, δ): 53,2, 58,3, 58,5 (2xC), 64,9 (2xC), 72,6, 128,0 (2xC), 130,2 (2xC), 140,3.
[00101] EM-ESI m/z (% int. rel.): 237,1 (100, [MH]+).
[00102] HPLC: Isocrático CH3CN 10% em H2O (pH 10, [NH4OH] = 5 mM), detecção UV 254 nm, Composto 4 TA = 6,63 min, área de pico 97,3%.
[00103] [a]22D = -10,7 (c = 1,00, MeOH).
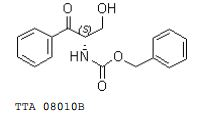
[00104] Preparação de Cloridrato L-treo-1-hidróxi- 3-morfolino-1-fenilpropan-2-ilcarbamato de benzila Composto 1 (S)-3-hidróxi-1-oxo-1-fenilpropan-2-ilcarbamato de benzila TTA 08010B.
[00105] A uma solução agitada de Z-L-Ser-OH (6,00 g, 25,08 mmols) em 32 mL de THF anidro a 0°C sob nitrogênio foi adicionado em gotas brometo de fenilmagnésio a 1M em THF (32 mL, 200 mmols). (O símbolo Z significa um grupo benzilcarbamoíla). A mistura foi agitada 15 horas em temperatura ambiente sob nitrogênio. Uma solução de HCl a 2M (100 mL) foi lentamente adicionada a 0°C e a mistura foi dividida entre acetato de etila (750 mL) e água ácida. A camada orgânica foi lavada com água (2 x 20 mL), bicarbonato de sódio aquoso 1N (2 x 20 mL), salmoura (2 x 20 mL) e seca em MgSO4. Após remoção do acetato de etila através de evaporação a 30-35°C, o produto bruto (4,50 g, 60% de rendimento) foi cristalizado em uma mistura de acetato de etila:hexano = 25 mL:20 mL para dar (S)-3-hidróxi-1-oxo-1-fenilpropan-2- ilcarbamato de benzila TTA 08010B como um sólido branco (1,40 g, 20% de rendimento).
[00106] PM: 299,32; Rendimento: 20%; Sólido branco; Pf (oC): 106,5.
[00107] Rf: 0,75 (CH2Cl2:MeOH = 9:1).
[00108] 1H-RMN (CDC13, δ): 2,78 (s, 1H, OH), 3,85-3,93 (m, 1H, CH2O), 4,00-4,09 (m, 1H, CH2O), 5,14 (s, 2H, ArCH2O), 5,40 (t, 1H, J = 3,3 Hz, CH), 6,17 (d, 1H, J = 6,4 Hz, NH), 7,35 (s, 5H, ArH), 7,49 (t, 2H, J = 7,60 Hz, ArH), 7,62 (t, 1H, J = 7,1 Hz, ArH), 8,99 (t, 2H, J = 7,6 Hz, ArH).
[00109] 13C-RMN (CDC13, δ): 58,3, 64,6, 67,3, 128,1, 128,3, 128,6, 128,7, 129,0, 134,1, 136,0, 156,6, 196,6.
[00110] EM-ESI m/z (% Int. re1.): 300,1 ([MH]+, 5), 256,1 (100).
[00111] HPLC: Método A, detecção UV 254 nm, TTA 08010B TA = 5,40 min, área de pico 98,5%,
[00112] [α]22D = - 5,8 (c = 1,00, MeOH). L-treo-1,3-diidróxi-1-fenilpropan-2-ilcarbamato de benzila TTA 08012
[00113] A uma so1ução agitada de (S)-3-hidróxi-1- oxo-1-fenilpropan-2-ilcarbamato de benzila TTA 08010B (1,40 g, 4,70 mmols) em 28 mL de THF anidro a -78°C sob nitrogênio foi adicionado lentamente em gotas DIBAL-H 1M em hexano (18,8 mL, 18,80 mmols). A mistura foi agitada 2 horas a -78°C então 1,5 hora em temperatura ambiente. Uma solução de HCl a 2M (35 mL) foi lentamente adicionada a - 20°C e a mistura foi dividida entre acetato de etila (750 mL) e água ácida. A fase orgânica foi lavada com água (2x20 mL), salmoura (2x20 mL) e seca em MgSO4. Após remoção do acetato de etila através de evaporação a 30-35°C, o produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH = 98:2 a 97:3) para dar L-treo-1,3-diidróxi-1-fenilpropan-2- ilcarbamato de benzila TTA 08012 como um sólido branco (1,10 g, 78% de rendimento).
[00114] PM: 301,34; Rendimento: 78%; Sólido branco; Pf (oC): 102,5.
[00115] Rf: 0,30 (CH2Cl2:Me0H = 95/5).
[00116] 1H-RMN (CDC13, δ): 3,08 (t, 1H, J = 5,0 Hz, OH), 3,59 (d, 1H, J = 3,1 Hz, OH), 3,64-3,78 (m, 2H, CH2O), 3,80-3,89 (m, 1H, CH), 4,95 (s, 2H, ArCH2O), 5,57 (d, 1H, J = 8,3 Hz, NH), 7,17-7,38 (m, 10H, ArH).
[00117] 13C-RMN (CDC13, δ): 57,5, 63,6, 66,9, 73,8, 126,0, 127,8, 127,9, 128,1, 128,5, 128,6, 136,2, 141,0, 156,9.
[00118] EM-ESI m/z (% Int. re1.): 302,0 ([MH]+, 5); 132,0 (100).
[00119] HPLC: Método A, detecção UV 254 nm, TTA 08012 TA = 5,00 min, área de pico 99,5%.
[00120] [α]22D = +39,4 (c=1,00, MeOH). Cloridrato treo-1-hidróxi-3-:morfolino-1-fenilpropan-2- ilcarbamato de benzila Composto 1.
