EP0832054B1 - Heterocyclische methylfreie radikale als biloverbesserungsmittel - Google Patents
Heterocyclische methylfreie radikale als biloverbesserungsmittel Download PDFInfo
- Publication number
- EP0832054B1 EP0832054B1 EP95931302A EP95931302A EP0832054B1 EP 0832054 B1 EP0832054 B1 EP 0832054B1 EP 95931302 A EP95931302 A EP 95931302A EP 95931302 A EP95931302 A EP 95931302A EP 0832054 B1 EP0832054 B1 EP 0832054B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- bis
- group
- mmol
- tetramethylbenzo
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 230000002708 enhancing effect Effects 0.000 title abstract description 4
- 239000002872 contrast media Substances 0.000 claims abstract description 17
- 239000002243 precursor Substances 0.000 claims abstract description 13
- 238000002595 magnetic resonance imaging Methods 0.000 claims abstract description 12
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 126
- 239000000203 mixture Substances 0.000 claims description 79
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 74
- 150000003254 radicals Chemical class 0.000 claims description 69
- -1 free radical compound Chemical class 0.000 claims description 56
- 238000000034 method Methods 0.000 claims description 29
- 125000000217 alkyl group Chemical group 0.000 claims description 25
- 239000011734 sodium Substances 0.000 claims description 25
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 23
- 150000001875 compounds Chemical class 0.000 claims description 20
- 229910052708 sodium Inorganic materials 0.000 claims description 19
- 125000003118 aryl group Chemical group 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 11
- 239000007983 Tris buffer Substances 0.000 claims description 11
- 229910052799 carbon Inorganic materials 0.000 claims description 10
- 150000003839 salts Chemical class 0.000 claims description 9
- 230000005291 magnetic effect Effects 0.000 claims description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 7
- 239000001301 oxygen Substances 0.000 claims description 7
- 229910052760 oxygen Inorganic materials 0.000 claims description 7
- 229910052717 sulfur Inorganic materials 0.000 claims description 7
- 150000001768 cations Chemical class 0.000 claims description 5
- 125000004423 acyloxy group Chemical group 0.000 claims description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 4
- 238000002496 oximetry Methods 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 230000003381 solubilizing effect Effects 0.000 claims description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 239000005864 Sulphur Substances 0.000 claims description 3
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 3
- 125000004429 atom Chemical group 0.000 claims description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 claims description 3
- 238000001983 electron spin resonance imaging Methods 0.000 claims description 3
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 3
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 3
- 125000005188 oxoalkyl group Chemical group 0.000 claims description 3
- 229910006074 SO2NH2 Inorganic materials 0.000 claims description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 2
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 2
- 125000002837 carbocyclic group Chemical group 0.000 claims description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 229910052710 silicon Inorganic materials 0.000 claims description 2
- 125000000565 sulfonamide group Chemical group 0.000 claims description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 2
- 230000002085 persistent effect Effects 0.000 claims 1
- 229940039231 contrast media Drugs 0.000 abstract description 6
- 238000004519 manufacturing process Methods 0.000 abstract description 5
- 125000005039 triarylmethyl group Chemical group 0.000 abstract description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 161
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 145
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 122
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 120
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 86
- 239000000243 solution Substances 0.000 description 81
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 72
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 54
- 238000005160 1H NMR spectroscopy Methods 0.000 description 48
- 239000000047 product Substances 0.000 description 48
- 238000006243 chemical reaction Methods 0.000 description 43
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 42
- 239000007832 Na2SO4 Substances 0.000 description 41
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 41
- 238000002953 preparative HPLC Methods 0.000 description 41
- 229910052938 sodium sulfate Inorganic materials 0.000 description 41
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 39
- 239000012074 organic phase Substances 0.000 description 36
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 35
- 238000003756 stirring Methods 0.000 description 33
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 32
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 32
- 125000005605 benzo group Chemical group 0.000 description 32
- 239000011541 reaction mixture Substances 0.000 description 32
- 239000012044 organic layer Substances 0.000 description 31
- 239000002904 solvent Substances 0.000 description 29
- 239000012300 argon atmosphere Substances 0.000 description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 25
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 24
- 238000001704 evaporation Methods 0.000 description 23
- 230000008020 evaporation Effects 0.000 description 23
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 22
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 22
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 21
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 20
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 20
- 229910000162 sodium phosphate Inorganic materials 0.000 description 20
- 238000000746 purification Methods 0.000 description 19
- 239000008346 aqueous phase Substances 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- 239000007787 solid Substances 0.000 description 17
- 230000007704 transition Effects 0.000 description 17
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 16
- 229910052786 argon Inorganic materials 0.000 description 16
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 16
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 16
- 230000007717 exclusion Effects 0.000 description 15
- 238000005259 measurement Methods 0.000 description 15
- 239000007864 aqueous solution Substances 0.000 description 14
- 238000001816 cooling Methods 0.000 description 14
- 235000011150 stannous chloride Nutrition 0.000 description 14
- 0 C1C*C*C1 Chemical compound C1C*C*C1 0.000 description 13
- 239000012298 atmosphere Substances 0.000 description 13
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 12
- 229920006395 saturated elastomer Polymers 0.000 description 12
- 238000003384 imaging method Methods 0.000 description 11
- 229910000027 potassium carbonate Inorganic materials 0.000 description 11
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 10
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 10
- 230000005855 radiation Effects 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 229910002092 carbon dioxide Inorganic materials 0.000 description 9
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 9
- 230000009467 reduction Effects 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 238000004108 freeze drying Methods 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 6
- 239000001569 carbon dioxide Substances 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical class C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 239000011591 potassium Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 5
- 238000002604 ultrasonography Methods 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- IVJFXSLMUSQZMC-UHFFFAOYSA-N 1,3-dithiole Chemical compound C1SC=CS1 IVJFXSLMUSQZMC-UHFFFAOYSA-N 0.000 description 4
- IXPVSRAWHLKCJB-UHFFFAOYSA-N 2,2,6,6-tetramethyl-[1,3]dioxolo[4,5-f][1,3]benzodioxole-8-carboxylic acid Chemical compound OC(=O)C1=C2OC(C)(C)OC2=CC2=C1OC(C)(C)O2 IXPVSRAWHLKCJB-UHFFFAOYSA-N 0.000 description 4
- CAQWNKXTMBFBGI-UHFFFAOYSA-N C.[Na] Chemical class C.[Na] CAQWNKXTMBFBGI-UHFFFAOYSA-N 0.000 description 4
- 239000002616 MRI contrast agent Substances 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- FFYPMLJYZAEMQB-UHFFFAOYSA-N diethyl pyrocarbonate Chemical compound CCOC(=O)OC(=O)OCC FFYPMLJYZAEMQB-UHFFFAOYSA-N 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- CTCKAWJFFJAAQS-UHFFFAOYSA-N methyl 2,2,6,6-tetramethyl-[1,3]dioxolo[4,5-f][1,3]benzodioxole-8-carboxylate Chemical compound COC(=O)C1=C2OC(C)(C)OC2=CC2=C1OC(C)(C)O2 CTCKAWJFFJAAQS-UHFFFAOYSA-N 0.000 description 4
- 230000005298 paramagnetic effect Effects 0.000 description 4
- 159000000000 sodium salts Chemical class 0.000 description 4
- VBLVGORCSZWNOH-UHFFFAOYSA-N 2,2,6,6-tetramethyl-[1,3]dioxolo[4,5-f][1,3]benzodioxole Chemical compound C1=C2OC(C)(C)OC2=CC2=C1OC(C)(C)O2 VBLVGORCSZWNOH-UHFFFAOYSA-N 0.000 description 3
- OOZLPQIJKNLNAP-UHFFFAOYSA-N 2,2,6,6-tetramethyl-[1,3]dithiolo[4,5-f][1,3]benzodithiole Chemical compound C1=C2SC(C)(C)SC2=CC2=C1SC(C)(C)S2 OOZLPQIJKNLNAP-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 150000001723 carbon free-radicals Chemical class 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- RXKJFZQQPQGTFL-UHFFFAOYSA-N dihydroxyacetone Chemical compound OCC(=O)CO RXKJFZQQPQGTFL-UHFFFAOYSA-N 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 3
- 229920002521 macromolecule Polymers 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 150000005691 triesters Chemical class 0.000 description 3
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- DNKLFQBAOVPJHX-UHFFFAOYSA-N 2,2,6,6-tetramethyl-[1,3]oxathiolo[4,5-f][1,3]benzoxathiole Chemical compound C1=C2SC(C)(C)OC2=CC2=C1SC(C)(C)O2 DNKLFQBAOVPJHX-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 229910004039 HBF4 Inorganic materials 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical class ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 229910006146 SO3M1 Inorganic materials 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- KIGALSBMRYYLFJ-UHFFFAOYSA-N chloro-(2,3-dimethylbutan-2-yl)-dimethylsilane Chemical compound CC(C)C(C)(C)[Si](C)(C)Cl KIGALSBMRYYLFJ-UHFFFAOYSA-N 0.000 description 2
- IIJREXIVDSIOFR-UHFFFAOYSA-N dichloromethane;heptane Chemical compound ClCCl.CCCCCCC IIJREXIVDSIOFR-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000002252 electron spin resonance oximetry Methods 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 238000011503 in vivo imaging Methods 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 238000006303 photolysis reaction Methods 0.000 description 2
- 230000015843 photosynthesis, light reaction Effects 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 description 2
- 238000001149 thermolysis Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- XLHUBROMZOAQMV-UHFFFAOYSA-N 1,4-benzosemiquinone Chemical compound [O]C1=CC=C(O)C=C1 XLHUBROMZOAQMV-UHFFFAOYSA-N 0.000 description 1
- MKVHKCQFSRHELX-UHFFFAOYSA-N 2,2-dibromo-3-ethoxy-3-oxopropanoic acid Chemical compound CCOC(=O)C(Br)(Br)C(O)=O MKVHKCQFSRHELX-UHFFFAOYSA-N 0.000 description 1
- RGHQKFQZGLKBCF-UHFFFAOYSA-N 2-bromoethyl acetate Chemical compound CC(=O)OCCBr RGHQKFQZGLKBCF-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- VXYJAQCWYHZLEA-UHFFFAOYSA-N CC(C)(Nc1c2)Oc1cc1c2NC(C)(C)O1 Chemical compound CC(C)(Nc1c2)Oc1cc1c2NC(C)(C)O1 VXYJAQCWYHZLEA-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- YIIMEMSDCNDGTB-UHFFFAOYSA-N Dimethylcarbamoyl chloride Chemical compound CN(C)C(Cl)=O YIIMEMSDCNDGTB-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- DBTDEFJAFBUGPP-UHFFFAOYSA-N Methanethial Chemical compound S=C DBTDEFJAFBUGPP-UHFFFAOYSA-N 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- HEMHJVSKTPXQMS-DYCDLGHISA-M Sodium hydroxide-d Chemical compound [Na+].[2H][O-] HEMHJVSKTPXQMS-DYCDLGHISA-M 0.000 description 1
- JFBZPFYRPYOZCQ-UHFFFAOYSA-N [Li].[Al] Chemical compound [Li].[Al] JFBZPFYRPYOZCQ-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000000862 absorption spectrum Methods 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- KVPDTCNNKWOGMZ-UHFFFAOYSA-N benzene-1,2,4,5-tetrathiol Chemical compound SC1=CC(S)=C(S)C=C1S KVPDTCNNKWOGMZ-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 210000003445 biliary tract Anatomy 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 239000012502 diagnostic product Substances 0.000 description 1
- 238000012631 diagnostic technique Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000001362 electron spin resonance spectrum Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000005294 ferromagnetic effect Effects 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 210000004051 gastric juice Anatomy 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 150000004668 long chain fatty acids Chemical group 0.000 description 1
- 238000005404 magnetometry Methods 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 238000006552 photochemical reaction Methods 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- IHSLHAZEJBXKMN-UHFFFAOYSA-L potassium nitrosodisulfonate Chemical compound [K+].[K+].[O-]S(=O)(=O)N([O])S([O-])(=O)=O IHSLHAZEJBXKMN-UHFFFAOYSA-L 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000002601 radiography Methods 0.000 description 1
- 210000005227 renal system Anatomy 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 125000001174 sulfone group Chemical group 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000009210 therapy by ultrasound Methods 0.000 description 1
- 150000003568 thioethers Chemical group 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 150000003628 tricarboxylic acids Chemical class 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
- C07B61/02—Generation of organic free radicals; Organic free radicals per se
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/20—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations containing free radicals, e.g. trityl radical for overhauser
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D497/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having oxygen and sulfur atoms as the only ring hetero atoms
- C07D497/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having oxygen and sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D497/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
- C07F7/0814—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring said ring is substituted at a C ring atom by Si
Definitions
- the present invention relates to certain novel triaryl methyl free radicals and their use as image enhancing agents in magnetic resonance imaging (MRI) as well as to contrast media containing such radicals and to the use of such radicals and their non-radical precursors in the manufacture of MRI contrast media.