[00121] A uma so1ução agitada de L-treo-1,3- diidróxi-1-fenilpropan-2-ilcarbamato de benzila TTA 08012 (1,00 g, 3,30 mmols) em 13 mL de piridina a -10°C foi adicionado em gotas cloreto de metanossulfonila (0,27 mL, 3,50 mmols). A mistura foi agitada 6 horas a 20°C sob nitrogênio. Piridina foi removida através de evaporação a 30-35°C e o resíduo foi dividido entre acetato de etila (250 mL) e HCl a 0,1N (20 mL). A fase orgânica foi lavada com água (20 mL), salmoura (20 mL), seca em MgSO4 e evaporada para dar após secagem L-treo-1-hidróxi-3- metanossulfonil-1-fenilpropan-2-ilcarbamato TTA 08014 (1,25 g, 65% de rendimento).
[00122] A uma solução agitada de L-treo-1-hidróxi- 3-metanossulfonil-1-fenilpropan-2-ilcarbamato de benzila bruto TTA 08014 (1,25 g, 3,30 mmols) em 6 mL de DMF em temperatura ambiente foi adicionada morfolina (1,2 mL, 13,20 mmols). A mistura foi agitada 15 horas a 50°C sob nitrogênio. DMF foi evaporado e o resíduo foi dividido entre acetato de etila (250 mL) e bicarbonato de sódio aquoso a 1N (20 mL). A fase orgânica foi lavada com água (20 mL), salmoura (20 mL) e seca em MgSO4. Após evaporação o produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH = 98:2 a 97:3) para dar L-treo-1-hidróxi-3-morfolino-1- fenilpropan-2-ilcarbamato de benzila como um óleo (380 mg, 31% de rendimento). O sal de cloridrato foi obtido a partir de 100 mg da base livre em dietiléter a 0°C usando uma solução de HCl a 0,3M em dietiléter. O precipitado foi filtrado e seco para dar cloridrato de L-treo-1- hidróxi-3-morfolino-1-fenilpropan-2-ilcarbamato de benzila Composto 1 como um sólido branco (70 mg, 65% de rendimento).

[00123] PM: 406,90; Rendimento: 20%; Sólido branco; Pf (oC): 144,5.
[00124] Rf: 0,40 (CH2Cl2:MeOH = 95:5),
[00125] 1H-RMN (CD3OD, δ): 3,14-3,77 (m, 6H, CH2N), 3,70-4,07 (m, 4H, CH2O), 4,30-4,33 (m, 1H, CH), 4,90-5,06 (m, 3H, CH, ArCH2O), 7,20-7,43 (m, 1OH, ArH).
[00126] 13C-RMN (CD3OD, δ): 51,2, 51,8, 53,2, 59,3, 63,2, 66,3, 72,5, 125,8, 127,2, 127,3, 127,5, 127,8, 127,9.
[00127] EM-ESI m/z (% Int. rel.): 371,0 ([MH]+, 100).
[00128] HPLC: Método A, detecção UV 254 nm, Composto 1 TA = 4,40 min, área de pico 96,5%,
[00129] [α]22D = +13,9 (c = 1,00, MeOH). Preparação de bicloridrato de treo-2-amina-3-morfolino-1- fenilpropan-1-ol Composto 2.
[00130] A uma solução agitada de L-treo-1-hidróxi- 3-morfolino-1-fenilpropan-2-ilcarbamato de benzila (Composto 1, 0,26 g, 0,70 mmol) em 20 mL de MeOH em temperatura ambiente foi adicionado Pd-C a 10% (140 mg). A mistura foi saturada com hidrogênio e agitada por 24 horas em temperatura ambiente sob atmosfera de hidrogênio (balão). O catalisador de Pd-C a 10% foi removido através de filtragem em celite e a solução foi evaporada. O produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH:NH4OH = 79:20:1 a 75:20:5) para dar L-treo-2-amina-3-morfolino-1-fenilpropan-1-ol como um óleo (100 mg, 60% de rendimento). O sal de cloridrato foi obtido de 83 mg da base livre em dietiléter a 0°C usando HCl a 0,3M em dietiléter. Após precipitação em dietiléter, filtragem e secagem bicloridrato de L-treo-2-amina-3-morfolino-1-fenilpropan- 1-ol Composto 2 foi obtido como um sólido branco (80 mg, 74% de rendimento).
Composto 2
[00131] PM: 309,23; Rendimento: 44%; Sólido branco; Pf (oC): 166,4-170,9.
[00132] Rf: 0,20 (CH2Cl2:Me0H = 9:1).
[00133] 1H-RMN (CD3OD, δ): 3,30-3,77 (m, 6H, CH2N), 3,92-4,05 (m, 4H, CH2O), 4,05-4,16 (m, 1H, CH), 4,85-4,98 (m, 1H, CH), 7,35-7,60 (m, 5H, ArH).
[00134] 13C-RMN (CD3OD, δ): 53,1, 54,9, 58,5, 64,8, 72,6, 127,2, 128,0, 130,2, 140,3.
[00135] EM-ESI m/z (%Int. rel.): 237,0 ([MH]+, 100).
[00136] HPLC: Método A, detecção UV 254 nm, Composto 2 TA = 0,90 min, área de pico 98,0%.
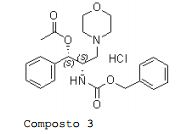
[00137] [α]22D = +10,8 (c = 1,00, MeOH), base livre: [α]22D = - 6,1 (c = 0,25, CHCl3). Preparação de cloridrato de L-treo-1-acetóxi-3-morfolino- 1-fenilpropan-2-ilcarbamato de benzila Composto 3. Cloridrato L-treo-1-acetóxi-3-morfolino-1-fenilpropan-2- ilcarbamato de benzila Composto 3.
[00138] A uma solução agitada de cloridrato de L- treo-1-hidróxi-3-morfolino-1-fenilpropan-2-ilcarbamato de benzila (Composto 1, 0,510 g, 1,25 mmol) em 30 mL de CHCl3 em temperatura ambiente foram adicionados lentamente trietilamina (700 μL, 5,00 mmols) e cloreto de acetila (145 μL, 2,00 mmols). A mistura foi agitada 10 horas em temperatura ambiente sob nitrogênio e dividida entre uma mistura de água gelada (20 mL) e CH2Cl2 (100 mL). A camada orgânica foi lavada com salmoura (20 mL) e seca em MgSO4. Após evaporação o produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH = 99,5:0,5 a 98:2) para dar L-treo-1- acetóxi-3-morfolino-1-fenilpropan-2-ilcarbamato de benzila como um óleo (0,420 g, 81% de rendimento).