- MRI magnetic resonance imaging
- MRI is a diagnostic technique that has become particularly attractive to physicians as it is non-invasive and does not involve exposing the patient under study to potentially harmful radiation, such as for example the X-radiation of conventional radiography.
- MR images are generated by manipulation of the MR signals detected from the sample, for example a human or animal body, placed in a magnetic field and exposed to pulses of radiation of a frequency (typically radiofrequency (RF)) selected to excite MR transitions in selected non-zero spin nuclei (the "imaging nuclei", which are generally water protons in body fluids) in the sample.
- RF radiofrequency
- the amplitude of the induced MR signals is dependent upon various factors such as the strength of the magnetic field experienced by the sample, the temperature of the sample, the density of the imaging nuclei within the sample, the isotopic nature and chemical environment of the imaging nuclei and the local inhomogeneities in magnetic field experienced by the imaging nuclei.
- the imaging parameters (nuclear density, T 1 and T 2 ) for tissues of interest may be altered and many proposals have been made for doing this by the administration of MRI contrast agents into patients under study (see for example US-A-4647447 (Gries/Schering), US-A-4925652 (Gries/Schering) and US-A-4863715 (Jacobsen/Nycomed)).
- MRI contrast agents are paramagnetic they produce significant reduction in the T 1 of the water protons in the body zones into which they are administered or at which they congregate, and where they are ferromagnetic or superparamagnetic (for example as suggested by Jacobsen) they produce a significant reduction in the T 2 of the water protons. In either case the result is enhanced (positive or negative) contrast in the MR images of such zones.
- contrast enhancement achievable by such agents in conventional MRI is relatively limited and it is generally not such as to allow a reduction in the image acquisition period or in the field strength of the primary magnet.
- This new technique for generating a MR image of the sample which is hereinafter termed Overhauser MRI (OMRI)
- OMRI Overhauser MRI
- RF radiation a frequency selected to excite nuclear spin transitions in selected nuclei in the sample
- MW or UHF radiation a frequency selected to excite electron spin transitions coupled to nuclear spin transitions for at least some of the selected nuclei
- MW or UHF radiation the MR images being generated from the resulting amplified MR signals (free induction decay signals) emitted by the sample.
- the paramagnetic substance which possesses the ESR transition which couples with the NMR transition of the image nuclei may be naturally present within the imaging sample or more usually may be administered as an OMRI contrast agent.
- OMRI contrast agents including for example nitroxide stable free radicals, chloranil semiquinone radical and Fremy's salt (US-A-4984573) and deuterated stable free radicals, in particular deuterated nitroxide stable free radicals (WO-A-90/00904).
- the present invention thus provides a radical compound of formula I .C(Ar 1 ) 3 (where each group Ar 1 , which may be the same or different is an optionally substituted aromatic group, preferably an optionally substituted 5-7 membered carbocyclic or heterocyclic aromatic ring optionally carrying one or more fused carbocyclic or heterocyclic rings, preferably a benzyl ring and at least one of said Ar 1 groups is a group Ar 3 of formula (wherein
- the optionally substituted aromatic group Ar 1 may be selected from any of the aromatic groups described for the triarylmethyl radicals of WO-A-91/12024.
- Ar 1 groups which are other than groups Ar 3 are groups Ar 1' of formula (wherein X, R 1 and R 7 are as defined above excluding the proviso that at least one X is S or S(O) n ).
- X is preferably an oxygen atom and R 7 is a hydrogen atom or an optionally hydroxylated alkyl, preferably methyl group.
- Preferred radical compounds of formula I are those in which each group Ar 1 is an aromatic group substituted by a solubilising group M and carrying two fused sulphur-containing heterocyclic rings each substituted by at least one, preferably two, solubilising groups M.
- Especially preferred radical compounds are those of formula I, wherein each of said Ar 1 groups is of formula: (wherein
- Especially preferred radical compounds of formula I according to the invention are tris(8-carboxy-2,2,6,6-tetrahydroxymethylbenzo[1,2-d:4,5-d ' ]bis(1,3)dithiole-4-yl)methyl and the salts (eg sodium salt) and bis-(8-sodium carboxylate-2,2,6,6-tetrakis-( 2 H 3 -methyl)benzo[1,2-d:4,5-d']-bis(1,3)dithiole-4-yl)-mono-(8-sodium carboxylate-2,2,6,6-tetrakis-( 2 H 3 -methyl)-benzo[1,2-d:4,5-d']-bis(1,3)dioxole-4-yl)methyl (herein referred to as perdeuterated trityl) and bis-(8-sodium carboxylate-2,2,6,6-tetrakis-(hydroxy- 2 H 2 -methyl)-benzo[1,2-d:4,5-
- Particularly preferred radical compounds of Formula I include those wherein at least one, preferably two, and more preferably all three Ar 1 groups are groups Ar 3 and any remaining Ar 1 groups are Ar 1' groups.
- each group Ar 3 or Ar 1' the two fused rings are the same.
- the solubilising groups M may be any of the solubilising groups conventionally used in diagnostic and pharmaceutical products.
- Particularly preferred solubilizing groups M include optionally hydroxylated, optionally alkoxylated alkyl or oxo-alkyl groups and groups of formulae R 5 , COOR 5 , OCOR 5 , CHO, CN, CH 2 S(O)R 5 , CONR 5 2 , NR 5 COR 5 , NR 5 2 , SO 2 NR 5 2 , OR 5 , PO 3 2 , SOR 5 , SO 2 R 5 , SO 3 M 1 , COOM 1 (where R 5 represents a hydrogen atom or an optionally hydroxylated, optionally aminated, optionally alkoxylated, optionally carboxylated alkyl, oxo-alkyl, alkenyl or alkaryl group and M 1 is one equivalent of a physiologically tolerable cation, for example an alkali or alkaline earth metal
- solubilizing groups M are groups of formula C(H) 3-n (CH 2 OH) n , R 9 , COR 9 , SR 9 , SOR 9 , SO 2 R 9 , CON(R 9 ) 2 , NR 2 , NHR 9 and CONHR 9 [where R 9 may represent a hydroxylated alkyl group such as a group (although any R 9 group attached to a sulphur, nitrogen or oxygen atom is preferably not hydroxylated at the ⁇ carbon)], and groups of formula SO 2 R 12 or SR 12 where R 12 is a group CH 2 COOR 13 , CH(COOR 13 ) 2 , CH 2 CONHR 9 , CH 2 CONR 9 2 , CR 5 (COOR 13 ) 2 , CH(CN)CO 2 R 13 , (CH 2 ) n SO 3 - M 1 , (CH 2 ) n COR 13 , CH(COR 9 )CH 2 COR 9 and CH(R 5 )COR 9 where
- any alkyl or alkenyl moiety preferably contains up to 6, especially preferably up to 4, carbon atoms and generally it is preferred for each of the three aryl monomers of the triaryl structure to be identical.
- group X is preferably selected from oxygen or sulphur atoms or SO 2 groups.
- each Ar 3 moiety preferably two and especially preferably all four X groups are sulphur atoms or S(O) n groups, preferably sulphur atoms or SO 2 groups.
- Suitable Ar 3 groups thus include for example those wherein the central aromatic ring carries fused rings of formula
- Preferred Ar 3 groups include
- R 7 is preferably a hydrogen atom or an optionally hydroxylated alkyl group, preferably a hydroxyC 1-3 -alkyl, especially a hydroxymethyl group.
- radical compounds of formula I preferred identities for the group R 1 include:
- Preferred radical structures of formula I include radical compounds of formulae Ia, Ib, Ic and Id: where R 11 and R 12 are selected from the list of preferred R 1 identities indicated above.
- R 11 and R 12 are selected from H, SCH 3 , SCH 2 CO 2 CH 2 CH 3 , SCH 2 COOH, SO 2 N(CH 3 )CH 2 (CHOH) 4 CH 2 OH, SO 2 NH 2 , SO 2 NCH 2 CH 2 OH and SO 2 NCH 2 CHOHCH 2 OH, and particularly preferably R 11 and R 12 are identical.
- the invention also provides a magnetic resonance imaging contrast medium composition
- a magnetic resonance imaging contrast medium composition comprising a radical compound of formula I together with at least one pharmacologically acceptable carrier or excipient.
- the radical compound should of course preferably be a physiologically tolerable radical, or one presented in a physiologically tolerable, e.g. encapsulated, form.
- the invention provides a method of magnetic resonance imaging wherein there is introduced into a human or non-human, preferably mammalian, subject an effective amount of a magnetic resonance signal amplifying agent and wherein an image of at least a part of said subject is generated, the improvement comprising introducing as said amplifying agent a radical according to the invention.
- novel triarylmethyl radicals of the invention have the advantages of the beneficial properties of stability at physiological pH, long half lives (at least one minute, and preferably at least one hour), long relaxation times, and surprisingly good relaxivity.
- the novel radical compounds of the invention exhibit surprising stability when compared with corresponding compounds lacking Ar 3 groups as defined above. Stability is of paramount importance when considering suitability of radical compounds for use as MRI contrast agents, and thus the radical compounds of the present invention represent a considerable advance in the art.
- free radicals which have relatively few transitions, e.g. less than 15, preferably less than 10, in their ESR spectra and radicals having narrow linewidth ESR transitions, e.g. up to 500 mG, preferably less than 150 mG, especially less than 60 mG and particularly less than 25mG, are especially preferred for use as OMRI contrast agents.
- the linewidths referred to are conveniently the intrinsic linewidths (full width at half maximum in the absorption spectrum) at ambient conditions).
- the hyperfine splitting constant is preferably very small.
- radicals having as few as possible non-zero spin nuclei, positioned as far away as possible from the paramagnetic centre are thus especially preferred.
- the triarylmethyl radicals may be coupled to further molecules for example to lipophilic moieties such as long chain fatty acids or to macromolecules, such as polymers, proteins, polysaccharides (e.g. dextrans), polypeptides and polyethyleneimines.
- the macromolecule may be a tissue-specific biomolecule such as an antibody or a backbone polymer such as polylysine capable of carrying a number of independent radical groups which may itself be attached to a further macromolecule. Coupling to lipophilic molecules or substitution of the radical with lipophilic groups is particularly useful since it may enhance the relaxivity of the radicals in certain systems such as blood.
- lipophilic and macromolecular derivatives of the radicals of formula I and salts thereof form a further aspect of the present invention.
- the linkage of a compound of formula I to the further molecule may be effected by any of the conventional methods such as the carbodiimide method, the mixed anhydride procedure of Krejcarek et al. (see Biochemical and Biophysical Research Communications 77 : 581 (1977)), the cyclic anhydride method of Hnatowich et al. (see Science 220 : 613 (1983) and elsewhere), the backbone conjugation techniques of Meares et al. (see Anal. Biochem. 142 : 68 (1984) and elsewhere) and Schering (see EP-A-331616 (Deutsch/Schering) for example) and by the use of linker molecules as described for in US-A-5208324 (Klaveness/Nycomed).
- novel triarylmethyl radicals of the invention may also be used as conventional MRI contrast agents, as ESR contrast agents or as ESR spin labels in ESR imaging or in magnetometry.
- the compounds according to the invention are especialily useful as MR signal enhancement agents for use in OMRI imaging of oxygen concentration (oximetry) or as spin labels in spin label oximetry.
- the present invention provides the use of compounds according to formula I, preferably perdeuterated compounds, in oximetry.
- the radical compounds of formula I may be prepared from their non-radical precursor compounds by conventional radical generation methods.
- Suitable non-radical precursor compounds include the corresponding triaryl methanes, triaryl methyl halides and triaryl methanols, and derivatives, e.g. ethers, of the triaryl methanols.
- the invention provides a process for the preparation of the radical compounds of formula I which comprises subjecting a radical precursor therefor to a radical generation step and optionally subsequently modifying the substitution on the aryl moieties, e.g. by oxidation or reduction.
- a radical precursor therefor to a radical generation step and optionally subsequently modifying the substitution on the aryl moieties, e.g. by oxidation or reduction.
- sulphide substituents e.g. -SCH 3 or -SCH 2 COOEt
- lipophilic substituents such as -SCH 2 COOEt
- hydrophilic substituents e.g. -SCH 2 CH 2 OH.
- the radical-precursor can be represented by formula II (Ar 1 ) 3 CLv where (Ar 1 ) 3 C is as previously defined and Lv is a group displaceable to produce a radical.
- Formula II embraces formulae such as (Ar 1 ) 3 COH (Ar 1 ) 3 CHal (Ar 1 ) 3 CH (Ar 1 ) 3 CCOOH (Ar 1 ) 3 C.CO.O.O.CO.C(Ar 1 ) 3 (Ar 1 ) 3 C.NN C(Ar 1 ) 3 (Where Hal represents halogen, e.g. Br or Cl).
- radical compounds of formula I may conveniently be prepared from corresponding triaryl methyl halides by reduction with a metal catalyst, such as copper, zinc or silver, or by electrolytic reaction on an electrode or by photochemical reaction in the presence of a chlorine radical scavenger, e.g. an olefin.
- a metal catalyst such as copper, zinc or silver
- the radicals may be prepared from the corresponding triaryl methanes by reaction with a base, e.g. in the presence of sodium hydride followed by a reaction with an oxidant, e.g. iodine in the presence of oxygen or a quinone such as chloranil, following for example the method described in US-A-3347941.
- radicals are prepared in situ via thermolysis or photolysis of an appropriate precursor, such as a peroxide or an azo compound.
- an appropriate precursor such as a peroxide or an azo compound.
- a further example of a method by which radical preparation may be effected is reaction of the corresponding triaryl methanols in the presence of an acid to form a carbonium ion followed by reduction to the free radical in the presence of a suitable reducing agent, such as metal ions e.g. Cr 2+ , Fe 2+ , or by electrochemical reduction.
- a suitable reducing agent such as metal ions e.g. Cr 2+ , Fe 2+
- the carbon free radicals may also be generated by a comproportionation reaction between cations and anions of a corresponding radical precursor.
- Triarylmethyl radicals may thus be prepared by mixing together a triarylmethyl radical precursor cation with a corresponding anion.
- Triarylmethyl radicals may also be prepared by thermolysis or photolysis or a corresponding dimeric triarylmethyl structure, for example an azobistriarylmethyl or a bis (triarylmethylcarboxylic acid) peroxide.
- An alternative method of preparation of triarylmethyl radicals is the electrochemical decarboxylation of a triarylmethylcarboxylate.
- Radicals with long half lives in aqueous solution for example at least one hour, preferably ten days, more preferably fifty days and especially preferably at least one year are particularly desirable for use in in vivo imaging.
- non-radical precursors may themselves be prepared by methods conventional in the art and a number of suitable methods are described in WO-A-91/12024.
- Perdeuterated trityl may be prepared by the method described for the preparation of its non-deuterated analogue in Examples 65 to 70 below but with the use of acetone-d 6 instead of acetone in the initial ketalisation step (described in Example 2 of WO-A-91/12024).
- Deuterated hydroxy trityl is prepared generally by successive steps of fused ring formation and deuterative reduction followed by analogous steps to those described for the preparation of the non-deuterated analogue in Examples 73 to 77 below.
- Contrast media manufactured or used according to this invention may contain, besides the inert free radicals (or the non-radical precursor where radical formation is to be effected immediately before administration), formulation aids such as are conventional for therapeutic and diagnostic compositions in human or veterinary medicine.
- the media may for example include solubilizing agents, emulsifiers, viscosity enhancers, buffers, etc.
- the media may be in forms suitable for parenteral (e.g. intravenous) or enteral (e.g.
- oral application for example for application directly into body cavities having external voidance ducts (such as the gastrointestinal tract, the bladder and the uterus), or for injection or infusion into the cardiovascular system.
- external voidance ducts such as the gastrointestinal tract, the bladder and the uterus
- solutions, suspensions and dispersions in physiological tolerable media will generally be preferred.
- Free radicals which are relatively unstable or insoluble in the sample environment may be encapsulated, e.g. in gastric juice resistant capsules containing a medium in which they are stable.
- the radical may be presented as an encapsulated freeze dried powder in a soluble capsule. Such formulations might conveniently be dissolved shortly before in viva use.
- the medium which preferably will be substantially isotonic, may conveniently be administered at a concentration sufficient to yield a 1 micromolar to 10 mM concentration of the free radical in the imaging zone; however the precise concentration and dosage will of course depend upon a range of factors such as toxicity, the organ targetting ability of the contrast agent, and the administration route.
- the optimum concentration for the free radical represents a balance between various factors. In general, optimum concentrations would in most cases lie in the range 0.1 to 100mM, especially 0.2 to 10mM, more especially 0.5 to 5mM.
- Compositions for intravenous administration would preferably contain the free radical in concentrations of 10 to 1000mM especially 50 to 500 mM.
- the concentration will particularly preferably be in the range 50 to 200mM, especially 130 to 170mM and for non-ionic materials 200 to 400mM, especially 290 to 330mM.
- compositions may perhaps be used having concentrations of for example 10 to 100mM for ionic or 20 to 200mM for non-ionic materials.
- concentration may conveniently be 0.1 to 100mM, preferably 5 to 25mM, especially preferably 6 to 15mM.
- 2,2,6,6-Tetramethylbenzo[1,2-d:4,5-d']bis(1,3)dithiole (2.86 g, 10 mmol; prepared according to WO-91/12024) was dissolved in anhydrous THF (75 mL) and cooled to -70 °C. n-Butyllithium (4.4 mL, 2.5 M in hexane) was added. The reaction mixture was allowed to reach ambient temperature. 4-Methoxycarbonyl-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d')-bis-(1,3)-dioxole (1.4 g, 5 mmol) was added as a solid.