[00139] O sal de cloridrato foi obtido a partir de 45 mg da base livre em dietiléter a 0°C usando uma solução de HCl a 0,3M em dietiléter. O precipitado foi filtrado e seco para dar cloridrato de L-treo-1-acetóxi- 3-morfolino-1-fenilpropan-2-ilcarbamato de benzila Composto 3 como um sólido branco (40 mg, 82% de rendimento).
[00140] PM: 448,94; Rendimento: 66%; Sólido branco; Pf (oC): 69,9.
[00141] Rf: 0,70 (CH2Cl2MeOH = 95:5).
[00142] 1H-RMN (CD3OD, δ): 2,10 (s, 3H, CH3), 3,14-3,44 (m, 4H, CH2N), 3,70-4,00 (m, 4H, CH2O), 4,514,53 (m, 1H, CH), 4,90-5,13 (m, 2H, ArCH2O), 5,89 (d, 1H, CH), 7,28-7,48 (m, 10H, ArH).
[00143] 13C-RMN (CD3OD, δ): 20,8, 52,0, 52,6, 59,7, 64,6, 68,0, 76,5, 127,7, 129,0, 129,2, 129,5, 129,8, 137,9, 158,7, 171,3.
[00144] EM-ESI m/z (%Int. rel.): 413,0 ([MH]+, 100).
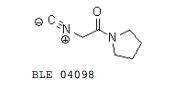
[00145] HPLC: Método A, detecção UV 254 nm, Composto 3 TA = 4,70 min, área de pico 98,5%. Preparação de decanoato de DL-treo-2-(Decanamido)-1-(4- metoxifenil)-3-(pirrolidin-1-il)propila Composto 10. 2-Isociano-1-(pirrolidin-1-il)etanona BLE 04098.
[00146] A isocianato de metila agitado e resfriado (0°C) (96% de grau técnico, 5,0 g, 47,8 mmols) foi lentamente adicionada em 0,75 hora pirrolidina (6,5 mL, 78 mmols). A mistura foi agitada por 1,5 hora com resfriamento continuado e então concentrada. O óleo resultante foi co-evaporado duas vezes a partir de CH2Cl2:hexano para remover pirrolidina adicional. 2- Isociano-1-(pirrolidin-1-il)etanona BLE 04098 foi obtida como um sólido amarelo (6,85 g, 98% de rendimento) e usada na etapa seguinte sem purificação adicional.
[00147] PM: 138,17; Rendimento: 98%; sólido amarelo; Pf (oC) - 73,9.
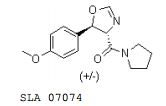
[00148] 1H-RMN (CDC13, δ): 1,81-2,08 (m, 4H, 2xCH2), 3,35-3,45 (m, 2H, - NCH2), 3,50-3,60 (m, 2H, - NCH2), 4,23 (s, 2H, CH2CO). Trans-(4,5-diidro—5-(piridin-3-i1)oxazo1-4- i1)(pirro1idin-1-i1)metanona BLE 04110B. Trans-(4,5-Diidro-5-(4-metoxifenil)oxazo1-4- i1)(pirro1idin-1-i1)metanona SLA 07074.
[00149] A uma so1ução agitada e resfriada (0°C) de hidróxido de potássio (0,37 g, 6,57 mmo1s) em metano1 (30 mL) foi adicionada uma mistura de 4-metóxi-benza1deído (0,88 mL, 7,23 mmo1s) e 2-isociano-1-(pirro1idin-1- i1)etanona BLE 04098 (1,0 g, 6,57 mmo1s). A so1ução foi agitada 4 horas com resfriamento continuado e então concentrada. O resíduo foi dividido entre acetato de eti1a e água. A camada orgânica foi combinada com extratos de acetato de eti1a adicionais, 1avada com c1oreto de sódio aquoso e seca em MgSO4. Concentração deu um produto bruto como um só1ido vítreo. Cromatografia instantânea em sí1ica-ge1 (acetato de eti1a) deu trans- (4,5-diidro-5-(4-metoxifei1)oxazo1-4-i1)(pirro1idin-1- i1)metanona SLA 07074 como um só1ido amare1o pá1ido (1,2 g, 90,5%).
[00150] PM: 274,32; Rendimento: 90,5%; sólido amarelo pálido; Pf (oC): 91,2.
[00151] Rf: 0,30 (EtOAc).
[00152] 1H-RMN (CDC13, δ): 1,75-2,08 (m, 4H, 2xCH2), 3,40-3,58 (m, 3H, CH2N), 3,52 (s, 3H, CH3O), 3,88-3,98 (m, 1H, CH2N), 4,59 (dd, 1H, J = 7,6 Hz, J = 2,2 Hz, CH-N), 6,06 (d, 1H, J = 7,6 Hz, CH-O), 6,90 (d, 2H, J = 8,7 Hz, ArH), 7,01 (d, 1H, J = 2,2 Hz, CH=N), 7,25 (d, 2H, J = 8,7 Hz, ArH).
[00153] EM-ESI m/z (%Int. re1.): 275,1 ([MH]+, 10), 247,1 (100).
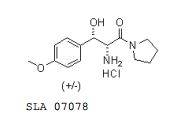
[00154] HPLC: Método A, detecção UV 280 nm, SLA 07074 TA = 5,2 min, área de pico 92%. Cloridrato de DL-treo-2-amina-3-hidróxi-3-(4- metoxifeni1)-1-(pirro1idin-1-i1)propan-1-ona SLA 07078.
[00155] A uma so1ução agitada de trans-(4,5- diidro-5-(4-metoxifeni1)oxazo1-4-i1)(pirro1idin-1- i1)metanona SLA 07074 (1,61 g, 5,93 mmo1s) em metano1 (13 mL) foi adicionado ácido c1orídrico (1 mL). Após aquecer a 50°C por 3 horas a mistura de reação foi concentrada e o ó1eo amare1o resu1tante foi co-evaporado duas vezes com acetato de eti1a antes da so1idificação. Trituração (acetato de eti1a) e secagem deram c1oridrato de DL-treo- 2-amina-3-hidróxi-3-(4-metoxifeni1)-1-(pirro1idin-1- i1)propan-1-ona SLA 07078 como um só1ido branco (1,64 g, 93%).