- t-Butyllithium 2.0 mL, 1.5 M in pentane was added followed by tetramethyl ethylendiamine (TMEDA) (0.447 mL, 3.0 mmol). The mixture was stirred for 5 min and then treated with ultrasound for 30 min. S8 (0.100 g, 3.12 mmol) was added and the ultrasound treatment was continued for 2 h. The reaction was quenched by addition of aqueous 0.2M KOH (50.0 mL). After washing with benzene (40 mL), the aqueous phase was collected, ether (60 mL) was added and the aqueous phase was acidified using 2M hydrochloric acid. The organic phase was separated, filtered and evaporated.
- TMGEDA tetramethyl ethylendiamine
- the pure acid (240 mg, 82 %) was isolated by preparative HPLC followed by lyophilization.
- the acid was converted into the corresponding sodium salt by the addition of water (50 mL) followed by adjustment of the pH to 7 with 1M aqueous NaOH and lyophilization.
- This radical was prepared using the procedure described in Example 7 from bis-(8-ethoxycarbonylmethylthio-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d']-bis(1,3)dithiole- 4-yl)-mono-(8-ethoxycarbonylmethylthio-2,2,6,6-tetramethyl-benzo[1,2-d:4,5-d']-bis (1,3)dioxole-4-yl) methyl (9 mg, 0.008 mmol) in methanol-d 1 (25 mL), D 2 O (5 mL) and 1M NaOD (48 ⁇ L, 0.048 mmol). The product was purified by preparative HPLC and the pure product was lyophilized.
- the aqueous phase was extracted with ether.
- the organic phase was separated, dried (MgSO 4 ) filtered and evaporated.
- the product was purified by preparative HPLC. The fractions were evaporated and water was added.
- the aqueous layer was extracted with ether.
- the organic layer was separated, dried (MgSO 4 ), filtered and evaporated.
- the product was dissolved by adding water and 1M KOH (0.387 mL, 0.387 mmol). The solution was lyophilized.
- reaction mixture was stirred for 2 min and then transferred to a separatory funnel containing degassed ether (20 mL) and water (20 mL). The organic layer was separated, dried (MgSO 4 ), filtered and evaporated. No further purification was done.
- 2,2,6,6-Tetramethylbenzo[1,2-d:4,5-d']-bis(1,3)dioxole (5.15 g, 23.2 mmol) was dissolved in dry ether (40.0 mL) in a dried, argon filled reaction vessel. The solution was cooled to 0 °C and n-butyllithium (9.29 mL, 2.5M in hexane) was added. After stirring for 15 min at ambient temperature, the mixture was cooled to 0 °C and 4-methoxycarbonyl-(2,2,6,6-tetramethylbenzo[1,2-d:4,5-d']-bis(1,3)dithiole)(4.0 g, 11.6 mmol) was added portionwise.
- Tin(II) chloride (5 mg, 0.022 mmol) was added followed by amalgamated zinc (2 mg, 0.030 mmol). The reaction was added to a mixture of degassed dichloromethane (40 mL) and water (30 mL). The organic layer was separated, dried (Na 2 SO 4 ) and evaporated. The radical was purified by preparative HPLC.
- the tricarboxylic acid was prepared as described in Example 19, however, after treatment with carbon dioxide overnight, the reaction mixture was filtered and the precipitate was transferred to a reaction flask and mixed with potassium carbonate (0.222 g, 1.61 mmol) in DMF (15 mL). After stirring at ambient temperature for 30 min methyl iodide (0.228 g, 1.61 mmol) was added and the reaction was stirred overnight. To the mixture was added hydrochloric acid (45 mL, 0.25M) and ether (45 mL). The ether phase was separated and the aqueous phase was extracted with ether (2*70 mL).
- the radical was synthesized as described in Example 22B from bis-(8-carboxy-2,2,6,6-tetramethylbenzo(1,2-d:4,5-d']-bis(1,3)dioxol-4-yl)-mono-(8-carboxy-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d']-bis(1,3)dithiole-4-yl)methanol (59.2 mg, 0.067 mmol) and BF 3 ⁇ Et 2 O (26.0 ⁇ L, 0.13 mmol) in acetonitrile (2.0 mL) and dichloromethane (0.5 mL). The yield was not determined.
- This compound was prepared from benzene-1,2,4,5-tetrathiole (20.0 g, 97 mmol), acetone-d 6 (48 mL, 0.65 mol) and HBF 4 (16.8 mL, 54 % in ether, 0.123 mol) using the procedure described for the corresponding protio compound in WO-91/12024.
- Tris-(2,2,6,6-tetrakis-( 2 H 3 -methyl)-benzo[1,2-d:4,5-d']bis(1,3)dithiole)methanol (10.8 g, 11.7 mmol) was dissolved in dry benzene (140 mL) together with TMEDA (17.6 mL, 118 mmol).
- t-BuLi 79 mL, 1.5M in pentane
- the nearly homogeneous solution was then transferred into a solution of diethyl pyrocarbonate (90 mL, 611 mmol) in benzene (76 mL). After 2 h, a saturated aqueous solution of NaH 2 PO 4 was added and the mixture was stirred for 10 min. The organic phase was dried (MgSO 4 ), evaporated and the product was recrystallized from acetonitrile.
- Tris-(8-ethoxycarbonyl-2,2,6,6-tetrakis-( 2 H 3 -methyl)-benzo[1,2-d:4,5-d']bis(1,3)dithiole)methanol (5.33 g, 4.6 mmol) was dissolved in CH 2 Cl 2 (40 mL) and a solution of trifluoromethane-sulfonic acid (1.5 mL, 17 mmol) in CH 2 Cl 2 (5 mL) was then added. After stirring for 7 min, a solution of tin(II) chloride (1.74 g, 9.2 mmol) in THF (6 mL) was added and the mixture was stirred for another 10 min. A saturated aqueous solution of NaH 2 PO 4 was added and, after stirring for a few minutes, the organic phase was separated, dried (MgSO 4 ) and evaporated. The radical was not purified, HPLC indicated 80 % pure product.
- Tris-(8-ethoxycarbonyl-2,2,6,6-tetrakis-( 2 H 3 -methyl)-benzo[1,2-d:4,5-d']bis(1,3)dithiole)methyl (4.72 g, 4.21 mmol) was dissolved in dioxane (82 mL) and 1M KOH (41 mL) was added. The solution was stirred at 50 °C for 2 h and then evaporated. Water (50 mL) was added and stirring was continued at 50 °C for another hour. The aqueous solution was acidified with 2M HCl and extracted with ether (2*150 mL). The organic phases were dried (MgSO 4 ) and evaporated. The product was purified by preparative HPLC.
- 1,2-dihydroxypropane-2-one 1,3-diacetate was prepared using the procedure described in the literature (Bentley and McCrae Org. Chem. 35 2082 (1970)).
- 1,3-Dihydroxyacetone 60 g was dissolved in pyridine (200 mL).
- pyridine 200 mL
- acetic anhydride 200 mL
- the pyridine, acetic acid and acetic anhydride were evaporated in vacuum.
- the residue was dissolved in ethyl acetate (400 mL), washed with 1M HCl (2*100 mL) and water (100 mL).
- the solution was dried (Na 2 SO 4 ) and evaporated.
- the crude product was recrystallized from ligroin.
- the solution was cooled to -22 °C and t-BuLi (14.4 mL, 1.5M in pentane) was added. After stirring for 3 h at -22 °C, the reaction mixture was transferred into a solution of diethyl pyrocarbonate (12.8 mL, 87 mmol) in heptane (23 mL) and dry benzene (23 mL) which was kept at -22 °C. The reaction mixture was then allowed to attain ambient temperature. After stirring for an additional hour, a saturated aqueous solution of NaH 2 PO 4 (40 mL) was added.
- the pure acid (240 mg, 82 %) was isolated by preparative HPLC followed by lyophilization.
- the acid was converted into the corresponding sodium salt by the addition of water (50 mL) followed by adjustment of the pH to 7 with 1M aqueous NaOH and lyophilization.
- 2,6-Dioxo-benzo[1,2-d:5,4-d']bis(1,3)oxathiole (1.0 g, 4.4 mmol), prepared according to the literature procedure (Fiedler, H. Berichte 95 , 1771 (1962)) was suspended in dry methanol (30 mL) and a solution of sodium methoxide in methanol (prepared from 20 mL methanol and 2.2 mmol sodium) was then added over a period of 15 minutes. After stirring for 15 minutes, the mixture was poured onto diethyl ether (50 mL) and 1 M aqueous HCl (25 mL). The aqueous phase was extracted twice with ether and the combined organic phases were dried (MgSO 4 ) and evaporated.
- reaction mixture was then quenched with diethyl ether/aq. NaH 2 PO 4 , the aqueous layer was extracted with ether and the combined organic layers were washed twice with water, dried (Na 2 SO 4 ), evaporated, chromatographed (silica gel; dichloro-methane:heptane 1:2) and finally triturated with ethanol to give the pure product as colorless crystals.
- Tris-(8-trimethylsilyl-2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methanol (0.62 g, 0.62 mmol) was dissolved in acetonitrile (150 mL).
- Sodium iodide (0.75 g, 6.0 mmol) and chlorotrimethylsilane (0.65 g, 6.0 mmol) was added in one portion. The mixture was stirred for 20 min and then poured onto diethyl ether/aq. NaHCO 3 .
- Tris-(2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']bis(1,3)-oxathiol-4-yl)methanol (0.40 g, 0.50 mmol) was suspended in dry diethyl ether (80 mL). The mixture was stirred and n-butyllithium (1.5 mL, 2.5M in hexane) was added dropwise over 10 min. After 10 min, the temperature was lowered to -78 °C and neat diethyl carbonate (5.25 g, 44.5 mmol) was added in one portion.
- Tris-(8-ethoxycarbonyl-2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methyl (0.050 g, 0.050 mmol) was dissolved in a premixed solution of trifluoroacetic acid (3 mL), acetic acid (3 mL), acetic anhydride (3 mL) and H 2 O 2 (1 mL, 35 % aqueous solution) and was left under an argon atmosphere for 80 h. The mixture was poured onto a saturated aqueous solution of NaCl and dichloromethane. The organic layer was washed with sat. NaCl and evaporated. After preparative HPLC, 0.008 g (16%) of the pure radical was isolated.
- Tris-(2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']bis(1,3)-oxathiol-4-yl)methanol (5.45 g, 53.5 mmol) was suspended in dry diethyl ether (800 mL). The mixture was stirred and n-butyllithium (33.5 mL, 2.5M in hexane) was added dropwise over 10 min. After 1 hour, the temperature was lowered to -78 °C and the mixture was rapidly transferred to a flask containing a large excess of solid carbon dioxide. The mixture was allowed to reach ambient temperature and was then poured onto water. The organic layer was removed and discarded and the aqueous layer acidified (pH 0) and extracted three times with ether. The combined organic layers were dried (Na 2 SO 4 ) and evaporated. The resulting yellow solid was purified by preparative HPLC.
- Tris-(8-carboxy-2,2,6,6-tetramethylbenzo-(1,2-d:5,4-d']-bis-(1,3)oxathiol-4-yl)methanol (0.92 g, 1.0 mmol) was dissolved in thionyl chloride (8 mL) and one drop of dimethylformamide was added. After 1 h, the mixture was placed on a rotary evaporator and evaporated with benzene (5*10 mL). A close to quantitative yield of orange crystals, which were not further purified, was obtained.
- a solution of sodium 1,2-O-isopropylidenglyceride (prepared from 0.28 g of sodium and 1,2-O-isopropylidenglycerine (5 mL) as described in Example 46) was stirred overnight with a solution of tris-(8-chlorocarbonyl-2,2,6,6-tetramethyl-benzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methanol (0.88 g, 0,88 mmol) in 1,2-O-isopropylideneglycerine (5 mL), and the resulting crude ketal was stirred with acetonitrile (100 mL) mixed with conc. HCl (25 mL).
- Tris-(8-chlorocarbonyl-2,2,6,6-tetramethyl-benzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methanol (0.80 g, 0.80 mmol) was dissolved in benzene (200 mL).
- a solution of of bis(2-hydroxyethyl)amine (8.0 g, 48 mmol) in water (200 mL) was added and after vigorous stirring overnight, the mixture was transferred to a separatory funnel. The aqueous layer was removed and the remaining solid plus the benzene layer was evaporated, dissolved in methanol and passed through a short column (neutral alumina).
- the amide was eluted with methanol. After evaporation, the product was stirred with water (50 mL) at 40 °C for 2 h and then isolated by filtration. After drying, 0.60 g (60%) of the pure amide was obtained.
- Tris-(8-di-(2-hydroxyethyl)amino-carbonyl-2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methanol (0.032 g, 0.027 mmol) was dissolved in dichloromethane (40 mL) and BF 3 ⁇ Et 2 O (0.15 mL, 1.19 mmol) was added. After stirring for 20 min, a solution of 15 mg SnCl 2 (15 mg, 0.079 mmol) in THF (10 mL) was added.
- Tris-(8-chlorocarbonyl-2,2,6,6-tetramethyl-benzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methanol (0.50 g, 0.50 mmol) was dissolved in benzene (20 mL).
- a solution of of dimethylamine (3.0 g, 67 mmol) in water (20 mL) was added and after treatment with ultrasound for 1h, the mixture was transferred to a separatory funnel. The aqueous layer was extracted with benzene and the combined organic layers were dried (Na 2 SO 4 ) and evaporated.
- Tris-(8-dimethylaminocarbonyl-2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methyl (0.020 g, 0.021 mmol) was dissolved in trifluoroacetic acid (2 mL) and H 2 O 2 (0.4 mL, 35 % in water) was added. After stirring overnight, the mixture was poured onto sat. NaCl/ dichloromethane. The organic layer was washed with sat. NaCl and evaporated. Purification by preparative HPLC gave the pure radical.
- Tris-2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methanol (1.0 g, 1.24 mmol) was dissolved in THF (25 mL).
- the mixture was cooled to -70 °C and n-butyllithium (5.8 mL, 1.6M in hexane) was added dropwise over 5 min, the cooling bath was removed and the mixture was left to attain ambient temperature over 30 min.
- the mixture was again cooled to -78 °C and sulfur (0.24 g, 7.5 mmol) was added.
- Tris-(ethoxycarbonylmethylthio-2,2,6,6-tetramethylbenzo[1,2-d:5,4-d']-bis(1,3)oxathiol-4-yl)methyl (0.032 g, 0.028 mmol) was treated with methanol (3 mL) and 1 % aqueous KOH (1 mL) for 30 min. The mixture was poured onto 1 M NaOH/ dichloromethane, the organic layer was discarded and the aqueous layer was carefully acidified and extracted with dichloromethane. The organic layer was washed with water, dried (Na 2 SO 4 ) and evaporated. Purification by preparative HPLC yielded 0.020 g (60 %) of the triacid radical.
- 2,2,6,6-Tetra(methoxycarbonyl)-4,8-dibromobenzo[1,2-d:4,5-d']bis(1,3)dithiole (6.76 g, 10.0 mmol) was dissolved in dry THF, the solution was cooled to 0 °C and a solution of DIBAL in toluene (17.8 ml, 100 mmol) was added dropwise. The solution was heated to reflux temperature for 3 h and then allowed to cool to room temperature. Methanol (20 ml) was added dropwise followed by water (60 ml) and the pH was adjusted to 2 using aqueous 6 M HCl.
- Tris(benzo[1,2-d:4,5-d']bis(1,3)dithiole-4-yl-2,6-dispiro-(4,4-dimethyl-3,5-dioxane))methanol (0.205 g, 0.156 mmol) was dissolved in dry benzene (12 ml) containing N,N,N',N'-tetramethylethylene diamine (0.33 ml, 2.18 mmol) under an atmosphere of argon.
- Tris(8-ethoxycarbonylbenzo[1,2-d:4,5-d']bis(1,3)dithiole-4-yl-2,6-dispiro-(4,4-dimethyl-3,5-dioxane))methanol 55 mg, 0.0359 mmol was dissolved in a mixture of glacial acetic acid (20 ml) and water (5 ml) and the solution was stirred at room temperature for 42 h. The solvents were removed by evaporation, traces of acid were removed by addition of benzene followed by evaporation. HPLC analysis indicated >98 purity of the product. Yield: 42.4 mg (91 %).
- Tris(8-ethoxycarbonyl-2,2,6,6-tetrahydroxymethylbenzo[1,2-d:4,5-d']bis(1,3)dithiole-4-yl)methanol (3.4 mg, 0.0026 mmol) was dissolved in acetonitrile (2 ml) and the solution was cooled to 0 °C.
- Trifluoromethanesulfonic acid (0.017 ml) was added and after 15 min, a solution of SnCl 2 (0.4 mg) in acetonitrile (1 ml) was added. After another 15 min, an aqueous NaH 2 PO 4 buffer was added and the solvents were removed by evaporation.
- 2,2,6,6-Tetramethylbenzo[1,2-d:4,5-d']bis(1,3)dithiole (2.86 g, 10 mmol; prepared according to WO-91/12024) was dissolved in anhydrous THF (75 mL) and cooled to -70 °C. n-Butyllithium (4.4 mL, 2.5 M in hexane) was added. The reaction mixture was allowed to reach ambient temperature. 4-Methoxycarbonyl-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d')-bis-(1,3)-dioxole (1.4 g, 5 mmol) was added as a solid.
- the aqueous phase was extracted with ether.
- the organic phase was separated, dried (MgSO 4 ) filtered and evaporated.
- the product was purified by preparative HPLC. The fractions were evaporated and water was added.
- the aqueous layer was extracted with ether.
- the organic layer was separated, dried (MgSO 4 ), filtered and evaporated.
- the product was dissolved by adding water and 1M KOH (0.387 mL, 0.387 mmol). The solution was lyophilized.
- a dry Soxhlet setup was provided with Benzo[1,2-d:4,5-d']bis(1,3)dithiole-2,2,6,6-tetracarboxylic acid tetraethyl ester (5.0g, 9.65mmol) in the upper compartment and a mixture of lithium aluminium deuteride (1.62g, 38.6mmol) and diethyl ether (300ml) in the lower, round-bottomed flask.
- the ether was heated to reflux temperature for 20h and the mixture was then allowed to cool.
- Methanol 150ml was added dropwise by water (50ml).
- the mixture was acidified with concentrated HCl. (20ml) and the solvent was reduced to 50ml by evaporation in vacuum.
- the white solid was filtered off, washed with water (2x25ml) and dried.
- the solution was cooled to -22 °C and t-BuLi (14.4 mL, 1.5M in pentane) was added. After stirring for 3 h at -22 °C, the reaction mixture was transferred into a solution of diethyl pyrocarbonate (12.8 mL, 87 mmol) in heptane (23 mL) and dry benzene (23 mL) which was kept at -22 °C. The reaction mixture was then allowed to attain ambient temperature. After stirring for an additional hour, a saturated aqueous solution of NaH 2 PO 4 (40 mL) was added.
- the pure acid (240 mg, 82 %) was isolated by preparative HPLC followed by lyophilization.
- the acid was converted into the corresponding sodium salt by the addition of water (50 mL) followed by adjustment of the pH to 7 with 1M aqueous NaOH and lyophilization.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Radiology & Medical Imaging (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Hydrogenated Pyridines (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Magnetic Resonance Imaging Apparatus (AREA)
- Pyridine Compounds (AREA)
- Cosmetics (AREA)
Claims (17)
- Beständige freie Radikalverbindung der Formel Isteht
 (worin die Gruppen X, die gleich oder verschieden sein können, jeweils für ein Sauerstoff- oder Schwefelatom oder eine Gruppe CO oder S(O)n (worin n für 1 bis 3 steht) stehen, mit der Maßgabe, daß wenigstens eine Gruppe X für ein Schwefelatom oder eine S(O)n-Gruppe steht;R1 für ein Wasserstoffatom oder eine Gruppe der Formel -M, -XM, -X-Ar2 oder -Ar2 steht, worin M eine wassersolubilisierende Gruppe ist,und Ar2 ein gegebenenfalls mit einer wassersolubilisierenden Gruppe M substituierter 5- bis 10-gliedriger aromatischer Ring ist; unddie Gruppen R7, die gleich oder verschieden sein können, jeweils für ein Wasserstoffatom oder eine Kohlenwasserstoffgruppe oder eine wassersolubilisierende Gruppe M steht, oder zwei Gruppen R7 zusammen mit dem Atom, an das sie gebunden sind, für eine Carbonylgruppe oder eine 5-bis 8-gliedrige Cycloalkyliden-, Mono- oder Dioxacycloalkyliden-, Mono- oder Diazacycloalkyliden- oder Mono- oder Dithiacycloalkyliden-Gruppe stehen, deren Ringanfügungskohlenstoff gegebenenfalls durch ein Siliciumatom ersetzt ist, und, falls R7 eine andere Bedeutung als Wasserstoff besitzt, R7 gegebenenfalls mit einer Hydroxylgruppe, einer gegebenenfalls alkoxylierten, gegebenenfalls hydroxylierten Acyloxy- oder Alkylgruppe oder einer wassersolubilisierenden Gruppe M substituiert ist))oder ein vorgegebenes Analogon oder Salz davon.