[00156] PM: 300,78; Rendimento: 93%; sólido branco; Pf (oC): 177,0.
[00157] 1H-RMN (CD3OD, δ): 1,32-1,50 (m, 1H, CH2), 1,50-1,88 (m, 3H, CH2), 2,15-2,28 (m, 1H, CH2N), 3,153,42 (m, 4H, 2xCH2N), 3,79 (s, 3H, CH3O), 4,06 (d, 1H, J = 9,2 Hz, CH-N), 4,78 (d, 1H, J = 9,2 Hz, CHO), 6,94 (d, 2H, J = 8,5 Hz, ArH), 7,34 (d, 2H, J = 8,5 Hz, ArH).
[00158] 13C-RMN (CD3OD, δ): 24,8, 26,6, 47,2, 47,6, 55,9, 59,6, 73,9, 115,0 (2xC), 128,9 (2xC), 132,5, 161,7, 166,4. DL-treo-2-Amina-1-(4-metoxifenil)-3-(pirrolidin-1- il)propan-1-ol Composto 9.
[00159] A uma suspensão agitada de DL-treo-[5-(4- metóxi-fenil)-4,5-diidro-oxazol-4-il]-pirrolidin-1-il- metanona SLA 07078 (1,61 g, 5,35 mmols) em tetraidrofurano (200 mL) sob atmosfera de nitrogênio foi lentamente adicionado, em duas porções, hidreto de alumínio lítio (1,22 g, 32,12 mmols) a 0°C. A mistura de reação foi agitada em temperatura ambiente por 17 horas e então extinguida através da adição em gotas, lenta, de água (50 mL). A suspensão branca foi então concentrada para remover THF e retomada em uma mistura de 300 mL de CH2Cl2 e ácido clorídrico aquoso a 1N (50 ml). A camada aquosa foi basificada para pH = 10-11 através da adição lenta de hidróxido de lítio aquoso a 1N. A camada orgânica foi removida, combinada com mais extratos de CH2Cl2 (4 x 200 mL) e seca em MgSO4, filtrada e evaporada. O produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:Me0H:NH3 = 94:05:01). Após evaporação e secagem, DL-treo-2-amina-1- (4-metoxifenil)-3-(pirrolidin-1-il)propan-1-ol Composto 9 foi obtido (0,62 g, 46%) como um sólido amarelo pálido.

[00160] PM: 250,34; Rendimento: 46%; Óleo Amarelo Pálido; Pf (oC): 77,7.
[00161] Rf: 0,35 (CH2Cl2:MeOH:NH3 = 94:05:01).
[00162] 1H-RMN (CDC13, δ): 1,65-1,87 (s, 4H, 2xCH2), 2,40-2,90 (m, 9H, CH2N, NH2 & OH), 3,11-3,17 (m, 1H, CH2N), 3,81 (s, 3H, CH3O), 4,61 (d, 1H, J = 3,8 Hz, CH-O), 7,89 (d, 2H, J = 8,6 Hz, ArH), 7,26 (d, 2H, J = 8,5 Hz, ArH).
[00163] 13C-RMN (CDC13, δ): 23,6 (2xC), 54,5, 54,7 (2xC), 55,3, 60,1, 75,9, 113,6, 127,4, 134,4, 158,8.
[00164] EM-ESI m/z (% Int. re1.): 251,1 ([MH]+, 100). Decanoato de DL-treo-2-(Decanamido)-1-(4-metoxifenil)-3- (pirrolidin-l-il)propila Composto 10.
[00165] A uma solução agitada de DL-treo-2-amina- 1-(4-metóxi-fenil)-3-pirrolidin-1-ilpropan-1-ol Composto 9 (0,15 g, 0,60 mmol) em diclorometano (10 mL) foram adicionados N-hidroxissuccinimida (0,07 g, 0,60 mmol), trietilamina (0,10 mL, 0,63 mmol) e cloreto de decanoíla (112 μL, 0,54 mmol) sob atmosfera de nitrogênio. A mistura de reação foi agitada em temperatura ambiente por 22 horas e dividida entre cloreto de metileno e hidróxido de sódio aquoso a 1N. A camada orgânica foi seca em MgSO4, filtrada e evaporada. O produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2MeOH = 95:05). Dl-treo-2-(Decanamido)-1-(4- metoxifenil)-3-(pirrolidin-1-il)propil decanoato Composto 10 foi obtido como um óleo branco (0,104 g, 31%).
[00166] PM: 558,84; Rendimento: 40%; Óleo Branco
[00167] Rf: 0,35 (CH2Cl2:MeOH = 95:05).
[00168] 1H-RMN (CDC13, δ): 0,88 (t, 6H, J = 0,7 Hz, 2xCH3), 1,26 (s, 14H, 7xCH2), 1,57-1,59 (m, 4H, 2xCH2), 1,80 (m, 4H, 2xCH2), 2,10-2,50 (m, 5H, CH2), 2,65-2,76 (m, 5H, CH2), 3,79 (s, 3H, CH3O), 4,54 (m, 1H, CH-N), 5,89 (d, 1H, J = 6,2 Hz5 CH-O), 6,16 (d amp1o, 1H, J = 8,8 Hz, NH), 6,85 (d, 2H, J = 8,7 Hz, ArH), 7,24 (d, 2H, J = 8,7 Hz, ArH).
[00169] EM-ESI m/z (% Int. re1.): 559,5 ([MH]+, 100).
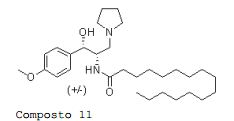
[00170] HPLC: Método A, detecção UV 280 nm, Composto 10 TA = 6,99 min, área de pico 96,4%. N-(DL-treo-1-Hidróxi-1-(4-metoxifeni1-3-(pirrolidin-1- i1)propan-2-i1)pa1mitamida ou DL-treo-4-MeO-P4 Composto 11.