(worin die Gruppen X, die gleich oder verschieden sein können, jeweils für ein Sauerstoff- oder Schwefelatom oder eine Gruppe CO oder S(O)n (worin n für 1 bis 3 steht) stehen, mit der Maßgabe, daß wenigstens eine Gruppe X für ein Schwefelatom oder eine S(O)n-Gruppe steht;R1 für ein Wasserstoffatom oder eine Gruppe der Formel -M, -XM, -X-Ar2 oder -Ar2 steht, worin M eine wassersolubilisierende Gruppe ist,und Ar2 ein gegebenenfalls mit einer wassersolubilisierenden Gruppe M substituierter 5- bis 10-gliedriger aromatischer Ring ist; unddie Gruppen R7, die gleich oder verschieden sein können, jeweils für ein Wasserstoffatom oder eine Kohlenwasserstoffgruppe oder eine wassersolubilisierende Gruppe M steht, oder zwei Gruppen R7 zusammen mit dem Atom, an das sie gebunden sind, für eine Carbonylgruppe oder eine 5-bis 8-gliedrige Cycloalkyliden-, Mono- oder Dioxacycloalkyliden-, Mono- oder Diazacycloalkyliden- oder Mono- oder Dithiacycloalkyliden-Gruppe stehen, deren Ringanfügungskohlenstoff gegebenenfalls durch ein Siliciumatom ersetzt ist, und, falls R7 eine andere Bedeutung als Wasserstoff besitzt, R7 gegebenenfalls mit einer Hydroxylgruppe, einer gegebenenfalls alkoxylierten, gegebenenfalls hydroxylierten Acyloxy- oder Alkylgruppe oder einer wassersolubilisierenden Gruppe M substituiert ist))oder ein vorgegebenes Analogon oder Salz davon. - Verbindung nach Anspruch 1, worin jede Ar1-Gruppe für einen gegebenenfalls substituierten 5- bis 7-gliedrigen, carbocyclischen oder heterocyclischen, aromatischen Ring steht, der gegebenenfalls wenigstens einen fusionierten carbocyclischen oder heterocyclischen Ring trägt.
- Verbindung nach Anspruch 1, worin jede Ar1-Gruppe, die keine Ar3-Gruppe ist, für eine Gruppe Ar1' der Formelsteht,
 worin die Gruppen X, die gleich oder verschieden sein können, jeweils für O, S, CO oder S(O)n stehen und n, R1 und R7 wie in Anspruch 1 definiert sind.
worin die Gruppen X, die gleich oder verschieden sein können, jeweils für O, S, CO oder S(O)n stehen und n, R1 und R7 wie in Anspruch 1 definiert sind. - Verbindung nach Anspruch 3, worin X in jeder Ar1'-Gruppe Sauerstoff ist und R7 Wasserstoff oder gegebenenfalls hydroxyliertes Alkyl ist.
- Verbindung nach Anspruch 1, worin die Ar3-Gruppe einen aromatischen Ring umfaßt, der fusionierte Ringe trägt, die ausgewählt sind unterworin R7 Wasserstoff oder gegebenenfalls hydroxyliertes Alkyl ist.

- Verbindung nach Anspruch 1, mit wenigstens einer Ar3-Gruppe, die ausgewählt ist unter
 worin R7 Wasserstoff oder gegebenenfalls hydroxyliertes Alkyl ist, und R1 wie in Anspruch 1 definiert ist.
worin R7 Wasserstoff oder gegebenenfalls hydroxyliertes Alkyl ist, und R1 wie in Anspruch 1 definiert ist.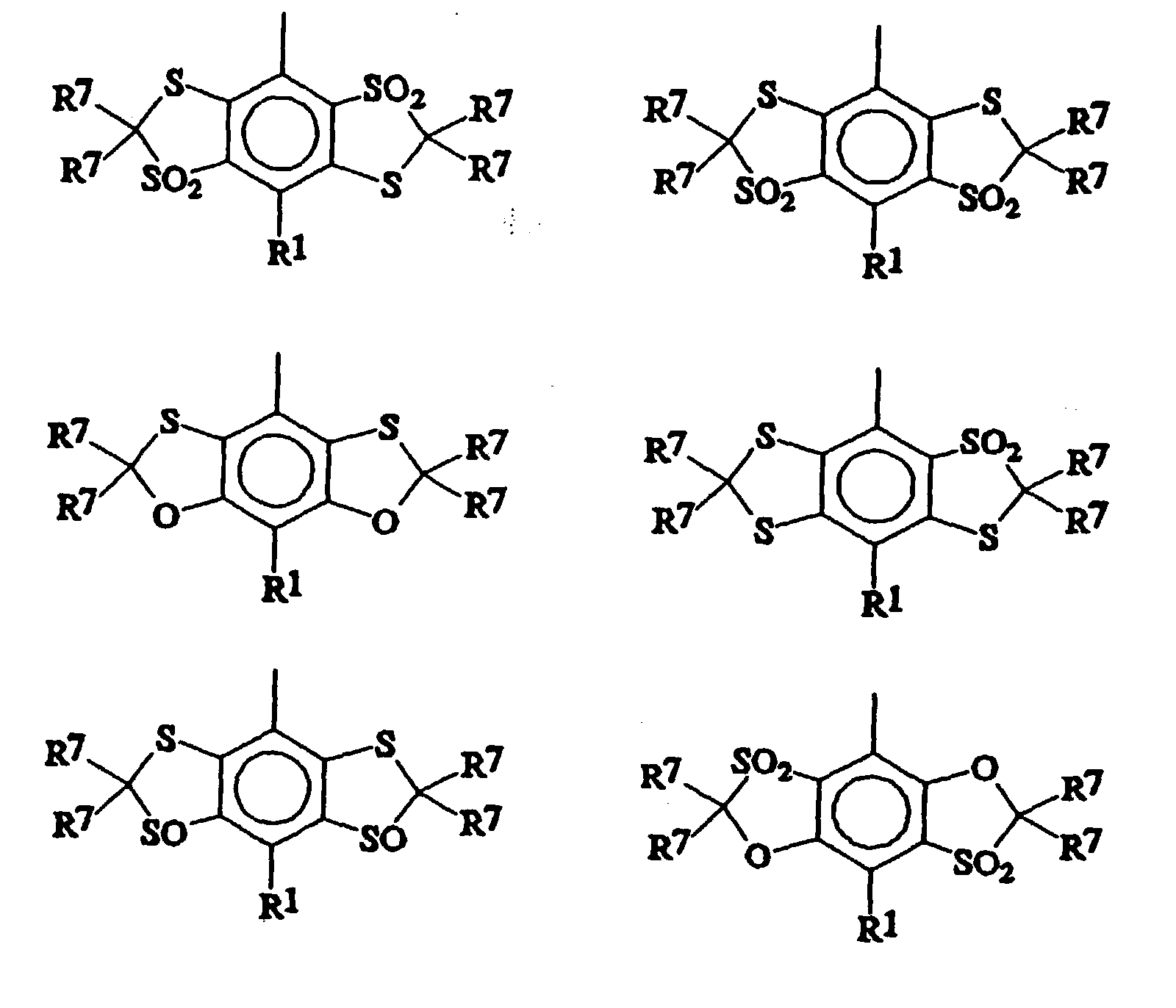
- Verbindung nach Anspruch 6, worin R1 ausgewählt ist unter Wasserstoff und(worin R2 Wasserstoff oder gegebenenfalls hydroxyliertes Alkyl ist).-SCH2COO-Na+-SO2R2-SR2-SCH2COOCH2CH3-SO2C(R2)2CH2CHOHCH2OH-SO2NR2 2-SO2CH2CON(R2)2
 -C-(CH2CH2OH)3-SO2-C(H)(COOCH2CH3)2-CH2CON(CH2CH2OH)2-COOH-CO2Me-CO2Et
-C-(CH2CH2OH)3-SO2-C(H)(COOCH2CH3)2-CH2CON(CH2CH2OH)2-COOH-CO2Me-CO2Et - Verbindung nach Anspruch 1 der Formel Ia, Ib, Ic oder Id
 (worin R11 und R12 Gruppen sind, die wie R1 in Anspruch 1 definiert sind)
(worin R11 und R12 Gruppen sind, die wie R1 in Anspruch 1 definiert sind)
oder ein Salz davon. - Verbindung nach Anspruch 8, worin R11 und, falls vorhanden, R12 ausgewählt sind unter Wasserstoff, SCH3, SCH2CO2CH2CH3, SCH2COOH, SO2N(CH3)CH2(CHOH)4CH2OH, SO2NH2, SO2NCH2CH2OH und SO2NCH2CHOHCH2OH.
- Zusammensetzung eines Kontrastmediums zur magnetischen Resonanzbildgebung, umfassend ein wie in Anspruch 1 definiertes physiologisch verträgliches Radikal zusammen mit wenigstens einem pharmakologisch akzeptablen Träger oder Exzipienten.
- Verfahren zur magnetischen Resonanzbildgebung, wobei eine wirksame Menge eines magnetische Resonanzsignale verstärkenden Agens, das ein wie in Anspruch 1 definiertes Radikal der Formel I oder ein Salz davon umfaßt, in ein menschliches oder nicht menschliches Individuum eingeführt und eine Abbildung von wenigstens einem Teil des Individuums erzeugt wird.
- Zusammensetzung eines Kontrastmediums zur ESR-Bildgebung, umfassend ein wie in Anspruch 1 definiertes physiologisch verträgliches Radikal zusammen mit wenigstens einem pharmakologisch akzeptablen Träger oder Exzipienten.
- Verfahren zur ESR-Bildgebung, wobei eine wirksame Menge eines ESR-Signale verstärkenden Agens, das ein wie in Anspruch 1 definiertes Radikal der Formel I oder ein Salz davon umfaßt, in ein menschliches oder nicht menschliches Individuum eingeführt und eine Abbildung von wenigstens einem Teil des Individuums erzeugt wird.
- Radikalverbindung nach Anspruch 1, worin jede Gruppe Ar1 eine aromatische Gruppe ist, die mit einer solubilisierenden Gruppe M substituiert ist und die zwei fusionierte, schwefelhaltige und jeweils mit wenigstens einer solubilisierenden Gruppe M substituierte heterocyclische Ringe trägt.
- Radikalverbindung nach Anspruch 14, worin jede der Ar1-Gruppen der Formelentspricht
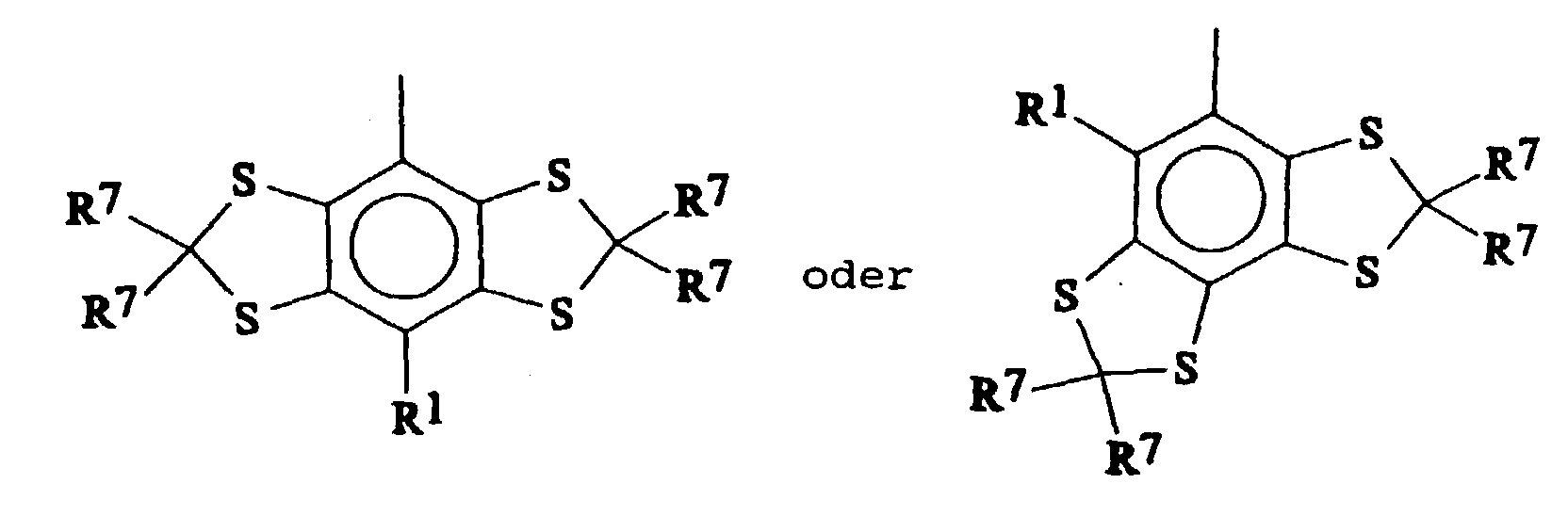 (worin R1 für eine Gruppe der Formel COOR5 oder COOM1 steht (worin R5 für ein Wasserstoffatom oder eine gegebenenfalls hydroxylierte, gegebenenfalls aminierte, gegebenenfalls alkoxylierte, gegebenenfalls carboxylierte Alkyl-, Oxoalkyl-, Alkenyl- oder Alkarylgruppe steht und M1 ein Äquivalent eines physiologisch verträglichen Kations ist);die Gruppen R7, die gleich oder verschieden sein können, jeweils für ein Wasserstoffatom oder eine Kohlenwasserstoffgruppe, wie eine Alkyl-, Hydroxyalkyl-, Alkoxyalkyl-, Alkoxycarbonyl- oder Carbamoylgruppe, stehen, und falls R7 eine andere Bedeutung als Wasserstoff besitzt, gegebenenfalls mit einer Hydroxylgruppe, gegebenenfalls alkoxylierten, gegebenenfalls hydroxylierten Acyloxy- oder Alkylgruppe substituiert sind; und wenigstens eine Gruppe R7 an jedem fusionierten Ring eine wassersolubilisierende Gruppe ist, d.h. eine andere Bedeutung als Wasserstoff und unsubstituiertes Alkyl besitzt),oder ein deuteriertes Analogon, eine Vorstufe oder ein Salz davon.
(worin R1 für eine Gruppe der Formel COOR5 oder COOM1 steht (worin R5 für ein Wasserstoffatom oder eine gegebenenfalls hydroxylierte, gegebenenfalls aminierte, gegebenenfalls alkoxylierte, gegebenenfalls carboxylierte Alkyl-, Oxoalkyl-, Alkenyl- oder Alkarylgruppe steht und M1 ein Äquivalent eines physiologisch verträglichen Kations ist);die Gruppen R7, die gleich oder verschieden sein können, jeweils für ein Wasserstoffatom oder eine Kohlenwasserstoffgruppe, wie eine Alkyl-, Hydroxyalkyl-, Alkoxyalkyl-, Alkoxycarbonyl- oder Carbamoylgruppe, stehen, und falls R7 eine andere Bedeutung als Wasserstoff besitzt, gegebenenfalls mit einer Hydroxylgruppe, gegebenenfalls alkoxylierten, gegebenenfalls hydroxylierten Acyloxy- oder Alkylgruppe substituiert sind; und wenigstens eine Gruppe R7 an jedem fusionierten Ring eine wassersolubilisierende Gruppe ist, d.h. eine andere Bedeutung als Wasserstoff und unsubstituiertes Alkyl besitzt),oder ein deuteriertes Analogon, eine Vorstufe oder ein Salz davon. - Radikalverbindung nach Anspruch 1, nämlich Tris-(8-carboxy-2,2,6,6-tetrahydroxymethylbenzo[1,2-d:4,5-d']bis(1,3)dithiol-4-yl)methyl, Bis-(8-natriumcarboxylat-2,2,6,6-tetrakis-(2H3-methyl)-benzo[1,2-d:4,5-d']-bis(1,3)dithio-4-yl)-mono-(8-natriumcarboxylat-2,2,6,6-tetrakis-(2H3-methyl)-benzo[1,2-d:4,5-d']bis(1,3)dioxol-4-yl)methyl oder Bis-(8-natriumcarboxylat-2,2,6,6-tetrakis-(hydroxy-2H2-methyl)-benzo[1,2-d:4,5-d']-bis(1,3)dithiol-4-yl)-mono-(8-natriumcarboxylat-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d']-bis(1,3)dioxol-4-yl)methyl.