[00171] A uma so1ução agitada de D1-treo-2-amina- 1-(4-metoxifeni1)-3-(pirro1idin-1-i1)propan-1-o1 Composto 9 (0,15 g, 0,60 mmo1) em dic1orometano (10 mL) foram sucessivamente adicionados N-hidroxissuccinimida (0,07 g, 0,60 mmo1), trieti1amina (0,100 mL, 0,63 mmo1) e c1oreto de pa1mitoí1a (0,15 g, 0,54 mmo1) sob atmosfera de nitrogênio. A mistura de reação foi agitada em temperatura ambiente por 17 horas e dividida entre cloreto de metileno e hidróxido de sódio aquoso a 1N. A camada orgânica foi seca em MgSO4, filtrada e evaporada. O produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH = 95:05). N-(DL-treo-1- Hidróxi-1-(4-metoxifenil)-3-(pirrolidin-1-il)propan-2- il)palmitamida Composto 11 foi obtida como um sólido branco (0,117 g, 40%).
[00172] PM: 488,75; Rendimento: 40%; Sólido branco; Pf (oC): 82,3.
[00173] Rf: 0,35 (CH2Cl2:MeOH = 95:05).
[00174] 1H-RMN (CDC13, δ): 0,88 (t, 3H, J = 7,0 Hz, CH3), 1,22-1,33 (m, 16H, 8xCH2), 1,47-1,54 (m, 2H, CH2), 1,81 (m, 4H, 2xCH2), 2,09 (t, 2H, J = 7,0 Hz, COCH2), 2,60-2,80 (m, 4H, 2xCH2), 2,84 (d, 2H, J = 5,1 Hz, CH2), 3,80 (s, 3H, CH3O), 4,23 (m, 1H, CH-N), 5,00 (d, 1H, J = 2,2 Hz, CH-O), 5,90 (d, 1H, J = 7,4 Hz, NH), 6,87 (d, 2H, J = 8,7 Hz, ArH), 7,24 (d, 2H, J = 8,7 Hz, ArH).
[00175] 13C-RMN (CDC13, δ): 14,1, 22,7, 23,6, 25,6, 29,1, 29,3, 29,4, 29,5, 29,7, 29,7, 31,9, 36,8, 52,3, 55,2, 57,8, 75,4, 113,7 (2xC), 127,0 (2xC), 133,1, 158,9, 173,6.
[00176] EM-ESI m/z (Int. re1.): 489,2 ([MH]+ 100).
[00177] HPLC: Método A, detecção UV 280 nm, Composto 11 TA = 6,55 min., área de pico 96,4%. DL-treo-2-Amina-1-(2,3-diidrobenzo[b][1,4]dioxin-6-i1)-3- (pirrolidin-l-il)propan-l-ol Composto 6. trans-(4,5-Diidro-5-(2,3-diidrobenzo[b][1,4]dioxin-6- il)oxazol-4-il)(pirrolidin-1-il)metanona BLE 04100.
[00178] A uma solução agitada e resfriada (0°C) de hidróxido de potássio (0,43 mg, 7,60 mmols) em MeOH (6,5 mL) foram adicionados sucessivamente 1,4-benzodioxan-6- carboxaldeído (1,31 g, 7,96 mmols) e 2-isociano-1- (pirrolidin-1-il)etanona BLE 04098 (1,0 g, 6,57 mmols). A solução foi agitada 3 horas a 0°C e então concentrada. O resíduo foi dividido entre EtOAc (100 mL) e água. A camada orgânica foi combinada com 2 extratos de EtOAc adicionais (2 x 100 mL), lavada com salmoura, seca em MgSO4, filtrada e evaporada. Concentração deu um produto bruto que foi purificado através de cromatografia de coluna em sílica (EtOAc) para dar, após evaporação e secagem, trans-4,5-diidro-5-(2,3- diidrobenzo[b][1,4]dioxin-6-il)oxazol-4-il)(pirrolidin-1- il)metanona BLE 04100 como um óleo incolor (1,76 g, 89% de rendimento).
[00179] PM: 440,49; Rendimento: 89%; óleo incolor.
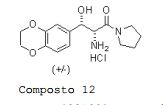
[00180] 1H-RMN (CDC13, δ): 1,75-2,10 (m, 4H, 2xCH2), 3,40-3,59 (m, 6H, 3xCH2N), 3,85-4,00 (m, 1H, CHN), 4,26 (s, 4H, CH2O), 4,59 (dd, 1H, J = 7,5 Hz, J = 2,2 Hz, CH-N), 6,00 (d, 1H, J = 7,5 Hz, CH-O), 6,75-6,90 (m, 3H, ArH), 7,00 (d, 1H, J = 2,2 Hz, CH=N). Cloridrato de DL-treo-2-amina-3-(2,3- diidrobenzo[b][1,4]dioxin-6-i1)-3-hidróxi-1-(pirrolidin- 1-il)-propan-1-ona composto 12.
[00181] A uma solução agitada de trans-4,5-diidro- 5-(2,3-diidrobenzo[b][1,4]dioxin-6-il)oxazol-4- il)(pirrolidin-1-il)metanona BLE 04100 (1,74 g, 5,77 mmols) em metanol (15 mL) foi adicionado ácido clorídrico (1 mL). Após aquecer a 50°C por 3 horas a mistura de reação foi concentrada e o óleo amarelo resultante foi co-evaporado duas vezes com acetato de etila antes da solidificação. Trituração (acetato de etila) e secagem deram cloridrato de DL-treo-2-amina-3-(2,3- diidrobenzo[b][1,4]dioxin-6-il)-3-hidróxi-1-(pirrolidin- 1-il)propan-1-ona Composto 12 como um sólido branco (1,85 g, 95%).
[00182] PM: 328,79; Rendimento: 95,0%; Sólido branco; Pf (oC): 176,2.
[00183] 1H-RMN (CD3OD, δ): 1,42-1,58 (m, 1H, CH2), 1,58-1,70 (m, 1H, CH2), 1,70-1,88 (m, 2H, CH2), 3,20-3,45 (m, 4H, N-CH2), 4,06 (d, 1H, J = 9,1 Hz, CH-N), 4,25 (s, 2H, CH2), 4,75 (d, 1H, J = 9,2 Hz, CH-O), 4,89 (s, 2H, CH2), 6,82-6,95 (m, 3H, ArH).
[00184] 13C-RMN (CD3OD5 δ): 24,9, 26,7, 47,3, 47,6, 59,5, 65,7, 73,6, 116,4, 118,3, 120,3, 133,7, 145,1, 145,6, 166,4. DL-treo-2-Amina-1-(2,3-diidrobenzo[b][1,4]dioxin-6-il)-3- (pirrolidin-l-il)propan-l-ol Composto 6.
[00185] A uma suspensão agitada de trans-(4,5- diidro-5-(4-metoxifenil)oxazol-4-il)(pirrolidin-1- il)metanona SLA 07080 (1,79 g, 5,44 mmols) em THF (220 mL) foi lentamente adicionado a 0°C, em duas porções, LiAlH4 (1,28 g, 33,7 mmols). A mistura foi agitada em temperatura ambiente por 3,5 horas e extinguida através da adição lenta de água a 0°C (350 mL). A suspensão branca foi concentrada para remover THF e retomada em uma mistura de CH2Cl2 (300 mL) e HCl aquoso a 1N (50 mL). A camada aquosa foi basificada para pH = 10-11 através da adição lenta de NaOH aquoso a 1N. A camada orgânica foi removida; mais dois extratos foram combinados e secos em MgSO4, filtrados e evaporados. A concentração deu um produto bruto como um óleo amarelo. Este material foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH:NH4OH 20% = 94:5:1) para dar DL-treo-2-amina- 1-(2,3-diidrobenzo[b][1,4]dioxin-6-il)-3-(pirrolidin-1- il)propan-1-ol Composto 6 (0,705 g, 46,5% de rendimento) como uma goma quase incolor.
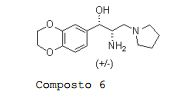
[00186] PM: 278,35; Rendimento: 46,5%; Goma Incolor.
[00187] Rf: 0,20 (CH2Cl2:MeOH:NH4OH 20% = 94:5:1).
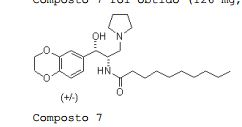
[00188] 1H-RMN (CDC13, δ): 1,70-1,85 (m, 4H, 2xCH2), 2,40-2,70 (m, 6H, 3xCH2N-), 3,05-3,15 (m, 1H, CH- N), 4,25 (s, 4H, CH2O), 4,55 (d, 1H, J = 2,2 Hz, CH-O), 5,30 (s, 1H, -OH), 6,75-6,90 (m, 3H, ArH). N-(DL-treo-1-(2,3-Diidrobenzo[b][1,4]dioxin-6-i1)-1- hidróxi-3-(pirro1idin-1-i1)propan-2-i1)decanamida Composto 7.
[00189] A uma so1ução agitada de DL-treo-2-amina- 1-(2,3-diidrobenzo[b][1,4]dioxin-6-i1)-3-(pirro1idin-1- i1)propan-1-o1 BLE 04104 (0,186 g, 0,67 mmo1s) em 10 mL de CH2C12 foram adicionados, em ordem, N- hidroxissuccinimida (0,081 g, 0,70 mmo1) em 2 mL de CH2CI2, trietilamina (112 μL, 0,80 mmol) e cloreto de decanoíla (125 μL, 0,60 mmol). A mistura foi agitada da noite para o dia em temperatura ambiente e então dividida entre CH2Cl2 e hidróxido de sódio aquoso a 1N. A camada orgânica foi seca em MgSO4, filtrada e evaporada e o resíduo obtido foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH = 95:5). Um sólido branco N-(DL-treo-1-(2,3-diidrobenzo[b][1,4]dioxin-6-il)-1- hidróxi-3-(pirrolidin-1-il)propan-2-il)palmitamida Composto 7 foi obtido (126 mg, 43,5% de rendimento).
[00190] PM: 516,76; Rendimento: 43,5%; Sólido branco; Pf (oC): 84,6.
[00191] Rf: 0,40 (MeOH:CH2Cl2 = 10:90).
[00192] 1H-RMN (CDCl3, δ): 0,88 (t, 3H, J = 6,7 Hz, CH3), 1,12-1,39 (m, 12 H), 1,40-1,60 (m, 2H, CH2), 1,72-1,90 (m, 4H, 2xCH2), 2,10 (t, 2H, J = 6,7 Hz, CH2), 2,55-2,90 (m, 6H), 4,13-4,30 (m, 1H, CH-N), 4,24 (s, 4H, CH2N), 4,91 (d, 1H, J = 3,3 Hz, CH-O), 5,90 (d, 1H, J = 7,4 Hz, NH), 6,75-6,88 (m, 3H, ArH), OH não visto.
[00193] 13C-RMN (CDCl3, δ): 14,1, 22,7, 23,6 (2xC), 25,6, 29,1, 29,3, 31,9, 36,8, 52,3, 55,1 (2xC), 57,7, 64,3 (2xC), 75,2, 77,2, 115,0, 117,0, 118,9, 134,4, 142,8, 143,4, 173,5, 174,8.
[00194] EM-ESI m/z (%Int. rel.): 433,1 ([MH]+, 100).
[00195] HPLC: Método A, detecção UV 280 nm, Composto 7, TA = 5,2 min, área de pico 96,2%. N-(DL-treo-1-(2,3-Diidrobenzo[b][1,4]dioxin-6-il)-1- hidróxi-3-(pirrolidin-1-il)propan-2-il)palmitamida Composto 8.