- Verwendung einer Verbindung nach Anspruch 1 in der Oximetrie.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/467,273 US5728370A (en) | 1993-04-02 | 1995-06-06 | Free Radicals |
| US467273 | 1995-06-06 | ||
| PCT/GB1995/002151 WO1996039367A1 (en) | 1994-03-31 | 1995-09-08 | Heterocyclic methyl free radicals as image enhancing agents |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0832054A1 EP0832054A1 (de) | 1998-04-01 |
| EP0832054B1 true EP0832054B1 (de) | 2000-12-27 |
Family
ID=23855069
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP95931302A Expired - Lifetime EP0832054B1 (de) | 1995-06-06 | 1995-09-08 | Heterocyclische methylfreie radikale als biloverbesserungsmittel |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US5728370A (de) |
| EP (1) | EP0832054B1 (de) |
| JP (1) | JP4054376B2 (de) |
| KR (1) | KR19990022477A (de) |
| CN (1) | CN1193952A (de) |
| AT (1) | ATE198313T1 (de) |
| AU (1) | AU709532B2 (de) |
| CA (1) | CA2222331A1 (de) |
| CZ (1) | CZ390197A3 (de) |
| DE (1) | DE69519752T2 (de) |
| ES (1) | ES2153046T3 (de) |
| HU (1) | HUT78020A (de) |
| NO (1) | NO975758L (de) |
| NZ (1) | NZ292582A (de) |
| PL (1) | PL323919A1 (de) |
| WO (1) | WO1996039367A1 (de) |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6063360A (en) * | 1993-04-02 | 2000-05-16 | Nycomed Imaging A/S | Free radicals comprising benzodithiole derivatives |
| GB9704669D0 (en) * | 1997-03-06 | 1997-04-23 | Nycomed Imaging As | Free radicals |
| ES2201462T3 (es) * | 1997-03-06 | 2004-03-16 | Amersham Health As | Radicales libres de triarilmetilo como agentes mejoradores de la imagen. |
| US7122736B2 (en) * | 2001-08-16 | 2006-10-17 | Midwest Research Institute | Method and apparatus for fabricating a thin-film solar cell utilizing a hot wire chemical vapor deposition technique |
| US7351402B2 (en) * | 2003-08-21 | 2008-04-01 | Massachusetts Institute Of Technology | Polarizing agents for dynamic nuclear polarization |
| NZ552898A (en) * | 2004-07-30 | 2011-02-25 | Ge Healthcare As | Method of producing a composition, composition and its use |
| ES2545377T3 (es) * | 2004-07-30 | 2015-09-10 | Ge Healthcare As | Radicales y su uso como agentes paramagnéticos en un proceso de polarización nuclear dinámica |
| JP5475995B2 (ja) | 2005-12-01 | 2014-04-16 | ジーイー・ヘルスケア・アクスイェ・セルスカプ | 動的核分極(dnp)法 |
| CN101325977A (zh) | 2005-12-08 | 2008-12-17 | 皇家飞利浦电子股份有限公司 | 使用Overhauser增强的NMR监测体内药物释放的系统和方法 |
| PL1962912T3 (pl) * | 2005-12-16 | 2013-03-29 | Ge Healthcare As | Sposób wytwarzania hiperpolaryzowanych karboksylanów amin organicznych |
| EP2018545A4 (de) | 2006-05-12 | 2011-03-09 | Massachusetts Inst Technology | Biradikale polarisierungsmittel für dynamische nuklearpolarisierung |
| KR20140144312A (ko) | 2006-08-30 | 2014-12-18 | 지이 헬스케어 에이에스 | 동적 핵 분극화 방법 및 이러한 방법에서 사용되는 화합물 및 조성물 |
| EP2072061A1 (de) | 2007-12-19 | 2009-06-24 | GE Healthcare Limited | Zusammensetzung und Verfahren zur Erzeugung eines metabolischen Profils bei Verwendung von 13C-MR |
| US8697034B2 (en) | 2008-10-10 | 2014-04-15 | The Board Of Regents Of The University Of Texas System | Hyperpolarized 89-yttrium and methods relating thereto |
| US8968703B2 (en) | 2009-09-10 | 2015-03-03 | Ge Healthcare Limited | 13C-MR detection using hyperpolarised 13C-fructose |
| JP5758982B2 (ja) | 2010-04-08 | 2015-08-05 | ブラッコ・イメージング・ソシエタ・ペル・アチオニBracco Imaging S.P.A. | 過分極基質の製造方法およびmriのための方法 |
| FR2967158A1 (fr) | 2010-11-08 | 2012-05-11 | Phosphoenix Sarl | Nouveaux radicaux triarylmethyle: leur preparation et application |
| EP2726458B1 (de) * | 2011-06-30 | 2017-01-04 | Godavari Biorefineries Ltd. | Synthese von cleistanthin a und derivaten davon |
| WO2013053839A1 (en) | 2011-10-12 | 2013-04-18 | Bracco Imaging Spa | Process for the preparation of hyperpolarized derivatives for use in mri analysis |
| US9693828B2 (en) | 2011-12-05 | 2017-07-04 | Bracco Imaging S.P.A. | Composition comprising acetic anhydride and a gadolinium complex, and method for the use in hyperpolarisation in MRI analysis |
| US8715621B2 (en) | 2012-03-15 | 2014-05-06 | Massachusetts Institute Of Technology | Radical polarizing agents for dynamic nuclear polarization |
| WO2013149935A1 (en) | 2012-04-02 | 2013-10-10 | Bracco Imaging Spa | Hyperpolarized amino acids |
| US9381257B2 (en) | 2012-07-13 | 2016-07-05 | Bracco Imaging S.P.A. | Triarylmethyl radicals |
| WO2014192894A1 (ja) * | 2013-05-29 | 2014-12-04 | 国立大学法人九州大学 | 生体における酸化還元反応を検出する方法 |
| CN103951613A (zh) * | 2014-04-21 | 2014-07-30 | 南开大学 | 用于蛋白质顺磁标记的dtpa类似物的设计与合成 |
| CN104610138A (zh) * | 2015-01-09 | 2015-05-13 | 南开大学 | 用于蛋白质顺磁标记的乙二胺四乙酸类探针的合成方法 |
| WO2019018655A1 (en) * | 2017-07-19 | 2019-01-24 | West Virginia University | SYNTHESIS OF BIOCOMPATIBLE TRITYLE RADICALS FOR APPLICATIONS AS HYPERPOLARIZATION AGENTS |
| DE102017122275B4 (de) * | 2017-09-26 | 2021-03-11 | Universität Potsdam | Fluoreszenzfarbstoffe basierend auf Benzo[1,2-d:4,5- d']bis([1,3]dithiol) |
| DE102018126261A1 (de) | 2018-10-22 | 2020-04-23 | Universität Bielefeld | Tritylverbindungen vom Finland-Typ |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3175940B2 (ja) * | 1990-02-12 | 2001-06-11 | ニコムド イノベーション アーベー | トリアリールメチルラジカルおよび磁気共鳴造影法における不活性炭素フリーラジカルの用途 |
| CA2102605A1 (en) * | 1991-05-23 | 1992-11-24 | Evan C. Unger | Liposoluble compounds for magnetic resonance imaging |
| AU668691B2 (en) * | 1991-08-09 | 1996-05-16 | Nycomed Innovation Ab | Use of persistent free-radicals in magnetic resonance imaging |
| GB9117258D0 (en) * | 1991-08-09 | 1991-09-25 | Hafslund Nycomed Innovation | Use of radicals |
| GB9307027D0 (en) * | 1993-04-02 | 1993-05-26 | Nycomed Innovation Ab | Free radicals |
-
1995
- 1995-06-06 US US08/467,273 patent/US5728370A/en not_active Expired - Fee Related
- 1995-09-08 PL PL95323919A patent/PL323919A1/xx unknown
- 1995-09-08 WO PCT/GB1995/002151 patent/WO1996039367A1/en not_active Ceased
- 1995-09-08 JP JP51933896A patent/JP4054376B2/ja not_active Expired - Lifetime
- 1995-09-08 AU AU34790/95A patent/AU709532B2/en not_active Ceased
- 1995-09-08 CA CA002222331A patent/CA2222331A1/en not_active Abandoned
- 1995-09-08 AT AT95931302T patent/ATE198313T1/de not_active IP Right Cessation
- 1995-09-08 ES ES95931302T patent/ES2153046T3/es not_active Expired - Lifetime
- 1995-09-08 EP EP95931302A patent/EP0832054B1/de not_active Expired - Lifetime
- 1995-09-08 NZ NZ292582A patent/NZ292582A/xx unknown
- 1995-09-08 KR KR1019970708958A patent/KR19990022477A/ko not_active Withdrawn
- 1995-09-08 DE DE69519752T patent/DE69519752T2/de not_active Expired - Lifetime
- 1995-09-08 HU HU9802729A patent/HUT78020A/hu unknown
- 1995-09-08 CZ CZ973901A patent/CZ390197A3/cs unknown
-
1996
- 1996-09-08 CN CN95197926A patent/CN1193952A/zh active Pending
-
1997
- 1997-12-05 NO NO975758A patent/NO975758L/no not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| PL323919A1 (en) | 1998-04-27 |
| ATE198313T1 (de) | 2001-01-15 |
| CA2222331A1 (en) | 1996-12-12 |
| CZ390197A3 (cs) | 1998-06-17 |
| EP0832054A1 (de) | 1998-04-01 |
| AU3479095A (en) | 1996-12-24 |
| JPH11505510A (ja) | 1999-05-21 |
| NZ292582A (en) | 1998-12-23 |
| JP4054376B2 (ja) | 2008-02-27 |
| HUT78020A (hu) | 1999-05-28 |
| CN1193952A (zh) | 1998-09-23 |
| DE69519752T2 (de) | 2001-08-02 |
| NO975758D0 (no) | 1997-12-05 |
| DE69519752D1 (de) | 2001-02-01 |
| US5728370A (en) | 1998-03-17 |
| ES2153046T3 (es) | 2001-02-16 |
| AU709532B2 (en) | 1999-09-02 |
| KR19990022477A (ko) | 1999-03-25 |
| NO975758L (no) | 1997-12-05 |
| WO1996039367A1 (en) | 1996-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0832054B1 (de) | Heterocyclische methylfreie radikale als biloverbesserungsmittel | |
| EP0515458B1 (de) | Triarylmethyl-radikale und die anwendung von inerten kohlenstofffreien radikalen in mri | |
| US5141740A (en) | Complexes and compositions for magnetic resonance imaging and usage methods | |
| US5530140A (en) | Free radicals | |
| EP0966414B1 (de) | Triarylmethylfreie radikale als bildverbesserungsmittel | |
| US6256527B1 (en) | Method for determining oxygen concentration | |
| US5384108A (en) | Magnetic resonance imaging agents | |
| EP0871896B1 (de) | Ein verfahren zur bestimmung der konzentration von sauerstoff in einer probe | |
| US6063360A (en) | Free radicals comprising benzodithiole derivatives | |
| US5217706A (en) | Complexes and compositions for magnetic resonance imaging | |
| US6013810A (en) | Free radicals | |
| CA2096543A1 (en) | Alkoxyamide derivatized chelates for mri | |
| WO1993002710A2 (en) | Use of persistent free radicals in magnetic resonance imaging | |
| KR19990044499A (ko) | 시료의 산소 농도 측정 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 19971201 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI LU MC NL PT SE |
|
| AX | Request for extension of the european patent |
Free format text: LT PAYMENT 971201;LV PAYMENT 971201;SI PAYMENT 971201 |
|
| 17Q | First examination report despatched |
Effective date: 19990413 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| ITF | It: translation for a ep patent filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI LU MC NL PT SE |
|
| AX | Request for extension of the european patent |
Free format text: LT PAYMENT 19971201;LV PAYMENT 19971201;SI PAYMENT 19971201 |
|
| LTIE | Lt: invalidation of european patent or patent extension | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 20001227 Ref country code: NL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20001227 Ref country code: LI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20001227 Ref country code: CH Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20001227 Ref country code: BE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20001227 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20001227 |
|
| REF | Corresponds to: |
Ref document number: 198313 Country of ref document: AT Date of ref document: 20010115 Kind code of ref document: T |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REF | Corresponds to: |
Ref document number: 69519752 Country of ref document: DE Date of ref document: 20010201 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2153046 Country of ref document: ES Kind code of ref document: T3 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20010327 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20010327 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20010330 |
|
| ET | Fr: translation filed | ||
| NLV1 | Nl: lapsed or annulled due to failure to fulfill the requirements of art. 29p and 29m of the patents act | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010908 Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010908 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20010910 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1004395 Country of ref document: HK |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: CD |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 69519752 Country of ref document: DE Representative=s name: J D REYNOLDS & CO., GB |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20140929 Year of fee payment: 20 Ref country code: ES Payment date: 20140926 Year of fee payment: 20 Ref country code: FR Payment date: 20140917 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IT Payment date: 20140924 Year of fee payment: 20 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20140929 Year of fee payment: 20 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R071 Ref document number: 69519752 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: PE20 Expiry date: 20150907 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20150907 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20151229 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20150909 |