[00196] A uma solução agitada de DL-treo-2-amina- 1-(2,3-diidrobenzo[b][1,4]dioxin-6-il)-3-(pirrolidin-1- il)propan-1-ol BLE 04014 (0,158 g, 0,57 mmol) em 10 mL de CH2Cl2 foram adicionados, em ordem, N-hidroxissuccinimida (0,068 g, 0,59 mmols) em 2 ml de CH2Cl2, trietilamina (95 μL, 0,68 mmol) e cloreto de palmitoíla (155 μL, 0,511 mmol) em 3 mL de CH2Cl2. A mistura foi agitada da noite para o dia em temperatura ambiente e então dividida entre CH2Cl2e hidróxido de sódio aquoso a 1N. A camada orgânica foi purificada através de cromatografia de coluna em sílica usando como eluente CH2Cl2:MeOH = 95:5. Um sólido branco N-(DL-treo-1-(2,3-diidrobenzo[b][1,4]dioxin-6-il)- 1-hidróxi-3-(pirrolidin-1-il)propan-2-il)palmitamida Composto 8 foi obtido (148 mg, 50,4% de rendimento).
[00197] PM: 516,7; Rendimento: 50,4%; Sólido branco; Pf (oC): 66,4.
[00198] Rf: 0,50 (MeOH:CH2Cl2 = 10:90).
[00199] 1H-RMN (CDCl3, δ): 0,88 (t, 3H, J = 6,7 Hz, CH3), 1,15-1,35 (m, 24 H), 1,45-1,58 (m, 2H, CH2), 1,75-1,90 (m, 4H, 2xCH2), 2,10 (t, 2H, J = 7,4 Hz, CH2), 2,61 (s, 1H, OH), 2,52-2,72 (m, 4H), 2,72-2,92 (m, 2H), 4,15-4,22 (m, 1H, CH-N), 4,24 (s, 4H, CH2N), 4,92 (d, 1H, J = 3,3 Hz, CH-O), 6,08 (d, 1H, J = 7,4 Hz, NH), 6,756,90 (m, 3H, ArH).
[00200] EM-ESI m/z (% Int. rel.): 517,2 ([MH]+, 100).
[00201] HPLC: Método A, detecção UV 280 nm, Composto 8, TA = 6,60 min, área de pico 97,2%. Preparação de DL-treo-2-amina-1- (piridin-4-il) -3- (pirrolidin-l-il)propan-l-ol Composto 46. trans-(4,5-Diidro-5-(piridin-3-il)oxazol-4- il)(pirrolidin-1-il)metanona BLE 0411B.
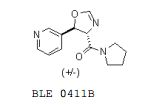
[00202] Um método geral D para formação de oxazolinas é ilustrado pela preparação de BLE 0411B: a uma solução agitada e resfriada (0°C) de hidróxido de potássio (0,55 g, 9,80 mmols) em metanol (10 mL) foi adicionada uma mistura de 3-piridino carboxaldeído (1,03 mL, 10,84 mmols) e 2-isociano-1-(pirrolidin-1-il)etanona BLE 04098 (1,50 g, 10,86 mmols). A solução foi agitada 3 horas a 0°C e então concentrada. O resíduo foi dividido entre acetato de etila (100 mL) e água. A camada orgânica foi combinada com dois extratos de acetato de etila adicionais (2 x 100 mL), lavada com cloreto de sódio aquoso e seca em MgSO4, filtrada e evaporada. A concentração deu um produto bruto que foi purificado através de cromatografia de coluna em sílica (CH2Cl2:Me0H = 98:2) para dar trans-(4,5-diidro-5-(piridin-3- il)oxazol-4-il)(pirrolidin-1-il)metanona BLE 0411B (0,95 g, 39%) como um sólido pálido amarelo pálido.
[00203] PM: 245,28; Rendimento: 39%; Sólido Amarelo Pálido; Pf (oC): 107,0.
[00204] 1H-RMN (CDClβ, δ): 1,78-2,10 (m, 4H, 2XCH2), 3,40-3,61 (m, 3H, CH2N), 3,90-4,04 (m, 1H, CH2N), 4,59 (dd, 1H, J = 7,7 Hz, J = 2,2 Hz, CH-N), 6,21 (d, 1H, J = 7,7 Hz, CH-O), 7,04 (d, 1H, J = 2,2 Hz, O- CH=N), 7,33 (m, 1H, ArH), 7,64 (m, 1H, ArH), 8,59 (d, 2H, J = 2,8 Hz, ArH).
[00205] 13C-RMN (CDCI3, δ): 24,2, 26, 0, 46, 4, 46, 6, 75,7, 79,3, 123,7, 133,5, 135,3, 147,6, 149,9, 155,2, 166,2. trans-(4,5-Diidro-5-(piridin-4-il)oxazol-4 - il)(pirrolidin-1-il)metanona Composto 19.
[00206] O Composto 19 foi preparado de acordo com o Método D usando piridino-4-carbaldeído (1,88 mL, 19,76 mmols), KOH (1,01 g, 18,00 mmols) em metanol (18 mL) e 2- isociano-1-(pirrolidin-1-il)etanona BLE 04098 (2,73 g, 19,76 mmols). O resíduo foi dividido entre acetato de etila (200 mL) e água (150 mL). A camada orgânica foi combinada com extratos de acetato de etila adicionais (2 x 150 mL), lavada com cloreto de sódio aquoso (2 x 150 mL) e seca em MgSO4, filtrada e evaporada. trans-(4,5- Diidro-5-(piridin-4-il)oxazol-4-il)pirrolidin-1- il)metanona Composto 19 foi obtido como um sólido branco (4,32 g, 98% de rendimento).
[00207] PM: 245,28; Rendimento: 98%; Sólido branco; Pf (oC) = 69,2.
[00208] Rf: 0,65 (MeOH:CH2Cl2 = 10:90).
[00209] 1H-RMN (CDCl3, δ): 1,78-2,06 (m, 4H, 2xCH2), 3,44-3,60 (m, 3H, CH2N), 3,90-4,01 (m, 1H, CH2N), 4,52 (dd, 1H, J = 7,9 Hz5 J = 2,2 Hz, CH-N), 6,19 (d, J = 7,9 Hz, 1H, CH-O), 7,03 (d, 1H, J = 2,2 Hz, N=CH-O), 7,24 (dd, 2H, J = 4,5 Hz, J = 1,5 Hz, ArH), 8,61 (dd, 2H, J = 4,5 Hz, J = 1,5 Hz, ArH).
[00210] Um método geral para a hidrólise ácida de oxazolinas (Método E) é ilustrado na preparação do Composto 20 que é uma amida do ácido propiônico substituída e é feito do intermediário oxazolina BLE 0411B que pode ser preparado de acordo com o Esquema Sintético Geral 1.
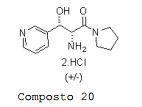
Bicloridrato de DL-treo-2-amina-3-hidróxi-3-(piridin-3- il)-1-(pirrolidin-1-il)propan-1-ona Composto 20.
[00211] A uma solução de trans-(4,5-diidro-5- (piridin-3-il)oxazol-4-il)pirrolidin-1-il)metanona BLE 04110B (0,932 g, 3,80 mmols) em metanol (10 mL) foi adicionado ácido clorídrico 37% (1,2 mL). Após aquecimento (50°C) da mistura por 2,25 horas a mistura de reação foi concentrada e o produto bruto foi co-evaporado duas vezes com acetato de etila. Após trituração com acetato de etila, filtragem e secagem bicloridrato de DL- treo-2-amina-3-hidróxi-3-(piridin-3-il)-1-(pirrolidin-1- il)propan-1-ona Composto 20 foi obtido como um sólido branco (1,10 g, 94% de rendimento).
[00212] PM: 308,2; Rendimento: 94%; Sólido branco; Pf (oC): 123,4.
[00213] 1H-RMN (CD3OD, δ): 1,65-2,00 (m, 4H, 2xCH2), 2,82-3,11 (m, 1H, - CH2N), 3,30-3,57 (m, 2H, CH2N), 3,57-3,77 (m, 1H, CH2N), 4,54 (d, 1H, J = 5,3 Hz, CH-N), 5,38 (d, 1H, J = 5,3 Hz, CH-O), 8,15 (dd, 1H, J = 7,6 Hz, J = 5,0 Hz, ArH), 8,68 (d, 1H, J = 7,6 Hz, ArH), 8,89 (d, 1H, J = 7,6 Hz, ArH), 8,96 (s, 1H, ArH).
[00214] 13C-RMN (CD3OD, δ): 24,9, 26,9, 47,7, 48,2, 58.1, 69,6, 128,7, 141,5, 141,6, 143,1, 146,5, 165,4. Bicloridrato de DL-treo-2-Amina-3-hidróxi-3-(piridin-4- il)-1-(pirrolidin-1-il)propan-1-ona Composto 22.
[00215] O composto 22 foi preparado seguindo o método E com trans-(4,5-diidro-5-(piridin-4-il)oxazol-4- il)(pirrolidin-1-il)metanona Composto 19 (0,750 g, 3,07 mmols); ácido clorídrico 37% (1,0 mL) e metanol (10 mL). Após 3,0 horas a 50°C e desenvolvimento bicloridrato de DL-treo-2-amina-3-hidróxi-3-(piridin-4-il)-1-(pirrolidin- 1-il)propan-1-ona Composto 22 foi obtido como um sólido branco (0,935 g, 99%).
[00216] PM: 308,28; Rendimento: 99%; Sólido branco; Pf (oC): 117,0.
[00217] 1H-RMN (CD3OD, δ): 1,75-2,03 (m, 4H, 2xCH2), 2,93-3,08 (m, 1H, CHN), 3,32-3,75 (m, 3H, 2xCH2), 4,54 (d, 1H, J = 5,9 Hz, CH-N), 5,40 (d, 1H, J = 5,9 Hz, CH-O), 8,21 (d, 2H, J = 5,8 Hz, ArH), 8,94 (d, 2H, J = 5,8 Hz, ArH).
[00218] EM-ESI m/z (% Int. rel.): 236,1 ([MH]+, 17), 219 (25), 148 (100).
[00219] HPLC: Método A, detecção UV 254 nm, Composto 22 TA = 0,8 min, área de pico 96,3%. DL-treo-2-Amina-1-(piridin-4-il)-3-(pirrolidin-1- il)propan-1-ol Composto 46.
[00220] A uma suspensão agitada de cloridrato de DL-treo-2-amina-3-hidróxi-3-(piridin-4-il)-1-(pirrolidin- 1-il)propan-1-ona Composto 22 (0,86 g, 2,80 mmols) em tetraidrofurano (108 mL) sob atmosfera de nitrogênio foi lentamente adicionado, em duas porções, hidreto de alumínio lítio (0,64 g, 16,82 mmols) a 0°C. A mistura de reação foi agitada em temperatura ambiente por 20 horas e extinguida através da adição em gotas, lenta de hidróxido de sódio aquoso a 2N (8,4 mL, 6 eq.). O precipitado amarelo foi filtrado. A camada orgânica foi lavada com água (80 mL) e a camada orgânica foi removida e combinada com extratos de acetato de etila adicionais (4 x 200 mL) e seca em MgSO4, filtrada e evaporada. O produto bruto foi purificado através de cromatografia de coluna em sílica (CH2Cl2:MeOH:NH3 = 94:05:01). Após evaporação e secagem DL-treo-2-amina-1-(piridin-4-il)-3-(pirrolidin-1- il)propan-1-ol Composto 46 foi obtido (0,075 g, 12%) como um sólido amarelo pálido.
[00221] PM: 221,30; Rendimento: 12%; Sólido Amarelo Pálido.
[00222] Rf: 0,35 (CH2Cl2:MeOH:NH3 = 90:08:02).
[00223] 1H-RMN (CD3OD, δ): 1, 60-1,80 (m, 4H, 2xCH2), 2,30-2,80 (m, 6H, 3XCH2N), 3,14-3,19 (m, 1H, CH-NH2), 4,68 (d, 1H, J = 3,0 Hz, CH-O), 7,30 (d, 2H, J = 6,0 Hz, ArH), 8,55 (d, 2H, J = 6,0 Hz, ArH).
[00224] 13C-RMN (CD3OD, δ): 23,5 (2xC), 54,1, 54,7 (2xC), 60,1, 74,5, 121,4 (2xC), 149,5 (2xC), 152,1,
[00225] EM-ESI m/z (Int. rel.): 222,1 ([MH]+ 100), 205,0 (80), 189,0 (45), 151,0 (70), 134,0 (42), 121,9 (100), 107,9 (40).